
Recherche de nouvelles cibles moléculaires dans les syndromes ...
146
UNIVERSITÉ DE LA MÉDITERRANÉE FACULTÉ DE MÉDECINE DE MARSEILLE École Doctorale des Sciences de la Vie et de la Santé Recherche de nouvelles cibles moléculaires dans les syndromes myélodysplasiques et leucémies aiguës myéloïdes T H È S E présentée et publiquement soutenue devant LA FACULTÉ DE MÉDECINE DE MARSEILLE le 29/11/2010 par M. Julien ROCQUAIN né le 7 octobre 1982 pour obtenir le grade de DOCTEUR de L’UNIVERSITÉ de la MÉDITERRANÉE SPÉCIALITÉ : ONCOLOGIE, PHARMACOLOGIE ET THÉRAPEUTIQUE Membres du Jury de la Thèse : Madame le Pr Diane BRAGUER Président Madame le Pr Dominique LEROUX Rapporteur Madame le Dr Ruth RIMOKH Rapporteur Monsieur le Dr Frédéric PINGUET Examinateur Monsieur le Dr Daniel BIRNBAUM Examinateur Madame le Dr Marie-Joëlle MOZZICONACCI Directeur
-
Upload
dangkhuong -
Category
Documents
-
view
231 -
download
1
Transcript of Recherche de nouvelles cibles moléculaires dans les syndromes ...

UNIVERSITÉ DE LA MÉDITERRANÉE
FACULTÉ DE MÉDECINE DE MARSEILLE
École Doctorale des Sciences de la Vie et de la Santé
Recherche de nouvelles cibles moléculaires dans les syndromes myélodysplasiques
et leucémies aiguës myéloïdes
T H È S E
présentée et publiquement soutenue devant
LA FACULTÉ DE MÉDECINE DE MARSEILLE
le 29/11/2010
par M. Julien ROCQUAIN
né le 7 octobre 1982
pour obtenir le grade de
DOCTEUR de L’UNIVERSITÉ de la MÉDITERRANÉE
SPÉCIALITÉ : ONCOLOGIE, PHARMACOLOGIE ET THÉRAPEUTIQUE
Membres du Jury de la Thèse :
Madame le Pr Diane BRAGUER Président
Madame le Pr Dominique LEROUX Rapporteur
Madame le Dr Ruth RIMOKH Rapporteur
Monsieur le Dr Frédéric PINGUET Examinateur
Monsieur le Dr Daniel BIRNBAUM Examinateur
Madame le Dr Marie-Joëlle MOZZICONACCI Directeur

1
UNIVERSITÉ DE LA MÉDITERRANÉE
FACULTÉ DE MÉDECINE DE MARSEILLE
École Doctorale des Sciences de la Vie et de la Santé
Recherche de nouvelles cibles moléculaires dans les syndromes myélodysplasiques
et leucémies aiguës myéloïdes
T H È S E
présentée et publiquement soutenue devant
LA FACULTÉ DE MÉDECINE DE MARSEILLE
le 29/11/2010
par M. Julien ROCQUAIN
né le 7 octobre 1982
pour obtenir le grade de
DOCTEUR de L’UNIVERSITÉ de la MÉDITERRANÉE
SPÉCIALITÉ : ONCOLOGIE, PHARMACOLOGIE ET THÉRAPEUTIQUE
Membres du Jury de la Thèse :
Madame le Pr Diane BRAGUER Président
Madame le Pr Dominique LEROUX Rapporteur
Madame le Dr Ruth RIMOKH Rapporteur
Monsieur le Dr Frédéric PINGUET Examinateur
Monsieur le Dr Daniel BIRNBAUM Examinateur
Madame le Dr Marie-Joëlle MOZZICONACCI Directeur

2
RÉSUMÉ
Au sein des hémopathies myéloïdes malignes, les syndromes myélodysplasiques
(SMD) et les leucémies aiguës myéloïdes (LAM) représentent des pathologies complexes et
hétérogènes résultant d’anomalies clonales des cellules souches médullaires. Elles sont
caractérisées par une hématopoïèse inefficace provoquant des cytopénies sanguines graves.
Les connaissances sur les anomalies moléculaires des SMD et des LAM, notamment
à caryotype normal, sont globalement pauvres et leur physiopathologie encore mal connue.
Une meilleure définition moléculaire est nécessaire pour une évaluation pronostique plus
précise de ces hémopathies et pour optimiser secondairement les stratégies thérapeutiques.
Cette thèse présente un panorama des classifications cytogénétiques et moléculaires
actuelles des SMD et LAM ainsi que l’étude de certaines altérations moléculaires
rencontrées dans ces maladies.
Grâce à l’apport des techniques d’analyse génomique à grande échelle, notamment la
CGH-array, notre laboratoire a identifié de nouvelles altérations génétiques, parmi lesquelles
les mutations du gène ASXL1, ainsi que des altérations des gènes codant les protéines de la
Cohésine et des régulateurs de la protéine CBL. Nous avons analysé une combinaison de
mutations de gène et émis l’hypothèse d’un modèle de leucémogenèse à 4 classes de
mutations, afin d’apporter des pistes dans la compréhension de la physiopathologie des SMD
et LAM.
Mots clés : syndrome myélodysplasique ; leucémie aiguë myéloïde, cibles moléculaires,
génomique, mutation de gène.

3
ABSTRACT
IDENTIFICATION OF NEW MOLECULAR TARGETS
IN MYELODYSPLASTIC SYNDROMES AND ACUTE MYELOID
LEUKEMIAS
Among myeloid malignancies, myelodysplastic syndromes (MDSs) represent a group
of complex diseases characterized by clonal abnormalities of bone marrow hematopoietic
precursor cells. They are defined by an ineffective hematopoiesis leading to peripheral
cytopenias. About 40% of MDSs secondarily evolve to acute myeloid leukemia (AML).
This risk of transformation is evaluated by several international prognostic scoring
systems like IPSS and WPSS. The WHO classification recognizes several classes of MDSs
essentially based on morphology and cytogenetics features, some with a high progression
risk, like refractory anemia with excess of blasts type 2, others with a low risk, like
refractory anemia with ringed sideroblasts. However, the classification of MDSs is still
unsatisfactory and relevant prognostic markers allowing earlier treatments for patients with a
high risk of transformation are still lacking. The physiopathology of SMDs and AMLs with
normal karyotype remains unclear. Currently, the only potentially curative treatment is
allogenic stem cell transplant, which is feasible for a restricted number of patients and can
display side effects and failures.
A better knowledge of the molecular biology of MDSs and AMLs is necessary for a
better understanding of these diseases and may provide new early prognosis indicators and
better strategies of treatments.
Keys words: Myelodysplastic syndrome, acute myeloid leukemia, molecular
targets, genomic, gene mutation.

4
Intitulé et adresse du laboratoire où la thèse a été préparée
Laboratoire d’Oncologie Moléculaire
Centre de Recherche en Cancérologie de Marseille
Unité Mixte de Recherche Inserm 891
Institut Paoli-Calmettes
232 boulevard Sainte Marguerite
13009 MARSEILLE
Directeur du Centre de Recherche : Dr Françoise BIRG
Directeur du Laboratoire : Dr Daniel BIRNBAUM
Directeur de thèse : Dr Marie-Joëlle MOZZICONACCI

5
REMERCIEMENTS
Madame le Pr Diane Braguer
Vous me faîtes l’honneur une deuxième fois cette année, de présider un jury de thèse et
je vous en remercie. Comme je vous l’avais déjà écrit dans la thèse de Pharmacie, c’est mon
premier semestre d’interne dans votre service, qui avait confirmé mon attirance pour l’étude
de la cancérologie. A travers ces quelques mots, je tenais, une nouvelle fois, à vous
remercier pour vos précieux conseils et encouragements lors de mon parcours «hospitalo-
universitaire» à Marseille.
Mesdames le Pr Dominique Leroux et le Dr Ruth Rimokh,
Je vous remercie d’avoir accepté de juger ce travail. J’espère que sa qualité, tant au
niveau hématologique que scientifique, sera conforme à vos attentes.
Monsieur le docteur Frédéric Pinguet,
C’est avec beaucoup de plaisir que j’ai appris votre participation à ce jury. C’est
avant tout un honneur pour moi, que vous ayez accepté de juger ce travail, ceci malgrè un
emploi du temps chargé. J’espère que ce travail sera au niveau de celui que l’on peut
attendre d’un pharmacien hospitalier-chercheur.
Monsieur le docteur Daniel Birnbaum,
Merci de m’avoir permis de concilier à la fois le DES de pharmacie hospitalière et
mon cursus M2/thèse de sciences dans votre laboratoire. Vous avez toujours été présent
pour me soutenir dans cette démarche hospitalo-universitaire et je tiens sincèrement à vous
remercier. J’ai découvert le monde de la recherche grâce à vous et j’ai beaucoup appris à
vos côtés. Votre passion pour la recherche est d’ailleurs « contagieuse » et j’espère pouvoir
très prochainement continuer à concilier activités hospitalières et travaux de recherche.

6
Madame le docteur Marie-Joelle Mozziconacci,
Marie-Joelle, je te remercie de m’avoir soutenu et dirigé tout au long de ces travaux
de thèse. Merci pour tes précieux conseils : j’ai énormément appris et ceci me servira tout
au long de ma vie professionelle. Je te remercie tout spécialement pour les « Maintenant,
remets-toi au travail », ces derniers mois, alors que j’étais débordé du côté hospitalier. La
rédaction de cette thèse aura été « laborieuse » mais riche d’enseignements. Ces « moments
de stress » ont sûrement eu un impact positif. En tout cas, j’espère que le résultat ici présent,
est convaincant.
Madame le Dr Véronique Gelsi-Boyer,
Véro, c’était un réel plaisir d’avoir travaillé à tes côtés. Je tenais tout particulièrement
à te remercier pour tes encouragements et tes conseils. Ils ont été essentiels dans
l’accomplissement de cette thèse.
Monsieur le Dr Max Chaffanet,
Max, même si tu as suivi de plus loin l’avancée de mes travaux de thèse, je souhaitais
te remercier pour les discussions que nous avons eues ensemble. Elles m’ont permis
d’avancer dans mes projets et je tenais à te remercier.
Ce temps partagé entre l’hôpital et le laboratoire me donne toujours l’impression d’un
temps trop vite passé !
J’espère que les liens professionnels tissés pendant ces 5 années entre l’hôpital et le
laboratoire perdureront et que je pourrai continuer à participer à certains projets de
recherche qui me sont chers.

7
A tous ceux avec qui j’ai eu le plaisir de travailler durant ces 5 années entre l’hôpital
et le laboratoire…
Un grand merci, avant tout, à toute l’équipe d’Oncologie Moléculaire !
Je ne peux citer chaque personne individuellement de crainte d’en oublier, la liste des gens
m’ayant permis de réaliser cette thèse est longue... Merci surtout pour cette bonne ambiance
au travail et en dehors !
Merci pour tous les soutiens et encouragements qui m’ont permis de surmonter les moments
difficiles…
A toutes ces précieuses rencontres au sein de l’Institut Paoli-Calmettes, au centre
régional de pharmacovigilance, à la pharmacie de l’hôpital Sainte Marguerite et de la
Timone… Je tiens à remercier toutes les personnes qui m’ont permis de concilier mes
activités hospitalières et de recherche.

8
A mes amis marseillais, néo-marseillais ou marseillais de passage,
A mes amis du « groupe »,
Aux nombreuses personnes que je n’ai pas citées mais qui me sont chers,
À mes grands parents, à toute ma famille,
Merci tout simplement d’être là…
À ma mère, mon père et mon petit frère,
Malgré la distance, vous avez toujours été présents pour me soutenir, dans les bons et
surtout les mauvais moments…
Je tenais tout particulièrement à vous dédier cette thèse, qui cloture ces 10 années
d’études universitaires.

9
SOMMAIRE
AVANT PROPOS 14�
I.� INTRODUCTION : LES HÉMOPATHIES MYÉLOÏDES 16
A.� Définition ..................................................................................................... 16
B.� Les syndromes myélodysplasiques (SMD) .................................................. 19�
Généralités ............................................................................................................. 19�
a)� Epidémiologie ......................................................................................... 19�b)� Facteurs de risque .................................................................................... 20�c)� Physiopathologie ..................................................................................... 21�d)� Bilan diagnostique ................................................................................... 22�e)� Evolution de la maladie et traitements .................................................... 22�
Classification des SMD .......................................................................................... 24
a)� Classification morphologique des SMD .................................................. 24�b)� Classification cytogénétique des SMD .................................................... 26�c)� Classification moléculaire des SMD ....................................................... 34�
SMD avec délétion 5q isolée ................................................................................. 39
a)� Quels sont les gènes impliqués dans la délétion ? ................................... 39�b)� Implication des micro-ARN ? ................................................................. 40�c)� Mécanisme d’action du lénalidomide ...................................................... 41
C.� Les leucémies aiguës myéloïdes (LAM) ...................................................... 42�
Généralités ............................................................................................................. 42
a)� Epidémiologie ......................................................................................... 44�b)� Facteurs de risque .................................................................................... 44�c)� Physiopathologie ..................................................................................... 45�d)� Bilan diagnostique ................................................................................... 45�e)� Traitements .............................................................................................. 46�
Classification des LAM ......................................................................................... 47
a)� Classification cytogénétique des LAM ................................................... 47�b)� Classification moléculaire des LAM ....................................................... 50
Synthèse des anomalies moléculaires des SMD et LAM-CN ............................... 57

10
II.� PRESENTATION DES TRAVAUX ET RESULTATS 61�
Fondements scientifiques du sujet de thèse ........................................................... 61
ARTICLE N°1 ....................................................................................................... 63�
Commentaires et discussion de l’article n°1 .......................................................... 69
ARTICLE N°2 ....................................................................................................... 71
Commentaires et discussion de l’article n°2 .......................................................... 83
ARTICLE N°3 ....................................................................................................... 86�
Commentaires et discussion de l’article n°3 .......................................................... 93
ARTICLE N°4 ....................................................................................................... 96�
Commentaires et discussion de l’article n°4 ........................................................ 102
Discussion générale .............................................................................................. 104
III.�CONCLUSION ET PERSPECTIVES 111
BIBLIOGRAPHIE 113
IV.�ANNEXES 132�
ARTICLE N°5 ..................................................................................................... 132Commentaires sur l’article n°5 ............................................................................ 144�

11
LISTE DES FIGURES ET TABLEAUX
Figure n°1 : Schéma de l’hématopoïèse 16�
Figure n°2 : Screening moléculaire des LAM 52�
Figure n°3 : Modèle de leucémogenése à 2 étapes selon Gilliland 58�
Figure n°4 : Modèle de leucémogenèse proposé par Rocquain et al, 2010 84�
Figure n°5 : Complexe protéique de la cohésine 94�
-----------------------------------------------------------------
Tableau n°1 : Classification OMS 2008 des hémopathies myéloïdes 17�
Tableau n°2 : Les SMD et LAM secondaires à l’administration de chimiothérapies
anticancéreuses 20�
Tableau n°3 : Classification FAB des SMD 25�
Tableau n°4 : Classification OMS des SMD 27�
Tableau n°5 : Principales anomalies chromosomiques des SMD de novo 28�
Tableau n°6 : Score IPSS 30�
Tableau n°7 : Evolution des SMD selon le score IPSS 30�
Tableau n°8 : Score WPSS 31�
Tableau n°9 : Valeur pronostique des anomalies cytogénétiques des SMD 32�
Tableau n°10 : Classification FAB des LAM 43�
Tableau n°11: Principales anomalies chromosomiques des LAM 47�
Tableau n°12 : Valeur pronostique des anomalies cytogénétiques des LAM 48�
Tableau n°13 : Classification OMS des LAM 49�
Tableau n°14 : Tableau récapitulatif selon le modèle de Gilliland des principales
altérations moléculaires des SMD et des LAM-CN 59�
Tableau n°15 : Tableau récapitulatif des principales altérations moléculaires
retrouvées dans les SMD 59�
Tableau n°16 : Tableau récapitulatif des principales altérations moléculaires
retrouvées dans les LAM-CN 60�
Tableau n°17 : Recapitulatif des mutations de gènes dans les HM 107�

12
LISTE DES ABREVIATIONS
ADN Acide DésoxyriboNucléique
AMM Autorisation de Mise sur le Marché
AR Anémie Réfractaire
AREB Anémie Réfractaire avec Excès de Blastes
ARN Acide Ribonucléique
ARS Anémie Réfractaire avec Sidéroblastes en couronne
ARS-T ARS avec Thrombocytose
ASXL1 Additional Sex Combs Like 1
c-GH comparative-Genomic Hybridization
CBF Core Binding Factor
CBL Casitas B-cell Lymphoma
CEBP� CCAAT/enhancer binding protein �
CRDM Cytopénie Réfractaire avec Dysplasie Multilignée
CRDU Cytopénie Réfractaire avec Dysplasie Unilignée
CSH Cellule Souche Hématopoïétique
del(5q) Délétion du bras long (q) du chromosome 5
GFM Groupe Français des Myélodysplasies
EPO Erythropoïétine
FAB French American British Group
FISH Fluorescenct In Situ Hybridization
FGFR1 Fibroblast Growth Factor Receptor 1
FLT3 FMS-like tyrosine kinase 3
G-CSF Granulocyte-Colony Stimulating Factor
HM Hémopathie Myéloïde
HDAC Histone Désacétylase
IDH Isocitrate Déshydrogénase
IPSS International Pronostic Scoring System
ITK Inhibiteur de Tyrosine Kinase
JAK2 Janus Kinase 2
LAM Leucémie Aiguë Myéloïde
LAM-CN LAM à Caryotype Normal

13
LMC Leucémie Myéloïde Chronique
LMMC Leucémie Myélomonocytaire Chronique
MLL Mixed Lineage Leukemia
NR Neutropénie Réfractaire
NPM1 Nucléophosmine
OMS Organisation Mondiale de la Santé
PDGFR Platelet-Derived Growth Factor Receptor
RT-PCR Reverse Transcriptase-Polymerase Chain Reaction
RTK Récepteur à activité Tyrosine Kinase
RUNX1 Runt-related transcription factor 1
SFH Société Française d’Hématologie
SMD Syndrome Myélodysplasique
SMD-I Syndrome Myélodysplasique Inclassable
SMP Syndrome Myéloprolifératif
SNP Single-Nucleotide Polymorphism
TET2 Ten Eleven Translocation-2
TIRAP Toll–Interleukin-1 Receptor domain–containing Adaptor Protein
TRAF6 Tumor necrosis factor Receptor–Associated Factor-6
TR Thrombopénie Réfractaire
TK Tyrosine Kinase
WT1 Wilms’s Tumor suppressor 1
WHO World Health Organisation
WPSS WHO classification-based Prognostic Scoring System

14
AVANT PROPOS
Les syndromes myélodysplasiques (SMD) et les leucémies aiguës myéloïdes (LAM)
sont les hémopathies myéloïdes les plus fréquentes. Ce sont deux groupes très hétérogènes
de pathologies de l’hématopoïèse définies par des anomalies clonales des cellules souches
médullaires. Près de 40% des SMD évoluent secondairement en leucémie aiguë myéloïde.
Les SMD, caractérisés par une hématopoïèse inefficace à l’origine de cytopénies
graves, sont particulièrement fréquents chez les personnes âgées, dont seule une minorité
peut recevoir le seul traitement actuellement potentiellement curatif, l’allogreffe de cellules
souches hématopoïétiques. Les cures de chimiothérapie sont rarement efficaces ou ne
peuvent être réalisées dans leur totalité et la plupart de ces patients décèdent des
conséquences de leurs cytopénies ou d’une transformation en LAM.
Alors que des cibles moléculaires ont été découvertes dans la majorité des cancers,
permettant l’avènement de thérapies ciblées, il n’existe pas actuellement de traitement ciblé
dans les SMD, faute de découverte d’anomalies moléculaires spécifiques. La prise en charge
thérapeutique demeure essentiellement symptomatique, visant à corriger les cytopénies.
Néanmoins, le traitement des SMD est actuellement en pleine évolution. Une nouvelle
classe thérapeutique, les agents hypométhylants représentés par l’azacytidine et la
décitabine, ont démontré un bénéfice de survie chez certains patients et permettent de
retarder la transformation en leucémie. Cependant, les marqueurs moléculaires pronostiques
et prédictifs de la réponse sont manquants pour mieux sélectionner les patients susceptibles
de répondre à ces nouveaux traitements.
De nouvelles altérations moléculaires (mutations des gènes ASXL1, TET2) ont
récemment été identifiées dans les SMD. D’autres études doivent être menées pour
comprendre leur impact dans la physiopathologie et dans l’évolution de la maladie. De plus,
de multiples anomalies génétiques sont à déceler dans les SMD sans anomalie actuellement
identifiable.
Les LAM représentent le type de leucémie aiguë le plus fréquent chez l’adulte. Elles
sont caractérisées par un blocage de la différenciation des progéniteurs myéloïdes et par leur
prolifération incontrôlée, provoquant l’accumulation de blastes dans la moelle osseuse et le
sang. Dans près de 60% des cas, des anomalies chromosomiques sont identifiables,

15
permettant de classifier les LAM en trois groupes pronostiques : favorable, intermédiaire et
défavorable.
Les LAM de bon pronostic sont traitées par des cures de chimiothérapie
conventionnelle alors que les patients avec LAM à haut risque sont orientés lorsque cela est
possible, vers l’allogreffe de cellules souches hématopoïétiques. Cependant, plus de 40% des
patients n’ont pas d’anomalies cytogénétiques détectables et sont classés dans le groupe de
pronostic intermédiaire. La stratégie thérapeutique optimale pour ces LAM n’est pas
clairement établie.
Récemment, plusieurs anomalies moléculaires, en particulier les mutations des gènes
FLT3 et NPM1, ont permis de créer des sous-groupes pronostiques et d’adapter plus
précocement et plus efficacement la prise en charge thérapeutique de ces patients. La valeur
pronostique de plusieurs autres anomalies moléculaires doit encore être confirmée et de
nouvelles altérations sont à découvrir dans les LAM dépourvues d’anomalies génétiques au
diagnostic.
L’identification des gènes mutés dans les hémopathies myéloïdes est devenue
incontournable pour une meilleure compréhension de la pathogenèse moléculaire des SMD
et des LAM. La découverte d’altérations moléculaires est également essentielle pour trouver
des marqueurs prédictifs de la réponse aux traitements et entrevoir de potentielles nouvelles
cibles thérapeutiques.
Cette thèse, tout en présentant le travail réalisé dans le laboratoire, se veut être un
panorama des classifications actuelles des SMD et LAM et de leurs anomalies moléculaires.
Les perspectives, issues des dernières découvertes de génomique seront discutées, ainsi que
les hypothèses sur la pathogenèse de ces maladies complexes et l’évolution de leur prise en
charge thérapeutique.
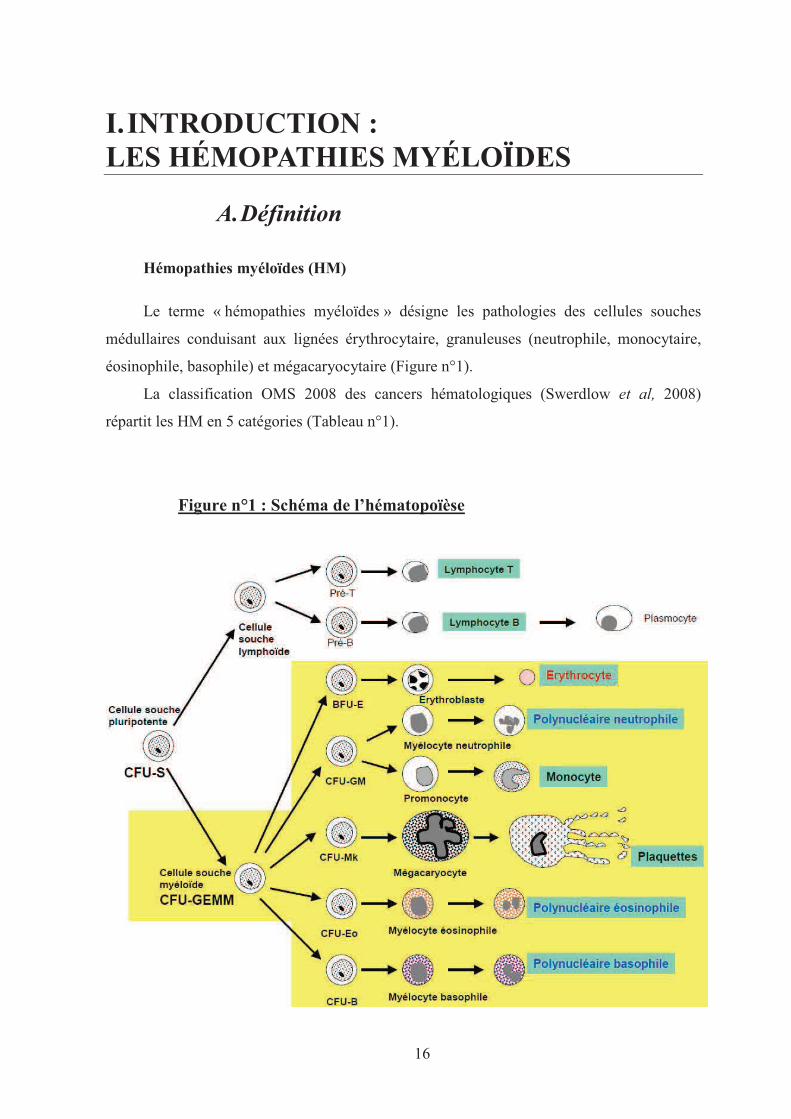
16
I.INTRODUCTION : LES HÉMOPATHIES MYÉLOÏDES
A.Définition
Hémopathies myéloïdes (HM)
Le terme « hémopathies myéloïdes » désigne les pathologies des cellules souches
médullaires conduisant aux lignées érythrocytaire, granuleuses (neutrophile, monocytaire,
éosinophile, basophile) et mégacaryocytaire (Figure n°1).
La classification OMS 2008 des cancers hématologiques (Swerdlow et al, 2008)
répartit les HM en 5 catégories (Tableau n°1).
Figure n°1 : Schéma de l’hématopoïèse

17
Tableau n°1 : Classification OMS 2008 des hémopathies myéloïdes
(D’après Swerdlow et al, 2008)
LAM et apparentés*
SMD
SMP**
Formes mixtes SMP/SMD***
Hémopathies myéloïdes et lymphoïdes avec hyperéosinophilie****
* Les LAM et SMD secondaires aux chimiothérapies appartiennent à cette catégorie.
** Les syndromes myéloprolifératifs (SMP) comprennent : - la leucémie myéloïde chronique (LMC) - la polyglobulie primitive (maladie de Vaquez) - la thrombocytémie essentielle - la myélofibrose primitive - la leucémie neutrophile chronique - la mastocytose systémique - la leucémie chronique à éosinophiles - les SMP inclassables.
*** Les formes mixtes SMP/SMD incluent : - la leucémie myélomonocytaire chronique (LMMC) - la leucémie myélomonocytaire juvénile - la LMC atypique (BCR-ABL négative) - les formes mixtes SMP/SMD inclassables dont les ARS-T (anémies réfractaires avec
sidéroblastes en couronne et thrombocytose).
**** Cette catégorie comprend des hémopathies avec réarrangement impliquant le platelet-derived growth factor receptor (PDGFR� ou �) ou le fibroblast growth factor receptor 1 (FGFR1).

18
Clonalité des hémopathies myéloïdes
Il est maintenant établi que l’origine des HM est une combinaison d’anomalies de la
prolifération et de la différenciation touchant un clone de cellules souches hématopoïétiques
(CSH) (Nowell, 1976 ; Boultwood et al, 2001). Toute la problématique de cette clonalité est
de savoir d’une part quels sont les événements nécessaires et suffisants à l’apparition et à
l’extension du clone malin et d’autre part, à quel niveau de l’hématopoïèse ils apparaissent.
La diversité phénotypique des HM est liée à différents niveaux de dérégulation du signal de
transduction dans la CSH. Ces dérégulations sont causées par plusieurs mécanismes
pathologiques comportant des altérations épigénétiques et génétiques, associées à des
dysfonctionnements immunitaires et des anomalies du stroma médullaire.
SMD/SMP
Les SMD sont caractérisés par une hématopoïèse inefficace provoquant des
cytopénies, alors qu’à l’inverse, les syndromes myéloprolifératifs (SMP) présentent une
surproduction en cellules sanguines matures. A la frontière de ces deux hémopathies se
trouvent les formes mixtes SMD/SMP, comme la leucémie myélomonocytaire chronique
(LMMC), qui présentent certaines des caractéristiques de chacune de ces deux maladies.
LAM
Les LAM peuvent être primaires ou résulter de la transformation aiguë d’une des
catégories d’hémopathies précitées (LAM secondaires). Elles sont définies par un blocage de
maturation des cellules myéloïdes à un stade précoce de différenciation, à l’origine de
l’accumulation d’éléments immatures dans la moelle osseuse et le sang.
Autre HM
Les hémopathies myéloïdes avec hyperéosinophilie constituent une nouvelle catégorie
d’HM dans la classification OMS 2008. Elles impliquent généralement un gène codant pour
une tyrosine kinase, le platelet-derived growth factor receptor (PDGFR) � ou � ou le
fibroblast growth factor receptor 1 (FGFR1) (Vardiman et al, 2009).

19
B. Les syndromes myélodysplasiques (SMD)
Généralités
Les SMD appartiennent à un groupe d’affections hétérogènes par leur expression
clinique et leur évolution. Ils sont caractérisés par une hématopoïèse inefficace à l’origine
de cytopénies sanguines contrastant avec une moelle osseuse généralement riche.
L’histoire naturelle de ces syndromes débute par une phase chronique évoluant plus
ou moins rapidement en LAM. Alors que certains patients restent stables cliniquement
pendant plusieurs années, la majorité d’entre eux (80%) présente des cytopénies qui
s’aggravent et qui sont à l’origine du décès du patient. Parallèlement, le nombre de blastes
augmentent plus ou moins rapidement selon les patients (Nimer, 2008).
a) Epidémiologie
L’incidence annuelle globale des SMD est de 4 à 5 cas pour 100.000 habitants, soit
en France, environ 2.500 nouveaux cas par an (GFM 2010). Sans cause actuellement
identifiée, les SMD sont près de deux fois plus fréquents chez les hommes que chez les
femmes (Sex ratio = 1,8) (Rollinson et al, 2008).
Les SMD sont des maladies du sujet âgé et sont rares avant l’âge de 50 ans (<1 cas
pour 100.000) : la médiane d’âge au moment du diagnostic se situe autour de 70 ans
(Catenacci et al, 2005). A partir de cet âge, l’incidence annuelle dépasse les 20 cas pour
100.000 habitants.
L’incidence des SMD est croissante du fait de l’augmentation de l’espérance de vie
et du vieillissement de la population (Guralnik et al, 2004 ; Corey et al, 2007).

20
b) Facteurs de risque
Un SMD est dit primaire quand son étiologie est inconnue, secondaire lorsque qu’une
cause a été identifiée.
Dans la grande majorité des cas de SMD, aucun facteur causal n’est retrouvé (Aul et
al, 1995 ; Strom et al, 2008), mais il est prouvé que l’exposition à certains toxiques
favorisent l’apparition d’un SMD :
La principale cause identifiée est l’exposition à une chimiothérapie anticancéreuse
(près de 20 % des cas), en particulier aux agents alkylants et inhibiteurs de topoisomérases II
(Leone et al, 2007), mais les radiations ionisantes ou certaines substances toxiques (tabac,
solvants organiques comme le benzène) augmentent également le risque de développer une
myélodysplasie (Strom et al, 2005).
En raison de leur mauvais pronostic, les SMD induits par les chimiothérapies (SMD-t)
occupent une entité à part entière avec les LAM-t dans la classification OMS 2008. La survie
médiane des SMD-t est d’environ 5 à 7 mois versus 20 mois pour les SMD de novo (Aul et
al, 1995). Les SMD-t et LAM-t présentent un délai d’apparition différent selon la classe de
chimiothérapies anticancéreuses (tableau n°2).
Tableau n°2 : Les SMD et LAM secondaires à l’administration de chimiothérapies anticancéreuses
(Larson, 2009)
Agents thérapeutiques Délai d’apparition
Alkylants 5-7 ans
Inhibiteurs de topoisomérase II 2-3 ans
Certains rares syndromes congénitaux sont également associés à un risque plus élevé
de SMD, comme les syndromes de Down, Bloom et Blackfan-Diamond ainsi que l’anémie
de Fanconi (Owen et al, 2008).

21
c) Physiopathologie
Les syndromes myélodysplasiques résultent d’anomalies moléculaires touchant un
clone de CSH provoquant une hématopoïèse inefficace (Boultwood et al, 2001).
La moelle osseuse est généralement riche, mais avec des anomalies qualitatives et
morphologiques des cellules (Tefferi et al, 2009). Elles sont caractérisées par des troubles
de la différenciation cellulaire et une apoptose importante d’une ou plusieurs lignées
hématopoïétiques à l’origine de cytopénies sanguines chez le patient (Parker et al, 2000).
Les évènements moléculaires initiaux et la voie de signalisation impliquée sont
encore inconnus. La complexité de ces hémopathies est en partie due à leur hétérogénéité :
les SMD sont un groupe d’entités distinctes caractérisées par des degrés variables
d’altération de l’hématopoïèse. Divers mécanismes pathologiques participent
simultanément ou successivement à l’établissement du phénotype de la maladie, tels que
des pertes ou gains de matériel génétique, des mutations de gènes, des modifications
épigénétiques, une réponse immunitaire déficiente ou une altération du stroma médullaire
(Heaney et al, 1999).
Contrairement aux LAM, les SMD sont rarement affectés par des translocations
chromosomiques équilibrées mais présentent surtout des pertes de matériel génétique à
l’origine de perte d’hétérozygotie par délétion hémizygote (perte d’un segment d’ADN sur
un des 2 chromosomes d’une même paire) ou par disomie uniparentale (lorsque le
segment d’ADN restant, provenant d’un même parent, est dupliqué). Les gains de matériel
génétique sont moins fréquents. Ces anomalies génétiques ne sont pas spécifiques aux
SMD et sont retrouvés dans d’autres HM (Bacher et al, 2009).
L’analyse des régions fréquemment affectées par des pertes d’hétérozygotie a permis la
découverte de plusieurs mutations de gènes potentiellement importants dans la
physiopathologies des SMD (Gondek et al, 2008 ; Dunbar et al, 2008).

22
d) Bilan diagnostique
L’interrogatoire et l’examen clinique sont essentiels afin de dater l’apparition des
cytopénies et de trouver l’existence d’un agent étiologique éventuel.
Sur le plan biologique, les examens suivants sont considérés comme indispensables :
- Hémogramme avec réticulocytes et examen des frottis sanguins.
- Myélogramme avec évaluation du pourcentage de cellules jeunes, de la
dysmyélopoïèse et coloration de Perls (visualisation des sidéroblastes).
- Caryotype médullaire.
(Ces examens seront répétés en cas d’évolutivité)
- Dosage sérique d’érythropoïétine (EPO) dans les formes à risque faible ou
intermédiaire.
- Ferritinémie et examens à visée de diagnostic différentiel ou pour éliminer une cause
associée d’anémie (carence en fer, carence en B12 ou en folates, insuffisance rénale,
hépatite, syndrome inflammatoire, hypothyroïdie, pic monoclonal).
Par ailleurs, lorsqu’un diagnostic de SMD de mauvais pronostic a été posé, le typage
HLA est recommandé chez tous les patients de moins de 65 ans dans l’hypothèse d’une
future allogreffe de CSH.
e) Evolution de la maladie et traitements
Les 2 principales causes de mortalité des patients atteins de SMD sont les infections et
les complications hémorragiques. Les infections sont à l’origine de près de 65% des décès
des patients (Kouides et al, 1997). Elles sont liées à la neutropénie, aux anomalies
qualitatives des neutrophiles ou à la transformation en leucémie aiguë. Les complications
hémorragiques sont responsables d’environ 20% des décès (Bowen et al, 2003). L’évolution
clinique des SMD est variable mais l’espérance de vie des patients atteints de SMD est
relativement faible : la survie à 3 ans est inférieure à 50 % (Rollinson et al, 2008).
Le traitement d’un SMD a pour but d’améliorer les cytopénies, de retarder la
transformation leucémique, de prolonger la survie et d’optimiser la qualité de vie du patient.
Le traitement symptomatique (Best Supportive Care) constitue souvent la seule prise en

23
charge, surtout pour les patients à faible risque ainsi que pour les patients à haut risque qui
sont inaptes à supporter un traitement intensif. Ces objectifs consistent à améliorer les
cytopénies et à maintenir la qualité de vie, but que permettent d’atteindre les transfusions de
culots globulaires et de plaquettes ainsi que les agents stimulant l’érythropoïèse (EPO) et les
facteurs de croissance granulocytaire (G-CSF) (Tefferi, 2009).
A l’exception du lénalidomide, qui est efficace dans les SMD avec délétion du bras
long du chromosome 5 [del(5q)] et des agents hypométhylants comme l’azacytidine, qui sont
utilisés dans les SMD avancés et permettent chez certains patients un gain de survie globale
de plusieurs années, il y a actuellement très peu de traitements efficaces dans les SMD
(Tefferi, 2010). De plus, lors d’une évolution en LAM, les leucémies secondaires répondent
rarement aux cures de chimiothérapies et sont souvent de mauvais pronostic.
Le taux de réponse aux agents déméthylants est du même ordre que pour la cytarabine
à faible dose et est relativement faible (autour de 15%) (Miller et al, 1992 ; Kantarjian et al,
2006). La greffe de CSH demeure la seule option thérapeutique curative, cependant celle-ci
ne peut s’effectuer que dans une minorité des cas (15 à 20 % des patients) et, en raison de sa
toxicité, elle n’est pas un choix raisonnable pour la plupart des patients présentant une
maladie à faible risque (Boehrer et al, 2009). Elle est associée à un taux relativement élevé
de décès (environ 39% à 1 an), de maladie du greffon contre l’hôte (environ 15% à 1 an)
pour une survie sans rechute estimée à environ 30% à 5 ans (Warlick et al, 2009).
L’allogreffe n’est donc proposée qu’aux patients de très mauvais pronostic (Cutler et al,
2004).
Le «Challenge» du traitement des SMD réside dans le choix d’une stratégie adaptée à
chaque patient, prenant en compte l’âge, l’état général, les comorbidités, le type de maladie,
le temps écoulé depuis le diagnostic et les scores pronostiques.

24
Classification des SMD
L’évaluation clinique des SMD repose sur diverses classifications. La première
classification, proposée en 1982 par le French-American-British Group (FAB), était basée
sur des critères exclusivement morphologiques (Bennett et al, 1982). Les classifications
suivantes proposées par l’OMS en 2001 et 2008 ont intégré des données biologiques à
haute valeur pronostique, grâce à l’apport de la cytogénétique et de la biologie
moléculaire. Cependant, chaque classification présente des insuffisances en termes de
spécificité ou de sensibilité vis-à-vis du pronostic clinique de la maladie.
A ces différentes classifications ont été associés plusieurs scores pronostiques tels
que l’International Pronostic Scoring System (IPSS) (Greenberg et al, 1997) et le WHO-
classification based Prognostic Scoring System (WPSS) (Malcovati et al, 2007).
a) Classification morphologique des SMD
En 1982, le groupe de coopération franco-americano-britanique (FAB) a publié une
classification morphologique distinguant 5 catégories de SMD (Tableau n°3). Un des critères
majeurs de cette classification est le taux de blastes sanguins et médullaires.

25
Tableau n°3 : Classification FAB des SMD
(Bennett et al, 1982)
Anémie Réfractaire (AR)
Moelle normale ou hypercellulaire < 1% de blastes dans le sang < 5% de blastes dans la moelle
Anémie Réfractaire avec Sidéroblastes en couronne (ARS)
< 1% de blastes dans le sang < 5% dans la moelle Sidéroblastes en couronne > 15% dans la moelle
Anémie Réfractaire avec Excès de Blastes (AREB)
Cytopénie > 2 lignées cellulaires du sang Dysplasie concernant les 3 lignées < 5% de blastes dans le sang 5-20% de blastes dans la moelle
Leucémie Myélomonocytaire Chronique (LMMC)
Mêmes caractéristiques que les AREB < 5% de blastes dans le sang 5-20% de blastes dans la moelle +/- corps d’Auer dans les blastes Monocytose > 1 G/L
Anémie Réfractaire avec Excès de Blastes en Transformation (AREB-T)
Mêmes caractéristiques que les AREB > 5% de blastes dans le sang 21-30% de blastes dans la moelle +/- corps d’Auer dans les blastes

26
b) Classification cytogénétique des SMD
La cytogénétique est depuis plus de vingt ans l’outil de diagnostic principal des
hémopathies myéloïdes (Yunis et al, 1988) et le principal facteur pronostique de ces
maladies (Pierre et al, 1990). Elle consiste en l’étude des anomalies chromosomiques
présentes sur le caryotype médullaire.
En 2001, les données cytogénétiques à haute valeur pronostique sont intégrées dans
une nouvelle classification des SMD réalisée par l’OMS. Les AREB-T, SMD avec un taux
de blastes >20% dans la moelle, sont désormais classés en LAM, les LMMC deviennent
une catégorie mixte SMD/SMP et les SMD avec délétion du bras long du chromosome 5
une entité distincte. La classification OMS 2001 a été réactualisée avec de nouvelles
données de cytogénétique et de biologie moléculaire en 2008 (Swerdlow et al, 2008).
(1) Classification OMS des SMD
La classification actuellement reconnue pour les SMD est la classification OMS 2008
(Swerdlow et al, 2008), qui classe les SMD en 6 catégories (Tableau n°4) selon le
pourcentage de blastes dans la moelle osseuse et le sang, la présence ou non d’une dysplasie
multilignée et le nombre de sidéroblastes en couronne.
La classification OMS 2001 a été la première à distinguer les SMD avec dysplasie
unilignée des SMD à dysplasie multilignée (Jaffe et al, 2001). Les SMD avec dysplasie
unilignée ont en effet un bien meilleur pronostic que ceux avec dysplasie multilignée, même
avec un pourcentage de blastes médullaires identique.
Depuis 2001, les leucémies myélomonocytaires chroniques (LMMC) ne sont plus classées
avec les SMD mais dans une entité mixte SMD/SMP.
L’ancien sous-groupe Anémie Réfractaire (AR) et certains SMD inclassables sont désormais
regroupés dans une entité appelée « Cytopénie Réfractaire avec Dysplasie Unilignée »
(CRDU), qui est sous divisée en 3 sous-groupes : Anémie Réfractaire (AR), Neutropénie
Réfractaire (NR) et Thrombopénie Réfractaire (TR). Le groupe «syndrome 5q-» a été
renommé «SMD avec del(5q) isolée».
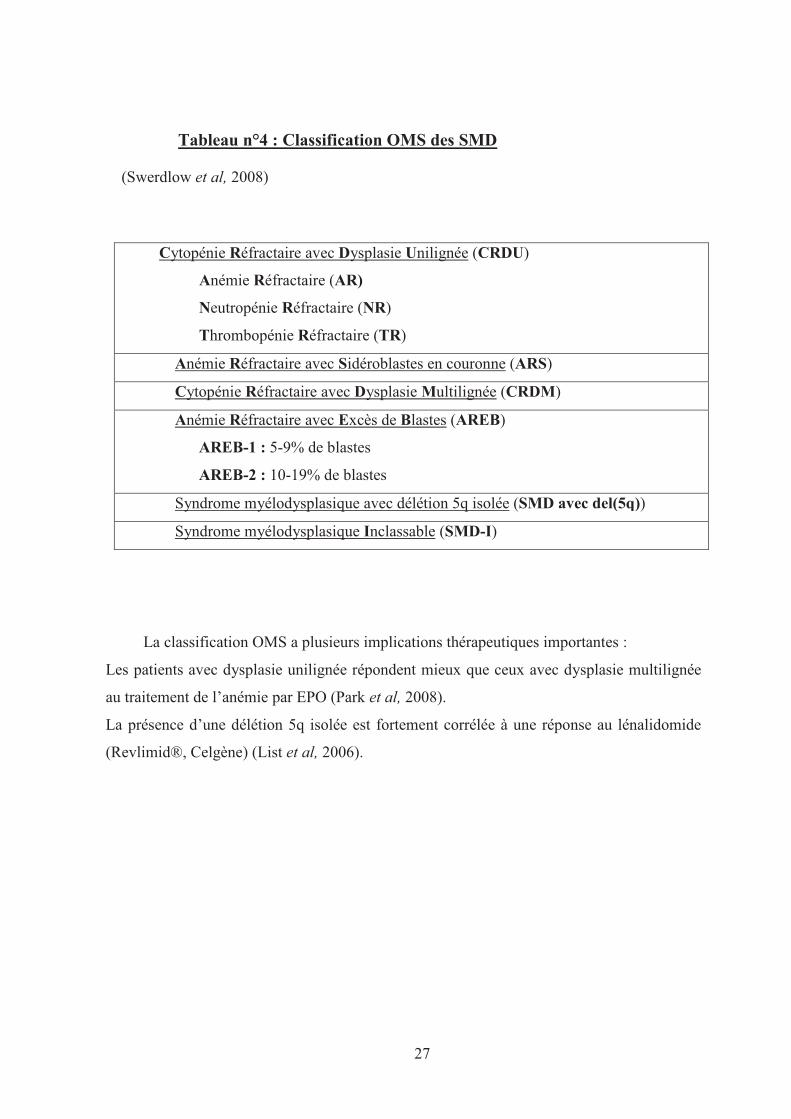
27
Tableau n°4 : Classification OMS des SMD
(Swerdlow et al, 2008)
Cytopénie Réfractaire avec Dysplasie Unilignée (CRDU)
Anémie Réfractaire (AR)
Neutropénie Réfractaire (NR)
Thrombopénie Réfractaire (TR)
Anémie Réfractaire avec Sidéroblastes en couronne (ARS)
Cytopénie Réfractaire avec Dysplasie Multilignée (CRDM)
Anémie Réfractaire avec Excès de Blastes (AREB)
AREB-1 : 5-9% de blastes
AREB-2 : 10-19% de blastes
Syndrome myélodysplasique avec délétion 5q isolée (SMD avec del(5q))
Syndrome myélodysplasique Inclassable (SMD-I)
La classification OMS a plusieurs implications thérapeutiques importantes :
Les patients avec dysplasie unilignée répondent mieux que ceux avec dysplasie multilignée
au traitement de l’anémie par EPO (Park et al, 2008).
La présence d’une délétion 5q isolée est fortement corrélée à une réponse au lénalidomide
(Revlimid®, Celgène) (List et al, 2006).

28
(2) Anomalies chromosomiques des SMD
Environ la moitié des SMD primaires montrent un caryotype altéré. Les SMD les
plus affectés par des anomalies chromosomiques sont les formes les plus avancées
(AREB1 et 2). Contrairement aux LAM qui présentent de nombreuses translocations
chromosomiques équilibrées, les anomalies chromosomiques des SMD sont
essentiellement représentées par des pertes de matériel génétique, sous forme de délétions
partielles ou totales d’un chromosome, ou sous forme de gains de matériel génétique
résultant de trisomies.
Une étude multicentrique internationale a confirmé la grande hétérogénéité des
SMD, en identifiant 684 types d’anomalies chromosomiques sur 1080 SMD à caryotype
anormal (Haase et al, 2007), la majorité de ces anomalies étant rares avec des fréquences
inférieures à 1%. Les principales anomalies sont par ordre décroissant : la perte partielle
ou totale d’un chromosome 5, la perte partielle ou totale d’un chromosome 7, la trisomie
8, la délétion 20q, la perte d’un chromosome X ou Y et la délétion 17p (Tableau n°5).
En général, les anomalies cytogénétiques ne sont pas spécifiques des SMD et sont
également retrouvées dans d’autres hémopathies myéloïdes.
Comparativement aux SMD primaires, les SMD secondaires à une chimiothérapie
(therapy-related) ont une fréquence plus élevée d’anomalies cytogénétiques, elles sont
retrouvées dans près de 80% des cas (Olney et al, 2001). Les délétions de chromosomes 5
et 7 qui concernent seulement 10-15% des SMD de novo représentent 50-60 % des SMD-t
(Pedersen-Bjergaard et al, 2008).
Tableau n°5 : Principales anomalies chromosomiques des SMD de novo
(D’après Haase et al, 2008)
Anomalie chromosomique Incidence (%)
Perte partielle ou totale d’un chromosome 5 15
Perte partielle ou totale d’un chromosome 7 10
Trisomie 8 8
Délétion 20q 4
Perte d’un chromosome X ou Y 2
Délétion 17p <1

29
La perte du chromosome Y peut être soit associée au SMD, soit physiologique, liée
à l’âge du patient (Wong et al, 2008).
Globalement, le type d’aberration chromosomique retrouvé dans les SMD (perte de
matériel chromosomique) laisse envisager qu’un des mécanismes moléculaires
prépondérant dans les SMD serait la perte d’expression de gènes suppresseurs de tumeur.
L’activation d’oncogènes serait plutôt l’apanage des LAM et pourrait être à l’origine de
l’acutisation des SMD.
(3) Scores IPSS et WPSS
Score IPSS
Le score International Prognostic Scoring System, publié en 1997, est toujours le score
pronostique le plus utilisé dans l’évaluation de la gravité des SMD.
L’IPSS est basé sur 3 critères (Greenberg et al, 1997) :
- Le pourcentage de blastes dans la moelle osseuse
- Le nombre de cytopénies sanguines
- Le caryotype.
Le score IPSS classe les SMD en 4 groupes selon le risque de transformation en
leucémie (Tableau n°6) :
- Faible risque
- Risque intermédiaire bas ou Int-1
- Risque intermédiaire élevé ou Int-2
- Haut Risque.
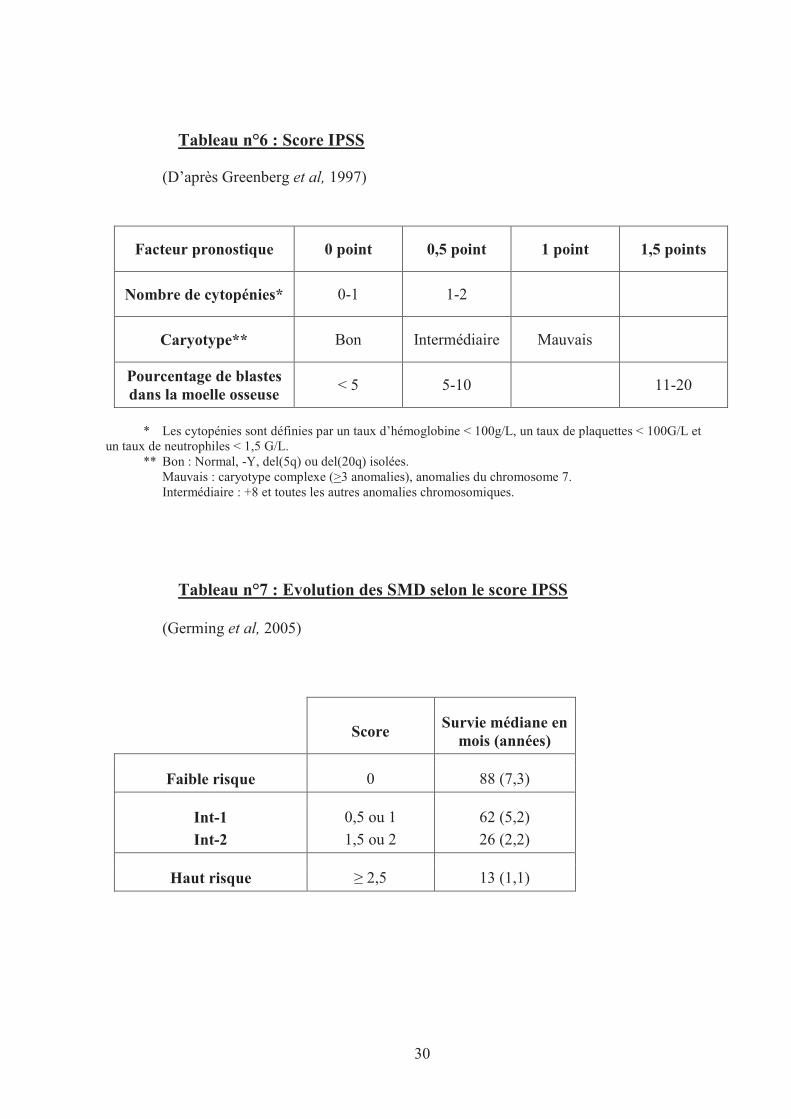
30
Tableau n°6 : Score IPSS
(D’après Greenberg et al, 1997)
Facteur pronostique 0 point 0,5 point 1 point 1,5 points
Nombre de cytopénies* 0-1 1-2
Caryotype** Bon Intermédiaire Mauvais
Pourcentage de blastes dans la moelle osseuse
< 5 5-10 11-20
* Les cytopénies sont définies par un taux d’hémoglobine < 100g/L, un taux de plaquettes < 100G/L et un taux de neutrophiles < 1,5 G/L.
** Bon : Normal, -Y, del(5q) ou del(20q) isolées. Mauvais : caryotype complexe (>3 anomalies), anomalies du chromosome 7. Intermédiaire : +8 et toutes les autres anomalies chromosomiques.
Tableau n°7 : Evolution des SMD selon le score IPSS
(Germing et al, 2005)
Score Survie médiane en
mois (années)
Faible risque 0 88 (7,3)
Int-1
Int-2
0,5 ou 1
1,5 ou 2
62 (5,2)
26 (2,2)
Haut risque � 2,5 13 (1,1)

31
Score WPSS
Bien que le score IPSS ait prouvé son efficacité dans les décisions cliniques et est
utilisé dans les études actuelles, il ne prend pas en compte l’impact négatif des transfusions
sanguines et le mauvais pronostic des dysplasies multiples. En 2007, Malcovati et al ont
proposé le score WHO-classification based Prognostic Scoring System qui intègre aux
critères de l’IPSS, des éléments de la classification OMS et le besoin transfusionnel.
Le besoin transfusionnel est inversement corrélé à la survie : les patients avec anémie sévère
dépendant de transfusions sanguines ont un pronostic de survie très sombre et un risque
élevé de transformation en LAM. Ce mauvais pronostic a plusieurs causes comme
notamment les effets délétères du surdosage en fer, avec des risques importants
d’insuffisance cardiaque, ainsi que la mauvaise oxygénation viscérale, due à l’anémie
(Cazzola et al, 2005).
Le score WPSS classe les SMD également en 4 groupes (différents de l’IPSS)
(Tableau n°8) :
- Faible risque (score = 0)
- Risque intermédiaire (score = 1)
- Risque élevé (score = 3 à 4)
- Risque très élevé (score = 5 à 6)
Tableau n°8 : Score WPSS
(Malcovati et al, 2007)
Variable \ Pondération 0 1 2 3
Catégories OMS AR, ARS, 5q– CRDM AREB-1 AREB-2
Caryotype Bon Intermédiaire Mauvais
Besoin transfusionnel Non Régulier
Abréviations : AR : Anémie Réfractaire ; ARS : Anémie réfractaire avec sidéroblastes en couronne ; 5q- : SMD avec del(5q) isolée ; CRDM : Cytopénie réfractaire avec dysplasie multilignée ; AREB : Anémie réfractaire avec excès de blastes.
Caryotype et pronostic : Bon : Normal, -Y, del(5q) ou del(20q) isolée ; Mauvais : complexe (� 3 anomalies), anomalies du chromosome 7 ; Intermédiaire : autres anomalies.
Besoin transfusionnel régulier : � 1 transfusion toutes les 8 semaines pendant une période de 4 mois.
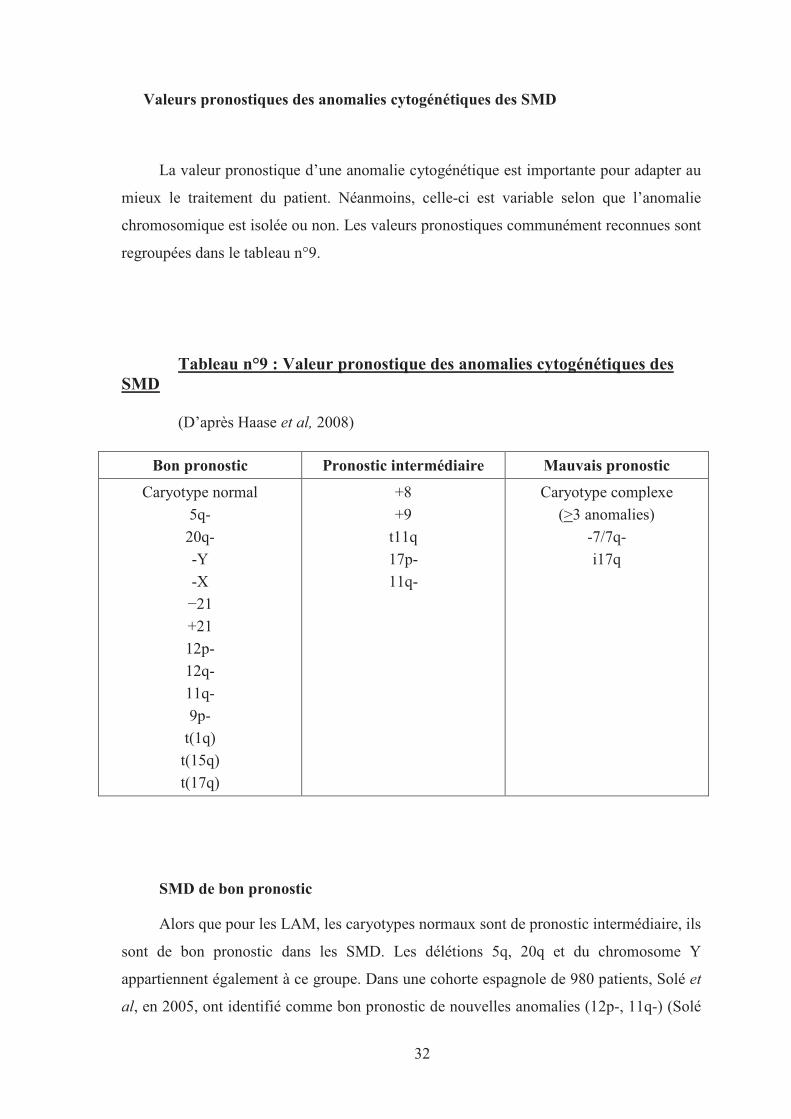
32
Valeurs pronostiques des anomalies cytogénétiques des SMD
La valeur pronostique d’une anomalie cytogénétique est importante pour adapter au
mieux le traitement du patient. Néanmoins, celle-ci est variable selon que l’anomalie
chromosomique est isolée ou non. Les valeurs pronostiques communément reconnues sont
regroupées dans le tableau n°9.
Tableau n°9 : Valeur pronostique des anomalies cytogénétiques des SMD
(D’après Haase et al, 2008)
Bon pronostic Pronostic intermédiaire Mauvais pronostic
Caryotype normal
5q-
20q-
-Y
-X
−21
+21
12p-
12q-
11q-
9p-
t(1q)
t(15q)
t(17q)
+8
+9
t11q
17p-
11q-
Caryotype complexe
(>3 anomalies)
-7/7q-
i17q
SMD de bon pronostic
Alors que pour les LAM, les caryotypes normaux sont de pronostic intermédiaire, ils
sont de bon pronostic dans les SMD. Les délétions 5q, 20q et du chromosome Y
appartiennent également à ce groupe. Dans une cohorte espagnole de 980 patients, Solé et
al, en 2005, ont identifié comme bon pronostic de nouvelles anomalies (12p-, 11q-) (Solé

33
et al, 2005). La grande étude multicentrique australo-germanique sur 2124 patients, a
récemment classé en plus dans ce groupe les délétions 5q-, 9p-, 12q-, 20q−, -X, −21, la
trisomie 21 (+21) et les translocations t(1q), t(15q) et t(17q). Ces altérations étaient de bon
pronostic lorsqu’elles étaient isolées au caryotype (Haase et al, 2007).
SMD de pronostic intermédiaire
Dans la majorité des études, la trisomie 8 constitue un groupe de pronostic
intermédiaire. Dans la base de donnée espagnole, Solé et al ont classé dans ce groupe la
trisomie 9, les translocation t(11q) et la délétion 17p et Haase et al dans la cohorte australo-
germanique, la délétion 11q- (Solé et al, 2005 ; Haase et al, 2007).
SMD de mauvais pronostic
Enfin, les SMD à caryotype complexe (� 3 anomalies) ainsi que les délétions du
chromosome 7 constituent un groupe de mauvais pronostic. Les patients avec SMD à
caryotype complexe montrent une survie médiane inférieure à un an. Solé et al, ont décrit
également une médiane de survie inférieure à un an pour les patients présentant un
isochromosome 17q (i17q) (Solé et al, 2005).

