
Post-doc (2011 2013) Financement régional et industriel
10
Post-doc (2011 – 2013) – Financement régional et industriel Sujet : « Synthèse totale d’archaeolipides tétraéthers – Vectorisation ciblée de peptides d’origine marine dans des archaeosomes pH-sensibles » Mots clé : archaeolipides, archaeosomes, poly(éthylène glycol), acide folique, vectorisation ciblée Collaboration industrielle sous charte de confidentialité Un des objectifs de l’innovation dans le domaine de la pharmacotechnie est d’élaborer des formes galéniques efficaces pour la vectorisation de molécules biologiquement actives. Toutefois, la vectorisation efficace et spécifique de principes actifs vers des cellules ou des tissus cibles reste actuellement un challenge important. Les archaeosomes, formulations liposomiales basées sur l’utilisation d’archaeolipides naturels ou synthétiques, semblent très prometteurs pour la vectorisation. En effet, les formulations à base d’archaeolipides naturels révèlent une stabilité in vitro et in vivo très supérieure à celle des liposomes conventionnels dans différentes conditions (hautes températures, milieux acides, présence de sérum animal, de phospholipases et/ou de sels biliaires). 1 Cependant, l’obtention d’archaeolipides naturels en quantité importante reste encore difficile. Par conséquent, toute une famille d’analogues de synthèse a été développée au sein de l’équipe « Chimie Organique Supramoléculaire » de l’UMR CNRS 6226. 2,3,4,5,6 L’incorporation de ces lipides dans des liposomes constitués de lipides conventionnels a permis d’améliorer sensiblement la stabilité in vitro des formulations dans des conditions compatibles avec une administration par voie orale. Ces propriétés remarquables résultent de la structure moléculaire atypique de ces lipides bipolaires et de leur mode d’organisation en monocouches au sein des membranes qui se distinguent très clairement du modèle classique en bicouche. Ces dernières années, de nouvelles modulations au niveau des têtes polaires d’archaeolipides de synthèse, de type diéther ou tétraéther, ont été apportés et permettent le greffage d’agents de ciblage cellulaire, notamment acide folique et sucre, ainsi que l’introduction de poly(éthylène glycol) en surface des formul ations. 1 Patel, G. B. A., B. J.; Deschatelets, L.; Fleming, L. P.; Sprott, G. D. Int. J. Pharma. 2000, 194, 39. 2 Brard, M. R., W.; Benvegnu, T.; Plusquellec, D. J. Am. Chem. Soc. 2004, 126, 10003. 3 Benvegnu, T.; Plusquellec, D.; Réthoré, G.; Sachet, M. Brevet FR0413028, WO Demand (PCT/EP 2005/056555), 2005. 4 Réthoré, G.; Montier, T.; Le Gall, T.; Delepine, P.; Cammas-Marion, S.; Lemiègre, M.; Lehn, P.; Benvegnu, T. Chem. Comm. 2007, 2054. 5 Brard, M.; Lainé, C.; Réthoré, G.; Laurent, I.; Neveu, C.; Lemiègre, L.; Benvegnu, T. J. Org. Chem. 2007, 72, 8267. 6 Benvegnu, T.; Lemiègre, L.; Cammas-Marion, S.; Eur. J. Org. Chem. 2008, 28, 4725.
Transcript of Post-doc (2011 2013) Financement régional et industriel

Post-doc (2011 – 2013) – Financement régional et industriel
Sujet : « Synthèse totale d’archaeolipides tétraéthers – Vectorisation ciblée de peptides d’origine marine
dans des archaeosomes pH-sensibles »
Mots clé : archaeolipides, archaeosomes, poly(éthylène glycol), acide folique, vectorisation ciblée
Collaboration industrielle sous charte de confidentialité
Un des objectifs de l’innovation dans le domaine de la pharmacotechnie est d’élaborer des formes galéniques
efficaces pour la vectorisation de molécules biologiquement actives. Toutefois, la vectorisation efficace et spécifique
de principes actifs vers des cellules ou des tissus cibles reste actuellement un challenge important.
