
THESE EN VUE DE L’OBTENTION DU DIPLOME DE ...3 I.2 Objectif Notre objectif est de rechercher le...
103
République Algérienne Démocratique et Populaire Ministère de l'Enseignement Supérieur et de la Recherche Scientifique Université d’Oran Faculté des Sciences Département de chimie THESE EN VUE DE L’OBTENTION DU DIPLOME DE DOCTORAT D’ETAT Spécialité : Chimie des matériaux CONTRIBUTION A L’ETUDE DE LA CONVERSION DU CHLOROMETHANE SUR LA H-ZSM5 Mécanisme et effet de structure Présenté par : M R Chikh BOUKRATEM Soutenue le 18 mai 2006 devant le jury composé de Président : Pr. Y. KHATIR (Université d’Oran) Examinateurs : Pr . Z DERRICHE (U.S.T.O) : Pr. O. CHERIFI (U.S.T.H.B) Rapporteur : Pr . Y. BENTAARIT (IRC/ LYON) Co- Rapporteur : Pr. A. BENGUEDDACH (Université d’Oran). Membre invitée : Pr. L. CHERIF (Université. Tlemcen)
Transcript of THESE EN VUE DE L’OBTENTION DU DIPLOME DE ...3 I.2 Objectif Notre objectif est de rechercher le...

République Algérienne Démocratique et Populaire Ministère de l'Enseignement Supérieur et de la Recherche Scientifique
Université d’Oran
Faculté des Sciences
Département de chimie
THESE EN VUE DE L’OBTENTION DU DIPLOME DE DOCTORAT
D’ETAT
Spécialité : Chimie des matériaux
CONTRIBUTION A L’ETUDE DE LA CONVERSION DU CHLOROMETHANE SUR LA H-ZSM5
Mécanisme et effet de structure
Présenté par :
MR Chikh BOUKRATEM
Soutenue le 18 mai 2006 devant le jury composé de
Président : Pr. Y. KHATIR (Université d’Oran)
Examinateurs : Pr . Z DERRICHE (U.S.T.O)
: Pr. O. CHERIFI (U.S.T.H.B)
Rapporteur : Pr . Y. BENTAARIT (IRC/ LYON)
Co- Rapporteur : Pr. A. BENGUEDDACH (Université d’Oran).
Membre invitée : Pr. L. CHERIF (Université. Tlemcen)

SOMMAIRE
CHAPITRE I : INTRODUCTION
I. Introduction…………………………………………………………………. 01
I.1 généralités sur la problématique………………………………………....... 02
I.2 Objectif……………………………………………………………………. 03
I.3 Etude Bibliographique…………………………………………………….. 03
I.3.1 Propositions diverses …………………………………………………… 03
I.3.1.1 Le Carbène……………………………………………………………. 03
I.3.1.2 L’hypothèse du carbonium……………………………………………. 04
I.3.1.3 L’ion Triméthyle Oxonium……………………………………………. 04
I.3.1.4 L’hypothèse de formation des radicaux………………………………. 05
Bibliographie…………………………………………………………………. 06
CHAPITRE II : PREPARATION DES CATALYSEURS ET
CARACTERISATION
II.1 Introduction…………………………………………………………......... 08
II.2 Structure et caractérisation des zéolites………………………………...... 08
II.2.1 Structure des zéolites……………………………………………………. 08
II.2.2 Principe de la synthèse………………………………………………….. 08
II.2.3 Laproprieté acide……………………………………………………….. 09
II.2.4 La ZSM5………………………………………………………………… 09
II.2.5 Préparation des échantillons……………………………….……………. 10
II.3 Techniques et caractérisation…………………………………..…………. 16
II.3.1 Spectroscopie infra rouge………………………………………………. 16
II.3.2 Appareillage…………………………………………………..…............ 17

II.3.3 L’analyse chimique………………………………………………………. 17
II.3.3.1 Spectre d’émission …………..………………………………………… 18
II.3.3.2 Spectre d’absorption ………………………………………………….. 18
II.3.4.1. RMN ………………………………………………………………….. 19
II.3.4.2. Le déplacement chimique ……………………………………………. 19
II.4. Détecteur par ionisation de flamme (DIF) ………………………………. 20
II.5 DTP (désorption à température programmée) ……………………………. 20
Bibliographie…………………………………………………………………… 23
CHAPITRE III : INFLUENCE DES PARAMETRES EXPERIMENTAUX
III.1. Partie Expérimentale ……………………………………………………. 25
III.1.1. Appareillage …………………………………………………………... 25
III.1.2. Etalonnage ……………………………………………………………. 26
III.1.2.1. Facteur de correspondance …………………………………………... 26
III. 1.2.2. Etalonnage en DTP …………………………………………………. 26
III.1.3. Conversion du chlorométhane …………………………………………. 27
III.2 Reproductibilité des résultats……………………………………..………. 27
III.3. Stabilité…………………………………………………………..………. 27
III.4. Facteur d’acidité………………………………………………….…........ 29
III.5. Vitesses de formation……………………………………………..……… 35
III.6. Dépôt de coke……………………………………………………..……… 35
III.7 Premiers produits de réaction…………………………………….………. 36
III.7.1 Distribution des oléfines ………………………………………………. 36
III.7.2 La température de réaction……………………………………..……… 41
III.8 Conclusion………………………………………………………………... 42
Bibliographie…………………………………………………………………… 43

CHAPITRE IV : RECHERCHE DU MECANISME
IV.1. Recherche du mécanisme…………………………...……………………. 45
IV.1.1 Liaison carbone-carbone ………………………………………….......... 45
IV.1.2 Investigation………………………………...………………………….. 45
IV.1.2.1 L’ion carbenium…………………………………….………………… 45
IV.1.2.2 Le Méthylène ………………………………………………………… 46
IV.1.2.2.1 Le premier produit……………………………………...………….. 46
IV.1.2.2.2 L’hypothèse de l’insertion………………………...……………….. 46
IV.1.2.2.3 H-ZSM5/Pt……………………………………………...……......... 50
IV.1.2.3 Les radicaux………………………………………………………….. 50
IV.2. Etude infra rouge……………………………………………………....... 51
IV.3 Rupture de la liaison carbone-chlore………………………...…………… 55
IV.4 Le Mécanisme………………………………………………………......... 58
IV.5. Déshydroxylation…………………………………………………........... 59
IV.6. Réaction intermoléculaire……………………………………………….. 63
IV.7. Le méthanol…………………………………………………...…………. 66
IV.8 Conclusion……………………………………………...………………… 67
Bibliographie…………………………………………………………………… 68
CHAPITRE V : CONSIDERATIONS CINETIQUES
V.1 Etude cinétique …………………………………………………................ 71
V.2 Distribution comparée des produits………………………...…………….. 71
V.3 La première étape de la réaction…………………………...……………… 72
V.3.2 Méthylation du benzène…………………………………………………. 74
V.6 Conclusion……………………………………...…………………………. 81
Bibliographie ……………………………………...………………………….. 82

CHAPITRE VI : EFFET DE STRUCTURE
V. Effet de structures ……………………………………………………..…. 84
VI.1 La ZSM12………………………………………………………………. 84
VI.2 effet de structure…………………………………...……………………. 85
VI.2.1 sur l’activité…………………………………………...………………. 85
VI.2.2 sur la sélectivité……………………………………...……………….. 87
Bibliographie…………………………………………………………………. 92
CONCLUSION GENERALE …………………………………………….. 93
ANNEXE ……………………………………………………………………. 96

1

2
I.1 Généralités sur la Problématique
Le méthane est inerte à toute transformation chimique usuelle à cause de la
stabilité des liaisons C-H. Des méthodes existent pour passer du méthane à la
molécule monosubstituée qu’on arrive à transformer .
Le passage par le chlorométhane ou le méthanol a fait l’objet de travaux de
recherche car la propriété catalytique des zéolites permet leur conversion en
hydrocarbures .La recherche du mécanisme de conversion du chlorométhane est
abordée dans ce travail. La conversion du méthanol donne des informations
complémentaires pour cette étude du mécanisme.
L’obtention du méthanol passe d’abord par la réaction du méthane avec l’eau à
haute température d’environ 900°C et en présence d’un catalyseur au nickel
aboutissant au gaz de synthèse (CO +3H2) accompagné d’autres réactions
comme (CO+H2O → CO2 +H2).
L’étape suivante (CO+2H2 →CH3OH) est conduite en phase gazeuse (1.2)
(250°C, 10 bar) sur un catalyseur au cuivre.
L’oxychloration du méthane (CH4+HCl+02) a été étudiée sur le catalyseur
CuCl imprégné au KCl, LaCl3 (3), mais aussi sur la zéolithe (4) Cu-Y.
Dans l’oxychloration du méthane, une partie de la réaction décrite par
DEACON(5).
2 HCl + ½ O2 → H2O +Cl2
permet de former de l’eau. Le chlore obtenu par rupture homolytique de
HCl va se substituer à l’hydrogène du méthane.
Ce même point est relaté dans l’utilisation du CuCl2/KCl fondu. Les sels
fondus transforment d’abord le méthane en quatre produits chlorés mais le gaz
chlorhydrique produit va, en présence d’air, accentuer la chloration de ces
différentes molécules.

3
I.2 Objectif
Notre objectif est de rechercher le mécanisme de conversion du
chlorométhane en expliquant le type de rupture de la liaison carbone-chlore et la
nature de l’intermédiaire réactionnel.
Puisque la transformation du méthanol présente des similitudes, nous
abordons certains aspects de la conversion du méthanol afin d’apporter des
éléments d’informations qui peuvent nous aider à mieux cerner la recherche du
mécanisme concernant le chlorométhane.
I.3 Etude bibliographique
I.3.1 Propositions Diverses.
Sachant que certaines réactions sur le méthane donnent des molécules
CH3X (CH3OH, CH3Cl), on peut dire que la transformation des molécules CH3X
sur la H-ZSM5, sert finalement d’intermédiaire pour la transformation de CH4.
Il y’a essentiellement trois propositions sur les intermédiaires réactionnels qui
sont le carbéne, l’ion carbénium et l’ion triméthyle oxonium.
I.3.1.1 Le carbène
Le carbène est proposé par Chang et Silvesti (6) ainsi que par Swabb et
Gates(7).
Lee et Wu (8) proposent la même hypothèse en envoyant sur le catalyseur
du diazométhane qui donne le méthoxyle qui lui –même réagit avec un excés de
méthyléne (carbène) . Celui-ci s’insére au niveau de C-H pour donner un
éthoxyl d’où C1 C2
(libérant par élimination de l’éthylène)
Ces auteurs expliquent que même si CH2 est stabilisé sur la surface,
l’élimination monomoléculaire de H2O dans CH3OH pour donner CH2 est
improbable parce que.
CH3OH :CH2 + H2O

4
est endothermique (349.3 Kcal /mole) que
2 CH3OH CH3OCH3 +H2O
est exothermique (-24.5 kcal/ mol) et que le diméthyle éther a été observé.
Chang et Silvestri (6) expliquent la formation de CH2 par élimination de H2O
dûe à l’action simultanée acide et basique des sites. Des calculs semi-empiriques
de mécanique quantique menés par Beran Jiru (9) suggèrent que le méthanol, en
présence d’un champ électrique fort tel qu’il en existe dans les zéolites, peut
donner les espèces :CH2 et CH2O .
Perot (10) et coll. ainsi que Cornerais et coll (4) expliquent la priorité de
l’éthylène ou du propène par la diffusion.
Pour le mécanisme, ils proposent l’insertion du méthylène sur le
diméthyle éther pour donner l’éthyle méthyle éther libérant l’éthylène.
I.3.1.2 L’hypothése du carbénium
Olah et Coll (12) tout comme Ono et Coll (13), proposent l’attaque du CH3
sur une liaison C-H du méthanol. Le carbénium qui a un rôle prépondérant a été
mis en évidence par étude infra-rouge dans ce mémoire ; de même que dans
l’étude menée par X-R Xia, et Y-L. Bi (14) ainsi que B-L Su et D. Jaumain (15).
Olah(16) le précise dans le cas de la polymérisation de CH4 avec SbF5-FSO3H et
l’alkylation des alcanes par le complexe CH3-SbF5 . Ono et coll.(13) observent,
en infra rouge , les bandes de vibration de C-D de CD3 et ceci pour montrer la
formation du méthoxyle.
I.3.1.3 L’ion triméthyloxonium
La formation du triméthyle oxonium, vient des travaux de Olah (16) et
Coll. ainsi que Sommer (17) et Coll. Ils traitent le sel de triméthyle oxonium par
une basse forte et établissent que le triméthyle oxonium réagit de manière
intermoléculaire pour donner l’alkyl diméthyle oxonium.
Il s’agit du triméthyle oxonium tétrafluoroborate (traité par une base) qui
perd un proton pour subir une méthylation (C1 C2). Le sel de
triméthyloxonium, (Me)30(+) BF4(-), après déprotonation est très actif pour une

5
alkylation par CH30H ou le diméthyle ether . Stevens (18) invoque le
réarrangement intramoléculaire de l’ion triméthyle oxonium.
Les mesures cinétiques, thermodynamiques et des considérations
théoriques présentées dans le travail de Vandenberg (19), proposent
l’intermédiaire triméthyle oxonium et déduisent que la formation de celui-ci est
l’étape limitante.
I.3.1.4 L’hypothèse de formation des radicaux
Selon Clarcke, K.A et Coll (20), la structure aluminosilicate contient des
défauts paramagnétiques localisés qui agissent sur le méthanol ou le diméthyle
éther pour enclencher une succession de radicaux menant aux produits.
L’analyse RPE montre le spectre des radicaux piégés par la α-phényl –N-
t-butyl nitrone
PhCH= N(+) (But) O(-) R° Ph ® CHN (But) O°
L’auteur propose la formation des radicaux au niveau du réactif à partir de
la présence, sur la surface, d’un radical S dont il ne précise pas la nature.

6
Bibliographie
1- M.L. POUTSMA ; L.F. ELECK ; D.A.IBARBIA ; P. RISCH et
J.A.RABO ; J. Catal. 52, 157-1978.
2- S.W.KAISER ; U.S. Patent 4, 289, 709-1981.
3- D.A DOWDEN ; C.R. SCHNELL et G.T .WALKER, Proceedings of the
4th. International congress on catalysis _ MOSCOW, P.201-1968.
4- Y.BENTAARIT et Coll. Brevet 1990.
5- DEACON,« Chim. Org. industrielle “by H.J. arpe-p.49, Ed Masson, 1981.
6- CHANG et SILESTI; J. Catal. 1977, 47, 249.
7- SWABB et GATES ; Ind. Eng. Chem. Fundam . 1972,11,540.
8- C.S.LEE, M.M.WU, J. Chem. Soc., Chem . comm.. 1985. 250
9- BERAN et JIRU, React. Kinet. Catal., lett, 9, 1978, 401.
10- G.PEROT et Coll., J Molecular . catal. 17, , 1982, 255.
11- F.X. CORMERAIS et Coll., J. Chem. Research, S. 1980, 362.
12- G.A.OLAH et coll., Chem. Soc. _1969, 91, 3261.
13- Y.ONA et Coll., Chem. Soc Faraday Trans. 1 1981, 77, 2209-2221.
14- Xin-Rui Xia, Yin-li BI, Tong-Hao Wu and Kai-Ji Zhen, catalysis letters,
1995, Vol 33, number 1-2, pp75-90.
15- Bao-Lian Su, Denis Jaumain, Proceedings of the 12th international
Zeolite conference, 1999, Vol 4, pp2689-2696.
16- G.A.OLAH et Coll., J. Chem. Soc._ chem.. communication 1986.
17- SOMMER et Coll., Chem. Soc._Chem. Communication, 1984, 1210.
18- T.S. STEVENS, van Nostrand Reihold Pub, Ca, LONDON, 1973.
19- a) J.P. VAN DEN BERG, J.P WOLTUIZEN; J.C.H VAN HOOFF; in
“Proceedings of the 5th international Conference on zéolites” REED,
L.V.; Ed. Heyden: LONDON, 1980; P.649.
b) VAN DEN BERG, J.P. Ph.D. Thesis, ETNDHOVEN, 1981.
20- J.K.A., CLARKE, J. of the chemical Society, 1986.

7

8
II-1 Introduction
Les zéolites présentent des propriétés telles que l’échange ionique , la
catalyse (12), le tamis moléculaire . Les zéolites requièrent de l’importance en
pétrochimie grâce aux propriétés catalytiques d’oligomérisation et de craquage
que possèdent ces solides. Ce qui permet d’étendre le domaine de recherche
dans le domaine .
A coté des zéolites naturelles dont la première a été découverte en 1756
par le baron CRONSTEDT (3), il y a celles qui sont synthétisées au laboratoire.
II- Structure et caractérisation des zéolites
II-2.1. Structure des zéolites
La caractéristique structurale de ces matériaux est la disposition
d’enchaînements aluminosilicates formés par des tétraèdres (SiO4) et (AlO4) liés
par les oxygènes.
La structure de la zéolite a une composition de la forme :
Mx
+ /n (ALO2)x (SiO2)y . z H2O.
M : cation d’échange de valence n (élément appartenant en général aux
alcalins et alcalino-terreux).
x + y = nombre total de tétraèdres.
y/x= rapport (variable selon la structure)
z = nombre de molécules d’eau.
Les Al3+ créent des charges négatives compensées par la présence de
cations dans les canaux. La charpente aluminosilicate est formée d’un
arrangement tridimensionnel de tétraèdres AlO4 et SiO4. Elle est anionique du
fait du défaut de charge de l’aluminium (+3) par rapport au silicium (+4).
II 2.2. Principe de la synthèse
La synthèse de la zéolite est réalisée par le mélange ternaire suivant :