34
c) Classification moléculaire des SMD
(1) Les enjeux d’une classification moléculaire
Actuellement, aucune anomalie moléculaire isolée n’a permis de reconstituer un
phénotype de SMD dans un modèle murin. L’hypothèse actuellement reconnue est que
plusieurs altérations seraient nécessaires pour le développement et la progression clinique
d’un SMD : des altérations moléculaires successives affecteraient un clone de CSH
provoquant sa multiplication anarchique et le remplacement progressif des CSH saines
dans la moelle (Tefferi, 2009).
Même si plusieurs mutations sur différents gènes ont récemment été identifiées, les
conséquences des mutations sur le phénotype demeurent en très grande partie incomprises.
La détermination du statut mutationnel de chaque type de SMD a pour enjeu de donner
des pistes dans la compréhension des mécanismes physiopathologiques de la maladie, de
trouver de nouvelles cibles moléculaires et, à terme, de mettre en place de nouvelles
stratégies thérapeutiques.
(2) Les gènes mutés dans les SMD
(a) NRAS
Les protéines RAS (NRAS, KRAS et HRAS) appartiennent à une famille de petites
protéines membranaires régulant les signaux de transduction de nombreux récepteurs
membranaires dont les RTK. Elles jouent un rôle important dans la régulation de la
prolifération, de la différenciation et de l’apoptose cellulaire. Les mutations des gènes RAS
ont été les 1ères mutations rapportées dans les hémopathies myéloïdes (Janssen et al, 1987 ;
Padua et al, 1988). Les mutations de NRAS, localisé en 1p13, sont de loin les plus
importantes.
Les mutations de l’oncogène NRAS sont présentes dans les SMD à une fréquence très
variable comprise entre 6 et 48% avec une moyenne de 10 à 15% (plus fréquente dans les
SMD à taux élevé de blastes comme les AREB). Elles sont corrélées à une transformation
accrue en LAM. Des études sur de grandes cohortes de patients sont nécessaires pour
confirmer que ces mutations constituent un facteur de risque indépendant de mauvais
pronostic (Padua et al, 1998 ; Bacher et al, 2007).

35
(b) TP53
Le gène TP53, localisé sur le bras court du chromosome 17 (17p13), représente le gène
le plus fréquemment muté dans tous les types de cancers. TP53 est un gène suppresseur de
tumeur essentiel dans la stabilité et l’intégrité du génome (Levine, 1997).
Dans les SMD de novo, TP53 est muté de façon hétérozygote chez environ 10% des
patients (Wattel et al, 1994). Les mutations de TP53 sont plus fréquemment rencontrées dans
les SMD secondaires à l’administration d’une chimiothérapie, à raison de 25 à 50% des cas
(Pedersen-Bjergaard et al, 2008). Les mutations de TP53 provoquent une résistance aux
chimiothérapies et sont généralement de mauvais pronostic notamment dans les SMD (Kita-
Sasai et al, 2001).
(c) TET2
Le gène TET2 (Ten Eleven Translocation-2) est situé sur le bras long du
chromosome 4 (4q24) et est fréquemment affecté par des pertes d’hétérozygotie. En 2009,
plusieurs équipes ont décrit des mutations du gène TET2 dans les SMD (Abdel-Wahad et
al, 2009 ; Jankowska et al, 2009 ; Mohamedali et al, 2009).
Environ 20 à 25% des SMD primaires présentent une ou plusieurs mutations de
TET2 (Delhommeau et al, 2009 ; Langemeijer et al, 2009) : TET2 constitue le gène le plus
fréquemment muté dans les SMD. Ces mutations sont rencontrées dans tous les types de
SMD et ne semblent pas associées à sous-groupe particulier de SMD.
Dans l’étude de Langemeijer et al portant sur 102 patients, les mutations de TET2
étaient plus fréquentes dans les SMD avec IPSS bas (41%) et intermédiaire-1 (27%) que
dans les SMD avec IPSS élevés (14%) et intermédiaire-2 (13%) (Langemeijer et al, 2009).
Les mutations de TET2 semblent de bon pronostic également dans l’étude menée par
Kosmider et al sur 96 patients (Kosmider et al, 2009), mais de plus grandes études doivent
encore préciser si les mutations de TET2 constituent un facteur moléculaire indépendant
de bon pronostic dans certains sous-types de SMD. Une étude a montré recemment
l’absence de valeur pronostique des mutations de TET2 dans 320 SMD (Smith et al,
2010).
Les mutations de TET2 sont considérées comme un événement précoce dans la
physiopathologie des SMD et les CSH avec mutation de TET2 montrent une croissance in
vitro plus importante que les CSH non mutées (Delhommeau et al, 2009). Les fonctions
cellulaires précises de TET2 ne sont pas connues. Il a été montré récemment qu’un gène
homologue, TET1, contrôlait la conversion de la 5-méthylcytosine en 5-

36
hydroxyméthylcytosine et qu’il était impliqué dans le contrôle épigénétique de la
transcription (Tahiliani et al, 2009). Ainsi, il est probable que les mutations de TET2
augmentent le potentiel prolifératif des cellules en interférant sur le degré de méthylation
de l’ADN. Ces résultats sont importants car de nombreux promoteurs de gènes
suppresseurs de tumeur seraient hyperméthylés dans les SMD et LAM et il a été montré
que les agents déméthylants pouvaient améliorer la survie de certains patients atteints de
SMD à haut risque (Fenaux et al, 2009).
(d) ASXL1
Le gène ASXL1 (Additional Sex Combs 1), localisé en 20q11, dont les mutations ont
été initialement identifiées dans le laboratoire de D.Birnbaum, chez des patients atteints de
SMD (Gelsi-Boyer et al, 2009) est actuellement reconnu comme le gène le plus
fréquemment muté dans les SMD avancés : environ 30% des AREB sont mutées (Boultwood
et al, 2010a). ASXL1 appartient à une famille de 3 membres encore peu connus, qui seraient
impliqués dans la régulation du remodelage de la chromatine. Les protéines ASXL possèdent
un domaine PHD (Plant Homeo Domain) C-terminal et appartiennent au complexe
polycomb et trithorax impliqué dans la programmation génétique des CSH (Fisher et al,
2006). Les mutations décrites conduisent fréquemment à la perte du domaine PHD de
ASXL1, responsable de la perte de ces fonctions régulatrices de la chromatine (Gelsi-Boyer
et al, 2009).
Dans le laboratoire de D. Birnbaum, 11% de SMD présentaient une mutation sur un
panel de 35 patients (Gelsi-Boyer et al, 2009) et Boultwood et al, ont ensuite retrouvé 15%
de patients mutés sur 182 SMD (Boultwood et al, 2010a). Les souris KO ASXL1 montrent
des altérations de la différenciation myéloïde et aussi lymphoïde, mais pas de
myélodysplasie ou de leucémie (Fisher et al, 2010). Les mutations d’ASXL1 ne sont pas
suffisantes à elles seules pour induire un SMD, ce qui confirme l’hypothèse que la maladie
serait la résultante de la combinaison de plusieurs anomalies moléculaires.
(e) RUNX1/AML1/CBFA
Le gène RUNX1 localisé en 21q22, code pour la sous-unité α du complexe
transcriptionnel CBF (Core Binding Factor) humain, et est exprimé dans toutes les lignées
hématopoïétiques. C’est un régulateur de nombreux gènes intervenant dans l’hématopoïèse
(Tenen, 2003) et dans la mégacaryocytopoïèse. Des mutations germinales de RUNX1,

37
transmises selon un mode autosomique dominant, sont responsables de la thrombopénie
familiale avec prédisposition aux LAM (Familial platelet disorder/Acute Myeloid Leukemia,
FPD/AML) (Jongmans et al, 2010).
Les mutations de RUNX1 sont retrouvées dans 5 à 10% des SMD primaires et leur
fréquence augmente proportionnellement au nombre de blastes (Harada et al, 2004). Elles
sont considérées comme de mauvais pronostic (Chen et al, 2007). Christiansen et al ont ainsi
retrouvé une fréquence élevée (41%) de mutations de RUNX1 dans les SMD-t (therapy-
related) (Christiansen et al, 2004).
(f) IDH1, IDH2
Les gènes codant pour les isocitrate déshydrogénases (IDH 1 et 2), localisés en 2q33
et 15q26 respectivement, ont été initialement trouvés mutés dans les gliomes puis
récemment dans 4 à 8 % des SMD (Kosmider et al, 2010a ; Rocquain et al, 2010a).
L’enzyme IDH convertit l’isocitrate en 2-hydroxyglutarate dans le cycle de Krebs. La
mutation d’IDH serait probablement une mutation gain de fonction du fait de son
association à une augmentation de la concentration de 2-hydroxyglutarate dans les cellules
mutées (Gross et al, 2010).
Les mutations d’IDH ne paraissent pas reliées à un type de SMD particulier
(Kosmider et al, 2010a). Une étude récente sur 193 patients a montré que les mutations
d’IDH1 mais pas celles d’IDH2 constituaient un facteur indépendant de mauvais pronostic
dans les SMD (Thol et al, 2010a). Ce résultat doit être confirmé sur de grandes cohortes
de patients.
Le fait qu’IDH soit retrouvé muté à tous les stades de la maladie et dans les LAM
permet de suggérer que les mutations de ce gène contribuent à la transformation
leucémique des SMD (Thol et al, 2010a).
(g) Les autres mutations décrites dans les SMD
Même si de nombreuses mutations sont décrites dans les SMD, leur incidence globale
toutes mutations confondues est moins élevée que dans les LAM et aucune de ces mutations
ne semble spécifique des SMD.
Les mutations de JAK2, gène localisé en 9p24, ont été découvertes en 2005 et sont
généralement retrouvées dans les syndromes myéloprolifératifs (50% des thrombocytémies
essentielles et des myélofibroses primitives et 95% des polyglobulies primitives). La

38
mutation la plus courante est la Val617Phe (remplacement de la valine en position 617 par
une phénylalanine). Les LMMC, ainsi que d’autres SMD/SMP peuvent présenter cette
mutation de JAK2, tout comme les anémies réfractaires sidéroblastiques avec thrombocytose
qui ont un taux de mutation d’environ 50% (Hellstrom-Lindberg, 2010). En revanche, les
mutations de JAK2 sont rares dans les SMD : <1% des patients (Steensma et al, 2005). Les
conséquences cliniques des mutations de JAK2 dans les SMD ne sont pas clairement
élucidées. Elles sont associées de façon significative à un taux élevé de plaquettes,
d’hémoglobine et de globules blancs, mais il n’a pas été montré de différence de survie entre
patients mutés et non mutés.
Récemment, des mutations concernant le gène CBL (Casitas B-cell Lymphoma),
localisé en 11q23, et codant pour une ubiquitine ligase, ont été identifiées dans les SMD. La
protéine CBL est impliquée dans la dégradation des récepteurs à activité tyrosine kinase en
augmentant leur ubiquinisation et leur élimination par le protéasome (Dunbar et al, 2008).
L’inactivation de CBL serait associée à un gain de croissance cellulaire. Les mutations de
CBL sont peu fréquentes dans les SMD, de l’ordre de 1% également mais seraient associées
à un mauvais pronostic (Reindl et al, 2009). Les patients avec mutation homozygote auraient
un pronostic de survie plus défavorable que ceux avec mutation hétérozygote, suggérant un
effet protecteur de l’allèle non muté restant (Makishima et al, 2009). Les études
fonctionnelles montrent que les mutations de CBL induisent une multiplication cellulaire
exacerbée, dépendante des facteurs de croissance via la voie FLT3 (Sanada et al, 2009).
Ainsi les inhibiteurs de FLT3 pourraient être efficaces dans les SMD avec mutation de CBL.
Certaines mutations initialement décrites dans les LAM ont ensuite été retrouvées dans
les SMD. Le gène codant pour la nucléophosmine NPM1 est l’un des gènes les plus mutés
dans les LAM, essentiellement les LAM à caryotype normal (cf. p51) mais il est rarement
muté dans les SMD : 0 à 1% des patients (Zhang et al, 2007 ; Rocquain et al, 2010a).
Le gène FLT3 est muté dans moins de 1% des SMD, KIT dans un peu plus de 1% et
les duplications partielles en tandem du gène MLL sont retrouvées dans 2,7% des patients
atteints de SMD (Bacher et al, 2007). Ces 3 derniers gènes sont plus fréquemment mutés
dans les LAM.

39
SMD avec délétion 5q isolée
Dans les SMD, les translocations chromosomiques et gènes de fusion sont rares. Le
seul sous-type de SMD dont la physiopathologie pourrait être définie par une anomalie
génétique simple est le syndrome 5q-.
Ce syndrome a été décrit en 1974 par Van den Berghe comme une entité clinique à
part entière, caractérisée par une délétion isolée du bras long d’un chromosome 5 (Van den
Berghe et al, 1974). Du fait de son bon pronostic, il occupe depuis 2001 une place distincte
dans la classification OMS des hémopathies myéloïdes. Les patients atteints de del(5q)
isolée répondent en grande partie au traitement par lénalidomide.
La biologie du syndrome 5q- est typique et retrouve :
- une anémie réfractaire macrocytaire souvent sévère,
- un degré variable de neutropénie,
- une hyperplaquettose,
- une dysplasie mégaryocytaire particulière dans une moelle de richesse normale ou
augmentée avec des mégacaryocytes hypolobés.
La transformation leucémique est faible, de l’ordre de 10% mais peut survenir à long
terme (Giagounidis et al, 2004).
a) Quels sont les gènes impliqués dans la délétion ?
En 2002, Boultwood et al ont décrit la région delétée localisée en 5q31-32,
comprenant 1,5 millions de bases et environ 40 gènes (Boultwood et al, 2002). Cette région
montre plusieurs gènes potentiellement suppresseurs de tumeur, cependant aucun de ces
gènes n’a été révélé muté ou inactivé.
En 2008, Ebert et al ont réalisé des expériences d’inactivation par une technique
d’interférence d’ARN sur chacun des 40 gènes de la région 5q32-33 et ont démontré que la
sous-expression du gène RPS14 provoquait une diminution de l’érythropoïèse et une
augmentation de l’apoptose des érythrocytes (Ebert et al, 2008). RPS14 est un gène codant

40
pour un composant de la sous-unité 40S du ribosome. D’autres gènes ribosomaux ont
récemment été décrits comme impliqués dans la survenue d’anémie, en particulier le gène
RPS19 dans l’anémie macrocytaire de Blackfan-Diamond (Draptchinskaia et al, 1999). Des
modèles animaux ont montré qu’une sous-expression de certains gènes ribosomaux pouvait
augmenter le risque de cancer (Amsterdam et al, 2004). La sous-expression de RPS14 serait
un des mécanismes principaux à l’origine de l’anémie du syndrome 5q-. Néanmoins, cela
n’explique pas le taux élevé de plaquettes et la multiplication incontrôlée des CSH.
b) Implication des micro-ARN ?
Les microARN (miARN) sont de petites chaines d’acide ribonucléique (ARN) non
codantes constitués d’environ 22 nucléotides. Elles ont la particularité de se fixer sur la
région 3’ non codante des ARN messagers (ARNm) pour induire leur dégradation ou une
interférence au niveau de leur traduction.
Plusieurs études récentes ont démontré que le syndrome 5q- est corrélé à la perte
d’expression de 2 microARN, abondants dans les CSH et progéniteurs hématopoïétiques, mi-
R145 et mi-R146a. Deux gènes de la famille de la voie de signalisation des récepteurs Toll-
like, Toll–interleukin-1 receptor domain–containing adaptor protein (TIRAP) et tumor
necrosis factor receptor–associated factor-6 (TRAF6), ont été identifiés comme les 2 cibles
respectives de mi-R145 et mi-R146a (Starczynowski et al, 2010).
Les modèles murins sous-exprimant ces 2 miARN ou ayant une surexpression de
TRAF6 montrent une hyperplaquettose, une neutropénie modérée et une dysplasie
mégaryocytaire, comme dans les SMD avec délétion 5q.
L’hypothèse actuelle sur la physiopathologie du syndrome 5q- est que les sous-
expressions conjointes de RPS14, miR-145 et miR-146a seraient à l’origine des principaux
signes de la maladie. Néamoins, il reste à démontrer que ces 3 altérations sont suffisantes
pour donner la maladie (modèle murin en cours d’étude). D’autres gènes sous-exprimés
pourraient être impliqués comme le gène SPARC, qui joue un rôle dans l’adhésion cellulaire,
l’apoptose et l’angiogenèse (Pellagatti et al, 2007) ou les gènes EGR1 et CTNNA1 qui
seraient étroitement impliqués dans la transformation leucémique (Ebert et al, 2008).

41
c) Mécanisme d’action du lénalidomide
Le lénalidomide (Revlimid®, Celgene), qui a actuellement comme unique AMM, le
traitement du myélome multiple, est par ailleurs particulièrement efficace dans le traitement
des patients atteints de syndrome 5q-. A l’inverse, il montre très peu d’efficacité sur les
cellules SMD non délétées en 5q (List et al, 2006).
Le lénalidomide a un mécanisme d’action complexe et très large. Il inhibe notamment
le TNF� et l’IL-6, induit l’apoptose et régule l’adhésion cellulaire, l’angiogenèse et l’activité
des lymphocytes NK.
Deux hypothèses tentent d’expliquer la plus grande sensibilité des cellules avec
del(5q) au lénalidomide:
- Le lénalidomide inhibe 2 phosphatases régulant le cycle cellulaire, Cdc25C et PP2Ac�, qui
sont localisé en 5q et délétées chez la majorité des patients avec syndrome 5q- (Wei et al,
2009).
- Le lénalidomide restaure l’expression du gène SPARC dans les progéniteurs avec délétion
5q (Pellagatti et al, 2007).
Le syndrome 5q- constitue un modèle particulièrement intéressant d’étude car il a pu
confirmer que les pertes d’hétérozygotie ainsi que la perte d’expression de miRNA étaient
des mécanismes importants dans l’apparition d’un SMD. De plus, l’action du lénalidomide
uniquement sur ce type de SMD confirme l’hétérogénéité des SMD. L’étude du mécanisme
d’action du lénalidomide constitue une bonne piste d’étude dans la compréhension de la
physiopathologie des SMD.

42
C.Les leucémies aiguës myéloïdes (LAM)
Généralités
Les LAM constituent comme les SMD un groupe très hétérogène d’hémopathies
malignes myéloïdes. Elles sont caractérisées par une prolifération clonale de précurseurs
myéloïdes anormaux provoquant une altération de l’hématopoïèse avec blocage précoce de
la différenciation cellulaire du clone malin.
Ce blocage de maturation est à l’origine de l’accumulation de blastes dans la moelle
osseuse et le sang. Depuis 2001 et la 1ère classification OMS, le pourcentage minimal de
blastes dans la moelle requis pour le diagnostic d’une LAM est passé de 30 à 20%.
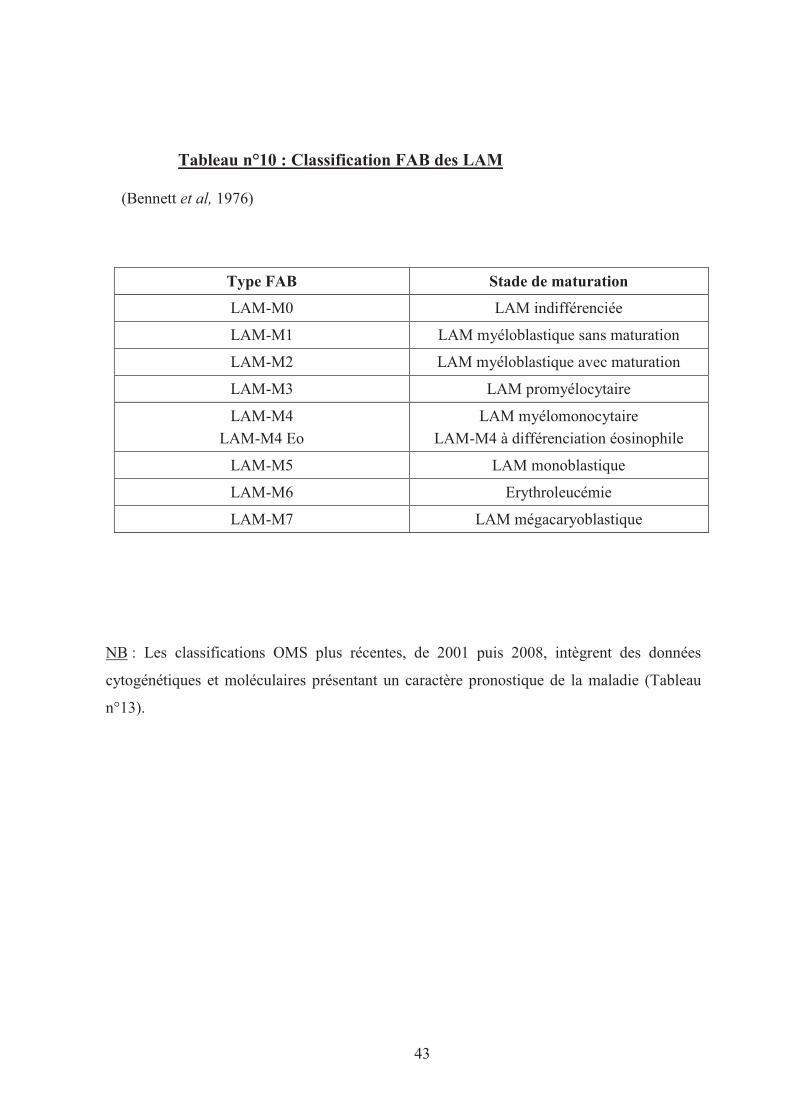
La classification FAB (Tableau n°10), établie il y a plus de 30 ans, demeure la base du
diagnostic morphologique des LAM (Bennett et al, 1976). Elle classe les LAM en 8 sous-
groupes de M0 à M7 en fonction des caractéristiques des blastes médullaires et de leur degré
de différenciation.
Cette classification ne présente qu’un intérêt morphologique car elle regroupe des
LAM d’aspect et d’évolution clinique très hétérogène et elle ne guide pas le clinicien dans le
choix d’une stratégie thérapeutique, à l’exception de la LAM-M3.
43
Tableau n°10 : Classification FAB des LAM
(Bennett et al, 1976)
Type FAB Stade de maturation
LAM-M0 LAM indifférenciée
LAM-M1 LAM myéloblastique sans maturation
LAM-M2 LAM myéloblastique avec maturation
LAM-M3 LAM promyélocytaire
LAM-M4
LAM-M4 Eo
LAM myélomonocytaire
LAM-M4 à différenciation éosinophile
LAM-M5 LAM monoblastique
LAM-M6 Erythroleucémie
LAM-M7 LAM mégacaryoblastique
NB : Les classifications OMS plus récentes, de 2001 puis 2008, intègrent des données
cytogénétiques et moléculaires présentant un caractère pronostique de la maladie (Tableau
n°13).

44
a) Epidémiologie
Les LAM sont les hémopathies myéloïdes les plus fréquentes, devant les SMD. Ce
sont aussi les leucémies les plus fréquentes, représentant plus du quart de toutes les
leucémies de l’adulte. L’incidence des LAM est destimée entre 5 et 8 cas pour 100.000
habitants (Deschler et al, 2006).
Elles sont majoritairement retrouvées chez les personnes âgées et rarement
diagnostiquées avant l’âge de 40 ans : l’âge médian au diagnostic est d’environ 65 ans.
L’incidence augmente avec l’âge, surtout après 50 ans et l'incidence annuelle à 70 ans est
de 20 pour 100.000 habitants. Les LAM sont rares chez l’enfant, les LA des jeunes enfants
étant à plus de 75% représentées par des leucémies aiguës lymphoïdes (Stiller, 2004).
Néanmoins, il existe un pic d’incidence de faible intensité pour les LAM entre 1 à 4 ans
(0,6 à 0,8 cas pour 100.000).
Comme pour les SMD, de façon inexpliquée, les hommes sont plus affectés que les
femmes (sex ratio = 1,5) (Estey et al, 2006).
b) Facteurs de risque
Comme pour les SMD, il existe de rares syndromes congénitaux prédisposant à une
plus grande fréquence de LAM tels que les syndromes de Bloom, Down, Kostmann et
Blackfan-Diamond (Deschler et al, 2006).
Les LAM secondaires par opposition aux LAM primaires ou de novo, regroupent les
LAM avec antécédents d’hémopathie myéloïde chronique (SMD, SMP, LMMC ou
hémoglobinurie paroxystique nocturne) ou avec antécédents d’exposition à un facteur de
risque. Plusieurs facteurs de risque on été recensés : exposition à un traitement par
chimiothérapie anticancéreuse, plus particulièrement par des agents alkylants ou des
inhibiteurs de topoisomérases II, exposition aux radiations, au benzène, aux herbicides ou à
la fumée de cigarette (Deschler et al, 2006).
Les LAM induites par les chimiothérapies (therapy-related) représentent 10 à 20% de
l’ensemble des LAM (Pedersen-Bjergaard et al, 2007).

45
c) Physiopathologie
Les LAM sont des proliférations malignes clonales des CSH associées à un blocage de
maturation à un stade précoce de la différenciation cellulaire. Il en résulte une accumulation
de cellules immatures, appelées blastes, dans la moelle osseuse, le sang, et éventuellement
dans d’autres organes. L’envahissement médullaire par les blastes perturbe l’hématopoïèse
normale et se traduit par des cytopénies avec leurs conséquences cliniques.
Comme dans les SMD, les infections représentent la cause majeure de décès des patients
atteints de LAM (Estey et al, 2006).
d) Bilan diagnostique
(1) Diagnostic biologique
Le diagnostic d’une LAM nécessite une numération–formule sanguine et un
myélogramme. Le bilan doit comporter une étude morphologique (critères FAB et signes de
dysplasie) et cytochimique (myéloperoxydases), un immunophénotypage des blastes
(permettant d’affirmer la nature myéloïde dans les cas difficiles sur le plan cytologique), une
étude cytogénétique de la moelle et une étude en biologie moléculaire (recherche de
transcrits de fusion issus de translocations chromosomiques).
(2) Evaluation pré-thérapeutique
L’interrogatoire précise les comorbidités antérieures et recherche le caractère
secondaire éventuel de l’hémopathie (exposition toxique, radio ou chimiothérapie
anticancéreuse, SMP ou SMD).
L’examen clinique apprécie l’indice de performance (performance status selon OMS,
Karnofsky), les manifestations tumorales, les signes infectieux.
Le bilan comporte une évaluation de la fonction cardiaque (fraction d’éjection
ventriculaire avant mise sous anthracycline), un bilan d’hémostase à la recherche d’une
coagulation intra-vasculaire disséminée (CIVD), une ponction lombaire en cas de signe
d’appel neurologique ou d’hyperleucocytose > 100G/l et un bilan pré-transfusionnel.

46
Toute fièvre doit être explorée avant traitement par hémocultures, prélèvements des
sites suspects, radiographie thoracique voire scanner en cas de signes d’appel pulmonaires.
Une recherche de donneur HLA compatible sera entreprise dès que possible pour les
patients candidats potentiels à une allogreffe de CSH.
e) Traitements
Classiquement, le schéma thérapeutique des LAM se déroule en en 3 étapes :
- L’induction qui consiste à induire une rémission hématologique complète (blastes < 5%,
polynucléaires neutrophiles >1G/L et plaquettes >100G/L) (Cheson et al, 2003). Le schéma
standard de la cure d’induction (protocole 3+7) est composé de 3 jours d’anthracycline
(daunorubicine ou idarubicine) suivis de 7 jours de cytarabine.
- La consolidation qui a pour but de prolonger la rémission par des cures à base de
cytarabine à haute dose tous les mois puis tous les 2 à 3 mois.
- La 3ème étape, destinée à prévenir les rechutes, qui varie selon l’âge et l’état clinique
du patient, le type de LAM et de la disponibilité d’un greffon. Il pourra s’agir d’une
allogreffe de cellules souches ou d’une chimiothérapie intensive (cures d’intensification).
Les LAM présentent des pronostics très variables. Avec le protocole d’induction à
base de cytarabine et anthracycline, la rémission complète est obtenue dans 60 à 80% des cas
pour les patients jeunes et dans 40 à 55% des cas pour les patients de plus de 60 ans (Farag
et al, 2005).
Cependant, malgré ces taux de rémission, 30 à 40% des patients rechutent et meurent de leur
maladie dans un délai de 2 ans et la survie globale à 5 ans reste inférieure à 50% chez les
adultes. La médiane de survie chez les plus de 65 ans est inférieure à 1 an et seulement 20%
de ces patients survivent 2 ans (Shipley et al, 2006).
Parmi les LAM, la LAM promyélocytaire (LAM-M3) constitue une entité particulière
caractérisée au niveau moléculaire par un réarrangement du récepteur α de l’acide rétinoïque
(RARA). L’utilisation du ligand de ce récepteur, l’acide tout-trans rétinoïque (ATRA),
comme thérapie ciblée, et sa combinaison à une chimiothérapie par idarubicine et aracytine
depuis les années 1990 a permis d’obtenir une survie à 5 ans proche de 75% (Wang et al,
2008).
47
Classification des LAM
a) Classification cytogénétique des LAM
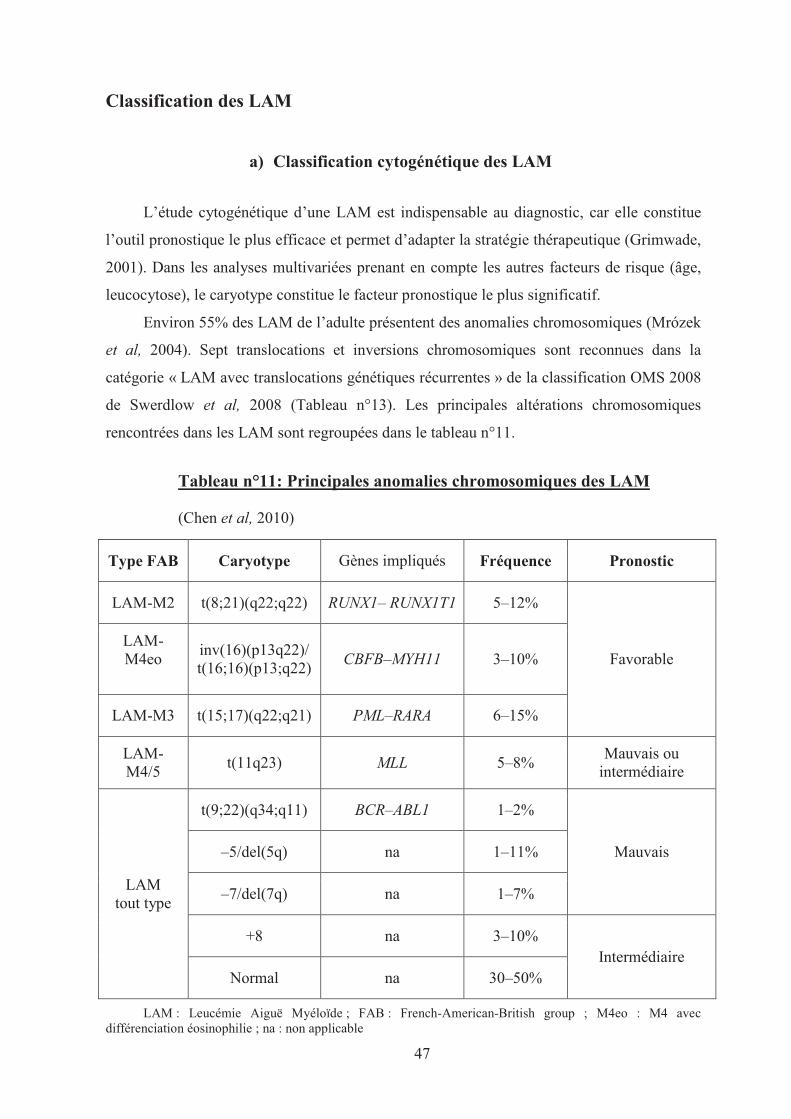
L’étude cytogénétique d’une LAM est indispensable au diagnostic, car elle constitue
l’outil pronostique le plus efficace et permet d’adapter la stratégie thérapeutique (Grimwade,
2001). Dans les analyses multivariées prenant en compte les autres facteurs de risque (âge,
leucocytose), le caryotype constitue le facteur pronostique le plus significatif.
Environ 55% des LAM de l’adulte présentent des anomalies chromosomiques (Mrózek
et al, 2004). Sept translocations et inversions chromosomiques sont reconnues dans la
catégorie « LAM avec translocations génétiques récurrentes » de la classification OMS 2008
de Swerdlow et al, 2008 (Tableau n°13). Les principales altérations chromosomiques
rencontrées dans les LAM sont regroupées dans le tableau n°11.
Tableau n°11: Principales anomalies chromosomiques des LAM
(Chen et al, 2010)
Type FAB Caryotype Gènes impliqués Fréquence Pronostic
LAM-M2 t(8;21)(q22;q22) RUNX1– RUNX1T1 5–12%
Favorable LAM-M4eo
inv(16)(p13q22)/ t(16;16)(p13;q22)
CBFB–MYH11 3–10%
LAM-M3 t(15;17)(q22;q21) PML–RARA 6–15%
LAM-M4/5
t(11q23) MLL 5–8% Mauvais ou
intermédiaire
LAM tout type
t(9;22)(q34;q11) BCR–ABL1 1–2%
Mauvais –5/del(5q) na 1–11%
–7/del(7q) na 1–7%
+8 na 3–10% Intermédiaire
Normal na 30–50%
LAM : Leucémie Aiguë Myéloïde ; FAB : French-American-British group ; M4eo : M4 avec différenciation éosinophilie ; na : non applicable

48
Les anomalies cytogénétiques sont classées en trois groupes selon leur valeur
pronostique en termes de survie globale et de risque de rechute : bon pronostic, pronostic
intermédiaire et mauvais pronostic. La faible fréquence de certaines anomalies (�10%) rend
impossible actuellement l’évaluation de leur valeur pronostique (Grimwade et al, 2009).
Les principales anomalies chromosomiques validées par le groupe Medical Research
Council (MRC), analysées sur 5635 patients de 16 à 59 ans avant mise sous traitement, sont
regroupées dans le tableau n°12 (Grimwade et al, 2010).
Le groupe de bon pronostic regroupe les patients avec t(15 ;17), t(8;21) et inv(16) ou
t(16 ;16).
Le groupe de mauvais pronostic comprend les caryotypes complexes (définis par au
moins 3 anomalies cytogénétiques) et les anomalies suivantes : -5/del(5q), -7/del(7q),
t(9;22), inv(3q) et les translocations impliquant la région 11q23 exceptée la t(9 ;11).
Les caryotypes complexes (� 3 anomalies) sont présents chez 10 à 12% des patients et sont
toujours associés à un très mauvais pronostic de survie (Mrózek et al, 2008).
Enfin, le groupe de pronostic intermédiaire rassemble les caryotypes normaux ainsi
que toutes les autres anomalies.
Tableau n°12 : Valeur pronostique des anomalies cytogénétiques des LAM
(Grimwade et al, 2010)
Valeur Pronostique Nombre de patient (n=5635)
Caryotype / anomalie chromosomique
Bon pronostic
t(15 ;17)
t(8 ;21)
inv(16)/t(16 ;16)
Mauvais pronostic
Complexe (�3 anomalies)
-5/del(5q)
-7/del(7q)
t(9;22)
inv(3q)
11q23 sauf t(9;11)
Pronostic intermédiaire
Normal
+6 ; +8 ; -Y ; t(9;11)
et toutes les autres anomalies

49
Tableau n°13 : Classification OMS des LAM
(Swerdlow et al, 2008)
LAM avec translocations génétiques récurrentes
LAM avec translocation t(8;21)(q22;q22); RUNX1-RUNX1T1
LAM avec inv(16)(p13q22) ou t(16;16)(p13;q22); CBFB-MYH11
LAM (promyélocytaire) avec translocation t(15;17)(q22;q21); PML-RARA
LAM avec translocation t(9;11)(p22;q23); MLLT3-MLL
LAM avec translocation t(6;9)(p23;q34); DEK-NUP214
LAM avec inv(3)(q21q26) ou t(3;3)(q21;q26); RPN1-EVI1
LAM (mégacaryoblastique) avec translocation t(1;22)(p13;q13); RBM15-MKL1
Entités provisoires : LAM avec mutation de FLT3
LAM avec mutation de NPM1
LAM avec mutation de CEBPA
LAM avec dysplasie multilignée
LAM et SMD thérapie-induits(therapy-related)
Alkylants
Inhibiteurs de topoisomérase II
Autres
Autres LAM
LAM indifférenciée
LAM sans maturation
LAM avec maturation
LAM myélo-monocytaire
LAM monoblastique
LAM érythrocytaire
LAM mégacaryoblastique
LAM basophile
LA panmyéloïde avec myélofibrose
Sarcome myéloïde
Proliférations myéloïdes liées au Syndrome de Down
Leucémie à cellules dendritiques blastiques plasmocytoïdes

50
b) Classification moléculaire des LAM
(1) Les enjeux d’une classification moléculaire
Les anomalies cytogénétiques constituent le principal élément pronostique des
LAM. Cependant, 50% à 60% des LAM ont un caryotype de pronostic intermédiaire
(Mrózek et al, 2004). Ce groupe rassemble des LAM hétérogènes au niveau de leur
présentation clinique et de leur pronostic de survie. Leurs anomalies moléculaires
demeurent en grande partie inconnues (Estey et al, 2006).
Le pronostic de survie des patients avec LAM à caryotype de pronostic
intermédiaire est difficile à évaluer. Des études récentes ont révélé plusieurs altérations
moléculaires permettant de mieux classer les patients et d’adapter plus précocement et
efficacement leur traitement (Schlenk et al, 2008). Récemment, les gènes TET2, ASXL1,
IDH1 et IDH2 sont venus s’ajouter aux gènes dont les mutations ont été décrites dans les
LAM tels que FLT3, NPM1, CEBP�, MLL, RUNX1, TP53, K/N-RAS, KIT et WT1.
(2) FLT3, NPM1 et CEBPA
Actuellement, l’OMS recommande l’analyse de trois gènes pour les LAM à
caryotype intermédiaire au diagnostic (Vardiman et al, 2009) : le gène FLT3, codant pour
un récepteur à activité tyrosine kinase (RTK), le gène NPM1 codant pour une protéine
multifonctionnelle régulant la transcription et le gène CEBP� codant pour un facteur de
transcription.
(a) FLT3
FLT3 (FMS-like tyrosine kinase 3), localisé en 13q12, est le gène le plus fréquemment
muté toutes LAM confondues (30 à 40%) et 30 à 35% des LAM à caryotype intermédiare
sont mutées (Levis et al, 2003). FLT3 code pour un RTK exprimé à la surface des
progéniteurs hématopoïétiques (Rosnet et al, 1996) qui joue un rôle important dans la
prolifération, la survie et la différenciation des CSH (Gilliland et al, 2002).
Il existe deux types de mutations de FLT3 (Mead et al, 2007) : La plus commune est
une duplication en tandem (ITD, Internal Tandem Duplication) dans les exons 14-15 à

51
l’origine de l’activation constitutive de la protéine FLT3 et de la prolifération incontrôlée des
CSH. Présente dans 15 à 30% des LAM à caryotype intermédiaire, cette mutation est
associée à un mauvais pronostic (Thiede et al, 2002). L’autre mutation de FLT3 est une
mutation ponctuelle située dans l’exon 20 du gène (codon 835), qui conduit au
remplacement d’un acide aspartique dans la boucle d’activation du domaine tyrosine kinase
(TKD). Elle est retrouvée dans approximativement 5 à 10% des LAM (10-15 % des LAM à
caryotype intermédiaire). La mutation TKD de FLT3 serait plutôt également de mauvais
pronostic (Yanada et al, 2005), mais son impact sur la survie n’est pas encore clairement
établi.
(b) NPM1
Les mutations du gène de la nucléophosmine NPM1, situé en 5q35, sont les mutations
les plus fréquemment décrites dans les LAM à caryotype intermédiaire (50 à 60%), et sont
retrouvées dans environ 25% des cas, toutes LAM confondues (Verhaak et al, 2005).
Le gène NPM1 est également impliqué dans des translocations chromosomiques dans
plusieurs hémopathies malignes. Il code pour une protéine nucléolaire hautement conservée
aux multiples fonctions dont notamment celle de protéine chaperone naviguant entre le
nucléole et le cytoplasme (Grisendi et al, 2006). Les mutations de NPM1 (insertions
localisées dans l’exon 12) provoquent une perte de fonction due à la délocalisation de la
protéine dans le cytoplasme (Falini et al, 2005). Elles sont rares dans les LAM secondaires et
sont associées à un bon pronostic dans les LAM à caryotype intermédiaire en l’absence de
mutation ITD de FLT3 (Döhner et al, 2005).
(c) CEBP�
CEBP� (CCAAT/enhancer binding protein �) est un facteur de transcription jouant un
rôle important dans la différenciation granulocytaire (Nerlov, 2004). Les mutations du gène
CEBP�, localisé en 19q13, induisent des troubles de la différenciation granulocytaire. Elles
ont été décrites à une fréquence de 8 à 15% dans les LAM et sont retrouvées dans 15 à 20%
des LAM intermédiaires (Pabst et al, 2001), majoritairement des LAM peu différenciées,
LAM-M1 et M2 de la classification FAB (Preudhomme et al, 2002).
Les mutations ont été associées à un pronostic favorable (Fröhling et al, 2004) mais il
a été montré récemment que seules les mutations homozygotes de CEBP� (2 allèles mutés)
constituaient un marqueur indépendant de bon pronostic dans les LAM, ce sous-groupe est

52
moins fréquent (environ 5%) (Pabst et al, 2009 ; Dufour et al, 2010). Les mutations de
NPM1 et les mutations homozygotes de CEBP� sont exclusives et constituent 2 groupes
distincts de bon pronostic dans les LAM à caryotype intermédiaire.
Les combinaisons des mutations de FLT3, NPM1 et CEBP� dans les LAM sont
reportées dans la figure n°2.
Figure n°2 : Screening moléculaire des LAM
(Renneville et al, 2008)

53
(3) Les autres gènes mutés dans les LAM
(a) MLL
Le gène MLL (Mixed Lineage Leukemia) localisé en 11q23, est fréquemment impliqué
dans des translocations chromosomiques dans les leucémies aiguës aussi bien myéloïdes que
lymphoïdes et possède de très nombreux gènes partenaires (Liu et al, 2009). MLL code pour
une histone méthyltransférase et ses mutations sont surtout retrouvées, à une fréquence de 5
à 11 %, dans les LAM à caryotype intermédiaire (Döhner et al, 2002).
Les mutations de MLL ont été les premières montrant un impact pronostique dans les
LAM à caryotype intermédiaire (Caligiuri et al, 1998). Elles sont associées dans 30 à 40%
des cas aux mutations ITD de FLT3 et sont considérées comme de mauvais pronostic
(Schnittger et al, 2000).
(b) RUNX1/AML1/CBFA
Le gène RUNX1, localisé en 21q22, constitue un des gènes les plus réarrangé dans les
LAM, par différents mécanismes : translocations chromosomiques, amplification et
mutations ponctuelles (Lutterbach et al, 2000).
6 à 13% des LAM de novo (environ 15% des LAM à caryotype intermédiaire) et
environ 40% des LAM secondaires à des chimiothérapies sont concernées par des mutations
ponctuelles de RUNX1 (Osato, 2004 ; Harada et al, 2003). Les LAM les moins différenciées
(LAM-M0) sont les plus affectées avec une fréquence de 15 à 50% et présentent souvent des
mutations bi-alléliques avec perte totale d’expression de RUNX1 (Preudhomme et al, 2000).
Tang et al ont montré récemment que les mutations de RUNX1 constituaient un
marqueur indépendant de mauvais pronostic dans les LAM de novo (Tang et al, 2009)
(c) TP53
Le gène TP53, localisé en 17p13, est un gène suppresseur de tumeurs. Il est muté dans
10% des LAM de novo et dans 30% des hémopathies myéloïdes induites pas les
chimothérapies (Haferlach et al, 2008 ; Christiansen et al, 2001). Ses mutations ont été
décrites dans de nombreuses tumeurs solides dans lesquelles elles provoquent une instabilité
génomique, une perte de l’apoptose cellulaire et causent une résistance aux chimiothérapies
anticancéreuses (Wattel et al, 1994). Les mutations de TP53 sont classiquement associées à
des caryotypes complexes et à un pronostic de survie très sombre (Bowen et al, 2009).

54
(d) NRAS
Deux grandes études ont montré que NRAS, localisé en 1p13, était muté dans 10 à 13%
des LAM (8-10% des LAM à caryotype intermédiaire). En revanche, ces mutations n’ont pas
montré clairement d’impact pronostique sur la survie des patients (Bowen et al, 2005 ;
Bacher et al, 2006).
(e) Autres gènes mutés
WT1 (Wilms’s tumor suppressor 1), localisé en 11p13, est un régulateur
transcriptionnel de gènes impliqués dans la croissance et le métabolisme de nombreux types
cellulaire (Scharnhorst et al, 2001). Les mutations du gène WT1 ont été décrites pour la
première fois en 1996 à une fréquence de 10 à 15% dans les LAM-CN (King-Underwood et
al, 1996), WT1 est également fréquemment surexprimé dans les LAM (Yang et al, 2007).
Les mutations de WT1 auraient un impact pronostique péjoratif (rechutes plus fréquentes,
survie globale diminuée) qui doit être confirmé dans de grandes études (Gaidzik V et al,
2009 ; Renneville et al, 2009) Une des raisons pourrait être leur association statistiquement
élevée aux mutations ITD de FTL3 (Becker et al, 2010).
Le proto-oncogène KIT, localisé en 4q12, code pour un RTK du stem cell factor (SCF)
et contrôle, particulièrement dans les progéniteurs myéloïdes, les voies de signalisation de la
prolifération, différenciation, migration et survie cellulaire (Lennartsson et al, 2005). Les
mutations de KIT ont été rapportées dans les LAM mais aussi dans d’autres cancers,
notamment dans les mastocytoses et les GISTs (GastroIntestinal Stroma Tumors). Elles sont
hautement spécifiques des LAM-M2 de la classification FAB : 70% des LAM mutées KIT
sont des LAM-M2. On les retrouve également dans les LAM-M1 et M4 mais rarement dans
les autres types de LAM. Elles sont souvent associées à la translocation t(8;21) et à
l’inversion du 16 qui impliquent les gènes CBFα et β, sous-unités du complexe
transcriptionnel CBF (Core Binding Factor). Ces anomalies perdent leur bon pronostic dans
les LAM avec mutation de KIT (Beghini et al, 2004, Boissel et al, 2006, Paschka et al,
2006).
D’autres gènes sont plus rarement mutés dans les LAM, comme une phosphatase
PTPN11 à raison de 2 à 4% des LAM (Hugues et al, 2005) et une autre tyrosine kinase,
JAK2, dans 1 à 2% des LAM de novo (Illmer et al, 2007). Des mutations du gène PU.1,

55
codant pour un facteur de transcription impliqué dans l’hématopoïèse (régulation des
récepteurs du granulocyte colony-stimulating factor (G-CSF), du M-CSF et du GM-CSF) ont
également été observées dans environ 3% des LAM (Mueller et al, 2002).
(4) Nouveaux gènes mutés dans les LAM
D’autres gènes présentant des mutations dans le LAM ont été récemment identifiés.
C’est le cas des gènes TET2, ASXL1, IDH1 et IDH2.
(a) TET2
Les mutations de TET2 ont été décrites dans 12 à 20 % des LAM de novo (Abdel-
Wahab et al, 2009 ; Rocquain et al, 2010 ; Nibourel et al, 2010). D’après une étude sur 111
LAM de novo, il n’y avait aucune différence clinique entre patients mutés et non mutés et les
mutations de TET2 ne constitueraient pas un facteur pronostique indépendant (Nibourel et al,
2010). Cette donnée doit être confirmée sur de plus larges cohortes de patients.
(b) ASXL1
Dans le laboratoire de D. Birnbaum, nous avons montré qu’ASXL1 était muté dans 11
LAM sur 63 (17,5%) (Carbuccia et al, 2010). Chou et al, ont récemment identifié 54 patients
mutés sur le gène ASXL1 sur une grande cohorte de patients atteints de LAM de novo
(n=501), soit une fréquence de 10,5% (Chou et al, 2010a). Les mutations étaient aussi
fréquentes dans les LAM à caryotype anormal que normal (12,9 vs. 8,9% ; p=0,179).
Les patients mutés ASXL1 présentent une survie globale moins élevée que les patients
non mutés, or les mutations d’ASXL1 ne constituaient pas un facteur indépendant de mauvais
pronostic dans l’étude de Chou et al. Elles étaient associées à un âge élevé ainsi qu’aux
mutations de RUNX1, qui constituent deux facteurs indépendants de mauvais pronostic
(Tang et al, 2009 ; Juliusson et al, 2009). Nous avons montré récemment que les mutations
de NPM1 et d’ASXL1 étaient mutuellement exclusives (cf. p 64 ; Carbuccia et al, 2010).

