Syndromes lymphoprolifératifs chroniques...Syndromes lympho-prolifératifs chroniques (SLPC)...
149
Syndromes lymphoprolifératifs chroniques Damien Roos-Weil [email protected] Service Hématologie Groupe Hospitalier Pitié-Salpêtrière
Transcript of Syndromes lymphoprolifératifs chroniques...Syndromes lympho-prolifératifs chroniques (SLPC)...

Syndromes lymphoprolifératifs chroniques
Damien Roos-Weil
Service Hématologie
Groupe Hospitalier Pitié-Salpêtrière
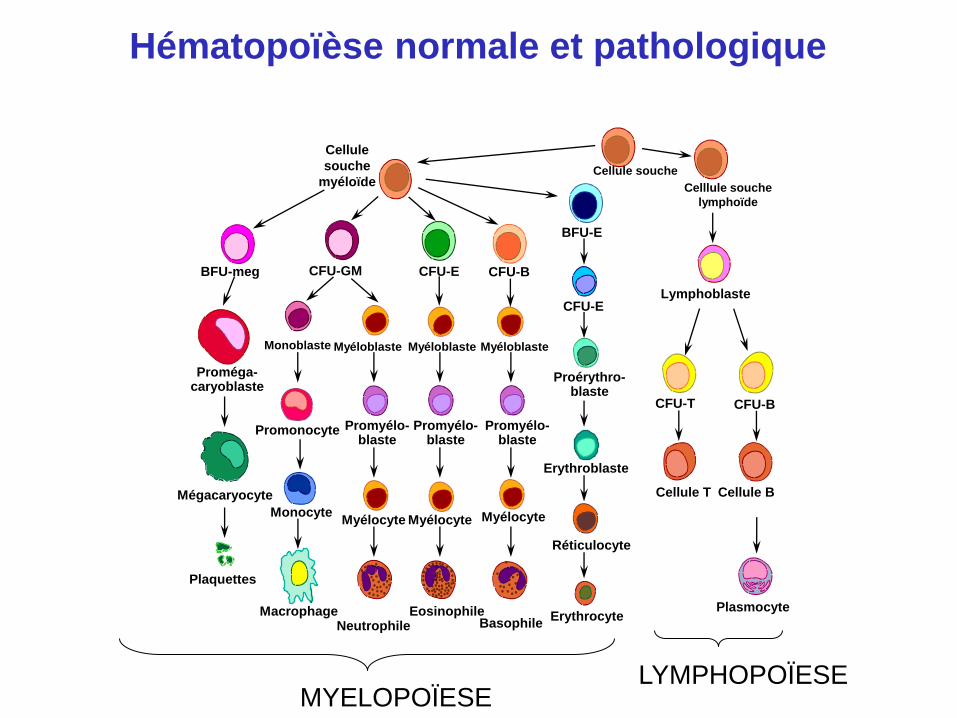
BFU-meg CFU-GM CFU-E CFU-B
BFU-E
CFU-E
CFU-T
Proérythro- blaste
Erythroblaste
Réticulocyte
Erythrocyte Basophile
Eosinophile Neutrophile
Macrophage
Plaquettes
Mégacaryocyte
Proméga- caryoblaste
Monocyte
Promonocyte
Myélocyte
Promyélo- blaste
Promyélo- blaste
Promyélo- blaste
Myélocyte Myélocyte
Monoblaste Myéloblaste Myéloblaste Myéloblaste
CFU-B
Cellule T Cellule B
Plasmocyte
Celllule souche
lymphoïde
Cellule souche
Cellule
souche
myéloïde
Lymphoblaste
LYMPHOPOÏESE MYELOPOÏESE
Hématopoïèse normale et pathologique
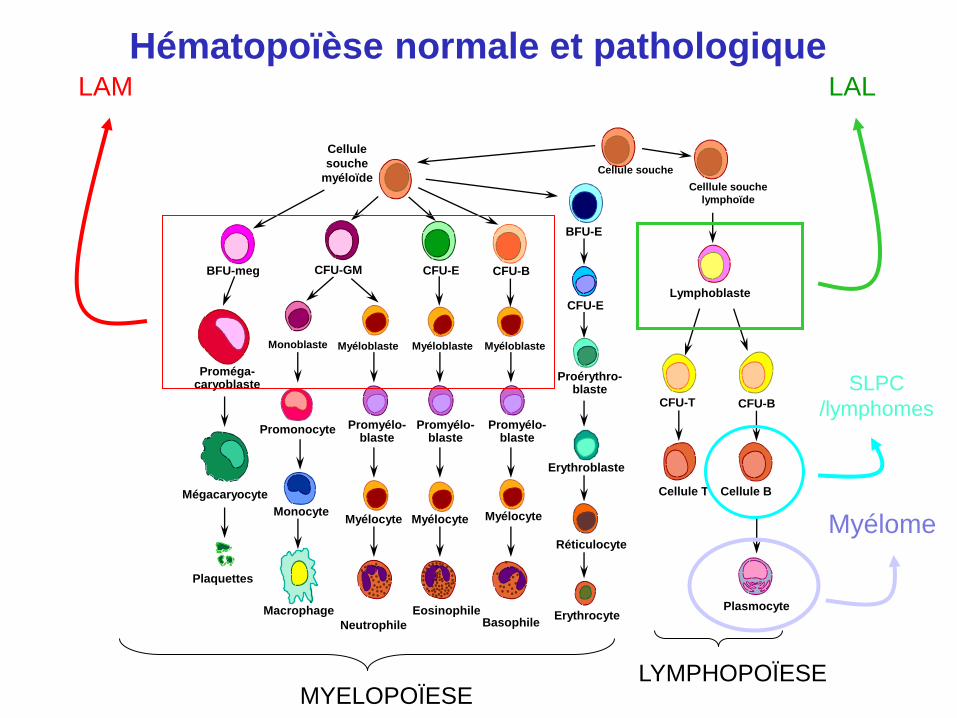
BFU-meg CFU-GM CFU-E CFU-B
BFU-E
CFU-E
CFU-T
Proérythro- blaste
Erythroblaste
Réticulocyte
Erythrocyte Basophile
Eosinophile
Neutrophile
Macrophage
Plaquettes
Mégacaryocyte
Proméga- caryoblaste
Monocyte
Promonocyte
Myélocyte
Promyélo- blaste
Promyélo- blaste
Promyélo- blaste
Myélocyte Myélocyte
Monoblaste Myéloblaste Myéloblaste Myéloblaste
CFU-B
Cellule T Cellule B
Plasmocyte
Celllule souche
lymphoïde
Cellule souche
Cellule
souche
myéloïde
Lymphoblaste
LYMPHOPOÏESE MYELOPOÏESE
LAL LAM
SLPC
/lymphomes
Myélome
Hématopoïèse normale et pathologique

Syndromes lympho-prolifératifs chroniques (SLPC)
(lymphomes de bas grade)
LYMPH-OME
cellule/tissu lymphoïde tumeur
Tumeur maligne du tissu lymphoïde : prolifération clonale de cellules lymphocytaires B
ou T matures à leurs différents stades de différenciation et d’activation
= groupe de maladies hétérogènes
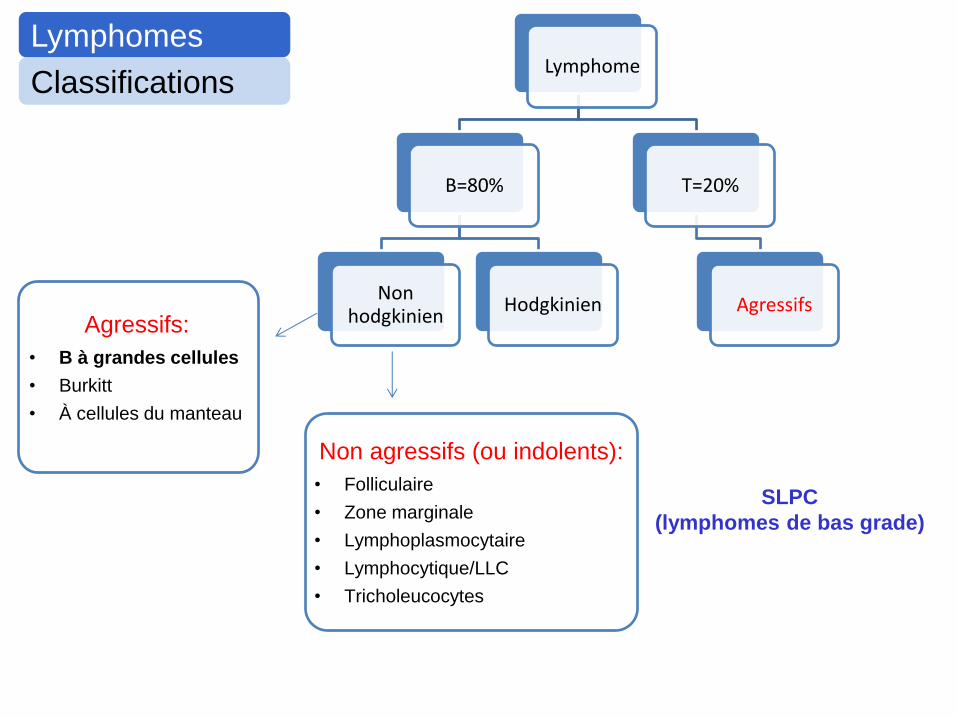
Lymphomes
Classifications Lymphome
B=80%
Non hodgkinien
Hodgkinien
T=20%
Agressifs Agressifs:
• B à grandes cellules
• Burkitt
• À cellules du manteau
Non agressifs (ou indolents):
• Folliculaire
• Zone marginale
• Lymphoplasmocytaire
• Lymphocytique/LLC
• Tricholeucocytes
SLPC
(lymphomes de bas grade)

Epidémiologie
Incidence des hémopathies(1)
36%
7%15%
12%
0%
1%
4%
9%
10%
4%
2%
0%
LNH
Hodgkin
Myélome
LLC/LL
LPL
Tricho
LAL/LL
Lymphoïde:
autres
LAM
LMC
Leucémies
autres
Divers
Hémopathies malignes

Epidémiologie
Lymphomes non hodgkiniens
Incidence des LNH(1)
51%
22%
7%
3%2%
2%2%4%0%2%2% 3%
DGCB
Folliculaire
Zone marginale
Manteau
Waldenström
Autres lymphoplasmocytaires
Burkitt
Mycosis/Sézary
Angio-immunoblastique
Anaplasique à grandes cellules
Autres T périphériques
T/NK

Classification OMS des
hémopathies lymphoïdes matures (2016)
Hémopathies lymphoïdes B
•Leucémie lymphoïde chronique/
lymphome lymphocytique
•Leucémie prolymphocytaire B
•Lymphome lymphoplasmocytaire
•Lymphome splénique à ly villeux
•Leucémie à tricholeucocytes
•Lymphome de la zone marginale
nodal ou extranodal
•Lymphome du MALT
•Lymphome folliculaire
•Lymphome à cellules du manteau
•Lymphome B diffus à grandes cellules
•….
Hémopathies lymphoïdes T
•Leucémie prolymphocytaire T
•Leucémie à LGL (grands
lymphocytes granuleux)
•Syndrome de Sezary
•Leucémie/lymphome T de l’adulte
•Lymphome T angioimmunoblastique
•Lymphome anaplasique à grandes
cellules
• …

Correspondance entre
stades de maturation lymphoïde B
et contrepartie lymphomateuse
Lymphome
du manteau
LLC
LAL
Myélome
?
Lymphome du MALT
SLVL
LLC
Lympho-plasmocytaire (MW)
Leucémie à tricholeucocytes ?
Lymphome folliculaire
Lymphome de Burkitt
Lymphome diffus à grandes
cellules
Plasmo-
cyte
MUTATIONS SOMATIQUES DES REGIONS VARIABLES DES IgH SANS MUTATION DES REGIONS VARIABLES DES IgH
Pro
B
Cellule
B naïve
Cellule
B CD5+
Cellule B
mémoire
ZONE DU
MANTEAU
CENTRE
GERMINATIF
Hypermutation
somatique
Commutation
de classe
Prolifération
Sélection Plasmocyte
Cellule B
du CG
Cellule B
du CG

Lymphomes
Etiologies
Infectieuses Autres
Immunodépression
Idiopathique+++
Formes familiales
VIH : risque 0,5% /an
EBV : Burkitt et
lymphome post-
transplantation
VHC : lymphome
splénique de la zone
marginale
HTLV1 : ATL
HHV8 : lymphome des
séreuses
Helicobacter Pylori
(estomac)
Campylobacter jejuni
(digestif)
Borrelia Burgdorferi
(peau)
Chlamydia Psittaci
(conjonctive)
Virus Bactéries
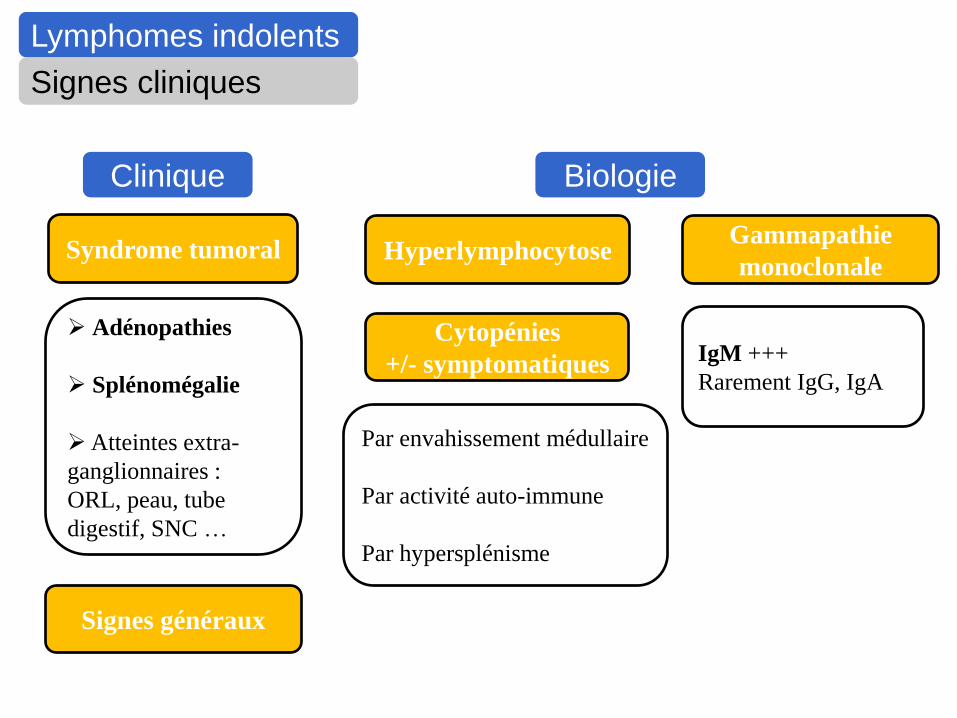
Lymphomes indolents
Signes cliniques
Syndrome tumoral
Adénopathies
Splénomégalie
Atteintes extra-
ganglionnaires :
ORL, peau, tube
digestif, SNC …
Signes généraux
Cytopénies
+/- symptomatiques
Par envahissement médullaire
Par activité auto-immune
Par hypersplénisme
Gammapathie
monoclonale
IgM +++
Rarement IgG, IgA
Clinique Biologie
Hyperlymphocytose

