
Protocoles de biologie moléculaire courants - UGMA experimentaux/Protocoles... · - Ajouter les...
56
Protocoles de biologie moléculaire courants 1
Transcript of Protocoles de biologie moléculaire courants - UGMA experimentaux/Protocoles... · - Ajouter les...

Protocoles de biologie moléculaire courants
1

BACTÉRIES ÉLECTROCOMPÉTENTES 1ÉLECTROPORATION 2TRANSFORMATION TYPE PHAGE M13 RECOMBINANT 4MESURE DE LA CONCENTRATION D’ADN 5PURIFICATION DE PLASMIDES, PREPARATIONS RAPIDES 6 1) PROTOCOLE TRÈS RAPIDE POUR ANALYSE DE RESTRICTION ET SÉQUENCE DOUBLE BRIN 6 2) PROTOCOLE AVEC LYSOZYME ET ÉBULLITION 7 3) CULTURE 4 À 20H DANS 2ML MILIEU MLS 7 4) EXTRACTION DE PLASMIDE SUR UNE COLONIE 8 5) PRÉPARATION RAPIDE DE COSMIDES 8PURIFICATION DE PLASMIDES, PREPARATION EN GRAND 9 1) MÉTHODE ALCALINE ET CHLORURE DE LITHIUM 9 2) LES COLONNES KIT MACHEREY-NAGEL 10 3) AUTRE MÉTHODE : EXTRACTION ALCALINE ET COLONNE SÉPHACRYL 10 Caractéristiques et préparation de la colonne 11PREPARATION DE MATRICES SIMPLE BRIN 12 DÉTERMINATION DE L'ORIENTATION DES CLONES 12PURIFICATION D’ADN DE PHAGES 13 1) PRÉPARATION DE PHAGES 13 2) MINI-PRÉPARATION D'ADN PHAGIQUE 13 a/ méthode 1 13 b/ méthode 2 14 c/ méthode 3 15PURIFICATION DES ADN GENOMIQUES 16 1) A PARTIR D'UN PRÉLÈVEMENT SANGUIN HUMAIN (MÉTHODE SANS PHÉNOL/CHLOROFORME) 16 2) À PARTIR DE CULTURE DE CELLULES 16 3) À PARTIR D'EXTRAITS VÉGÉTAUX 17 METHODE 17 MATERIEL 17 PROTOCOLE 18 4) À PARTIR DE TISSUS ANIMAUX 19 5) À PARTIR DE CULTURE DE LEVURE 19 Solutions pour l'Extraction d'ADN génomique 20 EXTRACTION ADN DE LEVURE 20PURIFICATION DE FRAGMENTS D’ADN 22 1) À PARTIR DE GEL LMP 22 2) PAR ÉLECTROÉLUTION 22 3) PAR ÉLECTROÉLUTION SUR PAPIER 22 4) PAR ADSORPTION SUR BILLES DE VERRE 23 a) Extraction avec JetSorb 23 b) Protocole adapté de Anal. Biochem. (1982), Marko et al. 23 1) Billes de verre 23 2) Iodure de sodium 6M 23 3) EtOH/lavage 23 5) CENTRIFUGATION SUR LAINE DE VERRE 24
2

6) GRADIENTS DE SEL 24 Solutions et matériel nécessaires : 24 Coulage du gradient : 24 7) RÉPARATION D'EXTRÉMITÉS 25 8) SOUS CLONAGE 25PREPARATION D’ARN 26 1) EXTRACTION DE RNA TOTAUX EN UNE SEULE ÉTAPE 26 2) PRÉPARATION D'ARN À PARTIR DE CULTURES CELLULAIRES 26 3) EXTRACTION ARN PHÉNOL CHAUD 27NORTHERN – GEL DENATURANT AU FORMALDEHYDE 29 1) GEL 1.2% 29 2) TAMPON DE MIGRATION 29 3) TAMPON DE CHARGE 29TRANSFERT DE COLONIES SUR FILTRES 30 APPLICATION AU CRIBLAGE D'UNE BANQUE DE COSMIDES 30DEPOT D’ADN SUR FILTRE DE TYPE "DOT BLOT" 31TRANSFERT DE SOUTHERN 32 1) PRÉPARATION DU GEL 32 2) HYDROLYSES DES ÉCHANTILLONS (ADN GÉNOMIQUE) ET DÉPÔTS 32 3) TRANSFERT DE L’ADN DU GEL 33 a/ Par le vide 33 b/ Sans vide 34MARQUAGE RADIOACTIF D'ADN 35 1) PRÉPARATION D'UN KIT DE MARQUAGE PAR 'RANDOM PRIMING': 35 2) PROTOCOLE DE MARQUAGE 35 3) SONDES D'EXTRÉMITÉS DE COSMIDES 36 4) ELIMINATION DES NUCLÉOTIDES NON INCORPORÉS SUR COLONNE G75 36 Préparation de la colonne 36 Comptage 37HYBRIDATIONS (SOUTHERN ET NORTHERN) 38 1) HYBRIDATION DANS UN FOUR 38 2) RENATURATION DE LA SONDE AVEC L'ADN TOTAL 39 3) DÉHYBRIDATION 39REACTIONS DE SEQUENCE 40 PRÉPARATION DE MATRICES À PARTIR DE PREP. RAPIDES DE PLASMIDE : 40SEQUENCAGE DE PRODUITS DE PCR 42 1) OBTENTION DE LA MATRICE SIMPLE BRIN 42 2) PRÉPARATION DES GELS DE SÉQUENCE 42 a) Prétraitement des plaques de verre 42 b) Solutions 43 c) Gels gradient (33 x 40cm) 43 1) Gel gradient à 6% 43 2) Simulation d'un gradient 44 d) Gels non gradient 44 e) Électrophorèse 45ANNEXES 46PRÉPARATION DES TAMPONS ET SOLVANTS 47SOLVANTS 47SOLUTIONS COURANTES 47
3

TAMPONS DE PRÉHYBRIDATION ET D’HYBRIDATION 50MILIEUX DE CULTURE POUR BACTÉRIES OU LEVURES 51SOLUTIONS STOCK D'ANTIBIOTIQUES SOLUTIONS STOCK D'IPTG ET D'X-GAL 53PRÉPARATION DES MINI-COLONNES 53
BACTÉRIES ÉLECTROCOMPÉTENTES
Différentes méthodes permettent d’introduire un fragment d’ADN dans une cellule. Des cellules ayant au préalable été rendues compétentes par traitement chimique sont incubées avec l’ADN puis un choc thermique est réalisé. L’alternative la plus courante est l’électroporation. Les cellules doivent être soigneusement lavées (dessalage) pour éviter les arcs (courts-circuits) lors du choc électrique. Les fragments d’ADN sont généralement portés par un vecteur contenant un système autonome de réplication et un système de sélection (le plus souvent un gène de résistance à un antibiotique qui permet à partir de culture sur milieu sélectif de sélectionner ensuite les clones qui ont intégré le fragment d’ADN, ou un gène supresseur). Les bactéries transformées vont ensuite se multiplier (1 colonie = 106 bactéries) lorsqu’elle seront mise en culture sur le milieu sélectif.
Un des facteurs clés de la préparation des bactéries compétentes est l'état des bactéries qui servent à ensemencer le milieu de culture. Pour avoir une bonne reproductibilité des résultats, Hanahan par exemple, recommande de faire l'étalement de la souche sur boîte à partir d'un stock conservé en glycérol à -80°C, puis de faire l'ensemencement du milieu à partir d'un étalement frais. On obtient des résultats plus variables quand on utilise des étalements repris de boîte en boîte. Seul le protocole d'électroporation sera présenté ici, se référer aux anciens recueils pour des traitements chimiques des bactéries (protocole PEG par exemple).
Matériel : - Eau refroidie (dans la glace),- Glycérol 10% refroidi (dans la glace),- A partir de l'étape 3, les bactéries doivent rester à 2°C.
• 1 - Inoculer 1litre de milieu LB avec 1/100 de volume d'une culture fraîche (i.e. de la nuit) de la souche utilisée.• 2 - Incuber à 37°C avec une agitation vigoureuse (300 rpm) jusqu'à DO : 0,5 - 0,6.Note : l'état des bactéries (pleine phase exponentielle) est un paramètre essentiel.• 3 - Refroidir dans la glace en agitant. Laisser 15 minutes (étape essentielle : sinon les bactéries risquent encore d'effectuer une division, 20 minutes pour des XL1).• 4 - Centrifuger à 4000g, 2°C dans des tubes prérefroidis et un rotor froid pendant 15 minutes.• 5 - Vider le surnageant immédiatement (culot peu compact) et suspendre les culots dans un litre au total d'eau (ou milieu faible force ionique tel que Hépès 1 mM pH : 7, le culot est alors plus compact). Centrifuger comme en 4.• 6 - Refaire un lavage avec 500 ml d’H2O et centrifuger.• 7 - Resuspendre les culots dans 20 ml de glycérol 10% froid ou GYT (tube Falcon).Centrifuger comme en 4.• 8 - Resuspendre l'ensemble des culots dans un volume final de 2 ml de milieu 10% glycérol froid ou GYT. La concentration cellulaire doit être d'environ 3x1010 cellules/ml.• 9 - Cette suspension peut être utilisée de suite (les fraîches marchent mieux) ou congelée par aliquots (130 µl) dans de la carboglace de préférence ou de l'azote liquide puis stockée à -80°C.
Référence : Current Protocols ... section 1.8.4-1.8.8Variante : selon addendum Current Protocols, l'utilisation de milieu GYT au lieu de glycérol 10% pour la resuspension finale (étape 8) améliore l'efficacité d'un facteur 4 à 10, en améliorant (peut-être) la survie des colonies.
4

GYT : Glycerol 10% Yeast extract 0.125% (p/v) Tryptone 0.25% (p/v)
5

ÉLECTROPORATION
Introduction de fragment d’ADN dans une cellule hôte.
Matériel : mettre dans la glace : les cuves à électroporation, tubes Eppendorfs froids, tubes contenant 1 ml de milieu LB (ou ML).
• 1 - Décongeler en douceur les cellules à T° ambiante ou sur de la glace.• 2 - Mettre 40 µl de bactéries dans un tube eppendorf pré-refroidi, ajouter 1 à 2 µl d'ADN (qui doit être dans une solution de faible force ionique, TE ou eau ; 0,5 µl de produit de ligation ne provoque pas d'arc).• 3 - Gene Pulser Biorad
Réglages standards bactéries et plasmides :voltage = 2,5 kVcapacitance = 25 µFrésistance = 200 ohms
On peut lire dans notes annexes Current Protocols une étude sur l’optimisation des paramètres en fonction notamment de la taille de l’ADN à introduire.
Utilisation de l’électroporateur :Mise en marche bouton face avant du module du bas.Capacitance et résistance : boutons en bas à droite et en haut à gauche.Voltage : appuyer (2 fois) simultanément sur les touches « raise » et « lower » module du bas jusqu’à affichage du préréglage 2,5 kV.
• 4 - Transférer le mélange dans une cuvette d'électroporation froide. La mettre dans la chambre d'électroporation et appliquer une pulsation en appuyant simultanément sur les 2 boutons (rouges) Pulse. (la constante de temps doit approcher le plus possible 5,2 msec).• 5 - Ajouter immédiatement 1 ml de milieu froid LB (ou SOC) aux cellules électroporées, et récupérer l'ensemble avec une pipette Pasteur.• 6 - Transférer les cellules dans un tube bouchon rouge 10 ml et incuber pendant une heure à 37°C (225 rpm).Entre chaque électroporation, ne pas oublier de remettre à 0 la constante de temps en appuyant sur Set Volt.• 7 - Avant étalement, centrifuger la culture bactérienne à 3000 rpm 5 minutes et éliminer le surnageant afin de garder environ 100 µl de culture, reprendre le culot dans le surnageant.• 8 - Etaler sur gélose les 100 µl de culture à l’aide d’une pipette râteau.
Note : extemporanément, on peut étaler sur la gélose 40 µl X-gal 2% et 40 µl IPTG 2%.
Réutilisation des cuves :Lavage :Eau du robinet abondammentEau distillée (avec pissette)Alcool (Avec pissette)- Conserver ouverture vers le bas par exemple dans un pot yaourt en verre (il en rentre 6 ou 7).- Remplir d'alcool jusqu'à mi-hauteur des cuvettes.- Fermer avec un Saran, garder à T° ambiante.Avant utilisation, mettre à sécher sous la hotte, ouverture vers le haut.
6

TRANSFORMATION TYPE PHAGE M13 RECOMBINANT
ATTENTION : ne pas oublier la préculture.
- Ajouter 50 µl de TG1 ou XL1 fraîches par boîte.
- Ajouter les bactéries et l'ADN à la gélose MGM (3 ml) préalablement maintenue à 45°C + 40 µl IPTG 2%+ 40 µl Xgal 2% (solutions stock conservées à -20°C).
- Vortexer rapidement et couler sur boîtes (les sortir à l'avance).
Stockage souches bactéries :Solution glycérol : 65% glycérol
0,1 M Mg SO40,025 M Tris pH8
- Mélanger 1ml de culture et 1ml de solution glycérol- Conserver à –80°C
7

MESURE de la CONCENTRATION d’ADNFluorimètre DyNa Quant 200
La mesure de la concentration d’ADN par fluorimétrie est bien adaptée lorsque l’ADN est contaminé par des ARNs ou des nucléotides libres (par rapport à une mesure d’absorbance UV, qui ne discrimine pas les acides nucléiques simple brin, double brin, nucléotides libres). HOESCHT 33258 (Bisbenzimide) a peu d’affinité pour l’ADN simple-brin ou les nucléotides libres. Dans le cas de très fortes contaminations cependant, faire une évaluation par comparaison avec un témoin sur gel.
MESUREPour plus de détails, voir classeur mode d'emploi.
1 - Mettre sous tension l'appareil, attendre 15 min (extinction automatique au bout d’une heure)2 - Préparer 20 ml de solution :
18 ml d’H2O2 ml de TNE 10x20 µl de stock H33258 (stock à 1mg/ml, dans eau milliQ,
conservée à 4°C à l’obscurité -bouteille teintée ou tube enveloppé dans papier aluminium. Conservation = 6 mois, peut perdre ses capacités assez brutalement)
3 - Mettre 2 ml de solution dans la cuve (la gravure G face à l’utilisateur)4 - Appuyer sur “ZÉRO”.5 - Ajouter 2 µl d'ADN de référence (ADN porc = 110 ng/µl)
La fluorescence varie en fonction de la concentration de l’ADN en GC, et de l’accessibilité de l’ADN à l’intercalant (pureté de l’ADN, topologie plasmide vs ADN génomique ; pour une évaluation précise, le témoin devrait donc être choisi en conséquence)
6 - Bien mélanger par brassage pipetman P1000 (éviter les bulles)7 - Appuyer sur “CALIB”8 - Saisir la valeur de la concentration du standard (100 ng/µl)9 - Appuyer sur ENTER10 - Ensuite pour les mesures : rajouter séquentiellement 2µl d’échantillon et vider la cuve lorsque l’affichage dépasse 2000 en cumulé.11 - A la fin, rincer la cuve à l’eau, la sécher, et la ranger dans la boîte dédiée.
Problèmes rencontrésLes problèmes qui peuvent être rencontrés sur cet appareil viennent de :
- stock Hoescht trop ancien,- échantillon d’ADN trop concentré ou non homogène,- échantillon mal mélangé.
8

