Comparaison des cinétiques de coagulation enzymatique et mixte ...

4 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
ProgrammeI - EXPLORATION DE L’ESPACE (5 TP, 10 heures en classe entière)
EXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Expériences mettant en évidence qualitativement des transformations lentes et rapides et les facteurs cinétiques, température et concentration à l’aide d’observations visuelles : H2O2 + I– et S2O3
2– + H+, tests caractéristiques utilisant le réactif de Fehling, le réactif de Tollens, par exemple.– d’un capteur de pression, d’une balance, d’un conductimètre, etc.Illustrations dans la vie courante : cuisson à l’auto-cuiseur, conservation des aliments par le froid, etc.
1. Transformations lentes et rapi-des– Mise en évidence expérimentale de transformations lentes et rapides.– Mise en évidence expérimentale des facteurs cinétiques : tempéra-ture et concentration des réactifs.– Rappels sur les couples oxydant/réducteur et sur l’écriture des équa-tions de réactions d’oxydoréduction.
– Écrire l ’équation de la réac-tion associée à une transformation d’oxydoréduction et identifier dans cette équation les deux couples mis en jeu.– Définir un oxydant et un réduc-teur.– Montrer, à partir de résultats expé-rimentaux, l’influence des facteurs cinétiques sur la vitesse de réaction.
Transformations lentes et rapides1
PARTIE 1
LA TRANSFORMATION D’UN SYSTÈME CHIMIQUE EST-
ELLE TOUJOURS RAPIDE ?
Cours
Découpage du cours1. Rappels d’oxydoréduction p. 142. Cinétique chimique p. 153. Transformations rapides p. 164. Transformations lentes p. 185. Facteurs cinétiques p. 196. Applications p. 20
Ce chapitre présente tout d’abord des rappels d’oxy-doréduction de la classe de 1re S, car la partie du programme « La transformation d’un système chimique est-elle toujours rapide ? » s’appuie essen-tiellement sur des transformations chimiques met-tant en jeu l’oxydoréduction.
♦ Les élèves n’ont jusqu’alors rencontré que des exemples de transformations rapides, par une approche simple s’appuyant largement sur des observations expérimen-tales : ils vont appréhender des systèmes chimiques dont les évolutions temporelles sont différentes.
1. Rappels d’oxydoréductionOn évoque l’oxydoréduction par le biais de rappels de définitions : oxydants, réducteurs, couples oxy-dant/réducteur, réaction d’oxydoréduction.
2. Cinétique chimiqueL’évolution temporelle des systèmes chimiques n’ayant jamais été traitée dans les classes précé-

5Chapitre 1 - Transformations lentes et rapides
décomposition d’une solution aqueuse de perman-ganate de potassium. Cette lente réduction des ions permanganate par l’eau est accélérée en milieu acide. Pour conserver le titre d’une solution aqueuse de permanganate de potassium, il est impératif de la préparer au dernier moment (solution fraîche) et surtout de ne pas l’acidifier lors de cette préparation (l’acidification avec de l’acide sulfurique s’effectue dans le vase réactionnel).
5. Facteurs cinétiquesLes grandeurs modifiant la durée d’évolution d’un système chimique sont abordées qualitativement dans cette fin de chapitre.Conformément au programme, seules la concentra-tion des réactifs et la température sont abordées. Mais dans l’activité 4, l’occasion est donnée d’évo-quer l’état de division des solides, un facteur cinéti-que largement mis à profit dans les expériences avec les métaux au collège.
6. ApplicationsIl s’agit d’illustrations prises dans la vie courante : diminution d’un temps de cuisson avec un autocui-seur, conservation des aliments par baisse de tem-pérature.Dans la classe, on évoque la diminution de la durée d’une transformation chimique avec un montage à reflux et le blocage d’une telle transformation par trempe chimique ou physique.
dentes (toutes les transformations étudiées étant quasi immédiates), le programme de TS aborde un nouveau domaine de la chimie.Pour la distinction entre transformations lentes et rapides, la persistance rétinienne constitue un réfé-rent temporel.
3. Transformations rapidesCes transformations chimiques, mettant en jeu des réactions de combustion, acido-basiques, de préci-pitation et surtout des réactions d’oxydoréduction très visuelles (iodométrie, manganimétrie), per-mettent de reprendre les bases de deux chapitres du cours de 1re S.
4. Transformations lentesLa formation de rouille, étudiée en classe de troi-sième, constitue une bonne introduction aux trans-formations lentes. Les réactions d’oxydoréduction sont en effet des supports très visuels pour les trans-formations chimiques à évolution lente. Ainsi l’ac-tion de l’eau oxygénée sur les ions iodure se traduit par l’apparition progressive de la couleur brune du diiode qui se forme.Le test au miroir d’argent s’appuie sur une transfor-mation chimique lente au cours de laquelle il y a réduction des ions argent (I) complexés en milieu basique.Transformation extrêmement lente, l’action des ions permanganate sur l’eau conduit lentement à la
Activités
2. L’état final dans les deux béchers se caractérise par une décoloration, la réduction des ions per-manganate conduit en milieu acide aux ions man-ganèse (II) incolores, les autres espèces étant également incolores.Généralement, la transformation dans le bécher 2 est terminée au bout de 5 min.3. La transformation dans le bécher 1 est quasi ins-tantanée ; elle est lente dans le bécher 2.4. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l) Fe2+(aq) = Fe3+(aq) + e–.Les deux demi-équations mettent en évidence un transfert d’électrons.5. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)C2O4H2(aq) = 2 CO2(aq) + 2 H
+(aq) + 2 e–.On retrouve ainsi l’équation de la réaction en mul-tipliant la première demi-équation par 2 et la seconde par 5 :
Transformations rapides ou lentes ? (page 22)
Cette activité permet d’aborder la cinétique des transformations à travers l’iodométrie et la manga-nimétrie, puis d’écrire des demi-équations des cou-ples utilisés. On présente également des espèces colorées (ion permanganate et diiode) qui permet-tent de bien appréhender visuellement les évolutions lentes ou rapides des systèmes chimiques étudiés. Il est intéressant d’observer qu’un même couple oxydo-réducteur peut, selon le partenaire, partici-per à des transformations lentes ou rapides.
A Manganimétrie1. La transformation immédiate se traduit par une décoloration dans le bécher 1.Dans le bécher 2, la réduction des ions permanga-nate n’est pas immédiate, la couleur rose peut per-sister plusieurs minutes.
ACTIVITÉ1
ACTIVITÉ1

6 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
2 MnO4–(aq) + 5 C2O4H2(aq) + 6 H
+(aq) 2 Mn2+(aq) + 10 CO2(aq) + 8 H2O(l).
B Iodométrie1. a) 2 S2O3
2–(aq) = S4O62–(aq) + 2 e–
I2(aq) + 2 e– = 2 l–(aq)
2 S2O32–(aq) + I2(aq) S4O6
2–(aq) + 2 l–(aq).
b)Oxydation
I2(aq) + 2 S2O32–(aq) 2 l–(aq) + S4O6
2–(aq).
Réduction
2. Il y a réduction du diiode (espèce colorée) par les ions thiosulfate avec formation de deux ions inco-lores. La transformation chimique se caractérise par une décoloration quasi immédiate.3. Les réactifs sont des espèces chimiques incolo-res : il se forme du diiode (espèce colorée) et des ions sulfate (incolores). La formation de diode per-met donc le suivi temporel de la transformation.4. La formation progressive de diiode se traduit par une intensification de la couleur jaune dans le bécher, dont la solution présente une teinte brune en fin de transformation.
Mise en évidence de facteurs cinétiques (page 23)
À l’aide d’expériences d’oxydoréduction, cette acti-vité permet de mettre en évidence, par une observa-tion qualitative et probante, la notion de facteur cinétique.
A Action de l’eau oxygénée H2O2(aq) sur les ions iodure I–(aq)
1. Le diiode (espèce chimique colorée) formé lors de la transformation chimique permet de suivre temporellement la transformation.2. a) Les six tubes côte à côte simulent des avance-ments différents de la réaction. Le contenu du tube 1 est plus foncé que celui des tubes 2, 3, 4, 5 et 6.Celui du tube 2 plus foncé que le 3, lui-même plus foncé que le 4, etc.b) Le suivi visuel du contenu des six tubes montre que la transformation est lente et que la formation progressive de diiode intensifie peu à peu la cou-leur du contenu du tube.
3. Les systèmes chimiques sont initialement iden-tiques ; l’état final de chaque système conduit à la formation de la même quantité de diiode. Les
ACTIVITÉ
2
ACTIVITÉ
2
contenus des six tubes sont identiques en fin de transformation.
B Modifier la durée d’évolution1. a) Le contenu de l’erlenmeyer (2) présente une teinte plus foncée que celui de l’erlenmeyer (1).b) Le système chimique contenu dans l’erlenmeyer (2) évolue plus rapidement que le (1). La couleur de la solution est liée à la formation progressive de diiode, laquelle est plus rapide dans le cas (2).2. a) n(iodure)introduit bécher (1) = 25 × 10
–3 × 0,10 = 2,5 × 10–3 mol.
n(iodure)introduit bécher (2) = 25 × 10–3 × 0,20 = 5,0 × 10–3 mol.
n(peroxyde d’hydrogène)introduit = 10 × 10–3 × 0,10
= 1,0 × 10–3 mol.xmax = 1,0 × 10
–3 mol : le peroxyde d’hydrogène est le réactif limitant dans les deux systèmes chimi-ques étudiés.Les ions iodure sont bien en excès, mais l’excès sert à solubiliser le diiode formé sous forme d’ions triio-dure I3
–.b) Le peroxyde d’hydrogène étant le réactif limi-tant dans les deux systèmes chimiques, il se forme la même quantité de diiode au final dans les deux systèmes. L’aspect des deux béchers est identique en fin de transformation.
3. La concentration de l’un des réactifs a été dou-blée : c’est le facteur cinétique mis en jeu dans ce protocole A.
4. La formation de diiode permettant le suivi tem-porel de la solution, il importe de s’assurer que le diiode formé soit solubilisé au fur et à mesure de sa production. Il n’est possible ici de jouer sur la concentration du réactif peroxyde d’hydrogène qu’à la condition qu’il reste le réactif limitant et que les ions iodure soient en excès suffisant.
Autre proposition :On ne change rien pour l’erlenmeyer (1) :10 mL d’eau oxygénée acidifiée de concentration C0 + 25 mL de solution aqueuse d’iodure de potas-sium, de concentration C2 en soluté apporté.
Pour l’erlenmeyer (2) :10 mL d’eau oxygénée acidifiée de concentration C0 + 10 mL d’eau + 25 mL de solution aqueuse d’iodure de potassium, de concentration C2 en soluté apporté.
5. Le contenu de l’erlenmeyer (3) présente une teinte plus foncée que celui de l’erlenmeyer (4) au
•

7Chapitre 1 - Transformations lentes et rapides
bout de 2 min. Le système chimique (3) évolue donc plus vite que le système (4).6. Le facteur cinétique mis en jeu dans ce protocole est la température du milieu réactionnel.
Suivi optiqued’une dismutation (page 24)
Malgré un temps de réponse voisin du dixième de seconde, l’œil constitue un outil performant pour suivre la formation lente de soufre colloïdal au cours de cette transformation chimique.
1. La formation de soufre colloïdal, S(s), permet un suivi visuel de la transformation chimique.2. La formation progressive de soufre colloïdal, S(s), opacifie la solution. La durée de disparition de la croix pour l’œil placé au-dessus du bécher peut constituer un outil de comparaison pour mettre en évidence les facteurs cinétiques (cf. exercice 21, page 30 du livre élève).3. a) S2O3
2– (aq) + 6 H+(aq) + 4 e– = 2 S(s) + 3 H2O(l)S2O3
2– (aq) + H2O(l) = 2 SO2(aq) + 2 H+(aq) + 4 e–.
On retrouve ainsi l’équation de la réaction en som-mant les deux demi-équations.b)
Réduction S2O3
2–(aq) + S2O32–(aq) + 4 H+(aq)
2 S(s) + 2 SO2(aq) + 2 H2O. Oxydation
4. L’ion thiosulfate est à la fois l’oxydant (réduc-teur conjugué : le soufre) et le réducteur (oxydant conjugué : le dioxyde de soufre) dans cette réac-tion d’oxydoréduction.5. Dans cette réaction d’oxydoréduction, l’ion thiosulfate s’oxyde et se réduit, ce qui justifie le terme de dismutation.
Suivi cinétique de transformations avec solides (page 24)
Cette activité permet d’aborder la cinétique des réactifs à l’état solide. Nous ne sommes pas dans le programme, mais bien pour comprendre expéri-mentalement pourquoi le chimiste, l’industriel pri-vilégient un tel état de division lorsqu’il utilise des solides.
1. a) Cu2+(aq) + 2 e– = Cu(s) Zn(s) = Zn2+(aq) + 2 e–.Les deux demi-équations mettent en évidence un transfert d’électrons.
ACTIVITÉ
3
ACTIVITÉ
3
ACTIVITÉ
4
ACTIVITÉ
4
b) L’oxydant est l’ion cuivre (II), de couleur bleue en solution aqueuse.
2. Les ions cuivre (II) étant consommés au cours de la transformation, la solution contenue dans le bécher s’éclaircit progressivement.
3. La solution s’éclaircit plus rapidement dans le bécher contenant le zinc en poudre.
4. La transformation chimique s’effectue plus rapi-dement lorsque l’état de division du solide est important. En utilisant de la poudre plutôt que de la grenaille, on augmente la surface de contact entre les réactifs : la transformation est plus rapide.L’état de division d’un solide modifie la durée d’évo-lution du système chimique : c’est un facteur ciné-tique.
Titrage d’une solution aqueuse de peroxyde d’hydrogène (page 25)
Ce TP ECE consiste à réaliser un titrage d’oxydoré-duction mettant en jeu la manganimétrie, il permet de vérifier les savoir-faire expérimentaux concer-nant :– la réalisation d’une dilution (choix du matériel, mode opératoire) ;– le prélèvement d’une solution (pipetage dans un bécher, rinçage de la pipette avec la solution à préle-ver, utilisation d’un pipeteur, repérage correct des niveaux) ;– la fiole jaugée (ajustement au trait de jauge et homogénéisation) ;– la burette graduée (rinçage de la burette avec la solution à prélever, ajustement du zéro) ;– l’agitateur magnétique (position et vitesse cor-rectes du barreau aimanté) ;– repérage de l’équivalence.
1 Transformation étudiée1. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)H2O2(aq) = O2(aq) + 2 H
+(aq) + 2 e–.
2. On retrouve l’équation de la réaction en multi-pliant la première demi-équation par 2 et la seconde par 5 :2 MnO4
–(aq) + 5 H2O2(aq) + 6 H+(aq)
2 Mn2+(aq) + 5 O2(g) + 8 H2O(l).
3. Le peroxyde d’hydrogène est le réducteur (oxy-dant conjugué : le dioxygène) dans cette réaction d’oxydoréduction.
ACTIVITÉ
5
ACTIVITÉ
5

8 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
2 Dosage de la solution diluée de peroxyde d’hydrogène
1. Le volume VE versé pour atteindre l’équivalence est de l’ordre de 22 mL.2. a) Le réactif limitant juste avant l’équivalence est l’ion permanganate.b) À l’équivalence, il y a changement de réactif limitant : le peroxyde d’hydrogène complètement consommé dans l’erlenmeyer devient le réactif limitant.
c) Lorsque l’équivalence est atteinte, la première goutte de réactif titrant versé colore la solution contenue en rose violacé.
3. a) C1 · V1 = 2 C2 · VE ; C1 = 2 ·2 E
1
C V
V.
Avec VE = 22,0 mL, on trouve :C1 = 8,8 × 10
–2 mol · L–1.b) C = 10 C1 = 8,8 × 10
–1 mol · L–1.Pour retrouver le titre, on multiplie par 11,2 :
8,8 × 10–1 × 11,2 = 9,9 volumes.
Corrigés des exercices (page 26)
1 DéfinitionsUne réaction d’oxydoréduction correspond à un transfert d’électrons, alors qu’une réaction acido-basique correspond à un transfert de protons.
2 Oxydoréduction1. Un oxydant est une espèce chimique susceptible de capter un ou plusieurs électrons.2. Un réducteur est une espèce chimique suscepti-ble de perdre un ou plusieurs électrons.
3 Vrai ou fauxa) Vrai.b) Faux : texte à venirc) Faux : texte à venird) Vrai.
4 Écrire une demi-équationC6H8O6(aq) = C6H6O6(aq) + 2 H
+(aq) + 2 e–.
5 Ajuster une demi-équationCr2O7
2–(aq) + 14 H+(aq) + 6 e– = 2 Cr3+(aq) + 7 H2O(l).
6 Écrire une réaction d’oxydoréductionÉcrire une réaction d’oxydoréductionMnO4
–(aq) + 4 H+(aq) + 3 Fe2+(aq) MnO2(s) + 2 H2O(l) + 3 Fe
3+(aq).
7 Vous avez dit titrer ! (vrai ou faux ?)a) Vrai.b) Vrai.c) Faux : avant l’équivalence, le réactif limitant est l’espèce chimique contenue dans la solution pré-sente dans le bécher.d) Vrai.e) Faux.
8 Rapide ou instantanée ?1. Sa durée est trop courte pour que l’évolution soit suivie à l’œil ou avec des appareils de mesure cou-rants. La durée d’évolution du système est infé-rieure ou égale à la durée de persistance rétinienne (1/10 s).2. Beaucoup de transformations rapides sont considérées comme instantanées, mais certaines transformations sont rapides sans être instanta-nées. On ne dispose pas de moyens pour étudier leur rapide évolution.
9 Trempe1. Il s’agit de bloquer une transformation chimique en jouant sur le facteur cinétique température.2. Un refroidissement brutal du milieu réactionnel (avec de la glace, par exemple) fige le système dans l’état où il se trouve et cela permet donc de l’analy-ser dans cet état.
10 Transformation ultrarapide1. L’apport d’énergie du détonateur permet d’en-clencher la transformation chimique, laquelle est très rapide.2. Il faut 130 g d’azoture de sodium.
11 Manganimétrie1. a) MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)
MnO4–(aq) + 4 H+(aq) + 3 e– = MnO2(s) + 2 H2O(l).
b) En milieu fortement acidifié par de l’acide sulfu-rique, le couple à prendre en compte est :
MnO4–(aq)/Mn2+(aq).
c) En milieu fortement acidifié, le réducteur conju-gué de l’ion permanganate est l’ion manganèse (II), incolore. Le passage d’une espèce chimique

9Chapitre 1 - Transformations lentes et rapides
colorée, l’ion permanganate, à une espèce chimi-que incolore, ion manganèse (II), favorise le repé-rage de l’équivalence lors d’un titrage.2. 4 MnO4
–(aq) + 4 H+(aq) 4 MnO2(s) + 2 H2O(l) + 3 O2(g).
12 Entretien des lentilles cornéennes1. Dans V = 100 mL, il y a m = 3,10 g de H2O2, soit :
n = m
M ; n =
3 1
34
, = 9,12 × 10–2 mol.
La concentration en H2O2 est donc :
[H2O2(aq)] = n
V ;
[H2O2(aq)] = 9 12 10
10
2
2
, –
–
× = 9,12 × 10–1 mol · L–1
2. Pour préparer 200 mL de solution S de concen-tration Cs = 9,1 × 10
–2 mol · L–1, il faut diluer 10 fois la solution S0.À la pipette, on prélève 20 mL de S0, que l’on verse dans une fiole jaugée de 200 mL. On complète à 200 mL avec de l’eau distillée et on homogénéise.
3. H2O2(aq) + 2 H+(aq) + 2 e– = 2 H2O(l)
H2O(l) = O2(g) + 2 H+(aq) + 2 e–
2 H2O2(aq) O2(g) + 2 H2O(l)
13 Transformations lentes ou rapides ?1. Lente. 4. Lente. 7. Rapide.2. Lente. 5. Rapide. 8. Lente.3. Rapide. 6. Lente.
14 La cocotte-minute1. L’augmentation de pression à l’intérieur de la cocotte entraîne l’élévation de la température d’ébullition d’eau (application de la loi des gaz par-faits). Si la température est plus élevée, le temps de cuisson est plus court.2. Avec les ultracuiseurs, la cuisson est accélérée, la durée de cuisson réduite. Tout dépassement de cuisson, ∆t, se traduit par une progression de la cuisson plus grande par rapport à une durée de cuisson en cocotte-minute.
15 Cinétique et cuisinea) La formation lente du dioxyde de carbone est favorisée par l’élévation de température : la pâte lève plus vite.b) Une température plus élevée permet de rendre la cuisson plus rapide (réaction de Maillard en sur-face).
c) L’état de division du chocolat favorise la prépa-ration.d) En abaissant la température, on ralentit les transformations chimiques liées à la décomposi-tion des aliments. La conservation est améliorée par rapport à celle des aliments conservés à la tem-pérature ordinaire.
16 Titrage d’une solution de diiode1. n(thiosulfate versé) = 1,0 × 10–4 mol · L–1.2. a) Les réactifs introduits dans le bécher sont dans des proportions stoechiométriques.b) Décoloration de la solution.c) n(diiode initial) = 0,5 n(thiosulfate) versé à l’équivalence.
3. C0 = 0 5
0
, ·.R EC V
V
4. C0 = 2,0 × 10–3 mol · L–1.
Masse de diiode = 0,51 g.
17 La liqueur de Fehling1. a) Les ions cuivre (II) sont réduits à l’état d’ions cuivre (I).b) L’aldéhyde réduit les ions cuivre (II) complexés de la liqueur de Fehling.
2. La transformation ne se fait pas à froid
3. n(Cu2O) = m
M ;
n(Cu2O) = 3 575
143 08
,
, = 2,50 × 10–2 mol.
4. a) n(aldéhyde) = 2,50 × 10–2 mol.
b) M = m
n ; M =
1 450
2 50 10 2
,
, –× = 58,0 g · mol–1.
18 Titrage d’un cachet de vitamine C1. Les deux demi-équations d’oxydoréduction sont : C6H8O6(aq) = C6H6O6(aq) + 2 H
+(aq) + 2 e–
I2(aq) + 2 e– = 2 I–(aq).
2. a) Dans le système chimique à l’équivalence, les quantités de matière des réactifs en présence sont donc dans des proportions stœchiométriques (les deux réactifs sont alors intégralement consom-més).Pour déterminer la relation existant entre les quantités de matière des réactifs introduits à l’équivalence, on utilise un tableau d’évolution du système chimique à l’équivalence ainsi que la pro-priété de simultanéité des réactifs limitants.

10 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
Équation de la réaction
I2(aq) + C6H8O6(aq) C6H6O6(aq) + 2 H
+(aq) + 2 I–(aq)
État du système
Av. Quantité de matière (mol)
Initial 0 n(I2)vers éq n(C6H8O6)présent 0 0 0
Final xmaxn(I2)vers éq– xmax = 0
n(C6H8O6)présent– xmax = 0
xmax 2 xmax 2 xmax
n(C6H8O6)présent – xmax = 0 et n(I2)vers éq – xmax = 0.xmax = n(C6H8O6)présent = n(I2)vers éq.
b) La solution devient bleu-noir.3. n(C6H8O6)présent = n(I2)vers éq= VE · C2
= VR · C(C6H8O6).
C(C6H8O6) = V C
VE
R
· 2 = 14 5 1 00 10
25 0
2, ,
,
–× ×
C(C6H8O6) = 0,58 × 10–2 mol · L–1
4. n = C(C6H8O6) · V ;n = 0,58 × 10–2 × 500,0 × 10–3 = 2,9 × 10–3 mol.m = n · M ;m = 2,9 × 10–3 × 176 = 510 × 10–3 g = 510 mg.
19 Titrage du dioxyde de soufre dans l’air1. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)SO2(aq) + 2 H2O(l) = SO4
2–(aq) + 4 H+(aq) + 2 e–
On retrouve ainsi l’équation de la réaction en mul-tipliant la première demi-équation par 2 et la seconde par 5.2 MnO4
–(aq) + 5 SO2(aq) + 2 H2O(l) 2 Mn2+(aq) + 5 SO4
2–(aq) + 4 H+(aq).2.
SolutionS0 (K
+(aq) +MnO4
– (aq)) Co
Solution S
Agitateur
3. a) Dans le système chimique à l’équivalence, les quantités de matière des réactifs en présence sont donc dans des proportions stœchiométriques (les deux réactifs sont alors intégralement consommés).b) À l’équivalence, avec la disparition des derniers ions permanganate, la solution devient incolore.c) Pour déterminer la relation existant entre les quantités de matière des réactifs introduits à
l’équivalence, on utilise un tableau d’évolution du système chimique à l’équivalence ainsi que la pro-priété de simultanéité des réactifs limitants.
Équation de la réaction
2 MnO4–(aq) + 5 SO2(aq) + 2 H2O(l)
2 Mn2+(aq) + 5 SO42–(aq) + 4 H+(aq)
État du système
Av. Quantité de matière (en mol)
Initial 0 n(MnO4–)E n (SO2)présent 0 0
Final xmaxn(MnO4
–)E – 2xmax
= 0n (SO2)présent
– 5 xmax = 02 xmax 5 xmax
n(MnO4–)E – 2 xm = 0 et n(SO2)présent – 5 xmax = 0
xmax = n(MnO )
24–
E = n(SO )
22–
initialement présents .
On retrouve donc la relation :5 n(MnO4
–(aq))E = 2 n(SO2(aq))initialement présents.
4. a) n(MnO4–(aq))E = CO · VO
n(MnO4–(aq))E = 1,00 × 10
–4 × 8,8 × 10–3 ;n(MnO4
–(aq))E = 8,8 × 10–7 mol.
b) n(SO2(aq))initialement présents = 5
2 (MnO )4–
En ;
n(SO2(aq))initialement présents = 5
2 × 8,8 × 10–7 ;
n(SO2(aq))initialement présents = 2,2 × 10–6 mol.
5. a) On a dosé 250 mL, ce qui signifie qu’il y a 2,2 × 10–6 mol dans ces 250 mL, lesquels provien-nent de 2 m3 d’air.Ce qui donne pour 1 m3 d’air : 1,1 × 10–6 mol, soit 1,1 × 10–6 × 64,5 = 71 × 10–6 g · m–3 = 71 µg · m–3.b) La teneur en dioxyde de soufre de l’air étudié ne respecte pas cette valeur limite.L’air est donc considéré comme pollué par rapport à la norme de l’O.M.S.
20 Eau de Javel
Exercice résolu dans le manuel de l’élève.
21 Une transformation opacifiante1. Quantité de matière = V · C ;Quantité de matière = 0,5 × 0,1 = 5 × 10–2 mol.Masse = 5 × 10–2 × 248,2 = 12,4 g.On pèse 12,4 g de solide dans une capsule sur une balance préalablement tarée. On verse, à l’aide d’un entonnoir à poudre, le solide dans une fiole jaugée de 500 mL contenant 1/3 d’eau ; on dissout le com-posé, puis on ajuste au trait de jauge avec de l’eau

11Chapitre 1 - Transformations lentes et rapides
distillée en tenant compte du ménisque. Enfin, on homogénéise le contenu de la fiole.2. Il s’agit d’effectuer une dilution par deux, autre-ment dit, on prélève 25,0 mL de la solution (S1) avec une pipette jaugée de 25 mL. On les verse dans une fiole jaugée de 50 mL et on complète au trait de jauge avec de l’eau distillée.
3. a)
Bécher [S2O32–] (mol · L–1) [H+] (mol · L–1)
1V C1 1
50
· =
45 0 1
50
× , = 0,090
V C1 1
50
· =
5 1
50
× = 0,10
2V C2 2
50
· = 45 0 050
50
× , = 0,045 0,10
b) La concentration en ions thiosulfate est deux fois plus faible dans le bécher 2 que dans le 1. La concentration d’un réactif est un facteur cinéti-que : la vitesse est d’autant plus grande (transfor-mation plus rapide) que la concentration en ion thiosulfate l’est également.Expérimentalement, la formation de soufre colloï-dal est plus rapide dans le bécher 1.Visuellement, la croix disparaît plus vite dans le bécher 1.c)• Bécher 1
Bécher 1 S2O32–(aq) + 2 H+(aq) = SO2(aq) + H2O(l) + S(s)
Initial 4,50 × 10–3 mol 5,00 × 10–3 mol 0 0
En cours 4,5 × 10–3 – x 5 × 10–3 – 2x x x
Fin 4,5 × 10–3 – xmax 5 × 10–3 – 2xmax xmax xmax
xmax = 2,50 × 10–3 mol,
n(soufre formé) = 2,50 × 10–3 molm(soufre formé) = 32 × 2,50 × 10–3 = 80 mg.• Bécher 2
Bécher 1 S2O32–(aq) + 2 H+(aq) = SO2(aq) + H2O(l) + S(s)
Initial 2,25 × 10–3 5,00 × 10–3 mol 0 0
En cours 2,25 × 10–3 – x 5 × 10–3 – 2x x x
Fin 2,25 × 10–3 – xmax 5 × 10–3 – 2xmax xmax xmax
xmax = 2,25 × 10–4 mol, acide en excès.
Masse de soufre : 32 × 2,25 × 10–3 = 72 mg.
22 Teinture d’iode officinale1. 1 L de teinture d’iode a une masse de 880 g et renferme donc 44 g de diiode :
C0 = n
V =
m
M V· ;
C0 = 44
254 1× = 1,73 × 10–1 mol · L–1.
2. La solution est préparée avec de l’éthanol comme solvant. Celui-ci facilite la solubilisation du diiode, espèce moléculaire, et permet de réaliser avec l’eau une solution homogène non saturée. Au laboratoire, on utilise les ions iodure pour faciliter la solubilisa-tion du diiode dans l’eau (formation d’ions triiodure).3. Pour effectuer une dilution au centième à partir de la solution initiale, il faut prélever 1,0 mL de la teinture d’iode, à l’aide d’une pipette jaugée de 1 mL munie d’une propipette, et les verser dans une fiole jaugée de 100 mL contenant une petite quan-tité d’eau. On ajuste ensuite au trait de jauge et on homogénéise.4. a) 2 S2O3
2–(aq) = S4O62–(aq) + 2 e–
I2(aq) + 2 e– = 2 I–(aq).
b)
Equation de la réaction
2 S2O32–(aq) + I2 (aq)
2 I–(aq) + S4O62–(aq)
État du système Av. Quantité de matière (en mol)
État initial 0 n(S2O32–)E n(I2)formé 0 0
État final xmaxn(S2O3
2–)E– 2 xmax = 0
n(I2)formé– xmax = 0
2 xmax xmax
n(S2O32–)E – 2 xmax = 0 et n(I2)formé – xmax = 0.
xmax = ½ n(S2O32–)E = n(I2)formé.
c) 2 n(I2)initial = n(S2O32–)E.
n(S2O32–)E = Cr · VE et n(I2)initial = C1 · V,
soit 2 C1 · V = Cr · VE.
d) C1 = 0 5, (C V
Vr E· )
:
C1 = 0 5 3 0 10 22 9
20 0
2, ( , , )
,
–× × = 1,72 × 10–3 mol · L–1.
e) C0 = 1,72 × 10–3 × 100 = 1,72 × 10–1 mol · L–1
f) ∆C = 1,73 × 10–3 – 1,72 × 10–3 = 0,01 × 10–3 mol · L–1.
Erreur relative : 0 01 100
1 73
,
,
× = moins de 1 %.
23 L’analyse de l’air par Lavoisier1. Il s’agit de l’oxyde de mercure (II), HgO(s).
2. n(mercure) = 22 3
200 6
,
, = 0,61 mol.

12 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
Le volume d’air initial confirme que le dioxygène est le réactif limitant.
3. a) n(oxyde de mercure formé) = 2 8
216 6
,
,= 1,3 × 10–2 mol.
b) xmax = 6,5 × 10–3 mol = 6,5 × 10–3 mol, soit un volume V(dioxygène) = 0,16 L.
c) V(oxygène) = 0 16 50
0 8
,
,
× = 10 pouces
On a consommé 10 pouces de dioxygène : il en reste donc 40 pouces.
4. a) Le fourneau permet de rendre plus rapide l’oxydation du mercure par le dioxygène (tempéra-ture facteur cinétique)b) L’expérience aurait duré beaucoup plus long-temps : la nuit, l’abaissement de température du
mélange réactionnel aurait ralenti la transforma-tion chimique.5. a) Le volume gazeux dans la cornue n’évoluant plus (l’air déphlogistiqué n’évolue plus), Lavoisier en conclut que la transformation chimique est ter-minée.b) Arrêter l’expérience avant terme (6 jours) n’aurait pas permis à Lavoisier d’obtenir la compo-sition exacte de l’air atmosphérique. Au bout de 20 jours (la transformation étant terminée après 12 jours), la composition du mélange réactionnel n’aurait pas évoluée et les conclusions de Lavoisier auraient été les mêmes.Dans tous les cas, le mérite de Lavoisier est d’avoir compris que tous les corps combustibles absorbent en brûlant uniquement la portion respirable de l’air en quantité égale à l’augmentation de poids.
Les compétences expérimentales
(page 32)
Cet exercice doit conduire l’élève à réfléchir sur la mise en œuvre d’expé-riences simples et à justifier ses choix.
Corrigé1. Observations visuelles de transformationLes réactions d’oxydoréduction utilisées dans ce chapitre sont faciles à mettre en œuvre, mais elles supposent une réflexion préalable, sur le choix du réactif limitant. Pour être probante, la transformation chimique doit conduire au final à la consommation totale du permanganate, afin d’obser-ver une décoloration immédiate. Il faut donc que le permanganate soit le réactif limitant.
• La proposition faite sur le dessin B est correcte.
2. Mise en évidence d’un facteur cinétiqueDans les deux démarches, les ions iodure sont en large excès permettant de solubiliser le diiode formé. La démarche A conduit au même état final, en jouant sur la concentration initiale des réactifs.
• La proposition faite sur le dessin A peut être considérée comme plus rigoureuse.

13Chapitre 1 - Transformations lentes et rapides
Exercices supplémentaires1. Mettre en jeu un facteur cinétique1. Comment rendre plus rapide une transformation chimique mettant en jeu des réactifs gazeux ?2. Même question avec une solution thermostatée dont les réactifs sont des ions.
Corrigé1. L’élévation de température permet d’accélérer la plupart des transformations chimiques mettant en jeu des réactifs gazeux.2. En utilisant des réactifs plus concentrés.
2. Demi-équationsÉcrire la demi-équation d’oxydoréduction relative à chacun des couples oxydant/réducteur suivants.1. MnO4
–(aq)/Mn2+(aq).2. CO2(aq)/HO2CCO2H(aq).3. NO3
–(aq)/NO(g).4. O2(aq)/H2O2(aq).5. S4O6
2–(aq)/S2O32–(aq).
6. H2O2(aq)/H2O(l).
Corrigé1. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l).2. 2 CO2(aq) + 2 H
+(aq) + 2 e– = HO2C–COOH(aq).3. NO3
–(aq) + 4 H+(aq) + 3 e– = NO(g) + 2 H2O(l).4. O2(g) + 2 H
+(aq) + 2 e– = H2O(l).5. S4O6
2–(aq) + 2 e– = 2 S2O32–(aq).
6. H2O2(aq) + 2 H+(aq) + 2 e– = 2 H2O(l).
3. Couples oxydant/réducteurMettre en évidence quatre couples oxydant/réduc-teur en associant les espèces de la liste suivante :a) Fe2+(aq) ; b) SO4
2–(aq) ; c) S2O32–(aq) ;
d) SO2(aq) ; e) S4O62–(aq) ; f) Fe(s) ;
g) Fe3+(aq) ; h) HSO3–(aq) ; i) SO3
2–(aq).
CorrigéS4O6
2–(aq)/S2O32–(aq). Fe2+(aq)/Fe(s).
Fe3+(aq)/Fe2+(aq). SO42–(aq)/SO2(aq).
4. Préparation de solution de diiodeLe diiode étant peu soluble dans l’eau, sa solubilisa-tion est favorisée par la présence d’ions iodure, avec lesquels il donne des ions triiodure, I3
–. On prépare une solution aqueuse de diiode en intro-duisant une masse m = 2,50 g de diiode dans une fiole jaugée de 500 mL contenant 10 g d’iodure de potassium : quelle est la concentration molaire en diiode de cette solution ?
Corrigén(diiode)introduit =
2 50
254
, = 1,0 × 10–2 mol.
[diiode(aq)] = 1 0 10
0 50
2,
,
–× = 2,0 × 10–2 mole · L–1.
Même préparée avec minutie, une telle solution de diiode n’est considérée comme titrée qu’après dosage par une solution titrée de thiosulfate de potassium (les ions iodure présents sont sensibles à l’oxydation).
5. Eau-forteUne eau-forte est une gravure obtenue en dessinant sur une plaque de cuivre recouverte d’un vernis protecteur à l’aide d’une pointe qui met le cuivre à nu. On verse alors de l’acide nitrique, H+(aq) + NO3
–(aq), lequel agit comme un burin chimique attaquant le cuivre non protégé. Au cours d’une opération de gravage, le volume de monoxyde d’azote dégagé est V = 240 mL. L’équation chimique de la réaction modélisant la transformation est :2 NO3
–(aq) + 3 Cu(s) + 8 H+(aq) 2 NO(g) + 3 Cu2+(aq) + 4 H2O(l).
1. Cette transformation chimique s’appuie-t-elle sur une réaction acido-basique ou d’oxydoréduc-tion ? Justifier la réponse en mettant en évidence les couples mis en jeu.2. Établir le tableau d’évolution du système tradui-sant les états initial, intermédiaire et final.3. Déterminer la quantité de matière de monoxyde d’azote formé.4. En déduire l’avancement maximal.5. Calculer la quantité de matière de cuivre qui a réagi et en déduire la masse de cuivre consommé. Corrigé1. Il s’agit d’une réaction d’oxydoréduction :
Cu(s) = Cu2+(aq) + 2 e–
NO3–(aq) + 4 H+(aq) + 3 e– = NO(g) + 4 H2O(l).
2.
Bécher 1 2 NO3–(aq) + 8 H+(aq) + 3 Cu(s)
= 3 Cu2+(aq) + 4 H2O(l) + NO(g)
Initial n0(nitrate) n0(Cu) 0
En cours n0 – 2x n0(Cu) – 2x x
Fin n0 – 2xmax n0(Cu) – 2xmax xmax
3. n(NO formé) = 240 10
24
3× –
= 10,0 × 10–3 mol.
4. xmax = n(NO formé) = 10,0 × 10–3 mol.
5. n(cuivre consommé) = 2 xmax ;n(cuivre consommé) = 20,0 × 10–3 mol.m(cuivre consommé) = 63,5 × 20,0 × 10–3 = 1,27 g.

14 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Suivi de l’évolution temporelle d’une transforma-tion :– par prélèvements successifs et titrage, par exemple réaction de H2O2 et de I–, dismutation de H2O2, réaction de S2O8
2– et de I–,– par utilisation d’un manomètre, d’un conductimè-tre, ou d’un spectrophotomètre.Tracé des courbes d’évolution de quantité de matière ou de concentration d’une espèce et de l’avancement de la réaction au cours du temps.Expériences qualitatives illustrant le phénomène d’absorption en lumière visible.Observation du spectre d’absorption d’une espèce colorée en solution.Étude expérimentale de la relation entre la concen-tration effective d’une espèce colorée en solution et l’absorbance pour une longueur d’onde donnée dans un domaine de concentration donné.Suivi d’une transformation chimique par compa-raison avec une échelle de teintes et/ou par spectro-photométrie.
2. Suivi temporel d’une transfor-mationTracé des courbes d ’évolution de quantité de matière ou de concentra-tion d’une espèce et de l’avancement de la réaction au cours du temps :ut i l isat ion du tableau descript i f d’évolution du système chimique, exploitation des expériences.– Une nouvelle technique d’analyse : la spectrophotométrie.L’absorbance A, grandeur mesurée par le spectrophotomètre.Relation entre l ’absorbance et la concentration effective d’une espèce colorée en solution pour une longueur d’onde donnée et pour une épaisseur de solution traversée donnée.Suivi de la cinétique d’une transfor-mation chimique par spectrophoto-métrie.
– Justif ier les différentes opé-rations réalisées lors du suivi de l ’évolution temporelle d’un système : exploiter les résultats expérimentaux.– Définir l’équivalence lors d’un titrage et en déduire la quantité de matière de réactif titré.– À partir de mesures expéri-mentales et du tableau descriptif de l’évolution du système, repré-senter, en fonction du temps, la variation des quantités de matière ou des concentrations d’un réactif ou d’un produit et de l ’avance-ment de la réaction.– Savoir utiliser, à une longueur d’onde donnée, la relation entre la concentration d’une espèce colo-rée en solution et l’absorbance.
2 Suivi temporel d’une transformation chimique
Cours
Découpage du cours1. Méthodes chimiques, les titrages p. 342. Suivi par conductimétrie p. 363. Suivi par pressiométrie p. 374. Spectrophotométrie p. 38
Dans ce chapitre, on a fait le choix de présenter tous les suivis possibles dans le respect du programme et de réserver l’exploitation quantitative des aspects cinétiques (vitesse de réaction, temps de demi-réaction) pour le chapitre 3.En préparation du chapitre 3 et de la définition de la vitesse de réaction, tous les suivis conduisent à l’éta-blissement de graphes x = f(t).
1. Méthodes chimiques, les titragesLa transformation rapide qui permet de suivre la transformation étudiée ne doit pas la perturber. Afin d’éviter toute confusion chez les élèves, on ne
♦ parle pas d’avancement de la réaction pour le titrage et on réserve l’avancement de la réaction x(t) pour celui de la transformation étudiée.
2. Suivi par conductimétriePeu de transformations chimiques peuvent être sui-vies par conductimétrie.L’hydrolyse du chlorure de tertiobutyle est considé-rée comme totale si l’on utilise un important excès d’eau.L’hydrolyse basique d’un ester (éthanoate d’éthyle par exemple) se prête également bien à un suivi conductimétrique.
3. Suivi par pressiométrieL’emploi d’un manomètre, abordé dès la classe de seconde, permet le suivi par pressiométrie d’une transformation chimique au cours de laquelle l’un des produits de la réaction est à l’état gazeux.

15Chapitre 2 - Suivi temporel d’une transformation chimique
Remarque : on peut réinvestir le facteur cinétique qu’est l’état de division d’un solide lors d’une trans-formation mettant en jeu un réactif solide et un réac-tif liquide (acide chlorhydrique et zinc, par exemple). L’utilisation de zinc en poudre réduit notablement la durée d’évolution de la transformation.
4. SpectrophotométrieAvant de pouvoir exploiter un suivi spectrophoto-métrique et en s’appuyant sur les connaissances antérieures des élèves, on introduit l’absorbance, la présentation du spectrophotomètre, la courbe d’analyse spectrale, la loi de Beer-Lambert.
Activités
Un suiviin situ (page 43)
Cette activité peut constituer un TP très intéressant dans lequel le suivi d’une transformation lente se fait par l’intermédiaire d’une transformation rapide.Le caractère in situ du titrage du diiode formé sup-pose une présentation minutieuse de l’expérience. Dès que la transformation lente est enclenchée (t = 0), il faut réagir à l’enchaînement des appari-tions, qui est rapide au début.Cette technique met bien en évidence le « ralentis-sement » de la transformation lors de l’évolution du système chimique. Il sera intéressant d’aller au moins jusqu’au temps de demi-réaction :
n(diiode formé) = n(diiode)max
2,
pour une exploitation ultérieure (facteurs cinétiques, par exemple).
1. a) L’introduction de la solution aqueuse de peroxodisulfate de potassium dans l’erlenmeyer, correspond au déclenchement de la transformation chimique (instant de date t = 0 s).b) Le diiode qui se forme lors de la transformation chimique étudiée est immédiatement réduit par les ions thiosulfate tant que ceux-ci sont en excès ; le thiodène ne détecte donc pas la présence de diiode.2. n(thiosulfate introduit) = 0,200 × 1,0 × 10–3 ; n(thiosulfate introduit) = 2,0 × 10–4 mol
3. n(diiode formé) = 0,5 n(thiosulfate introduit) ; n(diiode formé) = 1,0 × 10–4 mol.
4. La durée de formation de 1,0 × 10–4 mol de diiode n’est pas constante : elle augmente à chaque appa-rition. La transformation « ralentit ».5. La transformation « ralentit » au fur et à mesure de l’évolution du système chimique.
ACTIVITÉ
1
ACTIVITÉ
1 Suivipressiométrique (page 44)
L’expérience est volontairement limitée à une durée de 5 min, afin de réaliser plusieurs activités dans la même séance.On peut imaginer une autre version avec un seul protocole (la moitié de la classe avec de la poudre, l’autre moitié avec de la grenaille), mené jusqu’à l’évolution totale du système chimique, puis com-paraison des deux graphes x = f(t) et exploitation dans le cadre du chapitre 3.
1. Les pressiomètres étant généralement limités à 2 000 hPa, il importe de bien réfléchir à la masse initiale de zinc introduite. Le zinc, réactif limitant détermine la quantité de matière de dihydrogène formé et donc la pression finale.Dans l’hypothèse d’un protocole visant à obtenir x = f(t), il faut qu’en fin de transformation, Pfinal soit inférieure à 2 000 hPa (importance du volume du flacon). Peser d’abord le morceau de grenaille de zinc, puis avec la même précision, peser la même masse de poudre de zinc.Avec 730 mg de zinc, soit 11,2 × 10–3 mol :
État Zn(s) + 2 H3O+(aq)
Zn2+(aq) + H2(g) + 2 H2O(l)
Initial 11,2 × 10–3 nexcès 0 0
Intermé-diaire
11,2 × 10–3
– xnexcès – 2 xm x x 2 x
Final 11,2 × 10–3
– xmaxnexcès – 2 xmax xmax xmax 2 xmax
n(dihydrogène formé) (t) = x(t).
2. (P(t) – P0) · Vgaz = n(H2)(t) · R · T.
Or n(H2)(t) = x(t).
Il vient : x(t) = ( – ) ·
·( )P P V
T
t 0 gaz
R.
ACTIVITÉ
2
ACTIVITÉ
2
16 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
• Protocole 1 : x1(t) = ( – ) ·
·( )P P V
T
t1 0 gaz
R.
• Protocole 2 : x2(t) = ( – ) ·
·( )P P V
T
t2 0 gaz
R.
Les volumes de gaz mis en jeu et les températures d’expériences étant identiques, il vient en faisant le rapport des deux expressions précédentes :
P P
P P2 0
1 0
–
– =
x
x2
1
.
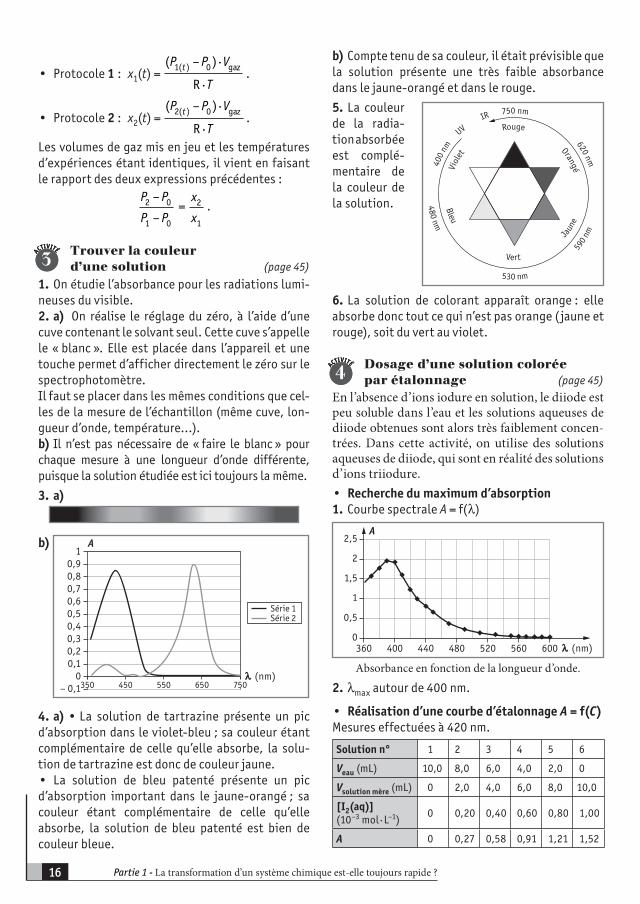
Trouver la couleurd’une solution (page 45)
1. On étudie l’absorbance pour les radiations lumi-neuses du visible.2. a) On réalise le réglage du zéro, à l’aide d’une cuve contenant le solvant seul. Cette cuve s’appelle le « blanc ». Elle est placée dans l’appareil et une touche permet d’afficher directement le zéro sur le spectrophotomètre.Il faut se placer dans les mêmes conditions que cel-les de la mesure de l’échantillon (même cuve, lon-gueur d’onde, température…).b) Il n’est pas nécessaire de « faire le blanc » pour chaque mesure à une longueur d’onde différente, puisque la solution étudiée est ici toujours la même.
3. a)
b)
– 0,10
0,10,20,30,40,50,60,70,80,9
1
450 550 650350 750l (nm)
A
Série 1Série 2
4. a) • La solution de tartrazine présente un pic d’absorption dans le violet-bleu ; sa couleur étant complémentaire de celle qu’elle absorbe, la solu-tion de tartrazine est donc de couleur jaune.• La solution de bleu patenté présente un pic d’absorption important dans le jaune-orangé ; sa couleur étant complémentaire de celle qu’elle absorbe, la solution de bleu patenté est bien de couleur bleue.
ACTIVITÉ
3
ACTIVITÉ
3
b) Compte tenu de sa couleur, il était prévisible que la solution présente une très faible absorbance dans le jaune-orangé et dans le rouge.
5. La couleur de la radia-tion absorbée est complé-mentaire de la couleur de la solution.
6. La solution de colorant apparaît orange : elle absorbe donc tout ce qui n’est pas orange (jaune et rouge), soit du vert au violet.
Dosage d’une solution colorée par étalonnage (page 45)
En l’absence d’ions iodure en solution, le diiode est peu soluble dans l’eau et les solutions aqueuses de diiode obtenues sont alors très faiblement concen-trées. Dans cette activité, on utilise des solutions aqueuses de diiode, qui sont en réalité des solutions d’ions triiodure.• Recherche du maximum d’absorption1. Courbe spectrale A = f(λ)
0
0,5
1
1,5
2
2,5
360 400 440 480 520 560 600 l (nm)
A
Absorbance en fonction de la longueur d’onde.
2. λmax autour de 400 nm.
• Réalisation d’une courbe d’étalonnage A = f(C)Mesures effectuées à 420 nm.
Solution n° 1 2 3 4 5 6
Veau (mL) 10,0 8,0 6,0 4,0 2,0 0
Vsolution mère (mL) 0 2,0 4,0 6,0 8,0 10,0
[I2(aq)](10–3 mol · L–1)
0 0,20 0,40 0,60 0,80 1,00
A 0 0,27 0,58 0,91 1,21 1,52
ACTIVITÉ
4
ACTIVITÉ
4
Viol
et
Rouge Orangé
Bleu Vert
J
aune
480 nm 530 nm
590
nm
400
nm
UV
IR 750 nm 620 nm

17Chapitre 2 - Suivi temporel d’une transformation chimique
1. à 4.
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
0 0,2 0,4 0,6 0,8 1
A
[diiode](mmol · L–1)
Si l’absorbance de la solution est A = 0,85, on trouve une concentration correspondante :
C0 = 0,56 × 10–3 mol · L–1.On remarquera l’importance du quadrillage et sur-tout la nécessaire linéarisation (avec outil de des-sin) du graphe.
5. Concentration massique = 0,56 × 10–3 × 254 ; concentration massique = 0,14 g · L–1.
Suivi cinétique d’une transformation par conductimétrie (page 46)
1 Préparation d’une solution alcooliquede 2-chloro-2-méthylpropane
1. Si l’on utilise de l’éthanol mélangé à l’eau, cette dernière peut hydrolyser le composé halogéné dès que le mélange est réalisé.Il faut donc utiliser de l’éthanol absolu.
2. Masse de RCl = 5,0 × 10–3 × 0,840 = 4,25 g.
n(RCl) = 4 25
92 5
,
, = 45,4 × 10–3 mol.
2 Suivi conductimétrique de la transformation
t (s) s (mS · cm–1)
0 0
30 1,49
60 2,9
90 3,98
120 4,96
150 5,8
180 6,46
210 7
240 7,43
270 7,72
300 7,98
ACTIVITÉ
5
ACTIVITÉ
5
330 8,16
360 8,27
390 8,34
420 8,39
450 8,45
480 8,52
510 8,53
540 8,53
570 8,53
3 Étude du système chimique mis en jeu
1. n(RCl)introduit = 45 4 10 5
50
3, –× × = 4,5 × 10–3 mol.
2.
États (CH3)3–CCl + 2 H2O(l) (CH3)3–C–OH + H3O
+(aq) + Cl–(aq)
Initial 4,5 × 10–3 nexcès 0 0 0
Intermé-diaire 4,5 × 10–3 – x nexcès – 2 x x x x
Final 4,5 × 10–3
– xmaxnexcès – 2 xmax xmax xmax xmax
xB théorique = 4,5 × 10–3 mol.
4 Suivi de la transformation1.
0
1
2
3
4
5
6
7
8
9
0 200 400 600 t (s)
s (mS · cm–1)
2. x(t) = n t0 · ( )σσ final(1)
.
n(RCl)restant (t) = n0 – x(t) ;
masse(RCl non hydrolysé) = (n0 – x(t)) × 92,5 ;
masse(RCl non hydrolysé) = n0 × 92,5 (1 – σσ
( )t
final
).
Par exemple, à t = 3 min (soit t = 180 s), on a :masse(RCl non hydrolysé) = 4,5 × 10–3
× 92,5 (1 – 6 46
8 53
,
,) = 1,71 g.

18 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
1 Mots manquants1. Visible. 3. Spectre.2. Blanc. 4. Solutions étalons.
2 Vrai ou faux ?1. Faux : Pour suivre une transformation chimique par conductimétrie, au moins l’une des espèces chimiques de la transformation doit être ionique.2. Faux : L’évolution temporelle d’un système chimique peut être réalisée en étudiant les varia-tions de la concentration.3. Faux : Il n’est pas indispensable que les réactifs soient dans les proportions stœchiométriques.
3 QCM1. c) Titrage.2. b) À un instant t, la quantité de matière de dihy-drogène formé est égale à l’avancement x(t).3. c) À l’équivalence, l’avancement de la réaction est : xmax = 0,5 n(thiosulfate)versé.4. b) Permanganate de potassium.
4 Le bleu de bromothymol1. La forme basique (pH = 12) absorbe dans le rouge – magenta : sa couleur est complémentaire de celle absorbée, soit le bleu vert.2. La forme acide présente un maximum d’absor-bance dans le visible autour de 450 nm (dans le bleu) : sa couleur est donc le jaune.
5 Suivi par conductimétrie1.
État (CH3)3–CCl + 2 H2O (CH3)3–C–OH + H3O
+ + Cl– (aq)
Initial 3,70 × 10–3 nexcès 0 0 0
Intermé-diaire 3,70 × 10–3 – x nexcès – 2 x x x x
Final 3,70 × 10–
– xmaxnexcès– 2 xmax xmax xmax xmax
2. xmax = 3,70 × 10–3 mol.
3. σ = λ(H3O+) · [H3O
+(aq)] + λ(Cl–(aq) · [Cl–(aq)] ;
σ = λ(H3O+) ·
x
V t( ) + λ(Cl–) ·
x
V t( ) ;
σ = x
V t( ) (λ(H3O
+) + λ(Cl–) ;
σ = x
V
(λ λ(H O ) (Cl ))3–+ +
(λ(H3O+) + λ(Cl–).
4. σ(t) en S · m–1, k en S · m–1 · mol–1, x(t) en mol.
5. σ(t) = k · x(t).σfinal = k · xmax.
σσ
( )t
final
= x t
x
( )
max
= x t
n
( )
0
, donc x(t) = n t0 · ( )σ
σ final
.
6. a) x2 = 5 10 3 7 10
9 10
3, ,
,
–× × = 2,1 × 10–3 mol.
n(RCl)restant = n0 – x2 ;n(RCl)restant = 3,7 × 10–3 – 2,1 × 10–3 = 1,6 × 10–3 mol.
b) m(RCl)non hydrolysé = (n0 – x2) ; m = 0,15 g.
6 Suivi par prélèvement1. a) S2O8
2–(aq) + 2 e– = 2 SO42–(aq).
2 l–(aq) = I2(aq) + 2 e– S2O8
2–(aq) + 2 l–(aq) 2 SO42–(aq) + I2(aq).
b)Oxydation
S2O82–(aq) + 2 l–(aq) 2 SO4
2–(aq) + I2(aq) .
Réduction
2. V1 = 500,0 mL ; C1 = 1,50 × 10–2 mol · L–1.n(S2O8
2–(aq)) = 0,5 × 1,50 × 10–2 = 7,50 × 10–3 mol.V2 = 500,0 mL ; C2 = 1,00 × 10–1 mol · L–1.n(I–) = 0,5 × 1,00 × 10–1 = 5,00 × 10–2 mol.
3. a)
S2O82–(aq) + 2 l–(aq) 2 SO4
2–(aq) + I2(aq)
t = 0 7,5 × 10–3 5,00 × 10–2 0 0
t 7,5 × 10–3 – x 5,00 × 10–2 – 2 x x 2 x
t final 7,5 × 10–3 – xmax 5,00 × 10–2 – 2 xmax xmax 2 xmax
b) x(t) = n(I2)formé = [I2(aq)] (V1 + V2).
c) xmax = 7,50 × 10–3 mol.
[I2(aq)]formé = x
V Vmax
1 2+ ;
[I2(aq)]formé = 7,50 × 10–3 mol · L–1.
7 Hydrolyse basique d’un composé halogéné1. a) n(hydroxyde)initial = 0,500 × 1,00 × 10–1
= 0,500 × 10–1 mol.
n(RCl)initial = 4 60
92 6
,
, = 49,6 × 10–3 mol.
Corrigés des exercices (page 47)

19Chapitre 2 - Suivi temporel d’une transformation chimique
b) RCl(aq) + HO–(aq) ROH(aq) + Cl–(aq)
Quantité de matière (mol)
t = 0 4,96 × 10–2 5,00 × 10–2 0 0
t 4,96 × 10–2
– x5,00 × 10–2
– xx x
t final – xmax5,00 × 10–2
– xmaxxmax xmax
T final 4,96 × 10–2 4,96 × 10–2 4,96 × 10–2
2. Le volume de la solution est 1,000 L[RCl(aq)]initial = 4,96 × 10–2 mol · L–1.[HO–(aq)]initial = 5,00 × 10–2 mol · L–1.
3. a) H3O+(aq) + HO–(aq) 2 H2O(l).
n(hydroxyde)restant = n(H3O+)versés à l’équivalence.
il vient [HO–(aq)] = C V
v1 · e ,
b) n(HO–)restant = C V V
v1 · ·e T .
4. a) Le tableau d’évolution montre :n(HO–)restant = n(hydroxyde)initial – x(t).
x(t) = n(hydroxyde)initial – n(HO–)restant ;x(t) = 5,00 × 10–2 – 1,55 × 10–2 = 3,45 × 10–2 mol.
b) La transformation n’est pas terminée :xmax = 4,96 × 10–2 mol.
8 Réduction du diiode par le zinc1. La concentration en diiode diminue, la couleur jaune s’éclaircit progressivement jusqu’à l’incolore.
2. V = 250,0 mL ; C0 = 20,0 × 10–3 mol · L–1;donc n0(I2) = 0,25 × 20,0 × 10–3 = 5,00 × 10–3 mol.
3. a)
États I2(aq) + Zn(s) 2 I–(aq) + Zn2+(aq)
Initial 5,00 × 10–3 nexcès 0 0
Final 5,00 × 10–3 – xmax 2 xmax xmax
b) x(t) = n(I2)consommé = 5,00 × 10–3 – x(t).
c) [I2(t)(aq)] = n
V
(I )2 ; [I2(t)(aq)] = 5,00 × 10–3 – x t
V
( ).
d) xmax = 5,00 × 10–3 mol.
4. n(I–)final = 2 xmax = 10,0 × 10–3 mol · L–1.
[I–(aq)]final = 10 0 10
0 25
3,
,
–× = 40,0 × 10–3 mol · L–1.
n(Zn2+)final = xmax = 5,00 × 10–3 mol · L–1.
[Zn2+(aq)]final = 20,0 × 10–3 mol · L–1.
5. [I2(aq)] · V = 5,00 × 10–3 – x(t),soit x(t) = 5,00 × 10–3 – [I2(aq)] × V.
t (s)0 200 400 600 800 1 000 1 200 1 4000
5
10
15
20
25[diiode] (mmol · L–1)
t (s)0 200 400 600 800 1 000 1 200 1 4000123456
x (mmol)
x = g(t).
9 Dosage d’une solution aqueuse de diiode par étalonnage
C (mmol · L–1)0 0,5 1 1,5 2 2,5 3 3,50
0,20,40,8
11,21,41,61,8
A
On trouve : Cdiluée = 1,9 × 10–3 mol · L–1.
2. Si la solution n’avait pas été diluée, le spectro-photomètre aurait été à saturation. Pour réaliser la dilution : fiole jaugée de 100 ml ; pipette jaugée de 10 ml ; solution mère ; eau distillée.3. C0 = 10 Cdiluée = 1,9 × 10–2 mol · L–1.
10 Utiliser la loi de Beer-Lambert pour un dosage
1. La solution de diiode absorbe dans le visible, plus particulièrement dans le bleu (couleur com-plémentaire de celle de la solution).2.V(S) mL 0,0 2,0 4,0 6,0 8,0 10,0
V’(eau) (mL)
10,0 8,0 6,0 4,0 2,0 0,0
[I2] (mol · L1)
0 0,0001 0,002 0,003 0,004 0,005
A 0,000 0,248 0,492 0,751 1,100 1,266

20 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
3. a)A
[I ]2
2,48× 103
2,46× 103
2,50× 103
2,75× 103
2,53× 103
b) Exceptée l’avant-dernière mesure, la valeur moyenne est : k = 2,50 × 103.A étant sans unité, k s’exprime en L · mol–1.
4. [I2]' = A
k
' ; [I2]' =
0 880
2 50 103
,
, × = 3,5 × 10–4 mol · L–1.
11 Dosage de la caféine
Exercice résolu dans le manuel de l’élève.
12 Oxydation des ions iodure1. Toutes les espèces chimiques sont incolores, sauf le diiode. Les solutions de diiode ayant une absorbance maximale voisine de 400 nm, il est inté-ressant de suivre par spectrophotométrie l’évolu-tion d’un système chimique au sein duquel se forme du diiode.2. S2O8
2–(aq) + 2 e– = 2 SO42–(aq)
2 l–(aq) = I2(aq) + 2 e– S2O8
2–(aq) + 2 l–(aq) 2 SO42–(aq) + I2(aq).
3.
État Avan-cement
S2O82–(aq) + 2 I–(aq)
SO42–(aq) + I2 (aq)
Initial 0,50 × 10–4 2,0 × 10–4
Intermé-diaire
0,50 × 10–4
– x2,0 × 10–4
– 2 xx x
Final 0,5 × 10–4
– xmax
2,0 × 10–4
– 2 xmaxxmax xmax
b) Les ions peroxodisulfate constituent le réactif limitant.0,50 × 10–4 – xmax = 0 donne x1m = 0,50 × 10–4mol.
4. n(diiode formé) = x1m = 0,50 × 10–4 mol.
5. a) Absorbance maximale = 1,65.b) A = k · [I2(aq)] · Amax = k · [I2(aq)]max.
k = Amax
2[I (aq)] =
A V
nmax
formé(diode)
· ; k = 50 L · mol–1.
6. a) t = 4 min : A1 = 1,18 et A2 = 1,52.b) Le facteur cinétique mis en évidence par la com-paraison de ces deux expériences est la concentra-tion initiale de l’un des réactifs.c) • Série 1 [I2(aq)]1(t = 4 min) =
A
k1 ;
[I2(aq)]1(t = 4 min) = 2,4 × 10–2 mol · L–1.
• Série 2 [I2(aq)]2(t = 4 min) =
A
k2 ;
[I2(aq)]2(t = 4 min) = 3,0 × 10–2 mol · L–1.
À l’instant de date t = 4 min, l’avancement de la réaction est plus important pour le système chimi-que 2, où la concentration initiale de l’un des réac-tifs est plus élevée.• Série 1 : x1(t = 4 min) = 3,6 × 10–5 mol.• Série 2 : x2(t = 4 min) = 4,6 × 10–5 mol.
13 Cinétique de la saponification du méthanoate d’éthyle
1. a) NaOH(s) = Na+(aq) + HO–(aq)Transformation totale.
D’après l’équation précédente, on a :[HO–(aq)] = [Na+(aq)] = C0 = 1,00 × 10–2 mol · L–1.
b) G0 = k · (λ(Na+) · [Na+(aq) + λ(HO–) · [HO–(aq)]) ; G0 = k · C0 · (λ(Na+) + λ(HO–) ;
G0 = k
V · n0 · (λ(Na+) + λ(HO–)). (1)
c) G0 = k · C0 · (λ(Na+) + λ(HO–)) Il faut convertir C0 en mol · m–3, donc multiplier C0 par 103.G0 = 0,01 × 1,00 × 10–2 × 103
× (5,01 × 10–3 + 19,9 × 10–3)G0 = 0,01 × 1,00 × 10–2 × (5,01 + 19,9) = 2,49 mS.
On conserve trois chiffres significatifs (C.S.), bien que k soit donnée avec un seul C.S., les valeurs de G du tableau 1 comportant trois C.S.2.
H–CO2–CH2–CH3 + HO= HCO2
– + CH3–CH2–OH
État Avanc.(mol)
Quantités de matière (mol)
Initial 0 n0 n0 0 0
Inter-médiaire x n0 – x n0 – x x x
3. a) Gt =k
V· [λ(Na+) · n0 + λ(HO–) · (n0 – x)
+ λ(HCO2–) · x]. (2)Gt = k · (λ(Na+) · [Na+(aq)] + λ(HO–) · [HO–(aq)]
+ λ(HCO2–) · [HCO2–(aq)].
Gt = k · (λ(Na+) · n
V0 + λ(HO–) ·
n x
V0 –
+ λ (HCO2–) · x
V.
21Chapitre 2 - Suivi temporel d’une transformation chimique
b) On développe l’expression (2) :
Gt = k
V · [λ(Na+) · n0] +
k
V · [λ(HO–) · n0]
– k
V · [λ(HO–) · x] +
k
V · [λ(HCO2–) · x].
Gt = k
V · x · [λ(HCO2–) – [λ(HO–)]
+ k
V · n0 · [λ(Na+) + λ(HO–)].
Gt = ax + b (3)
avec a = k
V · [λ(HCO2–) – [λ(HO–)]
et b = k
V · n0 · [λ(Na+) + λ(HO–)].
c) L’expression b correspond à G0, calculée à la question 1.b). λ(HO–) > λ(HCO2–) : on en déduit que la constante a est négative.d) La représentation de Gt en fonction de x serait une droite présentant un coefficient directeur négatif.
4. x = f(t).
t (min)0 20 40 60 80 100 1200
0,5
1
1,5
2
2,5x (mmol)
x (mmol) = f (t)t en min
14 Chimie et spéléologie
1. d = M( )CO2
29 ; d =
44
29 = 1,5.
Le gaz est plus dense que l’air : il est susceptible de s’accumuler dans les parties inférieures de la grotte.
2. Quantité de matière d’ions oxonium :n = C · VS ; n = 0,100 × 0,1 = 1 × 10–2 mol.
Quantité de matière de carbonate de calcium :
n = m
M( )CaCO3
; m = 2 0
100
, = 2,0 × 10–2 mol.
3.Équation chimique
CaCO3(s) + 2 H3O+(aq)
= Ca2+(aq) + CO2(g) + 3 H2O(l)
État Av. (mol) Quantités de matière (mol)
Initial xi = 0 2,0 × 10–2 1 × 10–2 0 0 Beaucoup
En cours de trans-formation
x 2,0 × 10–2 – x
1 × 10–2
– 2 x x x Beaucoup
Final(si totale) xmax
2,0 × 10–2
– xmax
1 × 10–2
– 2 xmaxxmax xmax Beaucoup
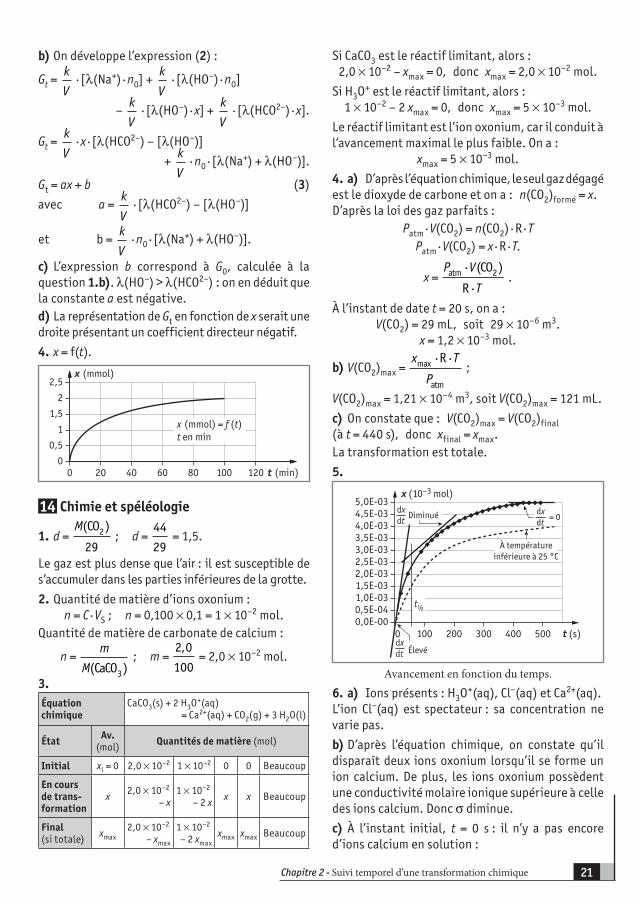
Si CaCO3 est le réactif limitant, alors :2,0 × 10–2 – xmax = 0, donc xmax = 2,0 × 10–2 mol.
Si H3O+ est le réactif limitant, alors :
1 × 10–2 – 2 xmax = 0, donc xmax = 5 × 10–3 mol.
Le réactif limitant est l’ion oxonium, car il conduit à l’avancement maximal le plus faible. On a :
xmax = 5 × 10–3 mol.
4. a) D’après l’équation chimique, le seul gaz dégagé est le dioxyde de carbone et on a : n(CO2)formé = x.D’après la loi des gaz parfaits :
Patm · V(CO2) = n(CO2) · R · TPatm · V(CO2) = x · R · T.
x = P V
Tatm 2(CO )
R
·
· .
À l’instant de date t = 20 s, on a :V(CO2) = 29 mL, soit 29 × 10–6 m3.
x = 1,2 × 10–3 mol.
b) V(CO2)max = x T
Pmax
atm
R· · ;
V(CO2)max = 1,21 × 10–4 m3, soit V(CO2)max = 121 mL.
c) On constate que : V(CO2)max = V(CO2)final(à t = 440 s), donc xfinal = xmax.La transformation est totale.
5.
0,0E-000,5E-041,0E-031,5E-032,0E-032,5E-033,0E-033,5E-034,0E-034,5E-035,0E-03
0 100 200 300 400 500 t (s)
x (10–3 mol)
dxdt
= 0
dxdt Élevé
dxdt
Diminué
À températureinférieure à 25 °C
t½
Avancement en fonction du temps.
6. a) Ions présents : H3O+(aq), Cl–(aq) et Ca2+(aq).
L’ion Cl–(aq) est spectateur : sa concentration ne varie pas.
b) D’après l’équation chimique, on constate qu’il disparaît deux ions oxonium lorsqu’il se forme un ion calcium. De plus, les ions oxonium possèdent une conductivité molaire ionique supérieure à celle des ions calcium. Donc σ diminue.
c) À l’instant initial, t = 0 s : il n’y a pas encore d’ions calcium en solution :

22 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
σini = λ(H3O+) · [H3O
+(aq)] + λ(Cl–) · [Cl–(aq)].
De plus : C = [H3O+(aq)]ini = [Cl–(aq)].
σini = [λ(H3O+) + λ(Cl–)] · C.
σini = [35,0 × 10–3 + 7,5 × 10–3] × 0,1 × 103.Attention : les concentrations sont à exprimer en mol · m–3.
σini = 4,25 S · m–1.
d) σ = λ(H3O+) · [H3O
+(aq)] + λ(Cl–) · [Cl–(aq)]+ λ(Ca2+) · [Ca2+(aq)].
σ = λ(H3O+) ·
C V x
V
· –S
S
2 + λ(Cl–) · C + λ(Ca2+) ·
x
VS
.
σ = λ(H3O+) · C ·
2x
V
· ( )λ H O3
S
+
+ λ(Cl–) · C + λ(Ca2+) · x
VS
.
σ = λ(H3O+) + λ(Cl–) · C ·
x
VS
· [– 2 λ(H3O+) + λ(Ca2+)].
σ = σini + x
VS
· [– 2 λ(H3O+) + λ(Ca2+)].
σ = 4,25 + x
0 100 10 3, –×× (– 2 × 35,0 × 10–3 + 12,0 × 10–3).
Attention : les volumes sont en m3.σ = 4,25 – 580 × x.
e) σmax = 4,25 – 580 × xmax × σmax = 1,35 S · m–1.
15 Suivi d’une transformation lente1. On procède à une dilution.– solution mère :C0 = 1,00 × 10–2 mol · L–1 ; V0, volume à prélever ;
– solution fille :C1 = 2,00 × 10–3 mol · L–1 ; V1 = 50,0 mL.
Au cours de la dilution, la quantité de matière de permanganate de potassium se conserve.Soit n0 = n1, donc C0 · V0 = C1 · V1.
V0 = C V
C1 1–
0
; V0 = 2 00 10 50 0
1 00 10
3
2
, – ,
,
–
–
××
= 10,0 mL.
• Protocole : On verse de la solution mère dans un bécher de 75 mL. À l’aide d’une pipette jaugée de 10 mL, on prélève V0 millilitres de la solution mère de ce bécher. On verse ce prélèvement dans une fiole jaugée de 50 mL. On ajoute de l’eau distillée jusqu’au trait de jauge en agitant au fur et à mesure de l’ajout, puis on homogénéise.
2. a) Demi-équation de réduction de l’ion perman-ganate :MnO4
–(aq) + 5 e– + 8 H
+(aq) = Mn2+(aq) + 4 H2O(l) × 2
Demi-équation d’oxydation de l’acide oxalique :H2C2O4(aq) = 2 CO2(aq) + 2 e– + 2 H+(aq) × 5Équation globale de la réaction :2 MnO4
–(aq) + 5 H2C2O4(aq) + 6 H+(aq)= 10 CO2(aq) + 2 Mn2+(aq)+ 8 H2O(l).
b) À l’instant t = 0 s, on a : n01 = C1 · V1 ; n01 = 2,00 × 10–3 × 20,0 × 10–3 ; n01 = 4,00 × 10–5 mol d’ions MnO4
–.
c) À l’instant t = 0 s, on a : n02 = C2 · V2 ; n02 = 5,00 × 10–2 × 20,0 × 10–3 ; n02 = 1,00 × 10–3 mol d’acide oxalique C2H4O2.
d) Attention : pour calculer l’avancement maximal, il faut tenir compte des coefficients stœchiométriques.
Si MnO4– est le réactif limitant, il est totalement
consommé. Alors :n01 – 2 xmax = 0,
soit xmax = n01
2 =
4 00 10
2
5, –× = 2,00 × 10–5 mol.
Si C2H4O2 est le réactif limitant, alors :n02 – 5 xmax = 0,
soit xmax = n02
5 =
1 00 10
5
3, –× = 2,00 × 10–4 mol.
Le réactif limitant est celui qui conduit à la valeur la plus faible de xmax. Il s’agit de l’ion permanga-nate :
xmax = 2,00 × 10–5 mol.
e) Les ions MnO4– colorent le mélange réactionnel
en violet. Au fur et à mesure de leur consommation, la coloration violette disparaît : le mélange devient progressivement incolore.
3. a) La seule espèce chimique colorée est MnO4–,
qui est responsable de l’absorbance de la solution.b) Les ions permanganate sont consommés au cours de l’évolution de la transformation chimique : en conséquence, l’absorbance de la solution diminue.Lorsque la transformation est terminée, tous les ions permanganate, réactif limitant, ont été consommés, l’absorbance de la solution tend vers zéro.
4. a) Nouvelle quantité de matière initiale d’acide oxalique : n02' = C2 · V2 ; n02' = 2,50 × 10–3 × 20,0 × 10–3 ; n02' = 5,00 × 10–5 mol.

23Chapitre 2 - Suivi temporel d’une transformation chimique
b) Pour MnO4–, d’après la question 2.d), on a :
xmax = 2,00 × 10–5 mol.
Pour C2H4O2 : n02' – 5 xmax = 0
soit n02
5
' =
5 00 10
5
5, –× = 1,00 × 10–5 mol,
avancement le plus faible des deux réactifs. Lors de cette nouvelle expérience, l’acide oxalique est le réactif limitant.
c) D’après la relation donnée à la question 3.b), on a :
x(t) = (2 × 10–5 – A(t) × 10 –5)A(t) × 10–5 = 2 × 10–5 – x(t) ;A(t) = [2 × 10–5 – x(t)] × 105.
Pour tfinale, la transformation étant supposée totale :
A(tfinale) = [2 × 10–5 – xmax] × 105 ;A(tfinale) = [2 × 10–5 – 1,00 × 10–5] × 105 = 1.
16 Transformation en double suivi1. Premier système
n(Mg) = 4,1 × 10–2 mol ;n(oxonium) = 3,0 × 10–3 mol.
Deuxième systèmen(Mg) = 4,1 × 10–2 mol ;
n(oxonium) = 1,0 × 10–2 mol.
2. x1m = 1,5 × 10–3 mol (ion oxonium, réactif limitant) ; x2m = 5,0 × 10–3 mol (ion oxonium, réactif limitant).
3. x1(t) = 3,0 10 –[H O (aq)]·
2
–33× + V
.
x1(t = 9 min) = 1,5 × 10–3 mol.
4. a) n(H2)(t) = x2(t).b) ∆P · V = n(H2) · R · T.c) x2(t) = 2,8 × 10–3 mold) ∆Pfinal = 398 hPa ; Pfinal = 1 408 hPa.
17 Un antiseptique courant, le Lugol®
1. La courbe représentative de A = f([I2(aq)]) (Fig. 1, p. XX) est une droite qui passe par l’ori-gine.L’absorbance étant proportionnelle à la concentra-tion en diiode, on écrit : A = k · [I2(aq)], avec k le coefficient de proportionnalité.
2. Amax = 2,00.L’abscisse du point d’ordonnée correspondante donne la valeur de [I2(aq)]max :
[I2(aq)]max = 8,0 × 10–3 mol · L–1.
00,5
11,5
22,5
3
0 0,002 0,004 0,006 0,008 0,01 0,012
A
[I2(aq)](mol · L–1)
3. La solution étant diluée dix fois, il faut une pipette jaugée de 10,0 mL et une fiole jaugée de 100,0 mL.
4. a) On prend l’abscisse du point d’ordonnée AS'0 = 1,00, soit [I2(aq)]S'0 = 4,0 × 10–3 mol · L–1.
b) CL = 10 × [I2(aq)]S'0 (solution S0 diluée dix fois) ; CL = 10 × 4,0 × 10–3 = 4,0 × 10–2 mol · L–1
c) Si on ne dilue pas la solution commerciale, l’ab-sorbance n’est pas mesurable, car
CL > [I2(aq)]max pour le spectrophotomètre.
5. Un oxydant est une espèce chimique capable de capter un ou plusieurs électrons.• Couple H2O2(aq)/H2O(l) : réduction, soit
H2O2(aq) + 2 H+(aq) + 2 e– = 2 H2O(l).Mais ici, compte tenu de l‘équation chimique pro-posée, on écrira plutôt :
H2O2(aq) + 2 H3O+(aq) + 2 e– = 4 H2O(l).
• Couple I2(aq)/I–(aq) : oxydation, soit2 I–(aq) = I2(aq) + 2 e–.
6. Relation stœ-chiométrique
H2O2(aq) + 2 I–(aq) + 2 H3O+(aq)
= I2(aq) + 4 H2O(l)
État du système Av. Bilan de matière (mol)
Initial 0 C2 · V2 Excès Excès 0 Solvant
Au cours de la transfor-mation
x C2 · V2 – x Excès Excès x Solvant
Final xfC2 · V2
– xfExcès Excès xf Solvant
Final si transfor-mation totale
xmaxC2 · V2– xmax = 0 Excès Excès xmax =
C2 · V2Solvant
7. [I2(aq)](t) = n t
V
(I )( )2
tot
.
Or d’après le tableau d’évolution :
n(I2)(t) = x(t) · [I2(aq)](t) = x
V
(t)
tot
,
soit x(t) = [I2(aq)](t) · Vtot.

