
ABREVIATIONS -...
219
ABREVIATIONS
-
Upload
truongkien -
Category
Documents
-
view
221 -
download
0
Transcript of ABREVIATIONS -...

ABREVIATIONS

AAN : anticorps anti nucléaires
ABEG : assez bon état général
AC : anticorps
ACR : american college of rhumatology
AEG : altération de l’état général
AG : antigène
AINS : anti inflammatoires non stéroïdiens
AMG : amaigrissement
ANCA : anticorps anti cytoplasme des polynucléaires neutrophiles
APS : anti paludéens de synthèse
AT : anti trypsine
ATCD : antécédents
ASLO : anticorps anti streptolysines O
BAAR : bacille alcoolo acido résistant
BAV : baisse de l’acuité visuelle
BK : bacille de koch
C-ANCA : les anticorps anti cytoplasme des polynucléaires neutrophiles à
fluorescence cytoplasmique
CD : cluster de différentiation
CH50 : complexe hémolytique 50%
CI : complexe immun
CRP : C réactive protein
DCD : décès
ECBU : examen cyto bactériologique des urines
ECG : électrocardiogramme
ELISA : enzyme linked immunosorbent assay
EMG : électromyogramme

EPP : électrophorèse des protides
FFS : five factor score
GB : globules blancs
GNRP : glomérulonéphrite rapidement progressive
HG : hémoglobine
HIV : virus de l’immunodéficience humaine
HLA : humain leucocyt antigen : les antigènes leucocytaires humaines
HTA : hypertension artérielle
HTAP : hypertension artérielle pulmonaire
HTIC : hypertension intra crânienne
HVB : virus de l’hépatite B
HVC : virus de l’hépatite C
IDM : infarctus de myocarde
IF : immunofluorescence
IFD : immunofluorescence directe
IFI : immunofluorescence indirecte
IG : immunoglobulines
IL : interleukine
INF gamma : interféron gamma
IRM : imagerie par résonance magnétique
KG : kilogramme
LCR : liquide céphalo -rachidien
LDH : lactate déshydrogénase
LEAD : lupus érythémateux aigu déssiminé
M.pneumoniae : mycoplasma pneumoniae
MK : maladie de Kawasaki
Mm hg : millimètres de mercure

MPA : micro poly angéite
MPO : myéloperoxydase
MW : maladie de Wegener
NFS : numération formule sanguine
OMI : oédeme des membres inférieurs
ORL : oto rhino laryngologie
P-ANCA : anticorps anti nucléaires des cytoplasmes des polynucléaires
neutrophiles à fluorescence péri nucléaire
PAN : péri artérite noueuse
PNE : polynucléaires éosinophiles
PNN : polynucléaires neutrophiles
PPR : pseudo polyarthrite rhizomélique
PR : polyarthrite rhumatoïde
PR3 : peroxydase de type 3
RAA : rhumatisme articulaire aigu
SCS : syndrome de churg et Strauss
SNC : système nerveux central
SPA : spondylarthrite ankylosante
TNF : tumor necrosing factor
VS : vitesse de sédimentation

SOMMAIRE

I -INTRODUCTION …………………………………………………………………………… 1
II-GENERALITES ……………………………………………………………………………… 3
1-définition des vascularites systémiques ……………………………………… 5
2-épidemiologie ……………………………………………………………………… 5
III- RAPPEL THEORIQUE ……………………………………………………………………… 13
1-Historique des vascularites ……………………………………………………… 15
2-Physiopathologie et le rôle des ANCA :……………………..………………… 18
a-Maladie de Horton ............................................................................... 18
b-Maladie de Takayasu …………………………………………………………… 21
c-les vascularites nécrosantes :………………………… ……………..………… 22
1-dépôts de CI circulants…………………………………..…………………….... 22
2-ANCA :……………………………………………………………………………… 23
*découverte des ANCA ……………………………………………………… 23
*détection des ANCA……………………………………………….…. ……. 23
*rôle pathogène des ANCA ………………………………………………… 26
*rôle des ANCA en pratique ……………………………………………… 27
3-activation lymphocytaire T ……………………………………………….. 28
3-Classification des vascularites et critères diagnostiques : ……………… 29
1- classification de Zeek ………………………………………………………. 30
2- classification de Fauci ……………………………………………………… 31
3 -classification de Wechsler…………………………………………………… 32
4-Classification de Chapel Hill………………………………………………… 34
5-Classification de lie …………………………………………………………… 48
6-Classification de Kahn et Peltier …………………………………………... 51
g-Les critères diagnostiques des vascularites systémiques de l’ACR …. 53

IV-MATERIEL ET METHODES ……………………………………………………………… 60
1-Objectif ……………………………………………………………………………… 62
2-Matériel et méthodes ……………………………………………………………… 62
V-RESULTATS ET ANALYSE ………………………………………………………………… 64
1-observations médicales …………………………………………………………… 66
2-données epidemiologiques ………………………………………………………. 123
3-antécédents ………………………………………………………………………… 124
4-motif d’hospitalisation …………………………………………………………… 125
5-données de l’examen clinique …………………………………………………… 125
6-examens complémentaires ……………………………………………………… 127
7-étiologies …………………………………………………………………………… 131
8-traitement et suivi ………………………………………………………………… 132
V- NOUVEAUTES THERAPEUTIQUES …………………………………………………… 152
1. les anti TNF alpha …………………………………………………………………154
2. les anti CD20……………………………………………………………………… 158
VI-CONCLUSION ……………………………………………………………………………… 162
VI-RESUMES …………………………………………………………………………………… 171
VII-REFERENCES BIBLIOGRAPHIQUES ……………………………………………………… 177

DDEEDDIICCAACCEE

TToouutteess lleess lleettttrreess
nnee ssaauurraaiieenntt ffaaiirree lleess mmoottss qquu’’iill ffaauutt ......
TToouuss lleess mmoottss nnee ssaauurraaiieenntt eexxpprriimmeerr
llaa ggrraattiittuuddee,, ll’’aammoouurr,, llee rreessppeecctt,, llaa rreeccoonnnnaaiissssaannccee......
AAuussssii,, cc’’eesstt ttoouutt ssiimmpplleemmeenntt qquuee......
ZZJJee ddééddiiee cceettttee tthhèèssee àà ……??

AA mmaa ttrrèèss cchhèèrree mmèèrree JJ eessppèèrree qquuee ttuu ttrroouuvveerraass ddaannss ccee ttrraavvaaiill llee ffrruuiitt ddee ttoonn ddéévvoouueemmeenntt,,eett ddee tteess lloonngguueess aannnnééeess ddee ssaaccrriiffiicceess ccoonnsseennttiiss ppoouurr mmeess ééttuuddeess ,,aaiinnssii qquuee ll’’eexxpprreessssiioonn ddee mmaa ggrraattiittuuddee eett ddee mmoonn pprrooffoonndd aammoouurr.. PPuuiissssee llee ddiieeuu,, llee ttoouutt ppuuiissssaanntt,, vvoouuss pprroottééggeerr eett vvoouuss aaccccoorrddeerr ssaannttéé eett bboonnhheeuurr..

A mon très cher père
Aucun mot ne saurait exprimer mon profond
respect, ma considération, mon profond amour et
toute ma gratitude pour tous les sacrifices que tu
as consenti pour mon éducation, mon instruction
et mon bien être.
Je te dédie ce travail comme témoignage de mon
respect et mon amour éternel. ..

AA mmoonn ttrrèèss cchheerr mmaarrii DDRRIISSSS
TTuu mm’’aass aaccccoommppaaggnnéé dduurraanntt cceettttee éépprreeuuvvee aavveecc
bbeeaauuccoouupp dd’’aammoouurr eett ppaattiieennccee..
CCeess qquueellqquueess mmoottss nnee ppeeuuvveenntt ttrraadduuiirree ttoouuss ccee qquuee
jjee rreesssseennss ppoouurr ttooii jjee ttee ddééddiiee ccee ttrraavvaaiill ccoommmmee ffrruuiitt
ddee ttoonn ddéévvoouueemmeenntt,, ddee ttoonn ssaaccrriiffiiccee eett ddee ttoouutt
ll’’aammoouurr qquuee jj aaii ppoouurr ttooii..

A mon très cher bébé J’attends impatiemment ton arrivée au monde Je te dédie cette thèse comme preuve d’amour et d’affection pour toi. Je t’aime très fort Que le bon dieu te garde pour moi.

A ma très chère sœur FATIMA J’espère que tu trouveras dans cette thèse l’expression de mon amour, ma grande sympathie et ma gratitude. . J’implore dieu qu’il t’apporte le bonheur et la réussite dans ta vie.

A ma très chère sœur HOUDA, son mari OMAR SEBTI et la petite MALAK Vous avez fait grande preuve de patiente et de sympathie, vous m’avez apporter beaucoup de soutien. Il n’est de mots susceptibles d’exprimer toute ma gratitude et mon affection pour vous.

A mon très cher frère SIDI MOHAMMED Tu m’as beaucoup aidé pour l’élaboration et la réussite de ce travail, tu m’as apporté conseil et grand soutien . Tu es le symbole de la patiente et de la sympathie. J’espère que tu trouveras dans ce travail le fruit de tous tes efforts. Aucun mot ne saura exprimer tout l’amour que j’ai pour toi.

A mes beaux parents :
Mr LOUDYI HAMID et Mme CHAMI BAHIA
JJee vvoouuss rreemmeerrcciiee ddee vvoottrree ggeennttiilllleessssee eett vvoottrree aammoouurr ..

AA mmeess bbeelllleess ssooeeuurrss eett lleeuurrss mmaarriiss
KKAAWWTTAARR eett SSOOUULLAAYYMMAANNEE LLBBEEKKKKAALLII
EEtt
ZZIINNEEBB eett RREEDDAA LLHHRRIICCHHII..

AA ttoouuttee llaa ffaammiillllee LLAAHHLLOOUU eett SSAAYYEERRHH
AA mmeess oonncclleess SSIIMMOOHHAAMMMMEEDD,, AARRBBII,, TTHHAAMMII,,
AABBDDEELLTTIIFF,, AABBDDEERRHHMMAANN,, TTAAHHRR,, eett AABBDDOOUU
AA mmeess ttaanntteess
NNAAJJAATT eett AATTIIQQUUAA
EEtt àà ttoouuss mmeess ccoouussiinnss eett ccoouussiinneess..

AA llaa mméémmooiirree ddee ggrraannddss ppaarreennttss
HHAAJJAA MMEEFFTTAAHHAA LLBBEEKKKKAALLII
HHAAJJAA FFAATTEEMMAA BBEENNNNAANNII
HHAAJJ LLAAHHLLOOUU MMOOHHAAMMMMEEDD
EETT
HHAAJJ SSAAYYEERRHH MMOOHHAAMMMMEEDD

AA llaa mméémmooiirree ddee mmaa ttaannttee FFAATTEEMMAA
QQuuee ccee ttrraavvaaiill ssooiitt uunnee pprriièèrree ppoouurr llee rreeppooss
ddee ttoonn ââmmee..

A tous mes amis HAJAR, KENZA, MERIEM, REDA, IKRAM, MAHA, ZINEB, LAMIAE,
GHIZLANE, CELIN, AMAL, KAWTAR, NISRINE, HAJAR, GHITA, SIHAM, et tous les autres que j ai oubliés.
A TOUS LES MEMBRES DU SERVICE DE MEDECINE INTERNE
DU CHU HASSAN II DE FES

REMERCIMENTS

AA mmoonn mmaaîîttrree eett pprrééssiiddeenntt ddee tthhèèssee
MMmmee llee PPrrooffeesssseeuurr AA..AAMMAARRTTII
PPrrooffeesssseeuurr dd’’aannaattoommoo ppaatthhoollooggiiee
CCHHUU HHAASSSSAANN IIII DDEE FFEESS
AA ll’’hhoonnnneeuurr qquuee vvoouuss nnoouuss ffaaiitteess eenn aacccceeppttaanntt ddee pprrééssiiddeerr llee
jjuurryy ddee nnoottrree tthhèèssee eesstt ppoouurr nnoouuss ll’’ooccccaassiioonn ddee vvoouuss
ttéémmooiiggnneerr nnoottrree pprrooffoonnddee rreeccoonnnnaaiissssaannccee ppoouurr vvooss qquuaalliittééss
hhuummaaiinneess..
VVeeuuiilllleezz ttrroouuvveezz iiccii,, ll’’eexxpprreessssiioonn ddee nnoottrree ggrraannddee
eessttiimmee..

A notre maître et rapporteur de thèse
MMmmee.. LLee pprrooffeesssseeuurr WW.. BBOONNOO
PPrrooffeesssseeuurr aaggrrééggéé ddee MMééddeecciinnee iinntteerrnnee
CCHHUU HHAASSSSAANN IIII FFEESS
VVoouuss mm’’aavveezz aaccccoorrddee’’ bbeeaauuccoouupp ddee vvoottrree tteemmppss pprréécciieeuuxx,, eett
vvoouuss mm’’aavveezz gguuiiddéé aavveecc rriigguueeuurr ppoouurr ll’’ééllaabboorraattiioonn ddee ccee
ttrraavvaaiill..
VVoouuss aavveezz ssuusscciittéé ttoouutt aauu ccoouurrss ddee mmoonn ppaassssaaggee ddaannss vvoottrree
sseerrvviiccee ttoouuttee ll’’aaddmmiirraattiioonn,, eenn mm’’iinncciittaanntt ssaannss hhééssiittaattiioonn àà
ffaaiirree ppaarrttiiee ddee vvoottrree ééqquuiippee ssoouuss vvoottrree gguuiiddee..
VVeeuuiilllleezz ttrroouuvveerr iiccii llee ttéémmooiiggnnaaggee ddee mmaa pprrooffoonnddee rreeccoonnnnaaiissssaannccee eett mmeess rreemmeerrcciieemmeennttss

AA nnoottrree mmaaîîttrree eett jjuuggee ddee tthhèèssee
MMrr llee PPrrooffeesssseeuurr MM..HHIIDDAA
PPrrooffeesssseeuurr ddee ppééddiiaattrriiee
CCHHUU HHAASSSSAANN IIII DDEE FFEESS
NNoouuss aavvoonnss ééttéé ttoouucchhééss ppaarr llaa bbiieennvveeiillllaannccee
eett llaa ccoorrddiiaalliittéé ddee vvoottrree aaccccuueeiill..
NNoouuss ssoommmmeess ttrrèèss sseennssiibblleess àà ll’’hhoonnnneeuurr qquuee
vvoouuss nnoouuss ffaaiitteess eenn aacccceeppttaanntt ddee jjuuggeerr
nnoottrree ttrraavvaaiill..
CC’’eesstt ppoouurr nnoouuss ll’’ooccccaassiioonn ddee vvoouuss ttéémmooiiggnneerr
eessttiimmee eett rreessppeecctt..

AA nnoottrree mmaaîîttrree eett jjuuggee ddee tthhèèssee
MMrr llee PPrrooffeesssseeuurr MM..FF..BBEELLEEHHSSSSEENN
PPrrooffeesssseeuurr ddee nneeuurroollooggiiee
CCHHUU HHAASSSSAANN IIII FFEESS
NNoouuss ssoommmmeess ppaarrttiiccuulliièèrreemmeenntt ttoouucchhééss ppaarr llaa ssppoonnttaannééiittéé
eett llaa ggeennttiilllleessssee aavveecc llaaqquueellllee vvoouuss aavveezz bbiieenn vvoouulluu
aacccceepptteerr ddee jjuuggeerr ccee ttrraavvaaiill..
NNoouuss VVoouuss rreemmeerrcciioonnss ccee ggrraanndd hhoonnnneeuurr qquuee vvoouuss nnoouuss
ffaaiitteess..
VVeeuuiilllleezz aacccceepptteerr,, cchheerr mmaaîîttrree,, ccee ttrraavvaaiill aavveecc ttoouuttee nnoottrree
eessttiimmee eett hhaauuttee vvéénnéérraattiioonn..

AA nnoottrree mmaaîîttrree eett jjuuggee ddee tthhèèssee
MMmmee llee PPrrooffeesssseeuurr SS..TTIIZZNNIITTII
PPrrooffeesssseeuurr ddee rraaddiioollooggiiee
CCHHUU HHAASSSSAANN IIII DDEE FFEESS
NNoouuss aavvoonnss ééttéé ppaarrttiiccuulliièèrreemmeenntt éémmuuss ppaarr llaa ggeennttiilllleessssee
aavveecc llaaqquueellllee vvoouuss aavveezz bbiieenn vvoouulluu aacccceepptteerr ddee jjuuggeerr ccee
ttrraavvaaiill..
NNoouuss nnee vvoouuss rreemmeerrcciieerroonnss jjaammaaiiss aasssseezz ddee ccee ggrraanndd
hhoonnnneeuurr qquuee vvoouuss nnoouuss ffaaiitteess..
VVeeuuiilllleezz aacccceepptteerr,, cchheerr mmaaîîttrree,, ccee ttrraavvaaiill aavveecc ttoouuttee nnoottrree
eessttiimmee eett rreessppeecctt..

A mes chers professeurs DR .SAMIRA RABHI DR AJDI FARIDA DR HARZY TAOUFIK ET Dr SQUALLI TARIK

1
INTRODUCTION

2
Le terme de vascularite ou d’angéite regroupe un champ immense de la
pathologie systémique qui est caractérisé sur le plan histo-pathologique par une
inflammation et une altération des vaisseaux sanguins.
Il en résulte une ischémie des tissus vascularisés par les vaisseaux ainsi
oblitérés. Ce processus inflammatoire peut atteindre tous les vaisseaux quelque soit
leur taille et leur localisation ; d’où l’existence d’un large spectre d’atteintes ;
regroupées dans le syndrome des vascularites.
Les vascularites représentent un champ très immense d’étude à l’échelle
nationale et internationale concernant les différentes présentations cliniques, le
groupement syndromique et surtout l’approche thérapeutique.
L’objectif de ce travail est d’étudier cette pathologie dans le service de
médecine interne du CHU HASSAN II de FES afin de déterminer les différentes
manifestations cliniques des vascularites systémiques, les critères retenus pour le
diagnostic, le traitement suivi et l’évolution.
Nous allons commencer notre travail par une étude épidémiologique des
vascularites, suivie d’un rappel historique et physiopathologique tout en signalant le
rôle actuel des ANCA. Nous allons rappelé les classifications des vascularites et les
critiquer, puis rapporter les observations de 20 patients présentant une vascularite
diagnostiquée et suivie au sein de service de médecine interne, puis nous allons
comparer notre série à la revue de la littérature en ce qui concerne la répartition
épidémiologique, le mode de révélation, les manifestations cliniques, les examens
complémentaires demandés, le traitement proposé et surtout les nouveautés
thérapeutiques en matière d’immunosuppresseurs.
3
GENERALITES

4
I-DEFINITION
II-EPIDEMIOLOGIE DES VASCULARITES

5
I-DEFINITION DES VASCULARITES :
Le terme de vascularite a longtemps été attribué à une atteinte artérielle
exclusive, et ce n’est que secondairement que l’on a reconnu la participation de
l’inflammation et de la thrombose veineuse dans certaines vascularites.[7]
C’est ainsi que l’on définit actuellement une vascularite systémique comme
une inflammation de la paroi vasculaire de tout type de vaisseaux, quelque soient sa
taille et son type. Cette inflammation aboutit à la destruction de la paroi vasculaire
et une sténose ou occlusion de la lumière du vaisseau.
Les vascularites peuvent être localisées, leur intérêt clinique est alors limité,
ou généralisées mettant en jeu le pronostic vital.
II –EPIDEMIOLOGIE DES VASCULARITES SYSTEMIQUES
Les efforts menés pour établir des systèmes de classification consensuels ont
largement bénéficié aux enquêtes épidémiologiques des vascularites et permis
d’exploiter une approche originale de l’étude des mécanismes physiopathologiques
en jeu.
Il existe en effet peu d’études épidémiologiques ayant étudié la fréquence et
la distribution des vascularites dans le monde. On ne dispose le plus souvent que
de données concernant une forme ou des constatations faites dans des centres
spécialisés, les études menés jusqu’à nos jours ont permis de reconnaître la
prévalence de certaines vascularites.
*C’est ainsi que la maladie de Horton est la plus fréquente des vascularites
systémiques primitives, l’incidence annuelle est estimée entre 15 et 35 cas /100 000

6
habitants des pays du nord de l’Europe ayant un âge supérieur à 50 ans et de moins
de 10 /100 000 habitants en Europe du sud [8].
Cette répartition géographique concorde d’ailleurs avec la distribution des
allèles HLA –DRB 04 et DRB 01 étroitement associée à la maladie de Horton et qui
prévalent dans les populations nordiques.
*Avec plus de 100 000 cas répertoriés à ce jour, la maladie de Kawasaki (MK)
est la première vascularite infantile, bien que décrite dans le monde entier, la MK est
caractérisée par une franche prédilection pour les enfants asiatiques.
Au japon où l’incidence annuelle est de 100 /100 000 enfants moins de 5
ans, la MK est d’environ 10 fois plus fréquente qu’en Europe et qu’en Amérique du
nord. Cette disparité ethnique a été confirmée au sein d’une population nord
américaine avec des incidences de 33, 23 et13/100 000 habitants moins de 5 ans
respectivement pour les enfants d’origine asiatique, les enfants noirs et les enfants
blancs. [8]

7
*La prévalence de la maladie de Wegener (MW) est estimée entre 26 et 95/100
000 habitants dans différentes régions de l’Amérique du nord et de l’Europe et
comme c’est le cas pour la maladie de Horton, ces donnés épidémiologiques
suggèrent une plus forte répartition de la MW dans les pays nordiques (figure1) [6].
Figure 1 : montrant la répartition de la granulomatose de Wegener et la nette
prédominance des pays nordiques [6].

8
*Pour ce qui est de la PAN, c’est une maladie rare et son incidence annuelle
est estimée à 4.6/million d’habitants en Angleterre. Il reste qu’il est difficile d’établir
précisément l’incidence de cette affection, car les descriptions de la PAN rapportent
de nombreuses observations qui correspondent en fait à d’autres maladies. Il s’agit
d’une maladie rare peu commune mais non grave, l’âge moyen de début est de 40
ans et le sex-ratio homme/femme est de 1.6/1 (fig2) [6].
Figure 2 : Répartition européenne de la PAN avec la nette prédominance de cette
vascularite dans les pays nordiques. [6]

9
*Pour ce qui est de la granulomatose de Churg et Strauss, l’incidence et la
prévalence sont difficiles à déterminer, mais on note selon les dernières études
publiées une prévalence plus importante dans les pays nordiques par rapport à une
étude française (fig3) [6] car les séries rapportées incluent de nombreux cas de
vascularites d’autres étiologies. La maladie de Churg et Strauss est cependant une
maladie rare, elle débute à tous les ages excepté chez l’enfant ; l’âge moyen de
début des symptômes est de 44 ans, le sex-ratio est de 1.3/1
Figure 3 ; répartition européenne de la maladie de Churg et Strauss ) [6]

10
*L’artérite de Takayasu est aussi une maladie rare, beaucoup moins fréquente
que la maladie de Horton, elle atteint de préférence les femmes jeunes ou les
adolescentes. Bien que la maladie soit plus fréquente en orient, elle peut être
rencontrée dans tous les pays et d’origine ethnique différente. Cette maladie semble
associée à des gènes HLA-DR2 .MB1 au japon et HLA-DR4 .MB3 aux états unis.
*Ces résultats et cette différence de répartition permettent de retenir quelques
remarques :
L’incidence globale des vascularites est en nette augmentation, et ceci peut
être du au progrès des techniques immunologiques, et à l’intérêt que lui accorde
certains centres spécialisés en la matière.
Les variations géographiques décrites pour certaines vascularites soulèvent
ainsi l’existence de facteurs génétiques prédisposants, et l’intervention d’un
mécanisme infectieux, ce qui peut expliquer l’augmentation de prévalence de
certaines vascularites en période hivernale,
L’origine infectieuse est discutée pour la PAN associée au virus de HBV (fig4) )
[6], et depuis la diffusion de la vaccination contre l’hépatite B, les études menées sur
le plan épidémiologique montrent une baisse de la recrudescence annuelle [4]. Cette
forme ne représente que 10% des formes cliniques de la PAN, elles sont souvent
associées à une hypertension artérielle, une orchite et des signes digestifs et moins
souvent un asthme. Les sérologies virales sont insuffisantes. Il est nécessaire de
rechercher l'ADN viral dans le sang. Les thérapeutiques antivirales destinées à
obtenir la séroconversion sont indiquées dans cette forme [5]. L’étude menée par
Guillevin et all montre que le virus de l’hépatite B responsable de la PAN est le même
virus de HVB responsable d’hépatites et de cirrhose de foie ; il est transmis par le
sang, les rapports sexuels et la salive. Il a une réplication importante, et il n’existe
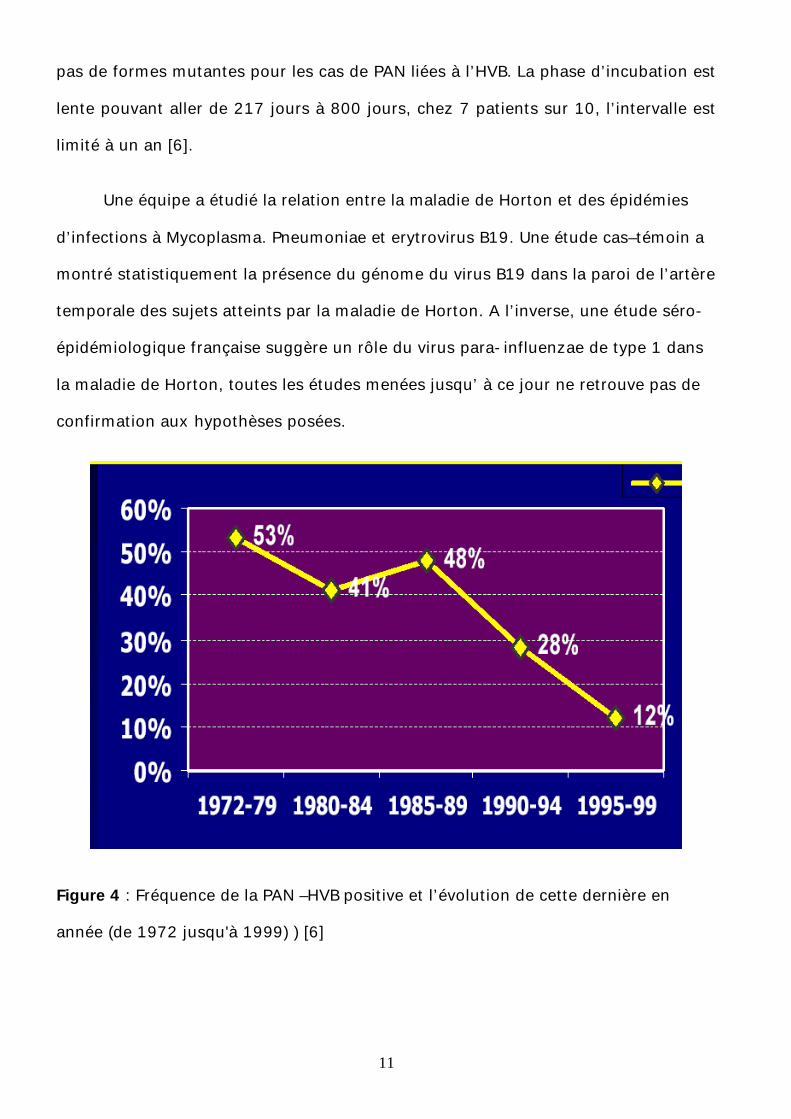
11
pas de formes mutantes pour les cas de PAN liées à l’HVB. La phase d’incubation est
lente pouvant aller de 217 jours à 800 jours, chez 7 patients sur 10, l’intervalle est
limité à un an [6].
Une équipe a étudié la relation entre la maladie de Horton et des épidémies
d’infections à Mycoplasma. Pneumoniae et erytrovirus B19. Une étude cas–témoin a
montré statistiquement la présence du génome du virus B19 dans la paroi de l’artère
temporale des sujets atteints par la maladie de Horton. A l’inverse, une étude séro-
épidémiologique française suggère un rôle du virus para-influenzae de type 1 dans
la maladie de Horton, toutes les études menées jusqu’ à ce jour ne retrouve pas de
confirmation aux hypothèses posées.
Figure 4 : Fréquence de la PAN –HVB positive et l’évolution de cette dernière en
année (de 1972 jusqu'à 1999) ) [6]

12
Une autre hypothèse repose sur l’intervention d’autres facteurs d’exposition
environnementale dans le déclenchement des vascularites. Pour la MK, il a été
incriminé un rôle favorisant d’un habitat à proximité de l’eau ou de l’exposition aux
produits de shampoing de tapis mais ceci reste controversé [8].
13
RAPPEL THEORIQUE

14
I –HISTORIQUE
II-PHISIOPATHOLOGIE DES VASCULARITES SYSTEMIQUES
1-Maladie de Horton
2-Maladie de Takayasu
3-Les vascularites nécrosantes
III-CLASSIFICATION DES VASCULARITES
1-classification de Zeek
2-classification de Fauci
3- classification de Wechsler
4-Classification de Chapel Hill
5-Classification de Kahn et Peltier
6-Classification de lie
7-Les critères diagnostiques des vascularites
systémiques de l’ACR

15
I-HISTORIQUE :
Les troubles vasculaires sont connus depuis la plus haute antiquité ; on trouve
déjà dans les anciens papyrus d’Égypte la description de l’anévrisme artériel et des
varices. La première description des artériopathies est faite en 1554 par Antoine
saporta, professeur à l’université de Montpellier mais ce n’est qu’à la fin du XVI
siècle et grâce à la levée de l’interdit des autopsies que les connaissances sur
vasculopathies s’élargissent et vers les années 1815, on évoque la nature
inflammatoire des troubles vasculaires ainsi on décrit qu’il existe de nombreuses
formes de vascularites suivant le type de vaisseau qui est touché et de la nature de
l’atteinte.
P.M.Zeek propose en 1952 la première classification des vascularites en 4
types: les vascularites d’hypersensibilité, les vascularites granulomateuses
allergiques, la périartérite noueuse et enfin, l’artérite temporale.
Par la suite, d’autres types de classifications ont été proposés. Plus
développées, celles-ci tiennent compte de la taille des vaisseaux atteints, de la
nature de l’atteinte, des critères histologiques et de la localisation préférentielle des
lésions.
*L’artérite temporale a était connue depuis 1436 mais ce n’est qu’à la fin du
XIX siècle qu’un chirurgien anglais décrit l’artérite temporale thrombotique du sujet
âgé.
En 1932, b.t. Horton et b.t. Magath rapportent l’observation de deux patients
présentant une fièvre, une faiblesse, une anorexie, une perte de poids et une
anémie, une zone douloureuse longe les vaisseaux de la tempe et le scalp. Une
biopsie des vaisseaux atteste les lésions typiques d’artérite et de périartérite

16
chronique avec granulome ainsi que des cellules géantes à l’origine du nom
d’artérite giganto-cellulaire qui lui sera attribué par certains auteurs. [1]
Fig5 Fig6
Figure 5 : Jan van eyck (1390-1441) le chanoine van der paele avec une artérite
temporale [1]
Figure6 : piero di Cosimo (1462-1521) francesco giamberti avec une artérite
temporale [1]

17
*L’aorto-artérite a été décrite pour la première fois en 1761, c’ était le cas d’
une femme de 40 ans hospitalisée à plusieurs reprises au cours des six dernières
années pour une insuffisance respiratoire et dont le pouls radial n’est pas
perceptible, la malade décède et son médecin Morgagni s’est empressé d’effectuer
une autopsie dont le résultat est revenu en faveur de lésions aortiques avec
anévrisme de la partie inférieure de l’aorte thoracique, mais ce n’est qu’en 1908 et
au cours du 12 ème congrés annuel de la société japonaise d’ophtalmologie que
Mikito Takayasu a décrit le cas d’ une patiente de 21 ans qui présente une
rétinopathie ischémique avec anastomose artério-veineuse .
*La périartérite noueuse a été décrite pour la première fois en 1866, les deux
auteurs kussmaul et Meier et au long de 35 pages ont rapporté le cas de deux
patientes dont la symptomatologie comprenait des myalgies, douleurs abdominales,
une neuropathie et une protéinurie, l’autopsie de la 1ère patiente a révélé des trajets
artériels indurés et nodulaires, l’examen microscopique met au jour des lésions du
glomérule rénal et des artères musculaires. Dans le deuxième cas, la biopsie
musculaire a été réalisée et l’étude histologique confirme l’atteinte de la paroi
artérielle accompagnée d’une réaction inflammatoire péri-artérielle. [1]
*En 1936, Friedrich Wegener réalise l’étude d’une forme frontière de la PAN. Il
s’agit d’une entité autonome comportant une granulomatose avec une rhinite et
néphropathie, les lésions intéressent les branches de l’artère rénale d’un diamètre
supérieur à 70 micromètres. En 1985, l’étude de la maladie de wegener connaît un
regain d’intérêt lorsque hall van der woude découvre dans le sérum des malades des
auto-anticorps réagissant avec le cytoplasme des polynucléaires neutrophiles ; les
ANCA. Ces anticorps sont dirigés contre la protéinase 3; un constituant essentiel des
granules alpha du cytoplasme des polynucléaires neutrophiles.

