
Activité vanessa lefebvre. Le français et les langues romanes.

Développement d’antioxydants pour les huiles de friture
et contribution à l’étude du mécanisme de formation des
monomères cycliques à partir d’acides gras oméga-3
Mémoire
Vanessa Perreault
Maîtrise en sciences et technologie des aliments
Maître ès sciences (M. Sc.)
Québec, Canada
© Vanessa Perreault, 2015


iii
Résumé
Les acides gras polyinsaturés oméga-3 sont reconnus pour leurs bienfaits sur la
santé. Cependant, lors d’un traitement thermique, tel que la friture, plusieurs dégradations
peuvent se produire, entre autres, l’oxydation, la polymérisation et la cyclisation. Ces
dégradations se produisent aux températures utilisées dans l’industrie pour la
désodorisation des huiles ainsi que par les consommateurs pour la friture. Ce projet avait
pour but, d’une part, de développer de nouveaux antioxydants naturels pour les huiles de
friture. Ces composés seraient des dérivés d’acides phénoliques. Le coumarate d’hexyle
s’est d’ailleurs avéré être un bon antioxydant potentiel pour l’utilisation en friture. D’autre
part, ce projet a permis de contribuer à la synthèse d’un acide gras oméga-3 marqué, l’acide
α-linolénique, afin de déterminer le mécanisme de formation des monomères cycliques. Un
schéma de synthèse pour l’acide gras marqué a été élaboré et la synthèse a été amorcée.


v
Abstract
Omega-3 polyunsaturated fatty acids are well known for their benefits on health.
Unfortunately, thermal processes, such as deep frying, lead to the formation of degradation
products such as cyclic fatty acid monomers, oligomers, polymers, trans and oxygenated
fatty acids. All these degradations occur at temperature used in industries for oil
deodorisation and by consumers in frying process. In this context, the first part of this
project was to develop novel natural antioxidants for frying oil. Different phenolic acid
derivatives were tested for their antioxidant properties. Amongst them, hexyl coumarate
have shown a good potential as an antioxidant in frying process. Second part of this project
was to synthesize deuterated omega-3 fatty acid, α-linolenic acid, to determine the
mechanism of formation of cyclic fatty acid monomers. A synthesis scheme of the
deuterated α-linolenic acid was elaborated and the synthesis was carry out.


vii
Table des matières
Résumé .................................................................................................................................. iii
Abstract ................................................................................................................................... v
Table des matières ............................................................................................................... vii
Liste des tableaux ................................................................................................................... xi
Liste des figures .................................................................................................................. xiii
Liste des abréviations ......................................................................................................... xvii
Remerciements ..................................................................................................................... xxi
Avant-Propos .................................................................................................................... xxiii
Introduction ............................................................................................................................. 1
Chapitre 1 Revue de littérature ............................................................................................... 5
1.1 Acides gras polyinsaturés oméga-3 .......................................................................... 7
1.1.1 L’acide α-linolénique ......................................................................................... 8
1.2 Altération des acides gras polyinsaturés ................................................................... 8
1.3 Dégradation oxydative des acides gras ..................................................................... 9
1.3.1 Mécanisme d’oxydation ..................................................................................... 9
1.3.2 Facteurs qui influence l’oxydation des acides gras ......................................... 12
1.3.3 Les antioxydants .............................................................................................. 13
1.3.3.1 Antioxydants primaires ............................................................................. 13
1.3.3.2 Antioxydants secondaires ......................................................................... 15
1.3.3.3 Antioxydants synthétiques ........................................................................ 15
1.3.3.4 Antioxydants naturels ............................................................................... 16
1.4 Monomères cycliques d’acide gras ......................................................................... 16
1.4.1 Monomères cycliques de l’acide α-linolénique ............................................... 17
1.4.2 Effets des MCAG sur la santé .......................................................................... 19
1.4.3 Cinétique de dégradation de l’acide α-linolénique .......................................... 19
1.4.4 Mécanisme de formation des MCAG .............................................................. 22
1.4.4.1 Formation des MCAG par cyclisation concertée ...................................... 22
1.4.4.2 Formation des MCAG passant par un intermédiaire radicalaire .............. 23
1.4.4.3 Formation des MCAG par cycloaddition intramoléculaire suivi d’une
migration prototropique [1,6] ............................................................................... 24

viii
Hypothèse et Objectifs ......................................................................................................... 27
Hypothèse ..................................................................................................................... 27
Objectifs généraux ........................................................................................................ 27
Chapitre 2 Antioxydants pour les huiles de friture .............................................................. 29
2.1 Introduction ............................................................................................................ 31
2.2 Hypothèse et objectifs spécifiques ......................................................................... 33
Hypothèse ................................................................................................................. 33
Objectifs ................................................................................................................... 33
2.3 Méthodologie ......................................................................................................... 34
2.3.1 Synthèse du vanillate d’hexyle ........................................................................ 34
2.3.2 Synthèse du vanillate de dodécyle .................................................................. 35
2.3.3 Synthèse du coumarate d’hexyle ..................................................................... 35
2.3.4 Analyse thermique différentielle ..................................................................... 36
2.3.5 Test d’efficacité des antioxydants dans l’huile de Canola .............................. 36
2.3.6 Test d’efficacité des antioxydants en condition de friture .............................. 37
2.3.7 Analyse des huiles ........................................................................................... 38
2.4 Résultats et Discussion ........................................................................................... 39
2.4.1 Synthèse du vanillate d’hexyle ........................................................................ 39
2.4.2 Synthèse du vanillate de dodécyle .................................................................. 40
2.4.3 Synthèse du coumarate d’hexyle ..................................................................... 41
2.4.4 Analyse en DSC .............................................................................................. 43
2.4.5 Test d’efficacité des antioxydants dans l’huile de Canola .............................. 45
2.4.5.1 Test antioxydant du vanillate d’hexyle .................................................... 45
2.4.5.2 Test antioxydant du vanillate de dodécyle ............................................... 46
2.4.5.3 Test antioxydant du coumarate d’hexyle ................................................. 47
2.4.6 Test d’efficacité des antioxydants en condition de friture .............................. 50
2.5 Conclusion .............................................................................................................. 52
Chapitre 3 Approche à la synthèse de l’acide α-linolénique marqué ................................... 53
3.1 Introduction ............................................................................................................ 55
3.2 Hypothèse et objectifs ............................................................................................ 57
Hypothèse ................................................................................................................. 57
Objectifs ................................................................................................................... 57

ix
3.3 Approche à la synthèse ........................................................................................... 58
3.3.1 Choix de l’atome marqueur ............................................................................. 58
3.3.2 Synthèse d’acides gras non marqués ou marqués ............................................ 59
3.3.3 Stratégie de synthèse ........................................................................................ 60
3.3.4 Synthèse de l’acide 14,14-[H2-H2]-Z,Z,E-octadéca-9,12,15-triénoïque .......... 63
3.4 Résultats et discussion ............................................................................................ 64
3.4.1 Mono-protection du nonane-1,9-diol ............................................................... 64
3.4.2 Oxydation de la fonction alcool ....................................................................... 67
3.4.3 Seconde partie de la synthèse .......................................................................... 69
3.4.3.1 Réduction/deutération du trans-pent-2-énoate de méthyle ....................... 69
3.4.3.2 Formation de la fonction nitrile ................................................................ 69
3.4.3.3 Réduction partielle du nitrile .................................................................... 70
3.4.4 Protection de l’alcool en éther de pyranyle ..................................................... 70
3.4.5 Réaction de Wittig ........................................................................................... 71
3.4.5.1 Préparation du sel d’ylure ......................................................................... 71
3.4.5.2 Formation de l’alcène ............................................................................... 72
3.5 Perspectives envisagées pour la synthèse de l’AAL marqué .................................. 74
3.5.1 Approche selon la réaction de Wittig .............................................................. 74
3.5.2 Approche selon un couplage d’alcynes ........................................................... 75
3.6 Conclusion .............................................................................................................. 76
Chapitre 4 Discussion générale, conclusions et perspectives ............................................... 79
4.1 Discussion générale ................................................................................................ 81
4.2 Conclusions et perspectives .................................................................................... 84
Bibliographie ........................................................................................................................ 87
Partie expérimentale ............................................................................................................. 95
Solvants et réactifs .................................................................................................... 97
Purification des composés ........................................................................................ 98
Appareils d’analyse pour la synthèse ........................................................................ 98
Chapitre 2 : Antioxydants pour les huiles de friture ................................................... 100
Synthèse du vanillate d’hexyle ............................................................................... 100
Synthèse du vanillate de dodécyle .......................................................................... 101
Synthèse du p-coumarate d’hexyle ......................................................................... 102

x
Chapitre 3 : Approche à la synthèse de l’acide α-linolénique marqué ....................... 103
Monoprotection du nonane-1,9-diol ....................................................................... 103
Oxidation du diol monoprotégé : synthèse du synthon (3.26) ............................... 104
Réduction/deutération du trans-pent-2-énoate de méthyle .................................... 105
Formation du nitrile ................................................................................................ 105
Réduction partielle du nitrile .................................................................................. 106
Protection de l’alcool en éther de pyranyle ............................................................ 106
Annexes .............................................................................................................................. 109

xi
Liste des tableaux
Tableau 1.1 : Identification des temps de rétention par GC et signaux en FT-IR des
insaturations pour les isomères de MCAG de l’AAL dérivés en esters méthyliques avec une
colonne BPX-70 (Dobson, et al., 1996a, Mossoba, et al., 1994, Sébédio, et al., 1987a). .... 18 Tableau 2.1 : Programme de température utilisé en GC pour la séparation des FAME. ..... 39 Tableau PE.1 : Programme de température utilisé en chromatographie en phase gazeuse
pour la caractérisation des composés de synthèse. ............................................................... 99


xiii
Liste des figures
Figure 1.1 : Structures de l'AAL, de l'AEP et de l'ADH, les principaux AGPI n-3. ............. 7 Figure 1.2 : Délocalisation du radical sur une structure d’AG insaturé (McClements et
Decker, 2007). ....................................................................................................................... 10 Figure 1.3 : Cycle d'oxydation des acides gras et mécanisme des antioxydants. ................ 11 Figure 1.4 : β-scission d’un hydroperoxyde lipidique (Frankel, 1980, McClements et
Decker, 2007). ....................................................................................................................... 12 Figure 1.5 : Résonance d’un électron dans la structure d’un groupement phénol
(McClements et Decker, 2007, Wanasundara et Shahidi, 2005). ......................................... 15 Figure 1.6 : Structures principales des MCAG formées par l'AAL (Christie et Dobson,
2000). .................................................................................................................................... 17 Figure 1.7 : Structure des 8 isomères géométriques de l’AAL ............................................ 20
Figure 1.8 : Cinétique de formation des MCAG à partir d’un mélange enrichi en isomères
mono-trans de l’AAL (A) et présentation des résultats pour chaque mono-trans (B)
(Desmarais, 2013). ................................................................................................................ 21 Figure 1.9 : Cinétique de formation des cycles à 5 carbones entre le C10 et le C14 (A) et
entre le C11 et le C15 (B) et des cycles à 6 carbones avec une double liaison en C8 (C) et
en C16 (D) (Desmarais, non publié). .................................................................................... 22 Figure 1.10 : Formation des MCAG à partir de l’AAL par la cyclisation concertée (adapté
de (Destaillats et Angers, 2005, Gast, et al., 1963)). ............................................................ 23 Figure 1.11 : Formation des MCAG à partir d’un AGPI par la voie radicalaire (adapté de
(Destaillats et Angers, 2005, Dobson, et al., 1996b)). .......................................................... 24 Figure 1.12 : Mécanisme général de formation des MCAG par cycloaddition
intramoléculaire. ................................................................................................................... 25
Figure 1.13 : Formation des MCAG à partir de l’AAL selon un réarrangement
sigmatropique [1,5] (adapté de (Destaillats et Angers, 2005)). ............................................ 25 Figure 2.1 : Schéma de synthèse du vanillate d’hexyle. ...................................................... 34 Figure 2.2 : Synthèse du vanillate de dodécyle. ................................................................... 35 Figure 2.3 : Synthèse du coumarate d’hexyle. ..................................................................... 36 Figure 2.4 : Réarrangement de McLafferty du vanillate d’hexyle. ...................................... 40 Figure 2.5 : Réarrangement de McLafferty du vanillate de dodécyle. ................................ 41
Figure 2.6 : Produits secondaires de la réaction d’estérification de l’acide p-coumarique. 42 Figure 2.7 : Réarrangement de McLafferty du coumarate d’hexyle. ................................... 42 Figure 2.8 : Structure de résonance de l’acide p-coumarique. ............................................. 43 Figure 2.9 : Comparaison des résultats du vanillate d’hexyle et de dodécyle et du
coumarate d’hexyle au DSC pour des températures de 50°C à 200°C. ................................ 44
Figure 2.10 : Analyse du vanillate de dodécyle par DSC de 20°C à 250°C. ....................... 44
Figure 2.11 : Teneur résiduelle en acides gras insaturés après 1 (A) et 2 (B) jours de
chauffage thermo-oxydatif à 120°C de l’huile de Canola avec différentes concentrations de
vanillate d’hexyle par rapport à l’huile de Canola seule. ..................................................... 46

xiv
Figure 2.12 : Teneur résiduelle en acides gras insaturés après 1 et 2 jours de chauffage
thermo-oxydatif à 120°C de l’huile de Canola avec le vanillate de dodécyle à une
concentration de 1,0% par rapport à l’huile de Canola seule. .............................................. 47 Figure 2.13 : Teneur résiduelle en acides gras insaturés après 1 (A) et 2 (B) jours de
chauffage thermo-oxydatif à 120°C de l’huile de Canola avec différentes concentration de
coumarate d’hexyle par rapport à l’huile de Canola seule. .................................................. 48 Figure 2.14 : Rapport des teneurs résiduelles en acide linoléique (A) et en acide α-
linolénique (B) à 180°C de l’huile supplémentée en coumarate d’hexyle (HC) ou en BHT à
une concentration de 0,1% par rapport à l’huile de Canola seule. ....................................... 49 Figure 2.15 : Teneur résiduelle des différents acides gras lors de la friture. ....................... 50 Figure 2.16 : Teneur résiduelle en acide linoléique (A) et en acide α-linolénique (B) de
l’huile supplémentée en coumarate d’hexyle (HC) ou en BHT à une concentration de 0,1%
par rapport l’huile de Canola seule en condition de friture. ................................................. 51
Figure 3.1 : Déplacement d’un atome de deutérium lors de la formation d’un cycle. ........ 58 Figure 3.2 : Mécanisme réactionnel pour un couplage d’alcyne suivi d’une hydrogénation
complète (A) et d’une hydrogénation partielle (B), ainsi qu’une réaction de Wittig (C). ... 59 Figure 3.3 : Mécanisme réactionnel d’une deutération à l’aide d’un dideutérium (A) et d’un
deutérure de lithium et d’aluminium (B). ............................................................................. 60 Figure 3.4 : Schéma rétrosynthétique I du 11,11-[2H,2H]-linolénate de méthyle (3.15) ..... 61 Figure 3.5 : Schéma rétrosynthétique II du 11,11-[2H,2H]-linolénate de méthyle (3.15) ... 61 Figure 3.6 : Schéma rétrosynthétique I de l'acide 14,14-[H2-H2]-Z,Z,E-octadéca-9,12,15-
triénoïque (3.40). .................................................................................................................. 62 Figure 3.7 : Schéma de synthèse de l’acide 15-trans-14,14-[2H,2H]-linolénique (3.40). ... 64 Figure 3.8 : Schéma réactionnel de la mono-protection du diol. ........................................ 65 Figure 3.9 : Fragmentation de la partie acétal du diol mono-protégé. ................................ 66 Figure 3.10 : Fragmentation de l’acétal. .............................................................................. 66
Figure 3.11 : Fragmentation du groupement acétal de l’aldéhyde ...................................... 68 Figure 3.12 : Fragmentation de McLafferty de l’aldéhyde ................................................. 68 Figure 3.13 : Schéma réactionnel de la réduction/deutération du trans-pent-2-énoate de
méthyle. ................................................................................................................................ 69 Figure 3.14 : Schéma réactionnel de la formation de la fonction nitrile via la réaction de
Mitsunobu-Wilk. .................................................................................................................. 70
Figure 3.15 : Schéma réactionnel de la réduction partielle de la fonction nitrile. ............... 70 Figure 3.16 : Réaction de protection de l’alcool en éther de pyranyle. ............................... 71 Figure 3.17 : Schéma réactionnel de la synthèse du sel d’ylure. ......................................... 72 Figure 3.18 : Réaction de Wittig avec le nonanal. .............................................................. 72 Figure 3.19 : (I) Chromatogramme des produits obtenus suite à la réaction de Wittig avec
le nonanal. (II) Spectre MS de l’alcène (3.35) obtenu suite à la réaction de Wittig. ........... 73
Figure 3.20 : Spectre MS de l’éther d’énol (3.50) obtenu lors de la réaction de Wittig. .... 73
Figure 3.21 : Hydrolyse de l’éther d’énol (3.50) pour former l’aldéhyde (3.51). ............... 74 Figure 3.22 : Schéma rétrosynthétique I via le couplage d’alcynes de l’acide 14,14-
[2H,2H]-Z,Z,Z-octadéca-9,12,15-triénoïque (Desmarais, 2013). ......................................... 76 Figure A.1 : Spectre MS du vanillate d’hexyle ................................................................. 110 Figure A.2 : Spectre IR du vanillate d’hexyle ................................................................... 110

xv
Figure A.3 : Spectre MS du vanillate de dodécyle ............................................................ 110
Figure A.4 : Spectre IR du vanillate de dodécyle .............................................................. 110 Figure A.5 : Spectre MS du coumarate d’hexyle ............................................................... 110 Figure A.6 : Spectre IR du coumarate d’hexyle ................................................................ 110 Figure A.7 : Spectre MS du mom-nonanol ........................................................................ 110 Figure A.8 : Spectre IR du mom-nonanol .......................................................................... 110 Figure A.9 : Spectre RMN 1H du mom-nonanol ............................................................... 110 Figure A.10 : Spectre RMN C13 du mom-nonanol ............................................................ 110 Figure A.11 : Spectre MS du synthon (3.12) ..................................................................... 110


xvii
Liste des abréviations
A• : Antioxydant radicalaire
AAL : Acide α-linolénique
AcOEt : Acétate d’éthyle
ADH : Acide docosahexaénoïque
AEP : Acide eicosapentanéoïque
AG : Acide gras
AGPI n-3 : Acide gras polyinsaturé oméga-3
AH : Antioxydant primaire
AL : Acide linoléique
AlCl3 : Trichlorure d’aluminium
AO : Acide oléique
ATR : de l’anglais «attenuated total reflectance», soit réflexion totale interne
BHA : Hydroxyanisole butylé
BHT : Hydroxytoluène butylé
bp : de l’anglais «boiling point», soit point d’ébullition 13C : Carbone 13
CCM : Chromatographie sur couche mince
CDCl3 : Chloroforme deutéré
CH2Cl2 : Dichlorométhane
CSA : Acide D-10-camphorsulfonique
δ : Déplacement chimique
D : Deutérium
DIAD : Diisopropyl azodicarboxylate
DIBAL-H : Hydrure de diisobutyl aluminium
DMF : N,N-diméthylformamide
DMOX : 4,4-diméthyloxazoline
DMSO : Diméthylsulfoxyde
DSC : de l’anglais «differential scanning calorimetry», soit colorimétrie
différentielle à balayage
équiv. : équivalent
Et2O : Éther diéthylique
EtOH : Éthanol
eV : Electronvolt
FAME : de l’anglais «fatty acid methyl ester», soit ester méthylique d’acide gras
FT-IR : Spectrométrie infrarouge à transformée de Fourier

xviii
GC : Chromatographie en phase gazeuse
GC-MS : GC couplé à un spectromètre de masse
GPr : Groupement protecteur 1H : Hydrogène ou proton 2H : Deutérium
H2O2 : Hydopéroxyde d’hydrogène
HCl : Acide chlorydrique
He : Hélium
HMPA : Hexaméthylphosphoramide
IE : Impact électronique
IR : Infrarouge
kcal : kilocalorie
KD : Vitesse de réaction du Deutérium
KH : Vitesse de réaction de l’Hydrogène
KHCO3 : Bicarbonate de potassium
KOH : Hydroxyde de potassium
KPa : Kilopascal
L• : Radical lipidique
LiAlD4 : Deutérure de lithium et d’aluminium
LiAlH4 : Hydrure de lithium et d’aluminium
LH : Acide gras
LO• : Radical alkoxyde
LOH : Alcool lipidique
LOO• : Peroxyde lipidique
LOOH : Hydropéroxyde lipidique
M : pic moléculaire
MCAG : Monomère cyclique d’acides gras
MCAG-D : Monomère cyclique d’acides gras marqué par le deutérium
MgSO4 : Sulfate de magnésium
MHz : Mégahertz
m/m : masse sur masse
MOM : méthoxyméthyle
MS : Spectrométrie de masse
m/v : masse sur volume
m/z : Rapport masse sur charge
n-BuLi : n-Butyllithium
NaCl : Chlorure de sodium
NaHCO3 : Bicarbonate de sodium

xix
NaOH : Hydroxyde de sodium
Na2S2O3 : Thiosulfate de sodium
O=PPh3 : Oxyde de triphénylphosphine
PDC : Dichromate de pyridinum
PPh3 : Triphénylphosphine
Psi : de l’anglais «pound per square inch», soit livre par pouce carré
RMN 13C : Résonance magnétique nucléaire du carbone 13
RMN 1H : Résonance magnétique nucléaire du proton
u.m. : Unité de masse
T : Tritium
t-BDMS : tert-Butyl diméthylsilyle
TBHQ : Butylhydroquinone tertiaire
THF : Tétrahydrofurane
TsOH : Acide p-toluènesulfonique
µm : Micromètre
νmax : Nombre d’onde


xxi
Remerciements
Je tiens tout d’abord à remercier Paul Angers, mon directeur de recherche, pour
m’avoir donné l’opportunité de réaliser mes deux années de maitrise au sein de son équipe
de recherche. Le fait de m’avoir permis de travailler seule sur certain projet lors de mon
baccalauréat m’a permis d’acquérir une autonomie qui m’a fait apprécier mon expérience
au maximum, tant lors de mon stage que durant ma maitrise. Merci pour les conseils, les
réponses à mes questions et les encouragements lorsque la synthèse ne semblait pas du tout
avancer. Son approche humaine et sincère n’aura fait que rendre cette expérience plus
agréable. Un énorme merci pour m’avoir permis de participer à un congrès international en
Turquie.
Je souhaiterais remercier Joseph Arul et Khaled Belkacemi pour avoir accepté
d’évalué ce mémoire.
Je tiens également à remercier Amélie Desmarais qui m’a accueilli dans le
laboratoire et m’a supervisé lors de mon stage il y a déjà quatre ans. Elle a su me
transmettre sa bonne humeur et sa persévérance.
Je tiens à remercier mes deux stagiaires, Laura Chevalier et Véronique Perreault,
qui m’ont aidé dans l’avancement des projets. Leur bonne humeur, leur persévérance et leur
initiative m’a beaucoup aidé à avancer. J’aimerais également remercier Ronan Corcuff pour
sa disponibilité, sa bonne humeur, son soutien et son aide pour les analyses. Je voudrais
également remercier Diane Gagnon, Pascal Lavoie, Gaétan Desnoyer et Pierre Côté.
Je tiens également à remercier mes collègues étudiants de l’ACCESTA et mes amis
pour leur soutien, leurs conseils et leurs encouragements et leur aide précieuse au quotidien.
Merci pour votre bonne humeur, les discussions et les rires échangés au cours des années.
Merci pour les sorties et les soupers de filles qui nous permettaient de décrocher. Merci

xxii
Amélie, Marie-Ève, Audrey-Anne, Laetitia, Marie-Christine, Elodie, Shyam, Sergei, Abdel,
Valérie, Alina, Cheslav et tous les autres.
Bien entendu, je souhaite remercier ma famille, mes parents et ma sœur, qui m’ont
supporté et encouragé tout au long de ce projet. Leur support moral a été d’une grande
importance durant ces deux années.
À tous ceux et celles que j’ai oublié : merci!

xxiii
Avant-Propos
Ce mémoire est composé de quatre chapitres. Le premier chapitre est une revue de
littérature traitant des dégradations oxydatives et thermiques des acides gras dans les huiles
de fritures. Enfin les problématiques qui en découlent seront présentées, ainsi que notre
hypothèse de recherche et les objectifs à atteindre afin de vérifier cette hypothèse.
Les deuxième et troisième chapitres présenteront les méthodologies et les résultats
obtenus pour les deux projets de recherche. Le deuxième sera sur l’efficacité des
antioxydants dans l’huile de friture et le troisième sera sur la synthèse de l’acide α-
linolénique marqué au deutérium.
Le dernier chapitre présentera une discussion et une conclusion générales, ainsi que
les perspectives pour les projets.


1
Introduction
L’industrie du fast-food est l’un des symboles de la malnutrition et de l’obésité en
Amérique du Nord. Ces facteurs peuvent d’ailleurs augmenter les risques de maladies
cardiovasculaires. À titre d’exemple, au Canada, en 2009, les maladies cardiovasculaires
étaient la cause d’environ 29% des décès, en partie dû à la mauvaise alimentation de la
population (Statistique Canada, 2009). En 2000, les coûts reliés à ces maladies étaient de
plus de 22 milliards de dollars au Canada (Agence de la santé publique du Canada, 2000).
Parmi les facteurs qui augmentent les risques de maladies cardiovasculaires, certains sont
indépendant de notre volonté, tel que l’âge et le sexe, alors que d’autres peuvent être
contrôlé, dont le diabète, les lipides anormaux et l’obésité (Aronne, et al., 2007, Dahlöf,
2010). Pour ces raisons, les consommateurs essaient d’inclure une alimentation plus saine
dans leurs habitudes de vie. Les bienfaits des acides gras polyinsaturés oméga-3 (AGPI
n-3) étant très souvent cités, cela incite donc les consommateurs à diminuer la quantité
d’acides gras (AG) saturés qu’ils consomment pour les remplacer par des AG insaturés.
L’utilisation de l’huile de Canola, riche en AG insaturés, comme huile de friture en est un
bon exemple.
Toutefois, les AGPI n-3 présent dans les huiles de friture tendent à se dégrader
lorsqu’ils sont en présence de certains facteurs, dont l’oxygène et les hautes températures.
En présence d’oxygène, l’oxydation sera la réaction prédominante. Les insaturations vont
alors former des peroxydes et des radicaux libres (Frankel, 1984), néfastes pour la santé. De
plus, lorsqu’on effectue un traitement thermique prolongé, les AGPI présents dans les
huiles peuvent subir des réactions de polymérisation, d’isomérisation en acides gras trans,
et peuvent également former des monomères cycliques d’acides gras (MCAG).
Inévitablement, toutes ces dégradations se produisent aux températures utilisées dans
l’industrie pour la désodorisation des huiles et pour la friture (Sébédio et Grandgirard,
1989).

2
Ainsi, les AGPI n-3 présents dans les huiles se détérioreront en AG trans, en
polymères et en MCAG sous l’effet de la chaleur (Sébédio et Grandgirard, 1989). Cela se
produit aux températures utilisées par l’industrie alimentaire pour la friture et la
désodorisation des huiles (environ 180°C), ainsi que par les consommateurs pour la friture
(Aladedunye et Przybylski, 2009). À titre d’exemple, les MCAG peuvent être retrouvés à
des teneur de 0,01 à 0,7% dans les huiles (Frankel, et al., 1984, Sébédio et Grandgirard,
1989). Plus le degré d’insaturation est élevé, plus l’AG aura tendance à se cycliser. Ainsi,
l’acide α-linolénique (AAL) sera l’acide qui formera le plus de MCAG dans les huiles
végétales. Les monomères de cet acide sont bien connus et leurs structures ont déjà été
caractérisées (Christie et Dobson, 2000, Dobson, et al., 1996a, Mossoba, et al., 1995).
Toutefois, leurs mécanismes de formation ne restent qu’hypothétique à ce jour.
Récemment, un mécanisme de formation des MCAG passant par une cycloaddition
intramoléculaire suivi d’une migration prototropique [1,6] a été proposé (Destaillats et
Angers, 2005).
Pour ce qui est de la dégradation oxydative, différents moyens peuvent être utilisés
afin de la limiter ou de la retarder. Parmi ceux-ci, les antioxydants sont parmi les plus
utilisés. Il existe deux types d’antioxydants, soit primaires et secondaires. Les premiers
vont réagir avec les radicaux formés afin d’arrêter la propagation des produits de
dégradation (Gutteridge, 1994, McClements et Decker, 2007), alors que les antioxydants
secondaires vont plutôt capter l’oxygène présent dans les huiles et ainsi retarder le
mécanisme d’oxydation (Gutteridge, 1994). Les antioxydants primaires sont ceux qui sont
le plus souvent utilisés dans les huiles de friture. Ce type d’antioxydants est principalement
de molécules possédant un groupement phénol. Le BHT, le BHA et le TBHQ sont les
antioxydants synthétiques les plus souvent retrouvés dans les huiles et les plus efficaces.
Certaines études ont toutefois démontré qu’ils pouvaient être potentiellement nocifs pour la
santé (Farag, et al., 2003, Shahidi et Zhong, 2005). Pour cette raison, les consommateurs
cherchent à les éviter.

3
La voie empruntée lors de la formation des MCAG n’a donc toujours pas été
élucidée. Une partie de ce projet visait donc à apposer des atomes marqueurs, soit le
deutérium, sur la structure de l’AAL. En les apposant sur un carbone impliqué dans la
réaction de cyclisation, le mécanisme emprunté pourra ainsi être élucidé. D’un autre côté,
ce projet visait également à développer des antioxydants pour les huiles de friture.
L’activité des antioxydants qui auront été synthétisés sera testée préalablement dans l’huile
de canola avant d’être testée en condition de friture.


Chapitre 1
Revue de littérature


7
1.1 Acides gras polyinsaturés oméga-3
Les acides gras polyinsaturés oméga-3 (AGPI n-3) sont reconnus pour leurs
nombreux bénéfices pour la santé. Ils permettent entre autre d’abaisser les risques de
maladies cardiovasculaires et contribueraient au développement de certaines fonctions du
cerveau (Kris-Etherton, et al., 2002, Ruxton, et al., 2004, Vancassel, 2004). De plus, les
AGPI n-3 aurait également un impact sur la santé oculaire, en particulier sur la rétine
(Jeffrey, et al., 2001, SanGiovanni et Chew, 2005). Pour tous ces bienfaits qui leurs sont
associés, les consommateurs tentent d’en inclure le plus possible dans leur diète, entre autre
par la consommation de poissons gras et d’huiles végétales riches en AGPI n-3, tel que
l’huile de Canola.
Les trois principaux AGPI n-3 sont l’acide α-linolénique (AAL), l’acide
eicosapentaénoïque (AEP) et l’acide docosahexaénoïque (ADH) (Figure 1.1). Le premier
est d’origine végétale, alors que l’AEP et l’ADH sont d’origine marine. L’AAL est dit
essentiel, car il ne peut être synthétisé par l’être humain. Son apport provient donc
essentiellement de l’alimentation. De plus, l’AAL sert de précurseur chez l’homme à la
synthèse des AGPI n-3 à longue chaîne, tels que l’AEP et l’ADH (Barcelo-Coblijn et
Murphy, 2009, Burdge et Calder, 2005).
HO
O
HO
O
HO
O
Acide -linolénique (AAL)
Acide eicosapentaénoique (AEP)
Acide docosahexaénoique (ADH)
9 12 15
5 8 11 14 17
4 7 10 13 16 19
Figure 1.1 : Structures de l'AAL, de l'AEP et de l'ADH, les principaux AGPI n-3.