24 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
Exercice supplémentaire
Hydrolyse basique du 2-bromobutane(D’après Bac Aix Marseille 1989)
Un composé halogéné, le 2-bromobutane, de for-mule CH3CHBr–CH2–CH3, réagit avec les ions hydroxyde HO– au cours d’une transformation lente, appelée hydrolyse basique, conduisant à la formation d’un alcool.L’équation associée à la réaction s’écrit :CH3CHBr–CH2–CH3 + HO–(aq)
CH3CHOH–CH2–CH3 + Br–(aq).Pour étudier la cinétique de cette hydrolyse basique, on prépare 5 échantillons initialement identiques en mélangeant à un instant de date t = 0 un volume V1 = 50,0 mL de solution alcoolique contenant 5,0 × 10–2 mol de 2-bromobutane et un volume V2 = 50,0 mL de solution alcoolique d’hydroxyde de potassium, K+(ol) + HO–(ol), de concentration en soluté c2 = 1,00 mol · L–1.À cinq instants différents, on réalise la trempe chimique d’un échantillon et on procède à un titrage
des ions bromure formés. Les résultats sont consi-gnés dans le tableau suivant.
Instant t (min) 0 15 30 45 60 90
[Br–(aq)] (mmol · L–1) 0 220 300 345 370 410
1. a) Déterminer la quantité de matière d’ions hydroxyde initialement présente dans le système chimique.b) Dresser un tableau d’évolution du système et en déduire la valeur de l’avancement maximal, xm.2. Montrer que la concentration en ions hydroxyde est donnée à chaque instant t par la relation :
[HO–(aq)] = 5,0 × 10–1 – 10x(avec x exprimé en mol · L–1).
3. a) À l’instant t5 = 90 min, la transformation est-elle terminée ?b) Quelle est la valeur de l’avancement de la réac-tion à la date t2 = 30 min ?
Les compétences expérimentales
(page 56)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de revenir sur des techniques de base que l’on retrouve de manière récurrente dans les sujets d’évaluation. Cet exercice doit conduire l’élève à justifier ses choix.
Corrigé1. Préparer d’une solution par dilutionUne dilution doit être réalisée avec la verrerie la plus précise possible. Les éprouvettes graduées sont donc à proscrire pour prélever les volumes de solution :– pour des volumes inférieurs à 50 mL, les prélèvements se font avec des pipettes jaugées munies de propipette ;– pour un prélèvement de 50,0 mL, la fiole jaugée paraît idéale, mais si l’on doit verser le prélèvement, il ne faut pas oublier qu’il s’agit d’un matériel « in ».
• La proposition faite sur le dessin B est correcte. Il faut en effet récupé-rer les eaux de rinçage de la fiole afin d’être certain que toute la quantité de matière prélevée se retrouve effectivement dans la solution finale.
2. Utilisation correcte d’un spectrophotomètreIl faut « étalonner » le spectrophotomètre préalablement à toute mesure.
• La proposition faite sur le dessin B est correcte.Toute mesure nécessite une référence (le « blanc ») généralement de l’eau distillée (le solvant) et les cuves utilisées doivent être toutes identiques.

25Chapitre 2 - Suivi temporel d’une transformation chimique
2. [HO–(aq)] = n
V
(HO )–
;
[HO–(aq)] = 5 00 10 2, ––× x
V = 5,0 × 10–1 – 10x.
3. À t5 : [Br–(aq)] = 10x,
soit :
x5 = 0 410
10
, = 0,041 mol = 4,11 × 10–2 mol < xmax.
La transformation n’est pas terminée.x30 = 3,00 × 10–2 mol.
4. Il y a consommation des ions hydroxyde, rempla-cés par les ions bromure.La conductivité σ diminue et tend vers une limite non nulle.
Corrigé1. a) n0(HO–) = C2 · V2 ;n0(HO–) = 50 × 10–3 × 1,00 = 50,0 × 10–3 mol.n(bromobutane) = 5,0 × 10–2 mol.
b)
États CH3CHBr–CH2–CH3 + HO–(aq) CH3CHOH–CH2–CH3 + Br–(aq)
Initial 50,0 × 10–3 5,00 × 10–2 0 0
En cours de transfor-mation
50,0 × 10–3 – x 5,00 × 10–2 – x x x
Final 50,0 × 10–3
– xmax
5,00 × 10–2
– xmaxxmax xmax
xmax = 5,00 × 10–2 mol.

26 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Utilisation d’un tableur-grapheur pour tracer la courbe x = f(t), par exemple, et déterminer la vitesse à différentes dates.Détermination de t1/2 à partir de résultats expé-rimentaux.Il lustration des évé-n e m e n t s a u n ive a u macroscopique.
– Vitesse de réactionDéfinition de la vitesse volumique de réaction exprimée en unité de quantité de matière par unité de temps et de volume.
v = 1V
xt
⋅ dd
,
où x est l’avancement de la réaction et V le volume de la solution.Évolution de la vitesse de réaction au cours du temps.– Temps de demi-réaction, noté t1/2Définition et méthodes de détermination.Choix d’une méthode de suivi de la transformation selon la valeur de t1/2.Interprétation de la réaction chimique en termes de chocs efficaces.Interprétation de l’influence de la concentration des entités réactives et de la température sur le nombre de chocs et de chocs efficaces par unité de temps.
– Savoir que la vitesse de réac-tion augmente en général avec la concentration des réactifs et avec la température.– Interpréter qualitativement la variation de la vitesse de réaction à l ’aide d ’une des courbes d’évolution tracées.– Connaître la définition du temps de demi-réaction t1/2.– Déterminer le temps de demi-réaction à l’aide de don-nées expérimentales ou en exploitant des résultats expé-rimentaux*.
3 Vitesse de réaction
Cours
Découpage du cours1. Vitesse volumique de la réaction p. 582. Détermination graphique de la vitesse volumique p. 583. Temps de demi-réaction p. 594. Facteurs influençant la vitesse et le temps de demi-réaction p. 605. Interprétation au niveau macroscopique p. 61
Dans le chapitre 2, les élèves ont étudié les différentes méthodes de suivi des transformations chimiques.Ce suivi temporel des transformations chimiques a conduit à une courbe traduisant l’évolution dans le temps de l’avancement x(t) de la réaction.
1. Vitesse volumique de la réactionElle est exprimée en unité de quantité de matière par unité de temps et de volume.Si le volume est souvent exprimé en L, le temps est avant tout adapté à l’évolution temporelle étudiée et peut être en secondes, minutes, heures ou jours.
2. Détermination graphique de la vitesse volumique
Cette partie se base exclusivement sur des déter-minations graphiques. L’élève doit être capable
♦ d’interpréter qualitativement l’évolution de la vitesse à partir de la courbe traduisant l’évolution de l’avancement x en fonction du temps.
3. Temps de demi-réactionAprès l’avoir défini, on s’intéresse à sa détermina-tion graphique dans le cas d’une transformation totale (la transformation limitée est traitée cha-pitre 12, p. 245).
4. Facteurs influençant la vitesse et le temps de demi-réaction
L’occasion est donnée ici, de revenir sur les facteurs cinétiques et d’observer leur influence sur la vitesse de la réaction ainsi que sur le temps de demi-réaction.
5. Interprétation microscopiqueCette partie permet d’expliquer au niveau micros-copique la réaction chimique. On revient encore sur l’influence des différents facteurs cinétiques afin de bien faire le lien entre macroscopique et microsco-pique.

27Chapitre 3 - Vitesse de réaction
Suivi temporel d’un système chimique par prélèvement (page 64)
Dans ce TP, le suivi d’une transformation lente se fait grâce à une transformation rapide. Il est possible d’élaborer des variantes, par exemple un TP collec-tif à partir d’un mélange réactionnel de volume plus important que celui proposé dans le protocole, cha-que binôme réalisant un ou plusieurs prélèvements à des instants de dates choisies par le professeur.Il est intéressant d’aller au moins jusqu’au temps de
demi-réaction (n(diiode)formé = n(diiode)max
2) pour
une exploitation ultérieure (facteurs cinétiques, par exemple).Pour l’exploitation, on peut également raisonner sur l’évolution du système chimique contenu dans le prélèvement.
1. a)n(H2O2)introduit = 2,0 × 10–3 × 1,5 = 3,0 × 10–3 mol.n(iodure)introduit = 100 × 10–3 × 1,0 × 10–1 n(iodure)introduit = 10,0 × 10–3 mol.Les ions iodure sont nécessairement en excès afin de solubiliser le diiode formé au cours de la trans-formation chimique sous forme d’ions triiodure,I3–(aq).
État H2O2(aq) + 2 I–(aq) + 2 H3O+(aq)= I2(aq) + 4 H2O(l)
Initial 3,0 × 10–3 10,0 × 10–3 0 0 0
En cours de trans-formation
3,0 × 10–3
– x10,0 × 10–3
– 2 xx x x
Final 3,0 × 10–3 – xmax
10,0 × 10–3 – 2 xmax
xmax xmax xmax
b) Deux valeurs possibles pour l’avancement maxi-mal : 3,0 × 10–3 mol ou 10,0 × 10–3 mol.L’avancement maximal à prendre en compte corres-pond à la plus petite de ces deux valeurs : le peroxyde d’hydrogène est bien le réactif limitant.c) xmax théorique = 3,0 × 10–3 mol.
d) x(t) = n(diiode)formé(t) = [I2(aq)] · VT,avec VT, le volume total.
2. a)n(diiode)formé = 0,5 n(thiosulfate)versé à l’équivalence.b) On raisonne sur le prélèvementn(diiode)formé(t) = [I2(aq)] · VE ;
ACTIVITÉ
1
ACTIVITÉ
1 n(diiode)formé(t) = 0,5 n(thiosulfate)versé à l’équivalence ;n(diiode)formé(t) = 0,5 · C0 · VE.
[I2(aq)](t) = 0 5, · ·C V
V0 E .
On raisonne sur le système chimique initial contenu dans le bécher (cf. tableau) :
x(t) = [I2(aq)](t) · VT.
On peut également raisonner sur le système chimi-que initial contenu dans le prélèvement (40 fois plus petit) :
x'(t) = [I2(aq)](t) · V.
t (min) VE1 (mL) [I2(aq)] x (t)
0 0 0 0
3 7,5 0,00375 0,00075
6 12,7 0,00635 0,00127
10 17,3 0,00865 0,00173
15 22,1 0,01105 0,00221
20 24,6 0,0123 0,00246
25 26,5 0,01325 0,00265
30 28,2 0,0141 0,00282
35 29,1 0,01455 0,00291
40 29,7 0,01485 0,00297
50 29,8 0,0149 0,00298
60 29,8 0,0149 0,00298
3. Avancement du système chimique contenu dans le bécher. (On retrouve pratiquement la valeur de xmax théorique si l’eau oxygénée utilisée est très « fraîche » ; elle peut se dégrader assez rapidement après ouverture.)
0,0005
0,001
0,0015
0,002
0,0025
0,003
0,0035
00 20 40 60 80 t (min)
x (mol)
4. On se place à xmax
2 et on détermine l’abscisse
correspondante : on obtient t1/2 = 20 min.
Activités

28 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
Suivi temporel d’un système chimique par titrage du diiode formé (page 65)
Dans ce TP collectif, le suivi d’une transformation lente se fait grâce à une transformation rapide. Chaque binôme part du même système chimique initial : seule la durée d’évolution est différente.Dans une deuxième expérience, on modifie le sys-tème chimique initial afin de mettre en évidence le facteur cinétique « concentration initiale de l’un des réactifs ».1. a) n(peroxo)introduit = 5,0 × 10–3 × 5,0 × 10–2 ;n(peroxo)introduit = 2,5 × 10–4 moln(iodure)introduit = 20,0 × 10–3 × 2,00 × 10–1 ;n(iodure)introduit = 4,00 × 10–3 mol.Dans l’expérience 2, la quantité de matière initiale d’iodure de potassium est doublée.Les ions iodure sont nécessairement en excès afin de solubiliser le diiode formé au cours de la trans-formation chimique sous forme d’ions triiodure, I3–(aq).
b) Pour les deux expériences :xmax théorique = 2,5 × 10–4 mol.
2. 2 S2O32–(aq) = S4O6
2–(aq) + 2 e– I2(aq) + 2 e– = 2 l–(aq) 2 S2O3
2–(aq) + I2(aq) S4O62–(aq) + 2 l–(aq).
3. La trempe permet de bloquer la transformation chimique étudiée à un instant t, afin de connaître la quantité de matière de diiode formé à cette date.4. a)n(diiode)formé = 0,5 n(thiosulfate)versé à l’équivalence.b) n(diiode)formé = 0,5 · C0 · VE.c) S2O8
2–(aq) + 2 I–(aq) 2 SO42–(aq) + I2(aq).
x(t) = n(diiode)formé ; x(t) = 0,5 · C0 · VE.
5. t (min) VE1 (mL) VE2 (mL) x1 (mmol) x2 (mmol)
0 0 0 0 0
3 3,5 6,1 0,0175 0,0305
5 4,7 11,2 0,0235 0,056
7 5,8 14,3 0,029 0,0715
10 8 19,2 0,04 0,096
15 11,7 24,9 0,0585 0,1245
20 14,1 29,2 0,0705 0,146
25 17,3 32,3 0,0865 0,1615
30 19,3 37,5 0,0965 0,1875
35 21,5 39,9 0,1075 0,1995
40 23,2 41,8 0,116 0,209
ACTIVITÉ
2
ACTIVITÉ
2 6. x1 = f(t) et x2 = g(t).
t (min)
x (mmol)
0,05
0,1
0,15
0,2
0,25
00 20 40 60
Série 1Série 2
x m2
7. a) Expérience 1 : t1/2 = 44 min.Expérience 2 : t1/2 = 15 min.b) L’évolution du système chimique au cours de l’expérience 2 est plus rapide que dans l’expé-rience 1 (temps de demi-réaction plus petit) ; on met en évidence le facteur cinétique « concentra-tion initiale de l’un des réactifs ».
8. On trace la tangente à l’un des graphes à plu-sieurs instants : on constate que le coefficient directeur de cette tangente diminue au fur et à mesure que le système chimique évolue.
Suivi temporeld’un système chimique par spectrophotométrie (page 66)
Pour une meilleure précision, la courbe d’étalon-nage et le suivi devraient être réalisés en tenant compte du λmax de la courbe d’analyse spectrale (ici autour de 400 nm).Cependant, travailler à λmax nécessite des solutions très diluées, car il faut que l’état final puisse être étudié avec le spectrophotomètre (absorbance maxi-male = 2 pour la plupart des appareils).Aussi travailler de 420 à 480 nm permet de se don-ner un peu de latitude dans le suivi.
1. a) Cuve 1n(peroxo)introduit = 5,0 × 10–4 × 5,0 × 10–2 ;n(peroxo)introduit = 2,5 × 10–5 mol.n(iodure)introduit = 2,0 × 10–3 × 2,0 × 10–1 ;n(iodure)introduit = 4,00 × 10–4 mol.
ACTIVITÉ
3
ACTIVITÉ
3
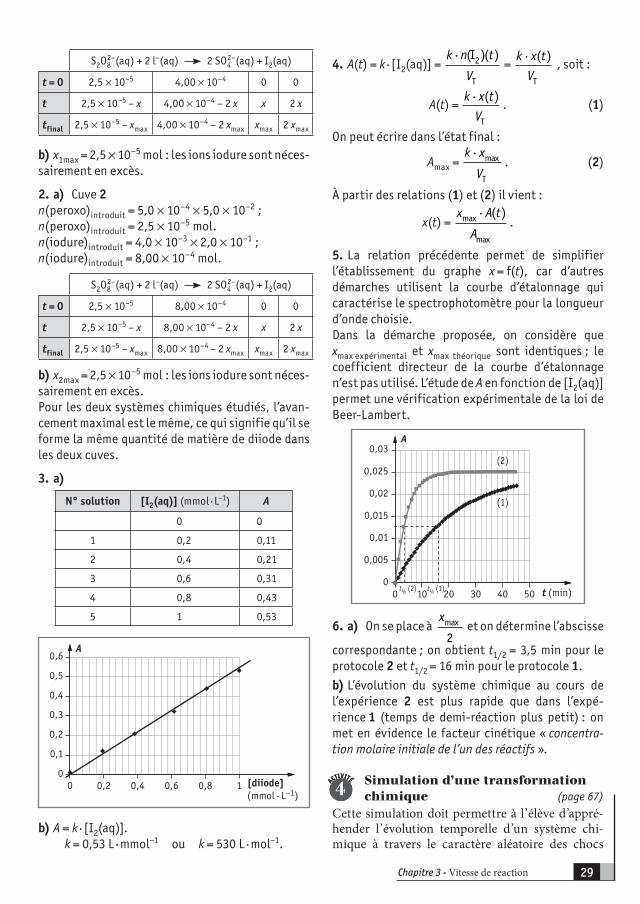
29Chapitre 3 - Vitesse de réaction
S2O82–(aq) + 2 l–(aq) 2 SO4
2–(aq) + I2(aq)
t = 0 2,5 × 10–5 4,00 × 10–4 0 0
t 2,5 × 10–5 – x 4,00 × 10–4 – 2 x x 2 x
tfinal 2,5 × 10–5 – xmax 4,00 × 10–4 – 2 xmax xmax 2 xmax
b) x1max = 2,5 × 10–5 mol : les ions iodure sont néces-sairement en excès.
2. a) Cuve 2n(peroxo)introduit = 5,0 × 10–4 × 5,0 × 10–2 ;n(peroxo)introduit = 2,5 × 10–5 mol.n(iodure)introduit = 4,0 × 10–3 × 2,0 × 10–1 ;n(iodure)introduit = 8,00 × 10–4 mol.
S2O82–(aq) + 2 l–(aq) 2 SO4
2–(aq) + I2(aq)
t = 0 2,5 × 10–5 8,00 × 10–4 0 0
t 2,5 × 10–5 – x 8,00 × 10–4 – 2 x x 2 x
tfinal 2,5 × 10–5 – xmax 8,00 × 10–4 – 2 xmax xmax 2 xmax
b) x2max = 2,5 × 10–5 mol : les ions iodure sont néces-sairement en excès.Pour les deux systèmes chimiques étudiés, l’avan-cement maximal est le même, ce qui signifie qu’il se forme la même quantité de matière de diiode dans les deux cuves.
3. a)
N° solution [I2(aq)] (mmol · L–1) A
0 0
1 0,2 0,11
2 0,4 0,21
3 0,6 0,31
4 0,8 0,43
5 1 0,53
0
0,1
0,2
0,3
0,4
0,5
0,6
0 0,2 0,4 0,6 0,8 1
A
[diiode](mmol · L–1)
b) A = k · [I2(aq)].k = 0,53 L · mmol–1 ou k = 530 L · mol–1.
4. A(t) = k · [I2(aq)] = k n t
V
· ( )(I )2
T
= k x t
V
· ( )
T
, soit :
A(t) = k x t
V
· ( )
T
. (1)
On peut écrire dans l’état final :
Amax = k x
V
· max
T
. (2)
À partir des relations (1) et (2) il vient :
x(t) = x A t
Amax
max
· ( ).
5. La relation précédente permet de simplifier l’établissement du graphe x = f(t), car d’autres démarches utilisent la courbe d’étalonnage qui caractérise le spectrophotomètre pour la longueur d’onde choisie.Dans la démarche proposée, on considère que xmax expérimental et xmax théorique sont identiques ; le coefficient directeur de la courbe d’étalonnage n’est pas utilisé. L’étude de A en fonction de [I2(aq)] permet une vérification expérimentale de la loi de Beer-Lambert.
0
0,005
0,01
0,015
0,02
0,025
0,03
0 10 20 30 40 50 t (min)
A
t½ (2) t½ (1)
(1)
(2)
6. a) On se place à xmax
2 et on détermine l’abscisse
correspondante ; on obtient t1/2 = 3,5 min pour le protocole 2 et t1/2 = 16 min pour le protocole 1.
b) L’évolution du système chimique au cours de l’expérience 2 est plus rapide que dans l’expé-rience 1 (temps de demi-réaction plus petit) : on met en évidence le facteur cinétique « concentra-tion molaire initiale de l’un des réactifs ».
Simulation d’une transformationchimique (page 67)
Cette simulation doit permettre à l’élève d’appré-hender l’évolution temporelle d’un système chi-mique à travers le caractère aléatoire des chocs
ACTIVITÉ
4
ACTIVITÉ
4

30 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
efficaces. Elle permet de bien faire comprendre la différence entre le mot réaction, qui traduit le résul-tat d’un tirage et d’un lancer de dé (nécessairement très rapide), et l’expression transformation chimi-que, qui se traduit au fil du temps par l’évolution de la composition du sac (niveau macroscopique).Le lancer de dé simule bien l’efficacité des chocs et permet d’introduire le facteur cinétique « tempéra-ture ».Cette simulation peut être utilisée pour présenter (dans les chapitres suivants) les transformations non totales : il suffit de considérer que le tirage d’une bille bleue et d’une bille verte conduit à une bille rouge et à une bille noire si le choc est efficace.Le nombre de tirages, volontairement limité à une centaine, ne permet pas d’obtenir des résultats par-faitement reproductibles dans les différents grou-pes, mais il met en évidence facilement les principales observations à faire. Seul un nombre de tirages très important permettrait d’obtenir des courbes exploitables.
1. a) Le nombre de chocs efficaces pour les élèves qui travaillent pourtant dans les mêmes conditions peut afficher une différence jusqu’à 5. Il peut être intéressant de sommer les résultats des élèves au sein d’un même groupe pour mieux obéir à la loi des grands nombres.Idem pour le groupe B.b) La transformation n’est pas terminée : il reste des boules rouges et des boules noires.
2. a) Le nombre de chocs efficaces est plus impor-tant pour les élèves du groupe B que pour ceux du groupe A.b) La transformation chimique simulée par le groupe B va plus vite que celle simulée par le groupe A.
3. Le professeur a choisi de simuler le facteur ciné-tique « température » : lorsque la température aug-mente, la probabilité de chocs efficaces croît.La simulation effectuée par le groupe B correspond à l’étude de la même transformation chimique que celle du groupe A, mais effectuée à une tempéra-ture plus élevée.
4. Si le groupe A travaille avec 20 billes rouges + 20 billes noires + 20 billes blanches, il suffit de donner au groupe B deux fois plus de billes blan-ches pour simuler une diminution de concentration des réactifs ou deux fois moins de billes blanches pour simuler une augmentation de la concentration des réactifs.Plus le nombre de molécules de solvant est grand et plus la probabilité de tirer une bille rouge et une bille noire est faible (on peut même, dans ce cas, s’affranchir du dé).
5. La réaction a lieu au niveau microscopique et résulte de chocs efficaces : le résultat est immé-diat. La transformation traduit l’évolution d’un ensemble d’entités réactives : cette évolution peut être rapide, lente et surtout dépend du système chimique en jeu (réactifs, température, pression).Pour donner une image permettant de bien diffé-rencier réaction et transformation, on peut imagi-ner une sortie de salle de cinéma. Chaque spectateur sort rapidement de la salle (réaction), mais l’écou-lement de l’ensemble des spectateurs hors de la salle dure plusieurs minutes. Cette durée pour vider la salle (transformation chimique) peut évoluer en fonction de divers paramètres :– diminuer (alerte incendie, intérêt à l’exté-rieur…) ;– augmenter (distribution, etc.).
Corrigés des exercices (page 68)
1 Compléter le résumé1. L’expression de la vitesse d’une réaction est
donnée par la relation v(t) = 1 ddV
xt
· , dans laquelle
V représente le volume de la solution et x(t) l’avan-cement de la réaction. La vitesse de réaction est maximale en début de transformation et tend vers zéro en fin de transformation. Pour déterminer la vitesse de réaction à un instant t sur un graphe x = f(t), il faut tracer la tangente au graphe à l’ins-
tant t, puis évaluer graphiquement le coefficient directeur de la tangente.
2. Pour déterminer graphiquement le temps de demi-réaction à partir d’un graphe x = f(t), il faut chercher graphiquement la valeur de l’avancement maximal xmax, qui correspond à l’ordonnée de l’asymptote horizontale du graphe. Puis il faut chercher sur le graphe, l’abscisse correspondant à xmax
2.

31Chapitre 3 - Vitesse de réaction
3. Une transformation chimique peut être modéli-sée par une ou plusieurs réactions chimiques. Acti-vées généralement par un apport d’énergie, les entités élémentaires appelées réactifs entrent en collision. Seuls les chocs efficaces conduisent à des entités nouvelles, appelées produits de la réaction.
2 Vrai ou faux1. a) Faux. b) Faux. c) Vrai. d) Faux. e) Faux.2. a) Vrai. b) Vrai. c) Faux. d) Vrai.
3 Exploiter un graphe1. b) 150 s.2. b) 4,1 × 10–3 mol · L–1.3. a) 8,2 × 10–6 mol.4. b) 2,1 × 10–5.
4 Facteur cinétique
Mélange
Solution aqueusede peroxodisulfate
de potassiumC0 = 5,0 × 10–2 mol · L–1
Solution aqueuse d’iodure de potassiumCR = 2,0 × 10–1 mol · L–1
A 50,0 mL 100,0 mL
B 60,0 mL 100,0 mL
C 50,0 mL100,0 mL + 50 mL
d’eau distillée
1. V3 > V2 > V1.2. Pour les trois mélanges :
n(iodure) = 20,0 × 10–3 mol.Pour les mélanges A et C :
n(peroxo) = 2,5 × 10–3 mol.Pour le mélange B : n(peroxo) = 3,0 × 10–3 mol.Pour les trois mélanges, les ions peroxodisulfate constituent le réactif limitant :– mélanges A et C : xmax = 2,5 × 10–3 mol ;– mélange B : xmax = 3,0 × 10–3 mol.
3. À l’instant t = 80 min, seule la transformation correspondant à la série 1 n’est pas terminée.
4. Les mélanges A et C présentant le même avance-ment maximal peuvent donc être comparés.Pour le mélange C, l’addition de 50 mL d’eau, diminue la concentration des réactifs, d’où une transforma-tion associée présentant un temps de demi-réaction plus grand : la durée de l’évolution de ce système chimique vers l’état final sera plus longue.
5. Les mélanges A et C présentent le même avance-ment maximal et correspondent aux séries 1 et 2.
Le graphe de la série 1 met en évidence un temps de demi-réaction plus grand que celui associé à la série : la série 2 correspond au mélange A, la série 1 au mélange C.Le mélange B avec un avancement final plus grand correspond à la série 3.
5 Oxydation des ions iodure1. L’absorbance n’a pas d’unité, aussi 500 s’exprime en L · mol–1.2. a)
État S2O82–(aq) + 2 I–(aq) SO4
2–(aq) + I2(aq)
Initial 1,0 × 10–5 4,0 × 10–4 0 0
Inter-médiaire 1,0 × 10–5 – x 4,0 × 10–4 – 2 x x x
Final 1,0 × 10–5 – xmax 4,0 × 10–4 – 2 xmax xmax xmax
b) Le réactif limitant est S2O82– (aq).
c) 1,0 × 10–5 – xmax = 0, d’où xmax = 10 × 10–6 mol.
3. a) A = 500 [I2(aq)], avec [I2(aq)] = x t
V V
( )
0 1+.
Il vient : x(t) = ( ) ·V V A0 1
500
+.
4. On se place sur le graphe à x = 4,6 × 10–6 mol : l’abscisse donne le temps de demi-réaction, soit environ 260 s.
5. On trace la tangente au graphe à l’instant t = 0 s. Le calcul du coefficient directeur donne :
∆∆
x
t = 2,5 à 3 × 10–8 mol · s–1.
En appliquant la relation v(t) = 1
V
x
t·d
d =
1
V
x
t·
∆∆
, on obtient :
v(t) = 1,0 à 1,2 × 10—5 mol · L–1 · s–1.
6.
0
0,005
0,01
0,015
0,02
0,025
0,03
0 10 20 30 40 50 t (min)
A
t½ (2) t½ (1)
(1)
(2)
L’évolution du système est beaucoup plus lente.
7. a) Absorbance.
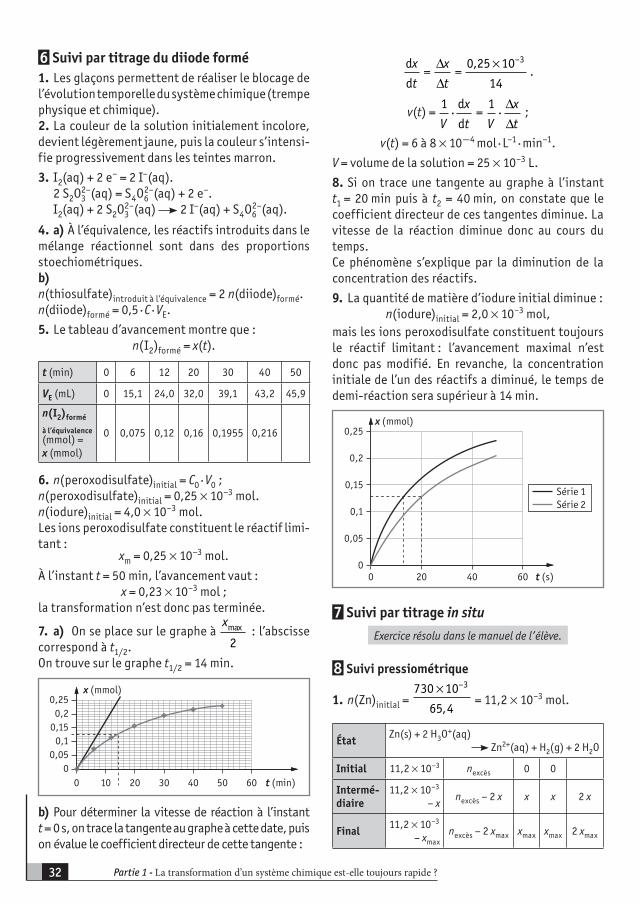
32 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
6 Suivi par titrage du diiode formé1. Les glaçons permettent de réaliser le blocage de l’évolution temporelle du système chimique (trempe physique et chimique).2. La couleur de la solution initialement incolore, devient légèrement jaune, puis la couleur s’intensi-fie progressivement dans les teintes marron.
3. I2(aq) + 2 e– = 2 I–(aq). 2 S2O3
2–(aq) = S4O62–(aq) + 2 e–.
I2(aq) + 2 S2O32–(aq) 2 I–(aq) + S4O6
2–(aq).
4. a) À l’équivalence, les réactifs introduits dans le mélange réactionnel sont dans des proportions stoechiométriques.b)n(thiosulfate)introduit à l’équivalence = 2 n(diiode)formé.n(diiode)formé = 0,5 · C · VE.
5. Le tableau d’avancement montre que :n(I2)formé = x(t).
t (min) 0 6 12 20 30 40 50
VE (mL) 0 15,1 24,0 32,0 39,1 43,2 45,9
n(I2)formé
à l’équivalence (mmol) = x (mmol)
0 0,075 0,12 0,16 0,1955 0,216
6. n(peroxodisulfate)initial = C0 · V0 ;n(peroxodisulfate)initial = 0,25 × 10–3 mol.n(iodure)initial = 4,0 × 10–3 mol.Les ions peroxodisulfate constituent le réactif limi-tant :
xm = 0,25 × 10–3 mol.
À l’instant t = 50 min, l’avancement vaut :x = 0,23 × 10–3 mol ;
la transformation n’est donc pas terminée.
7. a) On se place sur le graphe à xmax
2 : l’abscisse
correspond à t1/2.On trouve sur le graphe t1/2 = 14 min.
00,050,1
0,150,2
0,25
0 10 20 30 40 50 60 t (min)
x (mmol)
b) Pour déterminer la vitesse de réaction à l’instant t = 0 s, on trace la tangente au graphe à cette date, puis on évalue le coefficient directeur de cette tangente :
d
d
x
t =
∆∆
x
t =
0 25 10
14
3, –×.
v(t) = 1
V
x
t·d
d =
1
V
x
t·
∆∆
;
v(t) = 6 à 8 × 10—4 mol · L–1 · min–1.
V = volume de la solution = 25 × 10–3 L.
8. Si on trace une tangente au graphe à l’instant t1 = 20 min puis à t2 = 40 min, on constate que le coefficient directeur de ces tangentes diminue. La vitesse de la réaction diminue donc au cours du temps.Ce phénomène s’explique par la diminution de la concentration des réactifs.
9. La quantité de matière d’iodure initial diminue :n(iodure)initial = 2,0 × 10–3 mol,
mais les ions peroxodisulfate constituent toujours le réactif limitant : l’avancement maximal n’est donc pas modifié. En revanche, la concentration initiale de l’un des réactifs a diminué, le temps de demi-réaction sera supérieur à 14 min.
t (s)
x (mmol)
0
0,05
0,1
0,15
0,2
0,25
0 20 40 60
Série 1Série 2
7 Suivi par titrage in situ
Exercice résolu dans le manuel de l’élève.
8 Suivi pressiométrique
1. n(Zn)initlal = 7
65 4
30 10–3×,
= 11,2 × 10–3 mol.
État Zn(s) + 2 H3O+(aq)
Zn2+(aq) + H2(g) + 2 H2O
Initial 11,2 × 10–3 nexcès 0 0
Intermé-diaire
11,2 × 10–3
– xnexcès – 2 x x x 2 x
Final 11,2 × 10–3
– xmaxnexcès – 2 xmax xmax xmax 2 xmax

33Chapitre 3 - Vitesse de réaction
2. n(H2)(t) = x(t).
3. xmax = 11,2 × 10–3 mol.
4. La variation de pression, P(t) – P0 = ∆P, est due à la formation du dihydrogène :
∆P(t) · Vgaz = n(H2)(t) · R · T ;
Or n(H2)(t) = x(t), donc :
x(t) = ( ( )– ) ·
·
P t P V
T0 gaz
R.
5. À chaque instant t : x(t) = ∆P t V
T
( ) ·
·gaz
R.
À l’état final : xmax = ∆P V
T
( ) ·
·
final
Rgaz .
En faisant le rapport, on obtient :
x(t) = x P t
Pmax
final
· ( )
( )
∆∆
.
6. ∆P(15) = 520 hPa.
x(15) = x P
Pmax
final
· ( )
( )
∆∆
15.
x(15) = 520
543 × 11,2 × 10–3 = 10,7 × 10–3 mol.
7. On appelle temps de demi-réaction, t½, la durée au bout de laquelle l’avancement x de la réaction est égal à la moitié de sa valeur maximale :
x(t½) = 1/2 · xmax
Graphiquement, on trouve environ 4 min.
8. Si on utilise de l’acide chlorhydrique plus concen-tré, la vitesse de la réaction à t = 0 s augmente.L’évolution temporelle du système chimique est plus rapide si t½ inférieur à 4 min.
9 Suivi conductimétrie1. a) Pipette jaugée 5,0 mL ; éprouvette graduée.
2. a) n(RCl) = 5 0 0 85
92 5
, ,
,
× = 4,6 × 10–2 mol.
b) n0(RCl) = 4 6 10 3
50
2, –× × = 2,8 × 10–3 mol.
3.
État (CH3)3–CCl(aq) + 2 H2O(l) (CH3)3–C–OH(aq) + H3O
+(aq) + Cl–(aq)
Initial 2,8 × 10–3 Excès 0 0
Intermé-diaire 2,8 × 10–3– x n(excès) – 2x x x
Final 2,8 × 10–3– xm n(excès) – 2xm xm xm
4. Pour l’état final, on peut écrire σf = K · xm
K = 8 20
2 8 10 3
,
, –× = 2,9 × 103 mS · cm–1 · mol–1.
On pourra aussi donner K = 2,9 × 102 S · m–1 · mol–1.
5. À partir des deux expressions :σ(t) = K · x(t) et σ f inal = K · xm = K · n0(RCl),
on retrouve : x(t) = n t0 · σ
σ( )
final
.
6. x(t2) = 2 8 10 5 40
8 20
3, ,
,
–× × = 1,8 × 10–3 mole.
n(RCl)restant = n0(RCl) – n(RCl)consommé = n0(RCl) – x(t2)
n(RCl)restant = 2,8 × 10–3 – 1,8 × 10–3 = 1,0 × 10–3 mole.
Soit : m(RCl) = 1,0 × 10–3 × 92,5 = 9,3 × 10–2 g.
7. Compte tenu de la proportionnalité entre la conductivité et l’avancement, on détermine t1/2 sur le graphe σ(t) = f(t) en recherchant l’abscisse corres-pondant à σ(t1/2) = 4,10 mS · cm–1, soit environ 80 s.
8. Pour déterminer la vitesse de réaction, on trace la tangente au graphe à l’instant t = 0 s, puis on évalue le coefficient directeur de cette tangente :
d
d
σt
= ∆∆σt
; d
d
σt
= 9
80 = 0,11 mS · s–1.
v(t) = 1
V
x
t·d
d =
1
V
x
t·
∆∆
= 1
KV t·
∆∆σ
.
v(t) = 2 à 3 × 10–4 mol · L–1 · s–1.
Volume de la solution : V = 153 × 10–3 L.
10 Suivi de l’oxydation des ions iodure1. n(iodure) = n2 = C2 · V2 ;n(iodure) = 10,0 × 10–3 mol.n(eau oxygénée) = n1 = C1 · V1 ;n(eau oxygénée) = 4,5 × 10–3 mol.n(oxonium) = 0,5 × 0,02 × 2 = 20,0 × 10–3 mol.
2. Pour réaliser une trempe, qui permet de bloquer instantanément l’évolution de la transformation chimique dans le bécher. La transformation support du titrage totale et rapide n’est pas affectée par la basse température.
3. n(diiode)formé = 0,5 n(thiosulfate)versé à l’équivalence n(diiode)formé = 0,5 · C · VE.
[(I2)formé] · V = 0,5 · C · VE.
D’où : [(I2)formé] = 0 5, · ·C V
VE .
4. VE = [( ) ] ·
,
I formé2
0 5
V
C = 7,2 × 10–3 L.
34 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
5. a) x(t) = n(diiode)formé.b) x(t) = [(I2)formé] · V.
6. d
d
x
t =
∆∆
x
t = a, coefficient directeur de la tan-
gente au graphe à un instant t.Sur les trois exemples, on constate que le coeffi-cient directeur des tangentes successives diminue : la vitesse de la réaction diminue avec l’évolution temporelle du système.
00,050,1
0,150,2
0,250,3
0,350,4
0,45
0 10 20 30 40 t (min)
x (mmol)
7. On appelle temps de demi-réaction, t½, la durée au bout de laquelle l’avancement x de la réaction est égal à la moitié de sa valeur maximale :
x(t½) = 1/2 · xmax.Graphiquement, on obtient environ 5 min.
8. La concentration des ions iodure est deux fois plus élevée dans le mélange réactionnel.a) Le peroxyde d’hydrogène étant le réactif limi-tant, l’avancement maximal n’est pas modifié :– le temps de demi-réaction diminue ;– la vitesse de réaction à t = 0 s augmente.b) En tenant compte des réponses précédentes, on représente la courbe 2 (série 2) présentant une vitesse initiale plus grande et conduisant à la même asymptote horizontale.
00,050,1
0,150,2
0,250,3
0,350,4
0,45
0 10 20 30 40 t (min)
x (mmol)
Série 1Série 2
11 Saponification de l’éthanoate d’éthyle1. Toutes les espèces chimiques introduites ont la même concentration (dans un même volume V), les réactifs sont donc introduits dans les proportions stoechiométriques (pas de réactif limitant) :
C0 · V – xmax = 0, donc xmax = C0 · V.
Réaction C4H8O2(aq) + Na+(aq) + HO–(aq)= Na+(aq) + A–(aq) + B(aq)
Instant Av.
0 0 C0 · V C0 · V C0 · V C0 · V 0 0
t x(t)C0 · V– x(t)
C0 · VC0 · V– x(t)
C0 · V x(t) x(t)
• xmax 0 C0 · V 0 C0 · Vxma= C0 · V
xma= C0 · V
2.a) Les ions Na+(aq), HO–(aq) et A–(aq) assurent la conduction de la solution.b) Au cours de la transformation, on constate que lorsqu’un ion hydroxyde est consommé, il se forme un ion éthanoate (A–). La conductivité molaire ionique λ des ions hydroxyde étant supérieure à celle des ions éthanoate, la conductivité σ de la solution diminue donc.Les ions sodium interviennent dans la conductivité de la solution, mais pas dans son évolution.
c) σt = C0 · (λ(Na+) + λ(HO–)) + C0 · (λ(A–) – λ(HO–)).
σt = λ(Na+) · [Na+(aq)](t) + λ(HO–) · [HO–(aq)](t)+ λ(A–) · [A–(aq)](t).
σt = λ(Na+) · C V
V0 ·
+ λ(HO–) · C V x t
V0 – ( )·
+ λ(A–) · x t
V
( ).
σt = λ(Na+) · C0 + λ(HO–) · C0 – λ(HO–) · x t
V
( )
+ λ(A–) · x t
V
( ).
σt = C0 · (λ(Na+) + λ(HO–))
+ x t
V
( ) · (λ(A–) – λ(HO–)) (1)
d) À l’instant t = 0 s, on a : x(t = 0) = 0.Dans l’expression (1), le deuxième terme s’annule. Il reste : σ0 = C0 · (λ(Na+) + λ(HO–)) (2)
À l’instant t = ∞ :x(∞) = xmax = C0 · V.
σ∞ = C0 · (λ(Na+) + λ(HO–)) +C V
V0 ·
· (λ(A–) – λ(HO–)).
σ∞ = C0 · (λ(Na+) + λ(HO–)) + C0 · (λ(A–) – λ(HO–)).
Il reste : σ∞ = C0 · (λ(Na+) + (λ(A–)) (3)
e) À partir des expressions (1) et (2), on peut écrire :

35Chapitre 3 - Vitesse de réaction
σt = σ0 + x t
V
( ) · (λ(A–) – λ(HO–)).
σt – σ0 = x t
V
( ) · (λ(A–) – λ(HO–)).
Il vient :x(t) =
σ σ
λ λt 0
– –
–
(A )– (HO )· V ,
x(t) = σ σ
λ λ λ λt
– + + –A Na Na HO
–
( ) ( )– ( )– ( )·0
+V ,
x(t) = σ σ
λ λ λ λt
– + + –A Na Na HO
–
( ( ) ( ))–( ( ) ( ))·0
+ +V ,
x(t) = σ σ
λ λ λ λt
– + + –A Na Na HO
–
( ( ) ( ))–( ( ) ( ))· ·0 0
0+ +V
C
C.
x(t) =σ σ
λ λ λ λt
– + + –A Na Na HO
–
( ( ) ( )) · –( ( ) ( )) ·0
0 0+ +C C · V · C0,
x(t) = C0 · V · σ σσ σ
t 0
0
–
–∞.
En multipliant les numérateur et dénominateur par – 1, l’expression s’écrit alors :
x(t) = C0 · V · σ σσ σ
0 t
t
–
– ∞.
3. a) V, volume réactionnel (constant) en L,x(t), avancement en mol et t en s.La vitesse v(t) s’exprime en mol · L–1 · s–1.b) À instant t donné, il suffit de tracer la tangente à la courbe x = f(t) au point d’abscisse t.
d
d
x t
t
( ) =
∆∆x t
t
( ).
C’est le coefficient directeur de cette tangente au graphe à l’instant t.La vitesse est proportionnelle à ce coefficient directeur à chaque instant t.c) D’après la courbe x = f(t) fournie, les différents coefficients directeurs des tangentes à la courbe diminuent au cours du temps : la vitesse de la réac-tion diminue donc au cours du temps.Le facteur cinétique permettant d’expliquer cette diminution de vitesse est la « concentration des réactifs », qui diminue au cours du temps.d) xmax = C0 · V ;xmax = 1,0 × 10–2 × 0,1000 = 1,0 × 10–3 mol.Le temps de demi-réaction correspond à la durée nécessaire pour que l’avancement atteigne la moi-
tié de sa valeur maximale. La transformation étant totale :
xf = xmax.
t1/2 correspond à l’abscisse du point d’ordonnée :xmax
2 = 0,50 mmol, soit t1/2 = 16 min.
f) La « température » est un facteur cinétique. Avec une température plus basse, la vitesse volumique de réaction sera plus faible : le système atteindra moins rapidement l’état final (t1/2 sera plus grand).
00,10,20,30,40,50,60,7
0 5 10 15 20 25 30 t (min)
x (mmol)
Série 1Série 2
12 Étude cinétique par suivi spectrophotométrique
1. Un oxydant est une espèce chimique capable de capter un ou plusieurs électrons.Un réducteur est une espèce chimique capable de céder un ou plusieurs électrons.2. • Couple H2O2(aq)/H2O(l), réduction du peroxyde d’hydrogène :
H2O2(aq) + 2 H+(aq) + 2 e– = 2 H2O(l).
• Couple I2(aq)/I–(aq), oxydation des ions iodure :2 I–(aq) = I2(aq) + 2 e–.
3. a) n1 = n(I–)i = C1 · V1 ;n1 = 0,10 × 20,0 × 10–3 = 2,0 mmoln2 = n(H2O2)i = C2 · V2 ;n2 = 0,10 × 2,0 × 10–3 = 0,20 mmolLes proportions stœchiométriques montrent :
n(I–)i = 2 n(H2O2)i.Or ici, les ions iodure sont largement en excès, les réactifs ne sont pas dans les proportions stœchio-métriques.b)
Équation H2O2(aq) + 2 I–(aq) + 2 H3O+(aq)= I2(aq) + 4 H2O(l)
État initial n2 n1 Excès 0 Excès
État inter-médiaire n2 – x n1 – 2x Excès x Excès
État final n2 – xf n1 – 2xf Excès xf Excès
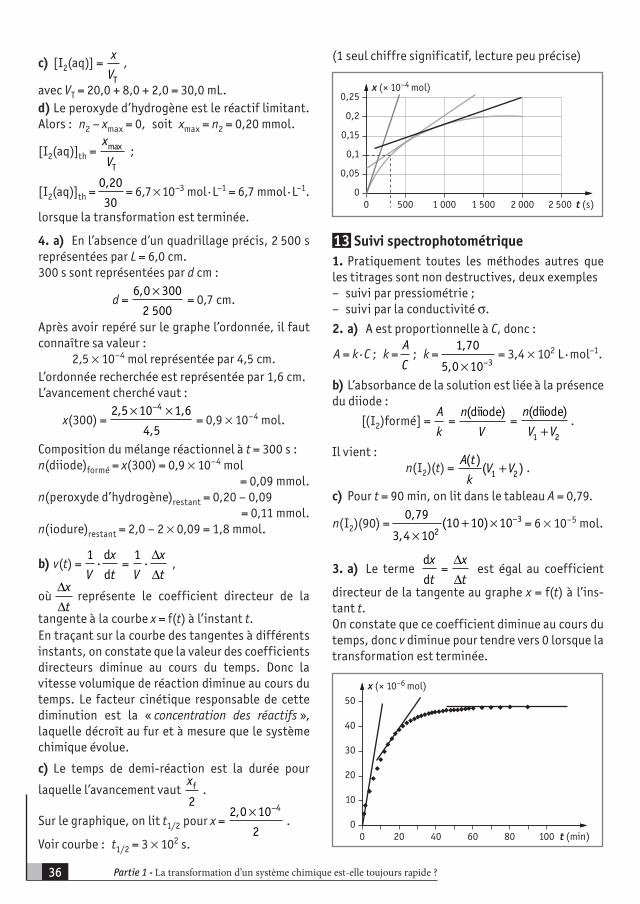
36 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
c) [I2(aq)] = x
VT
,
avec VT = 20,0 + 8,0 + 2,0 = 30,0 mL.d) Le peroxyde d’hydrogène est le réactif limitant. Alors : n2 – xmax = 0, soit xmax = n2 = 0,20 mmol.
[I2(aq)]th = x
Vmax
T
;
[I2(aq)]th =0 20
30
,= 6,7 × 10–3 mol · L–1 = 6,7 mmol · L–1.
lorsque la transformation est terminée.
4. a) En l’absence d’un quadrillage précis, 2 500 s représentées par L = 6,0 cm.300 s sont représentées par d cm :
d = 6 0 300
2 500
, × = 0,7 cm.
Après avoir repéré sur le graphe l’ordonnée, il faut connaître sa valeur :
2,5 × 10–4 mol représentée par 4,5 cm.L’ordonnée recherchée est représentée par 1,6 cm.L’avancement cherché vaut :
x(300) = 2 5 10 1 6
4 5
4, ,
,
–× × = 0,9 × 10–4 mol.
Composition du mélange réactionnel à t = 300 s :n(diiode)formé = x(300) = 0,9 × 10–4 mol
= 0,09 mmol.n(peroxyde d’hydrogène)restant = 0,20 – 0,09
= 0,11 mmol.n(iodure)restant = 2,0 – 2 × 0,09 = 1,8 mmol.
b) v(t) = 1
V
x
t·d
d =
1
V
x
t·
∆∆
,
où ∆∆
x
t représente le coefficient directeur de la
tangente à la courbe x = f(t) à l’instant t.En traçant sur la courbe des tangentes à différents instants, on constate que la valeur des coefficients directeurs diminue au cours du temps. Donc la vitesse volumique de réaction diminue au cours du temps. Le facteur cinétique responsable de cette diminution est la « concentration des réactifs », laquelle décroît au fur et à mesure que le système chimique évolue.
c) Le temps de demi-réaction est la durée pour
laquelle l’avancement vaut xf
2.
Sur le graphique, on lit t1/2 pour x = 2 0 10
2
4, –×.
Voir courbe : t1/2 = 3 × 102 s.
(1 seul chiffre significatif, lecture peu précise)
0
0,05
0,1
0,15
0,2
0,25
0 500 1 000 1 500 2 000 2 500 t (s)
x (× 10–4 mol)
13 Suivi spectrophotométrique1. Pratiquement toutes les méthodes autres que les titrages sont non destructives, deux exemples– suivi par pressiométrie ;– suivi par la conductivité σ.
2. a) A est proportionnelle à C, donc :
A = k · C ; k = A
C ; k =
1 70
5 0 10 3
,
, –× = 3,4 × 102 L · mol–1.
b) L’absorbance de la solution est liée à la présence du diiode :
[(I2)formé] = A
k =
n
V
(diiode) =
n
V V
(diiode)
1 2+.
Il vient :n(I2)(t) =
A t
kV V
( )( )1 2+ .
c) Pour t = 90 min, on lit dans le tableau A = 0,79.
n(I2)(90) = 0 79
3 4 1010 10 10
23,
,( ) –
×+ × = 6 × 10–5 mol.
3. a) Le terme d
d
x
t =
∆∆
x
t est égal au coefficient
directeur de la tangente au graphe x = f(t) à l’ins-tant t. On constate que ce coefficient diminue au cours du temps, donc v diminue pour tendre vers 0 lorsque la transformation est terminée.
0
10
20
30
40
50
0 20 40 60 80 100 t (min)
x (× 10–6 mol)

37Chapitre 3 - Vitesse de réaction
b) L’évolution temporelle du système chimique se traduit par la consommation des réactifs, leurs concentrations diminuent. Ceci explique la baisse de la vitesse de réaction au cours du temps.Pour t > 80 min, la composition du système chimi-que n’évolue plus.c) On pourrait, par exemple, augmenter la tempé-rature afin d’obtenir plus rapidement l’état final.
4. Montage à réaliser
Solution titrante2N2
+ (aq) +S2O32 –(aq)
de concentration C
Mélangeréactionnel
Pointe de spatulede thiodène
5. À l’équivalence, les réactifs introduits sont tota-lement consommés : ils ont été introduits dans les proportions stœchiométriques.
6. a) On raisonne sur l’échantillon prélevé (5,0 mL)n(I2)formé = 0,5 n(thiosulfate)versé à l’équivalence ;n(I2)formé = 0,5 (C' · V'E).
En travaillant sur 5,0 mL du mélange réactionnel, on n’a dosé qu’un quart du diiode formé :
n(I2)formé dans le mélange réactionnel = 2 (C' · V'E).
b) n(I2)formé dans le mélange réactionnel = 4,6 × 10–5 mol.
7.
Équation de la réaction
2 I–(aq) + S2O82–(aq)
I2(aq) + 2 SO42–(aq)
État du système Av. Quantité de matière (mol)
Initial x = 0 5 × 10–3 5,0 × 10–5 0 0
Intermé-diaire x 5 × 10–3
– 2 x5,0 × 10–5
– x x 2 x
Final xmax5 × 10–3
– 2 xmax
5,0 × 10–5 – xmax
xmax 2 xmax
S2O82–(aq) est le réactif limitant,
alors 5,0 × 10–5 – xmax = 0, donc xmax = 5,0 × 10–5 mol.
D’après le tableau :n(I2) = xmax = 5,0 × 10–5 mol, quantité maximale de diiode.
8. Dans la question 2.c), la spectrophotométrie nous a permis de déterminer n(I2) = 4,6 × 10–5 mol.Dans la question 6.b), le titrage a conduit à la même valeur.En théorie, dans le cas d’une transformation totale, on aurait pu obtenir : n(I2) = 5,0 × 10–5 mol.On calcule l’écart relatif entre ces deux valeurs :
n n
n
exp 2 th 2
th 2
(I ) (I )
(I )
− =
4 6 5 0
5 0
, ,
,
− = 0,080,
soit un écart relatif de 8 %.Deux explications possibles :– soit la transformation lente n’était pas terminée à t = 90 min ;– soit des erreurs expérimentales lors du titrage (erreur sur la détermination de V'E) ou lors du suivi spectrophotométrique ont été commises.
14 Corrosion des gouttières1.
Équation chimique
Zn(s) + 2 H3O+(aq)
= Zn2+(aq) + H2(g) + 2 H2O(l)
État du système
Av.(mol)
Quantités de matière (mol)
Initial 0 n(Zn)i n(H3O+)i 0 0 Excès
En cours de trans-formation
xn(Zn)i
– xn(H3O
+)i– 2 x
x x Excès
Final xmaxn(Zn)i
– xmax
n(H3O+)i
– 2 xmaxxmax xmax Excès
2. Si on suppose que le zinc est le réactif limitant, donc n(Zn)i – xmax = 0, alors :
xmax = n(Zn)i = m
M
(Zn)
(Zn)i ;
xmax = 0 50
65 4
,
, = 7,6 × 10–3 mol = 7,6 mmol.
Si on suppose que l’ion oxonium est le réactif limi-tant, alors :
n(H3O+)i – 2 xmax = 0,
soit xmax = n(H O )3
+i
2 =
[ ·H O (aq)]3+
i V
2 ;
xmax = 0 40 75 0 10
2
3, , –× × = 15 × 10–3 mol.
Le réactif limitant est le zinc, car il conduit à la valeur de l’avancement maximal la plus faible :
xmax = 7,6 × 10–3 mol = 7,6 mmol.

38 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
3. a) Le tableau d’évolution montre n(H2) = x(t) et d’après le texte :
(P – Pi) · Vgaz = n(H2) · R · T.
donc x(t) = ( – )
Ri gazP P V
T
·
·
b) Pour P = Pmax, alors x = xmaxD’après 3.a) :
x(t) = ( – )
Ri gazP P V
Tmax ·
·
x
xmax
= ( – )
R
R
( – )i gaz
i gaz
P P V
T
T
P P V
·
··
·
·max
;
x
xmax
= ( – )
( – )i
i
P P
P Pmax
, soit x = xmax · ( – )
( – )i
i
P P
P Pmax
.
c) L’échelle verticale de la figure est 0,5 cm 1 mmol.Pour t > 200 min : x = cte = xmax.On mesure 3,8 cm pour xmax, soit xmax = 7,6 mmol.Cette valeur est égale à celle calculée en 2.
d) Pour t = 50,0 min, P(50) = 1 452 hPa.Pi = 1 020 hPa et Pmax = 1 757 hPa.
Avec l’expression obtenue en 3.b) :
x = 7,6 × 10–3 × 1 452 1 020
1 757 1 020
–
– ;
x = 4,5 × 10–3 mol = 4,5 mmol
Remarque : ce calcul est effectué avec la valeur non arrondie de xmax.
4. a) v(t)= 1
V
x
t·d
d .
Le volume V de la solution étant constant, la vitesse volumique de réaction est alors proportionnelle à d
d
x
t.
Ce terme représente le coefficient directeur de la tangente à la courbe représentative de x(t) à l’ins-tant t.
b) d
d
x
tt
( )
est initialement élevé puis diminue au
cours du temps, jusqu’à tendre vers zéro pour t > 200,0 min. La vitesse initialement maximale, diminue progressivement jusqu’à zéro.
5. La « concentration initiale en ions oxonium » (réactif) est un facteur cinétique : plus elle est éle-vée et plus la vitesse initiale de réaction est éle-vée.
[H3O+(aq)]exp 1 > [H3O
+(aq)]exp 3 > [H3O+(aq)]exp 2,
donc v1 > v3 > v2
ou d
d
x
tt
=0
expé.1 > d
d
x
tt
=0
expé.3 > d
d
x
tt
=0
expé.2
Les tangentes à t = 0 étant difficiles à tracer conve-nablement, on peut s’intéresser au temps de demi-réaction :
x = xmax
2 ; x = 3,8 mmol
• Pour la courbe (a) : x = 3,8 mmol pour t = 33 min.• Pour la courbe (b) : x = 3,8 mmol pour t = 38 min.• Pour la courbe (c) : x = 3,8 mmol pour t = 50 min.
On peut donc dire :v(a) > v(b) > v(c).
On associe la courbe (a) à l’expérience 1, la courbe (b) à l’expérience 3 et la courbe (c) à l’expérience 2.
6. a) La poudre de zinc offre une plus grande sur-face de contact (état de division) avec la solution : plus la surface de contact est grande et plus la transformation est rapide.b) Pour l’expérience 6, l’avancement n’évolue qua-siment pas. On en déduit l’absence de réaction entre le zinc et la solution d’acide : la couche de carbonate de zinc protège le métal de l’attaque acide.
7. pH = – log[H3O+(aq)],
soit [H3O+(aq)] = 10–pH.
[H3O+(aq)] = 1 × 10–5 mol · L–1.
8. Trois facteurs se conjuguent pour expliquer la longévité des gouttières en zinc :– la concentration en ions oxonium (les eaux de pluies sont peu concentrées en ions H3O
+) ;– la surface du zinc en contact (une gouttière offre une faible surface de contact par rapport à la pou-dre de zinc) ;– la couche de carbonate de zinc (cette couche réduit fortement la surface de contact entre le zinc et les pluies acides).Pour chacun de ces facteurs, il y a très faible réacti-vité ou même empêchement à la réaction.

39Chapitre 3 - Vitesse de réaction
Les compétences expérimentales
(page 78)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de revenir sur des techniques de base que l’on retrouve de manière récurrente dans les sujets d’évaluation. Cet exercice conduit l’élève à justi-fier ses choix.
Corrigé
1. Suivi par titrageLe suivi temporel de cette transformation lente, nécessite de connaître la quantité de matière de diiode formée à chaque instant. Seule une trempe permet de figer le système chimique à un instant t ; on peut ensuite procé-der au titrage de diiode formé par la solution titrante de thiosulfate de sodium.
• Le dessin 2 est le seul à proposer une trempe (ajout de glaçons et d’eau glacée) : il représente la bonne démarche.
2. Utilisation d’un tableur grapheurAu cours d’une transformation chimique, l’avancement x(t) de la réaction croit et tend asymptotiquement vers xmax.
La vitesse de la réaction à un instant t est proportionnelle à d
d
x
t, que l’on
détermine graphiquement (coefficient directeur de la tangente au graphe
à un instant t). On accède également à d
d
x
t, et donc à v(t), avec un tableur.
Si les valeurs de x(t) sont connues pour des intervalles de temps ∆t petits,
on peut calculer d
d
x
t à un instant t en utilisant : [ ( ) – ( – )x t t x t t
t
+ ∆ ∆∆2
.
Le bon graphe est bien le B : il traduit la diminution de vitesse de réaction au cours de la transformation.
Exercice supplémentaire
Décomposition de l’eau oxygénéeLa décomposition du peroxyde d’hydrogène (eau oxygénée) est une transformation chimique lente, favorisée par la présence d’un catalyseur :
2 H2O2(aq) 2 H2O(l) + O2(g).On se propose de suivre la formation du dioxygène au cours de la décomposition du peroxyde d’hydro-gène en présence d’un catalyseur. Expérimentale-ment, on utilise un flacon parfaitement étanche équipé d’un bouchon à vis, dont la tubulure est reliée à un pressiomètre. On introduit dans le flacon un volume V0 = 20,0 mL de solution aqueuse de peroxyde d’hydrogène, de concentration C0 = 1,50 mol · L–1.On réalise une expérience à la température θ1 = 20 °C maintenue constante grâce à l’eau du cristallisoir dans lequel plonge le flacon. La pression initiale du
gaz dans le flacon est Pair = Patm = 1 005 hPa et le volume occupé par le gaz (air + oxygène formé) est V = 575 mL. À l’instant de date t = 0 s, on met en contact le contenu de la nacelle, contenant le catalyseur, avec la solution aqueuse de peroxyde d’hydrogène. On observe un fort dégagement gazeux et toutes les minutes, on note Pmesurée la pression du gaz (air + dioxygène formé) indiquée par le capteur. La pres-sion finale du gaz dans le flacon est Pf = 1 640 hPa.Les résultats de la série de mesures, traités par un tableur permettent d’obtenir le graphe PO2
= f(t), avec t exprimé en minutes.
1. Écrire les demi-équations électroniques des deux couples : H2O2(aq)/H2O(l) et O2(g)/H2O2(aq).2. a) Déterminer la quantité de matière initiale n(H2O2) de peroxyde d’hydrogène introduit.

40 Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
b) À l’aide d’un tableau d’évolution du système, cal-culer l’avancement maximal xmax attendu.3. a) Donner la relation liant la pression mesurée à la pression relative en dioxygène PO2
.b) En appliquant la loi des gaz parfaits, déduire la quantité de dioxygène formé au cours de l’expé-rience.4. a) Donner la relation liant n(O2)(g) théorique-ment formé et xmax .b) Montrer alors que le résultat expérimental est en accord avec la quantité de matière de peroxyde d’hydrogène initialement introduit.5. a) Montrer que l’avancement de la réaction x(t) est donné à chaque instant par la relation x(t) = xmax · PO2
/635 (avec PO2 en hPa)
b) En déduire la valeur de l’avancement de la réac-tion aux instants de date t1 = 3 min et t2 = 6 min. c) Comparer x(t1) et x(t2), puis conclure sur l’évolu-tion du système chimique au cours du temps.
t (min)0
100
200
300
400
500
600
700
0 1 2 3 4 5 6 7 8 9 1011121314151617181920
P(O2)
P(O2) = f(t).
6. Déterminer graphiquement le temps de demi-réaction.7. On rappelle la définition de la vitesse volumique de réaction :
v(t) = 1V
xt
·dd
.
Calculer la vitesse volumique initiale vt=0, en détaillant l’ensemble de la démarche.8. Si, dans le protocole, on ajoute 50 mL d’eau dis-tillée au volume V0 de peroxyde d’hydrogène, préciser comment les grandeurs suivantes sont modifiées :a) la vitesse volumique de réaction à t = 0 ?b) le temps de demi-réaction ?c) la quantité finale de dioxygène formé ?d) la pression finale dans le flacon ?Justifier les réponses.Donnée
R = 8,314 J · K–1 · mol–1.•
Corrigé1. H2O2(aq) + 2 H+(aq) + 2 e– = 2 H2O(l) O2(aq) + 2 H+(aq) + 2 e– = H2O2(aq).
2. n(H2O2)introduit = C0 · V0 ;n(H2O2)introduit = 1,50 × 20,0 × 10–3 = 30,0 × 10–3 mol.
État 2 H2O2(aq) 2 H2O(l) + O2(g)
Initial 30,0 × 10–3 Excès 0
Intermédiaire 30,0 × 10–3 – 2 x 2 x x
Final 30,0 × 10–3 – 2 xmax 2 xmax xmax
Il vient xmax = 15,0 × 10–3 mol.
3. Pmesurée = PO2 + P air
PO2 = Pmesurée – Patm.PO2 · V = n(O2) · R · T.
n(O2)formé = P V
T
(O )
R2 ⋅⋅
;
n(O2)formé = 63 500 575 10
831 293
6× ××
–
= 15,0 × 10–3 mol.
4. Le tableau d’évolution indique :xmax = n(O2)théorique.
n(O2)formé = 15,0 × 10–3 mol = n(O2)théorique.
5. n(O2)formé(t) = x(t) = P V
T
t(O )
R2 ( ) ·
·.
n(O2)formé(final) = xmax = P V
T
(O )
R2 (final) ·
·.
Il vient : x(t) = x P
P
tmax ( )· (O )
(O )2
2 (final)
.
x(t) = x P tmax 2 ( )(O )·
635 ;
x(t1) = 15 0 10 400
635
3, –× × = 9,45 × 10–3 mol.
x(t2) = 15 0 10 560
635
3, –× × = 13,2 × 10–3 mol.
x(t) augmente au cours du temps, mais ce n’est pas une évolution linéaire : il a moins progressé de 3 à 6 min que de 0 à 3 min.
6. Compte tenu de la proportionnalité de P(O2)( ft) et de x(t), on se place sur le graphe à
P(O )2 (final)
2 : l’abscisse correspond à t1/2.

41Chapitre 3 - Vitesse de réaction
0100200300400500600700
0 5 10 15 20 25 t (min)
P(O2) (hPa)
On obtient sur le graphe t1/2 = 2,5 min.
7. Pour déterminer la vitesse de réaction à l’instant t = 0 s, on trace la tangente au graphe à cette date, puis on évalue le coefficient directeur de cette tan-gente :
v(t) = 1
V
x
t·d
d =
1
V
x
t·
∆∆
;
v(t) = x
V
P
t
tmax ( )
··
( )
6352∆
∆
O .
v(t) = 175 15 0 10
635 2
3× ×× ×
, –
0,0 10–3.
∆
∆
P
t
t( )( )O2 =
700
4 = 175 hPa · min–1.
V(t) = 0,19 à 0,22 mol · L–1 · min–1
V = volume de la solution = 20,0 × 10–3 L.
8. L’ajout d’eau au mélange réactionnel diminue la concentration des réactifs :a) la vitesse de réaction à t = 0 s diminue ;b) le temps de demi-réaction augmente ;c) la quantité de matière finale de dioxygène formé reste inchangée ;d) la pression finale dans le flacon augmente, car l’ajout d’eau diminue le volume occupé par le gaz.

42 Partie 2 - La transformation d’un système chimique est-elle totale ?
ProgrammeB – LA TRANSFORMATION D’UN SYSTÈME CHIMIQUE EST-ELLE TOUJOURS TOTALE ? (4 TP, 9 HEURES EN CLASSE ENTIÈRE)1 – UNE TRANSFORMATION CHIMIQUE N’EST PAS TOUJOURS TOTALE ET LA RÉACTION A LIEU DANS LES DEUX SENS2 – ETAT D’ÉQUILIBRE D’UN SYSTÈME
EXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Mise en évidence par pH-métrie qu’une transforma-tion n’est pas toujours totale et que la réaction chimi-que qui lui est associée a lieu dans les deux sens : les exemples sont pris dans le domaine acido-basique.Modél i sat ion d ’un é tat d ’ équilibre dynamique à l’échelle microscopique.Mi s e e n é v i d e n c e p a r conductimétrie que, pour une réaction donnée, le quo-tient de réaction dans l’état d’équilibre du système est constant et ce, quel que soit l’état initial du système : les exemples sont pris sur des solutions d’acides carboxy-liques à différentes concen-trations.Détermination par conduc-timétrie du taux d’avance-ment final de la réaction de différents acides sur l’eau pour une même concentra-tion initiale.
1. Une transformation chimique n’est pas toujours totale et la réaction a lieu dans les deux sensIntroduction du pH et de sa mesure.Mise en évidence expérimentale sur une trans-formation chimique donnée, d’un avancement final différent de l’avancement maximal.Symbolisme d’écriture de l’équation de la réaction : le signe =.État d’équilibre d’un système chimique.Taux d’avancement final d’une réaction :
τ = xfinal/xmaximal.Interprétation à l’échelle microscopique de l’état d’équilibre en termes de cinétique : chocs efficaces entre entités réactives d’une part et et entités produites d’autre part.2. État d’équilibre d’un système chimiqueQuotient de réaction Qr : expression littérale en fonction des concentrations molaires des espè-ces dissoutes pour un état donné du système.Généralisation à divers exemple en solution aqueuse homogène ou hétérogène (présence de solides).Détermination de la valeur du quotient de réaction à l’équilibre du système noté Qr,éq.Constante d’équilibre K associée à l’équation d’une réaction, à une température donnée.Influence de l’état initial d’un système sur le taux d’avancement final d’une réaction.
Définir un acide ou une base selon Brønsted.Écrire l’équation de la réaction associée à une transformation acido-basique et identifier dans cette équation les deux couples mis en jeu.Connaître la définition du pH pour les solutions aqueuses diluées.Connaissant la valeur de la concentration et du pH d’une solution d’acide, calculer l’avancement final de la réaction de cet acide sur l’eau et le comparer à l’avancement maximal.Connaître la définition du taux d’avancement final et le déterminer à partir d’une mesure.Être capable de mesurer la valeur du pH d’une solution aqueuse avec un pH-mètre.Utiliser la relation liant la conductance G aux concentrations molaires effectives [Xi] des ions Xi en solution.Savoir que, lorsque l’état d’équilibre d’un système est atteint, les quantités de matière n’évoluent plus, et que cet état d’équilibre est dynamique.En disposant de l’équation d’une réaction, donner l’expression littérale du quotient de réaction Qr.Savoir que le quotient de réaction dans l’état d’équi-libre d’un système, Qr,éq, prend une valeur, indépen-dante de la composition initiale, qui est la constante d’équilibre associée à l’équation de la réaction.Savoir que, pour une transformation donnée, le taux d’avancement final dépend de la constante d’équilibre et de l’état initial du système.
Notion d’équilibre chimique4
PARTIE 2
LA TRANSFORMATION D’UN SYSTÈME CHIMIQUE
EST-ELLE TOTALE ?

43Chapitre 4 - Notion d’équilibre chimique
aisément d’expliquer le caractère limité de la trans-formation ;– le symbolisme de l’écriture de la réaction doit tenir compte de cette caractéristique. Le symbole = est donc dorénavant employé au lieu de la →, on ne peut présupposer du sens dans lequel se fait la réac-tion à l’équilibre (les deux en fait) ;– la notion d’état d’équilibre chimique dynamique est alors introduite.Ces notions sont abordées dans l’activité 2.
3. La constante d’équilibre, caractéristique de l’état d’équilibre
On recherche une grandeur caractéristique de l’état d’équilibre d’un système donné.On introduit, dans un premier temps, la grandeur « quotient de réaction », lié à l’écriture d’une réac-tion donnée et les règles relatives à son écriture : seules les espèces dissoutes en solution intervien-nent dans son expression et [espèce dissoute] est le nombre représentant la concentration effective de l’espèce dissoute exprimé en mol · L–1. On s’intéresse ensuite à la valeur Qr,f que prend ce quotient de réaction dans l’état final du système chimique. Par des mesures de conductimétrie (c’est l’occasion ici de réinvestir les connaissances acqui-ses en classe de première scientifique), on constate l’invariance de Qr,f et ce, quel que soit l’état initial. Le quotient de réaction à l’équilibre est alors appelé constante d’équilibre, notée K, et on indique que sa valeur est liée aux espèces chimiques mises en jeu lors de l’équilibre et qu’elle ne dépend que de la tem-pérature du système chimique.On insistera sur le fait que ce ne sont pas les concen-trations des espèces dissoutes qui fixent la valeur de K (comme le laisse supposer la démarche pour en déterminer la valeur), mais le contraire : la valeur de K (notamment) impose la composition du système chimique à l’état d’équilibre.L’activité 1 est consacrée à cette partie du chapitre.
4. Paramètres influençant le taux d’avancement
Enfin, on montre l’influence des conditions initia-les et de la constante d’équilibre sur la composition du système chimique à l’état d’équilibre et sur le taux d’avancement final de la réaction.L’activité 3, de type ECE, relative au contenu du cha-pitre, est proposée afin d’entraîner les élèves à ce type d’épreuve : elle permet à la fois une évaluation des compétences expérimentales et théoriques.
Découpage du cours1. Une transformation est-elle toujours totale ? p. 822. Notion d’équilibre chimique p. 853. La constante d’équilibre, caractéristique de l’état d’équilibre p. 874. Paramètres influençant le taux d’avancement p. 89
L’objectif de ce chapitre est de faire découvrir qu’une transformation chimique n’est pas toujours totale et que dans ce cas, le système chimique est le siège d’un équilibre chimique.Le chapitre est décliné en quatre parties.
1. Une transformation est-elle toujours totale ?Dans la première partie, après avoir rappelé les défi-nitions sur les acides, les bases, les couples acide-base et les transformations acidobasiques, on introduit la notion de pH comme indicateur des ions oxonium, H3O+(aq) : la valeur de la concentra-tion [H3O+(aq)] est déduite de la valeur du pH (la relation pH = – log [H3O+(aq] est vérifiée pour les solutions acides diluées et [H3O+(aq)] représente le nombre mesurant la concentration effective des ions oxonium exprimée en mol · L–1).
2. Notion d’équilibre chimiqueDans la deuxième partie, on s’intéresse à l’état final de systèmes chimiques obtenus lors de la mise en solution aqueuse de l’acide éthanoïque dans l’eau.Une solution aqueuse d’acide éthanoïque de concen-tration molaire en soluté C est un système chimique à l’état final dont la composition dans l’état initial est donnée par C (exemple : à une solution de concentra-tion C = 1,0 × 10–2 mol · L–1 correspond un état initial constitué de 1,0 × 10–2 mol d’acide éthanoïque et de la quantité d’eau nécessaire pour obtenir 1 litre de solu-tion). La détermination de l’avancement final réel de la réaction à partir de la mesure du pH de la solution montre qu’il est différent (inférieur) de l’avancement final maximal (qui s’obtient en considérant la réac-tion totale) : la transformation est limitée. La gran-deur « taux d’avancement final » est alors introduite pour caractériser cette transformation limitée et per-met d’en déduire le pourcentage de molécules d’acide éthanoïque qui se sont dissociées par réaction avec l’eau. Des expériences complémentaires de mesure de pH consécutives à l’addition modérée (sans variation notable de volume) d’acide éthanoïque pur ou d’étha-noate de sodium montrent que la réaction chimique mise en jeu s’effectue dans les deux sens :– des considérations cinétiques permettent alors
♦
Cours

44 Partie 2 - La transformation d’un système chimique est-elle totale ?
Étude du quotient de réaction à l’état d’équilibre (page 90)
Cette activité permet de mettre en évidence l’inva-riance, à température fixe, du quotient de réaction d’un système chimique à l’équilibre, que l’on nomme constante d’équilibre. C’est ici l’occasion de réinves-tir les connaissances acquises en 1re S sur la conduc-tance et la conductivité d’une solution ionique.
A Préparation de solutions aqueuses d’acide éthanoïque
1. Lorsque l’on veut diluer une solution x fois, le volume final de la solution diluée doit être égal à x fois le volume de solution initialement prélevée.
Concentration(mol · L–1)
1,0 × 10–3 2,0 × 10–3 5,0 × 10–3
Facteur de dilution 10 5 2
Pipette jaugée utilisée 10 mL 20 mL 25 mL
Fiole jaugée utilisée 100 mL 100 mL 50 mL
2. Prendre soin lors du prélèvement de réaliser ce dernier dans un bécher (et non dans le flacon !), de rincer les pipettes avec un peu de solution avant de faire les prises, de ne pas souffler dans la pipette pour faire tomber « la dernière goutte », d’ajuster correctement le niveau d’eau à la pipette compte-gouttes : le bas du ménisque formé par l’eau étant au niveau du trait de jauge.
B Mesure de la conductivité des solutionsLes conductimètres seront étalonnés avec des solu-tions standard de chlorure de potassium, on pren-dra soin de vérifier la température lors du réglage.1. Conversion : 1 ms · cm–1 = 10–1 S · m–1.
C (mol · L–1) 1,0 × 10–3 2,0 × 10–3 5,0 × 10–3 1,0 × 10–2
s (mS · cm–1) 4,9 × 10–2 6,9 × 10–2 11,4 × 10–2 16,2 × 10–2
s (S · m–1) 4,9 × 10–3 6,9 × 10–3 11,4 × 10–3 16,2 × 10–3
2.
Agitation
pH-mètre
ACTIVITÉ
1
ACTIVITÉ
1 C Étude de la transformation1. CH3COOH(aq) + H2O(l)
CH3COO–(aq)+ H3O
+(aq).2. Les deux couples mis en jeu sont les couples :CH3COOH(aq)/CH3COO
–(aq) et H3O+(aq)/H2O(l).
3. Pour un volume arbitraire V de solution :
CH3COOH(aq) + H2O(l) CH3COO
–(aq) + H3O+(aq)
État initial(x = 0) C · V Excès 0 0
État finalréel (x = xf)
C · V – xf Excès xf xf
4. Comme nf(H3O+) = xf et nf(CH3COO
–) = xf égale-ment, on a donc :
[H3O+(aq)]f = [CH3COO
–(aq)]f.
5. Qr,f = [H O (aq)] · [CH COO (aq)]
[CH COOH(aq)]3
+f 3
–f
3 f
.
6. [CH3COOH(aq)]f = C – [H3O+(aq)]f.
7. σ = λ(H3O+)
· [H3O+(aq)]f + λ(CH3COO
–) · [CH3COO–(aq)]f.
8. En utilisant la relation de la question 4, on obtient :
[H3O+(aq)]f =
σλ λ( ) ( )H O CH COO3
+3
–+.
9. Qr,f = [H O (aq)]
[H O (aq)]3
+f2
3+
fC −.
10. τ = x
xf
max
= V
V C
· [H O (aq)]
·3 f
+
= [H O (aq)]3 f
+
C.
D Exploitation des résultats1.C (mol ⋅ L–1) 1,0 × 10–3 2,0 × 10–3 5,0 × 10–3 1,0 × 10–2
s (S ⋅ m–1) 4,9 × 10–3 6,9 × 10–3 11,4 × 10–3 16,2 × 10–3
[H3O+(aq)]f(mol ⋅ L–1) 1,25 × 10–4 1,76 × 10–4 2,92 × 10–4 4,14 × 10–4
Qr,f 1,78 × 10–5 1,71 × 10–5 1,80 × 10–5 1,79 × 10–5
t 0,125 0,088 0,058 0,04
2. La valeur du quotient de réaction dans l’état final semble indépendante des conditions initiales.3. Le taux d’avancement final dépend des condi-tions initiales : plus la solution d’acide éthanoïque est diluée, plus le taux d’avancement final de la réaction de l’acide avec l’eau augmente.
Activités

45Chapitre 4 - Notion d’équilibre chimique
Avancement final et notion d’équilibre (page 92)
Dans la première partie, on montre que l’avance-ment final d’une transformation peut être différent de son avancement maximal : la grandeur « taux d’avancement final » est alors introduite et permet d’estimer le pourcentage d’espèce chimique en solu-tion ayant réellement réagi.Dans la deuxième partie, on recherche un paramè-tre influençant le taux d’avancement final pour une transformation donnée : la composition de l’état initial du système au travers de la concentration ini-tiale en soluté apporté.La troisième partie est une approche de l’équilibre chimique : on montre qu’une transformation peut être le siège de deux réactions antagonistes expli-quant de fait son caractère non total.
A Mise en solution de l’acide éthanoïqueLa valeur du pH mesuré pour une solution aqueuse d’acide éthanoïque de concentration molaire en soluté apporté C = 1,0 × 10–2 mol · L–1 est 3,4.1. Couples mis en jeu :CH3COOH(aq)/CH3COO
–(aq) ; H3O+(aq)/H2O(l).
L’acide éthanoïque intervient en tant qu’acide et l’eau en tant que base.CH3COOH(aq) + H2O(l) CH3COO
–(aq) + H3O+(aq).
2. a)CH3COOH(aq) + H2O(l)
CH3COO–(aq) + H3O
+(aq)
État initial(x = 0) 10–2 Excès 0 0
État finalhypothétique (x = xmax) 10–2 – xmax Excès xmax xmax
b) L’eau étant en excès, la transformation cesse lorsque :
10–2 – xmax = 0, soit xmax = 1,0 × 10–2 mol.
3. a) nf(H3O+) = [H3O
+(aq)]f · V = 10–pH ⋅ V ;
nf(H3O+) = 3,98 × 10–4 mol.
b) Dans l’état final réel :xf = nf(H3O
+) = 3,98 × 10–4 mol.4. a) On constate que xf ≠ xmax, avec xf < xmax.b) La mise en solution de l’acide éthanoïque dans l’eau n’est pas totale.5. a) τ = 3,98 × 10–2.b) Le pourcentage de molécules d’acide éthanoïque qui se sont dissociées est de l’ordre de 4 % seule-ment.
ACTIVITÉ
2
ACTIVITÉ
2 B Paramètre influençant le taux d’avancementPour la solution aqueuse S1, de concentration molaire en soluté apporté C1 = 1,0 × 10
–3 mol · L–1, la valeur du pH1 mesuré est 3,9.1. On considère un état initial constitué de 1,0 × 10–3 mol d’acide éthanoïque et de l’eau en excès de façon à obtenir un litre de solution de concentration C1.2. a)
CH3COOH(aq) + H2O(l) CH3COO
–(aq) + H3O+(aq)
État initialx = 0 10–3 Excès 0 0
État finalhypothétiquex = x'max
10–3 – x'max Excès x'max x'max
b) L’eau étant en excès, la transformation cesse lorsque 10–3 – x'max = 0, soit x'max = 1,0 × 10
–3 mol.
3. a) n'f(H3O+) = [H3O
+(aq)]f · V = 10–pH1 · V ;
n'f(H3O+) = 1,26 × 10–4 mol.
b) Dans l’état final réel :x'f = n'f(H3O
+) = 1,26 × 10–4 mol.4. a) τ' = 1,26 × 10–1.b) Le pourcentage de molécules d’acide éthanoïque dissociées est voisin de 12,6 %.5. Le taux d’avancement final de la mise en solu-tion de l’acide éthanoïque dans l’eau augmente lorsque dans l’état initial la concentration molaire en soluté apporté diminue. Plus la solution d’acide éthanoïque est diluée, plus le pourcentage de molécules d’acide éthanoïque dissociées est élevé.
C Les deux sens d’une transformation1. On observe une diminution du pH du milieu. Ceci se traduit par une augmentation de [H3O
+(aq)] dans le milieu, soit l’apparition d’ions oxonium supplé-mentaires (les variations de volumes étant négli-geables) selon la réaction 1 :CH3COOH(aq) + H2O(l) CH3COO
–(aq) + H3O+(aq).
2. On observe une augmentation du pH du milieu. Ceci se traduit par une diminution de [H3O
+(aq)] dans le milieu, soit une disparition d’ions oxonium. Les ions oxonium (acide) ne peuvent disparaître qu’en réagissant avec une base qui a été introduite dans le milieu, l’ion éthanoate selon la réaction 2 :CH3COO
–(aq) + H3O+(aq) CH3COOH(aq) + H2O(l).
3. Au sein du système constitué d’acide éthanoïque et d’eau, il y a formation d’ions éthanoate et d’ions oxonium selon la réaction 1. Cependant, les ions

46 Partie 2 - La transformation d’un système chimique est-elle totale ?
oxonium formés réagissent à leur tour avec les ions éthanoates également formés selon la réaction 2 : les deux réactions inverses l’une de l’autre se pro-duisent au sein du système chimique.4. La proposition d’écriture doit tenir en compte les deux sens possibles de la transformation : on n’utilise plus une flèche → (qui présuppose un sens particulier de la transformation) mais un = (qui signifie que les deux transformations inverses se produisent simultanément dans le milieu) :CH3COOH(aq) + H2O(l) = CH3COO
–(aq) + H3O+(aq).
5. Le caractère non total d’une transformation se justifie par l’existence au sein du système de deux transformations inverses. Des considérations ciné-tiques montrent que la vitesse v1 de réaction de la réaction 1 est plus grande que la vitesse v2 de la réaction 2 au départ.Au cours du temps, v1 diminue (la concentration des espèces réagissantes diminue, donc la probabilité d’avoir des chocs efficaces également) et v2 aug-mente (la concentration des espèces réagissantes augmente, donc la probabilité d’avoir des chocs efficaces aussi). Lorsque v1 = v2, les quantités de matières des espèces présentes ne varient plus macroscopiquement au cours du temps : on dit que le système est le siège d’un équilibre dynamique.
Constante d’équilibre pour deux transformations inverses (page 93)
Cette activité sert d’entraînement à l’épreuve d’éva-luation des capacités expérimentales. Outre les savoirs théoriques concernant les notions d’équili-bres chimiques, les compétences expérimentales éva-luées sont les suivantes : utiliser de la verrerie classique pour réaliser une dilution ; effectuer une pesée cor-rectement ; être capable de mesurer la valeur du pH d’une solution aqueuse avec un pH-mètre étalonné.
1 Transformation mettant en jeu l’acide éthanoïque et l’eau
Il s’agit de réaliser ici une dilution par 5. Le volume de solution S préparé étant de 50 mL, celui du pré-lèvement de solution S1 doit être égal à 10 mL.Le matériel à utiliser est donc :– un bécher pour verser la solution S1 à prélever ;– une pipette jaugée de capacité 10 mL ;– un pipeteur ;– une fiole jaugée de capacité 50 mL et son bouchon ;– une pipette compte-gouttes pour ajuster au trait de jauge et un petit bécher ;– une pissette d’eau distillée.
ACTIVITÉ
3
ACTIVITÉ
3
1. CH3COOH(aq) + H2O(l) = CH3COO–(aq) + H3O
+(aq).
2. K1 = [H O (aq)] [CH COO (aq)]
[CH COOH(aq)]3 f 3 f
3 f
+ −·.
3. La mesure du pH de la solution Cs donne 3,2.On en déduit :• [H3O
+(aq)]f = [CH3COO–(aq)]f = 10
–pH ; [H3O
+(aq)]f = 6,3 × 10–4 mol · L–1.
• [CH3COOH(aq)]f = CS – [CH3COO–(aq)]f ;
[CH3COOH(aq)]f = 1,94 × 10–2 mol · L–1.
• D’où la valeur K1 = 2,0 × 10–5.
2 Transformation mettant en jeu les ions éthanoate et oxonium
La mesure du pH de la solution S' donne pH = 5,2.1. m = C2 · V · M(CH3COONa) ; m = 0,88 g.Lors de la pesée, veiller à ce que le plateau de la balance soit protégé d’une feuille de papier, que la balance soit tarée correctement après avoir déposée la coupelle vie. Lors de la préparation, veiller à ce que la coupelle soit rincée, les eaux de rinçage récu-pérées, l’entonnoir également rincé et la dissolution par agitation faite avant ajustage au trait de jauge.2. CH3COO
–(aq) + H3O+(aq) CH3COOH(aq) + H2O(l).
3. K2 = [CH COOH(aq)]
[H O (aq)] [CH COO (aq)]3 f
3 f 3 f+ −·
.
4. a) & b) K2 = 1
1K.
5. ni(H3O+) = C3 · V3 ; ni(H3O
+) = 2,0 × 10–3 mol.6. n'0 = C2 · V2 ; n'0 = 8,0 × 10
–3 mol.7. nf(H3O
+) = [H3O+(aq)]f · V' = 10
–pH · V' ;nf(H3O
+) = 6,3 × 10–7 mol.8. xf = ni(H3O
+) – nf(H3O+) ≈ ni(H3O
+) ;xf = 2,0 × 10
–3 mol.9. On en déduit :• nf(H3O
+) = 6,3 × 10–7 mol, soit [H3O
+]f = 6,3 × 10–6 mol · L–1.
• nf(CH3COO–) = n'0 – xf ;
nf(CH3COO–) = 6,0 × 10–3 mol,
soit [CH3COO–(aq)]f = 6,0 × 10
–2 mol · L–1.• nf(CH3COOH) = xf = 2,0 × 10
–3 mol, soit [CH3COOH(aq)]f = 2,0 × 10
–2 mol · L–1.10. K2 = 5,3 × 10
4.
11. 1
2K = 1,89 × 10–5.
Aux erreurs liées à la manipulation de la valeur du pH, on peut considérer que cette valeur est celle de K1 : la relation établie à la question 4 est vérifiée.