Lymphomes indolents
Signes cliniques
Syndrome tumoral
Adénopathies
Splénomégalie
Atteintes extra-
ganglionnaires :
ORL, peau, tube
digestif, SNC …
Signes généraux
Clinique
Tous +++
LLC/lymphome lymphocytique (variante
prolymphocytaire B)
Lymphome folliculaire
Lymphome de la zone marginale
(splénique, ganglionnaire, MALT)
Lymphome lymphoplasmocytaire
(Waldenström)
Leucémie à tricholeucocytes
Lymphome à cellules du manteau

Lymphomes indolents
Signes cliniques
Gammapathie
monoclonale
IgM +++
Rarement IgG, IgA
Biologie
Hyperlymphocytose
LLC +++
Lymphome de la zone marginale
Lymphome folliculaire
Lymphome à cellules du manteau
Waldenström +++
LLC
Lymphome de la zone marginale
Lymphome à cellules du manteau
Autres lymphomes lymphoplasmocytaires

Diagnostic des syndromes lymphoprolifératifs
Un diagnostic multidisciplinaire …
– Clinique
– Biologique (hématologie)
. Cytologie
. Immunologie (phénotypage)
. Cytogénétique (translocations, anomalies de nombre)
. Moléculaire (gènes de fusion, mutations)
– Radiologique
– Anatomo-pathologique
Reposant souvent sur faisceau d’arguments

Examens biologiques diagnostiques des SLPC
1- Morphologie :
- Cytologie : valeur d’orientation
-sanguine : leucémies / lymphomes leucémisés
Hyperlymphocytose > 4 G/L et/ou lymphocytes pathologiques
- médullaire (ponctions, empreintes de BOM)
Infiltration lymphoïde > 30% et/ou lymphocytes pathologiques
-ganglionnaire (ponctions, empreintes de biopsie)
Valeur d’orientation des cytoponctions ganglionnaires, à confirmer
histologiquement +++
- liquide de ponctions …
- Histologie : indispensable au diagnostic de lymphome
- ganglionnaire, médullaire, autres tissus (digestifs, cutanés, ORL, …)

Leucémie lymphoïde chronique (LLC)
Frottis sanguin
MGG

Leucémie prolymphocytaire B
Frottis sanguin
MGG

Leucémie à tricholeucocytes
Frottis sanguin
MGG

Lymphome folliculaire
Frottis sanguin
MGG
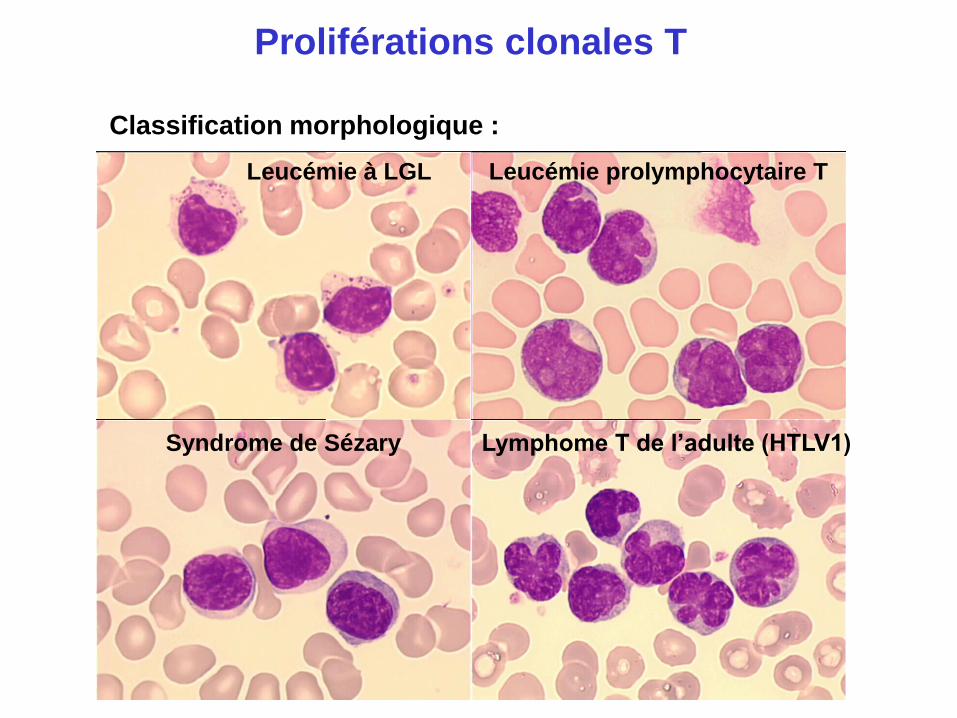
Proliférations clonales T
Classification morphologique :
Syndrome de Sézary Lymphome T de l’adulte (HTLV1)
Leucémie prolymphocytaire T Leucémie à LGL
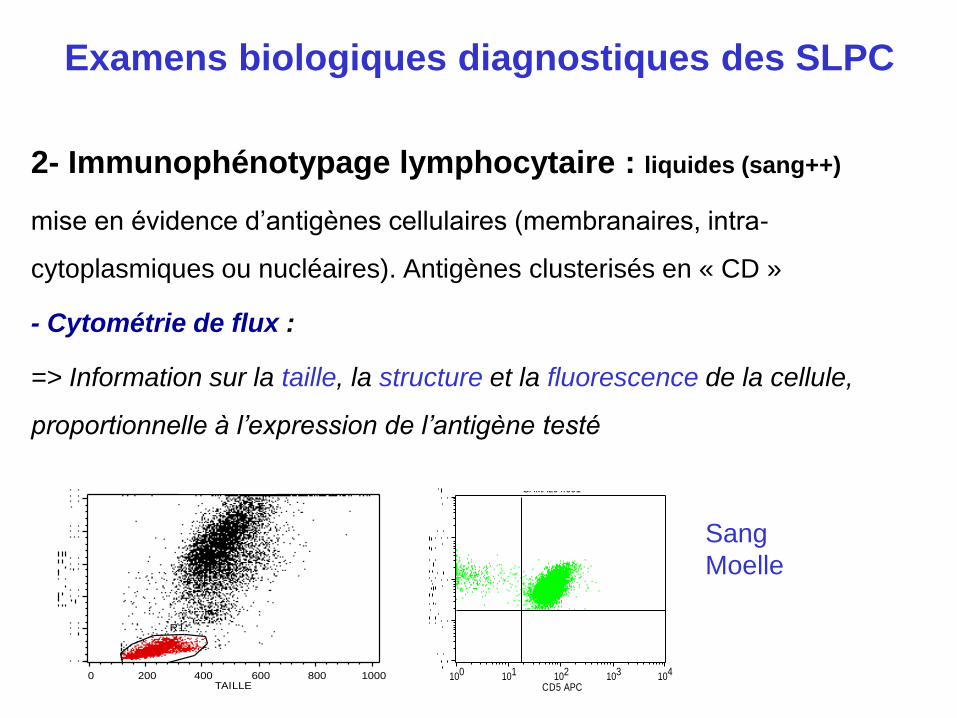
2- Immunophénotypage lymphocytaire : liquides (sang++)
mise en évidence d’antigènes cellulaires (membranaires, intra-
cytoplasmiques ou nucléaires). Antigènes clusterisés en « CD »
- Cytométrie de flux :
=> Information sur la taille, la structure et la fluorescence de la cellule,
proportionnelle à l’expression de l’antigène testé
GAMAL04.006
0 200 400 600 800 1000TAILLE
R1
GAMAL04.001
100 101 102 103 104
CD5 APC
Sang
Moelle
Examens biologiques diagnostiques des SLPC
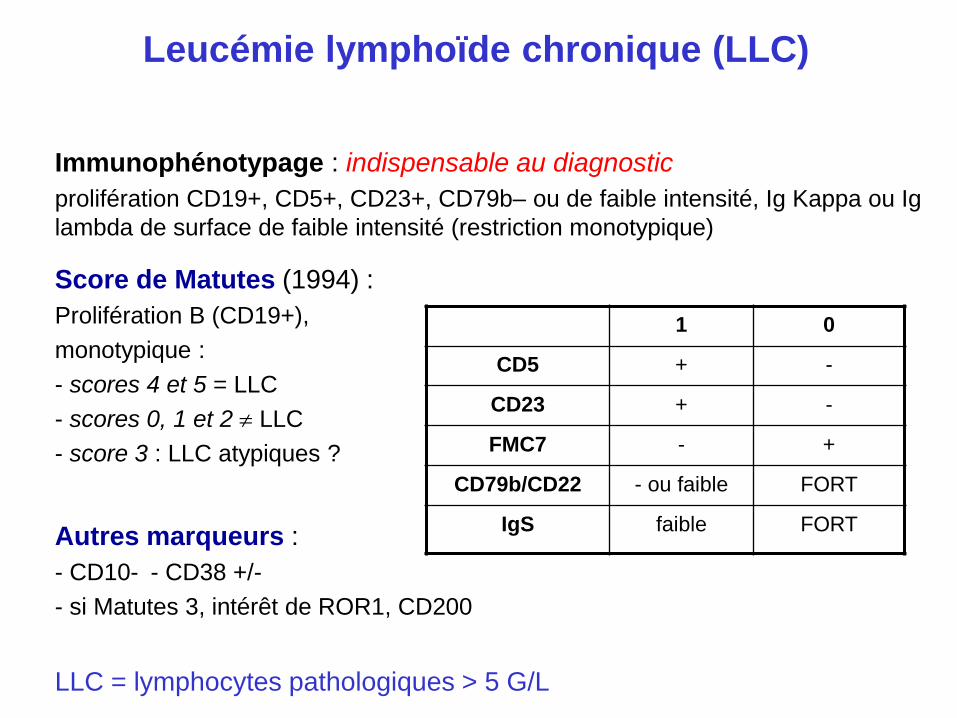
Leucémie lymphoïde chronique (LLC)
Immunophénotypage : indispensable au diagnostic
prolifération CD19+, CD5+, CD23+, CD79b– ou de faible intensité, Ig Kappa ou Ig
lambda de surface de faible intensité (restriction monotypique)
Score de Matutes (1994) :
Prolifération B (CD19+),
monotypique :
- scores 4 et 5 = LLC
- scores 0, 1 et 2 LLC
- score 3 : LLC atypiques ?
Autres marqueurs :
- CD10- - CD38 +/-
- si Matutes 3, intérêt de ROR1, CD200
LLC = lymphocytes pathologiques > 5 G/L
1 0
CD5 + -
CD23 + -
FMC7 - +
CD79b/CD22 - ou faible FORT
IgS faible FORT
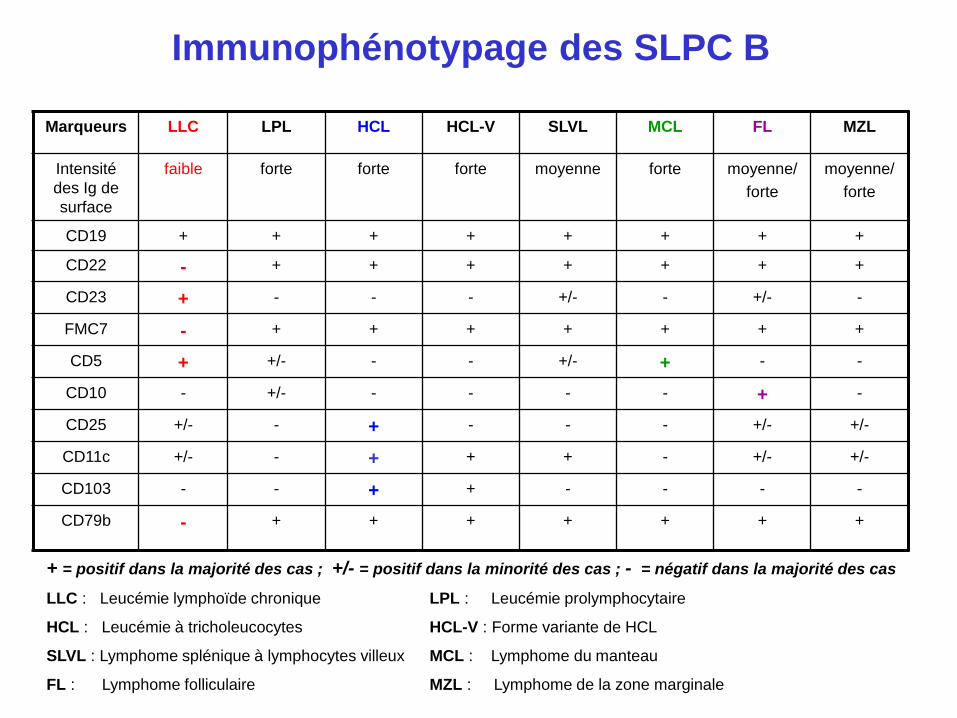
Immunophénotypage des SLPC B
Marqueurs LLC LPL HCL HCL-V SLVL MCL FL MZL
Intensité
des Ig de
surface
faible forte forte forte moyenne forte moyenne/
forte
moyenne/
forte
CD19 + + + + + + + +
CD22 - + + + + + + +
CD23 + - - - +/- - +/- -
FMC7 - + + + + + + +
CD5 + +/- - - +/- + - -
CD10 - +/- - - - - + -
CD25 +/- - + - - - +/- +/-
CD11c +/- - + + + - +/- +/-
CD103 - - + + - - - -
CD79b - + + + + + + +
+ = positif dans la majorité des cas ; +/- = positif dans la minorité des cas ; - = négatif dans la majorité des cas
LLC : Leucémie lymphoïde chronique LPL : Leucémie prolymphocytaire
HCL : Leucémie à tricholeucocytes HCL-V : Forme variante de HCL
SLVL : Lymphome splénique à lymphocytes villeux MCL : Lymphome du manteau
FL : Lymphome folliculaire MZL : Lymphome de la zone marginale

