
Session 13. Résistance aux antimicrobiens Les comités pharmaceutiques et thérapeutiques.

VETAGRO SUP CAMPUS VETERINAIRE DE LYON
Année 2014 - Thèse n° 041
L’IMMUNITE INNEE : BILAN DES CONNAISSANCES
ACTUELLES CHEZ LES RUMINANTS ET SUR LES
NOUVELLES CELLULES LYMPHOÏDES INNEES (ILC)
THESE
Présentée à l’UNIVERSITE CLAUDE-BERNARD - LYON I
(Médecine - Pharmacie)
et soutenue publiquement le 26 septembre 2014
pour obtenir le grade de Docteur Vétérinaire
par
SAVOYAT Céline
Née le 25 avril 1989
à Saint Martin d’Hères

2
3
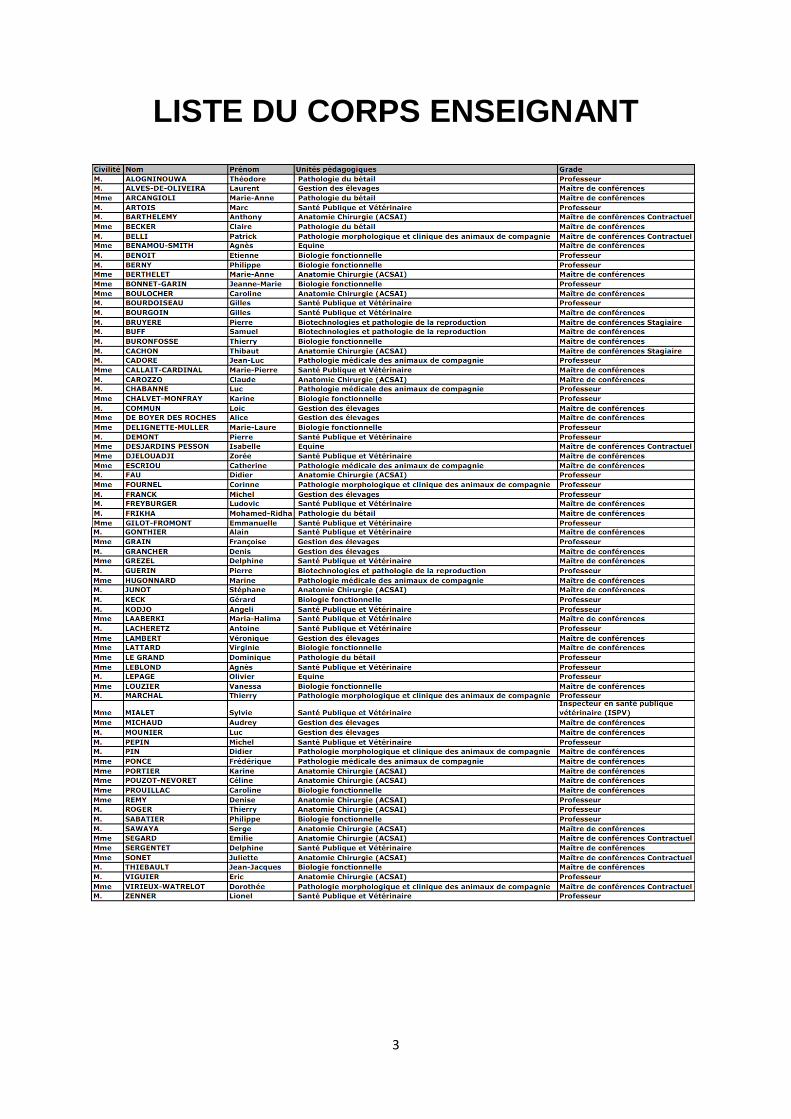
LISTE DU CORPS ENSEIGNANT

4

5
REMERCIEMENTS
A Madame le Professeur Alexandra TRAVERSE-GLEHEN,
Professeur à l’Université Claude Bernard de Lyon,
Vous qui nous avez fait l’honneur d’accepter la présidence de notre jury de thèse,
Veuillez trouver ici l’expression la plus distinguée de notre reconnaissance.
A Monsieur le Professeur Michel PEPIN,
Professeur à VetagroSup campus Vétérinaire de Lyon,
Pour nous avoir transmis votre passion pour l’immunologie,
Pour nous avoir encadré tout au long de ce travail,
Puissiez-vous trouver dans ce dernier l’expression de notre gratitude et le
témoignage de notre sincère respect.
A Madame le Maître de conférences Marie-Anne ARCANGIOLI,
Professeur à VetagroSup campus Vétérinaire de Lyon,
Pour nous avoir fait l’honneur de participer à ce jury de thèse
Veuillez accepter nos remerciements les plus sincères.

6
A mes parents,
Qui m’ont toujours soutenue, et ont fait de moi ce que je suis aujourd’hui
Pour m’avoir donné le gout des études,
Pour avoir toujours cru en moi, et m’avoir aidé dans les moments difficiles,
Pour toute l’aide qu’ils m’ont apportée pour ce travail, merci
A mon frère,
Pour toutes nos chamailleries passées et à venir
Pour tous les bons moments, merci
A mon parrain et ma marraine,
Pour avoir toujours été là, pour m’avoir toujours fait rire,
Pour toutes les bons moments passés ensemble, merci
Remets-toi bien
A toute ma famille,
Pour m’avoir toujours soutenue et pour tous les bons moments passés et les moments à
venir,
La distance ne nous empêchera pas de nous revoir et de passer de bonnes cousinades,
Merci pour ce cadre familial plein de convivialité
A Françoise,
Pour avoir répondu avec patience à toutes mes questions de français, même si je n’ai pas
toujours été claire…
Pour tout ce que tu m’apportes, ta gentillesse
Puisses-tu trouver ici, ma profonde reconnaissance
A Gaël,
Pour tous les bons moments que nous passons ensemble et tous les fous rires,
Pour avoir été une tornade qui a bouleversé ma vie,
Pour ta patience de tous les jours,
Pour tout le bonheur que tu m’offres,
Pour partir tous les deux à l’aventure l’an prochain, merci
Trouve ici l’expression de mon amour
A mon grand-père,
J’aurais tant aimé que tu sois là aujourd’hui,
Pour avoir toujours cru en moi,
Tu me manques énormément

7
Table des matières ..................................................................................................... 7
Table des illustrations ............................................................................................... 12
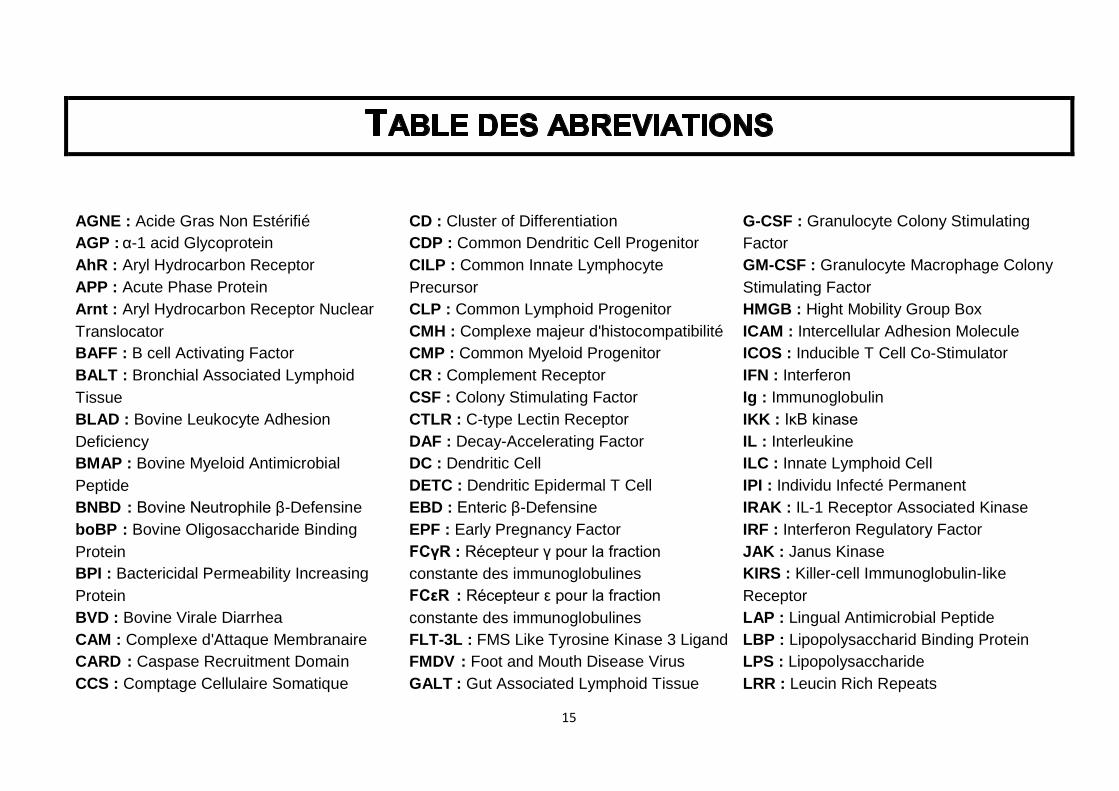
Table des abréviations ............................................................................................. 15
Introduction .............................................................................................................. 17
1ière PARTIE : Tour d’horizon de l’immunité innée..................................................... 18
I. Les différents niveaux de l’immunité innée .................................................. 19 A- Les barrières physiques .............................................................................. 19
Les épithéliums de recouvrement............................................................. 20 1)
a. La structure de l’épithélium ................................................................... 20
b. Les formations lymphoïdes associées aux muqueuses......................... 23
c. Le mucus produit par l’épithélium.......................................................... 25
La flore commensale ................................................................................ 25 2)
B- Les cellules de l’immunité innée .................................................................. 26
Les cellules myéloïdes ............................................................................. 27 1)
a. Les mastocytes ..................................................................................... 27
i. Origine des mastocytes ..................................................................... 28
ii. Fonction des mastocytes ................................................................... 28
b. Les granulocytes ................................................................................... 30
i. Les polynucléaires neutrophiles ......................................................... 31
ii. Les polynucléaires éosinophiles ........................................................ 37
iii. Les polynucléaires basophiles ........................................................... 39
c. Les monocytes - macrophages ............................................................. 40
d. Les cellules dendritiques ....................................................................... 42
i. Origine des cellules dendritiques ....................................................... 42
ii. Les différents types de cellules dendritiques ...................................... 45
iii. Fonctions ........................................................................................... 50
Les lymphocytes ...................................................................................... 53 2)
a. Les cellules Th17 .................................................................................... 53
b. Les cellules γδ T ................................................................................... 54
c. Les NKT ................................................................................................ 58
d. Les cellules T CD8+ .............................................................................. 61
C- Les facteurs humoraux ................................................................................ 62
Les lectines .............................................................................................. 62 1)
Le complément ........................................................................................ 63 2)
a. Historique ............................................................................................. 63
b. Les composants du complément .......................................................... 64
c. Cascades d’activation du complément .................................................. 65
i. Voie classique ................................................................................... 65
ii. Voie alterne ....................................................................................... 67
iii. Voie des lectines ............................................................................... 68
d. Rôles du complément ........................................................................... 69

8
e. Mécanismes de régulation du complément ........................................... 71
Les protéines de la phase aiguë de l’inflammation ................................... 72 3)
Les autres facteurs humoraux .................................................................. 74 4)
D- Les composants chimiques ......................................................................... 75
Les peptides antimicrobiens ..................................................................... 75 1)
Les réactifs oxygénés .............................................................................. 78 2)
II. Quand mettre en place une immunité innée ? ............................................. 79 Les signaux de danger reconnus par l’organisme ....................................... 80 A-
Les PAMPs .............................................................................................. 80 1)
Les DAMPs .............................................................................................. 81 2)
Les récepteurs ............................................................................................ 82 B-
Les Toll-Like Récepteurs ou TLR ............................................................. 83 1)
a. Structure ............................................................................................... 84
b. Activation du récepteur par son ligand .................................................. 84
c. Activation d’une cascade intracellulaire ................................................ 89
i. Les molécules impliquées .................................................................. 90
ii. La signalisation MyD88 dépendante .................................................. 91
iii. La signalisation indépendante du MyD88 .......................................... 92
d. Régulation des TLR .............................................................................. 92
e. Autres rôles des TLR ............................................................................ 93
Les NOD-Like Receptor ou NLR .............................................................. 94 2)
a. Les récepteurs NOD Like Receptor ...................................................... 94
b. La formation d’inflammasomes ............................................................. 95
Les C-Type lectin Receptor ou CTLR ....................................................... 96 3)
Les RIG Like Receptor ou RLR ................................................................ 97 4)
Les récepteurs au complément ................................................................ 97 5)
Les autres récepteurs .............................................................................. 99 6)
Un point important : la tolérance et l’homéostasie muqueuse .................... 100 C-
III. Initiation de la réponse innée ..................................................................... 108 L’inflammation locale ................................................................................. 108 A-
La phase vasculaire ............................................................................... 108 1)
La phase cellulaire ................................................................................. 110 2)
a. L’extravasation des cellules effectrices ............................................... 111
b. La phagocytose .................................................................................. 113
La phase de détersion ............................................................................ 114 3)
La phase de cicatrisation ....................................................................... 115 4)
L’inflammation systémique ........................................................................ 116 B-
Le langage chimique utilisé : les cytokines ................................................ 117 C-
Les chimiokines ..................................................................................... 117 1)
Les interleukines .................................................................................... 119 2)
a. Les cytokines pro-inflammatoires ........................................................ 119
b. Les cytokines anti-inflammatoires ....................................................... 123
Les hématopoïétines .............................................................................. 124 3)
La famille des interférons ....................................................................... 124 4)
Autres mécanismes effecteurs .................................................................. 124 D-
Initiation de la réponse adaptative ............................................................. 125 E-

9
Le prélèvement des antigènes ............................................................... 126 1)
Maturation des cellules dendritiques ...................................................... 128 2)
La migration des cellules dendritiques ................................................... 129 3)
Présentation de l’antigène ...................................................................... 129 4)
Une nouveauté : la mémoire innée ............................................................ 130 F-
IV. Evolution ................................................................................................... 134 2ième PARTIE : L’immunité innée chez les ruminants .............................................. 137
I. Particularités de l’immunité innée chez les ruminants ................................ 137 Chez les bovins ......................................................................................... 137 A-
Considérations anatomiques .................................................................. 137 1)
Les cellules ............................................................................................ 138 2)
a. Les cellules dendritiques ..................................................................... 138
b. Les NK ................................................................................................ 139
c. Les cellules γδ T ................................................................................. 139
d. Les granulocytes ................................................................................. 141
e. Les monocytes.................................................................................... 142
Composants humoraux et chimiques ..................................................... 142 3)
Les marqueurs de l’inflammation chez les ruminants ............................. 145 4)
Les problèmes liés à la sélection génétique ........................................... 145 5)
Exemples de maladies dans lesquelles l’action du système inné est 6)
importante .................................................................................................... 146
Les petits ruminants .................................................................................. 149 B-
Chez les ruminants sauvages ................................................................... 149 C-
II. Variation de cette immunité selon le stade physiologique .......................... 151 Mise en place de l’immunité innée pendant la vie fœtale ........................... 151 A-
Importance du colostrum ........................................................................... 152 B-
Mise en place de sa propre machinerie immunitaire .................................. 152 C-
Importance de la nutrition .......................................................................... 153 D-
Modifications du système immunitaire inné pendant la gestation .............. 155 E-
Modifications à la mise-bas ....................................................................... 159 F-
III. L’immunité de la mamelle .......................................................................... 160 Prévention de l’infection mammaire ........................................................... 161 A-
Mesures anatomiques ............................................................................ 161 1)
a. Le sphincter ........................................................................................ 163
b. Le canal et sa kératine ........................................................................ 163
c. La rosette de Fürstemberg .................................................................. 163
d. La compartimentation des quartiers .................................................... 163
e. Les jonctions serrées .......................................................................... 164
Les cellules impliquées .......................................................................... 164 2)
a. Les cellules épithéliales ...................................................................... 164
b. Les macrophages ............................................................................... 165
c. Les polynucléaires neutrophiles .......................................................... 165
d. Les NK ................................................................................................ 167
Mesures chimiques et humorales ........................................................... 167 3)
a. Les peptides antimicrobiens................................................................ 167
b. Les protéines de la phase aiguë ......................................................... 170
c. Le complément et les anticorps naturels ............................................. 170

10
d. Les composants chimiques ................................................................. 170
Les facteurs influençant les composants immuns du lait ........................ 171 4)
L’inflammation mammaire : 1er signe macroscopique d’une mammite ....... 171 B-
3ième PARTIE: Les nouvelles cellules de l’immunité innée : les cellules lymphoïdes
innées ou ILCs ....................................................................................................... 175
I. Les différents types d’ILCs ........................................................................ 176 Le groupe 1 des ILCs ................................................................................ 178 A-
Les cellules NK ...................................................................................... 178 1)
a. Différents types de cellules NK ........................................................... 178
b. Distribution .......................................................................................... 179
c. Développement des cellules NK ......................................................... 181
d. Mécanisme de la cytotoxicité des NK .................................................. 184
e. La mémoire des NK ............................................................................ 185
Les ILCs 1 .............................................................................................. 186 2)
Le groupe 2 des ILCs ................................................................................ 187 B-
Le groupe 3 des ILCs ................................................................................ 192 C-
Les cellules LTi ...................................................................................... 192 1)
Les ILCs 3 NCR+ .................................................................................... 194 2)
Les ILCs 3 NCR- .................................................................................... 196 3)
II. Le développement des ILCs ...................................................................... 197 Un précurseur commun avec le système immunitaire adaptatif : le CLP ... 197 A-
Du CLP au précurseur commun des ILCs : le CILP ................................... 198 B-
Différenciation des différents groupes d’ILC .............................................. 198 C-
Les signaux nécessaires au développement de chaque lignée ................. 199 D-
L’IL-7 ...................................................................................................... 199 1)
L’Id2 ....................................................................................................... 200 2)
Le RORγt ............................................................................................... 200 3)
Le GATA3 .............................................................................................. 200 4)
Le Notch signal ...................................................................................... 201 5)
Le ROR-α ............................................................................................... 201 6)
L’Aryl Hydrocarbon Receptor ou AHR .................................................... 201 7)
Le T-bet ................................................................................................. 202 8)
III. Les différents rôles des ILCs ..................................................................... 203 Les ILCs interviennent dans la régulation des tissus lymphoïdes .............. 203 A-
Mise en place des organes lymphoïdes secondaires par les LTi durant 1)
l’embryogénèse ............................................................................................ 204
Mise en place des organes lymphoïdes tertiaires par les LTi chez 2)
l’adulte… .............................................................. …………………………….205
Restauration des tissus lymphoïdes après un épisode viral par les LTi .. 207 3)
Les ILCs 3 stimulent l’immunité acquise ................................................. 207 4)
Les ILCs dans le tube digestif ................................................................... 208 B-
Expulsion des vers intestinaux grâce aux ILCs 2 ................................... 209 1)
Contrôle de la flore commensale intestinale par les ILCs 3 .................... 210 2)
....... Intervention dans l’immunité contre les bactéries intestinales : les ILCs 3 3)
……………………………………………………………………………………….211
Recrutement des monocytes par les NK ................................................ 212 4)

11
Les ILCs dans les poumons ...................................................................... 213 C-
Inflammation pulmonaire ........................................................................ 213 1)
Réparation du parenchyme pulmonaire.................................................. 214 2)
Des ILCs disséminées dans tout l’organisme : les cellules NK .................. 215 D-
Les NK dans la réponse antivirale .......................................................... 215 1)
Les NK dans la lutte contre les tumeurs ................................................. 216 2)
Les NK contre les protozoaires .............................................................. 216 3)
Les NK contre des bactéries .................................................................. 217 4)
Les NK contrôlent l’immunité .................................................................. 217 5)
E- Le dialogue des ILCs avec les autres cellules immunitaires ...................... 219
IV. Implication des ILCs dans des processus pathologiques ........................... 220
A- Les ILCs 2 sont impliquées dans l’asthme et les allergies ......................... 220
B- Les ILCs 3 et les maladies inflammatoires chroniques .............................. 224
C- Hyperréactivité pulmonaire due aux ILCs 2 suite à une infection virale ..... 225
CONCLUSION…………………………………………………………………………….226
Bibliographie .......................................................................................................... 227
Annexe I : Cytokines ....…………….………….………….……………………………..245
Annexe II : Chimiokines ..………………………………………………………………..250
Annexe III : Réponses types aux diférents agents pathogènes………..……..…....252

12
Figures
Figure 1 : Structures cellulaires de l’intestin grêle et du gros intestin. D’après
(McDermott & Huffnagle, 2013) et (Peterson & Artis, 2014). .................................... 21
Figure 2 : Représentation des GALT au niveau de l’intestin et de leur composition
cellulaire (Pearson, Uhlig, & Powrie, 2012). ............................................................. 25
Figure 3 : Hématopoïèse et lignées qui découlent de la cellule souche
hématopoïétique (Williams, 2012). ........................................................................... 27
Figure 4 : Neutrophiles observés avec la coloration de Wright-Giemsa (Harvey,
2001). ....................................................................................................................... 32
Figure 5 : Piège extracellulaire réalisé par les PNN (Delves, Martin, Burton, & Roitt,
2011). ....................................................................................................................... 35
Figure 6 : Mécanismes de défense des PNN (Theilgaard-Mönch et al., 2006). ........ 36
Figure 7 : PNE observés avec la coloration de Wright-Giemsa (Harvey, 2001). ....... 37
Figure 8 : Basophiles à la coloration de Wright-Giemsa (Harvey, 2001). .................. 39
Figure 9 : Monocytes à la coloration de Wright-Giemsa (Harvey, 2001). .................. 41
Figure 10 : Différenciation des cellules dendritiques depuis la cellule souche
hématopoïétique (HSC) (Kushwah & Hu, 2011). ...................................................... 43
Figure 11: Classification des sous types de cellules dendritiques. D’après (Kushwah
& Hu, 2011). ............................................................................................................. 46
Figure 12 : Structure du fragment C1q (Carroll & Sim, 2011). .................................. 64
Figure 13 : Voie classique d’activation du complément (Williams, 2012). ................. 66
Figure 14 : Voie alterne d’activation du complément (Williams, 2012). ..................... 68
Figure 15 : Voie des lectines pour l’activation du complément (Williams, 2012). ...... 68
Figure 16 : Les différents rôles du complément (Mastellos & Lambris, 2002). .......... 70
Figure 17 : Mécanisme de formation des réactifs oxygénés antimicrobiens (Beutler,
2003). ....................................................................................................................... 79
Figure 18 : Structure des parois des bactéries Gram+ et Gram- (Janeway et al.,
2009). ....................................................................................................................... 81
Figure 19 : Localisations cellulaires des différents PRR (Abbas et al., 2012). .......... 83
Figure 20 : Voies de signalisation possibles suite à l'activation des TLR dans
l’exemple de la reconnaissance de PAMPs viraux (Lester & Li, 2014). .................... 89
Figure 21 : Régulation de la tolérance intestinale (Kayama & Takeda, 2012). ........ 106
Figure 22 : Régulation des réponses immunes intestinales par les cellules innées
(Kayama & Takeda, 2012)...................................................................................... 107
Figure 23 : Mécanisme de la phagocytose par les macrophages. D’après(Janeway et
al., 2009). ............................................................................................................... 109

13
Figure 24: Modification des vaisseaux au début de l’inflammation D’après (Janeway
et al., 2009). ........................................................................................................... 110
Figure 25 : Mécanisme d'extravasation des leucocytes pendant la phase vacsulaire
D’après (Janeway et al., 2009). .............................................................................. 113
Figure 26 : La phagocytose est facilitée par l’opsonisation par le complément ou
d’autres opsonines. D’après (Janeway et al., 2009). .............................................. 114
Figure 27 : Mécanismes d'échantillonnage des antigènes par les cellules
dendritiques dans la lamina propria (S. C. Ng et al., 2010). .................................... 127
Figure 28 : Maturation des cellules dendritiques suite à la détection d'un PAMP
D’après (Janeway et al., 2009). .............................................................................. 128
Figure 29 : Mécanisme de mise en place de la mémoire immune innée (Quintin et al.,
2014). ..................................................................................................................... 133
Figure 30 : Granulocytes chez les bovins (Douglas & Jane Wardrop, 2010). ......... 142
Figure 31 : Anatomie du trayon de vache (Barone, 1978). ..................................... 162
Figure 32 : Classification des ILCs (Björkström, Kekäläinen, & Mjösberg, 2013). ... 177
Figure 33 : Cellule NK (Janeway et al., 2009)......................................................... 178
Figure 34 : Intermédiaires cellulaires du développement des NK murins (en haut) et
des NK humains (dans l’encadré) (J. Yu et al., 2013). ............................................ 182
Figure 35 : Natural helper à la coloration de Giemsa (Koyasu & Moro, 2011). ....... 188
Figure 36 : Fonctions spécifiques des tissus des ILCs (Björkström et al., 2013). .... 203
Figure 37 : Mise en place des organes lymphoïdes secondaires par les LTi (Neyt,
Perros, GeurtsvanKessel, Hammad, & Lambrecht, 2012). ..................................... 205
Figure 38 : Mécanisme de mise en place des tissus lymphoïdes tertiaires par les LTi
(Neyt et al., 2012). .................................................................................................. 206
Figure 39 : Influence des ILCs 3 sur le système immunitaire adaptatif (Hepworth &
Sonnenberg, 2014)................................................................................................. 208
Figure 40 : Rôles des ILCs dans l'homéostasie et la défense intestinales (M Cherrier
et al., 2012) . .......................................................................................................... 209
Figure 41 : Rôle des ILCs 2 dans l'inflammation pulmonaire (B. W. S. Li & Hendriks,
2013). ..................................................................................................................... 214
Figure 42 : Rôle de l'IL-33 dans l'hyperréactivité des voies respiratoires (H. Y. Kim et
al., 2011). ............................................................................................................... 222
Figure 43 : Mécanisme de l’asthme allergique (Deckers et al., 2013). .................... 224

14
Tableaux
Tableau I : Différents obstacles de l’épithélium pour empêcher la colonisation des
tissus par les pathogènes (D’après Janeway, Murphy, Travers, & Walport, 2009). .. 20
Tableau II : Caractéristiques principales des différents granules des PNN. D’après
(Paul, 2013). ............................................................................................................ 33
Tableau III : Principales protéines de la phase aiguë. .............................................. 73
Tableau IV : Les TLR et leurs caractéristiques. D’après (Abbas et al., 2012; De
Franco et al., 2009; Hopkins & Sriskandan, 2005; Paul, 2013; Turvey & Broide, 2010;
Uematsu & Fujimoto, 2010; Zhang et al., 2013). ...................................................... 86
Tableau V : Les récepteurs du complément à la surface des cellules (Janeway et al.,
2009). ....................................................................................................................... 98
Tableau VI : Parallèle entre les cellules Thelper et les ILCs. ...................................... 176
Tableau VII : Principales caractéristiques des Natural Helper, Nuocytes, Innate
Helper et MPP. Synthèse d’après (Koyasu & Moro, 2011; Nakae et al., 2013; Neill &
McKenzie, 2011; Pishdadian, Varasteh, & Sankian, 2012; Hergen Spits et al., 2013).
............................................................................................................................... 190
15
AGNE : Acide Gras Non Estérifié
AGP : α-1 acid Glycoprotein
AhR : Aryl Hydrocarbon Receptor
APP : Acute Phase Protein
Arnt : Aryl Hydrocarbon Receptor Nuclear
Translocator
BAFF : B cell Activating Factor
BALT : Bronchial Associated Lymphoid
Tissue
BLAD : Bovine Leukocyte Adhesion
Deficiency
BMAP : Bovine Myeloid Antimicrobial
Peptide
BNBD : Bovine Neutrophile β-Defensine
boBP : Bovine Oligosaccharide Binding
Protein
BPI : Bactericidal Permeability Increasing
Protein
BVD : Bovine Virale Diarrhea
CAM : Complexe d'Attaque Membranaire
CARD : Caspase Recruitment Domain
CCS : Comptage Cellulaire Somatique
CD : Cluster of Differentiation
CDP : Common Dendritic Cell Progenitor
CILP : Common Innate Lymphocyte
Precursor
CLP : Common Lymphoid Progenitor
CMH : Complexe majeur d'histocompatibilité
CMP : Common Myeloid Progenitor
CR : Complement Receptor
CSF : Colony Stimulating Factor
CTLR : C-type Lectin Receptor
DAF : Decay-Accelerating Factor
DC : Dendritic Cell
DETC : Dendritic Epidermal T Cell
EBD : Enteric β-Defensine
EPF : Early Pregnancy Factor
FCγR : Récepteur γ pour la fraction
constante des immunoglobulines
FCεR : Récepteur ε pour la fraction
constante des immunoglobulines
FLT-3L : FMS Like Tyrosine Kinase 3 Ligand
FMDV : Foot and Mouth Disease Virus
GALT : Gut Associated Lymphoid Tissue
G-CSF : Granulocyte Colony Stimulating
Factor
GM-CSF : Granulocyte Macrophage Colony
Stimulating Factor
HMGB : Hight Mobility Group Box
ICAM : Intercellular Adhesion Molecule
ICOS : Inducible T Cell Co-Stimulator
IFN : Interferon
Ig : Immunoglobulin
IKK : IκB kinase
IL : Interleukine
ILC : Innate Lymphoid Cell
IPI : Individu Infecté Permanent
IRAK : IL-1 Receptor Associated Kinase
IRF : Interferon Regulatory Factor
JAK : Janus Kinase
KIRS : Killer-cell Immunoglobulin-like
Receptor
LAP : Lingual Antimicrobial Peptide
LBP : Lipopolysaccharid Binding Protein
LPS : Lipopolysaccharide
LRR : Leucin Rich Repeats

16
LTC : Leucotriène
LTi : Lymphoid Tissue Inducer
MALT : Mucosa Associated Lymphoid Tissue
MAPKKK : Mitogen Activated Protein Kinase
Kinase Kinase
MARCO : Macrophage Receptor with
Collagenous Structure
MASP : MBL-Associated Serine Protease
MBL : Mannose Binding Lectin
MBP : Mannose Binding Protein
MCP : Mast Cell Progenitor
MDP : Macrophage Dendritic Cell Progenitor
MIP : Macrophage Inflammatory Protein
MyD88 : Myeloid Differenciation primary
response gene 88
NADPH : Nicotinamid Adenin Dinucleotid
Phosphate
NEMO : NF-κB Essential Modulator
NET : Neutrophil Extracellular Trap
NFAT : Nuclear Factor of Activated T Cells
NF-κB : Nuclear Factor κ B
NGF : Nerve Growth Factor
NIF : Neutrophil Inhibitory Factor
NK : Natural Killer
NLR : NOD-like Receptor
NOD : Nucleotide Oligomerization Domain
PAF : Platelet Activated Factor
PAMP : Pathogen Associated Molecular
Pattern
PDGF-β : Platelet Derived Growth Factor
PGD : Prostaglandine
PKR : Protein Kinase R
PLZF : Promyelocytic leukaemia Zinc Finger
PNB : Polynucléaire Basophile
PNE : Polynucléaire Eosinophile
PNN : Polynucléaire Neutrophile
PRR : Pattern Recognition Receptor
RANTES : Regulated on Activation Normal T
cell Expressed and Secreted
RIP : Receptor Interacting Protein
RLR : RIG-like Receptor
RORγt : RAR-related Orphan Receptor γ
SAA : Serum Amyloid A
SAP : Serum Amyloid Component
SBD : Sheep β-Defensine
SCF : Stem Cell Factor
SMAP : Sheep Myeloid Antimicrobial Peptide
SP : Surfactant Protein
STAT : Signal Transducer and Activator of
Transcription
STING : Stimulator of IFN genes
TAK : TGF-β Activated kinase
TAP : Tracheal Antimicrobien Peptide
TCF1 : T Cell Lineage Factor 1
TCR : T Cell Receptor
TGF : Transforming Growth Factor
Th : Lymphocyte T helper
TIR : Toll / IL Receptor
TLR : Toll Like Receptor
TNF : Tumor Necrosis Factor
Tollip : Toll Interacting Protein
TRAM : TRIF Related Adaptator Molecule
TRIF : TIR Related adaptator protein
Inducing Interferon
TSLP : Thymic Stromal Lymphopoietin
uNK : uterine NK
VCAM : Vascular Cell Adhesion Molecule
WC : Workshop Cluster
β-HB : β-Hydroxybutyrate

17
Tout être vivant est soumis à des agressions, qu’elles soient physiques (comme les
rayonnements solaires), chimiques ou biologiques. L’organisme doit donc être en
mesure de se défendre contre une multitude d’agents susceptibles de lui nuire. L’être
vivant a mis en place depuis des millénaires plusieurs systèmes de défenses. Le
système immunitaire a été pendant longtemps divisé en deux systèmes, inné et
adaptatif, et bien que ce clivage soit pratique sur un plan pédagogique, il est moins
marqué d’un point de vue physiologique. Le premier système est le système inné,
longtemps considéré comme mineur parce que mal connu, mais ces dernières
décennies ont permis de rectifier le rôle majeur de ce système dans l’initiation de la
réponse immunitaire. Il s’agit d’un système non spécifique : son mode d’action est le
même quel que soit l’agent incriminé. Le second est le système adaptatif, un
système de défense spécifique puisqu’il répond de manière spécifique à chaque
agent. Les dernières avancées en immunologie nous ont fait comprendre que ces
systèmes sont en fait intimement liés et travaillent en synergie. Le système
immunitaire est très répandu puisqu’il est présent chez tous les métazoaires et
certains ne possèdent pas de système adaptatif : ils n’ont alors pour seule défense
que le système inné. Ceci montre l’importance de ce système qui fait encore l’objet
de découvertes. En effet, en 2010, des cellules lymphoïdes innées (ou ILCs pour
Innate Lymphoid Cell) ont été découvertes. Ces cellules partagent des
caractéristiques lymphoïdes mais sont démunies de tout marqueur de lignée et ont
des fonctions très variées, notamment dans l’immunité innée, mais aussi dans le
maintien de l’homéostasie muqueuse. Le système immunitaire, indispensable à la
survie de tout être vivant continue donc à livrer ses secrets.
Nous débuterons cette thèse en fournissant quelques rappels sur le système
immunitaire inné, avec l’exemple des mammifères domestiques, en décrivant ses
composants et son mode de fonctionnement. Dans une seconde partie, nous
étudierons les particularités de ce système immunitaire inné chez les ruminants,
domestiques et sauvages, en insistant sur l’immunité de la mamelle, puisque les
ruminants sont souvent utilisés dans nos sociétés pour leur lait et la santé de la
mamelle a alors un enjeu économique non négligeable. Enfin, dans une troisième
partie, nous découvrirons les nouvelles cellules de l’immunité innée, les cellules
lymphoïdes innées. Nous décrirons les différentes cellules appartenant à cette
famille, leur différenciation et leurs fonctions au sein de l’organisme, qu’elles soient
bénéfiques ou délétères.

18
L’immunité innée est définie comme l’ensemble des mécanismes de résistance qui
permet la reconnaissance de différents agents pathogènes et la réponse pour faire
face à cette menace. Ils interviennent dans les phases précoces de la réponse de
l’hôte à l’infection. Cette immunité est constamment présente chez tous les individus
mais n’augmente pas avec l’exposition répétée à un pathogène donné.
Le système immunitaire inné constitue la première ligne de défense de l’organisme
contre des menaces de différentes natures. Il joue principalement trois fonctions :
C’est la première réponse face à l’invasion des microbes : il prévient, contrôle ou
élimine l’infection de l’hôte par de nombreux agents pathogènes (par la suite,
nommés « pathogènes »). S’il échoue, le système immunitaire adaptatif, plus
performant car plus ciblé, entre en jeu ;
Il reconnaît les produits de dommage et de mort cellulaires. Il doit éliminer ces
cellules et initier les réparations tissulaires ;
Il initie la réponse adaptative et peut l’influencer pour la rendre la plus optimale
possible face au pathogène engagé dans l’infection.
La plus importante avancée en immunologie ces 15 dernières années a été la
redécouverte de l’immunité innée, initialement mise en évidence grâce à Metchnikoff
au XIXème siècle (co-lauréat du prix Nobel en 1908 pour sa découverte de la
phagocytose). D’autres composants innés ont été découverts de façon fortuite,
comme le lysozyme par Fleming en 1932. Jusqu’en 1967, il n’était pas certain que
les cellules présentatrices d’antigènes soient nécessaires pour la présentation
d’antigène. Au milieu des années 1970, les cellules dendritiques ont été découvertes,
puis la découverte des cellules Natural Killer (par la suite, simplement notées NK) en
1975 constitue l’un des premiers lymphocytes innés mis en évidence. La nouvelle
ère de recherche du système immunitaire inné débute en 1989, grâce à Janeway,
qui découvre les PAMPs (pour Pathogen Associated Molecular Pattern) décris page
80. Depuis, les découvertes s’enchaînent : les composants du système du
complément, les lectines cellulaires de surface, les collectines comme les protéines
du surfactant, les protéines fixant le mannose, les récepteurs éboueurs et les
pentraxines. En 1994, un nouveau modèle de reconnaissance est découvert par
Matzinger : les cellules présentatrices d’antigènes peuvent être activées par des
signaux de danger libérés par les cellules endommagées (Paul, 2013). Enfin, en
2010, les cellules lymphoïdes innées (ILC) ont été découvertes, avec les LTi (pour

19
Lymphoid Tissu Inducer) puis les « natural helper » et nuocytes. Très rapidement, le
rapprochement est fait entre ces ILCs et les cellules T helper.
Le bras sensitif de ce système inné est constitué par toutes les cellules myéloïdes et
lymphoïdes qui expriment des récepteurs capables de détecter les pathogènes ou
des signaux de danger émis par l’organisme. D’autres cellules non immunes sont
également équipées de ces récepteurs, comme les cellules épithéliales. Les cellules
communiquent entre elles par des contacts cellule-cellule ou plus souvent par
l’intermédiaire de médiateurs moléculaires appelés cytokines. Enfin, le bras effecteur
de ce système est l’inflammation, qui quand elle est hors de contrôle peut causer des
dommages à l’hôte.
Nous allons tout d’abord étudier les différents acteurs de cette immunité innée (qu’ils
soient physiques, cellulaires, humoraux ou chimiques). Par défaut, l’étude est
réalisée chez l’homme, quand l’espèce concernée n’est pas mentionnée. Nous
verrons comment ce système détecte les pathogènes avant de rappeler les
différentes étapes de l’inflammation et le mode de communication employé par ce
système : les chimiokines, notamment pour activer le système adaptif. Enfin, nous
discuterons de la mémoire innée, découverte qui estompe la limite entre les
systèmes inné et adaptatif.
I. Les différents niveaux de l’immunité innée
Le système immunitaire regroupe des barrières physiques (formées par les
épithéliums), des cellules, des composants humoraux qui regroupent les produits de
sécrétions de ces cellules, le système du complément, des protéines et enzymes
antibactériennes et enfin, des composés chimiques. Cette diversité de moyens mise
en œuvre permet de cibler une grande panoplie de pathogènes et ces procédés sont
le plus souvent complémentaires mais aussi redondants.
A- Les barrières physiques
Les barrières anatomiques constituent la première ligne de défense de l’organisme.
Leur importance est mise en évidence par l’extrême susceptibilité des grands brulés
aux infections. Ces barrières physiques, constituées par les épithéliums de
recouvrement de la peau et des muqueuses sont complétées par des barrières
fonctionnelles comme les flores commensales, le mucus et d’autres sécrétions
antimicrobiennes, fournies par cet épithélium (Turvey & Broide, 2010).

20
Les épithéliums de recouvrement 1)
L’organisme est séparé du milieu extérieur par un épithélium de recouvrement. La
peau en est un exemple et sa différenciation est entièrement tournée vers la
protection contre l’extérieur. Cependant, tous les tractus digestif, respiratoire, génital
et urinaire sont également en contact direct avec le milieu extérieur. La particularité
des muqueuses est que cette séparation n’est réalisée qu’avec une seule couche
cellulaire, compatible avec les échanges inhérents à ces différents systèmes.
La peau constitue donc une barrière qui prévient l’entrée des microorganismes. La
kératinisation des cellules et surtout leur renouvellement constant accompagné de la
desquamation sont des moyens mis en œuvre pour empêcher les microorganismes
de pénétrer dans l’organisme. Les acides gras présents dans le sébum limitent
également la croissance bactérienne.
L’épithélium possède plusieurs moyens pour empêcher les microorganismes de
pénétrer dans les tissus sous-jacents. Ces moyens sont regroupés dans le Tableau I
ci-dessous. Les structures spécialisées comme l’appareil muco-ciliaire pulmonaire
permettent l’évacuation des microbes.
Tableau I : Différents obstacles de l’épithélium pour empêcher la colonisation des tissus par les pathogènes (D’après Janeway, Murphy, Travers, & Walport,
2009).
a. La structure de l’épithélium
La surface muqueuse respiratoire joue un rôle critique dans la détection et
l’élimination des pathogènes inhalés et les cellules épithéliales constituent la
première ligne de défense en fournissant une barrière physique mais aussi en
initiant, régulant et limitant ensuite la réponse immune innée et adaptative (Y. Li et
al., 2014).
Peau Intestin Poumon Œil / Nez
Mécanique
Cellules épithéliales liées par les jonctions serrées
Flux longitudinal d’air ou de
liquide
Déplacement
du mucus par
les cils
vibratoires
Larmes et cils
nasaux
Chimique Acides gras
pH bas
Enzymes
(pepsine)
Enzymes dans
les larmes
(lysozymes)
Peptides antibactériens
Microbiologique Flore normale

21
Le système immunitaire intestinal muqueux fait face à un défi unique : il est
constamment exposé à des antigènes étrangers, des bactéries commensales et
pathogènes, dont il est séparé par une seule couche de cellules épithéliales. Plutôt
que de créer une barrière imperméable, le principe de ce système est d’autoriser des
interactions contrôlées entre les microbes, les antigènes dérivés de la nourriture et
les cellules immunes sous-jacentes. De telles interactions sont permises par des
cellules spécialisées, représentées par les cellules M et les cellules dendritiques
transépithéliales. Elles sont nécessaires pour maintenir l’homéostasie tissulaire. Il est
maintenant admis que les cellules épithéliales intestinales produisent des cytokines
et des facteurs de croissance qui modifient la différenciation et la réponse du
système immun sous-jacent. Le maintien de l’homéostasie intestinale sera évoqué
page 100 (Sanos, Vonarbourg, Mortha, & Diefenbach, 2011).
Nous détaillerons la structure de l’épithélium intestinal (Figure 1, ci-dessous). Cette
muqueuse est la plus grande (environ 400 m² chez l’homme) et est particulièrement
adaptée à la colonisation par des bactéries commensales. Les autres épithéliums
sont construits sur le même modèle, avec des particularités propres à la fonction
primaire de l’organe dans lequel cet épithélium est localisé (comme par exemple les
pneumocytes à la place des entérocytes dans les poumons).
Figure 1 : Structures cellulaires de l’intestin grêle et du gros intestin. D’après
(McDermott & Huffnagle, 2013) et (Peterson & Artis, 2014).
IEL : lymphocyte intraépithélial.
La muqueuse de l’intestin grêle est construite avec des villosités et des cryptes. Ces
extensions permettent le prélèvement des nutriments et d’eau. Le principal type
cellulaire rencontré est l’entérocyte ou cellule épithéliale intestinale, recouverte de
microvillosités à la surface apicale et finement connectée à ses voisins par des
jonctions serrées grâce à des protéines d’adhésion. Ces jonctions serrées
connectent des cellules épithéliales adjacentes et sont liées au réseau d’actine et de
myosine cytoplasmique qui régule la perméabilité intestinale. Leur intégrité et donc la

22
perméabilité transépithéliale est régulée par des signaux dérivés des bactéries
commensales. Les jonctions serrées forment, avec les jonctions adhérentes et les
desmosomes, le complexe de jonction apicale intercellulaire. La surface épithéliale
est entièrement renouvelée tous les 2 à 5 jours. Ce turnover rapide participe
indirectement aux défenses de l’organisme en empêchant les microorganismes qui
ont réussi à pénétrer dans les cellules épithéliales de rentrer plus profondément dans
les tissus : ils sont éliminés en même temps que les cellules. Mais ce turnover est un
challenge supplémentaire pour le maintien de la continuité épithéliale. Les
entérocytes possèdent une fonction immunorégulatrice fondamentale qui influence le
développement et l’homéostasie des cellules immunes muqueuses. Enfin, les
cellules épithéliales transportent des immunoglobulines sécrétoires à travers la
barrière épithéliale. Après leur production par les plasmocytes (cellules B
complètement différenciées qui sécrètent de grandes quantités d’anticorps) de la
lamina propria, les complexes d’immunoglobulines de classe A (IgA) sont fixés par
leur récepteur à immunoglobuline sur la membrane basolatérale et sont transportés
activement vers la lumière intestinale (Geremia, Biancheri, Allan, Corazza, & Di
Sabatino, 2013; Lotz, Ménard, & Hornef, 2007; Peterson & Artis, 2014) .
Les cellules de Goblet sont dispersées sur toute la surface épithéliale et produisent
de grandes quantités de mucus. Cette production est régulée par les bactéries
commensales. Elles sont aussi capables de transporter des antigènes à travers la
couche épithéliale de façon active (Peterson & Artis, 2014).
Proches du fond des cryptes, à proximité des cellules souches, se trouvent des
cellules granuleuses, appelées cellules de Paneth, restreintes à l’intestin grêle. Ces
cellules sécrètent des peptides antimicrobiens comme les défensines, les
cathélicidines et le lysozyme, avec une demi-vie de 20 jours. Elles produisent aussi
des médiateurs inflammatoires comme le Tumor Necrosis Factor (ou TNF) et l’oxyde
nitrique et peuvent répondre aux stimuli inflammatoires (Cua & Tato, 2010; Lotz et
al., 2007).
La lamina propria décrit le tissu connectif sub-épithélial peuplé par des cellules
stromales et des cellules du système immun adaptatif formant le GALT (tissu
lymphoïde associé aux intestins) (Lotz et al., 2007; Peterson & Artis, 2014).
Les cellules entéroendocrines constituent un lien entre les systèmes
neuroendocrines entérique et central grâce à la sécrétion de nombreuses hormones
régulant la fonction digestive (Peterson & Artis, 2014).
Le prélèvement d’antigènes a lieu au niveau des plaques de Peyer. Ce sont des
agrégats lymphoïdes dans la sous-muqueuse de l’intestin grêle qui contiennent des
cellules B et T ainsi que des cellules dendritiques. Ces plaques sont des adaptations
spécialisées en conflit avec le concept de séparation complète entre les cellules
immunes de l’hôte et les microorganismes. Ces plaques sont recouvertes par des
cellules M (pour Microfold) qui ne possèdent pas de microvillosités mais une activité
de transcytose. Elles sont spécialisées dans le prélèvement spécifique ou non

23
d’antigènes dans la lumière et de microorganismes intacts pour les présenter au
système immun sous-jacent. Après la présentation d’antigènes, les lymphocytes des
plaques de Peyer se différencient en lymphocytes effecteurs et les cellules B
produisent des IgA, libérées dans la lumière intestinale, assurant un rôle dans les
défenses antibactériennes de l’hôte. Les cellules dendritiques échantillonnent des
antigènes microbiens grâce à leurs extensions cellulaires, les dendrites (Lotz et al.,
2007; Peterson & Artis, 2014).
L’ensemble des cellules est continuellement renouvelé par les cellules souches
épithéliales, pluripotentes, situées au fond des cryptes.
Nous verrons que des phagocytes mononucléaires subépithéliaux sont capables de
prélever des antigènes grâce à leurs dendrites transépithéliales.
Dans le poumon aussi, les cellules épithéliales sont la première ligne de défense
contre les pathogènes. Elles produisent un large éventail de récepteurs et de
composants antimicrobiens en plus de leur rôle physique de barrière. Par exemple,
lors de tuberculose, bien que les macrophages pulmonaires soient connus pour être
la cible des pathogènes microbiens comme Mycobacterium tuberculosis, les cellules
épithéliales pulmonaires peuvent, elles aussi, être atteintes par ces bactéries. La
voie Wnt est une voie de signalisation qui joue un rôle pivot dans la régulation du
destin cellulaire et dans l’organogénèse. La protéine ligand Wnt se fixe sur un
récepteur Fzd (frizzled) qui, après une cascade intracellulaire, mène à l’accumulation
de β-caténine dans le cytosol puis elle migre dans le noyau où elle interagit avec des
facteurs de transcription pour l’expression de gènes cibles comme la cycline D1 et
l’axine 2, impliquées dans la mise en place de l’inflammation. Cette signalisation a
lieu dans les cellules épithéliales pulmonaires et est impliquée dans le recrutement
des macrophages vers le site infectieux. Cependant l’activité Wnt/β-caténine peut
réguler négativement les réponses immunes comme l’activité des lymphocytes T
régulateurs (Treg) et la réponse tolérante des cellules dendritiques. Dans l’épithélium
pulmonaire, elle est liée à la fibrose pulmonaire et l’inflammation : son action pro- ou
anti-inflammatoire serait dépendante du contexte et des stimuli. La voie du Wnt/β-
caténine est bénéfique lors d’infection tuberculeuse puisqu’elle limite une
inflammation excessive. La voie de signalisation du Wnt est reliée à celle des TLR
(pour Toll Like Receptor, étudiés page 83) dans différents types de cellules. En effet,
elle est capable d’atténuer leur signalement et de permettre une réponse
inflammatoire dans les cellules épithéliales alvéolaires. Elle sert à maintenir un
équilibre entre la réponse inflammatoire et l’homéostasie tissulaire (Y. Li et al., 2014).
b. Les formations lymphoïdes associées aux muqueuses
L’organisme contient, outre la masse de tissu lymphoïde périphérique encapsulé
dans les nœuds lymphatiques et la rate, une quantité importante de tissu lymphoïde
non encapsulé situé dans les parois du tube digestif et des appareils respiratoire et
urogénital. Ce tissu constitue les tissus lymphoïdes associés aux muqueuses ou

24
MALT (pour Mucosa Associated Lymphoid Tissue). Il prend la forme d’infiltrats diffus
ou de nodules bien individualisés. Il s’agit d’un terme générique regroupant toutes les
muqueuses, mais le terme de MALT englobe :
Les formations lymphoïdes associées à l’appareil digestif ou GALT qui
comprennent les amygdales palatines, linguale et pharyngée, les follicules de
la muqueuse de l’œsophage, les plaques de Peyer de l’intestin grêle, les
follicules lymphoïdes isolés de l’intestin grêle et du colon, de l’appendice et
d’un très grand nombre de lymphocytes et plasmocytes dispersés dans le
chorion intestinal. L’anneau amygdalien est un tissu lymphoïde réparti en 4
groupes dont le plus volumineux est constitué par les amygdales palatines.
Celles-ci sont creusées de 12 à 15 cryptes profondes, revêtues d’un
épithélium pavimenteux pluristratifié, fréquemment occupées par des
bouchons de lymphocytes, de bactéries et de débris épithéliaux qui peuvent
se calcifier. Les plaques de Peyer contiennent des cellules B et T, appartenant
au système adaptatif et des cellules dendritiques. Ces plaques sont
uniquement présentes au niveau de l’intestin grêle alors que les follicules
lymphoïdes isolés sont présents dans l’intestin grêle et le colon. Les deux
structures contiennent des cellules M mais les follicules lymphoïdes isolés
contiennent surtout des cellules B et ne se développent qu’après la naissance.
Ces GALT sont schématisés sur la Figure 2, page 25, au niveau des
intestins ;
Les formations lymphoïdes associées aux bronches ou BALT (Bronchial
Associated Lymphoid Tissue) situées dans la muqueuse des grosses voies
aériennes. Elles présentent une analogie étroite avec les autres formes de
MALT mais sont plus petites. Il n’y a pas de vrais vaisseaux lymphatiques
afférents, comme dans l’intestin, mais des vaisseaux efférents drainent la
lymphe vers les nœuds lymphatiques régionaux.
Le tissu lymphoïde est disposé en follicules qui contiennent souvent des centres
germinatifs analogues à ceux des ganglions lymphatiques (Stevens & Lowe, 1993).
Nous verrons dans la 3ième partie que la mise en place de ces structures ainsi que
leur fonctionnement font appel aux ILCs.

25
Figure 2 : Représentation des GALT au niveau de l’intestin et de leur composition cellulaire (Pearson, Uhlig, & Powrie, 2012).
c. Le mucus produit par l’épithélium
La première barrière physique que rencontrent les bactéries intestinales et les
antigènes issus de la nourriture est la couche de mucus qui recouvre l’épithélium
intestinal. Le mucus est organisé en une couche inférieure stable et une couche
externe moins organisée. Il est produit par polymérisation de glycoprotéines
appelées mucines formant un gel et sécrétées par les cellules de Goblet. La couche
interne est ferme et dense, généralement stérile alors que la couche externe est plus
perméable et habitée par la flore commensale. Les microbes recouverts de mucus
adhèrent plus difficilement à l’épithélium. Dans le cas de l’appareil respiratoire, les
microorganismes sont entrainés avec le mucus et remontés grâce à l’appareil ciliaire.
Dans les intestins, le péristaltisme est un mécanisme indispensable à la progression
du bol alimentaire mais aussi à l’évacuation des bactéries. Des facteurs présents
dans cette couche de mucus agissent comme un signal qui stimule la réparation
épithéliale, la migration des cellules épithéliales et la résistance à l’apoptose
(Peterson & Artis, 2014).
La flore commensale 2)
La lumière intestinale est colonisée par une flore microbienne très variée et
dynamique. Sa composition est variable selon le segment intestinal considéré : le
jéjunum et l’iléum ont un grand nombre de bactéries Gram+ ; la valvule iléocæcale
constitue une barrière physique importante pour le cæcum et le colon qui sont

26
fortement colonisés par une flore Gram- anaérobie très diversifiée. L’âge de l’individu
a aussi une influence sur la composition de la flore. Cette flore a des rôles divers :
Elle permet de fermenter des substrats non digestibles pour l’hôte et de
produire des vitamines essentielles comme la vitamine K, B12, l’acide folique,
la biotine, ou des acides gras à chaîne courte ;
Elle prévient la colonisation et l’invasion par des bactéries pathogènes grâce à
la synthèse de substances bactéricides (bactériocines) et la compétition pour
les nutriments ;
Elle contribue au développement de l’intestin et à la différenciation cellulaire
de la surface épithéliale intestinale ;
Elle influence également le développement de l’appareil immun adaptatif des
intestins et elle contribue à la tolérance orale envers les antigènes de la
nourriture. Certains microbes stimulent l’inflammation et d’autres des
réponses anti-inflammatoires (Lotz et al., 2007; McDermott & Huffnagle, 2013;
Paul, 2013; Peterson & Artis, 2014).
Cependant, la colonisation bactérienne apporte avec elle le risque d’infection et
d’inflammation épithéliale ou la rupture de l’homéostasie des cellules immunes. Un
élément clé de la coexistence d’une communauté bactérienne commensale et des
cellules immunes muqueuses est le maintien de la ségrégation entre l’hôte et les
organismes, ce qui, à priori, n’est pas compatible avec l’existence des cellules M
(Peterson & Artis, 2014).
Le maintien de cette homéostasie est complexe et fait intervenir de nombreuses
cellules épithéliales et immunes. Il s’agit d’un équilibre entre des facteurs
inflammatoires qui limitent la flore et des signaux anti-inflammatoires qui limitent la
réponse de l’hôte. Cette régulation de l’homéostasie sera étudiée page 100.
B- Les cellules de l’immunité innée
Les cellules de l’immunité innée sont caractérisées par une réponse rapide à des
dangers étrangers en libérant une grande quantité de molécules effectrices qui
fournissent une protection immédiate contre ces microorganismes pathogènes. Elles
regroupent :
des cellules myéloïdes
o les granulocytes comme les polynucléaires neutrophiles (PNN), les
polynucléaires éosinophiles (PNE), les polynucléaires basophiles
(PNB)
o les mastocytes
o les monocytes-macrophages
o les cellules dendritiques

27
des cellules lymphoïdes comme les cellules γδ T, les cellules T helper (Th) 17,
et même les cellules T CD8+ (CD pour Cluster of Differentiation) qui
possèdent quelques fonctions innées (H Spits, 2011).
La Figure 3, ci-dessous, résume l’hématopoïèse et permet de bien cerner les
relations entre les différentes cellules.
Figure 3 : Hématopoïèse et lignées qui découlent de la cellule souche hématopoïétique (Williams, 2012).
Les cellules myéloïdes 1)
La lignée myéloïde se sépare de la lignée lymphoïde très précocement. Parmi ces
cellules, les thrombocytes et les érythrocytes ne concernent pas notre sujet et ne
seront donc pas traités. Nous aborderons d’abord les mastocytes, puis les
granulocytes issus du myéloblaste.
a. Les mastocytes
Les mastocytes sont des cellules peu granuleuses et sont donc difficilement
identifiables par les techniques courantes d’histochimie. Ce sont des cellules rondes
avec un noyau rond ou ovale mais non segmenté contrairement aux granulocytes.
Elles mesurent entre 7 et 20 μm de diamètre. Leur demi-vie est très longue

28
(plusieurs mois) et elles survivent à leur dégranulation. Les mastocytes matures,
complètement différenciés donc incapables de se diviser, ne sont pas présents dans
la circulation mais sont largement distribués dans l’organisme, très courants dans les
régions péri-vasculaires et muqueuses. Les mastocytes expriment des récepteurs
aux protéines du complément, aux neuropeptides et aux produits microbiens ainsi
que des récepteurs membranaires pour les immunoglobulines de type IgE (FCƐRI) et
IgG (FCγR). Quand les anticorps fixés sur les mastocytes ont capté leur antigène, un
signal intracellulaire permet de libérer les granulations cytoplasmiques dans l’espace
extracellulaire. Les granulations contiennent des médiateurs inflammatoires et
antimicrobiens. Ces granules fixent les colorants basiques grâce à leur
protéoglycanes, contiennent des cytokines, de l’histamine et provoquent des
changements dans les vaisseaux sanguins propres à l’inflammation (Abbas,
Lichtman, & Pillai, 2012; Dahlin & Hallgren, 2014; Paul, 2013).
i. Origine des mastocytes
L’origine des mastocytes est restée mystérieuse pendant des décennies. Ces
cellules sont issues de la moelle osseuse mais la maturation depuis le Mast cell
progenitor (MCP) a lieu dans les tissus périphériques. Le MCP circule vers ces tissus
périphériques dans un état immature et exprime le récepteur kit et le récepteur pour
la fraction Fc des IgE, le FCƐRI (comme les mastocytes matures). Cette migration est
régulée et augmentée lors d’inflammation. Parmi les facteurs de transcription, sont
nécessaires, dans l’ordre, le SCF (Stem Cell Factor), le STAT5 (Signal Transducer
and Activator of Transcription), le GATA2 puis le GATA1. Pour la maturation, le NGF
(Nerve Growth Factor) et la neurotrophine interviennent. Par contre, le GM-CSF
(Granulocyte Macrophage Colony Stimulating Factor), l’IFN-γ (Interféron) et le TGF-β
(Transforming Growth Factor) inhibent le développement des mastocytes ; et enfin
l’interleukine 3 (IL-3) et l’IL-4 inhibent les stades terminaux. Les MCP sont
néanmoins présents dans les intestins, de façon indépendante de la flore
microbienne mais cette présence est permise par les intégrines α4β7 et les molécules
d’adhésion des vaisseaux muqueux (MAdCAM1). Des précurseurs des mastocytes
sont également présents dans le péritoine, cette fois-ci grâce aux intégrines αMβ2 et
αIIbβ3. Dans les poumons, la présence régulière des MCP est dépendante des
cellules T et du VCAM1. L’IL-9 active les mastocytes et aide leur croissance. Le T-
bet exprimé par les cellules dendritiques permet d’attirer et de concentrer les
mastocytes sur le site de l’inflammation (Dahlin & Hallgren, 2014; V Kumar &
Sharma, 2010).
ii. Fonction des mastocytes
Les mastocytes ont de nombreux rôles, plus ou moins bénéfiques pour l’organisme :
Ils sont impliqués dans la défense de l’organisme contre les parasites ;
Ils jouent un rôle pivot dans l’immunité innée en réponse aux bactéries Gram-
en synthétisant du TNF-α qui recrute les PNN. Sa libération est activée par la

29
reconnaissance d’une molécule présente sur les pili des bactéries Gram- par
le CD48 des mastocytes ;
Ils stimulent l’inflammation aiguë et chronique notamment par la libération de
nombreuses cytokines comme l’IL-3, l’IL-4, l’IL-13, l’IL-15, l’IL-6 et le TNF-α
(décrites plus bas dans le paragraphe « Le langage chimique utilisé : les
cytokines », page 117). Le TNF-α, l’IL-1, et des chimiokines comme le CCL20
produits en réponse à l’activation du TLR4 (Toll Like Receptor) attirent les
cellules dendritiques, alors que l’histamine favorise l’expression de molécules
du Complexe Majeur d’Histocompatibilité de type II (CMH II). Par contre
l’activation des mastocytes par le TLR2, favorise la libération de cytokines de
type 2 comme l’IL-4 qui orientent la réponse adaptative vers une réponse Th2
et supprime la production d’IFN-γ et de TNF-α par les NK. Les mastocytes
libèrent de grandes quantités d’IL-13 et peu d’IL-4 ;
Ils sont responsables de l’hypersensibilité et des réactions inflammatoires
allergiques de type I à cause de la libération de cytokines actives (TNF-α, IL-1,
IL-6 et IL-10) de chimiokines (CCL-1, CCL-2, CCL-3, CXCL-1 et CXCL-2), des
médiateurs lipidiques (prostaglandines et leucotriènes) et de nombreuses
protéases (tryptase, chymase, cathepsine) ;
Ils modulent la réponse immunitaire en stimulant les cellules T et peuvent
ainsi agir sur l’intensité et sur la durée de l’inflammation ;
Ils présentent les antigènes aux cellules T via leurs molécules de CMH I et ils
induisent alors l’expansion clonale spécifique des cellules spécifiques de
l’antigène incriminé et peuvent polariser la réponse vers une réponse Th1 ou
Th2. Cependant, l’expression des molécules de CMH I n’est pas constitutive
mais induite par le TNF-α, l’IFN-γ et le LPS (lipopolysaccharide) ;
Ils ont récemment été identifiés comme des acteurs de la réparation tissulaire
et de la fibrose grâce à leur capacité de phagocytose pour nettoyer les lieux
de l’infection (Douglas & Jane Wardrop, 2010; Guo, Junttila, & Paul, 2012; V
Kumar & Sharma, 2010; Malaviya, Navara, & Uckun, 2001; Moretta et al.,
2005).
La Janus kinase 3 ou JAK3 est une tyrosine kinase intracellulaire qui intervient dans
le nettoyage bactérien par les mastocytes et le recrutement des PNN en régulant la
libération de TNF-α. Elle joue un rôle central dans l’initiation de la voie de
signalisation permettant la production de cytokines en activant les protéines STAT
via la phosphorylation de la tyrosine. JAK3 est ainsi un régulateur clé de la réponse
des mastocytes contre Escherichia coli (Malaviya et al., 2001).
Le rôle essentiel des mastocytes dans le développement de la réponse immune
innée effectrice est à mettre en relation avec leur localisation au niveau des
interfaces entre l’hôte et l’environnement, à proximité des vaisseaux sanguins et des
vaisseaux lymphatiques. En effet, ce sont d’excellentes cellules sentinelles puisqu’ils
sont capables d’activer les cellules T. Les mastocytes agissent sur les neurones via
l’histamine, sur les muscles lisses par les eicosanoïdes et augmentent la sécrétion

30
de mucus par les cellules épithéliales ce qui aide à immobiliser les pathogènes et les
éliminer dans les tractus digestif, respiratoire et urinaire (V Kumar & Sharma, 2010).
La sécrétion de nombreux facteurs par les mastocytes est activée par les IgE et leurs
antigènes, une anaphylatoxine, une hormone ou une neurohormone. La TSLP (pour
Thymic Stromal Lymphopoietin) active aussi les mastocytes en présence d’IL-1 et de
TNF. Une fois activés, ils sécrètent de nombreux médiateurs pro-inflammatoires et
vasoactifs. Ces molécules incluent l’histamine, la sérotonine, le TNF, les kinines, les
protéases stockés dans des granules. Les leucotriènes, les prostaglandines et le
PAF (pour Platelet Activated Factor) sont synthétisés lors de l’activation des
mastocytes. En présence de CSF, les mastocytes produisent essentiellement des
cytokines pro-inflammatoires alors qu’en présence de CSF et d’IL-4, ils produisent
principalement des cytokines de type Th2. Ils jouent donc un rôle important dans
l’immunité innée et acquise, lors d’infection bactérienne mais aussi lors d’auto-
immunité. Ils sont importants pour la maturation des cellules Th17 et sont reconnus
comme des cellules clés des désordres auto-immuns. Contrairement aux situations
d’allergie, les mastocytes dégranulent rarement lors des processus inflammatoires.
Le seul moyen d’expliquer l’implication des mastocytes dans des processus non
allergiques serait d’envisager une sécrétion de médiateurs sélective ou différentielle
sans dégranulation. Cette capacité est possible grâce à plusieurs mécanismes :
Les mastocytes peuvent sécréter le contenu d’un seul granule ;
Ils peuvent sécréter le contenu de certains granules grâce à des altérations
d’ultrastructure de leur noyau granuleux électron dense, ce qui indique une
sécrétion sans dégranulation ;
Les mastocytes subissent la libération sélective de médiateurs spécifiques
comme la sérotonine sans histamine. Ceci est possible par la séquestration
depuis les granules sécrétoires dans des vésicules contenant des protéines
de fixation de haute affinité de la sérotonine. La libération sélective
d’eicosanoïdes a aussi été observée. La libération sélective d’IL-6 est possible
en réponse au LPS bactérien (Theoharides et al., 2012).
La dégranulation nécessite l’exocytose des granules d’une façon calcium
dépendante mais la translocation des granules vers la surface ne dépend pas du
calcium (Theoharides et al., 2012).
Les mastocytes ne sont pas les seules cellules à posséder des granules : d’autres
cellules myéloïdes sont reconnaissables par la présence de granules dans leur
cytoplasme : les granulocytes.
b. Les granulocytes
Les granulocytes sont des leucocytes avec des noyaux polylobés et des granules
cytoplasmiques. Trois types de polynucléaires existent selon le type de coloration qui
permet de les identifier.

31
i. Les polynucléaires neutrophiles
Les granulocytes neutrophiles sont des phagocytes polynucléaires spécialisés dans
la destruction des pathogènes grâce à un large éventail de composants. Ce sont les
leucocytes les plus abondants (entre 40 et 70% du nombre total de leucocytes, ou
99% des granulocytes). Le nombre de PNN dans le sang est environ de 5000/mm3
chez l’homme mais leur nombre peut être multiplié par 5 à 10 lors d’une infection.
Chez les bovins, le nombre moyen de PNN est de 600 à 4000 X 106/L (ou 600-
4000/mm3). Nous verrons en seconde partie, les particularités de ces cellules dans
cette espèce. Il existe un équilibre dynamique entre un pool marginal séquestré dans
la micro-vascularisation et les PNN circulants (Becker & Arcangioli, 2014; Beutler,
2003).
Les neutrophiles sont produits dans la moelle osseuse de façon continue et sont
issus de la même lignée que les phagocytes mononucléaires. La différenciation des
neutrophiles est un processus complexe gouverné par un changement massif de
l’expression des gènes. Le passage du stade progéniteur myéloïde commun (MCP)
vers le progéniteur commun aux granulocytes et aux macrophages est régi par un
programme transcriptionnel qui diminue progressivement l’expression des gènes
communs à toutes les lignées. Par contre, en fin de différenciation, le passage du
stade promyélocyte au le stade myélocyte et métamyélocyte puis enfin au stade
neutrophile intra médullaire est dirigé par un programme transcriptionnel qui stimule
l’acquisition de fonctions spécifiques cruciales pour la réponse immune. Leur
production est stimulée par le G-SCF (Granulocyte Colony Stimulating Factor), le
GM-CSF, l’IL-3 et l’IL-6. Les neutrophiles matures sont des cellules pleinement
différenciées, incapables de se diviser. Ce sont des cellules à durée de vie courte
puisqu’elles circulent dans le sang pendant 7 à 12 heures chez l’homme (jusqu’à 90
heures) pendant lesquelles elles peuvent migrer vers les sites d’infections où elles
fonctionnent quelques heures puis meurent. Si le neutrophile n’est pas appelé dans
les 6 heures, il entre en apoptose et est phagocyté par les macrophages résidant
dans la rate et le foie. Cette durée de vie est considérablement augmentée lors
d’inflammation aiguë puisque le G-CSF, le GM-CSF, l’IFN-γ, le TNF-α, et le LPS
inhibent cette apoptose (Abbas et al., 2012; Paul, 2013; Theilgaard-Mönch, Porse, &
Borregaard, 2006).
Les PNN sont des cellules sphériques de 12-15 μm de diamètre avec de petits
pseudopodes à leur surface, augmentant ainsi la surface membranaire, ce qui est
crucial pour leur fonction de phagocytose. Elles sont reconnaissables par la forme
particulière de leur noyau polymorphe avec 3 à 5 masses de chromatine connectées
par de fins filaments. (Voir Figure 4, page 32)

32
Figure 4 : Neutrophiles observés avec la coloration de Wright-Giemsa (Harvey, 2001).
A : Chien / B : Vache / C : Cheval. Grossissement non précisé
Le cytoplasme contient des granulations de quatre types dont les caractéristiques
sont détaillées dans le Tableau II, page 33 :
Les granules primaires, ou azurophiles, sont les plus nombreux, et
contiennent des enzymes. En microscopie électronique, ce sont des organites
très denses ;
Les granules secondaires, ou spécifiques, servent à la destruction
microbienne mais aussi à la migration des cellules. Ils sont de taille plus petite
et apparaissent clairs en microscopie électronique ;
Les lysosomes tertiaires ou granules gélatinases, contiennent des
lysozymes et des enzymes servant à l’extravasation et à la migration des
cellules. Ces granules sont présents chez certaines espèces comme les
bovins, ovins, caprins, canins, félins, équins, mais aussi chez l’homme, le rat
et le lapin ;
Enfin, des vésicules sécrétoires servent à l’adhésion cellulaire.
Lors de la libération des granules, le contenu peut être déversé dans des
phagosomes (granules primaires et secondaires) ou dans le milieu extracellulaire
(granules secondaires et tertiaires). La formation des granules azurophiles a lieu au
stade prémyélocyte, celle des granules spécifiques au stade myélocyte et les
granules gélatinases se forment dans les PNN intra-médullaires. Dans ces trois
types de granules, il y a plus de 280 protéines (kinases, phosphatases,
transporteurs, protéines structurelles, transducteurs de signaux, et enzymes)
(Douglas & Jane Wardrop, 2010; Theilgaard-Mönch et al., 2006).

33
Tableau II : Caractéristiques principales des différents granules des PNN. D’après (Paul, 2013).
Granules peroxydases positifs Granules peroxydase négatifs
Granules primaires ou azurophiles Granules secondaires ou
spécifique
Granules tertiaires ou
gelatinase Vésicules sécrétoires
Contenu
(protéines
anti-
microbiennes
)
Myéloperoxydase => catalyse la formation
d’acide hypochloreux
Lysozyme
α-défensines => efficace contre des bactéries,
fungi et certains virus enveloppés
BPI (Bactericidal Permeability Increasing
Protein) => tue les bactéries Gram-
Lactoferrine
NGAL (Neutrophil Gelatinase
Associated Lipocalin)
Pentraxine 3
Lysozyme
B12-binding protein
Haptoglobine
Pro-défensines
Gp91phox/p22phox
Gp91phox /p22phox
Lysozyme Gp91phox/ p22phox
Récepteurs Thrombospondine Laminine R
CD14
CD16
CD35
Fml-R
Molécules
d’adhésion
CD11b CD18
CD66
CD67
CD11b/CD18
CD67
CD11b/CD18
CD67
Protéases
Cathepsine G
Elastase neutrophilique
Protéinase 3
=> Dégradation des composants de la matrice
extracellulaire
Collagénase
Arginase 1
Gelatinase => métalloprotéine qui
détruit les tissus
Leucolysine
Protéinase 3
Leucolysine
Particularités Les premiers granules mis en place lors du
développement des PNN
Plus petits et moins denses. Ne
contient qu’un seul constituant
parmi ceux en gras.
Encore plus petits, plus légers et
exocytose plus facile. Réservoir
d’enzymes dégradant les matrices
et de récepteurs nécessaires lors
de l’extravasation et la diapédèse
des PNN.
Pas de substance
toxique mais de
nombreuses protéines
plasmatiques et des
récepteurs

34
L’extravasation est le recrutement des PNN durant l’inflammation. Le mécanisme
précis de ce processus sera vu dans le paragraphe « L’extravasation des cellules
effectrices » page 111.
Les PNN amplifient l’inflammation en sécrétant des facteurs pro-inflammatoires
comme des leucotriènes, des prostaglandines, des cytokines et des chimiokines
(Douglas & Jane Wardrop, 2010; Paul, 2013).
Les neutrophiles sont connus pour être les premières lignes de défenses contre les
pathogènes, principalement les bactéries et les champignons, mais aussi les virus.
Ils forment, avec les macrophages et les cellules dendritiques, les phagocytes
professionnels et utilisent plusieurs mécanismes effecteurs pour détruire les
pathogènes. Parmi les mécanismes bactéricides, se trouvent :
Le mécanisme oxygéno-dépendant qui forme des ions superoxyde (O2-), du
peroxyde d’oxygène, des singulets d’oxygène… Ce mécanisme s’appelle
l’explosion respiratoire (ou « oxidative burst ») et est définit par
l’augmentation du métabolisme oxydatif indépendant des mitochondries. Il
mène à la génération d’O2- grâce au complexe NADPH oxydase (Nicotinamid
Adenin Dinucleotid Phosphate), un système enzymatique spécifique aux
phagocytes dans ces circonstances. Ce complexe se phosphoryle et se fixe
avec le cytochrome sur la membrane plasmatique, formant l’enzyme active qui
produit le O2-, converti par la superoxyde dismutase en peroxyde d’hydrogène
(H2O2) puis en acide hypochlorique (HOCl) par la myéloperoxydase. Ce
dernier constitue l’arme majeure des PNN et agit en synergie avec des
protéines des granules pour tuer les pathogènes dans le phagolysosome.
L’O2- peut aussi réagir avec l’oxyde nitrique pour former d’autres oxydants
cytolytiques comme le peroxynitrite ;
Le mécanisme indépendant de l’oxygène qui consiste à relarguer dans la
vacuole phagocytaire des enzymes lytiques et des polypeptides
antimicrobiens stockés dans les granules intracellulaires ;
La NETose, piège extracellulaire neutrophilique (ou Neutrophil Extracellulaire
Trap) qui est un mécanisme utilisé par les PNN pour capturer et détruire les
microbes dans l’espace extracellulaire. Ce mécanisme fait intervenir de la
chromatine nucléaire ornée de peptides antimicrobiens et des enzymes, le
tout sans membrane ni marqueur cytosolique. Voir Figure 5, page 35. Ce
mécanisme de mort intervient lors de stimulation neutrophilique intense.
Apparemment, l’enveloppe nucléaire, les granules et les membranes
cellulaires se dissolvent progressivement, permettant au contenu nucléaire de
se mélanger et de se condenser dans le cytoplasme avant d’être relâché dans
l’espace extracellulaire. Le NET se fixe alors à différentes bactéries Gram+ et
Gram- comme Staphylococcus aureus, Salmonella typhimurium,
Streptococcus pneumoniae et des streptocoques du groupe A, ainsi que des
champignons comme Candida albicans. De la même façon que ce qui est

35
observé dans le phagolysosome, la forte concentration locale de peptides
antimicrobiens et d’enzymes est responsable de la destruction du pathogène
piégé par le NET. Il semblerait que la formation du NET implique un signal ou
une cascade par les réactifs oxygénés. L’absence de NET serait impliquée
dans la pathogénie de maladies granulomateuses chroniques (Paul, 2013;
Theilgaard-Mönch et al., 2006).
Figure 5 : Piège extracellulaire réalisé par les PNN (Delves, Martin, Burton, & Roitt, 2011).
A : Staphylococcus aureus / B : Salmonella typhimurium / C : Shigella flexneri. La barre représente 500 nm.
A cause de leur équipement microbicide très puissant, les PNN sont des cellules
nuisibles capables de causer des dégâts aux tissus environnants lors de
l’inflammation aiguë. C’est pourquoi, les PNN initient un programme apoptique qui
facilite la résolution de l’inflammation et prévient les dommages tissulaires causés
par la lyse des cellules nécrotiques qui pourrait causer la libération des protéines
effectrices cytotoxiques et des réactifs oxygénés dans l’environnement extracellulaire
(Paul, 2013).
Les PNN possèdent de nombreux récepteurs aux cytokines et chimiokines, à des
médiateurs pro-inflammatoires, pour l’endothélium et aux protéines de la matrice
tissulaire, aux opsonines ainsi que de nombreux récepteurs pour détecter les
pathogènes qui seront décrits dans le paragraphe « Les récepteurs », page 82 (Paul,
2013).
La phagocytose est un élément clé de la réponse des PNN (détaillée dans le chapitre
« La phagocytose » page 113) mais les PNN sont aussi d’excellents producteurs de
médiateurs inflammatoires. Ils produisent notamment :
des cytokines pro-inflammatoires (TNF-β, IL-1α et β, IL-6, IL-7, IL-9, IL-16, IL-
17A/F, IL-18)
des cytokines anti-inflammatoires (IL-4, IL-10, et TGF-β)
des cytokines immunorégulatrices (IFN-α, IFN-β, IFN-γ, IL-12, IL-23)
des facteurs de croissance (G-CSF, M-CSF, GM-CSF)

36
des facteurs angiogéniques et fibrogéniques (VEGF, FGF2, TGF-β, HGF)
des chimiokines (CXCL-1, MIG, CXCL-9, CXCL-10, CXCL-11, CCL-3, CCL-4,
CCL-17, CCL-18, CCL-19, CCL20) (Paul, 2013).
Les PNN répondent aux stimuli inflammatoires par un changement intensif de
l’expression de leurs gènes. Juste après la phagocytose, les PNN surexpriment des
médiateurs inflammatoires comme des chimiokines et des cytokines, critiques pour le
recrutement des macrophages, des cellules T et d’autres PNN et des médiateurs
pour la modulation de leur réponse inflammatoire. Après cette phase précoce pro-
inflammatoire, les neutrophiles surexpriment des chimiokines et cytokines qui
stimulent l’angiogenèse, la prolifération des kératinocytes et fibroblastes et l’induction
de protéines effectrices antimicrobiennes dans les kératinocytes. Enfin, ils initient
une réponse transcriptionnelle qui stimule leur apoptose et leur dégradation par les
macrophages. La surexpression des gènes pro-apoptiques se fait en parallèle de la
diminution des récepteurs pour les médiateurs inflammatoires. Voir Figure 6 ci-
dessous (Theilgaard-Mönch et al., 2006).
Figure 6 : Mécanismes de défense des PNN (Theilgaard-Mönch et al., 2006).
Diapédèse schématisée en haut. 1 : phagocytose, destruction et dégradation des microorganismes. 2 : Exocytose des protéines des granules. 3 : Réponse pro-
inflammatoire. Ces 3 premiers stades correspondent à la phase effectrice sensu-stricto. 4 : Apoptose et phagocytose par les macrophages.