47Chapitre 4 - Notion d’équilibre chimique
1 Acide et base1. Un acide est une espèce chimique susceptible de céder un ou plusieurs protons.Une base est une espèce chimique susceptible de capter un ou plusieurs protons.2. Un couple acide base est un couple constitué d’un acide et d’une base lié par la relation formelle :
HA(aq) = H+(aq) + A–(aq).3. Couples : H3O
+(aq)/H2O(l) et H2O(l)/HO–(aq).
4. HA(aq) + H2O(l) = A–(aq) + H3O
+(aq).5. B(aq) + H2O(l) = BH
+(aq) + HO–(aq)
2 pH d’une solution1. Vrai.2. Vrai.3. Faux : pH = – log[H3O
+(aq)].4. Vrai.5. Faux : le pH augmente, car [H3O
+(aq)] diminue.6. Vrai à 25 °C.
3 Taux d’avancement d’une transformation1. Avancement maximal : avancement final de la transformation lorsqu’on la suppose totale.Avancement final : avancement final réel de la transformation.2. • Mesure de la conductivité du système chimi-que à l’état final lorsque la transformation met en jeu des espèces ioniques.• Mesure du pH du système chimique à l’état final lorsque la transformation met en jeu les ions oxo-nium.• Mesure de l’absorbance du système chimique à l’état final lorsque la transformation met en jeu une espèce chimique colorée.3. Taux d’avancement final : rapport entre l’avance-ment final et l’avancement maximal d’une transfor-
mation : τ = x
xf
max
.
4. Transformation totale : xf = xmax.Transformation limitée : xf < xmax.5. Oui.
4 Quotient de réaction
1. Qr = [D(aq)] [A(aq)]
[C(aq)] [B(aq)]
d d
c b
·
·.
2. Les espèces chimiques solides et l’eau dans le cas des transformations en solution aqueuse.3. Oui, car les valeurs des concentrations des espè-ces chimiques dépendent de l’avancement de la transformation.4. Des valeurs inverses.
5 Équilibre chimique1. Un système chimique est dans l’état d’équilibre lorsque toutes les espèces chimiques écrites dans l’équation modélisant la transformation sont pré-sentes et que leurs concentrations restent constan-tes au cours du temps.2. Existence simultanée de deux transformations inverses se produisant à la même vitesse.3. La constante d’équilibre est égale au quotient de réaction à l’équilibre.4. a) La constante d’équilibre est indépendante des conditions initiales.b) La constante d’équilibre ne dépend que de la température.5. Non : il ne reste pas de solide dans la solution.6. Oui : espèces ioniques et solide coexistent dans la solution.
6 Mise en solution d’acides et de bases1. Un acide est une espèce chimique susceptible de céder un ou plusieurs protons.Une base est une espèce chimique susceptible de capter un ou plusieurs protons.
2. a) Ion éthanoate : CH3COO–(aq).
Ion benzoate : C6H5COO–(aq).
Ammoniac : NH3(aq).Ion nitrite : NO2
–(aq).
b) • CH3COOH(aq) + H2O(l) = CH3COO–(aq) + H3O
+(aq).• C6H5COOH(aq) + H2O(l) = C6H5COO
–(aq) + H3O+(aq).
• NH4+(aq) + H2O(l) = NH3(aq) + H3O
+(aq).• HNO2(aq) + H2O(l) = NO2
–(aq) + H3O+(aq).
3. a) Acide méthanoïque : HCOOH(aq).Ion méthylammonium : CH3NH3
+(aq).Acide phosphorique : H3PO4(aq).Ion hydrogénocarbone : HCO3
–(aq).
b) • HCOO–(aq) + H2O(l) = HO–(aq) + HCOOH(aq).
• CH3NH2(aq) + H2O(l) = HO–(aq) + CH3NH3
+(aq).• H2PO4
–(aq) + H2O(l) = HO–(aq) + H3PO4(aq).
• CO32–(aq) + H2O(l) = HO
–(aq) + HCO3–(aq).
Corrigés des exercices (page 94)

48 Partie 2 - La transformation d’un système chimique est-elle totale ?
7 Ampholyte1. • HPO4
2–/PO43– (HPO4
2– est un acide). H2PO4
–/HPO42– (HPO4
2– est une base).• HSO4
2–/SO42– (HSO4
2– est un acide). H2SO4/HSO4
2– (HSO42– est une base).
• HS–/S2– (HS– est un acide). H2S/HS
– (HS– est une base).
2. • HPO42–(aq) + H2O(l) = H3O
+(aq) + PO43–(aq)
et HPO42–(aq) + H2O(l) = HO
–(aq) + H2PO4–(aq)
• HSO42–(aq) + H2O(l) = H3O
+(aq) + SO42–(aq)
et HSO42–(aq) + H2O(l) = HO
–(aq) + H2SO4(aq)• HS–(aq) + H2O(l) = H3O
+(aq) + S2–(aq) et HS–(aq) + H2O(l) = HO
–(aq) + H2S(aq).
8 pH de solutions1. pH = – log [H3O
+aq)] où [H3O+(aq)] est le nombre
représentant la valeur de la concentration en ions oxonium exprimée en mol · L–1. 2.Solutions A B C D
[H3O+(aq)] 5,0 × 10–4mol · L–1
2,5mmol · L–1
1,5 × 10–3mol · L–1
1,0 mol · m–3
pH 3,3 2,6 2,8 3
3. [H3O+(aq)] = 10–pH.
4.Solutions E F G H
pH 2,5 3,4 8,2 7
[H3O+(aq)] 3,2 × 10–3mol · L–1
4,0 × 10–4mol · L–1
6,3 × 10–9mol · L–1
1,0 × 10–7mol · L–1
9 Calcul de pH1. [H3O
+(aq)] = 1,6 × 10–3 mol · L–1.2. pH = 2,8.3. a) n(H3O
+) = 3,2 × 10–5 mol.b) [H3O
+(aq)] = 3,2 × 10–4 mol · L–1, donc pH = 3,5.
10 Détermination d’un avancement final1. a) HCl(g) + H2O(l) = H3O
+(aq) + Cl–(aq).b) xf = V · [H3O
+aq)]f = V · 10–pH ; xf = 6,3 × 10
–5 mol.
2. a) C6H5COOH(aq) + H2O(l)= C6H5COO
–(aq) + H3O+(aq).
b) [C6H5COO–(aq)]f = [H3O
+(aq)]f = x
vf .
σ = λ(H3O+) · [H3O
+(aq)]f+ λ(C6H5COO
–) · [C6H5COO–(aq)]f.
σ = x
vf · (λ(H3O
+) + λ(C6H5COO–).
d’où xf = σ
λ λ· v
(H O ) (C H COO )3+
6 5–+
.
c) Attention : V en m3 et σ en S · m–1.xf = 8,6 × 10
–6 mol.
11 Quotients de réaction
a) Qr = [H O (aq)] [C H COO (aq)]
[C H COOH(aq)]3
+6 5
–
6 5
·.
b) Qr = [HO (aq)] [C H NH (aq)]
[C H NH (aq)]
–2 5 3
2 5 2
· +.
c) Qr = 1
[Fe (aq)] [HO (aq)]3 – 3+ ·.
d) Qr = [S O (aq)] [I (aq)]
[I (aq)] [S O (aq)]4 6
2 – 2
2 2 32
−
−
·
· 22.
e) Qr = [Zn (aq)]
[Cu (aq)]
2+
2+.
f) Qr = [CH COOH(aq)] [NH (aq)]
[NH (aq)] [CH COO (3 3
4 3–
·
·+ aaq)].
g) Qr = [CH COOC H (l)] [H O(l)]
[CH COOH(l)] [C H O3 2 5 2
3 2 5
·
· HH(l)].
12 Constante d’équilibre de réactions inverses
1. NH4+(aq) + CH3COO
–(aq) = NH3(aq) + CH3COOH(aq).
2. K = [CH COOH(aq)] [NH (aq)]
[NH (aq)] [CH COO (3 3
4 3–
·
·+ aaq)].
3. NH3(aq) + CH3COOH(aq) = NH4+(aq) + CH3COO
–(aq).
4. K' = [CH COO (aq)] [NH (aq)]
[CH COOH(aq)] [NH (3
–4
3 3
·
·
+
aaq)].
5. a) & b) K' = 1
K ; K' = 2,51 × 104.
13 Ions du cuivre1. NH3(aq) + H2O(l) = HO
–(aq) + NH4+(aq).
2. a) Les ions Cu2+(aq) forment un précipité avec les ions HO–(aq) présents dans la solution aqueuse d’ammoniac.b) Cu2+(aq) + 2 HO–(aq) = Cu(OH)2(s).
c) K1 = 1
[Cu (aq)] [OH (aq)]2+ – 2·.

49Chapitre 4 - Notion d’équilibre chimique
3. a) Cu(OH)2(s) + 4 NH3(aq)= Cu(NH3)4
2+(aq) + 2 HO–(aq).
b) K2 = [HO (aq)] [Cu(NH ) (aq)]
[NH (aq)]
– 23 4
2
34
· +.
4. a) K3 = [Cu(NH ) (aq)]
[Cu (aq)] [NH (aq)]3 4
2
23
4
+
+ ·.
b) K3 = K1 · K2.
14 Étude d’une transformation
Exercice résolu dans le manuel de l’élève.
15 Taux d’avancement et dilution1. C2 = 1,00 × 10
–3 mol · L–1.2. On prélève 10 mL de solution S1 à la pipette jau-gée, que l’on introduit dans une fiole jaugée de 100 mL, et on étend avec de l’eau distillée jusqu’au trait de jauge.3. HCOOH(aq) + H2O(l) = HCOO
–(aq) + H3O+(aq).
4.HCOOH(aq) + H2O(l) = HCOO
–(aq) + H3O+(aq)
État initial C · V Excès 0 0
État finalthéorique C · V – xmax Excès xmax xmax
État finalréel C · V – xf Excès xf xf
xmax = C · V et xf = nf(H3O+) = [H3O
+(aq)] · V,
donc τ = [H O (aq)3
+f]
C.
5. a) τS1 = 0,125 = 12,5 %. τS2 = 0,398 = 39,8 %.b) Plus la concentration en soluté apportée est fai-ble, plus le taux d’avancement final augmente : la dilution favorise donc la dissociation du soluté.
16 Quotient de réaction à l’équilibre et dilution
1. CH3COOH(aq) + H2O(l) = CH3COO–(aq) + H3O
+(aq).
2. a) Qr'f = [H O (aq)] [C H COO (aq)]
[C H COOH(aq)]3
+f 6 5
–f
6 5 f
·.
3. CH3COOH(aq) + H2O(l)
= CH3COO–(aq) + H3O
+(aq)
État initial C · V Excès 0 0
État finalthéorique C · V – xmax Excès xmax xmax
État finalréel C · V – xf Excès xf xf
xf = nf (CH3COO–) = nf(H3O
+),donc [CH3COO
–(aq)]f = [H3O+(aq)]f
et nf(CH3COOH) = C · V – xf = C · V – nf(H3O+)
donc [CH3COOH(aq)]f = C – [H3O+(aq)]f.
4. Qr,f = [ (
]
H O aq)]
– [H O (aq)3
+f2
3+
fc0.
5.
Solution S1 S2 S3
Qr,f 1,65 × 10–5 1,62 × 10–5 1,64 × 10–5
6. La valeur du quotient de réaction à l’équilibre est indépendante de la concentration initiale en soluté apporté.
17 Solution aqueuse de fluorure d’hydrogène
1. HF(aq) + H2O(aq) = H3O+(aq) + F–(aq).
2.HF(aq) + H2O(aq) = H3O
+(aq) + F–(aq)
État initial 1,25 × 10–3 Excès 0 0
État finalthéorique 1,25 × 10–3 – xmax Excès xmax xmax
État final réel 1,25 × 10–3 – xf Excès xf xf
3. xmax = 1,25 × 10–3 mol.
4. xf = [H3O+(aq)]f · V = 10
–pH · V ;xf = 3,96 × 10
–4 mol.
5. a) τ = 0,32.b) τ < 1 : transformation non totale.6. [H3O
+(aq)]f = [F–(aq)]f = 1,59 × 10
–3 mol · L–1.[HF(aq)]f = 3,41 × 10
–3 mol · L–1.
7. K = 7,41 × 10–4.
18 Détermination d’une constante d’équilibre par conductimétrie
1. C2H5COOH(aq) + H2O(l)= C2H5COO
–(aq) + H3O+(aq).
2. C2H5COOH(aq) + H2O(l)
= C2H5COO–(aq) + H3O
+(aq)
État initial C · V Excès 0 0
État finalthéorique C · V – xmax Excès xmax xmax
État finalréel C · V – xf Excès xf xf

50 Partie 2 - La transformation d’un système chimique est-elle totale ?
3. [C2H5COO–(aq)]f = [H3O
+(aq)]f.[C2H5COOH(aq)]f = C – [H3O
+(aq)]f.
4. K = [ ]H O (aq)
– [H O (aq)]3
+f2
3+
fc0.
5. σ = λ(C2H5COO–) · [C2H5COO
–(aq)]f+ λ(H3O
+) · [H3O+(aq)]f.
Donc [H3O+(aq)]f =
σλ λ(C H COO ) (H O )2 5
–3+ +
;
[H3O+(aq)]f = 1,57 mol · m
–3 = 1,57 × 10–3 mol · L–1.6. K = 1,24 × 10–5.
19 Propriétés de l’acide propanoïque
Exercice résolu dans le manuel de l’élève.
20 Le comportement de deux acides dans l’eau
1. a) HClO4(aq) + H2O(l) = (aq) + H3O+(aq).
b)HClO4(aq) + H2O(l) = ClO4
–(aq) + H3O+(aq)
État initial CA · V Excès 0 0
État finalthéorique CA · V – xmax Excès xmax xmax
État finalréel CA · V – xf Excès xf xf
c) xmax = CA · Vet xf = nf(H3O
+) = V · [H3O+(aq)]f = V · 10
–pH.
d) τ = x
xf
max
= 10−pH
AC.
τ = 1 : la transformation est totale.2. a) C6H5COOH(aq) + H2O(l)
= C6H5COO–(aq) + H3O
+(aq).b)
C6H5COOH(aq) + H2O(l)= C6H5COO
–(aq) + H3O+(aq)
État initial C · V Excès 0 0
État finalthéorique C · V – xmax Excès xmax xmax
État finalréel C · V – xf Excès xf xf
c) τ = x
xf
max
= 10−pH
AC.
τ = 0,16 : la transformation est limitée.
3. De deux solutions acides de même concentra-tion, celle qui possède le taux d’avancement final le plus élevé est celle dont le pH est le plus faible.
21 À propos de l’aspirine1. m = Cs · Vs · MS ; m = 0,5 g.
2. [H3O+(aq)]f = 10
–pH = 1,3 × 10–3 mol · L–1.
3. AH(aq) + H2O(l) = A–(aq) + H3O
+(aq).
4. xf = nf(H3O+) = V · [H3O
+(aq)]f ; xf = 6,5 × 10
–4 mol.
5. xmax = Cs · Vs ; xmax = 2,78 × 10–3 mol.
6. a) τ = x
xf
max
; τ = 0,23.
b) La transformation est limitée (τ < 1).7. σ = λ(H3O
+) · [H3O+(aq)]f + λ(A
–) · [A–(aq)]f.
σ = λ(H3O+) ·
x
Vf
S
· (λ(H3O+) + λ(A–)),
d’où xf = σ
λ λ
·VS
(H O ) (A )3–+ +
.
8. xf = 5,7 × 10–4 mol.
9. • [H3O+(aq)]f = [A
–(aq)]f = x
Vf
S
;
[H3O+(aq)]f = 1,1 × 10
–3 mol · L–1.
• [AH(aq)]f = Cs – [A–(aq)]f ;
[AH(aq)]f = 4,45 × 10–3 mol · L–1.
10. K = [H O (aq)] [A (aq)]
[AH(aq)]3 f f
f
+ −· ;
K = 2,7 × 10–4.11. • Un écart de 0,1 unité de pH conduit à un écart relatif sur xf :
x x
xf f
f
(pH 2,9) (pH 2,8)
(pH 2,9)
= − ==
= 0,21 ≈ 21 %.
• Un écart de 1 mS · m–1 conduit à un écart relatif sur xf :
x x
xf f
f
( ) ( )
( )
σ σσ
= − ==
44 43
44 = 0,017 = 1,7 %.
La détermination de xf à partir de la mesure de la conductivité de la solution est nettement plus pré-cise que celle à partir du pH.

51Chapitre 4 - Notion d’équilibre chimique
Exercices supplémentaires1. Mélange de solutions acidesDans un bécher, on mélange un volume V1 = 10,0 mL de solution aqueuse acide S1, de pH1 = 2, et un volume V2 = 90,0 mL de solution aqueuse acide S2, de pH2 = 3.On obtient une solution S.1. Déterminer la concentration molaire puis la quantité de matière en ions oxonium de chacune des solutions S1 et S2.2. Quelle quantité de matière en ions oxonium est contenue dans la solution S ?3. En déduire la concentration molaire en ions oxo-nium puis le pH de la solution S.
Corrigé1. • [H3O
+(aq)]1 = 1,00 × 10–2 mol · L–1
et n1(H3O+) =1,00 × 10–4 mol.
• [H3O+(aq)]2 = 1,00 × 10
–3 mol · L–1 et n2(H3O
+) = 9,00 × 10–5 mol.2. nS (H3O
+) = 1,9 × 10–4 mol.3. [H3O
+(aq)]S = 1,9 × 10–3 mol · L–1 et pH = 2,7.
2. Taux d’avancement finalOn dissout une quantité de matière n = 1,00 × 10–3 mol de chlorure d’ammonium NH4Cl(aq) dans 100 mL d’eau.Le chlorure d’ammonium se dissocie totalement en ions ammonium, NH4
+(aq), et en ions chlorure, Cl–(aq).
Le pH de la solution obtenue est 5,1.1. L’ion ammonium est un acide : écrire l’équation de la réaction entre les ions ammonium et l’eau modélisant la transformation chimique se produi-sant.2. a) Construire le tableau de suivi de l’évolution de la transformation.b) En déduire l’avancement maximal xmax.3. L’avancement final de la transformation est :
xf = 7,94 × 10–7 mol.a) La transformation est-elle totale ?b) Déterminer la quantité de matière de chacune des espèces chimiques dans l’état final puis leur concentration molaire.c) Calculer le taux d’avancement final de la trans-formation.
Corrigé1. NH4
+(aq) + H2O(l) = NH3(aq) + H3O+(aq).
2.NH4
+(aq) + H2O(l) = NH3(aq) + H3O+(aq)
État initial 1,00 × 10–3 Excès 0 0
État final théorique 1,00 × 10–3 – xmax Excès xmax xmax
État final réel 1,00 × 10–3 – xf Excès xf xf
NH4+ est limitant, donc xmax = 1,00 × 10
–3 mol.
3. a) xf < xmax : la transformation est limitée.
Les compétences expérimentales
(page 100)
On présente dans cette évaluation des situations erronées couramment observées lors des manipulations des élèves et les manipulations correc-tes. L’objectif est de reconnaître les situations correctes et de justifier le choix en vue de la préparation à l’épreuve expérimentale du baccalauréat.
Corrigé1. Mesure correcte d’un pHLa représentation correcte est la B.La sonde est maintenue correctement par en support, est totalement immergée dans la solution et l’agitation est en marche.
2. Préparation d’une solutionLa représentation correcte est la A.La verrerie utilisée est de la verrerie de précision (pipette jaugée et fiole jaugée).

52 Partie 2 - La transformation d’un système chimique est-elle totale ?
b) nf(NH4+) ≈ 1,00 × 10–3 mol.
nf(NH3) = nf(H3O+) = 7,94 × 10–7 mol.
[NH4+(aq)]f = 1,0 × 10
–2 mol · L–1.[NH3(aq)]f = [H3O
+(aq)]f = 7,94 × 10–6 mol · L–1.
c) τ = 7,94 × 10–4.
3. Équilibre et calcul de pH1. Écrire l’équation de la réaction modélisant la transformation se produisant lors de la mise en solution aqueuse de l’acide éthanoïque.2. Donner l’expression de sa constante d’équilibre K, dont la valeur à 25 °C est K = 1,58 × 10–5.3. Une solution d’acide éthanoïque contient 3,16 × 10–6 mol d’ions éthanoate et 6,83 × 10–6 mol d’acide éthanoïque non dissocié.a) Déterminer la relation numérique entre [CH3COOH(aq)]f et [CH3COO–(aq)]f dans cette solution.b) En déduire la concentration molaire en ions oxonium de la solution ainsi que son pH.
Corrigé1. CH3COOH(aq) + H2O(l) = CH3COO
–(aq) + H3O+(aq).
2. K = Qr,f = [H O (aq)] [CH COO (aq)]
[CH COOH(aq)]3 f 3 f
3 f
+ −·.
3. a) [CH3COO–(aq)]f = 2,16 × [CH3COOH(aq)]f.
b) [H3O+(aq)] = 7,31 × 10–6 mol · L–1 et pH = 5,1.
4. Expérience du jet d’eauUn volume de chlorure d’hydrogène, HCl, gazeux a été introduit dans un ballon muni d’une tubulure fermée par une pince. Le ballon est retourné sur un cristallisoir rempli d’eau. Après avoir fait pénétrer quelques gouttes d’eau par la tubulure, on plonge cette dernière dans le cristallisoir, on observe alors un jet d’eau à l’intérieur du ballon. On recueille au final 100 mL de solution dans le ballon.1. Sachant que le chlorure d’hydrogène est un gaz très soluble dans l’eau, interpréter le phénomène.2. Quelques gouttes de nitrate d’argent introduites dans la solution contenue dans le ballon font appa-raître un précipité blanc, qui noircit à la lumière.a) Quel est ce précipité ?b) Quel type d’ions la solution aqueuse contient-elle ?3. Écrire l’équation de la réaction se produisant entre le chlorure d’hydrogène et l’eau.4. Le pH de la solution obtenue vaut 2,5.a) Calculer la concentration molaire des ions oxo-nium de la solution.b) En déduire l’avancement final de la réaction.
5. La transformation étant totale, déterminer la quantité de matière de chlorure d’hydrogène conte-nue initialement dans le ballon.
Corrigé1. La dissolution du gaz dans l’eau crée une dépres-sion à l’intérieur du ballon : l’eau du cristallisoir est alors aspirée dans le ballon.2. a) Le chlorure d’argent, AgCl(s).b) La solution contient donc des ions chlorure Cl–(aq).3. HCl(g) + H2O(l) = Cl
–(aq) + H3O+(aq).
4. a) [H3O+(aq)] = 3,16 × 10–3 mol · L–1.
b) xf = [H3O+(aq)] · V ; xf = 3,16 × 10
–4 mol.5. ni(HCl) = xf = 3,16 × 10
–4 mol.
5. Prévoir le pH d’une solutionOn prépare une solution d’acide éthanoïque, de concen-tration en soluté apporté C = 5,0 × 10–3 mol · L–1.1. Écrire l’équation de la réaction se produisant entre l’acide éthanoïque et l’eau.2. Construire le tableau de suivi de l’évolution de la transformation, en prenant une quantité de matière initiale d’acide éthanoïque égale à celle contenue dans un volume V de solution.

53Chapitre 4 - Notion d’équilibre chimique
3. En déduire l’expression des concentrations molaires à l’état final en acide éthanoïque et en ions éthanoate en fonction de la concentration molaire en ion oxonium et de la concentration en soluté apporté.4. a) Donner la constante d’équilibre, en l’expri-mant en fonction de c et de la concentration molaire en ions oxonium.b) Montrer que [H3O+(aq)]f est solution de l’équa-tion du second degré :
[H3O+(aq)]f2 + K · [H3O+(aq)]f – K · C = 0.
5. Sachant que K = 1,58 × 10–5, résoudre l’équation et déterminer [H3O+(aq)]f .6. Déterminer la valeur du pH pour cette solution.
Corrigé1. CH3COOH(aq) + H2O(l) = CH3COO
–(aq) + H3O+(aq).
2.C6H5COOH(aq) + H2O(l)
= C6H5COO–(aq) + H3O
+(aq)
État initial C · V Excès 0 0
État finalthéorique C · V – xmax Excès xmax xmax
État final réel C · V – xf Excès xf xf
3. [CH3COO–(aq)]f = [H3O
+(aq)]fet nf(CH3COOH) = C · V – xf = C · V – nf(H3O
+), donc [CH3COOH(aq)]f = C – [H3O
+(aq)]f.
4. a) K = Qr,f = [H O (aq)] [CH COO (aq)]
[CH COOH(aq)]3
+f 3
–f
3 f
·.
K = Qr,f = [H O (aq)]
– [H O (aq)]3
+f2
3+
fC.
b) K · C – K · [H3O+(aq)]f = [H3O
+(aq)]f2,
soit [H3O+(aq)]f
2 + K · [H3O+(aq)]f – K · C = 0.
5. La résolution de l’équation du second degré donne :
[H3O+(aq)]f = 2,73 × 10
–4 mol · L–1.6. pH = 3,6.
6. Détermination d’une solubilitéLe chlorure d’argent AgCl(s) est un solide ionique peu soluble dans l’eau. On prépare une solution aqueuse saturée de chlorure d’argent en dissolvant une masse m de chlorure d’argent solide, de façon à obtenir une solution saturée. La mesure de la
conductivité de la solution saturée obtenue donne σ = 180 µs · m–1 à 25 °C.On donne les valeurs de conductivité ioniques molaires λ à 25 °C suivantes :– ion chlorure : 7,5 × 10–3 S · m2 · mol–1 ;– ion argent : 6,2 × 10–3 S · m2 · mol–1.1. Écrire l’équation de dissolution du chlorure d’argent dans l’eau.2. Donner l’expression de la constante d’équilibre K ; sa valeur est 1,7 × 10–10.3. Dans la solution, quelle relation existe entre [Ag+(aq)]f et [Cl–(aq)]f ?4. En déduire les valeurs de [Ag+(aq)]f et de [Cl–(aq)]f.5. La solubilité s d’une substance chimique est la quantité maximale de cette substance que l’on peut dissoudre dans un litre de solvant.Déterminer la solubilité du chlorure d’argent en mol · L–1 puis en g · L–1.6. À 100 °C, la constante de l’équilibre vaut : K ' = 2,2 × 10–8.a) Calculer les valeurs de [Ag+(aq)]f et de [Cl–(aq)]f pour une solution saturée.b) Calculer la conductivité σ de la solution saturée.c) Conclure quant à l’influence de la température sur la solubilité du chlorure d’argent dans l’eau.
DonnéesMasse molaire de l’argent : 107,9 g · mol–1.Masse molaire du chlore : 35,5 g · mol–1.
Corrigé1. AgCl(s) = Ag+(aq) + Cl–(aq).2. K = [Ag+(aq)]f · [Cl
–(aq)]f.3. [Ag+(aq)]f = [Cl
–(aq)]f.4. [Ag+(aq)]f = [Cl
–(aq)]f = K = 1,3 × 10–5 mol · L–1.5. s = [Ag+(aq)]f = 1,3 × 10
–5 mol · L–1
s = 1,87 × 10–3 g.6. a) [Ag+ (aq)]f = [Cl
–(aq)]f = K= 1,48 × 10–4 mol · L–1.
b) σ = λ(Ag+) · [Ag+ (aq)]f + λ(Cl–) · [Cl–(aq)]f ;
σ = 2,0 × 10–3 S · m–1.c) Lorsque la température augmente, la solubilité du chlorure d’argent augmente.
7. La défense chimique des fourmis(D’après Bac Amérique du Nord, juin 2004.)
Pour se défendre, les fourmis utilisent deux moyens, leurs mandibules et la projection d’acide formique.
••

54 Partie 2 - La transformation d’un système chimique est-elle totale ?
Les mandibules immobilisent l’ennemi, tandis que l’acide formique brûle la victime. Une fourmi se sentant menacée se dresse donc sur les deux pattes arrière et peut projeter sur l’ennemi un jet d’acide formique à plus de 30 cm de son abdomen.L’acide formique (ou acide méthanoïque), soluble dans l’eau, a pour formule HCOOH. On se propose d’étudier quelques propriétés d’une solution aqueuse de cet acide.
DonnéesMasses molaires atomiques :M(C) = 12 g · mol–1 ;M(H) = 1 g · mol–1 ;M(O) = 16 g · mol–1.Conductivités molaires ioniques λ à 25 °C– H3O+ : 35,0 × 10–3 S · m2 · mol–1 ;– HCOO– : 5,46 × 10–3 S · m2 · mol–1.On rappelle l’expression de la conductivité σ d’une solution en fonction des concentrations molaires des espèces ioniques Xi dissoutes : σ = Σ λi · [Xi].
1. Dans une fiole jaugée de volume V0 = 100 mL, on introduit une masse m = 46,0 mg d’acide formique, puis on complète cette fiole avec de l’eau distillée jusqu’au trait de jauge et on homogénéise. On dis-pose alors d’une solution S0 d’acide formique, de concentration molaire en soluté apporté C0.a) Déterminer la valeur de la concentration C0.b) Écrire l’équation de la réaction associée à la transformation de l’acide formique en présence d’eau.c) Compléter le tableau 1 de l’évolution correspon-dant à cette transformation chimique, en fonction de C0, de V0, de xmax et de xf ; xmax et xf sont respec-tivement l’avancement maximal et l’avancement à l’équilibre.
Équation de la réaction …… + …… = …… + …….
États du système
Avanc. (mol)
Quantité de matière (mol)
État initial 0
État final (si transformation totale)
xmax
État d’équilibre xf
Tableau 1.
d) Exprimer le taux d’avancement final τ en fonc-tion de la concentration molaire des ions oxonium à l’équilibre, [H3O+(aq)]f, et de C0.
•
•
•
e) Donner l’expression du quotient de réaction à l’état d’équilibre, Qr,f .Montrer que ce quotient peut s’écrire sous la forme :
Qr, f = [H O (aq)]–[H O (aq)]
3+
f2
3+
fC0.
2. Exprimer la conductivité σ de la solution d’acide formique à l’état d’équilibre en fonction des conduc-tivités molaires ioniques des ions présents et de la concentration en ions oxonium à l’équilibre, [H3O+(aq)]f .3. La mesure de la conductivité à 25 °C de la solu-tion S0 donne σ = 0,050 S · m–1.En utilisant les relations obtenues précédemment, compléter le tableau 2.
Solution
S0 S1
Ci (mol · L–1) 0,010 0,1
s (S · m–1) 0,050 0,17
[H3O+(aq)]f (mol · m–3) 4,2
[H3O+(aq)]f (mol · L–1) 4,2 × 10–3
t (%) 4,2
Qr,f 1,8 × 10–4
Tableau 2.
4. On réalise la même étude en utilisant une solution S1 d’acide formique, de concentration C1 = 0,10 mol · L–1. Les résultats obtenus sont indiqués dans le tableau 2.En déduire l’influence de la concentration molaire en soluté apporté sur :a) le taux d’avancement final de la réaction ;b) le quotient de réaction dans l’état d’équilibre.
Corrigé1. a) C0 = ; C0 = 1,0 × 10
–2 mol · L–1.b) HCOOH(aq) + H2O(l) = HCOO
–(aq) + H3O+(aq).
c) Équation de la réaction
HCOOH(aq) + H2O(l)= HCOO–(aq) + H3O
+(aq)
Avanc. (mol)
État initial 0 C · V Excès 0 0
État finalthéorique xmax C · V – xmax Excès xmax xmax
État finalréel xf C · V – xf Excès xf xf

55Chapitre 4 - Notion d’équilibre chimique
d) xmax = C0 · V0 et xf = nf(H3O+) = V0 · [H3O
+(aq)]f
donc τ = x
xf
max
= [H O (aq)3
+f
0
]
C.
e) [HCOO–(aq)]f = [H3O+(aq)]f
et [HCOOH(aq)]f = C0 – [H3O+(aq)]f.
Donc : Qr,f = [H O (aq)] [HCOO (aq)]
[HCOOH(aq)]3
+f
–f
f
·
= [ ]
]
H O (aq)
– [H O (aq)3
+f2
0 3+
fC.
2. σ = λ(H3O+) · [H3O
+(aq)]f+ (λ(HCOO–) · [HCOO–(aq)]f
et [HCOO–(aq)]f = [H3O+(aq)]f.
Donc σ = (λ(H3O+) + (λ(HCOO–) · [H3O
+(aq)]f
on tire [H3O+(aq)]f =
σλ λ(HCOO ) (H O )–
3+ +.
3.Solution
S0 S1
Ci (mol · L–1) 0,010 0,1
s (S.m–1) 0,050 0,17
[H3O+]f (mol · m–3) 1,23 4,2
[H3O+]f (mol · L–1) 1,23 × 10–3 4,2 × 10–3
t (%) 12,3 4,2
Qr,f 1,7 × 10–4 1,8 × 10–4
4. a) Lorsque C0 augmente, le taux d’avancement final diminue.b) La valeur de Qr,f ne dépend pas de C0.

56 Partie 2 - La transformation d’un système chimique est-elle totale ?
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS CONNAISSANCES ET SAVOIR-FAIRE EXIGIBLES
Activités documen-taires et expérimen-tales autour du pH pour des produits de la vie courante et dans les milieux biologiques.
3. Transformations associées à des réactions acido-basiques en solution aqueuse– Autoprotolyse de l’eau ; constante d’équilibre appe-lée produit ionique de l’eau, notée Ke, et pKe.– Échelle de pH : solutions acide, basique et neutre.– Constante d’acidité, notée KA, et pKA.– Comparaison du comportement en solution, à concentration identique, des acides entre eux et des bases entre elles.– Constante d’équilibre associée à une réaction acido-basique.
– Savoir que Ke est la constante d’équilibre associée à l’équation de la réaction d’autopro-tolyse de l’eau.– Connaissant la valeur du pH d’une solu-tion aqueuse, dire si elle est acide, basique ou neutre.– À partir de la concentration molaire des ions H3O+ ou HO–, déduire la valeur du pH de la solution.– Associer la constante d’acidité KA à l’équation de la réaction d’un acide sur l’eau.
5 Les équilibres acido-basiques dans l’eau
Cours
Découpage du cours1. L’eau pure contient des ions p. 1022. Échelle de pH p. 1043. Constante d’acidité p. 1044. Comparaison des acides et des bases p. 1055. Diagrammes des espèces chimiques d’un couple p. 107
Dans ce chapitre, on aborde les transformations acido-basiques que les concepteurs du programme ont privilégiées pour illustrer la deuxième partie du programme : « La transformation d’un système chimique est-elle toujours totale ? ».Les élèves, qui jusqu’au chapitre précédent n’avaient rencontré que des exemples de transformations totales, vont approfondir ici cette nouvelle situation où la transformation s’effectue dans les deux sens.L’approche proposée est simple et s’appuie large-ment sur des observations expérimentales.
1. L’eau pure contient des ionsOn aborde l’autoprotolyse de l’eau en partant du constat que la conductivité de l’eau ultra-pure n’est pas nulle et que celle-ci contient des ions H3O+(aq) et HO–(aq) ; un calcul permet cependant de préciser que cette transformation est très limitée. On définit le produit ionique de l’eau et on montre sur des exemples son utilisation pour trouver la valeur du
♦ pH d’une solution à partir de la concentration molaire des ions HO–(aq), ou H3O+(aq), et vice-versa.
2. Échelle de pHCe paragraphe définit l’échelle de pH et les notions de solutions acide, neutre ou basique.Les travaux du danois Sørensen peuvent donner l’occasion de parler d’histoire des sciences et du lien entre chimie et biologie.
3. Constante d’aciditéOn définit la constante d’acidité d’un couple acide/base et le pKA. Sans unité, ces grandeurs dépendent uniquement de la température.L’exploitation des résultats de l’activité 2 montre que pour une température donnée, KA ne dépend pas de la composition initiale.Le professeur pourra préciser que la valeur du pKA d’un couple acide/base dépend du solvant dans lequel il est mesuré. On attribue une valeur négative, par exemple pKA = – 7 pour le couple HCl(aq)/Cl–(aq), ou supérieure à 14, par exemple, pKA = 16 pour le couple CH3CH2OH(aq)/CH3CH2O–(aq), au pKA d’un couple acide/base dont le KA n’est pas mesurable dans l’eau mais peut

57Chapitre 5 - Les équilibres acido-basiques dans l’eau
l’être dans un autre solvant. Les acides de pKA < 0 sont « nivelés » par l’eau, car ils réagissent avec l’eau selon une transformation totale ; il en est de même pour les bases de pKA > 14.
4. Comparaison des acides et des basesConformément au programme, il a été choisi d’in-troduire le taux d’avancement final pour comparer facilement les acides entre eux ou les bases entres elles, à concentration molaire identique.En partant des mesures du pH de deux solutions d’acides ou de bases courants, l’utilisation des tableaux descriptifs de l’évolution du système chimique permet de calculer τ et de conclure que la dissociation de l’acide ou la protonation de la base est plus ou moins grande selon la valeur de la constante KA du couple acide/base considéré.
5. Diagrammes des espèces chimiques d’un couple
On présente les diagrammes des espèces chimiques d’un couple acide/base.Le diagramme de prédominance est introduit à par-tir de la relation liant le pH, le pKA et les concentra-tions des espèces chimiques acide et basique du couple. Son application aux indicateurs colorés per-met de définir la zone de virage, notion reprise dans le chapitre 6 pour les dosages acido-basiques.Puis le diagramme de distribution donnant la répar-tition précise des deux espèces chimiques d’un cou-ple est détaillé.Le choix du bleu de bromothymol pour illustrer ces diagrammes résulte du fait que cet indicateur coloré est souvent utilisé en TP et dans les exercices.
Activités
Les concentrations effectives des espèces à l’équilibre sont indifféremment indiquées f ou éq.
De l’importanced’un bon pH ! (page 110)
Cette activité permet d’écrire des équations modé-lisant des transformations acide-base à partir d’un exemple connu des élèves : la surveillance du pH des piscines. L’apport dans l’eau d’ions H3O+(aq) ou HO–(aq), selon le cas, explique le recours à des pro-duits réducteur ou rehausseur de pH.On présente également dans cette activité l’utilisa-tion d’un indicateur coloré dans une application de la vie courante. L’introduction d’extraits de texte documentaire dans les sujets de baccalauréat per-met l’entraînement des élèves à l’extraction d’infor-mations d’un texte.
1. Le rouge de phénol est un indicateur coloré acide/base.2. CO2(aq) + 2 H2O(l) = HCO3
–(aq) + H3O+(aq).
Il y a formation d’ions H3O+(aq), donc le pH dimi-
nue.3. a) Na2CO3(s) = 2 Na
+(aq) + CO32–(aq).
CO32–(aq) + H2O(l) = HCO3
–(aq) + HO–(aq).b) Des ions HO–(aq) sont libérés, le pH augmente, d’où l’expression « rehausseur de pH ».4. NaHSO4(s) = Na
+(aq) + HSO4–(aq).
HSO4–(aq) + H2O(l) = SO4
2–(aq) + H3O+(aq).
Des ions H3O+(aq) sont libérés, donc le pH diminue,
d’où l’expression « réducteur de pH ».
ACTIVITÉ
1
ACTIVITÉ
1
Comparaison d’acidesen solution aqueuse (page 111)
La partie A de cette activité montre que la constante d’équilibre, qui dans le cas de la réaction des acides sur l’eau est la constante d’acidité KA, ne dépend pas pour un couple acide/base donné des conditions initiales.Le taux d’avancement final varie avec la concentra-tion molaire en soluté apporté.Dans la partie B, pour différents couples acide/base de même concentration molaire en soluté apporté, on vérifie que le taux d’avancement final augmente si KA augmente ou si pKA diminue.L’importance de la dissociation d’un acide peut être reliée à la valeur du pH de la solution ou à la gran-deur du KA du couple acide/base.
A Taux d’avancement et concentration1. Prélever 10 mL de la solution S0 à l’aide d’une pipette jaugée et les introduire dans une fiole jau-gée de 100 mL ; compléter avec de l’eau distillée.3. pH0 = 3,4 ; pH1 = 3,5 ; pH2 = 3,9.
4. τ = x
xf
max
= n
C V
(H O3+)
⋅ = [ ]H O (aq)3
+f ⋅
⋅V
C V
τ = [ ]H O (aq)3
+f
C.
5. a) KA = CH CO (aq) H O (aq)
CH CO H(aq)
3 2–
f3
f
3 2
⋅
+
f
.
ACTIVITÉ
2
ACTIVITÉ
2

58 Partie 2 - La transformation d’un système chimique est-elle totale ?
b) S0 S1 S2
pH 3,4 3,5 3,9
[H3O+(aq)]f (mol · L–1) 3,9 × 10–4 3,0 × 10–4 1,3 × 10–4
t 0,04 0,06 0,13
[CH3COO–(aq)]f(mol · L–1)
4,0 × 10–4 3,0 × 10–4 1,3 × 10–4
[CH3COOH(aq)]f(mol · L–1)
9,6 × 10–3 4,7 × 10–3 8,7 × 10–4
KA 1,8 × 10–5 1,9 × 10–5 1,9 × 10–5
6. KA est une constante qui ne dépend pas des conditions initiales.7. C’est dans la solution S3 que l’acide est le plus dissocié.8. Le taux d’avancement final augmente lorsque C diminue.
B Taux d’avancement et constante d’acidité2.
S0 S1 S2
pH 3,4 2,9 3,1
[H3O+(aq)]f (mol · L–1) 4,0 × 10–4 1,3 × 10–3 7,9 × 10–4
[A–(aq)]f (mol · L–1) 4,0 × 10–4 1,3 × 10–3 7,9 × 10–4
[HA(aq)]f (mol · L–1) 9,6 × 10–3 8,7 × 10–3 9,2 × 10–3
t 0,04 0,13 0,08
KA 1,7 × 10–5 1,9 × 10–4 6,8 × 10–5
3. a) L’acide le plus dissocié est celui pour lequel le pH est le plus petit.b) L’acide le plus dissocié est celui pour lequel le KA est le plus grand.
4. pKA0 = 4,8 ; pKA1 = 3,7 ; pKA2 = 4,2.
5. L’acide le plus dissocié est celui pour lequel le pKA est le plus petit.
6. Le taux d’avancement final augmente lorsque KA augmente ou pKA diminue.
C Conclusions1. a) À constante d’acidité fixe, le taux d’avance-ment final de la mise en solution d’un acide aug-mente lorsque la concentration molaire en soluté apporté diminue.
b) À concentration fixe, le taux d’avancement final de la mise en solution d’un acide augmente lorsque la constante d’acidité du couple dont fait partie le couple augmente.
2. Le taux d’avancement final dépend du rapport K
CA (réponse c).
Détermination du pKAd’un indicateur coloré (page 112)
Ce TP permet l’utilisation du pH-mètre et du spec-trophotomètre en travaillant sur l’exemple d’un indicateur coloré acide/base usuel, le bleu de bro-mothymol.Dans la partie A sont mis en évidence les différentes teintes prises par l’indicateur en fonction du pH.Les mesures d’absorbance conduisent dans la partie B au calcul du pKA du couple auquel appartient le BBT.Dans la partie C, l’exploitation des diagrammes de prédominance des espèces chimiques amène la notion de teinte sensible de l’indicateur.
A Les teintes du BBTLe BBT est jaune en milieu acide et bleu en milieu basique.En milieu neutre, le BBT est vert, puisque l’on a des entités jaunes et bleues en quantité très proches.
B pKA de l’indicateur coloré
1. C = C V
VBBT BBT
total
⋅ ; C = 4 × 10–5 mol · L–1.
2. HInd(aq) + H2O = Ind–(aq) + H3O
+(aq).
3. KA = Ind (aq) . H O (aq)
HInd(aq)
éq3
éq
é
− +
.
4. pH = pKA + log10 Ind (aq)
HInd(aq)
éq
éq
−
.
5. C = [HInd(aq)]éq + [Ind–(aq)]éq.
6. Dans S1 qui est très acide, on a [Ind–(aq)]eq ≈ 0,
donc [HInd(aq)]éq = C.Dans S3 qui est très basique, on a [HInd(aq)]éq ≈ 0, donc [Ind–(aq)]éq = C.
7. A1 = k1 · [HInd(aq)] = k1 · C.A3 = k2 · [Ind
–(aq)] = k2 · C.A2 = k1 · [HInd(aq)]éq + k2 · [Ind
–(aq)]éq.
8. A1 – A2 =k1 · C – (k1 · [HInd(aq)]éq + k2 · [Ind
–(aq)]éq).A1 – A2 = k1 · [Ind
–(aq)]éq – k2 [Ind–(aq)]éq.
ACTIVITÉ
3
ACTIVITÉ
3

59Chapitre 5 - Les équilibres acido-basiques dans l’eau
A1 – A2 = (k1 – k2) · [Ind–(aq)]éq.
A2 – A3 = k1 · [HInd(aq)]éq + k2 · [Ind–(aq)]éq – k2 · C.
A2 – A3 = k1 · [HInd(aq)]éq – k2 · [HInd(aq)]éq.A2 – A3 = (k1 – k2) · [HInd(aq)]éq.
A A
A A1 2
2 3
−−
= Ind (aq)
HInd(aq)
éq
éq
−
.
9. pKA = pH2 – log10 A A
A A1 2
2 3
−−
.
10. pKa = 7,1 # 7,3.
C Teinte sensible de l’indicateur coloré
1. pH = pKA + log10 B(aq)
A(aq)
éq
éq
.
Si [A(aq)]éq > 10 · [B(aq)]éq, on a : A(aq)
B(aq)
éq
éq
> 10 ;
log10
A(aq)
B(aq)
éq
éq
> 1 et log10
A(aq)
B(aq)
éq
éq
< – 1.
D’où pH – pKA < – 1, c’est-à-dire pH < pKA – 1 : le BBT est jaune.
2. Si [B(aq)]éq > 10 · [A(aq)]éq, on a :
B(aq)
A(aq)
éq
éq
> 10 et log10
B(aq)
A(aq)
éq
éq
> 1.
D’où pH – pKA > 1, c’est-à-dire pH > pKA + 1 : le BBT est bleu.3. À la fois jaune et bleu, le BBT est vert.4.
Teinte sensible :vert
Ind–(aq)prédomine
Couleur :bleu
Couleur :jaune
HInd(aq)prédomine pKA
pHpKA – 1 pKA + 1
Étude de quelques couplesacide/base (page 113)
Ce TP d’entraînement à l’épreuve d’évaluation des compétences expérimentales s’appuie sur les notions d’équilibres acido-basiques et de constante d’acidité pour vérifier les savoir faire concernant :– la burette graduée : rinçage de la burette avec la solution à prélever, ajustement du zéro ;
ACTIVITÉ
4
ACTIVITÉ
4
– l’agitateur magnétique : position et vitesse cor-rectes du barreau aimanté ;– le pH-mètre : rinçage des électrodes, immersion des électrodes, lecture d’une valeur stabilisée ;– la pipette jaugée : pipetage dans un bécher, rin-çage de la pipette avec la solution à prélever, utilisa-tion d’un pipeteur, repérage correct des niveaux ;– la fiole jaugée : ajustement au trait de jauge et homogénéisation.
1 pkA d’un indicateur coloré
1. Le BBT est jaune dans le bécher 1 et bleu dans le bécher 3.2. La teinte sensible du BBT est le vert. Lorsque la teinte sensible de la solution est obtenue, on a : pH = 7.3. HInd(aq) + H2O(l) = Ind
–(aq) + H3O+(aq).
4. K1 = Ind (aq) . H O (aq)
HInd(aq)
éq3
éq
− +
ééq
.
5. pH = pKA.6. pKA= 7.
2 Comparaison du comportement de deux acides dans l’eau
pH1 = 3,4 et pH2 = 2,9.1. Il faut prélever 5 mL de la solution S à l’aide d’une pipette jaugée (dilution 10).2. HA(aq) + H2O(l) = A
–(aq) + H3O+(aq).
3. KA = A (aq) . H O (aq)
HA(aq)
éq3
éq
− +
.
4. S1 S2
pH 3,4 2,9
[H3O+(aq)] (mol · L–1) 4,0 × 10–4 1,3 × 10–3
xf (mol) 2 × 10–5 6,3 × 10–5
xmax (mol) 5 × 10–4 5 × 10–4
t 0,04 0,13
5. Le taux d’avancement final est très inférieur à 1, donc les transformations ne sont pas totales.6. L’acide méthanoïque, qui a un taux d’avance-ment final supérieur, est le plus dissocié.7. L’acide méthanoïque, qui est le plus dissocié, possède la constante d’acidité la plus grande.
60 Partie 2 - La transformation d’un système chimique est-elle totale ?
Activité supplémentaire
Solutions acides et basiquesCf. Manuel de l’élève, p. 104
Rappels sur les acides et les basesDans la théorie de Bronsted, un acide est une espèce chimique susceptible de ………………........Une base, selon Bronsted, est une espèce chimique capable de …………………………………………Une réaction acido-basique est une réaction au cours de laquelle il y a un …………………………… entre un acide et une base.
1. Des deux réactions chimiques dont les équations sont données ci-après, laquelle modélise une trans-formation chimique acido-basique ? Justifier la réponse.
• Réaction 1Cl2(aq) + 2 HO–(aq) = ClO–(aq) + Cl–(aq) + H2O(l).
• Réaction 2CH3COOH(aq) + CO3
2–(aq) = CH3COO–(aq) + HCO3–(aq).
2. Identifier les deux couples acide/base en pré-sence.
pH d’une solution aqueuse3. Sachant que le pH d’une solution aqueuse est lié à sa concentration en ions oxonium, H3O+(aq), par la relation : pH = – log[H3O+(aq)]éq,où [H3O+(aq)]éq
représente la valeur sans dimension de cette concentration à l’équilibre et que, inverse-ment :
[H3O+(aq)]= 10–pH,compléter le tableau suivant.
pH [H3O+(aq)]éq (mol · L–1)
3,7 × 10–3
3,2
7,2 × 10–2
2,5
•
•
1 Conductivité de l’eau1. Dissociation très partielle de certaines molé-cules d’eau en ions oxonium et hydroxyde : c’est l’autoprotolyse.2. Ke = [H3O+(aq)]éq · [HO–(aq)]éq.Ke dépend seulement de la température.
2 Soude1. [HO–(aq)]éq = 10–3 mol · L–1,
d’où [H3O+(aq)]éq = Ke
–éq[HO (aq)]
;
[H3O+(aq)]éq = 10
10
14
3
–
– = 10–11 mol · L–1 ; pH = 11.
Corrigés des exercices (page 114)
4. Mesurer le pH des solutions aqueuses suivantes et préciser leur caractère acide ou basique.
Cf. Fiche pratique 8, p. 274 du Manuel de l’élève
Solution étudiée pHmesuré[H3O+(aq)]éq
(mol · L–1)
Eau du robinet
Eau distillée
Soda au cola
Corrigé1. La réaction 2 modélise une transformation acide/base car elle met en jeu un transfert de pro-ton de l’acide CH3COOH(aq) vers la base CO3
2–(aq).
2. Les couples acide/base sont CH3COOH(aq)/ CH3COO–(aq) et HCO3
–(aq)/CO32–(aq).
3.
pH [H3O+(aq)]éq (mol · L–1)
2,4 3,7 × 10–3
3,2 6,3 × 10–4
1,1 7,2 × 10–2
2,5 3,2 × 10–3
4.
Solution étudiée pHmesuré
[H3O+(aq)]éq(mol · L–1)
Milieu
Eau du robinet
7,5 selon région
3,2 × 10–8 Basique
Eau distillée 5,8 1,6 × 10–6 Acide
Soda au cola 3,0 1,0 × 10–3 Acide

61Chapitre 5 - Les équilibres acido-basiques dans l’eau
2. pH = 8, donc [H3O+(aq)] = 10–8,
d’où [HO–(aq)]éq = Ke
3+
éq[H O (aq)] ;
[HO–(aq)]éq = 10
10
14
8
–
– = 10–6 mol · L–1.
3 Échelle de pH1.
Jusd’orange
Jus detomate Lait Sang
Eaude mer
Eausavonneuse
pH
2. Solutions acides : jus d’orange, jus de tomate, lait.Solutions basiques : sang, eau de mer, eau savon-neuse.3. Solution de pH = 7.
4 Couple ion ammonium/ammoniac1. NH4
+(aq) + H2O(l) = NH3(aq) + H3O+(aq).
2. KA = NH (aq) H O (aq)
NH (aq)
3 éq 3
4+
éq
⋅
+
éq . pKA = – log KA.
3. pH = pKA + log10 NH (aq)
NH (aq)
3 éq
4+
éq
.
5 Comparer deux acidesHA1 et HA2 sont deux acides tels que KA1 > KA2.Ils sont introduits en solution aqueuse avec la même concentration.1. KA1 > KA2, donc A1 plus dissocié que A2 : le pH1 est plus faible que le pH2.2. A est prédominant sur B si [A] > [B].À pH = 6,4, on a pH > pKA1, donc A1
– est prédominant sur HA1.
6 Indicateur coloré1. Acides ou bases dont les deux formes ont des teintes différentes.2. La zone de virage est la zone de pH pour laquelle on passe d’une solution ayant la teinte de la forme acide à une solution ayant celle de la forme basique.On appelle teinte sensible la couleur de l’indicateur dans la zone de virage.
7 L’ammoniac en solution aqueuse
Exercice résolu dans le manuel de l’élève.
8 Des étiquettes décollées !1. A–(aq) + H2O(l) = HA(aq) + HO
–(aq).Couples acide/base mis en jeu :
HA(aq)/A–(aq) ; H2O(l)/HO–(aq).
2. n(A1–)0 = C0 · V ;
n(A1–)0 = 1,0 × 10
–3 × 0,200= 2,0 × 10–4 mol = n(A2
–)0.
3. • [H3O+(aq)]A = 10
–9,5 mol · L–1 ; [H3O
+(aq)]B = 10–10,3 mol · L–1.
• n(H3O+)A = [H3O
+(aq)]A · V ; n(H3O
+)A = 10–9,5 × 0,2 = 6,3 × 10–11 mol.
• n(H3O+)B = [H3O
+]B · V ; n(H3O
+)B = 10–10,3 × 0,2 = 1,0 × 10–11 mol.
• n(HO–)A = Ke
3+
A[H O (aq)] · V ;
n(HO–)A = 10–4,5 × 0,2 = 6,3 × 10–6 mol.
• n(HO–)B = Ke
3+
B[H O (aq)] · V ;
n(HO–)B =10–3,7 × 0,2 = 4,0 × 10–5 mol.
4. a) Avancement maximal :xmax = n(A
–)0 = 2,0 × 10–4 mol.
Avancement final : xf = n(HO–)f ;
xfA = 6,3 × 10–6 mol ; xfB = 4,0 × 10
–5 mol.
b) τ = x
xf
max
. τA = 6 3 10
2 0 10
6
4
,
,
–
–
××
= 0,03 = 3 % ;
τB = 4 0 10
2 0 10
5
4
,
,
–
–
××
= 0,2 = 20 %.
5. Plus la base est dissociée dans l’eau, plus la constante d’acidité du couple est petite et plus le pH est élevé, donc A = solution 1 et B = solution 2.
9 Comparaison de trois solutions acides1. HA(aq) + H2O(l) = H3O
+(aq) + A–(aq).xf = n(H3O
+) = [H3O+(aq)] · V ; xmax = n(HA) = C · V.
τ = x
xf
max
= [H O (aq)]3
+ ⋅⋅
V
C V = [H O (aq)]3
+
C =
10–pH
C.
2. τ1 = 4 % ; τ2 = 32 % ; τ3 = 13 %.
3. A plus dissocié que A', donc KA > KA'et KA1 < KA3 < KA2.
4. pKA1 = 4,75 > pKA3 = 3,75 > pKA2 = 2,86.
10 Désinfectant de l’eau de piscine1. [HClO(aq)] = [ClO–(aq)] au point d’intersection des courbes pour des pourcentages égaux à 50 %.

62 Partie 2 - La transformation d’un système chimique est-elle totale ?
Or pH = pKA + log [ClO (aq)]
[HClO(aq)]
–éq
éq
et [HClO(aq)]éq = [ClO–(aq)]éq,
donc pH = pKA, d’où pKA = 7,3.
2.
HClO(aq) ClO–(aq)
pKA3 = 7,3pH
3. Courbe rouge : HClO(aq) ; courbe bleue : ClO–(aq).
4. HClO(aq) + H2O(l) = H3O+(aq) + ClO–(aq),
donc : KA3 = [ClO (aq)] [H O (aq)]
[HClO(aq)]
–éq 3
+éq
éq
⋅.
5. pH = 8,3 > pKA. L’espèce prédominante est l’ion hypochlorite, ClO–(aq).
6. – log KA3 = pKA3 = – log [ClO (aq)]
[HClO(aq)]
–éq
éq
– log [H3O+(aq)]éq.
log [ClO (aq)]
[HClO(aq)]
–éq
éq
= pH – pKA3 ;
[ClO (aq)]
[HClO(aq)]
–éq
éq
= 10pH – pKA3.
[ClO (aq)]
[HClO(aq)]
–éq
éq
= 108,3 – 7,3 = 10.
7. Graphiquement : [ClO (aq)]
[HClO(aq)]
–éq
éq
= 85
8 5, = 10.
11 Diagramme de distribution des espèces de bleu de bromothymol1. [HInd(aq)] + [Ind–(aq)] = C.2. a) Ind–(aq).b) A = k · [Ind–(aq)].3. En milieu très basique : [HInd(aq)] = 0. Or [HInd(aq)] + [Ind–(aq)] = C, donc [Ind–(aq)] = C.L’absorbance est maximale :
A = Amax = k · [Ind–(aq)] = k · C.
4. A = k · [Ind–(aq)] et Amax = k · C.Donc :
A
Amax = [Ind (aq)]−
C et [Ind–(aq)] = C ·
A
Amax,
d’où [HInd(aq)] = C · (1 – A
Amax).
5.
pH
[HInd(aq)]
[Ind
– (aq)]
3,00E-05
2,00E-05
1,00E-05
0,00E+000,0 2,0 4,0 6,0 8,0 10,0
[Ind–(aq)]3,00E-05
2,00E-05
1,00E-05
0,00E+00
[HInd(aq)]
6. HInd(aq) Ind–(aq)
6,6pH
7. Le graphe montre qu’à partir de la valeur pH = 9, [HInd(aq)] = 0 : l’hypothèse est vérifiée.
8. pH = pKA + log [Ind (aq)]
[HInd(aq)]
− et
si [HInd(aq)] = [Ind–(aq)] (intersection des courbes), alors pH = pKA et KA = 10
–pKA .9. Intervalle de pH de la zone de virage voisin de pKA – 1 < pH < pKA + 1, soit ici 5,6 < pH < 7,6.
12 De la couleur des fleurs d’hortensia
Exercice résolu dans le manuel de l’élève.
13 Propriétés de l’acide benzoïqueA. 1. m0 = n0 · M = C0 · V0 · M ;
m0 = 1,0 × 10–2 × 0,100 × 122 = 0,12 g.
Calcul de la concentration massique.
t0 = m
V0
0
; t0= 0 12
0 100
,
, = 1,2 g · L–1.
La concentration massique est inférieure à la solu-bilité de l’acide benzoïque dans l’eau, donc la solu-tion n’est pas saturée.2. C6H5–COOH(aq) + H2O(l)
= C6H5–COO–(aq) + H3O
+(aq).3.
C6H5–COOH(aq) C6H5–COO–(aq)
pKA1 = 4,2pH = 3,1
pH
L’espèce prédominante à pH = 3,1 < pKA1 est l’acide benzoïque.4. C6H5–COOH(aq) + H2O(l)
= C6H5–COO–(aq) + H3O
+(aq)
État du système
Avanc. (mol)
Quantités de matière (mol)
État initial 0 C0 · V0 Excès0 0
État final xf C0 · V0 – xf xf xf

63Chapitre 5 - Les équilibres acido-basiques dans l’eau
5. Si la transformation est totale, l’acide benzoïque est totalement consommé, soit C0 · V0 – xmax = 0,donc xmax = C0 · V0 ;
xmax = 1,0 × 10–2 × 0,100 = 1,0 × 10–3 mol.
τ =x
xéq
max
= [H O (aq)]3 éq
+ ⋅
⋅
V
C V0
0 0
= [H O (aq)]3 éq
+
C0 = 10 pH–
C0;
τ = 10 3– ,
–,
1
21 0 10× = 10–1,1 = 7,9 %.
La réaction est très limitée : il y a peu d’ions ben-zoate formés. L’acide benzoïque est bien l’espèce prédominante.
6. Qr,éq = [C H –COO (aq)] [H O (aq)]
[C H –COOH(aq6 5 éq 3 éq
6 5
− +⋅
))]éq.
D’après l’équation chimique :[C6H5COO
–(aq)]éq = [H3O+(aq)]éq.
Qr,éq = [H O (aq)]
[C H –COOH(aq)]3 éq
2
6 5 éq
+
.
D’après la conservation de la matière :[C6H5COOH(aq)]i = C0 = [C6H5COOH(aq)]éq
+ [C6H5COO–(aq)]éq,
donc [C6H5COOH(aq)]éq = C0 – [C6H5COO–(aq)]éq
= C0 – [H3O+(aq)]éq.
On obtient :
Qr, éq = [H O (aq)]
–[H O (aq)]
3 éq2
0 3+
éq
+
C =
10
–10
2pH
0–pH
–
C ;
Qr, éq = 10 6– ,
– – ,, –
2
2 3 11 0 10 10× = 6,9 × 10–5
7. Qr, éq = KA1. pKA1 = – log KA1 = – log Qr, éq ; pKA1 = – log (6,9 × 10
–5) = 4,2.
B. En utilisant le diagramme de prédominance de la question A.3, on peut dire que l’espèce du couple acide benzoïque/ion benzoate prédominante est l’ion benzoate.
14 Des indicateurs colorés en cuisine1. Un indicateur coloré change de couleur en fonc-tion du pH.2. Le vinaigre est acide : le choux rouge devient violet (pH : 4 à 6). Le détergent est basique : l’eau de rinçage devient verte (pH : 9 à 12).3. HAInd(aq) + H2O(l) = A
–Ind(aq) + H3O
+(aq).
Ki = [A (aq)] [H O (aq)]
[HA (aq)]Ind–
éq 3+
éq
Ind éq
⋅
K i
3+
éq[H O (aq)] =
[A (aq)]
[HA (aq)]Ind éq
Ind éq
–
10
10
pK
pH
i−
− = 10pH pKi− =
[A (aq)]
[HA (aq)]Ind éq
Ind éq
–
.
4. [A (aq)]
[HA (aq)]Ind éq
Ind éq
–
= 106,5–11,5 = 10–5,
donc HA prédomine.
15 pH d’un mélangeA. 1.a) HNO2(aq) + H2O(l) = NO2
–(aq) + H3O+(aq).
KA1 = [H O (aq)] [NO (aq)]
[HNO (aq)]3 éq 2 éq
2 éq
+ −⋅.
b) HCOO–(aq) + H2O(l) = HCOOH(aq) + HO–(aq).
K = [HO (aq)] [HCOOH(aq)]
[HCOO (aq)]
éq éq
–éq
– ⋅.
2. a) Pour pH < pKA, l’acide conjugué du couple prédomine.b) La solution d’acide nitreux a un pH de 1,3 ; l’espèce prédominante est l’acide nitreux HNO2(aq).La solution de méthanoate de sodium a un pH de 8,7 ; l’espèce prédominante est l’ion méthanoate HCOO–(aq).
B. 1. HNO2(aq) + HCOO–(aq)
= NO2–(aq) + HCOOH(aq).
2.HNO2(aq) + HCOO
–(aq) = NO2–(aq) + HCOOH(aq)
État du système chimique
Avanc. (mol)
Quantités de matière (mol)
n(HNO2) n(HCOO–) n(NO2–) n(HCOOH)
État init. x = 0 n1 n2 0 0
État final x = xéq n1 – xéq n2 – xéq xéq xéq
• [HNO2(aq)]éq = n x
V1 éq
2
−
[HNO2(aq)]éq = 4 0 10 3 3 10
2 0 200
2 2, – ,
,
–× ××
−
= 1,8 × 10–2 mol · L–1
• [HCOO–(aq)]éq = n x
V2 éq
2
−
• [HCOO–(aq)]éq = 8 0 10 3 3 10
2 0 200
2 2, – ,
,
–× ××
−
= 12 × 10–2 mol · L–1

64 Partie 2 - La transformation d’un système chimique est-elle totale ?
• [HCOOH(aq)]éq = [NO2–(aq)]éq =
x
Véq
2 ;
[HCOOH(aq)]éq = [NO2–(aq)]éq
= 3 3 10
2 0 200
2,
,
–××
= 8,3 × 10–2 mol · L–1.
3. Pour le couple HNO2(aq)/NO2–(aq), on a :
KA1 = [H O (aq)] [NO (aq)]
[HNO (aq)]3 éq 2 éq
2 éq
+ −⋅, soit
– log KA1 = pKA1 = – log [H O (aq)] [NO (aq)]
[HNO (aq)]3 éq 2 éq
2 éq
+ −⋅
= – log [H O (aq)]3 éq+( ) – log [NO (aq)]
[HNO (aq)]2 éq
2 éq
−
.
pKa1 = pH – log [NO (aq)]
[HNO (aq)]2 éq
2 éq
−
ou pH = pKa1 + log [NO (aq)]
[HNO (aq)]2 éq
2 éq
−
;
pH = 3,3 + log 0 0825,
0,0175
= 4.
16 Détermination d’une constante d’équilibre1. a) Un acide est une espèce chimique capable de céder un proton H+.b) Couple acide éthanoïque/ion éthanoate :
CH3CO2H(aq)/CH3CO2–(aq).
Couple ion oxonium/eau : H3O+(aq)/H2O(l).
c) K = [CH CO (aq)] [H O (aq)]
[CH CO H(aq)]3 2 f 3 f
3 2 f
− +⋅.
2. a) Quantité de matière initiale d’acide étha-noïque : n1 = c1 · V1 ;
n1 = 2,7 × 10–3 × 0,100 = 2,7 × 10–4 mol.
b)
Avanc. CH3CO2H(aq) + H2O(l)= CH3CO2
–(aq) + H3O+(aq)
État initial x = 0 n1 Excès 0 0
État final théorique x = xmax n1 – xmax Excès xmax xmax
État final expérimental ou état d’équilibre
x = xf n1 – xf Excès xf xf
Si la transformation est totale, l’acide éthanoïque est totalement consommé, soit n1 – xmax = 0
xmax = n1, soit xmax = 2,7 × 10–4 mol.
c) [H3O+(aq)]f = 10
–pH, donc pH = – log[H3O+].
Or, d’après l’énoncé :log (2,0 × 10–4) = – 3,7.[H3O
+(aq)]f = 10–3,70 = 2,0 × 10–4 mol · L–1.
[H3O+(aq)]f =
x
Vf
1
, soit xf = [H3O+(aq)]f · V1.
xf = 2,0 × 10–4 × 0,100 = 2,0 × 10–5 mol.
d) τ1 = x
xf
max
; τ1 = 2 0 10
2 7 10
5
4
,
,
××
−
−
τ1 = 0,74 × 10–5 × 104 = 0,74 × 10–1 = 7,4 × 10–2.
La transformation est limitée, car τ1 < 1.e) D’après l’équation chimique :
[H3O+(aq)]f = [CH3CO2
–(aq)]f = x
Vf
1
.
[CH3CO2–(aq)]f = 2,0 × 10
–4 mol · L–1.
f) C1 = [CH3CO2H(aq)]0 = [CH3CO2H(aq)]f+ [CH3CO2
–(aq)]f.[CH3CO2H(aq)]f = C1 – [CH3CO2
–(aq)]f ;[CH3CO2H(aq)]f = 2,7 × 10
–3 – 2,0 × 10–4 = 27 × 10–4 – 2,0 × 10–4.
[CH3CO2H(aq)]f = 25 × 10–4 = 2,5 × 10–3 mol · L–1.
g) K1 = [CH CO (aq)] [H O (aq)]
[CH CO H(aq)]3 2 f 3 f
3 2 f
− +⋅.
K1 = 2 0 10 2 0 10
2 5 10
4 4
3
, ,
,
× × ××
− −
− =
4 0
2 510 108 3,
,× ×− ;
K1 = 1,6 × 10–5.
3. a) Présence d’ions éthanoate CH3CO2–(aq), d’ions
oxonium H3O+(aq) ; on néglige la présence des ions
hydroxyde, car le pH est très acide.[H3O
+(aq)]f = [CH3CO2–(aq)]f.
b) σ = λ(CH3CO2–) · [CH3CO2
–(aq)]f.+ λ(H3O
+) · [H3O+(aq)]f.
c) σ = λ(CH3CO2–) + λ(H3O
+) · [H3O+(aq)]f.
[H3O+(aq)]f = [CH3CO2
–(aq)]f = σ
λ λ( ) (CH CO H O )3 2–
3+ + ;
[H3O+(aq)]f = [CH3CO2
–(aq)]f = 5 00 10
4 1 10 35 9 10
2
3 3
,
, ,– –
×× + ×
−
= 5 00 10
40 10
2
3
,–
××
− = 5
4
10
10 10
2
3×
×
−
–.

65Chapitre 5 - Les équilibres acido-basiques dans l’eau
[H3O+(aq)]f = [CH3CO2
–(aq)]f = 1,25 mol · m–3
= 1,25 × 10–3 mol · L–1.d. 1. C2 = 1,0 × 10
–1 mol · L–1 = 100 × 10–3 mol · L–1 et [CH3CO2
–(aq)]f = 1,25 × 10–3 mol · L–1
C2
3 2 f[CH CO (aq)]− =
100
1 25, = 80,
donc [CH3CO2–(aq)]f =
C280
< C250
.
L’approximation 1 est justifiée.2. C2 = [CH3CO2H(aq)]ini = [CH3CO2H(aq)]f
+ [CH3CO2–(aq)]f
et C2 ≈ [CH3CO2H(aq)]f, donc [CH3CO2–(aq)]f est
négligeable face à [CH3CO2H(aq)]f.L’acide s’est très peu dissocié dans l’eau : la trans-formation peut être considérée comme très limitée.
3. K2 = [CH CO (aq)] [H O (aq)]
[CH CO H(aq)]3 2 f 3 f
3 2 f
− +⋅ = [H O (aq)]3 f
+
C2.
K2 = 1 25 10
0 10
32
,
,
×( )−
= 1 25 10
0 10
26,
,
( ) × −
= 1,56 × 10–5.
4. τ2 = [CH CO (aq)]3 2 f
−
C2 ;
τ2 = 1 25 10
0 10
3,
,
× − = 1,25 × 10–2.
5. a) Non : avec deux concentrations différentes, on obtient la même valeur de K.b) Oui : avec deux concentrations initiales différen-tes, on obtient deux taux d’avancement différents.
c) Affirmation 1 vraie : τ = x
xf
max
.
Plus l’acide est dissocié, plus xf est grand et plus τ l’est également (xmax constant).
Affirmation 2 fausse : C1 < C2.La solution 1 est plus diluée, mais τ1 > τ2, donc l’acide s’est davantage dissocié.
Les compétences expérimentales
(page 120)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de revenir sur des techniques de base que l’on retrouve de manière récurrente dans les sujets d’évaluation.Cet exercice doit conduire l’élève à justifier ses choix.
Corrigé1. Préparer correctement une solutionLa dilution devant être réalisée avec la verrerie la plus précise possible, les éprouvettes graduées sont à proscrire pour prélever les volumes de solu-tion. Il ne faut pas oublier non plus que l’utilisation du pipeteur doit être systématique pour les élèves.
• La proposition faite sur le dessin B est correcte.On doit employer une pipette jaugée de 2 mL pour prélever le BBT, les verser dans la fiole jaugée de 50 mL et compléter avec de l’eau distillée, avant d’homogénéiser.
2. Utiliser le spectrophotomètreIl faut étalonner le spectrophotomètre préalablement à toute mesure et garder à l’esprit que l’absorbance est une grandeur proportionnelle à la lon-gueur de solution traversée par la lumière.
• La proposition faite sur le dessin A est correcte.La mesure de référence (le « blanc ») nécessite l’emploi d’eau distillée (le solvant) et les cuves utilisées doivent être toutes identiques.

