Hémoglobinose C - Présentation - Le GFHC · Cas clinique Patiente de 20 ans . Origine...
112
Hémoglobinose C A propos d’un cas Galoisy A.(1), Mayeur Rousse C. (1), Ame S.(2), Christ C.(1), Lancelle D.(1), Sanjari F.(1), Mauvieux L.(1) (1) Laboratoire d’Hématologie, CHU Strasbourg (2) Service d’Oncohématologie, CHU Strasbourg
Transcript of Hémoglobinose C - Présentation - Le GFHC · Cas clinique Patiente de 20 ans . Origine...

Hémoglobinose C A propos d’un cas
Galoisy A.(1), Mayeur Rousse C. (1), Ame S.(2), Christ C.(1), Lancelle D.(1), Sanjari F.(1), Mauvieux L.(1)
(1) Laboratoire d’Hématologie, CHU Strasbourg (2) Service d’Oncohématologie, CHU Strasbourg

Cas clinique
Patiente de 20 ans Origine sénégalaise ATCD de crises palustres ATCD de lithiases biliaires ayant nécessité une cholécystectomie Examen clinique : splénomégalie

Hématies : 4,21.1012/L Hb : 11,1 g/dL VGM : 69,7 fL TCMH : 26 pg CCMH : 37,7 g/dL (fraction d’hématies ’hyperchromes’) CVGR : 18,6 % Réticulocytes : 236.109/L Plaquettes : 161.109/L
Leucocytes : 5,83.109/L P. Neutrophiles : 3,96.109/L P. Eosinophiles : 0,12.109/L P.Basophiles : 0,0.109/L Lymphocytes : 1,46.109/L Monocytes : 0,29.109/L (Advia 2120 Siemens)
Hémolyse : Bilirubine totale et non conjuguée augmentées Haptoglobine effondrée LDH limite supérieure de la normale Carence en fer et en folates au moment du diagnostic (Après supplémentation : Hb fluctuante entre 11,5 et 12 g/dL VGM à 75 fL)
Bilan Biologique
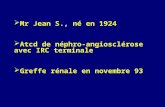
Cristaux
Cellules cibles
Frottis sanguin
=> Aspect des hématies très en faveur d’une hémoglobinose C
Codocytes Hématies rétractées denses
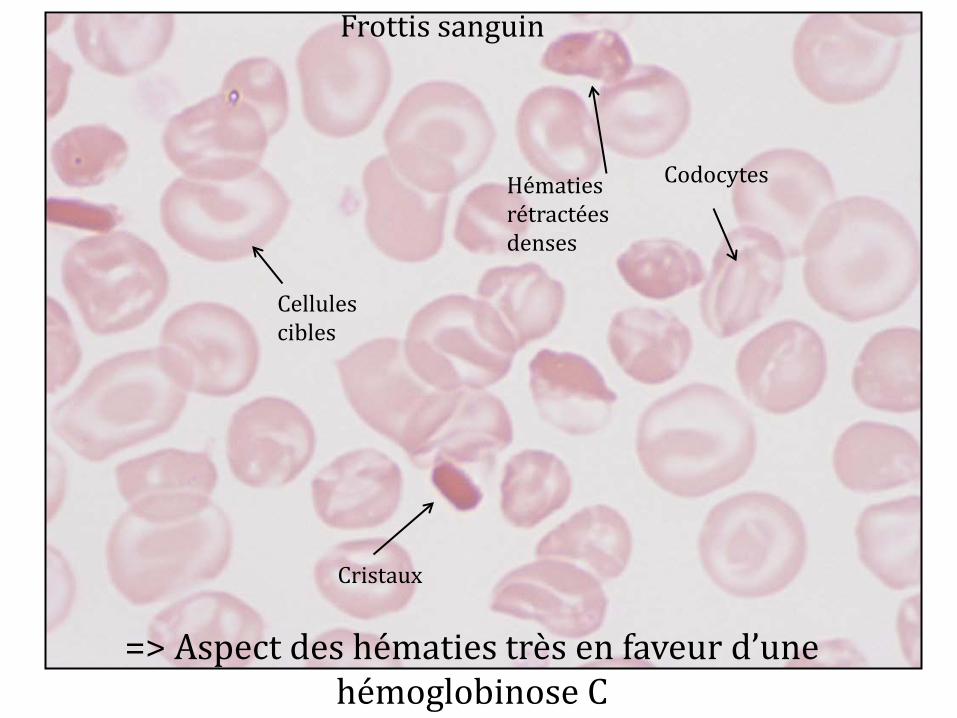
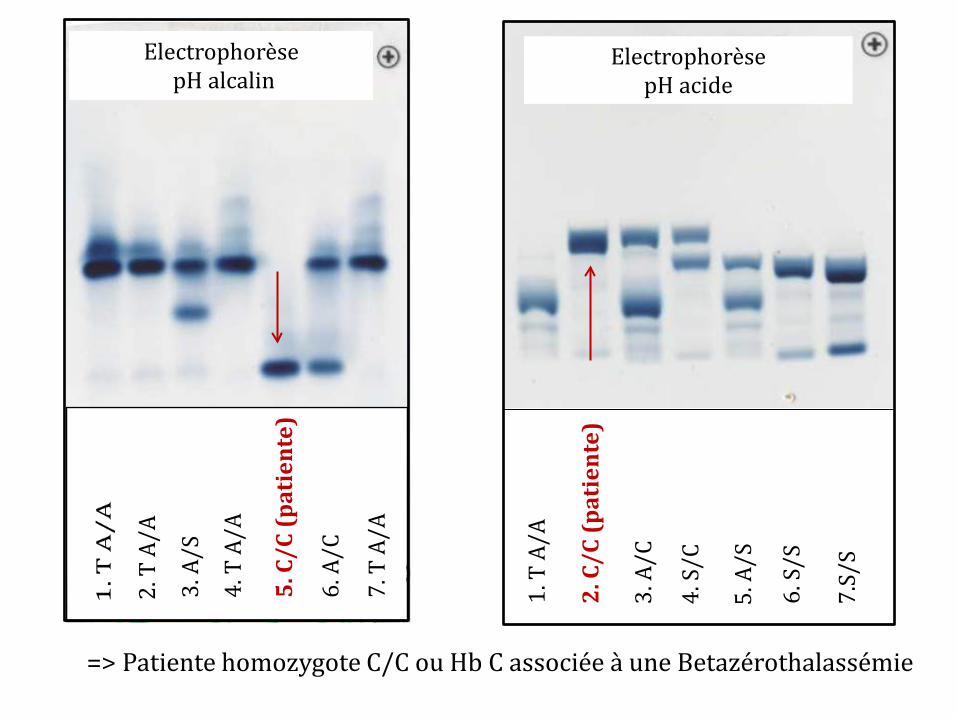
ISOELECTROFOCALISATION
Hb C (patiente)
HbA HbF
HbS
HbA2
HPLC
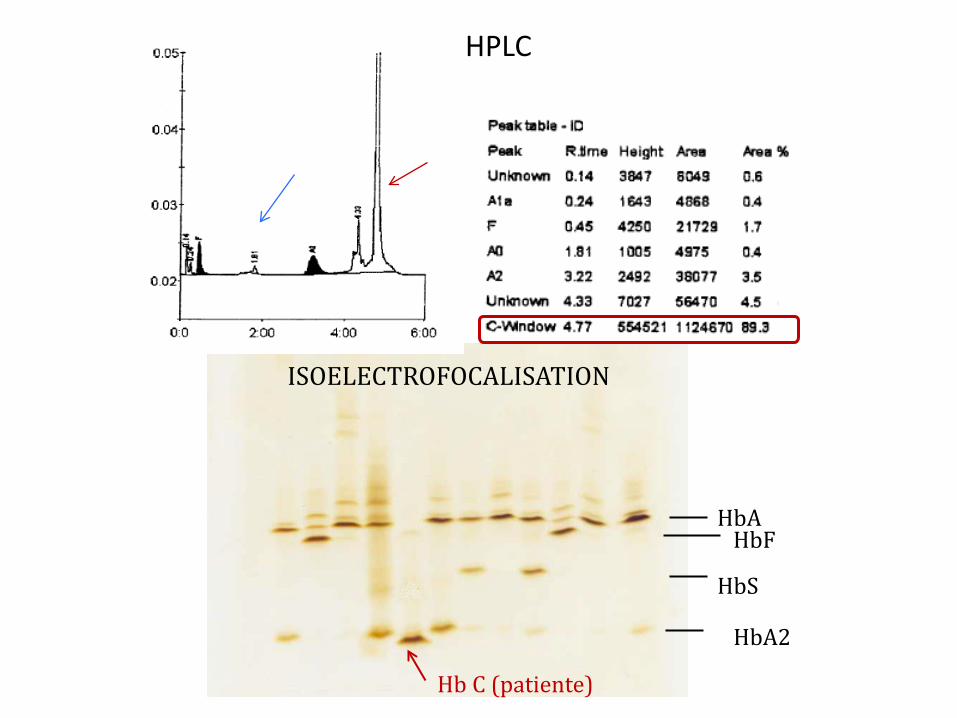
2. T
A/A
3. A
/S
4. T
A/A
5. C
/C (p
atie
nte)
6. A
/C
7. T
A/A
Electrophorèse pH alcalin
1. T
A/A
2. C
/C (p
atie
nte)
3. A
/C
4. S
/C
5. A
/S
Electrophorèse pH acide
6. S
/S
7.S/
S
=> Patiente homozygote C/C ou Hb C associée à une Betazérothalassémie
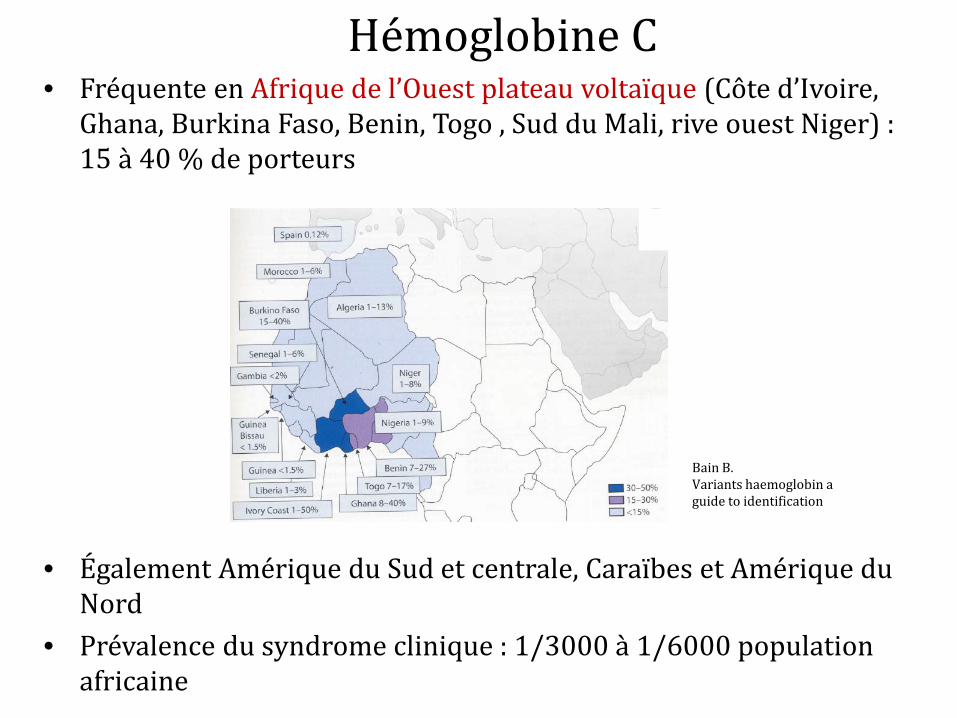
Hémoglobine C • Fréquente en Afrique de l’Ouest plateau voltaïque (Côte d’Ivoire,
Ghana, Burkina Faso, Benin, Togo , Sud du Mali, rive ouest Niger) : 15 à 40 % de porteurs
• Également Amérique du Sud et centrale, Caraïbes et Amérique du Nord
• Prévalence du syndrome clinique : 1/3000 à 1/6000 population africaine
Bain B. Variants haemoglobin a guide to identification

Protection vis-à-vis des formes graves du paludisme à P. falciparum
- Observation de la population Dogon au Mali (Agrawal, Blood, 2000, 96, (7) : 2358-63 )
- Etude sur 4000 habitants du Burkina Faso (Modiano, Nature 2001;414:305-8)
- In vitro : Incapacité des mérozoïtes de pénétrer les hématies contenant de l’HbC Non rupture des schizontes dans les hématies contenant de l’HbC Inhibition de la croissance du Plasmodium dans les hématies contenant l’HbC. Mais mécanisme probablement plus complexe
- Réduction du risque de forme grave
A/C = 29% C/C = 93%

• Mutation de la chaîne beta de la globine (acide glutamique en position 6 remplacé par une lysine)
• Physiopathologie
- Déshydratation avec perte d’eau et efflux de K+ => - Formation de cristaux d’hémoglobine C - Peu d’accidents occlusifs car cristaux labiles après déoxygénation de l’hémoglobine - Diminution affinité de l’oxygène (=> bonne oxygénation malgré l’anémie)
Physiopathologie

Clinique
• Homozygotes C/C : – Anémie hémolytique chronique modérée – Microcytose avec cellules déshydratées => CCMH élevée – Splénomégalie modérée mais constante – Complications :
• Lithiases biliaires nécessitant cholecystectomie, • Carence en folates • Anémie profonde par infection par parvovirus B19

• Hétérozygotes A/C : cliniquement asymptomatiques (parfois discrète microcytose et rares cellules cibles au frottis)
• Hétérozygotes S/C : syndromes drépanocytaires majeurs Avec quelques complications plus spécifiques que chez les homozygotes S/S : – Crises douloureuses ou syndrome thoracique aigu fréquence élevée
(2%) – Rétinopathie proliférative avec risque d’hémorragie du vitré ou
décollement de rétine – Ostéonécrose épiphysaire – Splénomégalie présente à l ’âge adulte
Hémoglobine C
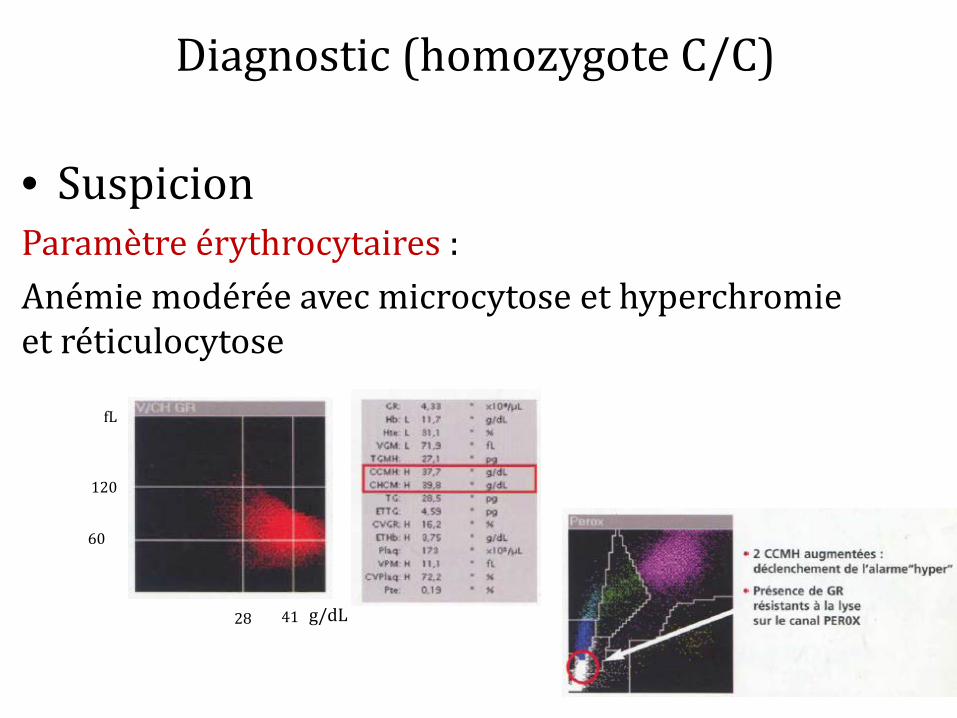
Diagnostic (homozygote C/C)
• Suspicion Paramètre érythrocytaires : Anémie modérée avec microcytose et hyperchromie et réticulocytose
41
60
120
28 g/dL
fL
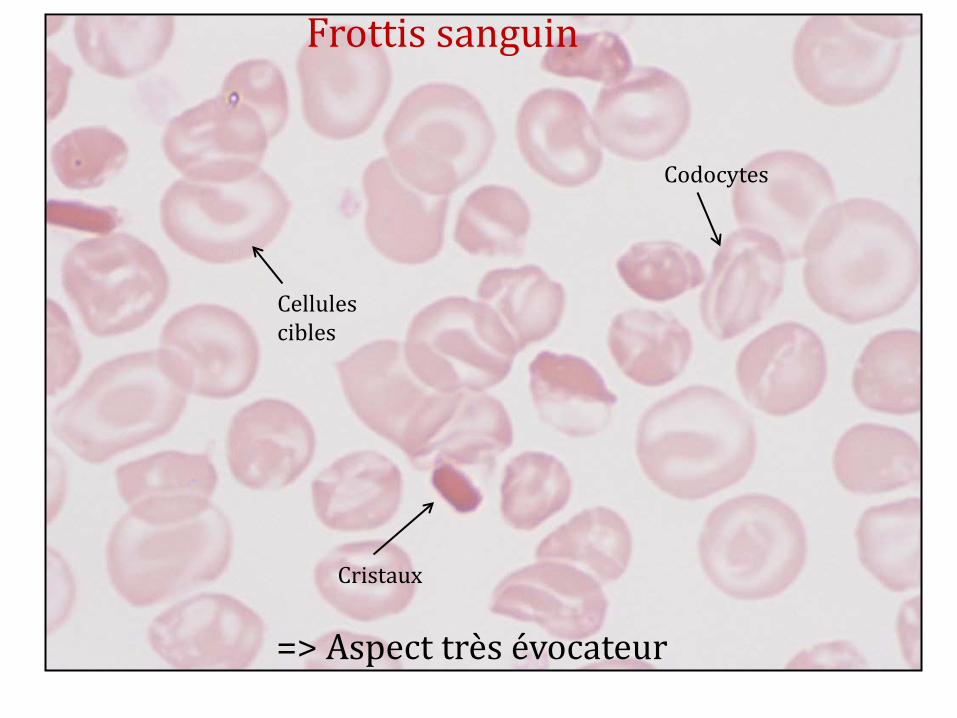
Cristaux
Codocytes
Cellules cibles
Frottis sanguin
=> Aspect très évocateur