18
*En 1914, J. G monckenberg et A. R Lamb décrivent le premier cas de PAN
associé à un asthme et à une hyperéosinophilie importante, mais l’établissement
définitif de l’entité est dû à Jacob Churg et lotte Strauss en 1950. Les manifestations
cliniques de la maladie sont l’asthme, l’hyperéosinophilie, des infiltrats pulmonaires,
une sinusite para-nasale et une mono-névrite multiple. On y observe souvent la
présence de C- ANCA, qui étonnamment réagissent avec la myélopéroxydase des
neutrophiles alors que cet aspect est habituellement dû à la présence d’ anti PR3.
*En 1879, le chirurgien viennois felix von Winiwater étudie le cas d’un patient
de 57 ans qui souffre depuis 12 ans de lésions ischémiques des membres inférieurs
ayant abouti à une gangrène spontanée. L’étude histologique de la pièce
d’amputation révèle la présence de nombreuses thromboses dans les artères et les
veines, cependant la structure de l’intima n’est pas touchée ce qui pousse Winiwater
à qualifier l’atteinte d’endoarteritis obliterans. Plusieurs cas analogues sont décrits
par Léo burger à new York en 1908. Ce dernier propose le nom de thromboangéite
oblitérante qui sera employé par la suite dans la littérature. [1]
II –PHISIOPATHOLOGIE DES VASCULARITES SYSTEMIQUES
La pathogénie des vascularites reste encore mal comprise. En effet, pour
faciliter cette étude, on va distinguer entre de types de groupes de vascularites sur
le point physiopathologique: les vascularites primitives nécrosantes et le 2éme
groupe des vascularites comprenant la maladie de Horton et la maladie de Takayasu.
A-la maladie de Horton :
L’étiologie de cette affection reste encore mal déterminée, on incrimine 2
facteurs : environnementaux et génétiques

19
L’observation de la maladie de Horton a incriminé le rôle éventuel d’un facteur
infectieux; on a noté une prévalence augmentée des anticorps anti virus para-
influenzae de type 1. Une relation étroite a été mise en évidence entre des pics
d’incidence de la maladie de Horton et les épidémies à mycoplasma pneumoniae,
parvovirus B19 et le chlamydia pneumoniae.
D’autres enquêtes épidémiologiques ont mis en évidence des fluctuations
cycliques entre l’incidence de la maladie de Horton et les infections à parvovirus
suggérant que le virus a un rôle pathogène dans cette maladie, néanmoins d’autres
études n’ont pas montré d’association entre cette infection et le début de la
maladie.
Par ailleurs, la maladie de Horton à biopsie positive semble survenir
préférentiellement en période hivernale, alors que l’incidence de la maladie à biopsie
négative semble plus importante en période estivale. Cependant, toutes les études
ne sont pas unanimes quand à l’aspect saisonnier de la maladie de Horton. Une
cause toxique et l’ensoleillement ont pu être évoqués mais aucune étude
épidémiologique récente n’a été convaincante.
Le stress aussi a été cité comme élément favorisant la survenue de cette
maladie.
Sur le plan génétique, l’étude de Barrier et coll. a permis de mettre en
évidence l’association de la maladie de Horton et l’antigène HLA DR4 présent chez
40% des patients et chez 20% des témoins. [9]
Plus récemment, a été mise en évidence une augmentation significative du
groupe HLA DR7, cependant sur le plan pratique, la présence ou l’absence d’allèles
du complexe majeur d’histocompatibilité chez les patients suspects de la maladie de

20
Horton ne modifie pas la conduite à tenir diagnostique et thérapeutique et n’a pas
d’incidence pronostique sur la maladie.
Une explication immuno-histologique de la maladie de Horton a été proposée
(fig 7) [3]. Dans cette étude proposé par Weyand and Goronzy, un antigène (Ag) va
être reconnu par les lymphocytes T de l’adventice, ces dernières vont traverser la
paroi artérielle via les vasa vasorum, et vont subir une expansion clonale ce qui
aboutit à la formation d’INF gamma (interféron gamma). Ces cellules stimulées par
l’INF vont entraîner la différentiation et la migration de macrophages et la formation
de cellules géantes.
Dans l’adventice, les macrophages vont produire des cytokines pro
inflammatoires (IL-1et IL-6) alors que dans la média et l’intima, ils contribuent aux
lésions artérielles par la production des métalloprotéines. La destruction artérielle
serait donc associée à des phénomènes de réparation avec la production de facteurs
angiogéniques par les cellules mononucléées et cellules géantes multinucléées.
Toutes ces anomalies vont aboutir à la dégradation de la limitante élastique interne
et à l’occlusion hyperplasique de la lumière.

21
Figure 7 : Explication immunopathologique de la maladie de HORTON [3].
B- La maladie de Takayasu :
C’est une panartérite caractérisée par une infiltration par des cellules
mononuclées inflammatoires et occasionnellement par des cellules géantes, on note
une prolifération et une fibrose de l’intima, une vascularisation et des lésions
séquellaires de la média, une fragmentation et une dégénérescence de la limitante
élastique, la lumière du vaisseau ainsi réduite peut être associé ou non à des
thromboses. L’atteinte de la vasa vasorum est fréquente, les modifications
pathologiques des différents organes au cours de la maladie de Takayasu sont le
reflet de l’ischémie des vaisseaux atteints.

22
Les mécanismes immuno-pathogéniques, de nature encore inconnue, sont
suspectés dans cette maladie, et comme c’est le cas pour les autres types de
vascularites, les complexes immuns sont présents mais leur signification
pathogénique reste obscure.
C-Les vascularites nécrosantes :
Le développement d’une lésion vasculaire inflammatoire et nécrosante semble
être un processus complexe incluant des réactions immunitaires non seulement
humorales mais aussi à médiation cellulaire.
1-Dépôt des complexes immuns circulants :
Ce mécanisme était longtemps considéré comme le principal mécanisme
physiopathologique des vascularites :
L’excès d’Ag et l’augmentation de la perméabilité vasculaire, induit au
passage des CI circulants et leur dépôt dans la paroi vasculaire, il se produit un
afflux des polynucléaires neutrophiles (PNN) facilité par le pouvoir chimiotactique de
certaines fractions du complément par le biais des molécules d’adhésion. [10,11]
Les PNN activés, libèrent leurs enzymes lysosomiales et induiraient la nécrose
de la paroi artérielle.
La localisation et l’intensité de ses lésions dépendent en effet de plusieurs
facteurs : [12]
*la nature de l’Ag qui les constitue.
*les qualités physico-chimiques des CI et la durée de leur demi-vie.
*les facteurs modifiant leur perméabilité vasculaire.

23
*la pression artérielle.
Ce mécanisme est retenu pour les vascularites nécrosantes notamment pour la
périartérite noueuse, en particulier lorsqu’elle est liée à une infection par le HVB, le
purpura rhumatoïde, la maladie de Wegener et les vascularites des
cryoglobulinémies.
2-Les anticorps anti cytoplasme des polynucléaires neutrophiles : les
ANCA
Les ANCA sont les auto-anticorps dirigés contre les Ag des PNN et des
monocytes.
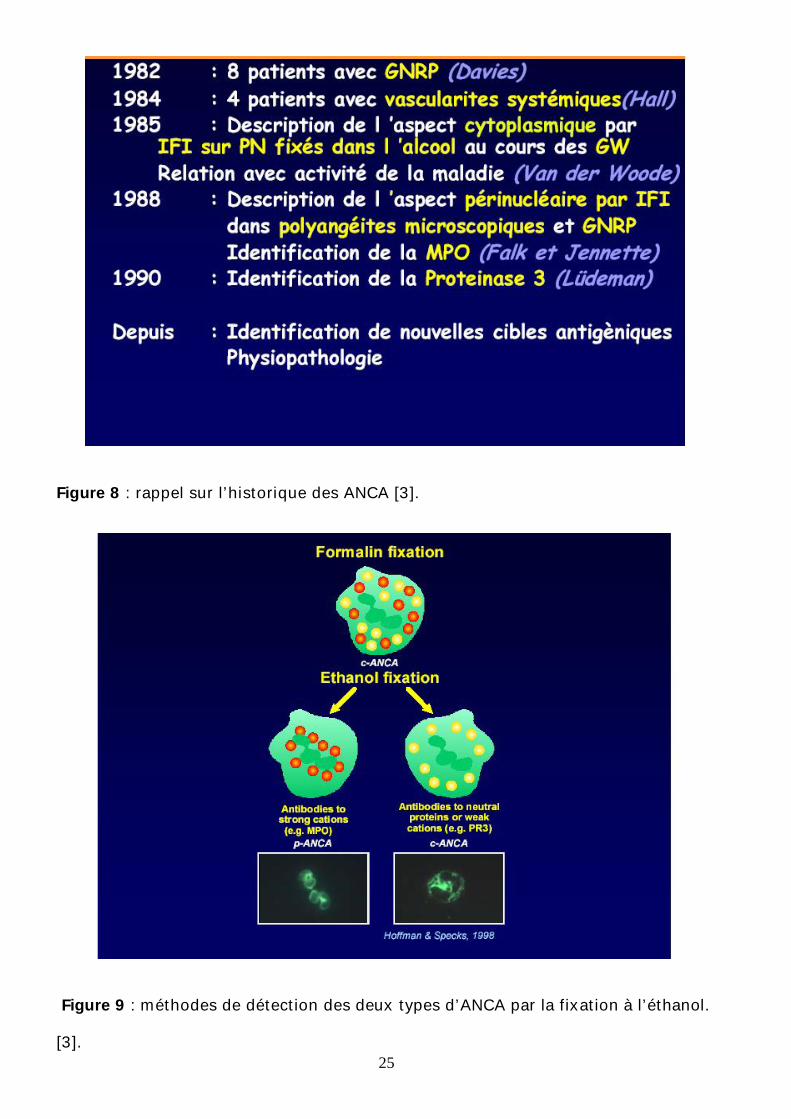
a-Découverte des ANCA :
Les ANCA connus depuis 1985 (fig 8) [3] représentaient un marqueur de
l’inflammation au cours de la maladie de Wegener. Ultérieurement, ils étaient utilisés
comme des marqueurs de l’inflammation au cours des vascularites touchant les
vaisseaux de petit calibre comme la polyangéite microscopique, le SCS et les
glomérulonéphrites rapidement progressives avec nécrose focale , sans dépôt
d’immunoglobulines, dites pauci-immunes [26]. Leur spécificité au cours des
vascularites nécrosantes est très importante surtout quand la spécificité aux ANCA
est de type anti-protéinase 3 (anti PR3) et que la recherche est positive tant en
immunofluorescence qu’en enzyme-linked immunosorbent assay (ELISA). [19]
b-La détection des ANCA :
La détection des ANCA se fait par IF indirecte avec 2 types de fluorescence
cytoplasmique qui sont observées :

24
Cet aspect est dû à la redistribution des Ag solubles du cytoplasme durant la
préparation des PNN et la fixation à l’alcool (fig 9) ,on distingue 2 types de
fluorescence; cytoplasmique diffuse des PNN et des monocytes dits C-ANCA, et une
fluorescence à renforcement péri-nucléaire des PNN dites P-ANCA (figures 10 et
11), [15,27].
Quand l’aspect en IF ne correspond ni à celui des P-ANCA ni à celui des C-
ANCA, on parle d’un aspect atypique (X-ANCA). Les ANCA sont souvent des
anticorps de classe IgG. [13]
Les P-ANCA et les C-ANCA réagissent avec les PNN et les monocytes, mais pas
avec les macrophages, les polynucléaires éosinophiles et les lymphocytes. [19]
Plusieurs enzymes lysosomiales contenues dans les granulations primaires et
secondaires des PNN ont été identifiées comme cible aux ANCA, les 2 principaux
antigènes connus sont la PR3 et la MPO (myélopéroxydase). La PR3 est l’enzyme
reconnue par la majorité des C-ANCA et la MPO est reconnue par les P-
ANCA [14].
25
Figure 8 : rappel sur l’historique des ANCA [3].
Figure 9 : méthodes de détection des deux types d’ANCA par la fixation à l’éthanol.
[3].
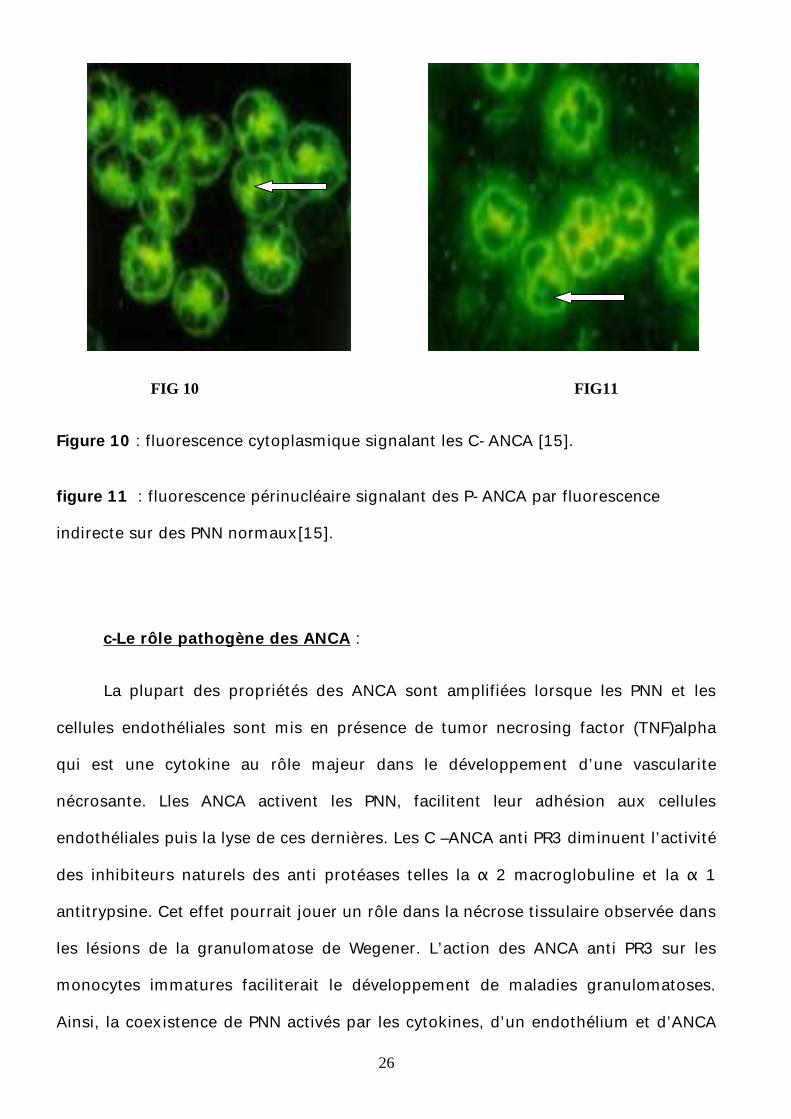
26
FIG 10 FIG11
Figure 10 : fluorescence cytoplasmique signalant les C-ANCA [15].
figure 11 : fluorescence périnucléaire signalant des P-ANCA par fluorescence
indirecte sur des PNN normaux[15].
c-Le rôle pathogène des ANCA :
La plupart des propriétés des ANCA sont amplifiées lorsque les PNN et les
cellules endothéliales sont mis en présence de tumor necrosing factor (TNF)alpha
qui est une cytokine au rôle majeur dans le développement d’une vascularite
nécrosante. Lles ANCA activent les PNN, facilitent leur adhésion aux cellules
endothéliales puis la lyse de ces dernières. Les C –ANCA anti PR3 diminuent l’activité
des inhibiteurs naturels des anti protéases telles la α 2 macroglobuline et la α 1
antitrypsine. Cet effet pourrait jouer un rôle dans la nécrose tissulaire observée dans
les lésions de la granulomatose de Wegener. L’action des ANCA anti PR3 sur les
monocytes immatures faciliterait le développement de maladies granulomatoses.
Ainsi, la coexistence de PNN activés par les cytokines, d’un endothélium et d’ANCA

27
pourrait déclencher une cascade de réactions menant à l’apparition de lésion de
vascularite (fig 12 et fig 13).
Figure 12 : Activation in vitro des neutrophiles après fixation de complexes immuns
solubles [3].
d-Le rôle des ANCA en pratique :
Habituellement, les patient atteints d’une vascularite systémique nécrosante
ont des ANCA positifs, les C-ANCA sont détectables chez 60 à 90% des patients
atteints d’une granulomatose de Wegener systémique et 50 à 75% de ceux
présentant une forme localisée. Les P-ANCA, par contre, sont plus caractéristiques
des glomérulonéphrites pauci-immunes (80% des patients sont P-ANCA +) de la
polyangéite microscopique (50 à 75% des patients) et du SCS (34 à 47% des
patients).
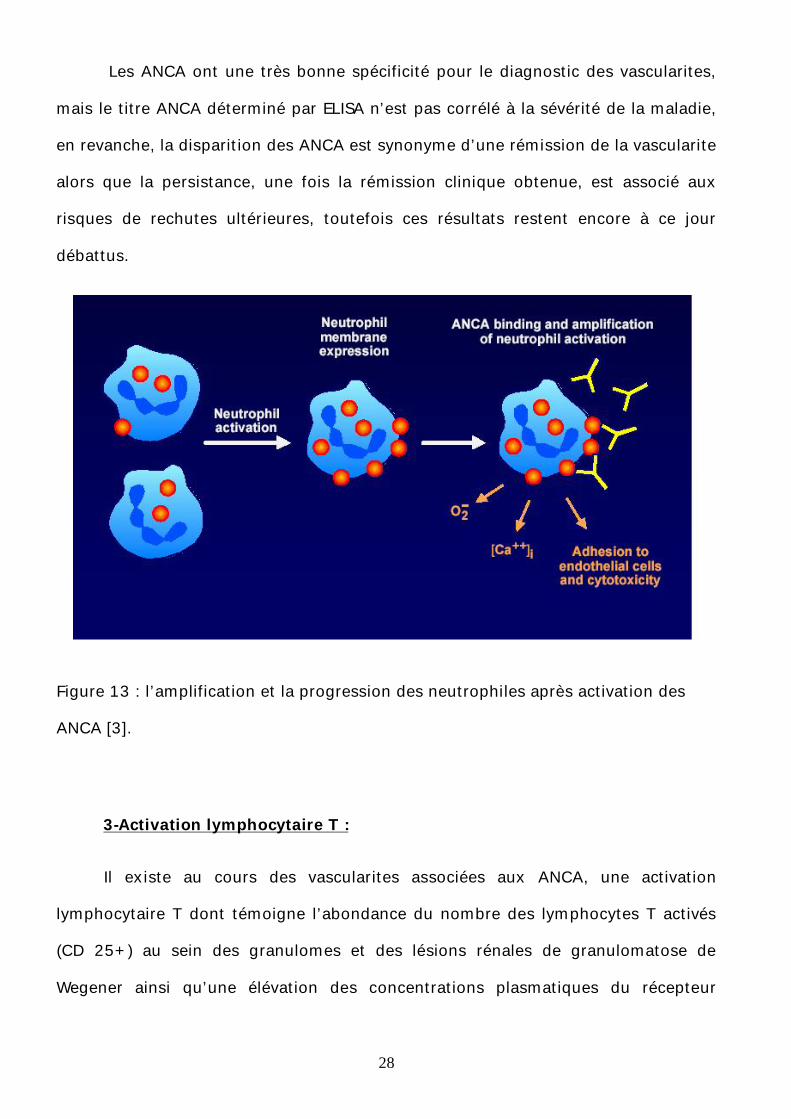
28
Les ANCA ont une très bonne spécificité pour le diagnostic des vascularites,
mais le titre ANCA déterminé par ELISA n’est pas corrélé à la sévérité de la maladie,
en revanche, la disparition des ANCA est synonyme d’une rémission de la vascularite
alors que la persistance, une fois la rémission clinique obtenue, est associé aux
risques de rechutes ultérieures, toutefois ces résultats restent encore à ce jour
débattus.
Figure 13 : l’amplification et la progression des neutrophiles après activation des
ANCA [3].
3-Activation lymphocytaire T :
Il existe au cours des vascularites associées aux ANCA, une activation
lymphocytaire T dont témoigne l’abondance du nombre des lymphocytes T activés
(CD 25+) au sein des granulomes et des lésions rénales de granulomatose de
Wegener ainsi qu’une élévation des concentrations plasmatiques du récepteur

29
soluble à l’interleukine 2, une production in situ de TNF alpha, d’ IL1béta et IL2r a
été démontré au sein des lésions rénales des glomérulonéphrites pauci immunes.
La mise en évidence récente de phénotypes différents du SCS va aussi dans le
sens de mécanismes cellulaires et humoraux différents au cours de la même
maladie.
III-CLASSIFICATION DES VASCULARITES
La vascularite et ses conséquences peuvent être la première et unique
manifestation de la maladie ; la vascularite peut être primitive, ou secondaire à une
autre affection ; dans les vascularites secondaires, les facteurs incriminés sont le
plus souvent des médicaments ; des infections bactériennes et virales, des maladies
de système ou des hémopathies malignes. Dans cette forme de vascularite,
l’existence de dépôts de CI sur des Ag plantés sur des organes rend vraisemblable
l’implication de phénomènes immunologiques.
Dans les vascularites dites primitives, c’est la découverte des ANCA qui a
permis de reconnaître les phénomènes étiopathogéniques mis en jeu.
Il n’existe pas de classification universellement reconnue des vascularites
systémiques, celle de Chapel Hill est la plus connue ; elle reste critiquable. Nous
allons ainsi rapporter respectivement les anciennes classifications des vascularites
abondonnées de nos jours et les trois principales classifications utilisées : celle de
Chapel Hill, de Lie et de Kahn Peltier. Nous allons apporter également un avis sur ces
différentes classifications.

30
1-Classification de Zeek :
C’est une classification histologique faite par Zeek en 1953, et qui introduit le
concept de vascularites nécrosantes. Elle permet de distinguer entre deux grands
groupes histologiques : les angéites nécrosantes et les artérites à cellules géantes.
Le groupe de vascularites nécrosantes regroupe les vascularites qui comprennent
obligatoirement trois éléments histologiques quelque soit la variété clinique : la
nécrose fibrinoïde de la paroi vasculaire, une réaction inflammatoire péri-vasculaire,
et une évolution vers la cicatrisation fibreuse. [16]
La plupart des angéites nécrosantes touchent les artères de moyen calibre
mais les artères de petit calibre et les veines peuvent également être intéressées.
Les artérites à cellules géantes comprennent la maladie de Horton et la
maladie de Takayasu, elles réalisent une infiltration pariétale de cellules
mononuclées et formation de cellules géantes.
Le troisième groupe des vascularites d’hypersensibilité regroupe le purpura
rhumatoïde, les vascularites d’hypersensibilité, les cryoglobulinémies mixtes et les
vascularites au cours des connectivites.
En effet, cette classification regroupe souvent des entités
anatomopathologiques et cliniques différentes (PAN et la granulomatose de
Wegener), inversement elle sépare des entités ayant le même substratum
pathogénique, mais réalisant des tableaux anatomo-cliniques différents comme il
est le cas de la vascularite d’hypersensibilité ou la PAN microscopique, et la PAN
classique. De plus, cette classification est étroitement tributaire de la qualité de la
biopsie.

31
2-Classification de Fauci :
Cette classification fut établie par Fauci en 1978, puis réactualisée en 1981 ;
elle se base en plus des critères de la classification de Zeek, sur des données
pathogéniques et sur certains regroupements syndromiques. [16].
1-Groupe de PAN :
- PAN classique
-vascularite de Churg et Strauss
-syndrome de chevauchement
2-Vascularites d’hypersensibilité :
- Maladie sérique et affections voisines
- purpura rhumatoïde
-vascularites des connectivites
-cryoglobulinémie essentielle
3-Maladie de Wegener
4-Granulomatose lymphomatoïde
5-Artérites à cellules géantes :
-maladie de Horton
- maladie de Takayasu
6-Angéite de Buerger
7-Maladie de Behçet
8-Vascularites du système nerveux central
9-Syndrome lymphoganglionnaire cutanéo-muqueux ou maladie de Kawasaki
10-Vascularites des affections malignes
11-Divers :
-syndrome de Cogan
-vascularite hypocomplémentémique

32
Cette classification garde quelques imperfections, on cite à titre d’exemple
que toutes les vascularites des connectivites sont regroupées dans les vascularites
d’hypersensibilité alors qu’elles ne sont pas toutes de ce type. Par contre, les
vascularites accompagnant les affections malignes sont classées à part alors qu’elles
sont souvent d’hypersensibilité.
3--classification de Wechsler
Cette classification revient sur les critères anatomopathologiques pour la
classification des vascularites malgré les inconvénients rapportés sur la classification
de Zeek, elle distingue entre :
1-Vascularites leucocytoclasiques :
▪vascularites cutanées pures
▪PAN microscopique
*aiguë :
- infectieuse
-toxique
-médicamenteuse
*subaiguë ou chronique :
- purpura rhumatoïde
-cryoglobulinémies mixtes
-connectivites
2-vascularites nécrosantes :
▪PAN classique
▪HTA maligne –athérosclérose
▪maladie de Kawasaki
▪LEAD-PR

33
3-vascularites nécrosantes et granulomateuses :
▪angéite allergique de Churg et Strauss
▪granulomatose de Wegener
▪formes frontières
4-vascularites granulomateuses :
▪maladie de Takayasu
▪maladie de Horton
5-vascularites thrombosantes :
▪maladie de Buerger
▪maladie de Behçet
6-vascularites fibrosantes :
▪périaortite ou anévrysmes inflammatoires aortiques
▪artérites et aortites infectieuses
7-vascularites à prédominance lymphocytaire :
▪infiltrats à lymphocytes normaux :
*LEAD
*rhumatismes inflammatoires : PR
▪infiltrats avec processus lymphomateux :
*papulose lymphomatoïde
*granulome malin centrofacial
*granulomatose lymphoïde de liebow
*LMNH-maladie de hodgkin
Cette classification garde donc beaucoup d’imperfections, les premières
remarques qui lui ont été attribuée sont la séparation artificielle d’aspect relevant
d’une même étiologie comme il est le cas de la vascularite lupique citée dans deux
entités différentes. Inversement, elle regroupe sous le même nom des vascularites

34
correspondant à des processus complètement différents comme il est le cas du
purpura rhumatoïde et des vascularites médicamenteuses.
4-La classification de Chapel Hill
Figure14 : classification de Chapel Hill montrant les 3 types de vaisseaux atteints.
[3].
Elle se base sur des données anatomopathologiques et cliniques, en
distinguant 3 groupes principaux de vascularite selon la taille des vaisseaux
atteints(fig14)[3]. : Vascularites des vaisseaux de gros calibre touchant l’aorte et ses
branches principales, les vascularites de moyen calibre touchant les artères de type
musculaire et les vascularites des petits vaisseaux atteignant les petites artères, les
artérioles et les capillaires. La taille des vaisseaux n’est pas toujours discriminante,
il existe par contre un chevauchement entre certaines formes.[29]

35
la classification de Chapel Hill (tableau 1)[2] par ailleurs n’intègre pas les
données immunologiques en particulier la présence d’ANCA ni les facteurs
étiologiques éventuels comme les infections, les médicaments, les hémopathies
malignes et certains cancers.
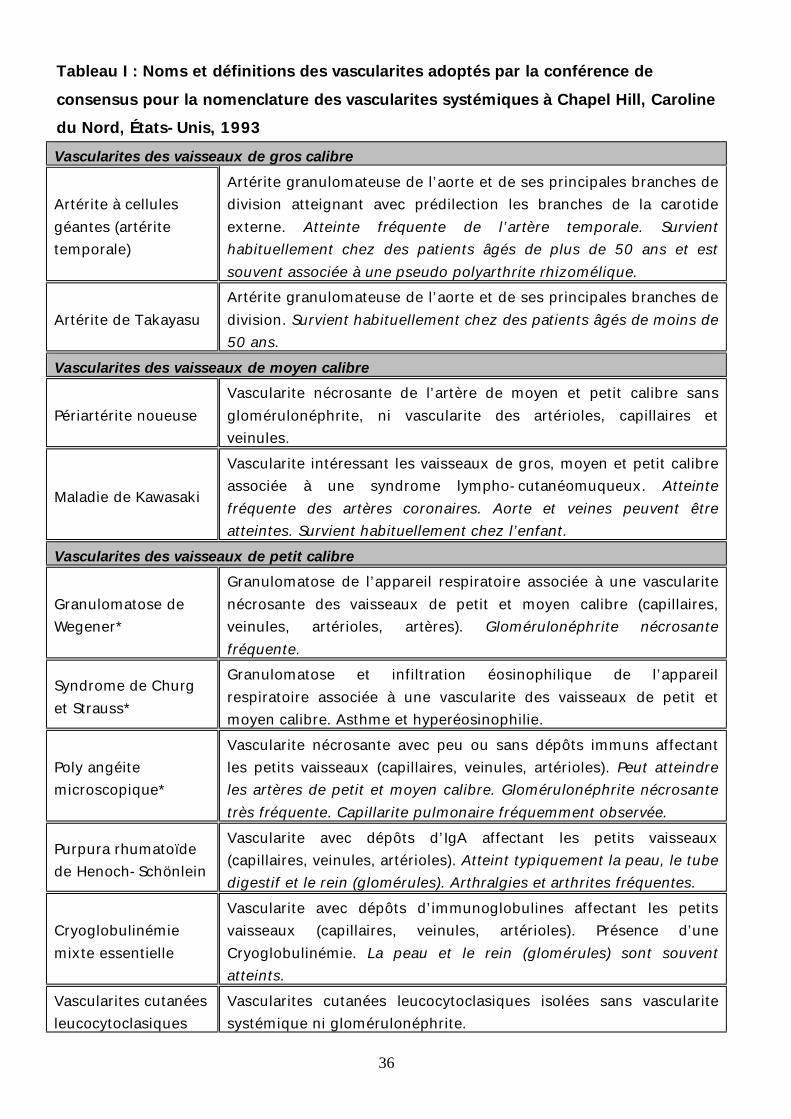
36
Tableau I : Noms et définitions des vascularites adoptés par la conférence de consensus pour la nomenclature des vascularites systémiques à Chapel Hill, Caroline du Nord, États-Unis, 1993 Vascularites des vaisseaux de gros calibre
Artérite à cellules géantes (artérite temporale)
Artérite granulomateuse de l’aorte et de ses principales branches de division atteignant avec prédilection les branches de la carotide externe. Atteinte fréquente de l’artère temporale. Survient habituellement chez des patients âgés de plus de 50 ans et est souvent associée à une pseudo polyarthrite rhizomélique.
Artérite de Takayasu Artérite granulomateuse de l’aorte et de ses principales branches de division. Survient habituellement chez des patients âgés de moins de 50 ans.
Vascularites des vaisseaux de moyen calibre
Périartérite noueuse Vascularite nécrosante de l’artère de moyen et petit calibre sans glomérulonéphrite, ni vascularite des artérioles, capillaires et veinules.
Maladie de Kawasaki
Vascularite intéressant les vaisseaux de gros, moyen et petit calibre associée à une syndrome lympho-cutanéomuqueux. Atteinte fréquente des artères coronaires. Aorte et veines peuvent être atteintes. Survient habituellement chez l’enfant.
Vascularites des vaisseaux de petit calibre
Granulomatose de Wegener*
Granulomatose de l’appareil respiratoire associée à une vascularite nécrosante des vaisseaux de petit et moyen calibre (capillaires, veinules, artérioles, artères). Glomérulonéphrite nécrosante fréquente.
Syndrome de Churg et Strauss*
Granulomatose et infiltration éosinophilique de l’appareil respiratoire associée à une vascularite des vaisseaux de petit et moyen calibre. Asthme et hyperéosinophilie.
Poly angéite microscopique*
Vascularite nécrosante avec peu ou sans dépôts immuns affectant les petits vaisseaux (capillaires, veinules, artérioles). Peut atteindre les artères de petit et moyen calibre. Glomérulonéphrite nécrosante très fréquente. Capillarite pulmonaire fréquemment observée.
Purpura rhumatoïde de Henoch-Schönlein
Vascularite avec dépôts d’IgA affectant les petits vaisseaux (capillaires, veinules, artérioles). Atteint typiquement la peau, le tube digestif et le rein (glomérules). Arthralgies et arthrites fréquentes.
Cryoglobulinémie mixte essentielle
Vascularite avec dépôts d’immunoglobulines affectant les petits vaisseaux (capillaires, veinules, artérioles). Présence d’une Cryoglobulinémie. La peau et le rein (glomérules) sont souvent atteints.
Vascularites cutanées leucocytoclasiques
Vascularites cutanées leucocytoclasiques isolées sans vascularite systémique ni glomérulonéphrite.

37
1- les vascularites des vaisseaux de gros calibre :
*La maladie de Horton est définie comme une pan-artérite inflammatoire à
cellules géantes, subaiguë segmentaire et plurifocale touchant avec prédilection les
artères de gros et de moyen calibre et les branches de la carotide externe en
particulier de l’artère temporale superficielle, elle représente la vascularite la plus
fréquente avec une incidence de 10 à 50 fois supérieure à l’incidence de la maladie
de Wegener. [24]
La maladie s’installe en général progressivement, par les symptômes
suivants : des céphalées temporales, uni ou bilatérales, d’intensité variable, sous
forme de brûlures superficielles, pulsatiles, permanentes avec des paroxysmes
spontanés ou provoqués par le moindre attouchement de la tête. [25]
Dans d’autres cas, il s’agit de douleur de la nuque, ou encore de douleurs
massétériennes, favorisées par la mastication, réalisant la très caractéristique
claudication intermittente de la mâchoire. Ces céphalées sont associées dans la
forme typique de la maladie à une AEG, une fièvre, et des douleurs rhumatismales,
ou un cordon à l’inspection de l’artère temporale (fig 14 et fig 15) [17]. Il s’agit de
douleurs scapulaires bilatérales, asymétriques, permanentes et invalidantes,
avec un enraidissement du rachis cervical . A l’heure actuelle, la biopsie d’artère
temporale reste le meilleur moyen de poser le diagnostic de MH. Mais au cours des
dix dernières années, l’utilité de nouvelles techniques d’imagerie pour poser le
diagnostic de vascularite des gros vaisseaux a été étudiée.[45] L’échographie
doppler de haute résolution et le signe du « halo » ont été décrits pour la première
fois par Schmidt et al. en 1995[47], [48], [49]. Il s’agit d’une zone hypoéchogène
siégeant dans la paroi de l’artère pathologique, témoin probable d’un œdème de la
paroi du vaisseau. Le diamètre de ce halo varie de 0,3 à 2 mm. Il disparaît dix à

38
14 jours après le traitement par corticoïdes. L’étude princeps [48] était très
séduisante. Tous les patients suspectés d’artérite temporale ont été suivis
prospectivement et ont bénéficié d’une échographie duplex. Trente patients avaient
une MH dont 21 avec une BAT positive, 37 une PPR avec une BAT négative et 15
étaient des témoins. Le signe du halo était présent chez 22/30 patients ayant une
MH et absents chez les patients n’ayant pas de MH soit une sensibilité de 73 %, une
spécificité de 100 %. La concordance entre les deux échographistes était de 100 %.
Les sténoses et les occlusions étaient des signes moins spécifiques de la MH.
Depuis, les résultats rapportés sont un peu moins attractifs et ne font parfois
pas mieux en terme de valeur diagnostique que les critères de l’ACR. Cette
technique reste néanmoins intéressante, car elle est non invasive et permet de
visualiser une plus grande longueur de la paroi. Elle pourrait dans les équipes
expérimentées, être une aide au diagnostic qui augmenterait la valeur diagnostique
des critères ACR.
L’IRM à haute résolution permet par contre, maintenant d’obtenir des images
d’une grande précision. L’IRM 3T permet de visualiser la lumière et la paroi des
artères et de mesurer l’œdème de la paroi [49], 50], [51] and [52]. Dans l’étude de Bley
et al. [52], 21 patients étaient suspectés de MH, dont neuf diagnostiqués sur les
critères de l’ACR et cinq avec une BAT positive. Il existait des anomalies
inflammatoires des artères crâniennes chez huit des neuf MH et chez tous ceux
ayant une BAT pathologique. Ces travaux sont donc également préliminaires mais
pourraient être intéressants pour diagnostiquer des formes inaccessibles à la
biopsie.
La tomographie par émission de positron (PET-scan) reste une méthode très
intéressante pour le diagnostic des localisations extratemporelles de la MH et dont

39
le diagnostic est très difficile, or les vaisseaux inflammatoires sont le siége d’un
métabolisme énergétique accru et peuvent donc être visualisés par la tomographie
par émission de positron marquée au 18-fluorodeoxyglucose (FDG) [53]. Il n’existe
actuellement que très peu d’études dans le cadre de la MH [54]. Trente-cinq patients
suspects de MH dont 33 avaient une BAT positive ont bénéficié d’un PET-scan initial
(réalisé après la BAT), puis à trois et six mois après l’instauration de la
corticothérapie s’il était initialement positif. Initialement, 29 patients (83 %) avaient
un marquage FDG positif sur au moins un site vasculaire. Par ordre de fréquence, les
artères sous-clavières sont les plus fréquemment atteintes (74 % des patients), puis
l’aorte abdominale (54 %) et thoracique (51 %). La diminution du marquage survient
surtout au cours des trois premiers mois. En revanche, il n’y a pas de corrélation
entre la persistance d’un marquage et le risque de récidive. Cet examen est très
onéreux et nécessiterait d’autres études pour être complètement validé dans la MH,
il est probablement intéressant dans les formes atypiques mais reste non spécifique.
De nouveaux examens d’imagerie sont donc actuellement à l’étude dans la MH
mais nécessitent d’être validés. L’échographie qui est un examen facilement
accessible pourrait apporter un argument supplémentaire au diagnostic de MH.
L’IRM et le PET-scan peuvent être utiles lors de diagnostics difficiles pour des sites
inaccessibles à la biopsie. Cependant, à l’heure actuelle, aucun d’entre eux ne peut
remplacer le Gold Standard qu’est la BAT.

