
Fonctions - CCDMD
29
FONCTIONS ORGANIQUES Introduction Les principales fonctions organiques permettent d'orienter efficacement l'analyse d'un produit organique. Les fonctions engendrent les familles de composés rencontrées en chimie organique et sont utilisées pour nommer ces dernières, selon les différentes règles de la nomenclature. Les fonctions organiques comportent des atomes de carbone, d’hydrogène, d’oxygène, de soufre, d’azote et d’halogènes, y compris l'agencement de ceux-ci, avec la présence de liens simples ou multiples, et avec des cycles ou non. La reconnaissance des principales fonctions d'une substance constitue un pas essentiel vers l'établissement de sa structure et son identification. Les fonctions retenues dans le cadre de cet ouvrage sont présentées dans l'ordre alphabétique : ACIDE RCOOH ALCANE RCH 2 CH 2 R ALCÈNE RCHCH 2 ALCOOL RCH 2 OH ALCYNE RCCR ALDÉHYDE RCHO ALKYLE TERTIAIRE R 3 CH AMIDE PRIMAIRE RCONH 2 AMIDE SECONDAIRE RCONHR AMINE PRIMAIRE RNH 2 AMINE SECONDAIRE RNHR AROMATIQUE C 6 H 5 R CÉTONE RCOR ESTER RCOOR ÉTHER ROR HALOGÉNURE RX NITRILE RCN PHÉNOL C 5 H 5 OH Pouvoir reconnaître la présence d'une ou de plusieurs fonctions sur un produit extrait, fabriqué ou analysé, est important : cette première étape dans l'étude d'un produit organique permet de confirmer le succès d'une synthèse ou d'une transformation. Lorsque l'on fait l'analyse d'un spectre infrarouge, la présence d'absorptions caractéristiques de groupes d'atomes particuliers permet de déduire la présence de différentes fonctions.
Transcript of Fonctions - CCDMD
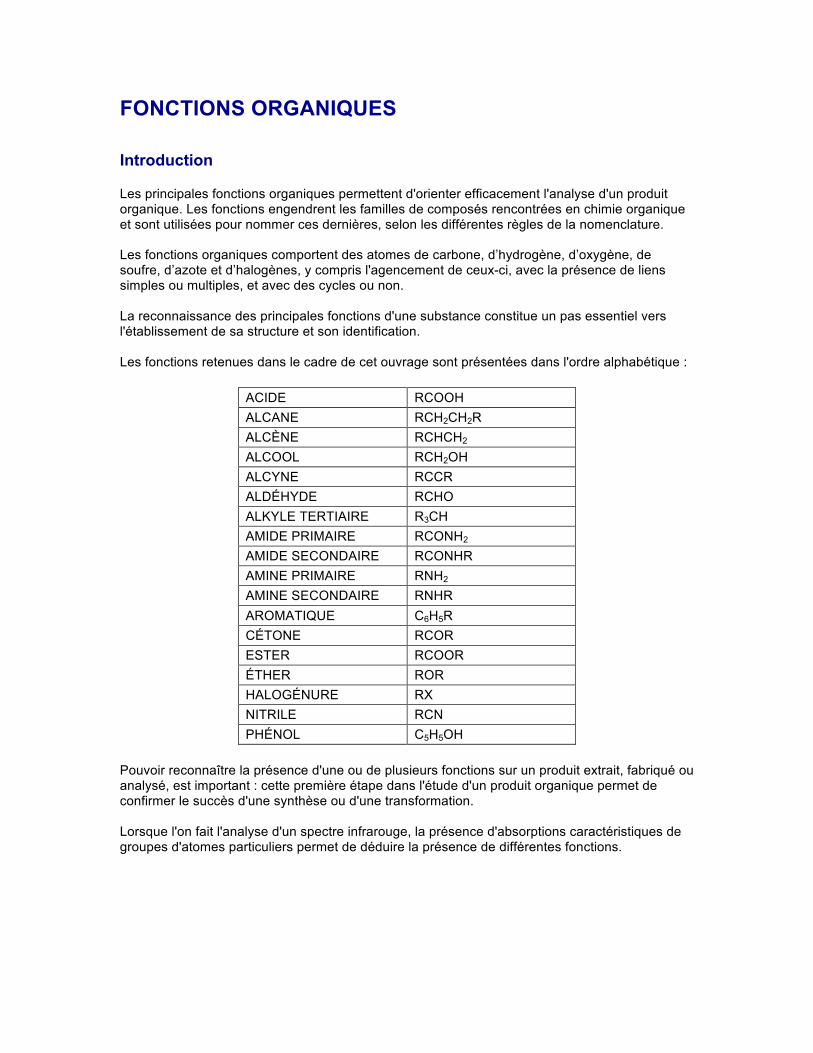
FONCTIONS ORGANIQUES Introduction Les principales fonctions organiques permettent d'orienter efficacement l'analyse d'un produit organique. Les fonctions engendrent les familles de composés rencontrées en chimie organique et sont utilisées pour nommer ces dernières, selon les différentes règles de la nomenclature. Les fonctions organiques comportent des atomes de carbone, d’hydrogène, d’oxygène, de soufre, d’azote et d’halogènes, y compris l'agencement de ceux-ci, avec la présence de liens simples ou multiples, et avec des cycles ou non. La reconnaissance des principales fonctions d'une substance constitue un pas essentiel vers l'établissement de sa structure et son identification. Les fonctions retenues dans le cadre de cet ouvrage sont présentées dans l'ordre alphabétique :
ACIDE RCOOH ALCANE RCH2CH2R ALCÈNE RCHCH2 ALCOOL RCH2OH ALCYNE RCCR ALDÉHYDE RCHO ALKYLE TERTIAIRE R3CH AMIDE PRIMAIRE RCONH2 AMIDE SECONDAIRE RCONHR AMINE PRIMAIRE RNH2 AMINE SECONDAIRE RNHR AROMATIQUE C6H5R CÉTONE RCOR ESTER RCOOR ÉTHER ROR HALOGÉNURE RX NITRILE RCN PHÉNOL C5H5OH
Pouvoir reconnaître la présence d'une ou de plusieurs fonctions sur un produit extrait, fabriqué ou analysé, est important : cette première étape dans l'étude d'un produit organique permet de confirmer le succès d'une synthèse ou d'une transformation. Lorsque l'on fait l'analyse d'un spectre infrarouge, la présence d'absorptions caractéristiques de groupes d'atomes particuliers permet de déduire la présence de différentes fonctions.

MODES DE VIBRATION 1. Élongation Appelé aussi vibration de valence ou « stretching », ce mode concerne la vibration de la molécule le long des liaisons. La fréquence de vibration est donnée par la relation ci-dessous, où k est la constante de force de la liaison (considérée ici comme un ressort), proportionnelle à l’énergie de liaison, et m la masse réduite des deux atomes reliés par cette liaison.
Ainsi, les liaisons multiples, plus énergétiques que les simples, auront une constante de force plus élevée, donc une fréquence de vibration (remplacée dans la pratique par le nombre d’ondes) plus élevée que celle des liaisons simples entre atomes identiques :
• C–C absorbe vers 1100 cm-1 • C=C vers 1600 cm-1 • CC triple vers 2100 cm-1
Par contre, les liaisons X–H, où X est un atome quelconque (C, N, O, ...), auront une fréquence d’élongation plus élevée que celle d’une liaison C–X, car la masse réduite, m, y est plus petite : pour C–H, m = 0,92 u.a.; pour C–C, m = 6 u.a.

2. Déformations dans le plan et en dehors du plan Considérons une structure qui comporte trois centres : en plus de la vibration de valence, l’angle des liaisons peut varier et il y aura flexion ou déformation. Ces déformations peuvent avoir lieu dans le plan des deux liaisons concernées ou en dehors du plan. Il y a aussi possibilité de déformations symétriques et asymétriques. Les mouvements de déformation dans le plan et hors du plan
Non seulement la nature des deux atomes vibrants intervient dans la valeur de la constante de force, mais aussi l’environnement électronique. Aussi chaque groupement fonctionnel aura-t-il des fréquences caractéristiques d’élongation et de déformation. Nous allons passer en revue les diverses fonctions grâce à l’étude de quelques spectres.
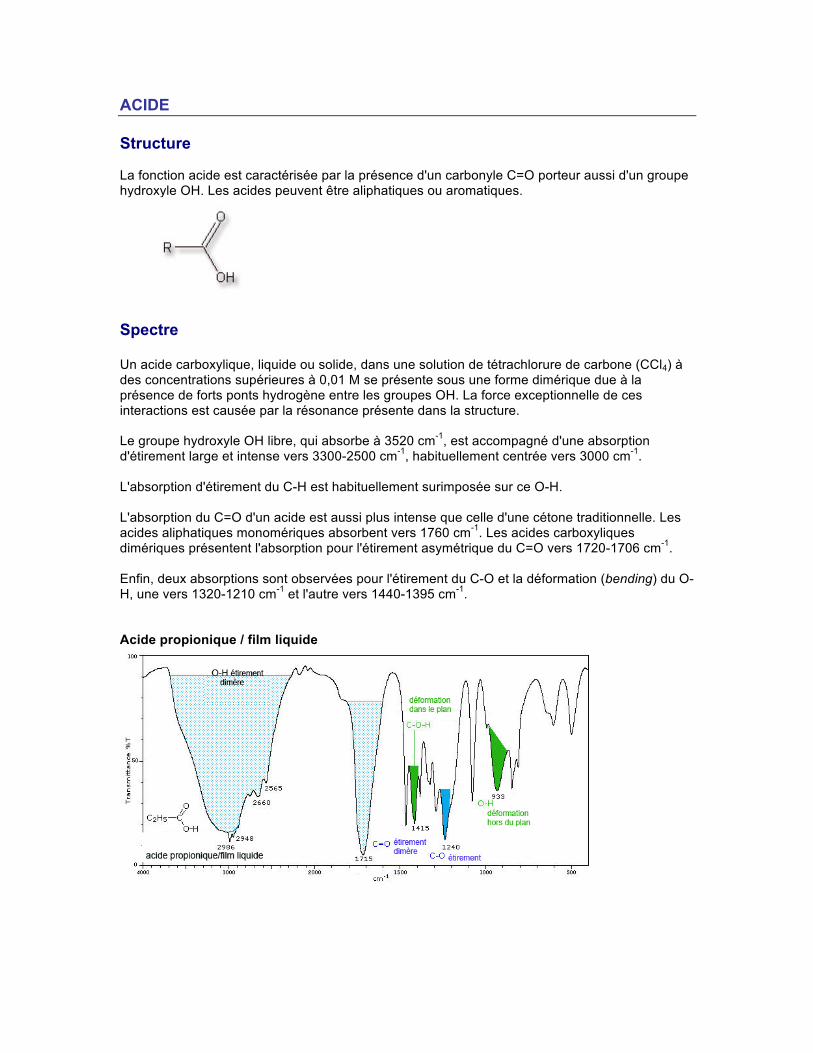
ACIDE Structure La fonction acide est caractérisée par la présence d'un carbonyle C=O porteur aussi d'un groupe hydroxyle OH. Les acides peuvent être aliphatiques ou aromatiques.
