La technique du recouplage en RMN solide : … · membranaires – mais il en va de même des...
44
Magistère Inter universitaire de Chimie Université Paris VII – Denis Diderot année 1999-2000 rapport présenté par Ludovic BERTHELOT Ludovic BERTHELOT La technique du recouplage en RMN solide : application aux lipides et aux peptides membranaires - RMN - U P R 90 5 2 - R M N - U P R 9 0 5 2 Travail effectué sous la direction du Pr. Philippe DEVAUX UPR 9052 Laboratoire de physico-chimie moléculaire des membranes biologiques IBPC – 13, rue Pierre et Marie Curie 75005 Paris
Transcript of La technique du recouplage en RMN solide : … · membranaires – mais il en va de même des...

Magistère Inter universitaire de Chimie Université Paris VII – Denis Diderot
année 1999-2000
rapport présenté par
Ludovic BERTHELOTLudovic BERTHELOT
La technique du recouplage en RMN solide :application aux lipides et aux peptides
membranaires
- RM
N -
UPR
9052
- RM
N -UP
R 90
52
Travail effectué sous la direction du Pr. Philippe DEVAUXUPR 9052
Laboratoire de physico-chimie moléculaire des membranes biologiquesIBPC – 13, rue Pierre et Marie Curie
75005 Paris

Sommaire
Introduction
Chapitre 1 : Présentation
Chapitre 2 : Recouplage de l’anisotropie du déplacement chimique
Étude de phases lipidiques
Chapitre 3 : Recouplage de l’interaction dipolaire
Mesures de distances dans les peptides membranaires
Conclusions et perspectives
Références
1
2
7
25
38
39


1
Introduction
armi les différentes molécules ou macromolécules du vivant, protéines solubles et acides nucléiques sont très
étudiés, et, à ce titre, de mieux en mieux décrits structurellement. Au contraire, les lipides et les protéines
membranaires – mais il en va de même des sucres, par exemple – sont encore sources de nombreuses interrogations. Si
leur rôle cellulaire ne fait aucun doute, si la description d’une membrane cellulaire en terme de bicouche lipidique est
acceptée, on ne s’explique toujours pas la grande diversité lipidique, pas plus que des processus comme la fusion
membranaire. Par ailleurs, les interactions entre lipides et protéines membranaires sont encore largement méconnues : il
est simplificateur de dire que les lipides auraient un rôle purement structural, alors que les protéines n’auraient qu’un
rôle fonctionnel au sein de la cellule. Ces différents aspects de présentation seront abordés dans notre premier chapitre.
Une classe importante de lipides est celle des phospholipides, qui possèdent tous un groupe phosphate. La résonance
magnétique nucléaire (RMN) du phosphore-31 (31P) est une méthode de choix dans la reconnaissance des phases de
phospholipides, et plus généralement dans l’étude des systèmes membranaires. Elle est complémentaire de diverses
autres techniques, comme la diffraction des rayons-X ou la résonance paramagnétique électronique (RPE). En solution
organique, on peut ainsi différencier chaque phospholipide selon le déplacement chimique du 31P, noyau de spin ½,
d’abondance naturelle 100%, ainsi que de sensibilité assez élevée.
Comme on le détaillera dans le deuxième chapitre, le spectre statique du phosphore-31 d’un phospholipide a une
allure caractéristique de sa phase. On parle de spectre large bande, car le pic d’absorption associé s’étend sur plusieurs
dizaines de ppm. L’asymétrie de ce spectre est alors le reflet de l’interaction dominante en RMN du 31P, l’anisotropie du
déplacement chimique. Mais dès que l’on a affaire à un mélange de lipides, la reconnaissance du profil du spectre est
compliquée par la superposition de chacun des profils des lipides. C’est pour séparer ces profils selon les lipides
présents dans le mélange –un spectre pour un lipide et non plus une superposition de spectres pour le mélange de
lipides– que la notion de recouplage est utile. Comme on le verra, les spectres obtenus par recouplage de l’anisotropie
du déplacement chimique ont deux dimensions, une selon laquelle on sépare les lipides présents selon leur tête polaire,
une autre selon laquelle on peut lire le profil large bande typique de la phase lipidique.
Par ailleurs, la RMN se trouve être, avec la cristallographie puis diffraction des rayons-X, une des méthodes de
choix dans l’étude structurale des protéines. Elle a même cette supériorité sur la cristallographie de pouvoir s’intéresser
à des paramètres dynamiques. Malheureusement, s’agissant de protéines membranaires, les deux techniques rencontrent
des difficultés importantes comparées au cas des protéines solubles. Le caractère hydrophobe du segment
transmembranaire oblige, afin d’éviter l’agrégation, à extraire les protéines de la membrane au moyen de détergents, ce
qui complique la cristallisation. Parallèlement, la viscosité des solutions de protéines membranaires a pour effet de
compliquer les spectres de RMN des liquides, en diminuant leur résolution. Tout ceci permet d’expliquer que, sur un
total de plus de 11.000 structures de protéines déposées à la PDB (http://www.rcsb.org/) il n’y ait que 186
structures de protéines membranaires, dont 138 déterminées par diffraction de rayons X et 35 par RMN.
Néanmoins, certaines structures de peptides membranaires sont connues à des basses résolutions ou calculées par
dynamique moléculaire par exemple. Il suffirait parfois de connaître quelques distances avec précision pour pouvoir
raffiner une structure ou mettre en évidence une altération. Le troisième chapitre s’attachera à montrer que des
techniques de recouplage similaires peuvent permettre de mesurer des distances entre résidus dans les peptides
membranaires.
P

2
Chapitre 1
Présentation
es deux années de recherche se sont écoulées à l’IBPC dans l’équipe de Physico-Chimie Moléculaire des
Membranes Biologiques. S’agissant d’un travail de biophysique, il convient dans ce premier chapitre d’en
présenter les deux aspects : tout d’abord la spécificité des objets membranaires étudiés, lipides et protéines, puis la
méthode d’étude, le recouplage d’interactions en spectroscopie de RMN. Les deux chapitres suivants montreront
successivement les résultats acquis en DEA et en première année de thèse, sur chacun de ces objets.
I. La membrane biologique et ses composants
A. Qu’est-ce qu’un lipide ?
Les lipides sont de petites molécules insolubles et amphiphiles. A ce titre, ils possèdent une tête hydrophile et une
queue hydrophobe, qui leur permettent de s’auto-assembler en structures en couches voire en bicouches. Cette structure
en bicouche est la base des membranes cellulaires. Ainsi, une caractéristique commune à tous les lipides est leur rôle
fonctionnel au sein des cellules, qui est d’abord un rôle de compartimentation et d'échange entre l'intérieur et l'extérieur.
Les détails ne seront pas abordés ici. On pourra par exemple se référer au chapitre 1 du Phospholipids Handbook,
par Gregor Cevc (1993), notamment pour la nomenclature des différents lipides.
B. Les phospholipides
Une famille importante de lipides est celle des phospholipides. Par ce terme, on désigne ici d’une manière abusive
ce qu’il serait plus convenable d’appeler les diacylphosphoglycérides, qui ne sont qu’un des membres de la famille des
phospholipides, mais l’un des principaux. Ces lipides sont les principaux constituants naturels des membranes
cellulaires. Ils constituent plus de 80% des lipides dans les membranes mitochondriale ou plasmique, et plus de 90%
dans les membranes nucléairesa.
Chimiquement, dans un phospholipide, une molécule de glycérol est estérifiée en position 1 et 2 par deux chaînes
d’acides gras saturées ou non, de 10 à 20 carbones de long. La position 3 est substituée par un groupement phosphate,
auquel est reliée une tête polaire noté X sur la figure 1.1. La configuration naturelle de l’atome de carbone 2 est (R), ce
que désigne généralement le symbole sn (pour stereospecific numbering).
OO
OR
O
R' O
P
O
O
O X_OH H
CH2OH
CH2OH1
3
2
(a) (b)
figure 1.1 : (a) molécule de sn-glycérol (le carbone prochiral 2 est en configuration (R))(b) : structure de base d’un diacylglycérophospholipide. X est la tête polaire et R et R’ sont les chaînes aliphatiques.
a Les lipides restant sont principalement des cardiolipides dans la membrane mitochondriale, et des sphingolipides dans
la membrane plasmique.
C

3
La nomenclature des phospholipides fait intervenir le tête polaire X, aussi bien que les chaînes d’acides gras R et R’.
La tête choline (-CH2CH2N+(CH3)3) est propre à la phophatidylcholine (PC). Les principales autres têtes polaires sont la
tête éthanolamine (-CH2CH2NH3+) pour la phospatidyléthanolamine (PE), et la tête sérine (-CH2CH(COO-)NH3
+) pour
la phosphatidylsérine (PS).
C. Le polymorphisme lipidique
Les molécules amphiphiles que sont les lipides ont une grande propension à s’organiser selon des assemblages
réguliers et ordonnés à deux ou trois dimensions. Au delà d’une concentration très faible en solution aqueuse, les lipides
vont s’associer pour mettre leurs têtes polaires en contact avec l’eau, et en isoler leurs chaînes hydrophobes. Une des
structures possibles est la bicouche, qui peut se refermer sur elle même pour former une vésicule. Mais d’autres formes
existent, comme la micelle (pour des détergents), la micelle inverse ou des formes intermédiaires que sont les phases
hexagonales ou cubiques. On passe d’une phase à l’autre par des variations des conditions du milieu : température,
pourcentage d’hydratation, pH, force ionique, présence d’agents extérieurs comme des protéines ou du cholestérol.
La figure 1.2 illustre les deux principales phases que l’on rencontrera par la suite : la phase hexagonale inverse, dite
HII, et la phase lamellaire (bicouche).
figure 1.2 : représentation schématique des phases hexagonale inverse (à gauche) et lamellaire (à droite) (d’après Gregor Cevc,Phospholipids Handbook , chapitre 2)
La phase HII fait apparaître un agencement hexagonal de “cylindres micellaires inverses”. Il existe aussi une phase
hexagonale (non inverse, dite HI), où les cylindres micellaires ne sont pas inverses (i.e. les lipides tournent alors leurs
têtes hydrophiles vers l'extérieur du cylindre).
La simple notion de bicouche est plus complexe que ne peut le faire apparaître la figure 1.2. Au sein de la phase
lamellaire, on fait encore la distinction entre phase cristal-liquide (ou fluide, dite Lá), et phase gel (dite Lâ) à plus basse
température. La différence entre les deux est surtout due à la flexibilité des chaînes hydrophobes, beaucoup plus grande
au sein de la phase fluide, comme l’illustre la figure 1.3, issue de simulations par dynamique moléculaire (Heller et al.,
1993). La diffusion latérale est également bien plus importante dans la phase fluide.
figure 1.3 : simulation de la dynamique de 5 molécules de POPC dans des membranes modèles(a) : en phase Lâ ; (b) : en phase Lá ; (d’après Heller et al., 1993)
D. La membrane biologique : exemple des érythrocytes
La membrane externe des globules rouges (érythrocytes) est l'exemple le mieux décrit de membrane cellulaire.
Composée en masse pour moitié de protéines membranaires, pour moitié de lipides, cette membrane peut être
facilement étudiée parce qu'on peut l'isoler du reste de la cellule de globule rouge. Par un procédé de lyse osmotique, le

4
globule éclate et une simple centrifugation permet d'éliminer l'hémoglobine qu'il renferme. On obtient alors des
stromas, ou fantômes percés (en anglais ghosts), constitués uniquement des membranes lipidiques et de leurs protéines
intrinsèques, ainsi que du cytosquelette. Les détails concernant la biophysique des membranes de globule rouge peuvent
par exemple être trouvés dans l'ouvrage de E. Shechter (1993). Pour mémoire on reportera simplement dans le tableau
1.4 la composition lipidique moyenne de ces membranes.
cholestérol 25phospholipides totaux 56
dont PC 23PE 20PS 11PI 2
sphingomyéline SM 18autres 1
tableau 1.4 : pourcentage de la quantité totale de lipides dans l'érythrocyte humain.
E. Les protéines membranaires
On estime que plus du quart des protéines du génome sont des protéines membranaires, qu’elles soient intégrales
(c’est à dire traversant la membrane) ou périphériques. Si l’on sait prédire d’après son profil d’hydrophobie qu’une
protéine est vraisemblablement membranaire, il est plus difficile de connaître sa structure, tant en diffraction des rayons
X qu’en RMN (Popot & Engelman, 2000).
Les deux principales structures de segments transmembranaires sont l’hélice-α et le tonneau-β. La figure 1.5 fournit
deux exemples parmi d’autres, de chacune de ces structures.
figure 1.5 : deux exemples de structures transmembranaires : à gauche, les deux hélices-α de la glycophorine A (Mac Kenzie etal., 1997), à droite, le tonneau-β (arrangement de feuillets-β pour former un pore) dans la porine
Les hélices transmembranaires présentent souvent des déformations structurales par rapport au cas idéal de l’hélice-
α. On reviendra au chapitre 3 sur l’une de ces déformations, la hernie-π, qui implique la torsion d’un acide aminé et a
pour conséquence de modifier les liaisons hydrogène entre résidus : alors que dans une hélice-α, la liaison se fait entre
résidus i et i+4, dans une hernie-π elle se fait entre résidus i et i+3 ou i+5. Des tels changements conformationnels sont
suspectés d’intervenir dans les associations entre hélices, ou d’avoir un rôle fonctionnel (Popot & Engelman, 2000). Or
une résolution inférieure à 2 Å est nécessaire pour conclure à l’existence de hernies-π. Lorsque la structure est connue à
une moins bonne résolution, une simple mesure précise de distance entre quelques résidus permettrait de trancher.

5
II. L’étude de composés membranaires par RMN
A. Rappels de RMN
On s’appuie sur les bases de RMN acquises en DEA. On suppose acquises quelques notions de mécanique
quantique, notamment la notion d’Hamiltonien H, dont les fonctions propres sont les fonctions d’onde ψ du système
pour la valeur propre E (l’énergie), selon l’équation de Schrödinger Hψ=Εψ. Des précisions théoriques peuvent être
trouvées dans de nombreux ouvrages. Citons notamment celui de K. Schmidt-Rohr et H. W. Spiess (1994), qui continue
de nous guider.
La RMN est une technique spectroscopique qui utilise les propriétés physiques de la matière placée dans un champ
magnétique non nul. Une description correcte en est faite au niveau quantique, et montre que, pour des noyaux
atomiques possédant un moment magnétique (donc de spin non nul), le champ magnétique, que l’on notera par la suite
B0 b, lève la dégénérescence des niveaux énergétiques. Cet effet Zeeman se traduit par un Hamiltonien :
HZ = −γ B0 Iz , où Iz est l’opérateur de spin en projection sur l’axe z, par définition confondu avec l’axe du champ
Bo.c
Toutes les autres interactions que peut subir le noyau considéré se traduisent par un Hamiltonien qui est une
perturbation au premier ordre de l’Hamiltonien Zeeman. Ce sont ces interactions qui sont intéressantes, car elles
expliquent la forme des raies d’absorption, leur déplacement, leur dédoublement ou leur élargissement. Elles se
traduisent localement par des fluctuations du champ magnétique ressenti par le noyau.
Faisons brièvement un inventaire des Hamiltoniens que l’on doit considérer pour notre système. Les expériences
étant menées sur le 31P, le 13C ou le 15N, on ne s’intéresse qu’aux spins ½.
• déplacement chimique : sous l’influence du champ B0, le nuage électronique génère un champ local autour du
noyau. Dans un cas isotrope (RMN liquide), ce champ local est proportionnel à B0, avec une constante de
proportionnalité σ de l’ordre de 10-6 (c’est la constante d’écran). Ici il faut multiplier B0 non plus par un scalaire, mais
par un tenseur de rang 2, i.e. par une matrice 3x3, notée $σ , le tenseur d’anisotropie du déplacement chimique. Alors on
a HCS = γ I $σB0 d.
• interaction dipolaire : lorsque deux noyaux i et j sont couplés par une interaction dipolaire, leur Hamiltonien
d’interaction se note : HD = I D Ii j$ ; $D est le tenseur d’interaction dipolaire. Un point important est que la constante
de couplage dipolaire, D exprimée en Hz, vaut :
D =h
2πγ I γ S
r3
où γI et γS sont les rapports gyromagnétiques respectifs des spins couplés I et S, et r est la distance entre ces noyaux.
• interaction scalaire : de même lorsque deux noyaux i et j sont couplés scalairement, on a un tenseur
d’interaction scalaire $J , et un Hamiltonien : HS = I J Ii j$ .
• interaction quadrupolaire : elle n’apparaît que pour des noyaux de spins plus grands que 1, donc on la
mentionnera sans la développer par la suite.
b On note les vecteurs en gras.c On remarquera que les Hamiltoniens seront dans la suite donnés à un facteur h près, i.e ils sont associés à une
pulsation ω = E / h et non plus à une énergie. d CS pour Chemical Shift

6
B. La RMN des solides en rotation à l’angle magique
Lorsque l’on travaille avec des solides ou des quasi-solides (comme c’est le cas pour des membranes), l’échantillon
a un nombre de degrés de liberté très réduit par rapport à la RMN liquide. Cela a pour conséquence de supprimer un
processus de moyennage de certains termes d’interactions, et donc de compliquer le spectre en l’élargissant. Par
exemple, le couplage dipolaire hétéronucléaire peut atteindre quelques centaines de Hz entre 13C et 15N, et l’anisotropie
du déplacement chimique quelques milliers de Hz. Il est alors difficile de tirer des informations de ce spectre. La
technique de la rotation à l’angle magique a pour but d’éliminer artificiellement ces interactions dans des échantillons
solides. Elle consiste à faire tourner l’échantillon dans le spectromètre, à une fréquence de quelques milliers de Hz dans
notre cas, autour d’un axe incliné par rapport à la verticale du champ d’un angle θ tel que3 1 02cos θ − = . On peut
alors montrer que, pour des vitesses de rotation suffisantes, les interactions telles que l’anisotropie du déplacement
chimique et le couplage dipolaire hétéronucléaire sont moyennées à zéro, et n’ont donc plus d’influence sur la
complexité du spectre. Il est ainsi plus aisé d’analyser le spectre obtenu, qui est dit spectre à haute résolution. Ce
phénomène sera expliqué plus en détails au chapitre 2.
figure 1.6 : schéma de principe d'une sonde MAS; sur une sonde Bruker, le “drive” arrive en haut car c'est le bouchonqui possède des pales. L’échantillon est contenu par le rotor.
C. Le recouplage
Après avoir supprimé toutes les interactions inhomogènes, il peut cependant être intéressant d’en réintroduire une
sélectivement. En effet, un spectre à haute résolution ne contient plus les informations d’ordre structurel et dynamique.
En revanche, pris séparément, l’anisotropie du déplacement chimique ou le couplage dipolaire hétéronucléaire peuvent
permettre de tirer des renseignements de structure ou de dynamique sur la molécule considérée. Cette réintroduction
sélective dans le spectre de l’interaction qui nous intéresse, après les avoir toutes supprimées par la rotation à l’angle
magique, porte le nom de recouplage. Il existe de nombreux moyens de recoupler des interactions. Beaucoup s’appuient
cependant sur une synchronisation d’impulsions radiofréquence avec la rotation de l’échantillon. C’est le cas des
méthodes développées tant au chapitre 2 qu’au chapitre 3.