37
ii. Les polynucléaires éosinophiles
Les éosinophiles sont des granulocytes sanguins qui possèdent des granulations
contenant des enzymes capables de détruire les membranes des bactéries, des
virus et des parasites mais elles peuvent aussi léser les tissus de l’hôte. Ce sont des
cellules granulées avec un noyau bilobé contenant une chromatine très condensée
et elles fixent fortement l’éosine. Voir Figure 7 ci-dessous. Elles mesurent entre 12
et 17 µm de diamètre et représentent entre 1 et 6% des leucocytes sanguins chez
l’homme et entre 0 et 20% chez les bovins (Abbas et al., 2012; Paul, 2013).
Figure 7 : PNE observés avec la coloration de Wright-Giemsa (Harvey, 2001). I : Chat / K : Chien / O : Cheval (les gros granules nombreux sont typiques de
l’espèce chevaline) / P : Vache (les nombreux petits granules sont spécifiques de l’espèce bovine).
Les PNE sont des cellules principalement issues de la moelle osseuse mais aussi du
thymus, de la rate et des poumons à partir du progéniteur myéloïde CD34+. Leur
production est régulée par les cellules Th2 et dure entre 2 et 6 jours. Leur
développement nécessite trois cytokines : le GM-CSF, l’IL-3 et l’IL-5. Les PNE
circulent ensuite dans le sang avec une durée de vie de 4 à 5 heures chez l’homme.
Quelques éosinophiles sont présents dans les tissus périphériques, notamment les
muqueuses respiratoire, gastro-intestinale, génitale et urinaire et leur nombre
augmente fortement en cas d’inflammation. Les éosinophiles sont activés par l’IL-3,
le GM-CSF, l’IL-5, les IgG et les IgA. L’IL-4 et l’IL-13 jouent un rôle central dans le
transfert des PNE vers les tissus muqueux. Ils migrent vers les sites inflammatoires
en réponse à la chimiokine éotaxine (CCL-11), synthétisée par les cellules Th2 et
expriment alors des intégrines qui permettent leur fixation puis leur extravasation

38
vers le milieu extravasculaire puis circulent entre les cellules vers le lieu de
l’inflammation de façon tout à fait similaire à ce qui a déjà été vu pour les PNN
(Abbas et al., 2012; Douglas & Jane Wardrop, 2010).
Comme les PNN, les PNE possèdent trois types de granules :
les granules primaires. Ils sont similaires à ceux des PNN et sont formés
précocement dans le développement des PNE. Ils contiennent la protéine
Crystal de Charcot-Leyden, protéine hydrophobique de rôle inconnu mais
spécifique des PNE ;
les granules secondaires ou spécifiques ou cristalloïdes. Ils contiennent des
protéines cytotoxiques. Il s’agit de la majorité des granules. Ces granules ont
une structure différente avec un noyau électron-dense et avec des protéines
cationiques. Ce sont essentiellement des protéines basiques (Myelin Basic
Protein), la protéine cationique E0 et la neurotoxine qui en dérive. La protéine
E0 est fortement cationique et sa toxicité découle de l’augmentation de la
perméabilité membranaire. Elle possède une activité contre les parasites ;
les petits granules denses. Ce sont de petits granules amorphes contenant de
l’aryl-sulfatase et de la phosphatase acide (Douglas & Jane Wardrop, 2010;
Paul, 2013).
Les PNE contiennent aussi des corps lipidiques qui sont des organites riches en
lipides non liés à des membranes, site majeur de synthèse des eicosanoïdes. Les
enzymes les plus importantes contenues dans les éosinophiles sont la protéine
basique majeure, la peroxydase éosinophilique, la protéine cationique éosinophilique
et la neurotoxine dérivée des éosinophiles (Douglas & Jane Wardrop, 2010; Paul,
2013).
Les PNE constituent la principale défense contre les parasites helminthiques en
sécrétant des médiateurs. Ils régulent aussi l’immunité en supprimant les réactions
d’hypersensibilité immédiate et sont capables de phagocyter des complexes immuns,
même s’ils sont moins efficaces dans ce rôle que les neutrophiles. Ils pourraient
aussi avoir un effet anti-tumoral (Douglas & Jane Wardrop, 2010).
Les PNE libèrent des médiateurs lipidiques comme des prostaglandines, du
thromboxane, LTC4 et du PAF, qui ont tous des activités pro-inflammatoires. Ils
produisent également de nombreuses cytokines comme le TGF-β, le GM-CSF, l’IL-3,
l’IL-4, l’IL-10, l’IL-12, l’IL-13, l’IL-16, l’IL-18, le TNF-α, le CXCL-8, CCL-5, SCF, NGF
et CCL-1, en partie préformées dans les granules et donc relâchées très rapidement
lors de la dégranulation (Paul, 2013).
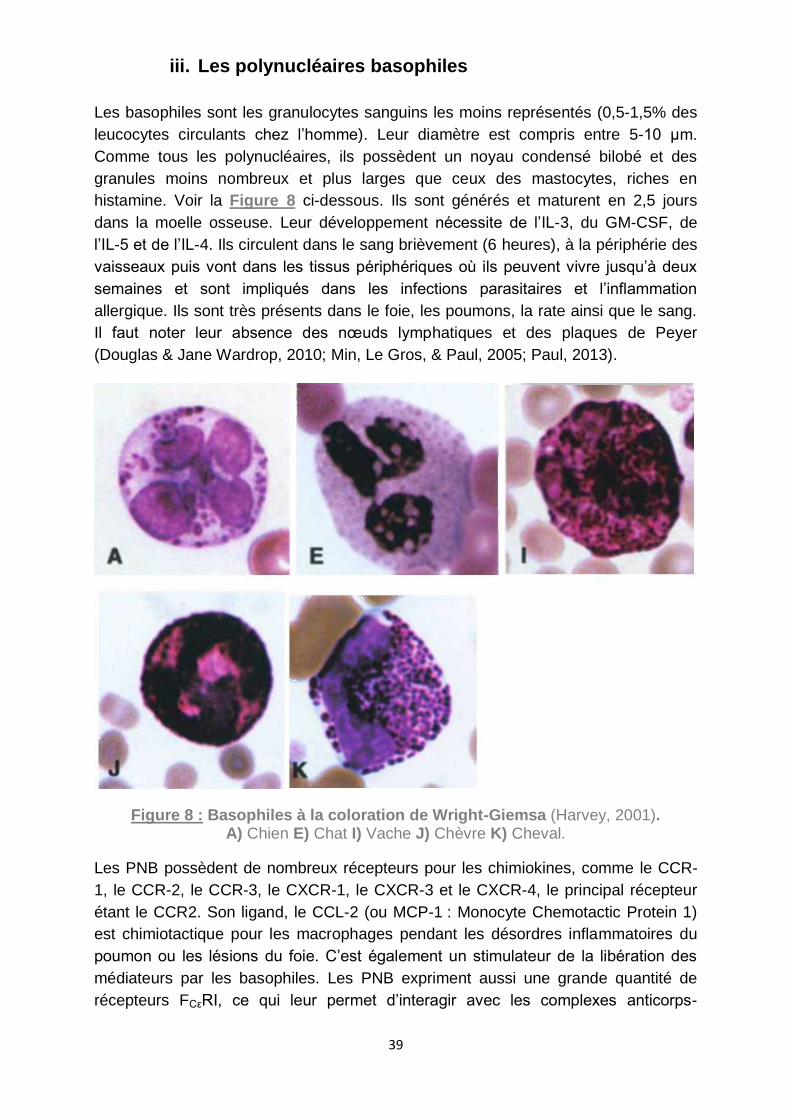
39
iii. Les polynucléaires basophiles
Les basophiles sont les granulocytes sanguins les moins représentés (0,5-1,5% des
leucocytes circulants chez l’homme). Leur diamètre est compris entre 5-10 μm.
Comme tous les polynucléaires, ils possèdent un noyau condensé bilobé et des
granules moins nombreux et plus larges que ceux des mastocytes, riches en
histamine. Voir la Figure 8 ci-dessous. Ils sont générés et maturent en 2,5 jours
dans la moelle osseuse. Leur développement nécessite de l’IL-3, du GM-CSF, de
l’IL-5 et de l’IL-4. Ils circulent dans le sang brièvement (6 heures), à la périphérie des
vaisseaux puis vont dans les tissus périphériques où ils peuvent vivre jusqu’à deux
semaines et sont impliqués dans les infections parasitaires et l’inflammation
allergique. Ils sont très présents dans le foie, les poumons, la rate ainsi que le sang.
Il faut noter leur absence des nœuds lymphatiques et des plaques de Peyer
(Douglas & Jane Wardrop, 2010; Min, Le Gros, & Paul, 2005; Paul, 2013).
Figure 8 : Basophiles à la coloration de Wright-Giemsa (Harvey, 2001). A) Chien E) Chat I) Vache J) Chèvre K) Cheval.
Les PNB possèdent de nombreux récepteurs pour les chimiokines, comme le CCR-
1, le CCR-2, le CCR-3, le CXCR-1, le CXCR-3 et le CXCR-4, le principal récepteur
étant le CCR2. Son ligand, le CCL-2 (ou MCP-1 : Monocyte Chemotactic Protein 1)
est chimiotactique pour les macrophages pendant les désordres inflammatoires du
poumon ou les lésions du foie. C’est également un stimulateur de la libération des
médiateurs par les basophiles. Les PNB expriment aussi une grande quantité de
récepteurs FCεRI, ce qui leur permet d’interagir avec les complexes anticorps-

40
antigènes. L’activation de ces récepteurs initie une cascade qui régule ensuite la
production et la libération de cytokines (Min et al., 2005).
Les basophiles interviennent dans les allergies, dans la dernière phase de
l’hypersensibilité immédiate. Ils ont également un rôle plus bénéfique dans la lutte
contre les helminthes où ils sont des chefs d’orchestre de la réponse immunitaire et
dans l’initiation de la réponse de type Th2 auprès des cellules T grâce à la
production très rapide et intense d’IL-4. Cette production est notamment initiée en
réponse à l’IL-3 produite par les lymphocytes T CD4+. L’IL-3 joue un rôle essentiel
dans la différenciation et la survie des PNB. Elle augmente leur production de
cytokines et favorise leur migration transendothéliale. Ce sont également des
producteurs de TNF-α (Douglas & Jane Wardrop, 2010; Neill & McKenzie, 2011).
Comme les mastocytes, les PNB possèdent de nombreux médiateurs dans leurs
granules :
des médiateurs préformés : histamine, protéases neutres des mastocytes
(tryptase, chymase…) stockés dans des granules cytoplasmiques, et relâchés
en quelques minutes ;
des médiateurs synthétisés comme le PAF, la PGD2 (prostaglandine) ;
des cytokines : IL-3, IL4, GM-CSF, IL-5 IL-6 IL-10, IL-13 (Paul, 2013).
c. Les monocytes - macrophages
Le système des phagocytes mononucléaires est constitué de cellules dont la fonction
primaire est la phagocytose. Ces cellules jouent un rôle central dans l’immunité innée
et adaptative (Abbas et al., 2012).
Dans le sang, la forme circulante est encore immature et s’appelle le monocyte. Ces
cellules restent 70 heures dans le sang chez les humains, 18 heures chez la souris
et 20 à 30 heures chez les bovins. Elles ont un diamètre compris entre 10 et 15 μm,
un noyau en forme de haricot et un cytoplasme contenant de fines granulations qui
sont des lysosomes, des vacuoles phagocytaires et des filaments squelettiques. Voir
Figure 9, page 41. Il existe deux types de monocytes. Le premier est appelé
inflammatoire car il est rapidement recruté du sang vers le site tissulaire
inflammatoire. Et le second peut être la source des macrophages résidents et de
certaines cellules dendritiques. Une fois que les monocytes entrent dans les tissus,
ils sont alors matures et sont appelés macrophages (Abbas et al., 2012).

41
Figure 9 : Monocytes à la coloration de Wright-Giemsa (Harvey, 2001). A) Chat B) Vache C) Cheval.
Les macrophages sont des phagocytes mononucléaires dérivés des monocytes
sanguins. Ils sont dispersés dans tout le corps et parfois dans le parenchyme de
certains organes. Il s’agit d’une population hétérogène, qui, selon le tissu hôte, prend
des noms différents :
les histiocytes,
les cellules de Küpffer (ou Kupffer ou Küpfer) dans le foie,
les macrophages alvéolaires des voies respiratoires,
la microglie (ou microgliocytes) du système nerveux central,
les ostéoclastes dans le tissu osseux.
Les macrophages ont des fonctions très variées :
Ils supervisent la réaction immunitaire grâce à l’élaboration de cytokines
chimiotactiques qui recrutent d’autres cellules comme les PNN sur le site de
l’inflammation ;
Ils peuvent engloutir et détruire les microbes ;
Ils ingèrent des cellules mortes ou apoptiques ;
Ils initient aussi la réponse adaptative en présentant les antigènes aux
lymphocytes T CD4+ via leur antigène CMH de classe II. Ils ne semblent pas
efficaces sur les cellules naïves mais attirent les cellules activées ;
Ils stimulent la réparation des tissus endommagés en favorisant l’angiogenèse
et la fibrose (Abbas et al., 2012; Beutler, 2003).
Les macrophages possèdent de nombreux récepteurs membranaires comme des
récepteurs pour la fraction Fc des immunoglobulines, pour le complément, pour les
intégrines, des TLR, des récepteurs au mannose et des récepteurs éboueurs
(Douglas & Jane Wardrop, 2010).
Des conditions inflammatoires affectent la différenciation des macrophages. Par
exemple, des macrophages exposés à l’IL-4 ou l’IL-13 ont un phénotype activé
caractérisé par une baisse de capacité à détruire les bactéries intracellulaires et une
hausse dans la participation à la résolution de l’infection. L’expression de certains
récepteurs de surface ou solubles est augmentée par des stimuli inflammatoires et
cela favorise la capacité à détruire les bactéries, cela altère les productions de

42
cytokines ou rend les macrophages réfractaires à d’autres stimulations (Bowdish,
Loffredo, Mukhopadhyay, Mantovani, & Gordon, 2007).
d. Les cellules dendritiques
Les cellules dendritiques sont des phagocytes mononucléaires très efficaces et
spécialisées dans la présentation d’antigènes aux cellules T naïves, ce qui fait de
ces cellules un lien entre l’immunité innée et l’immunité adaptative. Elles ont été
découvertes en 1973 par Ralph Steinman (co-lauréat du prix Nobel en 2011). Elles
sont capables d’initier une réponse immune primaire après avoir détecté des produits
microbiens. Elles jouent également un rôle dans l’induction de la tolérance
périphérique en agissant comme une police qui maintient la tolérance pour les
antigènes du soi. Enfin, elles régulent le type de réponse T et agissent donc comme
des chefs d’orchestre de la réponse immune adaptative (Clark et al., 2000; Liu, 2001;
Rowley & Fitch, 2012).
Les cellules dendritiques sont largement distribuées dans les tissus lymphoïdes, les
épithéliums muqueux et cutanés ainsi que dans les organes parenchymateux comme
le foie ou le poumon. Elles sont également présentes dans le derme, le cœur, ou
encore le rein (Abbas et al., 2012; Clark et al., 2000).
i. Origine des cellules dendritiques
Différentes voies possibles
Les cellules dendritiques sont issues de la moelle osseuse et circulent dans le sang
sous une forme immature. Elles migrent ensuite vers des tissus non lymphoïdes où
elles forment le réseau de cellules dendritiques interstitielles qui capturent les
antigènes puis migrent vers les zones T des nœuds lymphatiques drainant le
territoire. Dans le sang, se trouvent donc des précurseurs des cellules dendritiques
qui viennent de la moelle osseuse, la plupart sont des cellules immatures qui n’ont
pas encore rencontré d’antigène. Elles représentent 1% des leucocytes circulants
(Liu, 2001).
Les cellules dendritiques sont produites en continu à partir de cellules souches
hématopoïétiques de la moelle osseuse. Il existe plusieurs voies de différenciation ce
qui engendre les différents sous-types cellulaires décrits page 45. Les différentes
étapes de différenciation sont résumées dans la Figure 10, page 43.

43
Figure 10 : Différenciation des cellules dendritiques depuis la cellule souche hématopoïétique (HSC) (Kushwah & Hu, 2011).
CMP : Common Myeloid Progenitor / MDP : Macrophage-Dendritic Cell Progenitor / CLP : Common Lymphoid Progenitor /CDP : Common Dendritic Progenitor / DC :
Dendritic Cell
La voie la plus courante est la voie myéloïde, qui aboutit aux cellules dendritiques
conventionnelles. Ces dernières sont issues du Progéniteur Myéloïde Commun (ou
CMP sur la figure ci-dessus) qui se différencie ensuite en « Macrophage-Dendritic
Cell Progenitor » (MDP), précurseur commun aux macrophages, monocytes et
cellules dendritiques, défini par Lin-, CX3CR1+, CD11b-, CD115+, c-kit+, et CD135+. Il
se différencie ensuite probablement en un précurseur restreint aux cellules
dendritiques, appelé le « Common Dendritic Cell Progenitor » (CDP). Il s’agit du
dernier stade résidant dans la moelle osseuse. Le stade suivant, le pré-DC (pour
« pre-Dendritic Cell ») se trouve initialement dans la moelle osseuse puis dissémine
dans la rate et les nœuds lymphatiques via le sang. Ces pré-DC subissent une
prolifération et terminent leur différenciation en cellules dendritiques (Kushwah & Hu,
2011; Wu & Liu, 2007).
Une autre voie myéloïde possible passe par le stade monocyte : ces derniers
peuvent se différencier en cellule dendritique en présence de GM-CSF et d’IL-4 ou
suite à la phagocytose de bactéries. Ce processus a essentiellement lieu dans les
tissus sous-cutanés. Les précurseurs monocytes des cellules dendritiques (PréDC1)
expriment les antigènes myéloïdes CD11b, CD11c, CD13, CD14 et CD33 ainsi que
le récepteur au mannose. Elles expriment également le CD1a, CD1b, CD1c et CD1d.
Les cellules matures issues de ces précurseurs (les cellules dendritiques 1)

44
produisent de grandes quantités d’IL-12 et induisent donc une réponse de type Th1
et une réponse cytotoxique. Pendant plusieurs années, l’explication donnée à ce
phénomène était un terrain infectieux, mais en réalité, cette voie de différenciation a
lieu à l’état d’équilibre, et est amplifiée lors d’inflammation (Liu, 2001).
Une seconde voie de différenciation à partir du CDP est possible et fournit des
cellules dendritiques plasmacytoïdes. Le précurseur de ces cellules, le Pré-DC2
exprime des ARNm spécifiques aux lymphocytes comme le pré-Tα, l’Igλ-like14.1, et
le Spi-B. Ces cellules se différencient en cellules dendritiques plasmacytoïdes
immatures en présence d’IL-6 ou suite à une stimulation virale. Les cellules
dendritiques matures engendrées (les CD2) induisent une réponse de type Th2 et
inhibent la synthèse d’IL-10 par les cellules T tout en stimulant leur synthèse d’IFN-γ
et d’IL-10, aux propriétés antivirales (Liu, 2001).
Certaines cellules dendritiques ont une origine lymphoïde. C’est le cas notamment
des cellules dendritiques spléniques et thymiques. Il est important de noter que ces
cellules passent aussi par le stade pré-DC. Le phénotype de la cellule n’est pas le
reflet de sa lignée d’origine. Les cellules dendritiques dans les organes lymphoïdes
périphériques sont continuellement remplacées par des précurseurs sanguins et
celles de la rate et des nœuds lymphatiques peuvent subir un nombre limité de
divisions in situ et sont entièrement remplacées en 15 jours (Wu & Liu, 2007).
De nombreux facteurs influencent cette
différenciation
La voie lymphoïde est modulée par l’activation du TLR9 ou du TLR4 du CLP, ce qui
favorise la différenciation des cellules dendritiques. Le FLT-3L (pour (FMS Like
Tyrosine kinase 3 Ligand) joue un rôle dans le développement des cellules
dendritiques au niveau des tissus périphériques ainsi que dans leur homéostasie et
leur expansion. Il est particulièrement important pour le développement des cellules
dendritiques plasmacytoïdes et des cellules dendritiques CD8+ ou CD10+. Le CLP
peut donc donner des cellules dendritiques mais cette production n’a lieu que lors
d’inflammation ou d’infection. (Kushwah & Hu, 2011).
Les cellules dendritiques dérivées des monocytes ont besoin des cellules NK en
présence d’IFN-γ pour achever leur maturation. Les NK interviennent dans les stades
précoces de la différenciation des cellules dendritiques depuis les précurseurs
monocytes CD14+. Ils induisent la baisse de régulation du CD14 et la surexpression
du CD40, du CD80, du CD86, du DC-SIGN, et du DEC 205. Le processus requiert
un contact cellule-cellule entre les NK et les monocytes ainsi que des facteurs
solubles comme le GM-CSF et CD40L ou CD154 produit par les NK (Reschner,
Hubert, Delvenne, Boniver, & Jacobs, 2008).
Le facteur de transcription en doigt de zinc (ou « Zinc finger ») Ikaros joue un rôle
essentiel dans le développement des cellules dendritiques, qu’elles soient

45
conventionnelles ou plasmacytoïdes. En effet, il est nécessaire pour la génération du
progéniteur hématopoïétique précoce. RelB, un facteur de transcription membre de
la famille du NF-κB (Nuclear Factor κ B) est aussi important surtout pour les cellules
dendritiques conventionnelles CD8-. Enfin, les facteurs régulateurs de l’interféron,
IRF-1(Interferon Regulatory Factor), IRF-4 et IRF-8 sont importants pour les
différentes populations de cellules dendritiques. Id2, un autre facteur de transcription
« hélice-tour-hélice » inhibiteur est surexprimé pendant le développement des
cellules dendritiques CD8+ et des cellules de Langherans : il s’agit d’un inhibiteur du
développement des cellules plasmacytoïdes. De nombreuses cytokines sont aussi
impliquées dans ce développement. Le GM-CSF et Flt-3L sont tous les deux
indispensables pour la génération des cellules conventionnelles mais seul le Flt-3L
est critique pour les cellules plasmacytoïdes. Le TGF-β1 est nécessaire pour le
développement des cellules de Langherans (Wu & Liu, 2007).
D’autres facteurs de transcription sont spécifiques à chaque type de lignées et seront
détaillés lors de la description des sous-types de cellules dendritiques.
ii. Les différents types de cellules dendritiques
Nous avons vu qu’il existe plusieurs voies de différenciation pour les cellules
dendritiques et ces voies aboutissent à des sous-types cellulaires variés, résumés
dans la Figure 11, page 46.
Il a été décrit un groupe de cellules dendritiques exprimant des récepteurs NK,
nommées les IKDC (Interferon Producing Kill Dendritic Cell). Il s’est en fait avéré que
ces cellules sont des cellules NK et ne seront donc pas traitées dans ce paragraphe
(Reschner et al., 2008; Wu & Liu, 2007).
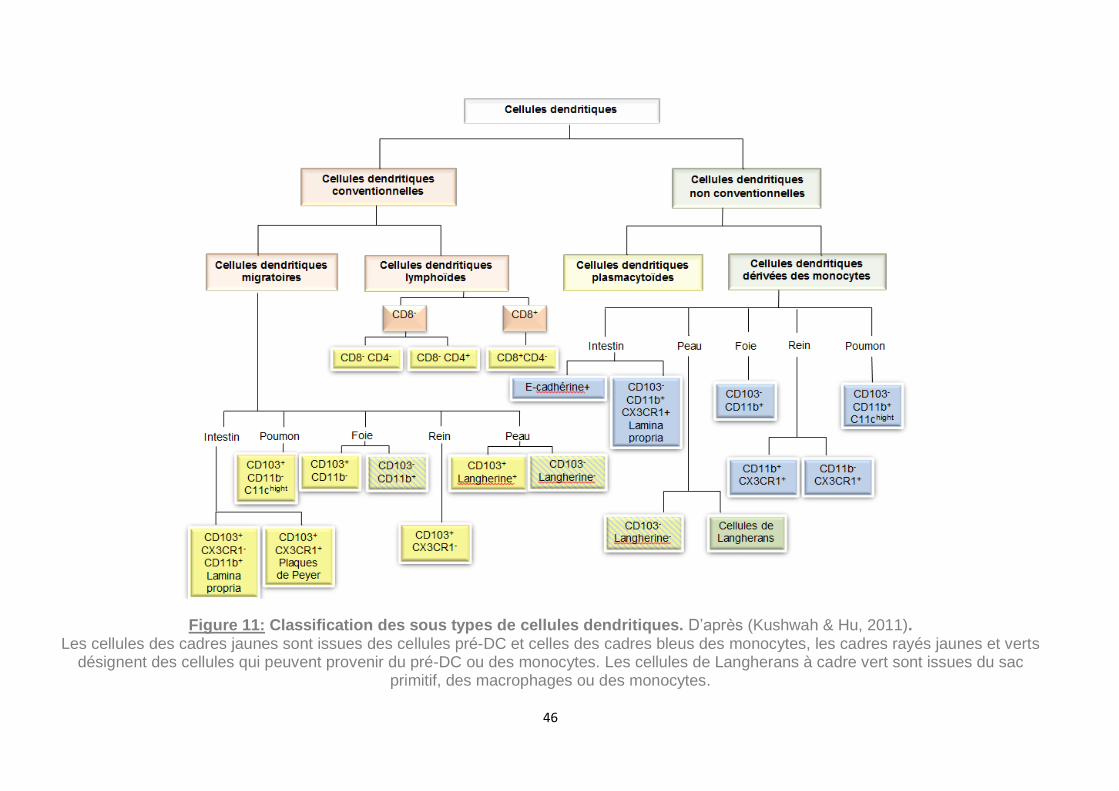
46
Figure 11: Classification des sous types de cellules dendritiques. D’après (Kushwah & Hu, 2011). Les cellules des cadres jaunes sont issues des cellules pré-DC et celles des cadres bleus des monocytes, les cadres rayés jaunes et verts
désignent des cellules qui peuvent provenir du pré-DC ou des monocytes. Les cellules de Langherans à cadre vert sont issues du sac primitif, des macrophages ou des monocytes.

47
Les cellules dendritiques conventionnelles
Les cellules dendritiques conventionnelles représentent la plupart des cellules
dendritiques. Elles sont localisées dans les tissus périphériques sous une forme
immature. La fin de la maturation a lieu lorsqu’elles rencontrent un antigène. Elles
migrent alors dans les nœuds lymphatiques pour la présentation antigénique et
servent de lien entre les deux immunités. Ce sont ainsi les principales cellules
activatrices des lymphocytes T. Leur demi vie est variable : courte dans la rate, les
nœuds lymphatiques, le foie et les reins (5-7 jours), elle peut aller jusqu’à 25 jours
dans les poumons (Abbas et al., 2012; Beijer, Kraal, & den Haan, 2013; Paul, 2013).
Les cellules dendritiques conventionnelles comprennent des cellules dendritiques
dérivées du pré-DC et elles peuvent ensuite être subdivisées en :
cellules dendritiques migratoires qui peuvent migrer des tissus
périphériques vers les organes lymphoïdes. Elles se situent dans la peau, les
poumons, le tractus intestinal, le foie et les reins (Kushwah & Hu, 2011) ;
Les cellules de Langherans et les cellules dendritiques dermiques sont
présentes dans la peau. Leur demi-vie est de plus de 21 jours pour les
premières et de 12 jours pour les secondes. Les cellules dendritiques
dermiques sont CD10+ langherine+ et sont les plus efficaces dans le
traitement des antigènes viraux via le CMH I, probablement par
présentation croisée. Ce type cellulaire joue aussi un rôle clé dans
l’initiation de la réponse Th1 et Th17 lors de sensibilisation sous
cutanée,
Dans le poumon, il y a plusieurs sous types de cellules dendritiques.
Les CD103+ CD11chigh CD11b- dans le réseau intraépithélial migrent
vers le nœud lymphatique drainant la zone et elles produisent de l’IL-
12. Ce sont les principaux initiateurs de la réponse T CD8+. Les CD103-
CD11chigh CD11b+ sont dans la lamina propria des voies respiratoires
(Kushwah & Hu, 2011; Paul, 2013),
Dans le tractus intestinal, les cellules dendritiques sont présentes dans
les plaques de Peyer, la lamina propria et les nœuds lymphatiques
mésentériques : (Kushwah & Hu, 2011; Paul, 2013)
Les CD103+ CX3CR1- CD11b+ de la lamina propria. Elles
dérivent du CDP sous le contrôle de Flt-3L et du GM-CSF. Dans
les plaques de Peyer où elles sont aussi présentes, le GM CSF
n’est pas nécessaire à leur développement. Ce sous-type
exprime de forts niveaux de CCR7 qui leur permet de suivre le
gradient chimiotactique vers les nœuds lymphatiques : ce sont
des cellules migratoires. Elles sécrètent des cytokines pro-
inflammatoires : ces deux qualités font de ces cellules les
initiatrices de la réponse adaptative dans le tractus intestinal. A
l’état d’équilibre, elles favorisent la différenciation des Treg via le

48
TGF-β et l’acide rétinoïque. Elles ont donc également un rôle
immunomodulateur. Ces cellules nécessitent l’IRF-4 et le Notch
2 pour leur différenciation (Beijer et al., 2013; Kushwah & Hu,
2011),
Les CD103- CX3CR1+ CD11b+ sont 4 à 5 fois plus nombreuses.
Elles ont une durée de vie courte et migrent de façon constitutive
ou lors d’inflammation vers les nœuds lymphatiques
mésentériques (S. C. Ng, Kamm, Stagg, & Knight, 2010) ;
Dans le foie, les CD11c+ B220- jouent un rôle dans la tolérance à l’état
d’équilibre et elles limitent les lésions d’ischémie ou de reperfusion
grâce à leur sécrétion d’IL-10. Elles peuvent encore être divisées en
CD103+ CD11b- et CD103- CD11b+, les premières exprimant le Flt-3L
de façon marquée, ce qui montre qu’il s’agit bien de cellules
conventionnelles issues de pré-DC. Elles ont également besoin de
l’IRF-8, de l’Id2 et du Batf3. Les CD103- peuvent être issues de pré-DC
ou de populations dérivées des monocytes (Beijer et al., 2013;
Kushwah & Hu, 2011; Paul, 2013) ;
cellules dendritiques lymphoïdes qui résident dans les organes et y restent.
Elles sont notamment présentes dans les nœuds lymphatiques, la rate et le
thymus et peuvent encore être divisées selon leur expression de CD4 et CD8.
A la naissance, la majorité des cellules dendritiques sont CD8-, et les CD8+
n’apparaissent qu’une semaine après la naissance pour devenir la population
dominante, comme chez l’adulte, en seulement 2 semaines. Par contre, chez
la souris à la naissance, seules les cellules CD8- sont présentes, puis à 3
semaines d’âge, ces cellules deviennent indétectables jusqu’à 6 semaines où
un changement de ratio se réalise et elles deviennent alors prédominantes.
Ces changements semblent indiquer que les cellules CD8- sont un stade
immature des CD8+ trouvés dans la rate des souris adultes et que les
différents sous-types de cellules dendritiques ont des cinétiques de
développement différentes durant l’ontogénèse (Wu & Liu, 2007) ;
Les CD8+ CD4+ sont issues des pré-DC CD24high. Ce sont les plus
grands producteurs d’IFN-α parmi tous les types de cellules
dendritiques et elles jouent donc un rôle dans l’immunité antivirale.
L’IFN-α augmente la cytotoxicité des NK et des cellules T et contribue
donc aussi à l’immunité antivirale. Elles sont également impliquées
dans l’induction de la tolérance (Kushwah & Hu, 2011) ;
Les CD8- CD4- produisent de l’IL12p70 lorsqu’elles sont activées. Ce
sont les cellules dendritiques qui produisent les plus grandes quantités
d’IFN-γ. Lorsqu’elles induisent la tolérance en inhibant la réponse T
cytotoxique, le TGF-β1 est impliqué alors que lorsqu’elles remplissent
un rôle inflammatoire c’est la signalisation du TLR9 qui est mise en jeu.
Pour leur différenciation, elles dépendent du ReIB, de l’IRF-2 et de
l’IRF-4 (Beijer et al., 2013; Kushwah & Hu, 2011) ;

49
Les CD8- CD4+ partagent de nombreuses similitudes avec le groupe
précédent et les deux réunis sont donc appelés cellules dendritiques
CD8-. Toutes ces cellules sont issues du pré-DC CD24low et expriment
le CD11b, le DCIR2 le Sirp-α et l’ESAM. L’acide rétinoïque est essentiel
pour entrer dans la lignée des CD8- CD11b+. Ce sont aussi des
inducteurs des réponses T CD4+. La seule différence fonctionnelle est
que les CD8- CD4+ ne peuvent pas produire d’IL12p70 (Kushwah & Hu,
2011).
Les cellules dendritiques non conventionnelles
o Les cellules dendritiques plasmacytoïdes
Les cellules dendritiques plasmacytoïdes interviennent précocement dans les
infections virales. Elles reconnaissent les acides nucléiques des virus intracellulaires
et produisent des protéines solubles appelées interféron de type 1 qui ont des
activités antivirales. Leur différenciation repose sur le Flt-3L. Elles sont issues du
précurseur myéloïde ou lymphoïde. La particularité de ces cellules est qu’elles ne
nécessitent pas la réplication virale pour produire de l’IFN, la présence seule du virus
suffit. L’IFN exerce ensuite un rétrocontrôle positif afin d’augmenter sa production.
Les cellules dendritiques plasmacytoïdes sont aussi critiques pour la différenciation
des cellules B activées en plasmocytes. Enfin, elles ont été associées au maintien de
la tolérance périphérique et à l’induction de réponses auto-immunes (Abbas et al.,
2012; Kushwah & Hu, 2011; Paul, 2013; Wu & Liu, 2007).
Le nombre absolu et la proportion des cellules dendritiques conventionnelles et
plasmacytoïdes n’atteignent pas les taux adultes avant l’âge de 5 semaines (Wu &
Liu, 2007).
Dans le poumon, le rôle des cellules dendritiques plasmacytoïdes est controversé
avec un rôle dans l’immunité et dans la tolérance. Elles orientent la réponse vers une
réponse Th2. Elles peuvent également réduire la maturation des cellules
conventionnelles et donc augmenter le ratio pDC/cDC, ce qui réduit l’inflammation
(Kushwah & Hu, 2011).
Une des particularités des cellules plasmacytoïdes concerne la présentation
d’antigène. Contrairement aux cellules dendritiques conventionnelles qui réduisent
leur production de CMH II lorsqu’elles sont activées, les cellules plasmacytoïdes
subissent également une maturation et présentent les antigènes mais la synthèse du
CMH II n’est pas réduite ce qui leur donne la possibilité de présenter de façon
continue les antigènes viraux endogènes lorsqu’elles sont dans un état activé
(Kushwah & Hu, 2011).

50
o Les cellules dendritiques dérivées des monocytes
Les monocytes Ly6hight se rendent au niveau des sites d’inflammation où elles se
différencient en cellules dendritiques E-cadhérine+. Ces dernières produisent de
grandes quantités d’IL-23 et d’IL-12 et exacerbent la colite T chez la souris, ce qui
montre leur importance dans l’inflammation (Kushwah & Hu, 2011).
Les cellules dendritiques dérivées des monocytes CD103- CX3CR1+ dérivent
également des monocytes Ly6hight, et sont capables de prélever des bactéries dans
la lumière intestinale via des dendrites transépithéliales. Ces cellules vivent plus
longtemps que les cellules dendritiques conventionnelles mais sont de faibles
stimulateurs de la prolifération des cellules T puisqu’elles migrent peu (Kushwah &
Hu, 2011).
Dans les poumons, les monocytes Ly6low donnent naissance aux CD103- CD11chight
CD11b+ dans des conditions normales. En réponse à des allergènes, les monocytes
sont recrutés depuis le sang vers le poumon où ils subissent une différenciation
massive en cellules dendritiques, les producteurs clés de CCL-17 et CCL-22,
responsables de l’infiltration des cellules Th2 et des PNE dans les voies respiratoires
(Kushwah & Hu, 2011).
iii. Fonctions
Les fonctions des cellules dendritiques sont liées : il s’agit tout d’abord de cellules
sentinelles sous leur état immature. Ensuite elles subissent une maturation avant
d’initier et d’orienter la réponse T adaptative.
Les cellules dendritiques sont des cellules
sentinelles
La capacité des cellules dendritiques à capturer des antigènes font de ces cellules
des sentinelles clés pour détecter les pathogènes qui ont franchi les barrières
périphériques et sont donc maintenant dans les tissus.
Les cellules dendritiques immatures ont aussi un rôle. Les cellules dendritiques
monocytaires Pré-DC1 ingèrent de nombreuses bactéries et champignons alors que
les cellules Pré-DC2 sont les cellules effectrices de la réponse immune innée
antivirale en produisant de grandes quantités d’IFN-α et -β. Contrairement aux autres
cellules du système immunitaire inné, les pré-DC1 et pré-DC2 ne meurent pas après
avoir accompli leur rôle mais se différencient en cellules dendritiques matures. Pour
les pré-DC2, cette différenciation se fait en absence de cytokines exogènes puisque
le TNF-α et l’IFN de type I agissent de façon autocrine (Liu, 2001).
Les cellules dendritiques reconnaissent un grand nombre de PAMPs (page 80), mais
elles sont également activées par le complément, et d’autres protéines comme le

51
MBP et des DAMPs (Damage Associated Molecular Pattern). Elles reconnaissent
tous ces signaux de danger grâce à une panoplie de récepteurs très variés, détaillés
page 82 (Clark et al., 2000).
Maturation des cellules dendritiques
Les cellules dendritiques répondent à un signal de danger par une série
d’événements : elles sont tout d’abord activées pour pouvoir prélever les antigènes
puis elles les présentent à leur surface. Pendant cette étape, ou juste après, elles
migrent vers les organes lymphoïdes où elles finissent leur maturation. Cette
maturation mène à une forte expression des molécules de CMH II et de molécules
de co-stimulation. Elle augmente également l’efficacité de la macropinocytose pour
optimiser le prélèvement d’antigènes associés aux stimuli inflammatoires (Clark et
al., 2000; Gleeson, 2014).
Seules les cellules dendritiques qui subissent une maturation optimale deviennent
réfractaires à la lyse des cellules NK et sont autorisées à promouvoir les réponses
Th1 après leur migration dans les nœuds lymphatiques. En effet, les cellules NK
peuvent lyser les cellules dendritiques immatures, principalement pour limiter la
réponse immune ou pour prévenir la tolérance envers les pathogènes. Mais les NK
peuvent également détruire les cellules dendritiques durant des infections
bactériennes, comme lors d’infection tuberculeuse, où les cellules dendritiques
infectées par Mycobacterium bovis expriment de nombreuses chimiokines comme
CCL-3, -4, -5, -20 et CXCL-8, qui attirent les cellules NK, alors activées par les
cellules dendritiques surtout par celles dérivées des monocytes. L’interaction des
cellules dendritiques avec les NK pourrait avoir lieu dans les zones inflammatoires
et/ou dans les nœuds lymphatiques. Les cellules dendritiques produisent de l’IL-18,
cytokine clé pour la mise en place d’une fente synaptique où la libération d’IL-12 par
les cellules dendritiques initie le dialogue. En synergie avec cette cytokine, les
cellules dendritiques expriment l’IL-15Rα, nécessaire pour l’activation des NK. Donc
l’interaction NK - cellules dendritiques nécessite non seulement un contact cellule-
cellule mais également des facteurs solubles comme l’IL-2, l’IL-18 et l’IFN de type I.
Les cellules dendritiques plasmacytoïdes activent les stades précoces des NK suivis
par les cellules dendritiques myéloïdes. Parmi les récepteurs des NK, le NKp30
semble être essentiel dans la maturation et le contrôle de l’apoptose de cellules
dendritiques. A un ratio NK/CD faible (1 : 5), l’interaction entre les deux cellules
aboutit à l’activation des cellules dendritiques. A l’inverse, a un ratio élevé (5 : 1), les
cellules NK tuent les cellules dendritiques immatures. De façon intéressante, lors
d’infection par Leishmania infantum, les cellules dendritiques immatures
surexpriment le CD1d et sont tuées par les cellules NKT (Natural Killer T cell) alors
qu’elles sont résistantes à la lyse par les cellules NK (Reschner et al., 2008; Toka &
Golde, 2013).
La maturation des cellules dendritiques par les lymphocytes innés est pertinente
dans les réponses précoces contre les cellules tumorales et certains virus, quand

52
l’absence d’inflammation et de PAMP ne permet pas la présentation d’antigènes. De
plus, de cette interaction découle la prolifération des lymphocytes innés, leur
production d’IFN-γ et leur activité cytolytique. Cette relation est donc essentielle dans
la réponse précoce contre les tumeurs, et certains pathogènes notamment viraux
avant qu’une réponse adaptative ne puisse être mise en place. Les cellules
dendritiques matures activent les NK pour produire de l’IFN-γ qui est nécessaire pour
la différenciation des cellules Th1 (Reschner et al., 2008; Ullrich et al., 2008).
Rôle dans la régulation de l’immunité innée
En plus de leur rôle d’induction d’une réponse adaptative, les cellules dendritiques
activent les cellules NK ainsi que les cellules NKT et γδ T via la synthèse de
chimiokines comme Mip-1a, IL-8, MCP-1, RANTES (Regulated on Activation Normal
T cell expressed and Secreted), MDC et DC-CK1. Toutes ces cellules participent à
l’expansion, à la sécrétion de facteurs solubles comme des cytokines et chimiokines
et à des activités cytolytiques (Reschner et al., 2008).
Les cellules dendritiques agissent également sur les granulocytes, comme lors
d’infection par Listeria monocytogenes, où les cellules dendritiques CD11c+ de la
rate orchestrent la réponse immune en produisant de l’IL-12 et de l’IL-18. Ces deux
cytokines entraînent la formation de clusters de PNN et de monocytes.
L’accumulation des NK dans ces zones permet la production d’IFN-γ, nécessaire
pour la maturation des monocytes en cellules dendritiques (S.-J. Kang, Liang, Reizis,
& Locksley, 2008).
Il existe aussi de nombreuses interactions entre les cellules γδ T et les cellules
dendritiques, qui font surexprimer le CD86 et le CMH I par les γδ T qui produisent
alors du TNF-α et de l’IFN-γ (Toka & Golde, 2013).
Induction et orientation de la réponse T
Les cellules dendritiques sont cruciales pour maintenir la tolérance envers les tissus
du soi (Wu & Liu, 2007).
Elles stimulent le développement des cellules Th1, Th2, Th17 et Treg grâce à 3 signaux :
le premier dérive de l’interaction d’un peptide microbien présenté par le CMH
II des cellules dendritiques qui se lie sur le récepteur de la cellule T,
le second nécessite des molécules de co-stimulation des cellules T,
le dernier dépend des molécules produites par les cellules dendritiques pour
orienter la polarisation de la cellule Th.
De façon simple, les cellules dendritiques exposées à un pathogène intracellulaire
stimulent une réponse Th1 alors que les helminthes conduisent à une réponse Th2.
Les cellules conventionnelles matures ont une prédisposition à stimuler des
réponses pro-inflammatoires avec de l’IL-6 et sont donc pro-Th1. Les cellules

53
immatures CD1b- stimulent une réponse Th2 (avec de l’IL-4) ou Th17 (avec l’IL-23).
Les cellules épithéliales intestinales produisent du TSLP qui inhibe la production d’IL-
12 par les cellules dendritiques en réponse aux bactéries et stimule la réponse Th2
(S. C. Ng et al., 2010; Olivier, Foret, Le Vern, & Guilloteau, 2012; Olivier, Foret, Vern,
Kerboeuf, & Guilloteau, 2013).
Dans des conditions inflammatoires, l’engagement des PRR (Pattern Recognition
Receptor, récepteurs détaillés page 82) régule la survie des cellules dendritiques et
la mort par apoptose. Une hausse initiale dans la survie des cellules dendritiques est
nécessaire pour garantir l’activation des cellules T, durant 2-3 jours. Cependant, les
cellules dendritiques doivent avoir une durée de vie limitée pour limiter les antigènes
disponibles pour les cellules T, contrôler les réponses immunes et prévenir l’auto-
immunité. Ceci est permis par le gène pro-apoptique Nur77, dont l’expression est
strictement régulée. Les cellules dendritiques décident si une réponse T doit être
activée ou supprimée. Cette capacité duale est contrôlée par le NF-κB (Zanoni &
Granucci, 2012).
Les lymphocytes 2)
Les lymphocytes sont les archétypes des cellules du système adaptatif avec les
lymphocytes T et B. Mais des lymphocytes participent aussi à l’immunité innée
comme les lymphocytes Th17, les cellules γδ T, les iNKT ou encore les ILCs
récemment découvertes et traitées en dernière partie de ce travail.
a. Les cellules Th17
Les cellules Th17 sont les nouveaux membres de la famille des cellules T helper.
Elles produisent de l’IL-17A, de l’IL-17F, de l’IL-22, et du CCL-20. Elles interviennent
dans l’immunité contre les bactéries extracellulaires et les champignons et pourraient
aussi agir dans les infections virales. Chez la souris, le terme Th17 réfère aux cellules
T CD4+ sécrétant de l’IL-17. Mais cela n’inclut pas les CD8+ et les γδ T qui
produisent aussi de plus grandes quantités d’IL-17 (Geremia et al., 2013; Louten,
Boniface, & de Waal Malefyt, 2009; Sutton et al., 2009; Sutton, Mielke, & Mills,
2012).
Rappelons qu’il existe plusieurs types de cellules T helper :
Les Th1, qui se développent en présence d’IL-12, produisent de l’IFN-γ, de l’IL-
2 et de la lymphotoxine. Elles sont impliquées dans l’immunité à médiation
cellulaire. Elles favorisent la présentation des antigènes intracellulaires et sont
donc essentielles à l’élimination des bactéries intracellulaires.
Les Th2 se différencient en présence d’IL-4 et produisent de l’IL-4, de l’IL-5 et
de l’IL-13. Elles sont critiques pour l’immunité humorale et interviennent donc
préférentiellement dans l’élimination des organismes extracellulaires.
Les Th17 sont fortement présentes lors de désordres auto-immuns. Elles
dépendent du RORγt (RAR-related Orphan Receptor γ qui sera étudié page

54
200 en dernière partie de ce travail) et sont induites par différentes cytokines
comme l’IL-1β, l’IL-6 et le TGF-β. L’IL-23 est important pour leur survie. Elles
produisent l’IL-22 grâce à l’AhR (Aryl Hydrocarbon Receptor) (Louten et al.,
2009; Verdier & Ruemmele, 2013).
Pour leur développement, le TGF-β (qui bloque la différenciation Th1 et Th2) et l’IL-6
sont nécessaires pour que la cellule T surexprime le RORγt et il s’ensuit l’activation
du STAT3 ainsi que l’expression de l’IL-23R. Ensuite, l’IL-21 et l’IL-23 permettent de
continuer la différenciation vers les Th17. Par contre, les cytokines Th2 comme l’IL-4,
et Th1 comme l’IFN-γ inhibent la différenciation des Th17. La production de TGF-β
lors de l’interaction T – cellule présentatrice d’antigènes est cruciale pour le
développement de cellules Th17 (Louten et al., 2009; Mars et al., 2009; Stockinger,
Veldhoen, & Martin, 2007).
Les Th17 interviennent dans l’immunité contre les bactéries et les levures notamment
dans l’immunité muqueuse:
Elles ont un rôle protecteur contre les infections cutanées par Candida
albicans et Staphylococcus aureus. Les muqueuses intestinales contiennent
un grand nombre de Th17 et l’expression d’IL-17A est fortement surexprimée
durant la colonisation post-natale par la flore microbienne sans causer de
dommage intestinal visible. Ceci montre le rôle important de cette cytokine
dans l’homéostasie muqueuse (Verdier & Ruemmele, 2013).
Elles jouent un rôle important dans la réponse de rappel contre
Mycobacterium tuberculosis. Les cellules Th17, peuvent amplifier et
coordonner les réponses immunes protectrices. En plus de l’IL-17A et IL-17F,
les cellules Th17 fabriquent de l’IL-22 et IL-10, production surexprimée par l’IL-
23. Cette dernière provoque chez les cellules Th1 la sécrétion d’IFN-γ et celle
d’IL-22 par les γδ T et T CD8+. L’IL-22 est pro-inflammatoire et induit
l’expression de protéines antimicrobiennes (Stockinger et al., 2007)
Les Th17 sont impliquées dans le développement de la dermatite de contact ou de
dermatite atopique. Elles sont également impliquées dans des maladies auto-
immunes comme les maladies inflammatoires chroniques des intestins et le psoriasis
(Louten et al., 2009; Mars et al., 2009).
Il est concevable que des cellules mémoires Th17 puissent survivre longtemps mais
ces cellules ne sont pas capables de sécréter de l’IL-17A sans un stimulus
inflammatoire constitué par l’IL-23 (Stockinger et al., 2007).
b. Les cellules γδ T
Les cellules γδ T représentent entre 3 et 5 % des lymphocytes totaux trouvés dans
les tissus lymphoïdes secondaires et le sang mais leur nombre augmente
considérablement dans le sang durant des infections virales et bactériennes (jusqu’à

55
50 % des cellules T). Elles constituent un petit pourcentage des cellules T circulantes
chez les adultes humains et murins mais constituent la majorité des lymphocytes
intra-épithéliaux dans les intestins et la muqueuse épithéliale. Nous verrons dans la
deuxième partie, page 139, que ces cellules sont beaucoup plus nombreuses chez
les bovins (Born, Reardon, & O’Brien, 2006; Komori, Meehan, & Havran, 2006;
Konigshofer & Chien, 2006; Reschner et al., 2008; Sutton et al., 2012).
Bien qu’elles expriment un TCR (T Cell Receptor), il n’est pas engagé dans les
complexes CMH-antigènes de la même manière que chez les cellules αβ T. Ces
cellules ont un répertoire de gènes Vγ et Vδ très limité : elles ne possèdent que
quelques paires de gènes Vγ et de Vδ. Dans la rate, les γδ T expriment le Vγ1 et Vγ4
et les lymphocytes intra-épithéliaux expriment le Vγ7. Le Vγ reflète donc l’origine
tissulaire et non pas une spécificité antigénique. Les TCR γδ sont donc des
récepteurs immuns typiquement adaptatifs mais avec des caractéristiques innées.
Les cellules γδ T diffèrent des lymphocytes classiques αβ T par un TCR avec une
structure et une fixation d’antigène uniques ce qui les rend indépendants des cellules
présentatrices d’antigènes pour reconnaître des épitopes étrangers. En fait, il
semblerait que ces cellules fixent les antigènes d’une manière plus similaire aux
immunoglobulines que comme les TCR αβ. Ce TCR apparait plus comme un PRR,
puisqu’il reconnaît des phosphoantigènes très répandus présents dans les voies
métaboliques bactériennes, ainsi que des produits de dommage cellulaire (Born et
al., 2006; Komori et al., 2006; Konigshofer & Chien, 2006; Reschner et al., 2008;
Sutton et al., 2012).
Les cellules αβ T et γδ T proviennent d’un même précurseur dans le thymus. Les
cellules αβ T CD4+ CD8+ (double positif), régulent la différenciation des cellules γδ T
par un mécanisme dépendant du RORγt et du récepteur à la lymphotoxine β
(Hayday & Tigelaar, 2003; Komori et al., 2006).
Ce sont les premières cellules T à migrer depuis le thymus et les premières cellules
immunes détectables chez le fœtus. Elles fournissent aux nouveaux-nés une
immunité avant l’activation complète du système adaptatif. Elles se concentrent dans
les sites épithéliaux et muqueux, notamment dans les intestins ou elles représentent
jusqu’à 50% des lymphocytes intra-épithéliaux (Born et al., 2006; Komori et al., 2006;
Konigshofer & Chien, 2006; Reschner et al., 2008; Sutton et al., 2012).
Chaque tissu a son propre type de cellules γδ T. Ces cellules constituent les
représentants cellulaires du système immunitaire inné le plus complexe et le plus
avancé. La plupart des ligands connus pour le TCR γδ sont des molécules du soi
capables d’indiquer un stress cellulaire comme le CD1, le MIC-A, le MIC-B ou le F1-
ATP synthase, exprimés à la surface des cellules tumorales. Le dernier est un
antigène mitochondrial normalement inaccessible : il s’agit d’une alarmine (Born et
al., 2006; Komori et al., 2006; Konigshofer & Chien, 2006; Reschner et al., 2008;
Sutton et al., 2012).

56
Leur stimulation par l’IL-1β et l’IL-23 induit la production d’IL-17A, d’IL-17F, d’IL-21,
et d’IL-22. Le profil de cytokine des cellules γδ T est prédéfini pendant la sélection
thymique par un mécanisme impliquant l’expression du CD27 (Guo et al., 2012).
Il existe plusieurs types de cellules γδ T définis par les gènes exprimés parmi ceux
du répertoire :
Si le TCR est le Vγ3Vδ1, ces cellules sont appelées cellules T épidermiques
dendritiques ou DETC (Dendritic Epidermal T Cell). Elles migrent vers la peau
et interagissent physiquement avec les kératinocytes. Bien qu’elles soient
capables de résider dans l’épiderme, elles ne sont pas capables de répondre
aux kératinocytes endommagés. La présence des TCR γδ qui peuvent
reconnaître un antigène exprimé dans la peau est cruciale. Dans l’épiderme
elles répondent en produisant de l’IFN-γ et dans le derme elles produisent de
l’IL-17A en réponse à l’IL-1β ou l’IL-23. Elles peuvent tuer des cellules
épithéliales stressées par un choc thermique ou par transformation tumorale
(Hayday & Tigelaar, 2003; Komori et al., 2006; Pang, Neves, Sumaria, &
Pennington, 2012) ;
Les cellules γδ T Vγ4+ sont présentes dans la langue et le tractus
reproducteur. Elles sont CD25+, CD44+ et produisent de l’IL-17A (Pang et al.,
2012) ;
Les γδ T Vγ5+ sont surtout localisées dans les intestins. La génération de ces
lymphocytes intra-épithéliaux intestinaux CD27+ et CD122+ dépend de l’IL-15
dans le thymus. Une fois que ces cellules quittent le thymus, d’autres facteurs
solubles sont nécessaires pour les attirer vers les intestins via le CCL-25 qui
interagit avec le CCR-9 exprimé à leur surface. Cette interaction augmente
l’expression de l’intégrine αEβ7 qui peut se fixer sur les E cadhérines
présentes à la surface des entérocytes intestinaux. D’autres précurseurs sont
les cellules des cryptoplaques dans la lamina propria. En fait, le thymus
envoie des précurseurs des cellules T pour former les cryptoplaques dans les
intestins qui subissent un réarrangement TCR et ensuite génèrent des
lymphocytes intra-épithéliaux αβ T et γδ T. Ces cellules produisent de l’IFN-γ
Ce sont des effecteurs cytolytiques avec un rôle immunoprotecteur, surtout
chez les jeunes. Ils sont présents dans les tissus lymphoïdes associés aux
intestins comme les cryptoplaques (Komori et al., 2006; Pang et al., 2012).
Les cellules γδ T jouent un rôle actif dans la régulation et la résolution des réponses
immunes puisqu’elles modulent l’inflammation, la lyse des cellules transformées et
augmentent l’efficacité de la réparation tissulaire. Elles ont une fonction régulatrice
dans la restauration et le maintien de l’homéostasie des tissus constamment
exposés aux challenges environnementaux. Elles interviennent notamment dans la
guérison des blessures, l’éventail de cytokines produites est cependant semblable à
celui des αβ T. Cependant différents types de γδ T auraient des fonctions différentes.
Les cellules γδ T CD8- lorsqu’elles sont activées, sont prolifératives et inflammatoires

57
alors que le type CD8+ supprime l’expression de certains gènes ce qui diminue le
chimiotactisme vers la muqueuse et inhibe la réponse immune (Komori et al., 2006;
Konigshofer & Chien, 2006; Skyberg et al., 2011).
Les cellules γδ T jouent un rôle essentiel dans la réponse immune précoce à un
large éventail d’agents infectieux incluant les champignons, les bactéries, les virus et
les parasites. Cela pourrait expliquer leur abondance au niveau des sites muqueux
ainsi que leur rapidité d’activation suite à leur exposition aux cytokines
inflammatoires ou aux pathogènes.
Les cellules γδ T activées acquièrent la capacité de présenter des antigènes mais
elles doivent être activées avant de commencer à prélever des antigènes. Elles
expriment alors le CCR-7, le récepteur qui leur permet de se diriger vers les nœuds
lymphatiques et des molécules de co-stimulation (CD40, CD80 et CD86) ainsi que
des molécules de CMH II. Elles préparent les réponses des cellules αβ T CD4+ et
CD8+ qui prolifèrent et se différencient en lymphocytes T cytotoxiques. Les cellules
γδ T engagent les interactions régulatrices avec les macrophages, les granulocytes
et les cellules dendritiques. Même dans des tissus sains, les interactions avec les
cellules myéloïdes sont fréquentes. A n’importe quel moment, la moitié des cellules
γδ T résidentes dans le poumon sont en contact étroit avec les cellules CMH II+, en
général des cellules dendritiques. Ces interactions suggèrent que les cellules γδ T
contrôlent les cellules myéloïdes (Born et al., 2006; Konigshofer & Chien, 2006).
Elles peuvent promouvoir le recrutement de cellules effectrices vers le site de
l’inflammation. En effet, elles recrutent les PNN, les macrophages et les cellules NK
et produisent de l’IFN-γ. De plus, l’IL-17 produite contribue à la résolution de
l’infection, surtout dans les premiers temps puisqu’elle augmente l’expression des
chimiokines et induit rapidement le recrutement des PNN. Cette cytokine est
notamment produite lors d’infection pulmonaire par Klebsiella pneumoniae ou par
Mycobacterium tuberculosis ou encore lors d’infection intestinale par Salmonella
enterica. Cette production d’IL-17 est détectable à peine quelques heures après
l’exposition à Escherichia coli et peut être maintenue pendant plusieurs jours,
suggérant que les γδ T sont requises pour augmenter la réponse antimicrobienne.
Enfin, les γδ T produisant de l’IL-17 sont trouvées dans le foie durant les trois
premiers jours lors d’une infection par Listeria monocytogenes ou Toxoplasma
gondii, tous deux des parasites intracellulaires. Beaucoup de cellules γδ T du sang
semblent être des cellules mémoires. La multiplication bactérienne est inhibée par
leur production d’IFN-γ et de TNF-α alors que l’exocytose de leurs granules réduit le
nombre de Brucella suis intracellulaire en lysant les macrophages infectés (Skyberg
et al., 2011). Elles apportent une protection contre Brucella abortus conférée par le
TNF-α qui agit de façon autocrine en augmentant l’expression du CD69. Les cellules
γδ T répondent initialement à l’infection par des signaux qui augmentent la fonction
des macrophages et avec une réponse accrue des γδ T aux signaux secondaires
comme les antigènes et les cytokines. Dans un second temps, les γδ T expriment

58
des signaux indicateurs d’un état effecteur (Cua & Tato, 2010; Komori et al., 2006;
Louten et al., 2009; Skyberg et al., 2011; Sutton et al., 2009, 2012).
Les γδ T diminuent l’intensité des réponses conduites par les cellules αβ T.
Cependant l’accumulation des cellules γδ T ne supprime pas toujours les réponses
αβ T : elles peuvent induire un changement de classe d’immunoglobulines. Cette
régulation des cellules αβ par les γδ T a lieu dans les épithéliums. Les γδ T
contiennent de hauts niveaux d’ARNm encodant pour des chimiokines comme
RANTES, MIP (Macrophage Inflammatory Protein 1α) MIP1β et la lymphotactine. En
plus, ces cellules ont de nombreux ARN de gènes encodant pour des effecteurs
cytolytiques comme le granzyme A et granzyme B, le Fas ligand (l’activation de ce
récepteur sur la cellule entraîne l’apoptose de cette cellule : la libération de ligand
Fas permet donc de lyser des cellules) et la lymphotoxine. L’IFN-γ relâché par les
cellules γδ T après la reconnaissance des cellules épithéliales induit l’expression du
ligand FAS par l’épithélium et qui cible les cellules activées systémiques infiltrant les
tissus (Hayday & Tigelaar, 2003).
Les cellules γδ T peuvent aussi se montrer cytotoxiques sous certaines conditions.
Dans les poumons, elles régulent l’inflammation durant l’hyperréactivité des voies
respiratoires (AHR) et de sepsis. Les cellules Vγ1.1+ stimulent l’expression de
l’hyperréactivité grâce à l’expression des cytokines Th2, alors que les Vγ2+
suppriment cet AHR (Komori et al., 2006; Konigshofer & Chien, 2006).
Les cellules γδ T s’accumulent dans le poumon durant le sepsis mais présentent peu
d’activité cytotoxique : elles diminuent l’expression d’IFN-γ et d’IL-10 et augmentent
l’expression du TNF-α. Les cellules γδ T surveillent les cellules épithéliales d’une
transformation maligne. Elles lysent les mélanomes et d’autres cellules épithéliales
cancéreuses grâce à la mise en place de porines (Komori et al., 2006).
c. Les NKT
Les cellules NKT sont des lymphocytes T innate like décrites pour la première fois
chez les rongeurs et les primates. Elles expriment un répertoire TCR restreint
(Vα14Jα18 chez la souris) et répondent aux glycolipides endogènes et exogènes
présentés par le CMH I-like CD1d des cellules dendritiques ce qui provoque la
production des cytokines de type 1 et de type 2 et active leur fonction cytolytique.
Après leur activation, ces cellules produisent très rapidement de hauts niveaux de
cytokines comme de l’IL-17 mais aussi de l’IL-22, et des cytokines
immunorégulatrices (Bourgeois et al., 2010; Cua & Tato, 2010; Diana & Lehuen,
2009; Vipin Kumar & Delovitch, 2014; Louten et al., 2009; Reschner et al., 2008;
Vomhof-Dekrey, Yates, & Leadbetter, 2014).
Ces cellules sont impliquées dans un grand nombre de fonctions immunes comme
l’auto-immunité, l’immunité contre les virus, les bactéries, les champignons, les
parasites et les tumeurs (Reschner et al., 2008).

59
Elles peuvent être divisées en plusieurs sous-types selon leur production de
cytokines :
Les iNKT conventionnelles ou NKT de type I, avec peu de variations dans
le TCR. Elles sont très présentes dans le foie, et répondent aux stimulations
par l’IL-12 et l’IL-18 ou l’IL-12 et l’IL-33 en produisant de l’IFN-γ. Elles
expriment le T-bet et le NK1.1. Elles dépendent du RORγt et du GATA3 pour
leur développement (tous les deux détaillés en troisième partie, page 200).
Ces cellules sont pro-inflammatoires et peuvent léser le foie. Lors de lésion
hépatique, ces cellules répondent très rapidement par reconnaissance directe
de lipides ou indirectement par les ligands des TLR présentés par les cellules
de Kupffer, les hépatocytes, et les cellules dendritiques myéloïdes. Par
exemple, suite à une ischémie ou une lésion induite par des toxines, la
sécrétion rapide de cytokines pro-inflammatoires par les iNKT se conclut par
l’accumulation de neutrophiles et macrophages dans le foie. Les iNKT
stimulent la fibrogenèse et elles peuvent tuer les hépatocytes directement ou
en activant les cellules NK ;
Les NKT de type II, dont les TCR sont plus variés sont surtout présentes
dans les nœuds lymphatiques périphériques mais très peu dans le foie. Ces
cellules seraient d’importance mineure chez la souris mais prédominantes
chez l’homme. Elles inhibent les lésions hépatiques causées par les iNKT.
Ces NKT reconnaissent plutôt les glycolipides endogènes comme le sulfatide,
abondant dans le système nerveux central, les reins et le foie. Stimulées par
l’IL-23 ou l’IL-1β, elles produisent de l’IL-17. Mais contrairement aux cellules
Th17, l’IL-6 n’est pas nécessaire à cette synthèse par les NKT ou par les γδ T.
D’autres cytokines comme l’IL-18 pourraient être impliquées dans cette
synthèse. Elles expriment le RORγt, l’IL-23R et l’IL-1R mais pas le T-bet ;
Le type IL-17RB+ CD4+, présent dans la rate et à peine détectable dans les
poumons. Elles répondent à l’IL-25 en produisant de l’IL-1, de l’IL-3 et de l’IL-4
(Cua & Tato, 2010; Guo et al., 2012; Vipin Kumar & Delovitch, 2014; Vipin
Kumar, 2013; Mars et al., 2009; Sutton et al., 2012; Van Kaer & Joyce, 2005).
Les NKT ont un rôle pivot dans la communication entre les deux immunités :
Les NKT semblent réguler la production d’IgE spécifiques d’un antigène. Elles
influencent ainsi les réponses immunes menées par les cellules T
conventionnelles CD4+, les cellules T CD8+ et les cellules B. Les iNKT aident
les cellules B à engager les C1d sur les cellules dendritiques, et augmentent
l’aide des cellules T CD4+ conventionnelles sur les cellules B en favorisant la
présentation d’antigène par les cellules dendritiques. Les iNKT sont capables
de promouvoir une réponse anticorps IgG robuste et primaire qui requiert l’IL-
21 et l’IFN-γ des iNKT et des molécules de costimulation comme le
CD40/CD40L et CD28/CD80 ou CD86 mais pas d’IL-4. L’aide des iNKT mène

60
au développement des plasmocytes et à une réponse mémoire humorale
(Nishimura et al., 2000; Vomhof-Dekrey et al., 2014).
Les cellules dendritiques CD1+ interagissent avec les NKT durant la phase
précoce de la réponse immune et cette interaction est critique pour la
séquence d’activation des cellules effectrices impliquées dans l’immunité
acquise. Les deux cellules communiquent par à une liaison CD40 – CD40L :
les cellules dendritiques produisent alors de l’IL-12 et les NKT le récepteur à
l’IL-12. Lorsque cette cytokine agit de façon paracrine sur le récepteur des
NKT, ces dernières produisent de l’IFN-γ, facteur clé de la maturation finale
des cellules T CD8+ CD69+ en lymphocytes T cytotoxiques fonctionnels
(Nishimura et al., 2000).
Les NKT sont donc au carrefour entre les réponses immunes innée et adaptative. Il
existe 5 différences entre les réponses des NKT et les réponses des T CD8+ :
Les NKT reconnaissent les lipides et glycolipides et les cellules CD8+ les
protéines ;
La charge de l’antigène sur le CD1 a lieu dans le compartiment
endosomal/lysosomal et non dans le réticulum endoplasmique ;
Les molécules de CD1 sont synthétisées et migrent vers le système
endosomal en utilisant des motifs trouvés dans leur domaine cytoplasmique ;
Le TCR semi invariant des NKT a une spécificité large ;
La réponse des NKT activés implique la sécrétion d’un grand nombre de
cytokines qui influencent les réponses innées et adaptatives (Gleeson, 2014).
Les iNKT régulent la réponse immunitaire :
L’activation des iNKT inhibe la différenciation de cellules T CD4+ naïves
autoréactives surtout dans la lignée Th17, mais également dans la lignée Th1.
La régulation de la lignée Th17 se fait par l’intermédiaire d’IL-4, d’IL-10 et
d’IFN-γ (Mars et al., 2009).
Les iNKT exercent un effet anti-inflammatoire en s’accumulant dans les
poumons lors de réaction inflammatoire surtout en présence d’IL-12. Cette
dernière stimule la libération d’IFN-γ par les iNKT en coordination avec l’IL-27
(Bourgeois et al., 2010).
Les NKT ont un rôle dans les réponses antivirales grâce au CD1d. Elles
interviennent dans la réponse contre le HSV-1 et HSV-2. Elles ne diminuent pas la
charge précoce en virus mais favorisent la clairance du virus (Diana & Lehuen,
2009).
Les iNKT stimulent la réponse antivirale au niveau de la rate et du pancréas en
stimulant ou inhibant les réponses des cellules T CD8+. Dans le pancréas, elles
stimulent la production d’IFN-α par les cellules dendritiques et inhibent la réplication
virale localement (Diana & Lehuen, 2009).

61
d. Les cellules T CD8+
La réponse des cellules T CD8+ aux pathogènes intracellulaires et aux tumeurs est
généralement attribuée au récepteur T et aux évènements qui découlent de son
activation. L’interaction spécifique du CMH avec le TCR sur ces cellules T mène aux
fonctions effectrices et la nature spécifique de cette reconnaissance est la
caractéristique des cellules B et T.
Cependant ces cellules T peuvent être activées de façon non spécifique par l’IL-12 et
l’IL-18, libérées lors des réponses innées et qui activent donc les fonctions effectrices
indépendamment du TCR. En effet, la fixation de ces cytokines entraîne la sécrétion
d’IFN-γ et de TNF-α par les cellules T CD8+. D’autres récepteurs innés transmettent
des signaux activateurs comme le NKG2D. L’engagement du CD94/NKG2D active
aussi la cytotoxicité, la prolifération et la sécrétion de TNF-α et d’IFN-γ. La production
de ce dernier peut être activée par de l’IFN-α et de l’IL-12, notamment produites en
réponse au LPS. En fait, les cytokines IL-21, IL-23 et IL-27 produites lors d’une
infection ou d’une inflammation peuvent mener à la production d’IFN-γ par les
cellules T CD8+ (Berg & Forman, 2006; Guo et al., 2012).
Par exemple, lors d’infection par Listeria monocytogenes, la réponse immune est
activée par l’action de l’IL-12 et de l’IL-18, sécrétées par les macrophages infectés ou
les cellules dendritiques et ces cytokines induisent chez les cellules T CD8+
mémoires ou effecteurs la production d’IFN-γ. De la même façon, les cellules T CD8+
interviennent dans la réponse innée contre Cryptosporidium parvum, Burkholderia
pseudomallei, Klebsiella pneumoniae et Shigella flexneri et agissent comme une
source précoce d’IFN-γ. Il semblerait que les cellules T CD8+ auto-réactives ne
créent pas d’auto-immunité mais jouent un rôle dans la lutte contre des pathogènes à
travers leur production de cytokines (Berg & Forman, 2006; Guo et al., 2012).
Ce qu’il faut retenir :
De nombreuses cellules interviennent dans l’immunité innée, avec des
origines, des localisations différentes, et surtout des fonctions et des modes
d’action différents mais très souvent imbriqués. Cet aperçu des cellules
impliquées dans l’immunité innée nous montre déjà la fragilité de la frontière
entre immunité innée et adaptative.

62
C- Les facteurs humoraux
L’immunité humorale est une immunité qui peut être transférée à des receveurs non
immunisés par transfert de sérum. L’exemple type de molécule humorale est
l’anticorps qui appartient au système adaptatif, bien que nous verrons qu’une petite
fraction de ces anticorps peut appartenir à l’immunité innée. Cette immunité
humorale vient compléter l’immunité cellulaire précédemment décrite. Le système
immunitaire inné emploie des protéines de petite taille qui interagissent avec un large
éventail de polysaccharides de la surface microbienne. Parmi ces molécules se
trouvent les collectines, les ficolines, le C1q, les galectines, les pentraxines, les
anticorps naturels et d’autres facteurs associés aux membranes cellulaires (Lillie,
Brooks, Keirstead, & Hayes, 2005).
Les lectines 1)
Les lectines sont des protéines avec des domaines de type collagène (triple hélice α)
et de type lectine, et ce dernier permet la fixation des glucides. L’unité de base est un
trimère, stabilisé par des ponts disulfures entre les 3 monomères. Parmi les
membres de cette famille, il y a :
les ficolines, des polypeptides homotrimériques divisées en 3 sous-familles :
le H-ficoline est le plus abondant et peut fixer des monosaccharides N-
acétylés, le LPS et les cellules apoptiques ;
le L-ficoline se fixe au glucosamine N-acétyl, au galactosamine N-
acétyl et à l’acide neuraminique N-acétyl, ainsi qu’à la protéine C
réactive, les peptidoglycanes et l’acide lipoteichoïque des bactéries ;
le M-ficoline se trouve à la surface des monocytes et dans les granules
de sécrétion. Il est très similaire au L-ficoline avec qui il partage 79% de
sa séquence d’acides aminés. Il active le complément. Une fois
phagocyté, le M-ficoline relâche le microorganisme après un
changement de conformation et est recyclé sur la membrane cellulaire.
les collectines, des lectines dépendantes du calcium. Il s’agit d’un groupe de
protéines qui partagent des points communs avec le collagène et des
propriétés fonctionnelles avec les lectines. Les collectines sont des PRR
sécrétoires qui peuvent agir comme des opsonines et causent la destruction
des microorganismes en stimulant les cellules phagocytaires. Les membres
les plus connus sont :
le Mannose Binding Protein ou Lectin (MBP ou MBL). L’interaction du
MBL avec le produit activateur est dépendante du calcium. Le MBL
reconnaît le mannose, le glucosamine et d’autres sucres terminaux des
bactéries. Comme les surfaces bactériennes ont de multiples sucres
répétitifs, cela permet au MBL de différencier le soi du non soi. Le MBL
peut activer le complément sans besoin d’immunoglobuline : il s’agit de
la voie d’activation purement innée du complément (décrite dans le
paragraphe suivant),

63
le SP-A et SP-D (Pulmonary Surfactant Protein A et D),
la conglutinine. La conglutinine est la protéine sérique avec les plus
hautes concentrations. Elle se fixe au N-acétylglucosamine, au
mannose et au fucose, et de manière Ca2+ dépendante au iC3b ce qui
contribue à son action antibactérienne. La conglutinine fixée stimule
l’explosion respiratoire. Par contre la conglutinine non fixée à un résidu
de sucre possède des effets inhibiteurs sur la production d’O2- et de
H2O2 par les PNN ;
La famille des pentraxines est une famille contentant la protéine C-réactive et
le SAP (pour Serum Amyloid Component). Elles se fixent sur les
polysaccharides de façon Ca2+ dépendante et activent la voie classique du
complément, là aussi, sans intervention d’anticorps (Dec, Wernicki, Puchalski,
Urban-Chmiel, & Wasko, 2012; Lillie et al., 2005; Tabel, 1996).
Une autre classification consiste à différencier les lectines selon leur forme :
Les lectines cruciformes agglutinent ou neutralisent les pathogènes, ce qui
augmente la clairance microbienne via la phagocytose et l’appareil
mucociliaire des poumons ;
Les lectines sertiformes interagissent avec les MASP (MBL-Associated Serine
Protease) qui activent la voie des lectines du complément (voir paragraphe
suivant) : cette interaction augmente la destruction des microbes et la
libération de médiateurs inflammatoires comme le C5a (Lillie et al., 2005).
Les ligands des lectines sont des constituants communs des enveloppes cellulaires
microbiennes comme le mannose, le peptidoglycane, la chitine et les acides
lipoteichoïques. Cette diversité de ligands les autorise à détecter des bactéries, des
virus, des champignons et des protozoaires. Elles activent la voie des lectines du
complément et donc, induisent des réponses inflammatoires et phagocytaires. Les
protéines de défense innée se fixent préférentiellement sur des domaines largement
répandus dans le règne microbien et sont donc des PRR (Pattern Recognition
Receptor). Cependant elles reconnaissent aussi des molécules de l’hôte comme des
cellules apoptiques. Ce sont des protéines multifonctionnelles qui interviennent dans
les défenses, avec d’importants rôles homéostatiques comme le nettoyage de
cellules apoptiques et des complexes immuns (Lillie et al., 2005).
Le complément 2)
a. Historique
Le système du complément a été découvert à la fin du XIXème siècle par Buchner
dans le sérum d’animaux en convalescence d’une infection bactérienne, capable de
lyser les bactéries responsables de cette infection. La lyse des bactéries était rendue
possible grâce à une substance thermostable spécifique et immune (les anticorps) et

64
une substance thermolabile, non spécifique et présente dans le sérum en dehors de
toute immunisation, capable de lyser les bactéries, mais détruite à 55°C (le
complément). Au début du XXème siècle, Bordet montre que l’action lytique du
complément peut s’exercer contre les globules rouges et il émet également
l’hypothèse qu’il s’agit d’un ensemble de constituants. En 1940, seuls 4 composants
sont connus (C1, C2, C3 et C4). Depuis, une trentaine de composants du
complément ont été découverts (Revillard & ASSIM, 1995).
b. Les composants du complément
Le complément est un ensemble de 35 protéines plasmatiques et membranaires qui
s’activent par différentes cascades après fixation de PAMPs (pour la voie alterne).
Beaucoup des facteurs du complément sont des protéases produites sous forme de
précurseurs inactifs et qui s’activent après la détection des PAMPs. Les composants
de la voie classique sont désignés par des chiffres accolés à la lettre C (de C1 à C9).
Lorsque le composant est clivé, les fragments sont alors désignés par une lettre en
plus (exemple : C3a et C3b). Les fragments qui possèdent la capacité de se fixer sur
une membrane cellulaire sont marqués d’un astérisque (comme le C3b*). Les
composants inactivés sont indiqués par la lettre i (ex : iC3b). Une barre au-dessus du
complexe dénote une activité enzymatique (Delves et al., 2011).
Le fragment C1q est une protéine constituée de
six sous-unités de trois chaînes polypeptidiques
homologues avec chacune une partie fibrillaire
N-terminale et une partie globulaire
périphérique. (Voir Figure 12 ci-contre) Les
différentes chaînes sont reliées par des ponts
disulfures au niveau de la région-N terminale
pour former les six sous-unités, elles-mêmes
reliées par d’autres ponts dissulfures au niveau
de leur partie globulaire pour former des
dimères. Les six chaînes polypeptidiques du
dimère sont associées en une triple hélice au
niveau de leur partie fibrillaire. Les trois dimères ainsi formés sont ensuite reliés de
façon non covalente pour former la structure globale héxamérique. Le C1q peut
reconnaître un site du fragment Fc des immunoglobulines de différentes classes
(IgM, IgG1, IgG2 et IgG3 mais pas les IgA, les IgE et les IgG4) (Carroll & Sim, 2011).
Les fragments C1r et C1s sont des sérines protéases possédant une structure
homologue. En présence de Ca2+, elles forment un complexe tétramérique C1s-C1r-
C1r-C1s activé par l’extrémité globulaire du fragment C1q ce qui libère leur site actif
de sérine protéase. C3 est le composant le plus concentré dans le sérum. C4 est
produit sous forme d’une longue chaîne qui est ensuite clivée en trois chaînes
polypeptidiques reliées par des ponts disulfures. Les fragments C6 à C8 sont formés
de trois chaînes polypeptidiques : α, β et γ ; reliées par des ponts disulfures (entre α
Figure 12 : Structure du fragment C1q (Carroll & Sim, 2011).

65
et γ) ou par des liaisons non covalentes (entre α et β). Le composant C9 est une
glycoprotéine monocaténaire de 70 kD qui possède une partie hydrophobe qui lui
permet de s’insérer dans une bicouche lipidique et une partie hydrophile, qui sera
située au centre du complexe d’attaque membranaire et qui permet le passage de
l’eau (Pastoret, Govaerts, & Bazin, 1990).
Le facteur B est proche du composant C2 et sont tous les deux clivés en deux
fragments : Ba / Bb et C2a / C2b, respectivement. Sur la partie C terminale des
fragments C2a et Bb se situe un site de sérine protéase (ces sites constituent le site
actif des C3-convertases de la voie classique et de la voie alterne).
Le facteur D circule dans le sang sous forme active. Il clive le facteur B grâce à son
activité sérine protéase.
La demi-vie des composants du complément varie entre quelques heures et 60
heures.
La plupart des protéines du complément sont synthétisées par le foie bien que
certains des composants soient synthétisés par les macrophages ou les cellules
épithéliales muqueuses.
c. Cascades d’activation du complément
Les protéines du complément circulent dans le sang comme des protéases actives
ou sous forme inactive mais activable. Elles clivent alors spécifiquement des cibles
ou interagissent avec d’autres protéines, initiant ainsi une cascade d’amplification
tout en relâchant des anaphylatoxines. L’activation du complément peut se faire par
la voie classique, la voie alterne ou la voie des lectines. Les 3 voies permettent
d’activer le C3 et ensuite de former les complexes d’attaque membranaire (CAM).
L’activation du complément permet également le dépôt de fragments de protéines
sur les cellules cibles ce qui favorise leur phagocytose par les macrophages.
i. Voie classique
L’activation du complément par la voie classique nécessite la fixation d’anticorps de
classe IgM ou IgG sur la surface bactérienne. Une IgM est suffisante pour activer la
voie classique alors que plusieurs IgG sont nécessaires. Grâce aux pentraxines, la
voie classique du complément peut être activée indépendamment de tout anticorps.
Elle peut également être initiée par des virus, la plasmine, la thrombine ou la protéine
C-réactive. Toutes les étapes sont résumées dans la Figure 13, page 66 (De
Franco, Robertson, & Locksley, 2009; Williams, 2012).