Les archaeosomes, formulations liposomiales basées sur l’utilisation d’archaeolipides naturels ou synthétiques,
semblent très prometteurs pour la vectorisation. En effet, les formulations à base d’archaeolipides naturels révèlent
une stabilité in vitro et in vivo très supérieure à celle des liposomes conventionnels dans différentes conditions (hautes
températures, milieux acides, présence de sérum animal, de phospholipases et/ou de sels biliaires).1 Cependant,
l’obtention d’archaeolipides naturels en quantité importante reste encore difficile. Par conséquent, toute une famille
d’analogues de synthèse a été développée au sein de l’équipe « Chimie Organique Supramoléculaire » de l’UMR
CNRS 6226.2,3,4,5,6 L’incorporation de ces lipides dans des liposomes constitués de lipides conventionnels a permis
d’améliorer sensiblement la stabilité in vitro des formulations dans des conditions compatibles avec une
administration par voie orale. Ces propriétés remarquables résultent de la structure moléculaire atypique de ces lipides
bipolaires et de leur mode d’organisation en monocouches au sein des membranes qui se distinguent très clairement
du modèle classique en bicouche.
Ces dernières années, de nouvelles modulations au niveau des têtes polaires d’archaeolipides de synthèse, de type
diéther ou tétraéther, ont été apportés et permettent le greffage d’agents de ciblage cellulaire, notamment acide folique
et sucre, ainsi que l’introduction de poly(éthylène glycol) en surface des formulations.
1 Patel, G. B. A., B. J.; Deschatelets, L.; Fleming, L. P.; Sprott, G. D. Int. J. Pharma. 2000, 194, 39. 2 Brard, M. R., W.; Benvegnu, T.; Plusquellec, D. J. Am. Chem. Soc. 2004, 126, 10003. 3 Benvegnu, T.; Plusquellec, D.; Réthoré, G.; Sachet, M. Brevet FR0413028, WO Demand (PCT/EP 2005/056555), 2005. 4 Réthoré, G.; Montier, T.; Le Gall, T.; Delepine, P.; Cammas-Marion, S.; Lemiègre, M.; Lehn, P.; Benvegnu, T. Chem.
Comm. 2007, 2054. 5 Brard, M.; Lainé, C.; Réthoré, G.; Laurent, I.; Neveu, C.; Lemiègre, L.; Benvegnu, T. J. Org. Chem. 2007, 72, 8267. 6 Benvegnu, T.; Lemiègre, L.; Cammas-Marion, S.; Eur. J. Org. Chem. 2008, 28, 4725.

Il a ainsi été montré que ces vecteurs innovant à base d’archaeolipides PEGylés ciblant et de phospholipides classique
permettaient, d’une part, d’encapsuler des peptides biologiquement actifs et, d’autre part, de conserver une bonne
réponse tout en diminuant les doses administrées avec une bonne protection du peptide encapsulé.
Toutefois, les chaînes de PEG, nécessaires pour minimiser la reconnaissance du nanovecteur par le système
immunitaire, peuvent gêner voire empêcher l’agent de ciblage de reconnaitre et d’interagir avec les récepteurs
cellulaires en raison de l’encombrement stérique résultant de la présence de ces polymères. Par ailleurs, si la
concentration en agent de ciblage en surface des nanovecteurs est trop importante, une reconnaissance non spécifique
peut être observée.
Afin de remédier à ces problèmes, Torchilin et al. ont proposé un concept astucieux et novateur : la double
vectorisation.7 Ces auteurs ont :
- Recouvert les vecteurs de PEG via une liaison hydrolysable à pH acide,
- Fonctionnalisé ces derniers à l’aide de ligands internalisables par la cellule (acide folique, par exemple).
Après injection par voie intraveineuse, les chaînes de PEG présentes en surface des vecteurs masquent les ligands
d’internalisation cellulaire tout en assurant un ciblage passif vers les tissus ou cellules tumoraux grâce à l’effet EPR
(Enhanced and Permeability Retention Effect). Lorsque la cible est atteinte, le pH y étant plus bas que celui du sang,
la liaison entre le PEG et le vecteur est hydrolysée, libérant le PEG et démasquant ainsi la fonction de pénétration
cellulaire. Une augmentation de la quantité de principe actif dans les cellules peut donc être observée.Erreur ! Signet non
défini.
7 Torchilin, V. Adv. Drug Del. Rel. 2006, 58, 1532.

Dans ce contexte, nous proposons d’utiliser ce concept de double vectorisation pour améliorer l’efficacité
thérapeutique de peptides d’origine marine, fournis par les partenaires du projet, en les encapsulant dans
des archaeosomes PEGylés à ligand masqué. Nous envisageons donc de concevoir des archaeosomes
composés de :
- Un tétraéther PEGylé ayant une liaison sensible au pH entre le tétraéther et un PEG assez long
(5000 Da),
- Un tétraéther PEGylé possédant un agent de ciblage cellulaire relié à un PEG assez court
(570 Da),
- Un co-lipide conventionnel commercial.
