
PROTÉOMIQUE STRUCTURALE DE … · Mise en contexte et biologie de Methanobacterium...
101
ISABELLE GIGNAC PROTÉOMIQUE STRUCTURALE DE METHANOBACTERIUM THERMOAUTOTROPHICUM; STRUCTURE ET FONCTION D’UNE PROTÉINE CLASSIFIÉE PRÉSERVÉE DANS LE GÉNOME Mémoire présenté à la Faculté des études supérieures de l'Université Laval dans le cadre du programme de maîtrise en biochimie pour l’obtention du grade de maître ès Sciences (M.Sc.) FACULTÉ DES SCIENCES ET DE GÉNIE UNIVERSITÉ LAVAL QUÉBEC AVRIL 2004 © Isabelle Gignac, 2004
Transcript of PROTÉOMIQUE STRUCTURALE DE … · Mise en contexte et biologie de Methanobacterium...

ISABELLE GIGNAC
PROTÉOMIQUE STRUCTURALE DE METHANOBACTERIUM
THERMOAUTOTROPHICUM; STRUCTURE ET FONCTION D’UNE PROTÉINE CLASSIFIÉE PRÉSERVÉE DANS LE GÉNOME
Mémoire présenté à la Faculté des études supérieures de l'Université Laval dans le cadre du programme de maîtrise en biochimie
pour l’obtention du grade de maître ès Sciences (M.Sc.)
FACULTÉ DES SCIENCES ET DE GÉNIE UNIVERSITÉ LAVAL
QUÉBEC
AVRIL 2004
© Isabelle Gignac, 2004

i
Résumé La fonction d’une protéine est ultimement déterminée par sa structure
tridimensionnelle. La compréhension complète de la fonction d’une protéine implique
donc la connaissance de sa structure. Le grand nombre de projet de séquençage de
génome résulte en plusieurs dizaines de milliers de séquences de protéines pour
lesquelles il n’existe aucune information fonctionnelle. La protéomique structurale
implique l’étude de la structure tridimensionnelle de toutes les protéines d’un protéome.
Comme la détermination de la structure d’une protéine est un processus laborieux, la
protéomique structurale est un des plus grands défis scientifiques du 21e siècle. Malgré
cela, plusieurs projets de protéomique structurale ont récemment débuté à travers le
monde.
Ce mémoire présente une étude qui s’inscrit dans le cadre d’un de ces projets à
grande échelle, la protéomique structurale de Methanobacterium thermoautotrophicum.
À l’aide de la résonance magnétique nucléaire, la structure tridimensionnelle de
MTH187, une protéine de Methanobacterium thermoautotrophicum, a été déterminée.
MTH187 est classifié comme ayant une séquence conservée, et sa fonction est inconnue.
La structure de MTH187 révèle que cette protéine de 111 acides aminés est composée de
six hélices α de 10 à 14 résidus. Par homologie structurale, il a été possible de classifier
MTH187 parmi une famille structurale de type « HEAT repeat ». Les protéines de cette
famille possèdent une fonction de reconnaissance protéines-protéines.

ii
Avant-Propos
Je tiens tout d’abord à remercier mon directeur de recherche, le Dr Stéphane Gagné
qui m’a accueilli dans son laboratoire et qui a eu confiance en moi tout au long de cette
maîtrise. Sa grande connaissance scientifique, sa présence et son dévouement m’ont
permis de franchir une étape des plus importante dans ma vie. Je le remercie également
pour sa franchise. Elle m’a permis de me connaître davantage. Je remercie mes collègues
de travail et amis, Pierre-Yves Savard, Jean-Baptiste Duvignaud, Leigh Willard, Katia
Lecours et Olivier Julien qui m’ont aidée et encouragée. Un grand merci en autre à
Olivier Julien, Etienne Noumen et Jonathan Pellicelli pour le travail supplémentaire
effectué sur mes résultats. Je tiens à remercier le laboratoire de Cheryl H. Arrowsmith de
l’Université de Toronto pour l’expression et la purification de la protéine. Également le
Centre National de RMN (NANUC) pour l’acquisition de certains spectres. Finalement,
un très gros merci à mon père et ma mère pour leurs amours, leurs aides et leurs
encouragements durant ces nombreuses années d’études. Vous m’avez permis de
persévérer dans ce que je rêvais de faire. Merci aussi à mon copain François pour son
soutien, son aide et sa compréhension dans les décisions prises depuis de nombreuses
années.

iii
Table des matières
CHAPITRE 1. INTRODUCTION...................................................................................... 1 1.1. Vue d’ensemble du mémoire ................................................................................... 1 1.2. Protéomique structurale ........................................................................................... 1 1.3. Cristallographie et RMN.......................................................................................... 2 1.4. Mise en contexte et biologie de Methanobacterium thermoautotrophicum ............ 3 1.5. Protéine MTH187 .................................................................................................... 5 1.6. Buts du projet de maîtrise ........................................................................................ 7
CHAPITRE 2. DÉTERMINATION DE LA STRUCTURE DES PROTÉINES PAR RÉSONANCE MAGNÉTIQUE NUCLÉAIRE ................................................................. 9
2.1. Niveaux de structure des protéines .......................................................................... 9 2.2. Principes de base en RMN..................................................................................... 11 2.3. Stratégies utilisées pour déterminer la structure des protéines par RMN.............. 14
2.3.1. Expression, marquage isotopique et purification des protéines...................... 14 2.3.2. Préparation des échantillons ........................................................................... 16 2.3.3. Acquisition des spectres RMN........................................................................ 17 2.3.4. Transformation des spectres en vue de leurs analyses.................................... 22 2.3.5. Contraintes structurales................................................................................... 23 2.3.6. Dynamique des protéines................................................................................ 28 2.3.7. Calcul de structures tridimensionnelles .......................................................... 29
CHAPITRE 3. MATERIELS ET METHODES............................................................... 32 3.1. Expression, marquage isotopique et purification de MTH187 .............................. 32 3.2. Préparation des échantillons .................................................................................. 33
3.2.1. Contenu des échantillons ................................................................................ 33 3.2.2. Concentration de la protéine dans l'échantillon .............................................. 34
3.3. Résonance magnétique nucléaire........................................................................... 34 3.4. Contraintes structurales.......................................................................................... 39
3.4.1. Contraintes NOEs ........................................................................................... 39 3.4.2. Contraintes d'angles dièdres............................................................................ 40
3.5. Génération de structures tridimensionnelles.......................................................... 41 3.6. Homologie structurale............................................................................................ 42
CHAPITRE 4. ATTRIBUTION DES SPECTRES RMN ET DÉTERMINATION DE LA STRUCTURE SECONDAIRE DE MTH187................................................................... 43
4.1. Attributions des spectres RMN.............................................................................. 43 4.1.1. Détermination des déplacements chimiques de la chaîne squelettique .......... 43 4.1.2. Attribution des déplacements chimiques des chaînes latérales...................... 47 4.1.3. Détermination des déplacements chimiques des résidus aromatiques........... 49
4.2. Synthèse de l’attribution ........................................................................................ 51 4.3. Structure secondaire de MTH187 .......................................................................... 53
CHAPITRE 5. STRUCTURE DE MTH187 ................................................................... 56 5.1. Calcul de structures de MTH187 ........................................................................... 56
5.1.1. Calculs avec CNS ........................................................................................... 56 5.1.2. Calcul avec ARIA/CNS .................................................................................. 57 5.1.3. Structures finales de MTH187........................................................................ 58

iv
5.1.4. Qualité des structures...................................................................................... 60 CHAPITRE 6. HOMOLOGIE STRUCTURALE ............................................................ 68 CHAPITRE 7. DISCUSSION .......................................................................................... 73
7.1. Attribution des déplacements chimiques de MTH187........................................... 73 7.2. Structure de MTH187 ............................................................................................ 74 7.3. Homologie structurale............................................................................................ 76
CHAPITRE 8. CONCLUSION ........................................................................................ 78 BIBLIOGRAPHIE............................................................................................................ 79 ANNEXE I........................................................................................................................ 82 ANNEXE II ...................................................................................................................... 83

v
Liste des équations Equation 1 : Équation de base en RMN. ν représente la fréquence de précession de
Lamor,(MHz), Bz est le champs magnétique. Le rapport gyromagnétique est une constante, qui dépend de la nature du noyau. ........................................................... 17
Equation 2: Relation entre le NOE (nuclear Overhauser effet) et la distance entre deux protons....................................................................................................................... 20
Equation 3 : Equation qui permet de connaître la constance de couplage 3JHNHα à partir du rapport d’intensité des pics et du délai d’acquisition des spectres (δ). ..................... 26
Equation 4: Équation de la RMSD (root mean square deviation). N est le nombre d’échantillon, i est la sommation sur chacun des échantillons. ri et ri
représente les ׳structure..................................................................................................................... 31
Equation 5 : I(t) est l’intensité après le délai t et I0 est l’intensité au t=0. ........................ 36

vi
Liste des tableaux
Table 1 : Table énumérant les paramètres d’acquisitions des différentes expériences RMN ......................................................................................................................... 38
Table 2 : Comparaison des méthodes de détermination de structures secondaires de MTH187.................................................................................................................... 55
Table 3 : Table des RMSD de la structure finale de MTH187 ......................................... 59 Table 4 : Statistiques structurales des structures RMN de MTH187 a.............................. 67 Table 5: Protéines identifiées par DALI comme étant structuralement homologues à
MTH187.................................................................................................................... 68 Table 6: Superposition des régions de MTH187 avec Aconitase B ................................. 70

vii
Liste des figures
Figure 1-1 : Histogramme représentant le nombre de gènes exclus de l’analyse de protéomique structurale (figure reproduite de ((Christendat et al., 2000))................. 4
Figure 1-2: Alignement de séquences des sous-unités α des phycocyanines phycocyanobilines lyases extraites de COG1413 généré avec ClustalW 1,74 (www.ch.embnet.org/software/ClustalW.html). La première colonne cite les abréviations des protéines de la famille COG1413. Les quatre premières protéines, dont la protéine étudiée MTH187, sont des protéines de Methanobacterium thermoautotrophicum d’où l’abréviation MTH. Le nombre à la suite est le numéro du gène. Les protéines suivantes proviennent des bactéries Pseudomonas aeruginosa, Bacillus subtilis, Bacillus halodurans, Mesorhizobium loti, Synechocystis sp. BOXSHADE (www.ch.embnet.org/software/BOX_form.html) est utilisé pour mettre en évidence les acides aminés identiques (boîtes noires) et semblables (boîtes grises). .......................................................................................... 6
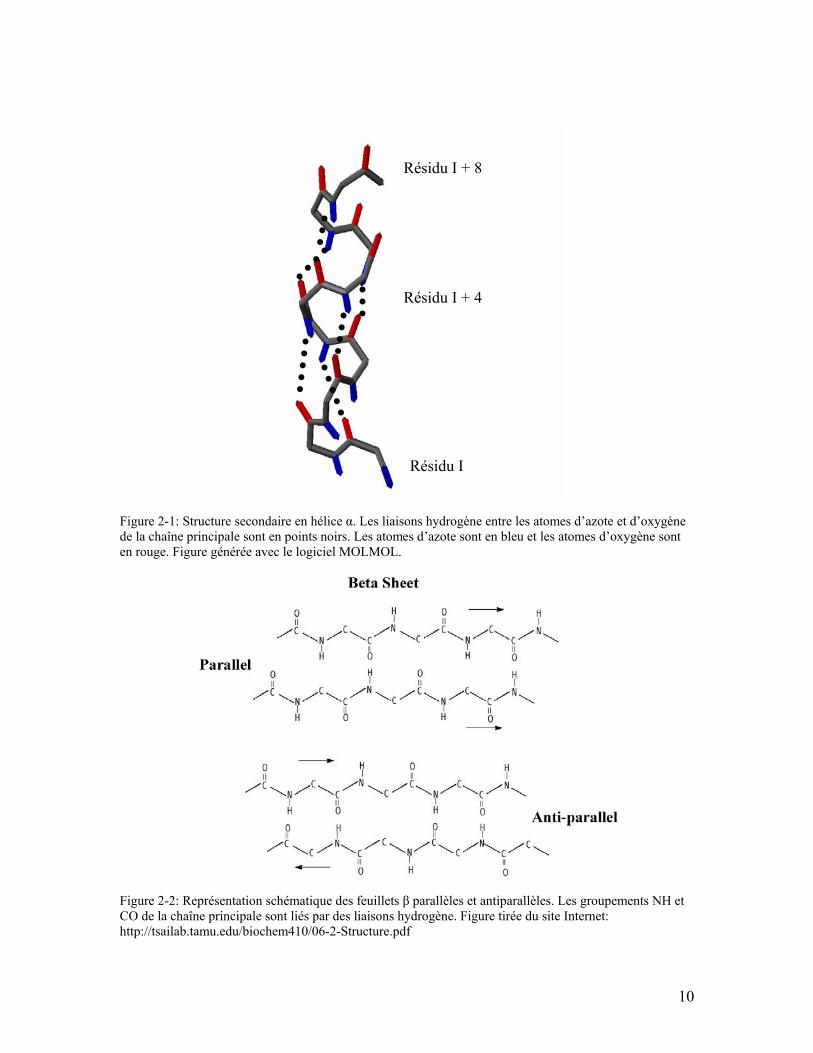
Figure 2-1: Structure secondaire en hélice α. Les liaisons hydrogène entre les atomes d’azote et d’oxygène de la chaîne principale sont en points noirs. Les atomes d’azote sont en bleu et les atomes d’oxygène sont en rouge. Figure générée avec le logiciel MOLMOL................................................................................................................. 10
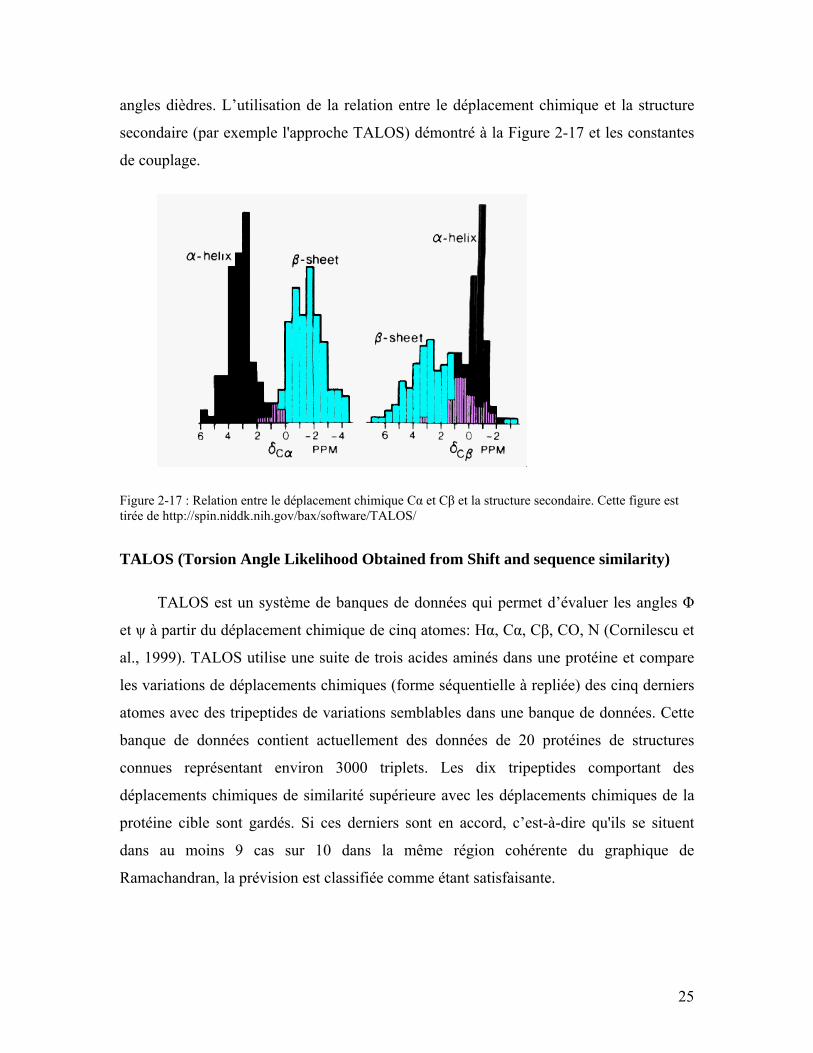
Figure 2-2: Représentation schématique des feuillets β parallèles et antiparallèles. Les groupements NH et CO de la chaîne principale sont liés par des liaisons hydrogène. Figure tirée du site Internet: http://tsailab.tamu.edu/biochem410/06-2-Structure.pdf................................................................................................................................... 10
Figure 2-3: Niveaux d’énergies et orientations des états de spins nucléaires du proton en absence et en présence d’un champ magnétique. La différence de population entre les deux niveaux d’énergies est beaucoup plus faible qu’illustré dans cette figure. (Figure tirée de www.rmn.uhp-nancy.fr/Mutzenhardt/RMNSV2CM1.pdf) ............ 12
Figure 2-4: Effets d’un champ magnétique externe sur un échantillon RMN. A) Transfert de la magnétisation le long des axes x et y. reproduite du livre ((Wüthrich, 1986). B) Rotation de la magnétisation autour de l’axe z.(reproduite du livre (Sanders, 1987).................................................................................................................................... 12
Figure 2-5: Retour à l’équilibre du moment magnétique avec la magnétisation orientée parallèlement à B0 (www.med.univ-rennes1.fr/cerf/edicerf/BASES/BA004_cv_rb_9.html). ........................................... 13
Figure 2-6: Signal RMN se traduisant par une décroissance du signal détecté en fonction du temps nommé FID (free induction decay). .......................................................... 13
Figure 2-7: Fonctions sinusoïdales FID où est appliquée une transformée de Fourier. (Figure tirée de www.rmn.uhp-nancy.fr/Mutzenhardt/RMNSV2CM1.pdf) ............ 14
Figure 2-8 : Diagramme qui résume les étapes de la détermination de structures tridimensionnelles d’une protéine par la méthode de résonance magnétique nucléaire (RMN)....................................................................................................................... 15
Figure 2-9: Déplacements chimiques observés pour les différents types de protons. Le spectre 1D a été enregistré dans une solution aqueuse. La FID est transformée par transformée de Fourier. Cette figure est reproduite de (Cavanagh et al., 1996)....... 18

viii
Figure 2-10: Corrélations obtenues lors de l’analyse d’un spectre 3D 15N-TOCSY-HSQC................................................................................................................................... 20
Figure 2-11: Corrélations obtenues lors de l’analyse d’un spectre 3D CBCA(CO)NH... 21 Figure 2-12: Corrélations obtenues lors de l’analyse d’un spectre 3D HNCACB. .......... 21 Figure 2-13: Corrélations obtenues lors de l’analyse d’un spectre 3D HNCO................. 21 Figure 2-14: Corrélations obtenues lors de l’analyse d’un spectre 3D HN(CA)CO. ....... 21 Figure 2-15 : Corrélations obtenues lors de l’analyse d’un spectre 3D HCCH-TOCSY . 22 Figure 2-16: Représentation d’une chaîne polypeptidique. L’angle de rotation autour de la
liaison N-Cα est l’angle phi(Φ) et celui autour de la liaison Cα-C’ est l’angle psi(ψ).Figure reproduite de http://www.ujf-grenoble.fr/BIO/Gurvan/phi_psi.html. 24
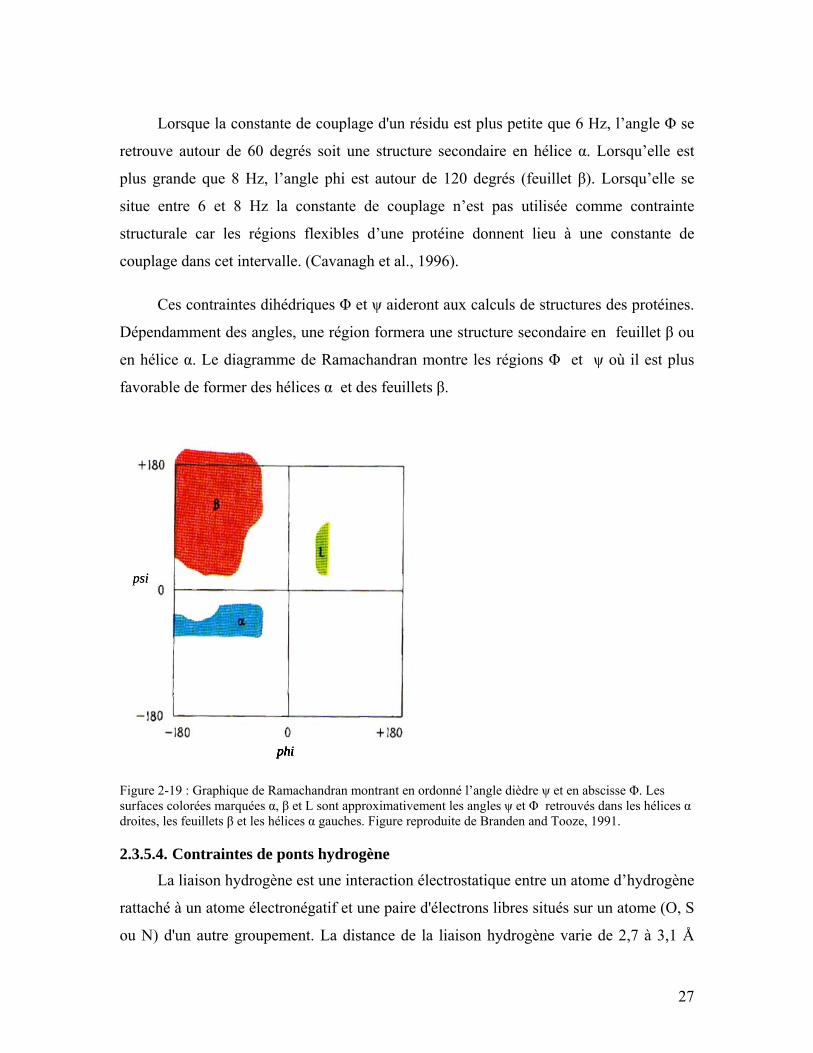
Figure 2-17 : Relation entre le déplacement chimique Cα et Cβ et la structure secondaire. Cette figure est tirée de http://spin.niddk.nih.gov/bax/software/TALOS/................ 25
Figure 2-18: Graphique de Karplus qui décrit la variation de la constante de couplage 3JHNHα avec l’angle dièdre Φ de la chaîne squelettique. ........................................... 26
Figure 2-19 : Graphique de Ramachandran montrant en ordonné l’angle dièdre ψ et en abscisse Φ. Les surfaces colorées marquées α, β et L sont approximativement les angles ψ et Φ retrouvés dans les hélices α droites, les feuillets β et les hélices α gauches. Figure reproduite de Branden and Tooze, 1991......................................... 27
Figure 2-20: Schéma qui représente la dynamique moléculaire d’un calcul de structures par recuit simulé (MDSA). a) représente le point de départ correspondant à un minimum d’énergies locales. b) montre qu’une augmentation de température permet de diminuer les barrières d’énergies et ainsi franchir une barrière plus facilement. c) représente un refroidissement lent de la température. En d) le minimum d’énergie globale est atteint. Figure tirée de www.bip.bham.ac.uk/osmart/bcm311_pef/slide27.html. ......................................... 30
Figure 3-1 : Vecteur d’expression pET-15b ..................................................................... 32 Figure 3-2: Région du vecteur d’expression pET-15b. (Novagen).................................. 33 Figure 3-3 : Script de transformation du spectre HCCH-TOCSY. Le symbole # désactive
la fonction qui suit et \ annonce une continuité de ligne.......................................... 37 Figure 4-1 : Spectres tridimensionnels de MTH187. 3D 15N-TOCSY-HSQC en beige, et
3D 15N-NOESY-HSQC en bleu. F1(1H), F3(1HN) montrés sur différent F2(15N). Les lignes blanches sur un même déplacement chimique en F1 relient les mêmes corrélations................................................................................................................ 43
Figure 4-2 : Spectres tridimensionnels de MTH187. CBCA(CO)NH en beige et HNCACB en bleu. F1(13C), F3(1HN) montrés sur différent F2(15N). Les lignes blanches sur un même déplacement chimique en F1 relient les mêmes corrélations. Le spectre HNCACB montre les corrélations Cα en noire et les Cβ en rouge. ........ 44
Figure 4-3: Spectres tridimensionnels de MTH187. HNCO en beige et HN(CA)CO en bleu. F1(CO), F3(1HN) montrés sur différent F2(15N). .............................................. 45
Figure 4-4: Plan 2D F1(13C), F3(1HN) du spectre tridimensionnel HN(CO)CA du résidu Phe 99 de la protéine MTH187. Le plan 15N est au déplacement chimique 126,86 ppm. .......................................................................................................................... 46
Figure 4-5 : Expansion du spectre 2D 13C-HSQC de MTH187 illustrant la région caractéristique des corrélations Hα-Cα. Les axes montrent les déplacements chimiques (δ) des atomes en partie par millions (ppm). Dans ce spectre à haute

ix
résolution où le couplage scalaire C-C est présent, les Cα des glycines donnent lieu à des doublets alors que les Cα des autres acides aminés donnent lieu à des triplets.. 47
Figure 4-6: Région sélectionnée F1(1H)-F3(1HN) du spectre 15N-TOCSY-HSQC de MTH187 situé à 118.7 ppm sur l’axe F2(15N).Les corrélations observées pour la leucine 31 sont indiquées. Les corrélations entre l’amide et les protons β sont très faibles, et les corrélations avec les protons γ et δ sont absentes dans ce spectre. ..... 48
Figure 4-7: Région sélectionnée F1(1H)-F3(1H) du spectre HCCH-TOCSY de MTH187 situé à 40.6 ppm sur l’axe F2(13C). Les corrélations observées pour la leucine 31 sont indiquées. Les corrélations entre les protons β et les protons γ sont très faibles dans ce spectre................................................................................................................... 48
Figure 4-8: Région sélectionnée F1(13C)-F3(1HN) du spectre CC(CO)NH de MTH187 situé à 108.8 ppm sur l’axe F2(15N). Les corrélations observées pour la leucine 31 sont indiquées. Les corrélations entre l’amide et les Cα, Cβ, Cγ et Cδ sont très bien visibles dans ce spectre. ............................................................................................ 49
Figure 4-9: Spectre 2D HCCH-COSY de MTH187, F3(1H)-F2(13C). La protéine MTH187 est dans une solution 100% D2O. Les corrélations positives (noires) correspondent au 13C ayant un nombre impair de carbone voisin et les corrélations négatives (bleu) au 13C ayant un nombre pair de carbone voisin. ...................................................... 50
Figure 4-10: Plan F1, F3 du spectre 3D HCCH-COSY de MTH187. MTH187 est dans une solution D2O. Les pics de la diagonale représentent les corrélations des protons et eux-même. Le proton Hδ corrèle avec le proton Hε du résidu 107. Les corrélations entre les protons Hζ2 et Hη2 du résidu 36 et Hδ-Hε du résidu 49 sont montré dans cette figure. ............................................................................................................... 51
Figure 4-11: : Spectre 2D 15N-HSQC de MTH187. Ce spectre montre les corrélations entre le proton (1H) et son 15N. Les pics reliés représentent les corrélations des deux protons avec leur azote des arginines et des asparagines.......................................... 52
Figure 4-12: Graphique montrant le résultat de l’analyse CSI pour la protéine MTH187. Les déplacements chimiques des 1Hα, 13Cα ,13Cβ ont été utilisés. La structure secondaire est résumée dans cette figure. Lorsque y =1, la structure secondaire favorisée est le feuillet β et lorsque y = -1 la structure secondaire favorisée est l’hélice. D’après l’analyse CSI, MTH187 est composée de 6 hélices α: Phe24-Ser30, Asn33-Trp44, Gly47-Leu61, Phe68-Ile80, Glu83-Ala93, Phe99-Glu109. Figure générée à partir du logiciel CSI (Wishart and Sykes, 1994)..................................... 53
Figure 4-13 : A) Graphique montrant le résultat du logiciel TALOS pour la protéine MTH187. Les déplacements chimiques des 1Hα , 13Cα ,13Cβ, CO et N ont été utilisés. Les régions vertes représentent les résidus où TALOS suggère une structure secondaire avec un niveau de confiance élevé. Les régions rouges représentent les régions où la structure secondaire est contradictoire (Bad). Les régions jaunes signifient des régions ambiguës et les régions grises correspondent aux régions non classées. L’analyse avec TALOS indique que MTH187 est composée de 6 hélices α: Glu25-Leu31, Arg37-Ile46, Val55-Glu63, Phe68-Ile80, Glu83-Ala93, Ala100-Glu109. B) Graphique de Ramachadran illustrant les angles dièdres du résidu central des dix tripeptides de la banque de données ayant des déplacements chimiques secondaires (secondary shifts) similaire à ceux du tripeptide R75-S76-L77. Cette analyse permet d’estimer les angles dièdres φ/ψ de S76 à –66 ± 9 / -37± 8............. 54

x
Figure 5-1 : Superposition de 30 structures de MTH187. 32ième lancement de calculs effectués par ARIA. La RMSD est de 0.66 Å pour la chaîne squelettique (résidus 24-109) et de 1.23 Å pour les atomes lourds entre les résidus 24 à 109 de la protéine.................................................................................................................................... 58
Figure 5-2 : Structure de MTH187 (haut : Vue en stéréo, bas : rotation de 90° de long de l’axe des X. Les calculs de structures tridimensionnelles ont été faits par ARIA et les structures ont été visualisées dans MOLMOL. Les six hélices α sont représentées. La structure montrée est la structure calculée dans l’eau où l’énergie est la plus basse parmi les 72 structures calculées............................................................................... 59
Figure 5-3 : Graphique de Ramachandran des 30 structures RMN des résidus 24-109 de MTH187. Les résidus Glycine sont représentés par des triangles. Les régions favorisées par les angles phi et psi sont les deux régions les plus foncées. Les régions moins permises mais acceptables sont d’une couleur grise intermédiaire et les régions non permises sont en gris très pâle. Le % des résidus situés dans les régions favorisées est de 90.7%. ............................................................................... 60
Figure 5-4 : (montrée dans les six pages suivantes) Graphique de Ramachandran des résidus 2 à 110 de la structure RMN de MTH187. Chaque point dans chaque graphique de Ramachandran représente les 30 structures générées. Chaque graphique de Ramachandran indique la conformation du squelette pour un résidu, et ce pour chacune des structures calculées. Les structures secondaires définies manuellement sur cette figure sont : hélice 1 (22-31), hélice 2 (36-49), hélice 3 (55-64), hélice 4 (68-80), hélice 5 (83-94), hélice 6 (100-109)....................................... 60
Figure 6-1 :Alignement des régions superposées entre l’Aconitase B (pdb ID 1l5j) et MTH187 obtenu par le programme DALI. L’identité entre les deux séquences pour les résidus 24-109 est de 21% et la similarité est de 35%. Les “ * ’’ signifient que les deux résidus alignés sont identiques. Les “.’’ signifient que les deux résidus sont semblables................................................................................................................. 69
Figure 6-2: Vue en stéréo de la superposition de MTH187 en bleu et de l’aconitase B (I.D.:1L5J) en rouge. Le logiciel utilisé pour visualiser est PYMOL. Le RMSD squelettique des résidus superposés est de 2.36Å..................................................... 69
Figure 6-3: Alignement des régions superposées entre AP2 (pdb1gw5) et MTH187. L’identité entre les deux séquences pour les résidus 24-109 est de 21% et la similarité est de 42%. Les “ * ’’ signifient que les deux résidus alignés sont identiques. Les “.’’ signifient que les deux résidus sont semblables....................... 70
Figure 6-4: Vue en stéréo de la superposition de MTH187 en bleu et de l’adapteur Ap2 (I.D.:1GW5) en rouge. Le logiciel utilisé pour la visualisation est MOLMOL. La RMSD de la chaîne squelettique de la région 24-109 de MTH187 et l’adapteur Ap2 est de 2.72Å. ............................................................................................................. 71
Figure 6-5: Structure tridimensionnelle de l’Aconitase B qui a tendance à former des dimères. Le jaune représente le motif de répétition qui ressemble au motif strcutral “Heat repeat’’. Le PDB ID de cette protéine est 1l5j-A. .......................................... 72
Figure 6-6: Structure tridimensionnelle de l’Aconitase B sous forme monomérique. La région en jaune représente la région ou la protéine MTH187 se superpose. Le PDB ID de cette protéine est 1l5j-A. ................................................................................. 72