66 Partie 2 - La transformation d’un système chimique est-elle totale ?
Exercices supplémentaires1. Réaction de l’acide éthanoïque et de l’eauOn introduit de l’acide éthanoïque pur CH3CO2H dans de l’eau. On obtient une solution aqueuse de volume V = 10 mL, de concentration molaire appor-tée en acide éthanoïque C = 2 × 10–2 mol · L–1. La mesure du pH de la solution donne 3,2.1. Écrire l’équation de la réaction de l’acide étha-noïque avec l’eau.2. Tracer le diagramme de prédominance du couple acide éthanoïque/ion éthanoate.En déduire l’espèce prédominante dans la solution.3. Déterminer l’avancement final de la réaction de l’acide éthanoïque avec l’eau (on pourra s’aider d’un tableau d’évolution du système) et le comparer à l’avancement maximal.4. Calculer le taux d’avancement final de cette réac-tion. Le résultat est-il cohérent avec celui de la ques-tion 2 ? Justifier la réponse.
Corrigé1. CH3CO2H(aq) + H2O(l) = CH3CO2
–(aq) + H3O+(aq).
2. Pour pH < pKA : l’acide du couple prédomine.Ici pH = 3,2 et pKA = 4,7, donc l’acide éthanoïque prédomine.3. xf = 10
–pH · V ; xf = 6,3 × 10–6 mol.
xmax = C · V ; xmax = 2 × 10–4 mol : la transformation
n’est pas totale.
4. a) t = x
xf
max
; t = 0,03, soit 3 %.
b) Seulement 3 % de l’acide éthanoïque a réagi : l’espèce qui prédomine est bien l’acide éthanoïque.
2. Identification d’un indicateur coloré(D’après Bac Centres étrangers 2003.)
On dispose d’un flacon d’indicateur coloré en solu-tion aqueuse avec comme seule indication, sa concentration molaire en soluté apportée :
C0 = 2,90 × 10–4 mol · L–1.On mesure son pH, qui vaut 4,18.Le couple acide/base présent dans cet indicateur coloré sera noté HInd(aq)/Ind–(aq).La solution d’indicateur coloré a été préparée à par-tir de la forme acide de l’indicateur : HInd(aq).L’équation de la réaction entre HInd(aq) et l’eau est :
HInd(aq) + H2O(l) = Ind–(aq).1. a) En considérant un volume V = 100 mL de solution aqueuse d’indicateur, déterminer le taux d’avancement final de la réaction de l’acide HInd(aq) avec l’eau.
b) Cet acide est-il totalement dissocié dans l’eau ? Justifier la réponse.2. Donner l’expression littérale de la constante d’acidité KA associée à l’équation de la réaction de l’acide HInd(aq) sur l’eau.3. Cette constante d’acidité vaut KA = 1,9 × 10–5 : calculer le pKA du couple HInd(aq)/Ind–(aq) et identifier l’indicateur à l’aide des données du tableau suivant.
Indicateur Couleur acide
Zone de virage
Couleur basique pKA
Hélianthine Jaune orangé
3,1 – 4,4 Rouge 3,7
Vert de bromocrésol Jaune 3,8 – 5,4 Bleu 4,7
Bleu de bromothymol Jaune 6,0 – 7,6 Bleu 7,0
Phénolphtaléine Incolore8,2
– 10,0Fuschia 9,4
Corrigé1. a) xmax = C0 · V ;xmax = 2,90 × 10
–4 × 100 × 10–3 = 2,90 × 10–5 mol.xf = [H3O
+(aq)]éq · V ;xf = 10
–4,18 × 100 × 10–3 = 6,6 × 10–6 mol.
b) τ = x
xf
max
; τ = 6 6 10
2 90 10
6
5
,
,
–
–
××
= 0,228 = 22,8 %.
Cet acide est peu dissocié dans l'eau
2. KA = [Ind (aq)] [H O (aq)]
[HInd(aq)]
–éq 3
+éq
éq
⋅.
3. pKA = – log KA = 4,7. D’après le tableau, il s’agit du vert de bromocrésol.
3. Étude de solutions aqueuses d’acide méthanoïque et d’acide benzoïque de même concentration
(D’après Bac Asie 2006.)
On dispose de solutions aqueuses d’acide méthanoï-que et d’acide benzoïque de même concentration molaire en soluté apporté C = 1,0 × 10–2 mol · L–1. La mesure du pH d’un volume V = 10 mL de chaque solution fournit les résultats suivants :– solution aqueuse d’acide méthanoïque : pH1 = 2,9 ;– solution aqueuse d’acide benzoïque : pH2 = 3,1.Donnée
Couple acide méthanoïque/ ion méthanoate :HCOOH(aq)/HCOO–(aq).
•

67Chapitre 5 - Les équilibres acido-basiques dans l’eau
1. Écrire l’équation de la réaction de l’acide métha-noïque avec l’eau.2. Calculer l’avancement final, l’avancement maxi-mal et en déduire le taux d’avancement final. (On pourra s’aider d’un tableau descriptif de l’évolution du système chimique.)3. Conclure sur le caractère total ou non de la trans-formation chimique modélisée par la réaction de l’acide méthanoïque avec l’eau.4. À partir de la comparaison des valeurs des pH des solutions aqueuses d’acides méthanoïque et benzoïque, indiquer pour quel acide la réaction avec l’eau est la plus avancée.
Corrigé1. HCOOH(aq) + H2O(l) = HCOO
–(aq) + H3O+(aq).
2. xéq = n(H3O+)éq = [H3O
+(aq)]éq · V = 10–pH · V ;
xéq = 10–2,9 × 10 × 10–3 = 1,3 × 10–5 mol.
HCOOH est le réactif limitant : xmax = C · V ;xmax = 1,0 × 10
–2 × 10 × 10–3 = 1,0 × 10–4 mol.
τ = x
xéq
max
= 10–pH ⋅
⋅V
C V = 10–pH
C ;
τ = 10
1 0 10
2 9
2
– ,
–, × = 10–0,9 = 0,13 = 13 %.
3. La transformation chimique mettant en jeu la réaction de l’acide méthanoïque et l’eau n’est pas totale, elle est limitée.
4. On a établi τ = 10–pH
C.
C étant la même pour les deux solutions, plus le pH est petit, plus τ est élevé.pH1 < pH2 : la réaction de l’eau avec l’acide métha-noïque est plus avancée que celle avec l’acide ben-zoïque.
4. Le vinaigreLe vinaigre est une solution aqueuse d’acide étha-noïque (ou acétique), de formule CH3COOH(aq). On mesure un pH de 3,4 pour une solution diluée de concentration molaire en soluté apporté 1,00 × 10–2 mol · L–1.Données
Couple acido-basique CH3COOH(aq)/CH3COO–(aq) : pKA = 4,75.Valeurs des conductivités molaires ioniques :– λ(H3O+) = 35,0 × 10–3 S · m2 · mol–1 ; – λ(CH3COO–) = 4,09 × 10–3 S · m2 · mol–1.
•
•
1. Écrire l’équation de la réaction modélisant la transformation chimique qui a lieu entre l’acide acétique et l’eau.2. Donner l’expression littérale de la constante d’équilibre associée à cette équation chimique.Calculer sa valeur numérique.3. À l’aide d’un tableau d’évolution du système chimique, calculer le taux d’avancement final de la réaction et conclure.4. On mesure la conductivité de la solution diluée d’acide acétique : on trouve σ = 15,5 × 10–3 S · m–1.Retrouver la valeur de son pH.
Corrigé1. CH3COOH(aq) + H2O(l) = CH3COO
–(aq) + H3O+(aq).
2. KA = [CH CO (aq)] [H O (aq)]
[CH CO H(aq)]3 2 éq 3 éq
3 2 éq
− +⋅.
KA = 10–pKA = 10–4,75 = 1,78 × 10–5.
3.CH3COOH(aq) + H2O(l) = CH3COO
–(aq) + H3O+(aq)
État du système
Av. (mol)
Quantités de matière (mol)
Étatinitial x = 0
n0 = C · Vn0 = 1,00
× 10–2 × 1,0Excès 0 0
En cours de trans-forma-tion
x 1,00 × 10–2 – x Excès x x
État final xf
nf = 1,00× 10–2 – xf
nf = 1,00 × 10–2
– 3,98 × 10–4nf = 9,60 × 10
–3
Excèsxf =3,98
× 10–4
xf = [H3O+(aq)]f · V
xf = 10–pH · V
xf = 10–3,40 × 1,0
xf = 3,98 × 10–4
τ =x
xéq
max
; τ =10
1 00 10
3 40
2
– ,
–, ×= 10–1,40 = 3,98 × 10–2 ≈ 4 %.
La transformation de l’acide éthanoïque avec l’eau n’est pas totale : elle est très limitée.4. σ = λ(H3O
+) · [H3O+(aq)]éq+ λ(CH3COO
–) · [CH3COO–(aq)]éq.
Or [H3O+(aq)]éq = [CH3COO
–(aq)]éq.σ = (λ(H3O
+) + λ(CH3COO–)) · [H3O
+(aq)]éq.
[H3O+(aq)]éq =
σλ λ(H O ) (CH COO )3 3
–+ + ;
[H3O+(aq)]éq =
15 5 10
35 4 4 09 10
3
3
,
( , , )
–
–
×+ ×
= 3,97 × 10–1 mol · m–3,soit [H3O
+(aq)]éq = 3,97 × 10–4 mol · L–1.
pH = – log [H3O+(aq)]éq = 3,40.

68 Partie 2 - La transformation d’un système chimique est-elle totale ?
ProgrammeB - LA TRANSFORMATION D’UN SYSTÈME CHIMIQUE EST-ELLE TOUJOURS TOTALE ? (4 TP, 9 heures en classe entière)3 - Transformations associées à des réactions acido-basiques en solution aqueuse
EXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Application des transfor-mations associées à des réactions acido-basiques : analyse d’une courbe pH = f(V) et choix d’un indi-cateur coloré pour repérer l’équivalence.Titrage d’un produit de la vie courante par pH-métrie, conductimétrie ou colori-métrie et applications.
3. Transformations associées à des réactions acido-basiques en solution aqueuse.Constante d’équilibre associée à une réaction acido-basiqueTitrage pH-métrique d’un acide ou d’une base dans l’eau en vue de déterminer le volume versé à l’équivalence et de choisir un indicateur coloré acido-basique pour un titrage.Qu’en est-il des transformations totales ?Détermination de taux d’avancement final d’une réaction sur un exemple de titrage acido-basique.
Déterminer la constante d’équilibre associée à l’équation d’une réaction acido-basique à l’aide des constantes d’acidités des couples en présence.Réaliser par suivi pH-métrique le titrage d’un acide ou d’une base en solution aqueuse.Déterminer, à partir des résultats d’une expé-rience, le volume versé à l’équivalence lors d’un titrage acide-base.Montrer qu’un indicateur coloré convenable-ment choisi permet de repérer l’équivalence.
6 Application aux titrages acido-basiques
Cours
Découpage du cours1. Réactions acido-basiques p. 1222. Applications aux titrages acido-basiques p. 1243. Suivi pH-métrique d’un titrage acido-basique p. 1254. Titrages acido-basiques colorimétriques p. 127
L’objectif du chapitre est de réinvestir les connais-sances acquises lors des précédents chapitres dans le but de réaliser des dosages acido-basiques.Dans la continuité de ce qui a été traité au chapi-tre 4, la constante d’équilibre relative à une réaction acido-basique mettant en jeu un acide et une base autres que l’eau : on montre que cette dernière s’ex-prime toujours en fonction des constantes d’acidités des couples mis en jeu. On montre ensuite, à partir de mesures de pH, que les transformations acido-basiques mettant en jeu les ions oxonium ou hydroxyde (en tant que réactifs) constituent bien souvent des réactions acido-basiques totales : ces dernières, aussi rapides, pourront donc servir de support lors de titrages.Après avoir effectué des rappels sur les titrages (abordés en classe de première scientifique) et tout particulièrement sur le caractère total de la trans-formation mise en jeu, on réalise un suivi du titrage acido-basique en effectuant la mesure du pH du milieu réactionnel lors d’ajouts consécutifs de solu-tion titrante : le suivi pH-métrique.
♦ La démarche proposée est celle recommandée par les programmes officiels : à partir du calcul prévi-sionnel du volume à verser à l’équivalence d’un titrage mettant en jeu un acide et une base de concen-trations connues, le suivi pH-métrique du même titrage est réalisé : le but est, pour l’élève, de repérer sur la courbe pH = f(Vtitrante) obtenue le point d’équi-valence E, lequel a pour abscisse la valeur du volume versé pour obtenir l’équivalence. Ce point singulier E est celui pour lequel le coefficient directeur de la tangente passe par un extremum.Deux méthodes de repérages sont alors proposées : celle des tangentes parallèles et celle de la dérivée, cette dernière exploitant l’outil informatique.De nombreux titrages acido-basiques peuvent être réa-lisés par les élèves en utilisant des produits commer-ciaux (vinaigre, lait, eaux minérales, boissons acidifiées, vins, médicaments (aspirine, vitamine C), produits ménagers tels les déboucheurs d’évier, etc.). L’objectif est alors de déterminer la concentration de l’espèce chimi-que acide ou basique qu’ils contiennent en réalisant un suivi pH-métrique et en exploitant la courbe pour déter-miner le volume versé afin d’obtenir l’équivalence.La dernière partie du chapitre montre comment le repérage de l’équivalence lors d’un titrage acido-basique et en l’absence de suivi pH-métrique peut se faire rapidement en utilisant un indicateur coloré acido-basique adapté.

69Chapitre 6 - Application aux titrages acido-basiques
Transformations lors d’un titrage acido-basique (page 128)
À partir de résultats expérimentaux et par le calcul du taux d’avancement final, on montre que certaines réactions acido-basiques peuvent être considérées comme totales (quasi totales). Ces transformations serviront de support lors de titrages acido-basiques.
A Dispositif expérimental
1. Le réactif titrant est l’ion hydroxyde, HO–(aq), contenu dans la soude (solution aqueuse d’hy-droxyde de sodium).Le réactif titré est l’acide éthanoïque.
2. Dispositif de titrage
Burette graduée
Sonde de pH
Barreaumagnétique
Agitateurmagnétique
Solution titrante
D˚C
pH-mètre
pH
3. CH3COOH(aq) + HO–(aq) = CH3COO
–(aq) + H2O(l).
B Courbe du dosage
1. VB < Vversé pour obtenir l’équivalence.L’acide éthanoïque est en excès.
2. Pour VB = 6 mL : pH = 5.
3. D’après le produit ionique de l’eau, on a :
[HO–(aq)]éq = Ke
3+[H O (aq)]
= 10
10
–pK
–pH
e
.
[HO–(aq)]éq = 1,0 × 10–9 mol · L–1.
ACTIVITÉ
1
ACTIVITÉ
1 C Tableau d’évolution du système1.
État CH3COOH(aq) + HO–(aq)= CH3COO
–(aq) + H2O(l)
– initial x = 0 CA · VA CB · V 0 Excès
– final réelx = xféq
CA · VA – xéq CB · V – xéq xéq Excès
2. Le réactif limitant est l’ion hydroxyde.3. L’avancement maximal est :
xmax = CB · V ; xmax = 6,0 × 10–4 mol.
4. n(HO–)éq = CB · V – xéq.
5. n(HO–)éq = [HO–(aq)]éq · (VA + V) ;
n(HO–)éq = 1,6 × 10–11 mol.
6. xéq = CB · V – n(HO–)éq ; xéq ≈ 6,0 × 10
–4 mol.
D Calcul du taux d’avancement final
1. t = x
xéq
max
; t = 1.
2. La transformation est totale.
E Dosage d’une base par un acide1. L’équation de la réaction acido-basique est :
NH3(aq) + H3O+(aq) = NH4
+(aq) + H2O(l).
Dans le système final :[H3O
+(aq)]éq = 10–pH = 2,5 × 10–9 mol · L–1.
Donc : n(H3O+)éq = [H3O
+(aq)]éq · (VA + VB) ;n(H3O
+)éq = 4,5 × 10–11 mol.
Le tableau descriptif de l’évolution montre que :xéq = CA · VA – n(H3O
+)éq ; xéq ≈ 8,0 × 10–4 mol.
L’avancement maximal de la transformation (l’acide étant le réactif limitant) est :
xmax = CA · VA ; xmax = 8,0 × 10–4 mol.
Le taux d’avancement est : t = x
xéq
max
; t = 1.
2. La transformation est totale.
Titrage pH-métrique d’un acide par une base (page 129)
A Retour sur la conductimétrie1. L’équivalence est repérée par le changement de pente de la courbe de dosage : on assimile la pre-mière partie de la courbe à une droite D1, la deuxième partie à une droite D2.
ACTIVITÉ
2
ACTIVITÉ
2
Activités
70 Partie 2 - La transformation d’un système chimique est-elle totale ?
Le point d’intersection de ces deux droites a pour abscisse la valeur du volume versé pour obtenir l’équivalence : VE.
2. On lit une valeur VE = 16,5 mL.
B Partie expérimentale1. Schéma du dispositif.
Agitation
2. & 3.CH3COOH(aq) + HO
–(aq) = CH3COO–(aq) + H2O(l).
4. Qr,éq = K = [CH COO (aq)]
[HO (aq)] [CH COOH(aq)]
3–
éq
–éq 3 éq·
;
K = [CH COO (aq)] [H O (aq)]
[CH COOH(aq)]
3–
éq 3 éq
3 éq
· +
·· ·[HO (aq)] [H O (aq)]–éq 3 éq
+ ;
K = K
KA
e
= 10(pK –pK )e A ; K = 1,58 × 109.
C Volume versé à l’équivalence1.
État CH3COOH(aq) + HO–(aq) CH3COO
–(aq) + H2O(l)
Initialx = 0 CA · VA CB · VB,E 0 Excès
Final(équivalence)x = xE
CA · VA – xE CB · VB,E – xE xE Excès
2. À l’équivalence, les quantités de matière des deux réactifs sont simultanément nulles, donc :
xE = CA · VA = CB · VB,E.
3. VB,E = C V
CA A
B
· ; VB,E = 17,0 × 10
–3 L.
Le point d’équivalence E est le point de la courbe dont l’abscisse est 17,0 mL.
4. Autour du point E, on observe une brusque aug-mentation du pH : c’est le saut de pH du dosage.
D Point d’équivalence
Utilisation de la dérivée1. pH
B
C
D
E
VB (mL)
dpHdVB
VBE
La courbe dérivée du pH par rapport au volume VB, dpH
d BV, présente un extremum.
2. On constate que la valeur de l’abscisse de l’extre-mum de la dérivée coïncide avec l’abscisse du point d’équivalence : sa valeur est donc V = 17,0 mL.La méthode pour déterminer le volume versé à l’équivalence consiste donc à projeter l’extremum de la courbe de la dérivée du pH sur l’axe des volu-mes versés, puis à en prendre la valeur coïncidente.
Méthode des tangentes parallèles3. pH
A B
C
D
E
F
VB(mL)VBE
Avec cette méthode, on trouve un volume équiva-lent VB,E = 17 mL. 4. Cette valeur est en accord avec celle obtenue par le calcul.5. Le pH à l’équivalence est : pHE = 8,2.L’indicateur approprié doit posséder une teinte sensible (zone de virage) qui contient la valeur du pH à l’équivalence. Ainsi, le changement de teinte correspondante du milieu réactionnel sera un indi-cateur visuel permettant de repérer l’équivalence. Parmi les indicateurs colorés proposés, le rouge de phénol et la phénolphtaléine conviennent.Remarque : de manière générale, un indicateur dont la zone de virage est entièrement contenue dans le saut de pH convient.
•
•

71Chapitre 6 - Application aux titrages acido-basiques
Dosage d’un vinaigre par conductimétrie (page 130)
1 Dilution du vinaigre1. CH3COOH(aq) + HO
–(aq) = CH3COO–(aq) + H2O(l).
2. Il s’agit de réaliser ici une dilution par 20. Le volume de solution S' préparé doit être 20 fois celui du prélèvement de la solution S.
3. Le matériel à utiliser est donc :– un bécher pour verser la solution S à prélever,– une pipette jaugée de capacité 5 mL,– un pipeteur,– une fiole jaugée de capacité 100 mL et son bouchon,– une pipette compte-goutte pour ajuster au trait de jauge et un petit bécher,– une pissette d’eau distillée.
2 Préparation du titrageDispositif de suivi du titrage
Burette graduée
Sondeconductimétrique
Barreaumagnétique
Agitateurmagnétique
Solution titrante
D˚C
pH-mètre
pH
ACTIVITÉ
3
ACTIVITÉ
3 3 Titrage conductimétrique1. L’équivalence est repérée par le changement de pente de la courbe de dosage : on assimile la pre-mière partie de la courbe à une droite D1, la deuxième partie à une droite D2.Le point d’intersection de ces deux droites a pour abscisse la valeur du volume versé pour atteindre l’équivalence VB,E. On obtient : VB,E = 13 mL.
2.
État CH3COOH(aq) + HO–(aq) CH3COO
–(aq) + H2O(l)
État initialx = 0 C'A · VA C'B · VB,E 0 Excès
État final(équivalence)x = xE
C'A · VA – xE C'B · VB,E – xE xE Excès
3. À l’équivalence, les quantités de matière des deux réactifs sont simultanément nulles, donc :
xE = C'A · VA = C'B · VB,E.
4. C'A = C V
VB BE
A
· ;
C'A = 6,5 × 10–2 mol · L–1.
5. CA = 20 C'A ; CA = 1,3 mol · L
–1.
6. mA = nA · MA = CA · V · MA ; mA = 7,8 g.
7. Avec une masse volumique de 1,0 g · mL–1, 100 g de vinaigre représentent 100 mL de vinaigre.D’après la définition du degré du vinaigre propo-sée, on trouve D = 7,8°.
Corrigés des exercices (page 131)
1 Constante d’équilibre1. HA1(aq) + A2
–(aq) = A–1(aq) + HA2(aq).
2. K = [A (aq)] [HA (aq)]
[HA ] [A (aq)]1–
2
1 2–
·
·.
3. K = K
KA1
A2
.
4. K = 10
10
pK
pK
A1
A2
−
− = 10(pK pK )A2 A1− .
2 Réactions acido-basique totales1.a) Le caractère total d’une transformation est lié à la constante d’équilibre (lorsque K augmente, la transformation a un caractère de plus en plus total).Pour une transformation totale : t = 1.2. Vrai.
3. Faux : elle doit être totale !

72 Partie 2 - La transformation d’un système chimique est-elle totale ?
3 Équivalence1. Lors du dosage, l’équivalence correspond à l’état du système pour lequel il y a changement du réactif limitant : à l’équivalence, les quantités de matière des réactifs s’annulent simultanément.2. • Observation d’un changement de couleur en présence d’un indicateur coloré.• Méthodes graphiques (des tangentes, de la déri-vée…) si on dispose d’une courbe de suivi du titrage.3.État HA(aq) + B(aq) = A–(aq) + BH+(aq)
Initial CA · VA CB · VBE 0 0
Final (équivalence)
CA · VA – xE CB · VBE – xE xE xE
À l’équivalence : CA · VA – xE = 0 ; CB · VBE – xE = 0.Donc xE = CA · VA et xE = CB · VBE.Donc : CA · VA = CB · VBE, relation à l’équivalence.
4. Lors du titrage entre un acide totalement disso-cié dans l’eau et une base, elle aussi totalement protonée dans l’eau.
4 Titrage colorimétrique1. La zone de virage d’un indicateur coloré est l’en-semble des valeurs du pH pour lesquelles les formes acide et basique de l’indicateur coexistent dans des proportions du même ordre de grandeur : aucune des deux formes ne prédomine sur l’autre.
2. L’indicateur coloré doit contenir dans sa zone de virage la valeur du pH du système lorsqu’il est à l’équivalence.
3. L’indicateur coloré est un acide (ou une base) : il consomme donc lui aussi de la solution titrante. Or dans un dosage, une seule transformation consom-mant le réactif titrant doit être mise en jeu : quel-ques gouttes suffisent pour ne pas trop fausser la valeur du volume à l’équivalence (il faut cependant que la couleur soit visible – on peut recommander de placer un papier blanc sous l’erlenmeyer lors du dosage pour distinguer plus nettement le change-ment de couleur).
5 Constante d’équilibre de réactions1. & 2. • H3O
+(aq) + HO–(aq) = 2 H2O(l).
K = 1
[HO (aq)] [H O (aq)]–éq 3 éq· +
= K
K
A
A
1
2
= 10
10
pK
pK
A1
A2
−
− ;
K = 1014.
• CH3COO–(aq) + H3O
+(aq) = CH3COOH(aq) + H2O(l).
K = [CH COOH(aq)]
[CH COO (aq)] [H O (aq)]
3 éq
3–
éq 3 éq· + =
1
A3K
= 10pKA3 ;
K = 6,3 × 105.
• NH4+(aq) + CH3COO
–(aq) = NH3(aq) + CH3COOH(aq).
K = [CH COOH(aq)] [NH (aq)]
[CH COO (aq)]
3 éq 3 éq
3–
éq
·
· [[NH (aq)]4+
éq
K = K
K
A
A
4
3
= 10
10
pK
pK
A4
A3
−
− ; K = 3,98 × 10–5.
• CH3COO–(aq) + HCOOH(aq)
= HCOO–(aq) + CH3COOH(aq).
K = [CH COOH(aq)] [HCOO (aq)]
[CH COO (aq)]
3 éq–
éq
3–
é
·
qq éq[HCOOH(aq)]·
K = K
K
A
A
5
3
= 10
10
pK
pK
A5
A3
−
− ; K = 10.
• HClO(aq) + NH3(aq) = ClO–(aq) + NH4
+(aq).
K = [NH (aq)] [ClO (aq)]
[NH (aq)] [HClO4+
éq–
éq
3 éq
·
· ((aq)]éq =
K
K
A
A
6
4
= 10
10
pK
pK
A6
A4
−
− ;
K = 100.
6 Comprimé effervescent1. NaHCO3(s) = Na
+(aq) + HCO3–(aq).
2. a) HA(aq) + HCO3–(aq) = A–(aq) + H2O,CO2(aq). (1)
Hcit(aq) + HCO3–(aq) = cit–(aq) + H2O,CO2(aq). (2)
b) K1 = 2 × 102 ; K2 = 2 × 10
3.c) Dégagement de dioxyde de carbone.
7 Acide malonique
1. K1 = [H O (aq)] [C H O (aq)]
[C H O (aq)]3 éq 3 3 4 éq
3 4 4 éq
+ −· ;
K2 = [H O (aq)] [C H O (aq)]
[C H O (aq)]
3 éq 3 2 4 éq
3 3 4–
é
+ −· 2
;
2. K1 = KA1 = 10
−pKA1 ; K1 = 1,26 × 10–3.
K2 = KA2 = 10
pKA2−
; K2 = 2,0 × 10–6.
3. a) K = [C H O (aq)]
[C H O (aq)] [HO (aq)]
3 2 4 éq
3 3 4–
éq–
éq
2−
· 22 ;
b) K = K K
K1 2
e2
·.
c) K = 2,5 × 1019.

73Chapitre 6 - Application aux titrages acido-basiques
8 Ammoniac et acide chlorhydrique
Exercice résolu dans le manuel de l’élève.
9 Réaction entre l’acide chlorhydrique et la soude
1. H3O+(aq) + HO–(aq) = 2 H2O(l).
2. K = 1
[HO (aq)] [H O (aq)]–éq 3 éq· +
= 1
Ke
; K = 1014.
3. [H3O+aq)]f = 10
–pH = 2,5 × 10–3 mol · L–1.nf(H3O
+) = [H3O+(aq)]f · (VA + VB) ;
nf(H3O+) = 5,0 × 10–5 mol.
xf = nf(H3O+) = 5,0 × 10–5 mol.
4. ni(HO–) = CB · VB ; ni(HO
–) = 5,0 × 10–5 mol.HO– est le réactif limitant, donc :
xmax = ni(HO–) = 5,0 × 10–5 mol.
5. t = 1 : la transformation est totale.6. Oui, car elle est totale (et rapide).
10 Réaction entre l’acide méthanoïque et les ions éthanoate
1. CH3COO–(aq) + HCOOH(aq)
= HCOO–(aq) + CH3COOH(aq).
2. K = [CH COOH(aq)] [HCOO (aq)]
[CH COO (aq)] [HCO3
–
3–
·
· OOH(aq)]
K = K
K
A
A
2
1
= 10
10
pK
pK
A2
A1
−
− ; K = 10.
3.
État CH3COO–(aq) + HCOOH(aq)
= HCOO–(aq) + CH3COOH(aq)
– initial C · V C · V 0 0
– final théorique C · V – xmax C · V – xmax xmax xmax
– final réel C · V – xf C · V – xf xf xf
4. [HCOO–(aq)]f = [CH3COOH(aq)]f = x
Vf
2 ·.
[HCOOH(aq)]f = [CH3COO–(aq)]f =
C V x
V
·
·
− f
2.
K = x
C V xf
f
2
2( · )− =
x
C V xf
f· −
2
.
5. x
C V xKf
f· −= ,
donc xf = C V K
K
· ·
1+ = 7,6 × 10–5 mol.
6. xmax = C · V ; xmax = 1,0 × 10–4 mol.
7. t = 0,76 : la transformation n’est pas totale.
11 Relations à l’équivalence
1. & 2. CH3COOH(aq) + HO–(aq)= CH3COO
–(aq) + H2O(l)
État initial CA · VA CB · VBE 0 Excès
État final(équivalence)
CA · VA – xE CB · VBE – xE xE Excès
À l’équivalence : CA · VA – xE = 0 et CB · VBE – xE = 0.xE = CA · VA et xE = CB · VBE.
D’où : CA · VA = CB · VBE, relation à l’équivalence.
C3H4O4(aq) + 2 HO–(aq)= C3H2O4
2–(aq) + 2 H2O(l)
État initial CA · VA CB · VBE 0 Excès
État final(équivalence)
CA · VA – xE CB · VBE – 2 xE xE Excès
À l’équivalence : CA · VA – xE = 0 et CB · VBE – 2 xE = 0.
xE = CA · VA et xE = C VB BE
2
·.
D’où : CA · VA = C VB BE
2
·, relation à l’équivalence.
12 Un conservateur alimentaire, l’acide benzoïque
1. a) Solution saturée = solution où soluté non dissous et soluté dissous sont en équilibre.b) m = s · M ; m = 0,5 g.2. a) C6H5COOH(aq) + HO
–(aq)= C6H5COO
–(aq) + H2O(l).b)
Burette graduée
Sonde de pH
Barreaumagnétique
Agitateurmagnétique
D˚C
pH-mètre
pH

74 Partie 2 - La transformation d’un système chimique est-elle totale ?
c) VBE = 14 mL. L’abscisse de l’extremum de la courbe dérivée donne la valeur du volume versé pour atteindre l’équivalence.d)
C6H5COOH(aq) + HO(aq)= C6H5COO
–(aq) + H2O(l)
État initial CA · VA CB · VBE 0 Excès
État final(équivalence)
CA · VA – xE CB · VBE – xE xE Excès
À l’équivalence :CA · VA – xE = 0 et CB · VBE – xE = 0.
xE = CA · VA et xE = CB · VBE.
Donc : CA · VA = CB · VBE, relation à l’équivalence.
D’où : CA = C V
VB BE
A
· ; CA = 1,75 × 10–2 mol · L–1.
e) s = CA · M ; s = 2,14 g · L–1.
3. a) pH = 4.
b) [HO–(aq)]f = KA
3 f
2
[H O (aq)]+ =
KA
pH2
10−.
nf(HO–) =
KA
pH2
10− · (VA + VB) ;
nf(HO–) = 2,2 × 10–12 mol.
ni(HO–) = CB · VB ; ni(HO
–) = 5,0 × 10–5 mol.Donc : xf = ni(HO
–) – nf(HO–) ≈ 5,0 × 10–5 mol.
xmax = ni(HO–) = 5,0 × 10–5 mol, donc xf = xmax : la
réaction est totale.
13 Canalisations bouchées1. m1.L = 1,2 kg = 1 200 g.
m(NaOH) = 20 % · m1.L ; m(NaOH) = 240 g.
n(NaOH) = m
M
(NaOH)
(NaOH) ; n(NaOH) = 6 mol.
Donc C1 = 6 mol · L–1.
2. a) H3O+(aq) + HO–(aq) = 2 H2O(l).
b)H3O
+(aq) + HO–(aq) = 2 H2O(l)
État initial CA · VAE C1 · V1 Excès
État final(équivalence)
CA · VAE – xE C1 · V1 – xE Excès
À l’équivalence :CA · VAE – xE = 0 et C1 · V1 – xE = 0.
xE = CA · VAE et xE = C1 · V1.
Donc : CA · VAE = C1 · V1, relation à l’équivalence.
c) VAE = C V
C1 1
A
·; VAE = 0,6 L = 600 mL.
Ce dosage n’est pas réalisable : il faut en pratique une seule descente de burette pour un dosage.Ce dosage nécessiterait donc 24 burettes de 25 mL !
3. Solution commerciale : C1 et prélèvement V1.Solution diluée : C'1 et volume préparé V'1.La dilution conserve la quantité de matière, donc :
pH
pHE
V (mL)
VE
0
5
10
10 20
E
C1 · V1 = C'1 · V'1, d’où C
C1
1' =
V
V
'1
1
= 40.
Le volume de solution fille préparée vaut 40 fois le volume du prélèvement : on utilise donc une pipette jaugée de contenance 5 mL et une fiole jaugée de 200 mL, ainsi qu’un pipeteur et un bécher pour réa-liser le prélèvement et ne pas pipeter directement la solution mère à prélever.
4. a) On a réalisé le montage A, puisque à V = 0 mL, le pH de la solution est supérieur à 7 (milieu basique).b) Méthode des tangentesLe point d’équivalence E a pour coordonnées :
VAE = 15,1 mL ; pHE = 6,7.
c) C'1 = C V
VA AE
1
· ; C'1 = 1,5 × 10
–1 mol · L–1
et C1 = 40 C'1 = 6,0 mol · L–1.
d) Les valeurs de C1 sont compatibles entre elles.e) La quantité d’ions hydroxyde n’est pas modifiée par la présence d’eau.
5. a) L’indicateur coloré qui convient est celui qui contient dans sa zone de virage la valeur du pH à l’équivalence.Ici pHE = 6,7 : c’est le bleu de bromothymol.b) Le milieu réactionnel passe d’une teinte bleue à une teinte verte (teinte sensible).

75Chapitre 6 - Application aux titrages acido-basiques
14 Dosage de l’acide citrique d’une limonade1. La limonade est débarrassée du dioxyde de car-bone dissous qu’elle contient. On effectue cette opération, car le dioxyde de carbone dissous est un acide en solution aqueuse ; ainsi, on ne dose que l’acide citrique au cours du dosage.
2. & 3.
0
2
4
6
8
10
0 1 2 3 4 5 6 7 8 9 VB (mL)
pH
8,276
5,787
Donc : VBE = 5,8 mL ; pHE = 8,3.4.
État H3A(aq) + 3 HO–(aq) = A–(aq) + 3 H2O(l)
– initial CA · VA CB · VBE 0 Excès
– final (équivalence)
CA · VA – xE CB · VBE – 3xE xE Excès
À l’équivalence : CA · VA – xE = 0 ; CB · VBE – 3xE = 0.
Donc xE = CA · VA et xE = C VB BE·
3.
D’où : CA · VA = C VB BE·
3, relation à l’équivalence.
5. CA = C V
VB BE
A
·
3 ; CA = 9,7 × 10
–3 mol · L–1.
6. L’indicateur coloré qui convient est celui qui contient dans sa zone de virage la valeur du pH à l’équivalence.Ici pHE = 8,3 : c’est la phénolphtaléine.
7. m = C · V · M ; m = 2,0 g.
15 Degré d’un vinaigre
Exercice résolu dans le manuel de l’élève.
16 Étude d’un produit ménager
1. C1 = C0
1 000.
Solution commerciale : C0 et prélèvement V0.Solution diluée : C1 et volume préparé V1.La dilution conserve la quantité de matière, donc :
C1 · V1 = C0 · V0,
donc C
C0
1
= V
V1
0
= 1 000.
Le volume de solution fille préparée vaut 1 000 fois le volume du prélèvement : on utilise donc une pipette jaugée de contenance 1 mL et une fiole jau-gée de 1 L, ainsi qu’un pipeteur et un bécher pour réaliser le prélèvement et ne pas pipeter directe-ment la solution mère.C’est le lot 3 qui convient.2. a) Lors du dosage, l’équivalence correspond à l’état du système pour lequel il y a changement du réactif limitant : à l’équivalence, les quantités de matière des réactifs s’annulent simultanément.b)
H3O+(aq) + (aq) = (aq) + H2O(l).
État initial CA · VAE C1 · V1 0 Excès
État final(équivalence)
CA · VAE – xE C1 · V1 – xE xE Excès
À l’équivalence :CA · VAE – xE = 0 et C1 · V1 – xE = 0
xE = CA · VAE et xE = C1 · V1.
D’où : CA · VAE = C1 · V1, relation à l’équivalence.
c) C1 = C V
VA AE
1
· ;
C1 = 1,07 × 10–2 mol · L–1 et C0 = 10,7 mol · L
–1.
d) L’indicateur coloré qui convient est celui qui contient dans sa zone de virage la valeur du pH à l’équivalence.Ici pHE = 5,7 : c’est le rouge de chlorophénol.
17 Injections alcalinisantes1. a) A–(aq) + H3O
+(aq) = HA(aq) + H2O(l).b) VAE = 19,5 mL. L’abscisse de l’extremum de la courbe dérivée donne la valeur du volume à l’équivalence.c)
H3O+(aq) + A–(aq) = HA(aq) + H2O(l)
État initial CA · VAE C1 · V1 0 Excès
État final(équivalence)
CA · VAE – xE C1 · V1 – xE xE Excès
À l’équivalence :CA · VAE – xE = 0 et C1 · V1 – xE = 0
xE = CA · VAE et xE = C1 · V1.
D’où : CA · VAE = C1 · V1, relation à l’équivalence,

76 Partie 2 - La transformation d’un système chimique est-elle totale ?
soit C1 = C V
VA AE
1
· ; C1 = 9,75 × 10
–1 mol · L–1.
2. a) HCOO3– + H3O
+(aq) = CO2,H2O(aq) + H2O(l).b) Augmentation du pH.
18 Étude de la vitamine C1. HA(aq) + HO–(aq) = A–(aq) + H2O(l)
2. [H3O+(aq)]f = 10
–pH = 1,0 × 10–4 mol · L–1.
3. HO–(aq) = Ke
3+[H O (aq)]
;
HO–(aq) = 1,0 × 10–10 mol · L–1.nf(HO
–) = [HO–(aq)]f · (VA + VB) ;nf(HO
–) = 2,5 × 10–12 mol.4. a)
HA(aq) + HO–(aq) = A–(aq) + H2O(l)
État initial CA · VA CB · VB 0 0
État finalthéorique CA · VA – xmax CB · VB – xmax xmax xmax
État finalréel CA · VA – xf CB · VB – xf xf xf
b) Donc nf(HO–) = CB · VB – xf,
soit xf = CB · VB – nf(HO–) ; xf ≈ 1,0 × 10
–4 mol.
5. xmax = CB · VB ; xmax = 1,0 × 10–4 mol.
Donc xf = xmax : la transformation est totale et peut servir de support pour le dosage.
6.
Burette graduée
Sonde de pH
Barreaumagnétique
Agitateurmagnétique
D˚C
pH-mètre
pH
b) Le point équivalent E a pour coordonnées :VBE = 14,4 mL et pHE = 8.
L’indicateur coloré qui convient est celui qui contient dans sa zone de virage la valeur du pH à l’équivalence.Ici pHE = 8 : c’est le rouge de crésol.
c) Lors du dosage, l’équivalence correspond à l’état du système pour lequel il y a changement du réactif limitant : à l’équivalence, les quantités de matière des réactifs s’annulent simultanément.
d)HA(aq) + HO–(aq) = A–(aq) + H2O(l)
État initial ni(HA) CB · VBE 0 Excès
État final(équivalence)
ni(HA) – xE CB · VBE – xE xE Excès
À l’équivalence :ni(HA) – xE = 0 et CB · VBE – xE = 0
xE = ni(HA) et xE = CB · VBE.
D’où : ni(HA) = CB · VBE ; ni(HA) = 2,88 × 10–4 mol.
5. Le comprimé contient :n(HA) = 10 ni(HA) ; n(HA) = 2,88 × 10–3 mol,
soit une masse :m = n · M ; m = 0,506 g = 506 mg.
L’indication « vitamine C 500 » indique que le cachet contient 500 mg de vitamine C.
19 Dosage de l’acide lactique contenu dans le lait
1. AH(aq) + HO–(aq) = A–(aq) + H2O(l).La transformation doit être totale.
2. K = [A ]
[HO ] [AH]
–
– · =
K
K
A
A
3
1
= 10(pK –pK )A1 A3 = 1,26 × 1010.
pH
AH prédomine
pH = 3,9
A– prédomine
3. À pH = 2,9, c’est la forme AH qui prédomine dans le lait.
4. Lorsque [AH] = [A–], on a : pH = pKA = 3,9. Le volume de soude versé est alors égal à 6 mL.
5.AH(aq) + HO–(aq) = A–(aq) + H2O(l)
État initial ni(AH) CB · VBE 0 Excès
État final(équivalence)
ni(AH) – xE CB · VBE – xE xE Excès
À l’équivalence :ni(AH) – xE = 0 et CB · VBE – xE = 0.

77Chapitre 6 - Application aux titrages acido-basiques
Donc : xE = ni(AH) et xE = CB · VBE.
D’où : ni(AH) = CB · VBE, soit ni(AH) = 6,0 × 10
–4 mol.
6. n(AH)1.L = 50 × n (AH) ; n(AH)1.L = 3,0 × 10–2 mol.
m = n · M ; m = 2,7 g. Le lait n’est pas frais.
20 Dosage de l’acide bromhydrique1. HBr(g) + H2O(l) = Br
–(aq) + H3O+(aq).
2. On travaille sur 1 L de solution :m1.L = d · reau ; m1.L = 1 470 g.mHBr = 46 % × m1.L ; mHBr = 676,2 g.
nHBr = m
M
(HBr)
(HBr) ; nHBr = 8,35 mol.
La concentration vaut : C = 8,35 mol · L–1.
3. a) H3O+(aq) + HO–(aq) = 2 H2O(l).
b) Lors du dosage, l’équivalence correspond à l’état du système pour lequel il y a changement du réactif limitant : à l’équivalence, les quantités de matière des réactifs s’annulent simultanément.c)
H3O+ + HO–(aq) = 2 H2O(l)
État initial CA · VA CB · VBE0 Excès
État final(équivalence)
CA · VA – xE CB · VBE0 – xE Excès
À l’équivalence :CA · VA – xE = 0 et CB · VBE0 – xE = 0.
xE = CA · VA et xE = CB · VBE0.
Donc : CA · VAE = CB · VBE0, relation à l’équivalence
VBE0 = C V
CA A
B
·; VBE0 = 1,67 L !
Ce dosage n’est pas réalisable en l’état. Une seule burette doit être utilisée pour obtenir l’équivalence et la capacité d’une burette est de 25 mL.
4. C' = CA100
.
Solution mère : concentration CA, volume VA à prélever.Solution fille : concentration C', volume V' à préparer.Conservation de la matière au cours de la dilution donc :
CA · VA = C' · V'.
D’où : C
CA
' =
V
V
'
A
= 100, donc V' = 100 VA.
Le volume de solution à préparer doit être 100 fois plus grand que celui du prélèvement.
On prélève S avec la pipette jaugée de capacité 10 mL à l’aide d’un pipeteur. On introduit le prélè-vement dans une fiole jaugée de capacité 1 000 mL et on étend jusqu’au trait de jauge avec de l’eau dis-tillée avant d’homogénéiser.
5. a) C' = V C
V
BE B
A
1·
' ; C' = 8,35 × 10–2 mol · L–1.
CA = 100 C' ; CA = 8,35 mol · L–1.
La valeur trouvée est en accord avec celle trouvée à la question 2.
b) C' = V C
V
BE B
A
2·
' ; C' = 8,55 × 10–2 mol · L–1.
CA = 100 C' ; CA = 8,55 mol · L–1.
c) Dans le cas de l’erreur 1, la quantité de matière en acide à doser du deuxième groupe est :
nA2 < nA1.
On obtiendrait un volume à l’équivalence VBE2 < VBE1, ce qui n’est pas le cas.
21 Dosage d’une solution d’acide chlorhydrique concentrée
1. H3O+(aq) + HO–(aq) = 2 H2O(l).
2. L’équivalence lors d’un dosage conductimétrique correspond au point d’intersection des deux droi-tes (cf. figure), soit VE = 11,2 mL.
V (mL)500
1 000
1 500
2 000
2 500
3 000
0 2 4 6 8 10 12 14 16 18 20
Conductance (µS)
3.H3O
+(aq) + HO–(aq) = 2 H2O(l)
État initial C1 · V1 CB · VE Excès
État final(équivalence)
C1 · V1 – xE CB · VE – xE Excès
À l’équivalence :C1 · V1 – xE = 0 et CB · VE – xE = 0
xE = C1 · V1 et xE = CB · VE.Donc :
C1 · V1 = CB · VE, relation à l’équivalence.

78 Partie 2 - La transformation d’un système chimique est-elle totale ?
D’où :C1 =
C V
VB E
1
· ; C1 = 11,2 × 10
–3 mol · L–1.
4. La solution S0 a été diluée 1 000 fois.C0 = 1 000 × C1 = 11,2 mol · L
–1
5. m0 = n0 · M(HCl) = C0 · V · M(HCl) ;m0 = 409 g.
6. m = r0 · V ; m = 1 160 g.
7. Le pourcentage massique (p) correspond à la masse de chlorure d’hydrogène présente dans 100 g de solution. Or pour 1 160 g de solution, on a 409 g de chlorure d’hydrogène, donc pour 100 g, on a p = 35,3 %.On trouve un pourcentage légèrement supérieur à celui donné par l’étiquette, mais celle-ci indique le pourcentage minimal en masse d’acide : l’indica-tion est donc correcte.
Les compétences expérimentales
(page 140)
On présente dans cette évaluation des situations erronées couramment observées lors des manipulations des élèves et les manipulations correctes.L’objectif est de reconnaître les situations correctes et de justifier le choix en vue de la préparation à l’épreuve expérimentale du baccalauréat.
Corrigé1. Suivi pH-métrique d’un titrage
• La représentation correcte est la B.La burette doit être correctement préparée (remplie jusqu’au 0). La sonde pH-métrique (fragile) doit être maintenue, ne pas être percutée par le tur-bulent lors de l’agitation et être totalement immergée (on peut donc ajou-ter de l’eau distillée dans le bécher : cela ne modifie pas la quantité de matière de l’espèce chimique à doser ni la valeur du volume à verser pour obtenir l’équivalence).
2. Repérage de l’équivalence
• La représentation correcte est la A.L’ajout de solution titrante doit cesser lorsque la teinte sensible de l’indi-cateur coloré est atteinte. Pour le BBT, cette teinte est verte.

ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
– Émergence d’un critère d’ évolution spontanée d’un système à partir de quel-ques expériences : mélange d’acide étha-noïque, d’éthanoate de sodium, d’acide méthanoïque, de méthanoate de sodium.– Exemples de transformations pris dans le domaine de l’oxydoréduction : mélange de solutions d’ions fer (II), d’ions fer (III), d’ ions iodure et de diiode ; mélange de solutions d’ions fer (II), d’ions cuivre (II), de poudre de fer et de poudre de cuivre.
1. Un système chimique évolue sponta-nément vers l’état d’équilibre– Quotient de réaction, Qr : expression lit-térale (rappel) et calcul de sa valeur pour un état quelconque donné d’un système.– Au cours du temps, la valeur du quo-tient de réaction Qr tend vers la constante d’équilibre K (critère d’évolution spon-tanée).– Illustration de ce critère sur des réac-tions acido-basiques et des réactions d’oxydoréduction.
– En disposant de l ’équation d’une réaction, donner l’expression littérale du quotient de réaction Qr, et calculer sa valeur dans un état donné du système.– Savoir qu’un système évolue sponta-nément vers un état d’équilibre.– Être capable de déterminer le sens d ’évolution d ’un système donné en comparant la valeur du quotient de réaction dans l’état initial à la constante d’équilibre, dans le cas de réactions acido-basiques et d’oxydoréduction.
Critère d’évolution spontanée7
PARTIE 3
Le sens « spontané » d’évolution d’un système
est-il prévisible ?
Cours
solides et du solvant, qui n’interviennent pas dans l’expression de Qr. Pour un gaz X, on ne considère que la concentration du gaz X dissous et on note [X(aq)]. Ce gaz n’apparaît plus comme un gaz !La constante Qr est sans dimension (en réalité, de dimension 1 !). Les expressions entre crochets qui apparaissent dans le quotient de réaction sont en fait les nombres exprimant les concentrations en mol · L–1. On peut également, en Terminale S, se contenter d’affirmer que Qr est sans dimension, sans le justifier.
Découpage du cours1. Rappels sur le quotient de réaction p. 1442. Prévision de l’évolution d’un système chimique p. 1453. Observe-t-on toujours une évolution
spontanée ? p. 147
1. Rappels sur le quotient de réactionConformément au programme, il a semblé utile de commencer ce chapitre par le rappel des définitions (données chapitre 4) du quotient de réaction (Qr, Qr,i, Qr,éq). On insiste particulièrement sur le cas des
♦
79Chapitre 7 - Critère d’évolution spontanée

80 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
• On peut également tracer Qr(x) sur calculette puisque l’on connaît l’expression de Qr(x) :
Qr(x) = ( )( – )11
2
2+ x
x.
Il suffit de taper cette expression dans la base de fonction de la calculette puis de définir les plages de variation (0 à 1 pour x et 0 à 12 pour Qr).Une fois l’évolution vers l’état d’équilibre établie, le critère d’évolution est alors aisément obtenu.
3. Observe-t-on toujours une évolution spontanée ?
• Certaines évolutions sont imperceptibles car cinétiquement très lentes (par exemple, l’oxydation des toits en zinc).• Certaines évolutions sont imperceptibles car très limitées (par exemple, les transformations inverses de transformations fortement favorisées dans un sens, comme « Cu(s) + Fe2+(aq) = Cu2+(aq) + Fe(s) » : τ = 1 × 10–24 %).Le calcul est développé page 148 du manuel de l’élève.• Certaines évolutions ne se produisent pas, car le système chimique est déjà à l’équilibre :
Qr,i = Qr,éq = K.• On a considéré qu’un système chimique n’évolue vers l’état d’équilibre chimique que « s’il le peut ».Par exemple, lorsque l’on mélange des ions argent (I) et chlorure de telle sorte que la solution aqueuse soit insaturée, le critère d’évolution spontanée per-mettrait de prévoir une évolution dans le sens de la dissolution du précipité de chlorure d’argent (Qr,i > K). Comme il n’existe pas de précipité, cette dissolution ne peut pas avoir lieu. L’évolution spon-tanée vers l’état d’équilibre n’a pas lieu.Remarque : Le calcul est développé page 149 du manuel de l’élève.
2. Prévision de l’évolution spontanée d’un système chimique
• Le critère d’évolution spontanée d’un système chimique n’a pas été présenté expérimentalement mais à l’aide d’un logiciel de simulation « Qr(x).xls » conçu et réalisé pour la circonstance. Ce logiciel (sous excel) est simple d’utilisation. On peut le télé-charger gratuitement sur le site Bordas :
« www.editions-bordas.fr/espaceTermChimie ».Il comporte des « macros », qu’il faut donc activer au lancement.Évidemment, cette démarche est critiquable puis-que les programmes qui pilotent le logiciel utilisent les lois que l’on veut établir ! Cette présentation nous a semblé intéressante, d’autant plus que l’expé-rience sera abondamment utilisée dans les activités de TP, pour confirmer la véracité du critère d’évolu-tion spontanée. En effet, la courbe Qr(x) matérialise bien le fait que Qr varie entre l’état final et l’état d’équilibre.Ce logiciel est de plus utilisé dans l’Activité 3 sur la loi de la modération.• Fonctionnement du logiciel :– on choisit les quantités de matière initiales dans les cellules A1, B2, B1, A2 (max : 10) du tableau d’évolution ;– on clique ensuite sur le bouton « pour commen-cer cliquer ici » ;– on choisit les deux pKA à la demande du logiciel (valeur la plus faible d’abord) ;– on fait avancer la transformation en cliquant sur la flèche droite de la barre de défilement. À chaque clic, les valeurs des quantités de matière des quatre espèces chimiques apparaissent dans le tableau d’évolution. Dès lors, la valeur de Qr est calculable pour la valeur de x considérée.Le logiciel calcule Qr et inscrit sa valeur dans le « tableau de valeurs » et sur le graphique.
Activités
Vérifier et appliquer le critère d’évolution en TP (page 150)
Cette activité permet de vérifier expérimentalement le critère d’évolution spontanée lors d’une transfor-mation modélisée par une réaction acide-base. Selon les conditions initiales, on aura une évolution spontanée dans le sens direct (mélange M) ou inverse (M'). On recherche ensuite comment réali-ser le mélange M'' pour qu’il n’y ait aucune évolu-tion.
ACTIVITÉ
1
ACTIVITÉ
1 A Réaction acide-base
1 Volumes (mL)
2 M M' M"
3 Solution aqueuse A V1 = 20 V1 = 5,0 V1 = 20
4 Solution aqueuse B V2 = 20 V2 = 50 V2 = αV1
5 Solution aqueuse A' V3 = 20 V3 = 50 V3 = αV4
6 Solution aqueuse B' V4 = 20 V4 = 5,0 V4 = 20
7 Qr, i calculé 1,0 1,0 × 102 α2

81Chapitre 7 - Critère d’évolution spontanée
8 Sens d’évolution prévu
Direct→
Inverse←
9
Sens de variation prévu du quotient[NH (aq)]
[NH (aq)]3
4+
↑ ↓
10[NH (aq)]
[NH (aq)]3 i
4+
i
calculé
1,0 10 α
11
[NH (aq)]
[NH (aq)]3 éq
4+
éq
expérimental
3 3
12 Sens d’évolutionobservé
Direct→
Inverse←
1. NH4+(aq) + CO3
2–(aq) = NH3(aq) + HCO3–(aq).
ac1 b2 b1 ac2
2. L’écriture de l’équation ne préjuge pas du sens dans lequel le système chimique évolue spontané-ment.
3 K = [NH (aq)] [HCO (aq)
[NH (aq)] [Co
3 éq 3–
éq
4+
éq 32–
·
· ((aq)]éq ;
K = K
KA1
A2
= 10
10
–
–
pK
pK
A1
A2
;
K = 10
10
20
10 3
– ,
– ,
9
= 101,1 = 12,6 ≈ 13.
4. a) & b) On pose VT = V1 + V2 + V3 + V4.
• Pour M :
Qr,i =
C V
V
C V
V
C V
V
C V
V
··
·
··
·
2 3
1 4
T T
T T
= V V
V V2 3
1 4
·
· ; Qr, i = 20 20
20 20
××
= 1,0.
• Pour M' :
Qr,i = V V
V V2 3
1 4
·
· ; Qr,i =
50 50
5 0 5 0
××, ,
= 1,0 × 102 ;
• Pour M" :
Qr,i = V V
V V2 3
1 4
·
· =
α αV V
V V1 4
1 4
·
· = α2.
5. • Sens d’évolution spontanée de MQr,i < K : évolution spontanée dans le sens direct de (1), →, d’après le critère d’évolution spontanée.
• Sens d’évolution spontanée de M'Qr,i > K : évolution spontanée dans le sens inverse ou indirect de (1), ←, d’après le critère d’évolution spontanée.
6. • Pour M, qui évolue dans le sens direct, d’après
l’équation de réaction, [NH (aq)]
[NH (aq)]3
4+
doit augmenter.
• Pour M', qui évolue dans le sens indirect, [NH (aq)]
[NH (aq)]3
4+
doit diminuer.
7. [NH (aq)]
[NH (aq)]3 i
4+
i
=
C V
V
C V
V
·
·
2
1
T
T
= V
V2
1
.
• Pour M :[NH (aq)]
[NH (aq)]3 i
4+
i
= V
V2
1
; [NH (aq)]
[NH (aq)]3 i
4+
i
= 20
20 = 1,0.
• Pour M' : [NH (aq)]
[NH (aq)]3 i
4+
i
= V
V2
1
; [NH (aq)]
[NH (aq)]3 i
4+
i
= 50
5 0, = 10.
• Pour M" : [NH (aq)]
[NH (aq)]3 i
4+
i
= V
V2
1
= α V
V1
1
= α.
8. On mesure le pH des mélanges :pH de M = 9,7 ; pH de M' = 9,7.
Comme pH = pKA + log [NH (aq)]
[NH (aq)]
3 éq
4+
éq
:
log [NH (aq)]
[NH (aq)]
3 éq
4+
éq
= pH – pKA1 ;
[NH (aq)]
[NH (aq)]
3 éq
4+
éq
= 10pH – pKA1.
• Pour M : [NH (aq)]
[NH (aq)]
3 éq
4+
éq
= 10pH – pKA1 ;
[NH (aq)]
[NH (aq)]
3 éq
4+
éq
= 109,7 – 9,20 = 100,5 = 3,2 ≈ 3.
• Pour M' :
[NH (aq)]
[NH (aq)]
3 éq
4+
éq
= 10pH – pKA1 = 109,7 – 9,20 = 100,5 = 3,2 ≈ 3 ;
9. • Pour M : [NH (aq)]
[NH (aq)]
[NH (aq)]
[NH (aq)
3 éq
4+
éq
3 i
4+
>]]i
.

82 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
Pour M' : [NH (aq)]
[NH (aq)]
[NH (aq)]
[NH (aq)
3 éq
4+
éq
3 i
4+
<]]i
.
Ces variations sont en accord avec le critère d’évo-
lution, car le rapport [NH (aq)]
[NH (aq)]3
4+
a augmenté de 1,0
à 3 pour M et diminué de 10 à 3 pour M'.Ces variations sont en accord avec les prévisions.
10. Pour qu’il n’y ait pas d’évolution, il faut :
Qr,i = K, soit α2 = K et donc α = K ;
α = 12 6, = 3,55 ≈ 3,6.
11. Pour préparer M" en réutilisant M :V2 = α · V1 ; V2 = 3,55 × 20 = 71 mL.V3 = α · V4 ; V3 = 3,55 × 20 = 71 mL.
Il suffit donc d’ajouter 51 mL de chacune des solu-tions B et A' au mélange M (contenant déjà 20 ml de chacune des solutions B et A'), avec une fiole jau-gée de 50 mL et une pipette graduée ou avec une burette (1 mL).
• On mesure le pH de M" : pH = 9,75.
[NH (aq)]
[NH (aq)]
3 éq
4+
éq
= 10pH – pKA1 ;
[NH (aq)]
[NH (aq)]
3 éq
4+
éq
= 109,75 – 9,20 = 100,55 = 3,55 ≈ 3,6 ;
[NH (aq)]
[NH (aq)]3 i
4+
i
= V
V2
1
= α ≈ 3,6.
À l’erreur expérimentale près, on peut considérer que le rapport n’a pas varié, donc pas d’évolution du système chimique.
B Réaction d’oxydoréductionL’objectif est le même que dans la partie A mais avec une réaction d’oxydoréduction.
1. a) & b) Demi-équations électroniques et équa-tion globale : Fe3+(aq) + e– = Fe2+(aq) (× 2) I2(aq) + 2 e– = 2 I–(aq) (× 1)
Donc : 2 Fe3+(aq) + 2 e– = 2 Fe2+(aq)2 I–(aq) = I2(aq) + 2 e–
2 Fe3+(aq) + 2 I–(aq) = I2(aq) + 2 Fe2+(aq).
Cette écriture ne préjuge pas du sens dans lequel évolue la transformation.
3.
État initial Fe3+(aq) I–(aq) Fe2+(aq) I2(aq)
Volume apporté (mL)
10 V 10 V V V
Concentrationapportée en ions
102 × C 102 × C C C
Quantité dematière apportée
103 × C · V 103 × C · V C · V C · V
4. VT = 10 V + 10 V + V + V = 22 V.
Qr,i = [Fe (aq)] [I (aq)]
[Fe (aq)] [I (aq)]
2+i2
2 i3+
i2 –
·
· ii2
;
Qr,i =
C V
V
C V
V
C V
V
×
× ×
× ×
22 22
10
22
2
3
× × ×
23
210
22
C V
V
;
Qr,i =
C V
V
C V
V
×
× ×
22
1022
3
12
4 =
22 10 12× –
C ;
Qr,i = 22 10
1 0 10
12
3
××
–
–, = 2,2 × 10–8.
5. Qr,i << K : évolution spontanée dans le sens direct, celui de la formation de I2 (et de Fe2+).
• Première manipulation (obtention du mélange M)– Couleur de A + B : jaune à cause de Fe3+, car le mélange est plus concentré en Fe3+ qu’en Fe2+.– Couleur de C + D : jaune clair à cause de I2 (mélange peu concentré en I2) ; I
– est incolore en solution aqueuse.– En mélangeant les contenus des béchers (1) et (2), on obtient une solution brune (couleur de I2).
6. Le système chimique M a réellement évolué dans le sens direct (formation de I2, validée par la colo-ration brune caractéristique).Cette évolution est en accord avec celle prévue à l’aide du critère d’évolution spontanée.

83Chapitre 7 - Critère d’évolution spontanée
7.État initial Fe3+(aq) I–(aq) Fe2+(aq) I2(aq)
Volume apporté (mL)
V V 10 V 10 V
Concentrationapportée en ions
C C 102 × C 102 × C
Quantité dematière apportée
C · V C · V 103 × C · V 10 × C · V
8. VT = 10 V + 10 V + V + V = 22 V.
Qr,i = [Fe (aq)] [I (aq)]
[Fe (aq)] [I (aq)]
2+i2
2 i3+
i2 –
·
· ii2
;
Qr,i =
10
22
10
22
32
3× ×
× × ×
×
C V
V
C V
V
C V
222 22
2 2
V
C V
V
× ×
;
Qr,i =
1022
22
7
3
4
× ×
×
C V
V
C V
V
= 22 107×
C ;
Qr,i = 22 10
1 0 10
7
3
××, –
= 2,2 × 1011.
9. Le sens d’évolution spontanée prévisible de M' est le sens inverse, celui de formation de Fe3+.
• Deuxième manipulation (obtention du mélange M')– Dans le ballon (3), le mélange E + F est légèrement jaune à cause de Fe3+ (solution peu concentrée).– Dans le bécher (3), le mélange C + G est légère-ment jaune à cause de I2 (solution peu concentrée).
10. Le système chimique a évolué dans le sens indi-rect (vers la formation de Fe3+) car le mélange a pris la couleur jaune caractéristique des solutions de ces ions.Cette évolution est en accord avec celle prévue par le critère d’évolution spontanée.
La loide la modération (page 152)
Traditionnellement, la loi de la modération est pré-sentée dans le chapitre « estérification ». On a anti-cipé sa présentation pour épuiser le sujet des
ACTIVITÉ
2
ACTIVITÉ
2
évolutions. L’évolution n’est pas ici spontanée mais résulte de l’ajout ou du retrait d’une espèce chimi-que réagissante. Une fois cet ajout ou ce retrait effec-tué, l’évolution du système chimique devient spontanée.
Remarque : Cette partie peut donc également être traitée dans le cadre du chapitre 10, L’équilibre estérification-hydrolyse.
1. a) & b) Qr,éq = K = [C(aq)] [D(aq)]
[A(aq)] [B(aq)]
éqc
éqd
éqa
éqb
·
· ;
Qr,éq =
n
V
n
V
n
V
n
V
( )·
( )
( )·
( )
C D
A BT T
T T
= n n
n n
( ) · ( )
( ) · ( )
C D
A B.
2. a) Q'r,i = n n
n n
( ) · ( )
( ) · ( ( ) )
C D
A B + α (le nouveau VT se sim-
plifie).
b) Comme n(B) + α > n(B) :
n n
n n
n n
n n
( ) · ( )
( ) · ( ( ) )
( ) · ( )
( ) · ( )
C D
A B
C D
A B+<
α, donc Q'r,i < K.
Comme Q'r,i < K, le système évolue dans le sens direct.c) Comme le système chimique évolue spontané-ment dans le sens direct : nf(A) < n(A). (Une quan-tité supplémentaire de A est consommée.)d) La loi de la modération est vérifiée, car l’addi-tion de B fait évoluer le système chimique dans le sens modérant cette augmentation de B : la consommation de B.La quantité finale de B sera plus importante, mais moins que s’il n’y avait pas eu évolution du système chimique.
3. a) Q"r,i = ( ( ) ) · ( )
( ) · ( )
n n
n n
C D
A B
+ γ.
Le nouveau VT se simplifie.
b) Comme (n(C) + γ) > n(C), on a : Q"r,i > K.Comme Q"r,i > K, le système chimique évolue sponta-nément dans le sens inverse lors de l’ajout de C.
d) nf(A) > n(A).
e) La loi de la modération est vérifiée, car l’addi-tion de C fait évoluer le système chimique dans le sens modérant cette augmentation de C : la consom-mation de C. La quantité de C sera plus importante, mais moins que s’il n’y avait pas eu évolution du système chimique.

84 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
4. L’élimination de D fait évoluer le système chimi-que dans le sens qui modère cette élimination de D, c’est-à-dire celui de la formation d’une quantité supplémentaire de D. L’évolution se fait spontané-ment dans le sens direct.Ceci peut être justifié par un raisonnement analo-gue aux précédents, en considérant « n(D) – z ».
5. a) D’après la loi de la modération, le système chimique réagit en consommant davantage d’ions Cl–(aq) pour modérer l’augmentation de la quantité d’ions chlorure. Le système évolue spontanément dans le sens inverse : celui de la précipitation de AgCl(s).b) La quantité de précipité de chlorure d’argent augmente.
Simuler pour comprendre l’évolution des systèmes (page 153)
Cette activité utilise le logiciel Qr(x).xls téléchar-geable gratuitement sur le site :
www.editions-bordas.fr/espaceTermChimieObjectif : ce logiciel s’avère ici très utile, car il donne la composition du système chimique à l’équilibre. La démarche est simple : on prévoit l’évolution par la loi de la modération, puis on vérifie avec le logiciel.
1. Valeur de la constante d’équilibre K :
ACTIVITÉ
3
ACTIVITÉ
3 A1(aq) + B2(aq) = B1(aq) + A2(aq)
K = [B (aq)] [A (aq)]
[A (aq)] [B (aq)]1 éq 2 éq
1 éq 2 éq
·
· ;
K = [B (aq)] [H O (aq)]
[A (aq)]
[A (aq)]1 éq 3+
éq
1 éq
2 é·· qq
2 éq 3+
éq[B (aq)] [H O (aq)]· ;
K = K
KA1
A2
= 10
10
–
–
pK
pK
A1
A2
; K = 104,75–3,75 = 10,0.
Activité supplémentaire
Loi de la modérationQuestions supplémentaires pour l’Activité 2
(Cf. page 152 du Manuel de l’élève.)
• À une nouvelle solution S1, on ajoute quelques gouttes d’une solution aqueuse concentrée d’am-moniac, NH3(aq).
1. Comment évolue S1 (Information) ?
2. a) Situer le pH de cette solution par rapport à 7. Justifier la réponse.b) Quelle évolution spontanée de S2 peut-on pré-voir lorsque l’on ajoute quelques gouttes d’acide chlorhydrique concentré ? Justifier la réponse.
c) Justifier la phrase : « Un composé solide ioni-que, à caractère basique, dans l’eau, sera plus solu-ble en milieu acide. »
Corrigé
1. La réaction d’équationAg+(aq) + 2 NH3(aq) = Ag(NH3)2
+(aq),consomme des ions Ag+(aq).Le système chimique S1, AgCl(s) ; Ag+(aq) ; Cl–(aq), réagit en formant des ions Ag+ pour modérer cette diminution ; le précipité de AgCl se dissout davantage.
2. a) L’ion Ag+(aq) est neutre ; en revanche, l’ion éthanoate CH3CO2
–(aq) est une base, donc le pH sera supérieur à 7.b) Les ions oxonium H3O
+(aq) de la solution acide ajoutée réagissent avec la base CH3CO2
–(aq) selon :H3O
+(aq) + CH3CO2–(aq) = H2O(l) + CH3CO2H(aq).
ac1 b2 b1 ac2
L’élimination des ions éthanoate provoque une pro-duction de ces ions d’après la loi de la modération.La quantité de précipité de AgCH3CO2 diminue.c) On a étudié un exemple à la question précédente.Le précipité de AgCH3CO2(s) se dissout mieux en milieu basique.
InformationLes ions argent réagissent avec l’ammoniac. Il se forme un ion complexe, de formule Ag(NH3)2
+(aq), selon :Ag+(aq) + 2 NH3(aq) = Ag(NH3)2
+(aq).• Soit une solution S2, réalisée en dissolvant de l’éthanoate d’argent solide, AgCH3CO2)(s), dans de l’eau distillée de telle sorte que cette solution soit saturée.L’équation de la réaction modélisant la transfor-mation chimique s’écrit :
AgCH3CO2(s) = Ag+(aq) + CH3CO2–(aq).
On rappelle que l’ion Ag+(aq) n’a pas de proprié-tés acido-basiques.

85Chapitre 7 - Critère d’évolution spontanée
2.Av. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 ni(A1) ni(B2) ni(B1) ni(A2)
E. int. x ni(A1) – x ni(B2) – x ni(B1) + x ni(A2) + x
E.F. xéqni(A1)
– xéq
ni(B2)– xéq
ni(B1)+ xéq
ni(A2)+ xéq
3. Qr =
n x
V
n x
V
n x
V
n x
V
i
T
i
T
i
T
i
T
B A
B A
( )·
( )
( )·
( )
1 2
2 1
+ +
− −
= n x n x
n x n xi i
i i
B A
B A
( ) · ( )
( ) · ( )1 2
2 1
+ +− −
.
4. Qr,i = Qr(x = 0) = n n
n ni i
i i
B A
B A
( ) · ( )
( ) · ( )1 2
2 1
et Qr,éq = Qr(x = xéq) = n x n x
n x n xi éq i éq
i éq i éq
B A
B A
( ) · ( )
( ) · ( )1 2
2 1
+ +
− − .
A Vérification du critère d’évolution spontanée
5. a) Dans cette question, il faut utiliser la partie précédent la partie A :
Qr,i = Qr(x = 0) = n n
n ni i
i i
B A
B A
( ) · ( )
( ) · ( )1 2
2 1
;
Qr,i = 1 00 1 00
1 00 1 00
, ,
, ,
××
= 1,00.
b) On est bien dans la situation souhaitée, car 1,00 < 10,0.c) L’évolution de ce système chimique s’effectue d’après le critère d’évolution spontanée dans le sens direct.
• Ouvrir le logiciel en « activant les macros ».
6. a) La dernière ligne du tableau d’évolution du logiciel confirme l’évolution prévue.Extrait du logiciel :
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,00 1,00 1,00
E.F. xéq = 0,52 0,48 0,48 1,52 1,52
Ces valeurs montrent que le système chimique a évolué dans le sens direct :
1,52 > 1,00 et 0,48 < 1,00.b) A2 est moins fort que A1 (pKA2 > pKA1).
7. a) Qr,i = Qr(x = 0) = n n
n ni i
i i
B A
B A
( ) · ( )
( ) · ( )1 2
2 1
;
Qr,i = 4 00 4 00
1 00 1 00
, ,
, ,
××
= 16,0.
On est bien dans la situation souhaitée, car 16,0 > 10,0.D’après le critère d’évolution spontanée, l’évolu-tion de ce système chimique s’effectue dans le sens inverse.
• Vérification du sens d’évolution spontanée prévu avec le logiciel.
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,00 4,00 4,00
E.F. xéq = – 0,20 1,20 1,20 3,80 3,80
La dernière ligne du tableau d’évolution du logiciel confirme l’évolution spontanée prévue, car :
3,80 < 4,00 et 1,20 > 1,00.A2 est moins fort que A1 (pKA2 > pKA1), mais les molécules de A2 sont quatre fois plus nombreuses !b) 1,00 – xéq = 1,00 – (– 0,20) = 1,20. Ceci montre que le système chimique évolue dans le sens inverse.
B Vérification de la loi de la modération1. On utilise le logiciel pour déterminer la compo-sition de ce mélange à l’équilibre.
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,00 1,00 1,00
E.F. xéq = 0,52 0,48 0,48 1,52 1,52
2. a) On ajoute de l’espèce chimique B2 : le sys-tème chimique doit donc évoluer dans le sens direct, de manière à modérer cette addition.b) Logiciel.
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,50 1,00 1,00
E.F. xéq = 0,67 0,33 0,83 1,67 1,67
c) La dernière ligne du tableau du logiciel confirme l’évolution spontanée prévue au 2.a.Il faut comparer la dernière ligne du tableau de la réponse 2.b à celle du 1. On voit que le système chimique a évolué vers la droite, car :
1,67 > 1,52 à droite et 0,33 < 0,48 à gauche.

86 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
3. Il faut comparer la dernière ligne du tableau à celle du 1.
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,00 1,00 1,00 + 3,00
E.F. xéq = 0,27 0,73 0,73 1,27 1,27
La loi de la modération prévoyait une évolution dans le sens inverse ; le logiciel également, car :
0,73 > 0,48 à gauche et 1,27 < 1,52 à droite.On peut aussi prendre l’état d’équilibre du 1 comme état initial en ajoutant 3 moles de A2.
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 0,48 0,48 1,52 1,52 + 3,00
E.F. xéq = 0,27 0,73 0,73 1,27 4,27
On parvient au même résultat.
4. Logiciel
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,00 1,00 – 0,50 = 0,50 1,00
E.F. xéq = 0,67 0,41 0,41 1,09 1,59
Il faut comparer la dernière ligne du tableau à celle du 1.La loi de la modération prévoyait une évolution dans le sens direct, le logiciel également, avec :
0,41 < 0,48 à gauche et 1,59 > 1,52 à droite.
5. Il doit donc rester 1 % de A1, c’est-à-dire :
1,00 × 1
100 = 0,01 mol de A1.
0,99 mol de A1 ont donc disparu et l’avancement final est xéq = 0,99.Par tâtonnement avec le logiciel, on arrive à :
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 8,84 0,10 0,10
E.F. xéq = 0,52 0,01 7,85 1,09 1,09
Il a donc fallu ajouter 7,84 mol de B2 au mélange donné (7,84 + 1,00 initial = 8,84).
Activité supplémentaire
Simuler pour comprendreQuestion supplémentaire pour l’Activité 3(page 153 du Manuel de l’élève, partie A).
• Cas où Qr,i = KOn mélange 1,00 mole de chacune des espèces chimiques A1 et B2 avec 2,00 moles de B1 et 5,0 moles de A2.Reprendre les questions 5 et 6 dans cette situation.
CorrigéOn a mélangé 1,00 mole de chacune des espèces chimiques A1 et B2 avec 2,00 moles de B1 et 5,0 moles de A2.
• Qr,i = 2 00 5 00
1 00 1 00
, ,
, ,
××
= 10,0.
• On est bien dans la situation souhaitée, car :Qr,i = K = 10,0.
• Le critère d’évolution spontanée ne prévoit pas d’évolution.• La dernière ligne du tableau d’évolution du logi-ciel confirme la prévision.
Avanc. A1(aq) + B2(aq) = B1(aq) + A2(aq)
E.I. 0 1,00 1,00 2,00 5,00
E.F. xéq = 0,00 1,00 1,00 2,00 5,00
État final identique à l’état initial, donc pas d’évo-lution.• L’effet de la « force » de A1 est juste compensé par celui du plus grand nombre de molécules de A2.

87Chapitre 7 - Critère d’évolution spontanée
1 QCM, plusieurs réponses possibles
1. c) Qr = [Cr (aq)] [Fe (aq)]
[Cr (aq)] [Fe (aq)]
3+ 2+
2+ 3+
·
·.
2. a) Le système évolue dans le sens direct si :Qr,i < K ;
d) Qr,i = 0 et K > 0.
3. c) On introduit uniquement les réactifs A et B : le quotient de réaction augmente.
4. a) & d) Le quotient de réaction est une grandeur positive et sans unité.5. a) & d) Le système est initialement à l’équilibre et évolue dans le sens direct après : ajout du com-posé A ; élimination du composé D.
2 Expression du quotient de réaction
1. Qr = [HO (aq)] [NH (aq)]
[NH (aq)]
–4+
3
·.
2. Qr = [Na+(aq)]·[Cl–(aq)].
4. Qr = [CH CO (aq)]
[CH CO H(aq)] [HO (aq)]3 2
–
3 2–·
.
5. Qr = [Fe (aq)]
[Cu (aq)]
2+
2+.
6. Qr = [CH CO (aq)] [NH (aq)]
[CH CO H(aq)] [NH (3 2
–4+
3 2 3
·
· aaq)].
7. Qr = [HCO (aq)] [H O (aq)]
[H O,CO (aq)]3–
3+
2 2
·.
8. Qr = [Fe (aq)] [SO (aq)]
[Fe (aq)] [S O (aq
3+42–
2+2 8
2–
·
· ))].
3 Calcul de Qr,i1. 2 Ag+(aq) + Cu(s) = 2 Ag(s) + Cu2+(aq).
2. Qr,i = [Cu (aq)]
[Ag (aq)]
2+
+ 2.
3. Qr,i = 0, car à l’état initial, il n’y a pas d’ions cui-vre (II).
4 Ion ammonium dans l’eau1. NH4
+ (aq) + H2O(l) = NH3(aq) + H3O+(aq).
2. K = [NH (aq)] [H O (aq)]
[NH (aq)]
3 éq 3+
éq
4+
éq
·
K = KA[NH (aq)]
[NH (aq)]4+
3
/KA([H O (aq)]
[H O(aq)]3
+
2
; K = 10–9,2.
3. Qr,i < K : le système évolue dans le sens direct.
5 Mélange acido-basique1. CH3CO2H(aq) + HCO2
–(aq)= CH3CO2
–(aq) + HCO2H(aq).
2. K = [CH CO (aq)] [HCO H(aq)]
[CH CO H(aq)]
3 2–
éq 2 éq
3 2 é
·
qq 2–
éq[HCO (aq)]· ;
K = [CH CO (aq)] [H O (aq)]
[CH CO H(aq)]3 2
–éq 3
+éq
3 2 éq
··
[HCO H(aq)]
[HCO (aq)] [H O (aq)]
2 eq
2–
éq 3+
éq· =
K
KA1
A2
; K = 0,10.
3. Qr,i = [CH CO (aq)] [HCO H(aq)]
[CH CO H(aq)] [3 2
–i 2 i
3 2 i
·
· HHCO (aq)]2–
i
.
4. Qr,i =
2 10 1 10
1 10 1 10
3 3
3 3
,0 ,0
,0 ,0
× × ×
× × ×
– –
– –V V
V V
= 2
1 0
,0
, = 2,0.
Qr,i > K : le système chimique évolue spontanément dans le sens inverse.
6 Réaction d’oxydoréduction1. Le volume du mélange réactionnel est 100,0 mL.
2. Qr = [Fe (aq)] [Ce (aq)]
[Fe (aq)] [Ce (
3+i2 3+
i2+
i2 4+
·
· aaq)]i2
Qr,i =
2 0 10 20 0 10
100 10
2 0 10 20 0 101 3
3
1, , , ,× × ××
× × × ×− −
−
− −−
−
− −
−
×× × ×
×× ×
3
3
3 3
3
100 102 0 10 30 0 10
100 10
2 0 1, , , 00 30 0 10
100 10
3 3
3
− −
−
× ××
,
Qr,i = 2 0 2 0
3 0 10 3 0 102 2
, ,
, ,
×× × ×− −
= 4,4 × 103.
3. Qr,i < K : le système chimique évolue spontané-ment dans le sens direct.
7 Réalisation d’un mélange
Exercice résolu dans le manuel de l’élève.
Corrigés des exercices (page 154)

88 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
8 Autour de l’ammoniac1. Une solution aqueuse S0 d’ammoniac, NH3(aq), de concentration initiale 3,0 × 10–3 mol · L–1, a un pH de 10,35 :
NH3(aq) + H2O(l) = NH4+(aq) + HO–(aq).
2. Qr =[NH (aq)] [HO (aq)]
[NH (aq)]4+ –
3
· .
Qr,i = [NH (aq)] [HO (aq)]
[NH (aq)]4+
i–
i
3 i
·.
3. NH3(aq) + H2O(l) = NH4
+(aq) + HO–(aq) solvant
État initial n0 / 0 0
En cours n0 – x / x x
État final n0 – xéq / xéq xéq
pH = 14,00 + log [HO–(aq)]. [HO–(aq)] = 10pH – 14,00 = 1010,35 – 14,00 = 10–3,65 ; [HO–(aq)] = 2,24 × 10–4 mol · L–1.
D’après le tableau : [NH4+(aq)] = [HO–(aq)].
[NH3(aq)] = C0 – [HO–(aq)] ; [NH3(aq)] = 3,00 × 10–3 – 2,24 × 10–4 ; [NH3(aq)] = 2,78 × 10–3 mol · L–1.
5. a) La nouvelle concentration initiale en ions ammonium est :
[NH4+(aq)] =
2 24 10 500 100 0100
53 5
500 10
4 3
3
,,
,× × × +
×
− −
− ;
[NH4+(aq)] = 5,98 × 10–4 mol · L–1.
b) Qr,i = 2 24 10 5 98 10
2 78 10
4 4
3
, ,
,
× × ××
− −
− = 4,82 × 10–5.
Qr,i > K : le système chimique évolue spontanément dans le sens inverse, donc le pH de la solution va diminuer.c) Nouvelle valeur du pH.
NH3(aq) + H2O(l) = NH4+(aq) + HO–(aq)
solvant
État initial
2,78 × 10–3 × 0,500 = 1,4 × 10–3
/5,98 × 10–4 × 0,500 = 2,99 × 10–4
2,24 × 10–4 × 0,500 = 1,12 × 10–4
En cours
1,39 × 10–3
+ x/
2,99 × 10–4
– x1,12 × 10–4
– x
État initial
1,39 × 10–3 + xéq
/2,99 × 10–4
– xéq
1,12 × 10–4 – xéq
On a :
1,8 × 10–5 =
1 12 10
0 500
2 99 10
0 500
1 39 1
4 4, –
,
, –
,
,
– –××
×
×
x xéq éq
00
0 500
3–
,
+ xéq
;
1,8 × 10–5 = 1 12 10 2 99 10
0 500 1 39 1
4 4, – , –
, ,
– –×( ) × ×( )× ×
x xéq éq
00 3– +( )xéq
.
On obtient une équation du second degré, dont les solutions sont :
xéq 1 = 3,6 × 10–4 mol ;xéq 2 = 5,8 × 10–5 mol.
La solution possible est xéq2 (la plus petite valeur).
pH = 14,00 + log 1 12 10 5 8 10
0 500
4 5, – ,
,
–× ×
− = 10.
9 Mélange acide/base1. HCO2H(aq) + CH3NH2(aq)
= HCO2–(aq) + CH3NH3
+(aq).
2. K = [CH NH (aq)] [HCO (aq)]
[CH NH (aq)]3 3
+éq 2
–éq
3 2 é
·
qq 2 éq[HCO H(aq)]· ;
K = [CH NH (aq)]
[CH CH (aq)] [H O (aq)]
3 3+
éq
3 2 éq 3+
éq· ·
[HCO (aq)] [H O (aq)]
[HCO H(aq)]2–
éq 3+
éq
2 éq
· =
K
KA1
A2
;
K = 10
10
3 75
10 56
– ,
– , = 106,81 = 6,45 × 106.
3. VT = V1 + V2 + V3 + V4.
Qr,i = [CH NH (aq)] [HCO (aq)]
[CH NH (aq)] [3 3
+i 2
–i
3 2 i
·
· HHCO H(aq)]2 i
;
Qr,i =
C V
V
C V
V
C V
V
C V
V
3 3 2 2
4 4 2 2
··
·
··
·T T
T T
;
Qr,i =
2 0 10 30 0 10
0 100
3 0 10 25 0 10
0
2 3 2 3, ,
,
, ,× × × × × × ×− − − −
,,
, ,
,
, ,
100
3 0 10 25 0 10
0 100
2 0 10 20 02 3 2× × × × × × ×− − − 110
0 100
3−
,
;
Qr,i = 1,5.
4. Qr,i < K : le système chimique évolue dans le sens direct.

