
Rhumatologie pédiatrique, quelles pathologies, FS AJI ... · Ostéomyélite chronique récurrente...
62
Rhumatologie pédiatrique, quelles pathologies, FS AJI , syndromes auto-inflammatoires maladies auto-immunes, interféronopathies, Quelle organisation de la prise en charge Pierre Quartier Unité d’Immuno-Hématologie et Rhumatologie pédiatrique Necker-Enfants Malades, Paris
Transcript of Rhumatologie pédiatrique, quelles pathologies, FS AJI ... · Ostéomyélite chronique récurrente...

Rhumatologie pédiatrique, quelles pathologies,
FS AJI , syndromes auto-inflammatoires
maladies auto-immunes, interféronopathies,
Quelle organisation de la prise en charge
Pierre Quartier
Unité d’Immuno-Hématologie et Rhumatologie pédiatrique
Necker-Enfants Malades, Paris

Rhumatologie pédiatrique : quelles maladies ?
Arthrites Juvéniles Idiopathiques (AJI)
– Formes non systémiques / Formes systémiques (“Still”)
Connectivites et vascularites “classiques”
– Lupus Erythémateux Disséminé, dermatomyosite juvénile, sclérodermie,
connectivites de chevauchement
– PAN de l’enfant, Kawasaki, vascularites à ANCA, Takayasu, …
Syndromes auto-inflammatoires avec participation prédominante de l’IL-1
– Fièvres récurrentes et assimilé : FMF, TRAPS, Sd hyper-IgD / MVK,
CINCA/Muckle Wells/Urticaire familial au froid, DIRA, DITRA …
– ± ostéomyélite multifocale et SAPHO, péricardites, myocartides récurrentes
– ± maladies granulomateuses inflammatoires de l’enfant (Blau, …)
Maladies monogéniques à expression mixte (inflammation, DI, auto-immunité, …) :
– Interferonopathies : AGS/Lupus engelure, SPENCD, STING, …
– CANDLE (lipodystrophie, inflammation), NLRC4 (auto-inflammation, SAM, …), …

Arthrites juvéniles idiopathiques
Début avant 16 ans, présence d’une arthrite ≥ 6 semaines
Après avoir éliminé toutes autre cause d ’arthrite
• infection +++
• Arthrite réactionnelle
• arthrite s’intégrant dans une pathologie connue
Diagnostics différentiels : MICIs, Lupus/Rhupus,
Hépatites auto-immunes, vascularite systémique, …
Rarement néoplasie a expression articulaire (plus souvent
arthralgies, douleurs métaphysaires) et systémique

AJI : principales entités
La forme systémique (« maladie de Still »)
L’oligoarthrite (< 4 articulations touchées au cours des 6
premiers mois, peut être étendue ensuite) ou la polyarthrite
débutant < 6 ans
– Souvent présence de FAN sans spécificité
– Risque d’uvéite à œil blanc = lampe à fente /3 mois 5 ans
L’arthrite non systémique du plus grand enfant :
– Tableaux d’ « oligoarthrite » révélant une spondylarthropathie
(± enthésite) ou liée au psoriasis (ongles / ATCDs familiaux)
– Polyarthrite rhumatoïde (facteur rhumatoïde, anti-CCP) ou non
– Polyarthrites sèches
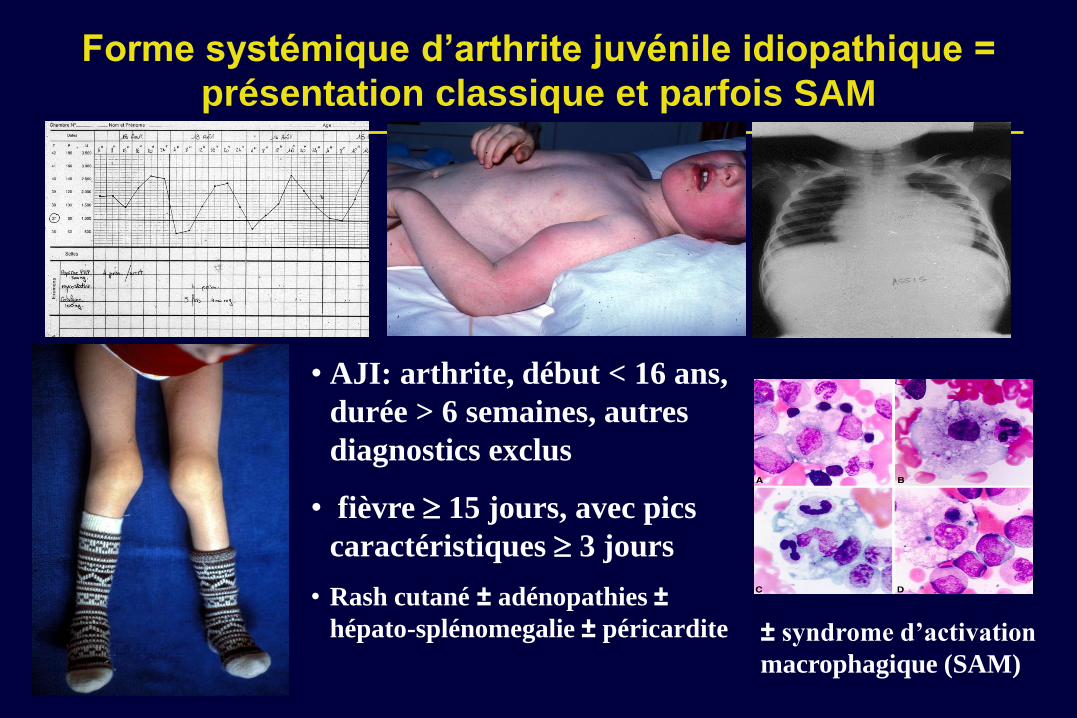
Forme systémique d’arthrite juvénile idiopathique =
présentation classique et parfois SAM
• AJI: arthrite, début < 16 ans,
durée > 6 semaines, autres
diagnostics exclus
• fièvre 15 jours, avec pics
caractéristiques 3 jours
• Rash cutané ± adénopathies ±
hépato-splénomegalie ± péricardite ± syndrome d’activation
macrophagique (SAM)

Phase initiale : prise en charge
Hospitalisation à décider en fonction de la gravité
Traitement anti-inflammatoire adapté (douleur, état général,
signes éventuel de SAM) :
– Anti-inflammatoire non stéroïdien (AINS) à fortes doses,
avec une efficacité particulière pour certains experts de
l’indométacine (3 mg/kg par jour en 2 ou 3 prises) …
attention tolérance, risque de SAM
– Parfois nécessité d’une corticothérapie d’emblée … ou
d’un traitement biologique précoce ? … *
*Contact précoce avec un centre expert

Formes évolutives, la vision « classique »
Formes monocycliques :
– Une seule grande poussée, après quelques mois rémission
complète définitive … 10-40% des patients
Formes polycycliques :
– Plusieurs poussées avec rémission complète entre deux
poussées … 2 à 35% dans la littérature … assez rare si l’on
exige une biologie intercritique normale
Formes persistantes :
– Activité de la maladie durable sur plusieurs années
– Classiquement > 50% des FSAJI, pronostic réservé
– Profil évolutif modifié par traitements récents ?

Formes non monocycliques, évolution
Phase initiale
Inflammatoire
± polyarthrite
Profil évolutif
« auto-inflammatoire »
Peu d’arthrite
Profil évolutif
systémique
et polyarthrite
Profil évolutif
polyarticulaire
sans signes
systémiques cliniques
REMISSION sans traitement ni séquelles

Formes particulières
Formes avec inflammation persistante sur la durée
– risque d’amylose secondaire
– risque d’HTAP
Formes évoluant d’un profil auto-inflammatoire à un profil
auto-immun
– apparition d’auto-ACs variés (FAN, FR, ANCAs, …)
– atteintes multi-organe possible (reins, poumons …)
Formes avec SAM et ferritine le plus souvent élevée
– ± présence de mutations/polymorphismes fonctionnellement
non neutres dans gènes de perforine, Munck 13.4, …
– Formes frontières avec des lymphohistiocytoses familiales ?
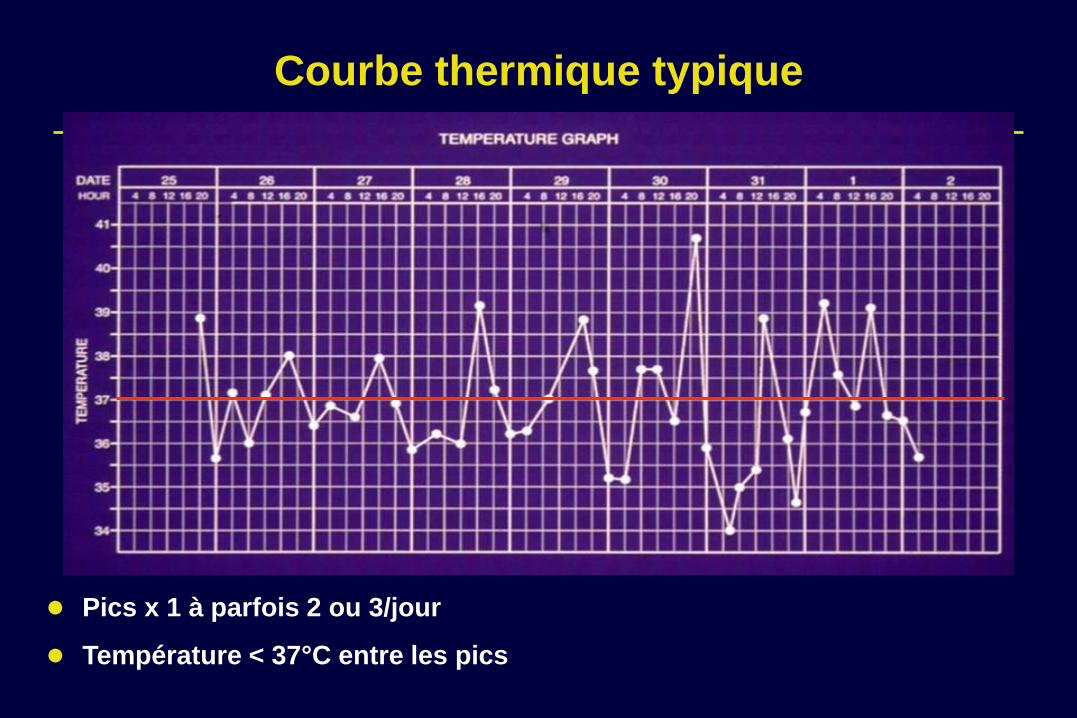
Courbe thermique typique
Pics x 1 à parfois 2 ou 3/jour
Température < 37°C entre les pics
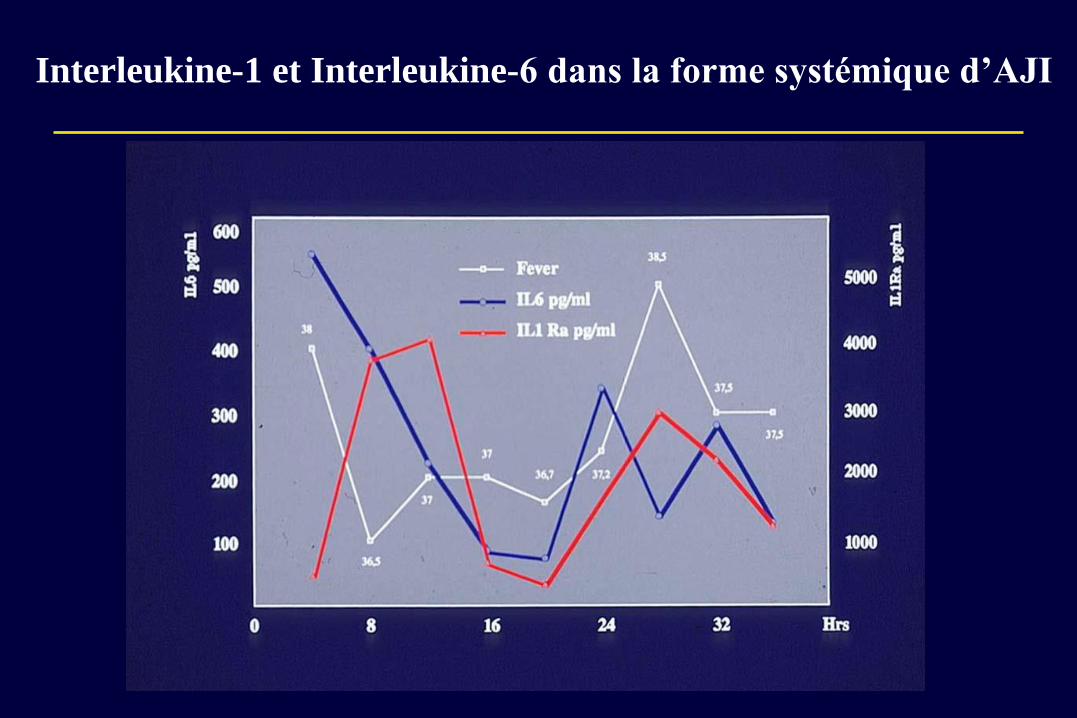
Interleukine-1 et Interleukine-6 dans la forme systémique d’AJI

