







Langages
Pages
Légal


N° d’ordre : 897
THESE
présentée devant
L'INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE TOUL OUSE
en vue de l'obtention du
DOCTORAT
Spécialité: Sciences Ecologiques, Vétérinaires, Agronomiques et Bioingénieries
Filière : Ingénieries Microbienne et Enzymatique
par
Stéphane EMOND
Ingénieur INSA Toulouse
EVOLUTION DIRIGEE DE L’AMYLOSACCHARASE PAR LA TECHN IQUE
MUTAGEN TM POUR L’OBTENTION DE VARIANTS THERMOSTABLES
Soutenue le 10 décembre 2007 devant la commission d’examen
Mme Birte Svensson Professeur, Université de Copenhague
Mme Magali Remaud-Siméon Professeur, INSA de Toulouse
Mr Denis Pompon Directeur de recherche, CNRS, CGM de Gif-sur-Yvette
Mr Philippe Mondon Directeur de laboratoire, MilleGen SA, Labège-Innopole
Mr Pierre Monsan Professeur, INSA de Toulouse
Cette thèse a été préparée au laboratoire d'Ingénierie des Systèmes Biologiques et des Procédés (UMR CNRS 5504, UMR INRA 792) de l'INSA de Toulouse et au laboratoire d'Evolution Moléculaire de la société MilleGen basée à Labège-Innopole dans le cadre de l'Ecole Doctorale SEVAB.

La sélection naturelle opère à la manière non d'un ingénieur, mais d'un bricoleur; un bricoleur qui ne
sait pas encore ce qu'il va produire, mais récupère tout ce qui lui tombe sous la main.
François Jacob , le jeu des possibles
Si le savoir peut créer des problèmes, ce n’est pas l’ignorance qui les résoudra.
Isaac Asimov , L’Univers de la Science

Je souhaiterais exprimer par les quelques mots suivants ma reconnaissance aux personnes qui m’ont accompagné aux cours de ces quatre années de thèse et qui, ainsi, ont contribué, de façon directe ou indirecte, à ce travail.
Merci à Pierre Monsan pour l’influence déterminante qu’il a jouée sur mon cursus universitaire et professionnel, pour la confiance qu’il m’a accordée en me proposant ce projet, pour la motivation, le dynamisme et l’enthousiasme (parfois bruyants !) qu’il transmet aux membres de son équipe. Merci à Magali Remaud-Siméon pour tout (et tout, c’est peu dire !) ce qu’elle m’a apporté au cours de ce travail qu’elle a profondément marqué par sa rigueur, sa curiosité et sa créativité (pas que) scientifiques, pour la motivation et le soutien (tant professionnels que psychologiques !) dont j’ai pu bénéficier à ses côtés. Merci à Isabelle André pour sa générosité, sa contribution structurante à ce travail (et aussi à moi-même !) et, en particulier, pour toute l’aide sans limite qu’elle m’a apportée pendant la rédaction du manuscrit et la préparation de la soutenance. Merci à Gabrielle Potocki-Véronèse également pour sa générosité, pour ses nombreux conseils, pour son aide importante lors des relectures et des corrections du manuscrit et pour avoir permis la finalisation de nombreux résultats sur ce projet. Merci à Cécile Albenne et Bart Van der Veen pour leur aide au tout début de cette thèse. Merci à tous les membres de l’EAD1 qui font du labo un endroit où j’ai aimé (et j’aime toujours) venir travailler. C’est difficile de citer tout le monde ici entre les arrivants, les permanents, les partants, ceux qui doivent partir et ne veulent jamais partir etc. Me viennent surtout à l’esprit : Elise, Claire, Emeline, Gilles et Gaëtan (Que de discussions sur ces maudites GS, DSR, ASR, AS et aussi sur tout un tas d’autres choses !) mais aussi David, Florence, Miguel, Romain, Sandra, Sophie Ba., Sophie G., Vincent, Yoann et j’en passe ! Un annuaire ne suffirait pas pour citer et remercier comme il se doit tous les membres de cette équipe ! Un grand merci général aussi à l’ensemble du personnel du LISBP et du GBA.
Cette thèse a été préparée en partie au sein de la société MilleGen dans le cadre d’une convention CIFRE. Je voudrais remercier Hakim Kharrat et Khalil Bouayadi pour m’avoir accueilli au sein de leur entreprise et pour leur confiance tout au long de ce projet. Merci à Philippe Mondon pour m’avoir intégré au sein de son équipe, pour m’avoir initié aux secrets et aux joies de la biomol et pour son flegme à toute épreuve ! Merci aux bio-informaticiens de MilleGen, Fabien Crozet, Laurent Douchy et Line Neil, pour leur disponibilité et pour leur aide cruciale sur l’analyse des résultats de mutagénèse aléatoire. Merci enfin à l’ensemble des personnels de MilleGen avec lesquels j’ai apprécié de travailler au quotidien : Christine, Nathalie, David, Marie-Julie, Olivier, Laurence, Céline, Patrick, Yann, Nadia, Marie, Suzanne, Ouafa etc.
Merci aux stagiaires pour leur grande contribution à ces travaux, pour leur enthousiasme et pour avoir partagé un bout de cette aventure avec moi: Pascale Klopp, Julien Grimoud, Clément Auriol, Sophie Mondeil et Kais Jaziri.
Merci aux personnes qui, avant la thèse, m’ont accueilli comme stagiaire dans leur labo et ont joué un rôle déterminant dans mes choix de formation: Nathalie Viguerie, Bernard Ducommun et Martine Defais.
Un grand merci à Birte Svensson et Denis Pompon pour avoir accepté d’évaluer ce manuscrit, pour l’intérêt qu’ils y ont porté et pour leurs conseils avisés lors de la soutenance..
Merci à mes amis.
Enfin, il n’existe tout simplement pas de mots justes et suffisants pour remercier ma famille, mes sœurs, mes parents et Sandra. Cette thèse leur est dédiée.

4
NOM : EMOND Prénom : Stéphane Titre : Evolution dirigée de l’amylosaccharase par la technique MutaGenTM pour l’obtention de variants
thermostables
Spécialité: Sciences Ecologiques, Vétérinaires, Agronomiques et Bioingénieries, Filière : Ingénieries
Microbienne et Enzymatique
Année : 2007 Lieu : INSA de Toulouse N° d’ordre : 897
RESUME :
L'amylosaccharase de Neisseria polysaccharea (AS) est une transglucosidase qui utilise le saccharose comme
donneurs d'unités glucosyle pour synthétiser un polymère de type amylose et glucosyler efficacement des
accepteurs comme le glycogène. Ce biocatalyseur est un outil industriel prometteur par sa capacité remarquable
à utiliser un substrat abondant et peu coûteux (le saccharose) pour la synthèse de polymères ou de
glucoconjugués d'intérêt. L'AS est cependant une enzyme peu adaptée aux contraintes industrielles du fait de son
faible niveau d'activité (kcat = 1 s-1), sa thermostabilité médiocre (t1/2 (30°C) = 21 h) et sa résistance limitée aux
solvants organiques. Pour améliorer ces propriétés, une approche d'ingénierie combinatoire (ou évolution
dirigée) reposant sur l’utilisation de la technique de mutagénèse aléatoire MutaGenTM brevetée par la société
Millegen a été appliquée à l’AS. Dans un premier temps, trois banques de variants comportant plus de 105 clones
ont été construites par réplication infidèle catalysée par les polymérases mutagènes d’origine humaine, pol β et
pol η. L’analyse des séquences d’un échantillon représentatif montre que la diversité accessible par cette
méthode (taux et types de mutation) dépend de la polymérase employée, ce qui démontre l’intérêt et la souplesse
de cette nouvelle approche. Un protocole d’isolement de variants thermostables ou résistants au DMSO
comprenant une première étape de sélection in vivo sur milieu solide suivi d’un criblage automatisé au format
microplaque a ensuite été optimisé. Le coefficient de variance du procédé a été réduit de 27 à 12.5%, ce qui
permet de minimiser le taux de faux positifs. Appliqué au criblage de plus de 7000 variants actifs, le protocole
développé a permis d’identifier 3 variants présentant une augmentation d’un facteur 3.5 à 10 de la
thermostabilité à 50 °C. Les modèles des variants thermostables ont été construits. Dans le cas du meilleur
variant (R20C/A451T), l’analyse structurale indique que l’augmentation de thermostabilité est liée à la
réorganisation du pont salin impliquant R20 et la création d’une liaison hydrogène médiée par T451. Ce variant
est la seule amylosaccharase active à 50°C, capable de produire des chaînes d’amylose de degré de
polymérisation moyen de 52 avec un rendement 3 fois plus élevé que celui observé pour l’enzyme sauvage à
30°C.
MOTS CLES :
Amylosaccharase, évolution dirigée, mutagénèse aléatoire, ADN polymérases infidèles, criblage à haut-débit,
thermostabilité, solvants organiques, polymère, amylose
Cette thèse a été préparée au laboratoire d'Ingénierie des Systèmes Biologiques et des Procédés (UMR CNRS
5504, UMR INRA 792) de l'INSA de Toulouse et au laboratoire d'Evolution Moléculaire de la société MilleGen
basée à Labège-Innopole.

5
SUMMARY:
Amylosucrase from Neisseria polysaccharea (AS) is a transglucosidase that uses sucrose as
glucosyl unit donor to synthesize an amylose-type polymer and to glucosylate efficiently
acceptor molecules like glycogen. Due to its ability to use sucrose which is a cheap and
readily available substrate, AS is a promising industrial biocatalyst for the synthesis of
amylose polymers or glucoconjugates of interest. However, the development of industrial
processes involving AS is limited by its low catalytic efficiency on sucrose alone (kcat = 1 s-1),
its thermostability (t1/2(50°C) = 3 min) and its weak resistance to organic solvents. A directed
evolution approach using MutaGenTM, a new random mutagenesis method patented by
MilleGen (Labège-Innopole, France), was applied to improve the properties of AS. First,
three AS variant libraries were constructed by replicating the DNA sequence encoding wild-
type AS with mutagenic human polymerases pol β and pol η. The sequence analysis of
randomly chosen clones showed that the error rate and the kind of mutations generated can be
monitored following the polymerases employed. A process enabling the identification of
variants displaying higher thermostability or increased resistance toward DMSO was then
optimized. It consists of two successive steps: (i) an in vivo selection which eliminates
inactive variants followed by (ii) automated screening of active variants to isolate mutants
displaying enhanced features. The coefficient of variance of the screening process was
decreased from 27% to 12.5%, minimizing false positive rate. Around 7,000 active AS
variants were screened for increased thermostability using this procedure. These experiments
led to the identification of three improved variants (two double mutants and one single
mutant) showing 3.5 to 10-fold increased half-lives at 50°C compared to the wild-type
enzyme. In silico comparative analysis between the wild-type AS structure and the modeled
variants allowed to highlight molecular features implicated in thermostability enhancement.
In the case of the most thermostable variant isolated (R20C/A451T), the observed
enhancement could be the result of a reorganization of salt bridges in the vicinity of C20 and
the introduction of a hydrogen bond between Thre451 and Asp488 in AS flexible loop 8. This
double mutant is the most thermostable amylosucrase known to date. When employed at
50°C, it produces amylose chains with a mean degree of polymerization of 52 with a yield up
to 3 times higher than that obtained for such amylose chain synthesis with the wild-type
enzyme at 30°C.

6
Publications
Glucansucrases from family 70 of the Glycoside-Hydrolases : What are the determinants of their
specifities ?
Fabre E., Joucla G., Moulis C., Emond S., Richard G., Potocki-Véronèse G., Monsan P. and Remaud-Siméon
M. (2006). Biocatalysis and Biotransformation, 24(1-2), pp. 137-145.
Optimized and Automated Protocols for High-Throughput Screening of Amylosucrase Libraries.
Emond S., Potocki-Véronèse G., Mondon P., Bouayadi K., Kharrat H., Monsan P. and Remaud-Siméon M.
(2007). Journal of Biomolecular Screening, 12(5), pp. 715-723.
A novel random mutagenesis process using human mutagenic DNA polymerases to generate enzyme
variant libraries.
Emond S., Mondon P., Pizzut-Serin S., Douchy L., Crozet F., Bouayadi K., Kharrat H., Potocki-Véronèse G.,
Monsan P. and Remaud-Siméon M. (2008). Protein Engineering, Design and Selection, 21(4), pp. 267-274.
Combinatorial engineering to enhance amylosucrase thermostability.
Emond S., André I., Jaziri K., Potocki-Véronèse G., Mondon P., Bouayadi K., Kharrat H., Monsan P. and
Remaud-Siméon M. (2008). Protein Science, 17(6):967-76.
Cloning, purification and characterization of a thermostable amylosucrase from Deinococcus
geothermalis.
Emond S., Mondeil S., Jaziri K., Andre I., Monsan P., Remaud-Simeon M., Potocki-Véronèse G. (2008). FEMS
Microbiology Letters, 285(1):25-32.
MutaGenTM : A random mutagenesis method providing a complementary diversity generated by human
error-prone DNA polymerases.
Mondon P., Grand D., Souyris N., Emond S., Bouayadi K. and Kharrat H. (2010). Book chapter in: In Vitro
Mutagenesis Protocols 3rd Edition (Editor: Jeff Braman), Methods in Molecular Biology vol. 634, Humana Press
(Totowa, NJ).

7
Communications orales
Amélioration de la thermostabilité de l’Amylosaccharase par ingénierie combinatoire.
Emond S., André I., Potocki-Véronèse G., Mondon P., Bouayadi K., Kharrat H., Monsan P. and Remaud-
Siméon M. (2008). 22èmes journées du Groupe Français des Glucides, Ax-Les-Thermes, France.
Communications par affiche
Combinatorial engineering of Amylosucrase using the MutaGen technology.
Emond S., Albenne C., Klopp P., Potocki-Véronèse G., Van der Veen B.A., Mondon P., Monsan P. and
Remaud-Simeon M. (2004). 8th European Training Course on Carbohydrates, Wageningen, Pays-Bas.
Rational and combinatorial engineering of Amylosucrase.
Albenne C., Potocki-Véronèse G., Van der Veen B.A., Emond S., Joucla G., Monsan P. and Remaud-Simeon
M. (2004). 2ème Symposium Aliment Santé, Toulouse, France.
Directed evolution of Amylosucrase using the MutaGen technology.
Emond S., Albenne C., Potocki-Véronèse G., Mondon P., Bouayadi K., Monsan P. and Remaud-Simeon M.
(2005). 6th Carbohydrate Bioengineering Meeting, Barcelona, Espagne.
Ingénierie combinatoire de l'Amylosaccharase de Neisseria polysaccharea.
Emond S., Albenne C., Potocki-Véronèse G., Mondon P., Bouayadi K., Monsan P. and Remaud-Siméon M.
(2006). 21èmes journées du Groupe Français des Glucides, Le Croisic, France.
Engineering Amylosucrase by directed evolution using MutaGenTM .
Emond S., Albenne C., Potocki-Véronèse G., Mondon P., Bouayadi K., Monsan P. and Remaud-Siméon M.
(2006). 21ème édition du Club de Bioconversion en Synthèse Organique, Ax-les-thermes, France.
Combinatorial engineering to enhance Amylosucrase thermostability.
Emond S., Potocki-Véronèse G., Mondon P., Bouayadi K., Monsan P. and Remaud-Siméon M. (2007). 7th
Carbohydrate Bioengineering Meeting, Braunschweig, Allemagne.
Modifiying glucansucrase regioselectivity to synthesize new oligosaccharides and glycoconjugates.
Champion E., Emond S., Bertrand A., André I., Morel S., Monsan P. and Remaud-Siméon M. (2007). Benzon
symposium 54 – Glycosylation : opportunities in drug development, Copenhague, Danemark.
Directed evolution to investigate and enhance Amylosucrase thermostability.
Emond S., André I., Jaziri K., Potocki-Véronèse G., Mondon P., Bouayadi K., Kharrat H., Monsan P. and
Remaud-Siméon M. (2007). 3rd Symposium on the alpha-amylase family, Smolenice, Slovaquie.

8
Sommaire

Sommaire
9
INTRODUCTION ......................................................................................................................................................................................... 12 CHAPITRE I : ETUDE BIBLIOGRAPHIQUE................. ......................................................................................................................... 17 PREMIERE PARTIE : LA FIDELITE DES ADN POLYMERASES .. .................................................................................................... 18
1. CLASSIFICATIONS DES ADN POLYMERASES.................................................................................................. 18 2. STRUCTURE GENERALE DES ADN POLYMERASES......................................................................................... 20 3. CONTROLE DE LA FIDELITE DE LA REPLICATION............................................................................................ 21
3.1 Sélection structurale des dNTPs............................................................................................................ 21 3.2. Extension des mésappariements et activité de correction des erreurs ................................................. 22 3.3 Décalage dans l'appariement des brins et génération d’addition ou de déletion.................................. 23
DEUXIEME PARTIE : L'EVOLUTION DIRIGEE DES ENZYMES .. ................................................................................................... 25
1. PRINCIPE ET OBJECTIFS.................................................................................................................................. 25 2. CREATION DE LA DIVERSITE : CONSTRUCTION DES BANQUES DE VARIANTS.................................................. 27
2.1 Banques de variants obtenues par mutagénèse aléatoire...................................................................... 27 2.1.1 Présentation des techniques de mutagénèse aléatoire ...................................................................................... 27 2.1.3 PCR à erreurs ou error-prone PCR .................................................................................................................. 30 2.1.4 MutaGenTM : réplication in vitro par des ADN polymérases à taux d’erreurs élevé ........................................ 31
2.2 Banques de variants obtenues par recombinaison aléatoire ................................................................. 32 2.2.1 Méthodes basées sur la recombinaison homologue ......................................................................................... 32
2.2.1.1 La PCR sexuelle ou "DNA shuffling" ..................................................................................................... 32 2.2.1.2 Les méthodes StEP et RPR...................................................................................................................... 35 2.2.1.3 Les méthodes RACHITT et L-Shuffling ................................................................................................. 37 2.2.1.4 Les méthodes de recombinaison in vivo.................................................................................................. 39
2.2.2 Recombinaison de séquences non homologues ............................................................................................... 40 2.2.2.1. Méthodes générant un point de recombinaison unique : ITCHY et SHIPREC....................................... 40 2.2.2.2. Méthode générant plusieurs points de recombinaison : NRR................................................................. 43
2.3 Banques de variants obtenues par des techniques de mutagénèse ou de recombinaison semi-aléatoires..................................................................................................................................................................... 45
2.3.1 Introduction de mutations ponctuelles à l'aide d’oligonucléotides dégénérés.................................................. 45 2.3.2 Techniques de recombinaison utilisant des oligonucléotides dégénérés.......................................................... 48
3. LE TRI DE LA DIVERSITE : SELECTION ET CRIBLAGE....................................................................................... 49 3.1 Techniques de sélection in vivo ............................................................................................................. 50 3.2 Techniques de sélection in vitro ............................................................................................................ 52
3.2.1 Le phage-display ............................................................................................................................................. 52 3.2.2 Méthodes de sélection par tri cellulaire utilisant la cytométrie en flux............................................................ 54 3.2.3 Le ribosome-display ........................................................................................................................................ 55 3.2.4 Création de compartiments in vitro par des émulsions artificielles ................................................................. 56
3.3 Applications du criblage à haut-débit dans l'évolution dirigée d'enzymes............................................ 58 TROISIEME PARTIE : L'AMYLOSACCHARASE DE NEISSERIA POLYSACCHAREA................................................................... 63
1. ORGANISMES PRODUCTEURS......................................................................................................................... 63 2. STRUCTURE DE L’AMYLOSACCHARASE DE NEISSERIA POLYSACCHAREA......................................................... 65
2.1 Structure tridimensionnelle de l'AS sauvage ......................................................................................... 65 2.2 Mécanisme catalytique de l’amylosaccharase ...................................................................................... 66 2.3 Facteurs structuraux responsables de la spécificité catalytique ........................................................... 69
3. MODE DE FORMATION DU POLYMERE............................................................................................................. 73 3.1. Paramètres cinétiques .......................................................................................................................... 73 3.2 Structure et morphologie du polymère insoluble synthétisé .................................................................. 74
4. LES REACTIONS EN PRESENCE D’ACCEPTEURS EXOGENES............................................................................. 75 4.1 Réactions d'accepteur sur le glucose et le maltose................................................................................ 75 4.2 Réaction d'accepteur sur les polysaccharides ....................................................................................... 76
4.2.1 Caractérisation des produits synthétisés à partir de saccharose et de glycogène.............................................. 76 4.2.2. Comportement cinétique................................................................................................................................. 77
5. APPLICATIONS INDUSTRIELLES DE L'AS ........................................................................................................ 78 6. INGENIERIE DE L'AS DE N. POLYSACCHAREA................................................................................................... 81
6.1 Ingénierie rationnelle ............................................................................................................................ 81 6.2 Ingénierie combinatoire ........................................................................................................................ 82
6.2.1 Génération d'une banque de première génération de l'AS................................................................................ 82 6.2.2 Mise au point de protocoles de sélection et de criblage................................................................................... 82 6.2.3 Premiers variants identifiés par ingénierie combinatoire................................................................................. 84

Sommaire
10
CHAPITRE II : CONSTRUCTION DE BANQUES DE VARIANTS D E L'AMYLOSACCHARASE PAR LA TECHNIQUE MUTAGEN .................................................................................................................................................................................................... 85 A NOVEL RANDOM MUTAGENESIS APPROACH USING HUMAN MUT AGENIC DNA POLYMERASES TO GENERATE ENZYME VARIANT LIBRARIES.............................................................................................................................................................. 86
1. SUMMARY ..................................................................................................................................................... 86 2. INTRODUCTION.............................................................................................................................................. 86 3. RESULTS AND DISCUSSION............................................................................................................................ 88
3.1 Construction of AS variant libraries using pol β and pol η .................................................................. 88 3.2 Mutation rates in AS variant libraries created with pol β and pol η .................................................... 91 3.3 Base substitution spectra ....................................................................................................................... 92 3.4 Insertion and deletion mutations ........................................................................................................... 93 3.5 Mutational impact at the protein level................................................................................................... 94
4. CONCLUSION AND FUTURE PERSPECTIVES..................................................................................................... 97 5. MATERIALS AND METHODS........................................................................................................................... 98
5.1 Bacterial strains and plasmids .............................................................................................................. 98 5.2 DNA manipulations ............................................................................................................................... 98 5.3 Site-directed mutagenesis ...................................................................................................................... 98 5.4 Construction of AS libraries following the MutaGenTM procedure ....................................................... 99 5.5 Construction of an AS variant library by epPCR ................................................................................ 100 5.6 Determination of amylose-producing clone fraction in a library of variants...................................... 100 5.7 Analysis of sequencing data ................................................................................................................ 101
6. SUPPLEMENTARY MATERIAL ....................................................................................................................... 102 6.1 Supplementary method: cloning, production and purification of recombinant human DNA polymerases β and η ...................................................................................................................................................... 102 6.2 Supplementary tables........................................................................................................................... 103
CHAPITRE III : OPTIMISATION DES PROTOCOLES DE SELEC TION ET DE CRIBLAGE DES BANQUES DE VARIANTS DE L'AMYLOSACCHARASE................................................................................................................................................................... 104 OPTIMIZED AND AUTOMATED PROTOCOLS FOR HIGH THROUGHP UT SCREENING OF AMYLOSUCRASE LIBRARIES ................................................................................................................................................................................................. 105
1. SUMMARY ................................................................................................................................................... 105 2. INTRODUCTION............................................................................................................................................ 105 3. RESULTS AND DISCUSSION.......................................................................................................................... 107
3.1 Choice of the recombinant AS expression system................................................................................ 107 3.2 Genetic selection for clones expressing active amylosucrase ............................................................. 108 3.3 Optimization of reproducible and automated screening conditions.................................................... 108
3.3.1 Definition of experimental conditions for normalized cell growth and recombinant AS expression............. 110 3.3.2 Conditions for screening thermostable AS variants....................................................................................... 114 3.3.3 Conditions for screening AS variants in organic cosolvents.......................................................................... 115
3.4 Screening a first generation AS variant library................................................................................... 116 4. CONCLUSION............................................................................................................................................... 118 5. MATERIALS AND METHODS......................................................................................................................... 119
5.1 Bacterial strains and plasmids ............................................................................................................ 119 5.2 DNA manipulations ............................................................................................................................. 119 5.3 Selection of active amylosucrase-expressing clones............................................................................ 119 5.4 Storage of individual active amylosucrase-expressing clones............................................................. 120 5.5 Growth conditions in microplate......................................................................................................... 120 5.6 Enzyme activity determination in microplate ...................................................................................... 120 5.7 Production, purification and thermostability comparison of wild-type and variant AS...................... 121
CHAPITRE IV : RECHERCHE ET CARACTERISATION DE VARIA NTS THERMOSTABLES DE L'AMYLOSACCHARASE....................................................................................................................................................................................................................... 122 COMBINATORIAL ENGINEERING TO ENHANCE AMYLOSUCRASE T HERMOSTABILITY................................................. 123
1. SUMMARY ................................................................................................................................................... 123 2. INTRODUCTION............................................................................................................................................ 123 3. RESULTS AND DISCUSSION.......................................................................................................................... 125
3.1 Isolation of thermostable amylosucrase variants................................................................................ 125 3.2 Thermal resistance of wild-type and variant amylosucrases............................................................... 127 3.3 Structural basis of thermostabilization mechanisms ........................................................................... 128 3.4 Amylose synthesis by thermostable amylosucrase variants................................................................. 132
4. CONCLUSION............................................................................................................................................... 134

Sommaire
11
5. MATERIALS AND METHODS......................................................................................................................... 136 5.1 Bacterial strains and plasmids ............................................................................................................ 136 5.2 DNA manipulations ............................................................................................................................. 136 5.3 Construction, selection and screening of AS random mutagenesis libraries....................................... 136 5.4 Site-directed mutagenesis .................................................................................................................... 137 5.5 Production, purification and activity assay of wild-type and variant AS ............................................ 138 5.6 Thermostability of wild-type and mutant amylosucrase ...................................................................... 138 5.7 Activity-temperature profile of wild-type and mutant amylosucrases ................................................. 139 5.8 In vitro synthesis of amylose and oligosaccharides............................................................................. 139 5.9 Carbohydrate analysis......................................................................................................................... 139 5.10 Iodine Binding Properties ................................................................................................................. 140 5.11 Computational modeling of AS variants............................................................................................ 140
CONCLUSION ............................................................................................................................................................................................ 142 REFERENCES BIBLIOGRAPHIQUES................................................................................................................................................... 151 ANNEXES.................................................................................................................................................................................................... 174
ANNEXE 1 : SEQUENCE NUCLEOTIDIQUE ET PROTEIQUE DE L’ASNP............................................................ 175 ANNEXE 2 : CARACTERISATION DE L’AMYLOSACCHARASE DE DEINOCOCCUS GEOTHERMALIS. .................. 177
TABLES DES ILLUSTRATIONS ............................................................................................................................................................. 186

12
Introduction

Introduction
13
Dans nos sociétés, la prise de conscience et les inquiétudes soulevées par les déchets
industriels et leurs effets sur l'environnement soulignent la nécessité de mettre au point des
procédés industriels propres et adaptés au développement durable. Ce contexte stimule
indéniablement la recherche en biocatalyse. En effet, qu'elle repose sur l'emploi de
microorganismes ou d'enzymes libres ou immobilisées, la biocatalyse industrielle peut
apporter des solutions efficaces et originales pour la production de molécules d'intérêt dans
les secteurs de l'agroalimentaire, de la chimie fine, de la santé, de l'environnement et des
bioénergies. Pourtant, le nombre et la diversité des applications mettant en œuvre des
enzymes restent modestes (Schoemaker et al., 2003). Bien que de nombreuses enzymes
naturelles possèdent des activités intéressantes sur le plan applicatif, elles ne sont souvent pas
adaptées aux conditions rencontrées dans les procédés industriels. Les paramètres limitants
sont en effet nombreux : niveaux de production insuffisants de l’enzyme, reconnaissance
médiocre (voire absence de reconnaissance) des substrats non naturels, faible résistance à la
température ou aux solvants organiques etc. Dans ce contexte, l’un des objectifs de
l’ingénierie enzymatique est de résoudre ces problèmes en modifiant les propriétés des
enzymes naturelles ou en créant de nouveaux biocatalyseurs (Bolon et al., 2002).
Parmi les méthodologies utilisées en ingénierie enzymatique, l’évolution dirigée vise à
modifier les caractéristiques d’une (ou de plusieurs) protéine(s) d’intérêt via la mise en
application des principes de l’évolution naturelle que sont la variation génétique et la
sélection phénotypique. Cette méthodologie expérimentale se distingue cependant de
l’évolution naturelle dans la mesure où son objectif est défini à l’avance et les moyens d’y
parvenir sont contrôlés par l’expérimentateur. La première étape d’une expérience d’évolution
dirigée consiste à générer de larges banques de variants de la protéine d’intérêt en utilisant des
techniques de mutagénèse et/ recombinaison aléatoires. Dans un second temps, ces banques
sont soumises à des tests de sélection et/ou de criblage permettant d’identifier les mutants qui
présentent un gain pour la propriété à améliorer. Plusieurs cycles combinant ces deux étapes
peuvent être enchaînés afin d’accumuler les mutations avantageuses jusqu’à ce que le niveau
de performance choisi par l’expérimentateur soit atteint. Cette approche, qui consiste à
explorer l'espace des variants de la (ou des) protéine(s) parentale(s), permet d'identifier des
mutants inédits possédant les caractéristiques désirées.
Les premiers exemples d’évolution de biomolécules dirigée (ou contrôlée) en laboratoire
datent de la fin des années 1960 avec les travaux du groupe de Spiegelman (Levisohn and
Spiegelman, 1969). C’est au cours des deux dernières décennies, avec le développement des

Introduction
14
outils du génie génétique et des technologies de sélection ou de criblage à haut-débit, que
l’évolution dirigée s’est imposée comme un outil expérimental efficace pour l’étude des
relations structure-activité des enzymes (et plus généralement des protéines) et leur
remodelage à des fins applicatives (Cherry and Fidantsef, 2003). Plusieurs techniques de
mutagénèse ou de recombinaison aléatoires ont été développées, parmi lesquelles la PCR à
erreurs (Cadwell and Joyce, 1992), le DNA shuffling (Stemmer, 1994a, b) et la méthode StEP
(Zhao et al., 1998) permettant la recombinaison de gènes parentaux mutés, etc. Ces
techniques sont, pour la plupart, brevetées. Leur efficacité a été démontrée pour la
construction d’enzymes plus efficaces, thermostables ou de spécificité modulée (Cherry and
Fidantsef, 2003; Yuan et al., 2005). Dans ce contexte, la méthode MutaGenTM brevetée par la
société française MilleGen (Bouayadi et al., 2002) apparaît comme un procédé alternatif
prometteur. Cette technique de mutagénèse aléatoire repose en effet sur l’utilisation in vitro
d’ADN polymérases possédant un taux d’erreur de réplication élevé (supérieur à 10-3, c'est-à-
dire plus d’une base erronée pour 1000 bases répliquées). En exploitant les spécificités de
différentes ADN polymérases de manière complémentaire, la technique MutaGenTM permet
une exploration large de l’espace des variants disponibles à partir à partir d’une séquence
parentale. Cette méthode a été récemment utilisée afin de générer des banques de fragments
variables simple chaîne d'anticorps ou d'augmenter la diversité initiale d'une banque native de
domaines variables (Mondon et al., 2007a; Mondon et al., 2007b). L'application de cette
technique à l'ingénierie d’une enzyme d’intérêt, l’amylosaccharase, a fait l'objet du présent
travail de thèse dans le cadre d’une convention CIFRE entre la société MilleGen et le groupe
de Catalyse et Ingénierie Moléculaire EnzymatiqueS (CIMES) de l’INSA de Toulouse.
L’amylosaccharase est une transglucosidase (E.C. 2.4.1.4.) capable de catalyser, à partir de
saccharose, la synthèse d’un homopolymère de type amylose composé d’unités gluycosyle
reliées par des liaisons α-1,4. Cette réaction de polymérisation s’accompagne de la libération
du fructose. L’amylosaccharase apparaît comme une enzyme remarquable du fait de sa
position à l’interface de plusieurs familles d’enzymes. En effet, elle appartient à la famille 13
des glycoside-hydrolases (GH13, aussi appelée famille des α-amylases), dans laquelle elle est
la seule enzyme dotée d’une activité polymérase. En outre, sa spécificité de synthèse des
liaisons α-1,4 s’apparente à celle des enzymes d’élongation de l’amidon et du glycogène.
L’amylosaccharase se distingue néanmoins de ces glycosyltransférases par sa capacité à

Introduction
15
utiliser le saccharose et non un sucre activé de type ADP ou UDP-glucose comme donneur
d’unités glucosyle.
Découverte en 1946 chez Neisseria perflava (Hehre and Hamilton, 1946), l’amylosaccharase
a été peu étudiée jusqu’au milieu des années 1990. De plus, elle a longtemps été décrite
comme une enzyme produite exclusivement par des bactéries du genre Neisseria. Cependant,
le développement des programmes de séquençage des génomes bactériens a permis de révéler
la présence de gènes codant des amylosaccharases putatives chez d’autres genres bactériens,
renforçant l‘importance physiologique de cette enzyme. L’intérêt porté à cette enzyme s’est
accru au cours des dix dernières années. D’une part, la capacité remarquable de
l’amylosaccharase à utiliser un substrat abondant, renouvelable et peu coûteux a motivé les
recherches en vue de son utilisation industrielle. En effet, l’amylosaccharase permet de
produire un polymère de type amylose qui présente des propriétés variées, notamment
mécaniques (rôle texturant) ou nutritionnelles (fibre alimentaire, prébiotique). Elle peut
également être utilisée en tant qu’outil de glucosylation pour former des glycoconjugués
possédant de nouvelles propriétés. D’autre part, l’amylosaccharase partage des caractères
structuraux et/ou fonctionnels avec les enzymes de la famille des α–amylases, les glucane-
saccharases et les enzymes d’élongation de l’amidon et du glycogène. La compréhension de
son mode d’action a constitué une avancée majeure dans le cadre de la connaissance et de la
maîtrise des enzymes agissant sur les sucres (Albenne et al., 2004).
L’étude de l’amylosaccharase de Neisseria polysaccharea (AS) a été entreprise depuis 1995
au sein de l’équipe CIMES. Ces travaux ont consisté dans un premier temps au clonage de
son gène, à l’analyse de sa séquence, à la surexpression de l’enzyme chez E. coli, à sa
purification et sa caractérisation cinétique (Potocki de Montalk et al., 2000a; Potocki de
Montalk et al., 1999; Potocki de Montalk et al., 2000b). La résolution de sa structure
tridimensionnelle en 2001 (Skov et al., 2001) a permis d’avancer dans la compréhension des
relations structure-activité de l’AS et d’entreprendre des travaux d’ingénierie rationnelle
visant à modifier son activité et sa spécificité (Albenne et al., 2002a; Albenne et al., 2004;
Albenne et al., 2007). Cependant, bien que l’AS possède des propriétés intéressantes sur le
plan technologique, sa mise en œuvre dans un procédé industriel nécessite une amélioration
de son faible niveau d'activité catalytique (kcat = 1 s-1), de sa thermostabilité (t1/2 = 21 heures à
30°C) et de sa résistance à la dénaturation par les solvants organiques. Des travaux
d’évolution dirigée ont donc été entrepris en complément de l’approche rationnelle, afin de
construire des variants plus adaptés aux contraintes industrielles (van der Veen et al., 2004;
van der Veen et al., 2006). Ces travaux ont consisté au développement de méthodes de

Introduction
16
sélection et de criblage dédiées à l’AS et ont conduit à l’identification de variants plus actifs
de l’AS à partir d’un échantillon de 150 mutants actifs provenant d’une banque de première
génération. Cependant, les protocoles de criblage développés initialement ne permettaient pas
le criblage fiable de banques (ou de sous-banques) de taille plus importante. Leur
inconvénient principal était en particulier lié au faible niveau de production des variants
obtenus à partir du vecteur d’expression utilisé pour le clonage des banques.
Les travaux exposés dans le présent rapport de thèse ont consisté dans un premier temps à
appliquer la technique MutaGenTM au gène codant l’amylosaccharase de N. polysaccharea et
à analyser la diversité générée avec cette méthode. Dans un second temps, nous avons
optimisé le procédé de criblage des banques de variants, afin de pouvoir analyser un plus
grand nombre de clones et d’identifier de manière fiable des mutants plus thermostables ou
plus résistants à la dénaturation par les solvants organiques. Enfin, nous avons entrepris
l’analyse des variants thermostables de l’AS isolés à partir des banques générées via la
technique MutaGenTM.
Le premier chapitre de ce manuscrit consiste en un exposé des éléments bibliographiques
permettant de situer la problématique de nos travaux. La première partie de ce chapitre sera
consacrée à une présentation succincte des ADN polymérases et de leurs caractéristiques, afin
de montrer l’intérêt que présente leur emploi dans un protocole de mutagénèse aléatoire. Une
seconde partie présentera les principales techniques de construction de banques, de sélection
et de criblage développées dans le domaine de l’évolution dirigée. Enfin, la troisième partie
de cette étude bibliographique sera consacrée à la description des connaissances et des travaux
concernant l’AS de N. polysaccharea.
Nous avons ensuite choisi de présenter les résultats obtenus au cours de cette thèse sous la
forme de trois publications rédigées en anglais, dans un esprit de concision et afin d’en
faciliter la divulgation. Le premier de ces chapitres décrit la construction et l’analyse de
banques de variants de l’AS générés par la technique MutaGenTM en utilisant deux ADN
polymérases humaines, pol β et pol η. Le second chapitre est consacré à l’optimisation du
procédé de criblage permettant d’identifier des variants de l’AS plus thermostables ou
résistants aux solvants organiques à partir des banques générées. Enfin, le troisième chapitre
décrit l’analyse des variants thermostables de l’AS isolés dans les banques générées par la
technique MutaGenTM et leur emploi à des températures réactionnelles non tolérées par
l’enzyme sauvage.

17
Chapitre I : Etude bibliographique

Chapitre I : Etude bibliographique
18
Première partie : la fidélité des ADN polymérases
La méthode de mutagénèse aléatoire que nous avons employée au cours de nos travaux repose
sur l'utilisation du caractère infidèle des ADN polymérases humaines β et η classées dans les
familles X et Y. Il nous a donc paru important de consacrer la première partie de cette étude
bibliographique à une présentation de cette famille d'enzymes et des mécanismes influençant
leur fidélité au cours de la réplication in vitro d'ADN. Notre objectif n'est donc pas de réaliser
un exposé exhaustif sur cette thématique qui a été l'objet d'intenses recherches depuis la
découverte de la première ADN polymérase chez E. coli dans les années 1950 (Kornberg,
1957). Il s'agit plutôt de présenter des données qui montrent l'intérêt d'élargir l'utilisation de
ces enzymes comme outils de mutagénèse aléatoire.
1. CLASSIFICATIONS DES ADN POLYMERASES
Le grand nombre d’ADN polymérases connues aujourd’hui illustre la complexité des
mécanismes cellulaires de réplication et de réparation du génome dans lesquels elles jouent un
rôle central. Cinq ADN polymérases sont dénombrées chez E. coli (nommées Ec Pol et
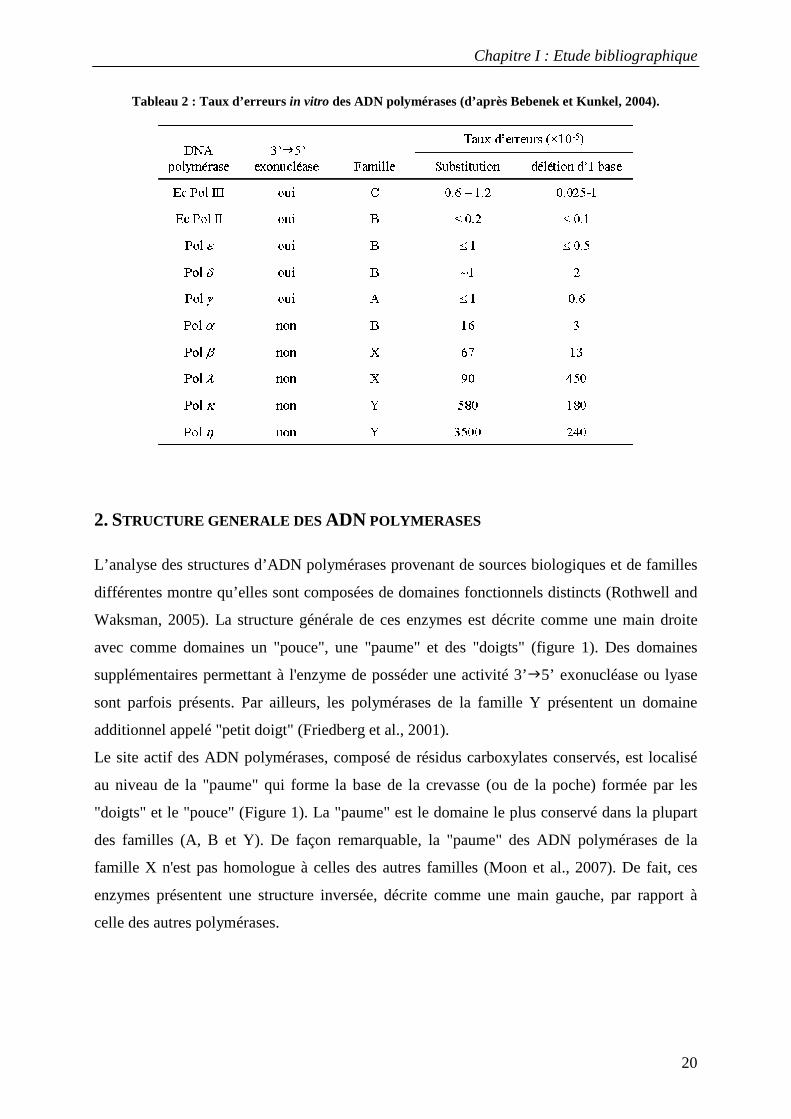
numérotées de I à V), neuf chez S. cerevisiae et seize chez l’homme (tableau 1) (Bebenek and
Kunkel, 2004). Elles sont regroupées par famille phylogénétique selon la proximité de leur
séquence primaire (Rothwell and Waksman, 2005). La famille A comprend l’ADN
polymérase I de E. coli, la Taq polymérase et les polymérases eucaryotes Pol γ, Pol θ et Pol
ν . La famille B regroupe l’ADN polymérase II de E. coli, les polymérases réplicatives
eucaryotes Pol α, Pol δ et Pol ε ainsi que Pol ζ. Les ADN polymérases classées dans la
famille C sont des enzymes bactériennes (ADN pol III de E.coli). La famille D n’a de
représentants que chez les archaebactéries. La famille X est formée par les polymérases
eucaryotes Pol β, Pol λ, Pol σ et Pol µ, ainsi que la Terminal Transférase (TdT). La famille Y
(Ohmori et al., 2001) regroupe des ADN polymérases spécialisées dans la réplication trans-
lésionnelle d’ADN endommagé (Kunz et al., 2000) : Pol IV et Pol V de E. coli et les
polymérases eucaryotes Pol η, Pol ι et Pol κ (Kunkel et al., 2003).
Les ADN polymérases peuvent également être différenciées selon leur taux d’erreurs, c’est-à-
dire leur propension à introduire un nucléotide erroné au cours de la réplication d’un ADN
non endommagé (tableau 2) (Kunkel, 2004). Les enzymes qui présentent les taux d’erreurs les
plus faibles sont les polymérases réplicatives appartenant aux familles A, B et C. Elles
Chapitre I : Etude bibliographique
19
catalysent la réplication in vitro d'ADN selon un mode processif et possèdent souvent une
activité 3’�5’ exonucléase (fonction de correction des erreurs ou proofreading). Les ADN
polymérases les moins fidèles sont les enzymes appartenant aux familles X et Y, intervenant
in vivo dans les mécanismes de réparation de l’ADN ou de réplication trans-lésionnelle. Elles
catalysent la réplication in vitro d'ADN non endommagé selon un mode distributif et sont
dénuées d’activité 3’�5’ exonucléase. Les erreurs générées par ces ADN polymérases
infidèles sont de tout type (substitution, insertion et délétion).
Tableau 1 : Les ADN polymérases identifiées chez Escherichia coli, chez Saccharomyces cerevisiae et chez
l’Homme (d’après Bebenek et Kunkel, 2004).
Chapitre I : Etude bibliographique
20
Tableau 2 : Taux d’erreurs in vitro des ADN polymérases (d’après Bebenek et Kunkel, 2004).
2. STRUCTURE GENERALE DES ADN POLYMERASES
L’analyse des structures d’ADN polymérases provenant de sources biologiques et de familles
différentes montre qu’elles sont composées de domaines fonctionnels distincts (Rothwell and
Waksman, 2005). La structure générale de ces enzymes est décrite comme une main droite
avec comme domaines un "pouce", une "paume" et des "doigts" (figure 1). Des domaines
supplémentaires permettant à l'enzyme de posséder une activité 3’�5’ exonucléase ou lyase
sont parfois présents. Par ailleurs, les polymérases de la famille Y présentent un domaine
additionnel appelé "petit doigt" (Friedberg et al., 2001).
Le site actif des ADN polymérases, composé de résidus carboxylates conservés, est localisé
au niveau de la "paume" qui forme la base de la crevasse (ou de la poche) formée par les
"doigts" et le "pouce" (Figure 1). La "paume" est le domaine le plus conservé dans la plupart
des familles (A, B et Y). De façon remarquable, la "paume" des ADN polymérases de la
famille X n'est pas homologue à celles des autres familles (Moon et al., 2007). De fait, ces
enzymes présentent une structure inversée, décrite comme une main gauche, par rapport à
celle des autres polymérases.

Chapitre I : Etude bibliographique
21
Figure 1 : Structure de l'ADN polymérase du phage T7 (d’après Beard et Wilson, 2003). Les ions Mg2+
(sphères magentas), essentiels à la catalyse, se fixent au niveau du site actif localisé dans la "paume". Le "pouce"
et les "doigts" interagissent respectivement avec l'ADN (les squelettes de l'amorce et de la matrice sont colorées
respectivement en rouge et rose) et la paire de bases en cours de formation (le dNTP entrant est coloré en rouge).
3. CONTROLE DE LA FIDELITE DE LA REPLICATION
3.1 Sélection structurale des dNTPs
La réaction catalysée par les ADN polymérases est le transfert du groupe nucléotidyle d'un
dNTP à l'hydroxyle 3' du résidu nucléotidyle terminal du brin d'ADN en cours d'élongation.
L'incorporation du nucléotide dépend de la présence de deux ions bivalents (Mg2+). Un
mécanisme général d'insertion des dNTPs au cours de la synthèse a été développé à partir de
l'analyse de la structure de différentes ADN polymérases (Rothwell and Waksman, 2005). Les
complexes binaires ADN polymérase/matrice présentent une conformation "ouverte" qui
semble se modifier lors de l'entrée d'un dNTP dans la crevasse. Le "pouce" (ou les "doigts"
chez les polymérases de la famille X) favorise l'interaction de l'enzyme avec l'ADN en cours
d'élongation et permet un positionnement correct de la matrice. Les "doigts" (ou le "pouce"
pour la famille X) stabilisent le dNTP entrant en se refermant sur la paire de bases néo-
formée. La structure de complexes ternaires ADN polymérase/matrice/dNTP possède une
conformation dite "fermée" mettant en évidence des contraintes stériques qui favorisent le
positionnement de dNTPs respectant les appariements Watson-Crick. La topologie du site

Chapitre I : Etude bibliographique
22
actif des ADN polymérases fidèles permet donc de sélectionner les dNTPs en fonction de leur
taille et de leur forme.
Récemment, la caractérisation de structures d'ADN polymérases de la famille Y (Ling et al.,
2001; Silvian et al., 2001; Trincao et al., 2001; Zhou et al., 2001) a mis en évidence un site
actif ouvert et accessible à des substrats volumineux (par exemple : une matrice endommagée
ou deux bases matrices intactes). L'analyse de complexes ternaires ADN/matrice/dNTP
montre également que ces polymérases ne subissent pas les modifications conformationelles
induites par l'entrée du dNTP. Par opposition avec les polymérases réplicatives fidèles, les
enzymes de la famille Y possèdent une structure capable de tolérer l'ADN matrice
endommagé et susceptible d'incorporer plus fréquemment des nucléotides erronés.
3.2. Extension des mésappariements et activité de correction des erreurs
Les ADN polymérases catalysent généralement moins efficacement l'extension à partir d'un
mésappariement qu'à partir d'une amorce correctement appariée. Cette réaction varie
cependant en fonction de la nature du mésappariement et des ADN polymérases. D'une part,
la synthèse à partir de mésappariements purine-pyrimidine est plus fréquemment observée
qu'à partir de mésappariements purine-purine ou pyrimidine-pyrimidine qui sont
structuralement moins adaptés au site actif des ADN polymérases (Goodman et al., 1993).
D'autre part, certaines polymérases comme Pol ζ et Pol κ, dont le rôle supposé est d'effectuer
la synthèse à partir d'une base insérée an face d'une lésion (Prakash and Prakash, 2002),
catalysent plus fréquemment l'incorporation d'un dNTP après un mésappariement que les
polymérases réplicatives (Pol δ, Pol γ et Pol ε). Pour ces dernières, la réplication à partir d'un
mésappariement est 10000 fois moins efficace qu'à partir d'une paire correctement appariée
(Goodman et al., 1993).
Les ADN polymérases réplicatives possèdent un domaine 3'�5' exonucléase (tableau 2),
localisée à proximité de la crevasse catalytique où se logent la matrice et le brin en cours
d'élongation (figure 1). Au cours de la réplication d'ADN, ces enzymes balancent
constamment entre leurs activités polymérase et exonucléase, l'équilibre entre ces deux
activités dépendant de leur compétition pour stabiliser l'extrémité 3'OH de l'amorce. L'activité
3'�5' exonucléase permet un mécanisme de correction des erreurs qui accroît
considérablement la fidélité des ADN polymérases réplicatives (Goodman et al., 1993). En
effet, la formation d'un mésappariement entraîne un blocage de la synthèse au cours duquel
l'activité exonucléase peut cliver le nucléotide erroné. La réplication peut alors reprendre à
partir d'une amorce correctement appariée. Ce mécanisme de correction peut être perturbé en

Chapitre I : Etude bibliographique
23
augmentant la concentration des dNTPs pour favoriser l'activité de polymérisation au
détriment de l'activité exonucléase (Goodman et al., 1993; Kunkel and Bebenek, 2000).
3.3 Décalage dans l'appariement des brins et génération d’addition ou de déletion
Parmi les erreurs de réplication, les mutations de type délétion ou addition résultent d'un
décalage dans l'appariement entre le brin matrice et le brin amorce (Streisinger et al., 1966).
Le taux d'additions et de délétions est très variable en fonction des ADN polymérases et du
contexte de séquences (Bebenek and Kunkel, 2000). Certaines ADN polymérases (comme Pol
λ ; tableau 2) possèdent des taux de délétion d'une base plus élevés que le taux de substitution
(Bebenek et al., 2003). Les mutations de type addition et délétion sont fréquemment
observées dans les séquences répétées. Cette observation a conduit à formuler l'hypothèse
classique du glissement de brin (figure 2) (Streisinger et al., 1966). Dans ce cas, la (ou les)
base(s) non appariée(s) peut (ou peuvent) être localisée(s) relativement loin du site actif de la
polymérase dans la mesure où le décalage entre le brin matrice et le brin amorce est stabilisé
par des appariements Watson-Crick corrects dont le nombre dépend de la taille de la séquence
répétée. Les délétions sont dues à la présence d'une ou plusieurs bases non appariées dans le
brin matrice. A l'inverse, les additions surviennent en raison de bases non appariées dans le
brin amorce en cours d'élongation.
Figure 2 : Génération d'additions et de délétions. Ce type de mutation survient lors du décalage d'un des brins
impliqués dans la synthèse. Le glissement du brin en cours d'élongation génère une addition. Le décalage du brin
matrice entraîne une délétion.

Chapitre I : Etude bibliographique
24
Ces décalages entre les brins peuvent apparaître au cours de translocations incorrectes de
l'ADN polymérase ou au cours de cycles dissociation/association de l'enzyme avec le
complexe amorce/matrice (Bebenek and Kunkel, 2000). Une délétion peut également se
produire lorsqu’une base incorrecte est insérée, ce qui peut conduire à un décalage de
l'amorce qui convertit ce mésappariement en appariement (avec une base non appariée en 5').
Les additions et les délétions peuvent également être provoquées par un défaut d'alignement
dans le site actif des ADN polymérases, en particulier pour les membres de la famille Y qui
possèdent un site actif très ouvert capable d’accueillir deux bases du brin matrice en même
temps (Ling et al., 2001). Dans ce cas, le dNTP entrant forme un appariement Watson-Crick
conforme mais pas avec la base matrice correcte.
De nombreux mécanismes sont donc susceptibles d'être à l'origine de la survenue de délétion
et/ou d'addition. Les mécanismes conduisant à la délétion ou l'addition d'une base sont
relativement bien documentés à l'heure actuelle (Garcia-Diaz and Kunkel, 2006). En
revanche, la genèse de délétions ou d'additions complexes, qui impliquent deux bases ou plus,
est encore peu étudiée et reste mal comprise.

Chapitre I : Etude bibliographique
25
Deuxième partie : l'évolution dirigée des enzymes
1. PRINCIPE ET OBJECTIFS
L’ingénierie combinatoire ou évolution dirigée est une approche expérimentale qui vise à
modifier les propriétés des protéines par la mise en pratique des principes de l’évolution
naturelle que sont la variation génétique et la sélection phénotypique. Elle consiste en deux
étapes schématisées sur la figure 3. Dans un premier temps, des banques de variants sont
construites par mutagénèse et/ou recombinaison aléatoires à partir de la (ou les) séquence(s)
ADN codant la (ou les) protéine(s) d'intérêt. Dans un second temps, ces banques sont
soumises à des protocoles de sélection et/ou de criblage de manière à isoler les mutants qui
présentent un gain pour la propriété ciblée. En utilisant les mutants améliorés pour la
construction de nouvelles banques, ce schéma expérimental peut être reproduit de façon
itérative jusqu’à ce que le niveau de performance choisi par l’expérimentateur soit atteint.
L'amélioration des propriétés d'une protéine par évolution dirigée consiste donc à explorer
l'espace des séquences possibles à partir de cette protéine. Ce dernier est théoriquement très
grand. Une protéine de N résidus est comprise dans un espace de 20N séquences. Le nombre
de variants qu'il est possible de générer en substituant simultanément M acides aminés de
Figure 3 : Principe de l’évolution dirigée des protéines.

Chapitre I : Etude bibliographique
26
cette séquence est de 19M × [ N! / [(N-M)! × M!] ]. A titre d'exemple, le nombre de mutants
substitués pour 5 résidus dans une protéine de 100 acides aminés est d'environ 2×1014. En
tenant compte de la dégénérescence du code génétique, il faudrait construire une banque dix
fois plus large (soit 1015 dans notre exemple) pour être assuré de sélectionner (ou de cribler)
l'ensemble des ces mutants. Ceci est en pratique impossible puisque les techniques de
sélection in vitro les plus performantes à l'heure actuelle permettent d'analyser au maximum
1012 variants (voir le paragraphe "Le tri de la diversité : sélection et criblage"). Le fait de
répéter les cycles d'évolution dirigée (construction de banques suivie de sélection et/ou
criblage) permet de contourner cette difficulté si on assume que les mutations bénéfiques
recherchées sont additives. Cette stratégie permet donc théoriquement de restreindre l'espace
des variants à explorer à chaque tour. En reprenant l'exemple de la protéine de 100 acides
aminés, le nombre de variants possibles à partir d'une substitution d'un résidu n'est plus que
d'approximativement 2000. En outre, la recherche du mutant le mieux adapté peut être
accélérée par l'emploi de techniques permettant de recombiner les variants positifs identifiés à
chaque cycle (voir le paragraphe "Banques de variants obtenus par recombinaison aléatoire").
Au cours des vingt dernières années, l'évolution dirigée s'est imposée comme une
méthodologie extrêmement efficace pour modifier les propriétés des enzymes : activité et
efficacité catalytiques, stabilité, activité en milieu non naturel, spécificité etc. Cette approche
permet notamment d'isoler des variants adaptés à des environnements réactionnels proches de
ceux des procédés industriels. A la différence des méthodes d'ingénierie rationnelle,
l'évolution dirigée ne requiert pas de connaissances préalables des relations structure-activité
de l'enzyme à modifier. Elle nécessite cependant de disposer du gène codant la protéine
d'intérêt cloné dans un vecteur d'expression recombinant.
Depuis les années 1980, le développement des outils du génie génétique et des technologies
de criblage à haut-débit a accompagné l'utilisation croissante de l'évolution dirigée dans les
laboratoires travaillant dans le domaine de l'ingénierie des protéines. Les méthodes de
création des banques sont relativement génériques dans la mesure où elles reposent sur la
manipulation des séquences ADN codant les enzymes d’intérêt. En revanche, les méthodes de
sélection ou de criblage destinées à l’identification des mutants améliorés nécessitent une
mise au point adaptée à chaque enzyme et à la propriété que l'expérimentateur a choisi
d'améliorer.

Chapitre I : Etude bibliographique
27
Nous commencerons dans un premier temps par décrire les différentes techniques dédiées à la
génération des banques de variants. La seconde partie de ce chapitre sera consacrée à la
description des méthodes développées pour l’isolement des variants d’intérêt.
2. CREATION DE LA DIVERSITE : CONSTRUCTION DES BANQUES DE VARIANTS
2.1 Banques de variants obtenues par mutagénèse aléatoire
2.1.1 Présentation des techniques de mutagénèse aléatoire
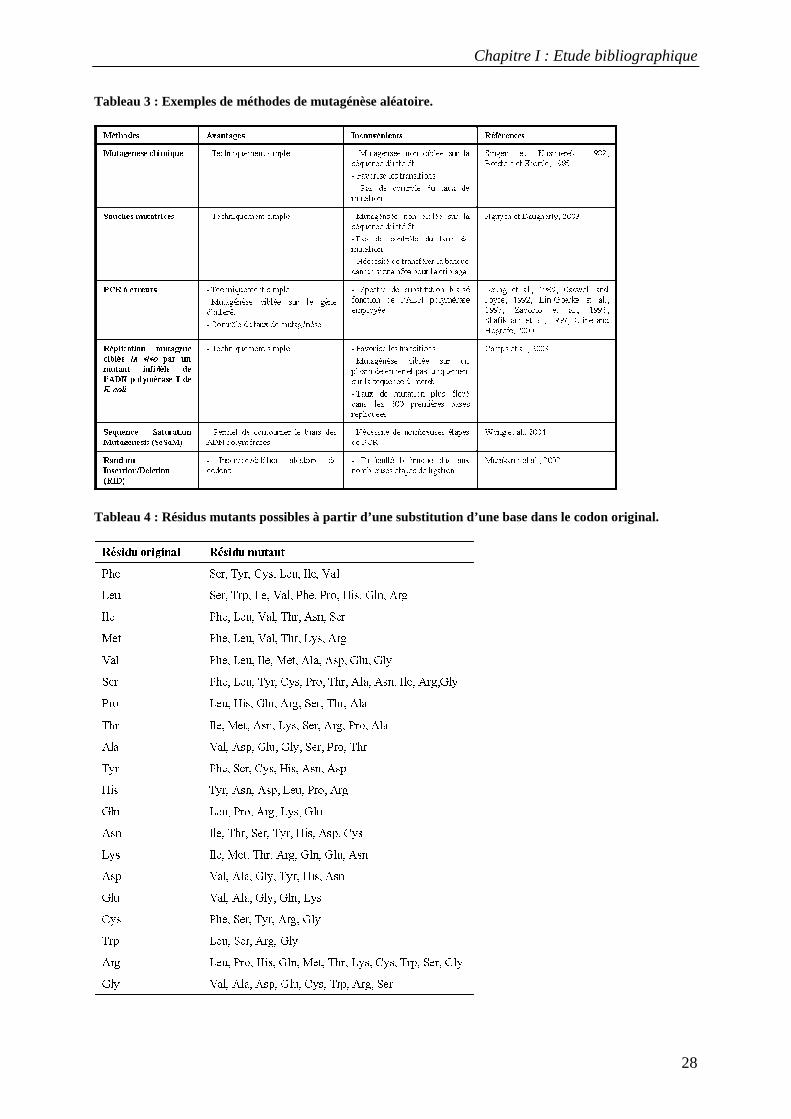
De nombreuses méthodes de mutagénèse aléatoire in vitro ou in vivo ont été développées et
employées dans le cadre de l'évolution dirigée des protéines (Tableau 3) (Neylon, 2004;
Wong et al., 2006). Elles permettent de générer tout type de mutation (substitution, insertion
et délétion). Leur efficacité a été démontrée dans de nombreux exemples d'évolution dirigée
de protéines. Cependant, la diversité générée par ces méthodes est biaisée dans la mesure où
les mutations au niveau nucléotidique et protéique ne sont pas équiprobables. La première
source de biais est liée à la nature du code génétique. La probabilité d'introduire par
mutagénèse aléatoire plus d'une mutation au sein d'un même codon est quasi nulle. Un acide
aminé donné ne peut donc être muté qu'en un nombre limité d'autres résidus (Tableau 4). Par
exemple, la substitution simple d’une base dans un codon valine permettra seulement de
générer des résidus phenylalanine, leucine, isoleucine, alanine, aspartate or glycine. Une
seconde source de biais est liée à la structure de l’ADN et son influence sur la représentation
des différents types de substitutions possibles. Les transitions (purine � purine ou pyrimidine
� pyrimidine) sont effet plus fréquemment observées que les transversions (purine �
pyrimidine ou inversement). La topologie du site actif des ADN polymérases est mieux
adaptée aux mésappariements purine-pyrimidine (G:T et A:C, qui génèrent des transitions)
qu'aux mésappariements purine-purine (G:G, A:A et G:A) et pyrimidine-pyrimidine (T:T,
C:C et T:C). Le déséquilibre entre transitions et transversions est plus ou moins marqué en
fonction de l'ADN polymérase utilisée et les conditions de sa mise en œuvre.
Chapitre I : Etude bibliographique
28
Tableau 3 : Exemples de méthodes de mutagénèse aléatoire.
Tableau 4 : Résidus mutants possibles à partir d’une substitution d’une base dans le codon original.