66
Figure 13 : Voie classique d’activation du complément (Williams, 2012).
Le fragment C1q se fixe, grâce à sa partie globulaire au fragment Fc des
immunoglobulines engagées dans un complexe antigène-anticorps ou d’autres
molécules activatrices (comme des acides nucléiques, de la chromatine, des
filaments intermédiaires cytoplasmiques, des membranes ou des protéines
mitochondriales, les polysaccharides capsulaires des bactéries Gram+, le lipide A,
des porines et des structures liposaccharidiques des Gram-, ou encore des
structures synthétiques comme les nanotubes de carbone…). Cette fixation
provoque un changement de conformation qui lui permet de s’associer à deux
molécules de C1r et de C1s en présence de calcium pour former la C1-estérase ou
C1qr2s2. Après sa fixation à la surface cellulaire cible, le C1q active les C1r qui
clivent alors les deux molécules de C1s pour former la protéase C1s sérine active
(Carroll & Sim, 2011).
Le C1s actif clive alors les composants C2 et C4 :
C4 est clivé en C4a et C4b ;
C2 vient se fixer sur la membrane cellulaire sur laquelle est fixée l’anticorps et
est alors clivé en C2a et C2b.
Les fragments C2b et C4a sont relâchés et agissent comme des protéines
chimiotactiques alors que les fragments C2a et C4b forment la C3-convertase ou
C4bC2a. La C3-convertase transforme de nombreux C3 en C3a et C3b. Le fragment
C3a est relâché. Le clivage expose un site thioester très actif sur le fragment C3b ce
qui lui permet de se lier à des surfaces cellulaires grâce à des liaisons amines ou
esters et ainsi participer à l’opsonisation des cibles. Le C3b se fixe également de
façon covalente à la C3-convertase pour former la C5-convertase ou C4bC2aC3b qui

67
clive le composant C5 en C5a (qui est relâché) et C5b, ce qui marque le début de la
voie commune. Le fragment C5b se lie à la membrane cellulaire sur laquelle est fixée
l’immunoglobuline initiale, ce qui permet d’initier la formation du complexe d’attaque
membranaire (CAM). En effet, le composant C6 se fixe au C5b, formant ainsi le
complexe C5bC6 sur lequel vient se fixer C7. Ce dernier subit alors un changement
de conformation entrainant l’exposition de son fragment hydrophobe qui s’insère
dans la membrane bactérienne, jouant ainsi le rôle d’ancre pour le CAM en
formation. C8 se fixe à son tour sur C7 et guide 18 molécules de C9, qui
s’assemblent en un anneau créant ainsi un pore dans la membrane bactérienne. C9
possède une surface hydrophobe qui permet son insertion dans la bicouche lipidique
ainsi qu’une surface hydrophile, qui se retrouve au centre du CAM, autorisant ainsi le
passage de l’eau. Le gradient osmotique entre l’intérieur de la bactérie et le milieu
extracellulaire entraîne une forte entrée d’eau dans la bactérie et provoque la lyse
cellulaire donc la mort de la bactérie. Le pore ainsi créé favorise la perte des
composants intracellulaires et l’entrée dans la cellule de médiateurs immunitaires. Le
CAM est surtout efficace contre les bactéries Gram- et les virus enveloppés (Carroll
& Sim, 2011; De Franco et al., 2009; Williams, 2012).
ii. Voie alterne
La voie alterne n’a pas besoin d’opsonine, c’est pourquoi elle est la principale voie
d’activation du complément intervenant dans la réponse innée. Dans le milieu
extracellulaire, la protéine C3 n’est pas très stable et est spontanément hydrolysée
pour former C3(H2O) surtout en présence d’eau ou d’ammoniac qui attaque le
groupement thioester du C3 inactif. La molécule résultante est très proche du C3b et
se lie en présence de magnésium au facteur B, alors clivé par le facteur D en Ba
(relâché) et Bb qui est une sérine protéase inactive. Le complexe C3bBb est une C3-
convertase qui hydrolyse alors des molécules de C3 : une grande quantité de C3b
est formée et se lie au complexe déjà présent et forme la C5-convertase
C3bBbC3b qui clive le C5 en C5a et C5b. La suite de l’activation est identique à la
voie classique et aboutit à la mise en place du CAM. Voir Figure 14, page 68 (Carroll
& Sim, 2011).
La voie alterne utilise trois molécules de reconnaissance pour ses cibles : le facteur
H, la properdine et le C3b. Le C3b se lie de façon covalente à la cible en
reconnaissant certains sucres et acides aminés. La C3-convertase C3bBb est
instable et présente une demi-vie de 2-3 minutes. Le facteur Bb se dissocie du C3b.
La properdine ralentie la dissociation du complexe et agit ainsi en tant que
stimulateur de l’activité du complément. La properdine se fixe à la surface du C3b et
accélère sa fixation au facteur B : le complexe C3bBP est alors formé, et, après
action du facteur D, devient C3bBbP, qui est une C3-convertase stabilisée. De
récentes études ont également montré que la properdine peut aussi se fixer
directement sur la membrane des cellules cibles (zymosan, érythrocytes de lapin,
cellules apoptiques ou nécrotiques) et recruter des C3b, jouant ainsi le rôle
d’initiateur de l’activation du complément (Carroll & Sim, 2011).

68
Figure 14 : Voie alterne d’activation du complément (Williams, 2012).
iii. Voie des lectines
La voie des lectines ressemble beaucoup à la voie classique mais a une capacité
d’activation moindre et l’opsonisation est réalisée par le MBL, la ficoline ou encore la
collectine 11 qui circulent sous forme complexée avec des MASP. Les MASP sont
similaires aux C1r et C1s mais le C1q fixe 4 protéases (2C1r et 2 C1s) dans la voie
classique alors que le MBL et le ficoline ne fixent qu’une protéase à la fois. La
fixation de ces complexes sur une surface activatrice entraîne un changement de
conformation qui provoque l’activation des MASP rendant possible le clivage de C4
et C2 qui autorise alors la formation de la C3-convertase. La formation de la C5-
convertase puis des CAM est identique à la voie classique. Voir Figure 15, ci-
dessous (Carroll & Sim, 2011).
Figure 15 : Voie des lectines pour l’activation du complément (Williams, 2012).

69
d. Rôles du complément
Le complément est essentiel dans la défense de l’organisme en reconnaissant les
microorganismes envahisseurs et il présente une activité cytolytique vis-à-vis des
bactéries, virus ou parasites grâce à la formation de CAM. Cette action directe est
complétée par le rôle d’opsonine des fragments C3b, et C5b. Le clivage du C3 en
C3b permet l’exposition d’un groupe thioester qui autorise sa fixation sur les
microorganismes et le clivage engendre également un changement de conformation
du C3b, qui rend possible la fixation du C3b sur le récepteur CR1 (Complement
Receptor 1) des macrophages, et des cellules dendritiques, facilitant ainsi la
phagocytose. D’autres opsonines sont créées dans la cascade du complément
comme le facteur iC3b qui interagit avec les CR3 et CR4 des macrophages ou le
C3g issu de la protéolyse du iC3b, reconnu par les CR2 des lymphocytes B
(Williams, 2012).
Le complément aide à la clairance des cellules lésées ou altérées de l’hôte comme
les cellules apoptiques ou nécrotiques. Il nettoie les dépôts anormaux de protéines
comme la substance amyloïde (Carroll & Sim, 2011; Williams, 2012).
Il contribue à la mise en place de l’inflammation en relâchant des médiateurs pro-
inflammatoires. Le C5a est une molécule chimiotactique pour les macrophages et les
fragments C5a, C6 et C7 attirent les PNN sur le lieu de l’inflammation. Les
anaphylatoxines C3a, C4a et C5a provoquent la dégranulation des mastocytes et
PNB, ce qui libère de l’histamine, qui crée alors une vasodilatation locale (Williams,
2012).
Le complément constitue également un lien vital entre l’immunité innée et
l’immunité acquise en augmentant la réponse humorale et stimulant les
lymphocytes B. Il provoque une hyperleucocytose par l’intermédiaire du fragment
C3e (Mastellos & Lambris, 2002).
Enfin, le complément intervient dans le développement des os puisque le C3 régule
la différenciation des ostéoclastes et de nombreux composants comme le C3, le C5,
le C9, le facteur H et la properdine interviennent dans la transformation du cartilage
en os (Mastellos & Lambris, 2002).
Pour la reproduction, le complément intervient lors de la fusion des gamètes
puisqu’il est clivé par les enzymes acrosomales du spermatozoïde et les fragments
servent alors de ligands pour des récepteurs du spermatozoïde et de l’oocyste. Lors
de la gestation, le complément est essentiel pour maintenir la tolérance fœto-
maternelle grâce au DAF, MCP et CR1 (Mastellos & Lambris, 2002).
Le complément intervient dans la régénération d’organes, notamment la
régénération hépatique chez les mammifères, où le fragment C5a stimule la
prolifération hépatique grâce à une action paracrine et en association avec l’IL-6. De
façon plus anecdotique, le complément est essentiel pour la régénération d’un

70
membre comme chez les amphibiens, il joue alors un rôle dans le développement
des muscles (Mastellos & Lambris, 2002).
Dans l’hématopoïèse et l’angiogenèse, le C1q et le C3a interviennent dans les
stades précoces (Mastellos & Lambris, 2002).
Le détail de ces fonctions est présenté dans la Figure 16, page 70.
Figure 16 : Les différents rôles du complément (Mastellos & Lambris, 2002).

71
e. Mécanismes de régulation du complément
Comme toute partie du système immunitaire, le complément est très finement régulé
avant d’éviter d’une part, son activation contre le soi et d’autre part, un emballement
du système qui deviendrait néfaste pour l’organisme.
Pour éviter l’attaque du soi, des protéines régulatrices plasmatiques (facteur H,
facteur I, C4bp, C1 inhibitor) et des protéines régulatrices liées aux cellules (DAF,
CR1, CD59, MCP et CRIg) préviennent l’attaque du complément.
Le facteur I se fixe sur le fragment C3b et le sépare du facteur Bb ce qui arrête le
rétrocontrôle positif.
Le facteur H est une protéine plasmatique de 155 kDa qui possède plusieurs sites de
fixation du C3b, du C3d et du iC3b. Il peut également se fixer sur les surfaces
bactériennes comme Borrelia burgdorferi, Streptococcus pneumoniae, Streptococcus
pyogenes, et Yersinia enterocolitica,... Sa principale fonction est de réguler
l’activation de la voie alterne en se liant préférentiellement au C3b non lié, ce qui
empêche la formation de C3bBb et donc stoppe l’activation du complément et permet
au facteur I de cliver le C3b formant le iC3b. Il dissocie aussi le Bb du composant
C3b mais aussi en se fixant au C3b. Il régule aussi la voie classique en entrant en
compétition avec le C1q pour les cibles. Le facteur H peut former des polymères
permettant de protéger une plus grande surface des cellules hôtes en inactivant le
C3b. Le facteur H peut parfois reconnaître des cellules de façon inadéquate comme
lors du syndrome hémolytique urémique, ou la dégénérescence maculaire liée à
l’âge. De nombreux microbes recrutent le facteur H pour échapper à l’activation du
complément (Carroll & Sim, 2011; Ferreira, Pangburn, & Cortés, 2010; Williams,
2012).
La protection des cellules de l’hôte est assurée par des régulateurs négatifs liés à la
membrane. Par exemple, le CR1 (ou CD35) induisent la dissociation des
convertases de C3 et recrutent le facteur I. De la même façon, le facteur accélérateur
de la dégradation (DAF) dissocie les convertases et le cofacteur membranaire de
protéolyse (ou CD46), recrute le facteur I et stimule la protéolyse du C3b. Enfin, le
CD59 ou protectine empêche la formation du CAM (De Franco et al., 2009).
L’importance de ces protéines régulatrices est montrée par le rejet lors de
xénotransplantation, ou lors de déficience en DAF et CD59 entraîne l’hémoglobinurie
nocturne paradoxale (PNH) qui est la conséquence d’une hémolyse intravasculaire
causée par le complément par manque de protéines régulatrices sur les globules
rouges. De plus, ces deux protéines empêchent l’apparition d’une anémie
hémolytique auto-immune. L’activation inappropriée ou excessive du complément
provoque des dommages tissulaires comme l’arthrite rhumatoïde, la dégénérescence
maculaire liée à l’âge, la myasthenia gravis, la sclérose multiple, la lésion de
reperfusion (dans ce dernier cas des protéines mitochondriales et des

72
phospholipides exposés servent de néo-antigènes) (Carroll & Sim, 2011; D. D. Kim &
Song, 2006).
A l’inverse, une activation insuffisante du complément prédispose aux infections et
au développement de maladies auto-immunes comme le lupus érythémateux
systémique par perturbation de la clairance des cellules apoptiques (Carroll & Sim,
2011).
Enfin, en cas d’activation prolongée du complément, l’organisme compense la
surproduction de C3b par la formation d’auto-anticorps contre la C3-convertase. Ces
auto-anticorps permettent d’éliminer des C3-convertases et donc de diminuer la
concentration sérique du C3b.
Les protéines de la phase aiguë de l’inflammation 3)
Les protéines de la phase aiguë ou APP (pour Acute Phase Proteins) sont une
famille de protéines dont la concentration est modifiée lors d’infection,
d’inflammation, de traumatismes (chirurgical ou autre), ou même lors de stress. Elles
participent aux défenses de l’organisme en neutralisant les agents inflammatoires, en
minimisant les dommages tissulaires et en participant à la réparation tissulaire. La
synthèse de ces protéines a principalement lieu dans le foie et est fortement
influencée par les cytokines notamment, par l’IL-6, l’IL-1 et le TNF sécrétées lors de
la réponse inflammatoire. La concentration des APP est proportionnelle à la sévérité
et l’extension des lésions tissulaires (Evans, O’Brien, & Watterson, 2009).
Il existe trois types de protéines de la phase aiguë :
Les protéines de la phase aiguë positives, dont la concentration augmente
très fortement (de 10 à 100 fois) en quelques heures. Leur action dure tout le
temps de l’inflammation. Exemple : protéine C-réactive, glycoprotéine acide
α1, macroglobuline α2, fibrinogène, haptoglobine, amyloïde A ou encore le
MBL. Ces protéines sont plus ou moins importantes selon les espèces : la
protéine dont la concentration augmente le plus est, respectivement, chez le
chien et les primates non humains, la protéine C-réactive, chez le rat, la
macroglobuline-α2 et chez la souris, l’amyloïde A ;
Les protéines à réponse retardée augmentent en une à trois semaines
comme les immunoglobulines et le complément ;
Les protéines de la phase aiguë négative comme l’albumine, dont la
concentration diminue due à l’hémodilution ou au transfert vers l’espace
extravasculaire secondaire à l’augmentation de la perméabilité vasculaire, ou
à l’augmentation de la pression osmotique ;
Les caractéristiques des principales protéines de la phase aiguë sont regroupées
dans le Tableau III, page 73.

73
Tableau III : Principales protéines de la phase aiguë.
Protéine Poids
moléculaire
Lieu de
synthèse
Demi-
vie Rôle
Amyloïde A 12 kDa
Foie,
rate,
testicules
Régulation du métabolisme
des lipides
Glycoprotéine
acide α1 41-43 kDa Foie 24 h
Transport d’hormones et de
xénobiotiques cationiques,
immuno-suppression non
spécifique
Haptoglobine 85-100 kDa Foie 3 jours Fixation de l’hémoglobine
libre
Macroglobuline-
α2 780 kDa Foie Défense de l’hôte
Mannose
Binding Protein
MBP
400-700
kDa Foie
1-2
jours
Active le complément,
opsonine
Protéine C-
réactive
(famille des
pentraxines)
100-140
kDa Foie
Norma-
lisation
en 1-2
sem
Opsonise, active le
complément Nettoyage des
débris cellulaires
Thiostatin 56 kDa Foie
La plupart des changements physiologiques de la réponse systémique est tournée
vers une augmentation du métabolisme : les ressources pour l’énergie sont issues
d’une augmentation du catabolisme, la baisse de synthèse hépatique de l’albumine
augmente le pool d’acides aminés disponibles mais une part consistante de ces
ressources est utilisée pour la synthèse des protéines de la phase aiguë,
modulatrices de la réponse inflammatoire (Ceciliani, Ceron, Eckersall, & Sauerwein,
2012).
Le sérum amyloid A (SAA) est produit par le foie après stimulation par l’IL-6 et le
TNF-α. Il agit comme apolipoprotéine : il fixe le cholestérol et l’enlève du site
inflammatoire. Il récupère ainsi le cholestérol des cellules mourantes, prévenant alors
son accumulation sous forme de plaques arthérosclérotiques. Le SAA agit comme
molécule chimiotactique et stimule ainsi la migration, l’adhésion et l’infiltration
tissulaire des monocytes et des PNN. Le SAA est également une opsonine pour un
grand nombre de bactéries Gram- grâce à son interaction avec une protéine de leur
membrane externe (OmpA) et stimule ainsi la phagocytose par les macrophages et
les neutrophiles des bactéries Gram- (Ceciliani et al., 2012).
L’haptoglobine est composée de deux peptides de 20 kDa et de deux autres
peptides de 35 kDa liés par des ponts dissulfures. L’haptoglobine se fixe sur

74
l’hémoglobine durant l’hémolyse intravasculaire et chaque molécule peut fixer 2
dimères d’hémoglobine α-β ; une fois formé, le complexe est stable et retiré de la
circulation par les cellules de Kupffer du foie. En fixant l’hémoglobine, l’haptoglobine
joue un rôle antioxydant supplémentaire de stabilisation du fer, ce qui entraîne une
baisse des dommages oxydatifs de l’hémoglobine à elle-même, à l’albumine
(puisque le libre-échange de l’hème de l’hémoglobine à l’albumine est inhibé), aux
lipides et aux tissus comme le rein. En effet, en plus de sa toxicité oxydative,
l’hémoglobine libre peut fixer l’oxyde nitrite, récupérant le NO et limitant sa
biodisponibilité. L’haptoglobine interagit directement avec les PNN et diminue leur
activité en inhibant l’explosion respiratoire (Ceciliani et al., 2012).
Les autres facteurs humoraux 4)
Le facteur BPI (pour Bactericidal Permeability Increasing) est une protéine riche en
lysine qui se fixe sur le LPS, ce qui évite l’activation des TLR4, et détruit certaines
bactéries Gram- comme Escherichia Coli et Salmonella typhimurium (Beutler, 2003;
Douglas & Jane Wardrop, 2010).
Les anticorps innés sont produits en absence de toute infection. Ils ont une large
spécificité pour des antigènes microbiens et du soi et peuvent donc réagir à de
nombreux pathogènes. La plupart de ces antigènes naturels sont de classe IgM, la
classe la plus efficace pour activer le complément. Ils sont codés par des gènes
réarrangés qui n’ont pas subi de diversification supplémentaire par le processus
d’hypermutation somatique. Leur affinité est faible pour de nombreux pathogènes et
ils réagissent de manière croisée avec de nombreux antigènes. Il reste à déterminer
s’ils sont produits en réponse à la flore normale ou en réponse au soi. Ils seraient
produits par les cellules B-1 B. Ces cellules possèdent de nombreux récepteurs
d’antigènes des cellules B (BCR) auto-réactifs. La majorité de ces cellules CD5+ sont
appelées B-1a, produites durant le développement fœtal. La principale cytokine
responsable de la croissance des cellules B est l’IL-7. Puis un autre facteur de
croissance intervient : la TSLP. Ces cellules peuvent produire des anticorps naturels
et ces derniers sont importants pour la mise en place du réseau de cellules
dendritiques folliculaires dans la rate (Hardy, 2006; Janeway et al., 2009).
La synthèse de fibrinogène cloisonne le foyer d’infection (Beutler, 2003).
Ce qu’il faut retenir :
Les facteurs humoraux sont donc de petites protéines qui favorisent l’action
des cellules en les attirant sur le lieu de l’inflammation (rôle chimiotactique) et
en favorisant la clairance des pathogènes en stimulant la phagocytose (rôle
d’opsonisation). Seul le complément, grâce à son complexe d’attaque
membranaire, peut directement lyser ses cibles, sans intervention de cellules.

75
D- Les composants chimiques
Le système immunitaire inné s’appuie donc sur les barrières physiques, puis, si le
pathogène réussi à pénétrer dans l’organisme, les facteurs humoraux et cellulaires
entrent en jeu. Une dernière défense innée existe : la défense chimique qui regroupe
les peptides antimicrobiens et les réactifs oxygénés. Ces armes peuvent être
présentes dans les tissus ou les sécrétions ou à l’intérieur des cellules
précédemment décrites.
Les peptides antimicrobiens 1)
Les peptides microbiens peuvent être classés dans les facteurs humoraux ou dans
les facteurs chimiques puisque certains de ces composants sont des enzymes.
Les peptides antimicrobiens au sens large se divisent en trois groupes :
les enzymes digestives clivant les structures microbiennes comme le
lysozyme ;
les peptides fixant un élément essentiel pour les bactéries comme le fer ou le
zinc (calprotectine, lactoferrine) ;
les peptides brisant la membrane microbienne (Linde et al., 2007).
Les peptides antimicrobiens sont définis comme des polypeptides antimicrobiens,
encodés par des gènes et synthétisés par les ribosomes. Les antibiotiques produits
par les bactéries ou les champignons utilisés en médecine pour combattre les
infections ne sont pas synthétisés par des ribosomes et ce sont des peptides
incorporant des acides aminés atypiques. C’est pourquoi ils ne font pas partie des
peptides antimicrobiens au sens strict et ne seront pas abordés dans ce travail
(Linde et al., 2007). Ce sont des peptides très répandus puisqu’ils sont présents chez
les drosophiles où plus de 20 gènes sont dénombrés. Il s’agit d’un mécanisme de
défense ancien puisqu’il est également trouvé dans les plantes et les insectes. Chez
les mammifères, seulement deux familles sont présentes : les cathélicidines et les
défensines, exprimées respectivement par les épithéliums et les PNN. Ils sont
produits comme propeptides qui nécessitent un traitement par des protéases pour
être actifs. Ces peptides tuent les microbes en détruisant leurs membranes
biologiques, leur action est spécifique aux membranes microbiennes. Ces peptides
sont amphipathiques et cationiques : ils peuvent donc s’insérer dans la membrane
pour y former des pores. La plupart des microbes ont une membrane riche en acide,
chargée négativement et les peptides antimicrobiens sont chargés positivement
grâce aux résidus d’arginine et de lysine et peuvent donc s’en approcher aisément. A
l’inverse, les cellules des eucaryotes possèdent une forte charge négative issue de
l’acide sialique, des glycoprotéines et des glycolipides ; cette charge positive tient les
peptides éloignés de la membrane. Le potentiel des membranes eucaryotes est de -
15 mV, ce qui est faible par rapport à celui des bactéries (-140 mV). L’efficacité de
ces peptides dépend de la bactérie, le spectre d’action est large mais certains

76
peptides ont des spécificités distinctes. Les peptides antimicrobiens sont présents,
dans les conditions physiologiques à des concentrations non antimicrobiennes. Les
concentrations physiologiques en sel, en particulier NaCl, et en ions Ca2+ et Mg2+
inhibent l’activité antimicrobienne des peptides. Par contre dans des conditions
particulières comme dans les phagosomes des leucocytes, les peptides
antimicrobiens participent activement à la destruction de la bactérie ingérée (Beutler,
2003; De Franco et al., 2009; Rainard & Riollet, 2006).
Ces peptides antimicrobiens rompent des composants très conservés et essentiels
pour la survie bactérienne comme les membranes de surface (ciblées par les
défensines et cathélicidines qui forment des pores) ou le peptidoglycane des Gram+
(ciblé par les lectines de type C). Cette stratégie permet de réguler les bactéries
pathogènes et commensales et limite la résistance des bactéries à ces peptides. Des
variations régionales dans la production de peptides antimicrobiens existent tout le
long de l’axe du tractus intestinal. Des interactions entre les peptides antimicrobiens
et les mucines permettent de concentrer les peptides antimicrobiens à la surface
épithéliale (Peterson & Artis, 2014).
Les peptides antimicrobiens ont la capacité de cibler tout organisme avec un
cholestérol libre et une membrane chargée négativement. Ainsi, ils sont actifs contre
les bactéries Gram+ et Gram-, les mycobactéries, les champignons, les parasites
intracellulaires et les virus enveloppés. Enfin, ils sont également capables de tuer
des cellules cancéreuses. Certaines études montrent également que les défensines
peuvent augmenter la cytotoxicité des NK. Enfin, ils interviennent dans la guérison
des blessures et la modulation des processus inflammatoires en inhibant l’activation
de la voie classique du complément (Linde et al., 2007).
Selon la cellule, ces peptides exercent une fonction intracellulaire (en participant à la
destruction oxygène-indépendante du microbe phagocyté) ou extracellulaire (ils
attaquent directement la paroi microbienne). Ils inhibent l’activité bactérienne,
fongique et virale. Ils sont stockés dans des granules dans les PNN et cellules de
Paneth ou sécrétés par les monocytes, macrophages, mastocytes, NK, kératinocytes
et cellules épithéliales. Ce sont les médiateurs du dialogue entre immunité innée et
adaptative (Auvynet & Rosenstein, 2009).
Le lysozyme, est présent dans les sécrétions et peut détruire la paroi des bactéries
Gram- et Gram+ grâce à un mécanisme enzymatique. Il est très concentré dans les
larmes et la salive et a été le premier peptide antimicrobien identifié (Beutler, 2003;
Paul, 2013).
La lactoferrine est une protéine de 80 kDa, de la famille des transferrines : elle
séquestre le fer. Elle diminue ainsi la disponibilité du fer pour les bactéries, altérant
alors leur métabolisme et elle compromet donc leur faculté à se répliquer. La
lactoferrine est trouvée dans les granules secondaires des PNN et dans le lait à de
fortes concentrations où elle provient alors des cellules épithéliales. La lactoferrine
peut aussi être traitée pour donner un peptide antimicrobien, la lactoferricine qui lyse

77
les membranes microbiennes. Cette protéine sera détaillée dans le paragraphe dédié
à l’immunité de la mamelle, page 167 (Paul, 2013).
Les défensines sont des peptides cationiques de 30 à 40 acides aminés constitués
de feuillets β à 3 brins avec 6 cystéines et 3 ponts disulfures. Il en existe plus d’une
centaine, regroupées en trois types, selon la position de ces ponts :
Les α-défensines. Les PNN en produisent et les stockent dans leurs
granules primaires. Les cellules de Paneth des cryptes intestinales
produisent une classe d’α-défensine appelées cryptidines. Elles sont aussi
produites par les leucocytes et les cellules épithéliales du tractus génital
femelle. Elles possèdent une activité antivirale (notamment contre le virus
de l’influenza et le HIV, l’herpès simplex virus, le cytomégalovirus et les
adénovirus…) ;
Les β-défensines. Elles sont synthétisées de manière constitutive par les
cellules épithéliales en contact direct avec l’environnement ou la flore
microbienne comme les poumons, les glandes salivaires, les glandes
mammaires, la prostate, ainsi que par les leucocytes. Son expression est
augmentée par le LPS et le peptidoglycane. Elles sont également
présentes dans le foie, le cœur, le placenta, et les testicules… Les β-
défensines 1 et 2 possèdent une activité contre les bactéries Gram- et
certains champignons alors que les β-défensines 3 tuent de nombreux
bactéries Gram+, Gram- et des levures opportunistes comme Candida
albicans. L’expression des β-défensines est stimulée par l’IFN-γ, le TNF-α,
l’IL-17 et l’IL-22 et inhibée par l’acide rétinoïque. Des concentrations
plasmatiques de β-défensines normales chez l’homme sont de l’ordre de
40 ng/mL et peuvent augmenter jusqu’à plus de 1µg/mL durant une
infection ;
Les θ-défensines. Ces dernières sont des molécules circulaires à 2
chaînes alors que les 2 autres catégories sont des feuillets β à 3 couches
plats. Elles ne sont pas présentes chez les humains, mais chez les autres
primates. Leur rôle antimicrobien est reconnu mais leur localisation précise
et leur mode d’action restent à déterminer (Auvynet & Rosenstein, 2009;
De Franco et al., 2009; Linde et al., 2007).
Les cathélicidines possèdent une structure en hélice α. Elles ressemblent aux α-
défensines mais ne possèdent pas de ponts disulfures. Elles sont produites par les
cellules épithéliales, les phagocytes et les kératinocytes et de façon constitutive dans
le thymus, la rate, la moelle osseuse, le foie, l’estomac, l’intestin et les testicules.
Dans les PNN, elles sont également produites sous forme de pro-peptides avec une
grande variété de séquences C-terminales puis traitées par des élastases dans les
granules secondaires avant d’être sécrétées en réponse à un stimulus microbien.
Les humains et la souris n’ont qu’un seul gène codant pour les cathélicidines,
contrairement aux porcins et aux bovins. Les cathélicidines tuent les bactéries

78
Gram+ et Gram-, ainsi que le trypanosome et fonctionnent comme des molécules
anti-inflammatoires ; elles abrogent aussi l’expression des molécules pro-
inflammatoires comme le TNF-α et IL-6 et la translocation nucléaire du NF-κB
(Auvynet & Rosenstein, 2009; Beutler, 2003; Paul, 2013).
Les histatines sont produites dans les glandes de la cavité orale (glandes parotide,
sublinguale et submandibulaire) et ciblent les membranes fongiques (Paul, 2013).
La phospholipase A2 (PLA2) est un autre peptide antimicrobien trouvé dans les
granules primaires des PNN, très similaires aux défensines. Il est également sécrété
par les cellules épithéliales et les cellules de Paneth. La PLA2 peut tuer directement
des bactéries Gram+ en hydrolysant les phospholipides de la membrane
microbienne. La distinction avec les membranes de l’hôte se fait grâce au BPI qui se
fixe au LPS des bactéries Gram-. Le BPI rompt l’intégrité membranaire bactérienne,
ce qui permet au PLA2 d’accéder au phospholipide (Paul, 2013).
La mauvaise régulation des peptides antimicrobiens est associée à des maladies
comme le psoriasis, la dermatite atopique ou la maladie de Crohn (Auvynet &
Rosenstein, 2009).
A ce jour, chez le chien, trois β-défensines et une cathélicidine ont été identifiées,
mais aucune n’a été découverte chez le chat. Chez les chevaux, deux peptides
antimicrobiens et trois cathélicidines ont été découverts dans les neutrophiles (Linde
et al., 2007).
Les réactifs oxygénés 2)
Des réactifs oxygénés sont produits par les neutrophiles et les macrophages pour
détruire les microbes. Cette synthèse est initiée par la NADPH-oxydase. Par une
réaction en chaîne, des ions superoxydes sont d’abord formés, ensuite transformés
en peroxyde d’hydrogène (H2O2) qui devient, grâce à la myéloperoxydase (MPO), de
l’acide hypochloreux (HOCl). Ce dernier est un antimicrobien très puissant, arme
centrale des neutrophiles et il peut également donner un oxygène singulet ou un
radical hydroxyle, tous deux très réactifs (Beutler, 2003).
Voici, page 79, Figure 17, l’ensemble des réactions qui permettent d’aboutir aux
réactifs oxygénés, encadrés en rouge dans le schéma.

79
Figure 17 : Mécanisme de formation des réactifs oxygénés antimicrobiens
(Beutler, 2003).
Après avoir rappelé les acteurs de l’immunité innée, nous allons étudier les
conditions dans lesquelles cette immunité se met en place, et celles où elle
n’intervient pas.
II. Quand mettre en place une immunité innée ?
Le système immunitaire doit s’activer lorsqu’un agent pathogène pénètre dans
l’organisme mais ne doit pas s’activer sans danger, car il risquerait de léser
l’organisme sain.
Ce qu’il faut retenir :
L’organisme possède donc un large éventail de défenses contre des
pathogènes variés. Ces moyens sont très efficaces mais peuvent s’avérer
dangereux pour l’organisme. C’est pourquoi, ils sont très finement régulés
(cette régulation concerne les cellules, comme les facteurs humoraux et
chimiques).

80
Les signaux de danger reconnus par l’organisme A-
L’immunité innée a trois approches pour détecter des microorganismes étrangers.
La détection des structures microbiennes répandues grâce à un nombre limité
de récepteurs : ce sont les PAMPs ;
La reconnaissance des conséquences métaboliques de l’infection ou de
l’inflammation : ce sont les DAMPs ;
Enfin, la détection du «missing self». Ce sont des molécules normalement
exprimées par les cellules mais qui ne le sont plus lorsque la cellule est
infectée par un microbe (Turvey & Broide, 2010).
On estime à 103, le nombre de molécules reconnues par le système immunitaire
inné. En comparaison, le système adaptatif peut reconnaître plusieurs milliards (109)
d’épitopes différents.
Les PAMPs 1)
Un PAMP (Pathogen-Associated Microbial Pattern) est un composant commun à un
large nombre de microbes mais pas aux cellules des mammifères. La plupart des
PAMPs sont des molécules essentielles à l’intégrité, au fonctionnement ou à la
réplication des microbes. De ce fait, l’agent infectieux ne peut pas échapper
facilement à la détection en les modifiant car toute mutation qui permettrait
d’échapper au système immunitaire est rarement viable. En effet, une mutation sur
les gènes des enzymes synthétisant le LPS des bactéries Gram- est en général
létale, de même que l’ARN double brin est une étape obligatoire pour la réplication
des virus à ARN. Un des premiers PAMPs identifié a été le lipopolysaccharide de la
paroi des bactéries Gram-. Les acides nucléiques typiques des microbes (ARN
double brin, ADN simple brin…) sont des PAMPs, mais les fragments de protéines
propres aux microbes sont aussi des PAMPs, comme la N-formylméthionine, le
lipopolysaccharide, l’acide lipoteichoïque et les oligosaccharides riches en mannose
(Abbas et al., 2012; De Franco et al., 2009).
Parmi les PAMPs, il y a de nombreux composants des parois bactériennes pour
plusieurs raisons. Tout d’abord, il s’agit de structures spécifiques aux bactéries et
cela permet donc de distinguer le soi du non soi. Ensuite, comme nous venons de
l’évoquer, des mutations sur les gènes de ces composants pariétaux sont rarement
viables. Voici présenté page 81, Figure 18, un rappel concernant les structures des
parois des bactéries Gram+ et Gram- afin de resituer les nombreux PAMPs évoqués
dans ce travail.

81
Figure 18 : Structure des parois des bactéries Gram+ et Gram- (Janeway et al., 2009).
Les DAMPs 2)
Un DAMP (pour Damage-Associated Molecular Pattern) est une molécule présente
dans l’hôte, surexprimée ou libérée pendant la lyse cellulaire et les dommages
tissulaires rencontrés lors d’infection virale ou de transformation. La libération de
cette molécule peut être la conséquence directe de l’infection ou être secondaire à
l’inflammation engendrée. L’ADN mitochondrial est un très bel exemple de DAMP,
puisqu’il n’est normalement pas en contact avec les cellules immunes. Les DAMPs
ne sont généralement pas relâchés par les cellules en apoptose (Abbas et al., 2012;
Turvey & Broide, 2010).
Les DAMPs sont des facteurs très nombreux et variés comme la protéine de haute
mobilité, la protéine de choc thermique (hsp90), l’acide hyaluronique et d’autres
composants de la matrice extracellulaire, le versican, la tenascine-C, des produits de
la réponse inflammatoire comme le serum Amyloid A, l’héparine, les sulfates, des β-
défensines ou des produits de coagulation comme le fibrinogène. Beaucoup de ces
DAMPs sont détectés par le TLR4, mais les cellules nécrotiques libèrent de l’ADN
double brin, détecté par le TLR3 (Paul, 2013).
La protéine de choc thermique ou protéine HMGB-1 (pour High Mobility Group Box)
est présente dans le noyau de toutes les cellules eucaryotes où elle stabilise le
nucléosome et régule la transcription de nombreux gènes. La protéine maintient
l’intégrité chromosomale et est considérée comme centrale dans l’inflammation. Elle
est sécrétée par les macrophages, les cellules dendritiques et les NK en réponse à
des blessures, à l’infection ou tout autre stimulus inflammatoire. La HMGB-1 est
exprimée durant le développement embryonnaire mais chez l’adulte, elle est
restreinte aux organes lymphoïdes et aux testicules. La HMGB-3 est exprimée chez

82
l’embryon et les cellules souches hématopoïétiques. Les HMGB sont impliquées
dans les réponses du système immun inné activé par les acides nucléiques. Elle est
requise pour l’activation complète du système immun inné induit par l’acide nucléique
par les PRR (Yanai, Ban, & Taniguchi, 2011).
L’acide urique est aussi un DAMP. Il provient de la dégradation des purines. Son
accumulation aboutit chez l’homme à des crises de goutte.
Les récepteurs B-
Un nombre important de récepteurs est dédié à la reconnaissance de molécules
microbiennes et ils ont été conservés à travers l’évolution puisqu’ils sont très
largement répandus. Les récepteurs de l’immunité innée sont appelés PRR ou
Pattern Recognition Receptor. Il s’agit d’une grande famille de récepteurs qui
reconnaissent les PAMPs ou les DAMPs. Une fois activés, les PRR modifient
l’expression des gènes de la cellule, ce qui permet au mécanisme effecteur de se
mettre en place.
Ces récepteurs sont présents sur les membranes cellulaires ou endosomales de
différents types de cellules et sont également présents dans le cytoplasme de ces
cellules comme indiqué sur la Figure 19, page 83. Ces localisations variées assurent
la réponse du système immunitaire aux microbes extracellulaires et intracellulaires
(Abbas et al., 2012; Paul, 2013).
Ces PRR incluent de nombreuses familles de molécules, comme les Toll Like
Receptor (TLR), les C-type Lectine Receptor (CTLR), les récepteurs éboueurs
(scavengers), tous des récepteurs transmembranaires ; ou encore les NOD-like
récepteurs (Nucleotide Binding Oligomerization Domain), RIG-like Receptor (RLR),
intra-cytoplasmiques (Geremia et al., 2013; Paul, 2013).
Ce qu’il faut retenir :
Le système immunitaire inné s’active en reconnaissant des motifs spécifiques
et bien définis, mais très répandus dans le règne microbien. Ceci permet de
couvrir un grand nombre de pathogènes en évitant également l’auto-immunité.
Enfin, lors d’inflammation stérile, le système immunitaire reconnait des
alarmines produites par les cellules en souffrance.

83
Figure 19 : Localisations cellulaires des différents PRR (Abbas et al., 2012).
Les Toll-Like Récepteurs ou TLR 1)
Les TLR sont une famille de récepteurs conservés au cours de l’évolution (ils sont
présents chez les plantes, insectes et mammifères), et exprimés par un large nombre
de types cellulaires. Treize TLR différents ont été identifiés chez les mammifères
dont neuf chez l’homme. Alors que le domaine cytoplasmique est très proche du
domaine intracellulaire du récepteur à l’IL-1, de fortes variations existent sur le
domaine extracellulaire (Beutler, 2003; De Franco et al., 2009; Lotz et al., 2007;
Paul, 2013).
La « spécificité » des TLR est augmentée par la formation de dimères. Par exemple,
le dimère formé par le TLR2 et le TLR6 détectent le peptidoglycane. La spécificité est
aussi augmentée grâce à des molécules accessoires comme le LBP (Liposaccharid
Binding Protein) sur lequel se fixe le LPS dans le sang ou le liquide extracellulaire ce
qui facilite sa fixation sur le TLR4 (Abbas et al., 2012; Hopkins & Sriskandan, 2005).
L’expression des TLR varie selon le stade de différenciation des leucocytes.
L’activation des cellules B par les TLR mène à la production d’anticorps naturels, qui
appartiennent aux réponses non spécifiques des lymphocytes B. L’expression des
TLR est dynamique et diminue avec l’âge et il existe une variation dans la réponse
TLR selon la dose de pathogène et le temps (Coutinho & Poltorack, 2003; Hopkins &
Sriskandan, 2005).

84
a. Structure
Les TLR sont des protéines dimériques. L’ectodomaine des TLR est composé d’un
domaine riche en leucine (ou Leucin Rich Repeats : LRR) qui permet l’interaction
avec le ligand et la composante cytoplasmique, globulaire, est appelée domaine TIR
(Toll/IL Receptor) et est impliquée dans la signalisation intracellulaire (Paul, 2013;
Uematsu & Fujimoto, 2010).
b. Activation du récepteur par son ligand
Chaque TLR reconnaît un ou plusieurs ligands. Ce sont donc des récepteurs
spécifiques d’un ligand, mais ce ligand est un PAMP et est donc très répandu dans le
monde microbien. C’est pourquoi ces récepteurs appartiennent à l’immunité innée.
La diversité des ligands de cette famille de récepteur est exceptionnelle : acides
nucléiques, protéines, lipides et polysaccharides. Ce sont donc des molécules non
spécifiques d’un pathogène mais très répandues (De Franco et al., 2009).
Les neutrophiles, les cellules NK, les mastocytes, les PNE, les cellules B et les
monocytes ont tous un modèle d’expression de TLR caractéristique avec des
conséquences immunologiques directes. L’expression des TLR discrimine les
cellules dendritiques plasmacytoïdes et myéloïdes (Hopkins & Sriskandan, 2005).
Les TLR1, 2, 4, 5, et 6 sont exprimés sur la membrane plasmatique et interviennent
dans la détection de produits bactériens, fongiques et protozoaires. A l’inverse, les
TLR3, 7, et 9 sont exprimés dans les membranes endosomales avec leur LRR dans
la lumière et leur TIR dans le cytosol : ces TLR intracellulaires détectent les acides
nucléiques viraux. Les TLR2 et 4 détectent des structures spécifiques aux virus et
aux bactéries. Tous les TLR ont une signalisation intracellulaire via le MyD88 vers le
NF-κB sauf le TLR3 qui signale via TRIF vers le NF-κB et d’autres facteurs de
transcription comme l’IRF3. Le TLR4 peut également signaler avec TRIF. Les
principales voies de signalisation seront vues dans le paragraphe « Activation d’une
cascade intracellulaire », page 89 (Novák, 2014; Paul, 2013).
La plupart des récepteurs reconnaissent directement leur ligand, d’autres ont besoin
de protéines accessoires. C’est le cas du TLR4 dont la reconnaissance du LPS
nécessite deux protéines accessoires et une protéine soluble de transfert lipidique.
En effet, la reconnaissance du LPS par TLR4 est très complexe : le LPS est un lipide
hexa-acylé et 5 des acyls sont entourés par le cofacteur MD-2 alors que le sixième
interagit avec le TLR4. Au total, trois molécules en plus du TLR4 sont nécessaires
pour reconnaître le LPS :
Le LBP, dans le sérum, se fixe sur les monomères de LPS et les transfère au
CD14. Il permet d’extraire les monomères des LPS des cellules bactériennes
pour les présenter aux cellules immunes ;
Le CD14 peut être une protéine soluble ou ancrée. Elle concentre le LPS et le
délivre au MD-2. C’est elle qui accepte le LPS apporté par le LBP ;

85
Le MD-2 se fixe au domaine extracellulaire du TLR et permet de lier la région
interne du LPS mieux conservée phylogénétiquement que sa région externe.
Cette reconnaissance complexe peut s’expliquer par le fait que l’activation du TLR4
par le LPS peut avoir des conséquences létales comme lors de choc septicémique,
c’est pourquoi plusieurs molécules sont nécessaires pour détecter le LPS (De Franco
et al., 2009; Paul, 2013).
La distinction entre le soi et le non-soi est faite indirectement par ces récepteurs : les
ligands sont spécifiques ou au moins très surexprimés, par les agents pathogènes et
ne sont pas ou peu présents chez l’hôte. Par exemple, l’ARN double brin reconnu
par le TLR3, est spécifique de la réplication des virus à ARN. Bien que des portions
d’ARN double brin soient présentes dans les ARN simples brins des cellules
apoptiques, ceux-ci restent rares. Les TLR7 et 8 reconnaissent l’ARN simple brin
mais riche en guanosine, le TLR9 reconnaît des motifs d’ADN avec le dinucléotide
CG dans lequel le C n’est pas méthylé (ADN CpG). Ce dinucléotide est sous
représenté environ 10 fois dans l’ADN des vertébrés. Par contre, la spécificité des
récepteurs intracellulaires qui détectent les acides nucléiques est assez faible mais
ils sont localisés dans les endosomes. Or l’ADN et l’ARN de la cellule hôte n’est pas
présent dans ces endosomes alors que les acides nucléiques microbiens le sont
(Abbas et al., 2012; Lester & Li, 2014).
Un microbe présente à sa surface plusieurs motifs reconnus, chacun étant capable
d’activer une réponse. De plus, chaque motif peut activer plusieurs TLR
simultanément. C’est pourquoi l’activation du système immunitaire par un pathogène
permet l’induction de la traduction d’un grand nombre de gènes dans des cellules
comme les macrophages ou les PNN. Cette diversité de TLR impliqués pour un seul
pathogène explique la complexité de la réponse immune. Ceci permet de coordonner
des réponses cellulaires avec la libération de cytokines pro-inflammatoires, la
destruction microbienne oxydative, et l’apoptose (Hopkins & Sriskandan, 2005).
Les TLR et leurs caractéristiques sont détaillés dans le Tableau IV, page 86.

86
Tableau IV : Les TLR et leurs caractéristiques. D’après (Abbas et al., 2012; De Franco et al., 2009; Hopkins & Sriskandan, 2005; Paul, 2013; Turvey & Broide, 2010; Uematsu & Fujimoto, 2010; Zhang et al., 2013).
TLR CELLULES LIGANDS PARTICULARITES VOIES DE
SIGNALISATION
RECEPTEURS SUR LA MEMBRANE CYTOPLASMIQUE
TL
R1
Tous les types cellulaires
Lipoprotéine
Lipopeptide triacylé des mycobactéries
Facteurs solubles bactériens
MyD88 ; IRAK ;
TRAF ;
NF-κB
TL
R2
Macrophages,
cellules dendritiques myéloïdes,
cellules B
Treg
Cellules NK
Cellules γδ T
Bactéries : Peptidoglycane, LPS,
lipoprotéines, lipopeptide, l’acide
lipoteichoïque et lipoarabinomannane
des mycobactéries, porines
Virus : protéines de la paroi virale
Levures : zymosan (composant
membranaire)
Fungi : phospholipomannane
Parasites : glycosylphosphoinositol
Endogène : défensines, HSP70, HSP60
Expression augmentée si
LPS et ARNdb, IL-2 IL-5 IL-
1β IFN-γ TNF-α
MyD88 ; IRAK ;
TRAF ;
NF-κB
TL
R4
Macrophages, monocytes,
mastocytes, cellules dendritiques,
PNN, endothélium : membrane
cytoplasmique
Cellules épithéliales, cellules γδ T,
cellules endothéliales : Golgi
Bactéries : LPS et acide lipoteichoïque
Virus : protéine F du VRS
Endogène : HSP70, HSP90 ;
fibronectine, héparine, acide
hyaluronique, fibrinogène
Dimère
Nécessite un cofacteur :
MD2, ainsi que le LBP et le
CD14
MyD88 ; IRAK ;
TRAF ;
NF-κB
Ou, dans les
endosomes : TRIF ;
NF-κB / IRF3

87
TLR CELLULES LIGANDS PARTICULARITES VOIES DE
SIGNALISATION
TL
R5 Cellules NK, mDC, monocytes,
surface basolatérale des cellules
épithéliales
Flagelline des flagelles bactériens
MyD88 ; IRAK ;
TRAF ;
NF-κB
TL
R6
Cellules B, mastocytes Lipopeptide diacylé du mycoplasme
MyD88 ; IRAK ;
TRAF ;
NF-κB
TL
R1 /
TL
R2
Lipopeptides triacétylés, Lipoprotéines,
Lipoarabinomannane, peptidoglycane
MyD88 ; IRAK ;
TRAF ;
NF-κB
TL
R2 /
TL
R6
Lipopeptide diacétylés Nécessité un cofacteur :
CD36
MyD88 ; IRAK ;
TRAF ;
NF-κB
RECEPTEURS SUR LES MEMBRANES ENDOSOMALES
TL
R3
Macrophages, cellules
dendritiques, T CD8+, NK, cellules
épithéliales rétiniennes,
cornéennes, intrahépatiques et
intestinales, kératinocytes
ARN double brin (virus) TRIF ; NF-κB / IRF3
TL
R7 Cellules dendritiques
plasmacytoïdes (+/- ? myéloïdes),
PNE,
Virus ; ARN simple brin Induit la synthèse d’IFN de
type I
MyD88 ; IRAK ;
TRAF ;
NF-κB / IRF7

88
TL
R8
Cellules NK, T ARN simple brin
Oligonucléotides riches en G
MyD88 ; IRAK ;
TRAF ;
NF-κB
TL
R9
Cellules dendritiques
plasmacytoïdes, macrophages,
épithélium, cellules NK, B
Motifs CpG non méthylés de l’ADN des
bactéries et virus, ADN simple ou double
brin
Production de TNF et IFN
de type I
Nécessite une protéine
Un93b pour l’insérer dans
les vésicules
intracellulaires
MyD88 ; IRAK ;
TRAF ;
NF-κB / IRF7
TL
R10
Cellules dendritiques
plasmacytoïdes, cellules B ?
MyD88 ; IRAK ;
TRAF ;
NF-κB
TLR CELLULES LIGANDS PARTICULARITES VOIES DE
SIGNALISATION

89
c. Activation d’une cascade intracellulaire
Les voies de signalisation des TLR passent par deux protéines adaptatrices MyD88
et TRIF, qui fournissent une plateforme centrale pour propager les signaux
complexes dérivés des TLR via une association cytoplasmique avec deux kinases
(IRAK et RIP) et des facteurs de transcription (Hopkins & Sriskandan, 2005).
Tous les TLR sauf le TLR3 ont une voie de signalisation qui implique MyD88 et
activent le NF-κB et donc une réponse inflammatoire. Pour le TRL 3, le signal passe
par le TRIF et active l’IRF-3 et donc l’expression des interférons de type 1. Le TLR4
peut agir avec MyD88 et TRIF. Le CD14 peut dicter l’adaptateur utilisé par le TLR4.
Quand il est fixé sur le LPS, il induit la voie du TRIF-TRAM et quand il est absent,
c’est la voie du MyD88 qui est utilisée. Il semblerait donc que les corécepteurs ne
soient pas seulement impliqués dans la reconnaissance du ligand, mais aussi dans
la signalisation (Abbas et al., 2012; Hou, Reizis, & DeFranco, 2008; O’Neill, 2006).
Voici dans la Figure 20 ci-dessous, les différentes voies de signalisation possibles
selon les récepteurs et les cytokines produites.
Figure 20 : Voies de signalisation possibles suite à l'activation des TLR dans l’exemple de la reconnaissance de PAMPs viraux (Lester & Li, 2014).

90
Pour plus de clarté, nous détaillerons d’abord les molécules impliquées dans ces
voies puis nous détaillerons les deux voies de signalisation.
La cinétique des voies de signalisation est inhérente à chaque voie. Par exemple,
l’activation d’IRF-3 par TRIF est plus rapide suite à l’activation de TLR3 que TLR4,
ce qui facilite une réponse antivirale à l’IFN-β alors que l’activation du NF-κB semble
être sous un contrôle à deux phases (signalisation MyD88-dépendante précoce, et
indépendante de MyD88 plus tardivement) (Hopkins & Sriskandan, 2005).
i. Les molécules impliquées
Les cascades intracellulaires permettent la transduction du message du TLR vers le
noyau pour initier la transcription des gènes impliqués dans la réponse. Le type de
cytokine produit est dépendant du TLR. Les TLR cytoplasmiques induisent la
production d’IL-6, de TNF et de la chimiokine IL-8 alors que les récepteurs
endosomaux induisent des interférons de type I.
Le MyD88 est une protéine adaptatrice qui capte le signal émis par le TLR lors de la
fixation du ligand.
Les IRAK (pour IL-1 Receptor Associated Kinases) sont des protéines kinases qui
peuvent s’activer les unes et les autres. Elles possèdent un domaine de mort
(« death domain »), capable d’interagir avec celui de MyD88. Ce domaine est un
module d’interaction entre des protéines, constitué de six hélices α. Les domaines de
mort de deux protéines interagissent entre eux ou avec d’autres domaines (comme
les LRR, ou les domaines TIR…). Ces motifs sont également impliqués dans la
régulation de l’apoptose et dans l’inflammation grâce à leur activation de caspases.
Ils sont connus pour leur rôle dans l’initiation de la cascade de signalisation des
récepteurs aux TNF.
Le NF-κB est un facteur de transcription hétérodimérique intervenant dans un grand
nombre de fonctions physiologiques et pathologiques comme l’inflammation les
réponses immunes et la survie cellulaire. Dans des cellules non stimulées, le NF-κB
se fixe sur des protéines inhibitrices IκB et est alors séquestré dans le cytoplasme.
Après stimulation, l’IκB est phosphorylé par la IκB kinase (IKK) qui est elle-même
phosphorylée et donc activée par la TAK1 (TGF-β activated kinase 1) (Chen & Chen,
2013).
L’ubiquitine est une petite protéine de 76 acides aminés très conservée chez tous
les eucaryotes, de la levure aux humains. L’acide carboxylique de la glycine de
l’ubiquitine s’attache de façon covalente à l’amine d’une lysine d’une autre protéine.
Cette fixation, favorisée par des ubiquitine-ligases comme le Cbl, est appelée
ubiquitinylation. Ensuite des protéines reconnaissent l’ubiquitine fixée sur la protéine
cible et dirigent les protéines ubiquitinylées vers les voies de dégradation.
L’ubiquitine permet donc de dégrader les récepteurs membranaires dans les
lysosomes et les protéines cytosoliques dans les protéasomes. Cependant, cette

91
ubiquitinylation peut être réversée par désubiquitinylation par des enzymes
spécifiques. L’ubiquitinylation joue un rôle essentiel dans la régulation des voies du
NF-κB, bien que ce rôle soit encore flou. Une molécule clé serait NEMO (pour NF-κB
Essential Modulator), qui contient deux ubiquitines, permettant de transformer le
signal de l’ubiquitinylation en activation de l’IKK (Chen & Chen, 2013; Janeway et al.,
2009).
Le Mal est le second adaptateur découvert. Il possède lui aussi un domaine TIR. Il
intervient avec le TLR4 et active lui aussi le NF-κB. Le TRIF est aussi une protéine
adaptatrice (pour TIR Related Adaptator Protein Inducing Interferon) recrutée par le
TLR3 et le TLR4, responsable de l’activation de l’IRF3 via une kinase appelée TBK-
1. Une 4ième molécule est le Trif Related Adaptator Molecule ou TRAM qui n’agit
qu’avec le TLR4. L’engagement du TRIF avec le TBK1 autorise la production d’IFN-
β. Le Mal et MyD88 sont requis pour l’activation précoce du NF-κB alors que TRIF et
TRAM sont nécessaires pour son activation plus tardive (O’Neill, 2006).
L’IRF-7 est un facteur de transcription clé pour l’induction de l’IFN de type I et il est
impliqué dans les voies MyD88 et TRIF. L’IRF-7 s’associe avec l’IRAK1 et l’IRAK4.
L’IRF-5 est activé par le MyD88 par de nombreux TLR. Il est essentiel pour
l’induction des gènes de protéines pro-inflammatoires comme l’IL-6, l’IL-12 et le TNF
mais pas pour l’IFN-α (O’Neill, 2006).
ii. La signalisation MyD88 dépendante
Dans la voie dépendant du MyD88, l’adaptateur est recruté directement à la queue
cytoplasmique du TRL. Après activation de son domaine TIR par l’interaction de son
domaine de mort avec celui du TLR, MyD88 recrute les IRAK qui fixent alors
l’adaptateur TRAF-6 activant ainsi une MAPKKK (Mitogen Activated Protein Kinase
Kinase Kinase) appelée TAK-1 qui phosphoryle et active le complexe iKK. Cette voie
fait intervenir les IRAK au nombre de 4 (1, 2, 4 et M). Ces kinases interagissent avec
le MyD88 grâce à leur domaine de mort, avec celui de MyD88. Un complexe se
forme avec six molécules de MyD88 et quatre molécules d’IRAK-4 puis quatre IRAK-
2 ou IRAK-4 sont recrutées. Le complexe obtenu est appelé MyDDosome, et il initie
le signal activé par les TLR. Ensuite l’IRAK-4 phosphoryle l’IRAK-1, une étape clé de
la signalisation. Les IRAK se dissocient alors du complexe pour aller interagir avec la
ligase TRAF-6 qui, en association avec Ubcl3 et UevlA polyubiquinisent TRAF-6.
Une cascade débute alors :
- TRAF-6 peut alors activer la kinase TAK1,
- TAK1 active l’IKK,
- L’IKK dégrade alors l’IκB (la protéine inhibitrice du NF-κB),
- Ceci libère le NF-κB qui peut alors se fixer sur l’ADN(Paul, 2013).
Le MyD88 est aussi impliqué dans l’activation de l’IRF-7, un autre facteur de
transcription pour l’IFN-α, lors d’activation du TLR7 ou TLR9. L’importance de cette

92
signalisation est montrée chez les souris déficientes en MyD88, incapables de
monter une réponse à de nombreux pathogènes (Paul, 2013).
iii. La signalisation indépendante du MyD88
La voie MyD88-indépendante implique le TRIF, utilisé par le TLR3 pour activer le NF-
κB via le TRAF-6 comme la voie du MyD88. Le TRIF peut également terminer par la
voie canonique du NF-κB mais par l’intermédiaire de kinases inhabituelles appelées
iκKƐ et la kinase TBK1. Elles activent alors des facteurs de transcription appelés IRF
(Interferon Regulatory Factors) comme l’IRF-3. Le TLR4 peut également activer le
TRIF, mais seulement dans ses endosomes, via le TRAM. Enfin, le SARM est un
régulateur négatif de la signalisation du TRIF (Paul, 2013).
d. Régulation des TLR
Ces cascades de signalisation offrent de nombreux points de contrôle pour la
régulation négative de l’activation du système immun. Quelques récepteurs négatifs
comme ST-2 séquestrent des protéines adaptatrices telles que MyD88, ou TIRAP, ce
qui limite leur disponibilité pour une signalisation. D’autres protéines adaptatrices
comme Tollip, IRAK-M ou SIGGR ciblent l’activité des kinases ou les
phosphorylations d’intermédiaires. La Toll Interacting Protein (ou Tollip) est un
régulateur négatif présent dans les tissus intestinaux et sa surexpression a été
observée dans les cellules épithéliales exposées au LPS. Il interagit directement
avec IRAK1 pour prévenir son autophosphorylation et donc une signalisation. Une
protéine cytoplasmique avec un domaine en doigt de zinc, A20 quant à elle, interfère
avec le TRAF-6, une molécule centrale dans de nombreuses voies de TLR,
notamment impliquée dans l’activation du NF-κB. C’est ainsi que l’A20 est un
inhibiteur large des TLR. La protéine SOCs a un effet suppressif très fort sur la
production d’interféron de type I alors qu’elle laisse intacte toute l’activité pro-
inflammatoire du NF-κB (Hopkins & Sriskandan, 2005; Lotz et al., 2007).
La caténine p120 régule la signalisation du TLR4. C’est un composant essentiel des
jonctions adhérentes des cellules endothéliales et d’autres cellules adhérentes
polarisées. Elle se lie directement sur les domaines cytoplasmiques des cadhérines
et contribue à la régulation de l’intégrité des jonctions entre les cellules. Elle joue un
rôle important dans l’adhésion cellule-cellule, le développement embryonnaire, la
prolifération cellulaire et leur polarité, la migration des cellules tumorales et la
progression des cancers. La caténine p120 régule la réponse inflammatoire dans la
peau via la régulation de l’activation de NF-κB et la migration des cellules immunes
en absence de tout stimulus inflammatoire. La surexpression de la caténine p120
inhibe la migration transendothéliale neutrophile. C’est une molécule critique pour
l’homéostasie endothéliale. La caténine p120 régule la voie de signalisation du TLR4
en inhibant la dégradation de l’IκB et donc elle inhibe aussi l’activation du NF-κB
induit par le LPS dans les poumons et les cellules endothéliales microvasculaires
pulmonaires (Hu, 2012)

93
Une autre régulation de la signalisation des TLR peut être réalisée par des ARN au
lieu des protéines. Les cellules du système inné peuvent subir des changements
drastiques dans leur programme de transcription pour rapidement exprimer les
gènes importants dans la défense. Les ARN non codants longs (lncRNA) sont des
longues molécules d’ARN qui possèdent des fonctions moléculaires différentes des
protéines encodées. Ils sont localisés dans diverses régions chromosomales. La
plupart de leur fonction est inconnue mais certains de ces ARN régulent différents
processus comme la transcription, la dégradation d’ARN et la traduction. De
nombreux lncRNA interagissent avec des modificateurs de chromatine qui altèrent la
structure de la chromatine et déterminent l’accessibilité de l’ADN aux facteurs de
transcription et aux ARN polymérases. Au moins deux lncRNA sont connus pour
réguler la traduction de protéines ; d’autres seraient des régulateurs de la réponse
immune. Les cellules dendritiques CD1c+ les expriment quand elles sont stimulées
par le LPS, les cellules T CD8+ en expriment des centaines, la plupart spécifiques
des lymphocytes. Le TNF induit l’expression de centaines de lncRNA dans les
fibroblastes, dont certains seraient des régulateurs post induction de la signalisation
du TNF pour tempérer la réponse immune (Fitzgerald & Caffrey, 2014).
Le développement de la tolérance de nombreuses cellules quand elles sont
exposées de façon répétée à une stimulation TLR serait généré grâce à des
modulations de tous ces contrôles plutôt que par une baisse d’expression des TLR
(Hopkins & Sriskandan, 2005).
En toute logique, les pathogènes ont développé de nombreuses stratégies pour
exploiter ces systèmes de signalisation. Par exemple, Salmonella typhimurium et
Yersinia spp utilisent une protéine effectrice pour supprimer l’activation de MAPK et
du NF-κB. Les virus semblent capables de cibler la signalisation des TLR de façon
encore plus spécifique, notamment grâce à une interaction directe avec IRAK-2 et
TRAF-6. D’autres virus, comme les Poxvirus ciblent le complexe kinase de l’IκB pour
rompre la cascade d’activation du NF-κB(Hopkins & Sriskandan, 2005).
e. Autres rôles des TLR
En plus de moduler la réponse pro-inflammatoire innée à travers la production de
cytokines et de chimiokines, les TLR participent à un grand nombre d’autres
processus innés comme la phagocytose, la production de métallo-protéases
matricielles, la séquestration de fer et la production de défensines. Ils sont aussi
cruciaux dans un large éventail de processus cellulaires fondamentaux comme la
polymérisation de l’actine, l’angiogenèse et l’induction de l’apoptose (Hopkins &
Sriskandan, 2005).
Les TLR interviennent dans le développement d’une réponse adaptative, puisqu’ils
sont impliqués dans l’activité des cellules B comme le changement de classe
d’anticorps. Dans les cellules dendritiques, ils interviennent dans la capture
d’antigènes, la différenciation et la migration. Les TLR4 et 9 sont associés à une

94
réponse Th1 alors que les TLR2 et 5 induisent une réponse Th2. La protéine
adaptatrice MyD88 joue un rôle complexe dans ce processus (Hopkins &
Sriskandan, 2005).
Les NOD-Like Receptor ou NLR 2)
a. Les récepteurs NOD Like Receptor
Les NOD Like Receptor (ou NLR) sont la deuxième principale famille de PRR. Ils
peuvent ensuite être divisés en trois sous-familles :
La famille des NLRC regroupe des récepteurs NLR contenant un domaine de
recrutement de caspase (CARD). Cette famille comprend les NOD1 et NOD2
ainsi que le NLRC4. Leur signalisation aboutit à l’activation du NF-κB de
façon similaire aux TLR, ce qui mène à l’induction des peptides
antimicrobiens et des cytokines inflammatoires ;
La famille des NLRP, qui contient un domaine pyrine, comprend le NLRP1 et
NLRP3, ce dernier étant un composant de l’inflammasome qui régule la
caspase 1 et mène à la production d’IL-1β ;
La famille des NAIP contient un domaine BIR.
Les NLR sont l’un des récepteurs intracellulaires les plus importants. Ils détectent les
peptidoglycanes. Leur activation induit une autophagie (voir paragraphe « Autres
mécanismes effecteurs » ; page 124) des pathogènes intracellulaires ainsi qu’une
production de cytokines pro-inflammatoires. Du polymorphisme dans les protéines
NOD est associé à la maladie inflammatoire chronique des intestins (Geremia et al.,
2013; Paul, 2013; Uematsu & Fujimoto, 2010).
Il existe plus de 20 NLR. Ces récepteurs recrutent une protéine associée à
l’apoptose qui recrute la caspase 1. Ensemble, ils forment un complexe
macromoléculaire appelé l’inflammasome (défini paragraphe suivant), qui permet la
sécrétion des formes zymogènes de la caspase 1 et des cytokines pro-
inflammatoires IL-18 et IL-1β (T. M. Ng, Kortmann, & Monack, 2013).
Tous les NLR sont intracellulaires : leur ligand doit être endocyté pour les activer.
Les domaines NOD (Nucleotide Oligomerization Domain) 1 et 2 détectent des
dérivés du peptidoglycane, et mènent à l’activation du NF-κB et à l’activation de
kinases (Paul, 2013).
Les NOD1 et NOD2 sont impliqués dans l’autophagie, notamment lors d’infection par
Salmonella typhimurium ou par Shigella flexneri : les NOD permettent la dégradation
des bactéries dans l’autophagosome et la préparation des antigènes pour qu’ensuite
ils soient présentés par les molécules de CMH II (Paul, 2013).

95
b. La formation d’inflammasomes
La famille des NLRP regroupe 14 protéines, toutes membres de l’inflammasome,
complexe multiprotéique constitué de NLRP et de la protéine chaperon Asc. Ce
complexe est nécessaire pour activer la caspase 1, enzyme chargée de traiter les
pro-formes de l’IL-1β et IL-18, cytokines importantes pour l’inflammation. L’interaction
du NLRP3 avec la protéine Asc est amplifiée par des agents phagocytés par les
macrophages (virus, bactérie, acide urique, silice, cristaux de cholestérol, …). Le
domaine CARD de l’Asc interagit quant-à-lui avec son homologue de la caspase, et
tout ceci concourt à l’activation de la caspase. De nombreuses bactéries, virus
(Influenza), champignons (Candida albicans ou Aspergillus fumigatus) et parasites
(Plasmodium falciparum, responsable de la malaria) activent l’inflammasome. Il
existe d’autres inflammasomes comme l’IPAF, qui répond aux flagelles bactériens
(activé par Salmonella, Shigella, Pseudomonas et Yersinia) ; l’inflammasome NLRP1
interviendrait lors d’infection par Bacillus anthracis. Enfin l’inflammasome contenant
l’AIM2 est activé par Listeria monocytogenes et Francisella tularensis (Paul, 2013).
La production d’IL-1β par l’inflammasome est donc réalisée en deux étapes. Un
premier signal doit activer le NF-κB pour initier la synthèse d’ARNm du pro-IL-1β et
un second signal active l’inflammasome et la libération d’IL-1β (Haneklaus, O’Neill, &
Coll, 2013).
Il existe plusieurs inflammasomes, impliqués dans la synthèse de cytokines pro-
inflammatoires.
L’inflammasome NLRP3 est le plus connu. Il est activé par des agents
phagocytés par les macrophages ou par des DAMPs ;
L’inflammasome NLRP12 est pro-inflammatoire contrairement à ce qui était
avancé il y a quelques années ;
La formation du complexe caspase1-ASC-NLRP7 mène à la formation d’IL-1β
mais pas à la pyroptose (forme de mort cellulaire programmée lors
d’inflammation dépendante de la caspase 1 : les cellules produisent des
cytokines, gonflent, éclatent et meurent) ;
Le NLRP6 implique des effets spécifiques du type cellulaire. Il semble
nécessaire, en coopération avec le NLRP3 pour le maintien de la microflore
intestinale. Il agit aussi pour tempérer les réponses inflammatoires non
désirées causées par l’activation des NLR dans les cellules hématopoïétiques
(T. M. Ng et al., 2013).
Une régulation fine de l’activité de la caspase 1 est cruciale puisqu’une réponse et
une mort cellulaire non contrôlées ont des conséquences négatives sévères. Un des
régulateurs est la Protéine Kinase R (ou PKR) dépendant de l’ARN double brin,
protéine impliquée dans la réponse aux infections virales mais aussi à de nombreux
PAMPs et DAMPs. Cependant elle peut se fixer sur l’inflammasome comme le
NLRP1, NLRP3 et NLRC4 et AIM2 (T. M. Ng et al., 2013).

96
Le NLRP3 est localisé dans l’espace périnucléaire dans le réticulum endoplasmique
et les mitochondries. Son activation est inhibée lorsque le métabolisme mitochondrial
est diminué. Ses ligands sont moins bien définis. Ils incluent des PAMPs viraux,
bactériens et fongiques, mais aussi des DAMPs comme les espèces oxygénées, des
cathepsines, l’ATP et des substances cristallines. L’activation de l’inflammasome
NLRP peut aboutir à la mort cellulaire par pyroptose grâce à la caspase 1. Pour cela,
il faut deux signaux : l’IFN-β et une réponse dépendante de l’infection (T. M. Ng et
al., 2013).
Un des inflammasomes les mieux étudiés contient l’AIM2, un membre de la
superfamille des protéines fixant l’ADN. Il est activé par l’ADN de bactéries
intracellulaires comme Listeria monocytogenes ou Francisella tularensis. L’ADN
mitochondrial (double brin, circulaire, 16,5 kb) peut aussi activer la caspase 1
lorsqu’il est relâché dans le cytosol, notamment dans les situations où la mitophagie
n’est pas fonctionnelle (la mitophagie est un mécanisme qui permet aux cellules de
recycler les mitochondries défectueuses). Dans ce cas, l’activation de la caspase 1
est dépendante de l’inflammasome du NLRP3. Après que l’inflammasome soit
exposé à l’ADN, la seconde partie de la signalisation consiste à réguler la
transcription de l’IFN de type I, l’IFN-α et -β et les gènes stimulés par l’IFN. L’IRF-3
est crucial pour cette réponse transcriptionnelle. Le NF-κB et le MAPK sont aussi
activés et permettent la transcription de cytokines inflammatoires comme l’IL-6 et le
TNF-α. Le STING (Stimulator of IFN genes) est une protéine localisée dans le
réticulum endoplasmique. Son activation entraîne son mouvement depuis le
réticulum endoplasmique vers des structures périnucléaires où il active l’IRF-3. Il est
aussi critique pour la production d’IFN suivant l’infection par des virus à ADN ou des
bactéries cytosoliques (Listeria monocytogenes) ou par un parasite (Plasmodium
falciparum). De nombreuses molécules interviennent dans la voie de signalisation de
STING (Holm, Paludan, & Fitzgerald, 2013).
Les C-Type lectin Receptor ou CTLR 3)
Les C-Type Lectin Receptor (ou CTLR) sont capables de détecter les cellules mortes
ou mourantes. Ces récepteurs, dont Ly49 et NKG2, sont présents sur les cellules
dendritiques et les macrophages (Paul, 2013; Sancho & Reis Sousa, 2013).
Les CTLR fixent les carbohydrates (ou lectines) d’une façon Ca-dépendante. Le
domaine de fixation du ligand, le CTLD, est un motif qui a évolué pour lier un grand
nombre de ligands, comme des glycanes, des protéines ou des lipides de manière
calcium indépendante cette fois-ci. Le récepteur Dectin-1 est le membre le plus
étudié. Il est présent sur les cellules dendritiques, macrophages, et certains
lymphocytes T et reconnaît les glycanes des parois fongiques, bactériennes et de
certaines plantes. Il reconnaît ainsi Candida, Aspergillus, Coccidioides,
Mycobacteria. La signalisation se fait via une tyrosine kinase qui active alors le
facteur de transcription NF-κB(Paul, 2013; Sancho & Reis Sousa, 2013).

97
Beaucoup de CTLR détectent des PAMPs mais ils peuvent aussi être impliqués dans
l’adhésion cellulaire et la communication ou encore lier des néoantigènes exprimés
par les cellules transformées. En fait, seul un petit groupe de CTLR peut fixer des
produits exposés ou relargués par les cellules mourantes ou mortes. C’est le cas de
DEC-205 qui se fixe sur les cellules apoptiques et nécrotiques. L’apoptose est
souvent perçue comme un processus silencieux ou anti-inflammatoire et les cellules
apoptiques qui ne sont pas rapidement nettoyées par leurs voisines ou les
phagocytes subissent un deuxième phénomène de désintégration appelé nécrose
secondaire. Il s’agit de la perte irréversible de l’intégrité de la membrane
plasmatique, qui est alors inflammatoire et immunogénique puisqu’elle permet le
relargage de constituants cellulaires, normalement protégés du système immunitaire
et qui agissent comme marqueurs de lésion tissulaire et leur qualité pro-
inflammatoire est probablement liée à la réparation tissulaire. Il est à noter que la
plupart des CTLR impliqués dans la reconnaissance des cellules mortes ont aussi un
ligand non soi (Sancho & Reis Sousa, 2013).
Les CTLR possèdent des motifs intracellulaires qui autorisent l’endocytose et dirigent
les récepteurs vers différents compartiments endosomaux. Ils peuvent modifier
l’expression des gènes des cellules myéloïdes en réponse à une rencontre avec une
cellule morte en induisant la libération de cytokines pro-inflammatoires et de
chimiokines permettant l’influx de PNN. Toutes ces actions sont nécessaires au
nettoyage des corps cellulaires et à la réparation tissulaire (Sancho & Reis Sousa,
2013).
Les RIG Like Receptor ou RLR 4)
Les RIG Like Receptor (ou RLR) sont des récepteurs cytosoliques qui détectent les
ARN viraux dans la plupart des cellules. Leur activation entraîne la production d’IFN
de type I. La signalisation se fait via JAK/STAT (Paul, 2013; Uematsu & Fujimoto,
2010).
Les récepteurs au complément 5)
Des récepteurs du complément (CR) sont présents sur les phagocytes et
reconnaissent de manière spécifique les composants du complément liés aux
pathogènes opsonisés. Ceci permet la fonction la plus importante du complément :
l’opsonisation qui consiste à faciliter la capture et la destruction des pathogènes par
les cellules phagocytaires. Parmi les composants du complément vus page 64, le
C3b est la principale opsonine, mais le C4b en est une aussi. La fixation du C3b sur
le CR1 ne stimule pas directement la cellule mais permet la phagocytose en
présence d’autres activateurs. Par contre la liaison entre iC3b et le CR3 est
suffisante à elle seule pour activer la phagocytose (Janeway et al., 2009).
Les principaux récepteurs aux compléments sont regroupés dans le Tableau V,
page 98.
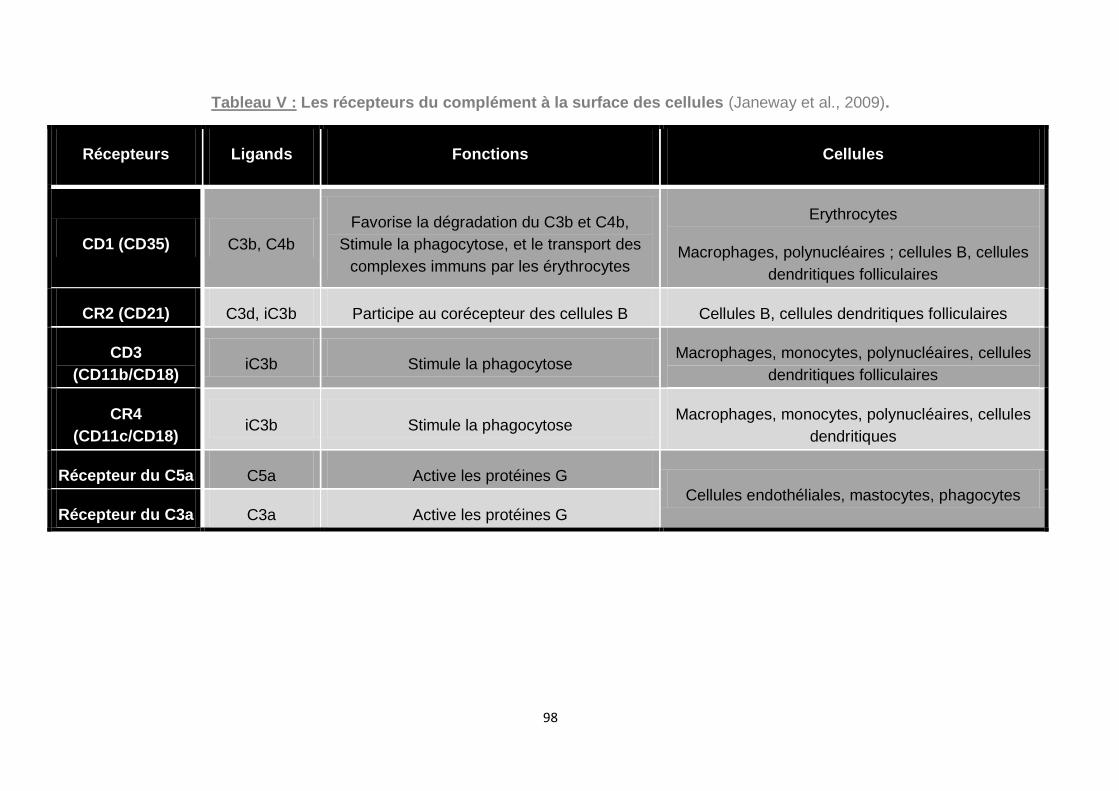
98
Tableau V : Les récepteurs du complément à la surface des cellules (Janeway et al., 2009).
Récepteurs
Ligands
Fonctions
Cellules
CD1 (CD35) C3b, C4b
Favorise la dégradation du C3b et C4b,
Stimule la phagocytose, et le transport des
complexes immuns par les érythrocytes
Erythrocytes
Macrophages, polynucléaires ; cellules B, cellules
dendritiques folliculaires
CR2 (CD21) C3d, iC3b Participe au corécepteur des cellules B Cellules B, cellules dendritiques folliculaires
CD3
(CD11b/CD18) iC3b Stimule la phagocytose
Macrophages, monocytes, polynucléaires, cellules
dendritiques folliculaires
CR4
(CD11c/CD18) iC3b Stimule la phagocytose
Macrophages, monocytes, polynucléaires, cellules
dendritiques
Récepteur du C5a C5a Active les protéines G
Cellules endothéliales, mastocytes, phagocytes
Récepteur du C3a C3a Active les protéines G

99
Les autres récepteurs 6)
Les récepteurs « scavengers » ou récepteurs éboueurs sont un groupe de
protéines conservées et anciennes. Ces récepteurs peuvent être insérés dans la
membrane plasmique ou être sécrétés. Ils sont bien connus pour leur rôle chez les
macrophages. Ils sont impliqués dans l’endocytose, la phagocytose et l’adhésion
cellulaire. Certains agissent comme des PRR qui fixent le LPS. Ils sont exprimés par
différentes cellules comme les macrophages et les cellules endothéliales. Il en existe
six différents, mais seuls deux sont bien connus :
le SR-A ;
le récepteur des macrophages avec une structure collagénique ou MARCO
(MAcrophage Receptor with COllagenous Structure) qui se lie aux bactéries
intactes, aux produits membranaires des bactéries, à l’ADN CpG bactérien,
ou à des protéines du soi modifiées. L’activation de certains TLR aboutit à
l’expression de MARCO grâce à MyD88. L’expression du MARCO modifie les
fonctions phagocytaires et de production de cytokines du macrophage (Paul,
2013) .
Les Macrophages Mannose Receptor sont des protéines trouvées dans la
membrane cytoplasmique des macrophages contenant des domaines riches en
cystéine et fibronectine et des domaines de la famille des lectine de type C. Ils
reconnaissent les carbohydrates riches en mannose des bactéries mais peuvent
aussi promouvoir la phagocytose des glycoprotéines riches en mannose de l’hôte.
Ce sont des récepteurs nécessaires pour la phagocytose de nombreuses bactéries,
virus et protozoaires (Paul, 2013).
Les Mannan-binding lectin ou MBL, de la famille des collectines. Elles se fixent sur
le mannose terminal et fucose des carbohydrates microbiens et reconnaissent
l’agencement spécifique spatial de ces sucres, qui diffère avec celui de l’hôte. Elles
reconnaissent toutes les classes de pathogènes, comme Staphylococcus,
Streptococcus, Pseudomonas, Klebsiella, Salmonella, Mycobacterium, Candida,
Influenza, et les trypanosomes. Leur principale fonction est d’activer le complément
via la voie de la lectine grâce à deux protéases sérines MASP-1 et 2 mais elles
peuvent aussi agir comme des opsonines (Paul, 2013).
Les pentraxines (déjà évoquées dans le paragraphe « Les lectines » page 62) ont
des ligands très variés comme l’ADN, des complexes immuns, des sucres, le
complément et la matrice extracellulaire. Elles se fixent à des surfaces bactériennes
et reconnaissent la phosphorylcholine. Ce sont des opsonines et elles activent la
voie classique du complément. Elles sont regroupées en deux types :
les pentraxines courtes (comme la protéine C réactive et le sérum amyloïde
P ; rapidement produits par le foie) ;
les pentraxines longues comme la pentraxine 3 (ou PTX3), impliquée dans la
lutte contre les bactéries et les champignons et exprimées par de nombreuses
cellules comme les cellules dendritiques, les macrophages, les PNN. La PTX3

100
se fixe sur des agents microbiens précis comme les levures et les composants
membranaires bactériens. Son expression est inductible dans les cellules
dendritiques immatures, les monocytes, les cellules endothéliales et les
fibroblastes en réponse à des cytokines pro-inflammatoires et des
composants bactériens. Sa concentration passe de <2 ng/ml à l’ordre du µg
dans le plasma sanguin puisque son expression est augmentée en réponse
aux composants fongiques comme le zymosan. Il est impliqué dans
l’opsonisation et donc l’élimination des champignons et des levures. La PTX 3
est aussi impliquée dans l’augmentation de la production d’oxyde nitrique.
Enfin la présence de la PTX3 contribue également au bras adaptatif de
l’immunité innée en affectant les propriétés fonctionnelles des cellules
présentatrices d’antigènes comme les macrophages ou les cellules
dendritiques (Bowdish et al., 2007).
Les protéines de reconnaissance du peptidoglycane sont une famille de PRR qui
fixent le peptidoglycane. Elles sont très conservées au cours de l’évolution
puisqu’elles sont présentes chez la drosophile. Certaines sont transmembranaires
mais la plupart sont sécrétées (Paul, 2013).
Un point important : la tolérance et l’homéostasie C-
muqueuse
Contrairement aux sites stériles où la reconnaissance d’un microorganisme étranger
initie une cascade inflammatoire, l’abondance de la flore commensale symbiotique
dans l’intestin nécessite la discrimination entre les flores commensale et pathogène.
La tolérance, observée dans l’organisme dans sa totalité, est le manque de réponse
inflammatoire. Il s’agit en fait d’une non réponse spécifique à un antigène et elle peut
Ce qu’il faut retenir :
Le système inné possède une grande variété de récepteurs (même si cette
diversité est négligeable, par rapport au système adaptatif) qui possèdent des
localisations variées afin de couvrir l’ensemble des pathogènes. Un même
antigène peut être reconnu par de nombreux récepteurs innés, avec des voies
de signalisation différentes ce qui engendre une multitude de réponses. Ceci
explique la grande variabilité et l’extrême complexité des réponses immunes
face à un pathogène entier comportant de nombreux antigènes. Cette
réponse multiple est bénéfique car les pathogènes ont parfois réussi à inhiber
certaines voies de signalisation des récepteurs.