Spectre Un acide carboxylique, liquide ou solide, dans une solution de tétrachlorure de carbone (CCl4) à des concentrations supérieures à 0,01 M se présente sous une forme dimérique due à la présence de forts ponts hydrogène entre les groupes OH. La force exceptionnelle de ces interactions est causée par la résonance présente dans la structure. Le groupe hydroxyle OH libre, qui absorbe à 3520 cm-1, est accompagné d'une absorption d'étirement large et intense vers 3300-2500 cm-1, habituellement centrée vers 3000 cm-1.
L'absorption d'étirement du C-H est habituellement surimposée sur ce O-H. L'absorption du C=O d'un acide est aussi plus intense que celle d'une cétone traditionnelle. Les acides aliphatiques monomériques absorbent vers 1760 cm-1. Les acides carboxyliques dimériques présentent l'absorption pour l'étirement asymétrique du C=O vers 1720-1706 cm-1. Enfin, deux absorptions sont observées pour l'étirement du C-O et la déformation (bending) du O-H, une vers 1320-1210 cm-1 et l'autre vers 1440-1395 cm-1. Acide propionique / film liquide
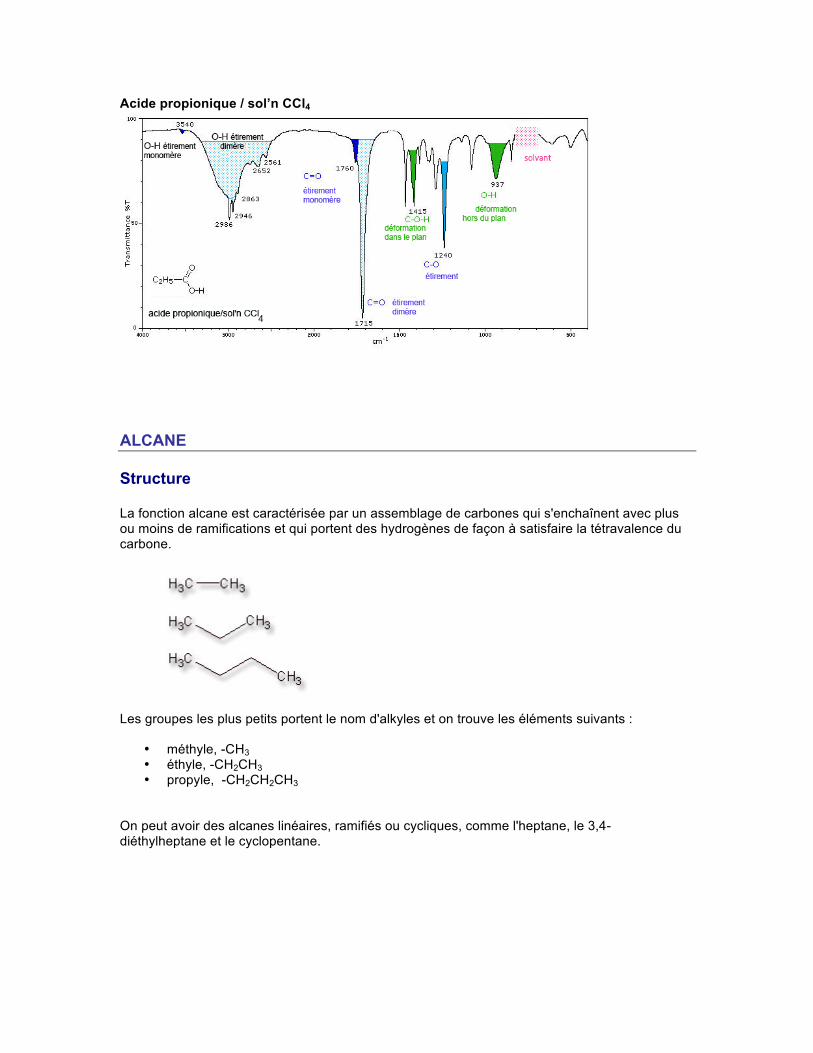
Acide propionique / sol’n CCl4
ALCANE Structure La fonction alcane est caractérisée par un assemblage de carbones qui s'enchaînent avec plus ou moins de ramifications et qui portent des hydrogènes de façon à satisfaire la tétravalence du carbone.
Les groupes les plus petits portent le nom d'alkyles et on trouve les éléments suivants :
• méthyle, -CH3 • éthyle, -CH2CH3 • propyle, -CH2CH2CH3
On peut avoir des alcanes linéaires, ramifiés ou cycliques, comme l'heptane, le 3,4-diéthylheptane et le cyclopentane.

Spectre Les alcanes cycliques ou acycliques sont constitués de groupes d'alkyles (souvent appelés paraffines) et sont interprétés selon quatre vibrations, l'étirement et la déformation (bending) du C-H et du C-C. Les vibrations du C-C se produisent vers 500 cm-1 et n'apparaissent pas dans le spectre. Les bandes assignées pour les vibrations d'étirement du C-C sont faibles et apparaissent vers 1200-800 cm-1; elles ne présentent pas d'intérêt diagnostique. Les vibrations les plus caractéristiques sont celles qui proviennent de l'étirement et de la déformation du C-H :
• le méthyle CH3 présente ses absorptions à 2960, 2870, 1460 et 1380 cm-1 • le méthylène CH2, à 2925, 2850, 1470 et 1250 cm-1 • le C-H du R3CH, à 2890 et 1340 cm-1 • le méthyle de -OCOCH3, à 1380 et 1365 cm-1 • le méthyle de -COCH3, à 1360 cm-1 • le méthyle de -COOCH3, à 1440 et 1360 cm-1
ALCÈNE Structure Les alcènes sont caractérisés par une structure plane pour les atomes portant la liaison double. L'alcène peut être monosubstitué, disubstitué cis ou trans, ou disubstitué sur le même carbone, trisubstitué et tétrasubstitué. Le lien double peut être conjugué avec un autre lien double C=C, C=O, C=N ou C=S. Un alcène peut faire partie d'une chaîne ouverte ou d'un cycle.
Spectre Les alcènes (appelés oléfines) cycliques ou acycliques, conjugués ou non, créent de la diversité du point de vue des sortes d'absorptions présentes dans ces molécules. Les alcènes linéaires non conjugués présentent une absorption d'étirement vers 1667-1640 cm-1. Le type de substitution fait varier quelque peu la position de celle-ci. La présence de symétrie dans les alcènes disubstitués trans et dans les alcènes tétrasubstitués diminue grandement l'intensité de l'absorption du C=C. On observera une valeur numérique anormalement élevée de l'onde pour un C=C difluoré ou trifluoré -CF=CF2, et -CH=CF2, soit environ 1754 cm-1. Par contre, la présence de chlore, de brome ou d'iode diminuera cette valeur.