Chapitre I : Etude bibliographique
29
Tableau 5 : Exemples et modes d'action d’agents mutagènes chimiques et physiques.
Les premières méthodes de mutagénèse aléatoire reposaient sur l'emploi d'agents
génotoxiques chimiques ou physiques (Tableau 5) (Botstein and Shortle, 1985; Singer and
Kusmierek, 1982). Dans ces approches, les agents mutagènes utilisés induisent dans un
premier temps des lésions dans la structure de l’ADN. Les mutations sont susceptibles de
survenir au moment de la réparation de l'ADN endommagé après son introduction dans la
cellule hôte (généralement E. coli). L’inconvénient principal de ces méthodes est lié au fait
qu’elles agissent sur tout matériel ADN (génome ou plasmide entier) sans cibler précisément
la séquence codant la protéine d’intérêt. Cette remarque est également valable pour les
techniques reposant sur l’utilisation de souches mutatrices (comme E. coli XL1-Red
commercialisée par Stratagene) dans lesquelles l’inactivation d’une ou plusieurs voies de
réparation de l’ADN augmente le taux d’erreurs au cours de la réplication (Nguyen and
Daugherty, 2003). Elles sont largement moins employées aujourd'hui que les techniques
utilisant des ADN polymérases in vitro, notamment la PCR à erreurs, qui permettent de
focaliser la création de diversité sur une séquence précise.
Dans la suite de ce chapitre, nous aborderons l'utilisation des ADN polymérases dans les
protocoles de mutagénèse aléatoire in vitro en décrivant la PCR à erreur et la méthode utilisée
au cours de notre travail, MutaGenTM.
Chapitre I : Etude bibliographique
30
2.1.3 PCR à erreurs ou error-prone PCR
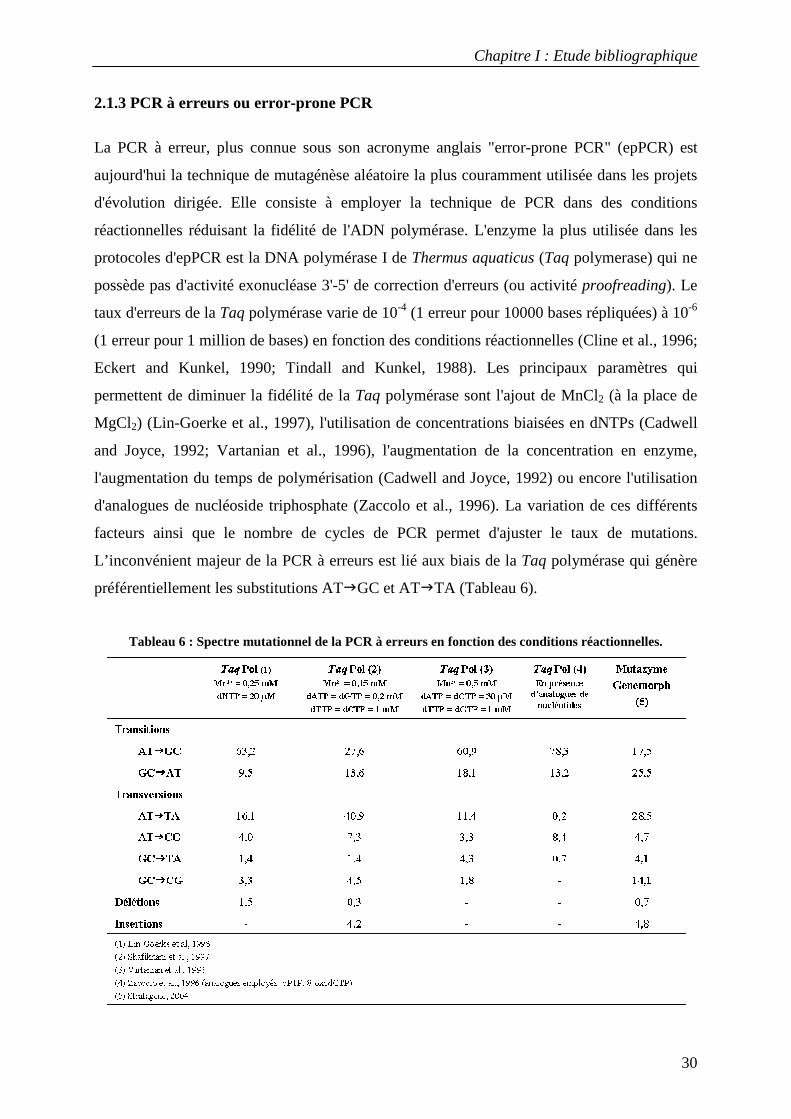
La PCR à erreur, plus connue sous son acronyme anglais "error-prone PCR" (epPCR) est
aujourd'hui la technique de mutagénèse aléatoire la plus couramment utilisée dans les projets
d'évolution dirigée. Elle consiste à employer la technique de PCR dans des conditions
réactionnelles réduisant la fidélité de l'ADN polymérase. L'enzyme la plus utilisée dans les
protocoles d'epPCR est la DNA polymérase I de Thermus aquaticus (Taq polymerase) qui ne
possède pas d'activité exonucléase 3'-5' de correction d'erreurs (ou activité proofreading). Le
taux d'erreurs de la Taq polymérase varie de 10-4 (1 erreur pour 10000 bases répliquées) à 10-6
(1 erreur pour 1 million de bases) en fonction des conditions réactionnelles (Cline et al., 1996;
Eckert and Kunkel, 1990; Tindall and Kunkel, 1988). Les principaux paramètres qui
permettent de diminuer la fidélité de la Taq polymérase sont l'ajout de MnCl2 (à la place de
MgCl2) (Lin-Goerke et al., 1997), l'utilisation de concentrations biaisées en dNTPs (Cadwell
and Joyce, 1992; Vartanian et al., 1996), l'augmentation de la concentration en enzyme,
l'augmentation du temps de polymérisation (Cadwell and Joyce, 1992) ou encore l'utilisation
d'analogues de nucléoside triphosphate (Zaccolo et al., 1996). La variation de ces différents
facteurs ainsi que le nombre de cycles de PCR permet d'ajuster le taux de mutations.
L’inconvénient majeur de la PCR à erreurs est lié aux biais de la Taq polymérase qui génère
préférentiellement les substitutions AT�GC et AT�TA (Tableau 6).
Tableau 6 : Spectre mutationnel de la PCR à erreurs en fonction des conditions réactionnelles.

Chapitre I : Etude bibliographique
31
Des kits basés sur le principe de l'epPCR ont été commercialisés par les sociétés Clontech
(DiversifyTM) ou Stratagene (GeneMorphTM). Le système développé par Clontech met en
œuvre la Taq polymérase et fournit des mélanges réactionnels prêts à l'emploi en fonction du
taux de mutation souhaité. A l'inverse, le kit GeneMorphTM de Stratagene repose sur
l'utilisation d'un mutant infidèle de la Taq polymérase baptisé MutazymeTM (tableau 6). Il
permet également de contrôler le taux de mutation en variant la concentration d'ADN matrice
et le nombre de cycles réactionnels.
2.1.4 MutaGenTM : réplication in vitro par des ADN polymérases à taux d’erreurs élevé
La technique MutaGenTM brevetée par la société française MilleGenTM (Bouayadi et al., 2002)
repose sur l'utilisation des ADN polymérases infidèles appartenant aux familles X et Y. Ces
ADN polymérases catalysent la synthèse de variants de la séquence ADN parentale au cours
d'une unique étape de réplication in vitro (Figure 4). Les copies mutées sont ensuite
amplifiées sélectivement par PCR puis clonées. L'emploi de différentes polymérases ayant
chacune leurs propres caractéristiques (taux d'erreurs, biais mutationnel) permet de varier le
taux de mutation et le spectre mutationnel appliqués aux banques de variants.
Figure 4 : Principe et étapes de la technique MutaGenTM .

Chapitre I : Etude bibliographique
32
La technique MutaGenTM a notamment été employée dans le cadre de projets d'ingénierie des
immunoglobulines afin de générer des banques de fragments variables simple chaîne
d'anticorps (scFv: single chain variable fragment) ou d'augmenter la diversité initiale d'une
banque native de domaines variables (chaînes lourdes et chaînes légères λ et κ). Ces travaux
ont été effectués dans la perspective d'identifier de nouveaux anticorps thérapeutiques dirigés
contre des cibles spécifiques et ont fait récemment l'objet d'une publication scientifique et
d'un brevet (Mondon et al., 2007a; Mondon et al., 2007b). Plusieurs banques de scFv ou de
domaines variables ont ainsi été générées avec un taux moyen de 1.8 mutations pour 100
acides aminés en utilisant les ADN polymérases β et η. L'application de la technique
MutaGenTM à l'ingénierie des enzymes a fait l'objet du présent travail de thèse.
2.2 Banques de variants obtenues par recombinaison aléatoire
Les techniques dont nous exposerons le principe dans cette partie permettent de construire des
banques de variants en recombinant plusieurs séquences parentales. Ces dernières peuvent
coder soit des variants de la même enzyme sauvage, soit des protéines homologues issues par
exemple d'espèces différentes. Les méthodes de "recombinaison homologue" reposent sur
l'hybridation et/ou l'élongation des brins ADN à recombiner et sont applicables lorsque les
séquences parentales possèdent un taux d'identité élevé (>60%). Les techniques de
"recombinaison non homologue" permettent de créer des hybrides à partir de séquences
parentales présentant un faible niveau d'identité (<50%).
2.2.1 Méthodes basées sur la recombinaison homologue
2.2.1.1 La PCR sexuelle ou "DNA shuffling"
La "PCR sexuelle" ou DNA shuffling est une méthode de recombinaison in vitro basée sur la
PCR (figure 5). Elle est brevetée par les sociétés américaines Affymax et Maxygen (Stemmer,
1994a, b; Stemmer and Crameri, 1995). La description de cette technique par Stemmer en
1994 a constitué une avancée majeure dans le domaine de l'évolution dirigée. En recombinant
des séquences parentales mutées, cette méthode permet de construire des banques de variants
hybrides. Les variants ayant accumulé les mutations bénéfiques pour la propriété à améliorer
pourront ensuite être isolés par sélection et/ou criblage et subir un nouveau cycle de
recombinaison/sélection/criblage (figure 5).

Chapitre I : Etude bibliographique
33
Techniquement, les séquences parentales à recombiner sont d'abord digérées aléatoirement
par la DNAse I. Une étape d'électrophorèse suivie d'une purification sur gel permet ensuite de
séparer les fragments générés et d'isoler ceux possédant la taille choisie au préalable. Ces
derniers sont rassemblés par des cycles successifs de PCR sans amorce qui sont répétés
jusqu'à l'obtention d'hybrides de même taille que les séquences parentales. Une PCR finale
avec amorce permet d'amplifier les variants et d'introduire les sites de restriction nécessaires
au clonage.
Figure 5 : Technique du DNA shuffling (d’après Patten et al., 1997). Une banque de variants est digérée par
la DNase I de façon à générer un ensemble de fragments de différentes tailles. Ces derniers sont ensuite
réassemblés par PCR, créant ainsi une banque de variants combinant les mutations initiales. Les points rouges et
verts représentent respectivement les mutations délétères et bénéfiques. Après sélection et/ou criblage pour la
propriété recherchée, les variants positifs peuvent être utilisés pour un nouveau tour de recombinaison.

Chapitre I : Etude bibliographique
34
Figure 6 : Technique du family shuffling (d’après Crameri et al., 1998).
Dans les cas où le point de départ de l'expérience d'évolution dirigée est une protéine unique,
les séquences parentales utilisées dans le DNA shuffling sont en général obtenues par PCR à
erreurs (ou par mutagénèse dirigée à saturation, voir paragraphe "2.3.1 Introduction de
mutations à l'aide d'oligonucléotides dégénérés"), recombinées, puis les mutants sélectionnés
et/ou criblés. Une variante du DNA shuffling, le family shuffling (Crameri et al., 1998),
permet de construire des banques de variants hybrides à partir de séquences codant des
protéines homologues, issues du même organisme ou d'espèces différentes (figure 6). Ces
séquences doivent présenter 60-70% d'identité pour pouvoir être recombinées. Dans la mesure
où les séquences parentales codent des protéines de structure tridimensionnelle similaire, le
family shuffling permet en principe de générer des hybrides actifs ou fonctionnels qui
adoptent le même type de repliement. La méthode décrite initialement (Crameri et al., 1998),
techniquement identique à celle décrite par Stemmer en 1994, a été appliquée à quatre gènes
de β-lactamases provenant de microorganismes différents et présentant de 60% à 82%
d'identité entre eux. En un cycle de recombinaison/sélection, cette stratégie a permis d'isoler
une chimère possédant une résistance de 270 à 540 fois plus élevée que les protéines
parentales. Cette nouvelle enzyme était composée de huit fragments (sept évènements de
recombinaison) provenant de trois des quatre gènes parentaux ainsi que 33 substitutions en
acides aminés. Les croisements qui ont permis de produire cette chimère sont survenus dans
des zones de 14 à 37 bp quasi identiques entre les séquences recombinées.
Des variantes du family shuffling ont été développées par l'équipe de Shigeaki Harayama
pour améliorer la fréquence de formation des protéines hybrides dans les cas où l'étape de

Chapitre I : Etude bibliographique
35
sélection/criblage ne permet pas de discriminer efficacement les protéines chimères des
protéines parentales. L'application de la procédure décrite initialement par Crameri et al.
(1998) à deux gènes homologues codant des catechol 2,3-dioxygenases (81% d'identité
nucléotidique et 84% d'identité protéique) générait moins de 1% de gènes hybrides (Kikuchi
et al., 1999, 2000). Deux approches ont été développées pour augmenter la fréquence de
formation d'hétéroduplexes et empêcher la reconstitution des séquences parentales par
formation d'homoduplexes. D'une part, le remplacement de la DNAse I par un jeu d'enzymes
de restriction au cours de l'étape de fragmentation a permis de générer des banques contenant
prés de 100% de gènes hybrides (Kikuchi et al., 1999). Bien qu'elle soit efficace, cette
approche n'est pas aléatoire puisque les croisements se produisent au niveau des sites de
restriction digérés. Une seconde stratégie consiste à utiliser les séquences parentales sous
forme d'ADN simple brin, ce qui permet d'augmenter le taux de gènes hybrides à 14%
(Kikuchi et al., 2000).
Le DNA shuffling présente l'avantage d'être relativement simple et flexible à mettre en œuvre.
De fait, elle est actuellement la technique de construction de banques par recombinaison la
plus largement utilisée. Cependant, elle reste difficile à appliquer avec des séquences longues
(> 2-3 kb) du fait de l'utilisation de la PCR. D'autre part, la modélisation informatique du
DNA shuffling (Maheshri and Schaffer, 2003; Moore et al., 2001) ainsi que l'analyse
expérimentale des séquences générées par cette méthode (Joern et al., 2002) ont montré que le
nombre de recombinaisons non silencieuses restait faible (1 à 4 recombinaisons/séquence). En
effet, les évènements de recombinaison se produisent plus fréquemment dans les régions
présentant des niveaux d'identité élevés. La diversité accessible par le DNA shuffling est donc
limitée par l'existence de "points chauds" de recombinaison.
2.2.1.2 Les méthodes StEP et RPR
Développées par le groupe de Frances H. Arnold, ces méthodes reposent comme le DNA
shuffling sur l'utilisation de la PCR et ont été brevetées par le Californian Institute of
Technology (Arnold et al., 1998).
L'acronyme anglais StEP (pour "Staggered Extension Process) (Zhao et al., 1998) désigne une
technique de PCR par laquelle les séquences recombinées sont générées en présence des
séquences parentales entières. Cette méthode (figure 7) consiste donc en une réaction de PCR
qui commence par l'hybridation d'un couple d'amorces défini sur les séquences parentales

Chapitre I : Etude bibliographique
36
Figure 7 : La technique StEP (d’après Zhao et al., 1998). Cette méthode de recombinaison in vitro repose sur
la PCR. A chaque cycle, le temps d’extension est très court. Ainsi, le fragment en cours d’élongation peut
s’hybrider sur un brin parental à chaque cycle. Seuls une amorce et un brin des deux matrices parentales sont
représentés sur cette figure.
suivie de cycles répétés de dénaturation et d'hybridation/élongation très courts. Ainsi, les
fragments en cours d'élongation peuvent s'apparier sur une matrice parentale différente à
chaque nouveau cycle. Des produits de PCR hybrides combinant des fragments de séquences
provenant des différentes matrices parentales sont générés au terme de cette procédure.
Cependant, cette approche présente un biais parce qu'elle favorise l'appariement des brins en
cours d'élongation sur les matrices parentales les plus ressemblantes. De la même façon que le
DNA shuffling, la StEP ne s'affranchit pas du problème des "points chauds" de
recombinaison.
La seconde méthode, baptisée RPR (pour Random Priming Recombination) (Shao et al.,
1998), consiste en la synthèse d'un très grand nombre de courts fragments d'ADN
complémentaires à différentes portions des séquences parentales à l'aide de petites amorces (6
bases) de séquence aléatoire (figure 8). Ces fragments contiennent un faible taux de mutations
ponctuelles dues aux mésappariements des amorces et à des erreurs d'incorporation des bases
lors de la synthèse. Une seconde étape de PCR sans amorce permet de recombiner et
rassembler les fragments produits au cours de la première PCR pour former des séquences de
même longueur que les matrices initiales parentales.

Chapitre I : Etude bibliographique
37
Figure 8 : La technique RPR (d’après Shao et al., 1998). (1) Synthèse de courts fragments d’ADN simples
brins à partir de petites amorces de séquences aléatoires. (2) Élimination des séquences matrices. (3)
Réassemblage des fragments par PCR. (4) Identification des variants positifs après clonage suivi de
sélection/criblage. (5) Ce processus peut être répété plusieurs fois jusqu’à obtention de l’amélioration
recherchée.
2.2.1.3 Les méthodes RACHITT et L-Shuffling
Ces deux techniques de recombinaison reposent sur une approche complètement différente
des méthodes décrites précédemment dans la mesure où l'étape de recombinaison n'est pas
réalisée par PCR mais par hybridation sur un brin d'ADN support (figure 9). Selon la méthode
RACHITT (pour Random Chimeragenesis on Transient Template) (Coco et al., 2001), les
séquences parentales sous forme simple brin complémentaire au brin support, sont digérées
par la DNase I pour générer des fragments de différentes tailles (figure 9). Ces derniers sont
ensuite appariés sur le brin support qui présente deux caractéristiques importantes. D'une part,
ce brin est synthétisé avec incorporation d'uracile ce qui le rend sensible à l'action de l'uracile-
ADN glycosylase. D'autre part, il est homologue mais non identique aux séquences parentales
fragmentées, ce qui empêche la reconstitution de ces dernières au cours de la procédure.
Après hybridation des fragments, les parties non appariées en 5' sont digérées par l'activité
endonucléase de la Taq polymérase. L'activité 3'-5' exonucléase de la Pfu polymérase permet
d'éliminer les parties non appariées en 3' puis son activité polymérase comble les brèches en
utilisant le brin support comme matrice. L'action d'une ligase permet de relier les différents

Chapitre I : Etude bibliographique
38
fragments entre eux. Le brin support est ensuite rendu non amplifiable par un traitement à
l'uracile-ADN glycosylase et une réaction finale de PCR permet d'amplifier les séquences
hybrides.
L'un des avantages de cette approche par rapport au DNA shuffling concerne le taux de
recombinaison : 14 croisements par gène en moyenne pour la technique RACHITT contre 1 à
4 pour le DNA shuffling. Cette différence peut être expliquée par deux raisons. D'une part, à
chaque cycle de DNA shuffling, la formation de produits non désirés obtenus par
mésappariement des fragments et des amorces interfère avec l'amplification des variants
recherchés; a contrario, l'unique étape d'hybridation dans la technique RACHITT limite cette
interférence. D'autre part, la technique RACHITT permet de recombiner des fragments de
plus petite taille que dans le DNA shuffling puisque les extrémités non appariées au cours de
l'étape d'hybridation sont éliminées. Par ailleurs, cette méthode ne génère pas de séquences
parentales à la différence du DNA shuffling et de la StEP. En revanche, elle est d'une mise en
œuvre pratique plus délicate que ces dernières à cause notamment de la synthèse du brin
support avec incorporation d'uracile.
Figure 9 : La technique RACHITT (d’après Coco et al., 2001).

Chapitre I : Etude bibliographique
39
La méthode L-shuffling brevetée par la société française Proteus (Dupret et al., 2002; Lefevre
et al., 2001) s'apparente à la technique RACHITT. Les séquences parentales sous forme
d'ADN double brin sont digérées par l'action d'un jeu d'enzymes de restriction. Les fragments
générés sont dénaturés, hybridés sur une matrice support (brin parental non clivé) puis reliés
entre eux par une ligase. Les séquences hybrides obtenues sont ensuite amplifiées puis
clonées. Elles peuvent ensuite être criblées ou soumises à un nouveau cycle de
digestion/hybridation/ligation avec un jeu différent d'enzymes de restriction. Cette approche a
récemment été employée dans le cadre d’un projet d’amélioration de l’affinité d’un anticorps
(Chodorge et al., 2008).
2.2.1.4 Les méthodes de recombinaison in vivo
L'expression "in vivo shuffling" a été employée pour désigner une technique de
recombinaison aléatoire réalisée dans la levure Saccharomyces cerevisiae (Cherry et al.,
1999). Dans le cadre de travaux sur l'amélioration de la stabilité d'une peroxydase fongique,
cette approche a été utilisée pour recombiner les mutations observées sur des variants obtenus
par différentes méthodes (mutagénèse aléatoire suivie de criblage ou mutagénèse dirigée). Les
séquences parentales à recombiner sont préparées par PCR de façon à ce que leurs extrémités
soient homologues aux zones du vecteur de clonage dans lequel elles doivent s'insérer. La
levure est transformée en même temps par les produits de PCR et le vecteur. La
recombinaison homologue des produits de PCR avec les extrémités du vecteur permet de
reconstituer un plasmide circulaire autonome. Cependant, des évènements de recombinaison
homologues surviennent également entre les produits de PCR ce qui permet de générer une
population de variants hybrides à partir des séquences parentales initiales. Cette technique de
recombinaison aléatoire est décrite par leurs auteurs comme étant plus simple à mettre en
œuvre que le DNA shuffling in vitro car elle ne nécessite pas de fragmentation aléatoire des
séquences parentales et pas d'étape de ligature avant transformation.
La méthode CLERY (Combinatorial Libraries Enhanced by Recombination in Yeast)
(Abecassis et al., 2000) est une approche apparentée à celle qui vient d'être décrite. En effet,
elle consiste en une première étape de DNA shuffling in vitro qui génère des variants hybrides
qui sont dans un second temps recombinés aléatoirement in vivo dans la levure qui est
également le système d'expression. Cette méthode qui vise à élargir la diversité initiale de
banques de variants construites in vitro a été utilisée avec succès pour générer des chimères à
partir de deux cytochromes P450 homologues humains partageant 74% d'identité

Chapitre I : Etude bibliographique
40
nucléotidique. L'analyse statistique et fonctionnelle réalisée à partir de variants choisis au
hasard dans les banques ou retenus après criblage confirme l'efficacité de cette méthode de
recombinaison aléatoire dans la levure.
2.2.2 Recombinaison de séquences non homologues
La génération d'évènements de recombinaison est difficile dans les cas où le niveau d'identité
entre les séquences parentales est faible (< 50%). Ceci a conduit au développement de
méthodes de "recombinaison non homologue" qui permettent de générer des variants hybrides
à partir de séquences parentales peu ou pas similaires. En effet, au contraire des stratégies
décrites précédemment, ces techniques ne reposent pas sur l'hybridation et/ou l'élongation des
brins à recombiner.
2.2.2.1. Méthodes générant un point de recombinaison unique : ITCHY et SHIPREC
La première méthode de ce type à avoir été décrite est la technique ITCHY (pour Incremental
Truncation for Creation of Hybrid Enzymes) (Ostermeier et al., 1999). Cette approche a fait
l’objet d’un dépôt de brevet par le Penn State Research Foundation (Benkovic et al., 2000).
La méthode ITCHY permet de construire des hybrides de deux séquences parentales
présentant un point unique de recombinaison (figure 10). Les séquences parentales sont
d'abord digérées séparément par une exonucléase III pour générer des banques de fragments
N- et C-terminaux de longueur variable. Ces différents fragments sont ensuite reliés par une
ligase pour former des hybrides de différentes tailles. Les évènements de recombinaison
générés par cette approche ne reposent donc pas sur l'identité des séquences parentales. Ainsi,
les points possibles de recombinaison sont situés aléatoirement sur la séquence, alors que dans
le cas du DNA shuffling ou de la StEP, ils se situent uniquement dans les régions à fort taux
d'identité. La technique ITCHY a été validée expérimentalement en construisant des
glycinamide ribonucléotide (GAR) transformylases hybrides entre l'enzyme de E. coli (PurN)
et son homologue humain (GART). Les séquences ADN codant ces protéines partagent
environ 50% d'identité. Cependant, les GAR transformylases sont modulaires en structure
comme en fonction : les résidus qui interragissent avec le substrat sont situés en N-terminal
alors que ceux qui contribuent à la catalyse sont en C-terminal. La méthode ITCHY est donc
une approche bien adaptée pour l'ingénierie combinatoire de ce type d'enzyme. Dans la
mesure où les GAR transformylases participent à la voie de biosynthèse des purines, une
méthode de sélection des hybrides actifs par complémentation génétique peut être envisagée
en utilisant une souche E. coli déficiente pour ces enzymes. Avec cette procédure, 9 et 111

Chapitre I : Etude bibliographique
41
hybrides actifs ont respectivement été isolés à partir de deux librairies de 1,3×105 et 1,2×106
clones, soit un hybride actif pour 10000 à 15000 hybrides initiaux.
La limite principale de la technique ITCHY est de générer des variants hybrides ne présentant
qu'un unique point de recombinaison. En recombinant ces variants par une étape de DNA
shuffling, il est possible d'augmenter le nombre de croisements (Lutz et al., 2001). Cette
méthode baptisée SCRATCHY permet d'accroître le taux de recombinaison initial à 2-3
recombinaisons par séquence.
Comme la technique ITCHY, la méthode SHIPREC (pour Sequence Homology-Independent
Protein Recombination) (Sieber et al., 2001), développée par le groupe de Frances Arnold et
brevetée par le Californian Institute of Technology (Arnold et al., 2001), permet de
recombiner deux séquences peu ou pas apparentées avec un point de croisement (figure 11).
Elle consiste à utiliser un gène constitué de 5' en 3' du gène codant la première protéine
parentale, suivi d'un linker contenant des sites de restriction uniques et du gène codant la
seconde protéine parentale. Cette séquence dimérique est fragmentée par l'action de la DNase
I puis traitée à la nucléases S1 pour produire des bouts francs. Les fragments correspondant à
la longueur d'un gène parental sont séparés du mélange par électrophorèse, purifiés puis
Figure 10 : La technique ITCHY (d’après Ostermeier et al., 2001). RE1 et RE2: enzymes de restriction ayant
un site unique dans le vecteur; P/O: promoteur; AntR: gène de résistance à un antibiotique.

Chapitre I : Etude bibliographique
42
circularisés par ligation intramoléculaire des bouts francs. Enfin, ces fragments sont linéarisés
par digestion avec une enzyme de restriction coupant dans le linker. La banque de gènes
hybrides ainsi générée peut être clonée puis transformée dans l'hôte d'expression approprié.
Cette méthode a été validée par ses auteurs en construisant une banque d'hybrides entre un
cytochrome membranaire P450 humain (1A2) et le domaine hème d'un cytochrome soluble
P450 de Bacillus megaterium (BM3) qui partagent seulement 16% d'identité protéique. Les
gènes hybrides ainsi créés ont ensuite été clonés en fusion C-terminale avec le gène codant la
chloramphenicol acétyltransférase (CAT), ce qui permet d'effectuer une présélection des
variants traduits dans le même cadre de lecture que cette dernière. D'autre part, il a été montré
que des bactéries exprimant des fusions d'une protéine insoluble avec la CAT étaient plus
faiblement résistantes au chloramphénicol que celles produisant des fusions avec une protéine
soluble (Maxwell et al., 1999). L'étape de présélection utilisée par Sieber et al. (2001) permet
donc aussi d'éliminer les hybrides mal repliés et insolubles. Environ 20% d'hybrides ont été
ainsi présélectionnés à partir d'une banque initiale de 250000 clones. Approximativement
2000 hybrides solubles ont ensuite été criblés pour une activité possédée par le cytochrome
P450-1A2 et non partagée par le cytochrome P450-BM3 (dééthylation de la 7-
éthoxyrésorufine). Deux hybrides fonctionnels ont ainsi été isolés, ce qui correspond à un taux
d'un hybride actif pour 1000 hybrides solubles (5000 hybrides initiaux). Les chimères isolés
possèdent une séquence correspondant majoritairement à la protéine parentale 1A2 possédant
10 à 15 résidus provenant de BM3.
Figure 11 : La technique SHIPREC (d’après Sieber et al., 2001). Construction d'une banque de protéines
hybrides à partir d'un gène dimèrique (correspondant ici aux cytochromes 1A2 et BM3) dans lequel les
séquences à recombiner sont séparées par un linker (contenant des sites de restriction uniques).

Chapitre I : Etude bibliographique
43
2.2.2.2. Méthode générant plusieurs points de recombinaison : NRR
Décrite plus récemment, la méthode NRR (pour Nonhomologous Random Recombination)
(Bittker et al., 2004; Liu et al., 2005) permet en revanche de construire en une étape des
variants hybrides avec plusieurs points de croisement (figure 12). La première étape consiste à
générer des fragments à bouts francs en soumettant les séquences parentales à un traitement à
la DNAse I puis à celui d'une ADN polymérase qui permet de générer des bouts francs. Ces
fragments sont ensuite recombinés par une réaction de ligature en présence de deux séquences
d'ADN double brin en forme d'épingle à cheveux. Ces dernières possèdent les sites de
restriction nécessaires au clonage des variants générés.
Cette méthode a été validée en construisant une librairie combinatoire à partir d'une enzyme
de relative petite taille (104 résidus), la chorismate mutase (CM) de Methanococcus
jannaschii. Cette protéine catalyse la conversion de chorismate en prephenate et participe à la
voie de biosynthèse de la tyrosine et de la phénylalanine. Les variants construits par NRR sont
clonés en fusion C-terminale avec le gène codant la chloramphenicol acétyltransférase, de
manière à effectuer une présélection des variants analogue à celle utilisée par Sieber et al.
(2001). Une seconde sélection est ensuite effectuée sur milieu minimum sans tyrosine pour
Figure 12 : La technique NRR (d’après Bittker et al., 2004). Une ou plusieurs séquences parentales sont
digérées par la DNase I. Les fragments sont traités par la T4 ADN polymérase pour générer des bout francs puis
assemblés de manière aléatoire par une ligase, en présence d’ADN en forme d’épingle à cheveux. Les extrémités
de ces dernières sont éliminées par des enzymes de restriction, ce qui permet de cloner les séquences chimères.

Chapitre I : Etude bibliographique
44
isoler les variants CM actifs produits dans une souche E. coli auxotrophe pour la tyrosine. A
l'issue de cette procédure, le nombre de colonies survivantes correspond à un taux de sélection
d'un variant actif pour 1500 variants présélectionnés à partir d'une banque initiale de 4,5×105
variants. Les variants CM ainsi sélectionnés présentent de nombreuses modifications
(délétions, répétitions, insertions et réarrangements) par rapport à la séquence parentale. De
manière à analyser le potentiel de leur méthode pour créer des hybrides à partir de séquences
non homologues, les auteurs ont ensuite construit une banque d'hybrides entre le gène codant
la CM et celui de la fumarase d'E. coli. Le choix de cette dernière était lié au fait qu'elle ne
possède aucune ressemblance en séquence et en activité avec la CM mais qu'elle présente le
même profil structural composé uniquement d'hélices α. L'étape de sélection sur milieu
minimum sans tyrosine permet d'isoler des hybrides possédant une activité CM avec un taux
d'un mutant actif pour une banque initiale de 2×105 clones. Au final, 90% des clones actifs
séquencés sont effectivement des chimères CM-fumarase, ce qui démontre l'efficacité de la
méthode NRR.
Les approches qui viennent d'être décrites permettent d'hybrider des séquences parentales sans
critère d'homologie ce qui peut conduire à la construction de nouveaux repliements
protéiques. Ceci constitue également une différence importante avec les techniques de
mutagénèse aléatoire ou de recombinaison homologue qui génèrent des variants possédant la
même structure globale que les protéines parentales. Les méthodes de "recombinaison non
homologue" permettent donc d'élargir l'exploration de l'espace des séquences et pourraient
favoriser ainsi le développement de nouvelles activités enzymatiques. Cependant, elles
présentent aussi l'inconvénient majeur de générer un très grand nombre de variants inactifs en
raison de la fréquence élevée de mutations non-sens, de décalages du cadre de lecture et de
repliements protéiques non fonctionnels. Ceci complique la recherche d'hybrides actifs dans la
mesure où il est nécessaire d'explorer des banques de taille élevée pour pouvoir les isoler. Il
est donc nécessaire d'associer ces approches à des procédures performantes de sélection et/ou
de criblage pour garantir leur efficacité.

Chapitre I : Etude bibliographique
45
2.3 Banques de variants obtenues par des techniques de mutagénèse ou de
recombinaison semi-aléatoires
Les méthodes qui seront présentées dans cette partie sont mises en œuvre dans le cadre de
projets combinant à la fois les techniques d'évolution dirigée (création de banques de variants,
sélection, criblage) et l'approche rationnelle (analyse de la structure tridimensionnelle,
alignements de séquences etc.). Elles visent à explorer la diversité dans des zones ciblées de la
(ou des) protéine(s) jouant (ou susceptibles de jouer) un rôle dans la propriété à modifier.
2.3.1 Introduction de mutations ponctuelles à l'aide d’oligonucléotides dégénérés
Diverses méthodes de mutagénèse dirigée mettant en œuvre des oligonucléotides synthétiques
dégénérés (mutagénèse à saturation) peuvent être employées pour introduire toutes les
mutations possibles sur un ou des codons choisis au préalable, voire sur l'ensemble des
codons de la séquence parentale. Ce type de stratégie permet de contourner la dégénérescence
du code génétique. La plupart des techniques de mutagénèse à saturation utilisées de nos jours
reposent sur l'emploi de la PCR.
L’approche la plus simple consiste à employer de façon équimolaire les quatre nucléotides
(NNN où N = A, C, G ou T) au niveau des codons à modifier (Oliphant et al., 1986).
Cependant, cette stratégie favorise la conservation des nucléotides originaux parce que les
oligonucléotides formant le plus grand nombre d’appariements de type Watson-Crick
s’hybrident préférentiellement sur le brin matrice. Ce biais peut être éliminé en réduisant le
nombre d’oligonucléotide possédant le (ou les) codon(s) original(aux) au cours de la synthèse
des amorces (Airaksinen and Hovi, 1998). Les mutations ne sont toutefois jamais introduites
de manière équiprobable. Par ailleurs, l’utilisation d’un mélange équimolaire des quatre
nucléotides entraîne également un autre biais en faveur des acides aminés possédant le plus
d’entrées dans le code génétique. Dans le cas d’une banque qui combine la mutagénèse de
plusieurs résidus, le rapport entre le nombre de gènes générés (43n où n est le nombre de
codons mutés) et le nombre de protéines attendues (20n) augmentera de façon exponentielle
avec le nombre de codons ciblés (tableau 7). Le nombre de variants génétiques dans une telle
banque sera donc largement plus élevé que le nombre effectif de protéines mutées. Pour
réduire ce biais, l’utilisation de codons alternatifs, NN(G/T) ou NN(G/C), dans lesquels la
troisième base est partiellement dégénérée, est souvent envisagée, car ils permettent de
représenter les 20 acides aminés par 32 codons différents (au lieu de 64) et ne génèrent qu’un
seul codon stop (UAG).

Chapitre I : Etude bibliographique
46
Tableau 7 : Mutagénèse à saturation. Nombre de variants générés selon la nature des codons dégénérés
L'emploi de la mutagénèse dirigée à saturation a fait l'objet de divers brevets. La technique
Gene Site Saturation MutagenesisTM (GSSM) (Short, 2003) a été développée par la firme
américaine Diversa (DeSantis et al., 2003; Kretz et al., 2004; Solbak et al., 2005). Elle
consiste à appliquer la mutagénèse dirigée à saturation à chaque codon du gène de la protéine
parentale, afin d'identifier les résidus ou les régions impliqués dans la propriété à modifier.
Les réactions de mutagénèse (une par résidu) sont effectuées par PCR inverse avec des
oligonucléotides dégénérés 32 ou 64 fois (figure 13A). Par cette approche, la taille théorique
d'une banque de mono-mutants construite à partir d'une protéine de 100 acides aminés sera de
32×100 = 3200 (ou 64×100 = 6400). La stratégie GSSM présente l'avantage de permettre
l'identification de substitutions non générées par les techniques de mutagénèse aléatoire. A
titre d'exemple, dans le cadre de travaux visant à améliorer la productivité et
l'énantiosélectivité d'une nitrilase, le meilleur variant isolé par DeSantis et al. (2003) possédait
une histidine (codon du type CAT/C) à la place d'une alanine (GCN), soit une substitution qui
requiert au moins le changement de deux bases dans le codon initial.
Développée par la société française Biométhodes, la stratégie Massive MutagenesisTM repose
également sur l'emploi de la mutagénèse dirigée à saturation (Delcourt and Blesa, 2002).
Contrairement à l'approche précédente, elle consiste à muter simultanément plusieurs résidus
au cours d'une réaction de PCR utilisant des amorces unidirectionnelles dégénérées (ou
permettant d'introduire un nombre limité de substitutions choisies au préalable) en présence
d'un mélange d'ADN polymérases et d'ADN ligases (figure 13B). Après digestion du vecteur
parental par Dpn I, les simples brins synthétisés à partir des amorces mutées sont transformés
dans E. coli. La banque de mono-mutants ainsi construite peut être directement analysée par
sélection et/ou criblage. Alternativement, l'ADN plasmidique des colonies obtenues après
transformation peut être extrait et utilisé comme matrice pour un nouveau cycle de
mutagénèse afin de générer des variants combinant plusieurs mutations. Cette méthode a

Chapitre I : Etude bibliographique
47
notamment été employée pour la construction d'une banque de variants de l'interleukine 15
humaine (Saboulard et al., 2005). L'objectif de cette étude était de muter trente-sept résidus de
la protéine sauvage en introduisant huit substitutions sur chaque position. Les amorces
unidirectionnelles utilisées pour effectuer ces travaux ont été synthétisées selon une approche
à haut-débit employant des puces à ADN (Lausted et al., 2004). Après trois cycles de
mutagénèse, la diversité de la banque résultante a été analysée en séquençant 96 clones
choisis au hasard. Parmi ces derniers, 58 étaient de type sauvage, 33 étaient des mono-mutants
et seulement 5 contenaient deux mutations. Ces résultats indiquent que prés de la moitié des
clones de la banque sont identiques à la protéine parentale. Les auteurs en attribuent la cause
au système de réparation des mésappariements de l'ADN chez E. coli. L'optimisation de
l'approche Massive MutagenesisTM est cependant nécessaire pour limiter le taux de clones
parentaux dans les banques.
Figure 13 : Techniques de mutagénèse dirigée à saturation. (A) Gene Site Saturation Mutagenesis :
méthode brevetée par Diversa. La réaction schématisée est effectuée pour chaque résidu de la protéine d'intérêt.
(B) Massive Mutagenesis : technique brevetée par Biométhodes.

Chapitre I : Etude bibliographique
48
2.3.2 Techniques de recombinaison utilisant des oligonucléotides dégénérés
Certaines techniques de recombinaison par PCR reposent sur l'emploi d'oligonucléotides
dégénérés. Elles sont considérées comme des approches semi-aléatoires dans la mesure où les
amorces sont dessinées à partir des informations données par l'alignement des séquences
parentales à recombiner. Ces dernières peuvent être des variants obtenus de façon rationnelle
ou combinatoire ou bien des protéines homologues provenant d'organismes différents. Les
approches utilisant des amorces dégénérées ont été développées dans le but d'augmenter le
taux de croisements et de limiter la production de séquences parentales.
La première approche de ce type à avoir été décrite et brevetée est la technique DOGS (pour
Degenerate Oligonucleotide Gene Shuffling ; figure 14) (Gibbs et al., 2002; Gibbs et al.,
2001). Les amorces utilisées dans cette méthode sont composées d'une partie centrale non
dégénérée correspondant à une zone conservée des séquences parentales; leurs extrémités en
5' et 3' sont en revanche dégénérées. Ces amorces sont utilisées pour amplifier des segments
de chaque séquence parentale. Ces produits sont ensuite mélangés dans une PCR par
recouvrement pour générer les variants hybrides. Le taux de recombinaison est contrôlé en
jouant sur les proportions des différents segments.
Figure 14 : Principe de la technique DOGS (d'après Gibbs et al., 2001).

Chapitre I : Etude bibliographique
49
Les oligonucléotides dégénérés ont également été employés pour construire des banques de
variants hybrides par des méthodes proches du DNA shuffling : le shuffling synthétique (Ness
et al., 2002) et la technique ADO (pour Assembly of Designed Oligonucleotides) (Zha et al.,
2003). Le shuffling synthétique a été breveté par la société Maxygen (Welch et al., 2001).
Dans ces deux approches, les gènes parentaux sont analysés de manière à dessiner un nombre
important d'oligonucléotides dégénérés dont la taille varie de 60 nucléotides dans le shuffling
synthétique à 140 pour la technique ADO. Les produits d'hybridation sont synthétisés par
PCR uniquement à partir du mélange des oligonucléotides dégénérés. Dans ces stratégies,
l'utilisation avantageuse d'amorces synthétiques permet le contrôle de la position des points de
recombinaison, l'introduction de mutations ponctuelles spécifiques et d'éviter la reformation
des gènes parentaux initiaux.
3. LE TRI DE LA DIVERSITE : SELECTION ET CRIBLAGE
L’identification de variants présentant un gain pour la propriété à améliorer constitue l’étape
la plus critique de l’évolution dirigée. D’une part, elle nécessite l'existence d'un lien, direct ou
indirect, entre la propriété protéique à améliorer (phénotype) et la séquence codante qui lui
correspond (génotype). Par ailleurs, elle repose sur des méthodes de sélection et/ou de
criblage propres à chaque enzyme et propriété à améliorer. Enfin, l’étape de construction des
banques permet de générer un nombre considérable de mutants qui peut varier de 104 à 1012 en
fonction des étapes expérimentales (clonage, transformation de microorganismes etc.). Par
conséquent, ces méthodes de sélection et/ou de criblage doivent être suffisamment rapides
(haut-débit) et sensibles pour détecter efficacement les variants positifs.
Les notions de "criblage" et de "sélection" sont souvent confondues. Nous appliquerons le
terme de "sélection" à un éventail d'approches qui permettent de tester simultanément un
grand nombre de variants et d’isoler directement ceux possédant la propriété désirée. Cette
définition regroupe donc les procédés classiques de sélection in vivo par complémentation
génétique, le phage display, la compartimentation in vitro et les méthodes utilisant la
cytométrie en flux. En revanche, le terme de "criblage" sera appliqué lorsque chaque variant
est analysé individuellement sur milieu solide, sur membrane ou au format microplaque. Dans
cette approche, les mutants améliorés sont extraits du reste de la banque par l'expérimentateur
en fonction des résultats à l'issue de la campagne de criblage.
Nous présenterons dans ce paragraphe les principales techniques de sélection et de criblage
développées et utilisées dans le cadre de l'évolution dirigée d'enzymes. Nous insisterons

Chapitre I : Etude bibliographique
50
surtout sur leur principe, leurs avantages et leurs limites sans rentrer dans le détail des
exemples cités.
3.1 Techniques de sélection in vivo
Les méthodes de sélection in vivo reposent sur un lien entre la survie cellulaire et l'activité de
l'enzyme d'intérêt. Historiquement, les techniques de sélection par complémentation génétique
ont d'abord été utilisées pour identifier des gènes ou des opérons impliqués dans des voies de
biosynthèse. Elles constituent la stratégie la plus rapide et la plus simple pour identifier les
clones recherchés dans une banque de variants. Leur principe est basé sur le fait que l'activité
de l'enzyme d'intérêt doit conférer à la cellule hôte un phénotype lui permettant de croître
dans des conditions de culture données. Ainsi, ces approches permettent de trier la diversité
des banques en favorisant le développement des clones exprimant les variants recherchés tout
en éliminant ceux qui ne sont pas désirés. Toutes ces techniques comportent une première
étape de transformation des cellules hôtes (bactéries ou levures) par les plasmides qui
constituent la banque. Les cellules bactériennes constituent donc les outils de choix pour le
développement d'un procédé de sélection in vivo. En effet, par comparaison avec les cellules
eucaryotes, leur efficacité de transformation est plus élevée, leur temps de génération plus
court et leur génome moins complexe donc plus facile à modifier si nécessaire.
Tableau 8 : Exemples de méthodes de sélection génétique employées en évolution d'enzyme.
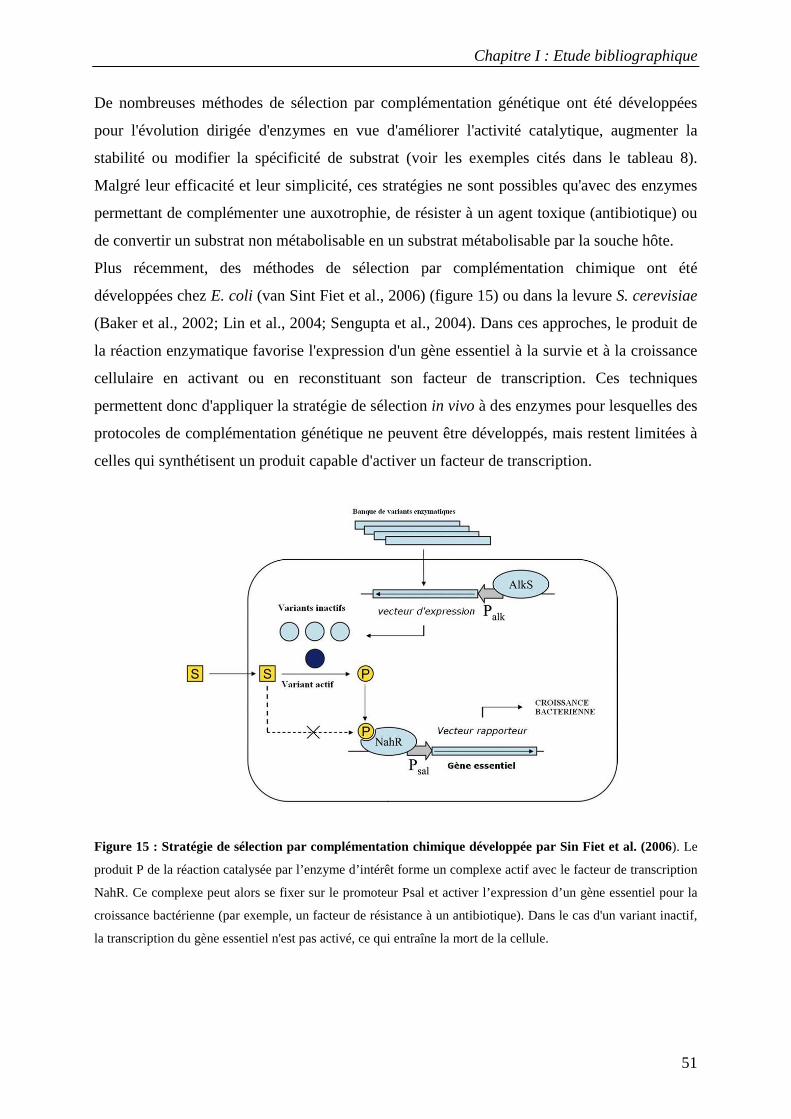
Chapitre I : Etude bibliographique
51
De nombreuses méthodes de sélection par complémentation génétique ont été développées
pour l'évolution dirigée d'enzymes en vue d'améliorer l'activité catalytique, augmenter la
stabilité ou modifier la spécificité de substrat (voir les exemples cités dans le tableau 8).
Malgré leur efficacité et leur simplicité, ces stratégies ne sont possibles qu'avec des enzymes
permettant de complémenter une auxotrophie, de résister à un agent toxique (antibiotique) ou
de convertir un substrat non métabolisable en un substrat métabolisable par la souche hôte.
Plus récemment, des méthodes de sélection par complémentation chimique ont été
développées chez E. coli (van Sint Fiet et al., 2006) (figure 15) ou dans la levure S. cerevisiae
(Baker et al., 2002; Lin et al., 2004; Sengupta et al., 2004). Dans ces approches, le produit de
la réaction enzymatique favorise l'expression d'un gène essentiel à la survie et à la croissance
cellulaire en activant ou en reconstituant son facteur de transcription. Ces techniques
permettent donc d'appliquer la stratégie de sélection in vivo à des enzymes pour lesquelles des
protocoles de complémentation génétique ne peuvent être développés, mais restent limitées à
celles qui synthétisent un produit capable d'activer un facteur de transcription.
Figure 15 : Stratégie de sélection par complémentation chimique développée par Sin Fiet et al. (2006). Le
produit P de la réaction catalysée par l’enzyme d’intérêt forme un complexe actif avec le facteur de transcription
NahR. Ce complexe peut alors se fixer sur le promoteur Psal et activer l’expression d’un gène essentiel pour la
croissance bactérienne (par exemple, un facteur de résistance à un antibiotique). Dans le cas d'un variant inactif,
la transcription du gène essentiel n'est pas activé, ce qui entraîne la mort de la cellule.

Chapitre I : Etude bibliographique
52
3.2 Techniques de sélection in vitro
3.2.1 Le phage-display
Le phage-display (Smith, 1985) constitue un outil très performant pour explorer les
interactions entre protéines, peptides et divers ligands. Dans cette technique, les variants qui
constituent la banque sont individuellement présentés à la surface de phages filamenteux non
lytiques (le plus souvent M13). Ceci est possible en créant une fusion génétique entre la
protéine d'intérêt et une protéine de la capside virale (en général pIII ou pVIII). Cette protéine
de fusion est sécrétée, via la séquence signal native de la protéine phagique, vers l'espace
périplasmique d'E. coli dans lequel les virions sont assemblés sans provoquer la lyse de la
cellule hôte. La protéine d'intérêt est ainsi exposée à la surface du phage et accessible à son
ligand spécifique. Ce dernier est fixé sur un support solide permettant la récupération
sélective des variants possédant l'affinité recherchée. Les phages portant les variants retenus
par la sélection sont récupérés et amplifiés par infection de nouvelles cellules bactériennes.
Plusieurs tours de cette procédure (appelée "biopanning") permettent d'enrichir la population
phagique en phages exposant le(s) variant(s) qui se lie(nt) le mieux au ligand spécifique.
La puissance du phage-display repose sur le lien physique entre la protéine d'intérêt et la
séquence génétique correspondante clonée dans l'ADN phagique. D'autre part, elle permet
d'explorer des banques de taille très importante (jusqu'à 107-8 variants). Cette technologie a
d'abord été utilisée comme une méthode générique de sélection de protéines à haute affinité
vis-à-vis d'un ligand spécifique : peptides liant la streptavidine (Devlin et al., 1990) ou des
anticorps (Cwirla et al., 1990), fragments d'anticorps ScFv (McCafferty et al., 1990) ou Fab
(Hoogenboom et al., 1991) etc. Son utilisation pour la sélection de variants enzymatiques sur
la base de leur activité catalytique est cependant plus complexe. En effet, la capacité d'une
enzyme à reconnaître un (nouveau) substrat et à catalyser une réaction doit être couplée à un
mécanisme de liaison. Différentes stratégies ont ainsi été développées, comprenant à la fois
des approches indirectes, reposant sur l'utilisation d'inhibiteurs, et des approches directes
mettant en jeu les substrats.
Les premières méthodes de phage-display appliquées à la sélection de variants enzymatiques
sont qualifiées d'indirectes, car elles reposent sur l'affinité de l'enzyme à différents types de
ligands : analogues de substrat (Vanwetswinkel et al., 2000), substrats (ou inhibiteurs)
suicides (Janda et al., 1994; Soumillion et al., 1994) ou analogues de l'état de transition
(Widersten and Mannervik, 1995). Après fixation dans le site actif, ces ligands empêchent la

Chapitre I : Etude bibliographique
53
libération de l'enzyme (figure 16A). Seuls les variants capables de lier le ligand immobilisé
sont ainsi sélectionnés. De façon générale, ces méthodes de sélection par affinité sont
efficaces pour la recherche de variants enzymatiques de spécificité modifiée. Cependant, elles
peuvent également entraîner l'isolement de variants moins actifs (voire inactifs) mais ayant
une affinité intacte pour le ligand utilisé. D'autre part, ce type d’approche ne permet pas
d’isoler des variants présentant seulement une augmentation de l'activité catalytique (kcat).
Le développement de méthodes de phage-display permettant de sélectionner directement les
variants enzymatiques sur la base de leur activité catalytique est plus difficile. En effet, les
produits étant susceptibles de s'éloigner du site catalytique après la réaction, il est nécessaire
de développer un lien physique entre le substrat et l'enzyme exposée à la surface du phage.
Ainsi, des approches consistant à produire des phages présentant à la fois l'enzyme et son
substrat ont été proposées (Demartis et al., 1999; Ponsard et al., 2001). En modifiant le
substrat, la réaction catalysée par l'enzyme provoque soit l'immobilisation du phage sur un
support solide (figure16B) soit son relarguage (figure 16C). Cette stratégie repose donc sur la
Figure 16 : Stratégie de sélection de banques de variants enzymatiques utilisant le phage display (d’après
Fernandez-gacio et al, 2003). (A) Une banque de phages exprimant les variants enzymatiques (en fusion avec
pIII) est sélectionnée par affinité pour un analogue de l’état de transition attaché par liaison covalente sur un
support solide. (B) Les fusions protéiques pIII-Calmoduline-variants enzymatique sont liées au substrat via un
peptide de liaison à la calmoduline. Après catalyse, le produit de la réaction se fixe sur un ligand spécifique
attaché par liaison covalente sur le support solide (d'après Demartis et al., 1999). (C) Sélection de
métalloenzyme par phage-display (Ponsard et al., 2001). Les fusions protéiques pIII-variants enzymatiques sont
d’abord inactivées après extraction du co-facteur métallique Zn2+ puis se lient à leur substrat attaché de manière
covalente sur le support. L’ajout du co-facteur entraîne la conversion du substrat en produit et conduit à l’élution
des phages exprimant un variant actif.

Chapitre I : Etude bibliographique
54
proximité entre l'enzyme, son substrat et le produit de la catalyse. Ce dernier peut donc être
utilisé pour isoler le variant d'intérêt. Il reste cependant difficile de généraliser ces méthodes
pour l'analyse de banques de variants enzymatiques puisqu'elles nécessitent de disposer de
ligands affins pour le produit (ou le substrat). Des techniques basées sur l'emploi de substrats
biotinylés pouvant se fixer sur la streptavidine ont été développées pour contourner ce
problème (Pedersen et al., 1998). Elles ont surtout été utilisées pour l'ingénierie d'ADN ou
d'ARN polymérases (Fa et al., 2004; Vichier-Guerre et al., 2006). Les méthodes de sélection
de variants enzymatiques par phage-display sont donc spécifiques des enzymes pour
lesquelles elles sont développées.
3.2.2 Méthodes de sélection par tri cellulaire utilisant la cytométrie en flux
L'utilisation de la cytométrie en flux (en anglais : FACS pour Fluorescence-Activated Cell
Sorting) permet d'identifier directement dans une population de cellules en culture liquide
celles qui expriment un variant possédant l'activité enzymatique recherchée. La cytométrie en
flux est une technologie de tri cellulaire à haut-débit qui permet d'analyser par marquage
fluorescent plus de 107 cellules par heure. La principale application de cette méthodologie
concerne l'analyse de phénotypes cellulaires tant en recherche qu'en diagnostic (cellules
sanguines, tumorales etc.).
L'emploi de la cytométrie en flux pour la sélection de variants enzymatiques présente
certaines difficultés pratiques. En effet, le substrat doit pénétrer dans la cellule et être converti
en produit fluorescent par la réaction enzymatique. De plus, ce produit doit être retenu par la
cellule afin que celle-ci puisse être analysée correctement par le cytomètre en flux. Le substrat
et le produit ne doivent donc pas affecter la viabilité cellulaire, être susceptibles d'être
modifiés par d'autres enzymes ou encore être peu solubles. Ces difficultés sont plus ou moins
marquées en fonction de l'activité enzymatique ciblée et ont conduit au développement
d'approches méthodologiques spécifiques. Par exemple, certaines consistent à exposer les
variants enzymatiques à la surface des bactéries gram négative hôtes (bacterial cell surface
display). La surface de ces dernières est naturellement chargée négativement, ce qui permet
l'ancrage d'un substrat fluorescent conjugué à une queue polycationique. Si le substrat est
converti en produit, la nature de la fluorescence change, ce qui permet de différencier les
cellules exposant un variant actif des autres. L'utilité de cette démarche a été démontrée pour
la sélection de variants de la protéase OmpA de E. coli (Olsen et al., 2000).

Chapitre I : Etude bibliographique
55
L'utilisation de la cytométrie en flux pour l'évolution dirigée d'enzymes a été décrite pour de
nombreux exemples que nous ne détaillerons pas ici (Aharoni et al., 2006; Becker et al., 2007;
Griswold et al., 2005; Kwon et al., 2004; Santoro and Schultz, 2002; Santoro et al., 2002;
Varadarajan et al., 2005; Wilhelm et al., 2007). Cette méthodologie a également été employée
en combinaison avec la technique de compartimentation in vitro par des émulsions
artificielles (voir le paragraphe "Création de compartiments in vitro par des émulsions
artificielles").
3.2.3 Le ribosome-display
A la différence des techniques précédentes, la technique du ribosome-display se déroule
entièrement in vitro et ne nécessite pas une étape de transformation de cellules. Dans cette
approche, le lien physique entre génotype et phénotype est réalisé à travers la formation au
cours de la traduction d'un complexe ternaire ARNm-ribosome-protéine qui peut être utilisé
pour la sélection (figure 17). Cette étape de traduction est effectuée in vitro à l'aide d'extraits
cellulaires procaryotes ou eucaryotes. La formation du complexe entre la séquence d'ARNm,
le ribosome et la protéine d'intérêt est possible grâce à l'absence de codon stop. Le dernier
résidu d'acide aminé reste fixé à son ARNt lui-même hybridé sur l'ARNm dans le ribosome,
ce qui bloque la libération de la protéine et l'ARNm. Pour permettre à la protéine d'intérêt
d'être repliée de manière fonctionnelle, celle-ci est généralement construite en fusion C-
terminale avec un peptide espaceur qui la sépare du ribosome. Après sélection, le complexe
ternaire est dissocié et l'ADNc correspondant aux variants positifs est préparé par RT-PCR.
A l'instar du phage-display, la technique de ribosome-display a surtout été appliquée pour la
sélection de peptides ou de fragments d'anticorps à haute affinité pour un ligand (de l'ordre de
10-9/-10 M) (Yan and Xu, 2006). Les premières utilisations de cette approche pour la sélection
de variants enzymatiques sont des méthodes indirectes reposant sur l'utilisation d'inhibiteurs
(Amstutz et al., 2002; Takahashi et al., 2002). Plus récemment, le ribosome-display a été
adapté pour sélectionner directement des variants actifs de la DNA ligase du phage T4
(Takahashi et al., 2005) ou pour isoler des RNA ligases à partir de banques de structures
aléatoires (Seelig and Szostak, 2007). A l'heure actuelle, relativement peu d'études rapportent
donc l'utilisation de cette technique pour la sélection de banques de variants enzymatiques.