• Confirmation
-Etude phénotypique de l’hémoglobine (intérêt du pH acide : Séparation des HbC, HbE et O arab qui migrent ensemble à pH alcalin) - Biologie moléculaire si nécessaire pour distinction homozygote avec association Betazerathalassémie



V Bardet et N Chapuis, Service d’Hématologie biologique, CHU Cochin, Hôpitaux Universitaires Paris Centre.
A propos de trois cas de déficit en G6PD diagnostiqués au décours
d’une chimiothérapie
A. Vincenot et E. André-Kerneïs, Service d’Hématologie biologique, CH Meaux, Groupe Hospitalier de l’Est
Francilien.
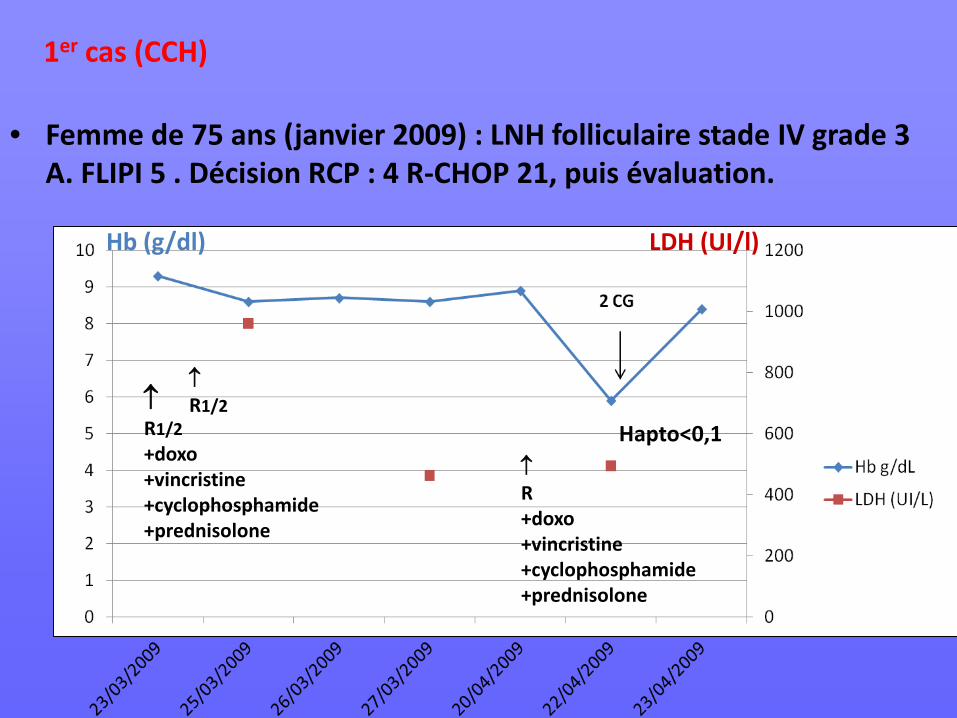
↑ R1/2 +doxo +vincristine +cyclophosphamide +prednisolone
↑ R1/2
↑ R +doxo +vincristine +cyclophosphamide +prednisolone
Hb (g/dl) LDH (UI/l)
Hapto<0,1
• Femme de 75 ans (janvier 2009) : LNH folliculaire stade IV grade 3 A. FLIPI 5 . Décision RCP : 4 R-CHOP 21, puis évaluation.
1er cas (CCH)

2ème cas (CCH) :
Homme de 22 ans, originaire du Surinam Diagnostic avril 2009 : LNH B diffus à grandes cellules stade IV osseux, IPI 2 suite à fracture du col fémoral Traité par 4 R-CHOP + 2 DHAP entre 06/09 et 10/09. 05/11/09 : PET-SCAN toujours positif, décision de RCP: arrêt DHAP, traitement par R –ACVBP.
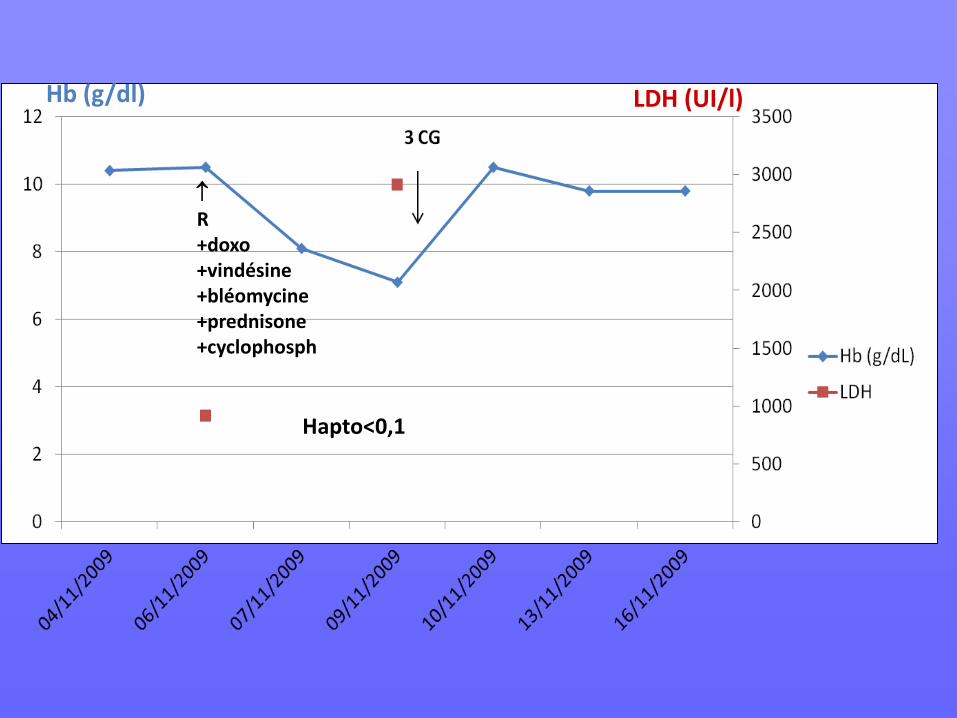
↑ R +doxo +vindésine +bléomycine +prednisone +cyclophosph
Hb (g/dl) LDH (UI/l)
Hapto<0,1
0
20
40
60
80
100
120
140
J1 J2 J3 J4 J5 J6 J90
200
400
600
800
1000
1200
1400
1600Hb (g/l) LDH (UI/l)
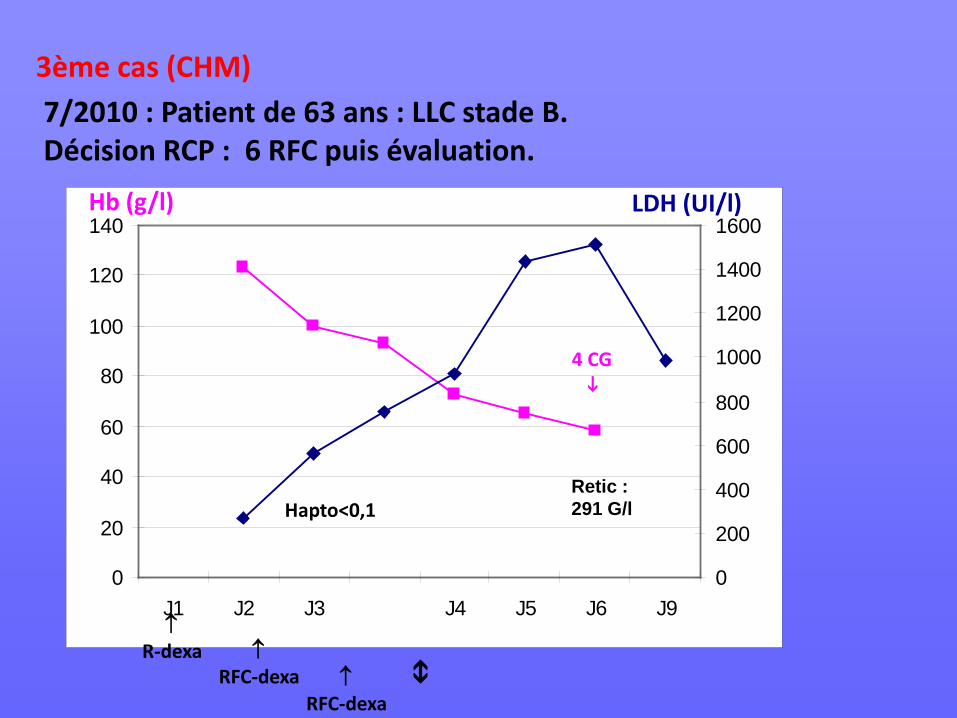
↑ R-dexa ↑
RFC-dexa ↑ RFC-dexa
Hapto<0,1 Retic : 291 G/l
4 CG
7/2010 : Patient de 63 ans : LLC stade B. Décision RCP : 6 RFC puis évaluation.
3ème cas (CHM)
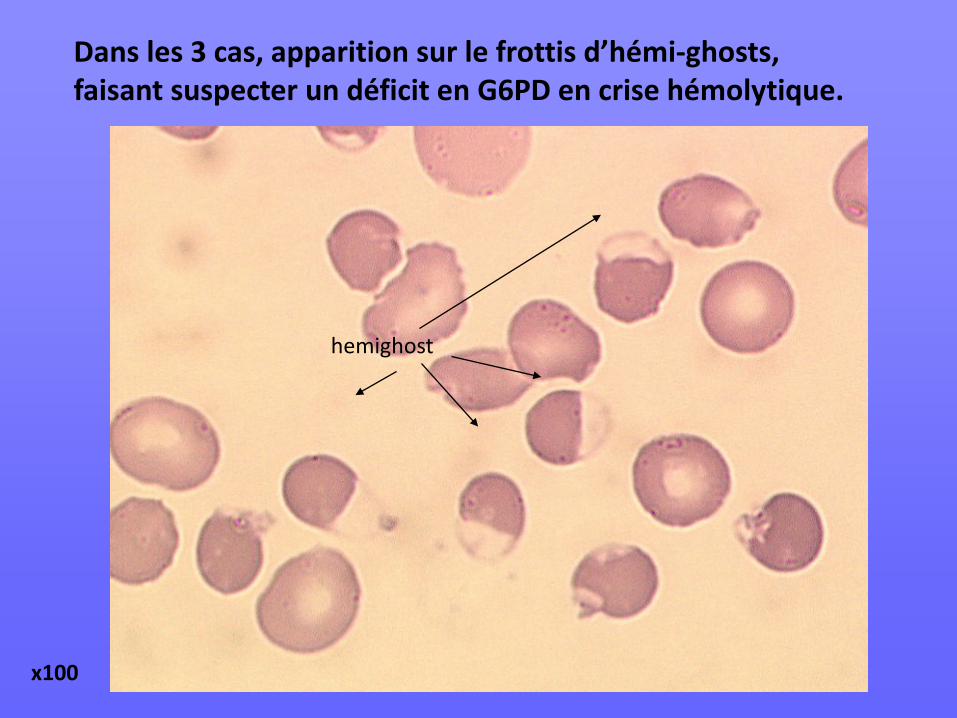
hemighost
Dans les 3 cas, apparition sur le frottis d’hémi-ghosts, faisant suspecter un déficit en G6PD en crise hémolytique.
x100
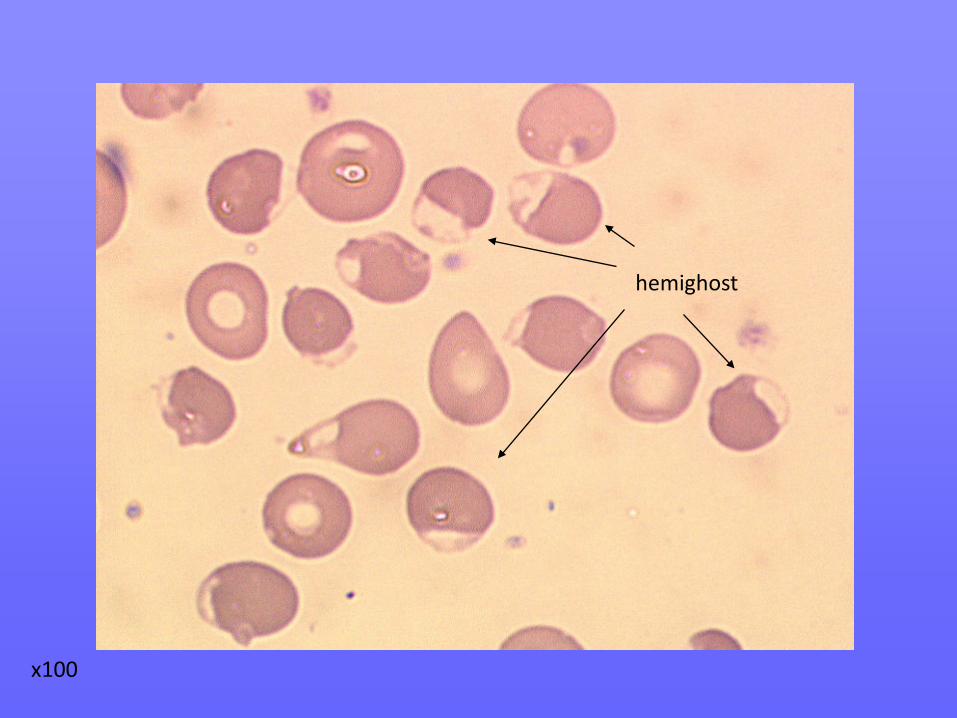
hemighost
x100
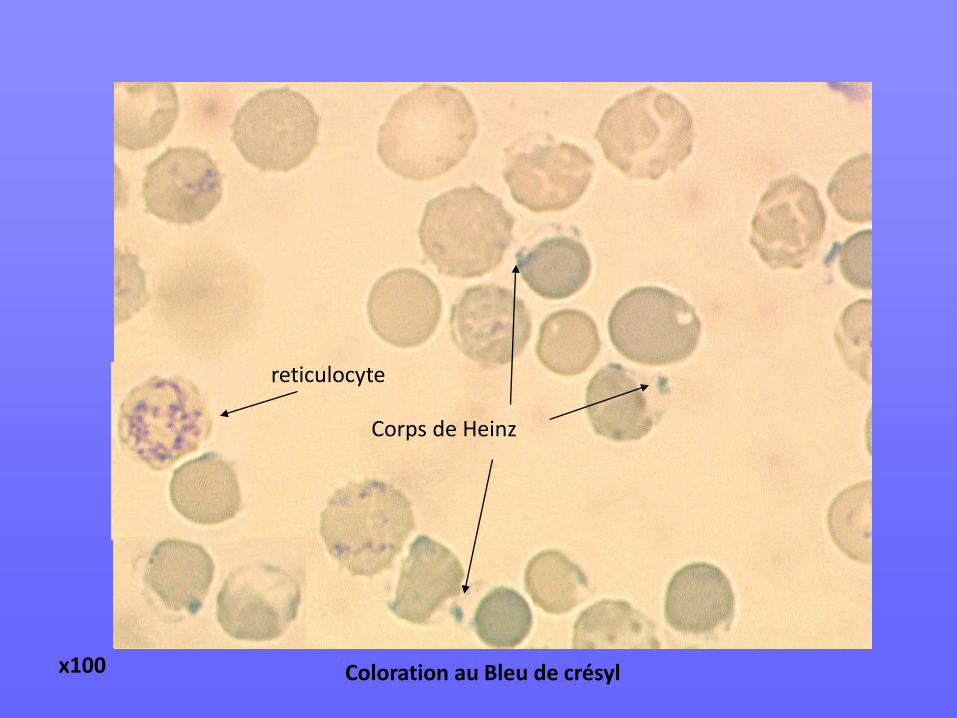
Corps de Heinz
reticulocyte
x100 Coloration au Bleu de crésyl

au CHU H Mondor : (N : 11-17 UI/g Hb) •1er cas : réalisé le 30/11/2009 : 5.5 UI/g Hb •2ème cas : réalisé le 30/11/2009 : 9.6 UI/g Hb •3ème cas : réalisé le 9/08/2010 : 7 UI/g Hb
Dans les 3 cas, le dosage est compatible avec un déficit hétérozygote (1ère patiente) ou hémizygote (2ème et 3ème patient).
Diagnostic confirmé par le dosage de G6PD :