101
se traduire par un échec du développement, ou par l’inhibition d’une réponse
inflammatoire contre un antigène spécifique. Il existe deux classifications : la
tolérance au soi (ce qui empêche le système immunitaire de s’attaquer aux tissus de
l’hôte) et la tolérance orale (le fait d’ingérer une substance diminue la réponse
systémique à cette substance). La présence d’un écosystème procaryote dense
dans un hôte eucaryote pose un paradoxe conceptuel puisque l’hôte dépend de la
présence symbiotique d’une population microbienne massive mais en même temps il
développe des mécanismes immuns innés pour nettoyer tous les sites non muqueux
des procaryotes pathogènes ou envahisseurs (Jump & Levine, 2004; Thaiss, Levy,
Suez, & Elinav, 2014).
Le système immunitaire inné favorise la tolérance naturelle en contrôlant la
croissance bactérienne dans la lumière intestinale et en tempérant la réponse
inflammatoire potentielle qui pourrait se mettre en place. L’épithélium intestinal est
responsable de certains de ces traits puisque les cellules de Goblet produisent des
mucines qui forment un film recouvrant la surface épithéliale apicale qui forme ainsi
une barrière physique entre le contenu intestinal et la surface épithéliale et aide à
contenir la flore commensale dans l’intestin en fournissant un site de liaison ample.
En retour, les mucines minimisent l’attachement de pathogènes comme Escherichia
coli. Les cellules épithéliales sécrètent également des peptides antimicrobiens qui
protègent les cryptes intestinales de la croissance bactérienne et protègent ainsi
l’intégrité épithéliale. Le plus abondant est l’α-défensine, sécrétée par les cellules de
Paneth stimulées uniquement par les bactéries. Les cellules épithéliales intestinales
sont aussi des cellules présentatrices d’antigènes non professionnelles puisqu’elles
portent des molécules de CMH II qui peuvent présenter des antigènes aux cellules T
CD4+. Elles peuvent présenter des peptides mais pas de protéines qui nécessitent
un traitement intracellulaire. La lamina propria intestinale est peuplée de cellules
stromales et de cellules présentatrices d’antigènes comme les macrophages et les
cellules dendritiques qui contribuent à la tolérance. Les cellules stromales expriment
des cyclo-oxygénases COX-2 leur permettant de produire de forts niveaux de
prostaglandine PGD2 possédant des fonctions immunorégulatrices. La PGD2 serait
essentielle pour l’absence de réponse inflammatoire au contenu luminal. Les
macrophages de la lamina propria n’expriment pas le CD89, le récepteur pour les
IgA, ce qui diminue la phagocytose initiée par les IgA. De même, les cellules
dendritiques dérivées des monocytes ou des myéloïdes n’expriment pas le CD89
(Jump & Levine, 2004).
Les cellules épithéliales produisent de nombreux signaux immuno-régulateurs
nécessaires pour les cellules immunes tolérantes, limitant ainsi l’état inflammatoire et
en dirigeant de façon appropriée les réponses immunes et adaptatives. Beaucoup de
ces réponses dépendent de la traduction des signaux bactériens commensaux par
les cellules épithéliales vers les cellules immunes. La production de cytokines
comme le TSLP, le TGF-β, l’IL-25 et le ligand induisant la prolifération des cellules B
est stimulée par les bactéries commensales via les PRR. Les trois cytokines et
l’acide rétinoïque produits par les cellules épithéliales en réponse aux signaux des

102
bactéries commensales stimulent le développement des cellules dendritiques et des
macrophages qui produisent de l’IL-10 et de l’acide rétinoïque. Certaines bactéries
commensales comme Bacteroides thetaiotaomicron stimulent l’export nucléaire
d’une sous-unité du NF-κB, ce qui limite son activation (Lotz et al., 2007; Peterson &
Artis, 2014).
Les acides gras à chaîne courte comme le butyrate sont produits par la fermentation
du microbiome et sont détectables dans le tractus intestinal. Le butyrate possède
aussi une activité anti-inflammatoire sur les cellules lymphoïdes et myéloïdes. Il agit
directement sur les leucocytes et inhibe la production d’IL-12, diminue l’expression
des molécules de co-stimulation et bloque la translocation du NF-κB dans le noyau
des cellules dérivées des monocytes et des macrophages. Les bactéries non
pathogènes du microbiome peuvent altérer l’état inflammatoire du système immun
intestinal pour le bénéfice de l’hôte. Les métabolites exogènes du tryptophane jouent
un rôle important dans l’homéostasie intestinale via l’AhR qui stimule la
différenciation des cellules Th17. Or le microbiome est une source de métabolites du
tryptophane (McDermott & Huffnagle, 2013).
Les cellules dendritiques sont critiques pour le maintien de la tolérance. Les cellules
dendritiques CD103+ CD11c+ expriment le CCR7 et sont considérées comme une
population majeure qui migre vers les nœuds lymphatiques mésentériques. Elles
stimulent la génération de cellules Treg grâce à la sécrétion d’acide rétinoïque avec
du TGF-β (Partie A de la Figure 21, page 106) alors que les CD103- aident le
développement de l’inflammation en augmentant l’expression des médiateurs
inflammatoires comme le TNF-α et l’IL-6. Les CXC3CR1+, CD11c+ CD11b+ sont
impliquées dans le nettoyage des pathogènes entéro-invasifs mais ne sont pas
retrouvées dans les nœuds lymphatiques. Leur expression des protéines des
jonctions serrées leur permet de former des dendrites transépithéliales qui pénètrent
dans la lumière de l’intestin pour prélever des antigènes exogènes. L’extension de
ces dendrites transépithéliales est initiée par une voie de signalisation des TLR dans
les cellules épithéliales et leur production d’IL-10 mène à la suppression des
cytokines inflammatoires et au développement des Treg (Partie B de la Figure 21,
page 106). Donc à l’état d’équilibre, la microflore prévient le transfert d’antigènes
commensaux vers les nœuds lymphatiques mésentériques par les CX3CR1+ sans
empêcher une réponse aux bactéries invasives par les DC CD103+ (Partie A de la
Figure 22, page 107) (Kayama & Takeda, 2012; McDermott & Huffnagle, 2013; S. C.
Ng et al., 2010; Peterson & Artis, 2014; Thaiss et al., 2014).
Les PRR sont connus pour leurs propriétés pro-inflammatoires dans des cellules
immunes, mais leur rôle dans la régulation de l’homéostasie tissulaire commence à
être considéré comme une fonction majeure dans les cellules épithéliales
intestinales. Les bactéries commensales délivrent des signaux qui contribuent à
l’homéostasie épithéliale et à la réparation tissulaire. Il y a donc un rôle bénéfique
pour la signalisation TLR dans les cellules épithéliales intestinales comme
l’expression de la protéine de choc thermique, des facteurs de croissance

103
épidermique et le TFF3. La proximité des cellules épithéliales avec les signaux
microbiens abondants de la lumière nécessite des mécanismes spécialisés pour
maintenir une hyporéactivité des PRR en réponse aux stimuli des bactéries
commensales. Les cellules épithéliales expriment des régulateurs négatifs des
intermédiaires de la voie de signalisation pro-inflammatoire délivrée par les PRR. La
production des réactifs oxygénés atténue l’activation du NF-κB, ce qui conduit les
cellules épithéliales à tolérer la flore commensale. Les macrophages résidents
intestinaux montrent une expression de PRR réduite et sont résistants à la sécrétion
de cytokines pro-inflammatoires après stimulation par des microbes. Par contre ils
sont toujours très phagocytaires. De la même façon, les cellules dendritiques du
colon possèdent peu de TLR. Récemment, de nombreux régulateurs négatifs de la
voie de signalisation des TLR ont été découverts (décrits page 92). Elles
comprennent la régulation d’expression des récepteurs et des co-récepteurs, la
présence d’antagonistes des récepteurs qui entrent en compétition avec le ligand et
des molécules de régulation tout le long de la voie de signalisation ainsi que la
diminution post transcriptionnelle d’intermédiaires essentiels dans la cascade de
signalisation. Ces circuits inhibiteurs affectent plusieurs TLR simultanément, ce qui
limite l’activation redondante de différents TLR (Lotz et al., 2007; Peterson & Artis,
2014).
La discrimination entre les microorganismes de la flore commensale et les
organismes pathogènes est nécessaire pour obtenir une réponse inflammatoire
appropriée. La polarisation de l’épithélium intestinal autorise la ségrégation des PRR.
Les bactéries commensales restent dans la lumière et ne sont en contact qu’avec la
membrane apicale des entérocytes alors que les bactéries pathogènes qui tentent de
pénétrer l’organisme peuvent passer par l’intérieur des cellules ou entre les cellules,
et se retrouvent donc en contact avec les récepteurs de la membrane basolatérale.
Par exemple, l’activation du TLR9 sur la face basolatérale de la cellule épithéliale
active la translocation nucléaire du NF-κB alors qu’une activation sur la face apicale
induit la stabilisation de l’IκB donc d’un effet inhibiteur. Le MDP est une protéine
chaperon qui stimule la localisation sur la membrane basolatérale. Contrairement
aux sites stériles, le contrôle de l’inflammation dans l’intestin doit reposer sur la
reconnaissance de signaux de dangers associés à la pathogénèse plutôt que sur la
présence de signaux microbiens seuls (Kayama & Takeda, 2012; Peterson & Artis,
2014).
La coévolution des PRR et de la colonisation microbienne mène probablement à un
réseau d’interdépendances.
Le NOD1 reconnaît les peptidoglycanes des bactéries Gram- et induit la genèse de
tissus lymphoïdes intestinaux, nécessaires pour maintenir une communauté
bactérienne stable dans les intestins. L’activation du NOD1 par des produits
microbiens stimule la capacité des PNN à tuer les pathogènes.
Le NOD2 reconnaît les dipeptides muramyl des bactéries et est impliqué dans la
maladie de Crohn. L’expression du NOD2, au niveau des cellules de Paneth, dépend
de la présence de bactéries commensales et crée un feedback dans lequel la

104
microflore commensale régule positivement la signalisation du NOD2 qui à son tour
régule négativement la flore commensale.
En plus des bactéries et des virus, la flore comprend de nombreuses espèces
fongiques. Les champignons commensaux sont reconnus par un récepteur C-type
lectine, le Dectin-1. L’hôte régule la microflore et cette dernière a un effet sur la
muqueuse de l’hôte et le développement du système immun systémique. Nous
verrons en 3ième partie de ce travail que les ILCs interagissent beaucoup avec la flore
microbienne. (Jump & Levine, 2004; S. C. Ng et al., 2010; Peterson & Artis, 2014)
L’inflammasome formé par la caspase 1 et les NLR a une influence complexe sur
l’inflammation et la réparation épithéliale. L’inflammasome NLRC4 induit la
production d’IL-1β par les phagosomes mononucléaires intestinaux après l’infection
avec des bactéries pathogéniques mais non commensales. Cette production d’IL-1β
dans les phagocytes représente une réponse immune innée qui peut discriminer les
bactéries commensales des pathogéniques (Thaiss et al., 2014).
Les réactifs oxygénés sont produits en réponse aux bactéries commensales et
pathogènes, et ont un rôle dans la signalisation au sein de la cellule épithéliale et ils
activent la réparation épithéliale indépendamment de leur effet antimicrobien. Grâce
à l’inactivation des phosphatases tyrosines sensibles au redox, les espèces
oxygénées stimulent la formation par les cellules épithéliales de molécules
d’adhésions focales, nécessaires pour la migration cellulaire et la guérison (Peterson
& Artis, 2014).
L’homéostasie intestinale est également régulée par les cellules du système
immunitaire adaptatif. Les cellules Th17 induites par l’IL-6 et le TGF-β expriment le
RORγt et se développent en réponse à l’IL-23. Lors de maladie de Crohn, les Th1 et
les Th17 sont augmentées dans la lamina propria. Les Treg CD4+ CD25+ contribuent à
la tolérance envers le soi en régulant les réponses immunes. Ces cellules peuvent
supprimer une réponse inflammatoire grâce à la production de cytokines anti-
inflammatoires comme l’IL-10 et le TGF-β, en plus d’inhiber la présentation
d’antigène (Partie F de la Figure 21, page 106). Les cellules Breg sont CD1dhigh et
sont aussi présentes dans les GALT d’où elles orchestrent la réponse immune
adaptative. Le CD1d est nécessaire pour la production d’IL-10, cytokine anti-
inflammatoire (Partie G de la Figure 21, page 106). Les cellules régulatrices du
système adaptatif aident à préserver l’équilibre intestinal grâce à l’IL-10 (Kayama &
Takeda, 2012; Peterson & Artis, 2014).
Le système immunitaire muqueux a la fonction essentielle de prévenir les infections
par les pathogènes intestinaux et de maintenir la tolérance envers la flore
bactérienne commensale. Dans l’intestin grêle, l’expression des molécules
d’adhésions MAdCAM-1 des cellules endothéliales avec la chimiokine CCL-25
permet de fixer les intégrines α4β7 et CCR-9 exprimées par les cellules B et T. Pour
le gros intestin, le « homing » est réalisé par les intégrines α4β7 et α4β1. En absence
d’inflammation, le système muqueux stimule la différenciation des cellules Treg alors

105
que des membres spécifiques de la microflore induisent la différenciation des cellules
Th17. L’acide rétinoïque active les Treg et inhibe les Th17. Les cellules dendritiques
muqueuses au contact de l’acide rétinoïque alimentaire, surexpriment le RALDH2 et
peuvent donc produire de l’acide rétinoïque. Cette boucle de régulation positive est
fonctionnelle dans l’intestin puisque l’expression du RALDH2 par les cellules
dendritiques est proportionnelle au niveau de vitamine A dans l’aliment (Beijer et al.,
2013).
Cependant, les effets protecteurs de la reconnaissance bactérienne par les cellules
épithéliales ont un coût. Bien que les signaux microbiens commensaux soient
protecteurs pour éviter une inflammation ou une infection, ils favorisent les tumeurs
et les cancers quand les réponses homéostasiques ne sont plus régulées
correctement. Les signalisations dans les cellules épithéliales comme ceux des TLR
(via MyD88 et NF-κB) ont toutes été impliquées dans le développement tumoral. La
convergence entre les voies de signalisation des PRR et les voies de signalisation
pro-oncogéniques peuvent expliquer l’effet cancérigène des stimulations
microbiennes. Par exemple, MyD88 contribue à stabiliser MYC, une protéine
oncogénique. Le NF-κB favorise le signal Wnt dans les cellules épithéliales ce qui
stimule leur dédifférenciation en cellules tumorales presque souches.
Paradoxalement, certains NLR protègent contre la cancérogénèse à travers la
régulation des cellules mortes et la surveillance de la prolifération des cellules
transformées ou endommagées (Peterson & Artis, 2014).
La perte de la tolérance, par dérégulation de l’expression des PRR ou des
défensines, ou des capacités de signalisation et des fonctions effectrices conduit à
des maladies comme la maladie inflammatoire chronique des intestins. Notamment,
l’expression du TLR4 est augmentée dans les cellules épithéliales intestinales lors de
maladie de Crohn ou de colite ulcérative (Jump & Levine, 2004).
Ce qu’il faut retenir :
L’homéostasie intestinale est donc un phénomène complexe qui fait intervenir
de nombreuses cellules et des médiateurs afin de maintenir l’équilibre entre la
tolérance envers la flore microbienne intestinale et la réponse inflammatoire
contre les bactéries pathogènes entéroinvasives. Un véritable dialogue
s’installe entre les bactéries commensales et les cellules épithéliales et
dendritiques.
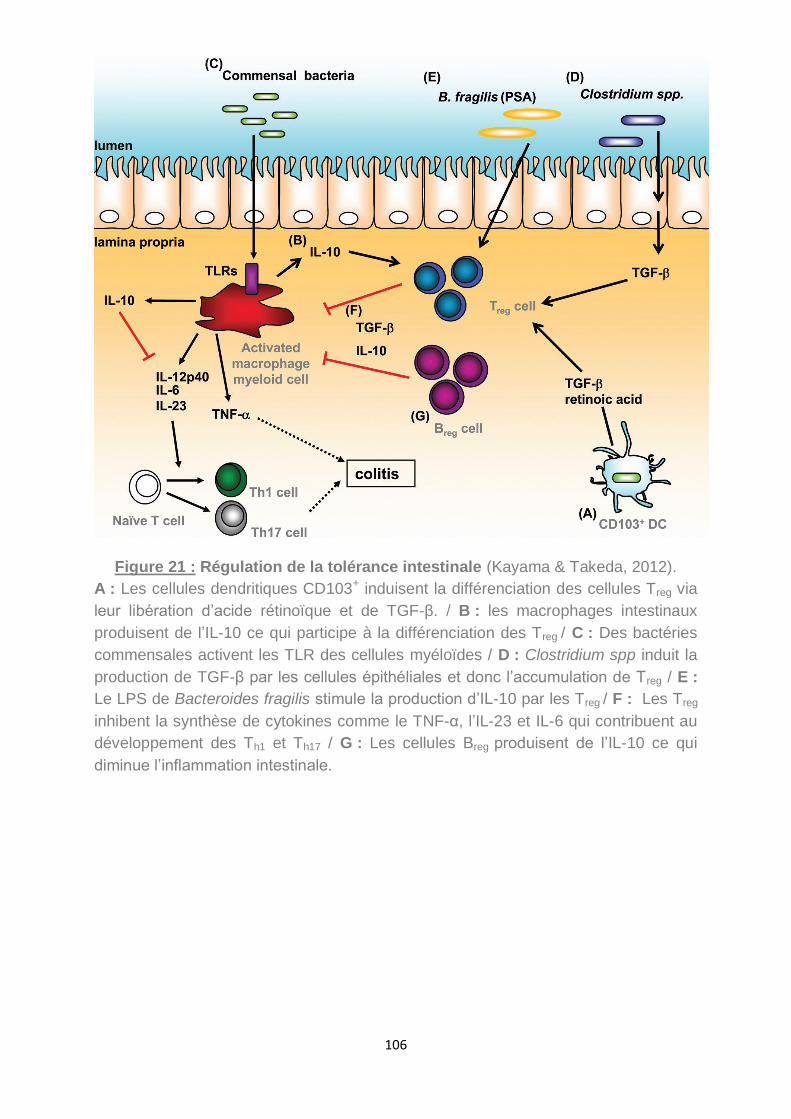
106
Figure 21 : Régulation de la tolérance intestinale (Kayama & Takeda, 2012).
A : Les cellules dendritiques CD103+ induisent la différenciation des cellules Treg via
leur libération d’acide rétinoïque et de TGF-β. / B : les macrophages intestinaux
produisent de l’IL-10 ce qui participe à la différenciation des Treg / C : Des bactéries
commensales activent les TLR des cellules myéloïdes / D : Clostridium spp induit la
production de TGF-β par les cellules épithéliales et donc l’accumulation de Treg / E :
Le LPS de Bacteroides fragilis stimule la production d’IL-10 par les Treg / F : Les Treg
inhibent la synthèse de cytokines comme le TNF-α, l’IL-23 et IL-6 qui contribuent au
développement des Th1 et Th17 / G : Les cellules Breg produisent de l’IL-10 ce qui
diminue l’inflammation intestinale.

107
Figure 22 : Régulation des réponses immunes intestinales par les cellules
innées (Kayama & Takeda, 2012). A : Les cellules dendritiques CD103+ activées via le TLR5 induisent la différenciation des Th1 et Th17. L’IL-23 qu’elles produisent induit la libération d’IL-22 et donc la sécrétion de peptide antimicrobien RegIIIγ par les cellules épithéliales intestinales / B : L’ATP de la flore commensale induit l’induction des cellules Th17 via les cellules dendritiques CX3CR1+ / C : Les bactéries filamenteuses initient la différenciation des Th17 / D : les cellules dendritiques E-cadhérines+ augmentent la différenciation des Th17 / E : Les cellules M régulent les cellules Th17 et Th1 en fonction de l’IL-10.

108
III. Initiation de la réponse innée
Après avoir décrit les acteurs du système inné et leur mode de détection de
l’agression, étudions leur rôle dans l’inflammation, réponse immédiate de
l’organisme.
L’inflammation locale A-
L’inflammation est le terme général pour l’accumulation locale de liquide, de
protéines plasmatiques et de globules blancs qui est initiée par une blessure, une
infection ou une réponse immune locale. Elle se divise en deux phases : une phase
précoce et transitoire, l’inflammation aiguë suivie de l’inflammation chronique si
l’infection persiste ou lors de maladie auto-immune. L’engagement des récepteurs
des cellules présentatrices d’antigènes comme les macrophages et les cellules
dendritiques avec des pathogènes entraîne l’ingestion et la lyse de la bactérie ainsi
que la production de cytokines et chimiokines qui déclenchent alors l’inflammation.
Ce processus permet de recruter les cellules et molécules immunes du sang et
d’augmenter le volume de lymphe exporté vers les nœuds lymphatiques afin d’initier
la réponse adaptative. L’inflammation est définie traditionnellement par les quatre
mots latins, calor, dolor, rubor et tumor (pour chaleur, douleur, rougeur et
tuméfaction) (Janeway et al., 2009).
L’inflammation est classiquement et schématiquement divisée en quatre phases que
nous verrons successivement.
La phase vasculaire 1)
La phase vasculaire correspond à la détection du pathogène par des cellules
sentinelles comme les macrophages ou les cellules dendritiques. Ces cellules
détectent les pathogènes par leur PRR et cela enclenche la production de
médiateurs et la phagocytose des microbes par ces cellules comme sur la
Figure 23, page 109 (Janeway et al., 2009).

109
Figure 23 : Mécanisme de la phagocytose par les macrophages.
D’après(Janeway et al., 2009).
Ces cellules produisent donc des facteurs qui initient l’inflammation en attirant les
cellules effectrices et en activant l’endothélium pour que ces cellules puissent
atteindre le tissu infecté comme indiqué sur la
Figure 23, ci-dessus. Parmi les cytokines produites, se trouvent :
l’IL-1β. Elle active localement l’endothélium vasculaire et les lymphocytes et
détruit localement les tissus. De façon systémique, elle induit la fièvre ;
l’IL-6. Elle active des lymphocytes ainsi que la production des protéines de la
phase aiguë ;
le TNF-α. Il active l’endothélium vasculaire et augmente sa perméabilité. Il est
également responsable de fièvre. Son importance est capitale : il permet la
mise en place de caillot dans la microcirculation, ce qui empêche le
pathogène d’entrer dans la circulation et de se répandre dans l’organisme. Par
Le macrophage exprime des récepteurs pour de
nombreux constituants bactériens
Le macrophage engloutit et digère la bactérie sur
laquelle il s’est fixé.
La fixation de la bactérie sur les récepteurs du
macrophage initie la libération de cytokines et de
petits médiateurs lipidiques de l’inflammation.

110
contre, une fois que le pathogène est dans le sang, la production de TNF-α
devient systémique et est responsable du choc associé au sepsie ;
l’IL-12. Elle active les cellules NK et induit la différenciation des Th1 ;
la chimiokine CXCL-8. Elle recrute les PNN et PNB dans le foyer infectieux
(Janeway et al., 2009).
La phase vasculaire se caractérise par des modifications importantes de la
microcirculation locale avec initialement, une constriction artériolaire très brève suivie
d’une dilatation des artérioles et des capillaires. Cette augmentation du débit
circulatoire explique la rougeur et la chaleur. Une vasoconstriction d’aval entraîne
une exsudation plasmatique importante qui aboutit à la formation d’un œdème,
responsable de la tumeur et de la douleur (par distension des tissus et hyperpression
sur les terminaisons nerveuses locales). Ces modifications ont lieu dans les minutes
qui suivent la blessure comme sur la Figure 24, ci-dessous (Genetet, 1997).
La formation d’œdème permet l’accumulation de médiateurs inflammatoires. Elle est
induite par des stimuli inflammatoires, ce qui initie l’activation de facteurs nucléaires
des cellules T activées (NFAT), facteur de transcription des cellules innées. Les
cellules dendritiques sont les principaux régulateurs de la formation d’œdème grâce
aux prostaglandines qu’elles produisent, régulées par le NFAT. Ces prostaglandines
entraînent une vasodilatation locale (Zanoni & Granucci, 2012).
Figure 24: Modification des vaisseaux au début de l’inflammation D’après (Janeway et al., 2009).
La phase cellulaire 2)
La phase cellulaire fait appel à la lignée thrombocytaire et celle des polynucléaires
puis des monocytes-macrophages. Enfin les lymphocytes sont concernés puis les
fibroblastes. Le rôle des plaquettes est essentiel par la libération de nombreux
médiateurs : protéases, enzymes lysosomiales, sérotonine, et eicosanoïdes.
L’adhérence des plaquettes à l’endothélium du site lésé suivi de la migration des
leucocytes est le résultat d’un système émetteur-récepteur (avec les ICAM et VCAM)
(Genetet, 1997).
Les cytokines produites par les macrophages
provoquent la dilatation des petits vaisseaux
sanguins locaux
Les leucocytes migrent vers la périphérie des vaisseaux sanguins par augmentation de
l’expression des molécules d’adhésion
Les leucocytes sortent au niveau du site de
l’infection
La formation de caillot de sang a lieu dans les
micro-vaisseaux

111
Les neutrophiles arrivent rapidement sur les lieux. L’adhésion aux parois des
veinules, puis la traversée de l’endothélium se font sous le contrôle d’un
chimiotactisme, afin d’arriver au niveau des tissus lésés pour y réaliser la
phagocytose.
Sans récepteur à la lymphotoxine β (LT-βR) sur les cellules endothéliales, les PNN
ne peuvent pas s’accumuler rapidement au site d’infection, réduisant ainsi les
capacités de l’hôte pour faire face à l’infection. Le recrutement des PNN se fait via un
mécanisme dépendant de CXCL-2 et/ou CXCL-1. L’infection locale des cellules
épithéliales induit des chimiokines qui attirent les cellules innées LT+ depuis les
follicules lymphoïdes organisés dans la couche épithéliale. L’expression de la
lymphotoxine sur les cellules RORγt+ active la signalisation LT-βR sur les cellules
épithéliales intestinales pour mobiliser la réponse immune innée précoce contre les
pathogènes bactériens. La signalisation par LT-βR active l’expression des
chimiokines CXCL-1 et CXCL-2 qui stimulent le recrutement vers le site d’infection
pour combattre le pathogène (Wang et al., 2010).
a. L’extravasation des cellules effectrices
La diapédèse est le mouvement des cellules du sang, surtout des leucocytes, qui
passent au travers des parois vasculaires pour migrer dans les tissus.
Dans les conditions normales, les leucocytes se répartissent au centre du flot
sanguin, là où le flux est le plus rapide. Dans les foyers inflammatoires, le
ralentissement du flux sanguin permet aux leucocytes de quitter le centre du
vaisseau et d’interagir avec l’endothélium. Même sans infection, les monocytes
migrent dans les tissus mais à un ratio plus faible (Douglas & Jane Wardrop, 2010;
Genetet, 1997; Paul, 2013; Theilgaard-Mönch et al., 2006).
La première chimiokine produite est le CXCL-8, sécrétée par les macrophages, elle
recrute les PNN. Cet afflux est maximal dans les 6 premières heures de la réponse
inflammatoire (Genetet, 1997).
La première étape de l’extravasation est le roulement grâce à l’expression des
sélectines P à la surface des cellules endothéliales. Il s’agit de glycoprotéines
membranaires exprimé suite à l’action du TNF-α libéré par les macrophages.
L’interaction des sélectines E et P avec les glycoprotéines permettent aux PNN et
monocytes d’adhérer de manière réversible à la paroi vasculaire, en roulant le long
de l’endothélium (Douglas & Jane Wardrop, 2010; Genetet, 1997; Paul, 2013;
Theilgaard-Mönch et al., 2006).
Ensuite, l’adhérence ferme est autorisée par les intégrines leucocytaires LFA-1 et
CR3 converties d’une forme inactive vers une forme active lorsque les PNN
rencontrent des molécules chimiotactiques comme le CXCL-8. Ces intégrines
interagissent avec des molécules endothéliales comme les « Intercellular Adhesion

112
Molécules » ICAM-1 et ICAM-2. Le roulement s’arrête et les leucocytes s’attachent
fermement à l’endothélium (Genetet, 1997).
Lors de la troisième étape, la diapédèse, les leucocytes traversent la paroi
endothéliale. Cette étape nécessite une interaction supplémentaire entre une
molécule apparentée aux Ig exprimée sur les leucocytes ainsi qu’au niveau des
jonctions entre les cellules endothéliales. Les leucocytes traversent la membrane
basale grâce à des enzymes qui lysent les protéines de la matrice extracellulaire
(Genetet, 1997).
La dernière étape est la migration des leucocytes sous l’influence de chimiokines
produites au niveau du foyer infectieux qui forment alors un gradient de
concentration. Les PNN peuvent alors commencer à interagir avec l’agent
étiologique. Ces médiateurs chimiotactiques sont des amines vasoactives, des
lipides pro-inflammatoires, des petits polypeptides, des facteurs chimiotactiques
(comme le C5a, le leucotriène B4, les chimiokines CXCL-8 aussi appelée IL-8,
CXCL-1/GROα, CXCL-5…) et des cytokines (TNF-α, IL-1β ou IL-17A) (Douglas &
Jane Wardrop, 2010; Genetet, 1997; Paul, 2013; Theilgaard-Mönch et al., 2006).
Toutes ces étapes sont schématisées dans la Figure 25, page 113.
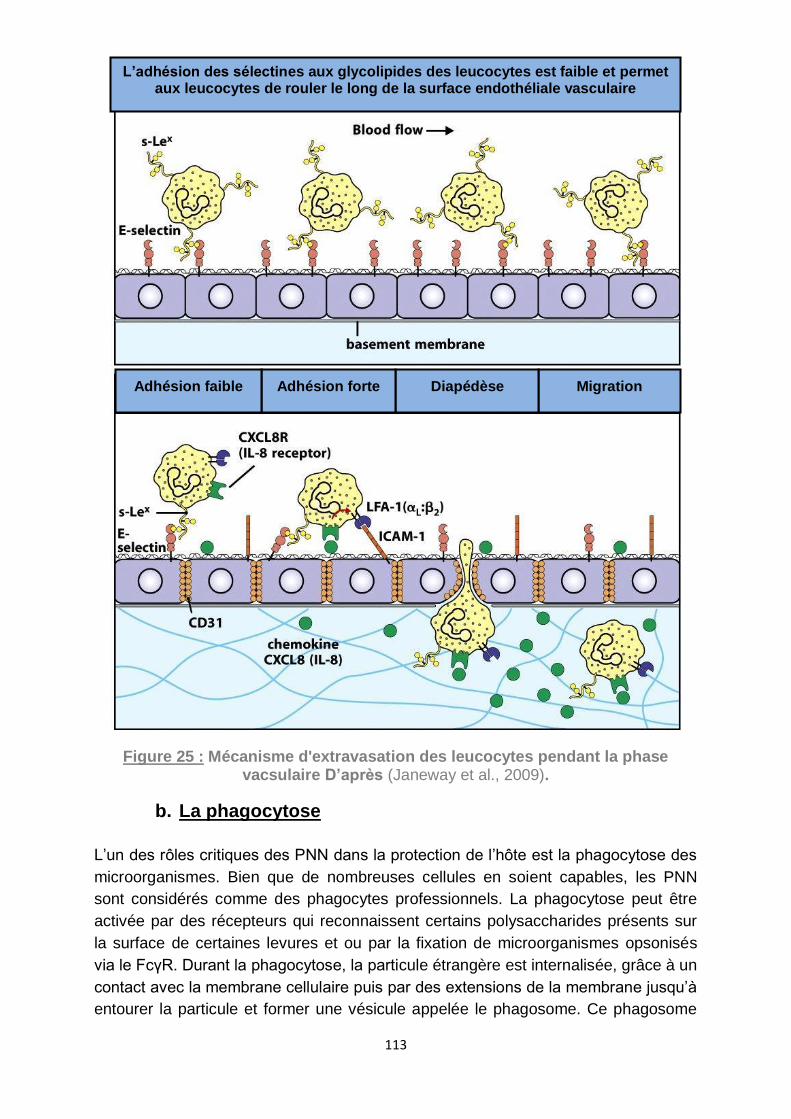
113
Figure 25 : Mécanisme d'extravasation des leucocytes pendant la phase vacsulaire D’après (Janeway et al., 2009).
b. La phagocytose
L’un des rôles critiques des PNN dans la protection de l’hôte est la phagocytose des
microorganismes. Bien que de nombreuses cellules en soient capables, les PNN
sont considérés comme des phagocytes professionnels. La phagocytose peut être
activée par des récepteurs qui reconnaissent certains polysaccharides présents sur
la surface de certaines levures et ou par la fixation de microorganismes opsonisés
via le FcγR. Durant la phagocytose, la particule étrangère est internalisée, grâce à un
contact avec la membrane cellulaire puis par des extensions de la membrane jusqu’à
entourer la particule et former une vésicule appelée le phagosome. Ce phagosome
L’adhésion des sélectines aux glycolipides des leucocytes est faible et permet aux leucocytes de rouler le long de la surface endothéliale vasculaire
Migration Diapédèse Adhésion forte Adhésion faible

114
fusionne ensuite avec des granules et donne le phagolysosome, un espace protégé
dans lequel des enzymes protéolytiques et d’autres composants bactéricides sont
déversés et la dégradation des pathogènes peut alors avoir lieu comme indiqué sur
le dernier cadre de la Figure 23, page 109. En même temps, la NADPH oxydase
s’assemble sur la membrane du phagosome et commence à générer des dérivés
oxygénés qui oxydent les lipides et les protéines microbiennes. L’activité de cette
enzyme conduit à l’acidification du phagosome qui active les composants
antimicrobiens dépendants du pH. Les PNN activés sur le site de l’infection
n’amplifient pas la réponse inflammatoire mais au contraire participent à sa
résolution (Paul, 2013).
La phagocytose est fortement activée par les opsonines comme le complément ou le
MBL, comme indiqué sur la Figure 26 ci-dessous.
Figure 26 : La phagocytose est facilitée par l’opsonisation par le complément ou d’autres opsonines. D’après (Janeway et al., 2009).
La phase de détersion 3)
C’est une phase de phagocytose intense, principalement par les PNN mais aussi par
les macrophages. Cette étape a pour but d’éliminer les tissus lésés (Janeway et al.,
2009).
Durant les dernières phases de l’inflammation, les PNN activés changent leur
synthèse d’eicosanoïdes en lipotoxines. La lipotoxine A4 et LXB4 stoppent le
chimiotactisme, l’adhésion et la transmigration à travers l’endothélium des PNN. Ils
synthétisent aussi de la résolvine et des protectines qui inhibent la migration
transendothéliale et l’infiltration tissulaire. Ils produisent même des cytokines anti-
inflammatoires comme le TGF-β et l’IL-1R agoniste. Enfin, les PNN doivent être
retirés du site inflammatoire. Pour cela ils entrent en apoptose et sont digérés par les
macrophages tissulaires (Paul, 2013).
La bactérie est recouverte par le complément dans
les voies alternative et des lectines
Quand seul le C3b est fixé sur le CR1, la bactérie n’est pas
phagocytée
Le C5a peut activer la phagocytose par les
macrophages via le CR1

115
La phase de cicatrisation 4)
Cette phase nécessite des cellules comme les fibrocytes et permet la production de
protéines matricielles des tissus intercellulaires comme le collagène, la fibronectine
et la laminine (Genetet, 1997).
Cette phase est plus ou moins complexe, et assez longue à se mettre en place. Elle
dépend du tissu concerné et sera donc plus ou moins fonctionnelle. Nous prenons
l’exemple d’une blessure musculaire puis d’une infection pulmonaire virale.
Une blessure musculaire entraîne l’activation rapide du système inné qui exerce des
effets pléiotropes sur la régénération musculaire. Dans les minutes qui suivent la
blessure, les PNN infiltrent le muscle lésé et relâchent des molécules réactives qui
aggravent les blessures du muscle. Ces dommages collatéraux causés par le
système inné sont ensuite suivis d’une phase de réparation réalisée par les
macrophages. Ceux qui infiltrent le tissu de façon précoce facilitent le nettoyage des
débris nécrotiques alors que des macrophages activés plus tardivement l’infiltrent
pour favoriser la croissance musculaire. Les cellules satellites qui résident sous la
lamina basale de chaque myofibre sont des cellules souches. Après une blessure,
ces cellules quiescentes deviennent actives et subissent une prolifération pour
donner des progéniteurs myogéniques qui se différencient ensuite en myofibres
matures. Pour cela, le signal Notch agit de façon autocrine et régule la prolifération
des cellules satellites alors que l’action paracrine de l’IL-6 et de l’IGF est impliquée
dans la différenciation des progéniteurs en myofibrilles. En plus des cellules
satellites, les FAP (Fibro/Adipo Progenitor) interviennent dans la régénération du
muscle et sa dégénération graisseuse. Ce sont des cellules bipotentielles qui
peuvent donner naissance à des fibroblastes et à des adipocytes. L’activation du
système immunitaire inné est le premier évènement dans la régénération tissulaire.
Les signaux Th2 comme l’IL-4 et l’IL-13 produits par les PNE régulent les fonctions
des FAP pour faciliter la régénération musculaire. En présence d’IL-4, les FAP
donnent des fibroblastes qui aident la myogenèse en facilitant le nettoyage des
débris nécrotiques. Par contre en absence d’IL-4, les FAP se différencient en
adipocytes et contribuent à la persistance de débris cellulaires et à la dégénération
grasse du muscle (Heredia et al., 2013).
Lors d’une infection pulmonaire virale, une fois que les cellules infectées par le virus
sont nettoyées et que l’inflammation pulmonaire est contrôlée, la réparation peut
commencer. La capacité du poumon à régénérer rapidement et correctement son
épithélium après l’infection détermine si la fonction pulmonaire sera rétablie
normalement ou si des complications vont avoir lieu. La restauration de la barrière
épithéliale respiratoire est divisée en trois étapes : les cellules épithéliales voisines
couvrent la zone dénudée grâce à une expansion et une migration locale. La
migration et la prolifération des cellules progénitrices permettent de reconstituer
l’épithélium et enfin, la différenciation des cellules épithéliales en type cellulaire bien
défini permet de restaurer les fonctions d’un épithélium respiratoire. Ce processus de

116
réparation est rapide. La barrière pulmonaire peut être largement si ce n’est
totalement reconstituée dans les jours qui suivent la clairance virale, selon la gravité
de l’infection. Les cellules mortes sont éliminées par l’appareil mucociliaire ou
retirées par les phagocytes et les cellules épithéliales. Les poumons contiennent des
cellules souches capables de former des bronchioles, alvéoles et des vaisseaux
pulmonaires (Yoo, Kim, Hufford, & Braciale, 2013).
L’inflammation systémique B-
Quand l’infection submerge les défenses locales, l’organisme répond en activant une
réponse systémique vaste. Ces évènements sont collectivement appelés la réaction
de la phase aigüe. Ces changements, distants du site d’inflammation, impliquent de
nombreux organes et des changements nutritionnels, biochimiques, physiologiques
et comportementaux. Les phénomènes les plus évidents sont la fièvre, la
leucocytose et la surexpression ou la sous-expression des protéines de la phase
aigüe. La réaction systémique est régulée par une séquence d’activation précise : les
PAMPs sont reconnus par les protéines sériques, le plus actif étant le LBP (LPS
Binding Protein qui délivre le LPS au récepteur CD14 des macrophages) puis après
l’activation du CD14, le complexe CD14-LPS interagit avec le TLR4 qui traduit le
signal inflammatoire à l’intérieur de la cellule et active une cascade intracellulaire qui
aboutit à la production de cytokines pro-inflammatoires et de chimiokines qui régulent
finement la réaction systémique : l’IL-1β, le TNF-α et l’IL-6 (Ceciliani et al., 2012).
Ce qu’il faut retenir :
L’inflammation locale est le mécanisme de base de l’immunité innée. Elle a
lieu dès qu’une blessure, septique ou non, apparaît dans l’organisme, et
permet, à elle seule, la résolution de nombreux débuts d’infections. Les 4
étapes de l’inflammation sont :
- la phase vasculaire qui permet l’arrivée de leucocytes,
- la phase cellulaire, avec l’extravasation des cellules puis la
phagocytose,
- la phase de détersion pour éliminer les débris leucocytaires du lieu
d’infection,
- la phase de cicatrisation pour restaurer la fonction du tissu d’origine
si cela est possible.
Une fois encore, la communication entre cellules par cytokines et
chimiokines est permanente.

117
Ces trois cytokines sont des pyrogènes endogènes grâce à l’induction de la
production de prostaglandine E2 par la cyclooxygénase-2. La prostaglandine agit sur
l’hypothalamus qui augmente la production de chaleur par le corps grâce à la graisse
brune. Ces cytokines sont aussi responsables de la production des protéines de la
phase aiguë, vues page 72. Ces protéines permettent de se fixer sur les pathogènes
et de remplir le même rôle que les anticorps, mais leur production n’est pas
spécifique d’un pathogène. Le dernier effet à distance des cytokines est la
leucocytose, c'est-à-dire une augmentation du nombre de PNN circulants. Ces PNN
proviennent de la moelle osseuse qui libère des cellules en grand nombre et de
certains sites des vaisseaux sanguins où une partie des PNN est marginalisée et
constitue un pool rapidement mobilisable (Janeway et al., 2009).
Après avoir étudié la réponse locale et systémique de l’organisme face à une
agression, nous allons décrire les molécules qui permettent aux cellules de
communiquer entre elles et autorisant, à partir d’une lésion locale, une réponse
systémique.
Le langage chimique utilisé : les cytokines C-
Nous avons déjà évoqué les cytokines produites par les cellules immunes pour
organiser la réponse immune. Les cytokines sont des protéines sécrétées par des
cellules et transmettent le signal via des récepteurs de surface spécifiques sur la
cellule productrice de la cytokine (action autocrine) ou sur des cellules cibles (action
paracrine). Le terme de cytokine réfère à des molécules douées d’activité biologique,
avec une action sur les leucocytes alors que les facteurs de croissance réfèrent à
des molécules qui ont une action sur les cellules somatiques, les interleukines
désignent des molécules de communication entre leucocytes. Les molécules
possédant une activité chimiotactique sont appelées des chimiokines.
Des tableaux récapitulant les cytokines et les chimiokines sont respectivement
présentés en annexe I et en annexe II.
Les chimiokines 1)
Les chimiokines sont un ensemble de polypeptides de 8 à 15 kDa. Depuis
l’identification des chimiokines CXCL-8 (ou IL-8) et CCL2 (MCP-1) à la fin des
années 1980, la superfamille des chimiokines s’est considérablement agrandie. Il en
existe plus de 50 connues à ce jour et plus de 20 récepteurs, couplés à des
protéines G. Ces chimiokines sont produites lors de réponse inflammatoire et
régulent l’hématopoïèse, le mouvement des leucocytes, l’angiogenèse et
l’organogénèse. Elles agissent principalement sur les PNN, les monocytes, les
lymphocytes et les PNE. La plupart, si ce n’est tous les gènes des chimiokines
proviennent probablement de la duplication d’un seul gène ancestral (Douglas &
Jane Wardrop, 2010; A Zlotnik & Yoshie, 2000).

118
La présence d’une cystéine sur l’extrémité N terminale permet de classer les
chimiokines en quatre familles :
Les chimiokines α possèdent deux cystéines séparées par un acide aminé :
elles sont également appelées chimiokines CXC ;
Les chimiokines β possèdent deux cystéines adjacentes : ce sont les
chimiokines CC ;
Les chimiokines γ ne possèdent qu’une seule cystéine : il s’agit de la sous-
famille des chimiokines C. Ces molécules appartiennent à la classe des
lymphotactine ;
Les chimiokines δ ont deux cystéines séparées par trois acides aminés : c’est
la sous-famille des chimiokines CX3C ou fractalkines (Douglas & Jane
Wardrop, 2010; Paul, 2013; A Zlotnik & Yoshie, 2000).
Une seule chimiokine peut se lier à plusieurs récepteurs alors qu’un récepteur peut
traduire un signal pour plusieurs chimiokines.
Pour la nomenclature, on utilise le type de sous famille suivi de L pour ligand et un
nombre (ex : CXCL-8 pour IL-8) (Douglas & Jane Wardrop, 2010; A Zlotnik & Yoshie,
2000).
Les chimiokines peuvent aussi être divisées selon leur fonction :
Les chimiokines homéostasiques, exprimées de façon constitutive dans un
tissu ou un organe. C’est le cas de CCL-21, nécessaire pour la migration de
nombreuses cellules T vers les nœuds lymphatiques. Cette cytokine semble
interagir avec le CCR7, exprimé par les cellules T naïves ;
Les chimiokines inflammatoires, surexprimées lors de stimuli immuns ou
inflammatoires par différents types cellulaires comprenant les macrophages,
les lymphocytes T et les fibroblastes…Les chimiokines plasmatiques comme
le CCL-14 et CCL-15, présentes en grande quantité dans le sérum, forment
une première sous-famille. La deuxième est constituée des chimiokines
plaquettaires : stockées à haute concentration dans les granules α des
plaquettes comme le CXCL-4, le CXCL-4L1, le CXCL-7 et le CCL-5. Elles sont
très rapidement relâchées lors de l’activation des plaquettes ;
Les chimiokines duales remplissent les deux fonctions précédemment décrites
(A Zlotnik & Yoshie, 2000).
Les chimiokines homéostatiques sont relativement anciennes en termes évolutifs et
bien conservées entre les espèces alors que les chimiokines inflammatoires sont
caractérisées par une évolution très rapide, probablement due à la forte pression de
sélection pour augmenter leur diversité quand l’individu doit faire face à un nouveau
pathogène qui représente une menace pour sa survie (Albert Zlotnik & Yoshie,
2012).
Les chimiokines sont parfois exprimées de façon spécifique et très précise. Les
cellules Th1 expriment le CCR-5 et CXCR-3 alors que les cellules Th2 expriment le
CCR-4 et le CCR-8. Les cellules Th17 expriment le CCR-6 et c’est le signal qui les

119
attire dans l’intestin grêle quand il est colonisé par le microbiome. Dans la peau, la
chimiokine la plus retrouvée est le CCL-27, spécifiquement exprimée par les
kératinocytes puis vient le CXCL-14. Dans les muqueuses, il y a deux principales
chimiokines CCL-25 et CCL-28. La première permet le recrutement de cellules T
dans la lamina propria de l’intestin. L’expression de certains récepteurs aux
chimiokines homéostatiques dans les cellules tumorales influence leur potentiel
métastatique. Les cellules dendritiques produisent plusieurs chimiokines, certaines
de façon très spécifique. Cela suggère un rôle dans l’initiation de la réponse immune
et certaines pourraient avoir des propriétés adjuvantes notamment dans le
recrutement de différentes cellules T. Les cellules dendritiques immatures expriment
le CCR-6. Une fois qu’elles ont trouvé un antigène, elles commencent leur
maturation et expriment alors le CCR7, essentiel pour leur migration vers les nœuds
lymphatiques locaux (Michel et al., 2012; A Zlotnik & Yoshie, 2000).
Les interleukines 2)
Une interleukine est une cytokine produite par un leucocyte et agissant sur un autre
leucocyte (Paul, 2013).
Les cytokines de type Th1 sont produites essentiellement par les lymphocytes T et
comprennent l’IL-2 et l’IFN-γ ; ou par les macrophages : l’IL-12. L’IL-15 aussi est une
cytokine Th1 produite par de nombreuses cellules. Ces cytokines participent au
développement des réponses cellulaires.
Les cytokines de type Th2 comprennent l’IL-4, l’IL-5, l’IL-13, et l’IL-10, produits par
des cellules variées. Elles interviennent dans la réponse humorale (Genetet, 1997).
a. Les cytokines pro-inflammatoires
Les membres de la famille de l’IL-1 (l’IL-18, l’IL-33, l’IL-6, le TNF-α et l’IL-8) sont les
prototypes des cytokines pro-inflammatoires puisqu’elles induisent une
vasodilatation, une perméabilité vasculaire augmentée et l’expression de molécules
de l’immunité innée (Paul, 2013).
L’IL-1 est responsable de la fièvre, tout comme l’IL-6 et l’IL-8, également des
cytokines pro-inflammatoires clés. L’IL-1 induit la synthèse de nombreuses cytokines
pro-inflammatoires comme les membres des CSF. Ses rôles sont très nombreux :
elle active les cellules T, B et NK, elle augmente les activités anti-tumorales
antimicrobiennes, la phagocytose, le chimiotactisme, l’adhérence des leucocytes….
(Cavaillon, 1996).
L’IL-7 contrôle la naissance, l’éducation, la persistance, la maturation et la mort des
cellules T. Chez les rongeurs, l’IL-7 est la seule cytokine absolument critique pour la
génération des lymphocytes innés (γδ, αβ NKT) et adaptifs (αβ T et B). L’IL-7 est
principalement produite par les cellules stromales dans la moelle osseuse, les

120
nœuds lymphatiques, le foie, les plaques de Peyer et les follicules lymphoïdes
isolés(J. Kang & Coles, 2012).
L’IL-11 provoque la formation des lymphocytes B, des plaquettes, des monocytes.
Elle active la synthèse des protéines de la phase aiguë (Cavaillon, 1996).
L’IL-12 augmente la cytotoxicité des NK, leur production d’IFN-γ et leur prolifération.
Elle agit de même sur les lymphocytes T. Elle oriente la réponse immune vers une
réponse Th1 et inhibe la réponse Th2 alors que l’IL-4 a les effets inverses (Cavaillon,
1996; Delves et al., 2011).
L’IL-15 est une cytokine pléiotrope avec un grand nombre de fonctions à cheval
entre l’immunité innée et adaptative. Elle permet la différenciation, la survie et/ou
l’activation des cellules NK, des cellules NKT, des lymphocytes intraépithéliaux et
des cellules T mémoires. Cependant, elle est aussi impliquée dans des pathologies
auto-immunes (arthrite rhumatoïde, maladie de Crohn) et dans le contrôle de la
réplication de certains parasites comme Toxoplasma gondii et contribue à
l’inflammation intestinale. Cette cytokine peut être produite par de nombreuses
cellules comme les entérocytes, les cellules dendritiques ou encore les
macrophages. L’IL-15 est nécessaire pour activer et attirer les monocytes sur les
lieux de l’inflammation (Schulthess et al., 2012).
L’IL-17 induit la maturation des neutrophiles ainsi que leur chimiotactisme. Ce rôle
est essentiel dans la lutte contre les bactéries. Les cellules γδ T ont été identifiées
comme la source principale de cette cytokine durant des infections bactériennes
différentes notamment par Listeria monocytogenes, et Escherichia coli Pour la
synthèse d’IL-17, différents signaux sont nécessaires dont le TCR, et le ROR-α.
D’autres cellules sont capables de fabriquer de l’IL-17A et de l’IL-17F comme les
cellules T CD8+ cytotoxiques, les cellules γδ 17 et les NKT. Les cellules épithéliales
sont la principale cible des cytokines des Th17. Il existe plusieurs types d’IL-17 (de A
à F)
L’IL-17A et l’IL-17F sont homologues et se fixent sur le même récepteur. Elles
sont toutes les deux impliquées dans le développement de l’inflammation et
les défenses de l’hôte contre les infections par l’expression de gènes codant
pour des cytokines pro-inflammatoires (TNF, IL-1, IL-6, G-CSF, GM-CSF) et
des chimiokines (CXCL-1, CXCL-5, IL-8, CCL-2 et CCL-7) ainsi que des
peptides antimicrobiens (des défensines) et des métalloprotéinases par les
fibroblastes, les cellules endothéliales et épithéliales. L’IL-17A et F stimulent la
production de mucus dans les voies respiratoires mais n’auraient pas le même
effet dans l’intestin ;
L’IL-17A, l’IL-17B, l’IL-17C et l’IL-17F (pas l’IL-17E) induisent l’expression de
cytokines pro-inflammatoires comme le TNF et l’IL-1β par les fibroblastes et
stimulent la migration des PNN. L’IL-17F est un plus faible inducteur des
cytokines pro-inflammatoires ;

121
L’IL-17E est produite par les cellules Th2, les cellules T CD8+ et CD4
+ des
cryptoplaques, par les mastocytes et les PNE. Elle est nécessaire pour
l’immunité adaptative. L’IL-17E active les cellules NKT, des macrophages et
les cellules épithéliales respiratoires et leur fait sécréter des cytokines Th2.
Les rôles de l’IL-17E dans l’inflammation des voies respiratoires sont
différents de ceux de l’IL-17A et IL-17F puisque la première induit une
éosinophilie grâce à des protéases alors que les deux derniers induisent une
neutrophilie ;
Enfin, l’IL-17B, l’IL-17C, et l’IL-17D peuvent avoir des activités similaires pour
induire des médiateurs inflammatoires et contribuent à la réponse
inflammatoire comme l’IL-17A et IL-17F (Iwakura, Ishigame, Saijo, & Nakae,
2011; Louten et al., 2009; Verdier & Ruemmele, 2013; Zenewicz & Flavell,
2013).
L’IL-22 est une cytokine sécrétée par les cellules immunes innées (cellules NK et LTi
like) et adaptatives (cellules T CD4+). Elle fait partie de la famille de l’IL-10 et induit
la sécrétion de protéines antimicrobiennes comme les RegIIIγ et RegIIIβ. L’IL-22 joue
un rôle important dans l’inflammation comme dans les maladies inflammatoires
chroniques et des maladies infectieuses. En maintenant l’intégrité des barrières
épithéliales, l’IL-22 aide à prévenir la dissémination des bactéries pathogènes
comme Klebsiella pneumoniae, Citrobacter rodentium et Salmonella enterica. L’IL-22
intervient dans la réparation tissulaire et est important pour la régénération du tissu
hépatique. Elle intervient dans la prolifération des kératinocytes et l’hyperplasie
épithéliale. Elle joue aussi un rôle protecteur dans l’inflammation. La sécrétion d’IL-
22 est stimulée par l’IL-23. L’IL-17, l’IL-17F et l’IL-22 agissent en synergie pour
combattre les infections bactériennes (Iwakura et al., 2011; Kinnebrew et al., 2012;
Louten et al., 2009; H Spits, 2011; Tumanov et al., 2011; Zenewicz & Flavell, 2013).
L’IL-22 est exprimée par différents types de lymphocytes :
Les cellules Th17 produisent de l’IL-22 sous la dépendance de l’IL-23. Cette
expression dépend de l’Ahr ;
Les cellules Th22 expriment le CCR-6, le CCR-4 et le CCR-10 mais pas l’IFN-γ,
l’IL-17A, le T-bet ou le RORγt. Des cytokines inflammatoires, comme l’IL-6 ou
le TNF-α stimulent la production d’IL-22 et l’activation de la vitamine D ;
Les cellules γδ T répondent immédiatement à l’IL-23 en exprimant l’IL-22 et
l’IL-17A. L’Ahr est un important facteur de transcription de l’expression d’IL-22
mais pas l’IL-17A ;
Les cellules NK peuvent être activées par deux cytokines en synergie : l’IL-12
et l’IL-18, probablement sécrétées par les macrophages ;

122
Nous verrons en 3ième
partie que les LTi produisent aussi de l’IL-22 (Zenewicz
& Flavell, 2013).
L’IL-23 est une cytokine clé orchestrant le dialogue entre l’immunité innée et
l’immunité adaptative. Elle joue un rôle central dans la réponse précoce aux
microbes. Elle est impliquée dans le maintien et l’amplification des cellules Th17 mais
aussi sur les cellules γδ T ou les iNKT qui répondent en produisant des cytokines
liées aux Th17. Elle agit aussi sur les ILCs (Geremia et al., 2013; Willems, Vollstedt, &
Suter, 2009).
Le ST2 est un récepteur transmembranaire avec des variants solubles dont le ligand
est l’IL-33. La fixation de l’IL-33 sur la forme transmembranaire du ST2 mène à
l’activation du NK-κB alors que sa fixation sur la forme soluble mène à l’inactivation
de cytokines. De plus le ST2 diminue l’expression des TLR de façon compétitive en
se fixant sur le MyD88. L’IL-33 apparait donc comme une cytokine traditionnelle mais
aussi comme un facteur nucléaire. L’IL-33 est exprimée par le système nerveux
central, le poumon et les intestins. Sa localisation et sa concentration sont variables :
Dans des cellules au repos, l’IL-33 est dans le noyau où elle interagit avec la
chromatine : elle agit comme répresseur transcriptionnel.
L’expression d’IL-33 est augmentée dans les tissus inflammatoires notamment
dans les cellules dendritiques, les macrophages et les fibroblastes, suite à la
stimulation de leur TLR. Elle est produite par les mastocytes, par des cellules
non immunes comme les cellules endothéliales, épithéliales et musculaires
lisses ainsi que par les fibroblastes.
De plus, l’IL-33 peut être libérée lors de nécrose cellulaire et agit donc comme
alarmine.
L’IL-33 induit la production de nombreuses cytokines, par l’intermédiaire du MyD88.
En effet, l’activation de l’expression de gènes passe par l’activation du NK-κB. L’IL-
33 agit sur de nombreuses cellules et la réponse dépend du type cellulaire :
Les cellules dendritiques expriment le CD80 ou l’OX40L et produisent des
cytokines inflammatoires.
Les mastocytes et basophiles produisent plus de cytokines pro-
inflammatoires.
Les cellules Th se polarisent dans un phénotype qui produit de l’IL-5, de l’IL-
13 mais pas d’IL-4. L’IL-33 régule ainsi le système immun adaptatif
Des cellules non immunes répondent également à l’IL-33 comme les cellules
endothéliales qui produisent du NO.
Les mastocytes produisent des cytokines et chimiokines comme l’IL-1β, l’IL-6,
l’IL-13, le TNF, le GM-CSF, le CXCL-8, le CCL-1 et le CCL-2.
Les PNB libèrent de l’IL-4, de l’IL-5, de l’IL-6, de l’IL-8, de l’IL-13, du GM-CSF,
du CCL-2, du CCL-3 et du CCL-4. L’IL-33 stimule la dégranulation par les IgE
ainsi que la migration.
Les PNE survivent plus longtemps, l’adhésion cellulaire est facilitée en
augmentant l’expression du CD11b et ICAM-1. La production de cytokines

123
augmente, notamment celle de l’IL-6, de l’IL-8, de l’IL-9, du CCL2 et des
superoxydes.
Les iNKT doublent leur nombre dans la rate et le foie. Ils augmentent leur
production d’IL-4 et d’IFN-γ après activation des TCR. Les iNKT participent
non seulement à l’immunité acquise mais aussi aux réponses immunes innées
puisque, comme les NK classiques, ils peuvent être complètement activés et
produire de grandes quantités d’IFN-γ sans l’activation de TCR, après
exposition par la cytokine pro-inflammatoire IL-12.
L’IL-33 est protectrice dans l’arthérosclérose en réduisant la formation de plaques
arthérosclérotiques. De même, dans les maladies cardiovasculaires, elle joue un rôle
protecteur. Elle a aussi un rôle homéostatique dans le métabolisme
musculosquelettique puisqu’elle prévient la résorption osseuse et affecte la
différenciation des ostéoclastes. (Bourgeois et al., 2009; Mirchandani, Salmond, &
Liew, 2012; Nakae et al., 2013)
L’IL-33 est activée par clivage par une caspase 1 et ensuite sécrétée par un
mécanisme non conventionnel. Cependant, l’IL-33, dans sa forme non raccourcie est
active, avant même le clivage par la caspase. Donc le clivage par la caspase durant
l’apoptose avec l’activation des inflammasomes n’est pas nécessaire pour l’activation
et la sécrétion d’IL-33. De plus, l’IL-33 est libérée par les cellules nécrotiques sans
clivage. Enfin, dans les PNN, l’IL-33 peut être clivée par une élastase et une
cathepsine G, et cette forme est 10 fois plus active que la forme longue. Certes, lors
d’infection fongique, bactérienne ou parasitaire, l’IL-33 participe à la clairance des
infections mais lorsque l’IL-33 est produite en trop grande quantité ou lorsqu’il n’y en
a pas besoin, elle conduit à la maladie de Crohn, la colite ulcérative ou encore
l’allergie par l’activation des macrophages, éosinophiles, cellules dendritiques ou
cellules Th2. (Bourgeois et al., 2009; Mirchandani et al., 2012; Nakae et al., 2013).
b. Les cytokines anti-inflammatoires
L’IL-10 est une cytokine anti-inflammatoire car elle inhibe l’activation des
macrophages et la différenciation des Th1. Elle inhibe aussi la libération des cytokines
par les macrophages comme l’IL-12 et diminue l’expression des molécules de CMH
II (Cavaillon, 1996).
L’IL-4 a un rôle complexe : elle active les macrophages et augmente leur capacité de
cellules présentatrices d’antigènes mais elle inhibe leur capacité cytotoxique
dépendante des anticorps. Une de ses fonctions les plus notables est l’inhibition de
la production des cytokines pro-inflammatoires comme l’IL-1, l’IL-6, l’IL-8, le TNF-α et
le GM-CSF. Sur les PNN, elle stimule la phagocytose et l’explosion respiratoire
(Cavaillon, 1996).
L’IL-13 ressemble à l’IL-4 : elle agit sur les monocytes, les lymphocytes B mais pas
sur les cellules T. Elle inhibe la production des cytokines pro-inflammatoires IL-6 et
IL-8 par les monocytes (Cavaillon, 1996).

124
Les hématopoïétines 3)
Les hématopoïétines sont des facteurs inducteurs de la différenciation et stimulent
l’hématopoïèse. Leur nom comprend CSF pour Colony Stimulatory Factor. Cette
famille englobe donc le G-CSF (Granulocyte) M-CSF (Monocyte), GM-CSF…. Ces
molécules peuvent aussi stimuler les cellules auxquelles elles ont donné naissance
(Genetet, 1997).
La famille des interférons 4)
Les IFN sont classés en 3 groupes :
Les IFN de type I (IFN-α, IFN-β, IFN-ω, IFN-τ…), connus pour leur protection
antivirale. Ils induisent la synthèse d’enzymes qui empêchent la production
des protéines virales ou bloquent la décapsidation des virus ou l’assemblage
des virions. Ils stimulent aussi l’activité des macrophages et des NK ;
L’IFN de type II (IFN-γ), avec un effet sur les macrophages et les
granulocytes : il augmente leur phagocytose, leur potentiel sécrétoire et
l’expression des molécules de CMH II donc la présentation d’antigènes
(Cavaillon, 1996) ;
L’IFN de type III (IFN-λ) joue également une part cruciale dans les défenses
antivirales de l’hôte au niveau des tractus gastro-intestinal et respiratoire
(Lester & Li, 2014).
Autres mécanismes effecteurs D-
La phagocytose est le principal moyen dont dispose le système inné pour détruire les
pathogènes extracellulaires (n’oublions pas cependant, tous les autres procédés déjà
évoqués comme la mise en place de complexes d’attaques membranaires, ainsi que
l’action des peptides antimicrobiens…et bien d’autres encore). Pour les microbes
Ce qu’il faut retenir :
Le système immunitaire est doté d’un véritable mode de communication entre
les cellules avec une panoplie de cytokines qui peuvent avoir des actions
autocrine ou paracrine. Chaque cytokine peut avoir plusieurs effets selon la
cible. De plus, lors d’une réponse inflammatoire, rappelons qu’un grand
nombre de cytokines différentes sont libérées, ce qui explique une fois encore
la complexité de la réponse immunitaire.

125
intracellulaires, les cellules NK sont impliquées mais un autre mécanisme existe pour
les éliminer : l’autophagie, déjà mentionnée plusieurs fois.
L’autophagie est un mécanisme homéostasique cellulaire où les cellules digèrent
leur propre cytoplasme pour la destruction ou le renouvellement des organites. Le
processus d’autophagie est très conservé phylogénétiquement puisqu’il est présent
chez les levures tout comme chez les mammifères. Ce phénomène recycle des
macromolécules cytosoliques à longue vie comme des protéines stables, pour aider
les besoins anaboliques cellulaires et la viabilité de l’organisme dans des conditions
d’affamement. Ce processus est impliqué dans le turnover normal des protéines et
organites et est central dans les activités cellulaires qui maintiennent un équilibre
entre la synthèse et la dégradation de différentes protéines. L’autophagie est aussi
impliquée dans le nettoyage des organites dégradés comme les mitochondries
compromises, protégeant ainsi les cellules de l’apoptose non prévue. Bien que ce
soit un mécanisme de maintien cellulaire, sous certaines conditions spécifiques, une
autophagie excessive peut entraîner une mort cellulaire programmée non apoptique.
Elle a été impliquée dans le cancer, les désordres dégénératifs comme l’Huntington,
Parkinson ou Alzheimer. Elle a un fort impact sur la présentation via le CMH II et est
contrôlée par des cytokines. L’autophagie séquestre des organites endommagés ou
une portion de cytosol dans une structure autophagosomale. Cette structure se
distingue des phagosomes conventionnels par la présence de la double membrane
qui la délimite et le contenu cytosolique intraluminal. La fusion entre
l’autophagosome et le lysosome aboutit à la formation d’un compartiment
dégradateur appelé l’autolysosome. Une induction physiologique de l’autophagie est
le manque d’acides aminés et en particulier en leucine ou l’absence de facteurs de
croissance qui autorisent normalement la cellule à prélever des nutriments.
L’autophagie est également un processus utilisé dans les défenses innées
puisqu’elle permet d’éliminer les pathogènes intracellulaires dans un processus
similaire à la capture et la digestion des organites intracellulaires. Ainsi, elle est
utilisée pour l’élimination des bactéries et virus intracellulaires, en plus de sa fonction
primaire de nettoyer le cytoplasme. Elle est induite par l’IFN-γ dans les macrophages
et des cellules non immunes et inhibée par l’IL-13 (Deretic, 2006; Peterson & Artis,
2014).
Initiation de la réponse adaptative E-
Une fois que les acteurs sont sur le site de l’infection et que la phagocytose est mise
en place, le système inné commence à préparer l’initiation du système adaptatif, au
cas où il serait dépassé par l’infection. Cette activation du système adaptatif
s’appelle la présentation d’antigène, effectuée principalement par les cellules
dendritiques, mais aussi par les macrophages.

126
Bien que les cellules présentatrices d’antigènes servent à initier l’immunité
adaptative spécifique d’un antigène, la préparation et la présentation des antigènes
sont des fonctions innées (Born et al., 2006).
Les cellules dendritiques sont connues pour être la principale cellule présentatrice
d’antigènes qui active les cellules T naïves. Nous prendrons donc leur exemple pour
décrire les différentes étapes de la présentation antigénique, bien que d’autres
cellules puissent faire de même.
Le prélèvement des antigènes 1)
L’antigène est prélevé dans le milieu par phagocytose, pinocytose et
macropinocytose (pour les antigènes solubles).
La pinocytose est très efficace pour prélever de petites molécules solubles.
Certaines cellules dendritiques peuvent prélever des molécules dans leur
environnement à un taux proche d’un volume cellulaire par heure. Les cellules
dendritiques peuvent ensuite présenter les antigènes ainsi prélevés aux lymphocytes
T. Les granulations de Birbeck des cellules dendritiques et les molécules langhérines
des cellules de Langherans sont impliquées dans la pinocytose (Clark et al., 2000).
La manière la plus efficace pour prélever des échantillons dans l’environnement pour
les cellules dendritiques est la macroendocytose ou prélèvement activé par des
récepteurs. Les cellules dendritiques n’expriment pas de récepteurs aux antigènes
spécifiques mais des PRR comme les récepteurs type lectine, les récepteurs
éboueurs, les récepteurs au complément ou les récepteurs aux immunoglobulines
(Clark et al., 2000).
L’expression des aquaporines 3 et 7 contribue à la macropinocytose. La
phagocytose est activée par l’attachement de particules extracellulaires sur des
récepteurs de surface (Paul, 2013).
Bien que les cellules dendritiques CD1b+ présentes dans la lymphe montrent des
traits de cellules dendritiques matures, elles maintiennent la capacité de prélever des
antigènes solubles efficacement et de phagocyter Salmonella. Ce sous-type est
capable à lui seul de présenter les antigènes à des cellules T CD4+ effectrices ou
mémoires mais aussi à des cellules T naïves et induit la production de l’IFN-γ et de
l’IL-10. Les cellules dendritiques de la lymphe expriment le récepteur CD14, pour le
LPS (Olivier et al., 2012).
Dans le tractus intestinal, plusieurs voies sont possibles pour que les cellules
dendritiques prélèvent les antigènes et les préparent pour leur présentation. Ces
différents moyens sont résumés dans la Figure 27, page 127 (S. C. Ng et al., 2010).
Traditionnellement, les cellules M, des cellules épithéliales spécialisées
prélèvent des macromolécules ou des microorganismes. Ces cellules sont

127
dispersées entre les cellules épithéliales conventionnelles et sont au sommet des
plaques de Peyer. Par exemple, les cellules M prélèvent Salmonella typhimurium
administré oralement. Les cellules dendritiques migrent en réponse aux bactéries
vivantes qui entrent dans la muqueuse via les cellules M ;
Les cellules dendritiques peuvent acquérir des antigènes indirectement par
l’internalisation de cellules épithéliales apoptiques ;
Les cellules dendritiques de la lamina propria peuvent ouvrir les jonctions
serrées entre les entérocytes et étendre des dendrites transépithéliales dans la
lumière intestinale. Une signalisation par les TLR depuis les cellules épithéliales
facilite l’extension des cellules dendritiques dans la lumière intestinale pour prélever
directement les bactéries ;
Le récepteur fœtal FcRn exprimé sur les tissus fœtaux et adultes, notamment
sur les cellules épithéliales, prélève des antigènes de la lumière et les transporte
vers les cellules dendritiques sous-épithéliales ce qui initie une réponse anticorps qui
aggrave l’inflammation intestinale ;
Les bactéries arrivent à avoir accès à la lamina propria, surtout lors de brèche
dans la barrière épithéliale ou lors d’inflammation. Les cellules dendritiques sont
présentes dans la couche même de l’épithélium et sont donc en compétition avec les
cellules épithéliales dans la préparation et la présentation des antigènes aux
lymphocytes intraépithéliaux (S. C. Ng et al., 2010).
Figure 27 : Mécanismes d'échantillonnage des antigènes par les cellules dendritiques dans la lamina propria (S. C. Ng et al., 2010).
1 : Transport des antigènes par les cellules M / 2 : extension des dendrites entre les cellules épithéliales / 3 : Une brèche intestinale donne un accès direct aux antigènes
/ 4 : le récepteur fœtal FcRn capte les antigènes pour les donner aux cellules dendritiques sous-jacentes / 5 : Les cellules dendritiques CD103+ CX3CR1- reçoivent
des antigènes conditionnés par les cellules épithéliales.

128
Maturation des cellules dendritiques 2)
Lorsqu’une cellule dendritique rencontre un antigène (cadre A de la Figure 28, ci-
dessous), elle change de phénotype et se spécialise, non plus dans le prélèvement
d’antigènes mais dans la présentation de ces antigènes. Elle acquiert ainsi la
capacité d’exprimer de grandes quantités de molécules de présentation du CMH I et
CMH II, ainsi que des molécules d’adhésion et de co-stimulation comme le CD40, le
CD54, le CD80 et le CD86. Enfin, lors de leur maturation, les cellules dendritiques
produisent des cytokines pro-inflammatoires comme l’IL-1, l’IL-6, l’IL-12, l’IL-18 et
l’IL-23 (Hou et al., 2008; Liu, 2001; Paul, 2013).
Seules les cellules de Langherans résidant dans l’épithélium expriment la E-
cadhérine qui permet leur attachement aux kératinocytes voisins. Dans des
conditions d’équilibre, les cellules de Langherans n’expriment pas le CMH II sur la
surface et la plupart de ces molécules sont intracellulaires. Après la rencontre de la
cellule avec l’antigène, le CMH II est rapidement transporté vers la surface cellulaire
et l’expression de l’E-cadhérine est réduite, ce qui permet aux cellules de
Langherans de se détacher des kératinocytes et de migrer vers les nœuds
lymphatiques. Après activation, leurs dendrites s’allongent pour échantillonner des
antigènes externes au stratum corneum (Kushwah & Hu, 2011).
Les cellules dendritiques sont capables de libérer des exosomes : des morceaux de
membranes cellulaires riches en molécules de CMH associées à des protéines de
co-stimulation capables de stimuler les lymphocytes T mais qui sont plus efficaces
lorsqu’ils sont « re-présentés » par des cellules dendritiques (Clark et al., 2000).
Figure 28 : Maturation des cellules dendritiques suite à la détection d'un PAMP D’après (Janeway et al., 2009).
Dans les tissus périphériques, les cellules dendritiques immatures rencontrent des
pathogènes et sont activées par les PAMPs
Les cellules dendritiques activent les cellules T naïves dans les zones T.
Le CCR7 dirige la migration vers les tissus lymphoïdes et augmente
l’expression des molécules de co-stimulation et du CMH
La signalisation du TLR induit la libération de CCR7 et favorise le traitement des
antigènes dérivés du pathogène

129
La migration des cellules dendritiques 3)
Les cellules dendritiques migrent tout d’abord vers un site inflammatoire, alors
qu’elles ne sont pas encore matures, puis lorsqu’elles ont rencontré un antigène, une
deuxième migration s’effectue, cette fois-ci, vers le nœud lymphatique.
Plusieurs molécules sont chimioattractives pour les cellules dendritiques immatures
vers les sites inflammatoires, comme le peptide N-formyl, un peptide bactérien ou les
β–défensines, détectées par les cellules dendritiques par le CCR-6. L’expression de
l’IL-12/23 p40 par les cellules dendritiques est cruciale pour leur migration dans des
sites cibles comme lors d’infection mycobactérienne. La fixation sur le récepteur à
l’IL-18 d’une cytokine de la famille de l’IL-1 est impliquée dans la migration des
cellules dendritiques et fournit un stimulus pro-inflammatoire pour continuer la
production d’IL-17 (Clark et al., 2000; Paul, 2013; Stockinger et al., 2007).
Très rapidement après leur activation, les cellules dendritiques expriment le
récepteur aux chimiokines CCR7 (cadre B et C de la Figure 28, page 128), et
cheminent dans les voies lymphatiques grâce à son ligand 6Ckine puis jusqu’aux
nœuds lymphatiques grâce à MIP-3β (Liu, 2001).
Présentation de l’antigène 4)
Bien que les cellules dendritiques soient d’excellentes cellules présentatrices
d’antigènes via le CMH II, ce ne sont pas les seules (les cellules B activées, les
monocytes en sont aussi capables).Contrairement aux macrophages qui réalisent
l’endocytose et digèrent rapidement l’antigène, les cellules dendritiques séquestrent
l’antigène et le préservent pour le présenter plus tard. La préservation des antigènes
est critique pour l’immunogénicité. Elle est permise par la faible activité des
protéases lysosomales et la faible acidité des lysosomes : tout ceci ralentit la
digestion de l’antigène pour pouvoir le charger sur le CMH ou le conserver durant la
migration des cellules dendritiques (Paul, 2013).
La voie de signalisation dépendante du MyD88 dans les cellules dendritiques joue un
rôle essentiel dans la production de cytokines innées et dans la polarisation Th1 des
cellules T CD4+ spécifiques de l’antigène. Le passage par MyD88 est
particulièrement important pour la production d’IL-12 qui peut alors stimuler les
cellules innées comme les cellules NK qui expriment l’IFN-γ et favoriser la
polarisation Th1 des cellules T CD4+. Le destin des cellules T n’est pas seulement
affecté par la cellule dendritique avec qui elle interagit mais aussi par des cellules
voisines comme d’autres cellules dendritiques et des cellules non dendritiques
présentes dans les tissus infectés (Hou et al., 2008).
L’endosome est un compartiment à l’intersection des voies endocytaires et
antérogrades. Les endosomes sont importants pour recycler les récepteurs
membranaires, pour la transduction des signaux, pour initier les réponses innées et

130
adaptatives mais aussi pour transporter des éléments nouvellement synthétisés à la
surface de la cellule. La sécrétion de cytokines et chimiokines par les cellules
activées est critique pour la phase effectrice de la réponse immune. La réponse
adaptative est initiée par la reconnaissance d’antigènes présentés par des molécules
de CMH II aux cellules T CD4+. Ces molécules de CMH II sont exprimées par les
cellules présentatrices d’antigènes comme les cellules dendritiques, macrophages et
cellules B. En tant que professionnels, les cellules dendritiques sont très efficaces
pour internaliser les antigènes par la macropinocytose ou l’endocytose via les
récepteurs. Ces deux voies convergent vers le système endosomal où les antigènes
internalisés sont hydrolysés par des protéases dans un environnement acide et les
peptides obtenus sont liés aux molécules de CMH II. Il existe deux voies de
présentation croisée. La voie cytosolique implique le transfert des protéines
exogènes depuis le compartiment endosomal vers le cytosol où ils sont préparés par
le protéasome et sont chargés sur le CMH I. Dans cette voie, les protéines
internalisées nécessitent d’être dépliées mais non dégradées dans la lumière du
compartiment endocytaire pour une translocation ultérieure dans le cytosol. Un autre
moyen de présenter les antigènes aux CD8+ est la voie vacuolaire, indépendante du
protéasome. L’antigène exogène est dégradé par les protéines lysosomales et
chargé sur des CMH I (Gleeson, 2014).
Une nouveauté : la mémoire innée F-
La présentation d’antigènes permet de sélectionner le clone lymphocytaire naïf
spécifique de l’antigène afin qu’il subisse une expansion clonale ou la réactivation
d’un lymphocyte mémoire, qui subira lui-aussi une expansion clonale. Jusqu’à
présent, la mémoire immunitaire était caractéristique de l’immunité adaptative mais
de récentes découvertes ont mis en évidence une mémoire au sein du système inné
Nous avons vu que les cellules du système inné comme les granulocytes, les
macrophages ou les NK sont rapidement disponibles pour combattre efficacement et
Ce qu’il faut retenir :
La présentation d’antigène est un rôle essentiel pour le système immunitaire
inné puisque cette étape active le système adaptatif. Cette présentation est
réalisée par les cellules dendritiques, spécialisées dans cette tâche, mais
aussi par les macrophages. Le prélèvement et la préparation de l’antigène
sont bien des fonctions innées, mais la présentation antigénique permet
d’initier la réponse adaptative : une fois encore, les deux immunités travaillent
ensemble et en continuité l’une de l’autre.