1
CHAPITRE 1. INTRODUCTION
1.1. Vue d’ensemble du mémoire Ce projet de maîtrise vise à connaître la structure et la fonction d’une protéine de la
bactérie Methanobacterium thermoautotrophicum. Pourquoi cette protéine me direz-
vous? En fait une grande partie des structures des protéines du génome de
Methanobacterium thermoautotrophicum sont analysées, dû premièrement à la
disponibilité du génome et dans le but de comprendre entre autre la tolérance de cette
bactérie à supporter des températures élevées.
Ce mémoire est séparé en huit chapitres. Le premier chapitre explique la
protéomique structurale suivi d’une introduction à la cristallographie et à la résonance
magnétique nucléaire. Par la suite nous présenterons la bactérie Methanobacterium
thermoautotrophicum et la protéine étudiée dans le cadre de ce projet de maîtrise:
MTH187. Comme ce mémoire utilise principalement la RMN, le chapitre 2 est dédié au
principe théorique qui entoure la détermination de la structures des protéines par
résonance magnétique nucléaire. Suivant les Matériels et méthodes au chapitre 3, les
résultats sont présentés aux chapitres 4 à 6. Le chapitre 4 montre l’attribution des spectres
RMN et la détermination de la structure secondaire de la protéine. Le chapitre 5 présente
la structure tertiaire de la protéine et l’homologie de structures est décrit dans le chapitre
6. Le chapitre 7 discute ensuite des résultats et le mémoire se termine par une conclusion
au chapitre 8.
1.2. Protéomique structurale L’aire de la protéomique structurale commença lorsqu’en 1936 Mirsky et Pauling
proposèrent que la fonction d'une protéine soit liée à sa conformation, c’est-à-dire à
l'arrangement tridimensionnel de ses atomes dans l’espace (Anfinsen, 1973). Ces paroles
eurent un très grand impact sur la compréhension de la biologie et sur la découverte et la
conception de nouveaux médicaments (Russell and Eggleston, 2000). Depuis, l’étude des
structures tridimensionnelles de toutes les protéines d’un protéome, nommée protéomique
structurale, fournit un nouveau mode de raisonnement pour la biologie structurale (Hol,

2
2000). L’étude expérimentale de la structure des protéines est un processus long et
coûteux nécessitant des équipements majeurs et des connaissances très pointues. Les
deux méthodes principales utilisées en protéomique structurale, soit la cristallographie et
la RMN, sont présentées dans la section qui suit. Une description détaillée de la RMN
sera présentée au chapitre 2.
1.3. Cristallographie et RMN Jusqu’en 1985, la seule méthode permettant de déterminer la structure
tridimensionnelle d'une macromolécule était la cristallographie par diffraction des rayons
X. Cette technique nécessite l’obtention de cristaux de protéine bien ordonnés. Malgré le
développement constant de nouvelles méthodes de cristallisation, il est parfois très
difficile et même impossible d`obtenir des cristaux utilisables. Depuis quelques années,
grâce aux progrès réalisés dans le domaine de la spectroscopie par résonance magnétique
nucléaire (RMN), il est possible de déterminer la structure tridimensionnelle en solution
de petites protéines, particulièrement des protéines de moins de 200 acides aminés
(Cavanagh et al., 1996). Ceci est dû à la fabrication de spectromètres à haut champ, la
mise au point de techniques de RMN multidimensionnelle et les progrès de
l’informatique. La RMN permet entre autre d'estimer la distance des protons, ainsi que
certains angles dièdres. À partir de la connaissance d'un nombre important de données
RMN, il est possible de déterminer la structure d’une protéine avec une résolution de
l’ordre de celle obtenue par les rayons X.
L’accomplissement récent de plusieurs projets de génomique ont fourni aux
scientifiques des informations sur la séquence de plusieurs génomes. Puisque chaque
protéine dans un organisme a une importance biochimique, la détermination de la
structure tridimensionnelle de toutes les protéines d’un protéome va permettre une
meilleure compréhension structure/fonction des protéines d’un organisme. Par
conséquent, des projets de protéomique structurale ont récemment débuté à plusieurs
endroits dans le monde, en outre au Japon, en Europe et en Amérique du Nord
(Heinemann, 2000; Terwilliger, 2000; Yokoyama et al., 2000).

3
1.4. Mise en contexte et biologie de Methanobacterium thermoautotrophicum
Un des projets de protéomique structurale concerne la bactérie Methanobacterium
thermoautotrophicum appelée également Methanothermobacter thermoautotrophicus
(Boone, 2000). Cette archaebactérie se retrouve dans des environnements à haute
concentration de sels, sans présence d’oxygène et où la température se situe entre 40 °C
et 70 °C (Smith et al., 1997). La température de croissance optimale est de 65 °C. Cette
bactérie se retrouve principalement dans les milieux riches en matière organique tel la
boue et dans les intestins des ruminants entre autre des bétails et des moutons. Ces
microorganismes sont capables de fermenter les déchets des animaux et des humains,
pour produire le méthane. Le méthane est utilisé à des fins de carburant dans plusieurs
pays en voie de développement où il assure la lumière et la chaleur (Pelczar et al., 1993).
Les protéines d’archaebactéries partagent beaucoup de séquences et caractéristiques
fonctionnelles avec les protéines eucaryotes, mais les archaebactéries sont souvent plus
petites et plus robustes. Cette robustesse est due à la composition de la paroi cellulaire qui
contient des glycoprotéines, des protéines et des polysaccharides comparativement aux
eubactéries dont la paroi cellulaire est principalement composée de peptidoglycanes
(Pelczar et al., 1993). De part ces caractéristiques mentionnées plus haut, les
archaebactéries servent d'excellents modèles pour des analyses structurales (Christendat
et al., 2000).
Le génome de Methanobacterium thermoautotrophicum a été séquencé en 1996
(Smith et al., 1997). En l’an 2000, l’étude de protéomique structurale a été lancée sur 424
protéines des 1871 gènes du protéome de Methanobacterium thermoautotrophicum par
des chercheurs de l’OCI (Ontario Cancer Institute) dont Cheryl H. Arrowsmith et Aled
M. Edwards (Christendat et al., 2000).
Deux critères d`exclusions ont été mis en application dans le choix des protéines.
Premièrement, les protéines associées à la membrane qui recouvraient 30% du protéome
de Methanobacterium thermoautotrophicum ont été rejetées de l’analyse, car les
structures de protéines membranaires ne peuvent être caractérisées rapidement.

4
Également, les protéines ayant un homologue dont la structure est connue ont été exclues.
Les protéines qui ont des possibilités de nouveaux repliements sont favorisées pour
l’analyse. Ce dernier groupe contient 27% des protéines de Methanobacterium
thermoautotrophicum (Christendat et al., 2000). La Figure 1-1 donne un aperçu du
nombre de gènes exclus par rapport au poids moléculaire de la protéine.
Figure 1-1 : Histogramme représentant le nombre de gènes exclus de l’analyse de protéomique structurale (figure reproduite de ((Christendat et al., 2000)).
Plusieurs autres protéines n’ont pas été sélectionnées pour le projet de protéomique
structurale, car leurs probabilités de former de nouveaux types de repliements étaient
restreintes et leurs pertinences biologiques semblaient limitées (Christendat et al., 2000).
Comme mentionné plus tôt, 424 protéines de Methanobacterium
thermoautotrophicum ont été choisies pour le clonage, l’expression et l’étude des
structures tridimensionnelles. 34% ont une annotation fonctionnelle, 54% sont classifiées
comme étant des protéines conservées dans le génome et 12% comme ayant une fonction
inconnue (Christendat et al., 2000).
La détermination de la structure tridimensionnelle d’une protéine demande un
certain temps. Elle peut prendre plusieurs mois dépendamment de la longueur de la
protéine. Pour augmenter la rapidité des découvertes des structures, les chercheurs de
l'OCI (Ontario Cancer Institute) collaborent avec d’autres laboratoires RMN pour
l’analyse des échantillons de protéines. Jusqu’à maintenant, seize structures ont été
résolues dans six différents laboratoires de cristallographie et dix-sept structures ont été

5
résolues dans sept différents laboratoires de RMN (Yee et al., 2003). Le nombre presque
identique de structures déterminées avec la cristallographie et la RMN montre que la
spectroscopie RMN peut apporter une contribution significative à la protéomique
structurale (Yee et al., 2003).
1.5. Protéine MTH187 Une des nombreuses séquences choisies pour l’analyse de la structure
tridimensionnelle est l’objet du projet présenté dans ce mémoire. La protéine MTH187
possède 111 acides aminés (12.4kDa) et son point isoélectrique est de 4.93. La séquence
de MTH187 est une séquence conservée dans les génomes à travers l’évolution et de
fonction encore inconnue. Basé sur une analyse bioinformatique (Tatusov et al., 1997),
MTH187 a été classée dans une banque de données COG. Cette classification
phylogénique contient des protéines homologues de la même organisation provenant
d’une duplication de gène (Tatusov et al., 2001). MTH187 est classé plus précisément
dans la famille COG1413, une famille de sous unité α d’une phycocyanine
phycocyanobiline lyase. La Figure 1-2 montre l’alignement de MTH187 avec quelques
protéines faisant partie de cette famille.
MTH187 1 --------------------------------------------MADE------------ MTH1806 1 -------MVNARLKEMRLMEPQKRMEAIEKLEPSEESVEILIAFLEDESHPVRFKAAEKL MTH1715 61 FPSEESLRAIEKFVDHPSEDIRRNSIESIAGLDPELGLRYASRALGDSSWMVRKTAAKVI MTH1378 1 ---------------------------MFFMEGKR--IDFLLEELRNP------------ PA2293 61 G-----RRLLPWLGHADAFVRASVLRALRELRLEESAVPAL-AALGDPQAAVRREAVAVL BS_ypgR_2 1 -------------------------AHLEQMDPKEEDIPVLQKALDDP------------ BH1718_2 1 -------------------------AALDKMDPTEEDLPVLEKALQDE------------ BH2232 61 WSTRMNTLHRILEFRMKDLLDDVLNMLNENKHYSDDEYLQIYRILAAFQYEDFLQHLLNP mlr6693 61 EKGNGAERIHAAEMLAALRSENAVASLLSALDRDRSREVRIAAAIALCDLGSLPPLDIAL mlr9194 60 A-----DSILPLVSHGRAFVRAAGFRGLKALRSRGALAPAL-AAMRDEDANVRAQALGVV slr1687 10 AMTALTLEQIASQLDSPNSRDRLIALASLRPYSSEEAVPLIKKVLDDDTLQVRSMAVFAL sll1663 1 --MSDSLTAIKALLGSDNFSDKVRGLNQLRALEPAEAFPLLKPLVNDANPRIRYAAVSQL slr1098 32 PPPPDPDEMLVLLTSREAPQRMLAARAFCEIADRR-AVEPLINLLGDSCPLVRVSAAYAL MTH187 5 ------------------------NKWVRRDVSTALSR-----------MGDE-AFEPLL MTH1806 54 --AEFGEASLEKLMEIM----DTAEGEIRRYATFALKK-----------IGDPRVTDHFI MTH1715 121 RRFGDKRCLEVLLDNLN-----DPDTEVRRHILLAVVN-----------MGEY-AVDPLL MTH1378 20 ------------------------DWVVREDAVELLAE-----------VADPRAVGPLI PA2293 115 GWLRHQPALAELARLAS----ADVDPEVRRAATGALG------------LSREATVLPAL BS_ypgR_2 24 ------------------------KVSIRRQAVVYLGM-----------IETP-DVLPLL BH1718_2 24 ------------------------KASIRRLATVYLGM-----------IEKP-VVLPLL BH2232 121 KIEMNEMEYRKLLFELEEKPFLNMVEHYQDFPSALKHT-----------VLDMIGVKHLV mlr6693 121 GKVGVVGQRSRRLIELFRRFPPARFEELRDHAARTDAVP----------FIRAAAIDALA mlr9194 114 AYLKLEETLPS-LIAAT----RDAEAIVRSVAVNALSF-----------TSQP-AAAAAV slr1687 70 GIKQTEECYPILVKLLET----DGDYGIRADAAGALGY-----------LEDERAFHPLC sll1663 59 DPVGKADLEQS-LQLLRDRLFNDPEIDVQSVAADVIGG-----------LKLTAAYPDLQ slr1098 91 GRNTDPTIVEPLIQSLQ----TDFNGYVRKGLVWALGN-----------CGDRRALAPLI

6
MTH187 29 ES-------LSNEDWRIR-----------GAAAWIIGNFQDER-----------AVEPLI MTH1806 97 EA-------LSDEDWGVR-----------KFAARSLGELGDLR-----------AVEPLI MTH1715 164 EK-------LSDPQWQTR-----------AMVVEALGEIGSRR-----------AVPRLK MTH1378 45 EA-------LDDEDYHVR-----------EAAALALATFDDRM-----------AVEPLR PA2293 159 CAA------LADAQWQVR-----------EEAATTLGKLGREE-----------AGEPLL BS_ypgR_2 48 YKA------LEDKAVSVR-----------RTAGDCLSDIGDPQ-----------AIPAMI BH1718_2 48 EKA------LKDPSVTVR-----------RTAGDCMSDIGDPA-----------AIPAMI BH2232 170 QMVPFLEERIHDEELEVR-----------VRSLKALSQLGIVQ-----------NEKIYY mlr6693 171 RAAG-----FHFADFFARATGDQ----SPEVAAAALRALGLTG--------HAEAPAILA mlr9194 157 TAA------LDDENWQVR-----------AAAADTLGRIGQTS-----------AVDGLT slr1687 115 RAF------YEDTEWLVR-----------FSAAVALGNLKDIR-----------AQTVLL sll1663 107 KAY------EETPEWLLQ-----------MSIVATLGEMGDRR-----------GFDLLK slr1098 136 HA-------LKTDISAVR-----------LWSASSLPNIAKLAYE-----DVIATIPPLI MTH187 60 KLLE-----DDSGFVRSGAARSLEQIGGE------------------------------R MTH1806 128 AALD-----DEDWGVKLAAVRSLGDLGDER---------------------------AIE MTH1715 195 GMLAGRRR-DENRYVRGKVAEALGRIGDPAALEDLHMALRDPYLFVRRKAREAIDIIDVE MTH1378 76 EHLS-----DDKPGVRYACALALGILGDE------------------------------D PA2293 191 KALA-----DDYWQVRLRAARALGRLRHRP---------------------------ARE BS_ypgR_2 80 KSLS-----DSSKLVRWRAAMFLYEVGDES---------------------------AIE BH1718_2 80 EALK-----DKNKLVRWRAAMFLYEVGDET---------------------------AID BH2232 208 PFVE-----SENWEERMMVAKILANVDSPE---------------------------AVS mlr6693 214 SAMA-----NGDWDVRAAAADAAGRLGLAG----------------------------LA mlr9194 189 AGLS-----DEYWQVRQKCLNALGKLKAQS---------------------------AVR slr1687 147 EALK-----SDEAVVQQAAIAALGEIGAVD---------------------------AVD sll1663 139 IALD-----SENSLIRTAAISALGELGNPE---------------------------ALP slr1098 173 EGLRR----DKMAAVRSNCAWAVGQLCRELPSN-------------------VVYATAID MTH187 85 V-RAAMEK--------------------------LAETGTGFARKVAVNYLETH------ MTH1806 156 P-IKKARR--------------------------KGDKDFKKAANKSLKKIQSGR----- MTH1715 254 PDLEEFHDGEISFRYPRFWNIFETRDGERLIDAWSDNLKIAIKRRRAEGLTADEFSEIIL MTH1378 101 S-IPDLEE--------------------------LLDDESPMVRRVAEVAISEIRKRAA- PA2293 219 A-LEALLG--------------------------HPIGNLRKEAALALGELADPASAQAL BS_ypgR_2 108 A-LRAAED--------------------------DPEFEVSLQVKMALERIEHGEEAKGS BH1718_2 108 A-LRLAED--------------------------DPEFEVAMQVKMALERIVGGEEAKGS BH2232 236 S-LQHLLK--------------------------DQSWWVRSQAAQSIVQQKNGKSVLRA mlr6693 241 APLSALMD--------------------------DEAWTVRYAAANALRSFGQPGERLLR mlr9194 217 Q-ISELLT--------------------------SDLASLRKESAAALGEIADPSSRAVL slr1687 175 A-ILAFAS--------------------------HEDWLIRQRLVEALGNLPCDQSRSAL sll1663 167 L-LASLVQ--------------------------DEDWQVRYRLALAVGHLEHPDRQSLL slr1098 210 ALLEALVE--------------------------DEDFGVKEDAKSALLRLGDARGLQMI
Figure 1-2: Alignement de séquences des sous-unités α des phycocyanines phycocyanobilines lyases extraites de COG1413 généré avec ClustalW 1,74 (www.ch.embnet.org/software/ClustalW.html). La première colonne cite les abréviations des protéines de la famille COG1413. Les quatre premières protéines, dont la protéine étudiée MTH187, sont des protéines de Methanobacterium thermoautotrophicum d’où l’abréviation MTH. Le nombre à la suite est le numéro du gène. Les protéines suivantes proviennent des bactéries Pseudomonas aeruginosa, Bacillus subtilis, Bacillus halodurans, Mesorhizobium loti, Synechocystis sp. BOXSHADE (www.ch.embnet.org/software/BOX_form.html) est utilisé pour mettre en évidence les acides aminés identiques (boîtes noires) et semblables (boîtes grises).
Une protéines portant une fonction de phycocyanine phycocyanobiline lyase se
trouvent généralement chez les cyanobactéries et les algues rouges (Fairchild et al.,
1992), bref, plusieurs organismes vivant dans les eaux peu profondes, tièdes et calmes.
Ces protéines participent à la photosynthèse (Voet and Voet, 1995). Jusqu’à maintenant,

7
neuf structures des phycocyanines ont été déterminées par cristallographie des rayons X
mais aucune par RMN.
Afin d’approfondir les connaissances de la protéine MTH187, nous avons
déterminé la structure tridimensionnelle de celle-ci. Étant donné le nombre de résidus
inférieurs à 200, la méthode choisie est la RMN.
1.6. Buts du projet de maîtrise Un des objectifs de ce projet est de déterminer, à l’aide de la RMN, la structure
tridimensionnelle de la protéine MTH187 qui pourrait servir de base pour déterminer sa
fonction. La fonction d’une nouvelle protéine peut potentiellement se déterminer par
homologie séquentielle c'est à dire par l’alignement de MTH187 avec des protéines
puisées dans une base de données qui contient des fragments de séquences semblables.
Les fonctions de ces protéines ont été déterminées soit expérimentalement ou par
homologie séquentielle. La fonction peut se déterminer également par homologie
structurale qui consiste à comparer la structure repliée de MTH187 avec d'autres
protéines repliées dont on connaît la fonction dans le but de repérer des structures
semblables dans l’espace et d’identifier le site actif de la protéine. C’est à ce moment que
la RMN intervient dans le projet.
Le travail effectué permettra d’obtenir une première structure tridimensionnelle de
protéine dans le laboratoire du Dr Gagné. Par la suite il sera plus facile de caractériser
d'autres protéines car les problèmes survenus au cours de ce projet pourront être évités.
Cette protéine, tout comme toutes les protéines de Metanobacterium
thermoautotrophicum, est stable à 65oC contrairement à plusieurs autres protéines,
retrouvées chez d’autres bactéries, qui se dénaturent à haute température. Les types
d’acides aminés retrouvés chez MTH187, les différents liens entre ceux-ci et l’analyse de
la conformation tridimensionnelle permettront de définir des règles générales, et de
potentiellement connaître les raisons pour lesquelles cette protéine tolère des
températures plus élevées.

8
La structure tridimensionnelle de MTH187 pourra également permettre
d’effectuer de la modélisation moléculaire pour d’autres protéines de Metanobacterium
thermoautotrophicum principalement les protéines retrouvées dans la famille COG1413:
MTH1806, MTH1715 et MTH1378. Cette modélisation permettra de soulever des
hypothèses sur les structures tridimensionnelles de ces trois protéines.

9
CHAPITRE 2. DÉTERMINATION DE LA STRUCTURE DES PROTÉINES PAR
RÉSONANCE MAGNÉTIQUE NUCLÉAIRE
La résonance magnétique nucléaire (RMN) est une technique spectroscopique très
utilisée. La très grande variété de spectres permet entre autre de déterminer la structure
tridimensionnelle de protéines en solution. La section suivante vise à présenter un survol
des concepts théoriques de la RMN et à introduire les stratégies utilisées pour déterminer
la structure d’une protéine par RMN.
2.1. Niveaux de structure des protéines Une protéine est composée d’une série d’acides aminés reliés par des liens
peptidiques. Cette chaîne primaire se replie et s’organise pour former des structures
secondaires. Parmi ces structures, il y a entre autre les hélices α, les feuillets β, les coudes
β et les structures désordonnées. L’interaction entre les atomes de chaque acide aminé
ainsi que l’encombrement stérique influence la structure secondaire.
L’hélice α demeure un élément classique de structure de protéines. L’hélice α a 3.6
acides aminés par tour d'hélice et des liaisons hydrogène entre les groupements CO d’un
acide aminé I et NH d’un acide aminé I+4 (Figure 2-1).
Le feuillet β est constitué de la combinaison de brins β. Ces brins sont alignés côte
à côte pour former des liaisons hydrogène entre les groupements CO d’un brin et les
groupements NH d’un autre brin. Lorsque l’orientation des brins sont dans la même
direction, il s’agit d’un feuillet β parallèle. Lorsque les acides aminés dans les brins
successifs ont des directions opposées, c’est un feuillet β antiparallèle (Figure 2-2)
(Branden and Tooze, 1991).
10
Figure 2-1: Structure secondaire en hélice α. Les liaisons hydrogène entre les atomes d’azote et d’oxygène de la chaîne principale sont en points noirs. Les atomes d’azote sont en bleu et les atomes d’oxygène sont en rouge. Figure générée avec le logiciel MOLMOL.
Figure 2-2: Représentation schématique des feuillets β parallèles et antiparallèles. Les groupements NH et CO de la chaîne principale sont liés par des liaisons hydrogène. Figure tirée du site Internet: http://tsailab.tamu.edu/biochem410/06-2-Structure.pdf
Résidu I + 8
Résidu I + 4
Résidu I

11
D’autres types de structures secondaires, tels les coudes, peuvent être rencontrés
dans les protéines. Les coudes permettent à la protéine de se replier afin de favoriser un
changement de direction. Enfin, beaucoup de protéines présentent des régions
désordonnées. Ces portions de protéines oscillent dans la solution, car peu de forces sont
présentes pour les maintenir en place (Voet and Voet, 1995).
Le repliement des structures secondaires forme la structure tertiaire. Ce repliement
est stabilisé par des interactions électrostatiques et/ou hydrophobes qui déterminent la
conformation de la protéine et son activité biologique. Beaucoup de protéines sont
constituées de plusieurs chaînes polypeptidiques (sous-unités) qui sont réunies par des
interactions non covalentes et quelques fois par des ponts disulfure. La structure
quaternaire désigne l’arrangement dans l’espace de ces sous-unités.
2.2. Principes de base en RMN La RMN repose sur l’analyse des propriétés magnétiques des noyaux atomiques.
Les noyaux des atomes engendrent l’existence d’un moment cinétique propre au noyau
appelé spin I. Le spin prend comme valeurs 0, 1/2, 1, 3/2, et ainsi de suite (I=0 signifie
qu’il n’y a pas de spin). Cette quantification du spin implique celle du moment
magnétique µ qui lui est associé (µ = γI). En effet, le fait d’avoir un spin non nul confère
aux particules les propriétés d'un dipôle magnétique. Une particule de spin 1/2 soumise à
un champ magnétique possède un moment magnétique qui peut prendre deux
orientations, dans le même sens que le champ B0 et dans le sens opposé. Le proton 1H,
les noyaux 13C, 19F ou 31P ont un spin de 1/2 et donc deux états de spins: +1/2 et –1/2.
Pour un spin égal à un, tel que celui du deutérium 2H ou le lithium 6Li, il y a trois
orientations possibles:-1, 0 et +1 (Evans, 1995). Plus généralement, il y a 2I+1
orientation pour un spin I.
Lorsque l’échantillon est soumis à un champ magnétique, à chacune de ces
orientations correspond un niveau d’énergie. Pour le proton dont le nombre de spin vaut
1/2, il existe deux niveaux d’énergies, avec un léger excès de population sur le niveau
d’énergie le plus bas (distribution de Boltzmann)(Figure 2-3). Ce sont les transitions

12
entre ces niveaux qui constituent le phénomène de résonance magnétique nucléaire
(Sanders and Hunter, 1987).
Figure 2-3: Niveaux d’énergies et orientations des états de spins nucléaires du proton en absence et en présence d’un champ magnétique. La différence de population entre les deux niveaux d’énergies est beaucoup plus faible qu’illustré dans cette figure. (Figure tirée de www.rmn.uhp-nancy.fr/Mutzenhardt/RMNSV2CM1.pdf)
Pour observer un signal, il faut rompre l’équilibre entre les deux niveaux
d’énergies, peuplant davantage le niveau supérieur. L’échantillon doit être soumis à un
deuxième champ magnétique (produit par une source de radiations électromagnétiques
appelées impulsion) perpendiculaire au premier champ produit par l’aimant. La
magnétisation globale sera transférée dans le plan x, y. Dès que l’impulsion est terminée,
le moment magnétique tourne autour de Bo et la magnétisation globale induit un courant
électrique dans la bobine du récepteur (“receiver coil’’) (Cavanagh et al., 1996) (Figure
2-4).
Figure 2-4: Effets d’un champ magnétique externe sur un échantillon RMN. A) Transfert de la magnétisation le long des axes x et y. reproduite du livre ((Wüthrich, 1986). B) Rotation de la magnétisation autour de l’axe z.(reproduite du livre (Sanders, 1987).