2- Immunohistochimie lymphocytaire : tissus
Sur coupes de tissu
Réalisées par les anatomo-pathologistes
CD20 CD3
Examens biologiques diagnostiques des SLPC

CD20
Bcl-2
CD3
Ganglion
lymphatique
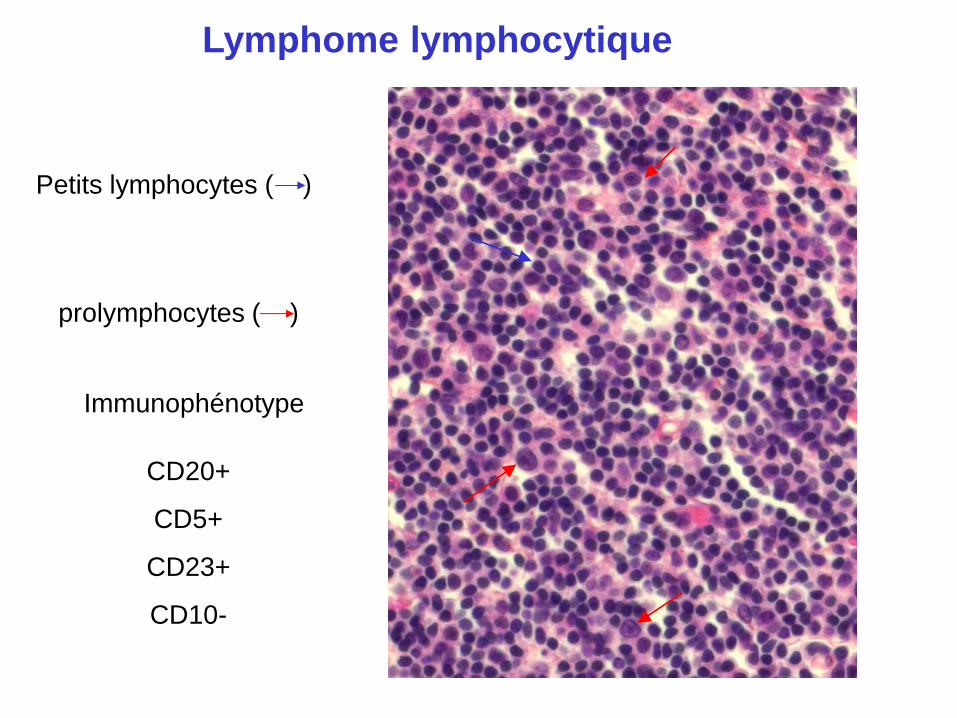
Petits lymphocytes ( )
prolymphocytes ( )
Lymphome lymphocytique
CD20+
CD5+
CD23+
CD10-
Immunophénotype

3- Cytogénétique :
- Conventionnelle : caryotype
analyse tout le génome
Ex. t(14;18)
- Moléculaire : FISH « Fluorescent In-Situ Hybridization »
ne détecte que les anomalies
recherchées
BCL2, 18q21, green
14q32.3, red,
Examens biologiques diagnostiques des SLPC
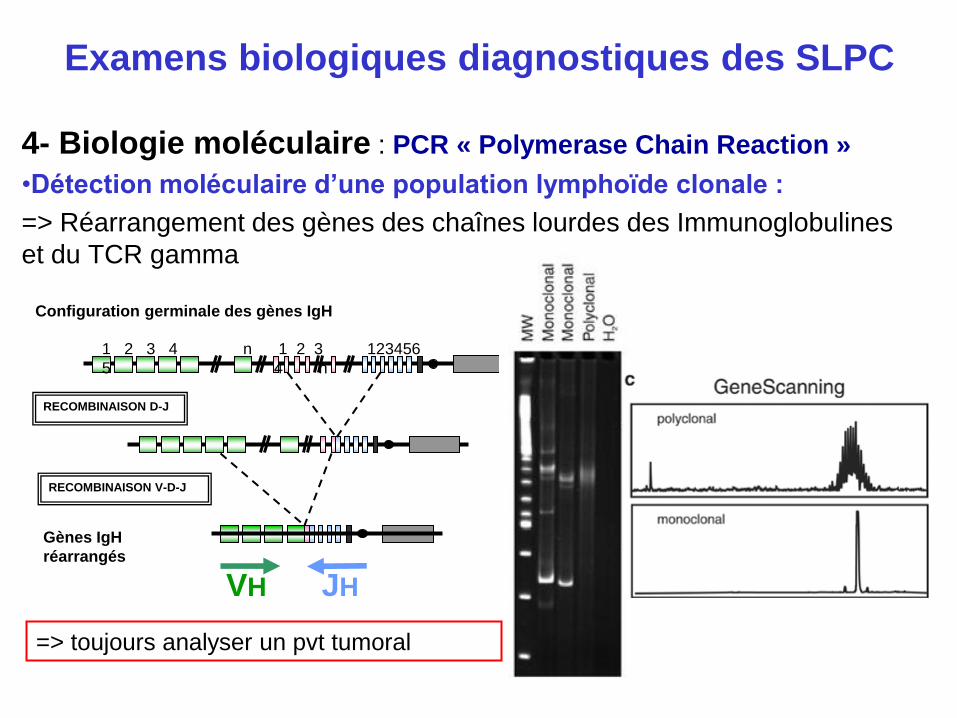
4- Biologie moléculaire : PCR « Polymerase Chain Reaction »
•Détection moléculaire d’une population lymphoïde clonale :
=> Réarrangement des gènes des chaînes lourdes des Immunoglobulines
et du TCR gamma
VH
Gènes IgH
réarrangés
RECOMBINAISON D-J
RECOMBINAISON V-D-J
1 2 3 4
5
n 1 2 3
4 n
123456
Configuration germinale des gènes IgH
JH
=> toujours analyser un pvt tumoral
Examens biologiques diagnostiques des SLPC

4- Biologie moléculaire : séquençage (PCR Sanger, PCR AS, NGS)
•Détection moléculaire de mutations ponctuelles de gènes:
•Visée diagnostique : MYD88 (Waldenström), BRAF (tricholeucocytes)
•Visée pronostique : TP53 ++
Examens biologiques diagnostiques des SLPC

Diagnostic des syndromes lymphoprolifératifs
Un diagnostic multidisciplinaire …
– Clinique
– Radiologique
– Anatomo-pathologique
– Biologique (hématologie)
. Cytologie
. Immunologie (phénotypage)
. Cytogénétique (translocations, anomalies de nombre)
. Moléculaire (gènes de fusion, mutations)
Reposant souvent sur faisceau d’arguments

Leucémie lymphoïde chronique

Epidémiologie
Une maladie de la personne âgée

> 50 ans = 90%
♂= 2 ♀
3 à 5 cas/100.000 par an
20 à 30 x plus en Europe, USA, Australie qu’en Asie
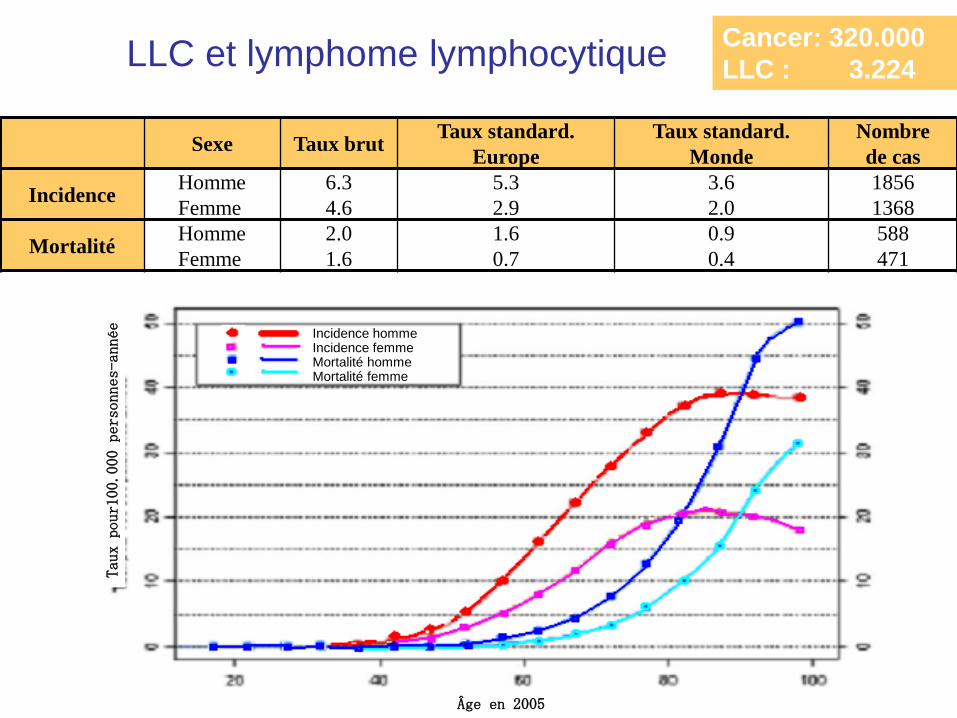
Sexe Taux brut Taux standard.
Europe
Taux standard.
Monde
Nombre
de cas
Incidence Homme
Femme
6.3
4.6
5.3
2.9
3.6
2.0
1856
1368
Mortalité Homme
Femme
2.0
1.6
1.6
0.7
0.9
0.4
588
471
Âge en 2005
Taux
pour100.000 personnes
-année
Incidence homme Incidence femme Mortalité homme Mortalité femme
Cancer: 320.000
LLC : 3.224 LLC et lymphome lymphocytique

Incidence des hémopathies(1)
36%
7%15%
0%
1%
4%
9%
10%
4%
2%
0%
12%
LNH
Hodgkin
Myélome
LLC/LL
LPL
Tricho
LAL/LL
Lymphoïde:
autres
LAM
LMC
Leucémies
autres
Divers

Rawstron et al., Blood 2002, 100 : 635-9
- 910 sujets > 40 ans
- hémogramme normal
- sang périphérique
- cytométrie en flux 4 couleurs
Lymphocytose monoclonale de type LLC
(MBL)
3.5%

LLC- Modes de révélation ?
Fortuit ++++
Syndrome tumoral (ADNP, SPM)
Complications infectieuses (pneumopathie)
Cytopénies



Evolution hétérogène : la LLC en tiers…
jamais besoin de
traitement
traitement après
une période
paisible
traitement
d'emblée

définition des
sites
ganglionnaires
1
2
3
5 4
LLC : classification de Binet
Binet et al., Cancer 1981, 48 : 198-206
stade A Hb > 100 g/l et plaquettes >100 x 109/l
< 3 aires ganglionnaires
Hb > 100 g/l et plaquettes > 100 x 109/l
≥ 3 aires ganglionnaires
Hb < 100 g/l et/ou plaquettes < 100 x 109/l
Quelque soit le nombre d’aire atteintes
stade B
stade C

stade A 63% > 10 ans (bon pronostic)
stade B 30% 5 ans 81 mois
(pronostic intermédiaire)
stade C 7% 2 ans 60 mois
(mauvais pronostic)
% des
patients
survie médiane
étude
LLC-76
étude
LLC-90
LLC : classification de Binet

Examens au diagnostic
• Clinique + hémogramme
• Immunophénotypage lymphocytaire sanguin
• Réticulocytes
• Haptoglobine
• Coombs direct
• Electrophorèse des protides sériques
• Imagerie : si adénopathies et traitement souhaité
Hémolyse
Hypogamma ? Pic ?

