Les Médicaments génériques - aip-marseille.org©dicaments... · - Renforcement de la...
13
UE INDUS 08.12.14 ;12.12.14 et 16.12.14 Pr. Andrieu 1 Les Médicaments génériques I) Définition : Copie d’un médicament original ≠ copie strictement identique Répertoire des groupes génériques (site ANSM) : - Médicaments génériques -> médicaments génériques associés - 7800 génériques A) Code santé publique : Même composition en substance active Même forme pharmaceutique (même biodisponibilité, même efficacité) Bioéquivalence avec la spécialité de référence Les différentes formes pharmaceutiques orales à libération immédiates sont considérées comme une même forme pharmaceutique. Les différentes sels, esters, éthers, isomères, mélanges d’isomères, complexes ou déridés d’un SA sont considérés comme une même SA, sauf s’ils présentent des propriétés sensiblement différentes au regard de la sécurité ou de l’efficacité = définition large de la substance active La présence des ENN excipients à effet notoire n’est pas spécifique aux génériques. Ils sont présents aussi bien dans les princeps que dans les génériques. B) EEN : Excipient à effet notoire : tout excipient dont la présence peut nécessiter des précautions d’emploi pour certaines catégories particulières de patient. La spécialité de référence est ou a été commercialisée : - En FR - Dans un état membre de l’UE II) Enjeu économique : Exception au principe général de liberté des prix Spécialités pharmaceutiques remboursables ne peuvent être vendues au dessus d’un prix plafond fixés par les pouvoirs publics Marges de distribution de ces médicaments réglementées Mécanismes incitatifs favorisants la délivrance des génériques pour maitriser les dépenses liées au remboursement de médicaments : - Prix des génériques significativement inférieur à celui de la référence. Dès la commercialisation du générique ou au moment de la perte de brevet : baisse de prix de 60% pour les génériques et 20% pour le princeps. Puis baisse de 12,5% après 18 mois d’exploitation du répertoire pour le princeps ou mise sur TRF (tarif forfaitaire de Responsabilité) - Mais sans mécanisme d’ajustement, pharmacien pas incité à délivrer de générique : marge en valeur absolue perçue par le pharmacie est toujours supérieurs pour un médicament onéreux - Par dérogation, la marge des génériques est, pour les pharmaciens, égale à celle de la spécialité de référence : neutralité des marges
Transcript of Les Médicaments génériques - aip-marseille.org©dicaments... · - Renforcement de la...

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 1
Les Médicaments génériques
I) Définition :
Copie d’un médicament original
≠ copie strictement identique
Répertoire des groupes génériques (site ANSM) :
- Médicaments génériques -> médicaments génériques associés
- 7800 génériques
A) Code santé publique :
Même composition en substance active
Même forme pharmaceutique (même biodisponibilité, même efficacité)
Bioéquivalence avec la spécialité de référence
Les différentes formes pharmaceutiques orales à libération immédiates sont considérées comme une
même forme pharmaceutique.
Les différentes sels, esters, éthers, isomères, mélanges d’isomères, complexes ou déridés d’un SA sont
considérés comme une même SA, sauf s’ils présentent des propriétés sensiblement différentes au regard
de la sécurité ou de l’efficacité = définition large de la substance active
La présence des ENN excipients à effet notoire n’est pas spécifique aux génériques. Ils sont présents aussi
bien dans les princeps que dans les génériques.
B) EEN :
Excipient à effet notoire : tout excipient dont la présence peut nécessiter des précautions d’emploi pour certaines
catégories particulières de patient.
La spécialité de référence est ou a été commercialisée :
- En FR
- Dans un état membre de l’UE
II) Enjeu économique :
Exception au principe général de liberté des prix
Spécialités pharmaceutiques remboursables ne peuvent être vendues au dessus d’un prix plafond fixés par
les pouvoirs publics
Marges de distribution de ces médicaments réglementées
Mécanismes incitatifs favorisants la délivrance des génériques pour maitriser les dépenses liées au
remboursement de médicaments :
- Prix des génériques significativement inférieur à celui de la référence. Dès la commercialisation du
générique ou au moment de la perte de brevet : baisse de prix de 60% pour les génériques et 20% pour
le princeps. Puis baisse de 12,5% après 18 mois d’exploitation du répertoire pour le princeps ou mise
sur TRF (tarif forfaitaire de Responsabilité)
- Mais sans mécanisme d’ajustement, pharmacien pas incité à délivrer de générique : marge en valeur
absolue perçue par le pharmacie est toujours supérieurs pour un médicament onéreux
- Par dérogation, la marge des génériques est, pour les pharmaciens, égale à celle de la spécialité de
référence : neutralité des marges

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 2
- Possibilités de remise par le fabricant beaucoup plus importante pour les génériques que pour les autres
médicaments remboursables.
C) Substitution du princeps :
Adhésion du patient indispensable, information par :
- ANSM garante de la sécurité et de l’autorisation des médicaments
- Organismes d’Assurance Maladie
Adhésion du médecin traitant à la substitution
D) EEN :
Peu de personnes sensibles aux EEN, mais ils doivent être pris en compte lors de la substitution !