9
- Un solvant qui est l’eau.
- Un mélange constitué de silice et d’alumine.
- Un agent organique comme le TPA+ qui a le rôle d’agent structurant ou
« template »(4,5) .
Le pH de cette solution doit garder une certaine valeur d’où l’introduction
de base. Le gel obtenu correspond à la composition de la zéolite synthétisée.
(la suite du procédé de synthèse est indiquée en II 2.5 : « préparation des
échantillons »)
II 2.3 La Propriété acide
L’activité catalytique de craquage, d’alkylation ou d’isomérisation vient de la
propriété acide du site .
L’acidité de Bronsted est obtenue par échange des cations alcalins initialement
présents comme Na(+) avec l’ion ammonium que l’on décompose à haute
température (6). (NH4+ NH3 + H+)
II.2.4 La ZSM5
La synthèse de la ZSM5 a été réalisée par la société Mobil (7) . Elle est
formée par cristallisation hydrothermale ; sa structure correspond à des mailles
élémentaires orthorhombiques.
Les dimensions de la maille élémentaire, obtenues à partir d’un
monocristal de ZSM5 (Si/Al=86) (8) sont :
a = 20,07 Å , b = 19,92 Å et c = 13,42 Å
Cette maille élémentaire a pour formule chimique :
Nax Alx Si96-x O192. 16H20
Où x est délimité par 0 et 12.
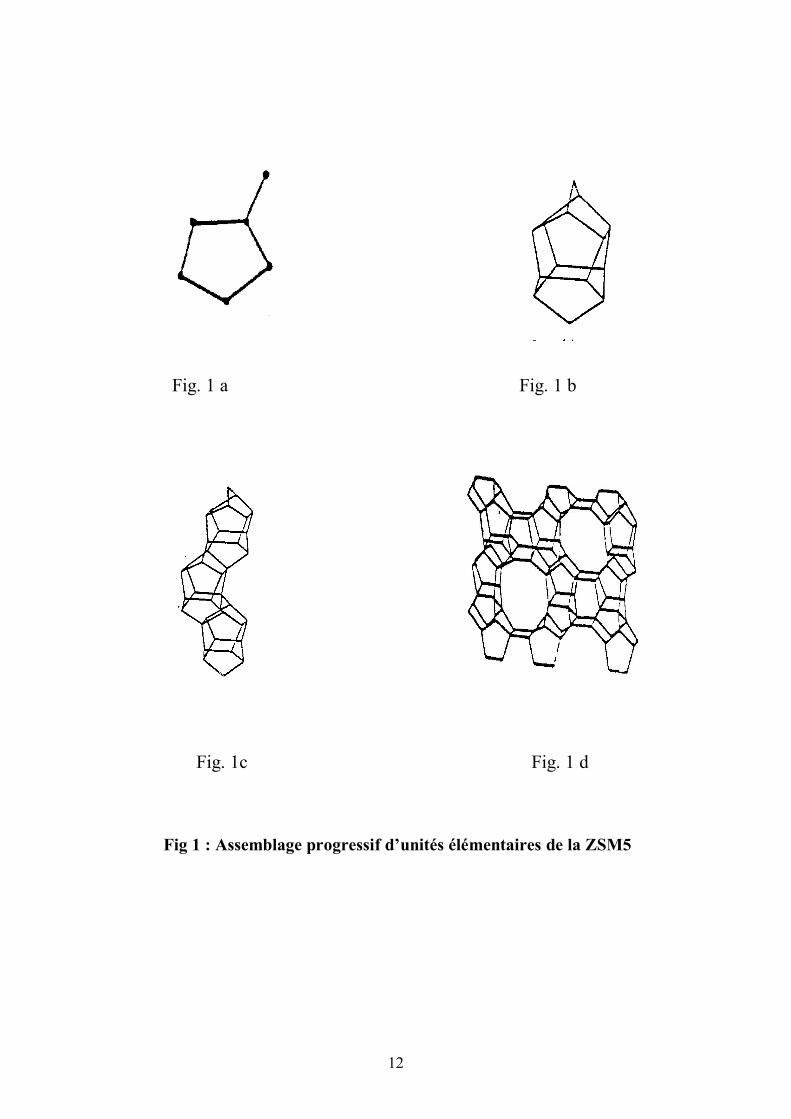
L’entité la plus simple dans l’édifice de la ZSM5 correspond à un
pentacycle lié à un tétraèdre (9) (fig. l a) . A partir de ce module générique 5-1
est construite l’unité pentasil (fig. 1b). Ces unités vont constituer des chaînes
(fig.1c) puis des feuillets (fig. 1d).
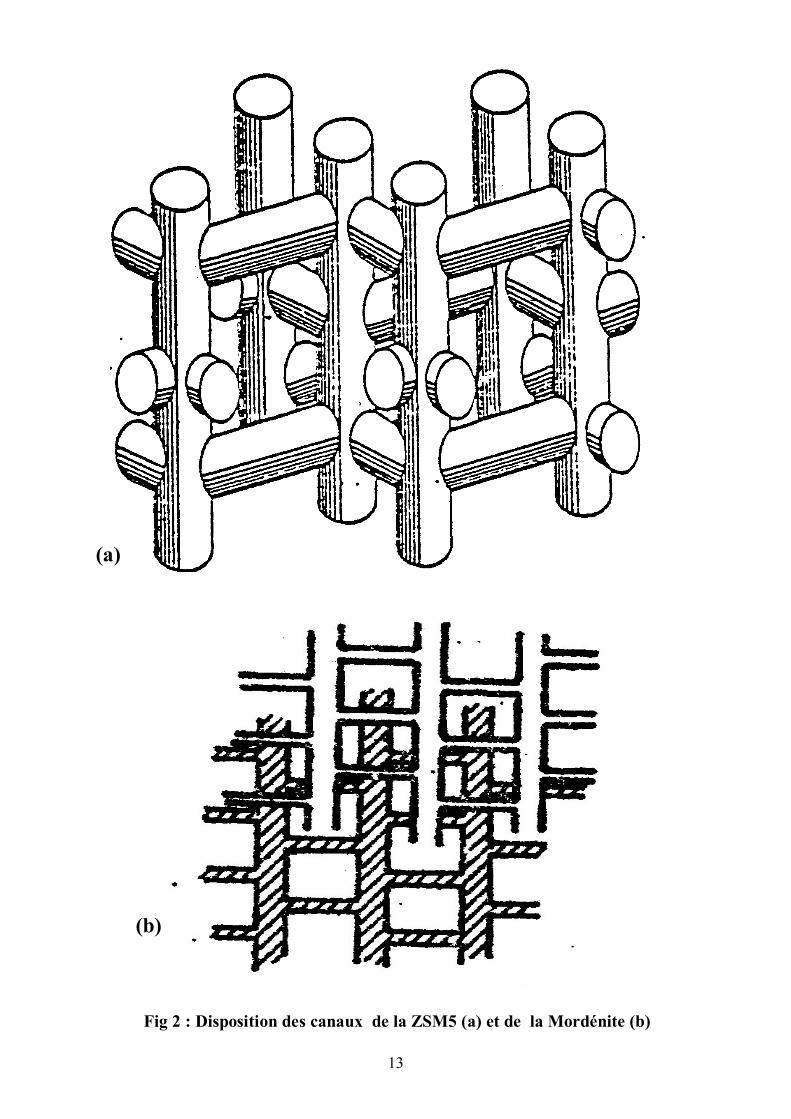
Les canaux sont constitués par une succession d’anneaux de 10 membres.

10
La ZSM5 est un solide de structure poreuse constituée de deux types distincts de
canaux intersectés sinusoïdalement. La section des canaux entrecroisés est de
forme quasi-circulaire de dimension 5.3 x 5.5 Å. Les canaux droits ont une
section dont la forme est elliptique avec les dimensions 5.1 x 5.5 Å.
Parmi les zéolites étudiées, on a la ZSM5, la ZSM12 et la mordénite. On
constate que ces solides disposent de canaux rectilignes
Les zéolites ont la propriété sélective (10).
Après analyse chimique (spectres d’absorption pour Al et pour Na) sur la ZSM5
,on a les pourcentages en Al et Na , ce qui nous permet de trouver le rapport
Si/Al à partir de Nax (Al
xSi 96-xO192) qui constitue la maille élémentaire.
L’aluminium a une valence inférieure à celle du Silicium
(Al3+/ Si4+) .Aussi lors de l’association des tétraèdres SiO4 AlO4, on a apparition
de charges négatives. L’électroneutralité est assurée par des ions métalliques
dans la proportion d’un cation monovalent ou d’un demi-cation divalent. La
taille du grain chez la ZSM5 varie entre 10 et 30 micron mètre .
La Mordénite se présente sous forme de :
- petits canaux constitués par une succession d’anneaux de 8 membres.
L’ouverture est également de forme elliptique de dimension 4,72 Å et 3,87 Å.
- gros canaux parallèles entre eux d’ouverture 6,95 Å et 5,81 Å . Ce
réseau est constitué de ces gros canaux qui vont déboucher sur les petits canaux.
La dimension du grain chez la Mordénite varie de 1 à 10 micron mètre .
II 2.5 Préparation des échantillons
La synthèse de plusieurs échantillons aux différentes teneurs en aluminium
permet de travailler en tenant compte de la variable acidité liée à des
concentrations distinctes en sites actifs par unité de surface. La synthèse de la
zéolite requiert une solution en Al2(SO4)3 , 18 H2O.

11
L’analyse en microscopie électronique présente une homogénéité des grains
pour les différents échantillons. La dimension du grain (fig. 3) croît avec le
rapport Si/Al.
12
Fig. 1 a
Fig. 1 b
Fig. 1c Fig. 1 d
Fig 1 : Assemblage progressif d’unités élémentaires de la ZSM5
13
Fig 2 : Disposition des canaux de la ZSM5 (a) et de la Mordénite (b)
(a)
(b)

14
Fig 3a : Forme et dimension du grain de ZSM5 (Si/Al=30)

15
Fig 3b : Forme et dimension du grain de ZSM5 (Si/Al=100)

16
Le procédé de synthèse est indiqué au paragraphe II.2.2. Le gel obtenu
est blanc. Il est mis dans un autoclave dont la température est portée à 165°C
pendant 5 jours.
De l’autoclave est retiré le flacon contenant le solide cristallisé. Après
décantation, on procède au lavage à l’eau permutée puis séchage à l’étuve
pendant la nuit à 100°C. L’étape de calcination qui a lieu en dynamique c’est à
dire sous un atmosphère et à 500°C, débarrasse le solide du template .
La forme ammonium de la zéolite est obtenue par échange au chlorure
d’ammonium ; cet échange a lieu sous agitation à la température ambiante ;
vient ensuite la centrifugation avec des lavages répètes à l’eau permutée puis on
sèche à l’étuve. L’analyse chimique donne la teneur en sodium résiduel.
La composition de la zéolite protonée est alors établie.
II.3 Technique de caractérisation
II 3.1 Spectroscopie Infra rouge
Cette technique permet la caractérisation de la zéolite synthétisée.
La réaction de conversion du chlorométhane a lieu dans la cellule
contenant la pastille de H-ZSM5.
Des changements sont observés sur les spectres avec l’apparition de
bandes relatives aux espèces intermédiaires de la réaction. Les divers
échantillons de ZSM5 préparés présentent des bandes de vibration
caractéristiques suivantes :
- Les bandes de vibration des groupes hydroxyles dans le domaine 3800-
3600 cm-1.
- Les bandes de vibration internes des tétraèdres à 1100 , 800 et
400 cm-1.
- Les bandes de vibration externes sont à 1220 cm-1 et 550 cm-1. cette
dernière est caractéristique de la vibration des anneaux à 5 tétraèdres.

17
Le rapport des intensités des bandes à 550cm-1 et 450cm-1 rend compte du
degré de cristallinité (12).
Lorsque la teneur en aluminium croît, les bandes de vibration internes à
TO4 se déplacent vers les basses fréquences parce que la liaison Al-O est plus
longue que la liaison Si-O (Al-O= 1.74 Å et Si-O = 1.62 Å).
En passant d’une silicalite à une ZSM5 où Si/Al est égal à 12, la vibration
antisymétrique du tétraèdre passe de 1100 à 1080 cm-1
II 3.2 Appareillage
Des pastilles de zéolites en présence de KBr d’un poids de 10 à 15 mg
sont obtenues par compression à 2.104 Pa ; la pastille est fixée sur le porte
échantillon et le tout est introduit dans la cellule. D’autres pastilles sont réalisées
par dilution dans du KBr à 1% en poids de la zéolite.
Deux appareils infra rouge sont utilisés. Le plus récent, le BRUCKER IFS
48, est un bifaisceaux. Dans le programme de cet appareil, le spectre est le
résultat de la différence entre l’analyse propre de la zéolite et une analyse
référentielle en atmosphère d’azote.
Le BRUCKER IFS 48 offre une variété d’utilisations dans la mesure où il
décèle, selon les besoins du manipulateur, les changements produits entre deux
spectres correspondants à deux traitement différents de l’échantillon, l’un des
deux spectres représentant la référence.
Le 2éme appareil, plus ancien, le PERKIN-ELMER 80, est un mono
faisceau. La rotation périodique d’un miroir dirige le faisceau incident suivant
une certaine fréquence, tantôt vers l’échantillon (pastille) tantôt vers une
atmosphère d’azote (référence).
II.3.3 L’analyse chimique
L’analyse est réalisée sur un Perkin–Elmer. Pour une synthèse
nouvellement réalisée ou après un échange, l’information donnée par l’analyse
chimique est nécessaire pour la déduction de la formule chimique de la zéolite.
On s’intéresse aux éléments sodium et aluminium . 100 mg d’échantillon sont

18
mis en solution par attaque fluorhydrique puis repris par l’acide chlorhydrique.
Les conditions de flamme et les longueurs d’onde utilisées pour le dosage sont
données pour chacun de ces éléments.
II.3.3.1 Spectre d’émission
Pour le cas du sodium, le spectre d’émission est obtenu lorsque le gaz est
soumis à l’action d’une décharge électrique . Ladite décharge est un brusque
apport d’énergie qui provoque le passage des atomes à des états excités .
Lorsque ces atomes retournent à leur état fondamental, les électrons retombent
sur les divers niveaux d’énergie autorisés et les photons émis donnent naissance
au spectre que l’on observe , spectre appelé spectre d’émission . Ce principe est
mis en pratique dans les tests de flamme que l’on exécute pour identifier les
métaux alcalins présents dans des échantillons à analyser. Chaque métal alcalin
donne lieu à une couleur de flamme caractéristique (jaune pour Na ). Les
couleurs sont dûes à des transitions électroniques dans les atomes excités du
métal. Les ions sont réduits dans la région située au centre inférieur de la
flamme en atomes du métal à l’état gazeux.
II.3.3.2. Spectre d’absorption
Dans le cas de l’aluminium, le spectre d’absorption s’observe lorsque
des atomes à l’état gazeux sont soumis à l’action d’une radiation
électromagnétique inçidente . De telles excitations électroniques peuvent être
interprétées comme étant des transitions au cours desquelles les électrons sautent
des niveaux d’énergie inférieurs vers des niveaux d’énergie supérieurs .Le
spectromètre dont on se sert pour étudier de telles transitions est muni d’un
sélecteur de longueurs d’onde qui permet au manipulateur de balayer la zone de
rayonnement électromagnétique qui convient . La radiation incidente provoque
une promotion d’électrons vers des états énergétiques excités . Seules les
radiations de fréquence bien définies sont absorbées par le gaz irradié.

19
II.3.4.1 RMN
Le spectre RMN de 27Al (50 ppm pour AlT et 0 ppm pour AlO ) est pris en
fonction de Al(H2O)63+ prise comme référence (T pour tétraédrique et O pour
octaédrique). Al extrait est déposé dans les canaux. En fait, l'aluminium est à
0 ppm parce que Al extrait se confond tout simplement avec la référence en
l'occurrence Al(H2O)63+ . Pour la désalumination des zéolites, on a la résonnance
à 50 ppm qui correspond à l'Aluminium du réseau (AlT) .La raie à 0 ppm
appartient à l'Aluminium extra réseau (AlO) . L'Aluminium du réseau se défait
des oxygènes qui forment de nouvelles liaisons OH . Al3+ va former Al(H2O)63+
extra réseau. Pour la RMN du 29Si , on a comme référence le tétramethylesylane
(TMS). II.3.4.2 Le Déplacement Chimique
Le déplacement chimique décrit la position d’un pic RMN .La plupart des
absorptions des hydrogènes en RMN du 1 H à 90 MHz se situent dans une zone
s’étalant sur 900 Hz . Plutôt que de mesurer et d’enregistrer la fréquence exacte
de chaque résonance (ce qui est difficile à réaliser avec précision sur le plan
téchnique ), on ajoute un standard interne par rapport auquel les positions des
pics dans le spectre sont mesurées . Ce composé est le Tétraméthylesylane
(CH3)4 Si (Point d’ébullition : 26,5°C) . Les 12 hydrogènes équivalents présents
dans ce composé sont blindés par rapport à ceux de la plupart des composés
organiques . De ce fait, ils résonnent à un endroit qui est opportunément
éloigné de la zone spectrale habituelle .(On a environ 1 % en (CH3)4Si
incorporé dans des solvants en RMN )
distance du pic par rapport à celui de (CH3)4 Si (en Hz)
fréquence du spectromètre (en MHz)
δ =

20
II.4. Détecteur par ionisation de flamme (DIF).
On a,pour le test catalytique, deux détecteurs à ionisation de flamme
consacrés aux linéaires et aux aromatiques , disposés dans un montage en série.
Puis d'un catharomètre disposé en série avec les détecteurs à ionisation de
flamme. Il est utilisé pour la détection du méthanol et du diméthyle éther ce qui
n'est pas possible sur le DIF ( voir annexe pour plus de détail ).
II .5 DTP (Désorption à température programmée)
Principe :
C’est le calcul du nombre de sites acides de la zéolite H-ZSM5.
Procédé :
- passage de O2 pendant la nuit avec élévation de température (5 °C/ mn)
jusqu’à 600°C pour éliminer les traces d’eau et autres molécules adsorbées.
- le lendemain ,on laisse refroidir le réacteur (où se trouve la zéolite) pour
faire l’adsorption à 100°C (on peut accélérer le refroidissement par le
ventilateur ).
- on chasse, par l’helium , l’oxygène restant (hélium qui passe en
permanence au niveau du catharomètre ).
- on bascule la vanne sur position « adsorption » alors qu’elle était en
position « traitement ».
- après refroidissement, on augmente la température jusqu’à 100°C.
- on fait manuellement des injections toutes les dix minutes (sinon on
procède en automatique ) .
(pour les premières injections, on n’attend pas dix minutes ; on peut faire
des injections rapides succéssives ).
- on voit apparaître des valeurs croissantes correspondant à la saturation .
- on met en place la désorption par la touche spécifique indiquée sur
l’appareil.
- attendre que la cellule soit purgée par le gaz vecteur .

21
- pour démarrer la désorption , on met l’enregisteur en marche après avoir
noté la température de départ . Car il faut mettre la programmation de
température.
- pour la désorption , attendre que la ligne de base soit d’une valeur de
200 ( quelques heures ).
- il faut une heure pour éliminer les NH3 physisorbés .
- on lance en même temps , l’enregistreur, l’intégrateur et le régulateur de
température (le 1er et le 2eme à 5 mm/mn pour le déroulement du papier) ; pour
la température, on règle pour 650 °C .
- après deux heures, pour 5 °C/mn , de 100 °C à 650 °C ,on arrête le
déroulement du papier de l’enregistreur .
- Traitement : O2
Inversion du gaz vecteur - Mesure : He
intégrateur Détecteur
Four
Réacteur Enregistreur de température
Injection automatique de NH3 (x)
Voie de référence
He O2
NH3
Fig 5 : appareil de désorption d’ammoniac à Température programmée

22
Après la saturation et l’évacuation des molécules physisorbées par le
balayage continu sous Hélium, la désorption est déclenchée à température
programmée.
Quand la température augmente, l’ammoniac désorbé est évacué par
l’hélium vers le détecteur qui donne des signaux proportionnels à la
concentration de matière évacuée .
Sur le chromatogramme figurent deux maxima correspondant à deux
types de sites de forces distinctes dont l’explication (13,14) est que le premier
pic de désorption d’ammoniac, se manifestant à environ 230°C, correspond aux
OH à 3740cm-1 et que le deuxième pic qui apparaît aux environs de 500 °C , est
relatif aux hydroxyles à 3600 cm-1(15).
La H-ZSM5, pour conserver ses sites acides, ne doit pas subir une
élévation de température très supérieure à 500 °C car dans une étude de l’effet
de température sur la zéolite, celle-ci voit le nombre de ses sites acides de
Bronsted diminuer et le nombre des sites de Lewis augmenter.

23
Bibliographie
1- D.W. BRECK, « Zéolite Molecular Sieves », Wiley, New Yark (1974).
2- J.A. RABO, « Zeolite chemistry and catalysis » ACS Monograph 171,
Washington, D.C. (1976).
3- A.F. CRONSTED, Akad. Handl. STOCKLM, 17, 120 (1756).
4- H. NAKAMOTO, TAKAMSI, Chem. Lett., (1981), 1739.
5- DM.BIBBY, N.B. MILESTONE, L.P. AMDRIDGE, Nature 285
(1980), 30.
6- H.W. HAYNES, catal. Rev., 17 (2) 273, (1978).
7- R.J. ARGAUER, G.R. LANDOLT, U.S. Pat. 3702886 (1972).
8- D.H. OLSON, G.T. KOKOTAILO, S.L. LAWTON et W.M. MEITER,
J.Phys. chem. 85 (1981) 2234-2243.
9- W.M.MEIER, “Molecular sieves” society of chemical industry
LONDON (1968) P.10.
10- P.B. WEISZ, PURE Appl. Chem., 52, 2091, (1980).
11- E.M. FLANINGEN dans “Zeolite chemistry and catalysis” ed
Rabo, J.A., ACS Monographe 171, WASHINGTON, D.C. (1976).
12- G.COUDURIER, C. NACCACHE, J.VEDRINE, J. Chem. Soc
chem. commun. (1982), 1413.
13- G.T. KERR et A.W. CHESTER, Thermochim. Acta, 3 (1971) 113.
14- N.Y. TOPSOE, K. PEDERSEN et E.G. DEROUANE, J. Catalysis
70 (1981) 41.
15- B-L.Su, D.Jaumain, Proceedings of the 12th international zéolite
conference, 1999, vol 4, pp2689-2696.