L’interleukine-6, cytokine clé dans la FS-AJI
Association de taux élevés d’IL-6 dans le sérum et le liquide
articulaire avec l’inflammation systémique, l’érosion
articulaires, le retard de croissance
Modèles murins transgéniques pour l’IL-6 ou son récepteur
– Arthrite érosive
– Inflammation systémique et petite taille
Traitements par un AC anti-récepteur de l’IL-6 (Tocilizumab) =
très efficace chez la plupart des patients sur manifestations
systémiques et articulaires
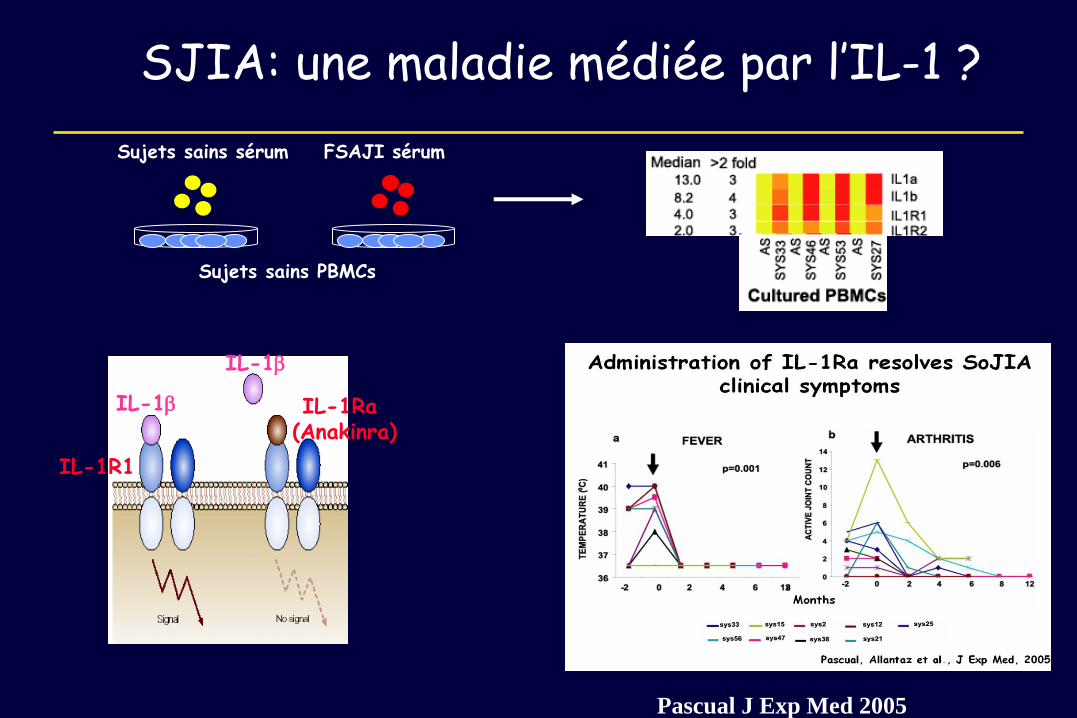
IL-1b
IL-1b
IL-1R1
IL-1Ra (Anakinra)
SJIA: une maladie médiée par l’IL-1 ?
Sujets sains PBMCs
Sujets sains sérum FSAJI sérum
Pascual J Exp Med 2005

AMM / essais en cours
FS-AJI évolutive, échec des AINS et corticostéroïdes :
– Anti-IL-6 :
• Tocilizumab : AMM voie IV 12 mg/kg toutes les 2 semaines
si poids < 30 kg, sinon 8 mg/kg toutes les 2 semaines
• Essai pédiatrique en cours avec le tocilizumab SC
– Anti-IL-1
• Utilisation hors AMM de l’anakinra
• Etudes récemment publiées pour le rilonacept
• AMM pour le canakinumab 4 mg/kg (maxi 300 mg) SC /4
semaines … mais prise en charge par l’assurance maladie
en attente (très cher)
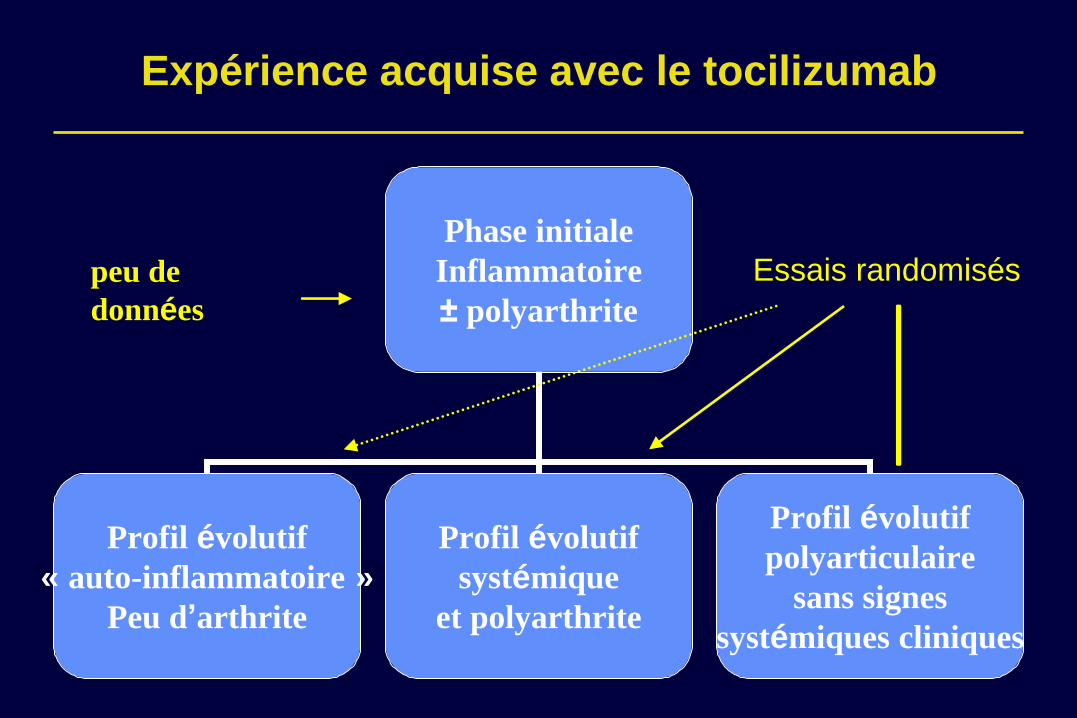
Expérience acquise avec le tocilizumab
Phase initiale
Inflammatoire
± polyarthrite
Profil évolutif
« auto-inflammatoire »
Peu d’arthrite
Profil évolutif
systémique
et polyarthrite
Profil évolutif
polyarticulaire
sans signes
systémiques cliniques
peu de
données
Essais randomisés
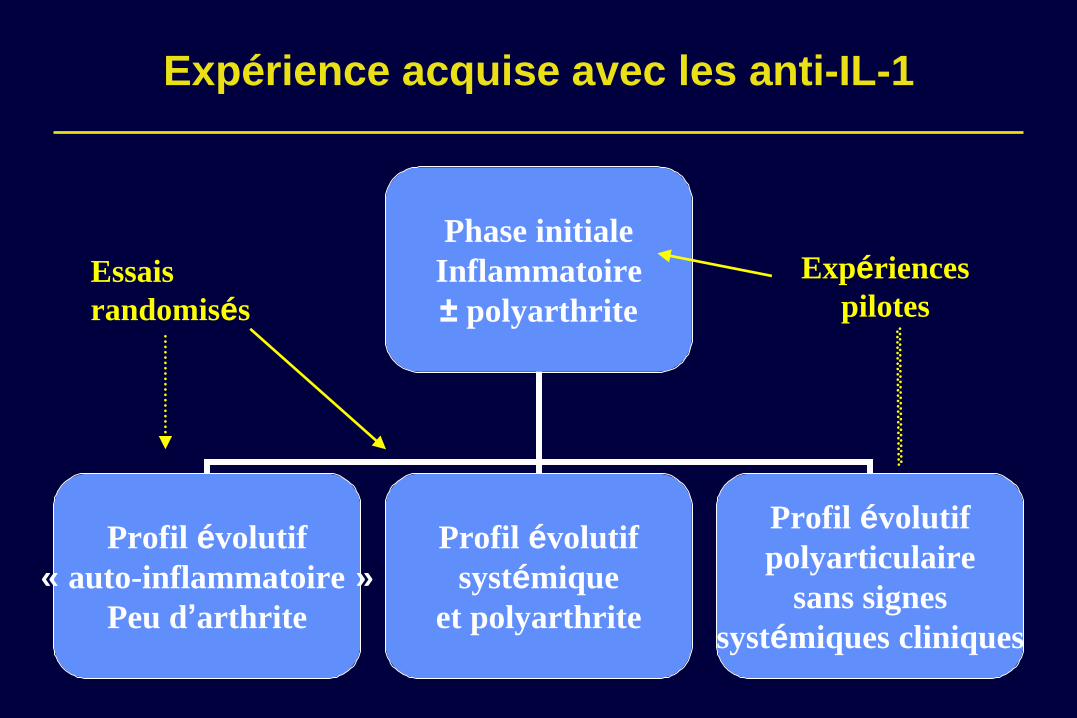
Expérience acquise avec les anti-IL-1
Phase initiale
Inflammatoire
± polyarthrite
Profil évolutif
« auto-inflammatoire »
Peu d’arthrite
Profil évolutif
systémique
et polyarthrite
Profil évolutif
polyarticulaire
sans signes
systémiques cliniques
Essais
randomisés
Expériences
pilotes