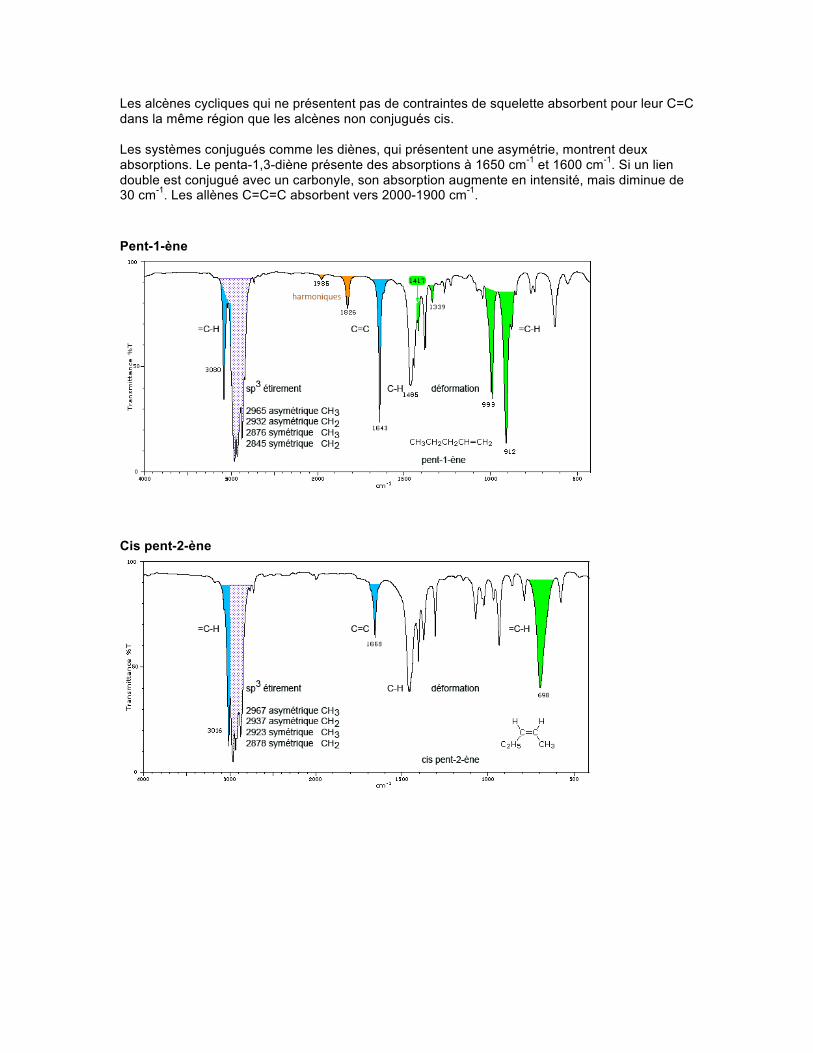
Les alcènes cycliques qui ne présentent pas de contraintes de squelette absorbent pour leur C=C dans la même région que les alcènes non conjugués cis. Les systèmes conjugués comme les diènes, qui présentent une asymétrie, montrent deux absorptions. Le penta-1,3-diène présente des absorptions à 1650 cm-1 et 1600 cm-1. Si un lien double est conjugué avec un carbonyle, son absorption augmente en intensité, mais diminue de 30 cm-1. Les allènes C=C=C absorbent vers 2000-1900 cm-1. Pent-1-ène
Cis pent-2-ène
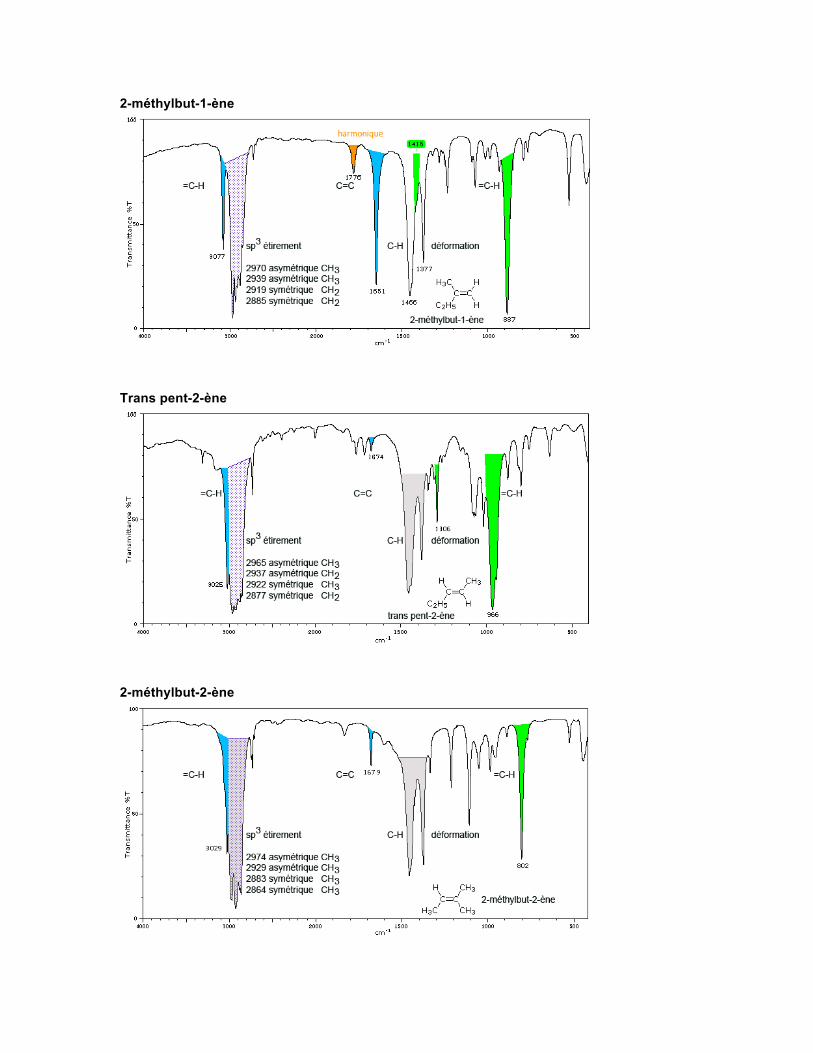
2-méthylbut-1-ène
Trans pent-2-ène
2-méthylbut-2-ène

ALCOOL ET PHÉNOL Structure La fonction alcool est caractérisée par la présence du groupe hydroxyle attaché au squelette organique, qui peut être aliphatique ou aromatique. Un alcool peut être primaire (RCH2OH), secondaire (RCHOHR) ou tertiaire (R3COH).
Spectre Les absorptions observées pour les alcools et phénols proviennent de l'étirement du O-H et du C-O. Ces vibrations sont sensibles à la formation de ponts hydrogène. De plus, l'étirement du C-O et les mouvements de déformation (bending) du O-H ne sont pas des modes de vibration indépendants parce qu'ils sont reliés aux mouvements de vibration des groupes adjacents. Le O-H non lié ou libre absorbe normalement fortement vers 3650-3584 cm-1. Une absorption forte et fine est observée si l'alcool est en solution diluée dans un solvant non polaire ou en phase gazeuse. La formation de ponts hydrogène intermoléculaires augmente au fur et à mesure que la concentration de l'alcool augmente, et d'autres absorptions commencent à apparaître sous 3550-3200 cm-1.
La vibration d'étirement du C-O produit une bande intense dans la région de 1260-1000 cm-1. La vibration de déformation du O-H produit habituellement une absorption dans la région de 1420-1330 cm-1. Dans les alcools primaires et secondaires, la déformation du O-H produit deux absorptions par association avec les vibrations de balancement (wagging) du C-H. Les mercaptans ayant un groupe SH, qui ressemble en réalité à un OH, présentent une absorption pour l'étirement du S-H vers 2600-2550 cm-1. Cette absorption est malheureusement faible et souvent non détectée dans le spectre de solutions diluées ou sous forme de film.
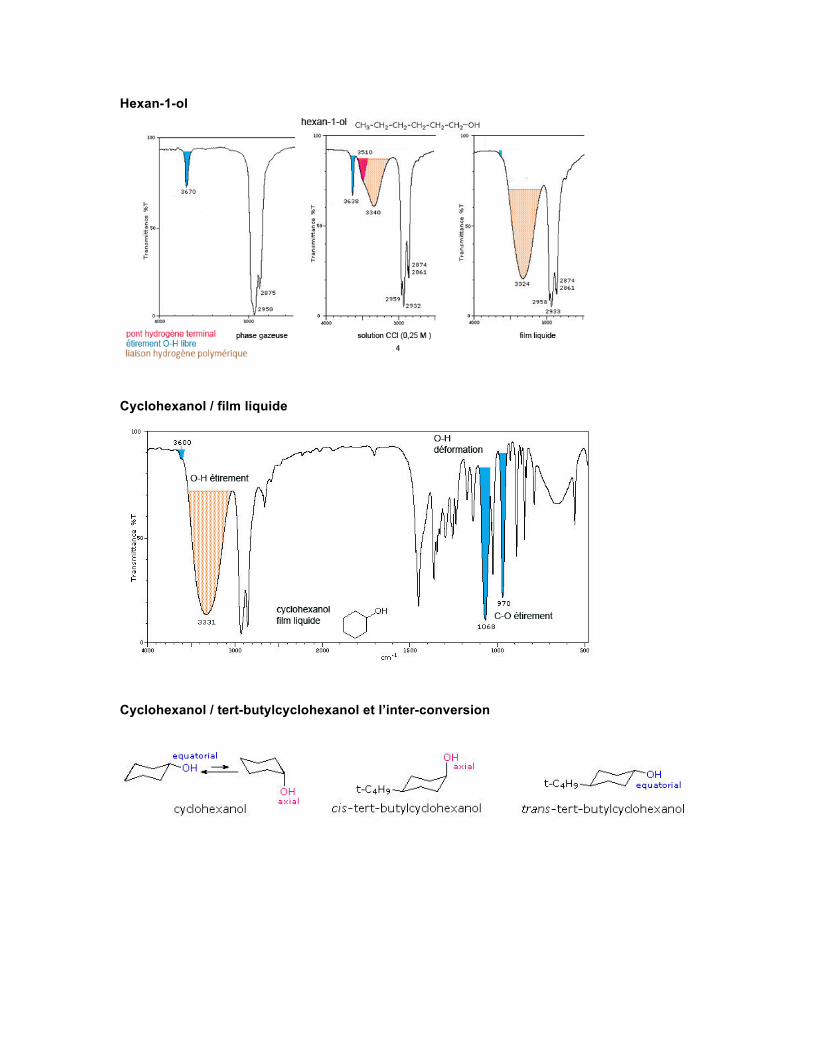
Hexan-1-ol
Cyclohexanol / film liquide
Cyclohexanol / tert-butylcyclohexanol et l’inter-conversion
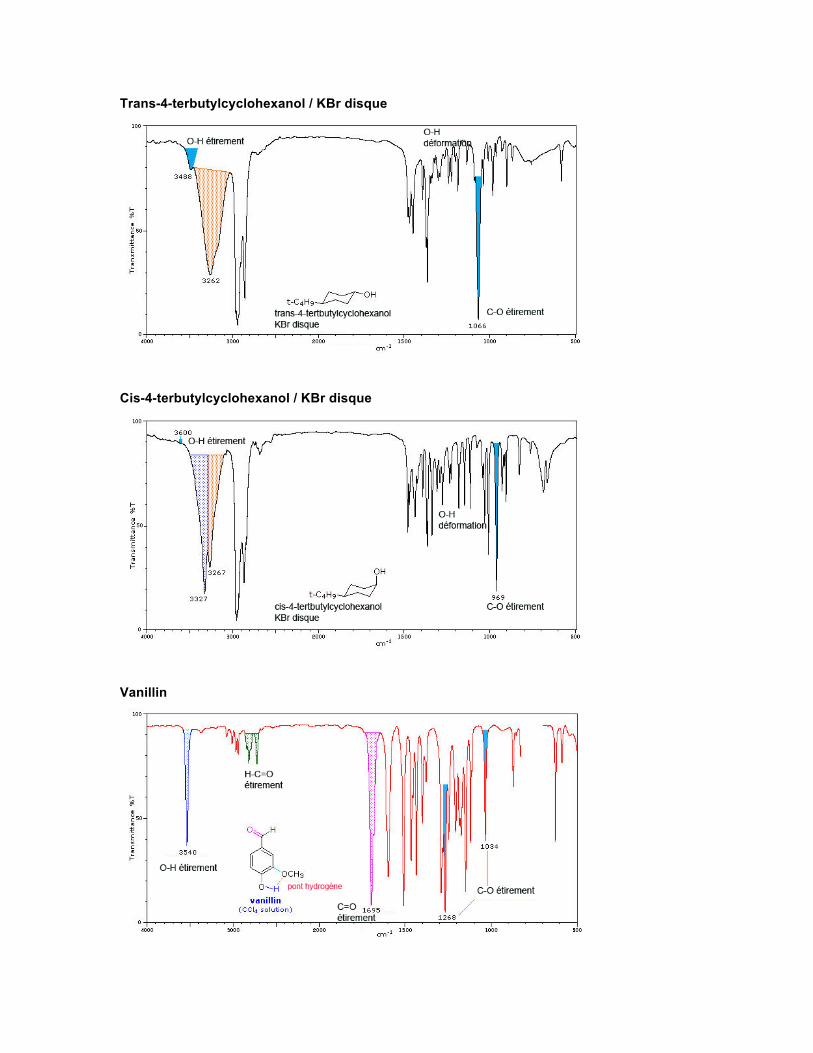
Trans-4-terbutylcyclohexanol / KBr disque
Cis-4-terbutylcyclohexanol / KBr disque
Vanillin

ALCYNE Structure La fonction alcyne est caractérisée par la présence d'un lien triple entre deux carbones. L'alcyne peut être monosubstitué RCCH, disubstitué symétrique RCCR ou asymétrique RCCR'.