La stratégie retenue implique d’abord la synthèse de la partie tétraéther des lipides. Cette synthèse, déjà
développée au laboratoire, a été optimisée et comprend les étapes suivantes :
- Couplage d’un cyclopentane diol avec un espaceur dichloré,
- Greffage d’un synthon phytanilé sur le cyclopentane dichloré,
- Débenzylation,
Le précurseur tétraéther diol, nécessaire à l’introduction des chaînes PEGylées, a ainsi été généré avec un
rendement global de 35 % sur 3 étapes.
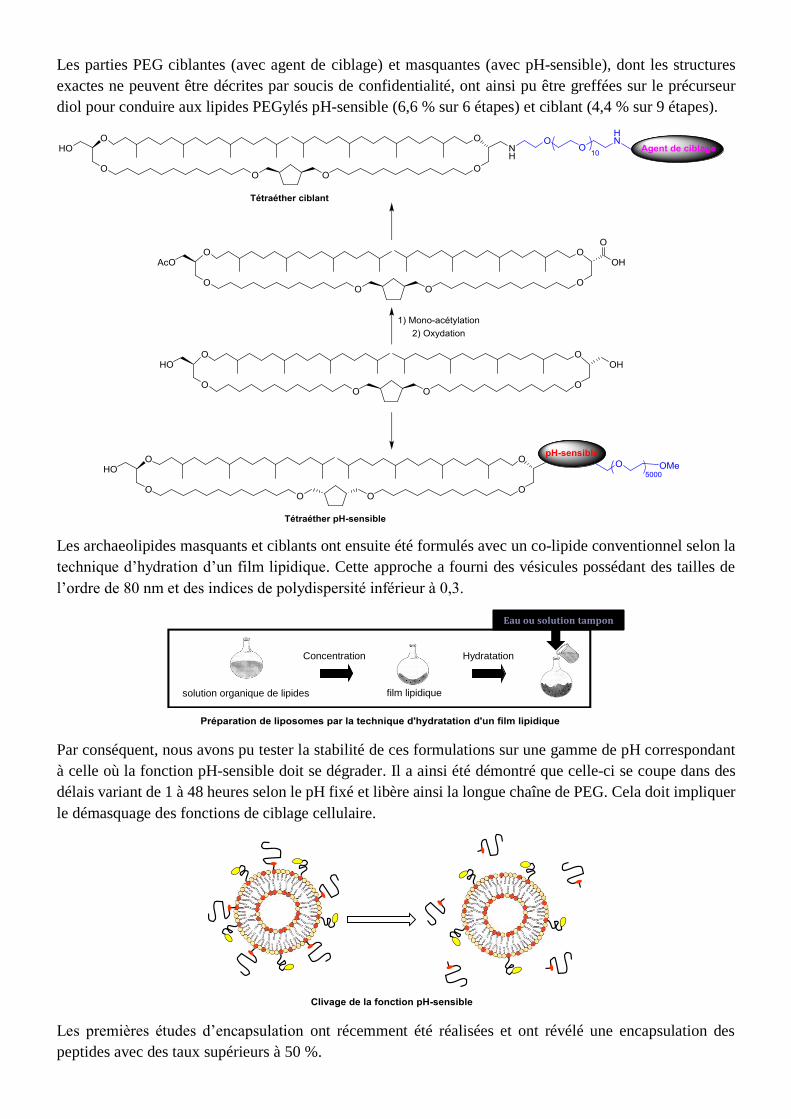
Les parties PEG ciblantes (avec agent de ciblage) et masquantes (avec pH-sensible), dont les structures
exactes ne peuvent être décrites par soucis de confidentialité, ont ainsi pu être greffées sur le précurseur
diol pour conduire aux lipides PEGylés pH-sensible (6,6 % sur 6 étapes) et ciblant (4,4 % sur 9 étapes).
