
David Geneviève - Sauramps Médical · Parmi les anomalies de la face, il est possible de distin-...
23
Chirurgie plastique de l’enfant et de l’adolescent 30 Introduction Le terme syndrome malformatif de la face regroupe de très nombreuses entités. Les malformations crâniofaciales sont fréquentes et touchent, toutes étiologies confondues, environ 1 à 2% de la population. Parmi les anomalies de la face, il est possible de distin- guer les malformations majeures telles les fentes facia- les ou les crâniosténoses, des malformations mineures qui sont nettement plus nombreuses (pertuis pré au- riculaire, oreilles mal ourlées ou mal implantées, etc…). Les causes sont très nombreuses : génétiques (ano- malies chromosomiques ou mutations dans des gènes), multifactorielles (combinaison de facteurs génétiques et environnementaux) ou purement environnementa- les (intoxication maternelle, déformation par contrain- tes externes). Ce chapitre permet d’introduire l’idée qu’il faut re- chercher systématiquement une forme syndromique associée à la malformation crâniofaciale (ou syndromes malformatifs de la face). En effet, pour un tiers à la moi- tié des cas, les malformations sont impliquées dans des Syndromes malformatifs de la face David Geneviève Plan du chapitre ● ● Introduction ● ● Génétique des crâniosténoses ● ● Génétique des fentes orofaciales ● ● Génétique des dysostoses mandibulofaciales formes syndromiques. Par ailleurs, une forme dite isolée (non syndromique) peut quand même récidiver, être héritée d’un parent ou transmis à sa descendance dans le cadre d’une pathologie liée à une mutation dans un gène signant une maladie Mendélienne. Ainsi l’identification de l’étiologie précise est une étape cruciale dans la prise en charge du patient et de sa famille dans l’optique d’une médecine de précision, personnalisée afin de dépister au plus tôt les autres complications génétiquement associées à la malfor- mation crâniofaciale (une malformation cardiaque par exemple). Par ailleurs, c’est l’étiologie précise qui per- mettra d’établir un conseil génétique adapté et éven- tuellement un diagnostic prénatal en cas d’acceptation de l’indication par le centre pluridisciplinaire de diag- nostic prénatal. Les nouveaux outils de génétique (analyse chromoso- mique par puces à ADN ou CGH-array et le séquençage nouvelle génération (ou NGS pour Next Generation Sequencing) nous permettent de porter de plus en plus souvent un diagnostic étiologique précis pour le pa- tient et sa famille. Nous ne traiterons ici que de l’orientation étiologiques de 3 grands groupes de malformations dites majeures, les crâniosténoses, les fentes orofaciales et les dysos- toses mandibulofaciales. Génétique des crâniosténoses Introduction Définition et incidence Les crâniosténoses (ou crâniosynostoses) sont définies par des déformations de la boite crânienne liée à des
Transcript of David Geneviève - Sauramps Médical · Parmi les anomalies de la face, il est possible de distin-...

Chirurgie plastique de l’enfant et de l’adolescent30
IntroductionLe terme syndrome malformatif de la face regroupe de
très nombreuses entités.
Les malformations crâniofaciales sont fréquentes et touchent, toutes étiologies confondues, environ 1 à 2% de la population.
Parmi les anomalies de la face, il est possible de distin-guer les malformations majeures telles les fentes facia-les ou les crâniosténoses, des malformations mineures qui sont nettement plus nombreuses (pertuis pré au-riculaire, oreilles mal ourlées ou mal implantées, etc…).
Les causes sont très nombreuses : génétiques (ano-malies chromosomiques ou mutations dans des gènes), multifactorielles (combinaison de facteurs génétiques et environnementaux) ou purement environnementa-les (intoxication maternelle, déformation par contrain-tes externes).
Ce chapitre permet d’introduire l’idée qu’il faut re-chercher systématiquement une forme syndromique associée à la malformation crâniofaciale (ou syndromes malformatifs de la face). En effet, pour un tiers à la moi-tié des cas, les malformations sont impliquées dans des
Syndromes malformatifs de la faceDavid Geneviève
Plan du chapitre
●● Introduction
●● Génétique des crâniosténoses
●● Génétique des fentes orofaciales
●● Génétique des dysostoses mandibulofaciales
formes syndromiques. Par ailleurs, une forme dite isolée (non syndromique) peut quand même récidiver, être héritée d’un parent ou transmis à sa descendance dans le cadre d’une pathologie liée à une mutation dans un gène signant une maladie Mendélienne.
Ainsi l’identification de l’étiologie précise est une étape cruciale dans la prise en charge du patient et de sa famille dans l’optique d’une médecine de précision, personnalisée afin de dépister au plus tôt les autres complications génétiquement associées à la malfor-mation crâniofaciale (une malformation cardiaque par exemple). Par ailleurs, c’est l’étiologie précise qui per-mettra d’établir un conseil génétique adapté et éven-tuellement un diagnostic prénatal en cas d’acceptation de l’indication par le centre pluridisciplinaire de diag-nostic prénatal.
Les nouveaux outils de génétique (analyse chromoso-mique par puces à ADN ou CGH-array et le séquençage nouvelle génération (ou NGS pour Next Generation Sequencing) nous permettent de porter de plus en plus souvent un diagnostic étiologique précis pour le pa-tient et sa famille.
Nous ne traiterons ici que de l’orientation étiologiques de 3 grands groupes de malformations dites majeures, les crâniosténoses, les fentes orofaciales et les dysos-toses mandibulofaciales.
Génétique des crâniosténoses
Introduction
Définition et incidenceLes crâniosténoses (ou crâniosynostoses) sont définies
par des déformations de la boite crânienne liée à des

