
Leucémie myéloïde chronique - · PDF fileHématologie biologique (Pr...
10
Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________ MAJ : janvier 2007 Page 1 sur 10 Leucémie myéloïde chronique Sommaire : - Définition, étiologie et signes cliniques - hémogramme - myélogramme - caryotype - biologie moléculaire - autres examens parfois réalisés - diagnostic différentiel - évolution : la phase blastique de la LMC : - la phase accélérée - la phase blastique (lymphoïde, myéloïde) - Aspects thérapeutiques généraux 1. Définition, épidémiologie et aspects cliniques Définition. Il s’agit d’une prolifération maligne et systématisée de la lignée granulocytaire sans blocage de maturation. C’est un syndrome myéloprolifératif chronique prédominant sur la lignée granuleuse, lié à un processus monoclonal affectant une cellule souche très primitive. Une anomalie cytogénétique est constamment associée à la prolifération : le chromosome Philadelphie (Ph1). Le diagnostic nécessite : hémogramme, myélogramme, caryotype et étude en biologie moléculaire L'évolution se fait en 3 phases : 1ère phase : chronicité 2ème phase : accélération (10% des pts se présentent d’emblée à ce stade) 3ème phase : transformation aiguë, ou acutisation ou phase blastique Epidémiologie Maladie rare : 600 – 1000 nouveaux cas / an en France (incidence = 1-2 nouveaux cas / 100 000 H/an) Survient surtout entre 30 à 50 ans mais peut se rencontrer à tous les âges (rare chez l'enfant) Discrète prédominance masculine : sex ratio = 1,1 à 1,2. Etiologie inconnue, mais dans 5% des cas elle est secondaire à une exposition chronique au benzène ou aux radiations ionisantes. Aspects cliniques généraux Dans 50% des cas : découverte fortuite sur un hémogramme anormal. Sinon : Symptomatologie liée à la splénomégalie (asthénie, sueurs nocturnes, perte de poids), quasi constante, de volume modéré à très important, indolore, mobile avec la respiration et isolée (l’échographie abdominale peut être utile).
Transcript of Leucémie myéloïde chronique - · PDF fileHématologie biologique (Pr...

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 1 sur 10
Leucémie myéloïde chronique Sommaire : - Définition, étiologie et signes cliniques - hémogramme - myélogramme - caryotype - biologie moléculaire - autres examens parfois réalisés - diagnostic différentiel - évolution : la phase blastique de la LMC : - la phase accélérée - la phase blastique (lymphoïde, myéloïde) - Aspects thérapeutiques généraux 1. Définition, épidémiologie et aspects cliniques Définition. Il s’agit d’une prolifération maligne et systématisée de la lignée granulocytaire sans blocage de maturation. C’est un syndrome myéloprolifératif chronique prédominant sur la lignée granuleuse, lié à un processus monoclonal affectant une cellule souche très primitive. Une anomalie cytogénétique est constamment associée à la prolifération : le chromosome Philadelphie (Ph1). Le diagnostic nécessite : hémogramme, myélogramme, caryotype et étude en biologie moléculaire L'évolution se fait en 3 phases : 1ère phase : chronicité 2ème phase : accélération (10% des pts se présentent d’emblée à ce stade) 3ème phase : transformation aiguë, ou acutisation ou phase blastique Epidémiologie Maladie rare : 600 – 1000 nouveaux cas / an en France (incidence = 1-2 nouveaux cas / 100 000 H/an) Survient surtout entre 30 à 50 ans mais peut se rencontrer à tous les âges (rare chez l'enfant) Discrète prédominance masculine : sex ratio = 1,1 à 1,2. Etiologie inconnue, mais dans 5% des cas elle est secondaire à une exposition chronique au benzène ou aux radiations ionisantes. Aspects cliniques généraux Dans 50% des cas : découverte fortuite sur un hémogramme anormal. Sinon : Symptomatologie liée à la splénomégalie (asthénie, sueurs nocturnes, perte de poids), quasi constante, de volume modéré à très important, indolore, mobile avec la respiration et isolée (l’échographie abdominale peut être utile).
Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 2 sur 10
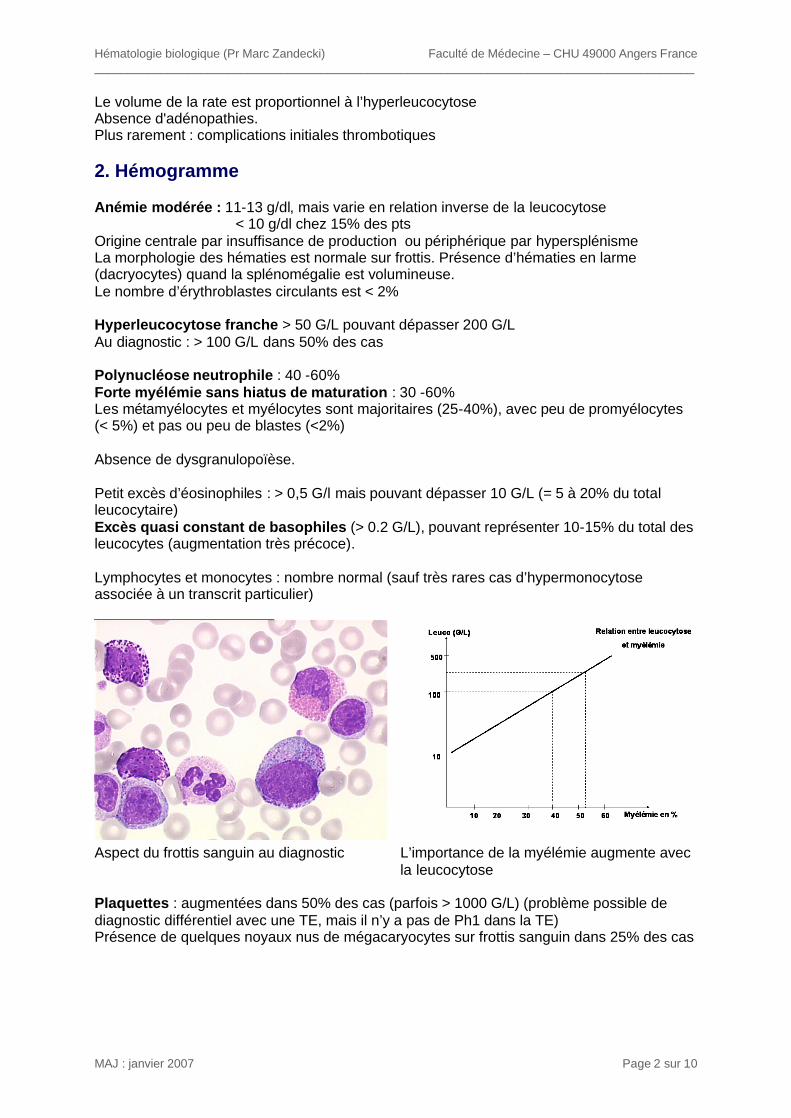
Le volume de la rate est proportionnel à l’hyperleucocytose Absence d'adénopathies. Plus rarement : complications initiales thrombotiques 2. Hémogramme Anémie modérée : 11-13 g/dl, mais varie en relation inverse de la leucocytose < 10 g/dl chez 15% des pts Origine centrale par insuffisance de production ou périphérique par hypersplénisme La morphologie des hématies est normale sur frottis. Présence d’hématies en larme (dacryocytes) quand la splénomégalie est volumineuse. Le nombre d’érythroblastes circulants est < 2% Hyperleucocytose franche > 50 G/L pouvant dépasser 200 G/L Au diagnostic : > 100 G/L dans 50% des cas Polynucléose neutrophile : 40 -60% Forte myélémie sans hiatus de maturation : 30 -60% Les métamyélocytes et myélocytes sont majoritaires (25-40%), avec peu de promyélocytes (< 5%) et pas ou peu de blastes (<2%) Absence de dysgranulopoïèse. Petit excès d’éosinophiles : > 0,5 G/l mais pouvant dépasser 10 G/L (= 5 à 20% du total leucocytaire) Excès quasi constant de basophiles (> 0.2 G/L), pouvant représenter 10-15% du total des leucocytes (augmentation très précoce). Lymphocytes et monocytes : nombre normal (sauf très rares cas d’hypermonocytose associée à un transcrit particulier)
Aspect du frottis sanguin au diagnostic L’importance de la myélémie augmente avec
la leucocytose Plaquettes : augmentées dans 50% des cas (parfois > 1000 G/L) (problème possible de diagnostic différentiel avec une TE, mais il n’y a pas de Ph1 dans la TE) Présence de quelques noyaux nus de mégacaryocytes sur frottis sanguin dans 25% des cas

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 3 sur 10
3. Myélogramme Indispensable, au moins pour définir le % blastes (début d'acutisation?) Ponction médullaire facile à réaliser, avec un os de dureté normale (à l’opposé de la splénomégalie myéloïde) Frottis très riche. Nombreux mégacaryocytes, de taille souvent réduite. Hyperplasie de la lignée granuleuse où tous les stades de maturation. Blastose < 5% (mais 5 – 19% dans les cas de diagnostic en phase accélérée) Erythroblastopénie : classiquement < 10% Excès d’éosinophiles et de basophiles, en parallèle de l’excès sanguin. Présence d’histiocytes surchargés de lipofuchsines, qui ont parfois une coloration bleue (histiocytes bleu-de-mer, appelés à tort de type Gaucher) dans > 30% des cas Biopsie ostéo-médullaire : moelle très riche avec disparition des adipocytes et absence de myélofibrose (à l’inverse de la splénomégalie myéloïde) Hyperplasie granulocytaire: rapport G/E = 10 – 30 (normale = 2-5)