7
Chapitre 2
Recouplage de l’anisotropie du déplacement chimique
Etude de phases lipidiques
e chapitre correspond au travail commencé lors de l’année de DEA. Ce travail avait pour but de différencier les
phases de chacun des lipides dans un mélange complexe, en recouplant l’anisotropie du déplacement chimique du31P dans les phospholipides.
Ce travail a pour l’instant donné lieu à deux posters, en septembre 1999 à Chamonix (Berthelot et al., 1999) et à
Florence en août 2000 (Warschawski et al., 2000).
I. La RMN du 31P dans les lipides
A. Pertinence de l’étude par RMN
Pour étudier et comprendre la membrane biologique, les méthodes d’étude sont multiples, et permettent chacune de
sonder des propriétés différentes. Après de premières études de phases lipidiques par diffraction de rayons X (Luzzatti,
1962), d’autres techniques ont été appliquées aux membranes ou aux modèles lipidiques, qui avaient toutes pour but
d’étudier une gamme de mouvements propres : ainsi, des méthodes de spectroscopie infra-rouge ou Raman peuvent
sonder des mouvements très rapides (107 à 1011 Hz), alors que les méthodes de RPE sondent des mouvements plus lents
(103 à 109 Hz). Des informations structurales peuvent également être obtenues par cryo-microscopie électronique, ou
par microscopie à force atomique.
Le but n’est pas dans cette présentation de comparer toutes ces méthodes en les développant, mais de situer la RMN
parmi elles. En effet, elle a un rôle bien particulier, parce qu’il s’agit d’une méthode qui peut couvrir une vaste gamme
de mouvements. Sa nature même lui permet de s’intéresser à un grand nombre de noyaux différents, de spin non nul :1H, 2H, 13C, 15N ou 31P principalement. Leurs propriétés spectroscopiques sont telles que les fréquences de mouvements
accessibles vont de 10-1 à 1010 Hz.
Nous avons choisi ici de mener une étude par RMN du 31P. C’est, avec 1H, le seul de ces isotopes abondant à l’état
naturel. Son rapport gyromagnétique est 2,5 fois plus faible que celui du proton, ce qui fait de lui un noyau tout de
même assez sensible1. Mais surtout, les spectres du 31P sont dominés par une interaction, le déplacement chimique, qui,
du fait des propriétés électroniques propres au noyau de phosphore, est fortement anisotrope (i.e. n’a pas la même
valeur suivant trois directions privilégiées de l’espace). Cette anisotropie du déplacement chimique a des conséquences
importantes sur le profil du spectre 31P, qui est caractéristique de la phase où se trouve le phospholipide.
Enfin, une membrane biologique est un objet mi-fluide, mi-solide, en fait un solide mou voire un cristal-liquide. On
entre alors dans le domaine de la RMN solide, avec son formalisme propre et surtout sa grande variété d’expériences
possibles. La rotation à l’angle magique et les techniques de recouplage sont alors des méthodes de choix dans l’étude
de ces membranes.
1 13C, avec un ã 4 fois plus faible que 1H, est encore moins sensible ; de plus, son abondance naturelle n’est que de 1%.
15N et 2H ont des ã respectivement 10 fois et 6,5 fois plus faibles que 1H, et nécessitent tous deux un enrichissementdu lipide.
C

8
C’est principalement ces deux aspects, anisotropie du déplacement chimique et effet de la rotation, que l’on va
tenter d’illustrer dans les paragraphes suivants.
B. La RMN en bande large du 31P dans les phospholipides
1. Les interactions dominantes
Toutes les interactions précédentes ne sont plus à considérer lorsque l’on s’intéresse au 31P dans les phospholipides.
• couplages homonucléaires : il n’y a qu’un 31P par phospholipide, donc il n’y a pas de couplage scalaire
homonucléaire 31P-31P. Il pourrait y avoir un couplage dipolaire 31P-31P, mais la diffusion latérale rapide (105-109 Hz)
des lipides au sein de la membrane empêche ce couplage d’être efficace (Warschawski, 1995).
• couplages hétéronucléaires : les couplages hétéronucléaires significatifs ne se font qu’entre 31P et 1H, car les
autres noyaux présents (13C voire 15N) ont une abondance négligeable dans des lipides non marqués. Le couplage
scalaire proton-phosphore est de l’ordre d’une dizaine de Hz. On verra que cette valeur est trop faible pour être
appréciable sur un spectre.
Le seul couplage à prendre en compte est donc le couplage dipolaire hétéronucléaire 31P-1H. La constante de
couplage associée reportée dans la littérature (Dufourc, 1986) varie de 1 à 5 kHz selon la liberté de mouvement de la
tête polaire contenant le groupe phosphate. Cette constante est bien sûr indépendante du champ B0.
• anisotropie du déplacement chimique (en anglais CSA pour Chemical Shift Anisotropy) : c’est l’interaction
prédominante, qui explique les profils des spectres statiques. On verra dans le paragraphe suivant que cette anisotropie a
une valeur typique de moins de 200 ppm, soit, sur un spectromètre 400 MHz 2, un écart en fréquence d’en général une
dizaine de kHz, et 30 kHz au maximum. Ainsi le couplage dipolaire 31P-1H, même plus faible, n’est pas vraiment
négligeable y compris à haut champ. On peut annuler ses effets par un simple découplage du proton, consistant en une
irradiation simultanée en large bande (Cullis & de Kruyff, 1976). Dans la pratique, on constate toutefois qu’il n’y a pas
d’incidence flagrante sur la forme du spectre statique avec ou sans découplage, si ce n’est des épaulements moins
marqués, et par conséquent on négligera dans la suite le couplage dipolaire hétéronucléaire, pour se concentrer
principalement sur le CSA et ses effets.
2. CSA et profil de raie
Le développement suivant peut être passé pour en arriver au résultat final. Des précisions pourront être trouvées
dans l'ouvrage de Schmidt-Rohr et Spiess (1994), ainsi que dans la thèse de E. Dufourc (1986). Sur ce point, ces deux
ouvrages reprennent les résultats initialement exposés par Seelig (Seelig, 1978). Le but est ici d'expliquer la forme des
spectres statiques en 31P pour les principales phases lipidiques.
Dans un repère lié au groupement phosphate, et appelé le système d'axes principaux (abrégé PAS en anglais, pour
Principal Axis System) la partie symétrique du tenseur d'anisotropie du déplacement chimique peut être diagonalisée:
$σσ
σσ
PASxx
yy
zz
=
0 0
0 0
0 0
Le fait que les 3 termes diagonaux soient différents traduit l'anisotropie de cette interaction, qui dépend de l'orientation ;
typiquement on donne: σxx=-80ppm, σyy=-25ppm et σzz=+110ppm (Seelig, 1978).
Posons B0=B0 k ; k est le vecteur unitaire orienté selon l'axe du champ. Soit ω γ0 = − B0 la pulsation de Larmor.
2 400 MHz est la fréquence de résonance du proton. Cela correspond à un champ de 9,4 T environ, et à une fréquence de
résonance en phosphore de 162 MHz (γΗ/γP�2,5).

9
L'Hamiltonien de déplacement chimique s'écrit: HCS CS= ω Iz
La pulsation de précession due au déplacement chimique peut s'exprimer par: ω ω σCSt= − 0 k k$ ; c'est donc une
forme bilinéaire symétrique. De ce fait, on ne tient compte dans son calcul que de $σ PAS , la partie symétrique de $σ , qui
est diagonale dans le système d'axes principaux. La partie antisymétrique du tenseur donne, quant à elle, une
contribution nulle.
Si l'on se place dans le repère PAS, k peut être exprimé au moyen de ses coordonnées polaires (θ, φ), et on a:
[ ]ω ω σ σ σCS xx yy zz= + +02 2 2(cos sin ) (sin sin ) (cos )φ θ φ θ θ
(ωCS dépend de θ et φ donc dépend bien de l'orientation de chaque molécule)
On fait alors intervenir la partie isotrope du tenseur, définie par 1/3 de sa trace: σ σ σ σiso xx yy zz= + +13
( ) ,
et on la soustrait aux valeurs principales pour définir: σ σ σα αα= − iso (α=x, y ou z)
Le résultat du calcul est une expression de la pulsation associée au CSA, en fonction des angles θ et φ. Elle fait
intervenir deux paramètres:
ησ σ
σ=
−y x
z
est le paramètre d'asymétrie
δ ω σ= − 0 z est le paramètre d'anisotropie.
De simples relations trigonométriques conduisent au résultat suivant (Schmidt-Rohr & Spiess, 1994):
[ ]ω ωδ
θ η θ φCS iso= + − −2
3 1 22 2cos sin cos( ) (1)
Mais on travaille avec des objets dont la symétrie n'est pas quelconque: en effet, la rotation rapide des
phospholipides autour d'un axe normal à la bicouche entraîne une symétrie axiale du tenseur de CSA dans un repère B
lié à la bicouche (i.e. η=0). Dans ce repère B, ce tenseur s'écrit :
$
/ /
σσ
σσ
B =
⊥
⊥
0 0
0 0
0 0
La direction de référence est ici n, la normale à la bicouche qui est aussi l'axe moyenné du phospholipide.
Les deux tenseurs diagonaux $σPAS et $σB peuvent être reliés au moyen d'un changement de bases faisant intervenir des
matrices de transformation d'angles d'Euler. Par conservation de la trace lors d'un changement de base, on a notamment:
σ σ σiso = +⊥13 2( )/ / .
En développant ce changement de bases, l'expression précédente de ωCS (équation (1)) peut être finalement transformée
en un dernier résultat (Seelig, 1978), ne faisant plus intervenir que l'angle β entre les directions n et k.

10
ω β ω σβ
CS iso( ) (cos
)= − − −−
0
2
123
3 12
∆σ
∆σ est l'anisotropie du déplacement chimique, définie par ∆σ σ σ= − ⊥/ / .
On note ω ω σiso iso= − −0 1( ) la pulsation isotrope, qui correspondrait au cas de la RMN liquide, et
∆ ∆ω ω σCSA =23 0 le facteur d'anisotropie (qui est égal à δ). Alors on a simplement:
ω ω ωβ
CS iso CSA= +−
∆ (cos
)3 1
2
2
(2)
Le résultat précédent fait apparaître une relation simple entre la pulsation de précession associée au CSA, et l'angle β
entre l'axe moyen du lipide (qui est aussi la normale à la bicouche) et l'axe du champ B0. On va voir que ce résultat
permet facilement de déduire l'allure du spectre large bande en 31P d'un phospholipide, selon la phase dans laquelle il se
trouve. La figure 2.1 illustre les différents profils de raies théoriques associés aux principales phases.
figure 2.1 : forme des spectres 31P statiques pour des phospholipides dans différentes phases; une fréquence nulle correspond àun déplacement chimique isotrope
(adapté d'après Dufourc, 1986)
Plaçons nous tout d'abord dans le cas d'une phase lamellaire. L'expression analytique des spectres (a) et (b) de la
figure 2.1 se déduit simplement de l'équation (2). Il faut pour cela comprendre que les normales n à la bicouche sont
distribuées dans l'espace d'une manière isotrope. En effet, ces bicouches forment des vésicules multilamellaires, de

11
forme sphérique. On doit alors calculer la probabilité dP pour un phospholipide donné d'avoir un axe moyen incliné
d'un angle β à dβ près par rapport à l'axe du champ.
On trouve facilement (Seelig, 1978) que : dP d p d= =12
sin ( )β β ω ω (c'est le calcul de l'angle solide pour un
cône de demi-angle au sommet β). p(ω), qui est la fonction de probabilité associée à la pulsation de résonance ω, vaut
donc : pd
d( )
(cos )ω
βω
=12
. En tirant cosβ de l'équation (2), et en dérivant, on peut montrer que :
pCSA
iso( )ωω ωω ω
=−−
⊥
⊥
1
6 3∆ω. Cette fonction de probabilité est proportionnelle à la hauteur du pic de pulsation
ω. En convoluant cette fonction par une fonction lorentzienne (pour rendre compte de la largeur de raie), on obtient un
profil tel que celui du spectre 2.1 (a). Ce spectre présente une singularité pour ω ω= ⊥ . Il est fortement dissymétrique,
et on dira de lui qu'il possède “un pic à droite”.
On peut par ailleurs remarquer sur la figure 2.1 que le spectre statique d'une phase lamellaire fluide Lα, s'il présente
la même asymétrie que celui d'une phase gel Lβ, puisqu'il s'agit également d'une phase lamellaire, en diffère cependant
sensiblement par la largeur : le spectre (b) est ainsi plus étroit que le spectre (a). Des arguments que nous ne reporterons
pas ici montrent que dans la phase fluide, l'ajout d'un mouvement d'oscillations au mouvement de rotation entraîne une
diminution des valeurs propres du tenseur du CSA. Ainsi, l'anisotropie du déplacement chimique ∆σ est plus faible en
phase fluide, et le spectre est plus fin.
Considérons maintenant le cas d'une phase hexagonale. Les lipides y sont arrangés en monocouches assemblées en
forme de cylindres. Ces lipides diffusent rapidement autour de l'axe du cylindre, si bien que le système cylindrique
présente désormais une symétrie axiale. Les directions des axes des cylindres sont isotropes. Pour un lipide dans un
cylindre donné, on peut définir deux valeurs propres du tenseur d'anisotropie: l'une, σ / /H , correspond au cas où le
cylindre a un axe colinéaire à B0 ; l'autre, σ⊥H , au cas où l'axe est normal à B0. Des considérations de symétrie
permettent de démontrer que, en comparant à une phase lamellaire fluide pour laquelle les valeurs propres sont notées
σ / /B et σ⊥
B , on a: σ σ/ /H B≈ ⊥ et σ σ σ⊥ ⊥≈ +H B B1
2 ( )/ / . Le calcul complet est notamment présenté par Seelig (Seelig,
1978). Dans la phase hexagonale, l'anisotropie apparente du déplacement chimique devient donc :
∆σ ∆σH H H B= − = −⊥σ σ/ /12 . En clair, le spectre statique 31P d'un lipide en phase hexagonale (1.7(c)) est inversé par
rapport à celui d'une phase lamellaire, et présente un CSA environ deux fois plus faible de celui d'un phase lamellaire
fluide (1.7(b)).
C. L’effet de la rotation
On n'a jusqu'à présent parlé que des spectres dits “statiques”, c'est à dire enregistrés pour un échantillon immobile au
cours du temps. Supposons maintenant que cet échantillon soit contenu dans un rotor tournant autour de lui même, et
dont la direction fasse avec la verticale du champ un angle bien déterminé, l’angle magique dont on a parlé au
chapitre 1. Alors expérimentalement on constate que, pour des vitesses de rotation suffisantes, le spectre du 31P n'a plus
l'asymétrie si particulière du paragraphe précédent, mais est au contraire un spectre à haute résolution, composé
simplement d'une raie fine, comme pour le spectre 2.1(d).
Cette technique de RMN solide, appelée la rotation à l'angle magique (en anglais MAS pour Magic Angle Spinning)
permet de moyenner l'anisotropie du déplacement chimique à zéro. C'est une méthode artificielle qui produit le même
résultat que celui obtenu pour des micelles, et également de très petites vésicules, où le CSA est moyenné