Bilan pré-thérapeutique (MTX ou biologique)
Viscéral à minima : biologie rénale, hépatique, NFS plaquettes
Infectieux
Optimiser la situation vaccinale :
– Compléter en amont vaccins vivants importants comme
ROR, varicelle
– Vacciner et faire rappel régulier pour DTP, hémophilus,
pneumocoque, méningo C, grippe
– Egalement conseillés : hépatite B, papillomavirus, autres
selon situation géographique/voyages
Et s’assurer de la bonne information/éducation
parents/patient

Le revers de la médaille des biologiques
Infections
Allergies (dont chocs anaphylactiques sous tocilizumab IV)
Atteintes hépatiques variées
Atteintes ± liées en tout ou partie à la maladie (SAM, HTAP, …)
Atteintes dysimmunes
– Atteintes psoriasiques
– Atteintes neurologiques démyélinisantes
– Atteintes intestinales inflammatoires (de type Crohn ou
colite indifférentiée
Risque de néoplasie ?

Syndromes auto-inflammatoires
Syndromes très IL-1 dépendants
– DIRA = déficit en antagoniste du récepteur de l’IL-1
– Cryopyrinopathies (Urticaire familial au froid, Muckle-Wells,
CINCA), Fièvre méditerranéenne familiale (en échec de
colchicine), autres dont des péri ou myocardites récurrentes
Syndromes plus complexes
– Parfois répondant à l’anti-IL-1 : déficit en mévalonate kinase,
forme systémique d’AJI, ± ostéomyélite multifocale, ± Blau
– Parfois autres abords thérapeutiques nécessaires

Présentation clinique
– Episodes récurrents de fièvre
– avec inflammation localisée aux niveaux de
différents organes (peau, articulations, séreuses..)
– Formes héréditaires et acquises

Formes « aquises »
– Maladie de Still
– PFAPA
– Maladie de Behçet
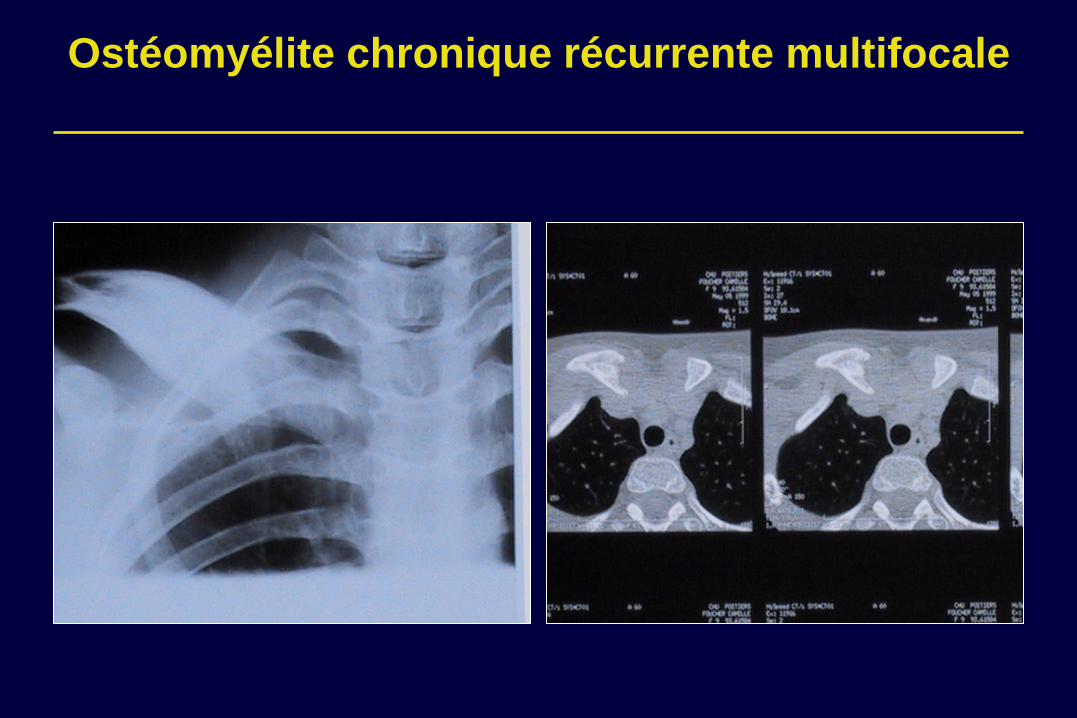
Ostéomyélite chronique récurrente multifocale

Formes héréditaires
• Transmission autosomique récessive: – FMF (pyrine/marenostrine)
• Population méditerranéenne • Épisode de fièvre et DOULEUR ADBO • Bref: 48h-72h
– Hyper IgD syndrome (mévalonate kinase) • 5 à 7 j • début précoce • Aphtes, adp, rash
– DIRA (Deficiency of interleukin 1 receptor antagonist)
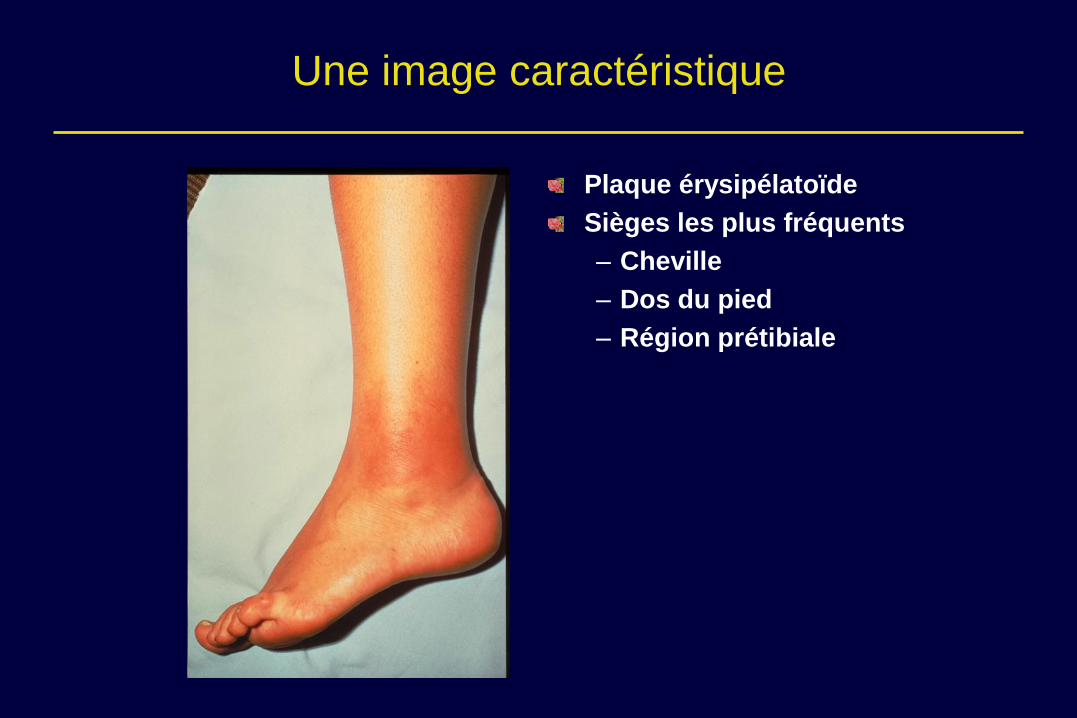
Une image caractéristique
Plaque érysipélatoïde
Sièges les plus fréquents
– Cheville
– Dos du pied
– Région prétibiale
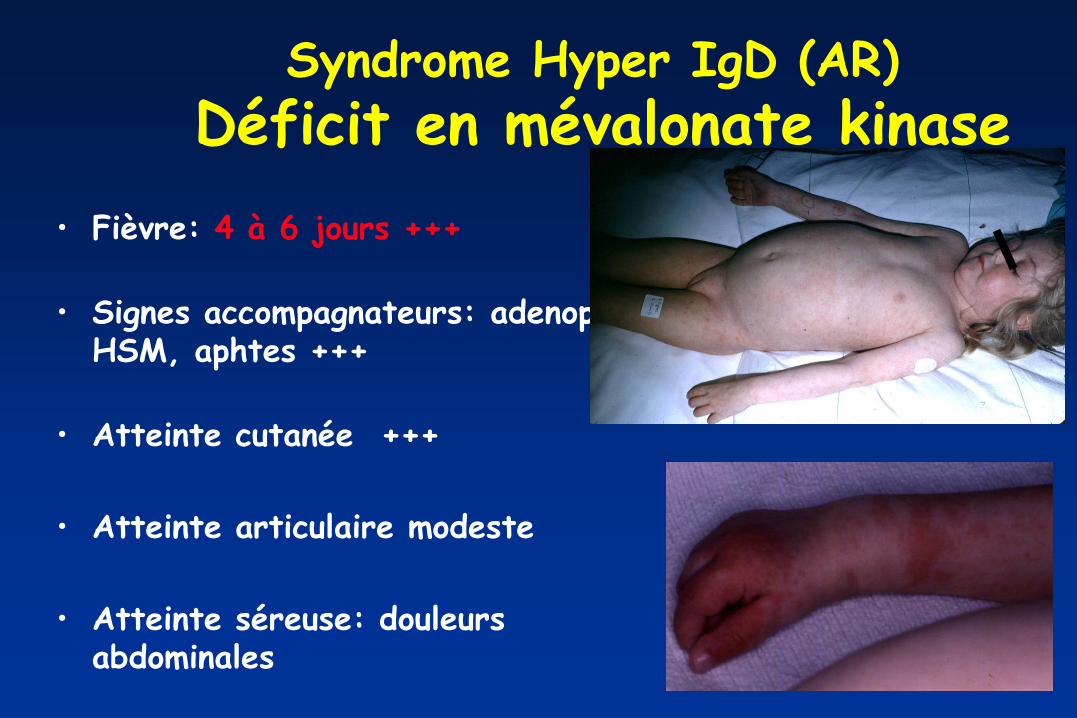
Syndrome Hyper IgD (AR)
Déficit en mévalonate kinase
• Fièvre: 4 à 6 jours +++
• Signes accompagnateurs: adenop,
HSM, aphtes +++
• Atteinte cutanée +++
• Atteinte articulaire modeste
• Atteinte séreuse: douleurs abdominales