31Syndromes malformatifs de la face
perturbations de sa croissance par fusion prématurée des sutures crâniennes (Figure 1). Il s’agit d’affections très hétérogènes tant sur le plan clinique que radiolo-gique ou génétique.
Toutes formes confondues, les crâniosténoses sont observées pour 0,4 à 0,66/1000 naissance soit environ 1 naissance sur 2000. Toutes les ethnies sont atteintes [1-2].
Formes cliniquesDeux types de classifications peuvent être appliquées.
Une classification anatomique en fonction de l’atteinte des sutures et une classification en fonction des symp-tômes cliniques associés ou non. Seule la classification en fonction des formes isolées ou syndromiques sera traitée ici.
Concernant la seconde classification, on définit des formes isoléeS où la crâniosténose est le seul symptôme du patient et des formes syndromiques où la crâniosté-nose est associée à d’autres symptômes cliniques (par-fois subtils) définissant un syndrome.
Les formes isolées sont les plus fréquentes et sont ob-servées approximativement dans 67% des cas. Les for-
mes syndromiques sont plus rares et sont observées dans 33% des cas environ [2].
GénétiqueIl est impératif de bien différencier les formes isolées
(c’est-à-dire non syndromiques) des formes syndromi-ques car la prise en charge du patient dans sa globalité et l’évaluation du risque pour une autre grossesse est très différente. Plusieurs modes d’hérédités existent : formes multifactorielles, monogéniques, formes chro-mosomiques, acquises, environnementales (contraintes externes, causes médicamenteuses par exemple).
Classification en fonction des symp-tômes associés ou non
Formes acquises●● Contrainte externes, in utéro ou post-natale
Il est indispensable de ne pas confondre une plagiocé-phalie dûe à une déformation par exemple et non liée à une crâniosténose.
●● Formes environnementales
Figure 1. Représentation schématique des crâniosténoses en fonction de la suture atteinte.
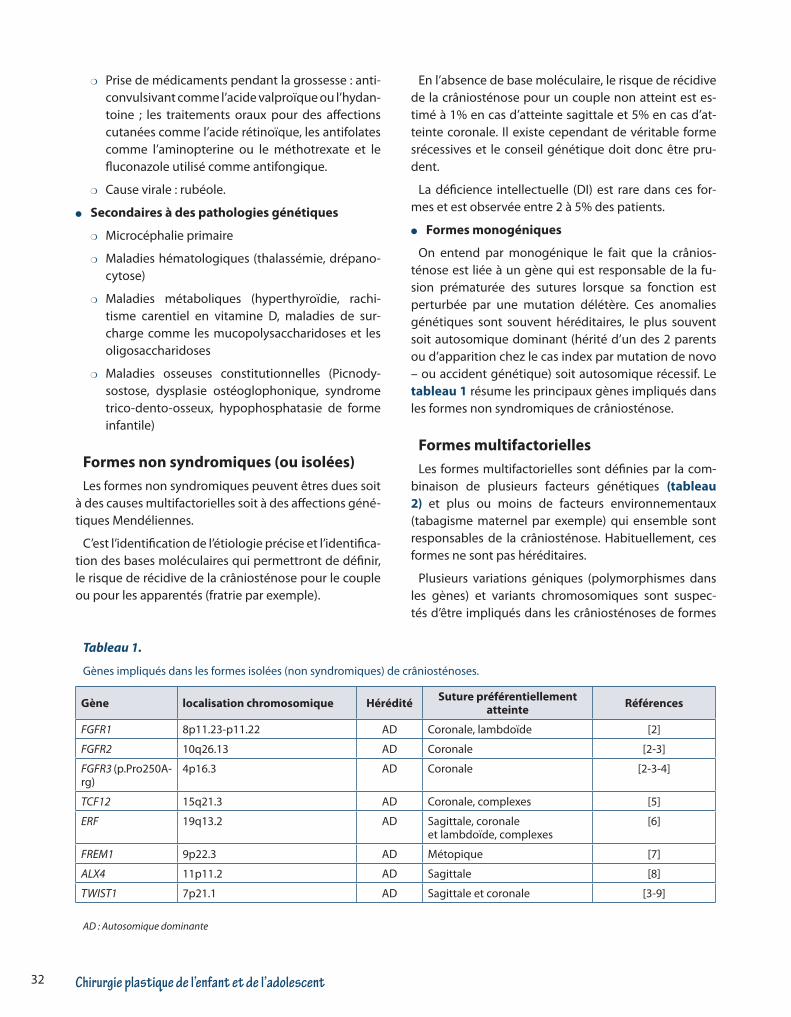
Chirurgie plastique de l’enfant et de l’adolescent32
●❍ Prise de médicaments pendant la grossesse : anti-convulsivant comme l’acide valproïque ou l’hydan-toine ; les traitements oraux pour des affections cutanées comme l’acide rétinoïque, les antifolates comme l’aminopterine ou le méthotrexate et le fluconazole utilisé comme antifongique.
●❍ Cause virale : rubéole.
●● Secondaires à des pathologies génétiques
●❍ Microcéphalie primaire
●❍ Maladies hématologiques (thalassémie, drépano-cytose)
●❍ Maladies métaboliques (hyperthyroïdie, rachi-tisme carentiel en vitamine D, maladies de sur-charge comme les mucopolysaccharidoses et les oligosaccharidoses
●❍ Maladies osseuses constitutionnelles (Picnody-sostose, dysplasie ostéoglophonique, syndrome trico-dento-osseux, hypophosphatasie de forme infantile)
Formes non syndromiques (ou isolées)Les formes non syndromiques peuvent êtres dues soit
à des causes multifactorielles soit à des affections géné-tiques Mendéliennes.
C’est l’identification de l’étiologie précise et l’identifica-tion des bases moléculaires qui permettront de définir, le risque de récidive de la crâniosténose pour le couple ou pour les apparentés (fratrie par exemple).
En l’absence de base moléculaire, le risque de récidive de la crâniosténose pour un couple non atteint est es-timé à 1% en cas d’atteinte sagittale et 5% en cas d’at-teinte coronale. Il existe cependant de véritable forme srécessives et le conseil génétique doit donc être pru-dent.
La déficience intellectuelle (DI) est rare dans ces for-mes et est observée entre 2 à 5% des patients.
●● Formes monogéniques
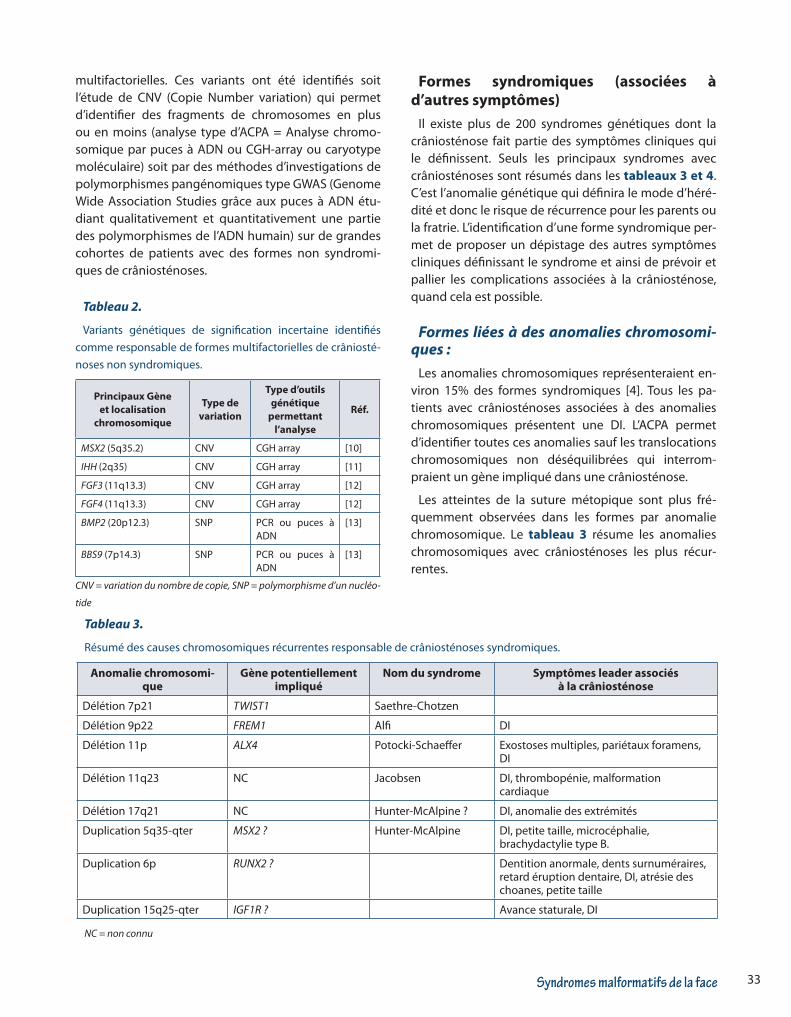
On entend par monogénique le fait que la crânios-ténose est liée à un gène qui est responsable de la fu-sion prématurée des sutures lorsque sa fonction est perturbée par une mutation délétère. Ces anomalies génétiques sont souvent héréditaires, le plus souvent soit autosomique dominant (hérité d’un des 2 parents ou d’apparition chez le cas index par mutation de novo – ou accident génétique) soit autosomique récessif. Le tableau 1 résume les principaux gènes impliqués dans les formes non syndromiques de crâniosténose.
Formes multifactoriellesLes formes multifactorielles sont définies par la com-
binaison de plusieurs facteurs génétiques (tableau 2) et plus ou moins de facteurs environnementaux (tabagisme maternel par exemple) qui ensemble sont responsables de la crâniosténose. Habituellement, ces formes ne sont pas héréditaires.
Plusieurs variations géniques (polymorphismes dans les gènes) et variants chromosomiques sont suspec-tés d’être impliqués dans les crâniosténoses de formes
Tableau 1.
Gènes impliqués dans les formes isolées (non syndromiques) de crâniosténoses.
Gène localisation chromosomique Hérédité Suture préférentiellement atteinte Références
FGFR1 8p11.23-p11.22 AD Coronale, lambdoïde [2]
FGFR2 10q26.13 AD Coronale [2-3]
FGFR3 (p.Pro250A-rg)
4p16.3 AD Coronale [2-3-4]
TCF12 15q21.3 AD Coronale, complexes [5]
ERF 19q13.2 AD Sagittale, coronale et lambdoïde, complexes
[6]
FREM1 9p22.3 AD Métopique [7]
ALX4 11p11.2 AD Sagittale [8]
TWIST1 7p21.1 AD Sagittale et coronale [3-9]
AD : Autosomique dominante
33Syndromes malformatifs de la face
multifactorielles. Ces variants ont été identifiés soit l’étude de CNV (Copie Number variation) qui permet d’identifier des fragments de chromosomes en plus ou en moins (analyse type d’ACPA = Analyse chromo-somique par puces à ADN ou CGH-array ou caryotype moléculaire) soit par des méthodes d’investigations de polymorphismes pangénomiques type GWAS (Genome Wide Association Studies grâce aux puces à ADN étu-diant qualitativement et quantitativement une partie des polymorphismes de l’ADN humain) sur de grandes cohortes de patients avec des formes non syndromi-ques de crâniosténoses.
Tableau 2.
Variants génétiques de signification incertaine identifiés comme responsable de formes multifactorielles de crâniosté-noses non syndromiques.
Principaux Gène et localisation
chromosomique
Type de variation
Type d’outils génétique
permettant l’analyse
Réf.
MSX2 (5q35.2) CNV CGH array [10]
IHH (2q35) CNV CGH array [11]
FGF3 (11q13.3) CNV CGH array [12]
FGF4 (11q13.3) CNV CGH array [12]
BMP2 (20p12.3) SNP PCR ou puces à ADN
[13]
BBS9 (7p14.3) SNP PCR ou puces à ADN
[13]
CNV = variation du nombre de copie, SNP = polymorphisme d’un nucléo-
tide
Formes syndromiques (associées à d’autres symptômes)
Il existe plus de 200 syndromes génétiques dont la crâniosténose fait partie des symptômes cliniques qui le définissent. Seuls les principaux syndromes avec crâniosténoses sont résumés dans les tableaux 3 et 4. C’est l’anomalie génétique qui définira le mode d’héré-dité et donc le risque de récurrence pour les parents ou la fratrie. L’identification d’une forme syndromique per-met de proposer un dépistage des autres symptômes cliniques définissant le syndrome et ainsi de prévoir et pallier les complications associées à la crâniosténose, quand cela est possible.
Formes liées à des anomalies chromosomi-ques :
Les anomalies chromosomiques représenteraient en-viron 15% des formes syndromiques [4]. Tous les pa-tients avec crâniosténoses associées à des anomalies chromosomiques présentent une DI. L’ACPA permet d’identifier toutes ces anomalies sauf les translocations chromosomiques non déséquilibrées qui interrom-praient un gène impliqué dans une crâniosténose.
Les atteintes de la suture métopique sont plus fré-quemment observées dans les formes par anomalie chromosomique. Le tableau 3 résume les anomalies chromosomiques avec crâniosténoses les plus récur-rentes.
Tableau 3.
Résumé des causes chromosomiques récurrentes responsable de crâniosténoses syndromiques.
Anomalie chromosomi-que
Gène potentiellement impliqué
Nom du syndrome Symptômes leader associés à la crâniosténose
Délétion 7p21 TWIST1 Saethre-Chotzen
Délétion 9p22 FREM1 Alfi DI
Délétion 11p ALX4 Potocki-Schaeffer Exostoses multiples, pariétaux foramens, DI
Délétion 11q23 NC Jacobsen DI, thrombopénie, malformation cardiaque
Délétion 17q21 NC Hunter-McAlpine ? DI, anomalie des extrémités
Duplication 5q35-qter MSX2 ? Hunter-McAlpine DI, petite taille, microcéphalie, brachydactylie type B.
Duplication 6p RUNX2 ? Dentition anormale, dents surnuméraires, retard éruption dentaire, DI, atrésie des choanes, petite taille
Duplication 15q25-qter IGF1R ? Avance staturale, DI
NC = non connu