PURIFICATION DE PLASMIDES, PREPARATIONS RAPIDES
Antibiotiques, concentration finale d’usage :Tétracycline :10 mg/l (500 µl d’un stock à 20 mg/ml par litre de milieu)Ampicilline : - pour les plasmides 50 à 150 mg/l (entre 0,5 et 1,5 ml d’un stock à 100 mg/ml
par litre de milieu)- pour les cosmides 50 à 80 mg/l
Kanamycine : 20 mg/l (1 ml d’un stock à 20 mg/ml par litre de milieu)
1) Protocole très rapide pour analyse de restriction et séquence double brinRéférences: Zhou et al., Biotechniques vol. 8, n°2, 172-173 (1990) également Jones et Schofield, NAR vol. 18 n° 24, p 7463-7464
• 1 - Faire pousser les clones dans 2 ml de milieu LB• 2 - Centrifuger la culture 10 secondes en tube Eppendorf• 3 - Eliminer le surnageant en gardant 50 -100 µl de milieu• 4 - Resuspendre en agitant (Vortex)• 5 - Ajouter 300 µl de solution B (soude)• 6 - bien agiter par retournement• 7 - Ajouter 150 µl de solution C (acétate de potassium ou de sodium)• 8 - Bien agiter par retournement• 9 - Centrifuger 2 minutes à 12000 rpm• 10 - Verser le surnageant dans des tubes eppendorf neufs et ajouter 0.9 ml (remplir les tubes) d'Ethanol (gardé à -20°C).• 11 - Centrifuger 2 minutes à 12000 rpm• 12 - Eliminer le surnageant et remplir les tubes avec éthanol 70% froid• 13 - Vider les tubes en égouttant et laisser sécher (ou Speed-vac 2 minutes)• 14 - Reprendre dans 40 µl de TE (ou TE+ RNase).
Remarques : ne pas s'arrêter au cours des étapes 1 à 11 (il est commode d'extraire les échantillons par douzaine).étape 12 : il peut être préférable de resuspendre les ADN dans 40µl de TE et de reprécipiter avec 100 µl d'EtOH, plutôt que de rincer à l'EtOH 70%.Congeler les ADN (déprotéinisation minime, risques de dégradation à 4°C)*option : étape 14 : reprendre dans 40 µl de TE + 0,5 µl de RNase à 10 mg/ml. Laisser 10' à 37°C.
• 15 - Ajouter 23 µl de PEG 6000 20%- NaCl 2,5M. Centrifuger 15’ à 14000 rpm.• 16 - Faire un rinçage à l'EtOH 70% froid. Centrifuger 5’ à 14000 rpm.• 17 - Vider les tubes en égouttant et laisser sécher.• 18 - Reprendre dans 40 µl de TE.
Solution AGlucose 50mM solA x 10 4,5gEDTA 10mM 10ml (0,5 M)Tris-HCl pH : 8,0 25mM 6,25ml (2 M)H2O qsp 50ml. Filtrer 0,2 micron
Solution B (extemporanément)NaOH 0,1M 0,5 ml (10 M)SDS 0,5% 1,25 ml (20%)
9

H2O qsp 50 ml(Pour obtenir 10 ml de solution 1, ajouter 100 µl de soude 10M et 250 µl de SDS 20% à 9,7 ml de T.E.)(2.5 ml de soude 0,4M et 250 µl de SDS 20% à 7,3 ml de T.E.)
Solution CAcétate de potassium (3M final) 150gH2O 167 mlAcide acétique qsp 500mlSolution finale pH 4.8
RNase A pour 1ml, peser 10 mg de RNase A(10 mg/ml) faire bouillir 10 min au bain-marieLe rendement est de 2 à 5 µg environ. Ce protocole donne de très bons résultats en séquençagedouble brin manuel (non testé sur séquenceur). Si les cultures sont faites en milieu tamponné typeMLS, il est préférable de l'éliminer complètement et de reprendre le culot bactérien dans du GET(glucose-EDTA-Tris, solution A annexes) puis de procéder comme décrit par Jones et Schofield.
2) Protocole avec lysozyme et ébullition (NAR, 1988, vol. 16, n° 20, p 9878) avantages : préparation en un seul tube ; pas de solvants
• 1 - Verser 1,5 ml de culture de la nuit dans un tube eppendorf et centrifuger 4’à 4°C.• 2 - Reprendre le culot en 200 µl de STET (8% m/vol saccharose, 0,1 % vol/vol Triton X-100, 50 mM EDTA, 50 mM Tris HCl pH 8).• 3 - Ajouter 4 µl de lysozyme (solution fraîche à 50 mg/ml ; stock en poudre conservé à 4°C ou -20°C).• 4 - Incuber 5 minutes à température ambiante.• 5 - Laisser 45 sec. dans un cristallisoir d'eau bouillante, centrifuger 10 min à 12000 rpm.• 6 - Enlever le culot avec un cure-dent, ajouter 8 µl de CTAB (Serva ; solution fraîche à 50 mg/ml), centrifuger 5 min. à température ambiante.• 7 - Le culot est repris dans 300 µl de NaCl 1,2 M par vortexage intensif et reprécipité par addition de 750 µl d'éthanol 100% froid et centrifugation 10 min.• 8 - Le culot final est rincé en 70% éthanol froid, séché 3 min. au dessiccateur ou laissé sur la paillasse, et resuspendu dans 40 µl de T.E.
Le rendement usuel pour des plasmides type pUC est de l'ordre de 5 µg. 1/2 préparation suffit à faire la séquence.
3) Culture 4 à 20h dans 2ml milieu MLS• 1 - centrifuger 2 ml dans un tube Eppendorf 8K 4min à 4°C• 2 - reprendre 200 µl sol. A ;• 3 - ajouter 200 µl de sol. B ; mélanger doucement par rotations multiples ; une fois que la solution est devenue visqueuse, vortexer à pleine puissance ; la solution doit être limpide (s'il reste un trouble, c'est que les bactéries ne sont pas toutes lysées)• 4 - ajouter 150 µl sol. C ; bien mélanger ; vortexer • 5 - centrifuger 14000 rpm 5 min.• 6 - surnageant + 350 µl phénol ; centrifuger 14000 rpm 5min• 7 - surnageant + 600 µl isopropanol (Eppendorf de 1,5 ml) ; centrifuger 14K 10 min• 8 - rincer le précipité avec 400 µl d’éthanol à 70%.• 9 - précipité repris dans 200 µl TE; analyser 1 à 5 µl
10

Note : pour des cosmides reprendre le précipité dans 50 µl et analyser 5 µl.
4) Extraction de plasmide sur une colonieSuffisante pour une digestion
• 1 - Récupérer (au cure-dent ou à l'anse de platine) la colonie (penser à la numéroter)et la resuspendre dans 100 µl de solution A ( voir au paragraphe 3) puis vortexer.• 2 - Ajouter 200 µl de sol. B (voir au paragraphe 3), mélanger par retournement puis rajouter 150 µl de NaOAc 3M pH 5.2 et vortexer vigoureusement environ 10 sec.• 3 - Centrifuger 5 min en eppendorf et récupérer le surnageant sur lequel on peut éventuellement faire un phénol avant de le précipiter à l'alcool.• 4 - Digérer la totalité de la préparation, repérer les clones positifs que l'on remettra en culture pour en faire des maxi-préparations.
5) Préparation rapide de cosmides• 1 - Faire une culture de 24h et 8ml de ML avec antibiotique en tube bouchon rouge de 10ml (bouchon percé) à 37°C avec agitation.• 2 - Centrifuger les tubes 10 minutes à 3000 rpm (Jouan)• 3 - Reprendre les culots dans 110 µl de solution A et transférer en tubes Eppendorf.• 4 - Ajouter 220 µl de solution B et bien agiter par retournement.• 5 - Ajouter 165 µl de solution C et bien agiter par retournement.• 6 - Centrifuger 5 minutes 12000 rpm.• 7 - Verser le surnageant dans un nouveau tube.• 8 - Ajouter un volume (450 µl) de phénol /chloroforme et bien agiter.• 9 - Centrifuger 2 minutes à 12000 rpm.• 10 - Transférer la phase aqueuse dans un nouveau tube et précipiter avec deux volumes d'éthanol 100% froid.• 11 - Mélanger par retournements et centrifuger 3 minutes à 12000 rpm.• 12 - Eliminer le surnageant et rincer le culot en remplissant le tube avec 1ml d'éthanol à 70% froid. Laisser 2 minutes et éliminer le surnageant.• 13 - Bien égoutter les tubes et laisser sécher les culots sur la paillasse.• 14 - Resuspendre dans 20 µl de TE (ou TE+RNase 1mg/ml).
11

PURIFICATION DE PLASMIDES, PREPARATION EN GRAND
Solution AGlucose 50mM solA x 10 4,5gEDTA 10mM 10ml (0,5 M)Tris-HCl pH : 8,0 25mM 6,25ml (2 M)H2O qsp 50ml. Filtrer 0,2 micron
Solution B (extemporanément)NaOH 0,2M 1 ml (10 M)SDS 1% 2,5 ml (20%)H2O qsp 50 ml(Pour obtenir 10 ml de solution B, ajouter 200 µl de soude 10M et 500 µl de SDS 20% à 9.5 ml de T.E.)
Solution CAcétate de potassium (3M final) 150 gH2O 167 mlAcide acétique qsp 500mlSolution finale pH 4.8
RNase A pour 1ml, peser 10 mg de RNase A(10 mg/ml) faire bouillir 10 min au bain-marie
1) Méthode alcaline et chlorure de lithiumPartir d'une boîte et de colonies fraîchement étalées : Selon Flint, préparations adaptées à cultures de
PACs, bon pour in situ. Rajout de extractions phénol-chloroforme pour nos besoins (hydrolyses de restriction, migrations sur gel). Quantités pour 200 ml de culture milieu 2YT.
• 1 - Piquer une colonie isolée. Inoculer milieu "Super" (MLS) ou 2YT + antibiotique adéquat dans un Erlen de volume égal à 5 fois le volume de culture (Erlen de 1 litre pour 200 ml de milieu 2YT). Cultiver sous agitation 300-400 rpm la nuit à 37ºC.• 2 - Centrifuger la culture 10 min 4000 rpm.• 3 - Resuspendre avec 10 ml sol. A (pipetter et vortexer vigoureusement) et transférer en tube Falcon 50 ml.+ 10 ml sol. B (fraîche) ; agiter doucement (mélanger par huit retournements, selon flint) ; la solution doit se prendre en gel. 5 minutes à température ambiante.+ 10 ml sol. C ; agiter doucement (huit retournements).• 4 - Centrifuger 4000 rpm, 4ºC, 10min.• 5 - Ajouter au surnageant (éventuellement filtré sur de la gaze –jetable- ou nylon type drain pour hybridation –récupérable-) 20 ml iso-propanol (propane-2-ol). Mélanger par retournements et centrifuger 4000 rpm 10min (on peut récupérer un peu plus d'ADN si on laisse précipiter 1h à -20°C avant de centrifuger). Laisser sécher le culot.• 6 - Rincer le culot avec Ethanol 70% froid, puis reprendre le culot dans 1ml TE ; transférer en tubes Eppendorf de 2 ml (réf 0030.120.094, de préférence à ceux de 2,2 ml qui ont tendance à fuire) ; ajouter 5 µl RNase A 10mg/ml ; 15min à 37°C.• 7 - Extraire avec 1 ml phénol, puis 1 ml phénol/chloroforme/alcool isoamylique Recommencer jusqu’à ce qu’il n’y ait plus d’interface (souvent trois extractions au total).
12

• 8 - Précipiter la phase aqueuse avec 0,3 ml NH4Ac (7,5M) et 2,5 ml éthanol 100% froid dans un tube 10 ml bouchon rouge. Mélanger par retournements et centrifuger 10 minutes à 4°C et 3000 rpm.• 9 - Eliminer le surnageant et dissoudre le culot dans 1ml TE. Précipiter à nouveau avec 0,1ml NaAc 3M pH : 5,2 et 2,5 ml éthanol 70% froid et centrifuger. Dissoudre le culot dans du TE stérile (300 µl).• 10 - Estimer la concentration par migration sur gel d’un aliquote de 5 µl digéré par EcoRI (dans un volume total de 10 µl).
Remarque : pour les PAC, dissoudre dans seulement 100 µl de TE.
2) Les colonnes Kit Macherey-NagelSemblent également bien adaptées au moins pour :
- séquençage- transfections de cellules- se présentent en trois formats : mini, 100 et 500- suivre protocole.
Remarque : faire parfois une deuxième élution
3) Autre méthode : extraction alcaline et colonne séphacrylLe milieu LS permet d'obtenir des rendements de l'ordre de 10 à 15 mg/litre pour des plasmides
de type pUC et des souches de type TG1, JM101.
• 1 - Ensemencer 250 ml de milieu avec antibiotique avec une colonie de bactéries.• 2 - Après 24 heures de culture pour des souches type TG1, 36 heures pour des souches type DH5 ou HB101, les bactéries sont centrifugées à environ 5000 rpm pendant 10 à15 min.• 3 - Le culot est resuspendu dans 5 ml de T.E. et laissé dans la glace 10 min.• 4 - Ajouter 12 ml d'un mélange de soude 0.2 N ; SDS 1% fait le jour même (sol. B).Mélanger doucement et laisser dans la glace 20 minutes environ.• 5 - Ajouter 6 ml d'acétate de potassium 5M pH : 4.8 (sol. C) et agiter fortement puis laisser 30 min. dans la glace.• 6 - Centrifuger pendant 15 min. à 5000 rpm.• 7 - Récupérer le surnageant en le filtrant sur de la gaze, ajouter 20 ml de chloroforme, agiter, laisser décanter ou centrifuger 5 min. à 3000 rpm.• 8 - Récupérer la phase aqueuse, ajouter 15 ml de propanol 2 et centrifuger à 10000 rpm pendant 10’.• 9 - Eliminer le surnageant, égoutter, laisser sécher et rependre le culot dans 2 ml de T.E.• 10 - Ajouter 20 µl de RNAse à 10 mg/ml et incuber pendant 30 minutes à 37 °C.• 11 - Ajouter 100 µl d'une solution de SDS à 10% et 20 µl d'une solution de protéinase K à 10 mg/ml et incuber à 50 °C pendant 1 heure.• 12 - Ajouter 1 ml de phénol, agiter, ajouter 1 ml de chloroforme : alcool isoamylique, agiter et centrifuger brièvement.
Commentaire : jusque-là, protocole classique lyse alkaline, avec légères variantes.
• 13 - Prélever la phase aqueuse, ajouter 200 µl de bleu de dépôt et déposer sur la colonne.
13

• 14 - Après environ 1 heure d'élution (premier bleu ayant parcouru le tiers de la colonne), prélever une quinzaine de fractions de 100 gouttes (3 à 5 ml).• 15 - Déposer 10 µl de chaque fraction sur un gel d'agarose. Habituellement, le plasmide est obtenu dans un volume d'une quinzaine de millilitres.• 16 - Précipiter avec propanol 2, rincer le culot à l'éthanol à 70% froid, laisser bien sécher, resuspendre dans 1 ml de T.E. et mesurer la concentration au fluorimètre.
Caractéristiques et préparation de la colonne.Utiliser, par exemple, une colonne d'un diamètre de 1 cm et d'une longueur de 50 cm.L'élution se fait par gravité avec un tampon Tris 20mM pH : 8 ; E.D.T.A. 1mM ; NaCl 1 M.
Dégazer le Séphacryl S-1000 pendant 30 min. avant le chargement.L'ADN chromosomique passe avant le plasmide, l'ARN après. Dans les conditions utilisées, les
premières fractions de plasmide sont contaminées par de l'ADN chromosomique.La colonne commence apparemment à saturer au delà d'une quantité de 2 mg de plasmide
environ.
14

PREPARATION DE MATRICES SIMPLE BRIN
• 1 - Inoculer 5 ml de milieu LB avec une colonie de JM101 (recA+) ou JM109 (recA-) cultivée sur milieu minimum (milieu M9 glucose de Maniatis et al. avec thiamine (vitamine B1)) et incuber à 37°C jusqu'en fin de phase exponentielle (D.0.1 environ). Cette préculture peut être conservée quelques jours au froid.• 2 - Diluer les cellules au 1/100 en milieu YT, et répartir 2 ml dans des tubes.• 3 - Piquer les colonies M13 (colonies bien séparées à partir de boites incubées moins de 12 heures à 37°C), et incuber avec agitation à 37°C pendant 5 à 8 heures• 4 - Transférer les cultures en tubes Eppendorf de 1.5 ml, centrifuger 5 min., verser le surnageant dans des tubes contenant 250 µl de PEG (20% PEG 6000, 2.5 M NaCl), mélanger, laisser 10 min. A température ambiante, centrifuger 5 min.• 5 - Le culot de phages doit être visible. Eliminer le surnageant avec une pipette Pasteur effilée et une trompe à eau, centrifuger 30 secondes, éliminer le surnageant résiduel.• 6 - Suspendre le culot dans 100 µl de T.E., ajouter 50 µl de phénol, agiter, ajouter 50 µl de chloroforme-isoamylalcool, agiter, centrifuger 5 min.• 7 - Prélever soigneusement la phase aqueuse (90-95 µl), ajouter 3 µl de chlorure de sodium 5 M et 250 µl d'éthanol, mélanger et laisser 30 min. dans la glace.• 8 - Centrifuger 10 min., éliminer le surnageant, laver avec de l'éthanol à 70% (remplir le tube, le laisser à température ambiante pendant 5-10 min., enlever l'éthanol). Laisser sécher pendant la nuit ou sous vide.• 9 - Suspendre dans 30 µl de T.E., et garder à -20°C.
NAR 1987 vol. 15, n°23 p 10047 propose un protocole très rapide avec CTAB et sans phénol. A essayer avec une solution fraîche de CTAB.
Détermination de l'orientation des clones- mélanger 1/20 de préparation de chacun des deux clones
0,5 µl de NaCl 5M.H2O qsp 10 µl
- incuber 10 min. à 60°C- déposer sur gel (avec un témoin)
15