Histoire du déficit en G6PD Dans l’Antiquité, Pythagore mettait en garde ses élèves contre les effets néfastes de l’ingestion de fèves. 1956 : Carson et col découvrent que les individus qui développent une anémie hémolytique lors de ttt par primaquine ont un taux abaissé de G6PD dans les GR. Carson PE et al, Science 1956, 124: 484-485.
1967 : standardisation des procédures pour l’étude de G6PD (WHO). Betke K. et al, WHO Tech Rep Ser 1967, 366: 1-53.
1966-86 : caractérisation de > 400 variants biochimiques 1986 : Séquençage du gène. Persico MG et al, Nucleic Acid Res 1986, 14: 2511-2522.
1986-2006 : identification de >140 variants moléculaires 1996 : développement d’un modèle structural de la protéine en 3 dimensions. Au SWN et al, Structure 2000, 8:293-203.
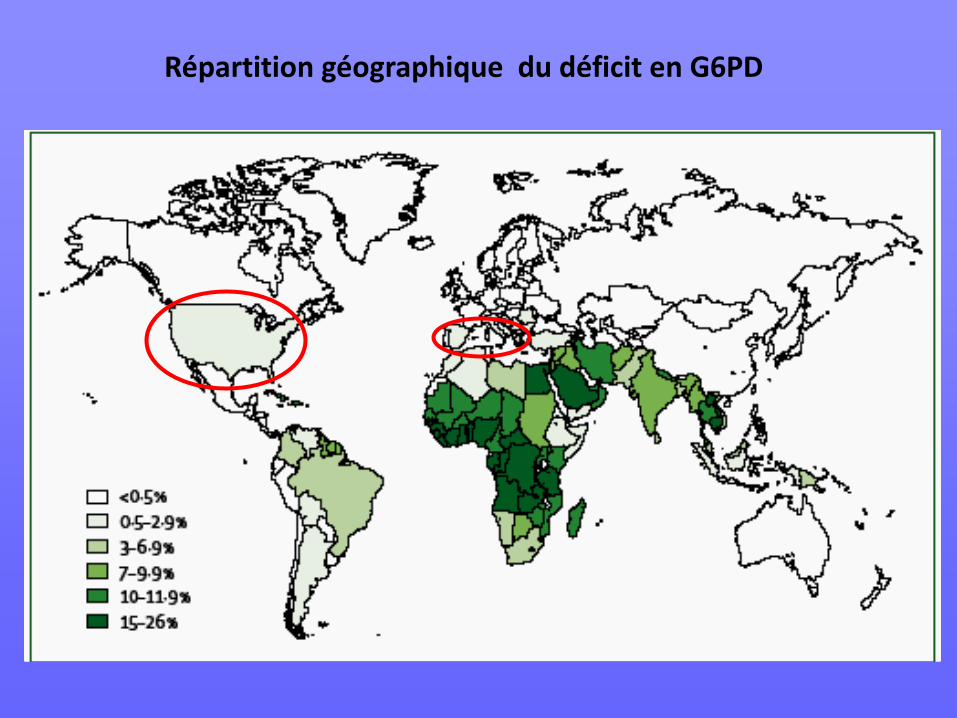
Répartition géographique du déficit en G6PD

Le + souvent, patients asymptomatiques, mais une anémie hémolytique aiguë, parfois grave, peut se produire : - lors de l'ingestion de certains aliments (comme les fèves), - lors de la prise de certains médicaments courants (certains antipaludéens, sulfamides, analgésiques cf liste établie par AFSSAPS en 2008 ) - au cours d'une infection.
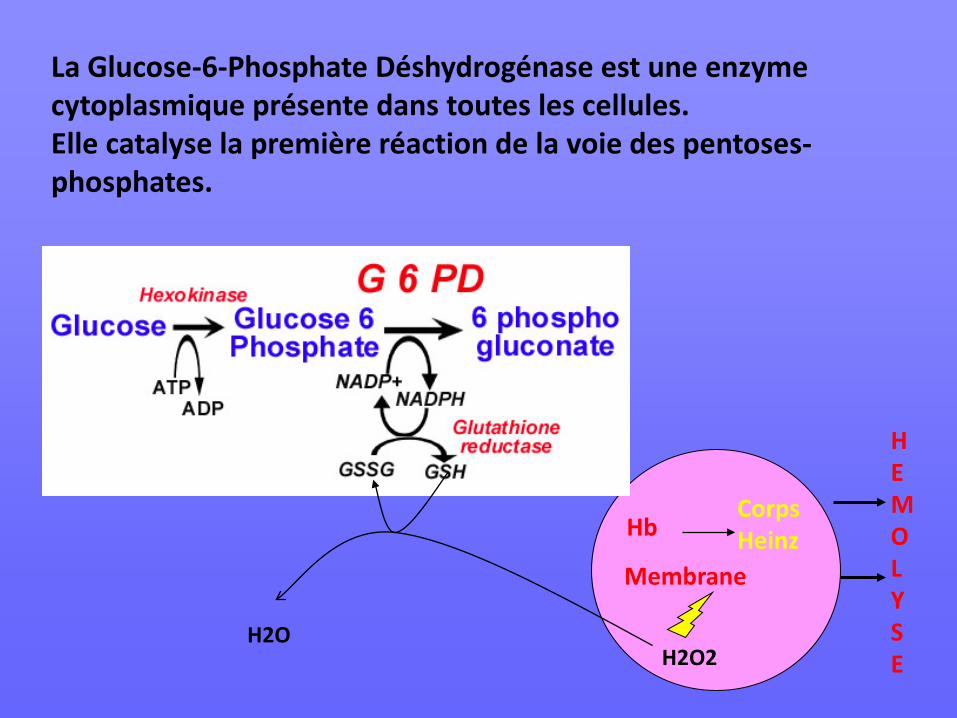
H2O2 H2O
La Glucose-6-Phosphate Déshydrogénase est une enzyme cytoplasmique présente dans toutes les cellules. Elle catalyse la première réaction de la voie des pentoses-phosphates.
Hb
Membrane
Corps Heinz
H E M O L Y S E

Diagnostic Par dosage spectrophotométrique de l’activité enzymatique du taux de production de NADPH à partir de NADP. Attention aux dosages faussement normaux en crise hémolytique ! (lié à l’augmentation d’activité enzymatique chez GR jeunes et rétic)

La classification OMS du déficit en G6PD se fonde sur le niveau d’activité érythrocytaire de l’enzyme : En 2008, l’ex-AFSSAPS a établi des recommandations contenant une liste de médicaments pouvant entraîner une crise hémolytique aiguë. Médicaments et déficit en G6PD : ansm.sante.fr/var/ansm.../2734cc2f7ab2d6484d492a34a3ccb67e.pdf
Classe Type déficit % activité enzymatique résiduelle
I sévère 1 à 2
II intermédiaire 3 à 10
III modéré 10 à 40
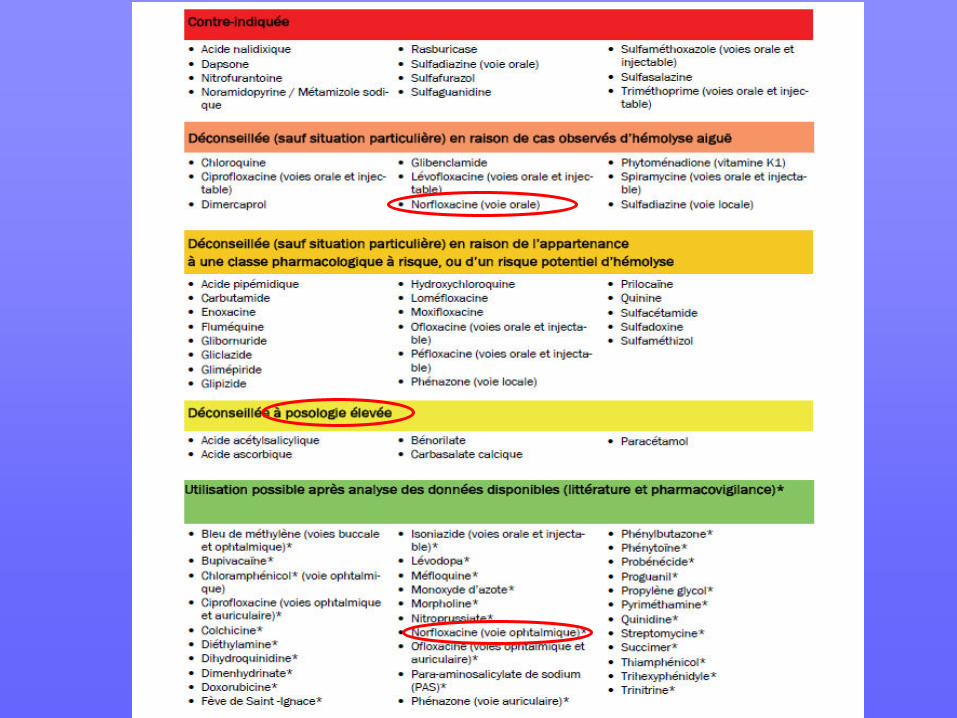

Gène de G6PD situé sur le ch X (Xq28), ⇒ déficit du à des mutations du gène, transmises sur le mode récessif lié à l'X. La sévérité des manifestations est fonction de la sévérité du déficit, les garçons hémizygotes et les filles homozygotes expriment pleinement le déficit, les filles hétérozygotes : expression variable, souvent absente ou modérée, fonction du % de GR déficitaires/non déficitaires qui dépend de l’inactivation du X ⇒ chez femme : dosage peut être faussement normal, seule analyse moléculaire est totalement fiable.

Sur le plan moléculaire, il existe de nombreux variants : • le + souvent : mutation ponctuelle avec substitution d’une base ⇒ remplacement d’un aa ⇒ niveau d’activité enzymatique variable. • ⇒ grande hétérogénéité clinique selon type de variant et degré activité enzymatique résiduelle • influencée par présence : autres anomalies génétiques telles anomalies de membrane, thal, déficit en PK, syndrome Gilbert, … • répartition géographique des variants : «Africain A-» : 90% des cas en Afrique, « méditerranéen » pour le pourtour méditerranéen : le + grave, (seul concerné par le «favisme » !!)

Suite de la prise en charge (1)
Enquête étiologique +++ : Hémolyse 24 à 72 h après administration
Patiente n°1 : Incrimination du Mabthéra. Pour les 2 cures suivantes de chimio sans R (CHOP) : hémolyse A noter : hémolyse pour la 1ère cure à dose fractionnée de R Patient n°2 : Incrimination du Fasturtec (prescrit de J0 à J4). Patient n°3 : Incrimination de la fludarabine. Pour les cures suivantes de chimio sans fluda (R-CHOP) : hémolyse

Les 3 médicaments incriminés : non inscrits encore sur la liste de l’AFSSAPS ⇒ ne pas oublier la déclaration à la pharmacovigilance permettant l’actualisation et l’exhaustivité de la liste. Mabthéra : incriminé dans le cas n°1, mais pas dans les 2 autres cas. Responsable d’une crise hémolytique à pleine dose, mais pas à ½ dose. Exemple concret de l’hétérogénéité clinique du déficit en G6PD, selon le variant moléculaire (à activité résiduelle de l’enzyme pourtant globalement similaire) et la dose.

Patiente n°1 : PET-SCAN en 07/09, réponse partielle 20% sur les aires ganglionnaires. Proposition de modification thérapeutique par DHAP, refus de la patiente. 09/09/09 décision de soins palliatifs. Patient n°2 : Splénectomie, autogreffe de CSP et radiothérapie en 01/2010. Toujours en RC depuis. Patient n°3 : Après 6 R-CHOP : en RP, refus de poursuite du ttt. Evolutif à l’heure actuelle.
Suite de la prise en charge (2)

Conclusion (1)
Déficit enzymatique le plus fréquent, touchant une population importante répartie dans le monde de façon superposable au paludisme. Hétérogénéité clinique importante, liée aux propriétés biochimiques des nombreux variants ⇒ hémolyse ou non face à un agent oxydant. Crise hémolytique inaugurale déclenchée à n’importe quel âge par : - Aliments : favisme essentiellement pour variant méditerranéen - Infections - Médicaments +++, cf liste ANSM.

Toute anémie hémolytique nécessite la réalisation d’un frottis sanguin, à la recherche d’anomalies cytologiques permettant d’orienter le diagnostic, telles les hémighosts pour le déficit en G6PD. Y penser si jaunisse néonatale avec bili > 150 µM (déficit en G6PD non recherché en systématique à la naissance). Sans oublier la déclaration à la pharmacovigilance +++.
Conclusion (2)

Anémie hémolytique inaugurale dans la maladie de Wilson
Atelier du 21/11/2012 – GFHC Mélanie Pannetier / Odile Fenneteau – Hôpital Robert Debré – AP HP

Anamnèse Jeune fille de 15 ans, originaire du Mali
Fin février : symptomatologie digestive (douleurs abdominales, diarrhée,
vomissements)
1er mars : fièvre ⇒ consultation médecin traitant ⇒ bilan biologique Hb : 7.3 g/dL, hyperleucocytose, bilan hépatique perturbé (cytolyse
hépatique, cholestase) Adressée aux Urgences de Robert Debré
Examen clinique :
Poids : 82kg Examen cardio-pulmonaire : normal hormis souffle systolique Examen abdominal : abdomen souple et dépressible, douloureux en
hypocondre droit. Ictère conjonctival. Urines foncées, selles plutôt claires. Ni vomissement, ni selle liquide.
Examen neurologique : normal Examen ORL : normal

Données biologiques (02/03/12)
• Hémoglobine : 7.3 g/dL
• VGM : 112 fL
• Réticulocytes : 658 G/L
• Leucocytes : 22.2 G/L
• Plaquettes : 203 G/L
• Anomalies GR :
Rares sphérocytes, hématies cibles
• TP : 32%
• Facteur V : 37%
• Fibrinogène : 3.24 g/L

Données biologiques (suite)
Bilan d’hémolyse:
• Haptoglobine : <0.10 g/L
• Bilirubine totale : 148 µM
• Bilirubine conjuguée : 83 µM
• LDH : 1916 UI/L
• Coombs direct : Négatif
• Electrophorèse Hb : Hétérozygote AS
• Dosages enzymatiques : Pas de déficit en G6PD Pas de déficit en PK
Bilan hépatique:
• ASAT : 79 UI/L
• ALAT : 19 UI/L
• PAL : 16 UI/L
• GGT : 294 UI/L
• Albumine : 28 g/L

Diagnostic de certitude
Anémie hémolytique associée à une insuffisance hépato-cellulaire
⇒ Hypothèse d’une maladie de Wilson
Bilan du métabolisme du cuivre: Cuprémie : 25.3 µM (N: 12.7-22.2 µM)
Cupriurie : 16 µM (N <0.8 µM)
Céruloplasmine : 0.56 mM (N: 2-4.5 mM)
Prise en charge Régime sans cuivre et traitement par D-pénicillamine (chélateur du Cu)
Suivi ultérieur dans un centre de référence de la maladie de Wilson
Enquête familiale à mener (auprès des parents et des 7 autres enfants de la fratrie)

Maladie de Wilson : généralités Affection héréditaire, transmission autosomique récessive
Surcharge tissulaire en Cu
foie, cerveau, œil
Gène Wilson (chromosome 13) code pour ATPase7B, protéine intracellulaire ayant pour fonctions : Le transport du Cu dans l’hépatocyte L’incorporation du Cu à la céruloplasmine L’élimination du Cu dans la bile
Maladie rare : incidence 1/30 000
Moins de 1500 cas en France Fréquence du portage hétérozygote : 1/90
Majorité des cas deviennent symptomatiques entre 5 et 35 ans
Symptômes hépatiques (45%) Symptômes neurologiques (35%) Troubles psychiatriques (10%) Manifestations hématologiques (2 à 10%)
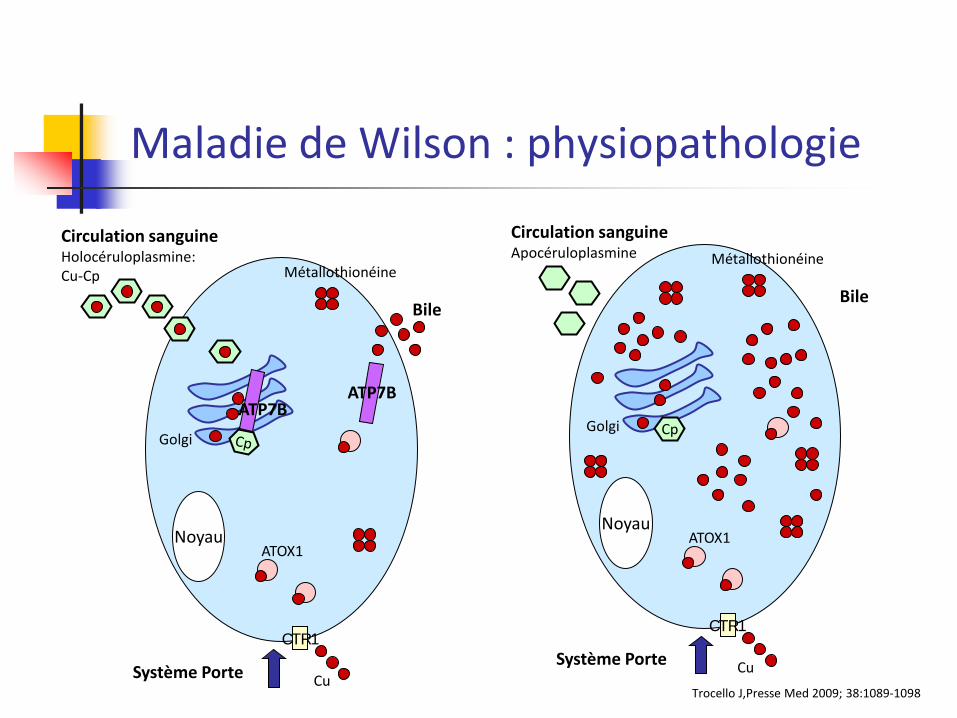
Maladie de Wilson : physiopathologie
Noyau
Système Porte
CTR1
Circulation sanguine Holocéruloplasmine: Cu-Cp
Bile
ATOX1
Métallothionéine
Cu
Golgi
ATP7B ATP7B
Trocello J,Presse Med 2009; 38:1089-1098
Noyau
Cp
Système Porte
CTR1
Bile
ATOX1
Métallothionéine
Cu
Golgi
Circulation sanguine Apocéruloplasmine