13
Comme présenté à la Figure 2-5 , l’aimantation globale M, va rejoindre sa position
initiale, c’est-à-dire parallèle à Bo. M revient à sa position initiale progressivement par un
phénomène de relaxation (Cavanagh et al., 1996).
Figure 2-5: Retour à l’équilibre du moment magnétique avec la magnétisation orientée parallèlement à B0 (www.med.univ-rennes1.fr/cerf/edicerf/BASES/BA004_cv_rb_9.html).
C’est dans cette période de retour à l’équilibre que les données sont enregistrées par
le spectromètre RMN. Les noyaux de la protéine vont résonner à différentes fréquences
dépendant en autre de l’environnement dans lequel ils se trouvent. Le signal enregistré est
une FID (free induction decay). On retrouve à la Figure 2-6 un exemple d’une FID.
Figure 2-6: Signal RMN se traduisant par une décroissance du signal détecté en fonction du temps nommé FID (free induction decay).
Ces signaux contiennent toutes les informations nécessaires, mais dans un
langage difficile à interpréter. Ils sont traités par la transformée de Fourier qui permet de

14
rendre visible un signal sinusoïdal qui est fonction du temps (y=f(t)) en le changeant en
un spectre RMN qui est fonction de la fréquence (y=f(ω))(Figure 2-7). Les fréquences de
résonance sont ensuite récupérées (Cavanagh et al., 1996).
Figure 2-7: Fonctions sinusoïdales FID où est appliquée une transformée de Fourier. (Figure tirée de www.rmn.uhp-nancy.fr/Mutzenhardt/RMNSV2CM1.pdf)
2.3. Stratégies utilisées pour déterminer la structure des protéines par RMN
Dans cette section, il sera question des étapes nécessaires à la détermination de la
structure d’une protéine. Ces étapes sont présentées à la page suivante (Figure 2-8).
2.3.1. Expression, marquage isotopique et purification des protéines Les analyses par RMN nécessitent de grandes quantités de protéines à l’état pur; il
est donc souvent nécessaire de surexprimer la protéine. À ce titre, en utilisant les
techniques standards de génétique, le gène d’intérêt est cloné dans un plasmide d`ADN
qui, une fois inséré dans les bactéries, est exprimé en très grande quantité.
Certains noyaux ne sont pas observables par la RMN faute de spins nucléaires
(I=0). Ainsi, pour des noyaux fondamentaux de la chimie organique tels 12C, et 16O nous
ne pourrons observer de phénomène de RMN. Ces atomes ont cependant des isotopes
naturels ayant un spin nucléaire non nul. Le 13C de spin 1/2 est retrouvé à 1.11% dans la
nature. Outre sa faible abondance naturelle, cet isotope possède un rapport
gyromagnétique quatre fois plus faible que celui du proton. Donc, ces deux facteurs
induisent une sensibilité très faible pour le 13C en spectroscopie RMN. Le 17O de spin 5/2
est beaucoup moins abondant que le 13C dont l’observation conduit à des raies spectrales
très larges et peu exploitables par une analyse RMN.

15
Les noyaux 14N, qui ont un spin de 1, sont visible par RMN, mais il est préférable
en RMN liquide de marquer l’azote afin de lui donner un état de spin 1/2. Ce marquage
diminue le nombre de niveaux d’énergies à deux au lieu de trois et facilite par conséquent
l'analyse. L’15N de spin 1/2 est moins abondant que le 13C dans la nature, il est retrouvé à
0.37 % (Cavanagh et al., 1996; Voet and Voet, 1995). Il donne de bons signaux RMN.
Son rapport gyromagnétique est dix fois plus petit que celui du proton.
Figure 2-8 : Diagramme qui résume les étapes de la détermination de structures tridimensionnelles d’une protéine par la méthode de résonance magnétique nucléaire (RMN).
Expression, marquage isotopique et purification des protéines
Préparation des échantillons
Attribution des spectres
Contraintes structurales
Calculs des structures tridimensionnelles
Famille de structures
Acquisition des spectres RMN
Expression, marquage isotopique et purification des protéines
Préparation des échantillons
Attribution des spectres
Contraintes structurales
Calculs des structures tridimensionnelles
Famille de structures
Acquisition des spectres RMN

16
Afin de contrer ces difficultés, il est possible de marquer uniformément les atomes
de carbone et d’azote aux 13C et 15N lors de l’expression de la protéine. Du chlorure
d'ammonium marqué (15NH4Cl) et du glucose marqué (13C-glucose) sont utilisés comme
seule source d'azote et de carbone dans le milieu de culture. Ce marquage isotopique des
protéines est essentiel afin de pouvoir analyser une multitude d’expériences RMN
multidimensionnelles.
Il peut parfois être utile de remplacer les protons par des deutériums afin d’alléger
les spectres des grosses protéines. Afin de produire une protéine deutérée, la croissance
des bactéries est effectuée dans le D2O en remplacement de H2O.
Les protéines recombinantes sont ensuite purifiées et leurs structures sont
caractérisées par RMN.
2.3.2. Préparation des échantillons La protéine dont la structure tridimensionnelle est analysée par RMN se doit d’être
dans une concentration et un environnement adéquat. La concentration de protéines pures
nécessaires dans un échantillon varie entre 0.5 et 2.0 mM. Cette concentration favorise un
signal par rapport au bruit satisfaisant et permet l’acquisition des spectres en un temps
approprié.
L’échantillon ne peut contenir d’impureté, du moins pas d’impureté contenant des
protons car ceux-ci interfèrent avec les protons de la protéine. Des sels et des ions
peuvent se retrouver dans la solution. Une concentration minimum de sels va offrir à la
protéine un environnement similaire à son milieu naturel, mais une concentration trop
élevée va nuire à celle-ci car les sels sont des éléments conducteurs et peuvent réchauffer
l’échantillon lors des impulsions.
La protéine peut se trouver à l’état de monomère ou de multimère. Les multimères
sont formés par l’agrégation de plusieurs molécules en solution. Lorsqu’il y a un échange
des deux conformations dans un échantillon étudié par RMN, les pics dans les spectres
deviennent plus larges. C’est pour cette raison qu’il est préférable de retrouver qu’une
seule conformation dans un échantillon.

17
La protéine doit être stable c’est-à-dire qu’elle doit demeurer active et garder sa
structure durant quelques semaines et ce à des températures parfois élevées. Le pH de
l’échantillon est très important pour observer tous les protons de la protéine. Le pH varie
généralement entre 5 et 7. Un pH trop élevé accélère l’échange des protons échangeables
avec le solvant.
Le solvant utilisé est généralement l’eau. 10% de D2O est ajouté à l’échantillon
pour verrouiller le spectromètre à une seule fréquence durant l’acquisition d’un spectre
(Cavanagh et al., 1996). En effet, étant donné que le champ magnétique varie
continuellement d’une infime valeur, une impulsion est lancée continuellement sur le
D2O afin d’ajuster la fréquence de référence des noyaux de la protéine. La fréquence de
résonance est référencée par une petite quantité (0.5mM) de DSS (sodium 2,2-dimétyl-2-
silapentane-5-sulfonate) ajoutée à l’échantillon.
2.3.3. Acquisition des spectres RMN L’acquisition de plusieurs spectres RMN permet d’identifier les fréquences de
résonance de tous les noyaux d’un composé. Cependant, la fréquence de résonance d’un
noyau est proportionnelle au champ magnétique appliqué. Il n’est donc pas commode de
repérer le déplacement chimique en fréquence puisque ce repère dépend du champ utilisé,
donc de l’appareil. La mesure utilisée est le déplacement chimique en ppm (partie par
million) (Cavanagh et al., 1996). En guise de simple exemple, un proton dans un champ
magnétique de 14 Tesla (T) résonne à 600MHz tandis que dans un champ magnétique dix
fois plus petit (1.4092 T), le proton résonne à 60 MHz. Le calcul peut être fait à partir de
l’Equation 1.
ν = γ Bz2л
Equation 1 : Équation de base en RMN. ν représente la fréquence de précession de Lamor,(MHz), Bz est le champs magnétique. Le rapport gyromagnétique est une constante, qui dépend de la nature du noyau.
Les noyaux d’un composé ne résonnent pas tous à la même fréquence. Les
protons résonnent typiquement entre –1 et 12 ppm (Figure 2-9), les carbones aliphatiques
résonnent typiquement entre 17 et 75 ppm et les azotes des amides typiquement entre 100

18
et 132 ppm. Le déplacement chimique est influencé par l’environnement local, c’est-à-
dire par les noyaux et les électrons qui se situent à proximité. Un élément électronégatif
tel un azote ou un oxygène déblinde un proton. Le déplacement chimique sera alors plus
élevé.
Figure 2-9: Déplacements chimiques observés pour les différents types de protons. Le spectre 1D a été enregistré dans une solution aqueuse. La FID est transformée par transformée de Fourier. Cette figure est reproduite de (Cavanagh et al., 1996)
Dans les spectres de résonance magnétique nucléaire de plus d’une dimension, un
pic est formé au déplacement chimique de la corrélation entre deux noyaux. Les
corrélations enregistrées entre les noyaux de la protéine peuvent être différentes d’un
spectre à l’autre. Elles peuvent s’effectuer entre les liens d’un même acide aminé ou à
travers l’espace. Certains spectres RMN aideront davantage à l’attribution squelettique,
c'est-à-dire la détermination des déplacements chimiques des noyaux sur la chaîne
principale, et d’autres types de spectres se consacreront davantage à déterminer les
déplacements chimiques sur les chaînes latérales.

19
Les expériences RMN homonucléaires sont basées sur un seul type d’atome: les
protons. Deux types d’expériences homonucléaires utilisées sont le COSY qui corrèle les
protons couplés et le TOCSY (Braunschweiler and Ernst 1983). où les corrélations sont
entre tous les protons faisant parties du même système de spin (Cavanagh et al., 1996).
Plusieurs acides aminés ont un système de spins bien à eux. Prenons exemple sur le
résidu alanine. Ce résidu est caractérisé par un pic très intense pour le groupement
méthyle situé en Hβ. En fait, ce pic est trois fois plus intense que le Hα. Il sera donc
possible d’associer plusieurs groupements de déplacements chimiques à un type d’acide
aminé. Par la suite, il sera important de relier ces acides aminés ensemble. Le spectre
NOESY est également une expérience RMN homonucléaire. Les outils pour attribuer une
séquence sont les NOEs qui seront détaillés un peu plus loin dans cette section.
Lors de l’analyse de plus grosses protéines, où il est nécessaire de marquer certains
résidus, des expériences RMN hétéronucléaires dont les spectres peuvent atteindre
jusqu’à quatre dimensions sont acquisitionnées.
Le spectre 15N-TOCSY-HSQC (Clore et al., 1991) de trois dimensions, dont la
protéine est marquée à 15N, révèle des corrélations à travers les liens d'un même acide
aminé (Figure 2-10). Cette expérience est acquisitionnée au spectromètre RMN en
induisant premièrement une impulsion de 90°. L’aimantation devient alors
perpendiculaire au champ de l’aimant. L’aimantation tourne autour de l’axe z et le
déplacement chimique des protons est alors mesuré au temps d’évolution t1. Ensuite, il y
a un temps de mélange où chaque proton transmet son aimantation à travers les liens aux
autres protons situés sur le même résidu. L’aimantation est transférée sur 15N par un
transfert INEPT et ainsi crée une deuxième dimension. Seulement les protons HN seront
transférés durant INEPT. Le déplacement chimique du 15N est enregistré durant le temps
d’évolution t2. Finalement, un INEPT inversé sera utilisé pour transférer l’aimantation sur
le 1HN et le déplacement chimique de celui-ci est enregistré au temps d'évolution t3
(Cavanagh et al., 1996).

Figure 2-10: Corrélations obtenues lors de l’analyse d’un spectre 3D 15N-TOCSY-HSQC
Comme mentionné plus haut, pour déterminer la structure d’une protéine il est
nécessaire de joindre tous les systèmes de spins des acides aminés. Les corrélations
NOEs (nuclear Overhauser effet) sont des corrélations dipolaires entre deux protons
situés à proximité dans l’espace (Cavanagh et al., 1996). Plus la distance entre deux
atomes est grande plus l’intensité de la corrélation NOEs est faible. Pour qu’il y ait
corrélation, la distance ne doit pas dépasser 5Å. Cette relation est inversement
proportionnelle et se réfère à la relation suivante.
Equatio
TOCS
d'hydr
très él
spectre
adjace
princip
corréla
D
squele
HNCA
spectre
précèd
NOE % 1/ r6
20
n 2: Relation entre le NOE (nuclear Overhauser effet) et la distance entre deux protons
L’expérience 15N-NOESY-HSQC (Ikura et al., 1990) est similaire au 15N-
Y-HSQC sauf que les spectres NOESY enregistrent des corrélations entre atomes
ogène près dans l’espace. En plus des corrélations entre les atomes d’hydrogène
oignés dans la séquence (distance maximum de 5Å une fois la protéine repliée), ce
RMN enregistre les corrélations entre les atomes d’hydrogène de résidus
nts, principalement les corrélations de l’hydrogène lié à l'azote de la chaîne
ale du résidu n avec les hydrogènes liés à N, Cα et Cß du résidu n-1. Ces
tions sont très utiles pour l’attribution séquentielle.
’autres spectres peuvent être acquisitionnés dans le but de déterminer la chaîne
ttique d’une protéine. L’expérience CBCA(CO)NH (Grzesiek et al., 1992) et
CB (Kay et al., 1990) utilisent la protéine marquée à 15N et au 13C. Ces deux
s donnent des informations concernant le déplacement chimique des Cα et Cβ qui
ent un acide aminé quelconque à partir du 1HN et 15N. Le HNCACB donne en plus

21
le déplacement chimique du 13Cα et du 13Cß de ce résidu. Un aperçu des corrélations sont
illustrées à la Figure 2-11 et Figure 2-12.
Figure 2-11: Corrélations obtenues lors de l’analyse d’un spectre 3D CBCA(CO)NH
Figure 2-12: Corrélations obtenues lors de l’analyse d’un spectre 3D HNCACB.
Une des expériences hétéronucléaires les plus sensibles acquisitionnées sur un
spectromètre RMN est le HNCO. Cette expérience pour laquelle la protéine doit être
marquée à 15N et au 13C donne les déplacements chimiques du groupement CO qui
précède un acide aminé à partir du HN (Figure 2-13). Même chose pour le HN(CA)CO
(Clubb R.T, 1992) qui, en plus, révèle les déplacements chimiques du groupement CO
sur un même acide aminé (Figure 2-14).
Figure 2-13: Corrélations obtenues lors de l’analyse d’un spectre 3D HNCO.
Figure 2-14: Corrélations obtenues lors de l’analyse d’un spectre 3D HN(CA)CO.

22
L’acquisition de tous ces spectres devrait résoudre en bonne partie l’attribution de
la chaîne squelettique d’une protéine. Pour attribuer les chaînes latérales de tous les
acides aminés d’une protéine, différents spectres peuvent être acquisitionnés. Certains
résidus contiennent une longue chaîne latérale difficilement attribuable à partir de
seulement l’analyse d’un 15N-TOCSY-HSQC.
Le spectre HCCH-TOCSY (Olejniczak et al., 1992) où la protéine est marquée au 13C donne les corrélations entre les protons d'un même acide aminé (Figure 2-15).
Contrairement au spectre 15N-TOCSY-HSQC, ces corrélations ne sont pas entre l’amide
et les protons d’un même acide aminé mais bien entre tous les CH situés sur un même
acide aminé. Celles-ci se répéteront sur différents plans autant de fois qu'il aura de CH.
Figure 2-15 : Corrélations obtenues lors de l’analyse d’un spectre 3D HCCH-TOCSY
Comme visualisé à la Figure 2-9, les composés aromatiques résonnent sensiblement
à la même fréquence que les amides de la chaîne squelettique. Il est difficile de les
analyser dans les spectres décrits précédemment. Par contre, ces protons aromatiques sont
non échangeables avec le solvant et les amides sont échangeables. Le milieu permettant
de caractériser les protons aromatiques est le D2O. En solution, les protons échangeables
seront deutérés. Les protons aromatiques seront présents sur les spectres appropriés.
2.3.4. Transformation des spectres en vue de leurs analyses Les signaux RMN émis lors de l’acquisition d’un spectre doivent subir quelques
modifications avant leurs analyses. La transformation des spectres implique plusieurs
fonctions mentionnées ci-dessous.

23
- Extraction du solvant
- Prédiction linéaire
- Multiplication du signal fid par une fonction mathématique
- Zéro filling
- Transformée de Fourier
L’extraction du solvant est une fonction qui supprime le signal émis par le solvant.
Cela permettra de visualiser un grand nombre de corrélations qui pouvaient se retrouver
sous les pics du solvant. La multiplication du signal fid par une fonction mathématique a
beaucoup de bienfait sur les spectres. Par exemple, l’application d’une fonction
sinusoïdale à la fid par rapport à l’application d’une fonction cosinus augmente la
résolution mais diminue le rapport signal sur bruit. L’application d’une fonction cosinus
comparativement à la fonction sinus augmente le rapport signal sur bruit mais donne des
raies plus larges. La prédiction linéaire ajoute des points qui sont prédits à partir des
points expérimentaux. La fonction suivante, Zéro filling, permet d’ajouter des zéros à la
fin du signal pour améliorer la résolution d’un spectre.
La transformée de Fourier convertit la fid en spectre de fréquence (Figure 2-7).
Cette fonction est exécutée sur toutes les dimensions d’un spectre. Les avantages de son
utilisation sont entre autres sa rapidité et le fait que les signaux s’additionnent, mais que
le bruit ne s’additionne pas.
D’autres fonctions sont utilisées afin d’améliorer davantage les spectres RMN. Une
fonction permet entre autre d’ajuster la phase, une autre permet d’appliquer une
correction polynomiale de la ligne de base. Les fonctions ajoutées, par essais et erreur à
la fid, permettent de générer des spectres de meilleures qualités.
2.3.5. Contraintes structurales Différentes contraintes structurales telles que les NOEs (Nuclear Overhauser
Effect), et les angles dièdres sont nécessaires afin de déterminer la structure des protéines

24
par RMN. Les contraintes de ponts hydrogène et bien d’autres contraintes non présentées
dans ce mémoire peuvent également être utilisées pour déterminer la structure de la
protéine.
2.3.5.1. Contraintes de distances Les contraintes NOEs fournissent des informations sur les distances entre atomes
voisins. Ces distances, à moins de 5Å, peuvent être intra-résidu ou inter-résidus
(séquentielle, moyenne portée, longue portée). L’expérience 15N-NOESY-HSQC permet
de connaître les corrélations entre amides et les protons à proximité dans l’espace.
L’expérience 13C-NOESY-HSQC donne les corrélations entre tous les protons attachés à
un carbone. Chaque proton lié à un carbone de chaque résidu aura des corrélations avec
tous les protons attachés sur un carbone près dans l’espace. Ce spectre n’est souvent pas
attribué entièrement, car plusieurs pics se chevauchent fréquemment.
L’interaction entre deux atomes, perçue à partir de l’intensité de la corrélation,
permet d’évaluer leurs distances à partir de la relation précédemment citée (Equation 2).
Cette calibration des NOEs est nécessaire pour le calcul des structures.
2.3.5.2. Contraintes d’angles dièdres Dans une protéine, les liaisons adjacentes à la liaison peptidique C-N peuvent
tourner autour de leurs axes. Ainsi, le trajet d'une protéine dans l’espace est défini par une
série de plans articulés entre eux au niveau des carbones α (Figure 2-16).
Figure 2-16: Représentation d’une chaîne polypeptidique. L’angle de rotation autour de la liaison N-Cα est l’angle phi(Φ) et celui autour de la liaison Cα-C’ est l’angle psi(ψ).Figure reproduite de http://www.ujf-grenoble.fr/BIO/Gurvan/phi_psi.html.
L’orientation de ces deux plans est définie par l'angle phi (Φ) et l'angle psi (ψ). Au
même titre que les distances NOEs, ces angles peuvent être utilisés comme contraintes
structurales. Deux méthodes sont généralement utilisées en RMN pour déterminer les
25
angles dièdres. L’utilisation de la relation entre le déplacement chimique et la structure
secondaire (par exemple l'approche TALOS) démontré à la Figure 2-17 et les constantes
de couplage.
Figure 2-17 : Relation entre le déplacement chimique Cα et Cβ et la structure secondaire. Cette figure est tirée de http://spin.niddk.nih.gov/bax/software/TALOS/
TALOS (Torsion Angle Likelihood Obtained from Shift and sequence similarity)
TALOS est un système de banques de données qui permet d’évaluer les angles Φ
et ψ à partir du déplacement chimique de cinq atomes: Hα, Cα, Cβ, CO, N (Cornilescu et
al., 1999). TALOS utilise une suite de trois acides aminés dans une protéine et compare
les variations de déplacements chimiques (forme séquentielle à repliée) des cinq derniers
atomes avec des tripeptides de variations semblables dans une banque de données. Cette
banque de données contient actuellement des données de 20 protéines de structures
connues représentant environ 3000 triplets. Les dix tripeptides comportant des
déplacements chimiques de similarité supérieure avec les déplacements chimiques de la
protéine cible sont gardés. Si ces derniers sont en accord, c’est-à-dire qu'ils se situent
dans au moins 9 cas sur 10 dans la même région cohérente du graphique de
Ramachandran, la prévision est classifiée comme étant satisfaisante.

26
La méthode de TALOS est empirique, les valeurs d’angles sont donc imprécises et
l’incertitude sur les valeurs est importante. TALOS donne de bien meilleurs résultats
lorsqu’il est utilisé en conjugaison avec la mesure des constantes de couplage.
2.3.5.3. Constantes de couplages La constante de couplage est la séparation en hertz des pics de protons couplés.
Pour déterminer la protéine en trois dimensions, la constante de couplage la plus
fréquemment utilisée est celle entre le HN et le Hα. L’expérience RMN HNHA (Geerten
W. Vuister, 1993) est utilisée pour définir ces constantes de couplage (3JHNHα). Le spectre
HNHA comporte trois dimensions (F1(15N), F2(1H), F3(1HN)(Cavanagh et al., 1996). Sur
ce spectre, le rapport des corrélations entre le pic transversal et le pic diagonal pour un
même résidu est proportionnel à 3JHNHα. À l’aide de l’équation mathématique présentée
ci-dessous, la valeur de 3JHNHα est déterminée.
= -tan2 (2π3 JHNHα δ)= -tan2 (2π3 JHNHα δ)
Equation 3 : Equation qui permet de connaître la constance de couplage 3JHNHα à partir du rapport d’intensité des pics et du délai d’acquisition des spectres (δ).
Une fois les constantes de couplages obtenues pour tous les acides aminés de la
protéine étudiée, il suffit grâce à la relation de Karplus (Harbison, 1993) présentée à la
Figure 2-18 de transposer les constantes de couplages en contraintes d’angles dièdres Φ
(phi).
Figure 2-18: Graphique de Karplus qui décrit la variation de la constante de couplage 3JHNHα avec l’angle dièdre Φ de la chaîne squelettique.
Scross Sdiagonal
27
Lorsque la constante de couplage d'un résidu est plus petite que 6 Hz, l’angle Φ se
retrouve autour de 60 degrés soit une structure secondaire en hélice α. Lorsqu’elle est
plus grande que 8 Hz, l’angle phi est autour de 120 degrés (feuillet β). Lorsqu’elle se
situe entre 6 et 8 Hz la constante de couplage n’est pas utilisée comme contrainte
structurale car les régions flexibles d’une protéine donnent lieu à une constante de
couplage dans cet intervalle. (Cavanagh et al., 1996).
Ces contraintes dihédriques Φ et ψ aideront aux calculs de structures des protéines.
Dépendamment des angles, une région formera une structure secondaire en feuillet β ou
en hélice α. Le diagramme de Ramachandran montre les régions Φ et ψ où il est plus
favorable de former des hélices α et des feuillets β.
phi
psi
phi
psi
phiphi
psipsi
Figure 2-19 : Graphique de Ramachandran montrant en ordonné l’angle dièdre ψ et en abscisse Φ. Les surfaces colorées marquées α, β et L sont approximativement les angles ψ et Φ retrouvés dans les hélices α droites, les feuillets β et les hélices α gauches. Figure reproduite de Branden and Tooze, 1991.
2.3.5.4. Contraintes de ponts hydrogène La liaison hydrogène est une interaction électrostatique entre un atome d’hydrogène
rattaché à un atome électronégatif et une paire d'électrons libres situés sur un atome (O, S
ou N) d'un autre groupement. La distance de la liaison hydrogène varie de 2,7 à 3,1 Å

28
(Van Holde and Mathews, 1990). Les contraintes de ponts hydrogène peuvent être
connues en mesurant la vitesse d’échange des amides lorsque la protéine est dissoute
dans le deutérium. Une série de spectres est acquisitionnée à de faibles intervalles de
temps. Le deutérium n’est pas visible dans un spectre RMN 1H donc plus le temps est
long avant la disparition du pic, plus le proton est difficilement échangeable avec le
solvant d’où la présence potentielle d’un pont hydrogène.
D’autres contraintes structurales peuvent être utilisées pour déterminer la structure
tridimensionnelle des protéines, mais ils n’ont pas été utilisés dans cette étude.
2.3.6. Dynamique des protéines La relaxation est le processus par lequel les spins des noyaux dans l'échantillon
reviennent à l'équilibre après avoir été excités. L’état de l'équilibre est l'état dans lequel
les populations des niveaux d’énergies correspondent à une distribution de Boltzmann. Le
taux de relaxation est influencé par les propriétés physiques et dynamiques de la
molécule. Ainsi, une étude de relaxation permettra de connaître les propriétés
dynamiques de la molécule. Afin de caractériser ces propriétés, certains paramètres sont
mesurés.
Tout d’abord, les temps de relaxation T1 (appelé parfois longitudinal ou spin-
réseau) caractérisent le retour à l’équilibre énergétique des noyaux après l’excitation.
Cette relaxation est due aux interactions avec le milieu environnant. Les temps de
relaxation transversale T2 (appelé aussi spin-spin) permettent également ce retour à
l’équilibre, mais le long des axes x et y (Levitt, 2001). Typiquement, 15N-T1 et 15N-T2
sont analysés pour la dynamique du squelette. De plus, {1H}15N-NOE est également dû à
la relaxation. Ces valeurs de temps de relaxation NOE sont mesurées.
La mesure de ces paramètres de relaxation permet d’extraire les paramètres de
dynamique suivant: le temps de corrélation globale (τc), le temps de corrélation local (τe),
et les paramètres d’ordre S2. Le temps de corrélation globale (τc) est le temps moyen dont
une molécule en solution a besoin pour accomplir une rotation d’un radian. Le temps de
corrélation local (τe) a la même signification que τc mais concernant un acide aminé. Les