Etalement médullaire richement cellulaire, avec nombreux mégacaryocytes (de taille normale ou réduite)
4. Caryotype Sur prélèvement de sang si myélémie nette, sinon sur prélèvement médullaire. Dans 95% des cas : présence du chromosome Philadelphie ou Ph1 qui correspond à un chromosome 22 raccourci, résultat de la translocation réciproque et équilibrée entre les bras longs des chr 9 et 22 : t(9;22)(q34;q11).

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 4 sur 10
Caryotype médullaire montrant un chromosome 22 raccourci correspondant au chromosome Philadelphie (image : F Brizard, Poitiers) Sur le bras long du chromosome 9 la région ABL (abelson) se coupe (souvent entre les régions Ib et a2) et sa partie télomérique (a2 …e19…) vient se localiser à la place de la partie télomérique du bras long du chromosome 22, après b2,b3,…, dans une région appelée BCR (breakpoint cluster region) Cette translocation aboutit à un chromosome 22 très court (= Ph1) sur lequel se trouve le gène chimérique BCR-ABL, formé du début de BCR et le la fin d’ABL
Dans 5 – 10% des cas le Ph1 n’est pas retrouvé tel quel, mais impliqué dans des translocations complexes (= Ph1 masqué) En général le Ph1 est isolé (= pas d’autre anomalie), mais parfois on retrouve déjà une ou quelques unes des anomalies apparaissant au cours de l’évolution = trisomie 8 ou 19, duplication du Ph1, anomalies du 17.

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 5 sur 10
Remarques : - Le Ph1 est présent dans toutes les lignées cellulaires issues du clone malin (toutes lignées myéloïdes et lymphocytaire B, rarement les lymphocytes T) - Il existe un chromosome Ph1 dans un tiers des LAL de l’adulte (mais le transcrit de fusion est différent) 5. Biologie moléculaire La translocation juxtapose une partie du gène bcr (breakpoint cluster région) du chr 22 e l'oncogène Abelson (c-abl) situé sur le chr 9 La conservation du cadre de lecture permet la synthèse d'ARN messagers hybrides dits chimériques comportant des séquences bcr en 5' et c-abl en 3'.