PURIFICATION D’ADN DE PHAGES
1) Préparation de phages• 1 - Cultiver les bactéries LA 101 jusqu’à environ 1 O.D. (5x108 bact/ml) suspendre dans 30 µl de T.E., et garder à -20°C.• 2 - Infecter 10 bactéries par 1 phage pendant 15 min à 37°C pour 1litre, il faut environ 109
phages et 1010 bactéries• 3 - Transférer dans 1litre de ML contenant 10mM MgCl2 et 0,2% de maltose• 4 - Incuber 7 heures au moins ou la nuit à 37°C• 5 - Ajouter 4 ml de chloroforme et agiter pendant 5 min• 6 - Centrifuger à 6000 rpm pendant 15 min à 4°C• 7 - Ajouter PEG 6000 1g/10ml
NaCl 0,584g/10ml• 8 - Précipiter à 4°C au moins une heure• 9 - Centrifuger à 2000 rpm pendant 30 min à 4°C• 10 - Suspendre le culot dans 8ml de tampon phage (Tris 10mM PH : 7.4, MgCl2 10mM)• 11 - Faire une extraction au chloroforme• 12 - Préparer 2 gradients de césium : 2ml de 1,7 ;1,5 ; 1,3 (Césium BRL ultrapur)
CsCl Tampon de phage1,3 10g 22,3 ml1,5 20g 25,2 ml1,7 20g 15,8 ml
• 13 - Centrifuger 3heures à 30.000 rpm ou 2heures 40.000 rpm• 14 - Prélever la bande et ajouter du CsCl 1,5 jusqu’à 3ml• 15 - Centrifuger dans le rotor SW 50-1 pendant 20 heures• 16 - Dialyser contre du Tris 10 mM, MgCl2 10 mM pH 7,4
2) Mini-préparation d'ADN phagiquea/ Méthode 1
• 1 - Inoculer 20 ml de milieu LB + 0,2% maltose avec une colonie de la souche bactérienne "phage spécifique" fraîchement étalée, et incuber la nuit à 37°C sous agitation.• 2 - Mesurer la DO à 600 nm (1UDO = 8.108 bactéries/ml, cependant la densité cellulaire en fonction de la DO varie selon les souches, cf. Sambrook 1.82), centrifuger à 4K 5min et reprendre le culot dans du milieu SM de façon à obtenir 4.109 bactéries/ml.• 3 - Etaler une boite avec le clone phagique à purifier, prélever 20 plages de lyse et les mettre à diffuser au moins 4h dans 200 µl de milieu SM.• 4 - Réaliser l'infection en incubant 15 min à 37°C 100 µl de bactéries, soit 4.108 bactéries, et 20 µl de l'inoculum phagique, soit 2.107 pfu).• 5 - Inoculer ces 120 µl dans 20ml de LB + 0,1% glucose + 10mM MgCl2 et incuber à 37°C sous agitation pendant 4 à 8H.• 6 - Lorsque la lyse est suffisante, ajouter 2 ml de CHCL3 et incuber encore 10min à 37°C sous agitation. A cette étape l'Erlen peut être conservé une nuit à 4°C.• 7 - Centrifuger 10 min à 12K et récupérer le surnageant.• 8 - Ajouter 10 µg/ml de RNAse DNAse free ainsi que 10 µg/ml de DNAse et incuber 30 min à 37°C.• 9 - Ajouter 4 ml de solution de lyse (EDTA 0,25M - Tris HCl pH : 8 ; 0,5M – SDS 2,5%), homogénéiser, et incuber 30min à 70°C.• 10 - Ajouter 6 ml d'acétate de K (8M), homogénéiser, et incuber 15min à 4°C.
16

• 11 - Centrifuger 30min à 10K et à 4°C. Conserver le surnageant auquel on ajoute 20ml d'isopropanol et incuber le tube 10 min à température ambiante.• 12 - Centrifuger 15 min à 10K et à 20°C. Réaliser trois lavages du culot avec 5 ml d'éthanol 70% sans centrifugation en faisant très attention au culot d'ADN.• 13 - Reprendre le culot dans 500 µl de TE 20:5 + 0,3M NaAc pH : 8, et transvaser l'échantillon dans un tube Eppendorf.• 14 - Extraire deux fois avec du phénol/chloroforme, puis deux fois avec un volume de chloroforme seul.• 15 - Précipiter l'ADN avec 2/3 de volume d'isopropanol, faire un lavage avec de l'éthanol 70%, sécher le culot et le reprendre dans environ 400 µl de TE 20:1.
On obtient environ 100 µg d'ADN phagique pur.
b/ Méthode 2• 1 - Inoculer 20ml de milieu LB + 0,2% maltose avec une colonie de LA101 fraîchement étalée.• 2 - Lorsque la DO600 est d'environ 1 (5 à 7h; 8.108 /ml), centrifuger 2000 rpm 10 min, reprendre dans 5ml SM• 3 - Ajouter à 1,5ml bactéries indicatrices 2. 108 pfu de phage. Incuber 30’ à 37°C.• 4 - Ajouter à 250 ml milieu LB + MgSO4 10mM préchauffé à 37°C dans un Erlenmeyer de 2litres. Cultiver 8 à 12 h sous agitation rapide (i.e. jusqu'à la lyse).• 5 - Ajouter 5 ml chloroforme. Incuber encore 30 min à 37°C sous agitation.• 6 - Refroidir à température ambiante sous un robinet d'eau froide.• 7 - Ajouter 250 µl DNAse I 1mg/ml (frais)
250 µl RNAse 1 mg/ml (frais). Incuber 30 min à température ambiante.• 8 - Ajouter 14,6 g NaCl. Dissoudre 5min sous agitation magnétique douce. Laisser 1h dans la glace.• 9 - Centrifuger 10 min 4°C 9000 rpm (Sorvall 6 x 500ml). Transférer le surnageant (en évitant le chloroforme du fond) dans une fiole propre et ajouter 25g PEG 6000.Dissoudre 5 min sous agitation magnétique douce. Laisser de 1h à la nuit dans la glace.• 10 - Centrifuger 10 min 4°C 9000 rpm. Bien ôter le surnageant (éventuellement centrifuger de nouveau). Reprendre le précipité dans 4 ml SM.Extraire avec 4ml chloroforme.• 11 - Centrifuger 30 000 rpm 2h ou 50 000 rpm 1h (godets oscillants).• 12 - Reprendre le culot de phage dans 2 ml SM.• 13 - Ajouter EDTA 0,5M 80 µl
protéinase K 50mg/ml 40 µlSDS 10 % 200 µl
Incuber à 65°C 30 min.• 14 - Extraire au phénol puis au phénol : chloroforme : alcool isoamylique (25:24:1).• 15 - Dialyser contre 3 x 2 litres TE
Solutions SMNaCl 100 mM pour 1 litre 5,8gMgSO4 ,7 H2O 8 mM 2gTris-HCl pH : 7,5 50 mM 50ml 1Mgélatine 0,1 mg/ml 5ml 2%
17

c/ Méthode 3(Grossberger, NAR Vol 15, 1987 p 6737)
• 1 - Choisir la plage désirée avec une pipette Pasteur stérile et la verser dans une tube de 18 ml contenant 0,3 ml de tampon d'adsorption et 0,2 ml d'une culture exponentielle de bactéries en milieu ML 0,4% maltose.
Tampon d'adsorption (10mM Mg Cl2 et 10mM Ca Cl2) :Dans 5 ml : 1ml 50 mM CaCl2
50 µl 1 M Mg Cl2
• 2 - Incuber le tube pendant 10 minutes à 37°C.• 3 - Ajouter 10 ml milieu ML contenant Mg Cl2 10mM et 0,1% glucose.
Dans 10 ml : 50 µl 20% glucose100 µl 1M Mg Cl2
• 4 - Incuber la nuit à 37°C avec agitation en inclinant à 45°. Les bactéries doivent être lysées.• 5 - Centrifuger les tubes à 5000 rpm pendant 10 minutes pour culotter les bactéries.• 6 - Centrifuger le surnageant à 30.000 rpm pendant 30 minutes.• 7 - Suspendre le culot dans 200 µl de milieu 63B1 dilué 10 fois, et le transférer dans un tube Eppendorf de 1,5 ml.Le milieu 63B1 1/10 peut-être remplacé par Tris 10mM pH 7.4, Mg Cl2 10mM• 8 - Préparer immédiatement avant usage une solution de protéinase K, 1 mg/ml, en milieu 63B1, et en ajouter 200 µl à chaque tube. Incuber à 37°C pendant 2 heures.• 9 - Faire une extraction phénol, suivie d'une extraction chloroforme.• 10 - Précipiter l'ADN avec NaCl 0,2M et 2 vol. d'éthanol 100%.• 11 - Centrifuger et suspendre le culot en 100 µl TE.• 12 – 10 µl sont utilisés pour une digestion de vérification.
18

PURIFICATION DES ADN GENOMIQUES.
1) À partir d'un prélèvement sanguin humain (Méthode sans phénol/chloroforme)Note : selon l’espèce considérée, la concentration en lymphocytes est variable (par exemple, plus élevée chez le porc). Dans d’autres cas (oiseaux, chameau), les globules rouges sont nucléés, et d’autres protocoles seront mieux adaptés.Travailler sur glace avec des gants.
• 1 - Congeler le prélèvement sanguin (sur EDTA) à –20°C dès réception (en général tube en verre de 5 ml).• 2 - Le jour de l'extraction, décongeler dans la glace. Si le prélèvement est encore dans le tube en verre, mettre le tube dans un tube 50 ml par précaution. En tout cas, éviter les chocs thermiques lors de la sortie du tube hors congélateur (le tenir par le bouchon caoutchouc, le mettre tout de suite dans la glace).• 3 - Laisser décongeler doucement et complètement, en tournant de temps en temps le(s) tube(s) posé (s) sur la glace.• 4 - Diluer avec environ 4 volumes de TE 20:5 froid (bouteille préalablement refroidie dans la glace).• 5 - Incuber dans la glace 15 min pour que la lyse des hématies ait lieu.• 6 - Centrifuger (godets oscillants) 20 min, à 4°C et 4000rpm. Eliminer doucement le surnageant.• 7 - Remettre en suspension les cellules avec 15 ml de TE 20:5 en vortexant et agitant si nécessaire. Le culot doit être complètement dissocié. Centrifuger 15 min à 4°C et 4000rpm.• 8 - Faire au minimum 2 lavages (jusqu'à ce que le culot soit translucide).• 9 - Reprendre le culot dans 1/2 volume de sang de départ de TE 20:5 (2,5 ml).• 10 – Ajouter : Sarcosyl (ou SDS) à 1% final, Protéinase K à 200 µg/ml final. (250 µl Sarcosyl ou SDS 10%, 25 µl de protéinase K à 20 mg/ml)• 11 - Incuber sous agitation (300 rpm) toute la nuit à 37°C ou 2h à 65°C .• 12 - Ajouter 1/3 volume d'acétate d'ammonium 7,5M (2,5 M final) et 2 volumesd'éthanol absolu froid.• 13 - Mélanger doucement par retournements jusqu'à apparition d'un flocon.• 14 - Récupérer l'ADN avec une pipette Pasteur boutonnée flambée et recourbée en hameçon ou centrifuger à 3000 rpm 4°C 10 min pour récupérer le culot.• 15 - Transférer le flocon dans 1ml de Tris 20mM pH : 8, EDTA 5mM, NaCl 0,2M.• 16 - Incuber à 37°C sous agitation très douce jusqu'à dissolution de l'ADN.• 17 - Ajouter 2 volumes d'éthanol absolu, mélanger par retournements, récupérer l’ADN avec pipette (flocon) ou par centrifugation comme à l’étape 14, rincer dans de l'éthanol 70%, bien égoutter le flocon (au besoin centrifuger et éliminer l'éthanol résiduel).• 18 - Reprendre dans 1ml de TE 20:1 pH : 8.• 19 - Mesurer la concentration au fluorimètre.
2) A partir de culture de cellules• 1 - Des cellules (quelques dizaines de millions) seront centrifugées et reprises dans 1 ml de PBS (Tampon Phosphate Salin).• 2 - Rajouter 5 ml de tampon S.E. (75 mM NaCl, 25 mM EDTA pH : 8), puis ajouter 125 µl de SDS 20% et 50 µl de protéinase K 20 mg/ml, et incuber une nuit à 37°C.• 3 - Ajouter 5 ml de phénol saturé neutralisé, agiter doucement pendant 5 min.• 4 - Ajouter 5 ml de chloroforme isoamylalcool, agiter doucement pendant 5 min, centrifuger à 4000 rpm pendant 10 min.
19

• 5 - Prélever la phase aqueuse (supérieure) et recommencer une extraction phénolchloroforme.• 6 - Ajouter 150 µl de chlorure de sodium 5 M (concentration finale de 150 mM) et 2 vol d'éthanol. Mélanger doucement. L'ADN doit précipiter en un long filament. Récupérer directement le filament avec une pipette pasteur, le rincer dans de l'éthanol 70 %, le laisser sécher avant de le suspendre dans 1 ml de T.E.• 7 - Mesurer la concentration au spectrophotomètre. Le rendement est habituellement de l'ordre de 400 µg.
3) A partir d'extraits végétauxMETHODE
- Série de traitements avec le CTAB (Bromure de Cétyl Triméhyl ammonium), détergent non ionique, pour lyser les cellules et purifier les Acides nucléiques (AN).- L’AN est récupéré par précipitation à l’éthanol ou à l’isopropanol inclus dans la dernière solution de CTAB.Remarque : Le CTAB libère les AN cellulaires totaux et forme un complexe insoluble avec eux quand la concentration initiale en NaCl est < à ,0,5 M. On élimine ainsi la majorité des contaminants cellulaires. Le complexe CTAB-AN est seulement soluble en présence de fortes concentrations en sel. Le détergent est alors éliminé et l’AN est récupéré par précipitation. Le CTAB résiduel est lavé par l’éthanol 80%.Pour diminuer l’activité nucléasique, le tissu pourra être congelé rapidement et décongelé seulement en présence du tampon d ‘extraction qui contient le détergent et une forte concentration en EDTA.Le rendement est de l’ordre de : 100 à 500 µg d’ADN par gramme de tissu frais de plantes.2 à 6 heures de purification selon la quantité de départ en matériel et la pureté et le rendement désiré.La méthode décrite ci-dessous peut s’appliquer à tous types de tissus végétaux. Quelques mg de tissus suffisent.
MATERIELSolution d’extraction CTABConservation à t° ambiante, plusieurs années2%(w/v) CTAB100 mM Tris HCl pH : 8 ,020 mM EDTA pH : 8,01,4 M NaCl
Solution de CTAB/NaCl10% de CTAB dans 0,7 M de NaClDissoudre 4,1 g de NaCl dans 20 ml d’eauAjouter lentement 10 g de CTAB, en chauffant et en remuantSi nécessaire chauffer à 65°C pour dissoudreAjuster à 100 ml avec de l’eau
Solution de précipitation CTABConservation à t° ambiante, plusieurs années.1%(w/v) CTAB50 mM Tris HCl pH : 8 ,010 mM EDTA pH : 8,0
2% (v/v) •-mercaptoéthanol (•-ME)
20