Chapitre I : Etude bibliographique
56
Figure 17 : Utilisation du ribosome-display pour la sélection de variants enzymatiques. La séquence ADN
codant le variant est transcrite in vitro en ARNm qui forme un complexe avec le ribosome. En fin de traduction
in vitro, l’absence de codon stop sur l’ARNm empêche le relargage du variant enzymatique. La sélection des
variants d’intérêt est effectuée en capturant le complexe par affinité. Après dissociation du complexe, une RT-
PCR permet d’isoler la séquence codant les variants sélectionnés.
3.2.4 Création de compartiments in vitro par des émulsions artificielles
A l'instar du ribosome-display, la technique de compartimentation in vitro (Tawfik and
Griffiths, 1998) ne nécessite pas la transformation d'un microorganisme hôte. Elle repose sur
l'utilisation d'émulsions d'eau dans de l'huile (E/H) formant des compartiments aqueux
microscopiques. Ces derniers dont le volume est proche de celui d'une bactérie (1 à 5 µm de
diamètre) peuvent servir de microréacteurs pour sélectionner des banques de variants. Il s'agit
de simuler un environnement cellulaire artificiel à l'intérieur duquel les variants de la protéine
d'intérêt seront synthétisés in vitro à partir de leur séquence ADN (un variant par
microémulsion) puis testés par rapport à la propriété que l'on cherche à améliorer. Cette
approche permet donc à la fois de miniaturiser le volume réactionnel (environ 5 femtolitres) et
de tester en même temps un très grand nombre de variants distincts (>1010 microémulsions
dans un tube de 1 mL). Différents modes de sélection utilisant cette technique ont été
développés en fonction de l'enzyme ciblée (Miller et al., 2007; Taly et al., 2007).
Une première possibilité consiste à créer un lien physique entre l'ADN codant le variant
enzymatique et le produit de l'activité recherchée (figure 18A). Ceci permet d'isoler
directement les séquences codant les variants positifs en utilisant une méthode de détection du
produit de la réaction. Cette stratégie est facilement applicable à la sélection d'enzymes de
modification de l'ADN : ADN méthyltransférases (Cohen et al., 2004; Lee et al., 2002;

Chapitre I : Etude bibliographique
57
Tawfik and Griffiths, 1998) et endonucléases de restriction (Doi et al., 2004). En effet, dans
ces exemples, une même molécule d'ADN combine à la fois la séquence codant un variant, le
substrat et par conséquent le produit de la réaction (si le variant est actif). L'utilisation de
microbilles sensibilisées à la streptavidine a permis d'étendre cette approche à d'autres types
d'enzymes (Griffiths and Tawfik, 2003; Levy et al., 2005) (figure 18B). Dans chaque
microémulsion, la séquence codant le variant à tester est biotinylée et fixée sur une microbille.
Après la synthèse du variant enzymatique, ce dernier (si il est actif) réagit avec le substrat
associé à un dérivé de biotine portant un groupe photosensible protecteur (empêchant la
fixation du substrat sur la microbille). L'irradiation des microémulsions aux UV permet au
produit (ou au substrat si le variant est inactif) de se fixer sur la microbille. Après rupture des
émulsions, les séquences codant un variant actif (reliées au produit de la réaction par le biais
des microbilles) peuvent être isolées par cytométrie en flux en utilisant un anticorps dirigé
contre le produit et conjugué à un fluorochrome.
Une seconde stratégie repose sur l'utilisation de substrats fluorophores pour isoler directement
par cytométrie en flux les microémulsions contenant les variants recherchés (figure
18C)(Mastrobattista et al., 2005). Dans une première microémulsion E/H, le variant
enzymatique est synthétisé in vitro puis réagit avec un substrat fluorophore. Après réaction,
les microémulsions E/H sont converties en doubles émulsions eau-huile-eau (E/H/E) qui sont
analysées par cytométrie en flux pour isoler celles qui sont fluorescentes. Dans ce cas de
figure, la microémulsion elle-même constitue le lien entre la séquence codant le variant
recherché et le produit de la réaction. L'utilisation d'émulsions E/H/E a également été décrite
pour l'encapsulation de bactéries exprimant une banque de variants enzymatiques (Aharoni et
al., 2006). Dans cet exemple, le substrat (et par conséquent le produit de la réaction
enzymatique) n'a pas besoin d'être internalisé à l'intérieur de la cellule ou d'être fixé à sa
surface.
Au cours des prochaines années, l'utilisation de la compartimentation in vitro comme outil de
sélection en évolution dirigée devrait bénéficier du développement des techniques de
microfluidiques qui permettront un meilleur contrôle de la formation et de l'utilisation des
microémulsions (Whitesides, 2006).
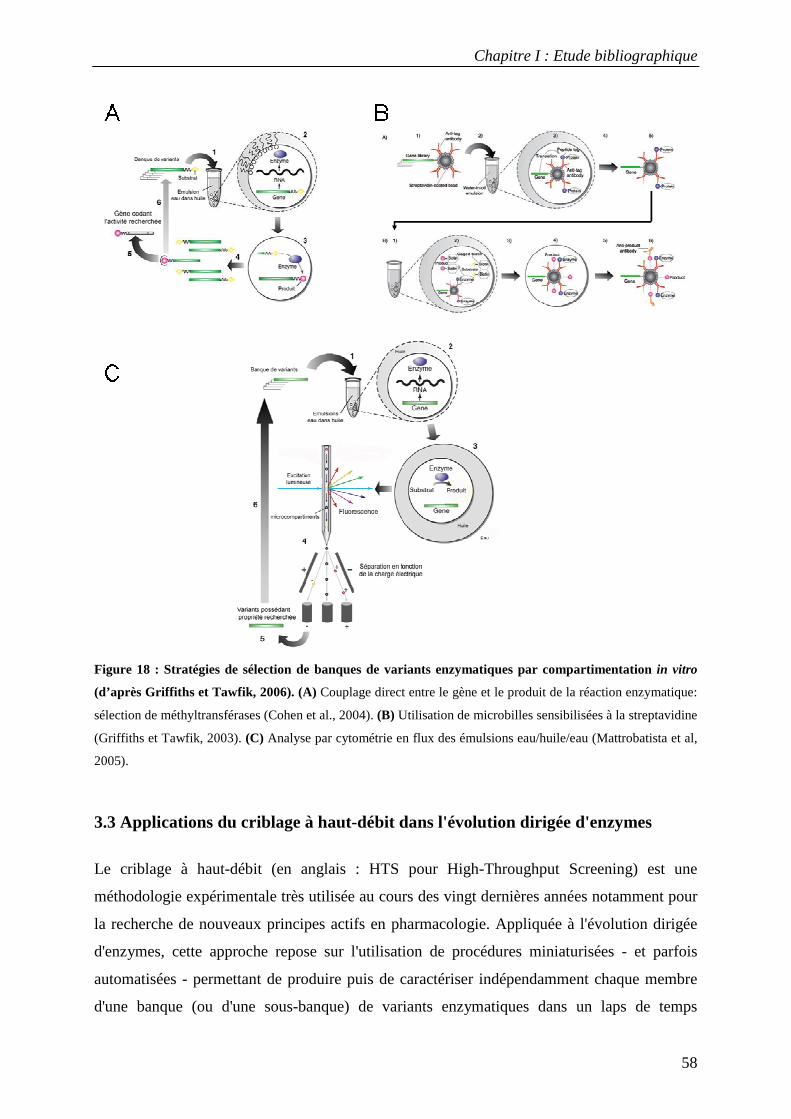
Chapitre I : Etude bibliographique
58
Figure 18 : Stratégies de sélection de banques de variants enzymatiques par compartimentation in vitro
(d’après Griffiths et Tawfik, 2006). (A) Couplage direct entre le gène et le produit de la réaction enzymatique:
sélection de méthyltransférases (Cohen et al., 2004). (B) Utilisation de microbilles sensibilisées à la streptavidine
(Griffiths et Tawfik, 2003). (C) Analyse par cytométrie en flux des émulsions eau/huile/eau (Mattrobatista et al,
2005).
3.3 Applications du criblage à haut-débit dans l'évolution dirigée d'enzymes
Le criblage à haut-débit (en anglais : HTS pour High-Throughput Screening) est une
méthodologie expérimentale très utilisée au cours des vingt dernières années notamment pour
la recherche de nouveaux principes actifs en pharmacologie. Appliquée à l'évolution dirigée
d'enzymes, cette approche repose sur l'utilisation de procédures miniaturisées - et parfois
automatisées - permettant de produire puis de caractériser indépendamment chaque membre
d'une banque (ou d'une sous-banque) de variants enzymatiques dans un laps de temps

Chapitre I : Etude bibliographique
59
raisonnablement court. Ces procédures doivent être robustes et reproductibles pour permettre
d'identifier de manière fiable le(s) variant(s) amélioré(s) dans les banques analysées et de
limiter le nombre de "faux-positifs". Leur mise au point est donc une étape préliminaire
essentielle pour définir et standardiser les conditions d'une campagne de criblage.
Dans les cribles utilisés en évolution dirigée, les variants enzymatiques sont généralement
produits de façon recombinante par un microorganisme hôte (le plus souvent E. coli ou S.
cerevisiae). Ainsi, l'individualisation des variants (étape préalable à toute campagne de
criblage) consiste en l'isolement des colonies bactériennes (ou de levure) après transformation
par les plasmides constituant la banque. Le criblage proprement dit est effectué à la suite de
cette phase préliminaire et son déroulement est fonction de l'enzyme d'intérêt et de la
propriété à améliorer. Les méthodes développées sont donc très nombreuses et peuvent être
distinguées selon qu’elles sont réalisées en milieu liquide ou sur support solide (tableau 9).
Le criblage par détection visuelle sur support solide est effectué directement à partir des
colonies en cours de croissance sur milieu de culture gélosé, soit après réplique des colonies
sur un/des filtre(s) membranaire(s) de nitrocellulose. La plupart des protocoles développés
repose sur l'utilisation de substrats chromophores ou fluorophores (Goddard and Reymond,
2004)), ce qui permet de repérer les clones exprimant les variants recherchés au cours du
criblage. Ces méthodes ne nécessitent pas d'étape de réarrangement des clones et sont souvent
directement appliquées après transformation des banques dans l'organisme hôte, permettant
ainsi d'analyser un grand nombre de variants simultanément. Elles sont fréquemment utilisées
comme crible préliminaire (ou primaire) pour éliminer les variants inactifs (contenant des
mutations délétères) présents dans les banques (tableau 9). Par ailleurs, l'utilisation de
réplique(s) sur filtre de nitrocellulose permet d'appliquer ces méthodes de détection visuelle
au criblage d'enzymes actives en conditions non physiologiques (Chen and Arnold, 1993;
Matsumura et al., 1999; You and Arnold, 1996). Dans certains exemples, l'utilisation de
systèmes d'imagerie digitale permet de rendre ce type d'approche plus quantitative (Delagrave
et al., 2001; Joern et al., 2001; Joo et al., 1999a).
La seconde stratégie regroupe les méthodes de criblage en milieu liquide. Au plan
expérimental, ces approches sont généralement plus complexes que les précédentes et
comportent plusieurs étapes que l'emploi d'automates et la miniaturisation des volumes
permettent de standardiser et d'accélérer. Après transformation de la banque dans l'hôte
d'expression, les colonies sont repiquées puis mises en culture au format microplaque (en
général 96 ou 384 puits). Cette étape peut éventuellement être précédée d'une sélection (ou
d'un criblage) primaire destinée à éliminer les colonies exprimant un variant enzymatique

Chapitre I : Etude bibliographique
60
inactif, ce qui permet de réduire la taille de la banque à cribler. Après croissance des
microorganismes et production des variants (suivies éventuellement d'étapes de lyse et
d'élimination des débris cellulaires), les réactions enzymatiques sont effectuées dans des
volumes réduits (de 5 à 200 µL). La consommation du substrat ou l'apparition du (ou des)
produit(s) sont mesurés le plus souvent par le biais de méthodes de détection colorimétriques,
fluorométriques ou chémoluminescentes (Reymond and Babiak, 2007). L'utilisation
d'instruments analytiques (Chromatographie en phase gazeuse, HPLC, spectrométrie de masse
etc.), qui permettent une meilleure précision dans les mesures, peut être envisagée,
notamment pour la recherche de variants enzymatiques présentant des modifications de
stéréosélectivité ou d'énantiosélectivité (Reetz, 2003; Reetz et al., 2000; Reetz et al., 2002;
Reetz et al., 2003; Reetz et al., 2004b). Cependant, elle requiert des étapes préalables de
préparation des extraits cellulaires (lyse, centrifugation des microplaques etc.) qui rendent les
protocoles de criblage plus complexes. De fait, les exemples de leur utilisation en évolution
dirigée sont relativement peu nombreux à l'heure actuelle (Fox et al., 2007; Funke et al., 2003;
Miller et al., 2007; Yoshikuni et al., 2006). Comme le montrent les exemples cités dans le
tableau 9, le criblage en milieu liquide permet d'analyser de nombreuses propriétés
enzymatiques : activité et/ou stabilité par rapport à la température et au pH, activité en solvant
organique, changement de spécificité de substrat etc.
La réussite d'une procédure de criblage repose sur sa fiabilité et sa vitesse d'analyse. Le
tableau 10 présente des exemples de tailles de banques analysées par ce type d'approche. Les
données rapportées dans la littérature montrent que la diversité explorée par les méthodes de
criblage varie en général entre 102 et 105 variants. Elles ont donc une capacité d'exploration de
la diversité moins importante que les techniques de sélection. Cependant, ces dernières sont
souvent difficiles à mettre en œuvre pour modifier des propriétés comme la thermostabilité, la
spécificité vis-à-vis de certains substrats ou l'activité en milieu non-naturel (solvants
organiques, modification du pH optimal etc.). Le criblage qui s'apparente à une forme
accélérée et miniaturisée de la procédure de caractérisation enzymatique est une méthodologie
plus flexible et mieux adaptée aux contraintes de certains projets d'évolution dirigée.
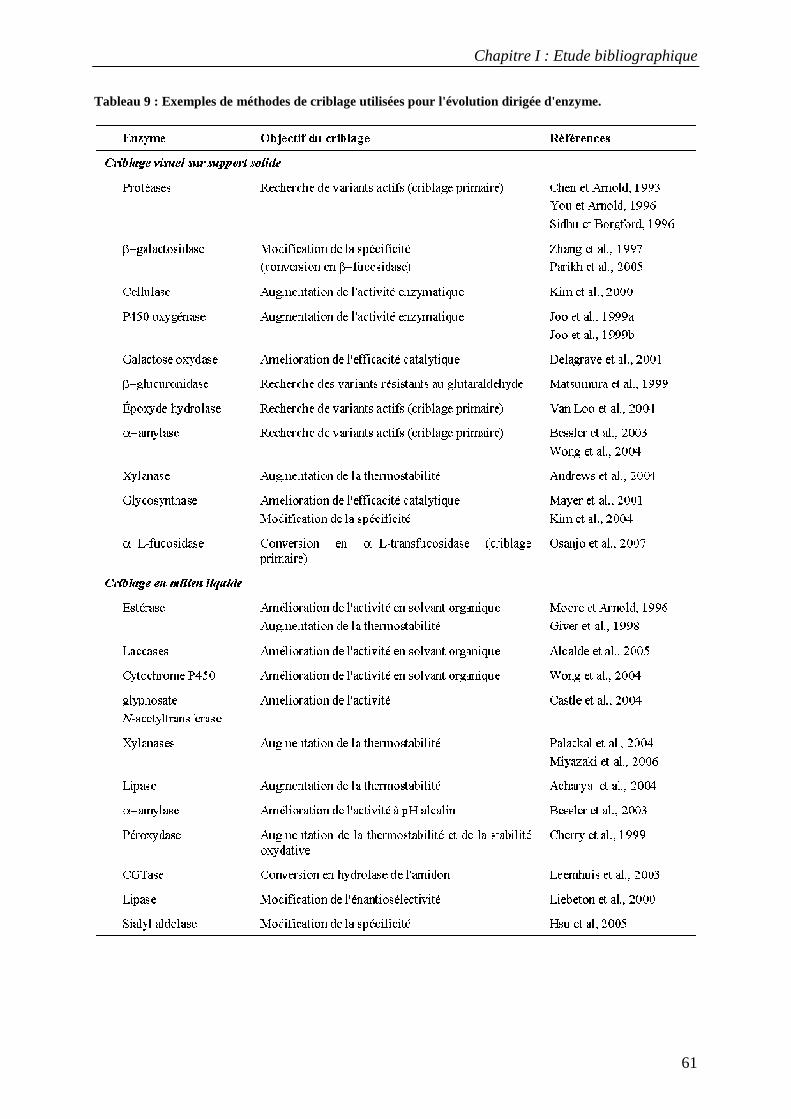
Chapitre I : Etude bibliographique
61
Tableau 9 : Exemples de méthodes de criblage utilisées pour l'évolution dirigée d'enzyme.
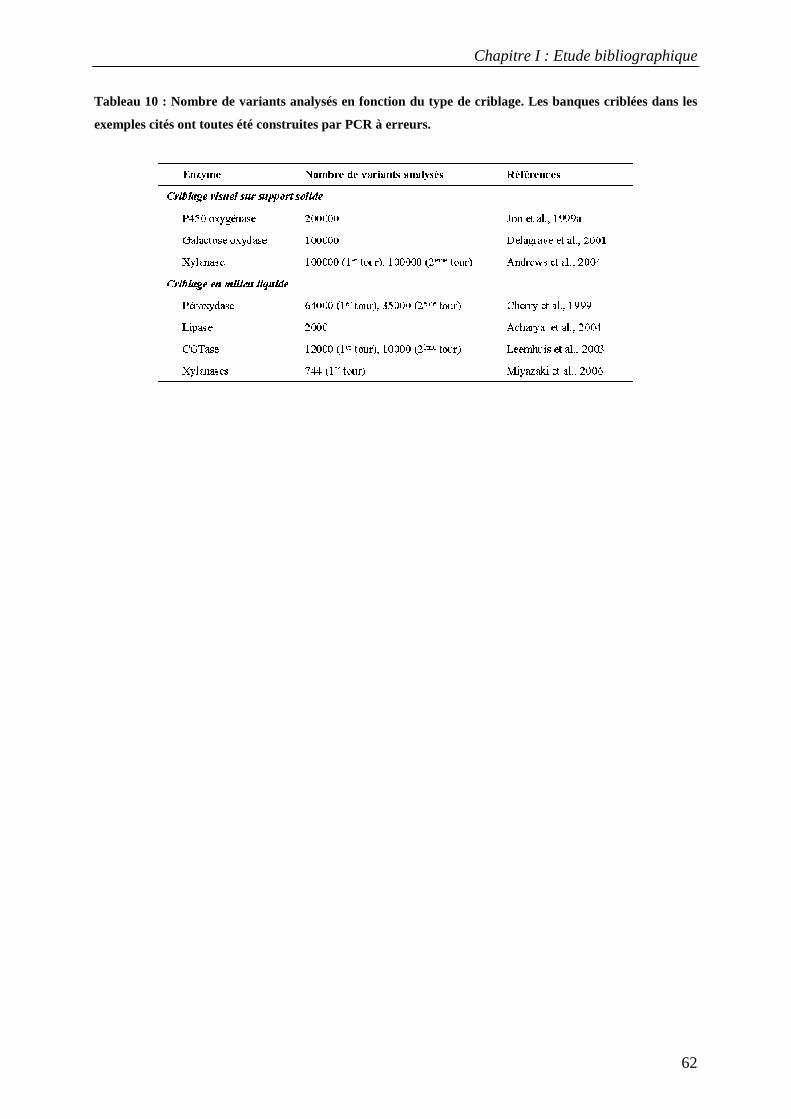
Chapitre I : Etude bibliographique
62
Tableau 10 : Nombre de variants analysés en fonction du type de criblage. Les banques criblées dans les
exemples cités ont toutes été construites par PCR à erreurs.

Chapitre I : Etude bibliographique
63
Troisième partie : L'amylosaccharase de Neisseria polysaccharea
Les amylosaccharases (E.C. 2.4.1.4) sont des transglucosidases qui utilisent le saccharose
comme substrat. Elles catalysent la formation d’un homopolymère de type amylose (dont les
unités glucosyle sont liées par des liaisons α-(1,4), en libérant du fructose (figure 19)(Okada
and Hehre, 1974). Sur la base des alignements de séquences, ces enzymes sont classées dans
la famille 13 des glycoside-hydrolases (GH13), qui regroupe majoritairement des enzymes
impliquées dans la modification de l’amidon. De par leur activité catalytique et leur
spécificité de substrat, les amylosaccharases sont uniques au sein de cette famille. Ce chapitre
fait la synthèse des caractéristiques biochimiques et structurales de ces enzymes et des travaux
d’ingénierie protéique s’y rapportant.
Figure 19 : Schéma des principales réactions catalysées par les amylosaccharases.
1. ORGANISMES PRODUCTEURS
Jusqu’à la fin des années 1990, les amylosaccharases recensées dans la littérature étaient
exclusivement produites par des bactéries non-pathogènes du genre Neisseria (genre connu
particulièrement pour ses deux espèces pathogènes N. meningitidis et N. gonorrhoeae). Hehre
et Hamilton ont été les premiers, en 1946, à découvrir une amylosaccharase intracellulaire
chez Neisseria perflava. Plus tard, la présence d’une amylosaccharase intracellulaire a été
révélée chez six autres espèces de Neisseriae (N. canis, N. cinerea, N. denitrificans, N. sicca
et N. subflava)(MacKenzie et al., 1978) appartenant à la flore de la cavité buccale et
potentiellement impliquées dans la formation de la plaque dentaire (Loesche, 1975). Enfin,
une amylosaccharase supposée extracellulaire a été identifiée chez l'espèce N. polysaccharea
qui provient du rhino-pharynx d'enfants sains d'Europe et d'Afrique (Riou et al., 1983). Cette
enzyme, dont le gène a été cloné puis séquencé en 1999, est la première amylosaccharase
recombinante caractérisée (Potocki de Montalk et al., 1999). Sa structure tridimensionnelle a
été décrite en 2002 (Skov et al., 2001).
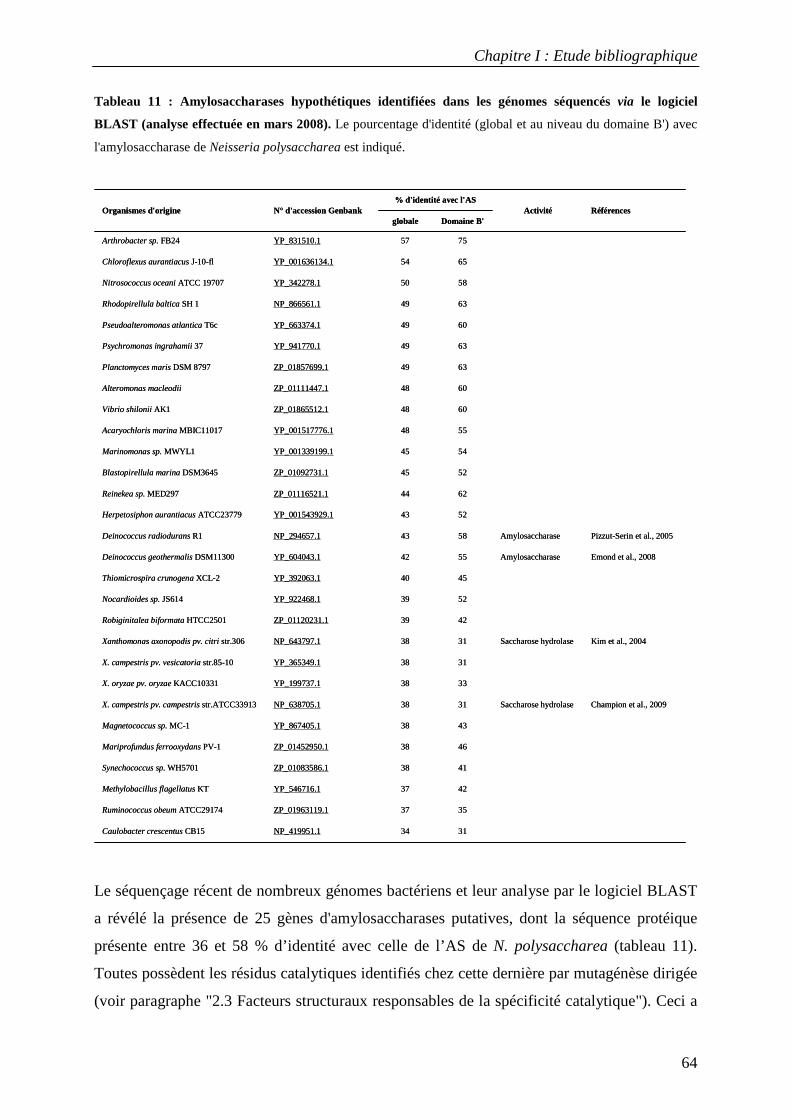
Chapitre I : Etude bibliographique
64
Tableau 11 : Amylosaccharases hypothétiques identifiées dans les génomes séquencés via le logiciel
BLAST (analyse effectuée en mars 2008). Le pourcentage d'identité (global et au niveau du domaine B') avec
l'amylosaccharase de Neisseria polysaccharea est indiqué.
Saccharose hydrolase
Saccharose hydrolase
Amylosaccharase
Amylosaccharase
Activité Références% d'identité avec l'AS
Organismes d'origine
31
35
42
41
46
43
31
33
31
31
42
52
45
55
58
52
62
52
54
55
60
60
63
63
60
63
58
65
75
Domaine B'
Champion et al., 2009
Kim et al., 2004
Emond et al., 2008
Pizzut-Serin et al., 2005
34NP_419951.1Caulobacter crescentusCB15
37ZP_01963119.1Ruminococcus obeumATCC29174
37YP_546716.1Methylobacillus flagellatusKT
38ZP_01083586.1Synechococcus sp. WH5701
38ZP_01452950.1Mariprofundus ferrooxydansPV-1
38YP_867405.1Magnetococcus sp. MC-1
38NP_638705.1X. campestris pv. campestrisstr.ATCC33913
38YP_199737.1X. oryzae pv. oryzaeKACC10331
38YP_365349.1X. campestris pv. vesicatoriastr.85-10
38NP_643797.1Xanthomonas axonopodis pv. citristr.306
39ZP_01120231.1Robiginitalea biformataHTCC2501
39YP_922468.1Nocardioides sp. JS614
40YP_392063.1Thiomicrospira crunogenaXCL-2
42YP_604043.1Deinococcus geothermalisDSM11300
43NP_294657.1Deinococcus radioduransR1
43YP_001543929.1Herpetosiphon aurantiacusATCC23779
44ZP_01116521.1Reinekea sp. MED297
45ZP_01092731.1Blastopirellula marina DSM3645
45YP_001339199.1Marinomonas sp. MWYL1
48YP_001517776.1Acaryochloris marina MBIC11017
48ZP_01865512.1Vibrio shilonii AK1
48ZP_01111447.1Alteromonas macleodii
49ZP_01857699.1Planctomyces marisDSM 8797
49YP_941770.1Psychromonas ingrahamii37
49YP_663374.1Pseudoalteromonas atlanticaT6c
49NP_866561.1Rhodopirellula balticaSH 1
50YP_342278.1Nitrosococcus oceaniATCC 19707
54YP_001636134.1Chloroflexus aurantiacusJ-10-fl
57YP_831510.1Arthrobacter sp. FB24
globaleN° d'accession Genbank
Saccharose hydrolase
Saccharose hydrolase
Amylosaccharase
Amylosaccharase
Activité Références% d'identité avec l'AS
Organismes d'origine
31
35
42
41
46
43
31
33
31
31
42
52
45
55
58
52
62
52
54
55
60
60
63
63
60
63
58
65
75
Domaine B'
Champion et al., 2009
Kim et al., 2004
Emond et al., 2008
Pizzut-Serin et al., 2005
34NP_419951.1Caulobacter crescentusCB15
37ZP_01963119.1Ruminococcus obeumATCC29174
37YP_546716.1Methylobacillus flagellatusKT
38ZP_01083586.1Synechococcus sp. WH5701
38ZP_01452950.1Mariprofundus ferrooxydansPV-1
38YP_867405.1Magnetococcus sp. MC-1
38NP_638705.1X. campestris pv. campestrisstr.ATCC33913
38YP_199737.1X. oryzae pv. oryzaeKACC10331
38YP_365349.1X. campestris pv. vesicatoriastr.85-10
38NP_643797.1Xanthomonas axonopodis pv. citristr.306
39ZP_01120231.1Robiginitalea biformataHTCC2501
39YP_922468.1Nocardioides sp. JS614
40YP_392063.1Thiomicrospira crunogenaXCL-2
42YP_604043.1Deinococcus geothermalisDSM11300
43NP_294657.1Deinococcus radioduransR1
43YP_001543929.1Herpetosiphon aurantiacusATCC23779
44ZP_01116521.1Reinekea sp. MED297
45ZP_01092731.1Blastopirellula marina DSM3645
45YP_001339199.1Marinomonas sp. MWYL1
48YP_001517776.1Acaryochloris marina MBIC11017
48ZP_01865512.1Vibrio shilonii AK1
48ZP_01111447.1Alteromonas macleodii
49ZP_01857699.1Planctomyces marisDSM 8797
49YP_941770.1Psychromonas ingrahamii37
49YP_663374.1Pseudoalteromonas atlanticaT6c
49NP_866561.1Rhodopirellula balticaSH 1
50YP_342278.1Nitrosococcus oceaniATCC 19707
54YP_001636134.1Chloroflexus aurantiacusJ-10-fl
57YP_831510.1Arthrobacter sp. FB24
globaleN° d'accession Genbank
Le séquençage récent de nombreux génomes bactériens et leur analyse par le logiciel BLAST
a révélé la présence de 25 gènes d'amylosaccharases putatives, dont la séquence protéique
présente entre 36 et 58 % d’identité avec celle de l’AS de N. polysaccharea (tableau 11).
Toutes possèdent les résidus catalytiques identifiés chez cette dernière par mutagénèse dirigée
(voir paragraphe "2.3 Facteurs structuraux responsables de la spécificité catalytique"). Ceci a

Chapitre I : Etude bibliographique
65
conduit en 2005 à la caractérisation d'une nouvelle amylosaccharase recombinante issue de D.
radiodurans (Pizzut-Serin et al., 2005). En revanche, la caractérisation des enzymes
identifiées chez les souches du genre Xanthomonas a montré qu'il s'agissait de saccharose
hydrolases (Champion et al., 2009; Kim et al., 2004; Kim et al., 2008). Ces enzymes
possèdent un domaine B’ présentant 35 % d’identité avec celui de l'amylosaccharase de
Neisseria polysaccharea (AS). Selon Pizzut-Serin et al. (2005), la structure du domaine B’
jouerait un rôle important dans l’activité polymérase. Seules les enzymes possédant un
domaine B’ partageant plus de 55 % d’identité avec celui de l’AS seraient aptes à catalyser la
réaction de polymérisation. Sur cette base, parmi les 25 gènes homologues à ceux de l’AS,
seuls 14 présentent un pourcentage d’identité supérieur à 55 % dans le domaine B’ et
coderaient donc pour des transglucosidases. A ce jour, l'AS est la plus étudiée et la mieux
caractérisée de ces enzymes.
2. STRUCTURE DE L ’AMYLOSACCHARASE DE NEISSERIA POLYSACCHAREA
2.1 Structure tridimensionnelle de l'AS sauvage
La séquence du gène rapportée par Potocki de Montalk et al. (1999) comporte 1911 paires de
bases, codant pour une protéine de 636 acides aminés (environ 70 kDa). L'optimisation du
procédé de purification a permis de purifier l'AS recombinante jusqu’à homogénéité, en
quantité suffisante pour la détermination par cristallographie et diffraction aux rayons X de sa
structure tridimensionnelle. Celle-ci a été obtenue en 2001 avec une résolution de 1,4 Å (Skov
et al., 2001). Elle présente une molécule de TRIS liée au site actif. C’est la seule structure
d’amylosaccharase disponible à ce jour. L'AS présente une organisation structurale en cinq
domaines nommés N, A, B, B' et C (figure 20). Les trois domaines A, B et C sont communs
aux α-amylases. Le domaine A qui comprend les résidus 98-184, 261-395, et 461-550,
correspond au domaine catalytique. Il est organisé en tonneau (β/α)8 comme pour toutes les
enzymes de la famille GH 13. La boucle 3 forme quant à elle le domaine B qui s’étend des
résidus 185 à 260. Il comporte deux feuillets β anti-parallèles incluant chacun une hélice α.
Enfin le domaine C (résidus 555 à 628) comporte exclusivement des brins β qui adoptent une
structure en sandwich ou en clé grecque.
L'AS possède également deux domaines supplémentaires qui lui sont propres : les domaines
N et B’. Le domaine B’ (résidus 395-460, boucle 7) qui forme une extension du domaine A,

Chapitre I : Etude bibliographique
66
comporte deux hélices α suivies d’un brin β et d’une autre hélice α. Le domaine N-terminal
(résidus 1 à 90) est composé de 6 hélices α. Deux de ces hélices soutiennent les hélices α3 et
α4 du tonneau et contribueraient au maintien de l’intégrité structurale. Par ailleurs,
l’amylosaccharase ne comporte pas de pont di-sulfure, malgré la présence de 6 résidus
cystéine dans sa séquence polypeptidique.
Figure 20 : Structure tridimensionelle de l'amylosaccharase de N. polysaccharea (d'après Skov et al.,
2001). Légendes des domaines: N (bleu sombre), A (bleu clair), B (jaune), B' (magenta) et C (rouge). Les résidus
catalytiques Asp286 et Glu328, respectivement localisés à la fin des feuillets β-4 et β-5 dans le domaine A, sont
également indiqués.
2.2 Mécanisme catalytique de l’amylosaccharase
A partir de saccharose, l'AS catalyse la formation (i) d'amylose insoluble, (ii) de
maltooligosaccharides, (iii) de fructose, (iv) de glucose issu de l'hydrolyse du saccharose et
(iv) de turanose et de tréhalulose (figure 21)(Albenne et al., 2004; Potocki de Montalk et al.,
2000b). Le milieu de synthèse est donc caractérisé par la présence de deux phases, l’une
soluble et l’autre insoluble.
Tableau 12 : Rendement des produits synthétisés par l'AS purifiée de N. polysaccharea à partir de 100
mM de saccharose (d'après Potocki-Véronèse et al., 2005).
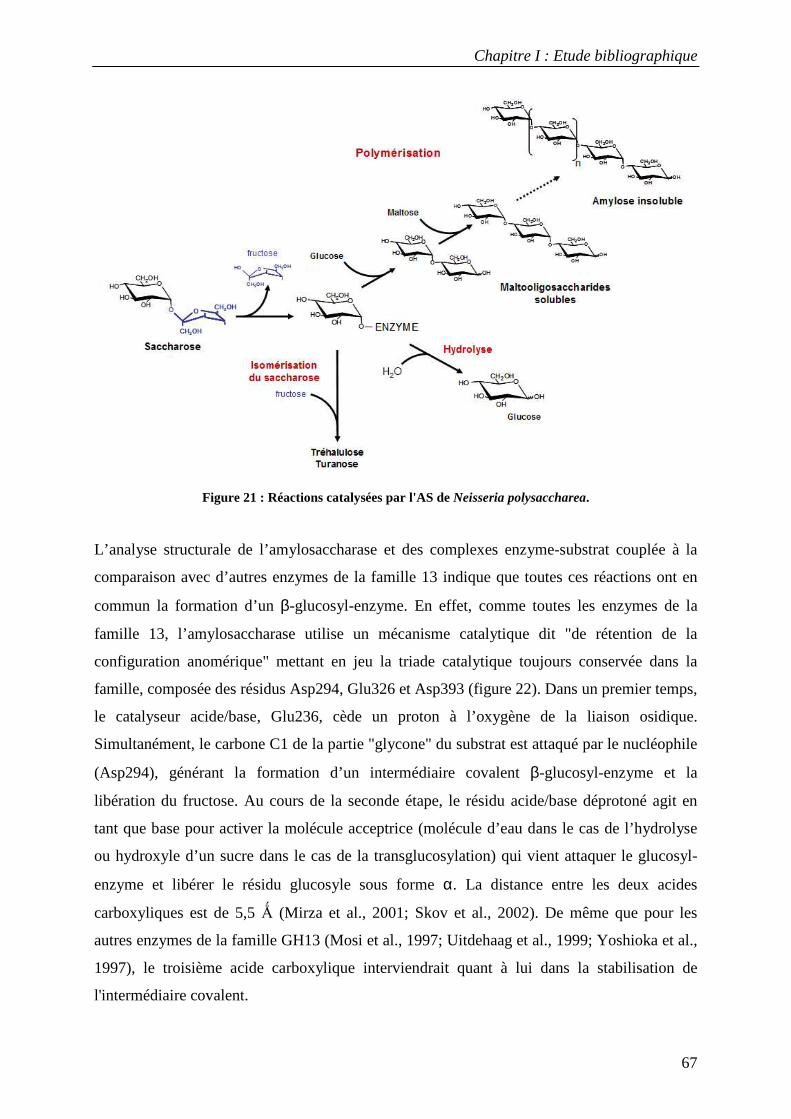
Chapitre I : Etude bibliographique
67
Figure 21 : Réactions catalysées par l'AS de Neisseria polysaccharea.
L’analyse structurale de l’amylosaccharase et des complexes enzyme-substrat couplée à la
comparaison avec d’autres enzymes de la famille 13 indique que toutes ces réactions ont en
commun la formation d’un β-glucosyl-enzyme. En effet, comme toutes les enzymes de la
famille 13, l’amylosaccharase utilise un mécanisme catalytique dit "de rétention de la
configuration anomérique" mettant en jeu la triade catalytique toujours conservée dans la
famille, composée des résidus Asp294, Glu326 et Asp393 (figure 22). Dans un premier temps,
le catalyseur acide/base, Glu236, cède un proton à l’oxygène de la liaison osidique.
Simultanément, le carbone C1 de la partie "glycone" du substrat est attaqué par le nucléophile
(Asp294), générant la formation d’un intermédiaire covalent β-glucosyl-enzyme et la
libération du fructose. Au cours de la seconde étape, le résidu acide/base déprotoné agit en
tant que base pour activer la molécule acceptrice (molécule d’eau dans le cas de l’hydrolyse
ou hydroxyle d’un sucre dans le cas de la transglucosylation) qui vient attaquer le glucosyl-
enzyme et libérer le résidu glucosyle sous forme α. La distance entre les deux acides
carboxyliques est de 5,5 Ǻ (Mirza et al., 2001; Skov et al., 2002). De même que pour les
autres enzymes de la famille GH13 (Mosi et al., 1997; Uitdehaag et al., 1999; Yoshioka et al.,
1997), le troisième acide carboxylique interviendrait quant à lui dans la stabilisation de
l'intermédiaire covalent.
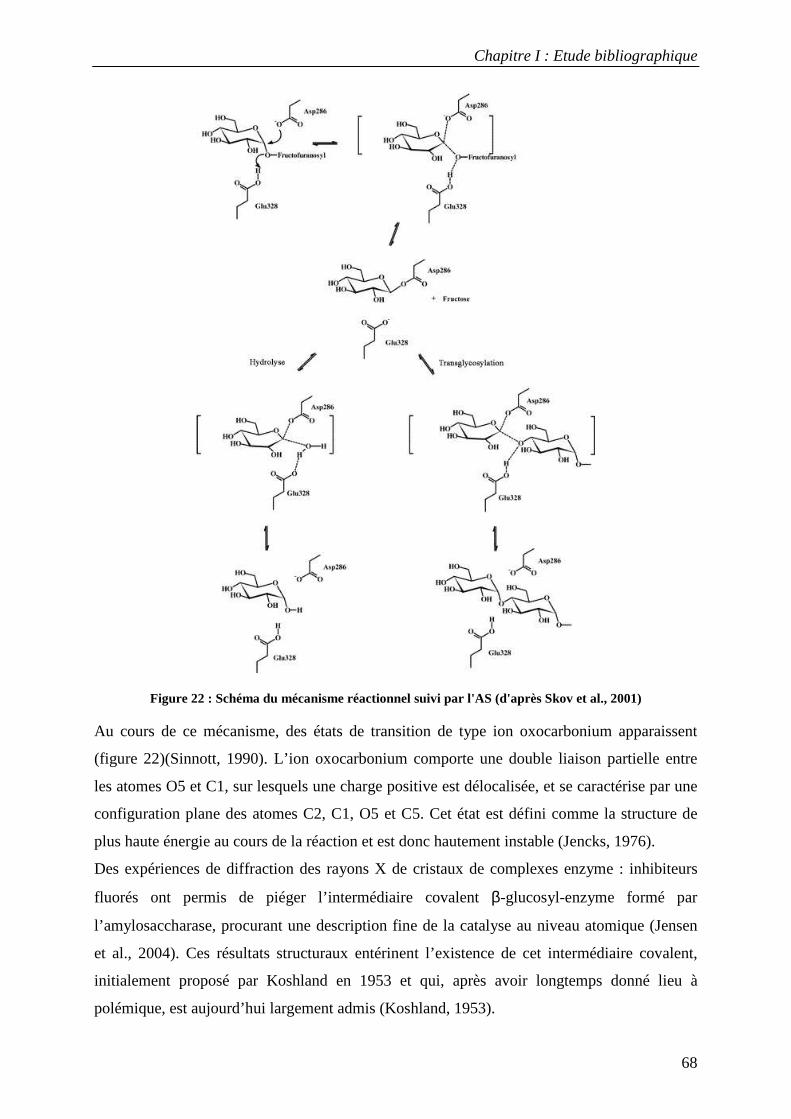
Chapitre I : Etude bibliographique
68
Figure 22 : Schéma du mécanisme réactionnel suivi par l'AS (d'après Skov et al., 2001)
Au cours de ce mécanisme, des états de transition de type ion oxocarbonium apparaissent
(figure 22)(Sinnott, 1990). L’ion oxocarbonium comporte une double liaison partielle entre
les atomes O5 et C1, sur lesquels une charge positive est délocalisée, et se caractérise par une
configuration plane des atomes C2, C1, O5 et C5. Cet état est défini comme la structure de
plus haute énergie au cours de la réaction et est donc hautement instable (Jencks, 1976).
Des expériences de diffraction des rayons X de cristaux de complexes enzyme : inhibiteurs
fluorés ont permis de piéger l’intermédiaire covalent β-glucosyl-enzyme formé par
l’amylosaccharase, procurant une description fine de la catalyse au niveau atomique (Jensen
et al., 2004). Ces résultats structuraux entérinent l’existence de cet intermédiaire covalent,
initialement proposé par Koshland en 1953 et qui, après avoir longtemps donné lieu à
polémique, est aujourd’hui largement admis (Koshland, 1953).

Chapitre I : Etude bibliographique
69
2.3 Facteurs structuraux responsables de la spécificité catalytique
L'arrimage d'un ligand à la surface d'une protéine ou à l'intérieur d'une crevasse catalytique
repose sur une complémentarité stéréospécifique et électronique mettant en jeu deux
principaux types d'interactions : les liaisons hydrogène et les interactions de Van der Waals
(Quiocho, 1986). Dans le cas d'un substrat glucidique, un phénomène important participant à
son orientation est le "stacking" (empilement ou sandwich). Il s'agit de l’ensemble des forces
mises en jeu (van der Waals et hydrophobes) entre un cycle aromatique (chaîne latérale de
résidus tels que la tyrosine, la phenylalanine ou le tryptophane) et les atomes de carbone du
cycle de la molécule de sucre, dont les plans sont disposés parallèlement. Ces résidus
aromatiques, encombrants, constituent des points d’ancrage clé pour le substrat, permettant
son arrimage ou pré-arrimage ou son orientation vers le site catalytique. Les interactions
mises en jeu au niveau de chaque unité de sucre sont considérées isolément. On parle de sous-
sites de fixation, nomenclature utilisée principalement pour les enzymes de dépolymérisation
comme les glycoside-hydrolases mais aussi les protéinases et les nucléases (Davies et al.,
1997; Suganuma et al., 1978; Thoma et al., 1971). Un sous-site est formé d’un ou de plusieurs
résidus et contribue plus ou moins fortement à la fixation de la molécule entière. Le nombre
de sous-sites et leur énergie d’interaction peuvent se mesurer expérimentalement et sont
intimement liés aux propriétés catalytiques des enzymes et à leur spécificité de substrat.
Une nomenclature a été proposée par Davies et al. (1997) pour les sous-sites de fixation des
glycoside-hydrolases. Leur nombre peut varier de deux, dans le cas des monoglycosidases et
dissaccharidases, à dix dans le cas de certaines α−amylases. Dans tous les cas, ces sites de
fixation correspondent à l'arrimage du substrat au niveau du site actif, permettant la rupture de
la liaison osidique. Ils sont à distinguer des sites secondaires de fixation du substrat pouvant
être localisés à la surface de la protéine. Les sous-sites sont numérotés de –n à +n, -n
représentant la partie non-réductrice et +n la partie réductrice (Figure 23). La rupture de la
liaison osidique s’opère entre les positions –1 et +1.
Figure 23 : Représentation schématique des sous-sites de liaison de l'AS (d'après la nomenclature
proposée par Davies et al., 1997).

Chapitre I : Etude bibliographique
70
Dans le cas de l'AS, le pont salin formé par les résidus Asp144 et Arg509 est localisé en
amont du sous-site -1, rendant inexistant les sites -2 à –n. L'analyse des complexes E328Q
(mutant inactif) : saccharose (figure 24A) et AS : glucose (figure 24B)(Mirza et al., 2001)
associée à des travaux de mutagénèse dirigée (Albenne et al., 2002a) a permis de mettre en
évidence la spécificité de l'enzyme envers son substrat. Celle-ci est assurée par des éléments
du sous-site -1, à savoir le pont salin (Asp144 et Arg509) et un double "stacking" impliquant
deux résidus aromatiques (Tyr147 et Phe250). Le pont salin, conférant au site actif une
topologie en forme de poche, semble essentiel à l'arrimage du substrat. La mutation des
résidus Asp144 et Arg509 en alanine conduit à une perte quasi-totale d'activité (Albenne et
al., 2002a). Le double stacking renforce la stabilisation de l'intermédiaire covalent. Les deux
acides aminés aromatiques sont situés de part et d'autre du cycle pyranosyle, assurant un
maintien de type "sandwich". Le cycle fructosyle est faiblement maintenu au niveau du sous-
site +1. Les résidus Asp394 et Arg446 assurent l'essentiel des interactions avec ce résidu mais
ne contribuent pas de façon majeure à la spécificité envers le saccharose. En effet, la
fragilisation du réseau de liaisons hydrogène au niveau du sous-site +1 entraînée par la
mutation de Asp394 ou Arg446 en alanine affecte l'activité envers le saccharose mais ne la
détruit pas (Albenne et al., 2002a). Après formation de l'intermédiaire covalent glucosyl-
enzyme, le cycle fructosyle est probablement facilement exclu du site actif, laissant ainsi de la
place pour l'arrimage des molécules acceptrices.
Figure 24: Représentation schématique des interactions (A) entre le mutant inactif E328Q et le saccharose
et (B) entre l'AS et le D-Glucose (d'apres Mirza et al., 2001). Les liaisons hydrogène sont représentées en
pointillées et les * représentent les molécules d'eau.
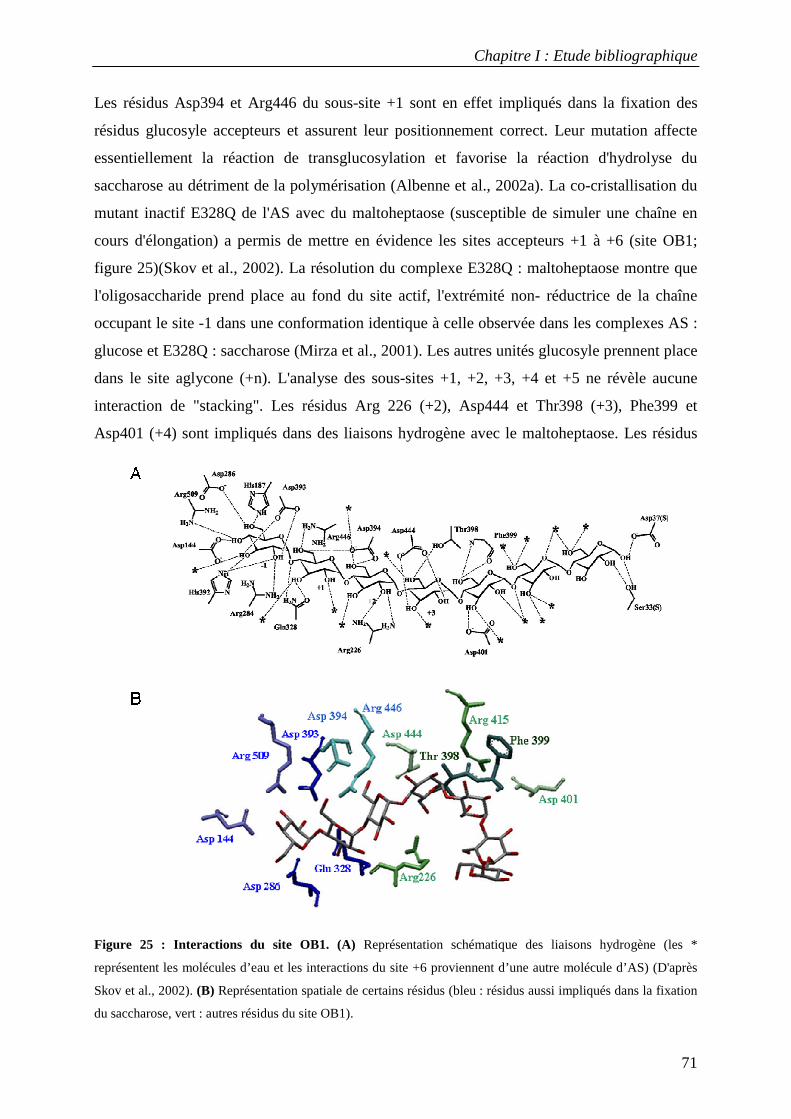
Chapitre I : Etude bibliographique
71
Les résidus Asp394 et Arg446 du sous-site +1 sont en effet impliqués dans la fixation des
résidus glucosyle accepteurs et assurent leur positionnement correct. Leur mutation affecte
essentiellement la réaction de transglucosylation et favorise la réaction d'hydrolyse du
saccharose au détriment de la polymérisation (Albenne et al., 2002a). La co-cristallisation du
mutant inactif E328Q de l'AS avec du maltoheptaose (susceptible de simuler une chaîne en
cours d'élongation) a permis de mettre en évidence les sites accepteurs +1 à +6 (site OB1;
figure 25)(Skov et al., 2002). La résolution du complexe E328Q : maltoheptaose montre que
l'oligosaccharide prend place au fond du site actif, l'extrémité non- réductrice de la chaîne
occupant le site -1 dans une conformation identique à celle observée dans les complexes AS :
glucose et E328Q : saccharose (Mirza et al., 2001). Les autres unités glucosyle prennent place
dans le site aglycone (+n). L'analyse des sous-sites +1, +2, +3, +4 et +5 ne révèle aucune
interaction de "stacking". Les résidus Arg 226 (+2), Asp444 et Thr398 (+3), Phe399 et
Asp401 (+4) sont impliqués dans des liaisons hydrogène avec le maltoheptaose. Les résidus
Figure 25 : Interactions du site OB1. (A) Représentation schématique des liaisons hydrogène (les *
représentent les molécules d’eau et les interactions du site +6 proviennent d’une autre molécule d’AS) (D'après
Skov et al., 2002). (B) Représentation spatiale de certains résidus (bleu : résidus aussi impliqués dans la fixation
du saccharose, vert : autres résidus du site OB1).

Chapitre I : Etude bibliographique
72
Figure 26 : Positionnement des sites OB1, OB2 et OB3. Deux molécules de maltheptaose occupent les sites
OB1 et OB2. Une molécule de maltotétraose occupe OB3. Le domaine B est coloré en orange, le domaine B' en
vert.
Asp394 et Arg446 du sous-site +1 jouent un rôle clé dans la réaction de transglucosylation,
étant responsables à la fois de la spécificité de produit et de substrat de l'AS (Albenne et al.,
2002a). Les sous-sites de +1 à +3 sont cependant considérés comme faibles, notamment du
fait de l'encombrement dû à Arg226. Le sous-site +4 serait en revanche considéré comme un
site d'ancrage fort pour les molécules acceptrices. L'arginine 415 assurant un soutien
hydrophobe au niveau de sous-site +4 pourrait être responsable de la sélectivité de la taille de
l'AS vis-à-vis des maltooligosaccharides. Une molécule doit donc être composée d'au moins
quatre cycles glucosyle pour jouer efficacement le rôle d'accepteur au moment de la réaction
de transfert. De façon remarquable, les résidus qui composent ces sous-sites accepteurs sont
situés dans le domaine B' qui est propre à l'AS. Par ailleurs, lors de la co-cristallisation du
mutant E328Q complexé avec des maltooligosaccharides, deux sites secondaires de fixation,
appelés OB2 et OB3, ont également été mis en évidence (figure 26). Ils sont localisés en
dehors du site actif et pourraient ainsi permettre un arrimage des chaînes d'amylose en cours
d'élongation ou promouvoir une fixation privilégiée des branchements du glycogène in vivo
(Albenne et al., 2007).

Chapitre I : Etude bibliographique
73
3. MODE DE FORMATION DU POLYMERE
3.1. Paramètres cinétiques
La production de l’AS et sa purification jusqu’à homogénéité a permis la caractérisation fine
des paramètres cinétiques. La vitesse de consommation du saccharose (au même titre que
l’évolution de la vitesse de formation du glucose et du polymère) par l'AS de N.
polysaccharea ne présente pas un profil Michaelien classique (Potocki de Montalk et al.,
2000b). Les auteurs ont proposé de modéliser le comportement de l’enzyme par deux
équations de Michaelis-Menten, l'une ou l'autre étant employée pour des concentrations
supérieures ou inférieures à une valeur seuil de la concentration en saccharose (20 mM). Ce
comportement atypique pourrait être dû à la présence d’un second site, non-catalytique, de
fixation du saccharose. Son occupation, aux fortes concentrations en saccharose, induirait des
modifications conformationnelles favorables à l’activité de l’enzyme, et en particulier à la
réaction de polymérisation. Les valeurs des constantes cinétiques apparentes sont présentées
dans le tableau 13. Elles révèlent que l’amylosacharase présente une faible efficacité
catalytique. Par ailleurs, une diminution de l'activité initiale a été observée pour des
concentrations en saccharose supérieures à 300 mM, ce qui peut être lié à une inhibition par
excès de substrat ou une limitation du transfert de matière dues à la viscosité du milieu
réactionnel (Albenne, 2002).
Le mécanisme de synthèse d'amylose a été élucidé grâce à l'étude de la structure
tridimensionnelle de l'AS et par l'analyse biochimique du suivi cinétique de la formation du
polymère (Albenne et al., 2004). L’étude des composés des fractions solubles et insolubles a
Tableau 13 : Valeurs des constantes cinétiques apparentes des vitesses initiales de consommation du
saccharose (ViS), d'hydrolyse du saccharose (ViG) et de polymérisation (ViGn) (d'après Potocki de
Montalk et al., 2000).

Chapitre I : Etude bibliographique
74
permis de révéler que la formation d’amylose résulte de l’élongation non-processive des
maltooligosaccharides produits par l’enzyme. La réaction de polymérisation est initiée par
l’hydrolyse du saccharose, qui permet de former du glucose, qui sert à son tour d’accepteur
pour former du maltose. Des réactions de transfert successives conduisant à la synthèse de
maltotriose puis de maltooligosaccharides de degré de polymérisation croissant se produisent
ensuite. L'orientation du maltoheptaose dans le site accepteur du complexe E328Q :
maltoheptaose montre que le transfert s'opère par l'extrémité non-réductrice de la molécule
acceptrice. Au délà d’une taille et d’une concentration critiques, certaines de ces chaînes
précipitent. La fraction insoluble est donc composée d’un mélange polydisperse de
maltooligosaccharides et d’amylose dont le DP moyen dépend de la concentration initiale en
saccharose. Les premiers produits formés (glucose, maltose et maltotriose) sont donc des
amorces nécessaires à la polymérisation. Ils sont les seuls à être accumulés dans le milieu. Les
maltooligosaccharides de taille supérieure sont, quant à eux, glucosylés beaucoup plus
efficacement, grâce notamment à la force d’arrimage du sous-site +4 (voir paragraphe "2.3
Facteurs structuraux responsables de la spécificité catalytique"). En absence de sacccharose,
les maltooligosaccharides de DP supérieur ou égal à 4 se comportent d’ailleurs comme des
donneurs d'unités glucosyle. Cette réaction n’intervient qu’après la consommation totale du
saccharose ou lorsque les maltooligosaccharides sont utilisés comme seul substrat. Ainsi, l'AS
coupe une liaison α-1,4 à l'extrémité non réductrice d'une molécule GN, libère un produit GN-1
et transfère le résidu glucosyle sur un autre GN pour former du GN+1 (Albenne et al., 2002b).
3.2 Structure et morphologie du polymère insoluble synthétisé
L’analyse de l’amylose insoluble synthétisée par l’AS par les techniques de chromatographie
d’exclusion de taille, de complexation de l’iode, de diffraction des rayons X et de microscopie
électronique à transmission et à balayage montre que la taille, la concentration et la
morphologie des chaînes d'amylose insoluble varient en fonction de la concentration en
saccharose initiale utilisée pendant la synthèse (tableau 14)(Potocki-Veronese et al., 2005).
Les chaînes les plus longues (synthétisées à partir de 100 mM saccharose initial) forment un
réseau semi-cristallin similaire à celui visualisé pour les gels d’amylose de plante, alors que
les chaînes plus courtes (synthétisées à partir de 300 ou 600 mM de saccharose initial)
s'organisent en agrégats poly-crystallins. L'étude par diffraction des rayons X de l’amylose
insoluble a permis de montrer qu'elle forme des cristaux de type B avec des degrés de
cristallinité allant de 45% (amylose formée à partir de 100 mM de saccharose) à 94%

Chapitre I : Etude bibliographique
75
(amylose formée à partir de 600 mM de saccharose) (tableau 14). La cristallinité des chaînes
peut être augmentée d'environ 30% après un traitement thermique (5 minutes à 90°C). De
manière remarquable, l’amylose synthétisée à partir de 600 mM de saccharose par l’AS et
traitée thermiquement est actuellement la forme la plus cristalline connue à ce jour. Cette très
forte organisation lui confère une extrême résistance aux enzymes digestives, quantifiée in
vitro à un niveau de 85 % d’amidon résistant.
Tableau 14 : Caractéristiques de l'amylose insoluble synthétisée par l'AS en fonction de la concentration
initiale en saccharose (d'après Potocki-Véronèse et al., 2005).
4. LES REACTIONS EN PRESENCE D’ACCEPTEURS EXOGENES
L'AS de N. polysaccharea est activée en présence d'amorces amylopolysaccharidiques.
Remaud-Siméon et al. (1995) ont, les premiers, démontré l'effet activateur du maltopentaose,
du maltoheptaose, de l'amidon et du glycogène, le fort pouvoir activateur de ce dernier
pouvant être corrélé au nombre d'extrémités de chaînes accessibles. Ces observations ont été
confirmées par la suite et les produits formés caractérisés (Potocki de Montalk et al., 2000a;
Potocki de Montalk et al., 1999; Putaux et al., 2006; Rolland-Sabaté et al., 2004).
4.1 Réactions d'accepteur sur le glucose et le maltose
Le glucose et le maltose ont été testés isolément comme accepteur à une concentration
équimolaire à celle du saccharose (50 mM). L'AS transfère des unités glucosyle sur ces
molécules au détriment de la polymérisation (Potocki de Montalk, 1999). Dans les deux
conditions, le produit formé majoritairement est celui résultant du transfert d'une seule unité
glucosyle sur la molécule acceptrice. Des transferts consécutifs ont ensuite lieu mais avec un
rendement plus faible. En présence de glucose et de maltose, l'activité initiale de l'AS est
augmentée respectivement d'un facteur 7,5 et 6,4.