Les points de cassure sont regroupés entre Ib et Ia sur abl, alors qu’il existe plusieurs régions de cassure sur bcr : - La région M bcr est majoritairement impliquée dans la LMC: le transcrit est b3a2 (60% des cas) ou b2a2 (35% des cas) (on parle de transcrits M bcr pour Major bcr) Remarque : on peut retrouver les 2 transcrits chez 5 à 10% des pts L'ARN chimérique bcr/abl est traduit en une protéine de fusion p210 bcr/abl ayant un pouvoir oncogénique avec très forte activité tyrosine kinase. - La région m bcr (minor bcr) est impliquée dans 0.4 % des LMC: le transcrit est e1a2, qui produit la protéine p190 (ce transcrit est fréquemment associé à l’existence d’une monocytose, une absence de basophilie, et une absence de splénomégalie; par ailleurs il est celui retrouvé dans environ 2/3 des LAL Ph+)

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 6 sur 10
- La région µ bcr (micro bcr) est impliquée dans < 0.1 % des LMC: le transcrit est e19a2, qui produit la protéine p230 (associée à la LMC à polynucléaires neutrophiles: expansion essentiellement de polynucléaires neutrophiles, thrombocytose parfois > 1000 G/L, évolution plus indolente que la LMC classique, une tendance moindre à transformer) Recherche de l’anomalie : On recherche le transcrit de fusion (ARN) par technique de RT-PCR (L’ARNm est transformé en ADN complémentaire puis amplifié par PCR). Actuellement on recherche et on quantifie le transcrit par technique RQ-PCR (rétrotranscription - quantification) 6. Autres examens parfois réalisés - Cytochimie des phosphatases alcalines leucocytaires (PAL) : absence d’enzyme. - Hyperuricémie - Augmentation des LDH - Augmentation de la vitamine B12 sanguine, par augmentation du transporteur transcobalamine 1 synthétisé par les granulocytes 7. Diagnostic différentiel de la LMC 7.1. Polynucléoses neutrophiles et myélémies Etats infectieux ou inflammatoires sévères (leucocytes rarement > 50 G/L, myélémie modérée < 15%, absence d’excès de polynucléaires basophiles), syndrome paranéoplasique par synthèse de cytokines stimulant la granulopoïèse (leucocytes parfois > 100G/L, sans myélémie) ; la situation clinique est différente. 7.2. Les autres syndromes myéloprolifératifs Dans quelques cas on observe au premier plan une polyglobulie (vraie) ou une hyperthrombocytose, avec myélémie discrète : la recherche du Ph1 est systématique dans les polyglobulies de Vaquez et les TE : la découverte d’un Ph1 (ou d’un remaniement bcr-abl) affirme le diagnostic de LMC, quelles que soit sa présentation biologique. La recherche de la mutation JAK2 est possible : elle n’est jamais présente dans la LMC. Par rapport à la splénomégalie myéloïde chronique, outre l’absence de Ph1, on retrouve sur le frottis sanguin des érythroblastes (5- 10%), une anisopoïkilocytose franche et de nombreuses hématies en larme, la ponction médullaire est de réalisation technique difficile (os très dur), et la BOM objective une myélofibrose nette. 7.3. Leucémie myélomonocytaire chronique: splénomégalie, hyperleucocytose avec polynucléose neutrophile, mais myélémie modérée, monocytose notable, pas de Ph1 7.4. Les exceptionnelles LMC atypiques : Ph1 négatives, avec souvent un aspect proche de celui d’une LMMC 8. Evolution de la LMC : la phase blastique La LMC comprend 3 étapes successives : phase chronique puis phase d’accélération puis phase blastique = transformation en un tableau de leucémie aiguë Jusqu’en 2000, les traitements de la phase chronique faisaient régresser les signes cliniques (splénomégalie) et corrigeaient plus ou moins l’hémogramme. Ils n’avaient soit