56
(c) IDH1 et 2
La prévalence des mutations d’IDH1 et 2 dans les LAM de novo semble similaire à
celle retrouvée dans les SMD, de l’ordre de 5 à 9% (Mardis et al, 2009 ; Paschka et al,
2010 ; Abbas et al, 2010 ; Marcucci et al, 2010) et d’environ 10% dans les LAM-CN.
Pas moins d’une quinzaine d’études ont été publiée en 2010 sur l’étude des mutations
d’IDH1 et 2 dans les LAM.
Les mutations d’IDH1/2 sont étroitement associées à un caryotype normal et à la
présence de mutations du gène NPM1 sans mutation ITD de FLT3 (Paschka et al, 2010 ;
Abbas et al, 2010 ; Marcucci et al, 2010). Paradoxalement, les mutations d’IDH sont
associées à un pronostic plutôt défavorable. Elles pourraient donc permettre de réajuster la
valeur pronostique des patients mutés NPM1 sans mutation ITD de FLT3 (Paschka et al,
2010).
La nouvelle mutation IDH2R140Q (Green et al, 2010) n’aurait pas de valeur
pronostique, seules les mutations IDH1R132 et IDH2R132 constitueraient un facteur
pronostique indépendant (Marcucci et al, 2010 ; Boissel et al, 2010). Une grande étude sur
1414 patients a montré récemment que la mutation IDH1R132 constituerait, également en
l’absence de mutation de NPM1, un facteur indépendant de mauvais pronostic chez les
patients de moins de 60 ans (Schnittger et al, 2010).

57
Synthèse des anomalies moléculaires des SMD et LAM-CN
Les anomalies moléculaires dans les SMD et LAM sont nombreuses. Cette diversité
est la résultante probable de la dérégulation des gènes impliqués dans la réparation de l’ADN
et dans la maintenance de l’intégrité génomique (Suela et al, 2007). L’hypothèse d’un
mécanisme de leucémogenèse en plusieurs étapes est en accord avec ce phénomène.
En 2001, Gilliland a proposé une association de deux types d’altérations moléculaires
comme modèle de leucémogenése (Gilliland et al, 2002) :
- Les mutations de classe I ont pour conséquence l’activation constitutive de tyrosine
kinases (TK) ou de leurs effecteurs, à l’origine d’une prolifération et d’un gain de survie des
progéniteurs hématopoïétiques. Lorsque ces mutations de classe I sont isolées, elles sont à
l’origine de syndromes LMC-like caractérisés par une leucocytose importante avec une
conservation de maturation et des fonctions des cellules myéloïdes.
- Les mutations de classe II provoquent la perte de fonction de facteurs de transcription
importants dans l’hématopoïèse. Ces mutations provoquent la perte des capacités de
différenciation des cellules myéloïdes. Lorsque les mutations de classe II sont isolées, elles
peuvent être à l’origine de signes cliniques retrouvés dans les SMD.
Chaque classe de mutations ne peut à elle seule être à l’origine d’une LAM. Ce sont
les patients affectés simultanément par des mutations de chacune des 2 classes (I et II) qui
développeraient un phénotype de LAM, caractérisé par une prolifération incontrôlée des
cellules myéloïdes et un blocage de la différenciation des CSH.

58
Figure n°3 : Modèle de leucémogenése à 2 étapes selon Gilliland
(Kelly and Gilliland, 2002)
Le modèle de Gilliland pose comme hypothèse que les LAM sont la conséquence de la combinaison de deux classes de mutations.
CML: Chronic myeloid leukemia; AML: Acute Myeloid Leukemia; MDS: Myelodysplastic Syndrome
Les anomalies moléculaires présentées dans cette thèse sont regroupées dans les
tableaux n°14, 15 et 16.
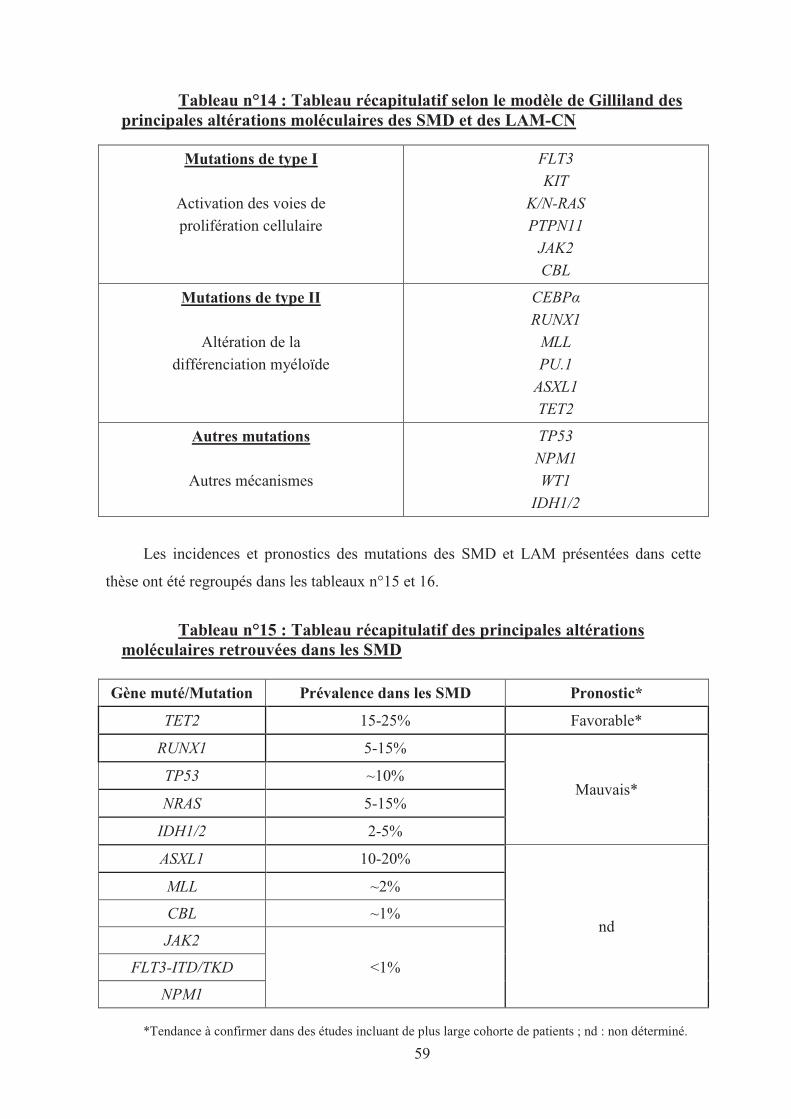
59
Tableau n°14 : Tableau récapitulatif selon le modèle de Gilliland des principales altérations moléculaires des SMD et des LAM-CN
Mutations de type I
Activation des voies de
prolifération cellulaire
FLT3
KIT
K/N-RAS
PTPN11
JAK2
CBL
Mutations de type II
Altération de la
différenciation myéloïde
CEBP�
RUNX1
MLL
PU.1
ASXL1
TET2
Autres mutations
Autres mécanismes
TP53
NPM1
WT1
IDH1/2
Les incidences et pronostics des mutations des SMD et LAM présentées dans cette
thèse ont été regroupés dans les tableaux n°15 et 16.
Tableau n°15 : Tableau récapitulatif des principales altérations moléculaires retrouvées dans les SMD
Gène muté/Mutation Prévalence dans les SMD Pronostic*
TET2 15-25% Favorable*
RUNX1 5-15%
Mauvais* TP53 ~10%
NRAS 5-15%
IDH1/2 2-5%
ASXL1 10-20%
nd
MLL ~2%
CBL ~1%
JAK2
<1% FLT3-ITD/TKD
NPM1
*Tendance à confirmer dans des études incluant de plus large cohorte de patients ; nd : non déterminé.
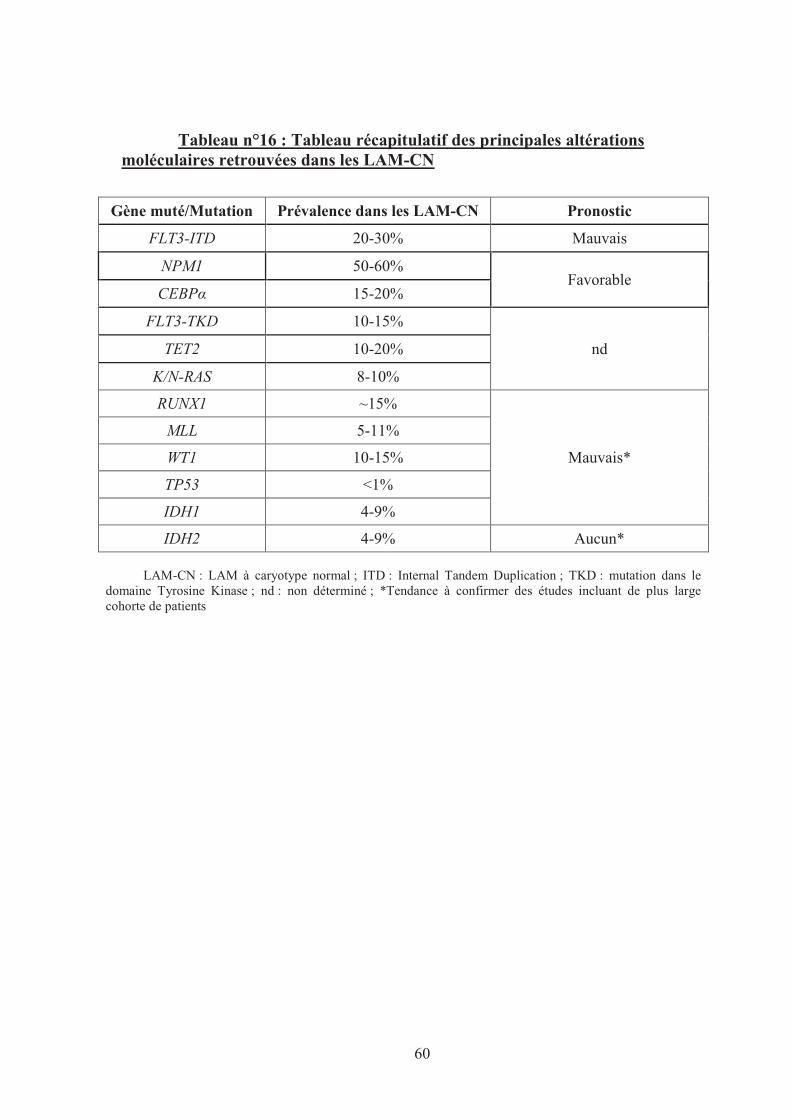
60
Tableau n°16 : Tableau récapitulatif des principales altérations moléculaires retrouvées dans les LAM-CN
Gène muté/Mutation Prévalence dans les LAM-CN Pronostic
FLT3-ITD 20-30% Mauvais
NPM1 50-60% Favorable
CEBP� 15-20%
FLT3-TKD 10-15%
nd TET2 10-20%
K/N-RAS 8-10%
RUNX1 ~15%
Mauvais*
MLL 5-11%
WT1 10-15%
TP53 <1%
IDH1 4-9%
IDH2 4-9% Aucun*
LAM-CN : LAM à caryotype normal ; ITD : Internal Tandem Duplication ; TKD : mutation dans le domaine Tyrosine Kinase ; nd : non déterminé ; *Tendance à confirmer des études incluant de plus large cohorte de patients

61
II. PRESENTATION DES TRAVAUX ET RESULTATS
Fondements scientifiques du sujet de thèse
La leucémogenèse est un processus complexe qui transforme une cellule souche
hématopoïétique en un clone cellulaire incapable de se différencier et dont la prolifération
est devenue incontrôlée. Plusieurs anomalies génétiques sont impliquées, notamment des
translocations chromosomiques, ainsi que des délétions, des amplifications et des mutations
de gènes. Les translocations chromosomiques générant des gènes de fusion jouent un rôle
important dans la genèse de certaines LAM. Cependant, elles sont rares dans les SMD et
plus de la moitié des LAM en sont dépourvues.
Les SMD se transforment dans près de 40% des cas en LAM. Les altérations
moléculaires provoquant l’acutisation sont globalement inconnues et actuellement, il est
difficile de prédire l’évolution clinique de ces hémopathies. La classification OMS permet de
classer les SMD et LAM selon leur pronostic clinique. Les scores IPSS et WPSS sont d’une
aide précieuse pour évaluer les SMD au diagnostic et lors de leur évolution mais ils sont
encore insuffisants pour que le clinicien puisse adapter précocement le traitement des
malades à haut risque de progression en LAM. La prise en charge des SMD reste de ce fait
majoritairement symptomatique.
Les anomalies cytogénétiques constituent actuellement le principal élément
pronostique des SMD et LAM, permettant de les classer en 3 sous-groupes : favorable,
intermédiaire et défavorable. Néanmoins, ces groupes rassemblent des cas d’évolution
clinique hétérogène.
L’évaluation du pronostic de survie et la stratégie thérapeutique optimale, notamment des
patients sans anomalies cytogénétiques au diagnostic, demeure un challenge. Récemment,
plusieurs altérations moléculaires (mutations de FLT3, CEBP� et NPM1) ont été identifiées

62
permettant de mieux classer les patients et d’adapter plus précocement et efficacement les
traitements des patients atteints de LAM à caryotype normal.
Néanmoins, de nombreuses LAM notamment à caryotype normal sont dépourvues de
marqueurs efficaces pour adapter la stratégie thérapeutique et des efforts dans la recherche
de nouveaux marqueurs doivent être poursuivis pour trouver de nouvelles altérations
moléculaires et confirmer les résultats obtenus dans les études précédentes.
Le travail de cette thèse a pour objectif l’étude des anomalies moléculaires rencontrées
dans les SMD et LAM. Grâce à l’apport des techniques d’analyse génomique à grande
échelle, notamment la CGH-array, notre laboratoire identifie de nouvelles altérations
génétiques qui pourraient expliquer les phénotypes cliniques hétérogènes de ces hémopathies
myéloïdes, dans l’optique d’optimiser secondairement les stratégies thérapeutiques de ces
maladies.

63
ARTICLE N°1
Mutual exclusion of ASXL1 and NPM1 mutations in a series
of acute myeloid leukemias.
Carbuccia N*, Trouplin V*, Gelsi-Boyer V, Murati A, Rocquain J,
Adélaïde J, Olschwang S, Xerri L, Vey N, Chaffanet M, Birnbaum D,
Mozziconacci MJ.
Leukemia 2010; 24(2): 469-73
* 1ers auteurs

5 Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MFet al. Nucleophosmin gene mutations are predictors of favorableprognosis in acute myelogeneous leukaemia with a normalkaryotype. Blood 2005; 106: 3733–3739.
6 Meloni G, Mancini M, Gianfelici V, Martelli MP, Foa R, Falini B.Late relapse of acute myeloid leukaemia with mutated NPM1 aftereight years: evidence of NPM1 mutation stability. Haematologica2009; 94: 298–300.
7 Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, ChaturvediA et al. Compassionate-use of sorafenib in Flt3-ITD positive acutemyeloid leukemia: sustained regression prior and post-allogeneicstem cell transplantation. Blood 2009; 113: 6567–6571.
8 Palmisano M, Grafone T, Ottaviani E, Testoni N, Baccarani M,Martineeli G. NPM1 mutations are more stable than FLT3 mutationsduring the course of disease in patients with acute myeloidleukaemia. Haematologica 2007; 92: 1268–1269.
Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias
Leukemia (2010) 24, 469–473; doi:10.1038/leu.2009.218;published online 29 October 2009
The majority of adult acute myeloid leukemias (AMLs)have normal cytogenetics and lack a balanced translocationwith a known fusion gene. They occur de novo or after a chronicphase (hereafter called primary and secondary, respectively).They are poorly understood and heterogeneous in termsof bioclinical presentation and prognosis, as well as in termsof molecular targets. The identification of mutations inseveral genes, such as NPM1 and FLT3, has revealed prognosissubgroups and modified their clinical management.1 A bettermolecular characterization should further improve both theunderstanding of the physiopathology and the treatment of theseleukemias.The additional sex comb-like 1 (ASXL1) gene, located in the
chromosomal region 20q11, belongs to a family of three identifiedmembers that encode poorly characterized proteins regulatingchromatin remodeling. The ASXL proteins contain a C-terminalPHD (Plant Homeo Domain) finger and belong to the polycomband trithorax complexes, which regulate the genetic program ofstem cells. We have recently shown that ASXL1 is mutated inroughly 10% of myelodysplastic syndromes,2 10% of myelo-proliferative neoplasms3 and 40% of chronic myelomonocyticleukemias.2 Using array comparative genomic hybridization(aCGH), we also observed a deletion of the ASXL1 locus in amyelodysplastic syndrome case.2 These observations suggest thatASXL1 is a tumor suppressor whose haploinsufficiency has a role inmyeloid diseases. The predicted truncated ASXL1 protein wouldlack its PHD finger, which could prevent it from bindingmethylated histone lysines and interacting with chromatin modi-fiers, thus enhancing self-renewal in hematopoietic progenitors.4
To determine whether ASXL1 is involved in AML, we usedDNA sequencing and aCGH to search for mutations anddeletions of the gene in 46 cases of AML with normal karyotypeand 17 cases with trisomy 8 (n¼ 14), 9q deletion (HD-0632),trisomy 11 (HD-0304) or 20q11–13 deletion (HD-0381) as asole karyotypic abnormality. The 63 AMLs were 46 primarycases and 17 transformations of a previous myeloid disease(of which seven have been previously reported2). We did notinclude therapy-related AMLs. All patients signed an informedconsent and the study was approved by our institutional reviewboard. We have previously described primers and conditions forASXL1 DNA amplification and sequencing.2 Fifty cases werealso studied by aCGH with high-density oligonucleotide arrays(Hu-244A, Agilent Technologies, Massy, France), as previouslydescribed.5 We also searched for mutations in CEBPA (exons1–8), FLT3 (internal tandem duplicationsFITDsFand pointmutation in exon 20-encoded kinase domainFTKD), JAK2
(exon 14), K and NRAS (exons 1 and 2), and NPM1 (exon 12), aswell as in CXXC5, FES and TAL1, three genes found altered bygain or loss in aCGH analysis.
In all, 41 out of the 50 cases studied by aCGH did not showany copy number aberration (isolated trisomy 8 was not takeninto account). The deletion of chromosomal region 20q11–13(HD-0381), which was visible on the karyotype, involved ASXL1(Table 1). We found heterozygous nonsense or frameshiftmutations of ASXL1 in 11 out of the 63 cases (B17.5%)(Table 1). We also found several cases of substitution, whichwe did not take into account because they may representpolymorphisms. Thus, in total, 12 cases out of 63 showed ASXL1alteration (19%). In two cases, we sequenced ASXL1 in DNAextracted from buccal smears of the patient with ASXL1mutation; ASXL1 was not mutated, showing that the mutationwas acquired.
In agreement with what is known for AML with normalkaryotype,5 almost half of the cases (28 out of 63, 44%) showedexon 12 NPM1 mutations. NPM1 mutations were mutuallyexclusive with ASXL1 alterations: none of the 28 NPM1-mutatedcases showed ASXL1 mutation or deletion, whereas 12 out ofthe 35 (34%) non-NPM1-mutated cases were mutated or deletedfor ASXL1 (P¼ 0.0018). Three ASXL1 missense mutations, forwhich we do not know whether they are acquired or not, werealso detected in cases without NPM1 mutations. FLT3 mutations(n¼ 6) and ITD (n¼ 14) were found in 19 cases (30%) andwere not observed with ASXL1 mutations except in one case(HD-0282). Three cases were mutated in either K or NRAS, andone case in JAK2. Data on CEBPA, available for 11 cases,revealed no mutation. Finally, we did not find any mutation ofCXXC5, FES and TAL1 genes.
Several tumor suppressors (for example, RB1, p53, RUNX1,5
TET2,2,3,6–13 ASXL12,3) are inactivated in various types ofmyeloid diseases. With respect to ASXL family members,deletions of ASXL1, ASXL2 and ASXL3 have been reported.2,11
Mutations of ASXL1 are found in myelodysplastic syndromes2
and myeloproliferative neoplasms,3 suggesting that additionalalterations are responsible for acute transformation; as the AMLsstudied by us were not associated with translocations, thesealterations are not well-known fusion genes but other mutations.They are not mutations of NPM1 as these were mutuallyexclusive with ASXL1 mutations. This could suggest that theNPM1 and ASXL1 mutations are epistatic. We could speculatethat ASXL1 and NPM1 have distinct functions but act at somemoment on the same cellular control. One such control couldbe the regulation of 9p INK4A–ARF–INK4B loci, whichconstitute an important node for the regulation of senescenceand self-renewal and are regulated by polycomb complexes aswell as NPM1, a histone chaperone.1 ASXL1 could have a rolein this regulation through its interaction with polycomb proteins.
Letters to the Editor
469
Leukemia

Tab
le1
MutationsofASX
L1,FLT3,NPM1an
dRASin
aseries
ofAMLs
Number
Sex/age
(years)
FAB
subtype
Secondary
toFLT
3ITD
FLT
3TKD
(D835)
NPM1
(exo
n12)
CEBPA
(exo
ns
1–8)
K,NRAS
(exo
ns1
and2)
JAK2
(V617F)
ASXL1a
(exo
n12)
Karyotype
aCGH
Genes
HD-0001
M/72
M5a
No
No
No
No
No
No
No
Trisomy8
Gain
3q26-qter
(162.7-terMb),
trisomy8
4100
HD-0003
M/70
M5a
No
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0008
M/66
M5a
CMML
No
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0009
F/68
M5b
No
No
Yes
No
na
No
No
Norm
al
NoCNA
HD-0010
M/73
M5b
No
No
Yes
No
No
No
No
Norm
al
NoCNA
HD-0011
F/47
M5b
Yes
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0012
M/21
M5a
No
Yes
No
na
No
No
No
Norm
al
NoCNA
HD-0016
M/78
M5a
No
Yes
No
na
No
No
No
Trisomy8
Trisomy8
HD-0019
M/67
M4
No
No
Yes
No
No
No
No
Norm
al
NoCNA
HD-0020
M/59
M4
No
No
No
na
No
No
c.3965C4G
p.Pro1322Arg
Norm
al
NoCNA
HD-0021
F/43
M4
No
Yes
Yes
No
No
No
No
Norm
al
NoCNA
HD-0024
M/61
M4
CMML
No
No
No
na
No
No
No
Norm
al
Loss7,
deletion8q24
(126.7–129.8
Mb)
3
HD-0026
F/39
M4
No
No
Yes
na
Kp.G
ly13Asp
No
No
Norm
al
NoCNA
HD-0027
M/62
M4
No
No
No
na
No
No
No
Norm
al
NoCNA
HD-0031
M/77
M5a
No
No
No
na
No
No
No
Norm
al
NoCNA
HD-0060
F/41
M5a
Yes
No
No
na
No
No
No
Trisomy8
Trisomy8
HD-0097
F/55
M4
No
Yes
No
na
No
No
No
Norm
al
na
HD-0098
M/77
M4
MDS
No
No
No
No
No
na
c.3306G4
Tp.G
lu1102Asp
Trisomy8
Gain
1p32.3-p35
(31.7–53.15Mb),
trisomy8,
lossXp11.3
4100(TAL1b)
UTX
HD-0099
F/61
M2
No
No
No
na
Np.G
ly12Asp
na
c.1934dupG
p.G
ly646TrpfsX12
Trisomy8
Trisomy8
HD-0100
M/78
M2
No
No
No
na
No
No
No
Trisomy8
Trisomy8
HD-0101
M/29
M6
No
No
No
na
No
No
No
Trisomy8
na
HD-0102
F/45
M4
No
No
Yes
No
No
No
No
Trisomy8
Trisomy8
HD-0103
M/60
na
PMF
No
No
No
na
No
No
c.1934dupG
p.G
ly646TrpfsX12
Trisomy8
na
HD-0104
M/79
M2
No
No
No
na
No
No
c.1934dupG
p.G
ly646TrpfsX12
(nc)
Norm
al
Loss
13q12.11
(19.6–20.0
Mb)
2(GJB6,
CRYL1)
HD-0105
F/68
M4
CMML
No
No
No
na
No
No
No
Norm
al
NoCNA
HD-0106
F/59
M5a
Yes
No
No
na
No
No
No
Norm
al
NoCNA
HD-0107
M/75
M4
No
No
Yes
No
No
No
No
Norm
al
NoCNA
HD-0108
M/46
M1
Yes
No
Yes
na
No
No
No
Norm
al
Gain
15q25.2-qter
(81.5-terMb)
4100(FESb)
HD-0109
M/60
M1
No
No
No
na
No
No
No
Norm
al
NoCNA
HD-0110
F/40
M4
No
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0111
M/71
M4
MDS
No
No
No
na
No
No
c.1900_1922del
p.G
lu635ArgfsX15
Norm
al
Loss5q31.2
(137.3–139.5
Mb)
15
(CXXC5b)
HD-0112
F/44
M1
No
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0113
F/49
M5b
Yes
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0114
F/69
na
Yes
No
Yes
na
No
No
No
Trisomy8
Trisomy8
HD-0115
M/71
M2
Yes
No
Yes
na
No
No
No
Trisomy8
Trisomy8
HD-0116
F/49
na
No
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0117
F/74
M2
Yes
No
Yes
na
No
No
c.2431A4G
p.Asn811Asp
Norm
al
NoCNA
HD-0118
M/69
M5b
No
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0119
M/57
M2
No
No
Yes
na
No
No
No
Norm
al
NoCNA
Letters to the Editor
470
Leukemia

Tab
le1(Continued
)
Number
Sex/age
(years)
FAB
subtype
Secondary
toFLT
3ITD
FLT3TKD
(D835)
NPM1
(exo
n12)
CEBPA
(exo
ns
1–8)
K,NRAS
(exo
ns1
and2)
JAK2
(V617F)
ASXL1a
(exo
n12)
Karyotype
aCGH
Genes
HD-0120
M/53
M6
No
No
No
No
No
No
No
Norm
al
LossXq25
(122.9–122.7
Mb)
STAG2
HD-0121
F/41
M1
Yes
No
Yes
na
No
No
No
Norm
al
NoCNA
HD-0122
M/64
M2
No
No
No
na
No
No
c.4127G4A
p.G
ly1376Asp
Norm
al
NoCNA
HD-0123
M/83
M1
Yes
Yes
Yes
na
No
No
No
Norm
al
NoCNA
HD-0124
F/71
M5b
No
Yes
No
na
No
No
No
Norm
al
NoCNA
HD-0125
M/74
M4
CMML
No
No
No
na
No
No
c.1772dupA
p.Tyr591Stop
Norm
al
NoCNA
HD-0126
F/59
M5b
No
No
No
na
No
No
No
Norm
al
NoCNA
HD-0127
F/74
M4
CMML
No
No
No
na
No
No
c.2423delC
p.Pro808LeufsX10
Norm
al
NoCNA
HD-0128
M/77
M4or5
No
No
Yes
No
No
No
No
Norm
al
NoCNA
HD-0186
M/79
M2
MDS
No
No
No
na
No
No
c.1934dupG
p.G
ly646TrpfsX12
Norm
al
NoCNA
HD-0198
M/73
M4
CMML
No
No
No
na
No
No
c.1851dupT
p.Lys618Stop(nc)
Trisomy8
Trisomy8,
losses
21q21
USP16-
RUNX13
HD-0282
M/61
M5
CMML
Yes
No
No
na
No
No
c.1934dupG
p.G
ly646TrpfsX12
Norm
al
na
HD-0304
M/69
M2
MDS
No
No
No
na
No
na
c.1934dupG
p.G
ly646TrpfsX12
Trisomy11
na
HD-0379
F/70
M4
PV
No
No
No
na
No
No
No
Norm
al
NoCNA
HD-0381
F/73
M4
CMML
No
No
No
na
No
No
No
Deletion20q
Loss20q11-q13.33
(30.4–59.4
Mb)
4100
(ASXL1b)
HD-0392
F/61
M4
No
No
Yes
na
No
Yes
No
Norm
al
NoCNA
HD-0402
M/70
M5
CMML
Yes
No
Yes
na
No
No
No
Norm
al
na
HD-0489
F/60
na
No
No
No
No
No
No
c.2057dupA
p.Cys687ValfsX31
Trisomy8
na
HD-0620
F/71
M5
No
No
No
na
No
No
No
Norm
al
na
HD-0630
M/61
M5a
CMML
No
No
No
na
No
No
No
Norm
al
na
HD-0632
F/66
M4
No
No
Yes
na
No
No
No
Deletion9q
na
HD-0649
M/78
M5b
Yes
No
Yes
na
No
No
No
Norm
al
na
HD-0693
M/74
M5
Yes
No
Yes
na
No
No
No
Norm
al
na
HD-0702
M/82
M4
CMML
No
No
No
na
Np.G
ly12Asp
No
No
Trisomy8
na
Abbreviations:aCGH,arraycomparative
genomic
hyb
ridization;CNA,copynumberaberration;CMML,chronic
myelomonocytic
leukemia;F,
female;ITD,internaltandem
duplication;MDS,
myelodysplasticsyndrome;M,male;na,notavailable;PMF,
primary
myelofib
rosis;PV,
polycythemia
vera.
aForASXL1,onlyamino-acid
variants
are
listed(in
gray:
nonsenseandframeshift
mutations;nc:confirmedasnonconstitutional).
bNotmutated.
Letters to the Editor
471
Leukemia

However, by aCGH, we did not find any loss of INK4 loci in ourseries of AML cases.
Actually, the difference between NPM1 and ASXL1 mutationsmay rather reflect two different routes of leukemogenesis thantwo alternate hits on the same route (Figure 1). NPM1 mutationsare uncommon in AMLs secondary to a chronic myeloiddisease.1,14 In contrast, 9 of the 17 secondary AMLs showedASXL1 mutation or deletion (vs 3 of 46 primary AMLs, includinga case with mixed lineage dysplasia, P¼ 0.0001) and ASXL1alterations were prominently observed in AMLs secondary to achronic myeloid disease (9 of 12), whereas it was not the casefor NPM1 mutations (2 of 28, P¼ 0.0038) (Table 2). Were thishypothesis validated by the study of more cases, the detection ofASXL1 mutations would help distinguish between primary andsecondary AMLs, and consequently help orient the prognosis,even in the absence of known chronic phases.
In contrast, TET2 mutations in a series of AML were found tocorrelate neither with the presence or absence of NPM1 or FLT3mutations nor with an antecedent of chronic myeloid disease.10
In another series, three of four TET2 mutations were found insecondary cases.8 Although TET2 and ASXL1 may function insimilar pathways of epigenetic regulation,4 there might be
differences in their relationship with NPM1 and in their windowof action during the course of the disease. In the case that hasboth ASXL1 and FLT3 alteration, we determined that the ASXL1mutation, but not the FLT3 ITD, was present at the chronic phase,suggesting that the ASXL1 and FLT3 mutations can cooperate inrare cases. Finally, the rare RAS mutations occurred indifferentlyin primary or secondary cases; one was found in an ASXL1-mutated case, which was not unexpected, as we have previouslyshown that RAS and ASXL1 mutations can co-occur.2 It will nowbe interesting to determine what other alterations can associatewith either the NPM1 or ASXL1 route of leukemogenesis.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the Inserm, Institut Paoli-Calmettesand grants from the Association pour la Recherche contre leCancer (AM, no. 4929) and Fondation de France (DB, ComiteLeucemie).
N Carbuccia1,5, V Trouplin1,5, V Gelsi-Boyer1,2,3, A Murati1,2
, J Rocquain1, J Adelaıde1, S Olschwang1,2, L Xerri2,3,N Vey2,3, M Chaffanet1, D Birnbaum1 and MJ Mozziconacci1,2
1Centre de Recherche en Cancerologie de Marseille,Laboratoire d’Oncologie Moleculaire, UMR891 Inserm,
Institut Paoli-Calmettes, Marseille, France;2Departement de BioPathologie, Institut Paoli-Calmettes,
Marseille, France;3Faculte de Medecine, Universite de la Mediterrane,
Marseille, France and4Departement d’Hematologie, Institut Paoli-Calmettes,
Marseille, France5These authors contributed equally to this work.
E-mail: [email protected]
References
1 Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S et al.Altered nucleophosmin transport in acute myeloid leukaemia withmutated NPM1: molecular basis and clinical implications.Leukemia 2009; 23: 1731–1743.
2 Gelsi-Boyer V, Trouplin V, Adelaıde J, Bonansea J, Cervera N,Carbuccia N et al. Mutations of polycomb-associated gene ASXL1in myelodysplastic syndromes and chronic myelomonocyticleukaemia. Br J Haematol 2009; 145: 788–800.
3 Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaıde J,Rey J et al. Mutations of ASXL1 gene in myeloproliferativeneoplasms. Leukemia 2009; 23: 2183–2186.
4 Acquaviva C, Gelsi-Boyer V, Birnbaum D. Myelodysplasticsyndromes: lost between two states? Leukemia 2009 (in press).
5 Gelsi-Boyer V, Trouplin V, Adelaıde J, Aceto N, Remy V, Pinson Set al. Genome profiling of chronic myelomonocytic leukemia:frequent alterations of RAS and RUNX1 genes. BMC Cancer 2008;8: 299–314.
6 Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S,Masse A et al. Mutation in TET2 in myeloid cancers. N Engl J Med2009; 360: 2289–2301.
7 Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel Jet al. TET2 mutations and their clinical correlates in polycythemiavera, essential thrombocythemia and myelofibrosis. Leukemia2009; 23: 905–911.
8 Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MMet al.Detection of mutant TET2 in myeloid malignancies other thanmyeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML.Leukemia 2009; 23: 1343–1345.
ASXL1
Other alteration (s)
NPM1
FLT3
Other alteration (s)
Chronic
phase
AML
sAML
FLT3
Other alteration (s)
First alteration
Figure 1 A model for a distinct role of ASXL1 and NPM1mutations inthe development of acute myeloid leukemia (AML) with normalkaryotype. A hematopoietic stem or progenitor cell acquires a firstmutation. Second hits, which target NPM1 and/or FLT3 and/or othermutation(s), induce strong reprogramming and lead to AML. Alter-natively, second hits in ASXL1 and/or other genes induce an overt orasymptomatic chronic phase that may progress to secondary AML(sAML) with the occurrence of additional mutation(s).
Table 2 Summary of the results
FLT3 alteration(ITD, TKD orboth) (n, %)
NPM1 mutation(exon 12)(n, %)
ASXL1 alteration(exon 12 nonsenseor frameshift,deletion) (n, %)
PrimaryAMLs
Yes: 17 (37) Yes: 26 (56.5) Yes: 3 (6)
(N¼ 46) No: 29 (63) No: 20 (43.5) No: 43 (94)SecondaryAMLs
Yes: 2 (12) Yes: 2 (12) Yes: 9 (53)
(N¼ 17) No: 15 (88) No: 15 (88) No: 8 (47)Total AMLs Yes: 19 (30) Yes: 28 (44.5) Yes: 12 (19)(N¼ 63) No: 44 (70) No: 35 (55.5) No: 51 (81)
Abbreviations: AML, acute myeloid leukemia; ITD, internal tandemduplication.
Letters to the Editor
472
Leukemia

9 Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh Jet al. Loss of heterozygosity 4q24 and TET2 mutations associatedwith myelodysplastic/myeloproliferative neoplasms. Blood 2009;113: 6403–6410.
10 Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J,Wadleigh M et al.Genetic characterization of TET1, TET2, and TET3alterations in myeloid malignancies. Blood 2009; 114: 144–147.
11 Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG,Massop M et al. Acquired mutations in TET2 are common inmyelodysplastic syndromes. Nat Genet 2009; 41: 838–842.
12 Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey Net al. TET2 gene mutation is a frequent and adverse event in chronicmyelomonocytic leukemia. Haermatologica 2009 (in press).
13 Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V,Picard F et al. TET2 mutation is an independent favorableprognostic factor in myelodysplastic syndromes (MDS). Blood2009; 114: 3285–3291.
14 Falini B. Cytoplasmic nucleophosmin in acute myelogenousleukemia with a normal karyotype. New Engl J Med 2005; 352:254–266.
PML–RARa ligand-binding domain deletion mutations associated with reduced diseasecontrol and outcome after first relapse of APL
Leukemia (2010) 24, 473–476; doi:10.1038/leu.2009.220;published online 29 October 2009
The promyelocytic leukemia retinoic acid receptor-a (PML–RARa)fusion gene is essential for both the initiation and perpetuationof acute promyelocytic leukemia (APL), and, consistent with itscentral pathogenic role, the effectiveness of all-trans retinoic acid(ATRA) and arsenic trioxide in the treatment of APL has beenlinked to the targeted destruction of PML–RARa.1 Over manyyears, adjustments of therapy with ATRA in combination withchemotherapy for the initial treatment of APL has achievedlong-term remission rates, apparent disease cure, inX85% of newAPL patients.2 In two North American phase III clinical trialsemploying ATRA/chemotherapy (E2491 and C9710), mutationsin the ligand-binding domain (LBD) of PML–RARa were found in30–40% of the minority of patients who relapsed (see Gallagheret al.3 and Gallagher et al.4 and manuscript in preparation). In fourof six patients from protocol E2491 who relapsed with LBDmutations, the APL clone harboring the mutation emerged andreplaced the previously predominant wild-type PML–RARa APLcell population long after the last treatment with ATRA.3 These
observations suggested that LBD mutation-harboring clones mightselectively acquire ATRA-independent mechanism(s) of increasedAPL cell proliferation/survival competitiveness, and that this mecha-nism(s) might be favored in late relapse patients. In a collaborativeeffort to further explore this possibility, samples from previouslyreported late relapse patients, that is, relapse after 44 years,5
were not available; however, eight relapse samples frompatients with a relatively long interval to first relapse (median,1015 days after treatment on French–Belgian–Swiss APL groupprotocol, APL2000, or off-protocol in a similar manner),2 wereavailable and were analyzed (Table 1). Notably, three patientsfound to have LBD mutations relapsed during ATRAmaintenance therapy after a median of 530 days (range,406–557 days), whereas five patients lacking LBD mutationsrelapsed long after the completion of ATRA therapy after amedian of 1072 days (range, 992–1249 days). The latter obser-vation suggests that the association of late-emerging LBD mutantclones, independent of proximate ATRA selection pressure,occurs less frequently than that suggested by the previous study(current, 0/5 cases versus reported, 4/6 cases3), which requiresevaluation in a larger study. Further study is also required to
Table 1 Vital data on eight relapse APL patients treated according to French–Belgian–Swiss APL group protocols: grouping by the absence orpresence of PML–RARa mutations at first relapse
Patient number Age Sex WBC�103/ul PML–RARatype
Cytogeneticabnormality
Protocol treatmenta Days to relapse ATRA maintenancetreatment
Patients without mutation (n¼5)1 42 M 2 L t(15;17) APL2000 w/o AraC 1037 Off2 56 M 0.6 L t(15;17)b BAPL2000 w AraCa 1072 Off3 38 F 1.2 L t(15;17)b APL2000 w AraC 1194 Off4 5 F NA L t(15;17) BAPL93c 1249 Nonec
5 11 F NA L t(15;17) BAPL2000 w AraCa,c 992 Nonec
Median or number 38 M-2; F-3 L-5; S-0 +8ÿ0 1072 On-0Off-5
Patients with mutation (n¼3)6 36 M 2.2 S +8, t(15;17) APL2000 w/o AraC 557 On7 57 M 4.6 L t(15;17) APL2000 w/o AraC 530 On8 NA M NA L +8, t(15;17) BAPL2000 w AraCa 406 OnMedian or number M-3; F-0 F L-2; S-1 +8–2 530 On-3
Off-0
Abbreviations: AraC, cytosine arabinoside; APL, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; NA, not available; PML–RARa,promyelocytic leukemia retinoic acid receptor-a fusion gene; WBC, white blood corpuscles.aB, same as cited protocol regimen, but treated off-protocol.
bTreatment-related APL.cNo ATRA administered during protocol maintenance therapy.
Letters to the Editor
473
Leukemia

69
Commentaires et discussion de l’article n°1
Près de 60% des LAM ont un caryotype de pronostic intermédiaire (Mrózek et al,
2004) et les anomalies moléculaires de ces LAM, surtout celles à caryotype normal,
demeurent en grande partie inconnues.
Dans le laboratoire de D. Birnbaum, de nouvelles mutations comme celles du gène
ASXL1 ont été décrites récemment (Gelsi-Boyer et al, 2009 ; Carbuccia et al, 2009) : elles
ont été retrouvées dans environ 10% des SMP, 10% des SMD et 40% des LMMC.
L’incidence de ces mutations dans les LAM était inconnue et nous avons réalisé le
séquençage d’ASXL1 par l’intermédiaire d’un séquenceur API 3130XL (Applied
Biosystems) sur 63 patients atteints de LAM à pronostique intermédiaire (46 LAM de novo
et 17 LAM secondaires à une hémopathie chronique). Nous avons ainsi constaté qu’ASXL1
était muté dans 17,5% de ces LAM avec une plus grande fréquence dans les LAM
secondaires (53%) par rapport aux LAM primaires (6%).
Les LAM primaires, notamment lorsque leur caryotype est normal, sont connues pour
être mutées sur le gène NPM1 dans 50 à 60% des cas (Falini et al, 2005) : Nous avons
retrouvé un pourcentage équivalent de mutations dans notre étude. De plus, nous avons
remarqué qu’aucun des 28 cas de LAM primaires et secondaires mutées sur NPM1 ne l’était
sur ASXL1, et qu’à l’inverse, parmi les 11 cas de LAM mutées sur ASXL1, aucun n’était
muté sur NPM1.
Cette étude suggère l’existence de deux voies distinctes de leucémogenèse, selon la
présence des mutations du gène ASXL1 ou du gène NPM1 : Les LAM primaires à caryotype
normal seraient caractérisées par une faible fréquence de mutations d’ASXL1 (3/46, 6.5%
dans notre étude) et une fréquence élevées de mutations de NPM1.
A l’inverse, les mutations d’ASXL1 seraient caractéristiques des LAM secondaires à une
hémopathie chronique. Autrement dit, les mutations de NPM1 sont rares dans les LAM
secondaires (Falini et al, 2009) alors qu’ASXL1 est fréquemment muté.
Au niveau des hémopathies chroniques, les mutations d’ASXL1 seraient plus fréquentes dans
les LMMC que dans les SMD et SMP.

70
Dans cet article, nous avons montré pour la première fois que les mutations de NPM1
et d’ASXL1 étaient exclusives. Cette donnée a été confirmée récemment sur une large
cohorte de 501 LAM (Chou et al, 2010), les patients avec ASXL1 muté présentaient un plus
mauvais pronostic.
Si notre hypothèse est vérifiée, la découverte de mutations ASXL1 dans les LAM
pourrait constituer une aide au diagnostic des LAM secondaires. Les LAM secondaires
présentent un pronostic plus défavorable que les LAM de novo et la découverte de mutations
d’ASXL1 chez un patient pourrait permettre d’adapter plus précocement la prise en charge
thérapeutique.

71
ARTICLE N°2
Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in
myelodysplastic syndromes and acute myeloid leukemias.
Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N, Birnbaum D, Gelsi-Boyer V,
Mozziconacci MJ. Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N, Birnbaum
D, Gelsi-Boyer V, Mozziconacci MJ.
BMC Cancer 2010; 10: 401.

RESEARCH ARTICLE Open Access
Combined mutations of ASXL1, CBL, FLT3, IDH1,IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 andWT1 genes in myelodysplastic syndromes andacute myeloid leukemiasJulien Rocquain1†, Nadine Carbuccia1†, Virginie Trouplin1†, Stéphane Raynaud1, Anne Murati1,2, Meyer Nezri3,
Zoulika Tadrist4, Sylviane Olschwang1,2, Norbert Vey5,6, Daniel Birnbaum1, Véronique Gelsi-Boyer1,2,5,
Marie-Joelle Mozziconacci1,2*
Abstract
Background: Gene mutation is an important mechanism of myeloid leukemogenesis. However, the number and
combination of gene mutated in myeloid malignancies is still a matter of investigation.
Methods: We searched for mutations in the ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and
WT1 genes in 65 myelodysplastic syndromes (MDSs) and 64 acute myeloid leukemias (AMLs) without balanced
translocation or complex karyotype.
Results: Mutations in ASXL1 and CBL were frequent in refractory anemia with excess of blasts. Mutations in TET2
occurred with similar frequency in MDSs and AMLs and associated equally with either ASXL1 or NPM1 mutations.
Mutations of RUNX1 were mutually exclusive with TET2 and combined with ASXL1 but not with NPM1. Mutations in
FLT3 (mutation and internal tandem duplication), IDH1, IDH2, NPM1 and WT1 occurred primarily in AMLs.
Conclusion: Only 14% MDSs but half AMLs had at least two mutations in the genes studied. Based on the
observed combinations and exclusions we classified the 12 genes into four classes and propose a highly
speculative model that at least a mutation in one of each class is necessary for developing AML with simple or
normal karyotype.
Background
Myeloid leukemogenesis is a complex process that
transforms a regulated hematopoietic stem or progenitor
cell into a proliferative cell unable to differentiate. Sev-
eral genetic alterations such as translocations, gene
mutations and deletions play a role in this process.
Balanced translocations generating gene fusion play a
major leukemogenic role in some classes of leukemias.
However, they are rare in myelodysplastic syndromes
(MDSs) and the majority of adult acute myeloid leuke-
mias (AMLs) have a normal karyotype (NK-AMLs).
MDSs are a heterogeneous group of clonal diseases
characterized by bone marrow dysplasia, various degrees
of cytopenia and a risk of progression to AML [1]. The
alterations that lead to these pre-leukemic disorders are
poorly defined [2]. Several genes have been identified
recently as mutated in MDSs and AMLs, including
ASXL1, located in chromosome arm 20q, CBL, located
in 11q, and TET2, located in 4q [3-11]. These mutations
are associated with biological and prognostic features
[9,11].
Similarly, gene mutations are likely to play a role in
the development of NK-AMLs. Indeed, the identification
of mutations in several genes such as NPM1 and FLT3
has revealed prognosis subgroups and modified the clin-
ical management of these leukemias [12,13]. A recent
whole-genome sequencing study of an AML case has
* Correspondence: [email protected]
† Contributed equally1Centre de Recherche en Cancérologie de Marseille; Laboratoire d’Oncologie
Moléculaire; UMR891 Inserm; Institut Paoli-Calmettes; Marseille, France
Full list of author information is available at the end of the article
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
© 2010 Rocquain et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the CreativeCommons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, andreproduction in any medium, provided the original work is properly cited.

revealed mutations in IDH1, which encodes the enzyme
isocitrate dehydrogenase [14]. AMLs occur de novo or
after a chronic phase (hereafter called primary and sec-
ondary, respectively). Such chronic phase can be an
MDS. We have recently shown that mutations in
ASXL1, in contrast to NPM1 mutations, are found in
MDSs and in secondary AMLs [15].
Mutation data are in line with the multi-hit model
proposed by Gilliland suggesting that many gene muta-
tions play a role in leukemogenesis [16]. Now that more
and more gene mutations are reported it is important to
determine when they occur in the various types of
malignant hematopoietic diseases and how they combine
in the development of leukemia. To begin to answer
these questions we searched for mutations in twelve
selected genes in a panel of 129 MDS and AML
samples.
Methods
Patients and samples
All patients signed an informed consent and the study
was approved by the institutional review board (“Com-
mission d’Orientation Scientifique”) of Institut Paoli-
Calmettes.
The 129 samples were selected on the absence of
known balanced translocation, and in the absence of
complex karyotype for the AML series (as defined by
more than three alterations). They included 65 cases of
MDS (Additional file 1Table S1), comprising, according
to the WHO criteria [17], 5 refractory anemia (RA), 13
refractory anemia with ring sideroblasts (RARS) (includ-
ing one with myelofibrosis), 7 refractory cytopenia with
multilineage dysplasia (RCMD), 16 refractory anemia
with excess of blasts type 1 (RAEB1), 19 refractory ane-
mia with excess of blasts type 2 (RAEB2) and 5 MDS-
unclassified (MDS-U) cases. Six cases were secondary to
hematopoietic or non-hematopoietic diseases (na in
IPSS column, Additional file 2Table S1). The majority
of MDS samples were collected at the time of diagnosis;
some were in therapeutic abstention of a known MDS
and some were under symptomatic treatment. Seventeen
cases were IPPS low risk (0), 23 were int-1 (0.5-1), 12
were int-2 (1.5-2) and 7 were high risk (≥2.5).
We studied 64 AMLs (Additional file 2Table S2) includ-
ing 46 primary cases and 18 transformations of a previous
myeloid disease (the same cohort of 64 AML patients,
listed in Additional file 2Table S2, had been analyzed for
mutations in the NPM1, FLT3, CEBPA, NRAS, KRAS,
JAK2, and ASXL1 genes and the results already reported
in ref. 15). The panel comprised 47 cases of NK-AML and
17 cases with either trisomy 8 (n = 14), 9q deletion (HD-
0632), trisomy 11 (HD-0304) or 20q deletion (HD-0381)
as a sole karyotypic abnormality.
Array-comparative genomic hybridization (aCGH) had
been performed on almost all samples and had allowed
the detection of deletions and breaks [9,10,15,18,19].
DNA sequencing
DNA sequencing of exon-coding sequences of ASXL1,
CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS,
RUNX1, TET2 and WT1 was done as follows. PCR ampli-
fications of bone marrow cell DNA were done in a total
volume of 25 μl PCR mix containing at least 5 ng tem-
plate DNA, Taq buffer, 200 μmol of each deoxynucleo-
tide triphosphate, 20 pmol of each primer and 1 unit of
Hot Star Taq (Qiagen). PCR amplification conditions
were as follows: 95°C 10 min; 95°C 30 sec, 55°C 30 sec,
72°C 30 sec to 1 min depending on PCR product length
for 35 cycles; 72°C 10 min. PCR products were purified
using Millipore plate MSNU030. One microliter of the
purified PCR products was used for sequencing using the
Big Dye terminator v1.1 kit (Applied Biosystems) includ-
ing the forward or reverse primer. After G50 purification,
sequences were loaded on an ABI 3130XL automat
(Applied Biosystems). The sequence data files were ana-
lyzed using both SeqScape and Phred/Phrap/Consed soft-
wares and all mutations were confirmed on an
independent PCR product. The exons studied were as
follows: ASXL1 exon 12, CBL exons 8 and 9, FLT3 exons
14, 15 and 20, IDH exon 4, JAK2 exon 14, RAS exons 1
and 2, NPM1 exon 12, RUNX1 exons 1 to 8, TET2 exons
3 to 11, WT1 exons 7 and 9. The primers for sequencing
are listed in [Additional file 3].
Results
Mutations in MDSs
We had previously shown that ASXL1 frameshift and
nonsense mutations occurred in exon 12 [10]. ASXL1
exon 12 frameshift mutations (10 times the same p.
Gly646Trpfsx12) were observed in 12 out of the 65
MDS cases (18.5%) including 1 out of 5 RA (20%), 2 out
of 16 RAEB1 (12.5%) and 9 out of 19 RAEB2 (47.4%)
(Additional file 1Table S1). We found 12 cases with
TET2 mutation (18.5%) and 4 with RUNX1 mutation
(6.2%). One patient (HD-0311) had two TET2 muta-
tions. TET2 mutations were frequent in RAEB1 (7/16,
43.8%). Mutations in RUNX1 and TET2 were mutually
exclusive but both could associate with ASXL1 muta-
tions: two cases showed both an ASXL1 and a TET2
mutation and three cases both an ASXL1 and a RUNX1
mutation. One case of ASXL1 deletion (HD-0190) and
one case of TET2 deletion (HD-0145) have been
reported [9,10]. One case (HD-0232) had a break in
RUNX1 detected by aCGH (not shown).
We did not find any FLT3, NPM1 or WT1 mutation.
One MDS-U had a JAK2 mutation and one RCMD case
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
Page 2 of 7
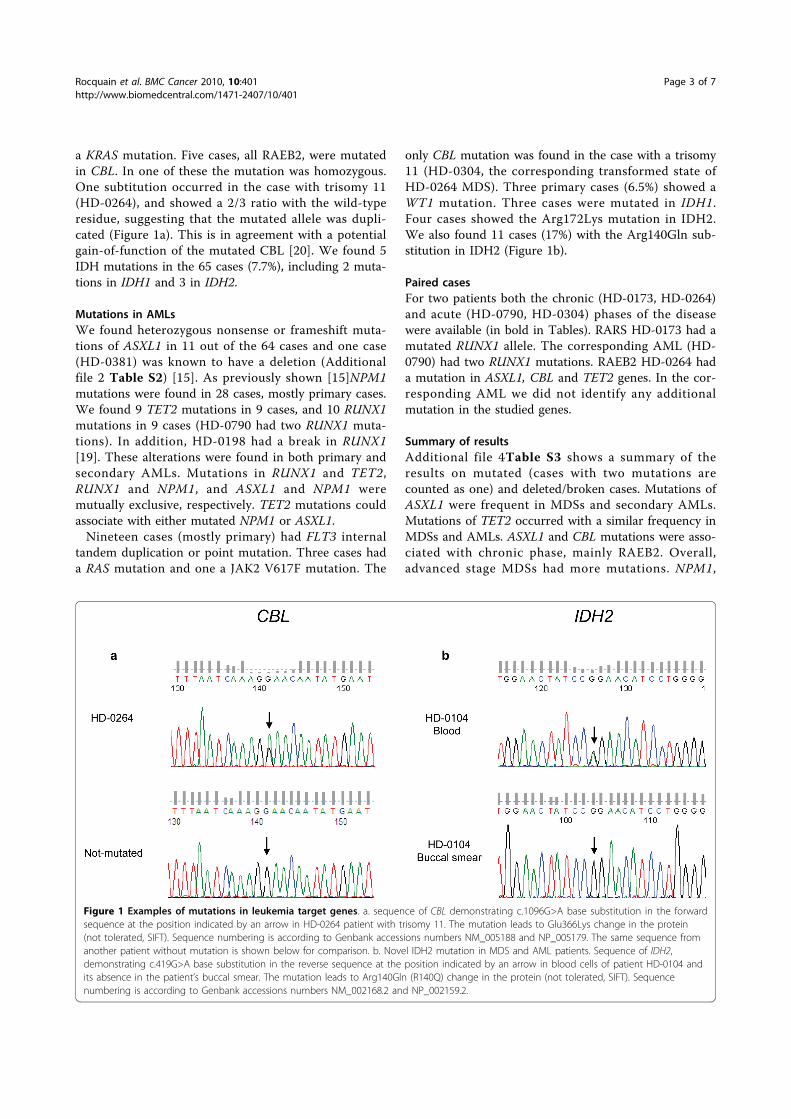
a KRAS mutation. Five cases, all RAEB2, were mutated
in CBL. In one of these the mutation was homozygous.
One subtitution occurred in the case with trisomy 11
(HD-0264), and showed a 2/3 ratio with the wild-type
residue, suggesting that the mutated allele was dupli-
cated (Figure 1a). This is in agreement with a potential
gain-of-function of the mutated CBL [20]. We found 5
IDH mutations in the 65 cases (7.7%), including 2 muta-
tions in IDH1 and 3 in IDH2.
Mutations in AMLs
We found heterozygous nonsense or frameshift muta-
tions of ASXL1 in 11 out of the 64 cases and one case
(HD-0381) was known to have a deletion (Additional
file 2 Table S2) [15]. As previously shown [15]NPM1
mutations were found in 28 cases, mostly primary cases.
We found 9 TET2 mutations in 9 cases, and 10 RUNX1
mutations in 9 cases (HD-0790 had two RUNX1 muta-
tions). In addition, HD-0198 had a break in RUNX1
[19]. These alterations were found in both primary and
secondary AMLs. Mutations in RUNX1 and TET2,
RUNX1 and NPM1, and ASXL1 and NPM1 were
mutually exclusive, respectively. TET2 mutations could
associate with either mutated NPM1 or ASXL1.
Nineteen cases (mostly primary) had FLT3 internal
tandem duplication or point mutation. Three cases had
a RAS mutation and one a JAK2 V617F mutation. The
only CBL mutation was found in the case with a trisomy
11 (HD-0304, the corresponding transformed state of
HD-0264 MDS). Three primary cases (6.5%) showed a
WT1 mutation. Three cases were mutated in IDH1.
Four cases showed the Arg172Lys mutation in IDH2.
We also found 11 cases (17%) with the Arg140Gln sub-
stitution in IDH2 (Figure 1b).
Paired cases
For two patients both the chronic (HD-0173, HD-0264)
and acute (HD-0790, HD-0304) phases of the disease
were available (in bold in Tables). RARS HD-0173 had a
mutated RUNX1 allele. The corresponding AML (HD-
0790) had two RUNX1 mutations. RAEB2 HD-0264 had
a mutation in ASXL1, CBL and TET2 genes. In the cor-
responding AML we did not identify any additional
mutation in the studied genes.
Summary of results
Additional file 4Table S3 shows a summary of the
results on mutated (cases with two mutations are
counted as one) and deleted/broken cases. Mutations of
ASXL1 were frequent in MDSs and secondary AMLs.
Mutations of TET2 occurred with a similar frequency in
MDSs and AMLs. ASXL1 and CBL mutations were asso-
ciated with chronic phase, mainly RAEB2. Overall,
advanced stage MDSs had more mutations. NPM1,
Figure 1 Examples of mutations in leukemia target genes. a. sequence of CBL demonstrating c.1096G>A base substitution in the forward
sequence at the position indicated by an arrow in HD-0264 patient with trisomy 11. The mutation leads to Glu366Lys change in the protein
(not tolerated, SIFT). Sequence numbering is according to Genbank accessions numbers NM_005188 and NP_005179. The same sequence from
another patient without mutation is shown below for comparison. b. Novel IDH2 mutation in MDS and AML patients. Sequence of IDH2,
demonstrating c.419G>A base substitution in the reverse sequence at the position indicated by an arrow in blood cells of patient HD-0104 and
its absence in the patient’s buccal smear. The mutation leads to Arg140Gln (R140Q) change in the protein (not tolerated, SIFT). Sequence
numbering is according to Genbank accessions numbers NM_002168.2 and NP_002159.2.
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
Page 3 of 7