24

25
III . 1. Partie Expérimentale:
III.1.1. Appareillage
L’analyse des produits de la réaction a montré qu’il est nécessaire de
disposer de 3 détecteurs distincts par le mode de détection (ionisation de flamme
ou catharométrie) ou bien par le type de colonne. On a donc deux détecteurs à
ionisation de flamme pour respectivement les produits linéaires et les produits
aromatiques. Le troisième détecteur, le catharomètre est utilisé pour le
méthanol lors de l’étude de la conversion de cet alcool.
La conversion des CH3X, qu’ils s’agisse de chlorométhane ou de
méthanol, donne des hydrocarbures linéaires et aromatiques suite à une
polymérisation des molécules de base. Pour avoir une bonne séparation des
différents produits, on a pris deux colonnes respectivement pour les linéaires et
les aromatiques. L’utilisation du catharomètre permet le suivi de la formation du
dimèthyle éther, molécule intermédiaire dans la transformation du méthanol.
Le montage en série de ces appareils permet de suivre par injection ou
poussée vers les colonnes du contenu des boucles d’injection. Le temps imparti
à la boucle pour libérer son contenu gazeux des produits est de 30 secondes ; on
attend quelques secondes pour que la concentration du flux gazeux soit
homogène pour produire l’injection au niveau du second appareil.
Les canalisations acheminant le flux gazeux sont maintenues à une
température suffisamment élevée pour éviter la condensation de certains
produits comme le benzène (76°C), le toluène (110°C), les xylènes (140°C) ou
le trimèthyle benzène (170°C).
Plusieurs séries de cordon chauffant de dimension de 12m chacune et
placées en parallèle permettent d’atteindre les 180°C. La chaleur émise est
maintenue constante . Cette température peut être régulée par un alternateur qui
réduit la tension .

26
Les mêmes précautions sont à respecter dans le réglage de la
température au niveau de la détection qui doit avoir lieu à plus de 170°C ainsi
que pour la température d’injection quand on injecte à la seringue.
III.1. 2. Etalonnage
III. 1.2.1 Facteur de correspondance
Les noms des produits et les temps de rétention sont introduits dans le
programme de l’intégrateur. On introduit aussi les facteurs de correspondance de
CH3Cl (ou CH3OH), facteurs qui sont obtenus par les rapports de surface d’1
torr de méthane pris comme étalon et d’1 torr de chlorométhane (ou de
méthanol). La finalité étant l’impression de la valeur exacte de pression du
réactif converti. L’évaluation des pressions, dans un premier temps, vient de
l’appréciation de visu des débits gazeux sur débitmètres à bulles.
Les courbes d’étalonnage sont établies pour les débitmètres massiques
dont le nombre et les limites d’utilisation conçus par le fabricant offrent les
possibilités de dilution par l’azote du réactif seul ou d’un mélange réactionnel.
Dans le cas de la transformation du méthanol, on dispose d’un
saturateur. Les vapeurs d’alcool sont évacuées par une arrivée d’azote, la
pression ne dépendant que de la température du bain dans lequel est plongé le
saturateur.
III.1.2.2 Etalonnage en D.T.P Quatre ou cinq boucles d’injection sont branchées distinctement à
l’appareil de désorption d’ammoniac.
Le volume de la boucle étant connu, on dresse une moyenne de surface
suite à plusieurs injections. A l’aide des surfaces moyennes des boucles , on
établit la courbe S= f (Vb).
La pente à cette courbe est nécessaire dés qu’on a fini d’intégrer la
surface totale de désorption à température programmée afin de calculer le
volume d’ammoniac désorbé. La surface totale a été obtenue par l’intégration
de portions de surface au niveau de l’intégrateur .

27
III. 1.3 Conversion du chlorométhane
La consommation en réactif peut être suivie soit par les valeurs affichées
par l’intégrateur qui donne les produits en PCH3Cl converti soit par les pressions
réelles des produits obtenus et ceci par PCH3Cl / n pour un produit n carboné.
Toutes les précisions seront basées sur la première définition, c’est à dire les
valeurs données en réactif converti. Par conséquent les sélectivités tiennent
compte du pourcentage de chaque produit consommé par rapport à l’ensemble.
Si on doit passer aux pressions réelles, les sélectivités des produits du mélange
donnent la même interprétation que dans le cas des valeurs de pressions
affichées.
III.2 Reproductibilité des résultats
Un vieillissement rapide ou bien une baisse d’activité de la zéolite
régénérée ne favorise pas l’étude de la réaction .
Dans le cas présent, il y a maintien d’activité après N régénérations sous air à
500°C de la H-ZSM5 (fig.1) (tableau III,1).
On pense que ni le sous produit de la réaction en l’occurrence l’acide
chlorhydrique ni la température de traitement qui est de 0,5°C/mn n’altérent le
solide. Chaque régénération ,faite sous air, permet d’éliminer le coke.
La température finale est de de 500°C. Au delà de cette valeur, la H-ZSM5
commence à perdre ses hydroxyles (1) .
III.3. Stabilité
C’est le suivi de la stabilité en fonction du temps de contact ou de la
température de réaction. Le réseau zéolitique de la ZSM5 permet un écoulement
fluide aux particules de la phase gazeuse.
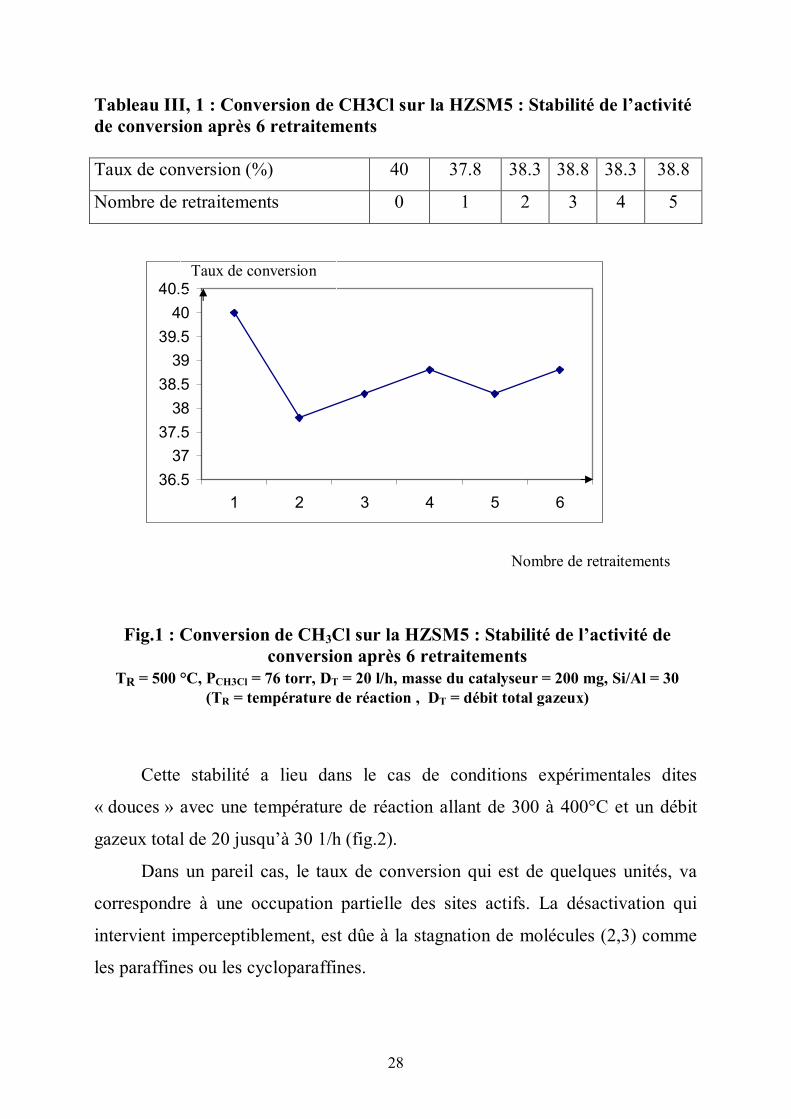
28
Tableau III, 1 : Conversion de CH3Cl sur la HZSM5 : Stabilité de l’activité de conversion après 6 retraitements Taux de conversion (%) 40 37.8 38.3 38.8 38.3 38.8
Nombre de retraitements 0 1 2 3 4 5
36.537
37.538
38.539
39.540
40.5
1 2 3 4 5 6
Cette stabilité a lieu dans le cas de conditions expérimentales dites
« douces » avec une température de réaction allant de 300 à 400°C et un débit
gazeux total de 20 jusqu’à 30 1/h (fig.2).
Dans un pareil cas, le taux de conversion qui est de quelques unités, va
correspondre à une occupation partielle des sites actifs. La désactivation qui
intervient imperceptiblement, est dûe à la stagnation de molécules (2,3) comme
les paraffines ou les cycloparaffines.
Taux de conversion
Nombre de retraitements
Fig.1 : Conversion de CH3Cl sur la HZSM5 : Stabilité de l’activité de conversion après 6 retraitements
TR = 500 °C, PCH3Cl = 76 torr, DT = 20 l/h, masse du catalyseur = 200 mg, Si/Al = 30 (TR = température de réaction , DT = débit total gazeux)

29
Inversement, une transformation du chlorométhane à 500°C, pour un
temps de contact assez long qui correspond à un faible débit (2,5 l/h par
exemple) , est caractérisée par une chute du taux de conversion suivie, au bout
de 5h de réaction, d’un « plateau » d’activité.
III .4. Facteur d’acidité
L’activité de la réaction de conversion croît avec le taux d’aluminium donné par
l’analyse chimique de la zéolite. La H-ZSM5 dispose d’un nombre de protons
tout au moins égal au nombre d’aluminium à quelque différence prés dûe à la
présence des Na(+).

30
Tableau III,2 : Stabilité de l’activité à faible conversion de CH3Cl
Taux de conversion (%) 6,6 5,7 4,9 4
Temps en heures 0 1 2 3
01234567
1 2 3 4
PCH3CL = 76 torr , TR= 500 °C DT = 20 l/h sur H-ZSM5 (Si/Al=30)
t(h)
Taux de conversion
Fig.2 : Stabilité de l’activité à faible conversion de CH3Cl

31
Le nombre des cations Na+ est d’autant plus restreint que le taux
d’échange, au chlorure d’ammonium, tend vers l’unité. On s’intéresse au
paramètre d’acidité parce qu’on a observé qu’une H-ZSM5 comportant 3
protons par maille et plus, subit l’influence du débit gazeux et que pour 1 proton
par maille, la vitesse de transformation reste inchangée quel que soit le débit
(fig.3)(tableau III,3). Dans une H-ZSM5 à 1 ou 2 protons par maille, le nombre
de sites actifs intervenant est le même indépendamment du temps de contact .
On retrouve cette constance dans la vitesse de conversion pour des
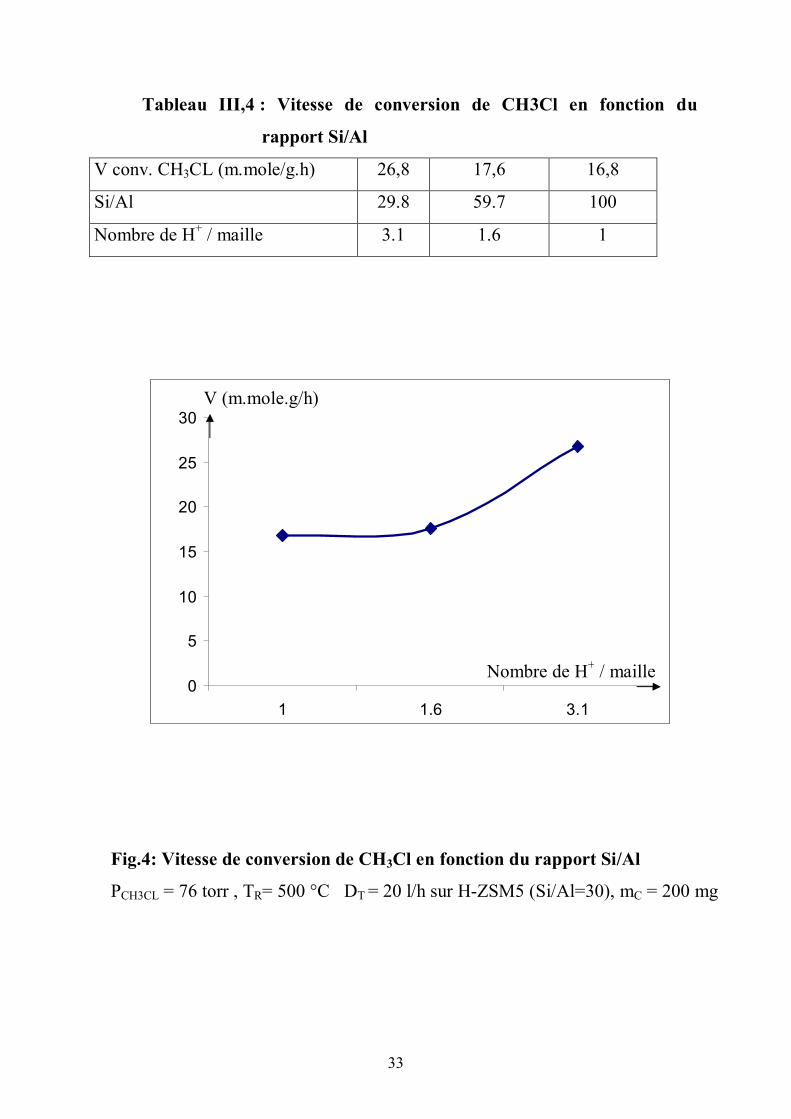
rapports Si/Al élevés. Pour des zéolites qui s’appauvrissent ( Si/Al tend de la
valeur 29.8 vers la valeur 100 ), la vitesse de conversion tend vers une valeur
constante (fig.4) (tableau III,4).
Le temps de contact ou W.H.S.V (Weight hourly space velocity) est
défini en g. mole/h. Il correspond au débit gazeux donné en l/h (litres / heure) à
la différence du temps de contact qui inclue la masse du catalyseur placée dans
le réacteur.
L’analyse quantitative de l’acidité des différents échantillons est suivie en
désorption d’ammoniac à température programmée.
On retrouve les valeurs des températures des deux extrémum de
désorption indiquées dans la littérature (4).
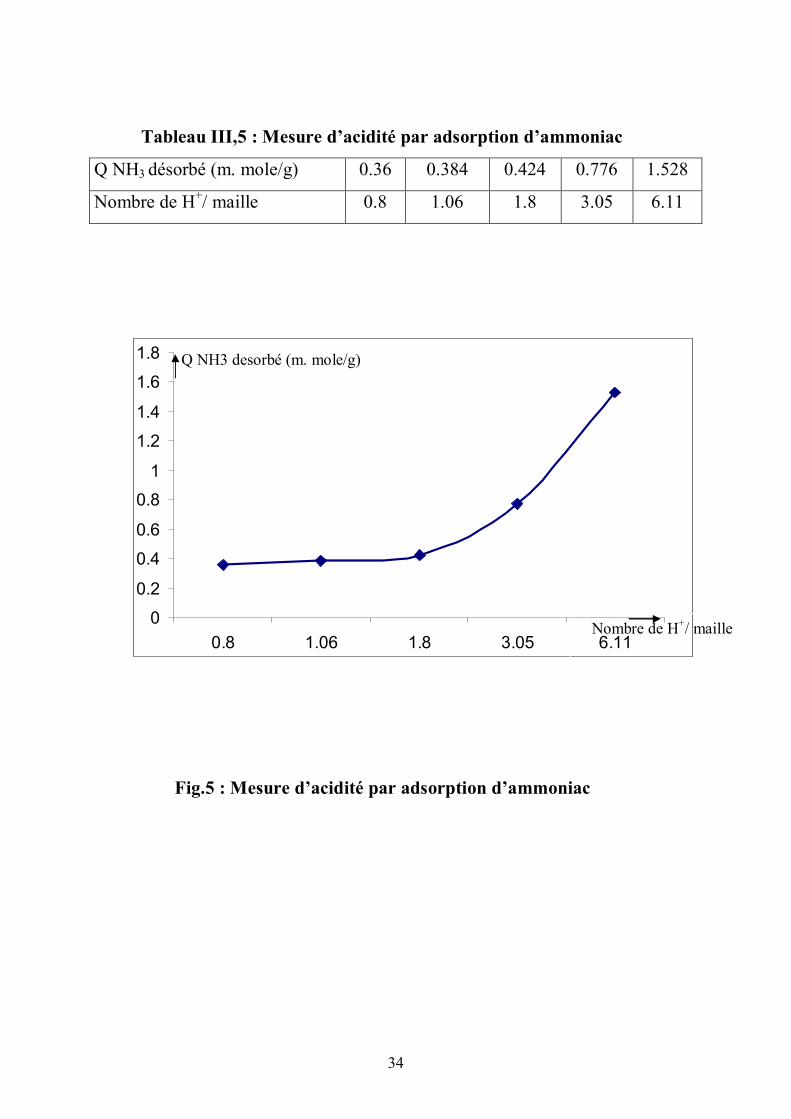
On a une bonne corrélation, une droite, entre le nombre de protons par
maille et la quantité des NH3 désorbés pour une acidité équivalente à plus de 2
protons par maille (fig.5) (tableau III,5). La rétention des molécules NH3
survient au niveau des OH terminaux et des OH pontés (chaque OH est lié a un
atome de silicium). Les OH pontés sont en interaction, par l’atome d’oxygène,
avec un atome d’aluminium voisin. A ce niveau on a un site actif. L’association
de NH3 avec le proton donne l’ion ammonium. Pour connaître le nombre de
protons par maille, il faut revenir au nombre d’aluminium par maille. Comme on
travaille souvent avec Si/Al et que pour (Si + Al) réunis , on a 96 atomes par
maille, on peut déduire le nombre de protons.
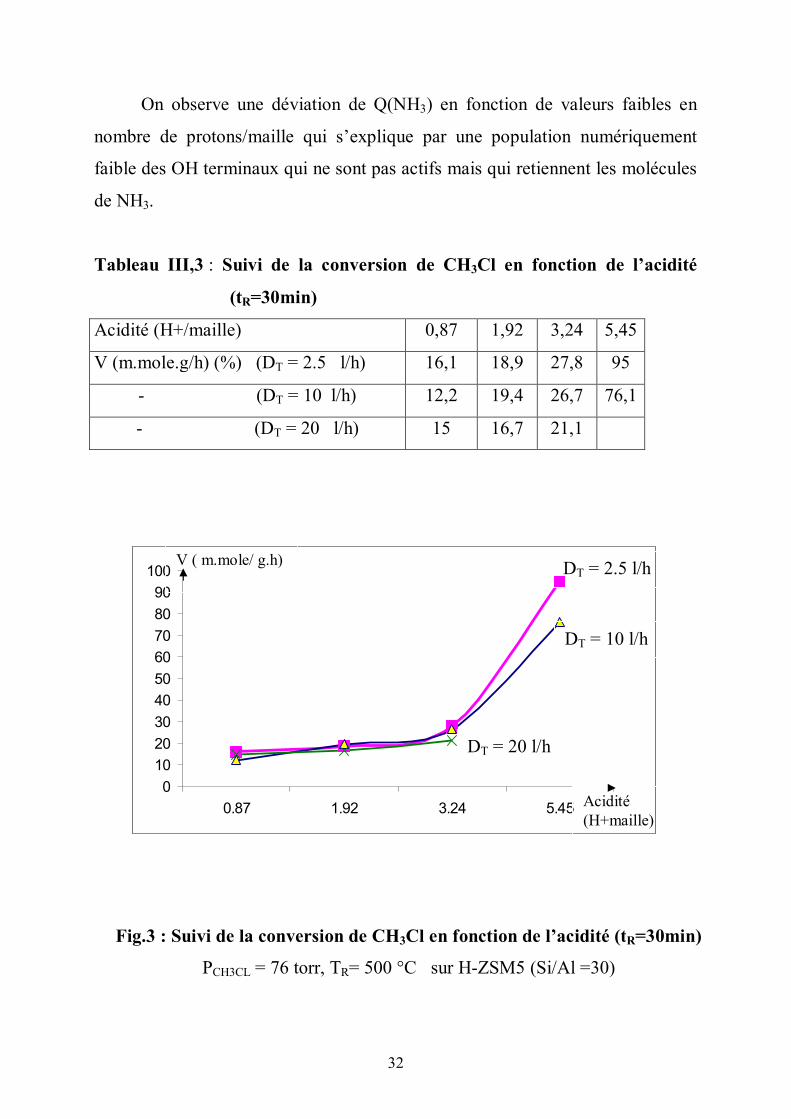
32
On observe une déviation de Q(NH3) en fonction de valeurs faibles en
nombre de protons/maille qui s’explique par une population numériquement
faible des OH terminaux qui ne sont pas actifs mais qui retiennent les molécules
de NH3.
Tableau III,3 : Suivi de la conversion de CH3Cl en fonction de l’acidité
(tR=30min)
Acidité (H+/maille) 0,87 1,92 3,24 5,45
V (m.mole.g/h) (%) (DT = 2.5 l/h) 16,1 18,9 27,8 95
- (DT = 10 l/h) 12,2 19,4 26,7 76,1
- (DT = 20 l/h) 15 16,7 21,1
Fig.3 : Suivi de la conversion de CH3Cl en fonction de l’acidité (tR=30min)
PCH3CL = 76 torr, TR= 500 °C sur H-ZSM5 (Si/Al =30)
0102030405060708090
100
0.87 1.92 3.24 5.45
V ( m.mole/ g.h)
Acidité (H+maille)
DT = 2.5 l/h
DT = 10 l/h
DT = 20 l/h
33
Tableau III,4 : Vitesse de conversion de CH3Cl en fonction du
rapport Si/Al
V conv. CH3CL (m.mole/g.h) 26,8 17,6 16,8
Si/Al 29.8 59.7 100
Nombre de H+ / maille 3.1 1.6 1
Fig.4: Vitesse de conversion de CH3Cl en fonction du rapport Si/Al
PCH3CL = 76 torr , TR= 500 °C DT = 20 l/h sur H-ZSM5 (Si/Al=30), mC = 200 mg
0
5
10
15
20
25
30
1 1.6 3.1
V (m.mole.g/h)
Nombre de H+ / maille
34
Tableau III,5 : Mesure d’acidité par adsorption d’ammoniac
Q NH3 désorbé (m. mole/g) 0.36 0.384 0.424 0.776 1.528
Nombre de H+/ maille 0.8 1.06 1.8 3.05 6.11
Fig.5 : Mesure d’acidité par adsorption d’ammoniac
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0.8 1.06 1.8 3.05 6.11
Q NH3 desorbé (m. mole/g)
Nombre de H+/ maille