29
paramètres d’ordre (S2) considèrent l’amplitude d’un acide aminé. Plus la valeur est
élevée, plus la région est rigide.
2.3.7. Calcul de structures tridimensionnelles La simulation de structures tridimensionnelles s’effectue à l’aide de toutes les
informations connues jusqu’à maintenant sur la protéine. La séquence de la protéine et les
contraintes structurales mentionnées ci-haut sont les éléments essentiels qui sont utilisés
dans les logiciels de calculs. Le but est de générer une famille de structures en accord
avec les contraintes expérimentales, tout en conservant la géométrie des acides aminés et
des liens peptidiques.
Un programme, nommé ARIA, développé au cours des dernières années est un des
programmes d'analyses automatisées des données RMN. Il accélère entre autre l'analyse
des données expérimentales en attribuant des corrélations dans les spectres NOESY.
ARIA possède également son propre système de calibration de NOE (Linge et al., 2001).
ARIA procède à 8 calculs itératifs dont la dernière itération subit une simulation
dans l’eau. Dans chacune de ces itérations, ARIA attribue, calibre et calcul un ensemble
de structures tridimensionnelles avec le logiciel CNS (Brunger et al., 1998; Linge et al.,
2001; Wishart and Sykes, 1994). Les simulations de structures peuvent se faire selon
deux méthodes : le mode Cartésien ou le mode Torsion. Le mode Cartésien tient compte
des coordonnées de chaque atome et les fait varier jusqu’à l’obtention de plus faibles
énergies où la structure est la plus probable en fonction des contraintes imposées. Le
mode torsion quant à lui varie des angles de torsion pour minimiser l’énergie présente
dans la structure.
La simulation des structures se fait à température élevée pour permettre le plus
grand nombre de mouvements structuraux qui seraient impossible à température
ambiante. Afin de ne pas rester emprisonné dans un minimum d’énergie locale c’est-à-
dire une énergie où il serait possible d’avoir des énergies encore plus basses, les calculs
de structures ont été faits par recuit simulé (simulated annealing) où la température
diminue palier par palier (Figure 2-20). Cette méthode permet aux structures générées de
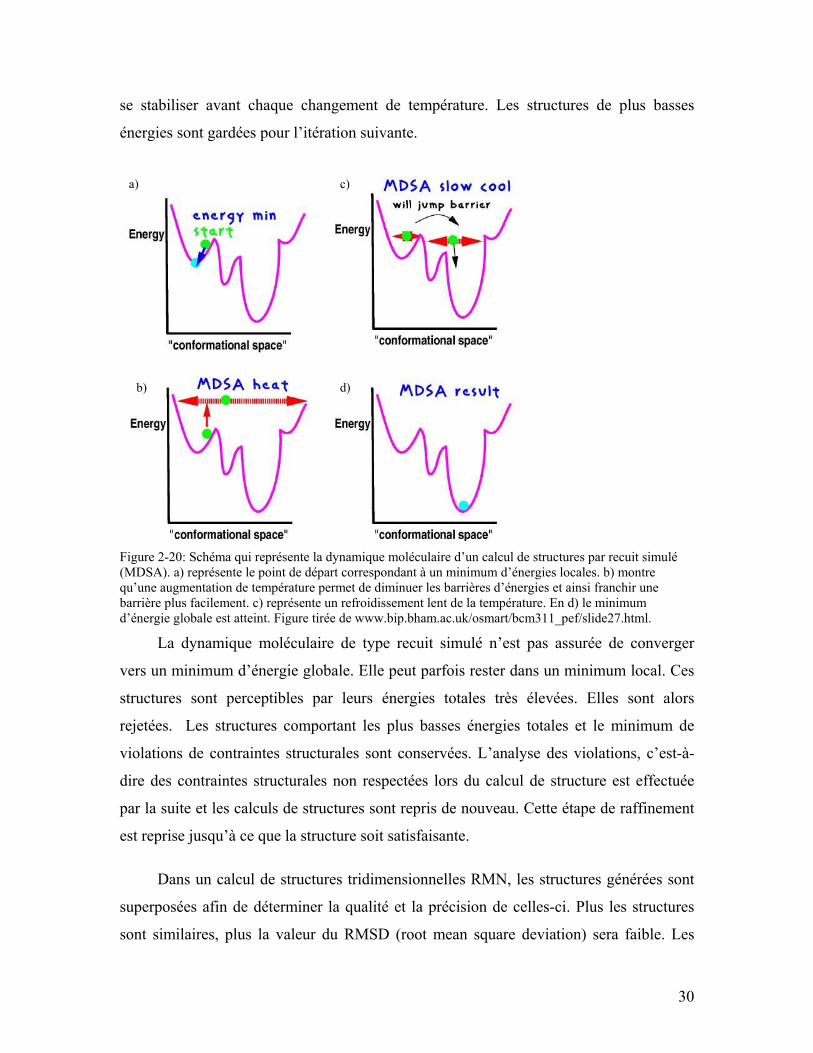
30
se stabiliser avant chaque changement de température. Les structures de plus basses
énergies sont gardées pour l’itération suivante.
a)
b) d)
c)a)
b) d)
c)
Figure 2-20: Schéma qui représente la dynamique moléculaire d’un calcul de structures par recuit simulé (MDSA). a) représente le point de départ correspondant à un minimum d’énergies locales. b) montre qu’une augmentation de température permet de diminuer les barrières d’énergies et ainsi franchir une barrière plus facilement. c) représente un refroidissement lent de la température. En d) le minimum d’énergie globale est atteint. Figure tirée de www.bip.bham.ac.uk/osmart/bcm311_pef/slide27.html.
La dynamique moléculaire de type recuit simulé n’est pas assurée de converger
vers un minimum d’énergie globale. Elle peut parfois rester dans un minimum local. Ces
structures sont perceptibles par leurs énergies totales très élevées. Elles sont alors
rejetées. Les structures comportant les plus basses énergies totales et le minimum de
violations de contraintes structurales sont conservées. L’analyse des violations, c’est-à-
dire des contraintes structurales non respectées lors du calcul de structure est effectuée
par la suite et les calculs de structures sont repris de nouveau. Cette étape de raffinement
est reprise jusqu’à ce que la structure soit satisfaisante.
Dans un calcul de structures tridimensionnelles RMN, les structures générées sont
superposées afin de déterminer la qualité et la précision de celles-ci. Plus les structures
sont similaires, plus la valeur du RMSD (root mean square deviation) sera faible. Les

31
RMSD peuvent être calculés selon le squelette, les atomes lourds (C, O, N,S) ou bien
différentes sections de la séquence protéique. Le RMSD est calculé suivant l’équation
suivante.
Equation 4: Équation de la RMSD (root mean square deviation). N est le nombre d’échantillon, i est la sommation sur chacun des échantillons. ri et ri
.représente les structure ׳
Les régions N-terminale et C-terminale de la protéine sont souvent non structurée et
libre en solution d’où le RMSD plus élevé. Les boucles (régions entre deux structures
secondaires) ont généralement un RMSD supérieur aux régions structurées telles les
hélices α et les feuillets β. Pour visualiser et manipuler les structures tridimensionnelles,
plusieurs logiciels sont disponibles. Swiss PDB Viewer, Inside II et Molmol (Koradi,
1996) ne sont que quelques exemples.

32
CHAPITRE 3. MATERIELS ET METHODES
3.1. Expression, marquage isotopique et purification de MTH187
La réaction en chaîne de la polymérase (PCR) a été utilisée pour amplifier la région
codante du gène MTH187. Ce gène a été cloné dans le vecteur d'expression procaryotique
pET-15b (Novagen) au site de restriction Nde1 et BamH1 (Figure 3-1). Au site N-
terminal de ce vecteur se trouve une séquence His-Tag suivie par un site de clivage de la
thrombine (Figure 3-2). Cette construction résulte en 20 résidus supplémentaires en N-
terminal (MGSSHHHHHHSSGLVPRGSH).
Figure 3-1 : Vecteur d’expression pET-15b

33
Figure 3-2: Région du vecteur d’expression pET-15b. (Novagen)
La protéine recombinante fut exprimée dans des cellules E. coli BL21(λDE3)
(Novagen). Les cellules ont poussé dans un milieu minimum M9 contenant des
suppléments de vitamines et de minéraux. La seule source d’azote et de carbone sont des
composés marqués. Le chlorure d'ammonium marqué: 15NH4Cl et/ou le 13C-Glucose.
Grâce à la séquence His-Tag, il a été possible de purifier la protéine à l’aide d'une
colonne de nickel.
Ces étapes d'expression et de purification ont été faites à Toronto dans le
laboratoire de Cheryl H. Arrowsmith. La méthode d’expression et de purification de la
protéine est décrite par (Christendat et al., 2000).
3.2. Préparation des échantillons
3.2.1. Contenu des échantillons Le premier échantillon a été préparé à Toronto dans le laboratoire de Cheryl H.
Arrowsmith. La protéine de cet échantillon est marquée uniquement à 15N. La protéine
est dissoute dans une solution de 600µl de tampon d’acétate de sodium 25mM à pH 6.2,
90% H2O : 10% D2O. La solution contient également 10mM de chlorure de sodium à une
concentration 10mM, 0,5mM de DSS(2,2-diméthyl-2-silapentane-5-sulfonate) et 22mM
de CHAPS. Le CHAPS ((3-[(Cholamidopropyl)diméthylammonio]-1-propane-sulfonate)
est utilisé pour que la protéine reste sous forme monomère dans l’échantillon. Un
échange entre monomère et dimère cause un élargissement les raies dans un spectre
RMN, donc le spectre devient plus difficile à interpréter.

34
Le deuxième échantillon dans lequel la protéine MTH187 est marquée à 15N et au 13C a été également préparé à Toronto. Les concentrations de CHAPS et de sels ont été
ajustées pour que les déplacements chimiques des atomes dans les spectres soient
semblables à ceux pris avec le premier échantillon.
Lors de l’analyse des résidus aromatiques, nous avons lyophilisé le deuxième
échantillon, ajouté 600µl de D2O à 99.996% et laissé reposer pour la nuit. Par la suite,
nous avons lyophilisé à nouveau pour enlever tout excès d'eau et redissous dans 600µl de
D2O 99.996%. 0.1% d’azide a également été ajouté à l’échantillon pour éviter la
croissance de bactéries. Le pH est vérifié à l’aide du pH-mètre et/ou l’imidazole. Pour
l’imidazole, une concentration dans l'échantillon de 2mM est ajoutée comme référence.
L’imidazole sert de référence de pH interne via la relation entre le pH et le déplacement
chimique d’un des pics de l’imidazole.
3.2.2. Concentration de la protéine dans l'échantillon Pour le deuxième échantillon, la concentration de MTH187 a été mesurée à l'aide
d'un dosage bicinchoninic acid (BCA) de la compagnie PIERCE.
3.3. Résonance magnétique nucléaire Comme le spectromètre RMN 600MHz du laboratoire n'était pas encore installé,
l'acquisition des spectres de l'échantillon dans lequel la protéine est marquée à 15N s'est
effectuée au centre national de résonance magnétique nucléaire (NANUC) situé à
Edmonton en Alberta. Le spectre 3D 15N-TOCSY-HSQC (Clore et al., 1991) a été
acquisitionné sur le spectromètre 500MHz et le spectre 3D 15N-NOESY-HSQC (Ikura et
al., 1990) sur le spectromètre 800MHz. Les autres spectres ont tous été acquisitionnés sur
le spectromètre RMN Varian INOVA 600MHz de l'université Laval à une température de
43.2°C. La température d’un échantillon fait varier le déplacement chimique donc, c'est
en ce référent au déplacement chimique des atomes des spectres acquisitionnés à
Edmonton que cette température nous ai apparu comme étant la meilleure.
L’attribution des déplacements chimiques des noyaux 1H, 15N, 13C a été réalisée en
utilisant plusieurs expériences RMN hétéronucléaires multidimensionnelles. Les
expériences suivantes ont été utilisées pour l'attribution de la chaîne squelettique: 3D 15N-

35
NOESY-HSQC, 3D 15N-TOCSY-HSQC, 3D CBCACONH (Grzesiek et al., 1992), 3D
HNCACB (Kay et al., 1990), 3D HNCO (Muhandiram, 1994), 3D HN(CA)CO (Clubb
R.T, 1992), 3D HCA(CO)N (Palmer et al., 1992), 3D HN(CO)CA (Bax and Ikura, 1991)
et le 2D 13C-HSQC (Muller, 1979). Les spectres 3D 15N-TOCSY-HSQC, 3D HCCH-
TOCSY (Olejniczak et al., 1992) et 3D CC(CO)NH (Lyons et al., 1993) ont été
acquisitionnés pour attribuer les chaînes latérales.
Ces derniers spectres ne permettent pas d’attribuer les déplacements chimiques des
groupements aromatiques. L’acquisition des spectres HCCH-COSY de deux et de trois
dimensions ont par conséquent été utilisés pour compléter l’attribution des Phe, Tyr et
Trp. Pour bien visualiser les corrélations entre les noyaux aromatiques, la protéine a été
dissoute dans le D2O.
Lors de la détermination de la structure tridimensionnelle de MTH187, des
contraintes d’angle dièdre ont été obtenues à partir de constantes de couplages 3JHNHα,
lesquelles ont été mesurées en utilisant l’expérience HNHA (Geerten W. Vuister, 1993).
Les contraintes NOEs ont été obtenues avec l’utilisation de deux expériences le 15N-
NOESY HSQC et le 13C-NOESY HSQC. Tous les deux ont un temps de mélange de
150ms.
L’échantillon dont la protéine est marquée à 15N et 13C a été utilisé pour les
expériences de relaxation dans une solution tampon 90% H2O et 10% D2O. Le pH a été
ajusté sur 6.2 avec HCl et/ou NaOH avant le transfert au tube RMN. Les expériences 15N-
T1, 15N-T2 et 15N-{1H}-15N ont été exécutées en utilisant les séquences d'impulsions de
(Farrow et al., 1994). Les T1 et T2 ont été acquisitionnés en utilisant les délais suivant.
Pour T1, des délais de [0,10,20,40,80,160,320,640 et 1280 ms] ont été utilisés et pour les
T2 ce sont des délais de [10,30,50,70,90,110,150 et 190 ms]. Après la transformation des
spectres, les intensités des pics ont été évaluées pour chaque délai donné pour calculer les
valeurs de T1 et de T2 de chaque résidu selon l’Equation 5. L’ajustement des données
(curvefitting) a été fait avec le logiciel curvefit (Mandel et al., 1995). L’incertitude sur les
valeurs de T1 et T2 a été calculée d’après la simulation de Jackknife. Elle consiste à

36
estimer des variances de manière à tenir compte de l’imputation de valeurs au moyen des
poids de rééchantillonnage (Shao, 1989).
I(t) = I0 e (–t/T1) et I(t) = I0 e (–t/T2)I(t) = I0 e (–t/T1) et I(t) = I0 e (–t/T2)
Equation 5 : I(t) est l’intensité après le délai t et I0 est l’intensité au t=0.
Pour le calcul des NOEs un spectre 15N-HSQC est acquisitionné avec effet NOE,
c’est à dire en saturant les protons, et un autre HSQC est acquisitionné sans effet NOE.
Par la suite, nous avons divisé les intensités du spectre avec NOE par celles du spectre
sans NOE.
Les T1, T2 et NOEs ont été calculés pour 78 acides aminés des 111 acides aminés
de MTH187. Ces acides aminés sont: 12,17-19, 24, 27, 30-56, 58-60, 63-66, 69-74, 76-
86, 88-98, 100, 101, 104-111.
C’est à l’aide du logiciel de Modelfree (Mandel et al., 1995) que l’on a pu calculer
le temps de corrélation de MTH187 dans l’eau. Le logiciel utilise des constantes
R1(1/T1), R2 (1/T2) et NOEs pour trouver le temps de corrélation global (τc). Pour obtenir
le temps de corrélation de la protéine MTH187 dans une solution de deutérium, nous
avons utilisé un rapport de viscosité D2O/H2O de 8.9 mpoise.
Les spectres ont tous été transformés à l’aide du logiciel NMRPipe (Delaglio et al.,
1995). Les fonctions typiques de transformation utilisées pour l’extraction de solvant sont
la fonction SOL, qui a été utilisée pour la grande majorité des spectres, et la fonction
POLY utilisée entre autre pour le spectre 13C-NOESY-HSQC. La fonction multiplicative
utilisée pour les fids des spectres est la fonction sinusoïdale ou cosinus. Le nombre de
point prédit lors de la prédiction linéaire dans une dimension donnée a toujours été la
quantité de points acquisitionnés dans cette même dimension. De plus, celle-ci a toujours
été effectuée après que deux dimensions aient été transformées. Le nombre de zéro ajouté
lors de la fonction zéro filling est le double du nombre de points acquisitionnés et prescrit
dans une dimension. De plus cette valeur doit être une puissance de deux. La Figure 3-3

37
montre un exemple typique de script de transformation en trois dimensions. Les
paramètres d’acquisitions de toutes les expériences sont mentionnés à la Table 1.
# Processing of F3 dimension (1H) # xyz2pipe -in data/pipe%03d.fid -x -verb \ | nmrPipe -fn POLY -time \ | nmrPipe -fn SP -off 0.3 -end 1.00 -pow 1 -c 1.0 \ | nmrPipe -fn ZF -auto \ | nmrPipe -fn FT \ | nmrPipe -fn PS -p0 90.00 -p1 0 -di \ | nmrPipe -fn EXT -x1 5.0PPM -xn -0.6PPM -sw \ | pipe2xyz -out DAT/3d%03d.DAT -x -ov # # Processing of F2 dimension (C) without LP # xyz2pipe -in DAT/3d%03d.DAT -z -verb \ | nmrPipe -fn SP -off 0.5 -end 0.95 -pow 1 -c 1.0 \ | nmrPipe -fn ZF -size 128 \ | nmrPipe -fn FT -di \ | pipe2xyz -out DAT/3d%03d.DAT -z -inPlace # # Processing of F1 dimension (1H) with LP # xyz2pipe -in DAT/3d%03d.DAT -y -verb \ | nmrPipe -fn LP -fb -pred 128 \ | nmrPipe -fn SP -off 0.3 -end 1.00 -pow 2 -c 1.0 \ | nmrPipe -fn ZF -size 512 \ | nmrPipe -fn FT \ | nmrPipe -fn PS -p0 0 -p1 0 -di \ | pipe2xyz -out DAT/3d%03d.DAT -y -inPlace # # Re-Processing of F2 dimension (C) with LP # xyz2pipe -in DAT/3d%03d.DAT -z -verb \ | nmrPipe -fn HT \ | nmrPipe -fn FT -inv \ | nmrPipe -fn ZF -inv \ | nmrPipe -fn SP -off 0.500 -end 0.95 -pow 1 -c 1.0 -inv \ | nmrPipe -fn LP -fb -pred 32 \ | nmrPipe -fn SP -off 0.5 -end 1.00 -pow 2 -c 1.0 \ | nmrPipe -fn ZF -size 128 \ | nmrPipe -fn FT \ | nmrPipe -fn PS -p0 -7.0 -p1 0 -di \ | pipe2xyz -out DAT/3d%03d.DAT -z -inPlace # # Base-line correct F1 (indirect 1H) dimension # xyz2pipe -in DAT/3d%03d.DAT -y -verb \ | nmrPipe -fn POLY -auto -ord 1 \ | pipe2xyz -out DAT/3d%03d.DAT -y -inPlace # # Base-line correct z dimension # xyz2pipe -in DAT/3d%03d.DAT -z -verb \ | nmrPipe -fn POLY -auto -window 5 -ord 1 \ | pipe2xyz -out DAT/3d%03d.DAT -z –inPlace
Figure 3-3 : Script de transformation du spectre HCCH-TOCSY. Le symbole # désactive la fonction qui suit et \ annonce une continuité de ligne.

38
Les spectres transformés ont été analysés en utilisant le programme NMRview
(Johnson B. A., 1994). Une fois tous les spectres attribués, un logiciel nommé Fillshifts
(Laboratoire du Dr Stéphane Gagné, Université Laval) activé à partir de NMRview a
permis de calculer une moyenne de tous les déplacements chimiques retrouvés dans les
spectres. Le fichier résultant regroupe les valeurs moyennes de déplacements chimiques
de tous les atomes de la protéine.
Table 1 : Table énumérant les paramètres d’acquisitions des différentes expériences RMN
dimensions nb. de points complexes
largeur spectrale
temps d'acquisition
prédiction linéaire
dimensions finales (nb de points finales)
15N TOCSY HSQC 1HN 512 8 000 aucune 1536
tm = 100ms 1H 141 5 997 115 768
15N 32 1 418 32 192 15N NOESY HSQC 1HN 768 12 000 aucune 1792
tm = 150ms 1H 200 10 000 200 912
15N 32 2 269 32 192 13C NOESY HSQC H1(F3) 512 10 000 179 h 30 aucune 1536
H2(F1) 128 7 676 128 768
13C 63 6 032 64 383
CBCACONH 1HN 750 10 000 71 h 30 aucune 1774
13C 70 12 064 70 268
15N 27 1 800 27 118
HNCO 1HN 750 10 000 20 h aucune 1774
13C 60 2 000 60 376
15N 32 1 800 32 192
HNCACB 1HN 750 10 000 69 h aucune 1774
13C 69 12 064 58 383
15N 27 1 800 32 187
HCCH-TOCSY 1H 640 10 000 aucune 1664
1HN 128 8 000 128 768
13C 32 12 066 32 192
HN(CA)CO 1HN 640 10 000 140 h aucune 1664
13C 60 2 000 60 376
15N 32 1 800 32 192
CC(CO)NH 1HN 750 10 000 aucune 1774
13C 128 12 064 128 512
15N 32 1 800 32 192
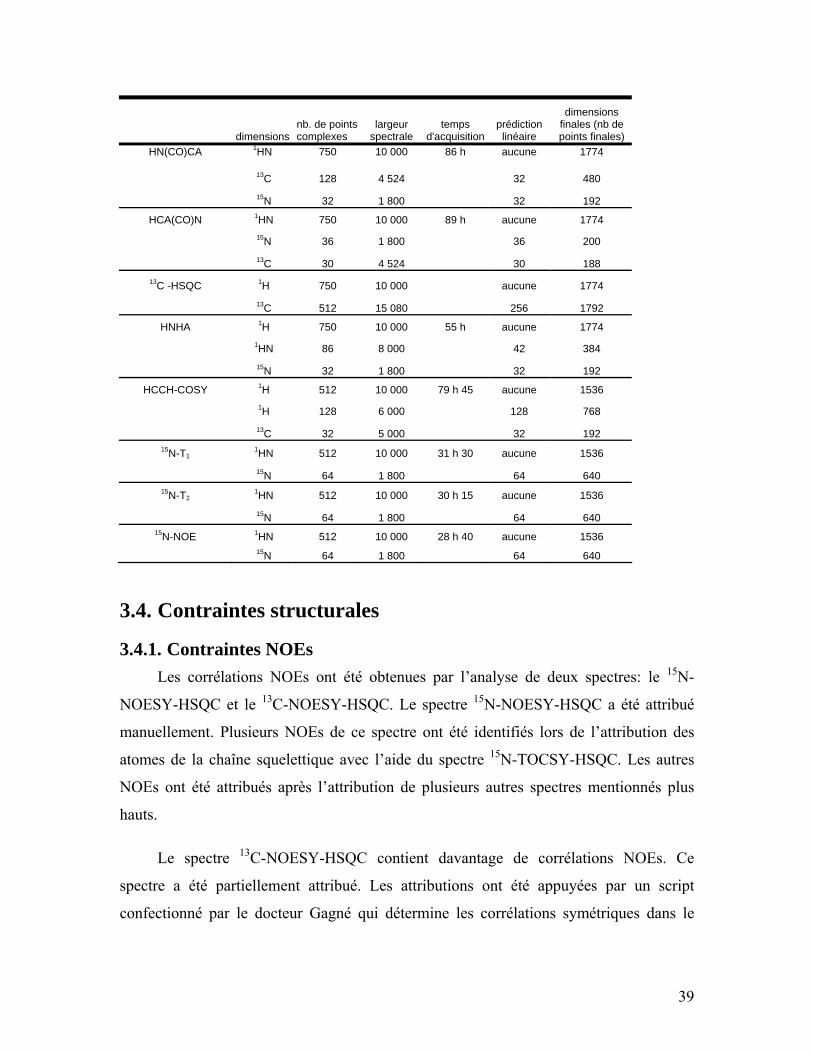
39
dimensions nb. de points complexes
largeur spectrale
temps d'acquisition
prédiction linéaire
dimensions finales (nb de points finales)
HN(CO)CA 1HN 750 10 000 86 h aucune 1774
13C 128 4 524 32 480
15N 32 1 800 32 192
HCA(CO)N 1HN 750 10 000 89 h aucune 1774
15N 36 1 800 36 200
13C 30 4 524 30 188 13C -HSQC 1H 750 10 000 aucune 1774
13C 512 15 080 256 1792
HNHA 1H 750 10 000 55 h aucune 1774
1HN 86 8 000 42 384
15N 32 1 800 32 192
HCCH-COSY 1H 512 10 000 79 h 45 aucune 1536
1H 128 6 000 128 768
13C 32 5 000 32 192 15N-T1 1HN 512 10 000 31 h 30 aucune 1536
15N 64 1 800 64 640 15N-T2 1HN 512 10 000 30 h 15 aucune 1536
15N 64 1 800 64 640 15N-NOE 1HN 512 10 000 28 h 40 aucune 1536
15N 64 1 800 64 640
3.4. Contraintes structurales
3.4.1. Contraintes NOEs Les corrélations NOEs ont été obtenues par l’analyse de deux spectres: le 15N-
NOESY-HSQC et le 13C-NOESY-HSQC. Le spectre 15N-NOESY-HSQC a été attribué
manuellement. Plusieurs NOEs de ce spectre ont été identifiés lors de l’attribution des
atomes de la chaîne squelettique avec l’aide du spectre 15N-TOCSY-HSQC. Les autres
NOEs ont été attribués après l’attribution de plusieurs autres spectres mentionnés plus
hauts.
Le spectre 13C-NOESY-HSQC contient davantage de corrélations NOEs. Ce
spectre a été partiellement attribué. Les attributions ont été appuyées par un script
confectionné par le docteur Gagné qui détermine les corrélations symétriques dans le

40
spectre et suggère différentes attributions. Le jugement d’un évaluateur est nécessaire
pour décider de l'attribution.
La calibration des NOEs de ces deux spectres a été faite pour les calculs de
structures tridimensionnelles. La majorité des calculs des structures de MTH187 ont été
calculés dans un mode de torsion avec un protocole recuit simulé utilisant le logiciel
ARIA version 1.2. Ce logiciel peut calibrer les NOEs. La calibration des NOEs dans le
spectre 13C-NOESY-HSQC a été réalisée uniquement à l’aide du système de calibration
contenu dans Aria. L’intensité du pic est calibrée en une distance et sur cette valeur une
erreur est ajoutée. Pour la calibration des HN, la conception d'un algorithme a été mise
aux points par Etienne Enoumen du laboratoire de Stéphane Gagné à l’université Laval.
La distance minimum est de 1.7 Å. Au fur et a mesure de sa conception, sa fiabilité a été
testée par rapport au système de calibration d'Aria.
3.4.2. Contraintes d'angles dièdres
3.4.2.1. TALOS Les angles phi et psi ont été estimés pour chaque résidu de MTH187 avec le
système de banques de données de TALOS (Cornilescu et al., 1999). Les déplacements
chimiques des atomes Hα, Cα, Cβ, CO, N furent utilisés et la comparaison des
déplacements chimiques s’est effectuée avec les 20 protéines dans la banque de données
de TALOS. La contrainte est acceptée si la corrélation est bonne dans au moins 9 cas sur
10. L’erreur minimum utilisée pour ces contraintes d’angles dièdres est de 14 degrés pour
le phi et 13 degrés pour le psi.
3.4.2.2. Constantes de couplages Les constantes de couplages entre les HN et les Hα des résidus de la protéine
MTH187 ont été mesurées à l’aide du spectre RMN HNHA. La période de couplage H-H
est de 25 ms.
Le spectre HNNA a été attribué à l’aide du logiciel NMRView. Les constantes de
couplages furent calculées avec l’outil d’analyse HNHA inclus avec NMRView. Les
paramètres nécessaires aux calculs des constantes sont le temps de mélange de 25 ms.
Une déviation standard de 0.02 et un facteur de correction de 0.86 furent utilisés. L’erreur

41
minimum utilisée pour les constantes de couplage est de ± 1,0 Hz. Les constantes de
couplage sont utilisées directement dans le logiciel CNS pour des calculs de structures.
3.4.2.3. Pont Hydrogène
L’échantillon dissous dans le D2O a été lyophilisé et redissout dans l'eau. Treize
spectres 15N-HSQC ont été acquisitionnés un à la suite de l’autre sur une période de 4
heures et comparés afin de déterminer quel est le temps d'échange de chaque groupement
NH.
Les contraintes de ponts hydrogène furent utilisées qu’à partir du dix-huitième
calcul de structures. Une contrainte de pont hydrogène fut définie pour les amides ayant
un échange lent avec le solvant et possédant un pont hydrogène potentiel dans le cycle de
calcul précédent. La contrainte de pont hydrogène a été définie sous forme de contrainte
de type NOE avec une distance de 2.1-3.2 Å.
3.5. Génération de structures tridimensionnelles La majorité des calculs de structures de MTH187 ont été calculés dans un mode de
torsion avec un protocole recuit simulé utilisant le logiciel ARIA version 1.2. Les calculs
de structures du logiciel ARIA se sont fait selon les valeurs de défauts offertes par le
logiciel. Dans la première étape, la température est augmentée à 10000 Kelvin. Par la
suite la température est passée de 10000 Kelvin à 1000 Kelvin en 5000 étapes de
dynamiques moléculaires. La deuxième étape de 1000 Kelvin à 50 Kelvin et ce, en
l’espace de 4000 étapes de dynamiques moléculaires. Les principaux paramètres utilisés
sont les contraintes de distances et les contraintes d’angles dièdres. Les contraintes de
distances, incluant les ponts hydrogènes, n’ont pas été utilisées à la première itération.
Lors du chauffage, 10 contraintes de distances ont été utilisées. Ensuite, au premier
refroidissement le nombre est monté à 50 et au deuxième refroidissement 50 contraintes
de distances ont également été utilisées. Pour les contraintes d’angles dièdres, 5
contraintes ont été utilisées lors du chauffage, 25 au premier refroidissement et 200 au
deuxième. Beaucoup d’autres paramètres interviennent dans les calculs de structures mais
les valeurs de défaut ont été utilisées.