89Chapitre 7 - Critère d’évolution spontanée
5.HCO2H(aq) + CH3NH2(aq)
= HCO2–(aq) + CH3NH3
+(aq)
État initial 4,00 × 10–4 7,50 × 10–4 7,50 × 10–4 6,00 × 10–4
En cours
4,00 × 10–4 – x
7,50 × 10–4 – x
7,50 × 10–4 + x
6,00 × 10–4 + x
État initial
4,00 × 10–4 – xéq
7,50 × 10–4 – xéq
7,50 × 10–4 + xéq
6,00 × 10–4 + xéq
6. K = [CH NH (aq)] [HCO (aq)]
[CH NH (aq)]3 3
+éq 2
–éq
3 2 é
·
qq 2 éq[HCO H(aq)]· ;
K =
6 00 10
0 100
7 50 10
0 100
4 00 1
4 4,
,
,
,
,
– –× +×
× +
×
x xéq éq
00
0 100
7 50 10
0 100
4 4– –
,
,
,
−×
× −x xéq éq
= 6,45 × 106.
On obtient une équation du second degré, dont les solutions sont :xéq1 = 7,50 × 10–4 mol (impossible) ;xéq2 = 4,00 × 10–4 mol (la plus petite valeur).
7. pH = pKA2 + log [CH NH (aq)]
[CH NH (aq)]
3 2 éq
3 3+
éq
;
pH = 10,56 + log 3 50 10
1 00 10
4
3
,
,
××
−
− = 10,1.
10 Mélange nickel/plomb
1. a) Qr,i = [Ni (aq)]
[Pb (aq)]
2+i
2+i
; Qr,i =
0 30
0 2000 70
0 200
,
,,
,
= 0,43.
b) Qr,i < K : le système chimique évolue spontané-ment dans le sens direct.
2.Pb2+(aq) + Ni(s) = Pb(s) + Ni2+(aq)
État initial 0,70 Excès Excès 0,30
En cours 0,70 – x Excès Excès 0,30 + x
État final 0,70 – xéq Excès Excès 0,30 + xéq
K = [Ni (aq)]
[Pb (aq)]
2+éq
2+éq
; K =
0 30
0 2000 70
0 200
,
,, –
,
+ x
x
éq
éq
= 2,4 × 10–3.
0 30
0 70
,
, –
+ x
xéq
éq
= 2,4 × 10–3, soit
xéq = 2 4 10 0 70 0 30
1 2 4 10
3
3
, , – ,
,
× ×+ ×
= 0,70 mol.
3. [Ni2+(aq)]éq = 0 30 0 70
0 200
, ,
,
+ = 5,0 mol · L–1.
[Pb2+(aq)]éq = 0 70 0 70
0 200
, ,
,
+ = 0 mol · L–1.
4. Perte de nickel :∆m(Ni) = 0,70 × 58,71 = 41 g.
Gain de plomb :∆m(Pb) = 0,70 × 207,19 = 1,5 × 102 g.
11 Régulation du pH de l’eau d’une piscine
Exercice résolu dans le manuel de l’élève.
12 À propos du zinc1. Réduction : Fe2+(aq) + 2 e– = Fe(s)Oxydation : Zn(s) = Zn2+(aq) + 2 e–.
2. n(Fe2+)i = C1 · V1 et n(Zn2+)i = C2 · V2 ;n(Fe2+)i = 1,00 × 10–1 × 0,100 = 1,00 × 10–2 mol.n(Zn2+)i = 1,00 × 10–1 × 0,200 = 2,00 × 10–2 mol.
3. Qr,i = [Zn (aq)]
[Fe (aq)]
2+
2+ =
c V
V V
c V
V V
2 2
1 2
1 1
1 2
·
·
+
+
= n
n
(Zn )
(Fe )
2+i
2+i
;
Qr,i = 2,00.L’histogramme correspondant à l’état initial est celui de la figure 2 (état E2). On retrouve les quan-tités de matière calculées à la question 2.
4. Qr < K : le système chimique évolue spontané-ment dans le sens direct de l’équation.Qr = K : système en état d’équilibre, pas d’évolution.Qr > K : évolution spontanée dans le sens inverse.Dans le cas de l’expérience :
Qr = 2,00 << K = 1,40 × 1011.Le système évolue dans le sens direct, avec forma-tion de Zn2+(s) et de Fe(s).
5. Ici : Qr = n
n
(Zn )
(Fe )
2+
2+ ;
Qr,1 = n
n
(Zn )
(Fe )
2+1
2+1
; Qr,1 = 0 025
0 005
,
, = 5.
Qr,3 = n
n
(Zn )
(Fe )
2+3
2+3
; Qr,3 = 0 015
0 015
,
, = 1,0.

90 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
6. Au cours de la transformation, le quotient de réaction évolue de façon à tendre vers K. Qr aug-mente donc (évolution en sens direct) et Qr,i = 2,00. Seul Qr,1 vérifie ces critères :
1,40 × 1011 > Qr,1 > 2,00.L’état E1 est le seul qui constitue un état intermé-diaire entre l’état initial et l’état final.7.
État Avanc.(mol)
Fe2+(aq) + Zn(s) = Zn2+(aq) + Fe(s)
Initial x = 01,0 × 10–2
m
M4
(Zn)
= 6 54
65 4
,
,= 1,0 × 10–1
1,0× 10–2
m
M3
(Fe)
= 5 56
55 6
,
,= 1,0 × 10–1
Final x = xf= 0,010
00,10 – 0,010
= 0,090,030 0,11
Les quantités de matières sont exprimées en mol.
8. Dans l’histogramme à compléter, la grandeur en ordonnée est la concentration.
[Fe2+(aq)]f = 0 mol · L–1 ; [Zn2+(aq)]f = n
V V
(Zn )
+
2+f
1 2
.
[Zn2+(aq)]f = 0 030
0 300
,
, = 0,10 mol · L–1.
0,05
0,1
0,15
0,2
0Ions fer (II) Ions zinc (II)
Concentration (mmol·L–1)
État d’équilibre
13 Exercice à caractère expérimental1. Cu2+(aq) + Zn(s) = Cu(s) + Zn2+(aq).
2. Qr,i = [ ( )]
[ ( )]
Zn aq
Cu aqi
i
2
2
+
+ ; Qr,i = = 1,0.
3. Qr,i < K : le système chimique évolue spontané-ment dans le sens direct, c’est-à-dire dans celui de la formation du cuivre solide. Il y a donc bien dispa-rition de la coloration bleue.
14 Autour d’une transformation dans le domaine d’oxydoréduction
1. • [Ag+(aq)]i = C V
V V1 1
1 2
·
+ ;
[Ag+(aq)]i = 1 0 10 20 10
40 10
1 3
3
, – –
–
× × ××
= 5,0 × 10–2 mol · L–1.
• [Cu2+(aq)]i = C V
V V2 2
1 2
·
+ ;
[Cu2+ (aq)]i = 5 0 10 20 10
40 10
2 3
3
, – –
–
× × ××
= 2,5 × 10–2 mol · L–1.
2. a) Qr = [Cu (aq)]
[Ag (aq)]
2+i
+i2
.
b) Qr,i = [Cu (aq)]
[Ag (aq)]
2+i
+i2
; Qr,i = 2 5 10
5 0 10
2
22
,
,
–
–
×
×( ) = 10.
c) Qr,i < K : d’après le critère d’évolution spontanée, le système chimique évolue dans le sens direct.d) On devrait observer un dépôt d’argent et une coloration bleue indiquant la présence d’ions cui-vre (II) dans la solution.
e) K = [Cu (aq)]
[Ag (aq)]
2+éq
+éq2
,
donc [Ag+(aq)]éq = [Cu (aq)]2+
éq
K ;
[Ag+(aq)]éq = 5,00 10–2×
×2 2 1015, = 4,8 × 10–9 mol · L–1.
Cette concentration est très faible : on peut parler de trace de Ag+. La transformation peut donc être considérée comme totale.
15 De l’or1. 3 Ag(s) + Au3+(aq) = 3 Ag+(aq) + Au(s).
2. • n(Ag) = m
M1
(Ag) ; n(Ag) =
3 0,
108 = 2,8 × 10–2 mol.
• n(Au) = m
M2
(Au) ;
n(Au) = 3 0 10 3, –×
197 = 1,5 × 10–5 mol.
• n(Ag+) = C3 · V3 ; n(Ag+) = 1,0 × 10–2 × 50,0 × 10–3 = 5,0 × 10–4 mol.
• n(Au3+) = C3 · V3 ; n(Au3+) = 1,0 × 10–2 × 50,0 × 10–3 = 5,0 × 10–4 mol.

91Chapitre 7 - Critère d’évolution spontanée
3. Qr,i =[Ag (aq)]
[Au (aq)]
+i3
3+i
; Qr,i =
5 0 10
0 1
5 0 10
0 1
43
4
,
,
,
,
–
–
×
×
= 2,5 × 10–5.
4. D’après le critère d’évolution spontanée, Qr,i < K : le système chimique évolue donc dans le sens direct.
5. La constante d’équilibre K étant très élevée, on est dans la même situation que dans l’exercice 15, donc la transformation sera considérée comme totale :
xf = xéq = xmax = 5,0 × 10–4 mol.
Le réactif limitant est Au3+.
3 Ag(s) + Au3+(aq) = 3 Ag+(aq) + Au(s)
État initial 2,8 × 10–2 5,0 × 10–4 5,0 × 10–4 1,5 × 10–5
En cours 2,8 × 10–2 – 3x
5,0 × 10–4 – x
5,0 × 10–4 + 3x
1,5 × 10–5 + x
État initial
2,8 × 10–2 – 3xf
5,0 × 10–4 – xf
5,0 × 10–4 + 3xf
1,5 × 10–5
+ xf
6. [Ag+(aq)]f = n
V
(Ag )f+
;
[Ag+(aq)]f = 5 0 10 4
0 100
4,
,
–× × = 2,0 × 10–2 mol · L–1.
7. m(Au) = n(Au) · M(Au) ;m(Au) = 5,0 × 10–4 × 197 = 99 × 10–3 g = 99 mg.
16 Mélange liquide solide1. Ca(CH3CO2)2(s) = Ca2+(aq) + 2 CH3CO2
–(aq).
• [CH3CO2–(aq)] =
2m
M
V
( )
( )
Ca(CH CO )
Ca(CH CO )3 2 2
3 2 2
solutio
nn
;
[CH3CO2–(aq)] =
239 5
158
128 6 10 3
×
× −
,
, = 3,89 mol · L–1.
• [NH4+(aq)] =
m
M
V
( )
( )
NH Cl
NH Cl4
4
solution
;
[NH4+(aq)] =
26 8
53 5
128 6 10 3
,
,
, × − = 3,89 mol · L–1.
• [CH3CO2H(aq)] =
ρ( ) · ( )
( )
CH CO H CH CO H
CH CO H3 2 3 2
3 2
solution
V
M
V
[CH3CO2H(aq)] =
28 6 1 05
60 0
128 6 10 3
, ,
,
,
×
× − = 3,89 mol · L–1.
• [NH3(aq)] = C V
V
( ) · ( )NH NH3 3
solution
;
[NH3(aq)] = 5 0 0 100
128 6 10 3
, ,
,
×× −
= 3,89 mol · L–1.
2. CH3CO2H(aq) + NH3(aq) = CH3CO2–(aq) + NH4
+(aq).
K = [CH CO (aq)] [H O (aq)]
[CH CO H(aq)]3 2
–éq 3
+éq
3 2 éq
·
· [NH (aq)]
[NH (aq)] [H O (aq)]
4+
éq
3 éq 3+
éq· ;
K = K
KA 3 2 3 2
–
A 4+
3
(CH CO H/CH CO )
(NH /NH ) ; K = 3,2 × 104.
4. Qr,i = [CH CO (aq)] [NH (aq)]
[CH CO H(aq)] [3 2
–i 4
+i
3 2 i
·
· NNH (aq)]3 i
;
Qr,i = 1, car toutes les espèces chimiques ont la même concentration initiale.
5. D’après le critère d’évolution, Qr,i < K : le système chimique évolue donc spontanément dans le sens direct.
6. K = [CH CO (aq)] [NH (aq)]
[CH CO H(aq)]3 2
–éq 4
+éq
3 2 é
·
qq 3 éq[NH (aq)]· ;
K = 0 500
0 500
2
2
,
,
+( )−( )
x
x
éq
éq
.
On obtient une équation du second degré dont les solutions sont :xéq = 0,494 mol ;xéq = 0,505 mol (impossible, puisque supérieure à xmax).
pH = 4,75 + log ( , , )
( , , )
0 500 0 494
0 500 0 494
+−
= 6,97.

92 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
Exercices supplémentaires
1. Formation d’un complexe du cobaltUne solution aqueuse de chlorure de cobalt (II), CoCl2, est rose. Elle est le siège d’une transforma-tion mettant en jeu :– l’ion tétrachlorocobaltate (II), de formule [CoCl4]
2–(aq) (bleu)– l’ion hexaaquacobalt(II), de formule [Co(H2O)6]
2+(aq) (rose).
1. Quel est l’ion majoritairement présent dans la solution de chlorure de cobalt (II) ?2. À 10 mL de solution chlorure de cobalt (II), on ajoute 3 g de chlorure de sodium : la solution devient bleue. Quel est l’ion formé ?3. Écrire l’équation de la réaction chimique corres-pondante.
Les compétences expérimentales
(page 160)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de revenir sur des techniques de base que l’on retrouve de manière récurrente dans les sujets d’évaluation. Cet exercice doit conduire l’élève à justifier ses choix.
Corrigé1. Réalisation d’une solutionOn réalise une dilution. La dilution conserve les quantités de matière.
Facteur de dilution : α = C
Cmère
fille
= V
Vfille
mère
.
Le volume à prélever est égal à :
Vmère = C V
Cfille fille
mère
· ; Vmère =
0 10 200
0 50
,
,
× = 40 mL.
2. Sens d’évolution spontanée1. Pour réaliser les prélèvements, on utilise une pipette et une propipette.
2. HCOOH(aq) + NH3(aq) = HCOO–(aq) + NH4+(aq).
3. a) K = [HCOO (aq)] [NH (aq)]
[HCOOH(aq)] [N
–éq 4
+éq
éq
·
· HH (aq)]3 éq
= K
KA
–
A 4+
3
(HCOOH/HCOO )
(NH /NH ) ;
K = 10
10
3 75
9 25
– ,
– , = 3,2 × 105.
b) Qr,i = [HCOO (aq)] [NH (aq)]
[HCOOH(aq)] [NH (
–i 4
+i
i 3
·
· aaq)]i ; Qr,i =
0 10 0 10
0 10 0 10
, ,
, ,
××
= 1,0.
4. [HCOO (aq)]
[HCOOH(aq)]
–i
i
= 1.
5. pH = pKA + log [HCOO (aq)]
[HCOOH(aq)]
–éq
éq
, soit [HCOO (aq)]
[HCOOH(aq)]
–éq
éq
= 10pH–pKA ;
pH = 5,6 × 102.Le système chimique évolue spontanément dans le sens direct.
7. D’après le critère d’évolution spontanée, Qr,i < K : le système chimique évolue dans le sens direct.La variation est bien en accord avec le critère d’évolution spontanée.

93Chapitre 7 - Critère d’évolution spontanée
4. On fait chauffer 5 mL de solution chlo-rure de cobalt (II). L’eau s’évapore : quelle cou-leur observe-t-on lors-que l’eau est évaporée ? Justifier la réponse.5. À 10 mL de solution chlorure de cobalt (II), on ajoute quelques gouttes d’acide chlo-rhydrique concentré. Quelles observations peut-on faire ? Justifier la réponse (cf. fig.).
Corrigé1. L’ion majoritairement présent dans la solution de chlorure de cobalt (II) est l’ion hexaaquacobalt(II), de formule [Co(H2O)6]
2+(aq).
2. L’ion formé est l’ion tétrachlorocobaltate (II), de formule [CoCl4]
2–(aq).
3. [Co(H2O)6]2+(aq) + 4 Cl–(aq)
= [CoCl4]2–(aq) + 6 H2O(l).
4. En chauffant, l’eau s’évapore. Le système chimi-que produit alors de l’eau, c’est-à-dire évolue dans le sens direct (en accord avec la loi de modération).
5. À l’état d’équilibre, si on ajoute des ions chlo-rure, le système chimique consomme alors des ions chlorure et évolue dans le sens direct : la solution devient bleue.
2. pH d’un mélange (D’après Bac Liban 2006.)
Dans cet exercice, on se propose de calculer la valeur du pH d’un mélange de deux solutions de pH connus.Données
pKA1 (HNO2(aq)/NO2–(aq)) = 3,3.
pKA2 (HCO2H(aq)/HCO2
–(aq)) = 3,8.pKe = 14,0.
A. Étude de deux solutionsLe pH d’une solution aqueuse d’acide nitreux HNO2(aq), de concentration en soluté apporté C1 = 0,20 mol·L–1, a pour valeur pH1 = 1,3 ; celui d’une solution aqueuse de méthanoate de sodium (HCOO–(aq) + Na+(aq)), de concentration en soluté apporté C2 = 0,40 mol · L–1, a pour valeur pH2 = 8,7.
•••
1. a) Écrire l’équation de la réaction entre l’acide nitreux et l’eau. Quelle est l’expression de sa constante d’équilibre ?b) Donner l’équation de la réaction entre l’ion méthanoate et l’eau, puis exprimer la constante d’équilibre associée à cette réaction.2. a) Sur l’axe des pH, suivant, placer les domaines de prédominance des deux couples acide/base en jeu.
0 147
Échelle des pH
b) Préciser l’espèce prédominante dans chacune des deux solutions précédentes.B. Étude d’un mélange de ces solutions3. On mélange un même volume V = 200 mL de chacune des deux solutions précédentes. La quan-tité de matière d’acide nitreux introduite dans le mélange est n1 = 4,0 × 10–2 mol, celle de méthanoate de sodium est n2 = 8,0 × 10–2 mol.a) Écrire l’équation de la réaction entre l’acide nitreux et l’ion méthanoate.b) Exprimer puis calculer le quotient de réaction Qr,i associé à cette équation dans l’état initial du sys-tème chimique.c) Exprimer le quotient de réaction dans l’état d’équilibre Qr,éq en fonction des constantes d’aci-dité des couples puis le calculer.d) Conclure sur le sens d’évolution de la réaction écrite en 1.a.4. a) Compléter le tableau d’évolution de la trans-formation entre l’acide nitreux et le méthanoate de sodium.
État Avanc. (mol)
Équation de la réaction
Initial n1 = n(HNO2(aq))
n2 = n(HCO–
2(aq))
Intermé-diaire
Équilibre
b) La valeur de l’avancement final, dans cet état d’équilibre est : xéq = 3,3 × 10–2 mol. Calculer les concentrations molaires des différentes espèces chimiques présentes à l’équilibre.c) En déduire la valeur de Qr,éq et la comparer à la valeur obtenue à la question 1.c.5. À l’aide de l’un des couples intervenant dans le mélange, vérifier que la valeur du pH du mélange est proche de la valeur pH3 = 4.

94 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
Corrigé1. a) HNO2(aq) + H2O(l) = NO2
–(aq) + H3O+(aq).
KA1 = [ )] · [ ]
(
H O (aq NO (aq)
[HNO aq)]3
+éq 2
–éq
2 éq
.
b) HCO2–(aq) + H2O(l) = HCO2H(aq) + HO–(aq).
K = [ )] · [ ]
(
HO (aq HCO H(aq)
[HCO aq)]
–éq 2 éq
2–
éq
.
2. a) Pour pH < pKA, l’acide conjugué du couple prédomine.
pH
pKA1 = 3,3pKA2 = 3,8
8,7 141,3
HCO2– (aq)HCO2H(aq)
NO2– (aq)HNO2(aq)
b) À pH 1,3, l’espèce chimique prédominante est l’acide nitreux, HNO2(aq), dans la solution d’acide nitreux.À pH 8,7, l’espèce chimique prédominante est l’ion méthanoate, HCOO–(aq), dans la solution de métha-noate de sodium.
3. a) HNO2(aq) + HCO2–(aq) = NO2
–(aq) + HCOOH(aq).
b) Qr,i = [ )] · [ ]
[ )] · [
HCO H(aq NO (aq)
HCO (aq HNO (2 i 2
–i
2–
i 2 aaq) i].
Dans l’état initial, l’acide méthanoïque et les ions nitrite NO2
–(aq) ne sont pas encore formés, donc Qr,i = 0.
c) Qr,éq = [ ] · [ )]
[ ] · [
NO (aq) HCO H(aq
HNO (aq) HCO
2–
éq 2 éq
2 éq 22–
éq(aq)]
Qr,éq = [ ] · [ )]
[ ] · [
NO (aq) HCO H(aq
HNO (aq) HCO
2–
éq 2 éq
2 éq 22–
éq
3 éq
3 éq(aq
H O aq)
H O aq))]·
(
(
+
+
.
On écrit l’équation de la réaction entre l’acide méthanoïque et l’eau :
HCO2H(aq) + H2O(l) = HCO2–(aq) + H3O
+(aq).
KA2 = [ )] · [ (
[ )]
HCO (aq H O aq)]
HCO H(aq2–
éq 3 éq
2 éq
+
.
donc : Qr,éq = [ )]
[ ( · [ )]
HCO H(aq
H O aq)] HCO (aq
2 éq
3 éq 2–
éq+
·[ )] · [ (
[ )]
NO (aq H O aq)]
HNO (aq2–
éq 3 éq
2 éq
+
= 1
KK
A2A1· .
Qr,éq = K
KA
A
1
2
= 10
10
pK
pK
A1
A2
−
− = 10
pKA1 pKA2− +;
Qr,éq = 10–3,3+3,8 = 100,5 = 3,2.
d) Qr,i < Qr,éq : le système chimique évoluera spon-tanément dans le sens direct.4. a)État HNO2(aq) + HCO2
–(aq) = NO2–(aq) + HCO2H(aq)
Initial n1 n2 0 0
Intermé-diaire n1 – x n2 – x x x
Équilibre n1 – xéq n2 – xéq xéq xéq
b) • [HNO2(aq)]éq = n x
V1
2
− éq ;
[HNO2(aq)]éq = 4 0 10 3 3 10
2 0 200
2 2, ,
,
× − ××
− − ;
[HNO2(aq)]éq = 1,8 × 10–2 mol · L–1.
• [HCO2–(aq)]éq =
n x
V2
2
− éq ;
[HCO2–(aq)]éq =
8 0 10 3 3 10
2 0 200
2 2, ,
,
× − ××
− − ;
[HCO2–(aq)]éq = 12 × 10–2 mol · L–1.
• [HCO2H(aq)]éq = [NO2–(aq)]éq = 8,3 × 10–2 mol · L–1.
c) Qr,éq = [ ] · [ )]
[ ] · [
NO (aq) HCO H(aq
HNO (aq) HCO
2–
éq 2 éq
2 éq 22–
éq(aq)] ;
Qr,éq = ( , )
, ,
0 083
0 018 0 12
2
× = 3,2.
Les valeurs obtenues sont les mêmes.
5. Pour le couple HNO2/NO2–, on a :
KA1 = [ ] · [ )]
[ ]
NO (aq) H O (aq
HNO (aq)2–
éq 3+
éq
2 éq
.
Soit – log KA1 = pKA1
= – log[ ] · [ )]
[ ]
NO (aq) H O (aq
HNO (aq)2–
éq 3+
éq
2 éq
= – log ([H3O+(aq)]éq) – log
[ ]
[ ]
NO (aq)
HNO (aq)2–
éq
2 éq
.
Soit pH = pKA1 + log[ ]
[ ]
NO (aq)
HNO (aq)2–
éq
2 éq
.
pH = 3,3 + log 0 0825
0 0175
,
,
= 4.
[H3O+(aq)] · V ;xf = 10–4,18 × 100 × 10–3 = 6,6 × 10–6 mol.

95Chapitre 7 - Critère d’évolution spontanée
3. SolubilitéOn introduit n0 = 1,0 ×10–3 mol de chlorure d’argent solide dans un volume V = 100 mL d’eau pure.On donne l’équation de dissolution du chlorure d’argent solide dans l’eau :
AgCl(s) = Ag+(aq) + Cl–(aq).Lorsque la solution est saturée, la constante d’équi-libre, notée Ks, a pour valeur Ks = 1,6 × 10–10.1. Écrire l’expression du quotient de réaction.2. Calculer la concentration en ions Ag+(aq) lors-que la solution est saturée.3. La solution ainsi préparée est-elle saturée ?4. On ajoute à la solution initiale 50,0 mL d’eau pure.a) Déterminer le quotient de réaction initial Qr,i lié à cette nouvelle situation.b) En déduire le sens d’évolution spontanée du sys-tème chimique.Données
On dispose de solutions aqueuses d’acide métha noïque et d’acide benzoïque, de même concentration molaire en soluté apporté C = 1,0 × 10–2 mol · L–1. La mesure du pH d’un volume V = 10 mL de chaque solution fournit les résultats suivants :– solution aqueuse d’acide méthanoïque : pH1 = 2,9 ;– solution aqueuse d’acide benzoïque : pH2 = 3,1.
Corrigé1. Qr = [Ag+(aq)] · [Cl–(aq)].Le système chimique saturé est à l’équilibre, donc :
Qr,éq = Ks ;[Ag+(aq)]éq · [Cl–(aq)]éq = Ks,
comme [Ag+(aq)]éq = [Cl–(aq)]éq.
2. À saturation :[Ag+(aq)]éq = [Cl–(aq)]éq = Ks = 1,3 × 10–5 mol · L–1.
3. Dans un litre d’eau, on peut dissoudre seulement 1,3 × 10–5 mol de AgCl ; dans 100 mL, on ne peut dissoudre que 1,3 × 10–6 mol de AgCl.La quantité de AgCl introduite est bien supérieure, donc la solution est saturée
4. On ajoute à la solution initiale 50,0 mL d’eau pure :Qr,i = [Ag+(aq)]i [Cl–(aq)]i ;Qr,i = (1,3 · 10–6 /0,15)² = 7,5 × 10–11.Qr,i < K : le système chimique évolue dans le sens direct de la dissolution.
•
•
4. Solubilité et formation d’un complexeOn introduit dans un bécher V = 100 mL d’eau et m = 0,0100 g de chlorure d’argent. Il se produit une dissolution, que l’on modélise par la réaction d’équa-tion :
AgCl(s) = Ag+(aq) + Cl–(aq).La constante d’équilibre associée à cette équation est : Ks = 1,6 × 10–10.1. La solution ainsi préparée est-elle saturée ? Justi-fier la réponse.2. Si on ajoute V1 = 100 mL d’eau, la solution est-elle encore saturée ?3. On introduit une solution aqueuse d’ammoniac : on observe une disparition du précipité. La solution est alors le siège de la formation d’un complexe [Ag(NH3)2]
+(aq), soluble dans l’eau. L’équation de cette réaction chimique est :
Ag+(aq) + 2 NH3(aq) = [Ag(NH3)2]+(aq).
Justifier l’augmentation de la solubilité du chlorure d’argent.
Corrigé1. n(AgCl) = m(AgCl )/M(AgCl ) ;n(AgCl) = 0,0100/(107,87 + 35,5) = 6,97 × 10–5 mol.Le système chimique saturé est à l’équilibre, donc :
Qr,éq = Ks ;
[Ag+(aq)]éq · [Cl–(aq)]éq = Ks,
comme [Ag+(aq)]éq = [Cl–(aq)]éq.
À saturation :[Ag+(aq)]éq = [Cl–(aq)]éq = Ks = 1,3 × 10–5 mol · L–1.
Dans un litre d’eau, on dissout seulement 1,3 × 10–5 mol de AgCl ; dans 100 mL, on ne peut dissoudre que 1,3 × 10–6 mol de AgCl.La quantité de AgCl introduite est bien supérieure, donc la solution est saturée.
2. D’après la constante Ks, on ne peut dissoudre dans 200 mL d’eau que 2,52 × 10–6 mol de AgCl.La quantité introduite est bien supérieure, donc la solution est saturée.
3. La formation du complexe [Ag(NH3)2]+(aq)
entraîne une diminution de la concentration en Ag+(aq).Pour la réaction liée à la dissolution de AgCl, il n’y a donc plus équilibre. On se retrouve dans la situa-tion Qr,i < K : le système chimique évolue dans le sens direct, c’est-à-dire celui de formation des ions Ag+.

96 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
5. Sens d’évolution d’une transformation acido-basique
On désire préparer une solution aqueuse de métha-noate de sodium par dissolution de méthanoate de sodium solide dans l’eau.1. Établir le protocole permettant de réaliser une solution de concentration molaire apportée C = 0,100 mol · L–1 et de volume V = 250 mL. Aide : la masse molaire du méthanoate de sodium ayant pour valeur M = 68,0 g · mol–1, calculer la masse m à dissoudre.2. On réalise la transformation associée à l’équa-tion chimique :HCO2H(aq) + CH3CO2
–(aq)= HCO2
–(aq) + CH3CO2H(aq).Pour cela, on prépare le mélange suivant :– S1 : solution d’acide méthanoïque, HCO2H(aq), de concentration molaire apportée C1 = 0,10 mol · L–1 et de volume V1 = 10,0 mL ;– S2 : solution aqueuse d’éthanoate (acétate) de sodium, CH3CO2
–(aq) + Na+(aq), de concentration molaire apportée C2 = 0,10 mol · L–1 et de volume V2 = 10,0 mL ;– S3 : solution aqueuse d’acide éthanoïque (acétique), CH3CO2H(aq), de concentration molaire apportée C3 = 0,10 mol · L–1 et de volume V3 = 10,0 mL ;– S4 : solution aqueuse de méthanoate de sodium, HCO2
–(aq) + Na+(aq), de concentration molaire apportée C4 = 0,10 mol · L–1 qui vient d’être prépa-rée, de volume V4 = 10,0 mL.Après agitation, le pH est égal à 4,2.a) Exprimer et calculer le quotient de réaction à l’état initial.b) Déterminer la valeur de la constante d’équilibre associée à cette équation.c) Comment évolue ce système chimique d’après le critère d’évolution spontanée ?3. Vérification expérimentaleCalculer les valeurs des quotients suivants :a) à l’état initial puis à l’état d’équilibre :
[ ]
[ ]
HCO (aq)
HCO H(aq)2–
2 et
[ ]
[ ]
CH CO (aq)
CH CO H(aq)3 2
–
3 2
b) à l’état d’équilibre en utilisant la valeur du pH.c) Comment évoluent ces quotients ? Dans quel sens a évolué le système chimique ?d) Le critère d’évolution spontanée est-il vérifié ?Données : Couples acide/base :
HCO2H(aq)/HCO2–(aq) :
KA1 = 1,8 × 10–4 ; pKA1 = 3,74.CH3CO2H(aq)/CH3CO2
–(aq) :KA2 = 1,8 × 10–5 ; pKA2 = 4,74.
•
•
Corrigé1. m(méthanoate de sodium) = C · V · M ;m(méthanoate de sodium)
= 0,100 × 250 × 10–3 × 68,0 = 1,70 g.Placer une coupelle sur la balance. Tarer puis peser 1,70 g de méthanoate de sodium. À l’aide d’un entonnoir, introduire le méthanoate de sodium dans une fiole jaugée de 250 mL. Avec de l’eau dis-tillée, rincer la coupelle et l’entonnoir. Continuer de verser de l’eau distillée jusqu’au ¾, puis agiter de façon à dissoudre le composé. Compléter d’eau distillée jusqu’au trait de jauge. Agiter pour homo-généiser.
2. a) Qr,i = ([ CH3CO2H(aq)]i× [HCO2
–(aq)]i)/([CH3CO2–(aq)]i × [HCO2H(aq)]i )
Qr,i =
1 0 10
4
1 0 10
41 0 10
4
1 0 10
4
3 3
3 3
, ,
, ,
– –
– –
× × ×
× × ×=V V
V V
11 0, .
b) K = ([ CH3CO2H(aq)]éq
× [HCO2–(aq)]éq)/([CH3CO2
–(aq)]éq × [HCO2H(aq)]éq).
K = ([ CH3CO2H(aq)]éq/([CH3CO2–(aq)]éq
× [H3O+(aq)]éq) × ([HCO2
–(aq)]éq
× [H3O+(aq)]éq)/[HCO2H(aq)]éq).
K = KA1/KA2 ; K = 10.c) Qr,i < K : le système chimique évolue dans le sens direct
3. Vérification expérimentalea) Calculer les valeurs des quotients suivants à l’état initial.
[HCO2−(aq)]i/[HCO2H(aq)]i = 1
[CH3CO2−(aq)]i/[CH3CO2H(aq)]i = 1.
Les espèces chimiques sont introduites avec les mêmes volumes et les mêmes concentrations.
b) Calculer les valeurs de ces mêmes quotients à l’état d’équilibre en utilisant la valeur du pH. On utilise la relation pH = pKA + log([A–(aq)]éq/[HA(aq)]éq), obtenue à partir de la constante KA.[HCO2
−(aq)]éq/[HCO2H(aq)]éq = 10pH–pKA1 = 2,9[CH3CO2
−(aq)]éq/[CH3CO2H(aq)]éq = 10pH–pKA2 = 0,29
c) Évolution des quotients :[HCO2
−(aq)]/[HCO2H(aq)] augmente, alors que [CH3CO2
−(aq)]/[CH3CO2H(aq)] diminue.On observe donc une évolution spontanée suivant le sens direct.Le critère d’évolution spontanée est vérifié.

97Chapitre 7 - Critère d’évolution spontanée
6. Transformation invisibleCf. Manuel de l’élève Cours § 3.1.b, p. 148
Dans un bécher, on introduit 20,0 mL d’une solution aqueuse de chlorure de fer (II) contenant des ions fer (II), Fe2+. La concentration en ions fer (II) est égale à 0,10 mol · L–1. On dépose alors dans le bécher du cuivre en poudre.
1. Écrire l’équation globale modélisant la réaction envisagée entre les ions fer (II) et le cuivre métal-lique. La constante d’équilibre de cette transfor-mation chimique est égale à K' = 10–26.2. Exprimer et calculer le quotient de réaction ini-tial Q'r,i. 3. Quel est le sens d’évolution spontanée du sys-tème chimique ?4. Établir le tableau d’évolution des espèces chimi-ques.5. Exprimer l’avancement x'éq à l’équilibre en fonc-tion de la constante K' et calculer sa valeur.6. Montrer que le taux d’avancement final t' est très faible.7. La réaction est-elle perceptible ?
DonnéesCouples oxydant/réducteur :Cu2+(aq)/Cu(s) ; Fe2+(aq)/Fe(s).
Corrigé1. Cu(s) + Fe2+(aq) = Cu2+(aq) + Fe(s).
2. Qr,i = [ ]
[ ]
Cu (aq)
Fe (aq)
2+i
2+i
; Qr,i = 0,
car à l’instant initial, il n’y a pas d’ion cuivre II.
3. D’après le critère d’évolution spontanée, Qr,i < K : le système chimique évolue donc dans le sens direct.
4.État Fe2+(aq) + Cu(s) = Cu2+(aq) + Fe(s)
Initial 2,00 × 10–3 Excès 0 Excès
En cours 2,00 × 10–3 - x Excès x Excès
Équilibre 2,00 × 10–3 – x'éq Excès x'éq Excès
5. K
x
V'[ ]
[ ] ,= =
× −
+
+ −
Cu (aq)
Fe (aq)
'
éq
éq
éq2
2 32 00 10 xx
Véq'
;
•
xéq' mol= × ×+
=− −
−−10 2 00 10
1 102 0 10
26 3
2629,
, · .
K
x
V'[ ]
[ ] ,=
+=
× −
+
−
Cu (aq)
Fe (aq)
'
éq
éq
éq2
32 2 00 10 xx
Véq'
;
xéq' mol= × ×+
=− −
−−10 2 00 10
1 102 0 10
26 3
2629,
, · .
6. t = x
xéq'
max
;
t = 2 0 10
2 00 10
29
3
,
,
××
−
− = 1,0 × 10–26 = 1 × 10–24 %.
t est très petit : la réaction ne sera pas perceptible.
7. Formation du cuivreUn erlenmeyer contient V = 25,0 mL de sulfate de cuivre (II), de concentration en soluté apporté C = 0,200 mol · L–1. On introduit m1 = 6,00 g d’étain (Sn) en poudre. On place l’erlenmeyer sur un agita-teur magnétique après avoir introduit un turbulent.
1. Établir le protocole pour réaliser les 25,0 mL de sulfate de cuivre (II) à partir du sulfate de cuivre (II) hydraté, CuSO4, 5 H2O.2. Montrer que l’étain est en excès.3. Exprimer et calculer le quotient de réaction ini-tial Qr,i.4. Comment évolue ce système chimique d’après le critère d’évolution spontanée ?5. Établir la relation liant la constante d’équilibre K associée à la réaction et l’avancement xéq.6. Calculer xéq ainsi que le taux d’avancement final.7. Que peut-on conclure sur cette réaction ?8. De quelle couleur sera la solution à l’état final ?
DonnéesSn2+(aq) incolore ; Cu2+(aq) bleu.Équation de réaction :
Sn + Cu2+(aq) = Cu + Sn2+(aq) ;K = 1,7 × 1016.
Corrigé1. On réalise une dissolution. Il faut déterminer la masse de sulfate de cuivre hydraté à peser :m(CuSO4, 5 H2O)= [CuSO4, 5 H2O] · V(CuSO4, 5 H2O) · M(CuSO4, 5 H2O).
••

98 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
m(CuSO4, 5 H2O)= 0,20 × 25,0 × 10–3 × 249,6 = 1,25 g.
On place une coupelle sur la balance. On tare, puis on pèse 1,25 g de CuSO4, 5 H2O. À l’aide d’un enton-noir, on place le solide dans une fiole jaugée de 25,0 mL, puis on ajoute de l’eau distillée (aux ¾). On agite, puis on complète avec de l’eau distillée jusqu’au trait de jauge avant d’homogénéiser.
2. n(Cu2+) = C · V ; n(Cu2+) = 5,00 × 10–3 mol.n(Sn) = m1/M ; n(Sn) = 6,00/118,7 = 5,05 × 10–2.D’après l’équation bilan et les quantités initiales de matière, le réactif en excès est bien l’étain.
3. Qr,i = [ ]
[ ]
Sn (aq)
Cu (aq)
2+i
2+i
= 0,
car à l’état initial, il n’y a pas d’ions étain (II).
4. D’après le critère d’évolution spontanée :Qr,i < K.
Le système chimique évolue donc dans le sens direct.
K
x
Vx
= =× −−
[ ]
[ ] ,
Sn (aq)
Cu (aq)
2+éq
2+éq
éq
é5 00 10 3qq
V
.
5 & 6.
xéq mo= × × ×+ ×
= ×−
−1 7 10 5 00 10
1 1 7 105 0 10
16 3
163, ,
,, ll .
τ =x
xéq
max
; τ = ××
=−
−
5 00 10
5 00 101 0
3
3
,
,, .

99Chapitre 8 - Transformations spontanées dans les piles
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Réalisation et étude de piles par exemple :Fe(s)/Fe2+(aq)//Cu2+(aq)/Cu(s) ;Cu(s)/Cu2+(aq)//Ag+(aq)/Ag(s) ;Zn(s)/Zn2+(aq)// Cu 2+(aq)/Cu(s)(pile Daniell),– à l’aide d’un ampèremètre (mise en évidence du sens de circulation du courant),– à l’aide d’un voltmètre (mise en évidence d’une f.é.m.).
Activités documentaires :– perspectives historiques,– comparaison des caractéris-tiques de piles usuelles.
2. Les piles, dispositifs mettant en jeu des transforma-tions spontanées permettant de récupérer de l’énergie– Transferts spontanés d’électrons entre des espèces chimiques (mélangées ou séparées) de deux couples oxydant/réducteur du type ion métallique/métal, Mn+ (aq)/ M(s). – Constitution et fonctionnement d’une pile :observation du sens de circulation du courant électrique, mouvement des porteurs de charges, rôle du pont salin, réactions aux électrodes. – La pile, système hors équilibre au cours de son fonc-tionnement en générateur.– Lors de l’évolution spontanée, la valeur du quotient de réaction du système chimique tend vers la constante d’équilibre. La pile à l’équilibre « pile usée » : quantité d’électricité maximale débitée dans un circuit.– Force électromotrice d’une pile (f.é.m.) E :mesure, polarité des électrodes, sens de circulation du courant (en lien avec le cours de physique).– Exemple de pile usuelle.
– Schématiser une pile– Utiliser le critère d’évolution spontanée pour déterminer le sens de déplacement des porteurs de charges dans une pile.– Interpréter le fonctionnement d ’une pi le en disposant d ’une information parmi les suivantes : sens de circulation du courant électrique, f.é.m., réactions aux électrodes, polarité des électrodes ou mouvement des porteurs de charges.– Écrire les équations des réac-tions modélisant les transforma-tions se produisant aux électrodes et relier les quantités de matière des espèces formées ou consommées à l’intensité du courant et à la durée de la transformation, dans une pile et lors d’une électrolyse.
8 Transformations spontanées dans les piles
Cours
Découpage du cours1. Transferts spontanés d’électrons
(entre espèces en contact ou pas (pile)) p. 1622. Le dispositif électrochimique appelé « pile » p. 1633. Étude du fonctionnement d’une pile p. 1644. Quantité d’électricité fournie par une pile p. 166
1. Transferts spontanés d’électrons (entre espèces en contact ou pas (pile))
Après avoir montré que la transformation « Cu2+ (aq) + Zn(s) » se fait par échange direct d’électrons, il paraît important de poser la question : « ne peut-on pas effectuer le transfert d’électrons à l’aide d’un fil lors de cet échange (qui deviendrait alors indi-rect) ? ». On construit pas à pas la pile, d’abord sans pont salin ni fil, puis avec un fil servant de pont, enfin avec un pont salin.
♦ 2. Le dispositif électrochimique appelé « pile »
La définition de la pile du bas de la page 163, est simple (deux électrodes métalliques différentes plongeant chacune dans une solution aqueuse contenant leur oxydant conjugué). Cette définition n’inclut, ni les piles de concentration, ni les piles avec électrodes de platine plongeant dans une solu-tion aqueuse contenant les ions oxydant et réduc-teur (ion fer (III) et ion fer (II) par exemple). Ces piles seront abordées dans les activités 2 et 4.Une pile n’existe que s’il y a dissymétrie, qu’elle soit dans le choix de la nature des électrodes ou des solu-tions ou de leur concentration ou encore de leur température (ex : thermo-couples). Nous n’avons pas retenu cette définition, plus exacte et plus com-plète mais plus complexe à ce niveau.

100 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
Les ponts salins doivent être remplis de nitrate de potassium ou nitrate d’ammonium dans l’agar agar, et non de chlorure de potassium, car les ions chlo-rure précipiteraient sous forme de chlorure d’argent dans les demi-piles contenant des ions argent.
3. Étude du fonctionnement d’une pileDans les schémas, on a préféré indiquer le sens du courant et des électrons par des flèches légendées et extérieures au circuit, pour éviter la confusion avec le sens positif de référence qui est généralement noté sur le conducteur (flèche avec i). Généralement, les condensateurs ont déjà été traités en physique à ce stade de l’année. Le sens positif de référence a donc déjà été introduit.Dans les piles, les concentrations des ions interve-nant dans l’expression de Qr sont calculées pour chaque compartiment, indépendamment du com-partiment voisin (donc pas d’effet de dilution).Remarque : Il faut absolument insister sur le fait qu’un voltmètre mesure algébriquement uVcom (comme un oscillographe visualise uYM).
4. Quantité d’électricité fournie par une pileLa clé de tous les problèmes quantitatifs est la double expression de la quantité d’électricité Q fournie par la pile : Q = I · ∆t = n(e–) · F, où n(e–) est la quantité de matière d’électrons échangés pendant la durée ∆t. Il paraît intéressant d’introduire le faraday (ou constante de Faraday) comme la valeur absolue de la charge d’une mole d’électrons :
F = NA · e = 9,65 × 104 C · mol–1.Le programme n’impose pas le faraday, mais son utilisation s’avère très pratique et tend à se générali-ser.La quantité n(e–) doit être reliée aux quantités de matière des espèces chimiques formées ou dispa-rues. Ces questions peuvent être résolues de plu-sieurs manières :– en dressant un tableau d’évolution concernant une demi-équation électronique ;– en utilisant un tableau concernant l’équation glo-bale. Cette méthode nécessite la création d’une colonne supplémentaire où figure n(e–). Attention, il n’est pas aisé d’exprimer n(e–) en fonction de l’avancement x dans ce cas. En effet, il faut pour cela écrire les deux demi-équations avec le même nom-bre d’électrons pour que l’avancement soit commun aux deux demi-équations et à l’équation globale.– en écrivant directement :
n(e–) = n(Ag+)consommé = n(Ag)formé
pour « Ag+(aq) + e– = Ag(s) »ou n(e–) = 2 × n(Cu2+)consommé = 2 × n(Cu)formé
pour « Cu2+(aq) + 2 e– = Cu(s) », par exemple.Les questions quantitatives sont également traitées dans le chapitre 9 suivant (électrolyse) page 184 et 185.La chaîne énergétique de la pile est réinvestie. Elle a été étudiée en 1re S.
Activités
Bilan des échangeset des déplacementsau sein d’une pile (page 168)
Cette activité est en fait une feuille de synthèse fort utile pour l’élève.Les mots cathode et anode ne sont pas utilisés dans ce chapitre. En effet le programme officiel préco-nise l’introduction de ces appellations dans le cha-pitre suivant sur l’électrolyse.Suite de déductions.On peut poser aux élèves quelques questions du type :– Comment déterminer le pôle + d’une pile à partir du critère d’évolution spontanée ?
ACTIVITÉ
1
ACTIVITÉ
1 – comment déterminer le pôle + d’une pile à l’aide d’un ampèremètre (ou d’un voltmètre) ?– etc.Échange d’électrons et polarités des électrodes.
1. Le voltmètre mesure algébriquementuVcom = uM'M = VM' – VM = – 1,0 V ;
VM > VM', donc M est le pôle +.
2. Le courant circulant dans la résistance de la gau-che vers la droite (sortie par le pôle +), les électrons arrivent sur l’électrode M. À l’interface métal M-solution, les électrons seront captés par un oxy-dant (M2+) pour former un réducteur (M) selon :
M2+(aq) + 2 e– = M(s).

101Chapitre 8 - Transformations spontanées dans les piles
M2+(aq)
M(s)+
M'3+(aq)
M'(s)
A
mA COM
RCourant
e–
+ –
Sur l’électrode de droite, les électrons quittent le métal réducteur M' pour former l’oxydant M'3+(aq) selon :
M'(s) = M'3+(aq)) + 3 e–.On multiplie la première demi-équation par 3 et la seconde par 2 :3 M2+(aq) + 6 e– = 3 M(s) et 2 M’(s) = 2 M’3+(aq)) + 6 e–.
On ajoute membre à membre, et on obtient :3 M2+(aq) + 2 M'(s) = 3 M(s) + 2 M'3+(aq)).
3. L’oxydation est la transformation d’un réducteur en oxydant :
M'(s) = M'3+(aq)) + 3 e–.
Cette oxydation se produit sur l’électrode de droite.
Facteurs d’influencede la f.e.m. E d’une pile (page 169)
Objectif. Il s’agit ici d’étudier les piles de concentra-tion par une démarche d’investigation.
1. L’hypothèse 1 sera réfutée car la photographie montre des piles de f.e.m. 1,5 V de grosseur différente.
2. L’électrode d’argent est toujours pôle + ; celle de cuivre ne l’est qu’associée avec le zinc ou le fer. Dans la pile fer-zinc, c’est l’électrode de fer qui est le pôle +.
3. Les valeurs expérimentales de la f.e.m. E mon-trent que E dépend de la nature des électrodes.
4. et 5. La pile cuivre-zinc a pour f.e.m. E = VCu– VZn. Si on dilue la solution de la demi-pile du côté de l’électrode de cuivre, le potentiel de l’électrode de zinc reste constant et celui de celle de cuivre dimi-nue, donc E diminue.E dépend donc de la concentration des solutions. (On peut se contenter de cette conclusion à ce niveau).Attention : le potentiel d’électrode augmente avec la concentration en oxydant de sa solution asso-ciée (loi de Nernst, hors programme en TS). Une diminution de concentration côté zinc aurait donc diminué VZn et augmenté E, car la différence de potentiel VCu– VZn aurait été plus grande.
ACTIVITÉ
2
ACTIVITÉ
2
6. E dépend de la nature des espèces chimiques qui interviennent et de leur concentration.7. C1 : 10 mL de solution mère dans 100 mL pour obtenir 1,0 × 10–1 mol · L–1 (binômes 1,3,5,7).C2 : 10 mL de solution mère dans 1 000 mL pour obtenir 1,0 × 10–2 mol · L–1 (binômes 2,8).C3 : 1,0 mL de solution mère dans 1 000 mL pour obtenir 1,0 × 10–3 mol · L–1 (binômes 4,6).Répartir le travail entre les binômes. Utiliser quel-ques fioles de 1 L pour gagner du temps.8. Cu(s) | Cu2+(aq) ; Ck || Cu2+(aq) ;1,0 mol · L–1| Cu(s).Le pôle + est toujours du côté de la solution la plus concentrée. Valeurs de Ek : 30 mV ; 60 mV ; 90 mV. Deux demi-piles identiques mais de concentrations différentes constituent une pile.
9. Ek = 0,030 × logC
C0
k
;
E = 0,030 × log1 0
1 0 10
,
, –× k = 0,030 × log 10k = 0,030 × k
en accord avec les valeurs trouvées à l’erreur expé-rimentale près.
10. a) Pour Ck = C0, logC
C0
k
= log 1 = 0. Donc E = 0.
b) Les piles du 8 sont appelées « piles de concentra-tion », car l’existence d’une f.e.m. non nulle est due à la différence de concentration des solutions de sulfate de cuivre (les électrodes étant identiques).11. Dans la première pile, on a la même concentra-tion pour chaque demi-pile et donc E = 0.
Dans la deuxième pile, E = 0,030 × log0 10
0 0010
,
,= 0,030 × log102,0 = 0,030 × 2,0 = 0,060 V.
Pourquoi une piles’use-t-elle ? (page 170)
A Étude de la pile Cu-Ag1. Schématiser et réaliser pratiquement cette pile.
Pont salin
Cu2+(aq)
Cu(s)
Ag+(aq)
Ag(s)
2. On trouve : E = 0,45 V.On mesure uAgCu = VAg – VCu = 0,45 V, donc VAg > VCu, donc le pôle + est l’électrode d’argent.
ACTIVITÉ
3
ACTIVITÉ
3

102 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
3. Mesure de l’intensité I du courant dans une résis-tance de valeur R = 10 Ω. La valeur trouvée (ici I = 0,246 mA) dépend de la pile réalisée (pont).
4. I = (établi par E–rI = RI en 1S).
I · (R + r) = E ; R + r = E
I ; r =
E
I – R.
On effectue le calcul : r = 0 45
0 246 10 3
,
, × − – 10 ;
r = 1,8 × 103 – 0,010 × 103 ; r ≈ 1,8 × 103 Ω.
5. G = 1
r = σ ·
S
L = (Σ(λi [Xi])) ·
S
L ;
r = L
S X· ( · [ ] )∑ λ i i
Dans la pile, r dépend de la surface des électrodes et de leur distance, de la concentration (par [Xi]) et de la nature des ions (par la conductivité molaire ionique λi).6. Sens du courant : de la lame d’Ag vers la lame de Cu dans les fils.Sens de déplacement : des électrons de l’électrode de Cu vers l’électrode d’Ag dans les fils.7. Cu(s) = Cu2+(aq) + 2 e–, puisque des électrons partent de l’électrode de cuivre (pôle –).Au pôle + : Ag+(aq) + e– = Ag(s) ;Cu(s) = Cu2+(aq) + 2 e– et 2 Ag+(aq) + 2 e– = 2 Ag(s). Donc 2 Ag+(aq) + Cu(s) = 2 Ag(s) + Cu2+(aq).8.
Av.(mmol)
2 Ag+(aq) + Cu(s) = 2 Ag(s) + Cu2+(aq) E(V)
E.I. 0 ni(Ag+) = c'V' = 1,00 × 10,0 × 10–3 = 10,0 × 10–3 mol= 10,0 mmol
Ex Ex ni(Cu2+) = cV = 0,500 × 10,0 × 10–3
= 5,00 × 10–3 mol= 5,00 mmol
* 0,45
t x n(Ag+) =10,0 × 10–3 – 2x
Ex Ex n(Cu2+) =5,00 × 10–3 + x
t ' x = 4,50
n(Ag+) = 10,0 × 10–3 – 2 × 4,50 × 10–3
= 1,0 × 10–3 mol= 1,0 mmol
Ex Ex n(Cu2+) = 5,00 × 10–3 + 4,50 × 10–3
= 9,50 × 10–3 mol= 9,50 mmol
0,40
t '' x = 4,95
n(Ag+) = 10,0 × 10–3
– 2 × 4,95 × 10–3
= 0,1 × 10–3 mol= 0,1 mmol
Ex Ex n(Cu2+) = 5,00 × 10–3 + 4,95 × 10–3
= 9,95 × 10–3 mol9,95 mmol
0,36
t ''' x = 4,995
n(Ag+) = 10,0 × 10–3
– 2 × 4,995 × 10–3
= 0,01 × 10–3 mol= 0,01 mmol
Ex Ex n(Cu2+) = 5,00 × 10–3 + 4,995 × 10–3
= 9,99(5) × 10–3 mol= 9,99(5) mmol
0,28
E.F. xéq = néq(Ag+) =10,0 × 10–3 – 2xéq
Ex Ex néq(Cu2+) =5,00 × 10–3 + xéq
0
E.F. si total
xmax = 5,0
nr(Ag+) =10,0 × 10–3 – 2xmax= 0
Ex Ex nmax(Cu2+) = 5,00 × 10–3 + xmax = 5,00 × 10–3 + 5,00 × 10–3 = 10,0 × 10–3 = 10,0 mmol
Dans ce tableau : Ex = excès ; Av = avancement. Le réactif limitant est Ag+ car le cuivre est en excès.
9. En utilisant la ligne « t » du tableau précédent, calculer les quantités de matière intermédiaires n(Ag+) et n(Cu2+) pour les trois les avancements x suivants : 4,50 mmol ; 4,95 mmol ; 4,995 mmol.On reporte les résultats dans les lignes colorées du tableau précédent.
B Usure d’une pileCette partie nécessite l’utilisation de la démarche d’investigation.– Situation déclenchante. Courbe I(t).– Question.Pourquoi l’intensité débitée par la pile décroît-elle au cours du temps ? Quel est le paramètre principal responsable de cette décroissance ?
Élaboration d’hypothèses argumentées– Hypothèse 1. L’examen de la formule I = E/(R + r) montre que si la résistance interne r de la pile aug-mente, l’intensité décroît (à R et E constants).– Hypothèse 2. L’examen de la formule I = E/(R + r) montre que si la f.e.m. E de la pile diminue, l’inten-sité décroît (à R et r constants).
Validation ou invalidation de l’hypothèse 11. Argumentation de la réponse en utilisant le A-5. (Voir A5).Dans la pile, la surface des électrodes et leur dis-tance ne change pas. La concentration des ions argent diminue, celle des ions cuivre (II) augmente. Globalement la résistance variera peu, car il y a toujours la même quantité d’ions, même si leur nature change un peu (il n’y a pas création d’ions oxonium ou hydroxyde qui auraient pu modifier la conductivité du fait de leur forte conductivité molaire ionique λ. Le pont sera plus résistant à cause des ions K+(aq) et NO3
–(aq) perdus ! Ceci est compensé par le fait que les solutions récupèrent ces ions !
2. La variation de résistance interne r de la pile (légère), ne peut pas expliquer à elle seule, la chute de E. L’hypothèse 1 est invalidée.
Validation ou invalidation de l’hypothèse 23. a) Pour vérifier l’hypothèse 2, il faut mesurer E au cours du temps.b) Peut-on travailler ainsi si l’on dispose de 30 minutes ? La décharge est trop longue pour être étudiée en 30 min.
•
•
•

103Chapitre 8 - Transformations spontanées dans les piles
c) Par exemple pour P4,995 :
C' = n
V
( )
'
Ag+ ; C' =
10 0 10 2 4 995 10
10 0 10
3 3
3
, ,
,
× − × ××
− −
−
C' = 1,0 × 10–3 mol · L–1 ;
C = n
V
( )
'
Cu2+ ; C =
5 00 10 4 995 10
10 0 10
3 3
3
, ,
,
× + ××
− −
−
C = 9,99(5) × 10–1 mol ≈ 1,0 mol · L–1.
xn(Ag+) mol
n(Cu2+) mol
C' = n(Ag+)/V' mol · L–1
C = n(Cu2+)/Ven mol · L–1
4,501,0× 10–3
9,50 × 10–3
1,0 × 10–3/ 10,0 × 10–3 = 1,0 × 10–1
9,50 × 10–3/9,5 × 10–3 = 1,0.On ajoutera 0,50 mL d’eau pour avoir un volume constant de 10,0 mL.
4,950,1× 10–3
9,95 × 10–3
1,0 × 10–29,95 × 10–3/10,0 × 10–3 = 0,995 ≈ 1,0
4,9950,01 × 10–3
9,99(5)× 10–3
1,0 × 10–39,99(5) × 10–3/10,0 × 10–3
= 0,999(5) ≈ 1,0
4. Les mesures donnent E = 0,40 V ; 0,36 V ; 0,28 V.5. C’est bien la chute de la f.e.m. E qui est respon-sable de la chute de I. Cette hypothèse est validée.6. Lorsque la pile débite un courant, sa f.e.m. E chute jusqu’à la valeur 0 dans l’état d’équilibre.
C Prolongements du B (facultatif)Cette partie permet (en particulier) de calculer la concentration en ions argent dans la pile usée donc à l’équilibre : [Ag+]éq ≈ 2,15 × 10–8 mol · L–1
1. a) et b) Montrer que lorsque la pile est déchar-gée (donc qu’à l’équilibre), [Ag+(aq)]éq ≅ 0.
Qr,éq = [ ]
[ ]
Cu
Ag
éq
éq
2
2
+
+ ; Qr,éq =
5 0 10
10 10 2
3
32
,
'
× +
× −
−
−
x
V
x
V
éq
éq
= K
K = 2,15 × 1015 ; V = V' = 10,0 × 10–3 L5,0 × 10–3 × V + xéq = 2,15 × 1015
× [(10,0 × 10–3 )2 + 4 x2éq – 40,0 × 10–3 × xéq ] ;
5,0 × 10–3 × 10,0 × 10–3 + xéq × 10,0 × 10–3
= 2,15 × 1015 × [(10,0 × 10–3 )2 + 4 x2éq
– 40,0 × 10–3 × xéq ] ….8,60 × 1015 x2
éq – 8,60 × 1013 xéq+ (2,15 ×1011 + 5,0 × 10–5) = 0
8,60 × 1015 x2éq – 8,60 × 1013 xéq + (2,15 × 1011 + e) = 0
8,60 × 1015 x2éq – 8,60 × 1013 xéq + (2,15 ×1011) ≈ 0,
car 5,0 × 10–5 << 2,15 × 1015.La solution valable physiquement estxéq ≈ 5,0 × 10–3 mol (on élimine la plus grande ou la négative).Ce qui donne : [Ag+]éq ≈ 0 mol · L–1.Conclusion : Lorsque la pile débite, le système évo-lue spontanément vers son état d’équilibre. La ten-sion à vide diminue pour s’annuler dans l’état d’équilibre. Dans cet état, on dit que la pile est usée : alors x = xéq, Qr = Qr, éq = K et Ieq = 0 A.« La pile s’use si l’on s’en sert. »
2. [Cu2+]éq = 5 10
10 10
3
3
× +
×
−
−
xéq ≅ 1,0 mol · L–1.
3. On peut alors chercher [Ag+]éq avec plus de préci-sion :
K = [ ]
[ ]
Cu
Ag
éq
éq
2
2
+
+ ; [Ag+]2
éq = [ ]Cu éq
2+
K ;
[Ag+]éq = [ ]Cu éq
2+
K ;
[Ag+]éq = 1 0
2 15 1015
,
, × = 2,15 × 10–8 mol · L–1 ≅ 0.
4. a) Qr = [ ]
[ ]
Cu
Ag
2
2
+
+ =
5 0 10
10 10 2
3
32
,/
× +
× −
−
−
x
VV
x
V
Qr = ( , )
( )
5 0 10
10 10 2
3
3 2
× + ×× −
−
−
x V
x ;
Qr = ( , ) ,
( )
5 0 10 10 0 10
10 10 2
3 3
3 2
× + × ×× −
− −
−
x
x.
b) Qr,i = Qr(0) = 0,50.
c) Compléter le tableau suivant sauf la colonne gri-sée. On prendra V = V' = 10,0 mL.
5. En ordonnées, on porte E et en abscisse, log Qr.
x (mol) Qr log Qr E
0 0,50 = Qr,i – 0,30 0,45
4,50 × 10–3 9,50 × 101 1,98 0,40
4,95 × 10–3 9,95 × 103 4,00 0,36
4,995 × 10–3 5,00 × 105 6,00 0,28
5,00 × 10–3 = xéq 2,15 × 1015 = K 15,3 0

104 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
6. a) et b) Modélisation du nuage de points obtenu par une droite (ordonnée E, abscisse log Qr).E = – 2,94 × 10–2 × log Qr + 0,46 (type y = a · x + b).
7. E = – 2,94 × 10–2
× log( , ) ,
( )
5 0 10 10 0 10
10 10 2
3 3
3 2
× + × ×× −
− −
−
x
x + 0,46.
8. Cette courbe est en accord avec les valeurs de E de la figure suivante car les croix sont sur la courbe.
0
– 0,1
0,10
0,20
0,30
0,40
x (mol)
E (V)
0 0,001 0,002 0,003 0,004 0,005 0,006
I = E/(R + r). À R et r constants, I est proportionnel à E, donc I(x) aurait exactement la même allure que E(x). E(t) ne varierait pas exactement comme E(x) car x n’est pas proportionnel au temps (x augmente plus vite au début et moins vite à la fin), mais l’al-lure de I(t) (ou de E(t)) est voisine de celle de E(x).
D’autres pilescourantes (page 172)
A Les piles du laboratoireLes piles courantes du commerce et du laboratoire ne correspondent pas exactement au schéma du cours :– M(s) | Mn+(aq) || M'n'+(aq) | M'(s) +.Certains exercices de bac sont posés sur ces piles courantes. Il convient donc de les traiter, pour évi-ter toute surprise, même si en principe toutes les informations qui dépassent le cours, devraient être données au baccalauréat.
1. Pt(s) | Fe2+(aq) ; Fe3+(aq) || Ag+(aq) | Ag(s).
Pont salin
Fe3+(aq)Fe2+(aq)
Pt(s)
Ag+(aq)
Ag(s)
2. Dans la pile Daniell, le vase poreux remplace le pont salin.
ACTIVITÉ
4
ACTIVITÉ
4
3. L’électrode de pH-mètre a une f.e.m. qui dépend du pH. Elle est donc sensible au pH et de plus, selon une loi connue. Moyennant un dispositif électroni-que approprié (suiveur, amplificateur de différence, amplificateur), elle permettra la mesure du pH.
4. Il n’y pas ici de séparation entre demi-piles.Pôle – (départ d’électrons) :
Zn(s) = Zn2+(aq) + 2 e– ;Pôle + (arrivée d’électrons) :
O2(aq) + 4 H+(aq) + 4 e– = 2 H2O(l).Le citron fournit naturellement le milieu acide.2 Zn(s) + O2(aq) + 4 H+(aq) = 2 Zn2+(aq) + 2 H2O(l).
B Les piles du commerceet la pile à combustible
5. Cette pile est qualifiée de saline car elle contient un composé qu’on appelait autrefois sel (NH4Cl).
6 & 7. Pôle – (départ d’électrons) :Zn(s) = Zn2+(gel) + 2 e– ;
Pôle + (arrivée d’électrons) :MnO2(s) + H+(gel) + e– = MnOOH(s).
Équation globale : MnO2(s) + 2H3O+(gel) + Zn(s)
= 2 MnOOH(s) + Zn2+ (gel) + 2H2O (gel).
8. L’acier assure la rigidité du système et permet d’assurer le contact électrique avec l’extérieur.
9. Lorsque le zinc de l’électrode négative est consommé, c’est le fer du boîtier en acier qui pour-rait le remplacer. Si le dioxyde de manganèse MnO2 est le réactif limitant, cette réaction ne pourra pas se produire. L’attaque de l’acier aurait pu permettre l’écoule-ment néfaste des espèces internes.
10. Cette pile est qualifiée d’alcaline car elle contient la base HO– (contenue dans la potasse KOH qui contient un élément de la colonne des alcalins : le potassium K).
11 & 12. Pôle – (départ d’électrons) :Zn (s) + 4 HO– (gel) = Zn(OH)4
2– (gel) + 2e– ; 2 HO–(aq) = 2 H2O (l) + 2 e– (× 2).Pôle + (arrivée d’électrons) : 2 MnO2 (s) + 2 H2O (gel) + 2 e– = 2 MnOOH (s) + 2 HO– (gel)2 MnO2 (s) + 2 HO– (gel) + 2 H2O (gel) + Zn (s) = 2 MnOOH (s) + Zn(OH)4
2– (gel).
13. Dans la pile, on utilise un solvant organique pour éviter la réaction du lithium sur l’eau.
14, 15 & 16. Pôle + (arrivée d’électrons) : H2(aq) + 2 HO–(aq) = 2 H2O(l) + 2 e– (× 2)

105Chapitre 8 - Transformations spontanées dans les piles
Pôle – (départ d’électrons) :O2(aq) + 2 H2O(l) = 4 HO–(aq) + 4 e–
En ajoutant membre à membre :2 H2(aq) + O2(aq) = 2 H2O(l).
17. Les piles usées doivent être ramenées chez le vendeur plutôt que jetées à la poubelle car elles contiennent des espèces toxiques (cadmium, mer-cure, argent, hydroxyde de potassium (potasse)…).
Corrigés des exercices (page 174)
1 QCM, plusieurs réponses possibles1. Une pile = deux demi-piles + un pont salin.2. Une pile est le siège d’une réaction d’oxydoré-duction.3. Lorsqu’une pile fonctionne : Qr < K.4. Si une pile débite, le système est hors équilibre.5. Porteurs de charges : ions dans l’électrolyte et électrons dans les conducteurs métalliques.
2 Schématiser une pile1.
Cu2+ ; SO42 – Zn2+ ; SO4
2 –
Zn
AA
+ –
I
Anions
Cations
Cu
e–
2. Par le circuit électrique les électrons vont de l’électrode de zinc à l’électrode de cuivre. Dans les solutions, les anions se dirigent vers l’électrode de zinc et les cations vers celle de cuivre.
3. Donner l’écriture conventionnelle de cette pile :(–) Zn(S)/Zn2+(aq), SO4
2–(aq) ;C = 0,020 mol · L–1//Cu2+ (aq), SO4
2– (aq) ;C = 0,020 mol · L–1/Cu(S) (+)
3 Pile cadmium-argent1. Qr,i = [Cd2+(aq)]/[Ag+(aq)]2 = (0,200)/(0,200)2
= 1/0,200 = 5,00
2. Qr,i < K : d’après le critère d’évolution, le sens spontané d’évolution de ce système est donc le sens direct.
3. Par le circuit électrique, les électrons vont de l’électrode de cadmium à l’électrode d’argent. Dans
les solutions, les anions se dirigent vers l’électrode de cadmium et les cations vers celle d’argent.
4. À l’extérieur de la pile, les électrons circulent de la borne négative vers la borne positive. La borne positive est donc l’électrode d’argent
5. (–) Cd(S)/Cd2+(aq), NO3– (aq) ;
C = 0,200 mol · L–1//Ag+(aq), NO3–(aq) ;
C = 0, 200 mol · L–1/Ag (S) (+).
4 Réalisation d’une pile fer-argent1. Le voltmètre mesure une tension négative lors-que sa borne « V » est reliée à l’électrode de fer : la borne positive de la pile est l’électrode d’argent.Écriture conventionnelle :(–) Fe(s)/Fe2+(aq), 2 Cl–(aq) ;
C = 0,10 mol · L–1//Ag+(aq), Cl–(aq) ;C = 0, 20 mol · L–1/Ag(s) (+).
2. À l’électrode d’argent se produit une réduction : Ag+(aq) + e– = Ag(s).
À l’électrode de fer se produit une oxydation :Fe(s) = Fe2+(aq) + 2 e–.
3. Par le circuit électrique, les électrons vont de l’électrode de fer à l’électrode d’argent.Dans les solutions, les anions se dirigent vers l’élec-trode de fer et les cations vers celle d’argent.
4. Assure l’électroneutralité des solutions.
5. Lorsque la pile débite, la concentration en ions argent diminue (cf. demi-équation à l’électrode d’argent). La masse de l’électrode de fer diminue (cf. demi-équation à l’électrode de fer).
5 Réalisation d’une pile zinc-fer1. a) Zn(s) + Fe2+(aq) = Fe(s) + Zn2+(aq).b) Qr,i = [Zn2+(aq)]/[Fe2+(aq)] ; Qr,i = 0,100/0,200 = 0,500.c) Qr,i < K : d’après le critère d’évolution, le système chimique évolue spontanément dans le sens direct.d) À l’électrode de zinc, il se produit une oxydation Zn(s) = Zn2+(aq) + 2 e– ; à l’électrode de fer, il se pro-duit une réduction Fe2+(aq) + 2 e– = Fe(s).

106 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
2. Pile en fonctionnement :
Fe2+ ; SO42 – Zn2+ ; SO4
2 –
Zn
VV
+ –
I
Anions
Cations
Fe
e–
Par le circuit électrique, les électrons vont de l’électrode de zinc à l’électrode de fer. Dans les solutions, les anions se dirigent vers l’électrode de zinc et les cations vers celle de fer.
3. a)
Zn(s) + Fe2+(aq) = Fe(s) + Zn2+(aq) Électrons échangés
E.I. m2/M(Zn) C1 · V1 m1/M(Fe) C2 · V2 0
t m2/M(Zn)– x
C1 · V1– x
m1/M(Fe)+ x
C2 · V2+ x
2 x
E.F. m2/M(Zn)– xf
C1 · V1– xf
m1/M(Fe)+ xf
C2 · V2+ xf
2 xf
Calcul de l’avancementOn sait que la quantité d’électricité débitée par la pile pendant 5,00 min est égale à Q = I · ∆t.La quantité d’électrons transférés est alors égale :
ne = Q/(NA · e) = (I · ∆t)/(NA · e).
L’avancement final est donné par la relation :ne = 2 xf soit xf = ½(I · ∆t)/(NA · e).
A.N. : xf = ½ × (965 × 10–3 × 5,00 × 60)/(6,02 × 1023
× 1,60 × 10–19) = 1,50 × 10–3 mol = 1,50 mmol.[Fe2+(aq)] = (C1 · V1 – xf)/V1
= (0,200 × 0,100 – 0,0015)/0,100 = 0,185 mol · L–1.
[Zn2+(aq)] = (C2 · V2 + xf)/V1= (0,100 × 0,100 + 0,0015)/0,100 = 0,115 mol · L–1.
3. b) m(Fe) = m1 + xf · M(Fe)= 71,0 + 0,00150 × 55,8 = 71,084 g.
m(Zn) = m2 – xf · M(Zn) = 103,0 – 0,00150 × 65,4= 102,902 g.
6 Durée de fonctionnement
Exercice résolu dans le manuel de l’élève.
•
7 Quantité maximale d’électricité1. n1 = n(Cu2+) = C · V et n2 = n(Pb2+) = C · V ;n1 = 2,00 × 10–2 × 0,0500 = 1,00 × 10–3 mol ;n2 = 2,00 × 10–2 × 0,0500 = 1,00 × 10–3 mol.
n3 = n(Pb) = m
M2
(Pb) ; n3 = 8 50
207 2
,
, = 0,041 0 mol.
n4 = n(Cu) = m
M1
(Cu) ; n4 = 5 70
63 5
,
, = 0,089 8 mol.
2.Cu2+(aq) + Pb(s) = Cu(s) + Pb2+(aq) Électrons
échangés
E.I. 1,00 × 10–3 0,0410 0,0898 1,00 × 10–3 0
En cours
1,00 × 10–3 – x
0,0410– x
0,0898+ x
1,00 × 10–3 + x
2 x
E.F. 1,00 × 10–3 – xf
0,0410– xf
0,0898+ xf
1,00 × 10–3 + xf
2 xf
3. xmax = 1,00 × 10–3 mol, car les ions cuivre (II) sont le réactif limitant.
4. Qmax = ne · NA · e = 2 xmax · NA · e ;Qmax = 2 × 1,00 × 10–3 × 6,02 × 1023 × 1,60 × 10–19 ; Qmax = 193 C.
5. La masse de l’électrode de cuivre augmente de : ∆m = xmax · M(Cu) ;∆m = 1,00 × 10–3 × 63,5 = 0,063 5 g.
6. Lorsque la pile sera usée : Qr = K.
8 Pile cobalt-cuivre1. Co(s) + Cu2+(aq) = Co2+(aq) + Cu(s)
2. Qr,i = [Co2+(aq)]/[Cu2+(aq)] = 0,25/0,20 = 1,3 < K, le système évolue bien dans le sens direct.
3. n(Co) = m/M(Co) = 0,30/58,9 = 0,0051 mol.n(Co2+) = C × V = 0,25 × 50,0 × 10–3 = 0,013 mol.n(Cu2+) = C × V = 0,20 × 50,0 × 10–3 = 0,010 mol.
Co(s) + Cu2+(aq) = Co2+(aq) + Cu(s) Électrons échangés
E.I. 0,0051 0,010 0,013 Excès 0
t 0,0051 – x 0,010 – x 0,013 + x Excès 2 x
E.F. 0,0051 – xf 0,010 – xf 0,013 + xf Excès 2 xf
Le réactif limitant d’après l’énoncé est le cobalt solide : xf = xmax = 0,0051 mol.Quantité d’électricité fournie par la pile :Q = ne · NA · e = 2 xmax · NA · e ; Q = 2 × 0,0051 × 6,02 × 1023 × 1,60
× 10–19 = 9,8 × 102 C.4. m(Cu) = xmax · M(Cu) ;
m(Cu) = 0,0051 × 63, 5 = 0,32 g.