12
“mécaniquement” du fait de leur faible temps de corrélation de rotation (τc ms≈ 1 ), et surtout de la rapide diffusion
de lipides à la surface.
1. Moyennage du CSA
Une explication simple de ce moyennage peut se faire ainsi : sous l'effet de la rotation du rotor autour de lui même,
toutes les molécules acquièrent une symétrie cylindrique, ou plutôt peuvent être remplacées par une pseudo-molécule de
symétrie cylindrique. Tous ces cylindres sont coaxiaux et leur axe commun fait un angle, noté α, avec la verticale du
champ; cette direction est également celle du rotor, et α est l'angle d'inclinaison du rotor par rapport à la verticale.
Alors, la pulsation de résonance associée au 31P de chaque molécule est donnée, d'après l'expression (2) par:
ω α ω ωα
CS iso CSA( ) (cos
)= +−
∆3 1
2
2
. Le facteur d'anisotropie ∆ωCSA dépend de chaque molécule, car il “garde
la trace” de la projection sur la pseudo-molécule. Néanmoins, pour un angle α α= m tel que 3 1 02cos αm − = , on
se débarrasse de cette contribution, et ω ωCS iso= . On n'a plus alors qu'une raie fine, centrée sur le pic isotrope
(comme si on était en RMN liquide, cf figure 2.1(d)). On dit que l'on a moyenné à zéro l'anisotropie du déplacement
chimique. L'angle αm = ≈ °arccos( ) '1
354 44 est appelé l'angle magique.
On ne rentrera pas dans la théorie du MAS, qui fait intervenir la mécanique quantique, et notamment la théorie de
l'Hamiltonien moyen, développée par Haeberlen (Haeberlen, 1968) puis Maricq et Waugh (Maricq & Waugh, 1979).
On peut cependant comprendre aisément que pour chaque molécule, la pulsation de résonance du CSA soit
désormais dépendante du temps, et même périodique (de période la durée de révolution du rotor). Le calcul de cette
pulsation, reporté dans la suite, dérive de celui présenté au §C.2.
On a pu montrer (§C.2.) que la pulsation de résonance associée au CSA se calculait par :ω ω σCSt= − 0 k k$ . Dans
un repère fixe par rapport au rotor (qui tourne à la pulsation ωr), le vecteur k a pour coordonnées :
k =
=
sin cos
sin sin
cos
cos
sin
α ωα ω
α
ωω
m r
m r
m
r
r
t
t
t
t2
312
, tandis que le tenseur de l'anisotropie du déplacement chimique est
( )$σ σ=≤ ≤ij i,j1 3
(il n'a pas de raison d'être diagonal dans le repère du rotor).
En développant le produit matriciel dans ce repère fixe par rapport au rotor, on fait apparaîtreσ σ σ σiso = + +( ) /11 22 33 3 , et l'expression finale de ωCS en fonction du temps peut se mettre sous la forme :
ω ω ω ω ω ωCS iso r r r rt C t C t S t S t( ) cos cos sin sin= + + + +1 2 1 22 2 (3)
Les valeurs des coefficients C et S qui dépendent des σij sont reportées dans la littérature (Schmidt-Rohr & Spiess,1994, p103). L'important est ici de faire apparaître la dépendance temporelle de la pulsation de résonance, qui estpériodique. Pour une vitesse de rotation grande devant la fréquence des interactions considérées, on peut moyenner
cette expression. Alors ω ωCS isot( ) = .
2. Impulsions de 180° synchronisées avec la rotation
Supposons que notre échantillon tourne dans un rotor incliné à l'angle magique à une fréquence suffisante pour
moyenner les interactions inhomogènes comme le CSA. Alors la pulsation de précession associée à ce CSA, calculée au
paragraphe précédent, peut être schématiquement représentée par le schéma 2.2(a). On comprend bien qu'en valeur
moyenne ω ωCS isot( ) = .

13
figure 2.2 : représentation schématique de la variation temporelle de ωCS−ωiso (a) en rotation à l'angle magique, à la pulsationde rotation ωr=2π/tr; (b) idem mais en appliquant 4 impulsions 180° synchronisées avec la rotation; la ligne pointillée symbolise une
valeur moyenne non nulle (adapté de Griffin, 1998).
Une technique utilisée pour réintroduire les interactions précédemment moyennées à zéro consiste à appliquer à
l'échantillon, durant sa rotation, une série d'impulsions radiofréquence 180° 3. Cette technique, initialement développée
par Alla (Alla et al., 1978), a ensuite été réutilisée dans le but de construire des spectres 2D de recouplage. La figure 2.3
permet de comprendre pourquoi on peut dire d’une impulsion 180° qu’elle inverse le sens de précession de
l'aimantation.
figure 2.3 : effet d’une impulsion 180° sur une aimantation qui précesse sous son déplacement chimique(on ne considère pas la relaxation)
Par conséquent une impulsion 180° réintroduit le CSA qui avait été auparavant moyenné par le MAS. Le CSA est
ainsi recouplé. Pour plusieurs impulsions 180° par période, la valeur moyenne de cette interaction est non nulle, comme
le montre la figure 2.2(b).
D. Le recouplage de l’anisotropie du déplacement chimique
1. Utilité de la technique de recouplage dans l’étude des lipides
Les paragraphes précédents ont tenté de mettre en évidence l’intérêt de la RMN du 31P dans l’étude des
phospholipides, notamment dans les attributions de phases, ou dans les mesures de paramètres d’ordre. Mais, comme

14
cela apparaîtra dans la partie expérimentale, les quantifications de phases sont délicates dans le cas de mélanges. Par
ailleurs, la rotation à l’angle magique, en produisant des raies fines et de grande résolution, est capable de “séparer” les
lipides selon la valeur de leur déplacement chimique isotrope. Il est donc naturel de tenter de leur appliquer les
techniques dites de recouplage.
2. La méthode de Tycko
Parmi toutes les méthodes de recouplage (du CSA ou de l’interaction dipolaire) qui existent, la méthode développée
par Robert Tycko (Tycko, 1989) joue un rôle particulier, parce qu’en théorie, la dimension indirecte du spectre (celle qui
correspond à l’incrémentation de t1) reproduit l’allure du spectre que l’on obtiendrait pour un échantillon statique.
Certaines méthodes donnent un profil non intuitif sur cette dimension, qu’il faut ensuite simuler in silico pour en
déduire les paramètres intéressants comme les éléments du tenseur d’anisotropie 4. Nous nous proposons d’expliquer
comment la séquence d’impulsions 180° appliquées durant un tour de rotor permet de recoupler le CSA et d’obtenir un
profil de raie identique à un spectre statique, à un facteur d’échelle près. Pour cela, les résultats exposés au cours des
paragraphes précédents vont être utiles. Une description de cette technique se trouve dans l’article original de Tycko,
mais aussi dans l’ouvrage de Schmidt-Rohr et Spiess (Schmidt-Rohr & Spiess, 1994, pp 195-198). Développée à
l’origine sur un noyau de 13C dans un sucre, cette méthode n’avait auparavant jamais été appliquée au 31P ni même dans
les phospholipides, bien que le CSA soit pour eux d’une grande importance.
Tout au long de l’expérience, l’échantillon tourne à une fréquence élevée, de l’ordre de 5kHz, et il est incliné à
l’angle magique par rapport à la verticale. Sous l’effet de ce MAS, et d’après l’équation (3), la pulsation de précession
ωCS a pour expression (à la constante ωiso près, que l’on ne mentionnera plus par la suite) :
ω ω ω ω ωCS r r r rt C t C t S t S t( ) cos cos sin sin= + + +1 2 1 22 2 . La valeur moyenne de ωCS sur une période de
rotation est nulle, i.e. ωCS t( ) = 0 . L’idée de base de la séquence décrite est d’appliquer un nombre pair
d’impulsions de 180° synchronisées avec la rotation, comme le détaille le paragraphe D.2, afin de rendre cette valeur
moyenne non nulle et proportionnelle au cas statique. Or si l’échantillon ne tournait pas, on aurait ωr=0, et donc
ω CSstat C C= +1 2 .
figure 2.4 : la fonction p(t) associée à une séquence d’impulsions de recouplage ;les impulsions sont de 180°, et ont une durée τ supposée négligeable devant tr.
Introduisons une fonction p(t), de module 1 et qui change de signe à chaque impulsion 180°. La figure 2.4 détaille
l’allure de cette fonction pour 4 impulsions 180°, dont la durée τ est supposée négligeable devant tr.
La proportionnalité imposée entre ωCS t( ) et ωCSstat se traduit mathématiquement par la formule :
3 on rappelle qu'en RMN impulsionnelle, une impulsion 180° fait tourner l'aimantation d'un angle 180° dans le
référentiel tournant (i.e. elle la change en son opposée si elle est orthogonale à sa direction). Elle a une duréeτ π γ= / B1 , pour un champ radiofréquence d'intensité Β1.

15
10t
p t t dtr
CS
t
CSstatr
( ) ( )ω χ ω∫ = (4)
En effet, la figure 2.3 montre comment une impulsion 180° fait changer de signe la fonction ωCS ; c’est ce que
traduit la fonction p(t). χ est le facteur d’échelle anisotrope.
L’équation (4) est en particulier satisfaite lorsque p(t) est une fonction paire, et que l’on a l’égalité
p t t dt p t t dtr
t
r
tr r
( )cos( ) ( )cos( )ω ω0 0
12
12 2∫ ∫= . La fonction sinus étant impaire, la première condition annule les
termes en facteur des coefficients S ; une conséquence est que les impulsions sont symétriques par rapport à une demi-
révolution du rotor (cf. figure 2.4). Par ailleurs, la deuxième condition sur les termes en facteur des coefficients C
impose que ces intégrales valent 12 χ tr .
Or chacune de ces intégrales se calcule aisément sur l’intervalle [τi, τi+1], où la fonction p(t) et constante et vaut ±1.
Ainsi pour k valant 1 ou 2, et pour 2N impulsions 180° par période de rotation :
p t k t dt k t dt k t dtk
kr
t
r r
t
r
n
n
N
r n
r
N
r
( ) cos( ) cos( ) ... cos( ) ( ) sin( )ω ω ωω
ω ττ
τ0 01
12 1
12 2
1∫ ∫ ∫ ∑= + + = −=
et finalement la condition sur les intégrales se ramène à : ( ) [sin( ) sin( )]− − ==
∑ 112
2 01
nr n
n
N
r nω τ ω τ (5)
Le résultat de ce calcul est une expression qui lie les inconnues τi à ωr. Nous venons d’établir une condition sur les
délais entre les impulsions de 180° telle que, si elle est vérifiée, la valeur moyenne de la pulsation de précession due au
déplacement chimique soit proportionnelle à la pulsation statique. La constante de proportionnalité est
χ ω= ∫1
0tp t t dt
rr
t r
( ) cos( ) . En fait, on a oublié de faire intervenir le déplacement isotrope ωiso, et le même calcul
que précédemment démontrerait que le facteur d’échelle isotrope est ξ = ∫1
0tp t dt
r
t r
( ) .
Alors ω ξ ω χ ωCS iso CSstatt( ) = + (6)
Il ne reste plus alors qu’à calculer les valeurs numériques qui vérifient l’équation (5). Dans son article, R. Tycko
dresse un tableau des résultats. Il y est notamment expliqué qu’il faut au minimum 4 impulsions 180° par tour de rotor
pour satisfaire cette équation. Il y a en particulier des solutions simples pour 4 et 6 impulsions, dont l’une d’elle est très
simple puisqu’elle n’utilise que 4 impulsions et donne un ξ=0. C’est cette solution qui est généralement la plus
employée, dans l’article original (Tycko et al., 1989) et y compris par la suite (Gross et al., 1997). Ainsi pour
ω τr 1 70 9= °. et ω τr 2 160 9= °. , on a les valeurs de ξ=0 et surtout χ=0,393 (soit des durées typiques de τ1=39µs et
τ2=89µs pour une fréquence de rotation de 5000Hz).
On est désormais en mesure de comprendre la séquence d’impulsions (le pulse program) nécessaire à mettre en
œuvre pour réaliser nos expériences de recouplage du CSA. Il s’agit bien sûr d’une séquence 2D, avec un temps
d’évolution t1 selon la dimension indirecte, et un temps d’acquisition t2 selon la dimension directe. Cette séquence est
illustrée sur la figure 2.5, et correspond à χ=0,393.
4 C’est le cas des séquences de Bax (Bax, 1983, 2 impulsions 180° par rotation) ou de Yarim-Agaev (Yarim-Agaev,
1982, 6 impulsions 180° par rotation)

16
figure 2.5 : séquence d’impulsions du recouplage du CSA par la méthode de Tycko (Tycko et al., 1989), correspondant àχ=0,393 et ξ=0 ; les phases sont les suivantes :
Φ1=Φ2=Φ4=(+x,-x,-x,+x,+y,-y,-y,+y) ; Φ3=(+y,-y,-y,+y,-x,+x,+x,-x,-y,+y,+y,-y,+x,-x,-x,+x)
En particulier, une des différences avec la séquence originale est que notre séquence ne nécessite pas de transfert de
polarisation par polarisation croisée pour amener l’aimantation transversale sur le 31P. C’est une conséquence de
l’abondance naturelle du 31P, ainsi que de son assez grand rapport gyromagnétique, qui permettent de débuter
directement la séquence par une impulsion de 90° sur le phosphore (à l’inverse de Tycko en 13C). Par ailleurs, le
cyclage de phases, détaillé dans la légende de la figure 2.5, ne sera pas expliqué ici. Nous mentionnerons simplement
que son effet principal sur la dimension indirecte (recouplée, donc large bande) est de rejeter aux deux extrémités du
spectres les artefacts expérimentaux, ainsi que de diminuer la sensibilité aux imperfections des impulsions qui ne
seraient pas exactement de 180°, et nous renverrons pour cela à l’article originel.
Enfin, on notera que l’on ne découple pas en 1H pendant la période d’évolution t1. Cela a inévitablement pour
conséquence de recoupler également le couplage dipolaire proton-phosphore, donc la coupe obtenue selon la dimension
indirecte ne sera pas purement due au CSA recouplé. Mais il a été vu que le couplage hétéronucléaire dipolaire était
faible ; de plus, il y aurait un risque d’interférences entre les impulsions 31P et 1H, ce qui rendrait le recouplage
inefficace. En revanche le canal proton est systématiquement découplé du canal phosphore durant t2, par l’application
d’une onde continue.
Tout ceci doit nous permettre en théorie d’obtenir des spectres 2D où l’on sépare sur une dimension les lipides selon
leur spectre haute résolution, et où l’on lit leur spectre recouplé du CSA, i.e. leur spectre statique multiplié par un
facteur d’échelle, sur l’autre dimension. Ces spectres seront présentés et commentés au paragraphe III, après avoir
auparavant présenté les méthodes expérimentales et le matériel employé.
II. Préparation des échantillons
A. Les préparations de lipides hydratés
Tous les lipides utilisés sont commerciaux, et de marque Sigma. Les solvants utilisés (chloroforme, méthanol et
éthanol) sont de marque Sigma, d'une pureté pour analyse. L'eau lourde (D2O), fournie par le CEA à Saclay, a une
pureté de 99,8%.
Les expériences ont été menées sur plusieurs échantillons de lipides. Les premières mises au point expérimentales se
sont faites sur des lipides uniques. Nous avons ainsi commencé par réaliser des échantillons de DMPC puis de DOPE.
Pour ce faire, 100mg de phospholipides sont mélangés à 100µL de D2O. Le mélange est homogénéisé par vortex, et la
dissolution des lipides est assurée par alternance de 3 cycles de congélation à –10°C – décongélation à +60°C. On forme
ainsi des vésicules multilamellaires hydratées à 50%. Après centrifugation (10min à 10.000g), le culot (120µL d'une
“pâte blanche”) est transféré dans un rotor MAS.