Tampon TE high saltConservation à t° ambiante, plusieurs années.100 mM Tris HCl pH : 8 ,00,1 mM EDTA pH : 8,01 M NaCl
24:1 (v/v) chloroform/alcool isoamyl
Ethanol 80% ou isopropanol
Mixeur ou pilon et mortier ou moulin à café
PROTOCOLEExtraction de l’ADN
• 1 – Ajouter le •-Me à la solution d’extraction CTAB pour obtenir une concentration finale de 2% (v/v). Environ 4 ml de solution d’extraction CTAB-•-Me par gramme de tissu frais. Dilution 1:1 de cette solution avec de l’eau stérile pour des tissus déshydratés. Utiliser le •-Me sous hotte chimique.• 2 – Chauffer cette solution et la solution de CTAB-NaCl à 65°C (0,4 à 0,5 ml par gramme de tissu frais).• 3 –Si possible, refroidir le broyeur (-80°C ou azote liquide).• 4 – Broyer le tissu de plante en une fine poudre et transférer dans tube résistant aux solvants organiques. Utiliser du tissu jeune et éviter les pédoncules et les veines les plus larges pour un rendement en ADN et une faible contamination en polysaccharides.• 5 – Ajouter la solution d’extraction CTAB-•-Me chaude sur le tissu broyé et mélanger.• 6 – Incuber 10 à 60 min à 65°C en mélangeant de temps en temps. Le rendement est fonction du temps d’incubation.• 7 – Extraire l’homogénat avec un volume égal de 24:1 chloroforme/alcool isoamyl.• 8 – Bien mélanger par retournement. Centrifuger 5 min à 4°C 7500 g. 10 000 rpm en microcentrifugeuse pour des petits échantillons < à 150 mg de tissu de départ.• 9 – Récupérer la phase aqueuse (phase supérieure).• 10 – Ajouter 1/10 vol. de la solution CTAB-Nacl à 65°C à la phase aqueuse récupérée.• 11 – Bien mélanger par retournement. Ajouter 1 vol. égal de chlroforme/alcool isoamyl pour extraire.• 12 –Mélanger. Centrifuger 5 min à 4°C à 7500g (ou 10 000 rpm si microcentrifugation) et récupérer la phase aqueuse (phase supérieure).
Précipitation des ADN• 13 – Ajouter exactement 1 vol. de solution de précipitation CTAB.• 14 – Bien mélanger par retournement. Si la précipitation est visible passer à l’étape suivante. Sinon incuber 30 min à 65°c.• 15 – Centrifuger 5 min à 500g 4°C (2700 rpm si microcentifugation). En absence de culot, rajouter de la solution de précipitation CTAB (jusqu’à 1/10 du vol. total), incuber entre 1 h et la nuit à 37°C. Centrifuger 5 min à 500g à 4°C.• 16 – Enlever mais ne pas jeter le surnageant. Resuspendre le culot dans du tampon TE high-salt (0,5 à 1 ml de TE-high salt par gramme de matériel de départ). Si le culot est difficile à suspendre, incuber 30 min à 65°C. Répéter jusqu’à dissolution.Lecture au fluorimètre du surnageant et jeter le culot si les AN sont dans le surnageant.• 17 – Ajouter 0,6 vol. éthanol 100% pour précipiter. Bien mélanger . Centrifuger 15 min 7500g 4°C
21

• 18 – Laver le culot avec l’éthanol 80%.• 19 – Sécher et resuspendre dans un volume min de TE (0,1 à 0,5 ml de TE par gramme de matériel de départ.
Purification plus approfondie ? ?• 20 – Protéinase K ou RNase A.
4) A partir de tissus animaux• 1 - L'échantillon, congelé dans l'azote, est broyé (marteau ou pistolet), puis repris dans du tampon de digestion à raison de 1g pour 6 ml, pour être incubé à 56°C pendant 12 à 18 heures.
Tampon de digestion : Tris HCl pH8 10 mMEDTA 25 mMNaCl 100 mMSDS 0,5%Protéinase K 0,1 mg/ml
• 2 - Faire ensuite une extraction au phénol et deux extractions au chloroforme.• 3 - Prélever la phase aqueuse et précipiter l'ADN.
Les rendements obtenus sont de l'ordre de plusieurs centaines de µg par gramme de tissu.
Également recommandé : tampon à l'urée.Tris 10 mMEDTA 10 mMNaCl 10 mMUrée 8M
puis Protéinase K : 150 µg/mlfaire des extractions phénol + chloroforme (pas phénol seul)
5) A partir de culture de levureCulture durée de préparation au moins 2h30Dans des plaques 24 puits, mettre 2,5 ml de YPG.Inoculer avec un rotofil en prélevant les cellules bien au centre de la colonie (1 colonie = 106 cellules). Incuber 48 h à 30°C ( souches ORD thermosensibles) avec agitation de 230 rpm.
Prélèvement de 1 ml pour extraction d'ADN en tubes repérés n°1 à 96 de 1,5 ml puis centrifugation 3000 rpm 10 min.Conservation des souches dans les plaques avec 1 ml de glycérol 60% mettre à -80°C
Solutions pour l'Extraction d'ADN génomiqueSolution de Lyse 5 ml pour 100 preparations
Sorbitol 3 ml de sol. 2M à 4°C 1,2 M finalEDTA 1 ml sol. 0,5 M 0,1 M finalß mercapto éthanol 50 µl sous hotte 1% finalZymoliase 10 mg à 20 U/mg 2 mg/ml finalH2O milliQ 950 µl (qsp 5 ml)EDTA 5 ml à 50 mM : 500 µl sol 0,5 M avec 4,5 ml H2OSDS 10 % : 1,5 mlProtéinase K 2 mg/ml : 500 µl
22

Acétate de potassium 5 M mis à 4°C : 2 mlIsopropanol : 25 mlTampon TE 5,5 ml : Tris 10 mM pH : 8 -EDTA 1 mM
Tampon TEN :Tris 150 µl sol 1 M pH : 8 10 mMEDTA 30 µl sol 0,5 M 1 mMNaCl 600 µl sol 5M 0,2 MH2O 14,220 ml (qsp 15 ml)RNAse A 250 à 1 mg/ml : dilution au dixième sol.10 mg/ml
Mélange Phénol/Chloroforme 15 ml :Phénol 7,5 mlChloroforme 24 vol / Alcool Isoamilique 1 vol 7,5 ml
EXTRACTION ADN DE LEVURE• 1 - Suspendre le culot dans 50 µl sol. de lyse.• 2 - Incuber 2 h à 37°C sur un agitateur à vibration. Centrifuger 8000 rpm 2 min.• 3 - Resuspendre le culot dans : 50 µl EDTA 50 mM
1,5 µl SDS 10 % bien mélanger5 µl protéinase K 20 mg/ml bien mélanger
• 4 - Incuber 30 min à 65°C en homogénéisant toutes les 5 min.• 5 - Ajouter 20 µl d'Acétate K froid bien mélanger.• 6 - Incuber 30 min (minimum) dans la glace. Centrifuger 12000 rpm 15 min 4°C.• 7 - Transvaser sans pipeter le surnageant dans un autre tube. Ajouter 75 µl d'Isopropanol, mélanger doucement jusqu'à formation complète du précipité et placer à 5 min - 80°C.• 8 - Centrifuger 12000 rpm à 4°C 10 min.• 9 - Suspendre le culot dans 25 µl de TE (Option OVN 4°C)• 10 - Ajouter 2,5 µl de RNAse A à 1 mg/ml. Incuber 1H à 37°C (agiter toutes les 5’)• 11 - Ajouter 50 µl Isopropanol, mélanger doucement jusqu'à formation complète du précipité. Placer à - 80°C pendant 5 min. Centrifuger 12000 rpm à 4°C 10 min.• 12 - Suspendre dans 100 µl de TEN. (Option OVN 4°C)• 13 - Ajouter sous hotte 100 µl de phénol/chloroforme, agiter, centrifuger 12000 rpm 10 min.• 14 - Prélever la phase supérieure aqueuse et ajouter 200 µl d’Ethanol 100 %, placer 5 min à - 80°C.• 15 - Centrifuger 12000 rpm à 4°C 10 min.• 16 - Laver le culot avec 200 µl d’Ethanol 70 %. Centrifuger 12000 rpm 4°C 10 min.• 17 - Vider le surnageant et sécher le culot sur paillasse.• 18 - Suspendre le culot dans 30 µl de TE (Option à 56°C 1 H puis vortex)Hydrolyse sur 10 µl d'ADN par HinfI (nuit à 37°C)
23

PURIFICATION DE FRAGMENTS D’ADN
Pour préparer des fragments de taille allant de quelques centaines à quelques milliers de nucléotides, des gels à 1% conviennent bien ; pour Nusieve GTG, 1,5% minimum. Un puits d'une vingtaine de microlitres permet de séparer des quantités d'ADN de l'ordre de quelques centaines de nanogrammes par bande. On colore au Bromure d'Ethidium après électrophorèse (évite de contaminer les cuves)NB : pour tous ces protocoles d'extraction, il est important d'utiliser de l'agarose très pur; l'agarose Seakem GTG (FMC; distribué par Tébu) donne de bons résultats de ligature.
1) A partir de gel LMP• 1- Visualiser les bandes sur une rampe ultra-violet (de préférence à 300 nm ou plus de longueur d'onde afin de réduire la photolyse de l'ADN), et les découper.• 2 - Compléter le volume à 500 µl environ avec du T.E. si nécessaire et laisser le tube Eppendorf 5 min. dans un bain marie à 65°C.• 3 - Extraire deux fois au phénol, deux fois au chloroforme.• 4 - Précipiter l'ADN avec 150 mM NaCl final, deux volumes d'éthanol 100% et laisser 30 min dans la glace.• 5 - Centrifuger 20 min.• 6 - Eliminer le surnageant et laver le culot en ajoutant 1ml d'éthanol à 70%. Laisser 5 min. à température ambiante, centrifuger 5 min, éliminer le surnageant, laisser sécher le culot, suspendre l'ADN dans du T.E.Les rendements d'extraction sont de l'ordre de 50 à 70%.
2) Par électroélution• 1 - Découper la bande d'agarose contenant le fragment comme précédemment puis le mettre dans un boudin de dialyse avec un volume de tampon d'électrophorèse suffisant (200 à 500 µl).• 2 - Fermer avec des pinces à dialyse, et déposer le boudin dans un bac à électrophorèse.• 3 - Faire migrer à 200 volts pendant 1/2 heure, inverser le courant 15 secondes, pipeter le tampon dans le boudin, rincer le boudin avec du T.E.• 4 - Précipiter l'ADN avec 0,15 M final de NaCl et 2,5 vol. d'éthanol 1/2 heure à -70°C (avec entraîneur acrylamide, cf annexe, si nécessaire).
Cf annexe pour préparation boudin de dialyse.
3) Par électroélution sur papier• 1 - Découper avec une lame de rasoir neuve une fente dans le gel, juste en dessous de la bande; y insérer un carré de papier (e.g. Whatmann 3MM) imbibé de TAE et adossé à un carré de membrane de dialyse, lui aussi imbibé de TAE (la membrane du côté de l'anode)• 2 - Remettre à migrer après avoir ôté un peu de tampon dans la cuve, afin que le gel ne soit pas submergé. Le temps de migration dépend de la concentration d'agarose; typiquement, pour un gel à 1%, il faut migrer 3 à 4 min à 100V (cuve Jewel Box).• 3 - Vérifier sous UV que tout l'ADN est rentré dans le papier. Retirer le papier et la membrane, et les introduire dans un tube Starstedt 0,5ml percé d'un trou d'aiguille (25G; 0,5mm). Introduire ce tube dans un Eppendorf 1,5ml sans bouchon.• 4 - Eluer avec 2 x 100 µl tampon d'élution, en centrifugeant 3min 5000rpm et 1min 14000rpm. Rassembler les éluats, éventuellement ajouter 10 µg ARNt, phénoler et précipiter avec 600 µl éthanol (sans rajouter de sel).• 5 - Reprendre dans 20 µl TE, et reprécipiter avec 2 µl NaAc 3M pH : 5,2 et 60 µl éthanol. Reprendre dans 10 à 100 µl TE.
24

Tampon d'élution : Acétate de sodium 0,3M pH : 7,0EDTA 1mMSDS 0,1 %
4) Par adsorption sur billes de verrea) Extraction avec JetSorbSuivre le protocole du kit, éventuellement en augmentant les temps de contact avec les billes de
verre (1 heure). Pour le rendement, le volume final d’élution est important, ainsi que le nombre de rinçages, puisque il y a un volume mort de liquide en fonction de la quantité de billes de silice utilisée (i.e. comme du sable).
b) Protocole adapté de Anal. Biochem. (1982) 121, 382-387, Marko et al.• 1 - Gel TAE ou TPE, pas TBE• 2 - Visualiser la bande sous UV ; la découper et ajouter 2,5 à 3 vol. NaI ; 55°C 3min (si la bande n'est pas complètement fondue remettre à 45°C).• 3 - Ajouter 10 µl billes de verre ; laisser à 4°C au moins une heure (ou la nuit), de préférence sous rotation pour éviter la sédimentation.• 4 - Centrifuger 15 sec. Rejeter le surnageant. Rincer 3 x 0,5ml EtOH/lavage. Après le dernier rinçage, recentrifuger 5sec pour éliminer les dernières traces d'éthanol.• 5 - Eluer 2 x 25 µl TE, 2 x 10min à 37°C.
Réactifsa) Billes de verre- Broyer en poudre la plus fine possible au mortier 20 filtres en fibre de verre Whatmann GF/A Ø47mm (1,80g). Reprendre 10 ml eau milli-Q. Centrifuger 3000rpm 10 min. Rejeter le surnageant.- Reprendre 10 ml eau m-Q et transférer dans une fiole de 50 ml. Sous la hotte, ajouter 10 ml HNO3 65%, et amener à 90°C sur une plaque chauffante. Laisser refroidir et centrifuger 5min 5000rpm (tubes Corex).- Rincer avec 30 ml eau m-Q en centrifugeant 5 min 5000rpm autant de fois que nécessaire pour amener le pH à celui de l'eau (au moins quatre fois)- Reprendre dans 3 ml de TE stérile (final : 4,5ml) 10 µl de cette préparation adsorbent de 5 à 10 µg d'ADN.
b) Iodure de sodium 6MNaI 90,8 g (dissoudre le NaI petit à petit dans l'eau, sinon il se prend en masse)Na2 SO3 1,5 geau qsp 100 ml filtrer;
ajouter : 0,5 g Na2 SO3 et garder à 4°C dans le noir.
c) EtOH/lavageEthanol 50% pour 100 ml 50 ml éthanol absolu (bouteille neuve)NaCl 100 mM 2 ml NaCl 5MTris pH 7.5 10mM 1ml Tris 1MEDTA 1mM 0.2 ml EDTA 0.5MEau 46.8ml
5) Centrifugation sur laine de verreD'après Heery et al (1990) Trends in Genetics vol.6, n°6 p173.
Après migration sur agarose, colorer le gel au BET et découper la bande intéressante dans un tube Eppendorf préalablement percé au fond et où on a disposé un peu de laine de verre siliconée sur une épaisseur de 2 à 3 mm (on rince la laine de verre au préalable avec 100 µl de TE et centrifugation
25

10 secondes). On coupe ensuite le bouchon de ce tube pour le placer dans un second tube Eppendorf et on centrifuge l'ensemble à 6000 rpm pendant 10 minutes.
Les fragments ainsi récupérés peuvent être directement marqués par Random Priming ou amplifiés par PCR, mais donnent des résultats peu reproductibles pour les clonages.
Ce protocole est clairement le plus rapide de tous, et permet le traitement en parallèle d’un grand nombre d’inserts. Cependant, son principe pénalise les grands fragments, et il est probablement déconseillé (rendements faibles) pour les fragments au delà de 4-5 kb.
6) Gradients de selPour un sous-clonage de type shot-gun notamment, il est commode de fractionner l'ADN par sa
taille sur un gradient. Cette méthode a l'avantage de ne pas faire perdre de matériel et de ne pas introduire d'inhibiteur de ligation.Solutions et matériel nécessaires :Solution 1 Acétate de potassium 5%, EDTA 2mMSolution 2 Acétate de potassium 20%, EDTA 2mMFiltrer ces solutions stock sur filtres 0.45 µm (bol inox).
Coulage du gradient :1 - Mettre 2.6 ml de solution 20% dans 'la chambre du fond' .2 - Ouvrir le robinet de communication et faire naviguer la solution de façon à supprimer l'air dans le passage.3 - Fermer le robinet.4 - Mettre 2.5 ml de solution 5% dans la chambre antérieure. Eventuellement s'il reste du liquide du gradient précédent dans le tube de sortie, faire avancer la solution dans le tube pour le chasser (ainsi que toute bulle) puis la faire retourner dans l'appareil à couler.5 - Mettre le barreau aimanté dans la chambre antérieure, placer l'ensemble sur agitateur magnétique, faire agiter le plus possible, ouvrir la connexion entre les chambres, ouvrir le robinet avant.6 - Remplir le tube polyalomère SW55 par le fond. Quand le tube est rempli, fermer le robinet (avant l'arrivée de la bulle), et retirer délicatement le tube.7 - Déposer l'ADN dans 100 µl (environ) à la surface.8 - Centrifuger 3 heures à 50000 rpm (ou 4.5 heures à 40000 rpm) à 22°C.9 - Collecter les fractions (par exemple 250 µl par fraction) en perçant à proximité du fond du tube.10 - Précipiter les fractions à l'éthanol en ajoutant de l'entraîneur (acrylamide linéaire, cf. annexes) à une concentration de 50 µg/ml.11 - Déposer un aliquote sur gel (par exemple une partie de 1 fraction sur 2).
Note : la taille de l'aiguille est importante : trop grosse, coule trop vite, contamination des fractions par le fond du tube i.e. grand fragments se manifeste par un smear de fragments hauts. Trop fine, temps de collecte élevé ou colmatage de l’aiguille.
Pour amorcer le coulage du gradient ou la collecte des fractions, on peut faire pression avec le doigt sur l'ouverture de l'appareil à gradient ou du tube. Inversement, en cas de problème, on peut stopper la collecte simplement en mettant le doigt sur le haut du tube.
7) Réparation d'extrémitésRemplissage à la Klenow dans le cas d'extrémités 5' débordantes (générées par enzymes de
restriction par exemple), remplissage à la T4 polymérase quand il y a des extrémités 3' débordantes (certains enzymes, tels que PstI, ou après sonication).
26