Déficit en mévalonate kinase
• Acidurie mévalonique en poussée
• Dosage des IgD: peu utile
• Activité mévalonate kinase sur lymphocytes ou fibroblastes
• Génétique: séquençage du gène de la mévalonate kinase

• 9 patients décrits (Aksentijevich et al, NEJM;360, 2009)
• Début très précoce
• Maladie à expression cutanée et articulaire
DIRA (Deficiency of interleukin 1 receptor antagonist)

Pustulose localisée à généralisée Biopsie cutanée: infiltration du derme et de l’épiderme par des neutrophiles, formation de pustules le long des follicules pileux, hyperkératose
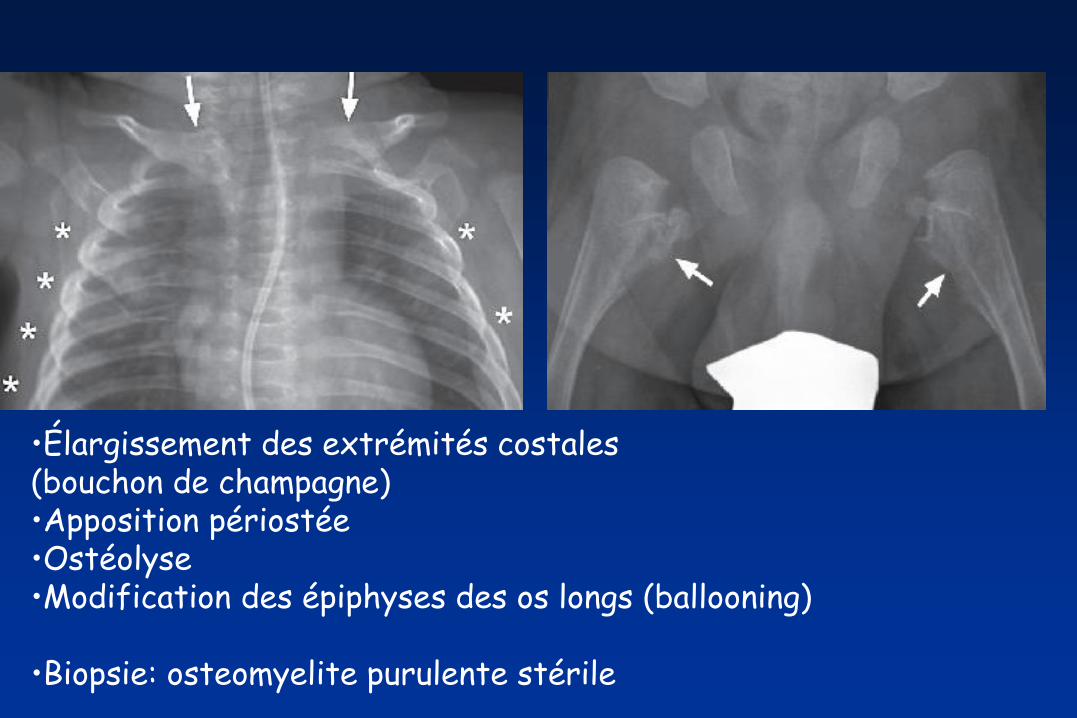
•Élargissement des extrémités costales (bouchon de champagne) •Apposition périostée •Ostéolyse •Modification des épiphyses des os longs (ballooning)
•Biopsie: osteomyelite purulente stérile
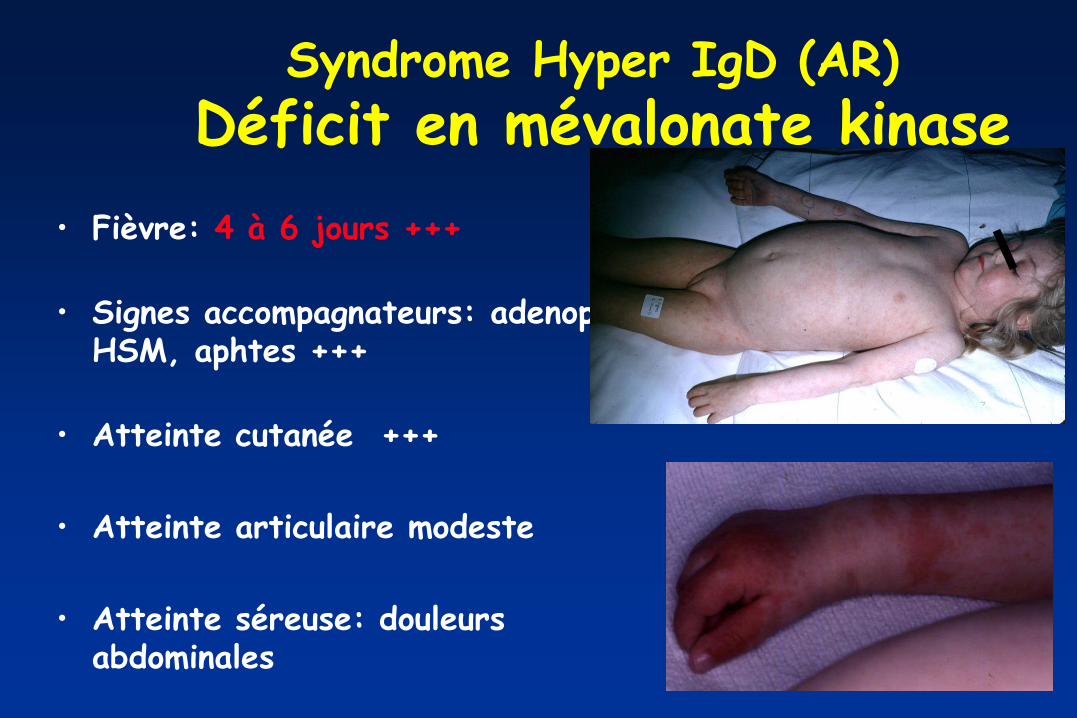
Syndrome Hyper IgD (AR)
Déficit en mévalonate kinase
• Fièvre: 4 à 6 jours +++
• Signes accompagnateurs: adenop,
HSM, aphtes +++
• Atteinte cutanée +++
• Atteinte articulaire modeste
• Atteinte séreuse: douleurs abdominales

Déficit en mévalonate kinase
• Acidurie mévalonique en poussée
• Dosage des IgD: peu utile
• Activité mévalonate kinase sur lymphocytes ou fibroblastes
• Génétique: séquençage du gène de la mévalonate kinase

• Transmission autosomique dominante: – TRAPS
– FCAS/MWS/CINCA (CIAS1-cryopyrine)
– Fièvre périodique associé à mutation de NALP12
– Syndrome de Blau (NOD2) – PAPA syndrome (Pyogenic arthritis pyoderma
gangrenosum acne) (CD2BP1, CD2 binding prot 1)