- Pour une spécialité sans EEN, choisir une spécialité sans EEN
- Pour une spécialité contenant un ou plusieurs EEN, choisir un générique contenant le ou les mêmes EEN
ou un générique partiellement ou totalement dépourvu de ces EEN
Substitution par un générique contenant des EEN que ne contient pas la spécialité prescrite, est possible
lorsqu’après avoir interrogé le patient, il apparait que pas de risque de survenue d’effets liés à ces EEN
Présences des EEN signalée dans le répertoire des génériques pour toutes les spécialités (générique et
princeps)
EEN signalés aux patients dans la notice du médicament (emballage)
III) Le marché des génériques :
Evolution de la part du marché des spécialités remboursables détenues par les génériques
Augmentation du marché grâce à :
- élargissement du répertoire
- Renforcement de la substitution dans groupe du répertoire (adhésion du patient)
- Incitations financière de l’Assurance Maladie généralisation et renforcement de la disposition « tiers
payants contre générique)
Remises consenties aux officines peuvent atteindre 17% pour les génériques -> diminution de la part en
valeur des génériques par rapport aux autres spécialités remboursables
Part de marché en valeur détenue par les génériques : poids dans l’économie d médicament
Génériques présents sur le marché non remboursable
Génériques présents sur le marché hospitalier, part de marché en valeur semble faible, mais :
- Forte pression sur les prix : labo exploitant la spécialité originale doit baisser son prix de vente pour
conserver une partie de son marché initial
- Effet modérateur incontestable sur les prix à l’hôpital
Part des génériques (en quantité) dans le marché pharmaceutique > 60% dans des pays comme
l’Allemagne, le RU et Pays Bas
Aux US, la part des génériques est très élevée : 80 des médicaments prescrits
Mais définition du générique différente : inclut les SA hors brevet
En FR : 46% en quantité
A) Intérêt économique :
Prix bas des génériques : économie sur Produits de Santé
Permettent de mieux rémunérer les efforts de recherche en octroyant aux médicaments véritablement
innovants un prix de vente supérieur à celui qui aurait pu être accordé en l’absence d’économies
engendrées par les génériques
B) Spécialités biologiques similaires :
Médicaments biologiques similaires : copies de médicaments biologiques dits de référence

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 3
Même composition qualitative et quantitative en substance active et même forme pharmaceutique qu’un
médicament biologique de référence
≠ génériques, en raison des différences liées à la variabilité de la matière première ou aux procédés de
fabrication
IV) Auto-générique :
Les auto-génériques : copies strictement identiques aux spécialités originales, obtenues avec l’accord des
titulaires de l’AMM de ces spécialités originales
Mécanisme utilisé par un labo titulaire d’une spécialité de référence en vue d’obtenir un auto-générique,
copie conforme de sa spécialité de référence en vue de l’exploiter lui-même ou de confier son exploitation
à un autre laboratoire
V) Cycle de vie du médicament :
Brevet : 20 ans de protection de la découverte
CPP : certificat complémentaire de protection : 5 ans de protection supplémentaire sans dépasser 15 ans
après obtention de la 1ère AMM dans un pays européen.
AMM : 8 ans de protection des données pharmaceutiques, non cliniques et cliniques du médicament
princeps
Une demande d’AMM pour un générique peut dont être déposée à l’ANSM au terme d’un délai de 8 ans à
partir de l’octroi de la 1ère AMM européenne du médicament de référence
A) Clause Bolar :
Législation avant 2004 :
- Données couvertes par la propriété intellectuelle des brevets consultables la fin effective de ces derniers,
c’est-à-dire dix années après leurs dépôts aux autorités, selon la loi française
- Pays européens ne bénéficiaient pas de la clause dite « Bolar » (≠US, Israël ou Inde). Impossible de réaliser
en Europe les études et essais de bioéquivalences nécessaires à l’obtention de l’AMM pour les
médicaments prochainement généricables, et encore sous la protection de leur brevet. Production de
lots cliniques également interdite.
- Production à l’échelle industrielle et stockage de génériques avant la date du princeps interdites
B) Notion d’AMM globale :
Depuis 2005 : AMM successivement accordées dans l’UE pour de nouvelles indications thérapeutiques,
voies d’administration ou dosages que ceux figurant dans l’AMM du produit initial = « extensions de
gamme », font partie d’une seule et même AMM et ne bénéficient pas de délai de protection
supplémentaire
- AMM nationalisée : en France (ANSM)
- AMM centralisée : en UE (EMA)
- AMM décentralisée : France ou autre états UE
C) Répertoire des groupes génériques
Regroupement d’une spécialité de référence et des spécialités qui en sont génériques
Directeur général de l’ANSM identifie les génériques pour l’inscription au répertoire. Il informe le titulaire
de l’AMM de la référence de l’octroi de l’AMM générique de se spécialité.