Chapitre I : Etude bibliographique
76
4.2 Réaction d'accepteur sur les polysaccharides
L’amylosaccharase peut aussi être utilisée pour la modification de polysaccharides variés.
Rolland-Sabaté et al. (2004) ont montré qu’elle glucosyle des glucanes contenant des liaisons
α-1,4 et α-1,6 (glycogène, amylopectine, limite-dextrines), en rallongeant leurs ramifications.
La teneur en amidon résistant des polysaccharides accepteurs est ainsi augmentée, et peut être
modulée en fonction de la structure de l’accepteur et du rapport donneur/accepteur utilisé pour
la glucosylation. Dans certains cas, l’élongation permet de conférer aux polymères de
nouvelles propriétés gélifiantes. C’est ce qui est observé lors de la glucosylation de
l’amylopectine (Rolland-Sabaté et al., 2004).
Un intérêt particulier a été porté à la glucosylation du glycogène. En effet, les bactéries du
genre Neisseria synthétisent un polymère de type glycogène selon une voie de biosynthèse
dans laquelle sont probablement impliquées les amylosaccharases (Buttcher et al., 1999). La
caractérisation de cette réaction et des produits de la glucosylation sont décrits ci-après.
4.2.1 Caractérisation des produits synthétisés à partir de saccharose et de glycogène
L’étude des réactions catalysées en présence de 100 mM saccharose et de concentrations
croissantes en glycogène a révélé la présence de glucose et d’oligosaccharides dans le milieu,
uniquement lorsque la concentration en glycogène n’excède pas 0,5 g/l. Dans ces conditions,
une partie du saccharose est utilisée pour former du glucose, des oligosaccharides et des
chaînes linéaires d’amylose de manière similaire à une réaction en présence de saccharose
seul. Le reste des résidus glucosyle issus du saccharose sont transférés sur le glycogène
accepteur (Potocki de Montalk et al., 2000a). Lorsque la concentration en glycogène
augmente, l’incorporation des résidus glucosyle dans le polymère est favorisée, au détriment
des autres réactions.
Le polymère insoluble synthétisé par l'AS en présence de 50 g/l de saccharose et de 1 g/l de
glycogène a été caractérisé par chromatographie d’exclusion de taille couplée à la diffusion de
la lumière, par déramification enzymatique et par complexation de l’iode (Potocki de Montalk
et al., 1999). La combinaison des méthodes utilisées a permis de conclure que l’AS rallonge
certaines ramifications du glycogène, par leur extrémité non-réductrice, de 12 à 75 unités
glucosyle. Cette réaction de rallongement a été analysée par cryo-microscopie électronique à
transmission (Putaux et al., 2006). Le diamètre des particules de glycogène augmente de 30 à
150 nm au cours de la réaction effectuée à partir de 100 mM de saccharose et 0.1 g/l de
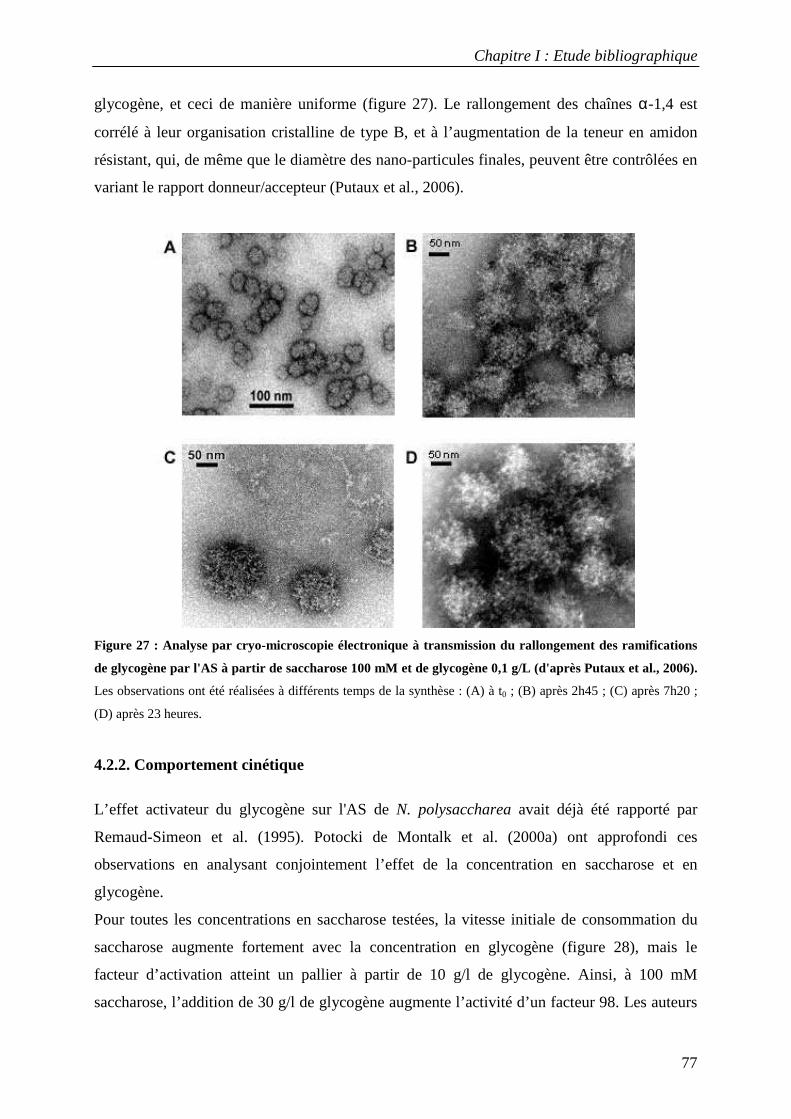
Chapitre I : Etude bibliographique
77
glycogène, et ceci de manière uniforme (figure 27). Le rallongement des chaînes α-1,4 est
corrélé à leur organisation cristalline de type B, et à l’augmentation de la teneur en amidon
résistant, qui, de même que le diamètre des nano-particules finales, peuvent être contrôlées en
variant le rapport donneur/accepteur (Putaux et al., 2006).
Figure 27 : Analyse par cryo-microscopie électronique à transmission du rallongement des ramifications
de glycogène par l'AS à partir de saccharose 100 mM et de glycogène 0,1 g/L (d'après Putaux et al., 2006).
Les observations ont été réalisées à différents temps de la synthèse : (A) à t0 ; (B) après 2h45 ; (C) après 7h20 ;
(D) après 23 heures.
4.2.2. Comportement cinétique
L’effet activateur du glycogène sur l'AS de N. polysaccharea avait déjà été rapporté par
Remaud-Simeon et al. (1995). Potocki de Montalk et al. (2000a) ont approfondi ces
observations en analysant conjointement l’effet de la concentration en saccharose et en
glycogène.
Pour toutes les concentrations en saccharose testées, la vitesse initiale de consommation du
saccharose augmente fortement avec la concentration en glycogène (figure 28), mais le
facteur d’activation atteint un pallier à partir de 10 g/l de glycogène. Ainsi, à 100 mM
saccharose, l’addition de 30 g/l de glycogène augmente l’activité d’un facteur 98. Les auteurs
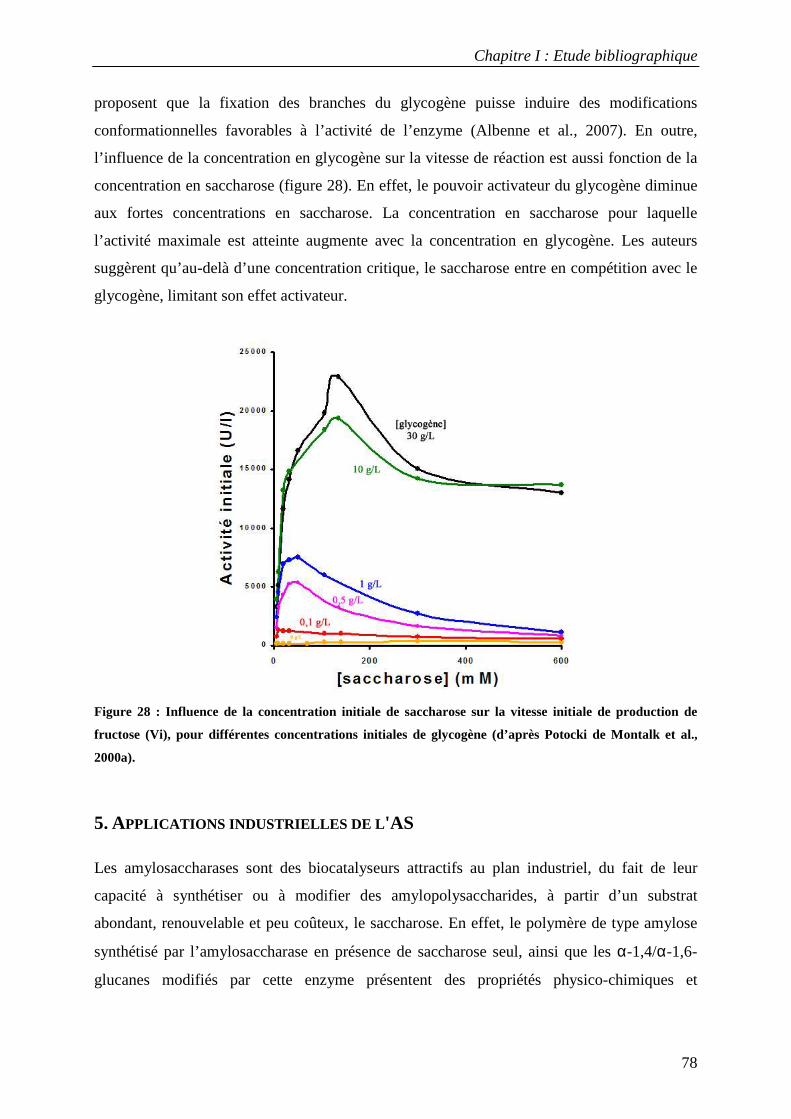
Chapitre I : Etude bibliographique
78
proposent que la fixation des branches du glycogène puisse induire des modifications
conformationnelles favorables à l’activité de l’enzyme (Albenne et al., 2007). En outre,
l’influence de la concentration en glycogène sur la vitesse de réaction est aussi fonction de la
concentration en saccharose (figure 28). En effet, le pouvoir activateur du glycogène diminue
aux fortes concentrations en saccharose. La concentration en saccharose pour laquelle
l’activité maximale est atteinte augmente avec la concentration en glycogène. Les auteurs
suggèrent qu’au-delà d’une concentration critique, le saccharose entre en compétition avec le
glycogène, limitant son effet activateur.
Figure 28 : Influence de la concentration initiale de saccharose sur la vitesse initiale de production de
fructose (Vi), pour différentes concentrations initiales de glycogène (d’après Potocki de Montalk et al.,
2000a).
5. APPLICATIONS INDUSTRIELLES DE L 'AS
Les amylosaccharases sont des biocatalyseurs attractifs au plan industriel, du fait de leur
capacité à synthétiser ou à modifier des amylopolysaccharides, à partir d’un substrat
abondant, renouvelable et peu coûteux, le saccharose. En effet, le polymère de type amylose
synthétisé par l’amylosaccharase en présence de saccharose seul, ainsi que les α-1,4/α-1,6-
glucanes modifiés par cette enzyme présentent des propriétés physico-chimiques et

Chapitre I : Etude bibliographique
79
biologiques remarquables qui permettent d’envisager des applications dans de nombreux
secteurs industriels.
Dans l’agro-alimentaire les amidons riches en amylose sont actuellement utilisés comme
texturants. Les propriétés gélifiantes des amylopectines ou du glycogène modifiés par l’AS
pourraient élargir la gamme des produits proposés. Une autre application possible dans ce
secteur concerne le domaine en fort développement des aliments fonctionnels. En effet,
l’amidon résistant n’est pas hydrolysé par les enzymes de l’estomac et de l’intestin grêle. Des
études réalisées chez le rat et le porc montrent qu’il arrive intact au niveau du gros intestin, où
il est utilisé comme substrat des bactéries fermentaires pour produire de l’hydrogène, du
dioxyde de carbone, du méthane, des acides organiques (comme l’acide lactique) et des acides
gras à courtes chaînes. L’amidon résistant joue donc le rôle de fibre alimentaire. De plus, il est
considéré comme prébiotique puisqu’il stimule dans le colon la croissance de bactéries
bénéfiques à l’organisme. Chez l’homme, les effets physiologiques potentiels sont la
prévention du diabète de type II, du cancer du colon, des maladies cardio-vasculaires et de
l’hypercholestérolémie, ainsi que le traitement de diarrhées infectieuses (Nugent, 2005). Les
amyloses sont aussi utilisées comme vecteur d’inclusion de molécules hydrophobes (arômes,
etc.)(Buleon and Colonna, 2007). En effet, la structure hélicoïdale de l’amylose définit un
domaine hydrophobe au coeur de la molécule capable d’inclure des molécules de même
nature.
Ces applications sont encore peu développées en raison du coût de production de l’amylose
obtenue par extraction à partir d’amidon. Dans ce contexte, la synthèse d’amylose à partir de
saccharose catalysée par l’AS pourrait concurrencer la voie de production actuelle. L’intérêt
potentiel de cette approche est confirmé par les nombreux brevets déposés depuis une
quinzaine d'années mentionnant l’utilisation de l’amylosaccharase recombinante de N.
polysaccharea en tant qu’outil de biosynthèse enzymatique (tableau 15).
Le deuxième secteur concerné est celui des semences. Les amylosaccharases sont des outils
de glucosylation performants pour la modification de la structure de polysaccharides ramifiés
telles que l’amylopectine. La modification in vivo de ce glucane, ainsi que l’augmentation du
rapport amylose / amylopectine de l’amidon synthétisé in vivo, constitue un enjeu pour la
production de nouveaux polymères végétaux (maïs, pommes de terre, tomates, ..) aux
propriétés nutritionnelles ou à la texture améliorées. Ainsi, la société Planttec Biotechnologie
a déposé un brevet décrivant l’introduction, dans des végétaux, du gène de l’AS couplé à celui
d’une enzyme de branchement. Les plantes transgéniques obtenues synthétisent de l’amidon
Chapitre I : Etude bibliographique
80
avec un rapport amylose / amylopectine différent de la plante sauvage, améliorant la qualité
des végétaux produits (Quanz, 2000).
Les applications envisagées pour l’AS sont donc nombreuses mais il apparaît qu’à ce jour
aucune valorisation industrielle n’ait encore été développée. Ceci est dû à la faible efficacité
catalytique de cette enzyme et à sa faible stabilité à la température. Ce constat est à l’origine
des travaux d’ingénierie.
Tableau 15 : Brevets déposés concernant une application des amylosaccharases.
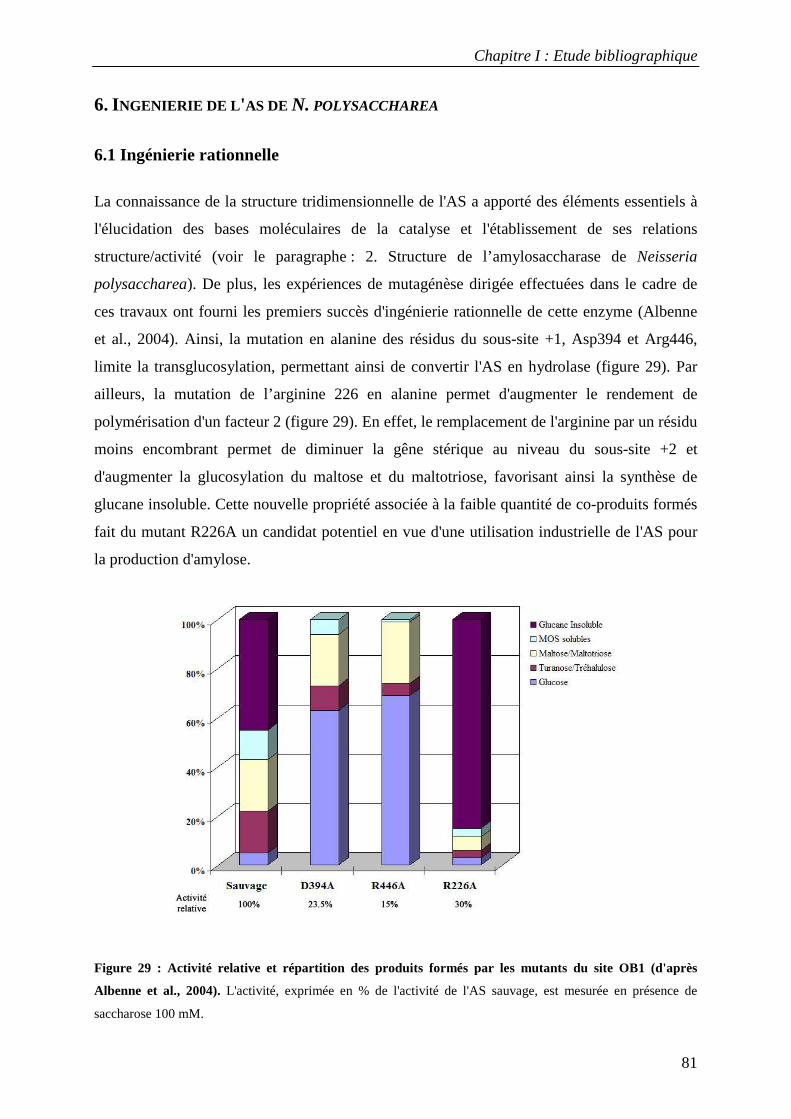
Chapitre I : Etude bibliographique
81
6. INGENIERIE DE L 'AS DE N. POLYSACCHAREA
6.1 Ingénierie rationnelle
La connaissance de la structure tridimensionnelle de l'AS a apporté des éléments essentiels à
l'élucidation des bases moléculaires de la catalyse et l'établissement de ses relations
structure/activité (voir le paragraphe : 2. Structure de l’amylosaccharase de Neisseria
polysaccharea). De plus, les expériences de mutagénèse dirigée effectuées dans le cadre de
ces travaux ont fourni les premiers succès d'ingénierie rationnelle de cette enzyme (Albenne
et al., 2004). Ainsi, la mutation en alanine des résidus du sous-site +1, Asp394 et Arg446,
limite la transglucosylation, permettant ainsi de convertir l'AS en hydrolase (figure 29). Par
ailleurs, la mutation de l’arginine 226 en alanine permet d'augmenter le rendement de
polymérisation d'un facteur 2 (figure 29). En effet, le remplacement de l'arginine par un résidu
moins encombrant permet de diminuer la gêne stérique au niveau du sous-site +2 et
d'augmenter la glucosylation du maltose et du maltotriose, favorisant ainsi la synthèse de
glucane insoluble. Cette nouvelle propriété associée à la faible quantité de co-produits formés
fait du mutant R226A un candidat potentiel en vue d'une utilisation industrielle de l'AS pour
la production d'amylose.
Figure 29 : Activité relative et répartition des produits formés par les mutants du site OB1 (d'après
Albenne et al., 2004). L'activité, exprimée en % de l'activité de l'AS sauvage, est mesurée en présence de
saccharose 100 mM.

Chapitre I : Etude bibliographique
82
6.2 Ingénierie combinatoire
L'analyse biochimique et structurale de l'AS a ouvert la voie à l'ingénierie rationnelle de cette
enzyme. Cependant, les potentialités d'application de l'AS nécessitent une amélioration de son
faible niveau d'activité catalytique (kcat = 1 s-1), de sa thermostabilité (t1/2 = 21 heures à 30°C)
et de sa résistance à la dénaturation par les solvants organiques. La modification de telles
propriétés résultent souvent des effets combinés de multiples mutations difficiles à prédire par
une approche rationnelle. L'évolution dirigée de l'AS a donc été entreprise afin d'identifier des
variants plus performants et adaptés aux contraintes industrielles.
6.2.1 Génération d'une banque de première génération de l'AS
Une banque de première génération de l'AS a été construite selon deux étapes (van der Veen
et al., 2004). Dans un premier temps, le gène de l'AS a été muté aléatoirement par des
réactions de PCR à erreurs utilisant la Taq polymérase ou le kit Mutazyme (Stratagene). Dans
un second temps, les produits d'amplification obtenus ont été fragmentés par digestion par des
enzymes de restriction, puis reassemblés par DNA shuffling. Après clonage dans un vecteur
d'expression et transformation dans E. coli, une librairie d'environ 50000 variants a été
obtenue.
Le vecteur employé pour le clonage des banques de variants de l'AS est le plasmide pZErO
commercialisé par la société Invitrogen. L'insertion d'une séquence au niveau du site multiple
de clonage de ce vecteur abolit la fusion de lacZ avec le gène ccdB (codant une toxine qui
entraîne la mort de la cellule en inhibant l'ADN gyrase). L'utilisation de ce système permet
une sélection positive des clones bactériens transformés par un plasmide contenant une copie
mutée du gène de l'AS.
6.2.2 Mise au point de protocoles de sélection et de criblage
Une méthode de sélection, basée sur la capacité des bactéries produisant une AS active à se
développer sur milieu minimum supplémenté en saccharose, a été mise au point (figure
30A)(van der Veen et al., 2004). Les souches E. coli de type K12 (la plupart de celles
usuellement employées en génie génétique) ne métabolisent pas le saccharose et ne sont pas
capables de croître sur un milieu minimum supplémenté par ce seul sucre. A l'inverse, les

Chapitre I : Etude bibliographique
83
cellules produisant une AS active acquièrent la capacité à se développer sur ce milieu, du fait
de libération de fructose associée à l'activité de l'enzyme. Cette technique de sélection
constitue une excellente stratégie pour réduire la taille des banques en ne conservant que les
variant actifs, lesquels pourront ensuite être criblés pour la propriété choisie. Ce test ne
permet pas de différencier les mutants les plus actifs de ceux dont l’expression a été
améliorée. De plus, les clones sélectionnés peuvent exprimer des variants de type hydrolase
ayant perdu l’activité polymérase parentale. Néanmoins, l’exposition des clones cultivés sur
ce milieu sélectif à la vapeur d’iode permet d’identifier ceux ayant produit un polymère de
type amylose (capable de complexer l’iode). Ces colonies sont alors colorées en bleu.
Les colonies exprimant une AS active sont mises en culture au format microplaque. Après
réaction enzymatique, l'activité de l'AS peut être révélée facilement par dosage colorimétrique
des produits réactionnels (figure 30B). La formation de sucres réducteurs peut être quantifiée
par la méthode au DNS (Sumner and Howell, 1935). La production de polymère d'amylose est
détectée par une coloration à l'iode qui permet en outre d'évaluer la taille moyenne des
chaînes produites en mesurant la longueur d’onde d’absorption maximale du complexe
amylose-iode (Banks et al., 1971). Ces méthodes, usuellement employées à l'échelle du
laboratoire pour la caractérisation de l'AS, ont été miniaturisées au format microplaque pour
la recherche de variants plus actifs et plus stables (van der Veen et al., 2004).
Figure 30 : Méthodes de sélection et de criblage de banques de variants de l'AS (d'après Van der Veen et
al., 2004). (A) Sélection positive de colonies productrices d'AS sur milieu sélectif. (1) Variant inactif, (2)
Variant actif à 25% et (3) AS sauvage. (B) Criblage colorimétrique de l'activité amylosaccharase. (1) Analyse de
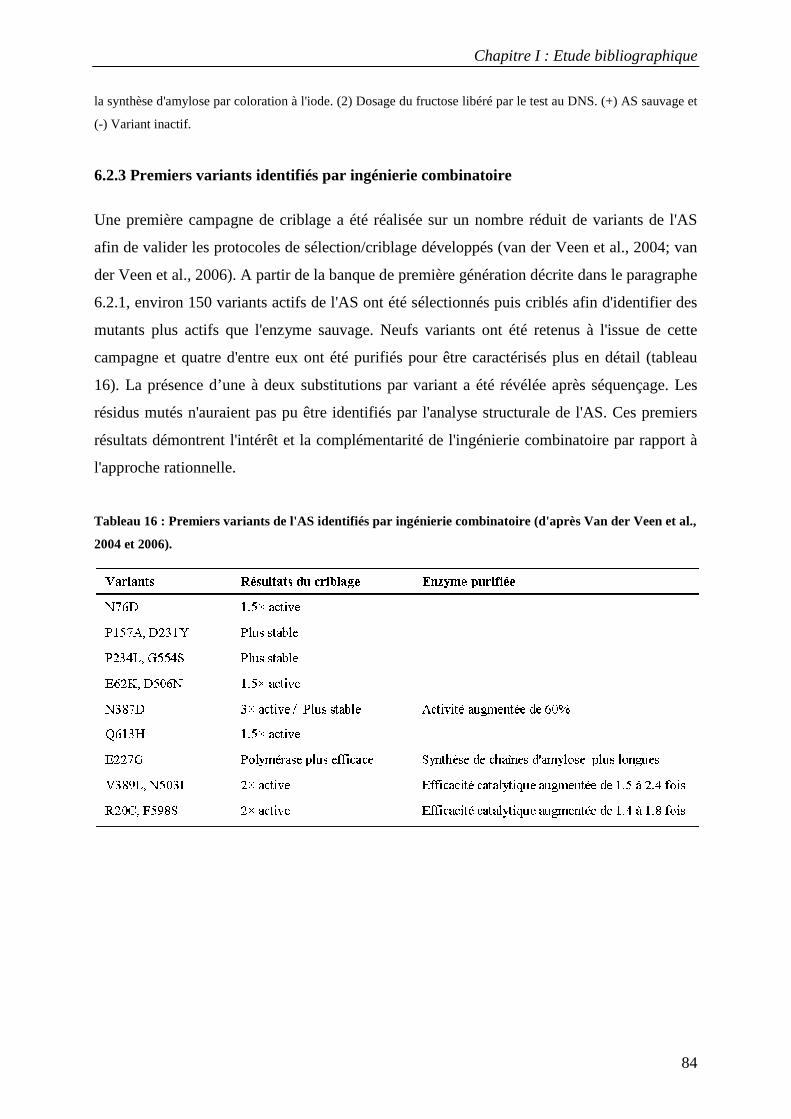
Chapitre I : Etude bibliographique
84
la synthèse d'amylose par coloration à l'iode. (2) Dosage du fructose libéré par le test au DNS. (+) AS sauvage et
(-) Variant inactif.
6.2.3 Premiers variants identifiés par ingénierie combinatoire
Une première campagne de criblage a été réalisée sur un nombre réduit de variants de l'AS
afin de valider les protocoles de sélection/criblage développés (van der Veen et al., 2004; van
der Veen et al., 2006). A partir de la banque de première génération décrite dans le paragraphe
6.2.1, environ 150 variants actifs de l'AS ont été sélectionnés puis criblés afin d'identifier des
mutants plus actifs que l'enzyme sauvage. Neufs variants ont été retenus à l'issue de cette
campagne et quatre d'entre eux ont été purifiés pour être caractérisés plus en détail (tableau
16). La présence d’une à deux substitutions par variant a été révélée après séquençage. Les
résidus mutés n'auraient pas pu être identifiés par l'analyse structurale de l'AS. Ces premiers
résultats démontrent l'intérêt et la complémentarité de l'ingénierie combinatoire par rapport à
l'approche rationnelle.
Tableau 16 : Premiers variants de l'AS identifiés par ingénierie combinatoire (d'après Van der Veen et al.,
2004 et 2006).

85
Chapitre II : Construction de banques de
variants de l'amylosaccharase par la
technique MutaGen
Ce chapitre a été publié dans Protein Engineering, Design and Selection.
A novel random mutagenesis process using human mutagenic DNA polymerases to generate enzyme variant libraries. Emond S., Mondon P., Pizzut-Serin S., Douchy L., Crozet F., Bouayadi K., Kharrat H., Potocki-Véronèse G., Monsan P. and Remaud-Siméon M. (2008). Protein Engineering, Design and Selection, 21(4), pp. 267-274.

Chapitre II : Construction de banques de variants de l'amylosaccharase
86
A novel random mutagenesis approach using human mutagenic DNA
polymerases to generate enzyme variant libraries
1. SUMMARY
The in vitro MutaGenTM procedure is a new random mutagenesis method based on the use of
low-fidelity DNA polymerases. In the present study, this technique was applied on a 2 kb
gene encoding amylosucrase, an attractive enzyme for the industrial synthesis of amylose-like
polymers. Mutations were first introduced during a single replicating step performed by
mutagenic polymerases pol β and pol η. Three large libraries (>105 independent clones) were
generated (one with pol β and two with pol η). The sequence analysis of randomly chosen
clones confirmed the potential of this strategy for the generation of diversity. Variants
generated by pol β were four to seven-fold less mutated than those created with pol η,
indicating that our approach enables mutation rate control following the DNA polymerase
employed for mutagenesis. Moreover, pol β and pol η provide different and complementary
mutation spectra, allowing a wider sequence space exploration than error-prone PCR
protocols employing Taq polymerase. Interestingly, some of the variants generated by pol η
displayed unusual modifications, including combinations of base substitutions and codon
deletions which are rarely generated using other methods. By taking advantage of the
mutation bias of naturally highly error-prone DNA polymerases, MutaGenTM thus appears as
a very useful tool for gene and protein randomization.
2. INTRODUCTION
Over the last two decades, directed evolution has become a powerful approach for tailoring
protein features to industrial needs and probing structure-activity relationships (Yuan et al.,
2005). Inspired by the Darwinian concept of natural evolution, this methodology consists in
constructing libraries of variant genes followed by screening and/or selection of their protein
products. In vitro creation of libraries enables the exploration of sequence space
independently from a selective pressure under physiological conditions and thus allows the
search for variants adapted to non-natural and harsh environment close to those found in

Chapitre II : Construction de banques de variants de l'amylosaccharase
87
industrial processes. One of the most effective strategies for library construction is to
randomly mutate the gene encoding the parental protein. This approach requires no structural
or mechanistic information and can uncover unexpected beneficial mutations. Several in vitro
or in vivo random mutagenesis methods have been described so far (Wong et al., 2006).
Among these, error-prone PCR (epPCR) techniques are the most popular due to their
simplicity (Cadwell and Joyce, 1992; Cline and Hogrefe, 2000; Leung et al., 1989; Lin-
Goerke et al., 1997; Shafikhani et al., 1997; Zaccolo et al., 1996).
DNA polymerases catalyse the template-directed incorporation of deoxynucleotides into a
growing primer terminus to generate a new complementary daughter strand. They have been
classified on the basis of their structural similarities into seven families. Among them, family
A, B, C, D and Y share a common structural organization that can be described as a right hand
with three distinct domains referred to as “palm”, “fingers” and “thumb” (Beard and Wilson,
2003). The active site is located within the palm domain. The fingers and thumb domains are
important for dNTP recognition/binding and correct positioning of the DNA substrate,
respectively. Crystallographic studies of Y family DNA polymerases (Ling et al., 2001;
Silvian et al., 2001; Trincao et al., 2001; Zhou et al., 2001) have revealed that these enzymes
differ from the other polymerases in having rather small fingers and thumb domains, which
lead to an open and solvent accessible active site in the palm domain. Moreover, Y family
polymerases possess an additional domain referred to as “little finger”, which determines the
catalytic efficiency and mutation spectra of each Y family member by influencing enzyme–
substrate interactions (Boudsocq et al., 2004). Beyond this common structural framework,
DNA polymerases from family X are described as “left-handed” because their palm domain is
not homologous with those of the other polymerase families (Beard and Wilson, 2003).
The fidelity of DNA polymerases, defined as the inverse of misinsertion frequency, is ensured
by base pair complementarity, by substrate-induced conformational changes and, in some
cases, by proofreading catalyzed by a 3’�5’ exonuclease domain (Kunkel, 2004). Such
fidelities span a wide range. Replicative DNA polymerases from structural families A, B and
C are high fidelity enzymes exhibiting error frequencies of around 10-6 (one error per million
nucleotides incorporated). The most commonly used polymerase in epPCR methods, Taq
DNA polymerase (Taq pol) from family A, displays an error rate that ranges between 10-4 and
10-5 (Eckert and Kunkel, 1990; Tindall and Kunkel, 1988). The least accurate DNA
polymerases belong to the X and Y families (Moon et al., 2007; Prakash et al., 2005). These
enzymes are involved in DNA damage repair mechanisms. Mammalian DNA polymerase β
(pol β), which is implicated in the base excision repair (BER) pathway (Matsumoto and Kim,

Chapitre II : Construction de banques de variants de l'amylosaccharase
88
1995), is the best known member of the X-family and exhibits an error frequency ranging
from 10-3 to 10-4 (Kunkel, 1985; Osheroff et al., 1999). Y-family polymerases, also known as
translesion synthesis (TLS) polymerases, replicate DNA in a distributive manner and lack any
exonucleolytic activity for proofreading (Kunkel, 2004). These enzymes display the highest
error rates measured among DNA polymerases (10-1 - 10-3)(Kunkel, 2004). In human, known
members of this family are DNA polymerases η (pol η), ι or κ (Kunkel et al., 2003).
The hypermutation profile of these repair DNA polymerases has been used to design a new in
vitro random mutagenesis technique, MutaGenTM (Bouayadi et al., 2002). This method
consists of a first single mutagenic replication step catalysed by a low-fidelity human DNA
polymerase, which is followed by a selective PCR amplification of the replicated mutated
sequences (figure 31). MutaGenTM was recently reported as an efficient method to enlarge the
initial diversity of human fragment antibody libraries (Mondon et al., 2007b). It has however
never been used on templates longer than 700 base pairs (bp). Here we report the use of the
MutaGenTM process either with human pol β or pol η to produce random variants of
amylosucrase from Neisseria polysaccharea (AS). This enzyme encoded by a 2 kb gene is an
attractive biocatalyst for the in vitro synthesis of amylose-like polymers using sucrose as sole
substrate (Potocki de Montalk et al., 1999). Three large amylosucrase variant libraries were
constructed using MutaGenTM. The generated diversity was analysed from sequencing results
of clones randomly chosen in the libraries. The advantages of this novel approach are
discussed on the basis of the provided results.
3. RESULTS AND DISCUSSION
3.1 Construction of AS variant libraries using pol ββββ and pol ηηηη
The MutaGenTM technique was applied to generate three libraries (named A, B and C) using
wild-type as gene as parental template. Libraries A and B are full-length gene libraries
constructed using pol β and pol η respectively. The initial procedure illustrated in figure 31
was modified to overcome pol β low processivity (Osheroff et al., 1999). As shown in figure
32, two parts of the as gene bearing an overlapping region were first replicated by pol β
during separate reactions. This step was performed in the presence of unbalanced dNTPs
concentrations to increase the polymerase in vitro error rate (Sinha and Haimes, 1981). The
replication products were then amplified by PCR and reassembled into a full-length gene

Chapitre II : Construction de banques de variants de l'amylosaccharase
89
during a final overlap-extension PCR step (figure 32). Conversely, the higher processivity of
pol η (Bebenek et al., 2001) allowed replicating the full-length as sequence during a single
reaction to generate library B (figure 31). Library C was constructed using pol η to focus the
introduction of mutations into a region of AS catalytic domain (from residues 242 to 470) that
includes many critical residues for catalytic activity and substrate specificity (Albenne et al.,
2004; Sarcabal et al., 2000; Skov et al., 2002). The cloning of the amplified replication
products into the pGEX vector resulted in libraries A, B and C of 8×105, 1×105 and 1×.105
independent clones, respectively. Prior to any selection, several clones from each library were
randomly chosen and sequenced in order to analyse the diversity generated by the MutaGenTM
approach (table 17).
Figure 31 : The MutaGenTM process. MutaGenTM consists of a first single mutagenic replication step followed
by selective PCR amplification of the replication products. This technique uses primers possessing a 5’ extension
(coloured in red) that is not complementary to the template. After template denaturation and primers
hybridisation, replication is carried out by a low-fidelity DNA polymerase. The mutated copies (coloured in
green) are then selectively amplified by a PCR procedure consisting of a first cycle with a low hybridisation
temperature followed by selective cycles (up to 25) with high hybridisation temperature disabling template
amplification. This methodology was applied to construct AS variant libraries B and C using pol η.
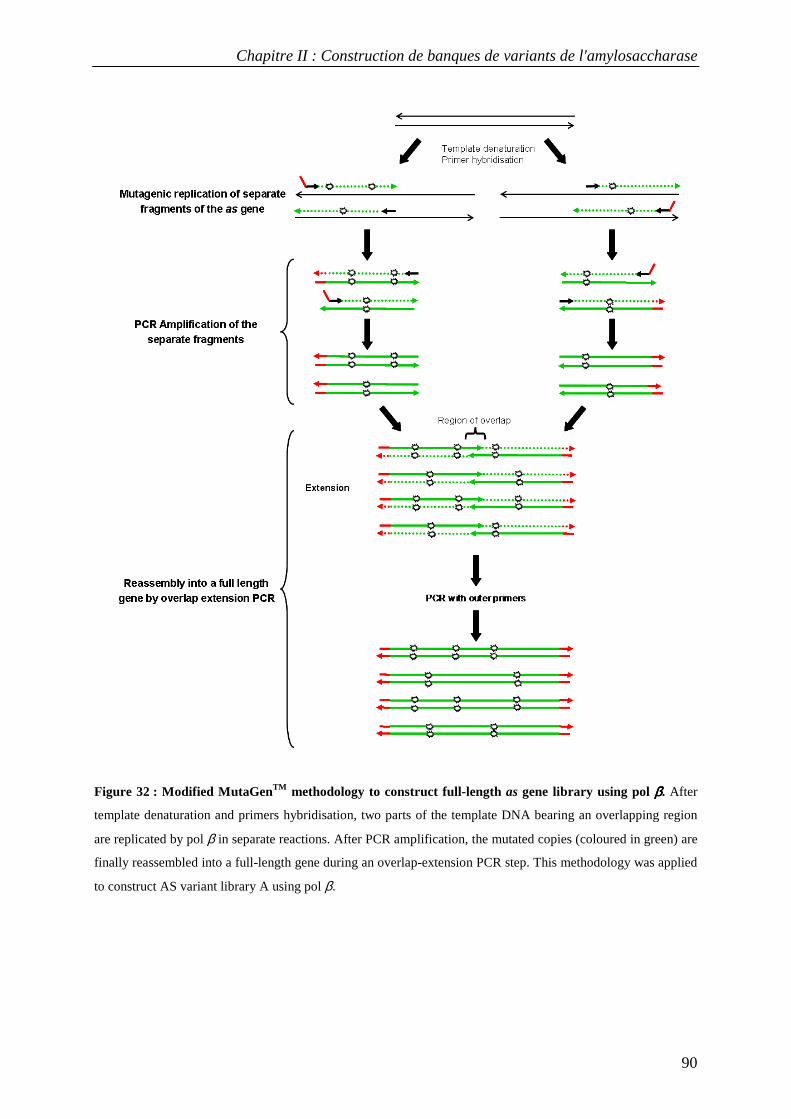
Chapitre II : Construction de banques de variants de l'amylosaccharase
90
Figure 32 : Modified MutaGenTM methodology to construct full-length as gene library using pol ββββ. After
template denaturation and primers hybridisation, two parts of the template DNA bearing an overlapping region
are replicated by pol β in separate reactions. After PCR amplification, the mutated copies (coloured in green) are
finally reassembled into a full-length gene during an overlap-extension PCR step. This methodology was applied
to construct AS variant library A using pol β.
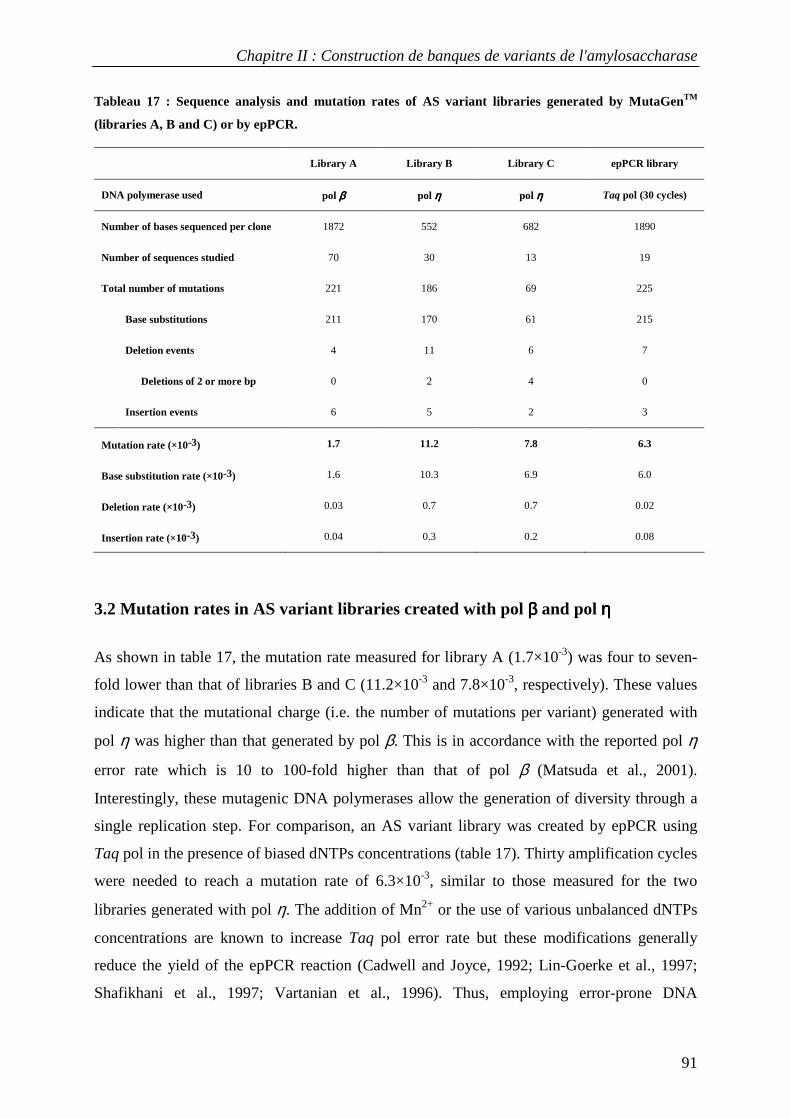
Chapitre II : Construction de banques de variants de l'amylosaccharase
91
Tableau 17 : Sequence analysis and mutation rates of AS variant libraries generated by MutaGenTM
(libraries A, B and C) or by epPCR.
Library A Library B Library C epPCR library
DNA polymerase used pol ββββ pol ηηηη pol ηηηη Taq pol (30 cycles)
Number of bases sequenced per clone 1872 552 682 1890
Number of sequences studied 70 30 13 19
Total number of mutations 221 186 69 225
Base substitutions 211 170 61 215
Deletion events 4 11 6 7
Deletions of 2 or more bp 0 2 4 0
Insertion events 6 5 2 3
Mutation rate (×10-3) 1.7 11.2 7.8 6.3
Base substitution rate (×10-3) 1.6 10.3 6.9 6.0
Deletion rate (×10-3) 0.03 0.7 0.7 0.02
Insertion rate (×10-3) 0.04 0.3 0.2 0.08
3.2 Mutation rates in AS variant libraries created with pol ββββ and pol ηηηη
As shown in table 17, the mutation rate measured for library A (1.7×10-3) was four to seven-
fold lower than that of libraries B and C (11.2×10-3 and 7.8×10-3, respectively). These values
indicate that the mutational charge (i.e. the number of mutations per variant) generated with
pol η was higher than that generated by pol β. This is in accordance with the reported pol η
error rate which is 10 to 100-fold higher than that of pol β (Matsuda et al., 2001).
Interestingly, these mutagenic DNA polymerases allow the generation of diversity through a
single replication step. For comparison, an AS variant library was created by epPCR using
Taq pol in the presence of biased dNTPs concentrations (table 17). Thirty amplification cycles
were needed to reach a mutation rate of 6.3×10-3, similar to those measured for the two
libraries generated with pol η. The addition of Mn2+ or the use of various unbalanced dNTPs
concentrations are known to increase Taq pol error rate but these modifications generally
reduce the yield of the epPCR reaction (Cadwell and Joyce, 1992; Lin-Goerke et al., 1997;
Shafikhani et al., 1997; Vartanian et al., 1996). Thus, employing error-prone DNA

Chapitre II : Construction de banques de variants de l'amylosaccharase
92
polymerases like pol β and pol η is clearly an advantage compared to epPCR approaches
which are based on serial replication steps to increase the mutation rate.
3.3 Base substitution spectra
The mutations generated by both pol β and pol η were predominantly single base
substitutions (95.5%, 91.4% and 88.4% of the total number of mutations in libraries A, B and
C, respectively). The ratio of transitions (Ts; base substitutions: purine to purine or pyrimidine
to pyrimidine) to transversions (Tv; base substitutions: purine to pyrimidine or pyrimidine to
purine) is a commonly used indicator to analyse the bias affecting the different types of base
substitutions produced by a random mutagenesis method. For instance, a mutational spectrum
with equiprobable base substitutions will exhibit a Ts/Tv ratio equal to 0.5. The rates of all
kinds of base substitutions were determined from the clones sequenced in the present study:
transitions represented 67.1 to 81.7% of mutations and transversions 18.3 to 32.9%. The
values of the Ts/Tv ratio for libraries A, B and C were 3.7, 4.5 and 2 respectively. For epPCR
using Taq pol, this ratio was shown to vary from 1.1 to 3.8 according to the reaction
conditions listed in table 18. The higher rate of transitions compared to that of transversions is
a common feature among random mutagenesis techniques that use DNA polymerases (Wong
et al., 2006). Indeed, purine:pyrimidine and pyridimidine:purine mispairs induce less
geometric distortions in DNA than purine:purine and pyrimidine:pyrimidine mispairs during
nucleotide incorporation by DNA polymerases.
As shown on table 18, transitions were generated more homogeneously by pol β (AT�GC:
43.9%; GC�AT: 34.8%) in comparison to pol η which shows a bias toward AT�GC
substitutions. The most frequent transversions produced by pol β were GC�TA (9%),
AT�TA (6.6%) and AT�CG (4.2%) whereas GC�CG mutations occurred with a rate of
1.4%. Using pol η, GC�TA (1.8% and 4.9% in libraries B and C, respectively) was the least
observed transversion while the most frequently generated were AT�TA (6.5% and 13.1%),
GC�CG (3% and 8.1%) and AT�CG (2.3% and 6.5%). In comparison, the use of various
epPCR conditions (table 18) does not modify the tendencies of Taq pol mutational spectrum,
which is biased toward AT�GC transitions (from 35% to 65.2%) and AT�TA transversions
(from 11.4% to 37.4%). The Mutazyme DNA polymerase which is a low-fidelity mutant of
Taq pol is less biased for transitions (AT�GC: 22.2%; GC�AT: 27.8%). This enzyme also
displays the same bias as Taq pol for AT�TA transversions (13.9%) but alternatively
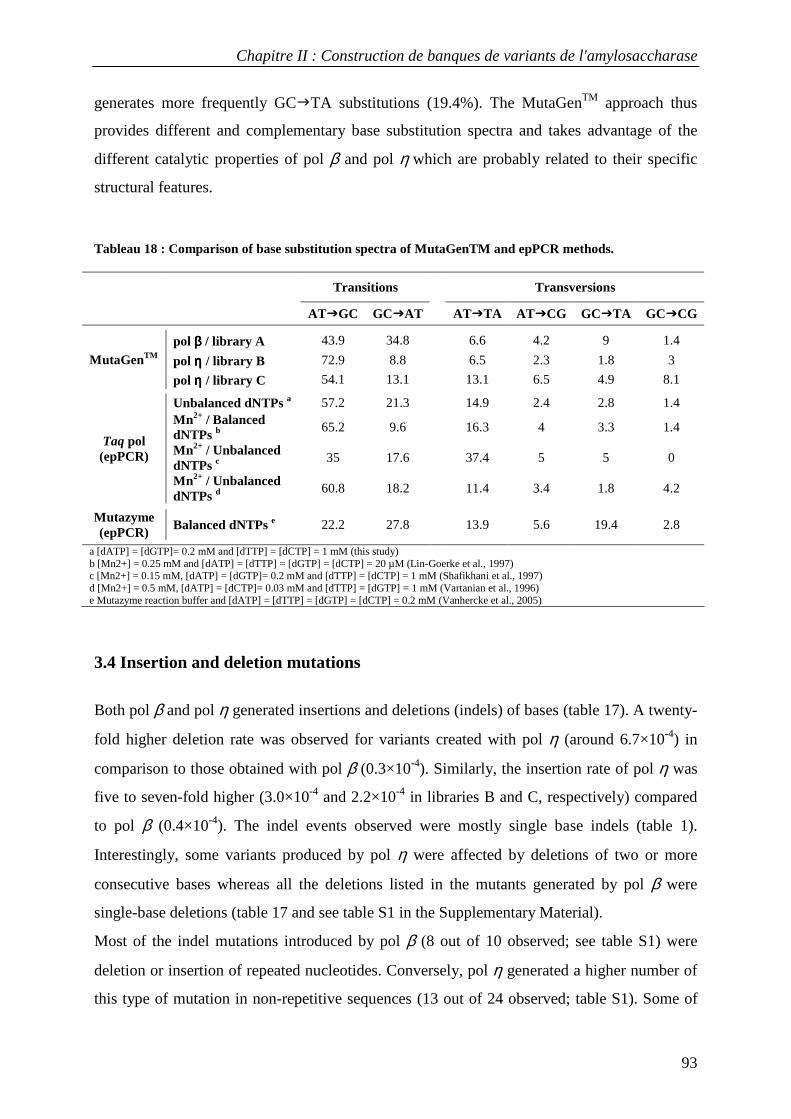
Chapitre II : Construction de banques de variants de l'amylosaccharase
93
generates more frequently GC�TA substitutions (19.4%). The MutaGenTM approach thus
provides different and complementary base substitution spectra and takes advantage of the
different catalytic properties of pol β and pol η which are probably related to their specific
structural features.
Tableau 18 : Comparison of base substitution spectra of MutaGenTM and epPCR methods.
Transitions Transversions
AT����GC GC����AT AT����TA AT����CG GC����TA GC����CG
pol β β β β / library A 43.9 34.8 6.6 4.2 9 1.4
pol η η η η / library B 72.9 8.8 6.5 2.3 1.8 3 MutaGenTM
pol η η η η / library C 54.1 13.1 13.1 6.5 4.9 8.1
Unbalanced dNTPs a 57.2 21.3 14.9 2.4 2.8 1.4 Mn 2+ / Balanced dNTPs b
65.2 9.6 16.3 4 3.3 1.4
Mn 2+ / Unbalanced dNTPs c
35 17.6 37.4 5 5 0 Taq pol (epPCR)
Mn 2+ / Unbalanced dNTPs d
60.8 18.2 11.4 3.4 1.8 4.2
Mutazyme (epPCR)
Balanced dNTPs e 22.2 27.8 13.9 5.6 19.4 2.8
a [dATP] = [dGTP]= 0.2 mM and [dTTP] = [dCTP] = 1 mM (this study) b [Mn2+] = 0.25 mM and [dATP] = [dTTP] = [dGTP] = [dCTP] = 20 µM (Lin-Goerke et al., 1997) c [Mn2+] = 0.15 mM, [dATP] = [dGTP]= 0.2 mM and [dTTP] = [dCTP] = 1 mM (Shafikhani et al., 1997) d [Mn2+] = 0.5 mM, [dATP] = [dCTP]= 0.03 mM and [dTTP] = [dGTP] = 1 mM (Vartanian et al., 1996) e Mutazyme reaction buffer and [dATP] = [dTTP] = [dGTP] = [dCTP] = 0.2 mM (Vanhercke et al., 2005)
3.4 Insertion and deletion mutations
Both pol β and pol η generated insertions and deletions (indels) of bases (table 17). A twenty-
fold higher deletion rate was observed for variants created with pol η (around 6.7×10-4) in
comparison to those obtained with pol β (0.3×10-4). Similarly, the insertion rate of pol η was
five to seven-fold higher (3.0×10-4 and 2.2×10-4 in libraries B and C, respectively) compared
to pol β (0.4×10-4). The indel events observed were mostly single base indels (table 1).
Interestingly, some variants produced by pol η were affected by deletions of two or more
consecutive bases whereas all the deletions listed in the mutants generated by pol β were
single-base deletions (table 17 and see table S1 in the Supplementary Material).
Most of the indel mutations introduced by pol β (8 out of 10 observed; see table S1) were
deletion or insertion of repeated nucleotides. Conversely, pol η generated a higher number of
this type of mutation in non-repetitive sequences (13 out of 24 observed; table S1). Some of

Chapitre II : Construction de banques de variants de l'amylosaccharase
94
our results are thus in agreement with the classical explanation that frameshift mutations
result from strand slippage during synthesis in repetitive DNA sequences (Streisinger et al.,
1966). Alternative mechanisms explaining indels of either repeated or non-repeated
nucleotides have also been proposed (Kunkel, 2004), including misalignment in the active site
which is supported by structural evidence for Sulfolobus solfataricus Dpo4, a family Y DNA
polymerase (Ling et al., 2001). However, the mechanisms by which complex indels involving
two or more nucleotides arise are still unknown (Garcia-Diaz and Kunkel, 2006).
3.5 Mutational impact at the protein level
The sequenced AS variants were analysed at the protein level to further evaluate the diversity
introduced by our method. Because of the high frameshift mutation rate of pol η, sequences
containing deletions and/or additions were more numerous in libraries B and C (40% and
30.8% respectively; table 19) than in library A (11.3%; table 19). Since clones from library B
were sequenced on a portion of 543 bp length (181 residues), this library should contain more
than 40% of AS variants affected by frameshift mutations. Table 20 summarizes the effect of
base substitutions observed in the sequenced variants at the protein level. Silent substitutions
represented 35.1%, 36.5% and 16.4% in libraries A, B and C respectively (table 20). Due to
the structure of the genetic code, most of these silent mutations were transitions occurring on
codon third positions (see table S2 in the Supplementary Material). These events lowered the
effective amino acid substitution rates which were 0.3%, 1.75% and 1.4% in libraries A, B
and C respectively (%: per 100 residues; table 20). Remarkably, most transversions (88.2%;
see table S2) lead to amino acid mutations. Among amino acid substitutions, stop codons
(nonsense substitution) were generated with relatively weak frequencies (table 20; library A:
3.3%; library B: 4.7%; library C: none observed).
The amount of variants retaining AS activity in each library was determined by detecting AS
variants expressing colonies able to produce amylose when grown on solid rich medium in the
presence of sucrose. These rates were 75%, 20% and 35% for libraries A, B and C
respectively. These values can be related to the mutation rates applied in the libraries. Indeed,
library A displayed the lowest mutation rate (1.7×10-3) and contained the highest amount of
clones retaining AS activity. Conversely, active AS clones were less numerous in libraries B
and C, for which higher mutation rates were measured. In addition, because of its high
frameshift mutation rate, pol η generates statistically more deleterious mutations than pol β.
Indeed, most of the indel events observed were single base indels which generally lead to
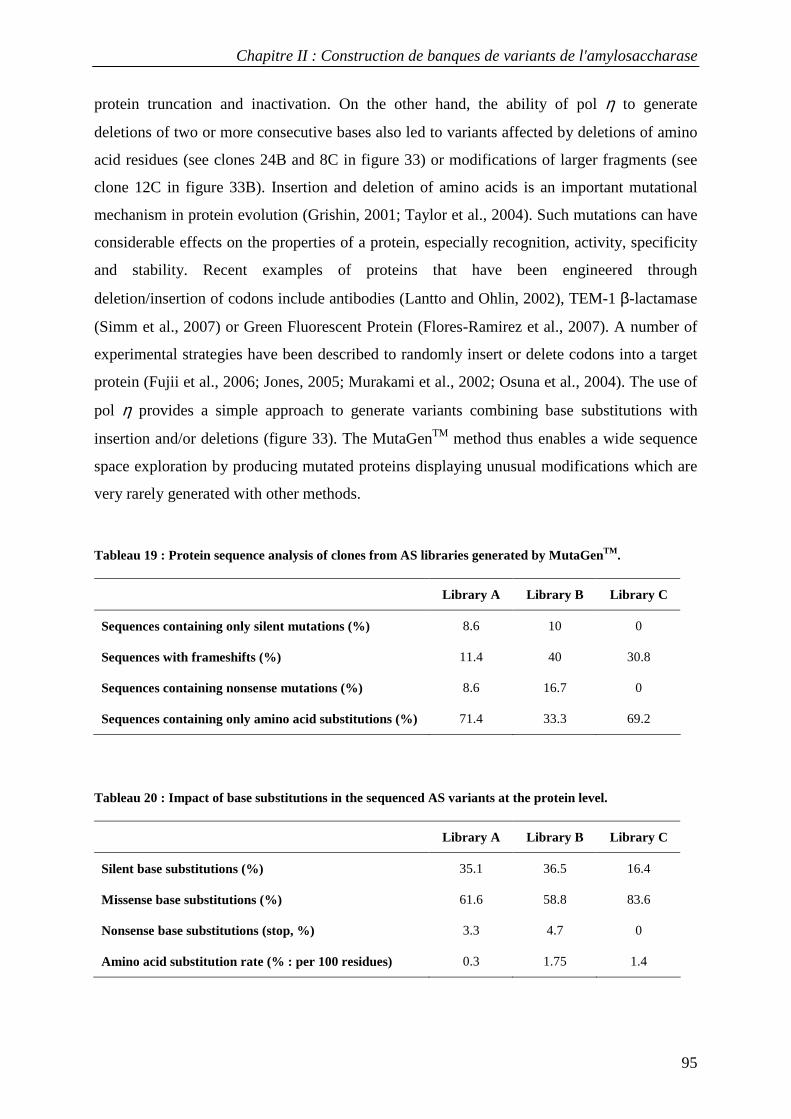
Chapitre II : Construction de banques de variants de l'amylosaccharase
95
protein truncation and inactivation. On the other hand, the ability of pol η to generate
deletions of two or more consecutive bases also led to variants affected by deletions of amino
acid residues (see clones 24B and 8C in figure 33) or modifications of larger fragments (see
clone 12C in figure 33B). Insertion and deletion of amino acids is an important mutational
mechanism in protein evolution (Grishin, 2001; Taylor et al., 2004). Such mutations can have
considerable effects on the properties of a protein, especially recognition, activity, specificity
and stability. Recent examples of proteins that have been engineered through
deletion/insertion of codons include antibodies (Lantto and Ohlin, 2002), TEM-1 β-lactamase
(Simm et al., 2007) or Green Fluorescent Protein (Flores-Ramirez et al., 2007). A number of
experimental strategies have been described to randomly insert or delete codons into a target
protein (Fujii et al., 2006; Jones, 2005; Murakami et al., 2002; Osuna et al., 2004). The use of
pol η provides a simple approach to generate variants combining base substitutions with
insertion and/or deletions (figure 33). The MutaGenTM method thus enables a wide sequence
space exploration by producing mutated proteins displaying unusual modifications which are
very rarely generated with other methods.
Tableau 19 : Protein sequence analysis of clones from AS libraries generated by MutaGenTM .
Library A Library B Library C
Sequences containing only silent mutations (%) 8.6 10 0
Sequences with frameshifts (%) 11.4 40 30.8
Sequences containing nonsense mutations (%) 8.6 16.7 0
Sequences containing only amino acid substitutions (%) 71.4 33.3 69.2
Tableau 20 : Impact of base substitutions in the sequenced AS variants at the protein level.
Library A Library B Library C
Silent base substitutions (%) 35.1 36.5 16.4
Missense base substitutions (%) 61.6 58.8 83.6
Nonsense base substitutions (stop, %) 3.3 4.7 0
Amino acid substitution rate (% : per 100 residues) 0.3 1.75 1.4
Chapitre II : Construction de banques de variants de l'amylosaccharase
96
(Légende sur la page suivante)

Chapitre II : Construction de banques de variants de l'amylosaccharase
97
Figure 33 : Distribution of amino acid mutations in AS variants generated with pol ηηηη. Sequenced variants
from libraries B and C are aligned with wild-type AS (AS WT). For the sake of clarity, only variants displaying
in-frame mutations are represented. Primary structure elements (N, B and B’ domains) are surrounded in green.
Secondary structure elements belonging to the (β/α)8 catalytic domain are surrounded in red (α-helixes) or blue
(β-strands). The residues (D286, E328 and D393) belonging to the catalytic triad are indicated by asterisks.
Amino acid mutations are surrounded in black. Panel A: Library B. Wild-type and variant AS sequences are
aligned from residues 5 to 186 which includes the N domain and the beginning of the (β/α)8 catalytic domain.
Panel B: Library C. Wild-type and variant AS sequences are aligned over the region targeted for random
mutagenesis (from residues 242 to 470) which includes the end of the B domain, a portion of the (β/α)8 catalytic
domain (from α-helix 3 to the beginning of α-helix 7) and the B’ domain.
4. CONCLUSION AND FUTURE PERSPECTIVES
Three large libraries of AS variants were constructed following the in vitro MutaGenTM
procedure using two human error-prone DNA polymerases, pol β and pol η. Unlike epPCR
methods, this technique allows the generation of protein variants through a single mutagenic
replication step. Moreover, sequence analysis of the libraries showed that the generated
diversity was related to specific features of the employed DNA polymerases. The use of either
pol β or pol η allowed the creation of libraries with different mutation rates and
complementary mutational spectra. The variants obtained through this approach combined all
types of mutations, including deletions of codons. Hence, MutaGenTM appears as a versatile
random mutagenesis tool since it takes advantage of the mutational bias of several error-prone
DNA polymerases. The potential of this method will be enlarged through the use of other
mutagenic polymerases like pol ι or pol κ.

Chapitre II : Construction de banques de variants de l'amylosaccharase
98
5. MATERIALS AND METHODS
5.1 Bacterial strains and plasmids
One Shot® E. coli TOP10 (Invitrogen) was used for transformation of ligation mixtures. E.
coli JM109 (Promega) was used to determine the fraction of active AS clones present in the
random mutagenesis libraries. Plasmid pGEX-6P3(Amersham Pharmacia Biotech) was used
for the cloning of libraries A and B. Plasmid pGST-AS, derived from pGEX-6P-3 (Potocki de
Montalk et al., 1999) was used as site-directed mutagenesis template to introduce the
restriction sites necessary for the cloning of library C. The AS encoding sequence inserted in
plasmid pCEASE01 (Van der Veen et al., 2004) was used as replication template for libraries
A and B.
5.2 DNA manipulations
Restriction endonucleases and DNA-modifying enzymes were purchased from New England
Biolabs and used according to the manufacturer’s instructions. DNA purification was
performed using QIAQuick (PCR purification and gel extraction) and QIASpin (miniprep and
maxiprep) (Qiagen). DNA sequencing was carried out using the dideoxy chain termination
procedure by MilleGen sequencing service (Labège, France).
5.3 Site-directed mutagenesis
Site-directed mutagenesis was carried out with the QuikChangeTM site-directed mutagenesis kit
(Stratagene) as previously described (Sarcabal et al., 2000) to introduce silent mutations
creating restriction sites HindIII and XhoI into the AS encoding sequence. Plasmid pGST-AS
was used as template. The pairs of primers used for each mutagenesis inverted PCR were
(restriction sites are underlined):
� AS-XhoI-for, 5’-GCGGCTTCTCGCAACTCGAGGACGGACGCTGGGTGT-3’ ;
� AS-XhoI-rev, 5’-ACACCCAGCGTCCGTCCTCGAGTTGCGAGAAGCCGC-3’ ;
� AS-HindIII-for, 5’-GCCGTTGACCGCATCAAGCTTTTGTACAGCATTGCTTTG-3’;
� AS-HindIII-rev, 5’-CAAAGCAATGCTGTACAAAAGCTTGATGCGGTCAACGGC-3’.