Exemple T4 polymérase (shot-gun) :Pour 10 µg d' ADN soniqué dans 90 µl total :
- tampon Boehringer L : 10 µl- dNTP stock 10 mM : 1 µl de chaque- T4 polymérase (Boehringer) : 10 UI
Laisser 15 minutes à température ambiante- Klenow (2.5 UI/µl, Boehringer) : 2,5 µl
Laisser 15 minutes à température ambianteEventuellement chloroforme avant de précipiter.
8) Sous clonageLes vecteurs Ready-To-Go de Pharmacia (existent en hydrolyse SmaI, EcoRI, BamHI) :
Vecteur (pUC, hydrolysé et déphosphorylé, 100 ng/tube), ligase et tampon, sont lyophilisés au fond d’un tube. Il suffit de rajouter l’insert dilué dans 20 µl d’eau.
Dans le cas où il faut hydrolyser le produit de ligature avant transformation (pour éliminer une sous population par exemple), précipiter avant l’hydrolyse (et reprécipiter avant l’électroporation). Pharmacia vend les mêmes vecteurs seuls.
Les produits PCR peuvent être sous clonés par kit Promega ou Invitrogen, qui tirent parti du rajout par la Taq polymérase classique (mais pas par les polymérases avec activité d’édition) d’un nucléotide (majoritairement un A) aux deux extrémités des produits synthétisés.
D’ailleurs, on peut en principe préalablement incuber un fragment d’ADN avec Taq et dATP afin d’utiliser ce kit.
27

PREPARATION D’ARN
1) Extraction de RNA totaux en une seule étapeAnalytical biochemistry 162, 156-159 (1987), Piotr Chomczynski and Nicoletta Sacchi.
Solution D (conservation 3 mois) Isothiocyanate de guanidium : 4 MCitrate de sodium pH : 7 : 25 mMSarkosyl : 0.5%
- Avant utilisation addition de 0.36ml de ß-mercapthoethanol 0.1M (manipulation sous hotte chimique) pour 50ml. (conservation 1 mois à T° ambiante)- Ajuster les volumes en fonction de la quantité de matériel.
100 µl à partir de 106 cellules1 ml à partir de 100 mg de tissus
PROTOCOLE POUR 10 millions (107) DE CELLULES1. Ajouter 1ml de sol. D. Homogénéiser (passer le lysat plusieurs fois dans une seringue)2. Ajouter 100 µl d’acétate de sodium 2M pH : 4. Agiter par inversion.3. Ajouter 1 ml de phénol saturé avec de l'eau (conservation 1 mois à 4°C) et 200 µl de chloroforme alcool isoamylique (49/1). Agiter 10s et refroidir sur glace pendant 15 min.4. Centrifuger 20 min à 8000rpm à 4°C.5. Transférer la phase aqueuse. Précipiter par 1ml d'isopropanol 100%. Placer à -20°C pendant 1 heure.6. Centrifuger 20 min à 13000 rpm à 4°C.7. Redissoudre dans 300 µl de sol. D.8. Précipiter par 1 vol d'isopropanol à - 20°C pendant 1 heure.9. Centrifuger. Rincer par de l'éthanol 70%.10. Séchage 15 min sous vide.11. Dissoudre dans 100 µl de SDS 0.5% à 65°C pendant 10 min.
2) Préparation d'ARN à partir de cultures cellulairesTous les tampons (sauf le Tris) sont traités par 0,1% diéthylpyrocarbonate (DEPC) la nuit puis
autoclavés 20 min à 121°C. Pour le traitement au DEPC, porter des gants en butyle ; se placer sous la sorbonne aspirante ; laisser l’aspiration en route toute la nuit. Le lendemain décontaminer éventuellement le plan de travail avec de l’eau de Javel. Les flacons de DEPC de 5 ml permettent de traiter 5 l de tampon ; une fois le flacon ouvert, l’utiliser entièrement; avant l’ouverture, percer le septum en butyle avec une aiguille pour relâcher l’éventuelle surpression due à la décomposition du DEPC. La verrerie est passée au four à 200°C 2 heures. Le plastique est autoclavé 30 min 121°C.Porter des gants en permanence.D’après Maniatis (2ème éd.) p. 7.10
1. Pour une boîte de Pétri de 10 cm ou un flacon de 75 cm2 (T75) à semi-confluence.2. Rincer deux fois avec 7 ml PBS froid.3. Lyser les cellules dans 2 ml tampon A (température ambiante).4. Gratter les cellules avec un grattoir stérile (très visqueux) ; transférer dans un tube en polypropylène stérile de 15 ml (Falcon).5. Rincer la boîte avec 2 ml de tampon B. Ajouter à l’extrait précédent.
28

6. Ajouter 4 ml de phénol (cf. infra). Agiter doucement en retournant. L’ADN chromosomique doit précipiter en un flocon blanchâtre. S’il ne précipite pas, c’est que le milieu n’est pas assez acide (pas de changement de couleur). Essayer alors de rajouter 50 µl de NaAc 2M pH : 4,0. Une fois que le flocon d’ADN est formé, on peut agiter plus fort (vortex). Agiter 2 min.7. Centrifuger 4000 rpm, 10min, 4°C.8. Ajouter au surnageant :
Tris 1M pH : 8,0 NaCl 5M Ethanol440 µl 180 µl 9 ml
9. Garder à 0°C au moins 30 min. Centrifuger à 4°C, 4000 rpm au moins 30 min.10. Reprendre dans 200 µl TE. Reprécipiter dans un tube Eppendorf 1,5 ml avec 20 µl NaAc 3M pH : 5,2 ; 600 µl éthanol.11. Resuspendre le précipité d'ARN dans 200 µl TE stérile. Dilution 1/80 et spectre. On obtient en général de 150 à 300 µg d’ARN total.(25 DO260 = 1mg/ml).12. Ajouter à l'ARN 600 µl EtOH et garder à -80°C. Au moment de l’emploi, prélever le volume désiré et ajouter 1/40 vol. NaAc 3M pH : 5,2. Centrifuger.
Solutions pour 50 ml :Tampon A
EDTA pH : 8,0 10mM 1 ml 0,5MSDS 0,5% 2,5 ml 10%
Tampon B NaAc pH : 5,2 100mM 1,6 ml 3MEDTA pH : 8,0 10mM 1 ml 0,5M
Phénol A partir de phénol (saturé en Tris 1M pH 7,5 ; •-mercaptoéthanol 1%; 8-hydroxyquinoléine 0,1%; gardé à 4°C ; plusieurs mois de conservation), préparer le jour même du phénol saturé en eau en ajoutant au phénol un demi volume d’eau et en vortexant (deux fois) ; si le pH final de l’extraction n’est pas assez acide, i.e. s’il reste trop de Tris du phénol original, l’ADN ne précipitera pas ; pour plus de sécurité, on peut saturer le phénol avec de l’eau une première fois, et avec NaAc pH : 5,2 (0,1M) la deuxième fois. Au moment de l’extraction de l’ADN, la phase phénol doit virer de l’orange vif au jaune verdâtre si les pH sont corrects.
3) Extraction ARN Phénol chaud1. Broyer les tissus dans l'azote le plus fin possible2. Transférer la poudre dans un tube contenant 2ml (ou plus) de tampon de lyse :
Na acétate pH 5 10mMNaCl 50mMSDS 0.5%
et 2ml de phénol BRL saturé avec de l'eau (le pH est acide).3. Agiter dans un bain marie à 65° pendant 5min.4. Refroidir dans la glace.5. Centrifuger 10-15 min à 3000 rpm.6. Prélever la phase aqueuse et extraire par 1 volume phénol/chloroforme.7. Centrifuger, prélever la phase aqueuse.8. Extraire par 1 volume de chloroforme.9. Précipiter avec 2 volumes d'éthanol et 200 mM de NaCl.10. Resuspendre dans l'eau.Optionnel: un inhibiteur de RNase l'aurintricarboxylic acid (ATA) peut être ajouté dans la
solution de lyse à 50 mM.
29

Note : RNASOL, de Bioprobe Systems, est également apprécié.
30

NORTHERN – GEL DENATURANT AU FORMALDEHYDE
Porter des gants. Faire tremper au préalable les cuves dans NaOH 0,1M.
1) Gel 1.2%agarose : 2,4 geau : 170 ml
Dissoudre sur plaque chauffante et sous agitation magnétique. Laisser refroidir à 50°C.Ajouter : Formaldéhyde 37% : 16,2 ml
MOPS x 10 : 20 ml
2) Tampon de migrationMOPS x 10 : 150 mlEau : 1,3 lFormaldéhyde 37% : 81 ml
3) Tampon de chargeFormamide désionisée : 1000 µlMOPS x 10 : 200 µlFormaldéhyde 37% : 400 µlBET 10 mg/ml : 8 µlBBP saturé : 100 µl
Filtrer 0,2 micronEchantillon 10 µl + tampon de charge 30 µl. Chauffer 5 min à 65°C. Glace 5 min. Migrer 100V,
2 à 4h selon la taille, en recirculant le tampon.
ATTENTION: gel fragile. Photographier.Transférer sur membrane Nylon (Hybond N+; Amersham) dans 20 mM NaOH, par transfert
capillaire la nuit.
SolutionsMOPS x 10
MOPS pH 7,0 200mM MOPS 41,8 gNa Acétate 50mM NaAc,3H2 O 6,8 gEDTA 10mM EDTA 0,5M 20 mlNaOH ajuster pH : 7,0
Eau qsp 1 litre
31

TRANSFERT DE COLONIES SUR FILTRES
(NAR 1989, vol 17, n° 1, p. 452)
1. Marquer le filtre nylon (identification, repérage), le poser sur l'agar 30 sec, repérer sa position et le retirer. On peut remettre les colonies à pousser.2. Poser le filtre sur papier Whatman imbibé de 2xSSC/5%SDS et laisser 2 minutes
ATTENTION à la précipitation du SDS en dessous de 20°C.3. Transférer les filtres sur le plateau dans un four micro-ondes et traiter pendant 2,5 min. à puissance maximum4. Mouiller les filtres pour les décoller. Une hybridation de 4 heures peut suffire avec une exposition de quelques heures. D'après les auteurs, c'est aussi applicable pour les phages.
Ne pas utiliser de nitrocellulose.On peut aussi traiter les filtres par :
Pose 2 minutes sur papier type Whatman imbibé de Soude 0,2M, SDS 1%Puis 2 minutes au micro-ondes puissance maximum.Puis membrane rincée 2 fois en TE
Application au criblage d'une banque de cosmidesOn utilise des boîtes 245x245 mm (Nunc réf. 166508). Compter 400 ml de milieu agar par
boîte, avec 50 µg ampicilline/ml. Les clones sont étalés à une concentration de quelques dizaines de milliers de clones par boîte selon l'objectif recherché. L'étalement se fait directement sur membrane (Hybond N+, colonies diluées dans 1 ml de milieu LB) et la boîte est mise à incuber à 37°C pendant 8 à 14 heures. Ensuite des répliques peuvent être obtenues par superposition d'une autre membrane (manipulations en dehors de la boîte ; repérer avec trous d'aiguille, et un trou au centre) et une incubation supplémentaire de quelques heures. Les membranes destinées à l'hybridation peuvent avoir été générées à partir de boites plastiques usagées. La première réplique, si elle doit être conservée, doit être faite à partir de boîtes neuves stériles.
32

DEPOT D’ADN SUR FILTRE DE TYPE "DOT BLOT"
Ce protocole permet de fixer de l'ADN sur membrane nitrocellulose ou nylon (genescreen, Hybond ...), sans séparation de taille, avec un appareil à filtrer (Schleicher et Schull) branché sur trompe à eau. Dans une tache circulaire de 3 mm de diamètre de Nytran chargé par exemple, il est possible de fixer jusqu'à 2 µg d'ADN. Au delà, il y a saturation de la membrane. Il n'est pas nécessaire d'utiliser de l'ADN complètement purifié : un lysat après incubation d'une nuit avec protéinase K à 37°C suffit. (Volume maxi par puit = 500 µl).
Pour les membranes nitrocellulose :1. A 50µl d’ADN (1 vol), ajouter 100 µl de NaOH 0,5 M, NaCl 1,5 M (2vol).2. 30 min à 0°C3. Ajouter 150 µl de Tris 0,5 M, NaCl 3M pH : 8,0 (1 vol)4. Imbiber la membrane dans 6xSSC5. Rincer les puits avec Tris 0,5 m, NaCl 3M pH : 8,06. Filtrer l’ADN (300 µl)7. 2 heures à – 80°C8. Hybrider comme indiqué dans le paragraphe ‘hybridation’
Pour les membranes nylon :1. Diluer l’ADN dans NaOH 0,4 M2. Imbiber la membrane de TE 1x3. Rincer les puits avec TE 1x4. Filtrer l’ADN5. Rincer les puits avec TE 1x6. Laisser sécher la membrane sur la paillasse en la protégeant dans du ‘papier filtre’7. Fixer l’ADN aux U.V. (facultatif)8. Hybrider comme indiqué dans le paragraphe ‘hybridation’
33

TRANSFERT DE SOUTHERN
1) Préparation du gelLes gels d'agarose sont réalisés à des concentrations pouvant varier de 0,4% à 1,5%. Des
concentrations de 0,8% à 1,2% conviennent bien pour les usages courants, et permettent de bons transferts de l'ADN. Le tampon d'électrophorèse est le Tris-acétate (TAE) ou TBE (voir annexe). L'électrophorèse est réalisée sans bromure d'éthidium dans le gel à un voltage de 3volts/cm environ (le voltage peut être porté à 10 volts/cm pour des migrations courtes, à condition de contrôler qu'un gradient de pH ne s'établisse pas entre les deux électrodes). Ici, l’exemple pris est la fabrication d’un blot d’ADN génomique à partir d’une migration sur le 40 cm transféré sur membrane de 30 cm, dans des conditions de migration permettant de conserver les fragments de taille allant de 450 paires de base à 25 kb. Ces conditions sont celles choisies par le laboratoire pour les projets type étude de population et retard mental. Le protocole peut être adapté à des situations plus simples (gels plus courts, quantité de matériel à détecter moindre). Dans le cas d’un ADN génomique humain, le dépôt de 5 µg d’ADN dans une loge équivaut au contenu en ADN de 1 million de cellules. Chaque fragment copie unique de 1 kilobase du génome est représenté par 1 picogramme d’ADN.
1. Gel 400ml 0,8% (3,2 g d’agarose) TBE 0,5x (TBE préparé en eau milliQ)2. Fondre le gel sur plaque chauffante dans Erlen de 1 litre3. Laisser refroidir (avec agitation, sur une plaque chauffante froide) jusqu’à 68°C (entre 55 et 60°C pour des gels de plus petite taille).4. Couler dans un moule mis à niveau, avec peigne 40 dents (orienté de façon à créer le rang de loge le plus près possible du début du gel).5. Laisser refroidir au moins une heure avant démoulage du peigne.
Il est indispensable de réaliser une recirculation du tampon pour éviter un gradient de pH et des distorsions de migration.
2) Hydrolyses des échantillons (ADN génomique) et dépôts(à titre indicatif) pour deux dépôts :
1. Hydrolyser 10 µg d'ADN dans 200 µl total (dans le cas d'échantillons de départ difficile à prélever (viscosité d'ADN génomique par exemple), il est recommandé de diluer les échantillons au préalable ou bien de mesurer la concentration au fluorimètre après hydrolyse).2. Utiliser 2,5 unité d'enzyme par µg d'ADN.3. Incuber 24 heures.4. Rajouter NaCl (Genapex) 5M de façon à ce que la concentration finale (compte-tenu du tampon d'hydrolyse) soit de 0,15 M NaCl (par exemple 6 µl de NaCl 5M pour une hydrolyse en 200 µl de tampon A de Boehringer (hydrolyses AluI, tampon sans NaCl)) et 2,5 volumes (500 µl) EtOH 100%5. Mélanger par retournement.6. Laisser 30 minutes dans la glace.7. Centrifuger 20 min. en rotor horizontal (Sigma) 12500 rpm 4°C (attention : certains tubes Eppendorf de mauvaise qualité cassent à partir de 12000 rpm, dans ce cas centrifuger à 11500).8. Eliminer le surnageant et bien égoutter (essuyer avec un papier absorbant sans toucher les culots).9. Laisser sécher sur la paillasse la nuit et reprendre dans 10 µl de TE.10. Laisser dissoudre plusieurs heures (éventuellement 2 heures à 56°C).
34