Puis, au terme d’un délai minimum de 60 jours, durant lequel le détenteur du brevet du princeps peut faire
valoir ses droits auprès du génériqueur, la spécialité générique est inscrite automatiquement au répertoire
Un générique peut être inscrit au répertoire avant l’expiration du brevet qui protège sa référence.
Néanmoins, il ne pourra pas être commercialisé avant que le brevet ne soit arrivé à échéance

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 4
Le répertoire des groupes génériques est l’outil sur lequel le pharmacien d’officine se base pour délivrer les
médicaments génériques.
D) Droit de substitution
- Depuis 1999, pharmacien « peut délivrer par substitution à la spécialité prescrite une spécialiste du même
groupe générique à condition que le prescripteur n’ait pas exclu cette possibilité, pour des raisons
particulières tenant au patient, par une mention express portée sur la prescription ».
- Pharmacien doit indiquer sur l’ordonnance le nom du médicament qu’il a substitué.
- Non substitution possible par le pharmacien si risque (ex : patient âgé et poly-médicamenté)
- Dispositif « tiers payant contre générique ». Si patient refuse la substitution, il doit faire l’avance des frais
des médicaments.
E) Le médicament générique
- Comme pour toute spécialité pharmaceutique, l’ANSM est chargée de l’évaluation, du contrôle en
laboratoire et de l’inscription des spécialités génériques.
- Une spécialité générique doit faire l’objet, avant sa commercialisation, d’une autorisation de mise sur le
marché délivrée par le directeur général de l’Agence, dans le cadre soit d’une procédure purement
nationale, soit d’une procédure européenne.
F) La demande d’AMM pour un générique comprend :
- Un dossier pharmaceutique : données apportant la preuve de la qualité du médicament ;
- Un dossier biopharmaceutique : données apportant la preuve de la bioéquivalence du générique par
rapport à la spécialité de référence.
Le dossier de demande est déposé versus un médicament de référence qui a un dossier d’AMM complet : données
pharmaceutiques, non cliniques et cliniques.
Le dossier est évalué par les évaluateurs de l’ANSM en utilisant des référentiels : recommandations des lignes
directrices de l’Agence Européenne des Médicaments et sur les monographies de la Pharmacopée Européenne.
VI) Qualité pharmaceutique SA :
- Pour les SA, similarité de la structure avec celle du produit de référence à démontrer par des tests
physicochimiques. Différences à argumenter en termes d’impact sur la sécurité et l’efficacité.
- Caractéristiques physico-chimiques des SA pouvant affecter la biodisponibilité du produit, doivent être
discutées : polymorphisme, taille des particules.
- Problème majeur de la qualité des médicaments génériques : la SA
Plusieurs fournisseurs de SA
Qualité des « Starting Materials » différente
Solvants différents
Synthèses différentes
- Hétérogénéité possible
- « Flow Chart » : suivi de la SA du site de synthèse à la libération du PF = sites de fabrication (stérilisation,
micronisation) + transport
Problème majeur de la qualité des médicaments génériques : La SA
- Plusieurs fournisseurs de SA - Qualité des « starting materials » différente - Solvants différents - Synthèses différentes
Hétérogénéité possible.
« Flow chart » : suivi de la sA du site de synthèse à la libération du PF = sites de fabrication (stérilisation,
micronisation) + transports.

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 5
Attestation de respect BPF du site de production de la SA par audit du pharmacien responsable de la libération du PF.
Responsabilité pharmaceutique tout le long de la chaine de production : SA + PF
Certificat BPF donné par une autorité européenne.
Si certificat BPF donné par une autre autorité attestation de l’équivalence des exigences BPF (ex : Chine).
A) Qualité pharmaceutique : forme solide orale
En complément des études de bioéquivalence, la similarité des profils de dissolution in vitro du générique et de la
référence doit être démontrée dans des milieux de dissolution appropriés simulant les milieux physiologiques (2
pour l’estomac, 4 à la sortie de l’estomac et et 6,8 pour l’intestin).
Des normes de dissolution in vitro sont fixées pour le contrôle de qualité du produit dans un milieu de dissolution
discriminant.
B) Qualité pharmaceutique : forme liquide injectable
Pour les solutions, les suspensions, les émulsions, les caractéristiques physico-chimiques pouvant impacter la BD
sont comparées (composition excipiendaire, viscosité, distribution granulométrique, propriétés de surface ect…).
C) Qualité pharmaceutique : forme cutanées semi-solides
Pour les préparations semi-solides pour application cutanée, la composition excipiendaire de la référence et du
générique sont comparées ainsi que les caractéristiques physico-chimiques, pharmaceutiques et rhéologiques.
Toute différence doit être argumentée en termes d’impact sur la sécurité et l’efficacité. Des études comparatives
ex-vivo de diffusion du PA sont également exigées.
VII) Développement pharmaceutique
De même que pour un nouveau produit, développement d’un générique doit être optimisé de manière à réduire les
impuretés à taux les plus bas possibles (surtout ceux liés à la SA).
Démontrer que les génériques présentent un profil d’impuretés qualitativement et quantitativement similaire à
celui de la référence.