Hémolyse dans la maladie de Wilson
Cu « libre » plasmatique
Inhibition d’enzymes érythrocytaires
Hexokinase Adénosine triphosphatase G6PD ⇒ Métabolisme du GR altéré
Système antioxydant du GR dépassé
[GSH] ↓ Superoxyde dismutase – Catalase - Glutathion peroxydase et réductase ↓ Peroxydation lipidique ↑
⇒ Stress oxydatif / production de radicaux libres
⇒ Hémolyse intravasculaire
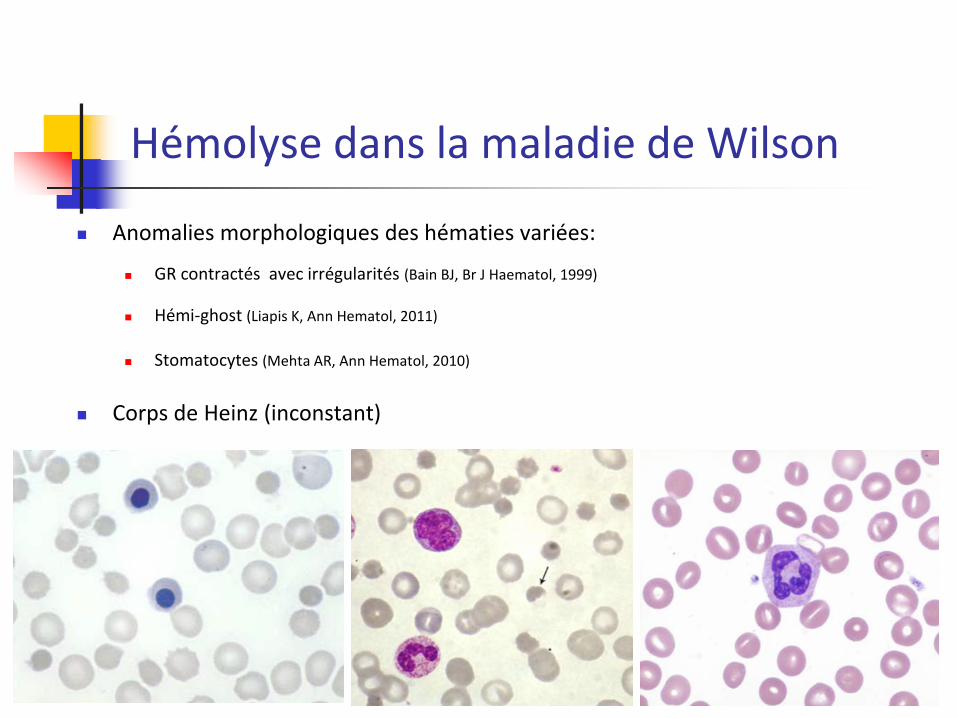
Hémolyse dans la maladie de Wilson
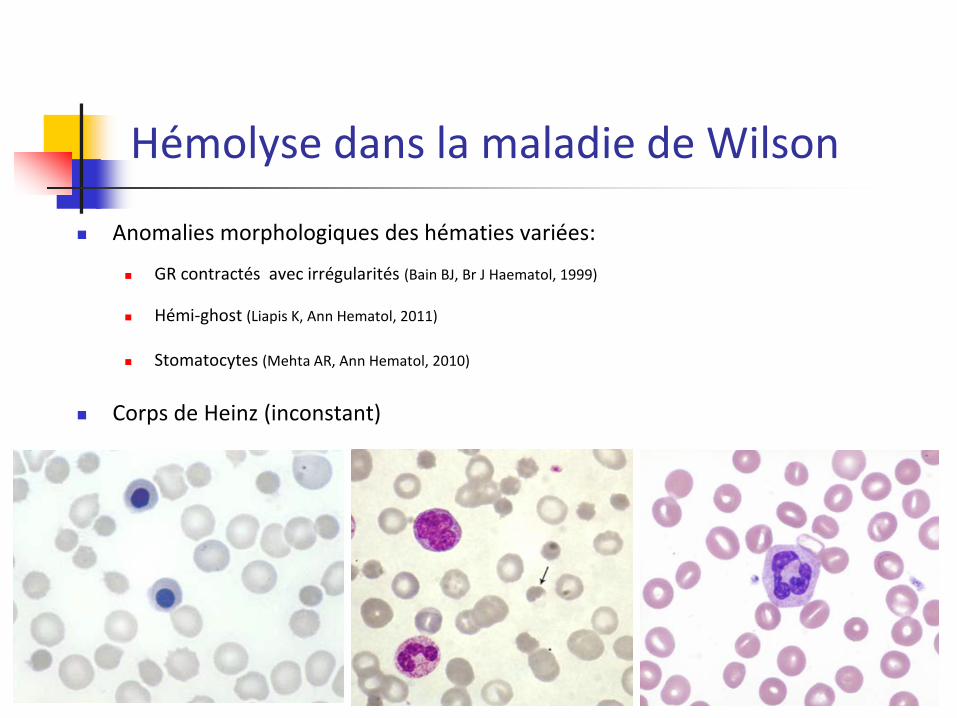
Anomalies morphologiques des hématies variées:
GR contractés avec irrégularités (Bain BJ, Br J Haematol, 1999)
Hémi-ghost (Liapis K, Ann Hematol, 2011)
Stomatocytes (Mehta AR, Ann Hematol, 2010)
Corps de Heinz (inconstant)

Conclusion
La survenue d’une anémie hémolytique aigüe associée ou non à une thrombopénie et des signes d’insuffisance hépato-cellulaire doit faire évoquer la possibilité d’une maladie de Wilson
La précocité du diagnostic et du traitement par les chélateurs du Cu et les sels de Zn sont essentielles pour
le pronostic de la maladie

Anémie hémolytique inaugurale dans la maladie de Wilson
Atelier du 21/11/2012 – GFHC Mélanie Pannetier / Odile Fenneteau – Hôpital Robert Debré – AP HP

Anamnèse Jeune fille de 15 ans, originaire du Mali
Fin février : symptomatologie digestive (douleurs abdominales, diarrhée,
vomissements)
1er mars : fièvre ⇒ consultation médecin traitant ⇒ bilan biologique Hb : 7.3 g/dL, hyperleucocytose, bilan hépatique perturbé (cytolyse
hépatique, cholestase) Adressée aux Urgences de Robert Debré
Examen clinique :
Poids : 82kg Examen cardio-pulmonaire : normal hormis souffle systolique Examen abdominal : abdomen souple et dépressible, douloureux en
hypocondre droit. Ictère conjonctival. Urines foncées, selles plutôt claires. Ni vomissement, ni selle liquide.
Examen neurologique : normal Examen ORL : normal

Données biologiques (02/03/12)
• Hémoglobine : 7.3 g/dL
• VGM : 112 fL
• Réticulocytes : 658 G/L
• Leucocytes : 22.2 G/L
• Plaquettes : 203 G/L
• Anomalies GR :
Rares sphérocytes, hématies cibles
• TP : 32%
• Facteur V : 37%
• Fibrinogène : 3.24 g/L

Données biologiques (suite)
Bilan d’hémolyse:
• Haptoglobine : <0.10 g/L
• Bilirubine totale : 148 µM
• Bilirubine conjuguée : 83 µM
• LDH : 1916 UI/L
• Coombs direct : Négatif
• Electrophorèse Hb : Hétérozygote AS
• Dosages enzymatiques : Pas de déficit en G6PD Pas de déficit en PK
Bilan hépatique:
• ASAT : 79 UI/L
• ALAT : 19 UI/L
• PAL : 16 UI/L
• GGT : 294 UI/L
• Albumine : 28 g/L

Diagnostic de certitude
Anémie hémolytique associée à une insuffisance hépato-cellulaire
⇒ Hypothèse d’une maladie de Wilson
Bilan du métabolisme du cuivre: Cuprémie : 25.3 µM (N: 12.7-22.2 µM)
Cupriurie : 16 µM (N <0.8 µM)
Céruloplasmine : 0.56 mM (N: 2-4.5 mM)
Prise en charge Régime sans cuivre et traitement par D-pénicillamine (chélateur du Cu)
Suivi ultérieur dans un centre de référence de la maladie de Wilson
Enquête familiale à mener (auprès des parents et des 7 autres enfants de la fratrie)

Maladie de Wilson : généralités Affection héréditaire, transmission autosomique récessive
Surcharge tissulaire en Cu
foie, cerveau, œil
Gène Wilson (chromosome 13) code pour ATPase7B, protéine intracellulaire ayant pour fonctions : Le transport du Cu dans l’hépatocyte L’incorporation du Cu à la céruloplasmine L’élimination du Cu dans la bile
Maladie rare : incidence 1/30 000
Moins de 1500 cas en France Fréquence du portage hétérozygote : 1/90
Majorité des cas deviennent symptomatiques entre 5 et 35 ans
Symptômes hépatiques (45%) Symptômes neurologiques (35%) Troubles psychiatriques (10%) Manifestations hématologiques (2 à 10%)
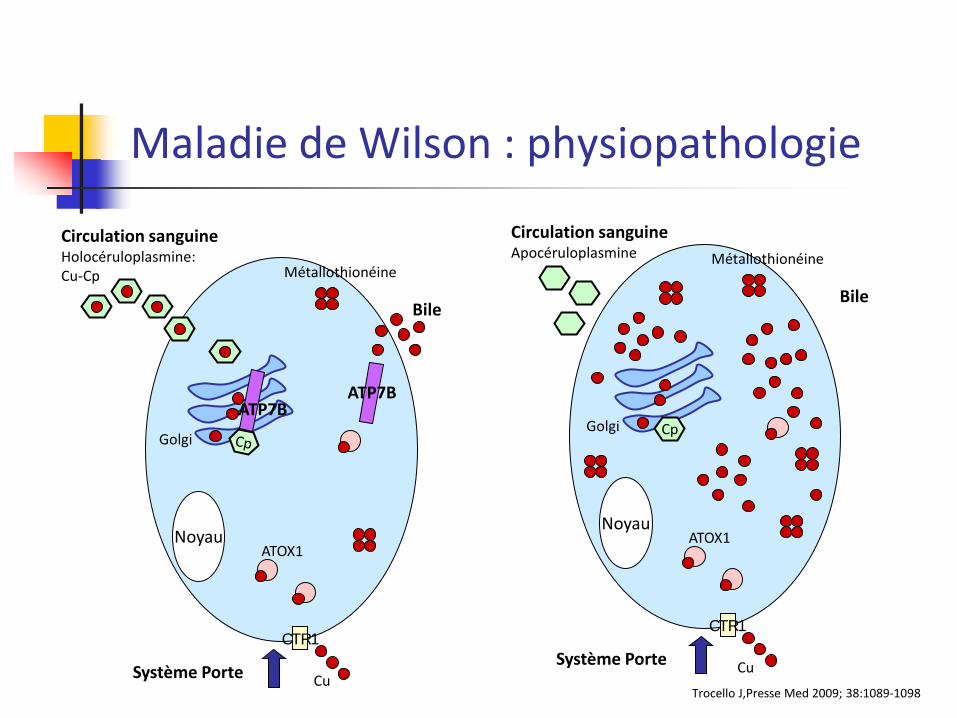
Maladie de Wilson : physiopathologie
Noyau
Système Porte
CTR1
Circulation sanguine Holocéruloplasmine: Cu-Cp
Bile
ATOX1
Métallothionéine
Cu
Golgi
ATP7B ATP7B
Trocello J,Presse Med 2009; 38:1089-1098
Noyau
Cp
Système Porte
CTR1
Bile
ATOX1
Métallothionéine
Cu
Golgi
Circulation sanguine Apocéruloplasmine

Hémolyse dans la maladie de Wilson
Cu « libre » plasmatique
Inhibition d’enzymes érythrocytaires
Hexokinase Adénosine triphosphatase G6PD ⇒ Métabolisme du GR altéré
Système antioxydant du GR dépassé
[GSH] ↓ Superoxyde dismutase – Catalase - Glutathion peroxydase et réductase ↓ Peroxydation lipidique ↑
⇒ Stress oxydatif / production de radicaux libres
⇒ Hémolyse intravasculaire
Hémolyse dans la maladie de Wilson
Anomalies morphologiques des hématies variées:
GR contractés avec irrégularités (Bain BJ, Br J Haematol, 1999)
Hémi-ghost (Liapis K, Ann Hematol, 2011)
Stomatocytes (Mehta AR, Ann Hematol, 2010)
Corps de Heinz (inconstant)

Conclusion
La survenue d’une anémie hémolytique aigüe associée ou non à une thrombopénie et des signes d’insuffisance hépato-cellulaire doit faire évoquer la possibilité d’une maladie de Wilson
La précocité du diagnostic et du traitement par les chélateurs du Cu et les sels de Zn sont essentielles pour
le pronostic de la maladie

Une drôle d’elliptocytose diagnostiquée chez un patient
de 60 ans
Sylvain Thépot1,Madeleine Feneant-Thibault2, Rebecca Kokode3, Véronique Picard3, Nathalie Auger4, Véronique Vergé1, Jean-Henri Bourhis5, Loïc Garçon6, Stéphane de Botton5, Véronique Saada1
1; Laboratoire d’Immuno-Hématologie, IGR, Villejuif
2; Laboratoire de Biochimie; Hôpital de Bicêtre, Kremlin-Bicêtre
3; Laboratoire d’Hématologie; Hôpital de Bicêtre, Kremlin-Bicêtre
4; Laboratoire de Pathologie Moléculaire, IGR, Villejuif
5; Service d’Hématologie Clinique, IGR
6; Laboratoire d’Hématologie, Hôpital St-Antoine, Paris

Histoire de la maladie Homme de 60 ans d’origine caucasienne
-ATCD médicaux: plusieurs IdM stentés; pas d’ATCD carcinologiques
- Absence d’ATCD familiaux significatifs
-Consulte à l’IGR en octobre 2010 pour bilan de bicytopénie (notion de thrombopénie et d’anémie en Avril 2010)
-Examen clinique:
-Bon EG
-Absence d’organomégalie
- Biologie:
-Anémie microcytaire régénérative: Hb: 9.9 g/dL; VGM: 77 fL; reticulocytes: 166 G/L; TCD négatif
-Hypochromie: CCMH: 30,3 g/dL ; % GR hypochromes : 53% (Nale <5%)
-GB: 5.4 G/L; PNN: 3 G/L; blastes 1%; signes de dysgranulopoïèse
-Plaquettes: 64 G/L
-Reste du bilan sans particularité: VS, hémostase, bilirubine totale, bilan hépato-cellulaire, mais sidérémie à la limite inférieure: 8.7 µM (Nale: 9 à 30 µM)
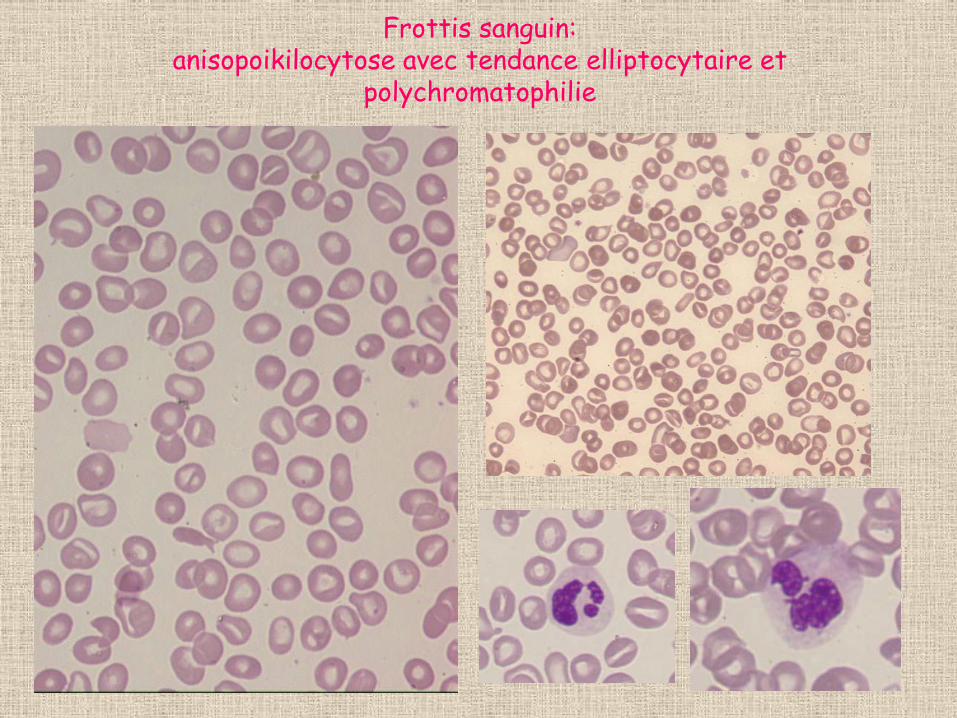
Frottis sanguin: anisopoikilocytose avec tendance elliptocytaire et
polychromatophilie