11. Vortexer, centrifuger brièvement, ajouter 2 µl de bleu (en laissant les tubes dans la centrifugeuse et déposant le bleu en haut de tube avec la P10), centrifuger et déposer 6 µl (5 µg) par piste (2 gels) avec P10.
L'ADN migre vers le pôle positif.
Dans les grandes cuves de migration :2,3 litres de tampon TBE 0,5X
Déposer 15 µl de bleu bromophénol seul très concentré dans la dernière loge.Voltage :110 VoltsTemps : 16 h 30 (par exemple de 17h30 à 10 heures le lendemain)Le milieu de la tache de BBP (bleu de bromophénol) doit être à 7,5 cm du bas du gel.
3) Transfert de l’ADN du gelEn général, l’ADN fractionné sur le gel est transféré sur un support manipulable sur lequel
l’ADN soit accessible. Pour des raisons essentiellement d’économie, les gels peuvent également être séchés, ils deviennent alors robuste et se conservent comme des feuilles. Cependant lors d’une hybridation par exemple, l’ADN piégé dans l’agarose n’est accessible qu’à de petites molécules (oligonucléotides). En outre l’argument financier perd de son importance lorsque on recycle les membranes.
Le recyclage s’effectue simplement en traitant une membrane usagée une nuit avec HCl 0,12 M, à température ambiante. Les membranes sont ensuite neutralisées au TE et conservées à 4°C jusqu’à utilisation.
a/ Par le videHabituellement, on souhaite prendre une photographie de la migration (après la migration,
colorer le gel pendant 20 min. dans une solution à 0.5 µg/ml de bromure d'éthidium). C’est le cas par exemple pour un gel d’hydrolyse de cosmides, plasmides, Pacs etc. C’est d’un moindre intérêt pour un ADN génomique car l’exposition aux UV fractionne l’ADN. Dans le cas présent (blots génomiques humains destinés à de nombreuses réhybridations) la photographie est à proscrire. De même, l’étape classique de traitement HCl (dont l’objectif est de faciliter le transfert des fragments de grande taille en les cassant en morceaux) est à bannir car elle pénalise les hybridations hétérologues nécessaires par exemple à la mise en évidence de profils multilocus avec des sondes minisatellites, sans améliorer pour autant l’hybridation avec des sondes monolocus.Préparer la cuve de transfert Pharmacia : Membrane 20x30 cm mouillée en eau milliQ.
1. Recouvrir le gel d'une solution soude 0,4 M. Laisser 1 à 2 heures en rajoutant de temps en temps du tampon. Ne pas laisser le gel sécher et ne pas laisser de soude autour du gel.2. Couper le gel à 8 cm du bord supérieur et mettre le gel en place directement.Positionner la partie supérieure du blotteur avec ses clips, mettre la pompe en route, le vide doit
s’établir, le fixer à 50 (après quelques minutes, vérifier par pincement du tuyau entre blotteur et bouteille qu’il n’y a pas de fuite de vide (l’aiguille ne doit pratiquement pas bouger). Souvent la fuite vient du blotteur lui-même, utiliser une pince de potence ; répéter ce contrôle du vide de temps en temps).
3. Recouvrir le gel d'une solution soude 0,4 M. Laisser 1 à 2 heures en rajoutant de temps en temps du tampon. Ne pas laisser le gel sécher et ne pas laisser de soude autour du gel.Essuyer la soude, enlever le gel et marquer la membrane (au crayon, côté face portant l’ADN)4. Rincer la membrane dans du 2xSSC puis du TE.5. Fixer aux U.V. avec le stratalinker : poser la membrane dans l’appareil face ADN vers le haut (lampes UV) bien à plat. Fermer la porte, appuyer sur autocrosslink, puis start. Le temps
35

d’exposition réglé automatiquement et mesuré par la petite cellule noire au fond de l’appareil est de l’ordre de 30 secondes.
NOTE : en principe, la fixation UV est inutile pour une membrane chargée. Elle ne semble pas gênante.
6. Conserver la membrane au sec à température ambiante jusqu’à la première hybridation, protégée par du papier filtre. Avant la première hybridation plonger la membrane dans un bain de soude et de TE.
b/ Sans videCeci est l’ancienne méthode plus traditionnelle, qui peut parfois présenter un intérêt (transfert
sur membranes non chargées par exemple)
1. Après la migration, colorer le gel pendant 20 min avec agitation dans une solution à 0,5 µg/ml de bromure d'éthidium puis le photographier.
Note : Pour faciliter le cas échéant le transfert des fragments de haut poids moléculaire, immerger le gel pendant 10 min. avec agitation dans une solution 0,12 M d'acide chlorhydrique.
2. Transférer le gel dans un bain dénaturant de soude 0,5 M, chlorure de sodium 1,5 M, pendant deux fois 20 min. avec agitation.3. Neutraliser le gel en deux fois 20 min. avec agitation dans un bain de Tris NaCl (Tris-HCl pH8, NaCl 1.5 M).4. Réaliser un pont au dessus d'un bac contenant une solution de 10 à 20xSSC (voir annexe) , à l'aide d'une plaque de verre. Couper une feuille de papier 3MM chevauchant la plaque de verre et baignant des deux côtés dans la solution de SSC. Bien imbiber la feuille de SSC, éliminer les bulles, déposer le gel, le recouvrir de la membrane préalablement trempée dans l'eau, couvrir la membrane d'une feuille de papier 3MM puis de papier absorbant et d'un poids (environ 0,5 kg pour un gel à 1% de 15 x 20 cm).Laisser transférer pendant la nuit à température ambiante.
Note : il faut s'assurer qu'il n'y a pas de communication directe entre le papier absorbant au-dessus du gel et le pont au dessous. Le cas échéant, isoler avec du parafilm ou du papier aluminium. Après une ou deux heures de transfert, il peut être utile de remplacer le papier absorbant déjà mouillé.
5. Après le transfert, enlever le papier absorbant, marquer le filtre( numéro, face portant l'ADN, orientation des dépôts) avec un marqueur noir à l'encre indélébile, rincer le filtre dans une solution de 2xSSC.6. Fixer l'ADN aux ultra-violets (254 nm) pendant une minute à 10 cm de la lampe (selon le type de lampe utilisée : le signal peut décroître très rapidement, il est donc nécessaire de déterminer empiriquement le temps d'exposition minimum).7. Rincer le filtre dans du TE avant de le laisser sécher sur du papier absorbant.
36

MARQUAGE RADIOACTIF D'ADN
Le marquage est souvent réalisé par initiation au hasard et synthèse avec la Klenow pour les marquages de routine. L'ADN est porté à ébullition pendant 5 minutes afin de le dénaturer avant le marquage, puis placé dans de la glace pour éviter sa renaturation. 10 microCi (1microl.) et 20 ng d'ADN permettent d'obtenir une sonde de l'ordre de 20.106 cpm . La séparation des nucléotides non incorporés se fait sur colonne Sephadex G75 et le comptage par dépôt de 1/100 ou 1/200 de la sonde sur filtre et estimation par compteur Geiger. Avant l’utilisation de la sonde, il est nécessaire de dénaturer une nouvelle fois l’ADN marqué (Ebullition + glace).
1) Préparation d'un kit de marquage par 'random priming':Il est très facile de les constituer soi-même à partir de Klenow (Boehringer, labelling grade ou
Euromedex MBI), d'oligonucléotides (Pharmacia Ref 27.2166 01),.Resuspendre les oligonucléotides dans 100 µl de T.E.
Mix x10 :pour 1 ml (permet de faire 10 kits)
Tris-HCl pH 7.2 0,5 M 500 µl stock 1MMgCl2 0,1 M 100 µl stock 1MDithioerythritol 1 mM 1 µl stock 1MBSA 2mg/ml 100 µl stock 20 mg/mlH2O 200 µlOligonucléotides 100 µl
100 µl de mix complet correspondant à 50 réactions faites dans 20 µl.Aliquotes de 100 µl, MixRP x10.
Nucléotides : pour marquage avec déoxycytidine:Faire un mélange 'GAT'
dATP 100 mM 1,5 µldTTP 100 mM 1,5 µldGTP 100 mM 1,5 µlH2O 900 µl
5 aliquotes de 180 µl, GAT
Marqueur de taille : faire une dilution à 100 pg/µl de 1kb ladder et 1 ng/µl de lambda HindIII.
Le kit est alors constitué de un tube de mix (100 µl), un tube GAT (180 µl), un tube de marqueur de taille, et un tube de Klenow à 2 u/µl.
2) Protocole de marquageLa réaction est faite dans 20 µl final (ou 10 µl, diviser par 2), avec l'ADN à marquer, 1 µl de
marqueur, 2 µl de mixRP, 3 µl de 'GAT', 1 ou 2 µl de [32P]dCTP (10-20 µCi), 1 µl de Klenow (2 u), et H2O.
1. Mélanger l'ADN, le marqueur et l'eau et faire bouillir 5 minutes au bain marie.2. Mettre dans la glace et centrifuger 5 secondes.3. Ajouter 2 µl Mix x10, 3 µl nucléotides GAT, 2µl radioactivité, 1 µl Klenow.4. Incuber 30 minutes (minimum ; 2 heures si possible) à 37°C dans le petit bain-marie derrière l’écran (vérifier la température).5. Passer sur colonne G75 pour éliminer les nucléotides non incorporés.6. Compter 3 µl de sonde déposée sur un papier filtre avec le compteur Geiger.
37

7. Faire bouillir la sonde 5 minutes au bain-marie avant l'hybridation.
3) Sondes d'extrémités de cosmidesPour cosmides en pWE15 ou Supercos1, utiliser les primers T3 et T7. Pour Lawrist, utiliser les
primers PL1 et PL2.La réaction se compose de :
ADN cosmide environ 500 à 1µgun primer 10 µM : 1 µlMix (Jeffreys)x10 : 2 µlGAT (idem kit RP) : 2 µlC 15µM : 1 µlTaq (type eurobio) : 0.5 µl ou moinsH2O : qsp 20µldCTP 32P : 3µl
PL1 ATACGACTCACTATAGGGAG Tm 58°CPL2 ACATACGATTTAGGTGACAC Tm 56°C
Le rendement de marquage (pour cette amplification linéaire) est linéairement proportionnel au nombre de cycles et à la quantité de matrice de départ.Programme indicatif : 94°C 20 secondes, 55°C 30 secondes 72°C 20 secondes ou moins.Finir à 10°C ou se débrouiller pour ne pas laisser un allongement long se faire en fin de cycle, car on veut des extensions très courtes (100-200 nucléotides).
L'amplification se fait en pièce radioactive, tubes 0.5 ml, sous huile. Mettre un plomb ou couvercle plexiglas sur le tube.
Après l'amplification, récupérer la sonde en rajoutant 130 µl de TE, et pipetant 150 µl. Il n'est pas gênant de prendre un peu d'huile en même temps. Passer sur colonne G75 et compter comme pour une sonde RP.
Il n'est pas nécessaire de faire bouillir ensuite puisque la sonde est simple brin.
4) Elimination des nucléotides non incorporés sur colonne G75Préparation de la colonne
1. Faire un stock de confettis à partir de filtres fibre de verre de type GF/C, à l'aide d'une perforeuse (prendre le classeur dédié en pièce radioactivité)2. Prendre une seringue 1ml (type insuline) et retirer le piston, et introduire au fond de la seringue un confetti GF/C, à l'aide du piston par exemple.3. Mettre la colonne sur portoir et la remplir de G75 (Pharmacia, préparée comme recommandé en TE). La colonne doit être remplie de G75 jusqu’à 1 ml (graduation), laisser le TE s’écouler.4. Déposer la sonde dans 300 µl de TE (par commodité, la reprendre dans 150 µl final, déposer, et rincer le tube avec 150 µl de TE).5. Laisser le TE s’écouler. Mettre un tube Eppendorf à bouchon à vis sous la colonne afin de réceptionner la sonde radioactive.6. Recharger 300 µl de TE (en deux fois 150 µl).La sonde passe avec les 300 µl.Laisser le TE s’écouler puis jeter la colonne, avec nucléotides non incorporés, dans la poubelle à déchets [32P].Comptage1. Déposer 1/100 de la sonde, soit 3 µl sur un papier absorbant (type benchcoat)2. Poser le papier sur le parafilm sur la sonde du compteur Geiger.3. 100 au compteur = 1.5 millions dpm dans la sonde. Par exemple, si 1/100 de la sonde donne 300 au compteur, on a 4.5 millions de dpm au total.
38

HYBRIDATIONS(Southern et Northern)
La sonde radioactive, dénaturée à la chaleur lorsque elle est double brin, est utilisée à une concentration finale de l'ordre de 106 cpm/ml (105 cpm/ml pour les minisatellites). Avant toute utilisation, les filtres sont déhybridés à la soude 0.4 N deux fois 20 min., puis neutralisés en T.E. Ils sont ensuite mis en tubes à hybridation deux par deux dos à dos et entre deux drains (sinon la sonde radioactive ne passera pas entre les deux filtres). Il est nécessaire de préincuber les filtres au moins 15 minutes avant d'ajouter la sonde (préhybridation). Pour la préparation du tampon de préhybridation et d'hybridation voir "Préparation des solvants et milieux" (ADN: 2% SDS; ARN: 5% SDS).
Pour un tube, prévoir entre 3ml (un filtre) 20 ml (8 à 10 filtres 20x30 dans tubes 35 cm) et 30 ml (30 ou 40 filtres taille Eurogem) de tampon .
Les températures d'hybridation, préhybridation, lavages sont fonctions des stringences cherchées.
La formule empirique permettant d'estimer la stabilité d'un duplex ADN:ADN en fonction de la longueur (L) des fragments, la composition en nucléotides, la concentration en ions Na+ (en M) et formamide (en %) du milieu d'hybridation est la suivante :
Tm = 81.5 + 0.41 (%G+C) + 16.6 log (Na+)- 0.63 (%form) - (300 + 2000 (Na+))/LTemps de préhybridation : 15 min. minimumTemps d'hybridation : la nuit
Pour des séquences parfaitement homologues, l'hybridation se fait à 65°C.Conditions stringentes de lavage: 68°C- 0,1 SSC (0,1% SDS)Les filtres sont ensuite mis en sac plastique soudable, et exposés avec un film Kodak XAR5 ou
Fuji RX avec écrans amplificateurs (nécessite alors une exposition à -70°C) ou sans écran amplificateur.
1) Hybridation dans un fourCette méthode apporte une nette amélioration par rapport à l'hybridation en sac, dès lors que les
lavages sont effectués classiquement dans un bain-marie.Les membranes sont interposées entre des drains, qui sont nécessaires à la bonne circulation du
tampon d'hybridation, l'ensemble est ensuite enroulé sur lui-même et disposé dans le tube.Il n'est pas nécessaire de renouveler le tampon après la préhybridation.
Dans certains cas, les sondes utilisées contiennent des séquences répétées en plus de séquences uniques. Il est alors nécessaire (et pas toujours suffisant), lors d'une hybridation sur filtre d'ADN génomique humain, par exemple, de neutraliser les séquences répétées. Une méthode utilisée consiste à ajouter un large excès (2000 fois) d'ADN génomique total soniqué. (protocole décrit par Blonden et al., dans NAR vol. 17, n° 14, p. 5611-562).
Deux choses changent par rapport au protocole habituel :1- Après marquage de la sonde, de l'ADN total est ajouté, le mélange est dénaturé puis préincubé à 65°C avant l'hybridation.2- Le tampon d'hybridation peut être différent.
2) Renaturation de la sonde avec l'ADN totalAprès marquage et séparation sur sephadex G 75, ajouter 100 µg d'ADN de placenta (Sigma;
soniqué à une taille de 500-800 pb ou cassé par autoclavage 110°C, 10 minutes) en 6xSSC final, dénaturer 10 min. dans l'eau bouillante et laisser 1h30 min. à 65°C.
39