131
tuer un large éventail de pathogène mais ne confèrent pas de spécificité ou de
mémoire aux défenses.
Les dernières années de recherche ont drastiquement modifié le dogme de
l’immunité innée non spécifique. En effet, les PRR sont responsables de l’activation
semi-spécifique des cellules du système inné grâce à la reconnaissance de PAMPs
(Quintin, Cheng, van der Meer, & Netea, 2014).
Il existerait en fait un bras adaptatif au système immunitaire inné qui augmente
considérablement le répertoire de reconnaissances et de réponses et qui peut,
partiellement, combler la limitation génétique inhérente au système inné. Les
invertébrés sont un excellent modèle pour le système inné puisqu’ils ne possèdent
pas de cellules lymphocytes-like qui expriment des récepteurs réarrangés
somatiques. L’exposition à un microbe ou à l’un de leur composant mène à une
réponse immune cellulaire et cette exposition est protectrice contre les infections
suivantes. Ce résultat peut être dû à une augmentation générale de l’immunité ou
peut être spécifique pour certaines classes de pathogènes. L’hôte reconnaît des
molécules étrangères modèles et initie une réponse immune, ce qui augmente la
résistance au microbe. L’immunité innée adaptative est une réponse à des molécules
générales et a des stimulations immunes générales alors que la réponse adaptative
est une réponse qui augmente la résistance au pathogène exposé. La composante
mémoire de la réponse innée consiste en une période de temps pendant laquelle il y
a une augmentation optimale suivie d’une réponse qui décline ou qui pourrait durer
plus longtemps (Bowdish et al., 2007).
L’immunité innée des plantes comporte une mémoire suivant une infection initiale. Le
système immunitaire des plantes, dénué de tout système adaptatif et sans les
cellules mobiles classiques comme les phagocytes ou les lymphocytes représente la
clé de voute de l’immunité animale. Les mécanismes de défense des plantes sont
des réponses immunes locales en plus d’effecteurs comme l’acide salicylique, le
jasmonate, l’éthylène ou l’oxyde nitrique. Ces médiateurs solubles sont exprimés en
réponse aux infections et sont de véritables molécules de signalisation qui
permettent de monter une défense efficace à travers les tissus de la plante. Après la
réinfection, ces mécanismes permettent de développer une réponse forte durable qui
représente une mémoire immunologique (Quintin et al., 2014).
On retrouve la même chose chez les invertébrés. Les macrophages peuvent
reprogrammer leur fonction pendant et après une infection, ce qui estompe la
distinction entre système inné et adaptatif. Il a été proposé le terme d’immunité
entrainée pour décrire l’augmentation des défenses innées de l’hôte quand une
seconde infection a lieu. L’immunité entrainée est définie comme une protection
contre une réinfection par le même agent ou un autre proche dans des organismes
qui ne possèdent pas de système adaptatif (Quintin et al., 2014).
Cependant, l’immunité entrainée est aussi importante chez les vertébrés chez qui
elle fournit une protection en parallèle de la réponse adaptative classique. La

132
découverte de caractéristiques de mémoire dans les cellules typiquement innées
comme les NK ou les monocytes représente un changement marquant. L’infection
virale latente, comme par un herpèsvirus, peut conférer une protection bénéfique
contre des pathogènes bactériens comme Listeria monocytogenes, ou Yersinia
pestis due à une augmentation du niveau basal de l’immunité innée comme
l’activation des macrophages ou l’augmentation de la production d’IFN-γ et de TNF-
α. Etrangement, cette protection requiert l’IFN-γ mais pas les fonctions effectrices
des cellules T CD4+ et CD8+ lors de la seconde infection. Des NK mémoires peuvent
rapidement dégranuler et produire des cytokines après réactivation, conférant une
immunité protectrice. La réponse immune innée a donc la capacité d’être entrainée.
Chez les mammifères, la mémoire immunologique innée a été découverte pour la
première fois il y a des décennies et a été attribuée aux macrophages. La mémoire
des NK dépend du récepteur Ly49H spécifique des virus alors que la mémoire pour
les haptènes se fait par le CXCR-6. La modulation des fonctions monocytaires et
macrophagiques est un composant essentiel de la réponse immune et elle peut
présenter des caractéristiques adaptatives : un premier contact des monocytes et
macrophages avec du LPS en grand quantité (comme lors de sepsis) les conduit à
un état tolérant durant lequel ils sont réfractaires à une seconde stimulation.
Cependant, des concentrations très faibles de LPS orientent les monocytes vers une
réponse inflammatoire. Cette réponse se fait via la kinase IRAK-1 et ne fait pas
intervenir le NF-κB. Ces cellules sont également capables de moduler l’expression
de leur PRR, notamment le MARCO, dectin-1 et pentraxin-3. Lors d’activation des
monocytes, des modifications de la chromatine ont lieu et des marqueurs
épigéniques sont alors détectables comme le H3K4me3 et H3K27ac (Figure 29,
page 133, 2ième ligne, T=4h). Cependant la plupart de ces marqueurs disparaissent
dans les heures qui suivent l’activation de la cellule. Alors que la méthylation des
histones et leur acétylation a lieu durant la stimulation cellulaire, l’acétylation des
histones disparait après retrait du stimulus. Seul H3K4 monoacéthylé persiste et
représente donc l’activateur latent. Le monocyte est alors amorcé. (Figure 29, page
133, 3ième ligne, T=24h, à gauche). Lorsqu’une deuxième stimulation par le LPS a
lieu (Figure 29, page 133, 3ième ligne, T=24h, à droite), cet activateur latent est
réveillé et met en place une réponse plus forte et plus rapide. Ainsi, cinq jours après
la stimulation initiale, le marqueur H3K4 est toujours observé. Le monocyte est alors
qualifié d’entrainé. Il peut, lui aussi réponse de façon plus forte et plus rapide grâce à
un facteur de transcription plus rapidement activé (Quintin et al., 2014).

133
Figure 29 : Mécanisme de mise en place de la mémoire immune innée (Quintin et al., 2014).
Les cellules vertes sont des macrophages ou des monocytes. H3H4me, H3K4me3 et H3K27ac sont des marqueurs du programme épigénétique.
Ce qu’il faut retenir :
Les monocytes et les macrophages semblent donc dotés d’une mémoire qui
leur permet de réagir plus rapidement et plus efficacement à une réinfection
par le même agent. Auparavant, la distinction entre le système adaptatif
spécifique, doté d’une mémoire et le système inné, non spécifique et sans
aucune mémoire était nette mais la découverte de l’immunité innée entraînée
continue à estomper cette distinction.

134
IV. Evolution
Nous avons étudié l’ensemble du système inné et son fonctionnement, nous allons
maintenant comparer ce système entre les différents règnes, animaux et végétaux,
avant d’étudier plus en détail, en deuxième partie, les particularités chez les
ruminants.
Tous les êtres vivants possèdent un système immunitaire pour se défendre contre
des agents infectieux. En effet, tous les organismes sont menacés par des
pathogènes et la sélection naturelle exercera nécessairement une pression en faveur
du développement des mécanismes de protection. Les bactéries se défendent contre
des parasites (plasmides) et des bactériophages en utilisant les enzymes de
restriction et des peptides antimicrobiens actifs contre les bactéries concurrentes.
Bien que des systèmes adaptatifs ou adaptatifs-like aient été découverts chez les
invertébrés et les poissons sans mâchoire (agnathes), l’immunité adaptative, basée
sur les immunoglobulines, les TCR et le CMH n’est trouvée que chez les vertébrés
(Janeway et al., 2009).
Le système immunitaire est comparé à la « reine rouge » dans « Alice au Pays des
Merveilles » puisqu’il doit continuellement évoluer pour éviter un retard : à cause du
conflit perpétuel avec les pathogènes, le système immunitaire est en flux constant.
Ceci se vérifie par les différences entre le système immunitaire d’animaux dans le
même groupe phylogénétique. Il s’agit du paradigme de Janus : des mécanismes
basiques sont obligatoires pour le bon fonctionnement du système immun mais ils
doivent évoluer rapidement pour éviter d’être surpassés par les pathogènes. Même
si le système immunitaire évolue vite, il y a également une large conservation de
mécanismes de défenses. Par exemple, les molécules du CMH I sont conservées
chez tous les gnathostomes. Le séquençage de l’ADN a révélé une similitude inouïe
entre les stratégies de l’immunité innée dans divers phylums (Paul, 2013).
Les peptides antimicrobiens sont le moyen de défense le plus ancien puisqu’ils sont
aussi présents chez les plantes. Même s’ils diffèrent par des détails de structure, il
est évident que les peptides des plantes, des insectes et des mammifères découlent
du même système ancestral (Janeway et al., 2009).
Les TLR sont eux, le mode de reconnaissance le plus ancien. Chez les plantes, ils
sont aussi présents et sont d’ailleurs associés à la production des peptides
antimicrobiens. Par contre, chez les vertébrés ils ont évolué et ont acquis de
nouvelles fonctions. Chez l’oursin, 222 gènes différents de TLR ont été identifiés et 4
protéines adaptatrices similaires au MyD88 sont codées. Par contre il semblerait
qu’ils n’aient qu’un seul NF-κB comme chez les mammifères : le résultat final de la
signalisation reste inchangé. Chez les mammifères les TLR permettent de détecter
les bactéries Gram- et Gram+ mais chez la drosophile, seules les bactéries Gram+
sont détectées par les TLR. Deux autres récepteurs ont été identifiés pour la

135
reconnaissance des bactéries Gram- et ils impliquent la voie des TNF (Janeway et
al., 2009).
Le complément est également très ancien mais les différentes voies d’activation sont
apparues plus tard et ont permis de cibler spécifiquement des surfaces microbiennes
et son rôle le plus ancestral est probablement l’opsonisation. Il est présent chez les
invertébrés avec notamment des homologues du C3 et du facteur B. Avec
l’émergence des chordés, les principaux constituants sont présents. La voie
d’activation par les lectines s’est développée chez les invertébrés avec, en premier
l’activation par la ficoline (Janeway et al., 2009).
La richesse des espèces et la diversité des habitats des invertébrés suggèrent que
l’absence d’immunité acquise n’a pas entravé leur succès.
L’hémocyanine est présente dans l’hémolymphe des arthropodes et des mollusques
et dans les stades larvaires de certains insectes et elle représente la protéine
majoritaire de l’hémolymphe. Elle transporte l’oxygène aux tissus et participe aux
processus physiologiques et homéostasiques comme la mue, le transport
d’hormones, l’osmorégulation et le stockage de protéines. C’est également un
précurseur pour les peptides antifongiques et antibactériens. Elle possède des rôles
antiviraux non spécifiques (Coates & Nairn, 2014).
Les vers de terre possèdent des produits humoraux et des molécules
antimicrobiennes uniques. Leurs leucocytes flottent librement dans le cœlome, ils
sont appelés cœlomocytes et sont divisés en deux sous-types : les petits et les
grands cœlomocytes qui peuvent phagocyter et lyser les cellules comme les cellules
NK. Ils sécrètent des facteurs humoraux et participent activement à l’inflammation en
moins de 24h. La cavité cœlomique remplie de fluide cœlomique contient des
cellules circulantes (cœlomocytes et chloragocytes) (Cooper, 2006).
Les invertébrés sans mâchoires ou agnathes constituent un groupe phylogénique
apparu il y a 500 millions d’années, il regroupe les lamproies ou les myxines parmi
d’autres et ces animaux possèdent un système adaptatif comme celui des vertébrés
avec mâchoire. Et de la même façon, l’activation du système adaptatif des vertébrés
sans mâchoires nécessite l’activation préalable du système inné (Kasamatsu, 2013).
L’importance de l’influence du système adaptatif sur le système inné est mise en
évidence en comparant la diversité des composants innés dans les organismes qui
possèdent un système adaptatif avec ceux qui en sont dépourvus. Les espèces sans
système adaptatif possèdent bien plus de PRR : 10 TLR sont présents chez les
humains, 21 chez les lamproies, 222 chez les oursins et 9 chez la drosophile. Il
existe deux hypothèses qui expliquent cette différence :
La perte du gnathostome : la diversité des récepteurs ancestraux abondants a
été perdue au cours de l’évolution chez ceux qui ont développé une immunité
adaptative. Un besoin réduit d’une telle variété a diminué la pression sélective
pour maintenir et diversifier les PRR ;

136
Le gain du deuterostome : les récepteurs innés ancestraux ont subi une
diversification marquée dans les organismes sans système immunitaire
adaptatif (Ward & Rosenthal, 2014).
Les TLR sont très fortement conservés parmi les métazoaires. Ils seraient toujours
sous une sélection positive ce qui rend l’hypothèse n°1 moins plausible. L’hypothèse
n°2 n’exclue pas que les PRR des vertébrés subissent aussi une sélection positive.
Du point de vue de l’évolution, l’immunité ne nécessite pas une défense parfaite
contre tous les pathogènes : pour proliférer un organisme a juste besoin d’avoir des
défenses plus efficaces comparativement parlant. Le moustique ne possède qu’un
récepteur à cytokine, le poisson zèbre en possède 36. Dans la cascade engendrée
par les cytokines, les molécules ont connu la même expansion. La drosophile ne
possède qu’un membre de la famille des JAK et un seul STAT alors que l’homme a 4
membres JAK et 7 membres STAT. Cette expansion de cytokines et de molécules
de signalisation en conjonction avec le développement d’un système adaptatif
semble plus consistent avec l’hypothèse n°2. Les vertébrés, à durée de vie plus
longue, ont découverts d’autres stratégies pour faire face aux mutations des
microbes qui ont des temps de doublement plus courts. Comme les mutations des
microbes sont non prédictibles, la meilleure option pour l’hôte est de trouver le
moyen le plus flexible et d’étendre la spécificité de leur système immunitaire
(Sirisinha, 2013; Ward & Rosenthal, 2014) .
Ce qu’il faut retenir :
Tous les êtres vivants possèdent un système immunitaire inné avec ses
récepteurs et ses molécules de communication. Ce système est souvent plus
développé dans les organismes qui n’ont pas de système adaptatif mais leur
survie montre l’importance et l’efficacité du système inné. Le système
adaptatif semble donc apparaître comme un système immunitaire « de luxe »,
acquis par les êtres supérieurs à durée de vie plus longue et qui sont donc
soumis à l’attaque de nombreux microbes en constante évolution.

137
Après avoir étudié le système immunitaire inné dans sa globalité, nous allons voir
ses spécificités chez les ruminants, animaux d’élevage dans le monde entier. Dans
les pays développés, les systèmes d’élevage se sont intensifiés afin d’optimiser les
productions. Ceci nécessite une sélection des animaux qui doivent toujours produire
plus. Avec cette intensification de l’élevage, de nouvelles pathologies sont apparues,
notamment à cause du surpassement des métabolismes des animaux. Ces grandes
variations dans leur métabolisme au cours de la vie influent aussi sur leur système
immunitaire. Ceci peut expliquer en partie la sensibilité particulière de nos animaux
domestiques très producteurs par rapport aux animaux sauvages, plus rustiques.
Après avoir décrit les particularités de l’immunité innée chez les ruminants, nous
étudierons ses variations selon le stade physiologique de la vache, depuis la vie
fœtale jusqu’à son vêlage. Enfin, nous nous concentrerons sur l’immunité de la
mamelle, organe particulièrement sollicité chez la vache laitière haute productrice,
avec l’importance financière des mammites, l’une des principales pathologies de la
vache laitière.
I. Particularités de l’immunité innée chez les
ruminants
Dans cette partie, nous verrons successivement les particularités de l’immunité innée
chez les bovins, les petits ruminants puis chez les ruminants sauvages, afin
d’essayer de comprendre la différence de sensibilité aux pathogènes partagés entre
toutes ces espèces.
Chez les bovins A-
Considérations anatomiques 1)
Les poils autour des narines fournissent une barrière physique à l’inhalation de
grosses particules. Le bétail a un arbre trachéobronchique long, ce qui augmente le
taux d’espace mort donc diminue le taux d’oxygène frais délivré aux poumons et
augmente le risque d’hypoventilation alvéolaire. L’appareil respiratoire peut être

138
colonisé par une grande variété de pathogènes bactériens : la trachée et les
poumons sont colonisés par des bactéries des genres Bacillus, Streptococcus,
Streptomyces, Micrococcus, et Pseudomonas. Cependant les alvéoles les plus
éloignées de la trachée restent relativement stériles chez le bétail sain. Le surfactant
a une activité antimicrobienne. Comme chez les autres espèces, il est constitué
d’une couche périciliaire proche des cellules apicales et d’une couche muqueuse en
contact avec la lumière aérienne. Le surfactant contient de nombreuses molécules
comme la lactoferrine et le lysozyme ainsi que des peptides antimicrobiens comme
les défensines, ou les cathélicidines. Dans les alvéoles, les macrophages alvéolaires
engloutissent les molécules inhalées (Ackermann, Derscheid, & Roth, 2010).
Les cellules 2)
Le développement des composants cellulaires du système immunitaire inné chez les
ruminants n’a pas été bien décrit. Il comprend les NK, les cellules dendritiques, les
cellules γδ T, les cellules B, les macrophages et les granulocytes. A ce jour, nous ne
connaissons rien d’éventuelles ILCs chez les ruminants, autres que les NK (Toka &
Golde, 2013).
On note une très grande variabilité dans les gènes TLR chez les bovins, témoin d’un
polymorphisme marqué. Ce polymorphisme peut expliquer la différence de
résistance à de nombreuses maladies. Etonnamment, les espèces sauvages n’ont
pas plus de diversité dans les gènes TLR que les espèces domestiques (Novák,
2014).
a. Les cellules dendritiques
Les cellules dendritiques représentent de 0,1 à 0,7% des cellules mononucléaires du
sang chez les bovins. Les cellules dendritiques plasmacytoïdes produisent de l’IFN-
α. Il existe deux sous-types de cellules dendritiques chez les bovins :
les cellules dendritiques CD172+ CD13- ;
les cellules CD172- CD13+ qui produisent plus d’IL-12.
Seules certaines populations de cellules dendritiques sont capables de produire de
l’IFN-γ comme les cellules dendritiques plasmacytoïdes (CD3- CD14- CD21- CD11c-
NK- TCRγ- CD4+ CMH II+ CD45RB+ CD172a+ CD32+), qui représentent 0,1% des
lymphocytes totaux. Ces cellules produisent de l’IFN de type I après stimulation avec
des agonistes du TLR9, à l’exemple des complexes immuns formés avec le virus de
la fièvre aphteuse : le virus possède une protéase leader (Lpro) capable de rompre
l’intégrité du NF-κB et donc de suspendre la transcription des gènes qui en
dépendent. Les cellules dendritiques sécrètent aussi de l’IFN de type III avec une
activité antivirale : l’IFN-λ3 induit la surexpression des gènes régulés par l’IFN
(González-Cano, Arsic, Popowych, & Griebel, 2014; Toka & Golde, 2013).

139
b. Les NK
Les NK bovins représentent entre 2 et 10% des lymphocytes et sont présents dans le
sang périphérique, la rate, le poumon, le foie, les nœuds lymphatiques et la moelle
osseuse. Ils expriment des récepteurs de cytotoxicité naturelle comme le CD335,
produisent de l’IFN-γ, lysent des cibles et ont un phénotype défini par NKp46+
CD335+ CD2+/- CD8+/- et CD3-. Ils expriment aussi une protéine D du groupe 2 des
NK, le NKG2D, un récepteur activateur des NK et des cellules T CD8+. Les NK sont
notamment activés :
en réponse au stress, comme durant le transport de bétail,
lors d’infections bactériennes, comme dans l’élimination des macrophages
infectés par Mycobacterium bovis. Ils interagissent avec les macrophages
infectés et augmentent leur capacité à produire de l’IL-12 et de l’oxyde nitrique
(NO). Ils induisent ainsi l’apoptose des cellules infectées et limitent donc la
réplication de la bactérie intracellulaire,
lors d’infections parasitaires. Par exemple, durant une infection par le
protozoaire Neospora caninum, les NK reconnaissent le parasite
intracellulaire, produisent de l’IFN-γ et détruisent les fibroblastes infectés. Lors
d’infection par Babesia bovis, un autre parasite protozoaire, les NK sont la
principale source précoce d’IFN-γ (Boysen & Storset, 2009; Toka & Golde,
2013).
Comme les bovins ne possèdent pas de CD1d, ils ne possèdent probablement pas
d’homologue des NKT (Boysen & Storset, 2009).
Il a longtemps été pensé que les gènes KIRS (Killer-cell Immunoglobulin-like
Receptors, codant pour des récepteurs régulant l’activité tueuse des NK via des
interactions avec les molécules de CMH I), étaient présents uniquement chez les
primates mais cette famille a été trouvée chez les bovins (Boysen & Storset, 2009).
c. Les cellules γδ T
Les cellules γδ T sont critiques pour la surveillance immune. Ce sont des cellules
résidentes dans de nombreux organes et tissus. Il est bien connu que ces γδ T
participent à l’immunité cellulaire et la protection contre les pathogènes importants
incluant les mycobactéries qui nécessitent une réponse organisée en granulomes. Le
paradigme émergent est qu’un PRR peut envoyer un signal activateur quand il est
engagé avec son ligand sur des cellules innées mais agir comme corécepteur sur
des cellules T. Cette différence serait due aux différences de molécules de
signalisation intracellulaire avec lesquelles ils sont associés (Rogers et al., 2005).
Alors que les γδ T constituent une minorité de lymphocytes dans la plupart des
espèces, elles constituent entre 20 et 50% des lymphocytes circulants chez les
jeunes ruminants et entre 5 à 30% chez l’adulte (contre seulement 5% chez l’homme
adulte). Le nombre de cellules γδ T augmente significativement dans la région des

140
plaques de Peyer durant le développement prénatal et diminue après la naissance.
Par contre, l’inverse est observé pour les villosités intestinales où quelques cellules
γδ T sont observées avant la naissance alors que leur nombre augmente après la
naissance (Baldwin et al., 2014; Boysen & Storset, 2009; Toka & Golde, 2013).
Chez les ruminants, il existe deux populations de γδ T basées sur des formes
moléculaires différentes du récepteur scavenger WC (Workshop cluster 1) Le WC1.1
est une glycoprotéine transmembranaire, membre de la famille des récepteurs
scavengers riches en cystéine, uniquement exprimé sur les cellules γδ T. Les γδ T
du sang expriment le WC contrairement à celles de la rate. Cette molécule est
importante pour la transduction du signal. Elle possède des sites de phosphorylation
de la thréonine ou de la sérine, ce qui lui permet le recrutement de différentes
kinases et phosphatases. Les deux populations de γδ T rencontrées chez les bovins
sont donc définies par :
WC1+ CD3+ CD5+ CD2- CD6- CD8- ; il s’agit de la sous-population
inflammatoire caractérisée par une production importante d’IFN-γ. Ce sont
aussi d’importants médiateurs de l’immunité cellulaire (type 1). Il existe 3
isoformes de WC1 : WC1.1, WC1.2 et WC1.3. Les deux premières sont
exprimées sur la plupart des cellules γδ T alors que la forme WC1.3 est
exprimée par une petite population des cellules WC1+. Parmi les WC1+ :
o les cellules γδ T WC1.1 représentent 10 à 20% du total. Elles
produisent de l’IFN-γ après stimulation par des cytokines grâce à la
présence du T-bet dans ces cellules. Ce sont des coordinateurs des
réponses inflammatoires chez les jeunes veaux. Elles possèdent un
rôle suppresseur des réponses αβ T en plus de leur rôle cytotoxique.
Enfin les cellules γδ T WC1+ peuvent prélever des antigènes et
exprimer certaines molécules de surface associées aux cellules
présentatrices d’antigènes comme le CMH II et ainsi induire la
prolifération des cellules T αβ CD4+,
o les WC1.2 ne produisent pas d’IFN-γ mais elles ont également un rôle
suppresseur des réponses αβ T et sont aussi cytotoxiques,
WC1- CD3+ CD5+ CD2+ CD6+ CD8+. Ce sous-type exprime la perforine chez le
bison américain (Born et al., 2006; Guzman, Price, Poulsom, & Hope, 2011;
Rogers et al., 2005; Toka & Golde, 2013).
Le répertoire de la chaîne δ du TCR chez les bovins est très divers : le TCR inclut 56
gènes V pour « variable » et 5 gènes D pour « diversity », 3 J pour « jonction » et 1 C
pour « constant ». Pour la chaîne γ, il y a 11 gènes V, 9 J et 6 gènes C. Le NKG2D,
un récepteur immunorégulateur des NK et des αβ T CD8+ est également présent sur
les γδ T : il interagit avec des molécules de stress et cette interaction active les
cellules γδ T WC1+ sans aucun autre stimulus, marquant leur appartenance à
l’immunité innée (Guzman et al., 2011; Rogers et al., 2005)

141
Ces cellules constituent une source précoce d’IFN-γ lors d’infection mycobactérienne
et lors d’autres infections intracellulaires (Boysen & Storset, 2009).
d. Les granulocytes
Les granules des éosinophiles des ruminants sont petits (voir Figure 30, page 142).
Les PNE sont impliqués dans la défense contre les parasites et lors d’allergie. C’est
pourquoi, lors d’infection par des endoparasites, la formule sanguine révèle une
éosinophilie même si elle est parfois absente. L’éosinopénie, quant à elle, peut être
le signe d’un stress, de toxine, de fibrose…(Harvey, 2001).
Contrairement aux autres espèces où les neutrophiles sont les leucocytes majeurs
du sang, chez les bovins, ils ne représentent que 25% des leucocytes sanguins. Ils
contiennent trois types de granulations. Les granules tertiaires sont plus nombreux,
larges, denses, et sans peroxydase (voir Figure 30, page 142). Ils sont donc très
différents des granules humains. Ils contiennent de la lactoferrine, des bacténécines
(de la famille des cathélicidines, elles perméabilisent la paroi des Escherichia coli),
des α-défensines et des β-défensines (actives contre les bactéries Gram+, Gram-,
les bactéries anaérobies, les champignons et les virus) mais il n’y a pas de lysozyme
dans les PNN bovins ou très peu. Les médiateurs inflammatoires PAF et TNF
induisent une augmentation de taille des PNN bovins, ce qui ralentit la diapédèse et
régule les mouvements des PNN vers le site inflammatoire. La demi-vie des PNN
dans la circulation sanguine est de 9 heures puis ils subissent spontanément
l’apoptose. Le foie, le poumon et la rate sont des sites importants de destruction des
PNN. En début de lactation, le taux d’apoptose est plus élevé, l’activité bactéricide et
de phagocytose des PNN sont diminuées. Durant l’infection, les cytokines pro-
inflammatoires et les produits bactériens peuvent retarder le moment de l’apoptose.
Les cytokines qui prolongent les PNN sont l’IL-1β, l’IL-2, l’IL-6, l’IL-8, l’IL-15, l’IFN-γ,
le G-CSF et le GM-CSF. Le LPS, molécule hautement pro-inflammatoire, inhibe
aussi l’apoptose des PNN. Une autre cytokine pro-inflammatoire relâchée lors de
mammite est le TNF-α, mais elle induit l’apoptose des PNN. Des variations existent
entre les vaches quant à l’efficacité de l’explosion respiratoire (permettant la
génération des réactifs oxygénés comme évoqué page 78) des PNN, ce qui peut
expliquer la différence de sensibilité aux infections intramammaires entre les
animaux. L’explosion respiratoire des bovins est influencée par les agents
thérapeutiques utilisés lors de traitement. Parmi les antibiotiques utilisés en
médecine vétérinaire, l’enrofloxacine augmente cette activité alors que la néomycine,
la lincomycine, la dihydrostreptomycine, l’oxytétracycline, la danofloxacine, la
pénicilline, le ceftiofur, la spiramycine, l’érythromycine et le chloramphénicol la
réduisent. Les glucocorticoïdes ne présentent aucun effet sur l’explosion respiratoire
des PNN. La vitamine E et le sélénium sont des antioxydants critiques et
l’alimentation des vaches est souvent déficiente sans complémentation. Une
neutrophilie est observable lors d’inflammation modérée ou après une inflammation
aiguë mais aussi lors de néoplasie, de stress ou lors de BLAD (pour Bovine
Leukocyte Adhesion Deficiency) caractérisée par l’absence d’expression d’intégrines

142
sur les leucocytes. Les animaux atteints ont alors des infections chroniques ou
récurrentes et des retards de cicatrisation. A l’inverse, une neutropénie est
fréquente lors d’inflammation aiguë mais aussi lors d’infection par le BVD (Bovine
Virale Diarrhea), la border disease ou le virus de la Blue Tongue. (Ackermann et al.,
2010; Paape, Bannerman, Zhao, & Lee, 2003).
Figure 30 : Granulocytes chez les bovins (Douglas & Jane Wardrop, 2010). PNE (à gauche), PNN (au centre) et PNB (à droite) à la coloration de Wright au
grossissement 1000.
e. Les monocytes
Les monocytes des ruminants domestiques mesurent entre 13 et 19 μm de diamètre.
Les granules sont petits et les vacuoles fréquentes et plus irrégulières (Harvey,
2001).
Composants humoraux et chimiques 3)
Les bovins comptent à ce jour 38 peptides antimicrobiens dont différentes
défensines et cathélicidines comme :
le « Bovine Myeloid Antimicrobial Peptide » (ou BMAP) ;
les bacténécines ;
les indolicidines ;
la « Bovine Oligosaccharide-Binding Protein » (ou BOBP), une protéine de
reconnaissance du peptidoglycane trouvée dans les PNN et PNE bovins : elle
pourrait participer aux défenses antiparasitaires ;
la lactoferrine et la lactoferricine, présentes dans le lait, décrites page 167 ;
les β-défensines possèdent une activité contre Escherichia coli, Klebsiella
pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus et Candida spp.
o le « Tracheal Antimicrobien Peptide », ou TAP, est une β-défensine de la
trachée. Il est présent tout au long des voies respiratoires et est induit par
des médiateurs pro-inflammatoires comme le TNF-α, l’IL-1β et le LTA. Son

143
spectre est large puisqu’il agit contre les bactéries Gram+ et Gram- ainsi
que contre des agents fongiques,
o le « Lingual Antimicrobial Peptide » ou LAP a été initialement découvert
dans la langue, d’où son appellation. En fait, il est plus répandu et se
trouve dans l’épithélium tout le long du tube digestif, dans le système
respiratoire, la glande mammaire et la cornée. Son expression est induite
lors d’infection à Mannheimia haemolytica et lors d’infection chronique à
Mycobacterium paratuberculosis,
o L’ « Enteric β-défensine » ou EBD, trouvé dans les intestins. Son niveau
d’expression est fortement augmenté lors de cryptosporidiose chez les
veaux ;
les cathélicidines sont bactéricides contre des bactéries comme Escherichia coli,
Salmonella typhimurium et Klebsiella pneumoniae et bactériostatiques contre
d’autres bactéries dont Enterobacter cloacae ;
le bBD-1, bovine β-défensine -1 de la glande mammaire ;
les granules denses des PNN contiennent des β-défensines (Bovine Neutrophile
β Defensin : BNBD 1 à 13) (Linde et al., 2007).
Bien que les peptides cationiques soient largement répandus dans le monde vivant,
des plantes aux insectes, les peptides anioniques ne sont présents que dans les
sécrétions pulmonaires des ruminants et des humains : ils possèdent des régions
riches en aspartate et nécessitent le zinc comme cofacteur. Ces peptides sont
exprimés constitutivement et sont actifs contre les bactéries Gram+ ou Gram-
(Ackermann et al., 2010; Meyerholz & Ackermann, 2005).
Les lectines bovines comme la conglutinine, et les collectines 43 et 46 fixent les
rotavirus et les virus influenza. La conglutinine est une lectine oligomérique présente
dans le sérum, le colostrum et le lait des bovins. Elle est capable de fixer du N-
acétylglucosamine terminal non réduit, des mannoses et des résidus de fucose. Les
collectines incluent aussi le MBP (Mannan Binding Protein), et les protéines du
surfactant A (SP-A) et D (SP-D). Les concentrations plasmatiques de certaines
lectines comme la conglutinine varient considérablement entre les individus d’un
groupe. Une concentration élevée de conglutinine plasmatique est héritable et
profitable pour les veaux (Lillie et al., 2005).
Concernant les protéines de la phase aiguë chez les bovins :
- la production de LBP est plus importante dans la mamelle, les glandes
salivaires, les épithéliums des pré-estomacs que dans le foie,
- l’haptoglobine peut être utilisée pour le diagnostic et le pronostic des entérites,
péritonites et endocardites ou lors de syndrome de la vache grasse,

144
- l’augmentation de SAA est plus importante lors d’inflammation aiguë que lors
d’inflammation chronique,
- l’haptoglobine, le SAA et le LBP sont des marqueurs sensibles des maladies
respiratoires. Des bovins avec des ulcères de la sole ou des abcès de la ligne
blanche ont des concentrations de SAA significativement plus élevées sans
que l’haptoglobine ne soit élevée,
- l’AGP bovine (α1-acid glycoprotein) est une glycoprotéine de la phase aiguë
des bovins de 20,5 kDa. Il s’agit d’une lipocaline : une protéine de transport de
petites molécules hydrophobes. Elle a ensuite été classée dans les
immunocalines, une sous-famille qui module la réaction inflammatoire. Elle
peut fixer plus de 300 molécules dans le plasma et sa concentration
augmente tellement lors de la phase aiguë qu’elle peut devenir la protéine la
plus abondante du sérum. C’est une puissante protéine anti-inflammatoire
puisqu’elle module tous les stades de la séquence d’attaque des PNN comme
le chimiotactisme, la production des réactifs oxygénés et leur dégranulation. A
forte concentration, elle inhibe la migration des monocytes et l’activité tueuse
des granulocytes en diminuant la libération des réactifs oxygénés. Elle stimule
la production d’IL-8 et inhibe l’exocytose des granules secondaires. Or elle est
stockée dans les granules secondaires : elle a donc une action paracrine et
endocrine pour tempérer l’attitude agressive des granulocytes dans les tissus
environnants. La concentration atteinte lors de la réaction de la phase aigüe
inhibe l’apoptose des monocytes, les cellules enlevant les cellules
inflammatoires, donc en allongeant leur durée de vie, l’AGP augmente l’effet
anti-inflammatoire des monocytes (Ceciliani et al., 2012).
Le BST2 est un facteur de restriction cellulaire qui bloque le relargage de virus
(comme des retrovirus, filovirus, arenavirus, herpesvirus, rhabdovirus). Ce facteur est
fortement induit par l’interféron de type I dans des types cellulaires variés. Le
génome bovin et ovin possède 2 gènes bBST2A et bBST2B (Takeda et al., 2012).
Chez les bovins, le rein était le seul site connu pour la production de 1,25-
dihydroxyvitamine D3 ou 1,25(OH)2D3 (chez les humains, cette conversion est aussi
possible par les monocytes). La principale fonction de la 1,25(OH)2D3 est de
maintenir l’homéostasie du calcium mais cette hormone régule aussi la réponse
immune en régulant plus de 1000 gènes. La 1,25(OH)2D3 régule la réponse immune
en augmentant la production d’oxyde nitrique, et en diminuant la production d’IFN-γ
dans les cellules mononucléaires du sang. Les récentes recherches ont montré que,
comme chez l’homme, la production d’1,25(OH)2D3 est possible dans les monocytes
où elle augmente l’expression du gène iNOS dans les monocytes activés et aussi
celle de RANTES, chimiokine pour les cellules Th et les monocytes. Cependant,
l’expression de la 1,25(OH)2D3 n’est pas modifiée dans le sérum lors d’une infection
(Nelson, Reinhardt, Thacker, Beitz, & Lippolis, 2010).

145
Les marqueurs de l’inflammation chez les ruminants 4)
L’interprétation de la réponse des cellules blanches sanguines chez les bovins est
essentielle dans le diagnostic, et le pronostic de nombreuses maladies. Les
ruminants ont le ratio neutrophiles/ lymphocytes le plus faible parmi les espèces
domestiques : approximativement 0,5 chez les bovins adultes. Ce ratio est fortement
modifié en cas d’inflammation. Le recrutement des cellules souches est plus lent que
chez les autres espèces, d’où la formule leucocytaire unique des ruminants lors
d’inflammation aiguë : les PNN deviennent majoritaires par rapport aux lymphocytes :
le rapport est alors supérieur à 1. Lors d’inflammation, dans les premières 24-48
heures, une forte neutropénie est présente, par recrutement des PNN sanguins vers
le site de l’inflammation et une mobilisation lente des cellules souches. Dès 24h, des
neutrophiles immatures commencent à apparaitre. En 3 à 5 jours, les ruminants
peuvent ainsi retrouver un nombre normal de neutrophiles sanguins. Par contre, en
cas d’inflammation chronique, le nombre de neutrophiles peut être normal ou
augmenté. Dans ce cas, le fibrinogène est un meilleur indicateur de l’inflammation.
Chez les bovins et ovins, ce fibrinogène peut monter jusqu’à 1,5 g/dL voire plus. Ce
phénomène est moins marqué chez la chèvre (Douglas & Jane Wardrop, 2010).
Les protéines de la phase aiguë peuvent aussi servir d’indicateurs de santé. Une
mammite entraîne une hausse de la concentration en APP dans le sérum et le lait
alors qu’une inflammation extra-mammaire n’augmente que la concentration sérique.
La protéine du surfactant A (SP-A) est produite en quantité beaucoup plus
importante dans le poumon fœtal proche du terme et montre donc le niveau de
maturation du poumon à la naissance. De plus, la production de SP-A augmente lors
de pneumonie virale et elle entre alors dans les vaisseaux lymphatiques pulmonaires
puis est conduit dans la circulation sanguine systémique. D’autres protéines de la
phase aiguë sont des marqueurs de pneumonie comme le SAA, l’haptoglobine,…
produites par le foie en réponse à l’IL-1 et au TNF-α. Dans l’appareil reproducteur, le
SAA est peu détectable dans des conditions physiologiques mais augmente
fortement en postpartum et reflète la sévérité de l’inflammation. Il en va de même
pour l’haptoglobine (Ackermann et al., 2010; Ceciliani et al., 2012).
Les problèmes liés à la sélection génétique 5)
La sélection génétique a eu pour but ces vingt dernières années de maximiser la
production de lait. Mais cette augmentation de production a une corrélation négative
sur l’immunité mammaire : la production de lait en quantité très largement supérieure
par rapport aux besoins de la progéniture provoque un stress physiologique et
augmente la dilution des défenses immunes solubles et cellulaires (Rainard & Riollet,
2006).
Pour sélectionner des vaches résistantes aux mammites, il convient de se focaliser
sur les gènes contrôlant les protéines du lait et parmi ces composants, il faut préférer

146
les peptides et protéines aux glyconjugates et lipides qui sont synthétisés par une
cascade enzymatique qui est plus difficilement manipulable. Les peptides de défense
sont des composants naturels de l’hôte et ils possèdent généralement une activité
antimicrobienne. Cependant, il ne faut pas que leur activité antimicrobienne soit trop
forte parce que sinon, ils pourraient altérer le processus de formation du fromage.
Les technologies transgéniques ont permis de créer des vaches sécrétant de la
lactoferrine humaine dans leur lait. La difficulté principale de ce procédé est
l’acceptation par le public (Rainard & Riollet, 2006).
Exemples de maladies dans lesquelles l’action du 6)
système inné est importante
Nous prenons maintenant quelques exemples de maladies bovines où le système
immunitaire inné joue un rôle important. Cette liste est évidemment non exhaustive et
le système inné intervient dans de nombreuses autres maladies avec une efficacité
plus ou moins marquée selon la pathogénie.
La maladie de la fièvre aphteuse est caractérisée par une forte fièvre et la formation
de vésicules sur la langue, les lèvres, les trayons et la mamelle, ainsi que sur les
pieds. Ce virus qui infecte les cellules épithéliales, est très contagieux et se réplique
très rapidement, à l’origine d’un temps d’incubation court et une importante quantité
de virus excrétée dans les aérosols. La protection est corrélée avec l’apparition dans
le sérum de forts niveaux d’anticorps neutralisants, 3-4 jours après l’infection. Le
virus est internalisé par les macrophages, et s’il n’est pas opsonisé par des
anticorps, cette internalisation est beaucoup plus lente et est probablement réalisée
par macropinocytose. Toutes les cellules dendritiques sont sensibles au virus même
s’il n’induit pas d’effet cytopathique : l’infection est abortive mais non inapparente.
Les cellules NK présentent une activité cytotoxique envers les cellules infectées par
le FMDV (Foot and Mouth Disease virus, ou virus de la fièvre aphteuse) grâce à la
perte rapide de CMH I que le virus engendre dans les cellules infectées. Les cellules
épithéliales produisent de l’IL-1α, du TNF-α et de l’IFN-α/-β/-γ de façon
proportionnelle à la quantité de virus. L’élément clé de la réponse immune contre le
FMDV serait l’IFN de type I. Une souche de virus est capable d’éteindre la synthèse
d’IFN de type I. La baisse de synthèse des protéines de l’hôte peut expliquer le
manque de synthèse d’IFN par les macrophages mais pas pour les cellules
dendritiques. Une autre source d’IFN est représentée par les cellules γδ T : elles
répondent aux antigènes de la fièvre aphteuse et montrent transitoirement un
phénotype activé par une augmentation de l’expression du CD25, une réduction de
celle du CD62L et du CD45RO, et une production accrue d’IFN-γ. Les γδ T WC1+
acquièrent la capacité des NK à tuer une cible, notamment en augmentant le
stockage de perforine et l’expression du CD335. Ces cellules peuvent servir de
cellule présentatrice d’antigènes en augmentant l’expression du CMH II et du CD13
(Summerfield, Guzylack-Piriou, Harwood, & McCullough, 2009; Toka & Golde, 2013).

147
Le Bovine Viral Diarrhea virus (BVDV) est un Pestivirus de la famille des flaviviridae.
Il s’agit d’un véritable fléau en élevage car la maladie est difficilement détectable
mais est responsable de mortalités embryonnaires et d’avortements. Le virus est
divisé en deux biotypes : la souche non cytopathogène (qui ne cause pas de mort
cellulaire) et la souche cytopathogène qui induit l’apoptose. Chez les adultes,
l’infection par une souche cytopathogène n’entraîne qu’une diarrhée passagère
parfois même inapparente. Par contre, si l’animal est gravide, la souche non
cytopathogène peut infecter le fœtus de façon permanente, apparemment en
inhibant la réponse interféron de type I par le fœtus en développement (Yamane,
Kato, Tohya, & Akashi, 2008). Il en résulte un Individu Infecté Permanent (IPI) qui
considère le virus comme du soi et ne monte donc pas de réponse immunitaire
contre lui (séronégatif), malgré une excrétion virale intense (viropositif). L’IPI est
donc une très forte source de virus dès la naissance, avant même qu’il ne présente
un retard de croissance. L’infection d’un IPI après la naissance par une souche
cytopathogène reliée à la souche non cytopathogène qui l’a contaminé in utero
conduit au développement de la maladie des muqueuses, indéniablement mortelle
avant l’âge de 3-4 ans. La défense antivirale initiale est innée et comprend la
synthèse d’IFN de type I comme l’IFN-α et l’IFN-β mais l’IFN n’est probablement pas
capable de franchir le placenta. Une autre source potentielle d’IFN serait le placenta
lui-même, au moins durant la fin de gestation. L’ARN du virus de la BVD est
détectable dans le sang fœtal 14 jours après l’inoculation du virus à la mère. A ce
moment-là, le virus est presque entièrement évacué de la circulation maternelle. Les
veaux IPI ont un niveau élevé d’ARNm des gènes de l’immunité innée (TNF-α, PKR,
OAS) ce qui indique une activation de la réponse IFN de type I (Yamane et al.,
2008). L’infection persistante par une souche non cytopathogène induit une réponse
de type I. En fait il semblerait que les cellules produisant de l’IFN dans les IPI soient
les seules cellules non infectées par le virus et sont donc stimulées par des voies
extracellulaires. Il semblerait que l’ARNdb, intermédiaire typiquement viral, stimule
l’apoptose de la cellule. Les IPI peuvent alors souffrir d’infections secondaires
causées par des pathogènes respiratoires ou entériques (Pépin & Laaberki, 2014;
Smirnova et al., 2012; Yamane et al., 2008).
Les cellules peuvent héberger un pathogène et ainsi le protéger du système
immunitaire. C’est le cas de l’arbovirus de la Blue Tongue chez les ruminants,
caractérisé par une nécrose, des hémorragies et un œdème secondaires à des
lésions vasculaires. Après la piqure d’un moucheron du genre Culicoïdes, vecteur du
virus, le virus est rapidement transporté par les cellules dendritiques vers les nœuds
lymphatiques régionaux. Les cellules dendritiques conventionnelles sont recrutées
en grand nombre vers la peau, ce qui facilite la réplication du virus au site initial
d’infection. La réplication a lieu dans le nœud lymphatique dans les cellules
dendritiques, les macrophages et les cellules endothéliales. L’infection des
macrophages mène à leur activation et à la synthèse de TNF-α, d’IL-1β et d’IL-8 et
induit la synthétase d’oxyde nitrique. Ensuite, le virus dissémine à travers de
nombreux organes où des réplications secondaires ont lieu dans les cellules

148
phagocytaires, les cellules endothéliales, les γδ T. Le virus induit la production d’IFN
de type I dans les cellules dendritiques plasmacytoïdes via une signalisation MyD88
dépendante (Maclachlan, Henderson, Schwartz-Cornil, & Zientara, 2013).
Lors de tuberculose bovine, dans les tissus infectés par Mycobacterium bovis, les
γδ T produisent des cytokines comme l’IL-2, l’IL-10, l’IL-12, l’IL-15 et l’IFN-γ mais pas
de chimiokines. Les γδ T WC1+ contribuent indirectement au chimiotactisme des
cellules T conventionnelles et non directement. L’interaction entre les γδ T WC1+ et
les cellules dendritiques leur confère un rôle de pont entre les deux immunités. Cette
interaction est contact-dépendante et provoque la libération d’IL-12 qui à son tour
augmente la sécrétion d’IFN-γ et la surexpression des molécules de surface des γδ
T. Lors de vaccination intranasale par le BCG, les γδ T WC1+ sont recrutées dans les
nœuds lymphatiques respiratoires et les tissus pulmonaires et un dialogue entre les
cellules dendritiques et les γδ T a alors lieu (Guzman et al., 2011).
L’infection par le virus de Schmallenberg, détecté pour la première fois en
novembre 2011, se caractérise chez les bovins adultes par une chute de lait, de la
fièvre et de la diarrhée. Il s’agit d’un orthobunyavirus émergeant, associé à des
avortements et malformations chez les moutons et les bovins. Cependant, chez les
animaux gravides, le passage de la barrière placentaire par le virus entraîne des
malformations fœtales du système nerveux et du système musculo-squelettique chez
les agneaux et les veaux entrainant de l’arthrogrypose-hydranencéphalie. Ce virus
se réplique dans les neurones cérébraux et spinaux. Les dommages musculaires
(hypoplasie) sont dus aux lésions du système nerveux puisque le virus infecte les
neurones de la matière grise de la moelle épinière. La virémie est courte (inférieure à
10 jours). La période critique se situe entre 30 et 150 jours de gestation chez la
vache. Lors de sa transcription, le virus inhibe la production d’IFN et le virus est donc
capable de moduler au moins indirectement les réponses immunes innées de l’hôte
(Pépin & Laaberki, 2014; Varela et al., 2013).
Ce qu’il faut retenir :
Le système immunitaire inné des bovins est sensiblement identique à celui de
l’homme. La numération formule sanguine, les dosages des protéines de la
phase aiguë et du fibrinogène sont tous des aides diagnostiques pour le
vétérinaire avec souvent une valeur pronostique très utile pour le choix de la
thérapeutique.

149
Les petits ruminants B-
Le mouton a, comme les bovins, un ratio neutrophiles/lymphocytes de 0,5. Mais chez
la chèvre adulte, ce ratio est de 1 ; chez la jeune chèvre de moins de 3 ans, ce ratio
peut être similaire aux bovins et ovins (Douglas & Jane Wardrop, 2010).
Chez les petits ruminants, l’haptoglobine est une protéine de la phase aiguë majeure,
dont la concentration peut être multipliée par 80 lors d’inflammation. L’AGP montre
une réponse modérée (concentration doublée) mais prolongée dans le temps : elle
serait plus le signe d’une inflammation chronique.
Chez le mouton, il existe deux β défensines, le SBD1 et SBD2 (Sheep Beta
Defensine). Le SBD1 est exprimé de façon constitutive dans les tissus pulmonaires
alors que le SBD2 n’est produit que de façon sporadique dans le tractus intestinal.
Les peptides anioniques sont augmentés par exemple lors d’infection chronique par
Mannheimia haemolytica et ils sont produits par l’épithélium réparé. Le SMAP
(Sheep Myeloid Antimicrobial Peptide) est une cathélicidine avec une activité
antimicrobienne à large spectre contre les Gram+ et Gram- (Ceciliani et al., 2012;
Linde et al., 2007; Meyerholz & Ackermann, 2005).
Chez les ruminants sauvages C-
Les données sur les animaux sauvages sont de plus en plus nombreuses ; il est
important de noter que ces données peuvent être l’objet de biais en raison des
conditions liées à la capture ou à la prise d’échantillonnage à cause du stress induit.
Nous allons comparer quelques espèces de ruminants sauvages.
Chez les ruminants sauvages, comme chez les chameaux, le ratio
neutrophiles/lymphocytes est plus élevé que chez les ruminants domestiques
puisqu’il est aux alentours de 1,5. Chez les chameaux, l’inflammation est
caractérisée par une neutrophilie, accompagnée ou non d’hyperfibrogénémie. Lors
d’inflammation aiguë, comme chez leurs homologues domestiques, les chameaux
présentent une neutropénie transitoire, mais celle-ci est plus courte : elle ne dure que
12 à 24 heures (Douglas & Jane Wardrop, 2010).
Chez les cervidés, les lymphocytes et les PNN sont les leucocytes les plus
nombreux et le ratio entre les deux populations est proche ou légèrement inférieur à
1 même si d’autres études ont pu montrer que les PNN prédominaient. Ceci illustre
la difficulté à obtenir des valeurs standards pour les ruminants sauvages (Douglas &
Jane Wardrop, 2010).
Chez le renne, le ratio neutrophiles / lymphocytes est proche de 0,5. La particularité
de cette espèce réside dans le fait qu’il existe deux populations de basophiles, une
semblable à celle décrite chez les autres espèces et l’autre appelée « basophiles

150
gris » car ils contiennent des granules intracytoplasmiques et périnucléaires qui
déforment le noyau (Douglas & Jane Wardrop, 2010).
Parmi les bovidés, chez le buffle, les monocytes sont plus larges que les
lymphocytes, contenant souvent des vacuoles. Le bison présente les mêmes
morphologies cellulaires et les mêmes formules que les ruminants domestiques
(Douglas & Jane Wardrop, 2010).
Chez l’ibex alpine (ou bouquetin), le SAA et AGP sont les principales protéines de la
phase aiguë et pourraient être utilisées pour identifier les animaux infectés de façon
subclinique, notamment pour éviter de les relâcher dans des populations saines
(Ceciliani et al., 2012).
Les températures froides et le manque de nourriture ont un impact négatif sur le
fonctionnement du système immunitaire des animaux sauvages. L’immunité est
diminuée durant les réponses de stress pour rediriger les ressources vers les
activités plus essentielles à la survie immédiate. Non seulement l’activité immunitaire
est réduite mais les cellules immunes et les tissus sont activement catabolisés pour
fournir des protéines et du glucose. Le problème est que le gain d’énergie devrait
être immédiat, or la répression du système immunitaire ne peut pas être rapide
puisqu’il s’agit d’un réseau de cellules et de tissus diffus. De plus l’élimination des
leucocytes se fait par apoptose, nécessitant une transcription de gènes donc du
temps et de l’énergie. Ainsi, sur de longues périodes, la suppression du système
immunitaire fournit de l’énergie mais à court terme la suppression immune
consomme plus d’énergie qu’elle n’en économise. Les hormones de stress
exacerbent l’inflammation puis l’activité des cellules T notamment Th1 et finalement
l’activité des cellules B. Une stimulation persistante par ces hormones supprime les
trois systèmes. Les organes lymphoïdes primaires et secondaires sont innervés par
le système sympathique et son activation induit le relargage de substances pro-
inflammatoires comme le CRH, la substance P, le CGRP. Ces molécules provoquent
l’expression de molécules d’adhésion (CXCL-8 et CCL-2), spécialement par les
macrophages. Les glucocorticoïdes augmentent la présentation antigénique et la
phagocytose par les macrophages.
En réponse à de telles conditions difficiles, les vertébrés activent l’axe hypothalamo-
pituitaire-adrénal et les sécrétions de glucocorticoïdes augmentent. Cependant, une
exposition à long-terme à des concentrations élevées de glucocorticoïdes a des
effets négatifs comme la suppression de l’immunité. La maturation, la différenciation
et la prolifération de toutes les cellules immunes sont observées ainsi que la
stimulation de l’apoptose des lymphocytes B et T, la suppression de la transcription
de l’IL-1, de sa translation et de sa sécrétion. Sont également présentes, une
réduction du chimiotactisme des lymphocytes, des monocytes et des granulocytes,
ainsi qu’une réduction du nombre de lymphocytes circulants et du nombre des
éosinophiles, basophiles et macrophages
Le stress aigu est immunoactivateur en augmentant la réponse inflammatoire pour
rapidement contenir le pathogène. Cependant, cette activation n’est pas présente
chez tous les animaux puisque chez les oiseaux, le stress aigu entraîne une

151
immunosuppression plutôt qu’une immunostimulation. Cette immunosuppression
touche particulièrement le système inné.
Le stress chronique diminue le nombre de leucocytes. Lors d’un stress aigu, le
nombre de leucocytes dans le sang diminue également mais cette diminution est due
à une redistribution : les leucocytes quittent la circulation et infiltrent le tissu de la
zone infectée (Chin, Quinn, & Burness, 2013; Martin, 2009).
Après cette brève comparaison des systèmes immunitaires innés des ruminants,
nous reprenons l’exemple du bovin. Nous allons à présent nous intéresser à la
variation de cette immunité innée tout le long de la vie du bovin.
II. Variation de cette immunité selon le stade
physiologique
Le système immunitaire évolue au cours de la vie de l’animal. Le système inné se
met en place dès la vie fœtale. A la naissance, la prise de colostrum est essentielle
afin d’assurer un apport en immunoglobulines suffisant, mais ce colostrum a aussi un
rôle local inné. Nous verrons ensuite l’importance de la nutrition, pilier de la santé de
l’élevage intensif puisqu’il devient difficile de fournir suffisamment d’énergie pour
couvrir les besoins toujours croissants des animaux fortement producteurs. Enfin,
nous verrons les changements qui affectent le système inné pendant la gestation et
à la mise bas et l’influence du stress.
Mise en place de l’immunité innée pendant la vie A-
fœtale
Pendant la vie fœtale, le foie est le principal organe d’hématopoïèse et il augmente
d’ailleurs considérablement de taille au fur et à mesure que le nombre de cellules
nucléées augmente. Le premier type cellulaire de l’immunité innée à apparaitre dans
la circulation fœtale est le couple monocyte/macrophage. La majorité de ces cellules
sont CMH II- mais une faible proportion est CMH II+. Seuls les macrophages du
cordon ombilical sont complètement fonctionnels à la naissance. Leur chimiotactisme
est réduit mais leur capacité de phagocytose est comparable à celle de l’adulte.
L’immunité muqueuse se met en place durant la vie fœtale, avec la présence de
l’amylase, du lysozyme et de la lactoferrine très tôt pendant la gestation (Holt &
Jones, 2000).

152
Importance du colostrum B-
En plus de fournir un repas complet avec tous les nutriments essentiels au veau
durant la phase initiale de sa vie, le colostrum fournit une protection immunologique
essentielle. Chez les ruminants, cette prise colostrale est indispensable puisqu’il n’y
a pas d’échanges de facteurs immuns in utero. Le colostrum via ses
immunoglobulines apporte au jeune une immunisation passive grâce à la
perméabilité de la barrière intestinale. Malheureusement cette barrière intestinale
s’imperméabilise très rapidement (48 heures) et le colostrum s’appauvrit en Ig mais
d’autres facteurs du colostrum et du lait sont importants pour les défenses du veau
(Stelwagen, Carpenter, Haigh, Hodgkinson, & Wheeler, 2009).
En effet, le colostrum apporte également de la vitamine A, de la lactoferrine qui
inhibe le développement des colibacilles pathogènes par compétition pour le fer, le
système lactoperoxydase-thiocyanate qui transforme le H2O2 en acide cyano-
sulfurique, avec un effet bactéricide et enfin, il apporte également du lysozyme,
bactériolytique sur les Gram+. Le colostrum apporte aussi des cytokines et des
leucocytes. Tous ces éléments sont disponibles directement in situ pour combattre
les virus et bactéries intestinales, véritable fléau pour les veaux (les gastro-entérites
néonatales ont une morbidité de plus de 20 % et une mortalité de plus de 15%). En
effet, ceci réduit le temps de réaction du système immunitaire, puisque les éléments
sont déjà sur le site de l’infection.
Mise en place de sa propre machinerie C-
immunitaire
Chez le veau à la naissance, le ratio neutrophiles / leucocytes est plus élevé que
chez les adultes et est supérieur à 1. A une semaine d’âge le ratio est similaire à
celui de l’adulte. Ce haut taux à la naissance pourrait être relié à la libération de
cortisol dû au stress puisqu’il n’est pas présent chez les veaux nés par césariennes
(Douglas & Jane Wardrop, 2010).
Les infections microbiennes sont une source majeure de mortalité néonatale. Un
système immunitaire immature (structures lymphoïdes incomplètes de la moelle
osseuse, un faible nombre de cellules B et T, ainsi que l’absence de cellules B et T
mémoires) est la cause de ce fort taux de mortalité. Chez le nouveau-né, les cellules
du système immunitaire inné sont en nombre inférieur (en absolu et en relatif) et
elles ne sont pas capables d’activer pleinement les réponses innées et adaptatives.
La proportion de cellules dendritiques dans la rate augmente de 2,5% en 5-6
semaines et le nombre de cellules dendritiques plasmacytoïdes adultes est atteint à
3 semaines. Dans le cordon ombilical les cellules dendritiques plasmacytoïdes et
myéloïdes sont dans un état plus immature que dans le sang périphérique de l’adulte

153
et elles expriment des niveaux inférieurs de CMH II, d’ICAM-1, de CD80 et de CD86.
Le passage par le NF-κB dans la signalisation de TLR4 est intact dans les cellules
dendritiques néonatales, compatible avec leur capacité à produire des cytokines pro-
inflammatoires. Le facteur de croissance hématopoïétique Flt-3L se fixe de façon
précoce aux précurseurs des cellules dans le foie fœtal, la rate ou la moelle osseuse
et double le nombre de cellules dendritiques conventionnelles et plasmacytoïdes
chez les jeunes traités avec cette protéine (Willems et al., 2009).
Chez le veau la capacité des PNN à phagocyter des levures recouvertes d’IgG est
moindre que chez la vache, probablement parce que les PNN des jeunes ont moins
de récepteurs Fc ce qui contribue à la susceptibilité des jeunes veaux aux bactéries
et virus (Paape et al., 2003).
Importance de la nutrition D-
La sélection génétique des 30-40 dernières années avec une nutrition qui ne cesse
de s’améliorer a permis aux vaches de produire de grandes quantités de lait,
excédant 8 500 litres de lait en 305 jours de lactation (jusqu’à 15 000 litres de lait par
an pour les meilleures). Cette augmentation de production laitière a été associée à
une baisse de fertilité (avec une réussite en 1ère insémination maintenant inférieure à
40%). L’augmentation de la production laitière cause également des troubles
métaboliques sévères, notamment en début de lactation où la lipomobilisation des
réserves corporelles est intense. La majorité des problèmes de santé de la vache
laitière a lieu autour de la mise bas et est liée à la difficulté des vaches à adapter leur
prise de nourriture à leurs besoins de lactation (Ingvartsen & Moyes, 2013; Morris et
al., 2009).
Une balance énergétique négative est un problème récurrent chez les vaches hautes
productrices en début de lactation. Les vaches sont en déficit énergétique jusqu’à 7-
9 semaines après le vêlage. Les acides gras volatiles à chaîne courte (acétate,
propionate et butyrate) sont des nutriments essentiels pour les ruminants et ils sont
formés au cours de la fermentation ruminale des fibres. Les acides gras sont
principalement incorporés dans les lipides cellulaires comme les triacylglycérols, les
phospholipides, les esters de cholestérols et les corps cétoniques ne sont pas
utilisés comme source d’énergie par les phagocytes. Les réserves énergétiques du
corps sont utilisées pour couvrir les besoins énergétiques nécessaires à la
production lactée, ce qui mène potentiellement à une augmentation des corps
cétoniques dans le lait. En effet, la mobilisation des acides gras conduit à la
production d’acides gras non estérifiés (AGNE) et les corps cétoniques comme le β-
hydroxybutyrate (β-HB) sont produits par le foie et leur concentration systémique
augmente au fur et à mesure de la mobilisation lipidique. L’oxydation des AGNE
dans le foie aboutit à la formation de réactifs oxygénés et donc à un stress oxygéné.
Ces AGNE sont ensuite prélevés par le foie et entrent dans la voie de β-oxydation
pour former du NADH et de l’acétyl-CoA. L’acétyl-CoA est ensuite oxydé par

154
condensation avec l’oxaloacétate et c’est aussi le principal substrat pour la
néoglucogenèse. Cette dernière est prioritaire en cas d’hypoglycémie. L’acétyl-CoA
s’accumule et est alors transformé en acétoacétyl-CoA puis en acétoacétate, ensuite
réduit par la mitochondrie en β-OH-butyrate : ce sont des corps cétoniques. De fortes
concentrations en corps cétoniques réduisent l’appétit, diminuent la mobilisation des
acides gras et aggravent donc le déficit énergétique (Ingvartsen & Moyes, 2013;
Morris et al., 2009; Suriyasathaporn, Heuer, Noordhuizen-Stassen, & Schukken,
2000).
Lors de balance énergétique négative (notamment le 1er mois de lactation), les gènes
codant pour des antioxydants sont surexprimés comme la catalase, la dismutase
superoxide (SOD), le peroxiredoxine1 (PRDX1) et l’oxydoréductase quinone NADPH
(NQO1). Les deux principaux produits de la rupture des acides gras lors de NEB sont
le palmitate et l’oléate. Seul le palmitate engendre un stress du réticulum
endoplasmique. Les stimuli proapoptiques peuvent interférer avec des fonctions
mitochondriales, une source significative de réactifs oxygénés. Pour redresser les
effets négatifs du stress oxydatif, les gènes impliqués dans la phosphorylation
oxydative et l’ubiquitinylation sont surexprimés et la dégradation des protéines
anormales dans le protéasome augmente. Pour répondre au stress oxydatif,
l’oxydation du NADPH est essentielle pour fournir des espèces réduites. Et pour
oxyder le NADPH la biosynthèse d’ubiquinine est nécessaire. L’échec pour redresser
les effets du stress oxydatif aboutit à la mort cellulaire (Morris et al., 2009).
Le déficit énergétique affecte l’immunité innée pour plusieurs raisons :
L’accumulation de corps cétoniques diminue l’efficacité de la phagocytose car
leur concentration est inversement proportionnelle à la génération d’anions
superoxydes donc à l’activité de l’explosion respiratoire ;
Le déficit énergétique est intimement lié à la lipidose hépatique et donc à un
taux d’anticorps plus faible : la cétose interfère avec la production
d’opsonines, qui sont nécessaires pour la phagocytose et les vaches
cétosiques ont un taux de cytokines produites par les lymphocytes comme les
interférons inférieur aux vaches témoins : la production de chimiokines est
également réduite chez les vaches cétosiques ;
La migration des leucocytes est glucose dépendant et ces cellules ne peuvent
pas utiliser les corps cétoniques comme source d’énergie supplémentaire.
C’est pourquoi les vaches cétosiques ont un risque accru de mammite et d’autres
infections péripartum, puisque le nombre de leucocytes est plus faible et la capacité
de produire des chimiokines est plus lente/plus faible donc un taux de migration des
leucocytes plus faible, ainsi qu’une capacité de production d’anions superoxyde
réduite : tout ceci concoure à baisser les défenses de l’organisme (Ingvartsen &
Moyes, 2013; Suriyasathaporn et al., 2000).

155
Les acides gras non estérifiés ont, de manière contradictoire, des propriétés
immunostimulatrices et immunosuppressives puisqu’ils agissent comme ligands pour
les TLR sur les macrophages résidents. Les AGNE augmentent l’activité d’explosion
respiratoire associée à la phagocytose et réduisent la viabilité des PNN : ils
pourraient être responsables de dommages cellulaires et de nécrose durant le début
de lactation. Parmi les acides gras à courte chaîne, le butyrate est un inducteur
potentiel de l’apoptose des PNN et un inhibiteur de leur prolifération cellulaire, leur
différenciation et leur mobilité. Les vaches avec une mobilisation rapide des graisses
peuvent subir un stress oxydatif augmenté causant des dommages tissulaires et
cellulaires. Un déficit énergétique diminue l’expression de certains gènes impliqués
dans la présentation d’antigène, l’explosion respiratoire, la réponse pro-inflammatoire
bien que d’autres gènes sont surexprimés par un déficit énergétique comme l’IL-1R,
l’IL-6 et des TLR (Ingvartsen & Moyes, 2013; Morris et al., 2009).
Modifications du système immunitaire inné E-
pendant la gestation
Les cycles sexuels et la gestation impliquent des processus inflammatoires stériles,
non pas en réponse à des signaux de danger, mais plutôt pour augmenter les
chances de reproduction. Les cellules immunitaires innées préparent la reproduction
et y répondent de la même façon qu’à des stress aigus. Il y a 60 ans, Peter Medawar
a émis trois hypothèses :
le système immun maternel est inactivé durant la gestation,
les antigènes du fœtus ne sont pas suffisamment développés pour être
reconnus par le système immunitaire,
la séparation entre le fœtus et le système immunitaire par le placenta protège
le fœtus.
Les trois hypothèses ont été depuis réfutées. Le rôle secondaire du système immun
est la propagation de l’espèce, dominant durant la conception et la gestation. Or
l’hypothèse de la femelle gestante immunodéprimée n’est pas compatible avec ce
rôle du système immunitaire, à savoir, aider le maintien de la gestation tout en
protégeant le fœtus et la mère des stress extérieurs. La gestation est une période
immunologique unique pour la mère puisque le système immunitaire de la mère doit
permettre le développement du conceptus sans mettre en danger la vie de la mère.
L’utérus doit être un site immunocompétent pour combattre les infections dès leur
mise en place. En cas d’infection, l’utérus, gravide ou non, doit être capable de
recruter des leucocytes et produire des cytokines en réponse aux microorganismes
(Oliveira et al., 2012; Schminkey & Groer, 2014).
Après l’ovulation, la quantité de mucus sécrété est augmentée. Ces mucines qui
couvrent le blastocyte entrant dans l’utérus l’empêchent de coller jusqu’à ce que la
caduque (couche formée par la muqueuse utérine au cours de la grossesse en

156
périphérie de la poche des eaux et expulsée au moment de la délivrance) soit prête.
En anticipant l’implantation, les neutrophiles et les cellules dendritiques commencent
à préparer les vaisseaux spiraux pour la gestation. Les œstrogènes suppriment
l’activité immune des cytokines pro et anti-inflammatoires comme l’IL-10 et le TNF-α,
le recrutement des PNN et l’activité des NK. Ils inhibent les réponses Th1 et
favorisent les réponses Th2. Le mucus fournit également un substrat nutritif pour le
sperme. La caduque est pleine de cellules immunes comme les macrophages, les
NK utérins (uNK), les T reg, et les cellules dendritiques. La présence des NK bovins a
été détectée dans l’endomètre dès le 16ième jour de la gestation. Les NK,
macrophages et PNN sont présents grâce aux cytokines pro-inflammatoires comme
l’IL-6 dont la fonction pendant le début de la gestation est de stimuler la croissance
plutôt que la défense. Les uNK ne sont pas cytotoxiques et quand ils sont activés par
l’IL-15 produite lors de la gestation, ils répondent en sécrétant de nombreuses autres
cytokines. Il existe deux types de NK : les CD56bright (non présents dans la circulation
sanguine mais dans les tissus lymphoïdes secondaires, avec pour activité principale
la production de cytokines) et les CD56dim. Les premiers sont recrutés par le
trophoblaste vers le placenta où ils sécrètent des cytokines. Le trophoblaste possède
des TLR et s’il reconnaît des pathogènes étrangers ou des cellules mortes, il attire
les monocytes. Le placenta agit comme un organe immun qui reconnaît les
changements dans son environnement et les signale aux autres cellules qui doivent
réagir (Schminkey & Groer, 2014).
Durant la période préimplantatoire, des protéines seconds messagers comme l’AKT1
ou la MAPKAPK2 sont surexprimées suggérant que l’endomètre devient plus réactif
aux facteurs dérivés de l’embryon avant son implantation. Au moment de
l’implantation, la surexpression du récepteur à l’interféron permet à l’endomètre de
mieux répondre à l’IFN-τ sécrété par le trophoblaste. Tout ceci suggère que pour
maintenir une gestation, une expression orchestrée des gènes immuns est
nécessaire et commence avant même l’implantation du conceptus (Oliveira et al.,
2012).
L’implantation simule une réaction similaire à un stress aigu de courte durée : les
macrophages sont activés pour faire le ménage et nettoyer toutes les cellules mortes
résultant d’un accouplement réussi (spermatozoïdes, cellules lymphocytaires). Cette
activation s’accompagne dans l’utérus de la sécrétion de fortes quantités d’IL-1,
d’interféron, de CSF1, de TNF…. Deux molécules inflammatoires sont
indispensables à la gestation : le LIF maternel et l’IL-11. Elles permettent la sécrétion
dans la matrice de métallo-protéases et l’expression de molécules d’adhésion par
l’ovocyte. Durant l’implantation, il y a suppression de l’immunité spécifique et
l’immunité Th1 est inhibée pendant la gestation (Chaouat et al., 2000; Schminkey &
Groer, 2014).
Enfin, la production de cytokines anti-inflammatoires est favorisée au détriment des
cytokines pro-inflammatoires. Au cours de la gestation la fonction endocrinienne des
gonades et de l’unité fœto-placentaire comprend la production de diverses protéines

157
et hormones, impliquées dans l’établissement de la gestation, le maintien du corps
jaune, la croissance fœtale et mammaire et enfin, l’immunotolérance du conceptus.
Le signal le plus précoce émis par l’embryon (juste après la fécondation) est la
zygotine, qui stimule dans les premières heures de développement de l’œuf, la
production par l’ovaire de l’EPF (Early Pregnancy Factor). Ce facteur existe chez la
souris, la femme, la brebis, la vache, la truie, la lapine et la jument. S’il y a
conception, l’action lutéolytique de l’utérus régulière est annulée. Il s’est alors posé la
question d’un facteur anti-lutéolytique produit par le conceptus ruminant. Le signal
produit a été initialement appelé trophoblastine puis interféron τ, interféron de type I.
En plus de sa fonction anti-lutéolytique, il joue un rôle important dans
l’immunotolérance du conceptus par sa mère. Chez les bovins, il est sécrété à partir
de 16 à 25 jours. Il inhibe les récepteurs à l’œstradiol, une réduction conséquente
des récepteurs à l’ocytocine, l’activation d’un inhibiteur de la cyclooxygénase et
d’une synthèse préférentielle de la PGE2 comparativement à la PGF2α. Chez la
brebis, cet interféron est produit de façon temporaire entre le 13ième et le 21ième jour
de gestation et il possède une activité immunosuppressive avérée. Chez la chèvre, la
fenêtre de production de l’IFN-τ est plus réduite, entre le 14 et le 17ième jour de
gestation. Chez toutes les espèces, la progestérone est nécessaire à l’établissement
de la gravidité. Elle induit la synthèse de molécules par l’endomètre comme
SERPINA14 qui, au moins chez le mouton, a des propriétés antiprolifératives sur les
lymphocytes et les cellules cancéreuses et elle induit aussi une augmentation
significative de la sécrétion de PGE2, connue pour son action immunosuppressive
(De Sousa, Figueiredo, El Amiri, Banga-Mboko, & Beckers, 2002; Oliveira et al.,
2012).
Après la placentation, qui nécessite un environnement pro-inflammatoire, l’axe
hypothalamo-pituitaire-adrénal est activé et produit du cortisol pour réguler la
synthèse de cytokines pro-inflammatoires afin de limiter la réponse innée avant
qu’elle ne cause des dommages physiologiques (Schminkey & Groer, 2014).
Le placenta représente une barrière anatomique entre les systèmes circulatoires de
la mère et du fœtus : les circulations utérines et fœtales ne sont jamais en
communication directe mais elles sont suffisamment contiguës pour que les
éléments nutritifs passent du sang maternel au sang fœtal et que les déchets fœtaux
passent dans le sens opposé. En dépit de cette proximité entre circulations utérine et
fœtale, grâce à des processus immunologiques particuliers, le fœtus et le placenta
ne sont pas rejetés par la mère. La protection de l’embryon contre le rejet
immunologique par les tissus maternels fait partie des processus physiologiques de
la gestation dont la coordination doit être particulièrement précise. L’un des
mécanismes probablement impliqués dans cette activité est la suppression non
spécifique locale de certaines classes de lymphocytes. Cette inhibition est le résultat
de l’action des facteurs produits par les lymphocytes T suppresseurs et d’un
mécanisme d’apoptose des lymphocytes T maternels activés présents au niveau de
l’interface materno-fœtale. Les cellules du trophoblaste expriment le Fas ligand
(FasL), également exprimé dans les testicules et la chambre antérieure de l’œil.

158
C’est un membre de la famille des TNF qui induit l’apoptose après son interaction
avec son récepteur spécifique essentiellement exprimé par les cellules
hématopoïétiques comme les lymphocytes T. L’expression des antigènes paternels
par le trophoblaste est supprimée chez les bovins, comme chez les chevaux et les
souris. Ceci permet la réduction de l’activation du système maternel aux antigènes
paternels mais fait du trophoblaste une cible idéale pour la lyse des NK. Chez la
vache, le tissu chorionique diminue l’expression du CMH I par le placentome durant
toute la gestation. Bien que les tissus embryonnaires soient étrangers à la mère, ils
s’organisent pour dissimuler leurs caractéristiques au moins pendant un certain
temps. L’embryon est aussi protégé par les cellules déciduales, étroitement jointives
qui l’emprisonnent totalement dès l’implantation du blastocyte. Cette barrière
protectrice arrête le drainage des lymphocytes vers les tissus maternels. De plus les
vaisseaux sanguins maternels ne pénètrent pas dans le trophoblaste, bloquant ainsi
un second mécanisme impliqué dans le rejet des greffes (De Sousa et al., 2002;
Oliveira et al., 2012).
Durant la gestation, l’expression des TLR2, 3, 4, 6 et 9 est plus importante dans
l’endomètre interplacentaire que dans le placentome. Les cellules épithéliales
endométriales sont stimulées par le LPS, expriment des protéines de la phase aiguë
et sécrètent des prostaglandines pro-inflammatoires ainsi que des cytokines pro-
inflammatoires comme l’IL-1β, l’IL-6 et l’IL-8. L’endomètre exprime aussi des β-
défensines et de la mucine 1. Il sécrète de nombreux facteurs immuns dont le CSF2
et le CSF3, l’IL-1β, l’IL-6, l’IL-8 et l’IL-10, ainsi que du TNF-α lors de la gestation
(Oliveira et al., 2012).
A partir du deuxième semestre de gestation, le placenta produit de grandes quantités
de progestérone qui inhibe le système immunitaire adaptatif qui pourrait reconnaître
le fœtus comme un envahisseur étranger. La progestérone agit aussi sur le système
inné en induisant la différenciation des NK en uNK qui ne sont pas cytotoxiques, et
qui agissent comme des régulateurs des cytokines de l’environnement utérin. La
progestérone inhibe les cytokines IL-6 et TNF-α ce qui contribue à la quiescence de
l’utérus pendant la gestation (Schminkey & Groer, 2014).
Dans l’utérus gravide, il y a une accumulation massive de macrophages dans
l’endomètre, présents dans le stroma des villosités maternelles du placentome. Les
macrophages les plus proches des tissus fœtaux expriment fortement les antigènes
du CMH II, alors que les macrophages plus profonds sont CD11b+ : il s’agit peut-être
de nouveaux arrivants qui ont migré depuis la circulation maternelle. L’endomètre
intercaronculaire exprime des récepteurs éboueurs (Oliveira et al., 2012).
Dans l’utérus gravide, le remodelage tissulaire est essentiel pour l’ajustement au
fœtus grandissant. Le PDGF-β (Platelet Derived Growth Factor) est très exprimé par
les macrophages, suggérant un rôle dans le remodelage tissulaire de ces derniers
puisque le PDGF-β stimule la prolifération et la différenciation des fibroblastes
(Oliveira et al., 2012).

159
A 34 jours de gestation, il n’y a aucun changement dans le nombre de cellules T CD4
CD8 ou γδ T par rapport aux animaux non gestants. Par contre, proches du terme,
les vaches voient leur nombre de cellules γδ T circulantes augmenter (Oliveira et al.,
2012).
Modifications à la mise-bas F-
Lors de la mise-bas, la rupture des membranes, les contractions utérines et les
modifications du col sont des processus inflammatoires. Le système inné mime une
réponse de stress voire de sepsie. Le contenu utérin, les membranes, les liquides
amniotiques sont inondés de cytokines pro-inflammatoires, de macrophages et de
PNN. Les métalloprotéases matricielles produites par les PNN, les macrophages
agissent sur l’utérus, le col et les tissus des membranes, et les remodèlent
(Schminkey & Groer, 2014).
Au moment de la parturition, les vaches présentent un leucogramme de stress avec
une neutrophilie, une lymphopénie, une éosinopénie ainsi qu’une monocytose. Les
mêmes phénomènes sont observables chez la chèvre et la brebis.
A la mise bas, l’immunosuppression est définie par une baisse de cellules T CD4+
CD8+ et γδ T ainsi que par une baisse de prolifération de ces cellules et de
production d’IFN-γ par les lymphocytes stimulés. D’autres études ont montré que
l’état d’immunosuppression ne serait pas dû à un nombre réduit de lymphocytes
mais une recirculation de certains sous-types de lymphocytes dont les Treg et les γδ
T. La rétention placentaire serait liée à un partage d’antigène de CMH I avec leur
conceptus puisque le détachement des membranes fœtales serait un processus
immun (Oliveira et al., 2012).
L’expression du CD18 des neutrophiles est maximale à la mise-bas mais diminue
ensuite alors que l’expression du CD62L par ces mêmes neutrophiles diminue 2
semaines avant la mise bas puis plus fortement encore à la mise bas. Ces
changements pourraient augmenter la susceptibilité aux mammites et aux autres
maladies. Les populations de leucocytes sanguins exprimant le CMH II sont
maximales en fin de gestation mais diminuent fortement au vêlage : il a été prouvé
qu’au vêlage, une dépression des fonctions immunes est observée (Morris et al.,
2009).
A la mise bas, la contamination de l’utérus par des bactéries est très courante (90%
des vaches). Cette contamination mène dans 40% des cas à une métrite dans la
semaine qui suit le vêlage et à une endométrite persistant pendant plusieurs
semaines dans 20% des cas. La première étape de la pathogénie des infections
utérines est la colonisation de l’endomètre par des bactéries Gram- comme
Escherichia coli : le LPS entraîne une réponse inflammatoire avec synthèse des
protéines de la phase aiguë. Dans l’endomètre, des transcrits d’ARN encodent pour

160
des protéines liées à l’inflammation comme l’IL-1β et l’IL-6 et l’IL-8, attractrice pour
les PNN et macrophages. Initiées par la fixation du LPS sur le TLR4 les cellules
épithéliales jouent normalement un rôle clé dans la détection des bactéries dans la
lumière. Cependant, au vêlage, la perte de la couche épithéliale expose les cellules
stromales aux bactéries présentes dans l’utérus. Les cellules stromales sont
beaucoup plus abondantes que les cellules épithéliales et plus proches des
vaisseaux : elles jouent donc un rôle important dans l’immunité contre les microbes
de l’endomètre. La signalisation passe par le TLR4 et MyD88 puis le NF-κB mais la
signalisation active aussi un grand nombre de kinases comme MEK, MAPK3/1 et
MAPK14. Les MAPK3/1 sont activés par phosphorylation après stimulation par le
LPS et ce mécanisme fait partie des voies de signalisation dans l’endomètre (Cronin,
Turner, Goetze, Bryant, & Sheldon, 2012).
III. L’immunité de la mamelle
L’immunité innée varie donc tout au long de la vie de l’animal, qui devient, dès l’âge
de 2-3 ans, très cyclique : à partir du premier vêlage, la vache laitière produit un veau
par an, afin d’avoir une lactation de dix mois et un tarissement de deux mois. Ainsi,
les deux premiers mois après le vêlage, l’involution utérine prépare l’utérus à une
nouvelle gestation pendant que la mamelle atteint le pic de lactation. Le plus tôt
possible, l’éleveur remet la vache à la reproduction. Après le pic de lactation, la
production de lait décroit lentement jusqu’au tarissement, deux mois avant le vêlage
suivant. La mamelle est donc très sollicitée pendant dix mois de l’année. Nous nous
intéressons donc à son immunité car cet organe est central pour de l’industrie
laitière.
Comprendre les défenses immunes de la mamelle est dans l’optique du
développement de mesures pour le contrôle des mammites, pathologie majeure des
ruminants laitiers. Notre connaissance de l’immunité innée de la glande mammaire
est encore limitée et incomplète mais elle progresse, notamment sur la
Ce qu’il faut retenir :
Les périodes critiques pour la santé animale correspondent toujours aux
périodes de stress ou de stress métabolique, circonstances qui perturbent le
bon fonctionnement du système immunitaire.
La complexité des moyens mis en œuvre pour permettre la tolérance du fœtus
par le système immunitaire montre l’efficacité du système inné à détecter tout
corps étranger dans l’organisme et l’importance de la détection du soi et du
non soi.