TRAPS: histoire naturelle
• Age de début plus tardif: enfance ou adolescence
• Fréquence des accès fébriles variables: 2-3/an
• Épisodes de fièvre prolongés: 14-21 jours
• Risque d’amyloidose à long terme
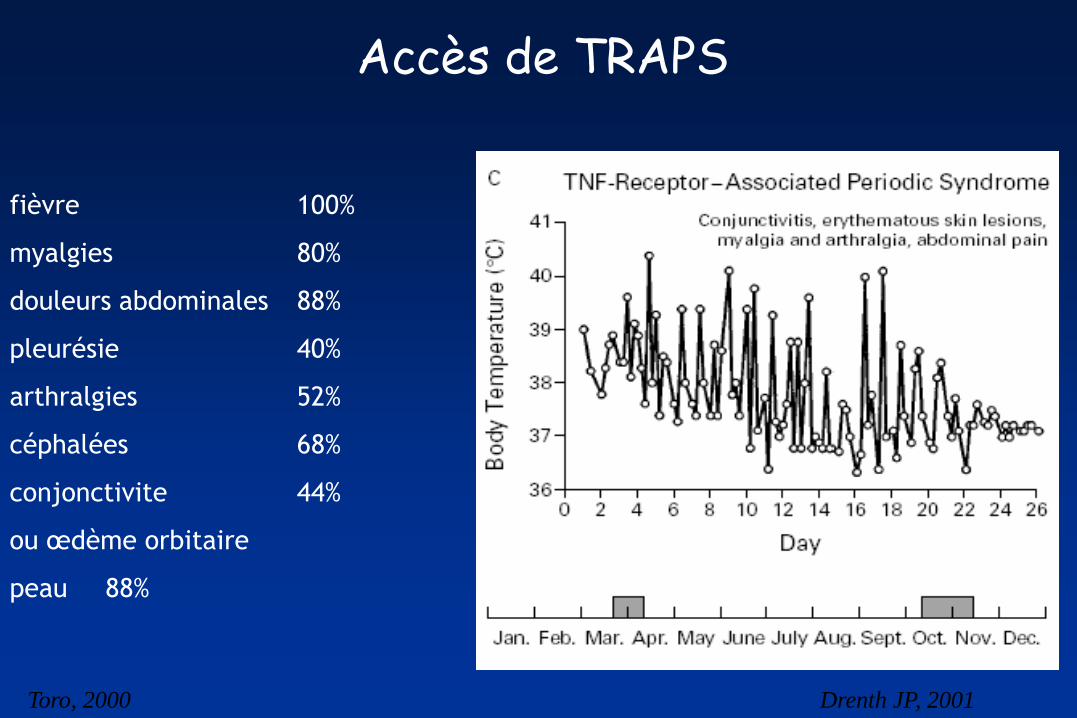
Accès de TRAPS
fièvre 100%
myalgies 80%
douleurs abdominales 88%
pleurésie 40%
arthralgies 52%
céphalées 68%
conjonctivite 44%
ou œdème orbitaire
peau 88%
Toro, 2000 Drenth JP, 2001
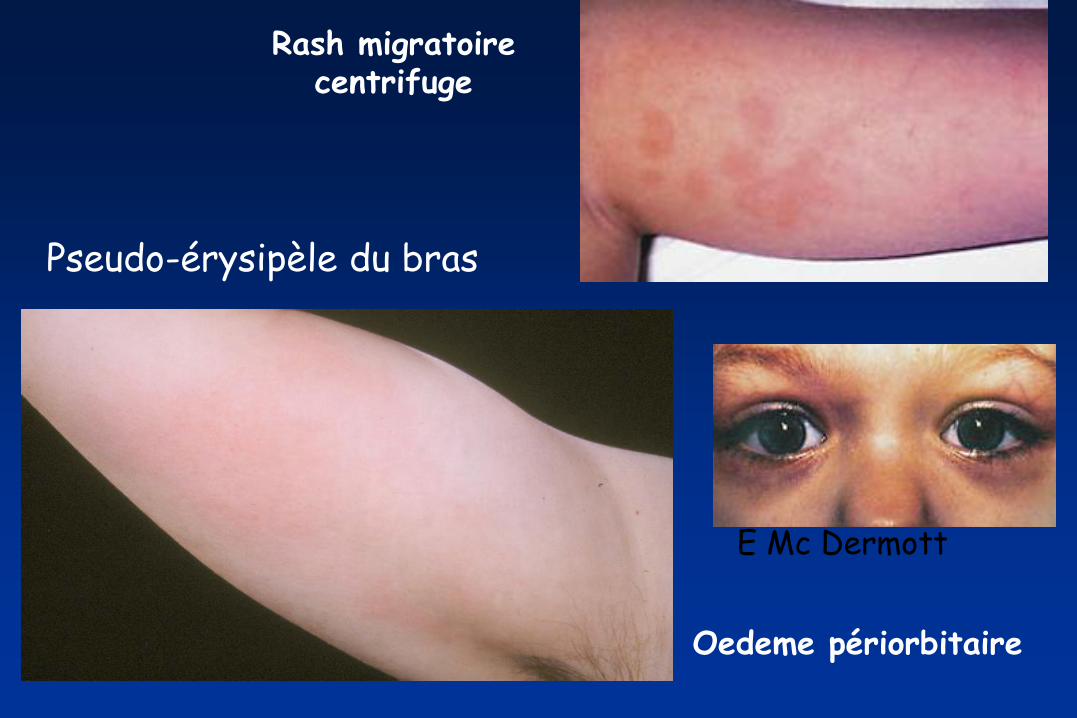
Pseudo-érysipèle du bras
E Mc Dermott
Rash migratoire centrifuge
Oedeme périorbitaire
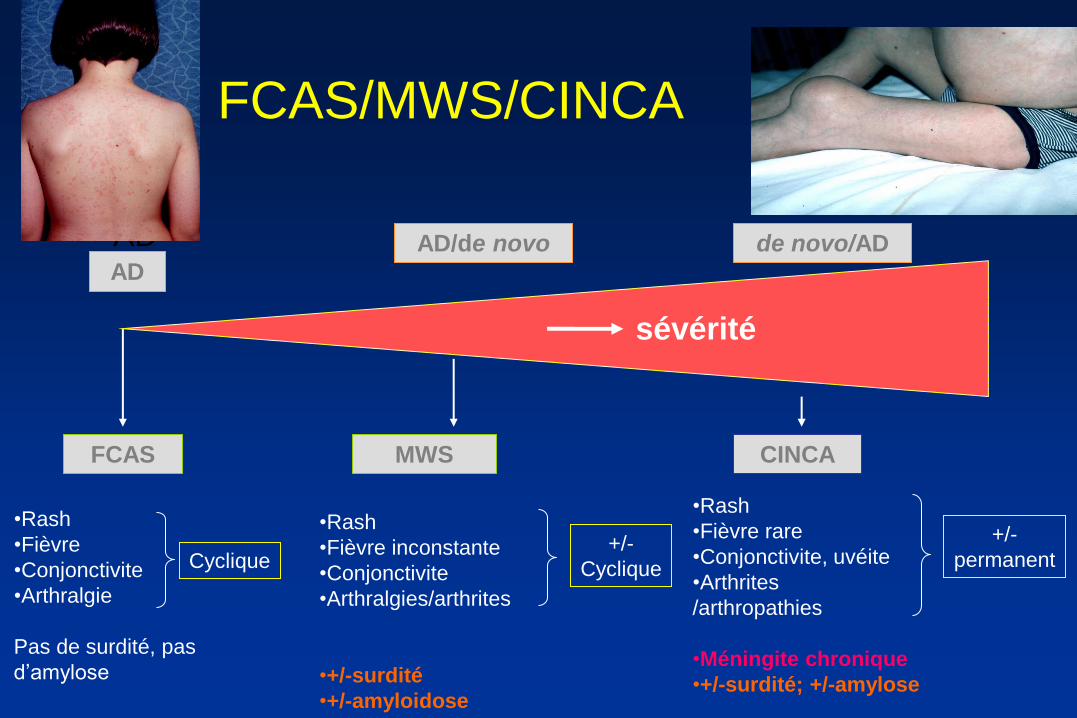
FCAS/MWS/CINCA
FCAS
•Rash
•Fièvre
•Conjonctivite
•Arthralgie
Pas de surdité, pas
d’amylose
MWS
•Rash
•Fièvre inconstante
•Conjonctivite
•Arthralgies/arthrites
•+/-surdité
•+/-amyloidose
CINCA
sévérité
•Rash
•Fièvre rare
•Conjonctivite, uvéite
•Arthrites
/arthropathies
•Méningite chronique
•+/-surdité; +/-amylose
+/-
Cyclique Cyclique
+/-
permanent
AD AD
AD/de novo de novo/AD
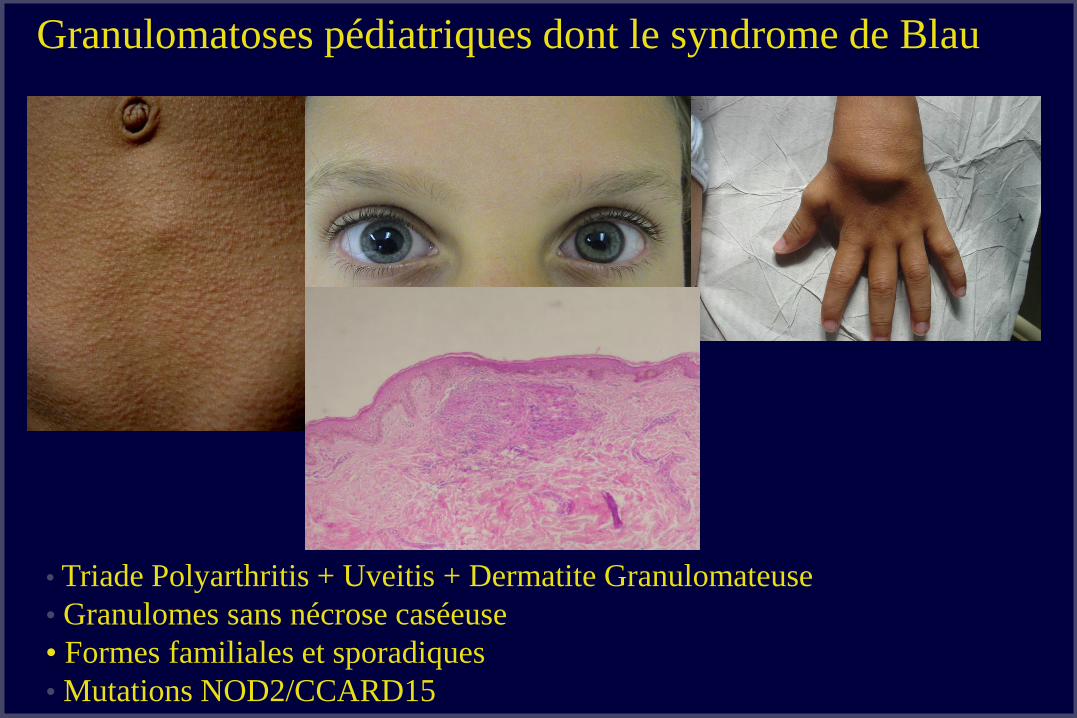
• Triade Polyarthritis + Uveitis + Dermatite Granulomateuse
• Granulomes sans nécrose caséeuse
• Formes familiales et sporadiques
• Mutations NOD2/CCARD15
Granulomatoses pédiatriques dont le syndrome de Blau
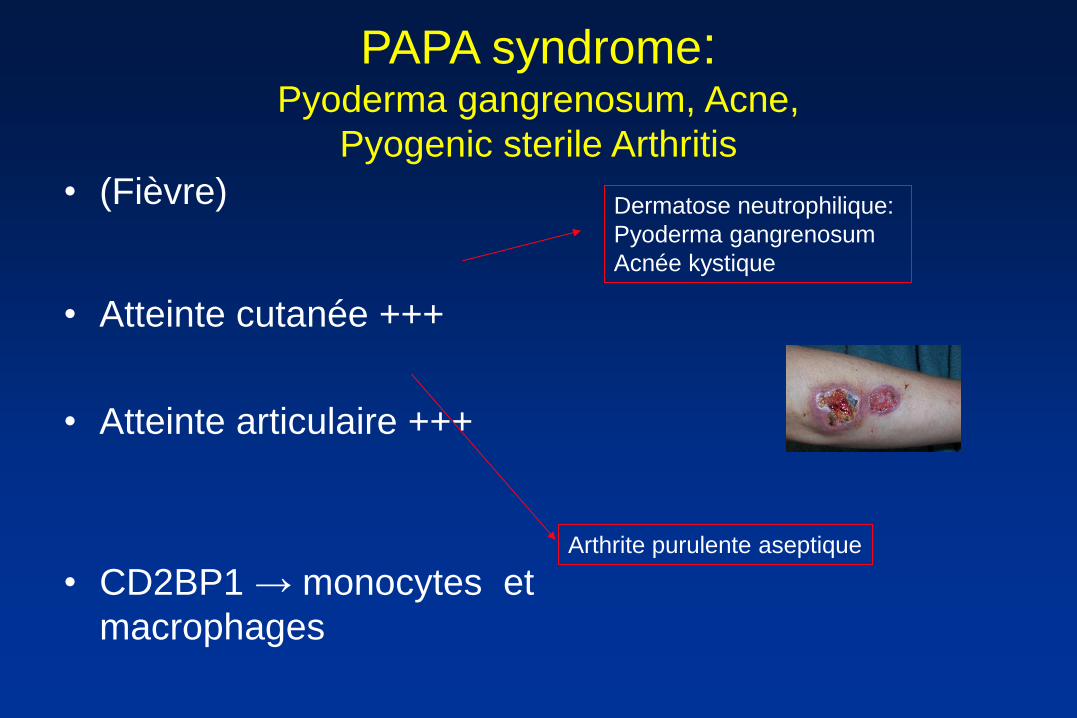
PAPA syndrome: Pyoderma gangrenosum, Acne,
Pyogenic sterile Arthritis
• (Fièvre)
• Atteinte cutanée +++
• Atteinte articulaire +++
• CD2BP1 → monocytes et
macrophages
Dermatose neutrophilique:
Pyoderma gangrenosum
Acnée kystique
Arthrite purulente aseptique