42
Les calculs ont été exécutés à l’aide d’un ordinateur DELL Précision 650 (deux
processeurs) sous le système d'exploitation Linux. Les structures sont visualisées avec le
logiciel MOLMOL. Parmi les structures possédant un RMSD bas, la structure possédant
la plus basse énergie globale s'est révélée être la structure choisie pour analyse plus
profonde, entre autre pour l'homologie structurale.
3.6. Homologie structurale Les logiciels utilisés pour la recherche d'homologues structuraux à MTH187 sont
DALI (distance matrix alignment) (Holm and Sander, 1996) et CE (Combinatorial
Extension) (Shindyalov, 1998). Les coordonnées de la meilleure structure en format PDB
ont été soumises aux programmes pour y trouver des structures homologues dans les
banques de données de structures connues.
Ces homologues ont été superposés un à un avec MTH187 à l’aide du logiciel
Swiss PDB Viewer (Guex, 1997). Les RMSD des atomes Cα de chaque superposition de
protéines ont été calculés et comparés pour identifier les structures les plus similaires à
MTH187.
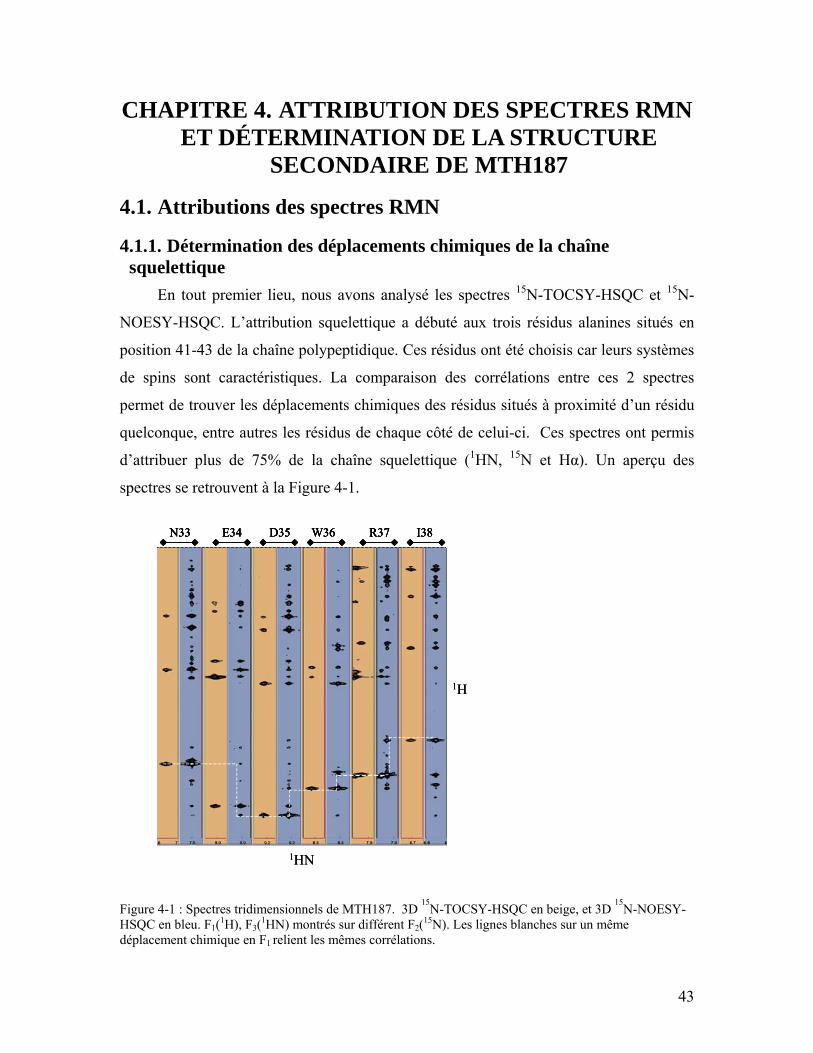
43
CHAPITRE 4. ATTRIBUTION DES SPECTRES RMN ET DÉTERMINATION DE LA STRUCTURE
SECONDAIRE DE MTH187
4.1. Attributions des spectres RMN
4.1.1. Détermination des déplacements chimiques de la chaîne squelettique
En tout premier lieu, nous avons analysé les spectres 15N-TOCSY-HSQC et 15N-
NOESY-HSQC. L’attribution squelettique a débuté aux trois résidus alanines situés en
position 41-43 de la chaîne polypeptidique. Ces résidus ont été choisis car leurs systèmes
de spins sont caractéristiques. La comparaison des corrélations entre ces 2 spectres
permet de trouver les déplacements chimiques des résidus situés à proximité d’un résidu
quelconque, entre autres les résidus de chaque côté de celui-ci. Ces spectres ont permis
d’attribuer plus de 75% de la chaîne squelettique (1HN, 15N et Hα). Un aperçu des
spectres se retrouvent à la Figure 4-1.
N33 E34 D35 W36 R37 I38N33 E34 D35 W36 R37 I38
1HN
1H
N33 E34 D35 W36 R37 I38N33 E34 D35 W36 R37 I38
1HN
1H
Figure 4-1 : Spectres tridimensionnels de MTH187. 3D 15N-TOCSY-HSQC en beige, et 3D 15N-NOESY-HSQC en bleu. F1(1H), F3(1HN) montrés sur différent F2(15N). Les lignes blanches sur un même déplacement chimique en F1 relient les mêmes corrélations.

44
Par la suite, l’obtention de l’échantillon doublement marqué (15N/13C) a permis
d’améliorer les attributions du squelettique à partir des spectres CBCA(CO)NH et
HNCACB (Figure 4-2). Les déplacements chimiques de la chaîne squelettique ont été
complétés, à l’exception de 12 résidus qui n’ont pu être attribués à partir de ces spectres.
Il est à noter que ces résidus sont majoritairement situés au début de la protéine. Ces
spectres ont permis de trouver les déplacements chimiques Cα et Cβ de plusieurs résidus.
N33 E34 D35 W36 R37 I38
1HN
13C
N33 E34 D35 W36 R37 I38
1HN
13C
Figure 4-2 : Spectres tridimensionnels de MTH187. CBCA(CO)NH en beige et HNCACB en bleu. F1(13C), F3(1HN) montrés sur différent F2(15N). Les lignes blanches sur un même déplacement chimique en F1 relient les mêmes corrélations. Le spectre HNCACB montre les corrélations Cα en noire et les Cβ en rouge.
Les spectres HNCO et HN(CA)CO (Figure 4-3) ont été analysés pour compléter les
attributions de la chaîne principale et également pour attribuer les déplacements
chimiques des C΄ de la chaîne squelettique. Après l’analyse de ces spectres, seulement
trois résidus n’ont pu être attribués. La méthionine située au début de la protéine, la
glycine 67 et la phénylalanine 99.

45
G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86
1HN
G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86
1HN
13CO
G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86
1HN
G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86G81 G82 E83 R84 V85 R86
1HN
13CO
Figure 4-3: Spectres tridimensionnels de MTH187. HNCO en beige et HN(CA)CO en bleu. F1(CO), F3(1HN) montrés sur différent F2(15N).
Quelques spectres additionnels ont été acquisitionnés dans le but d’attribuer les
déplacements chimiques des trois résidus non attribués. Le spectre HCA(CO)N a été très
utile pour confirmer certaines attributions déjà identifiées, mais aucune nouvelle
attribution n’a pu être trouvé. Le spectre HN(CO)CA (Figure 4-4), plus sensible que le
spectre CBCA(CO)NH, a cependant permis d’attribuer le déplacement chimique de
l’amide de la phénylalanine 99.
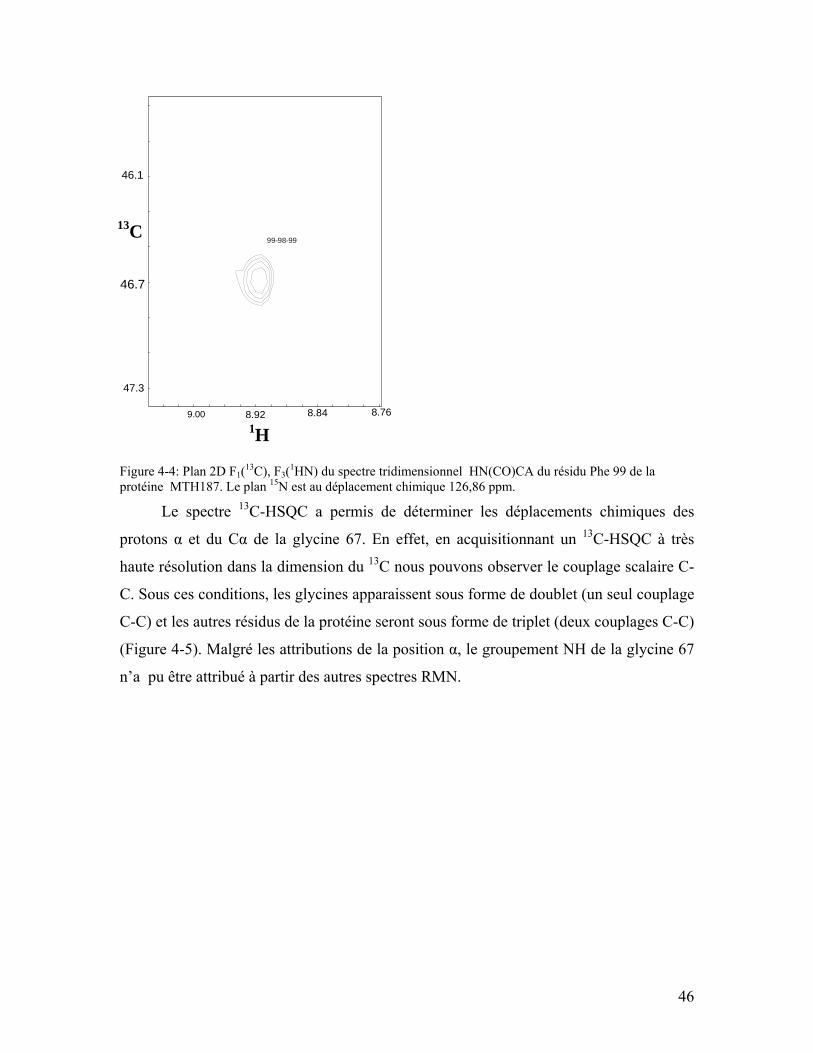
46
Figure 4-4: Plan 2D F1(13C), F3(1HN) du spectre tridimensionnel HN(CO)CA du résidu Phe 99 de la protéine MTH187. Le plan 15N est au déplacement chimique 126,86 ppm.
Le spectre 13C-HSQC a permis de déterminer les déplacements chimiques des
protons α et du Cα de la glycine 67. En effet, en acquisitionnant un 13C-HSQC à très
haute résolution dans la dimension du 13C nous pouvons observer le couplage scalaire C-
C. Sous ces conditions, les glycines apparaissent sous forme de doublet (un seul couplage
C-C) et les autres résidus de la protéine seront sous forme de triplet (deux couplages C-C)
(Figure 4-5). Malgré les attributions de la position α, le groupement NH de la glycine 67
n’a pu être attribué à partir des autres spectres RMN.
8.76 8.84 8.92 9.00
46.1
46.7
47.3
99-98-99
13C
1H

3.0 3.5 4.0
41.8
43.8
45.8
47.8
81
81
96
81
82
20
8296
20
47 47
4747
40
40
Figure 4-5 : Expansion du scorrélations Hα-Cα. Les axe(ppm). Dans ce spectre à hadonnent lieu à des doublets
4.1.2. Attribution dLe spectre 15N-T
de la chaîne latérale. L
et les autres protons à
pas très sensible, surto
loin du NH n’ont sou
HCCH-TOCSY a été n
de déplacements chim
l’aide du spectre CC(C
pu être identifiés. Ces
déplacements chimiqu
figures suivantes, (Figu
pour les trois derniers s
δ ( ppm)
13C
1H47
pectre 2D 13C-HSQC de MTH187 illustrant la région caractéristique des s montrent les déplacements chimiques (δ) des atomes en partie par millions ute résolution où le couplage scalaire C-C est présent, les Cα des glycines alors que les Cα des autres acides aminés donnent lieu à des triplets.
es déplacements chimiques des chaînes latérales OCSY-HSQC permet de déterminer les déplacements chimiques
e spectre 15N-TOCSY-HSQC donne les corrélations entre un NH
l’intérieur du même système de spins. Cependant, ce spectre n’est
ut pour les résidus dans une hélice-α. En effet, les protons situés
vent pas de corrélation avec celui-ci. C’est pourquoi le spectre
écessaire. Ce spectre a permis de trouver une très grande majorité
iques des protons des chaînes latérales ainsi que des carbones. A
O)NH les déplacements chimiques des carbones non attribués ont
derniers ont pu être retracés dans le spectre HCCH-TOCSY et les
es des protons sur ces carbones ont pu être attribués. Les trois
re 4-6, Figure 4-7, Figure 4-8) nous donne un exemple des plans
pectres cités plus haut où l’on retrouve la leucine 31.

48
2
6.5 6.9 7.37.7
2
4
6
8
HN31-HN31
HN31-Hβ131
HN31-Hβ231
Figure 4-6: Région sélectionnée F1(1H)-F3(1HN) du spectre 15N-TOCSY-HSQC de MTH187 situé à 118.7 ppm sur l’axe F2(15N).Les corrélations observées pour la leucine 31 sont indiquées. Les corrélations entre l’amide et les protons β sont très faibles, et les corrélations avec les protons γ et δ sont absentes dans ce spectre.
0.1 1.1 2.1 3.1
0.5
1.5
2.5
3.5
Hδ1131Hβ231Hδ2131Hβ131Hδ1131Hβ131
Hδ2131Hβ231
Hγ31Hβ231 Hγ31Hβ131
Hα31Hβ131Hα31Hβ231
0.1 1.1 2.1 3.1
0.5
1.5
2.5
3.5
Hδ1131Hβ231Hδ2131Hβ131Hδ1131Hβ131
Hδ2131Hβ231
Hγ31Hβ231 Hγ31Hβ131
Hα31Hβ131Hα31Hβ231
Figure 4-7: Région sélectionnée F1(1H)-F3(1H) du spectre HCCH-TOCSY de MTH187 situé à 40.6 ppm sur l’axe F2(13C). Les corrélations observées pour la leucine 31 sont indiquées. Les corrélations entre les protons β et les protons γ sont très faibles dans ce spectre.
1H
1H
1H
1H
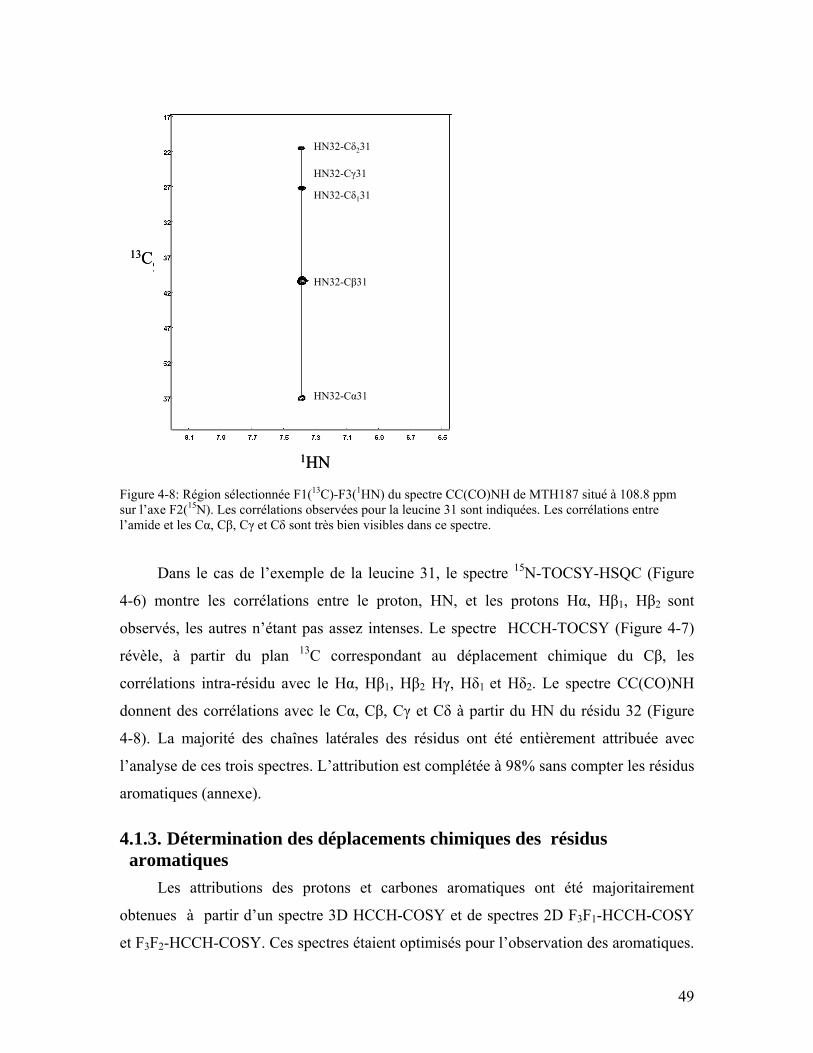
49
HN32-Cα31
HN32-Cβ31
HN32-Cδ131
HN32-Cδ231
C
HN32-Cγ31
HN32-Cα31
HN32-Cβ31
HN32-Cδ131
HN32-Cδ231
C
HN32-Cγ31
13C
1HN
HN32-Cα31
HN32-Cβ31
HN32-Cδ131
HN32-Cδ231
C
HN32-Cγ31
HN32-Cα31
HN32-Cβ31
HN32-Cδ131
HN32-Cδ231
C
HN32-Cγ31
13C
1HN
Figure 4-8: Région sélectionnée F1(13C)-F3(1HN) du spectre CC(CO)NH de MTH187 situé à 108.8 ppm sur l’axe F2(15N). Les corrélations observées pour la leucine 31 sont indiquées. Les corrélations entre l’amide et les Cα, Cβ, Cγ et Cδ sont très bien visibles dans ce spectre.
Dans le cas de l’exemple de la leucine 31, le spectre 15N-TOCSY-HSQC (Figure
4-6) montre les corrélations entre le proton, HN, et les protons Hα, Hβ1, Hβ2 sont
observés, les autres n’étant pas assez intenses. Le spectre HCCH-TOCSY (Figure 4-7)
révèle, à partir du plan 13C correspondant au déplacement chimique du Cβ, les
corrélations intra-résidu avec le Hα, Hβ1, Hβ2 Hγ, Hδ1 et Hδ2. Le spectre CC(CO)NH
donnent des corrélations avec le Cα, Cβ, Cγ et Cδ à partir du HN du résidu 32 (Figure
4-8). La majorité des chaînes latérales des résidus ont été entièrement attribuée avec
l’analyse de ces trois spectres. L’attribution est complétée à 98% sans compter les résidus
aromatiques (annexe).
4.1.3. Détermination des déplacements chimiques des résidus aromatiques
Les attributions des protons et carbones aromatiques ont été majoritairement
obtenues à partir d’un spectre 3D HCCH-COSY et de spectres 2D F3F1-HCCH-COSY
et F3F2-HCCH-COSY. Ces spectres étaient optimisés pour l’observation des aromatiques.
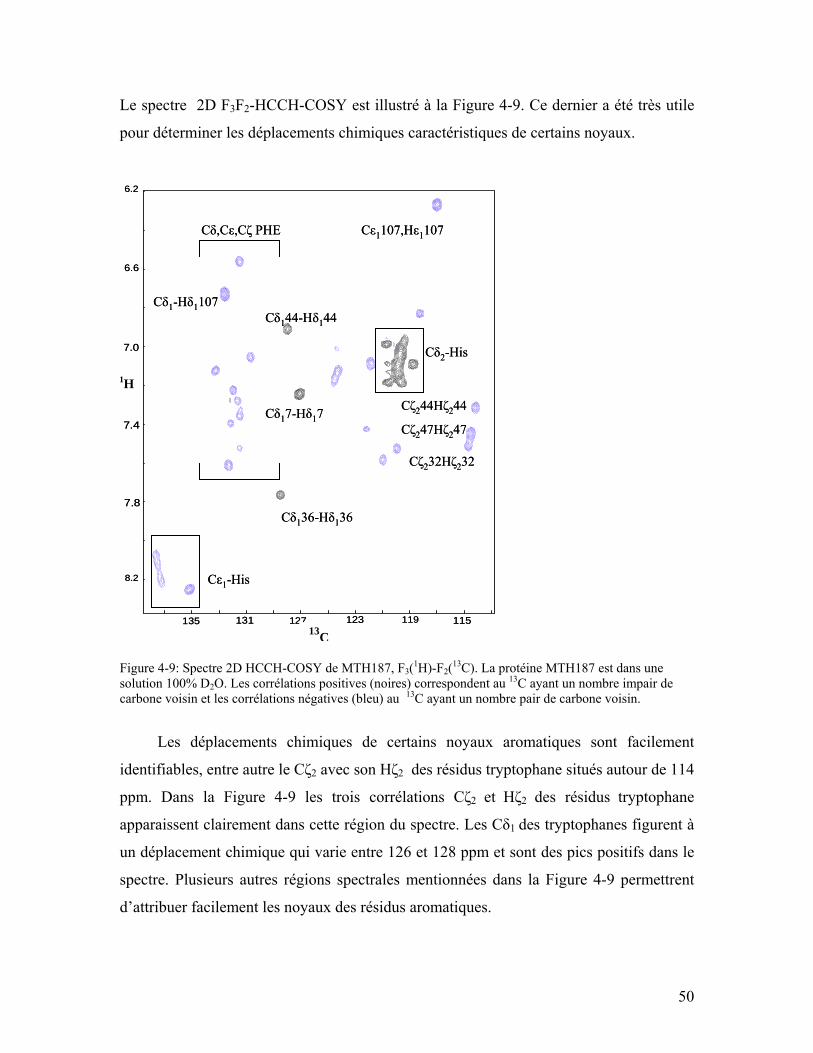
Le spectre 2D F3F2-HCCH-COSY est illustré à la Figure 4-9. Ce dernier a été très utile
pour déterminer les déplacements chimiques caractéristiques de certains noyaux.
11511912312131135
6.2
6.6
7.0
7.4
7.8
8.2
Cδ144-Hδ144
Cε1-His
Cδ136-Hδ136
Cδ1-Hδ1107
Cδ,Cε,Cζ PHE Cε1107,Hε1107
Cδ2-His
Cζ244Hζ244
Cζ247Hζ247
Cζ232Hζ232
Cδ17-Hδ17
11511912312131135
6.2
6.6
7.0
7.4
7.8
8.2
Cδ144-Hδ144
Cε1-His
Cδ136-Hδ136
Cδ1-Hδ1107
Cδ,Cε,Cζ PHE Cε1107,Hε1107
Cδ2-His
Cζ244Hζ244
Cζ247Hζ247
Cζ232Hζ232
Cδ17-Hδ17
Figure 4-9: Spectre 2D HCCH-COsolution 100% D2O. Les corrélatiocarbone voisin et les corrélations n
Les déplacements ch
identifiables, entre autre le C
ppm. Dans la Figure 4-9
apparaissent clairement dans
un déplacement chimique qu
spectre. Plusieurs autres rég
d’attribuer facilement les noy
1H
C137C13
713C
50
SY de MTH187, F3(1H)-F2(13C). La protéine MTH187 est dans une ns positives (noires) correspondent au 13C ayant un nombre impair de égatives (bleu) au 13C ayant un nombre pair de carbone voisin.
imiques de certains noyaux aromatiques sont facilement
ζ2 avec son Hζ2 des résidus tryptophane situés autour de 114
les trois corrélations Cζ2 et Hζ2 des résidus tryptophane
cette région du spectre. Les Cδ1 des tryptophanes figurent à
i varie entre 126 et 128 ppm et sont des pics positifs dans le
ions spectrales mentionnées dans la Figure 4-9 permettrent
aux des résidus aromatiques.

L’analyse du spectre 3D HCCH-COSY a permis d’identifier presque la totalité des
protons et carbones sur les cycles aromatiques. Le déplacement chimique des noyaux Cζ
et Hζ de la phénylalanine 49 n’ont pu être perçu ainsi que les Cζ3 et Hζ3 des trois
tryptophanes de MTH187. La Figure 4-10 montre un plan du spectre 3D HCCH-COSY.
16.36.7 7. 7.5
6.3
6.7
7.1
7.5
Hδ1107-Hε1107
Hε1107- Hδ1107
Hζ236-HH236
HH236- Hζ236
Hδ49-Hε49
Hε49-Hδ49
16.36.7 7. 7.5
6.3
6.7
7.1
7.5
Hδ1107-Hε1107
Hε1107- Hδ1107
Hζ236-HH236
HH236- Hζ236
Hδ49-Hε49
Hε49-Hδ49
Figure 4-10: Plan F1, F3 du spLes pics de la diagonale repréproton Hε du résidu 107. Les sont montré dans cette figure.
4.2. Synthèse de l’
Tous ces spectres o
protéine MTH187 excep
et de la Glycine 67. L’at15N-HSQC est illustré à l
H1
H1
1H
1H
51
ectre 3D HCCH-COSY de MTH187. MTH187 est dans une solution D2O. sentent les corrélations des protons et eux-même. Le proton Hδ corrèle avec le corrélations entre les protons Hζ2 et Hη2 du résidu 36 et Hδ-Hε du résidu 49
attribution
nt permis d’attribuer la chaîne squelettique des 111 résidus de la
tés les déplacements chimiques des HN et N de la Méthionine 1
tribution des chaînes latérales a été complétée à 98%. Le spectre
a (Figure 4-11) avec les attributions.

52
1H 6.5 6.7 6.9 7.1 7.3 7.5 7.7 7.9 8.1 8.3 8.5 8.7 8.9 9.1 9.3 9.5 9.7 9.9 10.1 10.3
104
106
108
110
112
114
116
118
120
122
124
126
128
130
M89S13
V105E25E56N48
N106R70
S76Q79R39
V55F24
E109
A74
L58
L62K91
K102L61
R75
V69E29A87
I38
L31I46
V103
E83 D51
S30
K60I80L108 V85R86M19
S66 F49V54
E22W7
100V8
I45
E90
S17W44 R101
A93A43 L27
E94
A23 A88L28
E4
D21
Q50
R84
S71
R37
E63
Y107 L92 D64R53 L77
G72
A73
G96
D35 N33R9D65
G47
A104A2 A41
E34
A15
T97
H111
S32
E52
G20
T95
W36
T110
G82G40 G81
G98
V12
R18
N5
D3
D11
Q50ε
N5δN48δ
N106δ
W36ε
Q33ε
W44εW7ε
Q79ε
Figure 4-11: : Spectre 2D 15N-HSQC de MTH187. Ce spectre montre les corrélations entre le proton (1H) et son 15N. Les pics reliés représentent les corrélations des deux protons avec leur azote des arginines et des asparagines.
15N

53
4.3. Structure secondaire de MTH187 Les données recueillies lors de l’attribution séquentielle du squelette de MTH187
ont permis de déterminer la structure secondaire de cette protéine. En effet l’utilisation de
l’indice de déplacement chimique (chemical shift index, CSI) (Wishart and Sykes, 1994)
indique que MTH187 possède une structure secondaire formée entièrement d’hélices α
(Figure 4-12).
-1
0
1
10 20 30 40 50 60 70 80 90 100 110
Phe24-Ser30
Asn33 -Trp44
Gly47-Leu61
Phe68-Ile80
Glu83-Ala93Phe99-Glu109
-1
0
1
10 20 30 40 50 60 70 80 90 100 110
Phe24-Ser30
Asn33 -Trp44
Gly47-Leu61
Phe68-Ile80
Glu83-Ala93Phe99-Glu109
Figure 4-12: Graphique montrant le résultat de l’analyse CSI pour la protéine MTH187. Les déplacements chimiques des 1Hα, 13Cα ,13Cβ ont été utilisés. La structure secondaire est résumée dans cette figure. Lorsque y =1, la structure secondaire favorisée est le feuillet β et lorsque y = -1 la structure secondaire favorisée est l’hélice. D’après l’analyse CSI, MTH187 est composée de 6 hélices α: Phe24-Ser30, Asn33-Trp44, Gly47-Leu61, Phe68-Ile80, Glu83-Ala93, Phe99-Glu109. Figure générée à partir du logiciel CSI (Wishart and Sykes, 1994).
L’analyse avec le logiciel TALOS indique également une structure secondaire sous
forme d’hélices α (Figure 4-13). Cependant les limites de ces hélices varient légèrement
comparées à l’analyse de type CSI. La Table 2 fait la comparaison entre les deux
méthodes de détermination de structures secondaires : CSI et TALOS.