Les archaeolipides masquants et ciblants ont ensuite été formulés avec un co-lipide conventionnel selon la
technique d’hydration d’un film lipidique. Cette approche a fourni des vésicules possédant des tailles de
l’ordre de 80 nm et des indices de polydispersité inférieur à 0,3.
solution organique de lipides film lipidique
Concentration Hydratation
Eau ou solution tampon
Par conséquent, nous avons pu tester la stabilité de ces formulations sur une gamme de pH correspondant
à celle où la fonction pH-sensible doit se dégrader. Il a ainsi été démontré que celle-ci se coupe dans des
délais variant de 1 à 48 heures selon le pH fixé et libère ainsi la longue chaîne de PEG. Cela doit impliquer
le démasquage des fonctions de ciblage cellulaire.
Les premières études d’encapsulation ont récemment été réalisées et ont révélé une encapsulation des
peptides avec des taux supérieurs à 50 %.

Thèse de doctorat (2006 – 2010) – Bourse régionale
Sujet : « Synthèse totale d’analogues de nucléosides à 4 et 6 chaînons et incorporation d’analogues
cyclobutyliques dans un motif oligonucléotidique antisens – Approche vers la synthèse de composés
galactosyl pyrrolo-pyridinones »
Mots clé : analogues de nucléosides, analogues d’oligonucléotides, stratégie antisens et anti-gène, synthèse
énantiosélective, cyclobutènes, cycloaddition [2+2] et [4+2], pyrrolo-pyridinones, -C-glycosylation,
régression cyclique
1 publication, 3 posters
1ère partie : Synthèse totale d’analogues de nucléosides à 4 chaînons et incorporation d’analogues
cyclobutyliques dans un motif oligonucléotidique antisens
Ce travail a porté sur la synthèse d’oligonucléotides à squelette cyclobutylique. En effet, l’intérêt pour ces structures
a été relevé ces dernières années par M. Yamaguchi et S. Katagiri.8 Nous avons donc souhaité accéder à des analogues
carbocycliques de l’Oxétanocine, une molécule découverte en 19869 qui a lancé l’intérêt pour ce type de structure.
Cependant, nous avons pensé qu’il serait plus judicieux d’utiliser des systèmes plus contraints sur la partie sucre (4’-
hydroxy au lieu de 4’-hydroxyméthyle) et plus flexibles sur la position pseudo-anomérique (1’-méthylène) pour
augmenter le degré de liberté au niveau de la position pseudo-anomérique et ainsi obtenir une meilleure
reconnaissance par la base nucléique. La fonction hydroxyle primaire serait protégée par une fonction 4,4’-
diméthoxytrityl (DMTr) indispensable pour envisager la synthèse oligonucléotidique.
La désymétrisation d’un diol méso racémique par une étape clé d’acétylation régiosélective et énantiosélective a
d’abord été effectué. La protection de l’hydroxyle primaire par la fonction DMTr, une désacétylation puis une étape
d’hydroboration-oxydation ont fourni un mélange d’isomères que nous ne sommes pas parvenu à séparer. Face à ces
difficultés inattendues, une voie plus lourde passant par une protection TBDPS a dû être utilisée, imposant une
déprotection de ce groupement seulement en fin de synthèse pour le remplacer par la fonction DMTr indispensable.
Même si cette étape s’est avérée plus délicate que prévu, nous avons finalement obtenus trois analogues
cyclobutaniques dont 2 en série thymine et 1 en série adénine avec des rendements globaux allant de 0,5 % à 5,3 %.
8 Katagiri, N.; Morishita, Y.; Osawa, I.; Yamaguchi, M. Tetrahedron Lett. 1999, 40, 6835. 9 (a) Shimada, N.; Hasegawa. S.; Harada. T.; Tomisawa, T.; Fujii, A.; Takita, T. J. Antibiot. 1986, 39, 1623 ; (b) Nakamura.
H.; Hasegawa, S.; Shimada, N.; Fujii. A.; Takita, T.; Iitaka, Y. Zbid 1986, 39, 1626.

Une première approche pour valider l’incorporation de ces motifs dans de nouveaux enchaînements
oligonucléotidiques originaux a ainsi pu être entreprise par l’équipe du Dr Rachid Benhida (Université de Nice
Sophia-Antipolis, Laboratoire de Chimie des Molécules Bioorganiques et des Arômes, UMR 6001 CNRS). Après
fonctionnalisation de son hydroxyle secondaire par un groupement phosphoramidite, un premier nucléoside a
finalement permis l’obtention de trois nouveaux enchaînements oligonucléotides originaux : deux 15-mères (ODN-
I et ODN-II) destinés à être testés dans un contexte antisens, et un 18-mère (ODN-III), qui sera testé pour la stratégie
anti-gène. Les évaluations sont encore en cours.