Cytologie
Matutes, Leukemia, 1994
Moreau, Am J Clin Path, 1997
1 0
CD5 + -
CD23 + -
FMC7 - +
CD79b/CD22 - ou faible fort
IgS faible fort
Cytométrie de flux
Homogénéité diagnostique vs Hétérogénéité clinique
Rai et al., Blood 1975
Binet et al., Cancer 1981
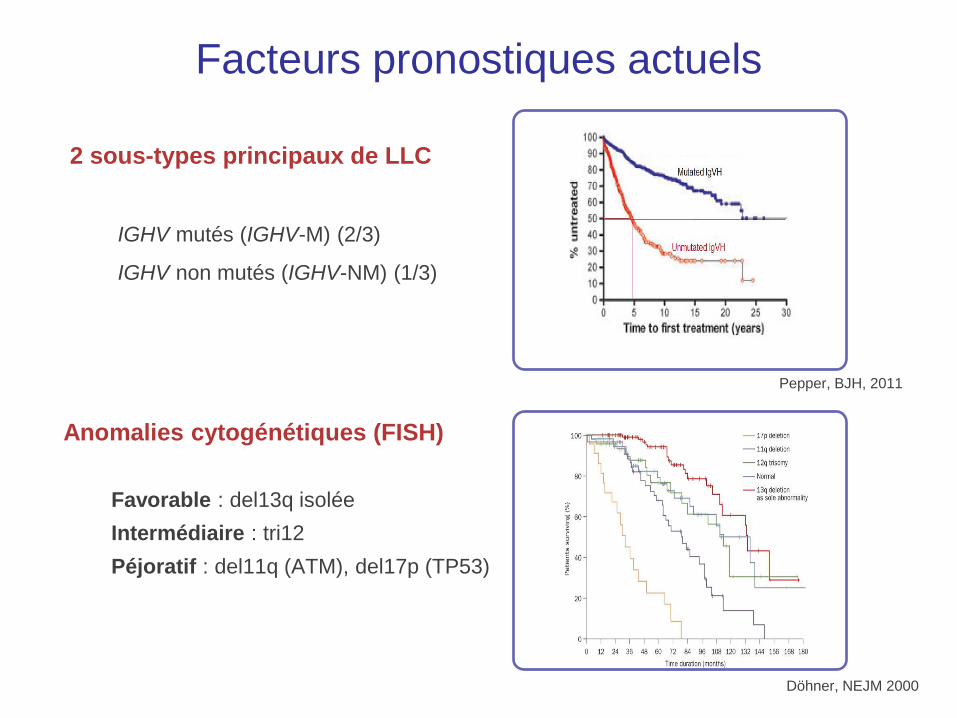
2 sous-types principaux de LLC
IGHV mutés (IGHV-M) (2/3)
IGHV non mutés (IGHV-NM) (1/3)
Döhner, NEJM 2000
Anomalies cytogénétiques (FISH)
Favorable : del13q isolée
Intermédiaire : tri12
Péjoratif : del11q (ATM), del17p (TP53)
Pepper, BJH, 2011
Facteurs pronostiques actuels

Statut mutationnel des IGHV
TTFT
Damle et al.
Blood, 1999
Hamblin et al.
Blood, 1999
Pepper, BJH, 2011 Baliakas et al. Lancet Haematol. 2015
OS
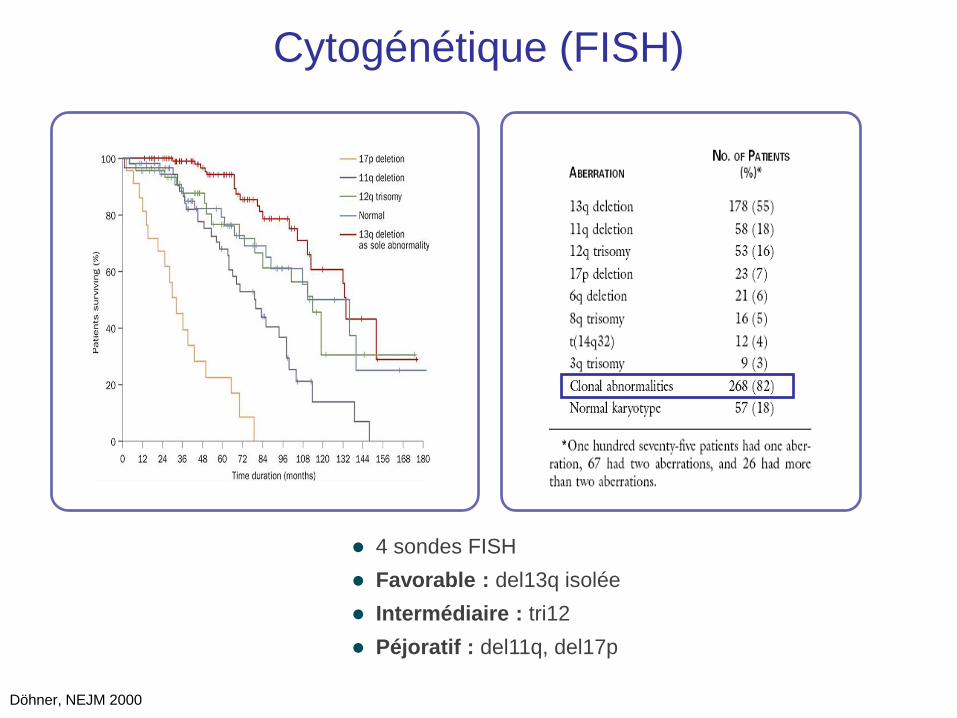
Cytogénétique (FISH)
Döhner, NEJM 2000
4 sondes FISH
Favorable : del13q isolée
Intermédiaire : tri12
Péjoratif : del11q, del17p

Futur ? Séquençage haut-débit (NGS)
Puente et al., Nat Genet 2011, Landau et al, Cell 2013, Puente et al., Nature 2015
Aucun événement unique ≠ Waldenström (MYD88), tricholeucocytes (BRAF)
Identification de nouvelles mutations récurrentes (NOTCH1, SF3B1, MYD88, BIRC3)
Peu de gènes mutés avec récurrence élevée, grand nombre avec récurrence faible
Fréquence < 25% des patients
Mutations les + fréquentes : SF3B1 (10-15%), NOTCH1 (10%), ATM (5-15%), TP53 (5-10%)
%

Rossi et al., Blood 2012
Impact pronostique
Intégration anomalies
cytogénétiques/mutations
Très favorable : del13q isolée
Favorable : tri12, pas d’anomalie
Intermédiaire : del11q, NOTCH1, SF3B1
Péjoratif : del17p, TP53, BIRC3
FISH FISH +
mutations
LLC – Pronostic (NGS)

Hétérogénéité inter-patient Hétérogénéité intra-patient
LLC – Hétérogénéité inter- et intra-clonale
Clonal Sous-clonal

Landau Cell
2015
LLC – Convergence / voies de signalisation

Malcikova et al., Blood 2009 Zenz et al., Nat Rev Cancer 2010
Anomalies de P53 – Délétion et/ou mutation
FISH Sanger/NGS
q arm
p arm
Normal Abnormal
TP53 wt TP53 mut TP53 mut
TP53 mut TP53 wt
TP53 variant del(17p)
TP53 wt
TP53 variant
del17p
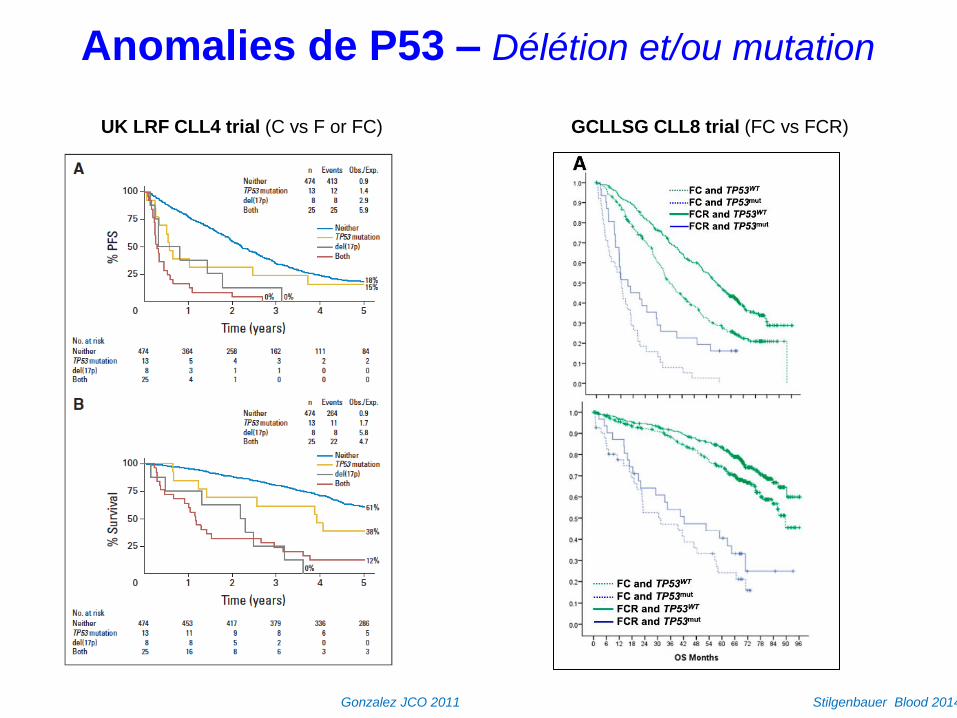
UK LRF CLL4 trial (C vs F or FC)
Gonzalez JCO 2011
GCLLSG CLL8 trial (FC vs FCR)
Stilgenbauer Blood 2014
Anomalies de P53 – Délétion et/ou mutation
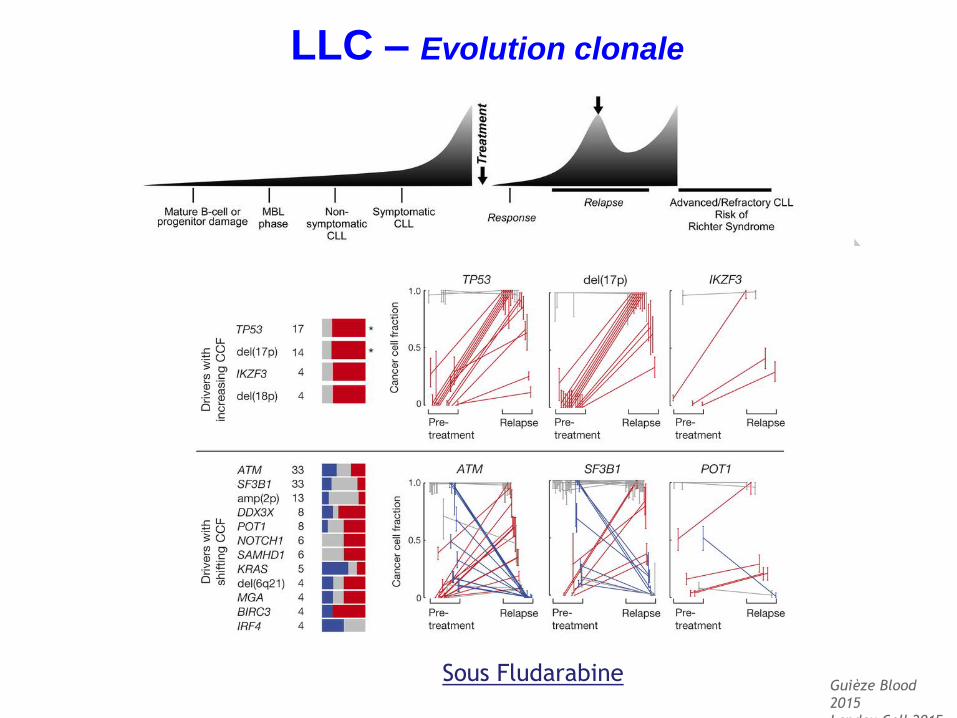
Guièze Blood
2015
Landau Cell 2015
Sous Fludarabine
LLC – Evolution clonale
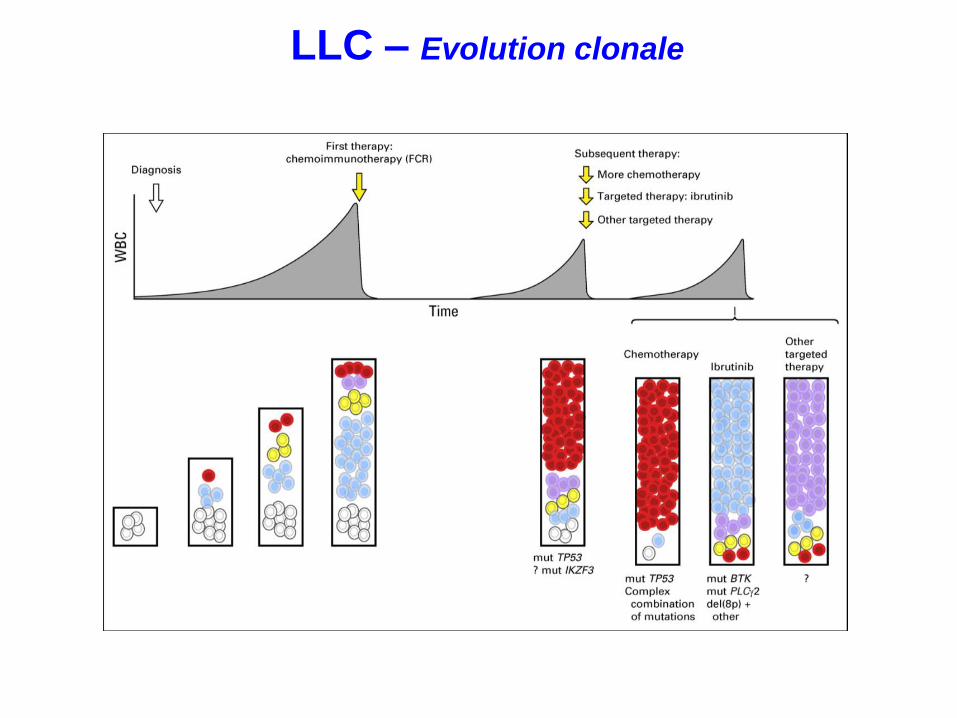
LLC – Evolution clonale

LLC – Evolution clonale
Rossi Leuk Lymphoma. 2017
Anomalies de TP53 à rechercher à chaque évolution/rechute

Prognostic Predictive
Age X X
CIRS X X
Stage X
β2microglobulin X
IGHV mutation X X
17p13 deletion X X
11q22-23 deletion X
Trisomy 12 X
13q deletion X
TP53 mutation X X
SF3B1 mutation X
NOTCH1 mutation X
Synthèse des facteurs pronostiques et prédictifs

Pronostic Predictive
Age X X
CIRS X X
Stage X
β2microglobulin X
CD49d X
CD38 X
ZAP70 X
IGHV mutation X X
17p13 deletion X X
11q22-23 deletion X
Trisomy 12 X
13q deletion X
TP53 mutation X X
SF3B1 mutation X
NOTCH1 mutation X
ATM mutation X
?
Clinique
. âge
. état général (CIRS)
. stade Binet
Biologie : créatinine
(fludarabine)
FISH (del17p, 11q, 13q, tri12)
Biologie moléculaire
. mutations TP53
. statut IGHV
Bilan pré-thérapeutique
Quels marqueurs et quand ?
Pour les patients nécessitant un traitement

Qui traiter ?