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 7 sur 10
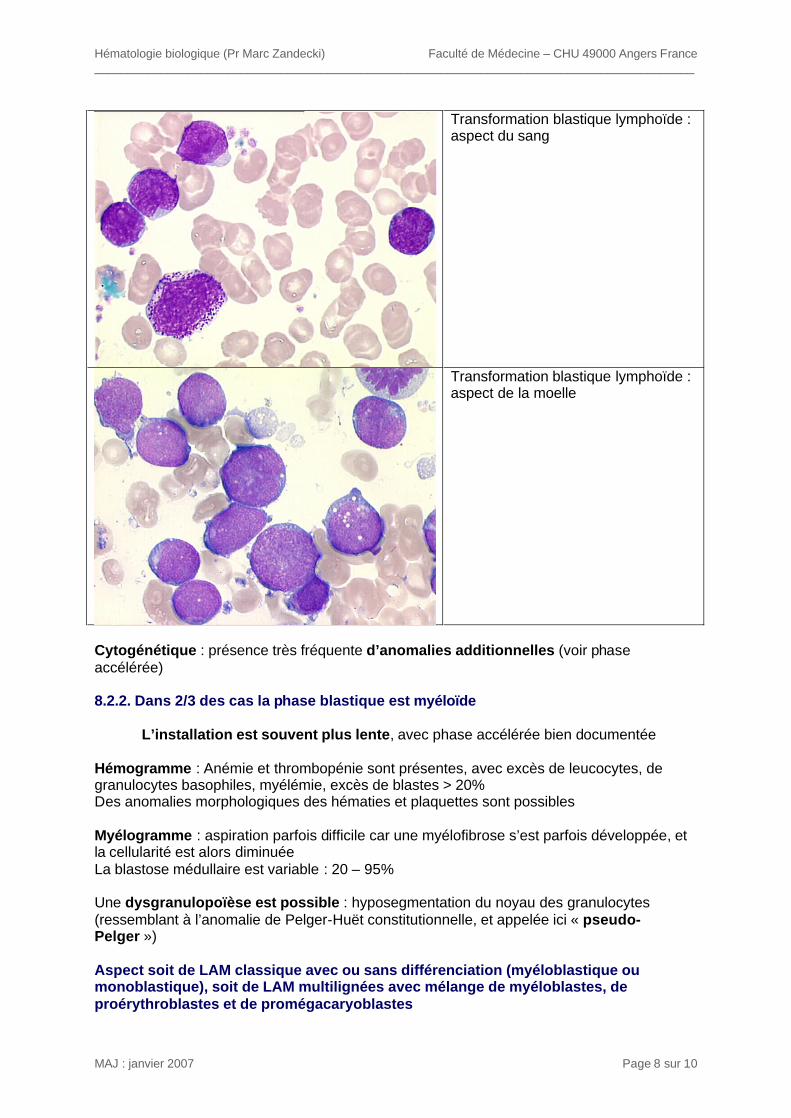
aucun effet sur le délai avant transformation blastique (busulfan), soit un effet retardateur modeste (hydrea) ou moyen (interféron alpha). La phase blastique survenait pour la majorité des pts 3 à 8 ans après le diagnostic. L’absence de ttt de la phase blastique marquait la fin de la maladie (survie < 6 mois). L’utilisation d’inhibiteurs de tyrosine kinase a permis d’allonger la durée de la phase chronique et de ramener le rythme de transformation blastique à environ 1-2 % par an. 8.1. Phase accélérée Présente chez 10% des pts dès le diagnostic, et présente chez 70% des pts avant la phase blastique : sa durée varie de 3 à 12 mois. On observe la modification d’un ou plusieurs paramètres cliniques et biologiques : Cliniquement : - sueurs nocturnes, perte de poids, réapparition de la splénomégalie, douleurs osseuses Hémogramme : - Réapparition d’une hyperleucocytose malgré le ttt - réapparition d’une myélémie comprenant : blastes + myéloblastes + promyélocytes : 7 à 20% ou blastes + myéloblastes > 10% -excès de polynucléaires basophiles > 20% (ou basophiles + éosinophiles > 20%) - Thrombopénie < 100 G/L ou > 1000 G/L malgré le ttt Moelle osseuse : - > 10% blastes - Basophiles + éosinophiles > 10% - apparition d’une dysgranulopoïèse - Fibrose réticulinique ou collagène à la BOM Evolution cytogénétique clonale chez 1/3 des pts : apparition d’anomalies complémentaires du génome, car le Ph1 induit une instabilité génomique : +8, dup Ph1, i(17q), +19. 8.2. Phase blastique Signes cliniques identiques à ceux de la phase accélérée ; des adénopathies et chloromes blastiques extramédullaires sont possibles Hémogramme : > 20% blastes Myélogramme : > 20- 30 % de blastes 8.2.1. Dans 1/3 des cas la phase blastique est lymphoïde Le plus souvent la phase d’accélération est discrète ou absente : chez un patient auparavant en bonne forme, avec maladie bien contrôlée, installation en quelques semaines d’une asthénie et d’une détérioration de l’hémogramme Hémogramme : anémie, thrombopénie, et hyperleucocytose d’importance variables ; l’excès de granulocytes basophiles n’est pas toujours retrouvé Blastose sanguine souvent franche, dépassant parfois 80% ; il peut persister une discrète myélémie Myélogramme : blastose franche souvent > 90% Pas de dysgranulopoïèse L’aspect morphologique et l’immunophénotype sont ceux d’une LAL commune BII, CD19+, CD22+, CD79a+, CD10+, cµ et SIg négatives. Cytochimie de la myéloperoxydase : négative. [exceptionnellement : LAL – T]
Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 8 sur 10
Transformation blastique lymphoïde : aspect du sang
Transformation blastique lymphoïde : aspect de la moelle
Cytogénétique : présence très fréquente d’anomalies additionnelles (voir phase accélérée) 8.2.2. Dans 2/3 des cas la phase blastique est myéloïde L’installation est souvent plus lente, avec phase accélérée bien documentée Hémogramme : Anémie et thrombopénie sont présentes, avec excès de leucocytes, de granulocytes basophiles, myélémie, excès de blastes > 20% Des anomalies morphologiques des hématies et plaquettes sont possibles Myélogramme : aspiration parfois difficile car une myélofibrose s’est parfois développée, et la cellularité est alors diminuée La blastose médullaire est variable : 20 – 95% Une dysgranulopoïèse est possible : hyposegmentation du noyau des granulocytes (ressemblant à l’anomalie de Pelger-Huët constitutionnelle, et appelée ici « pseudo-Pelger ») Aspect soit de LAM classique avec ou sans différenciation (myéloblastique ou monoblastique), soit de LAM multilignées avec mélange de myéloblastes, de proérythroblastes et de promégacaryoblastes