8
Bien que les AGPI n-3 soient bons pour la santé, ils sont tout de même sensibles aux
dégradations thermiques et oxydatives, produisant alors des produits nuisibles pour la santé.
En effet, les insaturations (doubles liaisons carbone-carbone) présentes sur les AG sont des
sites très susceptibles aux dégradations. Les AGPI n-3, contenant au moins trois
insaturations, sont donc très sensibles aux dégradations induites par la chaleur et
l’oxydation.
1.1.1 L’acide α-linolénique
L’AAL est présent dans certaines noix et certaines plantes. Son origine végétale en
fait l’AGPI n-3 le plus susceptible de se retrouver dans les huiles alimentaires. L’huile de
lin est celle contenant le plus d’AAL, soit 58,0g d’AAL pour 100g d’huile (Belitz, et al.,
2009a). Toutefois, cela la rend inappropriée pour une utilisation en alimentaire dû son haut
taux en AGPI n-3. Cette teneur en AAL la rend très susceptible aux réactions d’oxydation
et de polymérisation (Belitz, et al., 2009a, Kochhar, 2002). D’autres huiles très utilisées
pour la friture, comme l’huile de Canola et de soya, contiennent également des quantités
non négligeables d’AAL. Elles contiennent respectivement 9,2 et 7,8g d’AAL pour 100g
d’huile (Orthoefer et List, 2007c). Cela les rend donc moins adaptées pour la friture car
elles auront tendance à se dégrader plus facilement.
1.2 Altération des acides gras polyinsaturés
La friture est un mode de cuisson très employé autant dans les restaurants qu’à la
maison. L’huile de Canola est très prisée pour ce type de cuisson dû, entre autre, à son
faible coût. Toutefois, cette huile à un faible taux d’acides gras saturés et une forte teneur
en AGPI n-3. Ces derniers sont composés de trois insaturations et plus, ce qui les rend très
sensibles aux dégradations lorsqu’ils sont en présence de certains facteurs, tels que
l’oxygène, la vapeur d’eau ou les hautes températures. En présence d’oxygène, des
réactions d’oxydation se produisent, formant des composés volatiles et non-volatiles. Les
AGPI n-3 formeront alors des hydroperoxydes, des alcools, des aldéhydes et des polymères
(Dana et Saguy, 2001, Frankel, 1984, Marquez-Ruiz et Dobarganes, 2007). Tous ces

9
produits augmenteront la polarité de l’huile, ainsi que sa viscosité. En présence de vapeur
d’eau, les AGPI n-3, ainsi que les AG présents, formeront des AG libres, ainsi que des
mono- et di-glycérides. Ces derniers augmentent la polarité des huiles, alors que les AG
libres vont abaisser le point de fumée des huiles (Aladedunye et Przybylski, 2009, Dana et
Saguy, 2001, Perkins, 2007). Finalement, les hautes températures vont transformer les
AGPI n-3 en polymères, en AG trans et en monomères cycliques d’acides gras (MCAG)
(Billek, 2000, Frankel, et al., 1984, Sébédio et Grandgirard, 1989, Sébédio et Juaneda,
2007). Tous ces produits sont à éviter dans une saine alimentation, car ils ont des
répercussions néfastes sur la santé (Billek, 2000, Sébédio, et al., 2007).
1.3 Dégradation oxydative des acides gras
Lors de la friture des aliments, les AG présents dans l’huile, en particulier les AG
insaturés comme l’AAL, subiront une dégradation oxydative sous l’effet de la chaleur. Il
s’agit de la dégradation la plus importante se produisant dans les huiles de friture. Cette
dégradation mènera à la formation de produits nocifs pour la santé, tels que des radicaux
libres, des aldéhydes, des alcools et des peroxydes.
1.3.1 Mécanisme d’oxydation
L’oxydation est une réaction en chaîne radicalaire en trois étapes. Il y a tout d’abord
la formation de radicaux lipidiques, soit l’initiation, puis la propagation de la formation des
peroxydes et finalement la terminaison, soit la formation de produits non-radicalaires
(Figure 1.3). Ainsi, cette réaction peut se produire en continue jusqu’à ce qu’un substrat
l’arrête.
Pour ce qui est de l’initiation, cette réaction implique la perte d’un radical afin de
former un radical lipidique. Cela nécessite l’aide d’un agent externe afin de débuter la
réaction. Généralement, il s’agit de lumière, de chaleur, de radiation ionisante ou d’une
initiation chimique impliquant un ion métallique (Antolovich, et al., 2002, Kamal-Eldin et
Pokorny, 2005). Il est à noter que les AG insaturés sont plus faciles à convertir en radicaux

10
libres. En effet, un atome d’hydrogène est plus facilement extrait d’une molécule si une
double liaison se trouve sur le carbone adjacent. Ainsi, plus une molécule est insaturé, plus
il est facile d’extraire un atome d’hydrogène. À titre d’exemple, un lien C-H se situant sur
une chaîne aliphatique demande une énergie de 98 kcal/mol pour être dissocié. Cette
énergie diminue à 89 kcal/mol lorsque le lien C-H se trouve adjacent à une double liaison.
Lorsque ce lien se retrouve sur un groupement méthylène séparant deux doubles liaisons,
l’énergie de dissociation peut diminuer sous les 80 kcal/mol. Ainsi, plus un AG a de
doubles liens, plus il perdra un atome d’hydrogène facilement, et donc plus il s’oxydera
facilement (Kamal-Eldin et Pokorny, 2005, McClements et Decker, 2007).
De plus, dans les AGPI, le radical libre est stabilisé par la délocalisation d’un
double liens, ce qui entraînera la formation de doubles liens conjugués (Figure 1.2)
(McClements et Decker, 2007). Lorsque cette résonance se produit, la densité électronique
sera plus forte dans le centre (près du carbon 11) et plus faible sur les extrémités (près des
carbones 9 et 13). Lors de l’étape suivante, l’oxygène aura donc tendance à se fixer là où la
densité électronique est moindre, soit sur les extrémité du système de doubles liaisons
(Schaich, 2005).
H
H+
9 10
11
12 13 9 10
11
12 13
9 10
11
12 13
9 10
11
12 13
Figure 1.2 : Délocalisation du radical sur une structure d’AG insaturé (McClements et
Decker, 2007).
La propagation consiste en une oxydation. L’oxygène va réagir avec le radical libre
du lipide afin de former un radical peroxyde (LOO٠). Ce radical possède une grande
énergie, ce qui lui permet d’attirer un atome d’hydrogène d’une autre molécule d’AG.
Comme le lien C-H d’un AG insaturé est faible, il est donc facile pour un radical peroxyde

11
d’en extraire un hydrogène. Le radical de peroxyde est alors transformé en une molécule
d’hydroperoxyde lipidique (LOOH), en plus de former un autre radical lipidique. La
propagation des radicaux libres se fait donc ainsi (Kamal-Eldin et Pokorny, 2005;
McClements et Decker, 2007).
La terminaison se produit lorsque les radicaux sont présents en abondance. Ils
réagiront alors avec d’autres radicaux libres afin de former des espèces non radicalaires,
tels que des dimères. Dans les conditions atmosphériques, cela se fera entre des radicaux de
peroxyde et d’alkoxyde. Alors que dans un milieu où il y a une faible concentration en
oxygène, cela se fera entre deux radicaux d’alkyle (McClements et Decker, 2007). Les
produits obtenus à cette étape correspondent aux produits primaires d’oxydation.
LH L• LOO•
O2
LHLOOH(hydropéroxyde)
AH A•
LOOH
Produits secondaires(aldéhydes, cétones, alcools, acides courts, alcanes)
(chaîne d'acides gras)
Étape d'initiation Étape de propagation Antioxydant
H•
Figure 1.3 : Cycle d'oxydation des acides gras et mécanisme des antioxydants.
Des produits secondaires d’oxydation seront ensuite formés. Les hydroperoxydes,
formés précédemment, sont très instables dans les conditions de friture et se dégradent donc
très rapidement (Dobarganes, 2009, Lomanno et Nawar, 1982). Par exemple, à des
températures de 150°C, les peroxydes auront un temps de demi-vie d’une heure (Pryor,
1966). Ils se dégraderont donc en radicaux d’alkoxyle (LO٠). Il est à noter que ces radicaux
sont très réactifs. Or, ils ont assez d’énergie pour arracher un électron au lien covalent
adjacent et causer ainsi le clivage de la chaîne aliphatique. Cette réaction est appelée une β-
scission (Figure 1.4). Lors de celle-ci, un aldéhyde et un radical d’alkyle sont obtenus

12
après le clivage. Ce radical peut alors réagir avec de l’oxygène pour faire un
hydropéroxyde, un radical d’hydroxyle afin de former un alcool ou un radical d’hydrogène
pour former un hydrocarbure. Les produits formés dépendent de l’endroit où se situe
l’hydropéroxyde sur la molécule d’acide gras de départ (McClements et Decker, 2007). Il
peut donc y avoir plusieurs sortes de produits secondaires. Parmi ceux-ci, on en retrouve 3
types différents. Il y a des monomères, des composés à faible masse moléculaire qui
proviennent du clivage des chaînes d’hydropéroxyde et, finalement, des produits à haute
masse moléculaire qui sont formés par la polymérisation des radicaux libres des produits de
dégradation (Kamal-Eldin et Pokorny, 2005).
OOH
O
O
H
Figure 1.4 : β-scission d’un hydroperoxyde lipidique (Frankel, 1980, McClements et
Decker, 2007).
1.3.2 Facteurs qui influence l’oxydation des acides gras
Certains facteurs permettent aux acides gras polyinsaturés d’être convertis en
radicaux libres plus ou moins facilement. La présence de métaux lourds par exemple, en
particulier le fer, va accélérer l’oxydation (Perkins, 2007). Une grande concentration
d’oxygène, une température élevée et une grande surface de contact entre l’huile et l’air
peuvent également augmenter l’oxydation. L’eau, qui est générée par celle présente dans
les aliments, peut ralentir ou accélérer le processus d’oxydation. En effet, lorsque la
quantité d’eau commence à augmenter, cela diminue la mobilité de l’oxygène et des
métaux, ce qui diminue la vitesse de l’oxydation. D’un autre côté, lorsque l’activité de l’eau
devient plus élevé, l’eau libre présente dans l’huile augmentera la mobilité des
prooxydants, ce qui augmentera l’oxydation (Belitz, et al., 2009b, McClements et Decker,

13
2007). La présence de prooxydant ou d’antioxydant peuvent également modifier le taux
d’oxydation de l’huile (Kamal-Eldin et Pokorny, 2005, McClements et Decker, 2007). La
composition de l’huile a également un impact sur l’oxydation des acides gras. Comme il a
déjà été expliqué, plus il y a des AG insaturés dans l’huile par rapport aux AG saturés, plus
vite l’huile s’oxydera (Kamal-Eldin et Pokorny, 2005).
1.3.3 Les antioxydants
L’une des méthodes qui peut être utilisée pour retarder ou inhiber l’oxydation des
acides gras est l’utilisation d’antioxydants. Les antioxydants naturels, tel que les
polyphénols des plantes, sont de plus en plus prisés pour la conservation des produits
alimentaires, dû au développement d’un courant santé chez les consommateurs
(Antolovich, et al., 2002). Un antioxydant est une substance qui, lorsqu’elle se retrouve à
faible concentration dans un substrat oxydable, peut retarder ou inhiber, de façon
significative, l’oxydation de ce substrat (Gutteridge, 1994). En industrie alimentaire, les
antioxydants sont utilisés pour prévenir la détérioration de la qualité des produits et
maintenir leur valeur nutritive.
L’activité des antioxydants dépend des aliments dans lesquels ils sont ajoutés, de la
concentration utilisée, de la quantité d’oxygène présent et de la présence de catalyseur
d’oxydation (ions métalliques), ainsi que de la présence de molécules synergiques
(McClements et Decker, 2007). Leur activité dans les huiles dépend également de leur
stabilité thermique et de leur volatilité. Il existe deux sortes d’antioxydants, soit les
primaires et les secondaires. Chaque type possède un mode d’action différent. Les plus
souvent utilisés en alimentation sont les antioxydants primaires. Parmi ceux-ci, on peut
retrouver des composés synthétiques ainsi que naturels.
1.3.3.1 Antioxydants primaires
Les antioxydants primaires sont considérés comme des briseurs de chaîne ou des
intercepteurs de radicaux libres et comprennent principalement des composés phénoliques.

14
Ces molécules contiennent donc un cycle aromatique substitué par un groupement –OH ou
un groupement –OR. Ils sont efficaces à faibles concentrations, mais peuvent devenir
prooxydant à forte concentration. Ces antioxydants peuvent inhiber la réaction d’initiation
en réagissant avec un radical ou inhiber la propagation et la réaction de β-scission en
réagissant avec un des radicaux peroxydes ou alkoxydes. (Gutteridge, 1994, McClements et
Decker, 2007, Wanasundara et Shahidi, 2005, Yanishlieva et Marinova, 2001). Des
produits non radicalaires et plus stables sont alors obtenus. Ainsi, l’efficacité d’un
antioxydant est dépendante de sa capacité à donner un atome d’hydrogène à un radical
libre. Les réactions sont les suivantes (Antolovich, et al., 2002, Wanasundara et Shahidi,
2005) :
Réaction entre un antioxydant et un radical :
L• + AH → LH + A•
LOO• + AH → LOOH + A•
LO• + AH → LOH + A•
Réaction de terminaison :
A• + A• → AA
A• + LO• → LOA
A• + LOO• → LOOA
Les antioxydants radicalaires qui sont obtenus lors de ces réactions sont très stables
et n’induiront donc pas la propagation des radicaux libres, à moins qu’ils soient présents en
grande quantité. Ils vont plutôt réagir avec d’autres radicaux libres, comme les radicaux
peroxyde et les radicaux alkoxyde. L’efficacité antioxydante est donc également
dépendante de la stabilité de résonance de la molécule. En effet, lorsqu’un antioxydant
devient radicalaire, il est stable dû à la résonance de l’électron dans le cycle aromatique
(Figure 1.5) (McClements et Decker, 2007, Wanasundara et Shahidi, 2005).

15
OH
ROOH
O O O O
ROO
Figure 1.5 : Résonance d’un électron dans la structure d’un groupement phénol
(McClements et Decker, 2007, Wanasundara et Shahidi, 2005).
Les antioxydants primaires sont ceux qui sont le plus souvent retrouvés dans le
domaine alimentaire. Bien souvent, il s’agit d’antioxydants synthétiques, tels que le BHT,
BHA et TBHQ, mais ils peuvent également être d’origine naturelle. C’est le cas par
exemple pour le α-tocophérol et des dérivés de l’acide cinnamique.
1.3.3.2 Antioxydants secondaires
Les antioxydants secondaires sont considérés comme des antioxydants préventifs
pouvant agir en éliminant, ou du moins en diminuant la concentration d’oxygène présent
dans l’huile. Ils peuvent également agir en tant qu’agent séquestrant pour diminuer l’effet
des ions métalliques catalytiques, éliminer les espèces réactives de l’oxygène, tels que
H2O2 et O2-, ainsi que réagir avec l’oxygène singulet (1O2). Ce type d’antioxydants ne
permet donc pas de stabiliser les radicaux libres qui sont formés durant l’oxydation des AG
(Gutteridge, 1994, Wanasundara et Shahidi, 2005). Ces antioxydants sont principalement
composés de molécules possédants un système de liens π conjugués. C’est le cas par
exemple de l’acide ascorbique. Les antioxydants secondaires peuvent également augmenter
l’efficacité d’un antioxydant primaire lorsqu’ils sont ajoutés à ceux-ci (Wanasundara et
Shahidi, 2005).
1.3.3.3 Antioxydants synthétiques
Les antioxydants synthétiques comprennent très souvent un groupement phénolique.
Les plus souvent utilisés dans les huiles de friture sont le BHT, le BHA, le TBHQ, ainsi

16
que le gallate de propyle (Madhavi, et al., 1996, McClements et Decker, 2007). Ils
présentent tous un très bon effet antioxydant, mais plusieurs études ont démontré qu’ils
pouvaient avoir un effet néfaste sur la santé s’ils sont ingérés à forte dose (Farag, et al.,
2003, Shahidi et Zhong, 2005, Wanasundara et Shahidi, 2005). Pour cette raison, la teneur
de ces antioxydants est régie par la loi. Au Canada, la concentration de ces antioxydants
dans les huiles ne doit pas dépassée 0,02% (Santé Canada, 2013). La limitation est
également en vigueur aux États-Unis et en Europe. Toutefois, leur utilisation dans les
aliments commence à diminuer dû à leur effet cancérogène potentiel, ainsi qu’à un éveil de
la conscience santé chez les consommateurs (Shahidi et Zhong, 2005).
1.3.3.4 Antioxydants naturels
Afin de remplacer les antioxydants synthétiques, différents composés phénoliques
naturels peuvent être utilisés comme antioxydants. Les plus importants sont le α-
tocophérol, l’acide ascorbique et les dérivés de l’acide cinnamique (Madhavi, et al., 1996).
Bien que certains de ces antioxydants soient tout de même synthétisés, ils sont considérés
comme naturels puisqu’ils peuvent être retrouvés naturellement (Wanasundara et Shahidi,
2005). Le α-tocophérol est déjà retrouvé naturellement dans certaines huiles et est un
antioxydant efficace. D’autres molécules, tels que les acides caféique, férulique, vanillique
et p-coumarique, sont également retrouvés naturellement et ont déjà démontré des effets
antioxydants. Toutefois, la plupart des composés phénoliques, dû à leur polarité, sont
hydrosolubles et se dissolvent donc très mal dans les lipides (Choe et Min, 2009, Shahidi et
Zhong, 2010). C’est pour cette raison que ces composés n’ont jamais été utilisés dans les
huiles de friture. Une solution qui pourrait être envisagé est de rendre ces molécules plus
lipophiles, en ajoutant, par exemple, une chaîne hydrophobe sur ces molécules.
1.4 Monomères cycliques d’acide gras
Toutes les dégradations pouvant affectées les AG surviennent à des températures
entre 170°C et 195°C, soit celles utilisées dans l’industrie pour la transformation des huiles,
telles que la désodorisation, ainsi que pour la friture, qui est également utilisée comme

17
méthode de cuisson par les consommateurs (Belitz, et al., 2009b). Les monomères
cycliques d’acides gras (MCAG) sont présents en faible quantité dans les huiles de friture
polyinsaturées, soit à une teneur de 0,01 à 0,66% des acides gras totaux (Sébédio et
Grandgirard, 1989).
1.4.1 Monomères cycliques de l’acide α-linolénique
L’AAL peut former seize
isomères géométriques possédant quatre
structures principales (Figure 1.6). Deux
de ces isomères contiennent un
groupement cyclopentényle (cycle à 5
atomes de carbone) (1.3 et 1.4) et les
deux autres ont un groupement
cyclohexényle (cycle à 6 atomes de
carbone) (1.1 et 1.2) (Christie et
Dobson, 2000). Ces deux types
d’isomères se retrouvent
approximativement en même quantité
dans les huiles riches en AAL (Sébédio
et Grandgirard, 1989). Chacun d’eux
possède une double liaison dans le cycle, située entre les carbones 12 et 13, et une autre sur
la chaîne aliphatique. Pour ce qui est de la double liaison intra-cyclique, la configuration cis
est requise pour minimiser la tension dans le cycle. Les insaturations situées sur la chaîne
alkyle peuvent prendre la configuration Z ou E. Quant aux cycles, ils peuvent s’orienter cis
ou trans dans l’espace. Ce qui donne donc un total de seize isomères (Christie et Dobson,
2000).
Les temps de rétention en chromatographie en phase gazeuse (GC) des seize
isomères de MCAG ont été déterminés (Dobson, et al., 1996a). Une colonne capillaire
O
HO10 15
16
12
HO
O
8
10
12
15
HO
O
9
11 15
12
HO
O
10 14
15
12
1.1
1.2
1.3
1.4
Figure 1.6 : Structures principales des MCAG
formées par l'AAL (Christie et Dobson, 2000).
18
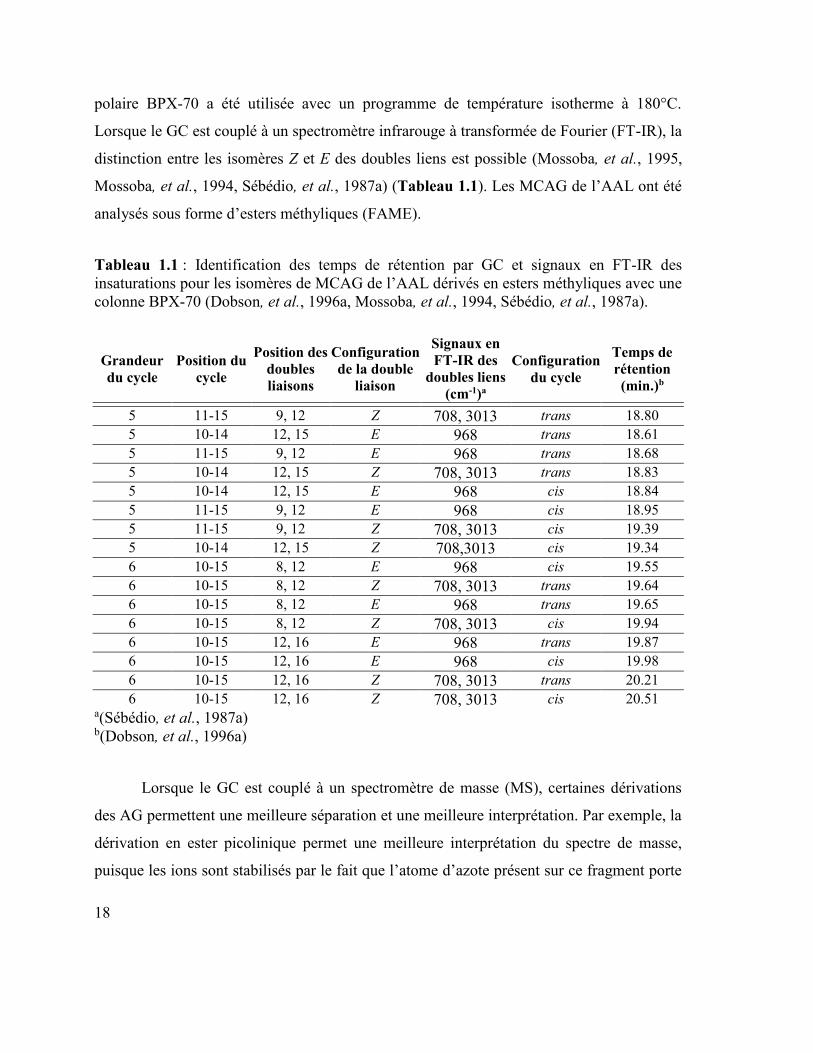
polaire BPX-70 a été utilisée avec un programme de température isotherme à 180°C.
Lorsque le GC est couplé à un spectromètre infrarouge à transformée de Fourier (FT-IR), la
distinction entre les isomères Z et E des doubles liens est possible (Mossoba, et al., 1995,
Mossoba, et al., 1994, Sébédio, et al., 1987a) (Tableau 1.1). Les MCAG de l’AAL ont été
analysés sous forme d’esters méthyliques (FAME).
Tableau 1.1 : Identification des temps de rétention par GC et signaux en FT-IR des
insaturations pour les isomères de MCAG de l’AAL dérivés en esters méthyliques avec une
colonne BPX-70 (Dobson, et al., 1996a, Mossoba, et al., 1994, Sébédio, et al., 1987a).
Grandeur
du cycle Position du
cycle
Position des
doubles
liaisons
Configuration
de la double
liaison
Signaux en
FT-IR des
doubles liens
(cm-1)a
Configuration
du cycle
Temps de
rétention
(min.)b
5 11-15 9, 12 Z 708, 3013 trans 18.80
5 10-14 12, 15 E 968 trans 18.61
5 11-15 9, 12 E 968 trans 18.68
5 10-14 12, 15 Z 708, 3013 trans 18.83
5 10-14 12, 15 E 968 cis 18.84
5 11-15 9, 12 E 968 cis 18.95
5 11-15 9, 12 Z 708, 3013 cis 19.39
5 10-14 12, 15 Z 708,3013 cis 19.34
6 10-15 8, 12 E 968 cis 19.55
6 10-15 8, 12 Z 708, 3013 trans 19.64
6 10-15 8, 12 E 968 trans 19.65
6 10-15 8, 12 Z 708, 3013 cis 19.94
6 10-15 12, 16 E 968 trans 19.87
6 10-15 12, 16 E 968 cis 19.98
6 10-15 12, 16 Z 708, 3013 trans 20.21
6 10-15 12, 16 Z 708, 3013 cis 20.51 a(Sébédio, et al., 1987a) b(Dobson, et al., 1996a)
Lorsque le GC est couplé à un spectromètre de masse (MS), certaines dérivations
des AG permettent une meilleure séparation et une meilleure interprétation. Par exemple, la
dérivation en ester picolinique permet une meilleure interprétation du spectre de masse,
puisque les ions sont stabilisés par le fait que l’atome d’azote présent sur ce fragment porte

19
la charge. La dérivation en 4,4-diméthyloxazoline (DMOX) a le même effet sur la
stabilisation des ions (Christie, 1998, Dobson, et al., 1995, Sébédio et Juaneda, 2007).
1.4.2 Effets des MCAG sur la santé
Les effets des MCAG sur la santé humaine sont étudiés depuis de nombreuses
années. Des études faites dans les années 50 suggéraient déjà que ces composés pouvaient
être parmi les plus toxiques retrouvés dans les huiles de friture dû à leur rôle dans la
lipogenèse (Sébédio et Grandgirard, 1989). Plusieurs études suggèrent d’ailleurs que les
MCAG peuvent être métabolisés comme les AG réguliers, c’est-à-dire qu’ils seront utilisés
pour la production d’énergie et ils seront incorporés dans les phospholipides des
membranes. En effet, des études suggèrent que la chaîne aliphatique présente sur les
MCAG pourrait participer à la production d’énergie de la même façon que les AG naturels
(Desmarais, 2013). Cela peut donc affecter le métabolisme des AG (Sébédio, et al., 2007).
Leurs effets sur le métabolisme intestinal dépendent de leurs positions dans le
triacylglycéride et la structure des MCAG peut également avoir un impact sur leur
récupération dans le système lymphatique s’ils sont absorbés comme des AG libres
(Martin, et al., 1997). De plus, une étude suggère que les MCAG pourraient avoir d’autre
effet sur la lipogenèse (Martin, et al., 2000). Des travaux réalisés dernièrement suggèrent
également leurs accumulations dans des organes cibles, tels que le foie et les tissus adipeux,
ainsi que leur élimination sous forme de glucuronides, de sulfates et/ou de nitrates. D’autres
sous-produits qui ont été observés suggèreraient la participation des MCAG dans le
mécanisme de β-oxydation (Desmarais, 2013).
1.4.3 Cinétique de dégradation de l’acide α-linolénique
L’AAL retrouvé naturellement contient trois insaturations de configuration cis. Lors
d’un chauffage, les insaturations auront tendance à s’isomériser en leur conformation trans,
cette conformation étant thermodynamiquement plus stable (Sonnet, 1980). L’AAL peut