Stades A ?

Critères « classiques »
• Binet stades B et C
• Stade A évolutif : temps de doublement < 12 mois

Comment traiter ?
• Immuno-chimiothérapie
• Thérapies ciblées
• Soins de support

Choix thérapeutiques
R = rituximab, F = fludarabine, B = bendamustine, C = Cyclophosphamide
TKI = inhibiteurs de tyrosine kinase, mAb = anticorps monoclonaux

Choix du traitement
• Age, état général du patient
• Espérance de vie
• Qualité de vie
• Présence d’une altération de la voie p53
(del17p / mutation TP53)

Sujet âgé ?
• Hématologie :
l’âge définit la capacité à recevoir un traitement
– Allogreffe myéloablative: 45-50 ans
– Allogreffe conditionnement réduit : 60-65 ans
– Autogreffe: 65-70 ans
– Chimiothérapie intensive: 60-70 ans

Sujet âgé ?
• Pour le reste : tous dans le même sac…
• Bilan et traitement effectués comme chez le sujet jeune
(< 60 ans) sauf chez sujet très âgé (> 80 ans)
• Mais est-ce la bonne conduite à tenir ?
• Différences avec le sujet jeune :
présence de vieillissement d’organes / maladie(s)
associée(s) compliquant la prise en charge

Raisonner en comorbidités et non en âge
• Patients sans comorbidité
But : rémission complète (Guérison ???)
• Patients avec comorbidité
But : contrôle de la maladie ? qualité de vie ?

R.Stauder et al., Ann Oncol 2016
SIOG position paper
Categorization according to risk assessment

CIRS
Score 0-60 Fit : CIRS < 7

Qui est inéligible à un traitement avec
analogue des purines ?

FCR résultats à long terme (Thompson Blood 2016)
R = rituximab, F = fludarabine, C = Cyclophosphamide

FCR traitement de référence mais qui peut en bénéficier ? médiane d’âge des essais 55 ans….
tAML/MDS (5.1%) and RT (9%) Benjamini O, Leuk Lymphoma 2014
Cytopenia 3mo (35%) and 9mo (12%) Strati P, Cancer 2013
Infections 9mo (38%) Strati P, Cancer 2013
Neutropenia (34%), infections (25%) Hallek M, Lancet 2010
Haematological toxicity >grade 3 (56%) Hallek M, Lancet 2010

Qui est inéligible à un traitement
par analogue des purines ?
•Altération de la fonction rénale Le débat existe : une clairance entre 30–70 ml/min nécessite
une réduction de dose
•Patients en mauvais état général (co morbidités, évaluation gériatrique)
•À risque élevé d’infection

Fludarabine
• Rein …diminuer les doses < 60 ml/mn
• Ne pas utiliser si clairance < 30ml/mn
• Infections, cytopénies Bactrim + Zélitrex (CD4 >200)
G-CSF
Même complications/précautions pour la bendamustine

Immuno-chimiothérapie
(antiCD20 + chimiothérapie)
Fit
RFC (rituximab, endoxan et fludarabine)
Unfit
R+ bendamustine
R+ chloraminophène
Nouveaux antiCD20 + chloraminophène

Tolérance chimiothérapie classique
Immuno-chimiothérapie
Myélosuppression
. Aplasie fébrile
. Transfusions
Infections
Asthénie
MDS/LAM secondaires
Prise LIMITEE

Traitements adjuvants (prévenir complications)
• Infectieuses
– Vaccination systématique de tous les patients contre le
pneumocoque et grippe quelque soit le taux d’Ig et le stade
– Antibioprophylaxie :
• Sulfaméthoxazole, Triméthoprime
• Valaciclovir
– IgIV hivernales après le premier épisode d’infection sévère
– G-CSF
• Myélosuppression (EPO, G-CSF)


G Meunier , et al Hematol Oncol 2016
Quels traitements ont été utilisés ?
Monothérapie :
• Chlorambucil seul (45,3 %)
Immunochimiothérapie (48,3 %) :
• Rituximab + Chlorambucil (22,7 %)
• Rituximab + Bendamustine (10,4 %)
• Rituximab + Cyclophosphamide + Dexaméthasone (5,5 %)
• Rituximab + Fludarabine + Cyclophosphamide (5,5 %)

G Meunier , et al Hematol Oncol 2016
First Line Chronic Lymphocytic Leukemia Treatment for the Elderly
Patients Over 79 Years: A FILO Retrospective Study
• Survie globale
médiane : 53,7 mois,
soit 4,5 ans
• Décès liés à la LLC :
60,8 % des patients
Temps écoulé depuis l’initiation du traitement (mois)
Survie Globale

Immuno-chimiothérapie
(antiCD20 + chimiothérapie)
Fit
RFC (rituximab, endoxan et fludarabine)
Unfit
R+ bendamustine
R+ chloraminophène
Nouveaux antiCD20 + chloraminophène

Choix thérapeutiques
R = rituximab, F = fludarabine, B = bendamustine, C = Cyclophosphamide
TKI = inhibiteurs de tyrosine kinase, mAb = anticorps monoclonaux

Patients avec altération de la voie TP53 :
quelles options thérapeutiques ?
Représente 10% des patients en première ligne
50% des patients en rechute
Inefficacité des traitements activant TP53
Nouvelles drogues (voie orale) inhibiteur du BCR (BTK = ibrutinib /PI3K = idelalisib)
inhibiteur BCL-2 = venetoclax

Choix thérapeutiques

Choix thérapeutiques
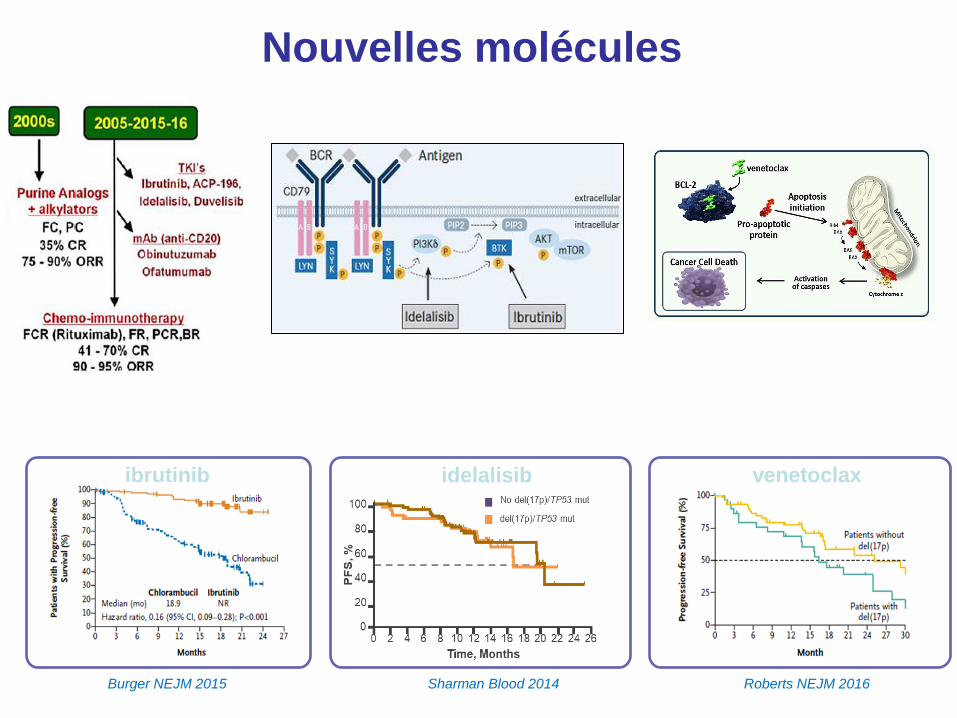
Burger NEJM 2015 Roberts NEJM 2016
ibrutinib venetoclax idelalisib
Sharman Blood 2014
Nouvelles molécules

Nouvelles molécules- Ibrutinib
ibrutinib
Ganglions +++

Nouvelles molécules- Venetoclax
Sang +++
Moelle +++

Ibrutinib
AMM dans 2 indications :
- LLC ayant reçu au moins un traitement antérieur,
- ou en 1ère ligne o en cas de del17p ou de mutation TP53 o chez les patients non éligibles à la fludarabine
85 patients R/R
391 patients R/R
(RESONATE)
269 patients (RESONATE-2)
1ère ligne
Byrd NEJM 2013 Byrd NEJM 2014 Burger NEJM 2015
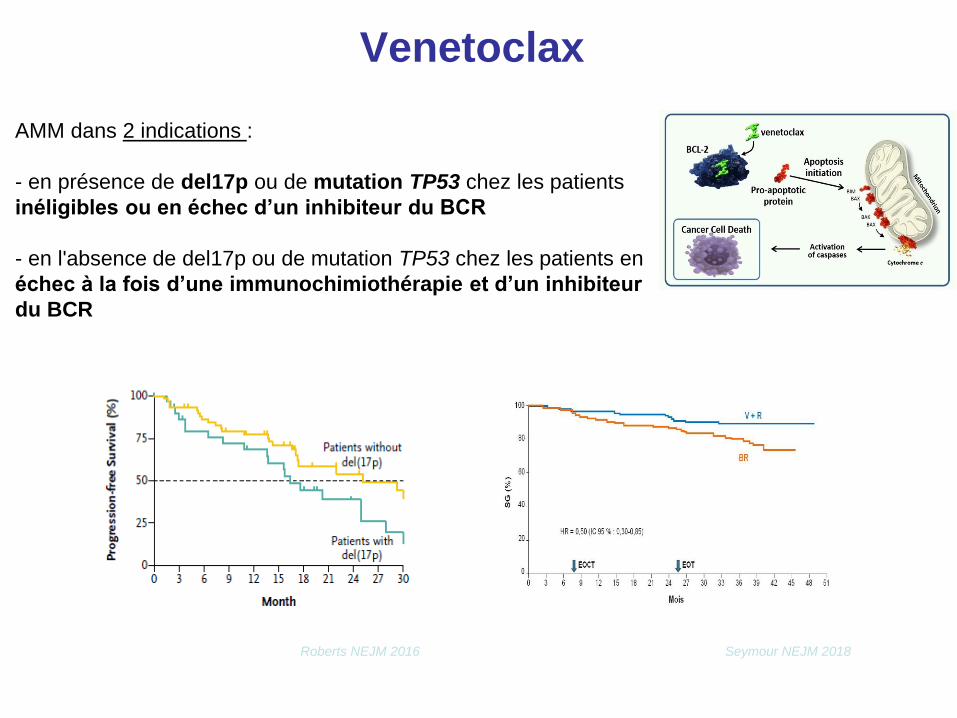
Venetoclax
AMM dans 2 indications :
- en présence de del17p ou de mutation TP53 chez les patients
inéligibles ou en échec d’un inhibiteur du BCR
- en l'absence de del17p ou de mutation TP53 chez les patients en
échec à la fois d’une immunochimiothérapie et d’un inhibiteur
du BCR
Roberts NEJM 2016 Seymour NEJM 2018

Nouvelles molécules - Tolérance
Effets secondaires Ibrutinib Idelalisib Venetoclax
Hémato
Neutropénie 15 30 40
Anémie 7 10 15
Thrombopénie 5 7 15
Non
hémato
Hémorragie 10 2 2
ACFA 5 2 2
HTA 10 5 4
Infections 5 10 1-16
Diarrhées 8 30 5
Hépatite 1 20 1
+ syndrome de lyse tumorale

Thérapie ciblée ≠ ciblée sur un type cellulaire !
Ibrutinib
Tec kinases (ITK, Tec…)
protection myocarde ischémique, activité autonome du
cœur (avec PLCg/PI-3Kg): activité voie calcique

Toxicité ibrutinib: myalgies/arthralgies et FA
• Tec kinases (ITK, Tec, pas Bmx/Btk/Rlk): protection myocarde ischémique,
activité autonome du cœur (avec PLCg/PI-3Kg): activité voie calcique
• Souvent retardées (> 3-6 mois de traitement)
• FA: poussée chez des pts déjà suivis: attention à l’ordonnance du
cardiologue:
– Amiodarone: inh.cytochrome = ↘ dose de 1-2 paliers de dose
– Changement d’AC: pas d’association plavix+aspirine, AVK sensés être interdits
avec l’ibrutinib en France (pas USA/Israël)
– Rechercher meilleure balance bénéfices/risques !
• Arthralgies: mains, genoux, cuisses, épaules
– Parfois gênant ADL/IADL
– Répondent bien à une baisse d’1 gélule d’ibrutinib