Si une nouvelle impureté est présente dans le générique ou si le % d’une impureté est significativement supérieur à
celui observé dans la référence, à justifier profil d’impuretés du générique à qualifier sur un plan toxicologique.
VIII) Dossier pharmaceutique : Eléments essentiels pour le générique
Données pour justifier de la qualité du médicament :
- Origine et spécification de la SA - Origine et spécification des excipients - Méthode de fabrication du PF - Méthode de contrôle du PF - Reproductibilité de la qualité dans un lot et entre lots : Validation des méthodes de fabrication et de
contrôle. - Maintient de la qualité : Etudes de la stabilité de la SA et du PF.
Développement pharmaceutique du générique doit justifier la similarité du générique à la référence in vitro.
D) Stabilité du PF
Péremption, conditions de conservation, stabilité en conditions d’utilisation à faire avec le générique.
UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 6
Peuvent être différentes de celles de la référence.
IX) Dossier biopharmaceutique / de bioéquivalence
BD/Bioéquivalence :
La BD décrit comment une SA devient disponible dans l’organisme pour produire son action biologique. Elle est
caractérisée par la quantité de SA disponible (qui atteint la circulation sanguine) et la vitesse de ce processus.
La bioéquivalence entre deux médicaments (même dose, même voie d’administration) = bioéquivalence de la BD.
A) Bioéquivalence :
Comparaison BD de deux formulations basée sur 2 paramètres pharmacocinétiques :
- Concentration maximale en SA (notée Cmax) dans le plasma après administration orale. - Aire sous la courbe des concentrations plasmatiques en SA au cours du temps (notée AUC).
Pour une même dose, si le profil pharmacocinétique identique du test T et de la référence R, in vivo chez l’homme
volontaire sain (au minimum chez 12 volontaires sains) :
Les formulations sont bio-équivalentes équivalentes sur le plan pharmacodynamique équivalence sur le
plan clinique.
B) Essai de bioéquivalence :
- Schéma expérimental classique : essai croisé (cross-over = 2 périodes séparées par une phase de « wash-out »), ou
le médicament est administré en dose unique à jeun, chez le volontaire sain, qui est son propre témoin.
- Groupe de sujets sains le plus homogène possible (âge, sexe, alcool…)
- Conclusion de l’étude de PK étendue à toute la population.
- Conditions d’administration standardisées : à jeun le + souvent pour éviter une interaction entre la prise de
nourriture et la formulation.
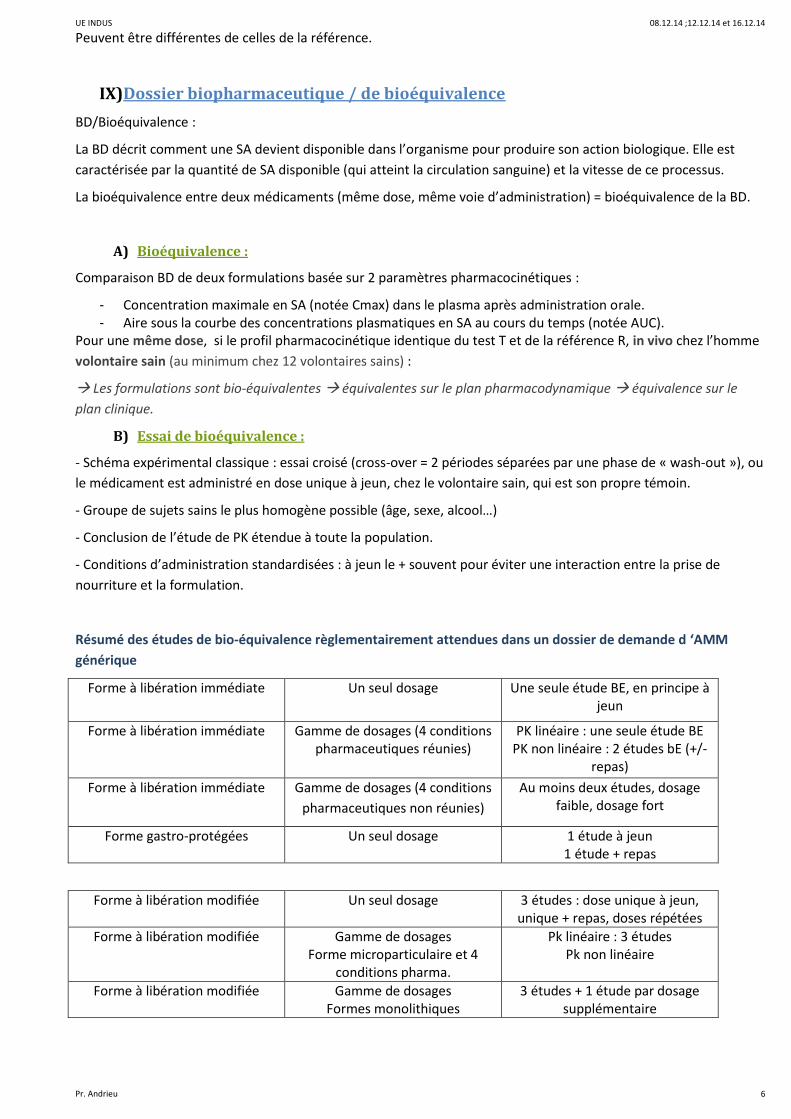
Résumé des études de bio-équivalence règlementairement attendues dans un dossier de demande d ‘AMM
générique
Forme à libération immédiate Un seul dosage Une seule étude BE, en principe à jeun
Forme à libération immédiate Gamme de dosages (4 conditions pharmaceutiques réunies)
PK linéaire : une seule étude BE PK non linéaire : 2 études bE (+/-
repas)
Forme à libération immédiate Gamme de dosages (4 conditions
pharmaceutiques non réunies)
Au moins deux études, dosage faible, dosage fort
Forme gastro-protégées Un seul dosage 1 étude à jeun 1 étude + repas
Forme à libération modifiée Un seul dosage 3 études : dose unique à jeun, unique + repas, doses répétées
Forme à libération modifiée Gamme de dosages Forme microparticulaire et 4
conditions pharma.