40
Figure 14(17) montre un cas d’artérite temporale se manifestant cliniquement par
une induration palpable et visible de l’artère temporale
Figure 15 aspect anatomopathologique de l’artèrite giganto cellulaire
*La maladie de Takayasu ou la maladie des « femmes sans pouls », frappe
surtout les jeunes femmes avant l’âge de 40 ans, les lésions inflammatoires
granulomateuses siégent sur l’aorte, en particulier l’arc aortique et ses branches (fig
16) [17], l’HTA due à une sténose de l’artère rénale ou à une coarctation de l’aorte
est présente dans plus de 70 % des cas et constitue une cause majeure de morbi-
mortalité. Elle se manifeste cliniquement, à sa phase occlusive, par une fièvre,
amaigrissement, altération de l’état général, avec des manifestations ischémiques
des troncs aortiques et supra aortiques : posture inhabituelle à type de flexion du
cou, amaurose, scotome. Aux membres supérieurs, on peut constater une
claudication intermittente des membres, une asymétrie tensionnelle avec abolition
ou diminution d’un ou de plusieurs pouls, un phénomène de Raynaud. [18]
L’atteinte des artères viscérales et abdominales est responsable d’une HTA
réno-vasculaire, de douleurs abdominales, d’épisodes diarrhéiques, claudication
intermittente des membres inférieurs, voire un anévrysme de l’aorte abdominale.
[25]

41
L’atteinte de l’artère pulmonaire ou de ses branches est retrouvée dans 50%
des cas, elle peut occasionner une symptomatologie faite de toux, d’hémoptysie, ou
d’HTAP.
Figure 16 : occlusion de l’artère sous clavière droite au cours de la maladie de
Takayasu [17]
2-Les vascularites de moyen calibre :
Ce groupe correspond à la PAN et la maladie de Kawasaki.
La PAN représente une inflammation nécrosante des artères de moyen calibre
et les petites artères mais qui par définition n’atteint pas les petits vaisseaux; la
localisation viscérale est principalement sous-diaphragmatique et dans moins de
20% des cas, la maladie est associée à une infection par le VHB.[18,23]
Les lésions siégent préférentiellement aux points de division vasculaire ; au
stade aigu, elle se caractérise par une nécrose fibrinoïde et inflammatoire de la paroi
vasculaire avec thrombose, accumulation des PNN, leucocytoclasie et infiltrat des

42
lymphocytes ; le stade de cicatrisation est marqué par la survenue de sténoses et de
micro-anévrismes bien visualisés sur l’artériographie, il en résulte une ischémie
parenchymateuse et formation d’infarctus.
Elle se manifeste cliniquement par un AMG et une AEG très importante, des
myalgies sont retrouvées dans la moitié des cas, elles sont diffuses et déclenchées
par la pression, le dosage des enzymes musculaires est habituellement normal, des
arthralgies prédominant sur les grosses articulations peuvent se voir.
L’atteinte du système nerveux périphérique met en jeu le pronostic
fonctionnel, il s’agit le plus souvent d’une polynévrite avec souvent une atteinte
distale et asymétrique, une neuropathie périphérique, des paresthésies
douloureuses, ou une paralysie.
Les signes sensitifs peuvent prédominer, une anesthésie douloureuse, des
troubles de la sensibilité superficielle et thermique sont fréquents, la sensibilité
profonde est le plus souvent respectée. L‘atteinte du SNC est rare mais de mauvais
pronostic. La présentation clinique varie selon la localisation cérébrale et le
mécanisme de l’atteinte vasculaire.
Une comitialité localisée ou généralisée, une hémiplégie voir des troubles
cognitifs parfois inauguraux, peuvent survenir. Des accidents vasculaires cérébraux
ischémiques ou hémorragiques peuvent aussi être la conséquence d’une HTA
maligne.
*La maladie de Kawasaki est une maladie aiguë fébrile du très jeune enfant et
se caractérise par un syndrome muco-cutané et lymphatique avec exanthèmes,
érythème polymorphe des pommes et des plantes ; œdème des extrémités,
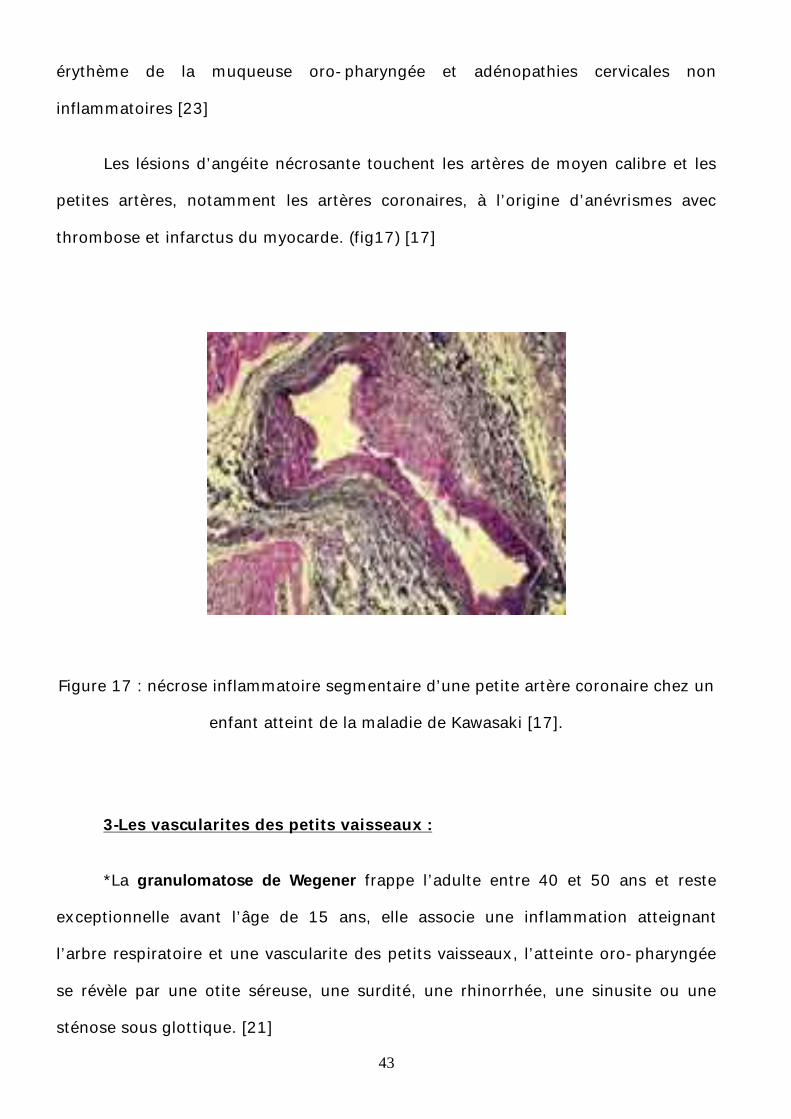
43
érythème de la muqueuse oro-pharyngée et adénopathies cervicales non
inflammatoires [23]
Les lésions d’angéite nécrosante touchent les artères de moyen calibre et les
petites artères, notamment les artères coronaires, à l’origine d’anévrismes avec
thrombose et infarctus du myocarde. (fig17) [17]
Figure 17 : nécrose inflammatoire segmentaire d’une petite artère coronaire chez un
enfant atteint de la maladie de Kawasaki [17].
3-Les vascularites des petits vaisseaux :
*La granulomatose de Wegener frappe l’adulte entre 40 et 50 ans et reste
exceptionnelle avant l’âge de 15 ans, elle associe une inflammation atteignant
l’arbre respiratoire et une vascularite des petits vaisseaux, l’atteinte oro-pharyngée
se révèle par une otite séreuse, une surdité, une rhinorrhée, une sinusite ou une
sténose sous glottique. [21]

44
L’atteinte pulmonaire entraîne des lésions denses nodulaires, alors que la
capillarite alvéolaire est responsable d’hémorragies alvéolaires et d’infiltrats. (fig 18)
[17,21]
L’évolution est marquée par l’apparition d’une glomérulonéphrite nécrosante
faisant révéler l’infection (20%), ces manifestations sont presque toujours associées
à des ANCA de type C avec une spécificité antigénique pour la PR3.
L’atteinte ophtalmologique se manifeste par une kératite, une épisclérite, une
uvéite, névrite optique, vascularite rétinienne, pseudo-tumeur de l’orbite sur
granulome pénoculaire, ou une exophtalmie. [21]
L’atteinte cutanée est présente dans 40% des cas, elle est sous forme d’un
purpura vasculaire infiltré, des ulcérations, des nodules sous-cutanés, des papules,
des vésicules siégeant en regard des grosses articulations notamment les coudes et
les poignets, un livedo, et une nécrose cutanée sur thrombose vasculaire.
L’atteinte articulaire représente par ailleurs 30 à 80% des cas, elle se manifeste
par des arthralgies voire des polyarthrites non érosives et non déformantes touchant
les petites et grosses articulations.
Le SCS associe un asthme ou une rhinite allergique sévère, une éosinophilie
sanguine importante et une mono ou multinévrite, des signes généraux, cutanés, et
articulaires sont présents chez plus de 50 %des malades. L’atteinte rénale est
beaucoup moins fréquente et moins sévère que dans les autres types de vascularites
des petits vaisseaux, elle se limite habituellement à une protéinurie et une
hématurie microscopique, [21,41]
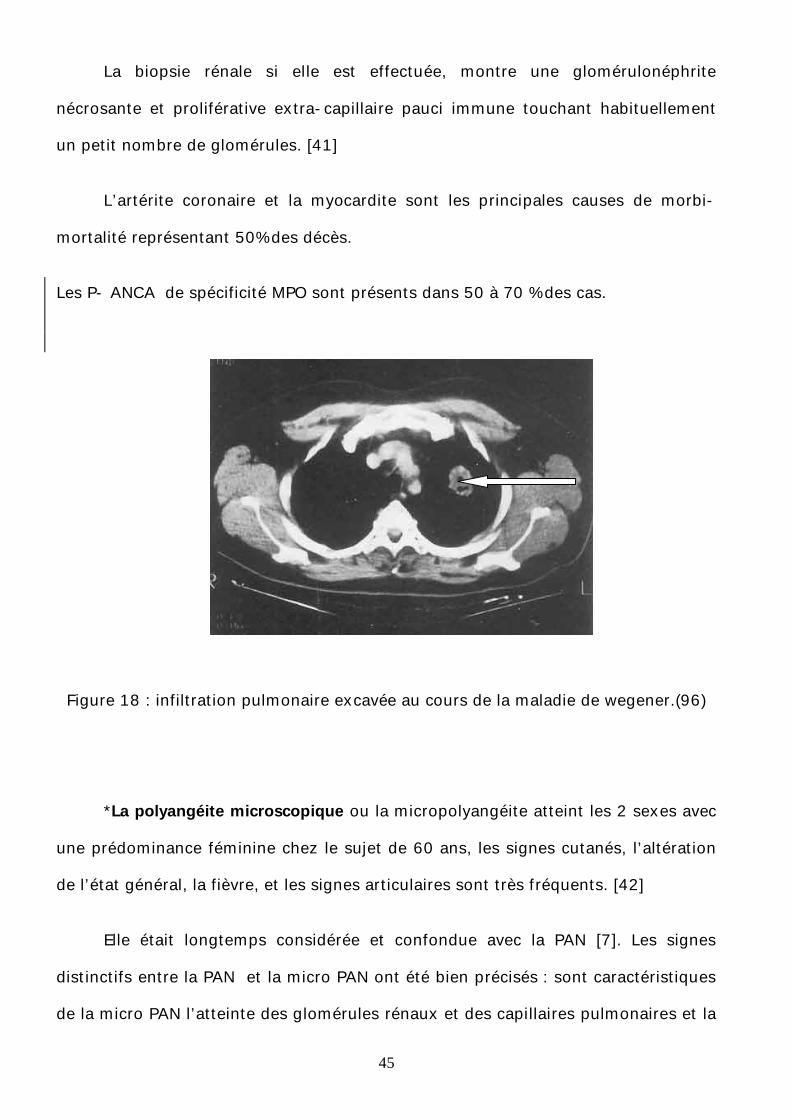
45
La biopsie rénale si elle est effectuée, montre une glomérulonéphrite
nécrosante et proliférative extra-capillaire pauci immune touchant habituellement
un petit nombre de glomérules. [41]
L’artérite coronaire et la myocardite sont les principales causes de morbi-
mortalité représentant 50% des décès.
Les P- ANCA de spécificité MPO sont présents dans 50 à 70 % des cas.
Figure 18 : infiltration pulmonaire excavée au cours de la maladie de wegener.(96)
*La polyangéite microscopique ou la micropolyangéite atteint les 2 sexes avec
une prédominance féminine chez le sujet de 60 ans, les signes cutanés, l’altération
de l’état général, la fièvre, et les signes articulaires sont très fréquents. [42]
Elle était longtemps considérée et confondue avec la PAN [7]. Les signes
distinctifs entre la PAN et la micro PAN ont été bien précisés : sont caractéristiques
de la micro PAN l’atteinte des glomérules rénaux et des capillaires pulmonaires et la

46
présence des ANCA, le sont pour la PAN l’association à une infection par le virus de
hépatite B, l’HTA réno-vasculaire et la présence de micro-anévrismes. (tableau 2)
Lorsque le processus d’angéite nécrosante intéresse électivement des artères
de moyen calibre, on est alors en droit d’évoquer la PAN cutanée ou cutanéo-
viscérale ou viscérale. Il existe cependant des formes avec intrication des deux types
lésionnels.
L’atteinte rénale est quasi-constante, elle se présente sous forme d’une
glomérulonéphrite nécrosante, l’atteinte pulmonaire avec hémorragies intra
alvéolaires ainsi que l’atteinte digestive, musculaire ou la neuropathie périphérique
sont présents dans 25 à 50 % des cas.
Fig 19 : lésions cutanées au cours de la périartérite noueuse.(97)
47
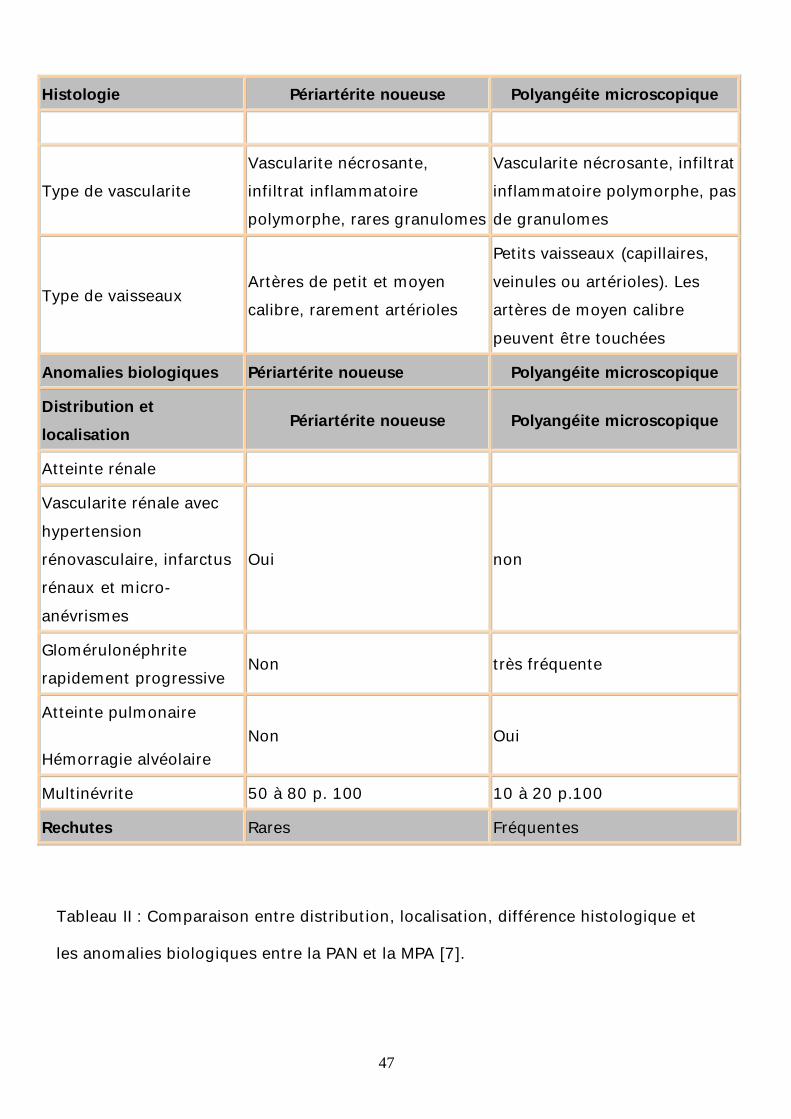
Histologie Périartérite noueuse Polyangéite microscopique
Type de vascularite Vascularite nécrosante, infiltrat inflammatoire polymorphe, rares granulomes
Vascularite nécrosante, infiltrat inflammatoire polymorphe, pas de granulomes
Type de vaisseaux Artères de petit et moyen calibre, rarement artérioles
Petits vaisseaux (capillaires, veinules ou artérioles). Les artères de moyen calibre peuvent être touchées
Anomalies biologiques Périartérite noueuse Polyangéite microscopique
Distribution et localisation
Périartérite noueuse Polyangéite microscopique
Atteinte rénale
Vascularite rénale avec hypertension rénovasculaire, infarctus rénaux et micro-anévrismes
Oui non
Glomérulonéphrite rapidement progressive
Non très fréquente
Atteinte pulmonaire
Hémorragie alvéolaire Non Oui
Multinévrite 50 à 80 p. 100 10 à 20 p.100
Rechutes Rares Fréquentes
Tableau II : Comparaison entre distribution, localisation, différence histologique et
les anomalies biologiques entre la PAN et la MPA [7].

48
*Le purpura rhumatoïde de Henoch-Schonlein est une maladie qui concerne
en premier lieu l’enfant, elle se caractérise cliniquement par la présence d’un
purpura vasculaire typique, influencé par l’orthostatisme, associé à des arthralgies
ou des arthrites migratrices des grosses articulations, sans destruction
radiologique, des douleurs abdominales constituent un critère diagnostique. La
localisation digestive de la vascularite peut être source de complications notamment
d’hémorragies digestives, d’invaginations intestinales, ou de perforations.
L’atteinte rénale met en jeu le pronostic de cette vascularite. En effet, il s’agit
de deux types d’atteintes; une néphropathie glomérulaire à dépôts mésangiaux d’Ig
A se manifestant cliniquement par une hématurie et une protéinurie, et dont
l’évolution est spontanément favorable, la deuxième est une néphropathie
glomérulaire proliférative et extra capillaire et dont la gravité est en fonction du
caractère segmentaire, focal ou diffus des lésions.
*Le terme de vascularite d’hypersensibilité leucocytoclasique regroupe les
vascularites d’origine infectieuse ou médicamenteuse surtout à manifestation
cutanée. Elle se manifeste cliniquement par un purpura vasculaire aigu, pétéchial,
parfois vésiculeux, nécrotique ou ulcéreux. Les autres manifestations extra-
cutanées sont rares: fièvre, arthralgies, myalgie, hématurie ou atteinte neurogéne
périphérique.
5– La classification de Lie :
Cette classification est effectuée en 1988. Elle propose d’inclure dans un
même cadre nosologique toutes les vascularites pulmonaires comprenant un
granulome sous le nom de vascularites granulomateuses et allergiques, elle offre au
praticien une bonne ligne de conduite pour la démarche diagnostique.

49
Elle permet de distinguer entre les vascularites infectieuses et non
infectieuses, mais en revanche, elle sépare les vascularites fréquemment considérées
comme très proches, comme c’est le cas de la PAN et du SCS. Elle inclut la maladie
de Takayasu dans les vascularites des gros, moyens et petits vaisseaux.
1-Vascularites infectieuses :
spirochètes
▪mycobactéries pyogènes : bactéries et champignons
▪rickettsies
▪virales
▪protozoaires
2-Vascularites non infectieuses :
▪lésions de gros, moyens et petits vaisseaux :
*maladie de Takayasu
*granulomatose et vascularites :
-vascularites céphaliques et extra céphaliques à cellules géantes -vascularites viscérales granulomateuses disséminées *artérites des rhumatismes chroniques et des
spondyloarthropathies
▪lésions prédominant au niveau des petites et moyens vaisseaux :
*thrombo-angéite oblitérante ou maladie de Léo burger
*polyartérites :
-PAN classique -PAN microscopique -PAN infantile -maladie de Kawasaki

50
*granulomatoses allergiques et vascularites :
-syndrome de churg et Strauss -sarcoïdose granulomateuse nécrosante
*vascularites des collagénoses :
-RAA -polyarthrite rhumatoïde -spondyloarthropathies -LEAD -poly myosites -dermatopolymyosites -poly chondrite atrophiante -syndrome de Gougerot Sjogrën -sclérodermie systémique -maladie de Behçet -syndrome de Cogan
▪lésions prédominant sur les petits vaisseaux :
*maladie sérique
*purpura rhumatoïde
*vascularites médicamenteuses
*cryoglobulinémie mixte
*vascularite d’hypocomplémentémie
*vascularites lymphocytaires
*vascularites et néoplasies
*fibrose rétro péritonéale
*colopathies inflammatoires chroniques
*cirrhose biliaire primitive
*syndrome de Goodpasture
*vascularites des transplantés

51
3-vascularites look alikes :
▪coarctation-hypoplasie—dysplasies artérielles
▪calcifications artérielles idiopathiques de l’enfance
▪vascularites des syndromes des anti phospholipides
▪ syndrome de Ehlers-danlos
▪pseudoxanthoma elasticium
§ ergotisme
▪embolies de cholestérol
▪embolies myxomateuses
§ neurofibromatoses
6 –La classification de Kahn et Peltier
Toutes les classifications pré-décrites n’ont pas pris en compte certaines
étiologies et n’avaient pas inclus la maladie de Behçet. La classification de Kahn et
Peltier a été plus complète. (Tableau III)
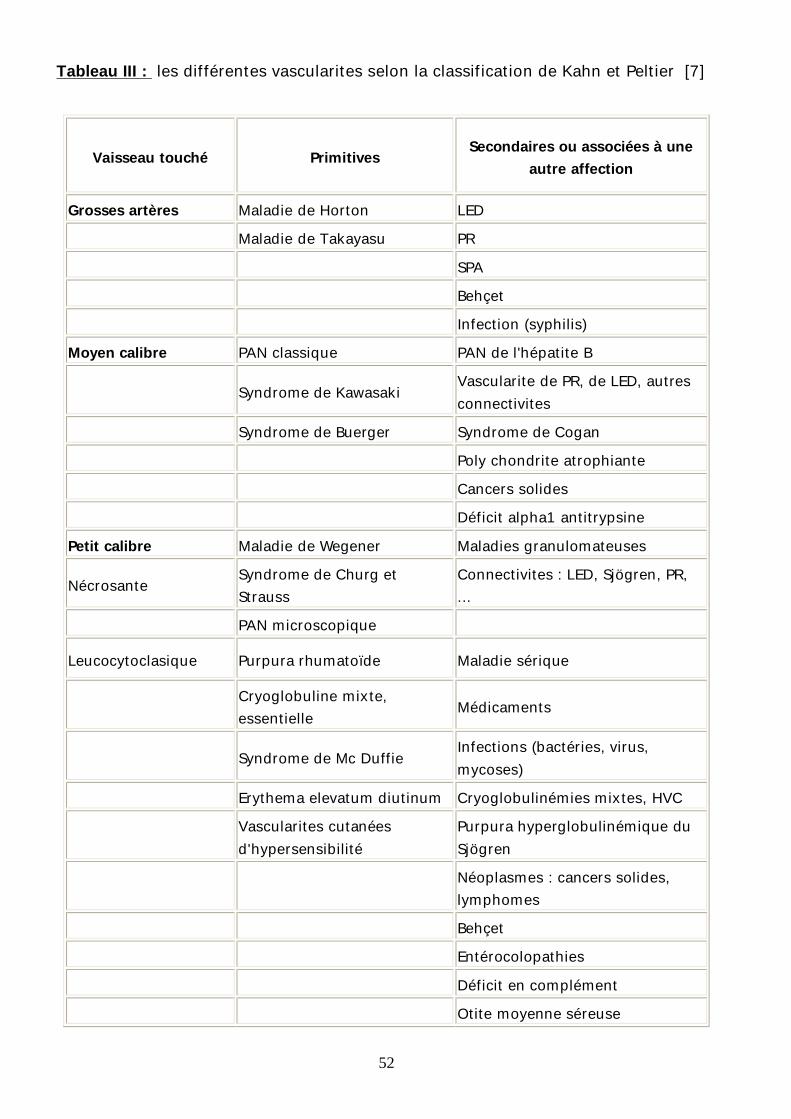
52
Tableau III : les différentes vascularites selon la classification de Kahn et Peltier [7]
Vaisseau touché Primitives Secondaires ou associées à une autre affection
Grosses artères Maladie de Horton LED Maladie de Takayasu PR SPA Behçet Infection (syphilis) Moyen calibre PAN classique PAN de l'hépatite B
Syndrome de Kawasaki Vascularite de PR, de LED, autres connectivites
Syndrome de Buerger Syndrome de Cogan Poly chondrite atrophiante Cancers solides Déficit alpha1 antitrypsine Petit calibre Maladie de Wegener Maladies granulomateuses
Nécrosante Syndrome de Churg et Strauss
Connectivites : LED, Sjögren, PR, ...
PAN microscopique
Leucocytoclasique Purpura rhumatoïde Maladie sérique
Cryoglobuline mixte, essentielle Médicaments
Syndrome de Mc Duffie Infections (bactéries, virus, mycoses)
Erythema elevatum diutinum Cryoglobulinémies mixtes, HVC
Vascularites cutanées d'hypersensibilité
Purpura hyperglobulinémique du Sjögren
Néoplasmes : cancers solides, lymphomes
Behçet Entérocolopathies Déficit en complément Otite moyenne séreuse

53
7-les critères diagnostiques des vascularites selon l’ACR : collège
américain de rhumatologie :
En 1990, l’ACR a proposé à partir d’une casuistique multicentrique de 1000
vascularites, une liste de critères dont le groupement permet d’établir une
classification. Certains critères ont fait l’objet de critiques légitimes, tel que le
critère histologique de la PAN puisque c’est la seule classification qui exige la
présence de cellules granuleuses ou mononuclées dans la paroi des vaisseaux de
petit et moyen calibre alors que l’image caractéristique est la nécrose fibrinoïde. Ces
critères ne distinguent pas entre la PAN et la micro PAN et ne tiennent pas compte
de la biologie, en particulier des ANCA. (Tableaux de IV à X)
Tableau IV : critères diagnostiques de la PAN d’après l’ACR 1990 [7]
• Amaigrissement = 4 kg depuis le début de la maladie, sans cause diététique
• Livedo reticularis • Orchite non infectieuse, non traumatique • Myalgies, faiblesse musculaire ou douleurs des membres • Mono-, multi- ou polynévrite • Pression artérielle diastolique > 90 mm Hg • Insuffisance rénale organique : urée > 0,40 g/l, créatinine > 15 mg/l sans
cause obstructive • Marqueurs sériques de l'infection par le virus de l'hépatite B (antigène ou
anticorps) • Anomalies de l'artériographie coelio-mésentérique ou rénale : présence de
sténoses artérielles ou d'anévrysmes n'étant pas d'origine athéromateuse ou en rapport avec une dysplasie fibromusculaire
• Infiltrats de granulocytes ou de cellules mononuclées dans la paroi de vaisseaux de petit et moyen calibre
• Diagnostic probable si 3 critères ou + (sensibilité = 82 %, spécificité : 87 %)

54
• Inflammation nasale ou buccale : ulcérations buccales douloureuses ou non, ou écoulement nasal purulent ou sanglant
• Anomalies radiographiques pulmonaires à type de nodules fixes, d'infiltrats ou d'images excavées
• Anomalies du sédiment urinaire : micro hématurie (> 5 hématies/champ) ou cylindres hématiques
• Inflammation granulomateuse histologique dans la paroi d'une artère ou péri, voire extravasculaire (artère de moyen calibre ou artériole)
• Diagnostic probable si 2 critères ou + (sensibilité = 88,2 %, spécificité = 92 %)
Tableau V : Critères diagnostiques de la maladie de Wegener selon l’ACR 1990 [7
• Asthme • Eosinophilie > 10% du nombre des leucocytes • Antécédents d'allergie, notamment rhinite allergique ou allergie
alimentaire ou autre (à l'exception des allergies médicamenteuses) • Mono-, multi-, polynévrite • Infiltrats pulmonaires mobiles • Sinusite aiguë ou chronique ou simple opacification des sinus aux
radiographies • Infiltrats éosinophiles extravasculaires d'une artère, artériole ou veinule • Diagnostic probable si 4 critères ou + (sensibilité = 85 %, spécificité : 99,7
%)
Tableau VI : critères diagnostiques de Churg et Strauss selon l’ACR 1990 [7]

55
• Age > 16 ans au début des symptômes • Prise médicamenteuse avant les premiers signes • Purpura vasculaire • Rash maculo papulaire • Atteinte artériolaire ou veinulaire avec granulocytes péri ou
extravasculaires • Diagnostic probable si 3 critères ou + (sensibilité = 71 %, spécificité =
84 %)
Tableau VII : critères diagnostiques des vascularites d’hypersensibilité selon l’ACR 1990 [7]
Tableau VIII : purpura rhumatoïde selon l’ACR 1990 [7]
• Purpura vasculaire • Age < 20 ans au début des symptômes • Douleurs abdominales diffuses ou saignements digestifs • Atteinte artériolaire ou veinulaire avec granulocytes dans la paroi des
vaisseaux • Diagnostic probable si 2 critères ou + (sensibilité = 87 %, spécificité = 88 %)

56
• Age > 50 ans au début des symptômes • Céphalées inhabituelles • Anomalies cliniques des artères temporales à type de douleurs provoquées
par la palpation ou diminution du pouls ou claudication de la mâchoire • Augmentation de la VS = 50 mm/h • Infiltrats de granulocytes ou de cellules mononuclées avec habituellement
présence de cellules géantes dans la paroi artérielle • Diagnostic probable si 3 critères ou + (sensibilité = 94 %, spécificité = 91 %)
Tableau IX : maladie de Horton d’après l’ACR 1990 [7]
• Age = 40 ans au début des symptômes • Claudication d'un membre, surtout membre supérieur • Diminution des pouls aux membres supérieurs • Asymétrie tensionnelle > 10 mm Hg pour la pression systolique aux bras • Souffles sous-claviers ou de l'aorte abdominale • Anomalies artériographiques à type de sténose ou occlusion de l'aorte • Diagnostic probable si 3 critères ou + (sensibilité = 91 %, spécificité = 98 %)
Tableau X : maladie de Takayasu selon l’ACR 1990[7]

57
D’autres vascularites ont également vu leurs critères diagnostiques précisés
par d’autres autorités, qu’il s’agisse de la maladie de Kawasaki avec les critères du
CDC d’Atlanta ou de la vascularite primitive du SNC selon les critères de Calabresse.
Tableau XI : critères du CDC pour le diagnostic de la maladie de Kawasaki [7]
• Fièvre durant 5 jours ou plus sans explication associée à 4 des signes suivants :
• Conjonctivite bilatérale • Atteinte des muqueuses : fissurations labiales, pharynx érythémateux ou
langue framboisée • Anomalie des extrémités : érythème des paumes et plantes, oedèmes des
mains et des pieds ou desquamation périphérique ou généralisée • Rash cutané, exanthème polymorphe • Adénopathies cervicales • * 80 % des cas ont < 4 ans ; rare après 8 ans

58
1. Vraie angéite primitive du SNC
Certaine Biopsie positive (leptoméninge, cortex cérébral ou névraxe) montrant une angéite avec ou sans granulome en l'absence de maladie systémique
Possible Angiographie montrant des rétrécissements segmentaires, des ectasies ou un aspect en chapelet associés à des signes cliniques : céphalées et/ou altérations neurologiques multifocales durant plus de 3 mois
et
Hypercellularité et protéinorrachie élevée dans le LCR
et
Exclusion de toute autre étiologie
2. Angéite bénigne du SNC
Angiographie typique et Atteinte aiguë focale ou multifocale neurologique et LCR normal (ou presque) et Exclusion de toute autre étiologie
Tableau XII : Critères diagnostiques de vascularite primitive du système nerveux
central (SNC) selon calabresse [7]

59
La classification de SAVAGE est utilisée aux états unis, et elle semble la plus
adaptée aux enfants, cette classification se base sur deux paramètres : la taille des
vaisseaux atteints, et la présence ou non du granulome.
Cette classification distingue donc la maladie de Horton et la maladie de
Takayasu comme une vascularite des gros vaisseaux avec granulome, la PAN et la
maladie de Kawasaki comme une vascularite des vaisseaux de moyen calibre sans
granulome, la granulomatose de Wegener et le SCS comme vascularite des petits
vaisseaux avec granulome, et enfin la MPA, les vascularites leucocytoclasiques, la
cryoglobulinémie essentielle comme des vascularites des petits vaisseaux sans
granulome.

60
MATERIEL ET
METHODES

61
I-OBJECTFS
II-PATIENTS ET METHODES
1-méthodes
2-critéres d’inclusion
3-données analysées

62
I-OBJECTIFS :
Le but de notre travail est d’effectuer une étude rétrospective sur les
vascularites systémiques colligées au sein du service de médecine interne durant la
période comprise entre janvier 2003 et décembre 2007. Nous Déterminerons ainsi
le profil de ces différentes entités en précisant les éléments du diagnostic positif et
leurs différents facteurs pronostiques. Nous allons ensuite discuter nos résultats en
nous reportant aux différentes classifications existantes et les différentes séries de
la littérature. Nous rapporterons également les différents progrès thérapeutiques.
II-PATIENTS ET METHODES :
1-Méthodes :
Il s’agit d’une étude rétrospective qui a concerné 20 observations médicales
sur les vascularites systémiques, rapportées au service de médecine interne du CHU
HASSAN II de FES en une période de 5 ans, allant de janvier 2003 à décembre 2007.
Les observations ont été sélectionnées à partir de l’étude des dossiers
d’hospitalisation.
2-Critéres d’inclusion :
Les patients inclus dans cette étude sont ceux qui présentent une vascularite
systémique diagnostiquée au sein du service selon les critères de l’ACR. Cette étude
n’inclue pas la maladie de Behçet et nous précisons qu’il s’agit d’une étude
monocentrique n’incluant pas les vascularites diagnostiquées et suivies en
ophtalmologie ou en neurologie ou en dermatologie. Nous avons exclus également
toutes les vascularites secondaires, il s’agit là de vascularites primitives.