Spectre Il y a deux absorptions possibles pour les alcynes, dues au lien triple CC et au C-H. L'absorption due à la fréquence d'étirement du lien triple CC est habituellement faible et apparaît dans la région de 2260-2100 cm-1. Si l'alcyne est disubstitué et symétrique, il n'y a pas d'absorption pour le lien triple CC. Si l'alcyne est monosubstitué, le lien triple CC absorbe vers 2140-2100 cm-1. Les alcynes disubstitués et asymétriques absorbent vers 2260-2190 cm-1. Cependant, si la masse des deux substituants est semblable ou que les deux substituants produisent des effets inductifs ou de résonance semblables, l'absorption est très faible. Pour des raisons de symétrie, un lien triple CC terminal produit une bande de plus forte intensité. L'absorption pour la fréquence d'étirement du C-H pour un alcyne monosubstitué apparaît vers 3333-3267 cm-1. ALDÉHYDE Structure La fonction aldéhyde est caractérisée par la présence d'un carbonyle situé en bout de chaîne et portant un hydrogène. Un aldéhyde peut être aliphatique ou aromatique si la fonction est portée par un noyau aromatique.
Spectre Le groupe C=O carbonyle des aldéhydes absorbe à des fréquences légèrement plus élevées que celles correspondant à un méthylcétone CH3COR. Les aldéhydes aliphatiques absorbent vers 1740-1720 cm-1. La conjugaison avec un lien double ou un aromatique diminue la fréquence d'absorption du carbonyle vers 1710-1685 cm-1. La formation de ponts hydrogène

intramoléculaires, comme dans le salicylaldéhyde, déplace l'absorption du C=O vers des valeurs inférieures de longueurs d'onde, soit 1666 cm-1. La majorité des aldéhydes présentent des absorptions pour la fréquence d'étirement du C-H aldéhydique RCHO dans la région de 2830-2695 cm-1. Deux bandes d'intensité moyenne sont observées pour ce C-H. La présence de deux bandes est due à la résonance de Fermi. Quelques aldéhydes aromatiques avec un groupe fortement électronégatif en ortho peuvent absorber jusqu'à une valeur aussi élevée que 2900 cm-1. ALKYLE TERTIAIRE Structure On traite l'alkyle tertiaire comme une fonction à part, ainsi qu'on l'a vu dans la section de la fonction alcane, parce que l'alkyle tertiaire ne porte pas d'hydrogènes.
Spectre Un alcane trisubstitué R3C-H présente deux absorptions pour le C-H, l'une à 2890 cm-1 et l'autre à 1340 cm-1. AMIDES PRIMAIRE ET SECONDAIRE Structure La fonction amide est relativement complexe : on trouve simultanément un carbonyle C=O et, attaché sur le carbone du C=O, un azote NH2 pour l'amide primaire ou un NHR' pour l'amide secondaire.
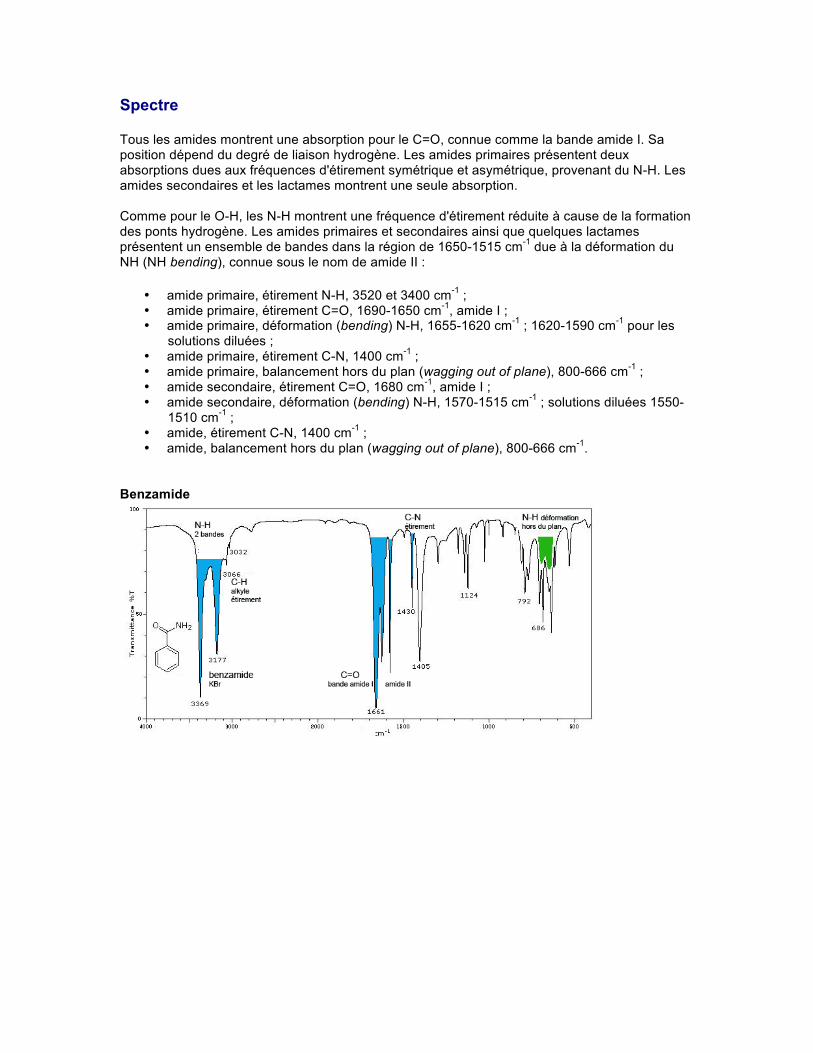
Spectre Tous les amides montrent une absorption pour le C=O, connue comme la bande amide I. Sa position dépend du degré de liaison hydrogène. Les amides primaires présentent deux absorptions dues aux fréquences d'étirement symétrique et asymétrique, provenant du N-H. Les amides secondaires et les lactames montrent une seule absorption. Comme pour le O-H, les N-H montrent une fréquence d'étirement réduite à cause de la formation des ponts hydrogène. Les amides primaires et secondaires ainsi que quelques lactames présentent un ensemble de bandes dans la région de 1650-1515 cm-1 due à la déformation du NH (NH bending), connue sous le nom de amide II :
• amide primaire, étirement N-H, 3520 et 3400 cm-1 ; • amide primaire, étirement C=O, 1690-1650 cm-1, amide I ; • amide primaire, déformation (bending) N-H, 1655-1620 cm-1 ; 1620-1590 cm-1 pour les
solutions diluées ; • amide primaire, étirement C-N, 1400 cm-1 ; • amide primaire, balancement hors du plan (wagging out of plane), 800-666 cm-1 ; • amide secondaire, étirement C=O, 1680 cm-1, amide I ; • amide secondaire, déformation (bending) N-H, 1570-1515 cm-1 ; solutions diluées 1550-
1510 cm-1 ; • amide, étirement C-N, 1400 cm-1 ; • amide, balancement hors du plan (wagging out of plane), 800-666 cm-1.