Pk linéaire : 3 études Pk non linéaire
Forme à libération modifiée Gamme de dosages Formes monolithiques
3 études + 1 étude par dosage supplémentaire

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 7
C) Doses en SA dans la biobatch = demande de biowaiver :
Pour plusieurs doses en SA dans le PF, linéarité à démontrer à partir de données bibliographiques.
Etudes de bio-équivalence avec le dose la plus sensible = PF avec dose la plus élevé.
Exonération (biowainer) pour les 3 autres doses si :
- Différents dosages fabriqués sur le même site de fabrication et selon le même procédé de fabrication. - Même composition qualitative des différentes formules. - Composition des différents dosages quantitativement proportionnelle (ratio entre quantité de chaque
excipient et de la SA est le même pour tous les dosages = formules homothétiques).
- Formules pseudohomothétiques : Quantité de SA < 5% du poids total du noyau du comprimé ou du poids du contenu de la gélule.
Quantités des excipients du noyau ou du contenu de la gélule les mêmes pour les différents dosages, seule quantité
de SA change.
Seule la quantité de l’excipient de remplissage est modifiée pour tenir compte de la quantité de SA).
- Les résultats de dissolution de tous les dosages (génériques et référence).
Si PK de la SA non linéaire, étude de bioéq avec la dose la plus sensible = PF avec dose la plus élevée.
Si AUC n’augmente pas proportionnellement à la dose de SA, une étude PK à dose la plus faible et une étude PK à dose la plus élevée
Bracketing possible si études nécessaires à différentes doses et à jeun/non à jeun.
D) Lots de médicaments pour essai de bioéquivalence
Médicament Référence acheté en FR (date de fabrication, date de péremption).
Médicament Générique : lot représentatif de la fabrication d’un lot industriel = « biobtach » - Origine de la SA précise = SA décrite dans le dossier AMM - Lots de 100 000 unités ou 10% de la taille industrielle : le plus grand des 2. - Lot de taille plus faible possible, à justifier - Lots à caractériser in vitro : dissolution
E) Essai de bioéquivalence
Prélèvement sanguins suffisamment nombreux pour mesurer la vitesse d’absorption (Cmax, Tmax). Il faut : - Prévoir au moins six prélèvements autour du Tmax - Prélever longtemps pour mesure la faction absorbée (AUC) = faire un suivi pendant au moins 4 à 5
demi-vies ; partie extrapolée d l’AUC ne devant pas excéder 20%
Echantillons prélevés sont analysés suivant des méthodes analytiques validées
Entité à doser molécule mère, que celle-ci soit le support de l’activité thérapeutique ou non.
F) Dosage analytique
Validation complète de la méthode analytique - Intérêt de la LC/MS/MS - Spécificité, précision, justesse, limite de quantification
Validation en deux phases - Pre-study phrase - Study phase
Cas des substances chirales : analyse des énantiomères
G) Analyse des résultats de PK :
ANOVA prenant en compte les principales sources de variabilité (traitement, période, séquence)
Transformation logarithmique des données rues
Statistique descriptive (moyenne, médiane, écart-type…)
Tests non paramétriques (Wilcoxon) pour les tmax

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 8
Pour démontrer la bioéquivalence, analyse de la variance (ANOA) sur les paramètres pharmacocinétiques (AUC et Cmax) obtenus avec les deux médicaments comparés.
Pour que deux formulation soient bioéquivalentes, il faut que les intervalles de confiance) 90% des paramètres pharmacocinétiques (AUC et Cmax) moyens (en échelle logarithmique) soient inclus dans l’intervalle [80,00% - 125,00%]
Si médicament à marge thérapeutique étroite (si écart entre concentrations efficaces et toxiques faible), l’intervalle d’acceptation de la bioéquivalence est resserré [90,00% - 111,11%]
Bilan : forme pharmaceutique - étude de bioéquivalence :
Même dose thérapeutique
Bioéquivalence Etude d’efficacité et de tolérance
Substitution
Génériques oui non oui
Syst. transdermiques oui non oui
Prép. pour inhalation Non représentatif de l’efficacité
oui non
Substances d’origine végétale
Non réalisable Traditionnellement utilisé non
Formes topiques Non réalisable Perméabilité oui
Pour une même fraction thérapeutique (SA), pour une même voie d’administration.