Chirurgie plastique de l’enfant et de l’adolescent34
Formes liées à des anomalies monogéni-ques
Il existe dans les bases de données d’aide au diag-nostic plus de 200 syndromes génétiques dont un des symptômes cliniques est une crâniosténose. Les formes
syndromiques dues à des mutations dans des gènes sont les formes les plus fréquentes.
Actuellement tous les gènes impliqués dans des for-mes syndromiques de crâniosténoses ne sont pas en-core identifiés.
Tableau 4.
Résumé des causes génétiques responsables de crâniosténoses syndromiques.
Nom du syndrome
Numéro OMIM
/incidence
Gène et localisation chromosomique
Héré-dité
Symptômes cliniques majeurs Symptômes neurologiques
Réf.
Apert 101200/1/50 000
FGFR2 (10q26.13) AD syndactylie osseuses et/ou cutanées des mains et des pieds
DI, surdité, malfor-mations cérébrales
variables
[14]
Crouzon 123500/1/50 000
FGFR2 (10q26.13);FGFR3 (4p16.3)IL11RA (9p13.3)
ADAD
AR
Proptose oculaire, prognathismeLa forme avec acanthosis nigricans est due à une mutation spécifique de FGFR3 (A391E)IL11RA : dents surnuméraires
Céphalée, épilepsie, surdité
[14][15]
Pfeiffer 101600/1/50 000
FGFR1 (8p11.23-p11.22)
AD hypertélorisme; pouce/gros orteil large;syndactylie partielle desdoigts/orteils
Hydrocéphalie,malformation d’Ar-
nold-Chiari
[14]
Saethre-Chotzen
101400/1/25 000 à 1/50 000
TWIST1 (7p21.1); AD Syndactylie cutanée entre les 2eme et 3eme doigts des mains,Malformation cardiaque congénitale
Hypertension Intracrâ-nienne,
foramens pariétaux
[14]
Muenke 602849/Inconnue
<1/100000
FGFR3 (4p16.3),mutation spécifique pro250arg (P250R)
AD Macrocéphalie Surdité, DI, retard moteur isolé
[14]
Beare-Stevenson cutis gyrata
123790/ <1/1000000
FGFR2 (10q26.13) AD cutis gyrata du dos et du cuir chevelu, acanthosis nigricans
DI;hydrocephalie;
agénésie du corps calleux
[14]
Antley-Bixler 207410/ <1/1000000
FGFR2 (10q26.13)POR (7q11.23)
CYP26B1 (2p13.2)
ARARAR
Synostose radiohumérale, Atrésie/sté-nose des choanes, fragilité osseuse.POR : Anomalie du déterminisme sexuel
DI variable [14]
Type Boston 604757/<1/1000000
MSX2 (5q35.2) myopie ou hypermétropie, premiers métatarsiens courts
Céphalée [14]
Carpenter 201000/<1/1000000
RAB23 (6p11.2)MEGF8 (19q13.2)
AR syndactylie des mains, polydactylie pré-axiale et syndactylie des pieds, obésité, malformations cardiaque et rénale, puberté précoce
DI variable, surdité, atrophie optique
[14][16]
Dysplasie crâniofronto-nasale (les femmes sont plus atteintes que les hommes)
304110<1/100000
EFNB1 (Xq13.3), Lié X Hypertélorisme, scoliose, hernie dia-phragmatique, gros orteil élargi, stria-tion unguéale
Hypotonie, hypoplasie du corps calleux, DI
[14]

35Syndromes malformatifs de la face
Shprintzen-Goldberg
182212/<1/100000
SKI (1p36.32)FBN1 (15q21.1)
ADAD
Syndrome marfanoïde, hyperlaxité et/ou contractures articulaires, pectus excavatum ou carinatum, scoliose, camptodactylie et arachnodactylie, pieds bots, hernie ombilicale, dilata-tion de l’aorte)
DI [14][17]
Baller-Gerold 218600<1/100000
RECQL4 (8q24.3) AR Petite taille, anomalie du rayon radial, malformations viscérales (cardiaque, rénale), poïkilodermie
DI [14]
Greig 155700 de1/100000 à
1/1000000
GLI3 (7p14.1) AD Polydactylie et syndactylie Hydrocéphalie, agéné-sie du corps calleux
[14]
Dysplasie crânioectoder-mique
218330<1/1000000
IFTI22 (3q21.3-q22.1);WDR35 (2p24.1)
AR ciliopathie; pathologie squelettique, néphro-nophtise, fibrose hépatique, rétinite pigmentaire
[14]
Bohring-Opitz (Opitz C trigo-nocéphalie)
605039<1/1000000
ASXL1 (20q11.21) AD naevus flammeus sur le front, cheveux bas implantés, hirsutisme, exoph-talmie, hypertélorisme, fente labio-pa-latine, rétrognathie, déformations en flexion des coudes et des poignets, camptodactylie, déviation cubitale des doigts
DI sévère [18]
3MC 208340<1/1000000
MASP1 (3q27.3)COLC11 (2p25.3)
ARAR
Synostose radiocubitale, fente la-bio-palatine, hernie ombilicale, mal-formations cardiaques ou rénales
DI [19]
Crâniosténose, Alopécie
605420<1/1000000
ALX4 (11p11.2) Alopécie, hypogonadisme, Agénésie corps calleux
DI [8]
Hypophospha-tasie de forme infantile
2415001/100000
ALPL (1p36.1) AR Petite taille, fragilité osseuse, néphro-calcinose, sclérotiques bleutées, chon-drodysplasie
[20]
DI = déficience intellectuelle, Adapté de Jezela-Stanek et al [14] ; AD : Autosomique dominante ; AR : Autosomique récessif
Conduite à tenir face à une crânios-ténose
L’identification de la cause précise de la crâniosténose est une étape indispensable dans la prise en charge du patient. En effet, afin de proposer une médecine per-sonnalisée et ainsi de prévenir d’éventuelles complica-tions, il est impératif, avant de proposer des études en génétique chromosomique ou moléculaire, de s’assurer si la crâniosténose du patient est isolée (non syndromi-que) ou fait partie d’un syndrome génétique.
Par ailleurs, l’indentification de l’étiologie précise per-mettra de donner un conseil génétique adapté et fiable qui permettra éventuellement de proposer un diagnos-tic prénatal en cas d’acceptation de l’indication par le Centre Pluridisciplinaire de Diagnostic Prénatal.
La Figure 2 est une proposition d’arbre décisionnel pour aider le clinicien dans le diagnostic de la forme syndromique de crâniosténose.
ConclusionLes étiologies des crâniosténoses sont très hétérogè-
nes tant sur le plan clinique que radiologique ou géné-tique. Les formes non syndromiques représentent 2/3 des causes pour 1/3 de formes syndromiques (c’est-à-dire faisant partie d’un syndrome avec d’autres manifes-tations que celle du massif crâniofacial). L’identification des bases moléculaires a révolutionné le diagnostic et le conseil génétique et a permis l’avènement de la mé-decine personnalisée et une amélioration de la prise en charge des formes syndromiques. L’arrivée du séquen-çage nouvelle génération va étendre ces connaissances et permettre de participer à une meilleure compréhen-sion de la physiopathologie des crâniosténoses et ainsi contribuer à une meilleure prise en charge des patients.
Enfin, le diagnostic de crâniosténose doit être impéra-tivement fait dans le contexte d’une hypophosphatasie ou d’un syndrome d’Antley-Bixler du fait de l’existence de traitements médicamenteux spécifiques à ces affec-tions.

Chirurgie plastique de l’enfant et de l’adolescent36
Génétique des fentes orofaciales
IntroductionLes fentes orofaciales sont les malformations faciales
les plus fréquentes et concernent une personne sur 700 naissances. Elles comprennent, les fentes labiales (FL), les fentes palatines (FP), les fentes labiopalatines (FLP) et les fentes faciales de Tessiers (Figure 3).
Tout confondu, les fentes dites isolées (ou non syn-dromiques, c’est-à-dire non associés à d’autres malfor-mations) sont observées dans un plus de 70% des cas. Parmi les fentes orofaciales, les fentes labiales sont les plus fréquentes. L’héritabilité des FLP est proche de 50% (étude de jumeaux). Les facteurs génétiques sont donc importants dans la genèse des FLP mais il existe égale-ment une part environnementale permettant de classer
les FLP dans les maladies complexes avec plusieurs mo-des d’hérédités dont l’hérédité multifactorielle.
Néanmoins, il existe également de très nombreux syndromes génétiques avec FLP. Afin de permettre une prise en charge précoce et adaptée des patients de donner un conseil génétique spécifique et de proposer une médecine personnalisée, il est crucial de détermi-ner le cadre étiologique de la FL/FP ou FLP. Ainsi, il faut considérer la FL/FP ou FLP comme un symptôme et dé-finir si ce symptôme est associé à d’autres symptômes (forme syndromique) ou isolé (forme non syndromique ou isolée).
Epidémiologie des FL/FP ou FLPParmi les fentes isolées, la proportion de la FL repré-
sente 25% des fentes, la proportion de la FP représente
Figure 2. Conduite à tenir face à un patient avec crâniosténose afin d’en déterminer l’étiologie.

37Syndromes malformatifs de la face
Figure 3. Différentes formes de fentes orofaciales (collection Dr Michèle Bigorre, CHRU Montpellier).
Figure 4. Répartition des fentes.
a : répartition des fentes labiale, palatine et labiopalatine parmi les fentes orofaciales. b : répartition des fentes isolées par rap-port aux fentes syndromiques parmi les fentes labiales.