Benzamide

N, N-diméthylacétamide
AMINES PRIMAIRE ET SECONDAIRE Structure La fonction amine est caractérisée par la présence d'un azote attaché à un carbone. Cet azote peut être porteur d'un ou de deux autres carbones. On trouve alors les appellations amine primaire (RNH2), amine secondaire (RNHR') et amine tertiaire (R3N), qui ne portent aucun hydrogène. Une amine peut être aliphatique ou aromatique si l'azote est lié à un noyau aromatique.
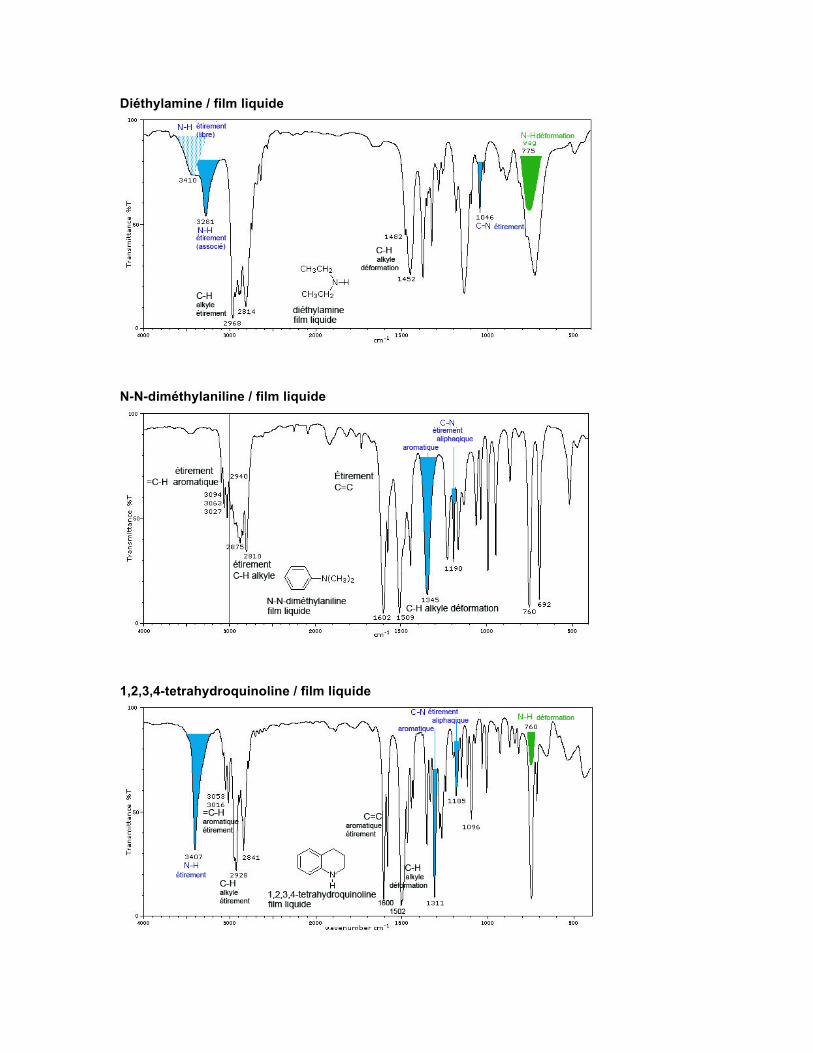
Spectre Les amines primaires, en solution diluée, présentent pour la fréquence d'étirement du N-H des absorptions à 3500 cm-1 et 3400 cm-1. Ces deux absorptions sont dues à l'étirement symétrique et asymétrique du N-H. Les amines secondaires présentent une seule absorption faible vers 3350-3310 cm-1. Les amines aromatiques absorbent plutôt vers des longueurs d'onde plus courtes (fréquences plus élevées).
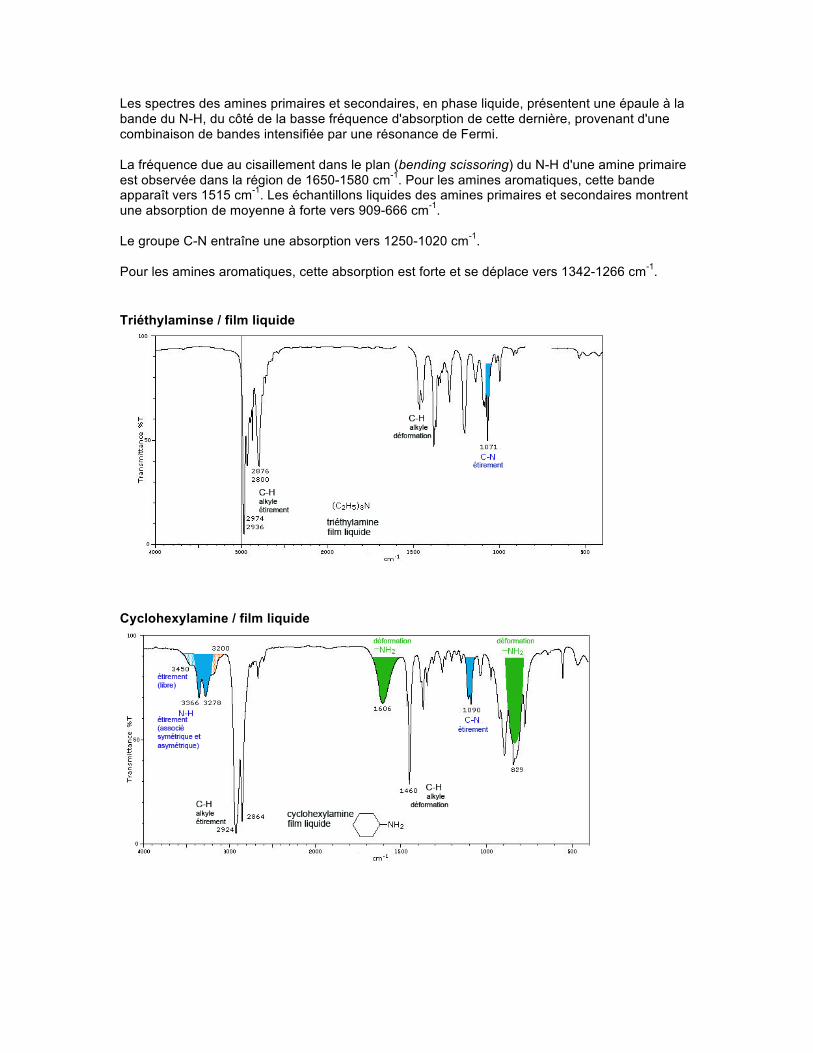
Les spectres des amines primaires et secondaires, en phase liquide, présentent une épaule à la bande du N-H, du côté de la basse fréquence d'absorption de cette dernière, provenant d'une combinaison de bandes intensifiée par une résonance de Fermi. La fréquence due au cisaillement dans le plan (bending scissoring) du N-H d'une amine primaire est observée dans la région de 1650-1580 cm-1. Pour les amines aromatiques, cette bande apparaît vers 1515 cm-1. Les échantillons liquides des amines primaires et secondaires montrent une absorption de moyenne à forte vers 909-666 cm-1. Le groupe C-N entraîne une absorption vers 1250-1020 cm-1. Pour les amines aromatiques, cette absorption est forte et se déplace vers 1342-1266 cm-1. Triéthylaminse / film liquide
Cyclohexylamine / film liquide
Diéthylamine / film liquide
N-N-diméthylaniline / film liquide
1,2,3,4-tetrahydroquinoline / film liquide
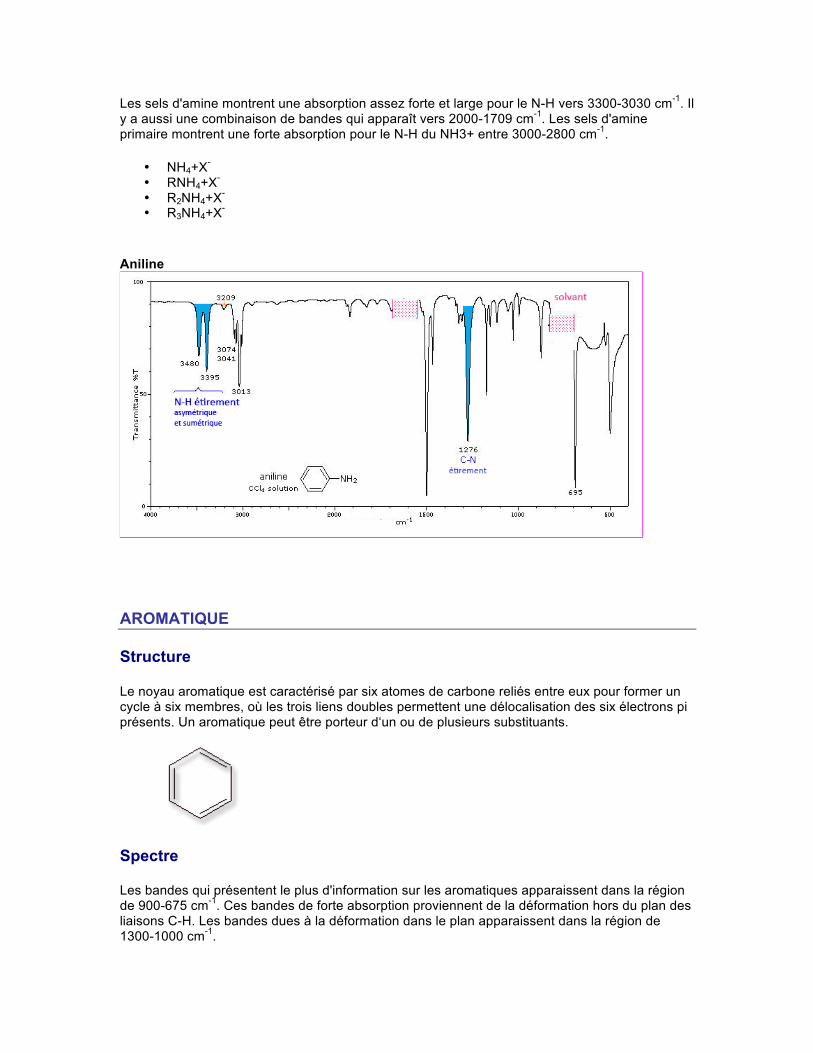
Les sels d'amine montrent une absorption assez forte et large pour le N-H vers 3300-3030 cm-1. Il y a aussi une combinaison de bandes qui apparaît vers 2000-1709 cm-1. Les sels d'amine primaire montrent une forte absorption pour le N-H du NH3+ entre 3000-2800 cm-1.