2ème partie : Synthèse totale d’analogues de nucléosides à 6 chaînons10
Ce travail a consisté à préparer de nouveaux analogues cyclohexéniques énantiopurs de nucléosides. L’intérêt pour
ce type de structure a débuté en 1993 quand une série de dérivés anhydrohexitol a démontré des propriétés
antivirales.11 Dans ce contexte, de nombreux analogues cyclohexéniques12 et cyclohexaniques13 optiquement purs
ont été synthétisés ces dernières années pour obtenir des propriétés anticancéreuses, notamment par incorporation
dans des oligonucléotides.14
La stratégie a d’abord impliqué l’ouverture stéréosélective d’un lactame cyclobuténique. Le diène (E,E) résultant a
ensuite réagit avec un diénophile portant un inducteur chiral selon une réaction de cycloaddition [4+2] totalement
régio- et endosélective qui a permis d’obtenir un adduit cyclohexénique énantiopur, dont la structure a été confirmée
par diffraction aux rayons X. Cette étape à l’avantage de créer 3 centres stéréogènes en 1 seule étape. Après une étape
de réduction qu’il nous a fallu optimiser pour obtenir un triol cyclohexénique, et l’introduction des bases nucléiques
par des méthodes classiques à partir d’une amine primaire, deux nouveaux analogues cyclohexéniques ont été obtenus
en série adénine et thymine avec des rendements globaux respectifs de 3,0 % sur 10 étapes et de 3,6 % sur 9 étapes.
Les résultats des évaluations biologiques de ces composés n’ont pas encore été enregistrés et seront complétés
ultérieurement dans le domaine anticancéreux.
10 Dalençon, S.; Ait-Youcef, R.; Pipelier, M.; Maisonneuve, V.; Dubreuil, D.; Huet, F.; Legoupy, S. J. Org. Chem. 2011, 76,
8059-8063. 11 (a) Verheggen, I.; Van Aerschot, A.; Toppet, S.; Snoeck, R.; Janssen, G.; Balzarini, J.; De Clercq, E.; Herdewijn, P. J. Med.
Chem. 1993, 36, 2033-2040; (b) Verheggen, I.; Van Aerschot, A.; Van Meervelt, L.; Rozenski, J.; Wiebe, L.; Snoeck, R.; Andrei,
G.; Balzarini, J.; Claes, P.; De Clercq, E.; Herdewijn, P. J. Med. Chem. 1995, 38, 826-835. 12 (a) Olivo, H. F.; Yu, J. J. Chem. Soc. Perkin Trans. 1 1998, 391-392; (b) Ferrer, E.; Alibes, R.; Busqué, F.; Figueredo, M.;
Font, J.; De March, P. J. Org. Chem. 2009, 74, 2425- 2432; (c) Wang, J.; Froeyen, M.; Hendrix, C.; Andrei, G.; Snoeck, R.; De
Clercq, E.; Herdewijn, P. J. Med. Chem. 2000, 43, 736-745; (d) Wang, J.; Herdewijn, P.; J. Org. Chem. 1999, 64, 7820-7827;
(e) Horvath, A.; Ruttens, B.; Herdewijn, P. Tetrahedron Letters 2007, 48, 3621-3623. 13 (a) Rosenquist, A.; Kvarnström, I. J. Org. Chem. 1996, 61, 6282-6288; (b) Mikhailov, S. N.; Blaton, N.; Rozenski, J.;
Balzarini, J.; De Clercq, E.; Herdewijn, P. Nucleosides & Nucleotides 1996, 15, 867-878; (c) Maurinsh, Y.; Schraml, J.; De
Winter, H.; Blaton, N.; Peeters, O.; Lescrinier, E.; Rozenski, J.; Van Aerschot, A.; De Clercq, E.; Busson, R.; Herdewijn, P. J.
Org. Chem. 1997, 62, 2861-2871; (d) Wang, J.; Busson, R.; Blaton, N.; Rozenski, J.; Herdewijn, P. J. Org. Chem.1998, 63,
3051-3058. 14 (a) Hendrix, C.; Rosemeyer, H.; Verheggen, I.; Seela, F.; Van Aerschot, A.; Herdewijn, P. Chem. Eur. J. 1997, 3, 110; (b)
Wang, J.; Herdewijn, P. J. Am. Chem. Soc. 2000, 122, 8595.