107Chapitre 8 - Transformations spontanées dans les piles
5. Calculer les concentrations en ions métalliques.[Cu2+(aq)] = (0,010 – 0,0051)/0,0500 = 0,098 mol · L–1
[Co2+(aq)] = (0, 013 + 0,0051)/0,0500 = 0,36 mol · L–1.
9 Pile Daniell1. D’après le branchement de l’ampèremètre, le cou-rant électrique va de l’électrode de cuivre à l’élec-trode de zinc par le circuit extérieur. Les électrons vont donc dans le sens opposé de l’électrode de zinc vers l’électrode de cuivre. Dans les solutions et le pont salin, Les anions vont donc vers l’électrode de zinc et les cations vers l’électrode de cuivre.
2. a) À l’électrode de cuivre, on a une réduction Cu2+(aq) + 2 e– = Cu(s) et à l’électrode de zinc, une oxydation Zn(s) = Zn2+(aq) + 2 e–.b) Cu2+(aq) + Zn(s) = Cu(s) + Zn2+(aq).
3. Qr = [Zn2+(aq)]/[Cu2+(aq)].Qr,i = [Zn2+(aq)]i/[Cu2+(aq)]i = C2/C1 = 1,0car C2 = C1.
4. Lorsque la pile fonctionne, Qr,i augmente et tend vers K.
5. Sachant que les métaux sont en excès, le réactif limitant est l’ion cuivre (II) donc xmax = 1,0 × 10–3 mol.Le nombre d’électrons transférés est ne = 2 xmax. La quantité maximale d’électricité que peut débi-ter cette pile est :Qmax = 2 xmax · NA · e ;Qmax = 2 × 1,0 × 10–3 × 6,02 × 1023 × 1,60
× 10–19 = 1,9 × 102 C.
10 Variation des concentrations1. a) Sachant que la masse de l’électrode de cad-mium diminue, à cette électrode, on a une oxyda-tion : Cd(s) = Cd2+(aq) + 2 e–.À l’électrode d’argent, on a donc une réduction :
Ag+(aq) + e– = Ag(s).b) Cd(s) + 2 Ag+(aq) = 2 Ag(s) + Cd2+(aq).
2. Quantités de matière initiales :n(Ag+(aq)) = c1 · V1 ; n(Ag+(aq)) = 0,20 × 10–2 × 25,0 × 10–3
= 5,0 × 10–5 mol.n(Cd2+(aq)) = c2 · V2 ; n(Cd2+(aq)) = 0,30 × 10–2 × 25,0 × 10–3
= 7,5 × 10–5 mol.n(Ag) = (r(Ag) · V(Ag))/M(Ag) ; n(Ag) = 3,0 × 10,50/107,9 = 0,29 mol.n(Cd) = (r(Cd) · V(Cd))/M(Cd) ; n(Cd) = 6,0 × 8,65/112,4 = 0,46 mol.
Cd(s) + 2 Ag+(aq) = 2 Ag(s) + Cd2+(aq) Électrons échangés
E.I. 0,46 5,0 × 10–5 0,29 7,5 × 10–5 0
t 0,46 – x5,0 × 10–5
– 2 x0,29 + 2 x
7,5 × 10–5
+ x2 x
E.F. 0,46– xéq
5,0 × 10–5
– 2 xéq
0,29+ 2 xéq
7,5 × 10–5
+ xéq2 xéq
Exprimons l’avancement au bout de 10 min.On sait que Q = I · ∆t.La quantité d’électrons transférés est alors égale
ne = Q/(NA · e) = (I · ∆t)/(NA · e) = 2 x.Soitx = (I · ∆t)/(2 · F) ;x = (10 × 10–3 × 60)/(2 × 96 500) = 3,1 × 10–6 mol.[Ag+(aq)] = (C1 · V1 – 2 x)/V1 = C1 – 2 x/V1 ;[Ag+(aq)] = 0,20 × 10–2 – 2 × 3,1 × 10–6/25,0
× 10–3 = 1,8 × 10–3 mol · L–1.[Cd2+(aq)] = (C2 · V2 + x)/V2 = C2 + x/V2 ;[Cd2+(aq)] = 0,30 × 10–2 + 3,1 × 10–6/25,0
× 10–3 = 3,1 × 10–3 mol · L–1.
3. m(Ag) = n(Ag) · M(Ag) ;m(Ag) = (0,29 + 2 x) × 107,9 = 31 g.m(Cd) = n(Cd) · M(Cd) ;m(Cd) = (0,46 – x) × 112,4 = 52 g.
11 Pile saline1. L’électrode de carbone est le siège d’une réduc-tion, c’est donc une électrode positive qui sert à collecter les électrons.
2. Zn(s) = Zn2+(aq) + 2 e–
3. (–) Zn(s)/Zn2+(aq)//MnO(OH)/MnO2,C (+)
4. L’électrolyte est acide car l’oxydation se fait en milieu acide.
5. On peut avoir comme réaction parasite l’oxyda-tion du zinc. Il faut utiliser du zinc amalgamé qui n’est pas attaqué par les ions oxonium. L’amalgame est un alliage mercure zinc dans ce cas.
6. On évite l’attaque du boîtier si tout le zinc n’est pas consommé. Il faut donc que l’oxyde de manga-nèse soit le réactif limitant.
7. Il faut associer trois piles de 1,5 V en série pour obtenir une pile plate de 4,5 V.
8. On parle de pile alcaline quand l’électrolyte contient un alcalin (le potassium par exemple dans l’hydroxyde de potassium). C’est une solution basi-que encore appelée solution alcaline.
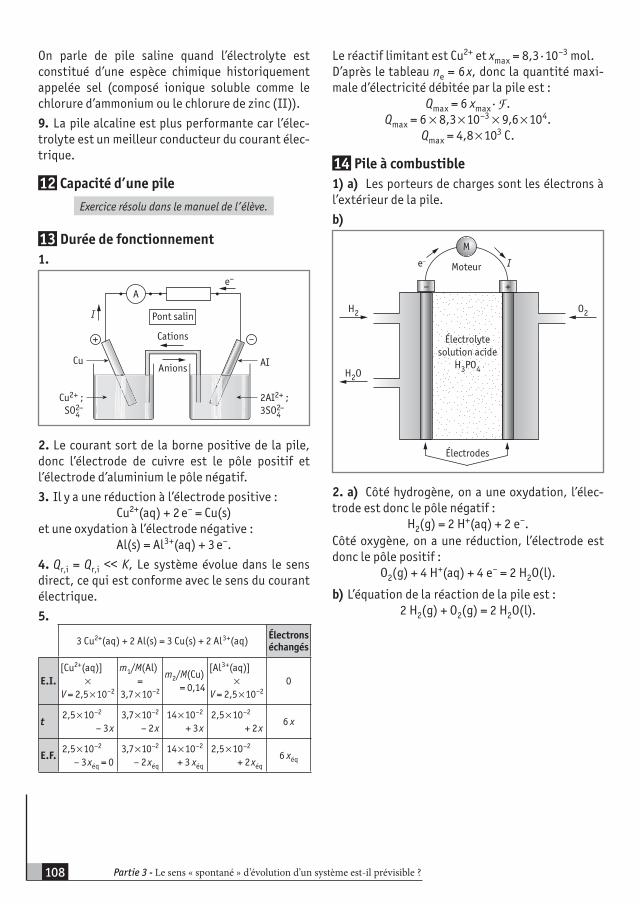
108 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
On parle de pile saline quand l’électrolyte est constitué d’une espèce chimique historiquement appelée sel (composé ionique soluble comme le chlorure d’ammonium ou le chlorure de zinc (II)).
9. La pile alcaline est plus performante car l’élec-trolyte est un meilleur conducteur du courant élec-trique.
12 Capacité d’une pile
Exercice résolu dans le manuel de l’élève.
13 Durée de fonctionnement1.
Cu2+ ;SO4
2 –2AI2+ ;3SO4
2 –
AI
A
+ –
I
Anions
Cations
Cu
Pont salin
e–
2. Le courant sort de la borne positive de la pile, donc l’électrode de cuivre est le pôle positif et l’électrode d’aluminium le pôle négatif.
3. Il y a une réduction à l’électrode positive :Cu2+(aq) + 2 e– = Cu(s)
et une oxydation à l’électrode négative : Al(s) = Al3+(aq) + 3 e–.
4. Qr,i = Qr,i << K, Le système évolue dans le sens direct, ce qui est conforme avec le sens du courant électrique.
5.
3 Cu2+(aq) + 2 Al(s) = 3 Cu(s) + 2 Al3+(aq) Électrons échangés
E.I.[Cu2+(aq)]
×V = 2,5 × 10–2
m1/M(Al)=
3,7 × 10–2
m2/M(Cu)= 0,14
[Al3+(aq)]×
V = 2,5 × 10–20
t2,5 × 10–2
– 3 x3,7 × 10–2
– 2 x14 × 10–2
+ 3 x2,5 × 10–2
+ 2 x6 x
E.F.2,5 × 10–2
– 3 xéq = 03,7 × 10–2
– 2 xéq
14 × 10–2
+ 3 xéq
2,5 × 10–2
+ 2 xéq6 xéq
Le réactif limitant est Cu2+ et xmax = 8,3 · 10–3 mol.D’après le tableau ne = 6 x, donc la quantité maxi-male d’électricité débitée par la pile est :
Qmax = 6 xmax · F .Qmax = 6 × 8,3 × 10–3 × 9,6 × 104.
Qmax = 4,8 × 103 C.
14 Pile à combustible1) a) Les porteurs de charges sont les électrons à l’extérieur de la pile.
b)
e– I
Électrodes
Électrolytesolution acide
H3PO4
H2 O2
H2O
Moteur
– +
M
2. a) Côté hydrogène, on a une oxydation, l’élec-trode est donc le pôle négatif :
H2(g) = 2 H+(aq) + 2 e–.Côté oxygène, on a une réduction, l’électrode est donc le pôle positif :
O2(g) + 4 H+(aq) + 4 e– = 2 H2O(l).
b) L’équation de la réaction de la pile est :2 H2(g) + O2(g) = 2 H2O(l).

109Chapitre 8 - Transformations spontanées dans les piles
Exercices supplémentaires
1. Étude d’une pile cuivre-argentD’après bac Liban 2003
À partir des couples oxydant/réducteur Cu2+(aq)/Cu(s) et Ag+(aq)/Ag(s) on peut envisager deux trans-formations dont les réactions peuvent être schéma-tisées par les équations suivantes : Cu(s) + 2 Ag+ (aq) = 2 Ag(s) + Cu2+(aq) (1) Cu2 (aq) + 2 Ag(s) = 2 Ag+(aq) + Cu(s) (2)Les constantes d’équilibres de ces réactions sont :
K1 = 2,1 × 1015 et K2 = 4,8 × 10–16.
A. Transformation chimique spontanée par transfert direct d’électrons
Un élève réalise l’expérience dont le protocole est donné ci-dessous :– Verser dans un bécher un volume V1 = 50 mL de solution aqueuse de sulfate de cuivre (II) de concen-tration molaire en soluté apporté C1 = 1,0 mol · L–1 et un volume V2 = 50 mL d’une solution aqueuse de nitrate d’argent de concentration molaire en soluté apporté C2 = 0,50 mol · L–1.
Les compétences expérimentales
(page 180)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de revenir sur des techniques de base que l’on retrouve de manière récurrente dans les sujets d’évaluation. Cet exercice doit conduire l’élève à justifier ses choix.
Corrigé1. Schéma
Cu2+ ; SO42 – Zn2+ ; SO4
2 –
Zn
VV COM
+ –
I
Anions
Cations
Cu
Pont salin
e–
2. Exploitation de mesures1. a) Qr,i = [Zn2+(aq)]/[Cu2+(aq)] = 0,5/0,5 = 1.Qr, i < K, d’après le critère d’évolution, La transformation évolue spontané-ment dans le sens direct.b) À l’électrode de cuivre il y a consommation d’électrons. Donc elle est chargée positivement. L’électrode de zinc est donc négative.2. Cf. schéma3. En diminuant la concentration en ions cuivre II la f.e.m. diminue, en diminuant la concentration en ions zinc, la f.e.m. augmente. Conclusion : Lorsqu’on modifie la concentration en ions des électrolytes, on modifie la f.e.m. de la pile.4. Si on ajoute de l’hydroxyde de sodium à la demi-pile côté cuivre, la concentration en ions cuivre II va diminuer, donc la f.e.m. va diminuer.5. f.e.m.A = 1,10 V l’électrode positive est l’électrode de cuivre, l’électrode négative est l’électrode zinc.f.e.m.E = – 0,46 V l’électrode positive est l’électrode d’argent, l’électrode négative est l’électrode cuivre.6. La polarité d’une demi-pile dépend du couple métal/ion métallique et de l’autre demi-pile avec laquelle elle est associée.

110 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
CorrigéA. 1. D’après les observations (intensification de la coloration bleue et dépôt gris), la réaction associée à la transformation chimique produit des ions cuivre II et de l’argent métallique. L’équation de cette réaction chimique est :
Cu(s) + 2 Ag+(aq) = 2 Ag(s) + Cu2+(aq).
2. Cf. cours chapitre 8 du Manuel de l’élève.Tout système qui subit une transformation chimique spontanée (donc sans apport extérieur d’énergie), évolue vers l’état d’équilibre chimique. Le quotient de réaction (Qr) de la réaction qui modélise cette transformation, évolue donc de Qr,i à Qr,éq = K.K est la constante d’équilibre associée à la réaction.• Si Qr,i < K, l’évolution spontanée se fait dans le sens direct de l’équation de réaction ;• Si Qr,i > K, l’évolution spontanée se fait dans le sens inverse (ou indirect) de l’équation de réaction ;• Si Qr,i = K, il n’y a aucune évolution.
Qr,i = [ ( )]
[ ( )]
Cu aq
Ag aqi
i
2
2
+
+ =
C V
V V
C V
V V
1 1
1 2
2 2
1 2
2
×+
×+
;
Qr,i =
1 0 50 10
0 100
0 50 50 10
0 100
3
32
,
,
,
,
× ×
× ×
−
− = 8,0.
3. On constate que Qr,i < K : le système chimique évolue spontanément dans le sens direct, ce qui est cohérent avec les observations
B. 1.
Cu2+ ; SO42 – Ag+ ; NO3
–
Ag
+ –
I
Anions
Cations
Cu
Circuit extérieure–
2. a) Par le circuit électrique, les électrons vont de l’électrode de cuivre à l’électrode d’argent. Dans les solutions, les anions se dirigent vers l’élec-trode de cuivre et les cations vers celle d’argent.
La solution de sulfate de cuivre est bleue, celle de nitrate d’argent incolore.– Plonger un fil d’argent et ajouter 3,0 g de poudre de cuivre de couleur rouge.– Agiter– Filtrer la solution obtenue et observer sa couleur.L’élève note dans son compte rendu de TP : « On observe un léger dépôt gris et une intensification de la coloration bleue ».
1. Parmi les deux réactions possibles, quelle est celle associée à la transformation chimique du sys-tème ?2. Rappeler le critère d’évolution spontanée.3. Calculer le quotient de réaction initial, puis, en appliquant le critère d’évolution spontanée, mon-trer que le sens d’évolution prévu est compatible avec les observations expérimentales de l’élève.
B. Constitution et fonctionnement de la pile cuivre-argent en circuit fermé
On dispose :– d’un fil de cuivre,– d’un fil d’argent,– d’une solution de sulfate de cuivre (II)
de volume V1 = 50 mL et de concentration molaire c1 = 1,0 mol · L–1,– d’une solution de nitrate d’argent
de volume V2 = 50 mL etde concentration molaire c2 = 0,50 mol · L–1,
– d’un papier imbibé de nitrate de potassium pou-vant constituer un pont salin.
1. Faire un schéma de la pile réalisable avec le maté-riel donné ci-dessus.2. On observe dans le circuit extérieur le passage d’un courant électrique de l’électrode d’argent vers l’électrode de cuivre.a) Préciser sur le schéma le sens de circulation des électrons dans le circuit et la polarité des électro-des.b) Écrire les demi-équations des réactions modéli-sant les transformations qui ont lieu à chaque élec-trode.c) Écrire l’équation de la réaction associée à la transformation ayant lieu dans la pile.d) La pile cuivre argent en fonctionnement est-elle un système chimique dans l’état d’équilibre ou hors d’équilibre ? Justifier la réponse en utilisant le cri-tère d’évolution spontanée.

111Chapitre 8 - Transformations spontanées dans les piles
b) D’après le mouvement des porteurs de charges à l’électrode d’argent, la demi-équation électroni-que est Ag+(aq) + e– = Ag(s) (oxydation) à celle de cuivre, on a Cu(s) = Cu2+(aq) + 2 e– (réduction).
c) Cu(s) + 2 Ag+(aq) = 2 Ag(s) + Cu2+(aq).
d) QC
Cr,i
i
i
Cu (aq)
Ag (aq)
=
= =+
+
2
22
2
1 0
( )
,
(00 504 0
2, ),= .
Qr,i < K : l’évolution spontanée se fait dans le sens direct de l’équation de la réaction. Le système est hors état d’équilibre.
2. Un système chimique(Extrait nouvelle Calédonie 2003)
On réalise une pile avec les couples Ag+/Ag et Fe3+/Fe2+.La constante d’équilibre associée à la réaction d’équation Ag+(aq) + Fe2+(aq) = Ag(s) + Fe3+(aq) a pour valeur K = 3,2.
Électrodede platine
Ag+(aq)Fe2+(aq)Fe3+(aq)
Électroded’argent
Récepteur
Schéma de la pile
1. Donner l’expression du quotient de réaction associé à l’équation de la réaction globale.2. Dans la pile, les concentrations molaires initiales des ions dans les solutions aqueuses sont :[Ag+(aq)]i = [Fe2+(aq)]i = [Fe3+(aq)]i
= 1,0 × 10–1 mol · L–1.Calculer le quotient de réaction à l’état initial.3. En déduire le sens d’évolution spontané de la réaction quand la pile fonctionne.4. Quelle est la polarité de chaque électrode ? Justi-fier à partir des demi-équations.5. On réalise une deuxième pile en modifiant les conditions initiales du système.Les concentrations initiales sont désormais :
[Ag+(aq)]i = [Fe2+(aq)]i = 1,0 × 10–1 mol · L–1
et [Fe3+(aq)]i = 1,0 × 10–2 mol · L–1.La polarité de chaque électrode reste-t-elle la même ? Justifier.
Corrigé1. Le quotient de la réaction associée à l’équation Ag+(aq) + Fe2+(aq) = Ag(s) + Fe3+(aq) a pour expres-sion : Qr = [Fe3+(aq)]/([Ag+(aq)] · [Fe2+(aq)]).
2. Quotient de réaction à l’état initial :Qr,i = [Fe3+]i/([Ag+(aq)]i · [Fe2+(aq)]i) ;Qr,i = 1,0 × 10–1/(1,0 × 10–1 )2 = 10.
3. Qr,i > K : l’évolution spontanée se fait dans le sens inverse de l’équation de la réaction.
4. D’après l’équation de la réaction de la pile :à l’électrode d’argent, on a une oxydation Ag(s) = Ag+(aq) + e– et à l’électrode de platine, la réduction Fe3+ + e– = Fe2+(aq). Donc l’électrode d’ar-gent est négative et celle de platine est positive.
5. Calcul du nouveau Qr,i :Qr,i = [Fe3+(aq)]i /([Ag+(aq)]i · [Fe2+(aq)]i) ;Qr,i = 1,0 × 10–2/(1,0 × 10–1)2 = 1,0.Qr,i < K, l’évolution spontanée se fait dans le sens direct de l’équation de réaction.D’après l’équation de réaction de la pile : à l’électrode d’argent, on a une réduction Ag+(aq) + e– = Ag(s) et à l’électrode de platine, l’oxydation Fe2+(aq) = Fe3+ + e–. Donc l’électrode d’argent est positive et celle de pla-tine est négative.
3. Pile à combustible(Suite de l’exercice 14, p. 179 du Manuel de l’élève.)
1. Le réactif qui est oxydé est appelé le « combustible » de la pile.a) Parmi les espèces chimiques présentes dans les couples, laquelle constitue le combustible ? Justifier la réponse en définissant la réaction de réduction.b) Préciser le nom de l’électrode où se produit la réduction. Cette électrode est-elle le pôle positif ou négatif de la pile ?2. Dans un véhicule motorisé fonctionnant grâce à une pile à combustible, on estime à 1,5 kg la masse de dihydrogène nécessaire pour parcourir 250 km.a) Calculer la quantité de matière de dihydrogène n(H2) correspondant à cette masse, puis le volume de dihydrogène V(H2) en mètre-cube (m3), dans les con-ditions où le volume molaire Vm est égal à 24 L · mol–1.b) Justifier le fait que les piles à combustible ne soient pas encore utilisées dans les voitures, en uti-lisant la réponse à la question 4.1.c) Rappelons la loi des gaz parfaits : P · V = n · R · T avec :P (pression du gaz) ; V (volume du gaz) ;

112 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
n (quantité de matière de gaz) ;R (constante des gaz parfaits) ;T (température du gaz).Proposer un moyen de réduire l’espace occupé par ce gaz, à température ambiante, pour la quantité de matière n de gaz calculée précédemment. Justifier la réponse à l’aide de la loi précédente.3. Dans la navette spatiale, les piles à combustibles débitent un courant d’intensité I = 200 A.a) Calculer la charge électrique Q libérée en 24 h.b) En déduire la quantité de matière nP des porteurs de charge, ayant circulé dans le circuit de la navette, pendant 24 heures et la quantité de matière n(H2) de dihydrogène consommée.
Corrigé1. a) Le combustible est le dihydrogène car c’est le réactif qui est oxydé.b) L’électrode où se produit la réduction est le pôle positif car les électrons y sont consommés.
2. a) n(H2) = m(H2)/M(H2) ; n(H2) = 1,5 × 103/2,0 = 7,5 × 102.V(H2) = n(H2) × Vm ;V(H2) = 7,5 × 102 × 24 = 1,8 × 104 L = 18 m3.b) L’espace occupé par le combustible pour parcou-rir 250 km est très important, donc on ne peut pas utiliser la pile à combustible pour une voiture.c) On sait que P × V = n × R × T. À une température donnée, pour une quantité de matière donnée, le produit n × R × T est constant. Donc P × V est aussi constant. Si on veut diminuer le volume du gaz V il faut augmenter la pression du gaz P.
3. a) Q = I · ∆t ; Q = 200 × 24 × 3 600 = 1,73 × 107 C.b) On cherche la quantité de matière d’électrons échangés :
Q = ne · F , soit ne = Q/F ; ne = 1,79 × 102 mol.D’après l’équation à l’électrode négative :
n(H2) = ½ ne = 89,5 mol.En 24 h, il a été consommé 89,5 mol de dihydrogène.
4. Pile zinc-cuivre France juin 2003.
On souhaite réaliser une pile au laboratoire. Pour cela, on dispose d’une lame de zinc et d’une lame de cuivre ainsi que d’un volume V1 = 100 mL d’une solution aqueuse de sulfate de zinc (II) de concen-tration molaire en soluté apporté c1 = 1,0 mol · L–1 et d’un volume V2 = 100 mL d’une solution aqueuse de sulfate de cuivre de concentration molaire en soluté apporté c2 = 1,0 mol · L–1 et d’un pont salin.
L’expérience est réalisée à la température de 25 °C. À cette température, la constante d’équilibre asso-ciée à l’équation :
Cu2+(aq) + Zn(s) = Zn2+(aq) + Cu(s)est K = 4,6 × 1036.La pile ainsi réalisée est placée dans un circuit électri-que comportant une résistance et un interrupteur. On ferme ce circuit électrique à l’instant de date t0 = 0 s.
1. Faire un schéma légendé de cette pile. Compléter le schéma avec la résistance et l’interrupteur.2. Déterminer le quotient de réaction Qr,i du système chimique ainsi constitué à l’instant de date t0. En déduire le sens d’évolution spontanée du système.3. Pour chaque électrode, écrire la demi-équation correspondant au couple qui intervient.4. En déduire, en justifiant la réponse, à quel métal correspond le pôle + de la pile et à quel métal corres-pond le pôle –.5. D’après la théorie, on considère que la pile s’ar-rête de fonctionner quand le réactif limitant, consti-tué soit par les ions Cu2+, soit par les ions Zn2+, a été complètement consommé.En utilisant l’équation de la réaction modélisant la transformation se produisant à l’une des électrodes, calculer la quantité maximale d’électricité que pourrait théoriquement débiter cette pile.On donne la constante d’Avogadro :
NA = 6,02 × 1023 mol–1,la charge électrique élémentaire e = 1,6 × 10 –19 C.
Corrigé1.
Cu2+ ; SO42 – Zn2+ ; SO4
2 –
Zn
+ –
I
Anions
Cations
Cu
Pont salin
e–
2. Qr,i = [Zn2+(aq)]/[Cu2+(aq)] = C1/c2 = 1,0.Qr,i < K : d’après le critère d’évolution, la transfor-mation évolue spontanément dans le sens direct.
3. À l’électrode de cuivre, on a une réduction : Cu2+(aq) + 2 e– = Cu(s).
À l’électrode de zinc, on a une oxydation : Zn(s) = Zn2+(aq) + 2 e–.

113Chapitre 8 - Transformations spontanées dans les piles
4. La lame de cuivre est le siège d’une réduction qui consomme des électrons, elle constitue donc le pôle positif. L’électrode de zinc constitue donc le pôle négatif.
5. Le réactif limitant est l’ion cuivre (II), le zinc est en excès.
xmax = C2 · V2.
Zn(s) = Zn2+(aq) + 2 e–
E.I. n 0,10 0
Réaction totale n – xmax 0,10 + xmax 2 xmax
On sait que : Q = ne · NA · e.D’après la demi-équation, on obtient :
Q = 2 x · NA · e.Donc Qmax = 2 · xmax · NA · e = 2 · C2 · V2 · NA · e ;Q = 2 × 1,0 × 0,100 × 6,02 · 1023 × 1,6 × 10–19
= 1,9 × 104 C.
5. Piles dentaires Exercice d’après bac
L’amalgame dentaire utilisé pour remplir les cavités provoquées par les caries, est un mélange constitué principalement d’un alliage argent-mercure de formule Ag2Hg3 et d’un alliage argent-étain de formule Ag3Sn. Pour confectionner les couronnes, on utilise des alliages précieux. La composition d’une couronne en or est la suivante :Au : 76 % ; Ag : 10 % ; Cu : 10 % ; Pd : 2 % ; Pt : 2 %.Le pH de la salive est voisin de 5.Données
H+(aq)/H2(g) ; Al3+(aq)/Al(s).1. Douleur provoquée par l’aluminium.Lorsqu’un morceau d’aluminium Al(s) provenant d’un emballage alimentaire vient au voisinage d’une carie soignée, on ressent une douleur. Celle-ci pro-vient de l’excitation des nerfs dentaires lors du court-circuit de la pile aluminium-salive-amal-game. Dans cette pile l’aluminium subit une oxyda-tion en ion Al3+.
AluminiumAmalgameNerf
Salive
•
a) Écrire l’équation de la réaction subie par l’alumi-nium.b) Quelle est la nature du pôle constitué par le mor-ceau d’aluminium dans cette « pile dentaire ».c) En déduire la nature du pôle constitué par l’amal-game.d) Dans la salive intervient le couple oxydoréduc-teur H+(aq)/H2(g). Écrire l’équation de la réaction qui se produit dans la salive.e) En déduire l’équation de la réaction modélisant la transformation spontanée qui a lieu dans cette pile.2. Lorsqu’une carie récemment obturée se trouve au voisinage d’une couronne en or, il se produit le court-circuit de la pile amalgame-salive-or. L’étain de l’amalgame est oxydé selon l’équation :
Ag3 Sn(s) = Sn2+(aq)+ 3 Ag(s) + 2 e–.a) Quelle est la nature du pôle constitué par l’amal-game.b) En déduire la nature du pôle constitué par la cou-ronne.c) Justifier le fait que l’on ressente un goût métalli-que lors du court-circuit de cette pile par la salive.d) La tension aux bornes de cette pile dentaire peut atteindre 500 mV pour un courant de quelques micro-ampères par cm2. À combien peut-on esti-mer la résistance du « circuit dentaire ».
Corrigé1. a) Al(s) = Al3+(aq) + 3 e–.b) L’aluminium s’oxyde et libère des électrons : il constitue l’électrode négative.c) L’amalgame constitue le pôle positif de la pile.d) Les ions oxonium se réduisent en dihydrogène à l’électrode positive :
2 H3O+(aq) + 2 e– = H2(g) + 2H2O(l)e) Sachant que les électrons ne doivent pas appa-raître, l’équation de la réaction modélisant la transformation spontanée est :2Al(s) + 6 H3O+(aq) = 2Al3+(aq) + 3 H2(g) + 6 H2O(l).
2. a) L’étain de l’amalgame est oxydé, des électrons sont libérés, l’amalgame constitue le pôle négatif.b) La couronne joue donc le rôle de pôle positif.c) D’après l’équation :
Ag3Sn(s) = Sn2+(aq)+ 3 Ag(s) + 2 e–,le goût métallique est du à la formation d’argent métallique à l’électrode négative.d) D’après la loi d’ohm, on a U = R · I
soit R = U/I ; R = 0 5
10 6
,−
= 5 × 105 Ω.

114 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
6. Pile cuivre-aluminium (D’après Bac Réunion, 2006.)
DonnéesLe faraday : valeur absolue de la charge d’une mole d’électrons de symbole F : F = 9,65 × 104 C · mol–1.Masse molaire de l’aluminium : 27 g · mol–1.
1. On introduit dans un bécher un volume V = 50 mL d’une solution aqueuse de chlorure d’aluminium (III), Al3+(aq) + 3 Cl–(aq)), de concentration en soluté apporté 0,10 mol · L–1, dans laquelle plonge une lame d’aluminium. Dans un second bécher, on introduit un volume V = 50 mL d’une solution de sul-fate de cuivre (II), Cu2+(aq) + SO4
2–(aq), de concen-tration molaire en soluté apporté 0,10 mol · L–1, dans laquelle plonge une lame de cuivre. On relie les deux bécher à l’aide d’un pont salin contenant du nitrate d’ammonium, NH4
+(aq) + NO3–(aq). Lors-
que l’on branche un voltmètre électronique avec sa borne COM reliée à l’électrode d’aluminium, on mesure une différence de potentiel U = + 1,8 V.a) Quelle est la polarité de la pile ?b) Quel est le rôle du pont salin ?
2. On relie la pile à un conducteur ohmique.a) Faire un schéma légendé en indiquant le sens du courant dans le circuit, et en représentant le dépla-cement des différents porteurs de charge à l’inté-rieur et à l’extérieur de la pile.b) Écrire et nommer les réactions qui se produisent aux électrodes.c) Montrer que la transformation entre les deux couples peut s’écrire :
3 Cu2+(aq)+ 2 Al(s) = 3 Cu(s) + 2 Al3+(s).La constante d’équilibre associée à la transforma-tion est K = 1020.Calculer le quotient de réaction initial.Montrer en appliquant le critère d’évolution spon-tanée que le sens d’évolution est cohérent avec le fonctionnement de la pile.
3. La pile fonctionne pendant 1 h 30 min en débi-tant un courant d’intensité constante I = 40 mA.a) Calculer la quantité d’électricité Q échangée pendant 1 h 30 min.b) Calculer la quantité de matière d’électrons n(e–) échangée pendant cette durée.c) Donner la relation entre n(e–) et n(Al), quantité de matière d’aluminium ayant disparu.d) Calculer la perte de masse de l’électrode d’alu-minium.
•
•
Corrigé1. a) Le voltmètre mesure une tension positive donc l’électrode de cuivre est le pôle positif et l’électrode d’aluminium est le pôle négatif.b) Le pont salin assure l’électroneutralité des solu-tions et permet le passage du courant électrique.
2. a)
Cu2+ ; SO42 – AI3+ ; 3CI–
AI
A
+ –
I
Anions
Cations
Cu
Pont salin
e–
b) Il y a une réduction à l’électrode positive :Cu2+(aq) + 2 e– = Cu(s)
et une oxydation à l’électrode négative : Al(s) = Al3+(aq) + 3 e–.
c) L’équation modélisant la transformation se produi-sant dans la pile ne doit pas faire apparaître d’électron :
3 Cu2+(aq) + 2 Al(s) = 3 Cu(s) + 2 Al3+(aq)
Qr,i = [ ( )]
[ ( )]
Al aq
Cu aqi
i
3 2
2 3
+
+ ; Qr,i =
( , )
( , )
0 10
0 10
2
3 = 10.
Qr,i << K : le système évolue dans le sens direct, ce qui est conforme avec les polarités de la pile.
3. a) Q = I · ∆t ; Q = 0,040 × 90 × 60 = 2,2 × 102 C.b) Quantité de matière d’électrons :
Q = n(e–) · F , d’où n(e–) = Q/F ; n(e–) = 2,2 × 102/96 500 = 2,3 × 10–3 mol.
c)
3 Cu2+(aq) + 2 Al(s) = 3 Cu(s) + 2 Al3+(aq) Électrons échangés
E.I.[Cu2+(aq)]
× V =5,0 × 10–3
n1 n2
[Al3+ (aq)]× V =
5,0 × 10–30
t 5,0 × 10–3
– 3 x n1 – 2 x n2 + 3 x 5,0 × 10–3
+ 2 x 6 x
E.F. 5,0 × 10–3
– 3 xéq
n1 – 2 xéq
n2 + 3 xéq
5,0 × 10–3
+ 2 xéq6 xéq
Le réactif limitant est Cu2+ et xmax = 1,7 × 10–3 mol.D’après le tableau, n(Al)disparu = 1/3 × n(e–).
d) m(Al)disparu = n(Al)disparu · M(Al)m(Al)disparu = 1/3 × n(e–) × M(Al) ;m(Al)disparu = 1/3 × 2,3 × 10–3 × 27 = 2,1 × 10–2 g.

115Chapitre 9 - Transformations forcées lors d’électrolyses
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
– Mise en évidence expérimentale de l’électrolyse sur un exemple.– Applications pratiques : exemples de l’accumulateur au plomb et de l’électro-lyse de la solution aqueuse de chlorure de sodium.– Applications à quelques systèmes chimiques pris dans le domaine de la vie : respiration, photosynthèse, par exemple.
3. Exemples de transformations forcées– Mise en évidence expérimentale de la possibilité, dans certains cas, de changer le sens d’évolution d’un système chimique en imposant un courant de sens inverse à celui observé lorsque le système chimi-que évolue spontanément (transformation forcée).– Réactions aux électrodes, anode et cathode.– Application à l’électrolyse : principe et exemples d’applications courantes et industrielles.
– Savoir que l’électrolyse est une transformation forcée.– Connaissant le sens du cou-rant imposé par le générateur, identifier l’électrode à laquelle se produit la réaction d’oxy-dation (anode) et l’électrode à laquelle se produit la réaction de réduction (cathode).
9 Transformations forcées lors d’électrolyses
Cours
Découpage du cours1. Transformation électrochimique forcée p. 1822. Autres transformations forcées p. 186
1. Transformation électrochimique forcée1.1. Transformations spontanées dans les pilesLe chapitre commence par la présentation de la pile Cu-Ag (déjà étudiée chapitre 8). À l’aide de l’équa-tion chimique globale, on prévoit déjà les concen-trations des diverses espèces chimiques dans chaque compartiment, en supposant la transformation chimique totale (sauf celles des ions nitrate et potas-sium arrivés par le pont).
1.2. Équilibre chimique dans la pile usée• On recherche les concentrations des diverses espèces chimiques dans l’état final d’équilibre, compte tenu de la nécessaire neutralité électrique des compartiments. On vérifie que la quantité de matière de charge positive est égale à la quantité de matière de charge négative pour chaque demi-pile.Pour l’ion argent, on récupère sa concentration à l’équilibre (cf. chap. 8). La concentration effective [Ag+(aq)] = 2,2 × 10–8 mol · L–1 correspond à la concentration en ions argent trouvée dans l’activité 3-C-3 de la page 171. Si cette activité n’a pas été trai-tée, il faudra déterminer cette concentration en exercice (avant ce paragraphe, pour ne pas le « hâcher »).
♦ • La pile usée est réalisée de la manière suivante :– compartiment argent : V mL de solution aqueuse de nitrate d’argent à 4,4 × 10–8 mol · L–1 + V mL de solution aqueuse de nitrate de potassium à 2 mol · L–1. Alors [K+(aq)] = [NO3
–(aq)] = 1 mol · L–1 et [Ag+(aq)] = 2,2 × 10–8 mol · L–1, en tenant compte de la dilution lors du mélange. On peut également préparer direc-tement une solution aqueuse dont les concentra-tions effectives en ion argent (I), nitrate et potassium sont respectivement :
2,2 × 10–8 mol · L–1, 1,0 mol · L–1, 1,0 mol · L–1 ;– compartiment cuivre : V mL de solution aqueuse de sulfate de cuivre (II) à 1 mol · L–1 + V mL de solu-tion aqueuse de nitrate de cuivre (II) à 1 mol · L–1. On aura ainsi, compte tenu de la dilution et de la double provenance des ions cuivre (II) :[Cu2+(aq)] = 1 mol · L–1, [SO4
2–(aq)] = 0,50 mol · L–1, [NO3
–(aq)] = 1 mol · L–1,en tenant compte de l’indice 2 affecté à l’ion nitrate dans la formule Cu(NO3)2.• En dehors de toute utilisation, cette pile usée sera maintenue en court-circuit, ne serait-ce que pour atteindre « parfaitement » l’état d’équilibre.Les données du manuel élève (cf. p. 183, lignes 12 à 14) indiquent les concentrations dans une même solution aqueuse. Si l’on réalise des mélanges, il fau-dra tenir compte de la dilution, comme indiqué pré-cédemment.

116 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
On donne ensuite la preuve théorique que ces concentrations conduisent à un état d’équilibre, en montrant que Qr,i = K. On montre expérimentale-ment que les grandeurs Iéq et Eéq sont nulles. On peut aussi prouver par un test au chlorure de sodium qu’il n’y pas d’ions argent (I) (prélèvement dans un tube à essai).
1.3. L’électrolyse une transformation forcée• On impose ensuite un courant de sens inverse à celui que débitait spontanément la pile (5 mA), à l’aide d’un générateur auxiliaire. Ayant choisi un sens positif de référence (celui du courant dans la pile), on constate que i = – 5 mA. On montre alors que les demi-équations et l’équation chimique globale sont inversées par rapport à celles écrites pour la pile.Cette expérience (qui fonctionne très bien) est très démonstrative et semble incontournable.Expérimentalement, si on laisse fonctionner le dis-positif 10 min, on peut mettre en évidence la forma-tion d’ions argent (I) par un test au chlorure de sodium (prélèvement dans un tube à essai). On valide ainsi l’écriture des équations de réaction modélisant cette transformation forcée.• L’aspect accumulateur peut être sommairement abordé : il sera repris en activité.
• Les problèmes quantitatifs n’apportent pas de nouveautés par rapport au chapitre précédent (chap. 8).• Les remarques déjà formulées au chapitre 8 du livre du professeur restent valables.
1.4. Caractéristiques du dispositif d’électrolyseLes définitions de la cathode et de l’anode sont don-nées, conformément au programme officiel.Les chaînes énergétiques de l’électrolyseur et de la pile, étudiées en 1re S, sont réinvesties et comparées.De manière générale, il semble qu’il faille maintenir la démarche qui consiste à :– recenser les espèces chimiques initialement pré-sentes dans l’électrolyseur ;– écrire les équations de toutes les réductions pos-sibles à la cathode et celles de toutes les oxydations possibles à l’anode (en n’oubliant pas le solvant et les électrodes). Les couples ox/red qui interviennent sont donnés ;– sélectionner les bonnes demi-équations électro-niques à partir des observations expérimentales (cf. Activité 3, p. 190).
2. Autres transformations forcéesL’exemple de la photosynthèse forcée est donné.
Activités
Applications industrielles de l’électrolyse (page 187)
1. a) +
A
mA COM
Courant e–
A C
b) Couples donnés : H2O(l)/H2(g) ; Cl–(aq)/Cl2(g).
– Réduction à la cathode C :2 H2O(l) + 2 e
– = H2(g) + 2 HO–(aq).
– Oxydation à l’anode A :2 Cl–(aq) = Cl2(g) + 2 e
–.– Équation globale :2 H2O(l) + 2 Cl
–(aq) = H2(g) + 2 HO–(aq) + Cl2(g).
ACTIVITÉ
1
ACTIVITÉ
1 c) En mélangeant les produits formés à l’anode et à la cathode, il se produit la transformation chimique d’équation : Cl2(aq) + 2 HO
–(aq) + 2 Na+(aq)= 2 Na+(aq) + Cl–(aq) + ClO–(aq) + H2O(l).
La partie entre accolades correspond à une solu-tion aqueuse d’eau de Javel.
2. a) +e– Courant
NiGlassmastermétallisé
Ni2+(aq)
NO3– (aq)
b) À l’anode (nickel) :Ni(s)an = Ni
2+(aq)an + 2 e–.
À la cathode (glassmaster), on a un dépôt métalli-que de nickel :
Ni2+(aq)cat + 2 e– = Ni(s)cat.
Équation globale :Ni(s)an + Ni
2+(aq)cat = Ni2+(aq)an + Ni(s)cat.

117Chapitre 9 - Transformations forcées lors d’électrolyses
c) L’anode passe en solution sous forme de Ni2+(aq), d’où le nom impropre « d’anode soluble ».
3. L’électrozingage consiste à déposer du zinc sur un métal pour le protéger de l’oxydation comme le sont les toits en zinc, par exemple. Le nickelage protège et embellit les objets.
Électrolyses de l’eau acidifiée et d’une solution aqueuse de sulfate de sodium (page 188)
A Électrolyse de l’eau acidifiéeCette manipulation donne d’excellents résultats quantitatifs, que l’on peut retrouver par la théorie. Elle gagne à être effectuée avec une alimentation stabilisée en courant (réglable). Elle peut être aussi réalisée avec une alimentation stabilisée en tension (réglable), voire non réglable mais associé à un rhéostat.Un générateur de courant se règle de la manière sui-vante :– placer le potentiomètre d’intensité à zéro et le potentiomètre de tension au maximum ;– mettre ce générateur en position « marche » ;– court-circuiter la sortie à l’aide d’un fil de connexion ;– régler l’intensité à la valeur I souhaitée ;– mettre le générateur en position « arrêt » et ôter le court-circuit. La valeur I de l’intensité est enregis-trée. Le générateur est prêt à être utilisé.
1. D’après la loi des gaz parfaits, la pression p, le volume V, la température et la quantité de matière n d’un gaz sont liés par la relation :
p · V = n · R · T ; donc V = n · R · T/p.On a travaillé à 22 °C et 1 000 hPa.À T = 22 + 273 = 295 Ket sous p = 1 000 hPa = 1 000 × 102 (Pa),le volume d’une mole est :
V = 1 00 8 32 295
1000 102
, ,× ××
= 0,024 5 L ;
Vm = 24,5 L · mol–1.
2. a) Espèces présentes :
H2O(l), SO42–(aq), H+(aq) ou H3O
+(aq) (= H+ hydraté).
– À l’anode doit se produire une oxydation. Deux oxydations sont possibles :
2 H2O(l) = O2(g) + 4 H+(aq) + 4 e–.
2 SO42–(aq) = S2O8
2–(aq) + 2 e–.
ACTIVITÉ
2
ACTIVITÉ
2
– À la cathode peut se produire une réduction. Deux réductions sont possibles :
2 H+(aq) + 2 e– = H2(g)(soit 2 H3O
+(aq) + 2 e– = H2(g) + 2 H2O(l))SO4
2–(aq) + 4 H+(aq) + 2 e– = SO2(aq) + 2 H2O(l).
b) Des dégagements de dioxygène à l’anode (une bûchette présentant un point incandescent se ravive) et de dihydrogène à la cathode (petite déto-nation à la flamme) sont observés et mis en évi-dence.Les réactions ayant effectivement lieu ont pour équation :– à l’anode (+) : 2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e– ;– à la cathode (–) : 2 H+(aq) + 2 e– = H2(g).
(soit 2 H3O+(aq) + 2 e– = H2(g) + 2 H2O(l)).
3. Quantité d’électricité totale qui a traversé l’élec-trolyseur en fonction de la durée ∆t de l’électrolyse et de l’intensité du courant imposé : Q = I · ∆t (1)– Expression de la quantité d’électricité totale qui a traversé l’électrolyseur en fonction de la quantité d’électrons : Q = n(e–) · F (2)où n(e–) est la quantité de matière d’électrons ayant circulé en ∆t.(On pose souvent NA · e = F , la constante de Faraday (ou faraday), soit la valeur absolue de la charge d’une mole d’électrons = 9,65 × 104 C · mol–1.)
– Quantité (de matière) d’électrons n(e–) ayant cir-culé dans le circuit pendant le même temps :(1) et (2) ⇒ I · ∆t = n(e–) · NA · e ;
donc : n(e–) = I t
e
·
·
∆NA
= I t
F
· ∆.
4.Équation 2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e–
E.I. n 0 n' 0
E.int. n – 2 x x n' + 4 x 4 x
E.F. n – 2 xfinal xfinal n' + 4 xfinal 4 xfinal
E.I = état initial ; E.F. = état final ; n = beaucoup.
5. Quantité de dioxygène formé en fonction de l’avancement : n(O2) = xfinal.Quantité d’électrons libérés en fonction de l’avan-cement : n(e–) = 4 xfinal.Résultats expérimentaux :
∆t = 8 min 30 s = 510 s ; I = 0,30 A ;V(O2)exp = 10,0 mL ; V(H2)exp = 20,2 mL.

118 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
n(O2) = xfinal = n( )–e
4 =
I t
e
·
·
∆4NA
;
n(O2) = 0 30 510
4 6 02 10 1 60 1023 19
,
, , –
×× × × ×
= 4,0 × 10–4 mol.V(O2)th = n(O2) · Vm ;V(O2)th = 4,0 × 10–4 × 24,5 = 9,8 × 10–3 L.V(O2)th = 9,8 mL,à comparer à V(O2)exp = 10,0 mL.L’écart relatif entre les deux valeurs est faible :
0 2
10
, × 100 = 2 %.
6. 2 H2O(l) = O2(g) + 2 H2(g).
7.Équation 2 H2O(l) = O2(g) + 2 H2(g)
E.I. n 0 0
E.int. n – 2 · x x 2 x
E.F. n – 2 xfinal xfinal 2 xfinal
n = beaucoup ; E.int. = état intermédiaire.
V
V
(H )
(O )2 exp
2 exp
= 20 2
10 0
,
, = 2,02
et V
V
(H )
(O )2 th
2 th
= 2 final
final
x
x = 2,00.
Il y a une bonne adéquation entre les rapports des volumes expérimentaux et théoriques.
B Électrolyse de la solution aqueuse de sulfate de sodium
Cette manipulation peut être réalisée sur le bureau, ou en binômes ou, mieux en quadrinômes.Elle peut être conduite par la démarche d’investiga-tion.Choisir au départ de l’eau distillée afin que le BBT soit vert. Ajouter, si nécessaire, quelques gouttes de solution aqueuse diluée d’hydroxyde de sodium (avant de séparer l’eau distillée dans les deux élec-trolyseurs).• Situation déclenchante.• Question.Établir les équations chimiques des réactions qui modélisent la transformation électrochimique observée à chaque électrode ainsi que globalement.
Hypothèses1. Espèces chimiques initialement présentes dans les solutions aqueuses : Na+(aq) ; SO4
2–(aq) ; H2O(l).
2. (H1) : 2 H2O(l) = O2(g) + 4 H+(aq) + 4 e–.
(H2) : 2 SO42–(aq) = S2O8
2–(aq) + 2 e–.
3. (H3) : 2 H2O(l) + 2 e– = H2(g) + 2 HO
–(aq) (H4) : SO4
2–(aq) + 4 H+(aq) + 2 e–
= SO2(aq) + 2 H2O(l). (H5) : Na
+(aq) + e– = Na(s).
4.Validation ou invalidation des hypothèses H1 et H2
À l’anode, le bleu de bromothymol vire au jaune, donc la solution aqueuse est acide. On observe aussi un dégagement gazeux.On valide donc l’hypothèse H1 :
2 H2O(l) = O2(g) + 4 H+(aq) + 4 e–.
Validation ou invalidation des hypothèsesH3, H4 et H5
À la cathode, le bleu de bromothymol vire au bleu, donc la solution aqueuse est basique. On observe aussi un dégagement gazeux. On valide donc l’hypothèse H3 :
2 H2O(l) + 2 e– = H2(g) + 2 HO
–(aq) (× 2)
Équation globaleOn ajoute membre à membre H1 et 2 H3 :2 H2O(l) + 4 H2O(l) + 4 e
–
= 2 H2(g) + 4 HO–(aq) + O2(g) + 4 H
+(aq) + 4 e– ;6 H2O(l) = 2 H2(g) + O2(g) + 4 HO
–(aq) + 4 H+(aq) ;6 H2O(l) = 2 H2(g) + O2(g) + 4 H2O(l) ;
soit 2 H2O(l) = 2 H2(g) + O2(g).
Prolongements5 & 6. On doit proposer une expérience qui mon-trera que les deux produits de la réaction électro-chimique globale ont été formés en quantité équivalente dans cette électrolyse.Réaliser cette expérience complémentaire en mélangeant dans un grand bécher, les deux solu-tions aqueuses jaune et bleu, on obtient une solu-tion aqueuse verte, donc neutre. Cela traduit le fait que les ions H+(aq) et HO–(aq) étaient produits en quantités équivalentes, comme le montre l’équa-tion chimique globale avant simplification.
•
•
•
•
•

119Chapitre 9 - Transformations forcées lors d’électrolyses
Exemplede transformation forcée (page 190)
Cette activité incontournable, peut être réalisée en cours pour la partie A et en cours ou en TP pour l’électrolyse proprement dite. On ne donne pas d’eau de dibrome concentrée aux élèves.
A Transformation spontanéeL’expérience
L’eau de dibrome utilisée doit être concentrée si l’on veut voir la couleur verte apparaître (mélange de l’excès de dibrome orangé et des ions cuivre (II) bleus apparus dans la solution aqueuse).L’extraction de l’excès de dibrome au cyclohexane permet d’obtenir une solution aqueuse plus ou moins bleue. On peut y caractériser les ions bro-mure par test au nitrate d’argent.Attention : Travailler en erlenmeyer bouché pour éviter de respirer les vapeurs de dibrome.
Si on part d’eau de dibrome peu concentrée, on ne verra pas la couleur bleue des ions cuivre (II).
Orangé
a b c
Orangé
Vert Bleu
Action du dibrome sur le cuivre métal : a, avant ; b, après : le cuivre a disparu et la solution a verdi ; c, test de Br2(aq)(couleur orangée) ; test de Br–(aq)
avec Ag+(aq) (précipité de AgBr(s)).
1. Oxydation : Cu(s) = Cu2+(aq) + 2 e–. Réduction : Br2(aq) + 2 e
– = 2 Br–(aq).
2. Cu(s) + Br2(aq) = Cu2+(aq) + 2 Br–(aq).
B Transformation forcéeL’expérience
+
A
mA COM
15 V
A C
Br– Cu2+
Courant e–
•
•
ACTIVITÉ
3
ACTIVITÉ
3 Cette expérience se réalise dans un tube en U, avec une solution aqueuse de bromure de cuivre (II), de concentration molaire apportée 0,50 mol · L–1. Cette solution est constituée d’un mélange à volumes égaux de solutions aqueuses de sulfate de cuivre (II) et de bromure de potassium (I). La tension utili-sée doit être voisine de 15 V pour obtenir un résultat rapide et, surtout, éviter de respirer du dibrome.
ObservationsAprès quelques minutes de fonctionnement :– du cuivre métal (de couleur rougeâtre) se dépose sur l’électrode de graphite reliée au pôle moins du générateur ;– autour de l’électrode reliée au pôle plus du géné-rateur apparaît une coloration verte. C’est la résul-tante de la couleur cyan, dû à l’ion cuivre (II), et du rouge orangé, dû au dibrome formé.
1. Espèces chimiques initialement présentes :Cu2+(aq) ; Br–(aq) ; solvant H2O(l) ; C (électrodes).
2. Dans les fils, les électrons vont de la borne néga-tive du générateur à sa borne positive.Dans la solution aqueuse, les ions bromure Br–(aq) vont dans le même sens, les ions cuivre (II) dans le sens inverse.
3. À l’électrode A (anode) : c’est de cette électrode, reliée au pôle plus du générateur, que partent donc les électrons vers le fil. Il s’y produit une oxydation, laquelle fournit ces électrons :
2 Br–(aq) = Br2(aq) + 2 e– ;
Intervention du solvant :2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e–.Intervention de l’électrode : elle est ici inatta-quable (graphite).
4. À l’électrode C (cathode).C’est par cette électrode, reliée au pôle moins du générateur, qu’arrivent les électrons dans l’élec-trolyseur. Il s’y produit une réduction, qui consomme ces électrons :
Cu2+(aq) + 2 e– = Cu(s).Intervention du solvant :
2 H2O(l) + 2 e– = H2(g) + 2 HO
–(aq).Intervention de l’électrode : elle est ici inatta-quable (graphite).
5. À l’anode : 2 Br–(aq) = Br2(aq) + 2 e–.
À la cathode : Cu2+(aq) + 2 e– = Cu(s).
6. 2 Br–(aq) + Cu2+(aq) = Br2(aq) + Cu(s).
•

120 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
Le sens de cette transformation est l’inverse de celui de la transformation chimique spontanée :
Cu2+(aq) + 2 Br–(aq) = Cu(s) + Br2(aq).Cette transformation n’est possible que grâce à l’apport d’énergie du générateur auxiliaire (par tra-vail électrique). Cette électrolyse est une transfor-mation forcée.
Les accumulateurs (page 191)
1. Pendant une conversation téléphonique, les accumulateurs de téléphone portable fonctionnent en pile : ils alimentent les circuits électroniques du téléphone.Pendant le temps de liaison à une prise secteur par l’intermédiaire d’un petit transformateur, les accu-mulateurs se rechargent grâce à l’énergie fournie par le secteur.
2. Le transformateur abaisse la tension du secteur (230 V) à une valeur de quelques volts.
3. Lorsque l’on actionne le démarreur de l’automo-bile, l’accumulateur fonctionne en pile. (On rap-pelle que le démarreur est un petit moteur électrique qui permet de lancer le moteur thermi-que de la voiture.)
4.
e– Courant
Pôle– Pôle+
SO42–(aq)
Pb(s) PbO2(s)
H3O+(aq)
Accumulateur au plomb qui débite dans une résistance.
5. a) Au pôle plus de cet accumulateur :PbO2(s) + SO4
2–(aq) + 4 H+(aq) + 2 e– = PbSO4(s) + 2 H2O(l).
b) Au pôle moins de cet accumulateur :Pb(s) + SO4
2–(aq) = PbSO4(s) + 2 e–.
c) Globalement :PbO2(s) + 2 SO4
2–(aq) + 4 H+(aq) + Pb(s) + 2 e–
= 2 PbSO4(s) + 2 H2O(l) + 2 e– ;
soit : PbO2(s) + 2 SO42–(aq) + 4 H3O
+(aq) + Pb(s)= 2 PbSO4(s) + 6 H2O(l).
ACTIVITÉ
4
ACTIVITÉ
4
6. a) Les accumulateurs d’une automobile sont rechargés lorsque la voiture roule : le moteur entraîne un alternateur qui produit un courant alternatif, lequel redressé, recharge la batterie.
b) e– Courant
Pôle– Pôle+
SO42–(aq)
Pb(s) PbO2(s)
H3O+(aq)
+
c) Le pôle positif est toujours l’électrode de PbO2(s), pendant la charge comme pendant la décharge.
7. a) À l’anode de l’accumulateur (électrode cou-verte de PbO2(s)), on a une oxydation :PbSO4(s) + 2 H2O(l)
= PbO2(s) + SO42–(aq) + 4 H+(aq) + 2 e–.
PbO2 est régénéré.À la cathode de plomb, on a une réduction :
PbSO4(s) + 2 e– = Pb(s) + SO4
2–(aq).Le plomb est régénéré.Globalement :2 PbSO4(s) + 6 H2O(l)
= PbO2(s) + 2 SO42–(aq) + 4 H3O
+(aq) + Pb(s).Toutes ces équations sont inversées par rapport au cas du fonctionnement en pile (décharge).
Charge
2 PbSO4(s) + 6 H2O(l) = PbO2(s) + 2 SO42–(aq) + 4 H3O
+(aq) + Pb(s).
Décharge
b) Cette transformation d’électrolyse est une transformation électrochimique forcée : c’est le générateur de charge qui force les électrons à cir-culer dans le sens observé.

121Chapitre 9 - Transformations forcées lors d’électrolyses
Activité supplémentaire
Les accumulateurs portablesnickel-métal hydrure (Ni-MH)
Cette activité fait suite à l’activitédu manuel élève (cf. p. 191).
Les accumulateurs portables « cadmium-nickel », qui représentaient le tiers du marché dans cette catégorie, sont appelés à disparaître dorénavant puisqu’une directive européenne de juillet 2006 interdit l’utilisation du cadmium. Cette interdiction devrait favoriser l’emploi d’accumulateurs Ni-MH, à moins que les accumulateurs « lithium-polymère » ou lithium-air ne deviennent prédominants. La demi-équation électronique qui intervient à la cathode lors de la charge est :
M(s) + H2O(l) + e– = MH(s) + HO–(aq).À l’anode intervient le couple NiOOH(s)/Ni(OH)2(s), en milieu basique. M représente un métal.
1. Établir la demi-équation électronique man-quante ainsi que l’équation chimique globale modé-lisant les transformations électrochimiques se produisant pendant la charge d’un accumulateur Ni-MH.2. Même question pendant la décharge.
CorrigéM(s) + H2O(l) + e
– = MH(s) + HO–(aq).NiOOH(s) + H2O(l) + e
– = Ni(OH)2(s) + HO–(aq).
• Pendant la charge (fonctionnement en électrolyseur)– À la cathode M, d’après le texte, la réduction :
M(s) + H2O(l) + e– = MH(s) + HO–(aq).
Cette électrode reçoit donc les électrons depuis le générateur auxiliaire ; elle est négative.– À l’anod e (positive), on a une oxydation :
Ni(OH)2(s) + HO– (aq) = NiOOH(s) + H2O(l) + e
–.– Équation globale :
M(s) + Ni(OH)2(s) = MH(s) + NiOOH(s).
• Pendant la décharge (fonctionnement en pile)
On inverse toutes les équations chimiques.– Sur M, on a une oxydation :
MH(s) + HO–(aq) = M(s) + H2O(l) + e–.
Les électrons quittent donc l’électrode M vers le circuit extérieur. Le métal M est le pôle moins de la cellule (qui fonctionne en pile).– Au pôle plus, on a une réduction :
NiOOH(s) + H2O(l) + e– = Ni(OH)2(s) + HO
–(aq).– Équation globale :
MH(s) + NiOOH(s) = M(s) + Ni(OH)2(s).
1 QCM, une seule réponse possible1. c) Lors d’une électrolyse, Qr s’éloigne de K.2. a) L’électrode siège de l’oxydation s’appelle l’anode.3. a) Dans une électrolyse, les électrons sont pro-duits à l’anode.4. a) Un électrolyseur est un récepteur.5. b) La réaction d’une électrolyse est forcée.6. b) Un électrolyseur transforme de l’énergie élec-trique en énergie chimique.
2 Électrolyse d’une solution de bromure de cuivre (II)
1. Br–(aq) ; Cu2+(aq) ; H2O(l) ; HO–(aq) ; H3O
+(aq).
2. Réactions d’oxydation à l’anode :2 Br–(aq) = Br2(l) + 2 e
–.2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e–.
Réactions de réduction à la cathode :Cu2+(aq) + 2 e– = Cu(s).2 H+(aq) + 2 e– = H2(g).
3. Cu2+(aq) + 2 Br–(aq) = Cu(s) = Br2(l).
3 Électrolyse d’une solution de chlorure de nickel (II)
1. G
+ –
Anode2 Cl–(aq) =Cl2(g) + 2 e
–
CathodeNi2+(aq)
+ 2 e– = Ni(s)
e–I
2. À la cathode, on observe la formation d’un dépôt de nickel.
Corrigés des exercices (page 192)
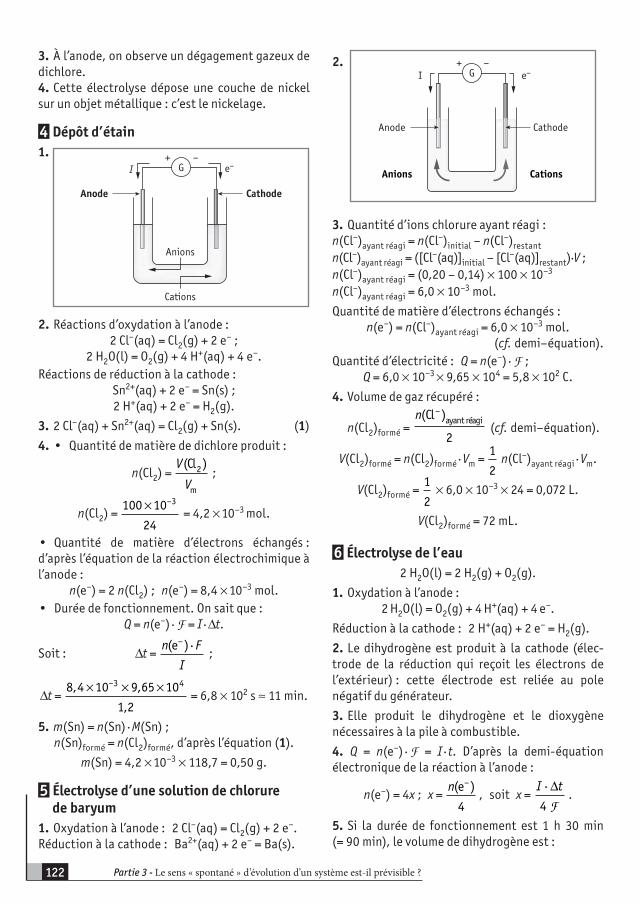
3. À l’anode, on observe un dégagement gazeux de dichlore.4. Cette électrolyse dépose une couche de nickel sur un objet métallique : c’est le nickelage.
4 Dépôt d’étain1.
G+ –
Anode
Anions
Cations
Cathode
e–I
2. Réactions d’oxydation à l’anode :2 Cl–(aq) = Cl2(g) + 2 e
– ;2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e–.Réactions de réduction à la cathode :
Sn2+(aq) + 2 e– = Sn(s) ;2 H+(aq) + 2 e– = H2(g).
3. 2 Cl–(aq) + Sn2+(aq) = Cl2(g) + Sn(s). (1)4. • Quantité de matière de dichlore produit :
n(Cl2) = V
V
(Cl )2
m
;
n(Cl2) = 100 10
24
3× –
= 4,2 × 10–3 mol.
• Quantité de matière d’électrons échangés :d’après l’équation de la réaction électrochimique à l’anode :
n(e–) = 2 n(Cl2) ; n(e–) = 8,4 × 10–3 mol.• Durée de fonctionnement. On sait que :
Q = n(e–) · F = I · ∆t.
Soit : ∆t = n F
I
(e )– · ;
∆t = 8 4 10 9 65 10
1 2
3 4, ,
,
–× × × = 6,8 × 102 s ≈ 11 min.
5. m(Sn) = n(Sn) · M(Sn) ;n(Sn)formé = n(Cl2)formé, d’après l’équation (1).
m(Sn) = 4,2 × 10–3 × 118,7 = 0,50 g.
5 Électrolyse d’une solution de chlorure de baryum
1. Oxydation à l’anode : 2 Cl–(aq) = Cl2(g) + 2 e–.
Réduction à la cathode : Ba2+(aq) + 2 e– = Ba(s).
2. –+G
Anions Cations
e–I
Anode Cathode
3. Quantité d’ions chlorure ayant réagi :n(Cl–)ayant réagi = n(Cl
–)initial – n(Cl–)restant
n(Cl–)ayant réagi = ([Cl–(aq)]initial – [Cl
–(aq)]restant)·V ;n(Cl–)ayant réagi = (0,20 – 0,14) × 100 × 10–3
n(Cl–)ayant réagi = 6,0 × 10–3 mol.
Quantité de matière d’électrons échangés :n(e–) = n(Cl–)ayant réagi = 6,0 × 10–3 mol.
(cf. demi–équation).Quantité d’électricité : Q = n(e–) · F ;
Q = 6,0 × 10–3 × 9,65 × 104 = 5,8 × 102 C.
4. Volume de gaz récupéré :
n(Cl2)formé = n( )Cl– ayant réagi
2 (cf. demi–équation).
V(Cl2)formé = n(Cl2)formé · Vm = 1
2 n(Cl–)ayant réagi · Vm.
V(Cl2)formé = 1
2 × 6,0 × 10–3 × 24 = 0,072 L.
V(Cl2)formé = 72 mL.
6 Électrolyse de l’eau2 H2O(l) = 2 H2(g) + O2(g).
1. Oxydation à l’anode :2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e–.
Réduction à la cathode : 2 H+(aq) + 2 e– = H2(g).
2. Le dihydrogène est produit à la cathode (élec-trode de la réduction qui reçoit les électrons de l’extérieur) : cette électrode est reliée au pole négatif du générateur.
3. Elle produit le dihydrogène et le dioxygène nécessaires à la pile à combustible.
4. Q = n(e–) · F = I · t. D’après la demi-équation électronique de la réaction à l’anode :
n(e–) = 4x ; x = n(e )–
4, soit x =
I t· ∆4 F
.
5. Si la durée de fonctionnement est 1 h 30 min (= 90 min), le volume de dihydrogène est :
122 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?

V(H2) = 2 x · Vm = I t V· ·∆ m
2 F ;
V(H2) = 10 10 90 60 24
2 9 65 10
3
4
× × × ×× ×,
= 6,7 × 103 L.
V(H2) = 6,7 m3.
7 Élaboration du cadmium
Exercice résolu dans le manuel de l’élève.
8 Électrode dite soluble1, 2 & 3
G+ –
AnodeCu(s) =
Cu2+(aq) + 2 e–
CathodeCu2+(aq)
+ 2 e– = Cu(s)
e–I
Anions
Cations
4. Au cours de l’électrolyse, la masse de l’anode diminue, car il se produit une oxydation du cuivre métallique.
5. De même la concentration en ions cuivre (II) ne varie pas au cours de l’électrolyse, car la même quantité est produite à l’anode et consommée à la cathode.
6. Il faut placer la lame de cuivre impur à l’anode pour la purifier : les impuretés tomberont au fond de l’électrolyseur.
7. Q = n(e–) · F = 2 n(Cu) · F = 2 (Cu)
(Cu)
m
M · F ;
Q = 3,6 × 104 C.
9 Accumulateur plomb-cuivre1. D’après l’écriture conventionnelle de cette pile, l’oxydation du plomb à l’électrode négative se pro-duit :
Pb(s) = Pb2+(aq) + 2 e–,
et à l’électrode positive, la réduction de l’ion cuivre II :
Cu2+(aq) + 2 e– = Cu(s).
Équation de la réaction chimique de la pile de constante d’équilibre K = 3,5 ×1015 :
Pb(s) + Cu2+(aq) = Pb2+(aq) + Cu(s).
2. Qr,i = [Pb (aq)]
[Cu (aq)]
2+
2+ ; Qr,i =
1 0
1 0
,
, = 1,0 < K.
Le système chimique évolue dans le sens direct.
3. & 4. +–
I
Pb
G
Cu
Électrolyte
CathodePb2+(aq)
+ 2 e– = Pb(s)
AnodeCu(s)
= Cu2+ (aq) + 2 e
–
5. Qr s’éloigne de K. 6. Ce dispositif agit aussi bien en générateur qu’en récepteur : c’est un accumulateur.
10 Corrosion1. Les ions zinc (II) doivent subir une réduction :
Zn2+(aq) + 2 e– = Zn(s).
2. La plaque d’acier, qui doit se recouvrir de zinc, correspond donc à la cathode.
3. Masse de zinc à déposer pour recouvrir une sur-face totale de 6,0 m² :
m(Zn) = 40,0 × 6,0 = 2,4 × 102 g.D’après la demi-équation électronique à la cathode (Zn2+(aq) + 2 e– = Zn(s)) :
n(Zn)formé = ½ n(e–).La quantité d’électricité est donc égale à :
Q = ne · F = 2 n(Zn)formé · F = 2m
M
(Zn)
(Zn)formé · F ;
Q = 2 × 2 4 10
65 4
2,
,
× × 9,65 × 104 = 7,1 × 105 C.
4. Q = I · ∆t, soit ∆t = Q/I ;Q = 7,1 × 105/(20 × 103) = 35,5 s.
11 Un coquetier en argent1. Le coquetier constitue la cathode.2. Équation de la réaction d’oxydation à l’anode :
2 H2O(l) = O2(g) + 4 H+(aq) + 4 e–.
Équations de réactions de réduction à la cathode :Ag+(aq) + e– = Ag(s).
2 H+(aq) + 2 e– = H2(g).
3. L’équation de la réaction électrochimique modé-lisant cette électrolyse est : 2 H2O(l) + 4 Ag
+(aq) = O2(g) + 4H+(aq) + 4 Ag(s).
123Chapitre 9 - Transformations forcées lors d’électrolyses

4. La concentration en ions argent (I) diminue au cours de cette électrolyse.5. a) Équation de la réaction électrochimique pouvant se produire à l’électrode d’argent :
Ag(s) = Ag+(aq) + e–.b) Équation modélisant la réaction de cette nou-velle électrolyse :
Ag(s) + Ag+ (aq) = Ag(s) + Ag+ (aq).c) La concentration en ions argent (I) ne varie donc pas.6. a) Quantité d’électricité : Q = I · ∆t = n(e–) · F .Quantité de matière d’électrons échangés :
n(e–) = I · ∆t/F.Masse d’argent déposée :m(Ag) = n(Ag) · M(Ag) = F n(e–) · M(Ag)
= (I · ∆t/F) · M(Ag) ;m(Ag) = 1,0 × 135 × 60 × 107,9/(9,65 × 104) = 9,1 g.b) L’électrolyse est plus longue, car une réaction électrochimique parasite doit avoir lieu.
12 Électrolyse d’une solution de chlorure de sodium
1. 2 Cl–(aq) = Cl2(g) + 2 e–.
2. La coloration rose de la phénolphtaléine met en évidence la présence d’ions hydroxyde, HO–(aq). Le « pop » caractérise la présence du dihydrogène.– L’eau est l’oxydant et les ions chlorure le réduc-teur de l’électrolyse.– Équation de réaction d’oxydation à l’anode, élec-trode A : 2 Cl–(aq) = Cl2(g) + 2 e
–.– Équation de réaction de réduction à la cathode, électrode B : 2 H2O(l) + 2 e
– = H2(g) + 2 HO–(aq).
– Équation de la réaction modélisant l’électrolyse : 2 Cl–(aq) + 2 H2O(l) = Cl2(g) + H2(g) + 2 HO
–(aq).
3.
État Av. 2 Cl–(aq) + 2 H2O(l)= Cl2(g) + H2(g) + 2 HO–(aq)
Électronséchangés
Initial 0 n1 n2 0 0 0 0
t x n1 – 2x n2 – 2 x x x 2x 2x
Final xf n1 – 2xf n2 – 2 xf xf xf 2xf 2xf
• n(Cl2) = V(Cl2)/Vm ;
n(Cl2) = 24 10
24
3× –
= 1,0 × 10–3 mol = 1,0 mmol.
• n(H2) = n(Cl2) = 1,0 mmol.• n(HO–) = 2 n(Cl2) = 2,0 mmol.• Quantité de matière d’électrons échangés : n(e–) = 2 n(Cl2) = 2,0 mmol.
• Quantité d’électricité : Q = n(e–) · F = I · t.
I = Q
t∆ =
n
t
(e )– ·F
∆ ;
I = 2 0 10 9 65 10
10 60
3 4, ,–× × ××
= 0,32 A.
4.
État Av. Cl2(g) + 2 HO–(aq)= ClO–(aq) + Cl–(aq) + H2O(l)
Électronséchangés
– initial 0n1 = 1,0
× 10–3n2 = 2,0
× 10–3 0 0 / 0
t x n1 – x n2 – 2x x x / 2x
– final xf n1 – xf n2 – 2xf xf xf / 2xf
On est dans les proportions stœchiométriques :
n1 = n2
2 · n(ClO–) = xf ; n1 = 1,0 × 10–3 mol.
[ClO–(aq)] = n
V
(ClO )– ;
[ClO–(aq)] = 1 0 10
0 200
3,
,
× = 5,00 × 10–3 mol · L–1.
13 Quel métal ?1. Quantité d’électricité : Q = I · ∆t = n(e–) · F .Quantité de matière d’électrons échangés :
n(e–) = I t
F
· ∆ ;
n(e–) = 0,350 × 3 600/96 500 = 1,31 × 10–2 mol.
2. Masse molaire du métal M(s).
État Avancement M2+(aq) + 2 e– = M(s)
– initial 0 n1 = 1,0 × 10–3 0 0
t x n1 – x 2 x x
– final xf n1 – xf 2 xf xf
n(M) = n(e )–
2 ; n(M) =
1 31 10
2
2, –× mol.
M(M) = m
n
(M)
(M) ; M(M) =
428 10
1 31 10
2
3
2
××
–
–, = 65,3 g.
3. Aux erreurs expérimentales près, le métal M est du zinc :
65 4 65 3
65 4 100
, ,
,
−×
= 0,15 %.
4. Volume de dichlore dégagé lors de cette électro-lyse, à P = 1 013 hPa et T = 18 °C.
124 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?

Équation chimique de la réaction d’électrolyse :2 Cl–(aq) + Zn2+(aq) = Cl2(g) + Zn(s).
n(Zn) = n(Cl2) = 1 31 10
2
2, –× mol et · V = n · R · T.
Soit : V = n T
P
(Cl ) R2 · · ;
V =
1 31 10
28 31 18 0 273 15
101 300
2,, ( , , )
–× × × + ;
V = 1,6 × 10–4 m–3 = 16 cL.
5. Concentration en ions chlorure à la fin de cette électrolyse.
[Cl–(aq)]f = n n
V
(Cl aq)) (Cl aq))–i
–f( (− ;
[Cl–(aq)]f = 060 0 5 1 31 10
0 5
2× − ×, ,
,
–
= 0,57 mol · L–1.
14 Électrolyse de l’acide iodhydrique1. Les espèces chimiques en solution sont :H3O
+(aq), I–(aq), H2O(l) et HO–(aq) (en quantité
négligeable).
2. Réactions d’oxydation à l’anode :2 I–(aq) = I2(g) + 2 e
– ;2 H2O(l) = O2(g) + 4 H
+(aq) + 4 e–.Réactions de réduction à la cathode :
2 H+(aq) + 2 e– = H2(g).
3. Équation chimique modélisant la réaction de cette électrolyse :
2 I–(aq) + 2 H+(aq) = I2(g) + H2(g).
4. a) Quantité d’électricité ayant traversé l’élec-trolyseur :Q = I · ∆t ; Q = 60 ×10–3 × 13 × 60 ; Q = 4,7 × 101 C.
b) Quantité de matière formée à l’anode :
n(I2) = n(e )–
2 =
Q
F2 ;
n(I2) = 4 7 10
2 9 65 10
1
4
,
,
×× ×
= 2,4 ×10–4 mol.
c) Le pH de la solution d’acide iodhydrique à l’ins-tant initial est :
pHi = – log ([H3O+(aq)]i) ;
pH = – log (1,0 × 10–2) = 2,0.d) Le pH de la solution d’acide iodhydrique à la fin de l’électrolyse :
[H3O+(aq)]f =
n n
V
(H O aq)) (H O aq))3+
i 3+
consommé( (− ;
= [H O aq)](H O aq))
3+
i3
+consommé(
(−
n
V
[H3O+(aq)]f = 1,0 × 10–2 – 2,4 × 10–4 ×
2
0 0500,
= 4,0 × 10–4 mol · L–1.
15 Déterminer la charge d’un ion
G
E1 E2
1. Les électrolyseurs E1 et E2 sont traversés par le même courant I, car ils sont montés en série.
2. Quantité d’électricité : qui a traversé l’électroly-seur E1 : Q1 = I · ∆t.La quantité d’électricité ayant traversé l’électroly-seur E2 est : Q2 = Q1 = I · ∆t.
3. La demi-équation électronique de la réaction modélisant la transformation électrochimique qui se produit à la cathode de l’électrolyseur E1 est :
Ag+(aq) + e– = Ag(s).
4. Quantité de matière d’électrons échangés dans l’électrolyseur E1 :
n(e–) = n(Ag, formé) = m
M
(Ag)
(Ag) ;
n(e–) = 1 80
107 9
,
, = 1,67 × 10–2 mol.
5. a) La quantité de matière d’électrons échangés dans l’électrolyseur E2 est la même que celle dans l’électrolyseur E1.b) Demi-équation électronique de la réaction modé-lisent la transformation électrochimique forcée qui se produit à la cathode de l’électrolyseur E2 :
Aun+(aq) + n e– = Au(s).c) Quantité de matière d’or déposée :
n(Au) = m
M
(Au)
(Au) ; n(Au) =
1 10,
197,0 = 5,58 × 10–3 mol.
d) Relation liant la quantité de matière d’électrons échangés et la quantité de matière d’or déposée :
n
n
(e )– = n(Au),
avec n charge de l’ion, donc nombre entier.e) La charge de l’ion or est :
n = n
n
(e )
(Au)
–
; n = 1,67 × 10–2/5,58 × 10–3 = 3,00.
L’ion or est l’ion or (III) : Au3+.
125Chapitre 9 - Transformations forcées lors d’électrolyses

126 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
16 Électroétamage1. Réactions d’oxydation à l’anode :
2 H2O(l) = O2(g) + 4H+(aq) + 4 e–.
Réactions de réduction à la cathode :Sn2+(aq) + 2 e– = Sn(s).
2. Surface à étamer :S = [(10 + 4,5) × 2] × 9,5 × 2 ;
S = 5,5 × 102 cm2 = 5,5 × 10–2 m2.(Il y a deux faces à étamer.)
3. Quantité d’étain à déposer :
n(Sn) = m
M
(Sn)
(Sn) ;
n(Sn) = 5 5 10 0 502, ,–× ×
118,7 = 2,3 × 10–4 mol.
Quantité de matière d’électrons échangés :n(e–) = 2 n(Sn) ; n(e–) = 4,6 × 10–4 mol.
Durée de l’électrolyse :
Q = I · ∆t, soit ∆t = n F
I
(e )– · =
2(Sn)
(Sn)
m
MF
I
·
.
∆t = 5 5 10 0 5 2 9 65 10
118 74 3 0
2 4, , ,
, ,
–× × × × ××
= 15 s.
4. La perte de masse de l’anode est égale à la masse déposée si l’étain de l’anode est pur, soit :
5,5 × 10–2 × 0,5 = 2,8 × 10–2 g.
5. La concentration ion étain (II) ne varie pas, car les ions étain consommés à la cathode sont pro-duits en même quantité à l’anode.
6. Lors de l’électrolyse, le système chimique ne res-pecte pas le critère d’évolution spontanée, car Qr s’éloigne de K.
17 Constante d’Avogadro
Exercice résolu dans le manuel de l’élève.
18 À propos du zinc
G+ –
Interrupteur K
Zinc Fer
Dépôt métallique
Dégagementde dihydrogène
Solution aqueusede sulfate de zinc
e–I
1. a) Les porteurs de charges sont les électrons. Dans les fils de connexion, ils circulent de l’élec-trode de zinc vers l’électrode de fer.En solution aqueuse, les cations, Zn2+(aq) et H3O
+(aq), sont attirés par l’électrode de fer (qui est négative) ; les anions, SO4
2–(aq) et HO–(aq), le sont attirés par l’électrode de zinc (laquelle est posi-tive).b) À l’électrode de fer, deux réductions sont pos-sibles :Zn2+(aq) + 2 e– = Zn(s) ; 2 H+(aq) + 2 e– = H2(g).
Ceci est en accord avec les observations expéri-mentales : le dégagement de dihydrogène et la for-mation du dépôt métallique.Cette électrode est le siège de réactions de réduc-tions : c’est la cathode.c) Le dépôt de zinc protège le fer de la corrosion.d) La masse de l’électrode de zinc diminue en rai-son de l’oxydation : Zn(s) = Zn2+(aq) + 2 e–.
2. a) Oxydation anodique : Zn(s) = Zn2+(aq) + 2 e–.Réduction cathodique : Zn2+(aq) + 2 e– = Zn(s).Bilan de l’électrolyse :
Zn(s) + Zn2+(aq) = Zn2+(aq)+ Zn(s).b) Quantité d’électricité :
Q = I · ∆t ; Q = 0,5 × 10 × 60 = 3,0 × 102 C.On sait que Q = n(e–) · F , soit :
n(e–) = Q/F ;n(e–) = 3,0 × 102/9,65 × 104 = 3,1 × 10–3 mol.
c) D’après la demi-équation électronique à l’anode :
n(Zn)disp = n(e )–
2.
d) La variation de masse de l’électrode de zinc est :∆m(Zn) = n(Zn)disp · M(Zn) = n(e–)/2 · M(Zn) ;
∆m(Zn) = ½ × 3,1 × 10–3 × 65,4 = 0,10 g.La masse de l’électrode de zinc diminue de 0,10 g.
19 Production de zinc1. Équation d’oxydation à l’anode :
2 H2O(l) = O2(g) + 4H+(aq) + 4 e–.
Équation de réduction à la cathode :Zn2+(aq) + 2 e– = Zn(s) ;2 H+(aq) + 2 e– = H2(g).
L’expérience montre un dégagement gazeux à une électrode et un dépôt à l’autre
2. Les porteurs de charges sont les électrons. Dans les fils de connexion, ils circulent de l’électrode positive vers l’électrode négative.

127Chapitre 9 - Transformations forcées lors d’électrolyses
En solution aqueuse, les cations, Zn2+(aq) et H3O
+(aq), sont attirés par l’électrode négative ; les anions, SO4
2–(aq) et HO–(aq), le sont par l’électrode positive.
+–e– I
Cathode Anode
AnionsCations
3. D’après l’expérience, il y a dégagement de dioxy-gène à l’anode et dépôt métallique à la cathode.Oxydation anodique :
2 H2O(l) = O2(g) + 4 H+(aq) + 4 e–.
Réduction cathodique : Zn2+(aq) + 2 e– = Zn(s) × 2Bilan de l’électrolyse :2 H2O(l) + 2 Zn
2+(aq) = O2(g) + 4 H+(aq) + 2 Zn(s).
4. Cette transformation électrochimique est une transformation forcée, car le générateur impose le sens du courant. Pour le vérifier, il faudrait calculer le quotient de réaction à plusieurs dates et montrer que ce quotient s’éloigne de la valeur de K.
5.Équation chimique
2 Zn2+(aq) + 2 H2O(l)= 2 Zn(s) + O2(g) + 4 H
+(aq)
– initial x = 0 n Excèsn(Zn)
= 0n(O2)
= 0Beaucoup
t x n – 2 x Excès 2 x x Beaucoup
– final xf n – 2 xf Excès 2 xf xf Beaucoup
L’eau est aussi le solvant : elle est présente en grande quantité, donc en excès. La solution de sul-fate de zinc (II) est acidifiée avec de l’acide sulfu-rique, qui dans l’état initial, apporte une grande quantité d’ions hydrogène H+(aq).
6. Quantité d’électricité : Q = I · ∆t = n(e–) · F .Quantité de matière d’électrons échangés :
n(e–) = 4 x, soit x = I t
F
· ∆2
.
7. D’après le tableau d’avancement :
n(Zn) = 2 x = I t
F
· ∆2
.
La masse obtenue après 48 h d’électrolyse est :
m(Zn) = n(Zn) · M(Zn) = 2 x · M(Zn) = I t
F
· ∆2
· M(Zn) ;
m(Zn) = 80 10 48 3 600
165
3× × ××
×2 05
= 4,5 × 106 g,
soit 4,5 tonnes, donc un ordre de grandeur de 106 g.
8. La masse de zinc réellement obtenue est plus faible.Il est possible que la seconde réduction à la cathode se produise ; on a donc une réaction parasite, ou l’intensité n’est pas constante au cours de l’électro-lyse.
9. Le rendement de la réaction étant η = 80 %, la quantité de dioxygène obtenue est :
n(O2) = η · x.
Or : n(O2) = V
V
(O )2
m
, soit x = V
V
(O )2
mη ·,
d’où I t
F
· ∆4
= V
V
(O )2
mη ·, donc V(O2) =
I t
F V
·
· ·
∆4 mρ
.
V(O2) = 80 10 48 3 600
40 80 24
3× × ××
× ×105
, ;
V(O2) = 20 × 48 × 36 × 0,80 × 24 ;V(O2) = 2 × 48 × 36 × 8 × 24 = 1,4 × 104 × 48 ;V(O2) = 6,6 × 105 L,donc un ordre de grandeur de 106 L.
20 Gravure d’un CD Lune1. L’électrode glassmaster constitue la cathode sur laquelle on dépose le nickel.L’électrode de nickel constitue l’anode soluble.
2. Électrode de nickel (anonde)
Ni(s) = Ni2+(aq) + 2 e–Glassmaster (cathode)Ni+(aq) + 2 e– = Ni(s)
Solution de sulfate de nickel
e– I
+
3. a) Volume du disque : V = π · (d/2)·e,avec d = diamètre et e = épaisseur.Masse du disque père :
ρ = m
V, soit m = ρ · V = ρ · π · d
2· e.