Chirurgie plastique de l’enfant et de l’adolescent38
30% des cas et la proportion de la FLP représente 45% des fentes (Figure 4). La FL ou la FLP peut être rarement observée dans le cadre de brides amniotiques (phéno-mène disruptif ) ou la FL/FLP peut s’intégrer dans une fente faciale. La fente faciale est une malformation crâniofaciale beaucoup plus rare.
Fente labialeL’incidence de la FL est de 1/700 à 1/1000 naissances
[21]. Cette incidence varie en fonction des ethnies. Elle est plus fréquente chez les indiens d’Amérique (3,6/1000 naissances) et les ethnies asiatiques (2,1/1000 naissan-ces au Japon et 1,7/1000 naissances en Chine). Elle est moins fréquente dans les ethnies africaines (0,3/1000 naissances) [22].
La FL est unilatérale dans 80% des cas et bilatérale dans 20% des cas. Elle est plus fréquemment observée à gauche (globalement 70% des FL unilatérales). Enfin, plus l’atteinte est sévère, plus le ratio homme/femme est élevé avec un excès de garçon suggérant des fac-teurs liés au chromosome X. La FL est le plus souvent non associée à d’autres symptômes (FL isolée ou non syndromique).
Fente palatineLa FP isolée doit être considérée comme une entité
différente de la FL. Plusieurs formes de FP existent : la FP complète avec défaut osseux et muqueux, la fente sous muqueuse et la luette bifide. Ces formes sont considé-rées par les généticiens comme un continuum malfor-matif.
L’incidence de la FP est d’approximativement 0,4/1000 naissances. Elle est plus fréquemment observée chez les femmes. La fente sous muqueuse du palais correspond à un défaut de fusion des muscles du palais mais avec une muqueuse intacte. L’incidence de la fente sous mu-queuse est estimée entre 1/1200 et 1/2000 naissances. L’incidence de la luette bifide (1/80 naissances chez les Caucasiens) est bien plus fréquente que l’incidence de la FP stricto sensu. Le sex ratio de ces 2 dernières formes est proche de 1.
Génétique des FL, FLP et FPPlusieurs formes d’hérédités sont possibles, quelle
que soit la forme de la FL, FLP ou FP (isolée ou syndro-mique) : maladie monogénique Mendélienne, multi-factorielle (c.à.d. coexistence de facteurs génétiques et de facteurs environnementaux, ou purement en-
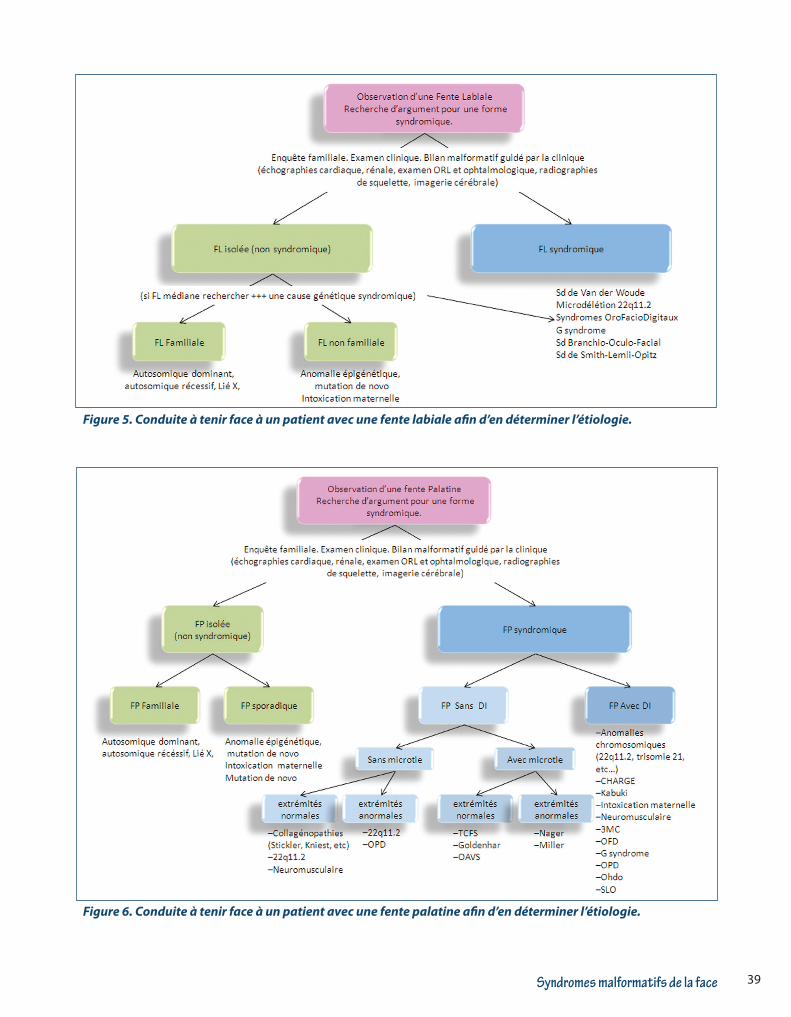
vironnemental comme dans certaines intoxications médicamenteuses pendant la grossesse). La Figure 5 schématise la conduite à tenir diagnostic face à une FL ou FLP et la Figure 6 schématise la conduite à tenir dia-gnostic face à une FP.
Génétique des FL, FLP et FP isoléesLes FL, FLP et FP isolées sont considérées comme étant
des pathologies génétiques multifactorielles, c’est-à-dire une combinaison de facteurs de susceptibilité génétique associée à des facteurs environnementaux. Néanmoins, en cas de transmission autosomique domi-nante clairement établie lors de la réalisation de l’arbre généalogique, une pathologie Mendélienne par muta-tions dans un gène spécifique est le plus probable.
Forme multifactorielle et facteurs géné-tiques
L’étude de jumeaux mono et dizygotes ont permis de déterminer que l’héritabilité dans les FL et les FP était importante. En effet, en cas de FL +/-P, le taux de concordance chez les jumeaux monozygotes était de 36% contre 4.7% chez les jumeaux dizygotes. En cas de FP le taux de concordance chez les monozygotes est de 22% contre 4.6% chez les jumeaux dizygotes. Des étu-des plus approfondies ont permis de déterminer que les facteurs génétiques dans les FP ne sont pas en faveur d’un modèle polygénique (association de plusieurs va-riants qui ensemble favorisent la genèse d’une FP) mais plutôt en faveur d’une hétérogénéité génétique (de très nombreux gènes mais responsables de peu de cas de FP pour chaque gène).
Les études d’associations pan genomiques (par ana-lyse de liaisons classiques ou étude génome entier - Genome Wide Association Studies, GWAS) ainsi que les études de gènes candidats ont permis d’identifier de nombreux gènes impliqués dans les fentes isolées. Le tableau 5 résume les gènes ou loci associés aux FL/FLP/FP isolées.
Facteurs environnementauxPlusieurs facteurs environnementaux ont également
été incriminés ou fortement suspectés comme fac-teurs de risque dans les fentes. Le tabagisme maternel augmente le risque de FL/FLP/FP de 1,3. Certains poly-morphismes dans les gènes GSTT1 (Glutathione S-trans-ferase theta) et NOS3 (nitric oxide synthase 3) semble influencer le risque de FL/FLP/FP lié à la consommation
39Syndromes malformatifs de la face
Figure 5. Conduite à tenir face à un patient avec une fente labiale afin d’en déterminer l’étiologie.
Figure 6. Conduite à tenir face à un patient avec une fente palatine afin d’en déterminer l’étiologie.
Chirurgie plastique de l’enfant et de l’adolescent40
de tabac maternel au cours de la grossesse [24]. Ce lien suggère fortement une interaction gène-environne-ment avec un effet métabolique par stress oxydatif res-ponsable de dégâts dans l’ADN responsable d’un phé-nomène d’apoptose en excès. Le rôle de l’exposition à l’alcool est moins évident, même si plusieurs études démontrent l’association entre consommation d’alcool au cours de la grossesse (en particulier l’alcoolisation aiguë) et risque augmenté de fentes. Le lien entre mé-tabolisme des folates et survenue des fentes n’est, là encore, pas totalement démontré. Les études sont dis-cordantes. Certaines études démontrent une diminu-tion des fentes chez les femmes enceintes prenant une supplémentation en acide folique alors que d’autres études ne démontrent pas d’effet de cette supplémen-tation. Cependant, un lien direct entre la prise d’acide valproïque (connu pour perturber le métabolisme des folates) au cours de la grossesse et la genèse d’un syn-drome malformatif est démontré.
Formes monogéniques mendéliennes des FL/FP et FLP isolées
Il existe des facteurs génétiques indéniables impliqués dans la genèse des FL/FP et FLP puisque, par exemple la concordance de FL chez 2 vrais jumeaux est entre 35% et 50% alors qu’elle n’est plus que de 4,7% chez des faux jumeaux. Le risque de récurrence empirique est variable en fonction du sexe de l’enfant atteint et de l’apparenté. De nombreuses études d’associations ont été réalisées, permettant d’identifier des facteurs génétiques de sus-ceptibilité pour les FL (Tableau 5). Cependant, il existe des véritables formes mendéliennes de FL par mutation causale dans plusieurs gènes différents. Plusieurs mo-des d’hérédités sont décrites. Le tableau 6 résume les gènes impliqués dans des formes mendéliennes isolées (non syndromiques) de FL/FP et FLP. Néanmoins ces gè-nes sont aussi impliqués dans des formes syndromiques de FL/FP et FLP. Enfin, il peut exister une coexistence de FL, FLP et/ou FP dans les familles avec mutations dans les gènes MSX1, FOXE1 et IRF6 [25].
Tableau 5.
Gènes ou locus de susceptibilité impliqués dans les formes isolées (non syndromiques) multifactorielles de FL/FLP/FP.
Locus/Gène Type d’analyse Locus/Gène Type d’analyse
Confirmation statistique Implication très probableIRF6 GWAS, GC, AL, M MSX1 GC, M
locus 8q24 GWAS, GC FOXE1 AL, GC, M VAX1 GWAS, GC MYH9 GC
MAFB GWAS locus ABCA4 GWAS locus 17q22 GWAS
BMP4 M FGFR2 M
GWAS = genome wide asssociation study, GC = gène candidat, AL = analyse de liaison, M = mutation, Adapté de [23]
Tableau 6.
Gènes impliqués dans les formes isolées FL, FLP et FP.
Gène localisation
chromosomique
Hérédité Type de fente Référence
IRF6 1q32.2 AD FL, FLP et FP [3-4]SATB2 2q33 AD FP [3-4]
SUMO1 2q33.1 AD FL, FLP et FP [3-4]MSX1 4p16.2 AD FL, FLP et FP [3-4]FOXE1 9q22 AD FP [3-4]FGF8 10q24 AD FL, FLP et FP [3-4]BMP4 14q22.2 AD FL, FLP et FP [3-4]TBX22 Xq12 Lié à l’X FP [3-4]