20
donc former 8 isomères géométriques (Figure 1.7). La configuration de l’AAL pourrait
donc avoir une influence sur la formation des monomères cycliques d’acides gras.
HO
O
HO
HO
HO
O
O
O
9 12 15
9-cis, 12-cis, 15-cis
9 12 15
9-cis, 12-cis, 15-trans
9 12
15
9
12 15
9
12
15
9 12
15
9-cis, 12-trans, 15-cis
9-cis, 12-trans, 15-trans
HO
HO
HO
HO
O
O
O
O9-trans, 12-trans, 15-cis
9
12 15
9
12
15
9-trans, 12-trans, 15-trans
9-trans, 12-cis, 15-cis
9-trans, 12-cis, 15-trans
Figure 1.7 : Structure des 8 isomères géométriques de l’AAL
Les acides gras les plus souvent retrouvés dans les huiles végétales utilisées pour la
friture sont l’acide palmitique (16:0), l’acide stéarique (18:0), l’acide oléique (AO) (18:1),
l’acide linoléique (AL) (18:2) et l’AAL (18:3). Ce dernier étant l’AG comportant le plus
d’insaturations, il sera donc le plus susceptible de subir des dégradations, oxydatives ou
thermiques, et donc l’acide gras qui formera le plus de MCAG (Sébédio et Grandgirard,
1989). D’ailleurs, les MCAG se formeraient 10 fois plus vite dans les huiles ayant une forte
teneur en AAL par rapport à celle ayant une forte teneur en AL (Sébédio, et al., 1987b),
bien que les AG mono- et di-insaturés forment également des MCAG (Christie et Dobson,
2000, Destaillats et Angers, 2005).
Afin d’optimiser la synthèse de l’AAL, des travaux ont été effectués afin de
déterminer l’effet des isomères sur la formation des MCAG (Desmarais, 2013). Ces travaux
suggèrent que la configuration des doubles liaisons, soit cis ou trans, aurait un impact sur la
cinétique de formation des MCAG. En effet, une double liaison trans permettrait de
diminuer l’encombrement et accélèrerait ainsi la formation des cycles. De plus, ces travaux
suggèrent que l’isomère mono-trans de l’AAL formerait des MCAG plus rapidement que
l’isomère di-trans. Lors du chauffage de l’AAL enrichi en mono-trans (Figure 1.8A), une
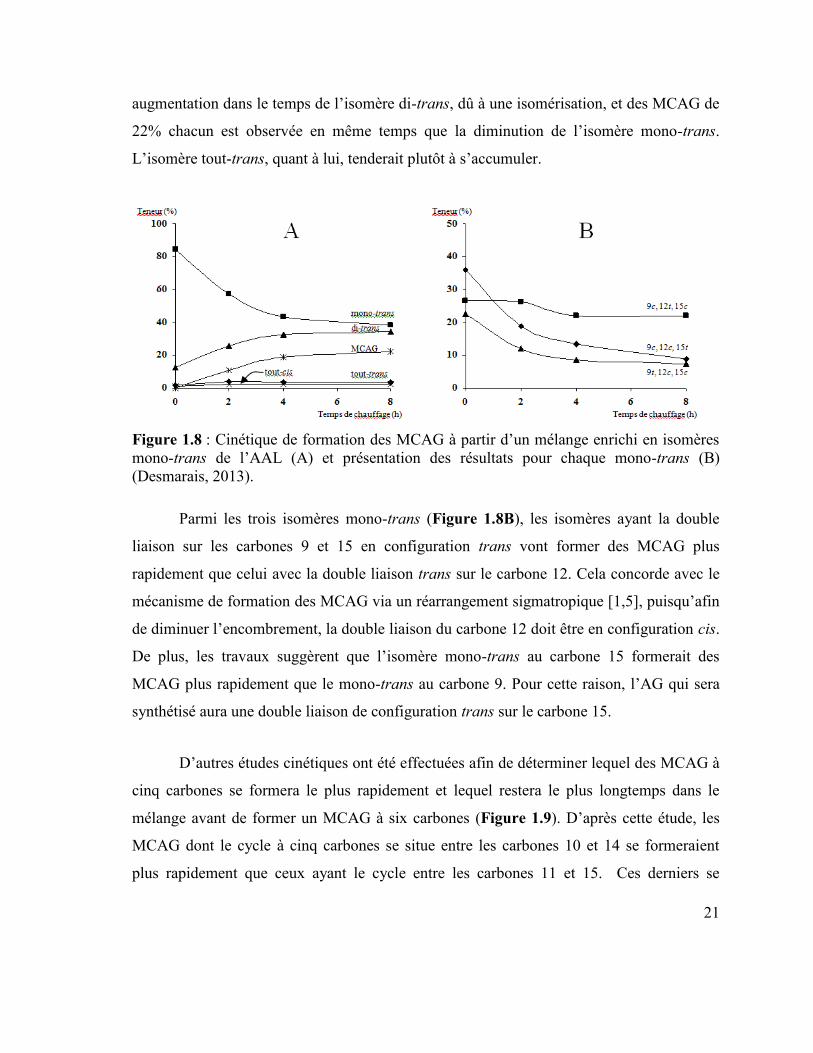
21
augmentation dans le temps de l’isomère di-trans, dû à une isomérisation, et des MCAG de
22% chacun est observée en même temps que la diminution de l’isomère mono-trans.
L’isomère tout-trans, quant à lui, tenderait plutôt à s’accumuler.
Figure 1.8 : Cinétique de formation des MCAG à partir d’un mélange enrichi en isomères
mono-trans de l’AAL (A) et présentation des résultats pour chaque mono-trans (B)
(Desmarais, 2013).
Parmi les trois isomères mono-trans (Figure 1.8B), les isomères ayant la double
liaison sur les carbones 9 et 15 en configuration trans vont former des MCAG plus
rapidement que celui avec la double liaison trans sur le carbone 12. Cela concorde avec le
mécanisme de formation des MCAG via un réarrangement sigmatropique [1,5], puisqu’afin
de diminuer l’encombrement, la double liaison du carbone 12 doit être en configuration cis.
De plus, les travaux suggèrent que l’isomère mono-trans au carbone 15 formerait des
MCAG plus rapidement que le mono-trans au carbone 9. Pour cette raison, l’AG qui sera
synthétisé aura une double liaison de configuration trans sur le carbone 15.
D’autres études cinétiques ont été effectuées afin de déterminer lequel des MCAG à
cinq carbones se formera le plus rapidement et lequel restera le plus longtemps dans le
mélange avant de former un MCAG à six carbones (Figure 1.9). D’après cette étude, les
MCAG dont le cycle à cinq carbones se situe entre les carbones 10 et 14 se formeraient
plus rapidement que ceux ayant le cycle entre les carbones 11 et 15. Ces derniers se
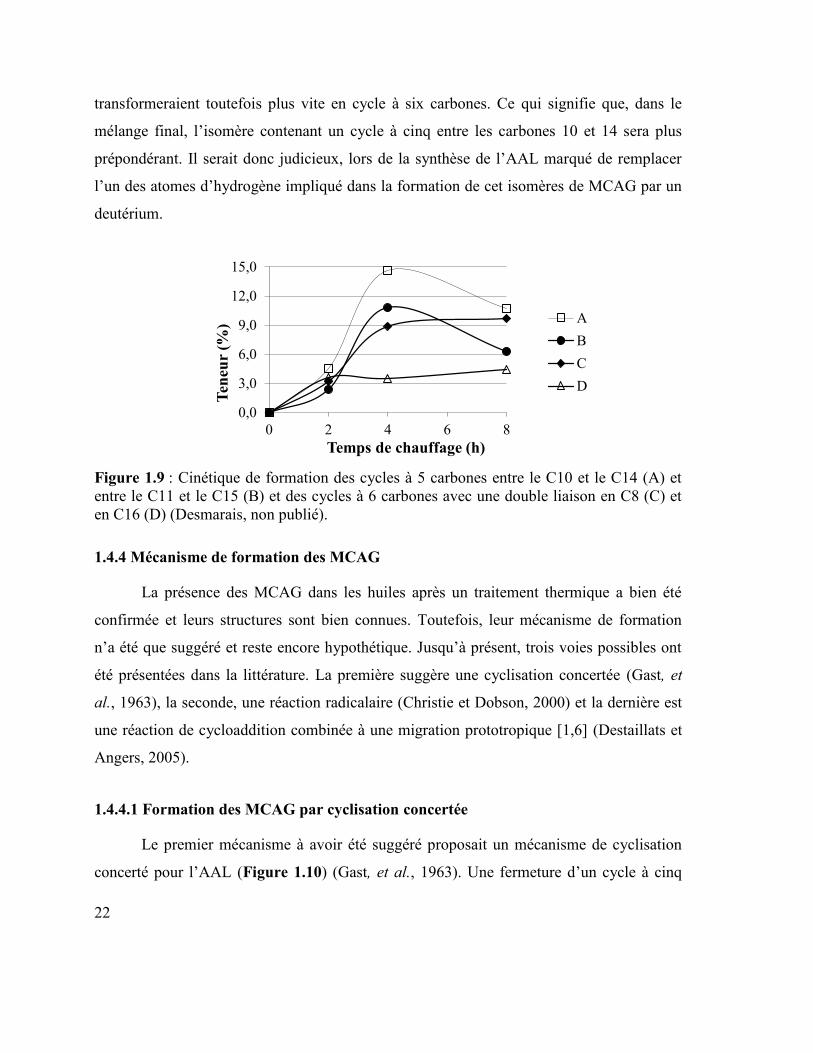
22
transformeraient toutefois plus vite en cycle à six carbones. Ce qui signifie que, dans le
mélange final, l’isomère contenant un cycle à cinq entre les carbones 10 et 14 sera plus
prépondérant. Il serait donc judicieux, lors de la synthèse de l’AAL marqué de remplacer
l’un des atomes d’hydrogène impliqué dans la formation de cet isomères de MCAG par un
deutérium.
Figure 1.9 : Cinétique de formation des cycles à 5 carbones entre le C10 et le C14 (A) et
entre le C11 et le C15 (B) et des cycles à 6 carbones avec une double liaison en C8 (C) et
en C16 (D) (Desmarais, non publié).
1.4.4 Mécanisme de formation des MCAG
La présence des MCAG dans les huiles après un traitement thermique a bien été
confirmée et leurs structures sont bien connues. Toutefois, leur mécanisme de formation
n’a été que suggéré et reste encore hypothétique. Jusqu’à présent, trois voies possibles ont
été présentées dans la littérature. La première suggère une cyclisation concertée (Gast, et
al., 1963), la seconde, une réaction radicalaire (Christie et Dobson, 2000) et la dernière est
une réaction de cycloaddition combinée à une migration prototropique [1,6] (Destaillats et
Angers, 2005).
1.4.4.1 Formation des MCAG par cyclisation concertée
Le premier mécanisme à avoir été suggéré proposait un mécanisme de cyclisation
concerté pour l’AAL (Figure 1.10) (Gast, et al., 1963). Une fermeture d’un cycle à cinq
0,0
3,0
6,0
9,0
12,0
15,0
0 2 4 6 8
Ten
eur
(%)
Temps de chauffage (h)
A
B
C
D

23
carbones entre les carbones 9 et 13 a donc lieu dû à un transfert d’un atome d’hydrogène.
La migration d’une double liaison se produit en même temps.
HOOC
H
9
13
EncombrementStérique
HOOC 9 13H
HOOCH
9 13
Acide -linolénique
(9-cis, 12-cis, 15-cis)
Acide -linolénique
(9-trans, 12-trans, 15-cis)
1.5
1.6
1.7
Figure 1.10 : Formation des MCAG à partir de l’AAL par la cyclisation concertée (adapté
de (Destaillats et Angers, 2005, Gast, et al., 1963)).
Toutefois, ce mécanisme n’explique pas la formation de MCAG à partir d’acide
gras monoinsaturé, comme l’acide oléique. De plus, la conformation tous-cis engendre un
encombrement stérique qui pourrait limiter la formation du monomère cyclique. Le
mécanisme n’explique la formation de cycle qu’entre les carbones 9 et 13, cet structure ne
correspondant à aucune de celle qui a précédemment été identifiée (Christie et Dobson,
2000, Dobson, et al., 1996a, Mossoba, et al., 1995).
1.4.4.2 Formation des MCAG passant par un intermédiaire radicalaire
Dans cette optique, un mécanisme impliquant un intermédiaire radicalaire (Figure
1.11) a été proposé (Dobson, et al., 1996b). Ce mécanisme consiste en l’abstraction d’un
hydrogène allylique, formant ainsi l’intermédiaire radicalaire. La fermeture du cycle a
ensuite lieu via le carbone radicalaire d’une double liaison. Le MCAG ainsi formé doit
alors soit acquérir ou perdre un hydrogène radicalaire afin de retrouver un état stable.

24
R R'
R , RO
9 12 15
R R'
9 12 15
R
9
12
15
R R'
R'
9
12
15
R
9
12
15
R' R
9
12
15
R' R
912
15
R'
RH
1.8 1.9 1.10
-H
Figure 1.11 : Formation des MCAG à partir d’un AGPI par la voie radicalaire (adapté de
(Destaillats et Angers, 2005, Dobson, et al., 1996b)).
Ce mécanisme permet effectivement d’obtenir tous les isomères des MCAG, autant
avec un cycle à cinq qu’à six carbones. Toutefois, si le MCAG radicalaire perd un
hydrogène radicalaire, une nouvelle double liaison sera formée. Des produits qui n’ont
jamais été identifiés (1.9 et 1.10) seront alors obtenus.
1.4.4.3 Formation des MCAG par cycloaddition intramoléculaire suivi d’une
migration prototropique [1,6]
L’hypothèse la plus récente suggère que les MCAG se formeraient d’abord selon
une cycloaddition intramoléculaire (Figure 1.12) (Destaillats et Angers, 2005). Ce
mécanisme permettrait la formation dans un premier temps des cycles à cinq carbones. Une
configuration cis de la double liaison entre les carbones 12 et 13 des AG de départ (1.14)
est requise pour que la cyclisation est lieu. Si ce n’est pas le cas, une tension de cycle trop
importante empêchera le cycle de se former.

25
C C
C H
C C
C H
C C
C H
H
R2
R1
H
R2
R1
( )n ( )n ( )n n = 1, 2
1.11 1.12 1.13
1.14 1.15
Figure 1.12 : Mécanisme général de formation des MCAG par cycloaddition
intramoléculaire.
Une migration prototropique [1,6] conduirait par la suite à la formation des MCAG
à six carbones (Figure 1.13). Le dernier réarrangement s’accompagne du déplacement
d’une double liaison, soit du carbone 15 au 16 (1.20) ou du 9 au 8 (1.21). Ce mécanisme
peut donc expliquer la cyclisation des AG mono-, di- et polyinsaturés. Il a également
l’avantage de se limiter à la formation des seize isomères observés de MCAG pour l’AAL
dans les huiles de friture (Dobson, et al., 1996b).
HOOC(H2C)7
CH3
H9
12
1415
HOOC(H2C)7
H CH3
9 1514
12
HOOC(H2C)7CH3
H
9
12
1516
10
1410
HOOC(H2C)6
H CH39
12
15
HOOC(H2C)6 H
CH39 15
12
10
HOOC(H2C)6
H
CH3
910 15
12
11
1.16 1.18 1.20
1.17 1.19 1.21
Figure 1.13 : Formation des MCAG à partir de l’AAL selon un réarrangement
sigmatropique [1,5] (adapté de (Destaillats et Angers, 2005)).
La synthèse d’un AG marqué au deutérium semble donc être un choix judicieux
pour vérifier l’implication du réarrangement sigmatropique [1,5] dans la formation des
MCAG. Le remplacement d’un des atomes d’hydrogène impliqués dans ce réarrangement,
soit celui sur les carbones 14 ou 11, par un atome marqueur, permettra d’élucider le
mécanisme de formation. Comme il s’agit d’un mécanisme en deux étapes, l’AG qui sera

26
synthétisé devra permettre de maximiser la formation des produits primaires, soir les
MCAG possédant un cycle à cinq carbones.
Ainsi, la friture induit différentes dégradations des AG. En présence d’oxygène,
l’oxydation sera la réaction principale. Celle-ci formera, entre autres, des peroxydes, des
hydroperoxydes et des radicaux libres. Des antioxydants peuvent être utilisés pour ralentir
l’oxydation. Ceux qui sont utilisés actuellement sont de nature synthétique et sont
potentiellement cancérigènes. Des antioxydants naturels, provenant d’acides phénoliques,
seraient une alternative à envisager. Toutefois, ces acides, dus à leur polarité, sont
hydrophile. L’ajout d’une chaîne lipophile sur les acides pourrait donc permettre
d’augmenter leur solubilité dans les huiles, et ainsi leur permettre d’être testé comme
antioxydant. D’un autre côté, la chaleur transformera les AG en polymère et en MCAG.
Ces derniers ont bien été identifiés dans les huiles de friture et leurs structures sont donc
bien connues. Toutefois, leur mécanisme de formation n’est encore qu’hypothétique. La
synthèse d’un isomère de l’AAL marqué à un endroit stratégique pourrait permettre
d’élucider le mécanisme.

27
Hypothèse et Objectifs
Hypothèse
D’un côté, l’oxydation des acides gras contribue à former des produits nuisibles
pour la santé. Des antioxydants peuvent être utilisés pour ralentir cette réaction. Les
antioxydants naturels sont toutefois trop hydrophiles et volatiles pour être utilisés dans les
huiles de friture. L’ajout d’une chaîne hydrophobe sur certains acides phénoliques
permettrait d’augmenter leur solubilité dans les lipides, tout en diminuant leurs volatilités.
Cela permettrait donc d’obtenir un meilleur effet antioxydant de ces molécules. D’un autre
côté, les hautes températures utilisé en friture engendrent la formation de MCAG. Ces
monomères ont bien été identifiés, mais leur mécanisme de formation est encore
hypothétique. Nous croyons qu’ils sont formés par une cycloaddition suivi d’une migration
prototropique [1,6]. La synthèse d’un isomère de l’AAL marqué d’atomes de deutérium sur
le carbone 14 pourrait permettre de démontrer le mécanisme de formation des MCAG.
Objectifs généraux
Synthétiser des dérivés d’acides phénoliques et testés leur efficacité antioxydante
dans l’huile de Canola et en conditions de friture.
Synthétiser un isomère de l’AAL marqué d’atomes de deutérium sur le carbone
14 et effectuer des traitements thermiques afin de cycliser l’AAL tout en suivant
le déplacement des atomes marqueurs.


Chapitre 2
Antioxydants pour les huiles de friture


31
2.1 Introduction
La friture est un mode de cuisson très apprécié en Amérique du Nord. Toutefois, les
huiles utilisées pour ce genre de cuisson contiennent très souvent une grande quantité d’AG
insaturés. Ainsi, lors d’une utilisation prolongée de l’huile de friture, diverses réactions de
dégradation se produisent. Parmi celles-ci, l’oxydation des AG en produits plus polaires,
tels que des peroxydes, des alcools et des aldéhydes, est la réaction de dégradation la plus
importante (Dana et Saguy, 2001, Frankel, 1984, Marquez-Ruiz et Dobarganes, 2007). Les
composés qui sont alors créés sont reconnus comme étant nocifs pour la santé. D’autres
produits, tous aussi nocifs, peuvent également être formés. Parmi ceux-ci, on retrouve les
AG trans et conjugués, ainsi que les MCAG (Orthoefer et List, 2007b).
Divers facteurs peuvent influencer l’oxydation des AG dans les huiles de friture.
Certains peuvent augmenter l’oxydation, comme la température et la concentration en
oxygène et en prooxydants, alors que d’autres, comme les antioxydants, vont la diminuer
(McClements et Decker, 2007). Deux sortes d’antioxydants existent, soit les primaires et
les secondaires (Gutteridge, 1994, Wanasundara et Shahidi, 2005). Les premiers sont
principalement des molécules qui contiennent un groupement phénolique. Leur mode
d’action consiste à inhiber l’initiation du mécanisme d’oxydation ou la propagation des
radicaux libres formés lors du mécanisme d’oxydation. Pour ce qui est des antioxydants
secondaires, ils consistent en des molécules capables de capter l’oxygène présent dans les
huiles. Ceux-ci sont généralement constitués de molécules ayant un système de liaisons π
conjuguées.
Les antioxydants primaires sont les plus utilisés dans les huiles alimentaires. Les
plus populaires sont des antioxydants synthétiques, tels que le BHT, le BHA et le TBHQ
(Madhavi, et al., 1996, McClements et Decker, 2007). Certaines études ont toutefois
démontré que ces produits pouvaient avoir un effet nocif sur la santé (Shahidi et Zhong,
2005). Pour cette raison, leur utilisation est régie par la loi dans la plupart des pays, dont le

32
Canada (Santé Canada, 2013). Malgré cela, et dû à un mouvement santé chez les
consommateurs, des antioxydants d’origine naturels sont recherchés afin de les remplacer.
Les composés naturels qui sont généralement utilisés sont des polyphénols, tel que
le α-tocophérol (Madhavi, et al., 1996). Plusieurs ont déjà démontré un effet antioxydant
significatif, toutefois, dû à leur groupement polaire, ces produits sont très peu solubles dans
les lipides (Choe et Min, 2009, Shahidi et Zhong, 2010). Ainsi, l’ajout d’une chaîne
hydrophobe à ces molécules en augmenterait la solubilité dans les lipides et ainsi
permettraient leurs usages dans les huiles de friture. Cet ajout pourrait également en
diminuer la volatilité et ainsi leur donner une plus grande stabilité à des températures
élevées, tels que celles utilisées en friture. Ce projet visait donc à développer des
antioxydants dérivant de produits naturels afin de pouvoir les utiliser en milieu de friture.

33
2.2 Hypothèse et objectifs spécifiques
Hypothèse
L’ajout d’une chaîne lipophile sur certains acides phénoliques permettra d’en
augmenter la solubilité dans les lipides, tout en diminuant leur volatilité. Cela permettra
donc de tirer profit de leur propriété antioxydante en friture.
Objectifs
Synthétiser et purifier des esters d’alcools gras d’acides phénoliques pour utiliser
comme antioxydants dans les huiles de friture.
Tester l’efficacité de ces molécules par des traitements thermo-oxydatifs dans
l’huile de Canola.
Tester l’efficacité de ces molécules en conditions de friture.

34
2.3 Méthodologie
2.3.1 Synthèse du vanillate d’hexyle
La méthode que nous avons utilisée afin d’estérifier les acides organiques consiste à
transformer les fonctions acides en sel de potassium pour ensuite les estérifier avec un
iodure d’alcane (Figure 2.1). Le protocole qui a été choisi (Pfeffer et Silbert, 1976)
consistait tout d’abord à faire un titrage de l’acide vanillique (2.1) avec une solution de
KOH afin de former le sel d’acide correspondant (2.2). Après l’isolation du sel, celui-ci
était ensuite mis en solution avec un iodure d’alcane, soit le 1-iodo-hexane, afin de former
l’ester final (2.3). Le vanillate d’hexyle est ainsi obtenu après une purification sur colonne
chromatographique de gel de silice. Une solution de 10% AcOEt / 90% hexane a été
utilisée comme éluant.
MeO
HO
O
OHKOH
MeO
HO
O
O-K+
EtOH
I-hexaneDMSO
85°C, 20h
MeO
HO
O
O
2.1 2.2 2.3
Figure 2.1 : Schéma de synthèse du vanillate d’hexyle.
Les conditions utilisées pour cette réaction ont été déterminée selon une étude
cinétique trouvée dans la littérature (Pfeffer et Silbert, 1976). Selon cette étude, le choix du
cation utilisé pour faire le sel, le solvant, ainsi que l’halogénure utilisé pour effectuer la
réaction ont un impact sur la vitesse de formation de l’ester final. Ainsi, le KOH a été
utilisé de préférence au NaOH afin de faire le sel d’acide. Un iodure a également été choisi
de préférence à un bromure, car il permettait d’obtenir le produit de l’estérification 17 fois
plus rapidement. Le DMSO a toutefois été choisi de préférence au HMPA, malgré son plus
faible rendement, pour des raisons de disponibilité et de toxicité.

35
2.3.2 Synthèse du vanillate de dodécyle
La synthèse du vanillate de dodécyle a d’abord été faite en suivant le même
protocole que pour le vanillate d’hexyle, mais après quelques essais, l’ester voulu (2.4)
n’était toujours pas formé. D’autres conditions réactionnelles ont été retenues. Elles
consistent à former le sel de l’acide, sans l’isoler, et à apposer la chaîne alkyle dans la
même étape (Moore, et al., 1979).
I-dodécane
K2CO3, acétone
, 16h
MeO
HO
O
OHMeO
HO
O
O
2.1 2.4
Figure 2.2 : Synthèse du vanillate de dodécyle.
L’acide vanillique (2.1) a été mis en solution dans l’acétone, puis du carbonate de
potassium a été ajouté. Après quelques minutes d’agitation, le 1-iodo-dodécane était ajouté.
La réaction a ensuite été maintenue à reflux pendant une nuit entière et l’ester (2.4) a été
obtenu après une purification sur colonne chromatographique de gel de silice en utilisant
une solution de 10% AcoEt / 90% hexane comme solvant d’élution.
2.3.3 Synthèse du coumarate d’hexyle
La synthèse du coumarate d’hexyle a été réalisée selon le même protocole que pour
le vanillate de dodécyle. Des ajustements ont toutefois dû être apportés, entre autre par
rapport au temps de réaction. Ainsi, l’acide p-coumarique (2.5) a été mis en solution dans
l’acétone avec le carbonate de potassium, puis le 1-iodo-hexane a été ajouté et le mélange
réactionnel a été maintenu à reflux pendant plusieurs nuits. L’ester (2.6) a été obtenu après
une purification sur colonne chromatographique de gel de silice en utilisant un gradient de
solvant pour l’élution : 35% AcOEt / 65% hexane, suivi de 50% AcOEt / 50% hexane, puis
de 65% AcOEt / 35% hexane.

36
HO
O
OH HO
O
O
I-hexane
K2CO3, acétone
, 16h
2.5 2.6
Figure 2.3 : Synthèse du coumarate d’hexyle.
2.3.4 Analyse thermique différentielle
La calorimétrie différentielle à balayage (DSC) est une technique que nous avons
utilisée afin de déterminer le comportement des différents composés utilisés comme
antioxydants sous l’effet de la chaleur. Cela pouvait donc nous permettre de déterminer la
température de fusion et la température de dégradation des produits. Un appareil DSC
Q1000 de TA Instrument à été utilisé. Le programme de température à consister en une
augmentation de températures de 10°C/min de 40°C à 200°C.
2.3.5 Test d’efficacité des antioxydants dans l’huile de Canola
Afin de vérifier l’effet antioxydants des différentes molécules, et préalablement aux
tests en condition de friture, des traitements thermo-oxydatif ont d’abord été fait dans
l’huile de Canola. Les antioxydants étaient alors dissouts dans l’huile de Canola à 1,0%
m/m. Des échantillons de 0,5g sont préparés dans des vials 2Dr. La moitié ne contient que
de l’huile de Canola et l’autre moitié contient l’huile supplémentée avec le composé testé.
Tous les échantillons sont alors mis dans un four à 120°C pour une période de 4h, 1 jour et
2 jours. Les vials étaient ouverts afin de permettre un contact avec l’air ambiant. Lorsque
les échantillons étaient sortis du four, ils étaient conservés sous atmosphère d’azote et à
-20°C jusqu’à ce qu’ils soient analysés. Lorsque ce test était concluant, c’est-à-dire lorsque
le composé montrait un effet antioxydant, d’autres tests étaient réalisés en diminuant la
concentration des antioxydants dans l’huile de Canola à 0,5%, puis à 0,1%. Si ces tests
étaient concluants, d’autres tests étaient faits en augmentant la température à 135°C, 150°C,
165°C, puis à 180°C. Des échantillons d’huile de Canola contenant du BHT aux mêmes
concentrations que le composé testé étaient également soumis aux différents tests. Ces

37
échantillons servaient de témoin positif et permettaient ainsi de comparer les résultats
obtenus avec un produit déjà reconnus comme un bon antioxydant.
Si un composé démontrait une efficacité antioxydante comparable à celle du BHT à
une température de 180°C et à une concentration de 0,1%, il était ensuite testé en milieu de
friture.
2.3.6 Test d’efficacité des antioxydants en condition de friture
Afin d’effectuer les tests en condition de friture, une friteuse de 2L (Hamilton
Beach) a été utilisée. Les frites et l’huile avaient un ratio 1:6 (m/v) tel qu’il a été
recommandé par Morton et Childley (1988), soit 1,2L d’huile pour 200g de frites. Les
analyses en milieu de friture ont été faites trois fois avec de l’huile de Canola, trois fois
avec de l’huile de Canola contenant 0,1% de BHT et une fois avec de l’huile de Canola
contenant 0,1% de notre antioxydant. Un seul test a été effectué avec le coumarate
d’hexyle, car nous ne disposions pas d’une quantité suffisante de produit pour effectuer
plusieurs tests. Les tests effectués avec le BHT servaient de contrôle positif. Les résultats
obtenus avec notre antioxydant ont été comparés à ceux obtenus avec le BHT et à l’huile de
Canola seule.
L’huile de la friteuse était tout d’abord chauffée pendant environ 30min. afin
d’obtenir une température d’environ 180°C. Les frites étaient alors plongées dans l’huile
pour une période de 10min. Une période d’attende de 20min. était effectué entre chaque lot
de frites afin de laisser l’huile reprendre sa température initiale. Quinze périodes de friture
ont été effectuées sur une période d’environ 8h. Un échantillon d’huile était prélevé avant
et après chaque lot de frites. Les huiles étaient ensuite gardées sous atmosphère d’azote à
une température de -20°C jusqu’à leur analyse. Tout au long des expériences de friture, la
température était mesurée à l’aide d’un thermomètre électronique. De plus, le niveau de
l’huile était constamment surveillé afin de maintenir le bon ratio de frites/huile (Morton et
Chidley, 1988).

38
2.3.7 Analyse des huiles
Différentes méthodes d’analyses peuvent être utilisées pour l’analyse du niveau
d’oxydation de l’huile de friture. La détermination des composés polaires, des AG oxydés,
des AG conjugués et la composition en AG de l’huile usée sont les méthodes standards les
plus utilisées. La mesure des peroxydes et le test de p-anisidine peuvent également être
réalisés (Orthoefer et List, 2007b). Toutefois, comme les peroxydes ont un temps de demi-
vie d’une heure à 150°C (Pryor, 1966) cette mesure ne parait donc pas appropriée pour des
huiles chauffées à 180°C pendant plusieurs heures. C’est donc pour cette raison que nous
avons choisi d’analyser la composition en acide gras résiduels des huiles de friture. Ainsi,
au lieu de mesurer ce qui c’est oxydé, nous analysons ce qui ne s’est pas dégrader.
L’analyse de l’efficacité se faisait en analysant la composition finale des huiles sous
leurs formes d’esters méthylés (FAME). Les cinq principaux acides qui ont été observés
sont l’acide palmitique (C16:0), l’acide stéarique (C18:0), l’acide oléique (C18:1), l’acide
linoléique (C18:2) et l’acide linolénique (C18:3). Les AG contenant le plus d’insaturations
seront ceux qui s’oxyderont le plus. L’acide palmitique, ne contenant pas d’insaturations ne
se détériorent pratiquement pas. Pour cette raison, il a été utilisé comme standard interne
lors du traitement des données. Ainsi, lors de l’analyse, en comparant les différentes
teneurs d’AG résiduels, s’il restait plus d’AG insaturés dans une huile contenant un
antioxydant, cela signifiait que le produit utilisé était efficace. Au contraire, si la teneur en
AG insaturés était plus faible dans une huile contenant un antioxydant que dans le contrôle,
cela signifiait que la substance utilisée agissait en fait comme un prooxydant.
La séparation par GC a été réalisée avec une colonne BPX-70 (60m x 0,25mm x
0,25 µm, dont le film intérieur de la phase stationnaire est composé de 70% Cyanopropyle
– 30% Polydiméthylsiloxane co polymère). Les échantillons ont été injectés en mode split
(ratio 1 : 50) et la température de l’injecteur a été fixée à 250°C. L’hydrogène (H2) a été
utilisé comme gaz vecteur avec un flux constant de 1,29mL/min et vitesse linéaire
constante de 32cm/sec. Pour la séparation par GC, un programme de gradient de

39
température a été utilisé (Tableau 2.1). Les FAME ont été identifiés en comparant leur
temps de rétention avec ceux de standards commerciaux.
Tableau 2.1 : Programme de température utilisé en GC pour la séparation des FAME.
Température
initiale
(°C)
Gradient
(°C/min)
Température
finale
(°C)
Maintien de la
température finale
(minutes)
60 0 60 1
60 10 190 15
190 5 200 14
2.4 Résultats et Discussion
2.4.1 Synthèse du vanillate d’hexyle
La synthèse du vanillate d’hexyle s’est faite en deux étapes (Pfeffer et Silbert,
1976). Tout d’abord, il y a eu la formation d’un sel de l’acide vanillique (2.2). Le sel a été
formé par titration de l’acide vanillique avec le KOH. La phénolphtaléine était utilisée
comme indicateur. Le sel est ensuite alkylé en utilisant un iodure d’alcane pour produire
l’ester final (2.3).
Le solide obtenu était une poudre ayant une teinte rosée dû à la phénolphtaléine. Un
rendement supérieur à 100% a souvent été obtenu, mais celui-ci peut s’expliquer par le
surplus de KOH présent dans le produit. La base était toutefois éliminée lors des lavages du
produit final avec de l’eau. Pour ce qui est de la couleur rosée du produit, elle provient de la
phénolphtaléine. Celle-ci était éliminée par un lavage du produit final avec une solution de
Na2S2O3. Un rendement après lavage et purification de 15% a été obtenu.
La présence de l’ester a été confirmée par le spectre MS (Annexe, Figure A.1).
L’ion moléculaire à m/z = 255 est bien présent. De plus, il est possible de voir le fragment
majoritaire à m/z = 168 (2.8). Ce fragment provient d’un réarrangement de McLafferty
(Figure 2.4). Les fragments à m/z = 123 et 151 proviennent du clivage des liaisons voisines
du lien C=O.