• NH4+X- • RNH4+X- • R2NH4+X- • R3NH4+X-
Aniline
AROMATIQUE Structure Le noyau aromatique est caractérisé par six atomes de carbone reliés entre eux pour former un cycle à six membres, où les trois liens doubles permettent une délocalisation des six électrons pi présents. Un aromatique peut être porteur d‘un ou de plusieurs substituants.
Spectre Les bandes qui présentent le plus d'information sur les aromatiques apparaissent dans la région de 900-675 cm-1. Ces bandes de forte absorption proviennent de la déformation hors du plan des liaisons C-H. Les bandes dues à la déformation dans le plan apparaissent dans la région de 1300-1000 cm-1.
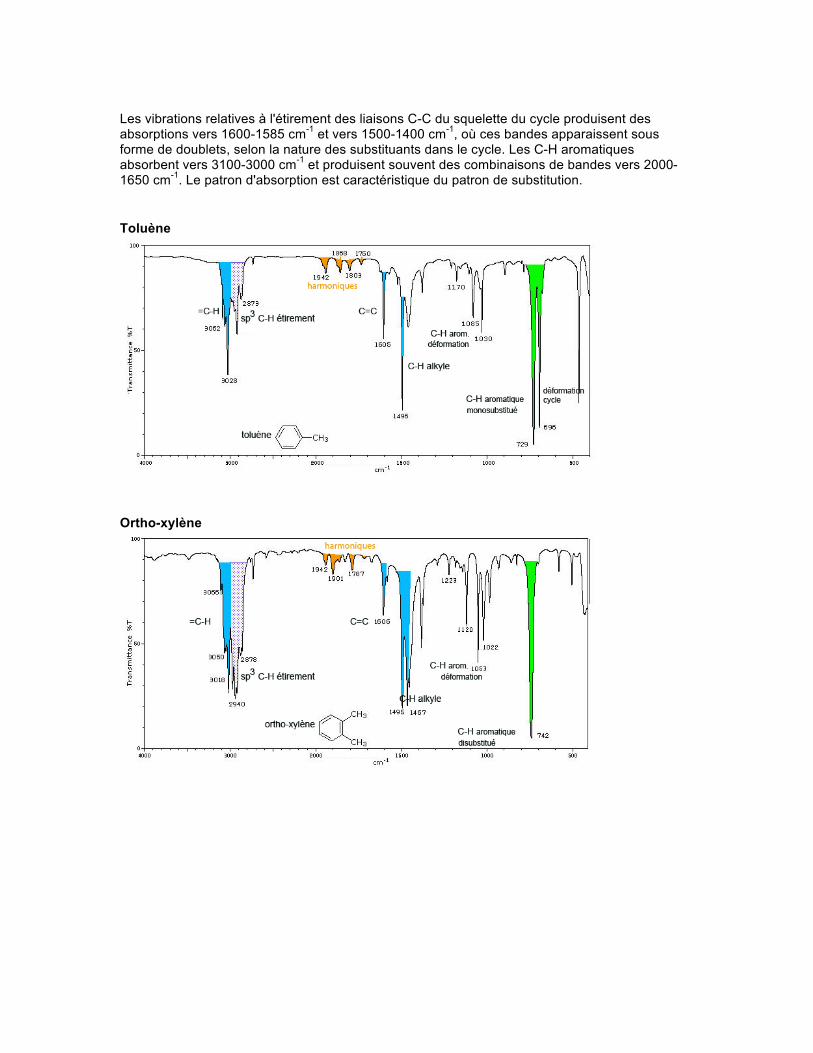
Les vibrations relatives à l'étirement des liaisons C-C du squelette du cycle produisent des absorptions vers 1600-1585 cm-1 et vers 1500-1400 cm-1, où ces bandes apparaissent sous forme de doublets, selon la nature des substituants dans le cycle. Les C-H aromatiques absorbent vers 3100-3000 cm-1 et produisent souvent des combinaisons de bandes vers 2000-1650 cm-1. Le patron d'absorption est caractéristique du patron de substitution. Toluène