Myélogramme • dysplasie portant sur les 3 lignées et excès
modéré de cellules immatures (5% blastes+ 2% myéloblastes)
• Lignée érythroide: 40% • Perls: absence de surcharge en hémosidérine
et de sidéroblastes en couronne • Caryotype médullaire: del (20)(q11q13) dans 12
mitoses associée à une trisomie 8 dans 9
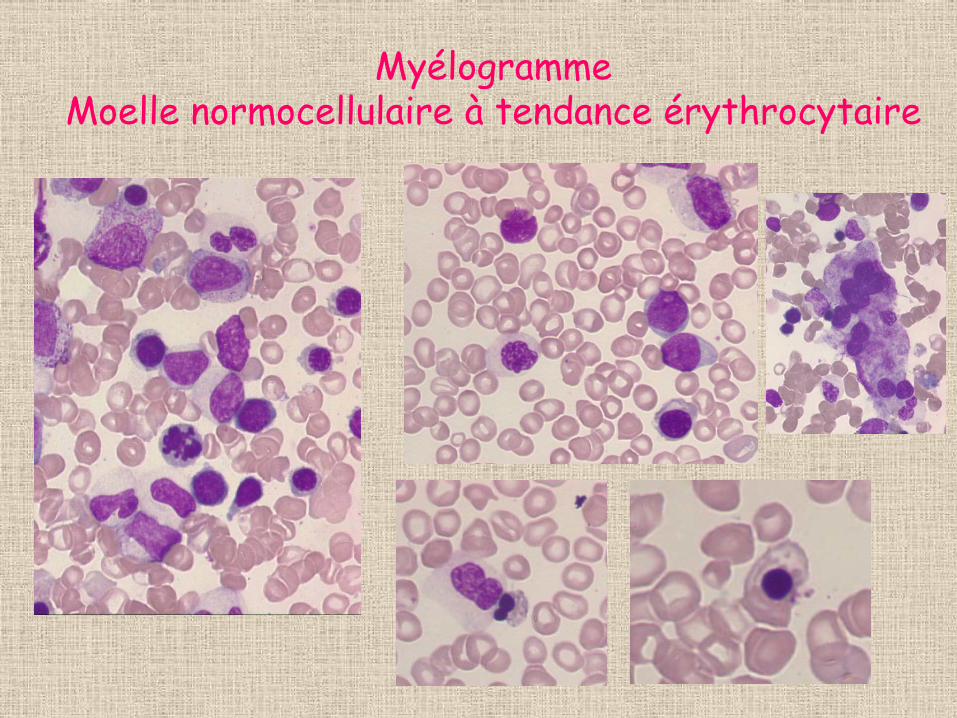
Myélogramme Moelle normocellulaire à tendance érythrocytaire
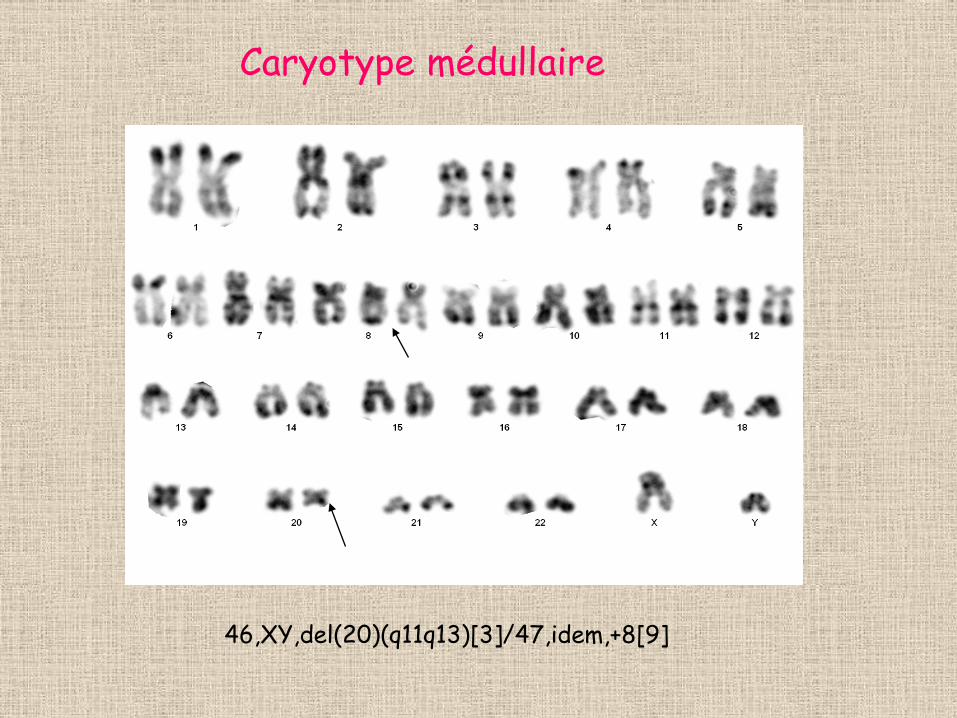
Caryotype médullaire
46,XY,del(20)(q11q13)[3]/47,idem,+8[9]
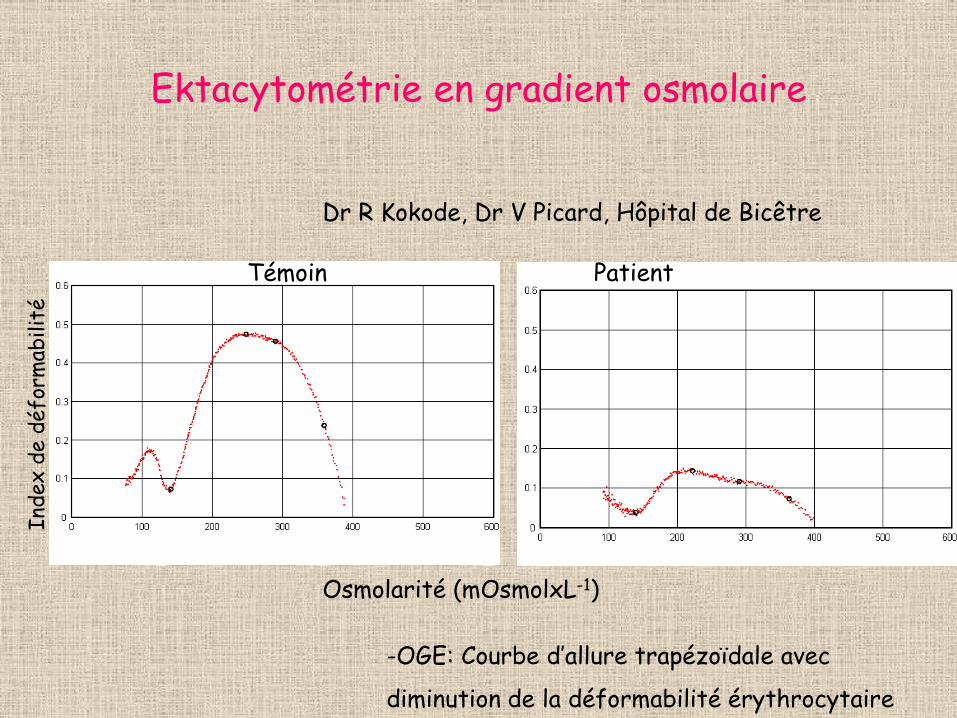
Ektacytométrie en gradient osmolaire
Dr R Kokode, Dr V Picard, Hôpital de Bicêtre
-OGE: Courbe d’allure trapézoïdale avec
diminution de la déformabilité érythrocytaire
Témoin Patient
Inde
x de
déf
orm
abili
té
Osmolarité (mOsmolxL-1)

EPP des protéines de membrane érythrocytaire
-Diminution significative de la protéine 4.1 T Gel Laemmli: diminution : -9% (significatif)
Protéine 4.1:
-phosphoprotéine du cytosquelette de la membrane du GR
-joue un rôle dans les propriétés mécaniques de la membrane en stabilisant les liaisons spectrine/actine
-gène EPB41: localisation chromosomique: 1p33-p34.2
Dr Madeleine Fénéant,
Hôpital de Bicêtre
T Gel Fairbanks: diminution : -12% (significatif)

L'ensemble des données permet d'évoquer le diagnostic d' elliptocytose acquise dans le cadre d'un syndrome myélodysplasique avec délétion 20q
Toutefois caractère constitutionnel non formellement éliminé (absence d’enquête familiale)
Bon EG, absence de cytopénies menaçantes: abstention thérapeutique et surveillance
8 mois plus tard: nouvelle consultation
- perte de poids de 6 Kg, absence de syndrome tumoral
- NFS: hyperleucoytose à 33 G/L: 37% de PNN, 14% de monocytes (4600/µL), Myélémie à 15%, blastes: 5%
anémie à 8.8 g/dL; VGM: 77 fL, CCMH: 29.6 g/dL réticulocytes: 155 G/L
érythroblastes circulants: 16%
thrombopénie stable: 56 G/L
LDH: 3N, TCD négatif
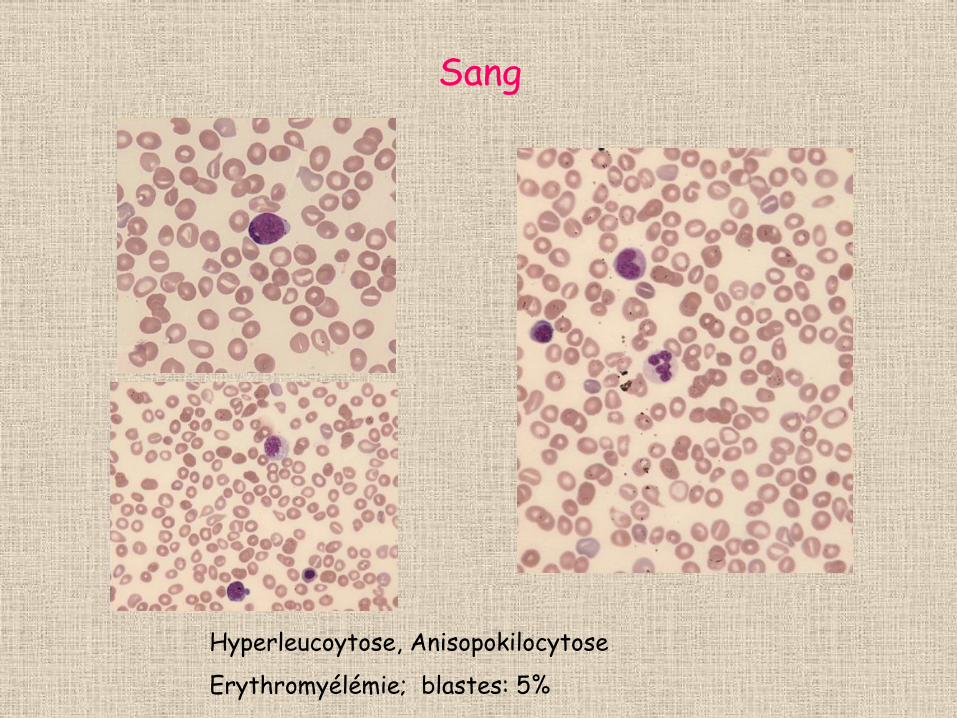
Sang
Hyperleucoytose, Anisopokilocytose
Erythromyélémie; blastes: 5%
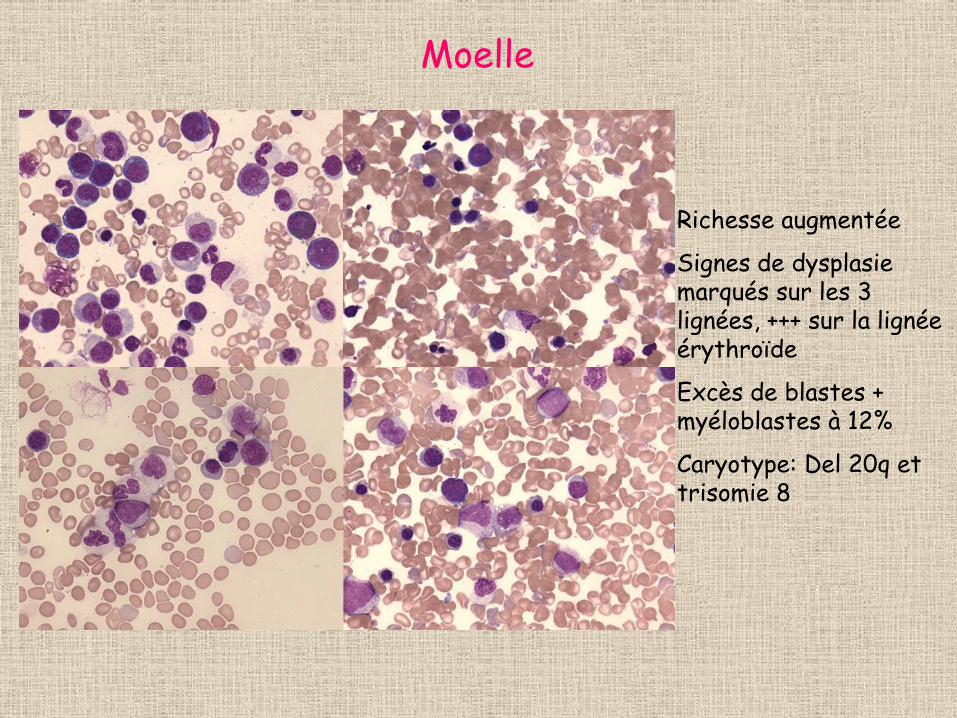
Moelle
Richesse augmentée
Signes de dysplasie marqués sur les 3 lignées, +++ sur la lignée érythroïde
Excès de blastes + myéloblastes à 12%
Caryotype: Del 20q et trisomie 8

• AREB II; score IPSS à 2.5
• Anémie mal tolérée: transfusions de CGR
• Discussion d’une allogreffe de CSP
• Traitement par 5 Azacytidine débuté le 01/08/2011
• Bilan à 4 cycles: diminution de la leucocytose mais persistance de 5% de blastes circulants avec myélémie à 9% + nyx nus +++de mégas, Hb: 8.4 (VGM normalisé par TRF), PLQ: 20 G/L; myélo: Hyperplasie érythroïde à 58%, 6% de blastes, dysplasie 3 lignées
• Patient perdu de vue
• Dernières nouvelles: DCD en 2012 d’une LAM réfractaire (hors IGR)

Elliptocytose acquise dans le cadre d’un syndrome myélodysplasique
1984: Hartz et col. rapportent un cas d’elliptocytose chez un patient de 78 ans avec syndrome « préleucémique », elliptocytose non présente 5 ans avant (Am J Clin Pathol 1984)
Association d’une MDS avec elliptocytose:
- 7 cas décrits entre 1984 et 2004: - hommes: 7/7 - âge: 59 à 81 ans (médiane: 66 ans) - 6 AR, 1 AREB -Elliptocytes++, +/- schizocytes sur le frottis, (absence d’ATCD suggérant une EH) - 4 analyses de la mb érythrocytaire: 2 déficits en protéine 4.1 - caryotype: réalisé chez 6 patients: - del 20q chez 4 - un caryotype complexe monosomal - un patient avec isochromosome 14q
D’après Hur, Clin Lab Haem, 2004

– Loïc Garçon: Analyse rétrospective de 6 cas de déficit acquis en protéine 4.1 (Am J Hematol, 2008)
• 4 H/2F • Âge médian: 72.5 ans (64 à 78 ans) • 3 AR, 1 SMD/SMP, 2 SMP (dont 1 MF) • Hb médiane 11g/dL (8.5 à 15 g/dL) • VGM médian: 96 fL (2 patients <80fL) • Réticulocytes: >110 G/L chez 5/6 (max: 195 G/L) • Elliptocytes: + à +++ • 5/6: EH exclue • Diminution de 4.1: de 11 à 45% par rapport au contrôle • OGE: diminution de l’index de déformabilité/ profil
trapézoïdal • % blastes MO: <5 chez 5 patients; 25% chez 1 patient • Caryotype: del 20q chez 4 patients, 1 patient avec del
1p sans del 20q

Association avec anomalie du chromosome 20q
-Anomalie cytogénétique de pronostic favorable: associée à un IPSS bas, un taux faible de TA et une survie médiane > à SMD sans del 20q
-Décrite ds SMD, SMP++, rarement dans les hémopathies lymphoïdes
- Observée chez 3 à 7% des SMD (isolée chez 1 à 2% des patients avec SMD) (Haase 2007; Braun, 2011)
-Délétion interstitielle : CDR: 6.6 à 10.4 Mb; perte probable de gènes suppresseurs, non clairement identifiés (gène candidat L3MBTL 1)
-Dysplasie constante des lignées érythroïde et mégacaryocytaire: anomalie touchant un progéniteur érythro/méga (Kurtin et al, Am J Clin Pathol, 1996)
-Réticulocytose moyenne plus élevée que chez les patients avec SMD sans del 20q: 72.5 G/L vs 51.7 G/L (Braun T, Leukemia Research, 2011)

Association avec anomalie du chromosome 20q
Douet- Guilbert N, April 2008 .http://AtlasGeneticsOncology.org/Anomalies/ider20qMMID1481.html
-A noter une variante cytogénétique: ider 20q; delétion d’une région 20q et duplication de la région délétée conduisant à un dérivé isochromosome 20
-Parfois difficile à voir en CG conventionnelle: FISH avec sonde 20q (D20S108) et sondes télomériques 20p et 20q
-Peut représenter une évolution clonale d’une délétion del 20q
-Rare: 0.5% des MDS; 0.26% des LAM (Smoley; 2007), sous-estimée??
-Valeur pronostique débattue
-Étude multicentrique franco-belge (F Mullier, S Daliphard, R Garand et al, Ann Hematol, 2011 (34 patients SMD: 21 del(20q) et 13 ider(20q))
-Cytologie +/- caractéristique ider(20q): PNN et PNEo hypogranulaires et vacuolisés, erythrophagocytose et thrombocytophagocytose par les PNN
-Elliptocytose observée chez 30% des patients avec MDS ider20q contre 5% des patients avec del 20q

Elliptocytose acquise • Rarement décrite, probablement sous estimée • Mécanisme moléculaire: pas connu
– Altération de l’expression de EPB41 dans le clone dysplasique:
– délétion de la région 1p? – altération de facteurs transactivateurs impliqués dans la transcription de
EPB41 et/ou ds le processing de son pré-ARNm?
• Association à del 20q ou ider 20q: pas d’explication
• Peu de données mais n’apparaît pas impacter sur le pronostic
• Y penser dans un tableau évocateur de SMD avec anémie régénérative

Anémie hémolytique chez une patiente de 40 ans : diagnostic
tardif d’anémie dysérythropoiétique congénitale de
type II
Dr C. Brumpt1, Dr T.Cynober2, Pr J.Delaunay2, Dr M.Fénéant-Thibaut2, Dr L.Ades3, Pr A.Bezeaud4
1Hopital Lariboisière, 2Hôpital Kremlin Bicêtre, 3Hôpital Avicenne, 4Hôpital Beaujon

Madame C. 40 ans adressée pour anémie.
•A l’interrogatoire : •Patiente algérienne •Anémie connue depuis 15 ans, jamais transfusée •Mariée, 6 enfants dont 2 en France, sans anémie connue •1 sœur anémique en Algérie
•A l’examen : •splénomégalie modérée sans ictère