1) Médicaments exonérés d’étude de bioéquivalence
Médicaments administrés par voie intravasculaire : - Pas de phase d’absorption : toute la quantité de médicament administré est biodisponible puisque
toute la dose est directement présente dans la circulation sanguine - Dossier pharmaceutique apporte la preuve de la similarité parfaite entre le générique et sa référence
2) Médicaments exonérés d’études de bioéquivalence = « biowaiver)
Génériques pouvant être exonérés d‘étude de bioéquivalence pour les SA : - Acétylscystéine - Ambroxol - Aspirine - Carbocystéine - Dextrométorphane - Ibuprofène
3) Dispense d’étude de bioéquivalence basée sur la
classification BCS :
Uniquement pour SA très solubles avec une absorption digestive chez l’homme connue et n’ayant pas un index thérapeutique étroit
Applicable aux formes à libération immédiate, aux produits solides pour administration orale
N’est pas applicable aux formulations à libération sublinguale, buccale, et modifiée
Formulations orodispersibles possibles que si l’absorption buccale est exclue.
Demande de biowaver :
Classification BSC biopharmaceutique pour une SA
- Kétoprofène - Naproxène - Paracétamol - Trimébutine - Trimétazide

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 9
Cette classification caractérise une substance active permet d’exonérer des études de pour bioéquivalence les médicaments génériques
Conséquences pour les variations des PF Dispense de bioéquivalence basée sur la classification BCS applicable si :
BCS classe 1 ET
Dissolution in trio très rapide (< 85% en 15min) ou aussi rapide (85% en 30min) que la référence ET
Les excipients qui pourraient affecter la biodisponibilité sont qualitativement et quantitativement les mêmes. En règle générale, l’utilisation des mêmes excipients en quantité similaire est recommandée
Mais aussi si :
BCS Classe III ET
Dissolution in vitro très rapide (< 85% en 15min) pour l’essai et la référence ET
Les excipients qui pourraient affecter la biodisponibilité sont qualitativement et quantitativement les mêmes.
H) Obligations réglementaires
Mêmes notes explicatives, recommandations en EU et USA
Quelque soit le type de médicaments (princeps, et/ou génériques), les fabricants et le exploitants localisés en FR doivent être autorisés par l’ANSM en tant qu’établissement pharmaceutique. La réglementation leur impose de pratiquer les mêmes conditions de fabrication et de contrôles.
Inspection des sites par ANSM ou Autre état membre de l’UE.
La pharmacovigilance a pour objet la surveillance, l’évaluation la prévention et la gestion du risques d’effet indésirable résultant de l’utilisation des médicaments avec AMM ; Référence ou Générique
Toute entreprise exploitant un médicament met en œuvre un système de pharmacovigilance (1 personne qualifiée par état membre) pour procéder au recueil et à l’évaluation scientifique des effets indésirables, dans un but de prévention et de réduction des risques, puis prendre des mesures appropriées
Obligation de déclarations des effets indésirables pour Référence et Test sur base européenne Eudravigilance
Dépôt de PSUR : rapport périodique actualisé de PV pour un générique si : - La soumission de PSUR constitue une condition de l’AMM - Sur demande des autorités lorsque le médicament soulève des préoccupations en matière de PV ou
lorsqu’il existe plus de rapport périodique actualisé de sécurité pour une SA après octroi de l’AMM
Mise en œuvre d’un système de gestion des risques pour chaque médicament dont l’AMM a été délivrée après le 21 juillet 2012
Surveillance des résultats des mesures de réduction des risques décrites dans le plan de gestion des risques
Mise à jour du système de gestion des risques et des données de pharmacovigilance afin de repérer des risques nouveaux, des changements existants -> ≠ rapport B/R
Notifications d’effets indésirables dans un EM de l’UE ou un état de l’espace économique européen ou un pays tiers dans Eudravigilance
X) Génériques et ANSM
Programme d’inspections des sites de développement et de fabrication (R ou générique) : - Contrôle de la qualité des médicaments, génériques ou non = respect des BPF, conformité des dossiers
de lots avec dossier d’AMM correspondant
Tous les sites fabricants français inspectés par les inspecteurs de l’ANSM au moins tous les 3 ans : - Inspection système : respect des BPF par les fabricants - Inspection produit : production d’un ou plusieurs médicaments génériques ou non
A) Inspections
Mondialisation au niveau de la production, de la distribution des substances actives, dans la fabrication et l’exploitation de médicaments, plus particulièrement en Inde, aux USA et en Chine
Constat de la mondialisation de la fabrication des médicaments en général.

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 10
Depuis 2008, programme d’inspection coordonné des fabricants de SA lancée en 2008 et associant EMA , TGA (Australie, US-FDA et EDQM (Direction Européenne de la qualité et du médicament).