17
Afin de modéliser un mélange biologique simple de lipides, nous avons également réalisé un mélange ternaire,
contenant proportionnellement environ 30% de DOPC, 40% de DOPE et 30% de cholestérol en moles. L'homogénéité
du mélange est assurée en dissolvant en tout 150mg de lipides dans 20mL de solvants organiques (mélange
chloroforme-méthanol 10:1). Après 3 cycles d'évaporation rotative sous vide puis redissolution dans 10mL de solvant,
100mg de poudre (contenant les 3 lipides) sont prélevés et dissous dans 100µL d’un tampon 150mM KCl, 10mM Tris,
0,2mM EDTA, pH=7,2. La préparation des vésicules est alors identique à celle précédemment exposée.
B. Les fantômes de globules rouges
Les échantillons de stromas sont préparés à partir d'un culot de globules rouges humains distribués par les
Établissements de Transfusion Sanguine de l'Assistance Publique/Hôpitaux de Paris. Il s’agissait d'un produit âgé de
trois jours au moment de la préparation, et non utilisable en transfusion pour cause de volume insuffisant. La
préparation de fantômes requiert deux tampons : un tampon de lavage A (145mM NaCl, 5mM KCl, 5mM HEPES,
0,5mM MgSO4, pH=7,4) et un tampon d'hémolyse B (5mM HEPES, 1mM EDTA, pH=8). Un maximum d'étapes sont
effectuées à 4°C, donc en chambre froide ou dans la glace. 40mL de globules rouges sont prélevés et lavés deux fois
avec le tampon A. La centrifugation dure 15min à 25.000g. On procède ensuite à la lyse osmotique en versant la
fraction lavée dans 40 volumes du tampon B. On laisse le mélange en agitation durant deux heures, avant de centrifuger
le tout à 26.000g pendant 20min à 4°C. On lave le culot de la sorte quatre autres fois, dans un volume de tampon B à
chaque fois. Après la première centrifugation, il faut enlever le “red button” rouge foncé (sous le culot), qui contient des
protéases. On récupère finalement un liquide blanc visqueux, constitué des membranes percées séparées de
l'hémoglobine : c'est le stroma. Pour concentrer l'échantillon en vue d'améliorer la qualité des spectres, il convient de lui
faire subir une ultra-centrifugation à 100.000g pendant 10min à 4°C. On peut alors remplir le rotor avec un volume de
120µL environ.
III. Utilisation de l’appareil de RMN
A. Description du matériel
Le spectromètre utilisé est un spectromètre de marque Bruker, modèle DMX 400 WB. Il s'agit d'un spectromètre à
large trou, dont la fréquence de résonance en proton est 400,13 MHz (et en 31P : 161,9 MHz). Son champ statique a une
valeur de 9,4T. La sonde qui l'équipe est une sonde-MAS 4mm à 2 canaux, un canal proton et un canal accordable
“large bande”, de marque Bruker. Le rotor en zirconium a un diamètre de 4mm, une hauteur de 1cm et est fermé par un
bouchon à ailettes en Kel-F. Son volume est de 120µL. Sa rotation à l'angle magique est assurée au moyen d'une unité
pneumatique qui délivre deux flux d'air, comme l'illustrait la figure 1.6.
La vitesse de rotation est contrôlée par stroboscopie, et est constante à moins de 1% près sur une durée de plus de 10
heures. La température de l'échantillon est maintenue constante au moyen d'une résistance chauffante qui règle la
température du gaz de lévitation (“bearing”).
L'ensemble du spectromètre est piloté par une station Indy Silicon Graphics, reliée à une console Bruker Avance
400.
B. Mise en œuvre des spectres de RMN
Le matériel décrit précédemment permet de réaliser les différents types de spectres du 31P mis en œuvre
expérimentalement : spectres haute résolution en MAS, spectres statiques et surtout spectres 2D recouplés.
18
Avec les caractéristiques propres au matériel, les durées des impulsions 90° sont typiquement de 4,5 à 5,5µs pour31P, et de 9 à 10µs pour 1H. Pour les spectres découplés sur le canal proton pendant l’acquisition, cela assure un
découplage de 25kHz environ.
La réalisation de spectres statiques (sans faire tourner le rotor) se fait paradoxalement au moyen de la sonde MAS
(qui possède un meilleur facteur de qualité qu'une sonde liquide), mais, comme il n'est pas possible de réguler la
température si l'on n'envoie pas d'air, il faut pour cela démonter la sonde pour y bloquer le rotor au moyen d'un papier
de Parafilm. On peut alors souffler de l'air chaud sans faire tourner le rotor. De plus, il faut adapter pour ces spectres
statiques une séquence d’impulsions avec écho en 31P. La décroissance du FID étant très rapide, on “récupère” les
premiers points en refocalisant l’aimantation au moyen d’une impulsion de 180°. Il faut pour cela étalonner au préalable
la durée de cette impulsion. Le canal proton est découplé pendant l’acquisition.
Les spectres 2D recouplés sont mis en œuvre avec des paramètres qui peuvent différer légèrement d’une expérience
à l’autre. Généralement les durées des expériences sont de 3 heures 30 avec 64 scans. Pour les fantômes de globules
rouges, il faut passer à 16 heures avec 256 scans.
IV. Mise au point sur un lipide, la DMPCAfin de mettre en application les techniques développées précédemment, et notamment de tester l’efficacité de la
méthode de recouplage, il nous est tout d’abord apparu nécessaire de nous intéresser à un cas simple et bien décrit dans
la littérature. La DMPC est connue pour présenter une transition de phase à 23°C, entre une phase lamellaire gel (Lβ) à
basse température et une phase lamellaire fluide (Lα) à haute température. On doit donc s’attendre à deux observations
sur les spectres statiques du 31P de la DMPC : ils doivent avoir une asymétrie caractéristique (un “pic à droite”), et se
rétrécir sensiblement lorsque l’on passe au dessus de la température de transition (cf. figure 2.1). Normalement, les
mêmes observations doivent être faites sur une coupe longitudinale (selon la dimension indirecte) d’un spectre 2D
recouplé de DMPC.
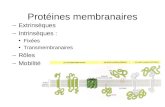
figure 2.6 : spectres statiques du 31P de la DMPC (a) à 10°C, en phase gel ; (b) à 30°C , en phase fluide.Les spectres sont réalisés avec un écho, et découplés du 1H pendant l’acquisition.
La figure 2.6 reproduit les spectres statiques (sans rotation de l’échantillon) obtenus pour la DMPC à 10°C (a) et à
30°C (b). La différence d’anisotropie entre les deux phases est aisément quantifiable. Les CSA des phases gel et fluide
(les écartements des raies ∆σ) ont des valeurs expérimentales de 75 ppm et 55 ppm respectivement, soit environ 12kHz

19
et 9kHz. Le rétrécissement du spectre observé en chauffant est d’environ 25%, et l’asymétrie est nettement marquée
dans les deux cas. Nous pouvions donc nous demander si la rotation à l’angle magique de l’échantillon, assortie de la
séquence développée précédemment, permettraient d’obtenir des résultats comparables. Bien sûr, l’utilisation d’une
telle méthode n’a ici qu’un intérêt formel, puisque la DMPC est le seul lipide présent et qu’il n’y a pas de nécessité à
faire apparaître une dimension supplémentaire au spectre. Mais par ailleurs, la réalisation de ces spectres a été
l’occasion d’optimiser leurs nombreux paramètres.
La figure 2.7 représente le spectre de recouplage obtenu avec la séquence de Tycko adaptée, pour un facteur
d’échelle anisotrope de 0,393 (et un facteur d’échelle isotrope nul), à une vitesse de rotation de 5000Hz. Le spectre est
reproduit sans être recadré. Les courbes de niveaux sont
d’intensité croissante vers le centre du spectre. L’axe
des abscisses (dimension directe, haute résolution à
5000Hz) est gradué en ppm, alors que l’axe des
ordonnées (dimension indirecte recouplée) est gradué en
Hz. On voit ainsi que la fenêtre spectrale dans la
dimension indirecte est égale à la fréquence de rotation
en Hz 5. La DMPC (ou plus simplement la PC) a un
déplacement chimique isotrope calibré par référence à 0
ppm.
Ce spectre met notamment en évidence l’efficacité
de notre séquence dans la dimension directe, toute
l’intensité étant concentrée dans une bande relativement
fine. Par ailleurs, le cyclage de phase a effectivement
pour conséquence de rejeter les artefacts à l’extérieur de
la dimension recouplée (ce sont les deux “pics” à
ω π/ 2 2500= ± Hz ).
De tels spectres ont été réalisés à différentes
températures (10°C et 30°C principalement). En
coupant le spectre 2.7 longitudinalement (i.e en suivant
l’axe de la bande fine et en passant par son maximum),
on peut extraire les profils de raies recouplés de la
DMPC. La figure 2.8 (en page suivante) dresse la comparaison entre ces profils à 10°C (a), et à 30°C (b), pour un
facteur d’échelle anisotrope de 0,2 , et en tournant à 5kHz. L’axe est désormais gradué en ppm. A nouveau deux points
sont à discuter : l’asymétrie et la largeur des profils de raies. Il est clair que les coupes dans la dimension recouplée ne
présentent pas la forme canonique des spectres statiques. On entrevoit ainsi déjà une faiblesse de la séquence choisie.
Les raisons en sont multiples, au premier rang desquelles se trouve la limitation intrinsèque de la résolution dans cette
dimension.
Néanmoins on reconnaît dans les deux spectres proposés une asymétrie lamellaire, même si le pic à droite est
fortement “aplati”. Par ailleurs, comme pour les spectres statiques, le passage de 10°C à 30°C s’accompagne d’un
rétrécissement de 20% également, qui marque la transition gel-fluide. Enfin, l’anisotropie du déplacement chimique
peut être mesurée dans chacune des phases; on mesure respectivement 14 ppm et 11 ppm pour les phases gel et fluide,
5 La séquence échantillonne un point toutes les 200µs (=2π/ωr), et ce dwell time est l’inverse de la fenêtre spectrale.
figure 2.7 : spectre 2D de recouplage réalisé sur la DMPC à 10°C ;la vitesse de rotation est de 5kHz, et la séquence correspond à χ=0,393

20
figure 2.8 : coupes longitudinales d’un spectre de recouplage de la DMPCà 10°C, (b) à 30°C; ωr/2π=5kHz, χ=0,2
qu’il faut diviser par le facteur d’échelle 0,2. On arrive à des résultats (respectivement 70 et 55 ppm) très proches de
ceux mesurés en statique, et cohérents avec la littérature.
Mentionnons pour finir que la réalisation de ces premiers spectres de recouplage sur la DMPC, ainsi que d’autres sur
la DOPE, mais qui ne seront pas reportés ici, nous a permis d’optimiser des paramètres de traitement des données, mais
également des valeurs importantes comme la vitesse de rotation du MAS. Trop rapide, on perd en résolution en ayant
une trop grande fenêtre spectrale, et la durée τ des impulsions 180° devient non négligeable devant τr (Ishii & Terao,
1998). Trop lente, on ne peut plus loger le spectre dans la fenêtre. Il est alors apparu optimal de tourner à une vitesse de
5kHz, qui est également la fréquence de rotation employée par Tycko. Mais toute adaptation est envisageable, puique ωr
et χ peuvent être modulés en fonction du CSA donc de la molécule
V. Les résultats sur le mélange ternaireLes expériences précédentes nous ont permis de faire déjà quelques remarques intéressantes concernant notre
séquence de recouplage. Mais elles n’ont jusqu’à présent pas démontré leur capacité à séparer les phospholipides selon
leur tête polaire. Par ailleurs, il n’a pas encore été observé de phase hexagonale, tant en statique que sur un spectre
recouplé.
Notre deuxième échantillon a donc été un mélange ternaire DOPC/DOPE/cholestérol, en pourcentage d’environ
30/40/30 en moles. C’est l’un des seuls décrits dans la littérature comme possédant une coexistence lamellaire-
hexagonal à une température accessible au sein d’un spectromètre (Moran & Janes, 1998, Tilcock et al., 1982). Comme
pour la PC seule, la première étude se fait en statique, afin d’observer la transition de phase. Les résultats sont reportés
sur la figure 2.9 par une évolution en température du profil de raies.
Ces spectres sont en tous points comparables avec ceux de Janes (Moran & Janes, 1998), bien que notre mélange ait
une composition légèrement différente. Bien sûr pour un mélange, on pourrait établir un diagramme binaire, ce qui
explique que chaque phospholipide passe progressivement d’une phase lamellaire fluide (a) à une symétrie hexagonale
(d). On remarque qu’à une température de coexistence des phases, le spectre ne peut pas aisément être décomposé en la
somme de ses composantes. La PC ayant un plus grand ∆σ que la PE (Cevc, 1993) 6, et leurs déplacements chimiques
isotropes étant respectivement 0 et 0,6 ppm, on peut attribuer les raies comme le montre la figure 2.9.
6 c’est ce que met en évidence l’astérisque sur le spectre 2.9(a).

21
figure 2.9 : évolution des spectres statiques du mélange ternaire avec la température; le spectre (a) a été augmenté d’un facteur 2.
Avant de comparer ces résultats avec les coupes longitudinales effectuées sur un spectre de recouplage du mélange
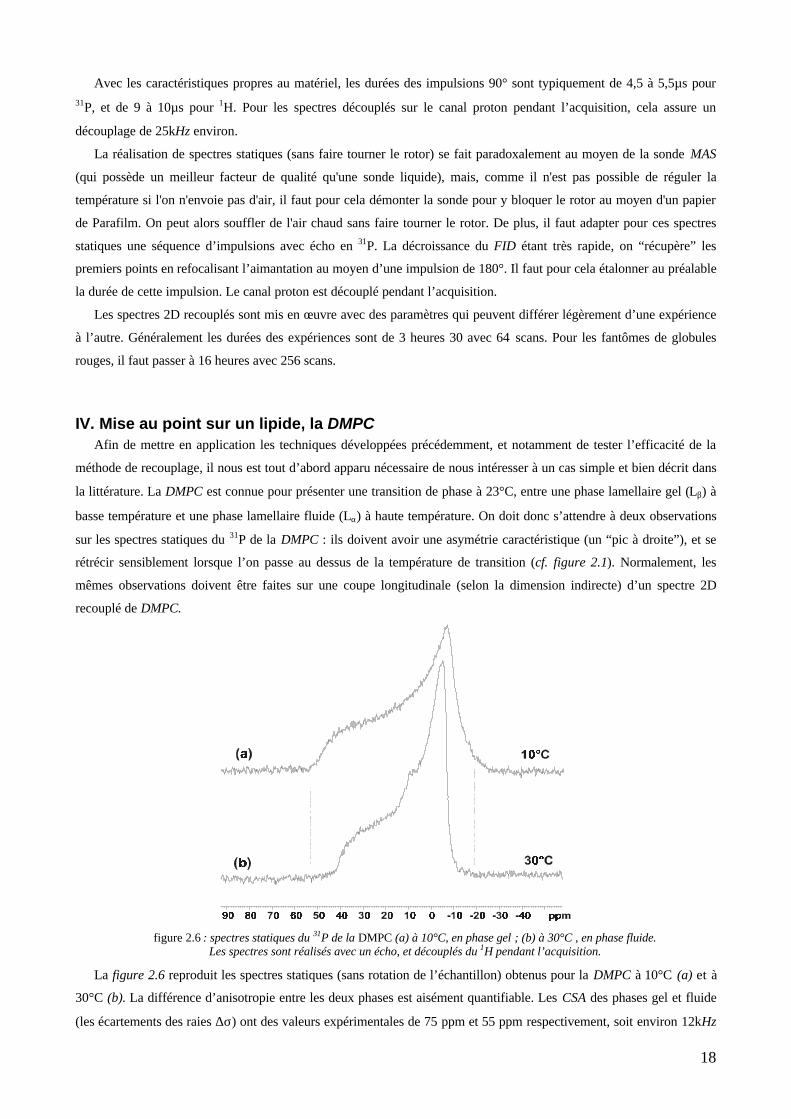
ternaire, présentons l’un de ces spectres. La figure 2.10 en page suivante reproduit un grossissement du spectre obtenu
pour un facteur d’échelle de 0,393 , à 37°C. On y distingue nettement les deux bandes séparées associées à la PC
(0 ppm) et à la PE (0,6 ppm). Sur de tels spectres on peut extraire les coupes longitudinales pour chacun des
phospholipides, à différentes températures. Les évolutions en fonction de la température sont retracées sur la figure
2.12. La dimension directe du spectre 2.10 est nettement résolue. La PC et la PE ont des déplacements chimiques
suffisament différents (0,6 ppm d’écart seulement) pour donner naissance à deux bandes parallèles mais distinctes. Cela
est confirmé par la coupe transversale de ces bandes, reproduite en figure 2.11, et qui est superposable à un simple
spectre MAS à 5000Hz. De plus, comme le montrent les coupes 2.12, ces bandes ont bien l’asymétrie attendue en
fonction de la phase lipidique. La PE est plus proche des formes canoniques. Mais dans les deux cas, la symétrie
hexagonale n’a pas un CSA moitié plus faible que celui d’une phase lamellaire. Le rétrécissement est visible, mais pas
quantitatif.
22
figure 2.10: gros plan d’un spectre de recouplage dumélange ternaire DOPE/DOPC/cholestérol à 37°C;
ωr/2π=5kHz, χ=0,393
figure 2.11: superpositon d’un spectre MAS à 5000Hz (a)et de la coupe transversale du spectre 3.5 selon la dimension
directe (b), pour le mélange ternaire à 37°C
figure 2.12 : coupes longitudinales de spectres de recouplage du mélange ternaire; seule la température varie;ωr/2π=5kHz ; χ=0,393