41Syndromes malformatifs de la face
Génétique des FL, FLP et FP syndro-miques
L’interrogation des bases de données de génétique re-trouve 581 syndromes avec une FP, 241 syndromes avec une fente labiale non médiane et 214 syndromes avec une FLP pour le logiciel LMD (london dysmorphology data base) et 548 syndromes avec une FP, 316 syndro-mes avec une FL non médiane et 269 syndromes avec
une FLP pour le logiciel Possum (Pictures of Standard Syndrome and Undiagnosed Malformations). Lorsque la FL est médiane, le logiciel LMD retrouve 49 syndro-mes et le logiciel Possum 62 Syndromes. La fente faciale médiane est évocatrice d’holoprosencéphalie ou de pa-thologie des cils (syndromes avec côtes courtes, poly-dactylie, syndromes orofaciaux digitaux ou OFD). Le ta-bleau 7 résume les étiologies des FL, FP et FLP dont les gènes ont été identifiés.
Tableau 7.
Formes syndromiques de FL/FP/FLP, dont les gènes responsables sont connus.
Type de fente
Groupe
de pathologiesSyndrome
Principaux symptômes
Gène et locali-sation chromo-
somiqueHérédité
Dans le cadre d’une fente faciale
Fente faciale de Tessier type 4
Fente facialeSPECC1L
(22q11.23)De novo
AD
Type de fente/Syndrome Gène
FL +/- FP Syndromes
malformatifs
Surdité, dystonie, hyper-télorisme
Surdité, dystonie, hypertélorisme
ACTB (7p22.1) AD
Syndrome hydrolethalus Pathologie létale
avec hydrocéphalie, polydactylie postaxiale
HYLS1 (11q24.2)
Lié à l’X
Syndrome de Van der Woude/ pterygium popli-
tés
Puits de la lèvre inférieure, ptérygium
IRF6 (1q32.2) AD
Retard mental liée à l’X associé à une FP ou une FL
DIPHF8
(Xp11.22)Lié à l’X
Syndrome de Gorlin Basocellulaires
multiplesPTCH1
(9q22.32)AD
Holoprosencéphalies Malformations
ligne médiane, DI, polydactylie
GLI2, (2q14.2)
SHH, (7q36.3)
SIX3, ( 2p21)
TGIF (18p11.31)
De novo AD
Syndrome de Greig/ Pallister Hall
Crâniosténose, polysyndactylie, DI
GLI3 (7p14.1) AD
Cancer gastrique familial et FLP
Cancer gastrique CDH1 (5q12) AD
Syndrome branchio-ocu-lo-facial (BOF)
Sinus branchial, lésions cutanées (tissus
thymique), pseudo-fente
TFAP2A (6p24.3)
AD

Chirurgie plastique de l’enfant et de l’adolescent42
Microphtalmie, agénésie du corps calleux
microphtalmie, agénésie du corps
calleuxVAX1 (10q25.3) AR
Associé à une atteinte
ectodermique
Dysplasie ectodermique et FLP
Agénésies dentaires multiples, sècheresse
cutanée
PVRL1 (11q23.3)
AR
EEC (ectrodactylie, dysplasie ectodermique et
fente)
Dysplasie ectodermi-que, agénésies den-
taires, ankyloblépharonTP63 (3q28) AD
Maladies os-seuses constitu-
tionnellesCrâniofrontonasal
Crâniosténose, nez bifide, hypertélorisme,
dysmorphie
EFNB1 (Xq13.1)
Lié à l’X
Roberts Phocomélie, crânios-
ténoseESCO2
(8p21.1)AR
Tetra-amelia with CLP Pathologie létale, hydrocéphalie
WNT3 (17q21.31)
AR
Fente labiale
médianeType de fente/Syndrome
Localisation chromo-somique
Gène
Opitz G/BBB malformation ligne
médiane, DIMID1 (Xp22.2) Lié à l’X
Oro-facial-digital type I Cilliopathie. Langue
bifide, anomalie extré-mité,
OFD1 (Xp22.3) Lié à l’X
FPType de fente/Syndrome
Localisation chromosomique
Gène
Syndromes
malformatifs Syndrome Oculofaciocar-diodental
Malformation chambre antérieure de l’œil, cardiaque, anomalie éruption
dentaire, oligodontie
BCOR (Xp11.4) Lié à l’X
Syndrome CHARGE
Malformation car-diaque, retard de crois-
sance, DI, anomalie OGE, oreille anormales asymétriques, agénésie canaux semi-circulaires
CHD7 (8q12.1)De novo,
AD
Syndrome d’Escobar (Le-thal multiple pterygium)
Pterygiums multiples, contractures articulai-res, hypomimie faciale
CHRNG (2q33) AR
Syndrome de Bamforth-Lazarus
Agénésie thyroïde, atrésie choane, ano-
malie cheveuxFOXE1 (9q22) AR
Syndrome de Van der Woude/popliteal pte-
rygium
Puits sur la lèvre inférieure
IRF6 (1q32.2) AD