Fabricants de PF qui utilisent des SA, responsables de la qualité des SA utilisées avec respect des BPF par des audits doivent assurer par des audits.
Juillet 2013 directive n°2011-62 : renforce les exigences vis-à-vis de l’importation de SA.
« Confirmation écrite » des autorités compétentes du pays d’exportation confirmant notamment le respect des BPF, sauf si SA proviennent d’un pays dont le système est reconnu par l’UE.
1) Inspection des essais de bioéquivalence
Dès 1994, programme spécifique d’inspections des essais de bioéquivalence pour les médicaments destinés au marchés français et européen.
Infirme proportion des essais de bioéquivalence soumis à l’ANSM réalisée en France, minorités d’essais réalisée en Europe : Espagne, Allemagne et Europe centrale
Plus de la moitié des essais pour les génériques : en Inde 05/12/14 : l’ANSM lance une procédure de suspension, à compter du 18 décembre, de 25 médicaments génériques commercialisés en France
2) Inspection des essais de PV
Depuis 2007, inspection des acticités de routine de PV et celles mises en place dans le cadre de PGR
10 à 15 inspections/an spécifiques et renforcées du système de PV des AMM
Entre 2007 et 2011, 42 inspections initiées à la suite d’un signal alertant sur des défaillances avérées ou potentielles d’un système de PV réalisées, dont 14 dans des établissements exploitant ayant une majorité de spécialités génériques
3) Programme de contrôle des PF
Contrôle de 200 spécialités du répertoire par an avec comparaison avec références
Choix des produits présentant des problèmes de qualité, ayant une influence sur la toxicité
Ex : adaptation posologique délicate (lévothyroxine), vérification du caractère gastro-résistant de molécules inefficaces à pH acide (spécialité à base de lansoprazole, oméprazole et pantoprazole) ou vérification de la stabilité en conditions d’utilisation de spécialités anticancéreuses (oxaliplatine).
4) Bilan des contrôles des PF
Analyses physico-chimiques ± microbiologiques (dosage SA et impuretés)
866 médicaments génériques inscrits au répertoire et 199 médicaments princeps qui ont été analysés ces 5dernières années
32 non conformités en 5 années de surveillance de marché correspondent à : - Détection de substances apparentées (ou impuretés) - Essais de sécabilité - Notices/ «étiquetages - Dosage en PA - Caractères organoleptiques - Essai de dissolution - Essai de désagrégation
En 2011, étude européenne sur les génériques de clopidogrel (PLAVIX) et ses 3 sels : 12 produits fins et 10matières premières en procédure européenne contrôlés par laboratoires nationaux participants (France, Allemagne et Luxembourg)
Etude FR avec 18 génériques ayant une AMM nationale
Bonne qualité de l’ensemble des spécialités génériques de ce groupe
5) Bilan des contrôles SA :
Teneurs en impuretés légèrement supérieures aux limites enregistrées
Teneures en eau supérieures aux spécifications
Echantillons contrôlés fournis par le titulaire de l’AMM

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 11
A partir de 2010, programme d’inspection, avec prélèvements effectués à l’initiative de l’ANSM en fonction des facteurs de risques potentiels
B) PV :
Circuit de notification des EI -> évaluation faite par l’ANSM des EI notifiés : même procédure pour médicament princeps ou générique
Evaluation des PSUR et des systèmes de gestion des risques des médicaments envoyés par les titulaires d’AMM à l’ANSM
1) PV : erreurs médicamenteuses avec les génériques
« guichets Erreurs médicamenteuses » de l’ANSM
Exemples de problématiques identifiées : similitude des conditionnements liée à l’utilisation d’une charte graphique des laboratoires génériqueurs (code couleur proche)
Confusions, abandons de traitement, frein à la prescription et non adhésion par les patients du fait d’un manque d’harmonisation de présentation pour une même molécule
Erreur de substituions : il existe le Répertoire des Médicaments Génériques de l’ANSM dont la consultation systématique permet aux professionnels de santé de vérifier que la substitution est adaptée
Cas particulier : antibiotique par voie injectable
Vancomycine et teicoplanine : processus de fabrication par voie de fermentation à partir de microorganismes -> compositions moins bien définies que les produits obtenues par synthèse chimique
Publication en 2010, d’une étude scientifique colombienne : différences d’activité antibactérienne importantes entre les génériques de la vancomycine et la spécialité de référence, sous leur forme injectable. En fait : spécialités non commercialisées en FR et modèle animal d’infection expérimentale de cuisse de souris critiquable
Efficacité de génériques d’antibiotiques obtenus par fermentation, en particulier pour la teicoplanine ?