Examens complémentaires (1)
NFS : •Hb 7,6g/dL, •VGM 94fL, TCMH 34,6pg, CCMH 36,6g/dL
•GB 6.109/L, Plaquettes 230.109/L
•Réticulocytes 145.109/L
•Erythroblastes 6%
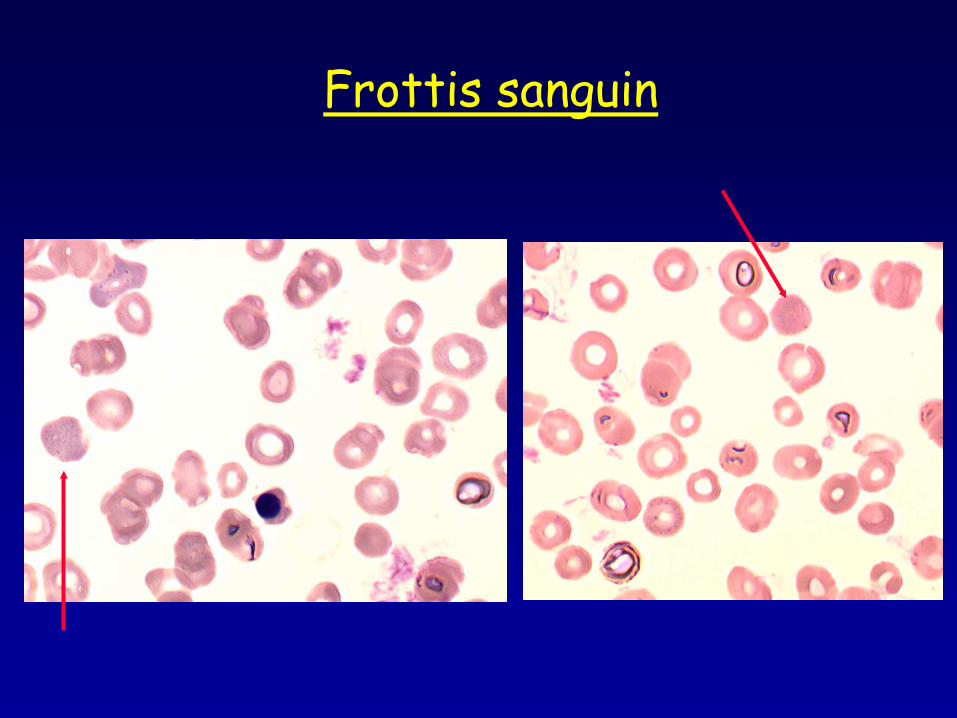
Frottis sanguin
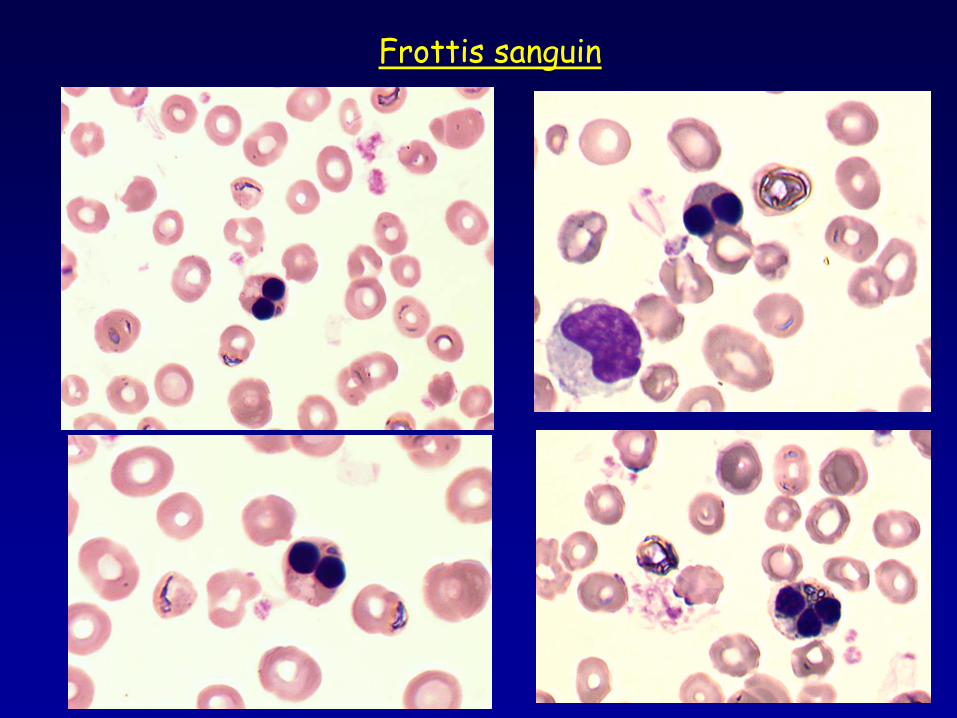
Frottis sanguin

Examens complémentaires (2)
Biochimie : • LDH 630 U/L, Haptoglobine <0.1g/L •Bilirubine totale 52µmol/L (<17), non conjuguée 47µmol/L
•Fer sérique 23µmol/L (10-30), tranferrine 1g/L (2-4), capacité totale de fixation 28µmol/L (50-80), coefficient de saturation de la transferrine 84%, Ferritine sérique 680µg/L (50-120)

Examens complémentaires (3)
•Test de Coombs : négatif
•Electrophorèse de l’hémoglobine : normale
•Dosages des enzymes érythrocytaires : absence de déficit en G6PD et en PK
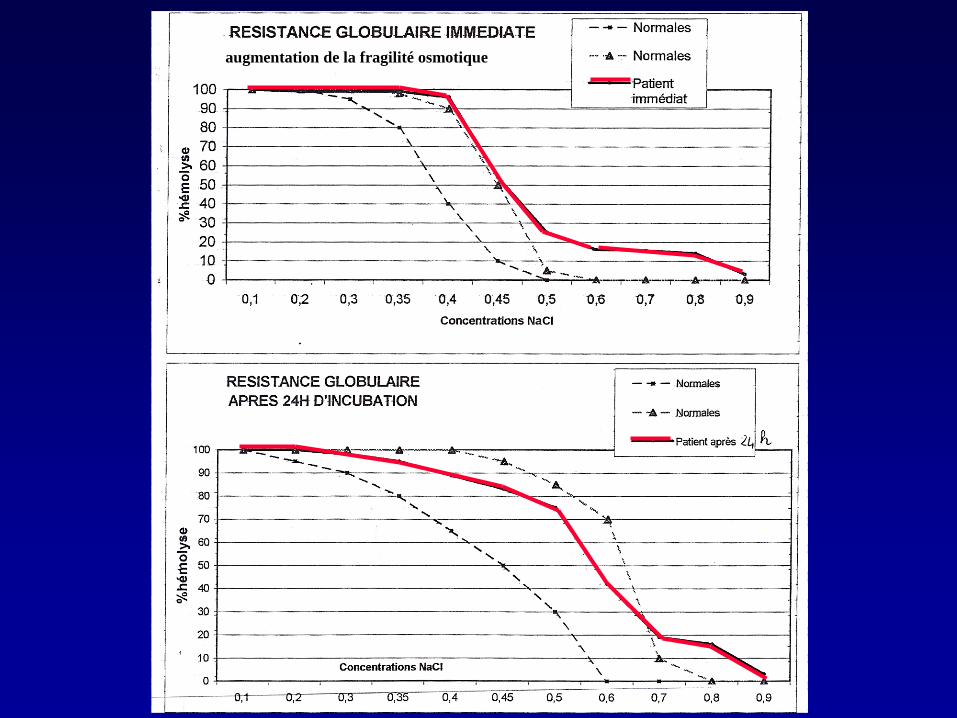
augmentation de la fragilité osmotique
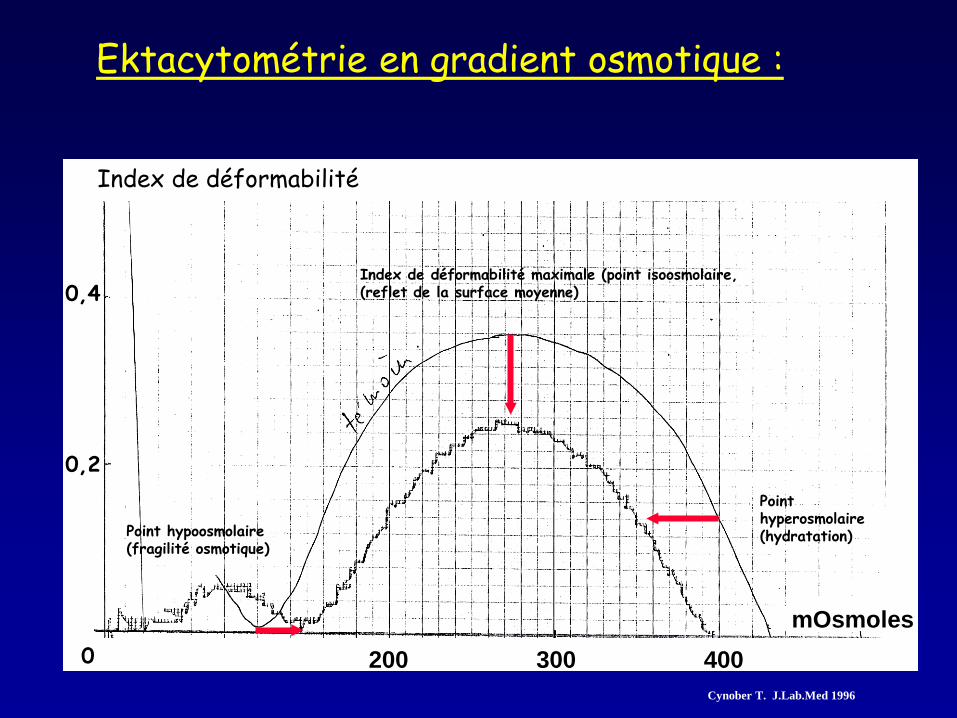
Ektacytométrie en gradient osmotique :
Index de déformabilité
mOsmoles 400 300 200
0,2
0
0,2
0,4
Point hypoosmolaire (fragilité osmotique)
Index de déformabilité maximale (point isoosmolaire, (reflet de la surface moyenne)
Point hyperosmolaire (hydratation)
Cynober T. J.Lab.Med 1996

Orientation vers une sphérocytose héréditaire (SH):
Avec des éléments inhabituels :
•Présentation clinique •NFS : Hb 8g/dL, 13% de cellules hyperdenses •Fragilité osmotique : augmentée •Ektacytométrie : compatible
•Réticulocytes : 145.109/L •Volume réticulocytaire : 115fL •Frottis sanguin •Hyperferritinémie
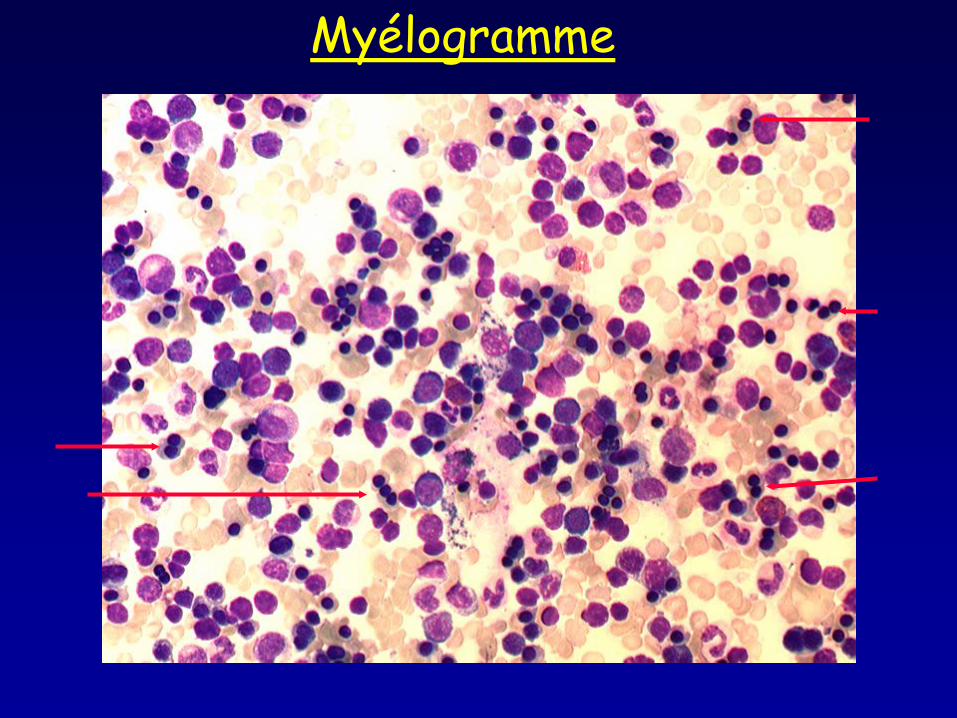
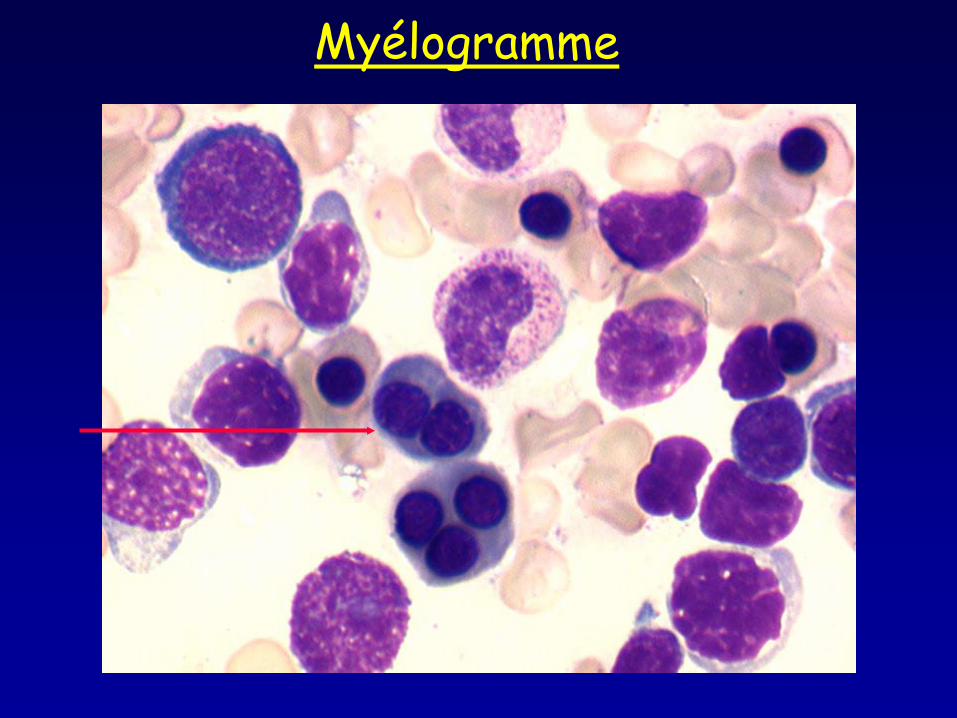
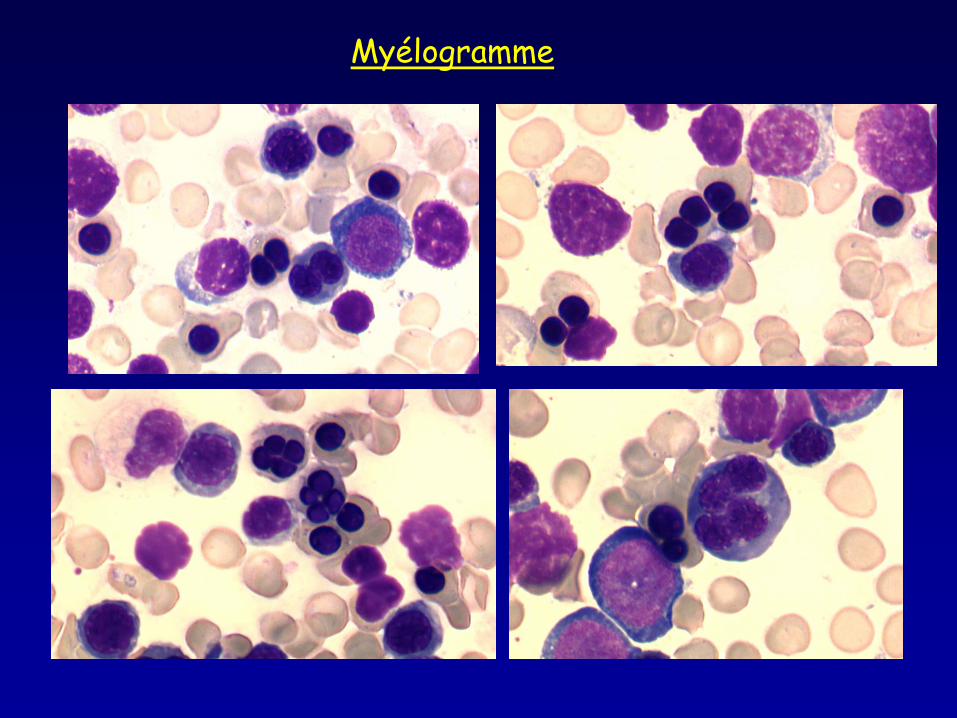
Myélogramme
Myélogramme
Myélogramme
P
T
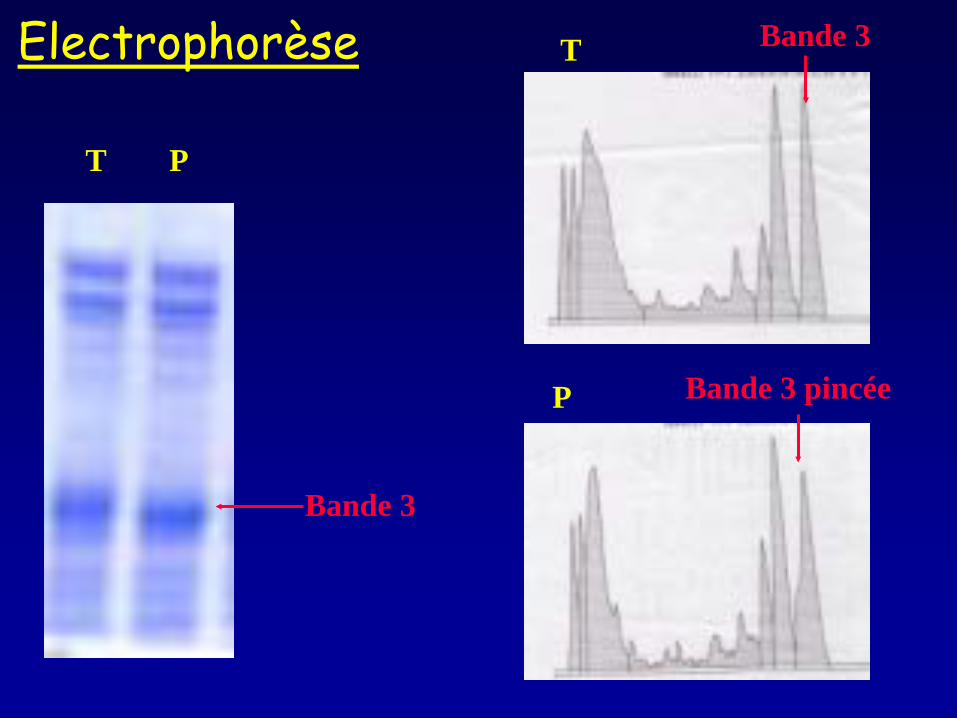
T P
Bande 3
Bande 3 pincée
Bande 3 Electrophorèse