63
3-Données analysées :
Diverses données cliniques et para cliniques ont été recueillies et analysées
pour chaque sujet étudié selon une fiche d’exploitation (voir annexe):
- Les données épidémiologiques (l’âge ; le sexe ; l’origine géographique ; les
antécédents particuliers notamment la prise médicamenteuse ; la notion de voyage à
l’étranger ; les antécédents rhino pharyngés)
- L’examen clinique et l’étude des différents appareils : peau et muqueuse;
ORL; poumons; rein; SNC et périphérique; cœur et vaisseaux; tube digestif; œil; les
articulations; les muscles et enfin les signes généraux.
- L’étude des examens paracliniques biologiques, radiologiques et
anatomopathologiques.
- Les modalités thérapeutiques, le suivi et la durée de suivi.

64
RESULTATS ET
ANALYSES

65
I- OBSERVATIONS II-DONNEES EPIDEMIOLOGIQUES III-ANTECEDENTS IV-MOTIF D’HOSPITALISATION V-DONNEES DE L’EXAMEN CLINIQUE VI-EXAMENS COMPLEMENTAIRES VII-ETIOLOGIES VIII-TRAITEMENT ET SUIVI IX-ANALYSES

66
I- OBSERVATIONS
Les résultats de notre étude sont exprimés en valeur absolue et en
pourcentage pour les variables qualitatives, en moyenne écart-type et valeurs
extrémités pour les variables quantitatives.
Observations ;maladie de Takayasu
Observation n°1
il s’agit de madame k .A âgée de 40 ans, célibataire, originaire et habitant fès,
femme au foyer, hospitalisée le 16/04/07 au service pour des douleurs du membre
supérieur gauche. Dans ses antécédents, c’est une patiente suivie pour une gastrite
antrale en 2005, sans autres antécédents pathologiques, qui présente depuis 03 ans
une claudication des membres supérieurs et inférieurs sans aucun signe associé
avec une aggravation de la symptomatologie 06 jours avant par l’apparition d’une
impotence fonctionnelle totale de l’hémicorps gauche plus importante au niveau du
membre supérieur gauche associée à une froideur du membre supérieur gauche, le
tout évolue dans un contexte de fièvre et d’amaigrissement non chiffrés.
L’examen à son admission trouve une patiente en ABEG, une TA à 130/70
mmHG au membre supérieur droit, imprenable à gauche, avec à l’examen
cardiovasculaire une abolition des pouls huméral, radial, cubital, pédieux et tibial
postérieur à gauche. Tous les pouls sont présents à droite, l’auscultation cardiaque
ne retrouve pas de souffle aux 4 foyers d’auscultation ni de signe d’insuffisance
cardiaque droite ou gauche.

67
L’examen locorégional du membre supérieur gauche retrouve une froideur.
L’examen cutanéo-muqueux trouve une plaque rouge et chaude en regard de la face
externe de la cheville gauche.
A l’examen neurologique, la station debout est impossible sans double aide, la
marche est impossible sans déficit sensitivo-moteur, sans troubles de la sensibilité
profonde, le reste de l’examen clinique notamment l’examen pleuro-pulmonaire est
sans particularité, l’examen abdominal ne retrouve pas de souffle para ombilical, ni
de douleurs à la palpation. L’examen ophtalmologique demandé à la recherche
d’une atteinte vasculaire rétinienne est normal.
Le diagnostic d’une maladie de Takayasu est suspecté ou d’une
thrombophlébite du membre supérieur gauche sur embole vasculaire.
L’Echodoppler artériel du membre supérieur gauche réalisé en urgence
montre une thrombose de l’artère axillaire gauche, l’étude détaillée de la paroi
vasculaire montre un épaississement pariétal et circonférentiel étendu en axillo-
huméral bilatéral avec une occlusion des artères axillaires et tibiale postérieure
gauche (fig 20-21-22). L’échographie trans-thoracique à la recherche d’une atteinte
valvulaire ou d’une coronarite est revenue normale, notamment pas d’HTAP.
Le bilan biologique effectué à la recherche d’un syndrome inflammatoire est
revenu normal.
Le diagnostic d’une maladie de Takayasu est retenu devant la présence de
plus de 3 critères selon les critères de diagnostic de l’ACR et la patiente est mise
sous :
- Anticoagulation curative.

68
- Corticothérapie à raison de 1mg/kg/jour de prednisone.
- Anti-agrégant plaquettaire : Aspirine à 100mg.
- et un inhibiteur calcique pour l’HTA.
L’évolution est marquée par l’apparition de douleur rétrosternale, un ECG
réalisé retrouve un tracé normal, le dosage des enzymes cardiaques est normal, on a
complété par une angio-TDM pour éliminer une dissection aortique qui était
normale. La corticothérapie est maintenue à de fortes doses pendant 03 mois puis
dégression.
L’évolution a montré la disparition des douleurs et de la claudication des
membres, la tension artérielle contrôlée à plusieurs reprises est de 140/90 mmHg
au bras droit et 140/80 mmHg au bras gauche, les pouls périphériques sont perçus
aux 2 membres supérieurs et aux 2 membres inférieurs d’une façon symétrique.
Nous avons préconisé donc de continuer la dégression de la corticothérapie.
Actuellement, la patiente est à 20 mg par jour sans complication.
69
Figures 20-21-22: Présence d’une occlusion totale et segmentaire de l’artère
axillaire gauche avec absence de flux et de reperméabilisation de l’artère humérale
en aval.

70
Observation n° 2
Il s’agit de madame B .A âgée de 36ans, femme au foyer, originaire et habitant
fès, mère d’un enfant, sans antécédents pathologiques particuliers qui présente
depuis 2 mois une claudication intermittente des 2 membres supérieurs, sans
froideur ni syndrome de Raynaud, ni autres signes associés. Le tout évolue dans un
contexte d’amaigrissement non chiffré sans fièvre ni anorexie.
L’examen à son admission trouve une patiente en assez bon état général, la
mesure de la tension artérielle aux 2 membres supérieurs retrouve 13O/80 mmHg
au membre droit, elle est imprenable à gauche. Le reste de l’examen clinique est
sans particularité.
Nous avons effectué un échodoppler des troncs supra-aortiques et des 2
membres supérieurs qui retrouve un épaississement de l’intima de la carotide
primitive droite de 0,9mm et de 1cm au niveau de l’artère carotide primitive gauche
avec une sténose de l’artère sous clavière homolatérale. Le reste du doppler est sans
anomalie notamment des membres inférieurs et des vaisseaux abdominaux.
Le bilan biologique est par ailleurs normal.
Le diagnostic d’une maladie de Takayasu est retenu et la patiente est mise
sous corticothérapie à raison de 1mg/kg/j avec traitement adjuvant.
L’évolution est marquée, par une disparition de la symptomatologie clinique
avec amélioration de la patiente. Nous avons commencé la dégression de la
corticothérapie en gardant une surveillance stricte de la patiente.

71
Un an après, la patiente a présenté une augmentation des chiffres tensionnels
à 18O /100 mmHG. L’échodoppler des 2 artères rénales montre des vitesses
systoliques normales mais un allongement du temps d’ascension systolique
traduisant une sténose de l’artère rénale gauche.
La patiente est mise sous trithérapie à base d’un inhibiteur de l’enzyme de
conversion, d’un diurétique, et un anti hypertenseur central associé à un régime
sans sel avec une bonne évolution tensionnelle.
Au cours de sa 2ème grossesse, la patiente a présenté une toxémie gravidique
pour laquelle elle était hospitalisée en obstétrique. Un traitement à base d’anti-
agrégant plaquettaire a été instauré de la 16ème à la 36ème semaine
d’aménorrhée.
La patiente était hospitalisée un seconde fois au cours de sa 3 ème grossesse
(14ème semaine d’aménorrhée) au service de médecine interne pour des céphalées
en casque et bourdonnement d’oreilles avec une baisse de l’acuité visuelle bilatérale
et progressive. La mesure de la tension artérielle aux 2 membres supérieur droit
puis gauche a retrouvé respectivement 150/80mmHg et 140/70 mmHg. Le reste de
l’examen clinique est sans particularité.
La protéinurie des 24 h était négative. L’ECBU était stérile. Un examen
ophtalmologique a retrouvé une rétinopathie hypertensive stade II. L’examen
gynécologique était normal avec une échographie obstétricale normale. L’évolution
était marquée par un avortement spontané chez la patiente.

72
OBSERVATION N°3
Il s’agit de madame H.L âgée de 36 ans, femme au foyer, mère de 3 enfants,
originaire et habitante fès, ayant comme antécédent un rhumatisme articulaire aigu
depuis 12 ans, sans cardite rhumatismale, thyroidectomisée depuis 5 ans,
actuellement sous antityroïdiens de synthèse, sans autres antécédents, hospitalisée
au service pour une claudication intermittente des membres.
Le début de sa symptomatologie remonte à 3 ans par l’apparition d’une
claudication intermittente des 2 membres supérieurs accompagnée de céphalées,
de sensation vertigineuse, des douleurs de la ceinture scapulaire irradiant vers les 2
membres supérieurs, et des douleurs de la hanche et du genou gauche, le tout
évolue dans un contexte d’asthénie.
L’examen clinique retrouve une tension artérielle à 140/60mmhg au membre
supérieur droit, la tension artérielle est imprenable à gauche, les pouls du membre
supérieur gauche sont tous abolis. Par ailleurs, ils sont tous présents au reste des
membres. L’auscultation cardiaque est sans particularité, l’examen articulaire ne
retrouve pas de douleurs provoquée à la mobilisation du genou, ni aux manœuvres
d’écartement des hanches, le reste de l’examen clinique est sans particularité.
Le bilan réalisé ne retrouve pas de syndrome inflammatoire, ni d’ anomalie
biologique à l’ionogramme, l’échographie des vaisseaux supra-aortiques montre
une inflammation bilatérale et diffuse des 2 carotides internes et externes,
l’échographie au doppler des vaisseaux abdominaux montre une sténose modérée
de l’artère rénale. Aux membres inférieurs, on a noté une discrète atteinte de
l’artère fémorale commune gauche ce qui peut expliquer les douleurs projetées en
regard de la hanche et du genou gauche.

73
Le diagnostic d’une maladie de Takayasu est retenu et la patiente est mise
sous corticothérapie à la dose de 1mg/kg/j avec le traitement adjuvant nécessaire,
la durée de suivi de cette patiente n’était malheureusement que de 2 mois.

74
OBSERVATION N°4
Il s’agit de madame S .F âgée de 44 ans, originaire et habitant fès, femme au
foyer, mariée et mère de 3 enfants, avec une notion d’une fausse couche en 1996 et
d’une mort foetale in utero en 1997, sans autres antécédents pathologiques
particuliers, qui a consulté aux urgences pour une dyspnée d’installation rapide
devenant une orthopnée avec des douleurs thoraciques, sans autres signes associés.
L’examen à son admission retrouve une patiente consciente, avec une tension
artérielle imprenable aux 4 membres, les pouls périphériques sont finement perçus
aux membres supérieurs et absents aux membres inférieurs, l’examen
cardiovasculaire retrouve un souffle systolique au foyer mitral, l’examen pleuro
pulmonaire est sans particularité, le reste de l’examen clinique est normal.
Une échographie cardiaque transthoracique réalisée en urgence, retrouve une
insuffisance cardiaque globale sur un rétrécissement aortique très serré, la patiente
est donc transférée au service de chirurgie cadio vasculaire en urgence où elle a
bénéficié d’une valvuloplastie aortique.
De retour à notre formation, des examens complémentaires sont réalisés
notamment un échodoppler des vaisseaux du cou qui retrouve un épaississement
pariétal proximal des 2 carotides sans retentissement hémodynamiques,
l’échodoppler des 2 membres supérieurs et inférieurs, ainsi que des vaisseaux
abdominaux est sans particularité.

75
Devant ce tableau clinique, une maladie de Takayasu est très probable, le
bilan biologique retrouve un syndrome inflammatoire avec une VS à 68 mm à la
première heure, et une CRP à 60 mg/l, la NFS retrouve une hyperleucocytose à 14
000/mm3 à prédominance PNN, le bilan rénal est normal, le reste du bilan
biologique est normal.
Le diagnostic d’une maladie de Takayasu est donc retenu et la patiente est
mise sous corticothérapie orale à raison 1 mg/kg/j, anticoagulant type héparine puis
anti vitamine K, avec un régime hyposodé, une supplémentation potassique et un
apport en calcium et vitamine D3 et bisphosphonates.
La patiente est ré hospitalisée il y a 5 mois pour une évaluation clinique,
biologique et échographique, l’examen à son admission retrouve une tension
artérielle imprenable aux membres supérieurs et inférieurs, mais elle ne rapporte
plus de claudication intermittente des membres, le bilan biologique retrouve une
persistance du syndrome inflammatoire avec une légère amélioration des chiffres
biologiques et la VS est à 42 min à la première heure et la CRP à 30 mg/l.
L’échographie des troncs supra aortiques retrouve le même aspect, une
échographie transthoracique est refaite, elle a retrouvé une prothèse valvulaire
aortique en place de bon fonctionnement, avec une fraction d’éjection à 62%, et des
valves mitrales épaissies, l’échographie rénale ainsi que le doppler des vaisseaux
abdominaux est sans particularité.
On a proposé à cette patiente donc une corticothérapie à raison de 0,5
mg/kg/j avec traitement adjuvant et l’introduction du méthotrexate à raison de
15mg/semaine avec de l’acide folique à 3 jours d’intervalle à 5mg/semaine.

76
L’évolution est marquée par l’apparition d’un diabète cortisonique, jugulé par
une insulinothérapie. La patiente est toujours suivie en consultation.

77
Observation : maladie de Léo buerger
Observation n°5
Il s’agit de Mr A.R âgé de 34 ans, originaire et habitant fès, tabagique
chronique à raison de un paquet par jour pendant 10 ans, traité pour une
tuberculose ganglionnaire en 1993, sans autres antécédents pathologiques
particuliers, notamment pas d’antécédents de phlébites des membres supérieurs ou
inférieurs, hospitalisé au service de médecine interne pour prise en charge d’une
nécrose distale du 5 ème orteil gauche.
Le début de sa symptomatologie remonte à un an par l’apparition de douleur
au niveau du 5 ème orteil gauche sans claudication intermittente des membres, ni
paresthésies, avec un syndrome de Raynaud au niveau des 2 membres supérieurs.
Le patient ne rapporte par ailleurs aucune symptomatologie cardiaque ou
neurologique particulière.
A l’examen, on retrouve une tension artérielle correcte de 120/70mmHg.
L’examen cutané retrouve une nécrose du 5 ème orteil gauche, un acrosyndrome
des 2 membres supérieurs type syndrome de Raynaud, associé à des troubles
trophiques, sans cyanose ni hémorragies péri–unguéales. L’examen cardio-
vasculaire ne retrouve pas de pouls au niveau du membre inférieur gauche.
L’auscultation cardiaque est sans anomalie, l’examen neurologique retrouve un
syndrome pyramidal, Le reste de l’examen clinique est sans particularité.
Devant ce tableau, nous évoquons une angiopathie diabétique, une artérite
par athérosclérose, une cardiopathie emboligène, une thrombophilie ou une
vascularite type Léo buerger.
Le bilan réalisé ne retrouve pas de syndrome inflammatoire, la numération
formule sanguine est sans particularité. L’ionogramme est correct notamment la

78
glycémie, et le bilan lipidique. Le bilan immunologique notamment les AC anti
nucléaires et les AC anti DNA natifs sont négatifs. L’échodoppler des membres
inférieurs retrouve une atteinte artérielle distale prédominant à gauche avec une
atteinte des artères radiales, sans lésion d’athérosclérose. Nous avons complété par
une échographie transthoracique, un doppler des troncs supra-aortiques qui sont
revenus normaux. L’échographie rénale et l’électrocardiogramme sont normaux.
Le diagnostic d’une maladie de Léo buerger est donc retenu et nous
recommandons chez ce patient un arrêt du tabac, un antiagrégant plaquettaire, un
anti-ischémique type PRAXILENE® sont prescrits en association à un antalgique.
L’évolution est marquée par une amélioration de la symptomatologie clinique,
une artériographie de contrôle est demandée à la sortie du patient, mais il a été
perdu de vue par la suite.

79
Observation : périartérite noueuse
Observation n°6
Il s’agit de Mr M.B âgé de 46 ans, marié et père de 4 enfants, originaire et
habitant Missour, tabagique chronique à raison de 3 cigarettes par jour pendant
16ans, sans autres antécédents pathologiques, hospitalisé au service pour des
nodules sous cutanés diffus associés à des arthralgies des grosses articulations des
membres inférieurs et des myalgies, un purpura pétéchial aux membres inférieurs,
et des paresthésies des membres inférieurs. Le patient ne rapporte pas
d’oligoanurie, ni d’hématurie, pas de douleurs abdominales, ni de rectorragies et
pas de symptomatologie cardiaque. Le tout évolue dans un contexte d’altération de
l’état général très importante et d’amaigrissement chiffré à 6 kg pendant 2 mois.
L’examen clinique trouve un patient très altéré, normotendu, l’examen
neurologique trouve un syndrome tétrapyramidal prédominant aux membres
inférieurs. L’examen cutané trouve des nodules sous cutanés diffus à tout le corps
épargnant le cuir chevelu. L’examen ostéo-articulaire ne retrouve pas
d’épanchement articulaire. Nous notons une légère limitation de la flexion des
articulations des membres inférieurs. Le reste de l’examen clinique est sans
particularité notamment l’examen cardio-vasculaire et pleuro-pulmonaire.
Devant ce tableau clinique, nous évoquons chez ce patient une atteinte
neurologique type mononévrite multiple ou atteinte systémique type vascularite
notamment une PAN.
Nous retrouvons un syndrome inflammatoire biologique avec une VS à 110
mm, une CRP à 80mg/l et une anémie inflammatoire à 10,6g/dl, avec une
hyperleucocytose à 17 000/mm3 à prédominance PNN. Le bilan immunologique
n’est pas effectué par manque de moyens chez ce patient, la fonction rénale est

80
normale, la sérologie d’hépatite B est négative. Une biopsie cutanée est revenue en
faveur d’une nécrose fibrinoïde au niveau des artères de petit et de moyen calibre,
l’électromyogramme pour l’exploration de l’atteinte neurologique parle d’une
mononévrite de type axonale.
Nous effectuons une électrophorèse des protides pour l’exploration du
syndrome inflammatoire, qui montre une hypoalbuminémie avec une courbe de
syndrome inflammatoire (alpha1 et alpha2 élevés), et à l’immunoélectrophorèse une
gammapathie monoclonale de type IgG kappa. Devant ces anomalies
électrophorétiques, nous complétons par un myélogramme, qui retrouve un taux de
plasmocytes à 10%, mais qui sont en augmentation par rapport au taux il y a 1 an
(5%). Le taux des LDH est très augmenté à 1308 UI/l pouvant être expliqué par une
hémolyse intramédullaire. Le reste du bilan biologique est normal. Sur le plan
radiologique, la radiographie standard du crâne profil montre la présence de
quelques images lytiques à l’emporte-pièce (fig 24).
L’ensemble de ces arguments cliniques, biologiques, radiologiques et
électrophysiogiques ont permis de retenir le diagnostic d’une PAN associée à un
myélome multiple au stade intermédiaire de Salmon et Durie.
Nous avons décidé de mettre le patient sous corticothérapie à la dose de
20mg/j vu les signes d’évolutivité mais sans index de sévérité, et pour le myélome ;
une chimiothérapie de type VAD (vincristine, adriblastine et dexaméthasone) était
proposée mais par manque de moyens, le patient a bénéficié de bolus de
dexaméthasone à la dose de 20mg/m² de surface corporelle par jour pendant 4
jours tous les 28 jours.

81
Une IRM médullaire (fig 25) a été demandé devant le syndrome tétrapyramidal,
elle montre une atteinte lytique de tous les corps vertébraux au niveau cervical et
dorso lombaire compatible avec une localisation myélomateuse vertébrale avec une
discarthrose cervicale C5-C6 et un canal lombaire étroit en L4-L5, ce qui a fait
évoluer la classification du myélome chez ce patient à un stade III de Salmon et
Durie. Un myélogramme de contrôle trouve une augmentation du taux des
plasmocytes à 26%, nous démarrons une chimiothérapie selon le schéma
ALEXANIAN: ALKERAN 0,25mg /kg/j + corticoïdes, associé à du chlodronate.
Le patient fut réhospitalisé pour un sepsis à point de départ pulmonaire pour
lequel il a bénéficié d’une double antibiothérapie par voie intraveineuse à base de
céphalosporine de 3ème génération associée à une fluoroquinolone type
ciprofloxacine. Une TDM thoracique réalisée à la fin du traitement a montré des
images de pleurésie enkystée. Le patient a bénéficié d’un drainage chirurgical
associé à une antibiothérapie à base de quinolone de 3 ème génération injectable
pendant 10 jours avec une nette amélioration clinique et radiologique. Le patient est
perdu de vue depuis 8 mois.
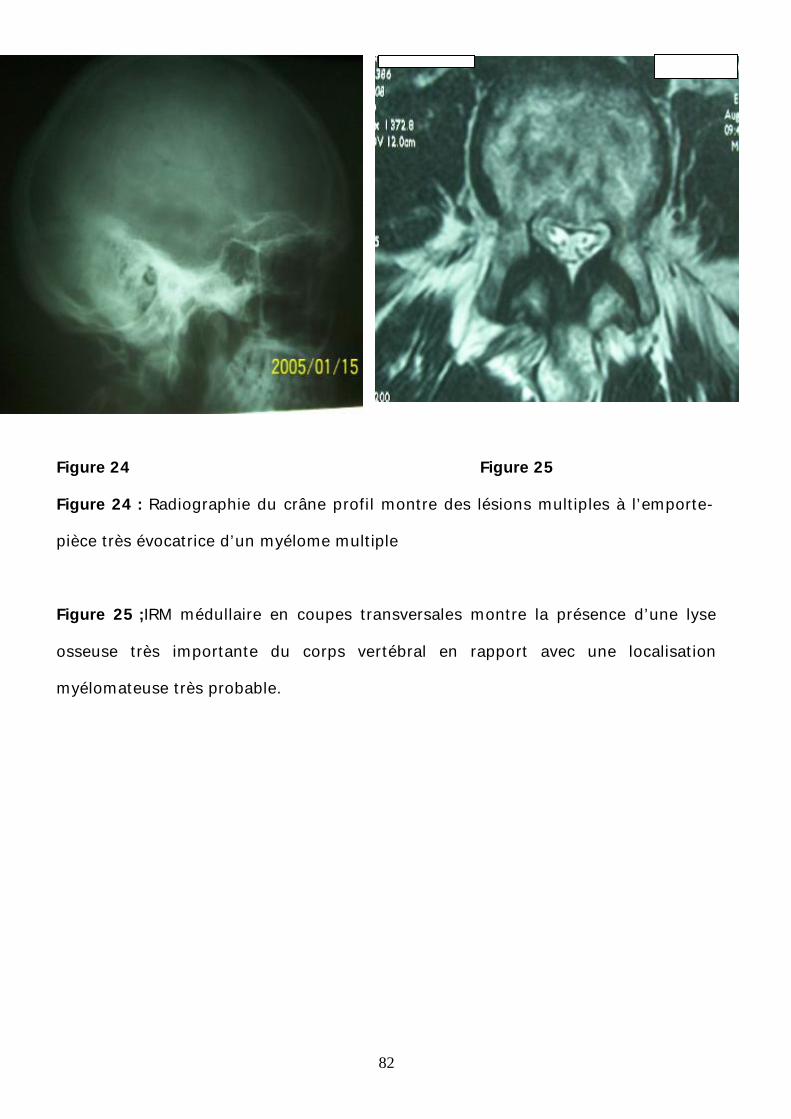
82
Figure 24 Figure 25
Figure 24 : Radiographie du crâne profil montre des lésions multiples à l’emporte-
pièce très évocatrice d’un myélome multiple
Figure 25 ;IRM médullaire en coupes transversales montre la présence d’une lyse
osseuse très importante du corps vertébral en rapport avec une localisation
myélomateuse très probable.

83
Observations ; maladie de Horton
Observation n°7
Il s’agit de madame Z.R, âgée de 64 ans, originaire et habitante Fès, mère de 7
enfants, ménopausée depuis 10 ans, ayant comme antécédent pathologique, une
fracture de l’extrémité inférieure du cubitus traitée orthopédiquement. Elle est suivie
depuis 10 ans pour des douleurs abdominales et troubles digestifs étiquetés comme
colopathie fonctionnelle sans antres antécédents pathologiques. Elle présente
depuis 03 mois des céphalées temporales avec une hyperesthésie du cuir chevelu et
une baisse de l’acuité visuelle bilatérale, sans symptomatologie cardio-respiratoire,
ni articulaire, ni de signes généraux .
L’examen à son admission trouve une patiente en bon état général, normo
tendue, apyrétique. L’examen de la tête retrouve un cordon variqueux sinueux
temporal bilatéral plus accentué à droite, sans abolition des pouls temporaux, tous
les pouls périphériques sont présents et symétriques, l’examen cardio vasculaire ne
retrouve pas de souffle à l’auscultation. L’examen ophtalmologique de notre
patiente retrouve une acuité visuelle de 1/10 à droite, et 2/10 à gauche.
Le tableau clinique est très évocateur d’une maladie de Horton, nous avons
complété par un bilan biologique qui retrouve un syndrome inflammatoire avec une
VS à 118 mm à la première heure, une CRP à 60 mg/l, une hyperleucocytose à
prédominance PNN avec une thrombocytose réactionnelle, sans anémie, le reste du
bilan biologique est tout à fait normal.
L’échodoppler des vaisseaux du cou retrouve un aspect remanié des carotides
avec thrombose de l’artère temporale droite, aux membres inférieurs. L’écho
doppler montre un remaniement pariétal de l’artère fémorale avec un amortissement
du flux en aval.

84
La biopsie de l’artère temporale retrouve une artérite temporale giganto
cellulaire. Le diagnostic d’une maladie de Horton est retenu et la patiente est mise
sous corticothérapie orale à la posologie de 0,75 mg/kg/j de prednisone avec
traitement adjuvant à base de potassium et de vitamine D3 supplémentation
potassique et bisphosphonate, de l’aspirine à la posologie de 250 mg/j vu la
présence de la thrombose artérielle fémorale.
L’évolution au bout de 3 mois est marquée par la disparition des céphalées,
et du syndrome inflammatoire biologique.
L’évolution à long terme est marquée chez cette patiente par l’apparition d’un
diabète cortisonique, équilibré sous antidiabétique oral, puis elle est perdue de vue.

85
Observation n°8
Il s’agit de Mr H.L, âgé de 68ans, originaire de Arfoud habitant fès, marié et
père de 6 enfants, ayant comme antécédents une hernie inguinale bilatérale pour
laquelle il s’est fait opéré, des varices du membres inférieurs droits non traitées, et
une néphrectomie droite en 1980 pour une pyonéphrose non documentée.
Le début de sa symptomatologie remonte à 2 mois par l’installation
progressive de dorsalgies puis de cervicalgies puis de céphalées en casque rebelles
à toute prise d’antalgiques, associée à des bourdonnements d’oreille, une
hyperesthésie du cuir chevelu, et une sensation vertigineuse, sans autres signes
associés ni digestifs, ni cardiaques. Le patient rapporte par ailleurs, une baisse de
l’acuité visuelle avec rougeur bilatérale, le tout évolue dans un contexte d’altération
de l’état général.
L’examen clinique trouve un patient en assez bon état général, normotendu
avec mesures tensionnelles comparatives aux 2 bras, l’examen cardio-vasculaire
retrouve un souffle systolique au foyer mitral avec des signes d’insuffisance
cardiaque droite, les pouls sont tous présents et symétriques. On retrouve une
cicatrice de néphrectomie à droite au niveau lombaire, une limitation de l’ouverture
de la cavité buccale avec des douleurs rachidiennes cervicales et dorsales à la
palpation des épineuses. L’examen ophtalmologique retrouve un oedème papillaire
bilatéral, avec au fond d’œil des vaisseaux grêles et signe de croisement, le reste de
l’examen clinque est sans particularité.
On retrouve un syndrome inflammatoire biologique avec une VS à 100 mm à la
première heure, une CRP à 65 mg/l, une hyperleucocytose à 16 800/mm3 à
prédominance PNN, une anémie inflammatoire à 10 ,6 g /dl et une thrombocytose
réactionnelle.

86
L’électrophorèse des protides sanguins montre une hypergammaglobulinémie
monoclonale à 19,8g/l, complétée par une immunoélectrophorèse qui a montré une
gammapathie monoclonale à IgG kappa. Devant ces anomalies biologiques, on a
complété par une ponction sternale et une biopsie ostéo médullaire qui sont
revenues normales.
L’ionogramme retrouve une légère perturbation de la fonction rénale. Le bilan
immunologique n’a pas pu être réalisé chez ce patient.
Le bilan radiologique retrouve à l’échographie abdominale des calcifications
sous capsulaires, hépatiques et inter hépato-pancréatiques avec un nodule
hypoéchogéne de la tête du pancréas, l’échographie trans-thoracique montre une
insuffisance mitrale minime, une insuffisance aortique minime, une bonne fonction
du ventricule gauche globale avec une contractilité segmentaire hétérogène
probablement en rapport avec une cardiopathie ischémique.
La radiographie standard du rachis cervical montre des calcifications diffuses
du rachis en rapport avec une cervicarthrose, de très bonne évolution sous
antalgique, anti inflammatoires et myorelaxant.
La biopsie de l’artère temporale retrouve un infiltrat inflammatoire avec
remaniement de la média, avec une limitante élastique fragmentée, il s’agissait
d’une artérite granulomatose avec foyer de nécrose fibrinoïde.

87
Le diagnostic d’une maladie de Horton associée à une gammapathie
monoclonale est retenu, le patient est mis donc sous corticothérapie orale à la
posologie de 1 mg/kg/j avec le traitement adjuvant nécessaire, l’ évolution est
favorable au bout de 3 mois, avec une amélioration des douleurs articulaires,
disparition des céphalées et de l’hyperesthésie du cuir chevelu, mais persistance des
douleurs thoraciques avec l’apparition d’une dyspnée stade III de la NYHA ce qui
nous a motivé à réaliser une échographie cardiaque transthoracique ayant montré
une surcharge athéromateuse sans sténose. Le patient est mis donc sous anti
agrégants plaquettaires type ASPIRINE pour prévenir l’apparition d’une
symptomatologie ischémique.

88
OBSERVATION 9 :
Il s’agit de madame I.H.S âgée de 70 ans, originaire de fès, habitant à
khémissate, femme au foyer, opérée en 1998 pour une cataracte de l’œil gauche
puis 6 mois après pour une cataracte de l’œil droit, hypertendue depuis 15ans sous
traitement non précisé, consultant pour des céphalées rebelles remontant à 1 année,
temporo-pariétales, avec des douleurs du cuir chevelu, ses douleurs ne cédant pas à
la prise du paracétamol, le tout évolue dans un contexte de conservation de l’état
général et d’apyrexie.
L’examen de notre patiente est sans particularité, on ne retrouve pas
d’abolition des pouls notamment au niveau temporal, l’examen cardio vasculaire est
sans particularité, l’examen pleuro pulmonaire est normal, l’examen ostéo
articulaire ne retrouve pas de douleurs ni d’enraidissement à la mobilisation des
différentes articulations, l’examen ophtalmologique retrouve un oedème papillaire,
avec une névrite optique unilatéral au fond d’œil.
Le bilan réalisé chez notre patiente retrouve une accélération de la vitesse de
sédimentation à 160 mm à la première heure, le reste du bilan est normal.
L’échodoppler des vaisseaux du cou est sans particularité, la biopsie de l’artère
temporale ne retrouve pas d’anomalies histologiques.
On a retenu le diagnostic de maladie de Horton sur les critères de l’ACR
suivants :
Début des symptômes après 50 ans.
Des céphalées localisées de début récent.
Accélération de la VS.

89
La patiente est mise sous corticothérapie orale à la posologie de 0.75 mg/kg/j
de prednisone avec traitement adjuvant à base d’un régime sans sel,
supplémentation potassique, calcique et de la vitamine D3 et bisphosphonates.
L’évolution au bout de 3 mois est marquée par la disparition des céphalées et
une diminution de la VS à 20 mm.
Par la suite la patiente a arrêté la corticothérapie d’elle-même, puis elle a été
perdue de vue.

90
OBSERVATION N°10 :
Il s’agit de MR B.M âgé de 61 ans, originaire et habitant fès, sans profession,
tabagique chronique à raison de 2 paquets par jour pendant 30 ans, sevré il y a 17
ans, traité par point de feu pour un ictère, traité orthopédiquement pour une
fracture de la jambe droite qui présente depuis dix mois des céphalées en casque
avec une hyperesthésie du Cuir chevelu et une baisse de l’acuité visuelle, le patient
rapporte par ailleurs des douleurs cervicales le tout évoluant dans un contexte de
sueurs nocturnes et d’asthénie .
L’examen clinique s’est révélé sans particularité en dehors d’une atrophie
optique de l’oeil droit, les pouls temporaux sont présents, et l’examen ostéo
articulaire est sans particularité.
Le bilan réalisé retrouve un syndrome inflammatoire avec une VS à 150mm,
une anémie inflammatoire à 8.8g/dl, le reste du bilan biologique est tout à fait
normal.
La biopsie de l’artère temporale droite retrouve un remaniement fibreux
discret sans lésion inflammatoire. On a complété par une échographie cardiaque et
rénale qui s’est révélée normale.
Le diagnostic de la maladie de Horton est donc retenu sur les critères de
l’ACR et le patient est mis sous corticothérapie orale à raison de 0.75mg/kg/j de
prednisone avec traitement adjuvant. L’évolution est marquée par la disparition des
douleurs.

91
OBSERVATION N°11
Il s’agit de madame A.F âgée de 64 ans, femme au foyer, diabétique sous
insuline, hypertendue depuis 20 ans sous PRETERAX , ayant une rhinite d’allure
allergique sans autres antécédents pathologiques particuliers, consultant pour une
fièvre prolongée depuis 3 mois, l’interrogatoire retrouve la notion de céphalées
temporales mais surtout à droite, avec une hyperesthésie du cuir chevelu, et un
brouillard devant les yeux, le tout évolue dans un contexte de fièvre prolongée à
39,5°C et d’altération de l’état général.
L’examen clinique retrouve les pouls temporaux en bilatéral, l’examen
ophtalmologique retrouve une cataracte bilatérale, le reste de l’examen clinique est
sans particularité, notamment cardiovasculaire, pleuro pulmonaire, abdominal et des
aires ganglionnaires.
Le bilan réalisé retrouve un syndrome inflammatoire biologique avec une VS à
110 mm à la première heure, une CRP à 259 mg/l, et une hyperleucocytose à
12 500 à prédominance PNN.
L’électrophorèse des protides sanguins retrouve une augmentation
polyclonale des gamma et de l’alpha 2 globuline, l’immunoélectrophorèse revient en
faveur d’une hypergammapathie inflammatoire.
Le bilan réalisé dans le cadre de fièvre au long cours retrouve un
épaississement discret des parois bronchiques à la radiographie thoracique de face,
l’échographie abdominale est sans particularité, la radiographie des sinus est
normale, l’échodoppler des vaisseaux du cou est sans particularité, la TDM
thoracique ne retrouve pas d’anomalies parenchymateuses ni des éléments
vasculaires médiastinaux.

92
La patiente est mise sou corticothérapie à la posologie de 0,75mg/kg/J de
prednisone avec traitement adjuvant, l’évolution est marquée par l’apyrexie dés le
lendemain, et une baisse de la CRP à 85mg après une semaine, le diagnostic d’une
maladie de Horton est donc retenu chez cette patiente qui est toujours suivie en
consultation.