Ortho-xylène
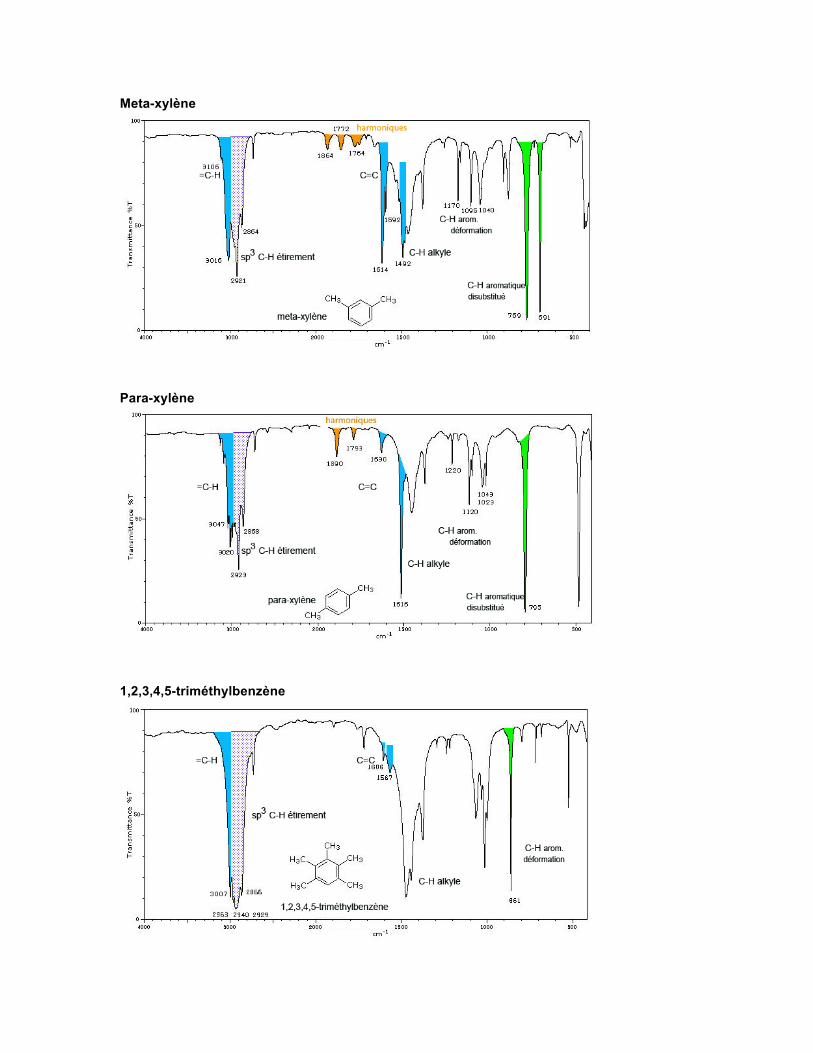
Meta-xylène
Para-xylène
1,2,3,4,5-triméthylbenzène
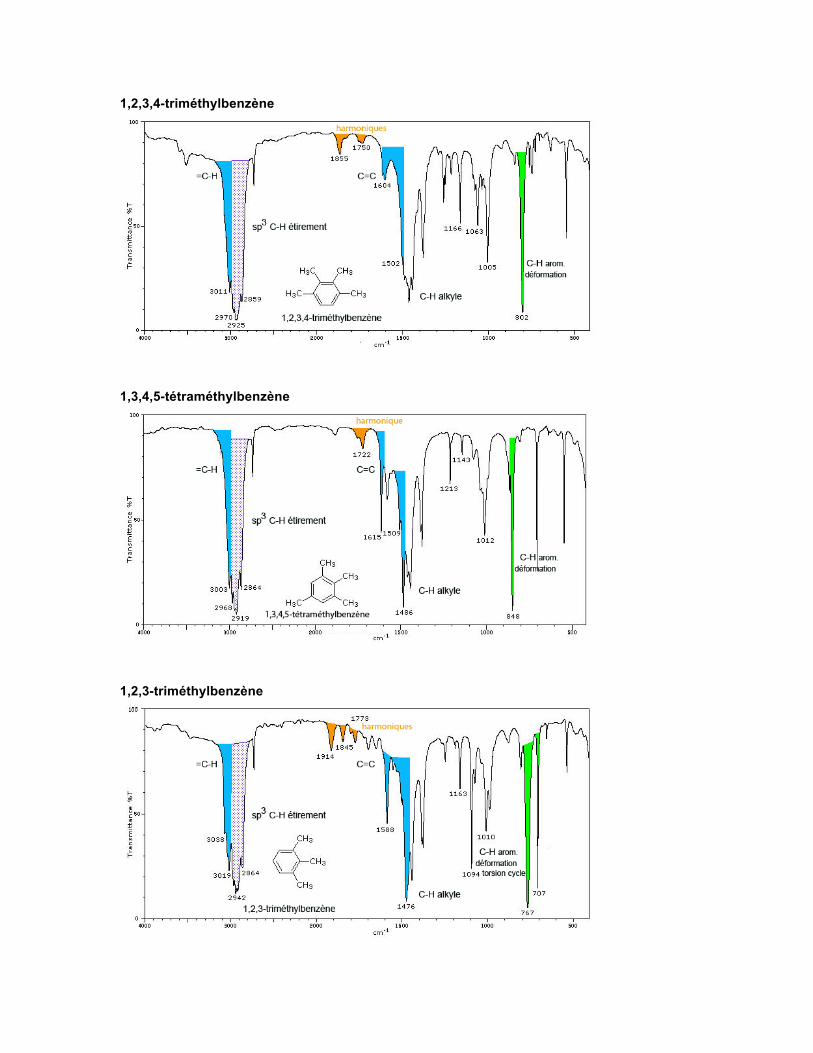
1,2,3,4-triméthylbenzène
1,3,4,5-tétraméthylbenzène
1,2,3-triméthylbenzène
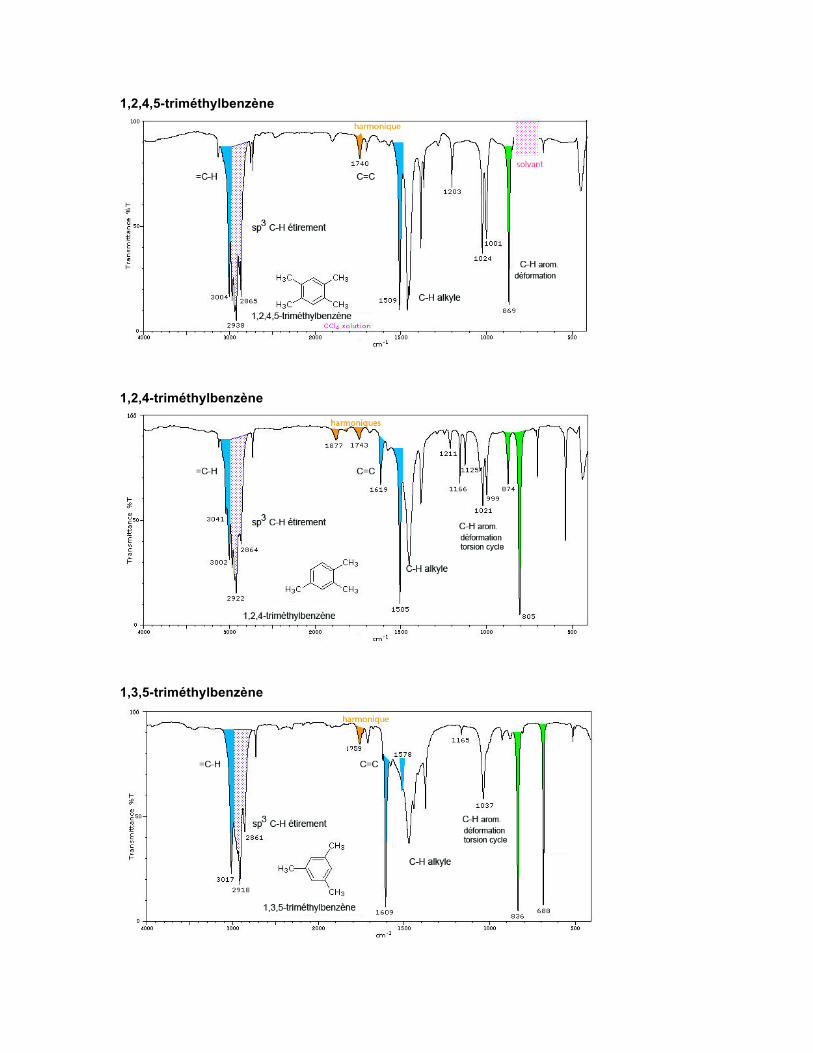
1,2,4,5-triméthylbenzène
1,2,4-triméthylbenzène
1,3,5-triméthylbenzène
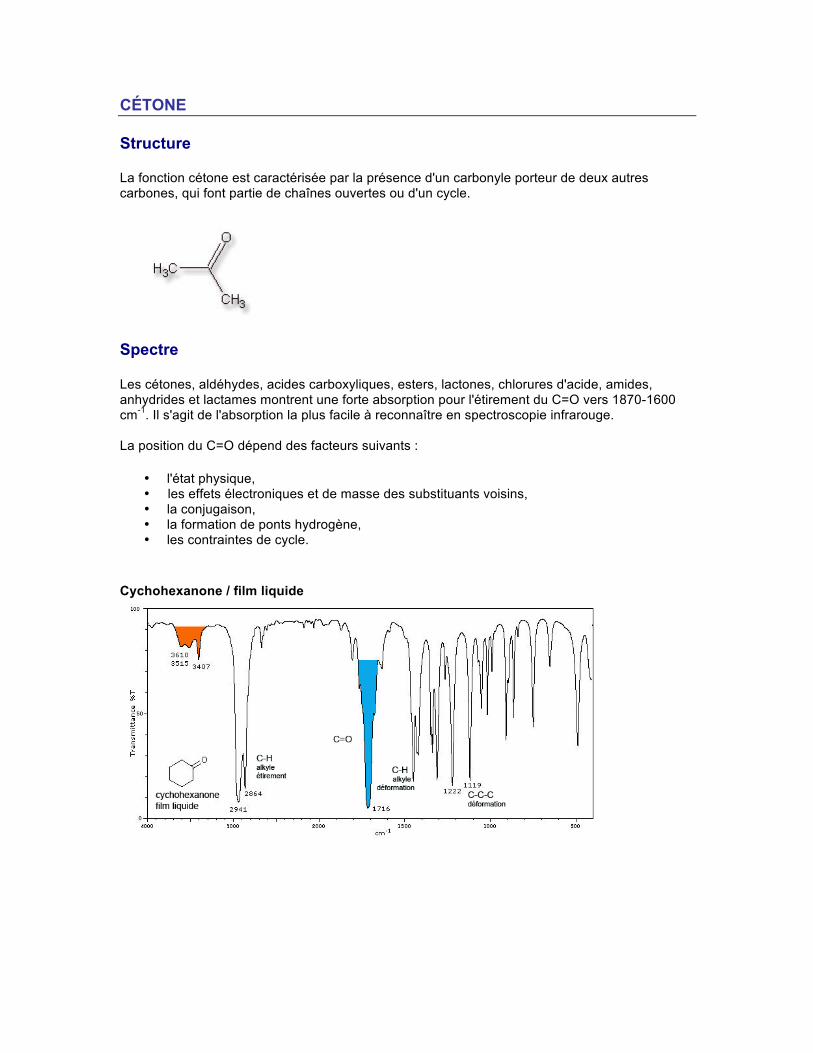
CÉTONE Structure La fonction cétone est caractérisée par la présence d'un carbonyle porteur de deux autres carbones, qui font partie de chaînes ouvertes ou d'un cycle.
Spectre Les cétones, aldéhydes, acides carboxyliques, esters, lactones, chlorures d'acide, amides, anhydrides et lactames montrent une forte absorption pour l'étirement du C=O vers 1870-1600 cm-1. Il s'agit de l'absorption la plus facile à reconnaître en spectroscopie infrarouge. La position du C=O dépend des facteurs suivants :
• l'état physique, • les effets électroniques et de masse des substituants voisins, • la conjugaison, • la formation de ponts hydrogène, • les contraintes de cycle.