54
A)
B)
Figure 4-13 : A) Graphique montrant le résultat du logiciel TALOS pour la protéine MTH187. Les déplacements chimiques des 1Hα , 13Cα ,13Cβ, CO et N ont été utilisés. Les régions vertes représentent les résidus où TALOS suggère une structure secondaire avec un niveau de confiance élevé. Les régions rouges représentent les régions où la structure secondaire est contradictoire (Bad). Les régions jaunes signifient des régions ambiguës et les régions grises correspondent aux régions non classées. L’analyse avec TALOS indique que MTH187 est composée de 6 hélices α: Glu25-Leu31, Arg37-Ile46, Val55-Glu63, Phe68-Ile80, Glu83-Ala93, Ala100-Glu109. B) Graphique de Ramachadran illustrant les angles dièdres du résidu central des dix tripeptides de la banque de données ayant des déplacements chimiques secondaires (secondary shifts) similaire à ceux du tripeptide R75-S76-L77. Cette analyse permet d’estimer les angles dièdres φ/ψ de S76 à –66 ± 9 / -37± 8.

55
Table 2 : Comparaison des méthodes de détermination de structures secondaires de MTH187
La structure secondaire des régions des acides aminés 33 à 63 de MTH187 est
moins bien définies. Les calculs des structures tridimensionnelles tenteront entre autre de
perfectionner ces régions.
CSI Hélices α TALOS Hélices α
24-30 25-31 33-44 37-46 47-61 55-63 68-80 68-80 83-93 83-93
99-109 100-109

56
CHAPITRE 5. STRUCTURE DE MTH187
5.1. Calcul de structures de MTH187
5.1.1. Calculs avec CNS Les contraintes structurales obtenues à partir des données expérimentales ont
permis de calculer la structure tridimensionnelle de MTH187. Plus de 25 lancements de
calculs ont été nécessaires pour obtenir des structures bien définies.
Tout d'abord un premier calcul de structures a été lancé avec les données NOEs
obtenus par l’attribution du spectre 15N-NOESY-HSQC. 670 contraintes NOEs ont été
utilisées pour calculer une famille de 40 structures. Le nombres de violations › 0.1 Å
pour ce calcul était de 321. La superposition de ces structures donne une RMSD de 8 Å
pour le squelette de la protéine. Les structures sont très différentes des unes des autres.
Plusieurs modifications ont été effectuées sur l’attribution du spectre 15N-NOESY-HSQC
car des attributions étaient erronées. Un nouveau calcul de structures a été effectué. Les 9
premières itérations de calculs ont porté entièrement sur des changements dans les
attributions du spectre 15N-NOESY-HSQC. La neuvième itération a été effectuée pour
100 structures avec 1079 contraintes NOEs dont 323 intra-résidus, 772 interactions à
moyennes portées et 34 interactions longues portées. Le nombre de violations de plus de
0.1 Å était de 202.
Le dixième calcul de structures s’est effectué avec les mêmes NOEs qu’au dernier
calcul mais en plus 50 contraintes d’angles phi, obtenues à partir des constantes de
couplage, ont été utilisées. Le nombre de violation de 0.1 Å a chuté à 41 violations.
Quelques modifications ont été faites sur les contraintes NOEs pour obtenir 1011
contraintes NOEs. Également 104 contraintes d’angles dièdres phi et psi sont ajoutées
grâce à l’analyse de TALOS. Les 50 contraintes d’angles phi sont toujours présentes dans
le calcul des 100 structures. À ce stade, la structure de MTH187 commence à montrer
quelques hélices α mais rien de très structurée. On peut compter 110 contraintes violées
dans ces structures.

57
Entre-temps, le spectre 13C-NOESY HSQC a été acquisitionné. Le spectre a été
partiellement attribué. En effet seulement 501 corrélations ont été identifiées dans ce
spectre. Ces attributions sont appuyées sur un script écrit par le docteur Gagné qui
détermine les corrélations symétriques dans le spectre et suggère différentes attributions.
5.1.2. Calcul avec ARIA/CNS Afin d’accélérer le processus de raffinement des structures, des structures ont été
calculées utilisant le logiciel ARIA. Dans chacun des calculs suivants 100 structures ont
été calculées. Au cours du 12ième lancement de calculs, les contraintes structurales
utilisées étaient les contraintes NOEs des spectres 15N-NOESY-HSQC et 13C-NOESY-
HSQC, les angles phi et psi obtenus avec TALOS et les constantes de couplage. Lors de
la calibration des NOEs, ARIA utilise UN temps de corrélation. Le temps de corrélation
utilisé à ce moment était estimé à 4.32 ns. sur la base du poids moléculaire de MTH187
lorsque la protéine est dans l'eau (15N-NOESY HSQC) et de 5.4 ns. dans le D2O (13C-
NOESY-HSQC). Ces temps de corrélation ont été utilisés jusqu'au dix-neuvième calcul
de structures avec ARIA. Au cours de ces calculs, grâce à l’analyse des violations,
plusieurs attributions ont été enlevées ou ajoutées. Également quelques angles dièdres ont
été modifiés. À partir du dix-huitième calcul de structures des contraintes de ponts
hydrogène ont été utilisées. À partir de cette itération, nous avons utilisé un temps de
corrélation déterminé expérimentalement par l’analyse des données de relaxation
(annexe) soit de 7.3 ns. dans l'eau et de 8.1 ns. dans le D2O.
La calibration des NOEs a été effectuée entièrement avec Aria jusqu'au dix-
huitième calcul de structure inclusivement. Par la suite, tout en modifiant certaines
contraintes, les autres calculs ont permis de comparer la calibration des NOEs obtenus
dans le spectre 15N-NOESY-HSQC effectuée avec Aria et avec la nouvelle méthode de
calibration conçu par Etienne Enoumen. Au cours des calculs, cette dernière méthode de
calibration a été perfectionnée et est devenue la méthode de choix pour la calibration des
NOEs dans le spectre 15N-NOESY-HSQC.

58
5.1.3. Structures finales de MTH187 Le 32ième calcul de structures a été basé sur 3087 contraintes NOEs, 94 contraintes
d’angles dièdres obtenus avec les constantes de couplage, 142 contraintes d’angles
dièdres obtenus avec TALOS et 9 contraintes de ponts hydrogène.
Les calculs complétés, l’attribution à la huitième itération de ARIA contient 1607
NOEs non ambiguës dont 318 dans le spectre 15N-NOESY-HSQC et 1246 NOEs dans le
spectre 13C-NOESY-HSQC. Des 1607 NOEs, 616 NOEs sont intra-résiduels, 219
séquentiels, 254 NOEs entre les protons situés à une distance moyenne et 518 sont à
longue distance. Certains NOEs sont classés comme étant ambiguës: 88 NOEs intra-
résiduels, 135 séquentiels, 170 à des distances moyennes et 349 à de longues distances.
L’énergie globale des 30 structures des 72 structures calculées varie entre 737 et 843
Kcal/mol. Les dix-huit dernières structures possèdent des énergies très élevées de l’ordre
de 1900 à 8587 Kcal/mol.
Les 30 structures de plus basses énergies ont subi une simulation dans l’eau par
ARIA. Ces structures sont considérées comme étant les structures finales de MTH187.
Celles-ci sont montrées à la Figure 5-1. Les RMSD de la superposition des 30 structures
finales sont résumées à la Table 3.
Figure 5-1 : Superposition de 30 structures de MTH187. 32ième lancement de calculs effectués par ARIA. La RMSD est de 0.66 Å pour la chaîne squelettique (résidus 24-109) et de 1.23 Å pour les atomes lourds entre les résidus 24 à 109 de la protéine.

59
Table 3 : Table des RMSD de la structure finale de MTH187
RMSD Chaîne squelettique
(Å) RMSD pour tous les atomes
lourds (Å) résidu 1-111 2.47 ± 0.67 2.98 ± 0.68 résidu 24-109 0.66 ± 0.1 1.23 ± 0.14
La structure de MTH187 possède trois paires d’hélices α. Les résidus sont placés de
sorte que ceux qui sont hydrophobes se retrouvent au centre de la protéine pour éviter
tout contact avec l’eau et les résidus hydrophiles se retrouvent vers l’extérieur (une étude
plus approfondie sera entreprise sur le sujet). Les résidus dans la région N-terminale, de 1
à 24, ne sont pas structurés. La Figure 5-2 illustre mieux les hélices α de la protéine
MTH187. Cette structure (Figure 5-2) possède l’énergie la plus basse des structures
simulées dans l’eau.
Figure 5-2 : Structure de MTH187 (haut : Vue en stéréo, bas : rotation de 90° de long de l’axe des X. Les calculs de structures tridimensionnelles ont été faits par ARIA et les structures ont été visualisées dans MOLMOL. Les six hélices α sont représentées. La structure montrée est la structure calculée dans l’eau où l’énergie est la plus basse parmi les 72 structures calculées.

60
5.1.4. Qualité des structures Afin de vérifier la qualité stéréochimique des structures de MTH187 satisfaisant les
contraintes structurales, le logiciel PROCHECK a été utilisé pour évaluer la géométrie
des résidus par rapport aux paramètres stéréochimiques dérivés des structures bien
structurées dans une banque de données (Laskowski, 1993). Les 30 structures de
MTH187 simulées dans l’eau ont été testées. Le tout est illustré à la Figure 5-3 et Figure
5-4.
Figure 5-3 : Graphique de Ramachandran des 30 structures RMN des résidus 24-109 de MTH187. Les résidus Glycine sont représentés par des triangles. Les régions favorisées par les angles phi et psi sont les deux régions les plus foncées. Les régions moins permises mais acceptables sont d’une couleur grise intermédiaire et les régions non permises sont en gris très pâle. Le % des résidus situés dans les régions favorisées est de 90.7%.
Figure 5-4 : (montrée dans les six pages suivantes) Graphique de Ramachandran des résidus 2 à 110 de la structure RMN de MTH187. Chaque point dans chaque graphique de Ramachandran représente les 30 structures générées. Chaque graphique de Ramachandran indique la conformation du squelette pour un résidu, et ce pour chacune des structures calculées. Les structures secondaires définies manuellement sur cette figure sont : hélice 1 (22-31), hélice 2 (36-49), hélice 3 (55-64), hélice 4 (68-80), hélice 5 (83-94), hélice 6 (100-109).

61
Figure 5-4 (partie 1 de 6)
-120
-60
0
60
120
1
2
3
45
6
7
8
9
10
11
12
13
14
15 16
17
18
19
20
21
22 23
24
25
26
27
28
29
30
ALA 2
1
23
4
56
7
8
9
10
11
12
13
1415
16
17
1819
20
21
22
23
24
2526
27
28
29
30
ASP 3
1
2
34
5
6
7
8
910
11
12
13
1415 1617
18
19
20
212223 24
25
26
27
28
2930
GLU 4
1
2
34
5
67
89
10
11
12
13
1415
1617
18
19
20
21
222324
25
2627
28
2930
ASN 5
-120
-60
0
60
120
1
2
3
45
678
9
10
11
1213
141516
17
18
19
20
21
22 23
24
25 26
27
28
2930
LYS 6
1
23
4
5
67
8
9
1011
12
13
1415
16
17
18
19
20
21
2223
24
2526
27
28
29
30
TRP 7
1
2 34
5
67
8
9
10
11
12
13
1415
1617
18
19
20
21
22
2324
25
26
27
28
29
30
VAL 8
1
2
34
56
78
910
11
12
13
14
15
16
17
18
19
20
21
22
23
2425262728
2930
ARG 9
-120
-60
0
60
120 1234 5678 91011121314
15161718
192021 2223
2425
26
2728
29
30
ARG 10
1
2
3456
78
910
11
12
13
14
15
16
1718
19
202122
23
24
2526
27
28
2930
ASP 11 1
2
3
4
5
6
7
8
91011
12
13
14
15
16
17
1819
2021
22
23
24
25
2627
28
29
30
VAL 12
1
2
3
4
5 67
8
9
10
11
12
1314 15
1617
18
19
20
21
22
2324
25
26
27
2829
30
SER 13
-120
-60
0
60
120 12
3
45
67
8910
11
12
13
14
15
16
17
1819
20
21
22 23
24
25
26
27
28
29
30
THR 14
1 23
456
7
891011
1213
14
151617
18 19
20
21
22
23
24
25 2627
28 29
30
ALA 15
12
3
45
6
789 1011
12
13
1415
1617
18
1920
21
22
2324
25
2627
28
29
30
LEU 16
123456 78 910
11 1213
141516
171819
20
212223
24
25 26
27
282930
SER 17
-180-120 -60 0 60 120 -180
-120
-60
0
60
1201
2
345 67
8
9
10
1112
1314
15
1617
18
19
20
21
22 23
24
2526
27
2829
30
ARG 18
-180-120 -60 0 60 120
1 2
3
4
567
89
1011
12
1314 15
16
17 1819
20
21
22
23 2425
26
27
28
29
30
MET 19
-180-120 -60 0 60 120
1
2
3
45
6789
10
11
12
13
14
15
16
17
18 19
20
21
22
23
24
25
26
2728
29
30
GLY 20
-180-120 -60 0 60 120 180
1 23
45
6
789
1011
12
13 1415 1617 181920 212223
2425
26
2728
2930
ASP 21
Phi (degrees)
Psi (
degr
ees)

62
Figure 5-4 (partie 2 de 6)
-120
-60
0
60
120
12
3
4
5
678910
11
12131415
1617
18
1920212223
24252627282930
GLU 22
12
3
4
5
678910
11
12
13141516
1718
19202122 23
24
25
2627282930
ALA 23
1234567891011121314151617181920212223
24252627282930
PHE 24
123456789101112131415161718192021222324252627282930
GLU 25
-120
-60
0
60
120
123456789101112
1314
15161718192021222324252627282930
PRO 26
12345678910
1112
131415161718
192021222324252627282930
LEU 27
12345678910111213
1415161718192021
2223242526272829
30
LEU 28
123
456789
10111213141516
1718192021
2223242526272829
30
GLU 29
-120
-60
0
60
120
123
456789
10
1112131415161718192021222324252627282930
SER 30
1234567
89
10111213141516171819202122
2324252627282930
LEU 31
1
2345678910
111213
14
15
16
17181920212223
24
25
2627282930
SER 32
1
234 56 78
9
10
11
12
131415
1617
1819202122
23
2425 26
27
28
29
30
ASN 33
-120
-60
0
60
120
12345678
9101112 13141516171819202122
23
242526
2728
29
30
GLU 34
123456789101112131415161718
19
2021222324
25
2627282930
ASP 35
1234
567
8
91011121314
151617181920
2122232425
26
2728
2930
TRP 36
12
345678910
11121314151617181920
21
222324252627282930
ARG 37
-180-120 -60 0 60 120 -180
-120
-60
0
60
120
1234567891011
1213 141516
1718192021 222324252627 282930
ILE 38
-180-120 -60 0 60 120
1234567891011121314151617181920212223242526272829
30
ARG 39
-180-120 -60 0 60 120
123456789101112131415161718192021222324252627282930
GLY 40
-180-120 -60 0 60 120 180
123456789101112131415161718192021222324252627282930
ALA 41
Phi (degrees)
Psi (
degr
ees)

63
Figure 5-4 (partie 3 de 6)
-120
-60
0
60
120
123456789101112131415161718192021222324252627282930
ALA 42
123456789101112131415161718192021222324252627282930
ALA 43
123456789101112131415161718192021222324252627282930
TRP 44
123456789101112131415
1617
18192021222324252627282930
ILE 45
-120
-60
0
60
120
123456789101112131415161718192021222324252627282930
ILE 46
123456789
1011121314
15161718192021222324252627282930
GLY 47
123456789101112131415161718192021222324252627282930
ASN 48
1234 5678910111213
141516
17
18192021222324252627282930
PHE 49
-120
-60
0
60
120
1234567891011
1213141516
17
18192021222324252627282930
GLN 50
1
2 34567891011
12
13
1415
1617 1819202122232425
2627282930
ASP 51
1
234567891011
12 131415
161718192021222324252627282930
GLU 52
123456789101112
131415
161718192021222324252627282930
ARG 53
-120
-60
0
60
12012 34567 891011
12131415
1617 18192021
2223242526
27282930
ALA 54
123456789101112131415
161718192021222324252627282930
VAL 55
123456789101112131415161718192021222324252627282930
GLU 56
123
4567891011121314151617
1819
20212223242526
27
282930
PRO 57
-180-120 -60 0 60 120 -180
-120
-60
0
60
120
123456789
101112131415
16171819 20212223242526
27 282930
LEU 58
-180-120 -60 0 60 120
12345678910111213
1415161718192021222324252627282930
ILE 59
-180-120 -60 0 60 120
12345678
910
11
1213
1415161718192021222324252627282930
LYS 60
-180-120 -60 0 60 120 180
1234
56
7891011121314
151617181920212223
24252627282930
LEU 61
Phi (degrees)
Psi (
degr
ees)
64
Figure 5-4 (partie 4 de 6)
-120
-60
0
60
120
1234
5
67891011
121314151617181920212223242526272829
30
LEU 62
123456
78
9101112
131415161718192021222324252627282930
GLU 63
1234
5
6789101112131415161718192021222324252627282930
ASP 64
12
3
4
5
6789
101112131415161718
192021
22
232425
262728
2930
ASP 65
-120
-60
0
60
120
12345
6
789101112131415161718
19
20
21
22
23
24
252627282930
SER 66
123
456
789101112131415161718 1920 21222324
252627282930
GLY 67
12345678 9101112131415161718192021222324252627282930
PHE 68
123456789101112131415161718192021222324252627282930
VAL 69
-120
-60
0
60
120
12345678910111213141516171819202122232425262728
2930
ARG 70
123456789101112131415161718192021222324252627282930
SER 71
123456789101112131415161718192021222324252627282930
GLY 72
123456789101112
131415161718
19202122232425262728
2930
ALA 73
-120
-60
0
60
120
123456789101112131415161718192021222324252627282930
ALA 74
123456789101112
1314151617181920
2122232425262728
2930
ARG 75
123456789101112
131415161718192021222324252627282930
SER 76
123456789
101112131415161718192021222324
252627282930
LEU 77
-180-120 -60 0 60 120 -180
-120
-60
0
60
120
1234567891011
1213
1415161718192021222324252627282930
GLU 78
-180-120 -60 0 60 120
123456789101112131415161718192021222324252627282930
GLN 79
-180-120 -60 0 60 120
1 2345 6789101112
1314151617 1819202122 2324 2526
2728
2930
ILE 80
-180-120 -60 0 60 120 18012
3
4
5
6789
10
1112 13
14
15
161718
19
20
21
22
23
24
25
262728
29
30
GLY 81
Phi (degrees)
Psi (
degr
ees)
65
Figure 5-4 (partie 5 de 6)
-120
-60
0
60
120
12
34
5
6789
10
11
12
13
14
15
16
17
1819
20
21
22
23
24
25
26
272829
30
GLY 82
123456789101112131415161718192021222324
252627282930
GLU 83
123
4
5 67891011121314
15161718192021 22
2324
2526272829
30
ARG 84
12345678910
111213141516171819
20212223
24252627282930
VAL 85
-120
-60
0
60
120
1234567
891011
1213
1415161718192021
222324
252627282930
ARG 86
123456 78 9101112 13
14151617181920212223242526272829 30
ALA 87
123456789
101112131415161718192021222324252627282930
ALA 88
123456789101112131415161718192021222324252627282930
MET 89
-120
-60
0
60
120
12345678910111213
141516171819202122232425
2627282930
GLU 90
123456 789101112131415
161718192021222324
252627
282930
LYS 91
1 23456
7891011
121314
15161718192021222324252627
282930
LEU 92
123
45678
9101112131415
1617
181920212223
242526
27
282930
ALA 93
-120
-60
0
60
120
12345678
9101112131415
161718192021222324252627282930
GLU 94
123
45678
9101112
13
14
15
16
1718 19
20 2122
23
2425
26
2728
29
30
THR 95
12
3
456
7
8
91011
1213
14
15
16
17
18
19
202122
23
24
25
26
27
28
29
30
GLY 96
12
3
4
5
67
8
9
1011
1213
1415
16 17
18
192021
22
232425
262728
2930
THR 97
-180-120 -60 0 60 120 -180
-120
-60
0
60
120
1
2
3
4
5
67
8
9
1011
12
13
14
15
16
17
18
19
20
21
2223
242526
2728
29
30
GLY 98
-180-120 -60 0 60 120
12
345
67
891011
1213
14
1516
17 18
19202122232425
2627282930
PHE 99
-180-120 -60 0 60 120
123456789101112131415161718192021222324252627282930
ALA 100
-180-120 -60 0 60 120 180
1234 5678910
111213141516
171819
2021
2223
242526
27282930
ARG 101
Phi (degrees)
Psi (
degr
ees)

66
Figure 5-4 (partie 6 de 6)
Pour chaque résidu de MTH187, plusieurs statistiques peuvent être émises. La
Table 4 donne une moyenne de certaines données lors du calcul de structures de
MTH187.
-120
-60
0
60
120
123456789101112131415161718
192021222324252627282930
LYS 102
12345678910111213141516171819202122232425 2627282930
VAL 103
123456789101112131415161718192021222324252627282930
ALA 104
12345678910111213
1415161718192021222324252627282930
VAL 105
-120
-60
0
60
120
123456789101112131415161718192021222324252627282930
ASN 106
123
456
7
89101112131415161718192021222324252627282930
TYR 107
-180-120 -60 0 60 120
1234567
8910111213141516171819202122232425262728
2930
LEU 108
-180-120 -60 0 60 120 180
12345
67
8910
111213141516171819202122
23
24
252627
282930
GLU 109
-180-120 -60 0 60 120 -180
-120
-60
0
60
120
12
3
4
5
67
89
1011
1213141516171819
20212223
24
25262728
2930
THR 110
Phi (degrees)
Psi (
degr
ees)

67
Table 4 : Statistiques structurales des structures RMN de MTH187 a
RMSD des contraintes structurales a
NOES 0.02 ± 0.0005
angles dièdres 0.619 ± 0.287 constante de couplage 0.723 ± 0.086
Énergies Kcal mol-1
E VDW -308.21 ± 4.2 E Bonds 26.685 ± 0.7 E cdih 4.5 ± 0.5
E TOTALES -3697.675 ± 94.277 E NOE 61.597 ± 3.719
chaîne squelettique Tous les atomes
RMSD atomique b
toute la protéine 2.47 ± 0.67 2.98 ± 0.68 résidus 24-109 0.66 ± 0.1 1.23 ± 0.14
région favorisée (%) c 90.70%
résidus situés dans régions permises 8.60% résidus situés dans régions non permises 0.60% a Les contraintes structurales et les énergies sont basées sur la moyenne des 10 premières structures du 32ième calcul. b RMSD (root mean square deviations) signifie la moyenne de la déviation de chacun des atomes par rapport aux structures. c Pourcentage des résidus 24 à 109 des 30 structures qui sont situés dans les régions les plus favorisées du graphique de Ramanchandran déterminés par le logiciel PROCHECK.
La région en N-terminale (résidus 1-21) est non structurée. Les angles phi et psi des
30 structures générées ne se concentrent pas dans une région donnée. Par contre pour les
régions formant les hélices α, les angles phi et psi sont presque tous situés à la même
position pour un résidu donné. Cela confère de belles hélices bien structurées. Les angles
de certains résidus pourraient être étudiés, en particulier la phénylalanine 99 où l’on
retrouve des angles Φ positifs. Également l’acide aspartique 65 et la glutamine 50 où les
angles Φ sont situés dans des régions plus ou moins permises. Malgré ces aspects
négatifs, la RMSD des 30 structures superposées est très satisfaisante. La structure en
générale de MTH187 est de bonne qualité

CHAPITRE 6. HOMOLOGIE STRUCTURALE
La structure de MTH187 a été utilisée afin d'identifier des homologues structuraux.
Les coordonnées de la protéine ont été soumises à un programme de recherche
d'homologues structuraux. Le programme utilisé ici se nomme DALI (distance matrix
alignment)(Holm and Sander, 1996). Le serveur DALI est un service automatique pour la
comparaison de structures en trois dimensions. Les coordonnées d'atomes ont été
soumises à DALI et nous avons obtenu les structures les plus semblables à la structure
soumise. Celles-ci sont énumérées dans le tableau suivant.
Table 5: Protéines identifiées par DALI comme étant structuralement homologues à MTH187
s
A
G
o
M
PDB ID RMSD (Å) nom de la protéine 1l5j-A 2.4 Aconitase B 3bct 5.6 β -Catenin fragment 1gw5-B 2.9 Adaptor-Related Protein Complex 1ee4-A 3.1 Karyopherin (Importin) α fragment 1bk5-A 2.7 Karyopherin α fragment 1b89-A 3.1 Clathrin Heavy Chain fragment 1hs6-A 3.2 Leukotriene A-4 Hydrolase 1qgr-A 2.8 Importin β Subunit 1dvp-A 2.8 Hepatocyte Growth Factor-Regulated Tyrosine Kinase Substrate 1bd8 2.6 P19Ink4D Cdk4/6 Inhibitor 1b3u-A 2.7 Protein Phosphatase Pp2A fragment 1qbk-B 2.6 Karyopherin β 2 fragment 1bpo-A 3.6 Clathrin Heavy-Chain Terminal Domain * La lettre après le PDB-ID est le nom de la chaîne. Le RMSD a été évalué entre les résidus 24 à 109 de MTH187 et la région homologue le l’autre protéine.
68
Dans le but de vérifier si d’autres protéines, ignorées par DALI, peuvent être
imilaires à la structure de MTH187, le logiciel CE (Shindyalov, 1998) a été utilisé.
ucune autre protéine n’a été rajoutée à la liste de protéine présentée à la Table 5.
énéralement, les mêmes protéines sont retrouvées entre DALI et CE, mais dans un
rdre différent.
Les protéines homologues ont été examinées une à une et superposées avec
TH187 à l’aide du logiciel SPDBviewer (recherche du domaine le plus semblable

69
automatiquement, vérification de la véracité de l’automatisation et obtention de la
meilleure superposition possible par alignement manuel). La superposition obtenue
manuellement est la même que celle obtenue avec DALI. La Figure 6-1 montre la région
séquentielle 63 à 151 de la protéine Aconitase B (1L5J) superposée avec la région 24 à
109 de MTH187.
pdb1l5j. 63 VKAGFLAAIA KGEAKSPLLT PEKAIELLGT MQGGYNIHPL IDALDDAK-- aria_19w 24 FEPLLESL SNEDW---RI RGAAAWIIGN FQDERAVEPL IKLLEDDSGF * * * ..* * . ** * *.* pdb1l5j. 111 LAPIAAKALS HTLLMFDNFY DVEEKAKAGN -EYAKQVMQSW AD aria_19w 69 VRSGAARSLE QIG-GERVRA AMEKLAETGT GFARKVAVNYL E . . **..* . * * .* * .
Figure 6-1 :Alignement des régions superposées entre l’Aconitase B (pdb ID 1l5j) et MTH187 obtenu par le programme DALI. L’identité entre les deux séquences pour les résidus 24-109 est de 21% et la similarité est de 35%. Les “ * ’’ signifient que les deux résidus alignés sont identiques. Les “.’’ signifient que les deux résidus sont semblables.
La superposition des structures de l’Aconitase B et de MTH187 sont illustrées à la
Figure 6-2. La RMSD (root mean square deviation) des régions semblables est présentée
dans la Table 6, une fois la meilleure superposition entre les deux structures obtenues.
Figure 6-2: Vue en stéréo de la superposition de MTH187 en bleu et de l’aconitase B (I.D.:1L5J) en rouge. Le logiciel utilisé pour visualiser est PYMOL. Le RMSD squelettique des résidus superposés est de 2.36Å.

Table 6: Superposition des régions de MTH187 avec Aconitase B
La
(Figure 6
l’Aconitas
de MTH1
à ces extré
MT
essais ont
semble y
De plus, l
selon DAL
La
Core’’ (1
alignemen
Figure 6-3: deux séquendeux résidu
MTH187 1L5J RMSD (Å)
F24-E34 A65-E75 3.07
R37-S66 L81-K110 1.95
V69-G81 L111-L123 1.43
G82-T97 M125-N140 2.78
VF99-E109 E141-A151 2.77
RMSD pour les résidus squelettiques de la région superposée est de 2.36Å
-2). Les six hélices α de MTH187 correspondent bien avec les hélices de
e B pour une RMSD de 2.27 Å. Comme on peut le constater, la région centrale
87 (les hélices 2, 3, et 4) est très bien superposée à l’aconitase comparativement
mités.
H187 a également été superposée avec la protéine β-Catenin (3bct). Plusieurs
été effectués, mais à l’endroit où MTH187 pourrait se superposer le mieux, il
avoir une coupure dans la chaîne entre les résidus 549 et 563 de la β-Catenin.
a superposition laissait à désirer d’après sa RMSD beaucoup trop haut (5.6 Å
I) Donc, l’analyse ne s’est pas poursuivie plus loin.
troisième superposition s’est faite avec la protéine “Ap2 Clathrin Adaptor
GW5). Selon Dali, la RMSD de la chaîne squelettique est de 2.9 Å. Un
t manuel a permis de mieux superposer les deux protéines (Figure 6-3).
MTH187 24 FEPLLESL S-NEDWRIRG AAAWIIGNFQ DE--RAVEPL IKLLEDDSGF pdb1gw5. 428 IIATLCENL DSLDEPDARA AMIWIVGEYA ERIDNADELL ESFLEGFHDE . * *.* .. *. * **.*.. . * * * .** MTH187 69 ---VRSGAAR SLEQIGGERV RAAMEKLAET GTGFARKVAV NYLE pdb1gw5. 477 STQVQLTLLT AIVKLFLKK- ---------- ----PSETQE LVQQVLS *. .. . . . .
70
Alignement des régions superposées entre AP2 (pdb1gw5) et MTH187. L’identité entre les ces pour les résidus 24-109 est de 21% et la similarité est de 42%. Les “ * ’’ signifient que les
s alignés sont identiques. Les “.’’ signifient que les deux résidus sont semblables.