En pratique…

• Anamnèse : ethnie, consanguinité, type de fièvre, … âge de début: – MVK/ CAPS: début précoce+++ < 1 an
– FMF : petite enfance
– TRAPS: enfance/adolescence
• Diagnostics différentiels: infection/tumeur
Examens complémentaires syndrome inflammatoire en poussée
normal en dehors dosage de l’acidurie mévalonique en poussée
génétique …

Connectivites / Vascularites
Lupus « classiques »
Sclérodermies
Myosites juvéniles
Maladies monogéniques à expression auto-immune /
inflammatoire

Myosite inflammatoire de l’enfant
Dermatomyosite: 85%
2 à 3 / 106 enfants (UK, USA)
Myosite de chevauchement:3-10%
Polymyosite: 2-5%

Diagnostic
Critères de Bohan et Peter
Signes musculaires
Déficit proximal
Elévation d’au moins un enzyme musculaire:
CPK, ASAT, LDH, aldolase
Atteinte myogène à l’EMG
Biopsie musculaire
+
Signes cutanés
Signes cutanés + ¾ critères: diagnostic certain

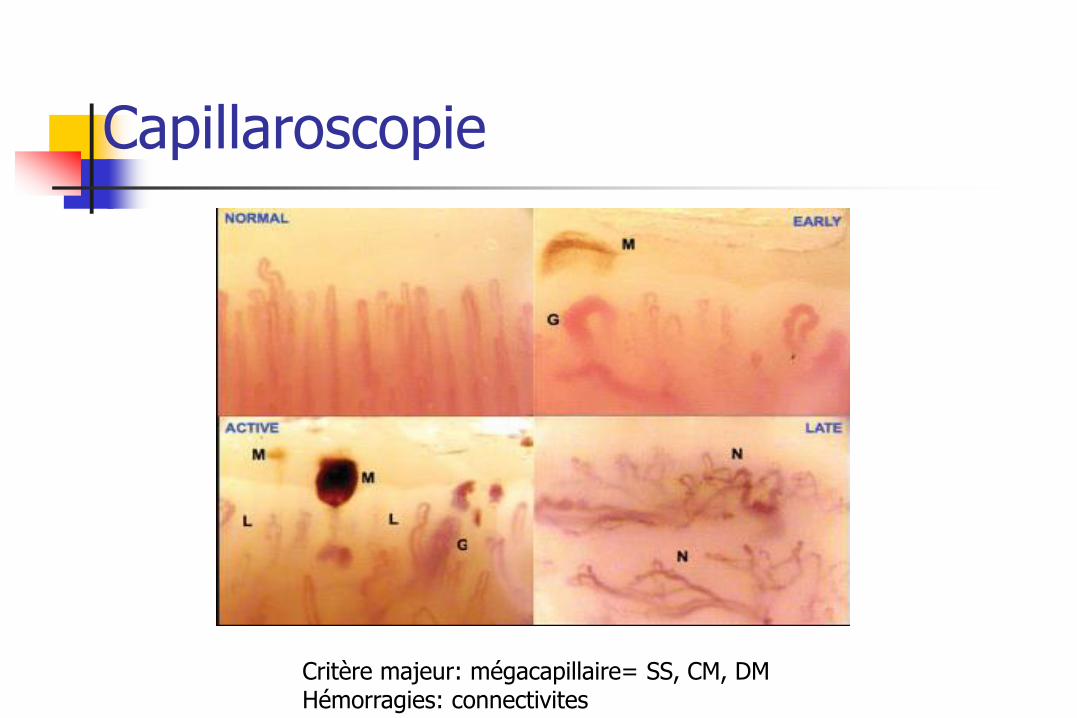
Capillaroscopie
Critère majeur: mégacapillaire= SS, CM, DM Hémorragies: connectivites
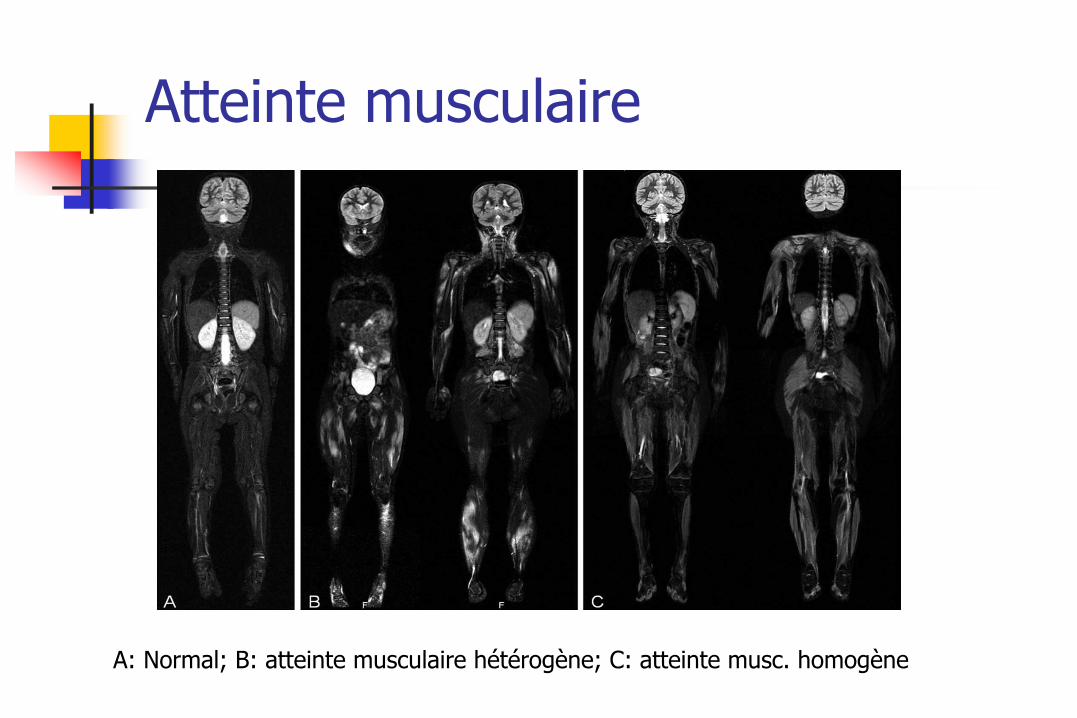
Atteinte musculaire
A: Normal; B: atteinte musculaire hétérogène; C: atteinte musc. homogène

Quand réaliser une biopsie musculaire ?
« facile »,
systématique en l’absence de signes cutanés
Deltoïde (à distance d’un site d’injection vaccinale++)
Expertise anapath nécessaire