23
VI. Discussion des résultats précédentsCes quelques résultats nous suffiraient presque pour commenter les avantages et les limites de notre technique de
recouplage. Alors qu’un spectre statique comme le 2.9(c) ne permet pas de déduire la proportion de lipide dans chaque
phase pour un point au milieu du fuseau (sur le diagramme de phases), une technique comme celle du recouplage sépare
effectivement nos deux lipides en leur associant un profil de raies cohérent. Il n’en reste pas moins que ce profil n’est
pas (encore) d’une précision suffisante pour permettre d’attribuer des proportions à chaque lipide dans chaque phase. En
théorie, le spectre 2.9(b) devrait par exemple être la somme des deux spectres 2.12(b). Les explications à ces limitations
sont multiples.
D’abord, on a vu que l’on recouplait également l’interaction dipolaire hétéronucléaire. On ne doit donc pas
s’attendre à obtenir des spectres aussi asymétriques que des spectres statiques. Un découplage continu durant la
séquence s’est avéré infructueux, probablement en raison d’interférences avec le recouplage.
De plus, les paramètres de l’expérience sont perfectibles. L’impulsion de 180°, notamment, n’est pas idéale, au sens
où elle n’est pas parfaitement carrée, pas parfaitement instantanée (τ µ≈ 11 s non négligeable devant τ µr s= 200 )
ou pas parfaitement reproductible. De même, les délais ne sont peut-être pas assez précis ou reproductibles et la
fréquence de rotation peut légèrement varier au cours de l’expérience. Bref, on le voit, les causes techniques sont
multiples pour justifier que l’expérience peut encore être améliorée.
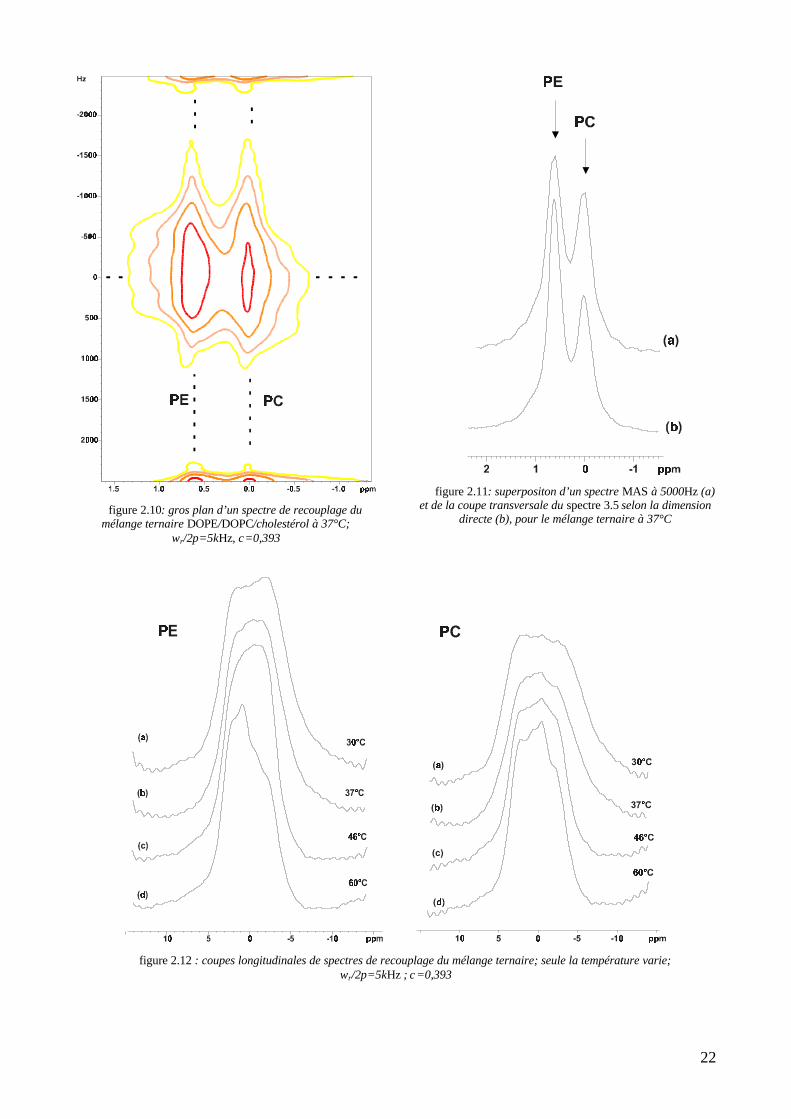
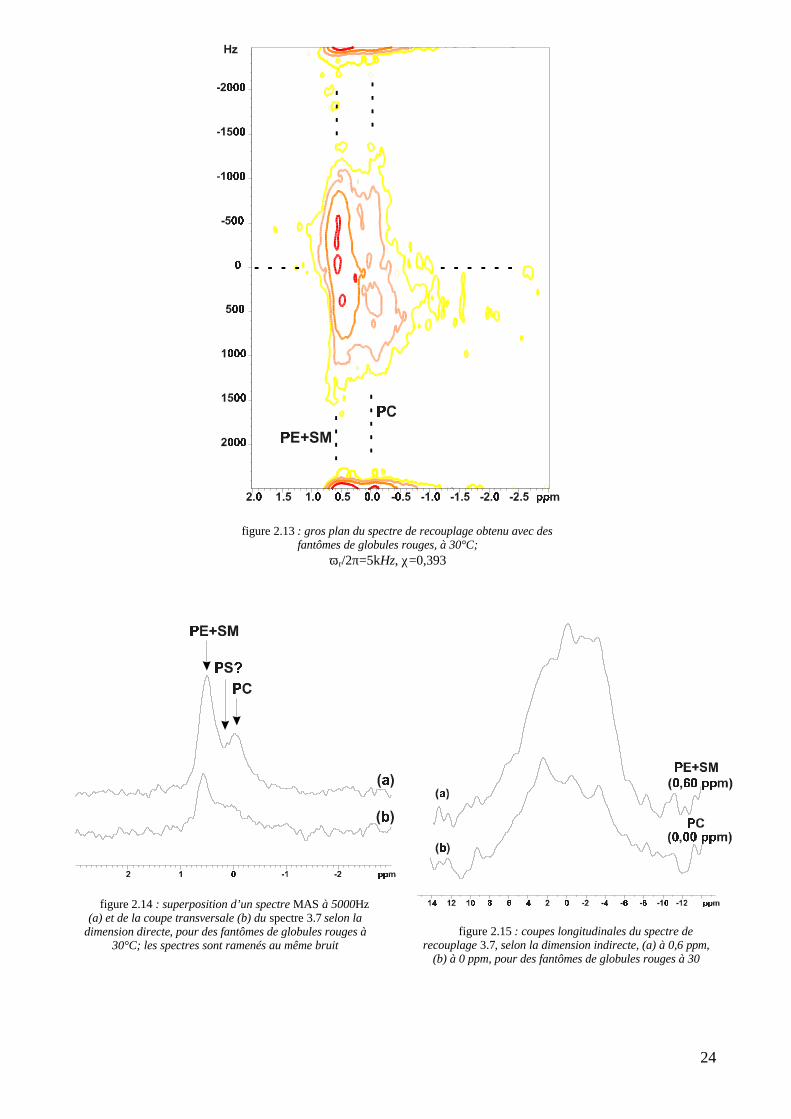
VII. Une extension à un système biologique : les fantômes de globules rougesIl s’est néanmoins avéré intéressant de tester cette expérience sur un échantillon biologique, en l’occurrence des
membranes d’érythrocytes isolées au sein de stromas. Si l’on ne doit vraisemblablement pas s’attendre à identifier des
phases lipidiques au moyen des coupes de leurs spectres de recouplage, la diversité lipidique de la membrane naturelle
et sa faible concentration dans notre échantillon posaient la question de la résolution dans la dimension directe, et
notamment du rapport signal/bruit. Il est à ce titre intéressant de noter que, pour être interprétable, un spectre 2D de
recouplage sur les stromas doit durer 16 heures au lieu de 3 heures 30 pour le mélange ternaire. Un gros plan de ce
spectre, obtenu à une température de 30°C, et pour un facteur d’échelle anisotrope de 0,393, est reproduit sur la figure
2.13. Comme on l’a souligné au chapitre 1, tableau 1.4, la membrane des érythrocytes recèle de nombreux lipides
possédant un atome de phosphore. Outre les phospholipides classiques, on compte notamment la sphingomyéline (SM),
dont le déplacement chimique isotrope vaut 0,6 ppm (comme la PE) (Warschawski, 1995). Une première vue sur le
spectre 2.13 nous indique déjà que la séquence de recouplage nous assure une résolution dans la direction directe
suffisante pour distinguer plusieurs bandes recouplées. La figure 2.14, qui représente la coupe transversale du spectre
2D pour une fréquence de 0Hz, corrobore ce fait. Pour cela, on le superpose avec un simple spectre MAS-haute
résolution à 5000Hz, ramené au même bruit. Le petit pic dû à la PS, à 0,15 ppm, déjà difficilement discernable sur un
spectre haute résolution, est invisible en recouplage, tant sur une coupe que sur la 2D. Le reste des spectres diffère peu.
L’attribution des bandes se fait donc telle qu’elle est précisée sur le spectre 2.13 : la PC à 0 ppm, la PE et la SM à
0.6 ppm. Les coupes longitudinales associées sont détaillées sur la figure 2.15. L’asymétrie y est difficile à interpréter,
surtout pour la PC, qui semble osciller. Il faut néanmoins souligner le fait que notre séquence de recouplage est capable
de séparer les lipides même en concentrations biologiques.
24
figure 2.14 : superposition d’un spectre MAS à 5000Hz(a) et de la coupe transversale (b) du spectre 3.7 selon la
dimension directe, pour des fantômes de globules rouges à30°C; les spectres sont ramenés au même bruit
figure 2.15 : coupes longitudinales du spectre derecouplage 3.7, selon la dimension indirecte, (a) à 0,6 ppm,
(b) à 0 ppm, pour des fantômes de globules rouges à 30
figure 2.13 : gros plan du spectre de recouplage obtenu avec desfantômes de globules rouges, à 30°C;
ωr/2π=5kHz, χ=0,393

25
Chapitre 3
Recouplage de l’interaction dipolaire
Mesures de distances dans les peptides membranaires
e chapitre reflète principalement le travail effectué depuis le début de ma thèse, en octobre 2000. Ce travail, bien
que dans la continuité du DEA, s’en distingue par la nouveauté des méthodes développées, ainsi que par la nature
des objets étudiés. Cette première année aura permis de mettre en œuvre les techniques choisies, de montrer leur
viabilité et leurs limites sur des premiers exemples, et de convaincre les biologistes du laboratoire qu’elles sont
complémentaires d’autres études structurales, et donc applicables à leurs échantillons. On présentera tout d’abord les
deux expériences de recouplage concernées, puis, dans le cadre d’une étude de peptides membranaires, les premiers
résultats expérimentaux sur un simple acide aminé, et leurs simulations informatiques.
I. Le recouplage de l’interaction dipolaire : REDOR et SFAM
A. Le contexte de l’étude de protéines membranaires par RMN
1. Les spins en présence
Les noyaux observables par RMN dans les protéines membranaires sont le proton 1H, le carbone-13, l’azote-15 et le
deutérium 2H. Chacun a ses caractéristiques et ses techniques propres, mais, parmi eux, le proton, qui possède le plus
fort rapport gyromagnétique, et une abondance naturelle de 100%, pourrait sembler le plus utile. Or il n’en est rien. Les
protons d’une protéine sont en effet couplés entre eux par un très fort couplage dipolaire, et, dans des systèmes orientés
ou statiques, cela a pour effet d’élargir considérablement les raies, rendant toute attribution impossible. Par ailleurs, ce
couplage, dit homogène selon la terminologie de Waugh (Maricq & Waugh, 1979) peut difficilement être moyenné à
zéro par la rotation à l’angle magique. Le plus souvent, une étude de protéine, a fortiori membranaire, passe donc par la
sélection des noyaux d’azote-15 et/ou de carbone-13. Le deutérium, noyau quadrupolaire de spin 1, donne lieu à des
études dont on ne parlera pas ici. 13C et 15N, tous deux de spins ½, ont pour caractéristiques d’avoir des spectres
dominés par deux types interactions : l’anisotropie de leurs déplacements chimiques respectifs, et le(s) couplage(s)
dipolaire(s) hétéronucléaire(s). Si l’abondance naturelle du 13C (1%), est parfois suffisante pour travailler sans
enrichissement isotopique, il faut par contre systématiquement enrichir sélectivement ou uniformément en azote-15a.
Enfin, on s’affranchit systématiquement du couplage hétéronucléaire avec les protons par irradiation continue de ceux-
ci durant l’expérience (c’est la méthode dite de découplage des protons).
2. Les expériences possibles
Il est important de garder à l’esprit que, pour être sûr de la fiabilité des informations structurales effectuées, le
peptide doit être inséré dans des membranes lipidiques, où il adopte sa conformation native. Sauf dans quelques cas
rares d’études en micelles (MacKenzie et al.,1997), on doit donc faire de la RMN sur des vésicules lipidiques. Leur
grande taille et leur fort taux de corrélation de rotation à l’échelle de la RMN interdit tout moyennage microscopique
propre à la RMN des liquides. Par la suite, les échantillons se prêtent à deux types d’études par RMN : une étude
a L’azote-15 possède par ailleurs un faible rapport gyromagnétique
C

26
statique, ou une étude en rotation à l’angle magique. Les informations accessibles sont différentes, voire
complémentaires. Pour un échantillon statique, il s’agit surtout d’informations d’angles relatifs de plans peptidiques ou
de liaisons entre atomes. On a pour cela recours à une orientation macroscopique du peptide dans l’aimant : le
distribution spatiale du peptide n’est alors plus isotrope, et la dépendance angulaire de l’interaction observée permet de
remonter aux orientations internes au peptide. Il existe plusieurs manières d’orienter les peptides membranaires dans
l’aimant, parmi lesquelles on ne mentionnera que l’orientation des bicouches lipidiques sur plaques de verre, ou
l’insertion dans des bicelles, sorte de disques lipidiques qui s’orientent dans le champs de l’aimant. Nous n’entrerons
pas ici dans le détail de ces études, et renverrons à la récente revue de J.H Davis et M. Auger (1999).
Notre travail s’inscrit dans le cadre de la RMN dite en rotation à l’angle magique, dont les bases ont pu être
présentées au chapitre 2. On a à cette occasion expliqué comment la rotation de tout l’échantillon autour d’un axe
incliné d’une valeur particulière, appelée l’angle magique, avait pour effet de moyenner à zéro l’anisotropie du
déplacement chimique. En fait, il en est de même pour toutes les interactions inhomogènes, c’est-à-dire dont
l’Hamiltonien commute avec lui même à chaque instant (Maricq & Waugh, 1979). Le couplage dipolaire
hétéronucléaire étant lui aussi de nature inhomogène, on peut obtenir un spectre fin, dit à haute résolution, d’un
échantillon de peptide membranaire en lui faisant subir une telle expérience de MAS (Magic Angle Spinning). Mais un
tel spectre, du 13C ou du 15N, s’il permet une attribution des résonances, ne comporte aucune information structurale.
Plusieurs techniques ont été développées dans les dix dernières années pour mesurer des couplages dipolaires faibles au
sein d’échantillons solides en rotation à l’angle magique. Et si l’on mesure une constante de couplage, on a accès à la
distance associée, connaissant la loi en 1/r3 rappelée au chapitre 1. La méthode de Résonance Rotationnelle (R2) a été
introduite par Griffin et Levitt (Levitt et al., 1990) pour mesurer des constantes homonucléaires, et est utilisée pour
mesurer des distances 13C-13C, par exemple. Il faut pour cela accorder la fréquence de rotation du MAS avec la
différence de fréquence de résonance des noyaux couplés ; la réintroduction du couplage dipolaire produit un échange
d’aimantation entre ces deux spins. D’autres séquences de recouplage homonucléaire existent, tels le DRAMA (Dipolar
Recovery at the Magic Angle) ou le RFDR (RF-Driven Recoupling). Les détails de ces techniques sont abordés dans la
revue de Bennett, Griffin et Vega (Bennett et al., 1994 ).
Les techniques de mesures de constantes de couplages hétéronucléaires sont moins nombreuses. Elles utilisent les
interférences entre le moyennage du MAS et l’application d’un train d’impulsions radiofréquences synchronisées. Ces
impulsions peuvent être discrètes et de 180° (cas du REDOR) ou continues et modulées en fréquence, en amplitude, ou
en phase (cas du SFAM).
B. Le REDOR
Le REDOR (Rotational Echo Double Resonance) est une méthode de recouplage dipolaire hétéronucléaire.
L’expérience a été conçue et réalisée pour la première fois en 1989 dans le laboratoire de Jack Schaefer (Gullion &
Schaefer, 1989). D’abord développée sur des échantillons simples (acides aminés en poudre), la méthode a été ensuite
appliquée à des échantillons d’intérêt biologique, généralement des peptides marqués 13C et 15N en membranes. Mais
détaillons tout d’abord le fonctionnement de la séquence.
1. La séquence REDOR
La séquence d’impulsions du REDOR est précisée en figure 3.1. C’est une séquence à trois noyaux, proche de la
séquence de Tycko développée au chapitre 2 dans son apparence. Le recouplage est ici aussi effectué au moyen
d’impulsions de 180° appliquées au noyau S (généralement l’azote), synchronisées avec la rotation. On a ici deux
impulsions par tour de rotor, une au début et une au milieu de chaque tour. Le canal du carbone ne contient qu’une
unique impulsion 180°, dont le but est de refocaliser le déplacement chimique isotrope du carbone.

27
1H
I= C13
S= N15
rotor
découplageCP
ramp-CP
90
180
180
0 4321
tr
(N)Nt =2tr 1
t2
t1
figure 3.1 : séquence d’impulsion de la version la plus commune de REDOR. Après un transfert de polarisation du proton aucarbone, un train d’impulsions de 180° synchronisées avec la rotation est appliqué sur l’atome d’azote. Le spectre du carbone estenregistré pour différentes valeurs incrémentées de N (N = nombre de tours de rotor par période REDOR). Le proton est découplé
par une séquence TPPM. Le cyclage de phase est de type XY-8.
La technique de REDOR s’appuie sur le déphasage de l’aimantation du noyau observé (le carbone-13) dû au
couplage dipolaire avec le second spin (l’azote-15). C’est une méthode de différence qui utilise des impulsions
radiofréquences en interférence avec le processus de moyennage du MAS. En clair, pour mesurer une distance carbone-
azote, il faut procéder à deux enregistrements, l’un en présence du train d’impulsions d’interférence sur l’azote, l’autre
en l’absence de ces impulsions. Les signaux/spectres associés, lorsque l’on incrémente la période de déphasage Ntr, sont
respectivement appelés S et S0. La différence des intensités (S - S0) ne dépend plus que du couplage dipolaire entre les
spins concernés, et permet une mesure de ce couplage.
Tentons d’expliquer ce point d’une manière analogue à celle du chapitre 2.
2. Le calcul de la séquence
Le calcul rigoureux de cette séquence doit faire intervenir une théorie telle que la théorie de Floquet, qui développe
l’Hamiltonien (périodique en fonction du temps) en une série de Fourier d’Hamiltoniens. On ne s’aventurera pas dans
ces calculs, et on renverra par exemple le lecteur à la revue de Bennett consacrée au recouplage (Bennett et al., 1994).
Il est cependant possible d’expliquer la séquence REDOR plus simplement, en s’appuyant sur la théorie de
l’Hamiltonien moyen. Evans développe notamment le calcul dans son livre consacré à la RMN biomoléculaire (Evans,
1995).
Considérons au sein d’un peptide une paire d’atomes 13C-15N séparés par une distance r. L’orientation du vecteur
entre ces atomes peut être repérée dans l’espace par les deux angles α et β, comme indiqué sur la figure 3.2. θm est
l’angle magique.