35
III.5. Vitesses de formation
En fonction du temps de contact, la vitesse de formation de l’éthylène
(fig.6) (tableau III,6) est constante contrairement à celle du propène qui diminue,
phénomène dû a la consommation de cette oléfine au profit des oligomères.
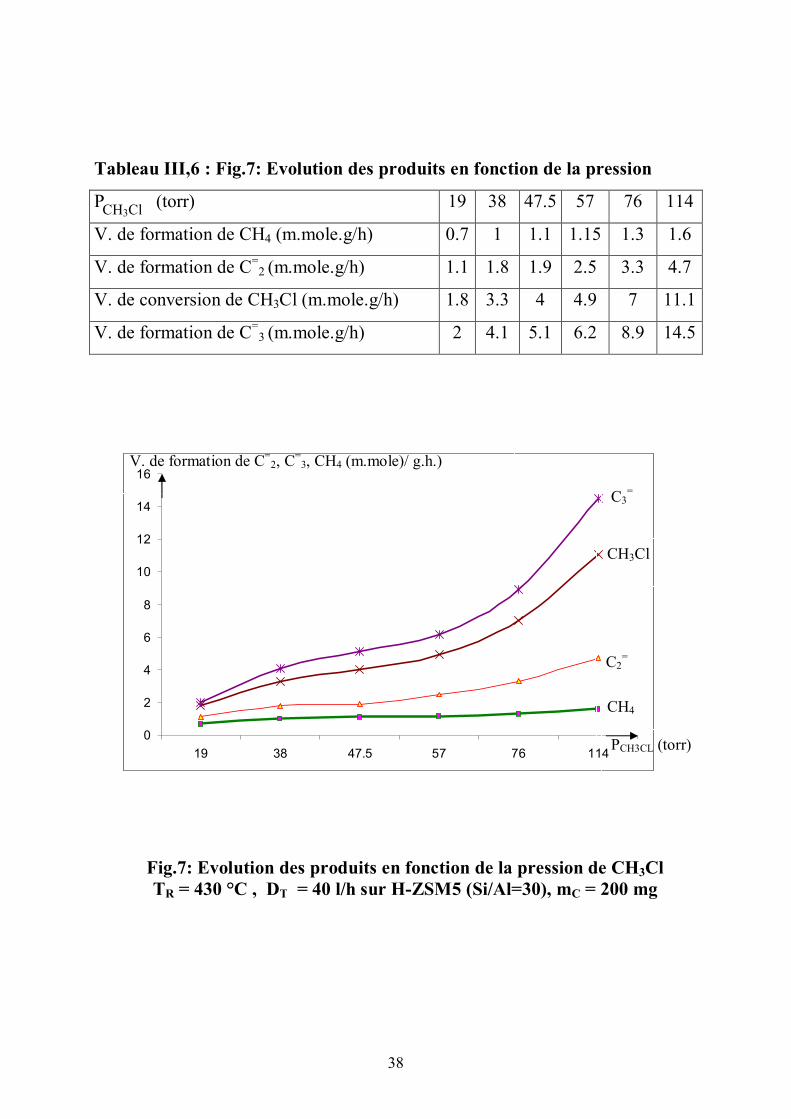
Le suivi des vitesses des produits (fig.7) (tableau III,7) montre que la
vitesse du méthane qui est quasi constante ne dépend pas de la pression du
réactif CH3Cl . La formation de CH4 ne dépend que des hydrures provenant des
cycloparaffines.
Le rapport V(C=3) /V(C=
2) passe de 3 à moins de 2 en passant d’une
température de réaction de 430°C (fig.7) à 500°C (fig.8) (tableau III,8). Cette
diminution dans le rapport des deux vitesses correspond à la conversion du
propène en d’autres molécules et non à l’augmentation de vitesse de formation
de l’éthylène qui varie très peu.
III.6. Dépôt de coke :
Le dépôt de Coke est à l’origine de la désactivation. On a remarqué que le
solide noircit avec la réaction ; le coke se dépose à l’intérieur mais à la surface
extérieure du catalyseur.
La formation du coke dans la H-ZSM5 durant la conversion du
chlorométhane a fait l’objet de diverses interprétations . Guisnet (5) et coll.
Expliquent qu’il provient des composés aromatiques bicycliques.
Zhongmin et coll. (2) pensent que la désactivation est dûe au blocage des
canaux et des pores par des cycloparaffines.
Quand on considère le test catalytique dans les conditions opératoires de
faible conversion, on retrouve le méthane au côté des premières oléfines
produites. Or le méthane est formé par transfert d’hydrures (3,6) du dépôt
hydrocarboné sur la surface (paraffines, cycloparaffines) vers les ions
carbénium.

36
X-R. Xia et Co (7) expliquent que lors du processus d’aromatisation, il y a
départ d’un atome hydrogène vers le méthoxyle qui désorbe pour donner le
méthane.
III.7. Premiers produits de la réaction
III.7.1. Distribution des oléfines
Dans les conditions d’une activité faible , on a uniquement l’éthylène et le
propène ; cela évite l’interférence de réactions secondaires qui compliquent le
raisonnement dans la recherche du mécanisme.
L’étude de la transformation de l’éthylène et du propylène sur la H-ZSM5,
d’après la littérature, fournit des informations sur le mode de conversion de ces
deux molécules . Dans ce chapitre, on accorde de l’intérêt à la sélectivité du
propène et de l’éthylène dans le mélange parce que certains auteurs ont travaillé
sur la nature de la formation de ces deux oléfines .
Des oligomères (8,9) sont formés à partir de propène marqué au C13
(10,11) ;l’éthylène est formé par craquage de ces oligomères .
Dans la conversion du chlorométhane, en faisant varier le temps de contact, on
note une diminution rapide de la sélectivité en propène (fig.9)(tableau III,9)
alors que celle de l’éthylène reste constante ce qui montre une participation
importante du propène dans les réactions secondaires. Comme par exemple
l’aromatisation (7) du propène. Le benzène qui dispose d’ électrons Л donne les
alkyles benzènes.

37
Tableau III,6 : vitesse des premiers produits en fonction du temps de
contact
(g.mole/h) 2,2 4,2 16,2
V.CH4 (m.mole.g/h) 2 1,65 1,2
V.c=2 (m.mole.g/h) 4,6 5,1 4,9
V.C3 = (m.mole.g/h) 9 8.2 5,9
0
2
4
6
8
10
2.2 4.2 16.2
V C= 2 , C=3 , CH4 (m.mole.g/h)
Fig. 6 : vitesse des premiers produits en fonction du temps de contact
PCH3CL= 76 torr , TR=500°C, Si/Al=60 , tR=30 min , DT = 10 l/h
V.C3 = V.C2
= V.CH4
(g.mole/h)
38
Tableau III,6 : Fig.7: Evolution des produits en fonction de la pression
P (torr) 19 38 47.5 57 76 114
V. de formation de CH4 (m.mole.g/h) 0.7 1 1.1 1.15 1.3 1.6
V. de formation de C=2 (m.mole.g/h) 1.1 1.8 1.9 2.5 3.3 4.7
V. de conversion de CH3Cl (m.mole.g/h) 1.8 3.3 4 4.9 7 11.1
V. de formation de C=3 (m.mole.g/h) 2 4.1 5.1 6.2 8.9 14.5
Fig.7: Evolution des produits en fonction de la pression de CH3Cl TR = 430 °C , DT = 40 l/h sur H-ZSM5 (Si/Al=30), mC = 200 mg
0
2
4
6
8
10
12
14
16
19 38 47.5 57 76 114
CH4
C2=
C3=
CH3Cl
V. de formation de C=2, C=
3, CH4 (m.mole)/ g.h.)
PCH3CL (torr)
CH3Cl

39
Tableau III,8 : Evolution des produits en fonction de la pression
PCH3CL (torr) 19 38 47.5 57 76 114
V. de formation de C=2, C=
3, CH4
(m.mole.g/h)
4.6 5.8 6.5 6.7 8.2 7.2
- 5.0 7.5 8.8 9.4 12 12.8
- 4.4 7.9 10.2 11.5 15.8 19.8
- 15 12.7 15 16.3 21.6 24
Fig.8 : évolution des produits en fonction de la pression
TR = 500 °C, DT = 40 l/h sur H-ZSM5 , (Si/Al=30), mC = 200 mg
0
5
10
15
20
25
19 38 47.5 57 76 114
CH4
CH3CL
C=3
C=2
Vitesse de formation de C=2, C=3, CH4 (m.mole/ g.h)
PCH3 CL (torr) CH3Cl

40
Tableau III,9 : sélectivités des oléfines en fonctions du temps de contact
(g.mole/h).
(g.mole/h) 2.1 4.4 17.8
Sélectivité de l’éthylène 37.7 38.7 36.7
Sélectivité du propène 41.7 36 29
Fig.9 ; sélectivités des oléfines en fonctions du temps de contact (g.mole/h) indiqué dans le tableau III,9. TR = 500 °C, DT = 20 l/h sur H-ZSM5 , (Si/Al=30), mC = 200 mg
g.mole/h 2.1 4.4 17.8
TR = 500 °C, DT = 20 l/h sur H-ZSM5 Si/Al=30 mC = 200 mg

41
III.7.2. La température de réaction
On suit l’évolution de l’éthylène et du propylène en présence de CH3Cl
(ou CH3OH) pour une température de réaction de 300°C pour éviter de
transformer l’une ou l’autre de ces oléfines.
Nous observons que le propène se transforme à température modérée
contrairement à l’éthylène qui nécessite une température beaucoup plus élevée.
Le mélange réactionnel initial constitué par (CH3Cl+C2H4) a bien donné
du propène par rapport aux références de (CH3Cl seul) et (C2H4 seul) où il ne
paraissait pas. Ce résultat a été observé par Lersch.P. et Bandermann.F.(4) .
Ils n’observent pas de propane qui doit se former à partir de la réaction
d’un hydrure avec l’ion C3H7(+) .

42
III . 8 Conclusion
Cette zéolite présente une configuration tridimensionnelle intéressante
dans la mesure où la disposition des canaux ainsi que leurs connexions,
contribue à garder une activité plus stable dans le temps pour des conditions
expérimentales modérées. L’influence de certains paramètres, dans le sens de
l’accroissement de l’activité, montre que la vitesse du propène diminue
comparativement à l’éthylène ou au méthane dont les vitesses de formation
restent constantes. Et ceci à cause d’une importante activité oligomérisante du
propène.
La production du méthane ne présente aucune sensibilité à la variation de
la pression partielle du chlorométhane. Cet alcane, formé par association d’un
hydrure et d’un ion carbénium, ne semble dépendre que des cycloparaffines
déposées .
On observe la diminution progressive des sites actifs à cause de la
formation de coke à l’intérieur et à l’extérieur du grain zéolitique. Cette
désactivation finit par bloquer l’activité au sein des canaux et des pores de la
zéolite.

43
Bibliographie
1- J.C. VEDRINE, A. AUROUX , C. NACCACHE, P. WIERZCHOWSKI,
E.G. DEROUANE, J.B. NAGY, Journal of catalysis 59, 248-262 (1979).
2- ZHONGMIN et Coll., Dalion institute of chemical physics, chnese
Aademy of science, Dalion, China, catalyst deactivation 1991, Elsevier
science Publiscers B. V. AMSTERDAM.
3- Xin-Rui Xia, Yin-li BI, Tong-Hao Wu and Kai-Ji Zhen, catalysis letters,
1995, Vol 33, number 1-2, pp75-90.
4- P.LERSCH et F. BDERMANN, Applied catalysis, 75 1991, 133-152.
5- M.GUISNET, P. MAGNOUS, C. CANOFF, “chemical reactions in
organic and inorganic constraint systems” D. Reidel Publ. Co.,
DORDRECHT 1986 P.131.
6- H.SHULZ, D.BARTH et Z.SIWEI. Studies in surface science and
catalysis vol. 68 CATALYST DEACTIVATION 1991 P.783.
7- X-R. Xia and Co, Catalysis letters, Vol 33, numbers 1-2, 1995, p.75-90.
8- DESSAU et LAPIERRE, Journal of Catlysis, 78, 1982, 136.
9- K.NGALULA. Thèse, 1997, Université de Namur, Belgique.
10- Y.W. BIRZEH et B.C.GATES, Journal of Catalysis 88, 240 (1984)
11- R.SHIGEISHI, A. GARFORTH, I. HARRIS et J.D. WYER (423) Journal
of Catalysis Vol. 130, number 2, August 1991.

44

45
IV.1. Recherche du Mécanisme
IV. 1. 1 Liaison carbone-carbone
La première liaison carbone-carbone peut être une insertion du
groupement méthyle sur une liaison C-H ou le résultat de l’insertion du
méthylène sur une liaison C-H.
IV . 1. 2. Investigation
IV.1. 2. 1. L’ion carbénium
L’ion carbénium mis en évidence par étude infra rouge provient de la
rupture de la liaison carbone-chlore.
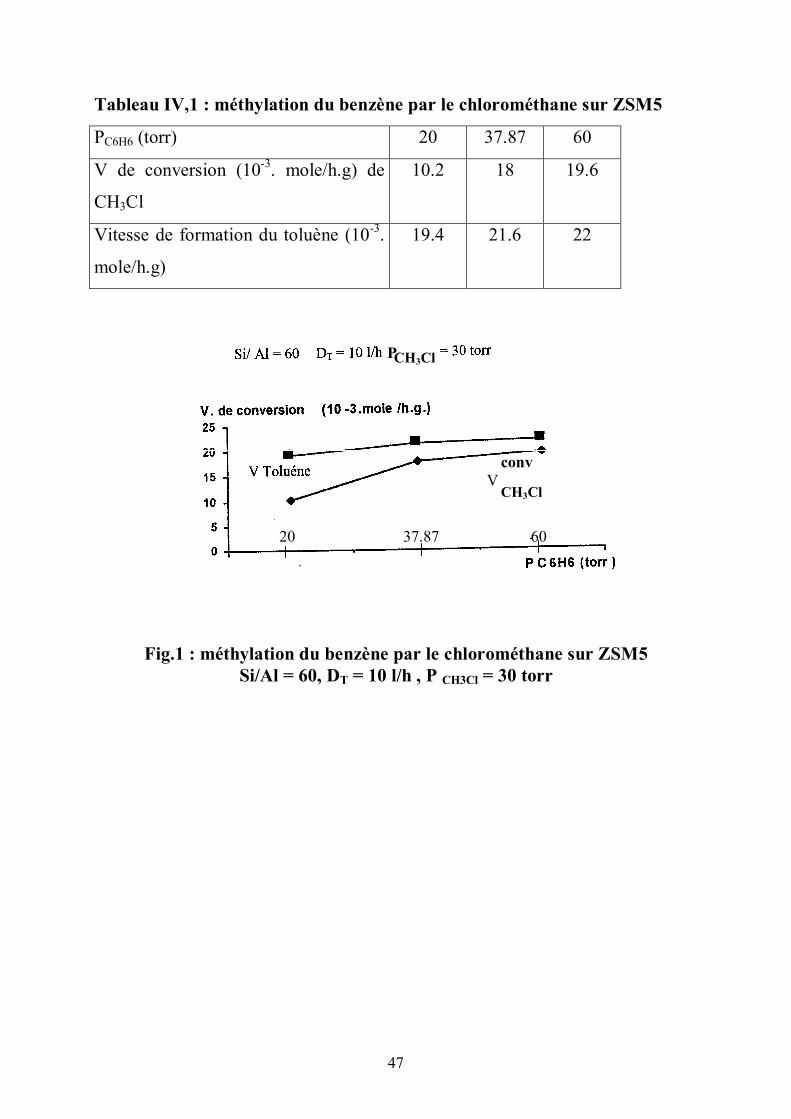
L’analyse chromatographique de la réaction du chlorométhane en
présence de benzène (fig.1) (tableau IV,1) montre l’activité du carbénium. Les
conditions opératoires choisies ne permettent pas, dans le cas du réactif seul ,
d’obtenir des aromatiques.
En présence de benzène apparaît le toluène par substitution électrophile
sur le benzène. Ce résultat montre la mobilité et la réactivité des CH3(+) en phase
gazeuse dans la zéolite.
Olah et collaborateurs(1), et Ono et Mori (2) proposent , dans le cas de
CH3OH, l’attaque de CH3(+) sur C-H du méthanol ou du diméthyle ether.
Ils observent le groupement CH3(+) en RMN du carbone 13 et en infra
rouge mais la proposition mécanistique n’est pas très clairement établie.
Par contre, sur un super acide, SbF5-FSO3H, Olah (1) aboutit aux
allongements de chaînes hydrocarbonées par l’insertion effective du groupement
CH3(+). Tout comme il explique, parallèlement, que CH3
(+) en présence
d’hydrogène moléculaire constitue l’intermédiaire carbone pentacoordiné CH5(+)
libérant le méthane qui lui-méme subit l’insertion du CH3(+).