40
O
O CH(CH2)3H
O
OMeO
HO
MeO
HO
H
m/z = 1682.7 2.8
m/z = 255
Figure 2.4 : Réarrangement de McLafferty du vanillate d’hexyle.
Le spectre IR (Annexe, Figure A.2) permet également de confirmer la
transformation de l’acide en ester. En effet, la fréquence de vibration de l’élongation du lien
C=O se trouve à 1707 cm-1 pour l’ester, alors qu’elle est à 1680 cm-1 pour l’acide vanillique
(Clavijo, et al., 2008). Les fréquences caractéristiques des acides et des esters se trouvant
normalement près de 1706-1720 cm-1 et 1735-1750 cm-1 respectivement, les fréquences qui
sont observés ici peuvent être expliquer par la présence d’un groupement aryle à proximité
qui induit une conjugaison des doubles liens (Silverstein, et al., 2007a). La bande
caractéristique de la vibration d’élongation du lien C-O est visible à 1278 cm-1 pour l’ester.
La fréquence d’élongation du lien CC(=O)-O est également visible à 1215 cm-1. À 3397
cm-1, il est possible de voir la fréquence d’élongation du lien O-H (Silverstein, et al.,
2007a). Cela confirme donc que l’ester possède toujours sa fonction phénol. Cela permet
donc de confirmer que l’ester a bien été obtenu.
2.4.2 Synthèse du vanillate de dodécyle
La synthèse du vanillate de dodécyle était tout d’abord faite selon le même
protocole que le vanillate d’hexyle. Toutefois, dû à un encombrement stérique dû à la
longueur de la chaîne alkyle, la réaction ne fonctionnait pas. Un protocole alternatif a donc
dû être trouvé. Ce dernier permet de former l’ester en une seule étape. Il consistait à mettre
en présence l’acide phénolique, le sel de potassium et l’iodure d’alcane (Moore, et al.,
1979). Ce protocole permettait donc de diminuer le temps de réaction en éliminant une
étape. Quelques ajustements ont dû être fait afin d’augmenter le rendement de la réaction et
de faciliter la purification. Ainsi, puisque l’acide ne semblait réagir en entier, nous avons
donc augmenté les équivalences du sel de potassium et de l’iodure de dodécane. De plus,

41
lors de l’arrêt de la réaction, un lavage supplémentaire avec une solution de Na2CO3 a été
fait afin d’éliminer l’acide vanillique restant. Suite à une purification à l’aide d’une colonne
de chromatographie, un solide blanc était obtenu.
La présence de l’ester a pu être confirmée par le spectre MS (Annexe, Figure A.3).
L’ion moléculaire m/z = 336 est bien présent. De plus, il est possible de voir le fragment à
m/z = 168 (2.10) qui est dû au réarrangement de McLafferty (Figure 2.5). Les fragments à
m/z = 123 et 151 proviennent du clivage des liaisons voisines du lien C=O.
O
O CH(CH2)9H
O
O
MeO
HO
MeO
HO
H
m/z = 1682.9 2.10
m/z = 336
Figure 2.5 : Réarrangement de McLafferty du vanillate de dodécyle.
Le spectre IR (Annexe, Figure A.4) confirme bien également la transformation de
la fonction acide en ester. En effet, il est possible de voir que la bande représentant la
vibration d’élongation du lien C=O est à 1685 cm-1 pour l’ester, alors qu’elle est à 1680 cm-
1 pour l’acide vanillique (Clavijo, et al., 2008). La fréquence qui est observée est également
plus faible que celle du vanillate d’hexyle dû à une chaîne aliphatique plus longue. La
fréquence de vibration de l’élongation du lien C-O est visible à 1281 cm-1. La fréquence
d’élongation du lien O-H est toujours visible à 3327 cm-1, confirmant que la fonction alcool
est toujours présente. Cela confirme donc que l’ester a bien été obtenu.
2.4.3 Synthèse du coumarate d’hexyle
La synthèse du coumarate d’hexyle a tout d’abord été optimisée. Le temps de
réaction, l’utilisation d’un chauffage et le sel utilisé, soit le KHCO3 ou le K2CO3, ont donc
été modifiés à plusieurs reprises. Les meilleurs résultats ont donc été obtenus en utilisant le
KHCO3 avec un chauffage à reflux pendant une semaine.

42
Nous avons également pu remarquer que lorsque la réaction était effectuée plus
longtemps, il arrivait que deux chaînes alkyles soit ajoutées au lieu d’une seule (2.11).
Puisque le pouvoir antioxydants de ces composés provient de leur fonction phénol, cette
molécule nous était donc d’aucune utilité. Ce produit était également très dur à séparer par
colonne chromatographique sur gel de silice.
O
OO
2.11
Figure 2.6 : Produits secondaires de la réaction d’estérification de l’acide p-coumarique.
Le coumarate d’hexyle s’est également avéré difficile à purifier. De bonnes
conditions ont été trouvées en colonne chromatographique sur gel de silice, mais très peu
de produits étaient récupérés suite à la purification. Le moyen le plus utile qui fut trouvé fut
d’évaporer l’essentiel de l’iodo-hexane restant dans le produit en laissant le produit final
plus longtemps sur le rotavapeur à une température plus élevée. Une colonne était donc
parfois nécessaire pour retirer l’iodo-hexane résiduel.
La présence de l’ester a été confirmé sur le spectre MS (Annexe, Figure A.5). L’ion
moléculaire à m/z = 248 est bien présent. Il est également possible de voir le fragment à
m/z = 164 (2.13) dû au réarrangement de McLafferty (Figure 2.7). Les fragments à
m/z = 91 et 65 sont caractéristiques d’un cycle aromatique substitué par un alkyle. Les
fragments à m/z = 119 et 147 proviennent du clivage des liaisons voisines du lien C=O.
HO
O
O
(CH2)3H
HO
O
O
H
m/z = 1642.132.12
m/z = 248
Figure 2.7 : Réarrangement de McLafferty du coumarate d’hexyle.

43
Le spectre IR (Annexe, Figure A.6) confirme bien également la transformation de
la fonction acide en ester. En effet, il est possible de voir que la bande représentant la
vibration d’élongation du lien C=O est à 1670 cm-1, alors qu’elle est aux environs de 1667
cm-1 pour l’acide p-coumarique. La valeur habituelle pour un ester est de 1735 cm-1 et celle
de l’acide est de 1710 cm-1 (Silverstein, et al., 2007a), mais les faibles valeurs obtenues
peuvent être expliqué par la proximité d’une double liaison suivie d’un cycle aromatique.
Cela permet donc la résonance des électrons (Figure 2.8). Ainsi, la résonance affaibli le
lien carbonyle, ce qui diminue sa fréquence d’absorption (Silverstein, et al., 2007a).
HO
O
OH
HO
O
OH
Figure 2.8 : Structure de résonance de l’acide p-coumarique.
De plus, la fréquence de vibration de l’élongation du lien C-O se trouve à 1273 cm-
1. Cela confirme donc que l’ester a bien été obtenu. Il est également possible de voir la
bande d’élongation du lien O-H à 3371 cm-1. Cela confirme donc que l’ester possède
toujours sa fonction phénol.
2.4.4 Analyse en DSC
Les résultats obtenus en DSC (Figure 2.9) montrent que, pour les trois composés,
aucun changement n’est visible entre 50 et 200°C. Puisque ces composés n’ont été utilisés
qu’à une température de 120°C, on est donc certain qu’aucune dégradation du composé ne
s’est produite lors des traitements thermo-oxydatifs, et donc qu’ils étaient bien présents
dans l’huile tout au long de l’expérience.
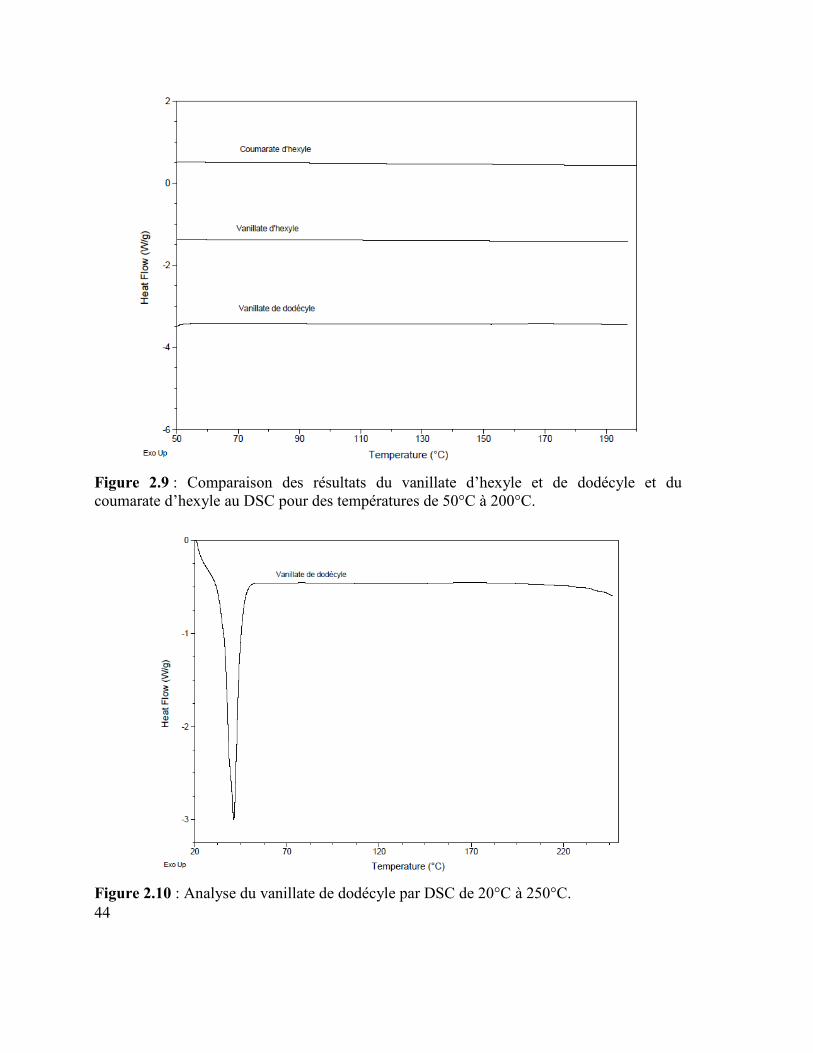
44
Figure 2.9 : Comparaison des résultats du vanillate d’hexyle et de dodécyle et du
coumarate d’hexyle au DSC pour des températures de 50°C à 200°C.
Figure 2.10 : Analyse du vanillate de dodécyle par DSC de 20°C à 250°C.

45
Pour le vanillate de dodécyle, puisqu’il s’agit du seul produit qui est sous forme
solide à température pièce, il était possible de voir son point de fusion lorsqu’on
commençait l’analyse à une température plus basse (Figure 2.10). Le solide fondait donc
au environ de 40°C, mais par la suite, aucun autre changement n’était visible.
2.4.5 Test d’efficacité des antioxydants dans l’huile de Canola
Les résultats présentés ci-dessous représentent la teneur résiduelle en AG de l’huile
supplémentée par rapport à l’huile de Canola seule. Ainsi, si la valeur obtenue est
supérieure à 1,00, cela signifie que le produit testé présente un effet antioxydant, puisqu’il
reste plus d’AG insaturés dans l’huile contenant le produit testé que dans l’huile seule.
2.4.5.1 Test antioxydant du vanillate d’hexyle
Comme il est possible de le voir (Figure 2.11), le vanillate d’hexyle a montré un
bon effet antioxydant lorsqu’il est utilisé à 1,0%. En effet, la teneur résiduelle en AG
insaturés des huiles contenant l’antioxydant est supérieure à celle de l’huile seule. Cette
différence est significativement différente pour l’AL (C18:2) et l’AAL (C18:3). Cet effet
est d’ailleurs plus visible après 2 jours de traitement thermo-oxydatif. Pour ce qui est de
l’AO (C18:1), très peu de changements sont observés, mais cela peut être expliqué par le
fait que cet acide ne contient qu’une seule insaturation et qu’il est de 10 à 40 fois moins
susceptible à l’oxydation que l’AL et l’AAL (Belitz, et al., 2009b, Scrimgeour, 2005). Il
aura donc moins tendance à se dégrader par rapport à l’AL et à l’AAL.
Ainsi, étant donné les résultats obtenus avec le vanillate d’hexyle à une
concentration de 1,0%, des tests avec ce composé ont donc été effectués a des
concentrations plus basses, soit 0,5% et 0,1%. À ces concentrations, il n’y a plus d’effet
antioxydant pour aucun des trois AG observés.
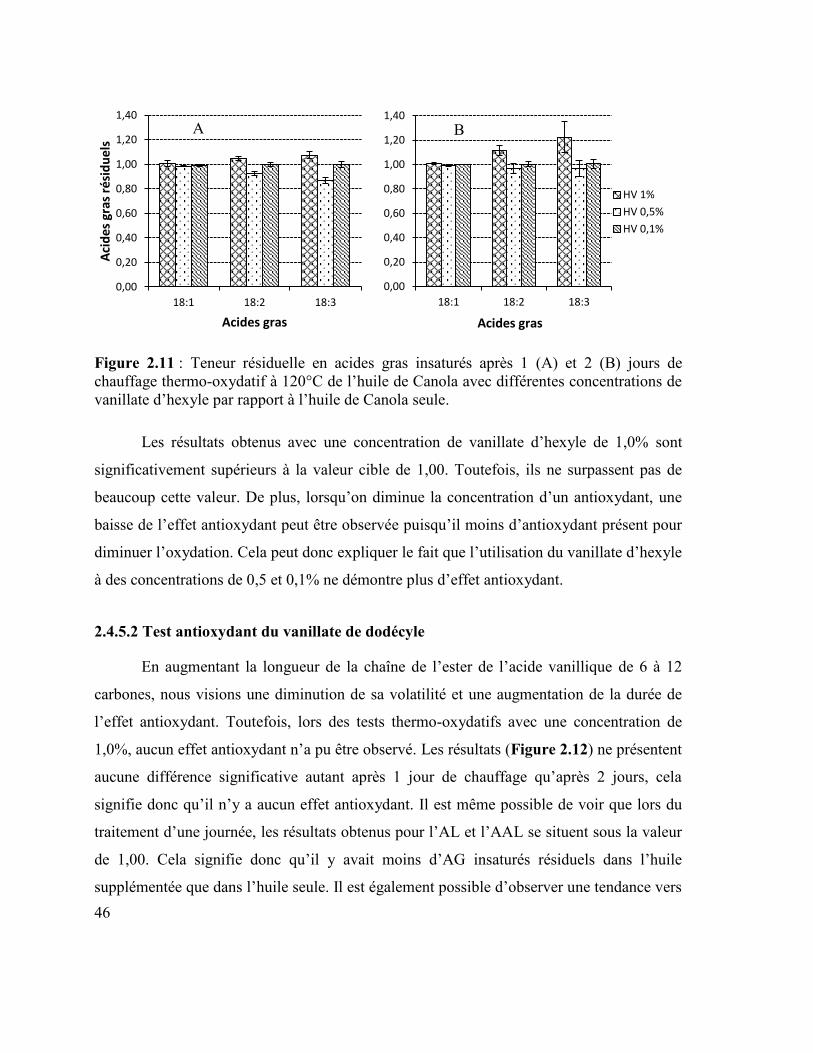
46
Figure 2.11 : Teneur résiduelle en acides gras insaturés après 1 (A) et 2 (B) jours de
chauffage thermo-oxydatif à 120°C de l’huile de Canola avec différentes concentrations de
vanillate d’hexyle par rapport à l’huile de Canola seule.
Les résultats obtenus avec une concentration de vanillate d’hexyle de 1,0% sont
significativement supérieurs à la valeur cible de 1,00. Toutefois, ils ne surpassent pas de
beaucoup cette valeur. De plus, lorsqu’on diminue la concentration d’un antioxydant, une
baisse de l’effet antioxydant peut être observée puisqu’il moins d’antioxydant présent pour
diminuer l’oxydation. Cela peut donc expliquer le fait que l’utilisation du vanillate d’hexyle
à des concentrations de 0,5 et 0,1% ne démontre plus d’effet antioxydant.
2.4.5.2 Test antioxydant du vanillate de dodécyle
En augmentant la longueur de la chaîne de l’ester de l’acide vanillique de 6 à 12
carbones, nous visions une diminution de sa volatilité et une augmentation de la durée de
l’effet antioxydant. Toutefois, lors des tests thermo-oxydatifs avec une concentration de
1,0%, aucun effet antioxydant n’a pu être observé. Les résultats (Figure 2.12) ne présentent
aucune différence significative autant après 1 jour de chauffage qu’après 2 jours, cela
signifie donc qu’il n’y a aucun effet antioxydant. Il est même possible de voir que lors du
traitement d’une journée, les résultats obtenus pour l’AL et l’AAL se situent sous la valeur
de 1,00. Cela signifie donc qu’il y avait moins d’AG insaturés résiduels dans l’huile
supplémentée que dans l’huile seule. Il est également possible d’observer une tendance vers
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
18:1 18:2 18:3
Aci
des
gra
s ré
sid
ue
ls
Acides gras
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
18:1 18:2 18:3
Acides gras
HV 1%
HV 0,5%
HV 0,1%
A B

47
la hausse lors du traitement de 2 jours, toutefois les résultats obtenus ne sont pas
significativement différent.
Figure 2.12 : Teneur résiduelle en acides gras insaturés après 1 et 2 jours de chauffage
thermo-oxydatif à 120°C de l’huile de Canola avec le vanillate de dodécyle à une
concentration de 1,0% par rapport à l’huile de Canola seule.
Il faut toutefois prendre en compte que les concentrations utilisées sont en masse sur
masse. Ainsi, lorsqu’on effectue la conversion de ces quantités en mole, il est possible
d’observer que pour une même quantité, il y a 1,3 fois plus de vanillate d’hexyle et près de
2 fois plus de BHT que de vanillate de dodécyle. Cela peut donc expliquer le fait que pour
une même concentration, on observe un effet antioxydant pour le vanillate d’hexyle et non
pas pour le vanillate de dodécyle.
2.4.5.3 Test antioxydant du coumarate d’hexyle
Les résultats après une journée de traitement (Figure 2.13A) montrent que seuls les
résultats à une concentration de 1,0% et de 0,5% ont un effet antioxydant. Pour ce qui est
des résultats avec le coumarate d’hexyle à une concentration de 0,1%, les résultats obtenus
ne démontrent aucune différence après une seule journée de traitement thermo-oxydatif.
0,00
0,20
0,40
0,60
0,80
1,00
1,20
18:1 18:2 18:3
Aci
des
gra
s ré
sid
ue
ls
Acides gras
DV 1j
DV 2j
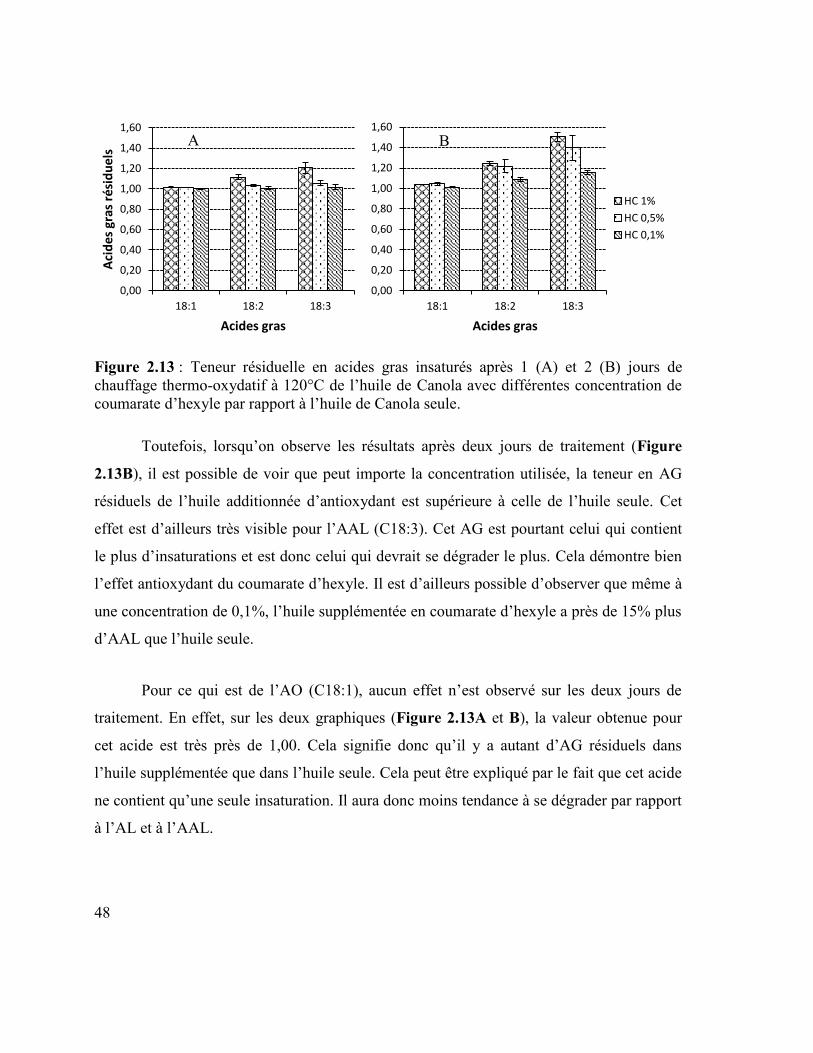
48
Figure 2.13 : Teneur résiduelle en acides gras insaturés après 1 (A) et 2 (B) jours de
chauffage thermo-oxydatif à 120°C de l’huile de Canola avec différentes concentration de
coumarate d’hexyle par rapport à l’huile de Canola seule.
Toutefois, lorsqu’on observe les résultats après deux jours de traitement (Figure
2.13B), il est possible de voir que peut importe la concentration utilisée, la teneur en AG
résiduels de l’huile additionnée d’antioxydant est supérieure à celle de l’huile seule. Cet
effet est d’ailleurs très visible pour l’AAL (C18:3). Cet AG est pourtant celui qui contient
le plus d’insaturations et est donc celui qui devrait se dégrader le plus. Cela démontre bien
l’effet antioxydant du coumarate d’hexyle. Il est d’ailleurs possible d’observer que même à
une concentration de 0,1%, l’huile supplémentée en coumarate d’hexyle a près de 15% plus
d’AAL que l’huile seule.
Pour ce qui est de l’AO (C18:1), aucun effet n’est observé sur les deux jours de
traitement. En effet, sur les deux graphiques (Figure 2.13A et B), la valeur obtenue pour
cet acide est très près de 1,00. Cela signifie donc qu’il y a autant d’AG résiduels dans
l’huile supplémentée que dans l’huile seule. Cela peut être expliqué par le fait que cet acide
ne contient qu’une seule insaturation. Il aura donc moins tendance à se dégrader par rapport
à l’AL et à l’AAL.
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
18:1 18:2 18:3
Aci
des
gra
s ré
sid
ue
ls
Acides gras
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
18:1 18:2 18:3
Acides gras
HC 1%
HC 0,5%
HC 0,1%
A B
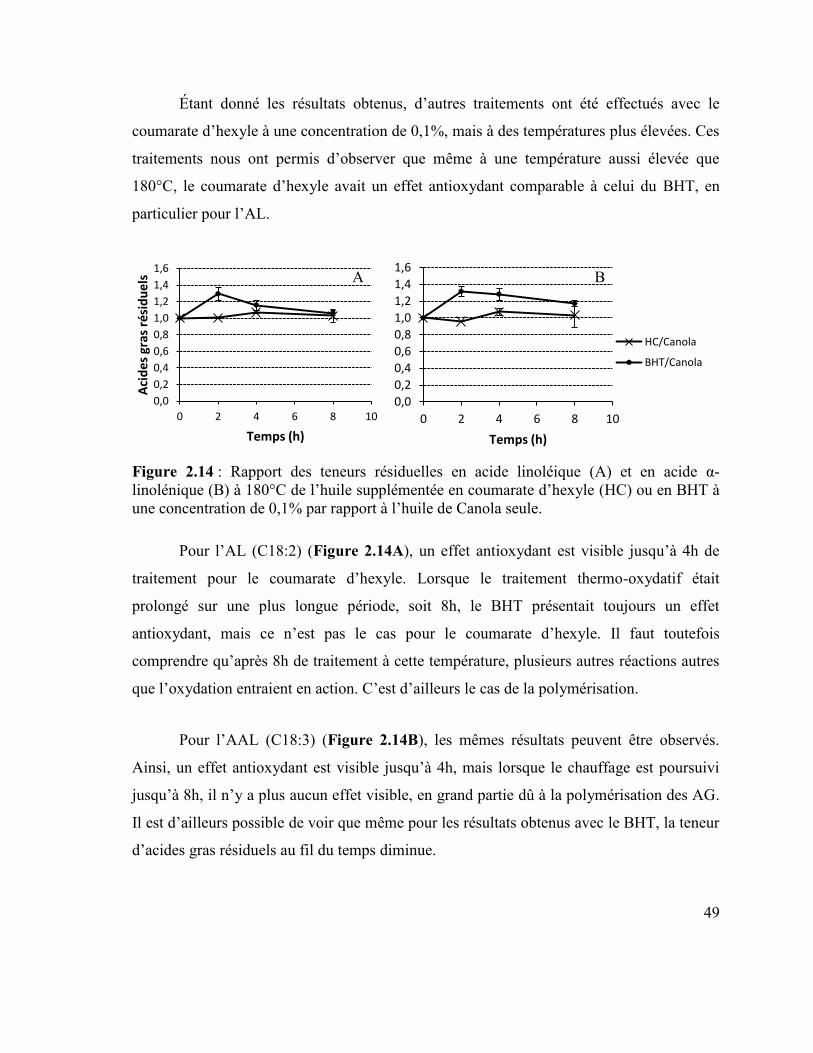
49
Étant donné les résultats obtenus, d’autres traitements ont été effectués avec le
coumarate d’hexyle à une concentration de 0,1%, mais à des températures plus élevées. Ces
traitements nous ont permis d’observer que même à une température aussi élevée que
180°C, le coumarate d’hexyle avait un effet antioxydant comparable à celui du BHT, en
particulier pour l’AL.
Figure 2.14 : Rapport des teneurs résiduelles en acide linoléique (A) et en acide α-
linolénique (B) à 180°C de l’huile supplémentée en coumarate d’hexyle (HC) ou en BHT à
une concentration de 0,1% par rapport à l’huile de Canola seule.
Pour l’AL (C18:2) (Figure 2.14A), un effet antioxydant est visible jusqu’à 4h de
traitement pour le coumarate d’hexyle. Lorsque le traitement thermo-oxydatif était
prolongé sur une plus longue période, soit 8h, le BHT présentait toujours un effet
antioxydant, mais ce n’est pas le cas pour le coumarate d’hexyle. Il faut toutefois
comprendre qu’après 8h de traitement à cette température, plusieurs autres réactions autres
que l’oxydation entraient en action. C’est d’ailleurs le cas de la polymérisation.
Pour l’AAL (C18:3) (Figure 2.14B), les mêmes résultats peuvent être observés.
Ainsi, un effet antioxydant est visible jusqu’à 4h, mais lorsque le chauffage est poursuivi
jusqu’à 8h, il n’y a plus aucun effet visible, en grand partie dû à la polymérisation des AG.
Il est d’ailleurs possible de voir que même pour les résultats obtenus avec le BHT, la teneur
d’acides gras résiduels au fil du temps diminue.
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
0 2 4 6 8 10
Aci
des
gra
s ré
sid
ue
ls
Temps (h)
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
0 2 4 6 8 10
Temps (h)
HC/Canola
BHT/Canola
A B