3ème partie : Approche vers la synthèse de composés galactosyl pyrrolo-pyridinones
Ce travail a porté sur l’accès à des dérivés galactosyl-pyrrolo-pyridinones selon une stratégie développée dans
l’équipe du Professeur Didier Dubreuil. Cette stratégie a impliqué une séquence rétrosynthétique de lactamisation
intramoléculaire / régression de cycle / cycloaddition [4+2] à partir de précurseur -galactosides 1C-acétyléniques.
Dans cet objectif, ma contribution s’est porté sur la préparation des précurseurs -galactosides 1-C-acétyléniques
portant en position 2 du sucre pyranosyle une fonction amine (ou amine masquée) R2. Cette fonction est destinée à
former ultérieurement le cycle lactamique de façon intramoléculaire en présence d’une fonction ester résiduelle.
Après diverses tentatives, la synthèse de 2-nitro--galactosides 1C-acétyléniques inconnus à ce jour a ainsi été
retenue et entreprise selon une méthode impliquant l’addition de Michael d’un acétylure de lithium sur un 2-
nitroglycal. Cette réaction a mené à des -galactosides 1C-acétyléniques diversement substitués, précurseurs des
dérivés galactosyl-pyrrolo-pyridinones.

Stage de Master 2 (2006)
Sujet : « Développement de nouvelles méthodologies de synthèse via des réactions de métathèse tandem
d’ouverture-fermeture de cycles à 4 chaînons »
Mots clé : métathèse d’alcènes et d’énynes, cyclobutènes
La formation de liaisons carbone-carbone par des méthodes fiables et efficaces est particulièrement importante en
chimie organique. Une de ces méthodes, la métathèse, a suscité beaucoup d'intérêt ces dernières années.15 Celle-ci
peut avoir lieu entre deux alcènes dans le cadre de la métathèse des oléfines, entre un alcène et un alcyne pour la
métathèse d'énynes et entre deux alcynes dans le cas de la métathèse d'alcynes.16
Les cyclobutènes étant de très bons substrats vis-à-vis des réactions de métathèse, l’objectif de mon stage de Master 2
a été la synthèse de différents bicycles adjacents insaturés par métathèse tandem d'ouverture et de fermeture
intramoléculaire. Après de nombreux essais, 2 bicycles ont été synthétisés par métathèse d’alcènes et 1 bicycle
comportant une fonction diénique a été préparé par métathèse d’énynes.
Ces synthons peuvent présenter un intérêt en tant que ligands pour la catalyse asymétrique17 mais également dans le
cadre de la préparation d’acétogénines (composés constituées par un cœur constitué d'un ou plusieurs hétérocycles
saturés oxygénés THF ou THP) présentant des propriétés antitumorales et insecticides. Le produit diénique peut,
quant à lui, se révéler intéressant dans le cadre de cycloadditions de type [4+2].
15 (a) Grubbs, R. H.; Chang, S. Tetrahedron 1998, 54, 4413-4450 ; (b) Blackwell, H. E.; O'Leary, D. J.; Chatterjee, A. K.;
Washenfelder, R. A.; Bussman, D. A.; Grubbs R. H. J. Am. Chem. Soc. 2000, 122, 58-71 ; (c) Jorgensen, M.; Hadwiger, P.;
Madsen, R.; Stutz, A. E.; Wrodnigg, T. M. Curr. Org. Chem. 2000, 4, 565-588. 16 Des catalyseurs à base de tungstène ou de molybdène ont été développés pour réaliser une métathèse de type yne-yne. Cette
chimie ne sera pas évoquée ici. On pourra consulter : (a) Fürstner, A. ; Angew. Chem. Int. Ed. 2000, 39, 3012-3043 ; (b)
Schrock, R. R. Chem. Rev. 2002, 102, 145-180. 17 Alexakis, A.; Tomassini, A.; Leconte, S. Tetrahedron 2004, 60, 9479-9484.