Chapitre II : Construction de banques de variants de l'amylosaccharase
99
The resulting plasmid pGST-AS(X-H) was used as replication template for library C and for
cloning the mutated replication products.
5.4 Construction of AS libraries following the MutaGenTM procedure
A mixture containing 1 µg of template plasmid, primers in equimolar concentration (200 nM)
and the appropriate replication buffer was treated for 5 min at 95°C and immediately cooled
down at 4°C to denature DNA strands. Following this step, mutagenic replications were
started by adding 50 µM dATP/dCTP, 100 µM dTTP/dGTP and 4 units of human pol β (for
library A) or 100 µM dNTPs and 4 units of pol η (for libraries B and C). ). One unit is defined
as the amount of enzyme required to catalyze the incorporation of 1 nmole of dNTP into an
acid-insoluble form in 1 hour at 37°C. These reactions were performed for 2 hours at 37°C.
Four primers were used for library A (italic characters indicate a tail that is not
complementary with the template):
� AmS-F, 5’-ACGATGCCTGCAGGTCGTGCAAGCTTGTAATGAATTCACAGTA-3’
(EcoRI site underlined) ;
� AmS-R, 5’-GAGGGCACATGGATCAGACCACTCTAGATGCATGCTCGAGAA-3’ (XhoI
site underlined) ;
� ASint-F, 5’–TACAGCAACCCGTGGGTATT–3’ ;
� ASint-R, 5’–AATACCCACGGGTTGCTGTA–3’.
The primers used for library B were AmS-F and AmS-R.
Two primers were used for library C:
� ASk7-F, 5’-TGCTCGTGATCGTGACGCTATAACTCGAGGACGGACGCT-3’ (XhoI site
underlined) ;
� ASk7-R, 5’- AGCTGCTGCACGCTCGTGACTACAAAAGCTTGATGCGGTCAA-3’
(HindIII site underlined).
Composition of pol β replication buffer composition was 50 mM Tris-Cl pH 8.8, 10 mM
MgCl2, 10 mM KCl, 1 mM DTT, 1% (v/v) glycerol. Replication buffer of pol η was 25 mM
Tris HCl pH 7.2, 1 mM DTT, 5 mM MgCl2, 2.5% (v/v) Glycerol. Human DNA polymerases β
and η were produced and purified as described in the supplementary material.
After the first step, two strategies were employed to amplify the mutated replication products
(figures 31 and 32). For library A, two groups of fragments generated by pol β (corresponding
to two separate parts of AS encoding sequence) were amplified with Platinum Taq polymerase

Chapitre II : Construction de banques de variants de l'amylosaccharase
100
(Invitrogen) in two different PCRs, one with primer pair AmsF/ASintR and the other with
ASintF/AmsR (figure 32). Due to their terminal complementarities, the two groups of
amplified replication products were extensively amplified with Platinum Taq in a first PCR
without primers (25 cycles of 20 sec at 94°C, 10 sec at 57°C and 2 min at 72°C) followed by a
second PCR with primers AmS-F and AmS-R (25 cycles of 20 sec at 94°C and 1 min 30 sec
at 72°C).
The fragments generated by pol η were amplified using Platinum Taq with primers AmS-
F/AmS-R (library B) or ASk7-F/ASk7-R (library C) through a single selective PCR process
(figure 1; first cycle: 2 min at 94°C, 10 sec at 58°C and 2 min at 72°C; then 25 cycles: 20 sec
at 94°C and 1 min 30 at 72°C).
The amplified replication products were cloned into EcoRI and XhoI restriction sites of
pGEX-6P3 (libraries A and B) or into XhoI and HindIII restriction sites of pGST-AS(X-H)
(library C). The ligation mixtures were transformed into electrocompetent E. coli TOP10 cells
subsequently plated on solid LB medium with 100 µg/ml ampicillin. After growth, the
number of colonies was determined to estimate the size of the libraries and some were
randomly chosen for DNA sequencing. For library storage, the colonies were scraped with
sterilized glycerol 10% (v/v) and conserved in this form at -80°C. Alternatively, the colonies
were used for plasmid extraction to store the libraries in the form of DNA solution.
5.5 Construction of an AS variant library by epPCR
Error-prone PCR using Taq DNA polymerase (New England Biolabs) was performed using
wild-type AS encoding sequence as template and the primer pair AmsF / AmsR. The reaction
was carried out from 5 ng of full pCEASE01vector with 5 U of Taq pol in 1X ThermoPol
buffer (New England Biolabs), 7mM MgCl2, 200 nM of each primer, 0.2 mM dATP/dGTP
and 1 mM dCTP/dTTP. The products were cloned into EcoRI and XhoI restriction sites of
pGEX-6P3. Ligation mixtures were transformed into electrocompetent E. coli TOP10 cells
subsequently plated on solid LB medium with 100 µg/ml ampicillin.
5.6 Determination of amylose-producing clone fraction in a library of variants
AS variant libraries were transformed into E. coli JM109 cells. The colonies were plated and
grown on solid LB medium containing 50 g/l sucrose, 1mM IPTG, 100 µg/ml ampicillin. The
colonies expressing active AS variants were detected by exposing the plates to iodine

Chapitre II : Construction de banques de variants de l'amylosaccharase
101
vapours, allowing an estimation of the amount of amylose-producing clones present in each
library.
5.7 Analysis of sequencing data
The sequences of clones randomly chosen in each library were analysed using MilleGen
proprietary software Mutanalyse 2.5. To characterize library diversity, various parameters
were determined from the alignment of the sequenced clones both at the nucleotide and the
protein levels. The mutation, base substitution, deletion and addition rates were calculated as
the ratio between the number of corresponding events and the number of bases sequenced.
The mutational spectrum consisted of listing the different mutation subtypes (additions,
deletions and substitutions). Mutanalyse 2.5 also determined the impact of base mutations at
the protein level. Clones from library A were sequenced over the entire encoding sequence
whereas those from library B were sequenced on a portion of 552 bp length. Clones from
library C were sequenced over the region targeted for random mutagenesis. Variants
generated by epPCR were sequenced over the entire encoding sequence.

Chapitre II : Construction de banques de variants de l'amylosaccharase
102
6. SUPPLEMENTARY MATERIAL
6.1 Supplementary method: cloning, production and purification of recombinant
human DNA polymerases ββββ and ηηηη
Full-length sequences encoding human pol β (GenBank accession number: NM002690) and
pol η (GenBank accession number: NM006502) were obtained by PCR amplification from
human brain and testis cDNA libraries (Clontech), respectively. PCR products were cloned
into the pMG20 vector (MilleGen). This plasmid allows the expression of recombinant
proteins with an N-terminal 6×His sequence tag under the control of the T7 RNA polymerase
promoter. Recombinant pol β or pol η were expressed in E.coli Origami BL21(DE3)
(Novagen). Cells were first grown under agitation at 37°C until 0.6 OD600. Expression of the
recombinant His-tagged DNA polymerases was then induced by IPTG (final concentration:
0.2 mM) for 3 hours at 20°C. Cells were harvested by centrifugation and concentrated to 80
OD600 in a lysis buffer (50 mM NaH2PO4 pH 8.0, 500 mM NaCl, 10 mM imidazole, 2%
Sigma antiprotease cocktail, 0.05% Triton X-100, 1 mg/mL lysozyme). After sonication and
centrifugation, the lysate supernatant was loaded onto a Ni2+-NTA resin (Qiagen) pre-
equilibrated with the lysis buffer. The column was thoroughly washed with 50 mM NaH2PO4
pH 8.0, 500 mM NaCl, 20 mM imidazole. Recombinant His-tagged DNA polymerases were
eluted with 50 mM NaH2PO4 pH 8.0, 500 mM NaCl, 250 mM imidazole. The proteins were
then dialysed against 20 mM Tris-HCl pH8.0, 50 mM NaCl, 1 mM DTT, 1 mM EDTA. After
addition of glycerol (final concentration: 50%), the purified polymerases were stored at -
20°C.
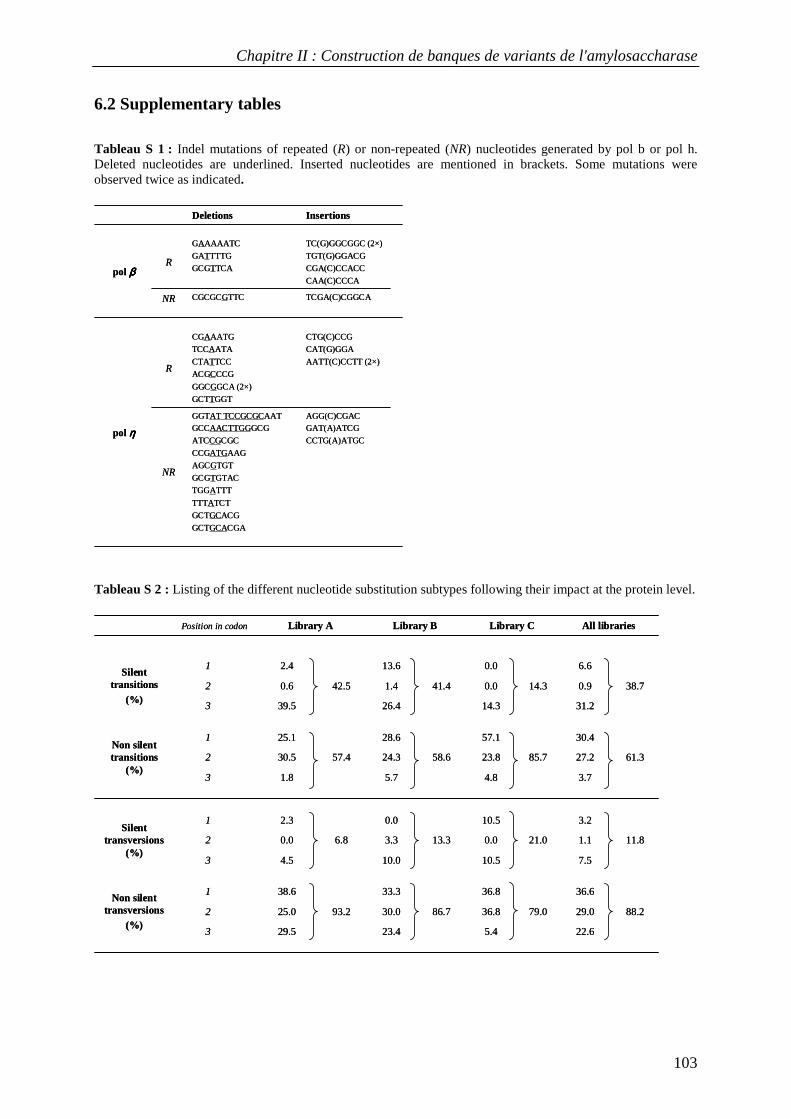
Chapitre II : Construction de banques de variants de l'amylosaccharase
103
6.2 Supplementary tables
Tableau S 1 : Indel mutations of repeated (R) or non-repeated (NR) nucleotides generated by pol b or pol h. Deleted nucleotides are underlined. Inserted nucleotides are mentioned in brackets. Some mutations were observed twice as indicated.
AGG(C)CGACGAT(A)ATCG
CCTG(A)ATGC
GGTAT TCCGCGCAATGCCAACTTGGGCG
ATCCGCGC
CCGATGAAG
AGCGTGT
GCGTGTAC
TGGATTT
TTTATCT
GCTGCACG
GCTGCACGA
NR
CTG(C)CCG
CAT(G)GGA
AATT(C)CCTT (2×)
CGAAATG
TCCAATA
CTATTCC
ACGCCCG
GGCGGCA (2×)
GCTTGGT
R
pol ηηηη
TCGA(C)CGGCACGCGCGTTCNR
TC(G)GGCGGC (2×)
TGT(G)GGACG
CGA(C)CCACC
CAA(C)CCCA
GAAAAATC
GATTTTG
GCGTTCAR
pol ββββ
InsertionsDeletions
AGG(C)CGACGAT(A)ATCG
CCTG(A)ATGC
GGTAT TCCGCGCAATGCCAACTTGGGCG
ATCCGCGC
CCGATGAAG
AGCGTGT
GCGTGTAC
TGGATTT
TTTATCT
GCTGCACG
GCTGCACGA
NR
CTG(C)CCG
CAT(G)GGA
AATT(C)CCTT (2×)
CGAAATG
TCCAATA
CTATTCC
ACGCCCG
GGCGGCA (2×)
GCTTGGT
R
pol ηηηη
TCGA(C)CGGCACGCGCGTTCNR
TC(G)GGCGGC (2×)
TGT(G)GGACG
CGA(C)CCACC
CAA(C)CCCA
GAAAAATC
GATTTTG
GCGTTCAR
pol ββββ
InsertionsDeletions
Tableau S 2 : Listing of the different nucleotide substitution subtypes following their impact at the protein level.
88.2
36.6
79.0
36.8
86.7
33.3
93.2
38.61Non silent
transversions(%)
29.036.830.025.02
22.65.423.429.53
4.5
0.0
2.3
1.8
30.5
25.1
39.5
0.6
2.4
Library A
6.8
57.4
42.5
7.510.510.03
1.10.03.32 11.8
3.2
21.0
10.5
13.3
0.01Silent
transversions(%)
3.74.85.73
27.223.824.32 61.3
30.4
85.7
57.1
58.6
28.61Non silent transitions
(%)
31.214.326.43
0.90.01.42 38.7
6.6
14.3
0.0
41.4
13.61Silent
transitions(%)
All librariesLibrary CLibrary BPosition in codon
88.2
36.6
79.0
36.8
86.7
33.3
93.2
38.61Non silent
transversions(%)
29.036.830.025.02
22.65.423.429.53
4.5
0.0
2.3
1.8
30.5
25.1
39.5
0.6
2.4
Library A
6.8
57.4
42.5
7.510.510.03
1.10.03.32 11.8
3.2
21.0
10.5
13.3
0.01Silent
transversions(%)
3.74.85.73
27.223.824.32 61.3
30.4
85.7
57.1
58.6
28.61Non silent transitions
(%)
31.214.326.43
0.90.01.42 38.7
6.6
14.3
0.0
41.4
13.61Silent
transitions(%)
All librariesLibrary CLibrary BPosition in codon

104
Chapitre III : Optimisation des protocoles
de sélection et de criblage des banques de
variants de l'amylosaccharase
Ce chapitre a été publié dans Journal of Biomolecular Screening.
Optimized and Automated Protocols for High-Throughput Screening of Amylosucrase Libraries. Emond S., Potocki-Véronèse G., Mondon P., Bouayadi K., Kharrat H., Monsan P. and Remaud-Siméon M. (2007). Journal of Biomolecular Screening, 12(5), pp. 715-723.

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
105
Optimized and automated protocols for high throughput screening of
amylosucrase libraries
1. SUMMARY
This work describes the design and validation of a general procedure for the high-throughput
isolation of amylosucrase variants displaying higher thermostability or increased resistance to
organic solvents. This procedure consists of two successive steps: (i) an in vivo selection
which eliminates inactive variants followed by (ii) automated screening of active variants to
isolate mutants displaying enhanced features. We chose an E. coli expression vector allowing
high production rate of the recombinant enzyme in miniaturized culture conditions. The
screening assay was validated by minimizing variability for various parameters of the
protocol, especially bacterial growth and protein production in cultures in 96-well
microplates. Recombinant amylosucrase production was normalized by decreasing the
coefficient of variance from 27% to 12.5%. Selective screening conditions were defined in
order to select variants displaying higher thermostability or increased resistance to organic
solvents. A first generation amylosucrase variants library, constructed by random
mutagenesis, was subjected to this procedure, yielding a mutant displaying a 25-fold
increased stability at 50°C compared to the parental wild-type enzyme.
2. INTRODUCTION
Directed evolution is a powerful approach for generating enzymes with desired properties,
such as improved catalytic activities, new substrate specificities and catalytic activities under
extreme conditions like high temperature or organic solvents (Yuan et al., 2005). This strategy
consists of isolating protein variants displaying an improvement for the property of interest
through iterative steps of random mutagenesis, DNA recombination, selection and/or
screening. Libraries of mutant genes can be generated by a large variety of generic methods,
such as chemical mutagenesis, error-prone PCR or DNA shuffling (Wong et al., 2006). In
contrast, selection and screening procedures need to be designed for each enzyme and
reaction. Moreover, the size of the libraries created through mutagenesis and/or recombination

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
106
processes is usually larger than the number of protein variants that can be screened. The same
limitation is applicable in the case of cDNA or genomic libraries derived from natural
sources, for which diversity is practically unlimited. Therefore, the design of reliable high-
throughput selection and screening methods that discriminate efficiently mutant enzymes
displaying desired properties among the available diversity without selecting false positives is
a critical point for the success of directed evolution experiments.
Amylosucrase from Neisseria polysaccharea (AS, EC 2.4.1.4) is a glucansucrase from
Glycoside Hydrolase (GH) family 13 that catalyses the synthesis of a water-insoluble
amylose-like polymer from sucrose, a readily available and low cost agroresource (Buttcher et
al., 1997; Potocki de Montalk et al., 1999). Contrary to starch or glycogen synthases (Preiss et
al., 1973), this enzyme does not require expensive nucleotide-activated sugar as glucosyl
donor. Furthermore, this enzyme is able to glucosylate various exogenous acceptors, mostly
oligo or polysaccharides, following a transglucosylation reaction which occurs at the cost of
sucrose hydrolysis and polymer synthesis (Potocki de Montalk et al., 2000b; Rolland-Sabaté
et al., 2004). AS is thus an attractive biocatalyst for the industrial synthesis of amylose-like
polymers, the modification of polysaccharides like glycogen or the production of
oligosaccharides (Potocki-Veronese et al., 2005; Putaux et al., 2006). Meanwhile, its use as an
industrial tool is limited by its low thermostability (t1/2 = 3 minutes at 50°C) and its weak
resistance toward organic solvents. In addition, further application developments such as
synthesis of longer amylose chains at higher temperatures or in the presence of organic
solvent clearly require adapting AS to these harsher conditions.
A directed evolution approach has already been initiated to optimize AS activity (van der
Veen et al., 2004; van der Veen et al., 2006). A zero background cloning strategy, the
pCEASE system, was developed and used to clone a first generation AS variant library
(containing approximately 50,000 clones). An in vivo selection protocol to discard inactive
variants was described. The DNS method for the measure of reducing sugar production
(Sumner and Howell, 1935) and the iodine staining to detect amylose formation (Banks et al.,
1971) were miniaturized in the 96-well microplate format to provide an activity screening
method. Preliminary small-scale screening assays on reduced sets of active variants (sixty or
ninety) allowed the isolation of mutants with 1.5 to 2-fold increased activity. However, this
approach was not convenient to screen wider libraries. The major drawback of this method
was the low level of recombinant enzyme produced using the pCEASE system which was not
suitable to develop accurate and fast screening assays.

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
107
Here we report the choice of a more efficient expression system to miniaturize recombinant
AS production in 96-well microplates. Using this system, we describe the optimization of an
automated screening procedure designed to isolate from AS variant libraries mutant enzymes
displaying higher thermostability or increased resistance to organic solvents. The screening
assay was validated by minimizing variability for various parameters of the protocol including
bacterial growth or protein production in microplate culture. Finally, the whole validated
procedure was tested by screening a first generation AS variant library created by random
mutagenesis.
3. RESULTS AND DISCUSSION
3.1 Choice of the recombinant AS expression system
To improve the sensitivity of the screening assay, we attempted to use a more efficient
expression system than pCEASE. The pGEX vector, enabling production of recombinant
protein with an N-terminal GST fusion, is routinely used for the production and the
purification of AS and its variants. Gene expression in this vector is under the control of the
synthetic tac promoter which allows higher protein production levels than that obtained when
the as gene is under the control of the lac promoter. Indeed, in flask culture (50 mL), cells
possessing pGST-AS produce 50-times more enzyme than those harboring pCEASE01 as
reflected by the difference in activity levels measured on sucrose at 30°C from sonicated cell
extracts (200 U/lculture versus 5 U/lculture). We compared the expression levels of both systems
in 96-well microplates to verify that this ratio was not affected when scaling down the culture
conditions. The difference in activity levels obtained from flask cultures was maintained in
96-well microplates. The mean activity per well from crude cell extracts harboring pGST-AS
was 143 U/lculture versus 19 U/lculture with pCEASE01. Therefore, we chose to use the pGEX
expression vector for further directed evolution experiments involving AS.
The general procedure designed to analyze AS variant libraries includes an in vivo selection
step eliminating clone expressing inactive mutants followed by an automated screen. This
procedure was optimized for the use of the chosen expression system, especially to reduce the
variability of the microplate activity assay.

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
108
3.2 Genetic selection for clones expressing active amylosucrase
Selection for E. coli cells harboring active AS variant is based on the fact that K12 derived
strains cannot metabolize sucrose (Reid and Abratt, 2005). Therefore, the use of a minimal
medium containing sucrose as sole carbon source allows the selection of E. coli clones
producing an active AS variant, because the enzyme cleaves sucrose, releasing fructose which
can be metabolized. AS variants displaying only hydrolytic activity would also be selected
following this method. The initial protocol developed using the pCEASE system (van der
Veen et al., 2004) was adapted for the use of the stronger pGEX expression system. Indeed,
pGST-AS E. coli cells did not grow when IPTG (inducer) was present in the selective media,
whereas cells containing pCEASE01 does. However, E. coli cells possessing pGST-AS were
able to develop on a selective medium without inducer. Actually, GST-AS overexpression
induced by IPTG seems to result in a toxic effect by limiting bacterial growth. Basal
expression of GST-AS without inducer is sufficient to complement E. coli cells on selective
sucrose medium. These results were validated by using an inactive AS variant (E328Q)
expressing clone as a negative control.
3.3 Optimization of reproducible and automated screening conditions
Our objective was to validate a reliable and fast high-throughput screening protocol.
Therefore, automation was systematically employed to standardize and accelerate different
stages of the screening process like bacterial inoculation, microplate filling up and the activity
assay. However, reproducibility is not only based on the use of robots. A screening procedure
consists of several steps, including cell growth, gene expression, bacterial lysis and the
activity assay. The errors within each step are reflected in the variability of the activity data
finally collected. This variability is linked to the ability of the screening procedure to identify
significantly improved variants and not false positives.
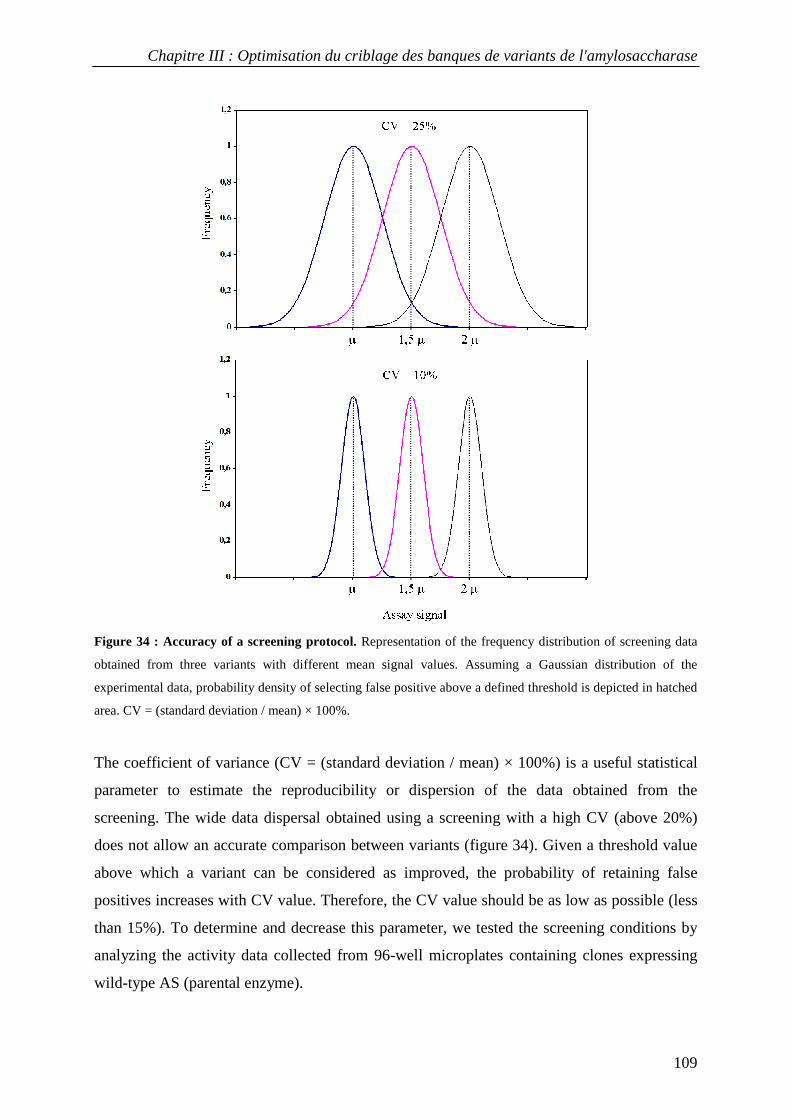
Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
109
Figure 34 : Accuracy of a screening protocol. Representation of the frequency distribution of screening data
obtained from three variants with different mean signal values. Assuming a Gaussian distribution of the
experimental data, probability density of selecting false positive above a defined threshold is depicted in hatched
area. CV = (standard deviation / mean) × 100%.
The coefficient of variance (CV = (standard deviation / mean) × 100%) is a useful statistical
parameter to estimate the reproducibility or dispersion of the data obtained from the
screening. The wide data dispersal obtained using a screening with a high CV (above 20%)
does not allow an accurate comparison between variants (figure 34). Given a threshold value
above which a variant can be considered as improved, the probability of retaining false
positives increases with CV value. Therefore, the CV value should be as low as possible (less
than 15%). To determine and decrease this parameter, we tested the screening conditions by
analyzing the activity data collected from 96-well microplates containing clones expressing
wild-type AS (parental enzyme).

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
110
3.3.1 Definition of experimental conditions for normalized cell growth and recombinant
AS expression
To assay the initial screening conditions (see Materials and Methods), cultures in 96-well
format were inoculated by replicating storage microplates containing E. coli clones harboring
pGST-AS. These cultures were grown under induction to allow recombinant GST-AS
expression. After bacterial lysis, crude cell extracts were assayed for AS activity on sucrose at
37°C. A CV of approximately 27% (n =288; three microplates) was determined from this
experiment. Figure 35B represents the AS activities measured in one representative
microplate. A wide dispersal of the activity levels and important differences between wells
were observed on this graph. This showed that the initial screening protocol was not
reproducible for each clone (well) assayed. To analyze growth conditions, cell density of the
microplate cultures was measured before lysis. Bacterial turbidity is a rather difficult
parameter to study at the microplate scale. Indeed, microplate cultures were read directly
without dilution or re-suspension; therefore, cell sedimentation in some wells during growth
cannot be circumvented and introduces more or less uncertainty in measurements. However,
the cell density profile of a representative microplate (figure 35A) was quite informative
about the fact that growth conditions were not reproducible between microplate wells. The
comparison of growth (figure 3(A) and activity (figure 35B) showed that in some wells, no
growth nor activity were measured (generally 3 to 10 wells per microplate) whereas in others,
activity was not detected even when growth was observed. Thus, the initial conditions of the
screening procedure needed to be modified in order to normalize bacterial growth and protein
expression levels. The study of these parameters was of importance to estimate the effect of
down-scaling a protocol we routinely perform in flask cultures.
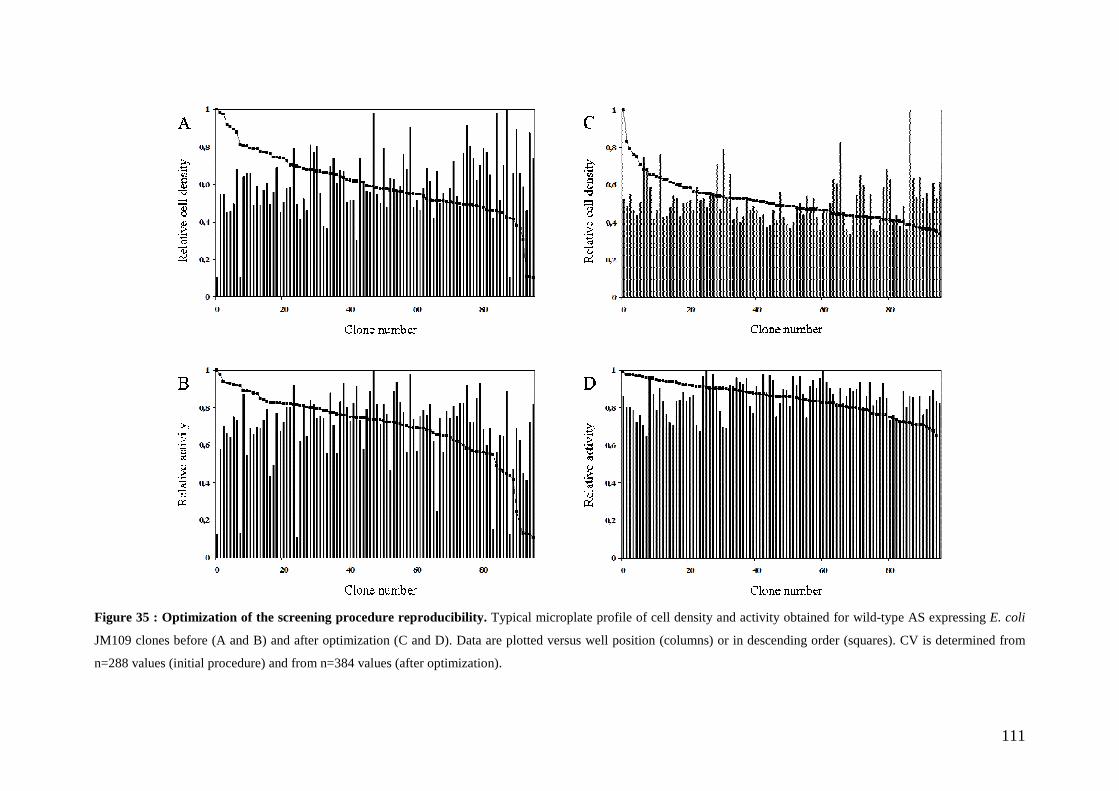
111
Figure 35 : Optimization of the screening procedure reproducibility. Typical microplate profile of cell density and activity obtained for wild-type AS expressing E. coli
JM109 clones before (A and B) and after optimization (C and D). Data are plotted versus well position (columns) or in descending order (squares). CV is determined from
n=288 values (initial procedure) and from n=384 values (after optimization).

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
112
The presence of inducer (IPTG) in culture medium is necessary to obtain high enzyme
production rates when using the pGEX expression system. However, it is also likely to limit
bacterial growth. According to the initial protocol, clones arranged in a storage microplate
(thawed cell suspensions) were replicated to inoculate a production microplate filled with LB
medium containing IPTG (see Material and Methods). Those drastic conditions can explain
the variability observed for bacterial growth and consequently for protein expression and
activity. Indeed, limited growth (or absence of growth) in some wells was not observed when
inoculating cultures in 96-well format without inducer but AS activity was not detectable in
those conditions (data not shown). To circumvent this problem caused by a combination of
inoculation and culture conditions, we considered the possibility of using fresh cultures in 96-
well format to inoculate the production microplates. Therefore, a step consisting of launching
starter 96-well cultures without inducer from storage microplates was added to the initial
procedure. A set of experiments using clones expressing wild-type AS was performed in order
to define new experimental conditions for this modified protocol and to analyze its
reproducibility.
As a first observation, the use of starter cultures in 96-well format improved the inoculation
efficiency of microplate cultures induced for protein production. More normalized growth
levels were obtained with this new procedure (figure 35C) compared to the initial protocol
(figure 35A). Moreover, absence of growth in some wells was not observed using this
method.
Growth kinetic was studied by analyzing a production culture in 96-well format replicated
from a starter microplate (figure 36A). From this experiment, growth rate in microplate
cultures was found to be much lower compared to that observed in routine flask cultures
(dotted line). Stationary phase was reached after 30 h of incubation. Indeed, miniaturized
culture conditions were more restrictive especially for aeration. To relate this growth profile
to enzyme expression, microplate cultures launched under the same conditions were incubated
for different times, subjected to lysis and then assayed for AS activity on sucrose at 37°C. In
agreement with the growth rate observed in microplates, the results presented in figure 36B
showed that nearly 24 h of culture were needed to reach GST-AS production levels (reflected
by activity calculation) similar to those obtained routinely in flask cultures (~ 200 U per liter
of culture). As liquid evaporation is not negligible for culture in 96-well format, growth above
24 h was not assayed. The CV values determined using the activity data collected from these
validation experiments were below 15 %, indicating that improving the inoculation efficiency
was needed to allow more reproducibility in the whole screening procedure. A standardized
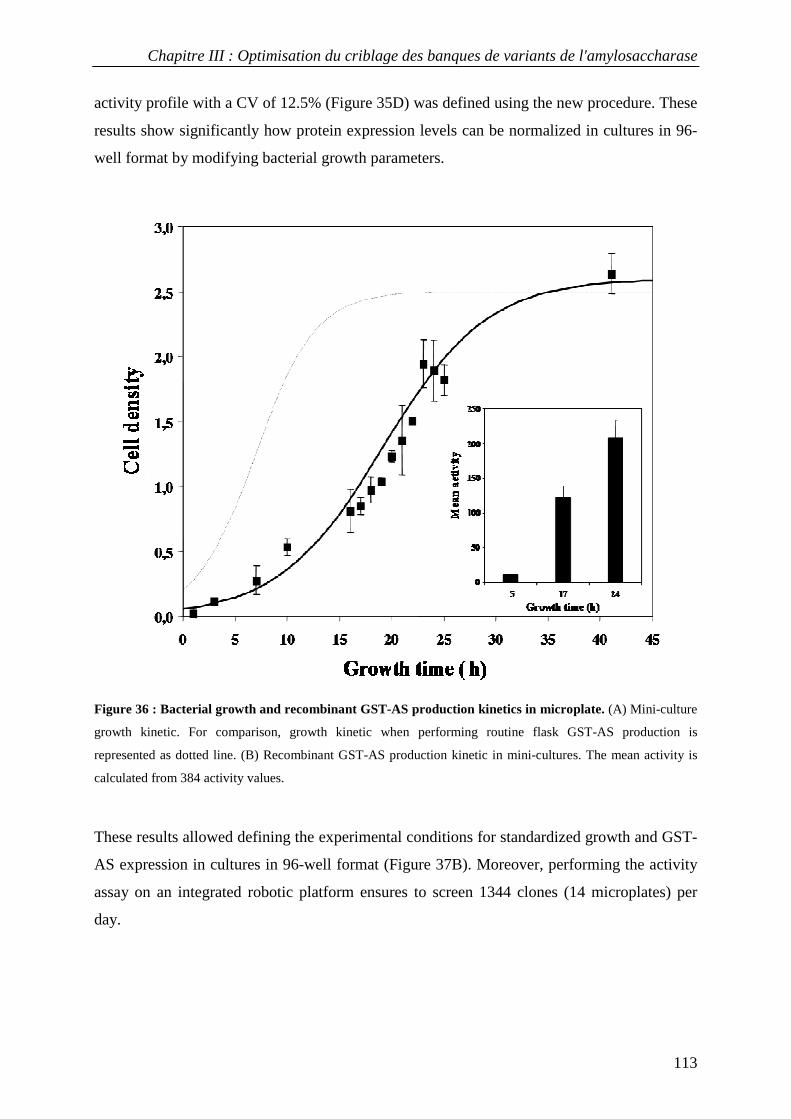
Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
113
activity profile with a CV of 12.5% (Figure 35D) was defined using the new procedure. These
results show significantly how protein expression levels can be normalized in cultures in 96-
well format by modifying bacterial growth parameters.
Figure 36 : Bacterial growth and recombinant GST-AS production kinetics in microplate. (A) Mini-culture
growth kinetic. For comparison, growth kinetic when performing routine flask GST-AS production is
represented as dotted line. (B) Recombinant GST-AS production kinetic in mini-cultures. The mean activity is
calculated from 384 activity values.
These results allowed defining the experimental conditions for standardized growth and GST-
AS expression in cultures in 96-well format (Figure 37B). Moreover, performing the activity
assay on an integrated robotic platform ensures to screen 1344 clones (14 microplates) per
day.
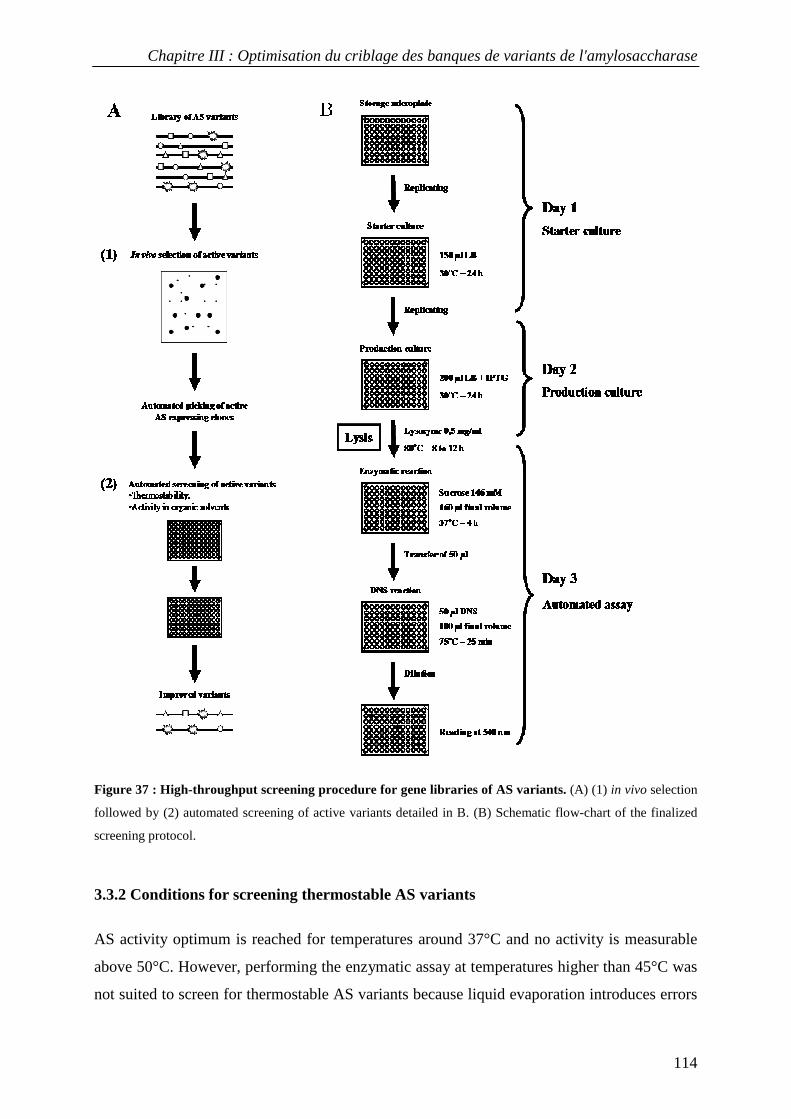
Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
114
Figure 37 : High-throughput screening procedure for gene libraries of AS variants. (A) (1) in vivo selection
followed by (2) automated screening of active variants detailed in B. (B) Schematic flow-chart of the finalized
screening protocol.
3.3.2 Conditions for screening thermostable AS variants
AS activity optimum is reached for temperatures around 37°C and no activity is measurable
above 50°C. However, performing the enzymatic assay at temperatures higher than 45°C was
not suited to screen for thermostable AS variants because liquid evaporation introduces errors
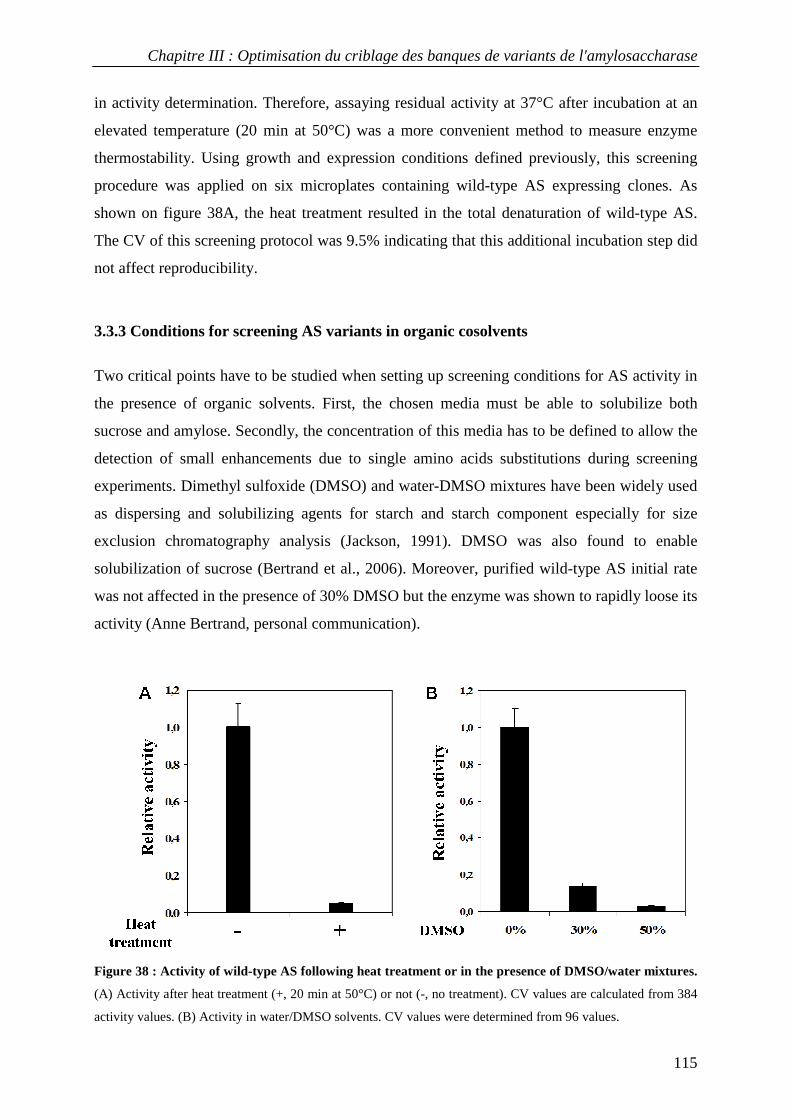
Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
115
in activity determination. Therefore, assaying residual activity at 37°C after incubation at an
elevated temperature (20 min at 50°C) was a more convenient method to measure enzyme
thermostability. Using growth and expression conditions defined previously, this screening
procedure was applied on six microplates containing wild-type AS expressing clones. As
shown on figure 38A, the heat treatment resulted in the total denaturation of wild-type AS.
The CV of this screening protocol was 9.5% indicating that this additional incubation step did
not affect reproducibility.
3.3.3 Conditions for screening AS variants in organic cosolvents
Two critical points have to be studied when setting up screening conditions for AS activity in
the presence of organic solvents. First, the chosen media must be able to solubilize both
sucrose and amylose. Secondly, the concentration of this media has to be defined to allow the
detection of small enhancements due to single amino acids substitutions during screening
experiments. Dimethyl sulfoxide (DMSO) and water-DMSO mixtures have been widely used
as dispersing and solubilizing agents for starch and starch component especially for size
exclusion chromatography analysis (Jackson, 1991). DMSO was also found to enable
solubilization of sucrose (Bertrand et al., 2006). Moreover, purified wild-type AS initial rate
was not affected in the presence of 30% DMSO but the enzyme was shown to rapidly loose its
activity (Anne Bertrand, personal communication).
Figure 38 : Activity of wild-type AS following heat treatment or in the presence of DMSO/water mixtures.
(A) Activity after heat treatment (+, 20 min at 50°C) or not (-, no treatment). CV values are calculated from 384
activity values. (B) Activity in water/DMSO solvents. CV values were determined from 96 values.
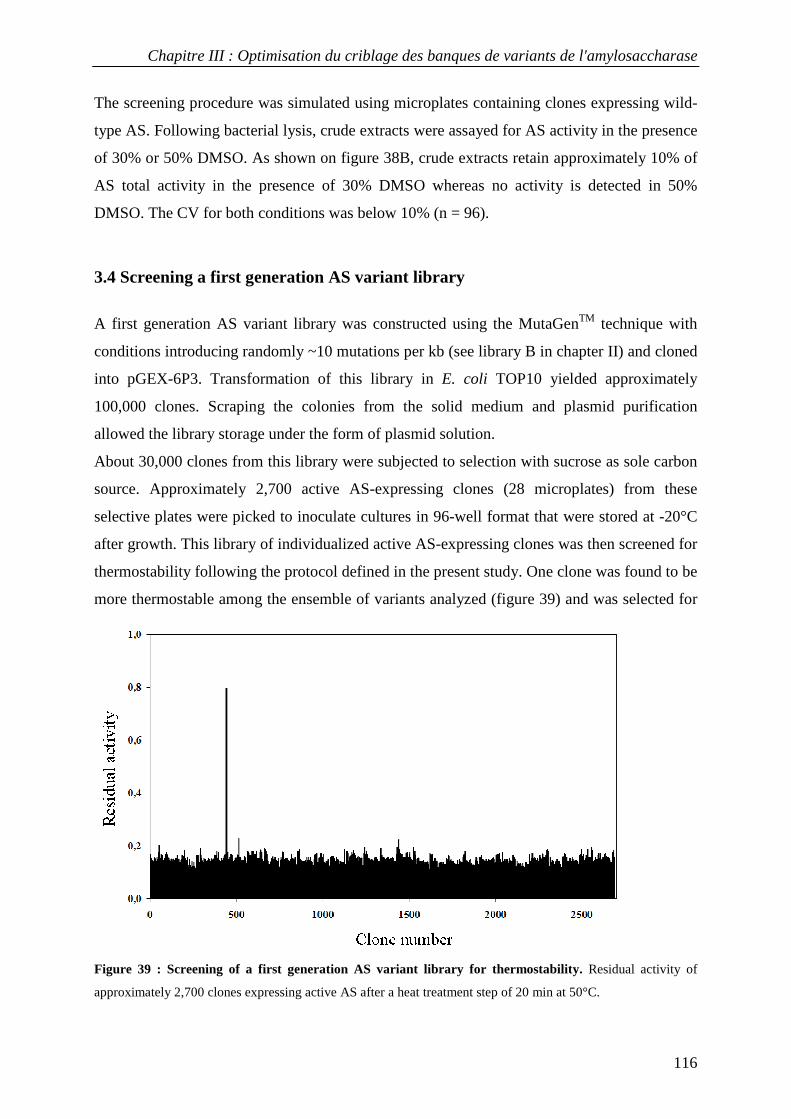
Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
116
The screening procedure was simulated using microplates containing clones expressing wild-
type AS. Following bacterial lysis, crude extracts were assayed for AS activity in the presence
of 30% or 50% DMSO. As shown on figure 38B, crude extracts retain approximately 10% of
AS total activity in the presence of 30% DMSO whereas no activity is detected in 50%
DMSO. The CV for both conditions was below 10% (n = 96).
3.4 Screening a first generation AS variant library
A first generation AS variant library was constructed using the MutaGenTM technique with
conditions introducing randomly ~10 mutations per kb (see library B in chapter II) and cloned
into pGEX-6P3. Transformation of this library in E. coli TOP10 yielded approximately
100,000 clones. Scraping the colonies from the solid medium and plasmid purification
allowed the library storage under the form of plasmid solution.
About 30,000 clones from this library were subjected to selection with sucrose as sole carbon
source. Approximately 2,700 active AS-expressing clones (28 microplates) from these
selective plates were picked to inoculate cultures in 96-well format that were stored at -20°C
after growth. This library of individualized active AS-expressing clones was then screened for
thermostability following the protocol defined in the present study. One clone was found to be
more thermostable among the ensemble of variants analyzed (figure 39) and was selected for
Figure 39 : Screening of a first generation AS variant library for thermostability. Residual activity of
approximately 2,700 clones expressing active AS after a heat treatment step of 20 min at 50°C.
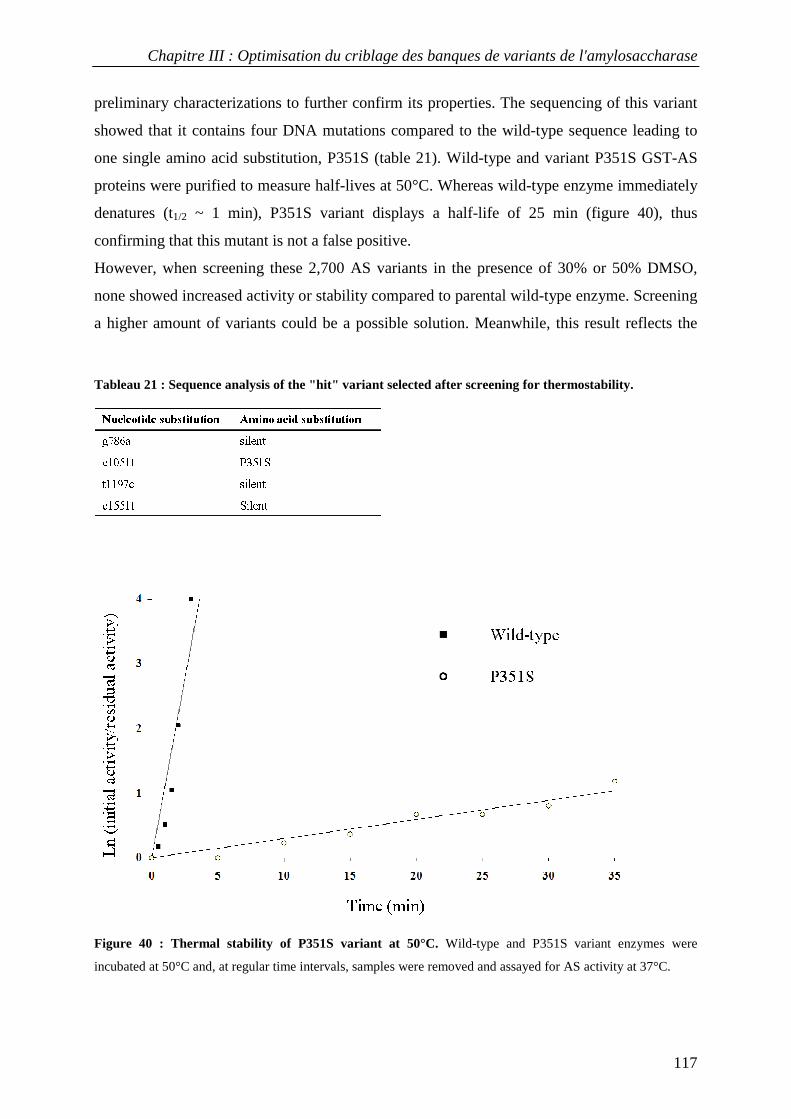
Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
117
preliminary characterizations to further confirm its properties. The sequencing of this variant
showed that it contains four DNA mutations compared to the wild-type sequence leading to
one single amino acid substitution, P351S (table 21). Wild-type and variant P351S GST-AS
proteins were purified to measure half-lives at 50°C. Whereas wild-type enzyme immediately
denatures (t1/2 ~ 1 min), P351S variant displays a half-life of 25 min (figure 40), thus
confirming that this mutant is not a false positive.
However, when screening these 2,700 AS variants in the presence of 30% or 50% DMSO,
none showed increased activity or stability compared to parental wild-type enzyme. Screening
a higher amount of variants could be a possible solution. Meanwhile, this result reflects the
Tableau 21 : Sequence analysis of the "hit" variant selected after screening for thermostability.
Figure 40 : Thermal stability of P351S variant at 50°C. Wild-type and P351S variant enzymes were
incubated at 50°C and, at regular time intervals, samples were removed and assayed for AS activity at 37°C.

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
118
difficulty of performing successful directed evolution experiments when using screening
conditions displaying strong stringency. Therefore, using less selective screening conditions
(below 30% DMSO for example) at the first round of the directed evolution process could
enable to detect mutants featuring small enhancements. A gradual increase of the screening
stringency during the next rounds would then make possible to find evolutionary trajectories
that lead to the desired enhancement of the property of interest. The critical point of this
strategy resides in the correct definition of the screening conditions for each round of the
directed evolution experiment.
4. CONCLUSION
A general selection and high-throughput screening procedure is provided for the analysis of
gene libraries of amylosucrase variants. As summarized in figure 36A, this procedure consists
of two successive steps: (i) an in vivo selection which eliminates inactive variants followed by
(ii) an automated screening of active variants to isolate mutants displaying better
thermostability or improved activity in organic cosolvents. In order to minimize variability,
each step of the procedure was standardized and automated when possible. A set of
experiments performed with wild-type AS expressing clones minimized the variability within
critical stages of the selection and screening procedure, thus reducing the probability of
selecting false positives when screening a variant library. Finally, the validated procedure was
used to analyze a first generation AS variant library, which led to the selection of a mutant
displaying improved thermostability. This standardized procedure will now be applied to
screen second and third generation libraries and provide even more thermostable AS.
Regarding the isolation of AS variants more stable in organic solvent, our preliminary
screening clearly indicates that less stringent conditions have to be employed during screening
of first generation libraries.

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
119
5. MATERIALS AND METHODS
5.1 Bacterial strains and plasmids
One Shot® E. coli TOP10 (Invitrogen) was used for transformation of ligation mixtures. E.
coli JM109 (Promega) was used to screen AS variants and large-scale production of the
selected mutants. Plasmid pGST-AS, derived from pGEX-6P-3 (Amersham Pharmacia
Biotech), was used for the construction, selection and screening of the AS random
mutagenesis library and for the production of glutathione S-transferase (GST)–amylosucrase
fusion proteins (expression of AS under control of the tac promoter)(Potocki de Montalk et
al., 1999). Plasmid pCEASE01, expressing amylosucrase under the control of the lac
promoter, was described elsewhere (van der Veen et al., 2004). Bacterial cells were grown on
LB (agar) containing 50 µg/mL kanamycin (when harboring pCEASE01), or 100 µg/mL
ampicillin (when harboring pGEX-6P-3-derived plasmid). For expression of amylosucrase in
E. coli JM109 media were supplemented with 1 mM isopropyl-β- -thiogalactose (IPTG).
5.2 DNA manipulations
A library of random mutants using AS as parent type was created following the MutaGen
random mutagenesis technique developed by MilleGen (Bouayadi et al., 2002) and described
elsewhere (see chapter II). Restriction endonucleases and DNA-modifying enzymes were
purchased from New England Biolabs and used according to the manufacturer’s instructions.
DNA purification was performed using QIAQuick (PCR purification and gel extraction) and
QIASpin (miniprep and maxiprep) (Qiagen). DNA sequencing was carried out using the
dideoxy chain termination procedure by MilleGen sequencing service (Labège, France).
5.3 Selection of active amylosucrase-expressing clones
Following transformation with pCEASE01, pGST-AS (wild-type or E328Q inactive mutant),
and the random mutagenesis library, E. coli JM109 cells were plated on solid LB containing
the appropriate antibiotic. After growth, the cells were scraped and diluted in physiological
water to an OD600 of 10−4. The clones were then subjected to selection pressure by plating
them onto solid M9 medium (42 Mm Na2HPO4, 24 mM KH2PO4, 9 mM NaCl, 19 mM
NH4Cl, 1 mM MgSO4, 0.5 µg/mL thiamine) containing 50 g/L sucrose as sole carbon source,

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
120
the appropriate antibiotic and 1 mM IPTG when needed. The plates were incubated for 3 to 5
days at 30°C, to obtain sufficient cell growth linked to expression of active amylosucrase.
5.4 Storage of individual active amylosucrase-expressing clones
Automated selective colony picking was carried out using colony picker QpixII (Genetix) that
can reach a cadence of 4,000 colonies per hour, according to five criteria: colony diameter,
roundness, proximity, contrast with the solid medium and colour. Colonies were transferred to
sterile 96-well microplates (NuncTM Brand Products, Denmark) filled with 150 µL LB
medium using a Biomek2000 pipettor (Beckman). After 24 h of growth at 30°C under
horizontal shaking (250 r.p.m) on an incubator-shaker (Kühner), microplates wells were filled
with 100 µL sterile Glycerol 30 % (v/v) and stored at -20°C. These microplates are referred as
storage microplates.
5.5 Growth conditions in microplate
Storage microplates were initially replicated using the QpixII automate to start cultures for
recombinant AS production in sterile 96-well microplates (NuncTM Brand Products; per well:
200 µL LB, 1 mM IPTG) grown for 18 h at 30°C under agitation. These microplates (referred
as production microplates) were then used for enzyme activity determination (see next
paragraph). After modifying the procedure, storage microplates were used to inoculate
cultures in 96-well microplates referred as starter microplates (150 µL LB per well). After
growth for 24 h at 30°C under agitation, these plates were duplicated using QpixII to start
production cultures in 96-well format grown at 30°C for 24 h under agitation (according to
the finalized protocol). Cell density levels at the end of growth incubation were measured by
recording the OD600 for each well using a TECAN Sunrise microplate reader. To analyse
growth during time incubation, a production microplate culture was grown and, at regular
time intervals, OD600 was measured for three randomly chosen wells.
5.6 Enzyme activity determination in microplate
Bacterial lysis was performed by adding lysozyme into production cultures to a final
concentration of 0.5 g/l and freezing the cells at -80°C for 8 to 12 h. After thawing for 1 h at
room temperature, the clones were screened using an integrated robotic TECAN Genesis

Chapitre III : Optimisation du criblage des banques de variants de l'amylosaccharase
121
RSP-200 platform, allowing liquid transfers, microplate and lid movements, barcode reading,
incubation, shaking and spectrophotometric reading of the microplates. After transfer of 80
µL of crude cell extracts in a non sterile 96-well microplate (Barloworld Scientific, UK),
enzymatic reaction was carried out by adding 80 µL of sucrose to a final concentration of 146
mM followed by incubation at 37°C for 4 h. Reducing-sugar production was measured by
adding 50 µL of the reaction mixture to 50 µL of dinitrosalicylic acid (DNS) reagent (Sumner
and Howell, 1935) in a polypropylene microplate (Evergreen Scientific, USA), incubating 25
min at 75°C on a microplate heater, adding 100 µL H2O, transferring of 120 µL in a
polystyrene flat bottom microtiter plate (Barloworld Scientific, UK), adding 120 µL H2O, and
measuring the absorbance at 540 nm with the Sunrise microplate reader. When screening for
thermostability, a heat treatment step (20 min, 50°C) was performed before adding sucrose.
For screening activity under organic solvents, dimethyl sulfoxide (DMSO) was added in the
reaction mixture to a final concentration of 30% or 50% (v/v; 160 µL final reaction volume).
Activity data collected under screening conditions were analyzed using Microsoft Excel. The
coefficient of variance (CV) was calculated using experimental data as follow: CV =
(standard deviation / mean) × 100%.
5.7 Production, purification and thermostability comparison of wild-type and
variant AS
Production and purification of wild-type and variant AS were performed as previously
described (Sarcabal et al., 2000). Since pure GST/AS fusion protein possesses the same
function and the same efficiency as pure AS (data not shown), enzymes were purified simply
to the GST/AS fusion protein stage (96 kDa). The enzymes (wild-type or mutated) were
obtained and stored in elution buffer (50 mM Tris-HCl, pH 7.0, 150 mM NaCl, 1 mM EDTA, 1
mM dithiothreitol). The protein content was determined by the micro-Bradford method, using
bovine serum albumin as a standard (Bradford, 1976). Half-lives of thermal inactivation were
determined for purified wild-type and variant GST-AS by incubating the enzymes (0.5
mg/mL) at 50°C for various time intervals. Initial and residual activities were measured at
37°C with 146 mM sucrose in PBS buffer at pH 7.3 using the DNS method to quantify the
released reducing sugar. Enzyme half-lives were deduced by fitting the neperian logarithm of
relative residual activity versus time curves.

122
Chapitre IV : Recherche et caractérisation
de variants thermostables de
l'amylosaccharase
Ce chapitre a été publié dans Protein Science.
Combinatorial engineering to enhance amylosucrase thermostability. Emond S., André I., Jaziri K., Potocki-Véonèse G., Mondon P., Bouayadi K., Kharrat H., Monsan P. and Remaud-Siméon M. (2008). Protein Science, 17(6):967-76.