Syndrome d’Andersen Petite taille, sca-
phocéphalie, dysmor-phie, QT long
KCNJ2 (17q23.1)
AD
Syndrome Kabuki
DI, syndrome malfor-matif, dysmorphie
KMT2D (12q12)
KD-M6A(Xp11.2)
De novo, AD
Lié X
Syndrome de Treacher Collins-Franceschetti et
apparentés
Hypoplasie malaire et mandibulaire, atrésie choane, dysmorphie.
Pas de DI
TCOF1, (5q32-q33.1)
POLR1D, (13q12.2)
POLR1C (6p21.1)
EFTUD2 (17q21.31)
AD
AD/AR
AR
AD
Syndrome de Miller Hypoplasie malaire et mandibulaire, ano-malie rayon cubital
DHODH (16q22)
AR
Syndrome de NagerHypoplasie malaire et mandibulaire, ano-malie rayon radial
SF3B4 (1q12-q21)
AD
Syndrome de Loeys-Dietz Aspect marfanoide,
dilatations vasculaires
TGFBR1 (9q22)
TGFBR2 (3p22)AD
Syndrome de Cornelia de Lange
Petite taille, ano-malie du rayon cubital,
microcéphalie, DI, dysmorphie
NIPBL, (5p13.1)
SMC1A (Xp11.22)
De novo, AD,
Lié à l’X
RMLX (sd de Rennepe-ning)
microcéphalie, DIPQBP1
(Xp11.23)Lié à l’X
Déficit intellectuel, épilepsie, dysmorphie,
ostéoporose.
épilepsie, dysmor-phie, ostéoporose
SATB2 (2q33) AD
FP et ankyloglossie ankyloglossie TBX22 (Xq12) Lié à l’X
Séquence de Pierre Robin (forme isolée ou
syndromique)
Voir tableau 4SOX9,
(17q24.3)
FAF1, (1p32.3)
RBM10 (Xp11.23)
De no-vo,AD, Lié
à l’X
Syndrome de Kallmann
Anosmie, difficultés de procréation, Hypo-gonadisme hypogona-
dotrope
FGFR1 (8p11.2)
FGF8 (10q24)
AD
AD
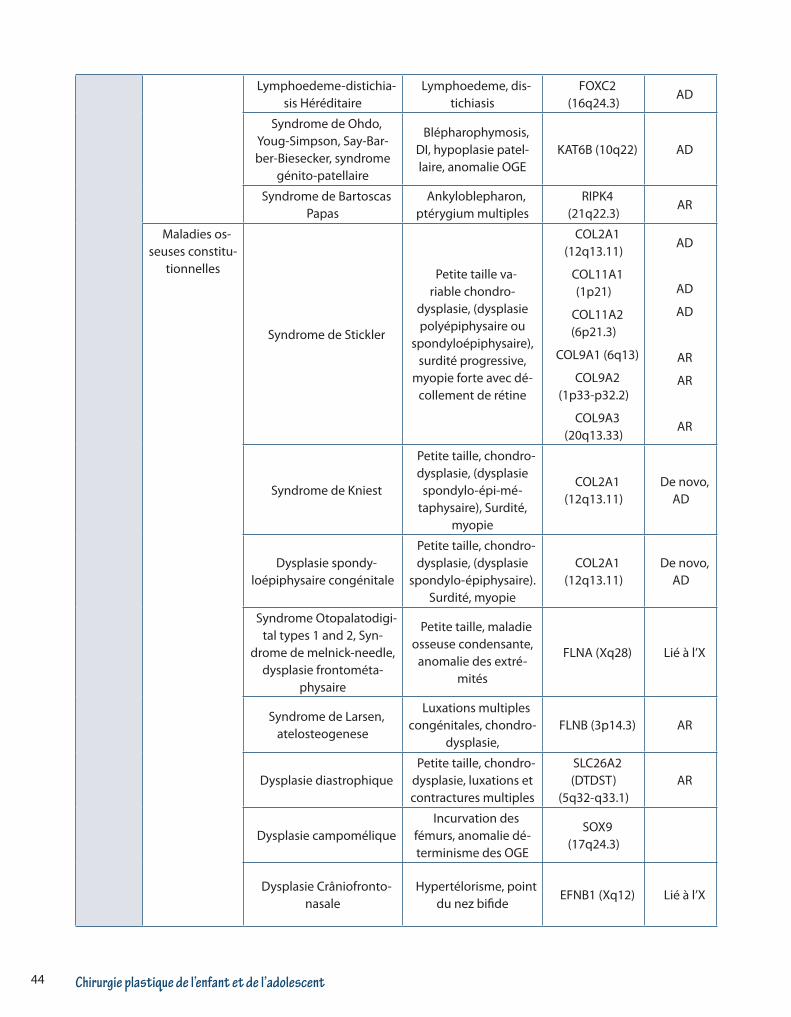
Chirurgie plastique de l’enfant et de l’adolescent44
Lymphoedeme-distichia-sis Héréditaire
Lymphoedeme, dis-tichiasis
FOXC2 (16q24.3)
AD
Syndrome de Ohdo, Youg-Simpson, Say-Bar-ber-Biesecker, syndrome
génito-patellaire
Blépharophymosis, DI, hypoplasie patel-laire, anomalie OGE
KAT6B (10q22) AD
Syndrome de Bartoscas Papas
Ankyloblepharon, ptérygium multiples
RIPK4 (21q22.3)
AR
Maladies os-seuses constitu-
tionnelles
Syndrome de Stickler
Petite taille va-riable chondro-
dysplasie, (dysplasie polyépiphysaire ou
spondyloépiphysaire), surdité progressive,
myopie forte avec dé-collement de rétine
COL2A1 (12q13.11)
COL11A1 (1p21)
COL11A2 (6p21.3)
COL9A1 (6q13)
COL9A2 (1p33-p32.2)
COL9A3 (20q13.33)
AD
AD
AD
AR
AR
AR
Syndrome de Kniest
Petite taille, chondro-dysplasie, (dysplasie
spondylo-épi-mé-taphysaire), Surdité,
myopie
COL2A1 (12q13.11)
De novo, AD
Dysplasie spondy-loépiphysaire congénitale
Petite taille, chondro-dysplasie, (dysplasie
spondylo-épiphysaire). Surdité, myopie
COL2A1 (12q13.11)
De novo, AD
Syndrome Otopalatodigi-tal types 1 and 2, Syn-
drome de melnick-needle, dysplasie frontométa-
physaire
Petite taille, maladie osseuse condensante,
anomalie des extré-mités
FLNA (Xq28) Lié à l’X
Syndrome de Larsen, atelosteogenese
Luxations multiples congénitales, chondro-
dysplasie,FLNB (3p14.3) AR
Dysplasie diastrophiquePetite taille, chondro-
dysplasie, luxations et contractures multiples
SLC26A2 (DTDST)
(5q32-q33.1)AR
Dysplasie campoméliqueIncurvation des
fémurs, anomalie dé-terminisme des OGE
SOX9 (17q24.3)
Dysplasie Crâniofronto-nasale
Hypertélorisme, point du nez bifide
EFNB1 (Xq12) Lié à l’X
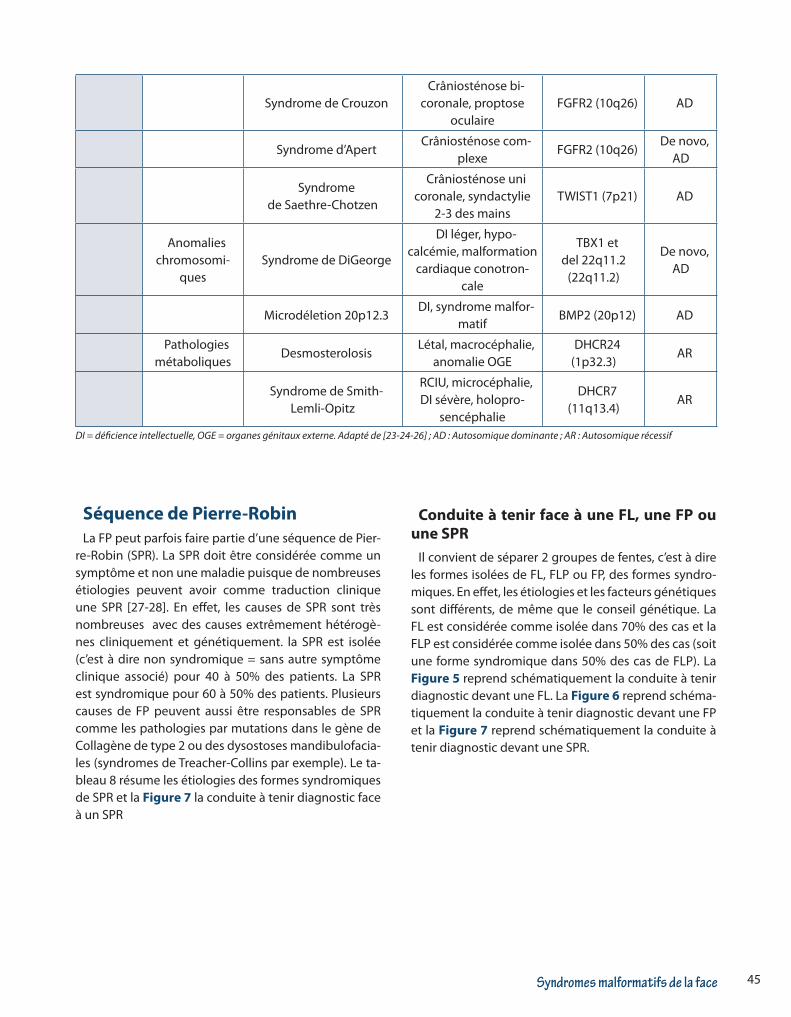
45Syndromes malformatifs de la face
Syndrome de Crouzon Crâniosténose bi-
coronale, proptose oculaire
FGFR2 (10q26) AD
Syndrome d’Apert Crâniosténose com-
plexeFGFR2 (10q26)
De novo, AD
Syndrome de Saethre-Chotzen
Crâniosténose uni coronale, syndactylie
2-3 des mainsTWIST1 (7p21) AD
Anomalies chromosomi-
quesSyndrome de DiGeorge
DI léger, hypo-calcémie, malformation
cardiaque conotron-cale
TBX1 et del 22q11.2
(22q11.2)
De novo, AD
Microdéletion 20p12.3DI, syndrome malfor-
matifBMP2 (20p12) AD
Pathologies métaboliques
Desmosterolosis Létal, macrocéphalie,
anomalie OGEDHCR24
(1p32.3)AR
Syndrome de Smith-Lemli-Opitz
RCIU, microcéphalie, DI sévère, holopro-
sencéphalie
DHCR7 (11q13.4)
AR
DI = déficience intellectuelle, OGE = organes génitaux externe. Adapté de [23-24-26] ; AD : Autosomique dominante ; AR : Autosomique récessif
Conduite à tenir face à une FL, une FP ou une SPR
Il convient de séparer 2 groupes de fentes, c’est à dire les formes isolées de FL, FLP ou FP, des formes syndro-miques. En effet, les étiologies et les facteurs génétiques sont différents, de même que le conseil génétique. La FL est considérée comme isolée dans 70% des cas et la FLP est considérée comme isolée dans 50% des cas (soit une forme syndromique dans 50% des cas de FLP). La Figure 5 reprend schématiquement la conduite à tenir diagnostic devant une FL. La Figure 6 reprend schéma-tiquement la conduite à tenir diagnostic devant une FP et la Figure 7 reprend schématiquement la conduite à tenir diagnostic devant une SPR.
Séquence de Pierre-RobinLa FP peut parfois faire partie d’une séquence de Pier-
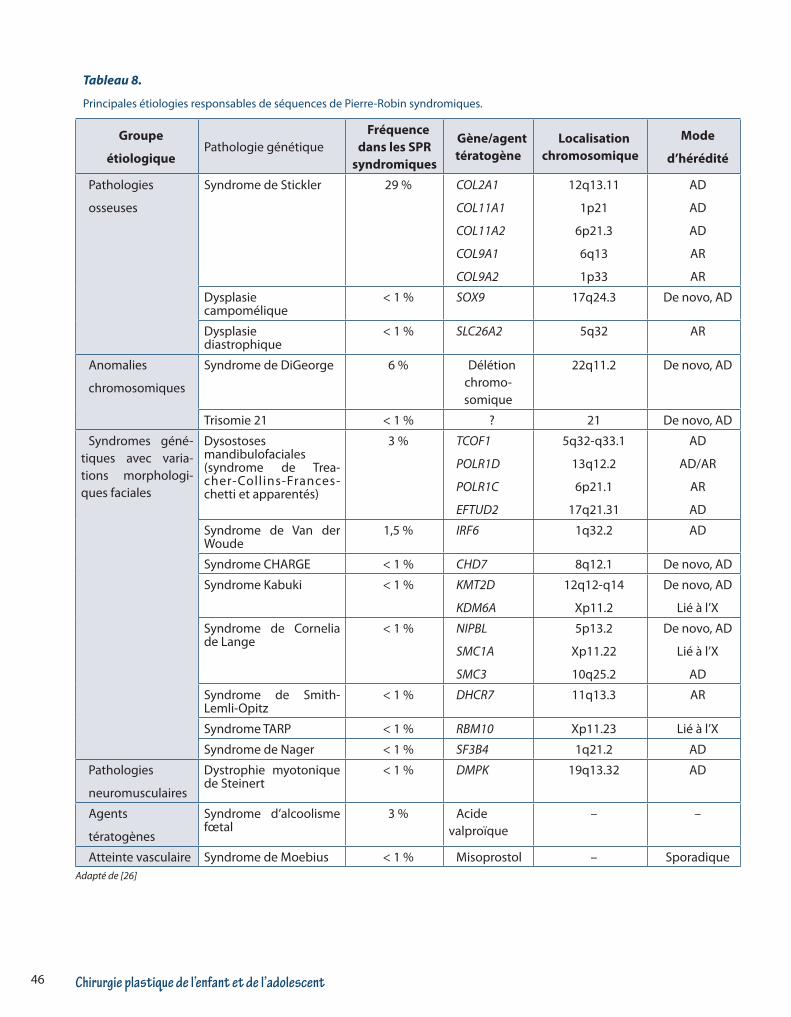
re-Robin (SPR). La SPR doit être considérée comme un symptôme et non une maladie puisque de nombreuses étiologies peuvent avoir comme traduction clinique une SPR [27-28]. En effet, les causes de SPR sont très nombreuses avec des causes extrêmement hétérogè-nes cliniquement et génétiquement. la SPR est isolée (c’est à dire non syndromique = sans autre symptôme clinique associé) pour 40 à 50% des patients. La SPR est syndromique pour 60 à 50% des patients. Plusieurs causes de FP peuvent aussi être responsables de SPR comme les pathologies par mutations dans le gène de Collagène de type 2 ou des dysostoses mandibulofacia-les (syndromes de Treacher-Collins par exemple). Le ta-bleau 8 résume les étiologies des formes syndromiques de SPR et la Figure 7 la conduite à tenir diagnostic face à un SPR
Chirurgie plastique de l’enfant et de l’adolescent46
Tableau 8.
Principales étiologies responsables de séquences de Pierre-Robin syndromiques.
Groupe
étiologiquePathologie génétique
Fréquence dans les SPR
syndromiques
Gène/agent tératogène
Localisation chromosomique
Mode
d’hérédité
Pathologies
osseuses
Syndrome de Stickler 29 % COL2A1
COL11A1
COL11A2
COL9A1
COL9A2
12q13.11
1p21
6p21.3
6q13
1p33
AD
AD
AD
AR
AR
Dysplasie campomélique
< 1 % SOX9 17q24.3 De novo, AD
Dysplasie diastrophique
< 1 % SLC26A2 5q32 AR
Anomalies
chromosomiques
Syndrome de DiGeorge 6 % Délétion chromo- somique
22q11.2 De novo, AD
Trisomie 21 < 1 % ? 21 De novo, AD
Syndromes géné-tiques avec varia-tions morphologi-ques faciales
Dysostoses mandibulofaciales(syndrome de Trea-cher-Collins-Frances-chetti et apparentés)
3 % TCOF1
POLR1D
POLR1C
EFTUD2
5q32-q33.1
13q12.2
6p21.1
17q21.31
AD
AD/AR
AR
AD
Syndrome de Van der Woude
1,5 % IRF6 1q32.2 AD
Syndrome CHARGE < 1 % CHD7 8q12.1 De novo, AD
Syndrome Kabuki < 1 % KMT2D
KDM6A
12q12-q14
Xp11.2
De novo, AD
Lié à l’X
Syndrome de Cornelia de Lange
< 1 % NIPBL
SMC1A
SMC3
5p13.2
Xp11.22
10q25.2
De novo, AD
Lié à l’X
AD
Syndrome de Smith-Lemli-Opitz
< 1 % DHCR7 11q13.3 AR
Syndrome TARP < 1 % RBM10 Xp11.23 Lié à l’X
Syndrome de Nager < 1 % SF3B4 1q21.2 AD
Pathologies
neuromusculaires
Dystrophie myotonique de Steinert
< 1 % DMPK 19q13.32 AD
Agents
tératogènes
Syndrome d’alcoolisme fœtal
3 % Acide valproïque
– –
Atteinte vasculaire Syndrome de Moebius < 1 % Misoprostol – SporadiqueAdapté de [26]