FLT3 and WT1 mutations were associated with primary
AMLs. Mutations of IDH1/2 were rare in chronic phase.
Not surprisingly, the number of mutated genes per case
was lower in MDSs than in AMLs: only 14% MDSs but
more than half AMLs had at least two gene mutations.
Discussion
We searched for mutations in a series of selected genes.
Mutations in some of these genes, such as ASXL1, CBL,
IDH and TET2, have been identified only recently in
myeloid diseases and have never been surveyed together
to date. A number of issues need be discussed.
The frequencies of mutations we observed are close to
what has been reported individually for each gene so
far. This is true for example for IDH1, NPM1, TET2
and WT1 [1,3-9,14,21,22]. In MDS we found slightly
more mutations of CBL than previously reported [11].
The IDH2 Arg140Gln was frequent in AMLs, rare in
MDSs. The mutation was not present in the buccal
smear DNA of a patient with AML (HD-0104), showing
it was acquired. The Arg140Gln mutation in IDH2 has
been reported in recent studies of myeloid diseases
[23,24]. A recent study has described ASXL1 as the
most frequently mutated gene in advanced MDSs [25],
which we confirmed here.
A repertoire of mutations
Based on the known functions of the proteins, on a pre-
vious model and classification [16], on where the muta-
tions were present (MDSs and/or secondary AMLs and/
or primary AMLs) and on how they combined (mutual
exclusion or association), we tentatively grouped the
genes in four classes. The first class includes RUNX1
and TET2. Mutations in these genes may cause clonal
dominance of hematopoietic stem cells [3]. ASXL1 and
NPM1 would constitute class II. Mutations in these
genes may promote a pathway leading towards either
primary or secondary AML [15]. Genes associated with
signaling pathways and proliferation [16] (CBL, FLT3,
JAK2, RAS) define class III. JAK2 mutation plays little
role in MDSs and NK-AMLs. We have shown that
FLT3 signaling is regulated by CBL [26]. However, the
two alterations may not be equivalent; if in our study
mutations in these genes were mutually exclusive and
were both more frequent in AMLs than in MDSs, CBL
but not FLT3 mutations were frequent in RAEB2 cases.
This is in agreement with recent studies that showed
alterations of CBL in chronic myeloid diseases [11,27].
Finally, for three reasons we grouped IDH1, IDH2, and
WT1 in a putative class IV. First, IDH and WT1 muta-
tions were exclusive but could co-occur with mutations
in genes from other classes. Second, they occurred pri-
marily in AMLs and were rare in MDSs. Third, muta-
tions of these genes could be associated with
modifications of the HIF1 and oxygen-sensing pathways
[28,29]. Class IV mutations are rather associated with
acute phase.
The existence of many cases with few or no mutations
in the selected genes, especially in some classes of
MDSs such as MDS-U, RA, RARS and RCMD but also
in primary AMLs (half of the cases with no or one
mutation), suggests that other genes remain to be stu-
died or discovered. Indeed, our study did not include
several target genes, such as CBFB [30], CDKN1A/B,
CEBPA, ETV6, KIT, NF1, NFIA [31], P53, RB1 and UTX
[32]; it did not include either a search for the recently
discovered mutations of the EZH2 gene [33,34]. More-
over, our aCGH analyses of the same series showed that
some cases without mutation in the studied genes do
have deletions of other cancer genes (for example
CDKN1B, ETV6 and UTX [32] are deleted in HD-0205)
[9,10,15]. Thus, the repertoire of altered genes in both
MDSs and AMLs is likely to include many genes and it
is probable that whole-genome sequencing studies will
confirm this. Because of this, and although no case had
four mutations, we propose that AML develops follow-
ing - at least - four cooperating mutations, one from
each class (Figure 2). This is highly speculative however;
the identification of new target genes and the study of
others will lead to a more precise picture. Also, because
we did not study AMLs with balanced translocation or
complex karyotype this model is proposed only for
AML with simple or normal karyotype.
Random or ordered accumulation?
An important question is whether the mutations occur
in a necessary order or randomly until differentiation of
the malignant hematopoietic clone is completely
blocked. RARS HD-0173 (one RUNX1 mutation) and
AML HD-0790 (two RUNX1 mutations) are instructive
samples from the same patient. It is tempting to inter-
pret this observation as a bi-allelic inactivation of the
RUNX1 gene. RUNX1 mutations are frequent in chronic
myelomonocytic leukemia [19] but rare in myeloproli-
ferative neoplasms except at the acute phase [35]. In our
series, mutations in TET2 were equally frequent in
MDSs and AMLs. Taken together this suggests that
inactivation of class I tumor suppressors (RUNX1,
TET2) can intervene at any stage, i.e. early or at pro-
gression. In studies of AMLs secondary to myeloproli-
ferative neoplasms (MPN) mutations of TET2 have
indeed been shown to occur early or late [30,36]. A
recent study showed that ASXL1 mutations were present
at the chronic stage and, in agreement with our findings,
that ASXL1, JAK2 and TET2 have non overlapping con-
tributions to myeloid transformation [36]. The order of
occurrence of ASXL1, TET2 and RUNX1 mutations may
vary in different diseases (MPNs and MDSs) and in
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
Page 4 of 7
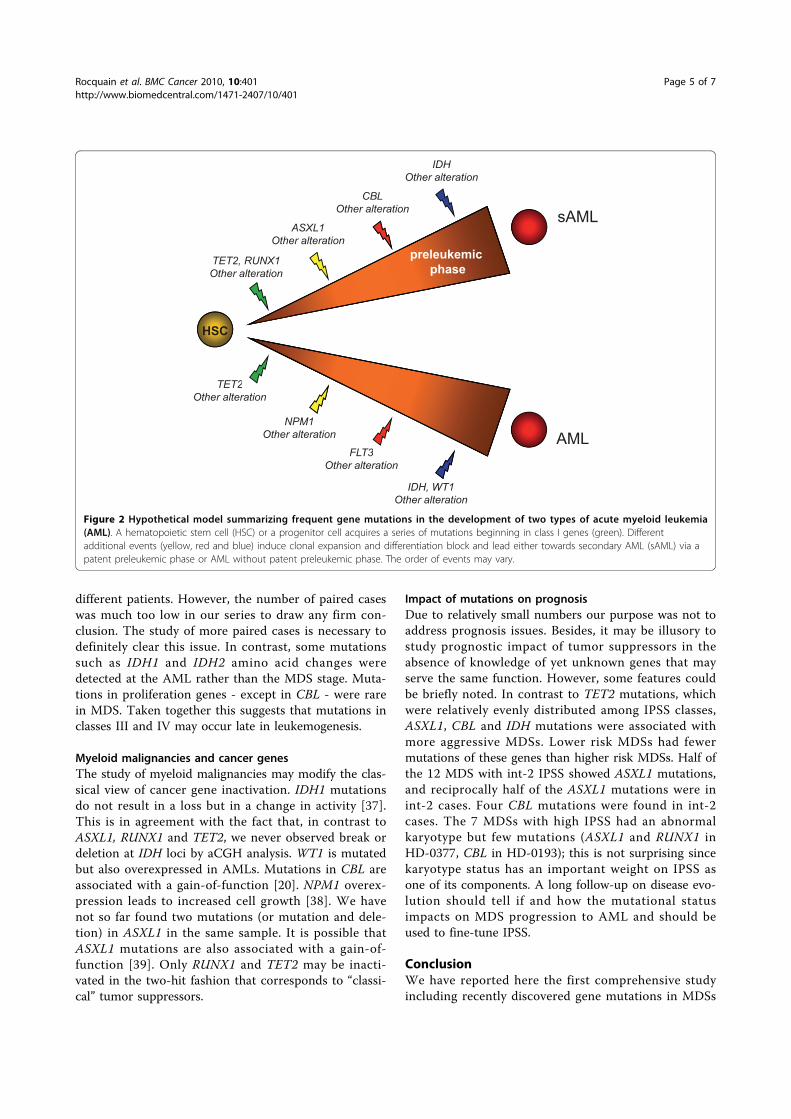
different patients. However, the number of paired cases
was much too low in our series to draw any firm con-
clusion. The study of more paired cases is necessary to
definitely clear this issue. In contrast, some mutations
such as IDH1 and IDH2 amino acid changes were
detected at the AML rather than the MDS stage. Muta-
tions in proliferation genes - except in CBL - were rare
in MDS. Taken together this suggests that mutations in
classes III and IV may occur late in leukemogenesis.
Myeloid malignancies and cancer genes
The study of myeloid malignancies may modify the clas-
sical view of cancer gene inactivation. IDH1 mutations
do not result in a loss but in a change in activity [37].
This is in agreement with the fact that, in contrast to
ASXL1, RUNX1 and TET2, we never observed break or
deletion at IDH loci by aCGH analysis. WT1 is mutated
but also overexpressed in AMLs. Mutations in CBL are
associated with a gain-of-function [20]. NPM1 overex-
pression leads to increased cell growth [38]. We have
not so far found two mutations (or mutation and dele-
tion) in ASXL1 in the same sample. It is possible that
ASXL1 mutations are also associated with a gain-of-
function [39]. Only RUNX1 and TET2 may be inacti-
vated in the two-hit fashion that corresponds to “classi-
cal” tumor suppressors.
Impact of mutations on prognosis
Due to relatively small numbers our purpose was not to
address prognosis issues. Besides, it may be illusory to
study prognostic impact of tumor suppressors in the
absence of knowledge of yet unknown genes that may
serve the same function. However, some features could
be briefly noted. In contrast to TET2 mutations, which
were relatively evenly distributed among IPSS classes,
ASXL1, CBL and IDH mutations were associated with
more aggressive MDSs. Lower risk MDSs had fewer
mutations of these genes than higher risk MDSs. Half of
the 12 MDS with int-2 IPSS showed ASXL1 mutations,
and reciprocally half of the ASXL1 mutations were in
int-2 cases. Four CBL mutations were found in int-2
cases. The 7 MDSs with high IPSS had an abnormal
karyotype but few mutations (ASXL1 and RUNX1 in
HD-0377, CBL in HD-0193); this is not surprising since
karyotype status has an important weight on IPSS as
one of its components. A long follow-up on disease evo-
lution should tell if and how the mutational status
impacts on MDS progression to AML and should be
used to fine-tune IPSS.
Conclusion
We have reported here the first comprehensive study
including recently discovered gene mutations in MDSs
IDH
Other alteration
sAML
CBL
Other alteration
Other alteration
ASXL1
Other alteration
preleukemic
phaseTET2, RUNX1
Other alteration
TET2
HSC
AML
NPM1
Other alteration
FLT3
TET2
Other alteration
FLT3
Other alteration
IDH, WT1
Other alteration
Figure 2 Hypothetical model summarizing frequent gene mutations in the development of two types of acute myeloid leukemia
(AML). A hematopoietic stem cell (HSC) or a progenitor cell acquires a series of mutations beginning in class I genes (green). Different
additional events (yellow, red and blue) induce clonal expansion and differentiation block and lead either towards secondary AML (sAML) via a
patent preleukemic phase or AML without patent preleukemic phase. The order of events may vary.
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
Page 5 of 7

and AMLs. We have proposed a speculative model of
cooperative leukemogenesis for AML with simple or
normal karyotype, which needs to be confirmed by the
study of more cases and completed by the discovery of
more genes. As this it represents a step towards the
necessary determination of the complete mutational
status of myeloid malignant diseases.
Additional material
Additional file 1: Table S1 Mutations of candidate genes in a series
of myelodysplastic syndromes.
Additional file 2: Table S2 Mutations of candidate genes in a series
of AMLs.
Additional file 3: Oligonucleotide primers used for sequencing the
coding regions of the selected genes.
Additional file 4: Table S3 Summary of results.
Acknowledgements
We are very grateful to the patients who participated to the study. This
work was supported by Inserm, Institut Paoli-Calmettes and grants from the
Fondation de France (DB, Comité Leucémie 2007-2009) and Association
pour la Recherche contre le Cancer (n° 4929; n° 4992).
Author details1Centre de Recherche en Cancérologie de Marseille; Laboratoire d’Oncologie
Moléculaire; UMR891 Inserm; Institut Paoli-Calmettes; Marseille, France.2Département de BioPathologie, Institut Paoli-Calmettes, Marseille, France.3Service de Médecine Interne, Centre Hospitalier Général, Martigues, France.4Service de Médecine Interne-Oncologie, Hôpital de Salon-de-Provence,
Salon-de-Provence, France. 5Faculté de Médecine, Université de la
Méditerranée, Marseille, France. 6Département d’Hématologie, Institut Paoli-
Calmettes, Marseille, France.
Authors’ contributions
JR, NC, VT, SR and SO obtained and analyzed gene sequencing data. AM,
MN, ZT, VGB; MJM and NV provided samples and bioclinical data. NV, DB,
VGB and MJM designed and supervised the study, analyzed the data and
wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Received: 22 February 2010 Accepted: 2 August 2010
Published: 2 August 2010
References
1. Tefferi A, Vardiman JW: Myelodysplastic syndromes. N Eng J Med 2009,
361:1872-1885.
2. Acquaviva C, Gelsi-Boyer V, Birnbaum D: Myelodysplastic syndromes: lost
between two states? Leukemia 2010, 24:1-5.
3. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A,
Kosmider O, Le Couedic JP, Robert F, Alberdi A, Plo I, Dreyfus FJ, Marzac C,
Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F,
Fontenay M, Vainchenker W, Bernard OA: Mutation in TET2 in myeloid
cancers. N Engl J Med 2009, 360:2289-2301.
4. Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM,
Hanson CA, Pardanani A, Gilliland DG, Levine RL: Detection of mutant TET2
in myeloid malignancies other than myeloproliferative neoplasms:
CMML, MDS, MDS/MPN and AML. Leukemia 2009, 23:1343-1345.
5. Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J,
O’Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP: Loss of
heterozygosity 4q24 and TET2 mutations associated with
myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113:6403-6410.
6. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J,
Wadleigh M, Malinge S, Yao JJ, Kilpivaara O, Bhat R, Huberman K, Thomas S,
Dolgalev I, Heguy A, Paietta E, Le Beau MM, Beran M, Tallman MS, Ebert BL,
Kantarjian HM, Stone RM, Gilliland DG, Crispino JD, Levine RL: Genetic
characterization of TET1, TET2, and TET3 alterations in myeloid
malignancies. Blood 2009, 114:144-147.
7. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M,
Stevens-Linders E, van Hoogen P, Geurts van Kessel A, Raymakers RAP,
Kamping EJ, Verhoef GE, Verburgh E, Hagemeijer A, Vandenberghe P, de
Witte T, van der Reijden BA, Janssen JH: Acquired mutations in TET2 are
common in myelodysplastic syndromes. Nat Genet 2009, 41:838-842.
8. Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, Westwood NB,
Strupp C, Gattermann N, Germing U, Mufti GJ: Novel TET2 mutations
associated with UPD4q24 in myelodysplastic syndromes. J Clin Oncol
2009, 27:4002-4006.
9. Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F,
Viguié F, Quesnel B, Beyne-Rauzy O, Solary E, Vey N, Hunault-Berger M,
Fenaux P, Mansat-De Mas V, Delabesse E, Guardiola P, Lacombe C,
Vainchenker W, Preudhomme C, Dreyfus F, Bernard OA, Birnbaum D,
Fontenay M: TET2 mutation is an independent favorable prognostic
factor in myelodysplastic syndromes (MDS). Blood 2009, 114:3285-3291.
10. Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N,
Lagarde A, Prébet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M,
Mozziconacci MJ, Vey N, Birnbaum D: Mutations of polycomb-associated
gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic
leukaemia. Br J Haematol 2009, 145:788-800.
11. Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H,
O’Keefe C, Hsi E, Paquette RL, Kojima S, List AF, Sekeres MA, McDevitt MA,
Maciejewski JP: Mutations of E3 ligase Cbl family members constitute a
novel common pathogenic lesion in myeloid malignancies. J Clin Oncol
2009, 27:6109-6116.
12. Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, Nicoletti N: Altered
nucleophosmin transport in acute myeloid leukaemia with mutated
NPM1: molecular basis and clinical implications. Leukemia 2009,
23:1731-1743.
13. Gaidzik V, Döhner K: Prognostic implications of gene mutations in acute
myeloid leukemia with normal karyotype. Semin Oncol 2008, 35:346-355.
14. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K,
Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP,
Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM,
Carmichael L, Elred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF,
Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK,
Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME,
Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH,
Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ,
Graubert TA, DiPersio JF, Wilson RK, Ley TJ: Recurrent mutations found by
sequencing an acute myeloid leukemia genome. N Eng J Med 2009,
361:1058-1066.
15. Carbuccia N, Trouplin V, Gelsi-Boyer V, Murati A, Rocquain J, Adélaïde J,
Olschwang S, Xerri L, Vey N, Chaffanet M, Birnbaum D, Mozziconacci MJ:
Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute
myeloid leukemias. Leukemia 2010, 24:469-473.
16. Gilliland DG: Molecular genetics of human leukemias: new insights into
therapy. Semin Hematol 2002, 39:6-11.
17. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A,
Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD: The
2008 revision of the World Health Organization (WHO) classification of
myeloid neoplasms and acute leukemia: rationale and important
changes. Blood 2009, 114:937-951.
18. Murati A, Gervais C, Carbuccia N, Finetti P, Cervera N, Adélaïde J, Struski S,
Lippert E, Mugneret F, Tigaud I, Penther D, Bastard C, Poppe B, Speleman F,
Baranger L, Luquet I, Cornillet-Lefebvre P, Nadal N, Nguyen-Khac F, Pérot C,
Olschwang S, Bertucci F, Chaffanet M, Lessard M, Mozziconacci MJ,
Birnbaum D: Genomic profiling of acute myelomonocytic leukemia:
alteration of the MYB locusin MYST3-linked cases. Leukemia 2009,
23:85-94.
19. Gelsi-Boyer V, Trouplin V, Adélaïde J, Aceto N, Remy V, Pinson S,
Houdayer C, Arnoulet C, Sainty D, Bentires-Alj M, Olschwang S, Vey N,
Mozziconacci MJ, Birnbaum D, Chaffanet M: Genome profiling of chronic
myelomonocytic leukemia: frequent alterations of RAS and RUNX1
genes. BMC Cancer 2008, 8:299-314.
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
Page 6 of 7

20. Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamasaki S, Tamura A,
Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J,
Gotoh N, Nakazaki K, Kawamata N, Onodera M, Nobuyoshi M, Hayashi Y,
Harada H, Kurokawa M, Chiba S, Mori H, Ozawa K, Omine M, Hirai H,
Nakauchi H, Koeffler HP, Ogawa S: Gain-of-function of mutated CBL
tumour suppressor in myeloid neoplasms. Nature 2009, 460:904-908.
21. Renneville A, Boissel N, Zurawski V, Llopis L, Biggio V, Nibourel O,
Philippe N, Thomas X, Dombret H, Preudhomme C: Wilms tumor gene
mutations are associated with a higher risk of recurrence in young
adults with acute myeloid leukemia: a study from the Acute Leukemia
French Association. Cancer 2009, 115:3719-3727.
22. Virappane P, Gale R, Hills R, Kakkas I, Summers K, Stevens J, Allen C,
Green C, Quentmeier H, Drexler H, Burnett A, Linch D, Bonnet D, Lister TA,
Fitzgibbon J: Mutation of the Wilm’s tumor 1 gene is a poor prognostic
factor associated with chemotherapy resistance in normal karyotype
acute myeloid leukaemia: the United Kingdom Medical Research Council
Adult Leukaemia Working Party. J Clin Oncol 2008, 26:5429-5435.
23. Green A, Beer P: Somatic mutations of IDH1 and IDH2 in the leukemic
transformation of myeloproliferative neoplasms. New Engl J Med 2010,
362:369-370.
24. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett B, Coller HA, Cross JR,
Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su S, Sharp KA,
Levine RL, Thompson CB: The common features of leukemia-associated
IDH1 and IDH2 mutations is a neomorphic enzyme activity converting
a-ketoglutarate to 2-hydoxyglutarate. Cancer Cell 2010, 17:1-10.
25. Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-
Santamaria C, Calasanz MJ, Larrayoz MJ, Garcia-Delgado M, Giagounidis A,
Malcovati L, Della Porta MG, Jädersten M, Killick S, Hellström-Lindbergh E,
Cazzola M, Wainscoat JS: Frequent mutations of the polycomb-associated
gene ASXL1 in the myelodysplastic syndromes and in acute myeloid
leukemia. Leukemia 2010, 24:1062-1065.
26. Lavagna-Sévenier C, Marchetto S, Birnbaum D, Rosnet O: FLT3 signaling in
hematopoietic cells involves CBL, SHC and an unknown P115 as
prominent tyrosine-phosphorylated substrates. Leukemia 1998,
12:301-310.
27. Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H,
Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP: 250K single
nucleotide polymorphism array karyotyping identifies acquired
uniparental disomy and homozygous mutations, including novel
missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res
2008, 68:10340-10357.
28. Pollard PJ, Ratcliffe PJ: Puzzling patterns of predisposition. Science 2009,
324:192-194.
29. Wagner KD, Wagner N, Wellman S, Schley G, Bondke A, Theres H, Scholz H:
Oxygen-regulated expression of the Wilm’s tumor suppressor Wt1
involves hypoxia-inducible factor-1 (HIF-1). FASEB J 2003, 17:1364-1366.
30. Couronné L, Lippert E, Andrieux J, Kosmider O, Radford-Weiss I, Penther D,
et al: Analyses of TET2 mutations in post-myeloproliferative neoplasm
acute myeloid leukemias. Leukemia 2010, 24:201-203.
31. Bernard F, Gelsi-Boyer V, Murati A, Giraudier S, Trouplin V, Adélaïde J, Rey J,
Olschwang S, Vainchenker W, Chaffanet M, Vey N, Mozziconacci MJ,
Birnbaum D: Alterations of NFIA in chronic malignant myeloid diseases.
Leukemia 2009, 23:583-585.
32. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C,
Edkins S, Hardy C, O’Meara S, Teague J, Butler A, Hinton J, Latimer C,
Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Cole J, Forbes S,
Jia M, Jones D, Kok CY, Leroy C, Lin ML, McBride DJ, Maddison M,
Maquire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L,
Pleasance E, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G,
Tarpey PS, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Collins VP,
Ichimura K, Law S, Wong J, Yuen ST, Leung SY, Tonon G, DePinho RA,
Tai YT, Anderson KC, Kahnoski RJ, Massie A, Khoo SK, Teh BT, Stratton MR,
Futreal PA: Somatic mutations of the histone H3K27 demethylase gene
UTX in human cancer. Nat Genet 2009, 41:521-523.
33. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV,
Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG,
Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NCP:
Inactivating mutations of the histone methyltransferase gene EZH2 in
myeloid disorders. Nat Genet 2010.
34. Nikoloski G, Langemeijer SMC, Kuiper RP, Knops R, Massop M,
Tönnissen ERLTM, van der Heijden A, Scheele TN, Vandenberghe P, de
Witte T, van der Reijden BA, Jansen JH: Somatic mutations of the histone
methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet
2010.
35. Ding Y, Harada Y, Imagawa J, Kimura A, Harada H: AML1/RUNX1 point
mutation possibly promotes leukemic transformation in
myeloproliferative neoplasms. Blood 2009, 114:5201-5205.
36. Abdel-Wahab O, Mansouri T, Patel J, Harris K, Yao J, Hedvat C, Hedvat C,
Heguy A, Bueso-Ramos C, Kantarjian H, Levine RL, Verstovsek S: Genetic
analysis of transforming events that convert chronic myeloproliferative
neoplasms to leukemias. Cancer Res 2010, 70:447-452.
37. Dang L, White DW, Gross S, Bennett BD, Bittinger MAA, Driggers EM,
Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE,
Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG,
Su SM: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature 2009, 462:739-744.
38. Grisendi S, Mecucci C, Falini B, Pandolfi PP: Nucleophosmin and cancer.
Nat Rev Cancer 2006, 494:493-505.
39. Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, Hess JL,
Humphries RK, Brock HW: Loss-of-function Additional sex coombs-like 1
mutations disrupt hematopoiesis but do not cause severe
myelodysplasia or leukemia. Blood 2010, 115:38-46.
Pre-publication history
The pre-publication history for this paper can be accessed here:
http://www.biomedcentral.com/1471-2407/10/401/prepub
doi:10.1186/1471-2407-10-401Cite this article as: Rocquain et al.: Combined mutations of ASXL1, CBL,FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genesin myelodysplastic syndromes and acute myeloid leukemias. BMCCancer 2010 10:401.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit
Rocquain et al. BMC Cancer 2010, 10:401
http://www.biomedcentral.com/1471-2407/10/401
Page 7 of 7
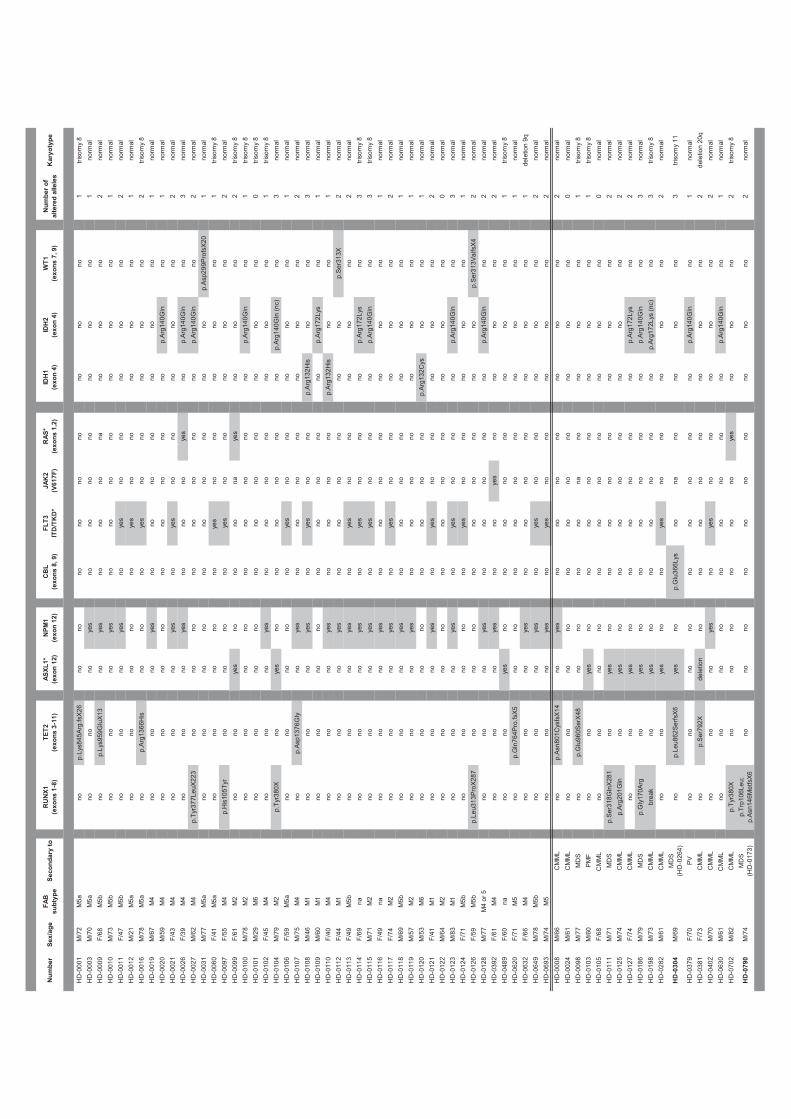
Nu
mb
erS
ex/a
ge
FA
B
sub
typ
eS
eco
nd
ary
toR
UN
X1
(exo
ns
1-8)
TE
T2
(exo
ns
3-11
)A
SX
L1*
(exo
n 1
2)N
PM
1(e
xon
12)
CB
L(e
xon
s 8,
9)
FL
T3
ITD
/TK
D*
JAK
2 (V
617F
)R
AS
*(e
xon
s 1,
2)ID
H1
(exo
n 4
)ID
H2
(exo
n 4
)W
T1
(exo
ns
7, 9
)N
um
ber
of
alte
red
alle
les
Kar
yoty
pe
HD
-00
01
M/7
2M
5a
no
p.L
ys8
45
Arg
.fsX
26
no
no
no
no
no
no
no
no
no
1tr
iso
my
8
HD
-00
03
M/7
0M
5a
no
no
no
yes
no
no
no
no
no
no
no
1n
orm
al
HD
-00
09
F/6
8M
5b
no
p.L
ys9
59
Glu
X1
3n
oye
sn
on
on
on
an
on
on
o2
no
rma
l
HD
-00
10
M/7
3M
5b
no
no
no
yes
no
no
no
no
no
no
no
1n
orm
al
HD
-00
11
F/4
7M
5b
no
no
no
yes
no
yes
no
no
no
no
no
2n
orm
al
HD
-00
12
M/2
1M
5a
no
no
no
no
no
yes
no
no
no
no
no
1n
orm
al
HD
-00
16
M/7
8M
5a
no
p.A
rg1
36
6H
isn
on
on
oye
sn
on
on
on
on
o2
tris
om
y 8
HD
-00
19
M/6
7M
4n
on
on
oye
sn
on
on
on
on
on
on
o1
no
rma
l
HD
-00
20
M/5
9M
4n
on
on
on
on
on
on
on
on
op
.Arg
14
0G
lnn
o1
no
rma
l
HD
-00
21
F/4
3M
4n
on
on
oye
sn
oye
sn
on
on
on
on
o2
no
rma
l
HD
-00
26
F/3
9M
4n
on
on
oye
sn
on
on
oye
sn
op
.Arg
14
0G
lnn
o3
no
rma
l
HD
-00
27
M/6
2M
4p
.Tyr
37
7L
eu
X2
23
no
no
no
no
no
no
no
no
p.A
rg1
40
Gln
no
2n
orm
al
HD
-00
31
M/7
7M
5a
no
no
no
no
no
no
no
no
no
no
p.A
sp
29
9P
rofs
X2
01
no
rma
l
HD
-00
60
F/4
1M
5a
no
no
no
no
no
yes
no
no
no
no
no
1tr
iso
my
8
HD
-00
97
F/5
5M
4p
.His
10
5T
yrn
on
on
on
oye
sn
on
on
on
on
o2
no
rma
l
HD
-00
99
F/6
1M
2n
on
oye
sn
on
on
on
aye
sn
on
on
o2
tris
om
y 8
HD
-01
00
M/7
8M
2n
on
on
on
on
on
on
on
on
op
.Arg
14
0G
lnn
o1
tris
om
y 8
HD
-01
01
M/2
9M
6n
on
on
on
on
on
on
on
on
on
on
o0
tris
om
y 8
HD
-01
02
F/4
5M
4n
on
on
oye
sn
on
on
on
on
on
on
o1
tris
om
y 8
HD
-01
04
M/7
9M
2p
.Tyr
38
0X
no
yes
no
no
no
no
no
no
p.A
rg1
40
Gln
(n
c)
no
3n
orm
al
HD
-01
06
F/5
9M
5a
no
no
no
no
no
yes
no
no
no
no
no
1n
orm
al
HD
-01
07
M/7
5M
4n
op
.Asp
13
76
Gly
n
oye
sn
on
on
on
on
on
on
o2
no
rma
l
HD
-01
08
M/4
6M
1n
on
on
oye
sn
oye
sn
on
op
.Arg
13
2H
isn
on
o3
no
rma
l
HD
-01
09
M/6
0M
1n
on
on
on
on
on
on
on
on
op
.Arg
17
2L
ysn
o1
no
rma
l
HD
-01
10
F/4
0M
4n
on
on
oye
sn
on
on
on
op
.Arg
13
2H
isn
on
o1
no
rma
l
HD
-01
12
F/4
4M
1n
on
on
oye
sn
on
on
on
on
on
op
.Se
r31
3X
2n
orm
al
HD
-01
13
F/4
9M
5b
no
no
no
yes
no
yes
no
no
no
no
no
2n
orm
al
HD
-01
14
F/6
9n
an
on
on
oye
sn
oye
sn
on
on
op
.Arg
17
2L
ysn
o3
tris
om
y 8
HD
-01
15
M/7
1M
2n
on
on
oye
sn
oye
sn
on
on
op
.Arg
14
0G
lnn
o3
tris
om
y 8
HD
-01
16
F/4
9n
an
on
on
oye
sn
on
on
on
on
on
on
o1
no
rma
l
HD
-01
17
F/7
4M
2n
on
on
oye
sn
oye
sn
on
on
on
on
o2
no
rma
l
HD
-01
18
M/6
9M
5b
no
no
no
yes
no
no
no
no
no
no
no
1n
orm
al
HD
-01
19
M/5
7M
2n
on
on
oye
sn
on
on
on
on
on
on
o1
no
rma
l
HD
-01
20
M/5
3M
6n
on
on
on
on
on
on
on
op
.Arg
13
2C
ysn
on
o1
no
rma
l
HD
-01
21
F/4
1M
1n
on
on
oye
sn
oye
sn
on
on
on
on
o2
no
rma
l
HD
-01
22
M/6
4M
2n
on
on
on
on
on
on
on
on
on
on
o0
no
rma
l
HD
-01
23
M/8
3M
1n
on
on
oye
sn
oye
sn
on
on
op
.Arg
14
0G
lnn
o3
no
rma
l
HD
-01
24
F/7
1M
5b
no
no
no
no
no
yes
no
no
no
no
no
1n
orm
al
HD
-01
26
F/5
9M
5b
p.L
eu
31
3P
roX
28
7n
on
on
on
on
on
on
on
on
op
.Se
r31
3V
alfsX
42
no
rma
l
HD
-01
28
M/7
7M
4 o
r 5
no
no
no
yes
no
no
no
no
no
p.A
rg1
40
Gln
no
2n
orm
al
HD
-03
92
F/6
1M
4n
on
on
oye
sn
on
oye
sn
on
on
on
o2
no
rma
l
HD
-04
89
F/6
0n
an
on
oye
sn
on
on
on
on
on
on
on
o1
tris
om
y 8
HD
-06
20
F/7
1M
5
no
p.G
ln7
64
Pro
.fsX
5n
on
on
on
on
on
on
on
on
o1
no
rma
l
HD
-06
32
F/6
6M
4n
on
on
oye
sn
on
on
on
on
on
on
o1
de
letio
n 9
q
HD
-06
49
M/7
8M
5b
no
no
no
yes
no
yes
no
no
no
no
no
2n
orm
al
HD
-06
93
M/7
4M
5n
on
on
oye
sn
oye
sn
on
on
on
on
o2
no
rma
l
HD
-00
08
M/6
6C
MM
Ln
op
.Asn
80
1C
ysfs
X1
4n
oye
sn
on
on
on
on
on
on
o2
no
rma
l
HD
-00
24
M/6
1C
MM
Ln
on
on
on
on
on
on
on
on
on
on
o0
no
rma
l
HD
-00
98
M/7
7M
DS
no
p.G
lu9
60
Se
rX4
8n
on
on
on
on
an
on
on
on
o1
tris
om
y 8
HD
-01
03
M/6
0P
MF
no
no
yes
no
no
no
no
no
no
no
no
1tr
iso
my
8
HD
-01
05
F/6
8C
MM
L
no
no
no
no
no
no
no
no
no
no
no
0n
orm
al
HD
-01
11
M/7
1M
DS
p.S
er3
18
Gln
X2
81
no
yes
no
no
no
no
no
no
no
no
2n
orm
al
HD
-01
25
M/7
4C
MM
Lp
.Arg
20
1G
lnn
oye
sn
on
on
on
on
on
on
on
o2
no
rma
l
HD
-01
27
F/7
4C
MM
Ln
on
oye
sn
on
on
on
on
on
op
.Arg
17
2L
ysn
o2
no
rma
l
HD
-01
86
M/7
9M
DS
p.G
ly1
70
Arg
no
yes
no
no
no
no
no
no
p.A
rg1
40
Gln
no
3n
orm
al
HD
-01
98
M/7
3C
MM
Lb
rea
kn
oye
sn
on
on
on
on
on
op
.Arg
17
2L
ys (
nc)
no
3tr
iso
my
8
HD
-02
82
M/6
1C
MM
Ln
on
oye
sn
on
oye
sn
on
on
on
on
o2
no
rma
l
HD
-030
4M
/69
MD
S
(HD
-02
64
)n
op
.Le
u8
62
Se
rfsX
6ye
sn
op
.Glu
36
6L
ysn
on
an
on
on
on
o3
tris
om
y 1
1
HD
-03
79
F/7
0P
Vn
on
on
on
on
on
on
on
on
op
.Arg
14
0G
lnn
o1
no
rma
l
HD
-03
81
F/7
3C
MM
Ln
o p
.Se
r79
2X
de
letio
nn
on
on
on
on
on
on
on
o2
de
letio
n 2
0q
HD
-04
02
M/7
0C
MM
Ln
on
on
oye
sn
oye
sn
on
on
on
on
o2
no
rma
l
HD
-06
30
M/6
1C
MM
L
no
no
no
no
no
no
no
no
no
p.A
rg1
40
Gln
no
1n
orm
al
HD
-07
02
M/8
2C
MM
Lp
.Tyr
38
0X
no
no
no
no
no
no
yes
no
no
no
2tr
iso
my
8
HD
-079
0M
/74
MD
S
(HD
-01
73
)
p.T
rp1
06
Le
u;
p.A
sn
14
6M
etf
sX
6n
on
on
on
on
on
on
on
on
on
o2
no
rma
l
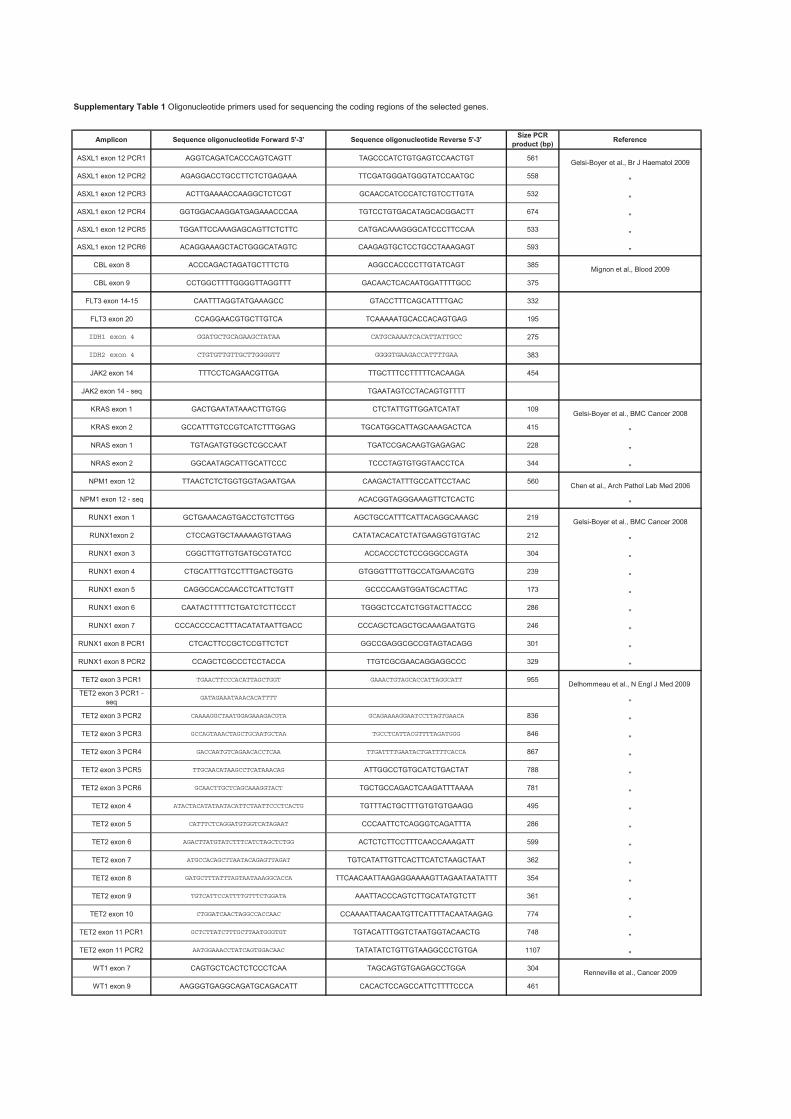
Amplicon Sequence oligonucleotide Forward 5'-3' Sequence oligonucleotide Reverse 5'-3'Size PCR
product (bp)Reference
ASXL1 exon 12 PCR1 AGGTCAGATCACCCAGTCAGTT TAGCCCATCTGTGAGTCCAACTGT 561Gelsi-Boyer et al., Br J Haematol 2009
ASXL1 exon 12 PCR2 AGAGGACCTGCCTTCTCTGAGAAA TTCGATGGGATGGGTATCCAATGC 558"
ASXL1 exon 12 PCR3 ACTTGAAAACCAAGGCTCTCGT GCAACCATCCCATCTGTCCTTGTA 532"
ASXL1 exon 12 PCR4 GGTGGACAAGGATGAGAAACCCAA TGTCCTGTGACATAGCACGGACTT 674"
ASXL1 exon 12 PCR5 TGGATTCCAAAGAGCAGTTCTCTTC CATGACAAAGGGCATCCCTTCCAA 533"
ASXL1 exon 12 PCR6 ACAGGAAAGCTACTGGGCATAGTC CAAGAGTGCTCCTGCCTAAAGAGT 593"
CBL exon 8 ACCCAGACTAGATGCTTTCTG AGGCCACCCCTTGTATCAGT 385Mignon et al., Blood 2009
CBL exon 9 CCTGGCTTTTGGGGTTAGGTTT GACAACTCACAATGGATTTTGCC 375
FLT3 exon 14-15 CAATTTAGGTATGAAAGCC GTACCTTTCAGCATTTTGAC 332
FLT3 exon 20 CCAGGAACGTGCTTGTCA TCAAAAATGCACCACAGTGAG 195
IDH1 exon 4 GGATGCTGCAGAAGCTATAA CATGCAAAATCACATTATTGCC 275
IDH2 exon 4 CTGTGTTGTTGCTTGGGGTT GGGGTGAAGACCATTTTGAA 383
JAK2 exon 14 TTTCCTCAGAACGTTGA TTGCTTTCCTTTTTCACAAGA 454
JAK2 exon 14 - seq TGAATAGTCCTACAGTGTTTT
KRAS exon 1 GACTGAATATAAACTTGTGG CTCTATTGTTGGATCATAT 109Gelsi-Boyer et al., BMC Cancer 2008
KRAS exon 2 GCCATTTGTCCGTCATCTTTGGAG TGCATGGCATTAGCAAAGACTCA 415"
NRAS exon 1 TGTAGATGTGGCTCGCCAAT TGATCCGACAAGTGAGAGAC 228"
NRAS exon 2 GGCAATAGCATTGCATTCCC TCCCTAGTGTGGTAACCTCA 344"
NPM1 exon 12 TTAACTCTCTGGTGGTAGAATGAA CAAGACTATTTGCCATTCCTAAC 560Chen et al., Arch Pathol Lab Med 2006
NPM1 exon 12 - seq ACACGGTAGGGAAAGTTCTCACTC"
RUNX1 exon 1 GCTGAAACAGTGACCTGTCTTGG AGCTGCCATTTCATTACAGGCAAAGC 219Gelsi-Boyer et al., BMC Cancer 2008
RUNX1exon 2 CTCCAGTGCTAAAAAGTGTAAG CATATACACATCTATGAAGGTGTGTAC 212"
RUNX1 exon 3 CGGCTTGTTGTGATGCGTATCC ACCACCCTCTCCGGGCCAGTA 304"
RUNX1 exon 4 CTGCATTTGTCCTTTGACTGGTG GTGGGTTTGTTGCCATGAAACGTG 239"
RUNX1 exon 5 CAGGCCACCAACCTCATTCTGTT GCCCCAAGTGGATGCACTTAC 173"
RUNX1 exon 6 CAATACTTTTTCTGATCTCTTCCCT TGGGCTCCATCTGGTACTTACCC 286"
RUNX1 exon 7 CCCACCCCACTTTACATATAATTGACC CCCAGCTCAGCTGCAAAGAATGTG 246"
RUNX1 exon 8 PCR1 CTCACTTCCGCTCCGTTCTCT GGCCGAGGCGCCGTAGTACAGG 301"
RUNX1 exon 8 PCR2 CCAGCTCGCCCTCCTACCA TTGTCGCGAACAGGAGGCCC 329"
TET2 exon 3 PCR1 TGAACTTCCCACATTAGCTGGT GAAACTGTAGCACCATTAGGCATT 955Delhommeau et al., N Engl J Med 2009
TET2 exon 3 PCR1 -
seqGATAGAAATAAACACATTTT
"
TET2 exon 3 PCR2 CAAAAGGCTAATGGAGAAAGACGTA GCAGAAAAGGAATCCTTAGTGAACA 836"
TET2 exon 3 PCR3 GCCAGTAAACTAGCTGCAATGCTAA TGCCTCATTACGTTTTAGATGGG 846"
TET2 exon 3 PCR4 GACCAATGTCAGAACACCTCAA TTGATTTTGAATACTGATTTTCACCA 867"
TET2 exon 3 PCR5 TTGCAACATAAGCCTCATAAACAG ATTGGCCTGTGCATCTGACTAT 788"
TET2 exon 3 PCR6 GCAACTTGCTCAGCAAAGGTACT TGCTGCCAGACTCAAGATTTAAAA 781"
TET2 exon 4 ATACTACATATAATACATTCTAATTCCCTCACTG TGTTTACTGCTTTGTGTGTGAAGG 495"
TET2 exon 5 CATTTCTCAGGATGTGGTCATAGAAT CCCAATTCTCAGGGTCAGATTTA 286"
TET2 exon 6 AGACTTATGTATCTTTCATCTAGCTCTGG ACTCTCTTCCTTTCAACCAAAGATT 599"
TET2 exon 7 ATGCCACAGCTTAATACAGAGTTAGAT TGTCATATTGTTCACTTCATCTAAGCTAAT 362"
TET2 exon 8 GATGCTTTATTTAGTAATAAAGGCACCA TTCAACAATTAAGAGGAAAAGTTAGAATAATATTT 354"
TET2 exon 9 TGTCATTCCATTTTGTTTCTGGATA AAATTACCCAGTCTTGCATATGTCTT 361"
TET2 exon 10 CTGGATCAACTAGGCCACCAAC CCAAAATTAACAATGTTCATTTTACAATAAGAG 774"
TET2 exon 11 PCR1 GCTCTTATCTTTGCTTAATGGGTGT TGTACATTTGGTCTAATGGTACAACTG 748"
TET2 exon 11 PCR2 AATGGAAACCTATCAGTGGACAAC TATATATCTGTTGTAAGGCCCTGTGA 1107"
WT1 exon 7 CAGTGCTCACTCTCCCTCAA TAGCAGTGTGAGAGCCTGGA 304Renneville et al., Cancer 2009
WT1 exon 9 AAGGGTGAGGCAGATGCAGACATT CACACTCCAGCCATTCTTTTCCCA 461
Supplementary Table 1 Oligonucleotide primers used for sequencing the coding regions of the selected genes.
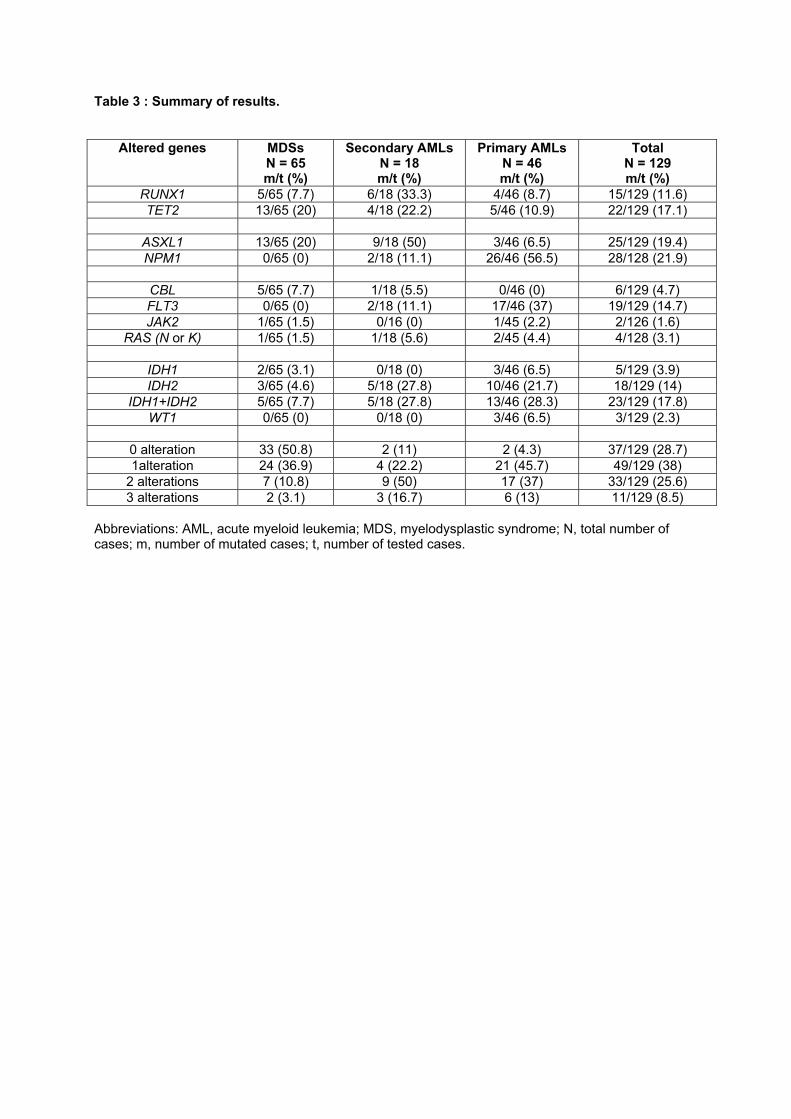
Table 3 : Summary of results.
Altered genes MDSs N = 65 m/t (%)
Secondary AMLs N = 18 m/t (%)
Primary AMLs N = 46 m/t (%)
Total N = 129 m/t (%)
RUNX1 5/65 (7.7) 6/18 (33.3) 4/46 (8.7) 15/129 (11.6)
TET2 13/65 (20) 4/18 (22.2) 5/46 (10.9) 22/129 (17.1)
ASXL1 13/65 (20) 9/18 (50) 3/46 (6.5) 25/129 (19.4)
NPM1 0/65 (0) 2/18 (11.1) 26/46 (56.5) 28/128 (21.9)
CBL 5/65 (7.7) 1/18 (5.5) 0/46 (0) 6/129 (4.7)
FLT3 0/65 (0) 2/18 (11.1) 17/46 (37) 19/129 (14.7)
JAK2 1/65 (1.5) 0/16 (0) 1/45 (2.2) 2/126 (1.6)
RAS (N or K) 1/65 (1.5) 1/18 (5.6) 2/45 (4.4) 4/128 (3.1)
IDH1 2/65 (3.1) 0/18 (0) 3/46 (6.5) 5/129 (3.9)
IDH2 3/65 (4.6) 5/18 (27.8) 10/46 (21.7) 18/129 (14)
IDH1+IDH2 5/65 (7.7) 5/18 (27.8) 13/46 (28.3) 23/129 (17.8)
WT1 0/65 (0) 0/18 (0) 3/46 (6.5) 3/129 (2.3)
0 alteration 33 (50.8) 2 (11) 2 (4.3) 37/129 (28.7)
1alteration 24 (36.9) 4 (22.2) 21 (45.7) 49/129 (38)
2 alterations 7 (10.8) 9 (50) 17 (37) 33/129 (25.6)
3 alterations 2 (3.1) 3 (16.7) 6 (13) 11/129 (8.5)
Abbreviations: AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; N, total number of cases; m, number of mutated cases; t, number of tested cases.