Cychohexanone / film liquide
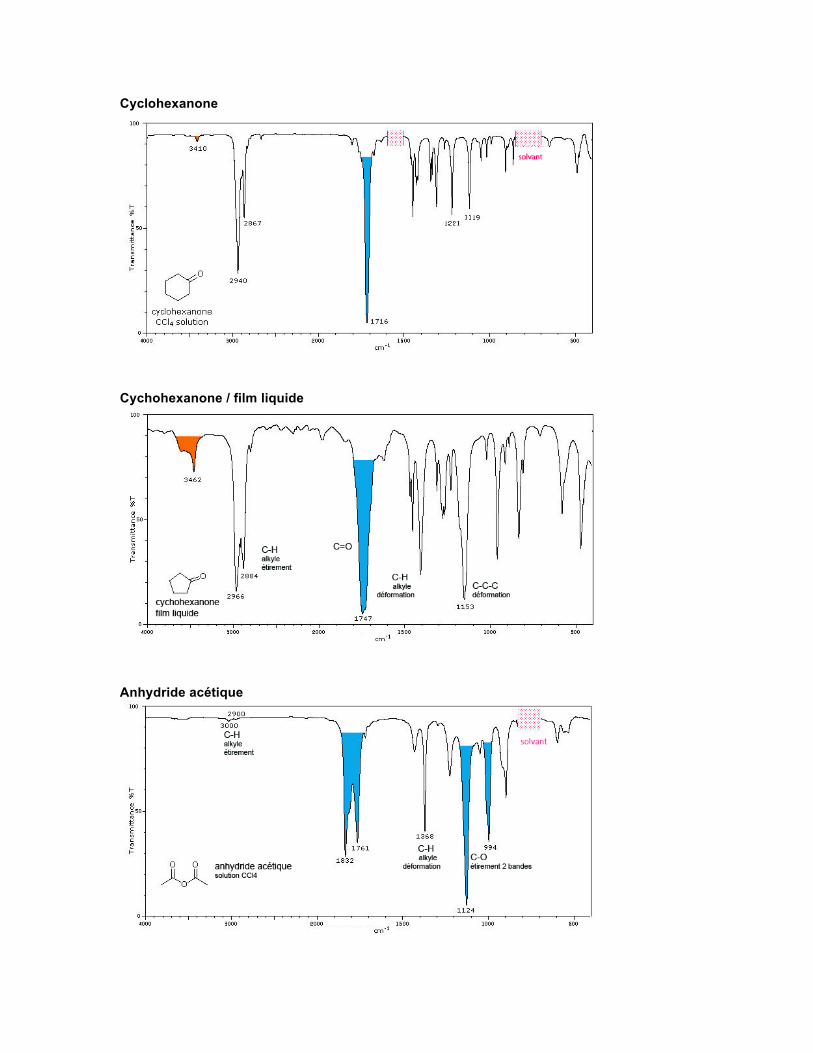
Cyclohexanone
Cychohexanone / film liquide
Anhydride acétique
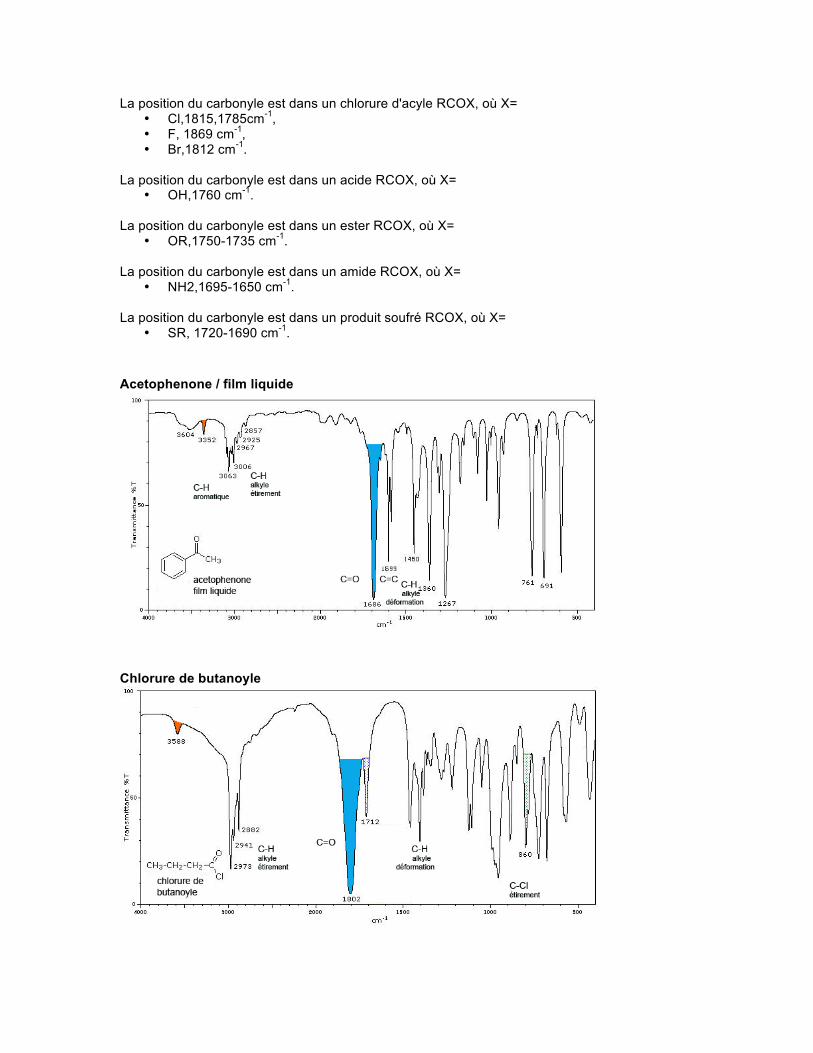
La position du carbonyle est dans un chlorure d'acyle RCOX, où X= • Cl,1815,1785cm-1, • F, 1869 cm-1, • Br,1812 cm-1.
La position du carbonyle est dans un acide RCOX, où X=
• OH,1760 cm-1. La position du carbonyle est dans un ester RCOX, où X=
• OR,1750-1735 cm-1. La position du carbonyle est dans un amide RCOX, où X=
• NH2,1695-1650 cm-1. La position du carbonyle est dans un produit soufré RCOX, où X=
• SR, 1720-1690 cm-1. Acetophenone / film liquide
Chlorure de butanoyle

Chlorure de benzoyle
ESTER Structure La fonction ester est caractérisée par la présence d'un carbonyle relié à un oxygène de type éther. Ces deux groupes sont emprisonnés entre deux carbones, comme dans RCOOR'.
Spectre Les esters et les lactones ont deux absorptions caractéristiques qui proviennent des fréquences d'étirement du C=O et du C-O. Le carbonyle absorbe à des fréquences habituellement supérieures à ce que l'on observe pour les cétones ordinaires. Les anhydrides montrent deux absorptions pour leurs deux carbonyles, soit à 1820 cm-1 et à 1750 cm-1.
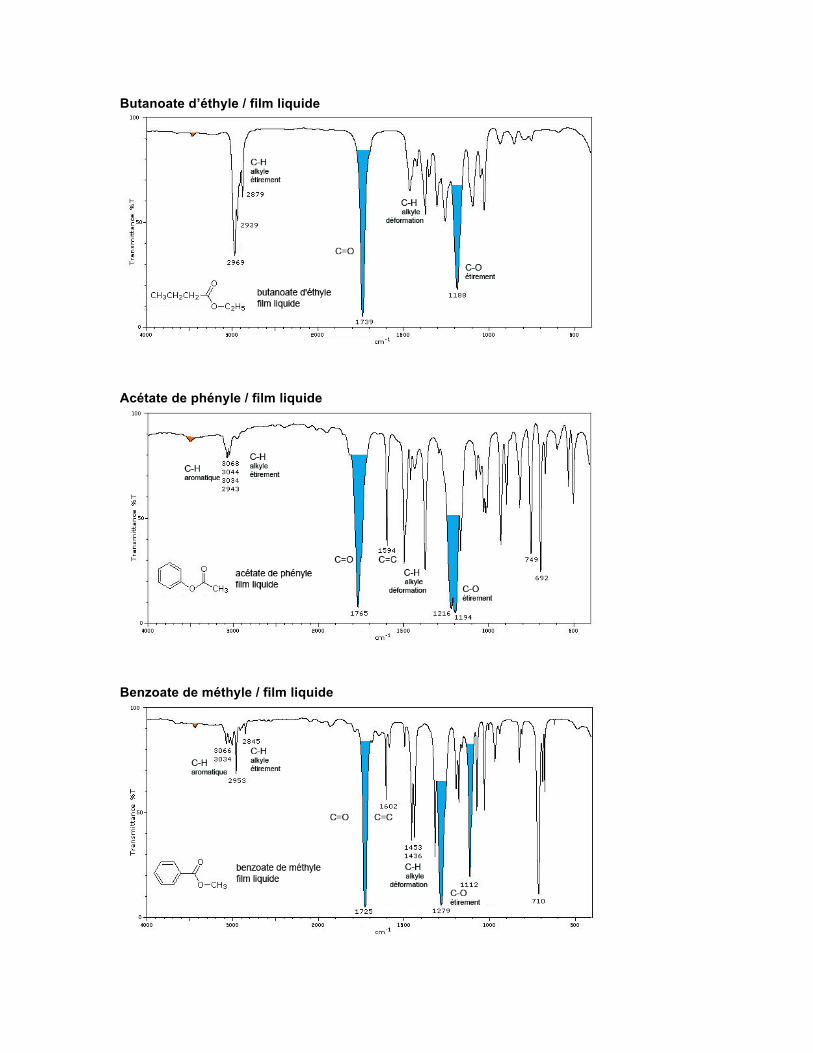
Butanoate d’éthyle / film liquide
Acétate de phényle / film liquide
Benzoate de méthyle / film liquide

ÉTHER Structure La fonction éther est caractérisée par la présence d'un seul oxygène emprisonné entre deux carbones. Un éther peut être aliphatique ou cyclique.
Spectre Les éthers présentent les mêmes caractéristiques de vibration que les liaisons C-C-C, mais, l'oxygène créant un changement dipolaire plus important, l'intensité des absorptions sera plus forte. L'absorption la plus caractéristique pour un éther aliphatique est observée à 1150-1085 cm-1, à cause de l'étirement symétrique du C-O-C. Le spectre des éthers du type aryle et alkyle montre une fréquence d'étirement asymétrique vers 1275-1200 cm-1. HALOGÉNURE Structure La fonction halogénure est caractérisée par une chaîne organique aliphatique, cyclique ou aromatique à laquelle est attaché un halogène comme le fluor, le chlore, le brome ou l'iode.
Spectre Il y a une forte absorption pour la fréquence d'étirement de la liaison carbone-halogène. La liaison C-Cl pour les halogénures aliphatiques produit une absorption assez large vers 850-550 cm-1. Le CCl4 produit une absorption intense à 797 cm-1. Les composés contenant du fluor absorbent sur une plage variant de 1400 à 730 cm-1. Un composé monofluoré montrera une forte bande vers 1100-1000 cm-1. Si le nombre d'éléments de fluor augmente, le spectre devient de plus en plus complexe. Les groupes CF3 et CF2 absorbent fortement dans la région de 1350-1120 cm-1. Les chlorobenzènes absorbent vers 1096-1089 cm-1.

NITRILE Structure La fonction nitrile est caractérisée par la présence d'un lien CN triple.
Spectre Les spectres des nitriles sont caractérisés par une absorption d'intensité de la liaison triple qui varie de faible à moyenne. Les nitriles aliphatiques absorbent vers 2260-2240 cm-1. La conjugaison diminue la fréquence d'absorption.


