50
Il faut toutefois prendre en compte que 8h de traitement thermo-oxydatif à 180°C
est beaucoup plus intense que 8h de traitement en friture. En effet, en friture, il y aura
différents facteurs, entre autres de la vapeur d’eau, qui seront présents et qui retarderont
l’oxydation des AG (Orthoefer et List, 2007a). Pour cette raison, afin de déterminer si nous
allions tester le coumarate d’hexyle en condition de friture, nous nous sommes surtout
attarder aux résultats obtenus après 4h de traitement thermo-oxydatif.
Ainsi, étant donnée les résultats obtenus, nous avons choisi de ne tester que le
coumarate d’hexyle en condition de friture, bien qu’un effet antioxydant ne soit pas visible
lors d’un traitement thermo-oxydatif de 8h.
2.4.6 Test d’efficacité des antioxydants en condition de friture
Après avoir effectué l’analyse des échantillons d’huiles recueillis lors de la friture,
nous avons remarqué que la teneur en AL (C18:2) ne diminuait presque pas (Figure 2.15),
contrairement à la teneur en AAL (C18:3) et en AO (C18:1). On peut expliquer ce manque
de changement par le fait que les frites que nous avons utilisées pour la friture avaient subi
un prétraitement dans l’huile de soja. Cette huile étant riche en AL (Orthoefer et List,
2007c), cela permet d’expliquer que la teneur de cet acide gras ne change pratiquement pas.
Figure 2.15 : Teneur résiduelle des différents acides gras lors de la friture.
75,00
80,00
85,00
90,00
95,00
100,00
105,00
110,00
0,00 2,00 4,00 6,00 8,00
Aci
de
s gr
as r
ési
du
els
(%
)
Temps (h)
C18:1
C18:2
C18:3
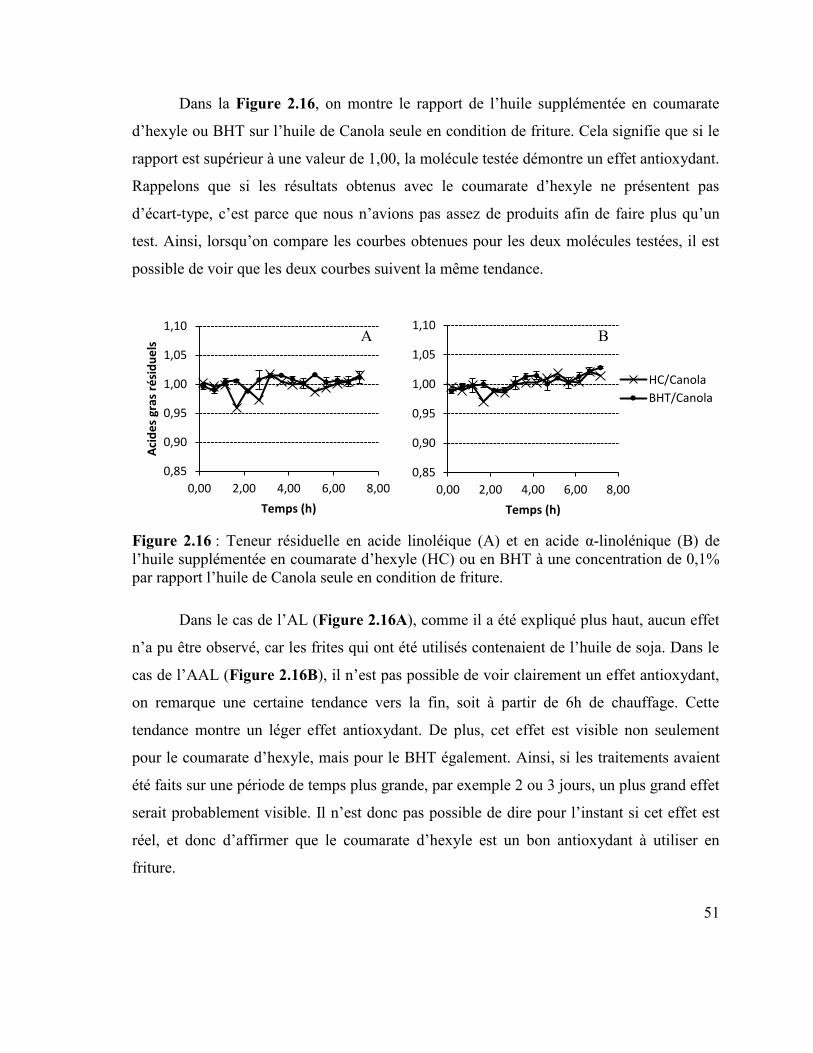
51
Dans la Figure 2.16, on montre le rapport de l’huile supplémentée en coumarate
d’hexyle ou BHT sur l’huile de Canola seule en condition de friture. Cela signifie que si le
rapport est supérieur à une valeur de 1,00, la molécule testée démontre un effet antioxydant.
Rappelons que si les résultats obtenus avec le coumarate d’hexyle ne présentent pas
d’écart-type, c’est parce que nous n’avions pas assez de produits afin de faire plus qu’un
test. Ainsi, lorsqu’on compare les courbes obtenues pour les deux molécules testées, il est
possible de voir que les deux courbes suivent la même tendance.
Figure 2.16 : Teneur résiduelle en acide linoléique (A) et en acide α-linolénique (B) de
l’huile supplémentée en coumarate d’hexyle (HC) ou en BHT à une concentration de 0,1%
par rapport l’huile de Canola seule en condition de friture.
Dans le cas de l’AL (Figure 2.16A), comme il a été expliqué plus haut, aucun effet
n’a pu être observé, car les frites qui ont été utilisés contenaient de l’huile de soja. Dans le
cas de l’AAL (Figure 2.16B), il n’est pas possible de voir clairement un effet antioxydant,
on remarque une certaine tendance vers la fin, soit à partir de 6h de chauffage. Cette
tendance montre un léger effet antioxydant. De plus, cet effet est visible non seulement
pour le coumarate d’hexyle, mais pour le BHT également. Ainsi, si les traitements avaient
été faits sur une période de temps plus grande, par exemple 2 ou 3 jours, un plus grand effet
serait probablement visible. Il n’est donc pas possible de dire pour l’instant si cet effet est
réel, et donc d’affirmer que le coumarate d’hexyle est un bon antioxydant à utiliser en
friture.
0,85
0,90
0,95
1,00
1,05
1,10
0,00 2,00 4,00 6,00 8,00
Aci
de
s gr
as r
ési
du
els
Temps (h)
0,85
0,90
0,95
1,00
1,05
1,10
0,00 2,00 4,00 6,00 8,00
Temps (h)
HC/Canola
BHT/Canola
A B

52
2.5 Conclusion
Lors du processus de friture, différentes dégradations affectent la qualité de l’huile.
La plus importante est l’oxydation. Certains moyens peuvent être pris afin d’éliminer, ou au
moins diminuer le niveau d’oxydation des AG. Parmi ceux-ci, on retrouve les antioxydants.
Ce projet visait donc à estérifier des acides phénoliques naturels afin d’obtenir des
antioxydants moins volatiles et plus solubles dans les huiles.
Différents produits ont donc pu être synthétisés et purifiés afin d’être testés comme
antioxydants dans les huiles de friture. Il s’agit du vanillate d’hexyle, du vanillate de
dodécyle et du coumarate d’hexyle. Ces trois composés ont d’abord été testés à l’aide de
traitements thermo-oxydatifs. Le vanillate d’hexyle a démontré un effet antioxydant, mais
seulement à haute concentration, soit 1,0%, car cette effet disparaissait lorsqu’on diminuait
sa concentration dans l’huile de Canola. Pour ce qui est du vanillate de dodécyle, même à
une concentration de 1,0%, il n’a démontré aucun effet antioxydant. Le coumarate d’hexyle
a toutefois démontré un effet antioxydant comparable à celui du BHT même à une
concentration de 0,1% et à une température de 180°C. Ce dernier a donc été testé en
condition de friture. Des résultats semblables au BHT ont également été obtenus lors des
tests en friture. Toutefois ces tests ne démontraient qu’un début d’effet antioxydant vers la
fin de la période de friture, soit après 6h de traitement. Ces résultats étaient les mêmes pour
le coumarate d’hexyle et pour le BHT. Les traitements de friture devraient donc être
réalisés sur une plus longue période, soit 2 ou 3 jours, afin de vérifier s’il y a bien un effet
antioxydant.

Chapitre 3
Approche à la synthèse de l’acide α-linolénique marqué


55
3.1 Introduction
Les AGPI n-3 sont reconnus pour les bienfaits qu’ils nous apportent. Ils
contribueraient entre autre à l’abaissement des risques de maladies cardiovasculaires, au
développement de certaines fonctions du cerveau (Kris-Etherton, et al., 2002, Ruxton, et
al., 2004, Vancassel, 2004) et ils auraient un impact positif sur la santé oculaire (Jeffrey, et
al., 2001, SanGiovanni et Chew, 2005). Toutefois, lors de traitement thermique intense, tel
que la friture, diverses réactions de dégradations peuvent se produire sur les AGPI n-3
contenu dans les huiles. Par exemple, lorsqu’ils sont en contact avec l’oxygène, ils
formeront des produits polaires qui sont nocifs pour la santé (Billek, 2000, Marquez-Ruiz et
Dobarganes, 2007). La chaleur formera entre autre des acides gras trans, des polymères et
des MCAG (Billek, 2000, Frankel, et al., 1984, Sébédio et Grandgirard, 1989, Sébédio et
Juaneda, 2007). Ces derniers peuvent être dommageables pour la santé (Sébédio, et al.,
2007, Sébédio et Grandgirard, 1989). De plus, puisque ces produits se forment dans les
huiles de friture, ils se retrouveront également dans les aliments frits et donc dans notre
alimentation.
Le présent chapitre traitera de la dégradation thermique des AGPI n-3 en MCAG.
En particulier ceux découlant de l’AAL. Étant donné qu’il s’agit de l’AG contenant le plus
de doubles liaisons dans les huiles végétales, il est donc l’acide qui subira le plus de
dégradation et celui qui formera le plus de MCAG. Les structures des MCAG de l’AAL
sont bien connues (Christie et Dobson, 2000, Dobson, et al., 1996a, Mossoba, et al., 1995).
Toutefois leur mécanisme de formation n’est encore qu’hypothétique. À ce jour, trois
mécanisme ont été proposés dans la littérature (Christie et Dobson, 2000, Destaillats et
Angers, 2005, Gast, et al., 1963). Le dernier à avoir été proposé suggère que les MCAG se
formeraient selon une cyclisation intramoléculaire suivi d’une migration prototropique
[1,6] (Destaillats et Angers, 2005). Contrairement aux deux autres mécanismes, ce dernier
explique non seulement la formation des MCAG à partir des AGPI, mais également à partir
des AG mono- et di-insaturés et il se limite à la formation des MCAG observés dans les
huiles.

56
Afin de vérifier cette hypothèse, la synthèse d’un isomère géométrique de l’AAL
marqué à un endroit impliqué dans la réaction de cyclisation sera faite. Les atomes
marqueurs permettront ainsi le suivi de la formation des MCAG à l’aide de la spectroscopie
RMN 1H et de la spectrométrie MS. Ce chapitre présentera donc l’approche qui a été
utilisée, les étapes qui ont été réalisées, ainsi que les problèmes rencontrés pour la synthèse
de cet acide qui sera utilisé pour étudier le mécanisme de formation des MCAG.

57
3.2 Hypothèse et objectifs
Hypothèse
Les MCAG sont des produits de dégradation des huiles utilisées en friture, autant
par l’industrie que par les consommateurs. Bien que leurs structures aient été bien
analysées, leur mécanisme de formation n’est encore qu’hypothétique. Nous croyons que
les MCAG sont formés selon un mécanisme de cycloaddition suivi d’une migration
prototropique [1,6]. La synthèse d’un AGPI n-3 marqué par des atomes de deutérium sur le
carbone 14 pourrait permettre de démontrer le mécanisme de formation des MCAG.
Objectifs
Déterminer une voie de synthèse pour l’AAL marqué par des atomes de
deutérium sur le carbone 14 et mono-trans en C-15.
Synthétiser, purifier et caractériser l’AAL marqué.
Effectuer des traitements thermiques afin de cycliser l’AAL marqué et suivre le
déplacement des atomes marqueurs par spectroscopie RMN 1H et MS.

58
3.3 Approche à la synthèse
Différentes approches étaient possibles afin de synthétiser un acide marqué. Les
approches peuvent varier selon la méthode utilisée pour apposer les atomes marqueurs,
ainsi que dans la méthode utilisée pour faire les différents couplages.
3.3.1 Choix de l’atome marqueur
Comme il a été mentionné, le remplacement d’un atome d’hydrogène sur la
structure de l’AAL menant à la formation du MCAG ayant un cycle à 5 membres entre les
carbones 10 et 14 est le choix le plus judicieux puisque ce sont ces cycles qui se forment le
plus rapidement et qu’ils se transformeront plus lentement en cycle à 6. Ainsi, les atomes
marqueurs seront sur le carbone 14 de la structure initiale de l’AAL. Le deutérium, un
isotope non-radioactif de l’hydrogène, sera utilisé comme atome marqueur. Cet atome est
un choix intéressant, car il réagira de la même façon que l’hydrogène. Toutefois, un effet
isotopique sera surement observable, soit que la vitesse de réaction du deutérium sera
inférieure à celle de l’hydrogène. En effet, le ratio KH/KD varie aux alentour de 7 dans la
plupart des réactions. Ce qui signifie que la rupture du lien C-H s’effectue environ 7 fois
plus vite que la rupture du lien C-D (Westheimer, 1961).
HOOC(H2C)7D
D
CH3
9
12
15
14 HOOC(H2C)7
D
CH3
D9
12
1415
3.1 3.2
Figure 3.1 : Déplacement d’un atome de deutérium lors de la formation d’un cycle.
Pour cette raison, l’AAL qui sera synthétisé comportera deux atomes de deutérium
sur le même carbone afin de s’assurer que ce sera bien un deutérium qui participera à la
réaction et non un hydrogène (Figure 3.1). De plus, le nombre de spin I du deutérium est
différent de celui de l’hydrogène (IH = ½, ID = 1). Donc, lorsque l’analyse par résonance
magnétique nucléaire du proton (RMN 1H) sera faite, le deutérium n’émettra pas de signal.

59
Finalement, le deutérium ayant une masse de plus que l’hydrogène, les fragments marqués
par les atomes de deutérium analysés par spectrométrie de masse (MS) présenteront donc
deux masses de plus que ceux qui ne seront pas marqués.
3.3.2 Synthèse d’acides gras non marqués ou marqués
Différentes synthèses d’AG saturés et insaturés ont déjà été proposées dans la
littérature. L’étape clé de ces synthèses est la création des doubles liaisons des acides
insaturés. Le couplage d’alcyne et la réaction de Wittig sont les réactions les plus souvent
employés. Le couplage d’alcyne se fait en présence d’un réactif de grignard, suivi d’une
hydrogénation de la triple liaison complète (Figure 3.2A) (DasGupta, et al., 1982) ou
partielle (Figure 3.2B) (Ahmad et Strong, 1948, Gensler et Thomas, 1951, Sgoutas, et al.,
1969, Stoffel, 1965). Cette dernière mènera à la formation d’insaturations dans la structure
(3.7). Il suffit alors de modifier les conditions de la réaction pour obtenir une double liaison
de configuration cis ou trans. D’autres synthèses utilisent des réactions d’oléfination, tel
que la réaction de Wittig (Figure 3.2C), pour former les doubles liaisons carbone-carbone
(Bergelson et Shemyakin, 1964, Eynard, et al., 1998, Gehring, et al., 1998). Ainsi, ces deux
méthodes peuvent être envisagées pour la synthèse de l’AAL marqué.
R Br R' R R'
R
R'
R
R'
R
O
H
Br R'
R R'
PPh3
base
A
B
C
3.3 3.4 3.5
3.6
3.7
3.8 3.9 3.10
Figure 3.2 : Mécanisme réactionnel pour un couplage d’alcyne suivi d’une hydrogénation
complète (A) et d’une hydrogénation partielle (B), ainsi qu’une réaction de Wittig (C).
Pour ce qui est du marquage des molécules avec des isotopes, il est possible de
retrouver dans la littérature des exemples utilisant des atomes de deutérium (D) et de
tritium (T). Afin de poser ces atomes sur les molécules, deux méthodes reviennent très

60
souvent, soit l’utilisation de dideutérium ou une réduction à l’aide d’un deutérure.
L’utilisation de dideutérium, comme il a été utilisé dans les travaux précédents (Sgoutas et
Kummerow, 1964, Sgoutas, et al., 1969, Stoffel, 1965), n’est pas une solution qui convient
dans les présents travaux, car cette réaction apposera les atomes de deutérium en position
vinylique, soit sur deux carbones différents (Figure 3.3A). Comme il a été mentionné
auparavant, il est préférable d’avoir deux atomes de deutérium sur le même carbone afin
d’éviter d’avoir un effet isotopique. Ainsi, une réduction à l’aide d’un deutérure de lithium
et d’aluminium (LiAlD4) (Figure 3.3B), comme il a été utilisé dans certains travaux
(DasGupta, et al., 1982, Maharvi, et al., 2010), serait une meilleure solution. De plus, dû à
la spécificité de la réaction de réduction, cela nous permet d’apposer les atomes de
deutérium à la bonne place.
R R'
D2
RR'
D
D
R
O
O
LiAlD4
R
D D
OH
A
B
3.11 3.12
3.13 3.14
Figure 3.3 : Mécanisme réactionnel d’une deutération à l’aide d’un dideutérium (A) et d’un
deutérure de lithium et d’aluminium (B).
3.3.3 Stratégie de synthèse
Lors de travaux préliminaires, différentes voies de synthèse ont été tentées. Ces
approches utilisaient le couplage d’alcyne suivi d’une hydrogénation catalytique sélective
avec un catalyseur de Lindlar (Figure 3.4 et Figure 3.5). Cela permettait d’obtenir des
doubles liens de configuration cis. Cette technique résultait en un AG final ayant des
doubles liens tout-cis. Ainsi, une réduction permettrait d’introduire les atomes de
deutérium, avant que l’alcool soit protégé d’un éther silylé (3.19). Deux couplages
d’alcynes via une réaction de Grignard à l’aide d’organomagnésiens entre les composés
3.18 et 3.19, ainsi qu’entre 3.16 et 3.17 permettra la formation de l’acide finale (3.15) dans
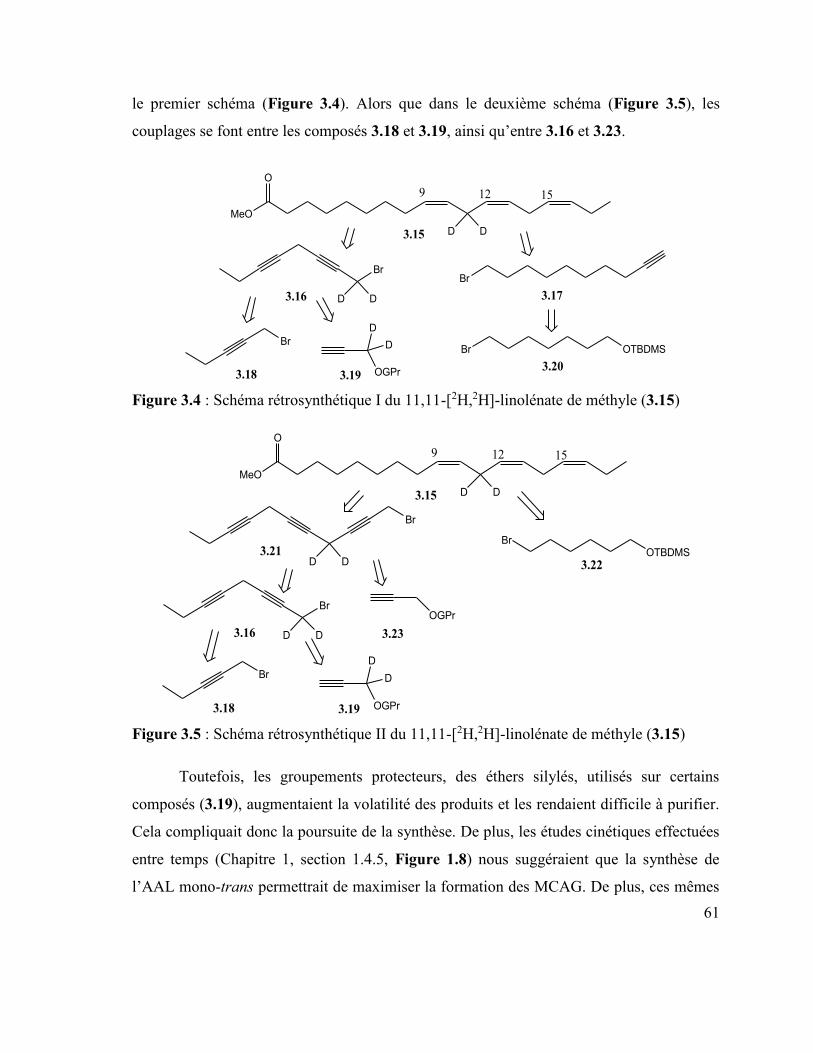
61
le premier schéma (Figure 3.4). Alors que dans le deuxième schéma (Figure 3.5), les
couplages se font entre les composés 3.18 et 3.19, ainsi qu’entre 3.16 et 3.23.
MeO
O
DD
9 12 15
3.15
BrBr
D D3.16 3.17
Br OTBDMSBr
D
D
OGPr3.18 3.193.20
Figure 3.4 : Schéma rétrosynthétique I du 11,11-[2H,2H]-linolénate de méthyle (3.15)
MeO
O
DD
9 12 15
3.15
Br
D
D
OGPr3.18 3.19
BrOTBDMS
Br
D D3.21
3.22
OGPrBr
D D3.16 3.23
Figure 3.5 : Schéma rétrosynthétique II du 11,11-[2H,2H]-linolénate de méthyle (3.15)
Toutefois, les groupements protecteurs, des éthers silylés, utilisés sur certains
composés (3.19), augmentaient la volatilité des produits et les rendaient difficile à purifier.
Cela compliquait donc la poursuite de la synthèse. De plus, les études cinétiques effectuées
entre temps (Chapitre 1, section 1.4.5, Figure 1.8) nous suggéraient que la synthèse de
l’AAL mono-trans permettrait de maximiser la formation des MCAG. De plus, ces mêmes

62
travaux (Chapitre 1, section 1.4.5, Figure 1.9) suggèrent également que les deutérium placé
sur le carbone 14 permettrait de maximiser la quantité de MCAG à 5 carbones qui serait
obtenu après les traitements thermiques (Desmarais, 2013).
Un autre schéma rétrosynthétique a déjà été proposé (Figure 3.6) (Desmarais,
2013). Celui-ci utilise la réaction de Wittig afin de former les doubles liens de
configuration cis. Dans ce schéma, les groupements R représentent un groupement alcool
ou un groupement protecteur. La structure finale est un isomère géométrique de l’AAL, soit
l’acide 14,14-[H2-H2]-Z,Z,E-octadéca-9,12,15-triénoïque. Ce schéma a également
l’avantage qu’une des trois doubles liaisons soit déjà synthétisée, se trouvant sur un des
composés de départ (3.27). Toutefois, un groupement t-BDMS, soit un éther silylé, était
utilisé afin de protéger la fonction alcool sur l’un des produits de départ (3.26). Ce
groupement étant imposant, il causait donc de l’encombrement stérique qui nuisait à la
réaction de Wittig subséquente et l’alcène (3.37) n’était donc pas formé.
HO
O
D D
O
DDH
RO PPh3
Br
MeO
O
HRO
O
OR'Ph3PBr
OR'Br
3.40
3.37 3.30
3.26 3.33
3.31
3.27
Figure 3.6 : Schéma rétrosynthétique I de l'acide 14,14-[H2-H2]-Z,Z,E-octadéca-9,12,15-
triénoïque (3.40).

63
3.3.4 Synthèse de l’acide 14,14-[H2-H2]-Z,Z,E-octadéca-9,12,15-triénoïque
En se basant sur le même schéma rétrosynthétique (Figure 3.6) qui a déjà été
présenté, nous avons suggéré un schéma de synthèse constitué de trois parties (Figure 3.7).
Par rapport à ce qui a déjà été proposé, seuls les groupements protecteurs et les conditions
de réaction ont été modifiés. Le schéma de synthèse prévoit la formation d’un aldéhyde
marqué par deux atomes de deutérium (3.30), la formation d’un aldéhyde non-marqué
(3.26) à partir du 1,9-nonanediol (3.24) et du couplage de ces deux structures. Ainsi, une
première réaction de Wittig aura lieu entre l’aldéhyde non-marqué (3.26) et un sel d’ylure
de phosphore (3.33). Le synthon obtenu (3.35) sera transformé de nouveau en sel d’ylure
(3.37) en vue d’une deuxième réaction de Wittig avec l’aldéhyde marqué. La structure
finale (3.40) sera obtenue suite à une oxydation.
1ère partie : formation de l’aldéhyde C1 à C9
O OHOOHHO
H O
O
O
3.24 3.25
3.26
1) (MeO)3CH, cat
CH2Cl2, t.p., 48h
2) DIBAL-H -78°C, 1h PDC, CH2Cl2
t.p., overnight
2e partie : Formation de l’aldéhyde C13 à C18
MeO
O
HO
DD
NC
DD
O
DDH
3.27 3.28
3.293.30
LiAlD4, AlCl3Et2O, -78°C, 2h
DIBAL-H, CH2Cl2
-78°C, 1,5h
, PPh3
DIAD, Et2O
CNHO
t.p., overnight

64
3e partie : Formation de la structure finale; deux réactions de Wittig
RO Br
3.31, R=H
3.32, R=THP
RO PPh3
BrRO
PPh3
3.33 3.34
O O OR
3.35
O O Br3.36
O O PPh3
Br3.37O O
PPh3
3.38
O OD D
HO
O
D D
3.39
3.40
PPh3
100°C, 1h
n-BuLi, THF
-78°C, 1h
3.26, THF -78°C, overnight
Ph3PBr2, CH2Cl2
t.p., 10min.
PPh3
90°C, 3h
3.30, THF
-78°C, overnight
n-BuLiTHF
-78°C, 1h
PDC, DMFt.p., overnight
Figure 3.7 : Schéma de synthèse de l’acide 15-trans-14,14-[2H,2H]-linolénique (3.40).
3.4 Résultats et discussion
3.4.1 Mono-protection du nonane-1,9-diol
Afin de s’assurer que les deux fonctions alcool du diol ne participent pas à la
réaction de Wittig, une mono-protection était de rigueur. Différents groupements
protecteurs pouvaient être utilisés. Dans les travaux qui ont été fait précédemment
(Desmarais, 2013), un groupement t-BDMS était utilisé. Toutefois, ce groupement était
imposant et causait donc de l’encombrement qui nuisant à la réaction de Wittig
subséquente. Pour cette raison, nous avons choisi de changer ce groupement protecteur afin
d’utiliser un groupement MOM. Ce groupement, tout comme le t-BDMS, résiste au clivage
oxydatif en présence du PDC, nécessaire pour la réaction d’oxydation subséquente (Wuts et
Greene, 2007).

65
La mono-protection d’un diol avec un groupement MOM se fait en deux étapes
(Figure 3.8). Il y a tout d’abord la formation d’un cycle (3.41) avec le
triméthylorthoformate, suivi de l’ouverture du cycle par l’utilisation du DIBAL-H (Takasu,
et al., 1988). Cette synthèse a dû subir plusieurs ajustements afin d’optimiser les conditions
de réaction. En effet, la mono-protection d’un diol utilisant un éther de MOM comme
groupements protecteurs était généralement fait avec des diols beaucoup plus courts. Le fait
que notre diol comporte 9 atomes de carbones entre chaque groupement alcool a joué un
rôle dans les faibles rendements obtenus.
OH OH
7
O O
OMe
7
OH OMOM
7(OMe)3CH DIBAL-H
3.24 3.25
3.41
Figure 3.8 : Schéma réactionnel de la mono-protection du diol.
Ainsi, différents catalyseurs ont été testés. Pour le catalyseur, les différentes
synthèses présentent dans la littérature utilisaient principalement deux catalyseurs, soit
l’acide D-10-camphorsulfonique (CSA) et l’acide p-toluènesulfonique (TsOH) (Friesen et
Vanderwal, 1996, Garcia, et al., 2007, He, et al., 2000). Comme les résultats étaient
similaires pour les deux catalyseurs, les réactions subséquentes ont tout de même était
faites avec le CSA de préférence au TsOH. Des ajustements ont également été faits par
rapport à la quantité de DIBAL-H qui était utilisé. Certains protocoles utilisaient 6,0
équivalents de DIBAL-H (He, et al., 2000), alors que d’autres en utilisaient 10,0
équivalents (Comin, et al., 2002, Friesen et Vanderwal, 1996, Takasu, et al., 1988). Ainsi,
pour des raisons d’économie de réactifs, différentes équivalences ont été utilisées afin de
déterminer la plus petite équivalence qui permettait l’ouverture complète du cycle. Il a donc
été déterminé que 4,0 équivalents de DIBAL-H serait suffisants.
La présence de l’alcool mono-protégé nous a été confirmée par le spectre MS
(Annexe, Figure A.7). Le pic moléculaire (M+) qui devrait se situer à m/z = 204 n’est pas

66
visible. Il est toutefois possible de voir le pic M+-1 à m/z = 203 (3.42). Celui-ci représente
la perte d’un atome d’hydrogène résultant de la fragmentation de l’acétal du groupement
MOM (Figure 3.9). Les fragments à m/z = 173 (3.43) et 45 (3.44) sont dû à la même
fragmentation (Budzikiewicz, et al., 1967).
H OCH3
H O OH
9H
OCH3
O OH
9
m/z = 203
H
H
OCH3
m/z = 45
H
H
O OH
m/z = 173
9
3.25
3.423.43
3.44
Figure 3.9 : Fragmentation de la partie acétal du diol mono-protégé.
Il est également possible de voir le fragment m/z = 159 correspondant à la perte de
l’acétal (Figure 3.10). Mais aussi, suite à la perte de se fragment, il est possible de voir la
perte d’une molécule d’eau à m/z = 141 caractéristique des spectres MS des alcools. En
FT-IR (Annexe, Figure A.8), nous remarquons la bande de 3473 cm-1 correspond à
l’élongation du lien O-H des hydroxyles. Une bande est également visible à 1029 cm-1. Elle
correspond à la fréquence d’élongation du lien C-O de l’alcool. Quant à la bande à 1105
cm-1, elle correspond à l’élongation asymétrique du lien C-O-C de l’acétal.
HO O O
m/z = 159
3.25
Figure 3.10 : Fragmentation de l’acétal.
Le spectre RMN 1H (Annexe, Figure A.9) révèle la présence d’un groupement
acétal par le déplacement chimique des protons à δ = 4,63 ppm, correspondant aux protons
sur le carbone 10, soit celui situé entre 2 atomes d’oxygène. Étant donné que la densité
électronique est plus faible près des atomes d’oxygène, les protons sont donc dans un

67
environnement qui est plus déblindé. C’est ce qui explique que son déplacement chimique
est plus élevé. Les déplacements chimiques se situant à δ = 3,51 et 3,34 ppm correspondent
aux protons se situant sur les carbones 9 et 11, soit ceux de chaque côté de l’acétal. La
fonction hydroxyle peut également être confirmée par les déplacements chimiques à δ =
5,03 et 3,62 ppm, correspondant respectivement au proton de l’hydroxyle et aux protons sur
le carbone 1, voisin de l’hydroxyle. Pour ce qui du spectre RMN 13C (Annexe, Figure
A.10), l’atome du carbone 10 est déblindé dû à la proximité de 2 atomes d’oxygènes. C’est
ce qui explique son déplacement chimique à δ = 93,3 ppm. Les déplacements chimiques à δ
= 67,9 et 63,1 ppm correspondent respectivement au carbone 9 et au carbone 1, soit le
carbone près de l’acétal et celui près de l’hydroxyle.
3.4.2 Oxydation de la fonction alcool
Cette réaction sert à obtenir l’aldéhyde, précurseur de la première réaction de
Wittig. Le PDC a été utilisé afin d’effectuer l’oxydation, car se produit permet d’obtenir le
niveau d’oxydation voulu en fonction du solvant utilisé pour les alcools primaires. Ainsi,
lorsque le DMF est utilisé comme solvant, le groupement alcool est oxydé en acide, alors
que si le CH2Cl2 est utilisé, un aldéhyde est obtenu (Corey et Schmidt, 1979, Piancatelli,
2001). Le CH2Cl2 a donc été utilisé comme solvant.
Ainsi, l’aldéhyde a bien été formé, sans poursuivre la réaction jusqu’à l’acide
carboxylique. La déprotection du groupement MOM n’est pas survenue, tel qu’attendu.
Seul l’aldéhyde protégé (3.26) était obtenu. Une colonne de chromatographie sur gel de
silice était nécessaire afin d’éliminer les produits de réduction du chrome. Toutefois le
produit n’a jamais pu être purifié correctement. Différentes conditions de solvants pour la
colonne de chromatographie ont été utilisées pour la purification de l’aldéhyde, mais cela
ne donnait jamais les résultats voulus.
Ainsi, la présence de l’aldéhyde a pu être confirmée grâce au spectre MS (Annexe,
Figure A.11), mais puisque nous ne sommes pas arrivés à le purifier, il n’a pas été possible