Le tampon d'hybridation est par exemple 0,125 M Na2HPO4 (pH : 7,2), 0,25 M NaCl, 1 mM EDTA, 2% SDS, 10% PEG 6000.
3) DéhybridationA faire juste avant l'hybridation. Les membranes se conservent mieux à 4°C en sac d'exposition
soudé, le SDS de la solution de lavage empêche le développement de champignons.1 ou 2 bains NaOH 0,4 N (20 minutes)2 ou trois bains TE 10:1 pH : 8 ou 7.5 (20 minutes)
Changer les membranes de bain en les transférant une par une. En particulier dans le cas de membranes neuves, éviter que des membranes se touchent sous peine de "décalcomanies".
Dans le cas de signaux récalcitrants sur blots de type hydrolyses de plasmides on peut faire le bains de soude à 42°C, ou plonger la membrane dans SDS 0.5% 100°C et laisser refroidir, puis 2 bains de TE. Ce traitement endommage la membrane. Pour un blot précieux, il sera préférable d’attendre que la radioactivité fixée se soit atténuée (qq semaines ou qq mois).
40

REACTIONS DE SEQUENCE
Préparation de matrices à partir de prep. rapides de plasmide :Référence.: Wong et al., NAR (1990) 18,5573.
1. Ajouter 15 µl de soude 0,4 N à 15 µl de préparation rapide. Incuber à température ambiante 5 min.2. Ajouter 120 µl d'éthanol froid, puis 15 µl d'acétate de potassium 1M pH : 5.3. Mélanger et laisser 5 min à température ambiante.4. Centrifuger 20 min. à 4°C et sécher le culot5. Ajouter 10 ng de primer puis le tampon d'annealing et incuber 45 min (et plus) à 37°C.6. Procéder comme indiqué dans les kits de séquençage, USB ou Pharmacia.
Le protocole et les réactifs proposés dans les kits du type"Sequanase" (néanmoins tendance à donner des "stop") ou pharmacia sont simples d'usage et donnent de bons résultats. C'est un aménagement du protocole initialement décrit par Sanger qui utilisait comme DNA polymérase l'enzyme Klenow. Le kit "séquanase" utilise la DNA polymérase de T7.
Les réactions finales peuvent être effectuées dans des plaques à micropuits (non traitées culture de cellules) maintenues à 37°C sur un bloc chauffant.
En fin de réaction et après 10 min. de dénaturation à 65°C en tampon formamide, les réactions sont dans un volume de 3 µl.
Modifications : - l'annealing peut être fait en incubant les tubes eppendorf 5 min à l'étuve à 65°C sur les portoirs gris puis 30 min sur la paillasse à température ambiante. C'est recommandé pour la séquence double brin et marche très bien pour le M13.
- on peut utiliser la moitié des volumes indiqués dans le kit sequanase.Actuellement, la technique préférée est :
-marquage du primer en 5' par la polynucléotide kinase et gammaATP p33-kit delta-Taq ou équivalent.
Marquage de primer en 5’ par la polynucléotide kinase et [32P] gamma ATP :Par matrice il faut prévoir 0,5 pmole de primer marqué.Pour marquer 10 pmoles :
Tampon x10 : 2 µlPrimer (si 10µM, i.e. 10pM/µl) : : 1 µlEnzyme T4 polynucléotide kinase : 1 µlGamma ATP[33P] : 3 µlH2O : 13 µlVolume total : 20 µl
L’enzyme Biolabs convient bien. Cette réaction est robuste, certains la font même à 95°C en quelques minutes. On peut vérifier le marquage par chromatographie élémentaire sur une chute de membrane nylon (bandelette 3x10 cm) comme suit :
Déposer un dot de bleu + 0,2 µl de réaction à par exemple 2cm d’un bout de la bandelette.Faire migrer en trempant le bas de la membrane dans soude 0,4N (bandelette posée sur plaque
inclinée)Compter sur compteur Geiger migré et non migré.Penser à inclure témoin gamma ATP.
41

Reconstitution d’un kit type delta taq pour séquençage manuel :Ce kit est bien adapté au séquençage manuel quand on a peu de matériel (quelques dizaines de
nanogrammes de produit PCR, ou séquençage direct sur cosmide par exemple).Sa reconstitution nécessite une polymérase spéciale (delta taq) commercialisée par Bioprobe
systems sous le nom de biotaq (conditionnement à 30 unités/µl).
Ensuite le kit se compose de tubes :1-Tampon (Reaction buffer ) 300 µl par kit pour 1 ml :
Tris HCl pH : 9,5 260 mM 2 60 µl de stock 1MMgCl2 65 mM 65 µl de stock 1MH2O 685 µl
2-Enzyme dilution buffer (200µl par kit) pour 1 ml :Tris HCl pH : 8 10mM 10 µl stock 1MBéta mercaptoéthanol 1mM 1µl de dilution1/14Tween 20 0.5% 50 µlNonidet P40 0.5% 50 µlH2O 890 µl
3-Termination mixes (500 µl par kit) :Mix dNTP* ddNTP (5mM) H2O
ddG 150 µl 5 µl 845 µlddA 150 µl 60 µl 790 µlddT 150 µl 90 µl 760 µlddC 150 µl 30 µl 820 µlmix dNTP* : 1 µl de chaque dNTP 100 mM dans 1 ml total
42

SEQUENCAGE DE PRODUITS DE PCR
1) Obtention de la matrice simple brinLe fragment d'ADN double brin amplifié par PCR doit être purifié pour éliminer les dNTP, les
amorces, ...Ceci est réalisé après migration sur un gel d'agarose NuSieve fait et migré dans le tampon TA
(Tris-HCl 40 mM pH : 7,4, acétate de sodium 30 mM). Le gel est coulé avec au préalable 1 µg/ml de BET (car le NuSieve se colore mal au BET).
A titre d'exemples :un fragment de 300-400 pb peut être déposé sur un gel 2%un fragment de 100-150 pb peut être déposé sur un gel 4%
Après migration, la bande d'agarose contenant le fragment d'ADN (100 ng ou plus) est découpée sous UV. On ajoute 100 µl à 200 µl d'eau (selon le pourcentage d'agarose du gel) et on fait fondre l'agarose à 65°C pendant au moins 10 min, en vortexant de temps en temps.
1 µl de cette solution d'ADN peut servir de matrice pour une PCR asymétrique. Cette PCR simple brin se déroule dans les mêmes conditions que la première amplification, exception faite pour la concentration d'un des primers dit "primer limitant". Ce primer doit être dilué dans des proportions allant de 1/30ème au 1/100ème (conditions à tester selon les primers) par rapport à la PCR double brin.
Une fraction aliquote (1/10) de la PCR asymétrique est analysée par électrophorèse sur gel d'agarose 2% (pour un fragment de 300 pb) ou sur gel d'acrylamide 6% (pour un fragment de 150 pb) afin de visualiser la quantité et la qualité de l'ADN amplifié. Le simple brin donne une bande de taille apparemment inférieure au double brin. Si l'amplification semble correcte, l'ADN simple brin doit ensuite être purifié par microcon.
Le type de microcon est choisi en fonction de la taille de l'ADN à purifier :Taille supérieure à 300 bases : centricon 100Taille inférieure à 300 bases : centricon 30.
Se conformer aux instructions du fabricant. Bien laver (au moins trois fois avec 400 µl d'eau).L'ADN est prêt à être séquencé.
2) Préparation des gels de séquenceLes gels avec échantillons marqués au soufre [35S] ou [33P] doivent être séchés avant d'être
exposés.
a) Prétraitement des plaques de verreNettoyer chacune des plaques avec une solution de SDS 1% en imbibant un kleenex et en
frottant vigoureusement. Opérer assez vite de façon à ce que le SDS ne sèche pas sur la plaque. Sécher la plaque avec des kleenex propres. On peut éventuellement optimiser le séchage en utilisant de l'alcool.
Après chaque utilisation, bien nettoyer les plaques à l'eau courante, en éliminant toute trace d'acrylamide résiduelle avec une éponge douce.
Avec des plaques suffisamment neuves, et surtout non rayées, il n'est pas nécessaire de siliconer les faces. En cas de problèmes de démoulages, et/ou de coulage, siliconer une ou les deux faces, avec un produit type Sigma coat ou analogue.
43

Exemple : Repelcoat /Dichlordiméthylsilane. Etaler le produit à la main (gantée), laisser sécher la plaque sous hotte et laver une fois avec de l'éthanol et des Kleenex, repasser une couche de silicone et laver 2-3 fois à l'éthanol. Cette plaque peut-être utilisée pour 2 ou 3 gels.
Repérer la face siliconée.
Note : il est sans doute moins coûteux et moins toxique de racheter un jeu de plaques (et de faire attention à ne pas le rayer avec des détergents ou éponges abrasives).
b) Solutions5x TBE (pour 10 litres) :
Tris base : 605 gAcide borique : 275 gNa-EDTA : 37 g
Ajuster à pH : 8.3 avec de l'acide borique
Acrylamide 38:2 pour 100 mlAcrylamide (Biorad) : 38 gBis-Acrylamide (Biorad) : 2 g
Conserver le TBE en bouteilles de verre, et surveiller que l'acide borique n'a pas cristallisé.L'acrylamide se conserve très bien à 4°C dans le noir. Manipuler avec des gants.Solutions d'acrylamides déjà faites : en général stock 40%, en proportion 29:1 acryl./bisacryl.
c) Gels gradient (33 x 40cm)L'intérêt du gel gradient est de ralentir la migration des petits fragments d'ADN en fin de
migration et ainsi de pouvoir lire plus de bases pour une même longueur de gel. Le principe du gradient est de diminuer le voltage dans la partie basse du gel, ce qui ralentis la migration. Cette baisse de voltage est réalisée par une augmentation de la concentration saline dans la partie basse (baisse de la résistance).
1) Gel gradient à 6%- pour 100 ml de solution à forte concentration saline (2.5 x TBE) (bas de gel) :
Urée 48 gSucrose 5 gBleu de bromophénol 5 mg10x TBE 25 mlAcrylamide 38:2 15 mleau 19 ml
Filtrer à travers un papier filtre. Cette solution peut être gardée à 4°C dans le noir pendant environ 10 jours. Dégazer avant usage.
- pour 200 ml de solution à faible concentration saline (0.5 x TBE) :Urée 96 g10 x TBE 10 mlAcrylamide 29:1 30 mlEau 88 ml
Filtrer et dégazer avant usage.
Couler les gels un par un :
44

12 ml mélange 2.5 x TBE24 µl TEMED24 µl 10% persulfate d'ammonium (frais)50 ml mélange 0.5 x TBE100 µl TEMED100 µl 10% persulfate d'ammonium
(conservation de15 jours environ)
L'acrylamide prend en 5 min. environ si les solutions sont à température ambiante.Mettre les solutions dans la glace quelques minutes avant d'ajouter Temed et Persulfate permet
de ralentir la polymérisation (mais les solutions trop froides risquent de cristalliser).Utiliser une pipette de 25 ml avec une "propipette", aspirer 10 ml de mélange incolore 0.5 x
TBE, puis les 12 ml de mélange bleu à 2.5 x TBE et établir un gradient en laissant monter quelques bulles d'air. Verser le gel entre les plaques légèrement inclinées, en commençant par le coté. Finir avec le reste de mélange 0.5 x TBE en versant par le milieu et en contrôlant que des bulles ne se forment pas. Introduire le peigne, le bloquer avec une pince à dessin. Laisser le gel polymériser à l'horizontale pendant au moins 30 min. Dès l'enlèvement du peigne, penser à bien rincer les loges une fois avec du tampon.
2) Simulation d'un gradientPar cette technique, le gradient se forme au cours de la migration et permet une meilleure
séparation des grands fragments. Mettre 150 ml d'une solution d'acétate de sodium 3M dans 350 ml de tampon TBE 1x, dans la cuve du bas. Cette solution sert de tampon de migration au niveau de la cathode (pôle négatif). Pour lire au delà de 400 pb, n'ajouter l'acétate de sodium qu’après 2 à 3 heures de migration. Penser à surveiller la température des plaques, baisser la puissance si nécessaire. La formation du gradient provoque une augmentation de l'ampérage en cours de migration, il faut alors réguler l'intensité débitée par le générateur.
d) Gels non gradientPour 500 ml de solution gel 6% (5 gels 30 x 40 cm) :
Urée : 225 gTBE 5x : 100 mlAcrylamide 40% : 75 mlEau qsp500 : 145 ml
- Pour couler le gel, il est pratique d'écarter légèrement les plaques avec une pointe de spatule, et d'utiliser une grosse seringue 50 ml.Pour un gel de 60 ml : APS 10% : 450 µl, TEMED : 45 µl- Pratique de couler le gel horizontal (pas de spacer en bas)
e) ElectrophorèseLe tampon d'électrophorèse est 0.5 x TBE en haut, 1xTBE en bas. La migration se fait en
limitant à 60 Watts. Préchauffer le gel en le mettant sous tension 15-60 min. avant de déposer les échantillons.
Déposer les échantillons juste après leur dénaturation à 65°C, avec un pipetman P20 et un cône spécial effilé.
Après la migration (environ 2 heures 30, 15 min. après la sortie du premier bleu), séparer les deux plaques.
45

Transférer le gel sur un papier type Whatmann, couvrir la face avec une feuille de Saran Wrap et sécher sur le sécheur : température maximale. Le séchage doit durer de l'ordre de 20 à 30 minutes. Le gel ne doit plus être collant. Ne pas laisser trop longtemps sous risque d'aspiration du signal (le cas échéant laisser sécher sur la paillasse).
46

ANNEXES
47

Préparation des tampons et solvants
SOLVANTSPhénol : Il est liquéfié à 65°C, bouchon débloqué, puis saturé et neutralisé tout d'abord avec une solution Tris pH 8 1 M (mélanger, laisser reposer, éliminer la phase aqueuse, recommencer une fois). Pour finir, remplacer la phase aqueuse par une solution Tris pH8, 10 mM, EDTA 1 mM (T.E.). Vérifier que le pH obtenu pour la phase aqueuse est supérieur à 7.6.Conserver le phénol dans des bouteilles de verre marron, à 4°C. Il est commode de colorer la phase phénolique (utiliser 0.1% de 8-hydroxyquinoline). Ce colorant jaune est un antioxydant et inhibe partiellement les RNases. Toujours manipuler le phénol sous hotte chimique avec des gants et des lunettes (en cas de contact avec la peau, laver abondamment à l'eau puis rincer à l'éthanol).Remarque : un grand nombre de fournisseurs vendent désormais le phénol déjà saturé.
Chloroforme-isoamylalcool : cette solution est obtenue directement par mélange de 24 volumes de chloroforme avec 1 volume d'isoamylalcool. Elle est conservée en bouteille fermée à température ambiante.
Butanol (1 ou 2) : le butanol peut être utilisé tel quel pour réduire les volumes de phase aqueuse (concentrer une solution d'ADN). Lorsqu'il est utilisé pour extraire le bromure d'éthidium d'une solution de chlorure de césium, il faut éviter de concentrer les sels. Pour ceci le butanol est préalablement saturé avec une solution de T.E. La solution saturée est conservée à température ambiante (armoire située sur le pallier).
SOLUTIONS COURANTES (eau ultrapure)Tris Acétate (TAE) 50 x:
Tris base 242 gAcide acétique 57.1 mlEDTA 0.5 M 100 mlH2O qsp 1 litre
Tris-Borate (TBE) 5 x: Tris base 540 gAcide borique 275 gEDTA Na2 37 gH20 qsp 10 litre
Donne pH 8.3 (sinon ajuster avec de l'acide borique)
Tris-Phosphate (TPE) 10 x: Tris base 108 gAcide phosphorique 15,5 mlNa2EDTA 7,4 gH2O qsp 1 litre
20 x SSC: NaCl 174 gCitrate de sodium 88.2 g
48

H20 800 mlAjuster le pH à 7.0 avec de la soude puis compléter le volume à 1 litre.
Tampon de dépôt d'ADN (10x) : d'après Sambrook et al.0.25% bleu de bromophénol0.25% xylène cyanol50% glycérolComplément en TE
Pour 20 ml :Dissoudre 50 mg de xylène et 50 mg de bleu de bromophénol dans 10 ml de TE et compléter à
20 ml avec du glycérol (en versant directement)
20% SDS Sodium Dodécyl Sulfate 400 gH20 1 litre
Chauffer à 68°CH20 qsp 2 litres
Manipuler sous hotte !En pratique :Flacon de 250 g, ajouter 1,025 l eau milliQ préchauffée, laisser dissoudre avec barreau magnétique et agitation la nuit.
Acétate d'ammonium 7,5 M Acétate d'ammonium 289 gH20 qsp 500 ml
Filtrer à la pompe à videPH : 7.5EDTA 0.5 M pH 8
EDTA di Sodium 186.1 gH20 800 ml
Agiter fortement la solution (ne se dissous qu'à partir de pH8)Ajuster le pH à 8 avec NaOH puis compléter le volume à 1 litre.
MgCl2 1M MgCl2, 6 H20 20,33 gH2O qsp 100 ml
Autoclaver la solution 20 min. à 120°C
MgSO4 1M MgSO4, 7H2O 24,6 gH2O qsp 100ml
Autoclaver la solution 20 min. à 120°C
Glucose 2M 36g/100 ml
Filtrer 0.22 µM
NaCl 5M NaCl (Genapex) 292.2 g
49