28
13C
15Nrθm β
α
figure 3.2 : schéma de l’orientation d’un vecteur entre deux spins 13C et 15N
La pulsation de transition ωD due au couplage dipolaire est fonction des angles α et β et de ωr, la pulsation de
rotation du rotor. Une impulsion de 180° sur le spin I change formellement le sens de précession de l’aimantation (voir
figure 2.3). Si l’on applique cette impulsion à un instant t1 après le début de la rotation, alors la valeur moyenne de la
pulsation ωD sur un tour de rotor Tr n’est plus nulle, mais vaut :
ω D =1Tr
ωD(α, β0
t1
∫ ;t)dt − ω D(α,β ;t)dtt1
Tr
∫
≠ 0
Après le transfert de polarisation du proton vers le carbone, l’aimantation est selon Ix.
Supposons que l’on ait qu’une impulsion de 180° sur le spin I (enregistrement de S0). Sous l’effet du couplage dipolaire,
cette aimantation devient du type )sin(2)cos( 11 tSItI DzyDx ωω + après un temps t1. Vient ensuite l’impulsion de
180°(x) sur le spin I, qui inverse le signe du second terme : )sin(2)cos( 11 tSItI DzyDx ωω − . Après une deuxième
période d’évolution sous le couplage dipolaire, on a donc à nouveau une aimantation selon Ix.
Si désormais on a en présence des impulsions de 180°(x) sur les deux spins I et S (enregistrement su spectre S), apparaît
alors le déphasage dû au recouplage dipolaire. Après t1, on a toujours une aimantation
)sin(2)cos( 11 tSItI DzyDx ωω + , mais les deux impulsions de 180° inversant chacune le signe du second terme,
cette aimantation reste )sin(2)cos( 11 tSItI DzyDx ωω + après les impulsions. La deuxième période d’évolution sous
le couplage conduit donc à une aimantation du type : )2sin(2)2cos( 11 tSItI DzyDx ωω + .
Pour une détection selon x, en remarquant que 2t1=NTr, on retiendra donc que :
)cos(et 0 rDxx NTISIS ωv∝∝
Par conséquent, pour une unique paire 13C-15N couplée dipolairement, )cos(1/ 0 rD NTSS ωv−=∆
Pour finir, dans l’échantillon solide, les vecteurs 13C-15N étant distribués isotropiquement, le signal macroscopique
enregistré est obtenu en sommant toutes les orientations possibles. Donc :
∆ S
S0
=
(1 − cos(ω D N Tr ))sin( β) dα dββ∫
α∫
sin( β ) dα dββ∫
α∫
(1)
L’équation (1) fait apparaître l’unique dépendance du rapport en fonction de la constante de couplage dipolaire D.
Historiquement, cette équation n’est intervenue qu’avec les premiers calculs de déphasage faits par Schaefer dans sa
revue sur le REDOR (Gullion & Schaefer, Advances in Magnetic Resonance, 1989), avec un formalisme d’Hamiltonien
moyen plus détaillé que nous ne l’avons présenté. Dans les premières parutions qui exploitaient ces résultats, l’équation
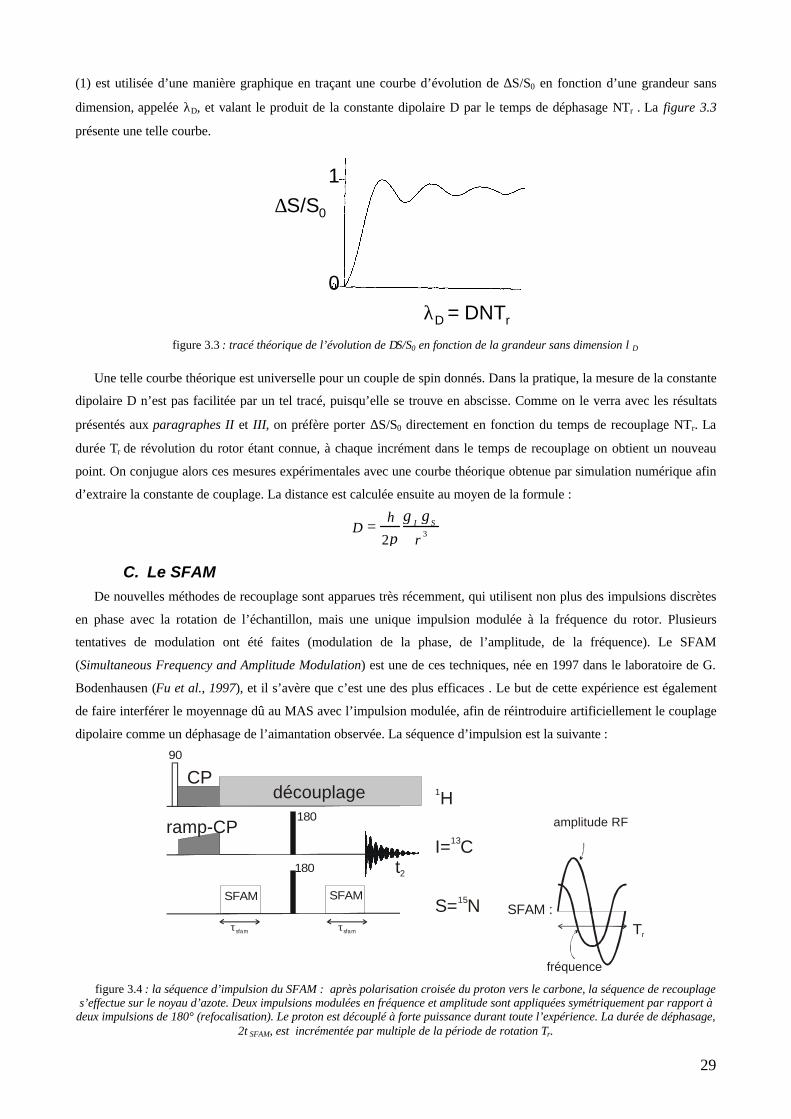
29
(1) est utilisée d’une manière graphique en traçant une courbe d’évolution de ∆S/S0 en fonction d’une grandeur sans
dimension, appelée λD, et valant le produit de la constante dipolaire D par le temps de déphasage NTr . La figure 3.3
présente une telle courbe.
1
0
λD = DNTr
∆S/S0
figure 3.3 : tracé théorique de l’évolution de ∆S/S0 en fonction de la grandeur sans dimension λD
Une telle courbe théorique est universelle pour un couple de spin donnés. Dans la pratique, la mesure de la constante
dipolaire D n’est pas facilitée par un tel tracé, puisqu’elle se trouve en abscisse. Comme on le verra avec les résultats
présentés aux paragraphes II et III, on préfère porter ∆S/S0 directement en fonction du temps de recouplage NTr. La
durée Tr de révolution du rotor étant connue, à chaque incrément dans le temps de recouplage on obtient un nouveau
point. On conjugue alors ces mesures expérimentales avec une courbe théorique obtenue par simulation numérique afin
d’extraire la constante de couplage. La distance est calculée ensuite au moyen de la formule :
D =h
2πγ
Iγ
S
r3
C. Le SFAM
De nouvelles méthodes de recouplage sont apparues très récemment, qui utilisent non plus des impulsions discrètes
en phase avec la rotation de l’échantillon, mais une unique impulsion modulée à la fréquence du rotor. Plusieurs
tentatives de modulation ont été faites (modulation de la phase, de l’amplitude, de la fréquence). Le SFAM
(Simultaneous Frequency and Amplitude Modulation) est une de ces techniques, née en 1997 dans le laboratoire de G.
Bodenhausen (Fu et al., 1997), et il s’avère que c’est une des plus efficaces . Le but de cette expérience est également
de faire interférer le moyennage dû au MAS avec l’impulsion modulée, afin de réintroduire artificiellement le couplage
dipolaire comme un déphasage de l’aimantation observée. La séquence d’impulsion est la suivante :
1H
I= C13
S= N15
découplageCP
ramp-CP
90
180
180
t2
SFAM SFAM
τsfam τsfam
amplitude RF
fréquence
SFAM :
Tr
figure 3.4 : la séquence d’impulsion du SFAM : après polarisation croisée du proton vers le carbone, la séquence de recouplages’effectue sur le noyau d’azote. Deux impulsions modulées en fréquence et amplitude sont appliquées symétriquement par rapport à
deux impulsions de 180° (refocalisation). Le proton est découplé à forte puissance durant toute l’expérience. La durée de déphasage,2τSFAM, est incrémentée par multiple de la période de rotation Tr.

30
L’impulsion de recouplage, notée SFAM sur le figure, est modulée sinusoïdalement en amplitude, et
cosinusoïdalement en fréquence. De cette manière, l’impulsion est de type ])sin[( 0 tSIS ωωω ∆+ . La porteuse de
pulsation ω0 est accordée à la fréquence de résonance de la raie irradiée. L’offset ∆ωS est lui même modulé en cosinus :
)cos( trS ωωω ∆=∆ et l’amplitude est modulée en sinus : )sin(max trISIS ωωω = , comme indiqué sur la figure 3.4.
Les fréquences de ces deux modulations sont égales à la fréquence de rotation rωπ /2 .
L’article original de Riqiang Fu montre comment cet artifice de modulation réintroduit la contribution du couplage
dipolaire. On ne rentrera pas dans les détails des calculs, et on ne parlera que du cas particulier où maxISωω =∆ , valeur
de quelques dizaines de kHz. Alors en décomposant la modulation en série de Fourier et en travaillant dans un
référentiel tilté propre à l’interaction, on peut monter que la pulsation de résonance associée au couplage dipolaire est
modifiée en étant multipliée par un facteur cos(ωrt). La valeur moyenne sur une période de rotation est alors non nulle,
comme dans le cas du REDOR, et introduit une composante de déphasage dépendante du temps de recouplage. En
portant la valeur de ∆S/S0 en fonction du temps de déphasage 2τSFAM , et en simulant l’expérience on en tire à nouveau
une valeur de couplage.
D. Comparaison des deux méthodes
Les avantages expérimentaux du SFAM sont a priori reliés aux inconvénients de la méthode de REDOR. On
l’approfondira dans la partie expérimentale, mais si l’on veut interférer avec une interaction périodique (la rotation de
l’échantillon), il semble a priori plus naturel d’appliquer une impulsion continue modulée à la même fréquence. Seuls
les progrès récents de l’électronique haute puissance permettent désormais de telles impulsions. Des méthodes telles
que le REDOR ou la méthode de Tycko du chapitre II, reposant sur l’application d’impulsions discrètes, ont le
désavantage de nécessiter des impulsions très courtes, et donc de très forte puissance. Si ce n’est pas le cas, si la rotation
est trop rapide ou si les impulsions ne sont pas assez courtes, alors la durée d’une impulsion rapportée à un tour de rotor
peut excéder 10%, ce qui n’est alors plus négligeable. Ces effets d’impulsions finies sont inexistants avec des séquences
continues comme le SFAM. Mais la difficulté de mise en œuvre expérimentale de telles séquences, surtout justifiée par
la nécessité d’une digitalisation suffisante de l’impulsion, explique le peu d’application du SFAM dans la littérature. Un
seul exemple d’application à un peptide membranaire a été publié depuis 1997 (Cotten et al., 1999), avec la contribution
de Riqiang Fu. La mesure est effectuée dans un dimère de gramicidine M, en membranes reconstituées et à la
température ambiante. Le SFAM s’avère alors apte à mesurer une constante d’une vingtaine de Hz, soit une distance de
4,5Å entre deux résidus spécifiquement marqués. Cela permet de trancher en faveur d’une association antiparallèle des
deux monomères.
La question de la sensibilité relative de ces deux méthodes a été peu abordée dans la littérature. Les constantes de
couplage que l’on peut espérer mesurer dans les deux cas sont d’une dizaine de Hz au minimum, ce qui correspond à
une distance maximale de l’ordre de 5Å entre carbone et azote, à 0,1Å près. Au delà de telles valeurs, les effets des
spins plus proches deviennent non négligeable, et la simulation doit introduire des corrections d’abondances naturelles
(Arshava et al., 1999). De telles mesures ont déjà été effectuées en REDOR, (Mueller et al., 1995), allant jusqu’à des
distances C-N de 6Å (Hing et al., 1994), mais jamais dans des conditions « biologiques » de peptides en membranes.
Par ailleurs, l’utilisation d’autres isotopes (avec d’autres rapports gyromagnétiques) permet d’accéder à d’autres ordres
de distances : les distances accessibles entre carbone-13 et fluor 19 sont de près de 13Å, par exemple. Néanmoins, il ne
faut pas perdre de vue que les distances carbone-azote de 3 à 4Å sont typiquement celles qui permettent d’affiner une
structure, comme on va le voir dans le paragraphe II.

31
II. Mise en évidence d’altérations structurales dans les hélices transmembranairesLes protéines membranaires sont au cœur des préoccupations de l’équipe de l’UPR 9052, que dirige Jean-Luc Popot.
Les méthodes d’étude physico-chimiques développées par l’équipe sont multiples, et même en RMN d’autres méthodes
sont mises en œuvre en parallèle, notamment au moyen de bicelles. La mesure de distances par recouplage dipolaire a
pour principal inconvénient sur les autres méthodes, notamment la cristallographie, de nécessiter un enrichissement
sélectif du peptide étudié. Quasiment autant de marquages sont nécessaires que l’on veut mesurer de distances. Pour
autant, dans une vaste gamme de distances, on peut effectuer une mesure à une très bonne précision. Il est des cas
cependant où quelques distances suffisent pour renforcer une hypothèse structurale.
Le modèle de repliement en deux étapes des protéines membranaires stipule que la formation des hélices est
découplée de l’association des domaines transmembranaires. Ce schéma est généralement accepté, et sa validité a été
démontrée expérimentalement (Borman & Engelman, 1992). Néanmoins, plusieurs études de la structure fine des
hélices transmembranaires tendent à montrer la possibilité de modulations structurales de type hernie-π, détectées au
sein de l’équipe par simulations numériques dans l’hélice transmembranaire d’une protéine appelée HER-2, sous la
forme de monomère et de dimère (sauvage et muté Glu) (Duneau et al., 1999). Ces déformations modifient fortement
l’empaquetage des résidus au voisinage des hélices, et sont observés dans certaines structures membranaires de
protéines connues. Récemment, la structure à haute résolution de la bactériorhodopsine confirme la présence de cette
déformation dans l’une des sept hélices, l’hélice G (Luecke et al., 1999).
figure 3.5 : simulations numériques du fragment de l’hélice transmembranaire du peptide HER-2. Les deux acides aminés engras sont la glycine-10 et l’isoleucine-5. En haut, la conformation suspectée en présence d’une hernie-pi ; en bas, les mêmes résidusen hélice-alpha. Le trait vert symbolise la distance de 4Å attendue entre carbonyle et azote dans une liaison hydrogène.En haut, cetteliaison se fait entre carbonyle de l’isoleucine-5 et NH de la glycine-10 (i-i+5) ; en bas entre le carbonyle de l’isoleucine-5 et le NH
de la glycine-9 (i-i+4). En bas, la distance attendue entre carbonyle de l’isoleucine-5 et azote de la glycine-10 devient 6Å.
(π)(π)
(α)(α)
N-term
C-term