68
de faire d’autres analyses pour confirmer la structure du produit. En MS, le pic moléculaire
(M+), qui devrait se situer à m/z = 202, n’est pas présent. Il est toutefois possible de voir le
pic à M+-1 à m/z = 201 (3.45). Ce pic est caractéristique de la perte d’un atome
d’hydrogène résultant de la fragmentation de la fonction acétal du groupement MOM
(Figure 3.11). Cela confirme donc que la fonction MOM n’a pas subi de clivage oxydatif.
Les fragments à m/z = 171 (3.46) et 45 (3.47) proviennent également de cette
fragmentation.
H OCH3
H O
8H
OCH3
O
8
m/z = 201
H
H
OCH3
m/z = 45
H
H
O
m/z = 171O
H
O
HH
O
8
3.26
3.453.46
3.47
Figure 3.11 : Fragmentation du groupement acétal de l’aldéhyde
Le fragment à M+-1 est également caractéristique de la fonction aldéhyde, ainsi que
le fragment à m/z = 157. Ce dernier correspond à un fragment obtenu suite à un
réarrangement de McLafferty (Figure 3.12). Ce réarrangement est très fréquent lorsqu’il y
a présence d’un carbonyle (Silverstein, et al., 2007b).
O
H CH2
CH2
CHH
O O
5
HC
CH2
O O
5
3.26m/z = 157
3.48
Figure 3.12 : Fragmentation de McLafferty de l’aldéhyde

69
3.4.3 Seconde partie de la synthèse
Cette partie est identique à celle présentée dans les travaux précédents (Desmarais,
2013). Il s’agit de la synthèse du composé deutéré. Aucune modification n’a été faite dans
les protocoles et les mêmes résultats ont été obtenus. Cette partie comprend la
réduction/deutération du trans-pent-2-énoate de méthyle, la formation du nitrile à partir de
l’alcool et la réduction partielle du nitrile en aldéhyde deutéré. Étant donné que l’aldéhyde
deutéré obtenu (3.30) se polymérise très rapidement et qu’il ne nécessite aucune
purification, il ne sera synthétisé que lorsqu’il sera utilisé.
3.4.3.1 Réduction/deutération du trans-pent-2-énoate de méthyle
Le composé deutéré (3.28) était donc obtenu suite à une réduction/deutération de
l’ester correspondant (3.27). L’ester était mis en solution dans l’Et2O, puis refroidi à -78°C.
Le AlCl3 et le LiAlD4 y étaient ensuite ajoutés (Wang, et al., 2003). Le produit a bien été
obtenu avec un rendement de 90% après purification. Celle-ci était réalisés sur une colonne
de chromatographie sur gel de silice en utilisant comme solvants d’élution une solution de
50% Et2O / 50% hexane, suivi de 60% Et2O / 40% hexane, puis de 70% Et2O / 30%
hexane.
MeO
O
HO
DD
3.27 3.28
LiAlD4, AlCl3Et2O, -78°C, 2h
Figure 3.13 : Schéma réactionnel de la réduction/deutération du trans-pent-2-énoate de
méthyle.
3.4.3.2 Formation de la fonction nitrile
Suite à la deutération, la fonction alcool du composé 3.28 était transformée en nitrile
(3.29) afin de permettre d’allonger le composé d’un atome de carbone. L’alcool était donc
mis en solution dans l’Et2O avec la PPh3, puis le DIAD était ajouté. Finalement la
cyanhydrine d’acétone était ajoutée (Aesa, et al., 1995, Boukouvalas et Wang, 2008). Le
produit a été obtenu avec un rendement de 31% après une purification sur une colonne de

70
chromatographie par gel de silice en utilisant un gradient de solvant : 100% pentane, suivi
de 1% Et2O / 99% pentane et finalement 3% Et2O / 97% pentane.
HO
DD
NC
DD
3.28 3.29
, PPh3
DIAD, Et2O
CNHO
t.p., overnight
Figure 3.14 : Schéma réactionnel de la formation de la fonction nitrile via la réaction de
Mitsunobu-Wilk.
3.4.3.3 Réduction partielle du nitrile
Une réduction partielle de la fonction nitrile du composé 3.29 a été effectuée en
utilisant une solution de DIBAL-H dans le CH2Cl2 (Figure 3.15). Un rendement de 97% a
été obtenu, sans purification. Toutefois, comme l’aldéhyde deutéré qui est obtenu (3.30) se
polymérise rapidement, cette réaction ne sera réalisée que quelques heures avant la réaction
de Wittig subséquente.
NC
DD
O
DDH
3.29 3.30
DIBAL-H, CH2Cl2
-78°C, 1,5h
Figure 3.15 : Schéma réactionnel de la réduction partielle de la fonction nitrile.
3.4.4 Protection de l’alcool en éther de pyranyle
Comme il avait été expliqué, la protection de la fonction alcool avec un groupement
t-BDMS induisait de l’encombrement lors de la formation du sel d’ylure et empêchait la
formation de l’alcène. Pour cette raison, un éther de pyranyle (3.32) a été sélectionné afin
de protéger l’alcool du 3-bromo-propanol (3.31). Cette réaction provient également des
travaux précédents (Desmarais, 2013). Aucune modification n’a été faite au protocole
(Figure 3.16) et les mêmes résultats ont été obtenus avec un rendement de 87% après
purification. Ainsi, l’alcool était dilué dans l’Et2O et le 3,4-dihydro-2H-pyrane, ainsi que
du p-TsOH à titre de catalyseur. Après une nuit d’agitation à température pièce, le produit
était purifié sur colonne chromatographique sur gel de silice en utilisant une solution de 5%
AcOEt / 95% hexane comme solvant d’élution.

71
HO Br O BrO
3.31 3.32
O , TsOH
Et2O, t.p.
Figure 3.16 : Réaction de protection de l’alcool en éther de pyranyle.
3.4.5 Réaction de Wittig
La réaction de Wittig se fait en deux étapes. Soit la préparation d’un sel d’ylure
(3.33) suivie par la formation de l’alcène (3.35). La première étape consiste à mélanger
ensemble le bromure protégé et la triphénylphosphine (PPh3). La deuxième se fait en
mettant en présence le sel (3.33) avec une base et l’aldéhyde (3.26). Un alcène (3.35) est
obtenu comme produit final. Différents protocoles pour cette réaction ont été présentés dans
la littérature, en utilisant différentes températures de réactions et différents réactifs (Eynard,
et al., 1998, Patil et Mavers, 1996, Perlman, et al., 2000).
Dans notre cas, puisque que l’aldéhyde provenant du diol mono-protégé (3.26) est
très difficile à purifier et que nous ne sommes pas parvenu à le purifier, les différents tests
pour la réaction de Wittig ont été faits en utilisant un nonanal disponible commercialement
comme aldéhyde. Ces tests servaient à optimiser la réaction de Wittig, ainsi que la
purification du produit final.
3.4.5.1 Préparation du sel d’ylure
Le sel d’ylure a été formé en suivant le protocole proposé (Perlman, et al., 2000). Il
suffisait de mettre en présence le bromure protégé (3.32) avec la PPh3 liquéfié, sans
l’utilisation de solvant (Figure 3.17). Après environ 1h de réaction, un produit ayant une
texture se situant entre un sel et une gomme était alors obtenu. Le sel obtenu n’a pas été
isolé et il a été utilisé directement à l’étape suivante.

72
THPO BrPPh3
100°C, 1h THPO PPh3
Br
3.32 3.33
Figure 3.17 : Schéma réactionnel de la synthèse du sel d’ylure.
3.4.5.2 Formation de l’alcène
Le sel d’ylure obtenu était alors dissous dans le THF. Du DMSO était parfois ajouté
afin de faciliter la solubilisation du sel. Un bain à ultra-son était très souvent utilisé afin de
dissoudre le sel. Différents protocoles ont été essayés pour la formation de l’alcène (3.49).
Certains étaient en présence de gel de silice (Patil et Mavers, 1996). Cela devait permettre
de faciliter l’élimination de l’oxyde de triphénylphosphine (O=PPh3). Cette réaction ne
s’est pas montrée très efficace. Un autre protocole suggéraient l’utilisation de HMPA en
combinaison avec le THF comme solvant (Eynard, et al., 1998). Toutefois le protocole qui
s’est montré le plus prometteur est celui qui, après la formation de l’ylure (3.34),
refroidissait le mélange à -78°C afin d’ajouter l’aldéhyde (Figure 3.18). La réaction était
ensuite maintenue à cette température pour 1h supplémentaire avant de la laisser revenir à
la température ambiante (Perlman, et al., 2000).
THPO PPh3
BrTHPO
PPh3
3.33
n-BuLi/THF
3.34
OTHP
3.49
nonanal/THF
Figure 3.18 : Réaction de Wittig avec le nonanal.
La présence d’un alcène (3.49) a été confirmée par le spectre MS (Figure 3.19).
L’ion moléculaire qui devrait se trouver à m/z = 268 n’est pas visible. Il est toutefois
possible de voir les fragments m/z = 85 et 183 dû au clivage du groupement THP. Suite à la
perte de ce groupement, il est possible de voir les fragments m/z = 57 et 126, associés au
clivage d’un alcène.
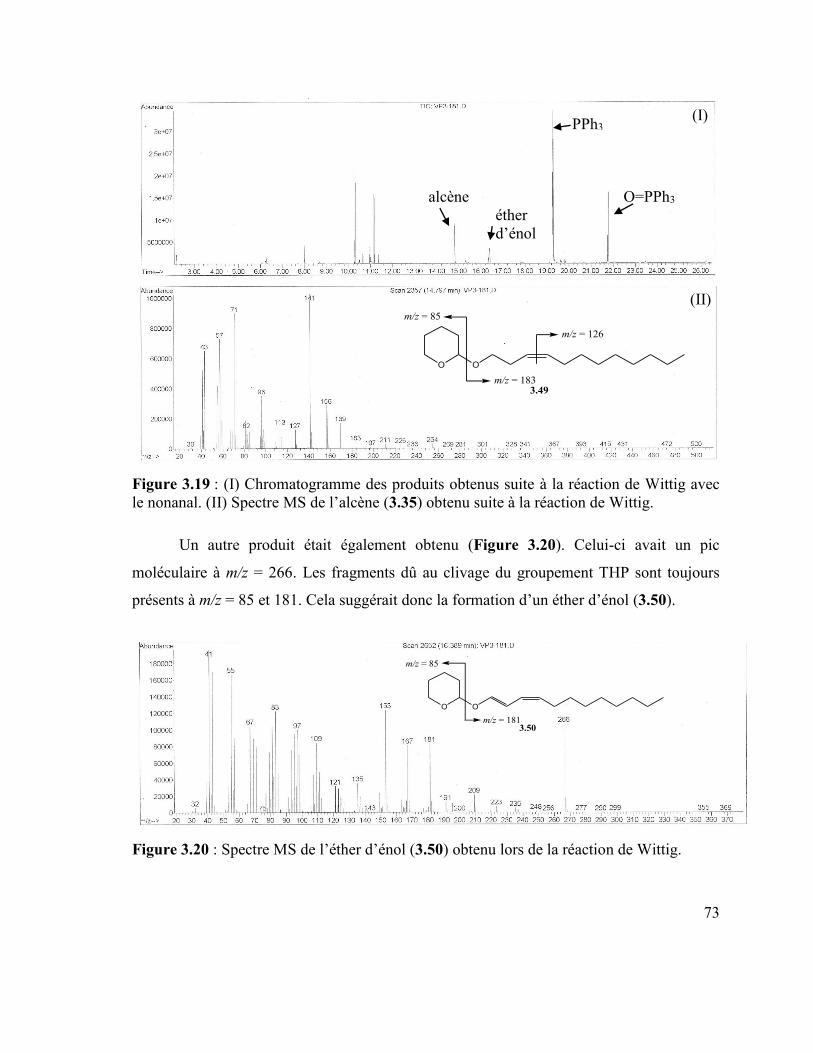
73
Figure 3.19 : (I) Chromatogramme des produits obtenus suite à la réaction de Wittig avec
le nonanal. (II) Spectre MS de l’alcène (3.35) obtenu suite à la réaction de Wittig.
Un autre produit était également obtenu (Figure 3.20). Celui-ci avait un pic
moléculaire à m/z = 266. Les fragments dû au clivage du groupement THP sont toujours
présents à m/z = 85 et 181. Cela suggérait donc la formation d’un éther d’énol (3.50).
Figure 3.20 : Spectre MS de l’éther d’énol (3.50) obtenu lors de la réaction de Wittig.
OO
m/z = 85
m/z = 183
m/z = 126
3.49
(I)
(II)
PPh3
O=PPh3 alcène
éther
d’énol
O O
m/z = 85
m/z = 1813.50

74
En supposant que ce produit serait isolé de préférence à l’alcène (3.49), il pourrait
également permettre la formation de l’AAL final. En effectuant une hydrolyse à l’aide du
DCl (Figure 3.21), cela nous permettrait d’effectuer l’insertion d’un atome de deutérium,
en plus de former un aldéhyde (3.51). Cet aldéhyde pourrait être utilisé pour la réaction de
Wittig subséquente. L’inconvénient de cette méthode est que l’atome de deutérium serait
alors sur le carbone 11 au lieu du 14 et il n’y aurait qu’un seul isotope au lieu de deux. Un
effet isotopique lors du chauffage de l’AAL pourrait alors être observé, comme il a déjà été
expliqué. Pour cette raison, nous n’avons pas retenu cette voie.
O O
3.50
DClO
H
D3.51
Figure 3.21 : Hydrolyse de l’éther d’énol (3.50) pour former l’aldéhyde (3.51).
3.5 Perspectives envisagées pour la synthèse de l’AAL marqué
D’autres approches peuvent également être envisagées pour la synthèse de l’AAL
marqué. La partie la plus difficile de la synthèse étant la formation des doubles liaisons de
configuration cis, différentes méthodes de synthèse peuvent donc être utilisées. Bien sûr, il
y a la réaction de Wittig, mais le couplage d’alcyne peut également être considéré.
3.5.1 Approche selon la réaction de Wittig
Cette approche est celle qui a été utilisée dans le présent projet. Bien que les essais
n’aient pas donné les résultats escomptés, le schéma de synthèse (Figure 3.7) utilisé semble
être l’approche la plus avantageuse. Dans l’éventualité où l’aldéhyde (3.26) serait purifié et
qu’une méthode efficace pour la synthèse et la purification de l’alcène (3.35) sera trouvé, il
s’agirait ensuite de déprotéger sélectivement l’éther de THP par rapport au groupement
MOM et d’effectuer une réaction de bromation afin d’obtenir le bromure (3.36) (Tanaka et

75
Oritani, 1997, Vankar et Shah, 1991). Un second sel d’ylure (3.37) serait alors formé afin
d’obtenir l’alcène (3.39) avec l’aldéhyde marqué (3.30). La déprotection de l’alcène
(Takasu, et al., 1988) suivi d’une oxydation devrait conduire à l’acide carboxylique final
(3.40).
D’autres alternatives à la réaction de Wittig, telles que la réaction de Peterson, de
McMurry et l’oléfination de Julia, peuvent également être utilisées. Toutefois, ces réactions
ne permettent pas d’obtenir la même sélectivité par rapport aux doubles liens cis que la
réaction de Wittig.
3.5.2 Approche selon un couplage d’alcynes
Une alternative à la présente approche serait d’utiliser un couplage d’alcynes via les
organocuprates, suivi d’une hydrogénation partielle grâce à un catalyseur de Lindlar, afin
de former les doubles liaisons. Cette voie de synthèse a déjà été envisagée, mais des
problèmes de volatilité des produits avaient été rencontrés. Une autre voie de synthèse
utilisant le couplage d’alcynes a également été proposée lors de travaux précédents
(Desmarais, 2013). Cette dernière se veut être une combinaison entre la réaction de Wittig
et le couplage d’alcynes (Figure 3.22).

76
HO
O
DD3.52
9 12 15
HO
D D
O
3.53
HO+
Br
DD3.54
3.55
HO+
Br
DD3.56 3.57
O
EtO
3.58
Figure 3.22 : Schéma rétrosynthétique I via le couplage d’alcynes de l’acide 14,14-
[2H,2H]-Z,Z,Z-octadéca-9,12,15-triénoïque (Desmarais, 2013).
Comme il est possible de le voir sur le schéma ci-dessus, l’acide ainsi formé
comporterait trois doubles liaisons cis et ne serait donc plus mono-trans. De plus, en
apposant les atomes marqueurs sur le carbone 14, cela pourrait permettre d’éviter les
problèmes de volatilités qui ont déjà été rencontrés. Il est a noté que les produits de départ
(3.54, 3.56 et 3.58) sont disponibles commercialement.
3.6 Conclusion
Une approche pour la synthèse de l’AAL marqué dans le but de vérifier le
mécanisme de formation des MCAG a été proposée. Cet acide devait être mono-trans et
avoir deux atomes de deutérium sur le carbone 14. Chaque étape de synthèse a été
optimisée afin de maximiser les rendements qui étaient obtenus. La réaction de Wittig a été
privilégiée pour la formation des doubles liens cis.

77
Une partie des protocoles utilisés pour la synthèse provenaient de travaux
précédents. Ces protocoles, qui permettaient la synthèse de l’aldéhyde marqué (3.30), n’ont
pas été modifiés. Pour ce qui est de la 1ere partie de la synthèse, le nonane-1,9-diol a été
mono-protégé par un acétal MOM. L’alcool mono-protégé (3.25) a bien été isolé et a pu
être caractérisé. L’oxydation de se produit pour former l’aldéhyde (3.26) a bien fonctionné,
mais le produit n’a pas pu être isolé, malgré l’essaie de différentes conditions de
purification. La structure de l’aldéhyde a toutefois pu être confirmée grâce aux analyses par
GC-MS. Puisque ce produit n’a pas été isolé, un aldéhyde commercial, soit le nonanal, a été
utilisé afin d’optimiser les conditions de réaction et de purification pour la réaction de
Wittig. L’alcène (3.49) a donc bien été obtenu, mais n’a toujours pas été isolé. Différentes
méthodes de purification ont été utilisées, dont une colonne de chromatographie, une
recristallisation et une chromatographie sur couche mince. La recristallisation a été utile
pour retirer la O=PPh3, mais l’alcène n’a jamais pu être isolé.
Les prochains travaux devraient se porter sur la purification de l’aldéhyde (3.26) et
de l’alcène (3.49), puis sur la continuation de la synthèse. S’il s’avère impossible de
purifier les différents produits, les diverses alternatives qui ont été proposées devraient être
envisagées pour la poursuite de la synthèse. Après la synthèse de l’AAL, des traitements
thermiques devront également être faits afin de déterminer le mécanisme de formation des
MCAG dans les huiles.


Chapitre 4
Discussion générale, conclusions et perspectives


81
4.1 Discussion générale
Lors du processus de friture, plusieurs dégradations se produisent au niveau des AG.
La température employée, les aliments qui y sont frits et la présence d’oxygène dans l’huile
sont les principaux facteurs qui induisent ces dégradations. Des AG libres, des mono- et
diglycérides, des polymères, des produits polaires, ainsi que des MCAG peuvent ainsi être
formés. Certains de ces produits étant néfastes pour la santé, il est donc important de
comprendre la formation de ces produits afin de pouvoir les inhiber ou, du moins, les
ralentir.
Du côté de l’oxydation, ses mécanismes sont déjà bien connus et des antioxydants
sont disponibles pour ralentir ce processus. Toutefois, les antioxydants commerciaux, tels
que le BHT, le BHA et le TBHQ, ont été reconnus pour avoir des effets nocifs sur la santé.
Pour cette raison, et également dû à la conscience des consommateurs pour leur santé, une
partie de ce projet consistait à trouver des antioxydants naturels à utiliser dans les huiles
destinées à la friture. Des acides phénoliques ont donc été choisis et dérivés en ester
d’hexyle et de dodécyle. Ces dérivations permettaient d’en diminuer la polarité afin de leur
procurer une meilleure solubilité dans l’huile. De plus, en allongeant la chaîne, nous avons
espéré diminuer la volatilité de l’ester, afin de leur permettre d’exprimer leur effet
antioxydant dans les huiles.
Trois composés ont donc été synthétisés et testés, avec différentes concentrations et
températures, dans l’huile de Canola sous des traitements thermo-oxydatifs, puis en
condition de friture. Le BHT a été utilisé comme témoin lors de tous ces tests. Des résultats
pour le vanillate d’hexyle, le vanillate de dodécyle et le coumarate d’hexyle ont donc été
obtenus. Pour ce qui est du premier, un effet antioxydant à pu être observé lorsqu’une
concentration de 1,0% était utilisé, mais il n’y avait plus aucun effet observable lorsque la
concentration était diminuée. Pour ce qui est du vanillate de dodécyle, nous avons espéré
obtenir un meilleur effet antioxydant qu’avec le vanillate d’hexyle, car en augmentant la
longueur de la chaîne, la volatilité du composé était sensé diminuer. Toutefois, aucun effet

82
antioxydant n’a pu être observé pour ce composé, même à une concentration de 1,0%. Cela
peut s’expliquer en partie par le fait que la concentration utilisée est en fait en masse par
masse, tel que prescrit par la législation, et non en mol. Ainsi, même si une même
concentration était utilisée, il y avait une moins grande quantité molaire de vanillate de
dodécyle que de vanillate d’hexyle.
Pour ce qui est du coumarate d’hexyle, un effet antioxydant a pu être observé à
toutes les concentrations utilisées et à différentes températures. Lorsque la température était
élevée, soit à 180°C, un effet antioxydant était visible jusqu’à 4h de chauffage. Lorsque le
traitement excédait ce temps, il n’y avait plus aucun effet visible. Toutefois, comme ces
traitements sont beaucoup plus intenses que ce que les huiles subiront lors d’un traitement
de friture, des tests supplémentaires en condition de friture ont été faits. Lors de ces tests, il
a été possible de voir qu’il n’y avait aucun changement dans le teneur en AL. Cela a pu être
expliqué par le fait que les frites qui ont été utilisées avaient subis un prétraitement dans
une huile de soya. Cette huile ayant une forte teneur en AL, a été relarguée dans l’huile de
friture, c’est ce qui explique que la teneur en cet AG ne changeait pas. Pour ce qui de
l’AAL, très peu de différence entre l’huile seule et l’huile supplémentée en coumarate
d’hexyle était visible. Il était toutefois possible de voir que, vers la fin de la période de
friture, soit après 6h, un léger effet antioxydant était visible. Cela laisse donc croire que si
la friture avait été faite sur une plus longue période, soit 2 ou 3 jours, un meilleur effet
antioxydant aurait pu être observé. Les mêmes tendances en friture ont pu être observées
pour le BHT.
Des tests en DSC ont été faits avec les trois composés afin de s’assurer qu’ils ne se
décomposaient pas dans la gamme de températures utilisées. Ces analyses ont montrées que
les produits étaient stables. De plus, nous avons pu déterminer le point de fusion du
vanillate de dodécyle, qui se trouve à environ 40°C.
D’un autre côté, les hautes températures utilisées en friture induisent des
modifications aux structures des AG, telle que la polymérisation ou la cyclisation. La

83
présence des MCAG dans les huiles a bien été reconnue et toutes les structures de MCAG
découlant de différents AG ont été caractérisées. Toutefois, leur mécanisme de formation
n’est encore qu’hypothétique à ce jour. L’AAL, étant l’AG avec le plus d’insaturations
dans les huiles végétales, est l’acide produisant le plus de MCAG. La deuxième partie de ce
projet visaient donc à synthétiser un AG marqué, soit l’AAL, afin de pouvoir suivre la
formation des MCAG par spectroscopie RMN 1H et MS.
En tenant compte des travaux cinétiques sur la formation des MCAG qui ont été fait
précédemment, nous en avons déduit le meilleur isomère de l’AAL que nous devions
synthétiser, ainsi que le meilleur emplacement des atomes marqueurs. Ainsi, un schéma de
synthèse de l’acide 14,14-[2H,2H]-Z,Z,E-octadéca-9,12,15-triénoïque a donc été élaboré en
s’inspirant de travaux faits précédemment. Ce plan de synthèse comprenait l’introduction
de deux atomes de deutérium via une réduction à l’aide d’un deutérure de lithium et
d’aluminium. La formation des deux doubles liens cis se faisait à l’aide de deux réactions
de Wittig.
La première partie de la synthèse impliquait la mono-protection d’un diol afin de
s’assurer qu’une seule des fonctions alcools ne participe à la réaction de Wittig. Nous avons
donc choisi de protéger l’un des hydroxyles à l’aide d’une fonction acétal MOM. Après
quelques ajustements du protocole de synthèse, et une purification, le produit mono-protégé
a bien été isolé. L’étape suivante consistait donc à oxyder la fonction alcool non-protégé
afin d’obtenir un aldéhyde. Le produit a bien été obtenu, mais il n’a jamais pu être isolé.
Différentes conditions de purification ont été essayées, mais l’aldéhyde n’était jamais pur.
Pour cette raison, un nonanal disponible commercialement a été utilisé afin de faire des
essais pour la réaction de Wittig subséquente.
Grâce aux travaux qui avaient déjà été fait sur cette synthèse, nous savions déjà que
les réactions de Wittig seraient problématiques. C’est donc pour cette raison que nous
avons décidé de faire des tests afin de déterminer les meilleures conditions de réaction et de
purification. Le 3-bromo-propanol protégé en éther de THP était utilisé avec le nonanal

84
commerciale. Ces essais nous ont donc permis d’obtenir un alcène, mais il n’a jamais pu
être isolé. Différentes méthodes, tels que les colonnes de chromatographie sur gel de silice,
les recristallisations et les chromatographies sur couche mince, ont été essayées. Les
recristallisations se sont montrées efficaces afin d’éliminer la O=PPh3, mais elles devaient
être combinées à une autre méthode afin d’isolé l’alcène.
Quant à la portion deutérée de l’AG, soit l’aldéhyde comportant les deux atomes de
deutérium, elle n’a pas été modifiée par rapport aux travaux précédents.
4.2 Conclusions et perspectives
Le projet portant sur le développement d’antioxydants pour les huiles de friture a
permis de testés trois dérivés d’acides phénoliques, soit le vanillate d’hexyle, le vanillate de
dodécyle et le coumarate d’hexyle. Les deux esters de l’acide vanillique n’ont pas
démontrés un assez bon effet antioxydant à 120°C pour poursuivre les traitements. Pour le
coumarate d’hexyle un bon effet antioxydant lors des traitements thermo-oxydatifs a été
observé. Pour cette raison, il a été testé en friture. Lors de ces traitements, aucune variation
n’a été observée pour l’acide linoléique, mais un début d’effet antioxydant était observable
pour l’acide α-linolénique aux environs de 6h de friture. La même tendance était observée
pour le BHT. Les prochains travaux sur ce projet devraient porter sur la continuation des
traitements de friture avec le coumarate d’hexyle et le BHT. Ces traitements devraient être
faits sur une plus grande période de temps, soit 2 à 3 jours. Cela pourrait permettre de
déterminer s’il y a vraiment un effet antioxydant qui peut être associé au coumarate
d’hexyle. De plus, d’autres dérivations d’acides phénoliques, tels que le coumarate de
dodécyle ou le cafféate d’hexyle, devraient également être testés comme antioxydant.
Pour le projet portant sur la synthèse de l’AAL marqué, un schéma de synthèse a été
élaboré et la synthèse a été amorcée. Les prochains travaux de ce projet devraient permettre
la continuation de la synthèse de l’AAL marqué. Si les réactions de Wittig causeraient
toujours des problèmes, des alternatives devraient être envisagées, tels l’oléfination de

85
Julia, la réaction de Peterson ou le couplage d’alcynes. Dans le cas où l’acide final serait
obtenu, des traitements thermiques devraient lui être appliqué afin de déterminer le
mécanisme de formation de MCAG.


87
Bibliographie
Aesa, M. C.; Baan, G.; Novak, L.; Szantay, C., Preparation of Unsaturated Nitriles by the
Modification of Mitsunobu-Wilk Procedure. Synthetic Communications 1995, 25,
(10), 1545-1550.
Agence de la santé publique du Canada, Le fardeau économique de la maladie au Canada,
2000, from http://www.phac-aspc.gc.ca/cd-mc/cvd-mcv/mcv_femc-cvd_ebic-
fra.php.
Ahmad, K.; Strong, F. M., The Synthesis of Unsaturated Fatty Acids. J. Am. Chem. Soc.
1948, 70, (5), 1699-1700.
Aladedunye, F.; Przybylski, R., Degradation and Nutritional Quality Changes of Oil During
Frying. J. Am. Oil Chem. Soc. 2009, 86, (2), 149-156.
Antolovich, M.; Prenzler, P. D.; Patsalides, E.; McDonald, S.; Robards, K., Methods for
testing antioxidant activity. Analyst 2002, 127, (1), 183-198.
Barcelo-Coblijn, G.; Murphy, E. J., Alpha-linolenic acid and its conversion to longer chain
n-3 fatty acids: Benefits for human health and a role in maintaining tissue n-3 fatty
acid levels. Prog. Lipid Res. 2009, 48, (6), 355-374.
Belitz, H. D.; Grosch, W.; Schieberle, P., Edible Fats and Oils. In Food Chemistry, 4th ed.;
Springer Berlin Heidelberg, 2009a, pp 640-669.
Belitz, H. D.; Grosch, W.; Schieberle, P., Lipids. In Food Chemistry, 4th ed.; Springer
Berlin Heidelberg, 2009b, pp 158-247.
Bergelson, L. D.; Shemyakin, M. M., Synthesis of Naturally Occurring Unsaturated Fatty
Acids by Sterically Controlled Carbonyl Olefination. Angew. Chem. Int. Ed. Engl.
1964, 3, (4), 250-260.
Billek, G., Health aspects of thermoxidized oils and fats. Eur. J. Lipid Sci. Technol. 2000,
102, (8-9), 587-593.
Boukouvalas, J.; Wang, J. X., Structure revision and synthesis of a novel labdane
diterpenoid from Zingiber ottensii. Org. Lett. 2008, 10, (16), 3397-3399.
Budzikiewicz, H.; Djerassi, C.; Williams, D. H., Mass Spectrometry of oganic compound.
Holden-Day, Inc.: San Francisco, Ca, 1967; p 690.