46
IV . 1.2. 2. Le Méthylène
IV. 1. 2.2.1. Le premier produit
Romanikov (3), trouve que l’éthylène est le premier produit dans la conversion
du chlorométhane ; il propose le méthylène comme intermédiaire. L’étude
infrarouge ,dans ce mémoire, met en évidence l’apparition des alkoxyles
(méthoxyles, éthoxyles) (4) d’où la formation de la liaison carbone-carbone .
D’autres travaux (5,6,7), proposent le propène comme premier produit dans
cette réaction.
Avec les échantillons de H/ZSM5 aux différentes teneurs en aluminium, et à
très faible conversion, on obtient du propène comme seul produit (fig. 2,3)
(tableaux : IV,2 ; IV,3). Or l’association des motifs CH2 doit normalement
passer par le dimère c’est à dire l’éthylène et ce avant la formation de toute autre
molécule.
IV.1. 2. 2. 2. L’hypothèse de l’insertion
Le méthylène peut être produit par décomposition du cétène ou du
diazomèthane. CH2 ,très actif , arrive à s’insérer sur les composés organiques.
Le diazomèthane gazeux délicat dans sa synthèse (8) à cause des risques
d’explosion, est condensé et conservé à –78°C.
Par décomposition du diazométhane, Lee et Wu (9) arrivent à obtenir l’éthylène.
Ils pensent que CH2 par association avec H(+) du site acide donne le méthoxyle
d’une part, et d’autre part l’association de deux méthylènes donne l’éthylène.
Dans la transformation du méthanol, l’insertion (10) du méthylène sur la liaison
C-H est proposée pour expliquer la formation de la liaison carbone-carbone.
47
Tableau IV,1 : méthylation du benzène par le chlorométhane sur ZSM5
PC6H6 (torr) 20 37.87 60
V de conversion (10-3. mole/h.g) de
CH3Cl
10.2 18 19.6
Vitesse de formation du toluène (10-3.
mole/h.g)
19.4 21.6 22
Fig.1 : méthylation du benzène par le chlorométhane sur ZSM5 Si/Al = 60, DT = 10 l/h , P CH3Cl = 30 torr
20 37.87 60
V
conv
CH3Cl
P CH3Cl

48
Tableau IV,2 : parution du premier produit selon la température de
réaction
TR (°C) 350 360 366 375 388 397
(%) Sc=2 0 0 2 5 9 14.5
(%) Sc=3 100 100 98 95 91 85.5
Fig.2: parution du premier produit selon la température de réaction
Si/Al = 60, DT = 40 l/h , P CH3Cl = 30 torr, mC = 200 mg.
TR (°C)
350 360 366 375 388 397
49
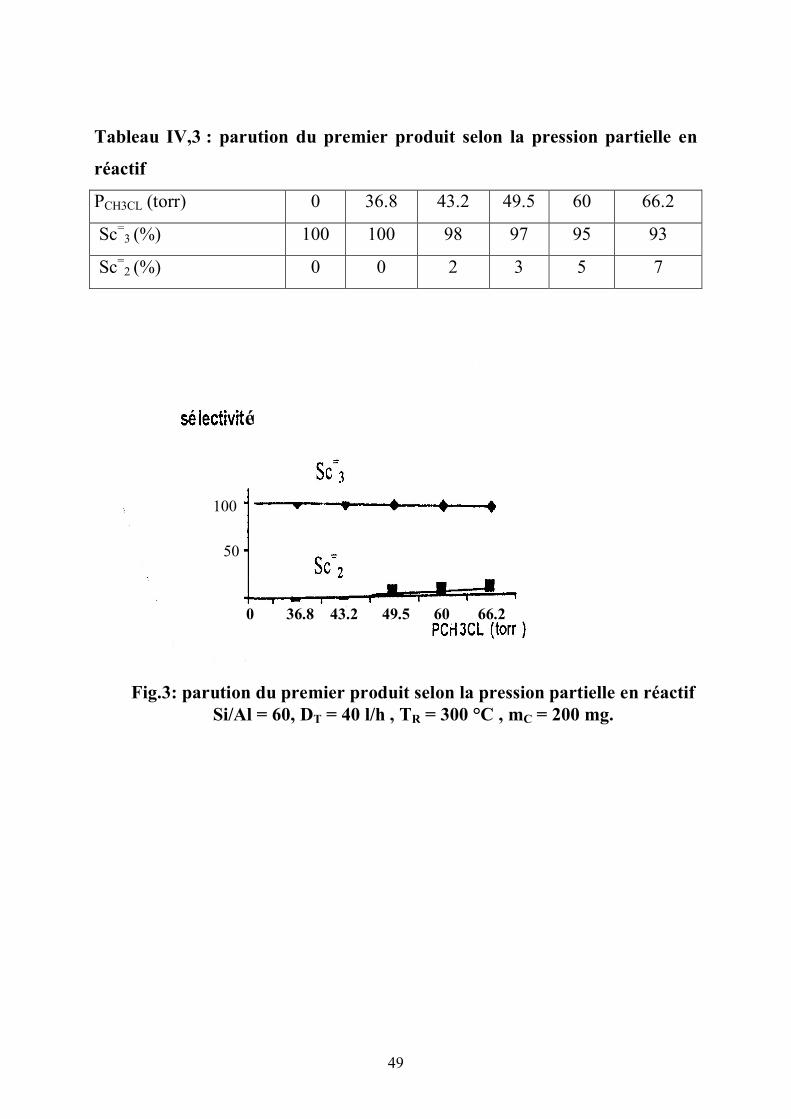
Tableau IV,3 : parution du premier produit selon la pression partielle en
réactif
PCH3CL (torr) 0 36.8 43.2 49.5 60 66.2
Sc=3 (%) 100 100 98 97 95 93
Sc=2 (%) 0 0 2 3 5 7
0 36.8 43.2 49.5 60 66.2
50
100
é
Fig.3: parution du premier produit selon la pression partielle en réactif Si/Al = 60, DT = 40 l/h , TR = 300 °C , mC = 200 mg.

50
Le méthylène est très actif ; il a une durée de vie de quelques dixièmes de
seconde (11). On a suivi le test avec le mélange gazeux chlorométhane/propane
pour voir si on obtient le butane et l’isobutane, preuve de l’existence et du
passage par le méthylène. Ce test est non concluant puisqu’aucun des produits
attendus n’est apparu. D’après le travail de Frey (12), le propane en présence de
CH2 donne les C4 ; le mode d’insertion de CH2 sur C-H est expliqué par
Meerwein (13).
IV. 1. 2.2. 3. H-ZSM5/Pt
On suppose que CH2 vient de CH3CL. On fait débiter le mélange gazeux
chlorométhane/ hydrogène sur une H-ZSM5 au platine traitée sous air jusqu’à
350°C, balayée sous azote puis sous H2 jusqu’à 450°C. Pour le test, on repasse à
une température de réaction de 300°C pour garder une assez faible activité et
éviter ainsi la complexité dûe à la présence de produits autres que l’éthylène et
le propène. On doit avoir de l’éthane résultant de l’hydrogénation de l’éthylène
et la baisse notable de la sélectivité en propène qui résulterait de l’interaction de
CH2 avec l’éthylène. On n’obtient pas d’éthane. Le méthane est le seul produit
du test.
IV.1. 2. 3. Les radicaux
Avec une température de 500°C, on pense à l’effet thermique du système sur la
liaison carbone-chlore.
En dépit des explications (14) selon lesquelles des défauts au niveau de la
structure zéolithique représentant des sites paramagnétiques seraient à l’origine
de la formation de radicaux piégés et mis en évidence en R.P.E, on n’a retenu
cette possibilité pour les raisons suivantes.

51
a) Si on doit tenir compte uniquement de l’effet thermique pour rompre la
liaison C-Cl et produire des radicaux, la réaction ne doit pas se produire
à basse température or elle a bien lieu.
b) On a fait passer CH3Cl sur le réacteur dépourvu de catalyseur à 500°C et
un faible débit de 0.5 l/h. Aucune transformation n’a lieu.
IV.2. Etude infra rouge
Parallèlement au suivi en dynamique de la réaction de conversion du
chlorométhane, on fait une étude spectroscopie infra rouge .
Expérimentalement, on réalise une pastille de H-ZSM5 d’environ 16 mg que
l’on traite sous oxygène puis sous vide dans une cellule jusqu’à 400°C. La
zéolite est ensuite mise en contact avec une pression de dix torr en
chlorométhane et le spectre infra rouge est alors enregistré.
Dans un premier spectre obtenu à la température ambiante, on voit nettement le
déplacement de la bande des OH pontés (fig.4,5), déplacement dû à l’interaction
de ces hydroxyles avec le réactif.
Cependant, la réaction n’a pas pour autant été entamée puisqu’en faisant le vide
pendant une heure, on retrouve exactement la même allure du spectre relatif à la
pastille vierge avant l’introduction du gaz. Il a fallu monter en température pour
observer des changements.
Une réintroduction de CH3Cl dans la cellule suivie d’un chauffage à 350°C
pendant une heure puis d’un pompage sous vide a permis d’observer (fig.6) les
bandes caractéristiques des vibrations C-H du groupement CH3 chimiquement
adsorbé sur la surface (SiOCH3), mais aussi de groupes alkoxyls comme cela a
été obsevé par d’autres auteurs (4,15). On retrouve ces mêmes bandes de
vibrations dans certaines analyses (16,17) infra rouge du méthanol.
On a pas pu mettre en évidence l’intermédiaire réactionnel envisagé, l’ion
Sillyle Diméthyl Oxonium à cause de son instabilité.
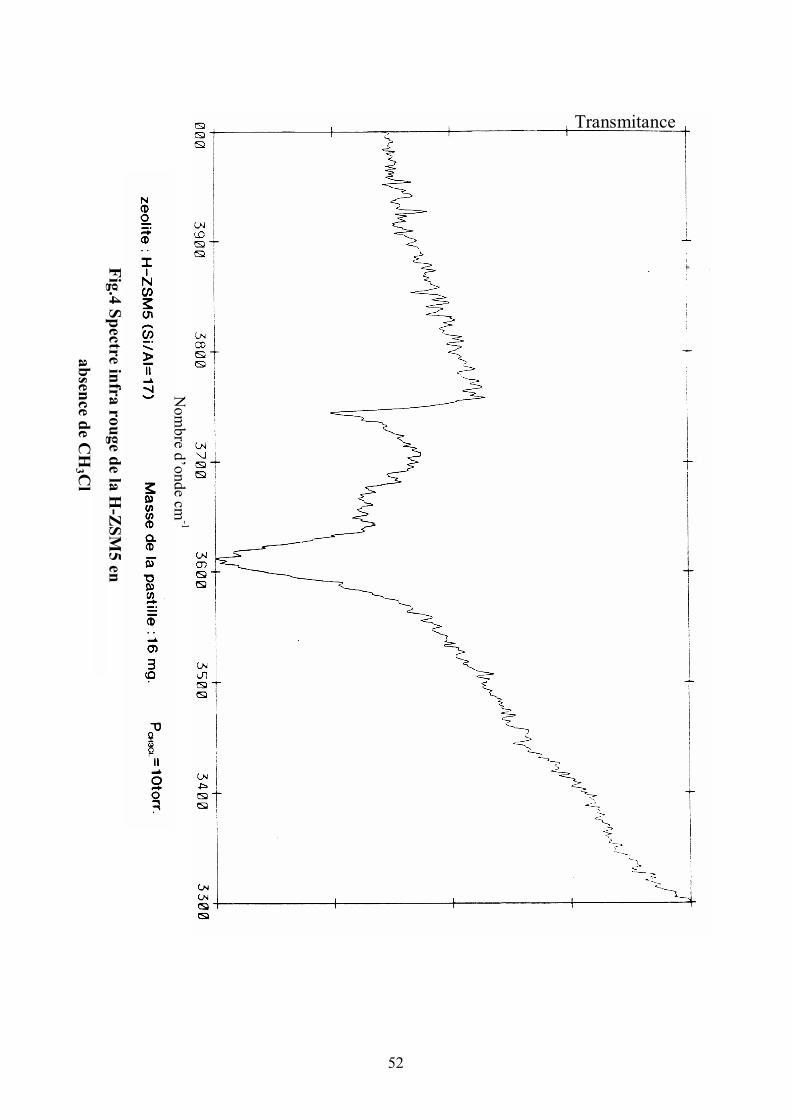
52
Nom
bre d’onde cm-1
Fig.4 Spectre infra rouge de la H-Z
SM5 en
absence de CH
3 Cl
Transmitance

53
Fig.5 : Déplacem
ent de la bande OH
la bande à 3600 cm
-1 se déplace vers les basses fréquences apparaissant comm
e une bande large entre 3400cm-1 et 3100 cm
-1 (observée par salvador et K
lading. J.Chem
. Soc. Faraday Trans, 1, 1977, 73, 1153.
Nom
bre d’onde cm-1
Transmitance

54
Fig.6 : Bandes de vibration CH
du groupement C
H3
Nom
bre d’onde cm-1
2980
2920
2852
2958
3207
3745
Transmitance

55
IV.3. Rupture de la liaison carbone-chlore
Dans cette partie sera abordée la réaction dans ses premières étapes. Pour le
mode de rupture de la liaison du groupement méthyle avec l’halogène, deux
voies sont possibles.
Une substitution électrophile où le proton de la zéolite arrive sur un doublet
électronique libre du chlore pour ensuite voir le départ du CH3(+). La mobilité
(19) du proton est confirmée en infra rouge par échange entre la structure HY
partiellement deutérée et CH3OH. La seconde voie de rupture est une
substitution nucléophile entre le chlore partant et l’oxygène du site par le biais
d’un de ses doublets électroniques libres.
Cette dernière explication est envisageable parce qu’elle est compatible avec
l’étude infra rouge que nous avons suivie (fig. 4 et fig. 5).
L’envoi, à la température ambiante (25°C), de CH3Cl sur une H-ZSM5 a
provoqué le déplacement de la banche à 3600 cm-1 vers les basses fréquences.
Cette bande relative aux hydroxyles en interaction avec l’aluminium voisin,
subit un abaissement qui va jusqu’à 3200 cm-1, déjà observé dans le cas du
méthanol (8), et provoqué par la forte interaction existant entre le doublet libre
de l’oxygène de l’hydroxyle et le groupement méthyle du chlorométhane.
H H
C
O+ Cl H
H
Si
-Al
CH3Cl + Al
OH
Si

56
Il est intéressant d’observer qu’il existe une corrélation entre l’activité du solide
dans le test catalytique et le déplacement de la bande à 3600cm-1.
On note d’après la figure 7, une nette différence d’interaction du réactif vis à vis
des deux zéolites. Cette différence est décrite par Jacobs (20).
Ceci confirme d’une part le type de rupture carbone-chlore et d’autre part une
activité catalytique étroitement liée à cette première étape de la réaction. Des
explications plus détaillées sont citées dans la partie (IV,3) sur le déplacement
de la bande OH de 3600cm-1 qui est différent chez la mordénite par rapport à la
H-ZSM5.

57
Fig.7 : Abaissem
ent de la bande à 3600 cm -1
le déplacement de la bande est plus im
portant dans le cas de la ZSM5 que dans la m
ordenite cette observation est en corrélation avec les activités respectivem
ent élevée et faible des deux zéolites
Transmitance
Nom
bre d’onde cm-1
ZSM5
Mordénite

58
IV.4. Le mécanisme
En admettant la première étape, une deuxième rupture est possible en
précisant que le premier groupement méthyle ne constitue pas de gène stérique
pour le second.
On assiste donc à la formation d’une espèce, le sillyldiméthyloxonium, un
peu semblable au triméthyle oxonium (20) trouvé dans la réaction de conversion
du méthanol.
La conversion de ce dernier passe par l’attaque du proton suivie du départ
de H2O. Le CH3(+) qui en découle attaque un deuxième CH3OH pour donner le
diméthyle éther avec restitution du proton à la zéolite.
Le diméthyle éther subit par son oxygène l’arrivée d’un autre méthyle
réalisant ainsi le triméthyle oxonium.
Le caractère instable du sillyl diméthyle oxonium ne permet pas à la
méthode physico-chimique de l’observer.
CH3
OH O OCH3
Si + CH3Cl H HCl Si
Al Al CL
Al
Si
(+)
(-)
CH3 +CH3Cl (+) O(CH3)2
Si Cl HCl + Si (-) CH2
(+) O
Al(-) Al

59
Certains (21) se sont penchés sur la synthèse, loin de la réaction, de
l’intermédiaire relatif à la transformation du méthanol.
Dans un autoclave, le diméthyle éther est mis en présence de PF5. La
réaction donne un produit de condensation, le sel de triméthyle oxonium.
PF5 3 (CH3)2 O 2 (CH3)3 (+)OPF6
(-) + POF3
Olah (22) a traité ce sel par une base forte pour provoquer une
déprotonation de manière à établir que l’alkylation a bien lieu et qu’elle est
intermoléculaire.
Nous trouvons une similitude entre le cheminement réactionnel de
transformation de CH3Cl et celui du CH3OH .Ces deux particules, une fois
débarrassées de X dans CH3X, vont évoluer de façon identique au sein de la
zéolite. Cependant, nous verrons plus loin que les activités sont différentes pour
des conditions expérimentales semblables du test de conversion
L’ion sillyl diméthyle oxonium perd un proton avant de donner naissance
à la liaison carbone-carbone (fig.10).
Cette liaison ne peut se former par réarrangement intra moléculaire.
C’est plutôt une réaction intermoléculaire (fig.10) comme c’est le cas du
méthanol (22).
IV. 5. Déshydroxylation
Le travail sur la zéolite déshydroxylée confirme la thèse du sillyl
diméthyle oxonium sachant qu’on arrive à convertir le chlorométhane sur ce
même solide dépourvu de sa propriété acide de Bronsted mais restant néanmoins
actif suite à une adsorption de diméthyle éther (fig. 9). Le traitement de la H-
ZSM5 se fait sous vide à 950°C (23). Après ce traitement, le solide est
déshydroxylé .

60
On envoie ensuite du diméthyle éther qui se fixe sur le site acide de
Lewis Si(+) auquel cas on retrouve exactement la forme prévalant dans cette
réaction.
L’analyse infra rouge montre que la forme obtenue Si –(+)O (CH3)2
donne des groupements méthoxyles par migration de l’un des méthyles sur un
site voisin, ce qui correspond aux vibrations caractéristiques (16) d’élongation et
de déformation (fig.8) observées.
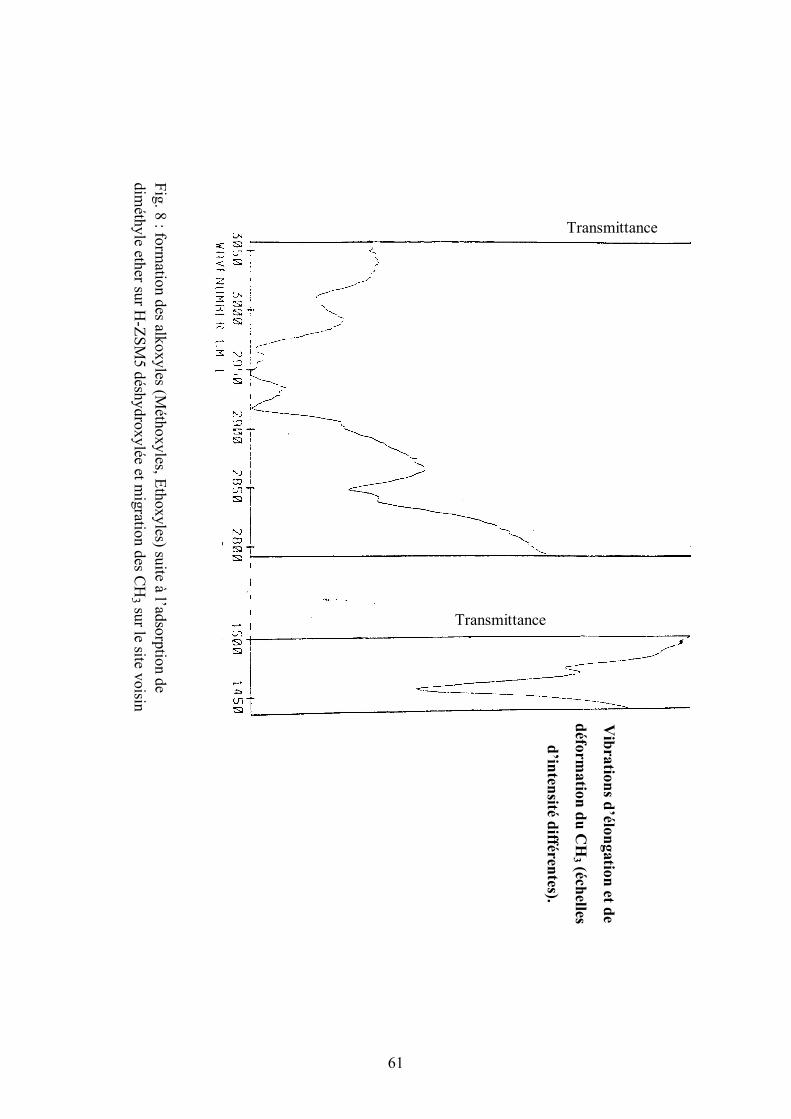
61
Fig. 8 : form
ation des alkoxyles (Méthoxyles, Ethoxyles) suite à l’adsorption de
diméthyle ether sur H
-ZSM5 déshydroxylée et m
igration des CH
3 sur le site voisin
Vibrations d’élongation et de
déformation du C
H3 (échelles
d’intensité différentes).
Transmittance
Transmittance