47Syndromes malformatifs de la face
Conseil génétique et conduite à tenir de-vant une FL, FLP ou FP
Le conseil génétique des FL, FLP et FP est différent si la fente est isolée ou syndromique. Il est donc capital, pour donner un conseil génétique adapté et pour proposer et coordonner une prise en charge précoce et spécifique des symptômes cliniques associés, de préciser le cadre étiologique de la FL, FLP ou FP. En cas de fente isolée, le conseil génétique est le plus souvent empirique. L’iden-tification des bases génétique des fentes isolées per-mettra probablement dans le futur de mieux préciser le conseil génétique et donc le risque de récidive pour le couple, la fratrie et les apparentés. Dans tous les cas, l’établissement de l’arbre généalogique doit être réalisé afin d’identifier des formes mendéliennes de FL, FP ou FLP. En cas de fente syndromique, c’est l’identification du syndrome qui permettra d’affiner le conseil géné-tique et de donner le risque de récidive.
Conseil génétique des FL isoléesLes données concernant les FL isolées sont peu nom-
breuses dans la littérature. Seules les données concer-nant les FLP sont disponibles. Globalement, en de-hors d’un arbre généalogique évocateur d’une forme mendélienne, le risque de récidive est assez faible pour les FL unilatérales (2 à 3%). Ce risque de récurrence peut, pour certains auteurs, être affiné par l’étude écho-graphique du muscle orbiculaire de la lèvre supérieure [29]. Ainsi, l’existence d’une hypoplasie de ce muscle augmente le risque de récurrence de FL pour un appa-renté. Les auteurs considèrent que l’hypoplasie du mus-cle orbiculaire de la lèvre supérieur doit être considérée comme étant dans le spectre de la FL.
Conseil génétique des FLP isoléesLe risque empirique (en dehors d’un arbre généalo-
gique évocateur d’une forme mendélienne) de récidive pour un couple avec un enfant avec une FLP non syn-dromique est estimé à 4%. Le risque de transmission à la descendance d’un parent avec FLP est estimé à 4,3%. Le risque pour les apparentés du second degré est de 0,6% et celui des apparentés du 3ème degré de 0,3% (risque population générale = 1/1000 soit 0.1%). Bien évi-demment plusieurs facteurs sont à prendre en compte comme l’ethnie et le sexe de la personne atteinte.
Conseil génétique des FP isoléesLe risque empirique (en dehors d’un arbre généalo-
gique évocateur d’une forme mendélienne) de récidive pour un couple avec un enfant avec une FP non syn-dromique est estimé à 1,8%. Le risque de transmission à la descendance d’un parent avec FP est estimé à 3% (risque population générale = 0,4/1000 soit 0.04%). Ce-pendant ce risque doit être modifié en fonction du sexe de la personne atteinte et de ses parents. Ainsi si l’enfant atteint est un garçon d’une fratrie, le risque pour un pro-chain garçon de la même fratrie est de 1,8% alors que si la personne atteinte est une mère, le risque de récidive pour sa fille est de 17,2%.
Conseil génétique des FL/FLP et FP syndro-miques.
Le conseil génétique est fonction de l’étiologie de la FL/FLP ou FP syndromique. Seule l’identification de l’étiologie de la FL/FLP ou FP permet de donner un conseil génétique adapté. Il sera donc nécessaire de mettre tous les moyens en œuvre pour identifier l’étio-logie précise. Le tableau 3 résume les causes génétiques avec gènes identifiés et les Figures 5 à 7 proposent une aide à l’identification de la cause étiologique.
ConclusionLes FL/FP et FLP sont très hétérogènes tant sur le plan
clinique que génétique. La part de formes non syndro-miques par rapport aux formes syndromiques dépend du type de fente. Les formes non syndromiques repré-sentent 75% à 50% des causes pour 25% jusqu’à 50% de formes syndromiques (c’est-à-dire faisant partie d’un syndrome avec d’autres manifestations que celle du massif crâniofacial). Les facteurs environnementaux sont très probablement impliqués dans les formes non syndromiques de FL et FP. Les FL, FLP et FP isolées doi-vent être considérées comme des maladies multifacto-rielles, une fois une forme familiale clairement exclue. Le diagnostic précis de l’affection reste cependant in-dispensable afin de permettre un diagnostic personna-lisé et une prise en charge ainsi qu’un conseil génétique adaptés pour le patient. L’arrivée du séquençage nou-velle génération va étendre ces connaissances et per-mettre de participer à une meilleure compréhension de la physiopathologie des FL/FP et FLP et ainsi contribuer à une meilleure prise en charge des patients.

Chirurgie plastique de l’enfant et de l’adolescent48
Génétique des dysostoses mandibulofaciales
Introduction
Définition et incidenceLe terme dysostose mandibulo-faciale est définie par
une atteinte symétrique des maxillaires supérieure et inferieure qui est très souvent associée à une microtie. Peuvent s’y associer d’autres malformations et/ou une anomalie des extrémités. On parle alors de dysostose acro-faciale ou d’acrodysostose faciale. Ce terme-là en-globe à nouveau, de nombreuses entités.
Toutes formes confondues, cette malformation crânio-faciale atteint 1 personne sur 35 000 environ.
Diagnostic et génétiqueIl existe de nombreux diagnostics différentiels des
dysostoses mandibulo-faciales. Ces diagnostics diffé-rentiels doivent être envisagés en cas d’atteinte très ou totalement asymétrique du visage.
Les dysostoses mandibulo-faciales sont très hétéro-gènes tant sur le plan clinique que génétique. Il existe de nombreuses affections responsables de ces patho-logies. Globalement, on distingue les formes sans ano-malies des extrémités des formes avec anomalies des extrémités.
Figure 7. Conduite à tenir face à un patient avec une séquence de Pierre-Robin afin d’en déterminer l’étiologie.

49Syndromes malformatifs de la face
ClassificationLes dysostoses mandibulo-faciales (DMF) sont clas-
sées en fonction des symptômes cliniques les accom-pagnant.
La cause la plus fréquente est le syndrome de Treacher Collins Franceschetti (STCF). Son incidence est estimée à 1/50 000 naissances. Il n’y a habituellement pas de DI. Les patients peuvent présenter une fente palatine, ou une atrésie des choanes. On peut observer un colo-bome de la paupière inférieure et une projection anor-male des cheveux sur la joue en avant de la microtie [26]. L’observation d’une DI ou d’une microcéphalie doit faire évoquer d’autres causes de DMF.
Les autres causes de DMF (outre la forme avec mi-crocéphalie par mutation dans le gène EFTUD2), sont beaucoup plus rares. Il faut savoir recherche une DMF par mutations dans le gène EFTUD2 en cas de microcé-phalie mais également d’autres malformations atypi-ques du STCF, comme une atrésie de l’œsophage, une agénésie des canaux semi-circulaires et un DI. Le ta-bleau 9 résume les différentes formes de DMF.
En cas d’observation d’une anomalie des extrémités en plus de la DMF, le cadre nosologique change, il s’agit alors de dysostose acro-faciale (DAF). Les DAF sont
beaucoup plus rares. Le tableau 10 répertorie les prin-cipales formes de DAF. L’étiologie la plus fréquente est le syndrome de Nager qui est dû à des mutations dans le gène SF3B4. Les symptômes cliniques sont très va-riables, en particulier concernant les extrémités. L’ano-malie du rayon radial va d’un pouce hypoplasique mal implanté à une véritable agénésie du rayon radial. La synostose radiocubitale est souvent présente.
Diagnostics différentielsEn cas d’atteinte très asymétrique, il faut envisager un
autre cadre nosologique : les microsomies hémifacia-les. Les microsomies hémifaciales sont plus fréquentes que les DMF. Là encore, cette entité est composée d’un groupe très hétérogène tant sur le plan clinique que radiologique, dont les syndromes Oculo Auriculo Verte-bral Syndrome (OAVS) et le syndrome de Goldenhar. Les bases moléculaires sont très mal connues.
Si l’atteinte est symétrique (micro-mandibulie et mi-crotie) mais que les os zygomatiques et maxillaires sont préservés, alors il sera possible d’envisager un syndrome auriculo-condylar qui est dû à des mutations dans les gènes EDN1, PLCB4 et GNAI3. Ce syndrome est hérité en récessif.
Tableau 9.
Classification des principales dysostoses mandibulo-faciales.
Type de dysostose
mandibulofacialeNuméro OMIM hérédité
Gène
et localisation
chromosomique
STCF type I 154500 AD TCOF1 (5q32)
STCF type II 613717 AD et AR POLR1D (13q12)
STCF type III 248390 AR POLR1C (6p22)
DMF avec microcéphalie – AD EFTUD2 (17q21)
DMF type Hutterite 248390 Probablement AR NC
DMF de Toriello 301950 Lié à l’X NC
DMF de Hedera-Toriello-Petty 608257 AD NC
DMF de Bauru 604830 AD NC
DMF de Verloes 602562 NC NC
DMF de Zhang – NC duplication1q21.2-q22
DMF de Burn-McKeown 608572 NC NC
STCF = syndrome de Treacher Collis-Franceshetti, DMF = Dysostose mandibulo-faciale, AD = autosomique dominant, AR = autosomique récessif, OMIM
= Online Mendelian Inheritance in Man; NC = Non connu, Adapté de [30]