32
La protéine HER-2 est un récepteur à activité tyrosine kinase, impliqué dans de nombreux cancers. Sa dimérisation
serait modulée par la hernie-π. En l’absence de structure 3D disponible pour le segment transmembranaire de HER-2,
l’existence expérimentale éventuelle d’une telle déformation, avec ses conséquences sur l’interface, est encore à
prouver. Or une mesure précise de distance permet de différencier les structures. Comme tendent à le prouver les
résultats de simulation numérique, certains résidus entre 5 et 15 s’associent en formant des liaisons hydrogène entre i
et i+5, et non plus entre i et i+4 (figure 3.5). On a donc choisi de travailler sur un segment transmembranaire
doublement marqué : la glycine-10 est doublement marquée 13Cα et 15N, tandis que l’isoleucine-5 est marquée 13CO. En
REDOR déphasé 15N – observé 13C, on a donc deux signaux du carbone, l’un correspondant au carbone α, à une
distance d’1,5Å de l’azote environ, l’autre correspondant au carbonyle. Les déplacements chimiques sont suffisamment
éloignés pour résoudre les deux pics. Quelque soit la structure, le couplage du Cα reste le même, et son déphasage
REDOR ou SFAM peut ainsi nous servir de référence. Par contre, dans l’hypothèse d’une hernie-π, le couplage 13CO-15N vaut une cinquantaine de Hz, alors que dans une hélice parfaite il est d’une quinzaine de Hz. On peut s’attendre à
voir un déphasage mesurable dans le premier cas, et pas dans le second…
De tels raisonnements ont conditionné la stratégie d’approche du problème. Il est envisagé de renforcer les résultats
en mesurant le couplage entre les deux carbones-13, par Résonance Rotationnelle par exemple. Le segment
transmembranaire (un 30-mère) est actuellement en cours de synthèse au sein du laboratoire partenaire de Pierre Hubert
(INSERM U338, Strasbourg), et sera prêt d’ici le mois de novembre. Après de premières mesures en poudre
lyophilisée, les techniques de réincorporation en membranes lipidiques reconstituées, développées au laboratoire,
permettront d’obtenir un échantillon en conditions biologiques.
Avant d’entamer une étude sur un produit biologique avec une méthode inédite, il convenait tout d’abord, pour en
optimiser les multiples paramètres, de mettre au point la méthode sur un échantillon simple.
III. Mise au point sur un échantillon d’alanine doublement marquée
A. Les nécessités techniques et matérielles
Les expériences de RMN sont réalisées sur le même spectromètre 400MHz que lors du DEA, et décrit au chapitre 2
paragraphe III. La principale différence est qu’il faut désormais l’équiper d’une sonde-MAS à trois canaux, un canal
proton et deux canaux accordables X et Y. Les rotors ont un diamètre de 4 mm, comme sur la sonde double. Toutes les
études ont été réalisées à une fréquence de rotation à l’angle magique de 5kHz et à la température ambiante.
Par ailleurs, les séquences développées ici nécessitent des puissances d’impulsion supérieures à la séquence de
Tyckob. Il faut travailler dans les zones de forte puissance des amplificateurs, sur les canaux X et Y en tout cas, et ce
afin d’avoir des impulsions les plus courtes possibles. Dans ces zones, les amplificateurs ne sont souvent pas linéaires.
Enfin, afin de préserver la pureté du signal en isolant chacun des circuits, et aussi afin de préserver l’électronique de la
sonde, sur chaque canal est branché un filtre passe-bande haute puissance en série, accordé à la fréquence du noyau
considéré. Seul le canal X, qui est détecté, passe par le préamplificateur. Outre le fait que des expériences telles que le
REDOR ou le SFAM n’ont jamais été menées en France sur des échantillons biologiques, il faut noter qu’elles ont
toujours été développées sur des spectromètres non-commerciaux, où l’électronique haute puissance, développée par le
laboratoire, supporte des courants plus intenses. Le premier objectif était donc conjointement de tester la faisabilité de
ces méthodes en termes de mesures de distances autant qu’en termes de puissance supportée. Il fallait pour cela un
échantillon simple, bien décrit structurellement, enrichi à 100% et concentré dans le rotor, afin d’obtenir des spectres
b Par la suite, on parle improprement de puissance d’impulsion en Hz. Cela correspond en fait à la fréquence de nutationdu spin sous l’effet de l’impulsion ; si τ est la durée d’une impulsion 180°, la puissance en Hz sera égale à 1/(2τ)

33
très rapidement. Notre choix s’est porté vers l’alanine marquée spécifiquement (13C2, 15N), donc sur le carbone alpha et
sur l’azote. On rappelle que la formule de l’alanine est : H2N*-C*H(CH3)-COOH (les étoiles symbolisent les atomes
marqués isotopiquement). Cet acide aminé en poudre fait parti des échantillons standards utilisés dans les premières
expériences de REDOR ou de SFAM. Les deux atomes marqués sont distants de 1,50Å , et donc couplés dipolairement
avec une constante de 907Hz.
B. Les premières optimisations
Les deux séquences que l’on se propose d’appliquer ont deux étapes communes : un transfert de polarisation du
proton vers le carbone (marqué CP sur les séquences 3.1 et 3.4), et un découplage du proton durant le déphasage et
l’acquisition. Un premier travail a consisté à l’optimisation de ces deux parties de la séquence, en utilisant à chaque fois
les techniques décrites dans la littérature comme étant les plus efficaces.
La polarisation croisée proton-carbone se fait à très forte puissance comparée à la RMN liquide, optimisée de l’ordre
de 50kHz sur les deux canaux. Une durée typique est de l’ordre quelques millisecondes. On travaille avec une durée de
2,5 ms par la suite. Pour optimiser cette étape, il a fallu construire des profils de polarisation croisée typiques des
solides (« finger pattern »). On renverra le lecteur à la revue sur le CP-MAS dans l’Encyclopédie de la RMN (Burum,
1993). On a choisi d’utiliser une version plus récente que le simple CP-MAS, le RAMP-CP (Metz et al., 1994).
L’amplitude de l’impulsion longue varie selon une rampe linéaire. Ainsi, le profil d’irradiation s’en trouve amélioré, et
le transfert est plus efficace. On a mesuré que l’utilisation d’une rampe sur le carbone entre 95% et 105% de l’amplitude
moyenne amélioré le signal d’un facteur 1,1.
Les techniques de découplage hétéronucléaire en solide sont multiples, et leurs fondements théoriques pas encore
bien compris. Le très fort couplage proton-proton étant homogène, la rotation de l’échantillon à des vitesses courantes
ne suffit pas à le moyenner. Par ailleurs, ce couplage entre protons rend difficile le moyennage entre protons et carbone,
tous les protons étant couplés entre eux. On a recours, en plus de la rotation, à des techniques de variation d’amplitude
ou de phase. Le TPPM (Two Pulses Phase Modulated) est une de ces techniques, peut-être la plus employée à l’heure
actuelle en solide. Inventée au sein de l’équipe de Griffin (Bennett et al., 1995), elle repose sur l’application continue en
proton d’une succession d’impulsions de 90° de phases opposées. Le découplage utilisé dans toutes les expériences de
REDOR ou de SFAM est de ce type, avec une phase optimum de 15°. La puissance appliquée sur le canal proton est de
60kHz. Sur un spectromètre commercial, on ne peut pas travailler à une puissance continue de 100kHz pendant près de
5ms sans faire prendre de risque à la sonde. L’utilisation du TPPM est une alternative intéressante lors des découplages
intenses.
C. Le REDOR
Expérimentalement, l’enregistrement de spectres de REDOR passe par une acquisition en carbone à chaque
incrémentation de la durée de déphasage. On doit donc programmer l’augmentation du nombre d’impulsions de 180°
appliquées sur l’azote, assortie d’une acquisition à chaque valeur du temps de déphasage. A une vitesse de rotation de
5kHz, le temps de déphasage est un multiple de 200µs. Une courbe REDOR complète est reconstruite pour un produit
(temps de déphasage) x (constante de couplage) de l’ordre de 4 ou 5. Notre alanine nous permet donc d’aller jusqu’à un
temps de déphasage de près de 5ms, ce qui est une durée de découplage en proton tout à fait supportable. Pour des
distances à mesurer plus grandes, ce temps-là augmente, et il faudra se contenter de moins de points pour éviter de
découpler trop longtemps.
Les spectres enregistrés se présentent comme sur le figure 3.6. Il s’agit d’un enregistrement à deux dimensions,
correspondant à la dernière version de REDOR programmée selon la séquence 3.1 : chaque signal est stocké dans la
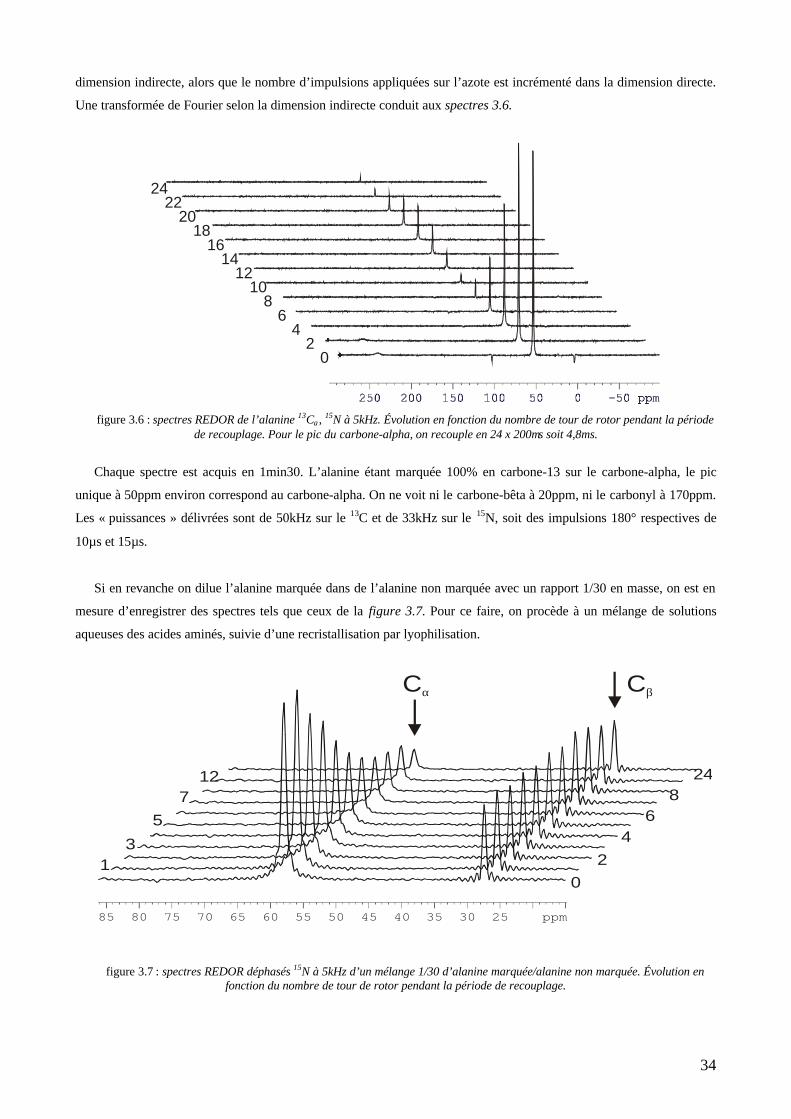
34
dimension indirecte, alors que le nombre d’impulsions appliquées sur l’azote est incrémenté dans la dimension directe.
Une transformée de Fourier selon la dimension indirecte conduit aux spectres 3.6.
02
46
810
1214
1618
2022
24
figure 3.6 : spectres REDOR de l’alanine 13Cα, 15N à 5kHz. Évolution en fonction du nombre de tour de rotor pendant la périodede recouplage. Pour le pic du carbone-alpha, on recouple en 24 x 200µs soit 4,8ms.
Chaque spectre est acquis en 1min30. L’alanine étant marquée 100% en carbone-13 sur le carbone-alpha, le pic
unique à 50ppm environ correspond au carbone-alpha. On ne voit ni le carbone-bêta à 20ppm, ni le carbonyl à 170ppm.
Les « puissances » délivrées sont de 50kHz sur le 13C et de 33kHz sur le 15N, soit des impulsions 180° respectives de
10µs et 15µs.
Si en revanche on dilue l’alanine marquée dans de l’alanine non marquée avec un rapport 1/30 en masse, on est en
mesure d’enregistrer des spectres tels que ceux de la figure 3.7. Pour ce faire, on procède à un mélange de solutions
aqueuses des acides aminés, suivie d’une recristallisation par lyophilisation.
25303540455055606570758085 ppm
0
86
4
2
24
1
127
5
3
Cα Cβ
figure 3.7 : spectres REDOR déphasés 15N à 5kHz d’un mélange 1/30 d’alanine marquée/alanine non marquée. Évolution enfonction du nombre de tour de rotor pendant la période de recouplage.

35
On ne présente que les évolutions comparées des pics des Cα et Cβ. Du fait de la dilution isotopique, chaque spectre
est désormais acquis en 12min. Cela permet aux résonances des carbones non enrichis d’apparaître. Mais la probabilité
de présence d’un couple 13Cβ−15N étant trop faible, aucun recouplage n’est observé pour le pic du carbone-bêta. Une
telle expérience nous place artificiellement dans des conditions similaires à celles d’un peptide doublement marqué.
HER-2 est précisément un 30-mère, et l’acquisition d’un spectre 13C nécessitera des temps longs. On voit aussi l’intérêt
de choisir des résonances suffisamment différentes entre elles.
Un dernière étape à notre expérience est de déduire à partir des spectres des points expérimentaux sur la courbe
∆S/S0=f(λD) aussi reproductibles que possible. Il faut tout d’abord enregistrer des spectres identiques à ceux présentés,
mais où l’on n’applique pas d’impulsions de déphasage sur l’azote (mesure de S0). Par ailleurs, les signaux doivent être
lus sur chaque spectre avec les mêmes paramètres. Il a fallu pour cela écrire une procédure d’automatisation des lectures
des signaux dans chaque spectre REDOR. Selon que l’on travaille avec les intensités de pics ou les aires sous les pics, il
est désormais possible de reporter d’un spectre à l’autre les paramètres décidés sur le premier de ces spectres.
De nombreuses variantes du REDOR ont été testées expérimentalement. On a notamment enregistré des spectres de
l’azote déphasés par le carbone (symétriques aux précédents, mais avec X=I=15N et Y=S=13C). L’azote étant un noyau
moins sensible que le carbone, il s’avère que la précision de mesure du signal est moins bonne. La littérature fait état de
ces difficultés, puisqu’il est admis qu’en observation 13C on peut accéder à des distances de 6Å, contre seulement 5Å en
observation 15N (Arshava et al., 1999). Pour cette raison, ces résultats ne seront pas discutés davantage.
D. Le SFAM
La version de SFAM mise en œuvre est celle présentée en figure 3.4. La durée totale entre le CP et l’acquisition en
carbone est de 10ms, avec un découplage à 60kHz en proton et un transfert de polarisation identiques à ceux du
REDOR. Cela laisse donc 5ms de part et d’autre des deux impulsions 180° pendant lesquels on applique une impulsion
SFAM modulée sur l’azote, de durée variable. D’une expérience à l’autre, cette durée est incrémentée par unité de
200µs (la rotation se fait à 5kHz), de 0 à 4ms (20 pas). Là encore, un programme de construction bidimensionnelle a été
écrit, avec la difficulté supplémentaire qu’à chaque incrément de temps, il faut recalculer l’impulsion modulée (le
« shape pulse »). Cela est réalisé au moyen d’une routine écrite en C qui crée à chaque pas une nouvelle impulsion.
Chaque impulsion est décrite dans le spectromètre avec un nombre de points croissant, et une digitalisation de 200ns (le
plus petit incrément de temps possible). Par ailleurs, on n’applique pas réellement une impulsion modulée en amplitude
et en fréquence, mais en amplitude et en phase (la variation d’offset cosinusoïdale est convertie en variation de phase
pour des raisons expérimentales). La version la plus récente du programme SFAM écrite s’avère capable de remplir
toutes ces conditions. Nous ne sommes néanmoins pas en mesure de présenter des spectres reproductibles de recouplage
en SFAM sur un échantillon d’alanine.
Si l’on pensait a priori que le canal le plus « fragile » serait le canal proton, qui supporte un découplage assez
intense durant près d’une dizaine de millisecondes, il n’en a rien été. Il s’avère que le canal Y, accordé dans nos
expériences à 15N, est le plus limitant. L’impulsion la plus courte délivrée à haute puissance est de l’ordre de 15µs. On
en applique deux par tour de rotor en REDOR, soit 30/200=15% du temps, ce qui n’est pas négligeable. Le SFAM
paraissait être une alternative, mais une partie de l’électronique 15N ne supporte pas de puissance supérieure à 35kHz
appliquée en continue pendant une durée de quelques millisecondes. Passé un seuil, l’électronique « se met dans le
rouge »et un voyant de surpuissance s’allume. On est alors pourtant en deçà des spécifications de la sonde. Après une
longue analyse des branchements, il semble que le problème vienne du filtre d’isolation du canal 15N, qui réfléchit une
part trop importante du signal-aller. C’est néanmoins une nouvelle encourageante, puisqu’un filtre plus performant doit
nous être très bientôt mis à disposition. En attendant, les spectres réalisés les jours où la haute puissance était supportée