62
Fig.9 Réaction sur solide traité

63
IV. 6. Réaction Intermoléculaire
Stevens (24) dans son étude du méthanol avait proposé un
réarrangement intramoléculaire .On note dans la figure 10 la mobilité de l’ion
carbénium. Le méthylène méthyle oxonium obtenu par déprotonation sera, avec
l’arrivée du (+)CH3 du site voisin, le siége de formation de la liaison carbone-
carbone.Le moyen de vérifier un processus intermoléculaire est de montrer la
dépendance entre la sélectivité du propène (ou de l’éthylène) et l’éloignement
des sites ce qui correspond en pratique à tester plusieurs échantillons de ZSM5
de teneurs différentes en aluminium.
Les valeurs retenues en sélectivité du propène sont celles correspondant
à un même taux de conversion pour les différents solides dans des tests
d’activité réduite . La représentation graphique (fig.11) nous montre quasiment
une droite établie entre le nombre d’aluminium par maille correspondant à la
distance inter-sites qualitative et la sélectivité du propène seul produit de
réaction avec l’éthylène . Ce résultat signifie que la réaction se déroule de
façon intermoléculaire. La corrélation trouvée montre qu’au lieu que SC3= (ou
SC2=) soit quelconque, celle-ci dépend de la teneur en aluminium, ce qui
physiquement correspond à la contribution commune de deux sites voisins dans
la formations de la liaison carbone-carbone.
Signalons qu’à raison de 3 aluminium par maille, la ZSM5 ne respecte
plus cette tendance à cause de la parution simultanée des paraffines (C3, ΣC4),
oléfines (C=2, C=
3).
La formation de la liaison carbone-carbone a été abordée (25) dans la
conversion du méthanol pour arriver à la même conclusion de contribution des
sites. L’auteur (25) part de diméthyle éther simplement marqué auC13. Le
résultat du test débouche sur des rapports en éthylène spécifiquement liés au
processus intermoléculaire de (+)CH3 intervenant sur l’intermédiaire réactionnel.
Le rapport en éthylène non marqué et simplement marqué est de 1 pour 2.
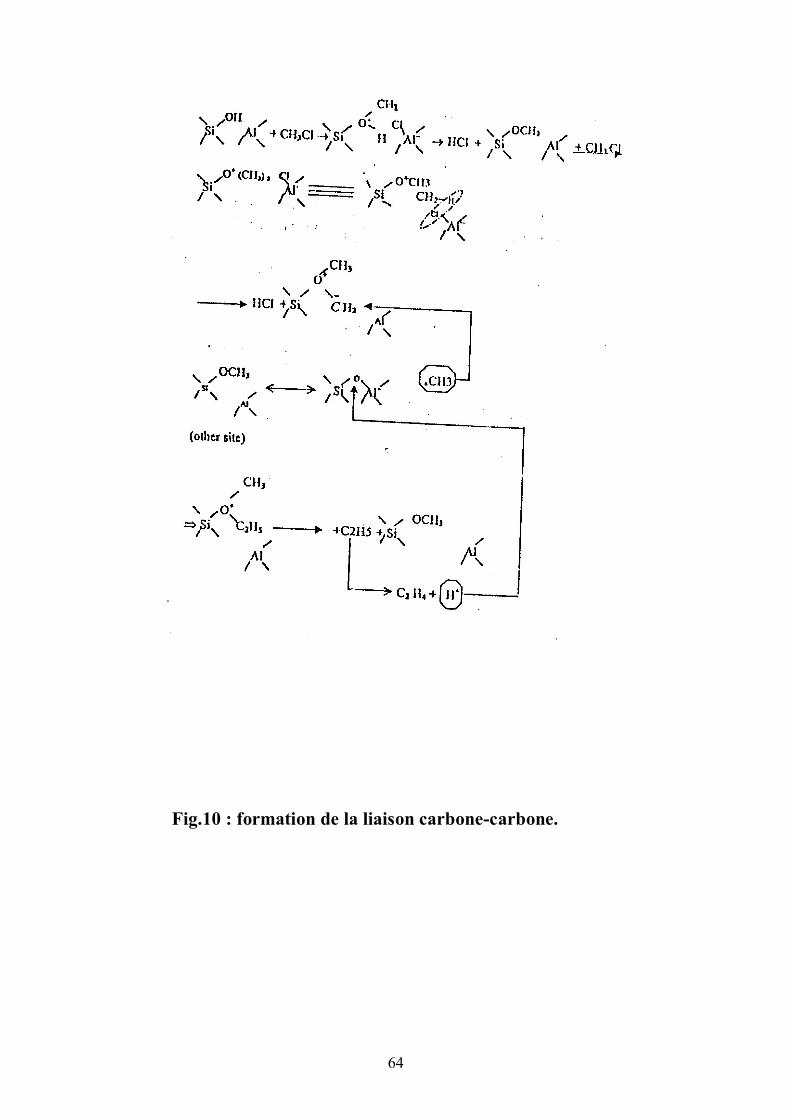
64
Fig.10 : formation de la liaison carbone-carbone.

65
Tableau IV,11 : Sélectivité du propène en fonction de la distance moyenne
inter-sites (ou nombre moyen d’Al par maille)
Nombre d’Al par maille 0,9 0,12 1,48
Sc=3(%) 9 70,4 92,8
Conversion de 2% en CH3CL sur H-ZSM5 à TR= 300°C
0
20
40
60
80
100
1 2 3
S C3=
Si/Al=60) (Si/Al=80)
(Si/Al=100)
Fig.11 : Sélectivité du propène en fonction de la distance moyenne inter-sites (ou nombre moyen d’Al par maille)

66
IV. 7. Le méthanol
S’il y a similitude de transformation de CH3Cl et de CH3OH de par la
nature de l’intermédiaire formé, l’ion oxonium, espèce fixe dans le cas du
chlorométhane et mobile en phase gazeuse dans le cas du méthanol, on note une
différence d’activité . Dans les mêmes conditions expérimentales, les activités
ou vitesses de conversion sont différentes. On explique cette différence par le
fait que les molécules CH3Cl doivent parvenir sur un site acide fixe pour
amorcer la transformation et donner forme à l’intermédiaire réactionnel alors
que dans le cas du méthanol, l’intermédiaire en question se retrouve dans la
phase gazeuse.
Lors du test de conversion du mélange équimolaire CH3Cl/CH3OH
(voir fig.7 chap .V), on assiste à une nette régression d’activité par rapport aux
états de référence des réactifs seuls. Ce résultat s’explique par l’inhibition de
CH3Cl sur CH3OH .

67
IV. 8. Conclusion
Le test catalytique spécifique à la présence et à l’insertion du méthylène
n’est pas concluant .
On a la formation d’une espèce , le sillyl diméthyle oxonium, semblable
à celle prévalant dans la transformation du méthanol en l’occurrence le
triméthyle oxonium.
La première étape de la réaction est une rupture de la liaison carbone-
chlore .
Le déplacement de la bande OH vers les basses fréquences est dû à
l’interaction du réactif avec le site, interaction qui correspond à une substitution
nucléophile d’ordre deux avec formation des méthoxyles et du gaz
chlorhydrique.
Le groupement méthylique passe d’une forme tétraédrique d’hybridation
sp3 à une forme planaire d’hybridation sp2 permettant à l’orbitale p axiale de
réaliser la substitution.
On a une corrélation entre les déplacements de bandes OH de deux
zéolites distinctes et leurs activités respectives.
En travaillant sur la ZSM5 déshydroxylée et sur laquelle on envoie,
après traitement, du diméthyle éther, on réalise la conversion effective du
chlorométhane.
Cette expérience sur le solide dépourvu de son acidité de Bronsted
rejoint à la fois l’analyse spectroscopique infra rouge et le test catalytique. Elle
permet de ressortir le rôle tenu par l’atome de chlore dans la déprotonation de
l’intermédiaire et de confirmer la nature de la transformation.

68
BIBLIOGRAPHIE
1- G.A. OLAH et Coll., J.An. chem. Soc. 1969, 91, 3261.
2- Y.ONO et Coll., chem. Soc. Faraday Trans. 1,1981, 77, 2209-2221.
3- V.N. ROMANIKOV et K.G.IONE, Kin. Et catal . (engl. Transl.) 25
(1984)75.
4- Xin-Rui Xia, Yin-li BI, Tong-Hao Wu and Kai-Ji Zhen, catalysis letters,
1995, Vol 33, number 1-2, pp75-90.
5- G.PEROT, F.X. CORMERAIS et M. GUISNET, J. Molec. Catal. 17 (1982)2
6- J.P. VAN DEN BERG, J.P. WOLTHUIZEN et J.H.C. VAN HOOFF, in
« proc. 5th Int. Conf. Zeolites”, L.V.C. REES, ed (Heyden and sons,
LONDON, 1980); p.649.
7- S. CECKIENCZ, J. coll. Interface sci, 90 (1982) 183.
8- G. WICKER, Thése de doctorat, 1966, Université LYON.
9- C.S. LEE et M.M.WU, J. Chem. Soc, chem. Commun. 1985, 250.
10- CHANG et SILVESTRI, J. catal. 1977, 47, 249.
11- TROZZOLO, MORRAY, WASSERMAN and YAGER, J am. Chem.. soc.
84, 3213 et 4990 (1962).
12- FREY ; Proc. Chem. Soc, 1959, 318.
13- MEERWEIN, RATHJEN et WERNER, Ber. 75, 1610 (1942) “ carbene
chemistry” p.p.21-26, 52-64, 251-258, Academic Press. Inc, NEW YORK,
1964.
14- John K.A. CLARKE, J. of the chemical society, 1986.
15- B-L.Su, D.Jaumain, Proceedings of the 12th international zéolite
conference, 1999, vol 4, pp2681-2688.
16- B.A. MORROW, J.C.S. FARADAY I, 1974, 8, 1527.
17- V. BOSACEK, Z. TVARUZKOVA, coll. C. Zech. Chem.. comm.., 1971,
36, 551.
18- P. SALVADOR et W.KLADNIG G, J. chem.. soc. Faraday Trans, 1, 1977,
73, 1153.

69
19- Jacobs, Catalysis review, 1982, p427-429.
20- T.S. STEVEN’S et W.E WATTS, in “ selected ” molecular
rearrangements” (Van Nostrand Reinhold Pub. Co., LONDON, 1973)
21- Salem, G. Ph. D. Thesis-University of Southern California LOS
ANGELES, 1980.
22- G.A. OLAH, DOGGWEILER et J.D. FELBERG, J. Org. Chem. Vol. 49.
n°12, 1984. 2112-2116.
23- J.C.VEDRINE, A. AUROUX, V BOLIS,, P. DEJAIFVE, C.
NACCACHE, WIERZCHOWSKI, E.G. DEROUANE, J.B. NAGY, J.P.
GILSON, J.H.C. VAN HOOFF, J.P. VAN DEN BERG and J.
WOLTHUIZEN. J.of catalysis 59, 248-262 (1979).
24- T.S. STEVEN ‘S et W.E.WTTS, in “” selected molecular
rearrangements” (van nostrand Reinhold Pub. Co, LONDON, 1973).
25- G.A. OLAH, H. DOGGWELLER, J.D. FELBERG, S. FROHLICH, M. J
GRDINA, R. KARPELES, T. KEUMT, Shin-ichi INABA, W.M. Ip., K.
LAMMER TSMA, G.SALEM et DERRICK C. TABOR.- J. Am. Chem.
Soc., Vol. 106, n° 7, 1974.

70

71
V-1 Etude Cinétique :
Des faibles pressions aux pressions élevées, on note que le réactif se
convertit suivant le modèle de Langmuir- Hinshelwood.
On observe que l’ordre par rapport au chlorométhane passe de 1 aux faibles
pressions à zéro à pression élevée (fig1) en reportant la vitesse de transformation
en fonction de la pression du réactif.
L’ordre zéro est atteint à 120 torr en précisant que la teneur en aluminium
de la ZSM5 correspond à Si / Al = 100 .
V-2 Distribution comparée des produits :
Le méthanol se transforme en diméthyle éther qui accède ,par le biais d’un
des deux doublets électroniques libres de son oxygène, au triméthyle
oxonium (1), celui –ci est déprotoné puis méthylé par réaction intermoléculaire.
Le chlorométhane passe par le siliciumdiméthyloxonium pour donner la
liaison carbone-carbone par déprotonation-méthylation. A travers deux tests
distincts respectifs à CH3Cl et CH3OH, pour des taux de conversion très proches
et des conditions expérimentales différentes, on réalise une même distribution
des produits formés (tableau 1). Ce qui montre la similitude dans la nature de la
transformation.

72
Tableau 1 : Distribution comparée des produits de la réaction (les valeurs du tableau sont des sélectivités données en pourcentage %)
C=2 C=
3 CH4 Cn (n >5) C4 Arom
* 21.9 29.4 2.3 8.6 3 19
** 23.4 26.9 0.5 10 1.6 23
* 76 torr CH3Cl, DT = 2.5 l/h, HZSM5 (Si/ Al= 30), TR= 401°C, α = 22.4%
** 76 torr de CH3OH, DT=20 l/h, HZSM5(Si/Al=30), TR= 331°C, α = 24.5%
(α = taux d’activité ou de conversion )
Les différences relevées sur les vitesses de conversion, plus grandes dans le
cas du méthanol ( v(CH3OH)= 100,2.10-3 mole/h.g) (v (CH3Cl) = 11,4.10-3 mole
/h.g), s’expliquent par une rupture de la liaison C-O de CH3OH plus facile que
celle de la liaison C-Cl de CH3Cl
V 3. La première étape de la réaction :
Le déplacement de la bande infra rouge des OH vers les basses fréquences
n'est pas le même pour deux zéolites distinctes en 1'occurence la ZSM5 et la
mordénite . Un tel abaissement est dû à 1'interaction de 1'hydroxyle avec le
réactif.
Il est plus accentué pour une ZSM5 qu'il ne 1'est pour la mordénite. La
rupture de la liaison carbone-chlore est l’étape lente .
Les différences entre l’hydroxyle et le réactif donnent une estimation de la
force acide. Cette interaction est évaluée par le déplacement Δυ (7) de υ OH
(frequency shift) :

73
Δυ = 3 q E / 4 e (2 μ )1/2 . D1/2
q = charge dipolaire.
E = champ électrique le long de l’axe OH
μ = masse réduite du groupe OH.
D = énergie de dissociation de la liaison OH
Δυ = est fonction de la force acide
Le test catalytique montre une activité plus importante pour la ZSM5 que
pour celle de la mordénite pour des conditions opératoires identiques .
Le résultat spectroscopique infra rouge et l’activité catalytique vont dans
le même sens en se référant aux déplacements respectifs aux deux solides.
Cette constatation se traduit par un mode de rupture de type SN2 (voir IV 4) .
Dans le test du mélange équimolaire (CH3Cl+CH3OH), la conversion du
méthanol est inhibée par le chlorométhane (fig 2).
Le méthanol emprunte le proton pour sa propre conversion contrairement
au chlorométhane dont la première étape de rupture nécessite une disposition
géométrique de la molécule telle qu'au niveau du site de Bronsted et du site de
Lewis a lieu la double interaction débouchant sur la rupture carbone-chlore .
Lors de la rupture carbone-chlore, le processus de substitution nucléophile
d'ordre 2 répond à la formulation
)()( xbxadtdx
où a et b représentent les concentrations des espèces nucléophiles entrant
dans la réaction.