Toxicité ibrutinib: thrombopénie et saignement
• Thrombopénie: évènement rare, OK ibrutinib si >50 000 :
– Rarement profondes <25 000 : surveillance NFP les 3 premiers mois de ttt
– Myélogramme chez patients LCM <100 000 plaq et multitraités
• Saignements modérés: « Sd de Willebrand acquis » chez 50-60% des pts:
– Inhibition de Tec/BTK plaquettes: voie agrégation au collagène (GpVI) et FvW
(GpIb-IX-V)
– Tests in vitro prédictifs du phénotype hémorragique clinique
– STOP ibrutinib: 3-7j avant le geste: 5j suffisent en général
(50% renouvellement plaquettaire)
– Si Ts plaq: idéalement >4-6h après dernière prise d’ibrutinib
• Souvent précoces (<2 mois de traitement) et transitoires

vWF/collagen
TxA2 ADP
Primary hemostasis
thrombin
Secondary hemostasis
Tissue Factor
0 Aspirine Plavix Aspirine
+ plavix Héparine
AVK
ADO
AVK/ADO+
aspirin
ibrutinib ✔ ✔ ✖ ✖ ✔ ✔
✓
✖
Platelet aggregation collagen
ibrutinib
aspirin plavix Heparin
VKA NAC 4-
Platelet
Recommandations

Diarrhea Bruising Musculoskeletal
pain Atrial fibrillation
100
%
AC/FA
Bruising /rash/diarrhea Ibrutinib
infections
0 3 6 12 24 months
Ibrutinib- Toxicité
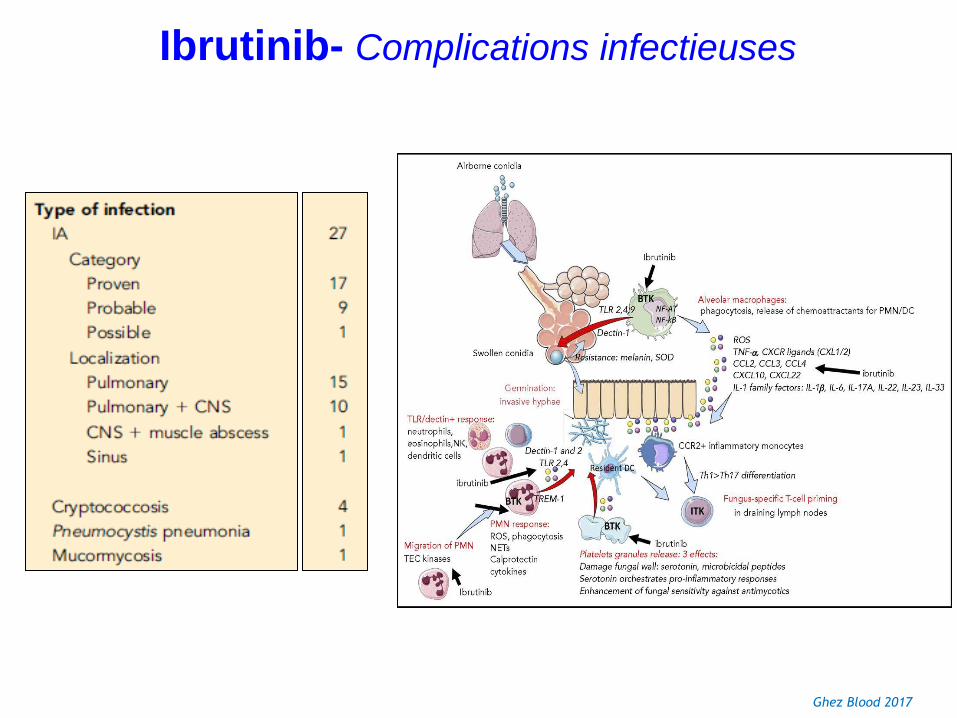
Ghez Blood 2017
Ibrutinib- Complications infectieuses
FIT
Ibrutinib
R-idelalisib
Venetoclax*
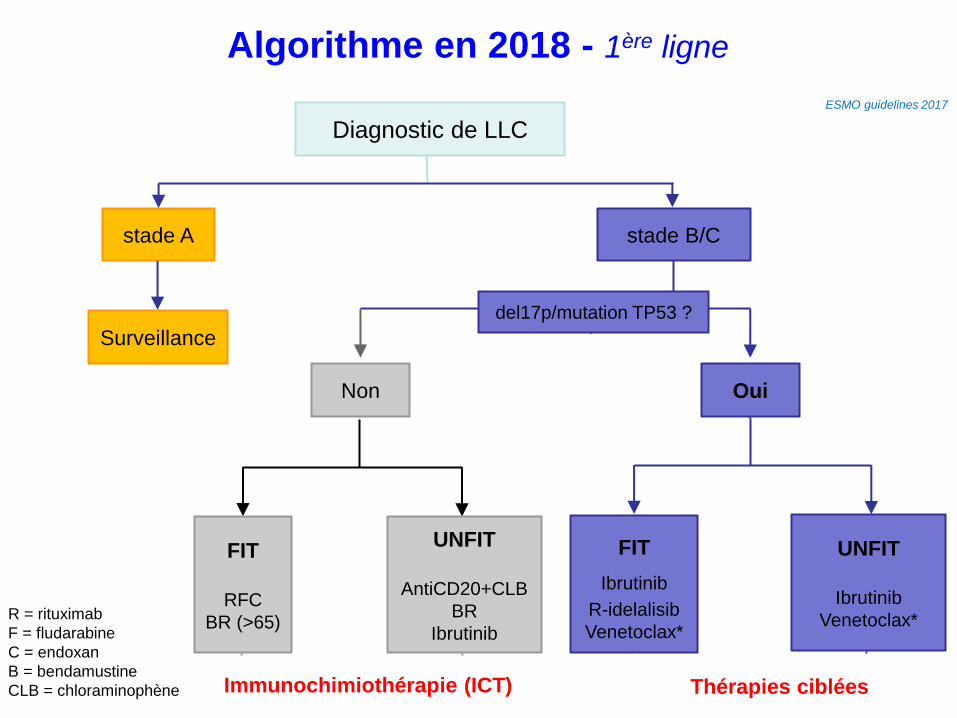
Algorithme en 2018 - 1ère ligne
Diagnostic de LLC
stade A stade B/C
Surveillance
Non Oui
del17p/mutation TP53 ?
FIT
RFC
BR (>65)
UNFIT
AntiCD20+CLB
BR
Ibrutinib
UNFIT
Ibrutinib
Venetoclax*
ESMO guidelines 2017
R = rituximab
F = fludarabine
C = endoxan
B = bendamustine
CLB = chloraminophène Immunochimiothérapie (ICT) Thérapies ciblées

FIT
Ibrutinib
R-idelalisib
Venetoclax*
2019 – Fin de l’ICT en 1ère ligne ?
Diagnostic de LLC
stade A stade B/C
Surveillance
Non Oui
del17p/mutation TP53 ?
FIT
RFC
BR (>65)
UNFIT
AntiCD20+CLB
BR
UNFIT
Ibrutinib
Venetoclax*
R = rituximab
F = fludarabine
C = endoxan
B = bendamustine
CLB = chloraminophène Immunochimiothérapie (ICT) Thérapies ciblées

Moreno Lancet Oncol 2018
• PFS médiane non atteinte vs. 19 mois (P<0.0001)
• RC/RCi: 41% vs. 16%
• MRD <10-4 sang et/ou moelle : 35% vs. 25%
• Nouveau traitement : 4% vs. 44%
Ibrutinib + Obinutuzumab (ibr-G) vs. Chlorambucil + Obinutuzumab
(clb-G)
• 1ère ligne chez > 65 ans
• 229 pts: ibr-G (n=113) ou clb-G (n=116)
• Del17p et/ou TP53 mut : 16 % (ibr-G) et 20% (clb-G)
PF
S
ILLUMINATE
2019 – Fin de l’ICT en 1ère ligne ?

Woyach NEJM 2018
Bendamustine-rituximab (BR) vs. ibrutinib (I) vs. ibrutinib-rituximab (IR)
• 1ère ligne > 65 ans
• 547 patients : BR (n = 183), I (n = 182), IR (n = 182)
• del17p 6%, TP53 mut 10%
0 6 12 18 24 30 36 42 48 52
0
10
20
30
40
50
60
70
80
90
100
Su
rvie
sa
ns
pro
gre
ss
ion
(%
)
Mois
Bras
BR
I
IR
n
176
178
170
24 mois
74 % (IC 95 % : 66-80 %)
87 % (IC 95 % : 8-92 %)
88 % (IC 95 % : 81-92 %)
I vs BR :
HR = 0,39
IC 95 % : 0,26-0,58
p < 0,001
IR vs BR :
HR = 0,38
IC 95 % : 0,25-0,59
p < 0,001
IR vs I :
HR = 1,00
IC 95 % : 0,62-1,62
p = 0,49
Avantage à l’ibrutinib en SSP (mais pas en survie globale)
Pas d’avantage dans le sous-groupe IGHV mutés
ALLIANCE
2019 – Fin de l’ICT en 1ère ligne ?

FIT
Ibrutinib
R-idelalisib
Venetoclax*
Diagnostic de LLC
stade A stade B/C
Surveillance
Non Oui
del17p/mutation TP53 ?
FIT
RFC
BR (>65)
UNFIT
AntiCD20+CLB
BR
UNFIT
Ibrutinib
Venetoclax*
R = rituximab
F = fludarabine
C = endoxan
B = bendamustine
CLB = chloraminophène Immunochimiothérapie Thérapies ciblées
2019 – Fin de l’ICT en 1ère ligne ?
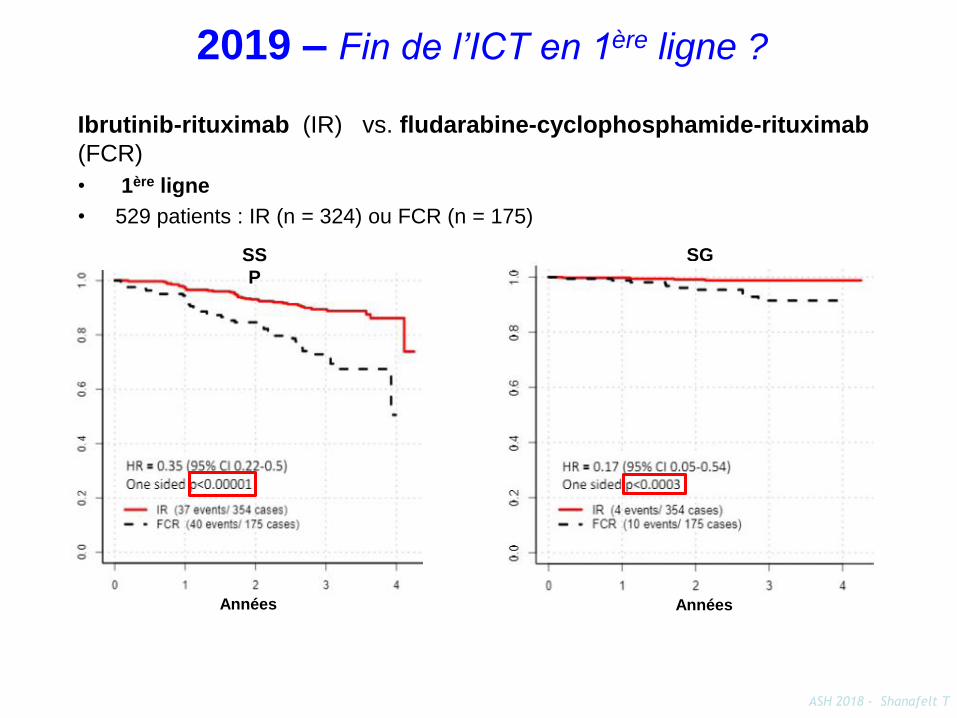
ASH 2018 - Shanafelt T
Ibrutinib-rituximab (IR) vs. fludarabine-cyclophosphamide-rituximab
(FCR)
• 1ère ligne
• 529 patients : IR (n = 324) ou FCR (n = 175)
Années
SS
P
SG
Années
2019 – Fin de l’ICT en 1ère ligne ?

FIT
Ibrutinib
R-idelalisib
Venetoclax*
Algorithme en 2018 - 1ère ligne
Diagnostic de LLC
stade A stade B/C
Surveillance
Non Oui
del17p/mutation TP53 ?
FIT
RFC
BR (>65)
UNFIT
AntiCD20+CLB
BR
Ibrutinib
UNFIT
Ibrutinib
Venetoclax*
ESMO guidelines 2017
R = rituximab
F = fludarabine
C = endoxan
B = bendamustine
CLB = chloraminophène Immunochimiothérapie (ICT) Thérapies ciblées

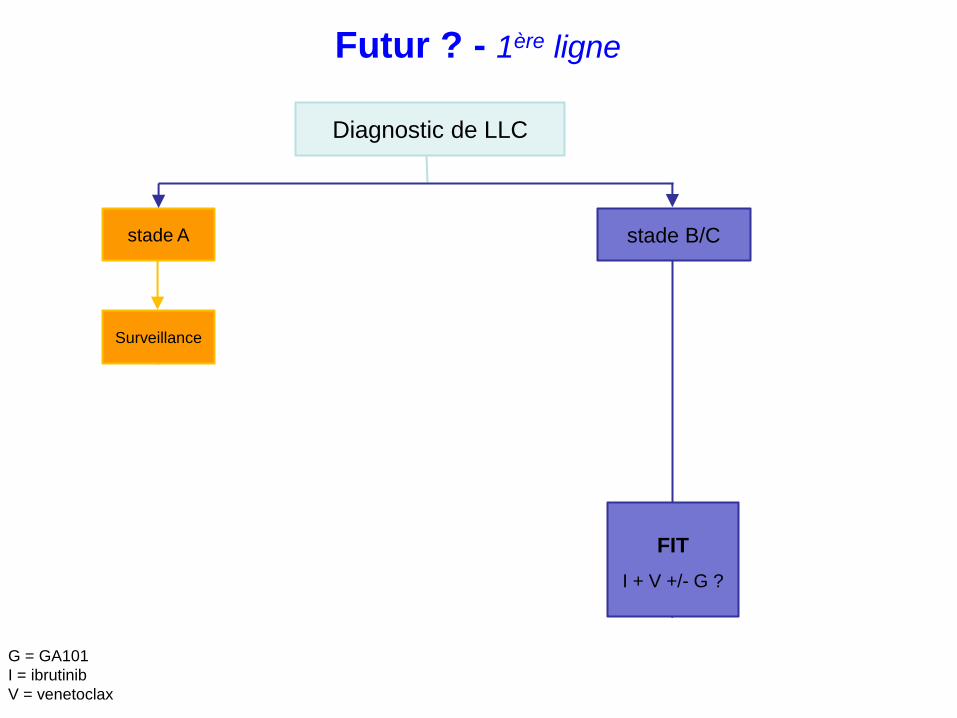
FIT
I + V +/- G ?
Futur ? - 1ère ligne
Diagnostic de LLC
stade A stade B/C
Surveillance
Non Oui
del17p/mutation TP53 ?
FIT
FCG ?
IbruFCG ?
UNFIT
Ibrutinib ?
R-Benda ?
UNFIT
Ibrutinib
I + V low dose ?
R = rituximab
F = fludarabine
C = endoxan
B = bendamustine
G = GA101
I = ibrutinib
V = venetoclax
IGHV non mutés ?
ICT Thérapies ciblées
Futur ? - 1ère ligne
Diagnostic de LLC
stade A stade B/C
Surveillance
G = GA101
I = ibrutinib
V = venetoclax
FIT
I + V +/- G ?