161
reconnaissance des pathogènes et l’induction des défenses locales, sur la
contribution des cellules épithéliales mammaires dans les défenses locales et sur la
mobilisation des leucocytes (Rainard & Riollet, 2006).
L’infection de la glande mammaire conduit à une mammite, induisant des troubles
mineurs ou majeurs, causant des pertes économiques dues à la chute de production
de lait, à la perte du lait impropre à la consommation humaine, aux usages massifs
d’antibiotiques et à la réforme prématurée de l’animal. La vaccination contre la
mammite est un champ d’investigation prioritaire de la recherche ces dernières
années mais les rares vaccins disponibles sont souvent d’efficacité limitée. Une autre
approche dans la prévention des mammites a alors été la sélection d’animaux plus
résistants. En effet, durant des années, la sélection des animaux pour leur forte
production de lait aurait également augmenté la sensibilité des animaux aux
mammites (Rainard & Riollet, 2006).
Il faut garder à l’esprit que les mammites sont très fréquentes (environ 30-35
mammites pour 100 vaches laitières par an) et entraînent une perte économique non
négligeable (1 milliards d’euros par an en France).
Prévention de l’infection mammaire A-
La lumière intramammaire est un milieu normalement aseptique. Contrairement à de
nombreux épithéliums (intestinal, buccal ou respiratoire), l’épithélium mammaire n’est
pas fréquemment stimulé par des composants bactériens et n’importe quelle bactérie
doit être traitée comme un intrus. Pour ce point, la glande mammaire ressemble au
système urinaire. La glande mammaire est très sensible aux infections car les
sécrétions mammaires fournissent une excellente source de nutriments pour les
pathogènes, la température du corps est idéale pour la croissance bactérienne et
l’ouverture du canal fournit un accès direct depuis l’environnement extérieur. Le
système immunitaire de la mamelle a donc évolué en une barrière très développée,
complexe et très protectrice contre les pathogènes envahisseurs, en intégrant des
réponses immunes acquises et innées. (Rainard & Riollet, 2006; Stelwagen et al.,
2009)
Mesures anatomiques 1)
La mamelle possède des défenses anatomiques naturelles très efficaces.
Cependant, avec l’usage des machines à traire, les contraintes mécaniques
appliquées sur le trayon ont tendance à réduire l’efficacité de ces mesures. C’est
pourquoi les éleveurs doivent apporter des soins quotidiens aux trayons, comme
l’usage d’un produit de post trempage après la traite. La hauteur entre le sol et la
mamelle est un facteur de risque pour la santé de la mamelle : les éleveurs
réforment les vaches avec une mamelle qui descend en dessous du jarret : plus la
mamelle est proche du sol, plus elle est sujette à des mammites.
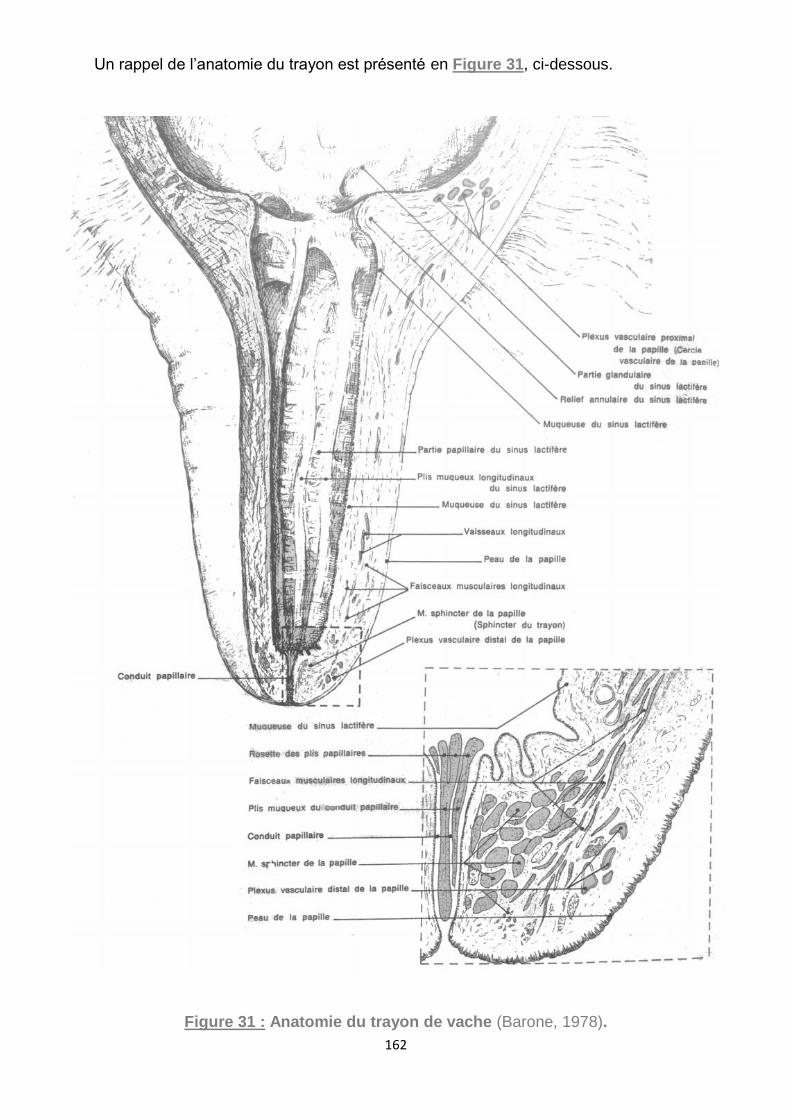
162
Un rappel de l’anatomie du trayon est présenté en Figure 31, ci-dessous.
Figure 31 : Anatomie du trayon de vache (Barone, 1978).

163
a. Le sphincter
L’extrémité du trayon contient un sphincter musculaire qui maintient le canal fermé
entre deux traites. Après la traite, deux heures sont nécessaires pour la contraction
de ce sphincter et donc pour la fermeture du trayon. Au moment de la traite,
l’ocytocine provoque le relâchement de ce muscle. La machine à traire peut induire
des désordres mécaniques et circulatoires sur les tissus du trayon, pouvant conduire
à une altération du fonctionnement correct du sphincter (Rainard & Riollet, 2006).
b. Le canal et sa kératine
Le canal du trayon est la première ligne de défense contre la mammite puisqu’il s’agit
de la principale voie d’entrée des pathogènes. Ce canal mesure entre 5 et 13 mm de
long et un diamètre de 0,4 mm. Rapporté à l’échelle humaine, le canal représente un
passage de 700-800 mètres de large pour un staphylocoque ou un colibacille. Des
moyens de protections sont donc présents pour prévenir leur pénétration dans la
mamelle. L’épithélium du canal du trayon est plus stratifié et pavimenteux que celui
du reste du trayon : la dégénérescence cornée est importante et l’accumulation
d’écailles de kératine sur les replis de la muqueuse forme un piège mécanique
contre l’entrée des bactéries. Le principal rôle de ce bouchon est de parfaire la
barrière physique en prévenant la pénétration des bactéries. La kératine peut aussi
fixer et immobiliser certaines souches de bactéries non encapsulées (on parle
d’adsorption). Lors de l’élimination des premiers jets à la traite, ce bouchon de
kératine est évacué et entraîne avec lui les germes qui ont commencé à remonter
dans le canal. Enfin, certains composants de cette kératine ont des propriétés
antimicrobiennes (Boudry, 2005; Rainard & Riollet, 2006).
La traite est très importante dans l’efficacité de ces barrières physiques puisqu’elle
rince le bouchon de kératine (Rainard & Riollet, 2006). Un des traitements les plus
efficaces pour soigner une mammite est d’ailleurs de traire la vache trois fois par jour
plutôt que deux.
c. La rosette de Fürstemberg
A la jonction entre le sinus et le conduit papillaire, un rétrécissement brusque du
diamètre forme une collerette de plis radiaires, appelée la rosette de Fürstemberg.
C’est une véritable barrière mécanique qui obstrue le conduit en dehors de la traite et
constitue ainsi la première ligne de défense.
d. La compartimentation des quartiers
Une lame fibreuse sépare complètement les quartiers droits et gauches : aucune
infection ne passe directement des quartiers droits aux quartiers gauches. Par
contre, les parenchymes des quartiers arrière et avant d’un même côté sont en

164
continu, l’infection peut donc s’étendre d’un quartier postérieur à un quartier
antérieur, toujours du même côté.
e. Les jonctions serrées
L’épithélium qui borde la glande mammaire sépare l’espace de collecte du lait et
l’espace interstitiel. Durant la lactation, cette séparation est imperméable aux petites
molécules comme le lactose ou les ions comme Na+ et Cl
-. Cette imperméabilité est
permise par les jonctions serrées, entre les cellules épithéliales, à leur pole apical :
ces jonctions forment une sorte de panier autour de chaque cellule épithéliale. Par
contre, pendant le tarissement, l’épithélium devient perméable aux grosses
molécules comme l’albumine et les Ig. Durant une mammite, il existe un passage de
molécules dans les deux sens (du lait vers le sang : lactose / du sang vers le lait :
albumine) : cette augmentation de perméabilité est essentielle pour l’immunité innée.
Elle est permise par les dommages cellulaires infligés par les toxines bactériennes et
par l’action de médiateurs pro-inflammatoires comme l’histamine, le TNF-α, et l’IFN-
γ. La perméabilité des jonctions serrées est sous contrôle hormonal (prolactine,
progestérone, et glucocorticoïdes) et de facteurs de croissance (Transforming
Growth Factor, TGF-β) et permet l’exsudation du plasma amenant de nombreux
effecteurs de l’immunité innée comme les anticorps naturels ou le complément
(Rainard & Riollet, 2006).
Les cellules impliquées 2)
Le lait d’une mamelle parfaitement saine ne devrait contenir que très peu de cellules
puisque la mamelle est un organe sécrétoire holocrine. En fait, Il y en a toujours un
peu à cause du renouvellement normal des cellules sécrétrices, des PNN et des
macrophages. Dans un quartier sain, le Comptage Cellulaire Somatique (CCS) est
inférieur à 100 000 cellules/mL, avec peu de PNN. Cette numération cellulaire est un
bon indicateur de santé de la mamelle : les éleveurs utilisent ces comptages fournis
par le contrôle laitier une à deux fois par mois pour évaluer les mammites cliniques
(mais en général, il y a des signes cliniques) et surtout les mammites subcliniques.
Au cours de la lactation, le CCS augmente et la proportion de PNN peut aller jusqu’à
40%. Au tarissement, la mamelle subit des changements physiologiques intenses.
Les concentrations cellulaires augmentent au début de la période sèche dans les 7
premiers jours jusqu’à 5 X 106 cellules/mL puis diminuent pour se stabiliser à 3 X 106
cellules/mL. Au vêlage, les CCS sont généralement supérieurs à 1 000 000
cellules/mL et diminuent jusqu’à 105 cellules/mL dans les 7 premiers jours (Rainard &
Riollet, 2006; Stelwagen et al., 2009; Suriyasathaporn et al., 2000).
a. Les cellules épithéliales
Les cellules épithéliales mammaires jouent un rôle essentiel dans cette réponse en
exprimant un large éventail de TLR capables de reconnaître des PAMPs à la surface

165
des pathogènes. La reconnaissance des PAMPs active une cascade (détaillée page
89) dans laquelle le NF-κB joue un rôle pivot pour coordonner des multiples signaux
et diriger l’expression des gènes de la réponse effectrice. Parmi ces récepteurs, se
trouvent le TLR2, le TLR4 et le TLR9. L’expression des deux premiers est
augmentée lors d’infection subclinique. Par contre, elles ne peuvent pas reconnaître
le LPS puisqu’elles n’expriment pas le CD14, nécessaire pour sa détection. Pourtant
le CD14 est présent dans le lait des mamelles saines, sa source pourrait être les
macrophages. Par contre, les cellules épithéliales produisent du LBP. Elles
synthétisent une large gamme de facteurs immuns comme la lactoferrine, les β-
défensines. Elles préparent les membranes entourant les globules gras, issues de la
membrane apicale des cellules épithéliales et libèrent de nombreux peptides,
protéines et lipides impliqués dans les défenses de l’hôte comme l’oxydase xanthine
et les sphingolipides, qui ont une activité antimicrobienne contre un grand nombre de
bactéries (Rainard & Riollet, 2006; Stelwagen et al., 2009).
b. Les macrophages
Les macrophages sont les cellules majeures du lait, des sécrétions de la mamelle
tarie et des tissus mammaires. Les macrophages du lait sont des cellules
phagocytaires capables d’ingérer la plupart des pathogènes responsables de
mammite. Ils sont moins efficaces que les PNN du lait et les deux sont moins
efficaces que leurs homologues sanguins. Ce sont des cellules présentatrices
d’antigènes et sont donc impliquées dans la détection des pathogènes envahisseurs
et dans l’initiation de l’inflammation et de l’immunité adaptative. Les capacités
fonctionnelles des macrophages de la mamelle sont diminuées après le vêlage, ce
qui est à mettre en relation avec l’incidence marquée des mammites à ce moment
(Rainard & Riollet, 2006).
La NOS est un élément clé dans l’activité des macrophages activés. Ce complexe
enzymatique catalyse la conversion de l’arginine en citrulline et en oxyde nitrique
(NO), un radical hautement réactif. Ce NO a une durée de vie très courte et réagit
avec l’oxygène pour donner du nitrite (NO2) et du nitrate (NO3) ou du peroxynitrite
lorsqu’il réagit avec l’ion superoxyde. La concentration de NO est très faible dans le
lait et le colostrum des vaches saines mais elle augmente temporairement après
infusion par le LPS d’Escherichia coli et à plus long terme et en plus grande quantité
lors d’infection par Escherichia coli ou Staphylococcus aureus (Rainard & Riollet,
2006).
c. Les polynucléaires neutrophiles
Les neutrophiles sont présents dans le lait normal mais à des concentrations trop
faibles pour permettre une phagocytose efficace en suspension. De plus, une partie
de ces neutrophiles ne sont pas viables, en apoptose, surtout dans la citerne. D’un
autre côté, la concentration des neutrophiles dans le lait sain semble inversement
proportionnelle au risque de mammite, donc ils doivent quand même être

166
suffisamment efficaces pour diminuer le risque de mammite (Rainard & Riollet,
2006).
Dans la glande infestée, 90% des PNN proviennent du recrutement depuis les
vaisseaux sanguins (les 10% restants sont déjà présents dans la mamelle saine).
Les signaux inflammatoires, notamment le C5a et le C3a, le LPS, l’IL-1, l’IL-2 et l’IL-8
guident les PNN vers le site de l’infection. De grandes quantités de C5a sont
détectées dans la mamelle lors d’infection à Escherichia coli alors que de faibles
quantités sont détectables lors d’infection à Staphylococcus aureus. Ceci explique
l’intensité de la réponse à ces deux agents de mammite et pourquoi Staphylococcus
peut établir des infections chroniques. En réponse aux signaux chimiotactiques
générés par les pathogènes, les PNN migrent du sang vers l’endothélium, la matrice
sous épithéliale, la membrane basale et l’épithélium mammaire puis vers le lait. Cette
migration est facilitée par les métalloprotéases matricielles libérées par les PNN lors
du passage de l’endothélium pour casser la matrice extracellulaire. Ces protéinases
dégradent le collagène, la laminine, les fibronectines, l’élastine et la protéine centrale
du protéoglycane (Paape et al., 2003; Suriyasathaporn et al., 2000).
Les PNN du lait sont peu efficaces dans la phagocytose. Ceci peut s’expliquer par
différentes raisons :
Quand les PNN entrent dans le lait, ils ingèrent des globules gras, et des
micelles de caséine et possèdent donc des vacuoles intracellulaires larges.
L’internalisation des membranes cellulaires mènent à la perte des
pseudopodes : il devient difficile pour les PNN d’ingérer des pathogènes par
manque de membrane. Les granules cytoplasmiques migrent et fusionnent
avec ces vacuoles pour former des phagolysosomes : même si les PNN
arrivent à internaliser des pathogènes, ils n’ont plus assez de granules pour
les détruire ;
La diapédèse des PNN à travers l’épithélium mammaire induit une baisse de
production de réactifs oxygénés par les PNN et donc contribue à la baisse
d’activité bactéricide ;
Les PNN du lait sont moins efficaces dans la phagocytose car ils possèdent
moins de réserves de glycogène donc moins d’énergie (3% de moins que les
PNN sanguins), pour la phagocytose ;
Les glucocorticoïdes contenus dans certains traitements intramammaires
réduisent la phagocytose des PNN dans le lait ;
Un défaut d’opsonine dans le lait (Paape et al., 2003; Suriyasathaporn et al.,
2000).
Bien que les PNN fournissent un effet bénéfique en éliminant les agents infectieux,
une exposition prolongée des tissus mammaires aux PNN aboutit à des blessures de

167
l’épithélium sécrétoire. Un des mécanismes passe par la libération de radicaux
oxygénés et de protéases, tous deux bactéricides et cytotoxiques pour les tissus de
l’hôte. Une inflammation prolongée lèse l’épithélium sécrétoire et mène donc à une
perte permanente de la production de lait. Une élimination rapide des PNN par les
macrophages après la neutralisation bactérienne est donc essentielle pour limiter les
dommages des PNN. L’apoptose des PNN est un facteur clé dans la résolution des
mammites (Paape et al., 2003).
d. Les NK
Les PNN et les macrophages sont efficaces pour éliminer les pathogènes
extracellulaires et les cellules NK sont présentes pour éliminer les pathogènes
intracellulaires. Elles sont aussi capables d’éliminer les bactéries en relâchant des
protéines bactéricides. Ils exercent une activité bactéricide contre Staphylococcus
aureus après stimulation par l’IL-2 (Rainard & Riollet, 2006).
Mesures chimiques et humorales 3)
Les composants humoraux regroupent les oligosaccharides, des gangliosides, des
espèces réactives oxygénées, des protéines de la phase aiguë, des facteurs
immunomodulateurs (dont de nombreuses cytokines pro et anti-inflammatoires) des
ribonucléases (ARNases) et une variété de protéines et peptides antimicrobiens.
Beaucoup de ces composants proviennent de cellules spécialisées de la glande
mammaire (Stelwagen et al., 2009).
a. Les peptides antimicrobiens
La lactoferrine (déjà évoquée page 75) est une protéine glycolysée qui appartient à
la famille des transferrines et fixe deux ions ferriques de façon réversible. Mais la
stabilité de fixation du fer dépend du pH. Cette chélation du fer lui confère une
propriété bactériostatique mais aussi une protection contre les radicaux libres
catalysés par le fer libre. Le citrate, produit en début de lactation est sécrété dans le
lait par les cellules épithéliales. Ce tampon chélate le fer et le rend indisponible pour
les bactéries. La fixation de fer et son relargage sont associés à des changements
de conformation importants dans lesquels la lactoferrine adopte un état ouvert ou
fermé. La forme saturée en fer est fermée et beaucoup plus compacte que la forme
apo. La lactoferrine peut fixer d’autres ions métalliques notamment Cu2+, Mn2+, Zn2+
mais avec une affinité moindre.
Elle possède un large spectre d’action antimicrobien puisqu’elle agit sur des
bactéries Gram-, des Gram+, des levures, des champignons, certains virus et
protozoaires. Parmi les agents pathogènes responsables de mammites, Escherichia
coli est le plus sensible à l’activité bactériostatique de la lactoferrine, suivi par
Staphylococcus aureus ; par contre les streptocoques sont résistants à cet effet.

168
Le lait de vache contient entre 20 et 200 μg/mL de lactoferrine, ce qui est faible par
rapport au lait humain (1-2 mg/mL). Les sécrétions de la glande pendant le
tarissement peuvent contenir jusqu’à 100 mg/mL de lactoferrine. La régulation de
son expression apparaît contraire à celle des autres protéines du lait : sa
concentration augmente au tarissement ou lors de mammite quand la production de
lait diminue. Il faut un délai de 24-48 heures pour que cet effet ait lieu. Elle est
exprimée dans l’épithélium bordant les canaux et les citernes, mais pas dans
l’extrémité du trayon. Elle est également présente dans d’autres sécrétions du corps
comme la salive, les larmes et le liquide broncho-alvéolaire ainsi que dans les
granules des leucocytes. Durant les mammites, la lactoferrine des granules des PNN
est relarguée et peut représenter jusqu’à 5% de la lactoferrine totale durant une
inflammation aiguë. Des médiateurs inflammatoires comme le PAF et l’IL-8 causent
aussi la libération de lactoferrine.
La lactoferrine a plusieurs actions dans le lait :
Elle est bactériostatique en entrant en compétition avec les bactéries pour la
fixation du fer, nécessaire à leur croissance. Cependant, dans le lait et le
colostrum le ratio citrate / lactoferrine empêche la lactoferrine d’être
bactériostatique mais pendant le tarissement ce ratio est plus favorable aux
propriétés chélatrices du fer de la lactoferrine. Les effets bactériostatiques et
bactéricides des protéines fixant le fer sont probablement peu efficaces
pendant la lactation mais pendant le tarissement, ils seraient essentiels. De
plus, les bactéries ont appris à détourner ce problème en synthétisant des
sidérophores, des petits chélateurs de fer à forte affinité et capables de le
transporter dans la bactérie grâce à un récepteur spécifique. D’autres
bactéries peuvent acquérir le fer directement via des récepteurs de surface
qui fixent la lactoferrine et transfèrent le fer dans la bactérie ;
Elle peut aussi être bactéricide en se fixant directement sur le LPS, l’acide
lipoteichoïque ou aux porines, où elle déstabilise alors la membrane
bactérienne, les fragilise et augmente leur perméabilité. Son activité
bactéricide est concentrée dans sa région aminoterminale, très basique qui
sous l’action d’une pepsine, libère la lactoferricine, protéine bactériolytique à
spectre large. La phase fluide du lait devient bactéricide pour Escherichia coli ;
La lactoferrine exerce une activité antivirale contre les virus à ADN et ARN
puisqu’elle inhibe l’attachement et l’entrée des virus en interagissant avec les
glucosaminoglycanes et les intégrines que certains virus utilisent pour
pénétrer dans les cellules. Elle semble aussi pouvoir inhiber la réplication
virale ;
Sur les levures, la lactoferrine peut déstabiliser leur paroi, mais aussi induire
l’apoptose ;
La lactoferrine est aussi anti-oxydante : l’activation des macrophages par le
LPS ou le TNF-α entraîne la production d’espèces oxygénées réactives,
amplifiée par la présence de fer libre. La lactoferrine limite ce processus et
donc le dommage causé aux membranes cellulaires ;

169
Elle possède des propriétés immunomodulatrices : elle a un effet négatif sur
la granulopoïèse et contrôle donc la production de PNN durant l’infection
aigue mammaire ;
La lactoferrine est anti-inflammatoire. Cette dernière fonction intervient
surtout dans un lait normal : la lactoferrine se fixe sur le lipide A du LPS
bactérien, ce qui le neutralise et l’empêche d’interagir avec le LBP. La
lactoferrine agit en synergie avec le complément puisqu’elle se fixe sur
Streptococcus agalactiae et active alors la voie classique du complément
grâce à l’opsonisation de la bactérie. Son inhibition des cytokines pro-
inflammatoires réduit la réponse inflammatoire systémique. Elle diminue
l’activation des neutrophiles dans le lait, prévenant ainsi les dommages
tissulaires que pourraient provoquer les neutrophiles ;
La lactoferrine est également pro-inflammatoire puisqu’elle stimule la
différenciation des cellules T, l’activité phagocytaire des cellules immunes et
potentialise la cytotoxicité des NK. Cette dualité dans les rôles de la
lactoferrine illustre l’ambivalence des composants du système immunitaire qui
se montrent tantôt anti-inflammatoires, tantôt pro-inflammatoires selon le
contexte du milieu et des cytokines et chimiokines présentes.
La lactoferrine a été l’un des premiers composants du lait à avoir été extraite
commercialement pour ses propriétés antimicrobiennes et antivirales. Elle est
présente dans la nourriture humaine et pour les animaux domestiques, dans les
produits cosmétiques, dans les dentifrices, les bains de bouche et les chewing-gums
(Berlutti et al., 2011; Linde et al., 2007; Paape et al., 2003; Pierce, Legrand, &
Mazurier, 2009; Rainard & Riollet, 2006; Stelwagen et al., 2009).
La transferrine est une autre protéine fixant le fer, mais contrairement au lait des
rongeurs et lagomorphes, celui des ruminants en contient très peu (1 mg/mL dans le
colostrum, 0,02-0,04 mg/mL dans le lait contre 4-5 mg/mL dans le sérum). La
transferrine n’est pas synthétisée par la glande mammaire des ruminants, elle
provient donc de la transcytose depuis le sang en conditions physiologiques, et par
exsudation du plasma lors de mammite. La transferrine constitue un agent chélatant
le fer et donc bactériostatique efficace avant même que la concentration de
lactoferrine n’augmente (Rainard & Riollet, 2006).
Le lysozyme est une protéine bactéricide qui clive le peptidoglycane des parois
cellulaires des bactéries Gram- et Gram+. Il agit en synergie avec le complément, la
lactoferrine ou les anticorps. Cependant le lait bovin n’en contient que 13 μg/100
mL : même s’il s’agit d’un moyen de défense efficace chez la femme, il n’est pas
considéré comme tel chez la vache. Le lysozyme est utilisé pour préserver la
nourriture dans l’industrie alimentaire (Rainard & Riollet, 2006; Stelwagen et al.,
2009).

170
b. Les protéines de la phase aiguë
Dans la mamelle, les cellules épithéliales produisent du sérum amyloid A3 dans des
conditions physiologiques, et lors de stimulation par le LPS ou la prolactine, cette
expression est fortement augmentée dans les tissus alvéolaire, ductal et citernal.
L’augmentation de concentration de SAA est présente dans les mammites aiguës
mais aussi subcliniques et chroniques. Ce SAA est alors produit par les cellules
épithéliales mammaires. Ce peptide a une activité antibactérienne contre Escherichia
coli, Streptococcus uberis et Pseudomonas aeruginosa. L’haptoglobine n’est
synthétisée par la mamelle qu’après stimulation par le LPS et uniquement dans les
régions alvéolaires. L’expression d’AGP est physiologique et lors d’infection elle
augmente dans les tissus ductal et citernal mais diminue dans les tissus alvéolaires
(Ceciliani et al., 2012; Rainard & Riollet, 2006).
c. Le complément et les anticorps naturels
Le système du complément contribue aux défenses de la mamelle. Il est présent
dans le lait des mamelles non enflammées à des concentrations basses mais
significatives. La voie classique n’est pas possible par manque de C1q mais la voie
alterne est fonctionnelle par dépôt d’opsonine C3b et iC3b sur les bactéries et la
génération de fragment C5a pro-inflammatoire. Les concentrations de C3 sont plus
hautes que ce qui est attendu sur la base d’un simple transsudat à partir du sang. Le
fragment C5a est chimioattracteur pour les PNN à travers l’épithélium mammaire. Il
intervient très rapidement après l’inoculation d’Escherichia coli ou Streptococcus
uberis, mais pas lors d’infection par Staphylococcus aureus, malgré la présence de
PNN (Rainard & Riollet, 2006).
Les anticorps naturels sont des anticorps produits en absence totale de stimulation
antigénique externe. Ce sont des anticorps-opsonines de classe IgM présents dans
le sérum. Ces anticorps sont en concentration suffisante en début de lactation,
notamment pour opsoniser Escherichia coli, mais en milieu et fin de lactation, leur
concentration devient insuffisante. Par contre, lors d’inflammation, l’exsudation du
plasma fournit des anticorps en grande quantité. Des anticorps naturels contre la
leucotoxine des staphylocoques sont trouvés dans le sérum de la plupart des vaches
adultes, sans aucun historique d’infection (Rainard & Riollet, 2006).
d. Les composants chimiques
La lactoperoxydase est une enzyme qui inhibe ou tue les bactéries en présence de
thiocyanate et de peroxyde d’hydrogène mais la faible teneur en oxygène du lait
limite son efficacité contre les pathogènes. La lactoperoxydase est extraite pour
conserver la nourriture, pour les soins oraux et comme fongicide pour plante
(Rainard & Riollet, 2006; Stelwagen et al., 2009).

171
L’oxydase xanthine, une enzyme de la membrane des globules gras du lait,
catalyse la formation d’oxyde nitrique à partir de nitrite inorganique, qui, sous des
conditions aérobies, mène à la génération de peroxynitrite, un agent bactéricide très
puissant. Le lait bovin possède une forte activité de cette enzyme et est
bactériostatique pour Escherichia coli. Le nitrite provient des bactéries ou de la NOS
(Nitric Oxide Synthase). Le peu d’oxygène du lait et le pH acide favorisent l’activité
de l’oxydase xanthine : son rôle protecteur est plausible (Rainard & Riollet, 2006).
Les facteurs influençant les composants immuns du 4)
lait
La présence et la concentration des composants du lait varient selon le stade de la
lactation : immédiatement post partum, de fortes concentrations en composants
immuns sont trouvées dans le colostrum (5% d’immunoglobulines, puis cette
concentration diminue pour réaugmenter en fin de lactation). De même pour la
lactoferrine, la concentration est minimale pendant le pic de lactation et augmente
progressivement ensuite avec la lactation. A l’inverse, la concentration de la
lactoperoxydase est plus cyclique tout au long de la lactation. Entre des vaches
saines, la concentration des composants immuns du lait varie énormément : une
différence d’un facteur 20 est possible pour la lactoferrine entre deux vaches saines,
il en va de même pour la lactoperoxydase. Cette différence naturelle notable entre
des animaux élevés dans les mêmes conditions est appelée variabilité individuelle.
Une telle disparité entre les animaux indique une opportunité de sélectionner les
animaux pour leur haut taux de facteurs immuns dans le lait (Stelwagen et al., 2009).
La pratique d’élevage influe aussi sur les concentrations des composants immuns du
lait. En effet, ne traire qu’une fois par jour, détend les jonctions serrées entre les
cellules épithéliales ce qui augmente les composants cellulaires et innés du lait. La
concentration en lactoferrine est doublée après seulement 3 jours de monotraite
(Stelwagen et al., 2009).
Les défenses mammaires sont parfois insuffisantes pour endiguer l’entrée des
pathogènes. Une inflammation mammaire s’instaure.
L’inflammation mammaire : 1er
signe B-
macroscopique d’une mammite
L’inflammation n’est pas initiée tant que la concentration bactérienne n’a pas atteint
un certain niveau dans le lait car cette détection bactérienne est le fruit de la collision
aléatoire des macrophages du lait avec les bactéries. La croissance bactérienne est
accompagnée de la libération de produits microbiens reconnus par l’hôte comme
signaux de danger. Cette détection se fait grâce aux TLR qui reconnaissent ces

172
PAMPs. Une phagocytose intense est alors entreprise : par exemple, les
macrophages sont activés par le LPS d’Escherichia coli et répondent en sécrétant de
l’IL-1. Les neutrophiles résidants contribuent aussi à la détection des envahisseurs
puisque les deux types cellulaires peuvent relâcher des médiateurs inflammatoires et
chimiotactiques suite à la détection d’une bactérie. Après stimulation de l’épithélium
par des bactéries, les cellules épithéliales peuvent générer un grand nombre de
médiateurs inflammatoires comme les cytokines (IL-1α, IL-1β IL-6 et TNF-α), les
chimiokines (IL-8, CXCL-5, CXCL-6 et CCL-5), des peptides antimicrobiens et des
métabolites de l’acide arachidonique. Ainsi, les cellules épithéliales agissent d’une
façon autocrine ou paracrine. Ces réponses sont très rapides, moins de 3 heures
après l’exposition au LPS ou l’infection bactérienne. Le TNF-α apparait très
rapidement comme médiateur central des fonctions inflammatoires et joue un rôle
important lors de mammite. Les différents médiateurs pro-inflammatoires peuvent
avoir une action autocrine, paracrine ou endocrine. Le but des étapes précoces de
l’inflammation est de localiser et éliminer l’infection grâce au recrutement de
leucocytes effecteurs comme les PNN vers le site de l’infection (Ballou, 2011)
(Rainard & Riollet, 2006). Les différentes étapes de l’inflammation déjà évoquées
page 108 sont alors mises en place.
Un des effets locaux est la vasodilatation des vaisseaux sanguins qui augmente le
flux sanguin et apporte plus de PNN vers le quartier mammaire infecté. Les jonctions
gap entre les cellules endothéliales sont perdues en réponse aux médiateurs
inflammatoires locaux, ce qui facilite la diapédèse des PNN et d’autres facteurs
immuns plasmatiques. L’augmentation du flux sanguin et de la perméabilité
vasculaire expliquent le gonflement et la chaleur visibles macroscopiquement
(Ballou, 2011).
Les leucocytes et surtout les macrophages, du lait libèrent du NO après stimulation
par des produits bactériens. En plus de son activité antimicrobienne, le NO est
impliqué dans l’hyperhémie de la glande mammaire qui suit le pic de fièvre. Le NO
agit comme modulateur de la cascade de l’acide arachidonique et réduit ainsi la
synthèse des leucotriènes, médiateurs importants de l’inflammation. Lors de
mammite subclinique, la concentration de lactoferrine augmente légèrement mais
lors de mammite aiguë, cette hausse peut atteindre les 6 mg/mL en 24-48 heures.
Dans ce dernier cas, la lactoferrine participe à l’activité bactériostatique du lait sans
cellules mais seulement quand la composition du lait a été profondément modifiée.
Plusieurs β-défensines sont exprimées par la mamelle bovine saine (LAP, TAP et
BNBD) ou après infection bactérienne (Rainard & Riollet, 2006).
Rappelons que les quatre quartiers de la mamelle d’une vache sont indépendants et
la réponse locale est donc limitée au quartier infecté. Les autres quartiers subissent
par contre les effets généraux alors que les effets locaux ne concernent que le
quartier infecté. Les effets systémiques sont l’hyperthermie et le changement de
partage des nutriments dans tout le corps. Un état catabolique est observé dans la
graisse et les muscles squelettiques concomitant avec la phase aiguë. Par contre,

173
dans le foie, un état d’anabolisme se met en place pour la synthèse des protéines de
la phase aiguë et la glucogenèse (Ballou, 2011).
Lors d’infection par Escherichia coli, qui provoque des mammites cliniques avec des
signes cliniques sévères et une perte de production en quelques heures, de grandes
quantités d’IL-1 et de TNF-α, cytokines pro-inflammatoires, sont produites ainsi que
l’IL-8. La concentration en neutrophiles augmente rapidement (en 3-12 heures) et
peut atteindre jusqu’à 107 cellules/mL. Un délai d’une heure pour le recrutement des
neutrophiles conduit à 8 fois plus d’Escherichia coli et encore plus d’endotoxines.
L’intensité et la rapidité du recrutement dépend du pathogène mais aussi de la
vache. La plupart du temps, la réponse de la phase aiguë suffit pour éliminer la
bactérie en quelques jours, ensuite les CCS retournent à des niveaux sains (Rainard
& Riollet, 2006).
Staphylococcus aureus est responsable de mammite moins sévère et souvent
subclinique : le recrutement des neutrophiles est modéré et retardé (24-48 heures) :
la bactérie survit à cette première vague de défenses ce qui conduit à la mise en
place d’une infection chronique durant laquelle l’inflammation et le recrutement des
leucocytes continuent pendant des mois. Deux mois après l’infection, les PNN
constituent toujours la majorité des cellules. Enfin, la diapédèse prolongée des
leucocytes finit par causer des dommages au tissu mammaire entrainant alors une
baisse de production (Rainard & Riollet, 2006).
L’infection mammaire par Mycoplasma bovis conduit à l’augmentation des
concentrations du BSA et LBP dans le lait ce qui montre que Mycoplasma bovis peut
induire une réponse inflammatoire qui altère la fonction de barrière vasculaire dans la
mamelle. Elle est aussi caractérisée par l’activation du complément et la production
de cytokines (TNF-α, IL-1β, TGF-α, IFN-γ, IL-12, IL-10, TGF-β1 et TGF-β2). Le délai
d’induction de cette réponse inflammatoire peut être attribué à la croissance lente de
Mycoplasma bovis dans la glande. (Kauf, Rosenbusch, Paape, & Bannerman, 2007).
La transition du tarissement à la lactation est abrupte et nécessite une coordination
homéostasique de l’organe pour supporter le nouvel état physiologique. La grande
majorité des nouvelles infections intramammaires apparaissent pendant le
tarissement ou pendant la colostrogenèse. Les vaches fraichement taries et les
vaches qui vont bientôt vêler produisent quand même du lait qui n’est pas trait : le lait
reste dans la mamelle et c’est un excellent milieu de culture pour les
microorganismes. Au tarissement, les concentrations en leucocytes assez élevées
pour être protectrices ne sont pas atteintes avant 8 jours après le tarissement. De
plus, la concentration de la lactoferrine est réduite donc le fer est plus disponible
pour les bactéries. Après la mise bas, les PNN ont une activité phagocytaire réduite
et sont en nombre réduit entrainant une sensibilité accrue de la mamelle à cette
période. Cependant, la vache en début de lactation a de hautes concentrations de
cytokines pro-inflammatoires et avec un rapide afflux de PNN dans la mamelle. Les
vaches après le vêlage ont une réponse pro-inflammatoire plus agressive que plus

174
tard dans la lactation. Ce phénomène est une compensation pour activer les
neutrophiles plus quiescents du post partum. Le statut redox altéré des vaches en
péripartum peut contribuer au dysfonctionnement de l’inflammation qui peut expliquer
l’incidence et la sévérité des mammites. Le nombre de lactations intervient aussi et le
risque de développer une mammite augmente avec la parité. Ce phénomène pourrait
s’expliquer par le fait que la détérioration des PNN est plus marquée sur les vaches
âgées, ou alors par le nombre de leucocytes circulant ou encore par les récepteurs
présents à leur surface (Ballou, 2011; Rainard & Riollet, 2006).
Enfin, il ne faut pas oublier que les cellules de l’immunité adaptative participent aux
défenses de la mamelle (lymphocytes T et B) mais leur recrutement ne fait pas l’objet
de ce travail (Rainard & Riollet, 2006).
Après avoir compris les mécanismes de l’immunité innée, certains ont cherché des
moyens naturels d’amplifier cette immunité, afin de limiter la consommation
d’antibiotiques. Le GM-CSF stimule l’activité antibactérienne des neutrophiles et des
macrophages. L’IL-1β et l’IL-2 induisent un influx de neutrophiles dans le lait quand
elles sont injectées dans le trayon. Le problème est que leur dose d’effet n’est pas
très loin de la dose toxique. L’IFN-γ recombinant bovin potentialise les réponses des
lymphocytes T, des macrophages, des neutrophiles et module l’immunité mammaire
en période péripartum. Cependant, les cytokines recombinantes injectées en
intramammaire ne sont pas aussi efficaces que les antibiotiques classiques
intramammaires (Rainard & Riollet, 2006).
Rappelons qu’une réponse inflammatoire excessive est délétère pour l’organisme et
peut provoquer la mortalité de l’hôte. L’inflammation peut causer des dommages
tissulaires locaux, et la cause en est généralement les PNN qui relâchent des réactifs
oxygénés ou des granules enzymatiques. Comme il existe un lien entre le statut
antioxydant réduit des vaches en péripartum et une réponse inflammatoire
exacerbée, pour limiter les mammites en péripartum, il faut supplémenter les vaches
en vitamine E et sélénium pour augmenter les capacités oxydatives et bactéricides
des PNN. Un autre moyen pour limiter l’interaction du LPS des Gram- et les
leucocytes inflammatoires de l’hôte est l’administration de CD14 ou de polymyxine B.
L’injection d’anti-inflammatoires non stéroïdiens a un effet limitant sur les signes
cliniques mais n’a pas d’effet sur les performances de production laitière (Ballou,
2011).
Ce qu’il faut retenir :
La mamelle possède donc de nombreux moyens de prévention et de défense
pour se protéger des microbes. Les mammites représentent néanmoins une
perte économique non négligeable et une importante consommation
d’antibiotiques qu’il faut réduire.

175
Les cellules lymphoïdes innées se caractérisent par trois traits :
L’absence de réarrangement pour un récepteur spécifique d’un antigène :
elles ne peuvent donc pas répondre de manière spécifique à un antigène ;
L’absence de marqueurs phénotypiques des cellules myéloïdes ou des
cellules dendritiques ;
Leur morphologie lymphoïde. (Hergen Spits et al., 2013; J. A. Walker,
Barlow, & McKenzie, 2013)
Les premières ILC découvertes en 1975 ont été les cellules NK (qui fournissent une
immunité précoce contre les virus et interviennent dans l’immunité contre les
cancers) puis, en 1997 les LTi (impliquées dans la formation des nœuds
lymphatiques durant l’embryogénèse). Ces cellules, même si elles ont des fonctions
différentes, partagent une partie de leur développement. Au cours des 5 dernières
années, il est devenu évident que les cellules NK ne sont pas les seules
représentantes des ILCs. Récemment, d’autres ILCs ont été découvertes et de façon
surprenante, ces nouvelles cellules lymphoïdes innées sécrètent des profils de
cytokines comparables à ceux produits par différents types de lymphocytes T helper.
En 2008, il a été montré que les LTi sont des membres d’une famille plus large d’ILC
LTi-like qui expriment des cytokines pro-inflammatoires comme l’IL-17A et l’IL-22,
jouant un rôle dans l’homéostasie muqueuse et des défenses similaires aux Th17.
Enfin, un dernier groupe a été découvert : ces cellules dupliquent les activités des Th2
avec la production d’IL-4, d’IL-5, d’IL-6 et d’IL-13 et sont impliquées dans les
défenses précoces contre les vers et les réponses allergiques. Ensemble, ces ILCs
peuvent être classées en trois groupes selon leur expression de cytokines reflétant
l’équivalent inné des Th1, Th2 et Th17. A l’exception des LTi, toutes les ILCs ont été
découvertes dans un contexte inflammatoire ou infectieux (Cording, Medvedovic,
Cherrier, & Eberl, 2014; Diefenbach, 2013; Hergen Spits et al., 2013).
Les ILCs ont été récemment reconnues comme une population majeure de cellules
effectrices immunes et les réponses précoces aux infections sont largement
dépendantes des cytokines qu’elles produisent. Elles sont définies par leur origine
depuis le progéniteur lymphoïde commun qui donne aussi naissance aux cellules B

176
et T. Cependant elles n’expriment pas de récepteur à l’antigène et ne subissent donc
pas de sélection clonale puis d’expansion comme les lymphocytes classiques. A
l’inverse, ces ILCs réagissent aux cytokines, et chimiokines produites par les tissus
lors de blessure ou d’infection et répondent en produisant des cytokines effectrices
(Cording et al., 2014).
I. Les différents types d’ILCs
Les ILCs peuvent classiquement être regroupées en trois groupes (Figure 32, page
177) selon le profil de cytokines qu’elles produisent et les facteurs nécessaires à leur
développement (Rankin, Groom, Mielke, Seillet, & Belz, 2013).
Le groupe des ILCs 1 comprend des cellules qui synthétisent de l’IFN-γ
comme les cellules NK et d’autres cellules nommées ILCs 1. Elles expriment
les facteurs de transcription T-box comme le T-bet et l’Eomes. Elles sont
l’équivalent inné des Th1 ;
Le groupe des ILCs 2 regroupe des cellules produisant les cytokines de type 2
comme l’IL-5 et l’IL-13. Ces cellules dépendent du GATA3 et du RORα pour
leur développement et leur fonctionnement. Elles correspondent aux Th2 du
système adaptatif ;
Le groupe des ILCs 3 inclut tous les types d’ILC produisant de l’IL-17 et/ou de
l’IL-22 comme les ILCs 3 ou ILCs RORγt+ et les cellules LTi. Ces cellules
dépendent du RORγt pour leur développement et leur fonction. Leur
équivalent adaptatif est le groupe des Th17 (Hepworth & Sonnenberg, 2014;
Vijay Kumar, 2013; B. W. S. Li & Hendriks, 2013; Montaldo, Vacca, Moretta, &
Mingari, 2014; Hergen Spits et al., 2013; Tait Wojno & Artis, 2012; J. A.
Walker et al., 2013; S. Yu, Kim, Chang, DeKruyff, & Umetsu, 2014).
Il existe un parallèle entre ces ILCs et les cellules T helper du système adaptatif,
résumé dans le Tableau VI, ci-dessous.
Tableau VI : Parallèle entre les cellules Thelper et les ILCs.
Groupe du
système
adaptatif
Cellule du
système
adaptatif
Cytokines
produites
Cellule du
système inné
Groupe du
système inné
LT helper
Th 1 IFN-γ ILC 1 ILC 1
Th 2 IL-13, IL-4, IL-
6, IL-15 ILC 2 ILC 2
Th17 IL-17, IL-22 NCR-
ILC 3 Th 22 IL-22
NCR+ ILCs 3
LTi
Treg TGF-β, IL-10,
IL-35 ???

177
Figure 32 : Classification des ILCs (Björkström, Kekäläinen, & Mjösberg, 2013).
Tout au long de cette partie, Citrobacter rodentium sera cité plusieurs fois. Il s’agit
d’une bactérie Gram-, du tractus gastrointestinal de la souris, qui s’attache à la paroi
intestinale avant d’effacer les microvillosités (« attaching and effacing pathogen »), à
la façon d’Escherichia coli : c’est un excellent modèle des EPEC ou des EHEC
(Escherichia coli entéropathogène ou entérohémorragique) alors que le colibacille
n’est que faiblement pathogénique chez la souris contrairement à Citrobacter
rodentium. De plus, il fournit un excellent modèle pour toutes les infections
restreintes à la lumière du tube digestif. C’est donc Citrobacter rodentium qui est
utilisé dans les études chez la souris. (Diefenbach, 2013)
Nous sommes conscients de la nouveauté de ces cellules et de nombreuses
informations sont régulièrement dévoilées, dont certaines sont réfutées par la suite.
Nous nous proposons de faire le bilan de ce qui est connu sur ces cellules à l’heure

178
actuelle. Nous utiliserons la nomenclature proposée par H. Spits en 2013 et
évoquerons des éventuels désaccords lorsque nous les rencontrerons.
Le groupe 1 des ILCs A-
Les cellules du groupe 1 produisent des cytokines de type 1 comme l’IFN-γ et le TNF
et sont incapables de produire des cytokines de type Th2 ou Th17. L’un des
membres phares de ce groupe est représenté par les cellules NK exerçant une
activité cytotoxique directe contre ses cellules cibles et produisant de l’IFN-γ. L’autre
membre de cette famille découvert beaucoup plus tardivement est constitué des ILCs
1 (Vijay Kumar, 2013; Hergen Spits et al., 2013).
Les cellules NK 1)
Les NK représentent 10% des lymphocytes sanguins chez l’homme et fournissent un
bras effecteur au système immun inné. Ils ont été ainsi nommés en rapport à leur
cytotoxicité naturelle (d’où leur nom Natural Killer) contre certaines tumeurs et
cellules infectées par des virus, mais ce sont également d’excellents producteurs
d’IFN-γ, de TNF-α, de GM-CSF, et d’autres cytokines et chimiokines. La production
de ces facteurs solubles par les cellules NK dans les phases précoces de l’immunité
innée influence significativement le recrutement et le fonctionnement des autres
cellules hématopoïétiques. Récemment, les cellules NK ont été rangées dans le
groupe 1 des ILCs puisqu’elles sécrètent l’IFN-γ (Campbell & Hasegawa, 2013; J. A.
Walker et al., 2013; J. Yu, Freud, & Caligiuri, 2013).
a. Différents types de cellules NK
Les cellules NK sont définies comme des lymphocytes dérivés de la moelle osseuse,
grands et granuleux. Voir Figure 33 ci-dessous.
Figure 33 : Cellule NK (Janeway et al., 2009).

179
Leur phénotype est le suivant :
- chez la souris : CD3- ; CD4- ; CD25-/+après activation ; CD16 (FCγRIII)-/+ ; CD 90- ;
CD56+ ; cKit (CD117)- ; CD122+ ; IL-7Rα (CD127)-/+ ; Sca1 (Ly6A) + ; ICOSlow ;
NKp46 (NCR1)+ ; IL-12Rβ2+ ;
- chez l’homme : CD3- ; CD4- ; CD25-/+après activation ; CD56+ ; cKit (CD117)- ; CD
16 (FCγRIII)-/+ ; IL-7Rα (CD127)-/+ ; CD161-/+ ; NKp44 (NCR2)-/+ ; ICOSlow ;
NKp46 (NCR1)+ ; IL-12Rβ2+ ; Les deux marqueurs en gras suffisent à définir
un NK. Le NKp44 n’est pas exprimé dans les cellules NK périphériques
sanguines mais est exprimé après leur activation. Le NKp46 et CD56 sont
aussi exprimés par des cellules ILCs 3 (Campbell & Hasegawa, 2013; Korbel,
Finney, & Riley, 2004; Paul, 2013; Sanos et al., 2011; Hergen Spits et al.,
2013).
On peut également séparer les cellules NK en plusieurs sous-groupes :
Les CD56bright CD16+ qui sont des cellules potentiellement cytotoxiques. Ce
sont les plus matures et les plus fréquentes. Elles peuvent libérer rapidement
de l’IFN-γ et d’autres cytokines de type I. Elles sont recrutées sur les tissus
enflammés puisqu’elles expriment les récepteurs CXCR-1 et CX3CR1, pour
des chimiokines produites lors d’inflammation ;
Les CD56bright CD16-, en plus petit nombre (entre 5 et 15% des NK sanguins),
sont peu cytolytiques mais grands producteurs d’IFN-γ. En fait, leur production
d’IFN-γ et de TNF dure dans le temps, surtout après stimulation par l’IL-2 ou
l’IL-15. Ce sont eux qui prolifèrent lors d’un contact avec les cellules
dendritiques. C’est le seul type de NK qui peut atteindre les nœuds
lymphatiques puisqu’ils peuvent facilement quitter les vaisseaux et ils
expriment les récepteurs aux chimiokines nécessaires, c’est-à-dire le CCR7,
le CXCR-3 et le CD62L (Campbell & Hasegawa, 2013; Marcenaro, Dondero,
& Moretta, 2006; Montaldo et al., 2014; Sanos et al., 2011).
b. Distribution
Les NK expriment essentiellement leur fonction dans les tissus et les organes
lymphoïdes secondaires où ils migrent durant l’inflammation en réponse à des
facteurs chimiotactiques. Les fonctions des NK sont grandement influencées par le
microenvironnement (contact cellule-cellule ou via des facteurs solubles). Les
interactions entre les cellules NK et les autres cellules immunitaires déterminent
l’issue finale de l’immunité innée et adaptative.
Les NK spléniques dégranulent plus que leurs homologues pulmonaires. Dans des
conditions physiologiques, le poumon est continuellement exposé aux antigènes
contrairement à la rate. Le poumon a donc développé des stratégies pour prévenir
l’inflammation locale avec, notamment, une réaction moindre des NK pulmonaires.

180
De plus, les NK spléniques expriment plus d’IL-13 et d’IFN-γ que les NK
pulmonaires. Donc la distribution des cellules NK semble conditionner leur rôle dans
les réponses immunes et cette différence pourrait être due à l’environnement
différent. Les NK mis en contact des macrophages montrent une cytotoxicité
augmentée et cet effet est plus marqué avec les NK spléniques, ce qui confirme leur
cytotoxicité plus importante. Par contre, les macrophages ne modifient pas la
sécrétion d’IL-12, de TNF-α ou encore d’IFN-γ (Michel et al., 2012).
Dans le foie, les cellules NK s’accumulent et représentent entre 30 et 50% des
lymphocytes hépatiques. Cette localisation leur permet d’être en contact avec les
produits bactériens et les antigènes dérivés des intestins. Les NK hépatiques sont
phénotypiquement et fonctionnellement différents de leurs homologues sanguins. Ils
jouent des rôles essentiels dans la lutte contre les infections virales, les métastases
tumorales, le carcinome hépatocellulaire et la prévention de la fibrose hépatique.
Comme les organes lymphoïdes secondaires, le foie est enrichi en cellules NK
CD56bright (Björkström et al., 2013; Moroso et al., 2011).
Dans le poumon, les cellules NK ne représentent que 10 % des lymphocytes et nous
venons d’aborder le fait qu’ils sont moins toxiques que leurs homologues sanguins
ou spléniques car ils expriment le récepteur inhibiteur NKG2A et ont donc un seuil
d’activation plus élevé. De plus, les macrophages alvéolaires produisent des
prostaglandines et des cytokines immunosuppressives, ce qui diminue leur
cytotoxicité. Les cellules épithéliales pulmonaires produisent de l’IL-15, indispensable
à la survie des NK. Ce sont ces cellules qui produisent de l’IL-22 pendant l’infection
virale par l’influenza et cette cytokine stimule la régénération épithéliale. Les cellules
NK pulmonaires ont une capacité de prolifération limitée car elles expriment moins
de CD122 que les NK spléniques (Björkström et al., 2013; Michel et al., 2012).
Dans l’utérus, le nombre de cellules NK augmente dans la muqueuse et le placenta
après l’implantation. Pendant la gestation, ils régulent les trophoblastes envahissants
et remodèlent les artères utérines. Une condition particulière qui montre l’influence
du microenvironnement sur les NK est la gestation : pendant le 1er trimestre, les
cellules NK représentent entre 50 et 70% des lymphocytes des tissus déciduaux.
Ces dNK (d pour décidual) sont faiblement cytotoxiques et relâchent des cytokines
particulières, différentes des NK sanguins (Björkström et al., 2013; Montaldo et al.,
2014).
Dans la moelle osseuse, les NK sont considérés comme les précurseurs des
populations NK matures présents dans la rate, le sang, le foie et les poumons
(Michel et al., 2012).
Dans les plaques de Peyer, les cellules NK expriment le NKp44 chez les humains et
le NKp46 chez la souris, et ne libèrent pas de perforines, ni d’IFN-γ comme les
autres sous-types mais produisent de l’IL-22. Cette cytokine initie un signal
transducteur et activateur de transcription, le STAT3, qui induit la production de
molécules antimicrobiennes comme les β-défensines. Elle stimule ainsi les cellules

181
épithéliales pour sécréter des cytokines anti-inflammatoires comme l’IL-10, sur les
hépatocytes elle induit la synthèse des protéines de la phase aiguë comme
l’amyloïde A et la protéine de fixation du LPS (LBP). Les cellules NK des intestins
expriment l’AhR, le RORα et le facteur de régulation de l’interféron (IRF). Il
semblerait que les microbes intestinaux influencent le développement des cellules
NK. Une des hypothèses serait que la microflore intestinale génère des métabolites
arylhydrocarboniques qui activent l’Ahr. Les cellules NK répondent à l’IL-23. Cette
cytokine provoque, chez les Th17 leur expansion et leur maintien mais chez les NK
muqueux, l’IL-23 est suffisante pour produire de l’IL-22 (Colonna, 2009).
Les cellules NK sont donc principalement trouvées dans le foie, la rate, le sang, et la
moelle osseuse et sont en nombre limité dans les nœuds lymphatiques. Cependant
ces rares NK dans les nœuds lymphatiques jouent un rôle clé en interagissant avec
les cellules dendritiques pour promouvoir les réponses IFN-γ par les cellules T et
empêcher la propagation des virus (Campbell & Hasegawa, 2013; J. Yu et al., 2013).
La distribution de ces cellules NK peut néanmoins varier selon les conditions
physiologiques. Par exemple, lors de restriction calorique, les NK sont réduits en
fréquence et en nombre dans la plupart des tissus périphériques. La génération des
NK dans la moelle osseuse semble intacte mais le maintien des NK dans les tissus
périphériques est affecté : la restriction calorique semble toucher l’homéostasie des
NK matures. Seuls certains types sont touchés comme les NK matures classiques
alors que les CD127+ ne le sont pas, ce qui augmente la fréquence de ces derniers,
qui ont par ailleurs un faible potentiel cytotoxique mais peuvent cependant produire
de grandes quantités de cytokines (Clinthorne, Beli, Duriancik, & Gardner, 2013).
c. Développement des cellules NK
Les cellules NK sont formées dans la moelle osseuse, mais aussi dans le thymus. Le
facteur de transcription T-bet coopère avec l’éomésodermine pour leur différenciation
(Hergen Spits et al., 2013).
Le développement des NK compte 5 stades de développement repris dans la Figure
34, page 182. Ce développement débute par la cellule souche hématopoïétique puis
par le passage par le CLP (Common Lymphoid Progenitor), encore commun à tous
les autres lymphocytes. Ensuite, les 5 stades suivants s’enchainent :
Stade 1 : les pro-NK : CD34+ CD117- CD94- CD56- ;
Stade 2 : pré-NK : CD34+ CD117+ CD94- CD56- CD161-/+ ;
Stade 3 : iNK : CD34- CD117+ CD94- CD56-/+ CD161+ ;
Stade 4 : CD56bright NK : CD34- CD3- CD94+ CD56 bright CD16-/dim ;
Stade 5 : CD56dim NK : CD34- CD3- CD94+ CD56dim CD16+ ;
Les deux derniers stades constituent les stades matures et les cellules sont
complètement différenciées bien que leur plasticité puisse leur permettre de passer
de CD56bright à CD56dim (Montaldo et al., 2014; Moroso et al., 2011).

182
Figure 34 : Intermédiaires cellulaires du développement des NK murins (en haut) et des NK humains (dans l’encadré) (J. Yu et al., 2013).
Seules les phases précoces de développement comme la génération du stade 1
depuis les cellules souches ont lieu dans la moelle osseuse alors que la suite a lieu
dans des tissus lymphoïdes secondaires. Tous les stades ont été observés dans la
moelle osseuse, le sang périphérique, les nœuds lymphatiques et les amygdales
alors que seuls les trois derniers stades sont détectés dans l’utérus humain. Durant
la vie fœtale, le foie est le principal organe d’hématopoïèse mais très rapidement
après la naissance, il réduit drastiquement cette activité, reprise par la moelle
osseuse. Cependant, le foie adulte maintient son potentiel de reprendre
l’hématopoïèse. Le foie adulte humain contient des précurseurs des cellules NK qui
se développent en cellules NK mature CD56bright et CD56dim fonctionnels. Tous les
stades de différenciation sont détectables et les stades 2 et 3 peuvent générer des
cellules NK cytotoxiques. Ces stades de précurseurs précoces (NKP) sont recrutés
depuis le sang périphérique et se différencient ensuite dans le foie ou les cellules
acquièrent des caractéristiques spécifiques du foie. L’éducation des NK (pour la
tolérance du soi) est possible dans n’importe quel tissu tant que le
microenvironnement soutient cette éducation. L’IL-15 est nécessaire pour le

183
développement des cellules NK. Dans le foie, elle est produite par les cellules de
Kupffer et l’IL-7, qui provoque la prolifération des cellules NK. Les stades 1 et 2 sont
encore multipotents et l’engagement définitif vers la lignée NK a lieu au stade 3 du
développement. Les cellules CD56bright ont une plus faible expression de CD127 et
une complète absence de CD117. Certaines affections virales peuvent accélérer ou
ralentir la différenciation des NK comme le CMV qui provoque un développement
plus rapide (Montaldo et al., 2014; Moroso et al., 2011).
Pour le développement des NK, de nombreux facteurs de transcription sont
essentiels. Parmi eux :
l’Id2, nécessaire pour le développement de toutes les ILCs. Il permet la perte
du potentiel B et T ;
le T-bet et l’Eomes appartiennent à la famille des T-box. Le T-bet dirige le
passage du stade pré-NK au stade iNK et stabilise le phénotype immature.
L’Eomes permet l’expression des récepteurs et maintient le phénotype
mature. Il intervient plus tardivement Enfin, les deux interviennent dans le
choix du sous-type de NK ;
L’E4BP4 ; ou NFIL3 pour « Nuclear Factor Interleukin 3 », facteur de
transcription qui régule la production d’IL-3. Il semblerait qu’il module
l’expression de l’Id2 et d’Eomes et il intervient donc plus précocement, dès le
stade CLP ;
l’Ets-1 est un autre facteur de transcription qui régule l’expression de l’Id2 et il
intervient également dans l’expression de récepteurs nécessaires au
fonctionnement des NK puisqu’il contrôle l’expression du CD122. Il intervient
entre les stades pré-NK et iNK. Il est également nécessaire à l’homéostasie
des NK matures. Le Tox (Thymocyte selection-associate Hight Mobility group
box) est important pour les NK mais aussi pour les LTi ;
Le BLIMP1 (pour B-Lymphocyte Induced Maturation Protein) interviendrait
dans la différenciation terminale des NK ;
Les IRF-1 et IRF-2 interviennent également puisque les NK sont des
producteurs d’IFN ;
Le GATA3 pourrait réguler l’expression du T-bet et d’Eomes (Hamerman,
Ogasawara, & Lanier, 2005; Rankin et al., 2013; Vosshenrich & Di Santo,
2013).
Le développement des cellules NK nécessite des interactions entre les cellules
progéniteurs NK et les cellules stromales de la moelle osseuse. Les précurseurs
expriment le CD122 (ou IL-15R) d’où la nécessité de l’IL-15 pour le développement.
D’autres cytokines comme l’IL-7, l’IL-21 (dans les derniers stades) et le SCF (Stem
Cell Factor) sont nécessaires. Ces cellules immatures expriment le NK1.1 et

184
acquièrent les récepteurs CD94-NKG2 et Ly49 avant la phase de prolifération.
Ensuite, la maturation finale est accompagnée par l’expression de CD11b et CD43
avec l’acquisition de la fonction cytolytique par les perforines ainsi que la production
d’IFN-γ. Les principales fonctions des NK matures (cytotoxicité et libération d’IFN-γ)
ne sont acquises qu’aux stades 4 et 5. Par contre les stades précoces ne libèrent
pas d’IFN-γ mais du TNF, du GM-CSF, de l’IL-22 et de l’IL-13 (Hamerman et al.,
2005; Montaldo et al., 2014).
L’IL-1β inhibe le développement des NK pour favoriser les ILCs 3. Enfin, l’IL-8 et le
MIP1α favorisent la mise en place de précurseurs de NK (Montaldo et al., 2014).
L’environnement des NK est très important pour moduler leur action : des NK
exposés peu de temps à l’IL-12 libèrent plus d’IFN-γ et de TNF-α et sont plus
cytolytiques. L’exposition à l’IL-4 a l’effet inverse. Enfin, seuls les NK exposés à l’IL-
12 peuvent induire la maturation des cellules dendritiques. Le microenvironnement
hépatique dirige préférentiellement les cellules vers le développement des NK
classiques alors que les MALT autorisent la production d’ILCs 3 et de NK
conventionnels (Moretta et al., 2005; J. Yu et al., 2013).
d. Mécanisme de la cytotoxicité des NK
La reconnaissance des cibles par les NK présente quelques particularités :
Les récepteurs sont portés par toutes les cellules NK (et non par un seul
clone) ;
Chaque cellule NK exprime des récepteurs activateurs et inhibiteurs pour la
reconnaissance des cibles ;
Les récepteurs sont souvent proches et peuvent avoir des spécificités de
ligands qui se recoupent ;
Les récepteurs NK fixent spécifiquement les molécules de CMH I mais sont
distincts fonctionnellement et structurellement des autres récepteurs qui fixent
le CMH I (TCR et CD8) (Paul, 2013).
Les NK disposent de plusieurs méthodes pour reconnaitre leurs cibles :
le « missing-self » : L’absence d’inhibition du NK a lieu lorsque les molécules
de CMH I sont absentes, ou sévèrement sous-exprimées (notamment lors
d’infection virale ou de transformation maligne). Cependant, les globules
rouges ou les cellules du tissu neural ne portent pas de CMH I et ne sont pas
lysées par les NK, ce qui montre l’importance de l’implication d’un récepteur
activateur. Il faut au moins deux récepteurs activateurs pour activer des
cellules NK au repos mais certains récepteurs activateurs sont suffisants seuls
lorsque la cellule NK a été activée préalablement par l’IL-2. Si un signal
inhibiteur (CMH I) est présent en même temps qu’un signal activateur, il ne se
passe rien : le signal inhibiteur est prioritaire

185
l’ « altered-self » : les NK peuvent être activés par les cellules qui expriment
un CMH de classe I normal si les récepteurs activateurs des NK sont
suffisamment engagés avec des néoantigènes induits par le stress, la
transformation tumorale ou encore l’infection (Campbell & Hasegawa, 2013;
Colonna, 2009; Korbel et al., 2004; Lodoen & Lanier, 2006; Moretta et al.,
2005; Paul, 2013).
Parmi les récepteurs inhibiteurs, un rôle important est joué par le « Killer
Immunoglobulin-like Receptor » (KIR) chez les humains et le Ly49 chez les souris.
Les autres récepteurs inhibiteurs sont le Ly49A, le CD94/NKG2. Des récepteurs
activateurs sont également présents comme le FcγRIIIA, NKG2D, et les « Natural
Cytotoxicity Receptors » (NCR) NKp30, NKp44, et NKp46. Le premier peut activer
une cytotoxicité dépendante des anticorps alors que les autres stimulent plutôt les
réponses contre les cellules (Korbel et al., 2004; Montaldo et al., 2014; Paul, 2013).
La façon de tuer ressemble à celles des cellules T CD8+ cytotoxiques alors que la
synthèse de cytokines est proche de celle des cellules T helper CD4+.
Plusieurs moyens cytotoxiques sont à disposition des NK pour tuer une cible :
Les cellules NK élaborent une synapse immunologique avec la cible et
libèrent des lysosomes cytotoxiques préformés, contentant des perforines ou
des granzymes ;
Les cellules NK induisent l’apoptose des cellules cibles grâce au ligand FAS ;
L’ADCC (Antibody Dependent Cell-mediated Cytotoxicity) est une lyse qui a
lieu après reconnaissance d’anticorps fixés sur la cellule cible.
L’amplitude de cette réponse des cellules NK contre les cellules malignes ou
infectées dépend de signaux d’alarmes relargués par les cellules immunes
sentinelles comme les cellules dendritiques ou les macrophages. Ces signaux
comprennent les interférons de type I. (IFN-α et IFN-β), l’IL-12, l’IL-18 et l’IL-15
(Colonna, 2009; Paul, 2013).
La plupart du temps, les tumeurs sont résistantes aux NK non activés du sang et
pour les tuer les NK doivent être exposés à des cytokines comme l’IL-2, l’IL-12, l’IL-
15 ou l’IFN-α qui augmentent leur cytotoxicité et qui sont relâchées par d’autres
types cellulaires. Un autre mécanisme qui active les NK est le TLR. L’IL-21, quant-à-
elle, stimule la cytotoxicité des NK et la production d’IFN-γ mais n’est pas impliquée
dans leur survie (Hamerman et al., 2005; Moretta et al., 2005).
e. La mémoire des NK
Comme ce qui a été vu dans le paragraphe « Une nouveauté : la mémoire innée »,
page 130, les NK, bien que définies comme cellules immunes innées sont capable
d’élaborer une réponse mémoire-like, plus caractéristique du système adaptatif. En
effet, sous certaines conditions, les NK peuvent donner une population qui peut
survivre et s’autorenouveler pendant plus de 6 mois et avec la capacité de monter

186
une réponse de rappel robuste et protectrice. Trois types de réponses mémoire ont
été mis en évidence par les cellules NK :
Certaines infections virales provoquent la multiplication des sous-populations
de NK exprimant des récepteurs « spécifiques » de ces virus. Ces cellules
peuvent survivre 70 jours et, après un challenge avec le même antigène viral,
elles peuvent proliférer de façon importante, d’une manière IL-12 dépendante.
Elles présentent alors une réponse cytotoxique et une production de cytokines
augmentées ce qui fournit une réponse accrue contre le virus ;
Des NK pré-stimulées par des cytokines (IL-12, IL-18 et IL-15) survivent au
moins plusieurs semaines ;
Les cellules NK dérivées du foie peuvent générer une réponse mémoire
spécifique d’un antigène. Ces cellules NK mémoires peuvent exprimer le
CXCR-6 et ce récepteur permet de moduler leur activité (Campbell &
Hasegawa, 2013; Moroso et al., 2011; J. Yu et al., 2013).
Les ILCs 1 2)
L’autre membre du 1er groupe est appelé ILC 1. Ces cellules ne possèdent aucun
marqueur cellulaire de surface l’associant à une lignée. Elles sont très proches des
ILCs 3 qui seront abordées page 192. Les scientifiques se demandent encore si les
ILCs 1 sont issues des ILCs 3 en perdant l’expression du RORγt (Hergen Spits et al.,
2013).
Les ILCs 1 expriment le T-bet et légèrement le RORγt (Björkström et al., 2013).
Selon les auteurs, le phénotype des ILCs 1 est différent. Il semblerait en fait qu’il y ait
plusieurs ILCs 1 :
Plusieurs auteurs mentionnent des cellules CD56- ; NKp44- et NKp46- (Fuchs
et al., 2013; Hergen Spits et al., 2013).
D’autres auteurs évoquent des cellules CD56+, NKp44+, NKp46+ qui expriment
le CD160. Elles nécessitent l’E4BP4 et le T-bet mais sont bien distinctes des
NK conventionnels (Fuchs et al., 2013; Vijay Kumar, 2013; J. A. Walker et al.,
2013).
Tous s’accordent à dire que les ILCs 1 ne sont pas cytotoxiques et qu’elles
produisent de l’IFN-γ après stimulation par de l’IL-12 et de l’IL-18 et qu’elles ne
dépendent pas de l’IL-15Rα pour leur développement, contrairement aux NK. Ces
cellules s’accumulent lors d’inflammation intestinale, ce qui laisse à supposer
qu’elles requièrent une colonisation bactérienne et des signaux inflammatoires pour
leur développement (Björkström et al., 2013; Fuchs et al., 2013; Hergen Spits et al.,
2013).

187
Le groupe 2 des ILCs B-
Les ILCs 2 ont été découvertes en 2011 comme des cellules non B non T qui
répondent à l’IL-25 en produisant des cytokines de type 2 comme l’IL-5 et l’IL-13,
notamment lors d’infestation par des helminthes. Elles produisent donc de l’IL-5, de
l’IL-9, et de l’IL-13 mais aussi de l’IL-6, de l’IL-10, du GM-CSF et un peu d’IL-4. En
fait, trois groupes différents ont été trouvés chez la souris. Pour l’instant, chez
l’homme un seul type d’ILC 2 a été découvert (Montaldo et al., 2014; Rankin et al.,
2013; J. a Walker & McKenzie, 2013; S. Yu et al., 2014).
Les ILCs 2 sont présentes dans le tractus gastro-intestinal, les FALC (Fat Associated
Lymphoid Cluster) localisés le long des vaisseaux sanguins dans le mésentère, les
nœuds lymphatiques, la rate, le foie, la moelle osseuse et les poumons. Elles
représentent moins de 0,2% des cellules de chaque tissu mais peuvent
considérablement augmenter après exposition à l’IL-25. Elles sont donc largement
répandues dans la muqueuse et se comportent comme des sentinelles innées dans
les tissus. Un de leurs rôles est de sécréter de larges quantités d’IL-13 et d’IL-5
lorsqu’elles sont stimulées par l’IL-33 et l’IL-25 produites par l’épithélium en réponse
à des dommages par des protéases ou des virus. Ces cellules régulent
l’homéostasie des PNE et leur cycle circadien en réponse à la prise de nutriments et
la régénération des muscles squelettiques après une blessure. Elles jouent un rôle
essentiel dans les réponses antihelminthiques et l’inflammation allergique des
poumons où elles stimulent une réponse Th2. Elles présentent des récepteurs aux
chimiokines comme le CXCR-6, le CXCR-4 et le CCR-9 qui sont impliquées dans la
distribution homéostasique des cellules lymphoïdes (Montaldo et al., 2014; Neill et
al., 2010; Vercelli, Gozdz, & von Mutius, 2013; J. A. Walker et al., 2013).
Les ILCs 2 résidantes dans le poumon restaurent l’intégrité épithéliale et la fonction
pulmonaire en produisant de l’amphiréguline, un régulateur de guérison des
blessures. L’infection des voies respiratoires par le H3N1 induit une hyperréactivité en
stimulant les macrophages alvéolaires qui produisent alors de l’IL-33 et donc activent
les ILCs 2 (Xue et al., 2013).
Ce qu’il faut retenir :
Tous les membres du groupe des ILCs 1 sont donc des producteurs d’IFN-γ qui
répondent à l’IL-12. Ils nécessitent l’E4BP4 et le T-bet pour leur développement.
De nombreuses recherches restent à faire au sujet des ILCs 1.
Seuls les NK sont également cytotoxiques, ce qui est nécessaire à leurs rôles
dans la lutte antivirale ou la surveillance des tumeurs, connus depuis
longtemps.

188
Là encore, les avis divergent selon les auteurs. Quatre types de cellules
correspondant aux critères ci-dessus ont été découverts en même temps : les natural
helper, les nuocytes (car « Nu » est la 13ième lettre de l’alphabet grec et ces cellules
sont les producteurs majeurs d’IL-13.), les innate helper et le progéniteur multipotent
(MPP). Les premières publications soulignent le caractère hétérogène de ces
groupes cellulaires mais depuis 2013, tous les articles s’accordent à dire qu’il s’agit
d’un seul type cellulaire, avec de légères différences de phénotypes selon la
localisation tissulaire. Nous utiliserons donc le terme d’ILCs 2 pour décrire les
caractéristiques de l’ensemble de ces cellules puis nous résumerons brièvement les
différences entre les 4 types cellulaires.
Voici ci-dessous, Figure 35, des natural helper. Les ILCs 2 sont caractérisées par :
- l’absence de marqueurs des lignées conventionnelles (lignées B, T,
myéloïde et érythroïde),
- l’expression de Sca1, cKit (CD117), IL-7Rα (CD127), Thy-1 (CD90), le CD25
et les récepteurs à l’IL-25 (IL-17BR), et à l’IL-33 (T1/ST2),
- la réponse à l’IL-25, l’IL-33 et à la lymphopoiétine stromale thymique (TSLP
pour (Thymic Stromal Lymphopoetin) en synthétisant des cytokines de type 2
(IL-4, IL-5, IL-9 et IL-13),
- leur dépendance au GATA3 et au RORα pour leur développement et leur
fonction (Guo et al., 2012; Koyasu & Moro, 2012; B. W. S. Li & Hendriks,
2013; Mjösberg & Spits, 2012; Neill & McKenzie, 2011; Pearson et al., 2012;
Hergen Spits et al., 2013; J. a Walker & McKenzie, 2013).
Figure 35 : Natural helper à la coloration de Giemsa (Koyasu & Moro, 2011).
La barre indique 20 µm
Le KLRG1, semble être un marqueur raisonnablement sélectif pour les ILCs 2. En
fait, lors de leur maturation, les ILCs 2 acquièrent l’expression du KLRG1 et perdent
celle de l’intégrine α4β7. Ce KLRG1 est un immunorécepteur inhibiteur qui se lie aux

189
E-cadhérines, hautement exprimées par les cellules épithéliales intestinales (Hoyler,
Klose, et al., 2013).
Les LSIG (Lin- Sca1high Id2high GATA3high) sont les progéniteurs spécifiques de la
lignée des ILCs 2. Elles se différencient en ILCs 2 grâce à la surexpression du
KLRG1 et l’augmentation de la sécrétion d’IL-5 et d’IL-13. Ces cellules ont été
précédemment identifiées comme LSK- (Lin- Sca1+ Kit-), précurseurs énigmatiques
de la moelle osseuse. Le groupe des ILCs 2 nécessite de nombreux facteurs et
cytokines pour leur développement comme l’ICOS (Inducible T Cell CO-Stimulator),
Tcf1 (T cell lineage factor 1, qui est impliqué dans la lignée T) ainsi que l’Id2, le
Notch signal, l’IL-7. Le GATA 3 intervient dans les dernières étapes de la
différenciation et dans l’acquisition des fonctions effectrices. Il régule aussi
l’homéostasie des ILCs 2 matures, et leur production de cytokines d’une façon dose-
dépendante. Le ROR-α est nécessaire pour les dernières étapes de la différenciation
(après l’Id2). Enfin, des cytokines interviennent également : l’IL-7, comme pour toute
ILC, mais aussi l’IL-33 (Hoyler, Klose, et al., 2013; Vijay Kumar, 2013; B. W. S. Li &
Hendriks, 2013; Mjösberg & Spits, 2012; Montaldo et al., 2014; Rankin et al., 2013;
Hergen Spits et al., 2013; Wong et al., 2012).
L’IL-9 produit par les ILCs 2 agit de manière paracrine et surexprime la production
d’IL-5 et d’IL-13 par ces cellules. Bien qu’il soit admis que les ILCs 2 produisent de
l’IL-9, aucune source n’évoque cette production chez l’un des quatre types cellulaires
décrits ci-dessus. La production d’IL-9 ne dépend pas de l’IL-33 comme pour les
autres cytokines produites par les ILCs 2 mais dépend de l’IL-2. La production des
cytokines de type 2 affecte le changement de classe d’anticorps, le recrutement de
cellules effectrices inflammatoires et l’hyperplasie des cellules de Goblet ce qui
conduit à une production de mucus augmentée (Vijay Kumar, 2013; Wilhelm, Hirota,
Stieglitz, Snick, & Tolaini, 2012; Xue et al., 2013).
Le MPP peut se différencier en mastocyte, basophile ou macrophage en présence
de SCF (stem cell factor) et d’IL-3. Ces cellules ont des capacités de présentation
d’antigènes et stimulent la prolifération des cellules T. Parmi les quatre types d’ILCs
2, les MPP sont les moins semblables puisqu’elles n’expriment pas l’IL-7R et pas ou
presque pas le T1/ST2. Leur caractère pluripotent fait qu’elles ne sont pas toujours
considérées comme ILCs 2. Les MPP ne sont d’ailleurs pas présentées par Spits
dans ses ILCs 2. Elles sont cependant indiquées dans le Tableau VII, page 190, car
elles sont souvent présentées avec les autres ILCs 2 par les auteurs. Le MPP diffère,
de façon évidente, des trois autres types cellulaires (IL-7Rα, CD69, IL-33R)
(Maclachlan et al., 2013; Neill & McKenzie, 2011).