93
OBSERVATION N°12
Il s’agit de madame F.F âgée de 55 ans, originaire et habitant fès, femme au
foyer, mariée et mère de 5 enfants, sans antécédents pathologiques particuliers,
présente depuis 5 mois des polyarthralgies des grosses articulations d’allure
inflammatoire, associées à des cervicalgies. La patiente rapporte aussi l’installation
de céphalées temporales rebelles aux antalgiques, associées à une hyperesthésie du
cuir chevelu, une claudication intermittente de la mâchoire et une baisse de l’acuité
visuelle bilatérale avec une rougeur oculaire, sans autres signes associés le tout
évolue dans un contexte d’altération de l’état général et d’apyrexie.
L’examen à son admission retrouve une patiente en assez bon état général,
normotendue, l’examen clinique notamment l’examen cardio vasculaire ne retrouve
pas d’abolition des pouls, les pouls temporaux sont présents et symétriques,
l’auscultation cardiaque ne retrouve pas de souffle, l’examen articulaire retrouve
une légère raideur à la mobilisation des poignets, sans limitation des mouvements,
l’examen de la charnière cervico occipitale ne retrouve pas de douleur à la
mobilisation du rachis cervical ni à la pression des épineuses, l’examen
neurologique est normal, l’examen pleuro pulmonaire est sans particularité, le reste
de l’examen clinique est tout à fait normal .
Devant ce tableau clinique, une vascularite type artérite de Horton est
fortement suspectée, on a complété le bilan par une vitesse de sédimentation qui est
revenue accélérée à 127mm à la première heure, et une CRP positive à 76mg/l, la
NFS retrouve une anémie normochrome normocytaire à 9,8 g/dl et une
thrombocytose réactionnelle. Le bilan radiologique notamment la radiographie des 2
mains en prenant les poignets et la radiographie du rachis cervical ne montre pas
d’anomalies.

94
L’examen ophtalmologique de cette patiente retrouve une acuité visuelle
conservée aux 2 yeux avec une papillite au fond d’œil.
Une corticothérapie orale à raison de 0,7mg/kg/j est démarrée en urgence
sans attendre une biopsie de l’artère temporale,la corticothérapie est associée à un
traitement adjuvant comprenant un régime sans sel,une supplémentation potassique
et en calcium vitamine D3.
L’évolution est marquée par la disparition des céphalées et des arthralgies
avec début de la dégression de la corticothérapie.
L’examen ophtalmologique de contrôle retrouve une hyperhémie papillaire
bilatérale, avec un bon réflexe maculaire, on a complété par une angiographie
rétinienne et un champ visuel qui n’ont pas retrouvé d’anomalies décelables.
L’évolution est marquée par une nette amélioration du syndrome
inflammatoire avec une vitesse de sédimentation à 10 mm et une CRP à 4 mg/l. La
corticothérapie a pu être dégréssé sans rechute de la symptomatologie jusquà 7
mg/j où il y a eu réapparition des arthralgies et de la rougeur oculaire ce qui nous a
poussé à reprendre une posologie supérieure momentanément.
.

95
Observations : maladie de Wegener
OBSERVATION N°13
Il s’agit de Mr D.L âgé de 45 ans, originaire de Taza habitant fès, cuisinier de
profession, marié et père de 3 enfants, transféré du service d’ORL pour prise en
charge d’une sinusite chronique compliqué d’ostéite.
Le patient a comme antécédents pathologiques un ulcère gastrique il y a 8 ans
traité, tabagique chronique à raison de un paquet par jour pendant 14 ans sevré il y
a 6 mois, sans autres antécédents pathologiques particuliers.
Le début de sa symptomatologie remonte à 2 ans, par l’installation d’une
symptomatologie ORL faite de céphalées frontales gauches intenses rebelles au
traitement antalgique associées à un écoulement nasal clair bilatéral au début
évoluant par la suite vers une obstruction nasale, le patient ne rapporte pas par
ailleurs une symptomatologie respiratoire ni rénale, le tout évolue dans un contexte
d’altération de l’état général et d’apyrexie
Devant cette symptomatologie, le patient est hospitalisé au service d’ORL où il
a bénéficié de 3 interventions chirurgicales avec multiples biopsies qui sont
revenues non spécifiques, la 4 ème intervention chirurgicale a consisté en une
séquestrectomie d’une pansinusite chronique avec ostéite des os propres du nez, le
résultat anatomopathologique est revenu en faveur d’une nécrose osseuse non
spécifique. Le patient rapporte par ailleurs une notion d’épistaxis, de rhinorrhées
postérieures et de sifflement de l’oreille gauche associé à un oedème de la joue
gauche, et des paresthésies de la joue, du nez et la région péribuccale gauche, sans
exophtalmie ni signes respiratoires ou autres signes associés.

96
L’examen à son admission trouve un patient en assez bon état général avec
une tension artérielle à 140/70 mmHg aux deux bras, apyrétique, l’examen cutané
trouve un oedème du visage exagéré au niveau de la joue gauche, chaud douloureux
à la palpation, étendu à la région jugulo carotidienne avec une nette déviation de la
cloison nasale (image 28).
L’examen ORL trouve un écoulement jaunâtre nasal nauséabond avec un
mauvais état bucco dentaire, une inflammation des amygdales, du voile du palais et
des piliers, sans lésion auditive, les aires ganglionnaires sont libres, l’examen pleuro
pulmonaire ne retrouve pas de râles crépitants à l’auscultation, l’examen
neurologique est normal, le reste de l’examen clinique est sans particularité.
Devant ce tableau clinique, on a évoqué une staphylococcie maligne de la face,
une granulomatose de Wegener, ou une cause néoplasique notamment un
lymphome nasal.
Le bilan réalisé retrouve une numération formule normale, un syndrome
inflammatoire fait d’une vitesse de sédimentation à 70 mm, une CRP à 60 mg /l,
l’ionogramme est correct, le prélèvement bactériologique réalisé au niveau de la face
est stérile, la protéinurie est de 0.3g/24h avec un ECBU stérile, par ailleurs la
radiographie du thorax est normal.
On a complété par un examen ophtalmologique qui a retrouvé un chémosis
conjonctival avec une rougeur diffuse et des sécrétions purulentes au niveau de
l’oeil gauche, la motilité oculaire ainsi que le fond d’œil sont par ailleurs normaux.
Devant ce tableau clinique de conjonctivite bactérienne de l’œil gauche, on a
démarré une antibiothérapie à base d’amoxicilline protégée à la posologie de 3
grammes par jour répartie en trois prises.

97
Le bilan biologique de contrôle à J5 du début de l’antibiothérapie montre une
hyperleucocytose 16 800/mm3 à prédominance PNN, et une thrombopénie à
40 000/mm3, associée à une ascension de la CRP à 70 mg/l, et devant l’altération
très importante de l’état général et le tableau clinique très évocateur de la maladie
de Wegener, une corticothérapie orale est démarrée en urgence à la posologie de
1mg/kg/j de prednisone avec traitement adjuvant sous couverture de
céphalosporines de 3éme génération injectable à raison de 2 grammes par jour.
L’examen ophtalmologique de contrôle retrouve une protrusion oculaire
irréductible indolore de l’œil gauche avec un chémosis et une hyperhémie
conjonctivale. Le reste de l’examen ophtalmologique est sans particularité, on a
complété donc par une TDM orbito-céphalique à la recherche d’un processus
oculaire responsable de l’exophtalmie, et thoracique à la recherche d’atteinte
pulmonaire secondaire à une vascularite de Wegener. La TDM orbitaire montre un
matériel de densité tissulaire hétérogène, comblant la totalité des fosses nasales et
des sinus maxillaires, frontaux, ethmoïdaux, et sphénoïdaux ;ce matériel tissulaire
est responsable de multiples foyers d’ostéolyses intéressant notamment l’ensemble
des cornets, la cloison nasale, la paroi des 2 sinus maxillaires, les 2 planchers de
l’orbite et le palais dur, cette ostéolyse présente un aspect ponctué par endroit, ce
matériel tissulaire présent a par ailleurs une extension en dehors des cavités naso
sinusiennes vers les 2 cavités orbitaires responsable d’une exophtalmie bilatérale
plus importante à gauche, vers la fosse fronto- maxillo- zygomatique gauche et les
parties molles jugales bilatérales, ces caractères sémiologiques de ce processus
lésionnel naso sinusien est très évocateur de la granulomatose de Wegener d’autant
plus qu’une pseudotumeur rétro-orbitaire peut exister au cours de cette maladie de
même qu’une ostéolyse. La TDM thoracique est par ailleurs normale. Le bilan
immunologique notamment le dosage des ANCA n’est pas réalisable vu l’urgence

98
thérapeutique et l’échographie cardiaque transthoracique retrouve une bonne
fonction systo-diastolique du ventricule gauche avec une HTAP modérée et des
cavités droites non dilatées. sur le plan thérapeutique, un traitement
immunosuppresseur était indiqué mais n’a pu être réalisé vu le contexte infectieux.
L’évolution s’est faite vers une aggravation de l’état clinique du patient avec
l’aggravation des épistaxis et l’installation de rectorragies de moyenne abondance.
Le bilan biologique retrouve une thrombopénie à 38 000 /mm3 avec un taux de
prothrombine effondré à 53%, évoquant un tableau clinique de coagulation intra
vasculaire disséminée probablement post-infectieuse ou même dans le contexte
d’une vascularite nécrosante.
Ces anomalies cliniques et biologiques nous ont poussé à démarrer des bolus
de méthylprédnisolone de 1g par jour pendant 3 jours, sous couverture
d’antibiothérapie, associés à des transfusions de plasma frais congelé . Malgré la
prise en charge thérapeutique du patient, il est décédé dans un tableau de choc
hémorragique.
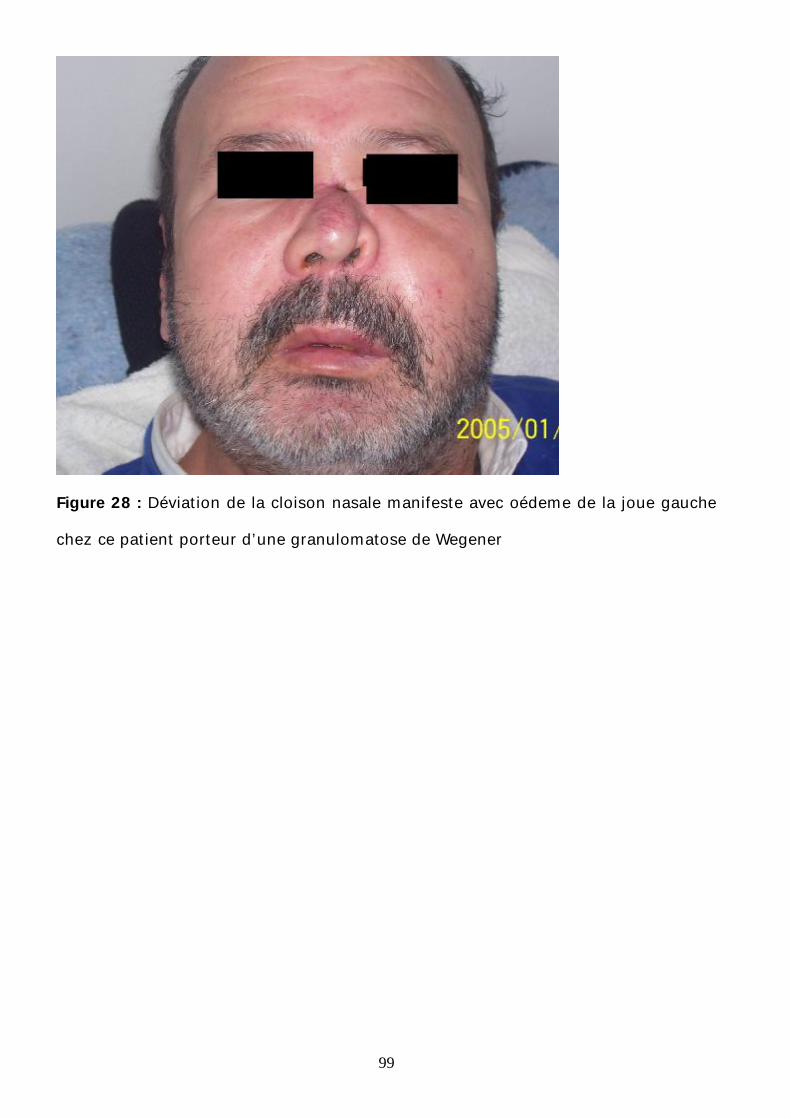
99
Figure 28 : Déviation de la cloison nasale manifeste avec oédeme de la joue gauche
chez ce patient porteur d’une granulomatose de Wegener

100
OBSERVATION N°14
Il s’agit de Mr A.M âgé de 33ans, originaire de Ktama et habitant Fès, fellah de
profession, tabagique chronique sevré il y a 15 ans, ayant comme antécédents
pathologiques, une bronchite asthmatiforme connue depuis 2000 sous traitement
non précisé, sans autres antécédents personnels ou familiaux particuliers,
hospitalisé au service pour des oedèmes des 2 membres inférieurs.
Le début de sa symptomatologie remonte à 2 mois par l’installation
progressive d’oedèmes siégeant le matin au niveau des paupières et arrivant à la fin
de la journée aux lombes et aux membres inférieurs.
Le patient rapporte par ailleurs des coliques néphrétiques associées à une
hématurie macroscopique, le tout évolue dans un contexte de conservation de l’état
général. Par ailleurs, le patient ne rapporte pas de symptomatologie ORL ni de
symptomatologie respiratoire.
L’examen clinique trouve un patient normotendu, bien portant, avec un
oedème généralisé, palpébral, au niveau des 2 membres inférieurs, blancs mous
prenant le godet à la pression, l’auscultation pulmonaire retrouve des râles sous
crépitants à la base des 2 hémichamps pulmonaires, l’examen ORL est sans
particularité, l’examen abdominal ne retrouve pas de contact lombaire, le reste de
l’examen clinique est sans particularité.
Devant ce tableau clinique, on a évoqué chez ce patient un syndrome
néphrotique, une néphropathie interstitielle, une maladie de système ou une
vascularite rénale.
Nous avons complété par un bilan biologique qui a retrouvé un syndrome
inflammatoire avec une VS à 115 mm à la première heure, une CRP à 144 mg/l, une

101
anémie inflammatoire à 8 ,8g/dl, avec à l’électrophorèse des protides sanguins une
hypoalbuminémie à 26g/l et une hyper alpha 2globulinémie, on a noté une petite
lymphopénie à la numération sanguine, le reste du bilan est revenu en faveur d’un
syndrome néphrotique avec une protéinurie de 24 h positive à la valeur de
3.68g/24h, une protidémie à 69g/l, un bilan rénal normal, et au compte d’addis une
leucocyturie à 20 800/mm3, une hématurie avec 11 020/mm3 d’hématies, et une
cylindiurie massive à 27 260/mm3. L’ECBU est stérile .
Le bilan immunologique réalisé chez notre patient est revenu négatif
notamment les AC anti nucléaires, les AC anti DNA natifs et les ANCA.
Sur le plan radiologique, la radiographie pulmonaire retrouve un
émoussement du cul de sac droit sans anomalie parenchymateuse, la TDM
thoracique retrouve un épanchement pleural bilatéral de faible abondance, on a
complété par une TDM du sinus qui a montré un épaississement muqueux diffus des
sinus avec un comblement quasi totale du sinus droit et des cellules ethmoïdales
avec une déviation du septum à droite.
La ponction biopsie rénale retrouve une glomérulonéphrite extra-capillaire
avec vascularite et des lésions d’hyalinose segmentaire et focale en faveur d’une
maladie de Wegener.
Le diagnostic d’une maladie de Wegener est retenu et le patient est mis sous
bolus de corticothérapie à la dose de 15mg/kg/j pendant trois jours relayé par voie
orale puis des bolus d’immunosuppresseurs type cyclophosphamide selon le
protocole GUILLEVIN des vascularites : J0 – J15 – J30-J60-J90-et J120 .
L’évolution au bout de 6 bolus est marquée par une nette amélioration
clinique, une disparition compléte du syndrome inflammatoire avec une amélioration

102
de la protéinurie, le compte d’addis de contrôle retrouve une leucocyturie à
33 000/mm3, une cylindrurie à 550 /mm3 et une hématurie à 4620/mm3.
Un relais thérapeutique par de l’azathioprine est proposé chez ce patient à la
posologie de 2mg/kg/j, à prendre au milieu des repas, en deux prises par jour pour
éviter une intolérance digestive et avec un contrôle biologique comprenant une NFS,
un bilan hépatique, un bilan rénal, une protéinurie de 24h et un compte d’addis.
Le bilan biologique de contrôle à 3 mois retrouve une protéinurie de 24h
toujours très positive à 3 ,35g/24h, et au compte d’addis, une aggravation des
données avec une hématurie à 21 645/mm3, une leucocyturie à 6600 /mm3 et une
cylindiurie à 6500/mm3 .On a décidé donc de mettre le patient sous BACTRIM à
raison de 2 comprimés par jour,et de reprendre les bolus de cyclophosphamide à
raison de 1g tous les 15 jours pendant 45jours (3bolus)puis un bolus toutes les 3
semaines avec une surveillance de la fonction rénale et de la protéinurie d’une façon
réguliére et rapprochée tous les 15 jours.

103
OBSERVATION N°15
Il s’agit de Mlle C.L âgée de 29 ans, célibataire, couturière de profession,
originaire et habitant fès, sans antécédents pathologiques particuliers, admise au
service de médecine interne pour prise en charge d’un syndrome anémique associé
à une bouffissure du visage.
La début de sa symptomatologie remonte à 3 mois par l’apparition d’un
syndrome anémique, associé à des arthralgies diffuses, d’allure mécanique et des
épistaxis de moyenne abondance, associées à des hémoptysies de faible à moyenne
abondance. Le tout évolue dans un contexte d’asthénie et d’anorexie.
L’évolution après 1mois est faite dans le sens d’une aggravation de la
symptomatologie clinique et l’apparition d’oedème des deux membres inférieurs
associé à une bouffissure du visage, évoluant par poussée sans douleurs lombaires,
ni hématurie macroscopique.
L’examen à son admission trouve une patiente consciente, normotendue à
10/06 mmHg, apyrétique, l’examen abdominal ne retrouve pas de contact lombaire
ni de matité déclive des flancs, l’examen pleuro pulmonaire ne retrouve pas de
syndrome d’épanchement pleural, l’examen cardio vasculaire est normal. Le reste de
l’examen clinique est sans particularité.
Devant ce tableau clinique de syndrome oedémato-ascitique, un syndrome
néphrotique dans le cadre d’un syndrome pneumorénal secondaire à une vascularite
est fortement suspecté, notamment une maladie de Wegener, un syndrome de Good
pasture ou une micropolyangéite.
Le bilan réalisé chez cette patiente montre une protéinurie de 24 h positive à
deux reprises à 4,28g/24h et à 4 ,80g/24h, la fonction rénale est correcte, l’ECBU

104
est stérile et le compte d’addis ne montre pas de leucocyturie ni d’hématurie.
Devant l’aggravation de la symptomatologie pulmonaire notamment l’aggravation
des hémoptysies, la radiographie pulmonaire de face réalisé en urgence a montré un
syndrome alvéolo interstitiel, probablement en rapport avec une hémorragie
alvéolaire secondaire à une vascularite, ou une cause infectieuse notamment une
tuberculose, ou une étiologie thrombo-embolique notamment un oedème
pulmonaire aigu de type lésionnel.
Une série de BK réalisé est revenue négative. La NFS montre un taux d’Hb à
3,3g/L. Une hémorragie intra alvéolaire est fortement suspectée, la décision a été
prise rapidement vu l’urgence de mettre la patiente sous bolus de
méthylprednisolone à raison de 15mg/kg/j pendant 3 jours, relayé au 4ème jour par
un bolus de cyclophosphamide.
Le reste du bilan notamment le bilan immunologique montre des ANCA
positifs au titre de 1/50 de type C-ANCA avec une spécificité enzymatique type PR,
Le diagnostic d’une maladie de Wegener est donc retenu. La TDM thoracique et
sinusienne sont faites n’ont pas montré d’anomalies particulières.. L’examen ORL ne
retrouve pas de lésions notamment de la muqueuse nasale mais la biopsie n’a pas
été réalisée.
L’évolution est marquée par une nette amélioration clinique avec une
disparition de la symptomatologie pulmonaire notamment les hémoptysies, et un
nettoyage radiologique spectaculaire. La décision thérapeutique est donc de
continuer les bolus de cyclophosphamide selon le protocole vascularite, relayé par
de l’azathioprine dès la 5 ème cure (J90).

105
Il s’agit donc d’une maladie de Wegener avec atteinte pulmonaire et rénale et
la biopsie rénale montre des lésions de glomérulonéphrite extra capillaire évoquant
en premier lieu une vascularite nécrosante, à ANCA de type maladie de Wegener …

106
Observation n°16:
Il s’agit de Mr Y.A âgé de 50 ans, médecin de profession, originaire et habitant
fès, marié et père de 03 enfants, hospitalisé le 06/06 pour prise en charge d’un œil
rouge associé à des arthralgies.
Dans ses antécédents, nous retrouvons la notion de rhinite intermittente
depuis l’enfance et d’une « colopathie fonctionnelle » depuis 02 ans. Le patient est
connu tabagique à raison de 27 paquets/ année sevré il y a 06 ans.
Le début de sa symptomatologie remonte à 2 mois et demi par l’installation
d’une rhinorrhée antérieure sanglante et croûteuse associée à une obstruction
nasale suivie de l’apparition depuis 02 semaines d’une rougeur oculaire bilatérale
sans douleur ni larmoiement, associées à des arthralgies de type inflammatoire,
intéressant les petites et les grosses articulations, sans signes digestifs, ni cardio -
respiratoires notamment pas de toux ni de dyspnée ni d’hémoptysie. Le tout évolue
dans un contexte d’amaigrissement non chiffré et d’apyrexie.
L’examen trouve un patient en ABEG, apyrétique, la tension artérielle est de
130/60 mmHg aux deux bras. L’examen ORL retrouve une narine gauche libre avec
une muqueuse ulcérée et présence au niveau de la narine droite d’une pseudo
tumeur en fromboise, sans perforation de la cloison nasale. Les 2 tympans sont
sans particularité, notamment, pas d’otorrhée, ni d’otite muqueuse séreuse.
L’examen de la cavité buccale est normale . L’examen ostéo-articulaire ne réveille
pas de douleurs à la mobilisation. Les articulations sont toutes libres avec une
mobilité active et passive conservée. L’examen ophtalmologique retrouve une
hyperhémie conjonctivale avec une kératite bilatérale. Le fond d’oeil est normal,
sans uvéite, ni névrite optique, ni signe de vascularite rétinienne.

107
L’examen cutané retrouve un livedo reticularis au niveau de la face interne du
genou gauche sans nodules sous-cutanés, ni d’ulcérations ou de nécrose. L’examen
neurologique est sans particularité de même que le reste de l’examen somatique.
Nous avons évoqué en premier chez ce patient essentiellement une vascularite
nécrosante type maladie de Wegener.
Le bilan réalisé retrouve une hyperleucocytose à 15 000 éléments/mm3 à
prédominance de PNN, une légère anémie à 10,8 g/dl, normochrome normocytaire
avec un syndrome inflammatoire biologique fait d’une CRP à 177 mg/l et d’une VS à
108 mm à la première heure.
L’ionogramme retrouve une légère augmentation de la créatinine à 12 mg /l,
une protidémie à 80 g/l, avec une protéinurie positive à 1,23g/24h. L’examen cyto-
bactériologique des urines ne montre pas de leucocyturie, ni de germes
décelables. La culture est négative. Nous avons complété par un compte d’Addis qui
retrouve une leucocyturie à 50 000/mm3 et une hématurie avec 100 000/ mm3. Le
bilan immunologique montre des ANCA positifs à 20 avec des C-ANCA très positifs
en IF à 80 ; les P- ANCA sont négatifs. La spécificité enzymatique est de type PR3.
Le scanner des sinus montre une déviation de la cloison nasale avec un
comblement sinusien et des images de lyse osseuse. Le scanner thoracique fait dans
le but de rechercher une atteinte pulmonaire asymptomatique a permis d’objectiver
la présence d’une pneumopathie interstitielle au niveau des bases.

108
Figure 23 ; montre un comblement sinusien avec déviation de la cloison nasale
La biopsie de la lésion narinaire a été en faveur d’une lésion inflammatoire
chronique non spécifique.
Le diagnostic d’une maladie de Wegener est tout de même retenu et le patient
est mis sous corticothérapie orale à raison de 1mg/kg/jour avec traitement
adjuvant. La protéinurie a été refaite à 15 jours d’intervalle et nous notons une
aggravation avec un chiffre à 3,40 g/24H, un compte d’addis avec une leucocyturie
qui s’est aggravée à 90000/mm3 et une fonction rénale qui s’est vite altérée avec un
chiffre méthylprednisolone de 1g/jour pendant 3 jours, avec une biopsie rénale qui
a par la suite montré une nécrose glomérulaire avec vascularite et glomérulopathie
avec prolifération extracapillaire, pas de dépôts immuns en IF ce qui est
parfaitement compatible avec une atteinte rénale dans le cadre d’une maladie de
Wegener. Nous avons instauré des bolus de cyclophosphamide IV à 1g à J0, J15, J30,

109
J60, J90, J120, relayés par de l’azathioprine à raison de 150 puis 200 mg par jour
avec une surveillance clinique, biologique portant sur le bilan rénal et le bilan
hépatique.
L’évolution est marquée par une nette amélioration clinique avec régression
des signes ORL et articulaires, ce qui nous a mené à débuter une dégression
progressive de la corticothérapie à partir de 3 semaines. La protéinurie s’est
négativée, l’hématurie et la leucocyturie ont nettement régressé et les chiffres de
créatinine se sont stabilisés à 15mg/L. Un mois après le passage à l’azathioprine, le
patient fait une rechute grave probablement favorisée par une infection broncho-
pulmonaire. La rechute était sous forme d’une altération de l’état général, fébrile à
40°C, une rhinite croûteuse manifeste et une hématurie qui réapparaît avec une
protéinurie à 2, 4 g/L et des chiffres de créatinine qui grimpent à 23 puis à 28
mg/L. Les ANCA faits à 2 reprises sont négatives. Nous entreprenons des bolus de
méthylprednisolone de 1g/J pendant 3 jours suivis par un bolus de
cyclophosphamide de 1g avec amélioration spectaculaire de l’état général et
apyrexie en 24 H, puis une baisse des chiffres de créatinine pour se stabiliser à 15
mg/L. nous avons mis en place 3 bolus de cyclophosphamide à 3 semaines
d’intervalles et une augmentation des doses d’azathioprine à 200mg/j et celles du
cotrimoxazol. Le patient a bien évolué et n’a pas fait de rechute à ce jour avec
actuellement 1 an de recul. Il continue à faire réapparaître de temps à autre des
croûtes nasales qui l’obligent à reprendre 20 à 25 mg/j de prednisone de manière
transitoire mais sans autre complication et disparition jusqu’à ce jour de la
protéinurie et stabilisation de la fonction rénale, sans hématurie, ni leucocyturie.

110
Observation n°17
Il s’agit de madame D.L âgée de 40 ans, mariée et mère de 2 enfants,
thyroidectomisée il y a 6 ans, et mise sous antithyroïdiens de synthèse, avec
antécédent de contage tuberculeux (sœur traitée pour une tuberculose pulmonaire
sous anti bacillaire depuis 2 mois) et d’une fausse couche il y a 1 mois, sans autres
antécédents pathologiques particuliers.
La patiente présente depuis 1 an des lésions cutanées à type de nodules
cutanés dermo-hypodermiques, siégeant au niveau de la face antérieure des deux
jambes avec une extension vers les deux avants bras, évoluant par poussées. Ces
lésions évoluent sans laisser de cicatrices, elles sont associées à des arthralgies
d’allure inflammatoire siégeant au niveau des grosses articulations, sans autres
signes associés, ni pulmonaire, digestif ou urinaire. Le tout évolue dans un contexte
d’altération de l’état général et d’amaigrissement chiffré à 6 kg. L’examen clinique
trouve une patiente en assez bon état général, avec à l’examen cutané des nodules
rouges et durs au niveau des ailes du nez et des paupières, des lésions d’érythème
noueux des 4 membres, l’examen pleuro pulmonaire est sans particularité, le reste
de l’examen clinique est normal.
Devant ce tableau clinique d’érythème noueux, on a évoqué une sarcoïdose,
une infection streptococcique malgré l’absence de signes infectieux précédant
l’apparition de la symptomatologie, une primo infection tuberculeuse vu les
antécédents de contage tuberculeux de la patiente ou d’autres affections
responsables d’un érythème noueux notamment des maladies de système ou une
vascularite.

111
Le bilan réalisé chez notre patiente dans le cadre d’un bilan étiologique d’un
érythème noueux montre une anémie normochrome normocytaire à 9g/dl avec une
vitesse de sédimentation accélérée à 95 mm à la première heure, les ASLO sont de
titre normal (167ui/ml), le dosage de l’enzyme de conversion est normal, l’intra
dermo -réaction à la tuberculine ainsi que la recherche de BAAR dans les
expectorations pendant 3 jours sont revenus négatifs.
L’ionogramme sanguin retrouve une perturbation du bilan rénal avec une
créatinine à 21,5mg/l et une urée à 0,88g/l, La protéinurie de 24 h est revenue
positive à 2,5g/24h, associée à une hypoalbuminémie et hyper gammaglobulinémie
polyclonale à l’électrophorèse des protides sanguins. L’ECBU est stérile, le compte
d’addis retrouve une leucocyturie à 4750/mm3 et une hématurie microscopique à
3100 hématies/mm3, sans cylindiurie, Le bilan hépatique est normal.
La biopsie des nodules cutanés est en faveur de lésions inflammatoires à
prédominance péri -vasculaire pouvant être compatible avec une vascularite.
Sur le plan radiologique, on a réalisé chez notre patiente, une radiographie
thoracique vu les antécédents familiaux de la patiente, cette dernière ne retrouve
pas d’anomalies parenchymateuses, on a complété par une TDM thoracique qui est
revenue normale.
L’échographie rénale réalisée dans le cadre d’une exploration de l’état
rénal.était normale. Nous avons également réalisé un bilan immunologique dans le
cadre d’une confirmation du diagnostic de vascularite. Ce dernier retrouve un taux
positif des AC anti nucléaires à 1/160, des AC anti DNA natif négatifs, des AC anti
antigènes solubles négatifs, et les ANCA type PR3 sont positifs à 14,3.
Les facteurs du complément C3 et C4 sont de valeurs normales.

112
Le diagnostic d’une vascularite type granulomatose de Wegener est retenue,
vu l’âge de la patiente, les lésions cutanées et articulaires, l’atteinte rénale et les
donnés du bilan immunologique.
Nous avons commencé une corticothérapie orale à la posologie de 1 mg/ kg/j
de prednisone vu le pronostic rénal en attendant les résultats d’une biopsie rénale
réalisée plus tard. L’histologie montrait une prédominance des lésions de néphrite
tubulo-interstitielle aiguë sur des séquelles de glomérulonéphropathie.
La protéinurie de 24 de contrôle était toujours positive à 1,37g/24 h après 2
mois de corticothérapie pleine dose, et vu les résultats anatomo-pathologiques,
nous avons réalisé un bolus de métylprednisolone de 15 mg/kg/j pendant 3 jours,
relayés par une corticothérapie orale, par mois pendant 6 mois.
L’ évolution était marquée par l’apparition de rougeurs oculaires au bout du 3
ème bolus de corticothérapie, d’une bouffissure du visage avec à l’examen clinique
une tension artérielle à 150/100 mmHg aux 2 bras, l’examen ophtalmologique a
retrouvé un chémosis conjonctival avec une exophtalmie , avec au fond d’œil, un
mauvais réflexe maculaire, présence de vaisseaux grêles et signe de croissant
vasculaire.
La patiente est mise sous inhibiteur calcique, avec une bonne évolution
tensionnelle, le bilan lipidique est normal. L’échographie rénale de contrôle retrouve
des reins qui sont de taille réduite et d’écho structure hyper-échogène,
dédifférenciés, ce qui nous a poussé à dégresser la corticothérapie et de proposer à
la patiente des bolus d’immunosuppresseurs type cyclophosphamide. La patiente a
présentée une aggravation de sa fonction rénale ,elle a bénéficié d’un seul bolus de
cyclophosphamide puis perdue de vue depuis 2005.

113
Observations :vascularite rénale non étiquetée
Observation n°18
Il s’agit de Mr N.H âgé de 47 ans, originaire et habitant Taza, père de 2
enfants, tabagique chronique à raison de 2 paquets par jour pendant 20 ans, avec
cannabisme et éthylisme occasionnel, suivi pour une schizophrénie depuis 1982
sous HALOPERIDOL et trihéxyphénydil, porteur d’un rhumatisme articulaire
chronique à type de polyarthrite rhumatoïde avec carpite, déminéralisation en
bandes et signes de destruction articulaire depuis 2003 mis sous salazopyrine à la
dose de 1G en 2 fois par jour et prednisolone à petites doses, hypertendu depuis
2006 sous un diurétique et inhibiteur calcique.
Le début de sa symptomatologie remonte à 8 mois par l’apparition de
vomissements alimentaires post-prandiaux précoces, sans douleurs abdominales, ni
pyrosis, ni troubles de transit, associés à une bouffissure du visage, une distension
abdominale et une augmentation de la taille des deux membres inférieurs, sans
dyspnée associée. Le patient rapporte par ailleurs des rectrorragies de faible
abondance, le tout évolue dans un contexte d’altération de l’état général.
L’examen clinique à son admission retrouve un patient en assez bon état
général, la tension artérielle est de 130/80 mmHg aux deux bras,une bouffissure du
visage, des oedèmes bilatéraux des membres inférieurs blancs moux, prenant le
godet, avec une matité déclive des flancs à l’examen abdominal, sans hépatomégalie
ni splénomégalie. Le toucher rectal retrouve des hémorroïdes externes expliquant
les épisodes de rectorragies. L’examen cardio vasculaire, pleuro pulmonaire est sans
particularité, l’examen des aires ganglionnaires retrouve des adénopathies
inguinales bilatérales lenticulaires douloureuses à la palpation, le reste des aires
ganglionnaires sont libres, le reste de l’examen clinique est sans particularité.