83
Commentaires et discussion de l’article n°2
Les SMD et les LAM sont la conséquence d’une succession d’altérations génétiques
touchant un clone de CSH. Les anomalies inaugurales, ainsi que leur ordre d’apparition, leur
nombre et leurs associations dans l’évolution de la maladie, sont inconnues. Plusieurs
mutations de gènes ont récemment été identifiées dans les SMD et les LAM. Mais, aucune
étude n’a analysé l’ensemble de ces mutations sur un panel de LAM et SMD dans le but
d’identifier leurs combinaisons.
Notre équipe a analysé par séquençage direct les combinaisons des mutations de 12
gènes connus pour être mutés dans les HM. Nous avons séquencé avec la même technique
que l’article précédent (séquenceur API 3130XL, Applied Biosystems), dans une série de
129 HM, les gènes ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1,
TET2 et WT1. Les 129 HM se répartissaient en 65 SMD et 64 LAM à caryotype
intermédiaire.
Le mécanisme de leucémogenèse actuellement reconnu est un processus multi-étapes.
Comme vu plus haut, le seul modèle actuellement décrit est le modèle de Gilliland, qui
présente deux classes de mutations (two-hit model), nécessaires au développement d’une
leucémie (Gilliland et al, 2002), la cellule souche ou progénitrice devant subir une mutation
de chaque étape pour devenir leucémogène.
Cependant, plusieurs mutations de gènes récemment décrites dont celles de WT1 et
d’IDH1 et 2, n’étant ni des facteurs ou cofacteurs de transcription ni en relation avec des TK,
ne peuvent être classées selon ce modèle alors qu’elles interviennent dans le processus
leucémogène. De plus les mutations de classe II concernent des gènes aux fonctions très
différentes. L’identification de certaines nouvelles mutations impliquées dans la
physiopathologie des SMD et de LAM suggère de les regrouper dans de nouvelles classes de
mutations.
Grâce aux combinaisons de mutations que nous avons identifiées, nous avons émis
l’hypothèse d’un modèle de leucémogenèse à quatre classes différentes de mutations dans
lequel la pathologie serait la résultante d’une combinaison de mutations de chaque classe
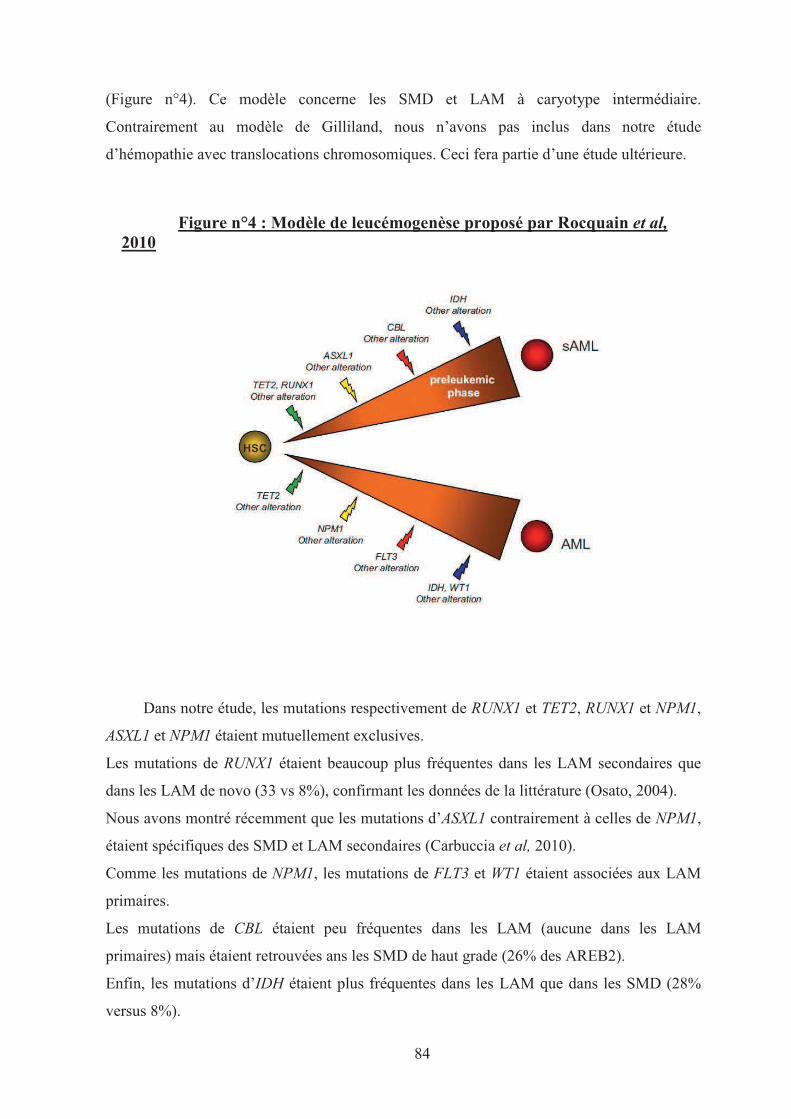
84
(Figure n°4). Ce modèle concerne les SMD et LAM à caryotype intermédiaire.
Contrairement au modèle de Gilliland, nous n’avons pas inclus dans notre étude
d’hémopathie avec translocations chromosomiques. Ceci fera partie d’une étude ultérieure.
Figure n°4 : Modèle de leucémogenèse proposé par Rocquain et al,
2010
Dans notre étude, les mutations respectivement de RUNX1 et TET2, RUNX1 et NPM1,
ASXL1 et NPM1 étaient mutuellement exclusives.
Les mutations de RUNX1 étaient beaucoup plus fréquentes dans les LAM secondaires que
dans les LAM de novo (33 vs 8%), confirmant les données de la littérature (Osato, 2004).
Nous avons montré récemment que les mutations d’ASXL1 contrairement à celles de NPM1,
étaient spécifiques des SMD et LAM secondaires (Carbuccia et al, 2010).
Comme les mutations de NPM1, les mutations de FLT3 et WT1 étaient associées aux LAM
primaires.
Les mutations de CBL étaient peu fréquentes dans les LAM (aucune dans les LAM
primaires) mais étaient retrouvées ans les SMD de haut grade (26% des AREB2).
Enfin, les mutations d’IDH étaient plus fréquentes dans les LAM que dans les SMD (28%
versus 8%).

85
A partir de ces différentes constatations, nous avons reclassé ces mutations en quatre
classes :
Les mutations de classe I (en vert sur le schéma) incluent des gènes à l’origine de la
clonalité de la maladie (TET2 et RUNX1) (Delhommeau et al, 2009).
Les mutations de classe II (NPM1 et ASXL1) permettent de différencier les LAM primaires
des LAM secondaires avec présence d’une phase chronique (en jaune sur le schéma)
(Carbuccia et al, 2010).
Les mutations de classe III (en rouge sur le schéma) correspondent à celles de la classe I de
Gilliland et concernent les gènes codant des protéines TK ou leur effecteurs (CBL, FLT3,
JAK2, RAS).
Les mutations de classe IV (en bleu sur le schéma) regroupent des mutations de gène
potentiellement impliqués dans l’angiogenèse tumorale comme celles d’IDH1/2 (Pollard et
al, 2009) ou dans d’autres mécanismes physiologiques, mais concernent surtout la phase
leucémique ou de transformation leucémique.
Il n’y a pas d’ordre établi dans la survenue de ces mutations hormis pour les mutations
de classe 4 qui surviendraient plutôt à la phase d’acutisation. Notre étude ne répertorie aucun
patient avec des mutations des quatre classes. Mais globalement, les mutations étaient
beaucoup plus fréquentes dans les LAM que dans les SMD, ce qui est en rapport avec les
données de la littérature :
Neuf (14%) des patients atteints de LAM et deux (10%) des patients atteints d’AREB2
présentaient des mutations de trois classes.
Seulement 10% des SMD mais près de la moitié des LAM présentaient des mutations de
deux classes.
Plusieurs mutations de gènes telles que celles de CEBP�, MLL, KIT ou P53 n’ont pas
été analysées dans notre étude. Celles-ci ainsi que de nombreuses mutations de gène non
connus pourront peut-être s’intégrer à ce modèle et le confirmer.
De plus, il serait intéressant d’étudier plus d’échantillons avec phase chronique et phase
aiguë chez un même patient (seulement deux dans notre étude) pour corroborer notre
modèle.
Notre étude représente néanmoins le premier modèle (à quatre classes de mutations)
permettant de différencier les LAM primaires des LAM secondaires à une hémopathie
chronique. Il constitue une première étape dans l’élaboration d’un statut mutationnel chez les
patients atteints de SMD et LAM.

86
ARTICLE N°3
Alteration of cohesin genes in myeloid diseases.
Rocquain J, Gelsi-Boyer V, Adélaïde J, Murati A, Carbuccia N, Vey N, Birnbaum D, Mozziconacci MJ, Chaffanet M.
Am J Hematol 2010; 85(9): 717-9.

Alteration of cohesin genes in myeloid diseases
Julien Rocquain,1 Veronique Gelsi-Boyer,1–3 Jose Adelaıde,1Anne Murati,1,2 Nadine Carbuccia,1
Norbert Vey,3,4Daniel Birnbaum,1Marie-Joelle Mozziconacci,1,2* and Max Chaffanet1
New genes involved in leukemogenesis, such as ASXL1 and TET2,
have been identified recently using genomic analyses of DNA from
patient samples. We have studied by array-comparative genomic
hybridization (aCGH) a series of 167 samples including myelodysplas-
tic syndromes, chronic myelomonocytic leukemias, and acute myeloid
leukemias. We found a deletion of the RAD21 and STAG2 genes, which
encode two components of the cohesin complex. We propose that
these alterations may compromise the cohesin complex and its regula-
tion of the transcription of genes.
Several new genes involved in myeloid leukemogenesis, including ASXL1,
CBL, and TET2, have been identified following genomics studies using
aCGH or single nucleotide polymorphism-arrays combined with sequence
analyses [1–3]. Mutation analyses have identified other genes, such as
IDH1 [4]. No alteration has been identified yet in many cases of malignant
myeloid disease and many other genes remain to be discovered.
Using aCGH, we searched for genome alterations in a series of 167
malignant myeloid diseases. We describe here two cases with loss of genes
encoding components of the same protein complex.
About two-thirds of the samples did not show any aCGH copy number aber-
rations (CNA): 41 of 63 MDSs (65%) (Supporting Information Table I), 37 of
53 CMMLs (�70%) (Supporting Information Table II), 35 of the 51 AMLs
(�69%) (Supporting Information Table III). This high proportion of no-CNA
profile in AMLs is probably due to our selection of cases with normal karyo-
type. In all cases, losses of whole or part of chromosome (e.g., 20q losses)
were found in agreement with the results of the karyotyping (except for HD-
0627 where the del20q could only be suspected on the aCGH profile).
Small deletions were more interesting. They were rare but indicative. We
found a deletion of TET2 in one myelodysplastic syndrome (MDS) and in one
chronic myelomonocytic leukemia (CMML), and a deletion of ASXL1 in one
MDS and in one acute myeloid leukemia (AML; Table I). We also found deletions
of NF1, RB1, RUNX1, and UTX. Some of these CNAs have been described in
our previous reports [1,5,6]. We here focused on two particular CNAs.
CMML HD-0259 was diagnosed in 2007 in a 70 year-old patient with no
particular antecedent. aCGH showed a small heterozygous deletion at 8q24
(chr8: 117768619-117969545 Mb), which comprised three genes including
RAD21 (Fig. 1A). The CMML evolved in an M5 FAB AML (HD-0402) in
2008. aCGH again revealed the RAD21 loss but no additional alteration.
The patient died in 2008, 6 months after acute transformation. AML HD-
0120 (M6 FAB) was diagnosed in 2005 in a 55 year-old patient without any
antecedent. aCGH detected a small deletion centered on the STAG2 gene,
at Xq25 (chrX: 122787860-122970717 Mb) (Fig. 1B). The patient died in
2007, 5 months after relapse. In both cases, the karyotype did not show any
abnormality and no other aCGH alteration was noticed.
HD-0120 had an IDH1 mutation (R132C). HD-0259 was the only CMML
case with an NPM1 exon 12 mutation; a FLT3-ITD was additionally detected
in the corresponding acute phase (HD-0402). There were no mutations in
the other studied genes associated with leukemogenesis. We searched for
mutations of RAD21 in 95 samples but did not find any.
At first look, deletions of RAD21 and STAG2 could appear as isolated
cases with limited interest. However, close examination revealed the involve-
ment of a new pathway of leukemogenesis. Indeed, the two genes happen to
encode subunits of the same nuclear complex, cohesin [7]. Cohesin is made
of four evolutionary-conserved core subunits, SMC1, SMC3, RAD21, and
STAG1/STAG2 (Fig. 2), and plays a major role in chromosome biology [7].
Abnormal functioning of cohesin compromises faithful segregation of sister
chromatids during cell division. Mutations in cohesin genes are responsible
for the Cornelia de Lange syndrome (CdLs), a multisystem anomaly disorder
with abnormal phenotypical and behavioral features, and may be associated
with chromosome instability in colorectal cancer [7,8]. However, we did not
detect any aneusomy in the two cases. But cohesin has other roles. Cohesin
participates in double-strand DNA break repair and long-distance regulation of
gene expression. In zebrafish, monoallelic Rad21 inactivation downregulates
Runx1 expression [9]. Cohesin is necessary for the function of the enhancer-
blocking transcriptional insulator CTCF, which is an interactor of nucleophos-
min and a positive regulator of tumor suppressor loci [10–12]. Cohesin can
also acts on transcription independently of CTCF [13]. Thus, loss of RAD21
or STAG2 could participate in leukemogenesis through enhancement of inacti-
vation of NPM1, RUNX1, and other tumor suppressor genes, such as RB1 or
CDKN2A/B. Loss of RAD21 co-occurred with an NPM1 mutation in case HD-
0259 showing that the two alterations can indeed cooperate. Abnormal
repression of transcription could be a general mechanism of leukemogenesis.
Alternatively, cohesin may silence oncogenes. The activity of cohesin/CTCF is
regulated by polo kinase 1, which interacts with CEP170 [5] and is coordi-
nated with epigenetic marks [10–12], which is in agreement with current views
of oncogenesis [14,15]. Thus, abnormal cohesin function may be involved in
the same pathway as TET2 or ASXL1.
To extend and validate our results, we surveyed published genomic stud-
ies of myeloid malignancies for alterations of cohesin genes. We found that
one similar small deletion at 8q24 including RAD21 [16] and one at Xq25
including STAG2 [17] have been found in independent series of 157 and 86
AMLs, respectively. The method used to detect these CNAs allowed the
conclusion that the deletions were acquired. In these studies, the involve-
ment of cohesin was not suggested.
In array studies of myeloid diseases [1,5,6,16,17], recurrence is overall
low but quite essential for identifying candidate target genes. Even if rare
(1%), alterations of cohesin genes are truly recurrent and found in three
independent series [16,17 this work]. These results point for the first time to
cohesin as a new player in leukemogenesis and contribute to establish the
comprehensive catalog of pathogenic genes. They provide an incentive to
look thoroughly for alterations (mutations?) in the genes encoding members
of the protein complex and its associated network (e.g., CTCF), as well as
to define the function of cohesin in hematopoiesis.
Materials and Methods
Panels of samples. We collected samples from 63 MDS, 53 CMML, and
51 AML. According to the WHO classification, the MDS panel comprised
five refractory anemia (RA), 13 RA with ring sideroblasts (RARS), six
refractory cytopenia with multilineage dysplasia (RCMD), 16 RA with
excess of blasts type 1 (RAEB1), 19 RA with excess of blasts type 2
(RAEB2), and 4 MDS-unclassified (MDS-U) cases. The 53 CMML panel
comprised 24 myelodysplastic (MD) forms (leucocytosis count <13 3 109/
L) and 29 myeloproliferative forms (leucocytosis count >13 3 109/L) (6).
The AML panel comprised 39 cases of primary AMLs and 12 of secondary
AMLs. It included 40 cases with normal karyotype and 10 cases with tris-
omy 8 and 1 with 20q deletion as a sole karyotypic abnormality. All
patients signed an informed consent and the study was approved by our
institutional review board.
TABLE I. Summary of Results
MDS CMML AML Total
Total number of studied samples 63 53 51 167
No CNA 41 (65%) 37 (69.8%) 35 (68.6%) 113 (67.7%)
ASXL1 loss 1 0 1 2
NF1 loss 0 2 0 2
RB1 loss 0 2 0 2
TET2 loss 1 1 0 2
Cohesin component loss 0 1 1 2
VVC 2010 Wiley-Liss, Inc.
American Journal of Hematology 1 http://wileyonlinelibrary.com/cgi-bin/jhome/35105
Letter
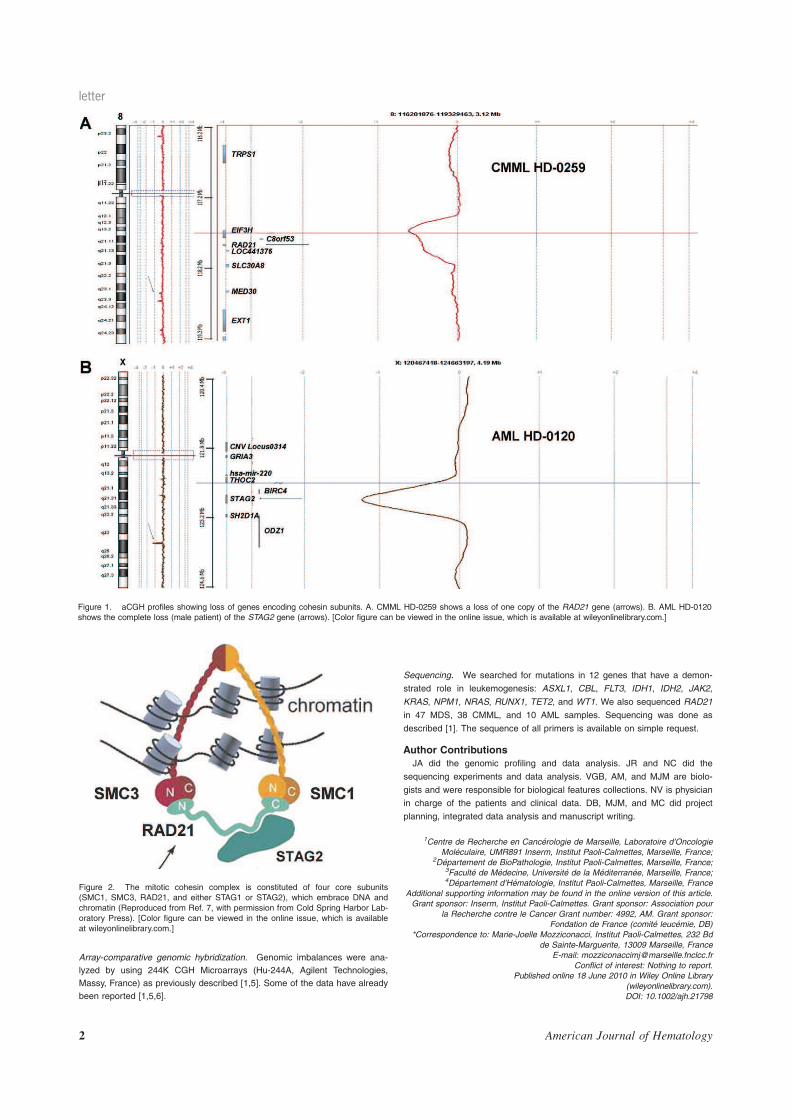
Array-comparative genomic hybridization. Genomic imbalances were ana-
lyzed by using 244K CGH Microarrays (Hu-244A, Agilent Technologies,
Massy, France) as previously described [1,5]. Some of the data have already
been reported [1,5,6].
Sequencing. We searched for mutations in 12 genes that have a demon-
strated role in leukemogenesis: ASXL1, CBL, FLT3, IDH1, IDH2, JAK2,
KRAS, NPM1, NRAS, RUNX1, TET2, and WT1. We also sequenced RAD21
in 47 MDS, 38 CMML, and 10 AML samples. Sequencing was done as
described [1]. The sequence of all primers is available on simple request.
Author Contributions
JA did the genomic profiling and data analysis. JR and NC did the
sequencing experiments and data analysis. VGB, AM, and MJM are biolo-
gists and were responsible for biological features collections. NV is physician
in charge of the patients and clinical data. DB, MJM, and MC did project
planning, integrated data analysis and manuscript writing.
1Centre de Recherche en Cancerologie de Marseille, Laboratoire d’Oncologie
Moleculaire, UMR891 Inserm, Institut Paoli-Calmettes, Marseille, France;2Departement de BioPathologie, Institut Paoli-Calmettes, Marseille, France;
3Faculte de Medecine, Universite de la Mediterranee, Marseille, France;4Departement d’Hematologie, Institut Paoli-Calmettes, Marseille, France
Additional supporting information may be found in the online version of this article.
Grant sponsor: Inserm, Institut Paoli-Calmettes. Grant sponsor: Association pour
la Recherche contre le Cancer Grant number: 4992, AM. Grant sponsor:
Fondation de France (comite leucemie, DB)
*Correspondence to: Marie-Joelle Mozziconacci, Institut Paoli-Calmettes, 232 Bd
de Sainte-Marguerite, 13009 Marseille, France
E-mail: [email protected]
Conflict of interest: Nothing to report.
Published online 18 June 2010 in Wiley Online Library
(wileyonlinelibrary.com).
DOI: 10.1002/ajh.21798
Figure 1. aCGH profiles showing loss of genes encoding cohesin subunits. A. CMML HD-0259 shows a loss of one copy of the RAD21 gene (arrows). B. AML HD-0120
shows the complete loss (male patient) of the STAG2 gene (arrows). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2. The mitotic cohesin complex is constituted of four core subunits
(SMC1, SMC3, RAD21, and either STAG1 or STAG2), which embrace DNA and
chromatin (Reproduced from Ref. 7, with permission from Cold Spring Harbor Lab-
oratory Press). [Color figure can be viewed in the online issue, which is available
at wileyonlinelibrary.com.]
letter
2 American Journal of Hematology

References
1. Gelsi-Boyer V, Trouplin V, Adelaıde J, et al. Mutations of polycomb-associated
gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leu-
kaemia. Br J Haematol 2009;145:788–800.
2. Makishima H, Cazzolli H, Szpurka H, et al. Mutations of E3 ligase Cbl family
members constitute a novel common pathogenic lesion in myeloid malignan-
cies. J Clin Oncol 2009;27:6109–6116.
3. Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid
cancers. N Engl J Med 2009;360:2289–2301.
4. Mardis ER, Ding L, Dooling DJ, et al. Recurrent mutations found by sequenc-
ing an acute myeloid leukemia genome. N Eng J Med 2009;361:1058–1066.
5. Gelsi-Boyer V, Trouplin V, Adelaıde J, et al. Genome profiling of chronic mye-
lomonocytic leukemia: frequent alterations of RAS and RUNX1 genes. BMC
Cancer 2008;8:299–314.
6. Carbuccia N, Trouplin V, Gelsi-Boyer V, et al. Mutual exclusion of ASXL1 and
NPM1 mutations in a series of acute myeloid leukemias. Leukemia 2010;24:
469–473.
7. Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chro-
mosome biology. Genes Dev 2008;22:3089–3114.
8. Barber TD, McManus K, Yuen KWY, et al. Chromatid cohesion defects may
underlie chromosome instability in human colorectal cancers. Proc Natl Acad
Sci USA 2008;105:3443–3448.
9. Horsfield JA, Anagnostou SH, Kuang-Hsien Hu J, et al. Cohesin-dependent
regulation of Runx genes. Development 2007;134:2639–2649.
10. Recillas-Targa F, De La Rosa-Velazquez IA, Soto-Teyes E, Benitez-Bribiesca
L. Epigenetic boundaries of tumour suppressor genes promoters: The CTCF
connection and its role in carcinogenesis. J Cell Mod Med 2006;3:552–566.
11. Zlatanova J, Caiafa P. CTCF and its protein partners; divide and rule? J Cell
Sci 2009;122:1275–1294.
12. Hakimi MA, Bochar DA, Schmiesing JA, et al. A chromatin remodelling com-
plex that loads cohesin onto human chromosomes. Nature 2002;418:994–998.
13. Schmidt D, Schwalie PC, Ross-Innes CS, et al. A CTCF-independent role for
cohesin in tissue-specific transcription. Genome Res 2010;20:578–588.
14. Acquaviva C, Gelsi-Boyer V, Birnbaum D. Myelodysplastic syndromes: Lost
between two states? Leukemia 2010;24:1–5.
15. Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify bio-
logically distinct subtypes in acute myeloid leukemias. Cancer Cell 2010;17:
13–27.
16. Bullinger L, Kronke J, Schon C, et al. Identification of acquired copy number
alterations and uniparental disomies in cytogenetically normal acute myeloid
leukemia using high-resolution single-nucleotide polymorphism analysis. Leu-
kemia 2010;24:438–449.
17. Walter MJ, Payton JE, Ries RE, et al. Acquired copy number alterations in
adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA 2009;106:
12950–12956.
letter
American Journal of Hematology 3
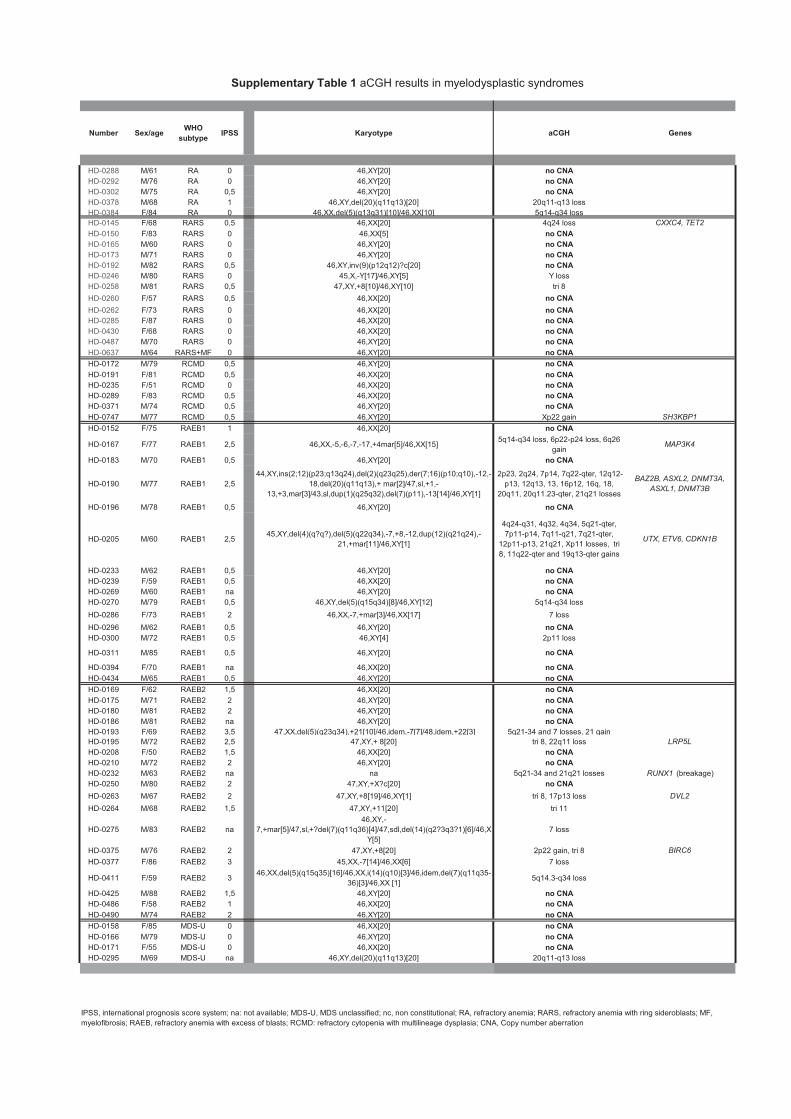
Number Sex/ageWHO
subtypeIPSS Karyotype aCGH Genes
HD-0288 M/61 RA 0 46,XY[20] no CNAHD-0292 M/76 RA 0 46,XY[20] no CNAHD-0302 M/75 RA 0,5 46,XY[20] no CNAHD-0378 M/68 RA 1 46,XY,del(20)(q11q13)[20] 20q11-q13 loss
HD-0384 F/84 RA 0 46,XX,del(5)(q13q31)[10]/46,XX[10] 5q14-q34 loss
HD-0145 F/68 RARS 0,5 46,XX[20] 4q24 loss CXXC4, TET2
HD-0150 F/83 RARS 0 46,XX[5] no CNAHD-0165 M/60 RARS 0 46,XY[20] no CNAHD-0173 M/71 RARS 0 46,XY[20] no CNAHD-0192 M/82 RARS 0,5 46,XY,inv(9)(p12q12)?c[20] no CNAHD-0246 M/80 RARS 0 45,X,-Y[17]/46,XY[5] Y loss
HD-0258 M/81 RARS 0,5 47,XY,+8[10]/46,XY[10] tri 8
HD-0260 F/57 RARS 0,5 46,XX[20] no CNA
HD-0262 F/73 RARS 0 46,XX[20] no CNAHD-0285 F/87 RARS 0 46,XX[20] no CNAHD-0430 F/68 RARS 0 46,XX[20] no CNAHD-0487 M/70 RARS 0 46,XY[20] no CNAHD-0637 M/64 RARS+MF 0 46,XY[20] no CNA
HD-0172 M/79 RCMD 0,5 46,XY[20] no CNAHD-0191 F/81 RCMD 0,5 46,XX[20] no CNAHD-0235 F/51 RCMD 0 46,XX[20] no CNAHD-0289 F/83 RCMD 0,5 46,XX[20] no CNAHD-0371 M/74 RCMD 0,5 46,XY[20] no CNAHD-0747 M/77 RCMD 0,5 46,XY[20] Xp22 gain SH3KBP1
HD-0152 F/75 RAEB1 1 46,XX[20] no CNA
HD-0167 F/77 RAEB1 2,5 46,XX,-5,-6,-7,-17,+4mar[5]/46,XX[15]5q14-q34 loss, 6p22-p24 loss, 6q26
gainMAP3K4
HD-0183 M/70 RAEB1 0,5 46,XY[20] no CNA
HD-0190 M/77 RAEB1 2,5
44,XY,ins(2;12)(p23;q13q24),del(2)(q23q25),der(7;16)(p10;q10),-12,-
18,del(20)(q11q13),+ mar[2]/47,sl,+1,-
13,+3,mar[3]/43,sl,dup(1)(q25q32),del(7)(p11),-13[14]/46,XY[1]
2p23, 2q24, 7p14, 7q22-qter, 12q12-
p13, 12q13, 13, 16p12, 16q, 18,
20q11, 20q11.23-qter, 21q21 losses
BAZ2B, ASXL2, DNMT3A,
ASXL1, DNMT3B
HD-0196 M/78 RAEB1 0,5 46,XY[20] no CNA
HD-0205 M/60 RAEB1 2,545,XY,del(4)(q?q?),del(5)(q22q34),-7,+8,-12,dup(12)(q21q24),-
21,+mar[11]/46,XY[1]
4q24-q31, 4q32, 4q34, 5q21-qter,
7p11-p14, 7q11-q21, 7q21-qter,
12p11-p13, 21q21, Xp11 losses, tri
8, 11q22-qter and 19q13-qter gains
UTX, ETV6, CDKN1B
HD-0233 M/62 RAEB1 0,5 46,XY[20] no CNAHD-0239 F/59 RAEB1 0,5 46,XX[20] no CNAHD-0269 M/60 RAEB1 na 46,XY[20] no CNAHD-0270 M/79 RAEB1 0,5 46,XY,del(5)(q15q34)[8]/46,XY[12] 5q14-q34 loss
HD-0286 F/73 RAEB1 2 46,XX,-7,+mar[3]/46,XX[17] 7 loss
HD-0296 M/62 RAEB1 0,5 46,XY[20] no CNAHD-0300 M/72 RAEB1 0,5 46,XY[4] 2p11 loss
HD-0311 M/85 RAEB1 0,5 46,XY[20] no CNA
HD-0394 F/70 RAEB1 na 46,XX[20] no CNAHD-0434 M/65 RAEB1 0,5 46,XY[20] no CNA
HD-0169 F/62 RAEB2 1,5 46,XX[20] no CNAHD-0175 M/71 RAEB2 2 46,XY[20] no CNAHD-0180 M/81 RAEB2 2 46,XY[20] no CNAHD-0186 M/81 RAEB2 na 46,XY[20] no CNAHD-0193 F/69 RAEB2 3,5 47,XX,del(5)(q23q34),+21[10]/46,idem,-7[7]/48,idem,+22[3] 5q21-34 and 7 losses, 21 gain
HD-0195 M/72 RAEB2 2,5 47,XY,+ 8[20] tri 8, 22q11 loss LRP5L
HD-0208 F/50 RAEB2 1,5 46,XX[20] no CNAHD-0210 M/72 RAEB2 2 46,XY[20] no CNAHD-0232 M/63 RAEB2 na na 5q21-34 and 21q21 losses RUNX1 (breakage)
HD-0250 M/80 RAEB2 2 47,XY,+X?c[20] no CNA
HD-0263 M/67 RAEB2 2 47,XY,+8[19]/46,XY[1] tri 8, 17p13 loss DVL2
HD-0264 M/68 RAEB2 1,5 47,XY,+11[20] tri 11
HD-0275 M/83 RAEB2 na
46,XY,-
7,+mar[5]/47,sl,+?del(7)(q11q36)[4]/47,sdl,del(14)(q2?3q3?1)[6]/46,X
Y[5]
7 loss
HD-0375 M/76 RAEB2 2 47,XY,+8[20] 2p22 gain, tri 8 BIRC6
HD-0377 F/86 RAEB2 3 45,XX,-7[14]/46,XX[6] 7 loss
HD-0411 F/59 RAEB2 346,XX,del(5)(q15q35)[16]/46,XX,i(14)(q10)[3]/46,idem,del(7)(q11q35-
36)[3]/46,XX [1]5q14.3-q34 loss
HD-0425 M/88 RAEB2 1,5 46,XY[20] no CNAHD-0486 F/58 RAEB2 1 46,XX[20] no CNAHD-0490 M/74 RAEB2 2 46,XY[20] no CNA
HD-0158 F/85 MDS-U 0 46,XX[20] no CNAHD-0166 M/79 MDS-U 0 46,XY[20] no CNAHD-0171 F/55 MDS-U 0 46,XX[20] no CNAHD-0295 M/69 MDS-U na 46,XY,del(20)(q11q13)[20] 20q11-q13 loss
IPSS, international prognosis score system; na: not available; MDS-U, MDS unclassified; nc, non constitutional; RA, refractory anemia; RARS, refractory anemia with ring sideroblasts; MF,
myelofibrosis; RAEB, refractory anemia with excess of blasts; RCMD: refractory cytopenia with multilineage dysplasia; CNA, Copy number aberration
Supplementary Table 1 aCGH results in myelodysplastic syndromes
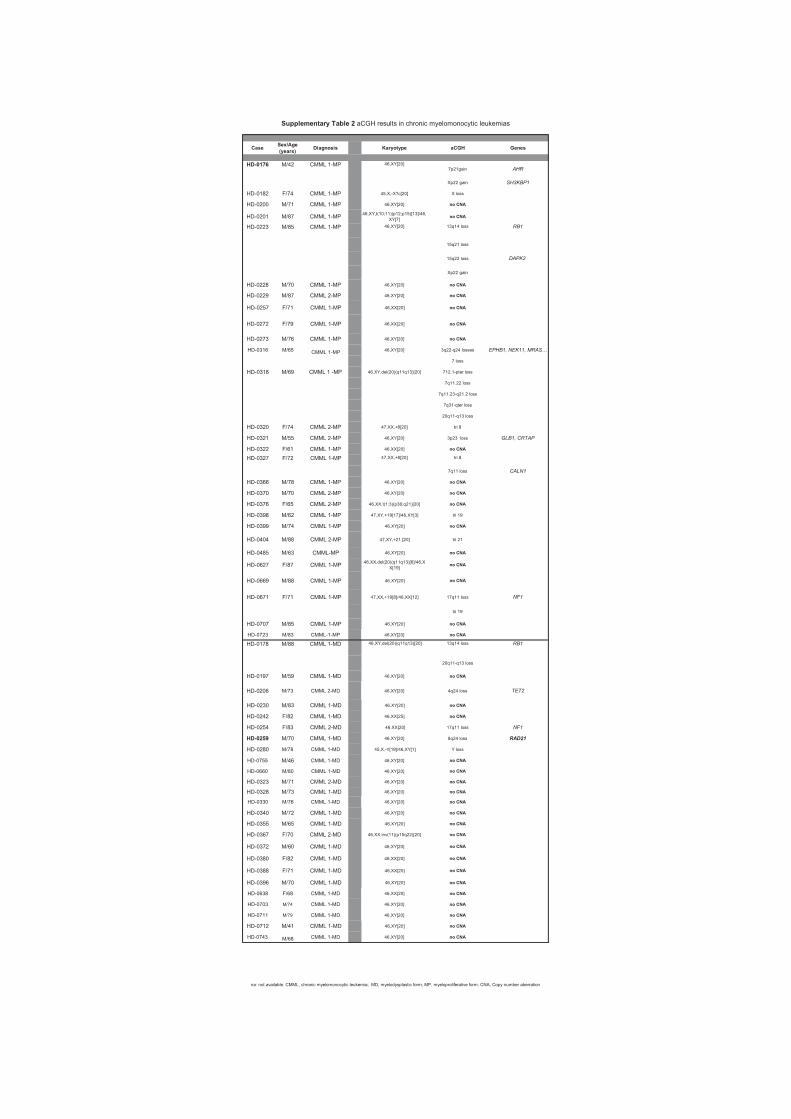
CaseSex/Age (years)
Diagnosis Karyotype aCGH Genes
7p21gain AHR
Xp22 gain SH3KBP1
HD-0182 F/74 CMML 1-MP 45,X,-X?c[20] X loss
HD-0200 M/71 CMML 1-MP 46,XY[20] no CNA
HD-0201 M/87 CMML 1-MP46,XY,t(10;11)(p12;p15)[13]/46,
XY[7]no CNA
13q14 loss RB1
15q21 loss
15q22 loss DAPK2
Xp22 gain
HD-0228 M/70 CMML 1-MP 46,XY[20] no CNA
HD-0229 M/87 CMML 2-MP 46,XY[20] no CNA
HD-0257 F/71 CMML 1-MP 46,XX[20] no CNA
HD-0272 F/79 CMML 1-MP 46,XX[20] no CNA
HD-0273 M/76 CMML 1-MP 46,XY[20] no CNA
HD-0316 M/65 CMML 1-MP 46,XY[20] 3q22-q24 losses EPHB1, NEK11, MRAS…
7 loss
HD-0318 M/69 CMML 1 -MP 46,XY,del(20)(q11q13)[20] 712.1-pter loss
7q11.22 loss
7q11.23-q21.2 loss
7q31-qter loss
20q11-q13 loss
HD-0320 F/74 CMML 2-MP 47,XX,+8[20] tri 8
HD-0321 M/55 CMML 2-MP 46,XY[20] 3p23 loss GLB1, CRTAP
HD-0322 F/61 CMML 1-MP 46,XX[20] no CNA
tri 8
7q11 loss CALN1
HD-0366 M/78 CMML 1-MP 46,XY[20] no CNA
HD-0370 M/70 CMML 2-MP 46,XY[20] no CNA
HD-0376 F/65 CMML 2-MP 46,XX,t(1;3)(p36;q21)[20] no CNA
HD-0398 M/62 CMML 1-MP 47,XY,+19[17]/46,XY[3] tri 19
HD-0399 M/74 CMML 1-MP 46,XY[20] no CNA
HD-0404 M/88 CMML 2-MP 47,XY,+21 [20] tri 21
HD-0485 M/63 CMML-MP 46,XY[20] no CNA
HD-0627 F/87 CMML 1-MP46,XX,del(20)(q11q13)[6]/46,X
X[19]no CNA
HD-0669 M/88 CMML 1-MP 46,XY[20] no CNA
HD-0671 F/71 CMML 1-MP 47,XX,+19[8]/46,XX[12] 17q11 loss NF1
tri 19
HD-0707 M/85 CMML 1-MP 46,XY[20] no CNA
HD-0723 M/83 CMML-1-MP 46,XY[20] no CNA
13q14 loss RB1
20q11-q13 loss
HD-0197 M/59 CMML 1-MD 46,XY[20] no CNA
HD-0206 M/73 CMML 2-MD 46,XY[20] 4q24 loss TET2
HD-0230 M/83 CMML 1-MD 46,XY[20] no CNA
HD-0242 F/82 CMML 1-MD 46,XX[25] no CNA
HD-0254 F/83 CMML 2-MD 46,XX[20] 17q11 loss NF1
HD-0259 M/70 CMML 1-MD 46,XY[20] 8q24 loss RAD21
HD-0280 M/78 CMML 1-MD 45,X,-Y[19]/46,XY[1] Y loss
HD-0755 M/46 CMML 1-MD 46,XY[20] no CNA
HD-0660 M/80 CMML 1-MD 46,XY[20] no CNA
HD-0323 M/71 CMML 2-MD 46,XY[20] no CNA
HD-0328 M/73 CMML 1-MD 46,XY[20] no CNA
HD-0330 M/76 CMML 1-MD 46,XY[20] no CNA
HD-0340 M/72 CMML 1-MD 46,XY[20] no CNA
HD-0355 M/65 CMML 1-MD 46,XY[20] no CNA
HD-0367 F/70 CMML 2-MD 46,XX,inv(11)(p15q22)[20] no CNA
HD-0372 M/60 CMML 1-MD 46,XY[20] no CNA
HD-0380 F/82 CMML 1-MD 46,XX[20] no CNA
HD-0388 F/71 CMML 1-MD 46,XX[20] no CNA
HD-0396 M/70 CMML 1-MD 46,XY[20] no CNA
HD-0638 F/68 CMML 1-MD 46,XX[20] no CNA
HD-0703 M/74 CMML 1-MD 46,XY[20] no CNA
HD-0711 M/79 CMML 1-MD 46,XY[20] no CNA
HD-0712 M/41 CMML 1-MD 46,XY[20] no CNA
HD-0743 M/68 CMML 1-MD 46,XY[20] no CNA
Supplementary Table 2 aCGH results in chronic myelomonocytic leukemias
na: not available. CMML, chronic myelomonocytic leukemia; MD, myelodysplastic form; MP, myeloproliferative form; CNA, Copy number aberration
CMML 1-MP 46,XY[20]
HD-0176 M/42 CMML 1-MP 46,XY[20]
HD-0223 M/85
HD-0327 CMML 1-MPF/72
46,XY,del(20)(q11q13)[20]CMML 1-MDM/88HD-0178
47,XX,+8[20]
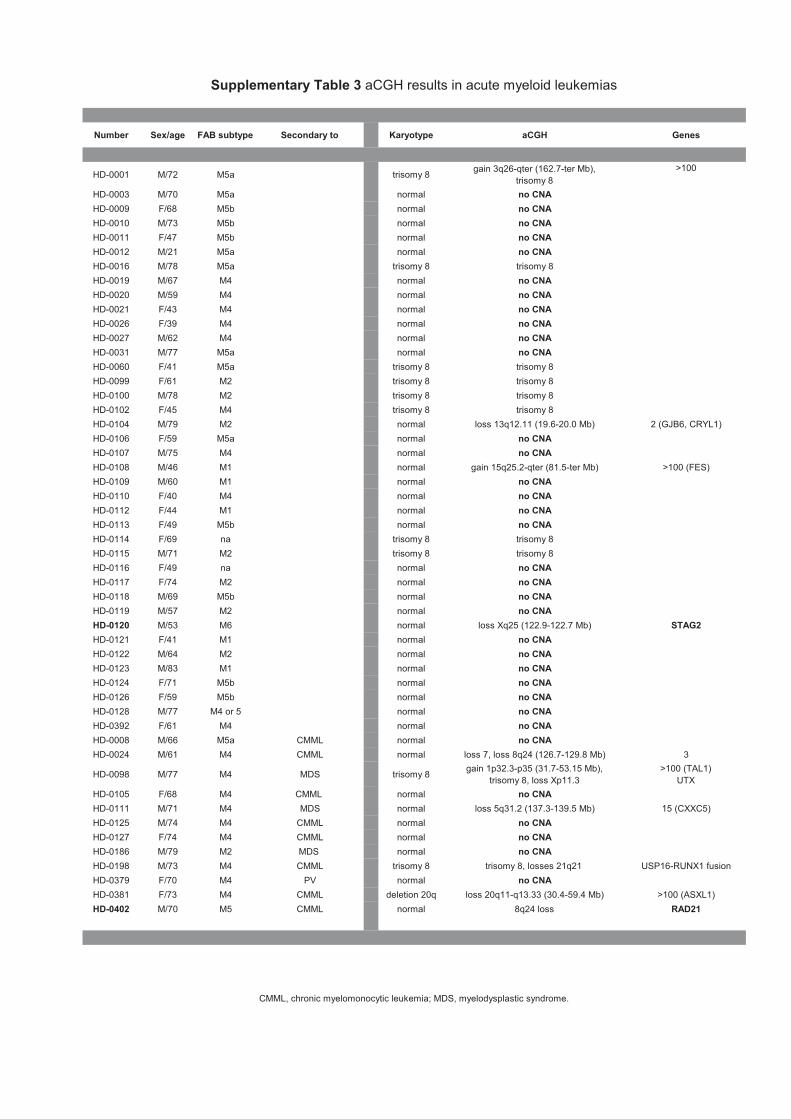
Number Sex/age FAB subtype Secondary to Karyotype aCGH Genes
HD-0001 M/72 M5a trisomy 8gain 3q26-qter (162.7-ter Mb),
trisomy 8
>100
HD-0003 M/70 M5a normal no CNA
HD-0009 F/68 M5b normal no CNA
HD-0010 M/73 M5b normal no CNA
HD-0011 F/47 M5b normal no CNA
HD-0012 M/21 M5a normal no CNA
HD-0016 M/78 M5a trisomy 8 trisomy 8
HD-0019 M/67 M4 normal no CNA
HD-0020 M/59 M4 normal no CNA
HD-0021 F/43 M4 normal no CNA
HD-0026 F/39 M4 normal no CNA
HD-0027 M/62 M4 normal no CNA
HD-0031 M/77 M5a normal no CNA
HD-0060 F/41 M5a trisomy 8 trisomy 8
HD-0099 F/61 M2 trisomy 8 trisomy 8
HD-0100 M/78 M2 trisomy 8 trisomy 8
HD-0102 F/45 M4 trisomy 8 trisomy 8
HD-0104 M/79 M2 normal loss 13q12.11 (19.6-20.0 Mb) 2 (GJB6, CRYL1)
HD-0106 F/59 M5a normal no CNA
HD-0107 M/75 M4 normal no CNA
HD-0108 M/46 M1 normal gain 15q25.2-qter (81.5-ter Mb) >100 (FES)
HD-0109 M/60 M1 normal no CNA
HD-0110 F/40 M4 normal no CNA
HD-0112 F/44 M1 normal no CNA
HD-0113 F/49 M5b normal no CNA
HD-0114 F/69 na trisomy 8 trisomy 8
HD-0115 M/71 M2 trisomy 8 trisomy 8
HD-0116 F/49 na normal no CNA
HD-0117 F/74 M2 normal no CNA
HD-0118 M/69 M5b normal no CNA
HD-0119 M/57 M2 normal no CNA
HD-0120 M/53 M6 normal loss Xq25 (122.9-122.7 Mb) STAG2
HD-0121 F/41 M1 normal no CNA
HD-0122 M/64 M2 normal no CNA
HD-0123 M/83 M1 normal no CNA
HD-0124 F/71 M5b normal no CNA
HD-0126 F/59 M5b normal no CNA
HD-0128 M/77 M4 or 5 normal no CNA
HD-0392 F/61 M4 normal no CNA
HD-0008 M/66 M5a CMML normal no CNA
HD-0024 M/61 M4 CMML normal loss 7, loss 8q24 (126.7-129.8 Mb) 3
HD-0098 M/77 M4 MDS trisomy 8gain 1p32.3-p35 (31.7-53.15 Mb),
trisomy 8, loss Xp11.3
>100 (TAL1)
UTX
HD-0105 F/68 M4 CMML normal no CNA
HD-0111 M/71 M4 MDS normal loss 5q31.2 (137.3-139.5 Mb) 15 (CXXC5)
HD-0125 M/74 M4 CMML normal no CNA
HD-0127 F/74 M4 CMML normal no CNA
HD-0186 M/79 M2 MDS normal no CNA
HD-0198 M/73 M4 CMML trisomy 8 trisomy 8, losses 21q21 USP16-RUNX1 fusion
HD-0379 F/70 M4 PV normal no CNA
HD-0381 F/73 M4 CMML deletion 20q loss 20q11-q13.33 (30.4-59.4 Mb) >100 (ASXL1)
HD-0402 M/70 M5 CMML normal 8q24 loss RAD21
Supplementary Table 3 aCGH results in acute myeloid leukemias
CMML, chronic myelomonocytic leukemia; MDS, myelodysplastic syndrome.

93
Commentaires et discussion de l’article n°3
La CGH-array est une technique de cytogénétique sur puces permettant d'analyser à
l’échelle pangénomique les variations du nombre de copies de gène dans l'ADN. Elle permet
la détection des gains ou des pertes de matériels génétiques non visibles par les techniques
traditionnelles de cytogénétique. Grâce à cette technique, notre laboratoire a identifié un
nouveau gène impliqué dans la leucémogenèse : ASXL1 (Gelsi-Boyer et al, 2009).
Dans le but d’identifier de nouvelles mutations de gène, nous avons analysé par CGH-
array une série de 167 HM (63 SMD, 53 LMMC et 51 LAM). Les 2/3 des échantillons ne
présentaient pas de variation du nombre de copie de gènes [41/63 SMD (65%) ; 37/53
LMMC (70%) ; 35/51 LAM (69%)]. Ceci est corrélé au fait que la majorité des patients de
l’étude présentaient un caryotype normal et cela suit les données publiées (Gondek et al,
2008). Nous avons sélectionné majoritairement des HM à caryotype normal car ce type
d’HM manque actuellement de marqueurs moléculaires.
Nous avons retrouvé de petites délétions déjà décrites dans ces HM comme celles du
gène TET2 dans un SMD et celles du gène ASXL1 dans un cas de LAM. Nous avons
également identifié des délétions des gènes NF1, RB1, RUNX1 et UTX. Nous nous sommes
plus précisément intéressés à deux délétions car elles impliquaient deux gènes codant pour
des protéines appartenant au complexe de la cohésine.
Le complexe de la cohésine comporte quatre sous-unités protéiques hautement
conservées (SMC1, SMC3, RAD21 et STAG1/STAG2, voir Figure n°5) et joue un rôle
important dans la biologie des chromosomes (Peters et al, 2007). Les anomalies de la
cohésine perturbent la ségrégation des chromatides sœurs lors de la division cellulaire
(Simmons et al, 2010). La cohésine participe également à la réparation des cassures double
brin de l’ADN et régule la transcription de gènes en s’associant à CTCF (Bauerschmidt et al,
2010).

94
Les deux gènes concernés dans les délétions étaient RAD21 et STAG2 :
- Le gène RAD21, localisé en 8q21, était délété chez un patient atteint de LMMC s’étant
rapidement transformée en LAM. Aucune mutation de RAD21 n’a été détectée sur l’autre
allèle. Ce patient avait également une mutation ITD de FLT3 et une mutation de NPM1.
- Le gène STAG2, situé en Xq25, était délété chez un patient atteint de LAM. Ce patient
présentait également une mutation d’IDH1.
Figure n°5 : Complexe protéique de la cohésine
(Peters et al, 2008)
Au vu des fonctions jouées par les protéines formant le complexe de la cohésine, les
altérations des gènes de la cohésine pourraient être à l’origine d’aberrations
chromosomiques, en particulier de délétions, et de perturbations de la transcription de gènes.
La perte de l’expression de RAD21 induit notamment la perte d’expression du gène RUNX1
(Horsfield et al, 2007).
Nous avons séquencé le gène RAD21 dans un panel de 95 HM majoritairement à caryotype
normal, mais nous n’avons pas trouvé de mutation. Dans l’hypothèse que des altérations
moléculaires de RAD21 provoqueraient des aberrations chromosomiques, il serait intéressant
de séquencer RAD21 et les autres gènes codant les sous unités de la cohésine, dans des HM à

95
caryotype complexe et d’étudier parallèlement en CGH-array les gènes codant le complexe
de la cohésine dans ces HM.
Dans la littérature, des délétions de gènes RAD21 et de STAG2 avaient déjà été
rapportées (Walter et al, 2009 ; Bullinger et al, 2010) sans que leur lien avec la cohésine
n’ait été établi. Les délétions impliquant les gènes de la cohésine ont une faible fréquence
(environ 1% dans notre étude). Les délétions de gène rapportés dans les HM ont toujours une
fréquence globalement faible (~1%), ce sont des fréquences qui ont été retrouvées pour les
délétions des gènes TET2 et ASXL1. Des mutations sur ces gènes ont ensuite été identifiées.
Notre étude montre pour la première fois l’implication de l’altération des gènes codant
la cohésine dans des hémopathies myéloïdes. Les altérations de la cohésine représentent un
nouveau mécanisme potentiellement impliqué dans la leucémogenèse et de nouvelles études
doivent montrer sa place dans la physiopathologie des HM.

96
ARTICLE N°4
Gain of CBL-interacting protein, a possible alternative to CBL
mutations in myeloid malignancies.
Adélaïde J, Gelsi-Boyer V, Rocquain J, Carbuccia N, Birnbaum DJ, Finetti P, Bertucci F, Mozziconacci MJ, Vey N, Birnbaum D,
Chaffanet M.
Leukemia 2010; 24(8): 1539-41.