Chirurgie plastique de l’enfant et de l’adolescent50
Le syndrome Question mark (malformation des oreilles leur donnant un aspect en forme de point d’interroga-tion) est allélique, c’est-à-dire dû aux mêmes gènes mais hérité en dominance.
Conseil génétique et CATL’identification de la cause précise de la dysostose
mandibulo-faciale est une étape indispensable dans la prise en charge du patient. En effet, afin de propo-ser une médecine personnalisée et ainsi de prévenir d’éventuelle complications, il est impératif de proposer des études en génétique moléculaire qui permettront d’affiner le diagnostic clinique.
Tableau 10.
Classification des principales dysostoses acro-faciale (dysostoses mandibulo-faciales avec anomalie des extrémités).
Type d’acrodysostose faciale Principaux signes cliniques Numéro OMIM HéréditéGene
et localisation chromosomique
Avec anomalie préaxiale
Nager Hypoplasie rayon radial 154400 AD SF3B4 (1q12-q21)
DAF de Guion-Almeida Pseudo CHARGEMicrocéphalie, atrésie de l’œsophage, agénésie des canaux semi circulaires
610536 AD EFTUD2 (17q21)
DAF de Kennedy-Teebi Microcéphalie, blépharo-phymosis
– AR –
DAF de Kelly Petite taille, anomalies génito-urinaires
– AR-lié à l’X
–
DAF de Reynolds Ptosis et absence de plis de flexion des interphalan-giennes
– AD Microdeletion 16p13.3?
Avec anomalie post axiale
Miller Hypoplasie rayon ulnaire 263750 AR DHODH (16q22
DAF avec anomalies vertébra-les
Anomalies vertébrales – Non connue
–
DAF de Weyers Cilliopathie, chondrodyspl-saie, petite taille, malforma-tion cardiaque
193530 AR EVC2 (4p16.2)
DAF type Arens – Non connue
–
Autres Voir [2] pour revue
DAF de Richieri-Costa Pierre Robin, anomalie ra-diale et cubiltale
260385 AR EIF4A3 (17q25.3)
ADF = dysostose acro-faciale, d’après [30] ; AD : Autosomique dominante ; AR : Autosomique récessif
La Figure 8 est une proposition d’arbre décisionnel pour aider le clinicien dans le diagnostic de la cause de la dysostose mandibulo-faciale ainsi que de ses diag-nostic différentiels.
Ces diagnostics différentiels devront être envisagés en cas d’atteinte très asymétrique ou d’absence d’atteinte majeure des malaires et/ou maxillaires supérieurs (mi-crosomies hémifaciales dont l’OAVS, le syndrome de Goldenhar les syndromes question mark ears et auricu-lo-condylar, l’anémie de Blackfan-Diamond et le syn-drome BOR - branchio-oto-rénal).

51Syndromes malformatifs de la face
Conclusion
Les dysostoses mandibulo-faciales font partie d’un groupe très hétérogène tant sur le plan clinique que radiologique. Certaines formes sont associées à une dé-ficience intellectuelle. Un diagnostic étiologique précis est indispensable à la bonne prise en charge du patient.
Par ailleurs, l’indentification de l’étiologie précise ren-dra possible un conseil génétique adapté et fiable qui permettra éventuellement de proposer un diagnos-tic prénatal en cas d’acceptation de l’indication par le Centre Pluridisciplinaire de Diagnostic Prénatal.
Bibliographie
1. Di Rocco F, Cormier-Daire V, Collet C, Le Merrer M, Renier D, Arnaud E. Syndromes avec crâniosténoses. p191. Syndromiques dysmorphi-ques, Doin éditeur
2. Roscioli T, Elakis G, Cox TC, et al. Genotype and clinical care corre-lations in crâniosynostosis: findings from a cohort of 630 Australian and New Zealand patients. Am J Med Genet C Semin Med Genet. 2013 Nov;163C(4):259-70.
3. Mulliken JB, Gripp KW, Stolle CA, Steinberger D, Muller U. Molecular analysis of patients with synostotic frontal plagiocephaly (unilateral coronal synostosis). Plast Reconstr Surg. 2004;113:1899–909.
4. Wilkie AO, Byren JC, Hurst JA, et al. Prevalence and complications of single-gene and chromosomal disorders in crâniosynostosis. Pedia-trics. 2010 Aug;126(2):e391-400.
5. Sharma VP, Fenwick AL, Brockop MS, et al. Mutations in TCF12, enco-ding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal crâniosynostosis. Nat Genet. 2013 Mar;45(3):304-7.
6. Twigg SR, Vorgia E, McGowan SJ, et al. Reduced dosage of ERF causes complex crâniosynostosis in humans and mice and links ERK1/2 signa-ling to regulation of osteogenesis. Nat Genet. 2013 Mar;45(3):308-13.
Figure 8. Conduite à tenir face à un patient avec une dysostose mandibulo-faciale afin d’en déterminer l’étiologie.

Chirurgie plastique de l’enfant et de l’adolescent52
7. Vissers LE, Cox TC, Maga AM, et al. Heterozygous mutations of FREM1 are associated with an increased risk of isolated metopic crâniosynos-tosis in humans and mice. PLoS Genet. 2011 Sep;7(9):e1002278.
8. Yagnik G, Ghuman A, Kim S, et al. ALX4 gain-of-function mutations in nonsyndromic crâniosynostosis. Hum Mutat. 2012 Dec;33(12):1626-9.
9. Seto ML, Hing AV, Chang J, et al. Isolated sagittal and coronal crânio-synostosis associated with TWIST box mutations. Am J Med Genet A. 2007 Apr 1;143(7):678-86.
10. Pelegrino Kde O, Sugayama S, Lezirovitz K, Catelani AL, Kok F, Chauffaille Mde L. MSX2 copy number increase and crâniosynostosis: copy number variation detected by array comparative genomic hybri-dization. Clinics (Sao Paulo). 2012 Aug;67(8):981-5.
11. Klopocki E, Lohan S, Brancati F, et al. Copy-number variations in-volving the IHH locus are associated with syndactyly and crâniosynos-tosis. Am J Hum Genet. 2011 Jan 7;88(1):70-5.
12. Grillo L, Greco D, Pettinato R, et al. Increased FGF3 and FGF4 gene dosage is a risk factor for crâniosynostosis. Gene. 2014 Jan 25;534(2):435-9. doi: 10.1016/j.gene.2013.09.120.
13. Justice CM, Yagnik G, Kim Y, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal crâniosynosto-sis near BMP2 and within BBS9. Nat Genet. 2012 Dec;44(12):1360-4. doi: 10.1038/ng.2463. Epub 2012 Nov 18.
14. Jezela-Stanek A, Krajewska-Walasek M. Genetic causes of syndro-mic crâniosynostoses. Eur J Paediatr Neurol. 2013 May;17(3):221-4.
15. Nieminen P, Morgan NV, Fenwick AL, et al. Inactivation of IL11 sig-naling causes crâniosynostosis, delayed tooth eruption, and supernu-merary teeth. Am J Hum Genet. 2011 Jul 15;89(1):67-81.
16. Twigg SR, Lloyd D, Jenkins D, et al. Mutations in multidomain pro-tein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am J Hum Genet. 2012 Nov 2;91(5):897-905.
17. Carmignac V, Thevenon J, Adès L, et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome. Am J Hum Genet. 2012 Nov 2;91(5):950-7.
18. Hoischen A, van Bon BW, Rodríguez-Santiago B, et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011 Jun 26;43(8):729-31.
19. Rooryck C, Diaz-Font A, Osborn DP, et al. Mutations in lectin com-plement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet. 2011 Mar;43(3):197-203.
20. Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007 Oct 4;2:40.
21. Stuppia L, Capogreco M, Marzo G, La Rovere D, Antonucci I, Gatta V, Palka G, Mortellaro C, Tetè S. Genetics of syndromic and nonsyndro-mic cleft lip and palate. J Crâniofac Surg. 2011 Sep;22(5):1722-6.
22. Lace B, Kempa I, Piekuse L, Grinfelde I, Klovins J, Pliss L, Krumina A, Vieira AR. Association studies of candidate genes and cleft lip and pa-late taking into consideration geographical origin. Eur J Oral Sci. 2011 Dec;119(6):413-7.
23. Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011 Mar;12(3):167-78
24. Blanton SH, Henry RR, Yuan Q, Mulliken JB, Stal S, Finnell RH, Hecht JT. Folate pathway and nonsyndromic cleft lip and palate. Birth De-fects Res A Clin Mol Teratol. 2011 Jan;91(1):50-60.
25. Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012 Jan;139(2):231-43.
26. Genevieve D, Captier G. Syndromes avec fentes labio-palatines. P261-282. Syndromes dysmorphiques, 2012. Doin éditeur.
27. Holder-Espinasse M, Abadie V, Cormier-Daire V, Beyler C, Manach Y, Munnich A, Lyonnet S, Couly G, Amiel J. Pierre Robin sequence: a series of 117 consecutive cases. J Pediatr. 2001 Oct;139(4):588-90.
28. Izumi K, Konczal LL, Mitchell AL, Jones MC. Underlying Genetic Diagnosis of Pierre Robin Sequence: Retrospective Chart Review at Two Children’s Hospitals and a Systematic Literature Review. J Pediatr. 2011 Oct 31.
29. Klotz CM, Wang X, Desensi RS, Grubs RE, Costello BJ, Marazita ML. Revisiting the recurrence risk of nonsyndromic cleft lip with or with-out cleft palate. Am J Med Genet A. 2010 Nov;152A(11):2697-702.
30. Wieczorek D. Human facial dysostoses. Clin Genet. 2013 Jun;83(6):499-510.











![SSPT.pdf · "pondre à butea cas éusnt'"iM8. Six niveaux de de décal.es sont distin- gués, donnant lieu à gil évaluztiona du taux d'inva]idité, avec](https://static.fdocuments.fr/doc/165x107/5aafe1e57f8b9a3a038dfc7c/ssptpdfpondre-butea-cas-usntim8-six-niveaux-de-de-dcales-sont-distin-gus-donnant.jpg)