114
Devant ce tableau clinique, une origine rénale, cardiaque ou hépatique du
syndrome oedémato-ascitique est évoquée.
Le bilan réalisé chez notre patient retrouve une anémie à 9,2g /dl normo
chrome normocytaire, avec un syndrome inflammatoire biologique fait d’une VS à 90
mm à la première heure, la protéinurie de 24 h est revenue très positive à 6,3 g /24
h, recontrôlée à 2 reprises, avec une hypoprotidémie à 40 g/l et une
hypoalbuminémie à 12 mg/l. Le bilan rénal est perturbé avec une créatinine à 12,4
mg/l et une urée à 0,5 g/l. Le bilan hépatique est normal ainsi que les sérologies
virales hépatiques qui sont revenues négatives, ce qui nous a permis d’éliminer une
origine hépatique. L’ECBU est stérile, le compte d’Addis montre une hématurie
microscopique avec une leucocyturie à plus de 10000/mm3, le diagnostic d’un
syndrome néphrotique impur est alors retenu.
Devant la persistance des adénopathies inguinales, des sérologies virales
notamment la sérologie syphilitique est effectuée, cette dernière est revenue
négative.
Le bilan immunologique notamment les AAN, les ANCA, les anti DNA natifs
sont négatifs.
L’échographie rénale ne retrouve pas d’anomalies de différentiation cortico
médullaire, ni de dilatation pyélo-calicielle. Le doppler des vaisseaux rénaux est
normal, le bilan de crase est normal et donc, une ponction biopsie rénale réalisée est
revenue en faveur d’une glomérulonéphrite proliférative et diffuse avec nécrose
fibrinoïde et signes de vascularites. ,la coloration au rouge Congo est négative ce
qui a pu éliminer la présence d’amylose. Nous avons complété par un examen ORL à
la recherche de signe en faveur d’une vascularite de Wegener mais il est revenu

115
normal. L’examen pleuro-pulmonaire ainsi que la radiographie thoracique de face
sont sans anomalies.
Nous avons conclu à une vascularite rénale dans le cadre d’une polyarthrite
rhumatoïde, nous avons traité par des bolus mensuels de cyclophosphamide en
induction sans méthylprednisolone à cause des antécédents psychiatriques, et en
gardant la même dose précedemment administrée de prednisolone en associant un
inhibiteur calcique, un inhibiteur de l’enzyme de conversion et un diurétique. Le
méthotrexate a été instauré à raison de 15 mg/semaine pour l’atteinte articulaire.
Après 3 bolus de cyclophosphamide, nous avons remarqué la disparition du
syndrome oedémato-ascitique, un équilibre tensionnel avec persistance des
oédémes palpébraux et d’une protéinurie positive, avec sur le plan biologique une
persistance de l’hématurie, de la leucocyturie et une aggravation de la fonction
rénale (créatinine à 15 mg/l).
Devant ce tableau clinique, nous avons décidé de passer à protocole
vascularite pour les bolus de cyclophosphamide. les administrations du
cyclophosphamide se font au rythme suivant : J0, J15, J30, J60, J90, et J120.
A la fin de la 6 ème cure de cyclophosphamide, nous avons proposé un relais
par mycophénolate mofétil à raison de 1g/jour (vu les moyens).
2 mois plus tard, le bilan biologique de contrôle retrouve une protéinurie de
24h de positive à 3.26g/24h avec au compte d’Addis une leucocyturie à 2700/mm3
et une hématurie à 57 000/mm3, sans cylindrurie, ces anomalies biologiques nous
ont poussé à augmenter la dose des immunosuppresseurs à 2 g/jour. L’évolution
est marquée par une stabilisation sur le plan clinique et biologique avec un bilan de
contrôle à 3 mois de traitement montrant une protéinurie qui a diminué à

116
1,5g/24h h avec au compte d’addis une leucocyturie à 47 500/mm3 et une
hématurie à 64 000/mm3 ,on a décidé donc de garder le même traitement
immunosuppresseur avec une surveillance clinique tous les 3mois,et biologique
notamment le bilan rénal tous les 15 jours au début,puis tous les 2 mois.
Figure 26 : radiographie comparative des deux mains montrant un aspect typique de
main rhumatoïde avec une déminéralisation en bandes et une carpite bilatérale plus
accentuée à gauche

117
OBSERVATION N°19
Il s’agit de madame H .B âgée de 56 ans, originaire et habitant taounate,
femme au foyer, sans antécédents pathologiques particuliers, admise au service
pour prise en charge d’un syndrome hémorragique.
Le début de sa symptomatologie remonte à deux semaines par l’apparition
d’une altération de l’état général très importante, et d’un syndrome fébrile, aggravé
il y a 5 jours par l’installation d’un syndrome hémorragique fait d’épistaxis de
moyenne à grande abondance et d’une hématurie.
L’examen à son admission retrouve une patiente altérée, consciente avec une
tension artérielle à 160/80 mmHg aux deux bras, fébrile à 38,8°C, l’examen trouve
des stigmates d’épistaxis après méchage postérieur, l’examen cardio vasculaire
retrouve un frottement péricardique diffus, l’examen pleuro pulmonaire retrouve des
râles sous crépitants à l’auscultation des bases pulmonaires, l’examen abdominal ne
retrouve pas de contact lombaire ni de douleur à la palpation, le toucher rectal ne
ramène pas de sang, le reste de l’examen clinique est sans particularité.
Devant ce tableau clinique de syndrome hémorragique, notre prise en charge
en urgence est de mettre la patiente en condition notamment une libération des
voies aériennes avec une oxygénothérapie en lunettes, prise d’une bonne voie
veineuse périphérique de bon calibre, et un sondage de la patiente avec
quantification des urines, le bilan réalisé en urgence montre une anémie à 8 ,9 g/dl
hypochrome microcytaire avec un taux de plaquettes normal et une
hyperleucocytose à 20 000/mm3 à prédominance PNN, le bilan rénal est très
perturbé avec une insuffisance rénale aiguë : urée à 3,48g/l et créatinine à 210
mg/l, la CRP est très augmentée à 100 mg/l .

118
Un bilan infectieux est réalisé en urgence notamment une radiographie
thoracique de face qui montre un syndrome interstitiel sans foyer individualisable,
un ECBU, 3 hémocultures, une échographie abdomino rénale qui a retrouvé des reins
de taille normale, bien différenciés, hyperéchogénes sans dilatation des cavités, on a
complété par un électrocardiogramme qui retrouve une onde T ample en V2 V3 et
une hypertrophie ventriculaire gauche.
Devant ce tableau clinique, une antibiothérapie est démarrée par voie intra
veineuse à base d’une céphalosporine de 3 ème génération et dont la posologie est
adaptée à la clairance de la créatinine, associée à un hémostatique type DYCINONE
,et un antiémétique.
La surveillance de la patiente est marquée par une anurie d’installation aiguë,
avec au reste du bilan des signes en faveur d’une insuffisance rénale organique, et
donc des séances d’hémodialyse sont proposées à cette patiente.
Au bout de la deuxième séance, on a remarqué une reprise de la diurèse, une
amélioration des chiffres rénaux avec une créatinine à 94mg/l et une urée à 1,67
g/l, mais une aggravation clinique avec un pic hypertensif à 180/80 mmHg et
l’installation de troubles de conscience associés à des méléna.
Le bilan biologique réalisé en urgence retrouve une anémie à 6,4 g/dl avec
une thrombopénie à 70 000 /mm3 et un TP à 64¨%, on a proposé à cette patiente
des transfusions en culots globulaires et en plaquettes.
Le diagnostic d’une insuffisance rénale aiguë organique compliquée d’un
syndrome hémorragique et d’une hypertension artérielle est donc retenu, la
néphropathie aiguë rapidement progressive d’origine vasculaire est discutée, ainsi
qu’une néphrite interstitielle aiguë post infectieuse ou immuno allergique.

119
Devant l’aggravation clinique de la patiente et l’installation de troubles de
conscience malgré l’amélioration biologique, une corticothérapie sous forme de
bolus de méthylprednisolone sous couverture d’antibiothérapie de type
ciprofloxacine a été proposée chez cette patiente en urgence. Nous avons associé
des diurétiques injectables pour l’amélioration de la symptomatologie rénale et
pulmonaire notamment du FUROSEMIDE spécial à la posologie de 250 mg en trois
fois par jour.
Nous avons complété le bilan étiologique par ailleurs par un bilan
immunologique notamment des ANCA qui sont revenus négatifs, une TDM
thoracique et un doppler des artères rénales sans anomalies.
L’amélioration clinique était remarquable avec une reprise de la conscience au
3 ème jour du bolus, une reprise partielle de la diurèse. Mais malheureusement la
patiente est décédée dans un tableau de coagulation intravasculaire disséminée ne
répondant pas aux transfusions de plasma frais congelé compliquée probablement
d’une fibrinolyse.
Il s’agissait probablement d’une vascularite rénale type Wegener ou
micropolyangéite ou good-pasture puisqu’il y avait cette atteinte rénale rapidement
progressive et d’une atteinte pulmonaire typer hémorragie alvéolaire. Nous n’avons
pas pu effectuer de biopsies vu l’état de la patiente et peut-être fallait-il les
effectuer en post-mortem mais cela est souvent très difficile à obtenir dans notre
contexte.

120
Observation :vascularite cérébrale
OBSERVATION N°20
Il s’agit de Mr F.A âgé de 49 ans, marié et père de 10 enfants, boucher de
profession, ayant comme antécédent pathologique une hypertension artérielle
depuis an, un diabète depuis 6 ans sous antidiabétiques oraux, tabagique chronique
depuis 30 ans, sevré il y a 4 mois, sans autres antécédents pathologiques
particuliers.
Le début de sa symptomatologie remonte à 1 an par l’installation brutale
d’une hémiplégie droite, le diagnostic d’un accident vasculaire cérébral
probablement ischémique transitoire est retenu chez ce patient, il était mis sous
antiagrégant plaquettaire type ASPIRINE 160 mg/J. L’évolution immédiate est
marquée par une nette amélioration clinique et une récupération du déficit, mais
une réinstallation brutale plus tard du déficit de l’hémicorps droit avec participation
faciale, l’apparition d’une aphasie, avec des crises convulsives tonicocloniques
généralisées, devant cette symptomatologie, le patient fut hospitalisé au service de
neurologie pour prise en charge.
Au cours de son hospitalisation en neurologie et dans le cadre du bilan, une
protéinurie de 24h est revenue positive à 3.23g/24h, d’où le transfert au service de
médecine interne pour prise en charge.
L’examen à son admission trouve un patient conscient, GCS à 15, bien orienté
dans le temps et dans l’espace, une tension artérielle est à 160/100, l’examen
neurologique trouve une station debout impossible sans double aide, avec un signe
de Romberg positif et une latéralisation à gauche, la marche est possible avec
double aide, avec un fauchage à droite, signe de babinski positif à droite, l’épreuve
doigt nez est perturbée des deux côtés, l’épreuve talon genou est impossible à

121
droite, le reste de l’examen neurologique est normal, l’examen cardio vasculaire
trouve un assourdissement des bruits du cœur, le reste de l’examen clinique est
sans particularité.
Devant ce tableau clinique, le diagnostic d’une vascularite avec atteinte rénale
et cérébrale est fortement suspecté, on a complété donc les examens
complémentaires; la vitesse de sédimentation est accélérée à 125mm et la CRP est
positive à 78mg/l ; la protéinurie de 24 heures est positive à 3.25g/24h ,l’ECBU est
stérile, l’échographie rénale est normale, l’examen doppler de l’aorte abdominale et
des deux artères rénales est sans particularité, le bilan immunologique notamment
les ANCA sont négatifs, l’IRM cérébrale montre de multiples lésions cérébrales sus et
sous tentorielles en hypo signal T1, et en hyper signal T2, non rehaussés après
l’injection du produit de contraste.
Les lésions sont multiples au niveau du tronc cérébral avec une lésion bulbaire
gauche, deux lésions pontiques dont la plus grande mesure 15mm et une lésion
mésencéphalique gauche, 2 lésions des 2 pédoncules cérébelleux et 2 lésions
cérébelleuses, une à droite et l’autre à gauche en sus tentoriel, les lésions sont
multiples et de petite taille au niveau de la substance blanche, la lésion la plus
grande est capsulo thalamique droite, et mesure 2 cm de diamètre, ces lésions sont
très évocatrices d’une vascularite et dont l’étiologie reste à déterminer.
Devant cette forte suspicion de vascularite, une ponction biopsie rénale est
fortement indiquée chez le patient, mais malheureusement le patient a refusé
catégoriquement et à plusieurs reprises le geste, et donc la décision de mettre le
patient sous bolus de méthyl prédnisolone relayé par une corticothérapie orale, suivi
de l’introduction d’immunosuppresseur type cyclophosphamide est posée (selon

122
protocole vascularite :J0-J15-J30-J60-J90-et J120), le patient est mis sous
insulinothérapie.
L’évolution est marquée par une nette amélioration clinique avec équilibre de
la marche, station debout équilibrée et possible sans aide, les manœuvres de
coordination ne sont pas perturbées, avec une amélioration du syndrome
inflammatoire mais persistance d’une protéinurie positive à 0 ,9g /24h.
A J 120, la biopsie rénale a été rediscutée mais toujours refusée par le patient
et le relais par azathioprine à 100 mg/j a été fait.

123
II-DONNEES EPIDEMIOLOGIQUES :
La série étudiée comprend la période de janvier 2003 à décembre 2007 ; 20
ou patients ont été hospitalisés pour diagnostic et prise en charge d’une vascularite
systémique. Tous nos patients sont d’origine marocaine.
L’âge moyen de nos patients au moment du diagnostic est de 47.63 patient
avec des extrêmes d’âge allant de 26 ans à 70 ans, la majorité de nos patients ont
un âge inférieur à 50 ans (68.42%) avec 31% au delà de 50 ans.
Le sex. Ratio est de 13 femmes pour 7 hommes soit 1.57(fig29)
SEXE FEMININSEXE MASCULIN
Fig 29 : répartition des vascularites selon le sexe
En ce qui concerne l’origine géographique, nous attribuons peu d’intérêt aux
résultats obtenus (13 de nos patients sont des habitants de Fès, 6 de nos patients
habitent la région de Taounate).

124
III--ANTECEDENTS :
Nos patients ont des antécédents divers ; cités comme suit :
v Une HTA suivie depuis plus de 10 ans sous monothérapie est retrouvée
dans 3 cas.
v Une bronchite asthmatiforme ou /et une rhinite allergique est retrouvée
dans 4 cas.
v Un antécédent de prise d’anti bacillaire pour une tuberculose
ganglionnaire est retrouvé dans 1 cas et pour une méningite tuberculeuse dans un
autre cas, avec un cas de contage tuberculeux sans manifestation clinique.
v 7 de nos patients sont des tabagiques chroniques, sevrés actuellement à
des durées différentes, équivalents de 36.8%.
v Aucun antécédent de pathologie auto immune n’est retrouvé de même
qu’aucun antécédent familial n’est retrouvé aussi.
v Un antécédent d’un état de mal convulsif étiqueté comme méningite
tuberculeuse est retrouvé dans un cas.
7 de nos patients n’avaient pas d’antécédents pathologiques particuliers.

125
IV –LE MOTIF D HOSPITALISATION : Les motifs d’hospitalisation sont très variables ;
v Les céphalées chroniques durant plus de 6 mois sont retrouvées chez 7 de
nos patients (36.8 %).
v Une abolition des pouls ou une claudication des membres est retrouvée
chez 3 patients.
v Les arthralgies sont retrouvées dans 5 cas (26.31%).
v Une fièvre prolongée inexpliquée avec syndrome inflammatoire est
retrouvée dans 2 cas.
v Les oédémes bilatéraux des deux membres inférieurs ; les nodules
cutanés ; une nécrose digitale du 5 éme orteil, un syndrome hémorragique ou un
tableau de troubles de conscience fébrile ne sont retrouvés que dans un seul cas.
V –LES DONNEES DE L’EXAMEN CLINIQUE :
Les données de l’examen clinique sont regroupées par appareil :fig 30
L’atteinte cutanée au cours des vascularites est retrouvée chez 8 de nos
patients (42.1%) elle est présente sous forme de nodules dermo -hypodermiques,
d’un érythème noueux, d’un livedo reticularis, de cordon variqueux au niveau
temporal, d’une nécrose distale d’un orteil, d’une lésion hyperchromique malaire
bilatérale dans un cas, et enfin d’une hyperesthésie du cuir chevelu chez 5 de nos
patients représentant 62.5% de la symptomatologie cutanée.
L’atteinte ORL n’est présente que dans 21 %, elle est présente sous forme
d’une rhinorrhée postérieure avec obstruction nasale ; d’épistaxis, et d’une
tuméfaction au niveau de la narine droite et dont la biopsie est revenue en faveur
d’une pseudo tumeur.

126
L’atteinte pulmonaire est retrouvée que chez 4 patients (20%); deux patients
présentent des épisodes de toux ashmatiforme depuis l’enfance, le diagnostic d’un
asthme du sujet jeune était retenu chez eux, et les deux autres avaient
respectivement une hémorragie alvéolaire et un syndrome interstitiel dans le cadre
d’une maladie de Wegener avec atteinte pulmonaire.
Il existe une symptomatologie rénale dans 15.8% sous forme d’une hématurie
macroscopique et d’un œdème généralisé prédominant au niveau des membres
inférieurs.
9 de nos patients ont une symptomatologie neurologique, équivalent de
47.36% dont plus de la moitié présentent des céphalées temporales
bilatérales. Nous avons objectivé chez un seul patient un syndrome pyramidal à
l’examen neurologique, et un seul cas de syndrome tétrapyramidal.
8 de nos patients présentent une atteinte cardiovasculaire (40%): une abolition
des pouls d’un membre avec une claudication intermittente des membres est
retrouvé chez 5 de ces derniers, l’examen des 3 autres patients révèlent un souffle
systolique au foyer mitral avec des signes d’insuffisance cardiaque droite chez l’un,
un frottement péricardique diffus chez une autre et une tachycardie paroxystique
chez le troisième patient. Un phénomène de Raynaud est retrouvé chez deux de nos
patients.
Tous nos patients ont bénéficié d’un examen ophtalmologique ; dont 21%
présentent une baisse de l’acuité visuelle; et 21% une rougeur oculaire ; on a
objectivé une atteinte ophtalmique, dans 63.15% sous forme de :
v Un chémosis conjonctival. v Une kératite bilatérale avec des ulcérations de cornée dans deux cas. v Une rétinopathie hypertensive stade II. v Une atrophie optique chez 3 de nos patients. v Un oedème papillaire chez deux patients. v Un signe de croisement au fond d’œil chez deux patients.

127
L’atteinte articulaire est présente chez 7 de nos patients dont 2 sont des
douleurs axiales ; les autres patients présentent des arthralgies d’allure
inflammatoire dont une est une Pseudo polyarthrite rhizomélique.
Les signes généraux notamment l’altération de l’état général est retrouvée
dans 73.68% et la fièvre dans 43%, Par ailleurs aucun de nos patients ne présentent
une atteinte digestive ; des myalgies sont rapportées chez un seul patiente mien
aussi.
0
10
20
30
40
50
60
70
cutanéeORLPulmorénalNeurocardioophtalmoarticulaire
Fig 30 pourcentage des atteintes en fonction des organes atteints
VI-LES EXAMENS COMPLEMENTAIRES : Nous avons réalisé chez nos patients 3 types d’examens complémentaires :
biologiques ; radiologiques et anatomopathologiques; les examens biologiques
comprennent 3 volets : bilan standard comprenant une numération formule
sanguine, un bilan rénal et hépatique, les sérologies virales et une protéinurie de 24
h. Un bilan inflammatoire essentiellement une vitesse de sédimentation, une C
réactive protéine (CRP) et une électrophorèse des protéines sériques et enfin un
bilan immunologique.

128
a- Les bilans biologiques :
L’hémogramme de nos patients présente les résultats suivants :
v Une anémie à tendance normo- chrome normocytaire chez 10 patients
(52.6%).
v Une hyperleucocytose à prédominance PNN chez 5 patients.
v Une neutropénie chez 1 patient.
v Une thrombopénie chez un patient et thrombocytose réactionnelle chez 3
patients.
La protéinurie de 24 h est positive dans 5 cas avec un syndrome néphrotique.
La sérologie syphilitique est positive dans 2 cas.
Le bilan rénal est perturbé chez 6 patients avec un cas d’insuffisance rénale aiguë
d’installation brutale.
Le bilan inflammatoire réalisé ; retrouve une VS accélérée à plus de 50 mm
dans 16 cas (84.2%) avec une moyenne de 86,62% (allant de 50 à 115 mm à la
première heure) avec à l’électrophorèse des protéines sériques une gammapathie
monoclonale à IgG chez 2 patients et un augmentation polyclonale d’allure
inflammatoire dans 3 cas.
Le bilan immunologique réalisé retrouve 2 cas de positivité des anticorps type
C ANCA avec spécificité enzymatique de type PR3 et deux cas de positivité des
anticorps anti nucléaires.

129
b- Les bilans radiologiques :
L’échodoppler réalisé chez la majorité de nos patients est pathologique chez
10 patients (52.6%) ; différents aspects sont retrouvés :
v Un épaississement pariétal circonférentiel avec ou sans remaniement
pariétal est retrouvé chez 5 patients.
v Une thrombose artérielle chez deux patients.
v Une sténose dans deux cas.
v Et une atteinte artérielle distale sans lésion athéromateuse ni inflammatoire
dans un cas.
Une TDM thoracique est réalisée dans 5 cas mais normale, elle retrouve un
épanchement pleural bilatéral de moyenne abondance dans un seul cas.
Une TDM sinusienne réalisée chez 5 patients retrouve :
v Un épaississement diffus avec comblement du sinus maxillaire et déviation
du septum dans trois cas.
v Des lésions d’ostéolyse dans un cas.
v Et une exophtalmie secondaire à un processus sinusien compressif dans un
cas.

130
c- Les examens anatomopathologiques :
Les biopsies réalisées sont de siége différent ; une biopsie de l’artère
temporale est réalisée chez 4 patients ; plusieurs aspects histologiques ont été
rencontrés :
v une artérite temporale giganto cellulaire dans deux cas.
v Et un remaniement fibreux sans lésions inflammatoires est retrouvé un seul
cas.
v une biopsie de l’artère temporale est revenue normale dans 1 cas.
Une ponction biopsie rénale est réalisée chez 4 patients ; elle a retrouvé des
lésions de glomérulonéphrite extra capillaire avec des signes d’hyalinose
segmentaire et focale dans un cas ; une glomérulonéphrite proliférative diffuse avec
nécrose fibrinoïde est retrouvé dans un cas ; et une néphrite tubulo interstitielle
aiguë chez un autre patient.
Une biopsie des glandes salivaires accessoires est réalisée chez 2 patients et
dans les deux cas elle est revenue normale.
Des biopsies cutanées sont réalisées chez 2 patients, elle a retrouvé un
histiocyto fibrome cutané dans un cas et des lésions inflammatoires très spécifiques
de vascularite dans le 2 ème cas.
Une biopsie d’une croûte nasale est réalisée dans deux cas, elle a retrouvé
des lésions inflammatoires chroniques non spécifiques dans un cas et des lésions de
nécrose osseuse dans le deuxième cas.

131
VII-ETIOLOGIES
En résumé, on peut conclure que pour nos patients, les vascularites
rencontrées sont les suivantes :
v Une maladie de Horton chez 6 patients.
v Une maladie de Takayasu chez 4 patients.
v Une granulomatose de Wegener chez 5 patients.
v Une vascularite rénale non étiquetée (Wegener ou PAN) est retrouvée chez 2
patients.
v Un cas de vascularite cérébrale.
v Une maladie de Léo burger était retrouvée chez un seul patient.
v Et une PAN dans un seul cas.
30%
25%25%
10%5% 5%
HortonTakayasuWegenerrénaleLéoBuergerPAN
Fig 31 : Fréquence des vascularites dans notre série

132
VIII- TRAITEMENT ET SUIVI :
Pour ce qui est de la prise en charge thérapeutique ; 5 de nos patients ont
bénéficié d’une corticothérapie orale à la dose de 0,75mg /kg/j de prednisone, 11
ont bénéficié d’une corticothérapie orale à la dose de 1mg/kg/j de prednisone
représentant 57.8% de l’ensemble des malades de notre série ; un bolus de
corticothérapie selon la posologie de 15mg/kg/j pendant 3 jours de
méthylprednisolone a été proposée chez 6 patients.
Tous nos patients ayant bénéficié d’une corticothérapie orale ont fait un bilan
infectieux comprenant systématiquement une NFS, une CRP, une radiographie
thoracique de face, un ECBU, des sérologies virales notamment la sérologie de HVB,
de l’HVC, de l’HIV ainsi qu’une série de recherches de BK dans les expectorations
pendant 3 jours de suite.
Les patients dont le traitement en première intention fait appel à des bolus de
corticothérapie ont bénéficié systématiquement d’un bilan infectieux, d’une kaliémie
et d’un électrocardiogramme.
Tous nos patients ayant bénéficié d’une corticothérapie prennent un
traitement adjuvant avec un régime désodé ; une supplémentation en potassium et
en calcium et vitamine D3 et bisphosphonate dès que c’est possible.
Des immunosuppresseurs à type de cyclophosphamide sont prescrits chez 3
patients cortico résistants, les bolus de cyclophosphamide sont prescrits au
protocole suivant : J0 -J15- J30- J60- J90 et J120 et à type d’azathioprine chez 3
patients en relais avec le cyclophosphamide.
Deux patients de notre série ont bénéficié d’un traitement associé à base de
méthotrexate pour des arthralgies, et pour une maladie de Takayasu cortico
résistante.

133
Un traitement à base d’ASPIRINE comme antiagrégant plaquettaire est prescrit
dans 3 situations où il existait un risque de thrombose artérielle.
L’évolution clinique est marquée par une amélioration clinique chez 89%des
patients, une amélioration biologique dans 52.6% des cas et une aggravation chez 5
de nos patients, cette aggravation s’est présentée comme suit :
v Une HTA avec une toxémie gravidique chez une patiente.
v Une rétinopathie hypertensive stade II chez une patiente.
v Un sepsis à point de départ pulmonaire chez un patient.
v Un chémosis conjonctival avec une exophtalmie dans un cas.
v Et un diabète cortisonique secondaire dans 2 cas.
Deux patients de notre série sont décédés, et dans les deux cas il s’agit d’une
coagulation intra vasculaire disséminée secondaire à une insuffisance rénale
décompensée dans un cas, et post sepsis dans le deuxième cas.
10 de nos patients sont toujours suivis et 7 de nos patients sont perdus de vue.
Nous allons ainsi rapporter les différentes expériences thérapeutiques et les
confronter à notre prise en charge.
1-la maladie de Horton :
Le traitement repose sur une corticothérapie dont la dose varie de
0.75mg/kg/j à 1mg/kg/j, lorsque les signes cliniques se sont amendés au bout de 4
à 6 semaines, une décroissance progressive est donc proposée, elle se fait d’une
façon progressive au rythme de 10%pour atteindre progressivement la dose
d’entretien minimum efficace qui est de 5 à 20 mg par jour. Des rechutes peuvent
survenir, elles sont malheureusement fréquentes :30 à 40%. La durée totale du
traitement est variable elle peut aller de 1 an jusqu'à 5 ans. [29,44]

134
Les mêmes posologies de corticothérapie sont proposées pour nos patients
au nombre de 6 : la posologie est de 0.75mg/kg/j chez 5 de nos patients, et de
1mg/kg/j chez un seul patient présentant une maladie de Horton avec un oédeme
papillaire bilatéral au fond d’œil[31].
Une telle corticothérapie est source d’effets secondaires, en particulier digestif
et ostéoporotique, d’où l’intérêt de prescrire des protecteurs gastriques et un
traitement vitamino calcique avec des bisphosphonates au cours d’une
corticothérapie prolongée, et c’est le cas de tous nos patients porteurs d’une
maladie de Horton.
Nous rapportons la série de L.Sailler (67) qui a évalué le traitement initial par
trois bolus de 500 mg de méthylprednisolone suivis de 20 mg/j d’équivalent
prednisone chez 15 patients. Après relais oral, deux patients ont présenté des
signes d’évolutivité clinique au cours du premier mois, nécessitant une majoration
de la corticothérapie. À un mois, 12 patients étaient asymptomatiques, dont neuf
avec normalisation de la vitesse de sédimentation et de la protéine C réactive. La
vitesse de sédimentation moyenne était de 23 mm (83mm avant le début du
traitement), la protéine C réactive de 13 mg/L (94mg/l avant le début du bolus). La
vitesse de sédimentation moyenne à 3 mois était à 12 mm.
Par ailleurs, le traitement des formes compliquées de la maladie de Horton
notamment en cas d’atteinte oculaire inaugurale, d’atteinte des vaisseaux de gros
calibre, de corticorésistance ou de corticodépendance. Le traitement fait appel à
d’autres molécules notamment l’ azathioprine, le méthotrexate, la dapsone ou
l’hydroxychloroquine[68].

135
Le méthotrexate peut être associé à la corticothérapie à la dose de 7.5mg à
15mg par semaine en cas de résistance ou de dépendance à des fortes doses.(45),il
a fait l’objet de quatre principales études contrôlées, randomisées (69)et (70) ,Celle
de van der Veen(69) comporte 40 patients ayant une pseudo polyarthrite
rhizomélique dont six seulement avec une MH), l’étude n’a pu démontrer le rôle
d’épargne du MTX. Quarante-deux patients ont été inclus dans l’étude de Jover et
al(71). Ces patients ayant une MH étaient randomisés pour recevoir 60 mg de
prednisone pendant 15 jours avec une décroissance en six mois associée à du MTX
(10 mg par semaine) ou à un placebo. Au terme du suivi, le MTX a significativement
permis une épargne cortisonique de 20 % sans majorer le nombre de rechute. En
revanche, il n’y a pas eu de différences en terme de qualité de vie puisque le nombre
d’événements fracturaires rapportés était identique dans les deux groupes.
Cependant, Hoffmann et al.[70] ont suivi pendant un an, 98 patients atteints d’une
MH évoluant depuis moins de six mois. Les patients recevaient une corticothérapie
de 1 mg/kg par jour pendant quatre semaines associée à 15 mg par semaine de
MTX ou à un placebo. La corticothérapie était diminuée progressivement, à jour
alterné pour obtenir un sevrage complet en six mois. Il n’y a pas eu d’impact sur la
qualité de vie des patients. Ainsi, si le MTX est probablement l’épargneur de
corticoïdes le plus fréquemment utilisé, son efficacité reste à confirmer.
Le pronostic global de la maladie de Horton traitée est bon, l’espoir de vie des
patients est identique à celui d’une population normale de même âge.
Nos quelques patients porteurs d’une maladie de Horton n’ont pas eu besoin
du méthotrexate.

136
2-la maladie de Takayasu :
Le traitement repose aussi sur la corticothérapie, elle permet la régression de
la symptomatologie clinique et la réapparition des pouls, elle est habituellement
utilisée à la posologie de 1mg/kg/j pendant quelques semaines, puis
progressivement diminuée.
La même posologie est utilisée chez nos 5 patients présentant la maladie de
Takayasu, un anti agrégant plaquettaire est utilisé chez 2 patientes présentant une
occlusion de l’artère axillaire. [44]
Les autres molécules notamment l’azathioprine, le cyclophosphamide, et le
méthotrexate sont utilisés en cas de corticodépendance ou de cortico-résistance,
[29] comme il est le cas d’une de nos patientes qui a bénéficié du méthotrexate à la
posologie de 15 mg/semaine avec une très bonne évolution clinique.
Le traitement au cours de la maladie de Takayasu repose aussi sur les anti
hypertenseurs, le traitement de l’insuffisance coronarienne et le traitement
chirurgical en cas de lésion sténosante menaçante.
3- la maladie de Léo burger :
Repose essentiellement sur l’arrêt du tabac, l’utilisation de vasodilatateurs,
d’antalgiques et de prostacyclines ou analogues type indométacine qui reste le
traitement de choix des poussées de la maladie [42]
Le traitement chirurgical notamment une revascularisation est très limitée en
raison du caractère distal de la lésion.
Il est rapporté dan notre série un seul cas de maladie de Léo burger, le tabac
est arrêté chez ce patient avec une bonne évolution.

137
4- Maladie de Kawasaki:
Le traitement repose sur l’aspirine et les gammaglobulines intra veineuses,
l’aspirine est prescrite, à la phase aigue à des doses anti inflammatoires allant de
100 à200 mg/kg/j puis à des doses anti agrégantes allant de 20 à 30mg/kg tous les
2 à3 jours en une seule prise pendant la phase de convalescence.
La corticothérapie au cours de la maladie de Kawasaki n’a pas de place elle est
par ailleurs contre indiquée,l’intérêt des gammaglobulines intra veineuses est
démontré d’une façon indiscutable avec une posologie de 2 g/kg en une seule
perfusion de 10 heures.
L’amélioration clinique et biologique est rapide notamment une régression des
anévrismes coronariens. Nous n’avons eu aucun cas de maladie de Kawasaki vu
qu’elle survient plutôt chez l’enfant.
5- La PAN :
L’utilisation des corticoïdes a considérablement modifié le pronostic de la
PAN. Le traitement de choix repose sur l’administration de 15mg/kg/j pendant 3
jours de méthyl prédnisolone relayé par une prise orale de corticoïdes à raison de 1
mg/kg/j, la durée du traitement d’attaque dépend de la réponse clinique. On
recommande une prise à pleine dose pendant 3 à 4 semaines, suivie d’une réduction
progressive dont le but est d’atteindre une mi dose de traitement de corticoïdes à 3
mois de traitement, puis 5 à 10 mg/jour au 12 ème mois. Le groupe français
d’etude des vascularites [72] recommande la corticothérapie comme traitement de
base au cours de la PAN, en l’absence d’une infection par le VHB, elle est prolongée
pendant environ 12 mois. Les bolus de méthylprednisolone sont habituellement
administrés à la posologie de 15 mg/kg en perfusion de 60 min, répétée toutes les
24 heures pendant 1 à 3 jours. Ils sont utilisés, particulièrement en cas de menace

138
immédiate du pronostic vital ou à la phase d'extension d'une polyneuropathie, en
raison de leurs efficacité rapide et relative innocuité [73]. La corticothérapie orale est
administrée à la posologie de 1 mg/kg/jour de prednisone ou son équivalent de
méthylprednisolone. Lorsque l'état clinique du patient s'est amélioré et que la
vitesse de sédimentation est normalisée, habituellement au bout d'un mois, la
décroissance de la corticothérapie peut débuter. Dans les essais thérapeutiques
menés par le Groupe Français d'Etude des Vascularites (74,75), la réduction des
posologies des corticoïdes était progressive avec une baisse initiale de 2,5 mg tous
les 10 jours pendant 1 mois, puis de 2,5 mg chaque semaine jusqu'à la moitié de la
dose initiale, alors poursuivie en plateau pendant 3 semaines. Une décroissance
hebdomadaire de 2,5 mg était ensuite préconisée jusqu'à la dose de 20 mg par jour.
Ensuite, la posologie était réduite plus progressivement d'1 mg par semaine ou
quinzaine jusqu'à 10 mg par jour. Après un nouveau palier de 3 semaines, la
décroissance était poursuivie d'1 mg chaque mois jusqu'au sevrage. Cette
thérapeutique est efficace et peut suffire à contrôler la maladie sans adjonction des
immuno-suppresseurs.
Le cyclophosphamide en association aux corticoïdes représente
l’immunosuppresseur de choix au cours des formes sévères de la PAN, il est
prescrit par voie injectable à raison de 0.6g/m2 toutes les 2 semaines pendant 4
semaines puis toutes les 4 semaines, la durée de traitement est de 4 à 10 mois.
Selon le groupe français d’étude des vascularites [72], Les doses initiales pour des
administrations par voie orale varient de 0,5 à 2,5 g à des intervalles d'une semaine
à un mois, le cyclophosphamide IV est administré à la posologie de 0,6 g/m2 par
perfusion mensuelle pendant un an [74]. La posologie est adaptée à la fonction
rénale. Les perfusions de cyclophosphamide permettent d'administrer une dose
cumulative inférieure et exposent le patient à une toxicité potentielle moindre que
lors de l'utilisation de la voie orale continue à efficacité égale

139
La décision du traitement par le cyclophosphamide doit tenir compte des
localisations viscérales, de leur sévérité et de l'activité de la maladie. Les corticoïdes
et le cyclophosphamide ne doivent pas être employés au delà d'un an de traitement.
Dans les formes graves de PAN, définies par un FFS au moins égal à 1, douze
perfusions de cyclophosphamide doivent être recommandées en première intention
[72].
Le FFS (five factor score) a été établi pour prédire le pronostic de la
périartérite noueuse, de la polyangéite microscopique et du syndrome de Churg-
Strauss au moment du diagnostic ou d'une poussée. Ce n'est pas un score de suivi
des vascularites. Les paramètres qui le composent sont les suivants :
* protéinurie supérieure à 1 g/24 h.
* créatinémie supérieure à 140 μmoles/l.
* cardiomyopathie spécifique.
* manifestations gastro-intestinales sévères.
* atteinte du système nerveux central.
Lorsque le FFS est à 0 la mortalité à cinq ans est de 12 %, lorsque le FFS égal 1
la mortalité est de 26 %, lorsque le FFS est égal à 2 la mortalité est de 46 %. Dans
une étude menée par l’équipe française de Guillevin (72), ils ont pu démontrer
l'intérêt de ce score pour prédire la mortalité à court terme et, a fortiori, dans les 24
premières heures.