Chapitre IV : Variants thermostables de l'amylosacharase
123
Combinatorial engineering to enhance amylosucrase thermostability
1. SUMMARY
Amylosucrase is a transglucosidase that catalyzes amylose-like polymer synthesis from
sucrose substrate. About 60,000 amylosucrase variants from two libraries generated by the
MutaGenTM random mutagenesis method were submitted to an in vivo selection procedure
leading to the isolation of more than 7,000 active variants. These clones were then screened
for increased thermostability using an automated screening process. This experiment yielded
three improved variants (two double mutants and one single mutant) showing 3.5 to 10-fold
increased half-lives at 50°C compared to the wild-type enzyme. Structural analysis revealed
that the main differences between wild-type amylosucrase and the most improved variant
(R20C/A451T) might reside in the reorganization of salt bridges involving the surface residue
R20 and the introduction of a hydrogen bonding interaction between T451 of B' domain and
D488 of flexible loop 8. This double mutant is the most thermostable amylosucrase known to
date and the only usable at 50°C. At this temperature, amylose synthesis by this variant using
high sucrose concentration (600 mM) led to the production of amylose chains twice longer
than those obtained by the wild-type enzyme at 30°C.
2. INTRODUCTION
Naturally available enzymes are usually not optimally suited for industrial applications. This
incompatibility is often related to their lack of stability under process conditions. The use of
high reaction temperatures may be advantageous regarding parameters like substrate and
product solubility, viscosity, process speed and microbial contamination. Moreover, high
stability is often considered as an economic advantage to reduce the enzyme quantity used in
industrial processes. For the past two decades, many methodologies describing protein
thermostability improvement through the introduction of mutations have been reported,
including comparison of naturally occurring homologous proteins, rational design and
directed evolution (Eijsink et al., 2004; Lehmann and Wyss, 2001; van den Burg and Eijsink,
2002). Sequence or structure comparisons of mesophilic enzymes with thermophilic relatives
has indeed shown to be an efficient approach to select thermostabilizing mutations (Ditursi et

Chapitre IV : Variants thermostables de l'amylosacharase
124
al., 2006; Haney et al., 1999; Hytonen et al., 2005; Miyazaki et al., 2001; Perl et al., 2000).
However, numerous protein sequences or reliable structural data are needed for this strategy
to be effective. Other rational strategies for stability engineering based on tertiary structure
analysis usually aim at improving the packing of the hydrophobic core, extending networks of
salt-bridges and hydrogen bonds that will favourably enhance the enthalpic contribution or
reduce the entropy of the unfolded state through salt or disulfide bridges, glycine substitutions
and introduction of prolines in turns (Eijsink et al., 2004; Vieille and Zeikus, 2001). However,
no general predictive rules enabling the design of proteins with increased stability can be
proposed despite numerous successful experimental reports. Elucidating the structural basis of
thermostability for a given protein and finding ways to improve it often appears as a specific
task depending strongly on the structural context.
Directed evolution appears as an efficient complementary approach to rational engineering.
This methodology consists of using random mutagenesis and recombination methods to
construct large libraries of protein variants which are subsequently selected or screened for
the desired trait. The identification of amino acids involved in specific functional features is
thus allowed through the exploration of the entire protein sequence without a priori
knowledge of its structure-activity relationships (Bloom et al., 2005; Petrounia and Arnold,
2000). This strategy has shown to be a powerful engineering approach to tailor the properties
of enzymes for a variety of industrial needs, such as increased thermostability, activity in
organic solvents and altered substrate specificity (Yuan et al., 2005). Noteworthy, the
outcome of directed evolution experiments is critically dependant on how libraries are
screened or selected. Therefore, the use of this approach is mainly limited by the development
of reliable high-throughput selection and screening systems enabling the isolation of the best
mutant(s) among the available diversity.
Amylosucrase from Neisseria polysaccharea (AS, EC 2.4.1.4) is a glucansucrase from
Glycoside Hydrolase (GH) family 13 that catalyses the de novo synthesis of a water-insoluble
amylose-like polymer from sucrose, a readily available and cheap agroresource (Potocki de
Montalk et al., 1999; Potocki de Montalk et al., 2000b). Noteworthy, the linear α-1,4 chains
formed during the reaction precipitate into semi-crystalline networks or aggregates when
reaching critical concentration and length, and cannot be further elongated. At 30°C, the
control of amylose chain precipitation is nicely monitored by sucrose initial concentration and
this enables the production of amylose with different morphologies and sizes (Potocki-
Veronese et al., 2005). Due to sucrose availability and low-cost, AS is an attractive
biocatalyst for amylose-like polymer synthesis. However, the development of industrial

Chapitre IV : Variants thermostables de l'amylosacharase
125
processes involving AS is limited by its low catalytic efficiency on sucrose alone (kcat = 1 s-1)
and thermostability (t1/2(50°C) = 3 min). Further application developments thus clearly
require adapting AS to harsher reaction conditions. Rational improvement of these properties
would benefit from comparisons with homologous proteins displaying different stability and
activity characteristics. Nevertheless, such an approach is not possible in the case of AS since
the only other described amylosucrase, isolated from mesophilic microorganism Deinococcus
radiodurans, possesses similar stability and activity properties (Pizzut-Serin et al., 2005).
Directed evolution techniques have already allowed the isolation of mutants displaying 1.5 to
4.2-fold increased activity compared to wild type AS (van der Veen et al., 2004; van der Veen
et al., 2006).
The present study describes the improvement of AS thermostability using directed evolution.
We previously reported the optimization of a two-step in vivo selection and automated
screening process enabling the search for AS variants with increased thermal stability (Emond
et al., 2007)(See also Chapter III). In the present study, we applied this procedure to screen
two different AS mutant libraries generated by MutaGenTM, an original random mutagenesis
technique based on the use of low-fidelity DNA polymerases (Bouayadi et al., 2002; Emond
et al., 2008b; Mondon et al., 2007b)(see also Chapter II). This work led to the isolation of
three improved AS variants. Their properties are described and discussed further.
3. RESULTS AND DISCUSSION
3.1 Isolation of thermostable amylosucrase variants
Wild-type AS whole encoding sequence was employed as template to introduce genetic
diversity using the MutaGenTM technique (Emond et al., 2008b)(See Chapter II). This
approach consists in generating random mutations by replicating the parental sequence with
human error-prone DNA polymerases. Two AS variant libraries, named A and B, were
constructed by utilizing two different polymerases, DNA polymerases β and η respectively.
Cloning and transformation of the mutagenized products to E. coli TOP10 yielded
approximately 8×105 clones for library A and 1×105 for library B. Plasmid DNA isolated
from these clones constituted the storage form of the libraries. The base mutation rates,
determined through DNA sequencing of randomly chosen clones, were 1.7 and 10.9

Chapitre IV : Variants thermostables de l'amylosacharase
126
mutations per kb for libraries A and B, respectively. These values were shown to be related to
the amounts of active AS variants which were 75% for library A and 20% for library B.
Transformation of library A to E. coli JM109 cells yielded 30,000 colonies which were then
subjected to selection with sucrose as sole carbon source. Approximately 4,500 active AS
expressing clones (47 microplates) were picked from these selective plates to inoculate small-
volume cultures in 96-well format that were stored at -20°C after growth. This library of
individualized active AS variants was then screened for increased thermostability after a heat
treatment step at 50°C for 20 minutes. The same procedure was followed to isolate and screen
2,700 active AS expressing clones from library B.
Among the variants screened, two clones from library A and one from library B were found to
be more thermostable compared to the wild-type AS and were thus retained for more detailed
characterization. The sequencing of the three selected variants revealed the mutations listed in
table 22. Both variants 1 and 2, isolated from library A, are double mutants, R20C/A451T and
A170V/Q353L respectively, each of them also contained an additional silent mutation.
Mutant 3 isolated from library B is a single mutant, P351S, enduring three silent mutations.
Tableau 22 : Nucleotide substitutions and resulting amino acid replacements of the variants selected after
screening for thermostability.
Mutant Library Nucleotide Amino acid Location
1 A c58t R20C N-terminus
g1351a A451T in B’ domain (loop 7)
a1834g** N612
2 A c285t** S95
c509t A170V in helix α2
a1058t Q353L in loop 6
3* B g786a** P262
c1051t P351S in loop 6
t1197c** F399
c1551t** N517
* Variant described previously (Emond et al., 2007) (See Chapter III)
** Silent mutation
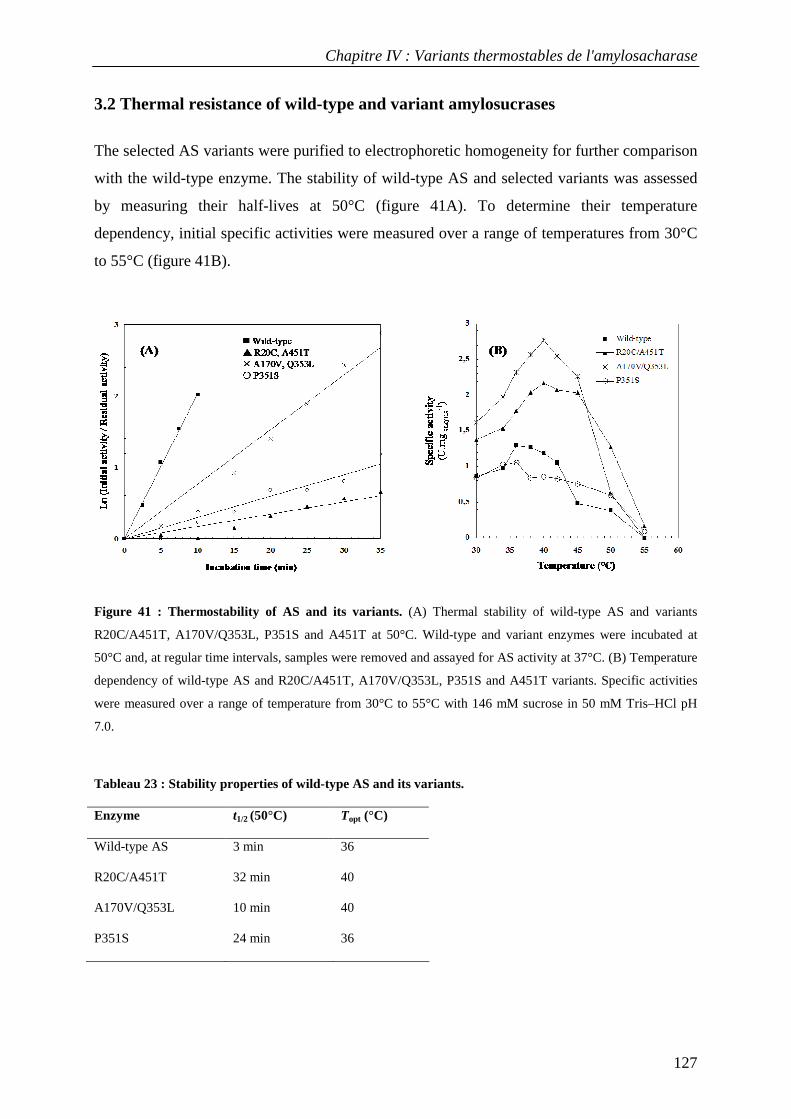
Chapitre IV : Variants thermostables de l'amylosacharase
127
3.2 Thermal resistance of wild-type and variant amylosucrases
The selected AS variants were purified to electrophoretic homogeneity for further comparison
with the wild-type enzyme. The stability of wild-type AS and selected variants was assessed
by measuring their half-lives at 50°C (figure 41A). To determine their temperature
dependency, initial specific activities were measured over a range of temperatures from 30°C
to 55°C (figure 41B).
Figure 41 : Thermostability of AS and its variants. (A) Thermal stability of wild-type AS and variants
R20C/A451T, A170V/Q353L, P351S and A451T at 50°C. Wild-type and variant enzymes were incubated at
50°C and, at regular time intervals, samples were removed and assayed for AS activity at 37°C. (B) Temperature
dependency of wild-type AS and R20C/A451T, A170V/Q353L, P351S and A451T variants. Specific activities
were measured over a range of temperature from 30°C to 55°C with 146 mM sucrose in 50 mM Tris–HCl pH
7.0.
Tableau 23 : Stability properties of wild-type AS and its variants.
Enzyme t1/2 (50°C) Topt (°C)
Wild-type AS 3 min 36
R20C/A451T 32 min 40
A170V/Q353L 10 min 40
P351S 24 min 36

Chapitre IV : Variants thermostables de l'amylosacharase
128
All the variants isolated during screening exhibited higher half-lives at 50°C than their wild-
type parent (figure 41A and table 23). Whereas the wild-type enzyme denatured very rapidly
(t1/2 = 3 min; table 26), R20C/A451T, A170V/Q353L and P351S mutants displayed half-lives
of 32, 10 and 24 min, respectively (figure 41A; table 23). As shown on figure 41B, the
activity-temperature profile of the double mutants R20C/A451T and A170V/Q353L was
broadened in comparison to the wild-type enzyme. Stabilization of these variants was
accompanied by an increase in the optimal temperature (40°C vs 36°C for wild-type AS; table
23 and figure 41B) and their activity between 40°C and 50°C was 1.5 to 2.5 times higher than
that of wild-type AS (figure 41B). Although no significant increase in optimal temperature
was observed for the P351S mutant (table 23), its specific activity between 45°C and 55°C
was higher than that of the wild-type enzyme (figure 41B), thus confirming the gain of
thermostability.
Double mutant R20C/A451T was the most stable enzyme at 50°C identified in this study.
Interestingly, a variant displaying the R20C substitution combined with the F598S mutation
was previously isolated during a screen for more active AS variants (van der Veen et al.,
2004; van der Veen et al., 2006). To analyze the contribution of the A451T substitution to the
improvement of AS thermostability, we constructed the A451T single mutant and the
resulting enzyme exhibited a half-life of 17 min at 50°C. In conclusion, the combination of
the R20C and A451T substitutions had clearly a beneficial effect on the enzyme stability.
3.3 Structural basis of thermostabilization mechanisms
AS is a (β/α)8-barrel enzyme, which possesses the three common domains found in GH
family 13: the characteristic barrel-catalytic A domain, a B domain between β-strand 3 and α-
helix 3, and a C-terminal Greek key domain. Beside these common features, AS structure also
contains two unique additional domains, namely the N-terminal α-helical domain, and a B’-
domain between β-strand 7 and α-strand 7 of the catalytic barrel (Skov et al., 2001). The five
mutations observed in the three thermostable variants isolated from screening are scattered all
over the protein structure (Table 23 and figure 42): three are located in the catalytic A domain
(A170V, P351S, Q353L), one in the specific B’ domain (A451T) and another one in the N-
terminal domain (R20C). Among these five point mutations, two of them (R20C and A170V)
are found on the protein surface, while the three remaining ones (P351S, Q353L, and A451T)
are located inside the enzyme structure, but do not affect the catalytically essential residues of

Chapitre IV : Variants thermostables de l'amylosacharase
129
AS previously identified (Sarcabal et al., 2000). Most of the exchanges observed are located
both in α-helices and in loops that connect β-strands with α-helices of the catalytic barrel and
none of them could have been predicted by rational analysis. To analyze the contribution of
these newly introduced residues to AS thermostabilization, the three variants identified during
screening were constructed in silico using the wild type enzyme structure as template.
Generated 3D-models of variants were subsequently subjected to an energy minimization
procedure. Although extensive molecular dynamics simulations would be needed to
investigate thoroughly protein conformation changes induced upon mutation, energy
minimization could still provide some hints regarding the possible structural determinants
likely to play a role on AS stabilization.
Double mutant R20C/A451T was the most thermostable variant. Wild-type AS structural
analysis revealed that R20 is a surface residue of the N domain, potentially involved in a salt-
bridge interaction with both D13 and E24 (Figure 43A). The electrostatic contribution to the
free-energy change upon such salt-bridge formation has been shown to vary significantly,
from being destabilizing to stabilizing to the protein structure depending on their geometry
location in the protein (Hendsch and Tidor, 1994; Kumar and Nussinov, 1999; Makhatadze et
al., 2003).
R20
A170
A451
P351
Q353
R20
A170
A451
P351
Q353
Figure 42 : Overview of mutation sites affecting amylosucrase thermostability. Secondary structure
elements are rendered in cartoon. Crystallographic maltoheptaose molecule is shown as a reference, coloured
in yellow in AS to highlight the position of the catalytic pocket (PDB 1MW0).
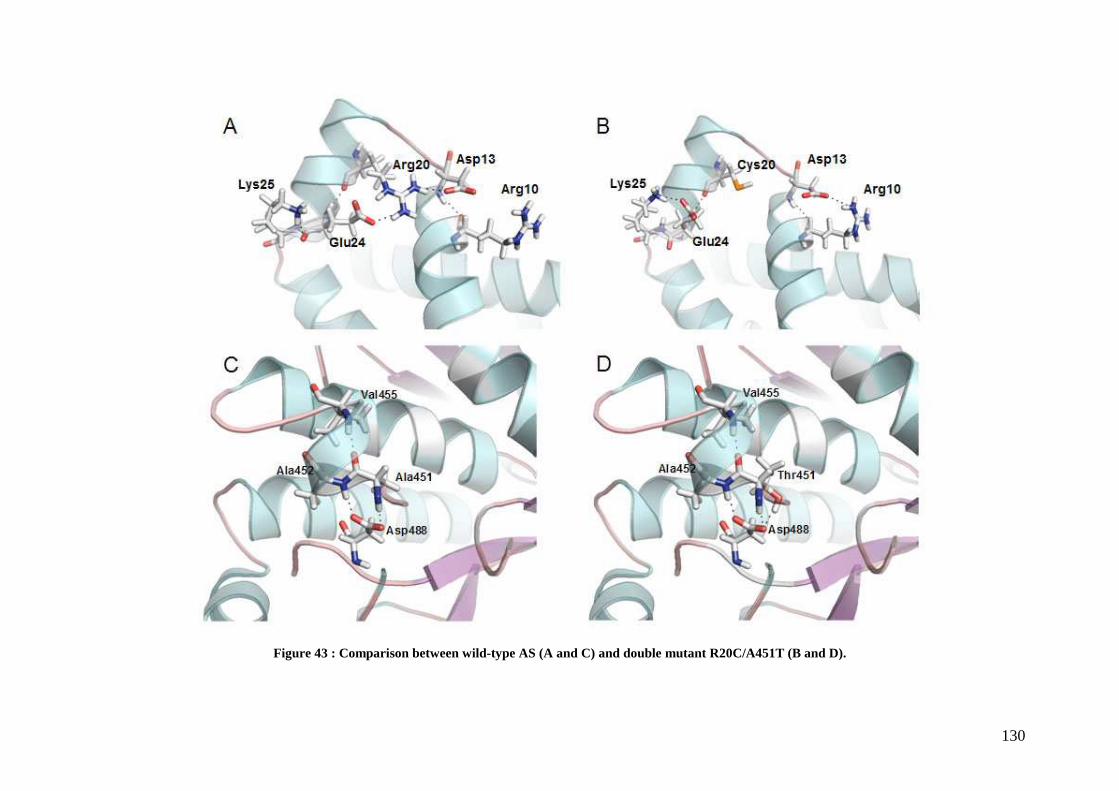
130
Figure 43 : Comparison between wild-type AS (A and C) and double mutant R20C/A451T (B and D).
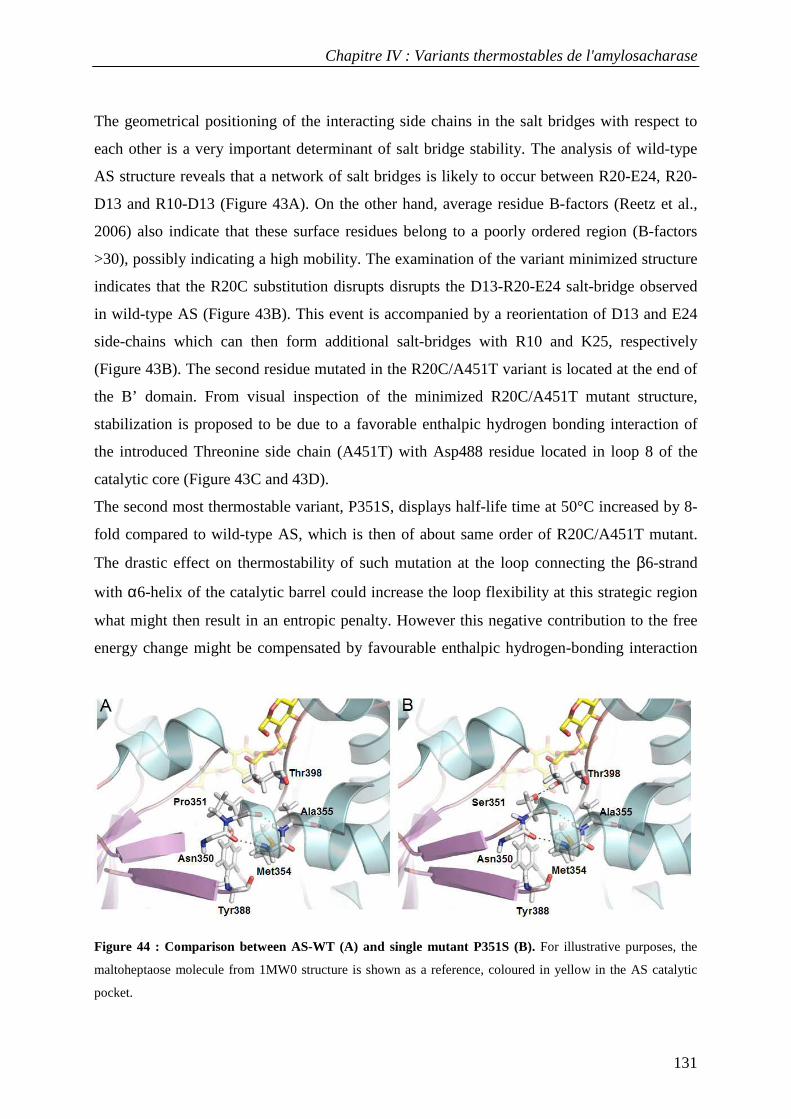
Chapitre IV : Variants thermostables de l'amylosacharase
131
The geometrical positioning of the interacting side chains in the salt bridges with respect to
each other is a very important determinant of salt bridge stability. The analysis of wild-type
AS structure reveals that a network of salt bridges is likely to occur between R20-E24, R20-
D13 and R10-D13 (Figure 43A). On the other hand, average residue B-factors (Reetz et al.,
2006) also indicate that these surface residues belong to a poorly ordered region (B-factors
>30), possibly indicating a high mobility. The examination of the variant minimized structure
indicates that the R20C substitution disrupts disrupts the D13-R20-E24 salt-bridge observed
in wild-type AS (Figure 43B). This event is accompanied by a reorientation of D13 and E24
side-chains which can then form additional salt-bridges with R10 and K25, respectively
(Figure 43B). The second residue mutated in the R20C/A451T variant is located at the end of
the B’ domain. From visual inspection of the minimized R20C/A451T mutant structure,
stabilization is proposed to be due to a favorable enthalpic hydrogen bonding interaction of
the introduced Threonine side chain (A451T) with Asp488 residue located in loop 8 of the
catalytic core (Figure 43C and 43D).
The second most thermostable variant, P351S, displays half-life time at 50°C increased by 8-
fold compared to wild-type AS, which is then of about same order of R20C/A451T mutant.
The drastic effect on thermostability of such mutation at the loop connecting the β6-strand
with α6-helix of the catalytic barrel could increase the loop flexibility at this strategic region
what might then result in an entropic penalty. However this negative contribution to the free
energy change might be compensated by favourable enthalpic hydrogen-bonding interaction
Figure 44 : Comparison between AS-WT (A) and single mutant P351S (B). For illustrative purposes, the
maltoheptaose molecule from 1MW0 structure is shown as a reference, coloured in yellow in the AS catalytic
pocket.

Chapitre IV : Variants thermostables de l'amylosacharase
132
between the introduced serine residue and T398 of the catalytic barrel that could also help to
stabilize the protein core (Figure 44).
The double mutant A170V/Q353L possesses a 3.5-fold increased half-life time at 50°C
compared to wild-type AS. Both mutations correspond to substitutions by more bulky
hydrophobic side chain residues. While A170 is a surface residue exposed to solvent of the
catalytic barrel α2 helix, Q353 belongs to loop 6 of the catalytic domain A and is located two
positions away in the sequence from P351. Given that single mutants have not been
constructed and characterized, it is difficult to assess the effect of individual mutations.
Nonetheless, Figure 45 suggests that L353 is nicely embedded in a hydrophobic protein core,
and such hydrophobic packing with nearby hydrophobic L357, A475 and I474 has been
shown to be a major stabilizing factor for thermophiles (Vieille and Zeikus, 2001).
Figure 45 : Representation of the hydrophobic region surrounding Q353L mutation in double mutant
A170V/Q353L.
3.4 Amylose synthesis by thermostable amylosucrase variants
Amylose productions by the wild-type and the two most thermostable variants, namely the
R20C/A451T and P351S mutants (table 24), were carried out over a 24 h period using either
100 or 600 mM sucrose at two different reaction temperatures, 30 or 50°C.
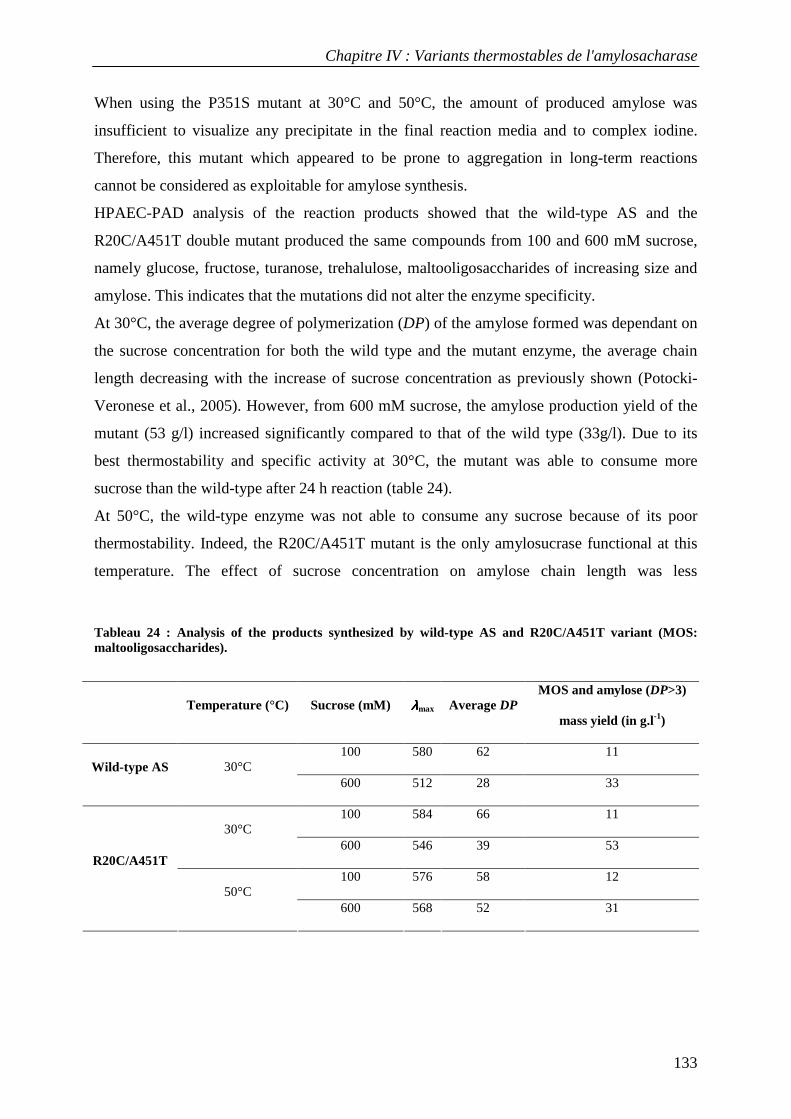
Chapitre IV : Variants thermostables de l'amylosacharase
133
When using the P351S mutant at 30°C and 50°C, the amount of produced amylose was
insufficient to visualize any precipitate in the final reaction media and to complex iodine.
Therefore, this mutant which appeared to be prone to aggregation in long-term reactions
cannot be considered as exploitable for amylose synthesis.
HPAEC-PAD analysis of the reaction products showed that the wild-type AS and the
R20C/A451T double mutant produced the same compounds from 100 and 600 mM sucrose,
namely glucose, fructose, turanose, trehalulose, maltooligosaccharides of increasing size and
amylose. This indicates that the mutations did not alter the enzyme specificity.
At 30°C, the average degree of polymerization (DP) of the amylose formed was dependant on
the sucrose concentration for both the wild type and the mutant enzyme, the average chain
length decreasing with the increase of sucrose concentration as previously shown (Potocki-
Veronese et al., 2005). However, from 600 mM sucrose, the amylose production yield of the
mutant (53 g/l) increased significantly compared to that of the wild type (33g/l). Due to its
best thermostability and specific activity at 30°C, the mutant was able to consume more
sucrose than the wild-type after 24 h reaction (table 24).
At 50°C, the wild-type enzyme was not able to consume any sucrose because of its poor
thermostability. Indeed, the R20C/A451T mutant is the only amylosucrase functional at this
temperature. The effect of sucrose concentration on amylose chain length was less
Tableau 24 : Analysis of the products synthesized by wild-type AS and R20C/A451T variant (MOS: maltooligosaccharides).
Temperature (°C) Sucrose (mM) λλλλmax Average DP MOS and amylose (DP>3)
mass yield (in g.l-1)
100 580 62 11 Wild-type AS 30°C
600 512 28 33
100 584 66 11 30°C
600 546 39 53
100 576 58 12 R20C/A451T
50°C 600 568 52 31
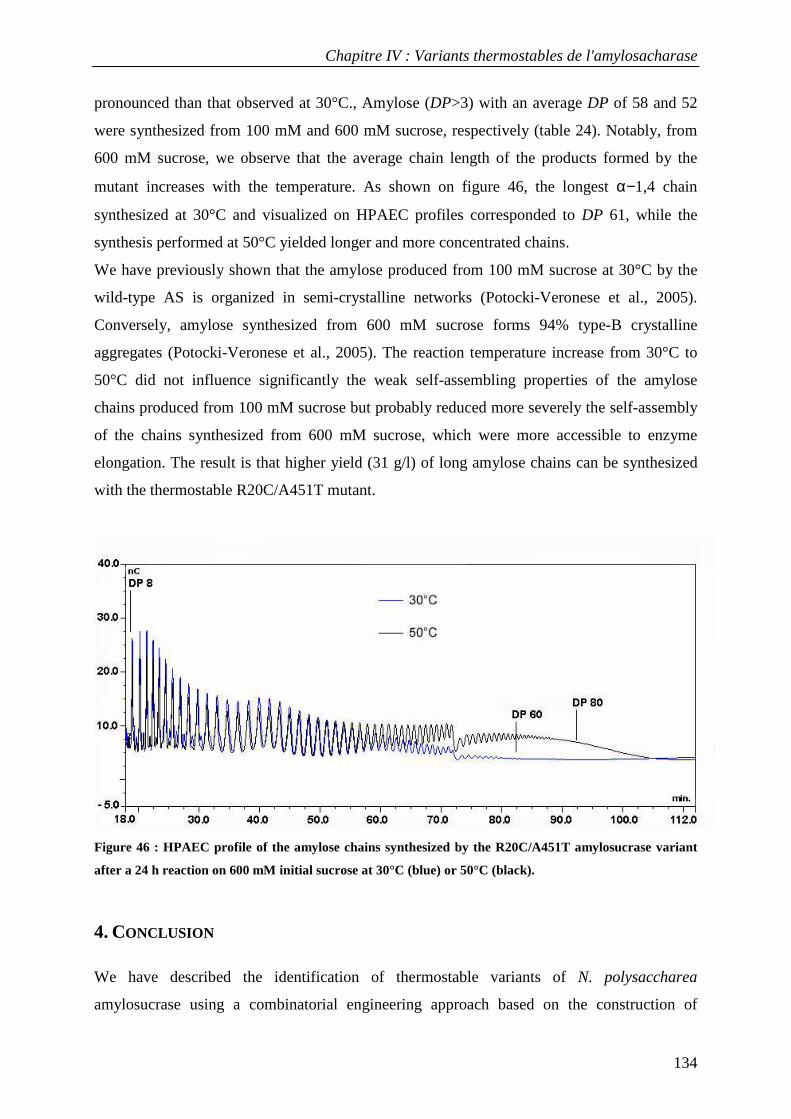
Chapitre IV : Variants thermostables de l'amylosacharase
134
pronounced than that observed at 30°C., Amylose (DP>3) with an average DP of 58 and 52
were synthesized from 100 mM and 600 mM sucrose, respectively (table 24). Notably, from
600 mM sucrose, we observe that the average chain length of the products formed by the
mutant increases with the temperature. As shown on figure 46, the longest α−1,4 chain
synthesized at 30°C and visualized on HPAEC profiles corresponded to DP 61, while the
synthesis performed at 50°C yielded longer and more concentrated chains.
We have previously shown that the amylose produced from 100 mM sucrose at 30°C by the
wild-type AS is organized in semi-crystalline networks (Potocki-Veronese et al., 2005).
Conversely, amylose synthesized from 600 mM sucrose forms 94% type-B crystalline
aggregates (Potocki-Veronese et al., 2005). The reaction temperature increase from 30°C to
50°C did not influence significantly the weak self-assembling properties of the amylose
chains produced from 100 mM sucrose but probably reduced more severely the self-assembly
of the chains synthesized from 600 mM sucrose, which were more accessible to enzyme
elongation. The result is that higher yield (31 g/l) of long amylose chains can be synthesized
with the thermostable R20C/A451T mutant.
Figure 46 : HPAEC profile of the amylose chains synthesized by the R20C/A451T amylosucrase variant
after a 24 h reaction on 600 mM initial sucrose at 30°C (blue) or 50°C (black).
4. CONCLUSION
We have described the identification of thermostable variants of N. polysaccharea
amylosucrase using a combinatorial engineering approach based on the construction of

Chapitre IV : Variants thermostables de l'amylosacharase
135
random mutant libraries, subsequently selected and screened for the desired trait. This
strategy led to the identification of three AS variants displaying 3.5 to 10-fold increased half-
lives at 50°C compared to the parent wild-type enzyme. Among them, the double mutant
R20C/A4551T is the most thermostable amylosucrase characterized to date. This new variant
was able to catalyze at 50°C the high yield synthesis of amylose chains probably displaying
different self-assembling properties than those produced at 30°C. Resulting physico-chemical
properties will be further investigated. In addition, a preliminary in silico comparative
analysis between the wild-type AS structure and the modeled variants allowed highlighting
molecular features possibly implicated in thermostability enhancement. To further optimize
amylosucrase, the combination of the mutations identified here with substitutions involved in
the enzyme efficiency and/or specificity obtained through rational and combinatorial
engineering will be investigated in a future study.

Chapitre IV : Variants thermostables de l'amylosacharase
136
5. MATERIALS AND METHODS
5.1 Bacterial strains and plasmids
One Shot® E. coli TOP10 (Invitrogen) was used for transformation of ligation mixtures. E.
coli JM109 (Promega) was used to screen AS variants and for large-scale production of the
selected mutants. Plasmid pGST-AS, derived from pGEX-6P-3 (GE Healthcare Biosciences),
was used for the construction, selection and screening of AS random mutagenesis libraries
and for the production of glutathione S-transferase (GST)–amylosucrase fusion proteins
(Potocki de Montalk et al., 1999). E. coli cells were grown on LB (agar) with 100 µg/mL
ampicillin. For expression of amylosucrase in E. coli JM109 media were supplemented with 1
mM isopropyl-β-D-thiogalactose (IPTG).
5.2 DNA manipulations
Restriction endonucleases and DNA-modifying enzymes were purchased from New England
Biolabs and used according to the manufacturer’s instructions. DNA purification was
performed using QIAQuick (PCR purification and gel extraction) and QIASpin (miniprep and
maxiprep) (Qiagen). DNA sequencing was carried out using the dideoxy chain termination
procedure by MilleGen sequencing service (Labège, France).
5.3 Construction, selection and screening of AS random mutagenesis libraries
Two libraries of AS variants, named A and B, were constructed by replicating wild-type AS
encoding sequence with human Pol β and Pol η respectively following the MutaGen
technique patented by MilleGen (Bouayadi et al., 2002; Mondon et al., 2007b). Detailed
description of the experimental procedure is reported elsewhere (Emond et al., 2008b)(See
also Chapter II). AS libraries were cloned into pGEX-6P-3. Ligation products were first
transformed in E. coli TOP10 to create the libraries. After plasmid extraction from the
resulting colonies, the library was then expressed in E. coli JM109 for selection and
screening.
A selection step was performed in order to isolate colonies expressing an active AS variant
(van der Veen et al., 2004). After transformation, E. coli JM109 cells were plated on solid LB
containing 100 µg/mL ampicillin. After growth, the colonies were scraped and diluted in

Chapitre IV : Variants thermostables de l'amylosacharase
137
physiological water to an OD600 of 10-4. The clones were then subjected to selection pressure
by plating them onto solid M9 medium (42 Mm Na2HPO4, 24 mM KH2PO4, 9 mM NaCl, 19
mM NH4Cl, 1 mM MgSO4, 0.5 µg/mL thiamine) containing 50 g/L sucrose as sole carbon
source and 100 µg/mL ampicillin. The plates were incubated for 3 to 5 days at 30°C, to obtain
sufficient cell growth linked to active amylosucrase production.
Colonies expressing active AS variants were picked using colony picker QpixII (Genetix) and
transferred to sterile 96-well microplates (NuncTM Brand Products, Denmark) filled with 150
µL LB medium. After 24 h of growth at 30°C under horizontal shaking (250 r.p.m) in an
incubator-shaker (Kühner), microplate wells were filled with 100 µL sterile Glycerol 30 %
(v/v) and stored at -20°C. These microplates are referred as storage microplates.
The screening procedure was performed as previously described (see chapter III). Storage
microplates were thawed and replicated to inoculate cultures in 96-well sterile microplates
referred as starter microplates (150 µL LB per well) grown for 24 h at 30°C under horizontal
shaking. These starter cultures were then replicated to start cultures for recombinant AS
production in sterile 96-well microplates (per well: 200 µL LB, 1 mM IPTG) which were
incubated at 30°C for 24 h under horizontal shaking. After growth, lysozyme was added to a
final concentration of 0.5 g/L and microplate cultures were frozen at –80°C for 8 to 12 h.
After thawing for 1 h at room temperature, AS variants were assayed for thermostability on an
integrated robotic TECAN Genesis RSP-200 platform. 80 µL of crude cell extracts were
transferred in a non sterile 96-well microplate (Barloworld Scientific, UK) and subsequently
submitted to a heat treatment step (20 min, 50°C). Enzymatic reaction was then performed by
adding 80 µL of sucrose to a final concentration of 146 mM followed by incubations at 37°C
for 4 h. Reducing sugar production was measured following a miniaturized dinitrosalicylic
acid (DNS) assay described previously (Emond et al., 2007)(See also Chapter III).
5.4 Site-directed mutagenesis
Site-directed mutagenesis was carried out with the QuikChangeTM site-directed mutagenesis kit
(Stratagene) as previously described (Sarcabal et al., 2000) to introduce the A451T
substitution into the wild-type AS encoding sequence. A KpnI restriction site was also
introduced silently to screen the correct clones. Plasmid pGST-AS (Potocki de Montalk et al.,
1999) was used as template. The pair of primers used for mutagenesis inverted PCR was
(KpnI restriction site is underlined and mutated bases are indicated in bold):
� AS-A451T-for, 5’-TGTCAGTGGTACCACCGCGGCATTGGTCGGCT-3’ ;

Chapitre IV : Variants thermostables de l'amylosacharase
138
� AS-A451T-rev, 5’-AGCCGACCAATGCCGCGGTGGTACCACTGACA-3’.
The resulting clones were confirmed by DNA sequencing.
5.5 Production, purification and activity assay of wild-type and variant AS
E. coli carrying the recombinant plasmid containing wild-type or mutated AS encoding
sequence was grown on LB containing 100 µg/mL ampicillin and 2 mM IPTG for 15 h. The
cells were harvested by centrifugation, resuspended and concentrated to an OD600 of 80 in
PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3). The
intracellular enzyme was extracted by sonication and the lysate supernatant was used as the
source for enzyme purification. Overexpression was verified by sodium dodecylsulfate–
polyacrylamide gel electrophoresis (SDS–PAGE).
Purification of wild-type and mutated enzymes was performed as previously described
(Potocki de Montalk et al., 1999) by affinity chromatography of the GST/AS fusion protein
on glutathione–Sepharose 4B (GE Healthcare Biosciences). The fusion protein solution was
subjected to proteolysis to remove the GST-tag, using the PreScission protease (GE
Healthcare Biosciences). The purified AS was finally eluted in PreScission buffer (50 mM
Tris–HCl pH 7.0, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol). The enzyme was
concentrated using centricon-10 (Amicon, Millipore).
The protein content was determined by the micro-Bradford method, using the Bio-rad reagent
(Bio-Rad laboratories, Hercules, CA) and bovine serum albumin as a standard (Bradford,
1976). Specific amylosucrase activity was measured at 30°C with 146 mM sucrose by HPLC
analysis using the procedure described below (carabohydrate analysis).
5.6 Thermostability of wild-type and mutant amylosucrase
Half-lives of thermal inactivation were determined for purified wild-type and variant GST-AS
by incubating the enzymes (around 500 mg/L) at 50°C for various time intervals. Initial and
residual activities were measured at 37°C with 146 mM sucrose in PBS buffer at pH 7.3 using
the DNS method to quantify the released reducing sugar. The first-order rate constant, kd, of
irreversible thermal denaturation was obtained from the slope of the plots of ln (initial activity
/ residual activity) versus time, and the half-lives (t1/2) were calculated as ln2/kd.

Chapitre IV : Variants thermostables de l'amylosacharase
139
5.7 Activity-temperature profile of wild-type and mutant amylosucrases
Initial specific activities of the purified wild-type and mutant amylosucrases were measured at
temperatures ranging from 30 to 55°C, in the presence of 146 mM sucrose and Tris 50 mM,
pH 7.0. Fructose and glucose concentrations were determined by HPLC using the procedure
described below. It was checked for all tested enzymes that the ratio between the glucose
(released by sucrose hydrolysis) and the fructose production rates (amount of free glucose or
fructose released versus time) was independent of the reaction temperature.
5.8 In vitro synthesis of amylose and oligosaccharides
Reactions were performed using the wild-type AS and the double mutant R20C/A451T at 1
U/mL (activity measured at 30°C on 146 mM sucrose with 0.1 g/L glycogen in 50 mM Tris-
HCl, pH 7.0). Reaction using the P351S variant was carried out at 0.5 U/mL since this
enzyme was prone to aggregate at higher concentrations. Synthesis reactions were performed
for 24 h at 30 and 50°C in 50 mM Tris-HCl, pH 7.0, containing 100 or 600 mM sucrose. For
carbohydrate analysis, samples collected at the beginning and at the end of the synthesis were
heated at 90°C during 5 min to stop the reaction.
5.9 Carbohydrate analysis
To determine the temperature dependency of wild-type and variant amylosucrases, glucose
and fructose concentrations were measured using a refractometer (Dionex), after separation
with a Ca2+ column (Biorad) and elution at 80°C with water at 0.6 mL/min flow rate.
After 24 h reaction using sucrose as substrate, the final media composition contains a soluble
fraction (monosaccharides and oligosaccharides) and a precipitate (longer α-1,4 linked linear
chains). Quantification of the products contained in the final reaction media was performed by
high-performance anion-exchange chromatography with pulsed amperometric detection
(HPAEC-PAD). For the determination of the reaction yields by quantification of
monosaccharides and oligosaccharides, the reaction media supernatants were diluted in water
to 17 mg/kg of total sugars. For analysis of the longer amylose chains, the final reaction
media (containing both soluble and insoluble products) was solubilized in 1 M KOH to a final
polymer content of 10 g/L. Separation was performed on a 4 × 250 mm Dionex Carbo-pack
PA100. The flow rate of 150 mM NaOH was 1.0 mL/min and an acetate gradient was applied

Chapitre IV : Variants thermostables de l'amylosacharase
140
as follows: 0-20 min, 6-196 mM; 20-70 min, 196-303.5 mM; 70-134 min, 303.5-450 mM;
135-140 min, 6 mM. Detection was performed by use of a Dionex ED40 module with a gold
working electrode and a Ag/AgCl pH reference. Amylose chain (DP>3) yields were
determined as the difference between the amount of sucrose consumed and the amount of
glucosyl residues incorporated into the soluble products (glucose, sucrose isomers, maltose
and maltotriose).
5.10 Iodine Binding Properties
The iodine reactivity of the amyloses produced by the wild-type AS and the double mutant
R20C/A451T was measured. The final reaction media (20 µL) were solubilized with 20 µL of
2 M KOH, neutralized with 200 µL 0.1 N HCl and mixed with 32 µl of aqueous iodine
solution [2% (w/v) KI and 0.2% (w/v) I2] and 8 µL of 0.1 N HCl in microtiter plates. The
maximum absorption wavelength (λmax) of the iodine/amylose complexes was determined by
recording spectra between 500 and 690 nm with 2 nm steps, on a Sunrise spectrophotometer
(Tecan, Switzerland). The corresponding average DP was calculated from the following
equation previously established for DP 28-70 amylose chains (John et al., 1983):
103/λmax = 11.6/DP+1.537
5.11 Computational modeling of AS variants
In silico construction of AS mutants was performed using the coordinates of the AS
crystallographic structure (Protein Databank entry: 1MW0). Hydrogens were added to each
atom of the AS protein using the InsightII molecular modelling package (Accelrys, San
Diego, CA, USA) running on a Silicon Graphics O2 workstation. The crystallographic water
molecules and bound substrate were removed from the structure. The AMBER9 software
(http://amber.scripps.edu/) was subsequently used to optimize the energy of the protein
structure in gas-phase using a combination of steepest descent and conjugated gradient
minimization steps.
To assess the contribution of the individual amino acids to the thermostability of AS, we
constructed the AS mutants, in which the amino acids were individually replaced in silico
with the corresponding amino acids observed in isolated improved mutants. Starting from the
optimized AS structure, site mutations were modeled using the Biopolymer module of

Chapitre IV : Variants thermostables de l'amylosacharase
141
InsightII software package (Accelrys). The conformation of the mutated residue side chain
was optimized by manually selecting a low-energy conformation from a side-chain rotamer
library. Steric clashes (van der Waals overlap) and non-bonded interaction energies
(Coulombic and Lennard-Jones) were evaluated for the different side-chain conformations.
All mutants were subsequently minimized following the same energy minimization procedure
described for AS and using the last available version of AMBER9 molecular mechanics force
field . All drawings were performed using PyMOL software (DeLano, 2002).

142
Conclusion

Conclusion
143
Les travaux qui viennent d’être présentés ont été réalisés dans le cadre de l’optimisation par
évolution dirigée de l’amylosaccharase, une enzyme d’intérêt pour la valorisation du
saccharose, agroressource abondante et peu coûteuse. Ils ont été axés autour de deux objectifs
généraux. Le premier consistait à appliquer la technique MutaGenTM pour la génération de
banques de variants enzymatiques et à démontrer l’utilité des ADN polymérases à fort taux
d’erreurs comme outils de mutagénèse aléatoire. Le second objectif était la construction par
ingénierie combinatoire de nouvelles amylosaccharases plus thermostables et résistantes à la
dénaturation par les solvants organiques.
Il convient ici de rappeler brièvement les données à notre disposition au début de nos travaux.
1- Un des projets internes à la société MilleGen visait à employer MutaGenTM pour
augmenter la diversité initiale de banques d’anticorps (ou de fragments d’anticorps). La
perspective principale de ces travaux, initiés dans le courant de l’année 2001, est
l’identification de nouveaux anticorps thérapeutiques. Les banques générées au cours de ce
projet ont fait récemment l’objet d’un dépôt de brevet et d’une publication scientifique
(Mondon et al., 2007a; Mondon et al., 2007b). La longueur des matrices parentales employées
au cours de ce projet était de 700 bp. Au début de nos travaux de thèse (octobre 2003), la
technique MutaGenTM n’avait jamais été appliquée à des séquences de plus de 1 kb en raison
de la faible processivité de certaines ADN polymérases, en particulier pol β. D’autre part, la
technique MutaGenTM n’avait pas encore été utilisée dans le cadre d’un projet d’évolution
dirigée d’enzyme d’intérêt industriel.
2- Des travaux d’évolution dirigée de l’AS de N. polysaccharea étaient depuis 2002 en
cours au sein de l’équipe CIMES. Ces travaux ont consisté au développement de méthodes de
sélection et de criblage dédiées à l’AS (et plus généralement aux glucane-saccharases) et ont
conduit à l’identification de variants plus actifs que l’AS sauvage à partir d’un échantillon de
150 mutants actifs provenant d’une banque de première génération générée par une approche
combinant la PCR à erreurs et le DNA shuffling (van der Veen et al., 2004; van der Veen et
al., 2006). Cependant, les protocoles de criblage développés au cours de ce projet n’étaient
pas optimisés pour le criblage fiable de banques (ou de sous-banques) de taille plus
importante. Leur inconvénient principal était en particulier lié au faible niveau d’expression
au format microplaque des variants obtenus à partir du vecteur d’expression (pZErO) utilisé
pour le clonage des banques.

Conclusion
144
Dans ce contexte, l’approche que nous avons choisie pour atteindre nos objectifs a été
décomposée en trois étapes :
1- Adapter la technique MutaGenTM à un gène de taille importante (2 kb), codant
l’amylosaccharase de N. polysaccharea et analyser la diversité générée avec cette méthode.
2- Optimiser le procédé de sélection et de criblage des banques de variants afin de
pouvoir analyser un plus grand nombre de clones et identifier de manière fiable des mutants
plus thermostables ou plus résistants à la dénaturation par les solvants organiques.
3- Analyser les propriétés des variants thermostables de l’AS isolés au cours du
criblage des banques générées via la technique MutaGenTM et identifier les déterminants
structuraux à l’origine de l’amélioration de la stabilité thermique chez ces variants.
Nous nous proposons à présent de faire le bilan de nos travaux et d’en dégager les
perspectives.
Construction et analyse des banques de variants de l’AS avec la technique MutaGenTM
Trois banques de variants de l’AS ont été construites en appliquant la méthode MutaGenTM à
la séquence codant l’AS sauvage. Ces banques ont été générées de façon à introduire
aléatoirement des mutations sur toute la séquence codante (banques A et B) ou sur un segment
codant pour une région de l’enzyme située entre les résidus 242 et 470 (dans laquelle sont
localisés des résidus clés pour l’activité catalytique de l’enzyme ; banque C). Les banques A
et B ont été construites en employant respectivement les ADN polymérases pol β et pol η. La
banque C a, quant à elle, été créée avec pol η. L’analyse de la diversité générée dans chaque
banque a montré que leurs caractéristiques (taux de mutations, substitutions, délétions et
additions) dépendent de l’ADN polymérase employée. Ainsi, les variants générés avec pol β
(banque A) présentent un taux de mutation quatre à sept fois inférieur à ceux générés avec pol
η (banques B et C). Ces résultats montrent que la charge mutationnelle moyenne des membres
d’une banque peut être modulée en fonction de l’ADN polymérase employée. D’autre part, la
représentation des différents types de mutations varie selon les banques. Ainsi, la technique
MutaGenTM permet d'accéder à une diversité différente en fonction de l'ADN polymérase
utilisée et de générer des variants combinant plusieurs types de mutations. En particulier,
certains variants obtenus avec pol η sont affectés par des délétions de plusieurs bases
successives qui peuvent entraîner l'élimination de résidus au niveau de la séquence protéique

Conclusion
145
primaire. La méthode MutaGenTM permet donc d'obtenir des mutants possédant des
modifications rarement observées en utilisant d'autres techniques de mutagénèse aléatoire.
Notre travail, ainsi que celui rapporté récemment par Mondon et al. (2007), démontre donc le
potentiel de la technique MutaGenTM pour la génération de diversité à travers l'emploi de deux
ADN polymérases présentant des propriétés très différentes, pol β et pol η. MilleGen
développe actuellement cette méthode via l'utilisation d'autres polymérases comme pol λ, pol
ι ou pol κ (voir tableau 2). Ces travaux devraient permettre d'élargir la diversité accessible
par cette approche.
Optimisation du procédé de sélection et de criblage des banques de variants de l’AS
Sur la base du travail décrit par Van der Veen et al. (2004), nous avons développé et optimisé
une procédure permettant d'identifier des variants de l'AS plus thermostables ou plus
résistants à la dénaturation par les solvants organiques. Elle comporte deux étapes
successives:
1- Un test de sélection in vivo permettant d'éliminer les variants inactifs de l'AS.
2- Le criblage automatisé des variants actifs permettant d'identifier ceux possédant une
amélioration pour la propriété désirée.
L'augmentation du niveau d’expression au format microplaque des variants de l'AS était
requise pour améliorer la sensibilité du protocole de criblage initial. Nous avons donc
remplacé le plasmide pZErO par le vecteur d'expression pGEX qui permet de produire 10 à
50 fois plus d'enzyme. La variabilité du protocole de criblage a ensuite été minimisée en
standardisant et automatisant chacune de ses étapes : mise en culture et croissance des
bactéries en microplaque, induction de l’expression de l’enzyme, lyse des cellules, réaction
enzymatique et dosage colorimétrique des sucres réducteurs. L'expression de l'AS
recombinante au format microplaque a été normalisée en diminuant le coefficient de variance
de 27 à 12.5% notamment grâce à l'utilisation d'une étape de pré-culture en plaque 96-puits
des clones bactériens. Des conditions sélectives ont ensuite été définies de manière à isoler
des variants de l'AS plus thermostables ou résistants au DMSO.
Nous avons utilisé cette procédure afin d'identifier des variants thermostables de l'AS dans les
banques A et B. Environ 7000 variants actifs ont été sélectionnés à partir de 30000 clones
provenant de chaque banque. Le criblage de ces variants a permis d'identifier trois mutants
plus thermostables: R20C/A451T, A170V/Q353L et P351S. Cependant, aucun mutant

Conclusion
146
résistant aux solvants organiques n'a été isolé après criblage en présence de DMSO (30 et
50%). Ce résultat est probablement dû à la trop forte stringence des conditions que nous avons
employées. Il reflète également la difficulté à définir les conditions d'une expérience
d'évolution dirigée. Le criblage de banques de taille plus grande dans ces conditions de forte
stringence permettrait d'augmenter la probabilité de détecter des variants présentant des
modifications importantes et originales par rapport à l'enzyme parentale. Alternativement,
l'utilisation de conditions moins sélectives au cours du (ou des) premier(s) cycle(s) d'évolution
dirigée pourrait conduire à l'identification de mutants présentant une amélioration minime de
la propriété ciblée. L'augmentation graduelle de la stringence du criblage au cours des cycles
de mutations/recombinaisons suivants permettrait d’identifier des variants possédant
l'amélioration recherchée.
Limitations du système d'expression utilisé
Le protocole de criblage que nous avons optimisé repose sur l'utilisation du système
d'expression pGEX qui permet la production de l'AS recombinante en fusion N-terminale
avec la GST. La présence de cette étiquette protéique n'affecte pas les propriétés catalytiques
de l'enzyme (Potocki de Montalk et al., 1999) mais semble en revanche avoir une influence
négative sur sa stabilité au stockage (Potocki de Montalk et al., 1999) et à la température
(t1/2(50°C) = 1 min pour la GST-AS, 3 min pour l'AS). Cette observation pourrait être due à
une plus forte tendance à l'agrégation pour la protéine de fusion, probablement liée à la
dimérisation de la GST (Tudyka and Skerra, 1997). L’influence positive ou négative de
l’étiquette GST sur la thermostabilité est cependant variable en fonction des variants de l'AS
identifiés au cours de notre travail. Ces variations pourraient être liées aux différences
structurales identifiées par modélisation moléculaire. Il est donc difficile de discuter
l'influence de l'étiquette GST dans le cadre du criblage des banques. Une meilleure
caractérisation des différences de stabilité entre les formes fusionnées et non fusionnées de
l'AS, en relation avec les modifications structurales induites lors de leur incubation à haute
température, pourrait être réalisée par dichroïsme circulaire, spectroscopie de fluorescence et
calorimétrie à titration isothermique.

Conclusion
147
Système de sélection ou de criblage alternatif
Un protocole de sélection génétique (ou de criblage visuel) in vivo permettrait d'identifier des
protéines hautement thermostables en explorant des banques de plus grande taille par rapport
aux techniques de criblage en milieu liquide (voir tableau 10). En général, ce type de stratégie
consiste à cultiver l'hôte d'expression sur milieu solide à des températures élevées non tolérées
par l'enzyme d'intérêt. Les variants thermostables peuvent ainsi être isolés par sélection
génétique ou par criblage visuel. Dans le cas d'Escherichia coli, la température maximale de
croissance est de 45°C. Cultivée à cette température, des colonies exprimant l'AS sauvage se
sont révélées capables de produire de l'amylose (détectée par coloration à l'iode). Escherichia
coli ne constitue donc pas un hôte d'expression adapté pour la recherche in vivo de variants
hyperthermostables de l'AS.
Le criblage visuel sur milieu solide d'AS thermostables peut être envisagé chez E. coli en
découplant les étapes de croissance et de test enzymatique. Dans un premier temps, les
colonies constituant la banque à cribler sont cultivées à 37°C afin de produire les variants de
l'AS. Dans un second temps, l'activité de ces derniers peut être analysée par coloration à l'iode
après ajout de top-agar contenant le substrat de l'enzyme et réaction à des températures plus
élevées (50, 60°C etc.). Le développement de cette approche est en cours au sein de l'équipe
CIMES en utilisant les variants identifiés au cours de cette thèse comme témoins positifs pour
valider les essais de mise au point.
Il est également possible de développer un protocole de sélection ou de criblage in vivo
d'enzymes thermostables en utilisant un hôte d'expression thermophile comme Thermus
thermophilus, dont la température optimale de croissance est de 70°C. En effet, cette souche a
été employée pour isoler par évolution dirigée des mutants thermostables de la 3-
isopropylmalate de S. cerevisiae (Tamakoshi et al., 2001) ou de la protéine de liaison à la
bléomycine (Brouns et al., 2005). Au cours de ces travaux de thèse, nous avons envisagé le
développement d’un protocole de sélection positive ou de criblage sur colonies (détection de
l'amylose produite par coloration à l'iode) en exprimant l’AS chez Th. thermophilus. La
séquence codant pour l’enzyme sauvage a été clonée dans le vecteur navette E. coli/Th.
thermophilus pMK18 (de Grado et al., 1999). Cependant, par manque de temps nous n'avons
pas pu finaliser le développement de cette stratégie.