Dans bases nationales et européenne de PV, pas de problème d’inefficacité de vancomycine et teicoplanine
Conformité à la monographie de la PE pour la teicoplanine : insuffisant pour conclure similarité entre Référence et Générique
Pas de teicoplanine Générique en FR
Cas particulier : forme buvables d’antibiotiques en pédiatrie
Les génériques d’antibiotiques en VO : difficultés d’acceptabilité. Défaut d’observance : absence d’efficacité thérapeutique + risque de résistances bactériennes
Ligne directive européenne sur les formulations pédiatriques, pas spécifique aux antibiotiques
Cas particulier : antiépileptiques
Alerte lancée par la ligue française contre l’épilepsie dans les années 2000
Etudes du CHU Rennes : sur 300 neurologues libéraux, 70% estiment que la substitution d’un antiépileptique s’était accompagnée : - D’appels téléphoniques - De signes d’inquiétudes de leurs patients, et - Problèmes lors de la substitution (récidive de crise, EI)
8 états européens ont pris des mesures : - Rétrécissement de la marge de bioéquivalence - Encadrement de la substitution - Interdiction de la substitution
Pas de démonstration scientifique d’une relation entre substitution et déséquilibre de la pathologie épileptique chez les patients
Mais : particularités de la maladie : nombreux facteurs favorisants ou provoquent une crise (situation anxiogènes)
Lettre aux professionnels de santé de l’ANSM : nécessité d’un dialogue entre médecin et patient pour substituer sans créer d’anxiété, sinon éviter la substitution
.

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 12
Cas particulier : lévothyroxine sodique
Lévothyroxine sodique : hormone thyroïdienne de synthèse à marge thérapeutique étroite
Nombre significatif de notification de perturbation de l’équilibre thyroïdien après substitution du princeps par un générique lors de l’arrivée sur le marché de spécialités génériques de lévothyroxine sodique
Recommandations aux professionnels de santé
Variations de l’exposition lors de changement de spécialités à base de lévothyroxine chez certains patients à risques
Recommandations de l’ANSM aux professionnels de santé : surveillance nécessaire en cas de changement entre deux spécialités à base de lévothyroxine - Spécialité générique vers spécialité de référence - Spécialité générique vers spécialité générique - Spécialité générique vers une autre spécialité générique
Chez patients à risque, évaluation clinique, voire biologique par un contrôle de la TSH après la substitution
Cas particulier : génériques de dispositifs transdermiques de fentanyl
Nécessiter de surveiller attentivement certains patients fébriles, patients âgés et enfants afin de prévenir tout risque de surdosage ou de sous dosage et les EI. Surveillance nécessaire en cas substitution :
- Spécialité référence vers spécialité de générique - Spécialité générique vers spécialité générique - Spécialité générique vers une autre spécialité générique
Cas particulier : génériques d’immunosuppresseurs :
Mesures pour mieux informer patients et professionnels de santé
ANSM travaille sur recherches pharmaco-épidémiologiques pour optimiser la traçabilité des prescriptions et de détecter des signaux = différence d’efficacité clinique.
Cas particulier : collyres
Référence : collyre avec conservateurs
Aujourd’hui, guideline pour limiter l’utilisation des consesrvateurs dans les médicaments
Doit-on mettre des conservateurs dans le générique ?
Le(s) même(s) conservateurs(s) ? en même quantité
Atteinte de l’intégrité des membranes cellulaires possibles avec conservateurs = tensio-actifs
C) Contrôle de la publicité des génériques :
Contrôle de la publicité des médicaments identiques
Doivent mentionner l’appartenance à la catégorie des spécialités générique et doivent préciser le nom de la réference et la mention de l’inscription au répertoire des génériques afin de guider le pharmacien dans la substitution
Droit de substitution ne doit pas être remis en cause par une publicité. Pas de différence entre plusieurs génériques ou entre le générique et la spécialité de référence qui iraient à l’encontre de la bioéquivalence et de la similarité reconnues entre les produits.
D) Effets négatifs des génériques :
Prescription en DCI peut être refusée, + NS
Report des prescriptions sur des molécules récentes non substituables
Moins d’information médicale
Peu de VM pour les Références génériques et les génériques
Ruptures d’approvisionnement
Qualité des SA -> Toxicité des impuretés des SA (MTH, carboplatine (allergies), gentamycine (décès)

UE INDUS 08.12.14 ;12.12.14 et 16.12.14
Pr. Andrieu 13
E) Conclusion :
Le médicament générique : même qualité pharmaceutique, mêmes propriétés toxicologiques et efficacité thérapeutique que la référence
Problème potentiel des délocalisations : SA et PF
Nombre de médicaments génériques élevé
Attention à la substitution pour des traitements chronique
Attention à la substitution en fonction de l’âge du patient
Le médicament générique se différencie de moins en moins des médicaments classiques
Ex : un médicament Référence qui a changé sa composition et son procédé de fabrication pour être fabriqué par un laboratoire génériqueur
Les « Big Pharma » ont créé des filiales pour développer leurs médicaments génériques











![[6] Le circuit de distribution des m dicaments … · Publique et de la Propriété Intellectuelle. a- Les différents systèmes de PDA à la pharmacie d’officine Cinq sociétés](https://static.fdocuments.fr/doc/165x107/5b9b5c5209d3f22d2a8d0de7/6-le-circuit-de-distribution-des-m-dicaments-publique-et-de-la-propriete.jpg)