LETTER TO THE EDITOR
Gain of CBL-interacting protein, a possible alternative to CBL mutations inmyeloid malignancies
Leukemia advance online publication, 17 June 2010;doi:10.1038/leu.2010.135
Alterations of several genes have a role in leukemogenesis.These genes may have distinct functions or may be involved inthe same complexes or pathways. Several recent studies haveshown that the CBL gene, located in chromosome region11q23.3, is mutated in chronic myeloid diseases.1–3 CBLencodes an E3 ubiquitin ligase that attenuates the proliferativesignal transduced by tyrosine kinase receptors such as FLT3 andKIT by enhancing their ubiquitination, downregulation, andlysosomal degradation. CBL mutations modify the proteinactivity4 and lead to an increased and/or prolonged signal.The two other members of the CBL family, CBLB and CBLC, arealso mutated in few cases of chronic myeloid diseases.3
Mutations of CBL can be heterozygous or homozygous. Patientswith homozygous mutations fare worse than patients withheterozygous ones.3 Most often homozygosity results fromacquired somatic uniparental disomy of chromosome arm 11q(11qUPD), which is detected by genome analyses usingsingle-nucleotide polymorphism arrays. Malignant genomescan also be studied by array-comparative genomic hybridization(aCGH).We used this latter technique to study 63 cases of
myelodysplastic (MD) syndromes (MDSs) and 53 casesof chronic myelomonocytic leukemias (CMMLs). According tothe WHO classification,5 the MDS panel comprised 5 cases ofrefractory anemia (RA), 13 cases of RA with ring sideroblasts,6 cases of refractory cytopenia with multilineage dysplasia(RCMD), 16 cases of RA with excess of blasts type 1 (RAEB1),19 cases of RA with excess of blasts type 2 (RAEB2) and 4 casesof MDS-unclassified. The 53 CMML panel comprised 25 MDforms (leucocytosis count o13� 109/l) and 28 myeloprolifera-tive (MP) forms (leucocytosis count 413� 109/l).6 All patientssigned an informed consent and the study was approved byour ethics committee. aCGH was done with high-densityoligonucleotide microarrays (Hu-244A, Agilent Technologies,Massy, France), as described previously.6
Two-thirds of the 63 MDS cases showed no copy numberalterations (CNAs) (that is, neither gain nor loss) (SupplementaryTable 1). Few cases had a single small CNA such as loss at 4q24(TET2), loss at 2p11 and gain at Xp22 (Figure 1). Gain at Xp22,found in RCMD HD-0747, contained the SH3KBP1 gene (SH3-domain kinase binding protein; chrX: 19 463 441–19 815640).Patient HD-0747, a 77-year-old man, has been followed upsince 2003 for RA in therapeutic abstention. In June 2009,a bone marrow aspiration showed evolution to RCMD. Thesample analyzed was taken at this period. Patient HD-0747is currently maintained in partial hematological responseunder high doses of erythropoietin. aCGH did not show anyother CNA in the HD-0747 genome.Out of the 53 CMMLs, 37 cases (68%) showed no CNA (some
of these cases have been reported previously7) (SupplementaryTable 2). Ten cases had a single or few small CNAs, includingloss of NF1, RB1 or TET2. One of these, HD-0176, showed again of SH3KBP1. Patient HD-0176 was a 46-year-old man
diagnosed with CMML1 in December 2006. Therapeuticabstention was decided. Ten months later, the patient died ofmeningitis. The HD-0176 genome did not have any otheralteration except a gain of the AHR gene at 7p21, the role ofwhich is unknown; it is noticeable, however, that AHR can actas CBL, as an E3 ubiquitin ligase,8 and that both AHR and CBLparticipate in the JUN and P38 kinase pathways.
In the same panel we searched for mutations in exons 8 and9 of CBL. DNA sequencing was done as described.7 Five MDScases, all RAEB2, were mutated in CBL (7.8%): p.Glu366Lys,p.Tyr368X, p.Leu380Pro, p.Lys381Arg and p.Pro417Leu(Supplementary Table 1). One substitution occurred in thecase with trisomy 11 (HD-0264). Mutations of CBL, includingtwo homozygous, were found in five CMML cases (B10%, fourin MP forms: p.Tyr317His, p.Cys396Tyr, p.Cys404Tyr andp.Phe418Ser; one in MD form: p.Cys404Tyr) (SupplementaryTable 2). The results are summarized in Table 1.
SH3KBP1 (SH3-domain kinase binding protein 1), alsocalled CIN85 (CBL-interacting protein of 85 kDa), codes fora cytoplasmic adaptor molecule that interacts and cooperateswith CBL to regulate signaling.9–11 RCMD HD-0747 andMP-CMML HD-0176, the two cases with Xp22 SH3KBP1 gain,occurred in males and displayed a normal karyotype. They werenot mutated in CBL exons 8 and 9. They were not mutatedeither in ASXL1, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS,RUNX1, TET2 and WT1 genes (not shown). As determined byaCGH, the SH3KBP1 gain was limited to an additional copy.This duplication and the CBL UPD11q suggest that limitedvariations of the pathway are sufficient to induce an effect onsignaling. Mutations of CBL have an effect on cell proliferation.4
SH3KBP1/CIN85 cooperates with CBL in the downregulationof signaling receptors.10 Because of UPD11q two copies of themutated CBL gene are often present in the malignant clone. In asimilar way, it could be possible that the SH3KBP1 duplicatedgene is mutated. We have looked in our Affymetrix microarraydata for the expression of SH3KBP1 in HD-0176, for which wehad mRNA. The level of expression was not overtly modified ascompared with normal bone marrow or other MDS/CMMLsamples.
In summary, we have found two examples of alteration of agene encoding a CBL interactor. This alteration may play a rolesimilar to that of CBL mutations in chronic myeloid diseases.Although the anomaly is rare (1.7% in our series), it is notinsignificant. First, the number of CNAs was globally low; incomparison we found only two deletions of TET2 andone deletion of ASXL1 in the same panel. Second, SH3KBP1gain increases by about 20% the cases with alteration of theCBL pathway (2 cases out of 116 tested; to be compared with10 mutated CBL). Third, it shows specificity, as we have notdetected such gain in 300 breast or colon cancers (not shown).This emphasizes the importance of CBL-associated deregulationof signaling in chronic myeloid diseases and constitutesan incentive to look for alterations in other members of theCBL/CIN85 pathway.12 In that respect we found a gain of thegene encoding MAP3K4, an interactor of CIN85,13 in a RAEB1with complex aCGH profile, and a gain of the gene encoding
Leukemia (2010), 1–3& 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00
www.nature.com/leu

the CBL interactor ARHGEF7/bPIX in an RCMD in a reportedstudy.14 Whether other E3 ubiquitin ligases (for example, AHRor SCF components) may have a role in these diseases alsoneeds to be investigated.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by Institut Paoli-Calmettes, Inserm, andgrants from the Association pour la Recherche contre le Cancer(no. 4992).
J Adelaıde1, V Gelsi-Boyer1,2,3, J Rocquain1, N Carbuccia1,DJ Birnbaum1, P Finetti1, F Bertucci1,3, MJ Mozziconacci1,2,
N Vey3,4, D Birnbaum1 and M Chaffanet11Centre de Recherche en Cancerologie de Marseille,
Laboratoire d’Oncologie Moleculaire, UMR891 Inserm,Institut Paoli-Calmettes, Marseille, France;
2Departement de BioPathologie, Institut Paoli-Calmettes,Marseille, France;
3Faculte de Medecine, Universite de la Mediterranee,Marseille, France and
4Departement d’Hematologie, Institut Paoli-Calmettes,Marseille, France
E-mail: [email protected]
References
1 Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS,Szpurka H et al. 250K single nucleotide polymorphism array
Table 1 Summary of results
MDS CMML Total
Number of cases studied 63 53 116aCGH CNA 40 (63.5%) 37 (69.8%) 77 (66.4%)SH3KBP1/CIN85 gain 1 (1.6%) 1 (1.9%) 2 (1.7%)CBL mutation 5 (7.9%) 5 (9.4%) 10 (8.6 %)Total alterations of pathway 6 (9.5%) 6 (11.3%) 12 (10.3%)
Abbreviations: aCGH, array-comparative genomic hybridization;CMML, chronic myelomonocytic leukemia; MDS, myelodysplasticsyndrome.
Figure 1 Chromosome X aCGH profiles showing SH3KBP1 gain. (a) RCMD HD-0747 shows a gain of one copy of the SH3KBP1 gene(arrows). Case HD-0637 represents the baseline profile and case HD-0411 (female) a profile corresponding to two chromosomes X. (b) CMMLHD-0176 shows one copy gain of SH3KBP1. CMML HD-0366 represents the baseline profile and HD-0367 a profile corresponding to twochromosomes X.
Letter to the Editor
2
Leukemia

karyotyping identifies acquired uniparental disomy and homo-zygous mutations, including novel missense substitutions of c-Cbl,in myeloid malignancies. Cancer Res 2008; 68: 10340–10357.
2 Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire Cet al. Frequent CBL mutations associated with 11q acquireduniparental disomy in myeloproliferative neoplasms. Blood 2009;113: 6182–6192.
3 Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J et al.Mutations of E3 ligase Cbl family members constitute a novelcommon pathogenic lesion in myeloid malignancies. J Clin Oncol2009; 27: 6109–6116.
4 Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamasaki S et al.Gain-of-function of mutated CBL tumour suppressor in myeloidneoplasms. Nature 2009; 460: 904–908.
5 Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ,Porwit A et al. The 2008 revision of the World HealthOrganization (WHO) classification of myeloid neoplasms andacute leukemia: rationale and important changes. Blood 2009;114: 937–951.
6 Gelsi-Boyer V, Cervera N, Bertucci F, Trouplin V, Remy V,Olschwang S et al. Genes expression profiling separateschronic myelomonocytic leukaemia in two molecular subtypes.Leukemia 2007; 21: 2359–2362.
7 Gelsi-Boyer V, Trouplin V, Adelaıde J, Aceto N, Remy V, Pinson Set al. Genome profiling of chronic myelomonocytic leukemia:
frequent alterations of RAS and RUNX1 genes. BMC Cancer 2008;8: 299–314.
8 Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H et al.Dioxin receptor is a ligand-dependent E3 ubiquitin ligase.Nature 2007; 444: 562–566.
9 Take H, Watanabe S, Takeda K, Yu ZX, Iwata N, Kajigaya S.Cloning and characterization of a novel adaptor protein, CIN85,that interacts with c-Cbl. Biochem Biophys Res Commun 2000;268: 321–328.
10 Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY,Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induceddownregulation of EGF receptors. Nature 2002; 416: 183–187.
11 Bezsonova I, Bruce MC, Wiesner S, Lin H, Rotin D, Forman-CayJD. Interactions between the three CIN85 SH3 domainsand ubiquitin: implications for CIN85 ubiquitination. Biochemistry2008; 47: 8937–8949.
12 Havrylov S, Rzheptskyy Y, Malinowska A, Drobot L, Redowicz MJ.Proteins recruited by the SH3 domains of Ruk/CIN85 adaptoridentified by LC-MS/MS. Protein Sci 2009; 7: 21.
13 Aissouni Y, Zapart G, Iovanna JL, Dikic I, Soubeyran P. CIN85regulates the ability of MEKK4 to activate the p38 MAP kinasepathway. Biochem Biophys Res Comm 2005; 338: 808–814.
14 Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG,Massop M et al. Acquired mutations in TET2 are common inmyelodysplastic syndromes. Nat Genet 2009; 41: 838–842.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
Letter to the Editor
3
Leukemia
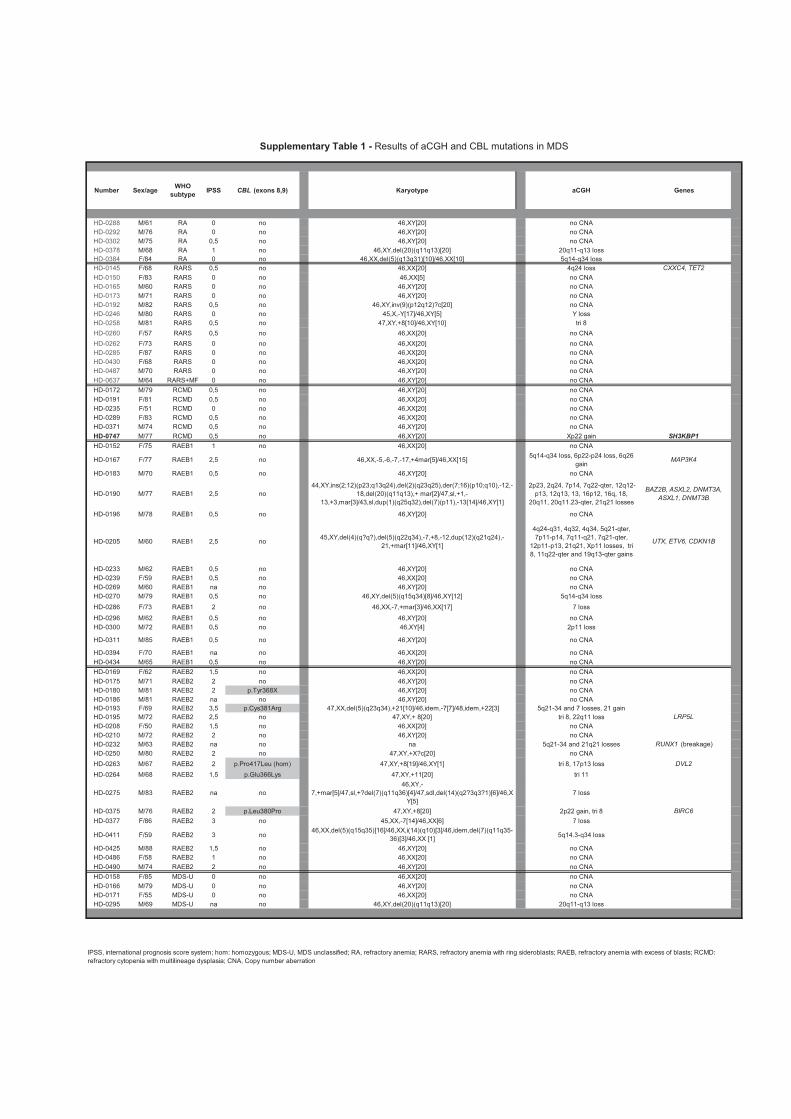
Number Sex/ageWHO
subtypeIPSS CBL (exons 8,9) Karyotype aCGH Genes
HD-0288 M/61 RA 0 no 46,XY[20] no CNA
HD-0292 M/76 RA 0 no 46,XY[20] no CNA
HD-0302 M/75 RA 0,5 no 46,XY[20] no CNA
HD-0378 M/68 RA 1 no 46,XY,del(20)(q11q13)[20] 20q11-q13 loss
HD-0384 F/84 RA 0 no 46,XX,del(5)(q13q31)[10]/46,XX[10] 5q14-q34 loss
HD-0145 F/68 RARS 0,5 no 46,XX[20] 4q24 loss CXXC4, TET2
HD-0150 F/83 RARS 0 no 46,XX[5] no CNA
HD-0165 M/60 RARS 0 no 46,XY[20] no CNA
HD-0173 M/71 RARS 0 no 46,XY[20] no CNA
HD-0192 M/82 RARS 0,5 no 46,XY,inv(9)(p12q12)?c[20] no CNA
HD-0246 M/80 RARS 0 no 45,X,-Y[17]/46,XY[5] Y loss
HD-0258 M/81 RARS 0,5 no 47,XY,+8[10]/46,XY[10] tri 8
HD-0260 F/57 RARS 0,5 no 46,XX[20] no CNA
HD-0262 F/73 RARS 0 no 46,XX[20] no CNA
HD-0285 F/87 RARS 0 no 46,XX[20] no CNA
HD-0430 F/68 RARS 0 no 46,XX[20] no CNA
HD-0487 M/70 RARS 0 no 46,XY[20] no CNA
HD-0637 M/64 RARS+MF 0 no 46,XY[20] no CNA
HD-0172 M/79 RCMD 0,5 no 46,XY[20] no CNA
HD-0191 F/81 RCMD 0,5 no 46,XX[20] no CNA
HD-0235 F/51 RCMD 0 no 46,XX[20] no CNA
HD-0289 F/83 RCMD 0,5 no 46,XX[20] no CNA
HD-0371 M/74 RCMD 0,5 no 46,XY[20] no CNA
HD-0747 M/77 RCMD 0,5 no 46,XY[20] Xp22 gain SH3KBP1
HD-0152 F/75 RAEB1 1 no 46,XX[20] no CNA
HD-0167 F/77 RAEB1 2,5 no 46,XX,-5,-6,-7,-17,+4mar[5]/46,XX[15]5q14-q34 loss, 6p22-p24 loss, 6q26
gainMAP3K4
HD-0183 M/70 RAEB1 0,5 no 46,XY[20] no CNA
HD-0190 M/77 RAEB1 2,5 no
44,XY,ins(2;12)(p23;q13q24),del(2)(q23q25),der(7;16)(p10;q10),-12,-
18,del(20)(q11q13),+ mar[2]/47,sl,+1,-
13,+3,mar[3]/43,sl,dup(1)(q25q32),del(7)(p11),-13[14]/46,XY[1]
2p23, 2q24, 7p14, 7q22-qter, 12q12-
p13, 12q13, 13, 16p12, 16q, 18,
20q11, 20q11.23-qter, 21q21 losses
BAZ2B, ASXL2, DNMT3A,
ASXL1, DNMT3B
HD-0196 M/78 RAEB1 0,5 no 46,XY[20] no CNA
HD-0205 M/60 RAEB1 2,5 no45,XY,del(4)(q?q?),del(5)(q22q34),-7,+8,-12,dup(12)(q21q24),-
21,+mar[11]/46,XY[1]
4q24-q31, 4q32, 4q34, 5q21-qter,
7p11-p14, 7q11-q21, 7q21-qter,
12p11-p13, 21q21, Xp11 losses, tri
8, 11q22-qter and 19q13-qter gains
UTX, ETV6, CDKN1B
HD-0233 M/62 RAEB1 0,5 no 46,XY[20] no CNA
HD-0239 F/59 RAEB1 0,5 no 46,XX[20] no CNA
HD-0269 M/60 RAEB1 na no 46,XY[20] no CNA
HD-0270 M/79 RAEB1 0,5 no 46,XY,del(5)(q15q34)[8]/46,XY[12] 5q14-q34 loss
HD-0286 F/73 RAEB1 2 no 46,XX,-7,+mar[3]/46,XX[17] 7 loss
HD-0296 M/62 RAEB1 0,5 no 46,XY[20] no CNA
HD-0300 M/72 RAEB1 0,5 no 46,XY[4] 2p11 loss
HD-0311 M/85 RAEB1 0,5 no 46,XY[20] no CNA
HD-0394 F/70 RAEB1 na no 46,XX[20] no CNA
HD-0434 M/65 RAEB1 0,5 no 46,XY[20] no CNA
HD-0169 F/62 RAEB2 1,5 no 46,XX[20] no CNA
HD-0175 M/71 RAEB2 2 no 46,XY[20] no CNA
HD-0180 M/81 RAEB2 2 p.Tyr368X 46,XY[20] no CNA
HD-0186 M/81 RAEB2 na no 46,XY[20] no CNA
HD-0193 F/69 RAEB2 3,5 p.Cys381Arg 47,XX,del(5)(q23q34),+21[10]/46,idem,-7[7]/48,idem,+22[3] 5q21-34 and 7 losses, 21 gain
HD-0195 M/72 RAEB2 2,5 no 47,XY,+ 8[20] tri 8, 22q11 loss LRP5L
HD-0208 F/50 RAEB2 1,5 no 46,XX[20] no CNA
HD-0210 M/72 RAEB2 2 no 46,XY[20] no CNA
HD-0232 M/63 RAEB2 na no na 5q21-34 and 21q21 losses RUNX1 (breakage)
HD-0250 M/80 RAEB2 2 no 47,XY,+X?c[20] no CNA
HD-0263 M/67 RAEB2 2 p.Pro417Leu (hom) 47,XY,+8[19]/46,XY[1] tri 8, 17p13 loss DVL2
HD-0264 M/68 RAEB2 1,5 p.Glu366Lys 47,XY,+11[20] tri 11
HD-0275 M/83 RAEB2 na no
46,XY,-
7,+mar[5]/47,sl,+?del(7)(q11q36)[4]/47,sdl,del(14)(q2?3q3?1)[6]/46,X
Y[5]
7 loss
HD-0375 M/76 RAEB2 2 p.Leu380Pro 47,XY,+8[20] 2p22 gain, tri 8 BIRC6
HD-0377 F/86 RAEB2 3 no 45,XX,-7[14]/46,XX[6] 7 loss
HD-0411 F/59 RAEB2 3 no46,XX,del(5)(q15q35)[16]/46,XX,i(14)(q10)[3]/46,idem,del(7)(q11q35-
36)[3]/46,XX [1]5q14.3-q34 loss
HD-0425 M/88 RAEB2 1,5 no 46,XY[20] no CNA
HD-0486 F/58 RAEB2 1 no 46,XX[20] no CNA
HD-0490 M/74 RAEB2 2 no 46,XY[20] no CNA
HD-0158 F/85 MDS-U 0 no 46,XX[20] no CNA
HD-0166 M/79 MDS-U 0 no 46,XY[20] no CNA
HD-0171 F/55 MDS-U 0 no 46,XX[20] no CNA
HD-0295 M/69 MDS-U na no 46,XY,del(20)(q11q13)[20] 20q11-q13 loss
IPSS, international prognosis score system; hom: homozygous; MDS-U, MDS unclassified; RA, refractory anemia; RARS, refractory anemia with ring sideroblasts; RAEB, refractory anemia with excess of blasts; RCMD:
refractory cytopenia with multilineage dysplasia; CNA, Copy number aberration
Supplementary Table 1 - Results of aCGH and CBL mutations in MDS
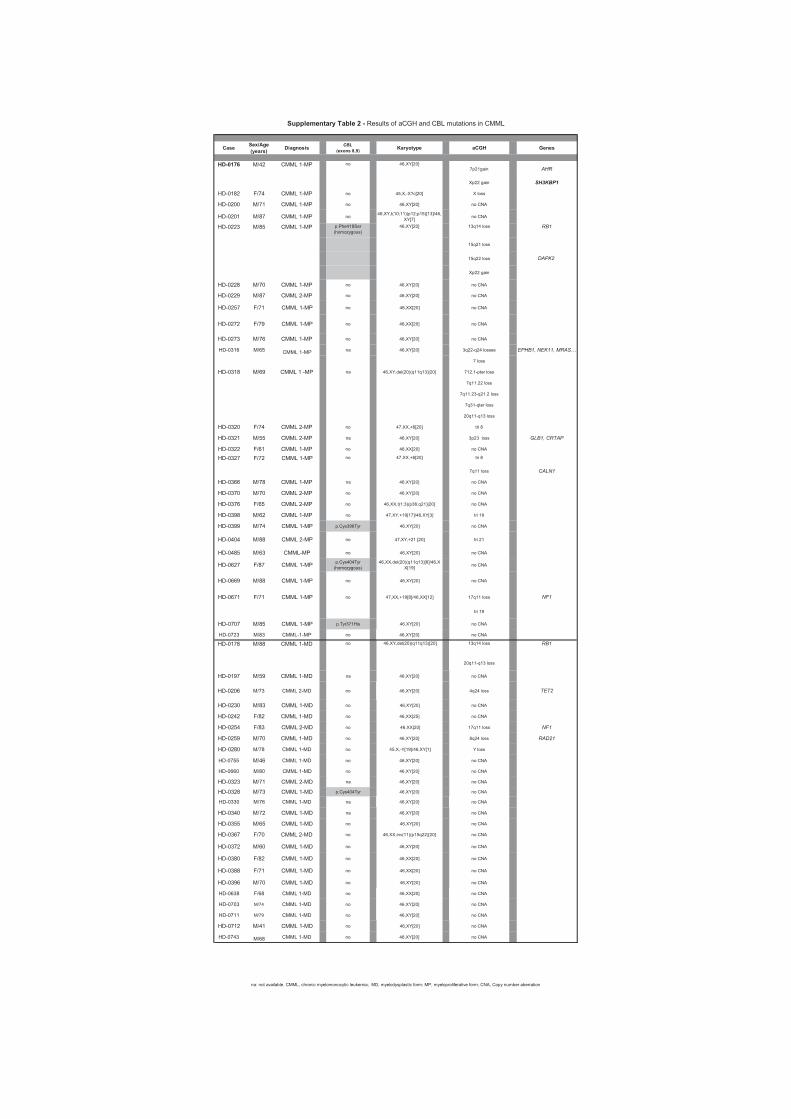
CaseSex/Age (years)
DiagnosisCBL
(exons 8,9)Karyotype aCGH Genes
7p21gain AHR
Xp22 gain SH3KBP1
HD-0182 F/74 CMML 1-MP no 45,X,-X?c[20] X loss
HD-0200 M/71 CMML 1-MP no 46,XY[20] no CNA
HD-0201 M/87 CMML 1-MP no46,XY,t(10;11)(p12;p15)[13]/46,
XY[7]no CNA
13q14 loss RB1
15q21 loss
15q22 loss DAPK2
Xp22 gain
HD-0228 M/70 CMML 1-MP no 46,XY[20] no CNA
HD-0229 M/87 CMML 2-MP no 46,XY[20] no CNA
HD-0257 F/71 CMML 1-MP no 46,XX[20] no CNA
HD-0272 F/79 CMML 1-MP no 46,XX[20] no CNA
HD-0273 M/76 CMML 1-MP no 46,XY[20] no CNA
HD-0316 M/65 CMML 1-MP na 46,XY[20] 3q22-q24 losses EPHB1, NEK11, MRAS…
7 loss
HD-0318 M/69 CMML 1 -MP na 46,XY,del(20)(q11q13)[20] 712.1-pter loss
7q11.22 loss
7q11.23-q21.2 loss
7q31-qter loss
20q11-q13 loss
HD-0320 F/74 CMML 2-MP no 47,XX,+8[20] tri 8
HD-0321 M/55 CMML 2-MP na 46,XY[20] 3p23 loss GLB1, CRTAP
HD-0322 F/61 CMML 1-MP no 46,XX[20] no CNA
tri 8
7q11 loss CALN1
HD-0366 M/78 CMML 1-MP na 46,XY[20] no CNA
HD-0370 M/70 CMML 2-MP no 46,XY[20] no CNA
HD-0376 F/65 CMML 2-MP no 46,XX,t(1;3)(p36;q21)[20] no CNA
HD-0398 M/62 CMML 1-MP no 47,XY,+19[17]/46,XY[3] tri 19
HD-0399 M/74 CMML 1-MP p.Cys396Tyr 46,XY[20] no CNA
HD-0404 M/88 CMML 2-MP no 47,XY,+21 [20] tri 21
HD-0485 M/63 CMML-MP no 46,XY[20] no CNA
HD-0627 F/87 CMML 1-MPp.Cys404Tyr
(homozygous)
46,XX,del(20)(q11q13)[6]/46,X
X[19]no CNA
HD-0669 M/88 CMML 1-MP no 46,XY[20] no CNA
HD-0671 F/71 CMML 1-MP no 47,XX,+19[8]/46,XX[12] 17q11 loss NF1
tri 19
HD-0707 M/85 CMML 1-MP p.Tyr371His 46,XY[20] no CNA
HD-0723 M/83 CMML-1-MP no 46,XY[20] no CNA
13q14 loss RB1
20q11-q13 loss
HD-0197 M/59 CMML 1-MD na 46,XY[20] no CNA
HD-0206 M/73 CMML 2-MD no 46,XY[20] 4q24 loss TET2
HD-0230 M/83 CMML 1-MD no 46,XY[20] no CNA
HD-0242 F/82 CMML 1-MD no 46,XX[25] no CNA
HD-0254 F/83 CMML 2-MD no 46,XX[20] 17q11 loss NF1
HD-0259 M/70 CMML 1-MD no 46,XY[20] 8q24 loss RAD21
HD-0280 M/78 CMML 1-MD no 45,X,-Y[19]/46,XY[1] Y loss
HD-0755 M/46 CMML 1-MD no 46,XY[20] no CNA
HD-0660 M/80 CMML 1-MD no 46,XY[20] no CNA
HD-0323 M/71 CMML 2-MD na 46,XY[20] no CNA
HD-0328 M/73 CMML 1-MD p.Cys404Tyr 46,XY[20] no CNA
HD-0330 M/76 CMML 1-MD na 46,XY[20] no CNA
HD-0340 M/72 CMML 1-MD na 46,XY[20] no CNA
HD-0355 M/65 CMML 1-MD no 46,XY[20] no CNA
HD-0367 F/70 CMML 2-MD no 46,XX,inv(11)(p15q22)[20] no CNA
HD-0372 M/60 CMML 1-MD no 46,XY[20] no CNA
HD-0380 F/82 CMML 1-MD no 46,XX[20] no CNA
HD-0388 F/71 CMML 1-MD no 46,XX[20] no CNA
HD-0396 M/70 CMML 1-MD no 46,XY[20] no CNA
HD-0638 F/68 CMML 1-MD no 46,XX[20] no CNA
HD-0703 M/74 CMML 1-MD no 46,XY[20] no CNA
HD-0711 M/79 CMML 1-MD no 46,XY[20] no CNA
HD-0712 M/41 CMML 1-MD no 46,XY[20] no CNA
HD-0743 M/68 CMML 1-MD no 46,XY[20] no CNA
47,XX,+8[20]no
CMML 1-MP no
na: not available. CMML, chronic myelomonocytic leukemia; MD, myelodysplastic form; MP, myeloproliferative form; CNA, Copy number aberration
HD-0327 CMML 1-MPF/72
Supplementary Table 2 - Results of aCGH and CBL mutations in CMML
46,XY,del(20)(q11q13)[20]noCMML 1-MDM/88HD-0178
46,XY[20]
HD-0223 M/85 CMML 1-MP p.Phe418Ser
(homozygous)
46,XY[20]
HD-0176 M/42

102
Commentaires et discussion de l’article n°4
Dans plus de la moitié des HM, aucune anomalie cytogénétique n’est identifiable au
caryotype et aucune mutation de gènes ayant une valeur pronostique telle que celles de FLT3
et NPM1 n’est décelable. De nouvelles mutations de gène sont encore à découvrir pour
comprendre la physiopathologie de ces maladies. La CGH-array est une technique efficace
pour l’identification de nouveaux gènes potentiellement impliqués dans la physiopathologie
de ces maladies.
Nous avons analysé par CGH-array une cohorte de 116 patients atteints d’HM
chroniques (63 SMD et 53 LMMC, les mêmes que l’article précédent) et nous avons
identifié un gain sur le chromosome X dans la région p22 sur deux échantillons, un de SMD
et un de LMMC. Cette région contient le gène SH3KBP1 (SH3-domain Kinase Binding
Protein 1), connu également sous le nom de CIN85 (CBL-interacting protein of 85 kDa).
SH3KBP1 est une protéine cytoplasmique régulant l’activité de la protéine CBL (Take et al,
2000 ; Soubeyran et al, 2002).
CBL est une ubiquitine-ligase E3 impliquée dans la régulation de la transduction du
signal des RTK tel que FLT3 et KIT, en induisant leur dégradation par le protéasome
(Schmidt et al, 2005). Le gène CBL est connu pour être muté dans de nombreuses HM à une
fréquence de 1 à 10% (Makishima et al, 2009).
Nous n’avons pas identifié d’altérations du nombre de copie du gène SH3KBP1 dans
300 échantillons de cancer du sein et du colon, ce qui montre que les gains en Xp12 d’une
copie du gène SH3KBP1 semblent spécifiques des HM.
Les deux échantillons présentant un gain d’une copie du gène SH3KBP1 ne
présentaient ni mutation de CBL, ni aucune mutation des gènes ASXL1, FLT3, IDH1, IDH2,
JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 et WT1. De nouvelles anomalies moléculaires
sont donc à rechercher dans ce type d’échantillon pour comprendre leur physiopathologie.

103
Par ailleurs, nous avons constaté deux autres gains de copies concernant deux gènes
proches de CBL :
- AHR qui code pour une autre ubiquitine-ligase comme CBL (Ohtake et al, 2007) présent
sur le même échantillon de LMMC contenant le gain de SH3KBP1.
- MAP3K4 qui code pour une protéine régulatrice du gène SH3KBP1 (Aissouni et al, 2005).
Nous émettons l’hypothèse du rôle potentiellement important de l’altération de gènes
codant les régulateurs de CBL dans la physiopathologie des hémopathies myéloïdes
chroniques. Aucune mutation n’a encore été décrite sur les gènes régulateurs de CBL mais le
fait que leur nombre de copie soit altéré nous laisse envisager des mutations. Il serait
intéressant de séquencer ces gènes, notamment le gène SH3KBP1 dans les HM, il est
possible qu’ils soient mutés.
Le rôle précis des ubiquitine-ligases E3 dans le développement des hémopathies
myéloïdes est encore inconnu. Néanmoins, ces protéines sont fréquemment altérées dans les
SMD et LAM et leur rôle devrait être étudié dans des modèles cellulaires et murins pour
déterminer si cette classe de protéine pourrait faire l’objet de potentielle cible thérapeutique.
Il serait également intéressant de montrer si les altérations génétiques des gènes des
ubiquitine-ligases sont de mauvais pronostic, comme l’ont montré Reindl et al, avec les
mutations de CBL (Reindl et al, 2009).

104
Discussion générale
De trop rares translocations à l’origine d’anomalie génétique « unique »
Une des premières étapes de la compréhension de l’oncogenèse, et plus
particulièrement de la leucémogenèse, a été la découverte des translocations
chromosomiques (Rowley, 2008). L’identification et l’étude des gènes situés au niveau des
points de cassure a permis de comprendre comment certaines translocations
chromosomiques pouvaient initier le processus leucémogène. Une révolution des
connaissances a été amenée par la découverte des gènes de fusion, en particulier BCR-ABL
dans la LMC et PML-RARA dans la LAM-M3 (de Klein et al, 1982 ; Goddard et al, 1991).
Ces découvertes ont conduit à l’avènement de thérapeutiques ciblées, qui dans le cas de la
LMC a fait passer cette hémopathie maligne du statut de maladie de mauvais pronostic à
celui de maladie potentiellement curable. Les patients atteints de LAM-M3 ont maintenant
une survie globale considérablement améliorée.
Malheureusement, ces deux situations font figure d’exception. Actuellement, aucune lésion
génétique «unique», à l’origine des SMD et des LAM n’a été identifiée. Ces hémopathies
résultent en fait de la combinaison complexe de multiples anomalies moléculaires.
Apport des techniques d’analyse génomique à grande échelle dans l’identification
de nouvelles altérations moléculaires
L’étude du caryotype est indispensable lors du diagnostic clinique des HM mais ne
permet que la détection de grandes délétions chromosomiques et la technique par FISH reste
limitée par la disponibilité de sondes spécifiques. Les études par hybridation génomique
comparative (CGH-array) et par puces pangénomiques Single Nucleotide Polymorphism
(SNP-array) permettent de détecter des pertes ou des gains de gènes ainsi que des pertes
d’hétérozygotie dans des régions chromosomiques d’aspect normal en cytogénétique
classique et concourent à révolutionner la génétique, notamment des SMD et LAM (Gondek
et al, 2007).
L’utilisation de la technique des SNP-array a montré que plus de la moitié (59%) des
patients atteints de SMD à caryotype normal présentaient des lésions génétiques cryptiques à
type de délétions ou gains d’ADN (Gondek et al, 2008). Ces nouvelles techniques d’analyse

105
montrent qu’il reste à découvrir de nombreuses régions délétées dans le génome ainsi que de
nouveaux gènes mutés (Mohamedali et al, 2007 ; Nowak et al, 2009). C’est grâce au SNP-
array que Delhommeau et al, ont identifié les mutations de TET2 dans les HM (Delhommeau
et al, 2009). Très récemment, Nikoloski et al, ont mis en évidence dans les SMD, des
mutations d’un nouveau gène, EZH2, codant pour une histone méthyltransférase, présent sur
le bras chromosomique 7q, souvent affecté par des délétions. Des délétions ou mutations
ponctuelles d’EZH2 étaient présentes chez environ 6% des patients atteints de SMD
(Nikoloski et al, 2010 ; Ernst et al, 2010).
Grâce aux données des analyses de CGH-array que notre laboratoire a colligées, nous
avons pu mettre en évidence de nouveaux gènes mutés ou susceptibles d’être altérés. Ainsi,
nous avons identifié des mutations du gène ASXL1 dans une série de SMD et LMMC, puis
dans des LAM, essentiellement secondaires. Nous espérons trouver prochainement de
nouvelles mutations dans certains gènes délétés, tels que ceux codant les protéines de la
cohésine ou ceux impliqués dans la régulation de la protéine CBL.
Les larges délétions monoalléliques constituent un challenge dans l’étude des lésions
moléculaires d’un cancer. Parfois, ces délétions importantes sont accompagnées sur l’autre
allèle, d’une mutation, microdélétion ou modification épigénétique d’un gène clé, ceci
confortant le modèle de la double inactivation des gènes suppresseurs de tumeurs (Knudson,
1971 et 2001). Parmi les gènes que nous avons étudiés, seuls TET2 et RUNX1 correspondent
à cette définition.
Néanmoins, dans certaines délétions de grande ampleur, l’autre allèle demeure entièrement
intact. Ce cas de figure pose l’hypothèse de l’haploinsuffisance d’un ou plusieurs gènes
comme un des mécanismes leucémogènes et des études fondamentales doivent venir appuyer
ces hypothèses.
Découvertes de nouveaux gènes aux fonctions encore inconnues
Les mutations de gènes ayant été découvertes récemment, les études fonctionnelles
n’ont pas encore été réalisées. Des modèles cellulaires et animaux doivent être élaborés afin
d’établir si certaines de ces mutations sont réellement impliquées dans le mécanisme
physiopathologique et si elles peuvent devenir à terme de potentielles cibles thérapeutiques.
Ces expériences sont par contre confrontées à la difficulté de culture des lignées SMD et à

106
créer des modèles de souris SMD, du fait du faible potentiel réplicatif des cellules
myélodysplasiques et de leur apoptose importante. (Campioni et al, 2005 ; Komeno et al,
2009 ; Martin et al, 2010).
Vers une analyse des combinaisons de gènes et du statut mutationnel des HM
Notre analyse par séquençage direct de 12 gènes dont les mutations avaient récemment
été décrites, a permis de poser l’hypothèse d’un modèle de leucémogenèse à quatre étapes.
Notre modèle doit être confirmé par de grandes études de statuts mutationnels des SMD et
LAM sans translocation chromosomique, mais il se pose en bon complément du modèle de
Gilliland qui ne pouvait inclure dans ces deux classes de mutations certains nouveaux gènes
mutés. Nous avons retrouvé des taux de mutations similaires à ceux décrits dans la littérature
à quelques différences près (Tableau n°17), notamment dans les LAM de novo. La littérature
reporte sutout des études incluant tous les types de LAM (sauf LAM-M3), contrairement à
notre étude qui se focalise sur les LAM à caryotype intermédiaire. Il y a ainsi des différences
d’incidence de mutations surtout au niveau de deux gènes : RAS et NPM1. En effet, les
mutations de RAS sont associées aux LAM à caryotype anormal (Auewarakul et al, 2007).
Les mutations de NPM1, comme vu plus haut, sont les mutations les plus fréquemment
décrites dans les LAM à caryotype intermédiaire (50-60% versus 20-30% toutes LAM
confondues). Dans les SMD, nous avons retrouvé un taux plus élevé de mutation de CBL,
ces mutations étaient présentes dans des SMD de stade avancé (5 cas décrits, tous dans des
AREB-2). Les mutations de CBL sont décrites à une plus grande fréquence dans les SMD
avancés et les LAM secondaires (Makishima et al, 2009), donc dans des HM à mauvais
pronostic (Reindl et al, 2009).
Les SMD et LAM constituent une mosaïque complexe d’altérations moléculaires.
Même si de nombreuses mutations de gène ont été décrites, de multiples altérations
moléculaires sont encore à découvrir. Certaines appartiennent aux classes I et II de Gilliland,
correspondant aux classes I, II, III de notre modèle, d’autres viendront certainement
compléter la classe IV. Les gènes codant pour des protéines régulant CBL peuvent être
regroupés dans la classe I de Gilliland ou la classe III de notre modèle et sont intéressants à
étudier pour caractériser leurs éventuelles anomalies moléculaires qui seraient à relier avec
les gains de copies que nous avons identifiés.
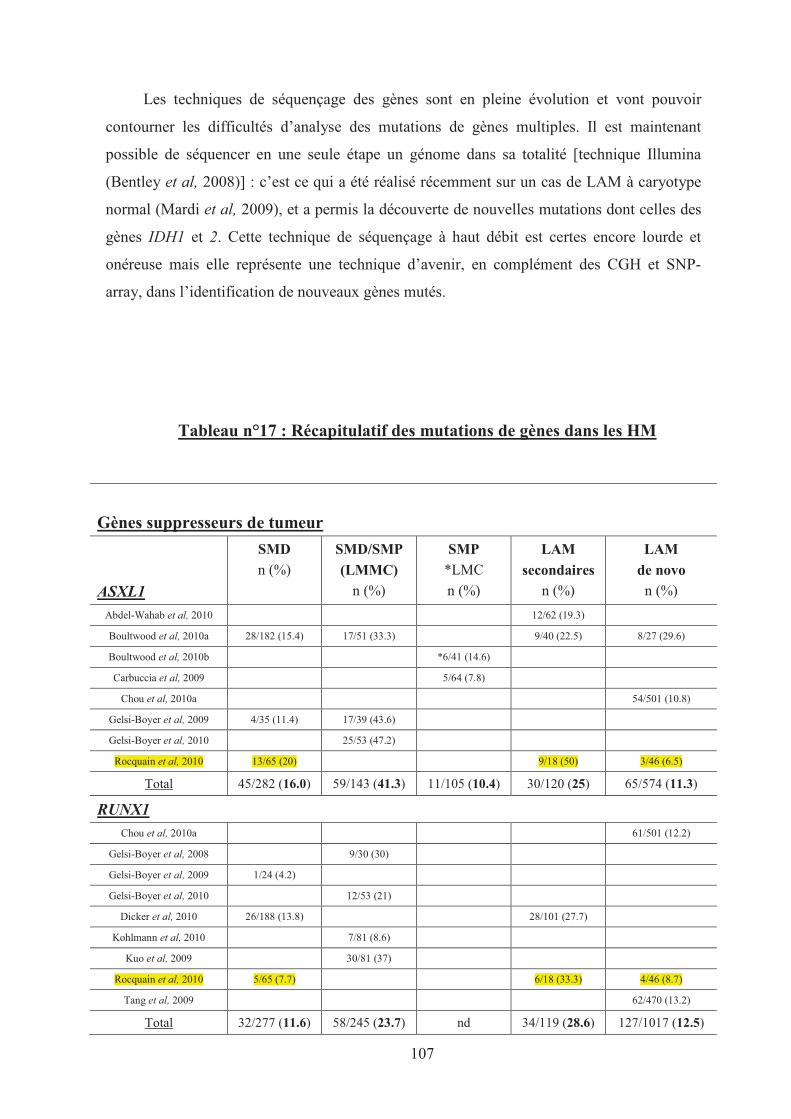
107
Les techniques de séquençage des gènes sont en pleine évolution et vont pouvoir
contourner les difficultés d’analyse des mutations de gènes multiples. Il est maintenant
possible de séquencer en une seule étape un génome dans sa totalité [technique Illumina
(Bentley et al, 2008)] : c’est ce qui a été réalisé récemment sur un cas de LAM à caryotype
normal (Mardi et al, 2009), et a permis la découverte de nouvelles mutations dont celles des
gènes IDH1 et 2. Cette technique de séquençage à haut débit est certes encore lourde et
onéreuse mais elle représente une technique d’avenir, en complément des CGH et SNP-
array, dans l’identification de nouveaux gènes mutés.
Tableau n°17 : Récapitulatif des mutations de gènes dans les HM
Gènes suppresseurs de tumeur
ASXL1
SMD
n (%)
SMD/SMP
(LMMC)
n (%)
SMP
*LMC
n (%)
LAM
secondaires
n (%)
LAM
de novo
n (%)
Abdel-Wahab et al, 2010 12/62 (19.3)
Boultwood et al, 2010a 28/182 (15.4) 17/51 (33.3) 9/40 (22.5) 8/27 (29.6)
Boultwood et al, 2010b *6/41 (14.6)
Carbuccia et al, 2009 5/64 (7.8)
Chou et al, 2010a 54/501 (10.8)
Gelsi-Boyer et al, 2009 4/35 (11.4) 17/39 (43.6)
Gelsi-Boyer et al, 2010 25/53 (47.2)
Rocquain et al, 2010 13/65 (20) 9/18 (50) 3/46 (6.5)
Total 45/282 (16.0) 59/143 (41.3) 11/105 (10.4) 30/120 (25) 65/574 (11.3)
RUNX1
Chou et al, 2010a 61/501 (12.2)
Gelsi-Boyer et al, 2008 9/30 (30)
Gelsi-Boyer et al, 2009 1/24 (4.2)
Gelsi-Boyer et al, 2010 12/53 (21)
Dicker et al, 2010 26/188 (13.8) 28/101 (27.7)
Kohlmann et al, 2010 7/81 (8.6)
Kuo et al, 2009 30/81 (37)
Rocquain et al, 2010 5/65 (7.7) 6/18 (33.3) 4/46 (8.7)
Tang et al, 2009 62/470 (13.2)
Total 32/277 (11.6) 58/245 (23.7) nd 34/119 (28.6) 127/1017 (12.5)
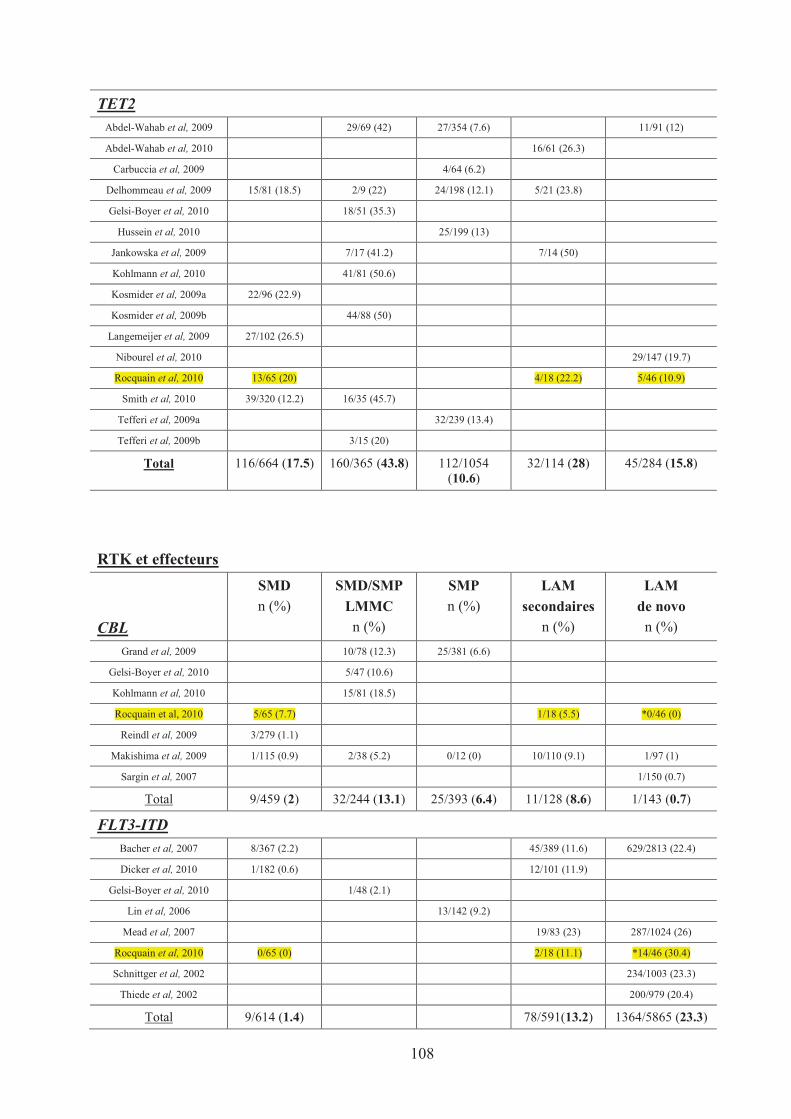
108
TET2
Abdel-Wahab et al, 2009 29/69 (42) 27/354 (7.6) 11/91 (12)
Abdel-Wahab et al, 2010 16/61 (26.3)
Carbuccia et al, 2009 4/64 (6.2)
Delhommeau et al, 2009 15/81 (18.5) 2/9 (22) 24/198 (12.1) 5/21 (23.8)
Gelsi-Boyer et al, 2010 18/51 (35.3)
Hussein et al, 2010 25/199 (13)
Jankowska et al, 2009 7/17 (41.2) 7/14 (50)
Kohlmann et al, 2010 41/81 (50.6)
Kosmider et al, 2009a 22/96 (22.9)
Kosmider et al, 2009b 44/88 (50)
Langemeijer et al, 2009 27/102 (26.5)
Nibourel et al, 2010 29/147 (19.7)
Rocquain et al, 2010 13/65 (20) 4/18 (22.2) 5/46 (10.9)
Smith et al, 2010 39/320 (12.2) 16/35 (45.7)
Tefferi et al, 2009a 32/239 (13.4)
Tefferi et al, 2009b 3/15 (20)
Total 116/664 (17.5) 160/365 (43.8) 112/1054 (10.6)
32/114 (28) 45/284 (15.8)
RTK et effecteurs
CBL
SMD
n (%)
SMD/SMP
LMMC
n (%)
SMP
n (%)
LAM
secondaires
n (%)
LAM
de novo
n (%)
Grand et al, 2009 10/78 (12.3) 25/381 (6.6)
Gelsi-Boyer et al, 2010 5/47 (10.6)
Kohlmann et al, 2010 15/81 (18.5)
Rocquain et al, 2010 5/65 (7.7) 1/18 (5.5) *0/46 (0)
Reindl et al, 2009 3/279 (1.1)
Makishima et al, 2009 1/115 (0.9) 2/38 (5.2) 0/12 (0) 10/110 (9.1) 1/97 (1)
Sargin et al, 2007 1/150 (0.7)
Total 9/459 (2) 32/244 (13.1) 25/393 (6.4) 11/128 (8.6) 1/143 (0.7)
FLT3-ITD
Bacher et al, 2007 8/367 (2.2) 45/389 (11.6) 629/2813 (22.4)
Dicker et al, 2010 1/182 (0.6) 12/101 (11.9)
Gelsi-Boyer et al, 2010 1/48 (2.1)
Lin et al, 2006 13/142 (9.2)
Mead et al, 2007 19/83 (23) 287/1024 (26)
Rocquain et al, 2010 0/65 (0) 2/18 (11.1) *14/46 (30.4)
Schnittger et al, 2002 234/1003 (23.3)
Thiede et al, 2002 200/979 (20.4)
Total 9/614 (1.4) 78/591(13.2) 1364/5865 (23.3)
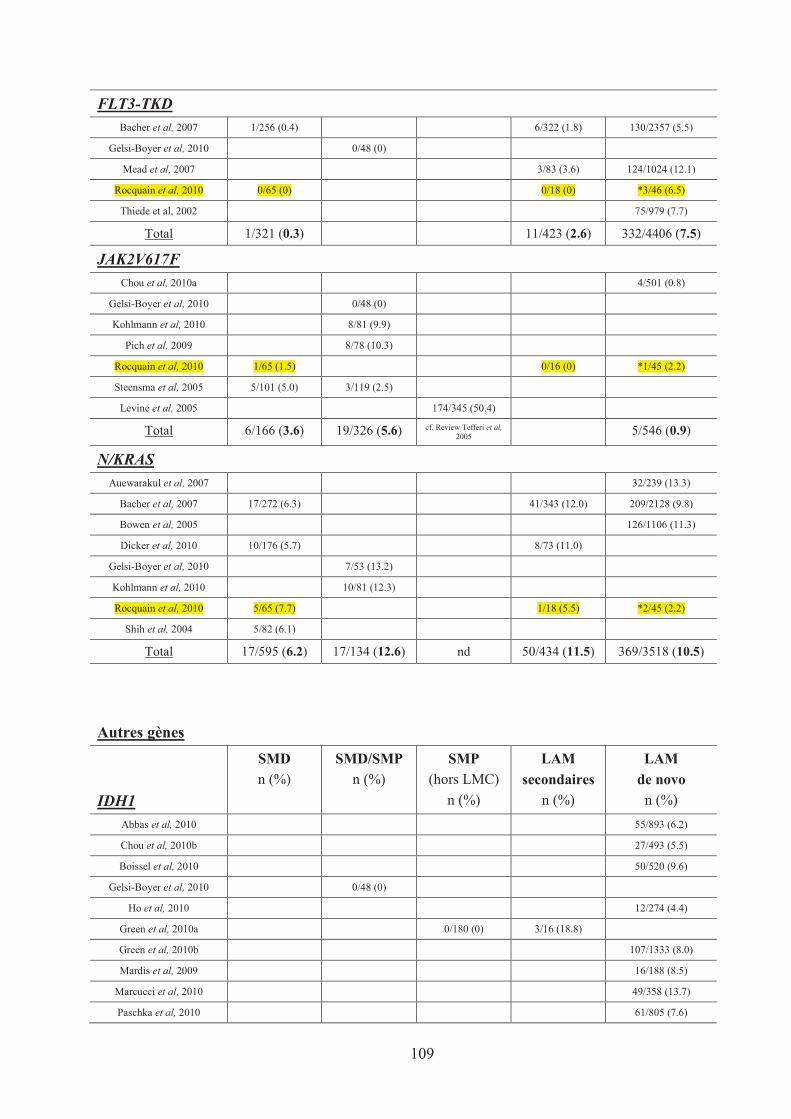
109
FLT3-TKD
Bacher et al, 2007 1/256 (0.4) 6/322 (1.8) 130/2357 (5.5)
Gelsi-Boyer et al, 2010 0/48 (0)
Mead et al, 2007 3/83 (3.6) 124/1024 (12.1)
Rocquain et al, 2010 0/65 (0) 0/18 (0) *3/46 (6.5)
Thiede et al, 2002 75/979 (7.7)
Total 1/321 (0.3) 11/423 (2.6) 332/4406 (7.5)
JAK2V617F
Chou et al, 2010a 4/501 (0.8)
Gelsi-Boyer et al, 2010 0/48 (0)
Kohlmann et al, 2010 8/81 (9.9)
Pich et al, 2009 8/78 (10.3)
Rocquain et al, 2010 1/65 (1.5) 0/16 (0) *1/45 (2.2)
Steensma et al, 2005 5/101 (5.0) 3/119 (2.5)
Levine et al, 2005 174/345 (50.4)
Total 6/166 (3.6) 19/326 (5.6) cf. Review Tefferi et al,2005
5/546 (0.9)
N/KRAS
Auewarakul et al, 2007 32/239 (13.3)
Bacher et al, 2007 17/272 (6.3) 41/343 (12.0) 209/2128 (9.8)
Bowen et al, 2005 126/1106 (11.3)
Dicker et al, 2010 10/176 (5.7) 8/73 (11.0)
Gelsi-Boyer et al, 2010 7/53 (13.2)
Kohlmann et al, 2010 10/81 (12.3)
Rocquain et al, 2010 5/65 (7.7) 1/18 (5.5) *2/45 (2.2)
Shih et al, 2004 5/82 (6.1)
Total 17/595 (6.2) 17/134 (12.6) nd 50/434 (11.5) 369/3518 (10.5)
Autres gènes
IDH1
SMD
n (%)
SMD/SMP
n (%)
SMP
(hors LMC)
n (%)
LAM
secondaires
n (%)
LAM
de novo
n (%)
Abbas et al, 2010 55/893 (6.2)
Chou et al, 2010b 27/493 (5.5)
Boissel et al, 2010 50/520 (9.6)
Gelsi-Boyer et al, 2010 0/48 (0)
Ho et al, 2010 12/274 (4.4)
Green et al, 2010a 0/180 (0) 3/16 (18.8)
Green et al, 2010b 107/1333 (8.0)
Mardis et al, 2009 16/188 (8.5)
Marcucci et al, 2010 49/358 (13.7)
Paschka et al, 2010 61/805 (7.6)
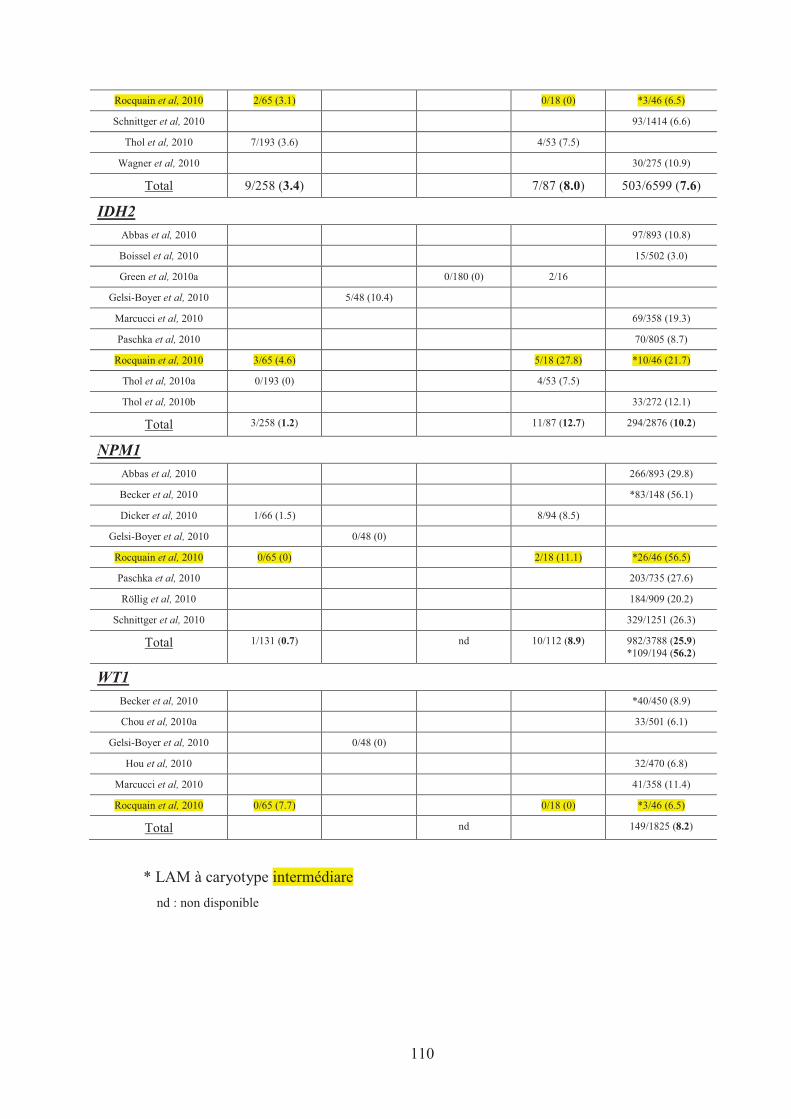
110
Rocquain et al, 2010 2/65 (3.1) 0/18 (0) *3/46 (6.5)
Schnittger et al, 2010 93/1414 (6.6)
Thol et al, 2010 7/193 (3.6) 4/53 (7.5)
Wagner et al, 2010 30/275 (10.9)
Total 9/258 (3.4) 7/87 (8.0) 503/6599 (7.6)
IDH2
Abbas et al, 2010 97/893 (10.8)
Boissel et al, 2010 15/502 (3.0)
Green et al, 2010a 0/180 (0) 2/16
Gelsi-Boyer et al, 2010 5/48 (10.4)
Marcucci et al, 2010 69/358 (19.3)
Paschka et al, 2010 70/805 (8.7)
Rocquain et al, 2010 3/65 (4.6) 5/18 (27.8) *10/46 (21.7)
Thol et al, 2010a 0/193 (0) 4/53 (7.5)
Thol et al, 2010b 33/272 (12.1)
Total 3/258 (1.2) 11/87 (12.7) 294/2876 (10.2)
NPM1
Abbas et al, 2010 266/893 (29.8)
Becker et al, 2010 *83/148 (56.1)
Dicker et al, 2010 1/66 (1.5) 8/94 (8.5)
Gelsi-Boyer et al, 2010 0/48 (0)
Rocquain et al, 2010 0/65 (0) 2/18 (11.1) *26/46 (56.5)
Paschka et al, 2010 203/735 (27.6)
Röllig et al, 2010 184/909 (20.2)
Schnittger et al, 2010 329/1251 (26.3)
Total 1/131 (0.7) nd 10/112 (8.9) 982/3788 (25.9) *109/194 (56.2)
WT1
Becker et al, 2010 *40/450 (8.9)
Chou et al, 2010a 33/501 (6.1)
Gelsi-Boyer et al, 2010 0/48 (0)
Hou et al, 2010 32/470 (6.8)
Marcucci et al, 2010 41/358 (11.4)
Rocquain et al, 2010 0/65 (7.7) 0/18 (0) *3/46 (6.5)
Total nd 149/1825 (8.2)
* LAM à caryotype intermédiare
nd : non disponible