Conclusion
148
Amylose synthétisée à 50°C
Parmi les variants thermostables identifiés, le double mutant R20C/A451T est à ce jour la
seule amylosaccharase active à 50°C. L'amylose synthétisée par ce variant à cette température
à partir de 600 mM de saccharose présente un degré de polymérisation moyen de 52, proche
de celui de l'amylose formée avec un rendement trois moins important par l’enzyme sauvage à
30°C à partir de 100 mM de substrat. De plus, en élevant la température réactionnelle de 30 à
50°C, le degré de polymérisation moyen des chaînes synthétisées à partir de 600 mM de
saccharose augmente de 39 à 52. Le passage de 30 à 50°C accroît probablement la solubilité
des chaînes d’amylose en cours de synthèse. Ces dernières restent ainsi plus longtemps
accessibles au site actif de l’enzyme, ce qui permet la production de chaînes plus longues qu’à
plus basse température. Les propriétés du polymère synthétisé à 50°C par le variant
R20C/A451T pourront être analysées en déterminant ses caractéristiques morphologiques (par
microscopie électronique à transmission) et cristallines (par diffraction des rayons X). Dans
une perspective d'application dans l’industrie agroalimentaire, il sera également intéressant de
mesurer sa teneur en amidon résistant et de la comparer avec les valeurs obtenues pour les
amyloses synthétisées par l'AS sauvage.
Déterminants structuraux à l’origine de l’amélioration de la thermostabilité
Les cinq mutations observées dans les trois variants thermostables identifiés après criblage
sont réparties sur l'ensemble de la structure de l'AS: trois sont situées dans le domaine
catalytique A (Ala170Val, Pro351Ser, Gln353Leu), une dans le domaine B' (Ala451Thr) et
une autre dans le domaine N (Arg20Cys). Deux de ces mutations (Arg20Cys et Ala170Val)
sont localisées à la surface de la protéine. Les trois autres mutations concernent des résidus
enfouis dans la structure de l'enzyme.
L'analyse structurale des variants construits par modélisation moléculaire nous a permis de
déceler et de discuter les mécanismes potentiellement impliqués dans la stabilisation de
l'enzyme. Les différences observées entre l'AS sauvage et le variant le plus thermostable
(R20C/A451T) résident essentiellement dans la réorganisation du pont salin impliquant le
résidu de surface Arg20 et la création d’une liaison hydrogène associée à la substitution de
Ala451 enfoui dans le domaine B' en Thr. Ces observations suggèrent l'importance de ces
déterminants structuraux dans la stabilisation thermique de l'enzyme. Toutefois, à ce stade et

Conclusion
149
malgré ces indications, la corrélation entre les mutations identifiées et le gain de stabilité
observé reste mal comprise.
A la différence de l'ingénierie rationnelle qui requiert une bonne compréhension des facteurs
impliqués dans le mécanisme d'inactivation thermique de l'enzyme d'intérêt (van den Burg
and Eijsink, 2002), l'évolution dirigée permet la recherche de variants améliorés en explorant
la diversité possible à partir de la séquence parentale. Elle ne requiert donc aucune
connaissance a priori des relations structure-activité de la protéine parentale, ce qui peut
constituer un avantage par rapport à l'ingénierie rationnelle lorsque l'objectif est d'améliorer
une propriété telle que la thermostabilité. La détermination du mécanisme d'inactivation
thermique de l'AS et des variants isolés au cours de notre travail permettra de mieux
comprendre le rôle des mutations identifiées dans la stabilisation de l'enzyme. Ceci sera
possible en analysant les propriétés de ces protéines (notamment la température de fusion) par
les techniques de dichroïsme circulaire et de calorimétrie différentielle.
Optimisation de l'amylosaccharase
La stratégie globale dans laquelle s'intègrent nos travaux a pour objectif d'optimiser les
potentialités de l'amylosaccharase comme biocatalyseur d'intérêt industriel. Les variants que
nous avons isolés constituent les amylosaccharases les plus stables identifiées à ce jour et l'un
d'entre eux (R20C/A451T) est capable de synthétiser de l'amylose à des températures
réactionnelles non tolérées par l'AS sauvage. Deux perspectives générales peuvent être
dégagées à partir de ces résultats.
La première perspective concerne la suite de nos travaux, à savoir l'amélioration de la
thermostabilité de l'amylosaccharase. Les mutants identifiés ici sont issus d'un premier cycle
d'évolution dirigée. Deux voies générales sont envisageables à partir de ces résultats. Une
première stratégie consistera à poursuivre l’évolution dirigée de l’AS sur la base des méthodes
développées au cours de cette thèse. Il s’agira de construire par mutagénèse et/ou
recombinaison aléatoire des banques de seconde (puis de troisième etc.) génération afin
d'isoler par criblage des variants de plus en plus thermostables en accumulant des mutations
bénéfiques. L'augmentation de la stringence du criblage à chaque cycle constituera le point
critique majeur de cette approche, dans la mesure où cela permettra d'identifier de nouveaux
variants, différents de ceux de la génération précédente. La seconde voie visera à mieux
comprendre le mécanisme d'inactivation thermique de l'enzyme (voir paragraphe précédent)
afin d’établir une approche d’ingénierie rationnelle (ou semi-rationnelle), complémentaire de

Conclusion
150
la précédente. Elle consistera à évaluer le rôle et l'additivité des mutations identifiées par
criblage à travers la construction (par mutagénèse dirigée) et l'analyse (par dichroïsme
circulaire et calorimétrie différentielle) des mutants combinant une à plusieurs substitutions
présentes dans les variants initiaux. Les deux approches envisagées pourront également
bénéficier de l'identification de nouvelles amylosaccharases provenant d'organismes
thermophiles (Voir annexe 2)(Emond et al., 2008a). Ces enzymes permettront, d'une part,
d'élargir l'analyse des paramètres structuraux impliqués dans la stabilité thermique de cette
famille de protéines et, d'autre part, d'entamer la construction d'amylosaccharases chimèriques
par le biais des techniques de family shuffling.
Enfin, une seconde perspective envisagée consiste en la construction de nouveaux outils de
glucosylation thermostables grâce à la combinaison des mutations identifiées au cours de nos
travaux avec celles identifiées par ingénierie rationnelle et impliquées dans la spécificité de
l'AS (Albenne et al., 2002a; Albenne et al., 2004). Cette approche permettra à terme d’obtenir
des variants de l’amylosaccharase capables de glucosyler des accepteurs non reconnus par
l’enzyme sauvage et dans des conditions réactionnelles non tolérées par cette dernière.
Ref tableaux+figures
(Acharya et al., 2004; Alcalde et al., 2005; Andrews et al., 2004; Bessler et al., 2003; Bittker et al., 2004; Camps et al., 2003; Castle et al.,
2004; Chen and Arnold, 1993; Cherry et al., 1999; Delagrave et al., 2001; Evnin et al., 1990; Fernandez-Gacio et al., 2003; Giver et al.,
1998; Griffiths and Tawfik, 2006; Griffiths et al., 2004; Hoseki et al., 1999; Hsu et al., 2005; Joo et al., 1999a; Joo et al., 1999b; Ju et al.,
2005; Kadonaga and Knowles, 1985; Kajiyama and Nakano, 1993; Kim et al., 2004; Kim et al., 2000; Lai et al., 2004; Leemhuis et al., 2003;
Liao et al., 1986; Liebeton et al., 2000; Matsumura et al., 1999; Matsumura and Aiba, 1985; Mayer et al., 2001; Miyazaki et al., 2006; Moore
and Arnold, 1996; Murakami et al., 2002; Myers et al., 1985; Osanjo et al., 2007; Ostermeier et al., 1999; Otten et al., 2002; Oue et al., 1999;
Palackal et al., 2004; Parikh et al., 2006; Parikh and Matsumura, 2005; Patten et al., 1997; Peng and Shaw, 1996; Pfeifer et al., 2005; Sidhu
and Borgford, 1996; Sio et al., 2002; Stratagene, 2004; van Loo et al., 2004; Wong et al., 2004a; Wong et al., 2004b; Wong et al., 2004c;
Yano et al., 1998; You and Arnold, 1996; Zhang et al., 1997)
(Arnold et al., 2000; Bengs and Braunagel, 1998a, b; Bengs et al., 1998a; Bengs et al., 1997; Bengs et al., 1998b; Bengs et al., 1999; Bengs
et al., 1998c; Bengs et al., 1998d; Bengs et al., 1998e; Bengs et al., 1998f; Bengs et al., 2000; Boersma et al., 2008; Delcourt and Blesa,
2002; Dupret et al., 2002; Hausmanns et al., 2000; Kossmann et al., 1994; Quanz, 1999; Quanz and Frohberg, 2003; Quanz and Provart,
1998; Quanz et al., 1998; Reetz et al., 2004a; Reetz et al., 2005; Short, 2001; Stemmer, 1997, 2003; Stemmer and Crameri, 1998) (Remaud-
Simeon et al., 1995; Weissmuller et al., 2000)

151
Références bibliographiques

Références bibliographiques
152
Abecassis, V., Pompon, D., and Truan, G. (2000). High efficiency family shuffling based on multi-step PCR and in vivo DNA recombination in yeast: statistical and functional analysis of a combinatorial library between human cytochrome P450 1A1 and 1A2. Nucleic Acids Res 28, e88.
Acharya, P., Rajakumara, E., Sankaranarayanan, R., and Rao, N.M. (2004). Structural basis of selection and thermostability of laboratory evolved Bacillus subtilis lipase. J Mol Biol 341, 1271-1281.
Aharoni, A., Thieme, K., Chiu, C.P., Buchini, S., Lairson, L.L., Chen, H., Strynadka, N.C., Wakarchuk, W.W., and Withers, S.G. (2006). High-throughput screening methodology for the directed evolution of glycosyltransferases. Nat Methods 3, 609-614.
Airaksinen, A., and Hovi, T. (1998). Modified base compositions at degenerate positions of a mutagenic oligonucleotide enhance randomness in site-saturation mutagenesis. Nucleic Acids Res 26, 576-581.
Albenne, C. (2002). Ingénierie rationnelle de l'amylosaccharase de Neisseria polysaccharea : de la structure tridimensionnelle aux bases moléculaire de la catalyse (INSA Toulouse, France).
Albenne, C., de Montalk, G.P., Monsan, P., Skov, L.K., Mirza, O., Gajhede, M., and Remaud-Simeon, M. (2002a). Site-directed mutagenesis of key amino acids in the active site of amylosucrase from Neisseria polysaccharea. Biologia, Bratislava 57, 119-128.
Albenne, C., Skov, L.K., Mirza, O., Gajhede, M., Feller, G., D'Amico, S., Andre, G., Potocki-Veronese, G., van der Veen, B.A., Monsan, P., et al. (2004). Molecular basis of the amylose-like polymer formation catalyzed by Neisseria polysaccharea amylosucrase. J Biol Chem 279, 726-734.
Albenne, C., Skov, L.K., Mirza, O., Gajhede, M., Potocki-Veronese, G., Monsan, P., and Remaud-Simeon, M. (2002b). Maltooligosaccharide disproportionation reaction: an intrinsic property of amylosucrase from Neisseria polysaccharea. FEBS Lett 527, 67-70.
Albenne, C., Skov, L.K., Tran, V., Gajhede, M., Monsan, P., Remaud-Simeon, M., and Andre-Leroux, G. (2007). Towards the molecular understanding of glycogen elongation by amylosucrase. Proteins 66, 118-126.
Alcalde, M., Bulter, T., Zumarraga, M., Garcia-Arellano, H., Mencia, M., Plou, F.J., and Ballesteros, A. (2005). Screening mutant libraries of fungal laccases in the presence of organic solvents. Journal of Biomolecular Screening 10, 624-631.
Amstutz, P., Pelletier, J.N., Guggisberg, A., Jermutus, L., Cesaro-Tadic, S., Zahnd, C., and Pluckthun, A. (2002). In vitro selection for catalytic activity with ribosome display. Journal of the American Chemical Society 124, 9396-9403.
Andrews, S.R., Taylor, E.J., Pell, G., Vincent, F., Ducros, V.M., Davies, G.J., Lakey, J.H., and Gilbert, H.J. (2004). The use of forced protein evolution to investigate and improve stability of family 10 xylanases. The production of Ca2+-independent stable xylanases. J Biol Chem 279, 54369-54379.

Références bibliographiques
153
Arnold, F.H., Shao, Z., Affholter, J.A., Zhao, H., and Giver, L. (1998). Recombination of polynucleotide sequences using random or defined primers. Patent WO/1998/42832.
Arnold, F.H., Shao, Z., Affholter, J.A., Zhao, H., and Giver, L. (2000). Recombination of polynucleotide sequences using random or defined primers. Patent US6153410
Arnold, F.H., Sieber, V., and Zhang, J.H. (2001). Production of functional hybrid genes and proteins. Patent WO/2001/30998.
Baker, K., Bleczinski, C., Lin, H., Salazar-Jimenez, G., Sengupta, D., Krane, S., and Cornish, V.W. (2002). Chemical complementation: a reaction-independent genetic assay for enzyme catalysis. Proc Natl Acad Sci U S A 99, 16537-16542.
Banks, W., Greenwood, C.T., and Khan, K.M. (1971). The interaction of linear, amylose oligomers with iodine Carbohydrate Research 17, 25-33.
Beard, W.A., and Wilson, S.H. (2003). Structural insights into the origins of DNA polymerase fidelity. Structure 11, 489-496.
Bebenek, K., Garcia-Diaz, M., Blanco, L., and Kunkel, T.A. (2003). The frameshift infidelity of human DNA polymerase λ. Implications for function. J Biol Chem 278, 34685-34690.
Bebenek, K., and Kunkel, T.A. (2000). Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harb Symp Quant Biol 65, 81-91.
Bebenek, K., and Kunkel, T.A. (2004). Functions of DNA polymerases. Adv Protein Chem 69, 137-165.
Bebenek, K., Matsuda, T., Masutani, C., Hanaoka, F., and Kunkel, T.A. (2001). Proofreading of DNA polymerase η-dependent replication errors. J Biol Chem 276, 2317-2320.
Becker, S., Michalczyk, A., Wilhelm, S., Jaeger, K.E., and Kolmar, H. (2007). Ultrahigh-throughput screening to identify E. coli cells expressing functionally active enzymes on their surface. Chembiochem 8, 943-949.
Bengs, H., and Braunagel, A. (1998a). Method of filtering UV-light (water insoluble linear poly-α-glucan. Patent WO0038645.
Bengs, H., and Braunagel, A. (1998b). Sun protection product with microparticles on the basis of water-insoluble linear polyglucan. Patent WO0038622.
Bengs, H., Brunner, A., and Schuth, S. (1998a). Use of α-1,4-D-glucan and/or resistant starch as flour substituent. Patent WO0038528.
Bengs, H., Grande, J., Schneller, A., and Boehm, G. (1997). Spherical microparticles containing linear polysacchaides. Patent WO9911695.
Bengs, H., Jacobasch, G., Schmiedl, D., Riesmeier, J., Quanz, M., Baeuerlein, M., and Provart, N. (1998b). α-amylase resistant polysaccharides, production method and use thereof and food products containing said polysaccharides. Patent WO0002926.

Références bibliographiques
154
Bengs, H., Polakowski, T., Held, A., and Gallert, K.C. (1999). Novel immobilized amylosucrase, the use thereof and method for producing poly (1,4)-α-glucan. Patent WO0125449.
Bengs, H., Schneller, A., Grande, J., Schuth, S., Boehm, G., and Braunagel, A. (1998c). Cosmetic or medical preparation for topical use (spherical microparticles). Patent WO0038623.
Bengs, H., Schuth, S., and Brunner, A. (1998d). Oral hygiene product containing a linear water-insoluble poly-α-glucan. Patent WO0038642.
Bengs, H., Schuth, S., Grande, J., and Boehm, G. (1998e). Oral hygiene product containing spherical microparticles on the basis of linear water-insoluble polyglucans. Patent WO0038643.
Bengs, H., Schuth, S., Grande, J., Boehm, G., and Schneller, A. (1998f). Controlled release tablet produced from linear polysaccharides. Patent WO9952558.
Bengs, H., Tomka, I., and Mueller, R. (2000). Gel comprised of a poly-α- (1,4)- glucan and starch. Patent WO0185836.
Benkovic, S.J., Ostermeier, M., Nixon, A.E., and Lutz, S. (2000). Construction of incremental truncation libraries. Patent WO/2000/72013.
Bertrand, A., Morel, S., Lefoulon, F., Rolland, Y., Monsan, P., and Remaud-Simeon, M. (2006). Leuconostoc mesenteroides glucansucrase synthesis of flavonoid glucosides by acceptor reactions in aqueous-organic solvents. Carbohydrate Research 341, 855-863.
Bessler, C., Schmitt, J., Maurer, K.H., and Schmid, R.D. (2003). Directed evolution of a bacterial α-amylase: toward enhanced pH-performance and higher specific activity. Protein Science 12, 2141-2149.
Bittker, J.A., Le, B.V., Liu, J.M., and Liu, D.R. (2004). Directed evolution of protein enzymes using nonhomologous random recombination. Proc Natl Acad Sci U S A 101, 7011-7016.
Bloom, J.D., Meyer, M.M., Meinhold, P., Otey, C.R., MacMillan, D., and Arnold, F.H. (2005). Evolving strategies for enzyme engineering. Curr Opin Struct Biol 15, 447-452.
Boersma, Y.L., Droge, M.J., van der Sloot, A.M., Pijning, T., Cool, R.H., Dijkstra, B.W., and Quax, W.J. (2008). A Novel Genetic Selection System for Improved Enantioselectivity of Bacillus subtilis Lipase A. Chembiochem 9, 1110-1115.
Bolon, D.N., Voigt, C.A., and Mayo, S.L. (2002). De novo design of biocatalysts. Curr Opin Chem Biol 6, 125-129.
Botstein, D., and Shortle, D. (1985). Strategies and applications of in vitro mutagenesis. Science 229, 1193-1201.
Bouayadi, K., Kharrat, H., Louat, T., Servant, L., Cazaux, C., and Hoffmann, J.S. (2002). Use of mutagenic polymerase for producing random mutations. Patent WO/2002/0238756.

Références bibliographiques
155
Boudsocq, F., Kokoska, R.J., Plosky, B.S., Vaisman, A., Ling, H., Kunkel, T.A., Yang, W., and Woodgate, R. (2004). Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J Biol Chem 279, 32932-32949.
Bradford, M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry 72, 248-254.
Brouns, S.J., Wu, H., Akerboom, J., Turnbull, A.P., de Vos, W.M., and van der Oost, J. (2005). Engineering a selectable marker for hyperthermophiles. J Biol Chem 280, 11422-11431.
Buleon, A., and Colonna, P. (2007). Physicochemical Behaviour of Starch in Food Applications. In The chemical physics of food, P. Belton, ed. (Oxford, Blackwell Publishing Ltd).
Buttcher, V., Quanz, M., and Willmitzer, L. (1999). Molecular cloning, functional expression and purification of a glucan branching enzyme from Neisseria denitrificans. Biochim Biophys Acta 1432, 406-412.
Buttcher, V., Welsh, T., Willmitzer, L., and Kossmann, J. (1997). Cloning and characterization of the gene for amylosucrase from Neisseria polysaccharea: production of a linear α-1,4-glucan. J Bacteriol 179, 3324-3330.
Cadwell, R.C., and Joyce, G.F. (1992). Randomization of genes by PCR mutagenesis. PCR Methods Appl 2, 28-33.
Camps, M., Naukkarinen, J., Johnson, B.P., and Loeb, L.A. (2003). Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proc Natl Acad Sci U S A 100, 9727-9732.
Castle, L.A., Siehl, D.L., Gorton, R., Patten, P.A., Chen, Y.H., Bertain, S., Cho, H.J., Duck, N., Wong, J., Liu, D., et al. (2004). Discovery and directed evolution of a glyphosate tolerance gene. Science 304, 1151-1154.
Champion, E., Remaud-Simeon, M., Skov, L.K., Kastrup, J.S., Gajhede, M., and Mirza, O. (2009). The apo structure of sucrose hydrolase from Xanthomonas campestris pv. campestris shows an open active-site groove. Acta Crystallogr D Biol Crystallogr 65, 1309-1314.
Chen, K., and Arnold, F.H. (1993). Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc Natl Acad Sci U S A 90, 5618-5622.
Cherry, J.R., and Fidantsef, A.L. (2003). Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol 14, 438-443.
Cherry, J.R., Lamsa, M.H., Schneider, P., Vind, J., Svendsen, A., Jones, A., and Pedersen, A.H. (1999). Directed evolution of a fungal peroxidase. Nat Biotechnol 17, 379-384.

Références bibliographiques
156
Chodorge, M., Fourage, L., Ravot, G., Jermutus, L., and Minter, R. (2008). In vitro DNA recombination by L-Shuffling during ribosome display affinity maturation of an anti-Fas antibody increases the population of improved variants. Protein Engineering, Design and Selection 21, 343-351.
Cline, J., Braman, J.C., and Hogrefe, H.H. (1996). PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 24, 3546-3551.
Cline, J., and Hogrefe, H.H. (2000). Randomize gene sequences with new PCR mutagenesis kit. Strategies 13, 157–161.
Coco, W.M., Levinson, W.E., Crist, M.J., Hektor, H.J., Darzins, A., Pienkos, P.T., Squires, C.H., and Monticello, D.J. (2001). DNA shuffling method for generating highly recombined genes and evolved enzymes. Nat Biotechnol 19, 354-359.
Cohen, H.M., Tawfik, D.S., and Griffiths, A.D. (2004). Altering the sequence specificity of HaeIII methyltransferase by directed evolution using in vitro compartmentalization. Protein Engineering, Design and Selection 17, 3-11.
Crameri, A., Raillard, S.A., Bermudez, E., and Stemmer, W.P. (1998). DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature 391, 288-291.
Cwirla, S.E., Peters, E.A., Barrett, R.W., and Dower, W.J. (1990). Peptides on phage: a vast library of peptides for identifying ligands. Proc Natl Acad Sci U S A 87, 6378-6382.
Davies, G.J., Wilson, K.S., and Henrissat, B. (1997). Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J 321, 557-559.
de Grado, M., Castan, P., and Berenguer, J. (1999). A high-transformation-efficiency cloning vector for Thermus thermophilus. Plasmid 42, 241-245.
Delagrave, S., Murphy, D.J., Pruss, J.L., Maffia, A.M., 3rd, Marrs, B.L., Bylina, E.J., Coleman, W.J., Grek, C.L., Dilworth, M.R., Yang, M.M., et al. (2001). Application of a very high-throughput digital imaging screen to evolve the enzyme galactose oxidase. Protein Eng 14, 261-267.
DeLano, W.L. (2002). The PyMOL Molecular Graphics System (2002) on World Wide Web http://www.pymol.org.
Delcourt, M., and Blesa, S. (2002). Method for massive mutagenesis. Patent WO/2002/16606.
Demartis, S., Huber, A., Viti, F., Lozzi, L., Giovannoni, L., Neri, P., Winter, G., and Neri, D. (1999). A strategy for the isolation of catalytic activities from repertoires of enzymes displayed on phage. J Mol Biol 286, 617-633.
DeSantis, G., Wong, K., Farwell, B., Chatman, K., Zhu, Z., Tomlinson, G., Huang, H., Tan, X., Bibbs, L., Chen, P., et al. (2003). Creation of a productive, highly enantioselective nitrilase through gene site saturation mutagenesis (GSSM). J Am Chem Soc 125, 11476-11477.

Références bibliographiques
157
Devlin, J.J., Panganiban, L.C., and Devlin, P.E. (1990). Random peptide libraries: a source of specific protein binding molecules. Science 249, 404-406.
Ditursi, M.K., Kwon, S.J., Reeder, P.J., and Dordick, J.S. (2006). Bioinformatics-driven, rational engineering of protein thermostability. Protein Engineering, Design and Selection 19, 517-524.
Doi, N., Kumadaki, S., Oishi, Y., Matsumura, N., and Yanagawa, H. (2004). In vitro selection of restriction endonucleases by in vitro compartmentalization. Nucleic Acids Res 32, e95.
Dupret, D., Lefevre, F., and Masson, J.M. (2002). Template-mediated, ligation-oriented method of nonrandomly shuffling polynucleotides. Patent WO/2002/086121.
Eckert, K.A., and Kunkel, T.A. (1990). High fidelity DNA synthesis by the Thermus aquaticus DNA polymerase. Nucleic Acids Res 18, 3739-3744.
Eijsink, V.G., Bjork, A., Gaseidnes, S., Sirevag, R., Synstad, B., van den Burg, B., and Vriend, G. (2004). Rational engineering of enzyme stability. J Biotechnol 113, 105-120.
Emond, S., Mondeil, S., Jaziri, K., Andre, I., Monsan, P., Remaud-Simeon, M., and Potocki-Veronese, G. (2008a). Cloning, purification and characterization of a thermostable amylosucrase from Deinococcus geothermalis. FEMS Microbiol Lett 285, 25-32.
Emond, S., Mondon, P., Pizzut-Serin, S., Douchy, L., Crozet, F., Bouayadi, K., Kharrat, H., Potocki-Veronese, G., Monsan, P., and Remaud-Simeon, M. (2008b). A novel random mutagenesis approach using human mutagenic DNA polymerases to generate enzyme variant libraries. Protein Engineering Design & Selection 21, 267-274.
Emond, S., Potocki-Veronese, G., Mondon, P., Bouayadi, K., Kharrat, H., Monsan, P., and Remaud-Simeon, M. (2007). Optimized and automated protocols for high-throughput screening of amylosucrase libraries. Journal of Biomolecular Screening 12, 715-723.
Evnin, L.B., Vasquez, J.R., and Craik, C.S. (1990). Substrate specificity of trypsin investigated by using a genetic selection. Proc Natl Acad Sci U S A 87, 6659-6663.
Fa, M., Radeghieri, A., Henry, A.A., and Romesberg, F.E. (2004). Expanding the substrate repertoire of a DNA polymerase by directed evolution. J Am Chem Soc 126, 1748-1754.
Fernandez-Gacio, A., Uguen, M., and Fastrez, J. (2003). Phage display as a tool for the directed evolution of enzymes. Trends Biotechnol 21, 408-414.
Flores-Ramirez, G., Rivera, M., Morales-Pablo, A., Osuna, J., Soberon, X., and Gaytan, P. (2007). The effect of amino acid deleltions and substitutions in the longest loop of GFP. BMC Chemical Biology 7.
Fox, R.J., Davis, S.C., Mundorff, E.C., Newman, L.M., Gavrilovic, V., Ma, S.K., Chung, L.M., Ching, C., Tam, S., Muley, S., et al. (2007). Improving catalytic function by ProSAR-driven enzyme evolution. Nat Biotechnol 25, 338-344.
Friedberg, E.C., Fischhaber, P.L., and Kisker, C. (2001). Error-prone DNA polymerases: novel structures and the benefits of infidelity. Cell 107, 9-12.

Références bibliographiques
158
Fujii, R., Kitaoka, M., and Hayashi, K. (2006). RAISE: a simple and novel method of generating random insertion and deletion mutations. Nucleic Acids Research 34, e30.
Funke, S.A., Eipper, A., Reetz, M.T., Otte, N., Thiel, W., Van Pouderoyen, G., Dijkstra, B.W., Jaeger, K.-E., and Eggert, T. (2003). Directed Evolution of an Enantioselective Bacillus subtilis Lipase. Biocatalysis and Biotransformation 21, 67 - 73.
Garcia-Diaz, M., and Kunkel, T.A. (2006). Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci 31, 206-214.
Gibbs, M.D., Bergquist, P.L., and Nevalainen, K.M. (2002). Degenerate oligonucleotide gene shuffling. Patent WO/2002/18629.
Gibbs, M.D., Nevalainen, K.M., and Bergquist, P.L. (2001). Degenerate oligonucleotide gene shuffling (DOGS): a method for enhancing the frequency of recombination with family shuffling. Gene 271, 13-20.
Giver, L., Gershenson, A., Freskgard, P.O., and Arnold, F.H. (1998). Directed evolution of a thermostable esterase. Proc Natl Acad Sci U S A 95, 12809-12813.
Goddard, J.P., and Reymond, J.L. (2004). Enzyme assays for high-throughput screening. Curr Opin Biotechnol 15, 314-322.
Goodman, M.F., Creighton, S., Bloom, L.B., and Petruska, J. (1993). Biochemical basis of DNA replication fidelity. Crit Rev Biochem Mol Biol 28, 83-126.
Griffiths, A.D., and Tawfik, D.S. (2003). Directed evolution of an extremely fast phosphotriesterase by in vitro compartmentalization. Embo J 22, 24-35.
Griffiths, A.D., and Tawfik, D.S. (2006). Miniaturising the laboratory in emulsion droplets. Trends Biotechnol 24, 395-402.
Griffiths, J.S., Cheriyan, M., Corbell, J.B., Pocivavsek, L., Fierke, C.A., and Toone, E.J. (2004). A bacterial selection for the directed evolution of pyruvate aldolases. Bioorg Med Chem 12, 4067-4074.
Grishin, N.V. (2001). Fold Change in Evolution of Protein Structures. Journal of Structural Biology 134, 167-185.
Griswold, K.E., Kawarasaki, Y., Ghoneim, N., Benkovic, S.J., Iverson, B.L., and Georgiou, G. (2005). Evolution of highly active enzymes by homology-independent recombination. Proc Natl Acad Sci U S A 102, 10082-10087.
Haney, P.J., Badger, J.H., Buldak, G.L., Reich, C.I., Woese, C.R., and Olsen, G.J. (1999). Thermal adaptation analyzed by comparison of protein sequences from mesophilic and extremely thermophilic Methanococcus species. Proc Natl Acad Sci U S A 96, 3578-3583.
Hausmanns, S., Kiy, T., Tomka, I., and Mueller, R. (2000). Soft capsules comprising a starch mixture having reduced branching degree. Patent WO0238132

Références bibliographiques
159
Hehre, E.J., and Hamilton, D.M. (1946). Bacterial synthesis of an amylopectin-like polysaccharide from sucrose. The Journal of Biological Chemistry 166, 777-778.
Hendsch, Z.S., and Tidor, B. (1994). Do salt bridges stabilize proteins ? A continuum electrostatic analysis. Protein Science 3, 211-226.
Hoogenboom, H.R., Griffiths, A.D., Johnson, K.S., Chiswell, D.J., Hudson, P., and Winter, G. (1991). Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res 19, 4133-4137.
Hoseki, J., Yano, T., Koyama, Y., Kuramitsu, S., and Kagamiyama, H. (1999). Directed evolution of thermostable kanamycin-resistance gene: a convenient selection marker for Thermus thermophilus. J Biochem (Tokyo) 126, 951-956.
Hsu, C.C., Hong, Z., Wada, M., Franke, D., and Wong, C.H. (2005). Directed evolution of D-sialic acid aldolase to L-3-deoxy-manno-2-octulosonic acid (L-KDO) aldolase. Proc Natl Acad Sci U S A 102, 9122-9126.
Hytonen, V.P., Maatta, J.A., Nyholm, T.K., Livnah, O., Eisenberg-Domovich, Y., Hyre, D., Nordlund, H.R., Horha, J., Niskanen, E.A., Paldanius, T., et al. (2005). Design and construction of highly stable, protease-resistant chimeric avidins. J Biol Chem 280, 10228-10233.
Jackson, D.S. (1991). Solubility Behavior of Granular Corn Starches in Methyl Sulfoxide (DMSO) as Measured by High Performance Size Exclusion Chromatography. Stärke 43, 422-427.
Janda, K.D., Lo, C.H., Li, T., Barbas, C.F., 3rd, Wirsching, P., and Lerner, R.A. (1994). Direct selection for a catalytic mechanism from combinatorial antibody libraries. Proc Natl Acad Sci U S A 91, 2532-2536.
Jencks, W.P. (1976). Binding energy, specificity, and enzymic catalysis: the Circe effect. Advances in Enzymology 43, 219-410.
Jensen, M.H., Mirza, O., Albenne, C., Remaud-Simeon, M., Monsan, P., Gajhede, M., and Skov, L.K. (2004). Crystal structure of the covalent intermediate of amylosucrase from Neisseria polysaccharea. Biochemistry 43, 3104-3110.
Joern, J.M., Meinhold, P., and Arnold, F.H. (2002). Analysis of shuffled gene libraries. J Mol Biol 316, 643-656.
Joern, J.M., Sakamoto, T., Arisawa, A., and Arnold, F.H. (2001). A versatile high throughput screen for dioxygenase activity using solid-phase digital imaging. Journal of Biomolecular Screening 6, 219-223.
John, M., Schmidt, J., and Kneifel, H. (1983). Iodine—maltosaccharide complexes: relation between chain-length and colour. Carbohydrate Research 119, 254-257.
Jones, D.D. (2005). Triplet nucleotide removal at random positions in a target gene: the tolerance of TEM-1 β-lactamase to an amino acid deletion. Nucleic Acids Res 33, e80.

Références bibliographiques
160
Joo, H., Arisawa, A., Lin, Z., and Arnold, F.H. (1999a). A high-throughput digital imaging screen for the discovery and directed evolution of oxygenases. Chem Biol 6, 699-706.
Joo, H., Lin, Z., and Arnold, F.H. (1999b). Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 399, 670-673.
Ju, J., Misono, H., and Ohnishi, K. (2005). Directed evolution of bacterial alanine racemases with higher expression level. J Biosci Bioeng 100, 246-254.
Kadonaga, J.T., and Knowles, J.R. (1985). A simple and efficient method for chemical mutagenesis of DNA. Nucleic Acids Res 13, 1733-1745.
Kajiyama, N., and Nakano, E. (1993). Thermostabilization of firefly luciferase by a single amino acid substitution at position 217. Biochemistry 32, 13795-13799.
Kikuchi, M., Ohnishi, K., and Harayama, S. (1999). Novel family shuffling methods for the in vitro evolution of enzymes. Gene 236, 159-167.
Kikuchi, M., Ohnishi, K., and Harayama, S. (2000). An effective family shuffling method using single-stranded DNA. Gene 243, 133-137.
Kim, H.S., Park, H.J., Heu, S., and Jung, J. (2004). Molecular and functional characterization of a unique sucrose hydrolase from Xanthomonas axonopodis pv. glycines. J Bacteriol 186, 411-418.
Kim, M.I., Kim, H.S., Jung, J., and Rhee, S. (2008). Crystal structures and mutagenesis of sucrose hydrolase from Xanthomonas axonopodis pv. glycines: insight into the exclusively hydrolytic amylosucrase fold. J Mol Biol 380, 636-647.
Kim, Y.S., Jung, H.C., and Pan, J.G. (2000). Bacterial cell surface display of an enzyme library for selective screening of improved cellulase variants. Appl Environ Microbiol 66, 788-793.
Kornberg, A. (1957). Enzymatic synthesis of deoxyribonucleic acid. Harvey Lect 53, 83-112.
Koshland, D.E. (1953). Stereochemistry and the mechanism of enzymatic reactions. Biological Reviews 28, 416-436.
Kossmann, J., Buttcher, V., and Welsh, T. (1994). DNA sequences coding for enzymes capable of faciliting the synthesis of α-1,4 glucans in plants, fungi and microorganisms. Patent WO9531553.
Kretz, K.A., Richardson, T.H., Gray, K.A., Robertson, D.E., Tan, X., and Short, J.M. (2004). Gene site saturation mutagenesis: a comprehensive mutagenesis approach. Methods Enzymol 388, 3-11.
Kumar, S., and Nussinov, R. (1999). Salt bridge stability in monomeric proteins. J Mol Biol 293, 1241-1255.
Kunkel, T.A. (1985). The mutational specificity of DNA polymerase-beta during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J Biol Chem 260, 5787-5796.

Références bibliographiques
161
Kunkel, T.A. (2004). DNA replication fidelity. J Biol Chem 279, 16895-16898.
Kunkel, T.A., and Bebenek, K. (2000). DNA replication fidelity. Annu Rev Biochem 69, 497-529.
Kunkel, T.A., Pavlov, Y.I., and Bebenek, K. (2003). Functions of human DNA polymerases η, κ and ι suggested by their properties, including fidelity with undamaged DNA templates. DNA Repair (Amst) 2, 135-149.
Kunz, B.A., Straffon, A.F., and Vonarx, E.J. (2000). DNA damage-induced mutation: tolerance via translesion synthesis. Mutat Res 451, 169-185.
Kwon, S.J., Petri, R., DeBoer, A.L., and Schmidt-Dannert, C. (2004). A high-throughput screen for porphyrin metal chelatases: application to the directed evolution of ferrochelatases for metalloporphyrin biosynthesis. Chembiochem 5, 1069-1074.
Lai, Y.P., Huang, J., Wang, L.F., Li, J., and Wu, Z.R. (2004). A new approach to random mutagenesis in vitro. Biotechnol Bioeng 86, 622-627.
Lantto, J., and Ohlin, M. (2002). Functional Consequences of Insertions and Deletions in the Complementarity-determining Regions of Human Antibodies. The Journal of Biological Chemistry 277, 45108-45114.
Lausted, C., Dahl, T., Warren, C., King, K., Smith, K., Johnson, M., Saleem, R., Aitchison, J., Hood, L., and Lasky, S.R. (2004). POSaM: a fast, flexible, open-source, inkjet oligonucleotide synthesizer and microarrayer. Genome Biol 5, R58.
Lee, Y.F., Tawfik, D.S., and Griffiths, A.D. (2002). Investigating the target recognition of DNA cytosine-5 methyltransferase HhaI by library selection using in vitro compartmentalisation. Nucleic Acids Res 30, 4937-4944.
Leemhuis, H., Rozeboom, H.J., Wilbrink, M., Euverink, G.J., Dijkstra, B.W., and Dijkhuizen, L. (2003). Conversion of cyclodextrin glycosyltransferase into a starch hydrolase by directed evolution: the role of alanine 230 in acceptor subsite +1. Biochemistry 42, 7518-7526.
Lefevre, F., Ravot, G., Nguyen, H.K., Ayrinhac, C., Pallas, S., Masson, J.M., and Dupret, D. (2001). Directed evolution and Phenomics screening of new biocatalysts. Mededelingen (Rijksuniversiteit Te Gent Fakulteit van de Landbouwkundige en Toegepaste Biologische Wetenschappen) 66, 249-256.
Lehmann, M., and Wyss, M. (2001). Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Curr Opin Biotechnol 12, 371-375.
Leung, D.W., Chen, E., and Goeddel, D.V. (1989). A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique 1, 11-15.
Levisohn, R., and Spiegelman, S. (1969). Further extracellular Darwinian experiments with replicating RNA molecules: diverse variants isolated under different selective conditions. Proc Natl Acad Sci U S A 63, 805-811.

Références bibliographiques
162
Levy, M., Griswold, K.E., and Ellington, A.D. (2005). Direct selection of trans-acting ligase ribozymes by in vitro compartmentalization. RNA 11, 1555-1562.
Liao, H., McKenzie, T., and Hageman, R. (1986). Isolation of a thermostable enzyme variant by cloning and selection in a thermophile. Proc Natl Acad Sci U S A 83, 576-580.
Liebeton, K., Zonta, A., Schimossek, K., Nardini, M., Lang, D., Dijkstra, B.W., Reetz, M.T., and Jaeger, K.E. (2000). Directed evolution of an enantioselective lipase. Chem Biol 7, 709-718.
Lin-Goerke, J.L., Robbins, D.J., and Burczak, J.D. (1997). PCR-based random mutagenesis using manganese and reduced dNTP concentration. Biotechniques 23, 409-412.
Lin, H., Tao, H., and Cornish, V.W. (2004). Directed evolution of a glycosynthase via chemical complementation. J Am Chem Soc 126, 15051-15059.
Ling, H., Boudsocq, F., Woodgate, R., and Yang, W. (2001). Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 107, 91-102.
Liu, D., Liu, J.M., and Bittker, J.A. (2005). Directed evolution of proteins. Patent WO/2005/116213.
Loesche, W.J. (1975). Bacterial succession in dental plaque : role in dental disease. In Microbiology, D. Schlessinger, ed. (Washington DC, American Society of Microbiology), pp. 132-136.
Lutz, S., Ostermeier, M., Moore, G.L., Maranas, C.D., and Benkovic, S.J. (2001). Creating multiple-crossover DNA libraries independent of sequence identity. Proc Natl Acad Sci U S A 98, 11248-11253.
MacKenzie, C.R., McDonald, I.J., and Johnson, K.G. (1978). Glycogen metabolism in the genus Neisseria: synthesis from sucrose by amylosucrase. Can J Microbiol 24, 357-362.
Maheshri, N., and Schaffer, D.V. (2003). Computational and experimental analysis of DNA shuffling. Proc Natl Acad Sci U S A 100, 3071-3076.
Makhatadze, G.I., Loladze, V.V., Ermolenko, D.N., Chen, X., and Thomas, S.T. (2003). Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J Mol Biol 327, 1135-1148.
Mastrobattista, E., Taly, V., Chanudet, E., Treacy, P., Kelly, B.T., and Griffiths, A.D. (2005). High-throughput screening of enzyme libraries: in vitro evolution of a β-galactosidase by fluorescence-activated sorting of double emulsions. Chem Biol 12, 1291-1300.
Matsuda, T., Bebenek, K., Masutani, C., Rogozin, I.B., Hanaoka, F., and Kunkel, T.A. (2001). Error rate and specificity of human and murine DNA polymerase η. J Mol Biol 312, 335-346.
Matsumoto, Y., and Kim, K. (1995). Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science 269, 699-702.

Références bibliographiques
163
Matsumura, I., Wallingford, J.B., Surana, N.K., Vize, P.D., and Ellington, A.D. (1999). Directed evolution of the surface chemistry of the reporter enzyme β-glucuronidase. Nat Biotechnol 17, 696-701.
Matsumura, M., and Aiba, S. (1985). Screening for thermostable mutant of kanamycin nucleotidyltransferase by the use of a transformation system for a thermophile, Bacillus stearothermophilus. J Biol Chem 260, 15298-15303.
Maxwell, K.L., Mittermaier, A.K., Forman-Kay, J.D., and Davidson, A.R. (1999). A simple in vivo assay for increased protein solubility. Protein Science 8, 1908-1911.
Mayer, C., Jakeman, D.L., Mah, M., Karjala, G., Gal, L., Warren, R.A., and Withers, S.G. (2001). Directed evolution of new glycosynthases from Agrobacterium β-glucosidase: a general screen to detect enzymes for oligosaccharide synthesis. Chem Biol 8, 437-443.
McCafferty, J., Griffiths, A.D., Winter, G., and Ch iswell, D.J. (1990). Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552-554.
Miller, O.J., Hibbert, E.G., Ingram, C.U., Lye, G.J., and Dalby, P.A. (2007). Optimisation and evaluation of a generic microplate-based HPLC screen for transketolase activity. Biotechnology Letters 29, 1759-1770.
Mirza, O., Skov, L.K., Remaud-Simeon, M., Potocki de Montalk, G., Albenne, C., Monsan, P., and Gajhede, M. (2001). Crystal structures of amylosucrase from Neisseria polysaccharea in complex with D-glucose and the active site mutant Glu328Gln in complex with the natural substrate sucrose. Biochemistry 40, 9032-9039.
Miyazaki, J., Nakaya, S., Suzuki, T., Tamakoshi, M., Oshima, T., and Yamagishi, A. (2001). Ancestral residues stabilizing 3-isopropylmalate dehydrogenase of an extreme thermophile: experimental evidence supporting the thermophilic common ancestor hypothesis. J Biochem (Tokyo) 129, 777-782.
Miyazaki, K., Takenouchi, M., Kondo, H., Noro, N., Suzuki, M., and Tsuda, S. (2006). Thermal stabilization of Bacillus subtilis family-11 xylanase by directed evolution. J Biol Chem 281, 10236-10242.
Mondon, P., Bouayadi, K., and Kharrat, H. (2007a). Highly diversified antibody libraries. Patent WO/2007/137616.
Mondon, P., Souyris, N., Douchy, L., Crozet, F., Bouayadi, K., and Kharrat, H. (2007b). Method for generation of human hyperdiversified antibody fragment library. Biotechnol J 2, 76-82.
Moon, A.F., Garcia-Diaz, M., Batra, V.K., Beard, W.A., Bebenek, K., Kunkel, T.A., Wilson, S.H., and Pedersen, L.C. (2007). The X family portrait: Structural insights into biological functions of X family polymerases. DNA Repair (Amst) 6, 1709–1725.
Moore, G.L., Maranas, C.D., Lutz, S., and Benkovic, S.J. (2001). Predicting crossover generation in DNA shuffling. Proc Natl Acad Sci U S A 98, 3226-3231.
Moore, J.C., and Arnold, F.H. (1996). Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nat Biotechnol 14, 458-467.

Références bibliographiques
164
Mosi, R., He, S., Uitdehaag, J., Dijkstra, B.W., and Withers, S.G. (1997). Trapping and characterization of the reaction intermediate in cyclodextrin glycosyltransferase by use of activated substrates and a mutant enzyme. Biochemistry 36, 9927-9934.
Murakami, H., Hohsaka, T., and Sisido, M. (2002). Random insertion and deletion of arbitrary number of bases for codon-based random mutation of DNAs. Nat Biotechnol 20, 76-81.
Myers, R.M., Lerman, L.S., and Maniatis, T. (1985). A general method for saturation mutagenesis of cloned DNA fragments. Science 229, 242-247.
Ness, J.E., Kim, S., Gottman, A., Pak, R., Krebber, A., Borchert, T.V., Govindarajan, S., Mundorff, E.C., and Minshull, J. (2002). Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently. Nat Biotechnol 20, 1251-1255.
Neylon, C. (2004). Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: library construction methods for directed evolution. Nucleic Acids Res 32, 1448-1459.
Nguyen, A.W., and Daugherty, P.S. (2003). Production of randomly mutated plasmid libraries using mutator strains. Methods in molecular biology 231, 39-44.
Nugent, A.P. (2005). Health properties of resistant starch. Nutrition Bulletin 30, 27-54.
Ohmori, H., Friedberg, E.C., Fuchs, R.P., Goodman, M.F., Hanaoka, F., Hinkle, D., Kunkel, T.A., Lawrence, C.W., Livneh, Z., Nohmi, T., et al. (2001). The Y-family of DNA polymerases. Mol Cell 8, 7-8.
Okada, G., and Hehre, E.J. (1974). New studies on amylosucrase, a bacterial alpha-D-glucosylase that directly converts sucrose to a glycogen-like alpha-glucan. J Biol Chem 249, 126-135.
Oliphant, A.R., Nussbaum, A.L., and Struhl, K. (1986). Cloning of random-sequence oligodeoxynucleotides. Gene 44, 177-183.
Olsen, M.J., Stephens, D., Griffiths, D., Daugherty, P., Georgiou, G., and Iverson, B.L. (2000). Function-based isolation of novel enzymes from a large library. Nat Biotechnol 18, 1071-1074.
Osanjo, G., Dion, M., Drone, J., Solleux, C., Tran, V., Rabiller, C., and Tellier, C. (2007). Directed evolution of the α-L-fucosidase from Thermotoga maritima into an α-L-transfucosidase. Biochemistry 46, 1022-1033.
Osheroff, W.P., Jung, H.K., Beard, W.A., Wilson, S.H., and Kunkel, T.A. (1999). The fidelity of DNA polymerase β during distributive and processive DNA synthesis. J Biol Chem 274, 3642-3650.
Ostermeier, M., Shim, J.H., and Benkovic, S.J. (1999). A combinatorial approach to hybrid enzymes independent of DNA homology. Nat Biotechnol 17, 1205-1209.

Références bibliographiques
165
Osuna, J., Yanez, J., Soberon, X., and Gaytan, P. (2004). Protein evolution by codon-based random deletions. Nucleic Acids Res 32, e136.
Otten, L.G., Sio, C.F., Vrielink, J., Cool, R.H., and Quax, W.J. (2002). Altering the substrate specificity of cephalosporin acylase by directed evolution of the β-subunit. J Biol Chem 277, 42121-42127.
Oue, S., Okamoto, A., Yano, T., and Kagamiyama, H. (1999). Redesigning the substrate specificity of an enzyme by cumulative effects of the mutations of non-active site residues. J Biol Chem 274, 2344-2349.
Palackal, N., Brennan, Y., Callen, W.N., Dupree, P., Frey, G., Goubet, F., Hazlewood, G.P., Healey, S., Kang, Y.E., Kretz, K.A., et al. (2004). An evolutionary route to xylanase process fitness. Protein Sci 13, 494-503.
Parikh, M.R., Greene, D.N., Woods, K.K., and Matsumura, I. (2006). Directed evolution of RuBisCO hypermorphs through genetic selection in engineered E.coli. Protein Eng Des Sel 19, 113-119.
Parikh, M.R., and Matsumura, I. (2005). Site-saturation mutagenesis is more efficient than DNA shuffling for the directed evolution of β-fucosidase from β-galactosidase. J Mol Biol 352, 621-628.
Patten, P.A., Howard, R.J., and Stemmer, W.P. (1997). Applications of DNA shuffling to pharmaceuticals and vaccines. Curr Opin Biotechnol 8, 724-733.
Pedersen, H., Holder, S., Sutherlin, D.P., Schwitter, U., King, D.S., and Schultz, P.G. (1998). A method for directed evolution and functional cloning of enzymes. Proc Natl Acad Sci U S A 95, 10523-10528.
Peng, W., and Shaw, B.R. (1996). Accelerated deamination of cytosine residues in UV-induced cyclobutane pyrimidine dimers leads to CC-->TT transitions. Biochemistry 35, 10172-10181.
Perl, D., Mueller, U., Heinemann, U., and Schmid, F.X. (2000). Two exposed amino acid residues confer thermostability on a cold shock protein. Nat Struct Biol 7, 380-383.
Petrounia, I.P., and Arnold, F.H. (2000). Designed evolution of enzymatic properties. Curr Opin Biotechnol 11, 325-330.
Pfeifer, G.P., You, Y.H., and Besaratinia, A. (2005). Mutations induced by ultraviolet light. Mutat Res 571, 19-31.
Pizzut-Serin, S., Potocki-Veronese, G., van der Veen, B.A., Albenne, C., Monsan, P., and Remaud-Simeon, M. (2005). Characterisation of a novel amylosucrase from Deinococcus radiodurans. FEBS Lett 579, 1405-1410.
Ponsard, I., Galleni, M., Soumillion, P., and Fastrez, J. (2001). Selection of metalloenzymes by catalytic activity using phage display and catalytic elution. Chembiochem 2, 253-259.

Références bibliographiques
166
Potocki-Veronese, G., Putaux, J.L., Dupeyre, D., Albenne, C., Remaud-Simeon, M., Monsan, P., and Buleon, A. (2005). Amylose synthesized in vitro by amylosucrase: morphology, structure, and properties. Biomacromolecules 6, 1000-1011.
Potocki de Montalk, G. (1999). L'amylosaccharase de Neisseria polysaccharea: aspects structuraux et caractérisation de son activité catalytique (INSA Toulouse, France).
Potocki de Montalk, G., Remaud-Simeon, M., Willemot, R.M., and Monsan, P. (2000a). Characterisation of the activator effect of glycogen on amylosucrase from Neisseria polysaccharea. FEMS Microbiol Lett 186, 103-108.
Potocki de Montalk, G., Remaud-Simeon, M., Willemot, R.M., Planchot, V., and Monsan, P. (1999). Sequence analysis of the gene encoding amylosucrase from Neisseria polysaccharea and characterization of the recombinant enzyme. J Bacteriol 181, 375-381.
Potocki de Montalk, G., Remaud-Simeon, M., Willemot, R.M., Sarcabal, P., Planchot, V., and Monsan, P. (2000b). Amylosucrase from Neisseria polysaccharea: novel catalytic properties. FEBS Lett 471, 219-223.
Prakash, S., Johnson, R.E., and Prakash, L. (2005). Eukaryotic translesion synthesis DNA polymerases: Specificity of Structure and Function. Annu Rev Biochem 74, 317-353.
Prakash, S., and Prakash, L. (2002). Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev 16, 1872-1883.
Preiss, J., Ozbun, J.L., Hawker, J.S., Greenberg, E., and Lammel, C. (1973). ADPG synthetase and ADPG- -glucan 4-glucosyl transferase: enzymes involved in bacterial glycogen and plant starch synthesis. Ann N Y Acad Sci 210, 265-278.
Putaux, J.L., Potocki-Veronese, G., Remaud-Simeon, M., and Buleon, A. (2006). α-D-glucan-based dendritic nanoparticles prepared by in vitro enzymatic chain extension of glycogen. Biomacromolecules 7, 1720-1728.
Quanz, M. (1999). Genetically modified plant cells and plants with increased activity of an poly (1,4)-α-glucan. Patent WO0073422.
Quanz, M. (2000). Genetically modified plant cells and plants with increased activity of an poly (1,4)-α-glucan. Patent WO/2000/73422.
Quanz, M., and Frohberg, C. (2003). Use of poly-α-(1,4)-glucans as resistant starch. Patent WO2005040223.
Quanz, M., and Provart, N. (1998). Nucleic acid molecules encoding an amylosucrase. Patent WO0014249.
Quanz, M., Provart, N., and Banasiak, R. (1998). Method for producing water-insoluble α-1,4-glucan. Patent WO9967412.
Quiocho, F.A. (1986). Carbohydrate-binding proteins: tertiary structures and protein-sugar interactions. Annu Rev Biochem 55, 287-315.

Références bibliographiques
167
Reetz, M.T. (2003). An overview of high-throughput screening systems for enantioselective enzymatic transformations. Methods in molecular biology 230, 259-282.
Reetz, M.T., Becker, M.H., Stoeckigt, D., and H.W., K. (2000). High-throughput-screening method for determining enantioselectivity. Patent WO/2000/58504.
Reetz, M.T., Becker, M.H., Stoeckigt, D., and H.W., K. (2004a). High-throughput-screening method for determining enantioselectivity. Patent US68824982B1.
Reetz, M.T., Carballeira, J.D., and Vogel, A. (2006). Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angewandte Chemie (International ed 45, 7745-7751.
Reetz, Manfred T., Eipper, A., Tielmann, P., and Mynott, R. (2002). A Practical NMR-Based High-Throughput Assay for Screening Enantioselective Catalysts and Biocatalysts. Advanced Synthesis & Catalysis 344, 1008-1016.
Reetz, M.T., Tielmann, P., Eipper, A., and Mynott, R. (2003). High-throughput screening method for determining the enantioselectivity of catalysts, biocatalysts, and agents. Patent WO/2003/075031.
Reetz, M.T., Tielmann, P., Eipper, A., and Mynott, R. (2005). High-throughput screening method for determining the enantioselectivity of catalysts, biocatalysts, and agents. Patent US2005153358.
Reetz, M.T., Tielmann, P., Eipper, A., Ross, A., and Schlotterbeck, G. (2004b). A high-throughput NMR-based ee-assay using chemical shift imaging. Chem Commun, 1366-1367.
Reid, S.J., and Abratt, V.R. (2005). Sucrose utilisation in bacteria: genetic organisation and regulation. Appl Microbiol Biotechnol 67, 312-321.
Remaud-Simeon, M., Albaret, F., Canard, B., Varlet, I., Colonna, P., Willemot, R., and Monsan, P. (1995). Studies on a recombinant amylosucrase. In Carbohydrate bioengineering, S.B. Petersen, B. Svensson, and S. Pedersen, eds. (Elsevier Science), pp. 313-320.
Reymond, J.L., and Babiak, P. (2007). Screening systems. Adv Biochem Eng Biotechnol 105, 31-58.
Riou, J.Y., Guibourdenche, M., and Popoff, M.Y. (1983). A new taxon in the genus Neisseria. Ann Microbiol (Paris) 134B, 257-267.
Rolland-Sabaté, A., Colonna, P., Potocki-Veronese, G., Monsan, P., and Planchot, V. (2004). Elongation and insolubilisation of α-glucans by the action of Neisseria polysaccharea amylosucrase. Journal of Cereal Science 40, 17-30.
Rothwell, P.J., and Waksman, G. (2005). Structure and mechanism of DNA polymerases. Adv Protein Chem 71, 401-440.
Saboulard, D., Dugas, V., Jaber, M., Broutin, J., Souteyrand, E., Sylvestre, J., and Delcourt, M. (2005). High-throughput site-directed mutagenesis using oligonucleotides synthesized on DNA chips. Biotechniques 39, 363-368.

Références bibliographiques
168
Santoro, S.W., and Schultz, P.G. (2002). Directed evolution of the site specificity of Cre recombinase. Proc Natl Acad Sci U S A 99, 4185-4190.
Santoro, S.W., Wang, L., Herberich, B., King, D.S., and Schultz, P.G. (2002). An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat Biotechnol 20, 1044-1048.
Sarcabal, P., Remaud-Simeon, M., Willemot, R., Potocki de Montalk, G., Svensson, B., and Monsan, P. (2000). Identification of key amino acid residues in Neisseria polysaccharea amylosucrase. FEBS Lett 474, 33-37.
Schoemaker, H.E., Mink, D., and Wubbolts, M.G. (2003). Dispelling the myths--biocatalysis in industrial synthesis. Science 299, 1694-1697.
Seelig, B., and Szostak, J.W. (2007). Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature 448, 828-831.
Sengupta, D., Lin, H., Goldberg, S.D., Mahal, J.J., and Cornish, V.W. (2004). Correlation between catalytic efficiency and the transcription read-out in chemical complementation: a general assay for enzyme catalysis. Biochemistry 43, 3570-3581.
Shafikhani, S., Siegel, R.A., Ferrari, E., and Schellenberger, V. (1997). Generation of large libraries of random mutants in Bacillus subtilis by PCR-based plasmid multimerization. Biotechniques 23, 304-310.
Shao, Z., Zhao, H., Giver, L., and Arnold, F.H. (1998). Random-priming in vitro recombination: an effective tool for directed evolution. Nucleic Acids Res 26, 681-683.
Short, J.M. (2001). Saturation mutagenesis in directed evolution. Patent US6171820.
Short, J.M. (2003). Saturation mutagenesis in directed evolution. Patent US/2003/175887.
Sidhu, S.S., and Borgford, T.J. (1996). Selection of Streptomyces griseus protease B mutants with desired alterations in primary specificity using a library screening strategy. J Mol Biol 257, 233-245.
Sieber, V., Martinez, C.A., and Arnold, F.H. (2001). Libraries of hybrid proteins from distantly related sequences. Nat Biotechnol 19, 456-460.
Silvian, L.F., Toth, E.A., Pham, P., Goodman, M.F., and Ellenberger, T. (2001). Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat Struct Biol 8, 984-989.
Simm, A.M., Baldwin, A.J., Busse, K., and Jones, D.D. (2007). Investigating protein structural plasticity by surveying the consequence of an amino acid deletion from TEM-1 beta-lactamase. FEBS Lett 581, 3904-3908.
Singer, B., and Kusmierek, J.T. (1982). Chemical mutagenesis. Annu Rev Biochem 51, 655-693.

Références bibliographiques
169
Sinha, N.K., and Haimes, M.D. (1981). Molecular mechanisms of substitution mutagenesis. An experimental test of the Watson-Crick and topal-fresco models of base mispairings. J Biol Chem 256, 10671-10683.
Sinnott, M.L. (1990). Catalytic mechanisms of enzymatic glycosyl transfer. Chemical Reviews 90 1171-1202.
Sio, C.F., Riemens, A.M., van der Laan, J.M., Verhaert, R.M., and Quax, W.J. (2002). Directed evolution of a glutaryl acylase into an adipyl acylase. European journal of biochemistry 269, 4495-4504.
Skov, L.K., Mirza, O., Henriksen, A., De Montalk, G.P., Remaud-Simeon, M., Sarcabal, P., Willemot, R.M., Monsan, P., and Gajhede, M. (2001). Amylosucrase, a glucan-synthesizing enzyme from the alpha-amylase family. J Biol Chem 276, 25273-25278.
Skov, L.K., Mirza, O., Sprogoe, D., Dar, I., Remaud-Simeon, M., Albenne, C., Monsan, P., and Gajhede, M. (2002). Oligosaccharide and sucrose complexes of amylosucrase. Structural implications for the polymerase activity. J Biol Chem 277, 47741-47747.
Smith, G.P. (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315-1317.
Solbak, A.I., Richardson, T.H., McCann, R.T., Kline, K.A., Bartnek, F., Tomlinson, G., Tan, X., Parra-Gessert, L., Frey, G.J., Podar, M., et al. (2005). Discovery of pectin-degrading enzymes and directed evolution of a novel pectate lyase for processing cotton fabric. J Biol Chem 280, 9431-9438.
Soumillion, P., Jespers, L., Bouchet, M., Marchand-Brynaert, J., Winter, G., and Fastrez, J. (1994). Selection of β-lactamase on filamentous bacteriophage by catalytic activity. J Mol Biol 237, 415-422.
Stemmer, W.P. (1994a). DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci U S A 91, 10747-10751.
Stemmer, W.P. (1994b). Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389-391.
Stemmer, W.P. (1997). Methods for in vitro recombination. Patent US5605793.
Stemmer, W.P. (2003). Shuffling polynucleotides by incomplete extension. Patent US6506603.
Stemmer, W.P., and Crameri, A. (1995). DNA mutagenesis by random fragmentation and reassembly. Patent WO/1995/22625.
Stemmer, W.P., and Crameri, A. (1998). DNA mutagenesis by random fragmentation and reassembly. Patent US5830721.
Stratagene (2004). Overcome mutational bias. Strategies 17, 20-21.

Références bibliographiques
170
Streisinger, G., Okada, Y., Emrich, J., Newton, J., Tsugita, A., Terzaghi, E., and Inouye, M. (1966). Frameshift mutations and the genetic code. Cold Spring Harb Symp Quant Biol 31, 77-84.
Suganuma, T., Matsuno, R., Ohnishi, M., and Hiromi, K. (1978). A study of the mechanism of action of Taka-amylase A1 on linear oligosaccharides by product analysis and computer simulation. J Biochem (Tokyo) 84, 293-316.
Sumner, J.B., and Howell, S.F. (1935). A method for determination of saccharase activity. J Biol Chem 108, 51-54.
Takahashi, F., Ebihara, T., Mie, M., Yanagida, Y., Endo, Y., Kobatake, E., and Aizawa, M. (2002). Ribosome display for selection of active dihydrofolate reductase mutants using immobilized methotrexate on agarose beads. FEBS Lett 514, 106-110.
Takahashi, F., Funabashi, H., Mie, M., Endo, Y., Sawasaki, T., Aizawa, M., and Kobatake, E. (2005). Activity-based in vitro selection of T4 DNA ligase. Biochem Biophys Res Commun 336, 987-993.
Taly, V., Kelly, B.T., and Griffiths, A.D. (2007). Droplets as Microreactors for High-Throughput Biology. Chembiochem 8, 263-272.
Tamakoshi, M., Nakano, Y., Kakizawa, S., Yamagishi, A., and Oshima, T. (2001). Selection of stabilized 3-isopropylmalate dehydrogenase of Saccharomyces cerevisiae using the host-vector system of an extreme thermophile, Thermus thermophilus. Extremophiles 5, 17-22.
Tawfik, D.S., and Griffiths, A.D. (1998). Man-made cell-like compartments for molecular evolution. Nat Biotechnol 16, 652-656.
Taylor, M.S., Ponting, C.P., and Copley, R.R. (2004). Occurrence and Consequences of Coding Sequence Insertions and Deletions in Mammalian Genomes. Genome Res 14, 555-566.
Thoma, J.A., Rao, G.V., Brothers, C., Spradlin, J., and Li, L.H. (1971). Subsite mapping of enzymes. Correlation of product patterns with Michaelis parameters and substrate-induced strain. J Biol Chem 246, 5621-5635.
Tindall, K.R., and Kunkel, T.A. (1988). Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry 27, 6008-6013.
Trincao, J., Johnson, R.E., Escalante, C.R., Prakash, S., Prakash, L., and Aggarwal, A.K. (2001). Structure of the catalytic core of S. cerevisiae DNA polymerase η: implications for translesion DNA synthesis. Mol Cell 8, 417-426.
Tudyka, T., and Skerra, A. (1997). Glutathione S-transferase can be used as a C-terminal, enzymatically active dimerization module for a recombinant protease inhibitor, and functionally secreted into the periplasm of Escherichia coli. Protein Sci 6, 2180-2187.
Uitdehaag, J.C., Mosi, R., Kalk, K.H., van der Veen, B.A., Dijkhuizen, L., Withers, S.G., and Dijkstra, B.W. (1999). X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat Struct Biol 6, 432-436.