88
Burdge, G. C.; Calder, P. C., Conversion of alpha-linolenic acid to longer-chain
polyunsaturated fatty acids in human adults. Reprod. Nutr. Dev. 2005, 45, (5), 581-
597.
Choe, E.; Min, D. B., Mechanisms of Antioxidants in the Oxidation of Foods.
Comprehensive Reviews in Food Science and Food Safety 2009, 8, (4), 345-358.
Christie, W. W., Mass spectrometry of fatty acids with methylene-interrupted ene-yne
systems. Chem. Phys. Lipids 1998, 94, (1), 35-41.
Christie, W. W.; Dobson, G., Formation of cyclic fatty acids during the frying process. Eur.
J. Lipid Sci. Technol. 2000, 102, (8-9), 515-520.
Clavijo, E.; Menéndez, J. R.; Aroca, R., Vibrational and surface-enhanced Raman spectra
of vanillic acid. J. Raman Spectrosc. 2008, 39, (9), 1178-1182.
Comin, M. J.; Leitofuter, J.; Rodriguez, J. B., Enantioselective synthesis of (+)-neplanocin
F. Tetrahedron 2002, 58, (16), 3129-3136.
Corey, E. J.; Schmidt, G., Useful procedures for the oxidation of alcohols involving
pyridinium dichromate in approtic media. Tetrahedron Lett. 1979, 20, (5), 399-402.
Dana, D.; Saguy, I. S., Frying of Nutritious Foods: Obstacles and Feasibility. Food Sci.
Technol. Res. 2001, 7, (4), 265-279.
DasGupta, S. K.; Rice, D. M.; Griffin, R. G., Synthesis of isotopically labeled saturated
fatty acids. J. Lipid Res. 1982, 23, (1), 197-200.
Desmarais, A. Contribution à l'étude du mécanisme de formation des monomères cycliques
d'acide gras à partir d'acides gras oméga-3 et leur métabolomique chez le rat. Ph.D.
Thesis, Université Laval, Québec, 2013.
Destaillats, F.; Angers, P., On the mechanisms of cyclic and bicyclic fatty acid monomer
formation in heated edible oils. Eur. J. Lipid Sci. Technol. 2005, 107, (10), 767-772.
Dobarganes, M. C., Formation of New Compounds during Frying - General Observations.
In Frying Oils - Chemistry, Harwood, J. L.; Weselake, R. J., Eds. The AOCS Lipid
Library: 2009.
Dobson, G.; Christie, W. W.; Brechany, E. Y.; Sebedio, J. L.; Le Quere, J. L., Silver ion
chromatography and gas chromatography-mass spectrometry in the structural
analysis of cyclic dienoic acids formed in frying oils. Chem. Phys. Lipids 1995, 75,
(2), 171-182.

89
Dobson, G.; Christie, W. W.; Sebedio, J. L., Gas chromatographic properties of cyclic
dienoic fatty acids formed in heated linseed oil. J. Chromatogr. A 1996a, 723, (2),
349-354.
Dobson, G.; Christie, W. W.; Sebedio, J. L., Monocyclic saturated fatty acids formed from
oleic acid in heated sunflower oils. Chem. Phys. Lipids 1996b, 82, (2), 101-110.
Eynard, T.; Poullain, D.; Vatèle, J.-M.; Noël, J.-P.; Chardigny, J.-M.; Sébédio, J.-L.,
Synthesis of methyl (5Z,8Z,11Z,14Z,17Z)- and (5Z,8Z,11Z,14Z,17E)-[18-14C]
eicosapentaenoate. J. Label. Compd Radiopharm. 1998, 41, (5), 411-421.
Farag, R. S.; El-baroty, G. S.; Basuny, A. M., Safety evaluation of olive phenolic
compounds as natural antioxidants. Int. J. Food Sci. Nutr. 2003, 54, (3), 159-174.
Frankel, E. N., Lipid oxidation. Prog. Lipid Res. 1980, 19, (1-2), 1-22.
Frankel, E. N., Lipid oxidation: Mechanisms, products and biological significance. J. Am.
Oil Chem. Soc. 1984, 61, (12), 1908-1917.
Frankel, E. N.; Smith, L. M.; Hamblin, C. L.; Creveling, R. K.; Clifford, A. J., Occurrence
of cyclic fatty acid monomers in frying oils used for fast foods. J. Am. Oil Chem.
Soc. 1984, 61, (1), 87-90.
Friesen, R. W.; Vanderwal, C., Total Synthesis of (±)-Dihydrokawain-5-ol. Regioselective
Monoprotection of Vicinal Syn-Diols Derived from the Iodocyclofunctionalization
of α-Allenic Alcohols. J. Org. Chem. 1996, 61, (26), 9103-9110.
Garcia, D.; Foubelo, F.; Yus, M., Cyclic Acetals as Precursors of Substitutes Isochromans
and Naphthoxepines. Heterocycles 2007, 74, 507-519.
Gast, L. E.; Schneider, W.; Forest, C. A.; Cowan, J. C., Composition of methyl esters from
heat-bodied linseed oils. J. Am. Oil Chem. Soc. 1963, 40, (7), 287-289.
Gehring, L.; Haase, D.; Habben, K.; Kerkhoff, C.; Meyer, H. H.; Kaever, V., Synthesis of
an unsaturated fatty acid analogue (18-(4'-azido-2'-hydroxybenzoylamino)-oleic
acid) and its interaction with lysophosphatidylcholine: acyl-CoA-O-acyltransferase.
J. Lipid Res. 1998, 39, (5), 1118-1126.
Gensler, W. J.; Thomas, G. R., Synthesis of Unsaturated Fatty Acids: Linoleic Acid. J. Am.
Chem. Soc. 1951, 73, (10), 4601-4604.
Gutteridge, J. M. C., Biological origin of free radicals, and mechanisms of antioxidant
protection. Chemico-Biological Interactions 1994, 91, (2-3), 133-140.

90
He, L.; Byun, H.-S.; Bittman, R., A Stereocontrolled, Efficient Synthetic Route to
Bioactive Sphingolipids: Synthesis of Phytosphingosine and Phytoceramides from
Unsaturated Ester Precursors via Cyclic Sulfate Intermediates. J. Org. Chem. 2000,
65, (22), 7618-7626.
Jeffrey, B.; Weisinger, H.; Neuringer, M.; Mitchell, D., The role of docosahexaenoic acid
in retinal function. Lipids 2001, 36, (9), 859-871.
Kamal-Eldin, A.; Pokorny, J., Lipid Oxidation Products and Methods Used for their
Analysis. In Analysis of Lipid Oxidation, Kamal-Eldin, A.; Pokorny, J., Eds. AOCS
Press: Urbana, Illinois, 2005.
Kochhar, S. P., Sesame, rice-bran and flaxseed oils. In Vegetable oils in food technology :
Composition, properties and uses, Gunstone, F. D., Ed. CRC Press: Boca Raton,
FL, 2002, pp 297-326.
Kris-Etherton, P. M.; Harris, W. S.; Appel, L. J., Fish consumption, fish oil, omega-3 fatty
acids, and cardiovascular disease. Circulation 2002, 106, (21), 2747-2757.
Lomanno, S. S.; Nawar, W. W., Effect of Heating Temperature and Time on the Volatile
Oxidative Decomposition of Linolenate. J. Food Sci. 1982, 47, (3), 744-746.
Madhavi, D. L.; Deshpande, S. S.; Salunkhe, D. K., Food Antioxidants : technological,
toxicological, and health perspectives. Marcel Dekker, Inc.: New York, 1996.
Maharvi, G. M.; Edwards, A. O.; Fauq, A. H., Chemical synthesis of deuterium-labeled and
unlabeled very long chain polyunsaturated fatty acids. Tetrahedron Letters 2010,
51, (49), 6426-6428.
Marquez-Ruiz, G.; Dobarganes, M. C., Nutritional and Physiological Effects of Used
Frying Oils and Fats. In Deep Frying, 2e ed.; AOCS Press: Urbana, 2007, pp 173-
203.
Martin, J. C.; Caselli, C.; Broquet, S.; Juaneda, P.; Nour, M.; Sebedio, J. L.; Bernard, A.,
Effect of cyclic fatty acid monomers on fat absorption and transport depends on
their positioning within the ingested triacylglycerols. J. Lipid Res. 1997, 38, (8),
1666-1679.
Martin, J. C.; Joffre, F.; Siess, M. H.; Vernevaut, M. F.; Collenot, P.; Genty, M.; Sebedio, J.
L., Cyclic fatty acid monomers from heated oil modify the activities of lipid
synthesizing and oxidizing enzymes in rat liver. J. Nutr. 2000, 130, (6), 1524-1530.
McClements, D. J.; Decker, E., Lipids. In Fennema's Food Chemistry, Fourth ed.;
Damodaran, S.; Parkin, K.; Fennema, O. R., Eds. CRC Press: New York, 2007.

91
Moore, G. G.; Foglia, T. A.; McGahan, T. J., Preparation of hindered esters by the
alkylation of carboxylate salts with simple alkyl halides. J. Org. Chem. 1979, 44,
(14), 2425-2429.
Morton, I. D.; Chidley, J. E., Methods and Equipement in Frying. In Frying in Food:
Principles, Changes, New Approaches, Varela, G.; Bender, A. E.; Morton, I. D.,
Eds. Ellis Harwood Ltd.: Chichester, 1988, pp 37-51.
Mossoba, M.; Yurawecz, M.; Roach, J. G.; Lin, H.; McDonald, R.; Flickinger, B.; Perkins,
E., Elucidation of cyclic fatty acid monomer structures. Cyclic and bicyclic ring
sizes and double bond position and configuration. J. Am. Oil Chem. Soc. 1995, 72,
(6), 721-727.
Mossoba, M. M.; Yurawecz, M. P.; Roach, J. A. G.; Lin, H. S.; McDonald, R. E.;
Flickinger, B. D.; Perkins, E. G., Rapid determination of double bond configuration
and position along the hydrocarbon chain in cyclic fatty acid monomers. Lipids
1994, 29, (12), 893-896.
Orthoefer, F. T.; List, G. R., Dynamics of Frying. In Deep Frying, 2e ed.; Erickson, M. D.,
Ed. AOCS Press: Urbana, 2007a, pp 253-275.
Orthoefer, F. T.; List, G. R., Evaluation of used frying oil. In Deep Frying, 2e ed.; AOCS
Press: Urbana, 2007b, pp 329-342.
Orthoefer, F. T.; List, G. R., Initial Quality of Frying Oil. In Deep Frying, 2e ed.; AOCS
Press: Urbana, 2007c, pp 33-48.
Patil, V. J.; Mavers, U., Wittig reactions in the presence of silica gel. Tetrahedron Letters
1996, 37, (8), 1281-1284.
Perkins, E. G., Volatile Odor and Flavor Components Formed in Deep Frying. In Deep
Frying, 2e ed.; AOCS Press: Urbana, 2007, pp 51-56.
Perlman, N.; Livneh, M.; Albeck, A., Epoxidation of Peptidyl Olefin Isosteres.
Stereochemical Induction Effect of Chiral Centers at Four Adjacent C Positions.
Tetrahedron 2000, 56, (11), 1505-1516.
Pfeffer, P. E.; Silbert, L. S., Esterification by alkylation of carboxylate salts. Influence of
steric factors and other parameters on reaction rates. J. Org. Chem. 1976, 41, (8),
1373-1379.
Piancatelli, G., Pyridinium Dichromate. In Encyclopedia of Reagents for Organic
Synthesis, Paquette, L. A., Ed. John Wiley & Sons, Ltd: 2001.

92
Pryor, W. A., Introduction to free radical chemistry. Prentice-Hall: 1966; p 110.
Ruxton, C. H. S.; Reed, S. C.; Simpson, M. J. A.; Millington, K. J., The health benefits of
omega-3 polyunsaturated fatty acids: a review of the evidence. J. Hum. Nutr. Dietet.
2004, 17, (5), 449-459.
SanGiovanni, J. P.; Chew, E. Y., The role of omega-3 long-chain polyunsaturated fatty
acids in health and disease of the retina. Prog. Retin. Eye Res. 2005, 24, (1), 87-138.
Santé Canada, Liste des additifs alimentaires autorisés, 2013, from http://www.hc-
sc.gc.ca/fn-an/securit/addit/list/11-preserv-conserv-fra.php.
Schaich, K. M., Lipid Oxidation : Theoretical Aspects. In Bailey's Industrial Oil and Fat
Products, Vol. 1, 6th ed.; Shahidi, F., Ed. John Wiley and Sons, Inc.: Hoboken,
New Jersey, 2005, pp 269-355.
Scrimgeour, C., Chemistry of Fatty Acids. In Bailey's Industrial Oil and Fats Products,
Vol. 1, 6th ed.; Shahidi, F., Ed. John Wiley and Sons, Inc.: 2005, pp 491-512.
Sébédio, J. L.; Chardigny, J. M.; Malpuech-Brugere, C., Physiological Effects of trans and
Cyclic Fatty Acids. In Deep Frying, 2e ed.; AOCS Press: Urbana, 2007, pp 205-
228.
Sébédio, J. L.; Grandgirard, A., Cyclic fatty acids: Natural sources, formation during heat
treatment, synthesis and biological properties. Prog. Lipid Res. 1989, 28, (4), 303-
336.
Sébédio, J. L.; Juaneda, P., Isomeric and Cyclic Fatty Acids as a Result of Frying. In Deep
Frying, 2e ed.; AOCS Press: Urbana, 2007, pp 57-86.
Sébédio, J. L.; LeQuéré, J. L.; Semon, E.; Morin, O.; Prevost, J.; Grandgirard, A., Heat
treatment of vegetable oils. II. GC-MS and GC-FTIR spectra of some isolated
cyclic fatty acid monomers. J. Am. Oil Chem. Soc. 1987a, 64, (9), 1324-1333.
Sébédio, J. L.; Prevost, J.; Grandgirard, A., Heat treatment of vegetable oils I. Isolation of
the cyclic fatty acid monomers from heated sunflower and linseed oils. J. Am. Oil
Chem. Soc. 1987b, 64, (7), 1026-1032.
Sgoutas, D. S.; Kummerow, F. A., Chemical Synthesis of Tritium-labeled Linoleic Acid.
Biochemistry 1964, 3, (3), 406-411.
Sgoutas, D. S.; Sanders, H.; Yang, E. M., Tritioboration and synthesis of tritium-labeled
polyunsaturated fatty acids. J. Lipid Res. 1969, 10, (6), 642-645.

93
Shahidi, F.; Zhong, Y., Antioxydants: Regulatory Status. In Bailey's Industrial Oil and Fats
Products, Vol. 1, 6th ed.; Shahidi, F., Ed. John Wiley and Sons, Inc.: 2005, pp 491-
512.
Shahidi, F.; Zhong, Y., Novel antioxidants in food quality preservation and health
promotion. Eur. J. Lipid Sci. Technol. 2010, 112, (9), 930-940.
Silverstein, R. M.; Webster, F. X.; Kiemle, D. J., Infrared Spectrometry. In Spectrometric
identification of organic compounds, Silverstein, Ed. 2007a.
Silverstein, R. M.; Webster, F. X.; Kiemle, D. J., Mass Spectrometry. In Spectrometric
identification of organic compounds, Silverstein, Ed. 2007b.
Sonnet, P. E., Olefin inversion. Tetrahedron 1980, 36, (5), 557-604.
Statistique Canada, Mortalité : liste sommaire des causes, 2009, from
http://www.statcan.gc.ca/pub/84f0209x/2009000/t001-fra.htm.
Stoffel, W., Chemical synthesis of H3- and 1-C14-labeled polyunsaturated fatty acids. J.
Am. Oil Chem. Soc. 1965, 42, (7), 583-587.
Takasu, M.; Naruse, Y.; Yamamoto, H., A convenient procedure for the regioselective
monoprotection of 1,n-diols. Tetrahedron Lett. 1988, 29, (16), 1947-1950.
Tanaka, A.; Oritani, T., A mild and efficient method for converting alcohols and
tetrahydropyranyl ethers to bromides with inversion of configuration. Tetrahedron
Letters 1997, 38, (11), 1955-1956.
Vancassel, S., Oméga-3 et neurotransmission cérébrale. Ol. Corps Gras Lipides 2004, 11,
(1), 58-65.
Vankar, Y. D.; Shah, K., A facile conversion of tetrahydropyranyl ethers to the
corresponding bromides and iodides. Tetrahedron Letters 1991, 32, (8), 1081-1084.
Wanasundara, P. K. J. P. D.; Shahidi, F., Antioxidants: Science, Technology, and
Applications. In Bailey's Industrial Oil and Fat Products, Vol. 1, 6th ed.; Shahidi,
F., Ed. John Wiley and Sons, Inc.: Hoboken, New Jersey, 2005, pp 431-489.
Wang, J. X.; Li, Y.; Zhang, C. X., The Efficient Synthesis of the Optically Active β-
Hydroxyl-γ-Butyrolactone Derivatives. J. Chin. Chem. Soc. 2003, 50, (6), 1183-
1187.

94
Wuts, P. G. M.; Greene, T. W., Protection for the Hydroxyl Group, Including 1,2- and 1,3-
Diols In Greene's protective groups in organic synthesis, 4th ed.; Hoboken, N. J.,
Ed. John Wiley and sons, Inc.: Hoboken, New Jersey, 2007, pp 16-366.
Yanishlieva, N. V.; Marinova, E. M., Stabilisation of edible oils with natural antioxidants.
Eur. J. Lipid Sci. Technol. 2001, 103, (11), 752-767.

Partie expérimentale


97
Solvants et réactifs
Pour réaliser les synthèses, des solvants de grade ACS ont été utilisés et achetés
chez Sigma-Aldrich (Milwaukee, WI, USA) ou chez Fisher Scientific (Ottawa, ON,
Canada). Tous les solvants utilisés pour les synthèses étaient préalablement séchés, comme
décrit ci-dessous, et conservés sur tamis moléculaire activé jusqu’à leur utilisation afin
d’éviter toutes réactions secondaires dues à la présence de traces d’humidité.
L’éther diéthylique (Et2O) a été conservé sur des tournures de sodium
métalliques fraichement préparées jusqu’à son utilisation.
Le tétrahydrofurane (THF) a été séché sur un mélange de benzophénone et de
tournures de sodium métalliques fraichement préparées. Le THF a été récupéré
par distillation (bp :66°C).
Le dichlorométhane (CH2Cl2), l’acétone, le DMSO et l’hexane ont été conservés
sur tamis moléculaire activé sans séchage.
Tous les réactifs utilisés pour les synthèses ont été achetés chez Sigma-Aldrich
(Milwaukee, WI, USA), sauf pour le deutérure de lithium et d’aluminium (LiAlD4) qui a
été acheté chez C/D/N Isotopes Inc. (Pointe-Claire, QC, Canada) et le trans-pent-2-énoate
de méthyle qui a été acheté chez Fisher Scientific (Ottawa, ON, Canada). Certains réactifs
ont été purifiés comme décrit ci-dessous. Les autres réactifs ont été utilisés tels quels pour
les différentes étapes de la synthèse.
La triphénylphosphine (PPh3) a été recristallisée dans l’EtOH anhydre froide,
puis les cristaux blancs ont été récupérés grâce à une filtration sous vide. Les
cristaux sont ensuite séchés toute une nuit dans un four à vide à une température
d’environ 40°C afin de retirer l’excédent d’EtOH. Cette purification a pour but
d’éliminer les traces d’oxyde de PPh3.

98
Purification des composés
Suite à leur synthèse, toutes les structures ont été purifiées par colonne sur gel de
silice. Une silice de taille de pores 60Å et de taille de particules de 40-60µm (230-
400mesh) a été utilisée. La silice a été achetée chez SiliCycle (Québec, QC, Canada). Pour
chacun des composés, les conditions d’élution qui ont été utilisées sont décrites dans leur
procédure expérimentale respective. Les purifications ont été suivies par chromatographie
sur couche mince (CCM) (20cm x 20cm, gel de silice 60 avec un révélateur UV F254) qui
ont été achetées chez SiliCycle (Québec, QC, Canada). Lorsque les rayons UV ne
parvenaient pas à révéler les CCM, d’autres révélateurs ont été utilisés. Des cristaux d’iode,
une solution de 2’,7’-dichlorofluorescéine diluée à 0,2% dans l’EtOH anhydre ou une
solution de molybdate d’ammonium sont les autres révélateurs à avoir été utilisés.
Appareils d’analyse pour la synthèse
Toutes les structures ont été caractérisées par la RMN 1H et 13C, par la spectroscopie
IR à transformée de Fourier (FT-IR) et par la MS selon les conditions mentionnées dans
cette section.
RMN 1H et 13C : Un appareil Bruker AC 300MHz a été utilisé. Les échantillons
étaient dilués dans le CDCl3. Les spectres ont été traités avec le logiciel
SpinWorks, version 2.5.5 (University of Manitba, MN, Canada).
FT-IR : Les échantillons ont été analysés à l’aide d’un spectromètre infrarouge à
transformée de Fourier (Nicolet™ 6700, Thermo Fisher Scientific, Madison, WI,
USA) par réflexion totale interne (ATR) sur un cristal de diamant. Le traitement
des données spectrales a été réalisé avec le logiciel OMNIC™ Software.
MS :Les échantillons ont été analysés à l’aide d’un appareil de GC de modèle HP
6890 de série I (Hewlett-Parckard) couplé à un spectromètre de masse de modèle
5973N (Agilent, Palo Alto, CA, USE) muni d’une source d’ionisation à impact
électronique (IE) et d’un quadrupôle. Le voltage de la source a été fixé à 70 eV et
sa température à 250°C. La séparation par GC a été réalisée avec une colonne

99
ZB-XLB (30m x 0,25µm x 0,25 µm, dont le film intérieur de la phase
stationnaire est composé de 5% Polysilarylène – 95% Polydiméthylsiloxane co
polymère). Les échantillons ont été injectés en mode split (ratio 1 : 50), la
température de l’injecteur a été fixée à 250°C et celle de l’interface à 170°C.
L’hélium (He) a été utilisé comme gaz vecteur avec un flux constant de
1,2mL/min et une pression de 9,34 psi. Pour la séparation par GC, un programme
de gradient de température a été utilisé (Tableau PE.1).
Tableau PE.1 : Programme de température utilisé en chromatographie en phase gazeuse
pour la caractérisation des composés de synthèse.
Température
initiale
(°C)
Gradient
(°C/min)
Température
finale
(°C)
Maintien de la
température
(minutes)
40 0 40 5
40 20 70 2
70 30 190 5
190 30 300 13

100
Chapitre 2 : Antioxydants pour les huiles de friture
Synthèse du vanillate d’hexyle
HO
O
O
O
L’acide vanillique (1,0 équiv.) est dissout dans de l’EtOH (0,4M). Cette solution est
titrée avec une solution de KOH 0,15M en utilisant de la phénolphtaléine comme
indicateur. Le solvant est ensuite évaporé sous pression réduite en chauffant le bain-marie à
85°C. Le sel obtenu est ensuite séché au four à vide à 50°C pendant 24h. Le sel (1,0 équiv.)
est dissout dans le DMSO (0,1M). Un peu d’EtOH peut être ajouté si nécessaire pour
faciliter la dissolution. L’iodo-hexane (1,0 équiv.) est ajouté au mélange réactionnel. Le
mélange est ensuite chauffé à reflux (85°C). Après 16h d’agitation, le ballon est refroidi à
température ambiante. De l’eau, ainsi qu’une solution de HCl 10% sont ajoutés afin
d’acidifier le mélange réactionnel. Le contenu du ballon est ensuite transvasé dans une
ampoule à décanter. La phase organique est extraite avec 3 portions d’éther de pétrole, puis
elle est lavée avec 4 portions d’une solution de HCl 10%, 4 portions d’eau, ainsi qu’une
portion d’une solution de Na2S2O3. La phase organique a été séchée sur du sulfate de
magnésium (MgSO4) anhydride et le solvant a été évaporé sous pression réduite. Le produit
obtenu a ensuite été purifié sur une colonne de chromatographie par gel de silice en
utilisant une solution de 10% AcOEt / 90% hexane comme solvant d’élution. Une fois le
solvant évaporé sous pression réduite, un liquide incolore a été récupéré avec un rendement
de 15% après la purification.
C14H20O4
Liquide incolore
IR : 3397, 2955, 2930, 2858, 1708, 1595, 1512, 1428, 1278, 1215,
νmax en cm-1 1100, 1030, 763.

101
MS (m/z) : 252 (18%, M+), 168 (100%), 151 (39%), 123 (6%), 108 (3%),
93 (2%), 65 (2%), 55 (2%), 41 (3%).
Synthèse du vanillate de dodécyle
HO
O
O
O
À une solution d’acide vanillique (1,0 équiv.) et d’acétone (0,13M), le KHCO3
(1,0 équiv.) est ajouté. Après 15 minutes d’agitation, l’iodo-dodécane (0,5 équiv.) est ajouté
au mélange réactionnel. Le mélange est ensuite porter à reflux (~70°C). Après 3 jours
d’agitation, le mélange réactionnel est refroidi à la température ambiante. Le mélange est
ensuite acidifié avec une solution de HCl 10%. Le mélange est par la suite transvasé dans
une ampoule à décanter. La phase organique est extraite avec 3 portions de CH2Cl2, puis
lavée avec une portion d’une solution de NaHCO3 saturée et une portion d’eau. La phase
organique a été séchée sur MgSO4 anhydride et le solvant a été évaporé sous pression
réduite. Le produit obtenu a ensuite été purifié sur une colonne de chromatographie sur gel
de silice en utilisant une solution de 10% AcOEt / 90% hexane comme solvant d’élution.
Le composé a été récolté avec un rendement de 11% après la purification sous la forme
d’un sel blanchâtre.
C20H32O4
Sel blanc
IR : 3327, 2922, 2848, 1685, 1595, 1514, 1427, 1281, 1229, 1196,
νmax en cm-1 1113, 1036, 965, 880, 765, 669.
MS (m/z) : 336 (15%, M+), 168 (100%), 151 (28%), 123 (5%), 108 (2%),
93 (2%), 83 (1%), 69 (3%), 55 (5%), 41 (5%).

102
Synthèse du p-coumarate d’hexyle
O
HO
O
À une solution d’acide p-coumarique (1,0 équiv.) et d’acétone (0,13M), le KHCO3
(1,0 équiv.) est ajouté. Après 15 minutes d’agitation, l’iodo-hexane (0,5 équiv.) est ajouté
au mélange réactionnel. Le mélange est ensuite porté à reflux (~70°C). Après 7 jours
d’agitation, le mélange réactionnel est refroidi à la température ambiante. Le mélange est
ensuite acidifié avec une solution de HCl 10%. Le mélange est par la suite transvasé dans
une ampoule à décanter. La phase organique est extraite avec 3 portions de CH2Cl2, puis
lavée avec une portion d’une solution de NaHCO3 saturée et une portion d’eau. La phase
organique a été séchée sur MgSO4 anhydride et le solvant a été évaporé sous pression
réduite. Lors de l’évaporation, le bain-marie était à une température supérieure à 60°C.
Cela permettait d’évaporer les résidus d’iodo-hexane restant dans le mélange. Une colonne
de chromatographie sur gel de silice a été préparée en utilisant un gradient de solvant
d’élution : 35% AcOEt / 65% hexane, suivi de 50% AcOEt / 50% hexane, puis de 65%
AcOEt / 35% hexane. Le composé a été récolté avec un rendement de 6% après la
purification sous la forme d’un liquide incolore.
C15H20O3
Liquide incolore
IR : 3371, 2953, 2924, 2855, 1669, 1601, 1584, 1514, 1306, 1272,
νmax en cm-1 1209, 1167, 1018, 979, 832, 637.
MS (m/z) : 248 (60%, M+), 164 (100%), 147 (91%), 119 (43%), 107 (23%),
91 (39%), 65 (23%).