128 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
Toutes les longueurs sont en centimètre, car la masse volumique est en g · cm–3 :
m = 8,90 × π × 23 0
2
, × 3,05 × 10–2 = 113 g.
3. b) n(Ni) = m
M
père
(Ni) ; n(Ni) =
113
58 7, = 1,93 mol.
4. Q = n(e–) · F = I · ∆t.D’après l’équation de réduction cathodique :
n(e–) = 2n(Ni), soit I = 2 (Ni)n
t
·F
∆ ;
donc : I = 2 1 93 96
2 3
× ××
, 500
600 = 51,7 A.
21 Traitement de l’eau d’une piscine1.a) Les espèces chimiques présentes sont les ions Na+(aq) et Cl–(aq) (solution aqueuse de chlorure de sodium) et l’eau (H2O, H3O
+(aq) et HO–(aq)). Or, il se forme du dichlore et du dihydrogène. Les couples mis en jeu sont : H2O(l)/H2(g) et Cl2(g)/Cl
–(aq) .b) L’espèce chimique oxydée est l’ion chlorure, Cl–. c) Une oxydation se produit à l’anode qui est l’élec-trode reliée à la borne positive du générateur, ici l’électrode B. On obtient un dégagement gazeux de dichlore à cette électrode.
2. a)
Av. 2 H2O(l) + 2 Cl–(aq)
= H2(g) + Cl2(g) + 2 HO–(aq)
Quantité d’électrons échangés
(mol)
E.I. 0 Excès n(Cl–)i 0 0 0 0
t x Excès n(Cl–)i – 2 x x x 2 x 2 x
E.F. xf Excès n(Cl–)i – 2 xf xf xf 2 xf 2 xf
b) D’après le tableau :n(e–) = 2 x et n1(Cl2) = x, donc n(e–) = 2 n1(Cl2).
2. Équation chimique.
Cl2(g) + 2 HO–(aq) = H2O(l) + Cl
–(aq) + ClO–(aq).
D’après l’équation :n2(Cl2)consommé = n(ClO
–)formé.
4. La transformation associée à l’équation (2) étant considérée comme totale et rapide, tout le dichlore formé est consommé, donc :
n1(Cl2) = n2(Cl2)consommé,
soit n1(Cl2) = n(ClO–)formé.
5. Q = I · ∆t = n(e–) · F ,
soit n(e–) = I t· ∆
F = 2 n(ClO–)formé
d’après la question précédente.On obtient donc :
n(ClO–(aq)formé) = I t· ∆2F
;
n(ClO–(aq)formé) = 20 3600
2 1 0 105
×× ×,
= 10 3600
1 0 105
××,
n(ClO–(aq)formé) = 3 6 10
1 0 10
4
5
,
,
××
= 0,36 mol.

129Chapitre 9 - Transformations forcées lors d’électrolyses
Exercices supplémentaires
1. Accumulateur au plombLes accumulateurs au plomb sont très utilisés dans l’industrie automobile.Un accumulateur au plomb est constitué d’une élec-trode en plomb et d’une électrode en plomb recou-verte d’oxyde de plomb ; l’électrolyte est une solution d’acide sulfurique, 2 H+(aq) + SO4
2–(aq), de concen-
tration C = 6,0 mol · L–1 contenant un précipité de sulfate de plomb (PbSO4).Données
Couples oxydant/réducteur :PbO2(s)/PbSO4(s) ; PbSO4(s)/Pb(s).Masse molaire : M(PbO2) = 239,2 g · mol–1.
•
•
Les compétences expérimentales
(page 200)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de revenir sur des techniques de base que l’on retrouve de manière récurrente dans les sujets d’évaluation. Cet exercice doit conduire l’élève à justifier ses choix.
Corrigé1. RéalisationMasse à dissoudre : m(ZnSO4) = C · V · M(ZnSO4) ;
m(ZnSO4) = 0,50 × 50 × 10–3 × (65,4 + 32,1 + 4 ×16,0) = 4,0 g.– Placer une coupelle sur la balance.– Tarer puis peser 4,0 g de sulfate de zinc (II).– À l’aide d’un entonnoir, introduire le sulfate de zinc dans une fiole jaugée de 50 mL.– Avec de l’eau distillée rincer la coupelle et l’entonnoir.– Continuer de verser de l’eau distillée jusqu’au ¾ puis agiter de façon à dissoudre le composé.– Compléter d’eau distillée jusqu’au trait de jauge.– Agiter pour homogénéiser.
2. Exploitation1.
+–e– I
CathodeZn
A G
AnodeZn
AnionsCations
2. Oxydation anodique : Zn(s) = Zn2+(aq) + 2 e–. Réduction cathodique : Zn2+(aq) + 2 e– = Zn(s).3. Masse de zinc attendue :4. m(Zn)expérimentale < m(Zn)attendue.
5. L’écart s’explique soit par l’existence d’une réaction parasite, soit du fait d’une intensité non constante.
6. La masse de zinc perdue à l’anode sera égale à celle formée à la cathode si l’anode est en zinc pur ; elle sera inférieure si l’électrode n’est pas en zinc pur.

130 Partie 3 - Le sens « spontané » d’évolution d’un système est-il prévisible ?
1. Lorsqu’il fonctionne en générateur, l’équation de la réaction se produisant à l’électrode recouverte d’oxyde de plomb est :PbO2(s) + 2 SO4
2–(aq) + 4 H+(aq) + 2 e–
= PbSO4(s) +2 H2O(l).a) Indiquer les polarités des électrodes.b) Donner l’équation de la réaction qui se produit à l’électrode de plomb.c) En déduire l’équation de la réaction se produi-sant lorsque l’accumulateur débite.2. Donner l’écriture conventionnelle de cet accu-mulateur.3. Montrer que la capacité de l’accumulateur est proportionnelle à la quantité d’ions oxonium.4. Au cours de sa charge, l’accumulateur est monté en opposition avec le générateur qui impose le sens du courant.a) Faire un schéma en indiquant le nom des électro-des.b) Donner l’équation de la réaction correspondant à la charge de l’accumulateur. Cette réaction est-elle spontanée ?5. En considérant les couples donnés, quelle autre électrolyse réalise-t-on pendant la charge de l’accu-mulateur ?6. Si on réalise plusieurs charges et décharges, quel inconvénient suscite cette électrolyse parasite ? Comment y remédier ?7. Expliquer pourquoi, lors de la charge d’un accu-mulateur, ce dernier doit être ouvert.8. Pour être rechargé, un accumulateur doit fournir une quantité d’électricité de 110 A.h. Quelle sera la durée de la recharge si le courant a une intensité constante de 12 A ?9. En réalité, la durée de cette recharge est plus lon-gue : pourquoi ?10. Quelle masse d’oxyde de plomb forme-t-on si l’on ne tient pas compte de la réaction parasite ?
Corrigé1. a) L’électrode recouverte d’oxyde de plomb est le siège d’une réduction (elle reçoit donc les élec-trons) ; elle constitue donc la borne positive de la pile.L’électrode de plomb est le siège d’une oxydation : elle constitue la borne négative.
b) L’équation de la réaction qui se produit à l’élec-trode de plomb :
Pb(s) + SO42–(aq) = PbSO4(s) + 2 e
–.
c) L’équation de la réaction se produisant lorsque l’accumulateur débite :PbO2(s) + Pb (s) + 4 H
+(aq) + 2 SO42–(aq)
= 2 PbSO4(s) + 2 H2O(l).
2. Pb(s)/PbSO4(s)//électrolyte acide sulfurique//PbSO4(s)/PbO2(s).
3. C = Qmax = n(e–) · F =
n
F
(H )+
2.
La capacité est bien proportionnelle à la quantité d’ions oxonium H+(aq).
4. En charge :a)
+–
I
Pb
G
PbO2
Électrolyte
CathodePb2+(aq)
+ 2 e– = Pb(s)
AnodePb2+(aq) + 2 H2O= PbO2 + 4H
+ + 2 e–
b) 2 PbSO4(s) + 2 H2O(l)= PbO2(s) + Pb(s) + 4 H
+(aq) + 2 SO42–(aq).
Cette réaction est forcée, car le générateur impose le sens du courant.
5. Il se produit également l’électrolyse de l’eau :2 H2O(l) = O2(g) + 2 H2(g).
6. Si cette électrolyse parasite survient, on doit constater une diminution de la quantité d’eau. Il faut donc ajouter de l’eau au bout de plusieurs recharges.7. L’accumulateur doit être ouvert pour éliminer les gaz produits par l’électrolyse parasite :
O2(g) + 2 H2(g).
8. Quantité d’électricité :Q = 110 A.h = 110 × 3 600 = 3,96 × 105 A.s ;
Q = 3,96 × 105 C.Durée de la recharge si le courant a une intensité constante de 12 A :
∆t = Q
I ;
I · ∆t = 3 96 10
12
5, × = 3,3 × 104 s.
9. La durée de la recharge est plus longue, soit du fait de la réaction parasite, soit parce que l’inten-sité qui n’est pas constante.

131Chapitre 9 - Transformations forcées lors d’électrolyses
10. m(PbO2) = n(PbO2) · M(PbO2) = Q
F2 · M(PbO2) ;
m(PbO2) = 3 96 10
2
5, ×× 96 500
× 239,2 + 2 × 16,0) ;
m(PbO2) = 5,5 × 102 g.
2. Pièce de monnaieI. Dans l’industrie monétaire, on cuivre une ron-delle d’acier, appelée « flan », pour obtenir certaines pièces de monnaie comme les pièces de 1, 2 et 5 cen-times d’euros.Après avoir subi plusieurs dégraissages chimiques et électrolytiques, suivis de différents rinçages, le cui-vrage du « flan » s’effectue par électrolyse d’une solu-tion de nitrate de cuivre (II), Cu2+(aq) + 2 NO3
–(aq).1. L’électrolyse est-elle une transformation chimi-que forcée ou spontanée ? Justifier la réponse.2. Écrire la demi-équation électronique modélisant la réaction qui a lieu au niveau de la rondelle métallique.3. À quelle borne du générateur est reliée la rondelle ?4. Ce « flan » constitue-t-il l’anode ou la cathode de l’électrolyseur ?5. Pour maintenir constante la concentration en ions cuivre (II), Cu2+(aq), dans l’électrolyte, que doit-on faire ?II. En fait, le cuivrage s’effectue, à 60 °C, sur un tonneau dans lequel peuvent se trouver 80 kg de ron-delles d’acier, soit environ 18 000 rondelles.Pour une pièce de 5 centimes d’euros, la surface totale (les deux faces incluses !) à cuivrer est d’envi-ron 9,2 cm2. On souhaite que l’épaisseur du dépôt soit d’au moins 25 µm ± 5 µm.Données
Masse volumique du cuivre : ρ = 8 960 kg · m–3.Masses molaires atomiques :M(N) = 14,0 g · mol–1 ; M(O) = 16,0 g · mol–1 ; M(Cu) = 63,5 g · mol–1.Charge d’une mole d’électrons : 1 F = 96 500 C · mol–1.
6. Déterminer la masse de cuivre à déposer sur une rondelle d’acier.7. Pour un lot de 80 kg, quelle quantité de cuivre d’environ faut-il ?8. Quelle quantité d’électricité doit circuler pour réaliser ce dépôt ?9. L’intensité du courant est constante et égale à 1 200 A : quelle est la durée de l’opération ?
••
•
Corrigé1. Le générateur impose le sens de déplacement des électrons. L’apport d’énergie électrique du générateur permet à la transformation chimique de se produire. C’est une transformation forcée.
2. On réalise un dépôt de cuivre sur le « flanc ». La demi-équation électronique est :
Cu(s) = Cu2+(aq) + 2 e–.
3. L’ion cuivre (II) est réduit, la réduction néces-site un apport d’électrons. La rondelle est donc reliée à la borne – du générateur.
4. Au niveau du « flanc », il se produit une réduc-tion des ions cuivre (II). Le flanc constitue donc la cathode de l’électrolyseur.5. Pour maintenir la concentration en ions cuivre (II), il faut produire ces ions à l’anode en oxydant des atomes de cuivre. À l’anode, il se produirait l’oxydation des atomes de cuivre :
Cu(s) = Cu2+(aq) + 2 e–.Chaque fois qu’un ion Cu2+(aq) est consommé à la cathode, un ion Cu2+(aq) est formé à l’anode. La concentration en ion Cu2+(aq) reste alors constante.
6. Le volume de cuivre à déposer est : V = e · S.La masse de cuivre est :m = ρ · V, soit m = ρ · e · S ;m = 8 960 × 25 × 10–6 × 9,2 × 10–4 ;m = 2,06 × 10–4 kg = 0,206 g = 206 mg.
7. Pour le lot de 80 kg, soit 18 × 103 rondelles, il faudra :
m' = 18 × 103 × m = 3,71 kg.
8. Pour qu’un atome de cuivre se dépose sur la ron-delle, il faut 2 électrons. Donc :
Q = 2 n(Cu) · F = 2 m
M
'(Cu)
(Cu) · F ;
Q = 2 × 3 71 10
63 5
3,
,
× × 96 500 = 1,13 × 107 C.
9. Q = I · ∆t, soit ∆t = Q
I ;
∆t = 1 13 10
1 200 60
7, ××
= 157 min.

132 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
D é c ouv r i r qu e l e s transformations fai-sant intervenir des réactions d ’estérif i-cation et d’hydrolyse sont lentes, qu’elles conduisent à un état d’ équilibre chimique et qu’ il est possible de modifier la vitesse et/ou le taux d’avan-cement f inal de ces réactions.
1. Les réactions d’estérification et d’hydrolyse– Formation d’un ester à partir d’un acide et d’un alcool, écriture de l’équation chimi-que de la réaction modélisant cette transfor-mation, appelée réaction d’estérification.– Hydrolyse d’un ester, écriture de l’équa-tion chimique de la réaction correspon-dante.– Mise en évidence expérimentale d’un état d’équilibre chimique lors des trans-formations faisant intervenir des réactions d’estérification et d’hydrolyse.– Définition du rendement d’une trans-formation– Définition d’un catalyseur– Contrôle de la vitesse de réaction : tem-pérature et catalyseur– Contrôle de l’état final d’un système : excès d ’un réactif ou élimination d’un produit.
– Reconnaître dans la formule d’une espèce chimique organique les groupes caractéristiques :
–OH, –CO2H, –CO2R– Écrire l’équation des réactions modélisant l’estérifica-tion et l’hydrolyse.– À partir de la formule semi-développée d’un ester, retrouver les formules de l’acide carboxylique et de l’al-cool correspondants.– Savoir nommer les esters comportant cinq atomes de carbone au maximum.– Savoir que les réactions d’estérification et d’hydrolyse sont inverses l’une de l’autre et que les transformations associées à ces réactions sont lentes.– Savoir qu’un catalyseur est une espèce qui augmente la vitesse d’une réaction chimique sans figurer dans l’équa-tion de la réaction et sans modifier l’état d’équilibre du système chimique.– Savoir que l’excès de l’un des réactifs et/ou l’élimi-nation de l’un des produits déplace l’état d’équilibre du système chimique dans le sens direct.– Calculer le rendement d’une transformation
L’équilibre estérification-hydrolyse10
PARTIE 4
Comment contrôler l’évolution d’un
système chimique ?
Ce chapitre est le premier portant sur la chimie orga-nique, domaine déjà rencontré en première. L’obten-tion d’un ester à partir d’acide carboxylique et d’alcool, découverte par Marcellin Berthelot en 1860 et étudiée dans les années suivantes, constitue un exemple supplémentaire de passage à un nouveau
Découpage du cours1. Composés organiques oxygénés p. 2042. Synthèse des esters p. 2053. Hydrolyse des esters p. 2064. L’équilibre estérification-hydrolyse p. 2075. Rendement de la synthèse d’un ester p. 208
♦
Cours

133Chapitre 10 - L’équilibre estéri: cation-hydrolyse
groupe d’atomes caractéristiques (programme de première S). On retrouve la notion d’équilibre chimi-que (présentée chapitre 4) et celle de facteur cinéti-que (cf. chapitre 2). On peut mettre en place un suivi cinétique de la transformation grâce à des dosages acido-basiques (cf. chapitre 7). On découvre la notion de catalyseur et celle du déplacement d’équilibre.
1. Composés organiques oxygénésOn rappelle les composés organiques déjà rencontrés et on introduit une nouvelle famille, qu’il convient de bien distinguer des acides carboxyliques.L’activité 1 permet aux élèves de préparer ce para-graphe en révisant les composés oxygénés rencon-trés en première et utiles en terminale.
2. Synthèse des estersL’obtention d’un ester à partir d’acide carboxylique et d’alcool est étudiée de manière proche de celle de nos précurseurs. L’activité 3 constituée de morceaux de textes écrits par Marcellin Berthelot montre en effet à l’élève que les caractéristiques de cette trans-formation étaient connues avant même que l’on puisse écrire correctement l’équation de la réaction modélisant cette transformation.On retrouve un facteur cinétique déjà rencontré (température) et on découvre l’existence d’une espèce chimique capable d’augmenter significative-ment la vitesse d’une réaction qu’on appelle un cata-lyseur (sans modifier l’état final de la transformation). L’activité 2 permet de retrouver ce facteur cinétique température par l’intermédiaire de dosages acido-basiques.
3. Hydrolyse des estersOn présente l’hydrolyse comme la réaction inverse de la précédente, que l’on étudie de la même manière que
la précédente. On amène l’élève à découvrir que ces réactions présentent des caractéristiques analogues.
4. L’équilibre estérification hydrolyseOn retrouve une notion rencontrée précédemment : celle de l’équilibre chimique. Dans l’aspect micros-copique de l’état d’équilibre chimique, les chocs efficaces ont toujours lieu, mais ces chocs concer-nent :– le catalyseur et l’acide carboxylique lors de l’esté-rification ;– l’ester et le catalyseur lors de l’hydrolyse.Le calcul de la valeur du quotient de réaction à l’équilibre chimique permet de montrer que l’eau (lorsqu’elle n’est pas solvant) doit apparaître dans Qr. C’est l’occasion d’insister sur l’utilité de l’indi-cation (aq) concernant la nature des espèces chimi-ques d’un système.
5. Rendement de la synthèse d’un esterAprès avoir présenté le dispositif classique de syn-thèse, on présente le mode de calcul d’un rendement expérimental. La question qui vient ensuite naturel-lement est de savoir comment augmenter ce rende-ment sans changer les réactifs.Le déplacement d’équilibre est abordé de manière qualitative pour permettre à l’élève de comprendre l’utilité d’utiliser un réactif en excès. L’activité sup-plémentaire (synthèse d’un ester à l’arôme de banane) constitue un exemple expérimental de ce déplacement.Enfin, en fonction des températures d’ébullition des différentes espèces, on utilise dans certains cas deux autres montages permettant d’éliminer l’un des produits au fur et à mesure de sa formation : distillation fractionnée (cas de l’ester) ou appareil de Dean-Stark (cas de l’eau).
À la découverte d’une nouvelle famille (page 211)
Cette activité permet d’écrire des formules semi-développées de composés organiques et de revoir ce qu’est la valence d’un atome.À partir d’exemples proposés, on cherche à recon-naître les alcools et les acides carboxyliques, puis on déduit les caractéristiques de la nouvelle famille.
ACTIVITÉ
1
ACTIVITÉ
1 1. a) Formules semi-développées1 CH3–CH2–OH 2 H–COOH
3 CH3–CH2–CO–O–CH3 4 (CH3)3–C–OH
5 CH3–CO–O–CH2–CH3 6 CH3–CH2–CH2–OH
7 CH3–CH(CH3)–CH2–OH 8 CH3–CH2–CH(CH3)–COOH
9 CH3–COOH 10 H–CO–O–CH2–CH3
11 CH3–CH(OH)–CH3 12 H–CO–O–CH3
13 (CH3)2–CH–CH(OH)–CH3 14 CH3–OH
15 CH3–CH2–COOH 16 CH3–CH2–CH2–CH2–OH
Activités

134 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
b) La valence d’un atome de carbone est 4, celle d’un atome d’oxygène 2 et celle d’un atome d’hy-drogène 1.
2. a) & b)1 CH3–CH2–OH Alcool primaire Éthanol
4 (CH3)3–C–OH Alcool tertiaire2-méthylpropan-2-ol
6 CH3–CH2–CH2–OH Alcool primaire Propan-1-ol
7CH3–CH(CH3)–CH2–OH
Alcool primaire2-méthylbutan-1-ol
11 CH3–CH(OH)–CH3 Alcool secondaire Propan-2-ol
13(CH3)2–CH–CH(OH)–CH3
Alcool secondaire3-méthylbutan-2-ol
14 CH3–OH Alcool primaire méthanol
16CH3–CH2–CH2–CH2–OH
Alcool primaire Butan-1-ol
3. a) & b)2 H–COOH Acide méthanoïque
8 CH3–CH2–CH(CH3)–COOH Acide-2-méthylbutanoïque
9 CH3–COOH Acide éthanoïque
15 CH3–CH2–COOH Acide propanoïque
4. a) Les esters présentent le groupe d’atome carac-téristique C O R
O
, avec R radical carboné.
b)3 C2H5–CO–O–CH3 Propanoate de méthyle
5 CH3–CO–O–C2H5 Éthanoate d’éthyle
10 H–CO–O–C2H5 Méthanoate d’éthyle
12 H–CO–O–CH3 Méthanoate de méthyle
L’estérificationet l’hydrolyse (page 212)
Cette activité doit être commencée par l’élève à la maison pour pouvoir espérer la mener à son terme…Cette activité comporte plusieurs objectifs :– faire comprendre à l’élève que cette transforma-tion s’effectue avec des réactifs purs (et non en solu-tion aqueuse) d’où les conséquences lors de l’écriture du quotient de réaction ;– rappeler les relations entre masse volumique et volume d’un réactif pur à l’état liquide ;– revoir l’équivalence d’un titrage acido-basique ;– revoir les facteurs cinétiques présentés chapitre 2 ;– montrer le caractère limité de cette transforma-tion.La partie A de l’activité constitue un rappel sur les caractéristiques physico-chimiques des composés
ACTIVITÉ
2
ACTIVITÉ
2
organiques (densité, masse volumique, températu-res de changement d’état). À partir de ces données, on tente d’élaborer un protocole réalisable en une séance de travaux pratiques.La partie B constitue le protocole à suivre au cours duquel des titrages acido-basiques sont effectués.La partie C constitue un rappel des conditions expé-rimentales à respecter lors d’un suivi cinétique.La partie D constitue l’exploitation avec une aide pour les calculs et une analyse de résultats expéri-mentaux.
A Réfléchir avant d’agir !1. a) La densité est un nombre sans unité qui dans le cas d’un liquide est égal au quotient de la masse volumique du liquide par la masse volumique de l’eau.b) La masse volumique de l’acide éthanoïque pur est de 1 050 g · L–1.c) La quantité de matière
n = m
M =
ρ(acide) ·VM
, donc V = n M·
ρ(acide) ;
V = 1 60 05
1 050
× , = 57,2 × 10–3 L.
d) La même formule donne V' = 91,5 mL pour le butan-1-ol.
2. a) CH3COOH + CH3–CH2–CH2–CH2–OH= CH3–CO–O–CH2–CH2–CH2–CH3 + H2O.
b) L’acide et l’alcool devant être en quantités éga-les, les calculs précédents montrent un volume total de 150 mL pour un mélange constitué d’une mole de chacun des réactifs.En divisant les proportions par deux, on obtient un volume total de 75 mL environ (28,6 mL d’acide éthanoïque et 45,8 mL de butan-1-ol).c) Ce mélange contient 0,5 mol d’acide carboxyli-que et 0,5 mol d’alcool.
3. a) CH3COOH(aq) + HO–(aq)
= CH3COO–(aq) + H2O(l).
b) Le prélèvement de 3 mL de mélange réactionnel
représente 3
75 =
1
25 du volume total, soit aussi
1
25de la quantité de matière initiale de chacun des réactifs ; d’où n(acide) = 0,02 mol.Pour doser cet acide il faut qu’à l’équivalence :
n(base) = n(acide) = 0,02 mol.Or n(base) = CB · VBE,
donc VBE = n
C
(base)
B
; VBE = 0,2 L = 200 mL.

135Chapitre 10 - L’équilibre estéri: cation-hydrolyse
c) Il n’existe pas dans les laboratoires de lycée des burettes graduées ayant un tel volume.d) Il faut prendre une solution aqueuse d’hydroxyde de sodium 10 fois plus concentrée (donc molaire) pour une chute de burette de 20 mL.
4. a) On peut agir sur la température du milieu réactionnel, sur la présence d’un catalyseur, sur l’agitation du mélange réactionnel.b) Les volumes de solution aqueuse d’hydroxyde de sodium, versés pour obtenir l’équivalence, vont diminuer car l’acide carboxylique est un réactif amené à disparaître (pas totalement…) au cours de la transformation.c) Il faut choisir la phénolphtaléine.d) Le virage sera de l’incolore au rose pâle.
B Protocole expérimental1. D’après la formule ci-dessus
n(acide) = 1 050 0 020
60 05
× ,
, = 0,35 mol ;
n(alcool) = 810 0 032
74 12
× ,
, = 0,35 mol.
2. Ce mélange est équimolaire.3. L’acide sulfurique est un catalyseur, il augmente la vitesse de la réaction sans en modifier l’état final.4. Il faut une pipette graduée et une propipette pour effectuer ce prélèvement.5. Ce tube mis à part permet de déterminer la quan-tité totale d’acide présent initialement.
C Suivi cinétique1. a) On place le contenu des tubes dans un erlen-meyer d’eau glacée pour bloquer la réaction (fac-teur température et concentration des réactifs)b) Cette opération se nomme une trempe.2. On rince les tubes pour récupérer avant le dosage tous les acides qui y sont contenus.3. C’est toujours le facteur cinétique température qui est en jeu.
D Exploitation1. a) Na(0) = CB · V1E.b) Na(t) = CB · ViE.c) Ne(t) = CB · V1E – CB · ViE.d) On prélève 3 mL d’un mélange réactionnel qui contenait au départ 54 mL, soit 1/18e du mélange total. D’où NESTER(t) = 18 × Ne(t).e) Tableau d’évolution x(t) = NESTER(t).
2. a) Cf. Fig. 3, p. 213.b) Pour un même instant de date t :
x (40 °C) < x (70 °C).c) On calcule le rendement expérimental au bout d’une demi-heure :
– à 40 °C : ρ = 0 18
0 35
,
, = 50 % ;
– à 70 °C : ρ = 0 22
0 35
,
, = 63 %.
3. a) À 70 °C, la courbe de l’avancement stagne : la réaction semble parvenue à son terme.b) Équilibre chimique.c) Ce même état sera atteint plus tard à 40 °C.d) La courbe représentative de x(t) serait toujours située en dessous de la courbe à 40 °C en l’absence de catalyseur.
La synthèse d’un esteren 1860 (page 214)
Avec cette activité, l’élève est amené à réaliser le chemin parcouru par les scientifiques pour com-prendre les phénomènes mis en jeu dans une trans-formation. Comment appeler un composé nouveau ? Quelle formule lui donner ? Comment le distinguer de ceux que l’on connaît déjà ?….
1. Les éthers composés sont aujourd’hui des esters.2. a) Ligne 13 : l’union ne se réalise que d’une manière lente. Ligne 19-20 : la formation exige le concours du temps.b) Lignes 15-16 : une portion de l’acide et une de l’alcool demeurent libres simultanément.Lignes 17-18 : destruction de la combinaison avec une difficulté semblable à celle qui a précédé à son accomplissement.c) Lignes 21-22 : intervention d’agents auxiliaires qui semblent provoquer le phénomène sans y concourir par une affinité directe.3. Les facteurs cinétiques sont la température et l’agitation du mélange réactionnel.4. CH3COOH + CH3–CH2–OH =
CH3–CO – O–CH2–CH3 + H2O. acide éthanoïque + éthanol = éthanoate d’éthyle + eau.
5. Les esters sont pratiquement insolubles dans l’eau ; ils sont en revanche solubles dans les sol-vants organiques.Ils sont en général odorants (pas toujours de manière agréable) et volatils (faible température d’ébullition).
ACTIVITÉ
3
ACTIVITÉ
3

136 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
Suivi cinétiqued’une estérification (page 215)
Cette activité constitue un TP d’entraînement à l’épreuve d’évaluation des capacités expérimentales Elle est constituée d’une dilution préalable, d’un démarrage d’étude cinétique et de titrages acido-basiques pour vérifier les savoir-faire concernant :– le démarrage d’une étude cinétique (mise en tem-pérature simultanée, et déclenchement du chrono-mètre) ;– la burette graduée (rinçage de la burette avec la solution à prélever, ajustement du zéro) ;– l’agitateur magnétique (position et vitesse cor-rectes du barreau aimanté) ;– la pipette jaugée (pipetage dans un bécher, rin-çage de la pipette avec la solution à prélever, utilisa-tion d’un pipeteur, repérage correct des niveaux) ;– la fiole jaugée (ajustement au trait de jauge et homogénéisation).– l’arrêt correct à l’équivalence.
1 Dilution préalable de la solution1. Il faut utiliser une fiole jaugée de 100 mL et une pipette jaugée de 20 mL (équipée d’une propi-pette).2. Réalisation pratique.
ACTIVITÉ
4
ACTIVITÉ
4 2 Réalisation du suivi cinétique
3 Dosage par titrage acido-basique
4 Questions et exploitation1. C’est un réfrigérant à air, destiné à condenser les éventuelles vapeurs.2. Pour disposer d’eau distillée glacée lors des rin-çages des tubes à essais.3. Il faut doser tout l’acide présent dans le tube à essai.4. C’est une trempe ou blocage cinétique : on joue sur la température et la concentration des réactifs.5. VE1 < VE2 < VE3, car on dose l’acide qui est un réac-tif (dont la quantité de matière diminue au cours du temps).6. La quantité de matière d’ester obtenu augmente au cours du temps.7. L’ester est en général peu soluble dans l’eau ; l’agi-tation due au dosage donne un mélange trouble.8. Le volume équivalent par dosage acido-basique ne sera jamais nul pour deux raisons :– le mélange réactionnel contient de l’acide sulfu-rique, catalyseur qui ne disparaît pas ;– la réaction étant limitée, il restera toujours de l’acide éthanoïque dans le mélange réactionnel.
Activité supplémentaire
Synthèse d’un ester à l’arôme de bananeDans certains produits manufacturés, on peut lire sur l’étiquette : arôme de banane. Cette activité per-met de réaliser la synthèse d’un ester dont l’odeur rappelle celle de la banane en utilisant le dispositif d’un chauffage à reflux, l’extraction de l’ester est effectuée à l’aide d’une ampoule à décanter. La com-paraison entre théorie et pratique permettra de jus-tifier certaines étapes du protocole.
A PrésentationPour réaliser la synthèse de l’acétate d’isoamyle dont le nom en nomenclature officielle est l’éthanoate de 3-méthylbutyle, il faut de l’acide éthanoïque et du 3-méthylbutan-1-ol.
1. Écrire la formule semi-développée de l’alcool.2. Indiquer la classe de cet alcool.3. Écrire l’équation de la réaction modélisant la transformation permettant d’obtenir cet ester.
4. Pour réaliser cette synthèse, on utilise un dispo-sitif de chauffage à reflux.a) Comment doit s’effectuer la circulation de l’eau dans le réfrigérant à boules ?b) Quel est l’intérêt du réfrigérant à boules ?
5. On ajoute en plus dans le ballon de l’acide sulfu-rique.a) Quel est le rôle de cet acide ?b) Est-il nécessaire d’en ajouter une grande quan-tité ?c) Faut-il mesurer cette quantité avec précision ?
6. Pourquoi place-t-on également quelques billes de verre dans le ballon ?
B Préparation de l’esterDans le ballon du dispositif de chauffage à reflux, introduire à l’aide d’éprouvettes graduées :– V1 = 11 mL d’alcool isoamylique,– V2 = 23 mL d’acide éthanoïque,

137Chapitre 10 - L’équilibre estéri: cation-hydrolyse
– V3 = 4 mL d’acide sulfurique commercial,– quelques billes de verre ou grains de pierre ponce.Adapter le ballon au dispositif, puis faire circuler l’eau dans le réfrigérant à boules.Commencer à chauffer : lorsque l’ébullition est atteinte, régler le chauffe-ballon pour que l’ébulli-tion soit douce.Le reflux doit durer environ 30 min. Remplacer alors le chauffe-ballon par un bain de glace (sans arrêter la circulation de l’eau dans le réfrigérant).
1. Sachant que pour l’alcool isoamylique, la masse molaire moléculaire M1 = 88 g · mol–1 et la densité d1 = 0,81, déterminer la quantité de matière initiale d’alcool, n1, introduite dans le ballon.2. Sachant que pour l’acide éthanoïque, la masse molaire moléculaire M2 = 60 g · mol–1 et la densité d2 = 1,05, calculer la quantité de matière d’acide éthanoïque, n2 , introduite dans le ballon.3. En déduire le réactif en excès.4. Expliquer la raison pour laquelle l’un des réactifs a été placé en excès par rapport à l’autre.5. Quelle sera la valeur de l’avancement maximal ?
C Extraction de l’esterAprès refroidissement, placer le contenu du ballon dans une ampoule à décanter contenant déjà 50 mL de solution aqueuse saturée de chlorure de sodium, Na+(aq) + Cl–(aq), en évitant que les billes ou grains aillent dans l’ampoule.Agiter et purger régulièrement puis éliminer la phase aqueuse (inférieure) et recommencer l’opéra-tion avec 50 mL de solution aqueuse saturée de chlorure de sodium.Éliminer la phase aqueuse puis ajouter 50 mL de solution aqueuse saturée d’hydrogénocarbonate de sodium, Na+(aq) + HCO3
–(aq).Purger régulièrement l’ampoule à décanter et après avoir éliminé la phase aqueuse, recommencer avec la solution d’hydrogénocarbonate de sodium.Récupérer la phase organique dans un erlenmeyer et introduire un peu de sulfate de magnésium anhydre.Mesurer à l’aide d’une éprouvette graduée le volume V4 d’ester obtenu.
1. Pourquoi doit-on refroidir le ballon et son contenu avant d’ajouter la solution saturée de chlo-rure de sodium ?2. La densité de la solution aqueuse saturée de chlo-rure de sodium est voisine de 1,1, celle de l’ester non miscible à l’eau est d4 = 0,87.a) Dessiner l’ampoule à décanter et justifier les posi-tions respectives des phases aqueuse et organique.
b) Que contient la phase aqueuse après la première extraction ?c) Que contient la phase organique après cette pre-mière extraction ?3. Pourquoi utilise-t-on une solution saturée de chlorure de sodium ?4. L’ion hydrogénocarbonate HCO3
–(aq) fait partie de deux couples acide/base :
CO2,H2O/HCO3–(aq) et HCO3
–(aq)/CO32–(aq).
a) Identifier le dégagement gazeux se produisant lors de l’addition de la solution d’hydrogénocarbo-nate de sodium dans l’ampoule à décanter.b) Écrire les équations des réactions modélisant les transformations qui ont lieu dans l’ampoule à décanter.c) Quelle est la particularité de l’ion hydrogénocar-bonate ?d) Comment le qualifie-t-on ?5. Quel est le rôle du sulfate de magnésium anhydre ?
D Rendement de l’opérationAprès filtration, la mesure du volume d’ester obtenu donne V4 = 12,5 mL. L’ester obtenu a pour densité d4 = 0,87 et pour masse molaire moléculaire M4 = 130 g · mol–1.
1. Déterminer la quantité de matière n4 d’ester effectivement obtenu.2. Calculer le rendement de cette synthèse3. Commenter la valeur obtenue.4. Comment peut-on vérifier la pureté de l’ester ?5. Compte tenu des températures d’ébullition des espèces chimiques intervenant dans la transforma-tion, y a-t-il un autre moyen de synthétiser l’ester en grande quantité ?6. Pourquoi a-t-on choisi de travailler avec un excès d’acide carboxylique par rapport à l’alcool ?
Expèces chimiques
Acide éthanoïque
Alcool isoamylique
Éthanoate d’isoamyle
Densité 1,05 0,81 0,87
Masse molaire moléculaire 60 g · mol–1 88 g · mol–1 130 g · mol–1
Température d’ébullition 118 °C 131 °C 142,5 °C
Solubilité dans l’eau
En toutes proportions
26,7 g · L–1 1,6 g · L–1
Solubilité dans l’eau salée
En toutes proportions
< 26,7 g · L–1 < 1,6 g · L–1
Tableau des principales caractéristiques des composants de la manipulation.

138 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
E Comparaison avec la théorieOn rappelle que la constante d’équilibre lors de l’estérification avec un alcool primaire prend pour valeur K = 4. On cherche à calculer la valeur à l’équilibre de l’avancement pour le système chimique précédent constitué initialement de 0,10 mol d’alcool et 0,40 mol d’acide carboxylique.1. Dresser le tableau d’évolution des espèces chimi-ques au cours de l’estérification à l’aide de l’avance-ment, noté x.2. Écrire l’expression littérale du quotient de réac-tion en fonction des quantités de matière pour la réaction d’estérification.3. Que vaut le quotient de réaction à l’équilibre ?4. Déterminer la valeur de l’avancement à l’équilibre : on aura à résoudre une équation du second degré.5. La valeur calculée est-elle en accord avec celle obtenue lors de la manipulation ?6. Rechercher les causes de la différence entre la théorie et la pratique.
Corrigé
A. Présentation1. CH3–CH(CH3)–CH2–CH2 - OH.2. Alcool primaire.3. CH3–COOH + HO–(CH2)2–CH(CH3)2
= CH3–CO–O–(CH2)2–CH(CH3)2 + H2O.4. a) Circulation de l’eau ascendante.b) Condenser les vapeurs (éviter les pertes).5. a) Catalyseur.b) Une faible quantité suffit.c) Grande précision inutile.6. Elles sont destinées à régulariser l’ébullition.
B. Préparation de l’ester1. La quantité initiale : n1 = 0,10 mol d’alcool.2. La quantité initiale d’acide carboxylique : n2 = 0,40 mol.3. L’acide éthanoïque est en excès.4. On cherche à déplacer l’équilibre en faveur de l’ester.5. L’avancement maximal est donné par le réactif en défaut, donc xmax = 0,10 mol.
C. Extraction de l’ester1. On ne peut pas toucher le ballon : il est trop chaud.2. a) La phase aqueuse est la phase inférieure car sa densité est plus importante que celle de l’eau.
b) Dans la phase aqueuse, il y a les acides (éthanoï-que et sulfurique) et une partie de l’alcool.c) La phase organique contient l’ester et de l’alcool.3. Solution saturée, car l’ester et l’alcool y sont très peu solubles. De plus, cela augmente la différence de densité entre les phases organique et aqueuse, d’où une meilleure séparation.4. a) C’est du dioxyde de carbone CO2.b) HCO3
–(aq) + H3O+(aq) = CO2(g) + 2 H2O(l) et
HCO3–(aq) + CH3COOH(aq) = CO2(g) + CH3COO
–(aq).c) Il peut jouer le rôle d’un acide ou d’une base.d) C’est un ampholyte (ou espèce amphotère).5. Le sulfate de magnésium anhydre permet d’enle-ver toute trace d’eau dans la phase organique.
D. Rendement de l’opération1. La quantité de matière : n4 = 0,084 mol d’ester.2. Rendement = 84 %.3. Rendement supérieur à 67 % grâce à la présence d’un réactif en excès par rapport à l’autre.4. Mesure de la densité, chromatographie, tempé-rature d’ébullition.5. On ne peut pas extraire l’ester au fur et à mesure de sa formation, car sa température d’ébullition est la plus élevée. On aurait pu extraire l’eau avec un montage de Dean-Stark.6. L’acide étant soluble dans l’eau en toutes pro-portions, il était plus facile à séparer de l’ester dans la phase aqueuse.
E. Comparaison avec la théorie
1. Acide + alcool = ester + eau
État Avanc. Quantité de matière (mol)
Initial x = 0 0,4 0,1 0 0
En cours x 0,4 – x 0,1 – x x x
Final xf 0,4 – xf 0,1 – xf xf xf
2. Qr,éq = ( )
( , – ) ( , – )
x
x xf2
f f0 4 0 1×.
3. Qr,éq = 4.
4. xf2 = 4(0,4 – xf) · (0,1 – xf).
Il faut résoudre l’équation : 3 xf2 – 2 xf + 0,16 = 0.
∆ = 2,08 ; xf 1 = 0,093 mol et xf 2 = 0,57 mol.Seule solution acceptable : xf 1 < xmax.5. Valeur obtenue en accord, même si plus élevée.6. Elles sont multiples : pertes lors de l’extraction, durée trop courte du chauffage à reflux.
139Chapitre 10 - L’équilibre estéri: cation-hydrolyse
1 Donner un nom
Composé Famille Nom
CH3–CH2–OHAlcool primaire Éthanol
CH3–(CH2)2–COOHAcide carboxylique Acide butanoïque
CH3–COO–C2H5 Ester Éthanoate d’éthyle
CH3–CH(OH)–CH3Alcool secondaire Propan-2-ol
H–COO–(CH2)2–CH3 Ester Méthanoate de propyle
CH3–CH(CH3)–COOHAcide carboxylique
Acide-2-méthylpropanoïque
CH3–CH(CH3)–CH2OHAlcool primaire 2-méthylpropan-1-ol
CH3–CH2–COO–C2H5 Ester Propanoate d’éthyle
2 Écriture de formules semi-développées1.
Composé Nom
CH3–OH Méthanol
CH3–COOH Acide éthanoïque
CH3–COO–CH3 Éthanoate de méthyle
CH3–C(OH)–CH3 CH3
2-méthylpropan-2-ol
C2H5–COO–C2H5 Propanoate d’éthyle
H–COO–C2H5 Méthanoate d’éthyle
CH3–CH(OH)–C2H5 Butan-2-ol
H–COOH Acide méthanoïque
2. Famille Nom
Alcool primaire Méthanol
Acide carboxylique Acide éthanoïque
Ester Éthanoate de méthyle
Alcool tertiaire 2-méthylpropan-2-ol
Ester Propanoate d’éthyle
Ester Méthanoate d’éthyle
Alcool secondaire Butan-2-ol
Acide carboxylique Acide méthanoïque
3. Écriture topologique Nom
OH Méthanol
OH
OAcide éthanoïque
O
OÉthanoate de méthyle
OH 2-méthylpropan-2-ol
O
OPropanoate d’éthyle
O
OMéthanoate d’éthyle
OH Butan-2-ol
OH
OAcide méthanoïque
3 La réaction d’estérification1. H–COOH + CH3–CH2–CH2–OH
= H–COO–(CH2)2–CH3 + H2O.2. Acide méthanoïque + propan-1-ol
= méthanoate de propyle + eau.3. Cette réaction est lente, limitée et athermique.4. Le rendement : ρ = 0,67 = 67 %.5. Le catalyseur permet d’atteindre plus rapide-ment l’état d’équilibre décrit sans le modifier.6. En ajoutant l’un des réactifs en large excès par rapport à l’autre, ou en éliminant au fur et à mesure de sa production, l’un des produits (ester ou eau) de la réaction.
4 La réaction d’hydrolyse1. CH3–COO–CH3 + H2O = CH3–COOH + CH3–OH.2. Éthanoate de méthyle + eau = acide éthanoïque + méthanol.3. Cette réaction est lente, limitée et athermique.4. Le rendement est ρ = 0,33 = 33 %.5. Il y aura 40 mmol d’acide et 40 mmol d’alcool ; il restera 80 mmol d’eau et 80 mmol d’ester.
5 L’équilibre chimique1. R–COOH + R'–CH2–OH = R–COO–CH2–R' + H2O.
Corrigés des exercices (page 216)

140 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
2. Qr = n n
n n
(ester) (eau)
(acide) (alcool)
·
·.
Dans l’état initial : n(ester) = n(eau) = 0, n(acide) = n(alcool) = 1 mol, donc Qr,i = 0.
3. Comme Qr,i < K, le système évolue dans le sens gauche-droite : il y a formation d’ester et d’eau.
4. Équation acide + alcool = ester + eau
État Avanc. Quantité de matière (mol)
Initial x = 0 1 1 0 0
En cours x 1 – x 1 – x x x
Équilibre xéq 1 – xéq 1 – xéq xéq xéq
5. Dans l’état final du système, à l’équilibre chimique, les quantités de matière vérifient :
x
x
éq2
éq( )1 2− = 4, donc
x
xéq
éq1− = 2 et xéq =
2
3 mol.
État final du système : il y a 0,67 mol d’ester, 0,67 mol d’eau, 0,33 mol d’acide et 0,33 mol d’alcool.6. Une augmentation de température entraîne une arrivée plus rapide du système à l’état d’équilibre chimique.7. On effectue un déplacement d’équilibre, qui a pour conséquence d’augmenter le rendement de cette transformation.
6 Rose ou géranium1. A(l) + H2O(l) =
—COOH(l) + HO–CH2–CH(CH3)2.
2. Benzoate d’isobutyle + eau= acide benzoïque + 2-méthylpropan-1-ol.
3. Réaction lente, limitée et athermique.4. a) Fausse : le catalyseur permet d’atteindre plus rapidement l’état d’équilibre.b) Fausse : le catalyseur augmente la vitesse des réactions (estérification et hydrolyse).c) Fausse : cf. réponse b).
7 Pomme ou ananas ?1. a) CH3–COO–CH2–CH2–CH2–CH3.b) CH3–COOH + HO–(CH2)3– CH3
= CH3–COO–CH2–CH2–CH2–CH3 + H2O.c) Acide éthanoïque et butan-1-ol.d) Lente, limitée, athermique.
2. a) CH3–(CH2)2–COO–CH2–CH3.b) CH3–(CH2)2–COO–CH2–CH3 + H2O
= CH3–(CH2)2–COOH + HO– CH2–CH3.c) Acide butanoïque et éthanol.d) Lente, limitée, athermique.3. C6H12O2 pour ces deux esters.
4. Ils sont isomères.5. CnH2nO2.
8 Étude d’une estérification
Exercice résolu dans le manuel de l’élève.
9 Obtention d’un ester à l’odeur d’abricot1. a) A = acide butanoïque : CH3–(CH2)2–COOH.B = pentan-1-ol, de formule HO–(CH2)4–CH3.b) B est un alcool primaire.2. La réaction de synthèse se nomme estérification et a pour équation :CH3–(CH2)2–COOH + HO–(CH2)4–CH3
= CH3–(CH2)2–COO–(CH2)4–CH3 + H2O.
3. Cette réaction est lente, limitée et athermique.4. Le rendement est de 67 % si les réactifs sont au départ dans les proportions stœchiométriques.
5. En présence de catalyseur (deuxième expé-rience), la réaction se fait plus vite, l’état d’équili-bre est atteint plus rapidement. Si de plus, la température est plus élevée, la réaction se fait encore plus vite, sans pour autant modifier l’état d’équilibre.
10 Arôme de la poire1. CH3–COOH + CH3–(CH2)4–OH
= CH3–COO–(CH2)4–CH3 + H2O.2. À l’équivalence, lors du changement de réactif limitant : nA = CA · VA = CB · VBdonc nA = 2 × 24,5 × 10
–3 = 49 × 10–3 mol.La quantité d’ester est : n = 50 – 49 = 1 mmol.
Rendement : ρ = 150
= 2
100 = 2 %.
3. a) État d’équilibre atteint.b) Pour cette même durée, le rendement sera supé-rieur à 2 % : la température, facteur cinétique, augmente la vitesse d’une réaction, la quantité de matière d’ester formé est plus grande.c) Le taux d’avancement maximal d’une estérifica-tion lorsque l’alcool est primaire et que les réactifs sont au départ dans les proportions stœchiomé-

141Chapitre 10 - L’équilibre estéri: cation-hydrolyse
triques est de 67 %. On ne peut donc pas attendre un rendement de 100 %.
4. L’acide sulfurique est un catalyseur qui permet d’augmenter la vitesse d’une réaction, donc le ren-dement est identique à celui du 3.
11 Préparation d’un parfum à odeur de lavande
1. a) Le groupe d’atomes caractéristique –OH est celui de la fonction alcool.b) C’est un alcool tertiaire.2. C’est un ester.3. a) CH3COOH + linalol = acétate de linalyle + eau.b) Cette réaction est lente, très limitée et athermique.4. Le rôle du catalyseur est d’augmenter la vitesse de la transformation, permettant d’atteindre plus rapidement l’état d’équilibre sans le modifier.
5. a) Qr,i = n n
n n
(ester)i
(eau)i
(acide)i
(alcool)i
·
·.
Le quotient de réaction dans l’état initial vaut :Qr,i = 0.
b) Comme Qr,i < K, le système chimique évolue spon-tanément dans le sens direct de la formation d’ester.
6. a) 40 mL de linalol correspondent à :m = µ · V ; m = 34,8 g,
donc
n(linalol) = m
M ; n(linalol) =
34 8
154
, = 226 mmol.
Comme le mélange de départ est équimolaire, il y avait aussi 226 mmol d’acide éthanoïque, donc :
m(acide) = 13,6 g.Finalement :
V(acide éthanoïque) = m
µ' ;
V(acide éthanoïque) = 13 6
1 05
,
, = 13 mL.
b) ρ = n
n
(ester)
(ester)obtenu
max
; ρ = 0 011
0 226
,
, = 0,05 = 5 %.
c) Le résultat n’est pas surprenant, car l’estérifica-tion d’un alcool tertiaire donne un rendement de 5 %.
7. a) Mettre un réactif (l’acide éthanoïque) en large excès par rapport à l’autre.b) Éliminer l’eau au fur et à mesure de sa fabrica-tion avec un appareil de Dean-Starck.
12 Estérification1. a)Équation Acide + alcool = ester + eau
État Avanc. Quantité de matières (mol)
Initial x = 0 0,20 0,20 0 0
En cours x 0,20 – x 0,20 – x x x
Équilibre xéq 0,20 – xéq 0,20 – xéq xéq xéq
b) Qr,éq = x
x
éq
éq
2
2 20 20( , – ) = 4.
c) xéq = 2(0,20 – xéq), soit xéq = 0 40
3
, = 0,13 mol.
d) τ = x
xéq
max
; τ = 0 13
0 20
,
, = 0,67, soit τ = 67 %.
e) Le mélange final contient 0,13 mol d’ester, 0,13 mol d’eau, 0,07 mol d’acide carboxylique et 0,07 mol d’alcool.
2. a) On a alors :x
x xéq
éq éq
2
0 20 0 50( , – )( , – ) = 4,
soit 3 x2éq – 2,8 xéq + 0,4 = 0.∆ = 3,04 donne :– xéq1 = 0,176 mol– xéq2 = 0,76 mol (à rejeter car supérieur à xmax = 0,20 mol).b) Taux d’avancement final :
τ' = x
xéq1
max
= 0,88, soit τ' = 88 %.
c) L’ajout d’un réactif en excès par rapport à l’autre déplace l’équilibre dans le sens direct de la forma-tion des produits.
13 Les phéromones1. A : Groupe caractéristique ester (en noir) :
CH3–CH(CH3)–CH2–CH2–O–CO–CH3.C : un groupe caractéristique des alcools secondaires (en noir), un autre des alcènes (en gris) :
(CH3)2–C=CH–CH2–CH2–CH(OH)–CH3.
2. a) E = acide éthanoïque : CH3–COOH.D = 3-méthylbutan-1-ol :
CH3–CH(CH3)–CH2–CH2–OH.b) CH3–COOH + CH3–CH(CH3)–CH2–CH2–OH = A + H2O.c) Affirmations : 1, fausse ; 2, fausse ; 3, vraie.3. a) R' = CH3.b) CH3–OH : méthanol.

142 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
4. a) C = n
V =
m
M V· ; C =
10
128 1
15−
×C = 7,8 × 10–18 mol · L–1.b) Quantités utilisées beaucoup plus faibles ; com-posés employés non toxiques pour la nature.
14 Synthèse d’un arôme
Exercice résolu dans le manuel de l’élève.
15 Composé à odeur de framboise1. C’est un ester2. a) H–COOH + CH3–CH(CH3)–CH2–OH
= H–COO–CH2–CH(CH3)–CH3 + H2O.b) L’acide carboxylique A est l’acide méthanoïque, l’alcool B est le 2-méthylpropan-1-ol.3. a) Montage de chauffage à reflux.b) Chauffe-ballon et réfrigérant à boules.c) Chauffage pour accélérer la réaction ; le reflux permet de ne rien perdre.4. a) Le graphe donne :
00,050,10,150,20,250,30,35
0 20 40 60 80
Instantt (min)
n (ester) (mol)
Quantité de matière d’ester en fonction du temps.
b) Équation Acide + alcool = ester + eau
État Avanc. Quantité de matières (mol)
Initial x = 0 0,50 0,50 0 0
En cours x 0,50 – x 0,50 – x x x
Équilibre xéq 0,50 – xéq 0,50 – xéq xéq xéq
c) On constate que x = n.d) La vitesse volumique de réaction v(t) est donnée par l’expression :
v(t) = 1
V
x
t·d
doù V est le volume du mélange réactionnel.
e) Comme x = n, alors v(t) = 1
V
n
t·d
d.
f) La vitesse est nulle à cet instant, car la tangente à la courbe est horizontale, donc son coefficient directeur est nul.
g) Rendement ρ = n
n
( )
( )max
ester
esterobtenu ;
ρ = 0 33
0 50
,
, = 0,66 = 66 %.
16 Synthèse d’un additif alimentaire à odeur de rhum
1. HCOOH + HO–CH2–CH3 = HCOO–CH2–CH3 + H2O.2. Y = méthanoate d’éthyle3. L’état final est un état d’équilibre : coexistence des réactifs et produits en proportions constantes4. n(ester) = n(eau) = 0,8 mol ;n(acide) = n(alcool) = 0,4 mol, donc K = 4.5. Le rendement ρ = 0,8/1,2 = 67 %.6. Équation A + B = Y + W
État Avanc. Quantité de matières (mol)
Initial x = 0 2,4 1,2 0 0
Intermé-diaire x 2,4 – x 1,2 – x x x
Final xf = 1 mol 2,4 – xf = 1,4 1,2 – xf xf xf
7. xmax = 1,2 mol et ρ2 = x
xf
max
; ρ2 = 1
1 2, = 83 %.
8. ρ1 < ρ2 prévisible : introduction d’un réactif en excès par rapport à l’autre ; déplacement d’équilibre dans le sens direct.9. Le distillat est constitué par l’ester dont la tem-pérature d’ébullition est la plus faible.
10. On obtient n3 = m
M ;
n3 =85 5
74
,= 1,16 mol d’ester, soit ρ3 =
1 16
1 2
,
,= 96 %.
11. ρ1 < ρ3 prévisible, car on élimine l’ester au fur et à mesure de sa formation (le système chimique n’est jamais à l’équilibre).
17 Synthèse des constituants de l’essence de jasmin
1. Accélération de la réaction (élévation de tempé-rature) sans perte de réactifs (reflux des vapeurs liquéfiées dans le ballon).2. a) Estérification.b) Lente, limitée, athermique.3. CH3–COOH + C6H5–CH2–OH
= CH3–COO–CH2–C6H5 + H2O.

143Chapitre 10 - L’équilibre estéri: cation-hydrolyse
4. n(acide) = m
M ; n(acide) =
10
60 = 0,17 mol.
Pour que le mélange soit équimolaire, il faut que l’on ajoute 0,17 mol d’alcool benzylique, soit une masse : m' = n · M' ; m' = 0,17 × 108 = 18 g.
Donc un volume V' = m'
'µ, soit V' = 17,3 mL.
5. ρ = n
n
(ester)
(ester)obtenu
max
; ρ = 0 11
0 17
,
, = 0,67 = 67 %.
V2 = 15,8 mL correspondent à m2 = 16,7 g, donc :
n(ester)obtenu = m
M2
2 ;
n(ester)obtenu = 16 7
150
, = 0,11 mol.
6. a) Le rendement ne change pas en prolongeant la durée du chauffage, puisque la réaction est limi-tée et que le rendement maximal a été atteint avec la précédente manipulation.b) La quantité de catalyseur utilisée ne change pas le rendement de la réaction. Le catalyseur permet d’atteindre plus rapidement cette limite.c) En augmentant la quantité de matière initiale d’acide éthanoïque, le rendement sera amélioré par déplacement d’équilibre.
18 Synthèse de l’éthanoate de benzylea) Le cyclohexane est un gaz vecteur qui forme avec l’eau un composé gazeux qui monte jusque dans le réfrigérant droit. Au contact des parois froides, ce composé redevient liquide et tombe par gravité dans le tube gradué situé sous le réfrigérant droit. L’eau et le cyclohexane n’étant pas miscibles à froid, les deux liquides se séparent, le cyclohexane moins dense surnage, l’excédent retourne dans le ballon.b) La quantité d’eau obtenue est :
n = 2 5
18
, = 0,14 mol,
ce qui donne un rendement ρ = 0 14
0 17
,
, = 83 %.
c) Quantité d’ester obtenue : n' = 19
150 = 0,13 mol,
pour un rendement final ρ' = 76 %.Des pertes sont inévitables lorsqu’il faut séparer l’ester des autres constituants du mélange réac-tionnel.
19 Le benzoate de benzyle1. C6H5–COOH + C6H5–CH2–OH
= C6H5–COO–CH2–C6H5 + H2O.
2. C’est une estérification qui est lente, limitée et athermique.3. a) L’emploi d’un catalyseur permet d’atteindre plus rapidement l’état d’équilibre chimique sans le modifier.b) On utilise couramment l’acide sulfurique.4. a) Le chauffe-ballon (repéré par la lettre a) contient le ballon (repéré par la lettre b), lequel est surmonté d’un réfrigérant à boules (repéré par la lettre c). L’eau doit rentrer par la partie inférieure et ressortir par la partie supérieure du réfrigérant.b) Chauffage pour accélérer la réaction ; le reflux permet de ne rien perdre.
5. a) • n(acide benzoïque) = m
M ;
n(acide benzoïque) = 24
122 = 0,20 mol.
• n(alcool) = m
M ; n(alcool) =
15 6
108
, = 0,14 mol.
• m = µ · V ; m = 15,6 g pour l’alcool benzylique.L’acide benzoïque est donc le réactif en excès, puis-que les nombres stœchiométriques sont identiques dans l’équation chimique.
b) Rendement ρ = n
n
(ester)
(ester)obtenu
max
;
ρ = 0 094
0 14
,
, = 0,65 = 65 %.
n(ester obtenu) = m
M
'
' ;
n(ester obtenu) = 20
212 = 0,094 mol.
20 Hydrolyse d’un esterA. 1. L’éthanoate de 3-méthylbutyle, dont la for-mule est donnée ci-après, présente la fonction ester (en gras) :
CH3–CO–O–CH2–CH2–CH(CH3)2.2. Ester + eau = CH3–COOH + HO–CH2–CH2–CH(CH3)2.3. On obtient de l’acide éthanoïque (dont le groupe caractéristique est en gras) appartenant à la famille des acides carboxyliques et du 3-méthylbutan-1-ol (dont le groupe caractéristique est en italique) appartenant à la famille des alcools.
B. 1. La réaction support du dosage est :CH3COOH(aq) + HO
–(aq) = CH3COO–(aq) + H2O(l).
2. K = [CH COO (aq)]
[CH COOH(aq)] [HO (aq)]
3–
eq
3 éq–
éq· =
K
KA
e
;

144 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
K = 1,8 × 109
3. a) L’équivalence correspond au changement de réactif limitant.b) À l’équivalence, les réactifs ont été introduits dans les proportions stœchiométriques, d’où :
nA = CB · VBE.c) Le mélange initial de volume 50 mL a été réparti dans 10 béchers, donc chaque bécher contient un dixième du mélange réactionnel. D’où : n(quantité de matière d’acide qui se serait formé dans le milieu réactionnel initial) = 10 nA = 10 × CB × VBE.d) Les calculs donnent :
Instant t (min) 0 10 20 30 40 50 60 90 120
n (mmol) 0 19 34 45 54 61 68 78 84
Le graphe donne :
0
20
40
60
80
100
0 20 40 60 80 100 120 140
Instantt (min)
n (mmol)
Évolution de la quantité de matière d’acide au cours du temps.
C. 1. C’est une transformation lente, limitée et athermique.
2. n(ester) = µ ·V
M ; n(ester) = 0,10 mol.
n(eau) = 35
18 = 1,9 mol.
3. Le tableau d’évolution des espèces chimiques à l’aide de l’avancement donne :
Équation Ester + eau = acide + alcool
État Avanc. Quantité de matières (mol)
Initial x = 0 0,10 1,9 0 0
En cours x 0,10 – x 1,9 – x x x
Équilibre xéq 0,10 – xéq 1,9 – xéq xéq xéq
L’ester est le réactif limitant : xmax = 0,10 mol.
4. Le taux d’avancement final de cette réaction d’hydrolyse est :
τ1 = x
xéq
max
.
D’après l’énoncé, on considère : xéq = 84 mmol,
donc τ1 = 0 084
0 10
,
, = 0,84 = 84 %.
21 Sur les traces…1. CH3–COOH + C2H5–OH = CH3–COO–C2H5 + H2O(l).L’ester formé est de l’éthanoate d’éthyle.2. On refroidit les ampoules avant chaque dosage pour bloquer la réaction d’estérification : cette opération s’appelle une trempe.3. a) Le tableau d’évolution des espèces chimiques à l’aide de l’avancement donne :
Équation Acide + alcool = ester + eau
État Avanc. Quantité de matières (mmol)
Initial x = 0 100 100 0 0
En cours x 100 – x 100 – x x x
Équilibre xéq 100 – xéq 100 – xéq xéq xéq
Si la réaction était totale :xmax = 0,1 mol = 100 mmol.
b) Sachant que n = quantité de matière d’acide étha-noïque, on peut voir d’après le tableau ci-dessus que : n = 100 – xf, donc xf = 100 – n,d’où les tableaux suivants.
Instant t (h) 0 4 10 20 40 100 150 200 250 300
Avancement final (mmol) 0 25 36 48 56 64 65 66 67 67
Taux d’avan-cement (%) 0 25 36 48 56 64 65 66 67 67
4. Par définition : τ = x
xf
max
.
Les valeurs sont regroupées dans le tableau précédent.5.
0
20
40
60
80
100
0 100 200 300
Instantt (h)
Avancement (%)
À 100 °C À une température supérieure
Évolution du taux d’avancement final.
6. La transformation, lente, dure 300 h.Elle est limitée : son taux d’avancement maximal n’atteint pas 100 %.7. Cf. graphe précédent : la température est un fac-teur cinétique uniquement.

145Chapitre 10 - L’équilibre estéri: cation-hydrolyse
Exercices supplémentaires
1. La synthèse du benzoate de méthyle(D’après Bac Amérique du Nord, 2006.)
Le benzoate de méthyle, ester utilisé en parfumerie, est un des constituants de diverses huiles essentiel-les (essence de Niobé, d’œillet ou d’ylang-ylang).On prépare le benzoate de méthyle par réaction entre l’acide benzoïque, de formule C6H5COOH, et le méthanol, de formule CH3OH.Pour réaliser cette transformation, on mélange une masse m1 = 12,2 g d’acide benzoïque avec un volume
V2 = 30 mL de méthanol, quelques gouttes d’acide sulfurique concentré et quelques grains de pierre ponce.On chauffe à reflux pendant 60 min. Après traite-ment de la phase contenant l’ester, on isole une masse égale à 9,52 g de benzoate de méthyle.1. Déterminer les quantités de matière initiales n1 d’acide benzoïque et n2 de méthanol.2. Au vu du mode opératoire, identifier les facteurs cinétiques sur lesquels on a joué pour minimiser la durée d’évolution de cette transformation.
Les compétences expérimentales
(page 224)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est utile de connaître les différents montages possibles pour une synthèse organique. De plus, il est nécessaire de savoir les « monter » et les faire fonctionner. Cette activité doit conduire l’élève à justifier ses choix expé-rimentaux.
Corrigé1. Montages pour synthèse
1 Support élévateur 4 Colonne de Vigreux 7 Erlenmeyer
2 Chauffe-ballon 5 Réfrigérant à boules 8 Réfrigérant droit
3 Ballon 6 Thermomètre 9 Support ou potence
2. Synthèse du méthanoate d’éthyle1. La température d’ébullition du méthanoate d’éthyle étant de loin la plus faible, c’est le montage C qu’il faut choisir.2. Ce montage est celui de la distillation fractionnée.3. On peut espérer un rendement proche de 100 %, car l’extraction de l’es-ter au fur et à mesure de sa fabrication ne permet pas au système d’être à l’équilibre chimique.
3. Synthèse du méthanoate de butyle1. La température d’ébullition de l’ester proche de celle des autres consti-tuants oblige à choisir le montage A.2. C’est un montage de chauffage à reflux3. On peut espérer un rendement de 67 % si les réactifs sont introduits en proportions stœchiométriques.
4. Utilisation des montages précédents1. Il faut ajouter des billes de verre ou des grains de pierre ponce.2. La circulation de l’eau dans un réfrigérant doit être ascendante3. Il ne faut pas arrêter la circulation de l’eau lorsque cesse le chauffage, le milieu réactionnel encore chaud est toujours susceptible d’émettre des vapeurs qu’il faut refroidir.

146 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
3. On a effectué un chauffage à reflux du mélange réactionnel : quelle en est l’utilité ?4. Écrire l’équation de la réaction modélisant la synthèse du benzoate de méthyle.5. Déterminer le réactif limitant.6. En déduire la quantité de matière théorique d’es-ter que l’on pourrait obtenir si la transformation était totale.7. Définir et calculer le rendement expérimental de cette synthèse.8. D’après les températures d’ébullition des consti-tuants, quelle est la seule façon pour le chimiste d’augmenter le rendement de cette synthèse ?
Données
ComposésMasse
molaire(g · moL–1)
Masse volumique(g · mL–1)
Température d’ébullition
(°C)
Acide benzoïque 122 1,3 249
Méthanol 32 0,80 64,65
Benzoate de méthyle 136 1,1 199,6
Corrigé1. La quantité de matière d’acide benzoïque est :
n1 = m
M1
1
; n1 = 0,100 mol.
La quantité de matière de méthanol est :
n2 = m
M2
2
; n2 = 24
32 = 0,75 mol.
Avec m2 = µ · V2 ; m2 = 24 g.
2. Les facteurs cinétiques sont la température (le chauffage à reflux) et la présence d’un catalyseur (acide sulfurique).
3. Éviter les pertes en réactifs grâce au reflux.
4. C6H5–COOH + CH3–OH = C6H5–COO–CH3 + H2O.
5. Le réactif limitant est l’acide benzoïque, en plus faible quantité compte tenu des nombres stœchio-métriques.
6. On peut obtenir au maximum 0,100 mol d’ester.
7. On obtient 9,52 g d’ester, soit 0,07 mol.Le rendement est de 70 %.
8. La seule façon pour le chimiste d’augmenter le rendement de cette synthèse consiste à mettre un des réactifs en excès par rapport à l’autre.
2. Obtention du méthanoate de méthyle(D’après Bac Inde, 2000.)
On étudie la réaction entre l’acide méthanoïque et le méthanol. Dans plusieurs récipients, on mélange une masse m1 = 2,3 g d’acide carboxylique et une masse m2 = 1,6 g d’alcool. Les récipients sont scellés et placés dans une étuve à 30 °C. Au bout de 24 h, on constate que la masse restante d’acide est constante et vaut m = 0,76 g.
1. Écrire l’équation de la réaction modélisant la transformation chimique entre l’acide méthanoïque et le méthanol en utilisant leurs formules semi-développées.2. Quelles sont les caractéristiques de cette transfor-mation ?3. Déterminer les quantités de matières initiales des réactifs.4. Dresser un tableau d’évolution du système chimi-que à l’aide de l’avancement de la réaction.5. À partir de la masse restante d’acide, déterminer l’avancement de la réaction.6. Définir et calculer le rendement de la synthèse de l’ester.7. Par quel procédé pourrait-on augmenter nota-blement le rendement de cette synthèse ?Données
Composé Masse molaire (g · mol–1)
Température d’ébullition (°C)
Acide carboxylique 46 100,7
Alcool 32 64,7
Ester 60 31,5
Corrigé1. H–COOH + CH3 – OH = H–COO–CH3 + H2O.2. Cette transformation est lente, limitée et ather-mique.
3. n(acide) = m
M1
1
; n(acide) = 2 3
46
, = 0,05 mol.
n(alcool) = m
M2
2
; n(alcool) = 1 6
32
, = 0,05 mol.
4. Équation Acide + alcool = ester + eau
État Avanc. Quantité de matières (mol)
Initial x = 0 0,05 0,05 0 0
En cours x 0,05 – x 0,05 – x x x
Équilibre xéq 0,05 – xéq 0,05 – xéq xéq xéq

147Chapitre 10 - L’équilibre estéri: cation-hydrolyse
5. n(acide)restant = 0 76
46
, = 0,017 mol,
donc d’après le tableau ci-dessus :0,017 = 0,05 – xéq.
On trouve :xéq = 0,05 – 0,017 = 0,033 mol = n(ester)apparu.
6. Rendement : ρ = n
n
(ester)
(ester)apparu
max
;
ρ = 0 033
0 05
,
, = 0,66 = 66 %.
7. On peut augmenter le rendement de cette syn-thèse en utilisant un montage de distillation frac-tionnée : le système ne sera jamais à l’équilibre chimique car l’ester de température d’ébullition la plus faible sera extraite du mélange au fur et à mesure de sa formation.
3. Autre suivi cinétique (D’après Bac Inde, 2003.)
On étudie la cinétique de la formation d’un ester à partir d’acide éthanoïque et de propan-1-ol.On maintient, à la température constante θ, sept erlenmeyers numérotés 1, 2, 3, …, 7, contenant cha-cun un mélange de 0,500 mol d’acide éthanoïque et de 0,500 mol de propan-1-ol.Tous les tubes sont préparés à l’instant de date t = 0 s et on dose d’heure en heure l’acide restant dans le mélange. On peut ainsi en déduire la quantité de matière d’ester formé :– à t = 1 h, dosage de l’erlenmeyer 1 ;– à t = 2 h, dosage de l’erlenmeyer 2, etc.
A. La réaction d’estérification1. En utilisant les formules semi-développées, écrire l’équation de la réaction modélisant l’estérifi-cation et nommer l’ester formé.2. On dispose d’un flacon de propan-1-ol pur : quel volume de cet alcool doit-on verser dans chacun des sept erlenmeyers ?3. Exprimer la quantité de matière d’ester formé dans un erlenmeyer à un instant de date t en fonc-tion de la quantité de matière d’acide restant.
B. Titrage de l’acide restant• Mode opératoireÀ l’instant de date t considéré, le contenu de l’erlen-meyer est versé dans une fiole jaugée puis dilué avec de l’eau distillée pour obtenir 100 mL de solution. On en prélève 5 mL que l’on verse dans un bécher.
On titre cette solution par une solution aqueuse d’hydroxyde de sodium (soude), Na+(aq) + HO–(aq), de concentration molaire en soluté apporté CB = 1,0 mol · L–1. On en déduit la quantité de matière d’acide restant dans le bécher puis dans les 100 mL de départ, ce qui permet de déterminer la quantité d’ester à l’instant de date t dans les 100 mL de départ.4. Écrire l’équation de la réaction support du titrage.5. Rappeler la définition de l’équivalence.6. Pour l’erlenmeyer 1 (t = 1 h), le volume de solu-tion de soude versé pour atteindre l’équivalence est 14,2 mL. En déduire la quantité de matière d’acide restant dans l’erlenmeyer et la quantité de matière d’ester formé à cet instant.
C. Cinétique de la réaction d’estérificationLe titrage des solutions contenues dans les sept erlenmeyers précédents a permis le tracé de la courbe donnée ci-après.
00,050,100,150,200,250,300,350,40
0 1 2 3 4 5 6 7 8
t (h)
x ester (mol)
L’avancement de la réaction est défini par la quan-tité de matière x d’ester formé.7. a) Dresser le tableau descriptif de l’évolution du système chimique.b) Déterminer l’avancement maximal xmax ainsi que l’avancement à l’équilibre xéq.c) Comparer ces deux valeurs et déterminer le ren-dement ρ de la transformation.8. Rappeler l’expression de la vitesse volumique v d’une réaction. Quelle interprétation géométrique ou graphique peut-on en donner ? Comment cette vitesse évolue-t-elle au cours de la transformation ? Justifier.9. Calculer la constante d’équilibre K ' de cette réac-tion d’estérification.Données
Masses molaires atomiques en g · mol–1 : M(H) = 1,0 ; M(C) = 12 ; M(O) = 16.
Masse volumique du propan-1-ol : 0,80 g · cm–3.
•
•

148 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
Corrigé1. CH3–COOH + CH3–(CH2)2–OH
= CH3–COO–(CH2)2–CH3 + H2O.L’ester est l’éthanoate de propyle.
2. On veut 0,500 mol de propan-1-ol, soit une masse :
m = n · M ; m = 0,5 × 60 = 30 g.
On sait que µ = m
V, donc V =
m
µ ;
V = 30
0 80, = 37,5 mL.
On doit verser 37,5 mL de cet alcool dans chacun des erlenmeyers.
3. On sait qu’à un instant t, x(t) = avancement et que x(t) = n(ester) à cet instant.
Par ailleurs : 0,5 – x(t) = n(acide restant),donc x(t) = 0,5 – n(acide restant),d’où n(ester) = 0,5 – n(acide restant).
4. CH3COOH(aq) + HO–(aq) = CH3COO
–(aq) + H2O(l).
5. L’équivalence correspond au changement de réactif limitant.
6. n(acide restant) = CBVBE ;n(acide restant) = 1,42 × 10–2 mol à l’instant de date t = 1 h dans les 5 mL de solution dosée.
Donc dans les 100 mL de la fiole jaugée, il y a 20 fois plus d’acide, soit 0,284 mol.
D’où : n(ester) = 0,5 – 0,284 = 0,216 mol.
7. a)Équation Acide + alcool = ester + eau
État Avanc. Quantité de matières (mmol)
Initial x = 0 0,5 0,5 0 0
En cours x 0,5 – x 0,5 – x x x
Équilibre xéq 0,5 – xéq 0,5 – xéq xéq xéq
b) Donc xmax = 0,5 mol.D’après le graphe fourni : xéq = 0,33 mol.c) On constate que xéq < xmax, ce qui signifie que la réaction est limitée. Son rendement vaut :
ρ = x
xéq
max
; ρ = 0 33
0 5
,
, = 0,66 = 66 %.
8. La vitesse volumique d’une réaction est égale à la dérivée de l’avancement par rapport au temps divisée par le volume du mélange réactionnel :
v = 1
V
x
t·d
d.
La valeur de cette vitesse se calcule à un instant de date t en divisant la valeur du coefficient directeur de tangente à la courbe x(t) à cet instant t par le volume du mélange réactionnel.La vitesse de la réaction diminue au cours du temps car les tangentes sont de plus en plus horizontales, la vitesse est nulle à t = 6 h.
9. K' = n n
n n
(ester) (eau)
(acide) (alcool)éq éq
éq éq
·
· ;
K' = 0 33 0 33
0 17 0 17
, ,
, ,
××
= 3,8.

149Chapitre 11 - Contrôle du rendement par le choix des réactifs
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS CONNAISSANCES ET SAVOIR-FAIRE EXIGIBLES
Activités documentaires et expérimentales :Synthèse de l’aspirine à par-tir d’un anhydride d’acide. Identification par CCM.Synthèse et propriétés d’un savon.Choix des conditions expé-r imentales permet tant , lorsque plusieurs réactions chimiques interviennent dans une transformation, d’en privilégier une.Exemple d ’application : titrage direct de l’aspirine.
2. Des exemples de contrôle de l’évolution de systèmes chimiques pris dans l’in-dustrie chimique et dans les sciences de la vie– Changement d’un réactif.Synthèse d’un ester à partir d ’un anhydride d ’acide et d’un alcool.Hydrolyse basique des esters : applications à la saponif i-cation des corps gras (pré-parations et propriétés des savons, relations structure-propriétés).
– Calculer le rendement d’une transformation.– Mettre en œuvre au laboratoire, en justifiant le choix du matériel à utiliser : chauffage à reflux, distillation fractionnée, cristallisa-tion, filtration sous vide, chromatographie sur couche mince.– Respecter les consignes de sécurité.– Justifier les étapes d’un protocole.– Écrire l’équation de la réaction d’un anhydride d’acide sur un alcool et de l’hydrolyse basique d’un ester.– Savoir que l’action d’un anhydride d’acide sur un alcool est rapide, qu’elle donne un ester et que l’avancement maximal est atteint.– Savoir que l’hydrolyse basique d’un ester est rapide et que l’avancement maximal est atteint.– Identifier la partie hydrophile et la partie hydrophobe d’un ion carboxylate à longue chaîne.
11 Contrôle du rendement par le choix des réactifs
Cours
Découpage du cours1. Rappel sur l’estérification et l’hydrolyse p. 2262. Préparation industrielle des esters p. 2263. Hydrolyse basique des esters p. 2284. Propriétés des savons p. 230
Dans le chapitre 10, les élèves ont étudié l’équilibre estérification-hydrolyse de l’ester et les rendements correspondants.Le chapitre 11 permet de montrer :– d’une part, que l’on parvient à produire une même espèce chimique, soit l’ester en modifiant l’un des réactifs, soit l’ion carboxylate en modifiant le pH du milieu ;– d’autre part, que le rendement de ces transforma-tions est nettement amélioré grâce à ces modifica-tions.L’approche proposée est simple et s’appuie large-ment sur des observations expérimentales.
1. Rappel sur l’estérification et l’hydrolyseRappel du chapitre précédent et introduction des thèmes abordés dans le chapitre.
2. Préparation industrielle des estersPrésentation des anhydrides d’acide, nomenclature, propriétés et sécurité.
♦ L’action d’un anhydride d’acide sur un alcool est rapide : elle donne un ester et l’avancement maxi-mal est atteint.• Exemples :– préparation de l’éthanoate de butyle (Activité exploitation d’expériences, p. 232) ;– préparation de l’aspirine (Activité TP, p. 234-235).
3. Hydrolyse basique des estersL’hydrolyse basique d’un ester est rapide et l’avance-ment maximal est atteint.• Exemple : hydrolyse basique des corps gras, appe-lée saponification (Activité TP, p. 233).
4. Propriétés des savonsPour illustrer les limites d’utilisation des savons, il est commode de préparer une solution concentrée d’eau savonneuse à partir d’eau distillée et de savon de Marseille râpé. On montre facilement la forma-tion des précipités dans les quatre milieux propo-sés.Remarque : il est aisé de montrer que cette précipitation n’a pas lieu en utilisant un détergent à la place du savon.