Nouvelles thérapies : nouvelles problématiques…
Immuno-chimiothérapie Thérapies ciblées
Myélosuppression
. Aplasie fébrile
. Transfusions
Infections
Asthénie
MDS/LAM secondaires
Prise LIMITEE
Cardio-pulmonaires
. HTA
. AC/FA
Digestifs : diarrhées
Hépatiques, articulaires…
Interactions médicamenteuses +++
(co-morbidités)
Prise CONTINUE (jusqu’à progression)
Observance

Concertation +++
Hématologue
Pharmacien
Médecin traitant
Gériatre
Cardiologue
Infirmière de coordination +++

Objectifs
Meilleure adéquation patient / traitement
Améliorer tolérance / compliance / qualité de vie
Modifications du protocole thérapeutique
Adaptation de doses
Interventions * traitement des comorbidités
* dépistage et exploration de troubles cognitifs
* prise en charge de la dénutrition
* traitement anti-dépresseur
* mise en place d’aides pour prévenir les
hospitalisations pour perte d’autonomie ...

LLC- Conclusion
• Place de l’ICT bousculée par les thérapies ciblées en 1ère ligne
• Nouveau standard de traitement en 1ère ligne ?
• ibrutinib +/- antiCD20, ibrutinib + venetoclax +/- anti-CD20 ?
• place pour l’ICT +/- « améliorée » chez patients fit, IGHV mutés ?
• Thérapies ciblées pour tout le monde ?
1) Guérison ? Arrêt de traitement (MRD) ?
2) Tolérance ?
• Durée limitée ?
• Réduction de doses ?
• Que proposer en cas de résistance ?
• Hétérogénéité +++ (1/3 patients ne nécessitant jamais de traitement)
• Formes graves (anomalies de P53) restent graves
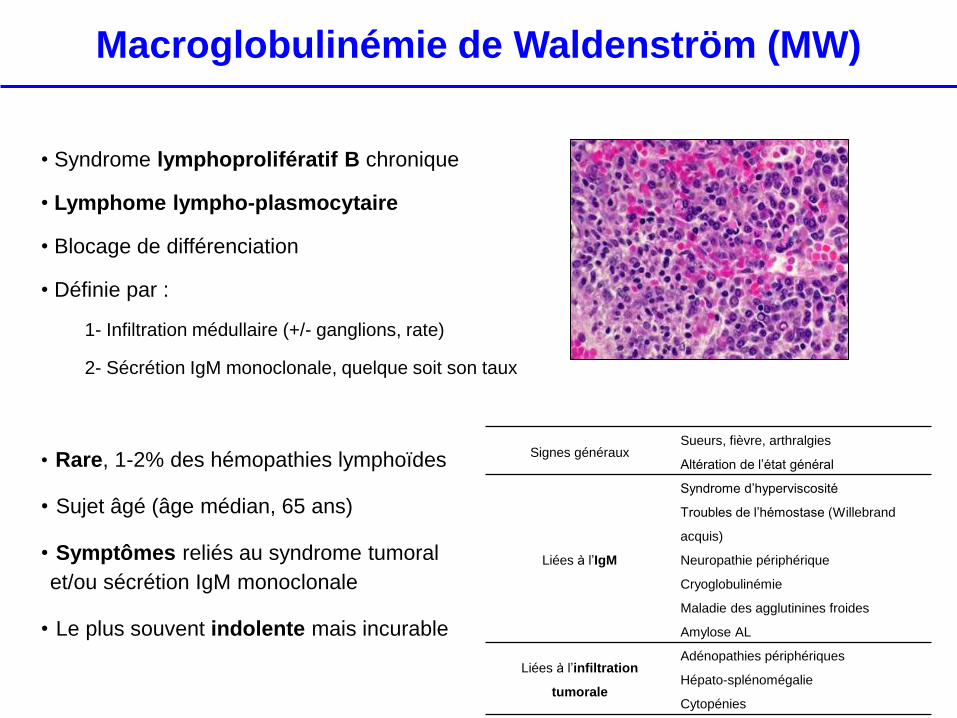
Macroglobulinémie de Waldenström (MW)
• Syndrome lymphoprolifératif B chronique
• Lymphome lympho-plasmocytaire
• Blocage de différenciation
• Définie par :
1- Infiltration médullaire (+/- ganglions, rate)
2- Sécrétion IgM monoclonale, quelque soit son taux
• Rare, 1-2% des hémopathies lymphoïdes
• Sujet âgé (âge médian, 65 ans)
• Symptômes reliés au syndrome tumoral
et/ou sécrétion IgM monoclonale
• Le plus souvent indolente mais incurable
Signes généraux Sueurs, fièvre, arthralgies
Altération de l’état général
Liées à l’IgM
Syndrome d’hyperviscosité
Troubles de l’hémostase (Willebrand
acquis)
Neuropathie périphérique
Cryoglobulinémie
Maladie des agglutinines froides
Amylose AL
Liées à l’infiltration
tumorale
Adénopathies périphériques
Hépato-splénomégalie
Cytopénies

Cadre nosologique
• Lymphoprolifération + IgM:
– MW
– LZM
– LLC
– MM (très rare)
• LNH lymphoplasmocytaire:
– + IgM= MW
– + IgG ou A ou non sécréteur

Treon NEJM 2012
Xu Blood 2013
• MYD88 L265P (> 95%), hétéro- ou homozygote
• Substitution nucléotidique unique (T pour C)
• Autres : domaine TIR (1-2%)
• Activation constitutive voies NF-kB, JAK/STAT
• Evénement le plus souvent clonal (précoce)
GMSI IgM (60-80%), LBDGC-ABC (30%), LZM (5-10%), LLC (2-5%)
MYD88 Chr. 3p22
Exons 3-4
309 1 DD TIR domain
I22
0T
L2
65
P
M232T
S
243N
MW – Mutations de MYD88
Clinique
• Infiltration tissulaire: – cytopénies
– adénopathies/ HSMG (15-20%)
– autres: rein, SNC (Bing Neel), tube digestif …
• Hyperviscosité (rare): – troubles neurosensoriels
– syndrome hémorragique
• Activité de l'IgM (10%):
– anti-MAG
– agglu froides (10%), Schnitzler, cryoglobuline de type 2
• Cryoglobuline 1 (5%) ou dépôts amylose (rare)

Hyperviscosité
• Moins de 15% des patients
• Clinique:
– troubles neuro-sensoriels
– syndrome hémorragique (intéraction IgM/ facteurs de coagulation)
– défaillance cardiaque
• Fond d’œil
• Traitement en urgence :
– EP: très efficaces
– chimio

Critères de traitement
• Cytopénies (Hb inf 10 g/dL, plq inf 100 000/mm3)
• Signes généraux
• Syndrome tumoral important (+ 5 cm ou symptomatique)
• Complications de l’IgM
• Urgences thérapeutiques: – hyperviscosité
– AHAI
– cryoglobuline
– …
• Surveillance très attentive si plus de 50 g/L

Choix du traitement
• Âge + comorbidités
• Quel délai d’action nécessaire ?
• Quelles manifestations (immunologique ? tumorale ?)
• Âge + c

RCD et R-Bendamustine = gold standard
• Rituximab 375 mg/m2 J1 IV
• Cyclophosphamide 200 mg/m2 J1 à J5 PO
• Dexamethasone 20 mg J 1 PO
• Toutes les 3 semaines; 6 cures
• Rituximab 375 mg/m2 J1 IV
• Bendamustine 70-90 mg/m2 J1 et J2 IV
• Toutes les 4 semaines; 6 cures

RCD
• ORR 83%
• Faible toxicité
• Délai réponse 4,1 mois
• PFS médiane: 35 mois
• OS médiane: 95 mois
Dimopoulos, 2007, JCO, ; Kastritis, Blood, 2015

Ibrutinib
• Rationnel: mutation MyD88 activatrice
de la signalisation BTK
• Excellents résultats, 63 patients:
– ORR: 90,5 %
– PFS: 69% à 2 ans
– OS: 95% à 2 ans
– Délai réponse: 4 semaines
– Durée moyenne tt: 19 mois
– Toxicité acceptable: cytopénies/ FA/ saignements

Ganglionnaire
Splénique
MALT (estomac, oculaire, peau)
Thérapeutique :
Cas particulier du MALT gastrique : éradication H. Pylori
Immunochimiothérapie : R-Chlorambucil, R-Bendamustine
Lymphome de la zone marginale
Rôle de stimulation antigénique chronique
agents infectieux (H.Pylori)
maladies auto-immunes (Sjögren)
Immunophénotypage lymphocytaire (sang, moelle) :
Matutes < 4, pas de marqueur spécifique
Diagnostic anatomopathologique +++
Cytogénétique, biologie moléculaire
Pronostic : très bon

Leucémie à tricholeucocytes
Mode de révélation
. Fortuit
. Asthénie
. Splénomégalie
. Infections (légionnellose, tuberculose, MAC)
Cytopénies (pancytopénie, monocytopénie)
Frottis (sang, moelle)
Immunophénotypage lymphocytaire (sang, moelle), BOM :
pop lymphoïde B CD19+/20+/22+, CD103+, CD25+
Biologie moléculaire BRAF V600E (90%)
Thérapeutique :
analogue des purines (cladribine) +++ en 1ère ligne
rituximab
inhibiteur de BRAF
Complications infectieuses +++

Situation 1
Femme de 68 ans
Pas d’ATCD notable hormis HTA contrôlée sous monothérapie
Hyperlymphocytose à 17 G/L, isolée
Examen clinique normal
Examens ?

Matutes 5

Situation 1
Femme de 68 ans
Pas d’ATCD notable hormis HTA contrôlée sous monothérapie
Hyperlymphocytose à 17 G/L, isolée
Examen clinique normal
Examens ?
Diagnostic de LLC
Quel est le stade de cette LLC ? Quel traitement préconisez-vous ?

Situation 2
Femme de 73 ans
Pas d’ATCD notable hormis HTA contrôlée sous monothérapie
LLC stade A depuis 5 ans. Surveillance, vaccinations.
Apparition d’adénopathies cervicales, axillaires et inguinales de 3 cm de diamètre
Hémoglobine 120 g/L, plaquettes 90 G/L, lymphocytes 100 G/L
Quel est le stade de cette LLC ?
Quel(s) examen(s) préconisez-vous ?

Situation 2
Femme de 73 ans
Pas d’ATCD notable hormis HTA contrôlée sous monothérapie
LLC stade depuis 5 ans. Surveillance, vaccinations.
Apparition d’adénopathies cervicales, axillaires et inguinales de 3 cm de diamètre
Hémoglobine 120 g/L, plaquettes 90 G/L, lymphocytes 100 G/L
LLC stade C
Evaluation état général, co-morbidités : CIRS = 3
Créatinine (MDRD 70 mL/mn)
Cytogénétique (FISH) : del13q monoallélique, pas de del11q/tri12 ou del17p
Biologie moléculaire : pas de mutation de TP53
Quel traitement ?
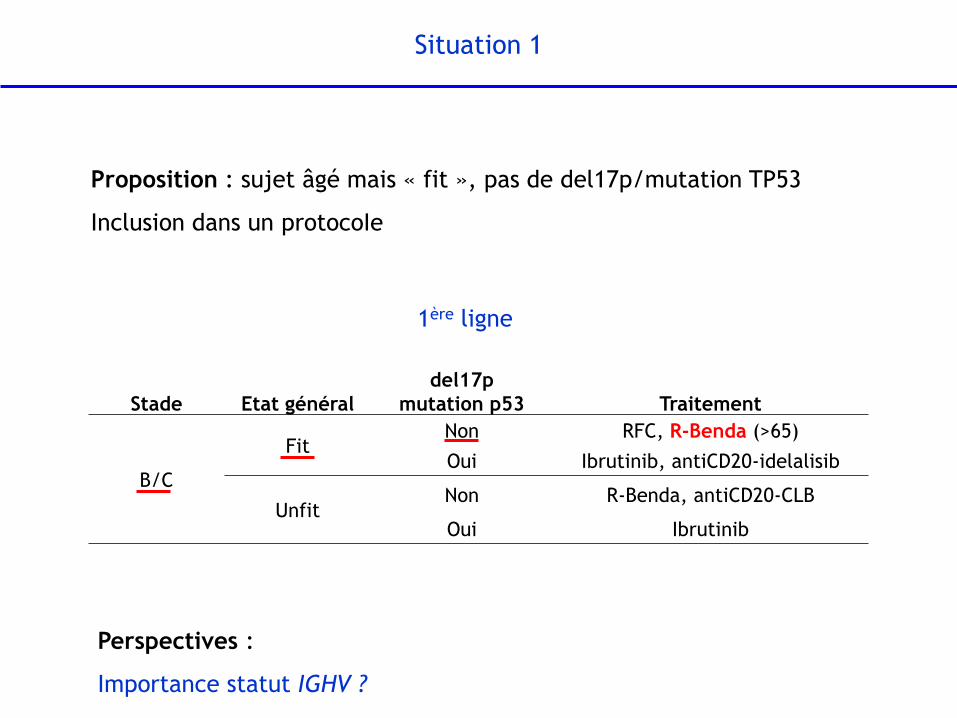
Situation 1
Proposition : sujet âgé mais « fit », pas de del17p/mutation TP53
Inclusion dans un protocoIe
Stade Etat général
del17p
mutation p53 Traitement
B/C
Fit Non RFC, R-Benda (>65)
Oui Ibrutinib, antiCD20-idelalisib
Unfit Non R-Benda, antiCD20-CLB
Oui Ibrutinib
1ère ligne
Perspectives :
Importance statut IGHV ?