140
C’est un donc un score pronostique mesuré lors de la prise en charge du
patient au moment du diagnostic, ainsi il n’est permis de donner de la
cyclophosphamide qu’aux patients avec un score FFS supérieur ou égal à 1, le
traitement par corticoïdes est réservé aux patients dont le score est égal à 0, et en
cas d’échec on a recours aux immunosuppresseurs.
Les échanges plasmatiques sont réservés actuellement aux formes de la PAN
liée au virus de l’hépatite B.
Le pronostic de la PAN dépend des complications de la vascularite notamment
les perforations intestinales et l’atteinte cardiaque, la survie à 10 ans atteint 80%, et
les décès précoces sont l’apanage des formes fulminantes.
Nous rapportons un seul cas de PAN deux dans notre série qui présente une
association avec un myélome multiple, une corticothérapie orale est prescrite chez
lui à la posologie de 1mg/kg/j pendant 6 semaines avec début de la dégression de
la corticothérapie, l’évolution est marquée par une nette amélioration clinique et
biologique.
6-la maladie de Wegener :
L’introduction du cyclophosphamide associé à la corticothérapie est
actuellement le traitement de référence de la granulomatose de Wegener.[37]
La posologie de la corticothérapie est de 1mg/kg/j avec une dégression rapide
de la posologie au bout de la 4éme semaine pour atteindre une durée totale de 1an ,
Elle est parfois précédée d'un ou plusieurs bolus de méthylprednisolone (15 mg par
kilo et par jour)(76), Hoffman(77) propose un passage rapide à une corticothérapie à
jours alternés afin de réduire le risque d'effets secondaires.

141
Le contrôle de la maladie repose alors essentiellement sur le
cyclophosphamide. L'European systemic vasculitis trial group (EUVAS) recommande
des doses intermédiaires à celles employées dans les études américaines ou
françaises. Objectivement, les résultats cliniques sont comparables, quelque soit le
mode de décroissance de la corticothérapie mais le risque iatrogène incite
aujourd'hui à réduire au maximum les doses de corticoïdes. Il est souhaitable dès la
fin du premier mois de réduire massivement la dose de corticoïdes et d'essayer
d'atteindre une demi-dose au bout de 3 à 6 mois.
Le cyclophosphamide est administré par perfusion intra veineuse courte de
0.5à 0.7mg/m2 toutes les 2 semaines pendant 1 mois puis toutes les 3 semaines
jusqu’à la rémission [39,40].
Les rechutes sont fréquentes et touchent moins de 50% des patients après une
rémission complète survenant dans les 2 premières années du traitement.
L’azathioprine se positionne aujourd’hui comme un traitement de référence
pour maintenir une rémission de la maladie de wegener. Aprés un traitement de 3 à
6 mois par le cyclophosphamide, il est maintenant démontré que l’azathioprine peut
se substituer au cyclophosphamide.(74) . La dose thérapeutique initiale est de 2 à 3
mg par kilo et par jour.
L’association méthotrexate-corticoïdes a donné des résultats encourageants
chez des patients atteints d’une maladie de Wegener de gravité moyenne, (76-77)La
dose hebdomadaire est de 0,3 mg/kg/semaine. Son efficacité est inférieure à celle
du cyclophosphamide mais de bons résultats ont été obtenus et ce médicament est
l'objet de divers protocoles prospectifs (NIH, EUVAS).

142
Certains auteurs ont attiré l’attention sur l’efficacité du cotrimoxazole dans le
traitement de la maladie de Wegener, il doit de plus être prescrit systématiquement
pour la prophylaxie de la pneumocystose pulmonaire qui peut survenir sous
immunosuppresseurs. [40]
Les échanges plasmatiques [79] peuvent être indiqués lorsqu’il existe une
insuffisance rénale progressive et notamment en cas de glomérulonéphrite extra
capillaire grave,un des essais thérapeutiques conduit par un groupe européen
montre le bénéfice des échanges plasmatiques qui permettent de réduire le nombre
de malades dialysés à 3 et 12 mois,ce que le bolus de méthyl prédnisolone ne
permet pas d’obtenir ,de même que les échanges plasmatiques peuvent être
prescrits en cas d’hémorragie alvéolaire par analogie avec le syndrome de Good-
Pasture.[34]
Dans notre série, nous rapportons le cas de 4 patients suivis pour une
granulomatose de Wegener et dont le traitement repose sur une corticothérapie
orale avec des bolus de cyclophosphamide au rythme d’un protocole vascularite
proposé au patient, un relais par l’azathioprine est proposé dans notre série.
Le mycophénolate mofétil semble prometteur en traitement d'entretien.
D'autres médicaments (deoxyspergualine, leflunomide) ont été essayés chez un
nombre limité de malades(80). Ils ont toujours été prescrits en traitement d'entretien
ou chez des malades qui rechutent ou qui présentent une forme réfractaire à une
association de corticoides et de cyclophosphamide. La ciclosporine paraît efficace
chez quelques malades mais ne devrait pas être utilisée en première ou en seconde
ligne thérapeutique. Chez les malades réfractaires à tout traitement, l'EUVAS
propose de traiter par les antithymoglobulines et on peut aussi tenter d'utiliser les
anti-TNF [80].

143
D'autres auteurs proposent également une chimiothérapie intensive suivie
d'autogreffe de moelle. Il faudra en analyser les résultats lorsque le recul sera
suffisant.
7-Le syndrome de Churg et Strauss :
Le traitement repose comme il est pour la PAN sur la corticothérapie orale à la
dose de 1mg/kg/j.
La survie à 10 ans est de l’ordre de 80%, la principale cause de décès est
l’atteinte cardiaque, les rechutes sont plus fréquentes qu’au cours de la PAN,
l’asthme peut persister ou réapparaître après une guérison clinique de la vascularite,
restant souvent cortico dépendant, et empêchant le sevrage définitif de la
corticothérapie. [33]
8- La MPA :
Le traitement de la MPA est comparable à celui de la PAN, pour les formes
dont Le FFS est nul, et superposable à celui de la granulomatose de Wegener pour
les formes présentant des éléments de mauvais pronostic (FFS égal ou supérieur à
1).
Comme pour les autres formes de vascularites à ANCA, les rechutes sont
fréquentes, évalués à un tiers des patients. Les échanges plasmatiques pourraient en
revanche avoir une place privilégiée dans l’arsenal thérapeutique au cours de la MPA
avec atteinte rénale, ou pulmonaire sévère .

144
IX-ANALYSES
L’étude analytique de ses données retrouve une grande ressemblance clinique
entre notre série et d’autres séries européennes, nous avons comparé notre série à
celle des vascularites de l’enfant à l’échelle locale (12), et à une série de 27 cas de
vascularites diagnostiquées au Sénégal (58) et ceci pour des raisons de gradient de
fréquence qui est communément connu comme étant inversé par rapport à la
fréquence de ces vascularites. Nous avons remarqué une grande différence entre les
deux séries étudiées. La vascularite la plus fréquente dans la série infantile est le
purpura rhumatoïde, puis en deuxième position la maladie de Kawasaki ce qui est
très différent de notre série. Ceci est normal quand on connaît la répartition des
vascularites par rapport à l’âge. Dans la série sénégalaise (58), il rapporte le cas de
27 patients porteurs de vascularites et dont seulement 7 sont primitives, la
vascularite prédominante est la maladie de Horton par 3 cas suivie de la
granulomatose de Wegener par 2 cas (fig34)
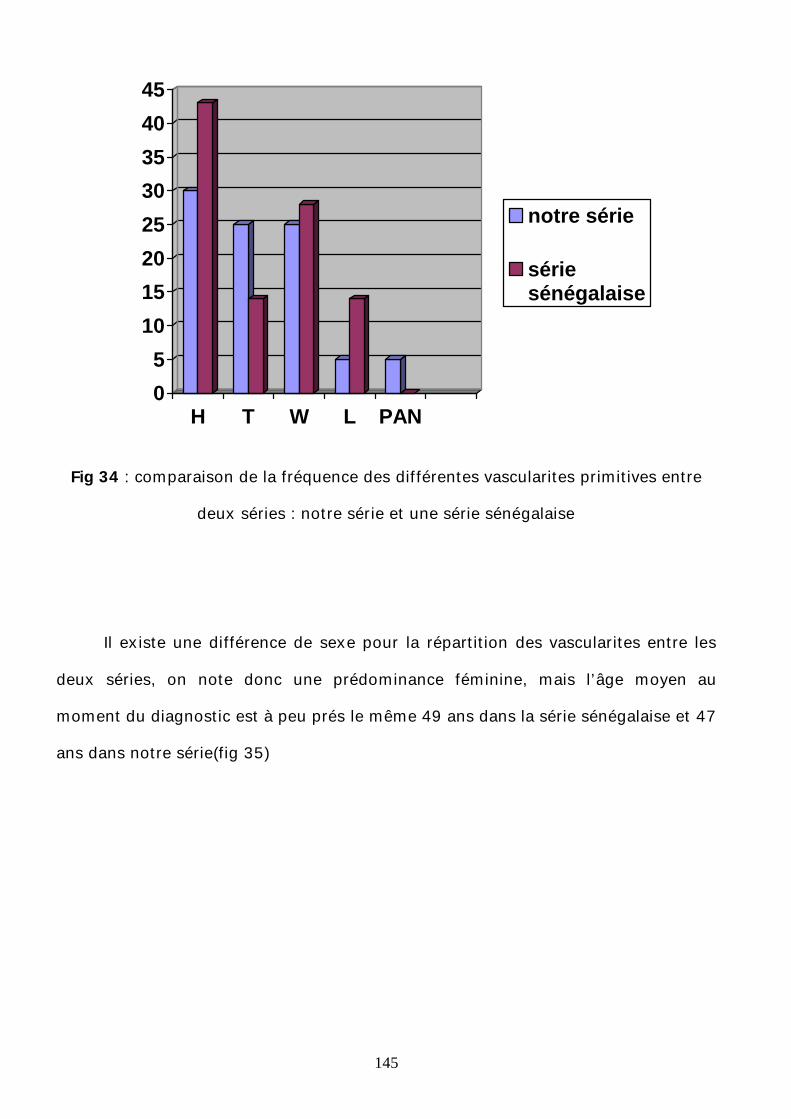
145
05
1015202530354045
H T W L PAN
notre série
sériesénégalaise
Fig 34 : comparaison de la fréquence des différentes vascularites primitives entre
deux séries : notre série et une série sénégalaise
Il existe une différence de sexe pour la répartition des vascularites entre les
deux séries, on note donc une prédominance féminine, mais l’âge moyen au
moment du diagnostic est à peu prés le même 49 ans dans la série sénégalaise et 47
ans dans notre série(fig 35)
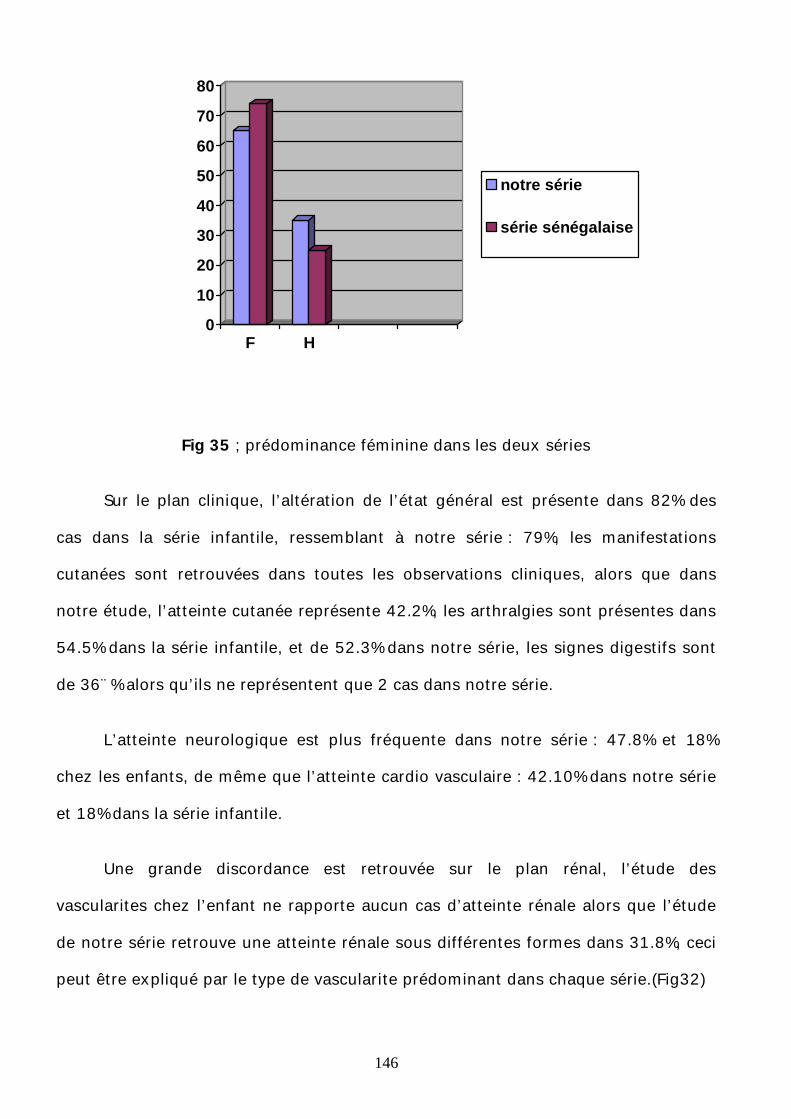
146
0
10
20
30
40
50
60
70
80
F H
notre série
série sénégalaise
Fig 35 ; prédominance féminine dans les deux séries
Sur le plan clinique, l’altération de l’état général est présente dans 82% des
cas dans la série infantile, ressemblant à notre série : 79%, les manifestations
cutanées sont retrouvées dans toutes les observations cliniques, alors que dans
notre étude, l’atteinte cutanée représente 42.2%, les arthralgies sont présentes dans
54.5% dans la série infantile, et de 52.3% dans notre série, les signes digestifs sont
de 36¨% alors qu’ils ne représentent que 2 cas dans notre série.
L’atteinte neurologique est plus fréquente dans notre série : 47.8% et 18%
chez les enfants, de même que l’atteinte cardio vasculaire : 42.10% dans notre série
et 18% dans la série infantile.
Une grande discordance est retrouvée sur le plan rénal, l’étude des
vascularites chez l’enfant ne rapporte aucun cas d’atteinte rénale alors que l’étude
de notre série retrouve une atteinte rénale sous différentes formes dans 31.8%, ceci
peut être expliqué par le type de vascularite prédominant dans chaque série.(Fig32)
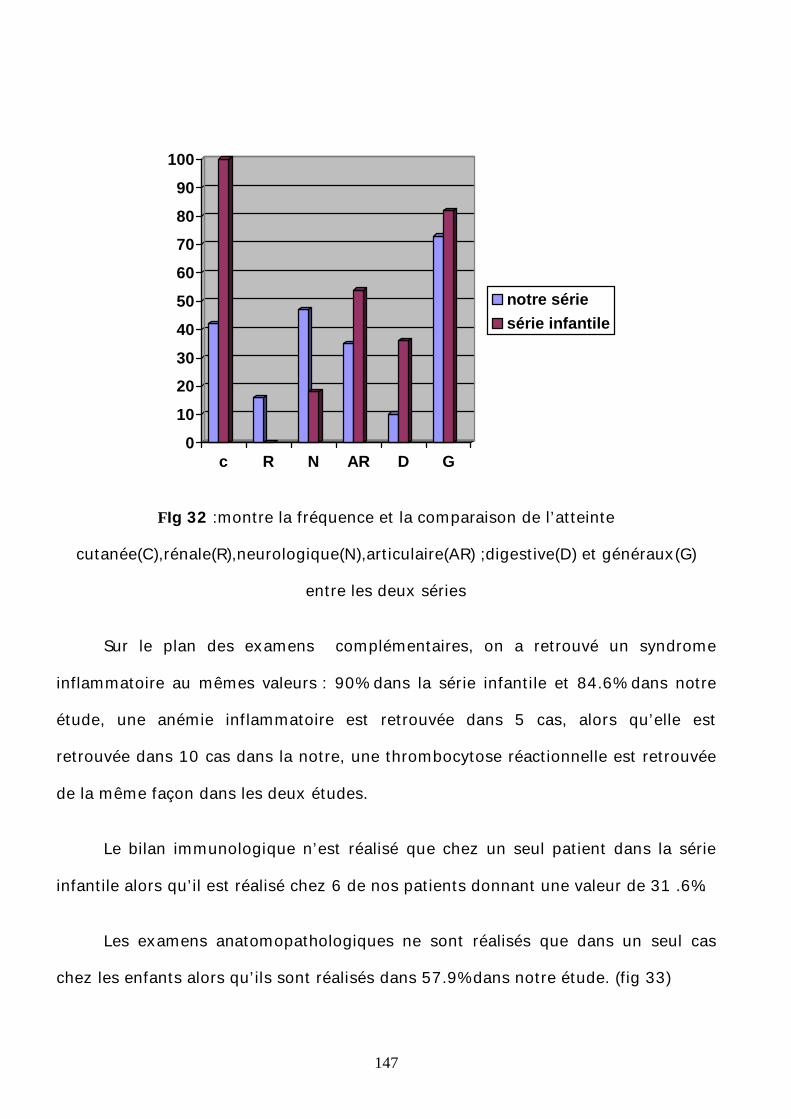
147
0
10
20
30
40
50
60
70
80
90
100
c R N AR D G
notre série série infantile
FIg 32 :montre la fréquence et la comparaison de l’atteinte
cutanée(C),rénale(R),neurologique(N),articulaire(AR) ;digestive(D) et généraux(G)
entre les deux séries
Sur le plan des examens complémentaires, on a retrouvé un syndrome
inflammatoire au mêmes valeurs : 90% dans la série infantile et 84.6% dans notre
étude, une anémie inflammatoire est retrouvée dans 5 cas, alors qu’elle est
retrouvée dans 10 cas dans la notre, une thrombocytose réactionnelle est retrouvée
de la même façon dans les deux études.
Le bilan immunologique n’est réalisé que chez un seul patient dans la série
infantile alors qu’il est réalisé chez 6 de nos patients donnant une valeur de 31 .6%.
Les examens anatomopathologiques ne sont réalisés que dans un seul cas
chez les enfants alors qu’ils sont réalisés dans 57.9% dans notre étude. (fig 33)

148
0102030405060708090
i I A
notre sériesérie infantile
Fig 33 : Comparaison des examens complémentaires demandés dans les deux
séries, bilans inflammatoires (i),bilan immunologique(I) ;et bilan
anatomopathologique(A)
Sur le plan thérapeutique, un traitement symptomatique à base
d’antipyrétiques et d’antalgiques est préconisés chez tous les enfants, des
antibiotiques sont prescrits dans 82% et des immunoglobulines par voie intra
veineuse dans un cas.
Pour ce qui est de la corticothérapie, elle est prescrite à la posologie de 2
mg/kg/j chez 18% des malades, alors qu’elle est prescrite par voie orale à des
posologies allant de 0.7 et 1mg/kg/j dans 94% dans notre série.
Les immunosuppresseurs n’ont jamais été utilisés chez les enfants alors qu’ils
sont utilisés dans 4 cas de notre étude.
Un taux de guérison est retrouvé à 45% dans la série infantile et de 89% dans
notre série.

149
Nous rapportons également le cas d’une étude marocaine menée au service de
médecine interne au CHU IBN SINA de RABAT, c’est une étude comportant 47 cas de
maladie de Takayasu [59], les observations ont retrouvé une prédominance de
l’atteinte féminine :43 femmes pour 4 hommes seulement, avec un âge moyen de 24
ans. La maladie de Takayasu est une affection touchant principalement la femme
(deux à 24 fois plus) et cette donnée est largement vérifiée dans cette série puisque
le sex-ratio est de 11/1. L’âge de découverte de la maladie est de 26,2 ans. Ce
chiffre rejoint les séries asiatiques [60-61] et sud-américaines [62-63], alors que la
médiane d’âge de diagnostic y est de dix ans plus élevée dans les séries françaises
[64, 65]. 29 patients (62 %) présentaient des signes généraux. Les signes
fonctionnels vasculaires étaient présents chez 37 patients (79 %). L’examen clinique
retrouvait une anomalie des pouls périphériques chez 45 patients (96 %) et au moins
un souffle vasculaire chez 36 malades (76 %). L’étude a donc montré la
prédominance de l’atteinte des troncs supra aortiques par rapport à l’atteinte de
l’aorte abdominale : 83%La corticothérapie était instaurée chez 41 patients (87 %) à
une dose variant entre 0,5 et 1 mg/kg/j. Un traitement par immunosuppresseurs
était instauré chez deux patientes ayant une maladie très évolutive et très
inflammatoire. Il était donné sous forme de cyclophosphamide injectable mensuel au
départ, puis de méthotrexate (20 mg par semaine). Le graphique suivant illustre la
grande ressemblance entre notre série et la série de Rabat (fig34).

150
0102030405060708090
100
1 2 3 4 5 5
notre série série de Rabat
Fig34 ;montrant une grande ressemblance clinique,radiologique,et
thérapeutique des deux séries marocaines,avec une comparaison qui a concerné et
de droite à gauche ,la fréquence de l’atteinte féminine, l’âge, l’abolition clinique des
pouls, les anomalies de l’écho doppler et l’utilisation de la corticothérapie orale.
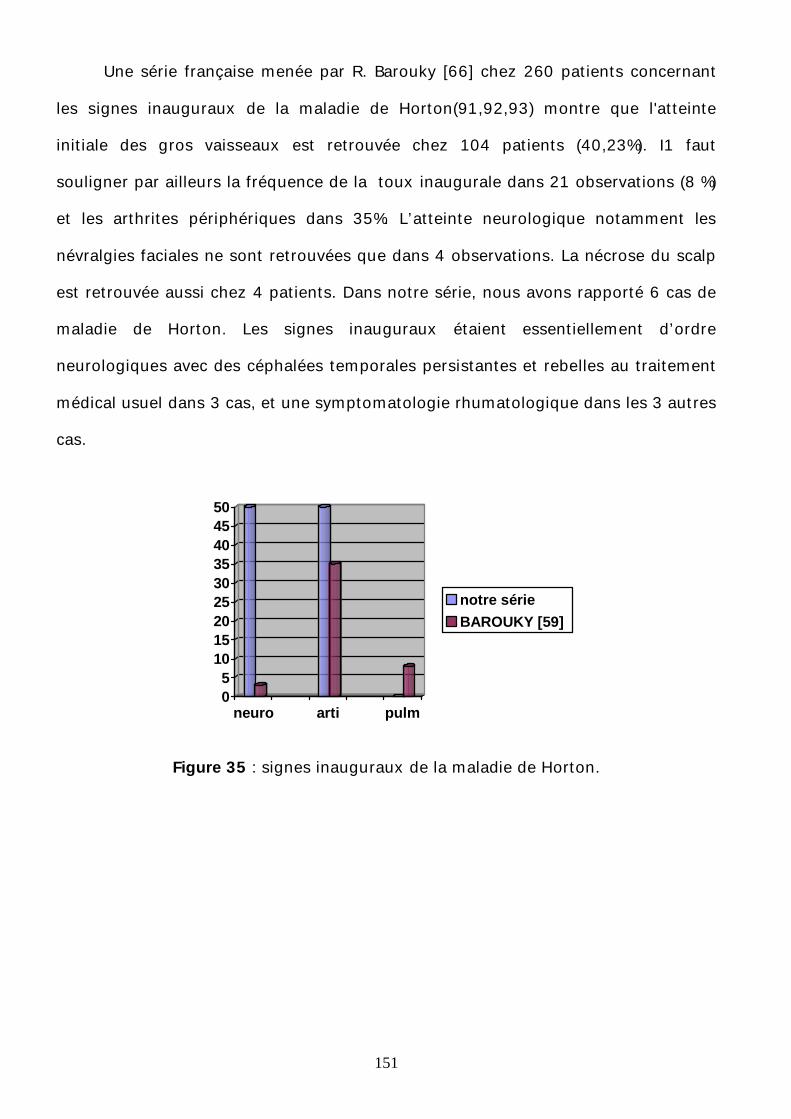
151
Une série française menée par R. Barouky [66] chez 260 patients concernant
les signes inauguraux de la maladie de Horton(91,92,93) montre que l'atteinte
initiale des gros vaisseaux est retrouvée chez 104 patients (40,23%). I1 faut
souligner par ailleurs la fréquence de la toux inaugurale dans 21 observations (8 %)
et les arthrites périphériques dans 35%. L’atteinte neurologique notamment les
névralgies faciales ne sont retrouvées que dans 4 observations. La nécrose du scalp
est retrouvée aussi chez 4 patients. Dans notre série, nous avons rapporté 6 cas de
maladie de Horton. Les signes inauguraux étaient essentiellement d’ordre
neurologiques avec des céphalées temporales persistantes et rebelles au traitement
médical usuel dans 3 cas, et une symptomatologie rhumatologique dans les 3 autres
cas.
05
101520253035404550
neuro arti pulm
notre série BAROUKY [59]
Figure 35 : signes inauguraux de la maladie de Horton.

152
NOUVEAUTES THERAPEUTIQUES

153
I-ANTI TNF ALPHA
II-LES ANTI CD20

154
Dans cette partie consacrée aux nouveautés thérapeutiques, nous allons
comparer deux traitements immunosuppresseurs qui ont marqué un très grand
intérêt dans le traitement des vascularites nécrosantes ANCA positives, en rechute
ou en échec thérapeutique sous corticoïdes et immunosuppresseurs.
I-les anti TNF alpha:
1-TNF alpha et vascularites ANCA positives :
Les ANCA sont en effet, des éléments de la chaîne conduisant à l’agression
vasculaire, ce sont des immunoglobulines de type G, et certains grains du
cytoplasme des polynucléaires peuvent être à l’origine de la production des ANCA.
Les ANCA sont utilisés comme marqueur de l’évolution des vascularites,et en
particulier de la maladie de Wegener, l’élévation de leur titre est synonyme d’une
rechute, mais il reste toujours une discordance entre l’évolution clinique et le titre
des ANCA ne permettant pas d’en faire un marqueur d’adaptation thérapeutique.
Les inhibiteurs du TNFα représentent une révolution thérapeutique en rhumatologie
dans le traitement de la polyarthrite rhumatoïde (PR) et des spondylarthropathies.
Aujourd’hui, ces nouveaux médicaments pourraient également être utiles dans
d’autres maladies auto-immunes et dans les vascularites.
2-le mode d’action de l’infliximab : REMICADE ®:
C’est un anticorps anti monoclonal chimérique anti TNF alpha, dont la partie
variable est d’origine murine et la partie constante humaine d’iso type IgG1.

155
L’infliximab se lie au TNF alpha trans. membranaire, et neutralise ses effets, il
diminue le taux des cytokines pro inflammatoires comme IL6, le taux de
l’antagoniste du récepteur de l’IL1, les taux d’haptoglobine et de fibrinogène, le
taux de GCSF, et les métalloprotéases, l’expression des molécules d’adhésion est
également diminué.
3-les contre indications :
Le REMICADE est contre indiqué dans les situations suivantes :
*Sujet moins de 17 ans.
*Pathologie infectieuse évolutive.
* antécédent d’hypersensibilité à l’influximab.
*Grossesse, et absence de contraception efficace durant la phase de traitement et
6 mois après son arrêt.
*Association avec un autre immunosuppresseur que le méthotrexate.
4-les effets secondaires de l’infliximab :
Les effets secondaires remarqués sont fréquents mais reste bénins :céphalées
,nausées,diarrhées,vertiges,douleurs thoraciques,dyspnée,
toux,rhinite,fièvre,frissons,asthénie,sueurs, ou douleurs abdominales .
L’infliximab comme étant un anti TNF alpha expose plus au risque
infectieux,une étude menée(82) montre que les infections à des germes intra
cellulaires et en particulier la listeria et les mycobactéries sont fréquemment
rencontrés chez des sujets traités par l’infliximab,ainsi que des cas de
tuberculose,et d’aspergillose.

156
Une augmentation des taux des anticorps anti DNA natifs et des auto
anticorps anti nucléaires sans lupus clinique ont été décrits(82),les auto anticorps
disparaissent progressivement après l‘arrêt du traitement . Leur signification
demeure jusqu’à ce jour inconnue, leur fréquence est évaluée à 12% en technique
radio immunologique par le test de Farr.
Les autres effets secondaires du traitement sont une toxicité hématologique
avec des cas de pancytopénie sévère,une atteinte neurologique comme une
aggravation des maladies démyélinisantes à type de sclérose en plaques,des effets
secondaires cardiaques à type d’aggaravtaion d’une insuffisance cardiaque pré
existante,une atteinte respiratoire d’ordre infectieux essentiellement
D’autres troubles méritent d’être citer : infections du tractus urinaire,
pyélonéphrite, vaginites, réactions anaphylactiques, chocs anaphylactiques,
dépression, confusion ou agitation.
5-l’expérience de l’infliximab dans le traitement des vascularites :
L’infliximab est administré en perfusion intra veineuse de 2 heures, il se
distribue dans le compartiment intra vasculaire, sa demie vie est de 8 à 10 jours, le
taux d’infliximab demeure détectable dans le sang 12 semaines après une
administration unique.
Il est utilisé dans les vascularites nécrosantes réfractaires ou en rechutes [82]
après corticothérapie et plusieurs lignes d’immunosuppresseurs, diverses équipes
[82] qui travaillent sur les vascularites systémiques commencent à utiliser ce
médicament avec des résultats encourageants mais à long terme les modalités de
traitement ne sont pas bien définies.

157
6-protocole thérapeutique de l’infliximab
Le protocole utilisé est le suivant :une perfusion de 3mg/kg d’infliximab sont
administrés à J1 et J15,si avant la troisième perfusion à J45 le patient est en
rémission complète ,le traitement sera poursuivi à la même dose à J45,J73 ,J101,et
J129 .
Par contre, si la réponse est partielle, la posologie de l’infliximab passera à
5mg/kg à J45, puis toutes les 4 semaines jusqu’à J353.
Enfin, en cas d’absence de réponse clinique à J45, la posologie de l’infliximab
sera de 5mg/kg et une nouvelle évaluation sera faite à J73 (4semaines plus tard), si
une réponse partielle est obtenue, le traitement sera poursuivi d’une façon
mensuelle jusqu’à maximum de 12 mois, à J353, dans le cas contraire, le traitement
par infliximab sera abandonné.
Si la rémission complète est obtenue plus tôt, le traitement par infliximab
pourra être interrompu et le traitement immunosuppresseur et corticoïdes sera
poursuivi.[32]
Ainsi pour une durée de 12 mois au maximum ,à raison d’une perfusion
toutes les 4 semaines,le malade pourra recevoir 14 injections d’infliximab :J1, J15,
J45 ;J73, J101, J129, J157, J185, J 213, J 241, J 269, J 297, J 325,et J 353(Fig36)

158
.
Infliximab 3mg/kg à J 1 et J15
*
rémission compléte rémission partielle absence de réponse
3mg/kg à J45 ,J 73,J 101 5mg/kg à J45puis / 5mg/kg J 129 4 sem jusqu ’a J 353
évaluation à J73
réponse partielle abscence
influximab/mois jusqu’à abondonnéJ 353
Fig36 : résumé du protocole thérapeutique d’utilisation l’infliximab.
II-le rituximab : anti CD20 : MABTHERA®
1-le mode d’action du rituximab :
C’est un anticorps monoclonal chimérique capable de se lier à la molécule
membranaire lymphocytaire B, le CD20. Cette fixation est à l’origine d’une
destruction lymphocyte B et de mécanismes complexes notamment une activation
du signal d’apoptose, une stimulation de la cytotoxicité à médiation cellulaire
antigène dépendante, et une activation du complément impliquant le C1q.
2-contre indications :
L’influximab sera contre indiqué chez les patients présentant une allergie ou
une hypersensibilité à l’un des composants du produit ou aux protéines murines
.

159
3-les effets indésirables :
Les effets indésirables attendus à notifier sont les suivants :
Des infections virales notamment à cytomégalovirus,méningo encéphalite à
entérovirus,et des infections bactériennes ,des manifestations cardio vasculaires
notamment une aggravation des pathologies cardiaques,une atteinte hématologique
sous forme de thrombopénie,ou d’anémie hémolytique,des manifestations
respiratoires notamment une insuffisance respiratoire.
Autres types d’atteintes ont été décrites : neuropathies, insuffisance rénale,
éruptions bulleuses, syndrome de Stevens Johnson, myalgies, arthralgies, et
hypertonie musculaire.
4-l’expérience du rituximab dans le traitement des vascularites :
Son expérience reste encore modeste, mais prometteuse, c’est notamment le
cas des cryoglobulinémies et des vascularites associées aux ANCA.
Dans une étude italienne [83] menée sur 20 patients atteints de vascularite
cryoglobulinémique avec une infection par le virus de l’hépatite C résistant à
l’interféron alpha ont été traités par 4 perfusions hebdomadaires du rituximab [84-
85].
Une réponse complète clinique et biologique a été observée avec un recul
moyen de plus de 12 mois, le taux des cryoglobulines a nettement diminué alors
que la virémie s’est majorée, sans complication hépatique à long terme.