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 9 sur 10
Acutisation myéloïde : aspect du myélogramme Présence d’un excès de grands blastes d’allure myéloblastique
Excès de micromégacaryocytes et de blastes peu différenciés
Cytogénétique : les anomalies caryotypiques additionnelles sont quasi constantes : habituellement : trisomie 8, iso(17q), doublement du Ph1, et/ou trisomie 19 quelques patients : monosomie 17 ou del(17p), anomalie du chromosome 11, monosomie ou réarrangement du chromosome 7 Plus rarement une anomalie cytogénétique comparable à celle des LAM de novo est observée : t(15;17) ou inv(16) [Les anomalies morphologiques sont identiques à celles de la LA de novo ayant ces anomalies cytogénétiques] Remarques : - Les mécanismes moléculaires impliqués dans la progression sont mal connus : ils font sans doute intervenir une augmentation d’activité oncogénique (duplication du Ph1, c-myc, Rb, AML-evi1) et une perte d’activité suppressive de tumeur (perte de p53, instabilité accrue du génome). - l’anomalie 17p est associée à l’aspect morphologique pseudo-Pelger

Hématologie biologique (Pr Marc Zandecki) Faculté de Médecine – CHU 49000 Angers France __________________________________________________________________________________________
MAJ : janvier 2007 Page 10 sur 10
9. Aspects thérapeutiques généraux Le ttt est nécessaire (décès en < 2 ans sans ttt). Définition de la réponse au traitement (A. Kantarjian, NEJM 2002;346:645-652) : Réponse hématologique complète = normalisation de la numération globulaire (leucocytes < 10 G/L, plaquettes < 450 G/L, et formule leucocytaire sans blastes ni promyélocytes, et < 5% de myélocytes + métamyélocytes) Réponse cytogénétique : majeure (complète) = absence de Ph1 sur 100 métaphases étudiées au caryotype partielle si on retrouve entre 1 et 34% de métaphases avec Ph1 mineure si on retrouve 35 - 90 % de métaphases anormales Le seul traitement curatif : allogreffe de moelle osseuse. Limitée aux patients < 50 – 55 ans, et avec donneur HLA identique, apparenté ou non (concerne potentiellement 30% des pts). Chimiothérapie. But du ttt : diminuer le plus possible le nombre de cellules Ph1+, car la présence du Ph1 entraîne une instabilité du génome, l’apparition d’anomalies complémentaires du génome et à terme la phase blastique. Les protocoles thérapeutiques utilisent tous un inhibiteur de l’activité tyrosine kinase excessive (imatinib), souvent associé à une autre molécule (interféron alpha, aracytine). On obtient 98% de rémissions hématologiques , des réponses cytogénétiques dans > 50% cas (majeures dans 31% des cas), et même des réponses moléculaires (transcrit non retrouvé en biologie moléculaire). Actuellement la transformation blastique survient à raison de 1% par an. Autres traitements utilisables : hydroxyurée (Hydréa) en traitement court pour diminuer rapidement l’hyperleucocytose, avant le ttt spécifique ; interféron alpha ; aracytine ; homoharringtonine. Le traitement de la phase blastique utilise des polychimiothérapies incluant un inhibiteur de tyrosine kinase. Les succès sont limités mais des rémissions sont observées. (Revu par M Zandecki, janvier 2007)


