111
III. CONCLUSION ET PERSPECTIVES
La découverte récente de nombreuses altérations moléculaires dans les SMD et LAM
nous ouvre les portes d’une nouvelle ère de compréhension de la biologie de ces maladies.
Le développement important ces dernières années des techniques d’analyse pangénomique a
permis de façon très rapide, l’identification de nouvelles lésions génétiques et épigénétiques
et a fait exploser le nombre de données sur la biologie moléculaire de ces maladies.
Apports de l’étude du transcriptome
Des études d’expression des gènes effectuées sur des progéniteurs hématopoïétiques
ont confirmé l’hétérogénéité des SMD et des LAM (Hoffmann et al, 2002 ; Ueda et al,
2003). Des études complémentaires sont encore nécessaires pour mieux classer ces maladies
et prédire leur évolution clinique par l’intermédiaire des signatures d’expression génique
(Sridhar et al, 2009). Les études d’expression des gènes permettent également d’identifier
des gènes sous ou surexprimés pour lesquels leur contribution à la maladie n’est pas
clairement élucidé. Par exemple dans les SMD, les gènes surexprimés DLK1 (codant delta-
like 1 homologue, une protéine aux fonctions hématopoïétiques encore inconnues) et BMI1
(codant une protéine du complexe polycomb impliquée dans l’auto-renouvellement
cellulaire) apparaissent intéressants (Chen et al, 2004 ; Pellagatti et al, 2004).
Il est intéressant de coupler à l’analyse du statut mutationnel des études d’expression
des gènes par puce à ADN. Boultwood et al, ont montré par exemple que les mutations
d’ASXL1 étaient reliées à une activation de la voie des récepteurs à l’acide rétinoïque (RAR),
confirmant 2 études montrant qu’ASXL1 pouvait agir soit comme co-activateur, soit comme
co-répresseur des RAR (Cho et al, 2006 ; Lee et al, 2010). Notre laboratoire maîtrise bien
l’étude du trancriptome par puces à ADN et l’analyse des nombreux nouveaux échantillons
que nous avons collectés, croisée avec les statuts mutationnels que nous avons établis,
pourrait apporter de précieuses informations.
Epigénétique
Les modifications épigénétiques par méthylation de l’ADN et acétylation des histones
altèrent la transcription des gènes (Feinberg et al, 2004). Une hyperméthylation anormale

112
des sites promoteurs des gènes est fréquente dans les SMD et particulièrement importante
dans les SMD de mauvais pronostic ainsi que lors de leur transformation en LAM (Galm et
al, 2006). Ces modifications épigénétiques provoquées par des mutations de gènes (comme
UTX par exemple, Van Haaften et al, 2009) altèrent à leur tour l’expression de certains gènes
suppresseurs de tumeurs. Le gène FZD9, situé sur le chromosome 7, est le gène dont le
promoteur semble le plus hyperméthylé lors de délétion du chromosome 7 : il constituerait
un nouveau gène suppresseur de tumeur important dans la transformation des SMD en LAM
(Jiang et al, 2009).
L’étude des altérations épigénétiques est d’autant plus essentielle que des agents
déméthylants [Décitabine (Dacogen®, Janssen Cilag), Azacitidine (Vidaza®, Celgene)] ont
montré leur efficacité dans certains SMD et LAM (Fenaux et al, 2009 et 2010). Leur
association aux inhibiteurs des histones désacétylases comme l’acide valproïque, est une
stratégie séduisante qui a montré des résultats intéressants, notamment dans les LAM mutées
MLL (Whitman et al, 2005 ; Gore et al, 2006). L’utilisation des agents déméthylants semble
prometteuse, particulièrement chez les personnes âgées qui ne peuvent tolérer des
traitements lourds (Shen et al, 2010).
Notre laboratoire a acquis la méthode et va prochainement étudier les niveaux de
méthylation des différents gènes mutés que nous avons étudiés. L’étude du méthylome est
très récente et de nombreuses anomalies des degrés de méthylation des gènes sont à
découvrir.
Quel avenir pour la prise en charge des LAM et SMD ?
En 2010, les SMD et LAM demeurent des maladies incurables en absence d’allogreffe
de moelle osseuse. La principale raison des difficultés à élucider la physiopathologie de ces
hémopathies est leur grande hétérogénéité au niveau génétique et moléculaire. Contrairement
au modèle de la LMC, les cibles moléculaires sont multiples et leurs altérations complexes
(combinaison de mutations, délétions, gains et méthylations).
De réels progrès ont été réalisés ces dernières années pour mieux classer ces
pathologies et mieux comprendre certaines voies impliquées dans leur physiopathologie. Les
nouvelles techniques d’analyse génomique telles que le séquençage pangénomique et
l’analyse du méthylome, voire la protéomique, vont très prochainement améliorer de façon
importante notre vision de la physiopathologie des SMD et LAM. L’accumulation très
rapide de nouvelles connaissances moléculaires va permettre dans un avenir proche, de
révolutionner les stratégies thérapeutiques de ces maladies.

113
BIBLIOGRAPHIE
(241 références)
Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders J, Zeilemaker A, van Putten WJ, Rijneveld A, Löwenberg B, Valk PJ. Acquired mutations in the genes encoding IDH1 andIDH2 both are recurrent aberrations in acute myeloid leukemia (AML): prevalence and prognostic value. Blood 2010 (in press).
Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, Huberman K, Thomas S, Dolgalev I, Heguy A, Paietta E, Le Beau MM, Beran M, Tallman MS, Ebert BL, Kantarjian HM, Stone RM, Gilliland DG, Crispino JD, Levine RL. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114; 144-147.
Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C, Heguy A, Bueso-Ramos C, Kantarjian H, Levine RL, Verstovsek S. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res 2010; 70(2): 447-52.
Adélaïde J, Gelsi-Boyer V, Rocquain J, Carbuccia N, Birnbaum DJ, Finetti P, Bertucci F, Mozziconacci MJ, Vey N, Birnbaum D, Chaffanet M. Gain of CBL-interacting protein, a possible alternative to CBL mutations in myeloid malignancies. Leukemia 2010; 24(8): 1539-41.
Aissouni Y, Zapart G, Iovanna JL, Dikic I, Soubeyran P. CIN85 regulates the ability of MEKK4 to activate the p38 MAP kinase pathway. Biochem Biophys Res Comm 2005; 338: 808-814.
Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol 2004; 2(5): 139.
Auewarakul CU, Lauhakirti D, Tocharoentanaphol C. Frequency of RAS gene mutation and its cooperative genetic events in Southeast Asian adult acute myeloid leukemia. Eur J Haematol2006; 77(1): 51-6.
Aul C, Gattermann N, Schneider W. Epidemiological and etiological aspects of myelodysplastic syndromes. Leuk Lymphoma 1995; 16: 247-62.
Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica 2007; 92: 744-52.
Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood 2006; 107(10): 3847-53.
Bacher U, Schnittger S, Kern W, Weiss T, Haferlach T, Haferlach C. Distribution of cytogenetic abnormalities in myelodysplastic syndromes, Philadelphia negative myeloproliferative neoplasms, and the overlap MDS/MPN category. Ann Hematol 2009; 88(12): 1207-13.
Bauerschmidt C, Arrichiello C, Burdak-Rothkamm S, Woodcock M, Hill MA, Stevens DL, Rothkamm K. Cohesin promotes the repair of ionizing radiation-induced DNA double-strand breaks in replicated chromatin. Nucleic Acids Res 2010; 38(2): 477-87.
Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D, Whitman SP, Paschka P, Holland KB, Schwind S, Wu YZ, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Moore JO, Caligiuri MA, Larson RA, Bloomfield CD. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood 2010; 116(5): 788-92.

114
Beghini A, Ripamonti CB, Cairoli R, Cazzaniga G, Colapietro P, Elice F, Nadali G, Grillo G, Haas OA, Biondi A, Morra E, Larizza L. KIT activating mutations: incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica 2004; 89: 920-925.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 1976; 33(4): 451-8.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara E Catenazzi M, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008; 456: 53-9.
Boehrer S, Ades L, Tajeddine N, Hofmann WK, Kriener S, Bug G, Ottmann OG, Ruthardt M, Galluzzi L, Fouassier C, Tailler M, Olaussen KA, Gardin C, Eclache V, de Botton S, Thepot S, Fenaux P, Kroemer G.. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene 2009; 28: 2205-2218.
Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, Raffoux E, Leblanc T, Thomas X, Hermine O, Quesnel B, Baruchel A, Leverger G, Dombret H, Preudhomme C. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 2006; 20: 965-970.
Boissel N, Nibourel O, Renneville A, Gardin C, Reman O, Contentin N, Bordessoule D, Pautas C, de Revel T, Quesnel B, Huchette P, Philippe N, Geffroy S, Terre C, Thomas X, Castaigne S, Dombret H, Preudhomme C. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol 2010; 28(23): 3717-23.
Boultwood J, Fidler C, Strickson AJ, Watkins F, Gama S, Kearney L, Tosi S, Kasprzyk A, Cheng JF, Jaju RJ, Wainscoat JS. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood 2002; 99: 4638-4641.

115
Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, Larrayoz MJ, Garcia-Delgado M, Giagounidis A, Malcovati L, della Porta MG, Jadersten M, Killick S, Hellstrom-Lindberg E, Cazzola M, Wainscoat JS. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 2010a, 24: 1062-1065.
Boultwood J, Perry J, Zaman R, Fernandez-Santamaria C, Littlewood T, Kusec R, Pellagatti A, Wang L, Clark RE, Wainscoat JS. High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia 2010b; 24(6): 1139-45.
Boultwood J, Wainscoat JS. Clonality in the myelodysplastic syndromes. Int J Hematol 2001; 73: 411-5.
Bowen D, Culligan D, Jowitt S, Kelsey S, Mufti G, Oscier D, Parker J; UK MDS Guidelines Group. Guidelines for the diagnosis and therapy of adult myelodysplastic syndromes. Br J Haematol 2003; 120: 187-200.
Bowen D, Groves MJ, Burnett AK, Patel Y, Allen C, Green C, Gale RE, Hills R, Linch DC. TP53
gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 2009; 23(1): 203-6.
Bowen DT, Frew ME, Hills R, Gale RE, Wheatley K, Groves MJ, Langabeer SE, Kottaridis PD, Moorman AV, Burnett AK, Linch DC. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 2005; 106: 2113-2119.
Bullinger L, Krönke J, Schön C, Radtke I, Urlbauer K, Botzenhardt U, Gaidzik V, Carió A, Senger C, Schlenk RF, Downing JR, Holzmann K, Döhner K, Döhner H. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia 2010; 24: 438-449.
Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, Knuutila S, Mrózek K, Oberkircher AR, Marcucci G, de la Chapelle A, Elonen E, Block AW, Rao PN, Herzig GP, Powell BL, Ruutu T, Schiffer CA, Bloomfield CD. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res 1998; 58: 55-59.
Campioni D, Secchiero P, Corallini F, Melloni E, Capitani S, Lanza F, Zauli G. Evidence for a role of TNF-related apoptosis-inducing ligand (TRAIL) in the anemia of myelodysplastic syndromes. American Journal of Pathology 2005; 166: 557–563.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, Vainchenker W, Bernard OA, Chaffanet M, Vey N, Birnbaum D, Mozziconacci MJ. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009; 23(11): 2183-6.
Carbuccia N, Trouplin V, Gelsi-Boyer V, Murati A, Rocquain J, Adélaïde J, Olschwang S, Xerri L, Vey N, Chaffanet M, Birnbaum D, Mozziconacci MJ. Mutual exclusion of ASXL1 and NPM1
mutations in a series of acute myeloid leukemias. Leukemia 2010; 24(2): 469-73.
Catenacci DV, Schiller GJ. Myelodysplastic syndromes: a comprehensive review. Blood Rev2005; 19: 301-19.
Cazzola M, Malcovati L. Myelodysplastic syndromes – coping with ineffective hematopoiesis. N Engl J Med 2005; 352: 536–8.
Chen CY, Lin LI, Tang JL , Ko BS, Tsay W, Chou WC, Yao M, Wu SJ, Tseng MH, Tien HF. RUNX1 gene mutation in primary myelodysplastic syndrome – the mutation can be detected

116
early at diagnosis or acquired during disease progression and is associated with poor outcome. Br J Haematol 2007; 139: 405-14.
Chen G, Zeng W, Miyazato A , Billings E, Maciejewski JP, Kajigaya S, Sloand EM, Young NS. Distinctive gene expression profiles of CD34 cells from patients with myelodysplastic syndrome characterized by specific chromosomal abnormalities. Blood 2004; 104: 4210-8.
Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer2010; 10(1): 23-36.
Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH, Schiffer CA, Doehner H, Tallman MS, Lister TA, Lo-Coco F, Willemze R, Biondi A, Hiddemann W, Larson RA, Löwenberg B, Sanz MA, Head DR, Ohno R, Bloomfield CD. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol 2003; 21: 4642-49.
Cho YS, Kim EJ, Park UH, Sin HS, Um SJ. Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J Biol Chem 2006; 281: 17588-17598.
Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010a (In press).
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 2010b; 115(14): 2749-54.
Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations of AML1 are common in therapy-related myelodysplasia following therapy with alkylating agents and are significantly associated with deletion or loss of chromosome arm 7q and with subsequent leukemic transformation. Blood 2004; 104: 1474-1481.
Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity ofp53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol 2001; 19: 1405-1413.
Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer 2007; 7(2): 118-29.
Cutler CS, Lee SJ, Greenberg P , Deeg HJ, Pérez WS, Anasetti C, Bolwell BJ, Cairo MS, Gale RP, Klein JP, Lazarus HM, Liesveld JL, McCarthy PL, Milone GA, Rizzo JD, Schultz KR, Trigg ME, Keating A, Weisdorf DJ, Antin JH, Horowitz MM. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 2004; 104: 579-85.
Delhommeau F, Dupont S, Della Valle V , James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F, Fontenay M, Vainchenker W, Bernard OA. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360: 2289-301.
Della Porta MG, Malcovati L, Travaglino E, Pietra D, Pascutto C, Passamonti F, Invernizzi R, Castello A, Magrini U, Lazzarino M, Cazzola M. Clinical relevance of bone marrow fibrosis

117
and CD34-positive cell clusters in primary myelodysplastic syndromes. J Clin Oncol 2009; 27: 754-62.
Deschler B, Lübbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer 2006; 1; 107(9): 2099-107.
Dicker F, Haferlach C, Sundermann J, Wendland N, Weiss T, Kern W, Haferlach T, Schnittger S. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia 2010; 24(8): 1528-32.
Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, Bullinger L, Fröhling S, Döhner H: Mutant nucléophosmine (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 2005, 106(12): 3740-3746.
Döhner K, Tobis K, Ulrich R, Fröhling S, Benner A, Schlenk RF, Döhner H. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol 2002, 20(15): 3254-3261.
Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B, Dahl N. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet 1999; 21: 169-175.
Dufour A, Schneider F, Metzeler KH, Hoster E, Schneider S, Zellmeier E, Benthaus T, Sauerland MC, Berdel WE, Büchner T, Wörmann B, Braess J, Hiddemann W, Bohlander SK, Spiekermann K. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol.2010; 28(4): 570-7.
Dunbar AJ, Gondek LP, O'Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP. 250 K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res 2008; 15; 68(24): 10349-57.
Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, Golub TR. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008; 451(7176): 335-9.
Ebert BL. Deletion 5q in myelodysplastic syndrome: a paradigm for the study of hemizygous deletions in cancer. Leukemia 2009; 23(7): 1252-6.
Ernst T, Chase A, Zoi K, Waghorn K, Hidalgo-Curtis C, Score J, Jones A, Grand F, Reiter A, Hochhaus A, Cross NC. Transcription factor mutations in myelodysplastic/myeloproliferative neoplasms. Haematologica 2010; 95(9): 1473-80.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42(8): 722-6.
Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006; 368: 1894-907.
Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, Nicoletti I. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 2009; 23: 1731-1743.

118
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005, 352(3): 254-266.
Farag SS, Ruppert AS, Mrózek K, Mayer RJ, Stone RM, Carroll AJ, Powell BL, Moore JO, Pettenati MJ, Koduru PR, Stamberg J, Baer MR, Block AW, Vardiman JW, Kolitz JE, Schiffer CA, Larson RA, Bloomfield CD. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: a cancer and leukemia group B study. J Clin Oncol 2005; 23(3): 482-93.
Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4(2): 143-53.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10: 223-232.
Fenaux P, Mufti GJ, Hellström-Lindberg E, Santini V, Gattermann N, Germing U, Sanz G, List AF, Gore S, Seymour JF, Dombret H, Backstrom J, Zimmerman L, McKenzie D, Beach CL, Silverman LR. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 2010; 28: 562-9.
Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, Hess JL, Humphries RK, Brock HW. Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood 2010; 115: 38-46.
Fisher CL, Randazzo F, Humphries RK, Brock HW. Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 2006; 369: 109-118.
Fröhling S, Schlenk RF, Stolze I, Bihlmayr J, Benner A, Kreitmeier S, Tobis K, Döhner H, Döhner K. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 2004; 22(4): 624-33.
Gaidzik VI, Schlenk RF, Moschny S, Becker A, Bullinger L, Corbacioglu A, Krauter J, Schlegelberger B, Ganser A, Döhner H, Döhner K; German-Austrian AML Study Group. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 2009; 113(19): 4505-11.
Galm O, Herman JG, Baylin SB. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev 2006; 20: 1-13
Gelsi-Boyer V, Trouplin V, Adélaïde J, Aceto N, Remy V, Pinson S, Houdayer C, Arnoulet C, Sainty D, Bentires-Alj M, Olschwang S, Vey N, Mozziconacci MJ, Birnbaum D, Chaffanet M. Genome profiling of chronic myelomonocytic leukemia: frequent alterations of RAS and RUNX1 genes. BMC Cancer 2008; 8: 299.
Gelsi-Boyer V, TrouplinV , Adelaide J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 145: 788-800.

119
Gelsi-Boyer V, Trouplin V, Rocquain J, Adélaïde J, Carbuccia N, Esterni B, Finetti P, Murati A, Arnoulet C, Zerazhi H, Fezoui H, Tadrist Z, Nezri M, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D . ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol 2010 (In press).
Germing U, Gattermann N, Strupp C, Aivado M, Aul C. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res 2000; 24: 983-92.
Germing U, Hildebrandt B, Pfeilstöcker M, et al. Refinement of the International Prognostic Scoring System (IPSS) by including LDH as an additional prognostic variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia 2005; 19: 2223-31.
Giagounidis AA, Germing U, Haase S, Hildebrandt B, Schlegelberger B, Schoch C, Wilkens L, Heinsch M, Willems H, Aivado M, Aul C. Clinical, morphological, cytogenetic, and prognostic features of patients with myelodysplastic syndromes and del(5q) including band q31. Leukemia 2004; 18: 113-119.
Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002; 100(5):1532-42.
Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell 2002; 1(5):417-20.
Gondek LP, Tiu R, Haddad AS, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Single nucleotide polymorphism arrays complement metaphase cytogenetics in detection of new chromosomal lesions in MDS. Leukemia 2007; 21:2058-2061.
Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008; 111:1534–1542.
Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, Rudek MA, Zhao M, Smith BD, Manning J, Jiemjit A, Dover G, Mays A, Zwiebel J, Murgo A, Weng LJ, Herman JG. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res 2006; 66(12): 6361-9.
Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, Cross NC. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009; 113(24): 6182-92.
Green A, Beer P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 2010; 362(4):369-70.
Green CL, Evans CM, Hills RK, Burnett AK, Linch DC, Gale RE. The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 2010 (In press).
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, Ohyashiki K, Toyama K, Aul C, Mufti G, Bennett J. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079-88.
Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK; National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116(3): 354-65.

120
Grimwade D, Hills RK. Independent prognostic factors for AML outcome. Hematology Am Soc Hematol Educ Program 2009; 385-95.
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood 1998; 92(7): 2322-2333.
Grimwade D. The clinical significance of cytogenetic abnormalities in acute myeloid leukaemia. Best Pract Res Clin Haematol 2001; 14(3): 497-529.
Grisendi S, Mecucci C, Falini B, Pandolfi PP: Nucleophosmin and cancer. Nat Rev Cancer 2006; 6(7): 493-505.
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010; 207(2): 339-44.
Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood 2004; 104: 2263-2268.
Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol 2008; 87: 515-26.
Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, Kundgen A, Lübbert M, Kunzmann R, Giagounidis AA, Aul C, Trümper L, Krieger O, Stauder R, Müller TH, Wimazal F, Valent P, Fonatsch C, Steidl C. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood2007; 110: 4385-4395.
Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53
gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008; 22(8): 1539-41.
Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T. High incidence of somatic mutations in the AML1 ⁄ RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 2004; 103: 2316-24.
Harada H, Harada Y, Tanaka H, Kimura A, Inaba T. Implications of somatic mutations in theAML1 gene in radiation-associated and therapy-related myelodysplastic syndrome/acute myeloid leukemia. Blood 2003; 101(2): 673-80.
Heaney ML, Golde DW. Myelodysplasia. N Engl J Med 1999; 340(21): 1649-60.
Hellstrom-Lindberg E, Malcovati L. Supportive care, growth factors, and new therapies in myelodysplastic syndromes. Blood Rev 2008; 22: 75-91.
Hellstrom-Lindberg E. Significance of JAK2 and TET2 mutations in myelodysplastic syndromes. Blood Rev 2010; 115: 1779-1784.
Ho PA, Alonzo TA, Kopecky KJ, Miller KL, Kuhn J, Zeng R, Gerbing RB, Raimondi SC, Hirsch BA, Oehler V, Hurwitz CA, Franklin JL, Gamis AS, Petersdorf SH, Anderson JE, Reaman GH, Baker LH, Willman CL, Bernstein ID, Radich JP, Appelbaum FR, Stirewalt DL, Meshinchi S. Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 2010; 24(5): 909-13.
Hofmann WK, de Vos S, Komor M, Hoelzer D, Wachsman W, Koeffler HP. Characterization of gene expression of CD34+ cells from normal and myelodysplastic bone marrow. Blood 2002; 100: 3553-60.

121
Horsfield JA, Anagnostou SH, Hu JK, Cho KH, Geisler R, Lieschke G, Crosier KE, Crosier PS. Cohesin-dependent regulation of Runx genes. Development 2007; 134(14): 2639-49.
Hugues L, Cavé H, Philippe N, Pereira S, Fenaux P, Preudhomme C. Mutations of PTPN11 are rare in adult myeloid malignancies. Haematologica 2005; 90(6): 853-4.
Hussein K, Abdel-Wahab O, Lasho TL, Van Dyke DL, Levine RL, Hanson CA, Pardanani A, Tefferi A. Cytogenetic correlates of TET2 mutations in 199 patients with myeloproliferative neoplasms. Am J Hematol 2010; 85(1): 81-3.
Illmer T, Schaich M, Ehninger G, Thiede C; DSIL2003 AML study group. Tyrosine kinase mutations of JAK2 are rare events in AML but influence prognosis of patients with CBF-leukemias. Haematologica 2007; 92(1): 137-8.
Jaffe ES, Harris NL, Stein H, Vardiman JW. WHO classification of tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon IARC Press 2001.
Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, O'Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009; 113: 6403-6410.
Janssen JW, Steenvoorden AC, Lyons J, Anger B, Böhlke JU, Bos JL, Seliger H, Bartram CR. RAS gene mutations in acute and chronic myelocytic leukemias, chronic myeloproliferative disorders, and myelodysplastic syndromes. Proc Natl Acad Sci USA 1987; 84: 9228-9232.
Jiang Y, Dunbar A, Gondek LP Mohan S, Rataul M, O'Keefe C, Sekeres M, Saunthararajah Y, Maciejewski JP. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009; 113: 1315-25.
Jongmans MC, Kuiper RP, Carmichael CL, Wilkins EJ, Dors N, Carmagnac A, Schouten-van Meeteren AY, Li X, Stankovic M, Kamping E, Bengtsson H, Schoenmakers EF, van Kessel AG, Hoogerbrugge PM, Hahn CN, Brons PP, Scott HS, Hoogerbrugge N. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: clues for improved identification of the FPD/AML syndrome. Leukemia 2010; 24(1): 242-6.
Juliusson G, Antunovic P, Derolf A, Lehmann S, Möllgård L, Stockelberg D, Tidefelt U, Wahlin A, Höglund M. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood 2009; 113(18): 4179-4187.
Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, Helmer R 3rd, Shen L, Nimer SD, Leavitt R, Raza A, Saba H. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006; 106: 1794-803.
Kantarjian H, O'Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, List A, Fenaux P, Sanz G, Issa JP, Freireich EJ, Garcia-Manero G. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer 2008; 113: 1351-61.
Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet 2002; 3: 179-98.
King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms’ tumor gene WT1
in leukemias. Blood 1996; 87: 2171-2179.
Kita-Sasai Y, Misawa S, Kaneko H, Kobayashi M, Nakao M, Nakagawa H, Fujii H, Taniwaki M. International prognostic scoring system and TP53 mutations are independent prognostic indicators for patients with myelodysplastic syndrome. Br J Haematol 2001; 115: 309-12.

122
Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68(4): 820-3.
Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer 2001; 1(2): 157-62.
Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, Dicker F, Schnittger S, Dugas M, Kern W, Haferlach C, Haferlach T. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 2010; 28(24): 3858-65.
Komeno Y, Kitaura J, Kitamura T. Molecular bases of myelodysplastic syndromes: lessons from animal models. J Cell Physiol 2009; 219(3): 529-34.
Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, Hunault-Berger M, Slama B, Vey N, Lacombe C, Solary E, Birnbaum D, Bernard OA, Fontenay M. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia 2010a; 24(5): 1094-6.
Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie JN, Beyne-Rauzy O, Stamatoulas A, Dreyfus F, Ifrah N, de Botton S, Vainchenker W, Bernard OA, Birnbaum D, Fontenay M, Solary E; Groupe Francophone des Myélodysplasies. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009b; 94(12): 1676-81.
Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguie F, Quesnel B, Beyne-Rauzy O, Solary E, Vey N, Hunault-Berger M, Fenaux P, Mansat-De Mas V, Delabesse E, Guardiola P, Lacombe C, Vainchenker W, Preudhomme C, Dreyfus F, Bernard OA, Birnbaum D, Fontenay M, TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 2009a; 114: 3285-3291.
Kouides PA, Bennett JM. Understanding the myelodysplastic syndrome. Oncologist 1997; 2: 389-401.
Kuo MC, Liang DC, Huang CF, Shih YS, Wu JH, Lin TL, Shih LY. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia 2009; 23(8): 1426-31
Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, Kamping EJ, Verhoef GE, Verburgh E, Hagemeijer A, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 2009; 41: 838-42.
Larson RA. Therapy-related myeloid neoplasms. Haematologica 2009; 94(4):454-9.
Lee SW, Cho YS, Na JM, Park UH, Kang M, Kim EJ, Um SJ. ASXL1 represses retinoic acid receptor-mediated transcription through associating with HP1 and LSD1. J Biol Chem 2010; 285: 18-29.
Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells 2005; 23: 16-43.
Leone G, Pagano L, Ben-Yehuda D, Voso MT. Therapy-related leukemia and myelodysplasia: susceptibility and incidence. Haematologica 2007; 92: 1389–98.
Levine AJ. P53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Fröhling S,

123
Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7(4): 387-97.
Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia 2003; 17(9): 1738-52.
Lin P, Jones D, Medeiros LJ, Chen W, Vega-Vazquez F, Luthra R. Activating FLT3 mutations are detectable in chronic and blast phase of chronic myeloproliferative disorders other than chronic myeloid leukemia. Am J Clin Pathol 2006; 126(4): 530-3.
List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, Powell B, Greenberg P, Thomas D, Stone R, Reeder C, Wride K, Patin J, Schmidt M, Zeldis J, Knight R; Myelodysplastic Syndrome-003 Study Investigators. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med 2006; 355(14):1456-65.
Liu H, Cheng EH, Hsieh JJ. MLL fusions: pathways to leukemia. Cancer Biol Ther 2009; 8(13): 1204-11.
Lutterbach B, Hiebert SW. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene 2000; 245: 223–235.
Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H, O'Keefe C, Hsi E, Paquette RL, Kojima S, List AF, Sekeres MA, McDevitt MA, Maciejewski JP. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. Clin Oncol 2009; 27: 6109–6116.
Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, Giagounidis A, Hildebrandt B, Bernasconi P, Knipp S, Strupp C, Lazzarino M, Aul C, Cazzola M. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007; 25: 3503-10.
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010; 28(14):2348-55.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med2009; 10; 361(11):1058-66.
Martin MG, Welch JS, Uy GL, Fehniger TA, Kulkarni S, Duncavage EJ and Walter MJ. Limited engraftment of low-risk myelodysplastic syndrome cells in NOD/SCID gamma-Cchain knockout mice. Leukemia 2010 (In press).
Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 2007; 110(4):1262-70.

124
Mehrotra B, George TI, Kavanau K, Avet-Loiseau H, Moore D 2nd, Willman CL, Slovak ML, Atwater S, Head DR, Pallavicini MG. Cytogenetically aberrant cells in the stem cell compartment (CD34+lin-) in acute myeloid leukemia. Blood 1995; 86(3):1139-47.
Miller KB, Kim K, Morrison FS, et al. The evaluation of low-dose cytarabine in the treatment of myelodysplastic syndromes: a phase-III intergroup study. Ann Hematol 1992; 65:162-8.
Mohamedali A, Gäken J, Twine NA, Ingram W, Westwood N, Lea NC, Hayden J, Donaldson N, Aul C, Gattermann N, Giagounidis A, Germing U, List AF, Mufti GJ. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood 2007; 110: 3365-73.
Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, Westwood NB, Strupp C, Gattermann N, Germing U, Mufti GJ. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. J Clin Oncol 2009; 27: 4002–4006.
Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev 2004; 18: 115-36.
Mrózek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol 2008; 358(4):365-377.
Mueller BU, Pabst T, Osato M, Asou N, Johansen LM, Minden MD, Behre G, Hiddemann W, Ito Y, Tenen DG. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood 2002; 100: 998-1007.
Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer 2004; 4: 394–400.
Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, Dombret H, Dreyfus F, Quesnel B, Geffroy S, Quentin S, Roche-Lestienne C, Cayuela JM, Roumier C, Fenaux P, Vainchenker W, Bernard OA, Soulier J, Fontenay M, Preudhomme C. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood 2010; 116(7): 1132-5.
Nikoloski G, Langemeijer SMC, Kuiper RP, Knops R, Massop M, Tönnissen E, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetic2010 (in press).
Nimer SD. Myelodysplastic syndromes. Blood 2008; 111: 4841-4851.
Nisse C, Lorthois C, Dorp V, Eloy E, Haguenoer JM, Fenaux P. Exposure to occupational and environmental factors in myelodysplastic syndromes: preliminary results of a case-control study. Leukemia 1995; 9:693-9.
Nowak D, Nolte F, Mossner M, Nowak V, Baldus CD, Hopfer O, Noll S, Thiel E, Wagner F, Hofmann WK. Genome-wide DNA-mapping of CD34+ cells from patients with myelodysplastic syndrome using 500K SNP arrays identifies significant regions of deletion and uniparental disomy. Exp Hematol 2009; 37: 215-24.
Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960; 132:1497.
Nowell PC. The clonal evolution of tumor cell populations. Science 1976; 194:23-8.
Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii-Kuriyama Y, Kato S. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007; 444: 562-566.

125
Olney HJ, Le Beau MM. The cytogenetics of myelodysplastic syndromes. Best Pract Res Clin Haematol 2001; 14: 479-495.
Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene 2004; 23(24):4284-96.
Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia--a review. Br J Haematol 2008; 140(2):123-32.
Pabst T, Eyholzer M, Fos J, Mueller BU. Heterogeneity within AML with CEBPA mutations; only CEBPA double mutations, but not single CEBPA mutations are associated with favourable prognosis. Br J Cancer 2009; 100(8): 1343-6.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001 Mar;27(3):263-70.
Padua RA, Guinn BA, Al-Sabah AI, Smith M, Taylor C, Pettersson T, Ridge S, Carter G, White D, Oscier D, Chevret S, West R. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10- year follow-up. Leukemia 1998; 12: 887–92.
Park S, Grabar S, Kelaidi C, Beyne-Rauzy O, Picard F, Bardet V, Coiteux V, Leroux G, Lepelley P, Daniel MT, Cheze S, Mahé B, Ferrant A, Ravoet C, Escoffre-Barbe M, Adès L, Vey N, Aljassem L, Stamatoullas A, Mannone L, Dombret H, Bourgeois K, Greenberg P, Fenaux P, Dreyfus F; GFM group (Groupe Francophone des Myélodysplasies). Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008; 111: 574-82.
Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000; 96: 3932-8.
Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA, Vukosavljevic T, Perrotti D, Vardiman JW, Carroll AJ, Kolitz JE, Larson RA, Bloomfield CD. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol 2006; 24: 3904-3911.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, Späth D, Kayser S, Zucknick M, Götze K, Horst HA, Germing U, Döhner H, Döhner K. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 2010; 28(22): 3636-43.
Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2008; 22: 240-8.
Pedersen-Bjergaard J, Andersen MT, Andersen MK. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2007: 392-7.
Pellagatti A, Esoof N, Watkins F, Langford CF, Vetrie D, Campbell LJ, Fidler C, Cavenagh JD, Eagleton H, Gordon P, Woodcock B, Pushkaran B, Kwan M, Wainscoat JS, Boultwood J. Gene expression profiling in the myelodysplastic syndromes using cDNA microarray technology. Br J Haematol 2004; 125: 576-83.
Pellagatti A, Jädersten M, Forsblom AM, Cattan H, Christensson B, Emanuelsson EK, Merup M, Nilsson L, Samuelsson J, Sander B, Wainscoat JS, Boultwood J, Hellström-Lindberg E. Lenalidomide inhibits the malignant clone and up-regulates the SPARC gene mapping to the

126
commonly deleted region in 5q- syndrome patients. Proc Natl Acad Sci USA 2007; 104(27): 11406-11.
Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev 2008; 22: 3089-3114.
Pich A, Riera L, Sismondi F, Godio L, Davico Bonino L, Marmont F, Francia di Celle P. JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol 2009; 62(9): 798-801.
Pierre R, Catovsky D, Mufti G, Swansbury G. Clinical cytogenetic correlations in myelodysplasia (preleukemia). Cancer Genet Cytogenet 1990; 44: 15.
Pollard PJ, Ratcliffe PJ: Puzzling patterns of predisposition. Science 2009, 324:192-194.
Preudhomme C, Sagot C, Boissel N, Cayuela JM, Tigaud I, de Botton S, Thomas X, Raffoux E, Lamandin C, Castaigne S, Fenaux P, Dombret H; ALFA Group. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood 2002; 100: 2717-2723.
Preudhomme C, Warot-Loze D, Roumier C, Grardel-Duflos N, Garand R, Lai JL, Dastugue N, Macintyre E, Denis C, Bauters F, Kerckaert JP, Cosson A, Fenaux P. High incidence of biallelic point mutations in the Runt domain of the AML1/PEBP2 alpha B gene in Mo acute myeloid leukemia and in myeloid malignancies with acquired trisomy 21. Blood 2000; 96: 2862–2869.
Reindl C, Quentmeier H, Petropoulos K, Greif PA, Benthaus T, Argiropoulos B, Mellert G, Vempati S, Duyster J, Buske C, Bohlander SK, Humphries KR, Hiddemann W, Spiekermann K. CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/ myelodysplastic syndrome subtypes. Clin Cancer Res 2009; 15: 2238-2247.
Renneville A, Boissel N, Zurawski V, Llopis L, Biggio V, Nibourel O, Philippe N, Thomas X, Dombret H, Preudhomme C. Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 2009; 115(16): 3719-27.
Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, Preudhomme C. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia2008; 22(5): 915-31.
Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, Tadrist Z, Olschwang S, Vey N, Birnbaum D, Gelsi-Boyer V, Mozziconacci MJ. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 2010a; 10: 401.
Rocquain J, Gelsi-Boyer V, Adélaïde J, Murati A, Carbuccia N, Vey N, Birnbaum D, Mozziconacci MJ, Chaffanet M. Alteration of cohesin genes in myeloid diseases. Am J Hematol2010b; 85(9):717-9.
Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001- 2004, using data from the NAACCR and SEER programs. Blood 2008; 112:45-52.
Rosnet O, Bühring HJ, Marchetto S, Rappold I, Lavagna C, Sainty D, Arnoulet C, Chabannon C, Kanz L, Hannum C, Birnbaum D. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 1996; 10(2): 238-48.
Rowley JD. Chromosomal translocations: revisited yet again. Blood 2008; 112(6): 2183-9.

127
Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M, Thiessen C, Tickenbrock L, Schwäble J, Brandts C, August B, Koschmieder S, Bandi SR, Duyster J, Berdel WE, Müller-Tidow C, Dikic I, Serve H. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 2007; 110(3): 1004-12.
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N, Onodera M, Nobuyoshi M, Hayashi Y, Harada H, Kurokawa M, Chiba S, Mori H, Ozawa K, Omine M, Hirai H, Nakauchi H, Koeffler HP, Ogawa S. Gain-of-function of mutated C-CBL
tumour suppressor in myeloid neoplasms. Nature 2009; 460(7257):904-8.
Scharnhorst V, van der Eb AJ, Jochemsen AG. WT1 proteins: functions in growth and differentiation. Gene 2001; 273(2):141-61.
Schlenk RF, Benner A, Krauter J, Büchner T, Sauerland C, Ehninger G, Schaich M, Mohr B, Niederwieser D, Krahl R, Pasold R, Döhner K, Ganser A, Döhner H, Heil G. Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: a survey of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol 2004; 15(18):3741-3750.
Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, Habdank M, Späth D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Döhner H; German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008; 358(18):1909-18.
Schmidt MH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol 2005; 6(12): 907-18.
Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 2010 (In press).
Schnittger S, Kinkelin U, Schoch C, Heinecke A, Haase D, Haferlach T, Büchner T, Wörmann B, Hiddemann W, Griesinger F: Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia 2000; 14(5):796-804.
Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Löffler H, Sauerland CM, Serve H, Büchner T, Haferlach T, Hiddemann W. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002; 100:59-66.
Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, Berry D, Ahmed S, Zhu W, Pierce S, Kondo Y, Oki Y, Jelinek J, Saba H, Estey E, Issa JP. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 2010; 28(4):605-13.
Shih LY, Huang CF, Wang PN, Wu JH, Lin TL, Dunn P, Kuo MC. Acquisition of FLT3 or N-ras
mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia 2004 Mar; 18(3): 466-75.
Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol 2009; 37(6): 649-58.
Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR, Rowe JM, Forman SJ, Appelbaum FR. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a

128
Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000; 15; 96(13): 4075-83.
Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gäken J, Lea NC, Przychodzen B, Mian SA, Nasser EE, Shooter C, Westwood NB, Strupp C, Gattermann N, Maciejewski JP, Germing U, Mufti GJ. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 2010 (In press).
Solé F, Luno E, Sanzo C, Espinet B, Sanz GF, Cervera J, Calasanz MJ, Cigudosa JC,Milla F, Ribera JM, Bureo E,MarquezML, Arranz E, Florensa L. Identification of novel cytogeneticmarkers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica 2005; 90:1168–1178.
Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 2002; 416: 183-187.
Sridhar K, Ross DT, Tibshirani R, Butte AJ, Greenberg PL. Relationship of differential gene expression profiles in CD34+ myelodysplastic syndrome marrow cells to disease subtype and progression. Blood 2009; 114(23): 4847-58.
Starczynowski Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, Buckstein R, Lam W, Humphries RK, Karsan A. Identification of miR-145
and miR-146a as mediators of the 5q– syndrome phenotype. Nature 2010; 16(1): 49-58.
Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, Gilliland DG, Tefferi A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood 2005; 106: 1207-9.
Stiller CA. Epidemiology and genetics of childhood cancer. Oncogene 2004; 23(38): 6429-44.
Strom SS, Gu Y, Gruschkus SK, Pierce SA, Estey EH. Risk factors of myelodysplastic syndromes: a case–control study. Leukemia 2005; 19: 1912–8.
Strom SS, Velez-Bravo V, Estey EH. Epidemiology of myelodysplastic syndromes. Semin Hematol 2008; 45: 8–13.
Suela J, Alvarez S, Cifuentes F, Largo C, Ferreira BI, Blesa D, Ardanaz M, García R, Marquez JA, Odero MD, Calasanz MJ, Cigudosa JC. DNA profiling analysis of 100 consecutive de novo acute myeloid leukemia cases reveals patterns of genomic instability that affect all cytogenetic risk groups. Leukemia 2007; 21: 1224–1231.
Swerdlow SH, Campo E, Harris NL, Jaffe ES., Pileri SA, Stein H, Thiele J, Vardiman JW. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324: 930-935.
Take H, Watanabe S, Takeda K, Yu ZX, Iwata N, Kajigaya S. Cloning and characterization of a novel adaptor protein, CIN85, that interacts with c-Cbl. Biochem Biophys Res Commun 2000; 268: 321-328.
Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, Huang CF, Lee FY, Liu MC, Yao M, Huang SY, Ko BS, Hsu SC, Wu SJ, Tsay W, Chen YC, Lin LI, Tien HF. AML1/RUNX1

129
mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood 2009; 114 (26): 5352-61.
Tefferi A, Gilliland DG. The JAK2V617F tyrosine kinase mutation in myeloproliferative disorders: status report and immediate implications for disease classification and diagnosis. Mayo Clin Proc 2005; 80(7): 947-58.
Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, Hanson CA, Pardanani A, Gilliland DG, Levine RL. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009b; 23: 1343-1345.
Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A, Li CY, Hanson CA, Mesa R, Bernard O, Delhommeau F, Vainchenker W, Gilliland DG, Levine RL. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009a; 23(5): 905-11.
Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med 2009; 361(19):1872-85.
Tefferi A. Myelodysplastic Syndromes--Many New Drugs, Little Therapeutic Progress. Mayo Clin Proc 2010 (In press).
Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer 2003; 3: 89-101.
Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M, Neubauer A, Ehninger G, Illmer T. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002; 99: 4326-4335.
Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M, Goehring G, Schumann C, Bug G, Ottmann O, Hofmann WK, Schlegelberger B, Ganser A, Heuser M. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis.Haematologica 2010a; 95(10): 1623-7.
Thol F, Damm F, Wagner K, Göhring G, Schlegelberger B, Hoelzer D, Lübbert M, Heit W, Kanz L, Schlimok G, Raghavachar A, Fiedler W, Kirchner H, Heil G, Heuser M, Krauter J, Ganser A. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 2010b; 116(4): 614-6.
Ueda M, Ota J, Yamashita Y, Choi YL, Ohki R, Wada T, Koinuma K, Kano Y, Ozawa K, Mano H. DNA microarray analysis of stage progression mechanism in myelodysplastic syndrome. Br J Haematol 2003;123:288-96.
Van de Loosdrecht AA, Westers TM, Westra AH, Drager AM, van der Velden VH, Ossenkoppele GJ. Identification of distinct prognostic subgroups in low- and intermediate-1-risk myelodysplastic syndromes by flow cytometry. Blood 2008; 111:1067-77.
Van den Berghe H, Cassiman JJ, David G, Fryns JP, Michaux JL, Sokal G, Distinct haematological disorder with deletion of long arm of no. 5 chromosome, Nature 1974; 251: 437-438.
van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague J, Butler A, Hinton J, Latimer C, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Cole J, Forbes S, Jia M, Jones D, Kok CY, Leroy C, Lin ML, McBride DJ, Maddison M, Maquire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, Pleasance E, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Collins VP, Ichimura K, Law S, Wong J, Yuen ST, Leung SY, Tonon G, DePinho RA, Tai YT, Anderson KC, Kahnoski RJ, Massie A, Khoo SK, Teh BT,

130
Stratton MR, Futreal PA. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 2009; 41(5): 521-3.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114(5): 937-51.
Verhaak RG, Goudswaard CS, van Putten W, Bijl MA, Sanders MA, Hugens W, Uitterlinden AG, Erpelinck CA, Delwel R, Löwenberg B, Valk PJ: Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood2005, 106(12): 3747-3754.
Wagner K, Damm F, Gohring G, Gorlich K, Heuser M, Schäfer I, Ottmann O, Lübbert M, Heit W, Kanz L, Schlimok G, Raghavachar AA, Fiedler W, Kirchner HH, Brugger W, Zucknick M, Schlegelberger B, Heil G, Ganser A, Krauter J. Impact of IDH1 R132 mutations and an IDH1
single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 2010; 28(14): 2356-64.
Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, Baty J, Heath S, Westervelt P, Watson MA, Tomasson MH, Nagarajan R, O'Gara BP, Bloomfield CD, Mrózek K, Selzer RR, Richmond TA, Kitzman J, Geoghegan J, Eis PS, Maupin R, Fulton RS, McLellan M, Wilson RK, Mardis ER, Link DC, Graubert TA, DiPersio JF, Ley TJ. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA 2009; 106: 12950-12956.
Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood2008; 111(5): 2505-15.
Warlick ED, Cioc A, Defor T, Dolan M, Weisdorf D. Allogeneic stem cell transplantation for adults with myelodysplastic syndromes: importance of pretransplant disease burden. Biol Blood Marrow Transplant 2009; 15: 30-8.
Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994; 84: 3148-57.
Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, Byrd J, Sokol L, Lawrence N, Pireddu R, Dewald G, Williams A, Maciejewski J, List A. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci USA 2009; 106(31): 12974-9.
Whitman SP, Liu S, Vukosavljevic T, Rush LJ, Yu L, Liu C, Klisovic MI, Maharry K, Guimond M, Strout MP, Becknell B, Dorrance A, Klisovic RB, Plass C, Bloomfield CD, Marcucci G, Caligiuri MA. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 2005; 106(1): 345-52.
Wong AK, Fang B, Zhang L, Guo X, Lee S, Schreck R. Loss of the Y chromosome: an age-related or clonal phenomenon in acute myelogenous leukemia/ myelodysplastic syndrome?Arch Pathol Lab Med 2008; 132: 1329-32.
Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia 2005;19: 1345-1349.
Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia 2007; 21(5): 868-76.

131
Yunis JJ, Lobell M, Arnesen MA, Oken MM, Mayer MG, Rydell RE, Brunning RD: Refined chromosome study helps define impact of age and gender and failure to identify a very-low-risk prognostic subgroups in most patients with primary myelodysplastic and acute myelogenous leukaemia. Br J Haematol 1988; 68:189.
Zhang Y, Zhang M, Yang L, Xiao Z. NPM1 mutations in myelodysplastic syndromes and acute myeloid leukemia with normal karyotype. Leuk Res 2007; 31: 109-11.