74
Entre CH3Cl et la surface du catalyseur s'établit un complexe de
substitution faisant intervenir 1'oxygene du site et le réactif .On a autant de sites
(actifs) que de molécules CH3Cl se rapprochant de la surface et rentrant en
réaction . On a une bijection ou binôme (site actif, molécule CH3Cl) de façon à
avoir a=b dans1'expression ci-dessus. Et on ne peut tenir compte ni des
particules restant dans la phase gazeuse loin de la surface ni des sites
n'intervenant pas dans la réaction à 1'instant t.
La détermination de la pression P0 du réactif se fait par extrapolation au
temps initial de la pression de chlorométhane converti au temps t=0 (fig.3a),
)²()()( xaxbxadtdx
=> aktxa
xaxa
ktxadtdx
11)²(
où a = P0 : pression de réactif converti au temps t.=0.
x = P : pression de réactif converti au temps t.
k = constante (obtenue par intégration)
On obtient effectivement une droite (fig.3b) ce qui confirme 1'explication
donnée sur la nature de la conversion du chlorométhane .
V . 4. Méthylation du benzène
Dans un test de faible conversion du chlorométhane, on a uniquement des
produits linéaires. En présence de benzène,le chlorométhane, par ses
groupements CH3(+), donne du toluène, et suivant les conditions expérimentales,
apparaissent d'autres dérivés polysubstitués.
A partir des pressions d'aromatiques formés, on sait la quantité de réactif
convertie sachant qu’on a un méthyle pour le toluène, 2 pour le xylène et 3 pour
le triméthyle benzène.
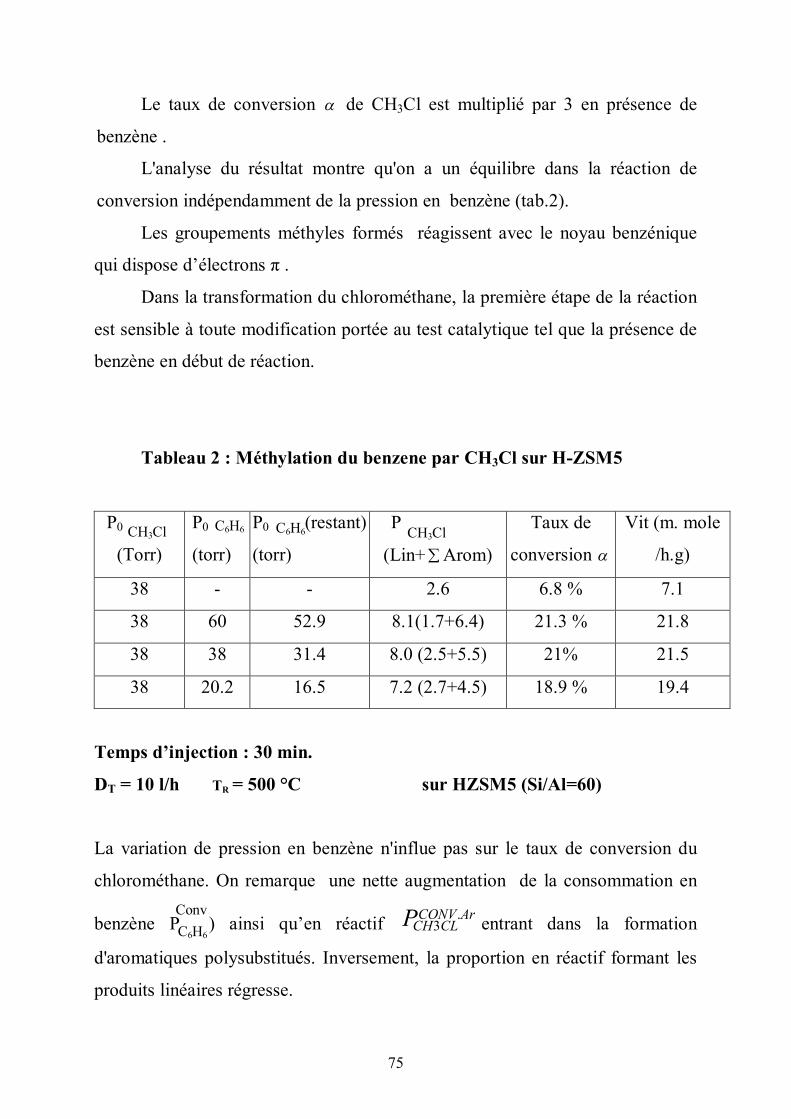
75
Le taux de conversion de CH3Cl est multiplié par 3 en présence de
benzène .
L'analyse du résultat montre qu'on a un équilibre dans la réaction de
conversion indépendamment de la pression en benzène (tab.2).
Les groupements méthyles formés réagissent avec le noyau benzénique
qui dispose d’électrons π .
Dans la transformation du chlorométhane, la première étape de la réaction
est sensible à toute modification portée au test catalytique tel que la présence de
benzène en début de réaction.
Tableau 2 : Méthylation du benzene par CH3Cl sur H-ZSM5
P0
(Torr)
P0
(torr)
P0 (restant)
(torr)
P
(Lin+Arom)
Taux de
conversion
Vit (m. mole
/h.g)
38 - - 2.6 6.8 % 7.1
38 60 52.9 8.1(1.7+6.4) 21.3 % 21.8
38 38 31.4 8.0 (2.5+5.5) 21% 21.5
38 20.2 16.5 7.2 (2.7+4.5) 18.9 % 19.4
Temps d’injection : 30 min.
DT = 10 l/h TR = 500 °C sur HZSM5 (Si/Al=60)
La variation de pression en benzène n'influe pas sur le taux de conversion du
chlorométhane. On remarque une nette augmentation de la consommation en
benzène P ) ainsi qu’en réactif ArCONVCLCHP .
3 entrant dans la formation
d'aromatiques polysubstitués. Inversement, la proportion en réactif formant les
produits linéaires régresse.
CH3Cl C6H6 C6H6 CH3Cl
Conv C6H6
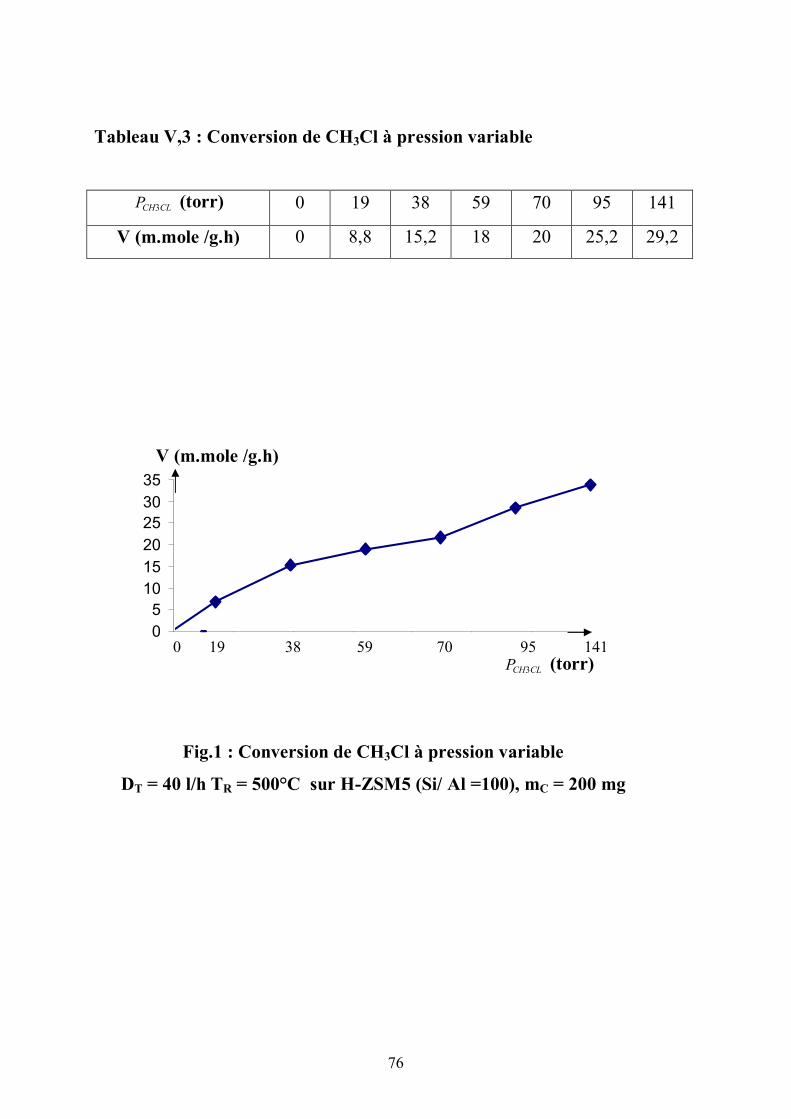
76
Tableau V,3 : Conversion de CH3Cl à pression variable
CLCHP 3 (torr) 0 19 38 59 70 95 141
V (m.mole /g.h) 0 8,8 15,2 18 20 25,2 29,2
05
101520253035
1 2 3 4 5 6 7
PCH3Cl (torr)
Fig.1 : Conversion de CH3Cl à pression variable
DT = 40 l/h TR = 500°C sur H-ZSM5 (Si/ Al =100), mC = 200 mg
0 19 38 59 70 95 141
V (m.mole /g.h)
CLCHP 3 (torr)

77
Tableau V,4 : Conversion du mélange équimolaire : CH3Cl +CH3OH
t (h) 0,5 2 3
V . de conv de CH3Cl ( m.mole/g.h ) 20 18 17
V . de conv de CH3Cl + CH3OH ( m.mole/g.h ) 95 90 86
V . de conv de CH3OH ( m.mole/g.h ) 126 118 114
Fig 2 : Conversion du mélange équimolaire : CH3Cl +CH3OH P CH3Cl = 38 torr , P CH3OH = 38 torr DT = 40 l/h , TR = 500°C , Si/Al = 100 Pour CH3Cl et CH3OH testés séparément, On a : P CH3Cl = 38 torr , DT = 40 l/h , TR = 500°C , Si/Al = 100 P CH3OH = 38 torr, DT = 40 l/h , TR = 500°C , Si/Al = 100
0.5 2 3
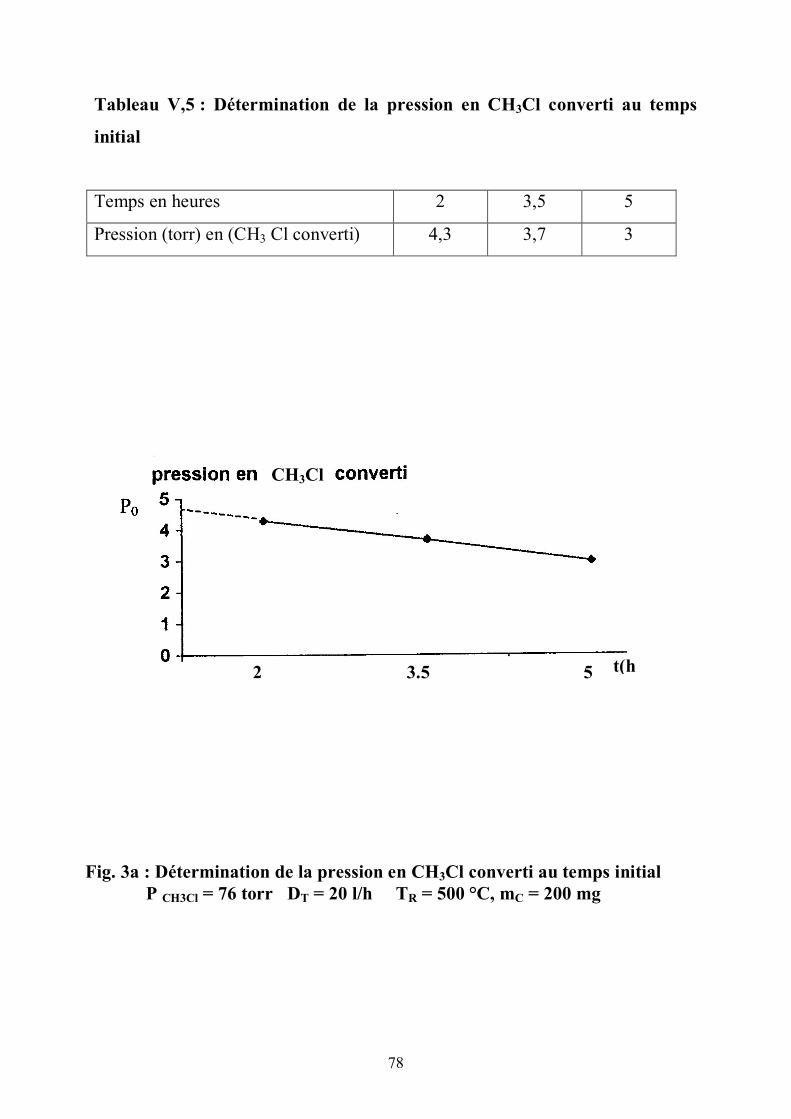
78
Tableau V,5 : Détermination de la pression en CH3Cl converti au temps
initial
Temps en heures 2 3,5 5
Pression (torr) en (CH3 Cl converti) 4,3 3,7 3
Fig. 3a : Détermination de la pression en CH3Cl converti au temps initial P CH3Cl = 76 torr DT = 20 l/h TR = 500 °C, mC = 200 mg
2 3.5 5 t(h
CH3Cl
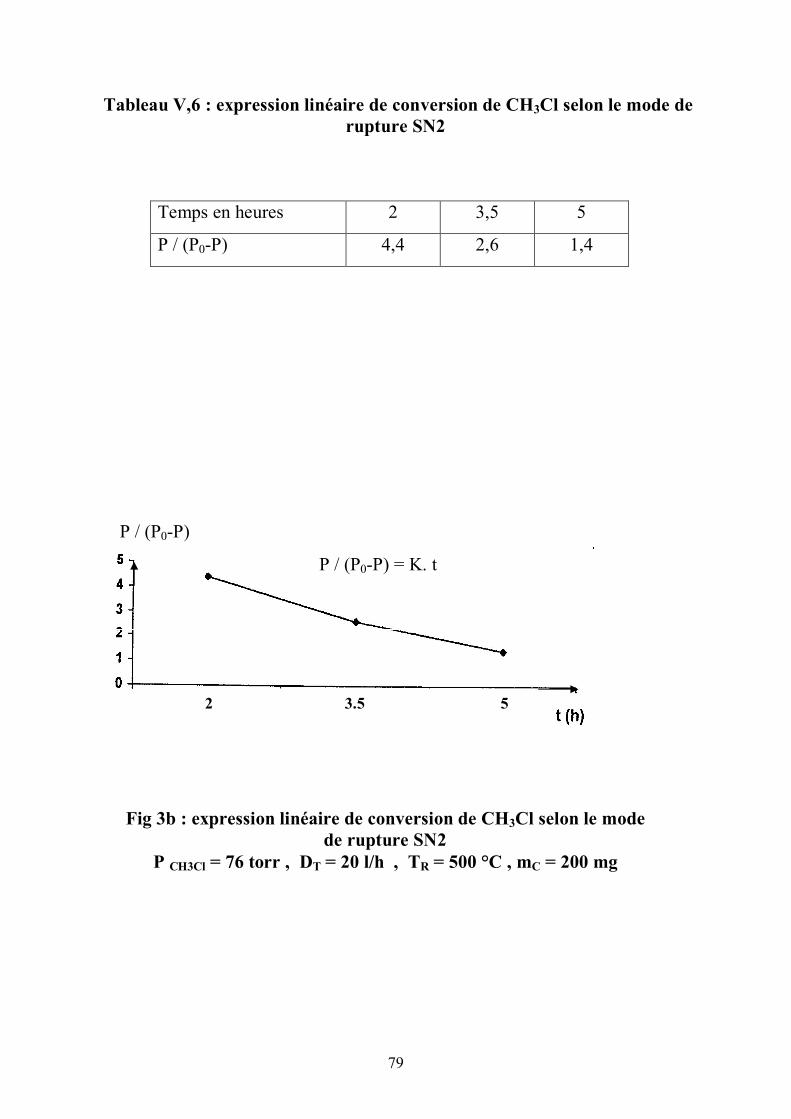
79
Tableau V,6 : expression linéaire de conversion de CH3Cl selon le mode de rupture SN2
Temps en heures 2 3,5 5
P / (P0-P) 4,4 2,6 1,4
Fig 3b : expression linéaire de conversion de CH3Cl selon le mode de rupture SN2
P CH3Cl = 76 torr , DT = 20 l/h , TR = 500 °C , mC = 200 mg
2 3.5 5
P / (P0-P) = K. t
P / (P0-P)
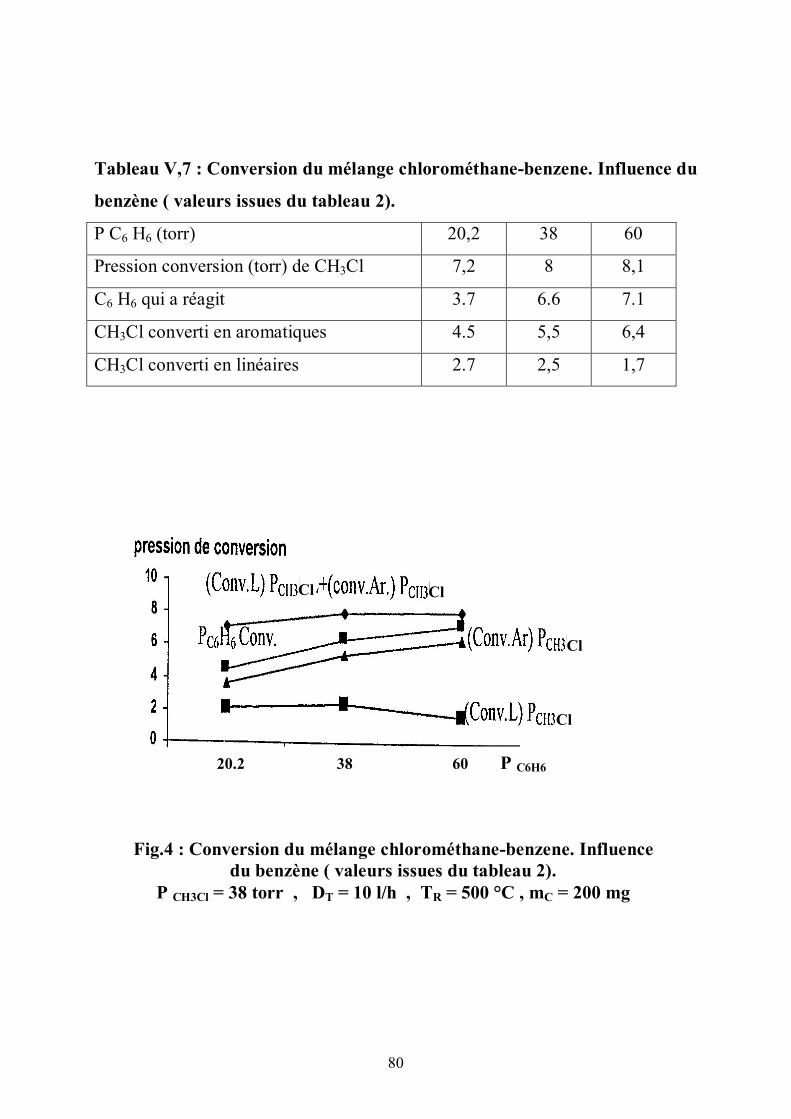
80
Tableau V,7 : Conversion du mélange chlorométhane-benzene. Influence du
benzène ( valeurs issues du tableau 2).
P C6 H6 (torr) 20,2 38 60
Pression conversion (torr) de CH3Cl 7,2 8 8,1
C6 H6 qui a réagit 3.7 6.6 7.1
CH3Cl converti en aromatiques 4.5 5,5 6,4
CH3Cl converti en linéaires 2.7 2,5 1,7
Fig.4 : Conversion du mélange chlorométhane-benzene. Influence du benzène ( valeurs issues du tableau 2).
P CH3Cl = 38 torr , DT = 10 l/h , TR = 500 °C , mC = 200 mg
20.2 38 60 P C6H6
Cl
Cl
Cl
Cl

81
V. 5. Conclusion .
Le méthanol et le chlorométhane présentent des activités bien
différentes . Le méthanol a un taux de conversion plus élevé par rapport au
chlorométhane dans les mêmes conditions opératoires. On note une similitude
dans la nature de la transformation de ces deux réactifs.
Lors du test catalytique de faible activité où ne paraissent que le propène et
l’éthylène, la question relevée dans la littérature de voir paraître l’une ou l’autre
de ces deux oléfines, tient plus du choix des conditions de travail que du type
d'intermédiaire réactionnel .
Le résultat spectroscopique infra rouge explique le mode de rupture de la
liaison carbone_chlore .

82
Bibliographie
1. 5-T.S.STEVEN'S et W.E.WATTS, in “selected molecular
rearrangements”
(Van Nostrand Reinhold Pub.co,LONDON,1973).
2. Jacobs, catalysis review, 1982, p427 – 429
3. D. Jaumin, B.L.Su, studies in surface science and catalysis, 2000, vol 130,
p1607-1612.
4. B.L.Su, D. Jaumin, J. of molecular calalysis. A. chemical, vol 197, N°1 pp
263-273, 2003.

83

84
VI. Effet de structure
VI. 1. la ZSM12
L’effet de structure de la ZSM12 présente des différences d'activité, de
sélectivité , de stabilité et de régénérabilité du solide . Le spectre infra rouge de
la ZSM12 donne une bande à 575cm-1 caractéristique de la vibration à 6
tétraèdres.
Pour une même teneur en aluminium des zéolites équivalent à une
même quantité d'ammoniac désorbée, la mesure de désorption à
température programmée de la ZSM12 présente, par rapport à la ZSM5, une
différence relevée sur la deuxième température de désorption ( 2eme pic du
chromatogramme : tableau1) relative à la rétention des NH3 sur les OH pontés ,
située à 426 °C. Celle-ci est plus faible pour la ZSM12 que celle de la ZSM5
qui se situe à 500°C. Ceci explique les différences des forces acides donc
d'activité des deux solides dans la conversion du chlorométhane. La structure de
la ZSM12 est semblable à celle de la mordénite avec cependant des canaux de
plus grande section d’environ 9 Å, alors que pour la modénite, la section est de
6 Å .
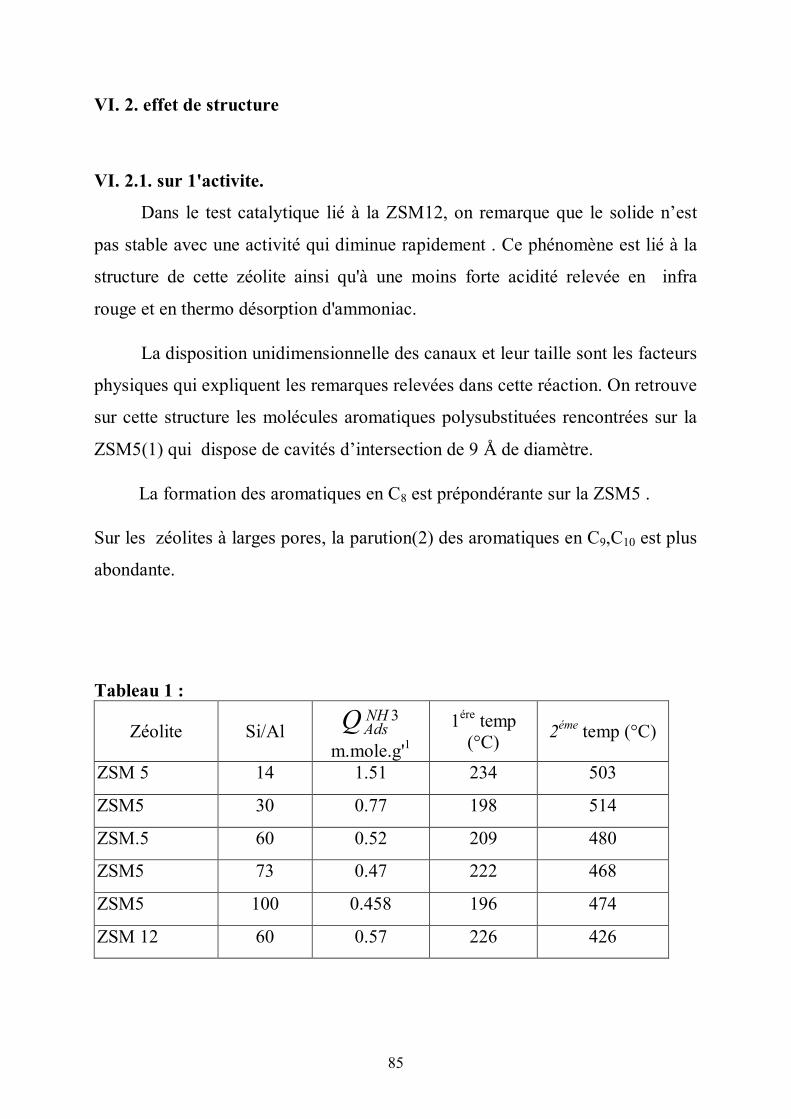
85
VI. 2. effet de structure
VI. 2.1. sur 1'activite.
Dans le test catalytique lié à la ZSM12, on remarque que le solide n’est
pas stable avec une activité qui diminue rapidement . Ce phénomène est lié à la
structure de cette zéolite ainsi qu'à une moins forte acidité relevée en infra
rouge et en thermo désorption d'ammoniac.
La disposition unidimensionnelle des canaux et leur taille sont les facteurs
physiques qui expliquent les remarques relevées dans cette réaction. On retrouve
sur cette structure les molécules aromatiques polysubstituées rencontrées sur la
ZSM5(1) qui dispose de cavités d’intersection de 9 Å de diamètre.
La formation des aromatiques en C8 est prépondérante sur la ZSM5 .
Sur les zéolites à larges pores, la parution(2) des aromatiques en C9,C10 est plus
abondante.
Tableau 1 :
Zéolite Si/Al 3NH
AdsQ m.mole.g'1
1ére temp (°C) 2éme temp (°C)
ZSM 5 14 1.51 234 503
ZSM5 30 0.77 198 514
ZSM.5 60 0.52 209 480
ZSM5 73 0.47 222 468
ZSM5 100 0.458 196 474
ZSM 12 60 0.57 226 426