71
Figure 6-4: Vue en stéréo de la superposition de MTH187 en bleu et de l’adapteur Ap2 (I.D.:1GW5) en rouge. Le logiciel utilisé pour la visualisation est MOLMOL. La RMSD de la chaîne squelettique de la région 24-109 de MTH187 et l’adapteur Ap2 est de 2.72Å.
Comme montré à la Figure 6-4, seulement quatre des six hélices de MTH187 se
superposent adéquatement. Malgré les nombreux essais, les deux dernières hélices de
MTH187 ne se superposent pas avec l’adapteur Ap2, sans perdre la superposition des
quatre premières.
La superposition de MTH187 a été tentée avec toutes les autres protéines issues de
la recherche avec DALI (Table 5). La structure se rapprochant le plus à la structure de
MTH187 reste celle de l’Aconitase B.
La région de l’Aconitase B qui a été superposée, plus particulièrement la région N-
terminal (1-160) de celle-ci, ressemble fortement à la famille structurale des “Heat
repeat’’(Huntingtin-Elongation-A subunit-TOR)(Williams et al., 2002). Cette famille est
caractérisée par une répétition de repliements de 3 à 36 paires d’hélices α antiparallèles
formant des supers-hélices appelés “HEAT’’. Les protéines possédant cette forme de
structures ont une fonction commune de reconnaissance protéine-protéine dans plusieurs
procédés cellulaires (Andrade et al., 2001). Les deux figures qui suivent montre la
structure de l’Aconitase B.

72
Figure 6-5: Structure tridimensionnelle de l’Aconitase B qui a tendance à former des dimères. Le jaune représente le motif de répétition qui ressemble au motif strcutral “Heat repeat’’. Le PDB ID de cette protéine est 1l5j-A.
Figure 6-6: Structure tridimensionnelle de l’Aconitase B sous forme monomérique. La région en jaune représente la région ou la protéine MTH187 se superpose. Le PDB ID de cette protéine est 1l5j-A.
Une autre famille de supers-hélices sont les “Armadillo repeat’’. Ces protéines ont
également une implication dans les interactions protéine-protéine (Andrade et al., 2001).
En analysant le contenu de cette famille, plusieurs protéines se retrouvant dans la liste des
protéines similaires à MTH187 donnée par DALI (Table 5), font parties de cette famille
des "Armadillo repeat''. En guise d’exemple, la protéine β-Catenin identifiée dans la
banque de donnée Protein Data Bank (PDB) par 3bct (Huber et al., 1997). Également ces
autres protéines, inscrites dans la Table 5, ont ce même motif: la karyopherin (1bk5) et
son fragment α (1ee4a), les fragments de chaîne de la clathrin (clathrin heavy chain
fragment)(1b89-A) et la sous-unité β de l’importin (1qgr).

73
CHAPITRE 7. DISCUSSION
Grâce aux travaux présentés dans ce mémoire, il a été possible de déterminer la
structure tridimensionnelle d’une des nombreuses protéines de la bactérie
Methanobacterium thermoautotrophicum et de classer celle-ci dans une famille de
structure.
7.1. Attribution des déplacements chimiques de MTH187 L’attribution des déplacements chimiques de la protéine s’est déroulée relativement
bien. Il aurait été cependant plus facile de débuter par l’attribution des spectres
CBCA(CO)NH et HNCACB car ces spectres sont plus sensibles, moins encombrés et les
systèmes de spin des carbones Cα et Cβ sont plus caractéristiques que les spins des
résidus dans les spectres 15N-TOCSY-HSQC et 15N-NOESY-HSQC. Le spectre HNCO
est également un spectre très sensible. D’ailleurs, les corrélations de tous les carbonyles
sont présents dans ce spectre; même le carbonyle de la glycine 67 dont le groupement
NH est le seul à ne pas avoir été attribué. Le NH de la Méthionine 1 manque également
dans l’attribution, mais les résidus situés en début de chaîne sont souvent difficiles à
attribués car le NH interagit avec le solvant.
En amont de la Méthionine 1 de MTH187 se trouve une chaîne de 20 résidus
correspondant au tag de poly-histidines. Plusieurs résidus de ce segment sont perceptibles
dans les spectres, en autre le CBCA(CO)NH, HCA(CO)N et le HNCO. Des erreurs se
sont souvent glissées dans l’attribution des déplacements chimiques dues à des
corrélations entre les résidus du vecteur. Les déplacements chimiques de ces résidus
interféraient avec l’attribution des corrélations des résidus de la protéine. Ceci a contribué
énormément à augmenter le temps d’analyse des spectres. Pour ma part, il aurait été
préférable de couper ce segment après la purification en procédant à une digestion
enzymatique à l’aide de la trombine. Cette étape aurait engendré par contre de purifier à
nouveau la protéine et donc de diminuer le rendement de celle-ci.

74
Les spectres choisis pour l’attribution des chaînes latérales ont tous été essentiels
pour connaître les déplacements chimiques des différents atomes de la protéine. En
évaluant l’attribution des résidus, nous pouvons constater que quelques déplacements
chimiques des résidus arginines à partir des Nε n’ont pas été complétés. Ces
déplacements chimiques auraient pu être déterminés à l’aide des deux premiers spectres
acquisisionnés soit le 15N-TOCSY-HSQC et 15N-NOESY-HSQC mais les corrélations
entre ces protons et les atomes à proximité se faisaient rare.
7.2. Structure de MTH187 Le nombre de structures tridimensionnelles calculées influencent la RMSD des
structures. Calculer un plus grand nombre de structures donne de meilleures statistiques.
Un calcul de 25 structures avait été entrepris par ARIA et seulement dix d’entre elles
étaient simulées dans l’eau. LA RMSD était très bonne mais moins significative. C’est
pourquoi, un plus grand nombre de structures a été calculées par la suite. 72 structures
dont 30 structures calculées dans l’eau permettent d’avoir une meilleure idée de la famille
de structures de MTH187 (Figure 5-1).
La structure de MTH187 est composée de six hélices α. Lors du calcul des
structures secondaires de MTH187 selon les déplacements chimiques avec le logiciel CSI
et TALOS, une légère contradiction est survenue quant aux régions des hélices α. Le
logiciel CSI suggère des hélices α des résidus 33 à 44 et des résidus 47 à 61. Les hélices α
sont préférées des résidus 37 à 46 et 55 à 63 lors de l’analyse des déplacements
chimiques avec le logiciel TALOS. La structure tridimensionnelle a permis d’évaluer la
performance de l’approche CSI et TALOS à évaluer la structure secondaire. La structure
tridimensionnelle de MTH187 contient entre autre des hélices dans les régions 36 à 49 et
55 à 64 (Figure 5-4). Les régions sous forme d’hélices α dans la structure tertiaire sont
davantage comme l’avait prédit TALOS. Dans notre cas l’approche de TALOS a été
supérieur à CSI. Peut-être que pour d’autres protéines l’approche de CSI serait de
meilleure qualité. Pour les deux approches, l’attribution de la structure secondaire à partir
des déplacements chimiques de MTH187 a été très précise. Les hélices dans la structure
tertiaire sont en générale toutes au même endroit.

75
La distribution des angles dièdres phi et psi dans le graphique de Ramachandran
(90.7% dans les régions les plus favorisées définies par le logiciel Procheck) montre la
géométrie favorable des structures de MTH187. Les résidus qui posent potentiellement
un problème sont principalement la glutamine 50, l’acide aspartique 65 et la
phénylalanine 99. Les angles phi de ces résidus sont dans des régions positives. Des
angles phi dans cette région sont très rares puisque qu’il pourrait y avoir des
encombrements stériques. Par contre ces résidus sont situés dans des boucles très courtes
entre deux hélices. Il peut donc être possible que les deux hélices empêchent tout
mouvement dans la boucle. Ces données au niveau de la structure de MTH187 doivent
être vérifiées. D’autres calculs de structures seront générés afin de perfectionner
davantage la structure de MTH187.
Des contraintes structurales pourraient être modifiées afin d’obtenir un plus grand
nombre de structures superposées et une meilleure superposition de ces structures. Les
angles dièdres obtenus à l’aide de TALOS sont des données théoriques c’est-à-dire que
les déplacements chimiques des peptides sont comparés à d’autres afin de connaître les
angles Φ et ψ. De leur coté, les constantes de couplage sont des données expérimentales
qui sont prises directement dans le spectre HNHA. Ces contraintes peuvent parfois ne pas
être en accord l’une envers l’autre pour un résidu donné. Pour ma part, lorsqu’un cas
comme celui-ci survient, il serait logique d’éliminer ces contraintes d’angles dièdres afin
que le calcul de structures soit repris seulement avec les contraintes de constantes de
couplages.
Malgré l’amélioration de la structure qui peut-être fait, nous pouvons interpréter
que la protéine MTH187 contient six hélices dont trois parallèles d’un coté et trois
parallèles de l’autre. Les résidus sont placés de sorte que ceux qui sont hydrophobes se
retrouvent au centre de la protéine pour éviter tout contact avec l’eau et les résidus
hydrophiles se retrouvent vers l’extérieur. Les résidus dans la région N-terminale, de 1 à
24, ne sont pas structurés mais peut-être est-ce dû au segment his-tag en amont de celle-ci
qui empêche la région de se repliée.

76
La protéine MTH187 a tendance à dimériser. La structure de MTH187,
particulièrement la région N-terminale, peut être structurée différemment lorsqu’elle est
sous forme de dimère. Des études RMN de la protéine sous cette conformation seraient
intéressantes afin de vérifier cette dernière hypothèse et également de regarder les
interactions entre les deux dimères.
7.3. Homologie structurale La structure de MTH187 a été comparée avec des milliers de structures de protéines
dans les banques de données. Peu importe le logiciel utilisé pour la comparaison, les
mêmes protéines y ressortent mais peuvent être dans un ordre différent. En comparant
l’alignement des régions superposées entre l’Aconitase B (pdb ID 1l5j) et MTH187 et
entre AP2 (pdb1gw5) et MTH187, nous pouvons remarquer que le pourcentage d’identité
entre les deux groupes de séquences est de 21% dans les deux cas et le poucentage de
similarité est de 35% lors de la comparaison de MTH187 avec l’Aconitase B et de 42%
lorsque la protéine est comparée avec AP2. Malgré la plus grande similarité de séquence
entre MTH187 et AP2, la protéine où l’on retrouve la plus grande homologie structurale
avec MTH187 est l’Aconitase B. La différence de plusieurs acides aminés entre ces deux
protéines n’empêche pas la grande similarité de leurs structures.
Ces protéines homologues à MTH187, en particulier l’Aconitase B, ressemble au
motif structural “Heat repeat’’(Huntingtin-Elongation-A subunit-TOR). Les protéines qui
possèdent cette forme de structures ont une fonction commune de reconnaissance
protéine-protéine dans plusieurs procédés cellulaires (Andrade et al., 2001). Plusieurs
protéines ont également un motif structural "Armadillo repeat'' qui ressemble beaucoup
au model “Heat repeat’’. La fonction de ce type de structure est également une fonction
de reconnaissance protéine-protéine.
La principale différence structurale entre la protéine étudiée MTH187 et les
familles structurales “HEAT repeat’’ et “Armadillo repeat’’ est la longueur des hélices α.
Dans le cas des “HEAT repeat’’, les longueurs des hélices varies entre 37 et 47 résidus et
pour les “Armadillo repeat’’, le nombre de résidus est d’environ 40 (Andrade et al.,
2001). La protéine MTH187 atteint un maximum de 14 résidus dans une hélices α. C’est

77
la principale raison pour laquelle il est sujet d’un domaine ressemblant au motif de
répétition HEAT et non pas d'un ''vrai'' motif HEAT.
La protéine Aconitase B, contenant une région semblable à la région de répétition
HEAT, contient des hélices α beaucoup moins longues. En effet, la superposition de
l’Aconitase B avec MTH187 se révèle de bonne qualité. La superposition des régions
(Table 6) révèle des RMSD acceptables, soit de 1.43 Å à 3.07 Å. Également, la position
des six hélices α une par rapport à l’autre est similaire à celle de l’Aconitase B, le RMSD
est de 2.36 Å pour la chaîne squelettique. Les autres protéines similaires à MTH187,
tirées du logiciel Dali, avaient souvent des difficultés à être superposées correctement ou
bien si la superposition était possible, la RMSD était plus élevée.
Avant la détermination de la structure tridimensionnelle de MTH187, l’homologie
séquentielle révélait que MTH187 ressemblait à une sous-unité α d’une phycocyanine
phycocyanobiline lyase. Une fois la structure tridimensionnelle de MTH187 obtenue, la
comparaison de celle-ci avec les neufs structures des phycocyanines peut être faite. Les
phycocyanines sont toutes composées entièrement d’hélice α. MTH187 est également
composée uniquement d’hélice α mais celle-ci ne sont pas disposées de la même façon.
Le motif structural des phycocyanines n’est pas de la forme “HEAT repeat’’. Lorsque la
structure est différente, nous pouvons supposer une fonction différente, donc MTH187
n’est fort probablement pas une phycocyanine. L’homologie séquentielle doit être une
erreur.
Pour soutenir l'hypothèse que MTH187 appartient à la famille « Heat repeat », une
recherche des domaines conservés a été faite. MTH187 semble avoir une séquence
similaire aux séquences de la famille « Heat repeat ».

78
CHAPITRE 8. CONCLUSION
L’attribution des déplacements chimiques a été faite pour plus de 98% des 111
acides aminés de la protéine MTH187. Dans la chaîne squelettique, seule la glycine 67
n’a pu être attribuée. Par de nombreux calculs, la structure tridimensionnelle de MTH187
a été résolue. La structure est composée de six hélices α positionnées deux à deux dont
les résidus hydrophobes sont regroupés au centre de la protéine. La fonction de MTH187
n’est toujours pas confirmée. Par contre la structure de MTH187 est comparable à une
famille structurale appelée « HEAT repeat ». Cette famille est caractérisée par des
repliements d’un grand nombre de paires d’hélices α antiparallèles formant des supers-
hélices. Cette famille ne contient que de grosses protéines. Par contre MTH187 a
tendance à dimériser. La forme dimérique de MTH187 pourrait davantage ressembler à la
famille « HEAT repeat ». Ces travaux ont grandement contribué à l’amélioration des
connaissances sur la structure et la fonction de MTH187. La structure tridimensionnelle
de MTH187 sera étudiée davantage et sera utilisée entre autre pour la modélisation
moléculaire par homologie structurale de d’autres protéines, plus particulièrement
d’autres protéines de Methanobacterium thermoautotrophicum.

79
BIBLIOGRAPHIE Andrade, M.A., Petosa, C., O'Donoghue, S.I., Muller, C.W. and Bork, P. (2001)
Comparison of ARM and HEAT protein repeats. J. Mol. Biol., 309, 1-18. Anfinsen, C.B. (1973) Principles that govern the folding of protein chains. Science, 181,
223-230. Bax, A. and Ikura, M. (1991) An efficient 3D NMR technique for correlating the proton
and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J. Biomol. NMR, 1, 99-104.
Boone, D.R. (2000) Bergey,s Manuel of Systematic bacteriology, New York. Branden, C. and Tooze, J. (1991) Introduction to protein structure. Garland Publishing,
Inc, New York. Braunschweiler, L. and Ernst , R.E. (1983) J. Magn. Reson., 53, 313-340. Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve,
R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T. and Warren, G.L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr., 54 ( Pt 5), 905-921.
Cavanagh, J., Fairbrother, W.J., Palmer, A.G., 3rd and Skelton, N.J. (1996) Protein NMR Spectroscopy Principles and Practice. Academic Press, New York.
Christendat, D., Yee, A., Dharamsi, A., Kluger, Y., Savchenko, A., Cort, J.R., Booth, V., Mackereth, C.D., Saridakis, V., Ekiel, I., Kozlov, G., Maxwell, K.L., Wu, N., McIntosh, L.P., Gehring, K., Kennedy, M.A., Davidson, A.R., Pai, E.F., Gerstein, M., Edwards, A.M. and Arrowsmith, C.H. (2000) Structural proteomics of an archaeon. Nat. Struct. Biol., 7, 903-909.
Clore, G.M., Bax, A. and Gronenborn, A.M. (1991) Stereospecific assignment of beta-methylene protons in larger proteins using 3D 15N-separated Hartmann-Hahn and 13C-separated rotating frame Overhauser spectroscopy. J. Biomol. NMR, 1, 13-22.
Clubb R.T, T.V., Wagner G. (1992) J.Magn. Reson., 97, 213-217. Cornilescu, G., Delaglio, F. and Bax, A. (1999) Protein backbone angle restraints from
searching a database for chemical shift and sequence homology. J. Biomol. NMR, 13, 289-302.
Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J. and Bax, A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 6, 277-293.
Evans, J.N.S. (1995) Biomolecular NMR Spectroscopy. Oxford University Press, Oxford. Fairchild, C.D., Zhao, J., Zhou, J., Colson, S.E., Bryant, D.A. and Glazer, A.N. (1992)
Phycocyanin alpha-subunit phycocyanobilin lyase. Proc Natl Acad Sci U S A, 89, 7017-7021.
Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D. and Kay, L.E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry, 33, 5984-6003.
Geerten W. Vuister, B.A. (1993) Quantitative J Correlation:Anew Approach for Measuring Homonuclear Three-Bond J(HNHA) Coupling Constants in 15N-Enriched Proteins. J.Am.Chem.Soc., 115, 7772-7777.

80
Grzesiek, S., Dobeli, H., Gentz, R., Garotta, G., Labhardt, A.M. and Bax, A. (1992) 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-gamma. Biochemistry, 31, 8180-8190.
Guex, P. (1997) SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis, 18, 2714-2723.
Harbison, G.S. (1993) J.Am. Chem. Soc., 115, 3026-3027. Heinemann, U. (2000) Structural genomics in Europe: slow start, strong finish? Nat.
Struct. Biol., 7 Suppl, 940-942. Hol, W.G. (2000) Structural genomics for science and society. Nat. Struct. Biol., 7 Suppl,
964-966. Holm, L. and Sander, C. (1996) Mapping the protein universe. Science, 273, 595-603. Huber, A.H., Nelson, W.J. and Weis, W.I. (1997) Three-dimensional structure of the
armadillo repeat region of beta-catenin. Cell, 90, 871-882. Ikura, M., Kay, L.E. and Bax, A. (1990) A novel approach for sequential assignment of
1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry, 29, 4659-4667.
Johnson, B. A., Blevins, .R.A. (1994) NMRView: A computer program for the visualization and analysis of NMR data. J. Biomolecular NMR, 4, 603-614.
Kay, L.E., Iruka, M., Tschudin, R. and Bax, A. (1990) J.Magn. Reson., 89, 496-514. Koradi, R., Billeter, M., and Wüthrich, K. (1996) MOLMOL: a program for display and
analysis of macromolecular structures. J. Mol. Graphics, 14, 51-55. Laskowski, R.A. (1993) Procheck: a program to check the stereochemical quality of
protein structures. J.Appl.Cryst., 26, 283-291. Levitt, M.H. (2001) Spin dynamics, Basics of Nuclear Magnetic Resonance. Wiley, New
York. Linge, J.P., O'Donoghue, S.I. and Nilges, M. (2001) Automated assignment of
ambiguous nuclear overhauser effects with ARIA. Methods Enzymol, 339, 71-90. Lyons, B.A., Tashiro, M., Cedergren, L., Nilsson, B. and Montelione, G.T. (1993) An
improved strategy for determining resonance assignments for isotopically enriched proteins and its application to an engineered domain of staphylococcal protein A. Biochemistry, 32, 7839-7845.
Mandel, A.M., Akke, M. and Palmer, A.G., 3rd. (1995) Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol., 246, 144-163.
Muhandiram, D.R., Kay, L.E. (1994) J.Magn. Reson., 103, 203-216. Muller. (1979) L.Am.Chem. Soc, 101, 4481-4484. Olejniczak, E.T., Xu, R.X. and Fesik, S.W. (1992) A 4D HCCH-TOCSY experiment for
assigning the side chain 1H and 13C resonances of proteins. J. Biomol. NMR, 2, 655-659.
Palmer, A.G., 3rd, Fairbrother, W.J., Cavanagh, J., Wright, P.E. and Rance, M. (1992) Improved resolution in three-dimensional constant-time triple resonance NMR spectroscopy of proteins. J. Biomol. NMR, 2, 103-108.
Pelczar, M.J.Jr., Chan, E.C.S. and Krieg, N.R. (1993) Microbiology, Concepts and Applications. McGraw-Will, Inc, New York.

81
Russell, R.B. and Eggleston, D.S. (2000) New roles for structure in biology and drug discovery. Nat. Struct. Biol., 7 Suppl, 928-930.
Sanders, J.K.M. and Hunter, B.K. (1987) Modern NMR Spectroscopy. Oxford University Press, Oxford.
Shao, J.a.W., C.F.J. (1989) A general theory for jackknife variance estimation. Ann. Statist, 17, 1176-1197.
Shindyalov, B. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Engineering, 11, 739-747.
Smith, D.R., Doucette-Stamm, L.A., Deloughery, C., Lee, H., Dubois, J., Aldredge, T., Bashirzadeh, R., Blakely, D., Cook, R., Gilbert, K., Harrison, D., Hoang, L., Keagle, P., Lumm, W., Pothier, B., Qiu, D., Spadafora, R., Vicaire, R., Wang, Y., Wierzbowski, J., Gibson, R., Jiwani, N., Caruso, A., Bush, D., Reeve, J.N. and et al. (1997) Complete genome sequence of Methanobacterium thermoautotrophicum deltaH: functional analysis and comparative genomics. J Bacteriol, 179, 7135-7155.
Tatusov, R.L., Koonin, E.V. and Lipman, D.J. (1997) A genomic perspective on protein families. Science, 278, 631-637.
Tatusov, R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D. and Koonin, E.V. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res., 29, 22-28.
Terwilliger, T.C. (2000) Structural genomics in North America. Nat. Struct. Biol., 7 Suppl, 935-939.
Van Holde, K.E. and Mathews, C.K. (1990) Biochemistry. The Benjamin/Cummings Plublishing Compagny,Inc., Oregon.
Voet, D. and Voet, J. (1995) Biochemistry, Second Edition. DeBoeck Université, NewYork.
Williams, C.H., Stillman, T.J., Barynin, V.V., Sedelnikova, S.E., Tang, Y., Green, J., Guest, J.R. and Artymiuk, P.J. (2002) E. coli aconitase B structure reveals a HEAT-like domain with implications for protein-protein recognition. Nat. Struct. Biol., 9, 447-452.
Wishart, D.S. and Sykes, B.D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR, 4, 171-180.
Wüthrich, K. (1986) NMR of Proteins and Nucleic acids. Wiley Interscience, New York. Yee, A., Pardee, K., Christendat, D., Savchenko, A., Edwards, A.M. and Arrowsmith,
C.H. (2003) Structural proteomics: toward high-throughput structural biology as a tool in functional genomics. Acc. Chem. Res., 36, 183-189.
Yokoyama, S., Hirota, H., Kigawa, T., Yabuki, T., Shirouzu, M., Terada, T., Ito, Y., Matsuo, Y., Kuroda, Y., Nishimura, Y., Kyogoku, Y., Miki, K., Masui, R. and Kuramitsu, S. (2000) Structural genomics projects in Japan. Nat. Struct. Biol., 7 Suppl, 943-945.

82
ANNEXE I
Le temps de corrélation (τc) de la protéine dans l’eau a été obtenue par des calculs
de relaxation soit les valeurs de T1, T2 et NOEs pour certains résidus de MTH187. Les
résultats de ces calculs sont présentés à la figure suivante.
a
b
c
a
b
c
a
b
c
15N-T1 des résidus de MTH187 (a), 15N-T2 (b), et {1H}-15N-NOE (c) à 600 MHz. Seulement 78 résidus ont été utilises pour les calculs de relaxation. Les incertitudes ont été obtenues en utilisant la simulation de Jack Knife. Les résultats indiquent que MTH187 a un τc= 7.3 ns dans l’eau. La structure secondaire est montrée en bleu.

83
ANNEXE II Table des déplacements chimiques de MTH187 1.CA 55.776 1 1.HA 4.423 1 1.CB 33.124 1 1.HB2 2.058 2 1.HB1 1.953 2 1.CG 32.132 1 1.HG2 2.446 2 1.HG1 2.506 2 1.C 175.845 1 2.N 124.471 1 2.HN 8.209 1 2.CA 52.857 1 2.HA 4.318 1 2.CB 19.399 1 2.HB1 1.392 1 2.C 177.419 1 3.N 119.139 1 3.HN 8.130 1 3.CA 54.654 1 3.HA 4.527 1 3.CB 41.350 1 3.HB2 2.628 2 3.HB1 2.700 2 3.C 176.586 1 4.N 120.858 1 4.HN 8.306 1 4.CA 57.486 1 4.HA 4.141 1 4.CB 30.095 1 4.HB2 2.045 2 4.HB1 1.955 2 4.CG 36.249 1 4.HG2 2.236 2 4.HG1 2.236 2 4.C 176.574 1 5.N 118.298 1 5.HN 8.280 1 5.CA 53.839 1 5.HA 4.614 1 5.CB 38.821 1 5.HB2 2.740 2 5.HB1 2.741 2 5.ND2 112.635 1 5.HD21 7.528 2 5.HD22 6.819 2 5.C 175.501 1 6.N 120.257 1 6.HN 7.900 1 6.CA 57.214 1 6.HA 4.143 1
6.CB 32.755 1 6.HB2 1.600 2 6.HB1 1.600 2 6.CG 24.550 1 6.HG2 1.170 2 6.HG1 1.170 2 6.CD 29.137 1 6.HD2 1.561 2 6.HD1 1.561 2 6.CE 42.313 1 6.HE2 2.869 2 6.HE1 2.879 2 6.C 176.454 1 7.N 119.922 1 7.HN 7.883 1 7.CA 57.316 1 7.HA 4.683 1 7.CB 29.600 1 7.HB2 3.221 2 7.HB1 3.329 2 7.CD1 127.073 3 7.HD1 7.244 1 7.NE1 129.151 1 7.HE1 10.117 3 7.CZ2 114.500 3 7.HZ2 7.450 3 7.CH2 124.476 1 7.HH2 7.165 1 7.C 176.127 1 8.N 119.956 1 8.HN 7.626 1 8.CA 62.583 1 8.HA 4.029 1 8.CB 32.810 1 8.HB 2.009 1 8.CG2 20.919 1 8.HG21 0.870 2 8.CG1 21.800 1 8.HG11 0.870 2 8.C 175.733 1 9.N 123.474 1 9.HN 8.054 1 9.CA 56.403 1 9.HA 4.270 1 9.CB 30.901 1 9.HB2 1.759 2 9.HB1 1.882 2 9.CG 27.255 1 9.HD2 3.200 2 9.HD1 3.200 2
9.C 176.356 1 10.N 122.164 1 10.HN 8.245 1 10.CA 56.570 1 10.HA 4.314 1 10.CB 30.949 1 10.HB2 1.850 2 10.HB1 1.793 2 10.CG 27.165 1 10.HG2 1.680 2 10.HG1 1.680 2 10.CD 43.220 1 10.HD2 3.150 2 10.HD1 3.150 2 10.C 176.089 1 11.N 120.824 1 11.HN 8.358 1 11.CA 54.739 1 11.CB 41.032 1 11.HB2 2.688 2 11.HB1 2.688 2 11.C 176.755 1 12.N 119.657 1 12.HN 7.994 1 12.CA 63.424 1 12.HA 4.058 1 12.CB 32.435 1 12.HB 2.155 1 12.CG2 21.148 1 12.HG21 0.895 2 12.CG1 20.700 1 12.HG11 0.919 2 12.C 176.468 1 13.N 117.339 1 13.HN 8.268 1 13.CA 60.086 1 13.HA 4.324 1 13.CB 63.523 1 13.HB2 3.930 2 13.HB1 3.938 2 13.C 175.869 1 14.N 116.157 1 14.HN 8.058 1 14.CA 63.656 1 14.HA 4.227 1 14.CB 69.413 1 14.HB 4.272 1 14.CG2 21.835 1 14.HG21 1.228 1 14.C 175.265 1