132
IV. ANNEXES
ARTICLE N°5
ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia.
Gelsi-Boyer V*, Trouplin V*, Rocquain J*, Adélaïde J, Carbuccia N, Esterni B, Finetti P, Murati A, Arnoulet C, Zerazhi H, Fezoui H,
Tadrist Z, Nezri M, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D.
Br J Haematol 2010; 151(4): 365-75
* 1ers auteurs

ASXL1 mutation is associated with poor prognosis and acute
transformation in chronic myelomonocytic leukaemia
Chronic myelomonocytic leukaemia (CMML) is a haemato-
poietic disease currently classified by the WHO organization in
a mixed category called MDS/MPN (myelodysplastic syn-
dromes/myeloproliferative neoplasm) (Vardiman et al, 2002).
Despite recent molecular improvements in the classification of
myeloid neoplasms, the diagnosis of CMML is still based on
cytological criteria including persistent peripheral monocytosis
>1 · 109/l, <20% blasts in the blood or bone marrow (BM)
and bone marrow dysplasia in one or more myeloid lineage.
The only molecular criterium is the absence of BCR-ABL1
transcript (Bennett et al, 1982; Vardiman et al, 2002). Because
the blast number is a prognostic factor, CMML is divided in
Veronique Gelsi-Boyer,1,2,3* Virginie
Trouplin,1* Julien Roquain,1* Jose
Adelaıde,1 Nadine Carbuccia,1 Benjamin
Esterni,4 Pascal Finetti,1 Anne Murati,1,2
Christine Arnoulet,2 Hacene Zerazhi,5
Hacene Fezoui,6 Zoulika Tadrist,7 Meyer
Nezri,8 Max Chaffanet,1 Marie-Joelle
Mozziconacci,1,3 Norbert Vey3,9 and
Daniel Birnbaum1
1Laboratoire d’Oncologie Moleculaire, Centre de
Recherche en Cancerologie de Marseille, UMR891
Inserm, Institut Paoli-Calmettes, 2Universite de la
Mediterranee Aix-Marseille II, 3Departement de
BioPathologie, Institut Paoli-Calmettes, 4Bureau
d’etudes cliniques, Biostatistiques, Institut Paoli-
Calmettes, Marseille, 5Service de Medecine Interne
Onco-Hematologie, Centre Hospitalier General
d’Avignon, Avignon, 6Service d’Onco-
Hematologie, Hopital Font-pre, Centre
Hospitalier Intercommunal de Toulon, Toulon,7Service de Medecine Interne
Onco-Hematologie, Centre Hospitalier General de
Salon de Provence, Salon de Provence, 8Service de
Medecine Interne, Centre Hospitalier General,
Martigues, and 9Departement d’Hematologie,
Institut Paoli-Calmettes, Marseille, France
Received 22 June 2010; accepted for publication
12 July 2010
Correspondence: Dr. Veronique Gelsi-Boyer,
MD, PhD, Laboratoire d’Oncologie Moleculaire,
Centre de Recherche en Cancerologie de
Marseille, UMR891 Inserm, 27 Bd. Leı Roure,
13009 Marseille, France.
E-mail: gelsiv@marseille. fnclcc.fr
*Equal contributors.
Summary
Chronic myelomonocytic leukaemia (CMML) is a haematological disease
currently classified in the category of myelodysplastic syndromes/
myeloproliferative neoplasm (MDS/MPN) because of its dual clinical and
biological presentation. The molecular biology of CMML is poorly
characterized. We studied a series of 53 CMML samples including 31 cases
of myeloproliferative form (MP-CMML) and 22 cases of myelodysplastic
forms (MD-CMML) using array-comparative genomic hybridisation
(aCGH) and sequencing of 13 candidate genes including ASXL1, CBL,
FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, PTPN11, RUNX1, TET2
and WT1. Mutations in ASXL1 and in the genes associated with proliferation
(CBL, FLT3, PTPN11, NRAS) were mainly found in MP-CMML cases.
Mutations of ASXL1 correlated with an evolution toward an acutely
transformed state: all CMMLs that progressed to acute phase were mutated
and none of the unmutated patients had evolved to acute leukaemia. The
overall survival of ASXL1 mutated patients was lower than that of unmutated
patients.
Keywords: array-CGH, ASXL1, chronic myelomonocytic leukaemia,
mutations, gene mutation, prognosis.
research paper
First published online 29 September 2010
ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375 doi:10.1111/j.1365-2141.2010.08381.x

two types: type 1 with fewer than 5% blasts in the blood and
10% blasts in the bone marrow, and type 2 with between 5%
and 19% in the blood or 10% and 19% in the bone marrow
(Vardiman et al, 2002, 2009).
Classification of CMML has always been a matter of debate
based on the two distinct clinical presentations: dysplastic
with symptomatic complications of cytopenia, and prolifer-
ative with splenomegaly or hepatomegaly. In 1994, on the
basis of an arbitrary threshold of peripheral leucocyte count
(WBC), a dysplastic (<13 · 109/l) and a proliferative (>13 ·
109/l) form were defined by the French-American-British
(FAB) group (Bennett et al, 1994), which are designated as
MD-CMML and MP-CMML, respectively, in this report.
Until recently, neither biological nor prognostic data sup-
ported of this distinction except that MP-CMML seemed to
have more RAS mutations (Padua et al, 1988, 1998). Recently,
new molecular alterations have been described in CMML. We
and others have showed a high frequency of RUNX1 and
TET2 mutations (Gelsi-Boyer et al, 2008; Kosmider et al,
2009a; Kuo et al, 2009). Mutations of TET2 and CBL
occurred in 50% (Kosmider et al, 2009a) and 5% (Makishima
et al, 2009) of CMML cases, respectively. Recently, we
described exon 12 mutations of ASXL1 with a frequency
about 44% (Gelsi-Boyer et al, 2009). Currently, the impact of
these alterations remains to be determined and the biological
and clinical presentations are still heterogeneous and benefit
from different therapies: cytotoxic in the MP form, haemat-
opoietic growth factors in the MD form.
We have performed a comprehensive study of molecular
alterations in a series of 53 CMMLs using genome-wide, high-
density array-comparative genomic hybridization (aCGH) and
exon-coding sequencing of 13 candidate genes. ASXL1 muta-
tions were shown to be associated with MP-CMML and
MP-CMMLs with overall more alterations. Most importantly,
mutations in ASXL1 correlated with acute transformation of
CMML and the overall survival rate was lower in ASXL1
mutated patients than in unmutated patients.
Patients and methods
Patients and samples
A series of consecutive bone marrow samples obtained from 53
patients, who all signed an informed consent, were collected.
Among these were 31 cases of MP-CMML and 22 cases of MD-
CMML, as initially defined by the FAB group with a WBC > or
<13 · 109/l, respectively. The median age and WBC were
74 years and 30 · 109/l, respectively, in the MP-CMML group
and 76Æ5 years and 9 · 109/l, respectively, in the MD-CMML
group. The main clinical and biological data of the 53 samples
are presented in Supplementary Table SI. Most of the samples
(39 cases) were obtained from newly diagnosed patients.
Fourteen were known CMMLs and not receiving any treatment
(nine cases) or under symptomatic treatment (five cases). Most
patients of the latter group (12/14) had been diagnosed more
of 1 year previously. Six patients had received prior chemo-
therapy or radiotherapy for an independent solid tumour.
A normal karyotype was observed in 40 patients (20 MP-
CMMLs and 20 MD-CMMLs); a del(20q)(q11q13) was found
in three patients (2 MP-CMMLs and 1 MD-CMML); a trisomy
of a commonly affected chromosome (8, 19, 21) was encoun-
tered in 4 MP-CMMLs. One MD-CMML had an 11q
inversion, one MP-CMML had a t(10;11)(p12;p15) and one
MP-CMML had a t(1;3)(p36;q21). Nucleic acid extraction was
done as previously described (Gelsi-Boyer et al, 2008).
Array comparative genomic hybridization (aCGH)
aCGH was done on all samples but two (HD-0397, HD-0715).
Some results have been published for 38 cases (Gelsi-Boyer
et al, 2008, 2009). Genomic imbalances were analysed by
aCGH using 244K CGH Microarrays (Hu-244A; Agilent
Technologies, Massy, France) as previously described
(Adelaıde et al, 2007; Etienne et al, 2007). Scanning was done
with an Agilent Autofocus Dynamic Scanner (G2565BA;
Agilent Technologies). Data analysis was done as previously
described (Murati et al, 2003; Barrett et al, 2004) and visual-
ized with CGH analytics 3.4 software (Agilent Technologies).
Extraction data (log2 ratio) was done with CGH analytics while
normalized and filtered log2 ratios were obtained from
‘feature extraction’ software (Agilent Technologies). Copy
number changes were characterized as reported (Adelaıde et al,
2007; Etienne et al, 2007).
DNA sequencing
Polymerase chain reaction (PCR) and direct sequencing were
done using standard conditions with gene-specific primers
designed to amplify coding sequence of ASXL1 (exon 12), CBL
(exons 8, 9), FLT3 (exons 14, 15, 20), IDH1/IDH2 (exon 4),
JAK2 (exon 14), KRAS/NRAS (exons 1, 2), NPM1 (exon 12),
PTPN11 (exons 3, 11), RUNX1 (exons 1–8), TET2 (exons
3–11) and WT1 (exons 7, 9) as previously described (Gelsi-Boyer
et al, 2008, 2009; Kosmider et al, 2009a,b; Carbuccia et al,
2010). Primers were described by Gelsi-Boyer et al (2008) and
Rocquain et al (2010a). Most PCR amplifications were done in
a total volume of 25 ll PCR mix containing at least 10 ng
template DNA, Taq buffer, 200 lmol of each deoxynucleotide
triphosphate, 20 pmol of each primer and 1 unit of Hot Star
Taq (Qiagen, Courtaboeuf, France). PCR amplification con-
ditions were as follows: 95�C 10 min; 95�C 30 s, variable
temperature 30 s, 72�C 45 s for 30 cycles; 72�C 10 min. PCR
products were purified using Millipore plate MSNU030. The
purified PCR products (2 ll) were used for sequencing using
the Big Dye terminator v1.1 kit (Applied Biosystems, Cour-
taboeuf, France). After G50 purification, sequences were
loaded on an ABI 3130XL automat (Applied Biosystems).
The sequence data files were analysed using the seqscape
software and all mutations were confirmed on an independent
PCR product.
V. Gelsi-Boyer et al
366 ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375

Statistical analysis
Clinical and biological factors were taken at the time of
sampling whose date corresponds to the search for molecular
alterations. The different quantitative biological variables were
compared by the Student test (t-test) and prevalence of
mutations or alterations detected by karyotype or aCGH by the
Chi-square test. Overall survival was defined by the time from
the date of diagnosis to the date of death. Patients still alive
were right censored at the date of last contact. ‘Time-to-acute
myeloid leukaemia (AML)’ was defined by the time from the
date of sampling to the date of acute transformation. Patients
still alive were right censored at the date of last contact. In
time-to-AML analysis, deaths occurring before AML transfor-
mation were treated as a competing event. To investigate the
association between outcome and the different covariates,
univariate analysis was done using Log-Rank, Wald, and Gray
tests. Cox Proportional Hazard and Fine and Gray models
were used in a multivariate approach. Cumulative incidences
of AML transformations were estimated by the Prentice
method. All statistical tests were two-sided at the 5% level of
significance.
Results
aCGH profiles of CMML
aCGH profiles were established in 51 of the 53 CMML cases
(Table I). aCGH identified alterations that were observed by
conventional cytogenetics (9/51) except for three patients
with balanced translocations (HD-0201, HD-0316, HD-0178),
and for case HD-0367 with a del(20)(q11q13), for which
aCGH did not show a frank deletion at 20q probably due to
the low number of affected cells. In nine cases (17%) aCGH
detected rare and limited losses or gains, not visible on the
karyotype. They affected very few genes, as previously
described (Gelsi-Boyer et al, 2008; Adelaıde et al, 2010;
Rocquain et al, 2010b), including some with known tumour
suppressor function and leukaemogenic activity (NF1, RB1
and TET2). No copy number aberrations were observed in
70% of cases (36/51).
Mutations in candidate genes are frequent in CMML
We studied the coding sequences of 13 genes from the 53
cases (Table I). Some results have been previously published
(Gelsi-Boyer et al, 2008, 2009; Kosmider et al, 2009a). In 25
cases (49%) we found 20 frameshift (including p.Gly646-
Trpfsx12 in 7 cases) and five nonsense mutations in ASXL1
exon 12. Three patients (HD-0228, HD-0328, HD-0370), had
an acquired mutation because it was absent from buccal
smear DNA (not shown). CBL exon 8 mutations were found
in about 10% of cases (5/47); one case (HD-0223) had a
homozygous mutation. One case (HD-0367) had an internal
tandem duplication of FLT3. We found five IDH mutations
in 48 cases (10%); all were in IDH2 (four had
p.Arg140Gln). Seven patients out of the 53 cases (13%)
had a KRAS or NRAS mutation. We previously examined
the sequence of exons 3 and 13 of the PTPN11 gene and
mutations were found in two cases (Gelsi-Boyer et al, 2008).
Twelve out of 53 patients (21%) were mutated for RUNX1
and 36% of patients were mutated for TET2. No mutation
was found in NPM1, JAK2 and WT1. On the basis of
small deletions detected by aCGH and spanning only few
genes, we searched for mutations in an additional set of
candidate genes: ASXL2, BRAF, CBFB, CTNNB1, KDM3B,
KDM6A, KDM6B, NFIA, PYGO1, SPI1, RAF1, and TET3
but failed to find any mutations (V. Trouplin, J. Rocquain)
except in one patient (HD-0398) for which a mutation in
NFIA exon 2 was found and previously reported (Bernard et al,
2008).
MP- and MD-CMMLs show quantitative differences in
molecular alterations
Genomic alterations detected by conventional cytogenetics or
aCGH were different in MP- and MD-CMML (Table I): 15 out
of 31 MP-CMMLs and four out of 22 MD-CMMLs showed
alterations. MP-CMMLs had more genomic alterations than
MD-CMMLs (P = 0Æ049).
We compared the prevalence of the mutated genes in MP-
and MD-CMML. Nineteen of the 25 ASXL1 mutations were
found in 30 MP-CMMLs vs. 6 in 22 MD-CMMLs. Mutations
in ASXL1 but not in RUNX1 or TET2 were more frequent in
MP than in MD-CMMLs (P = 0Æ03) (Table I). No difference
was observed between the two forms for CBL, FLT3, IDH1,
IDH2, PTPN11, RAS, RUNX1 or TET2. No RAS pathway
mutation (RAS and PTPN11) was observed in MD-CMMLs.
Mutations in proliferation-associated genes CBL, FLT3,
KRAS, NRAS and PTPN11 were mutually exclusive and,
taken as a whole, significantly associated with MP-CMML
(14/123 vs. 1/91; P = 0Æ008). Overall, the number of muta-
tions (ASXL1 and proliferation genes) was higher in MP than
in MD-CMMLs (P = 0Æ018). Altogether, molecular events
(mutations and genomic alterations) were more frequent in
MP-CMMLs (69/273 events) than in MD-CMMLs (26/172)
(P = 0Æ0018).
Correlations between ASXL1 mutation and clinical and
biological features in CMML
The main clinical and biological features of 51 CMML cases
were examined with respect to ASXL1 mutations (Table II).
The presence of an ASXL1 mutation was associated with higher
WBC (30 · 109/l vs. 15 · 109/l) (P = 0Æ006), higher peripheral
blood (P = 0Æ005) and bone marrow monocytosis (P = 0Æ04)
and with lower haemoglobin levels (P = 0Æ03). No difference
was noted in mean cell volume (MCV), neutrophil and platelet
counts, or bone marrow blast levels. In MP-CMMLs, ASXL1
mutation correlated with a lower level of haemoglobin
Gene Mutations in CMML
ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375 367

Table
I.Mutationsofcandidategenes
inaseries
ofCMML.
Case
Sex/age
(years)
Diagnosis
RUNX1
(exons1–8)
TET2
(exons3–11)
ASXL1
(exon12)
NPM1
(exon12)
WT1
(exons7,
9)
CBL
(exons8,9)
FLT3
ITD/TKD
JAK2
(V617F)
KRAS
(exons1,
2)
NRAS
(exons1,2)
PTPN11
(exons3,13)
IDH1
(exon4)
IDH2
(exon4)
Number
ofaltered
genes
Karyotype
aCGH
HD-0176M/42
MP-C
MML1no
no
no
no
no
no
no
no
no
no
no
no
no
046,XY[20]
gain
7p21
(AHR),
Xp22
(SH3
KBP1)
HD-0182F/74
MP-C
MML1no
no
no
no
no
no
no
no
no
p.Gly12Asp
no
no
no
145,X,
-X?c[20]
loss
X
HD-0200M/71
MP-C
MML1no
no
no
no
no
no
no
no
p.Ala146
Val
no
no
no
no
146,XY[20]
noCNA
HD-0201M/87
MP-C
MML1no
no
p.Leu1266
HisfsX9
no
no
no
no
no
no
p.Ala157
Gly
no
no
no
246,XY,t(10;11)
(p12;p15)
[13]/46,XY[7]
noCNA
HD-0223M/85
MP-C
MML1no
splicing
defect
p.Arg1068X
no
no
p.Phe418
Ser(hom)
no
no
no
no
nd
no
no
346,XY[20]
loss
13q14
(RB1),
15q21,
15q22
(DAPK2),
gain
Xp22
HD-0228M/70
MP-C
MML1no
p.Trp1198X
p.Thr836
LeufsX2(nc)
no
no
no
no
no
p.Gly
12Ser
no
no
no
no
346,XY[20]
noCNA
HD-0229M/87
MP-C
MML2no
p.Gln1191X
no
no
no
no
no
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0257F/71
MP-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
p.Arg140
Gln
146,XX[20]
noCNA
HD-0272F/79
MP-C
MML1no
p.Arg1452X
p.Tyr1560
LeufsX18
p.Gly646
TrpfsX12
no
no
no
no
no
no
p.Gly
13Asp
na
no
no
346,XX[20]
noCNA
HD-0273M/76
MP-C
MML1no
p.Gly1361Serp.Lys888
GlufsX6
no
no
no
no
no
no
no
na
no
p.Arg140
Gln
346,XY[20]
noCNA
HD-0316M/65
MP-C
MML1p.Pro
425L
eu
na
na
no
na
na
na
no
no
no
p.Asp61Tyr
na
na
246,XY[20]
losses
3q22–24
(EPHB1,
NEK11,
MRAS…)
HD-0318M/69
MP-C
MML1p.Arg
166X
no
p.Gln768X
na
na
na
na
no
no
no
no
na
na
246,XY,del(20)
(q11q13)[20]
loss
20
q11-q13
V. Gelsi-Boyer et al
368 ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375

Table
I.(C
ontinued).
Case
Sex/age
(years)Diagnosis
RUNX1
(exons1–8)
TET2
(exons3–11)
ASXL1
(exon12)
NPM1
(exon12)
WT1
(exons7,
9)
CBL
(exons8,9)
FLT3
ITD/TKD
JAK2
(V617F)
KRAS
(exons1,
2)
NRAS
(exons1,2)
PTPN11
(exons3,13)
IDH1
(exon4)
IDH2
(exon4)
Number
ofaltered
genes
Karyotype
aCGH
HD-0320F/74
MP-C
MML2splicing
defect
p.Arg1404X
p.Gly646
TrpfsX12
no
no
no
no
no
no
no
p.Ala72Thr
no
no
447,XX,+8[20]
tri8
HD-0321M/55
MP-C
MML2p.Arg166X
no
p.H
is630
ProfsX66
no
no
na
no
no
no
no
no
no
no
246,XY[20]
loss
3p23
(GLB1,
CRTAP)
HD-0322F/61
MP-C
MML1no
no
no
no
no
no
no
no
no
no
no
no
no
046,XX[20]
noCNA
HD-0327F/72
MP-C
MML1p.Tyr377
LeufsX223
p.Cys1193Trp
p.Gly646
TrpfsX12
no
no
no
no
no
no
p.Gly12Asp
no
no
no
447,XX,+8[20]
tri8,loss
7q11
(CALN1)
HD-0366M/78
MP-C
MML1no
no
p.Ser846
GlnfsX5(nc)
na
na
na
na
no
no
no
no
na
na
146,XY[20]
noCNA
HD-0367F/70
MP-C
MML2no
no
no
no
no
no
ITD
no
no
no
no
no
no
146,XX,inv(11)
(p15q22)[20]
noCNA
HD-0370M/70
MP-C
MML2no
no
p.Thr1271
LysfsX10
(nc)
no
no
no
no
no
no
no
no
no
no
146,XY[20]
noCNA
HD-0376F/65
MP-C
MML2no
no
no
no
no
no
no
no
no
no
no
no
no
046,XX,t(1;3)
(p36;q21)[20]
noCNA
HD-0397F/71
MP-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
no
046,XX[20]
na
HD-0398M/62
MP-C
MML1no
no
p.Gly646
TrpfsX12
no
no
no
no
no
no
no
na
no
no
147,XY,+19[17]/
46,XY[3]
tri19
HD-0399M/74
MP-C
MML1p.Ser
141L
eu
no
p.Gly646T
rpfsX12
no
no
p.Cys396T
yrno
no
no
no
no
no
no
346,XY[20]
noCNA
HD-0404M/88
MP-C
MML2p.Arg201X
p.Asn1581
IlefsX17
p.H
is630P
rofsX66
no
no
no
no
no
no
no
na
no
no
347,XY,+21[20]tri21
HD-0485M/63
MP-C
MML2p.Arg320X
p.Leu1252Pro
p.Gly646T
rpfsX12
no
no
no
no
no
no
no
na
no
no
346,XY[20]
noCNA
HD-0627F/87
MP-C
MML1no
no
p.Arg1068X
no
no
p.Cys404
Tyr
(hom)
no
no
no
no
na
no
no
246,XX,del(20)
(q11q13)
[6]/46,XX[19]
noCNA
HD-0669M/88
MP-C
MML1no
no
p.Ala611A
rgfsX8
no
no
no
no
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0671F/71
MP-C
MML1p.Lys110A
rgno
p.Tyr591X
no
no
no
no
no
no
no
na
no
no
247,XX,+19[8]/
46,XX[12]
loss
17q11
(NF1),
tri19
HD-0707M/85
MP-C
MML1no
no
no
no
no
p.Tyr371H
isno
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0715M/69
MP-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
no
046,XY[20]
na
HD-0723M/83
MP-C
MML1no
no
p.1213IlefsX3
no
no
no
no
no
no
p.Gly12Val
na
no
p.Arg140Gln3
46,XY[20]
noCNA
Gene Mutations in CMML
ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375 369

Table
I.(C
ontinued).
Case
Sex/age
(years)Diagnosis
RUNX1
(exons1–8)
TET2
(exons3–11)
ASXL1
(exon12)
NPM1
(exon12)W
T1
(exons7,
9)
CBL
(exons8,9)
FLT3
ITD/TKD
JAK2
(V617F)K
RAS
(exons1,2)N
RAS
(exons1,2)P
TPN11
(exons3,13)ID
H1
(exon4)ID
H2
(exon4)
Number
ofaltered
genes
Karyotype
aCGH
HD-0178M/88
MD-C
MML1no
no
no
no
no
no
no
no
no
no
no
no
p.Arg140
Gln
146,XY,del(20)
(q11q13)[20]
loss
13q14
(RB1),
20q11-q13
HD-0197M/59
MD-C
MML1p.Arg320X
no
no
na
na
na
na
no
no
no
no
na
na
146,XY[20]
noCNA
HD-0206M/73
MD-C
MML2no
deletion
p.Asp879
GlufsX7
no
no
no
no
no
no
no
na
no
p.Arg172
Lys
346,XY[20]
loss
4q24
(TET2)
HD-0230M/83
MD-C
MML1no
no
p.H
is630P
rofsX66
no
no
no
no
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0242F/82
MD-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
no
046,XX[25]
noCNA
HD-0254F/83
MD-C
MML2no
no
no
no
no
no
no
no
no
no
na
no
no
046,XX[20]
loss
17q11
(NF1)
HD-0271F/82
MD-C
MML1no
no
p.Pro1263GlnfsX17
no
no
no
no
no
no
no
na
no
no
146,XX[20]
noCNA
HD-0280M/78
MD-C
MML1no
p.Ser1189
ValfsX37
no
no
no
no
no
no
no
no
na
no
no
145,X,-Y[19]/
46,XY[1]
loss
Y
HD-0328M/73
MD-C
MML1no
p.Gly355A
sp
(hom)
p.Gly646T
rpfsX12
no
no
p.Cys404T
yrno
no
no
no
no
no
no
346,XY[20]
noCNA
HD-0330M/76
MD-C
MML1p.Gly50
GlnfsX4
na
na
na
na
na
na
no
no
no
no
na
na
146,XY[20]
noCNA
HD-0355M/65
MD-C
MML1no
p.Cys1289Phe
no
no
no
no
no
no
no
no
no
no
no
146,XY[20]
noCNA
HD-0372M/60
MD-C
MML1no
no
no
no
no
no
no
no
no
no
no
no
no
046,XY[20]
noCNA
HD-0380F/82
MD-C
MML1no
p.Gln1414X
no
no
no
no
no
no
no
no
no
no
no
146,XX[20]
noCNA
HD-0388F/71
MD-C
MML1p.Met133Ilep.Leu1721PhefsX8
no
no
no
no
no
no
no
no
na
no
no
246,XX[20]
noCNA
HD-0396M/70
MD-C
MML1no
p.Ser354X
no
no
no
no
no
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0638F/68
MD-C
MML1no
p.Leu1394TrpfsX54
no
no
no
no
no
no
no
no
na
no
no
146,XX[20]
noCNA
HD-0660M/80
MD-C
MML1no
no
p.Arg693X
no
no
no
no
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0703M/74
MD-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
no
046,XY[20]
noCNA
HD-0711M/79
MD-C
MML1no
p.Gln649X
no
no
no
no
no
no
no
no
na
no
no
146,XY[20]
noCNA
HD-0712M/41
MD-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
no
046,XY[20]
noCNA
HD-0743M/68
MD-C
MML1no
no
no
no
no
no
no
no
no
no
na
no
no
046,XY[20]
noCNA
HD-0755M/46
MD-C
MML1no
p.Arg1359Cys
p.Thr822AsnfsX11
no
no
no
no
no
no
no
na
no
no
246,XY[20]
noCNA
Sampleswereseparated
accordingto
thewhitebloodcellcount(W
BC):MP-C
MMLwithWBC>13
·10
9/landMD-C
MMLwithWBC<13
·109/l.
Hom,homozygous;nc,verified
asnotconstitutional;na,
nonavailable.
V. Gelsi-Boyer et al
370 ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375

(P = 0Æ03) and platelet count (P = 0Æ002) and with a higher
monocytosis (P = 0Æ04). In MD-CMMLs, no correlations with
ASXL1 mutation were observed.
Among ASXL1 mutated cases (25/51), 11 (9 MP and 2 MD)
had evolved to acute transformation (Table II). DNA from the
acute phase was available in one case (HD-0398); we looked
for the ASXL1 mutation and found the same alteration (not
shown). In contrast, no acute transformation was observed in
the unmutated cases. In other words, all transformed cases had
an ASXL1 mutation but not all ASXL1 mutated cases had
progressed to AML. ASXL1 mutation and acute transforma-
tion were correlated (P = 0Æ0005). Patient HD-0316 had
progressed to acute phase but was not included in the 51
patients as we could not determine the ASXL1 status because
of lack of material at both stages.
Overall survival (median follow-up of 29Æ5 months) was
analysed for the 53 patients. Median overall survival was
27Æ6 months (Fig 1A). ASXL1 status could be determined at the
time of diagnosis for 51 patients. Kaplan–Meier analysis showed
a lower overall survival rate in the ASXL1 mutated patients
(Fig 1B). With respect toMP/MD forms, only a trend to a better
survival of the MD patients was observed (not shown). Within
MP-CMML patients, cases mutated for ASXL1 had a poorer
survival than the unmutated cases (not shown). The cumulative
incidence of AML (Fig 1C) showed that all the patients who
developed AML did so within 2 years and that ASXL1 mutation
Table II. Clinical and biological features of ASXL1 mutated and unmutated CMML patients.
Number of patients ASXL1 mutated ASXL1 unmutated P value
CMML
Sex ratio 51 3Æ16 1Æ27 0Æ23
Age (years): median (range) 51 74 (46–88) 71 (41–88) 0Æ39
Leucocyte count (·109/l): median (range) 51 30 (6–188) 15 (2Æ8–26Æ4) 0Æ006
Haemoglobin level (g/l): median (range) 50 100 (81–141) 125 (74–162) 0Æ03
MCV (fl): median (range) 35 92 (86–106) 93 (79–101) 0Æ33
Neutrophil count (·109/l): median (range) 41 15 (1Æ5–41) 2 (0Æ1–12Æ9) 0Æ07
Blood monocyte count (·109/l): median (range) 50 4Æ65 (1–60) 2 (1–46) 0Æ005
Platelets count (·109/l): median (range) 50 117Æ5 (34–420) 208 (39–660) 0Æ07
Bone marrow blasts (%): median (range) 51 7 (2–18) 6 (1–14) 0Æ11
Bone marrow monocytes (%): median (range) 50 20 (6–35) 15 (3–45) 0Æ04
MP-CMML 30 19 11 0Æ03
MD-CMML 21 6 15
No acute transformation 51 14 26 0Æ0005
Acute transformation 51 11 0
MP-CMML
Sex ratio 2Æ8 0Æ8 0Æ25
Age (years): median (range) 30 74 (55–88) 70Æ5 (42–87) 0Æ13
Leucocyte count (·109/l): median (range) 30 35 (6–188) 17 (15–26Æ5) 0Æ08
Haemoglobin level (g/l): median (range) 29 110 (81–125) 115 (85–152) 0Æ03
MCV (fl): median (range) 21 92 (80–106) 88Æ5 (74–101) 0Æ11
Neutrophil count (·109/l): median (range) 26 25 (7–41) 10 (7–12Æ9) 0Æ54
Blood monocyte count (·109/l): median (range) 30 10 (1–60) 4Æ5 (1Æ2–46) 0Æ04
Platelets count (·109/l): median (range) 29 118 (34–420) 272 (57–660) 0Æ002
Bone marrow blasts (%): median (range) 30 7 (2–16) 6 (2–14) 0Æ54
Bone marrow monocytes (%): median (range) 29 25 (6–35) 14 (3–45) 0Æ16
MD-CMML
Sex ratio 21 5 1Æ8 0Æ75
Age (years): median (range) 21 76Æ5 (46–83) 75 (41–88) 0Æ9
Leucocyte count (·109/l): median (range) 21 8Æ9 (4–12) 7Æ1 (2Æ8–12Æ8) 0Æ6
Haemoglobin level (g/l): median (range) 21 120 (89–141) 140 (74–162) 0Æ77
MCV (fl): median (range) 13 89 (86–100) 93Æ5 (91–101) 0Æ46
Neutrophil count (·109/l): median (range) 19 5 (1Æ5–7) 7Æ1 (0Æ1–7Æ7) 0Æ15
Blood monocyte count (·109/l): median (range) 21 1Æ3 (1–4Æ5) 1Æ4 (1–2Æ6) 0Æ55
Platelets count (·109/l): median (range) 21 122 (43–414) 130 (39–399) 0Æ5
Bone marrow blasts (%): median (range) 21 7 (4–18) 5 (4–13) 0Æ2
Bone marrow monocytes (%): median (range) 21 16 (10–20) 15 (10–13) 0Æ83
MCV, mean cell volume.
In bold: significant P value.
Gene Mutations in CMML
ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375 371
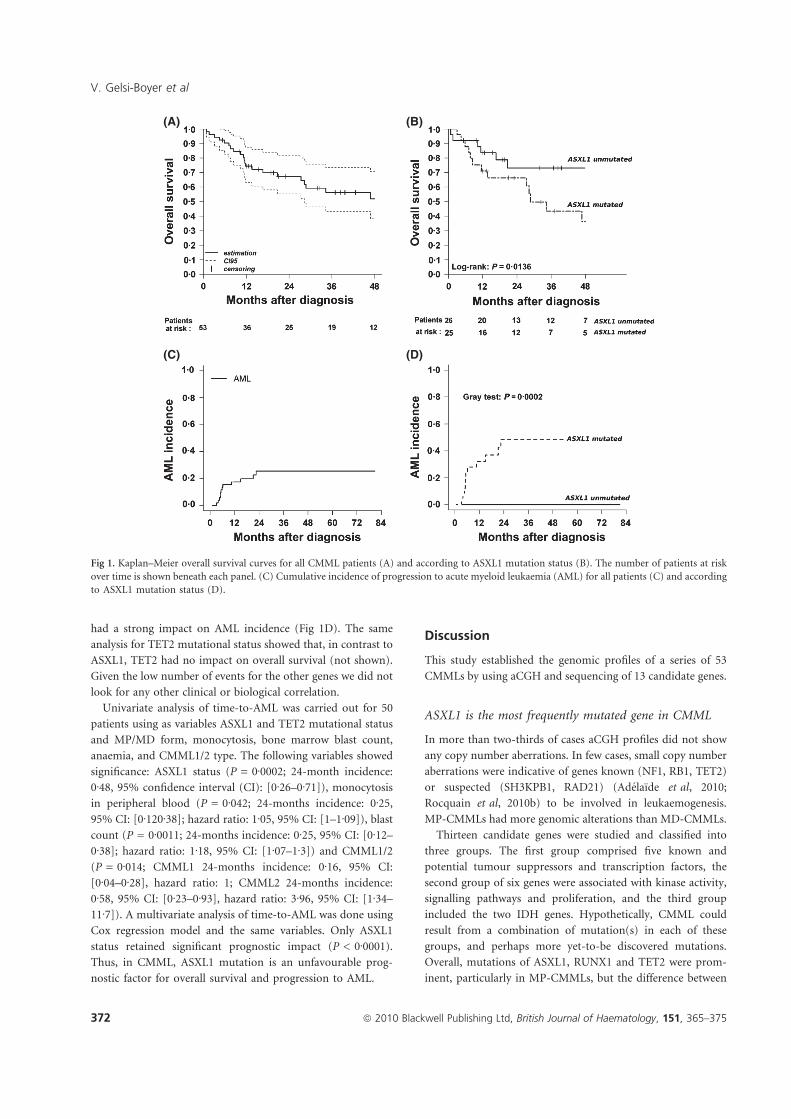
had a strong impact on AML incidence (Fig 1D). The same
analysis for TET2 mutational status showed that, in contrast to
ASXL1, TET2 had no impact on overall survival (not shown).
Given the low number of events for the other genes we did not
look for any other clinical or biological correlation.
Univariate analysis of time-to-AML was carried out for 50
patients using as variables ASXL1 and TET2 mutational status
and MP/MD form, monocytosis, bone marrow blast count,
anaemia, and CMML1/2 type. The following variables showed
significance: ASXL1 status (P = 0Æ0002; 24-month incidence:
0Æ48, 95% confidence interval (CI): [0Æ26–0Æ71]), monocytosis
in peripheral blood (P = 0Æ042; 24-months incidence: 0Æ25,
95% CI: [0Æ120Æ38]; hazard ratio: 1Æ05, 95% CI: [1–1Æ09]), blast
count (P = 0Æ0011; 24-months incidence: 0Æ25, 95% CI: [0Æ12–
0Æ38]; hazard ratio: 1Æ18, 95% CI: [1Æ07–1Æ3]) and CMML1/2
(P = 0Æ014; CMML1 24-months incidence: 0Æ16, 95% CI:
[0Æ04–0Æ28], hazard ratio: 1; CMML2 24-months incidence:
0Æ58, 95% CI: [0Æ23–0Æ93], hazard ratio: 3Æ96, 95% CI: [1Æ34–
11Æ7]). A multivariate analysis of time-to-AML was done using
Cox regression model and the same variables. Only ASXL1
status retained significant prognostic impact (P < 0Æ0001).
Thus, in CMML, ASXL1 mutation is an unfavourable prog-
nostic factor for overall survival and progression to AML.
Discussion
This study established the genomic profiles of a series of 53
CMMLs by using aCGH and sequencing of 13 candidate genes.
ASXL1 is the most frequently mutated gene in CMML
In more than two-thirds of cases aCGH profiles did not show
any copy number aberrations. In few cases, small copy number
aberrations were indicative of genes known (NF1, RB1, TET2)
or suspected (SH3KPB1, RAD21) (Adelaıde et al, 2010;
Rocquain et al, 2010b) to be involved in leukaemogenesis.
MP-CMMLs had more genomic alterations than MD-CMMLs.
Thirteen candidate genes were studied and classified into
three groups. The first group comprised five known and
potential tumour suppressors and transcription factors, the
second group of six genes were associated with kinase activity,
signalling pathways and proliferation, and the third group
included the two IDH genes. Hypothetically, CMML could
result from a combination of mutation(s) in each of these
groups, and perhaps more yet-to-be discovered mutations.
Overall, mutations of ASXL1, RUNX1 and TET2 were prom-
inent, particularly in MP-CMMLs, but the difference between
(A)
(C) (D)
(B)
Fig 1. Kaplan–Meier overall survival curves for all CMML patients (A) and according to ASXL1 mutation status (B). The number of patients at risk
over time is shown beneath each panel. (C) Cumulative incidence of progression to acute myeloid leukaemia (AML) for all patients (C) and according
to ASXL1 mutation status (D).
V. Gelsi-Boyer et al
372 ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375

the two forms was not statistically significant for the three
genes taken as a whole (P = 0Æ19). Mutations in these three
genes were not mutually exclusive. This is in contrast to what
we observed in myelodysplastic syndromes (MDS) and AML
where RUNX1 and TET2 mutations were mutually exclusive
but could each be associated with ASXL1 mutations (Rocquain
et al, 2010a). We have no explanation for this observation.
ASXL1 mutations are found in all myeloid neoplasms (Car-
buccia et al, 2009, 2010; Gelsi-Boyer et al, 2009; Kosmider
et al, 2009b; Boultwood et al, 2010a,b; Sugimoto et al, 2010;
Tefferi, 2010). In the present study ASXL1 appeared to be the
most frequently mutated gene in CMML, as it is in MDS
(Boultwood et al, 2010a; Rocquain et al, 2010a). ASXL1
mutations are found in high-risk cases of MDS.
As previously observed (Gelsi-Boyer et al, 2008), mutations
in the second group of ‘proliferation’ genes were mutually
exclusive. The frequency of mutations of CBL was in the range
of that observed recently (Makishima et al, 2009). With two
exceptions (a CBL mutation and an NF1 deletion detected by
aCGH), mutations in these genes were a hallmark of MP-
CMML and may explain the proliferative features of this form.
In agreement, it has recently been shown that JAK2 mutations
are associated with MP-CMML (Ricci et al, 2010). However, in
our series we did not find any JAK2 mutation. Overall, the
number of mutations (ASXL1 and proliferation genes) was
higher in MP-CMML than in MD-CMML. This provides a
molecular basis for the further classification of CMML into
MP and MD forms initially defined by the FAB group.
However, the equal number of mutations of RUNX1 and TET2
in both forms suggests a common mechanism of oncogenesis.
ASXL1 mutation is associated with poor prognosis and
acute transformation of CMML
The MP and MD classification may have a molecular basis
(Gelsi-Boyer et al, 2007; this work) but had no significant
impact on prognosis in our series. Several studies have
similarly found that this classification does not by itself offer
enough prognostic information (Germing et al, 1998;
Nosslinger et al, 2001; Gonzalez-Medina et al, 2002). We have
here shown that, in contrast, the ASXL1 mutation is associated
with both poor overall survival and acute transformation. In
our series, no unmutated cases had evolved to acute phase
whereas the cases that had progressed to acute leukaemia were
all mutated (except for one case for which the ASXL1 status
could not be determined). A multivariate analysis of time-to-
AML including ASXL1 mutation and other factors revealed
that only ASXL1 status has significant impact. However,
ASXL1 status was not significant in multivariate analysis of
overall survival (not shown), perhaps because of the limited
number of cases or the existence of confounding factors, such
as death by other causes. Some mutated cases had not
progressed to acute phase. This may be because patients had
died before experiencing acute leukaemia, and may have been
the case for patients HD-0230 and HD-0660, whose ASXL1-
mutated CMML progressed from CMML1 to CMML2. For
mutated patients still alive it could be a question of time. If
this were the case, the ASXL1 mutation could be used to
predict the risk of progression of CMML to acute leukaemia.
In contrast, TET2 mutations had no impact. A previous
study had found prognostic impact of TET2 mutation but
only in CMML1 (Kosmider et al, 2009a).
ASXL1 mutation is associated with aggressive forms of other
myeloid disorders, including high-risk MDS, primitive mye-
lofibrosis (Carbuccia et al, 2009) and secondary acute myeloid
leukaemia (Carbuccia et al, 2010). If confirmed on a larger
series the data would endow ASXL1 with such a prognostic
value that its mutational status may be systematically deter-
mined in the clinical setting and that the management of
patients with mutation may be adapted. It could also be
interesting to classify CMMLs studied in clinical trials
according to ASXL1 status.
Authors’ contributions
AM, CA, HZ HF, ZT, MN, MJM, and NV selected the cases,
and provided and reviewed the clinical and biological data. JA
and MC generated and analysed the aCGH data. VT, JR, and
NC generated and analysed the sequencing data. BE and PF did
the statistical analyses. VGB and DB supervized the study and
wrote the manuscript. All authors approved the study and the
manuscript.
Acknowledgements
We are grateful to the patients who accepted to participate in
this study. The work was supported by Inserm and Institut
Paoli-Calmettes.
References
Adelaıde, J., Finetti, P., Bekhouche, I., Repellini, L., Geneix, J., Sir-
coulomb, F., Charafe-Jauffret, E., Cervera, N., Desplans, J., Parzy,
D., Schoenmakers, E., Viens, P., Jacquemier, J., Birnbaum, D.,
Bertucci, F. & Chaffanet, M. (2007) Integrated profiling of basal and
luminal breast cancers. Cancer Research, 67, 11565–11575.
Adelaıde, J., Rocquain, J., Gelsi-Boyer, V., Carbucci, N., Birnbaum,
D.J., Mozziconacci, M.J., Vey, N., Birnbaum, D. & Chaffanet, M.
(2010) Gain of CBL-interacting protein: a possible alternative to
CBL mutations in myelodysplastic syndromes and chronic myelo-
monocytic leukemia. Leukemia, 24, 1539–1541.
Barrett, M.T., Scheffer, A., Ben-Dor, A., Sampas, N., Lipson, D.,
Kincaid, R., Tsang, P., Curry, B., Baird, K., Meltzer, P.S., Yakhini, Z.,
Bruhn, L. & Laderman, S. (2004) Comparative genomic hybridiza-
tion using oligonucleotide microarrays and total genomic DNA.
Proceedings of the National Academy of Sciences of the United States of
America, 101, 17765–17770.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A.,
Gralnick, H.R. & Sultan, C. (1982) Proposals for the classification of
the myelodysplastic syndromes. British Journal of Haematology, 51,
189–199.
Gene Mutations in CMML
ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375 373

Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A.,
Gralnick, H., Sultan, C. & Cox, C. (1994) The chronic myeloid
leukaemias: guidelines for distinguishing chronic granulocytic,
atypical chronic myeloid, and chronic myelomonocytic leukaemia.
Proposals by the French-American-British Cooperative Leukaemia
Group. British Journal of Haematology, 84, 746–754.
Bernard, F., Gelsi-Boyer, V., Murati, A., Giraudier, S., Trouplin, V.,
Adelaıde, J., Rey, J., Olschwang, S., Vainchenker, W., Chaffanet, M.,
Vey, N., Mozziconacci, M.J. & Birnbaum, D. (2008) Alterations of
NFIA in chronic malignant myeloid diseases. Leukemia, 23, 583–535.
Boultwood, J., Perry, J., Pellagatti, A., Fernandez-Mercado, M.,
Fernandez-Santamaria, C., Calasanz, M.J., Larrayoz, M.J., Garcia-
Delgado, M., Giagounidis, A., Malcovati, L., Della Porta, M.G.,
Jadersten, M., Killick, S., Hellstrom-Lindberg, E., Cazzola, M. &
Wainscoat, J.S. (2010a) Frequent mutation of the polycomb-asso-
ciated gene ASXL1 in the myelodysplastic syndromes and in acute
myeloid leukemia. Leukemia, 24, 1062–1065.
Boultwood, J., Perry, J., Zaman, R., Fernandez-Santamaria, C., Little-
wood, T., Kusec, R., Pellagatti, A., Wang, L., Clark, R.E. & Wains-
coat, J.S. (2010b) High-density single nucleotide polymorphism
array analysis and ASXL1 gene mutation screening in chronic mye-
loid leukemia during disease progression. Leukemia, 24, 1139–1145.
Carbuccia, N., Murati, A., Trouplin, V., Brecqueville, M., Adelaıde, J.,
Rey, J., Vainchenker, W., Bernard, O.A., Chaffanet, M., Vey, N.,
Birnbaum, D. & Mozziconacci, M.J. (2009) Mutations of ASXL1
gene in myeloproliferative neoplasms. Leukemia, 23, 2183–2186.
Carbuccia, N., Trouplin, V., Gelsi-Boyer, V., Murati, A., Rocquain, J.,
Adelaıde, J., Olschwang, S., Xerri, L., Vey, N., Chaffanet, M., Birn-
baum, D. & Mozziconacci, M.J. (2010) Mutual exclusion of ASXL1
and NPM1 mutations in a series of acute myeloid leukemias.
Leukemia, 24, 469–473.
Etienne, A., Carbuccia, N., Adelaıde, J., Bekhouche, I., Remy, V., Sohn,
C., Sainty, D., Gastaut, J.A., Olschwang, S., Birnbaum, D., Mozz-
iconacci, M.J. & Chaffanet, M. (2007) Rearrangements involving 12q
in myeloproliferative disorders: possible role of HMGA2 and SOCS2
genes. Cancer Genetics and Cytogenetics, 176, 80–88.
Gelsi-Boyer, V., Cervera, N., Bertucci, F., Trouplin, V., Remy, V.,
Olschwang, S., Chaffanet, M., Vey, N., Mozziconacci, M.J. & Birn-
baum, D. (2007) Gene expression profiling separates chronic
myelomonocytic leukemia in two molecular subtypes. Leukemia, 21,
2359–2362.
Gelsi-Boyer, V., Trouplin, V., Adelaıde, J., Aceto, N., Remy, V., Pinson,
S., Houdayer, C., Arnoulet, C., Sainty, D., Bentires-Alj, M., Ols-
chwang, S., Vey, N., Mozziconacci, M.J., Birnbaum, D. & Chaffanet,
M. (2008) Genome profiling of chronic myelomonocytic leukemia:
frequent alterations of RAS and RUNX1 genes. BMC Cancer, 8, 299–
315.
Gelsi-Boyer, V., Trouplin, V., Adelaıde, J., Bonansea, J., Cervera, N.,
Carbuccia, N., Lagarde, A., Prebet, T., Nezri, M., Sainty, D., Ols-
chwang, S., Xerri, L., Chaffanet, M., Mozziconacci, M.J., Vey, N. &
Birnbaum, D. (2009) Mutations of polycomb-associated gene
ASXL1 in myelodysplastic syndromes and chronic myelomonocytic
leukaemia. British Journal of Haematology, 145, 788–800.
Germing, U., Gattermann, N., Minning, H., Heyll, A. & Aul, C. (1998)
Problems in the classification of CMML – dysplastic versus prolif-
erative type. Leukemia Research, 22, 871–878.
Gonzalez-Medina, I., Bueno, J., Torrequebrada, A., Lopez, A., Vallespi,
T. & Massague, I. (2002) Two groups of chronic myelomonocytic
leukaemia: myelodysplastic and myeloproliferative. Prognostic
implications in a series of a single center. Leukemia Research, 26,
821–824.
Kosmider, O., Gelsi-Boyer, V., Ciudad, M., Racoeur, C., Jooste, V.,
Vey, N., Quesnel, B., Fenaux, P., Bastie, J.N., Beyne-Rauzy, O.,
Stamatoulas, A., Dreyfus, F., Ifrah, N., de Botton, S., Vainchenker,
W., Bernard, O.A., Birnbaum, D., Fontenay, M. & Solary, E. (2009a)
TET2 gene mutation is a frequent and adverse event in chronic
myelomonocytic leukemia. Haematologica, 94, 1676–1681.
Kosmider, O., Gelsi-Boyer, V., Cheok, M., Grabar, S., Della-Valle, V.,
Picard, F., Viguie, F., Quesnel, B., Beyne-Rauzy, O., Solary, E., Vey,
N., Hunault-Berger, M., Fenaux, P., Mansat-De Mas, V., Delabesse,
E., Guardiola, P., Lacombe, C., Vainchenker, W., Preudhomme, C.,
Dreyfus, F., Bernard, O.A., Birnbaum, D. & Fontenay, M. (2009b)
TET2 mutation is an independent favorable prognostic factor in
myelodysplastic syndromes (MDSs). Blood, 114, 3285–3291.
Kuo, M.C., Liang, D.C., Huang, C.F., Shih, Y.S., Wu, J.H., Lin, T.L. &
Shih, L.Y. (2009) RUNX1 mutations are frequent in chronic myel-
omonocytic leukemia and mutations at the C-terminal region might
predict acute myeloid leukemia transformation. Leukemia, 23,
1426–1431.
Makishima, H., Cazzolli, H., Szpurka, H., Dunbar, A., Tiu, R., Huh, J.,
Muramatsu, H., O’Keefe, C., Hsi, E., Paquette, R.L., Kojima, S., List,
A.F., Sekeres, M.A., McDevitt, M.A. & Maciejewski, J.P. (2009)
Mutations of e3 ubiquitin ligase cbl family members constitute a
novel common pathogenic lesion in myeloid malignancies. Journal
of Clinical Oncology, 27, 6109–6116.
Murati, A., Adelaıde, J., Popovici, C., Mozziconacci, M.J., Arnoulet, C.,
Lafage-Pochitaloff, M., Sainty, D., Birnbaum, D. & Chaffanet, M.
(2003) A further case of acute myelomonocytic leukemia with inv(8)
chromosomal rearrangement and MOZ-NCOA2 gene fusion.
International Journal of Molecular Medicine, 12, 423–428.
Nosslinger, T., Reisner, R., Gruner, H., Tuchler, H., Nowotny, H.,
Pittermann, E. & Pfeilstocker, M. (2001) Dysplastic versus prolif-
erative CMML–a retrospective analysis of 91 patients from a single
institution. Leukemia Research, 25, 741–747.
Padua, R.A., Carter, G., Hughes, D., Gow, J., Farr, C., Oscier, D.,
McCormick, F. & Jacobs, A. (1988) RAS mutations in myelodys-
plasia detected by amplification, oligonucleotide hybridization, and
transformation. Leukemia, 2, 503–510.
Padua, R.A., Guinn, B.A., Al-Sabah, A.I., Smith, M., Taylor, C., Pet-
tersson, T., Ridge, S., Carter, G., White, D., Oscier, D., Chevret, S. &
West, R. (1998) RAS, FMS and p53 mutations and poor clinical
outcome in myelodysplasias: a 10-year follow-up. Leukemia, 12,
887–892.
Ricci, C., Fermo, E., Corti, S., Molteni, M., Faricciotti, A., Cortelezzi,
A., Lambertenghi Deliliers, G., Beran, M. & Onida, F. (2010) RAS
mutations contribute to evolution of chronic myelomonocytic leu-
kemia to the proliferative variant. Clinical Cancer Research, 16,
2246–2256.
Rocquain, J., Carbuccia, N., Trouplin, V., Raynaud, S., Murati, A.,
Nezri, M., Tadrist, Z., Olschwang, S., Vey, N., Birnbaum, D., Gelsi-
Boyer, V. & Mozziconacci, M.J. (2010a) Combined mutations of
ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS,
RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and
acute myeloid leukemias. BMC Cancer, 10, 401.
Rocquain, J., Gelsi-Boyer, V., Adelaıde, J., Murati, A., Carbuccia, N.,
Vey, N., Mozziconacci, M.J., Birnbaum, D. & Chaffanet, M. (2010b)
Alteration of cohesin genes in myeloid diseases. American Journal of
Hematology (in press).
V. Gelsi-Boyer et al
374 ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375

Sugimoto, Y., Muramatsu, H., Makishima, H., Prince, C., Jankowska,
A.M., Yoshida, N., Xu, Y., Nishio, N., Hama, A., Yagasaki, H.,
Takahashi, Y., Kato, K., Manabe, A., Kojima, S. & Maciejewski, J.P.
(2010) Spectrum of molecular defects in juvenile myelomonocytic
leukaemia includes ASXL1 mutations. British Journal of Haematology,
150, 83–87.
Tefferi, A. (2010) Novel mutations and their functional and clinical
relevance in myeloproliferative neoplasms: JAK2, MPL, TET2,
ASXL1, CBL, IDH and IKZF1. Leukemia, 24, 1128–1138.
Vardiman, J.W., Harris, N.L. & Brunning, R.D. (2002) The world
health classification (WHO) of myeloid neoplasms. Blood, 100,
2292–2302.
Vardiman, J.W., Thiele, J., Arber, D.A., Brunning, R.D., Borowitz,
M.J., Porwit, A., Harris, N.L., Le Beau, M.M., Hellstrom-Lindberg,
E., Tefferi, A. & Bloomfield, C.D. (2009) The 2008 revision of the
WHO classification of myeloid neoplasms and acute leukemia:
rationale and important changes. Blood, 114, 937–951.
Supporting information
Additional Supporting Information may be found in the
online version of this article:
Table SI. Clinical and biological features of a series of
CMML.
Please note: Wiley-Blackwell are not responsible for the
content or functionality of any supporting materials supplied
by the authors. Any queries (other than missing material)
should be directed to the corresponding author for the article.
Gene Mutations in CMML
ª 2010 Blackwell Publishing Ltd, British Journal of Haematology, 151, 365–375 375

144
Commentaires sur l’article n°5
Nous avons établi les profils CGH-array ainsi que les combinaisons de mutations de 13
gènes, les 12 gènes présents dans l’article 2 (Rocquain et al, BMC Cancer 2010) plus le gène
PTPN11, dans une série de 53 patients atteints de leucémie myélomonocytaire chronique
(LMMC). Cette pathologie n’a pas été discutée dans ce travail et cet article est présenté en
annexe car la LMMC, hémopathie anciennement classée dans les SMD, est depuis 2001
reclassée dans les formes mixtes SMD/SMP par l’OMS.
Les LMMC sont divisées en formes myéloprolifératives (MP) et formes myélodysplasiques
(MD) selon le taux de leucocytes dans le sang (respectivement inférieur ou supérieur à
13G/L). Notre étude a été réalisée sur 31 LMMC-MP et 22 LMMC-MD.
Nous avons montré que les mutations d’ASXL1 et des gènes impliqués dans la
prolifération cellulaire étaient essentiellement retrouvées dans les cas de LMMC-MP. Les
mutations d’ASXL1 étaient corrélées à une acutisation de la LMMC. Toutes les LMMC qui
se sont transformées en leucémie aiguë étaient mutées sur ASXL1 et aucun des patients non
mutés n’a évolué en leucémie aiguë. De plus, nous avons mis en évidence un impact négatif
sur la survie des mutations du gène ASXL1 dans les LMMC. Ces résultats sont en accord
avec les données récemment publiées dans les LAM primaires (Chou et al, 2010).
L’ensemble des travaux du laboratoire depuis 2009 sur le gèneASXL1 a permis de
montrer que les mutations d’ASXL1 :
- sont retrouvées dans de nombreuses hémopathies myéloïdes chroniques et aiguës mais sont
plus fréquentes dans les LMMC (taux de mutation proche de 50%) que dans les autres HM
chroniques (SMD, SMP respectivement 16% et 10%),
- représentent les mutations les plus fréquentes au sein des LMMC,
- constitueraient un facteur de risque de transformation des LMMC en LAM, et plus
généralement de mauvais pronostic dans les LMMC,
- pourraient permettre de distinguer parmi les LAM les formes primaires des formes
secondaires.

145
Il serait ainsi intéressant d’étudier l’impact pronostique des mutations d’ASXL1 dans
les SMD et de montrer si elles constituent ou non un facteur de risque d’acutisation (ce n’est
pas le cas dans les LMMC-MD). Les mutations d’ASXL1 sont en effet retrouvées plus
fréquemment dans les SMD avancés (Rocquain et al, 2010 ; Boultwood et al, 2010),
néanmoins leur valeur pronostique demeure inconnue.

RÉSUMÉ
Au sein des hémopathies myéloïdes malignes, les syndromes myélodysplasiques
(SMD) et les leucémies aiguës myéloïdes (LAM) représentent des pathologies complexes et
hétérogènes résultant d’anomalies clonales des cellules souches médullaires. Elles sont
caractérisées par une hématopoïèse inefficace provoquant des cytopénies sanguines graves.
Les connaissances sur les anomalies moléculaires des SMD et des LAM, notamment à
caryotype normal, sont globalement pauvres et leur physiopathologie encore mal connue. Une
meilleure définition moléculaire est nécessaire pour une évaluation pronostique plus précise
de ces hémopathies et pour optimiser secondairement les stratégies thérapeutiques.
Cette thèse présente un panorama des classifications cytogénétiques et moléculaires
actuelles des SMD et LAM ainsi que l’étude de certaines altérations moléculaires rencontrées
dans ces maladies.
Grâce à l’apport des techniques d’analyse génomique à grande échelle, notamment la
CGH-array, notre laboratoire a identifié de nouvelles altérations génétiques, parmi lesquelles
les mutations du gène ASXL1, ainsi que des altérations des gènes codant les protéines de la
Cohésine et des régulateurs de la protéine CBL. Nous avons analysé une combinaison de
mutations de gène et émis l’hypothèse d’un modèle de leucémogenèse à 4 classes de
mutations, afin d’apporter des pistes dans la compréhension de la physiopathologie des SMD
et LAM.
Mots clés : syndrome myélodysplasique ; leucémie aiguë myéloïde, cibles moléculaires, génomique, mutation de gène.


