150 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
Préparation de l’éthanoate de butyle (page 232)
Cette préparation a déjà été réalisée au chapitre 10. L’activité proposée ici, sous forme d’exploitation de résultats expérimentaux, illustre que le changement de réactif modifie grandement le rendement de la transformation.
A Réactifs et produits de la réaction1.
H3C C
O
C
O
H3C
O
+ CH3CH2CH2CH2OH = CH3COO(CH2CH2CH2CH3)+ CH3COOH.
2. Butan-1-ol• R 10-22-37/38-41 : inflammable ; nocif en cas d’ingestion ; irritant pour les voies respiratoires et la peau ; risque de lésions oculaires graves.• S 7/9-13-26-37/39-46 : conserver le récipient bien fermé et dans un endroit bien ventilé ; conser-ver à l’écart des aliments et boissons ; en cas de contact avec les yeux, laver immédiatement et abondamment avec de l’eau et consulter un spécia-liste ; porter des gants appropriés et un appareil de protection des yeux ; en cas d’ingestion, consulter un médecin.
Anhydride éthanoïque• R 10-20/22-34 : inflammable ; nocif par inhala-tion et par ingestion ; provoque des brûlures.• S 26-36/37-39/45 : en cas de contact avec les yeux, laver immédiatement et abondamment avec de l’eau et consulter un spécialiste ; porter un vête-ment de protection et des gants appropriés ; porter un appareil de protection des yeux et du visage ; en cas d’accident ou de malaise, consulter immédiate-ment un médecin.
Éthanoate de butyle• R 10 : inflammable.• S 25 : éviter le contact avec les yeux.
Acide éthanoïque• R 10/35 : inflammable ; provoque de graves brû-lures.• S 23-26-36/37/39-45 : ne pas respirer les vapeurs ; en cas de contact avec les yeux, laver immédiate-ment et abondamment avec de l’eau et consulter un spécialiste ; porter un vêtement de protection
ACTIVITÉ
1
ACTIVITÉ
1 approprié, des gants et un appareil de protection des yeux et du visage ; en cas d’accident ou de malaise, consulter immédiatement un médecin.a) Chauffage loin de toute flamme : au moins l’un des corps est inflammable (ici tous).b) Chauffage sous la hotte ou à reflux : pour éviter de respirer les vapeurs (qui sont irritantes pour les voies respiratoires) et prévenir tout contact des vapeurs avec les yeux et la peau.
B Mode opératoire1. a) Quantités de matières :– butan-1-ol : n1 = 5 × 10
–2 mol ;– anhydride éthanoïque : n2 = 5 × 10
–2 mol.b) Pas de réactif limitant.2. a) On veut éliminer l’acide éthanoïque formé.b) Le gaz qui se dégage est du dioxyde de carbone.c) HCO3
–(aq) + CH3COOH(aq)= CO2(g) + H2O(l) + CH3COO
–(aq).d) Expérimentalement, le rajout d’une petite quan-tité d’eau permet de localiser la phase aqueuse.
C Exploitation des résultats1. a) n(ester) = 5 × 10–2 mol. D’où m(ester) = 5,8 g.b) (m((ester))obtenu = 6,5 × 0,88 = 5,7 g.2. a) Rendement de la préparation :
R = 5 7
5 8
,
, = 0,98.
b) Ce résultat doit être comparé à 0,66 (cf. chap. 10).
Préparationd’un savon (page 233)
Ce TP permet aux élèves de se familiariser avec la mise en œuvre expérimentale des opérations uni-taires demandées dans le programme :– chauffage à reflux,– relargage,– filtration sous vide.
A Réactifs et solvant1. a)
H2C O
HC
H2C
O
O
C
C
C
C17H33
C17H33
C17H33
O
O
O
ACTIVITÉ
2
ACTIVITÉ
2
Activités

151Chapitre 11 - Contrôle du rendement par le choix des réactifs
b) R– = –C17H33, correspond à CnH2n–1, donc à un radical insaturé.
2. a) Les deux réactifs sont solubles dans l’éthanol.La réaction se produit dans la masse des réactifs, et non seulement à l’interface des deux.b) Pour éviter l’évaporation de l’éthanol, on utilise un montage à reflux.L’inflammabilité de l’éthanol conduit à travailler hors de toute flamme, d’où l’emploi d’un chauffe-ballon.
B Mode opératoire1. La pierre ponce régularise l’ébullition.2. Le chauffage à reflux permet d’augmenter la durée de contact entre les réactifs à chaud, sans perdre de produits.3. C’est dans l’eau salée que la solubilité du savon est la plus faible, d’où sa précipitation.4. Il est préférable de verser d’abord le liquide, car le savon « encrasse » le filtre en bouchant les pores du papier filtre et empêche ensuite un bon essorage.
C Exploitation des résultats1.
H2C O
HC
H2C
O
O
C
C
C
C17H33
C17H33
C17H33
O
O
O+ 3(Na++HO–) =
H2C OH
HC
H2C
OH
OH
+ 3(C17H33 C O–+ Na+).
O
2. n(huile d’olive) = 13 0 8
884
× , = 0,012 mol.
n(base) = 0 040 100
40
, × = 0,1 mol.
L’équation de réaction montre que les proportions stœchiométriques sont telles que :
n(huiled’olive)
1 =
n(base)
3 ;
n(base)
3 =
0 1
3
, = 0,033 mol.
L’huile d’olive est donc le réactif limitant.
3. a) mth = 3 n(huile d’olive) · M(savon).mth = 3 × 0,012 × 304 = 10,9 g.
b) Comparaison avec le résultat expérimental.
D Coloration du savon1. Si le savon prend la couleur rose violacé en pré-sence de phénolphtaléine, c’est que son pH est supérieur à 9,8, donc basique.2. Il n’est pas prudent d’utiliser ce savon, car il pré-sente un pH trop élevé pour respecter la peau.
Préparation de l’aspirine et contrôle de sa pureté (page 234)
Ce TP répond à la lettre au libellé officiel du pro-gramme. Il permet aux élèves de se familiariser avec la mise en œuvre expérimentale des opérations uni-taires demandées :– chauffage à reflux,– filtration sous vide,– purification par recristallisation,– chromatographie sur couche mince.
A Quelques informations1. Précautions des personnes : ne pas respirer les vapeurs ; protéger les yeux et la peau.Précautions générales :– éviter la présence de flammes ou d’étincelles ;– travailler avec du matériel bien sec.2. L’anhydride éthanoïque réagit violemment avec l’eau.
B Préparation de l’aspirine1.
COOH
OH
+H3C C
O
O
CH3CO
=COOH
O C CH3
O
+ H3C COH
O.
2. a) n(acide salicylique) = 0,087 mol.n(anhydride éthanoïque) = 0,190 mol.b) L’équation de réaction montre que la réaction se fait mole à mole. Le réactif limitant est celui qui est en plus petite quantité de matière ; la valeur de l’avancement maximal sera égale à la quantité de ce réactif.Soit xmax = n(acide salicylique) = 0,087 mol.m(aspirine) = 0,087 × 180,2 = 15,6 g.
3. a) Rendement : RB = mB
15 6,.
b) L’interprétation du résultat est fonction de l’état de l’aspirine brute préparée.(Régulièrement, on obtient des rendements supé-rieurs à 100 %, liés à une préparation très humide et pouvant renfermer un certain nombre d’impuretés.)4. L’abaissement de température et l’agitation énergique permettent de faire précipiter l’aspi-rine.5. Le produit brut peut contenir des impuretés (l’acide salicylique, l’acide sulfurique et l’anhydride éthanoïque qui n’ont pas réagi).6. Le lavage à l’eau froide élimine les corps solubles dans l’eau non éliminés précédemment.
ACTIVITÉ
3
ACTIVITÉ
3

152 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
C Purification de l’aspirine
1. RR = m
mP
B
2
.
2. RP = 2
15 6
mP
,.
3. RP = RB · RR.
D Contrôle de pureté par C.C.M.1. Calcul des Rf.
2. Interprétation du chromatogramme :A : aspirine brute. Il apparaît deux taches, donc le corps n’est pas pur ; les rapports frontaux sont tels qu’il s’agit d’un mélange d’aspirine et d’acide sali-cylique.B : aspirine recristallisée. Il n’apparaît qu’une seule tache, il s’agit donc d’un produit pur, identique à l’aspirine de référence (C).
3. La recristallisation a permis d’éliminer une impureté. Son rôle est donc de purifier le produit brut préparé.
Activité supplémentaire
Contrôle de pureté de l’aspirine préparée par dosage
Ce dosage acido-basique indirect (ou en retour) permet de déterminer la pureté de l’aspirine recristallisée.La solution aqueuse d’hydroxyde de sodium (B) a pour concentration en soluté apporté :
CB = 0,700 mol · L–1.La solution aqueuse de chlorure d’hydrogène (A) a pour concentration en soluté apporté :
CA = 0,100 mol · L–1.
A Première phaseDans un erlenmeyer, introduire une quantité connue de solution (B) en excès VB0 = 10,0 mL avec une masse m = 500 mg d’aspirine à doser et chauffer l’ensemble au bain-marie pendant quelques minu-tes pour que la transformation d’hydrolyse du groupe caractéristique ester se produise en plus de la réaction acido-basique classique.
1. Pourquoi chauffer quelques minutes les deux réactifs ?
L’équation de réaction (1) modélisant la transfor-mation entre la solution d’hydroxyde de sodium et l’aspirine à chaud s’écrit :
OCOCH3
COOH
+ 2HO– =
OH
COO–
+ CH3COO– + H2O.
2. Calculer la quantité de matière d’hydroxyde de sodium nB0 (mol) initialement introduite.
3. Calculer la quantité de matière apparente d’aspi-rine naspirine (mol) initialement introduite.
B Seconde phaseRéaliser le dosage acido-basique de la solution aqueuse d’hydroxyde de sodium non utilisée dans la première phase par la solution (A).4. Quel indicateur coloré est-il possible d’utiliser pour repérer l’équivalence ? Repérer le volume à l’équivalence, VAE.5. Pourquoi utilise-t-on l’expression dosage en retour ou dosage indirect ?6. Calculer la quantité de matière d’acide chlorhydri-que nAE (mol) versée pour atteindre l’équivalence.7. En déduire :a) la quantité de matière de soude nB1 (mol) consommée dans la première phase ;b) la quantité de matière réelle n'aspirine d’aspirine contenue dans le prélèvement ;c) le pourcentage de pureté de l’aspirine préparée.
Corrigé1. Le chauffage des deux réactifs pendant quel-ques minutes permet à la réaction d’hydrolyse de l’ester d’être totale.2. nB0 = CB · VB0 ; nB0 = 0,700 × 10,0 × 10
–3 = 7,0 × 10–3 mol.
3. n(aspirine) = m
M ;
n(aspirine) = 0 500
180 2
,
, = 2,77 × 10–3 mol.
4. Le bleu de bromothymol permet de repérer l’équivalence.5. Ce terme est utilisé ici, car on ne dose pas direc-tement l’aspirine, mais la partie de la solution d’hydroxyde de sodium initialement introduite qui n’a pas réagi avec l’aspirine.
153Chapitre 11 - Contrôle du rendement par le choix des réactifs
6. nAe = CA · VAE = …… mol.
7. a) La quantité d’hydroxyde de sodium consom-mée dans la première phase est donc :
nB1 = nB0 – nAE ;nB1 = 7 × 10–3 – ….. = ….. mol.
b) L’équation de réaction (1) montre que :
n'aspirine = nB12
; n'aspirine = .....
2 = … mol.
c) Pourcentage de pureté :
p = naspirine'
–,2 77 10 3× = 0, … soit … %.
1 Nomenclature
Form
ule
sem
i-dé
velo
ppée
CH3C OH
O O CCH3
OC
CH3
O
O CC2H5
OC
CH3
O
CH2OH
CHOH
CH2OH
Fonc
-ti
on Acide carboxylique
Anhydride d’acide
Anhydride d’acide
Poly-alcool
Nom Acide
éthanoïqueAnhydride éthanoïque
Anhydride éthanoïque
et propanoïque
Glycérol
Form
ule
topo
logi
que
OH
O
OO
OO
O
O
OHOH
OH
2 Équations de réactions1. CH3CH2OH(aq) + CH3COOH(aq)
= CH3COOC2H5(aq) + H2O(l).
2. CH3CH2OH(aq) + CH3COOCOCH3(aq)= CH3COOC2H5(aq) + CH3COOH(aq).
3. CH3CH2OCOCH3(aq) + Na+(aq) + HO–(aq)
= CH3COO–(aq) + Na+(aq) + CH3CH2OH(aq).
3 Parallèle entre méthodes de production
Transformations Caractères
a) Estérification directe : acide + alcool
Réaction lente, limitée et athermique
b) Production à partir d’un anhydride d’acide et d’un alcool
Réaction rapide et totale
4 Hydrolyse basique d’un ester1. Hydrolyse : réaction chimique de l’eau sur une substance au cours de laquelle une liaison chimique est brisée.
2. R C
O
O
R'
HO–
–
R C
O
O
+ R'OH
ester +
+
base =
=
ion carboxylate + alcool
3.
HA(aq)prédomine
A–(aq)prédomine
pKA + 1pKA
HA(aq)/A–(aq)pKA – 1
pH
5 Aspirine1. Aspirine ou acide acétylsalicylique ou acide ortho-acétyl benzoïque.2.
COOH
OH
+H3C C
O
O
CH3CO
=COOH
O C CH3
O
+ H3C COH
O
3. Estérification.4. Antipyrétique : médicament dont le rôle est de combattre la fièvre.Analgésique (ou antalgique) : médicament qui pré-vient ou diminue la sensation de douleur.Anti-inflammatoire : médicament qui lutte contre l’inflammation.
6 Vocabulaire1. Phile : aimer, rechercher.Phobe : avoir peur de, détester.2. a) R–COO–.b) & c) Schématisé par :
CO–
O
Rqueue hydrophobe
COO–
tête hydrophile
3. a) Tête hydrophile : partie chargée électrique-ment, qui peut s’unir facilement avec l’eau.Queue lipophile : partie apolaire, pouvant se lier avec les matières graisseuses.
Corrigés des exercices (page 236)

154 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
b) Tête hydrophile : tête lipophobe.Queue lipophile : queue hydrophobe.
4. Cf. schéma Fig. 8, p. 230 du Manuel de l’élève.
7 Préparation d’un ester1. M(anhydride propanoïque) = 130 g · mol–1.M(butan-1-ol) = 74 g · mol–1.n(anhydride propanoïque) =
6 5
130
, = 0,05 mol.
n(butan-1-ol) = 0,05 mol.n(butan-1-ol) = m(butan-1-ol) · M(butan-1-ol) ;n(butan-1-ol) = 0,05 × 74 = 3,7 g.2. a) C2H5COOCOC2H5 + C4H9OH
= C2H5COOC4H9 + C2H5COOH.b) Réaction rapide et totale.3.
Équation de réaction C2H5COOC4H9 + C2H5COOH= C2H5COOC4H9 + C2H5COOH.
État Avanc.(mol)
Quantités de matières (mol)
Initial 0,05 0,05 0 0
Intermé-diaire x
0,05 – x
0,05 – x
x x
Final xmax = 0,05 0 0 xmax = 0,05 xmax = 0,05
M(ester) = 130 g · mol–1.m(ester) = n(ester) · M(ester) ;m(ester) = 0,05 × 130 = 6,5 g.
8 Rendement d’une transformation chimique
1. R = n
nobtenue
théoriqueespérée
= m
mobtenue
théoriqueespérée
.
2. La réaction se fait mole à mole, donc l’alcool (en plus petite quantité) est le réactif limitant.La quantité maximale d’ester espérée est de 0,20 mol, d’où le rendement :
R = 0 19
0 20
,
, = 0,95, soit 95 %.
9 Préparation d’un savon1. Solvant commun aux deux réactifs (non miscibles).2. a) Pour faire précipiter le savon.b) Cette opération porte le nom de relargage.
10 Efficacité d’un savon1. Eau dure : eau riche en ions calcium, Ca2+(aq), et/ou magnésium, Mg2+(aq).2. Une partie du savon précipite avec les deux ions présents et ne participe pas au lavage.
3. Le dispositif généralement installé sur le réseau d’alimentation est un adoucisseur, qui permet de passer d’une eau dure à une eau douce.
11 Sécurité au laboratoire1. & 2.Anhydride acétique• R 10-20/22-34 : inflammable ; nocif par inhala-tion et par ingestion ; provoque des brûlures.• S 26-36/37/39-45 : en cas de contact avec les yeux, laver immédiatement et abondamment avec de l’eau et consulter un spécialiste ; porter un vête-ment de protection et des gants appropriés ; porter un appareil de protection des yeux et du visage ; en cas d’accident ou de malaise, consulter immédiate-ment un médecin.
Méthyl-3 butanol-1• R 10-20 : inflammable ; nocif par inhalation.• S 24-25 : éviter le contact avec la peau et les yeux.
Cf. schéma d’un montage à reflux sur un chauffe-ballon dans les activités (Fig. 1, p. 234 du manuel).
12 Préparation artisanale du savon1. Ce sont des sels d’acides gras.2. Mélange eau-glycérol.3. Relargage : méthode de séparation d’un composé en solution aqueuse par diminution de sa solubi-lité, provoquée par l’addition d’un autre composé. Dans cette phrase, le terme de neutralisation est pris au sens général (élimination de l’excès de soude) pour se soustraire à ses effets corrosifs.4. a) La neutralisation parfaite correspond à pH = 7.b) Il s’agit d’un abus de langage, car le savon (sel d’un acide gras, acide faible, et d’une base forte, soude) présente un caractère basique.5. Les lavages successifs à l’eau entraînent l’excès de soude et de sel (composés solubles dans l’eau).6. a) Solidification : passage état liquide à solide.b) À la température de 50-60 °C, une partie de l’eau s’évapore, provoquant la précipitation totale du savon.
13 Bizarre, bizarre
Exercice résolu dans le manuel de l’élève.
14 Préparation de l’aspirine1. Anhydride éthanoïque ou anhydride acétique, de formule chimique :

155Chapitre 11 - Contrôle du rendement par le choix des réactifs
H3C OC C
CH3
O O
2. Montage à reflux
Sortie de l’eau
Arrivée d’eau
Mélangeréactionnel
+ pierre ponce
Chauffe-ballon
Ballon
Réfrigérantà boules
3. Compte tenu des coefficients stoechiométriques de l’équation de réaction (1 mole pour 1 mole), le réactif limitant est celui dont la quantité de matière est la plus petite.• Quantité d’acide salicylique :
n1 = m
M ; n1 =
5 00
138
, = 0,0362 mol.
• Quantité d’anhydride A :
n2 = m
M2
2
, or m2 = µ · V2 et µ = d · µeau
avec µeau = 1 g · cm–3, donc n2 =
µ ·V
M2
2
= d V
M
· 2
2
;
n2 = 1 08 7 0
102
, ,× = 0,0741 mol.
Le réactif limitant est l’acide salicylique
4. a) Si le rendement était de 100 %, le réactif limitant serait totalement consommé, on aurait :
n(aspirine) = nlimitant = n(ac.salicylique).
Donc : m(aspirine)max = n · M ;m(aspirine)max = 0,0362 × 180 = 6,52 g.
b) Rendement effectif :
ρ = m
m
(aspirine)
(aspirine)formée
max
; ρ = 4 20
6 52
,
, = 0,64, soit 64 %.
15 Interprétation d’une C.C.M.1. CH3COOCOCH3 + C3H7CH2OH
= CH3COOC4H9 + CH3COOH.2. Produit brut : directement issu de la préparation, contient des impuretés (sous-produits de préparation).Produit pur : ayant subi une purification dans le but d’éliminer les impuretés présentes.3. Dans le produit brut, on peut trouver : de l’anhy-dride éthanoïque, du butan-1-ol de l’ester et de
l’acide éthanoïque. Tous constituent des impure-tés, sauf l’ester que l’on cherche à préparer.4. A et B sont deux références.C est le produit brut préparé, il présente trois taches : une est à la hauteur de l’ester de référence, il s’agit donc de cet ester ; une autre est à la hau-teur du butan-1-ol de référence, donc présent dans le produit brut ; une troisième, correspondant à un autre composé, n’est pas identifiable.D est le produit pur : il ne présente qu’une seule tache, ne correspondant qu’à un seul composé qui a migré à la même hauteur que l’ester de référence. Il s’agit donc de cet ester.
16 Interprétation d’un graphe1. a) À pH = 3 en milieu acide, la vitesse d’hydro-lyse de l’ester augmente au fur et à mesure de l’évo-lution de la transformation : la réaction produit de l’acide carboxylique, lequel fait diminuer le pH.b) À pH = 7 en milieu neutre, la vitesse d’hydrolyse de l’ester diminue au fur et à mesure de l’évolution de la transformation : la réaction produit de l’acide carboxylique, lequel fait diminuer le pH vers 4,5.c) À pH = 10 en milieu basique, l’acide carboxylique formé réagit avec la base présente. Dans ce cas, l’hydrolyse basique reste rapide.2. Déplacement de l’équilibre : l’équilibre est déplacé si l’un des produits de la transformation est éliminé au fur et à mesure de sa formation, ce qui est le cas à pH = 10. Cette hydrolyse basique porte alors le nom de saponification.
17 Mal de tête
Exercice résolu dans le manuel de l’élève.
18 Saponification d’une huile1. a) Le propan-1,2,3-triol est couramment appelé glycérol.b) Les trois fonctions esters sont repérées ci-après.
H2C O
HC
H2C
O
O
C
C
C
C17H33
C17H33
C17H33
O
O
O
c) L’hydrolyse basique des esters porte le nom de saponification.d) Les produits de la transformation sont le glycé-rol et l’oléate de sodium.

156 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
Exercices supplémentaires1. L’arôme de bananeLes questions 1, 2, 3, 4 et 5 sont indépendantes.L’arôme de banane est dû :– soit à la présence d’extraits naturels de banane,– soit à la présence d’un composé artificiel, l’acétate de butyle (ou éthanoate de butyle).1. Donner l’une des raisons qui font qu’un indus-triel puisse plutôt avoir recours à l’utilisation du composé artificiel.Questions préliminaires2. L’acétate de butyle a pour formule semi-dévelop-pée :
CH3CO
O
CH2CH2CH2CH3
a) À quelle famille de composés organiques appar-tient cette espèce chimique ?b) La synthèse de l’acétate de butyle E peut être réa-lisée à partir d’un acide carboxylique A et d’un alcool B.L’équation associée à la réaction modélisant la syn-thèse de E s’écrit :
A + B = E + H2O.
Les compétences expérimentales
(page 242)
Dans l’optique de l’épreuve d’évaluation des capacités expérimentales, il est indispensable de maîtriser l’opération de distillation fractionnée. Ce type d’opération conduit l’élève à identifier les risques encourus et à choi-sir correctement les techniques pour travailler dans les meilleures condi-tions de sécurité.
Corrigé1. Rectification : distillation fractionnée d’un liquide pour le purifier.2. Risque : R11, facilement inflammable.3. Éléments du montage :
1, support élévateur ; 2, chauffe-ballon ; 3, ballon bicol ou bouilleur ; 4, ampoule de coulée ; 5, colonne à distiller ; 6, tête de colonne ; 7, thermomètre ; 8, réfrigérant droit ; 9, allonge coudée ; 10, erlen-meyer pot de recette.
4. Le pot de recette est plongé dans un mélange réfrigérant eau + glace pour éviter l’évaporation de l’ester, très volatil.5. a) Les vapeurs d’ester puis le distillat dans les deux tiers du réfrigérant droit descendent, tandis que l’eau de réfrigération passe dans la double enveloppe, entrant par le bas et ressortant par le haut.b) Un tel mode de circulation croisé est dit à contre-courant.6. On utilise une ampoule de coulée pour introduire progressivement le mélange à distiller dans le bouilleur lorsque le volume du mélange à dis-tiller est supérieur aux deux tiers de la capacité du bouilleur.7. C’est la valeur de la température en tête de colonne qui permet de conduire l’opération de distillation et de repérer la fraction qui passe en tête de colonne.8. a) On peut contrôler la nature du distillat obtenu en mesurant son indice de réfraction.b) À l’aide d’un réfractomètre.

157Chapitre 11 - Contrôle du rendement par le choix des réactifs
Reconnaître A et B parmi les composés suivants.
Acides carboxyliques Alcools
Acide méthanoïque
HCOOHButan-1-ol
CH3(CH2)3 OH
Acide acétique(ou acide éthanoïque)
CH3COOH Éthanol CH3CH2OH
Acide butanoïque
CH3CH2CH2COOHPropan-1-ol
CH3CH2CH2OH
Synthèse de l’acétate de butyle au laboratoireOn se propose de synthétiser au laboratoire l’acétate de butyle E à partir des composés A et B et de réali-ser un suivi cinétique de cette synthèse. Pour cela, on introduit dans un bécher placé dans un bain d’eau glacée :– un volume VA = 5,8 mL d’acide carboxylique A ;– un volume VB = 9,2 mL d’alcool B (soit 0,10 mol) ;– quelques gouttes d’acide sulfurique concentré.
Données
Masse molaire M (g · mol–1)
Masse volumique m (g · mL–1)
Température d’ébullition sous
pression normale qéb (°C)
A 60 1,05 118,2
B 74 0,81 117,7
E 116 0,87 126,5
Eau 18 1,00 100,0
3. a) Indiquer pourquoi il est nécessaire de placer initialement le bécher dans un bain d’eau glacée.b) Justifier succinctement l’intérêt d’ajouter de l’acide sulfurique, sachant qu’il ne participe pas à la transformation chimique étudiée.c) Le mélange initial acide + alcool est équimo-laire : la quantité d’acide introduit est égale à 0,10 mole. En utilisant les données, écrire l’expression littérale permettant de calculer la quantité de matière d’acide carboxylique A introduite dans un volume VA.d) Déterminer l’avancement maximal de la réaction dans ces conditions. (Pour la résolution de cette question, l’utilisation ou non d’un tableau d’évolu-tion est laissée au choix du candidat.)Suivi de la synthèse par titrage de l’acide restantOn agite le mélange initial et on répartit avec préci-sion le mélange dans 10 tubes à essais placés préala-blement dans un bain d’eau glacée ; chaque tube contient ainsi un dixième du volume du mélange initial. On munit chaque tube d’un réfrigérant.
On place ensuite simultanément tous les tubes dans un bain thermostaté à 80 °C et on déclenche alors le chronomètre (instant de date t0 = 0 s).Afin de réaliser un suivi temporel de la synthèse de l’acétate de butyle, on dose, à des instants détermi-nés, les acides restants (acide sulfurique et acide car-boxylique A) dans chacun des tubes par une solution aqueuse d’hydroxyde de sodium (soude), de concen-tration apportée C = 1,0 mol · L–1, en présence d’un indicateur coloré. Avant chaque titrage, on plonge le tube dans un bain d’eau glacée.Une étude préalable a permis de connaître le volume de soude nécessaire au titrage de l’acide sulfurique présent dans chacun des tubes. Les résultats expéri-mentaux des titrages successifs sont donnés ci-après. On désigne par Véq le volume de soude nécessaire au titrage de l’acide carboxylique seul.
t (min) 0 5 10 15 20 30 45 60 75 90
Véq (mL)
10,0 6,3 5,0 4,4 4,0 3,7 3,4 3,3 3,3 3,3
4. Quel est le rôle de l’indicateur coloré ?5. Justifier, sans calcul, l’évolution au cours du temps du volume de soude à verser pour atteindre l’équivalence.6. L’équation chimique associée au titrage de l’acide carboxylique seul par la soude est la suivante :
RCO2H(aq) + HO–(aq) = RCO2–(aq) + H2O(l).
Définir l’équivalence correspondant à ce titrage.7. En raisonnant sur le contenu d’un tube (c’est-à-dire sur un volume égal au dixième du volume du mélange réactionnel initial), exprimer la quantité de matière d’acide carboxy lique présent dans un tube à un instant de date t en fonction de C et de Véq (on peut utiliser un tableau d’évolution).8. Pour la totalité du mélange initialement préparé 5,8 mL d’acide carboxylique A et 9,2 mL d’alcool B :a) préciser la relation existant entre l’avancement de la réaction de synthèse de l’ester et la quantité de matière d’ester formé (pour la résolution de cette question, l’utilisation ou non d’un tableau d’évolu-tion est laissée au choix du candidat) ;b) montrer qu’à un instant de date t donné, l’avan-cement de cette réaction de synthèse de l’ester est donné par la relation : x = 0,10 – 10 C · Véq.
Évolution temporelle de l’avancement de la synthèse organiqueÀ partir des résultats expérimentaux, il est donc possible de tracer la courbe donnant l’évolution
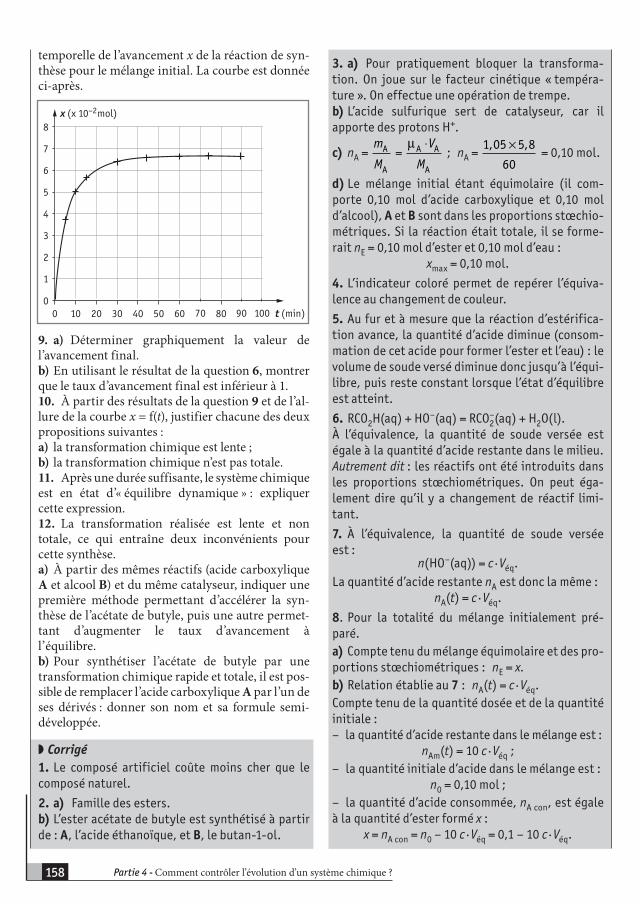
158 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
temporelle de l’avancement x de la réaction de syn-thèse pour le mélange initial. La courbe est donnée ci-après.
t (min)
x (x 10–2mol)
7
6
8
5
4
3
2
1
00 10 20 30 40 50 60 70 90 10080
9. a) Déterminer graphiquement la valeur de l’avancement final.b) En utilisant le résultat de la question 6, montrer que le taux d’avancement final est inférieur à 1.10. À partir des résultats de la question 9 et de l’al-lure de la courbe x = f(t), justifier chacune des deux propositions suivantes :a) la transformation chimique est lente ;b) la transformation chimique n’est pas totale.11. Après une durée suffisante, le système chimique est en état d’« équilibre dynamique » : expliquer cette expression.12. La transformation réalisée est lente et non totale, ce qui entraîne deux inconvénients pour cette synthèse.a) À partir des mêmes réactifs (acide carboxylique A et alcool B) et du même catalyseur, indiquer une première méthode permettant d’accélérer la syn-thèse de l’acétate de butyle, puis une autre permet-tant d’augmenter le taux d’avancement à l’équilibre.b) Pour synthétiser l’acétate de butyle par une transformation chimique rapide et totale, il est pos-sible de remplacer l’acide carboxylique A par l’un de ses dérivés : donner son nom et sa formule semi-développée.
Corrigé1. Le composé artificiel coûte moins cher que le composé naturel.
2. a) Famille des esters.b) L’ester acétate de butyle est synthétisé à partir de : A, l’acide éthanoïque, et B, le butan-1-ol.
3. a) Pour pratiquement bloquer la transforma-tion. On joue sur le facteur cinétique « tempéra-ture ». On effectue une opération de trempe.b) L’acide sulfurique sert de catalyseur, car il apporte des protons H+.
c) nA = m
MA
A
= µA A
A
⋅VM
; nA = 1 05 5 8
60
, ,× = 0,10 mol.
d) Le mélange initial étant équimolaire (il com-porte 0,10 mol d’acide carboxylique et 0,10 mol d’alcool), A et B sont dans les proportions stœchio-métriques. Si la réaction était totale, il se forme-rait nE = 0,10 mol d’ester et 0,10 mol d’eau :
xmax = 0,10 mol.
4. L’indicateur coloré permet de repérer l’équiva-lence au changement de couleur.
5. Au fur et à mesure que la réaction d’estérifica-tion avance, la quantité d’acide diminue (consom-mation de cet acide pour former l’ester et l’eau) : le volume de soude versé diminue donc jusqu’à l’équi-libre, puis reste constant lorsque l’état d’équilibre est atteint.
6. RCO2H(aq) + HO–(aq) = RCO2
–(aq) + H2O(l).À l’équivalence, la quantité de soude versée est égale à la quantité d’acide restante dans le milieu. Autrement dit : les réactifs ont été introduits dans les proportions stœchiométriques. On peut éga-lement dire qu’il y a changement de réactif limi-tant.
7. À l’équivalence, la quantité de soude versée est :
n(HO–(aq)) = c · Véq.La quantité d’acide restante nA est donc la même :
nA(t) = c · Véq.
8. Pour la totalité du mélange initialement pré-paré.a) Compte tenu du mélange équimolaire et des pro-portions stœchiométriques : nE = x.b) Relation établie au 7 : nA(t) = c · Véq.Compte tenu de la quantité dosée et de la quantité initiale :– la quantité d’acide restante dans le mélange est :
nAm(t) = 10 c · Véq ;– la quantité initiale d’acide dans le mélange est :
n0 = 0,10 mol ;– la quantité d’acide consommée, nA con, est égale à la quantité d’ester formé x :
x = nA con = n0 – 10 c · Véq = 0,1 – 10 c · Véq.

159Chapitre 11 - Contrôle du rendement par le choix des réactifs
9. a)
012345678
0 10 20 30 40 50 60 70 80 90 100
x (en 10–2 mol)
t(en min)
Sur le graphe, l’asymptote horizontale donne la valeur maximale de l’avancement :
xf = 6,7 × 10–2 mol.
b) Le taux d’avancement final est donné par la rela-tion :
τ = x
xf
max
, soit τ = 0 067
0 10
,
, = 0,67, donc τ < 1.
10. a) « La transformation chimique est lente » : sur le graphique, l’équilibre est atteint en 50 minu-tes environ (x n’augmente plus).b) « La transformation chimique n’est pas totale », car τ < 1.11. Le système chimique est dit en équilibre, car sa composition chimique n’évolue plus du point de vue macroscopique.Au niveau microscopique, les réactions d’estérifi-cation et d’hydrolyse de l’ester ont encore lieu, mais elles produisent autant d’ester qu’elles en consomment.12. a) Pour accélérer la réaction d’estérification, on peut modifier un facteur cinétique, par exemple la température : une élévation de température accélère la synthèse de l’ester.Pour augmenter le taux d’avancement à l’équilibre, il faut modifier l’équilibre dans le sens de formation de l’ester. On peut :– utiliser l’un des réactifs en excès ;– éliminer l’un des produits au fur et à mesure de sa formation (l’eau dans ce cas, car sa température d’ébullition est la plus faible).b) Si on veut une transformation rapide et totale, il faut utiliser un anhydride d’acide : l’anhydride éthanoïque.
H3C OC
CH3C
O O
2. Préparation de l’aspirine(Publication scientifi que : extrait du Vögel.)
De nombreuses publications scientifiques sont publiées en anglais : traduire ce texte.Phenols, unlike amines, cannot be acetylated satisfactorily in aqueous solution : acetylation proceeds readily with ace-tic anhydride in the presence of a little concentrated sulphu-ric acid as catalyst. Salicylic acid (o-hydroxy-benzoïc acid) upon acetylation yields acetylsalicylic acid or aspirin :
OH
COOH+ (CH3CO)2O
H2SO4
OCOCH3
COOH+ CH3COOH.
salicylic acid aspirin
Place 10 g of dry salicylic acid and 15 g (14 mL) of acetic anhydride in a small conical flask, add 5 drops of concen-trated sulphuric acid, and rotate the flask in order to secure thorough mixing. Warm on a water bath to about 50-60 °C, stirring with the thermometer, for about 15 minu-tes. Allow the mixture to cool and stir occasionally. Add 150 mL of water, stir well and filter at the pump. Recrystal-lise the crude acetylsalicylic acid from a mixture of equal volumes of acetic acid and water.The following is an alternative method of purifying the crude aspirin. Dissolve the solid in about 30 mL of hot alcohol and pour the solution into about 75 mL of warm water : if a solid separates at this point, warm the mixture until solution is complete and then allow the clear solution to cool slowly. Beautiful needle-like crystals will separate. The yield is 13 g. The air-dried crude product may also be recrystallised from benzene or from ether-light petroleum (b.p. 40-60 °C)Acetylsalicylic acid decomposes when heated and does not possess a true, clearly-defined m.p. Decomposition points ranging from 128 °C to 135 °C have been recorded ; a value of 129-133 °C is obtained on a electric hot plate. Some decomposition may occur if the compound is recrystallised from a solvent of high boiling point or if the boiling period during recrystallisation is unduly prolonged.
Corrigé• Les phénols, contrairement aux amines, ne don-nent pas d’acétylation satisfaisante en solution aqueuse. L’acétylation s’effectue facilement avec l’anhydride acétique en présence d’un peu d’acide sulfurique comme catalyseur.L’acide salicylique (acide orthohydroxybenzoïque) fournit, au cours de l’acétylation, de l’acide acétyl-salicylique ou aspirine :
OH
COOH+ (CH3CO)2O
H2SO4
OCOCH3
COOH+ CH3COOH.
salicylic acid aspirin

160 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
• Placer 10 g d’acide salicylique sec et 15 g (soit 14 mL) d’anhydride acétique dans un erlenmeyer, ajouter 5 gouttes d’acide sulfurique concentré et agiter l’erlenmeyer tout en mélangeant.Mettre l’erlenmeyer dans un bain-marie sous agita-tion en veillant à maintenir une température de 50 à 60 °C dans l’erlenmeyer pendant 15 min. Laisser refroidir le mélange tout en agitant de temps en temps. Ajouter 150 mL d’eau, bien mélanger et fil-trer sous vide.Recristalliser l’acide acétylsalicylique brut dans un mélange d’acide acétique et d’eau en volumes égaux.• Il existe une autre méthode pour purifier l’aspi-rine brute.Dissoudre l’aspirine dans environ 30 mL d’alcool chaud et verser la solution dans environ 75 mL d’eau tiède. S’il reste du solide, faire chauffer jus-
qu’à ce qu’il disparaisse. Laisser refroidir lentement la solution limpide : de beaux cristaux en forme d’aiguilles vont apparaître. Le produit obtenu a pour masse 13 g.Le produit brut séché à l’air peut également être recristallisé dans du benzène ou dans de l’éther de pétrole (θeb : 40-60 °C).L’acide acétylsalicylique se décompose quand on le chauffe et ne possède en aucun cas un point de fusion bien défini. Des températures de décompo-sition s’étalant de 128 à 135 °C ont été enregis-trées. Une valeur entre 129 et 133 °C est obtenue avec une plaque électrique chaude (banc de Köfler).Une décomposition peut survenir si le composé est recristallisé dans un solvant à haut point d’ébulli-tion ou si le temps d’ébullition pendant la recristal-lisation est prolongé excessivement.

161Chapitre 12 - Contrôle de la vitesse par catalyse
ProgrammeEXEMPLES D’ACTIVITÉS CONTENUS CONNAISSANCES ET SAVOIR-FAIRE EXIGIBLES
Expériences qualitatives sur la catalyse.Recherche documen-taire sur la catalyse et ses applications.
2. Des exemples de contrôle de l’évolution de systèmes chimiques pris dans l’industrie chimique et dans les sciences de la vie.– Utilisation de la catalyseCatalyse homogène, hétérogène, enzymatique.Sélectivité des catalyseurs.
– Savoir qu’un catalyseur agit sélective-ment lors d’une transformation.
12 Contrôle de la vitesse par catalyse
Cours
Découpage du cours1. Mise en évidence de la catalyse p. 2442. Qu’est-ce que la catalyse ? p. 2453. Caractéristiques de la catalyse p. 2464. La catalyse dans l’industrie p. 248
Ce chapitre conclut l’ensemble du programme : en effet, la catalyse trouve des applications dans tous les domaines de la chimie. L’importance actuelle des recherches sur les catalyseurs et les perspectives industrielles justifient un chapitre consacré à ce sujet.Les catalyseurs sont bien souvent des composés extrêmement complexes. Toutefois, le choix des exemples retenus est volontairement simple, mais il permet aux élèves de percevoir les différents modes de catalyses au programme et les propriétés des catalyseurs.
1. Mise en évidence de la catalyseOn aborde la catalyse sur un exemple expérimental, que l’on visualise aisément par des changements de couleurs et que l’on interprète en révisant les notions
♦ d’oxydoréduction étudiées dans le début du pro-gramme.
2. Qu’est-ce que la catalyse ?Ce paragraphe permet d’énoncer les définitions des trois types de catalyses ; il est en lien étroit avec les activités.
3. Caractéristiques de la catalyseUne approche microscopique du phénomène de la catalyse permet l’interprétation des phénomènes macroscopiques observés. Mécanisme, propriétés, sélectivité et spécificité sont abordés à partir d’exem-ples, toujours en lien avec les activités.
4. La catalyse dans l’industrieCe paragraphe pourrait à lui seul constituer un livre tant les études actuelles sur l’intervention des cata-lyseurs dans l’industrie prennent une place impor-tante.L’utilisation des ressources informatiques permet aux élèves de compléter et de répondre aux diverses questions qui se posent.
Activités
À la découvertede la catalyse (page 250)
Ce TP répond aux exigences du programme : expé-riences qualitatives sur la catalyse, il permet mettre en évidence les trois types de catalyses : homogène, hétérogène et enzymatique.
ACTIVITÉ
1
ACTIVITÉ
1 A Oxydation de l’acide éthanedioïque par l’eau oxygénée
1. & 2. H2O2(aq) + 2 H3O+(aq) + 2 e– = 4 H2O(l)
H2C2O4(aq) + 2 H2O(l) = 2 CO2(g) + 2 H3O+(aq) + 2 e–.
L’équation globale est donnée par addition des deux précédentes.

162 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
3. Co2+(aq), rose, donne Co3+(aq), vert, puis rede-vient Co2+(aq), rose.4. Les ions cobalt (II) jouent le rôle de catalyseur, car ils accélèrent la transformation et subissent eux-mêmes une transformation avant d’être régé-nérés.Toutes les espèces chimiques sont dans la même phase liquide : il s’agit d’une catalyse homogène.
B Action du diiode sur l’aluminium1. Al(s) = Al3+(aq) + 3 e–
I2(s) + 2 e– = 2 I–(aq).
2. 2 Al(s) + 3 I2(s) = 2 Al3+(aq) + 6 I–(aq).
3. L’équation de réaction montre que :n(Al)
2 =
n(I2)
2, donc n(I2) =
3
2 n(Al).
n(Al) = 1
27 = 0,037 mol,
donc n(I2) = 3
2 × 0,037 = 0,055 5 mol.
m(I2) = 0,055 5 × 127 = 14,1 g.
4. Manipulation du diiode solideRisques :• R 20/21 : nocif par inhalation et par contact avec la peau.Sécurité :• S 23-24-25 : ne pas respirer les vapeurs ; éviter le contact avec la peau et les yeux.5. a) Les deux réactifs sont à l’état solide ; le cata-lyseur est l’eau à l’état liquide.b) La catalyse est donc hétérogène.
C Hydrogénation catalytique d’un alcène1. a) Les deux réactifs sont à l’état gazeux, le catalyseur est à l’état solide.b) Il s’agit d’une catalyse hétérogène.
2. Étape 1 : réactif proche du catalyseur.Étape 2 : phénomène d’adsorption.Étape 3 : adsorption du second réactif.Étape 4 : réactifs modifiés réagissant à la surface du catalyseur.Étape 5 : produit de la transformation du catalyseur.Étape 6 : phénomène de désorption.
3. L’industrie agroalimentaire cherche à produire des corps gras à chaîne saturée à partir de corps gras à chaîne insaturée, et ceci par hydrogénation catalytique.
D Hydrolyse de l’amidon1.
Test à l’eau iodée
Durée (min) 0 3 6 9
T1 + + + +
T2 + + + –
Test à la liqueur de Fehling
Durée (min) 0 3 6 9
T1 – – – –
T2 – – + +
2. L’amylase catalyse l’hydrolyse de l’amidon.
La chimieverte (page 252)
Les catalyseurs ont pris une importance considéra-ble dans les nouvelles orientations de la chimie, au point de figurer parmi les grands principes de la chimie verte. Cette activité répond aux recomman-dations du programme : recherche documentaire sur la catalyse et ses applications.
1. Les grandes orientations qui permettent de titrer : « La catalyse pilier de la chimie verte »– économie d’énergie, d’atomes et de temps ;– catalyseurs plus sélectifs (moins de produits secondaires) ;– synthèse plus propre de produits élaborés ;– catalyseurs plus efficaces pour éliminer les oxy-des d’azote ;– solutions curatives permettant d’éliminer les substances toxiques.2. Propriétés d’un catalyseur :– il accélère la vitesse de réaction en abaissant la barrière énergétique ;– il n’est pas détruit dans la transformation ;– il a la propriété d’être sélectif.3. Les deux types de catalyse étudiés en cours, homogène et hétérogène, sont suggérés dans les exemples de cet article. La catalyse enzymatique peut être perçue au niveau du traitement des bio-carburants ; la photocatalyse est citée mais n’est pas au programme de terminale.4. Économie d’énergie, d’atomes et de temps.5. a) Les catalyseurs les plus prometteurs sem-blent être les catalyseurs enzymatiques.b) Lors de la synthèse de l’aspartame, il se forme plusieurs isomères, dont l’un a un goût amer et est à éliminer.
ACTIVITÉ
2ACTIVITÉ
2

163Chapitre 12 - Contrôle de la vitesse par catalyse
On utilise une biocatalyse pour terminer la synthèse. L’enzyme employée est appelée thermolysine : elle provient de Bacillus proteolicus/thermoproteolyticus des mers chaudes du Japon.
6. Dans les pots catalytiques des voitures, les prin-cipaux métaux permettant de traiter les gaz d’échappements sont le platine, le rhodium et le palladium.
7. Photocatalyse : catalyse par la lumière.
Dismutation catalytiquede l’eau oxygénée (page 254)
Le but de ce TP est de familiariser les élèves avec le déroulement des TP d’évaluation des compétences expérimentales. Il met en évidence qualitativement les trois modes de catalyses (homogène, hétérogène et enzymatique) sur un même exemple : la dismuta-tion de l’eau oxygénée.Le travail expérimental, très simple, permet d’ap-précier la curiosité scientifique des élèves et leur degré de réflexion.
A Étude préliminaire1. H2O2(aq) + 2 H3O
+(aq) + 2 e– = 4 H2O(l) H2O2(aq) + 2 H2O(l) = O2(g) + 2 H3O
+(aq) + 2 e–.
2. L’équation globale est obtenue par addition des deux demi-réactions précédentes.
3. Le dioxygène dégagé peut être mis en évidence en présentant une tige de bois incandescente qui a tendance à se rallumer.
B Différentes catalyses• Dismutation de l’eau oxygénée
catalysée par les ions fer (III)Observations :– le gaz dégagé en (a) et (b) est du dioxygène ;– le dégagement gazeux dans le tube (a) est beau-coup plus important que dans le tube (b) ;– dans le tube (a), la solution initialement jaune rouille devient vert très pâle pour redevenir jaune rouille. On observe la même évolution de couleur mais très peu soutenue dans le tube (b) ;– (a) et (c) ont rigoureusement la même couleur finale.
1. a) Les ions fer (III) jouent le rôle de catalyseur.b) Ce processus, dans lequel tous les intervenants sont dans un même état physique dans une seule phase, est une catalyse homogène.
ACTIVITÉ
3
ACTIVITÉ
3
2. La concentration élevée du catalyseur dans le tube (a) accélère plus efficacement la transforma-tion de l’eau oxygénée.3. L’évolution de la concentration molaire en ions fer (III) est telle qu’elle diminue dans un premier temps, pour augmenter ensuite et redevenir identi-que à sa valeur initiale.
4. a) & b)• H2O2(aq) + 2 H2O(l) = O2(g) + 2 H3O
+(aq) + 2 e–. Fe3+(aq) + e– = Fe2+(aq). D’où l’équation globale : H2O2(aq) + 2 H2O(l) + 2 Fe3+(aq)
= O2(g) + 2 H3O+(aq) + 2 Fe2+(aq). (1)
• H2O2(aq) + 2 H3O+(aq) + 2 e– = 4 H2O(l).
Fe2+(aq) = Fe3+(aq) + e–
D’où l’équation globale : H2O2(aq) + 2 H3O
+(aq) + 2 Fe3+(aq)= 4 H2O(l) + 2 Fe3+(aq). (2)
• (1) + (2) donne :2 H2O2(aq) = O2(g) + 2 H2O(l).
Le catalyseur a participé à la transformation : dans une première étape, il est consommé ; dans un second temps, il est régénéré. Il n’apparaît dans le bilan global.
• Dismutation de l’eau oxygénée catalysée par le platine
Observations :– il apparaît un important dégagement gazeux dans le mélange réactionnel ;– le gaz dégagé est du dioxygène.5. a) Le platine joue le rôle de catalyseur.b) Le processus mis en œuvre lors de la décomposi-tion de l’eau oxygénée est une catalyse hétéro-gène.6. Le phénomène de catalyse se localise au niveau de la surface du platine.7. Plus l’aire de la surface de contact sera impor-tante plus le catalyseur sera efficace.
• Dismutation de l’eau oxygénée catalysée par une matière d’origine animale ou végétale
Observations :– il apparaît une effervescence au contact du mor-ceau de foie.8. a) Le morceau de foie joue le rôle de catalyseur.b) Le processus mis en jeu dans cette expérience est une catalyse enzymatique.9. Le phénomène de catalyse se localise au niveau du morceau de foie.

164 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
Activité supplémentaire
La fabrication de la bièreLa brasserie est l’industrie de la fabrication de la bière, laquelle est fabriquée essentiellement à partir d’orge.La France est le quatrième producteur européen de bière. Cette activité présente un exemple de phéno-mène de catalyse enzymatique, utilisé pour cette importante fabrication industrielle en termes éco-nomiques.
A Principe de fabricationLa fabrication de la bière comporte trois phases principales.• Le maltage est une germination limitée de l’orge, qui a pour but de développer le potentiel enzymati-que du grain. Il comporte les opérations de trempe, de mise en germoir et de touraillage.• Le brassage, pendant lequel l’amidon du malt est hydrolysé en sucres fermentescibles (maltose et glu-cose) par les enzymes du grain. Il comprend les opé-rations de concassage du malt, de brassage proprement dit, de filtration (séparation des drê-ches et du moût), de cuisson et d’aromatisation par du houblon. Cette cuisson correspond à l’ébullition du moût : elle permet d’extraire les composés parfu-més du houblon responsables de l’amertume de la bière mais aussi de stopper l’action des enzymes.• La fermentation est obtenue par ensemencement de levures. Les sucres fermentescibles sont alors transformés en alcool et en dioxyde de carbone au
cours de l’étape de fermentation principale et celle de maturation en cave de garde.D’autres produits minoritaires issus des réactions métaboliques de la levure jouent un rôle important dans le goût final de la bière. Ce sont des alcools à chaînes plus longues, comme l’alcool amylique, l’alcool isoamylique et l’alcool phényléthylique. Les acides à chaînes courtes excrétés par les levures, tels l’acide éthanoïque, l’acide butyrique et leurs esters, sont également responsables de la saveur de la boisson.
B Exploitation du document
DonnéesÉthanol : masse volumique µ = 0,79 g · mL–1 ; M = 46,1 g · mol–1.Amidon : (C6H10O5)n.Maltose : C12H22O11.Glucose : C6H12O6.Alcool amylique : CH3CH2CH2CH2CH2OH.Alcool isoamylique : CH3CH(CH3)CH2CH2OH.
1. Sur le schéma récapitulatif des différentes opéra-tions de fabrication de la bière, repérer les zones correspondant aux trois phases principales : mal-tage, brassage, fermentation.2. Pourquoi « développer le potentiel enzymatique » lors de l’opération de maltage ?3. a) Quelles transformations sont catalysées par les enzymes créées lors du maltage ?
•
•••••
1. Cuve à tremper 3. Touraille 5. Cuve matière 9. Cuve de garde 11. Soutirage7. Chaudière à moût
2. Germoir 4. Concasseur à malt 6. Cuve filtre 10. Filtre 12. Étiquetage8. Cuve de fermentation
Les différentes étapes de la fabrication de la bière.

165Chapitre 12 - Contrôle de la vitesse par catalyse
1 Définitions1. Catalyseur : espèce chimique ou mélange d’espèces chimiques accélérant ou orientant une réaction chimique sans être consommée.
2. & 3.Type de catalyse • Homogène • Hétérogène • Enzymatique
Carac-téris-tique
• Le catalyseur appartient à la même phase que les réactifs.
• Le catalyseur n’appartient pas à la même phase que les réactifs.
• Le catalyseur est une enzyme.
Cataly-seurs
• Acido-basique• Redox
• À l’état solide• De nature protéique
2 Esters et catalyse1. a) Vrai ; b) faux ; c) faux.2. a) Vrai ; b) faux ; c) faux.3. a) Faux ; b) vrai ; c) faux ; d) faux.4. a) Faux ; b) faux ; c) faux.
3 Classement1. Catalyse hétérogène. 3. Catalyse hétérogène.2. Catalyse hétérogène. 4. Catalyse homogène.
4 Catalyse hétérogèneLe but recherché est d’augmenter le plus possible la surface de contact entre le catalyseur et les réactifs.
Corrigés des exercices (page 255)
b) Écrire les équations des réactions correspondant aux transformations amidon/maltose et maltose/glucose.4. Pour quelles raisons porte-t-on le moût à ébulli-tion ?5. a) Quel est le rôle des levures ?b) Écrire l’équation de réaction modélisant la transformation alcoolique du glucose en éthanol et en dioxyde de carbone.6. Quelle espèce chimique est responsable de l’effet moussant de la bière ?7. Donner le nom en nomenclature officielle des alcools amylique et isoamy lique.8. Degré alcoométrique : le titre alcoométrique volumique d’une bière est égal au nombre de litre d’éthanol contenu dans 100 L de bière. Son sym-bole est : « % vol ».Soit une bière à x = 6 % vol : exprimer littéralement la concentration molaire en éthanol de cette bière en fonction de x, de µ et de M, puis la calculer.
Corrigé1. Maltage : opérations 1, 2 et 3 sur le schéma.Brassage : opérations 4, 5, 6 et 7 sur le schéma.Fermentation : opérations 8 et 9 sur le schéma.2. Dans cette phase, on développe le potentiel enzymatique du grain pour permettre ensuite l’hy-drolyse de l’amidon du malt.3. a) Les transformations catalysées par les enzy-mes sont l’hydrolyse du maltose et celle du glucose.b) • Hydrolyse de l’amidon en maltose :
2 (C6H10O5(aq))n + n H2O(l) = n C12H22O11(aq).
• Hydrolyse du maltose en glucose : C12H22O11(aq) + H2O(l) = 2 C6H12O6(aq).4. On porte le moût à ébullition pour extraire les composés parfumés du houblon et pour stopper l’action des enzymes.5. a) Les enzymes permettent la fermentation du moût.b) Transformation alcoolique du glucose en étha-nol et dioxyde de carbone :
C6H12O6(aq) = 2 C2H5OH(aq) + 2 CO2(g).6. L’effet moussant de la bière est lié à son « déga-zage » de CO2(g).7. Alcool amylique : pentan-1-ol.Alcool isoamylique : 3-méthyl butan-1-ol.8. En appelant v le volume d’alcool et V celui de bière :– volume d’alcool dans 1 L de bière :
v = x · V ; v = 0,06 × 1 = 0,06 L = 60 mL ;– masse d’alcool correspondante :
m = µ · v = µ · x · V ; m = 0,79 × 60 = 47,4 g ;– quantité de matière correspondante :
n = m
M =
µ ⋅ ⋅x V
M ; n =
47 4
46 1
,
, = 1,028 mol.
Soit une concentration molaire : C = 1,028 mol · L–1.
Concentration molaire :
C = n
V =
µ ⋅ ⋅⋅x V
M V =
µ ⋅ x
M ;
C = 0 79 0 06
46 1 10 3
, ,
, –
××
= 1,028 mol · L–1.

166 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
5 Efficacité d’un catalyseur solide1. Volume de fer correspondant à une masse de 1 g :
m = µ · V ; V = m
µ, soit V =
1
8 7, = 0,115 cm3.
a) Le volume étant connu, le rayon de la sphère et la surface latérale de la sphère sont calculables :
V = 4
3 π · r3, soit r =
3
4
1 3V
π
/
; r = 0,30 cm,
d’où l’aire de la surface latérale :S = 4π · r2 ; S = 4π × (0,30)2 = 1,14 cm2.
La surface massique est :
Sm = S
m, soit Sm =
1 14
1
, = 1,14 cm2 · g–1.
b) Le volume étant connu, l’arête du cube et la sur-face latérale du cube sont calculables :
V = a3, soit a = (V)1/3 ; a = 0,486 cm,d’où l’aire de la surface latérale :
S' = 6a2, soit S' = 6 × 0,4862 = 1,42 cm2.La surface massique est :
S'm = S
m, soit S'm =
1 42
1
, = 1,42 cm2 · g–1.
c) Il faut ajouter l’aire des deux faces planes à l’aire de la surface latérale :
S" = S + 2π · r2 ;S" = 1,14 + 2π × (0,30)2 = 1,14 + 0,565 = 1,71 cm2.
La surface massique est :
S"m = S
m
", soit S'm =
1 71
1
, = 1,71 cm2 · g–1.
2. S"m > S'm > Sm.C’est sous la forme la plus divisée d’hémisphères que le catalyseur présente la plus grande surface par unité de masse et est donc le plus efficace.
6 Pot catalytique1. Le catalyseur ne transforme pas : en présence du catalyseur, la transformation des composants nocifs contenus dans les rejets d’échappements est de 99 %.2. a) Matières actives : platine et rhodium.b) Il ne s’agit pas du catalyseur mais de la masse de contact.3. a) C’est une catalyse de type hétérogène.b) C’est la surface d’échange importante (trois ter-rains de football) :4. 2 CO + O2 = 2 CO2 ; C2H6 +
7
2 O2 = 2 CO2 + 3 H2O.
La définition primitive d’oxydation (association avec l’oxygène) permet directement de conclure à une réaction d’oxydation.
5. L’effet secondaire de la présence d’une trop grande quantité de dioxyde de carbone dans l’atmosphère est lié au fait qu’il constitue un gaz à « effet de serre ».6. Il s’agit d’une réduction, car la transformation cor-respond à une perte d’oxygène (définition primitive).
7 Oxydation des ions iodure par l’eau oxygénée
1. L’équation de réaction modélisant la transfor-mation montre que les réactifs sont dans les pro-portions stœchiométriques lorsque :
n(H2O2) = n(I )–
2 =
n(H O )3+
2.
Cette condition est remplie et les ions oxonium sont en large excès. On note aussi que :
n(H2O2) = n(I2), d’où [I2] = [H2O2].
Expériences 1 et 3 : [I2]max = 0,003 mol · L–1.Expériences 2 et 4 : [I2]max = 0,006 mol · L–1.
2. Expériences 1 et 3 : tracés (c) et (d) ; la limite atteinte est 0,003 mol · L–1. L’expérience 3 est réalisée à une température supérieure à celle de l’expérience 1, donc le graphe (c) correspond à l’expérience 3.(a) et (b) atteignent une limite de 0,006 mol · L–1, donc correspondent aux expériences 2 et 4.Il est possible de les différencier par la durée néces-saire pour atteindre la valeur maximale de la concen-tration : plus courte pour le graphe (a), c’est dans ce cas que le catalyseur est présent, donc expérience 4.
8 Hydrolyse de l’urée1. La vitesse volumique instantanée de formation de l’ion ammonium est donnée par le coefficient directeur de la tangente à la courbe c = f(t) à l’ins-tant considéré :
v = d
d
c
t,
• Cas 1C1 = 0,000 1 mol · L–1 : v = 2 × 10–4 mol · L–1 · s–1.
• Cas 2C2 = 0,000 4 mol · L–1 : v = 5 × 10–4 mol · L–1 · s–1.
2. Le calcul précédent et l’allure des deux courbes montrent que l’influence de la concentration du catalyseur enzymatique n’est pas négligeable.3. Plus la concentration du catalyseur enzymatique est élevée, plus son efficacité est grande.4. Un catalyseur n’est pas consommé, donc la concen-tration en uréase en fin de transformation sera :
C1 = 1 × 10–4 mol · L–1 ; C2 = 4 × 10–4 mol · L–1.

167Chapitre 12 - Contrôle de la vitesse par catalyse
9 Estérification1. L’écriture de l’équation de réaction modélisant l’estérification sans ou avec catalyseur donne :
sans ou avec H3O+
CH3CH2OH(aq) + CH3COOH(aq)= CH3COOC2H5(aq) + H2O(l).
2.
100
200
300
400
500
600
2 4 5 8 10 12 14
n2 (mmol)
t (j)
3. a) La détermination de la vitesse initiale de forma-tion de l’ester se fait par calcul du coefficient directeur de la tangente à la courbe à l’instant de date t = 0.Cette détermination donne les résultats suivants :– cas 1 : v1 = 0,10 mol · h–1 ;– cas 2 : v2 = 0,40 mol · h–1.b) Les ions oxonium H3O
+(aq) jouent le rôle de cata-lyseur, car en leur présence, toutes choses identi-ques par ailleurs, la vitesse de transformation est augmentée.
4. a) La vitesse d’estérification diminue au cours du temps : cela s’observe sur les deux graphes, car le coefficient directeur de la tangente aux graphes diminue au fur et à mesure de l’évolution de la transformation.b) Le facteur cinétique responsable de cette diminu-tion est la concentration des réactifs, laquelle dimi-nue au fur et à mesure que la transformation avance.
5. a) Le mélange équimolaire d’un alcool primaire avec un acide carboxylique conduit à une limite de 66 %, donc la limite de cette estérification sera 0,66 mole.b) Les ions oxonium n’ont aucune influence sur cette limite, car le catalyseur augmente la vitesse d’une transformation équilibrée sans en modifier l’état d’équilibre.
10 Le parfum de poire1. Le groupe caractéristique présent (pointillés) correspond à la fonction ester.
H3C OC
CH2H2C
CH2H2C CH3
O
2. a) La fonction ester résulte de :acide carboxylique + alcool = ester + eau.
Si A = acide carboxylique, B contient la fonction chimique alcool : CH3–CH2–CH2–CH2–CH2–OH.b) CH3CO2H(l) + C5H11OH(l) = C7H14O2(l) + H2O(l).c) A, acide éthanoïque (ou acétique) ; B, pentan-1-ol. L’autre produit formé est l’eau.d) Réaction d’estérification.
3. Le mélange réactionnel est préparé et maintenu à basse température pour ralentir fortement la trans-formation d’estérification. Des fractions de volumes égaux de ce mélange réactionnel sont placées dans une série de tubes à essais. Tous introduits dans un bain thermostaté à un instant t = 0 s, à laquelle on déclenche simultanément un chronomètre. La trans-formation chimique débute. À différents instants, on retire un tube : on le verse dans un volume d’eau glacée, afin de bloquer la réaction d’estérification (opération dite de trempe). Deux facteurs cinéti-ques bloquent la transformation : la dilution et l’abaissement de température.On titre l’acide éthanoïque restant dans le tube à essais par une solution aqueuse d’hydroxyde de sodium de concentration molaire connue. À la tem-pérature du mélange, seule la réaction acide/base a lieu, celle de saponification ester/base est négli-geable. On détermine ainsi la quantité de matière d’acide éthanoïque restant dans un tube à chaque instant t. On en déduit la quantité de matière d’acide ayant réagi dans le mélange réactionnel, donc la quantité d’ester formé.
4. a) L’acide sulfurique joue le rôle de catalyseur : la transformation très lente devient plus rapide. L’état d’équilibre est plus vite atteint sans être modifié.b) L’acide sulfurique n’intervient pas dans l’équation de la réaction (une caractéristique d’un catalyseur).5. a)Équation modéli-sant l’estérification A + B = éthanoate de pentyle + eau
État Avanc. (mol)
Quantités de matière (mol)
Initial 0 nA(0) = 0,50 nB(0) = 0,50 0 0
Intermé-diaire x nA(0) – x nB(0) – x x x
À t = 60 min
x = n(60)= 0,33
0,17 0,17 0,33 0,33

168 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
b) n(éthanoate de pentyle) = n = x.
6. a) v = 1
V ·
d
d
x
t =
1
V ·
d
d
n
t.
b) v = 1
V ·
d
d
n
t ;
d
d
n
t est le coefficient directeur de
la tangente, à l’instant t, à la courbe représentative de n = f(t). Ce coefficient directeur diminue au cours du temps d’où la vitesse de réaction diminue au cours du temps. La concentration des réactifs est le facteur cinétique qui justifie cette évolution.c) Le système a atteint l’état d’équilibre à partir de t = 50 min, car : n = nf = 0,33 = constante.
7. L’état final d’équilibre du système est identique à celui obtenu avec catalyseur( il ne modifie pas l’état d’équilibre final) : la même limite de 0,33 mol serait atteinte dans un temps beaucoup plus long.
0
0,1
0,2
0,3
0 10 20 30 40 50 60 70
n (mol)
t (min)
11 Entretien des lentilles cornéennes
Exercice résolu dans le manuel de l’élève.
12 La catalyse homogène
Exercice résolu dans le manuel de l’élève.
13 Pot catalytique et pollution1. Catalyseur : substance réduisant la durée d’une transformation et retrouvée en fin de transformation.Se retrouve en fin de réaction : … métaux nobles, bien que non consommés.Réduit la durée d’une transformation : ces métaux facilitent trois réactions.
2. Nature du catalyseur : platine, rhodium, palladium.
3. Catalyse hétérogène : catalyseur = solide ; réac-tifs = gaz de combustion rejetés par le moteur.
4. • Obligation de consommer une essence sans plomb (poison du catalyseur).• Fonctionnement satisfaisant à une température éle-vée donc après quelques minutes de fonctionnement.• Réglage du moteur parfait pour renvoyer un mélange résiduel dans des proportions telles que les réactions soient aussi complètes que possible.
5. La température élevée est responsable de la for-mation des oxydes d’azote.
6. v(N2)t = d N
d
n
t
( )2 .
7. Cette vitesse est déterminée graphiquement en traçant la tangente à la courbe à l’instant consi-déré. La valeur numérique est celle du coefficient directeur de cette tangente. Sur le graphe :
v(N2)0 = 0,0125 mol · s–1.
8. La vitesse diminue au cours du temps (notam-ment car la concentration des réactifs diminue).
14 Exploitation expérimentale1. a) On doit rincer l’erlenmeyer à l’eau distillée et introduire les eaux de rinçage dans la fiole jaugée.b) L’homogénéisation de la solution S2 s’obtient par agitation de la fiole jaugée fermée avec un bou-chon.L’agitation d’une fiole jaugée s’effectue par retour-nement de celle-ci.c) • La protection des yeux demande le port obli-gatoire des lunettes.• La protection des voies respiratoires implique de manipuler sous la hotte aspirante.• La protection de la peau s’effectue par utilisa-tion d’une paire de gants.• La protection des vêtements est réalisée par le port d’une blouse fermée.2. a) Le réglage de l’absorbance zéro du spectro-photomètre permet de compenser l’absorbance de la cuve et celles des produits autres que la matière dont on veut étudier l’absorbance.b) La manipulation des cuves demande le plus grand soin :– ne pas toucher les faces transparentes des cuves avec les doigts ;– ne remplir les cuves qu’au trois-quarts ;– vérifier l’absence de bulles d’air dans les cuves.
Question supplémentaire pour cet exercicePourquoi les deux absorbances sont-elles identiques ?
CorrigéLes deux absorbances sont identiques, car la quan-tité de matière de cuivre (II) n’a pas varié entre le témoin et la solution aqueuse de peroxyde d’hydro-gène dans laquelle les ions cuivre (II) ont joué le rôle de catalyseur.

169Chapitre 12 - Contrôle de la vitesse par catalyse
Les compétences expérimentales
(page 264)
Cet exercice d’évaluation des compétences expérimentales permet de cou-vrir l’ensemble des connaissances à acquérir sur la spectrophotométrie :dilution, traitement des cuves, choix judicieux des conditions expérimen-tales et exploitation des résultats.Ici cette technique est utilisée pour mettre en évidence la conservation du catalyseur dans une transformation catalysée.
Corrigé1. Réalisation d’une gamme étalon1. La conservation de la quantité de matière permet d’écrire :
C0 · V0 = Cf · Vf, avec Vf = 100 mL.
D’où : V0 = C
Cf
0
· Vf, soit V0 = Vf
10.
2. Le ménisque est correctement ajusté sur le schéma A.
2. Position des cuves de spectrophotométrieLa lumière doit traverser les faces transparentes de la cuve : schéma B. La proposition faite sur le dessin B est donc correcte.
3. Réglage initial1. La longueur d’onde doit être réglée de façon que l’absorbance de la substance soit maximale.Par lecture sur le graphe : λ = 640 nm.2. L’initialisation du spectrophotomètre demande de régler l’absorbance zéro.3. La cuve spectrophotométrique doit être préparée avec tous les produits sauf le réactif à doser.
4. Exploitation des résultats expérimentaux1. Par lecture sur le graphe : CX = 0,78 mmol · L–1.2. Si A'X = 0,22, alors : C'X = 0,16 mmol · L–1.3. Compte tenu de la dilution (10 mL de catalyseur pour un volume total de 50 mL), la concentration molaire initiale était de :
C"X = 5 C'X ; C"x = 5 × 0,16 = 0,80 mmol · L–1.Aux erreurs de lecture près sur la courbe, on retrouve la concentration de la solution initiale en catalyseur.Cela confirme le fait que le catalyseur intervient puis est régénéré dans le processus de catalyse.

170 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
Exercice supplémentaireFermentation dans le vin• « Le vin est une boisson provenant exclusivement de la fermentation du raisin frais ou du jus de raisin frais ». Telle est la définition légale du vin, mais der-rière le terme « fermentation » se cachent des trans-formations que les chimistes ont mis des années à découvrir.• Dans les années 1960, on commença à s’intéresser à une autre fermentation qui se produit générale-ment après la fermentation alcoolique et à laquelle on attachait peu d’importance jusqu’alors, car on pensait qu’il s’agissait d’un achèvement de la fer-mentation alcoolique.Il s’agit en fait de la fermentation malolactique, laquelle consiste en une transformation totale de l’acide malique présent dans le jus de raisin en acide lactique sous l’action de bactéries. Cette fermenta-tion, longtemps ignorée, a une influence reconnue sur la qualité gustative de certains vins, à condition de la conduire convenablement.• Les techniques actuelles de suivi de ces fermen-tations mettent en œuvre un dosage enzymatique : elles consistent essentiellement à doser l’alcool contenu dans le vin.• Principe du dosage– Étape 1 : on effectue une distillation du vin de telle façon que l’on recueille une solution incolore contenant tout l’éthanol présent dans le vin.– Étape 2 : l’éthanol est oxydé par la nicotinamide-adénine-dinucléotide (NAD+(aq)) dans une réac-tion catalysée par une enzyme spécifique. La réaction produit de la nicotinamide-adénine-dinu-cléotide réduite (NADH) en quantité de matière égale à celle de l’éthanol dosé selon l’équation :CH3CH2OH(aq) + NAD+(aq)
= CH3CHO(aq) + NADH + H+(aq). (1)– Étape 3 : on mesure l’absorbance de la NADH par spectrophotométrie à la longueur d’onde 340 nm lors du dosage.
A. 1re partie :détermination du degré alcoolique d’un vin
« On appelle degré alcoolique d’une boisson alcoo-lisée le volume (exprimé en mL) d’éthanol contenu dans 100 mL de cette boisson, les volumes étant mesurés à 20 °C ». On l’exprime en % vol.• Questions préliminaires1. Montrer que la réaction (1) est bien une réaction d’oxydo-réduction, en faisant apparaître le transfert d’électrons entre les deux couples donnés.
DonnéeCouples d’oxydoréduction :CH3CHO(aq)/CH3CH2OH(aq) ;NAD+(aq)/NADH(aq).
2. Quel est le rôle du catalyseur ?
• Étalonnage du spectrophotomètreOn réalise une gamme de quatre solutions étalons. Chaque solution étalon contient :– NAD+(aq) en excès,– le catalyseur,– une solution aqueuse de concentration massique connue en éthanol.On mesure l’absorbance de chaque solution étalon et on obtient les résultats suivants.
Solution étalon S1 S2 S3 S4
Concentration massique Cm en éthanol (mg · L–1)
50 100 200 300
Absorbance A 0,08 0,16 0,32 0,48
3. Lors du réglage initial, quelle valeur doit-on don-ner à l’absorbance de la solution de référence avant toute mesure ?4. Tracer la courbe A en fonction de la concentra-tion massique.5. a) Montrer que la représentation graphique est en accord avec la loi de Beer-Lambert A = k · Cm.b) Déterminer la valeur de k en L · mg–1.
• Préparation et dosage de l’éthanol contenu dans le vin
On distille 20 mL de vin ; le distillat est ensuite ajusté à 200 mL avec de l’eau distillée pour obtenir une solution, appelée D.On prépare l’échantillon à doser par spectrophoto-métrie en introduisant dans une fiole jaugée de 50 mL, que l’on complétera avec de l’eau distillée :– 1 mL de solution D,– le catalyseur,– NAD+(aq) en excès.L’absorbance mesurée pour cet échantillon vaut :
Ae = 0,30.6. Montrer que l’échantillon préparé correspond à une dilution au 1/50e de la solution D.7. Par une méthode de votre choix à préciser, déter-miner à partir de l’absorbance mesurée Ae la concen-tration massique en éthanol de l’échantillon étudié.8. En déduire la concentration massique en étha-nol :
•

171Chapitre 12 - Contrôle de la vitesse par catalyse
Dans une réaction limitée, le temps de demi-réaction,
t½, et celui pour lequel x = xf
2.
Annexe : Fermentation dans le vin x = f(t).
25
20
15
10
5
5 10 15 20 25
x (mmol)
t (j)
Évolution de l’avancement en fonction du temps.
Corrigé1. Les demi-équations électroniques associées aux couples oxydant/réducteur CH3CHO(aq)/CH3CH2OH(aq) et NAD+(aq)/NADH(aq) s’écrivent :
CH3CH2OH(aq) = CH3CHO(aq) + 2 H+ + 2 e–(aq)NAD+(aq) + H+(aq) + 2 e– = NADH(aq)
En additionnant les deux demi-équations précé-dentes, on obtient l’équation chimique (1) modéli-sant la transformation :CH3CH2OH(aq) + NAD+(aq)
= CH3CHO(aq) + NADH(aq) + H+(aq).L’éthanol cède deux électrons, que fixe NAD+(aq).
2. Le catalyseur accélère ou oriente une transfor-mation chimique.
3. L’initialisation du spectrophotomètre se fait par mise à zéro de l’absorbance pour la solution de réfé-rence (cuve contenant tous les produits de la solu-tion à doser sauf le réactif à doser, ici l’éthanol).
4. Graphe A = f(Cm) :
100150200250300350400450
100 150 200 250
A (× 10–3)
Cm (mg · L–1)
Courbe d’étalonnage de l’absorbancede la solution aqueuse d’éthanol.
•a) de la solution D,b) du vin.9. Déterminer alors le degré alcoolique du vin.DonnéeMasse volumique de l’éthanol supposée constante dans le domaine de concentration considéré :
ρ = 0,80 kg · L–1.
B. 2e partie :cinétique de la fermentation malolactique
L’équation de la fermentation malolactique est :COOH–CH2–CHOH–COOH(aq)
= CH3–CHOH–COOH(aq) + CO2(g). Acide malique Acide lactique
Le dosage enzymatique de l’acide malique restant dans le vin a donné les résultats suivants pour une température de fermentation maintenue à 20 °C.
Concentration massique Cm(t) en acide malique (g · L–1)
3,5 2,3 1,6 0,8 0,5 0,27 0
Instant t (j) 0 4 8 12 16 20 28
10. a) Montrer que la concentration molaire en acide malique restant dans le vin à l’instant t s’ex-prime par :
[acide malique] ( ) =( )mt
C t134
.
b) En déduire la quantité de matière d’acide mali-que initiale dans un litre de vin, n(acide malique) (t = 0).
11. À l’aide d’un tableau descriptif de l’évolution du système, montrer que l’avancement à l’instant t de la réaction pour un litre de vin se met sous la forme :
x(t) = 2,6 × 10–2 – n(acide malique) (t).
12. La courbe représentant les variations de x en fonction du temps t est donnée en annexe.a) Comment peut-on, à partir du graphe, évaluer la vitesse volumique de réaction à l’instant t ? (Aucun calcul n’est demandé.)b) Commenter l’évolution de la vitesse volumique de la transformation au cours du temps.
13. Définir et déterminer le temps de demi-réac-tion.DonnéesMasses molaires atomiques (g · mol–1) :
M(H) = 1,0 ; M(C) = 12 ; M(O) = 16.
•
•

172 Partie 4 - Comment contrôler l’évolution d’un système chimique ?
5. a) Le graphe est une droite qui passe par l’ori-gine. L’absorbance A est donc proportionnelle à la concentration massique Cm, soit : A = k · Cm, avec k coefficient directeur de la droite.
b) k = ∆∆
A
Cm
; k = 0 48 0 08
300 50
, – ,
– = 1,6 × 10–3 L · mg–1.
6. Au cours de la dilution, la loi de conservation de la matière est respectée. En notant avec l’indice D le prélèvement issu de la solution étendue du dis-tillat et avec l’indice e pour l’échantillon, il vient :
CD · VD = Ce · Ve ⇒ Ce = CD · V
VD
e
= CD · 1
50.
Dans cette relation, le coefficient de dilution est bien 1/50.
7. Connaissant l’absorbance mesurée pour l’échan-tillon, Ae = 0,30, on détermine la concentration massique dans l’échantillon par lecture directe sur la courbe d’étalonnage ou par application numéri-que à partir de la relation :
A = k · Cm ⇒ Cme = A
k ;
Cme = 0 30
1 6 10 3
,
, –× = 1,9 × 102 mg · L–1.
8. a) La solution D a été diluée 50 fois. Le calcul de la concentration massique de D peut s’écrire :CD = 50 Cme ⇒ CD = 50 × 1,9 × 102 = 9,4 × 103 mg · L–1,soit 9,4 g · L–1.b) La solution D correspond à 200 mL de solution obtenue par dilution du distillat issu de la distilla-tion de 20 mL de vin. Cela correspond à une dilution par 10 du vin initial. Sa teneur massique en éthanol est donc :
CmVin = 10 CD ; CmVin = 10 × 9,4 = 94 g · L–1.
9. La définition du degré alcoométrique du vin per-met de conduire le calcul suivant.Masse d’éthanol m contenu dans 100 mL de vin :
m = CmVin · V ; m = 94 × 0,1 = 9,4 g.
Volume d’éthanol correspondant dans 100 mL de vin :
m = ρ · V ⇒ V = m
ρ ;
V = 9 4 10
0 8
3,
,
–× = 1,2 × 10–3, soit V = 12 mL.
Soit dans 100 L de vin : 12 L d’éthanol.Le degré alcoométrique arrondi est donc 12°.
10. a) La relation entre les concentrations massi-que et molaire s’écrit :
C C Mmassique molaire= ·
⇒ = =CC
Mmolairemassiqueacidemalique[ ]
La masse molaire de l’acide malique est :M = (4 × 12) + (5 × 16) + (6 × 1) = 134 g · mol–1.
[ ]( )( )
acidemalique .
À l’inst
massiquetC t
=134
aant initial = 0 s,
acidemalique
t
[ ],
03 5
1340= = ,, ·
( ,( )
026
0 026 1 0
1
0
mol L
acidemalique)
−
= = × =n t ,,026mol.
11.Avanc. Acide
maliqueAcide
lactique CO2
Instant initial t = 0 0 0,026 0 0
Instant t x(t) 0,026 – x(t) x(t) x(t)
Donc : n(acide malique)(t) = 0,026 – x(t),soit x(t) = 0,026 – n(acide malique)(t).
12. a) Par définition, la vitesse instantanée volu-mique de formation d’un corps est donnée par la relation :
v = 1
V
x
t·d
d;
d
d
x
t représente le coefficient directeur de la tan-
gente à la courbe à l’instant considéré.V correspond au volume du milieu réactionnel.b) Le coefficient directeur diminue au cours du temps, donc la vitesse volumique diminue. Le fac-teur cinétique responsable de cette décroissance est la concentration, car au fur et à mesure de l’évo-lution de la transformation, la concentration des réactifs diminue.
13. Le temps de demi-réaction est la durée au bout de laquelle l’avancement de la réaction a atteint la moitié de sa valeur maximale.L’avancement final est xf = 0,026 mol, donc le temps de demi-réaction correspond à la durée au bout de laquelle :
x = xf
2, soit x =
0 026
2
, = 0,013 mol.
Par lecture directe sur la courbe de l’avancement en fonction du temps, on trouve :
t1
2
= 8 jours.