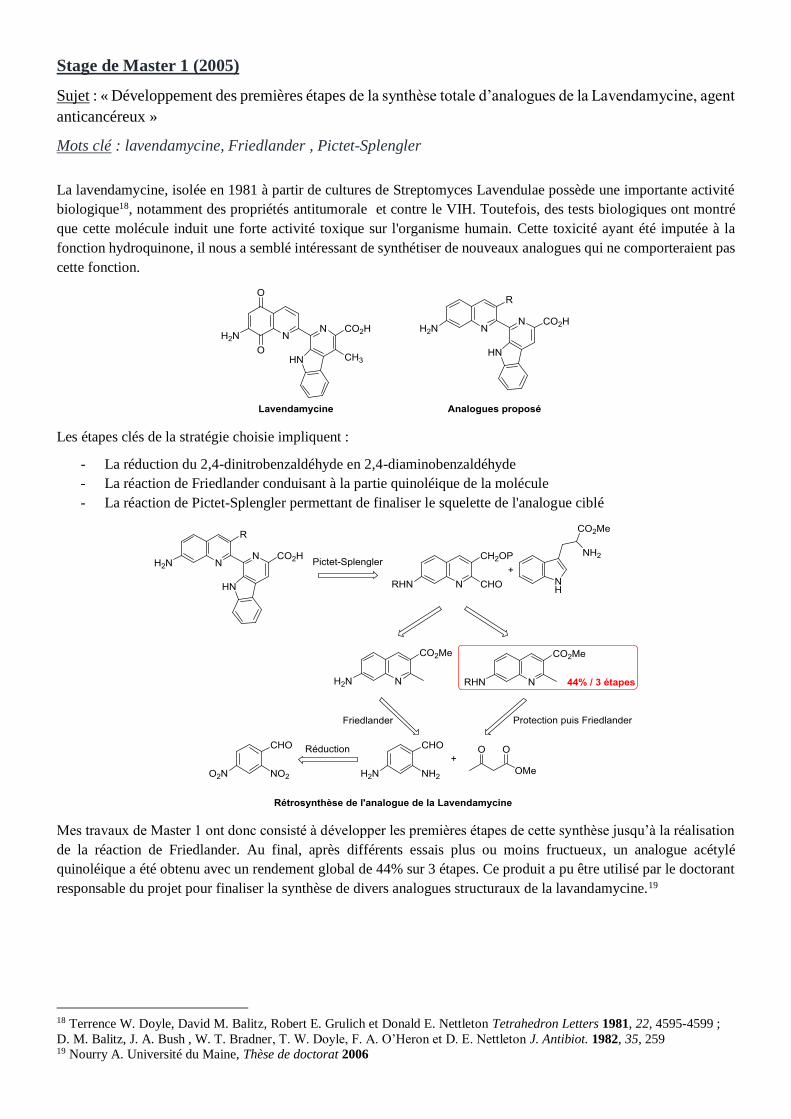
Stage de Master 1 (2005)
Sujet : « Développement des premières étapes de la synthèse totale d’analogues de la Lavendamycine, agent
anticancéreux »
Mots clé : lavendamycine, Friedlander , Pictet-Splengler
La lavendamycine, isolée en 1981 à partir de cultures de Streptomyces Lavendulae possède une importante activité
biologique18, notamment des propriétés antitumorale et contre le VIH. Toutefois, des tests biologiques ont montré
que cette molécule induit une forte activité toxique sur l'organisme humain. Cette toxicité ayant été imputée à la
fonction hydroquinone, il nous a semblé intéressant de synthétiser de nouveaux analogues qui ne comporteraient pas
cette fonction.
Les étapes clés de la stratégie choisie impliquent :
- La réduction du 2,4-dinitrobenzaldéhyde en 2,4-diaminobenzaldéhyde
- La réaction de Friedlander conduisant à la partie quinoléique de la molécule
- La réaction de Pictet-Splengler permettant de finaliser le squelette de l'analogue ciblé
Mes travaux de Master 1 ont donc consisté à développer les premières étapes de cette synthèse jusqu’à la réalisation
de la réaction de Friedlander. Au final, après différents essais plus ou moins fructueux, un analogue acétylé
quinoléique a été obtenu avec un rendement global de 44% sur 3 étapes. Ce produit a pu être utilisé par le doctorant
responsable du projet pour finaliser la synthèse de divers analogues structuraux de la lavandamycine.19
18 Terrence W. Doyle, David M. Balitz, Robert E. Grulich et Donald E. Nettleton Tetrahedron Letters 1981, 22, 4595-4599 ;
D. M. Balitz, J. A. Bush , W. T. Bradner, T. W. Doyle, F. A. O’Heron et D. E. Nettleton J. Antibiot. 1982, 35, 259 19 Nourry A. Université du Maine, Thèse de doctorat 2006


