160
Un autre travail italien mené par Marco Massari porte sur 15 patients ayant
une vascularite cryoglobulinémique mixte associée à une hépatite C ou à un
lymphome non Hodgkinien (85), une réponse complète clinique et biologique a été
obtenu avec un recul allant de 9 à 31mois. Les taux des facteurs rhumatoïdes et de
cryoglobulines étaient très abaissés alors que la fraction C4 du complément était
remontée. Une thrombose de l’artère centrale de la rétine et une infection cutanée
ont été notés.
Au cours de la maladie de Wegener et plus généralement des vascularites
associées aux ANCA, les premiers résultats confirment l’intérêt du rituximab.
Specks et coll(83) avaient été les premiers à décrire le cas d’un patient atteint
de la maladie de Wegener réfractaire en rémission complète clinique et biologique
après une cure de rituximab, l’augmentation des taux des ANCA au 11éme mois a
justifié une deuxième cure suivie là encore d’une négativation prolongée des ANCA.
Les équipes de mayo clinic et de Cambridge [89] ont rapporté 2 courtes séries
de 11 et 6 patients atteints d’une vascularite avec ANCA, dans tous les cas une
rémission complète a été obtenue, une forte déplétion lymphocytaire B, une
disparition des ANCA, a pu être noté chez 9 des 11 patients de mayo clinic. 2 des
11patients américains ont rechuté aux 6éme et 12éme mois, leur taux des
lymphocytes B et des ANCA sont revenus au valeur de départ, une seconde cure de
rituximab a été indiquée suivie d’une nouvelle mise en rémission.
Ainsi l’ensemble de ces publications plaide en faveur d’un effet bénéfique du
rituximab dans le traitement des vascularites réfractaires : épargne cortisonique,
obtention d’une rémission dans un délai rapide, rémission clinique et biologique
prolongée et faible risque iatrogène en dépit d’une déplétion lymphocytaire B
prolongée et profonde. Le risque de rechutes n’est toutefois pas négligeable, mais il

161
demeure tout à fait acceptable compte tenu de la sévérité et de la gravité des
affections ainsi traitées.
5-protocole thérapeutique :
Le rituximab sera administré à la dose de 375mg/m2 /j à J1, J8, J15 et J22, si
la réponse évaluée à J60 est complète ou partielle, une perfusion sera administrée
tous les 4 mois durant 1 an : M4, M8, M12. Ce traitement espacé est justifié par les
données pharmacologiques et par l’expérience hématologique qui montre que des
injections tous les 4 mois préviennent la survenue de rechutes.
Au terme du traitement, le traitement immunosuppresseur et corticoïdes sera
poursuivi, en cas d’absence de réponse à J60, le traitement par rituximab sera
abandonné, ainsi seuls les patients qui ont répondu aux 4 premières perfusions
recevront un traitement d’entretien.
La corticothérapie pourra être majorée dans un premier temps, mais la dose
sera ensuite ramenée après l’arrêt du rituximab à la posologie qu’elle était avant la
rechute. L’introduction des immunosuppresseurs peut se faire à J28, le choix se
portera sur le cyclophosphamide, le méthotrexate, l’azathioprine ou le
mycophénolate mofétil(MMF).
Nous recommandons de ne pas prescrire d’immunosuppresseur au moment
où la rechute est survenue, et de changer de lignée thérapeutique.
Le méthotrexate, si il est choisi, sera administré à la dose de
0.3mg/kg/semaine, si c’est l’azathioprine, la dose sera de 2mg/kg/j, si c’est le
MMF, la dose sera de 2g/j, si c’est le cyclophosphamide en bolus, la dose intra
veineuse sera de 0.6g/m2 toutes les 3 semaines.
162
CONCLUSION

163
Les vascularites forment un groupe d’affections différentes par leurs
présentations cliniques, leurs aspects histologiques, leurs critères diagnostiques et
leurs pronostics.
Nous avons tenté à travers ce travail de passer en revue les différents concepts
dans les domaines étiopathogéniques, et nosologiques des vascularites, ce qui nous
a permis en revanche d’avoir une nouvelle vision dans l’approche des vascularites
systémiques. Cette étude était illustrée par les 20 cas colligés au service de
médecine interne du CHU HASSAN II de FES .
Nous avons pu déduire à travers ce travail, la difficulté qui s’est opposée au
long des années pour aboutir à une classification univoque, les vascularites
systémiques ainsi représentent une entité pathologique très diverse sur le plan
clinique et histologique mais la reconnaissance des critères de diagnostic valables
pour chaque vascularite a pu améliorer la conception de cette classification.
Dans notre étude, les tableaux cliniques sont très polymorphes, avec une
atteinte cutanée quasi constante, et des signes généraux prédominants. L’étude
para clinique biologique est dominée par la présence d’un syndrome inflammatoire,
le marquage immunologique notamment le dosage des ANCA ainsi que l’étude
anatomopathologique ont une place très importante pour le diagnostic confirmatif.
Dans notre étude, nous avons remarqué une prédominance des vascularites
des gros vaisseaux notamment de la maladie de Horton et de la maladie de
Takayasu. Aucune explication étiopathogénique ne peut être donné pour le moment
vu le manque d’études comparatives à l’échelle nationale en matière de répartition
géographique

164
La prise en charge thérapeutique de nos malades se fait en fonction de la
forme de la vascularite, une corticothérapie orale ou en bolus est toujours proposé,
deux types d’immunosuppresseurs sont souvent utilisés : le cyclophosphamide et
l’azathioprine qui ont donné d’excellent résultat à l’échelle internationale et dont
l’évaluation à l’échelle locale est en cours avec des résultats préliminaires qui sont
très encourageants. Notre attitude thérapeutique dans le service est donc conforme
aux différents protocoles utilisés notamment par le Groupe Français d’Etude sur les
Vascularites.

165
La fiche d’exploitation pour étude des vascularites systémiques :
Nom et prénom :
Age :
Sexe : Origine : N de dossier :
Type de vascularite :
Motif d’hospitalisation :
Date d’hospitalisation :
Date de début des symptômes :
INTERROGATOIRE :
Athérosclérose : Tuberculose :
néoplasie : Profession :
voyages : Tabagisme :
prise de drogues : Habitudes sexuelles :
Prise médicamenteuse :
ATCD rhino pharyngés :
ATCD chirurgicaux :
ATCD gynéco obstétricaux :
ATCD familiaux :
SYMPTOMATOLOGIE ET SIGNES CLINIQUES :
Peau et muqueuse :
Urticaire ulcérations :
Érythème : gangrènes :
Nodules : purpura :
Aphtes : prurit :

166
Hyperesthésie : autres :
ORL :
Ulcérations : polypes :
Croûtes : rhinites :
Surdité : Autres :
Poumons :
Asthme : nodules :
Infiltrations : hémorragie alvéolaire :
Rein
OMI : hématurie :
Oedéme palpébral : autres :
Système nerveux central :
Convulsions : céphalées :
Signes d’HTIC : déficit moteur ou sensitif :
Pseudo tumeur : vascularite cérébrale
Système nerveux périphérique :
Cœur et vaisseaux :
Insuffisance cardiaque : HTA :
IDM : douleurs thoraciques :
Douleurs abdominale : nécrose cutanée :
Artérite des membres inf autres :
Tube digestif :
Douleurs abdominales : hémorragie digestive :
Troubles du transit : autres :
Œil :
BAV : névrite optique :
Oedème papillaire : vascularite rétinienne :

167
Uvéite postérieure : autres :
Articulaires
Arthralgies : PPR :
Musculaires :
Signes généraux :
BIOLOGIE :
VS CRP :
NFS : GB : PNN :
PNE : Hb ;
plaquettes : EPP sérique :
Bilan rénal :
Urée : créatinine :
Protéinurie de 24 h : Compte d’addis :
ECBU :
Glycémie :
COMPLEXES IMMUNS ET COMPLEMENTS :
CH50 : C3 :
C4 : Facteurs rhumatoïdes :
SEROLOGIES VIRALES ET BACTERIENNES :
HVB: HVC:
HIV: Parvovirus B12:
Streptocoques : rickettsies :
ASLO : autres :

168
AUTOANTICORPS :
AAN :
Anti DNA natif :
AC Anti AG solubles: - AC anti RO SSA
-SSB
ANCA :- C ANCA:
- P ANCA:
AC anti phospholipides :
AC anti endothélium vasculaire :
IgGantiC1q :
IMAGERIE :
RX pulmonaire :
Echodoppler des Vx :
-Cou et supra aortique :
-Membre supérieur :
-Vx abdominaux :
-Membre inférieur :
Echographie cardiaque :
Echographie rénale :
AngioTDM thoracique ou abdominale :
Angio IRM :
Artériographie :
TDM cérébrale :
TDM sinus :
Angio IRM cérébrale :
EMG :

169
BIOPSIES :
Ponction biopsie rénale :
Biopsie des glandes salivaires accessoires :
Biopsie Cutanée ou des nodules sous cutanés :
Biopsie neuromusculaire :
Biopsie de l’artère temporale :
Biopsie d’une croûte nasale :
DIAGNOSTIC RETENU :
DATE DE DIAGNOSTIC :
CRITERES DE L’ACR :
SIGNES DE GRAVITE : Rénale :
Neurologique :
TRAITEMENT INSTAURE :
Protocole :
Corticothérapie : par voie orale :
Par bolus :
Traitement adjuvant :
Immunosuppresseurs :
Traitement associé :
Médical :Antalgiques
AINS
Chirurgical :

170
EVOLUTION :
A court terme : 3 mois
Favorable :
Défavorable :- Clinique
-Biologiques
-Radiologique
A long terme : favorable
Complications : de la maladie
Du traitement
SUIVI :
Toujours : w
Perdu de vue :
DCD :

171
RESUMES

172
RESUME Les vascularites sont définies comme un groupe hétérogène d’affections
regroupant sous ce nom l’ensemble des manifestations cliniques secondaires à une
inflammation des vaisseaux quelque soient leurs types, et leurs tailles. Cette
inflammation aboutit à la destruction de la paroi vasculaire et une sténose ou
occlusion de la lumière du vaisseau.
L’objectif de ce travail est de faire une étude rétrospective sur les vascularites
systémiques colligées au sein du service de médecine interne du CHU HASSAN II
FES ; dans la période comprise entre janvier 2003 et décembre 2007, de les définir
en précisant les éléments du diagnostic positif, d’illustrer et discuter les différentes
classifications des vascularites systémiques, et de rapporter les progrès
thérapeutiques.
Cette étude a concerné 20 observations médicales; elles ont été sélectionnées
à partir de l’étude des dossiers d’hospitalisation.Seuls les patients dont le diagnostic
de vascularite a été posé au service ont été retenu au cours de notre étude, en
dehors de la maladie de Behçet.
Les résultats, retrouvent une nette prédominance féminine avec un age moyen
de diagnostic de 47 ans, les tableaux cliniques sont très polymorphes, avec une
atteinte cutanée quasi constante, et des signes généraux prédominants, l’étude para
clinique biologique est dominée par la présence d’un syndrome inflammatoire, le
marquage immunologique notamment le dosage des ANCA ainsi que l’étude
anatomopathologique ont une place très importante pour le diagnostic confirmatif.

173
Nous avons remarqué par ailleurs, une prédominance des vascularites des
gros vaisseaux notamment de la maladie d’Horton et de la maladie de
Takayasu, avec 6cas de maladie d’Horton et 4cas de maladie de Takayasu,nous
avons rapporter 5cas de granulomatose de wegener,et deux cas de vascularites
rénales,des cas sporadiques de maladie de Léo Buerger,de périartérite noueuse et de
vascularite cérébrale sont rapportés .
La prise en charge thérapeutique de nos malades passe par l’instauration de
corticothérapie ou d’immunosuppresseurs, comme il est le cas pour les autres
études menées, nous rapportons enfin les nouveaux médicaments utilisés en
matière d’immunosuppresseurs notamment les anti CD 20 et les anti TNF alpha et
dont les études dans ce domaine sont en cours d’essai. Notre attitude
thérapeutique dans le service est donc conforme aux différents protocoles utilisés
notamment par le Groupe Français d’Etude sur les Vascularites.

174
SUMMARY The vascularites are defined as a very mixed group of affections gathering
under this name the whole of the secondary clinical demonstrations to an ignition of
the vessels that they that are their types, and their tailles.this ignition leads to the
destruction of the vascular wall and a stenose or occlusion of the light of the vessel.
The objective of this work is to make a retrospective study on the systemic
vascularites colligées within the service of internal medicine during the time ranging
between January 2003 and December 2007, to define them by specifying the
elements of the positive diagnosis, to illustrate and discuss various classifications of
the systemic vascularites, and to bring back therapeutic progress.
For that, one made a retrospective study which related to 20 medical
observations; colligées within the service of internal medicine of the CHU HASSAN II
of Fès; the observations were selected starting from the study of the files of
hospitalization. Only the patients whose diagnosis of vascularite was posed with the
service were retained during our study, apart from the disease of behçet,
The results, find a clear female prevalence with an average age of 47 years
diagnosis, the clinical pictures are very polymorphic, with a quasi constant
cutaneous attack, and prevalent general signs, the biological study clinical para is
dominated by the presence of an inflammatory syndrome, immunological marking in
particular the proportioning of the ANCA as well as the anatomopathologic study
have a very important place for the confirmative diagnosis we noticed in addition, a
prevalence of the vascularites large vessels in particular of the disease of Horton and
disease of Takayasu.

175
The therapeutic assumption of responsibility of our patients passes by the
introduction of corticothérapie or immunosuppresseurs, as it is the case for the
other undertaken studies, we bring back finally the new drugs used as regards
immunosuppresseurs in particular the anti cluster of differencition 20 and the anti
TNF alpha and whose studies in this field are in the course of test.

176
vvصــملخvv
يعتبر التهاب األوعية مجموعة غير متجانسة من األمراض التي تتضمن مجموعة من
.األعراض السريرية، الناتجة عن التهاب األوعية كيف ما كان نوعها وحجمها
.ينتج عن هذا االلتهاب تدمير الجدار الوعائي وضيق الوعاء-
ة حول التهاب األوعية المنتقاة في مصحة الطب يهدف هذا العمل إلى قيام بدراسة رجعي-
، كما يهدف إلى التعريف 2007إلى دجنبر 2003الباطني خالل الفترة الممتدة من يناير
.بعناصر التشخيص ومناقشة مختلف التصنيفات مع توضيح مستجدات العالج في هذا الميدان
بمصلحة الطب الباطني قمنا لهذا الهدف بدراسة رجعية لعشرين تقرير طبي، منتقاة
بالمستشفى الجامعي الحسن الثاني بفاس، تم األخذ بعين االعتبار الحاالت التي تم تشخيصها في
.مصلحتنا، مع استثناء مرض بهجة
47أوضحت النتائج المحصل عليها ارتفاع نسبة النساء مع معدل السن المتوسط يساوي
دائم لإلصابات الجلدية، كما وضحت الدراسة سنة، األعراض السريرية كانت مختلفة، مع حضور
البيولوجية وتقدير كمية المضادات الجسمية لسيتوبالزم الخاليا الدموية المتعددة النوايا تحتل مكانة
الحظنا من خالل هذه الدراسة ارتفاع عدد حاالت التهاب األوعية ذات الحجم الكبير خاصة . مهمة
".تاكاياشو"و" هورتون"مرض
.هذا المرض اعتمدنا على دواء الكورتيك والمواد المحبطة للمناعةلعالج
20وفي ختام دراستنا نقدم كذلك األدوية المحبطة للمناعة الجديدة خاصة مضادات س د
.لألورام ألفا والتي دراستها في طور اإلنجاز ومضادات العوامل

177
BIBLIOGRAPHIE

178
1-Histoire des vascularites : René louis humbel, édition vignette historique pages
341-357.www.ssm.lu/pdfs/03_05_341-358.pdf
2-Noms et définitions des vascularites adoptés par la conférence de consensus
pour la nomenclature des vascularites systémiques à Chapel Hill, Caroline du Nord,
États-Unis, 1993 L. Guillevin et al. La revue de médecine interne 24 (2003) 172–
182.
3-chapel Hill nomenclature ;classification,épidémiologie ,pathogénie et traitement
des vascularites nécrosantes ,groupe français d’étude des vascularites ;GFEV Laure
Hélène Noël et Loïc Guillevin hôpitaux Necker et
Cochinwww.vascularite.com/get_pdf.php?id=147
4-périarthérite noueuse et polyangéite microscopique ;ERIC HACHULLA,médecine
interne de B.DEVULDER,PIERRE IVES HATRON , connaissances II,les pathologies
systémiques,pages 142-149. Www/nephrohus.org
5-périarthérite noueuse ; diagnostic et évolution, Service de Rhumatologie
Professeur Olivier MEYER, Cours DCEM3Pathologie de l'appareil locomoteur –
rhumatologie, DCEM3, Internat n° 329.
6-pathogénie et traitement des vascularites nécrosantes, pathogénie de la PAN
pages 120-126, GFEV, laure héléne noël, loic Guillevin
www.vascularite.com/get_pdf.php?id=147.
7-aspects cliniques pratiques des vascularites ; proposition d’un arbre décisionnel
pour le praticien,O .MEYER,service de rhumatologie,professeur OLIVIER MEYER
pagesperso-orange.fr/corine.bensimon/vascularites.htm.

179
8-les vascularites nécrosantes systémiques ; classification et stratégies actuelles de
traitement,revue de médecine interne24(2003)172-
182,L.GUILLEVIN,A.MAHR,P.COHEN
9-maladie d’Horton : étude analytique,rappel physiopathologique ,pages 8-12.
10-immunologie de JEAN PIERRE REVILLARD,pages 261-262. books.google.co.ma/books?isbn=2804138054
11-les vascularites systémiques d’étiologie déterminée,aspects cliniques et
thérapeutiques,L.Guillevin,diagnostics difficiles en médecine interne,sous la
direction de H.Roussel,D.Vital Durand,J-L Dupond,2eme édition,maloine,1999,pages
991-1008.
12-les vascularites de l’enfant,revue de la littérature illustrée par 11 cas,thèse N
316, 2003M.siddat,pages 20-21.
13-Les vascularites systémiques, séance de la société médicale des hôpitaux de
paris, 26mars 1999, rédacteur/ invité ; loic Guillevin, annales de médecine interne
2000, 151, n3, 178-183 ; les auto anticorps ANCA.description et rôle
immunopathologique.
14-la découverte des ANCA ,une avancée majeure pour la prise en charge des
vascularites,dossier thématique :vascularites nécrosantes systémiques associées
aux ANCA,presse médicale Vol 36 - N° 5-C2 - Mai 2007.
15-vascularites : interprétation des ANCA, T .Hannedouche, nephrohus.org, le 5
octobre 2001.
16-les vascularites systémiques : revue de la littérature –nouvelle classification,
thèse N 355, Z.tazi mezalek année 1992, page 39.

180
17-vascularites : classification, T.Hannedouche, w .ww.nephoHUS.org Le 5 octobre
2000 .
18-les vascularites : classification des vascularites, Patrick cherin, Christian
pagnoux, réflexions rhumatologiques, N65, tome8, janvier 2004, pages 4-9 .
19-les vascularites : les anticorps anti cytoplasme des neutrophiles (ANCA) rôle et
intérêt clinique dans les vascularites, phillipe lesavre, réflexions rhumatologiques,
N65, tome8, janvier 2004, pages13-16.
20-vascularites : syndrome de good pasture, T.Hannedouche, Septembre 2000
www.nephrohus.org
21-les vascularites : manifestations pulmonaires des vascularites à ANCA, phillipe
brissaud, réflexions rhumatologiques, N65, Tome8, janvier 2004, pages17-24.
22-manifestations abdominales et digestives au cours des vascularites systémiques,
Christian pagnoux, annales de médecine interne, vol154, N7, novembre 2003.
23-les vascularites systémiques, avant propos,L .Guillevin,la revue du
praticien,2000,50,pages247-248.
24-vascularites systémiques, pour la pratique, MF.Kahn, la revue du praticien, 2000,
50, pages295-297.
25-collége français des enseignants de rhumatologie avec la collaboration de
X.chevalier,RM.filipeau,P.Goutille ,J.Sibilia,les vascularites,in
rhumatologie(abrégé)connaissances et pratiques,édition Masson,2002.
26-systemic vascularitis : epidemiology, classification and environnemental factors,
leader, ann rheum dis 2000,59.

181
27-causes et mécanismes des vascularites, lersavre, LH Noël, la revue du praticien,
2000 ,50.pages 255,260 .
28-vascularites,davidson,in medecine interne,principes et pratique,édition
2000,dirigés par C.Haslett,traduit de la 18éme édition anglaise,maloine,113.159.
29-traitement des vascularites systémiques, F.Lhote, la revue du praticien 2000.
2000, vol. 50, no3, pp. 285-294 (37 ref.)
30-purpura rhumatoïde et immunoglobulines intra veineuses,revue de médecine
interne,A.Ruellan,M.Khatibi,T .staub,1997,n18,pages 727-729.
31-groupe français d’étude des vascularites,RATTAP,traitement par influximab
versus rituximab des vascularites nécrosantes systémiques ANCA positives,en
rechutes ou réfractaires aux corticoïdes et aux immunosuppresseurs,essai pilote
prospectif multi centrique,version 2,21/06/2004,investigateur et coordinateur du
groupe français d’étude des vascularites :professeur loic Guillevin
www.vascularite.com/get_pdf.php?id=12 -
32-vascularites systémiques, syndromes auto immuns et inhibiteurs du TNF alpha,
M.de bandt, la revue de médecine interne, volume 27, issue5, mai 2006, pages 363-
365 .
33-protocole randomisé de traitement des vascularites systémiques, choix du
traitement d’entretien par azathioprine et prédnisone après obtention d’une
rémission complète (REMAIN), investigateur principal pour la France : Pr.py .Hatron,
CHU de Lille, octobre 2004. www.vascularite.com/get_pdf.php?id=16

182
34-les vascularites nécrosantes systémiques ;classification et stratégies actuelles de
traitement,la revue de médecine interne,24,2003,pages 172-
182,L.Guillevin,A .mahr,P.Cohen.
35-les vascularites pulmonaires, jean François cordier, hôpital cardio vasculaire et
pneumologie luis pradel, université Claude Bernard, 69394, Lyon, cedex 03, France,
EMC consulte. www.emc-consulte.com/afficher-
article?item=22921&cap=1&filetype=1
36-les cryoglobulinémies mixtes, P.Cacoud, L.Musset, la revue du praticien2000, vol
50, pages 276-280.
37-la granulomatose de Wegener, JF Cordier, la revue du praticien 2000,50, pages
271-275.
38-a prospective randomized trial comparing steroids with pulse cyclophosphamide
versus steroids and oral cyclophasphamide in the treatement of generalized
Wegener ‘s granulomatis, L.Guillevin, JF.Cordier, F.Lhote et al, arthris rhem
1997,40 ,2187.
39-a straged approach to the treatement of wegener’s granulomatis ;induction of
remission with glucocorticoids and daily cyclophosphamide switching to
methotrexate for remission maintenance,CA Langford,C Taler williams,KS Barron,MC
Scheller,arthris rheum 1999,42,2666.
40-the treatement of wegener’s granulomatis with tremetroprim/sulfamethaxazole :
illusion or vision, RA Deramee, arthris rheum, 1988, 31,1068.
41-Maladies rares des vaisseaux ; le syndrome de churg et Strauss, P.Cohen, STV
(sang-thrombose-vaisseaux) n 8, vol 12, octobre 2000, pages 515-518.

183
42-périarérite noueuse,L Mouton,P Cohen,STV(sang-thrombose-vaisseaux)n
4,vol12,avril 2000,pages 237-241.
43-que va-t-il se passer demain dans le domaine des vascularites, L.Guillevin,
annales de médecine interne, 1994, 145, pages 514- 515.
44-epidemiologie et facteurs étiologiques des artérites à cellules géantes(maladie
de Horton et maladie de Takayasu)J Emmerich,JN Fissinger,annales de médecine
interne,1998,149,N 7,pages 425 -432.
45-La maladie de Horton en 2007 : épidémiologie, imagerie et traitement,Giant cell
arteritis in 2007: Epidemiology, imaging and treatment Valérie Devauchelle-
Pensec,Sandrine Jousse, Claire Destombe et Alain Sarauxa ,revue du rhumatisme ,27
septembre 2007
46-W.A. Schmidt, H.E. Kraft and L. Volker et al., Colour Doppler sonography to
diagnose temporal arteritis, Lancet 345 (1995), p. 866.
47-W.A. Schmidt, H.E. Kraft and K. Vorpahl et al., Color duplex ultrasonography in
the diagnosis of temporal arteritis, New Engl Journal Med 337 (1997), pp. 1336–
1342.
48-F.B. Karassa, M.I. Matsagas and W.A. Schmidt et al., Meta-analysis: test
performance of ultrasonography for giant-cell arteritis, Ann Intern Med 142 (2005),
pp. 359–369.
49-T.A. Bley, O. Weiben and M. Uhl et al., Assessment of the cranial involvement
pattern of giant cell arteritis with 3T magnetic resonance imaging, Arthritis Rheum
52 (2005), pp. 2470–2477.

184
50-M. Markl, M. Uhl and O. Wieben et al., High resolution 3T MRI for the assessment
of cervical and superficial cranial arteries in giant cell arteritis, J Magn Reson
Imaging 24 (2006), pp. 423–427.
51-T.A. Bley, T. Ness and K. Warnatz et al., Influence of corticosteroid treatment on
MRI findings in giant cell arteritis, Clin Rheumatol 26 (2007), pp. 1541–1543.
52-T.A. Bley, M. Uhl and N. Venhoff et al., 3-T MRI reveals cranial and thoracic
inflammatory changes in giant cell arteritis, Clin Rheumatol 26 (2007), pp. 448–450.
53-A. Turlakow, H.W. Yeung and J. Pui et al., Fludeoxyglucose positron emission
tomography in the diagnosis of giant cell arteritis, Arch Intern Med 161 (2001), pp.
1003–1007
54-D. Blockmans, L. de Ceuninck and S. Vanderschueren et al. Repetitive 18F-
fluorodeoxyglucose positron emission tomography in giant cell arteritis: a
prospective study of 35 patients, Arthritis Rheum 55 (2006), pp. 131–137.
55-PM.ZEEK, CC.SMITH, JC WEETER:studies on periarteritis nodosa,the difference
between the vascular lesions of PAN and of hypersensybility.ann J path,1948,vol
24,p 889.
56-RH ZAX,SJ.HODGE,JP LALLEN:cutaneous leucocytoclastic vascularites,serial
histopathologic evaluation demonstrates dynamic nature of the infiltrate,arch
derm,1990,vol 126,pages 69-72.
57-lesvascularites, classification histopathologique, J.WESCHSLER, P.BRUNEVAL,
F.CAPRON, JP CAMILLERI, revue de médecine interne, 1988, N 9, pages 507-515.

185
58 -Vascularites systémiques : étude de 27 cas Sénégalais S. Dialloa, S. Diallob, I.B.
Diopc, A. Talld, S. Ndongoe and T.M. Diope ,aMédecine Interne, CHU A le Dantec,
Dakar, Sénégal,bMedecine Interne, C.H.U. le Dantec, Dakar, Sénégal,cCardiologie,
C.H.U. le Dantec, Dakar, Sénégal,,Orl, CHU A le Dantec, Dakar, Sénégal,Médecine
Interne, C.H.U. le Dantec, Dakar, Sénégal
Available online 18 October 2007.
59- La maladie de Takayasu au Maroc. À propos de 47 observationsA. El Asri1, Z.
Tazi-Mezalek1*, M. Aouni1, M. Adnaoui1, A. Mohattane1,Y. Bensaid2, A. Maaouni1
1Service de médecine interne, hôpital Ibn-Sina, 10000 Rabat, Maroc ; 2service de
chirurgie vasculaire, hôpital Ibn-Sina,10000 Rabat, Maroc. Rev Méd Interne 2002 ;
23 : 9-20
60- Hotchi M. Pathological studies onTakayasu’s arteritis. HeartVessels 1992 ; 7
(suppl) : 11-7.
61- Park JH, Hong SK, Choi KJ, Sohn DW, Or BH, Luc MM, et al. Takayasu arteritis in
Korea: clinical and angiographic features. Heart vessels 1992 ; 7 (suppl) : 55-9.
62- Lupi-Herrera E, Sanchez-Torres G, Marcushamer J, Mispireta J,Horwitz S, Vela JE.
Takayasu’s arteritis: clinical study of 107 cases. Am Heart J 1977; 93: 94-103.
63- Dabague J, Reyes PA. Takayasu’s arteritis in Mexico: a 38-year clinical
perspective through litterature review. Int J Cardiol 1996; 5 (suppl): 103-9.

186
64- Fiessinger JN, Tawfik-Taher S, Capron L, Laurian C, Cormier JM, Camilleri JP, et
al. Maladie de Takayasu critères diagnostiques. Nouv Presse Méd 1982 ; 11 : 583-6.
65-Blétry O, Kieffer E, Thomas D. L’artérite de Takayasu. San Thrombose Vaisseau
1990 ; 2 : 245-52.
66- Symptômes inauguraux de la maladie de Horton sur une série de 260 patients
R. Barouky, C. Becourt-Vedomme, C. Alexandre, R. Gonthier, I. Durieu, D. Vital
Durand, H. Laurent, H. Rousset I Service de mddecine inteme, CH Lyon=Sod, 69495
Pierre-Bdnite cedex; 2service de rhumatologie, CHU de Saint-Etienne, 42055 Saint-
~-_tJenne cedex 2; aservice de gdriatde, hOpital de la ChadM, 42000 Saint-Etienne;
~service de mddecine interne, CH de Saint-Chamond, 42403 Saint-Chamond, La
Revue de Médecine Interne, Volume 22, Issue 7, July 2001, Pages 631-637
67-Maladie de Horton non compliquée : traitement initial par trois bolus de 500 mg
de méthylprednisolone suivis de 20 mg/j d’équivalent-prednisone. Évaluation chez
15 patients L. Sailler*, M. Carreiro, S. Ollier, H. Juchet, P. Delobel, D. Ferrand, M.
Klinger, P. Arlet ,Service de médecine interne, hôpital de Rangueil-Larrey, CHU,
31998 Toulouse cedex, France La Revue de Médecine Interne, Volume 22, Issue
11, October 2001, Pages 1032-1038
68-La maladie de Horton compliquée : modalités thérapeutiques Complicated giant
cell arteritis: therapeutic modalities , Christian Agardand Jacques Henri Barrier
Service de médecine interne B, Hôpital Hôtel-Dieu, CHU de Nantes
La Presse Médicale, Volume 33, Issue 1, January 2004, Pages 51-59

187
69-M.J. van der Veen, H.J. Dinant and C. van Booma-Frankfort et al., Can
methotrexate be used as a steroid sparing agent in the treatment of polymyalgia
rheumatica and giant cell arteritis?, Ann Rheum Dis 55 (1996), pp. 218–223.
70- G.S. Hoffman, M.C. Cid and D.B. Hellmann et al., A multicenter, randomized,
double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant
cell arteritis, Arthritis Rheum 46 (2002), pp. 1309–1318.
71-J.A. Jover, C. Hernandez-Garcia and I.C. Morado et al., Combined treatment of
giant-cell arteritis with methotrexate and prednisone: a randomized, double-blind,
placebo-controlled trial, Ann Intern Med 134 (2001), pp. 106–114.
72-le groupe français d’etude des vascularites(GFEV) périartérite noueuse,traitement
,www.vascularites.com.
73- Renoux M, Hilliquin P, Menkes C. Les bolus de méthylprednisolone en
rhumatologie. Ann Méd Interne (Paris). 1994;145:133-9
74-Guillevin L, Lhote F, Jarrousse B, Fain O. Treatment of polyarteritis nodosa and
Churg-Strauss syndrome. A meta-analysis of 3 prospective controlled trials
including 182 patients over 12 years. Ann Méd Interne (Paris). 1992;143:405-16
75-Guillevin L, Fain O, Lhote F, et al. Lack of superiority of steroids plus plasma
exchange to steroids alone in the treatment of polyarteritis nodosa and Churg-
Strauss syndrome. A prospective, randomized trial in 78 patients. Arthritis Rheum.
1992;35:208-15

188
76-groupe français d’étude des vascvularites, traitement de la granulomatose de
wegener www/vacularites.com
77- . Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al.
Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med
1992;116(6):488-98
78 . Reinhold-Keller E, Beuge N, Latza U, De Groote K, Rudert H, Nölle B, et al. An
interdisciplinary approach to the care of patients with Wegener's granulomatosis.
Long-term outcome in 155 patints. Arthritis Rheum 2000;43:1021-32
79 . Matteson EL, Gold KN, Bloch DA, Hunder GG. Long-term survival of patients
with Wegener's granulomatosis from the American College of Rheumatology
Wegener's Granulomatosis Classification Criteria Cohort. Am J Med
1996;101(2):129-34
80 . Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener's
granulomatosis. A clinical and imaging study of 77 cases. Chest 1990;97(4):906-12
81 . Walton E. Giant-cell granuloma of respiratory tract (Wegener's granulomatosis).
Br. Med. J. 1958; 2:26
82-les anti TNF dans les maladies auto immunes par X. MARIETTE Service de
Rhumatologie, INSERM EMI 0109, Hôpital de Bicêtre, Université Paris XI,

189
83-Rituximab in mixed cryoglobulinemia: increased experience and perspectives
Salvatore De Vita*, Luca Quartuccio, Martina Fabris Rheumatology Clinic, DPMSC,
University of Udine, Udine, Italy Digestive and Liver Disease, Volume 39, Supplement
1, September 2007, Pages S122-S128
84- Ferri C, Sebastiani M, Giuggioli D, Cazzato M, Longobardo G, Antonelli A, et al.
Mixed cryoglobulinemia: demographic, clinical, and serological features and survival
in 231 patients. Semin Arthritis Rheum 2004; 33:355 74.
85- Alric L, Plaisier E, Thebault S, Peron JM, Rostaing L, Pourrat J, et al. Influence of
antiviral therapy in hepatitis C virus-associated cryoglobulinemic MPGN. Am J Kidney
Dis 2004;43:617 23.
86- Cacoub P, Saadoun D, Limal N, Sene D, Lidove O, Piette JC, et al. PEGylated
interferon alfa-2b and ribavirin treatment in patients with hepatitis C virus-related
systemic vasculitis. Arthritis Rheum 2005; 52:911 5.
87- Mazzaro C, Zorat E Caizzi M, Donada C, Di Gennaro G, Maso LD, et al.
Treatment with peg-interferon alfa-2b and ribavirin of hepatitis C virus-associated
mixed cryoglobulinemia: a pilot study. J Hepatol 2005;42:632 8.
88- Mazzaro C, Franzin E Tulissi R Pussini E, Crovatto M, Carniello GS,et al.
Regression of monoclonal B-cell expansion in patients affected by mixed
cryoglobulinemia responsive to a-interferon therapy. Cancer 1996;77:2604 13.

190
89- Patriarca E Silvestri E Fanin R, Zaja E Sperotto A, Baccarani M. Long-lasting
complete remission of hepatitis C virus (HCV) infection and HCV-associated
immunocytoma with alpha-interferon treatment. Br J Haematol 2001;112:370 2.
91-. Petursdottir, H. Johansson and E. Nordborg et al., The epidemiology of biopsy-
positive giant cell arteritis: special reference to cyclic fluctuations, Rheumatology
(Oxford) 38 (1999), pp. 1208–1212.
92- C. Salvarani, S.E. Gabriel and W.M. O’Fallon et al., The incidence of giant cell
arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern, Ann
Intern Med 123 (1995), pp. 192–194.
94-C. Nordborg, H. Johansson and V. Petursdottir et al., The epidemiology of
biopsy-positive giant cell arteritis: special reference to changes in the age of the
population, Rheumatology (Oxford) 42 (2003), pp. 549–552.
95-S. Kobayashi, T. Yano and Y. Matsumoto et al., Clinical and epidemiologic
analysis of giant cell (temporal) arteritis from a nationwide survey in 1998 in Japan:
the first government-supported nationwide survey, Arthritis Rheum 49 (2003), pp.
594–598.
96-glomérulonéphrites extra capillaires –micro vascularites à ANCA ,ECN ,items
264,134par Thierry Hanndouche, www.nephrohus.org/s/spip.php?article102
97-www.leucemie-espoir.org


