103
Chapitre 3 : Approche à la synthèse de l’acide α-linolénique marqué
Monoprotection du nonane-1,9-diol
O OHO
Dans un ballon anhydre et sous atmosphère d’azote, le nonane-1,9-diol (1,0 équiv.)
est dissout dans le CH2Cl2 (0,07M). Le catalyseur (acide D-10-camphorsulfonique) (0,2
équiv.) et l’orthoformiate de triméthyle (2,0 équiv.) sont ajoutés dans le mélange
réactionnel. L’agitation est ensuite maintenue pendant 16 heures. Le ballon est ensuite
refroidi à -78°C (bain glace sèche / acétone) et une solution de DIBAL-H (1M dans le
CH2Cl2) (4,0 équiv.) est ensuite ajouté goutte-à-goutte. L’agitation est ensuite maintenue à
cette température pendant 1 heure supplémentaire. Le bain de glace est par la suite retiré
pour que la température revienne à la température ambiante. Dans une ampoule à décanter,
une solution de NaOH 2N y est versée. Le mélange réactionnel est ensuite transvasé dans
l’ampoule à décanter, puis la phase organique est extraite 3 fois avec de l’Et2O. La phase
organique est ensuite lavée avec une portion d’eau et une portion d’eau saturée en NaCl. La
phase organique a été séchée sur du sulfate de magnésium (MgSO4) anhydride et le solvant
a été évaporé sous pression réduite. Le produit obtenu a ensuite été purifié sur une colonne
de chromatographie par gel de silice en utilisant un gradient de solvant : 50% Et2O / 50%
hexane, suivi de 60% Et2O / 40% hexane et finalement 70% Et2O / 30% hexane. Une fois le
solvant évaporé sous pression réduite, un liquide incolore a été récupérée avec un
rendement de 37%.
C11H24O3
Liquide incolore
IR : 3473, 2926, 2854, 1683, 1595, 1517, 1432, 1281, 1202, 1105,
νmax en cm-1 1029, 918, 765.
O OHO
1
2
3
4
5
6
7
8
9 10 11

104
RMN 1H (CDCl3) : 5,03 (1H, s, OH)
δ en ppm 4,63 (3H, d, CH3-10)
3,62 (2H, t, CH2-1)
3,51 (2H, t, CH2-9)
3,34 (2H, d, CH2-11)
1,70 (2H, s, CH2-8)
1,55 (2H, m, CH2-2)
1,31 (10H, m, CH2-3,4,5,6,7)
RMN 13C (CDCl3) : 95,3 (C-10), 67,9 (C-9), 63,1 (C-1), 51,1 (C-11), 32,8 (C-2),
δ en ppm 29,7 (C-8), 29,5 (C4-5-6), 26,2 (C-7), 25,7 (C-3).
MS (m/z) : 203 (1%, M+-1), 189 (3%), 173 (6%), 159 (7%), 141 (21%),
123 (25%), 111 (19%), 97 (44%), 81 (58%), 69 (67%),
55 (65%), 45 (100%), 41 (50%).
Oxidation du diol monoprotégé : synthèse du synthon (3.26)
H O
O
O
Dans un ballon anhydre et sous atmosphère inerte, le diol monoprotégé (1,0 équiv.)
et le PDC (1,5 équiv.) sont dissouts dans le CH2Cl2 (2,5mL par gramme de PDC).
L’agitation est ensuite maintenue pendant 16 heures. De l’Et2O est ensuite versé dans le
mélange réactionnel. Le contenu du ballon est ensuite filtré dans une ampoule à décanter.
La phase organique est ensuite lavée avec 4 portions d’eau. La phase organique a été séchée
sur du sulfate de magnésium (MgSO4) anhydride et le solvant a été évaporé sous pression
réduite. Un rendement de 60% était obtenu avant purification.
C11H22O3
Liquide brunâtre

105
MS (m/z) : 201 (0,3%, M+-1), 187 (0,3%), 171 (3%), 157 (11%), 139 (12%),
123 (21%), 111 (14%), 97 (37%), 81 (47%), 71 (22%), 67
(34%), 55 (48%), 45 (100%), 41 (37%).
Réduction/deutération du trans-pent-2-énoate de méthyle
HO
DD
Dans un ballon anhydre et sous atmosphère inerte, l’ester (1,0 équiv.) était dissout
dans l’Et2O (0,1M). Le mélange était ensuite refroidi à -78°C (bain glace sèche / acétone).
Le AlCl3 (1,1 équiv.), puis le LiAlD4 (1,1 équiv.) étaient ajoutés par petite portion. Après
2h d’agitation et toujours à -78°C, de l’eau (10% du volume de l’Et2O) était ajoutée. Dans
une ampoule à décanter, une solution de HCl 10% était ajoutée, ainsi que de la glace
concassée (faire une couche d’environ 1cm). Le mélange réactionnel était versé dans
l’ampoule à décanter. La phase organique est ensuite extraite avec 2 portions d’Et2O, puis
lavée avec une portion d’eau et une portion de d’eau saturée en NaCl. La phase organique a
été séchée sur du sulfate de magnésium (MgSO4) anhydride et le solvant a été évaporé sous
pression réduite. Le produit obtenu a ensuite été purifié sur une colonne de
chromatographie sur gel de silice en utilisant comme solvant d’élution une solution de 50%
Et2O / 50% hexane, suivi de 60% Et2O / 40% hexane, puis de 70% Et2O / 30% hexane. Une
fois le solvant évaporé sous pression réduite, un rendement de 90% était obtenu.
Formation du nitrile
NC
DD
Dans un ballon anhydre et sous atmosphère inerte, l’alcool deutéré (1,0 équiv.) et la
PPh3 (1,5 équiv.) son dissouts dans l’Et2O (0,15M). Le mélange est ensuite refroidi à 0°C
(bain eau / glace). Le DIAD (1,5 équiv.) est ajouté goutte-à-goutte. Dans une ampoule à
additionner, la cyanhydrine d’acétone (1,5 équiv.) est diluée dans l’Et2O (0,2M), puis
ajoutée au mélange réactionnel. L’agitation est maintenue toute la nuit. Le mélange est

106
filtré puis rincer avec du pentane. Le solvant est ensuite évaporé sous pression réduite. Le
mélange est à nouveau filtré et le solvant évaporé. Le produit obtenu a ensuite été purifié
sur une colonne de chromatographie sur gel de silice en utilisant un gradient de solvant :
100% pentane, suivi de 1% Et2O / 99% pentane et finalement 3% Et2O / 97% pentane. Une
fois le solvant évaporé sous pression réduite, un liquide incolore a été récupéré avec un
rendement de 31%.
Réduction partielle du nitrile
H
D D
O
Dans un ballon anhydre et sous atmosphère inerte, le nitrile marqué (1,0 équiv.) est
dilué dans le CH2Cl2 (0,1M). Le mélange réactionnel est refroidi à -78°C (bain de glace
sèche / acétone). Une solution de DIBAL-H 1M dans le CH2Cl2 (1,1 équiv.) est ensuite
ajoutée goutte-à-goutte. Une agitation vigoureuse est ensuite maintenue pendant 90
minutes. Toujours à -78°C, une solution de HCl 10% (10% du nombre de mol de DIBAL-
H) est ajoutée dans le mélange. La réaction est ensuite revenue à la température ambiante
sur une période de 1h. Le contenu du ballon a été transvasé dans une ampoule à décanter
contenant une solution de HCl 10%. La phase organique a été extraite avec 2 portions de
CH2Cl2, puis lavée avec une portion d’eau et une portion de d’eau saturée en NaCl. La
phase organique a été séchée sur du sulfate de magnésium (MgSO4) anhydride et le solvant
a été évaporé sous pression réduite. Un rendement de 97% était obtenu sans purification.
Protection de l’alcool en éther de pyranyle
O O Br
Dans un ballon anhydre et sous atmosphère inerte, l’alcool (1,0 équiv.) est dilué
dans l’Et2O (0,05M). Le 3,4-dihydro-2H-pyrane (1,3 équiv.) est ajoutée lentement. Le
TsOH (0,03 équiv.) est ensuite ajoutée dans le mélange. L’agitation est maintenue toute la

107
nuit. Le mélange est dilué avec de l’Et2O, puis il est transvasé dans une ampoule à décanter.
La phase organique est lavée avec une portion de solution saturée en NaHCO3, puis avec
une portion d’eau. La phase organique a été séchée sur du sulfate de magnésium (MgSO4)
anhydride et le solvant a été évaporé sous pression réduite. Le produit obtenu a ensuite été
purifié sur une colonne de chromatographie sur gel de silice en utilisant une solution de 5%
AcOEt / 95% hexane comme solvant d’élution. Une fois le solvant évaporé sous pression
réduite, un liquide incolore a été récupéré avec un rendement de 87%.


Annexes

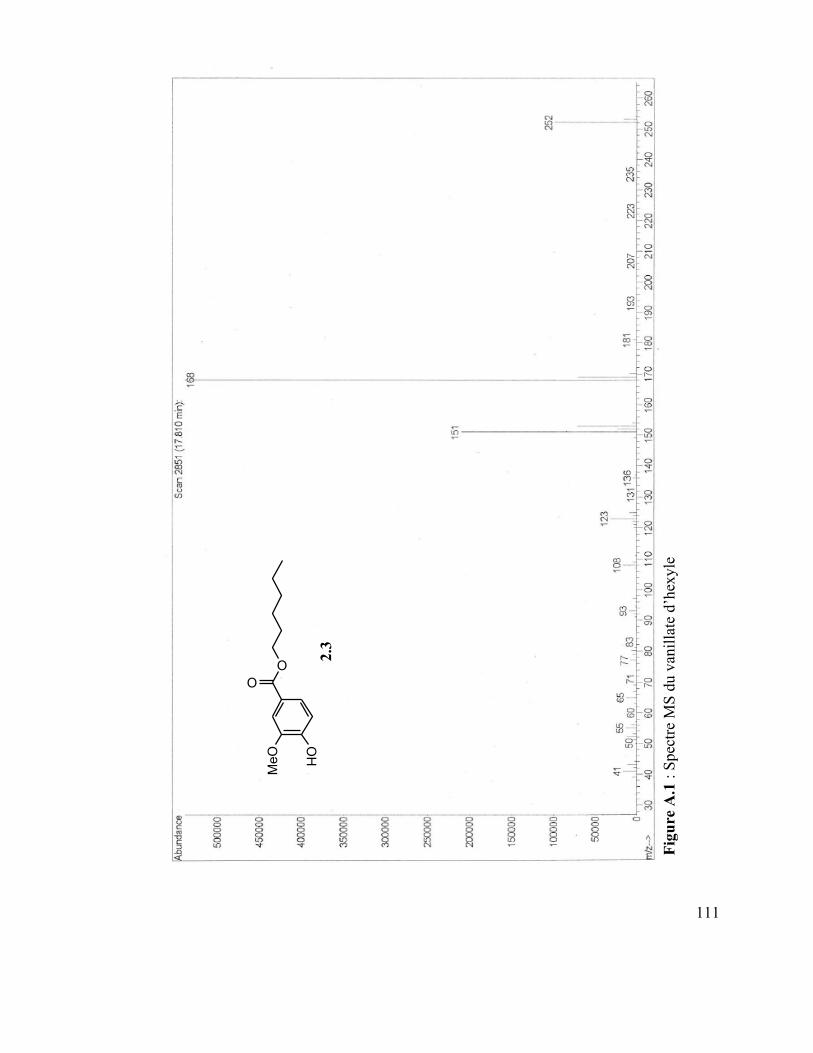
111
MeO
OH
O
O
2.3
Fig
ure
A.1
: S
pec
tre
MS
du v
anil
late
d’h
exyle
112
Fig
ure
A.2
: S
pec
tre
IR d
u v
anil
late
d’h
exyle

113
Fig
ure
A.3
: S
pec
tre
MS
du v
anil
late
de
dodéc
yle
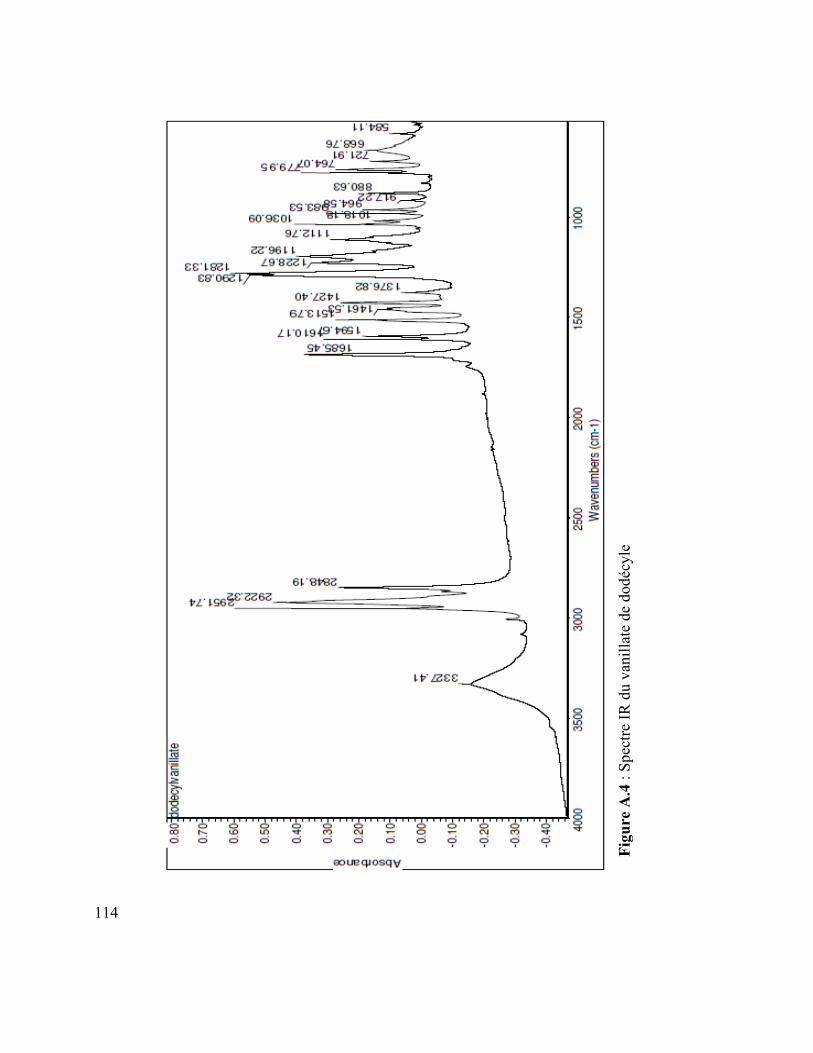
114
Fig
ure
A.4
: S
pec
tre
IR d
u v
anil
late
de
dodéc
yle
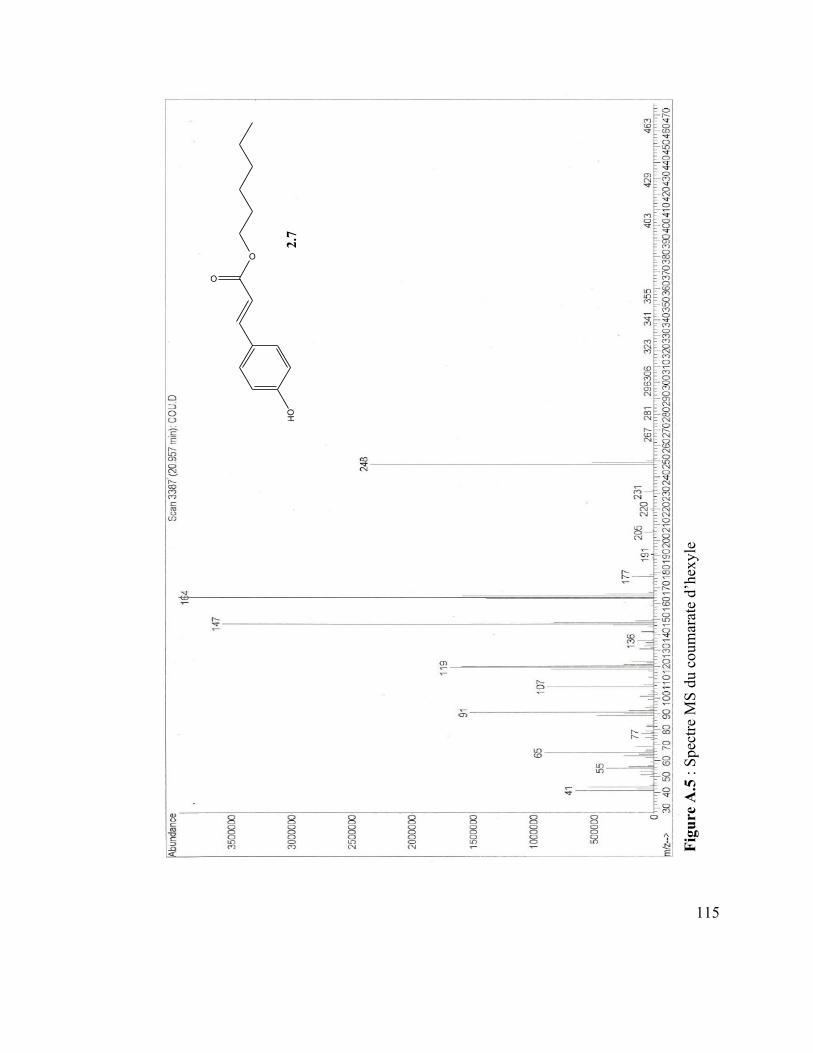
115
OH
O
O
2.7
Fig
ure
A.5
: S
pec
tre
MS
du c
oum
arat
e d
’hex
yle
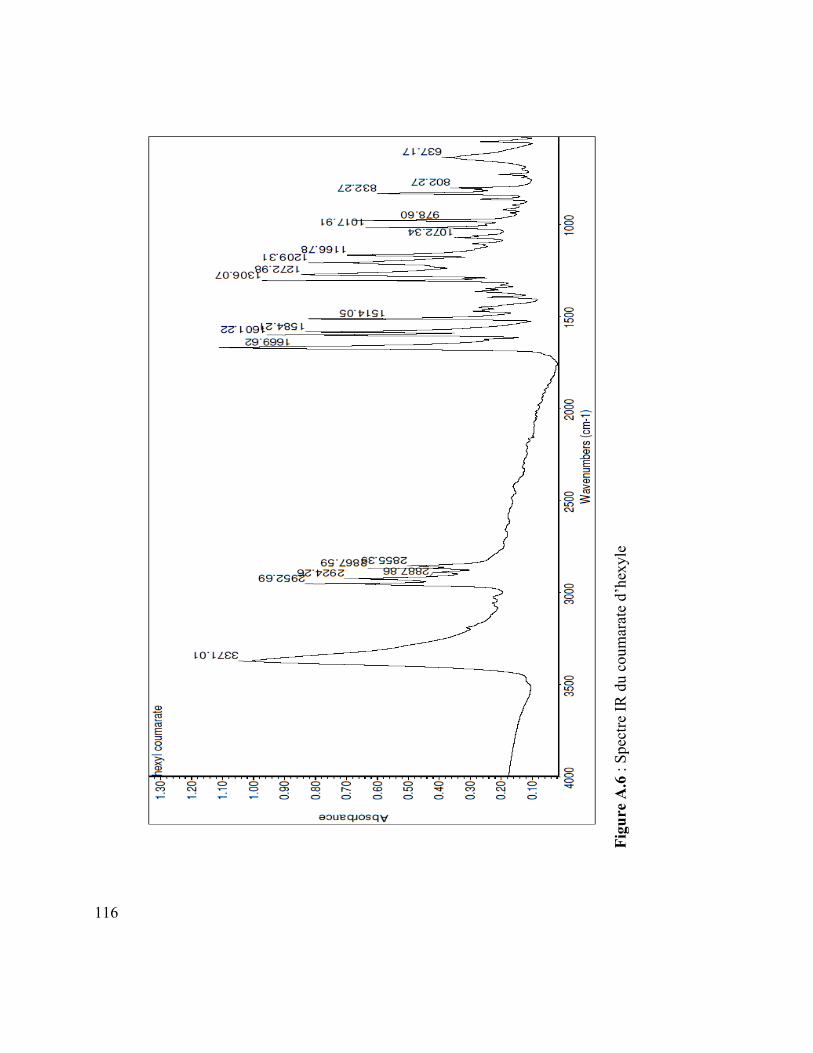
116
Fig
ure
A.6
: S
pec
tre
IR d
u c
oum
arat
e d
’hex
yle
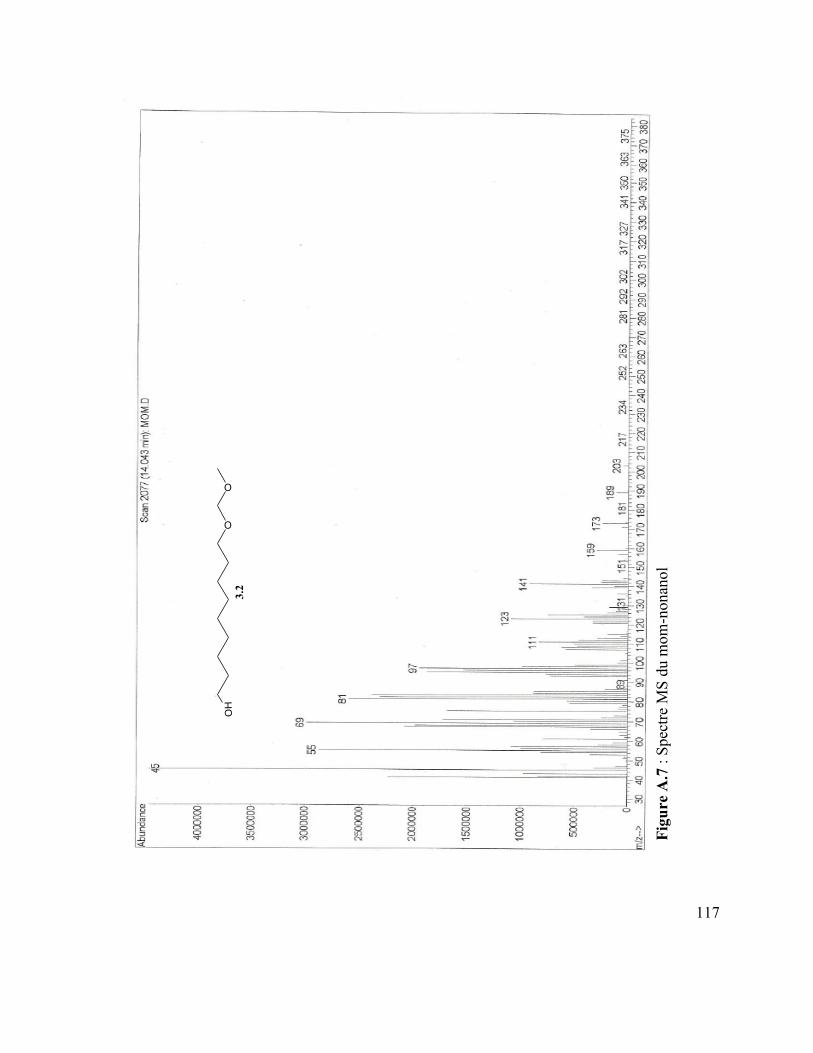
117
OH
O
O
3.2
Fig
ure
A.7
: S
pec
tre
MS
du m
om
-nonan
ol
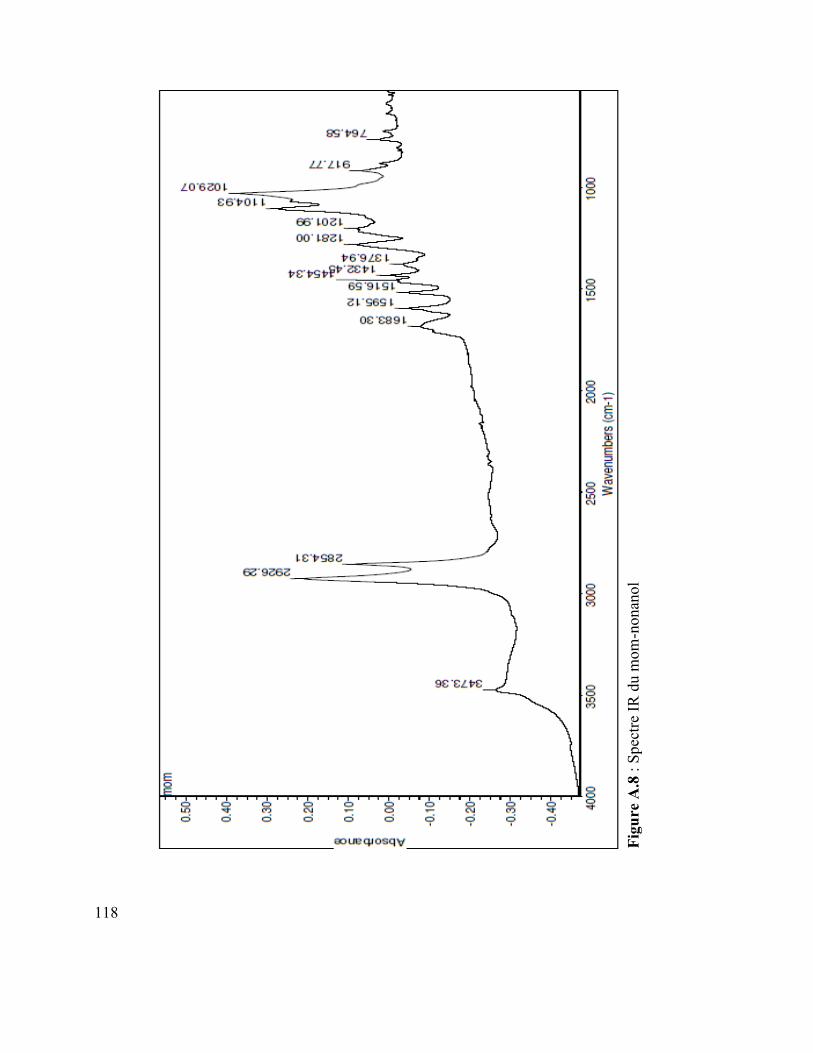
118
Fig
ure
A.8
: S
pec
tre
IR d
u m
om
-nonan
ol
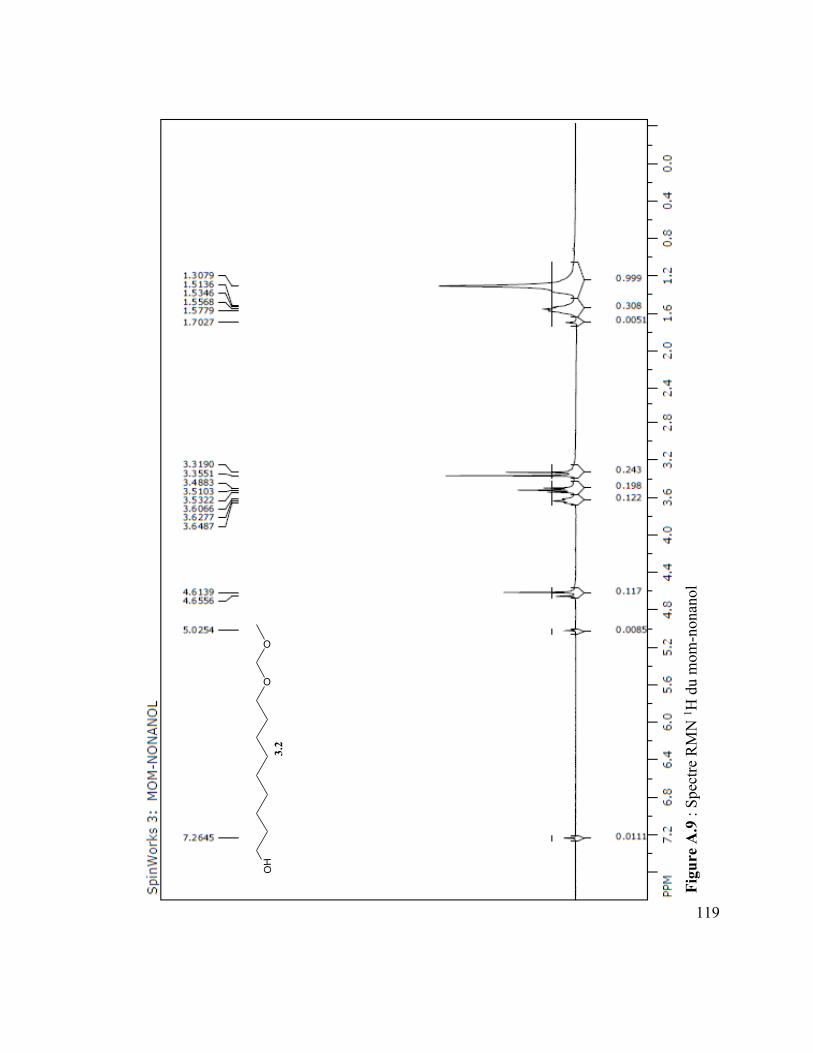
119
OH
O
O
3.2
Fig
ure
A.9
: S
pec
tre
RM
N 1
H d
u m
om
-nonan
ol
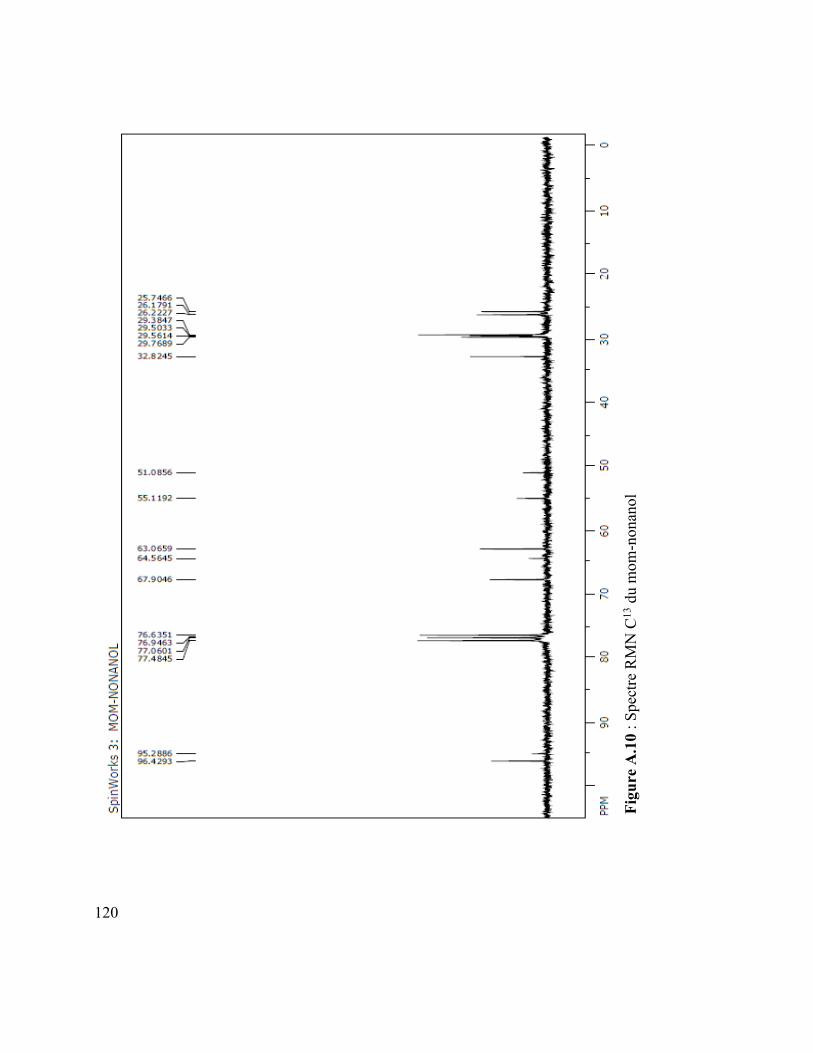
120
Fig
ure
A.1
0 :
Spec
tre
RM
N C
13 d
u m
om
-nonan
ol
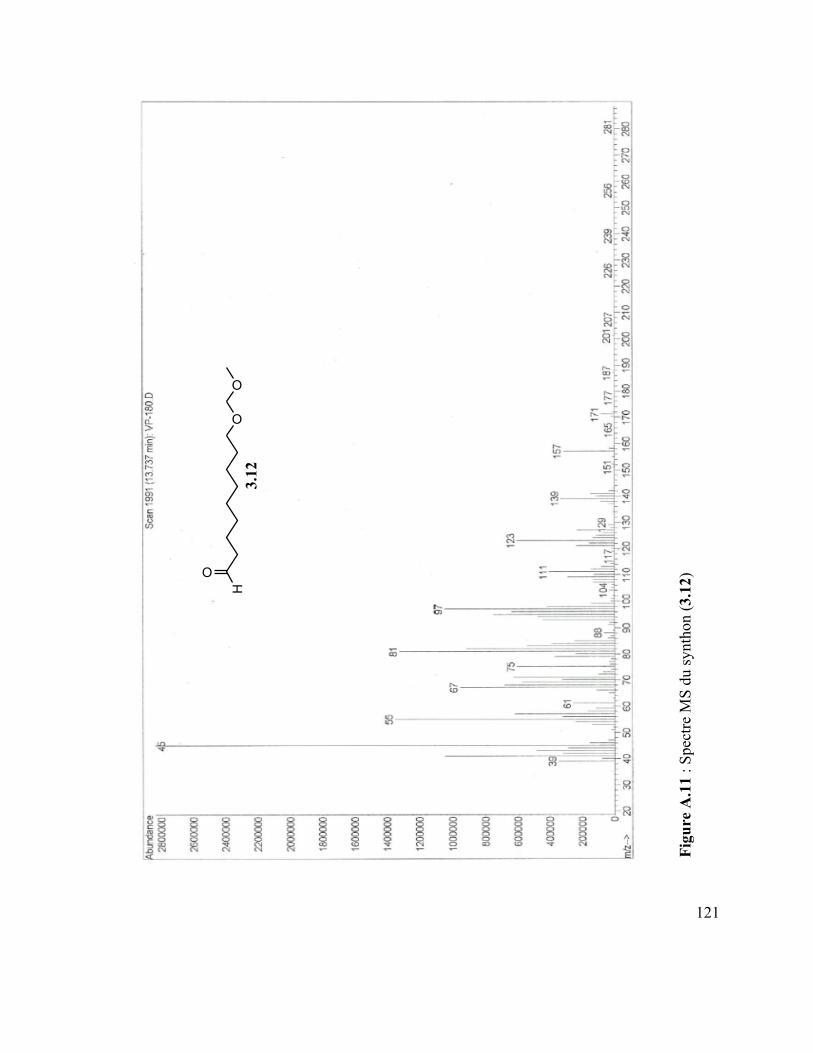
121
Fig
ure
A.1
1 :
Spec
tre
MS
du s
ynth
on (
3.1
2)
H
O
O
O
3.1
2


