Références bibliographiques
171
van den Burg, B., and Eijsink, V.G. (2002). Selection of mutations for increased protein stability. Curr Opin Biotechnol 13, 333-337.
van der Veen, B.A., Potocki-Veronese, G., Albenne, C., Joucla, G., Monsan, P., and Remaud-Simeon, M. (2004). Combinatorial engineering to enhance amylosucrase performance: construction, selection, and screening of variant libraries for increased activity. FEBS Lett 560, 91-97.
van der Veen, B.A., Skov, L.K., Potocki-Veronese, G., Gajhede, M., Monsan, P., and Remaud-Simeon, M. (2006). Increased amylosucrase activity and specificity, and identification of regions important for activity, specificity and stability through molecular evolution. Febs J 273, 673-681.
van Loo, B., Spelberg, J.H., Kingma, J., Sonke, T., Wubbolts, M.G., and Janssen, D.B. (2004). Directed evolution of epoxide hydrolase from A. radiobacter toward higher enantioselectivity by error-prone PCR and DNA shuffling. Chem Biol 11, 981-990.
van Sint Fiet, S., van Beilen, J.B., and Witholt, B. (2006). Selection of biocatalysts for chemical synthesis. Proc Natl Acad Sci U S A 103, 1693-1698.
Vanhercke, T., Ampe, C., Tirry, L., and Denolf, P. (2005). Reducing mutational bias in random protein libraries. Analytical biochemistry 339, 9-14.
Vanwetswinkel, S., Avalle, B., and Fastrez, J. (2000). Selection of β-lactamases and penicillin binding mutants from a library of phage displayed TEM-1 β-lactamase randomly mutated in the active site omega-loop. J Mol Biol 295, 527-540.
Varadarajan, N., Gam, J., Olsen, M.J., Georgiou, G., and Iverson, B.L. (2005). Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc Natl Acad Sci U S A 102, 6855-6860.
Vartanian, J.P., Henry, M., and Wain-Hobson, S. (1996). Hypermutagenic PCR involving all four transitions and a sizeable proportion of transversions. Nucleic Acids Res 24, 2627-2631.
Vichier-Guerre, S., Ferris, S., Auberger, N., Mahiddine, K., and Jestin, J.L. (2006). A population of thermostable reverse transcriptases evolved from Thermus aquaticus DNA polymerase I by phage display. Angewandte Chemie (International ed) 45, 6133-6137.
Vieille, C., and Zeikus, G.J. (2001). Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiology and Molecular Biology Reviews 65, 1-43.
Weissmuller, M., Quanz, M., and Provart, N. (2000). Polysaccharides containing α-1,4-glucan chains and method for producing the same. Patent WO/2000/39321.
Welch, M., Ness, J.E., Gustafsson, C., Stemmer, W.P., and Minshull, J. (2001). Use of codon-varied oligonucleotide synthesis for synthetic sequence recombination. Patent WO/2001/23401.
Whitesides, G.M. (2006). The origins and the future of microfluidics. Nature 442, 368-373.

Références bibliographiques
172
Widersten, M., and Mannervik, B. (1995). Glutathione transferases with novel active sites isolated by phage display from a library of random mutants. J Mol Biol 250, 115-122.
Wilhelm, S., Rosenau, F., Becker, S., Buest, S., Hausmann, S., Kolmar, H., and Jaeger, K.E. (2007). Functional cell-surface display of a lipase-specific chaperone. Chembiochem 8, 55-60.
Wong, D.W., Batt, S.B., Lee, C.C., and Robertson, G.H. (2004a). High-activity barley α-amylase by directed evolution. Protein J 23, 453-460.
Wong, T.S., Arnold, F.H., and Schwaneberg, U. (2004b). Laboratory evolution of cytochrome P450 BM-3 monooxygenase for organic cosolvents. Biotechnol Bioeng 85, 351-358.
Wong, T.S., Tee, K.L., Hauer, B., and Schwaneberg, U. (2004c). Sequence saturation mutagenesis (SeSaM): a novel method for directed evolution. Nucleic Acids Res 32, e26.
Wong, T.S., Zhurina, D., and Schwaneberg, U. (2006). The diversity challenge in directed protein evolution. Comb Chem High Throughput Screen 9, 271-288.
Yan, X., and Xu, Z. (2006). Ribosome-display technology: applications for directed evolution of functional proteins. Drug Discov Today 11, 911-916.
Yano, T., Oue, S., and Kagamiyama, H. (1998). Directed evolution of an aspartate aminotransferase with new substrate specificities. Proc Natl Acad Sci U S A 95, 5511-5515.
Yoshikuni, Y., Ferrin, T.E., and Keasling, J.D. (2006). Designed divergent evolution of enzyme function. Nature 440, 1078-1082.
Yoshioka, Y., Hasegawa, K., Matsuura, Y., Katsube, Y., and Kubota, M. (1997). Crystal structures of a mutant maltotetraose-forming exo-amylase cocrystallized with maltopentaose. J Mol Biol 271, 619-628.
You, L., and Arnold, F.H. (1996). Directed evolution of subtilisin E in Bacillus subtilis to enhance total activity in aqueous dimethylformamide. Protein Eng 9, 77-83.
Yuan, L., Kurek, I., English, J., and Keenan, R. (2005). Laboratory-directed protein evolution. Microbiology and Molecular Biology Reviews 69, 373-392.
Zaccolo, M., Williams, D.M., Brown, D.M., and Gherardi, E. (1996). An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J Mol Biol 255, 589-603.
Zha, D., Eipper, A., and Reetz, M.T. (2003). Assembly of designed oligonucleotides as an efficient method for gene recombination: a new tool in directed evolution. Chembiochem 4, 34-39.
Zhang, J.H., Dawes, G., and Stemmer, W.P. (1997). Directed evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc Natl Acad Sci U S A 94, 4504-4509.

Références bibliographiques
173
Zhao, H., Giver, L., Shao, Z., Affholter, J.A., and Arnold, F.H. (1998). Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat Biotechnol 16, 258-261.
Zhou, B.L., Pata, J.D., and Steitz, T.A. (2001). Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol Cell 8, 427-437.

174
Annexes

Annexes
175
ANNEXE 1 : SEQUENCE NUCLEOTIDIQUE ET PROTEIQUE DE L ’ASNP
Domaine N fS P N S Q Y L K T R I L D I Y T P Ef 18 TCC CCG AAT TCA CAG TAC CTC AAA ACA CGC ATC TTG GAC ATC TAC ACG CCC GAA 54 fQ R A G I E K S E D W R Q F S R R Mf 36 CAG CGC GCC GGC ATC GAA AAA TCC GAA GAC TGG CGG CAG TTT TCG CGC CGC ATG 108 fD T H F P K L M N E L D S V Y G N Nf 54 GAT ACG CAT TTC CCC AAA CTG ATG AAC GAA CTC GAC AGC GTG TAC GGC AAC AAC 162 fE A L L P M L E M L L A Q A W Q S Yf 72 GAA GCC CTG CTG CCT ATG CTG GAA ATG CTG CTG GCG CAG GCA TGG CAA AGC TAT 216 fS Q R N S S L K D I D I A R E N N Pf 90 TCC CAA CGC AAC TCA TCC TTA AAA GAT ATC GAT ATC GCG CGC GAA AAC AAC CCC 270 ββββ1 D W I L S N K Q V fG G V Cf Y V D L F 108 GAT TGG ATT TTG TCC AAC AAA CAA GTC GGC GGC GTG TGC TAC GTT GAT TTG TTT 324 αααα1 A G D L fK G L K D K I P Y F Q E L Gf 126 GCC GGC GAT TTG AAG GGC TTG AAA GAT AAA ATT CCT TAT TTT CAA GAG CTT GGT 378 β β β β2 L T Y dL H L Md P L F K C P E G K S D 144 TTG ACT TAT CTG CAC CTG ATG CCG CTG TTT AAA TGC CCT GAA GGC AAA AGC GAC 432 G G Y A V S S Y R D V N P A L G T I 162 GGC GGC TAT GCG GTC AGC AGC TAC CGC GAT GTC AAT CCG GCA CTG GGC ACA ATA 486 αααα2 ββββ3 fG D L R E V I A A L H Ef A G I fS A Vf 180 GGC GAC TTG CGC GAA GTC ATT GCT GCG CTG CAC GAA GCC GGC ATT TCC GCC GTC 540
Domaine B fV D Ff I fF N H T S N E H E W A Q R Cf 198 GTC GAT TTT ATC TTC AAC CAC ACC TCC AAC GAA CAC GAA TGG GCG CAA CGC TGC 594 fA A G D P L F D N F Y Y I F P D R Rf 216 GCC GCC GGC GAC CCG CTT TTC GAC AAT TTC TAC TAT ATT TTC CCC GAC CGC CGG 648 fM P D Q Y D R T L R E I F P D Q H Pf 234 ATG CCC GAC CAA TAC GAC CGC ACC CTG CGC GAA ATC TTC CCC GAC CAG CAC CCG 702 fG G F S Q L E D G R W V W T T F N Sf 252 GGC GGC TTC TCG CAA CTG GAA GAC GGA CGC TGG GTG TGG ACG ACC TTC AAT TCC 756 αααα3 fF Q W D L N Y Sf N fP W V F R A M A Gf 270 TTC CAA TGG GAC TTG AAT TAC AGC AAC CCG TGG GTA TTC CGC GCA ATG GCG GGC 810 ββββ4 fE M L F L A N Lf G V D fI L R M Df A V 288 GAA ATG CTG TTC CTT GCC AAC TTG GGC GTT GAC ATC CTG CGT ATG GAT GCG GTT 864 A F I W K Q M G T S C E N L P fQ A Hf 306 GCC TTT ATT TGG AAA CAA ATG GGG ACA AGC TGC GAA AAC CTG CCG CAG GCG CAC 918 αααα4 fA L I R A F N A V M R If A A P A V F 324 GCC CTC ATC CGC GCG TTC AAT GCC GTT ATG CGT ATT GCC GCG CCC GCC GTG TTC 972 ββββ5 αααα5 F fK S Ef A I V H P fD Q V V Q Yf I G Q 342 TTC AAA TCC GAA GCC ATC GTC CAC CCC GAC CAA GTC GTC CAA TAC ATC GGG CAG 1026 ββββ6 D E C Q fI G Yf N P L Q M A L L W N T 360 GAC GAA TGC CAA ATC GGT TAC AAC CCC CTG CAA ATG GCA TTG TTG TGG AAC ACC 1080 αααα6 L A T R E V fN L L H Q A L T Yf R H N 378 CTT GCC ACG CGC GAA GTC AAC CTG CTC CAT CAG GCG CTG ACC TAC CGC CAC AAC 1134

Annexes
176
ββββ7 L P E H T fA W V N Yf V R S H D D I fGf 396 CTG CCC GAG CAT ACC GCC TGG GTC AAC TAC GTC CGC AGC CAC GAC GAC ATC GGC 1188 Domaine B' fW T F A D E D A A Y L G I S G Y D Hf 414 TGG ACG TTT GCC GAT GAA GAC GCG GCA TAT CTG GGC ATA AGC GGC TAC GAC CAC 1242 fR Q F L N R F F V N R F D G S F A Rf 432 CGC CAA TTC CTC AAC CGC TTC TTC GTC AAC CGT TTC GAC GGC AGC TTC GCT CGT 1296 fG V P F Q Y N P S T G D C R V S G Tf 450 GGC GTA CCG TTC CAA TAC AAC CCA AGC ACA GGC GAC TGC CGT GTC AGT GGT ACA 1350 fA A A L V G L A Q Df D P H A fV D R If 468 GCC GCG GCA TTG GTC GGC TTG GCG CAA GAC GAT CCC CAC GCC GTT GAC CGC ATC 1404 αααα7 ββββ8 fK L L Y S I A L Sf T G G L fP L I Yf L 486 AAA CTC TTG TAC AGC ATT GCT TTG AGT ACC GGC GGT CTG CCG CTG ATT TAC CTA 1458 G D E V G T L N D D D W S Q D S N K 504 GGC GAC GAA GTG GGT ACG CTC AAT GAC GAC GAC TGG TCG CAA GAC AGC AAT AAG 1512 S D D S R W A H R P R Y N E A L Y A 522 AGC GAC GAC AGC CGT TGG GCG CAC CGT CCG CGC TAC AAC GAA GCC CTG TAC GCG 1566 αααα8 Q R N D P S T A fA G Q I Y Q G L R Hf 540 CAA CGC AAC GAT CCG TCG ACC GCA GCC GGG CAA ATC TAT CAG GGC TTG CGC CAT 1620 fM I A V R Q Sf N P R F D G G fR L V Tf 558 ATG ATT GCC GTC CGC CAA AGC AAT CCG CGC TTC GAC GGC GGC AGG CTG GTT ACA 1674 Domaine C fF N T N N K H I I G Y I R N N A L Lf 576 TTC AAC ACC AAC AAC AAG CAC ATC ATC GGC TAC ATC CGC AAC AAT GCG CTT TTG 1728 fA F G N F S E Y P Q T V T A H T L Qf 594 GCA TTC GGT AAC TTC AGC GAA TAT CCG CAA ACC GTT ACC GCG CAT ACC CTG CAA 1782 fA M P F K A H D L I G G K T V S L Nf 612 GCC ATG CCC TTC AAG GCG CAC GAC CTC ATC GGT GGC AAA ACT GTC AGC CTG AAT 1836 fQ D L T L Q P Y Q V M W L E I Af 628 CAG GAT TTG ACG CTT CAG CCC TAT CAG GTC ATG TGG CTC GAA ATC GCC 1884
Les éléments de structure secondaire sont colorés en jaune pour les feuillets β et en rouge
pour les hélices α. Les domaines N, B, B' et C sont grisés. Les résidus de la triade catalytique
sont indiqués en violet. Les acides aminés mutés chez les variants thermostables identifiées au
cours de cette étude sont entourés. La séquence est numérotée d'après la séquence déposée
dans la Protein Data Bank (numéro d'accés : 1G5A).

Annexes
177
ANNEXE 2 : CARACTERISATION DE L ’AMYLOSACCHARASE DE DEINOCOCCUS
GEOTHERMALIS .
Cloning, purification and characterization of a thermostable amylosucrase from
Deinococcus geothermalis.
Emond S., Mondeil S., Jaziri K., Andre I., Monsan P., Remaud-Simeon M., Potocki-Veronese
G. (2008).
FEMS Microbiology Letters, 285(1):25-32

R E S E A R C H L E T T E R
Cloning,puri¢cationand characterizationofa thermostableamylosucrase fromDeinococcusgeothermalisStephane Emond1,2,3, Sophie Mondeil1,2,3, Kais Jaziri1,2,3, Isabelle Andre1,2,3, Pierre Monsan1,2,3,Magali Remaud-Simeon1,2,3 & Gabrielle Potocki-Veronese1,2,3
1Universite de Toulouse; INSA, UPS, INP; LISBP, Toulouse, France; 2INRA, UMR792 Ingenierie des Systemes Biologiques et des Procedes, Toulouse, France;
and 3CNRS, UMR5504, Toulouse, France
Correspondence: Gabrielle Potocki-
Veronese, Laboratoire d’Ingenierie des
Systemes Biologiques et des Procedes, Institut
National des Sciences Appliquees de
Toulouse, 135 Avenue de Rangueil, F-31400
Toulouse, France. Tel.: 133 561 559 446;
fax: 133 561 559 400; e-mail:
Received 18 March 2008; accepted 17 April
2008.
First published online 3 June 2008.
DOI:10.1111/j.1574-6968.2008.01204.x
Editor: Marco Moracci
Keywords
amylosucrase; Deinococcus geothermalis ;
recombinant expression; amylopolysaccharide;
amylose synthesis; thermostability.
Abstract
Amylosucrase is a transglucosidase that catalyses the synthesis of an amylose-type
polymer from sucrose, an abundant agro-resource. Here we describe a novel
thermostable amylosucrase from the moderate thermophile Deinococcus geother-
malis (DGAS). The dgas gene was cloned and expressed in Escherichia coli. The
encoded enzyme was purified and characterized. DGAS displays a specific activity
of 44 U mg�1, an optimal temperature of 50 1C and a half-life of 26 h at 50 1C.
Moreover, it produces an a-glucan at 50 1C, with an average degree of polymeriza-
tion of 45 and a polymerization yield of 76%. DGAS is thus the most active and
thermostable amylosucrase known to date.
Introduction
Amylosucrase [Enzyme Commission (EC) number 2.4.1.4]
is a glucansucrase belonging to glycoside-hydrolase (GH)
family 13. This transglucosidase catalyses the de novo synth-
esis of an amylose-like polymer from sucrose (Potocki de
Montalk et al., 2000), a cheap agro-resource. Unlike starch
or glycogen synthases (Preiss et al., 1973), this enzyme does
not require any expensive nucleotide-activated sugar as
glucosyl donor and thus can be seen as an attractive
biocatalyst for the industrial synthesis and modification of
amylopolysaccharides (Rolland-Sabate et al., 2004, Quanz &
Frohberg, 2005). To date, only two recombinant amylosu-
crases, from Neisseria polysaccharea (NPAS) (Potocki de
Montalk et al., 1999) and Deinococcus radiodurans (DRAS)
(Pizzut-Serin et al., 2005), have been characterized. NPAS
has been the most extensively studied. In particular, crystal-
lographic studies have shown that its structure consists of
five domains. Like all GH family 13 enzymes, NPAS pos-
sesses the characteristic (b/a)8-barrel folded catalytic
domain A domain, the B domain between b-strand 3 and
a-helix 3 and the C-terminal greek key domain (Skov et al.,
2001). Besides these common features, the NPAS structure
also contains two unique additional domains, namely the
N-terminal a-helical domain and a B0 domain between
b-strand 7 and a-helix 7 of the catalytic barrel. NPAS
catalysis proceeds through an a-retaining mechanism invol-
ving the formation of a covalent glucosyl enzyme inter-
mediate (Uitdehaag et al., 1999; Jensen et al., 2004). It
initializes polymer formation by releasing, through sucrose
hydrolysis, a glucose molecule that is subsequently used as
the first acceptor molecule (Albenne et al., 2004), to produce
soluble maltooligosaccharides. These maltooligosaccharides
are then elongated from their nonreducing ends through a
nonprocessive mechanism until precipitation of amylose
chains occurs. The average size, morphology and crystalline
properties of the formed insoluble polymer can be better
monitored by varying sucrose initial concentration
FEMS Microbiol Lett 285 (2008) 25–32 Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works

(Potocki-Veronese et al., 2005). However, the use of amylo-
sucrase as an industrial tool is limited by its low thermo-
stability. Indeed, the two amylosucrases characterized to
date present very low half-lives at both 30 and 50 1C
[t1/2(30 1C) = 21 and 15 h for NPAS and DRAS, respectively;
t1/2(50 1C) = 3 min for NPAS].
Here we describe the search for hypothetical thermostable
amylosucrases by exploring the microbial natural diversity
through a BLAST search focused on thermophilic microorgan-
ism genomes. This led to the identification of a locus
annotated as encoding an ‘a-amylase (catalytic region)’ in
the moderate thermophile Deinococcus geothermalis (DGAS)
(optimum growth temperature between 45 and 50 1C)
(Ferreira et al., 1997). This locus was cloned and expressed
in Escherichia coli. The properties of the resulting protein
were investigated to study its activity and thermostability
in comparison with other previously characterized
amylosucrases.
Materials and methods
Database search and sequence analysis
To identify putative amylosucrases, a BLAST search was
performed on available microbial genomes using the NCBI
BLAST tool (http://www.ncbi.nlm.nih.gov/sutils/genom
_table.cgi) and as query the protein sequence of NPAS
amylosucrase (GenBank accession no. AJ011781). Protein
sequences were analyzed using ALIGNX (Vector NTI Advance
10 suite, Invitrogen) and the JALVIEW multiple alignment
editor (Clamp et al., 2004). Amino acid alignments over the
NPAS B0 domain were made between residues 384 and 418.
Bacterial strains and cloning vectors
DGAS DSM11300 (Deutsche Sammlung von Mikroorganis-
men und Zellkulturen GmbH, Braunschweigh, Germany)
was grown at 47 1C in Thermus 162 medium (1 g L�1 yeast
extract, 1 g L�1 tryptone, 100 mg L�1 nitriloacetic acid,
40 mg L�1 CaSO4 � 2H2O, 200 mg L�1 MgCl2 � 6H2O, 0.5 mM
iron citrate, 4 mM KH2PO4, 12 mM Na2HPO4, trace
elements) at pH 7.2.
Plasmid pBAD-TOPO-TA was used for cloning of the
PCR product corresponding to the dgas gene. Plasmid
pGEX-6P3 (Amersham Biosciences) was used for the ex-
pression of the fusion gst-dgas gene. Escherichia coli TOP10
(Invitrogen) and JM109 (Promega) were used for recombi-
nant pBAD-TOPO-TA plasmid amplification and for
gst-dgas gene expression, respectively.
DNA purification and manipulations
DGAS genomic DNA was purified as described (Phalip et al.,
1994). Escherichia coli plasmid isolation was performed
using the QIASpin miniprep kit (Qiagen). PCR fragments
(digested or not) extraction from agarose gel was carried out
using the UltraClean GelSpin DNA purification kit
(MoBio). Restriction endonucleases and DNA-modifying
enzymes were purchased from New England Biolabs and
used according to the manufacturer’s instructions. DNA
sequencing was carried out using the dideoxy chain termi-
nation procedure (v3.1 Cycle Sequencing Kit; Applied
Biosystems) by MilleGen sequencing service (Labege,
France).
Cloning of dgas gene
Locus YP_604043.1 (dgas) was amplified by PCR using
genomic DNA from DGAS as template. The forward and
reverse primers (synthesized by Eurogentec, France) were
designed to match the 50 and the 30 ends of the dgas gene.
BamHI and XhoI restriction sites (underlined) were also
introduced at their 50 termini to allow cloning into vector
pGEX-6P3 in fusion with the glutathione-S-transferase
(GST) encoding gene.
Forward primer: 50-GGATCCATGCTGAAAGACGTGC-30.
Reverse primer: 50-CTCGAGTTATGCTGGAGCCTCC-30.
A 2-kb DNA fragment was amplified by PCR with a
Biorad MyCycler thermal cycler from the following PCR
mixture: 50 ng genomic DNA, 1 mM of each primer, 0.2 mM
of each dNTP, 1� polymerase buffer, 5% dimethyl sulph-
oxide and 1 U DyNAzyme II DNA polymerase (Finnzymes).
The program used consisted of 30 cycles of 94 1C for 1 min,
55 1C for 45 s and 72 1C for 3 min. The PCR product was
purified and cloned into the pBAD-TOPO-TA vector. The
resulting plasmid was then digested using BamHI and XhoI
and the DNA fragment corresponding to the dgas gene was
subcloned into pGEX-6P3 using T4 DNA ligase. The final
construct was named pGST-DGAS.
DGAS production and purification
Escherichia coli JM109 cells containing pGST-DGAS were
first grown at 37 1C in 2XYT medium containing
100 mg mL�1 ampicillin until OD600 nm reached 0.5. Gene
expression was then induced by adding isopropyl b-D-1-
thiogalactopyranoside (IPTG) up to a final concentration of
10 mM and the culturing was continued for 15 h at 30 1C.
The cells were harvested by centrifugation (8000 g, 15 min,
4 1C), and the pellet was resuspended to a final OD600 nm of
80 in phosphate-buffered saline (140 mM NaCl, 2.7 mM
KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3). After
sonication, the extract was centrifuged (15 000 g, 40 min,
4 1C) and the supernatant was harvested for subsequent
purification. DGAS was purified by affinity chromatography
using Glutathion-Sepharose-4B support (Amersham Bios-
ciences) as described previously (Potocki de Montalk et al.,
1999). The protein content was determined by the micro
FEMS Microbiol Lett 285 (2008) 25–32Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
26 S. Emond et al.

Bradford method (Bradford, 1976) using the Bio-Rad
reagent (Bio-Rad laboratories, Hercules, CA) and bovine
serum albumin as standard.
Amylosucrase assay
One unit of amylosucrase activity corresponds to the
amount of enzyme that catalyzes the release of 1 mmol of
reducing sugars per minute in the assay conditions. As for
NPAS (Potocki de Montalk et al., 1999) and DRAS, DGAS
and GST-DGAS specific activity was determined at 30 1C
using 50 g L�1 sucrose and 0.1 g L�1 glycogen in 50 mM Tris-
HCl buffer, pH 7.0. The concentration of reducing sugars
was determined using the dinitrosalicylic method (Sumner
& Howell, 1935) and fructose as standard.
Thermostability and temperature dependencyof DGAS
Thermal inactivation at 30 and 50 1C was analyzed for
purified GST-DGAS and DGAS by incubating the enzymes
for various time intervals. Initial and residual activities were
measured at 50 1C with 50 g L�1 sucrose and 0.1 g L�1 glyco-
gen in 50 mM Tris-HCl buffer, pH 7.0, using the dinitro-
salicylic method to quantify the released reducing sugars. As
it was not possible to calculate the half-lives of irreversible
thermal denaturation following a first-order rate model,
half-lives were determined as the incubation time corre-
sponding to 50% inactivation.
DGAS temperature dependency was tested by measuring
initial specific activities of the purified enzyme with 50 g L�1
sucrose in 50 mM Tris-HCl buffer, pH 7.0, at temperatures
ranging from 30 to 65 1C.
In vitro synthesis of amylose
Reactions were carried out using DGAS at 1 U mL�1 (activity
measured at 30 1C from 50 g L�1 sucrose and 0.1 g L�1
glycogen in 50 mM Tris-HCl, pH 7.0). Synthesis reactions
were performed for 24 h at 30 and 50 1C in 50 mM Tris-HCl,
pH 7.0, containing 100 or 600 mM sucrose. For carbohy-
drate analysis, samples collected at the beginning and at the
end of the synthesis were heated at 90 1C during 5 min to
stop the reaction.
Carbohydrate analysis
During a 24-h reaction using sucrose as substrate, a soluble
fraction (monosaccharides and oligosaccharides) and a
precipitate (oligosaccharides and longer a-1,4 linked linear
chains) were formed. For qualitative analysis, the final
reaction media (containing both soluble and insoluble
products) were solubilized in 1 M KOH to a final polymer
content of 10 g L�1 and analyzed by high-performance
anion-exchange chromatography with pulsed amperometric
detection (HPAEC-PAD). For the determination of reaction
yields by quantification of monosaccharides and oligosac-
charides, reaction media supernatants (corresponding to the
soluble fractions) were diluted in water to 17 mg kg�1 total
sugars. Separation was performed on a 4� 250 mm Dionex
Carbo-pack PA100. The flow rate of 150 mM NaOH was
1.0 mL min�1 and an acetate gradient was applied as follows:
0–20 min, 6–196 mM; 20–70 min, 196–303.5 mM;
70–134 min, 303.5–450 mM; 135–140 min, 6 mM. Detection
was performed using a Dionex ED40 module with a gold
working electrode and a Ag/AgCl pH reference. HPAEC-
PAD does not enable one to directly quantify the amount of
oligosaccharides for which standards are not available [mal-
tooligosaccharides of degree of polymerization (DP)4 7
and oligosaccharides of unknown structure]. Consequently,
polymerization yields (corresponding to the production of
DP4 3 chains contained in the total reaction media, which
comprise soluble and insoluble fractions) were determined
as the difference between the molar amount of sucrose
consumed and the molar amount of glucosyl residues
incorporated into glucose, sucrose isomers, maltose and
maltotriose.
Iodine binding properties
The iodine reactivity of the amylose chains produced by
DGAS was measured. The final reaction media (20 mL) were
solubilized with 20 mL of 2 M KOH, neutralized with 200 mL
0.1 N HCl and mixed with 32 mL of aqueous iodine solution
[2% (w/v) KI and 0.2% (w/v) I2] and 8mL of 0.1 N HCl in
microtitre plates. The maximum absorption wavelength
(lmax) of the iodine/amylose complexes was determined by
recording spectra between 500 and 690 nm, with 2 nm steps,
on a Sunrise spectrophotometer (Tecan, Switzerland). The
corresponding average DP was calculated from the following
equation previously established for DP 28–70 amylose
chains (John et al., 1983):
103=lmax ¼ 11:6=DP þ 1:53728
Three-dimensional (3D) modelling
3D modelling of DGAS was performed with
ESYPRED3D (http://www.fundp.ac.be/sciences/biologie/urbm/
bioinfo/esypred/) (Lambert et al., 2002), which utilizes the
MODELLER program (Sali & Blundell, 1993), using the crystal
structure of amylosucrase from NPAS (PDB: 1G5A) as
template. Molecular graphics were performed using SYBYL7.3
(Tripos, St. Louis).
FEMS Microbiol Lett 285 (2008) 25–32 Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
27Thermostable amylosucrase from Deinococcus geothermalis
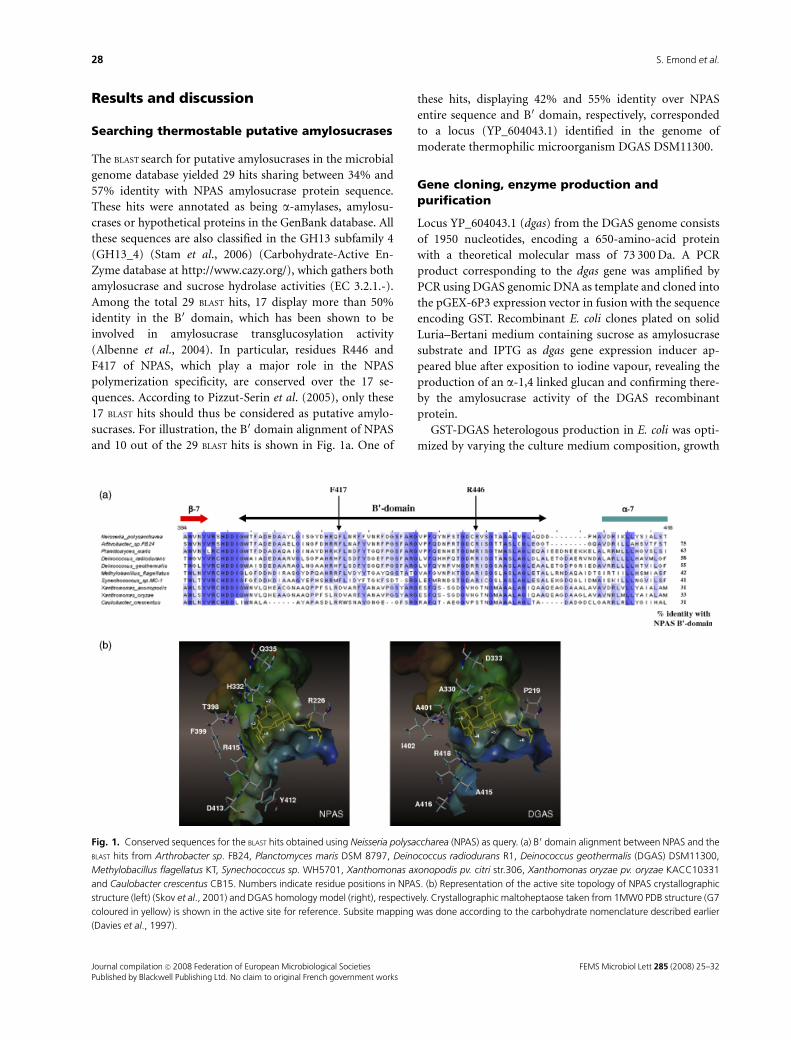
Results and discussion
Searching thermostable putative amylosucrases
The BLAST search for putative amylosucrases in the microbial
genome database yielded 29 hits sharing between 34% and
57% identity with NPAS amylosucrase protein sequence.
These hits were annotated as being a-amylases, amylosu-
crases or hypothetical proteins in the GenBank database. All
these sequences are also classified in the GH13 subfamily 4
(GH13_4) (Stam et al., 2006) (Carbohydrate-Active En-
Zyme database at http://www.cazy.org/), which gathers both
amylosucrase and sucrose hydrolase activities (EC 3.2.1.-).
Among the total 29 BLAST hits, 17 display more than 50%
identity in the B0 domain, which has been shown to be
involved in amylosucrase transglucosylation activity
(Albenne et al., 2004). In particular, residues R446 and
F417 of NPAS, which play a major role in the NPAS
polymerization specificity, are conserved over the 17 se-
quences. According to Pizzut-Serin et al. (2005), only these
17 BLAST hits should thus be considered as putative amylo-
sucrases. For illustration, the B0 domain alignment of NPAS
and 10 out of the 29 BLAST hits is shown in Fig. 1a. One of
these hits, displaying 42% and 55% identity over NPAS
entire sequence and B0 domain, respectively, corresponded
to a locus (YP_604043.1) identified in the genome of
moderate thermophilic microorganism DGAS DSM11300.
Gene cloning, enzyme production andpurification
Locus YP_604043.1 (dgas) from the DGAS genome consists
of 1950 nucleotides, encoding a 650-amino-acid protein
with a theoretical molecular mass of 73 300 Da. A PCR
product corresponding to the dgas gene was amplified by
PCR using DGAS genomic DNA as template and cloned into
the pGEX-6P3 expression vector in fusion with the sequence
encoding GST. Recombinant E. coli clones plated on solid
Luria–Bertani medium containing sucrose as amylosucrase
substrate and IPTG as dgas gene expression inducer ap-
peared blue after exposition to iodine vapour, revealing the
production of an a-1,4 linked glucan and confirming there-
by the amylosucrase activity of the DGAS recombinant
protein.
GST-DGAS heterologous production in E. coli was opti-
mized by varying the culture medium composition, growth
Fig. 1. Conserved sequences for the BLAST hits obtained using Neisseria polysaccharea (NPAS) as query. (a) B0 domain alignment between NPAS and the
BLAST hits from Arthrobacter sp. FB24, Planctomyces maris DSM 8797, Deinococcus radiodurans R1, Deinococcus geothermalis (DGAS) DSM11300,
Methylobacillus flagellatus KT, Synechococcus sp. WH5701, Xanthomonas axonopodis pv. citri str.306, Xanthomonas oryzae pv. oryzae KACC10331
and Caulobacter crescentus CB15. Numbers indicate residue positions in NPAS. (b) Representation of the active site topology of NPAS crystallographic
structure (left) (Skov et al., 2001) and DGAS homology model (right), respectively. Crystallographic maltoheptaose taken from 1MW0 PDB structure (G7
coloured in yellow) is shown in the active site for reference. Subsite mapping was done according to the carbohydrate nomenclature described earlier
(Davies et al., 1997).
FEMS Microbiol Lett 285 (2008) 25–32Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
28 S. Emond et al.
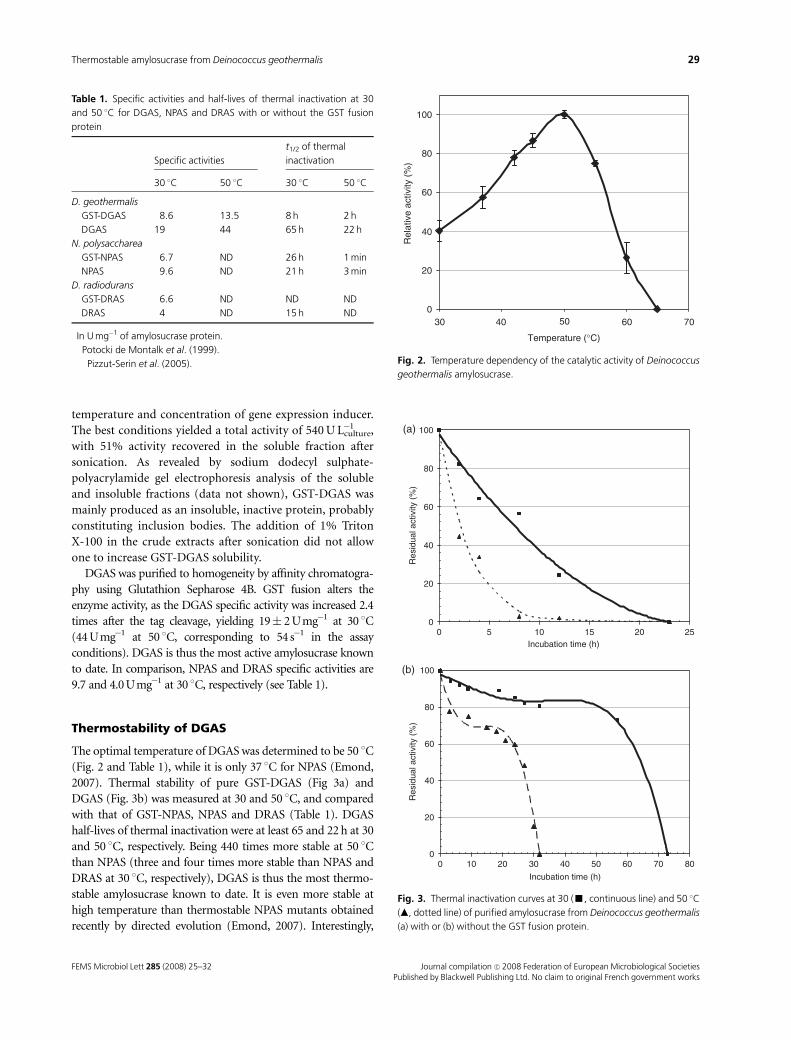
temperature and concentration of gene expression inducer.
The best conditions yielded a total activity of 540 U Lculture�1 ,
with 51% activity recovered in the soluble fraction after
sonication. As revealed by sodium dodecyl sulphate-
polyacrylamide gel electrophoresis analysis of the soluble
and insoluble fractions (data not shown), GST-DGAS was
mainly produced as an insoluble, inactive protein, probably
constituting inclusion bodies. The addition of 1% Triton
X-100 in the crude extracts after sonication did not allow
one to increase GST-DGAS solubility.
DGAS was purified to homogeneity by affinity chromatogra-
phy using Glutathion Sepharose 4B. GST fusion alters the
enzyme activity, as the DGAS specific activity was increased 2.4
times after the tag cleavage, yielding 19� 2 Umg�1 at 30 1C
(44 Umg�1 at 50 1C, corresponding to 54 s�1 in the assay
conditions). DGAS is thus the most active amylosucrase known
to date. In comparison, NPAS and DRAS specific activities are
9.7 and 4.0 Umg�1 at 30 1C, respectively (see Table 1).
Thermostability of DGAS
The optimal temperature of DGAS was determined to be 50 1C
(Fig. 2 and Table 1), while it is only 37 1C for NPAS (Emond,
2007). Thermal stability of pure GST-DGAS (Fig 3a) and
DGAS (Fig. 3b) was measured at 30 and 50 1C, and compared
with that of GST-NPAS, NPAS and DRAS (Table 1). DGAS
half-lives of thermal inactivation were at least 65 and 22 h at 30
and 50 1C, respectively. Being 440 times more stable at 50 1C
than NPAS (three and four times more stable than NPAS and
DRAS at 30 1C, respectively), DGAS is thus the most thermo-
stable amylosucrase known to date. It is even more stable at
high temperature than thermostable NPAS mutants obtained
recently by directed evolution (Emond, 2007). Interestingly,
Table 1. Specific activities and half-lives of thermal inactivation at 30
and 50 1C for DGAS, NPAS and DRAS with or without the GST fusion
protein
Specific activities�t1/2 of thermal
inactivation
30 1C 50 1C 30 1C 50 1C
D. geothermalis
GST-DGAS 8.6 13.5 8 h 2 h
DGAS 19 44 65 h 22 h
N. polysaccharea��
GST-NPAS 6.7 ND 26 h 1 min
NPAS 9.6 ND 21 h 3 min
D. radiodurans���
GST-DRAS 6.6 ND ND ND
DRAS 4 ND 15 h ND
�In U mg�1 of amylosucrase protein.��Potocki de Montalk et al. (1999).���Pizzut-Serin et al. (2005).
0
20
40
60
80
100
30 40 50 60 70
Temperature (°C)
Rel
ativ
e ac
tivity
(%
)
Fig. 2. Temperature dependency of the catalytic activity of Deinococcus
geothermalis amylosucrase.
0
20
40
60
80
100
0 10 20 30 40 50 60 70 80
0
20
40
60
80
100
0 5 10 15 20 25Incubation time (h)
Incubation time (h)
Res
idua
l act
ivity
(%
)R
esid
ual a
ctiv
ity (
%)
(a)
(b)
Fig. 3. Thermal inactivation curves at 30 (’, continuous line) and 50 1C
(m, dotted line) of purified amylosucrase from Deinococcus geothermalis
(a) with or (b) without the GST fusion protein.
FEMS Microbiol Lett 285 (2008) 25–32 Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
29Thermostable amylosucrase from Deinococcus geothermalis
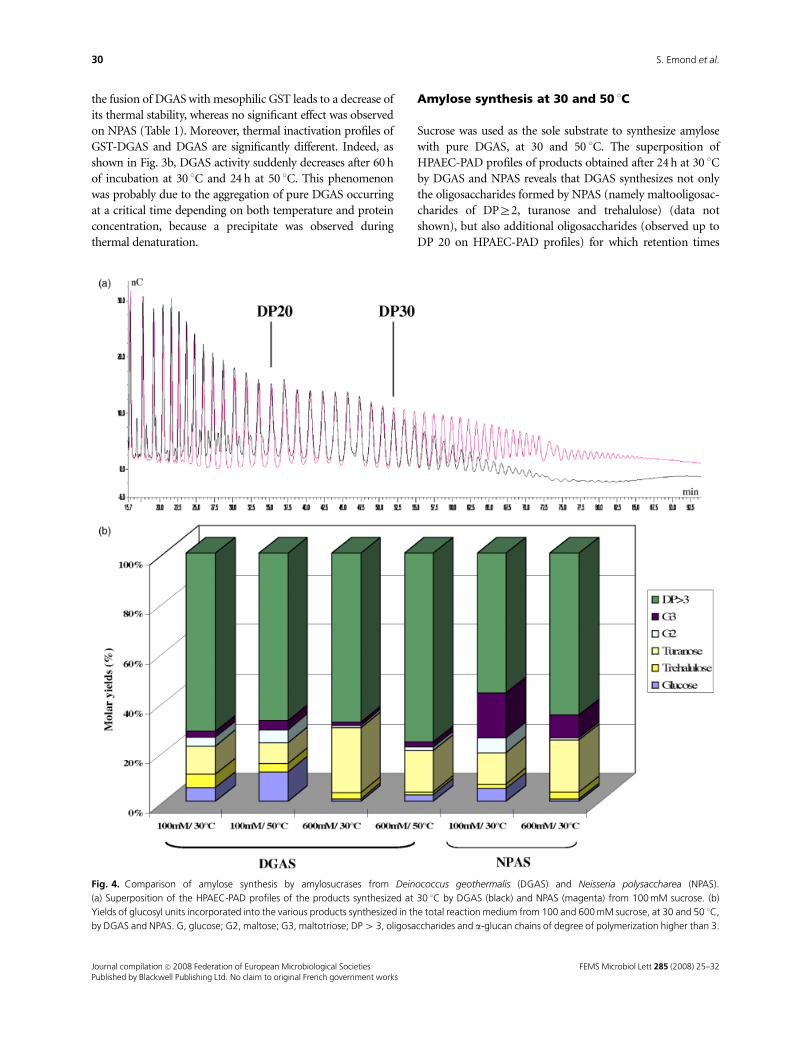
the fusion of DGAS with mesophilic GST leads to a decrease of
its thermal stability, whereas no significant effect was observed
on NPAS (Table 1). Moreover, thermal inactivation profiles of
GST-DGAS and DGAS are significantly different. Indeed, as
shown in Fig. 3b, DGAS activity suddenly decreases after 60 h
of incubation at 30 1C and 24 h at 50 1C. This phenomenon
was probably due to the aggregation of pure DGAS occurring
at a critical time depending on both temperature and protein
concentration, because a precipitate was observed during
thermal denaturation.
Amylose synthesis at 30 and 50 1C
Sucrose was used as the sole substrate to synthesize amylose
with pure DGAS, at 30 and 50 1C. The superposition of
HPAEC-PAD profiles of products obtained after 24 h at 30 1C
by DGAS and NPAS reveals that DGAS synthesizes not only
the oligosaccharides formed by NPAS (namely maltooligosac-
charides of DPZ2, turanose and trehalulose) (data not
shown), but also additional oligosaccharides (observed up to
DP 20 on HPAEC-PAD profiles) for which retention times
Fig. 4. Comparison of amylose synthesis by amylosucrases from Deinococcus geothermalis (DGAS) and Neisseria polysaccharea (NPAS).
(a) Superposition of the HPAEC-PAD profiles of the products synthesized at 30 1C by DGAS (black) and NPAS (magenta) from 100 mM sucrose. (b)
Yields of glucosyl units incorporated into the various products synthesized in the total reaction medium from 100 and 600 mM sucrose, at 30 and 50 1C,
by DGAS and NPAS. G, glucose; G2, maltose; G3, maltotriose; DP4 3, oligosaccharides and a-glucan chains of degree of polymerization higher than 3.
FEMS Microbiol Lett 285 (2008) 25–32Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
30 S. Emond et al.
corresponded neither to sucrose isomers, nigerose, palatinose,
trehalose, maltooligosaccharides, isomaltooligosaccharides
nor to a-1,2 branched glucooligosaccharides (Fig. 4a). These
products might result from successive glucosylations of su-
crose isomers or sucrose itself, as these disaccharides are
present in significant concentration in the reaction medium.
Given the ability of DGAS to glucosylate a larger diversity of
oligosaccharides, it is thus seen as being more polyspecific
than NPAS. Also, Fig. 4a illustrates the high polydispersity of
a-glucans produced by both DGAS and NPAS, suggesting that
these enzymes probably use a similar nonprocessive polymer-
ization mechanism (Albenne et al., 2004).
The average sizes of polymers produced from 100 mM
sucrose at 30 1C by DGAS and NPAS, respectively, are
comparable (Table 2). Nevertheless, as indicated in Fig. 4a,
the size distribution of the a-glucan chains produced by
NPAS and DGAS from 100 mM sucrose slightly differs.
Indeed, chains of DP4 30 are less concentrated in the
reaction medium of DGAS in comparison to that of NPAS
(Fig. 4a). This correlates fairly well with the lower values of
mean DP calculated on the basis of the maximum absorp-
tion wavelength of iodine–amylose complexes (Table 2).
Differences were also observed in the product synthesis
yields of both enzymes (Fig. 4b). The production yields of
turanose, trehalulose and maltotriose significantly differed
between the enzymes, together with the polymerization
yield, which was slightly higher for DGAS than for NPAS.
These data, as well as the high polyspecificity of DGAS,
indicate that its oligosaccharide binding site OB1 (Skov et al.,
2002) is likely to be structurally different from that of NPAS,
allowing the accommodation of a larger variety of acceptor
molecules (fructose to form sucrose isomers, maltooligosac-
charides to be elongated and other oligosaccharides yielding
to the synthesis of products with unknown structure). The
sequence identity with NPAS (42%) allowed a homology
modelling of DGAS using NPAS as template (root mean
square deviation of 1.25 A over all atoms; Fig. 1b). A detailed
inspection of the DGAS active site revealed a high conserva-
tion of amino acid residues forming the � 1 and 11 subsites
(Fig. 1b) (Davies et al., 1997), in particular the catalytic triad
conserved for all members of the GH13 family. Most
significant changes were observed at subsites 12 to 15,
where key residues known to participate in oligosaccharide
elongation in NPAS are not conserved in DGAS. It has been
shown previously that mutation R226A in NPAS could
drastically increase the polymer formation (Albenne et al.,
2004). The corresponding residue in DGAS is P219. Conse-
quently, significant differences in the active site topology are
expected, which could explain the distinct products formed
by each enzyme.
Importantly, DGAS was also able to synthesize amylose at
50 1C, from 100 and 600 mM sucrose, whereas NPAS
was inactive in those conditions. At both 30 and 50 1C,
DGAS synthesized the same oligosaccharides, and as re-
vealed by HPAEC-PAD (data not shown) and by iodine
complexation (Table 2), the average amylose chain
length remained unmodified by the reaction temperature
increase.
Finally, using 600 mM initial sucrose, the DGAS poly-
merization yield slightly increased from 68% to 76% when
switching temperature from 30 to 50 1C. Taken together, the
present results demonstrate that DGAS is so far the most
attractive enzymatic tool for in vitro synthesis of amylopoly-
saccharides in industrial processes.
Acknowledgements
The authors wish to thank Sandra Pizzut-Serin and Benoıt
Bordignon for helpful technical assistance.
References
Albenne C, Skov LK, Mirza O et al. (2004) Molecular basis of the
amylose-like polymer formation catalyzed by Neisseria
polysaccharea amylosucrase. J Biol Chem 279: 726–734.
Bradford MM (1976) A rapid and sensitive method for the
quantitation of microgram quantities of protein utilizing the
principle of protein-dye binding. Anal Biochem 72: 248–254.
Clamp M, Cuff J, Searle SM & Barton GJ (2004) The Jalview Java
alignment editor. Bioinformatics 20: 426–427.
Davies GJ, Wilson KS & Henrissat B (1997) Nomenclature for
sugar-binding subsites in glycosyl hydrolases. Biochem J 321:
(Part 2): 557–559.
Emond S (2007) Directed evolution of amylosucrase using the
MutaGenTM technology toward the identification of
thermostable variants. Ph.D. Thesis, Institut National des
Sciences Appliques, Toulouse.
Ferreira AC, Nobre MF, Rainey FA et al. (1997) Deinococcus
geothermalis sp. nov. and Deinococcus murrayi sp. nov., two
extremely radiation-resistant and slightly thermophilic species
from hot springs. Int J Syst Bacteriol 47: 939–947.
Table 2. Maximum absorption wavelength of the iodine–amylose com-
plexes obtained from the products synthesized at 30 and 50 1C by the
amylosucrases from Neisseria polysaccharea and Deinococcus geother-
malis from 100 and 600 mM sucrose, and corresponding average DP
Temperature
( 1C)
Sucrose
(mM) lmax
Average
DP
NPAS� 30 1C 100 576� 4 58�4
600 546� 4 39�2
DGAS 30 1C 100 562� 6 48�4
600 552� 4 42�2
50 1C 100 546� 8 39�4
600 558� 4 45�3
� Potocki-Veronese et al. (2005).
FEMS Microbiol Lett 285 (2008) 25–32 Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
31Thermostable amylosucrase from Deinococcus geothermalis

Jensen MH, Mirza O, Albenne C, Remaud-Simeon M, Monsan P,
Gajhede M & Skov LK (2004) Crystal structure of the covalent
intermediate of amylosucrase from Neisseria polysaccharea.
Biochemistry 43: 3104–3110.
John M, Schmidt J & Kneifel H (1983) Iodine–maltosaccharide
complexes: relation between chain-length and colour.
Carbohydr Res 119: 254–257.
Lambert C, Leonard N, De Bolle X & Depiereux E (2002)
ESyPred3D: prediction of proteins 3D structures.
Bioinformatics 18: 1250–1256.
Phalip V, Dartois V, Schmitt P & Divies C (1994) Cloning of the
D-lactate dehydrogenase gene from Leuconostoc mesenteroides
subsp. cremoris. Biotechnol Lett 16: 221–226.
Pizzut-Serin S, Potocki-Veronese G, van der Veen BA, Albenne C,
Monsan P & Remaud-Simeon M (2005) Characterisation of a
novel amylosucrase from Deinococcus radiodurans. FEBS Lett
579: 1405–1410.
Potocki de Montalk G, Remaud-Simeon M, Willemot RM, Planchot
V & Monsan P (1999) Sequence analysis of the gene encoding
amylosucrase from Neisseria polysaccharea and characterization of
the recombinant enzyme. J Bacteriol 181: 375–381.
Potocki de Montalk G, Remaud-Simeon M, Willemot RM,
Sarcabal P, Planchot V & Monsan P (2000) Amylosucrase from
Neisseria polysaccharea: novel catalytic properties. FEBS Lett
471: 219–223.
Potocki-Veronese G, Putaux JL, Dupeyre D, Albenne C, Remaud-
Simeon M, Monsan P & Buleon A (2005) Amylose synthesized
in vitro by amylosucrase: morphology, structure, and
properties. Biomacromolecules 6: 1000–1011.
Preiss J, Ozbun JL, Hawker JS, Greenberg E & Lammel C (1973)
ADPG synthetase and ADPG- -glucan 4-glucosyl transferase:
enzymes involved in bacterial glycogen and plant starch
synthesis. Ann NY Acad Sci 210: 265–278.
Quanz M & Frohberg C (2005) Use of poly-a-(1,4)-glucans as
resistant starch. Patent WO2005040223.
Rolland-Sabate A, Colonna P, Potocki-Veronese G, Monsan P &
Planchot V (2004) Elongation and insolubilisation of
a-glucans by the action of Neisseria polysaccharea
amylosucrase. J Cereal Sci 40: 17–30.
Sali A & Blundell TL (1993) Comparative protein modelling by
satisfaction of spatial restraints. J Mol Biol 234: 779–815.
Skov LK, Mirza O, Henriksen A et al. (2001) Amylosucrase, a
glucan-synthesizing enzyme from the alpha-amylase family.
J Biol Chem 276: 25273–25278.
Skov LK, Mirza O, Sprogoe D et al. (2002) Oligosaccharide and
sucrose complexes of amylosucrase. Structural implications
for the polymerase activity. J Biol Chem 277: 47741–47747.
Stam MR, Danchin EG, Rancurel C, Coutinho PM & Henrissat B
(2006) Dividing the large glycoside hydrolase family 13 into
subfamilies: towards improved functional annotations of
alpha-amylase-related proteins. Protein Eng Des Sel 19:
555–562.
Sumner JB & Howell SF (1935) A method for determination of
saccharase activity. J Biol Chem 108: 51–54.
Uitdehaag JC, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L,
Withers SG & Dijkstra BW (1999) X-ray structures along the
reaction pathway of cyclodextrin glycosyltransferase elucidate
catalysis in the alpha-amylase family. Nat Struct Biol 6:
432–436.
FEMS Microbiol Lett 285 (2008) 25–32Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd. No claim to original French government works
32 S. Emond et al.

186
Tables des illustrations

187
FIGURES
Figure 1 : Structure de l'ADN polymérase du phage T7 ................................................................................. 21 Figure 2 : Génération d'additions et de délétions............................................................................................. 23 Figure 3 : Principe de l’évolution dirigée des protéines................................................................................... 25 Figure 4 : Principe et étapes de la technique MutaGenTM . .............................................................................. 31 Figure 5 : Technique du DNA shuffling. ........................................................................................................... 33 Figure 6 : Technique du family shuffling. ......................................................................................................... 34 Figure 7 : La technique StEP. ............................................................................................................................36 Figure 8 : La technique RPR..............................................................................................................................37 Figure 9 : La technique RACHITT. .................................................................................................................. 38 Figure 10 : La technique ITCHY....................................................................................................................... 41 Figure 11 : La technique SHIPREC. ................................................................................................................. 42 Figure 12 : La technique NRR. .......................................................................................................................... 43 Figure 13 : Techniques de mutagénèse dirigée à saturation............................................................................ 47 Figure 14 : Principe de la technique DOGS...................................................................................................... 48 Figure 15 : Stratégie de sélection par complémentation chimique développée par Sin Fiet et al. (2006).... 51 Figure 16 : Stratégie de sélection de banques de variants enzymatiques utilisant le phage display. ........... 53 Figure 17 : Utilisation du ribosome-display pour la sélection de variants enzymatiques.. ........................... 56 Figure 18 : Stratégies de sélection de banques de variants enzymatiques par compartimentation in vitro. 58 Figure 19 : Schéma des principales réactions catalysées par les amylosaccharases...................................... 63 Figure 20 : Structure tridimensionelle de l'amylosaccharase de N. polysaccharea....................................... 66 Figure 21 : Réactions catalysées par l'AS de Neisseria polysaccharea. ........................................................... 67 Figure 22 : Schéma du mécanisme réactionnel suivi par l'AS ........................................................................ 68 Figure 23 : Représentation schématique des sous-sites de liaison de l'AS. .................................................... 69 Figure 24: Représentation schématique des interactions (A) entre le mutant inactif E328Q et le saccharose
et (B) entre l'AS et le D-Glucose............................................................................................................... 70 Figure 25 : Interactions du site OB1.................................................................................................................. 71 Figure 26 : Positionnement des sites OB1, OB2 et OB3................................................................................... 72 Figure 27 : Analyse par cryo-microscopie électronique à transmission du rallongement des ramifications
de glycogène par l'AS à partir de saccharose 100 mM et de glycogène 0,1 g/L.................................... 77 Figure 28 : Influence de la concentration initiale de saccharose sur la vitesse initiale de production de
fructose (Vi), pour différentes concentrations initiales de glycogène.................................................... 78 Figure 29 : Activité relative et répartition des produits formés par les mutants du site OB1...................... 81 Figure 30 : Méthodes de sélection et de criblage de banques de variants de l'AS. ........................................ 83 Figure 31 : The MutaGenTM process.. ............................................................................................................... 89 Figure 32 : Modified MutaGenTM methodology to construct full-length as gene library using pol ββββ............. 90 Figure 33 : Distribution of amino acid mutations in AS variants generated with pol ηηηη. .............................. 97 Figure 34 : Accuracy of a screening protocol. ................................................................................................ 109 Figure 35 : Optimization of the screening procedure reproducibility.......................................................... 111 Figure 36 : Bacterial growth and recombinant GST-AS production kinetics in microplate...................... 113 Figure 37 : High-throughput screening procedure for gene libraries of AS variants. ................................ 114 Figure 38 : Activity of wild-type AS following heat treatment or in the presence of DMSO/water mixtures
................................................................................................................................................................... 115 Figure 39 : Screening of a first generation AS variant library for thermostability. ................................... 116 Figure 40 : Thermal stability of P351S variant at 50°C................................................................................. 117 Figure 41 : Thermostability of AS and its variants.. ...................................................................................... 127 Figure 42 : Overview of mutation sites affecting amylosucrase thermostability.. ....................................... 129 Figure 43 : Comparison between wild-type AS (A and C) and double mutant R20C/A451T (B and D). . 130 Figure 44 : Comparison between AS-WT (A) and single mutant P351S (B). .............................................. 131 Figure 45 : Representation of the hydrophobic region surrounding Q353L mutation in double mutant
A170V/Q353L. ......................................................................................................................................... 132 Figure 46 : HPAEC profile of the amylose chains synthesized by the R20C/A451T amylosucrase variant.
................................................................................................................................................................... 134

188
TABLEAUX
Tableau 1 : Les ADN polymérases identifiées chez Escherichia coli, chez Saccharomyces cerevisiae et chez l’Homme (d’après Bebenek et Kunkel, 2004). ........................................................................................ 19
Tableau 2 : Taux d’erreurs in vitro des ADN polymérases (d’après Bebenek et Kunkel, 2004). ................. 20 Tableau 3 : Exemples de méthodes de mutagénèse aléatoire. ......................................................................... 28 Tableau 4 : Résidus mutants possibles à partir d’une substitution d’une base dans le codon original. ...... 28 Tableau 5 : Exemples et modes d'action d’agents mutagènes chimiques et physiques. ................................ 29 Tableau 6 : Spectre mutationnel de la PCR à erreurs en fonction des conditions réactionnelles. ............... 30 Tableau 7 : Mutagénèse à saturation. Nombre de variants générés selon la nature des codons dégénérés 46 Tableau 8 : Exemples de méthodes de sélection génétique employées en évolution d'enzyme. .................... 50 Tableau 9 : Exemples de méthodes de criblage utilisées pour l'évolution dirigée d'enzyme. ....................... 61 Tableau 10 : Nombre de variants analysés en fonction du type de criblage. Les banques criblées dans les
exemples cités ont toutes été construites par PCR à erreurs. ................................................................ 62 Tableau 11 : Amylosaccharases hypothétiques identifiées dans les génomes séquencés via le logiciel
BLAST (analyse effectuée en mars 2008). Le pourcentage d'identité (global et au niveau du domaine B') avec l'amylosaccharase de Neisseria polysaccharea est indiqué....................................................... 64
Tableau 12 : Rendement des produits synthétisés par l'AS purifiée de N. polysaccharea à partir de 100 mM de saccharose (d'après Potocki-Véronèse et al., 2005).................................................................... 66
Tableau 13 : Valeurs des constantes cinétiques apparentes des vitesses initiales de consommation du saccharose (ViS), d'hydrolyse du saccharose (ViG) et de polymérisation (ViGn) (d'après Potocki de Montalk et al., 2000). ................................................................................................................................. 73
Tableau 14 : Caractéristiques de l'amylose insoluble synthétisée par l'AS en fonction de la concentration initiale en saccharose (d'après Potocki-Véronèse et al., 2005)............................................................... 75
Tableau 15 : Brevets déposés concernant une application des amylosaccharases......................................... 80 Tableau 16 : Premiers variants de l'AS identifiés par ingénierie combinatoire (d'après Van der Veen et al.,
2004 et 2006)............................................................................................................................................... 84 Tableau 17 : Sequence analysis and mutation rates of AS variant libraries generated by MutaGenTM
(libraries A, B and C) or by epPCR. ........................................................................................................ 91 Tableau 18 : Comparison of base substitution spectra of MutaGenTM and epPCR methods. ................... 93 Tableau 19 : Protein sequence analysis of clones from AS libraries generated by MutaGenTM . ................. 95 Tableau 20 : Impact of base substitutions in the sequenced AS variants at the protein level. ..................... 95 Tableau 21 : Sequence analysis of the "hit" variant selected after screening for thermostability. ........... 117 Tableau 22 : Nucleotide substitutions and resulting amino acid replacements of the variants selected after
screening for thermostability.................................................................................................................. 126 Tableau 23 : Stability properties of wild-type AS and its variants. .............................................................. 127 Tableau 24 : Analysis of the products synthesized by wild-type AS and R20C/A451T variant (MOS:
maltooligosaccharides). ........................................................................................................................... 133
Top Related