190
Tableau VII : Principales caractéristiques des Natural Helper, Nuocytes, Innate Helper et MPP. Synthèse d’après (Koyasu & Moro, 2011; Nakae et al., 2013; Neill & McKenzie, 2011; Pishdadian, Varasteh, & Sankian, 2012; Hergen Spits et al., 2013).
Marqueur de
surface
Natural
helper Nuocyte Innate helper MPP
Lineage - - - -
IL-7Rα (CD127) + + + -
IL-25R / + + +
IL-33R
(T1/ST2) + +/- + -/low
CKit (CD117) + +/- + / - / Low +
Sca-1(LY6A) + + - +
CD25 (IL-2Rα) + - + /
CD45 + + + +
Thy-1 (CD90) + + + /
CD34 - - / -
CD44 + + + /
CD69 + / + -
CMH II - + /
+ si culture
avec IL-3 et
SCF
CD4 - - -
ICOS (CD278) / + + /
Localisation
Cluster
lymphoïde
mésentérique
Moelle
osseuse, rate,
nœuds
lymphatiques
mésentériques,
intestin, sang
Nœuds
lymphatiques
mésentériques,
rate, foie
GALT, plaque
de Peyer,
nœuds
lymphatiques
mésentériques,
cryptoplaques
Expression
GATA3 + + + -
Expression
RORα / + / /
Cytokines
produites
IL-4 IL-5 IL-6
IL-13 IL-2
IFN-γ
IL-4 IL-6 IL-5
IL-13 IL-10 IL-2 IL-4 IL-5 IL-13 /
Le moment précis de leur mise en place n’est pas encore connu, mais les ILCs 2
sont présentes dans le sang du cordon à la naissance. Le GATA3 est exprimé de
façon plus intense par ces ILCs 2 du cordon. Les garçons ont une proportion plus
élevée d’ILCs 2 dans le cordon que les filles nouveau-nées bien qu’aucune
différence ne soit détectable chez l’adulte. Pourtant les filles ont une réponse Th1
plus marquée que les hommes avec des marqueurs inflammatoires plus élevés et

191
une clairance de l’infection plus rapide. Ceci offre une meilleure protection contre les
infections mais augmente considérablement la susceptibilité aux auto-immunités
(Forsberg et al., 2014).
Les ILCs 2 sont sensibles aux dérivés de l’acide arachidonique :
- Les leucotriènes cystéines (CysLT) sont des médiateurs lipidiques dérivés de
l’acide arachidonique produits par les mastocytes, PNE et macrophages et
les cellules dendritiques. Le CysLT1R (Cysteinyl Leukotriene Receptor1) est
un récepteur de haute affinité pour le leucotriène D4 (LTD4), exprimé sur les
cellules structurelles comme les cellules musculaires lisses, ou des cellules
immunes comme les PNE, macrophages, et les cellules dendritiques. Le
CysLT1R est exprimé de façon constitutive par les ILCs du poumon et de la
moelle osseuse et reste exprimé par les ILCs 2 après un challenge.
L’activation de ce récepteur aboutit à une production intense d’IL-4, d’IL-5 et
d’IL-13 (Doherty et al., 2013).
- Les prostaglandines D2 (PGD2), principaux métabolites de l’acide
arachidonique. Elles sont libérées par les mastocytes lors de réponse
allergique et induisent la migration des ILCs 2 et la production de cytokines
Th2 comme l’IL-4, l’IL-5 et l’IL-13 et de nombreuses autres cytokines pro-
inflammatoires (IL-3, IL-8, IL-9, l’IL-21 et GM-CSF). Les effets pro-
inflammatoires des PGD2 sont initiés par le CRTH2, une protéine couplée au
récepteur pour les PGD2, également abondante dans les PNE, PNB, et
cellules Th2. L’activation du CRTH2 augmente fortement l’expression du
récepteur à l’IL-33. Or cette dernière est responsable de la migration de ces
ILCs 2 de façon dose-dépendante. Le CRTH2 est donc essentiel pour la
migration des ILCs 2 vers les PGD2 et est donc un récepteur
chimioattractant. A travers la surexpression des récepteurs à l’IL-33 et à l’IL-
25 et les interactions synergiques de ces récepteurs, le CRTH2 joue un rôle
pivot dans le lien entre l’immunité innée et adaptative dans les ILCs 2. Les
PGD2, l’IL-25 et l’IL-33 agissent de concert dans la réponse immune des
ILCs 2. Ces cellules sont enrichies dans les sites de l’inflammation après une
infection parasitaire ou une réaction allergique (Xue et al., 2013).
Ce qu’il faut retenir :
Les ILCs 2 répondent à l’IL-25 et l’IL-33 en libérant des cytokines de type 2
(IL-4, IL-5, IL-9 et IL-13). Elles nécessitent le GATA3 et le RORα pour leur
développement. Elles sont présentes dans les FALC et seraient impliquées
dans la lutte contre les parasites ainsi que dans les réactions allergiques.

192
Le groupe 3 des ILCs C-
Les groupe des ILCs 3 est défini par la production d’IL-17A et/ou d’IL-22. Le RORγt
est la caractéristique des ILCs 3 : il est fondamental pour leur développement et leur
maintien. Cependant, le RORγt dirige également les cellules T effectrices vers des
cellules Th17 produisant de l’IL-17 et de l’IL-22.
L’exemple type des ILCs 3 est les cellules LTi, cruciales pour le développement des
organes lymphoïdes secondaires pendant l’embryogénèse. Ces cellules ont été
découvertes en 1997 (Hergen Spits et al., 2013).
Plus récemment, un autre type d’ILC 3 a été découvert. Il s’agit des ILCs 3 qui
diffèrent des LTi par leur expression du NCR NKp46. Ces cellules produisent de l’IL-
22 mais pas d’IL-17A. Nous les appellerons NCR+ ILCs 3. Ces cellules n’expriment
pas le récepteur CCR-6 et émergent en période post natale sous le contrôle de
l’AHR (ArylHydrocarbon Receptor). Ces cellules sont indispensables pour la réponse
immunitaire contre certaines bactéries dans l’intestin (Hergen Spits et al., 2013).
Enfin, un autre type d’ILC 3 est responsable de colite et ce type cellulaire n’exprime
pas le NKp46 et sécrète de l’IL-17A en plus de l’IL-22. Elles expriment le CCR-6,
mais pas le T-bet et sont la principale population pendant la vie fœtale. Ces cellules
sont nommées NCR- ILCs 3 (Hergen Spits et al., 2013).
Le RORγt est nécessaire pour la différenciation des ILCs 3 mais pas pour les NK et
les cellules ayant exprimé le RORγt ont perdu leur potentiel NK. Cependant, il
semblerait que le stade III des NK soit hétérogène et contiendrait des ILCs 3 et des
NK immatures (Montaldo et al., 2014).
Comme les cellules Th17, les ILCs 3 dépendent du facteur de transcription RORγt
pour leur développement et leur fonction. De plus, leur développement nécessite
aussi de l’IL-7Rα comme les ILCs 2 (Hergen Spits et al., 2013).
Les cellules LTi 1)
Ces cellules, découvertes à la fin des années 1990 expriment des molécules
nécessaires au développement des tissus lymphoïdes comme le LTβ et jouent donc
un rôle crucial dans la mise en place des Plaques de Peyer et des nœuds
lymphatiques. Ce sont des agents majeurs de l’organogénèse lymphoïde pendant
l’embryogénèse et après la naissance, elles régulent l’architecture des tissus
lymphoïdes ainsi que le maintien de la mémoire des lymphocytes T. Ces cellules
produisent aussi de l’IL-17A et de l’IL-22, ce qui leur confère un rôle dans la
médiation de l’immunité contre les pathogènes entériques (Björkström et al., 2013; J.
A. Walker et al., 2013).
La demi-vie des LTi est de 3 semaines (M Cherrier & Eberl, 2012).

193
Leur phénotype est le suivant : Lin- RORγt
+ CD4
-/+ CD56
- CD127
+ CD117
+ NKp44
-
NKp46- IL-23R+ et IL-1R+ cKit+ CCR-6+ CCR-7+ et CXCR-5+ (Colonna, 2009; Koyasu
& Moro, 2012; Montaldo et al., 2014; Hergen Spits et al., 2013).
Les LTi, peuvent être divisées en CD4+ et CD4-. Les deux sous-types expriment la
lymphotoxine LTα1β2, qui provoque des changements au niveau des cellules
stromales pour recruter de plus amples LTi et des lymphocytes B et T. Les cellules
stromales LTβR+ produisent des cytokines comme le CXCL-1 et expriment des
molécules d’adhésion ICAM et VCAM. Les LTi sont beaucoup plus rares chez
l’adulte, ce qui reflète la moindre nécessité pour l’adulte de créer de nouvelles
structures lymphoïdes. Elles ne produisent pas d’IFN-γ mais de l’IL-22 en réponse à
l’IL-23 mais ce n’est pas un message suffisant : un message de co-stimulation est
nécessaire. Par exemple la stimulation de l’IL-23R et du TLR2 mène à une
production massive d’IL-22. L’IL-23 peut être produite par des monocytes activés,
des macrophages et des cellules dendritiques. La lymphotoxine joue un rôle
essentiel dans le contrôle de la production d’IL-22 par les ILCs RORγt+. Le signal qui
passe par le LTβR est critique dans la défense contre Citrobacter rodentium, en
contrôlant cette production d’IL-22. En fait, lors d’infection par C. rodentium,
l’expression de LTα1β2 dans les ILCs augmente, ce qui déclenche chez les cellules
LTβR, (principalement cellules dendritiques CD11c+) la production de cytokines d’IL-
23. Une cascade est alors initiée puisque cet IL-23 active les ILCs qui produisent des
molécules effectrices comme l’IL-22 (Cording et al., 2014; Cornelissen, Aparicio
Domingo, Reijmers, & Cupedo, 2011; Pearson et al., 2012; Sanos et al., 2011; H
Spits, 2011; Strober, 2010).
Après la naissance, les LTi sont présentes dans la rate, les nœuds lymphatiques et
les intestins (dans la lamina propria, notamment au niveau des cryptoplaques et des
plaques de Peyer) : ce sont d’importants producteurs d’IL-17A et d’IL-22 qui
préservent l’intégrité de l’épithélium et induisent la production de facteurs
cytotoxiques contre des envahisseurs potentiels. La lymphotoxine α1β2 est produite
par les ILC RORγt+ fœtales et est essentielle pour l’induction du développement des
nœuds lymphatiques et des plaques de Peyer mais elle est également impliquée
pour la clairance de Citrobacter rodentium, en recrutant des neutrophiles grâce à des
chimiokines sécrétées par l’épithélium activé (Björkström et al., 2013; Cornelissen et
al., 2011; Koyasu & Moro, 2012; Romera-Hernandez, Aparicio-Domingo, & Cupedo,
2013; Strober, 2010).
Chez l’adulte, les LTi CD4+ stimulent la maturation des tissus lymphoïdes tertiaires
après la naissance ainsi que la restauration des tissus lymphoïdes secondaires
après une infection virale. Il est intéressant de noter que le développement des LTi
adultes n’est pas sous la dépendance du Notch signal, contrairement à leurs
homologues fœtaux (Koyasu & Moro, 2012).
La différenciation des LTi est sous le contrôle des facteurs de transcription RORγt et
Id2 et, à une étape plus précoce, le Notch signal est indispensable pour produire les

194
précurseurs des LTi : les cellules α4β7+. Leur survie est assurée par l’IL-7 qui régule
aussi leur activité (M Cherrier & Eberl, 2012; Marie Cherrier, Sawa, & Eberl, 2012;
Koyasu & Moro, 2012).
Grâce à l’expression de CXCR-5, les LTi matures sont recrutés vers les bourgeons
de nœuds lymphatiques et des plaques de Peyer exprimant le CXCL-13. Le CXCR-5
stimule aussi l’intégrine α4β1, ce qui permet la fixation des LTi sur les VCAM1 des
plaques de Peyer et nœuds lymphatiques. Les LTi expriment le TRANCE et son
récepteur TRANCE-R, nécessaires pour le développement des nœuds lymphatiques
mais pas pour les plaques de Peyer. Les LTi peuvent donc être vues comme des
« bombes à cytokines » mobiles qui induisent une micro-inflammation dans
l’environnement stérile du fœtus, qui se développe ensuite en nœud lymphatique
(Vijay Kumar, 2013).
Les ILCs 3 NCR+ 2)
Elles sont également appelées cellules NK22, cellules NCR22, cellules NKR-LTi,
NCR+ ILC 3 et ILC 22. Elles expriment les marqueurs de surface NK mais ne
possèdent pas de granzymes ou de perforines et ne sont pas cytotoxiques. Ces
cellules sont CCR-6-. Elles dépendent de l’IL-7 mais pas de l’IL-15. Elles se trouvent
préférentiellement dans le colon et l’intestin grêle. Elles expriment le NKp46 chez les
souris et le NKp44 chez l’homme. La production d’IL-22 est régulée, via le récepteur
AHR par des composants alimentaires (Björkström et al., 2013; Diefenbach, 2013;
Pearson et al., 2012; Rankin et al., 2013)
Les ILCs 3 NCR+ produisent de l’IL-22 et de l’IFN-γ mais pas d’IL-17A ou d’IL-17F.
Elles sont localisées dans les organes lymphoïdes secondaires, les amygdales et la
lamina propria intestinale où elles contribuent à l’homéostasie épithéliale et aux
défenses de l’hôte contre les pathogènes extracellulaires. Elles expriment fortement
le T-bet. Elles n’apparaissent qu’après la naissance sous contrôle de l’AHR, un
facteur de transcription activé par des ligands endogènes ou environnementaux. La
microflore et l’IL-23 interviennent aussi dans leur développement. Il en existe
quelques-unes à la naissance mais leur nombre augmente considérablement dans
les 3-4 premières semaines de vie. Cette production d’IL-22 semble dépendre de la
présence de la flore commensale et elle engendre une surexpression de gènes
impliqués dans les défenses antimicrobiennes. Il s’agit de la source principale d’IFN-
γ en réponse à Salmonella enterica subsp enterica serovar Typhimurium et l’IFN-γ
stimule la libération de mucus par les cellules de Goblet. Elles peuvent en fait
sécréter un grand nombre de cytokines comme le TNF, le GM-CSF, l’IL-17A, l’IL-8,
l’IL-2 et le CCL20 mais elles ne sont pas cytotoxiques (Diefenbach, 2013; Hoyler,
Connor, Kiss, & Diefenbach, 2013; Vijay Kumar, 2013; Leavy, 2013; Montaldo et al.,
2014; Rankin et al., 2013; Sanos et al., 2011; J. A. Walker et al., 2013).
Elles présentent plusieurs stades de développement dans la lamina propria.
L’expression du T-bet est corrélée avec la baisse d’expression du cKit (gène de la

195
protéine Kit). Puis l’expression du NKp46 débute et enfin celle du RORγt. A chaque
étape l’expression du T-bet augmente, et donc, la production d’IFN-γ aussi. Ces
cellules produisent également de l’IL-22 mais pas d’IL-17. La portion d’ILCs 3 T-bet+
augmente avec l’âge (Hoyler, Connor, et al., 2013; Vijay Kumar, 2013).
Certains auteurs (Hergen Spits et al., 2013) affirment que les NCR+ expriment le cKit
mais d’autres (Diefenbach, 2013; Hoyler, Connor, et al., 2013) pensent qu’elles ne
l’expriment pas.
Elles sont impliquées dans le maintien de la fonction de barrière intestinale et
l’homéostasie immune. Elles semblent avoir besoin de la flore pour se multiplier mais
cependant, les changements dans la population après un traitement antibiotique ne
les affectent pas. Les études récentes ont montré que les ILCs 3 ne dépendent pas
de signaux dérivés des bactéries commensales mais que le phénotype, la structure
des populations, les capacités fonctionnelles de ces cellules changent pour
s’accommoder des altérations physiologiques. Les interactions avec la flore
commensale sont importantes pour l’expression du RORγt et la production d’IL-22.
Cependant il semblerait que la flore commensale diminue la quantité d’IL-22 produite
par les ILCs 3 RORγt+, peut-être par augmentation de la quantité d’IL-25 produite par
les cellules épithéliales qui à son tour diminue les productions d’IL-23 et d’IL-22. Les
interactions entre la flore commensale et les ILCs sont critiques dans la modulation
de la production d’IL-22 et dans le maintien de l’homéostasie intestinale. Des
dommages à l’intestin provoquent une extension de la population des ILCs 3 NCR+
et augmentent la production d’IL-22. Ceci initie l’inflammation intestinale qui dépasse
l’effet suppresseur anti-inflammatoire des bactéries commensales. L’IL-22 dérivée
des ILCs 3 NCR+ est le facteur clé dans la résolution de l’inflammation locale dans
l’intestin et prévient l’inflammation systémique. Les ILCs 3 expriment des molécules
de co-stimulation comme le CD30L et l’OX40L leur permettant ainsi d’interagir avec
les cellules T. En plus, elles contribuent au maintien de la mémoire des cellules T
CD4+ après une infection par Listeria monocytogenes (Tait Wojno & Artis, 2012).
La production d’IFN-γ est notamment importante pour l’immunité contre les
infections. En effet, il provoque la libération du mucus par les cellules de Goblet pour
protéger les cellules. Lors d’infection par Salmonella enterica, les ILCs 3 NKp46+
CCR-6- T-bet+ sont la source de cet IFN-γ et cette production est contrôlée par l’IL-12
(médiateur de la réponse Th1) et non par l’IL-23 (Hoyler, Connor, et al., 2013).
Le signal de l’IL-22 passe par la kinase janus 1 (Jak-1) et le STAT1, 3 et 5 (Signal
Transducer and Activator of Transcription). Le récepteur IL-22R n’est exprimé que
par des cellules non-hématopoïétiques : l’IL-22 est une cytokine produite par les
cellules immunes pour moduler la fonction des cellules épithéliales. La production
d’IL-22 par les LTi et les ILCs NCR+ dépend de la présence de la microflore
commensale et cet IL-22 est nécessaire pour atteindre l’état d’équilibre entre les
protéines antimicrobiennes produites par les cellules épithéliales et la flore
commensale Les cellules dendritiques modulent cette expression d’IL-22 par les

196
ILCs d’une façon positive et négative. Les cellules épithéliales détectent des
changements dans la colonisation par les microbes et sécrètent alors de l’IL-25 qui
se fixe sur l’IL-25R des cellules dendritiques. Ces dernières répriment alors les ILCs
RORγt+ qui produisent moins d’IL-22. En revanche, les cellules dendritiques peuvent
détecter les produits bactériens et produisent alors de l’IL-23 qui engage l’IL-23R des
ILCs RORγt+ ce qui stimule la production d’IL-22. L’IL-22 stimule la prolifération
épithéliale et maintient ainsi la seule couche de cellules épithéliales qui protège du
contenu intestinal riche en microbes (Romera-Hernandez et al., 2013; Sanos et al.,
2011).
Il semblerait que les ILCs 3 puissent donner des ILCs 1 en perdant l’expression du
RORγt en absence d’IL-7 (J. A. Walker et al., 2013).
Il est maintenant clair que les NKR+ RORγt+ sont les cellules issues d’un précurseur
NKR- RORγt+ et non pas des NK. Il s’agit d’une différenciation en deux étapes : les
RORγt+ acquièrent l’expression du NKR. Dans une étape secondaire, certains NKR+
RORγt+ diminuent l’expression du RORγt pendant que l’expression du NKR est
stable et deviennent des NKR+ « ex-RORγt ». La perte du RORγt est dépendante de
l’environnement. Alors que dans l’intestin grêle, seules quelques cellules diminuent le
RORγt, la perte du RORγt est plus complète dans le colon ou la rate. La perte de
l’expression du RORγt par les NKR+ RORγt+ illustre la plasticité dans la lignée des
ILC RORγt+. Les RORγt+ ILC et les NKR+ RORγt+ produisent des cytokines comme
l’IL-22 et l’IL-17. Ces deux cytokines fournissent une protection contre les infections
intestinales et assurent le maintien de l’homéostasie épithéliale intestinale durant
l’état d’équilibre. Par contre, les NKR+ « ex-RORγt » ne produisent pas d’IL-22 et
d’IL-23 mais sont des producteurs d’IFN-γ en réponse à l’IL-12 et l’IL-23 (Vonarbourg
& Diefenbach, 2012).
Les ILCs 3 NCR- 3)
Les ILCs 3 CCR-6+ sont Kithigh et T-bet-. Un sous-type exprime le CD4 et l’autre non.
Elles apparaissent pendant la vie fœtale sans nécessiter l’Ahr pour leur
développement. Ces cellules produisent de grandes quantités d’IL-22 et peuvent être
activées pour produire de l’IL-17A ou de l’IL-17F. Certains auteurs confirment qu’elle
peuvent produire de l’IFN-γ (Hergen Spits et al., 2013), d’autres pensent qu’elles
n’en sont pas capables (Diefenbach, 2013; Hoyler, Connor, et al., 2013).

197
Après avoir découvert les différents types d’ILCs, nous allons nous concentrer sur
leur développement, puisque les groupes d’ILCs sont déterminés selon le profil de
cytokines produites, mais aussi selon les facteurs de transcription nécessaires à leur
développement.
II. Le développement des ILCs
Les différents groupes d’ILCs partagent un grand nombre de caractères ce qui laisse
à penser à un ancêtre commun et des voies de développement proches. Plusieurs
schémas de différenciation des différentes ILCs ont été proposés mais, à ce jour,
aucun n’explique parfaitement la formation de toutes ces cellules à partir d’un
précurseur commun.
Un précurseur commun avec le système A-
immunitaire adaptatif : le CLP
Toutes les ILC dérivent d’un progéniteur lymphoïde commun qui se met en place
depuis des cellules souches hématopoïétiques du foie fœtal ou de la moelle
osseuse. Le Common Lymphoid Progenitor (CLP), dans la moelle osseuse, est le
précurseur des lignées lymphoïdes en incluant les cellules B, les cellules T et les
ILCs. Ces cellules sont caractérisées par leur absence de marquage conventionnel
(Lin-) et l’expression des récepteurs à l’IL-7, la FLT3 (FMS-related Tyrosine Kinase 3)
et le Kit. Ce précurseur est présent dans le foie fœtal (B. W. S. Li & Hendriks, 2013;
Mjösberg & Spits, 2012; Pearson et al., 2012; Rankin et al., 2013; J. A. Walker et al.,
2013).
Ce qu’il faut retenir :
Le groupe des ILCs 3 comprend des cellules qui dépendent du RORγt pour
leur développement :
- Les cellules LTi, intervenant dans la mise en place des tissus
lymphoïdes secondaires. Elles produisent de l’IL-22 en réponse à l’IL-
23,
- Les ILCs 3 NCR+, qui nécessitent l’Ahr pour leur développement. Elles
interviennent dans l’immunité intestinale contre les bactéries. Elles
produisent de l’IL-22. Elles sont T-bet+,
- Les ILCs 3 NCR-, sécrétant de l’IL-17 et de l’IL-22.

198
Du CLP au précurseur commun des ILCs : le B-
CILP
Les ILCs doivent tout d’abord perdre le potentiel B et T grâce à l’inhibiteur de
transcription Id2. Il permet d’inhiber la transcription de protéines E, nécessaires pour
la différenciation des cellules B et T. Le signal Notch intervient également pour
inhiber la voie B, notamment dans la lignée T, et la cellule obtenue est alors un
précurseur commun à toutes les ILC que l’on appelle le Common Innate Lymphocyte
Precursor (CILP) (Rankin et al., 2013; J. A. Walker et al., 2013).
Le précurseur des ILCs est caractérisé par une forte expression du facteur de
transcription Promyelocytic Leukaemia Zinc Finger (PLZF) et est présent dans le foie
fœtal et la moelle osseuse adulte. Les cellules B et T n’expriment pas le PLZF au
cours de leur développement : le stade qui l’exprime est donc ultérieur au CLP. Bien
que les ILCs matures ne l’expriment pas, les 3 groupes l’expriment durant leur
développement. Il semblerait que les LTi ne soient pas issues de ce précurseur
(Bordon, 2014; Cording et al., 2014).
Différenciation des différents groupes d’ILC C-
A partir du précurseur commun aux ILC, différents signaux vont orienter la
différenciation vers les trois groupes d’ILCs :
- Le E4BP4 (ou NFIL3) est nécessaire pour la formation des cellules NK et le
T-bet pour les ILCs 1. Le passage CLP-NKP implique une baisse
d’expression du CD135 puis l’expression du CD122 et enfin la suppression
de l’expression du CD127 ;
- Le GATA3 et le RORα sont nécessaires pour engager la voie de
différenciation des ILCs 2 et permettent d’obtenir un précurseur des ILCs 2,
le pré-ILC 2 qui est très similaire dans son phénotype aux ILCs 2 matures, si
ce n’est, le manque de cKit et de KLRG1. Ce précurseur est localisé dans la
moelle osseuse et seuls les ILCs 2 matures sont présents dans les tissus
périphériques (J. a Walker & McKenzie, 2013) ;
- La présence de RORγt oriente la différenciation vers les ILCs 3. Le
précurseur des LTi exprime l’intégrine α4β7 alors que celui des ILCs 3 ne
l’exprime pas. Ce précurseur α4β7+ qui donnera les LTi exprime ensuite le
RORγt (M Cherrier, Ohnmacht, Cording, & Eberl, 2012; Cording et al., 2014;
Vosshenrich & Di Santo, 2013; J. A. Walker et al., 2013).
A l’exception des cellules NK conventionnelles, toutes les ILC requièrent l’IL-7 pour
leur développement ainsi que le Notch signal (J. A. Walker et al., 2013).

199
Les facteurs de transcription spécifiques d’une lignée sont exprimés dans les Th et
les ILCs et sont donc utilisés comme marqueurs spécifiques. Cependant, une
plasticité des lignées a été observée dans les cellules T comme dans les ILC. Par
exemple un environnement inflammatoire induit chez les ILCs 3, la baisse
d’expression du RORγt et la surexpression du T-bet et de l’IFN-γ. Il reste cependant
à déterminer s’il s’agit d’un sous-type mature des ILCs 3 ou d’un précurseur RORγt+
immature qui est capable de donner un phénotype d’ILCs 1. Le développement des
ILCs 3 ne serait pas aussi linéaire que ce que l’on pensait et un précurseur
exprimant le PLZF peut donner naissance à des ILCs 1, ILCs 2 et ILCs 3 (Cording et
al., 2014).
L’impact de la microflore intestinale sur la génération des ILCs 3 reste controversé.
La génération des LTi dans les fœtus est totalement indépendante de la microflore,
mais la génération des ILCs 3 après la naissance peut être induite par certaines
bactéries. L’AHR semble être requis pour le maintien des sous-types d’ILCs 3 après
la naissance. Les ligands de l’AHR se retrouvent dans la nourriture grâce aux
légumes de la famille des crucifères comme les brocolis et les choux de Bruxelles.
Un autre nutriment nécessaire pour la génération d’ILCs 3 est la vitamine A (Cording
et al., 2014).
Le CCL20, une chimiokine produite par les cellules épithéliales contrôle la migration
des cellules exprimant le CCR-6, un récepteur important pour le homing des cellules
ILCs 3 qui migrent vers certains microenvironnements intestinaux (Qiu et al., 2012).
Les ILCs 2 matures expriment de hauts niveaux de récepteurs à la chimiokine CCR-
9 qui permet leur migration de la moelle osseuse à la lamina propria (Hoyler, Klose,
et al., 2013).
Les signaux nécessaires au développement de D-
chaque lignée
L’IL-7 1)
Contrairement aux NK, les ILCs 2 et ILCs 3 expriment le CD127 ou IL-7Rα et l’IL-7
est donc impliquée dans leur développement, leur différenciation, leur maintien et
leur fonction. L’IL-7 n’est pas impliquée dans le développement des NK (Vonarbourg
& Diefenbach, 2012).
L’IL-7 stabilise l’expression du RORγt dans les ILCs RORγt+. La microflore contrôle
l’expression de l’IL-7 par les cellules épithéliales intestinales. L’IL-7 est essentielle
pour le développement des cellules T et son expression dans le thymus n’est donc
pas surprenante. La microflore exacerbe l’expression de l’IL-7 dans les intestins.

200
L’IFN-γ stimule aussi la production d’IL-7. Le mécanisme par lequel l’IL-7 stabilise le
RORγt reste obscur (Vonarbourg & Diefenbach, 2012).
L’Id2 2)
Il s’agit d’un facteur de transcription doté d’une structure en hélice-tout-hélice qui
inhibe un autre facteur de transcription : les protéines E-box en formant un
hétérodimère avec ce dernier. Il est nécessaire au développement de toutes les
ILCs car il entraîne la perte du potentiel B et T (M Cherrier et al., 2012; Rankin et al.,
2013; Sanos et al., 2011).
L’Id2 n’est pas exprimé par les cellules souches hématopoïétiques, ni par le
progéniteur multipotent ou encore par le CLP. Il apparait au stade CILP (Common
Innate Lymphoid Progenitor) (Hoyler, Klose, et al., 2013).
Le RORγt 3)
Le RORγt ou RAR-Related Orphan Receptor γ est encodé par le gène RORC chez
les humains. Il s’agit d’un facteur de transcription de la famille des récepteurs
nucléaires. L’expression du RORγt est nécessaire pour le développement des LTi et
des ILCs 3 mais il est également indispensable pour le développement des Th17, les
NKT invariants (Marie Cherrier et al., 2012; Rankin et al., 2013; Sanos et al., 2011).
L’expression du RORγt est organe-spécifique et est influencée par des facteurs
environnementaux comme la flore commensale. En effet elle est plus marquée sur
les ILCs de l’intestin grêle que sur celles du colon, de la rate ou des nœuds
lymphatiques (J. a Walker & McKenzie, 2013).
Seuls les mammifères expriment le RORγt et ce sont les seuls à développer des
structures lymphoïdes très organisées comme les nœuds lymphatiques et les
plaques de Peyer (M Cherrier & Eberl, 2012).
Le GATA3 4)
Le GATA 3 est un facteur de transcription protéique qui possède une structure en
doigts de zinc. C’est un important régulateur du développement des cellules T et il
est aussi impliqué dans la différenciation des cellules épithéliales, la maturation des
ILCs 2 et le maintien des ILCs 2 matures. Il est exprimé très faiblement chez les
ILCs RORγt+ et les cellules NK : c’est pourquoi de forts niveaux d’expressions du
GATA 3 sont des caractéristiques des ILCs 2 qui l’expriment continuellement quel
que soit leur stade de développement ou d’activation. De plus, en absence de
GATA3, les ILCs 2 ne sont pas capables de migrer dans l’intestin (Hoyler, Klose, et
al., 2013).
Il stimule la production d’IL-13 par les ILCs 2 et la sécrétion d’IL-4, d’IL-5 et d’IL-13
par les cellules Th2 (J. A. Walker et al., 2013).

201
Le Notch signal 5)
Durant la lymphogénèse, le signal via le récepteur Notch modifie l’expression des
gènes et a des rôles cruciaux dans le développement des cellules T en inhibant la
différenciation des lymphocytes B. Il est également requis pour la formation des ILCs
3 et des ILCs 2 (M Cherrier et al., 2012; J. A. Walker et al., 2013).
Les protéines Notch sont des récepteurs transmembranaires capables de détacher
leur domaine intracellulaire qui peut alors se diriger vers le noyau et entraîner la
transcription de gènes. Le Notch 2 est impliqué dans le développement des cellules
RORγt+ (Rankin et al., 2013).
Le ROR-α 6)
Le RAR-related Orphan Receptor-α, aussi connu sous NR1F1 est un récepteur
nucléaire. Sa fixation à des hormones ou des cytokines surexprime plusieurs gènes.
Il est impliqué dans le développement des ILCs 2 mais on ne sait pas encore s’il est
restreint à cette lignée ou s’il est exprimé par d’autres cellules (Hoyler, Connor, et al.,
2013).
L’Aryl Hydrocarbon Receptor ou AHR 7)
Ce récepteur intervient dans la différenciation des cellules T régulateurs, les cellules
Th17 et les cellules γδ T qui sont dans l’épithélium intestinal. Il intervient aussi dans le
développement et la fonction des cellules LTi et ILCs 3 (M Cherrier et al., 2012; J. A.
Walker et al., 2013).
C’est un facteur de transcription hélice-tour-hélice dépendant d’un ligand. Il est
présent dans le cytoplasme de nombreuses cellules à l’intérieur de complexes
protéiques incluant la protéine de choc thermique 90 (HSP90). Différents ligands
sont connus comme le TCDD (tetrachlorobenzo-p-dioxine) ou le FICD (6-
formylindolo-[3,2-b]-carbazole, un photoproduit du tryptophane) ou des ligands
alimentaires notamment dans les végétaux de la famille des brassicacées. Ces
ligands rentrent dans la cellule et se lient à l’AHR : l’AHR subit alors une
translocation vers le noyau et se dimérise avec son partenaire l’Arnt (Aryl
Hydrocarbon receptor nuclear translocator). Ce complexe peut alors induire la
transcription d’un certain nombre de gènes (Rankin et al., 2013).
Il est très connu pour son effet sur les toxines environnementales comme les
dioxines. Alors que ces dernières stimulent les cellules T régulatrices, et donc
améliorent l’auto-immunité, les métabolites du tryptophane stimulent les fonctions
Th17 et donc aggravent les maladies auto-immunes (Vonarbourg & Diefenbach,
2012).

202
L’AHR intervient dans de nombreuses fonctions des ILCs 3. La mise en place des
structures lymphoïdes isolées après la naissance nécessite l’Ahr alors que pendant
l’embryogénèse la formation des plaques de Peyer n’en nécessite pas. La réponse
des ILCs 3 à l’IL-23, passe par l’Ahr et induit la production d’IL-22.
Les avis divergent concernant l’effet de la flore sur l’AHR.
Certains affirment que la flore microbienne n’est pas un ligand pour l’AHR,
contrairement à ce qui a longtemps été pensé. Ceci indique alors que la microflore
n’est pas un régulateur du développement des ILCs 3. Par contre, l’AHR intervient
bien dans la survie des ILCs 3 en augmentant l’expression de la protéine
antiapoptique Bcl-2 et de l’IL-7R (ce qui a pour conséquence là encore une
augmentation du Bcl-2) (Hergen Spits & Mjösberg, 2012).
D’autres affirment au contraire que la flore microbienne peut réguler la fonction de
l’AHR. Il est tentant de dire que des ligands endogènes générés par des bactéries
résidentes activent l’AHR et stimulent ainsi un développement minimum d’ILCs
RORγt+. Pour étoffer cette hypothèse, il a été montré que des métabolites du
tryptophane générés par des bactéries intestinales activent l’AHR au moins in vitro
(Qiu et al., 2012).
Le T-bet 8)
Le T-bet est un facteur de transcription de la famille très conservée des T-box,
impliqué dans les processus de développement. Il est connu pour être impliqué dans
la mise en place de l’immunité Th1, mais aussi pour le contrôle de la production de
l’IFN-γ.
Il est très exprimé par les cellules ILCs RORγt+ NKp46+ mais pas pour les ILCs
NKp46- RORγt+ (Rankin et al., 2013).
Ce qu’il faut retenir :
Les ILCs proviennent d’un précurseur commun avec les lymphocytes
conventionnels. La perte des potentiels B et T est très précoce et implique le
facteur de transcription Id2. De nombreux facteurs de transcription
interviennent ensuite dans les différents groupes, et le schéma de
différenciation n’est pas entièrement connu.
Le T-bet et l’E4BP4 sont nécessaires pour les ILCs 1.
Le GATA3 et le RORα sont indispensables pour obtenir des ILCs 2.
Le RORγt dirige la différenciation vers les ILCs 3.

203
III. Les différents rôles des ILCs
Nous avons vu que les ILCs sont disséminées dans l’organisme. Nous allons
maintenant étudier, le rôle de ces ILCs dans les principaux organes où elles se
trouvent, comme résumé très brièvement dans la Figure 36 ci-dessous.
Figure 36 : Fonctions spécifiques des tissus des ILCs (Björkström et al., 2013).
Les ILCs interviennent dans la régulation des A-
tissus lymphoïdes
Les ILCs sont des cellules innées mais elles agissent beaucoup sur le système
adaptatif. Les LTi interviennent lors de la mise en place des organes lymphoïdes
secondaires et tertiaires et sont responsables de la réparation de ces organes après
un épisode viral. Enfin les ILCs 3 interagissent avec les cellules B et T. Les ILCs sont
donc essentielles pour le système immun adaptatif.

204
Mise en place des organes lymphoïdes secondaires 1)
par les LTi durant l’embryogénèse
Les organes lymphoïdes secondaires comprennent les nœuds lymphatiques et les
plaques de Peyer. Leur mise en place est programmée génétiquement (Rankin et al.,
2013).
Le développement fœtal des organes lymphoïdes secondaires est initié par
l’interaction des LTi avec les cellules stromales du tissu lymphoïde. Ce processus
nécessite l’interaction entre l’intégrine α4β7 des LTi avec l’adressine MadCAM-1
exprimée par l’endothélium des veinules des bourgeons lymphoïdes.
L’acide rétinoïque est produit par les neurones et recrute les LTi à l’emplacement des
futurs nœuds lymphatiques. Le TRANCE, fournit par les bourgeons de nœuds
lymphatiques est requis pour la maturation finale des LTi et l’expression de la
lymphotoxine LTα1β2 par les LTi. Cette lymphotoxine active les cellules stromales via
le récepteur LTβR. Ces dernières expriment alors des molécules d’adhésion comme
l’ICAM-1 et le VCAM1 ainsi que les chimiokines structurales comme le CCL-21, le
CCL-19 (attirant les cellules T) et le CXCL-13 (attirant les cellules B) comme indiqués
sur la Figure 37, page 205. Ces facteurs sont cruciaux pour le développement des
tissus lymphoïdes (Marie Cherrier et al., 2012; Hepworth & Sonnenberg, 2014;
Rankin et al., 2013; Sonnenberg, Monticelli, Elloso, Fouser, & Artis, 2011; Upadhyay
& Fu, 2014).
Pour la formation des plaques de Peyer, ce n’est pas le TRANCE qui intervient pour
l’expression de la lymphotoxine mais l’IL-7 (M Cherrier & Eberl, 2012; Marie Cherrier
et al., 2012).
Il est à noter que les LTi ne sont pas responsables de la mise en place des FALC
puisqu’ils sont présents chez les souris déficientes en RORγt donc en LTi (Koyasu &
Moro, 2012).

205
Figure 37 : Mise en place des organes lymphoïdes secondaires par les LTi (Neyt, Perros, GeurtsvanKessel, Hammad, & Lambrecht, 2012).
Mise en place des organes lymphoïdes tertiaires par 2)
les LTi chez l’adulte
Les organes lymphoïdes tertiaires se développent en réponse à l’environnement
notamment à la microflore. Les follicules lymphoïdes isolés et les cryptoplaques sont
des organes tertiaires (Rankin et al., 2013).
Contrairement aux plaques de Peyer et des nœuds lymphatiques qui sont finalisés à
la naissance, de nombreux clusters intestinaux dans la lamina propria de l’intestin et
du colon qui se développent dans la première semaine de vie. Après la naissance,
les LTi stimulent la maturation des tissus lymphoïdes tertiaires (Sonnenberg et al.,
2012; Vonarbourg & Diefenbach, 2012).
Les récepteurs aux chimiokines CXCR-5 et CXCL-13 des LTi sont impliqués dans la
formation des tissus lymphoïdes tertiaires (Vijay Kumar, 2013).

206
L’IL-7 est aussi impliquée dans le développement des clusters lymphoïdes après la
naissance. Ces clusters se développent à proximité des cryptes intestinales qui
hébergent des cellules souches épithéliales et des cellules de Paneth. Le
recrutement des ILCs RORγt+ vers la lamina propria est la première étape et elles
s’accumulent dans les cryptes intestinales. Il semblerait que ces cellules soient déjà
là à la naissance. Ces cellules se multiplient vers les 10-14ième jours après la
naissance. Le signal qui initie cette seconde étape est inconnu mais implique
l’expression de LTα1β2 par les LTi et son interaction avec les cellules stromales qui
expriment alors l’ICAM1 et le VCAM1. Enfin, les cellules B sont recrutées dans
certains cryptoplaques grâce notamment à la microflore. Ils sont alors appelés
follicules lymphoïdes isolés. Les chimiokines impliquées pour le recrutement des
lymphocytes sont les mêmes que pour la mise en place des organes lymphoïdes
secondaires. Ces différents médiateurs sont schématisés dans la Figure 38 ci-
dessous (Hepworth & Sonnenberg, 2014; Vonarbourg & Diefenbach, 2012).
Figure 38 : Mécanisme de mise en place des tissus lymphoïdes tertiaires par les LTi (Neyt et al., 2012).

207
Restauration des tissus lymphoïdes après un épisode 3)
viral par les LTi
Après un épisode viral, la destruction des nombreuses cellules infectées présentes
dans les organes lymphoïdes par les lymphocytes T CD8+ peuvent endommager
fortement la structure de ces tissus lymphoïdes. Ceci survient au cours de certaines
infections virales comme avec le LCMV (virus de la chorioméningite lymphoïde) : il y
a rupture de l’intégrité des organes lymphoïdes notamment avec la perte des
cellules stromales des zones T. Une fois que le virus est éliminé, les LTi rétablissent
la séparation correcte des zones T et B, la réparation de la zone marginale, et la
formation de la trame de cellules réticulaires fibroblastiques, qui sont des cellules
multifonctionnelles puisqu’elles produisent des chimiokines, et génèrent des
structures de soutien pour les cellules T et cellules dendritiques en migration. Ces
cellules forment un système de conduits qui aident à distribuer les antigènes solubles
à travers tout le parenchyme des organes lymphoïdes secondaires. Les LTi jouent
donc un rôle essentiel dans la réparation des dommages tissulaires observés suite à
une infection aiguë par le LCMV (Hepworth & Sonnenberg, 2014; Vijay Kumar,
2013).
Les ILCs 3 stimulent l’immunité acquise 4)
Les ILCs modifient la réponse adaptative B et T grâce à des interactions cellules-
cellules. Les ILCs 3 NCR+ chez les humains coproduisent le facteur activant les
cellules B (BAFF), un régulateur positif des fonctions des cellules B (à gauche sur la
Figure 39, page 208). Ceci suggère que les ILCs localisées dans les tissus
lymphoïdes peuvent directement stimuler les réponses B (Hepworth & Sonnenberg,
2014; Pearson et al., 2012).
Bien que les ILCs 3 RORγt+ CMH II+ soient capables de prélever efficacement, de
traiter et de présenter des antigènes exogènes in vitro elles sont incapables d’induire
la prolifération T CD4+ : elles peuvent inhiber plutôt que de stimuler les cellules T.
Elles régulent l’homéostasie des cellules T CD4+ dans les intestins à travers
l’expression de CMH II (à droite sur la Figure 39, page 208) (Hepworth &
Sonnenberg, 2014; Pearson et al., 2012).
L’expression de l’hétérotrimère LTα1β2 par les ILCs 3 est nécessaire pour la
production d’IgA indépendante des cellules T via la modulation de la fonction des
cellules dendritiques. Ensuite la sécrétion de trimères de LTα3 par les ILCs 3 est
requise pour la production d’IgA par les cellules B dépendante des cellules T de la
lamina propria grâce à la régulation de la migration des cellules T vers les intestins
(Hepworth & Sonnenberg, 2014).
Les ILCs 3 interagissent avec les cellules mémoires CD4+ dans les nœuds
lymphatiques par l’intermédiaire de leur expression de haut niveau d’OX40L. Elles

208
augmentent ainsi la durée de vie des cellules mémoires (en haut sur la Figure 39, ci-
dessous (Pearson et al., 2012; Romera-Hernandez et al., 2013).
Figure 39 : Influence des ILCs 3 sur le système immunitaire adaptatif (Hepworth & Sonnenberg, 2014).
Les ILCs dans le tube digestif B-
Le long du tractus digestif, toutes les ILCs coopèrent. En effet, les ILCs 2
interviennent pour l’expulsion des vers intestinaux, les ILCs 3 contrôlent la flore
commensale et combattent la flore pathogène et les ILCs 1 et ILCs 3 combattent les
infections bactériennes comme le montre la Figure 40, page 209.
Ce qu’il faut retenir :
Le groupe des ILCs 3 est donc en lien étroit avec l’immunité adaptative :
- Les LTi permettent la mise en place des organes lymphoïdes
secondaires (nœuds lymphatiques et plaques de Peyer) et tertiaires
(cryptoplaques et follicules lymphoïdes isolés)
- Les ILCs 3 régulent les lymphocytes conventionnels et notamment les
lymphocytes mémoires, cependant ce ne sont pas des cellules
présentatrices d’antigènes.

209
Figure 40 : Rôles des ILCs dans l'homéostasie et la défense intestinales (M Cherrier et al., 2012) .
Les NK22 du schéma sont les ILCs 3 NCR+ et les ILCs 17 sont les NCR-.
Expulsion des vers intestinaux grâce aux ILCs 2 1)
L’expulsion efficace des vers helminthiques intestinaux passe par la libération d’une
grande quantité d’IL-13 qui stimule l’hyperplasie des cellules à mucus et la
contraction des muscles lisses. Les cytokines de type Th2 sont induites très
précocement après l’infection helminthique, avant même que les cellules Th2
spécifiques du pathogène ne soient activées. Bien que l’expulsion finale des vers
nécessite les Th2, une réponse immune précoce innée est importante pour
restreindre l’invasion des helminthes avant que la réponse immune adaptative ne
commence (Diefenbach, 2013; J. a Walker & McKenzie, 2013; J. A. Walker et al.,
2013).
En effet, lors d’infestation parasitaire, les cellules épithéliales synthétisent de l’IL-25,
de l’IL-33 et de la TSLP, lesquels induisent la prolifération des ILCs 2 qui expriment
alors de l’IL-5, de l’IL-6 et de l’IL-13 comme le montre la Figure 40, ci-dessus. Ces
cytokines orientent la réponse inflammatoire vers une réponse de type 2, en incluant

210
une hypersécrétion de mucus, la prolifération des lymphocytes B, l’activation des
éosinophiles et des macrophages. Les ILCs 2 sont indispensables au recrutement
des PNE au site de l’infection helminthique grâce à leur production d’IL-13, régulée
par le GATA3 (Diefenbach, 2013; J. a Walker & McKenzie, 2013; J. A. Walker et al.,
2013).
La TSLP produite par les cellules épithéliales est une cytokine qui induit les Th2. Elle
provoque la différenciation des cellules T CD4+ naïves en cellules Th2 via les cellules
dendritiques. Ces dernières produisent des chimiokines qui attirent les PNN et PNE
(CXCL-8 et CCL-24 respectivement) ainsi que des chimiokines attirant les Th2
comme le CCL-17 et le CCL-22 (Koyasu & Moro, 2011).
L’IL-33 induit également la production d’IL-5 et d’IL-13 par les mastocytes. Les PNB
répondent à l’IL-33 en produisant de l’IL-4, de l’IL-5, de l’IL-6 et de l’IL-13. L’IL-33 est
donc bien impliquée dans les réponses inflammatoires Th2 puisqu’elle induit la
production de cytokines Th2, l’hyperplasie des cellules de Goblet l’éosinophilie et la
production d’IgE (Koyasu & Moro, 2011).
Contrôle de la flore commensale intestinale par les 2)
ILCs 3
Chez les mammifères sains, les bactéries commensales se cantonnent à la lumière
intestinale, la surface épithéliale et les tissus lymphoïdes associées aux intestins
(GALT). L’IL-22 joue un rôle dans le contrôle de l’équilibre précaire entre le système
immunitaire intestinal et la flore commensale intestinale, l’inflammation et la
réparation des tissus lésés. Cette cytokine est produite par les cellules T CD4+ et par
les ILCs 3. Ces dernières jouent un rôle capital dans la restriction des bactéries
commensales et la prévention de l’inflammation systémique. Elles jouent ainsi un
rôle critique dans le maintien de l’homéostasie des muqueuses grâce à des signaux
échangés avec la couche épithéliale et avec les bactéries intestinales. Les cellules
épithéliales régulent les ILCs 3 en réponse aux signaux bactériens. Par exemple,
elles produisent de l’IL-25 qui supprime la production d’IL-23 par les macrophages et
diminue donc la production d’IL-22 par les ILCs 3. En contraste, les bactéries
commensales stimulent la production d’IL-7 par les cellules épithéliales, ce qui
augmente la production d’IL-22 par les ILCs 3 (Peterson & Artis, 2014; Rankin et al.,
2013; Sonnenberg et al., 2012).
La présence d’ILCs sécrétant de l’IL-22 est indépendante de la présence des
bactéries commensales mais les ILCs produisent une grande quantité d’IL-22 en
réponse à l’IL-23. L’IL-22 produite stimule la synthèse d’IL-25 par les cellules
épithéliales qui régule alors les cellules dendritiques (Sonnenberg et al., 2012; J. A.
Walker et al., 2013).
Cette homéostasie immune intestinale est fragile et peut être altérée par des troubles
métaboliques comme une carence en vitamine A, par exemple. Cette carence est en

211
fait associée à des modifications des ILCs. Le nombre des ILCs 3 est réduit et leur
production d’IL-17 et d’IL-22 se retrouve elle aussi diminuée. Cette carence en
vitamine A prédispose donc aux infections bactériennes gastro-intestinales. Elle
entraîne également une augmentation du nombre d’ILCs 2 et donc de l’IL-4, de l’IL-5
et de l’IL-13. Cette hausse est peut être due à une réponse accrue à l’IL-7. En fait, le
déficit en nutriments n’est pas associé à une immunodépression globale mais peut
résulter dans l’activation spécifique d’une branche de l’immunité (Kugelberg, 2014).
Intervention dans l’immunité contre les bactéries 3)
intestinales : les ILCs 3
Même si l’organisme est tolérant envers sa flore commensale, il doit rester vigilant et
surveiller les bactéries pathogènes. L’IL-17, l’IL-22 et l’IFN-γ sont importants pour la
protection des surfaces muqueuses. Ces cytokines sont produites par le groupe des
ILCs 3 (LTi, ILCs 3 NCR+ et ILCs 3 NCR-) (Di Santo, 2010).
Les LTi expriment le RORγt et la voie de la lymphotoxine est utilisée pour initier les
défenses de l’hôte. L’IL-22 est notamment importante pour éliminer Citrobacter
rodentium, Klebsiella pneumoniae et cette production fait intervenir la lymphotoxine
qui se fixe sur le LTβR des cellules dendritiques, qui sont à proximité des LTi dans
les follicules lymphoïdes isolés, régulant ainsi leur production d’IL-23 et de l’IL-22 par
cascade. Les cellules dendritiques peuvent aussi détecter l’infection via la
signalisation d’un TLR puis du MyD88 qui provoque la sécrétion d’IL-23. Cet IL-23
est reconnu par l’IL-23R des LTi adultes qui produisent alors de l’IL-22. Le LTβR est
essentiel dans la formation de synapses immunologiques innées entre les LTi et les
cellules dendritiques (Björkström et al., 2013; Cording et al., 2014; Tumanov et al.,
2011; Upadhyay & Fu, 2014).
Dans des conditions normales, ce sont les ILCs 3 NCR+ qui produisent de l’IL-22
dans la lamina propria de l’intestin grêle (voir paragraphe précédent). Par contre, en
début d’infection, ce sont les LTi qui sont la source principale d’IL-22. Lors
d’infection, leur nombre augmente dans le colon et les nœuds lymphatiques
mésentériques. L’IL-22 est indispensable pour l’immunité dans les six premiers jours
de l’infection et pendant cette période les ILCs en sont la principale source. Il existe
d’autres sources d’IL-22 comme les cellules dendritiques, les cellules NK
conventionnelles, mais les LTi CD4+ en restent la principale source. Passés les
premiers jours, l’immunité adaptative prend le relais (Sonnenberg et al., 2011;
Tumanov et al., 2011).
L’IL-22 agit sur les cellules épithéliales qui surexpriment des peptides antimicrobiens
via l’activation du facteur de transcription STAT3. Parmi ces peptides se trouvent la
famille des REG (RegIIIβ, RegIIIγ), des lectines de type C et des β-défensines. Plus
important, l’IL-22 maintient l’intégrité des cellules épithéliales, prévenant ainsi la
dissémination des bactéries pathogènes en augmentant les signaux anti-apoptiques
ce qui maintient l’intégrité de l’épithélium qui joue alors son rôle de barrière physique

212
(M Cherrier et al., 2012; Cording et al., 2014; Koyasu & Moro, 2012; Rankin et al.,
2013; Sonnenberg et al., 2011; Tumanov et al., 2011; Upadhyay & Fu, 2014; J. A.
Walker et al., 2013).
Les ILCs 3 NCR+ interviennent également dans la réponse aux bactéries intestinales.
Salmonella enteridis ou typhimurium causent chez l’homme une gastroentérite
impliquant des nausées, des vomissements, de la fièvre, de la diarrhée et des
crampes. Elle est acquise par ingestion de nourriture contaminée. Une telle
entérocolite est caractérisée par une inflammation diffuse du cæcum et du colon,
avec de larges lésions épithéliales et des infiltrats inflammatoires, principalement par
des PNN. Une cytokine clé dans la résolution de cette infection est l’IFN-γ. En effet, il
active les programmes antimicrobiens des phagocytes mononucléaires ainsi que les
réponses T. De plus, il contrôle la dispersion systémique du pathogène et la charge
microbienne dans les tissus muqueux. Cependant, une production excessive d’IFN-γ
contribue à l’inflammation de la muqueuse. En plus de son action sur les cellules
hématopoïétiques, l’IFN-γ agit sur les cellules stromales pour augmenter l’excrétion
de mucus par les cellules de Goblet, ce qui fortifie la barrière épithéliale. L’origine de
cet IFN était jusqu’à présent mal connue, mais la découverte des ILCs a permis de
découvrir que 80% de l’IFN-γ proviennent des ILCs 3 NCR+ et seulement 20%
proviennent des NK conventionnels. Cette production est induite par l’IL-12 et l’IL-23
a seulement un effet négligeable. Salmonella exploite la réponse inflammatoire pour
établir une infection de la muqueuse (Diefenbach, 2013).
Les infections muqueuses peuvent aussi être causées par des champignons, et ce
sont alors les ILCs 3 NCR- qui interviennent. Les infections fongiques constituent un
danger grave pour la santé, particulièrement chez les sujets immunodéprimés.
Candida albicans peut causer des infections superficielles au niveau de la peau, des
cavités orales ou vaginales ou disséminer dans tout l’organisme et être mortel. L’IL-
17 représente un mécanisme de défense de l’hôte crucial contre ces infections
fongiques. Les Th17 sont donc évidemment acceptés comme acteurs importants dans
l’immunité antifongique et sont considérés comme la principale source d’IL-17 au
cours d’une infection fongique. Cependant cette source d’IL-17 n’est pas très rapide.
Il existe donc une autre source d’IL-17 lors de l’interaction précoce entre le
champignon et l’hôte. Les ILCs 3 NCR-, productrices d’IL-17, sont alors la principale
source d’IL-17 dans la muqueuse à ce moment crucial (Gladiator, Wangler,
Trautwein-Weidner, & LeibundGut-Landmann, 2013).
Recrutement des monocytes par les NK 4)
Lors d’infestation par Toxoplasma gondii, les NK de la lamina propria (NKp46+,
NK1.1+ CD127-) se différencient grâce à l’IL-15 fournie par les cellules stromales,
probablement les cellules épithéliales. Ces cellules constituent une importante
source de CCL-3 en réponse à l’IL-18 et ce CCL-3 permet le recrutement de
monocytes inflammatoires (Schulthess et al., 2012).

213
Les ILCs dans les poumons C-
Inflammation pulmonaire 1)
Les ILCs 2 pulmonaires stimulent l’inflammation pulmonaire en réponse à de
nombreux stimuli antigéniques comme indiqué sur la Figure 41, page 214.
Lors d’infection virale par l’influenza, le nombre d’ILCs 2 présentes dans le poumon
augmente considérablement et atteint un pic 5-6 jours après le début de l’infection,
moment qui correspond également au pic d’IL-5 et d’IL-13 et donc d’hyperréactivité
pulmonaire. En effet, l’IL-5 attire les éosinophiles, cellules effectrices majeures de
l’inflammation. L’IL-4 et l’IL-13 agissent sur les plasmocytes qui produisent alors de
grandes quantités d’IgE (B. W. S. Li & Hendriks, 2013).
Les ILCs 2 mènent à une augmentation de la quantité d’IL-13 produite, d’une façon
indépendante des antigènes, lors de leur activation par les prostaglandines PGD2
produites par les mastocytes et qui agissent seules ou en synergie avec les
cytokines produites par les cellules épithéliales pulmonaires, c’est-à-dire l’IL-25 et
l’IL-33. La lipotoxine A4 mène à une augmentation de l’apoptose des éosinophiles
via les cellules NK et une diminution de la production d’IL-13 par les ILCs 2 (Vijay
Kumar, 2013).
Il a été montré que, lors d’une infection intestinale par des helminthes, les ILCs 2
pulmonaires produisent de l’IL-5 et de l’IL-13, comme leurs homologues intestinaux.
Ce qu’il faut retenir :
Les ILCs sont donc nécessaires pour :
- le maintien de l’homéostasie intestinale grâce à la production d’IL-22
par les ILCs 3 NCR+,
- la lutte contre les bactéries pathogènes par l’IFN-γ produit par les ILCs
3 NCR+ et l’IL-22 des LTi,
- la réponse aux helminthes grâce aux ILCs 2 et leur production d’IL-13
- la lutte contre les protozoaires où les NK attirent les monocytes via le
CCL-3,
- les défenses contre des champignons grâce à la synthèse d’IL-17 par
les ILCs 3 NCR-.
Il faut bien sûr garder à l’esprit que d’autres cellules et cytokines interviennent
dans ces réponses du système inné

214
De nombreux helminthes migrent à travers les poumons où ils causent des
dommages tissulaires avant d’être toussés et avalés pour finir leur cycle dans les
intestins. L’hypothèse la plus probable est que l’IL-5 produite par les ILCs 2 recrute
des éosinophiles dans les poumons, ce qui favorise l’expulsion des parasites. Par
contre le rôle de l’IL-13 n’est pas encore déterminé dans ce contexte. C’est un
inducteur potentiel de l’hyperplasie des cellules épithéliales, ce qui est utilisé lors de
réparation tissulaire ou lors de fibrose pulmonaire chronique (Monticelli, Sonnenberg,
& Artis, 2012; Yasuda et al., 2012).
Figure 41 : Rôle des ILCs 2 dans l'inflammation pulmonaire (B. W. S. Li & Hendriks, 2013).
Réparation du parenchyme pulmonaire 2)
L’analyse génotypique des ILCs 2 a révélé qu’elles ont une signature
transcriptionnelle riche en gènes qui régulent les voies de réparation tissulaire
comme le gène qui code pour le facteur de croissance épidermal (EGF), de la famille
de l’amphiréguline. Cette dernière est reconnue pour intervenir dans le remodelage
tissulaire et la réparation des blessures épithéliales aiguës dues à l’asthme ou après
une infection par l’influenza. Les ILCs 2 produisent de l’amphiréguline en réponse à
l’IL-2, l’IL-7 et à l’IL-33. Elle permet la réparation de l’intégrité épithéliale mise à mal
lors d’infection virale. Cependant, les facteurs cruciaux pour la restauration de
l’homéostasie comme l’hyperplasie des cellules épithéliales, la sécrétion de mucus
ou encore le dépôt de collagène peuvent être délétères s’ils sont mis en place en

215
absence de tout dommage tissulaire (B. W. S. Li & Hendriks, 2013; Mjösberg & Spits,
2012; Monticelli et al., 2012; J. a Walker & McKenzie, 2013).
Des ILCs disséminées dans tout l’organisme : les D-
cellules NK
Lors de leur découverte, les cellules NK ont été identifiées comme pouvant
spontanément lyser les cellules tumorales sans immunisation préalable, puis elles
ont été considérées comme des cellules éliminant toutes les cellules manquant de
marqueurs du soi. La plupart des recherches se tournent alors vers leur capacité
cytotoxique mais peu de chercheurs se sont tournés vers leur rôle de chef
d’orchestre de la réponse immune. Les NK sont notamment capables d’interagir avec
les cellules dendritiques et elles polarisent les cellules T vers les cellules Th1 dans
les nœuds lymphatiques. Enfin, les cellules NK jouent un rôle précoce dans la
défense antivirale mais aussi antiparasitaire, et antibactérienne. Les NK sont
présentes dans l’utérus vide et gestant et elles interviennent dans la physiologie de
la reproduction puisqu'elles interviennent dans le remodelage tissulaire des
vaisseaux lors de l’implantation fœtale (Boysen & Storset, 2009).
Les NK dans la réponse antivirale 1)
La cytotoxicité des NK et la production d’IFN-γ jouent un rôle crucial dans la
résolution des infections virale, comme par des herpesvirus, des papillomavirus et
des virus influenza.
Nous avons vu page 184, comment les NK reconnaissent leur cible grâce à leurs
récepteurs inhibiteurs et activateurs. Les NK possèdent des récepteurs aux
antigènes viraux comme la protéine hémagglutinine du virus influenza qui induit
directement la cytotoxicité des NK via le récepteur NKp46. Mais l’activation des NK
dans les maladies virales peut aussi être indirecte et est réalisée par des cytokines,
principalement l’IL-12 et les IFN-α et -β sécrétées par les macrophages ou les
cellules dendritiques (Korbel et al., 2004).
Lors d’une infection virale par des virus à ADN, les cellules dendritiques sont
activées via le TLR9 qui reconnaît l’ADN CpG non méthylé. Ceci enclenche le signal
Ce qu’il faut retenir :
Les ILCs 2 interviennent dans l’inflammation pulmonaire grâce à leur
production d’IL-4, d’IL-5 et d’IL-13 et aussi dans la réparation tissulaire grâce
à l’amphiréguline sécrété.

216
d’activation cellulaire via le MyD88 et les cellules dendritiques produisent de l’IFN de
type I et de l’IL-12. Ces cytokines stimulent les cellules NK qui produisent de l’IFN-γ
et sont cytotoxiques, ce qui permet le contrôle de l’infection virale(Lodoen & Lanier,
2006).
Rappelons que les NK ont deux principaux mécanismes effecteurs : la sécrétion
d’IFN-γ et la lyse directe des cellules infectées par exocytose des granules qui
contiennent des perforines et des granzymes. Les NK produisent aussi de la LTαβ et
du TNF, ce qui permet d’induire de l’IFN-β. La lymphotoxine et l’IFN-β sont impliqués
dans le contrôle non cytolytique de certaines infections virales humaines (Colonna,
2009; Lodoen & Lanier, 2006).
Les NK dans la lutte contre les tumeurs 2)
Nous avons déjà évoqué la surveillance des tumeurs par les NK grâce à la détection
du CMH altéré ou de la perte de CMH par les cellules transformées.
Les cellules NK interviennent dans la surveillance des tumeurs et, pour illustrer
l’importance de cette surveillance, après transfert de cellules tumorales, les cellules
NK ont éliminé plus de 90% des cellules tumorales en moins de 24 heures (Paul,
2013).
Elles possèdent une activité cytolytique contre les cellules tumorales. Elles libèrent
rapidement des cytokines pro-inflammatoires et des chimiokines impliquées dans les
réponses précoces inflammatoires (Montaldo et al., 2014).
Les NK contre les protozoaires 3)
Les cellules NK fournissent la base pour la résistance précoce à la leishmaniose
mais ce rôle semble n’être que peu lié à la cytotoxicité mais plutôt à leur production
de cytokines, notamment l’IFN-γ. Les cellules NK ne s’activent pas directement mais
sont activées par des cytokines, notamment IL-12 et IL-18 (Korbel et al., 2004).
De la même façon, lors de trypanosomose, les cellules NK interviennent, stimulées
par l’IL-12 en contrôlant la parasitémie grâce à l’IFN-γ. L’activation directe des NK
par des glycolipides parasitaires ainsi que la cytotoxicité par les NK restent encore
incertaines (Korbel et al., 2004).
Les cellules NK montrent une activité cytolytique envers les macrophages infectés
par Toxoplasma gondii ainsi que contre le tachyzoïte extracellulaire. Les cellules
dendritiques reconnaissent le parasite puis activent les cellules NK (Korbel et al.,
2004).
Dans le cas de la malaria (provoquée par Plasmodium falciparum), les cellules NK
sont activées dans les premières heures alors que les cellules γδ T et les NKT
répondent après 24 à 48 heures. Cette activation précoce des NK implique une fois

217
encore l’IL-12 et légèrement l’IL-18, délivrées par les cellules accessoires. Les
globules rouges sanguins parasités subissent de nombreuses altérations
structurelles comme l’exposition membranaire anormale de spectrine et Band3 ainsi
que des néoantigènes parasitaires. Il semblerait que les NK reconnaissent ces
érythrocytes altérés et la cytotoxicité ne s’exercerait que contre ces globules rouges
là (Korbel et al., 2004; Lodoen & Lanier, 2006).
Les NK contre des bactéries 4)
Outre leur rôle antiviral et de surveillance des tumeurs, les NK interviennent
également dans l’immunité contre des bactéries. Les cellules NK peuvent modifier
leur expression des récepteurs activateurs comme le NKp30, le NKp46 et le NKG2D.
Ainsi les NK interviendraient aussi lors d’infections par Mycobacterium tuberculosis
(via NKG2D et NKp46), ou par Listeria monocytogenes (Lodoen & Lanier, 2006).
Les NK contrôlent l’immunité 5)
Grâce à la sécrétion de nombreuses cytokines pro-inflammatoires comme l’IFN-γ, le
TNF-α, la lymphotoxine α, le GM-CSF, et des chimiokines comme RANTES, la
lymphotactine, MIP-1α, MIP-1β le CCL-3 les NK peuvent orchestrer l’immunité et
alerter les autres types cellulaires d’un danger. L’IFN-γ active les macrophages, et
dans une moindre mesure, les neutrophiles et stimule la différenciation des
lymphocytes Th1 CD4+, ainsi que la présentation d’antigènes et le changement de
classe des immunoglobulines produites par les lymphocytes B (passage aux IgG)
(Korbel et al., 2004; Paul, 2013).
La production de ces facteurs solubles par les cellules NK dans les phases précoces
de l’immunité innée influence significativement le recrutement et le fonctionnement
des autres cellules hématopoïétiques. Par exemple, les cellules NK augmentent les
réponses T spécifiques des antigènes si les titres en virus sont modestes mais elles
limitent une activation excessive des cellules T si les titres viraux sont élevés. A
travers des contacts physiques et la production de cytokines, les cellules NK sont
centrales dans la régulation des cellules dendritiques et des PNN pour promouvoir
ou restreindre certaines réponses immunes (Campbell & Hasegawa, 2013).
Rappelons que la plupart des cellules aberrantes ou déficientes en CMH de classe I
sont sensibles à la lyse par les cellules NK alors que les cellules normales sont
protégées par les molécules de classe I. Cependant les cellules dendritiques
immatures sont une exception puisqu’elles sont sensibles à la lyse des NK jusqu’à
leur maturation où elles deviennent résistantes car les cellules matures expriment
plus de molécules de CMH I qui inhibent la lyse des NK comme nous l’avons évoqué
dans le paragraphe « Fonctions des cellules dendritiques », page 50 . Ceci permet
de contrôler la qualité des cellules dendritiques. Les cellules dendritiques qui
échouent dans l’expression du CMH I sont supprimées. De telles cellules
défectueuses pourraient, après expression du CCR7 et migration vers le nœud

218
lymphatique induire une réponse T inappropriée de faible affinité et éventuellement
conduire à une réponse Th2 ou un état de tolérance (Moretta et al., 2005).
En fait, les deux types cellulaires peuvent mutuellement s’activer lors d’une réponse
immune :
Les cellules dendritiques peuvent induire chez les cellules NK la production
d’IFN-γ (via un message codé par l’IL-2), la cytotoxicité, l’expression de CD69,
et leur prolifération (par l’intermédiaire de l’lL-12 et de l’IL-18). Cette activation
nécessite des facteurs solubles mais implique aussi un contact direct. Chez la
souris, le contact semble plus important que les facteurs solubles, alors que
chez l’homme, c’est le contraire qui est observé. Les cellules dendritiques
apparaissent comme les activateurs de la réponse NK dans les phases
précoces de la réponse immune, avant qu’une réponse adaptative immune ne
soit initiée. Cette interaction a lieu :
dans les nœuds lymphatiques secondaires, puisque c’est là où les
cellules dendritiques activées migrent. Elles rencontrent alors des NK
non cytolytiques mais fortement producteurs de cytokines puisque ce
sont des NK CD56bright CD16- perforine-. Les cellules dendritiques
entraînent alors la production d’IFN-γ grâce à l’IL-12 qu’elles libèrent.
Cet IFN-γ est nécessaire pour orienter la réponse immune vers le type
Th1. Les deux types de cellules dendritiques peuvent activer les
cellules NK : les cellules dendritiques myéloïdes et plasmacytoïdes
mais selon des stimuli différents ;
dans les tissus enflammés. Cette fois-ci, ce sont les NK cytotoxiques
CD56dim CD16+ qui migrent dans les tissus enflammés grâce à leurs
récepteurs à l’IL-8 et la fractalkine, chimiokines induites par des
cytokines inflammatoires comme l’IL-1 et TNF-α. Une fois sur le site
de l’inflammation, les NK rencontrent des cellules dendritiques ;
Les cellules dendritiques sont activées par le contact avec les NK et du TNF-
α. La maturation des cellules dendritiques par les NK est nécessaire dans les
infections causées par les virus, habituellement incapables d’initier cette
maturation. Cela conduit à la production de TNF-α et d’IL-12 et à la
surexpression de molécules stimulatrices comme le CD86 par les cellules
dendritiques. Rappelons que si le ratio NK/cellules dendritiques est faible, les
cellules dendritiques prolifèrent, si le ratio NK/cellules dendritiques est élevé,
les NK tuent les cellules dendritiques ;
Les cellules NK sont nécessaires pour la production d’IL-12 par les cellules
dendritiques, cette interaction est indispensable pour la génération d’une
réponse T CD8+ (Ferlazzo, 2005; Hamerman et al., 2005; Marcenaro et al.,
2006).

219
L’interaction entre les cellules NK et les cellules dendritiques est pertinente pour la
génération de lymphocyte T cytotoxique (CTL) efficace contre les virus cytopathiques
(Moretta et al., 2005).
D’un autre côté, les NK activées pourraient devenir des cellules présentatrices
d’antigènes. Après activation les cellules NK peuvent surexprimer le CMH de classe
II le CD80 et le CD86 et acquérir des mécanismes uniques de capture et de
présentation d’antigènes en impliquant des récepteurs activateurs comme le NKp46,
le CNp30 et le NKG2D. Les cellules NK acquièrent des fonctions de cellules
présentatrices d’antigènes après avoir tué une cellule cible (Reschner et al., 2008).
Le dialogue des ILCs avec les autres cellules E-
immunitaires
Des preuves récentes ont montré que les ILCs sont des régulateurs critiques de
l’immunité innée et de l’inflammation aux surfaces muqueuses des mammifères,
comme les voies respiratoires et le tractus gastro-intestinal. Les ILCs agissent
comme l’homologue inné des cellules Th CD4+ qui appartiennent au bras adaptatif du
système immunitaire (Hepworth & Sonnenberg, 2014).
Le groupe 3 des ILCs peut aussi jouer un rôle significatif en modulant le système
adaptatif en stimulant la génération, l’organisation et le maintien des tissus
lymphoïdes secondaires, maintenant la fonction de barrière épithéliale et avec des
interactions directes avec les cellules immunes adaptatives (Hepworth &
Sonnenberg, 2014).
Les ILCs 3 qui produisent l’IL-22 apparaissent comme régulateurs indirects de la
réponse T CD4+ et de l’homéostasie intestinale en modulant la composition et
l’endiguement des bactéries commensales (Hepworth & Sonnenberg, 2014).
Les ILCs 2 peuvent induire la prolifération des cellules B stimuler la production d’IgA
via la sécrétion d’IL-5 et d’IL-6 et dans certaines circonstances, elles peuvent
exprimer le CMH II (Hepworth & Sonnenberg, 2014).
Enfin, nous venons de voir le contrôle de l’immunité par les NK, rôle qui implique un
dialogue précis avec de nombreuses cellules immunitaires.
Les ILCs 2 relâchent de l’IL-5 en réponse à un peptide intestinal vasoactif, lui-même
libéré après un repas. Cette relation entre l’IL-5 et la prise de nourriture pourrait
expliquer le cycle circadien du nombre d’éosinophiles en régulant l’éosinophilopoïèse
et l’accumulation des PNE dans les tissus (S. Yu et al., 2014).

220
IV. Implication des ILCs dans des processus
pathologiques
Les ILCs ont donc de nombreux rôles dans la protection de l’organisme, mais elles
ont également des rôles pathologiques, notamment dans l’asthme et les allergies et
dans les maladies inflammatoires intestinales chroniques.
Les ILCs 2 sont impliquées dans l’asthme et les A-
allergies
L’asthme allergique est une maladie inflammatoire chronique des voies respiratoires
dues à une réponse immunologique inappropriée à des stimuli environnementaux.
Il existe plusieurs types d’asthme :
- un phénotype d’asthme allergique activé par une exposition à des
allergènes et associé à la présence de PNE dans les voies respiratoires. Cet
asthme fait intervenir une sensibilisation allergique et aussi le système
immunitaire adaptatif ;
- Des phénotypes d’asthme non allergiques existent associés à une exposition
à des facteurs environnementaux comme la pollution de l’air, la fumée de
cigarette, l’obésité, les infections virales et le stress. Cette forme est souvent
associée à la présence de PNN dans les voies respiratoires et d’une
immunité innée indépendante des Th2 ;
- Enfin, il existe également de l’asthme intrinsèque (S. Yu et al., 2014).
L’asthme est caractérisé par une hyperréactivité des voies respiratoires (AHR) qui se
traduit par la production de mucus, le recrutement des mastocytes et des PNE, la
contraction des muscles lisses et le remodelage des voies respiratoires. Tout ceci
concourt à une bronchoconstriction et est initié par l’IL-25, l’IL-33 et le TSLP. L’IL-33
est produit par les cellules épithéliales activées via leur TLR4 qui fixent un allergène
ou par les macrophages alvéolaires activées par les NKT (H. Y. Kim et al., 2011;
Vijay Kumar, 2013; J. a Walker & McKenzie, 2013; J. A. Walker et al., 2013).
L’asthme est couramment associé à une inflammation éosinophilique, qui lorsqu’elle
est persistante, mène à un remodelage du tissu pulmonaire par des processus de
réparation successifs, avec notamment, de la fibrose subépithéliale, l’hyperplasie des
muscles lisses, la métaplasie des cellules muqueuses et une angiogenèse
augmentée (B. W. S. Li & Hendriks, 2013).
Lorsque que les récepteurs TLR des cellules épithéliales sont activés, de
nombreuses cytokines sont produites comme le GM-CSF, l’IL-1α, l’IL-1β, l’IL-25 et
l’IL-33. Ces cytokines orientent les cellules dendritiques vers une réponse de type

221
Th2 via l’IL-4 qui permet ensuite de produire du GATA3. Ce dernier est suffisant pour
induire la différenciation des Th2 (B. W. S. Li & Hendriks, 2013).
L’hyperréactivité des voies respiratoires est induite par les NKT, activés par détection
de glycolipides. Ils induisent directement chez les macrophages alvéolaires la
production d’IL-33, qui active à son tour les NKT ainsi que les ILCs 2 pour augmenter
la production d’IL-13. Le rôle central de l’IL-33 est résumé dans la Figure 42, page
222. De plus le niveau d’IL-33 est proportionnel à la sévérité de l’asthme. Différentes
études ont montré que les ILCs 2 jouent un rôle prépondérant dans l’asthme grâce à
leur production d’IL-5 et d’IL-13 et ces cellules seraient même impliquées dans
l’induction de l’inflammation allergique du poumon. Les iNKT sont activés par l’IL-12,
l’IL-18, l’IL-33 et les interférons de type I produits par les cellules qui amplifient de
faibles signaux. Les iNKT activés par des cytokines inflammatoires, un antigène ou
une infection virale peuvent stimuler le remodelage des voies respiratoires. Leur rôle
est dépendant de l’IL-13 et leur action passe par leur production d’IFN-γ (Barrett &
Austen, 2009; H. Y. Kim et al., 2011; B. W. S. Li & Hendriks, 2013; Monticelli et al.,
2012; Pishdadian et al., 2012; Vercelli et al., 2013; J. A. Walker et al., 2013).
Enfin, les ILCs 2 sont également activées par les leucotriènes grâce au récepteur à
leucotriène cystéinyle 1 (CysLT1R). La production d’IL-4 par les ILCs 2 ne dépend
pas de l’IL-33 mais de LTD4, un leucotriène. Les ILCs 2 expriment le récepteur pour
la lipoxine A4, un facteur qui diminue la production d’IL-13 par les ILCs 2 (Deckers,
Branco Madeira, & Hammad, 2013; Vercelli et al., 2013).
Les ILCs 2 produisent également de l’IL-9, une cytokine reliée à l’inflammation
allergique des voies aériennes, très exprimée dans les poumons des patients
asthmatiques. La production de cette cytokine est sous la dépendance de l’IL-12
sécrétée par les cellules de l’immunité adaptative (Koyasu & Moro, 2012).
Les cytokines effectrices, responsables de l’asthme sont donc :
- L’IL-4 qui induit la production d’IgE par les lymphocytes B et l’expression de
VCAM1 sur les cellules endothéliales ;
- L’IL-5 qui induit le développement des éosinophiles, leur recrutement vers
les poumons et leur activation. Ensuite, les PNE produisent un large éventail
de cytokines et chimiokines et sont une source potentielle de leucotriène C4
et de PAF (Platelet Activating Factor) qui induit la production de mucus et la
contraction des muscles lisses. ;
- L’IL-9 qui stimule en retour la production des cytokines de type 2 par les
ILCs. Elle est responsable de production de mucus, d’hyperplasie des
cellules de Goblet, donc une surexpression d’IL-9 est responsable du
remodelage des voies aériennes qui cause l’asthme ;
- L’IL-13 qui induit le remodelage des voies respiratoires et l’hyperréactivité ;
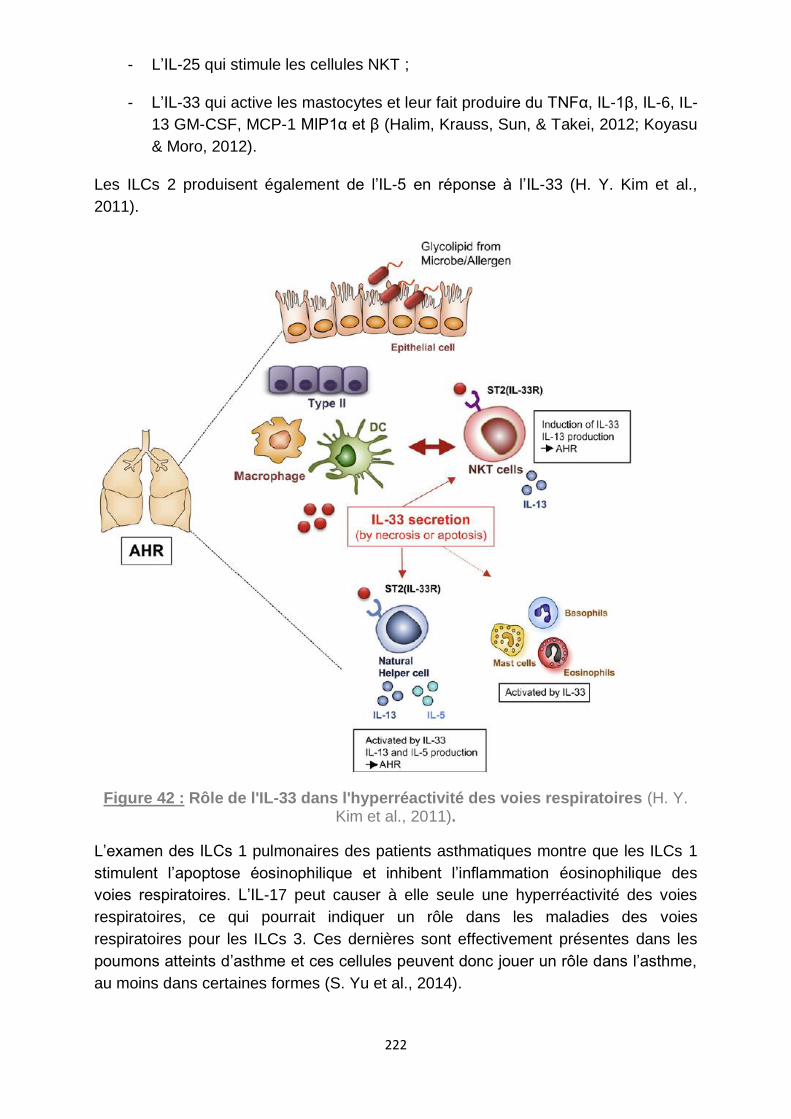
222
- L’IL-25 qui stimule les cellules NKT ;
- L’IL-33 qui active les mastocytes et leur fait produire du TNFα, IL-1β, IL-6, IL-
13 GM-CSF, MCP-1 MIP1α et β (Halim, Krauss, Sun, & Takei, 2012; Koyasu
& Moro, 2012).
Les ILCs 2 produisent également de l’IL-5 en réponse à l’IL-33 (H. Y. Kim et al.,
2011).
Figure 42 : Rôle de l'IL-33 dans l'hyperréactivité des voies respiratoires (H. Y. Kim et al., 2011).
L’examen des ILCs 1 pulmonaires des patients asthmatiques montre que les ILCs 1
stimulent l’apoptose éosinophilique et inhibent l’inflammation éosinophilique des
voies respiratoires. L’IL-17 peut causer à elle seule une hyperréactivité des voies
respiratoires, ce qui pourrait indiquer un rôle dans les maladies des voies
respiratoires pour les ILCs 3. Ces dernières sont effectivement présentes dans les
poumons atteints d’asthme et ces cellules peuvent donc jouer un rôle dans l’asthme,
au moins dans certaines formes (S. Yu et al., 2014).