Statut IGHV : facteur pronostique et prédictif après RFC
Lin YH, Blood. 2009. Thompson PA, Blood. 2016. Rossi D, Blood. 2015
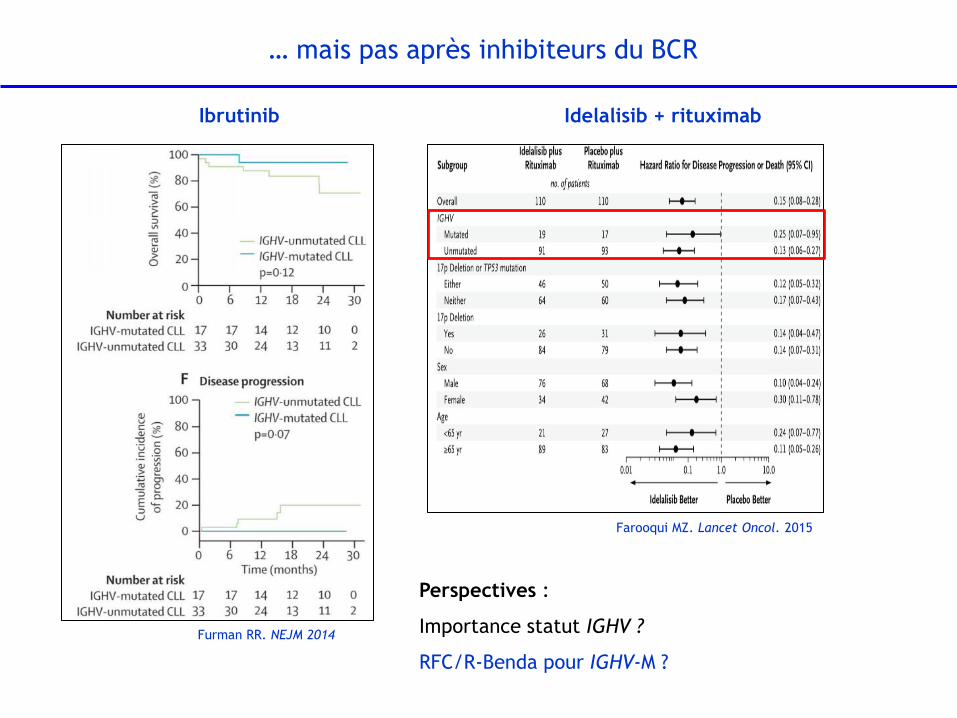
Ibrutinib Idelalisib + rituximab
Furman RR. NEJM 2014
… mais pas après inhibiteurs du BCR
Farooqui MZ. Lancet Oncol. 2015
Perspectives :
Importance statut IGHV ?
RFC/R-Benda pour IGHV-M ?

Situation 3
Même patiente
Après 4 mois de traitement par R-Bendamustine, la patiente est hospitalisée
pour une pneumopathie interstitielle fébrile
Quel diagnostic faut-il redouter ?
Pneumocystose

Situation 3
Même patiente (76 ans)
Rechute 3 ans post-R-Bendamustine (lymphocytose, adénopathies axillaires
et thrombopénie)

Situation 3
Même patiente (76 ans)
Rechute 3 ans post-R-Bendamustine (lymphocytose, adénopathies axillaires
et thrombopénie)
Rechute
Traitement
Prog > 2-3 ans Même traitement
Prog < 2-3 ans
Ibrutinib Lenalidomide
R-Benda antiCD20-idelalisib, alemtuzumab
Venetoclax (allogreffe ?)
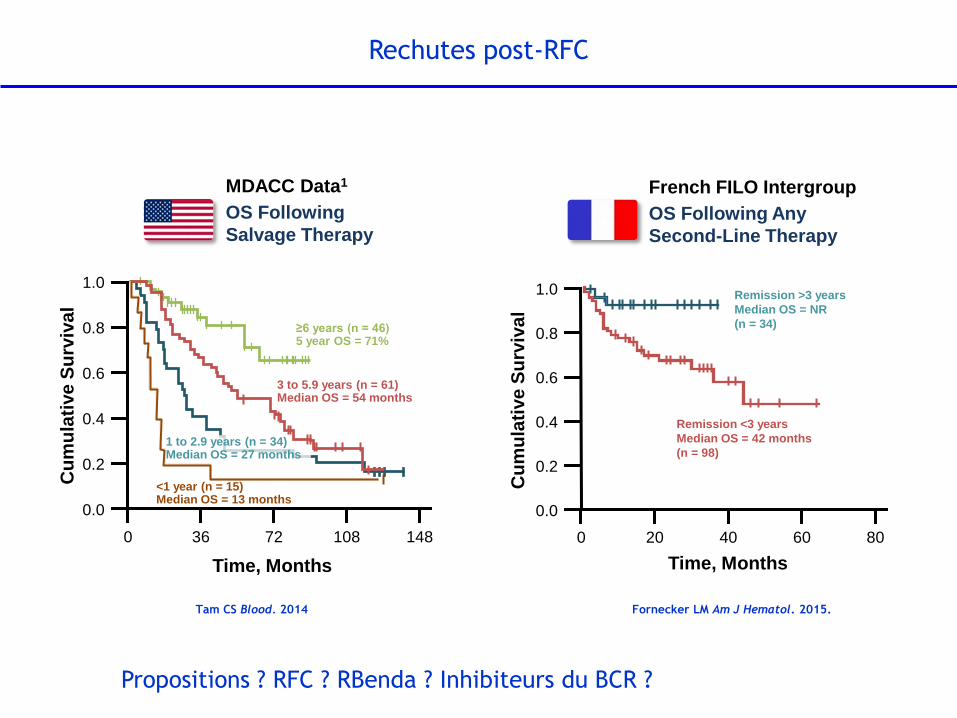
1 to 2.9 years (n = 34) Median OS = 27 months
MDACC Data1
OS Following
Salvage Therapy
Time, Months
3 to 5.9 years (n = 61) Median OS = 54 months
P<.001
≥6 years (n = 46) 5 year OS = 71%
<1 year (n = 15) Median OS = 13 months
1.0
0.8
0.6
0.4
0.2
0.0
0 36 72 108 148
Cu
mu
lati
ve
Su
rviv
al
Cu
mu
lati
ve
Su
rviv
al
French FILO Intergroup
OS Following Any
Second-Line Therapy
Time, Months
Remission <3 years
Median OS = 42 months
(n = 98)
Remission >3 years
Median OS = NR
(n = 34)
1.0
0.8
0.6
0.4
0.2
0.0
0 20 40 60 80
Rechutes post-RFC
Tam CS Blood. 2014 Fornecker LM Am J Hematol. 2015.
Propositions ? RFC ? RBenda ? Inhibiteurs du BCR ?

Situation 3
Même patiente (76 ans)
Rechute 3 ans post-R-Bendamustine (lymphocytose, adénopathies axillaires
et thrombopénie)
Nouvelle évaluation +++
Gériatrique : HTA sous bithérapie, AVC ischémique sylvien droit/ACFA…
CIRS = 12
MDRD 50 mL/mn
Caryotype/FISH
Recherche mutation TP53

Rossi Leuk Lymphoma. 2017
LLC- Evolution clonale
Exemple de TP53

Situation 3
Caryotype normal, FISH : pas de del17p
Présence d’une mutation TP53 R175H (exon 5) en biologie moléculaire
Même patiente
Rechute 3 ans post-R-Bendamustine (lymphocytose, adénopathies axillaires
et thrombopénie)
Sous analogues puriques/fludarabine, évolution clonale est la règle (57/59 patients)

Situation 3
Proposition :
Inhibiteurs du BCR: ibrutinib
Mais attention HTA, ACFA, antiagregants/anticoagulants
Si inéligible, venetoclax

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5 g/dl, hyperlymphocytose 30 G/L
Examens ?

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5 g/dl, hyperlymphocytose 30 G/L
ECG, troponine
Immunophénotypage lymphocytaire : Matutes 4
VGM 105 fl, réticulocytes : 375 G/L

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5 g/dl, hyperlymphocytose 30 G/L
Immunophénotypage lymphocytaire : Matutes 4
Réticulocytes : 375 G/L
Quel mécanisme de l’anémie suspectez-vous ?
Comment complétez-vous le bilan ?

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5 g/dl, hyperlymphocytose 30 G/L
Immunophénotypage lymphocytaire : Matutes 4
Réticulocytes : 375 G/L
AHAI
Bilan hémolyse (haptoglobine, LDH, bilirubine)
Test de Coombs direct

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5g/dl, hyperlymphocytose 30 G/L
Immunophénotypage lymphocytaire : Matutes 4
Réticulocytes : 375 G/L
Comment traitez-vous l’anémie ?

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5g/dl, hyperlymphocytose 30 G/L
Immunophénotypage lymphocytaire : Matutes 4
Réticulocytes : 375 G/L
Transfusion lente de culots globulaires
Corticothérapie 1 à 2 mg/kg/j

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5g/dl, hyperlymphocytose 30 G/L
Immunophénotypage lymphocytaire : Matutes 4
Réticulocytes : 375 G/L
Il n’y a pas de syndrome tumoral. Les plaquettes sont normales.
Quel est le stade de la LLC ?

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
. Altération rapide de l’état général
. Asthénie +++, douleurs thoraciques
. Anémie à 5g/dl, hyperlymphocytose 30 G/L
Immunophénotypage lymphocytaire : Matutes 4
Réticulocytes : 375 G/L
Faut-il traiter la LLC ?
Si oui, comment ?
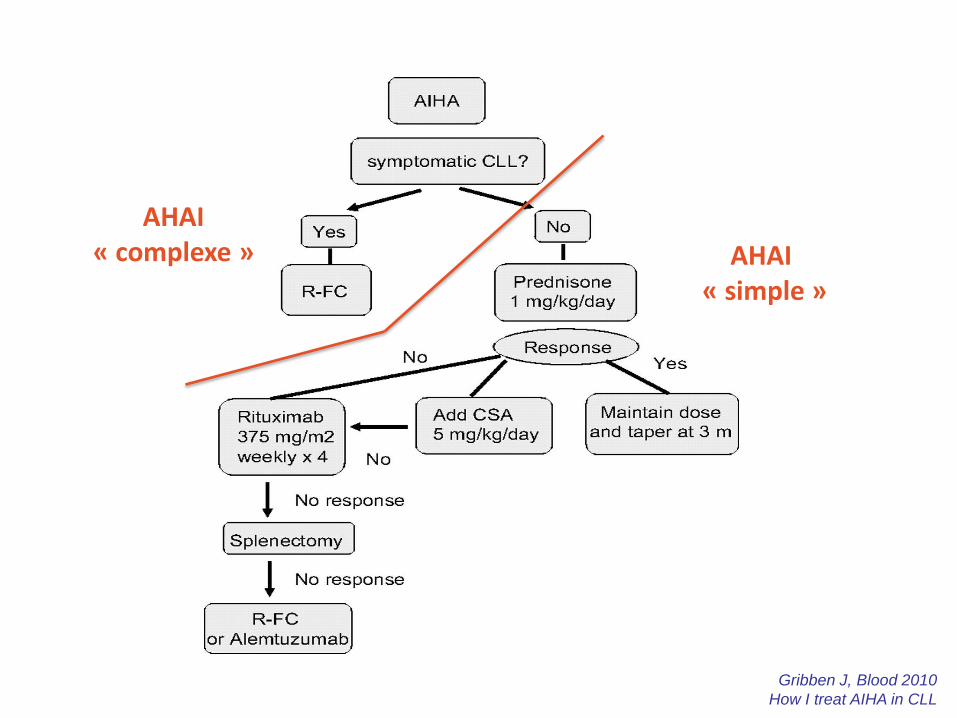
Gribben J, Blood 2010
How I treat AIHA in CLL
AHAI « simple »
AHAI « complexe »

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
Anémie à 5 g/dl, hyperlymphocytose 30 G/L, Réticulocytes : 375 G/L
Immunophénotypage lymphocytaire : Matutes 4
Traitement de la LLC si cortico-résistance
Si nécessité de traitement,
. Évaluation gériatrique
. Cytogénétique/biologie moléculaire (TP53)
. Rituximab/Bendamustine, R-Endoxan
. Ibrutinib (si inéligible alkylants, si anomalie TP53)

Situation 5
M. Sylvain C
85 ans, ATCD : BPCO, cardiopathie ischémique
Anémie à 5 g/dl, hyperlymphocytose 30 G/L, Réticulocytes : 375 G/L
Immunophénotypage lymphocytaire : Matutes 4
Corticothérapie seule
Le patient est perdu de vue
Il revient 3 ans plus tard avec une nette altération de l’état général,
des sueurs profuses et une adénopathie axillaire de 6 cm
Quel diagnostic suspectez-vous ?
Comment en faire le diagnostic ?