H20 qsp 1 litreAutoclaver la solution 20 min. à 120°C
Tris-NaCl pH 7.5 Tris 121.1 gNaCl 87 gH20 600 ml
Ajuster le pH à 7.5 puis compléter le volume à 1 litre.Autoclaver la solution 20 min à 120°C.
Tampon Ficoll pour Southern génomiqueEntraineur acrylamide
Plusieurs types d'entraîneurs existent, les plus connus sont le glycogène, dextran T40, et acrylamide. Ce dernier est le moins coûteux. Il est constitué par polymérisation d'acrylamide sans le bisacrylamide.
Solution d'acrylamide 5%Rajouter TEMED et APS (ammonium persulfate) comme pour un gel d'acrylamide
Exemple :0,5 g acrylamide ultrapure10 ml H2O20 µl temed20 µl APS 10%
Laisser polymériser.L'entraîneur s'utilise à concentration de l'ordre de 50 µg/ml de phase aqueuse à précipiter (ou
qté suffisante pour voir un culot). Le stock 5% est donc 1000x concentré.
SOLUTIONS A B C ET RNase POUR EXTRACTION DE PLASMIDES ET COSMIDESSolution A
Glucose 50mM 4,5gEDTA 10mM 10ml 0,5MTris-HCl pH8 25mM 6,25ml 2MH2O qsp 50ml.
Filtrer 0,2micron
Solution B (à faire le jour même) NaOH 0,2M 1ml 10MSDS 1% 2.5ml 20%H2O qsp 50ml
(Pour obtenir 10 ml de solution B, ajouter 100 microl. de soude 10M et 250 microl. de SDS 20% à 9.5 ml deT.E.)
Solution C Acétate de potassium (5M final) 150gH2O 167 mlAcide acétique qsp 500ml
50

Solution finale pH 4.8
RNAse A Pour 1ml, peser 10 mg de RNAse A (10 mg/ml). Faire bouillir 10 min au bain-marie
Tampons de préhybridation et d’hybridationCHURCH (d'après Church et Gilbert , PNAS 1984, vol. 81, p.1991-1995)
2% SDS (ADN) ou 5% SDS (ARN)0.45 M Na2HPO4 pH 7,20.5% lait écrémé1 mM EDTA
Pour 1 litre: SDS 20% 100 ml ou 250 mlNa2HPO4 1M 450 mlLait 5 g (le dissoudre préalablement dans de l’eau)EDTA O.5M 2 mlH20 qsp 1 litre
BLONDEN (NAR volume 17 n°14)Na2HPO4 0.125MNaCL 0.25MSDS 2%PEG 6000 10%EDTA 1mM
Pour 1 litre: Na2HPO4 1M 125 mlNaCL 5M 50 mlSDS 20% 100 mlPEG 6000 100 gEDTA 1M 2 mlH2O qsp 1 litre
Solution de Na2HP04 (pH7,3 1M)Pour 1 litre : Na2HPO4, 12 H2O 358 gDissoudre dans 800 ml d'eau en chauffantPuis amener à pH : 7,3 avec de l'acide phosphorique (environ 10 ml)Puis qsp 1 litre avec H2O
Milieux de culture pour bactéries ou levuresMilieu minimum M9 glucose avec thiamine:
Ce milieu permet la propagation de bactéries TG1 (ou JM101) sans perte du pilus sexuel nécessaire à l'infection par M13.
Na2HPO4, 6H20 14,3 g (si 2 H2O, 10.2 g)KH2PO4 3 gNaCl (Genapex) 0.5 g
51

NH4Cl 1 gAgarose 15 gH2O qsp 1 litre
Ajuster le pH à 7.4, passer à l'autoclave (15 minutes à 110°C), laisser refroidir, et ajouter :MgSO4 1M 2 mlGlucose 20% 10 mlCaCl2 1 M 0.1 mlThiamine 0.01% (vitamine B1)
(Solutions stériles)
Milieu riche (LB):Tryptone 10 gExtrait de levure 5 gNaCl (Genapex) 10 gH2O 800 ml
Ajuster le pH à 7.5 avec de la soude et compléter le volume à 1 litreAutoclaver 30 min à 120°C
Milieu "super" (MLS ou TB) :Pour une préparation de 2 litresSolution I
Tryptone 24gYeast extract 48gGlycerol 10 mlH2O qsp 1800 ml
Solution IIK2HPO4 25gKH2PO4 4,6 gH2O qsp 200 mlStériliser à l'autoclave dans des bouteilles séparées. Pour un flacon contenant 900 ml de solution
I en faire un contenant 100 ml de solution II, etc… On obtient alors par exemple 500 ml de milieu en mélangeant une bouteille de 450 ml de solution I et une de 50 ml de solution II.
Pour couler des boites de Petri, ajouter à ces milieux, avant de passer à l'autoclave, 16 g/l d'agar. Pour de la gélose molle (étalement de phage M13), n'utiliser que 7 g/l d'agar.
Milieu SOBBacto-tryptone 20 gExtrait de levure 5 gNaCl 5M (Genapex) 2 mlKCl 1M 2.5 mlH2O qsp 800 ml
Ajuster à un pH : 6.9Autoclaver la solution 30 min à 120°C, puis ajouter:
MgCl2 1M 10 mlMgSO4 1M 10 mlH2O qsp 1 litre
Ces solutions sont stériles
52

Milieu SOCTryptone 4 gExtrait de levure 1gNaCl 5M 0.1 mlH2O 160 mlKCl 250 mM 2 mlNaOH pour pH : 7H2O qsp 200 ml
Milieu YTBacto- tryptone 8 gExtrait de levure 5 gNaCl 5 gH2O qsp 1 litre
Autoclaver 20 min à 120°C
Milieu 2YTBacto- tryptone 16 gExtrait de levure 10 gNaCl 5 gH2O qsp 1 litre
Autoclaver 20 min à 120°C
Gélose MGMBacto-tryptone 10 gExtrait de levure 5 gNaCl 10 gAgar 7 gH2O 800 ml
Ajuster le pH à 7.5 et compléter le volume à 1 litre.
Milieu YPD (Levure) pour 500 ml:5 g yeast extract10 g bacto peptone10 g glucose500 ml H2O MilliQ
Ajouter d'abord un peu des 500 ml d'eau, bien mélanger jusqu'à homogénéisation, puis ajouter le restedes 500 ml. Autoclaver 30 min à 120°C.Pour couler environ 25 boites YPD, rajouter 10 g d'Agar pour 500 ml.
SOLUTIONS STOCK D'ANTIBIOTIQUES SOLUTIONS STOCK D'IPTG ET D'XGALAmpicilline: 100 mg/ml à dissoudre dans l'eau.Utiliser à des concentrations finales variant de 50 µg/ml (cosmides) à 150 µg/ml (Blue Scribe).
Tetracycline: 20 mg/ml à dissoudre dans l’eau additionnée d’éthanol ou d'acide chlorhydrique. La tétracycline est sensible à la lumière, conserver les stocks ou les boites à l'obscurité.Utiliser à des concentrations finales allant de 7.5 µg/ml à 15 µg/ml.
53

Kanamycine : 25 mg/ml à dissoudre dans l'eau. Utiliser à concentration de 10 à 35 µg/ml. (Pour les PACs : 35µg /ml final)
Les solutions Xgal et IPTG s'utilisent avec certains plasmides et phages M13 et permettent de distinguer les clones porteurs du vecteur seul de ceux qui contiennent un insert.
Le vecteur seul donne des colonies bleus.IPTG (isopropylthiogalactoside): stock 2 % dans l'eauX-gal (5-bromo-4-chloro-3-indolyl-b-D-galactoside): stock 2% (20mg/ml) en diméthylformamide (tube en verre).Ces solutions stock peuvent être conservées plusieurs semaines à -20°C dans l'obscurité.Utiliser 40 µl. de chaque solution pour une boite ou un étalement M13.
Préparation des mini-colonnesSEPHACRYL S300 (Elimination de l'ARN)Préparation de la colonne:
- Retirer le piston de la seringue, et l'utiliser pour placer un tampon de coton de verre siliconé au fond- Remplir la seringue de Séphacryl S300, centrifuger à 2000 rpm pendant 2 min. en centrifugeuse à portoirs balançant.- Compléter à 0,5 ml et conserver la colonne à +4°C en la bouchant avec la partie en caoutchouc du piston.- Centrifuger, par exemple, dans des tubes Falcon 2059 de 15 ml.
Utilisation:- Retirer le bouchon- Laver 2 à 3 fois la seringue avec de l'eau en centrifugeant à 2000 rpm pendant 2 min.- Centrifuger une dernière fois à 2000 rpm pendant 2 min pour vider la colonne.- Déposer 40 µl de TE dans la seringue, centrifuger 2 min. à 2000 rpm- Vérifier que le volume élué est de l'ordre de 40 µl., sinon recommencer.- Déposer les 40 µl de préparation. rapide, après avoir fait une RNAse (40 µl d'ADN + 1µl de RNAse à 10 mg/ml et incuber 15 min. à 37°C), et mettre au fond du tube falcon un tube eppendorf après en avoir coupé le bouchon.- Centrifuger 2 min. à 2000 rpm.
Eventuellement, recommencer l'opération sur une 2ème seringue.
SEPHADEXPréparation du séphadex :
- Peser 10 g de poudre séphadex G75 et la verser dans 300 à 400 ml du tampon souhaité (T.E., eau ...)-Laisser le séphadex gonfler selon les instructions du fournisseur: (G25, G50: 3 heures à 20°C, 1 heure à 90°C; G100: 72 heures à 20°C, 5 heures à 90°C). Quand on veut de manière plus générale utiliser un sephadex pour un dessalage, et si la quantité de sels est importante, il peut être nécessaire de dégazer le séphadex.
Préparation d'une colonne pour centrifugation- Retirer le piston de la seringue, et l'utiliser pour placer un tampon de coton de verre siliconé au fond.
54

- Remplir la seringue de G50, centrifuger à 2000 tpm pendant 2 min. en centrifugeuse à portoirs balançants, compléter de nouveau avec du G50 et recommencer jusqu'à ce que le G50 arrive à 5 mm environ du haut de la seringue après centrifugation. Conserver la colonne à 4 °C en la bouchant avec la partie en caoutchouc du piston. centrifuger par exemple dans des tubes Falcon 2059 de 15 ml.
Utilisation:- Retirer le bouchon, déposer 100 µl. de tampon dans la seringue, centrifuger 2 min. à 2000 rpm, vérifier que le volume élué est de l'ordre de 100 µl. sinon recommencer.- Déposer la sonde marquée diluée à 100 µl. au haut de la seringue, et mettre au fond du tube Falcon un tube eppendorf après en avoir coupé le bouchon. Centrifuger 2 min. à 2000 rpm. Les nucléotides libres restent dans la colonne
Il peut arriver des problèmes de lot de G75 (se manifeste par non coulage de la colonne).
Boudins de dialyse :10 mM bicarbonate de sodium (ou Tris 10mM, EDTA 10 mM).Laisser hydrater 1 heure 60°C ou bouillir.Rincer.Conserver en 10 mM EDTA à 4°C.
TNE x10 pour fluorimètre :Tris 0.1 M,EDTA 10 mM,NaCl 2 M, pH 7.4 (pH avec HCl).
Filtrer, et garder à 4°C jusqu'à 3 mois.Pour 1 litre : Tris 1M pH7.4 100 ml
EDTA 0.5M 2mlNaCl 5M 400 mlH2O qsp 1 litre
55
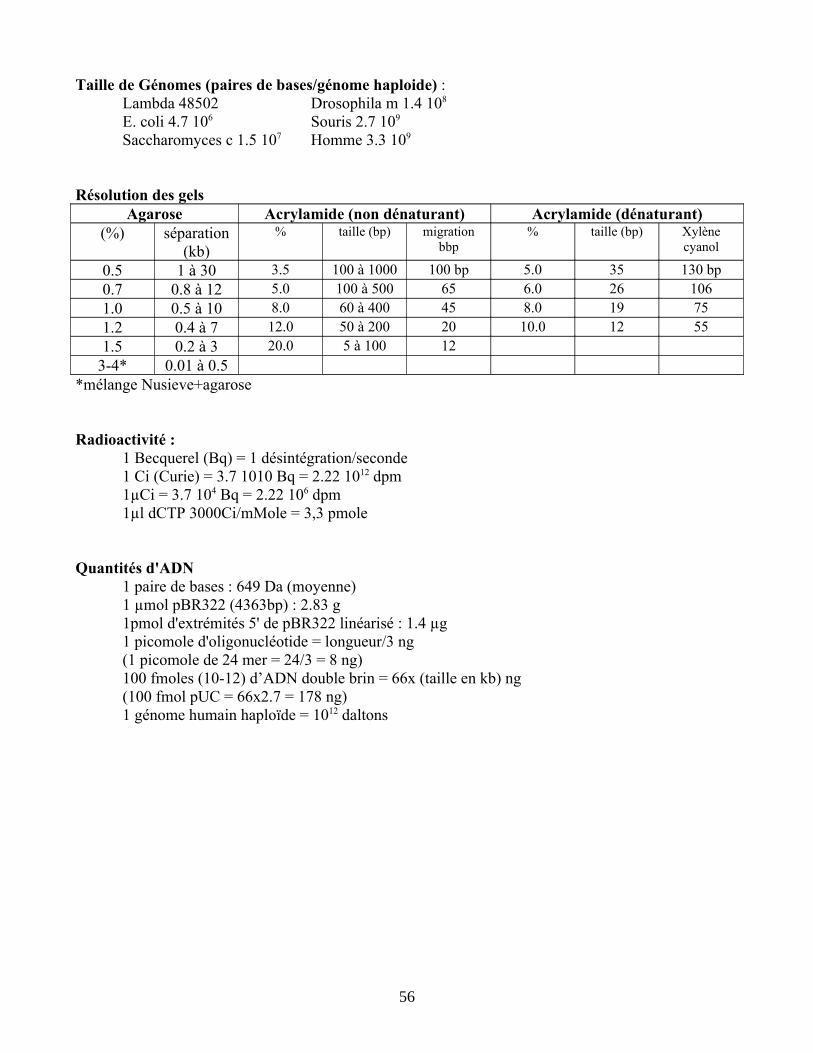
Taille de Génomes (paires de bases/génome haploide) :Lambda 48502 Drosophila m 1.4 108
E. coli 4.7 106 Souris 2.7 109
Saccharomyces c 1.5 107 Homme 3.3 109
Résolution des gelsAgarose Acrylamide (non dénaturant) Acrylamide (dénaturant)
(%) séparation (kb)
% taille (bp) migration bbp
% taille (bp) Xylène cyanol
0.5 1 à 30 3.5 100 à 1000 100 bp 5.0 35 130 bp0.7 0.8 à 12 5.0 100 à 500 65 6.0 26 1061.0 0.5 à 10 8.0 60 à 400 45 8.0 19 751.2 0.4 à 7 12.0 50 à 200 20 10.0 12 551.5 0.2 à 3 20.0 5 à 100 12
3-4* 0.01 à 0.5*mélange Nusieve+agarose
Radioactivité :1 Becquerel (Bq) = 1 désintégration/seconde1 Ci (Curie) = 3.7 1010 Bq = 2.22 1012 dpm1µCi = 3.7 104 Bq = 2.22 106 dpm1µl dCTP 3000Ci/mMole = 3,3 pmole
Quantités d'ADN1 paire de bases : 649 Da (moyenne)1 µmol pBR322 (4363bp) : 2.83 g1pmol d'extrémités 5' de pBR322 linéarisé : 1.4 µg1 picomole d'oligonucléotide = longueur/3 ng(1 picomole de 24 mer = 24/3 = 8 ng)100 fmoles (10-12) d’ADN double brin = 66x (taille en kb) ng(100 fmol pUC = 66x2.7 = 178 ng)1 génome humain haploïde = 1012 daltons
56


















![medicine.careers360.com · Roll No.Name of Candidate MGM INSTITUTE OF HEALTH SCIENCES - 2014 [MGM UG CET -2014 RESULT ] Sr. No. PHYCHEMBIO TOTAL 05, June 2014 1010001AALIYA IMTIYAJ](https://static.fdocuments.fr/doc/165x107/5e7bdaca8958d45c3253ebca/roll-noname-of-candidate-mgm-institute-of-health-sciences-2014-mgm-ug-cet-2014.jpg)