Atteinte inflammatoire
Infiltrat CD3, macrophage endomysial, périmysial, périvasculaire
Atteinte vasculaire
Perte capillaire ++
Epaississement, oblitération artérielle, infarctus
Atteinte musculaire
Hyperexpression HLA I
Atrophie périfasciculaire
Dégérérescence/
régénération
Atteinte tissu conjonctif
Fibrose endomysiale, périmysiaale
Atteintes histologiques
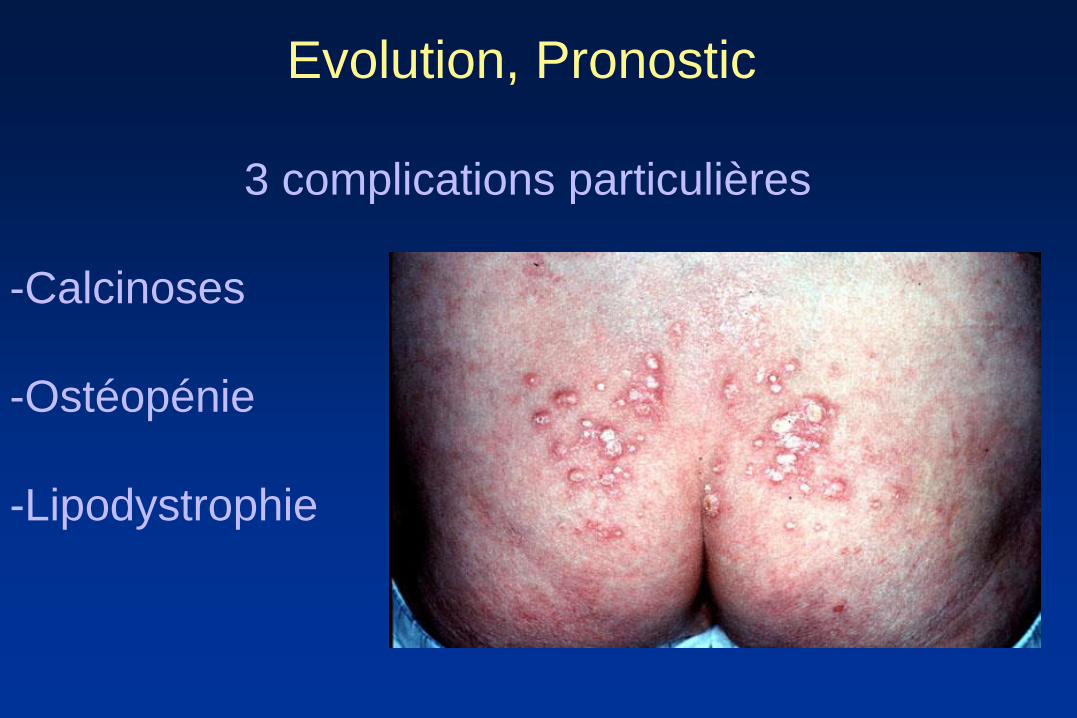
Evolution, Pronostic
3 complications particulières
-Calcinoses
-Ostéopénie
-Lipodystrophie
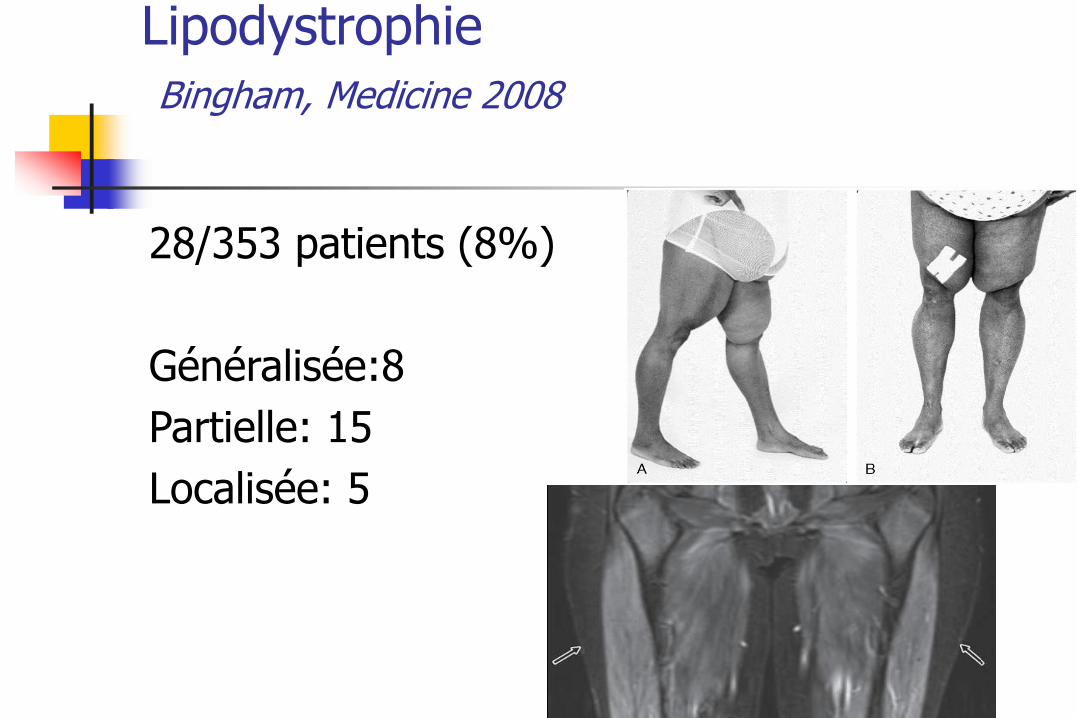
Lipodystrophie Bingham, Medicine 2008
28/353 patients (8%)
Généralisée:8
Partielle: 15
Localisée: 5

Anticorps spécifiques des myosites chez l’enfant
AC anti-synthétase (cytoplasmic aminoacyl-tRNA synthetase (ARS) enzymes): anti-JO1, PL7, PL12: 1-3% Mains de « mécanicien », Gottron, fièvre, Raynaud, arthrite, PI. Patients plus âgés AC antiTIF1-g (p155)—nuclear transcription cellular differentiation: 25-30%: atteinte cutanée sévère? Evolution chronique Anti-Mi-2 (Helicase protein—nuclear transcription): <10%; atteinte cutanée classique, atteinte musculaire modérée, bonne réponse au tt Anti-MJ: crampes, dysphonie, monocyclique AC anti MDA5 (Melanoma differentiation-associated gene 5): ulcérations cutanée, papules palmaire, ulcérations buccales . fréquence et gravité de l’atteinte pulmonaire
Hétérognéité

Prise en charge (centre expert +++)
Formes classiques : corticothérapie, méthotrexate, kinésithérapie
Formes partiellement réfractaires ou trop corticodépendantes : MMF, rituximab, …
Formes particulières, digestives, ulcératives … parfois échanges plasmatiques, IGIV, ritux

Lupus syndromiques
Brigitte Bader-Meunier
Yannick Crow

LES systémique pédiatrique
• Rare : 15-20% LES
• Plus sévère que chez l’adulte : atteintes rénales, neurologiques
• Implications plus forte de facteurs génétiques ? – Porteurs d’allèles de risque de développer un LES
– Déficits en complément (fractions précoces de la voie classique)
– Autres
• LES syndromiques

LES syndromique
1. Mutations
• TREX (syndrome d’Aicardi-Goutières)
• TRAP
2. Anomalies chromosomiques
• Klinefelter et autres
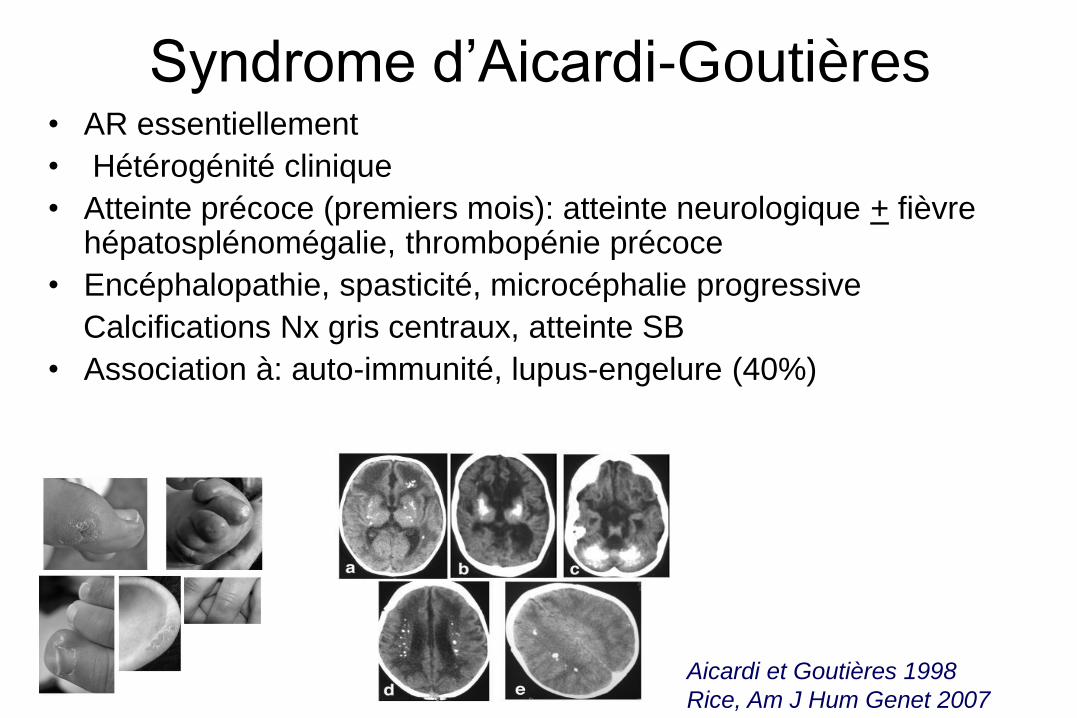
Syndrome d’Aicardi-Goutières • AR essentiellement
• Hétérogénité clinique
• Atteinte précoce (premiers mois): atteinte neurologique + fièvre hépatosplénomégalie, thrombopénie précoce
• Encéphalopathie, spasticité, microcéphalie progressive
Calcifications Nx gris centraux, atteinte SB
• Association à: auto-immunité, lupus-engelure (40%)
Aicardi et Goutières 1998
Rice, Am J Hum Genet 2007