Anémie dysérythropoïétique congénitale de type II (CDAII):
• Forme la plus fréquente d’anémie dysérythropoïétique • 450 cas recensés dans le monde (Iolascon A. Haematologica 2012)
•Autosomique récessive •Age moyen à la présentation 5 ans (1mois-25ans) ; au diagnostic 16 ans (4mois- 65ans) (98 patients Iolascon A. Blood 2001 ; 48 patients Heimpel H. Blood 2003)
• Anémie le plus souvent modérée avec érythropoïèse inefficace et durée de vie raccourcie des globules rouges • Réticulocytose modérée • Ictère, splénomégalie • Surcharge martiale avec complications fréquentes
• Frottis sanguin : aniso-poikilocytose, ponctuations basophiles • Myélogramme : moelle érythroblastique avec >10% érythroblastes binucléés, macrophages pseudo Gaucher
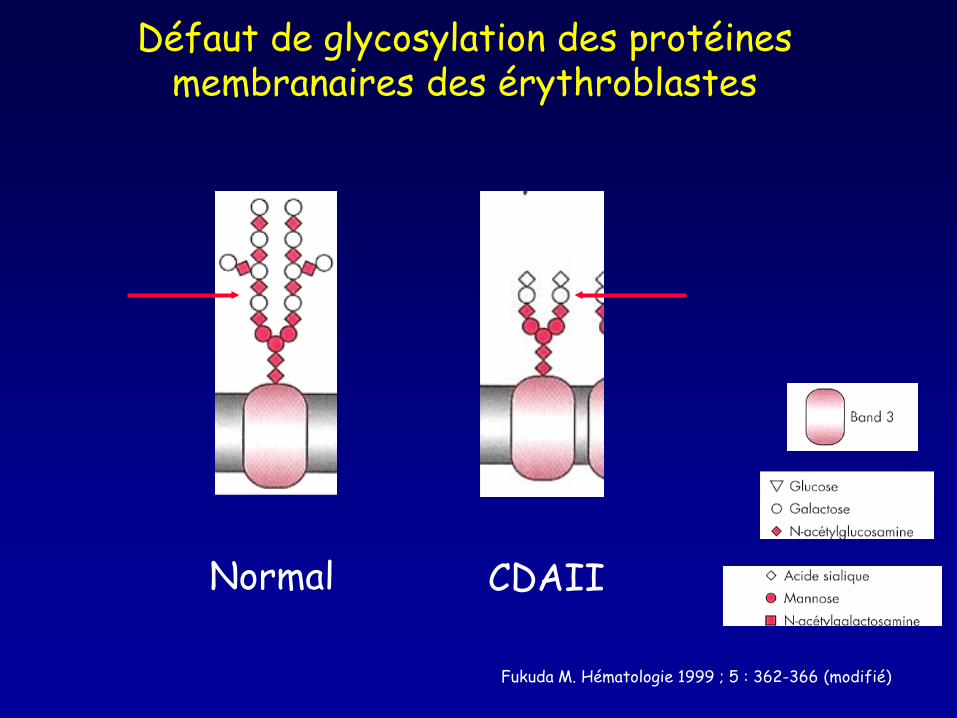
Défaut de glycosylation des protéines membranaires des érythroblastes
Normal CDAII
Fukuda M. Hématologie 1999 ; 5 : 362-366 (modifié)
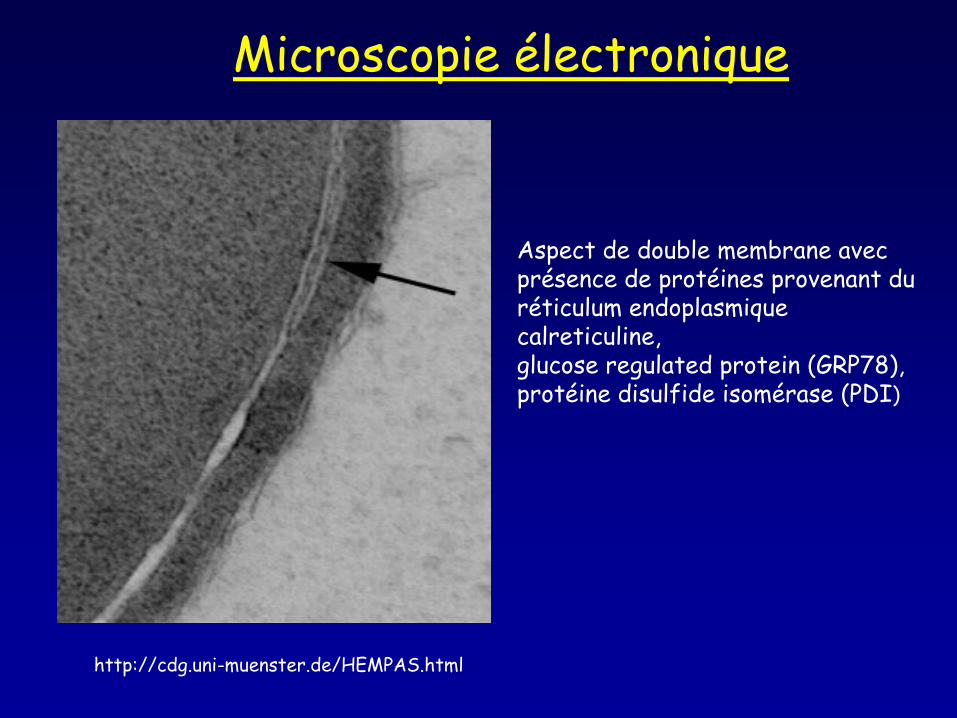
http://cdg.uni-muenster.de/HEMPAS.html
Microscopie électronique
Aspect de double membrane avec présence de protéines provenant du réticulum endoplasmique calreticuline, glucose regulated protein (GRP78), protéine disulfide isomérase (PDI)
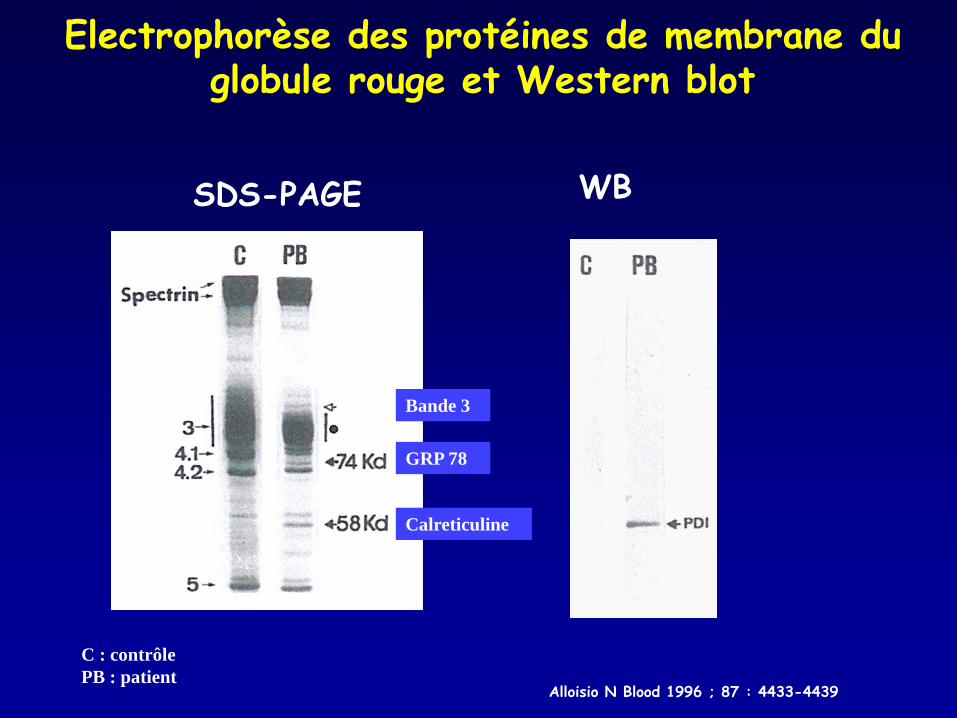
Electrophorèse des protéines de membrane du globule rouge et Western blot
SDS-PAGE WB
Alloisio N Blood 1996 ; 87 : 4433-4439
GRP 78
Calreticuline
C : contrôle PB : patient
Bande 3

Mutation du gène SEC23B • Gène situé en (20p11.23) 54kb, 20 exons (Bianchi P. Human Mutation 2009),
•Code pour une protéine SEC23B : composant essentiel du complexe protéique COPII (coat protein complex II) vésicules coatées qui transportent les protéines secrétées du RE vers le Golgi.
• > 50 mutations décrites ((Iolascon A. Haematologica 2012)
• R14W, E109K, R497C et I318T représentent 50% des mutations
• Pas de mutations non sens homozygote (léthal?) • Patients avec mutations non sens et faux sens : phénotype
plus sévère (que faux sens homozygote) (Iolascon A. Haematologica 2010)

Traitement symptomatique
• Transfusions • Chélation fer • Exceptionnellement splénectomie
(Heipel H. Blood 2003)

La CDAII peut partager avec la SH certains signes
• Anémie hémolytique constitutionnelle avec ictère et splénomégalie
Eléments de pondération : •Taux faible de réticulocytes pour le degré d’anémie •Macrocytose érythrocytaire et réticulocytaire (>110fL) •Hyperferritinémie
Signes trompeurs en faveur de la SH : •Augmentation du nombre de cellules hyperdenses •Augmentation de la fragilité osmotique •Tracé ektacytométrique compatible

•Moelle riche érythroblastique avec au moins 10% de précurseurs érythroblastiques binuclées
•Electrophorèse des protéines de la membrane érythrocytaire : pincement sans diminution de la bande 3 •+/- Immunoblot : présence anormale de la protein disulfide isomérase •Séquençage du gène SEC23B
Propositions de critères diagnostiques pour les CDA II :
•Anémie hémolytique constitutionnelle avec ictère et splénomégalie
•Erythropoïèse inefficace