86
Si la ZSM5 a des résultats reproductibles, la ZSM12 présente une baisse
d'activité après chaque retraitement (fig.1a) . Cette baisse d'activité est dûe à la
désalumination de cette zéolite (fig.1b) où on voit 1'augmentation d'aluminium
extra réseau. On a fait le même constat pour la mordénite.
Des mesures calorimétriques ont montré que les chaleurs d'adsorption
d'ammoniac sont plus importantes (150-163 kj/mole) pour la ZSM5 que dans le
cas d'autres zéolites(113-117 kj/mole). Celles-ci ayant une force catalytique
acide plus faible.
Ce résultat est en accord avec les mesures de DTP qu'on a effectuées et où
on observe des différences de température de désorption d'ammoniac à partir des
sites acides. Celle relative à la ZSM5 est voisine de 500°C alors que celle de la
ZSM12 ou la mordénite est proche de 420°C . Si la force acide de la ZSM12
est moins importante, sa base conjuguée favorise la désalumination de la
ZSM12,ce qui explique la fig.1b. Revenons à la partie II,3.4.1 pour avoir plus
d’explications . Il s’agit de l’aluminium qui ne fait plus partie du réseau où il se
présentait en Al tétraédrique et se retrouve en Al octaédrique. Ainsi, au niveau
de la figure 1b , on a un pic (ordonnée) voisin de 0 ppm (plus grand après
désalumination) et qui correspond à Al extra réseau.
L'activite des mordénite,ZSM12, chute brutalement au cours du temps de
réaction (fig.2) . Sur cette même figure est représentée, à titre comparatif, la
courbe relative à la ZSM5.
La mordénite avec sa structure unidimensionnelle comparable à la ZSM12
mais de section d'ouverture moins grande , évaluée à 6Å, est beaucoup moins
active. Ces deux solides ont une fluidité moins importante que chez la ZSM5.
Ce qui explique la désactivation rapide observée. Cette chute brutale d’activité
est dûe à 1'empoisonnement des sites et des pores obstrués .
Au niveau de la ZSM5, 1'enchevêtrement des canaux circulaires
rectilignes avec les canaux d'ouverture elliptique offre une plus grande fluidité
des molécules permettant une activité stable (fig.2).
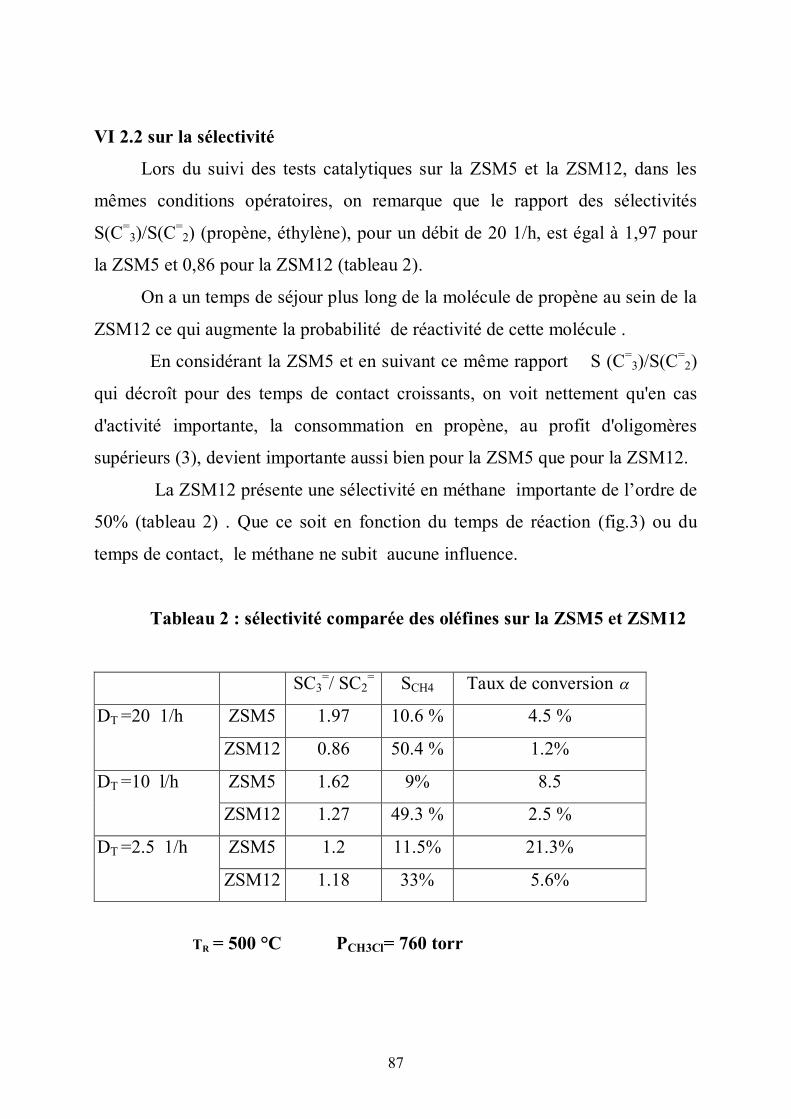
87
VI 2.2 sur la sélectivité
Lors du suivi des tests catalytiques sur la ZSM5 et la ZSM12, dans les
mêmes conditions opératoires, on remarque que le rapport des sélectivités
S(C=3)/S(C=
2) (propène, éthylène), pour un débit de 20 1/h, est égal à 1,97 pour
la ZSM5 et 0,86 pour la ZSM12 (tableau 2).
On a un temps de séjour plus long de la molécule de propène au sein de la
ZSM12 ce qui augmente la probabilité de réactivité de cette molécule .
En considérant la ZSM5 et en suivant ce même rapport S (C=3)/S(C=
2)
qui décroît pour des temps de contact croissants, on voit nettement qu'en cas
d'activité importante, la consommation en propène, au profit d'oligomères
supérieurs (3), devient importante aussi bien pour la ZSM5 que pour la ZSM12.
La ZSM12 présente une sélectivité en méthane importante de l’ordre de
50% (tableau 2) . Que ce soit en fonction du temps de réaction (fig.3) ou du
temps de contact, le méthane ne subit aucune influence.
Tableau 2 : sélectivité comparée des oléfines sur la ZSM5 et ZSM12
SC3=/ SC2
= SCH4 Taux de conversion
DT =20 1/h ZSM5 1.97 10.6 % 4.5 %
ZSM12 0.86 50.4 % 1.2%
DT =10 l/h
ZSM5 1.62 9% 8.5
ZSM12 1.27 49.3 % 2.5 %
DT =2.5 1/h
ZSM5 1.2 11.5% 21.3%
ZSM12 1.18 33% 5.6%
TR = 500 °C PCH3Cl= 760 torr

88
tableau VI, 1 : Baisse d’activée à chaque régénération
H-ZSM 12 (Si/Al = 40 )
Nombre de
régénérations 1 2 3 4 5 6
% 37 33,3 32,2 29,4 27,8 27,2
Fig 1a : Baisse d’activée à chaque régénération
H-ZSM 12 (Si/Al = 40 )
PCH3Cl = 76 torr , DT = 2.5 l/h , TR = 500 °C
1 2 3 4 5 6
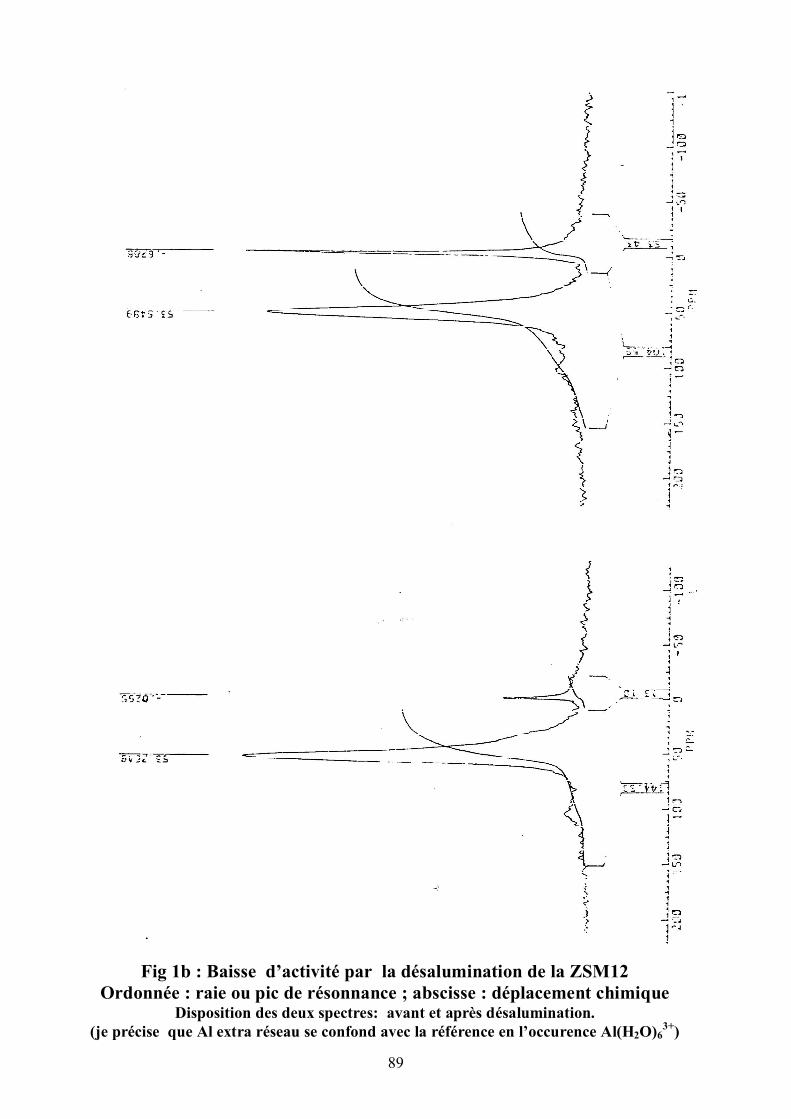
89
Fig 1b : Baisse d’activité par la désalumination de la ZSM12 Ordonnée : raie ou pic de résonnance ; abscisse : déplacement chimique
Disposition des deux spectres: avant et après désalumination. (je précise que Al extra réseau se confond avec la référence en l’occurence Al(H2O)6
3+)

90
Tableau VI,2 : Chute brutale d’activité sur la ZSM 12
t(mn) 30 120 180
V (mmole/g) 0,24 0,12 0,12
1,06 0,08 0,08
3,44 3,28 3,1
30 120 180
Fig.2 chute brutale d’activité sur la ZSM 12 TR = 500 °C , DT = 200 l/h , PCH3Cl = 76 torr
91
Tableau VI,3 : Formation du méthane au cours du temps
Temps en heures 0,5 2 3,5 5
V CH4 (mmole/g) (2.5 l/h) 1,78 1,7 1,42 1,3
V CH4 (mmole/g) (10 l/h) 1,88 1,8 1,7 1,6
V CH4 (mmole/g) (20 l/h) 2,48 2,2 1,9 1,6
0 l/h
)
Fig.3 Formation du méthane au cours du temps P CH3Cl = 38 torr , TR = 500 °C , Si/Al = 60
4

92
BIBLIOGRAPHIE
1- Eric G.DEROUANE.P.DEJAIFVE et Zelimir GABELICA. "Molecular
Shape Selectivity of ZSM5,modified ZSM5 and ZSM1J types zeolites”
331,1981,Faraday.
2- K. NGAWLA, Thèse 1997, Namur, Belgique.
3- X.R.Xia, Y.L. BI and co, Catalysis letters, 1995, vol33, p75-90.

93

94
La ZSM5 est stable avec des résultats reproductibles . On note une
regénérabilité de ses sites aprés traitement. On ne retrouve pas ces qualités sur
la mordénite ni sur la ZSM12 au niveau desquelles 1'activité régresse très
rapidement à un taux assez faible de conversion .Les résultats du test catalytique
n’étant plus les mêmes après retraitement . Ce fait s’explique par la disposition
unidimensionnelle des canaux .
Dans le cas d'une forte activité, la situation de la multitude des produits
avec des réactions d'oligomérisation et de craquage, ne nous aide pas à expliquer
le mécanisme. Par conséquent, on travaille dans les conditions expérimentales
où seules les premières molécules produites puissent rendre compte des toutes
premières étapes de la réaction. La parution de l'éthylène exclusivement ne
constitue pas un argument suffisant pour expliquer le mécanisme par la présence
du méthylène .
Le test réalisé sur le mélange chlorométhane / propane n'a pu,en dehors de
la conversion propre au CH3Cl, confirmer 1'existence du méthylène qui aurait
donné, par sa propriété d’insertion sur une liaison C-H ,du butane et de
l’isobutane.
D'autres hypothèses comme le passage par les radicaux et autres
propositions, suite à des tests appropriés, n'ont pas été concluants.
Par la spectroscopie infra rouge, on a des informations sur le mode de
rupture carbone-chlore .

95
La première étape correspond à une substitution nucléophile faisant
intervenir les sites acides de Bronsted et Lewis dans un rôle commun de cassure
de la liaison carbone chlore.
On observe une corrélation entre le déplacement de la bande OH de deux
solides distincts,suite a 1'interaction réactif/site, et leurs activités catalytiques
respectives. Le site qui est le siège de la formation du groupement méthoxyle
assure la fixation d'un second groupe méthyle . L'intermediaire réactionnel
formé est 1'ion silicium diméthyle oxonium.
La formation d'éthylène a lieu suite a une déprotonation-methylation sur
l’intermédiaire . Dans cette réaction, on note que pour la ZSM5,la sélectivité en
méthane qui est de 1'ordre de 10% reste constante quelles que soient les
conditions de 1'experience .
Les autres zéolites testées,en plus de leur désactivation rapide, présentent
une forte sélectivité en méthane d'un ordre de 50%.

BIBLIOGRAPHIE
D.Jaumain et B.L.Su (1),(2) ont réalisé une étude infra rouge concernant la
réaction de conversion du chlorométhane sur la ZSM5. L'étude de cette réaction
a été étendue par K. Ngalula (3) vers l'aspect sélectif des produits obtenus en
rapport avec les structures étudiées. L’influence des paramètres opératoires du
test catalytique et de structure a aussi été abordée (4). La spectroscopie infra
rouge a été l'essentiel du travail réalisé par les équipes de X-R. Xia et Co (5) et
B-L. SU et Co (6) où ils mettent en évidence les sites actifs . X-R. Xia et Co (5)
démontrent que le chlorométhane est réversiblement adsorbé sur les sites acides
à la température ambiante et qu'à 100°C , CH3Cl est adsorbé irréversiblement
sur le site acide .L'auteur explique qu'il se produit une dissociation avec
formation du groupement méthoxyle dont la vibration caractéristique C-H du
CH3 est de 2960et 2870 cm -1 . Des alkoxyles sont formés sur la surface
caractérisés par la vibration relative au groupement méthylène -CH2- dont les
valeurs sont 1460 et 2930 cm -1 .Ils expliquent qu'à 100°C , le méthoxyle et les
alkoxyles sont des espèces formées sur la surface .
X-R. Xia et Co (5) évoquent aussi la présence d'une faible proportion de
produits aromatiques détectés par une absorption caractéristique à 1510 cm -1 .
Entre 100 et 200°C ,les éspèces adsorbées en l'occurrence les méthoxyles et les
alkoxyles sont convertis en aromatiques. Au-delà de 300°c, les aromatiques
désorbent dans la phase gazeuse. Le même auteur propose, pour la formation de
la première liaison C-C ,une polarisation du groupement méthoxyle et un
affaiblissement de la liaison C-H au sein de ce groupement . Les alkoxyles
fournissent les aromatiques via un transfert d'hydrogène et l'association des ions
carbénium avec les oléfines .Il explique que l'atome d'hydrogène qui vient du
phénomène d'aromatisation stimule la désorption des alkoxyles qui vont donner
des alcanes.

Cette réaction a été suivie sur une série de zéolites échangées avec
différents cations comme le lithium, le sodium,, le potassium et le césium.
Les auteurs Jaumain D. et Su B-L.(7) expliquent qu'il y a une différence
significative entre les activités et les selectivités du Li- et Na-ZSM5 d'une part et
K- et Cs-ZSM5 d'autre part. Sur les échantillons en Li- et Na- , les selectivités
en C5-C6 sont plus élevées. Sur les échantillons K-ZSM5 et Cs-ZSM5 ,les
produits en C4 sont favorisés.
D'autres études comparatives ont été menées sur la ZSM5 , la ZSM12 et la
Mordénite (8) . Deux faits importants ressortent concernant la force acide
nettement supérieure sur la ZSM5 comparée à la mordénite .Le spectre infra
rouge montre un déplacement de bande à 3600 cm -1 ,relatif aux OH du site
acide, plus important que celui de la mordénite (spectre réalisé à la température
ambiante en présence de 10 torr de réactif CH3Cl ) .
A partir de cette étude bibliographique, nous essayons d'apporter notre
modeste contribution pour expliquer par spectroscopie infra rouge des faits qui
peuvent aider à comprendre le mécanisme. Les investigations portent sur la
nature de la rupture de la liaison C-Cl ,la formation de HCl et la formation de la
première liaison carbone-carbone. Le choix de la ZSM5 ,dans cette étude, se
justifie par sa stabilité dans le test catalytique et des résultats fiables.

1- D.Jaumain , Bao-Lian Su . Studies in surface science and catalysis, vol.
130, p.1607-1612, 2000.
2- Bao-Lian Su , Denis Jaumain, Kabanga Ngalula, Marguerite Briend .
Proceedings of the 12 th International zeolite conference , vol. 4, p.2681-2688,
1999.
3- K. Ngalula, Thèse, 1997.
4- C. Boukratem, roumanian review of chemistry, 1997.
5- X-R. Xia, Y-L . Bi, T-H . Wu, K-J. Zhen . Catalysis Letters, vol. 33,
numbers 1-2, 1995.
6- B-L. Su, D.Jaumain, Proceedings of the 12 th International zeolite
conference, vol. 4., p.2689-2696., 1999.
7- D. Jaumain , B-L Su, Journal of Molecular Catalysis A: Chemical,
vol.197., n°1, p. 263-273, 2003.
8- C. Boukratem, Journal of Chineese Chemical Society., 2003.






![DIPLOME - "Coup de théâtre" [mémoire]](https://static.fdocuments.fr/doc/165x107/568c577e1a28ab4916cabee5/diplome-coup-de-theatre-memoire.jpg)