36
par les appareils montrent un recouplage qui semble comparable au REDOR. Ces points expérimentaux n’ont cependant
pas encore été simulés, contrairement aux données REDOR.
IV. Simulation des résultatsLa simulation numérique en RMN a deux vocations : elle permet de concevoir de nouvelles séquences en
modélisant la manipulation des spins que l’on peut écrire sur le papier ; et elle permet d’extraire des informations
cachées derrière des données expérimentales, en modélisant l’expérience et en ajustant le paramètre cherché. C’est dans
cette deuxième optique que nous utilisons la simulation.
S’agissant de RMN des solides, la simulation est confrontée à des problèmes spécifiques que ne connaît pas la
simulation en RMN des liquides. Dans les deux cas, la simulation vise à évaluer numériquement la solution de
l’équation de Liouville-von Neumann :
)](),([)( ttitdtd
ρρ Η−=
où ρ(t) est l’opérateur densité décrivant l’état du système et H(t) l’Hamiltonien dépendant du temps.
On néglige pour cela toute considération de relaxation du système, et on résout cette équation en faisant apparaître un
propagateur U(t), tel que )()0()()( tUtUt += ρρ . Ce propagateur se calcule au moyen d’une intégrale, et une
méthode simple de calcul consiste à considérer l’Hamiltonien constant sur des intervalles de temps infinitésimaux. Par
ailleurs l’Hamiltonien, qui fait intervenir les contributions de toutes les interactions présentes (champ rf, déplacement
chimique isotrope et anisotrope, couplage scalaire, interaction dipolaire voire quadripolaire) est périodique dans le
temps pour des raisons de rotation de l’échantillon dans le rotor. Donc en RMN des solides, il est uniquement nécessaire
de calculer l’Hamiltonien sur une période de rotation du rotor. Néanmoins, ce calcul est dépendant du cristallite (de
l’orientation que l’on donne a priori aux vecteurs qui caractérisent notre spin). La majeure partie du temps de calcul est
donc utilisée à sommer toutes les orientations de spins possibles, de façon à refléter l’isotropie de la poudre. Des
méthodes de calcul ont été développées pour effectuer plus complètement ce que l’on appèle le moyennage de poudre.
Ces méthodes ne sont pas intégrées au logiciel standard C++ de simulation de la RMN des liquides, GAMMA
(http://gamma.magnet.fsu.edu/) . C’est la raison principale de notre utilisation d’un autre logiciel de
simulation très récent, SIMPSON (http://nmr.imsb.au.dk/simpson), développé au sein de l’équipe de Niels
Nielsen. Cette plate-forme de programmation en C et Tcl permet d’avoir recours à de nombreuses méthodes de
moyennage de poudre, sur un nombre variable de cristallites.
Nous avons programmé numériquement le REDOR en utilisant les paramètres de l’expérience, à savoir une rotation
à 5kHz, des puissances respectives de 50 kHz et 33 kHz sur 13C et 15N. On n’a utilisé que deux spins, un 13C de CSA
100ppm et résonnant à 50ppm, et un 15N couplé dipolairement avec une constante ajustable. On suppose dans un
premier temps le transfert de polarisation parfait, et tous les protons découplés. On utilise 320 cristallites moyennés
selon la méthode REPULSION (Bak & Nielsen, 1997), et 18 angles gamma. Le temps de calcul est de trois minutes sur
un Pentium III 500.
La figure 3.8 reproduit les résultats de telles simulations menées à différentes constantes de couplage dipolaire, pour
des distances carbone-azote autour de 1,50Å. Les courbes simulées sont portées en valeur absolue. Elles sont construites
par segment entre chacun des points simulé, les points étant nécessairement distants d’un tour de rotor (l’étape suivante
sera de tracer les courbes mathématiquement au moyen de leur expression analytique, afin de lisser chaque courbe). Les
points expérimentaux sont extraits de spectres identiques aux spectres 3.6, selon la méthode développée au paragraphe
III C, et par des mesures d’intégrales sous les pics.
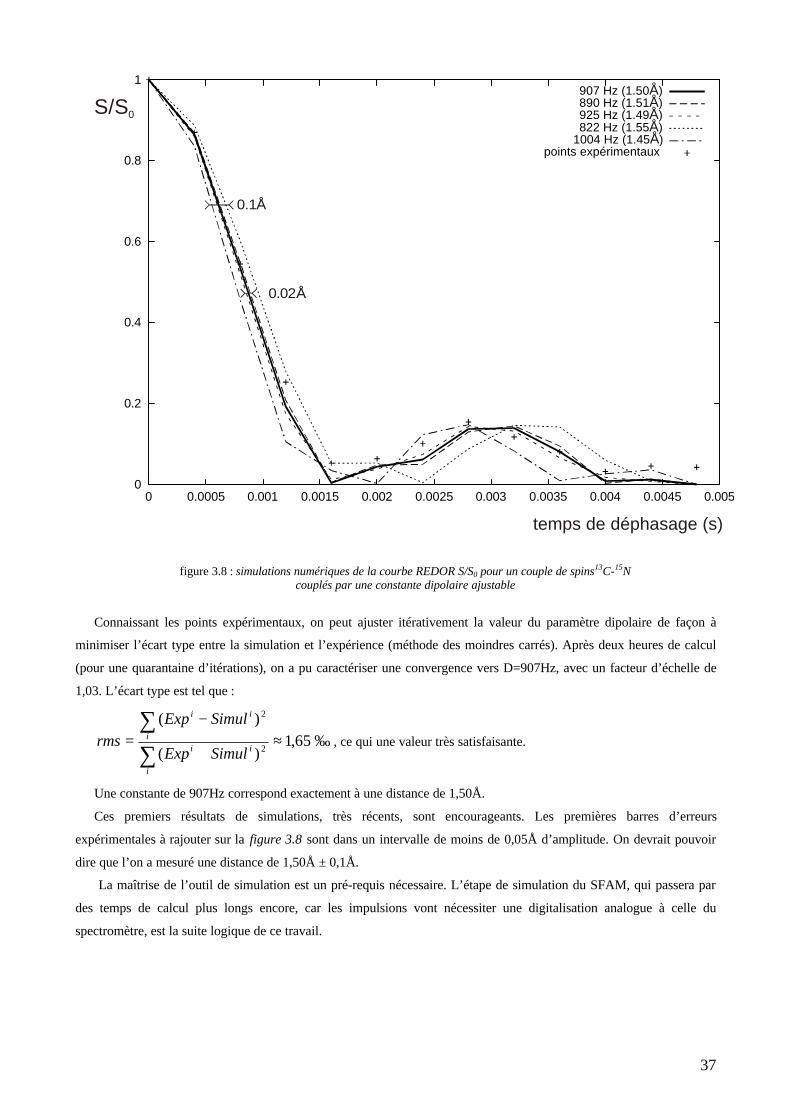
37
0
0.2
0.4
0.6
0.8
1
0 0.0005 0.001 0.0015 0.002 0.0025 0.003 0.0035 0.004 0.0045 0.005
S/S0
temps de déphasage (s)
907 Hz (1.50 )Å890 Hz (1.51 )Å925 Hz (1.49 )Å822 Hz (1.55 )Å
1004 Hz (1.45 )Åpoints expérimentaux
0.1Å
0.02Å
figure 3.8 : simulations numériques de la courbe REDOR S/S0 pour un couple de spins13C-15Ncouplés par une constante dipolaire ajustable
Connaissant les points expérimentaux, on peut ajuster itérativement la valeur du paramètre dipolaire de façon à
minimiser l’écart type entre la simulation et l’expérience (méthode des moindres carrés). Après deux heures de calcul
(pour une quarantaine d’itérations), on a pu caractériser une convergence vers D=907Hz, avec un facteur d’échelle de
1,03. L’écart type est tel que :
‰65,1)(
)(
2
2
≈+
−=
∑∑
i
iii
ii
SimulExp
SimulExprms , ce qui une valeur très satisfaisante.
Une constante de 907Hz correspond exactement à une distance de 1,50Å.
Ces premiers résultats de simulations, très récents, sont encourageants. Les premières barres d’erreurs
expérimentales à rajouter sur la figure 3.8 sont dans un intervalle de moins de 0,05Å d’amplitude. On devrait pouvoir
dire que l’on a mesuré une distance de 1,50Å ± 0,1Å.
La maîtrise de l’outil de simulation est un pré-requis nécessaire. L’étape de simulation du SFAM, qui passera par
des temps de calcul plus longs encore, car les impulsions vont nécessiter une digitalisation analogue à celle du
spectromètre, est la suite logique de ce travail.

38
Conclusion et perspectives
l me paraissait nécessaire de rappeler longuement le contenu des expériences de mon DEA. Ces
expériences ont continué de m’occuper au début de ma thèse, et devraient prochainement faire naître un
article. Par ailleurs, même si le sujet de ma thèse n’est pas dans le continuité directe du DEA, les techniques de
RMN en sont un prolongement naturel.
Le travail de DEA s’inscrit dans une problématique plus vaste que celle du polymorphisme lipidique. On
continue de penser que le recouplage pourrait s’avérer un outil intéressant pour mettre en évidence l’existence de
zones rigides au sein des membranes biologiques. La conception de la membrane en terme de « mosaïque
fluide »due à Singer et Nicholson (1972) a ainsi récemment évoluée au profit d’une interprétation en termes de
« radeaux » (« rafts ») rigides associant des sphingolipides et du cholestérol, et qui « flotteraient » au sein de la
bicouche fluide (Simons & Ikonen, 1997). Ces domaines rigides se formeraient préférentiellement à proximité de
protéines spécifiques (Denisov et al., 1998). S’ils impliquent majoritairement un certain lipide, on peut espérer
mettre en évidence sa rigidité sur des spectres de recouplage tels que ceux montrés au chapitre 2.
L’autre volet, plus récent, d’application des méthodes de recouplage en RMN solide, concerne la mesure de
constantes de couplage dipolaire entre atomes de carbone-13 et d’azote-15 introduits sélectivement au sein d’une
protéine membranaire. Cette année passée aura surtout permis de définir la stratégie d’étude afin de vérifier une
hypothèse, celle de l’existence des hernies-pi. Elle aura également été l’occasion de mettre en œuvre les
techniques d’étude sur un composé simple, afin d’en optimiser les multiples paramètres. Les premiers résultats
sont encourageants, et les difficultés d’ordre expérimental sont en passe d’être résolues. Les calculs de
simulations sont poursuivis, en attendant de disposer de l’échantillon biologique souhaité.
I

39
Références
Chapitre 1
Cevc G. (1993) « Phospholipids handbook », Marcel Dekker, inc
Heller H., Schaefer M. & Schulten K. (1993) « Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid-crystal phases », J. Phys. Chem. 97 : 8343-8363
MacKenzie K. R., Prestegard J. H., & Engelman D. M. (1997) Science 276 : 131-133
Popot J.-L. & Engelman D. M. (2000) « Helical membrane protein folding, stability, and evolution », Annu. Rev. Biochem. 69 :881-922
Schechter E. (1993) « Biochimie et biophysique des membranes - Aspects structuraux et fonctionnels », Masson
Schmidt-Rohr K. & Spiess H. W. (1994) « Multidimensional solid-state NMR and polymers », Academic Press
Chapitre 2
Alla M.A., Kundla E.I. & Lippmaa E.T. (1978) « Selective determination of anisotropic interactions from powders », JETP Lett. 27 :94
Bax A, Szeverenyi N. M. & Maciel G. E. (1983) J. Magn. Reson. 43 : 400
Berthelot L., Warschawski D. E. & Devaux P. (2000) « 31P NMR in lipid membranes. CSA recoupling » poster session, AlpineConference on Solid State NMR, Chamonix Mt-Blanc
Cullis P. R. & de Kruyff B. (1976) « 31P NMR studies of unsonicated aqueous dispersions of neutral and acidic phospholipids.Effects of phase transitions, p2H and divalent cations on the motion in the phosphate region of the polar headgroup », Biochim.Biophys. Acta 436 : 523-540
Dufourc E. J. (1986) « La résonance magnétique nucléaire du deutérium et du phosphore dans les milieux organisés. Théorie etapplications à l’étude des interactions lipides-protéines dans les membranes biologiques », thèse de doctorat es sciences del’Université de Bordeaux I
Griffin R. G. (1998) « Dipolar recoupling in MAS spectra of biological solids », Nature Struct. Biol.-NMR supplement july 1998 :508-512
Gross J. D., Warschawski D. E. & Griffin R. G. (1997) « Dipolar recoupling in MAS NMR : a probe for segmental order in lipidsbilayers », J. Am. Chem. Soc. 119 : 796-802
Haeberlen U. & Waugh J. S. (1968) « Coherent averaging effects in magnetic resonance », Phys. Rev. 175 : 453-467
Ishii Y. & Terao T. (1998) « Manipulation of nuclear spin hamiltoniens by rf-field modulations and its applications to observation ofpowder patterns under magic-angle spinning », J. Chem. Phys. 109 :1366-1374
Maricq M. M. & Waugh J. S. (1979) « NMR of rotating solids », J. Chem. Phys. 70 : 3300-3316
Moran L. & Janes N. (1998) « Tracking phospholipid populations in polymorphism by sideband analyses of 31P magic anglespinning NMR », Biophys. J. 75 : 867-879
Schmidt-Rohr K. & Spiess H. W. (1994) « Multidimensional solid-state NMR and polymers », Academic Press
Seelig J. (1978) « 31P nuclear magnetic resonance and the head group structure of phospholipids in membranes », Biochim. Biophys.Acta 515 : 105-140
Tilcock C. P. S., Bally M. B., Farren S. B. & Cullis P. R. (1982) « Influence of cholesterol on the structural preference ofdioleylphosphatidylethanolamine-dioleylphosphatidylcholine systems : a phosphorus-31 and deuterium nuclear magnetic resonancestudy », Biochemistry 21 : 4596-4601
Tycko R., Dabbagh G. & Mirau P. A. (1989) « Determination of chemical-shift-anisotropy lineshapes in a two dimensional magic-angle-spinning NMR experiment », J. Magn. Reson. 85 : 265-274

40
Warschawski D. E. (1995) « Membranes biologiques, membranes modèles : nouvelles approches utilisant la RMN solide en rotationà l’angle magique », thèse de doctorat en physique de l’Université Paris 7
Warschawski D. E., Traikia M., Devaux P. F. & Bodenhausen G. (1998) « Solid-state NMR of membrane systems : the use ofanisotropic interactions », Biochimie 80 : 437-450
Warschawski D. E., Berthelot L. & Devaux P. (2000) « 31P CSA recoupling in lipids » poster session XIX ICMRBS, Florence, Italy
Yarim-Agaev Y., Titunjian P. N. & Waugh J. S. (1982) J. Magn. Reson. 47 : 51
Yeagle P. L. (1993) « Membranes : phosphorus-31 NMR », Encyclodedia of Nuclear Magnetic Resonance, John Wiley & Sonseditor, 5 : 3015-3022
Chapitre 3
Arshava B., Breslav M. Antohi O., Stark R. E., Garbow J. R., Becker J. M & Naider F. (1999) « Long-distance rotational echodouble resonance measurements for the determination of secondary structure and conformational heterogeneity in peptides » SolidState NMR 14 : 117-136
Bak M. & Nielsen N. (1997) « REPULSION : a novel approach to efficient powder averaging in solid-state NMR » J. Magn. Reson.125 : 132-139
Bennett A. E., Griffin R. G., & Vega S. (1994) « Recoupling of homo- and heteronuclear dipolar interactions in rotating solids »NMR Basic Principles and Progress 33 : 1-77
Bennett A. E., Rienstra C., Auger M., Lakshmi K. V. & Griffin R. G. (1995) « Heteronuclear decoupling in rotating solids » J.Chem. Phys. 103 : 6951-6958
Borman B. J. & Engelman D. M. (1992) Annu. Rev. Biophys. Biomol. Struct. 21 : 232-242
Burum D.G. « Cross polarizationin solids » Encyclodedia of Nuclear Magnetic Resonance, John Wiley & Sons editor, 3 : 1535-1542
Cotten M. , Fu R. &Cross T. A. (1999) « Solid state NMR and hydrogen-deuterium exchange in a bilayer-solubilized peptide :structural and mechanistic implications » Biophys. J. 76 : 1179-1189
Davis J. H. & Auger M. (1999) « Static and magic angle spinning NMR of membrane peptides and proteins » Prog. NMR Spect.35 :1-84
Duneau J. P., Crouzy S., Garnier N., Chapron Y. & Genest M. (1999) « Molecular dynamics simulations of the ErbB-2transmembrane domain within an explicit domain environment. Comparison with vacuum simulations » Biophys. Chem. 76 : 35-53
Evans J. N. S. (1995) « Biomolecular NMR spectroscopy », Oxford University Press
Fu R., Smith S. A. & Bodenhausen G. (1997) « Recoupling of heteronuclear dipolar interactions in solid state magic-angle spinningNMR by simultaneous frequency anf amplitude modulation » Chem. Phys. Lett. 272 : 361-369
Gullion T. & Schaefer J. (1989) « Rotational-echo double resonance NMR » J. Magn. Res. 81 : 196-200
Gullion T. & Schaefer J. (1989) « Detection of weak heteronuclear dipolar coupling by rotational echo double resonance nuclearmagnetic resonance » Advances in Magnetic Resonance 13 : 57-83
Hing A. W., Tjandra N., Cottam P. F., Schaefer J. & Ho C. (1994) « An investigation of the ligand-binding site of the glutamine-binding protein of Escherichia Coli using rotational-echo double resonance NMR » Biochemistry 33 : 8651-8661
Levitt M. H., Raleigh D. P., Creuzet F. & Griffin R. G. (1990) J. Chem. Phys. 92 : 6347-6364
Luecke H., Schobert B., Richter H. T., Cartailler J. P. & Lanyi J. K. (1999) J. Mol. Biol. 291 : 899-911
MacKenzie K. R., Prestegard J. H., & Engelman D. M. (1997) Science 276 : 131-133
Maricq M. M. & Waugh J. S. (1979) « NMR of rotating solids », J. Chem. Phys. 70 : 3300-3316
Metz G., Wu X. & Smith S. O. (1994) « Ramped-amplitude cross polarization in magic-angle spinning NMR » J. Magn. Reson.Series A 110 : 219-227
Mueller D. D., Schmidt A., Pappan K. L., McKay R. A. & Schaefer J. (1995) « Activator carbamino carbon to inhibitor phosphorusinternuclear distances in ribulose-1,5-bisphosphate carboxylase/oxygenase. A solid state NMR study » Biochemistry 34 : 5597-5603

41
Conclusion
Denisov G., Wanaski S., Luan P. Glaser M.& McLaughlin S. (1998) « Binding of basic peptides to membranes produces lateraldomains enriched in the acidic lipids phosphatidylserine and phosphatidylinositol 4,5-bisphosphate : an electrostatic model andexperimental results » Biophys. J. 74 : 731-744
Simons K. & Ikonen E. (1997) « Functionnal rafts in cell membranes » Nature 385 : 569-572