Mutations de TREX 1
(3’-repair DNA exonuclease 1)
• Syndrome d’Aicardi-Goutières
AGS1 3p21 TREX1/DNaseIII 35%
AGS2 13q14 RNASEH2B 45%
AGS3 11q13 RNASEH2C 15%
AGS4 19p13 RNASEH2A <5%
AGS5 20q11 SAMHD1/DCIP ~10%
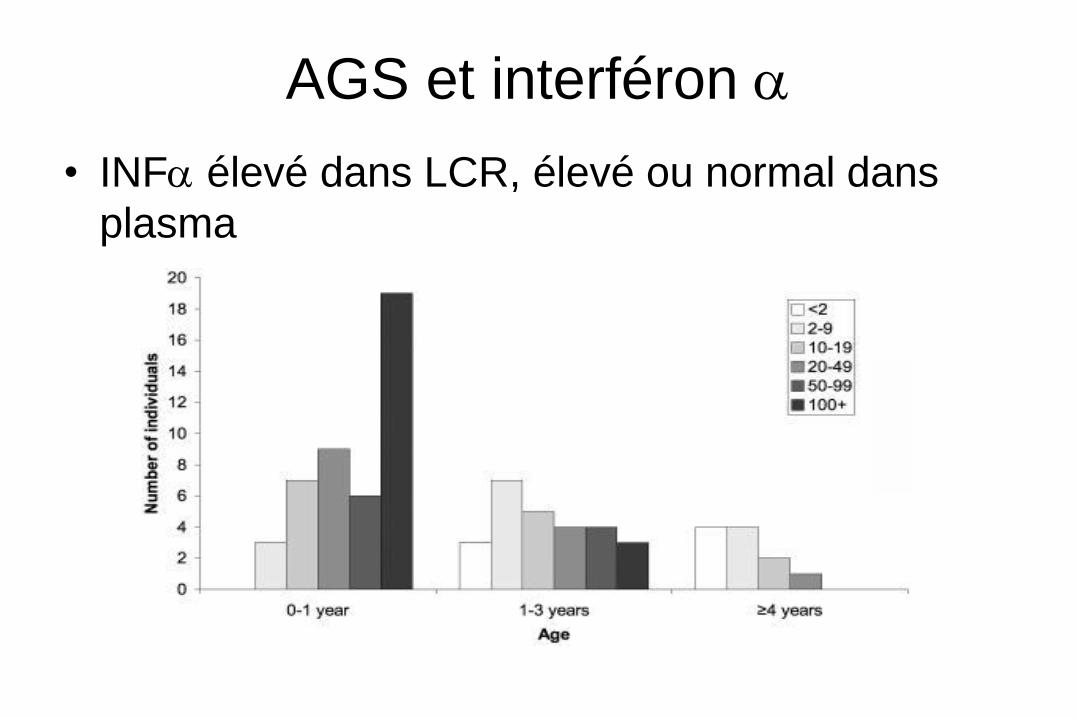
AGS et interféron a
• INFa élevé dans LCR, élevé ou normal dans
plasma
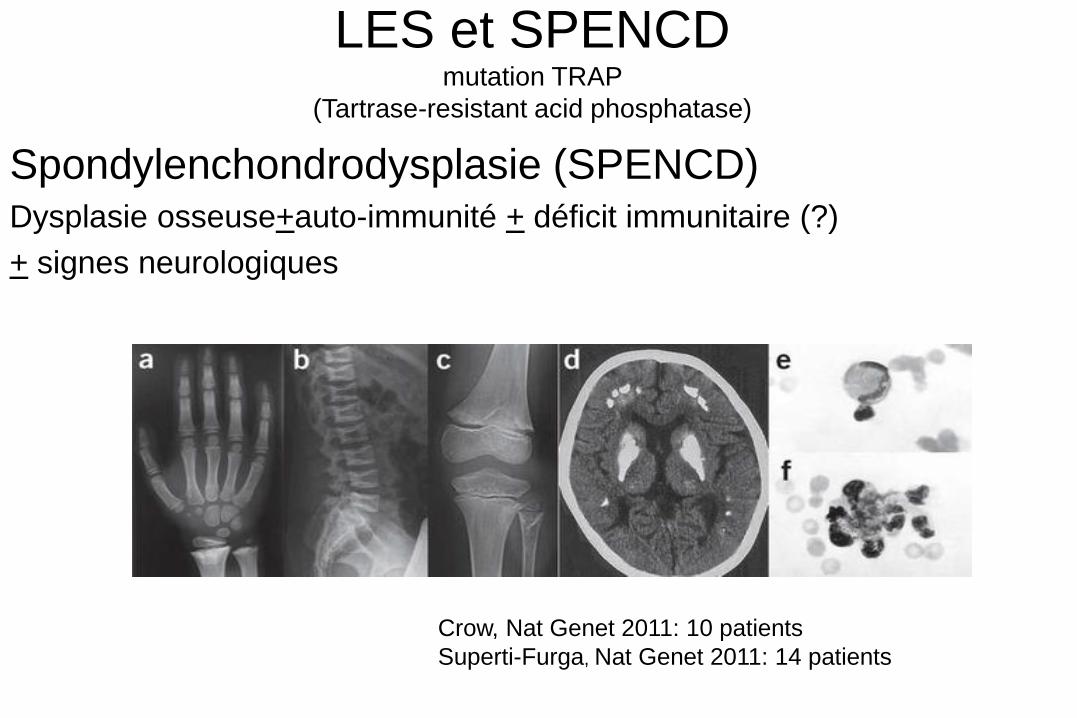
LES et SPENCD mutation TRAP
(Tartrase-resistant acid phosphatase)
Spondylenchondrodysplasie (SPENCD) Dysplasie osseuse+auto-immunité + déficit immunitaire (?)
+ signes neurologiques
Crow, Nat Genet 2011: 10 patients
Superti-Furga, Nat Genet 2011: 14 patients
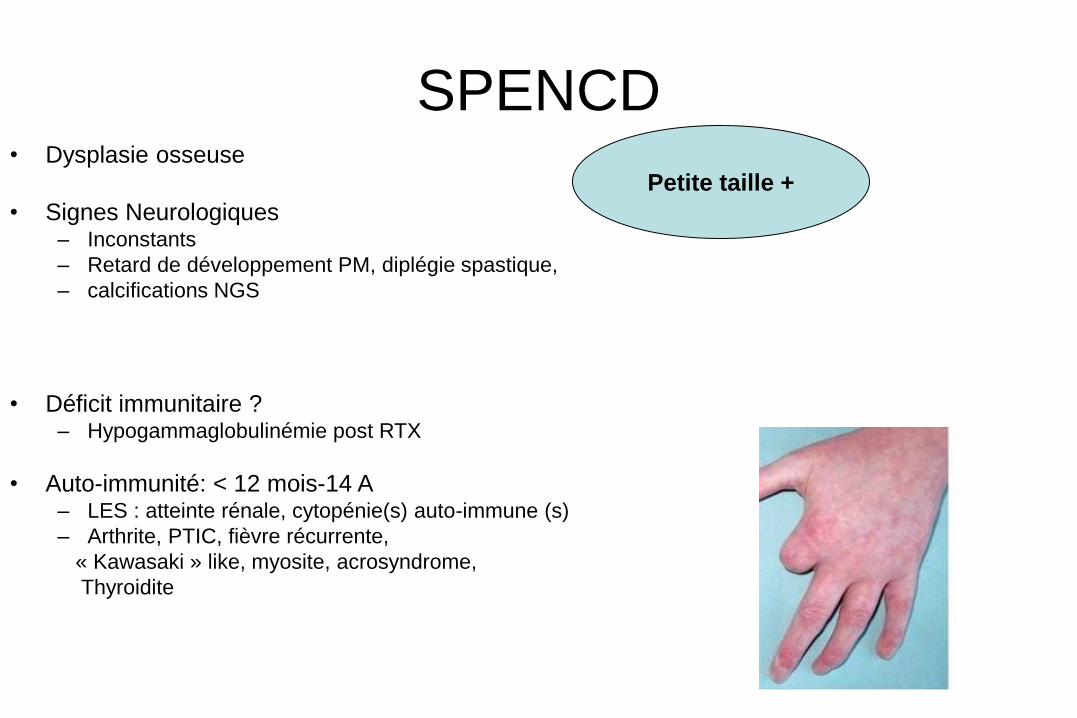
SPENCD • Dysplasie osseuse
• Signes Neurologiques – Inconstants
– Retard de développement PM, diplégie spastique,
– calcifications NGS
• Déficit immunitaire ? – Hypogammaglobulinémie post RTX
• Auto-immunité: < 12 mois-14 A – LES : atteinte rénale, cytopénie(s) auto-immune (s)
– Arthrite, PTIC, fièvre récurrente,
« Kawasaki » like, myosite, acrosyndrome,
Thyroidite
Petite taille +
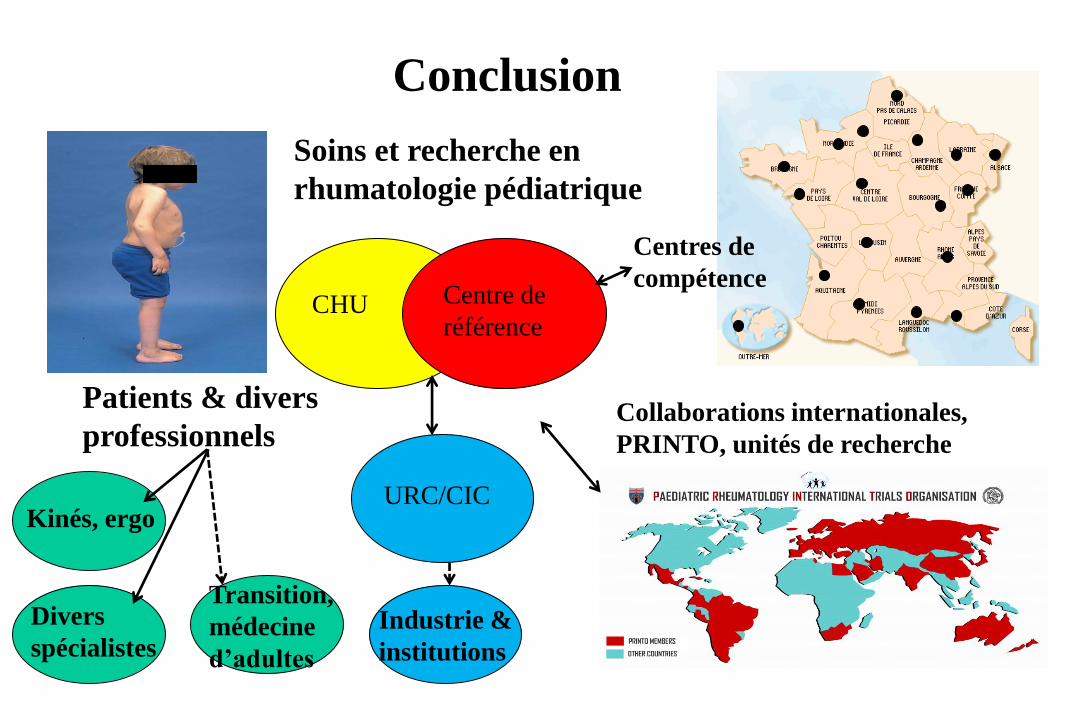
Conclusion
CHU
Collaborations internationales,
PRINTO, unités de recherche
Centre de
référence
URC/CIC Kinés, ergo
Soins et recherche en
rhumatologie pédiatrique
Patients & divers
professionnels
Divers
spécialistes
Transition,
médecine
d’adultes
Industrie &
institutions
Centres de
compétence

La rhumatologie pédiatrique France / International
En France :
– 2 CMR, 18 centres de compétences (certains multi-sites :
www.cerhumip.fr)
– une filière maladies rares avec 5 CMR d’adultes
– une société savante, la SOFREMIP, avec congrès annuel
– un DIU de rhumatologie pédiatrique Paris-Lyon
International :
– une société savante, la PReS, avec congrès annuel
– Young Investigator meeting, sessions pour « trainees »
– Réseau PRINTO pour recherche / enseignement des jeunes


