84
15.N 124.964 1 15.HN 7.990 1 15.CA 54.042 1 15.HA 4.214 1 15.CB 18.875 1 15.HB1 1.409 1 15.C 178.677 1 16.N 118.397 1 16.HN 8.012 1 16.CA 56.556 1 16.HA 4.232 1 16.CB 42.070 1 16.HB2 1.520 2 16.HB1 1.700 2 16.CG 25.543 1 16.HG 1.551 1 16.CD1 25.172 1 16.HD11 0.875 2 16.CD2 23.820 1 16.HD21 0.875 2 16.C 178.073 1 17.N 114.395 1 17.HN 7.957 1 17.CA 59.920 1 17.HA 4.284 1 17.CB 63.625 1 17.HB2 3.898 2 17.HB1 3.921 2 17.C 175.280 1 18.N 120.801 1 18.HN 7.913 1 18.CA 56.635 1 18.HA 4.381 1 18.CB 30.658 1 18.HB2 1.946 2 18.HB1 1.819 2 18.CG 27.222 1 18.HG2 1.670 2 18.HG1 1.670 2 18.CD 43.626 1 18.HD2 3.192 2 18.HD1 3.192 2 18.C 176.781 1 19.N 119.516 1 19.HN 8.011 1 19.CA 56.812 1 19.HA 4.405 1 19.CB 33.752 1 19.HB2 2.110 2 19.HB1 2.110 2 19.CG 32.646 1 19.HG2 2.562 2 19.HG1 2.674 2 19.CE 20.972 1 19.HE1 0.940 1 19.C 176.725 1 20.N 108.541 1
20.HN 8.321 1 20.CA 45.958 1 20.HA2 4.095 2 20.HA1 4.092 2 20.C 174.742 1 21.N 121.101 1 21.HN 8.380 1 21.CA 56.358 1 21.HA 4.684 1 21.CB 41.237 1 21.HB2 2.822 2 21.HB1 2.730 2 21.C 177.589 1 22.N 119.884 1 22.HN 8.662 1 22.CA 58.274 1 22.HA 4.300 1 22.CB 29.430 1 22.HB2 2.110 2 22.HB1 2.108 2 22.CG 36.578 1 22.HG2 2.345 2 22.HG1 2.437 2 22.C 176.580 1 23.N 120.608 1 23.HN 8.005 1 23.CA 52.616 1 23.HA 4.427 1 23.CB 20.170 1 23.HB1 1.607 1 23.C 176.909 1 24.N 118.125 1 24.HN 7.900 1 24.CA 62.556 1 24.HA 3.349 1 24.CB 39.806 1 24.HB2 2.750 2 24.HB1 2.549 2 24.CD1 131.500 3 24.HD1 6.560 3 24.CE1 130.690 3 24.HE1 7.055 3 24.CZ 128.112 1 24.HZ 6.790 1 24.CE2 130.690 3 24.HE2 7.055 3 24.CD2 131.500 3 24.HD2 6.560 3 24.C 175.992 1 25.N 116.972 1 25.HN 8.605 1 25.CA 62.242 1 25.HA 3.808 1 25.CB 26.375 1 25.HB2 2.182 2 25.HB1 2.055 2 25.CG 37.435 1
25.HG2 2.380 2 25.HG1 2.433 2 25.C 176.638 1 26.N 104.666 1 26.CA 65.712 1 26.HA 4.324 1 26.CB 31.220 1 26.HB2 2.284 2 26.HB1 1.702 2 26.CG 28.078 1 26.HG2 2.133 2 26.HG1 1.977 2 26.CD 50.012 1 26.HD2 3.542 2 26.HD1 3.640 2 26.C 180.227 1 27.N 120.309 1 27.HN 7.241 1 27.CA 58.244 1 27.HA 3.863 1 27.CB 42.076 1 27.HB2 1.843 2 27.HB1 1.067 2 27.CG 27.478 1 27.HG 1.744 1 27.CD1 24.796 1 27.HD11 0.896 2 27.CD2 24.310 1 27.HD21 0.894 2 27.C 177.504 1 28.N 120.825 1 28.HN 8.016 1 28.CA 57.876 1 28.HA 3.498 1 28.CB 41.559 1 28.HB2 0.759 2 28.HB1 1.607 2 28.CG 26.545 1 28.HG 1.273 1 28.CD1 25.416 1 28.HD11 0.784 2 28.CD2 24.306 1 28.HD21 0.697 2 28.C 180.914 1 29.N 118.776 1 29.HN 8.080 1 29.CA 58.995 1 29.HA 3.947 1 29.CB 29.528 1 29.HB2 2.025 2 29.HB1 2.026 2 29.CG 36.154 1 29.HG2 2.203 2 29.HG1 2.324 2 29.CD 0.000 1 29.C 179.593 1 30.N 115.608 1

85
30.HN 7.661 1 30.CA 62.301 1 30.HA 4.324 1 30.CB 63.746 1 30.HB2 3.722 2 30.HB1 4.143 2 30.HG 0.000 1 30.C 174.835 1 31.N 118.715 1 31.HN 7.356 1 31.CA 56.920 1 31.HA 4.135 1 31.CB 40.483 1 31.HB2 1.949 2 31.HB1 1.507 2 31.CG 25.671 1 31.HG 2.181 1 31.CD1 27.315 1 31.HD11 0.853 2 31.CD2 21.715 1 31.HD21 0.698 2 31.C 176.178 1 32.N 108.905 1 32.HN 7.384 1 32.CA 57.243 1 32.HA 4.612 1 32.CB 64.095 1 32.HB2 3.919 2 32.HB1 3.995 2 32.C 174.322 1 33.N 123.005 1 33.HN 7.501 1 33.CA 55.455 1 33.HA 4.346 1 33.CB 41.361 1 33.HB2 2.535 2 33.HB1 2.926 2 33.ND2 114.090 1 33.HD21 8.057 2 33.HD22 6.959 2 33.C 175.248 1 34.N 124.677 1 34.HN 8.916 1 34.CA 58.841 1 34.HA 4.055 1 34.CB 29.886 1 34.HB2 2.085 2 34.HB1 2.155 2 34.CG 36.314 1 34.HG2 2.380 2 34.HG1 2.380 2 34.C 176.577 1 35.N 122.980 1 35.HN 9.222 1 35.CA 53.704 1 35.HA 4.816 1 35.CB 42.270 1
35.HB2 2.572 2 35.HB1 3.011 2 35.C 177.487 1 36.N 128.634 1 36.HN 8.322 1 36.CA 59.831 1 36.HA 4.264 1 36.CB 28.018 1 36.HB2 3.538 2 36.HB1 3.655 2 36.CD1 128.510 3 36.HD1 7.763 1 36.NE1 130.318 1 36.HE1 10.268 2 36.CZ2 114.690 3 36.HZ2 7.515 3 36.CH2 124.400 1 36.HH2 7.005 1 36.C 177.127 1 37.N 121.988 1 37.HN 7.856 1 37.CA 59.683 1 37.HA 3.444 1 37.CB 29.378 1 37.HB2 1.239 2 37.HB1 1.380 2 37.CG 24.945 1 37.HG2 0.047 2 37.HG1 -0.393 2 37.CD 43.058 1 37.HD2 2.603 2 37.HD1 2.680 2 37.C 178.013 1 38.N 117.623 1 38.HN 6.703 1 38.CA 64.370 1 38.HA 3.617 1 38.CB 38.221 1 38.HB 1.887 1 38.CG1 28.934 2 38.HG12 1.510 1 38.HG11 0.998 1 38.CD1 11.829 1 38.HD11 0.869 1 38.CG2 17.346 1 38.HG21 0.979 2 38.C 177.109 1 39.N 118.119 1 39.HN 8.199 1 39.CA 59.596 1 39.HA 4.281 1 39.CB 31.761 1 39.HB2 1.893 2 39.HB1 2.100 2 39.CG 28.625 1 39.HG2 1.798 2 39.HG1 1.655 2
39.CD 43.871 1 39.HD2 3.564 2 39.HD1 3.240 2 39.C 178.720 1 40.N 104.188 1 40.HN 8.620 1 40.CA 48.500 1 40.HA2 3.780 2 40.HA1 3.780 2 40.C 174.383 1 41.N 124.556 1 41.HN 7.918 1 41.CA 56.244 1 41.HA 4.134 1 41.CB 18.462 1 41.HB1 1.577 1 41.C 178.859 1 42.N 117.124 1 42.HN 8.135 1 42.CA 55.533 1 42.HA 3.926 1 42.CB 18.941 1 42.HB1 1.403 1 42.C 178.571 1 43.N 120.433 1 43.HN 8.288 1 43.CA 55.647 1 43.HA 3.704 1 43.CB 17.820 1 43.HB1 1.303 1 43.C 178.524 1 44.N 114.350 1 44.HN 8.060 1 44.CA 61.433 1 44.HA 3.916 1 44.CB 30.065 1 44.HB2 3.193 2 44.HB1 3.326 2 44.CD1 127.997 3 44.HD1 6.910 1 44.NE1 129.236 1 44.HE1 8.765 3 44.CE2 0.000 2 44.CZ2 114.138 3 44.HZ2 7.310 3 44.CH2 124.260 1 44.HH2 7.127 1 44.CZ3 118.228 3 44.HZ3 6.834 3 44.CD2 0.000 2 44.C 177.816 1 45.N 114.570 1 45.HN 7.794 1 45.CA 63.736 1 45.HA 4.046 1 45.CB 39.820 1 45.HB 1.890 1

86
45.CG1 29.247 2 45.HG12 1.952 1 45.HG11 1.229 1 45.CD1 14.258 1 45.HD11 0.878 1 45.CG2 19.095 1 45.HG21 1.055 2 45.C 179.210 1 46.N 119.132 1 46.HN 8.490 1 46.CA 65.955 1 46.HA 4.226 1 46.CB 38.609 1 46.HB 2.118 1 46.CG1 31.000 2 46.HG12 1.529 1 46.HG11 1.573 1 46.CD1 14.761 1 46.HD11 0.474 1 46.CG2 18.097 1 46.HG21 1.038 2 46.C 177.556 1 47.N 110.660 1 47.HN 8.444 1 47.CA 47.288 1 47.HA2 3.848 2 47.HA1 3.997 2 47.C 174.417 1 48.N 116.622 1 48.HN 8.065 1 48.CA 55.675 1 48.HA 4.415 1 48.CB 39.068 1 48.HB2 2.610 2 48.HB1 2.609 2 48.ND2 113.156 1 48.HD21 7.627 2 48.HD22 7.117 2 48.C 175.992 1 49.N 115.037 1 49.HN 7.659 1 49.CA 59.938 1 49.HA 4.224 1 49.CB 39.247 1 49.HB2 3.280 2 49.HB1 3.280 2 49.CD1 132.385 3 49.HD1 7.620 3 49.CE1 131.509 3 49.HE1 7.358 3 49.CE2 131.509 3 49.HE2 7.358 3 49.CD2 132.385 3 49.HD2 7.620 3 49.C 175.167 1 50.N 113.502 1 50.HN 8.088 1
50.CA 57.507 1 50.HA 3.617 1 50.CB 27.320 1 50.HB2 2.363 2 50.HB1 2.184 2 50.CG 34.939 1 50.HG2 2.178 2 50.HG1 2.321 2 50.NE2 111.421 1 50.HE21 7.482 2 50.HE22 6.795 2 50.C 172.751 1 51.N 119.167 1 51.HN 8.306 1 51.CA 53.124 1 51.HA 4.814 1 51.CB 44.501 1 51.HB2 1.835 2 51.HB1 2.689 2 51.C 177.763 1 52.N 125.785 1 52.HN 9.091 1 52.CA 58.639 1 52.HA 4.029 1 52.CB 29.658 1 52.HB2 2.130 2 52.HB1 2.130 2 52.CG 36.834 1 52.HG2 2.396 2 52.HG1 2.240 2 52.C 178.109 1 53.N 122.531 1 53.HN 9.549 1 53.CA 59.466 1 53.HA 4.168 1 53.CB 30.997 1 53.HB2 1.975 2 53.HB1 1.975 2 53.CG 27.511 1 53.HG2 2.020 2 53.HG1 1.920 2 53.CD 43.815 1 53.HD2 3.190 2 53.HD1 3.190 2 53.C 177.470 1 54.N 114.993 1 54.HN 7.767 1 54.CA 52.107 1 54.HA 4.213 1 54.CB 19.945 1 54.HB1 1.447 1 54.C 176.546 1 55.N 117.083 1 55.HN 7.545 1 55.CA 68.699 1 55.HA 3.261 1 55.CB 31.371 1
55.HB 2.149 1 55.CG2 20.644 1 55.HG21 0.695 2 55.CG1 25.351 1 55.HG11 1.044 2 55.C 175.840 1 56.N 117.032 1 56.HN 8.713 1 56.CA 62.191 1 56.HA 4.045 1 56.CB 26.649 1 56.HB2 2.081 2 56.HB1 2.080 2 56.CG 36.851 1 56.HG2 2.226 2 56.HG1 2.301 2 56.C 177.068 1 57.N 104.392 1 57.CA 65.733 1 57.HA 4.246 1 57.CB 31.265 1 57.HB2 2.239 2 57.HB1 1.661 2 57.CG 27.840 1 57.HG2 1.980 2 57.HG1 1.940 2 57.C 179.260 1 58.N 116.630 1 58.HN 7.519 1 58.CA 58.219 1 58.HA 4.012 1 58.CB 41.936 1 58.HB2 2.137 2 58.HB1 1.129 2 58.CG 26.268 1 58.HG 2.006 1 58.CD1 22.729 1 58.HD11 0.932 2 58.CD2 27.239 1 58.HD21 0.621 2 58.C 178.747 1 59.N 118.799 1 59.HN 8.290 1 59.CA 63.733 1 59.HA 3.568 1 59.CB 36.515 1 59.HB 2.147 1 59.CG1 28.225 2 59.HG12 1.536 1 59.HG11 1.261 1 59.CD1 12.950 1 59.HD11 0.622 1 59.CG2 17.422 1 59.HG21 0.851 2 59.C 180.037 1 60.N 118.820 1 60.HN 7.259 1

87
60.CA 59.066 1 60.HA 4.094 1 60.CB 31.968 1 60.HB2 1.930 2 60.HB1 1.931 2 60.CG 25.066 1 60.HG2 1.584 2 60.HG1 1.478 2 60.CD 28.941 1 60.HD2 1.685 2 60.HD1 1.680 2 60.CE 42.330 1 60.HE2 2.958 2 60.HE1 2.958 2 60.C 179.579 1 61.N 118.257 1 61.HN 7.492 1 61.CA 56.494 1 61.HA 4.168 1 61.CB 41.199 1 61.HB2 1.997 2 61.HB1 1.520 2 61.CG 26.935 1 61.HG 0.970 1 61.CD1 22.938 1 61.HD11 1.016 2 61.CD2 22.500 1 61.HD21 1.024 2 61.C 178.146 1 62.N 118.214 1 62.HN 7.548 1 62.CA 57.410 1 62.HA 4.208 1 62.CB 41.825 1 62.HB2 2.150 2 62.HB1 1.462 2 62.CG 25.861 1 62.HG 2.059 1 62.CD1 23.026 1 62.HD11 0.900 2 62.CD2 26.202 1 62.HD21 0.820 2 62.C 176.768 1 63.N 112.580 1 63.HN 7.257 1 63.CA 54.945 1 63.HA 4.512 1 63.CB 30.129 1 63.HB2 1.924 2 63.HB1 2.399 2 63.CG 36.662 1 63.HG2 2.265 2 63.HG1 2.271 2 63.C 176.196 1 64.N 122.241 1 64.HN 7.325 1 64.CA 55.619 1
64.HA 4.238 1 64.CB 44.384 1 64.HB2 2.945 2 64.HB1 2.654 2 64.C 175.721 1 65.N 123.636 1 65.HN 8.340 1 65.CA 56.509 1 65.HA 4.364 1 65.CB 41.572 1 65.HB2 2.615 2 65.HB1 2.614 2 65.CG 0.000 1 65.C 177.074 1 66.N 115.390 1 66.HN 9.679 1 66.CA 56.826 1 66.HA 4.440 1 66.CB 62.980 1 66.HB2 3.719 2 66.HB1 4.087 2 66.C 176.840 1 67.CA 47.400 1 67.HA2 3.570 2 67.HA1 3.930 2 67.C 176.070 1 68.N 122.900 1 68.HN 8.130 1 68.CA 61.055 1 68.HA 4.625 1 68.CB 40.281 1 68.HB2 3.523 2 68.HB1 3.030 2 68.CD1 131.500 3 68.HD1 7.525 3 68.CE1 131.970 3 68.HE1 7.220 3 68.CZ 129.277 1 68.HZ 6.860 1 68.CE2 131.970 3 68.HE2 7.220 3 68.CD2 131.500 3 68.HD2 7.525 3 68.C 176.783 1 69.N 118.134 1 69.HN 7.222 1 69.CA 66.188 1 69.HA 3.435 1 69.CB 32.305 1 69.HB 2.349 1 69.CG2 23.823 1 69.HG21 1.046 2 69.CG1 22.681 1 69.HG11 1.403 2 69.C 177.341 1 70.N 116.594 1 70.HN 8.126 1
70.CA 59.951 1 70.HA 4.045 1 70.CB 30.703 1 70.HB2 1.610 2 70.HB1 1.789 2 70.CG 27.076 1 70.HG2 1.703 2 70.HG1 1.406 2 70.CD 43.936 1 70.HD2 2.560 2 70.HD1 3.220 2 70.C 178.511 1 71.N 113.249 1 71.HN 7.924 1 71.CA 62.379 1 71.HA 4.074 1 71.CB 63.191 1 71.HB2 4.027 2 71.HB1 4.027 2 71.C 175.895 1 72.N 111.973 1 72.HN 7.033 1 72.CA 44.297 1 72.HA2 1.498 2 72.HA1 1.963 2 72.C 174.044 1 73.N 122.869 1 73.HN 8.069 1 73.CA 55.131 1 73.HA 3.900 1 73.CB 18.205 1 73.HB1 1.371 1 73.C 178.814 1 74.N 118.240 1 74.HN 7.807 1 74.CA 55.718 1 74.HA 3.777 1 74.CB 18.535 1 74.HB1 1.484 1 74.C 179.299 1 75.N 118.288 1 75.HN 7.514 1 75.CA 58.996 1 75.HA 4.011 1 75.CB 29.137 1 75.HB2 1.626 2 75.HB1 1.629 2 75.CG 26.733 1 75.HG2 1.827 2 75.HG1 1.720 2 75.CD 43.531 1 75.HD2 3.283 2 75.HD1 3.077 2 75.C 179.769 1 76.N 118.062 1 76.HN 8.645 1 76.CA 63.509 1

88
76.HA 4.292 1 76.CB 63.502 1 76.HB2 4.090 2 76.HB1 3.346 2 76.HG 0.000 1 76.C 176.300 1 77.N 122.775 1 77.HN 8.333 1 77.CA 58.939 1 77.HA 3.834 1 77.CB 41.857 1 77.HB2 0.965 2 77.HB1 1.866 2 77.CG 27.476 1 77.HG 1.695 1 77.CD1 26.166 1 77.HD11 0.591 2 77.CD2 25.136 1 77.HD21 0.861 2 77.C 178.070 1 78.N 117.352 1 78.HN 7.427 1 78.CA 59.581 1 78.HA 3.962 1 78.CB 29.221 1 78.HB2 2.317 2 78.HB1 2.264 2 78.CG 36.522 1 78.HG2 2.300 2 78.HG1 2.470 2 78.C 178.079 1 79.N 117.851 1 79.HN 7.787 1 79.CA 58.187 1 79.HA 4.014 1 79.CB 29.410 1 79.HB2 2.052 2 79.HB1 2.178 2 79.CG 33.823 1 79.HG2 2.332 2 79.HG1 2.386 2 79.NE2 109.998 1 79.HE21 7.312 2 79.HE22 6.565 2 79.C 177.843 1 80.N 119.280 1 80.HN 7.875 1 80.CA 65.778 1 80.HA 3.525 1 80.CB 39.874 1 80.HB 1.702 1 80.CG1 29.757 2 80.HG12 2.180 1 80.HG11 2.180 1 80.CD1 15.536 1 80.HD11 0.713 1 80.CG2 16.698 1
80.HG21 0.833 2 80.C 177.155 1 81.N 104.195 1 81.HN 7.857 1 81.CA 45.496 1 81.HA2 3.806 2 81.HA1 4.019 2 81.C 172.964 1 82.N 104.719 1 82.HN 8.027 1 82.CA 45.039 1 82.HA2 3.988 2 82.HA1 4.473 2 82.C 176.157 1 83.N 119.107 1 83.HN 8.810 1 83.CA 59.285 1 83.HA 4.093 1 83.CB 29.947 1 83.HB2 2.087 2 83.HB1 2.089 2 83.CG 36.237 1 83.HG2 2.380 2 83.HG1 2.380 2 83.C 179.137 1 84.N 121.215 1 84.HN 8.819 1 84.CA 59.081 1 84.HA 4.165 1 84.CB 29.371 1 84.HB2 1.849 2 84.HB1 2.051 2 84.CG 27.907 1 84.HG2 1.754 2 84.HG1 1.650 2 84.CD 43.134 1 84.HD2 3.229 2 84.HD1 3.218 2 84.C 179.943 1 85.N 119.126 1 85.HN 7.368 1 85.CA 67.302 1 85.HA 3.298 1 85.CB 32.159 1 85.HB 1.654 1 85.CG2 21.888 1 85.HG21 0.306 2 85.CG1 25.374 1 85.HG11 0.982 2 85.C 177.136 1 86.N 119.381 1 86.HN 7.793 1 86.CA 61.235 1 86.HA 3.563 1 86.CB 29.318 1 86.HB2 2.128 2 86.HB1 1.871 2
86.CG 28.085 1 86.HG2 1.576 2 86.HG1 1.751 2 86.CD 42.995 1 86.HD2 3.270 2 86.HD1 3.276 2 86.C 177.761 1 87.N 118.779 1 87.HN 8.044 1 87.CA 55.051 1 87.HA 4.144 1 87.CB 18.225 1 87.HB1 1.484 1 87.C 180.738 1 88.N 120.572 1 88.HN 7.611 1 88.CA 54.881 1 88.HA 4.114 1 88.CB 18.271 1 88.HB1 1.393 1 88.C 180.645 1 89.N 117.325 1 89.HN 8.471 1 89.CA 57.088 1 89.HA 4.197 1 89.CB 31.355 1 89.HB2 1.830 2 89.HB1 2.285 2 89.CG 32.902 1 89.HG2 2.550 2 89.HG1 1.750 2 89.C 178.298 1 90.N 120.174 1 90.HN 8.566 1 90.CA 60.206 1 90.HA 3.922 1 90.CB 29.637 1 90.HB2 2.050 2 90.HB1 2.218 2 90.CG 37.214 1 90.HG2 2.550 2 90.HG1 2.228 2 90.C 179.225 1 91.N 117.813 1 91.HN 7.260 1 91.CA 58.876 1 91.HA 4.223 1 91.CB 31.980 1 91.HB2 1.930 2 91.HB1 1.930 2 91.CG 24.816 1 91.HG2 1.446 2 91.HG1 1.532 2 91.CD 28.756 1 91.HD2 1.699 2 91.HD1 1.699 2 91.CE 42.436 1
89
91.HE2 2.960 2 91.HE1 2.960 2 91.C 179.804 1 92.N 122.334 1 92.HN 8.070 1 92.CA 57.399 1 92.HA 3.967 1 92.CB 43.464 1 92.HB2 1.249 2 92.HB1 1.767 2 92.CG 27.401 1 92.HG 1.610 1 92.CD1 24.283 1 92.HD11 0.833 2 92.CD2 24.973 1 92.HD21 0.833 2 92.C 179.013 1 93.N 120.383 1 93.HN 8.423 1 93.CA 54.860 1 93.HA 3.962 1 93.CB 18.448 1 93.HB1 1.371 1 93.C 178.020 1 94.N 114.261 1 94.HN 7.300 1 94.CA 58.086 1 94.HA 4.358 1 94.CB 31.292 1 94.HB2 2.189 2 94.HB1 2.189 2 94.CG 36.161 1 94.HG2 2.251 2 94.HG1 2.430 2 94.C 178.529 1 95.N 106.487 1 95.HN 8.077 1 95.CA 61.889 1 95.HA 4.613 1 95.CB 71.092 1 95.HB 4.390 1 95.CG2 21.812 1 95.HG21 1.215 1 95.C 175.615 1 96.N 111.590 1 96.HN 8.387 1 96.CA 44.885 1 96.HA2 3.697 2 96.HA1 4.383 2 96.C 172.647 1 97.N 109.639 1 97.HN 8.306 1 97.CA 60.477 1 97.HA 4.654 1 97.CB 71.819 1 97.HB 4.244 1 97.CG2 21.316 1
97.HG21 1.207 1 97.C 175.108 1 98.N 108.542 1 98.HN 8.481 1 98.CA 46.653 1 98.HA2 4.070 2 98.HA1 3.858 2 98.C 175.352 1 99.N 126.862 1 99.HN 8.915 1 99.CA 61.662 1 99.HA 4.169 1 99.CB 39.348 1 99.HB2 3.300 2 99.HB1 3.165 2 99.CD1 132.160 3 99.HD1 7.400 3 99.CE1 131.660 3 99.HE1 7.276 3 99.CZ 129.660 1 99.HZ 7.070 1 99.CE2 131.660 3 99.HE2 7.276 3 99.CD2 132.160 3 99.HD2 7.400 3 99.C 176.274 1 100.N 119.980 1 100.HN 8.581 1 100.CA 55.619 1 100.HA 3.781 1 100.CB 19.246 1 100.HB1 1.511 1 100.C 178.285 1 101.N 114.769 1 101.HN 6.848 1 101.CA 59.275 1 101.HA 3.677 1 101.CB 29.899 1 101.HB2 1.235 2 101.HB1 1.599 2 101.CG 27.787 1 101.HG2 1.554 2 101.HG1 1.679 2 101.CD 43.190 1 101.HD2 3.166 2 101.HD1 3.102 2 101.C 177.194 1 102.N 118.290 1 102.HN 7.484 1 102.CA 59.628 1 102.HA 3.858 1 102.CB 32.316 1 102.HB2 1.810 2 102.HB1 1.810 2 102.CG 24.979 1 102.HG2 1.353 2 102.HG1 1.510 2
102.CD 29.314 1 102.HD2 1.666 2 102.HD1 1.666 2 102.CE 42.321 1 102.HE2 2.980 2 102.HE1 2.980 2 102.C 179.505 1 103.N 119.101 1 103.HN 8.106 1 103.CA 65.942 1 103.HA 3.423 1 103.CB 31.723 1 103.HB 1.901 1 103.CG2 23.380 1 103.HG21 0.443 2 103.CG1 20.791 1 103.HG11 0.747 2 103.C 178.067 1 104.N 124.074 1 104.HN 7.740 1 104.CA 55.641 1 104.HA 4.451 1 104.CB 20.032 1 104.HB1 1.595 1 104.C 179.323 1 105.N 117.214 1 105.HN 8.552 1 105.CA 66.852 1 105.HA 3.586 1 105.CB 31.909 1 105.HB 2.071 1 105.CG2 21.123 4 105.HG21 0.955 4 105.CG1 22.885 4 105.HG11 1.099 4 105.C 178.822 1 106.N 116.863 1 106.HN 7.804 1 106.CA 56.838 1 106.HA 4.458 1 106.CB 39.260 1 106.HB2 2.783 2 106.HB1 2.787 2 106.ND2 112.785 1 106.HD21 7.808 2 106.HD22 6.866 2 106.C 177.909 1 107.N 122.295 1 107.HN 8.771 1 107.CA 62.561 1 107.HA 3.963 1 107.CB 38.923 1 107.HB2 3.107 2 107.HB1 3.229 2 107.CD1 132.590 3 107.HD1 6.740 3 107.CE1 116.970 3

90
107.HE1 6.280 3 107.CE2 116.970 3 107.HE2 6.280 3 107.CD2 132.590 3 107.HD2 6.740 3 107.C 179.078 1 108.N 119.384 1 108.HN 8.399 1 108.CA 57.405 1 108.HA 4.121 1 108.CB 41.459 1 108.HB2 1.632 2 108.HB1 1.976 2 108.CG 26.877 1 108.HG 2.058 1 108.CD1 26.000 1 108.HD11 0.830 2
108.CD2 22.497 1 108.HD21 0.986 2 108.C 178.603 1 109.N 116.503 1 109.HN 7.838 1 109.CA 58.477 1 109.HA 4.216 1 109.CB 30.457 1 109.HB2 2.111 2 109.HB1 2.221 2 109.CG 36.750 1 109.HG2 2.307 2 109.HG1 2.533 2 109.C 178.095 1 110.N 105.927 1 110.HN 7.363 1 110.CA 61.402 1
110.HA 4.370 1 110.CB 70.714 1 110.HB 4.163 1 110.CG2 21.774 1 110.HG21 1.135 1 110.HG1 4.200 1 110.C 173.594 1 111.N 125.239 1 111.HN 7.341 1 111.CA 57.596 1 111.HA 4.390 1 111.CB 30.002 1 111.HB2 2.278 2 111.HB1 3.189 2 111.C 178.435 1


