Xérocytose Stomatocytose héréditaire à globule rouge déshydraté
A propos d’une famille
Marion Eveillard GFHC 21 novembre 2012
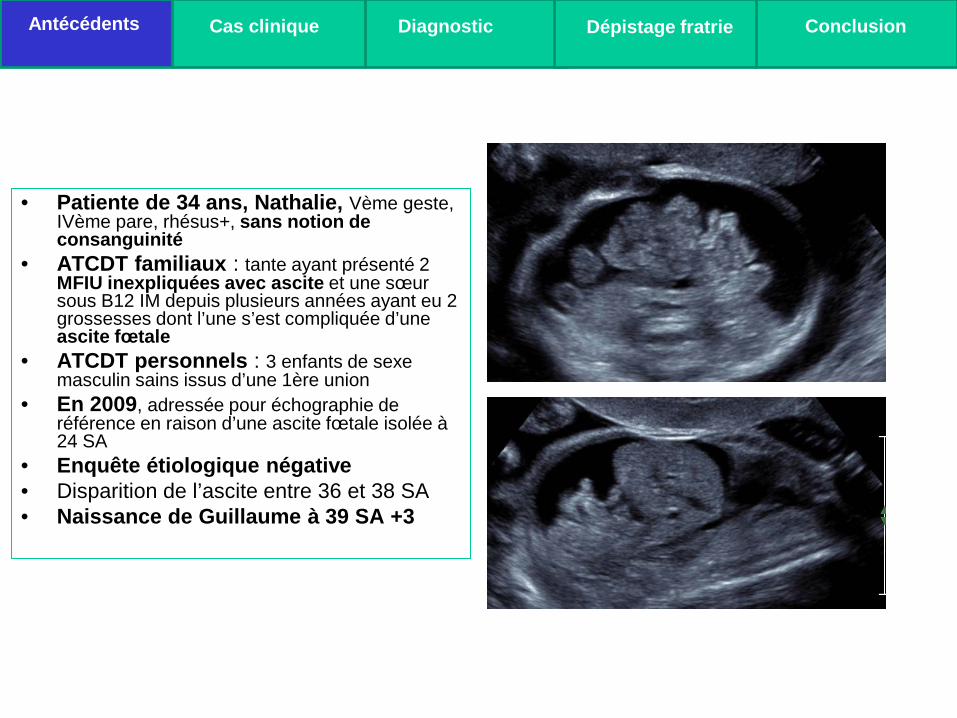
• Patiente de 34 ans, Nathalie, Vème geste, IVème pare, rhésus+, sans notion de consanguinité
• ATCDT familiaux : tante ayant présenté 2 MFIU inexpliquées avec ascite et une sœur sous B12 IM depuis plusieurs années ayant eu 2 grossesses dont l’une s’est compliquée d’une ascite fœtale
• ATCDT personnels : 3 enfants de sexe masculin sains issus d’une 1ère union
• En 2009, adressée pour échographie de référence en raison d’une ascite fœtale isolée à 24 SA
• Enquête étiologique négative • Disparition de l’ascite entre 36 et 38 SA • Naissance de Guillaume à 39 SA +3
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
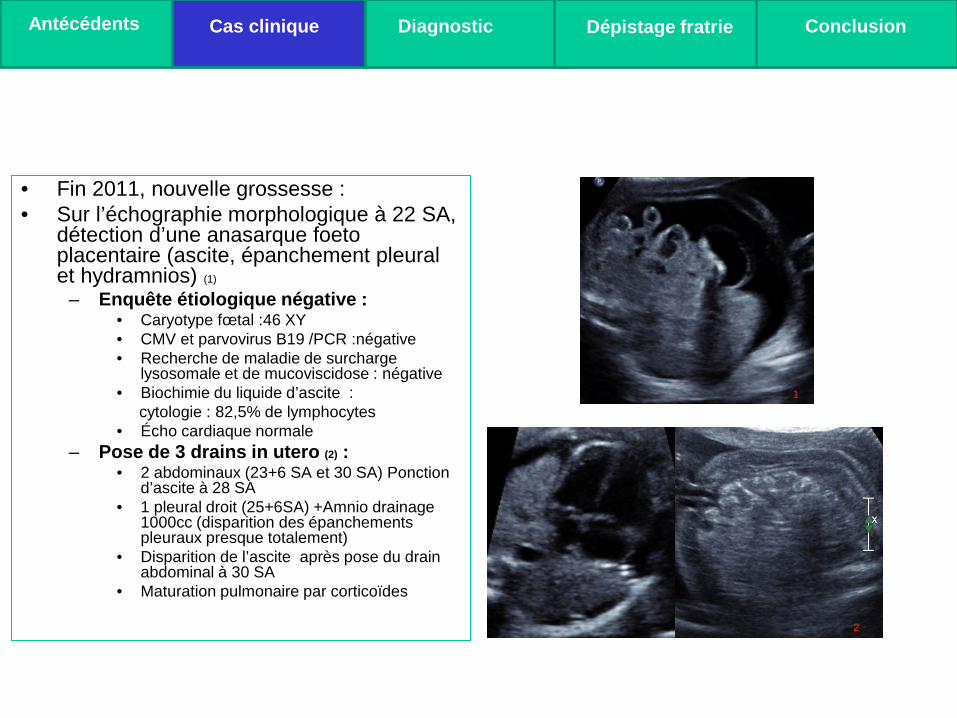
• Fin 2011, nouvelle grossesse : • Sur l’échographie morphologique à 22 SA,
détection d’une anasarque foeto placentaire (ascite, épanchement pleural et hydramnios) (1)
– Enquête étiologique négative : • Caryotype fœtal :46 XY • CMV et parvovirus B19 /PCR :négative • Recherche de maladie de surcharge
lysosomale et de mucoviscidose : négative • Biochimie du liquide d’ascite : cytologie : 82,5% de lymphocytes • Écho cardiaque normale
– Pose de 3 drains in utero (2) : • 2 abdominaux (23+6 SA et 30 SA) Ponction
d’ascite à 28 SA • 1 pleural droit (25+6SA) +Amnio drainage
1000cc (disparition des épanchements pleuraux presque totalement)
• Disparition de l’ascite après pose du drain abdominal à 30 SA
• Maturation pulmonaire par corticoïdes
1
2
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
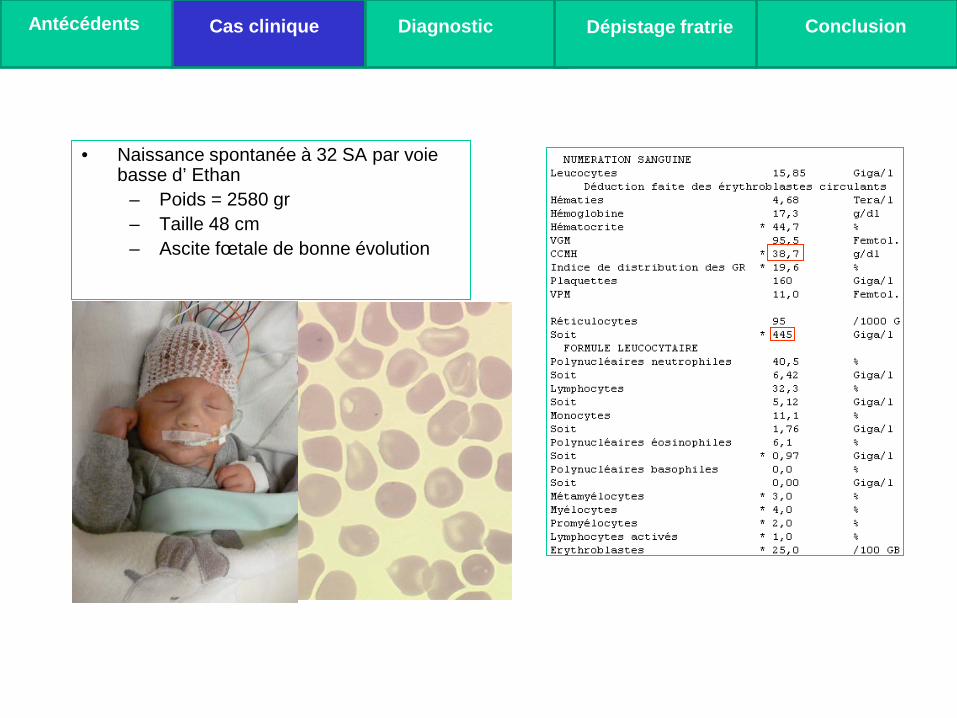
• Naissance spontanée à 32 SA par voie basse d’ Ethan
– Poids = 2580 gr – Taille 48 cm – Ascite fœtale de bonne évolution
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
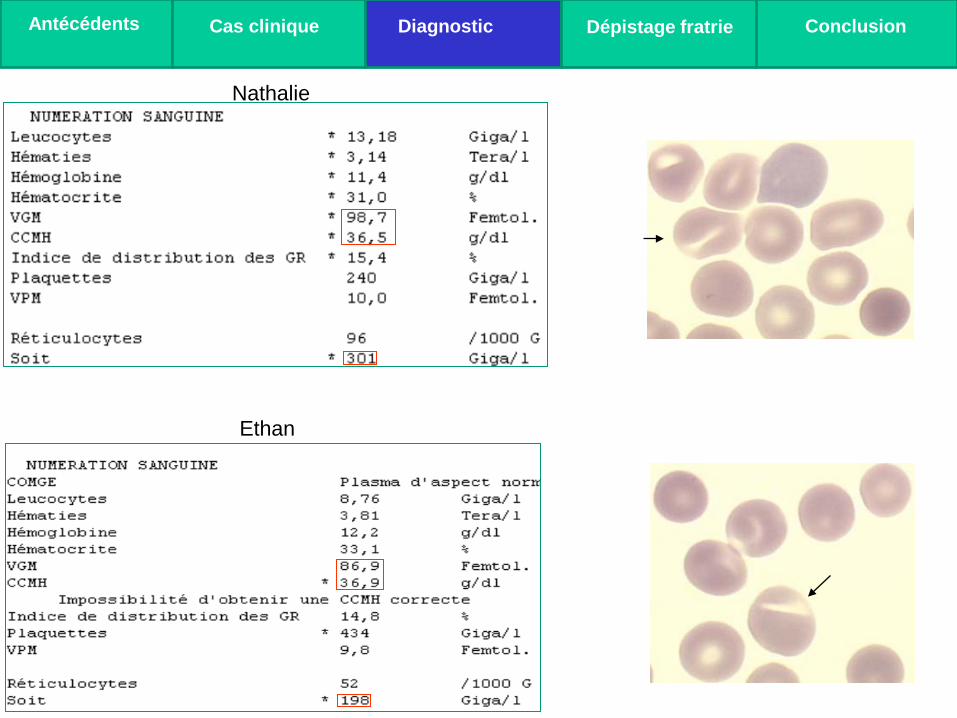
Nathalie
Ethan
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
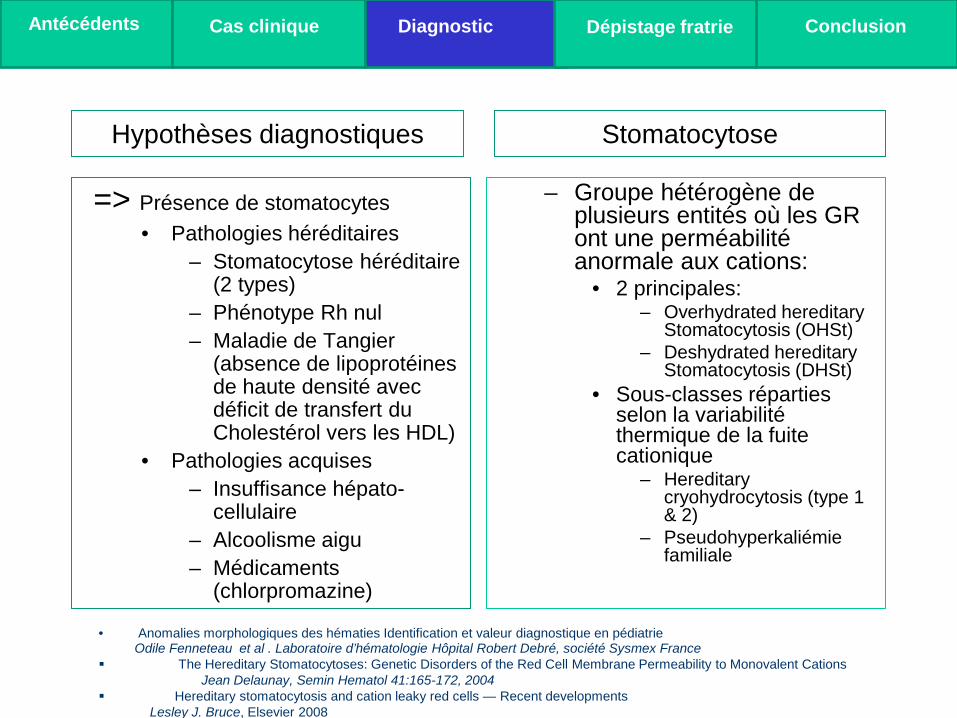
Hypothèses diagnostiques
=> Présence de stomatocytes • Pathologies héréditaires
– Stomatocytose héréditaire (2 types)
– Phénotype Rh nul – Maladie de Tangier
(absence de lipoprotéines de haute densité avec déficit de transfert du Cholestérol vers les HDL)
• Pathologies acquises – Insuffisance hépato-
cellulaire – Alcoolisme aigu – Médicaments
(chlorpromazine)
– Groupe hétérogène de plusieurs entités où les GR ont une perméabilité anormale aux cations:
• 2 principales: – Overhydrated hereditary
Stomatocytosis (OHSt) – Deshydrated hereditary
Stomatocytosis (DHSt) • Sous-classes réparties
selon la variabilité thermique de la fuite cationique
– Hereditary cryohydrocytosis (type 1 & 2)
– Pseudohyperkaliémie familiale
• Anomalies morphologiques des hématies Identification et valeur diagnostique en pédiatrie Odile Fenneteau et al . Laboratoire d’hématologie Hôpital Robert Debré, société Sysmex France The Hereditary Stomatocytoses: Genetic Disorders of the Red Cell Membrane Permeability to Monovalent Cations
Jean Delaunay, Semin Hematol 41:165-172, 2004 Hereditary stomatocytosis and cation leaky red cells — Recent developments Lesley J. Bruce, Elsevier 2008
Stomatocytose
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
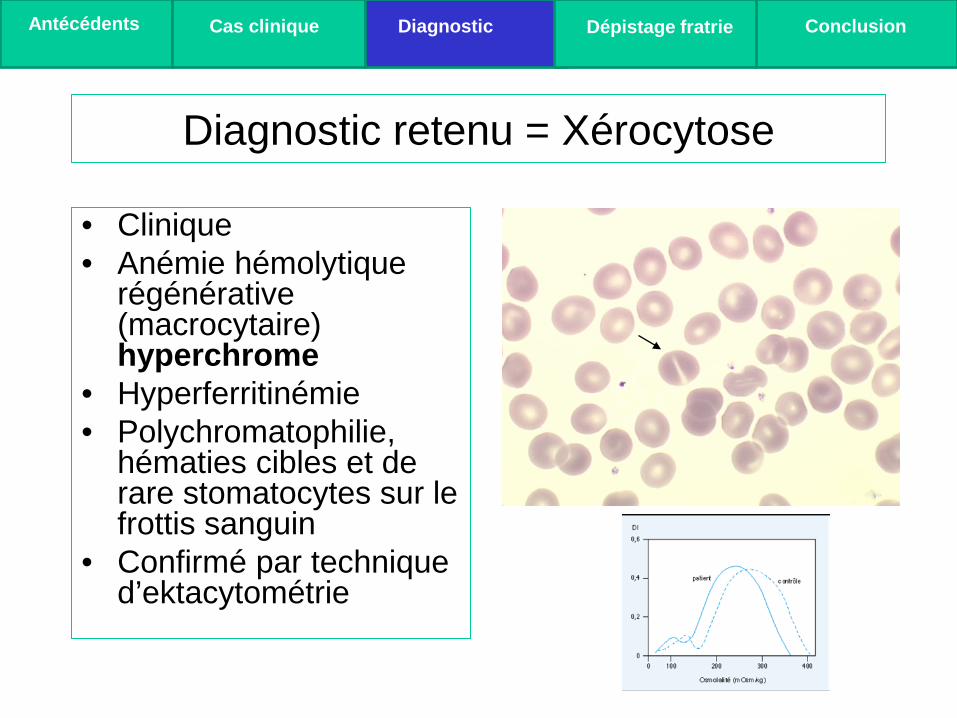
Diagnostic retenu = Xérocytose
• Clinique • Anémie hémolytique
régénérative (macrocytaire) hyperchrome
• Hyperferritinémie • Polychromatophilie,
hématies cibles et de rare stomatocytes sur le frottis sanguin
• Confirmé par technique d’ektacytométrie
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
• 1 famille/50 000 • Transmission autosomique dominante • Clinique: hydrops foetalis, parfois résolutif spontanément en
péri ou pré-natal/ pseudo-hyperkaliémie • Biologique : anémie parfois sévère – macrocytaire (VGM = 95 – 115 fl) et – Hyperchrome (CCMH > 35 g / dl) – Sur frottis : morphologie souvent normale, mais on peut
observer des GR contractés et spiculés, des hématies cibles, des échinocytes et de rare stomatocytes
– Ektacytométrie : courbe décalée à gauche, technique de référence
– Analyse génétique
Diagnostic de la xérocytose
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
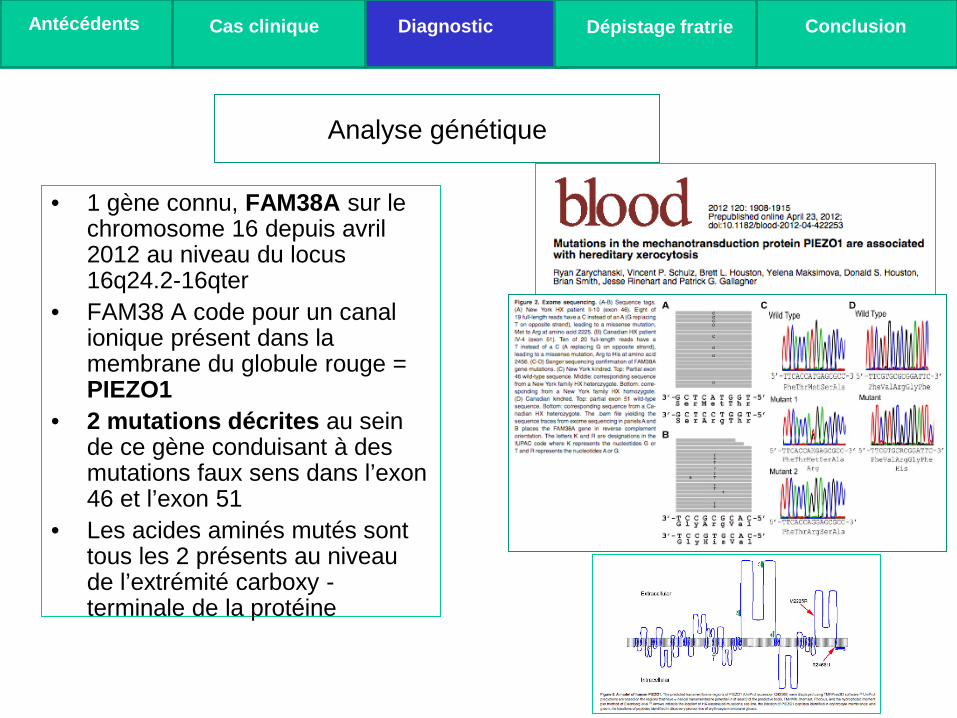
• 1 gène connu, FAM38A sur le chromosome 16 depuis avril 2012 au niveau du locus 16q24.2-16qter
• FAM38 A code pour un canal ionique présent dans la membrane du globule rouge = PIEZO1
• 2 mutations décrites au sein de ce gène conduisant à des mutations faux sens dans l’exon 46 et l’exon 51
• Les acides aminés mutés sont tous les 2 présents au niveau de l’extrémité carboxy - terminale de la protéine
Analyse génétique
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
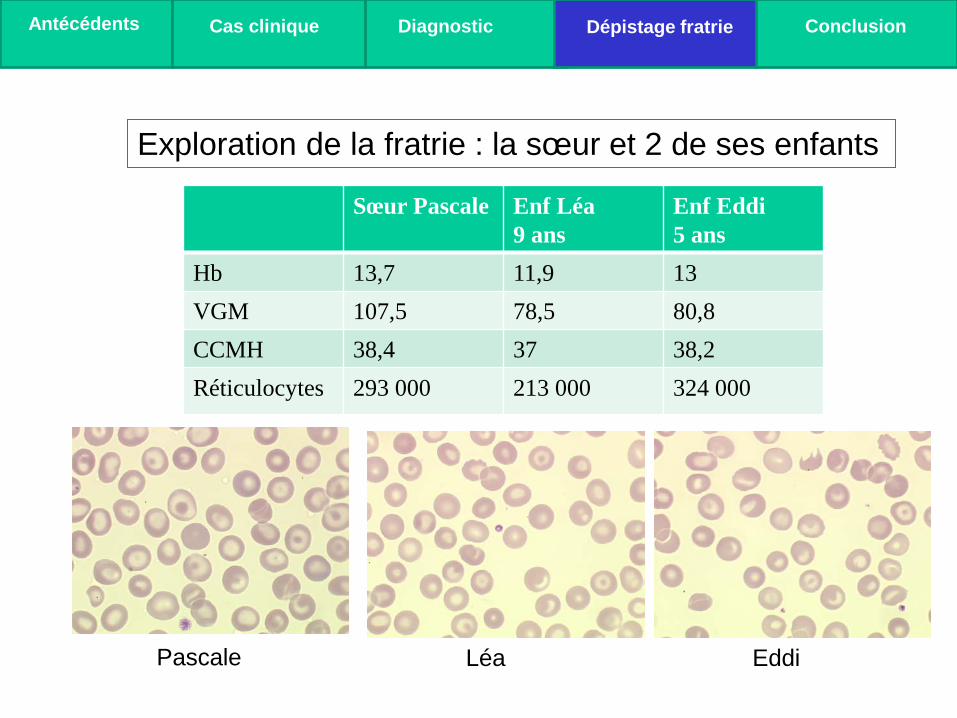
Sœur Pascale Enf Léa 9 ans
Enf Eddi 5 ans
Hb 13,7 11,9 13 VGM 107,5 78,5 80,8 CCMH 38,4 37 38,2 Réticulocytes 293 000 213 000 324 000
Exploration de la fratrie : la sœur et 2 de ses enfants
Pascale Léa Eddi
Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
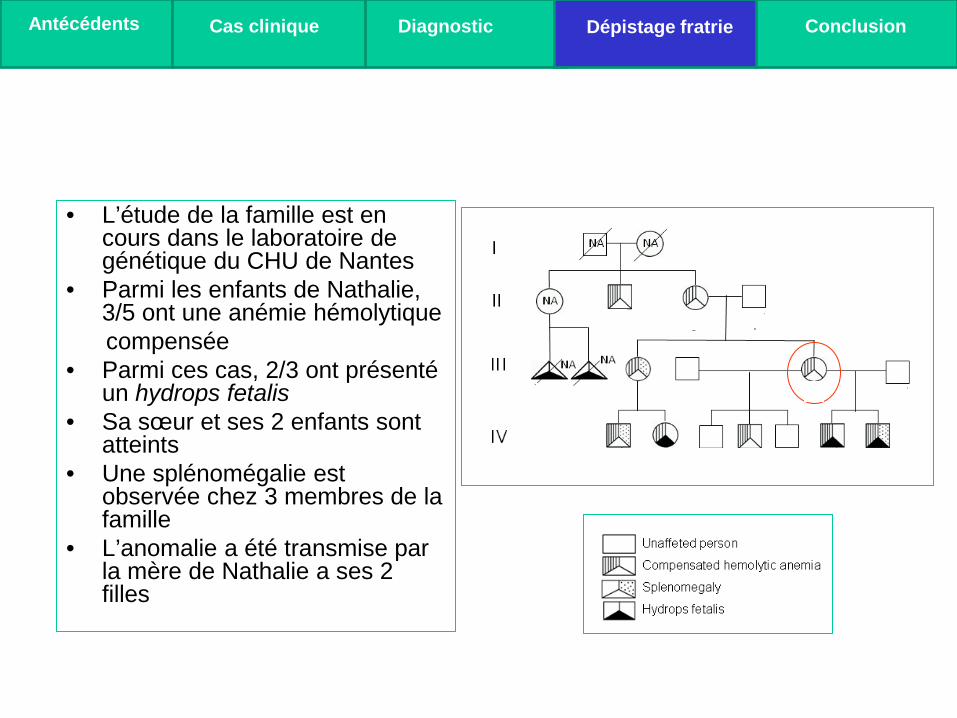
• L’étude de la famille est en cours dans le laboratoire de génétique du CHU de Nantes
• Parmi les enfants de Nathalie, 3/5 ont une anémie hémolytique
compensée • Parmi ces cas, 2/3 ont présenté
un hydrops fetalis • Sa sœur et ses 2 enfants sont
atteints • Une splénomégalie est
observée chez 3 membres de la famille
• L’anomalie a été transmise par la mère de Nathalie a ses 2 filles

Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
• Des questions ne sont toujours pas résolues malgré la découverte du gène :
La grande variabilité des phénotypes observés : L’hydrops fetalis n’est pas toujours présent La pseudo hyperkaliémie familiale avec/sans anomalie hématologique
Le rôle exact de la protéine PIEZO 1 n’est pas encore établi de façon précise au sein du globule rouge ainsi que sur les autres cellules de l’organisme et il reste à le déterminer
• La résolution spontanée de ces cas d’ascite fœtale nous a amenés à monter un projet d’étude intra - hospitalier pour l’analyse du gène FAM38A au sein des familles ayant présenté des cas d’anasarque fœtale inexpliqués

Introduction article
Antécédents Cas clinique Diagnostic Dépistage fratrie Conclusion
• Laboratoire de génétique Dr C. Beneteau G. Thierry Pr C.Le Caignec • Laboratoire d’hématologie Dr R. Garand Benoit Tessoulin Nelly Robillard Myriam Chevalier Pr M.C.Béné • Le service de génétique obstétrique Dr C.Le vaillant • Le service de pédiatrie Dr M.L. Couec