223
Durant l’asthme allergique, la principale fonction des cellules dendritiques est de
capturer des allergènes inhalés et de les transporter via le CCR7 vers la zone T du
nœud lymphatique drainant la zone. Ainsi, les cellules dendritiques sont importantes
durant la phase de sensibilisation mais aussi durant l’inflammation et elles
contribuent au recrutement des cellules inflammatoires. L’épithélium joue aussi un
rôle dans la phase de sensibilisation puisqu’il fournit la première ligne de défense
contre les invasions environnementales comme les allergènes, les virus et les
polluants. Bien qu’il représente une barrière physique protégeant les tissus sous-
jacents, l’épithélium sécrète également des chimiokines et cytokines qui attirent et
activent les cellules immunes. L’IL-25 potentialise l’induction des cellules Th2 dans la
phase de sensibilisation, elle amplifie cette réponse inflammatoire dans la phase de
challenge. Les neutrophiles, les mastocytes et les basophiles et les cellules
épithéliales produisent de l’IL-25 en réponse aux allergènes comme Aspergillus
fumigatus…. L’IL-25 recrute ensuite les cellules hématopoïétiques et les cellules
résidentes pulmonaires et stimule ainsi la différenciation des cellules Th2 d’une
manière IL-4 dépendante et STAT 6 dépendante. Elle induit aussi la production des
cytokines Th2 et des métalloprotéases matrix 7. Les premières augmentent le niveau
d’IL-25 et les secondes le clivent en une forme plus active. Ainsi, l’IL-25 potentialise
l’induction de l’inflammation Th2, l’amplifie et peut induire une hyperréactivité des
voies respiratoires en absence de toute cytokine 2 (Barrett & Austen, 2009; Deckers
et al., 2013).
Durant la phase effectrice, l’IL-33 produite par les cellules dendritiques en réponse à
des complexes immuns contribue donc au maintien de l’immunité Th2. Après
réexposition à l’allergène, les monocytes sont recrutés dans les poumons et
produisent des chimiokines qui attirent les PNE, les mastocytes, les PNB et les
cellules Th2. Sur les éosinophiles, l’IL-33 augmente leur expression du CD11b et leur
survie, mais également leur production d’IL-8 en réponse à l’IL-3, l’IL-5 et au GM-
CSF. Dans le même temps, des cytokines produites par l’épithélium comme l’IL-33 et
le GM-CSF contribuent au développement des ILCs 2, à l’activation et à la
différenciation des cellules dendritiques pulmonaires, à l’activation et la survie des
PNE, des mastocytes et des basophiles. Les mastocytes, armés des IgE produits
durant la phase de sensibilisation, fixent l’allergène et libèrent de l’histamine, de l’IL-6
et du TNF-α, renforçant ainsi la migration des cellules dendritiques vers les nœuds
lymphatiques. Les mastocytes interagissent aussi avec les ILCs 2 et augmentent la
production d’IL-5 par les ILCs 2. Le mécanisme qui aboutit à l’asthme allergique est
détaillé dans la Figure 43, page 224 (Deckers et al., 2013).

224
Figure 43 : Mécanisme de l’asthme allergique (Deckers et al., 2013).
Les ILCs 3 et les maladies inflammatoires B-
chroniques
Les maladies inflammatoires chroniques de l’intestin regroupent chez l’homme la
maladie de Crohn (inflammation discontinue de tout le tractus intestinal) et la colite
ulcérative (inflammation de la muqueuse superficielle et de la sous muqueuse du
colon). Les principales cytokines impliquées dans la pathogénèse de ces maladies
sont l’IL-17A, l’IL-17F, l’IL-22 et l’IFN-γ. Ces cytokines sont produites par les
lymphocytes T helper, mais aussi par les cellules γδ T, les NK et les lymphocytes
NKT ainsi que par les ILCs. En effet, les ILCs 3 produisent notamment de l’IL-17A et
de l’IFN-γ en réponse à l’IL-23 (J. A. Walker et al., 2013).
Les ILCs produisant de l’IL-17 sont responsables de colite chronique chez la souris.
L’expression de l’IL-17, induite par l’IL-23 est potentialisée par le TNF-α produit par
les cellules dendritiques. Le T-bet est nécessaire pour l’expression optimale d’IFN-γ
par les ILCs. L’IBD (Inflammatory Bowel Disease) est indirectement causée par les
ILCs. L’IBD dépend de l’axe IL-23 / IL-17 et Helicobacter typhlonius joue aussi un

225
rôle clé. Le T-bet contrôle un programme transcriptionnel dans le système
immunitaire inné intestinal qui impacte profondément la balance entre la
maintenance ou la perte de l’homéostasie. Lors d’IBD, les ILCs 3 NCR- sont la
source primaire d’IL-17 et cette production est suffisante pour induire la maladie.
Avant que les ILCs ne soient découvertes, les cellules dendritiques étaient
supposées responsables des IBD, caractérisées par une dérégulation de la réponse
immune à la flore microbienne (S. C. Ng et al., 2010; Powell et al., 2012).
Dans la maladie de Crohn, l’IL-15 et l’IL-18 coopèrent pour promouvoir la production
de CCL-3 par les NK intestinaux et donc le recrutement des monocytes qui
produisent alors du TNF-α en excès. L’administration d’anticorps anti-TNF-α permet
de limiter cette production de TNF en excès et constitue une thérapie efficace
(Schulthess et al., 2012).
Hyperréactivité pulmonaire due aux ILCs 2 suite C-
à une infection virale
Les ILCs 2 sont présentes dans les poumons, surtout pendant une infection par
l’influenza et sont responsables de l’hyperréactivité respiratoire. Le virus affecte les
macrophages alvéolaires qui relâchent de l’IL-33 activant les ILCs 2 qui se
développent et produisent de l’IL-5 et IL-13. Le rôle des ILCs 2 dans l’infection par le
virus de l’influenza peut expliquer le mécanisme d’apparition de l’asthme.
Cependant, lors d’infection par l’influenza, les ILCs 2 produisent aussi de
l’amphiréguline, requise pour réparer les tissus pulmonaires et leur homéostasie : les
ILCs 2 interviennent dans le maintien de l’intégrité épithéliale pulmonaire et
améliorent les fonctions pulmonaires après un épisode viral mais aussi dans les
maladies pulmonaires inflammatoires (Deckers et al., 2013; Monticelli et al., 2012; S.
Yu et al., 2014).
Ce qu’il faut retenir :
Les ILCs sont également impliquées dans des processus pathologiques. Leur
découverte a ainsi permis de mieux comprendre ces dérèglements
immunologiques mais les modèles proposés jusqu’alors ne doivent pas être
oubliés. Tout comme la réponse aux microbes, la pathogénie de ces maladies
est un phénomène complexe qui fait intervenir un grand nombre de cellules et
de cytokines.

226

227
Abbas, A. ., Lichtman, A. ., & Pillai, S. (2012). Cellular and molecular immunology. 7th edition (Saunders E., p. 545). Philadelphia, Edinburgh.
Ackermann, M. R., Derscheid, R., & Roth, J. A. (2010). Innate immunology of bovine respiratory disease. The Veterinary Clinics of North America. Food Animal Practice, 26(2), 215–28. doi:10.1016/j.cvfa.2010.03.001
Auvynet, C., & Rosenstein, Y. (2009). Multifunctional host defense peptides: antimicrobial peptides, the small yet big players in innate and adaptive immunity. The FEBS Journal, 276(22), 6497–508. doi:10.1111/j.1742-4658.2009.07360.x
Baldwin, C. L., Hsu, H., Chen, C., Palmer, M., McGill, J., Waters, W. R., & Telfer, J. C. (2014). The role of bovine γδ T cells and their WC1 co-receptor in response to bacterial pathogens and promoting vaccine efficacy: a model for cattle and humans. Veterinary Immunology and Immunopathology, 159(3-4), 144–55. doi:10.1016/j.vetimm.2014.02.011
Ballou, M. A. (2011). Inflammation: Role in the etiology and pathophysiology of clinical mastitis in dairy cows. Journal of Animal Science, 90(5), 1466–78. doi:10.2527/jas.2011-4663
Barone, R. (1978). Anatomie comparée des mamifères domestiques. Tome 3 Splanchnologie II. Appareil uro-génital. Foetus et ses annexes. Péritoine et topographie abdominale. 1ième édition. (Vigot, Ed.) (p. 951).
Barrett, N. A., & Austen, K. F. (2009). Innate cells and T helper 2 cell immunity in airway inflammation. Immunity, 31(3), 425–37. doi:10.1016/j.immuni.2009.08.014
Becker, C., & Arcangioli, M.-A. (2014). Utilisation de l’hématologie en pathologie bovine. In SNGTV (Ed.), Journée nationale des GTV : Les examens complémentaires : atouts du diagnostic et de la prescritpion raisonnée. (pp. 117–122). Reims.
Beijer, M. R., Kraal, G., & den Haan, J. M. M. (2013). Vitamin A and dendritic cell differentiation. Immunology, 142(1), 39–45. doi:10.1111/imm.12228
Berg, R. E., & Forman, J. (2006). The role of CD8 T cells in innate immunity and in antigen non-specific protection. Current Opinion in Immunology, 18(3), 338–43. doi:10.1016/j.coi.2006.03.010
Berlutti, F., Pantanella, F., Natalizi, T., Frioni, A., Paesano, R., Polimeni, A., & Valenti, P. (2011). Antiviral properties of lactoferrin-a natural immunity molecule. Molecules (Basel, Switzerland), 16(8), 6992–7018. doi:10.3390/molecules16086992

228
Beutler, B. (2003). Innate immunity: an overview. Molecular Immunology, 40(12), 845–859. doi:10.1016/j.molimm.2003.10.005
Björkström, N. K., Kekäläinen, E., & Mjösberg, J. (2013). Tissue-specific effector functions of innate lymphoid cells. Immunology, 139(4), 416–27. doi:10.1111/imm.12098
Bordon, Y. (2014). Innate lymphoid cells: on the origin of ILCs. Nature Reviews. Immunology, 14(3), 133. doi:10.1038/nri3629
Born, W. K., Reardon, C. L., & O’Brien, R. L. (2006). The function of γδ T cells in innate immunity. Current Opinion in Immunology, 18(1), 31–8. doi:10.1016/j.coi.2005.11.007
Boudry, B. (2005). Traire un lait de qualité : une attention de tous les jours. In Qualité du lait et gestion du troupeau (p. 13). Henri Chapelle.
Bourgeois, E. A., Levescot, A., Diem, S., Chauvineau, A., Bergès, H., Milpied, P., … Herbelin, A. (2010). A natural protective function of invariant NKT cells in a mouse model of innate-cell-driven lung inflammation. European Journal of Immunology, 41(2), 299–305. doi:10.1002/eji.201040647
Bourgeois, E. A., Van, L. P., Samson, M., Diem, S., Barra, A., Roga, S., … Herbelin, A. (2009). The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-γ production. European Journal of Immunology, 39(4), 1046–55. doi:10.1002/eji.200838575
Bowdish, D. M. E., Loffredo, M. S., Mukhopadhyay, S., Mantovani, A., & Gordon, S. (2007). Macrophage receptors implicated in the “adaptive” form of innate immunity. Microbes and Infection / Institut Pasteur, 9(14-15), 1680–7. doi:10.1016/j.micinf.2007.09.002
Boysen, P., & Storset, A. K. (2009). Bovine natural killer cells. Veterinary Immunology and Immunopathology, 130(3-4), 163–77. doi:10.1016/j.vetimm.2009.02.017
Campbell, K. S., & Hasegawa, J. (2013). Natural killer cell biology : an update and future directions. The Journal of Allergy and Clinical Immunology, 132(3), 536–44. doi:10.1016/j.jaci.2013.07.006
Carroll, M. V, & Sim, R. B. (2011). Complement in health and disease. Advanced Drug Delivery Reviews, 63(12), 965–75. doi:10.1016/j.addr.2011.06.005
Cavaillon, J.-M. (1996). Les cytokines. 2ième édition (Masson., p. 589). Paris.
Ceciliani, F., Ceron, J. J., Eckersall, P. D., & Sauerwein, H. (2012). Acute phase proteins in ruminants. Journal of Proteomics, 75(14), 4207–31. doi:10.1016/j.jprot.2012.04.004
Chaouat, G., Zourbas, S., OStojic, S., Lapprée-Delage, G., Ledée, N., Mairowitzi, V., … Martal, J. (2000). Placenta et immunité. In Immunité et gestation (p. 108). Paris.

229
Chen, J., & Chen, Z. J. (2013). Regulation of NF-κB by ubiquitination. Current Opinion in Immunology, 25(1), 4–12. doi:10.1016/j.coi.2012.12.005
Cherrier, M., & Eberl, G. (2012). The development of LTi cells. Current Opinion in Immunology, 24, 1–6.
Cherrier, M., Ohnmacht, C., Cording, S., & Eberl, G. (2012). Development and function of intestinal innate lymphoid cells. Current Opinion in Immunology, 24(3), 277–83. doi:10.1016/j.coi.2012.03.011
Cherrier, M., Sawa, S., & Eberl, G. (2012). Notch, Id2, and RORγt sequentially orchestrate the fetal development of lymphoid tissue inducer cells. The Journal of Experimental Medicine, 209(4), 729–40. doi:10.1084/jem.20111594
Chin, E. H., Quinn, J. S., & Burness, G. (2013). Acute stress during ontogeny suppresses innate, but not acquired immunity in a semi-precocial bird (Larus delawarensis). General and Comparative Endocrinology, 193, 185–92. doi:10.1016/j.ygcen.2013.08.007
Clark, G., Angel, N., Kato, M., Lopez, A., Macdonald, K., Vuckovic, S., & Hart, D. N. (2000). The role of dendritic cells in the innate immune system. Microbes and Infection, 257–272.
Clinthorne, J. F., Beli, E., Duriancik, D. M., & Gardner, E. M. (2013). NK cell maturation and function in C57BL/6 mice are altered by caloric restriction. Journal of Immunology (Baltimore, Md. : 1950), 190(2), 712–22. doi:10.4049/jimmunol.1201837
Coates, C. J., & Nairn, J. (2014). Diverse immune functions of hemocyanins. Developmental and Comparative Immunology, 45(1), 43–55. doi:10.1016/j.dci.2014.01.021
Colonna, M. (2009). Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity, 31(1), 15–23. doi:10.1016/j.immuni.2009.06.008
Cooper, E. L. (2006). Comparative immunology. Integrative Zoology, 1(1), 32–43. doi:10.1111/j.1749-4877.2006.00010.x
Cording, S., Medvedovic, J., Cherrier, M., & Eberl, G. (2014). Development and regulation of RORγt(+) innate lymphoid cells. FEBS Letters. doi:10.1016/j.febslet.2014.03.034
Cornelissen, F., Aparicio Domingo, P., Reijmers, R. M., & Cupedo, T. (2011). Activation and effector functions of human RORC+ innate lymphoid cells. Current Opinion in Immunology, 23(3), 361–7. doi:10.1016/j.coi.2011.03.002
Coutinho, A., & Poltorack, A. (2003). Innate immunity: from lymphocyte mitogens to Toll-like receptors and back. Current Opinion in Immunology, 15(6), 599–602. doi:10.1016/j.coi.2003.09.020

230
Cronin, J. G., Turner, M. L., Goetze, L., Bryant, C. E., & Sheldon, I. M. (2012). Toll-like receptor 4 and MYD88-dependent signaling mechanisms of the innate immune system are essential for the response to lipopolysaccharide by epithelial and stromal cells of the bovine endometrium. Biology of Reproduction, 86(2), 51. doi:10.1095/biolreprod.111.092718
Cua, D. J., & Tato, C. M. (2010). Innate IL-17-producing cells: the sentinels of the immune system. Nature Reviews. Immunology, 10(7), 479–89. doi:10.1038/nri2800
Dahlin, J. S., & Hallgren, J. (2014). Mast cell progenitors: Origin, development and migration to tissues. Molecular Immunology, 1–9. doi:10.1016/j.molimm.2014.01.018
De Franco, A., Robertson, M., & Locksley, R. (2009). Immunité. La réponse immunitaire dans les maladies infectieuses et inflammatoires. (De Boeck U., p. 365). Bruxelles.
De Sousa, N., Figueiredo, J. R., El Amiri, B., Banga-Mboko, H., & Beckers, J. (2002). Influence potentielle des hormones et protéines synthétisées au cours de la gestation sur l’état immunitaire de la mère. Ann. Méd. Vét., 146(2), 71–83.
Dec, M., Wernicki, A., Puchalski, A., Urban-Chmiel, R., & Wasko, A. (2012). The effect of conglutinin on production of reactive oxygen species in bovine granulocytes, 13(May 2011), 33–38.
Deckers, J., Branco Madeira, F., & Hammad, H. (2013). Innate immune cells in asthma. Trends in Immunology, 34(11), 540–7. doi:10.1016/j.it.2013.08.004
Delves, P. J., Martin, S. J., Burton, D. R., & Roitt, I. M. (2011). Roitt’s essential immunology. 12th edition (Wiley Blac., p. 546). Singapour.
Deretic, V. (2006). Autophagy as an immune defense mechanism. Current Opinion in Immunology, 18(4), 375–82. doi:10.1016/j.coi.2006.05.019
Di Santo, J. P. (2010). An IL-1β-dependent switch in innate mucosal immunity? Immunity, 32(6), 734–6. doi:10.1016/j.immuni.2010.06.012
Diana, J., & Lehuen, A. (2009). NKT cells: friend or foe during viral infections? European Journal of Immunology, 39(12), 3283–91. doi:10.1002/eji.200939800
Diefenbach, A. (2013). Innate lymphoid cells in the defense against infections. European Journal of Microbiology & Immunology, 3(3), 143–51. doi:10.1556/EuJMI.3.2013.3.1
Doherty, T. A., Khorram, N., Lund, S., Mehta, A. K., Croft, M., & Broide, D. H. (2013). Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. The Journal of Allergy and Clinical Immunology, 132(1), 205–13. doi:10.1016/j.jaci.2013.03.048
Douglas, J., & Jane Wardrop, K. (2010). Schalm’s Veterinary Hematology. Sixth Edition (Wiley Blac., p. 1232). Singapore.

231
Evans, G. O., O’Brien, P. ., & Watterson, C. L. (2009). Animal Clinical chemistry : A practical guide for toxicologists and biomedical researchers. Second edition (CRC Press., p. 320).
Ferlazzo, G. (2005). Natural killer and dendritic cell liaison: recent insights and open questions. Immunology Letters, 101(1), 12–7. doi:10.1016/j.imlet.2005.04.015
Ferreira, V. P., Pangburn, M. K., & Cortés, C. (2010). Complement control protein factor H: the good, the bad, and the inadequate. Molecular Immunology, 47(13), 2187–97. doi:10.1016/j.molimm.2010.05.007
Fitzgerald, K. A., & Caffrey, D. R. (2014). Long noncoding RNAs in innate and adaptive immunity. Current Opinion in Immunology, 26, 140–6. doi:10.1016/j.coi.2013.12.001
Forsberg, A., Bengtsson, M., Eringfält, A., Ernerudh, J., Mjösberg, J., & Jenmalm, M. C. (2014). GATA binding protein 3(+) group 2 innate lymphoid cells are present in cord blood and in higher proportions in male than in female neonates. The Journal of Allergy and Clinical Immunology, (C). doi:10.1016/j.jaci.2014.01.027
Fuchs, A., Vermi, W., Lee, J. S., Lonardi, S., Gilfillan, S., Newberry, R. D., … Colonna, M. (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity, 38(4), 769–81. doi:10.1016/j.immuni.2013.02.010
Genetet, N. (1997). Immunologie. 3e édition (Editions m., p. 604). Cachan.
Geremia, A., Biancheri, P., Allan, P., Corazza, G. R., & Di Sabatino, A. (2013). Innate and adaptive immunity in inflammatory bowel disease. Autoimmunity Reviews, 13(1), 3–10. doi:10.1016/j.autrev.2013.06.004
Gladiator, A., Wangler, N., Trautwein-Weidner, K., & LeibundGut-Landmann, S. (2013). Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. Journal of Immunology (Baltimore, Md. : 1950), 190(2), 521–5. doi:10.4049/jimmunol.1202924
Gleeson, P. A. (2014). The role of endosomes in innate and adaptive immunity. Seminars in Cell & Developmental Biology, 31C, 64–72. doi:10.1016/j.semcdb.2014.03.002
González-Cano, P., Arsic, N., Popowych, Y. I., & Griebel, P. J. (2014). Two functionally distinct myeloid dendritic cell subpopulations are present in bovine blood. Developmental and Comparative Immunology, 44(2), 378–88. doi:10.1016/j.dci.2014.01.014
Guo, L., Junttila, I. S., & Paul, W. E. (2012). Cytokine-induced cytokine production by conventional and innate lymphoid cells. Trends in Immunology, 33(12), 598–606. doi:10.1016/j.it.2012.07.006
Guzman, E., Price, S., Poulsom, H., & Hope, J. (2011). Bovine γδ T cells: cells with multiple functions and important roles in immunity. Veterinary Immunology and Immunopathology, 148(1-2), 161–7. doi:10.1016/j.vetimm.2011.03.013

232
Halim, T. Y. F., Krauss, R. H., Sun, A. C., & Takei, F. (2012). Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity, 36(3), 451–63. doi:10.1016/j.immuni.2011.12.020
Hamerman, J. a, Ogasawara, K., & Lanier, L. L. (2005). NK cells in innate immunity. Current Opinion in Immunology, 17(1), 29–35. doi:10.1016/j.coi.2004.11.001
Haneklaus, M., O’Neill, L. a J., & Coll, R. C. (2013). Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Current Opinion in Immunology, 25(1), 40–5. doi:10.1016/j.coi.2012.12.004
Hardy, R. R. (2006). B-1 B cells: development, selection, natural autoantibody and leukemia. Current Opinion in Immunology, 18(5), 547–55. doi:10.1016/j.coi.2006.07.010
Harvey, J. . (2001). Atlas of veterinary hematology. Blood and bone marrow of domestic animals (Saunders C., p. 228). China.
Hayday, A., & Tigelaar, R. (2003). Immunoregulation in the tissues by γδ T cells. Nature Reviews. Immunology, 3(3), 233–42. doi:10.1038/nri1030
Hepworth, M. R., & Sonnenberg, G. F. (2014). Regulation of the adaptive immune system by innate lymphoid cells. Current Opinion in Immunology, 27, 75–82. doi:10.1016/j.coi.2014.01.013
Heredia, J. E., Mukundan, L., Chen, F. M., Mueller, A. a, Deo, R. C., Locksley, R. M., … Chawla, A. (2013). Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell, 153(2), 376–88. doi:10.1016/j.cell.2013.02.053
Holm, C. K., Paludan, S. R., & Fitzgerald, K. a. (2013). DNA recognition in immunity and disease. Current Opinion in Immunology, 25(1), 13–8. doi:10.1016/j.coi.2012.12.006
Holt, P. G., & Jones, C. A. (2000). The development of the immune system during pregnancy and early life. Allergy, 55(8), 688–97. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10955693
Hopkins, P. A., & Sriskandan, S. (2005). Mammalian Toll-like receptors: to immunity and beyond. Clinical and Experimental Immunology, 140(3), 395–407. doi:10.1111/j.1365-2249.2005.02801.x
Hou, B., Reizis, B., & DeFranco, A. L. (2008). Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity, 29(2), 272–82. doi:10.1016/j.immuni.2008.05.016
Hoyler, T., Connor, C. A., Kiss, E. A., & Diefenbach, A. (2013). T-bet and Gata3 in controlling type 1 and type 2 immunity mediated by innate lymphoid cells. Current Opinion in Immunology, 25(2), 139–47. doi:10.1016/j.coi.2013.02.007

233
Hoyler, T., Klose, C. S. N., Souabni, A., Turqueti-Neves, A., Pfeifer, D., Rawlins, E. L., … Diefenbach, A. (2013). The transcription factor GATA3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity, 37(4), 634–648. doi:10.1016/j.immuni.2012.06.020.The
Hu, G. (2012). p120-catenin : a novel regulator of innate immunity and inflammation. Crit Rev Immunol, 32(2), 127–138.
Ingvartsen, K. L., & Moyes, K. (2013). Nutrition, immune function and health of dairy cattle. Animal : An International Journal of Animal Bioscience, 7 Suppl 1, 112–22. doi:10.1017/S175173111200170X
Iwakura, Y., Ishigame, H., Saijo, S., & Nakae, S. (2011). Functional specialization of interleukin-17 family members. Immunity, 34(2), 149–62. doi:10.1016/j.immuni.2011.02.012
Janeway, C. A., Murphy, K., Travers, P., & Walport, M. (2009). Immunobiologie. 3e édition (De Boeck U., p. 889). Bruxelles.
Jump, R. L., & Levine, A. D. (2004). Mechanisms of natural tolerance in the intestine: implications for inflammatory bowel disease. Inflammatory Bowel Diseases, 10(4), 462–78. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15475760
Kang, J., & Coles, M. (2012). IL-7: the global builder of the innate lymphoid network and beyond, one niche at a time. Seminars in Immunology, 24(3), 190–7. doi:10.1016/j.smim.2012.02.003
Kang, S.-J., Liang, H.-E., Reizis, B., & Locksley, R. M. (2008). Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity, 29(5), 819–33. doi:10.1016/j.immuni.2008.09.017
Kasamatsu, J. (2013). Evolution of innate and adaptive immune systems in jawless vertebrates. Microbiology and Immunology, 57(1), 1–12. doi:10.1111/j.1348-0421.2012.00500.x
Kauf, A. C. W., Rosenbusch, R. F., Paape, M. J., & Bannerman, D. D. (2007). Innate immune response to intramammary Mycoplasma bovis infection. Journal of Dairy Science, 90(7), 3336–48. doi:10.3168/jds.2007-0058
Kayama, H., & Takeda, K. (2012). Regulation of intestinal homeostasis by innate and adaptive immunity. International Immunology, 24(11), 673–80. doi:10.1093/intimm/dxs094
Kim, D. D., & Song, W.-C. (2006). Membrane complement regulatory proteins. Clinical Immunology (Orlando, Fla.), 118(2-3), 127–36. doi:10.1016/j.clim.2005.10.014
Kim, H. Y., Chang, Y.-J., Subramanian, S., Lee, H.-H., Albacker, L. A., Matangkasombut, P., … Umetsu, D. T. (2011). Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. The Journal of Allergy and Clinical Immunology, 129(1), 216–27.e1–6. doi:10.1016/j.jaci.2011.10.036

234
Kinnebrew, M. A., Buffie, C. G., Diehl, G. E., Zenewicz, L. A., Leiner, I., Hohl, T. M., … Pamer, E. G. (2012). Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity, 36(2), 276–87. doi:10.1016/j.immuni.2011.12.011
Komori, H. K., Meehan, T. F., & Havran, W. L. (2006). Epithelial and mucosal γδ T cells. Current Opinion in Immunology, 18(5), 534–8. doi:10.1016/j.coi.2006.06.001
Konigshofer, Y., & Chien, Y.-H. (2006). γδ T cells - innate immune lymphocytes? Current Opinion in Immunology, 18(5), 527–33. doi:10.1016/j.coi.2006.07.008
Korbel, D. S., Finney, O. C., & Riley, E. M. (2004). Natural killer cells and innate immunity to protozoan pathogens. International Journal for Parasitology, 34(13-14), 1517–28. doi:10.1016/j.ijpara.2004.10.006
Koyasu, S., & Moro, K. (2011). Type 2 innate immune responses and the natural helper cell. Immunology, 132(4), 475–81. doi:10.1111/j.1365-2567.2011.03413.x
Koyasu, S., & Moro, K. (2012). Role of innate lymphocytes in infection and inflammation. Frontiers in Immunology, 3(May), 101. doi:10.3389/fimmu.2012.00101
Kugelberg, E. (2014). Innate lymphoid cells: nutrients direct immune balance. Nature Reviews. Immunology, 14(3), 137. doi:10.1038/nri3626
Kumar, V. (2013). Innate Lymphoid cells: New Paradigm in Immunology of Inflammation. Immunology Letters, 1–15. doi:10.1016/j.imlet.2013.11.003
Kumar, V. (2013). NKT-cell subsets : promoters and protectors in inflammatory liver disease. Journal of Hepatology, 59(3), 618–20. doi:10.1016/j.jhep.2013.02.032
Kumar, V., & Delovitch, T. L. (2014). Different subsets of natural killer T cells may vary in their roles in health and disease. Immunology, 142(Table 1), 321–336. doi:10.1111/imm.12247
Kumar, V., & Sharma, A. (2010). Mast cells: emerging sentinel innate immune cells with diverse role in immunity. Molecular Immunology, 48(1-3), 14–25. doi:10.1016/j.molimm.2010.07.009
Kushwah, R., & Hu, J. (2011). Complexity of dendritic cell subsets and their function in the host immune system. Immunology, 133(4), 409–19. doi:10.1111/j.1365-2567.2011.03457.x
Leavy, O. (2013). Innate-like lymphocytes : Will the real ILC1 please stand up? Nature Reviews. Immunology, 13(2), 67. doi:10.1038/nri3397
Lester, S. N., & Li, K. (2014). Toll-like receptors in antiviral innate immunity. Journal of Molecular Biology, 426(6), 1246–64. doi:10.1016/j.jmb.2013.11.024

235
Li, B. W. S., & Hendriks, R. W. (2013). Group 2 innate lymphoid cells in lung inflammation. Immunology, 140(3), 281–7. doi:10.1111/imm.12153
Li, Y., Shi, J., Yang, J., Ma, Y., Cheng, L., Zeng, J., … Liu, X. (2014). A Wnt/β-catenin negative feedback loop represses TLR-triggered inflammatory responses in alveolar epithelial cells. Molecular Immunology, 59(2), 128–35. doi:10.1016/j.molimm.2014.02.002
Lillie, B. N., Brooks, A. S., Keirstead, N. D., & Hayes, M. A. (2005). Comparative genetics and innate immune functions of collagenous lectins in animals. Veterinary Immunology and Immunopathology, 108(1-2), 97–110. doi:10.1016/j.vetimm.2005.07.001
Linde, A., Ross, C. R., Davis, E. G., Dib, L., Blecha, F., & Melgarejo, T. (2007). Innate immunity and host defense peptides in veterinary medicine. Journal of Veterinary Internal Medicine / American College of Veterinary Internal Medicine, 22(2), 247–65. doi:10.1111/j.1939-1676.2007.0038.x
Liu, Y. J. (2001). Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell, 106(3), 259–62. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11509173
Lodoen, M. B., & Lanier, L. L. (2006). Natural killer cells as an initial defense against pathogens. Current Opinion in Immunology, 18(4), 391–8. doi:10.1016/j.coi.2006.05.002
Lotz, M., Ménard, S., & Hornef, M. (2007). Innate immune recognition on the intestinal mucosa. International Journal of Medical Microbiology : IJMM, 297(5), 379–92. doi:10.1016/j.ijmm.2007.03.010
Louten, J., Boniface, K., & de Waal Malefyt, R. (2009). Development and function of TH17 cells in health and disease. The Journal of Allergy and Clinical Immunology, 123(5), 1004–11. doi:10.1016/j.jaci.2009.04.003
Maclachlan, N. J., Henderson, C., Schwartz-Cornil, I., & Zientara, S. (2013). The immune response of ruminant livestock to bluetongue virus : From type I interferon to antibody. Virus Research, 182, 71–7. doi:10.1016/j.virusres.2013.09.040
Malaviya, R., Navara, C., & Uckun, F. M. (2001). Role of Janus kinase 3 in mast cell-mediated innate immunity against gram-negative bacteria. Immunity, 18(2), 313–21. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11520465
Marcenaro, E., Dondero, A., & Moretta, A. (2006). Multi-directional cross-regulation of NK cell function during innate immune responses. Transplant Immunology, 17(1), 16–9. doi:10.1016/j.trim.2006.09.019
Mars, L. T., Araujo, L., Kerschen, P., Diem, S., Bourgeois, E., Van, L. P., … Herbelin, A. (2009). Invariant NKT cells inhibit development of the Th17 lineage. Proceedings of the National Academy of Sciences of the United States of America, 106(15), 6238–43. doi:10.1073/pnas.0809317106

236
Martin, L. B. (2009). Stress and immunity in wild vertebrates : timing is everything. General and Comparative Endocrinology, 163(1-2), 70–6. doi:10.1016/j.ygcen.2009.03.008
Mastellos, D., & Lambris, J. D. (2002). Complement : more than a “guard” against invading pathogens? Trends in Immunology, 23(10), 485–91. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12297420
McDermott, A. J., & Huffnagle, G. B. (2013). The microbiome and regulation of mucosal immunity. Immunology, 142(1), 24–31. doi:10.1111/imm.12231
Meyerholz, D. K., & Ackermann, M. R. (2005). Antimicrobial peptides and surfactant proteins in ruminant respiratory tract disease. Veterinary Immunology and Immunopathology, 108(1-2), 91–6. doi:10.1016/j.vetimm.2005.08.003
Michel, T., Poli, A., Domingues, O., Mauffray, M., Thérésine, M., Brons, N. H. C., … Zimmer, J. (2012). Mouse lung and spleen natural killer cells have phenotypic and functional differences, in part influenced by macrophages. PloS One, 7(12), e51230. doi:10.1371/journal.pone.0051230
Min, B., Le Gros, G., & Paul, W. E. (2005). Basophils : a potential liaison between innate and adaptive immunity. Allergology International : Official Journal of the Japanese Society of Allergology, 55(2), 99–104. doi:10.2332/allergolint.55.99
Mirchandani, A. S., Salmond, R. J., & Liew, F. Y. (2012). Interleukin-33 and the function of innate lymphoid cells. Trends in Immunology, 33(8), 389–96. doi:10.1016/j.it.2012.04.005
Mjösberg, J., & Spits, H. (2012). Type 2 innate lymphoid cells-new members of the “type 2 franchise” that mediate allergic airway inflammation. European Journal of Immunology, 42(5), 1093–6. doi:10.1002/eji.201242549
Montaldo, E., Vacca, P., Moretta, L., & Mingari, M. C. (2014). Development of human natural killer cells and other innate lymphoid cells. Seminars in Immunology, 26(2), 107–113. doi:10.1016/j.smim.2014.01.006
Monticelli, L. A., Sonnenberg, G. F., & Artis, D. (2012). Innate lymphoid cells : critical regulators of allergic inflammation and tissue repair in the lung. Current Opinion in Immunology, 24(3), 284–9. doi:10.1016/j.coi.2012.03.012
Moretta, A., Marcenaro, E., Sivori, S., Della Chiesa, M., Vitale, M., & Moretta, L. (2005). Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends in Immunology, 26(12), 668–75. doi:10.1016/j.it.2005.09.008
Moroso, V., Famili, F., Papazian, N., Cupedo, T., van der Laan, L. J. W., Kazemier, G., … Kwekkeboom, J. (2011). NK cells can generate from precursors in the adult human liver. European Journal of Immunology, 41(11), 3340–50. doi:10.1002/eji.201141760
Morris, D. G., Waters, S. M., McCarthy, S. D., Patton, J., Earley, B., Fitzpatrick, R., … Wathes, D. C. (2009). Pleiotropic effects of negative energy balance in the

237
postpartum dairy cow on splenic gene expression : repercussions for innate and adaptive immunity. Physiological Genomics, 39(1), 28–37. doi:10.1152/physiolgenomics.90394.2008
Nakae, S., Morita, H., Ohno, T., Arae, K., Matsumoto, K., & Saito, H. (2013). Role of interleukin-33 in innate-type immune cells in allergy. Allergology International : Official Journal of the Japanese Society of Allergology, 62(1), 13–20. doi:10.2332/allergolint.13-RAI-0538
Neill, D. R., & McKenzie, A. N. J. (2011). Nuocytes and beyond : new insights into helminth expulsion. Trends in Parasitology, 27(5), 214–21. doi:10.1016/j.pt.2011.01.001
Neill, D. R., Wong, S. H., Bellosi, A., Flynn, R. J., Daly, M., Langford, T. K. A., … Mckenzie, A. N. J. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature, 464(7293), 1367–1370. doi:10.1038/nature08900.Nuocytes
Nelson, C. D., Reinhardt, T. A., Thacker, T. C., Beitz, D. C., & Lippolis, J. D. (2010). Modulation of the bovine innate immune response by production of 1α,25-dihydroxyvitamin D(3) in bovine monocytes. Journal of Dairy Science, 93(3), 1041–9. doi:10.3168/jds.2009-2663
Neyt, K., Perros, F., GeurtsvanKessel, C. H., Hammad, H., & Lambrecht, B. N. (2012). Tertiary lymphoid organs in infection and autoimmunity. Trends in Immunology, 33(6), 297–305. doi:10.1016/j.it.2012.04.006
Ng, S. C., Kamm, M. A., Stagg, A. J., & Knight, S. C. (2010). Intestinal dendritic cells : their role in bacterial recognition, lymphocyte homing, and intestinal inflammation. Inflammatory Bowel Diseases, 16(10), 1787–807. doi:10.1002/ibd.21247
Ng, T. M., Kortmann, J., & Monack, D. M. (2013). Policing the cytosol-bacterial-sensing inflammasome receptors and pathways. Current Opinion in Immunology, 25(1), 34–9. doi:10.1016/j.coi.2012.11.009
Nishimura, T., Kitamura, H., Iwakabe, K., Yahata, T., Ohta, A., Sato, M., … Koda, T. (2000). The interface between innate and acquired immunity : glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. International Immunology, 12(7), 987–94. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10882410
Novák, K. (2014). Functional polymorphisms in Toll-like receptor genes for innate immunity in farm animals. Veterinary Immunology and Immunopathology, 157(1-2), 1–11. doi:10.1016/j.vetimm.2013.10.016
O’Neill, L. A. J. (2006). How Toll-like receptors signal : what we know and what we don ’ t know. Current Opinion in Allergy and Clinical Immunology, 18, 3–9. doi:10.1016/j.coi.2005.11.012

238
Oliveira, L. J., Barreto, R. S. N., Perecin, F., Mansouri-Attia, N., Pereira, F. T. V, & Meirelles, F. V. (2012). Modulation of maternal immune system during pregnancy in the cow. Reproduction in Domestic Animals = Zuchthygiene, 47 Suppl 4, 384–93. doi:10.1111/j.1439-0531.2012.02102.x
Olivier, M., Foret, B., Le Vern, Y., & Guilloteau, L. A. (2012). Capacities of migrating CD1b+ lymph dendritic cells to present Salmonella antigens to naive T cells. PloS One, 7(1), e30430. doi:10.1371/journal.pone.0030430
Olivier, M., Foret, B., Vern, Y. Le, Kerboeuf, D., & Guilloteau, L. A. (2013). Plasticity of migrating CD1b + and CD1b - lymph dendritic cells in the promotion of Th1 , Th2 and Th17 in response to Salmonella and helminth secretions. PloS One, 8(11), 1–11. doi:10.1371/journal.pone.0079537
Paape, M. ., Bannerman, D. D., Zhao, X., & Lee, J.-W. (2003). The bovine neutrophil : Structure and function in blood and milk. Veterinary Research, 34, 597–627. doi:10.1051/vetres
Pang, D. J., Neves, J. F., Sumaria, N., & Pennington, D. J. (2012). Understanding the complexity of γδ T-cell subsets in mouse and human. Immunology, 136(3), 283–90. doi:10.1111/j.1365-2567.2012.03582.x
Pastoret, P. P., Govaerts, A., & Bazin, H. (1990). Immunologie animale (Flammarion., p. 740). Paris.
Paul, W. E. (2013). Fundamental immunology. seventh edition (Lippincott., p. 1283). Philadelphia.
Pearson, C., Uhlig, H. H., & Powrie, F. (2012). Lymphoid microenvironments and innate lymphoid cells in the gut. Trends in Immunology, 33(6), 289–96. doi:10.1016/j.it.2012.04.004
Pépin, M., & Laaberki, M. H. (2014). Les anomalies congénitales d’origine infectieuse chez les ruminants. Le Point Vétérinaire - Expert Rural, 342, 2–7.
Peterson, L. W., & Artis, D. (2014). Intestinal epithelial cells : regulators of barrier function and immune homeostasis. Nature Reviews. Immunology, 14(3), 141–53. doi:10.1038/nri3608
Pierce, A., Legrand, D., & Mazurier, J. (2009). La lactoferrine : une protéine multifonctionnelle. Médecine Sciences, 25, 361–9.
Pishdadian, A., Varasteh, A.-R., & Sankian, M. (2012). Type 2 innate lymphoid cells : friends or foes-role in airway allergic inflammation and asthma. Journal of Allergy, 2012, 130937. doi:10.1155/2012/130937
Powell, N., Walker, A. W., Stolarczyk, E., Canavan, J. B., Gökmen, M. R., Marks, E., … Lord, G. M. (2012). The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity, 37(4), 674–84. doi:10.1016/j.immuni.2012.09.008

239
Qiu, J., Heller, J. J., Guo, X., Chen, Z. E., Fish, K., Fu, Y.-X., & Zhou, L. (2012). The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity, 36(1), 92–104. doi:10.1016/j.immuni.2011.11.011
Quintin, J., Cheng, S.-C., van der Meer, J. W., & Netea, M. G. (2014). Innate immune memory: towards a better understanding of host defense mechanisms. Current Opinion in Immunology, 29C, 1–7. doi:10.1016/j.coi.2014.02.006
Rainard, P., & Riollet, C. (2006). Innate immunity of the bovine mammary gland. Veterinary Research, 37, 369–400.
Rankin, L., Groom, J., Mielke, L. A., Seillet, C., & Belz, G. T. (2013). Diversity, function, and transcriptional regulation of gut innate lymphocytes. Frontiers in Immunology, 4(March), 22. doi:10.3389/fimmu.2013.00022
Reschner, A., Hubert, P., Delvenne, P., Boniver, J., & Jacobs, N. (2008). Innate lymphocyte and dendritic cell cross-talk : a key factor in the regulation of the immune response. Clinical and Experimental Immunology, 152(2), 219–26. doi:10.1111/j.1365-2249.2008.03624.x
Revillard, J. ., & ASSIM. (1995). Immunologie générale : 2ième édition (De Boeck U., p. 367). Paris.
Rogers, A. N., VanBuren, D. G., Hedblom, E., Tilahun, M. E., Telfer, J. C., & Baldwin, C. L. (2005). Function of ruminant γδ T cells is defined by WC1.1 or WC1.2 isoform expression. Veterinary Immunology and Immunopathology, 108(1-2), 211–7. doi:10.1016/j.vetimm.2005.08.008
Romera-Hernandez, M., Aparicio-Domingo, P., & Cupedo, T. (2013). Damage control : Rorγt+ innate lymphoid cells in tissue regeneration. Current Opinion in Immunology, 25(2), 156–60. doi:10.1016/j.coi.2013.01.007
Rowley, D. a, & Fitch, F. W. (2012). The road to the discovery of dendritic cells, a tribute to Ralph Steinman. Cellular Immunology, 273(2), 95–8. doi:10.1016/j.cellimm.2012.01.002
Sancho, D., & Reis Sousa, C. (2013). Sensing of cell death by myeloid C-type lectin receptors. Current Opinion in Immunology, 25(1), 46–52. doi:10.1016/j.coi.2012.12.007
Sanos, S. L., Vonarbourg, C., Mortha, A., & Diefenbach, A. (2011). Control of epithelial cell function by interleukin-22-producing RORγt+ innate lymphoid cells. Immunology, 132(4), 453–65. doi:10.1111/j.1365-2567.2011.03410.x
Schminkey, D. L., & Groer, M. (2014). Imitating a stress response: A new hypothesis about the innate immune system’s role in pregnancy. Medical Hypotheses, 82(6), 721–9. doi:10.1016/j.mehy.2014.03.013
Schulthess, J., Meresse, B., Ramiro-Puig, E., Montcuquet, N., Darche, S., Bègue, B., … Cerf-Bensussan, N. (2012). Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity, 37(1), 108–21. doi:10.1016/j.immuni.2012.05.013

240
Sirisinha, S. (2013). Evolutionary insights into the origin of innate and adaptive immune systems : different shades of grey. Asian Pac Journal Allergy Og Immunology, 32, 3–15.
Skyberg, J. A., Thornburg, T., Rollins, M., Huarte, E., Jutila, M. A., & Pascual, D. W. (2011). Murine and bovine γδ T cells enhance innate immunity against Brucella abortus infections. PloS One, 6(7), e21978. doi:10.1371/journal.pone.0021978
Smirnova, N. P., Webb, B. T., Bielefeldt-Ohmann, H., Van Campen, H., Antoniazzi, A. Q., Morarie, S. E., & Hansen, T. R. (2012). Development of fetal and placental innate immune responses during establishment of persistent infection with bovine viral diarrhea virus. Virus Research, 167(2), 329–36. doi:10.1016/j.virusres.2012.05.018
Sonnenberg, G. F., Monticelli, L. a, Alenghat, T., Fung, T. C., Hutnick, N. A., Kunisawa, J., … Artis, D. (2012). Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science, 336, 1321–1325.
Sonnenberg, G. F., Monticelli, L. A., Elloso, M. M., Fouser, L. A., & Artis, D. (2011). CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity, 34(1), 122–134. doi:10.1016/j.immuni.2010.12.009
Spits, H. (2011). Another armament in gut immunity : lymphotoxin-mediated crosstalk between innate lymphoid and dendritic cells. Cell Host & Microbe, 10(1), 3–4. doi:10.1016/j.chom.2011.07.002
Spits, H., Artis, D., Colonna, M., Diefenbach, A., Di Santo, J. P., Eberl, G., … Vivier, E. (2013). Innate lymphoid cells--a proposal for uniform nomenclature. Nature Reviews. Immunology, 13(2), 145–9. doi:10.1038/nri3365
Spits, H., & Mjösberg, J. (2012). The aryl hydrocarbon receptor : a sentinel safeguarding the survival of immune cells in the gut. Immunity, 36(1), 5–7. doi:10.1016/j.immuni.2012.01.004
Stelwagen, K., Carpenter, E., Haigh, B., Hodgkinson, a, & Wheeler, T. T. (2009). Immune components of bovine colostrum and milk. Journal of Animal Science, 87(13 Suppl), 3–9. doi:10.2527/jas.2008-1377
Stevens, A., & Lowe, J. (1993). Histologie (Pradel Ed., p. 378). Paris.
Stockinger, B., Veldhoen, M., & Martin, B. (2007). Th17 T cells : linking innate and adaptive immunity. Seminars in Immunology, 19(6), 353–61. doi:10.1016/j.smim.2007.10.008
Strober, W. (2010). The LTi cell, an immunologic chameleon. Immunity, 33(5), 650–2. doi:10.1016/j.immuni.2010.11.016
Summerfield, A., Guzylack-Piriou, L., Harwood, L., & McCullough, K. C. (2009). Innate immune responses against foot-and-mouth disease virus : current understanding and future directions. Veterinary Immunology and Immunopathology, 128(1-3), 205–10. doi:10.1016/j.vetimm.2008.10.296

241
Suriyasathaporn, W., Heuer, C., Noordhuizen-Stassen, N. E., & Schukken, Y. H. (2000). Hyperketonemia and the impairment of udder defense : a review. Veterinary Research, 31, 397–412.
Sutton, C. E., Lalor, S. J., Sweeney, C. M., Brereton, C. F., Lavelle, E. C., & Mills, K. H. G. (2009). Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity, 31(2), 331–41. doi:10.1016/j.immuni.2009.08.001
Sutton, C. E., Mielke, L. A., & Mills, K. H. G. (2012). IL-17-producing γδ T cells and innate lymphoid cells. European Journal of Immunology, 42(9), 2221–31. doi:10.1002/eji.201242569
Tabel, H. (1996). Alternative pathway of complement in ruminants : role in infection. Veterinary Immunology and Immunopathology, 54(1-4), 117–21. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8988854
Tait Wojno, E. D., & Artis, D. (2012). Innate lymphoid cells : balancing immunity, inflammation, and tissue repair in the intestine. Cell Host & Microbe, 12(4), 445–57. doi:10.1016/j.chom.2012.10.003
Takeda, E., Nakagawa, S., Nakaya, Y., Tanaka, A., Miyazawa, T., & Yasuda, J. (2012). Identification and functional analysis of three isoforms of bovine BST-2. PloS One, 7(7), e41483. doi:10.1371/journal.pone.0041483
Thaiss, C. A., Levy, M., Suez, J., & Elinav, E. (2014). The interplay between the innate immune system and the microbiota. Current Opinion in Immunology, 26, 41–8. doi:10.1016/j.coi.2013.10.016
Theilgaard-Mönch, K., Porse, B. T., & Borregaard, N. (2006). Systems biology of neutrophil differentiation and immune response. Current Opinion in Immunology, 18(1), 54–60. doi:10.1016/j.coi.2005.11.010
Theoharides, T. C., Alysandratos, K.-D., Angelidou, A., Delivanis, D.-A., Sismanopoulos, N., Zhang, B., … Kalogeromitros, D. (2012). Mast cells and inflammation. Biochimica et Biophysica Acta, 1822(1), 21–33. doi:10.1016/j.bbadis.2010.12.014
Toka, F. N., & Golde, W. T. (2013). Cell mediated innate responses of cattle and swine are diverse during foot-and-mouth disease virus (FMDV) infection: a unique landscape of innate immunity. Immunology Letters, 152(2), 135–43. doi:10.1016/j.imlet.2013.05.007
Tumanov, A. V, Koroleva, E. P., Guo, X., Wang, Y., Kruglov, A., Nedospasov, S., & Fu, Y.-X. (2011). Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host & Microbe, 10(1), 44–53. doi:10.1016/j.chom.2011.06.002
Turvey, S. E., & Broide, D. H. (2010). Innate immunity. The Journal of Allergy and Clinical Immunology, 125(Suppl 2), S24–32. doi:10.1016/j.jaci.2009.07.016

242
Uematsu, S., & Fujimoto, K. (2010). The innate immune system in the intestine. Microbiology and Immunology, 54(11), 645–57. doi:10.1111/j.1348-0421.2010.00267.x
Ullrich, E., Ménard, C., Flament, C., Terme, M., Mignot, G., Bonmort, M., … Zitvogel, L. (2008). Dendritic cells and innate defense against tumor cells. Cytokine & Growth Factor Reviews, 19(1), 79–92. doi:10.1016/j.cytogfr.2007.10.009
Upadhyay, V., & Fu, Y.-X. (2014). Lymphotoxin organizes contributions to host defense and metabolic illness from innate lymphoid cells. Cytokine & Growth Factor Reviews, 25(2), 227–33. doi:10.1016/j.cytogfr.2013.12.007
Van Kaer, L., & Joyce, S. (2005). Innate immunity : NKT cells in the spotlight. Current Biology : CB, 15(11), 429–431. doi:10.1016/j.cub.2005.05.032
Varela, M., Schnettler, E., Caporale, M., Murgia, C., Barry, G., McFarlane, M., … Palmarini, M. (2013). Schmallenberg virus pathogenesis, tropism and interaction with the innate immune system of the host. PLoS Pathogens, 9(1), e1003133. doi:10.1371/journal.ppat.1003133
Vercelli, D., Gozdz, J., & von Mutius, E. (2013). Innate lymphoid cells in asthma: when innate immunity comes in a Th2 flavor. Current Opinion in Allergy and Clinical Immunology, 14(1), 29–34. doi:10.1097/ACI.0000000000000023
Verdier, J., & Ruemmele, F. M. (2013). Molecular mechanisms and cell targets of Th17 cells in the gastrointestinal tract : an innate sense of adaptivity. International Reviews of Immunology, 32(5-6), 475–92. doi:10.3109/08830185.2013.829471
Vomhof-Dekrey, E. E., Yates, J., & Leadbetter, E. A. (2014). Invariant NKT cells provide innate and adaptive help for B cells. Current Opinion in Immunology, 28C(Dc), 12–17. doi:10.1016/j.coi.2014.01.007
Vonarbourg, C., & Diefenbach, A. (2012). Multifaceted roles of interleukin-7 signaling for the development and function of innate lymphoid cells. Seminars in Immunology, 24(3), 165–74. doi:10.1016/j.smim.2012.03.002
Vosshenrich, C. A. J., & Di Santo, J. P. (2013). Developmental programming of natural killer and innate lymphoid cells. Current Opinion in Immunology, 25(2), 130–8. doi:10.1016/j.coi.2013.02.002
Walker, J. a, & McKenzie, A. N. J. (2013). Development and function of group 2 innate lymphoid cells. Current Opinion in Immunology, 25(2), 148–55. doi:10.1016/j.coi.2013.02.010
Walker, J. A., Barlow, J. L., & McKenzie, A. N. J. (2013). Innate lymphoid cells-how did we miss them? Nature Reviews. Immunology, 13(2), 75–87. doi:10.1038/nri3349
Wang, Y., Koroleva, E. P., Kruglov, A. A., Kuprash, D. V, Nedospasov, S. A., Fu, Y.-X., & Tumanov, A. V. (2010). Lymphotoxin β receptor signaling in intestinal

243
epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity, 32(3), 403–13. doi:10.1016/j.immuni.2010.02.011
Ward, A. E., & Rosenthal, B. M. (2014). Evolutionary responses of innate immunity to adaptive immunity. Infection, Genetics and Evolution, 21, 492–6. doi:10.1016/j.meegid.2013.12.021
Wilhelm, C., Hirota, K., Stieglitz, B., Snick, J. Van, & Tolaini, M. (2012). Interleukin 9 fate reporter reveals induction of innate IL-9 response in lung inflammation, 12(11), 1071–1077. doi:10.1038/ni.2133.Interleukin
Willems, F., Vollstedt, S., & Suter, M. (2009). Phenotype and function of neonatal DC. European Journal of Immunology, 39(1), 26–35. doi:10.1002/eji.200838391
Williams, A. . (2012). Immunology : Mucosal and body surface defenses (Wiley-Blac., p. 380). Oxford.
Wong, S. H., Walker, J. A., Jolin, H. E., Drynan, L. F., Camelo, A., Barlow, J. L., … Andrew, N. J. (2012). Rorα is essential for nuocyte development. Nature Immunology, 13(3), 229–236. doi:10.1038/ni.2208.Ror
Wu, L., & Liu, Y.-J. (2007). Development of dendritic-cell lineages. Immunity, 26(6), 741–50. doi:10.1016/j.immuni.2007.06.006
Xue, L., Salimi, M., Panse, I., Mjösberg, J. M., McKenzie, A. N. J., Spits, H., … Ogg, G. (2013). Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. The Journal of Allergy and Clinical Immunology, 133(4), 1184–94. doi:10.1016/j.jaci.2013.10.056
Yamane, D., Kato, K., Tohya, Y., & Akashi, H. (2008). The relationship between the viral RNA level and upregulation of innate immunity in spleen of cattle persistently infected with bovine viral diarrhea virus. Veterinary Microbiology, 129(1-2), 69–79. doi:10.1016/j.vetmic.2007.11.004
Yanai, H., Ban, T., & Taniguchi, T. (2011). Essential role of high-mobility group box proteins in nucleic acid-mediated innate immune responses. Journal of Internal Medicine, 270(4), 301–8. doi:10.1111/j.1365-2796.2011.02433.x
Yasuda, K., Muto, T., Kawagoe, T., Matsumoto, M., Sasaki, Y., Matsushita, K., … Nakanishi, K. (2012). Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proceedings of the National Academy of Sciences of the United States of America, 109(9), 3451–6. doi:10.1073/pnas.1201042109
Yoo, J.-K., Kim, T. S., Hufford, M. M., & Braciale, T. J. (2013). Viral infection of the lung: Host response and sequelae. The Journal of Allergy and Clinical Immunology, 1–14. doi:10.1016/j.jaci.2013.06.006
Yu, J., Freud, A. G., & Caligiuri, M. A. (2013). Location and cellular stages of natural killer cell development. Trends in Immunology, 1–10. doi:10.1016/j.it.2013.07.005

244
Yu, S., Kim, H. Y., Chang, Y.-J., DeKruyff, R. H., & Umetsu, D. T. (2014). Innate lymphoid cells and asthma. The Journal of Allergy and Clinical Immunology, 133(4), 943–50; quiz 51. doi:10.1016/j.jaci.2014.02.015
Zanoni, I., & Granucci, F. (2012). Regulation and dysregulation of innate immunity by NFAT signaling downstream of pattern recognition receptors (PRRs). European Journal of Immunology, 42(8), 1924–31. doi:10.1002/eji.201242580
Zenewicz, L. a, & Flavell, R. a. (2013). Recent advances in IL-22 biology. International Immunology, 23(3), 159–63. doi:10.1093/intimm/dxr001
Zhang, S.-Y., Herman, M., Ciancanelli, M. J., Pérez de Diego, R., Sancho-Shimizu, V., Abel, L., & Casanova, J.-L. (2013). TLR3 immunity to infection in mice and humans. Current Opinion in Immunology, 25(1), 19–33. doi:10.1016/j.coi.2012.11.001
Zlotnik, A., & Yoshie, O. (2000). Chemokines: a new classification system and their role in immunity. Immunity, 12(2), 121–7. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10714678
Zlotnik, A., & Yoshie, O. (2012). The chemokine superfamily revisited. Immunity, 36(5), 705–16. doi:10.1016/j.immuni.2012.05.008

245
Cytokine Source majeure Action Signalisation
INT
ER
LE
UK
INE
S
IL-1 Macrophages,
cellules épithéliales Fièvre, activation des cellules T et macrophages
IL-2 Th1
Facteur de croissance des cellules T, des cellules B, de la production
d’anticorps, augmente l’activité des NK, stimule les macrophages, effet
antitumoral, stimule le développement des Treg, stimule la différenciation Th1
et Th2 et inhibe la différenciation Th17
STAT1, STAT3,
STAT5
IL-3
Cellules T, cellules
épithéliales et
thymiques
Stimule la prolifération, la survie et le développement des cellules
progénitrices hématopoïétiques, production d’IL-4 par basophiles, stimule la
phagocytose et la cytotoxicité
IL-4 Th2 Prolifération des cellules B, changement de classe d’Ig, augmente le CMH
II, différenciation des cellules Th2, effet antitumoral STAT6, STAT5
IL-5 Cellules T Stimule la lignée éosinophilique, production d’Ig par les cellules B
IL-6
Cellules T,
macrophages et
cellules endothéliales
Croissance des cellules B et T, induction des protéines de la phase aiguë,
fièvre STAT3
IL-7 Cellules stromales Croissance des thymocytes, croissance des cellules T, survie et croissance
des cellules T périphériques, homéostasie des cellules T CD8+ et CD4+
STAT1, STAT3,
STAT5
L’IL-8 est une chimiokine (CXCL-8)
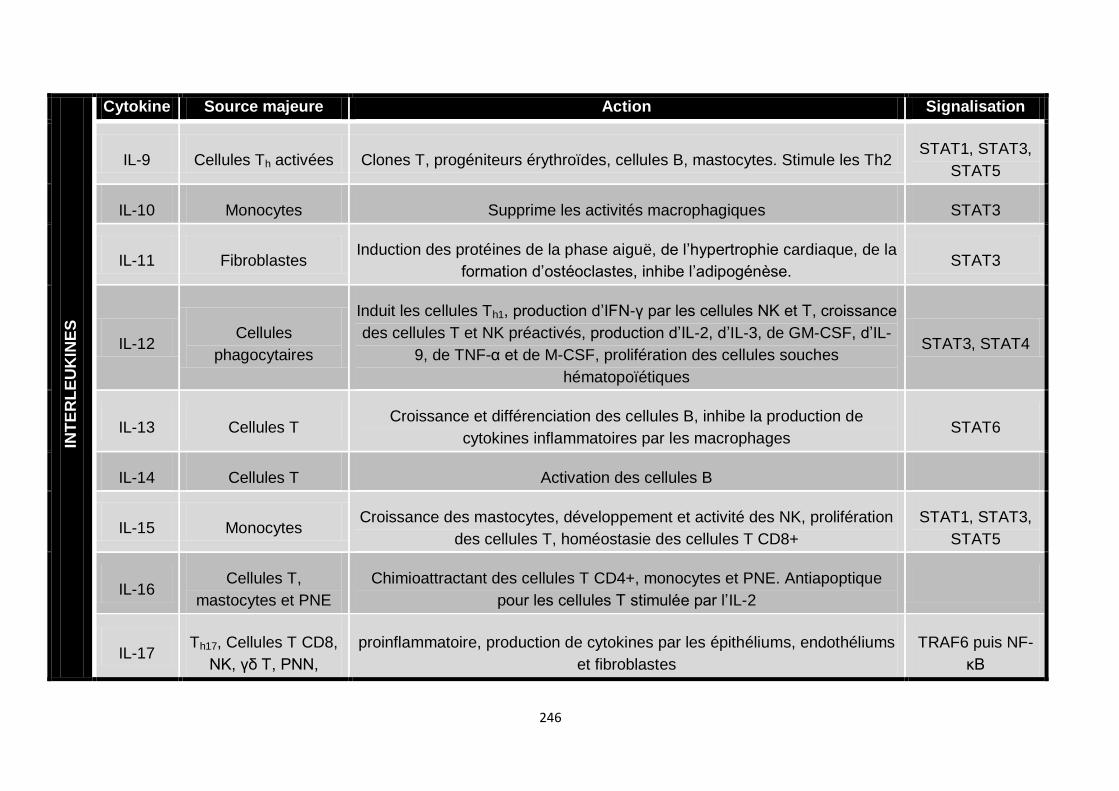
246
INT
ER
LE
UK
INE
S
Cytokine Source majeure Action Signalisation
IL-9 Cellules Th activées Clones T, progéniteurs érythroïdes, cellules B, mastocytes. Stimule les Th2 STAT1, STAT3,
STAT5
IL-10 Monocytes Supprime les activités macrophagiques STAT3
IL-11 Fibroblastes Induction des protéines de la phase aiguë, de l’hypertrophie cardiaque, de la
formation d’ostéoclastes, inhibe l’adipogénèse. STAT3
IL-12 Cellules
phagocytaires
Induit les cellules Th1, production d’IFN-γ par les cellules NK et T, croissance
des cellules T et NK préactivés, production d’IL-2, d’IL-3, de GM-CSF, d’IL-
9, de TNF-α et de M-CSF, prolifération des cellules souches
hématopoïétiques
STAT3, STAT4
IL-13 Cellules T Croissance et différenciation des cellules B, inhibe la production de
cytokines inflammatoires par les macrophages STAT6
IL-14 Cellules T Activation des cellules B
IL-15 Monocytes Croissance des mastocytes, développement et activité des NK, prolifération
des cellules T, homéostasie des cellules T CD8+
STAT1, STAT3,
STAT5
IL-16 Cellules T,
mastocytes et PNE
Chimioattractant des cellules T CD4+, monocytes et PNE. Antiapoptique
pour les cellules T stimulée par l’IL-2
IL-17 Th17, Cellules T CD8,
NK, γδ T, PNN,
proinflammatoire, production de cytokines par les épithéliums, endothéliums
et fibroblastes
TRAF6 puis NF-
κB

247
Cytokine Source majeure Action Signalisation
IL-18 Macrophages Induit la production d’IFN-γ par les cellules T et NK, favorise la
différenciation des Th1
IL-19 Monocytes Induit l’expression de l’IL-6 et du TNF-α par les monocytes
IL-20 Th1 Stimule la prolifération des kératinocytes et la production de TNF-α
IL-21 Cellules T CD4+
activées
Prolifération des cellules T, inhibe la prolifération des cellules B, effet
antitumoral, proapoptose pour les cellules V et NK, stimule la prolifération
des Tfh et Th17
STAT1, STAT3,
STAT5
IL-22 NK Production des protéines de la phase aiguë STAT3
IL-23 Cellules dendritiques Maintien et prolifération des Th17, augmente la production d’IFN-γ
INT
ER
LE
UK
INE
S
IL-24
Monocytes et
cellules T Inhibe la croissance tumorale
IL-25 Cellules Th2,
mastocytes Favorise la production des cytokines Th2
IL-26 Cellules T et NK
IL-27
Monocytes,
macrophages,
cellules dendritiques
Induit l’expression du T-bet

248
INT
ER
LE
UK
INE
S
Cytokine Source majeure Action Signalisation
L’IL-28 et L’IL-29 sont des IFN-λ
IL-31 Cellules T activées
IL-33 MyD88
IL-35 Cellules T reg Immunosuppression
Fa
cte
urs
de
sti
mu
lati
on
de
s
co
lon
ies
M-CSF
Cellules T, cellules
stromales de la
moelle
Développement de la lignée monocytaire
G-CSF Fibroblastes,
monocytes Développement des PNN STAT3
GM-CSF Cellules T,
macrophages
Développement de la lignée myélomonocytaire, surtout les cellules
dendritiques STAT5
Inte
rféro
ns
IFNα Leucocytes, cellules
dendritiques Action antivirale, augmente l’expression du CMH I
STAT1, STAT2,
STAT4
IFNβ Fibroblastes Action antivirale, augmente l’expression du CMH I STAT1, STAT2,
STAT4
IFNλ Propriétés antivirales

249
Cytokine Source majeure Action Signalisation
IFNτ Reconnaissance de la gestation
IFN-γ Cellules T et NK
Activation des macrophages, augmentation de l’expression du CMH et de
l’apprêtement antigénique, commutation de classe des Ig, supprime la
réponse Th2
STAT1
TN
F
TNF-α
Th, Macrophages,
cellules dendritiques,
mastocytes, NK,
cellules B
Cytotoxicité tumorale, cachexie, induit la sécrétion de cytokines, active les
macrophages, propriétés antivirales
TNF-β
Lympho-
toxine
Th et cellules T
cytotoxique
Cytotoxicité tumorale, augmente la phagocytose des PNN et macrophages,
antiviral, impliqué dans le développement des organes lymphoïdes.

250
Classe Chimiokine Cellule productrice Cellule attirée Principaux effets
CXC
CXCL-8
(IL-8)
Monocytes, macrophages,
fibroblastes, kératinocytes, cellules
endothéliales PNN, cellules T naïves
Mobilise, active et dégranule les PNN,
angiogenèse
CXCL-7 Plaquettes PNN Active les PNN, angiogenèse
CXCL-1
CXCL-2
CXCL-3
Monocytes, fibroblastes,
endothéliums
PNN, cellules T naïves,
fibroblastes Active les PNN, fibroplasie, angiogenèse
CXCL-10 Kératinocytes, monocytes, cellules
T, fibroblastes, endothéliums
Cellules T au repos, cellules
NK, monocytes
Immunostimulant, anti-angiogenèse, favorise
l’immunité Th1
CXCL-12 Cellules stromales Cellules T naïves,
progéniteurs, cellules B
Développement des cellules, écotaxie des
lymphocytes
CXCL-13 Cellules stromales Cellules B Ecotaxie des lymphocytes
CC
CCL-3
(MIP-1α)
Monocytes, cellules T, mastocytes,
fibroblastes
Monocytes, cellules NK et T,
PNB, cellules dendritiques Défense antivirale, favorise l’immunité Th1
CCL-4
(MIP-1β)
Monocytes, macrophages,
neutrophiles, endothéliums
Monocytes, cellules NK et T,
cellules dendritiques
CCL2
(MCP-1)
Monocytes, macrophages,
fibroblastes, kératinocytes
Monocytes, cellules NK et T,
PNB, cellules dendritiques
Activation des macrophages, libération
d’histamine par les PNB, favorise l’immunité
Th2

251
Classe Chimiokine Cellule productrice Cellule attirée Principaux effets
CC
CCL-5
(RANTES) Cellules T, endothéliums, plaquettes
Monocytes, cellules NK et T,
PNB, PNE, cellules
dendritiques
Dégranulation des PNB, activation des
cellules T, inflammation chronique
CCL11
(Eotaxine)
Endothéliums, monocytes,
épithéliums, cellules T PNE, monocytes, cellules T Allergie
CCL-18 Cellules dendritiques Cellules T naïves Activation des cellules T naïves
C
XCL-1
(lymphotacti
ne)
Cellules T CD4+ CD8+ Thymocytes, cellules
dendritiques, cellules NK Trafic et développement des lymphocytes
CX3C CX3CL-1 Monocytes, endothélium, cellules
microgliales Monocytes et cellules T Adhérence leucocyte-endothélium

252
REPONSE A UNE BACTERIE EXTRACELLULAIRE : Orientation vers une réponse Th2
Pathogénie : destruction des tissus par l’inflammation et action de toxines (endotoxines : composants de la paroi bactérienne comme le LPS / exotoxines : sécrétées
activement par les bactéries, beaucoup plus cytotoxiques)
Réponse de l’organisme : activation du complément (lyse moins efficace sur les bactéries Gram+, à paroi plus fine), inflammation et phagocytose puis réponse
adaptative de type humoral.
Récepteurs impliqués : TLR2, 4, 5 et 9, récepteur au mannose, récepteur scavenger.
Cellules Chimiokines Mode d’activation Cytokines produites Actions
Macrophages
Cellule sentinelle qui détecte le
pathogène : pas de
chimiokines ;
Puis attirés par les produits
libérés par les granules des
PNN
Récepteurs scavenger et au
mannose ; TLR TNF-α ; IL-1 ; IL-6 ; IL-8
Phagocytose, attraction des
PNN
Cellules dendritiques Cellule sentinelle qui détecte le
pathogène
IL-23
TGF-β
Phagocytose
Attraction des Th17 (IL-23)
Présentation d’antigène
PNN Attirés par CXCL-8 ; CXCL-7 Récepteurs aux opsonines
(complément et anticorps)
Phagocytose +++ ;
NETose
Mastocytes Attirés par les granules des
PNN TNF-α Attraction des PNN
Th17 Attirés par IL-23 et TGF-β des
cellules dendritiques TLR4
IL-17 ; IL-22
IL-2 ; IL-8 ; CXCL-1 ;
CXCL-2
Induit une forte production de
peptides antimicrobiens (IL-17)
et d’un facteur antiapoptique
pour maintenir l’intégrité
épithéliale (IL-22)
ILCs 3 NCR- IL-23 des cellules dendritiques IL-17 ; IFN-γ ?; IL-22
LTi IL-23 des cellules dendritiques
Activation par les cellules
dendritiques (nécessité de
LTβR)
IL-22
Moyens d’échappement : variation antigénique, inhibition de l’activation du complément, résistance à la phagocytose

253
REPONSE A UNE BACTERIE INTRACELLULAIRE : Orientation vers une réponse Th1
Pathogénie : survie et réplication à l’intérieur des cellules
Réponse de l’organisme : Réponse Th1 mise en place par PNN puis macrophages et NK. + réponse adaptative
Récepteurs impliqués : TLR et NLR intracellulaires
Cellules Chimiokines Mode d’activation Cytokines produites Actions
Macrophages Cellule infectée TLR et NLR intracellulaire (si
macrophage est infecté) TNF, IL-1β, IL-12 et IL-15
Active les NK
Autophagie
PNN Cellule activée par l’IL1β des
macrophages infectés
Destruction directe des
bactéries dans l’espace
extracellulaire et cytotoxictié
pour les cellules infectées
NK
Cellule sentinelle (pas de
chimiokines pour l’attirer)
Sécrétion de chimiokines
activée par l’le TNF et l’IL-12
des macrophages infectés
Détecte des néoantigènes sur les
cellules infectées IFN-γ
Active les macrophages et la
phagocytose
Cellules dendritiques Cellule infectée, ou attirée par
CCL-2 TLR et NLR intracellulaire TNF, NO, IL-12 et IL-15
Active les NK
Autophagie
Présentation d’antigènes
Moyens d’échappement : inhibition de la formation du phagolysosome, inactivation des réactifs oxygénés, rupture du phagolysosome, inhibition de l’apoptose

254
REPONSE A UN VIRUS
Pathogénie : Virus dans une capside +/- bicouche lipidique si virus enveloppé.
Phase extracellulaire : virus sensible aux Ac, à la phagocytose et à l’action du complément.
Phase intracellulaire : virus protégé. Infections latentes
Réponse de l’organisme : action lytique des NK, production d’IFN-γ et réponse adaptative (cellules T CD8+)
Récepteurs impliqués : TLR, CLR, NLR.
Cellules Chimiokines Mode d’activation Cytokines produites Actions
Cellule infectée / Production des protéines
virales /
Présentation des particules
virales via le CMH I aux
cellules T CD8+=> lyse de la
cellule
Cellule dendritique
plasmacytoïdes PRR IFN-α
Active la transcription de gènes
empêchant la sortie du virus de
la cellule : permet aux cellules
voisines de ne pas être
permissives
NK IFN
CMH I absent ou altéré
Les IFN-α et –γ augmentent la
cytotoxicité
Lyse des cellules infectées
Cellule γδ T Cytotoxicité
Macrophages Détectent les particules virales
dans le milieu extracellulaire
IFN-β, IFN-γ ;
IL-1 ; IL-12 ; TNF-α
Rend les cellules voisines non
permissives
Proinflammatoire : active les
cellules NK, T et PNN
Moyens d’échappement : Modification des antigènes de surface, dégradation des composants des voies de signalisation des TLR ou inhibition de la transcription des
gènes antiviraux.

255
REPONSE A UN HELMINTHE : orientation vers une réponse Th2
Pathogénie : Vers adultes présents dans le tube digestif. Parfois nécessité de migration trachéale.
Réponse de l’organisme : Réponse Th2 : phagocytose non possible mais libération de substances lytiques et augmentation de la production de mucus et du
péristaltisme. Immunité adaptative.
Récepteurs impliqués : TLR, CLR, NLR.
Cellules Chimiokines Mode d’activation Cytokines produites Actions
Mastocytes PRR IL-4, IL-5 et IL-13
Histamine ; héparine ;
protéases libérées, active les
cellules T
PNE IL-13, CCL-11 PRR Libération des granules avec
des protéines cationiques
ILCs 2 IL-25, IL-33 des cellules
épithéliales IL-4, IL-5 et IL-13
Augmentation de la production
de mucus, prolifération des
lymphocytes B, activation es
PNN et macrophages
Cellules dendritiques PRR IL-4, IL-5 et IL-13 Polarisation des Th2 et
activation des ILCs 2
Moyens d’échappement : Changement de cuticule à chaque stade, surface riche en carbohydrates amorphes.
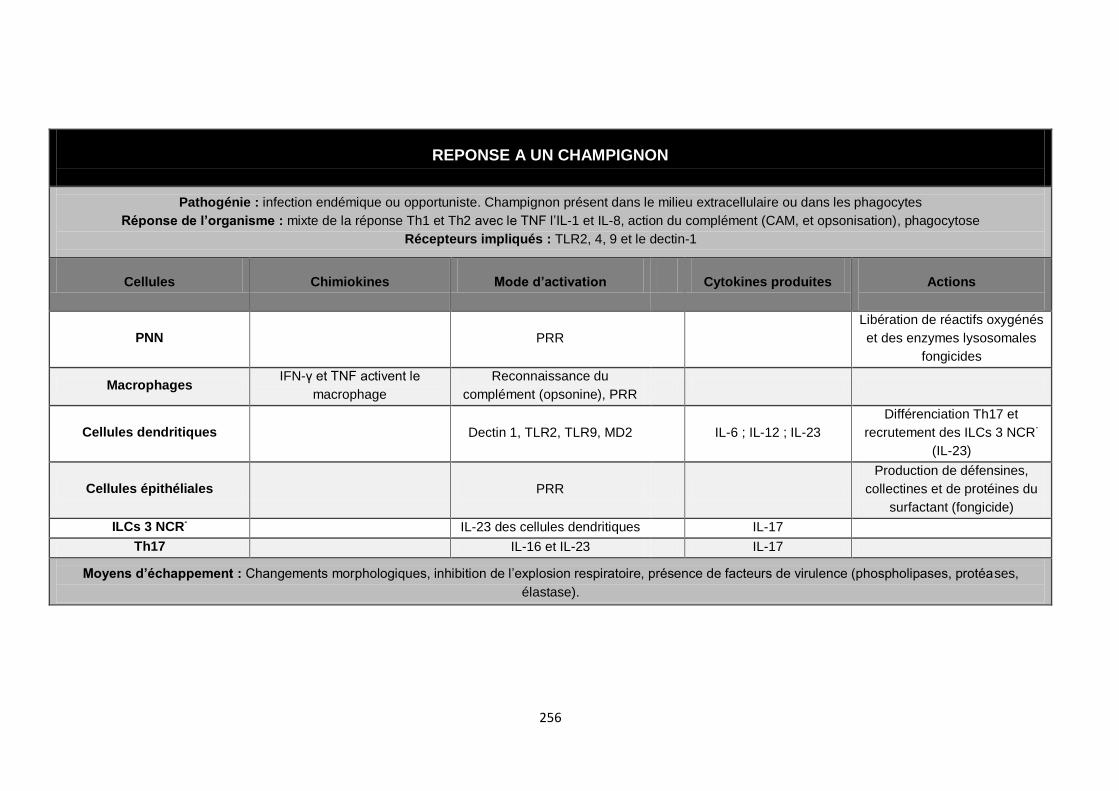
256
REPONSE A UN CHAMPIGNON
Pathogénie : infection endémique ou opportuniste. Champignon présent dans le milieu extracellulaire ou dans les phagocytes
Réponse de l’organisme : mixte de la réponse Th1 et Th2 avec le TNF l’IL-1 et IL-8, action du complément (CAM, et opsonisation), phagocytose
Récepteurs impliqués : TLR2, 4, 9 et le dectin-1
Cellules Chimiokines Mode d’activation Cytokines produites Actions
PNN PRR
Libération de réactifs oxygénés
et des enzymes lysosomales
fongicides
Macrophages IFN-γ et TNF activent le
macrophage
Reconnaissance du
complément (opsonine), PRR
Cellules dendritiques Dectin 1, TLR2, TLR9, MD2 IL-6 ; IL-12 ; IL-23
Différenciation Th17 et
recrutement des ILCs 3 NCR-
(IL-23)
Cellules épithéliales PRR
Production de défensines,
collectines et de protéines du
surfactant (fongicide)
ILCs 3 NCR- IL-23 des cellules dendritiques IL-17
Th17 IL-16 et IL-23 IL-17
Moyens d’échappement : Changements morphologiques, inhibition de l’explosion respiratoire, présence de facteurs de virulence (phospholipases, protéases,
élastase).

257
NOM PRENOM : SAVOYAT CELINE
TITRE : L’immunité innée : bilan des connaissances actuelles chez les
ruminants et sur les nouvelles cellules lymphoïdes innées (ILC)
Thèse d’Etat de Doctorat Vétérinaire : Lyon, 26 septembre 2014
RESUME :
Ce travail démontre l’importance du système immunitaire inné. Il recense les cellules impliquées, leur
description, leurs rôles et leurs relations.
L’inflammation joue un rôle central dans cette immunité innée. Elle est très souvent suffisante pour
stopper les infections débutantes. Elle constitue une étape indispensable à l’activation du système
adaptatif.
Le système inné est présent chez tous les êtres vivants. Son importance dans les défenses de la mamelle de
la vache et la variabilité qui existe entre les individus offrent des possibilités de sélection des animaux pour
prévenir les mammites.
Les nombreuses collaborations entre les systèmes immunitaires inné et adaptatif, et la présence de cellules
intervenant dans les deux systèmes estompent cette limite.
La découverte récente des cellules lymphoïdes innées (ILC pour Innate Lymphoid Cell) perturbe cette
vision dichotomique de l’immunité puisque les lymphocytes étaient jusqu’à présent classés parmi les
cellules de l’immunité adaptative. L’étude de ces cellules a révélé leur implication non spécifique dans
l’immunité contre les virus, les bactéries, et les tumeurs, mais aussi dans l’homéostasie intestinale, la mise
en place des tissus lymphoïdes et la réparation tissulaire. Les ILCs interviennent également dans certains
dérèglements du système immunitaire responsables de maladies chroniques.
MOTS CLES : - Immunité
- Ruminants
- Tissu lymphoïde
JURY :
Président : Madame le Professeur TRAVERSE GLEHEN Alexandra
1er Assesseur : Monsieur le Professeur PEPIN Michel
2ème Assesseur : Madame le Professeur ARCANGIOLI Marie-Anne
DATE DE SOUTENANCE : 26 septembre 2014
ADRESSE DE L’AUTEUR : 4 rue Lafontaine
38170 SEYSSINET


















