Khadidja Sidelarbi Implication du chaperome de la protéine ...
226
THÈSE Pour l'obtention du grade de DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées Laboratoire Signalisation et transports ioniques membranaires - STIM (Poitiers) (Diplôme National - Arrêté du 25 mai 2016) École doctorale : Biologie-santé - Bio-santé (Limoges) Secteur de recherche : Biomolécules, pharmacologie, thérapeutique Présentée par : Khadidja Sidelarbi Implication du chaperome de la protéine F508del-CFTR dans son transport intracellulaire et/ou sa dégradation : rôle des lectines EDEMs et la mannosidase du RE Directeur(s) de Thèse : Frédéric Becq, Caroline Norez Soutenue le 24 novembre 2017 devant le jury Jury : Président Thierry Bergès Professeur, Université de Poitiers Rapporteur Valérie Chappe Professeur associé, Université de Dalhousie, Halifax, Canada Rapporteur Thomas Falguières Chargé de recherche INSERM, Saint-Antoine, Paris Membre Frédéric Becq Professeur, STIM, Université de Poitiers Membre Caroline Norez Maître de conférences, STIM, Université de Poitiers Membre Christelle Coraux Chargé de recherche INSERM, Université de Reims Pour citer cette thèse : Khadidja Sidelarbi. Implication du chaperome de la protéine F508del-CFTR dans son transport intracellulaire et/ou sa dégradation : rôle des lectines EDEMs et la mannosidase du RE [En ligne]. Thèse Biomolécules, pharmacologie, thérapeutique. Poitiers : Université de Poitiers, 2017. Disponible sur Internet <http://theses.univ-poitiers.fr>
Transcript of Khadidja Sidelarbi Implication du chaperome de la protéine ...

THÈSE
Pour l'obtention du grade deDOCTEUR DE L'UNIVERSITÉ DE POITIERS
UFR des sciences fondamentales et appliquéesLaboratoire Signalisation et transports ioniques membranaires - STIM (Poitiers)
(Diplôme National - Arrêté du 25 mai 2016)
École doctorale : Biologie-santé - Bio-santé (Limoges)Secteur de recherche : Biomolécules, pharmacologie, thérapeutique
Présentée par :Khadidja Sidelarbi
Implication du chaperome de la protéine F508del-CFTR dansson transport intracellulaire et/ou sa dégradation : rôle des
lectines EDEMs et la mannosidase du RE
Directeur(s) de Thèse :Frédéric Becq, Caroline Norez
Soutenue le 24 novembre 2017 devant le jury
Jury :
Président Thierry Bergès Professeur, Université de Poitiers
Rapporteur Valérie Chappe Professeur associé, Université de Dalhousie, Halifax, Canada
Rapporteur Thomas Falguières Chargé de recherche INSERM, Saint-Antoine, Paris
Membre Frédéric Becq Professeur, STIM, Université de Poitiers
Membre Caroline Norez Maître de conférences, STIM, Université de Poitiers
Membre Christelle Coraux Chargé de recherche INSERM, Université de Reims
Pour citer cette thèse :Khadidja Sidelarbi. Implication du chaperome de la protéine F508del-CFTR dans son transport intracellulaire et/ousa dégradation : rôle des lectines EDEMs et la mannosidase du RE [En ligne]. Thèse Biomolécules, pharmacologie,thérapeutique. Poitiers : Université de Poitiers, 2017. Disponible sur Internet <http://theses.univ-poitiers.fr>

THESE
Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées) (Diplôme National - Arrêté du 25 mai 2016)
Ecole Doctorale : Biosanté
Secteur de Recherche : Biomolécules, pharmacologie, thérapeutique
Présentée par :
Khadidja SIDELARBI
************************
Implication du chaperome de la protéine F508del-CFTR dans son transport
intracellulaire et/ou sa dégradation : rôle des lectines EDEMs et de la mannosidase du RE
************************
Directeur de Thèse : Pr. Frédéric BECQ et Dr. Caroline NOREZ
************************
Soutenue le 24 novembre 2017 devant la Commission d’Examen
************************
JURY
Valérie Chappe Professeur associé, Université de Dalhousie, Halifax Rapporteur Thomas Falguières Chargé de recherche INSERM, Saint-Antoine, Paris Rapporteur Christelle Coraux Chargé de recherche INSERM, Université de Reims Examinateur Thierry Berges Professeur des Universités, Université de Poitiers Examinateur Caroline Norez Maître de Conférences, Université de Poitiers Examinateur Frédéric Becq Professeur des Universités, Université de Poitiers Examinateur

Sommaire
1
Sommaire Sommaire .................................................................................................................................. 1
Abréviations .............................................................................................................................. 4
Liste des Figures ....................................................................................................................... 6
Liste des tableaux ..................................................................................................................... 9
Introduction ............................................................................................................................ 10
I. Le contrôle qualité des glycoprotéines ........................................................................ 10
A. La voie ERAF ................................................................................................................. 11
1) Les modifications co- et post-traductionnelles .......................................................... 13
2) Les interactions avec les protéines chaperonnes ........................................................ 18
B. La voie ERAD ................................................................................................................ 33
1) ERMnI et son rôle dans ERAD .................................................................................. 40
2) EDEMs et leurs rôles dans ERAD ............................................................................. 43
3) Régulation des régulateurs d’ERAD « ERAD tuning » ............................................. 45
4) Réponse à l’accumulation des protéines mal repliées UPR ....................................... 47
II. Exemple du contrôle qualité : la protéine CFTR vs F508del-CFTR ........................ 49
A. Du gène à la protéine CFTR ........................................................................................... 49
1) Le gène CFTR ............................................................................................................ 49
2) Structure de la protéine CFTR ................................................................................... 50
3) Les fonctions de la protéine CFTR ............................................................................ 52
4) Les mutations du CFTR ............................................................................................. 53
B. Biosynthèse et repliement : F508del-CFTR vs WT-CFTR ............................................ 55
C. Le contrôle qualité de CFTR dans le RE et sa dégradation par ERAD .......................... 58
1) Les principales chaperonnes et lectines ..................................................................... 58
2) Motif de rétention dans le RE : AFT (Arginine-Framed Tripeptides) ....................... 61
3) Code de sortie du RE : le code diacide ...................................................................... 62
D. F508del-CFTR et la réponse UPR .................................................................................. 62

Sommaire
2
III. Le système de contrôle qualité du RE comme cible thérapeutique .......................... 64
Contexte, problématiques et objectifs .................................................................................. 70
Matériels et méthodes ............................................................................................................ 74
I. Matériel cellulaire ......................................................................................................... 74
Fiche technique N°1 : ............................................................................................................ 74
Lignées cellulaires, culture et entretien ............................................................................ 74
Fiche technique N°2 : ............................................................................................................ 76
Congélation et décongélation des lignées cellulaires ....................................................... 76
Fiche technique N°3 : ............................................................................................................ 77
Isolement des cellules épithéliales bronchiques des patients CF ..................................... 77
II. Techniques de biologie moléculaire et de biochimie .................................................. 79
Fiche technique N°4 ............................................................................................................... 79
Mesure de l’activité du protéasome ................................................................................. 79
Fiche technique N°5 : ............................................................................................................ 80
Transfection de siRNA (small interfering RNA) ............................................................. 80
Fiche technique N°6 : ............................................................................................................ 82
Western blot ..................................................................................................................... 82
Fiche technique N°7 : ............................................................................................................ 87
Biotinylation de surface ................................................................................................... 87
Fiche technique N°8 : ............................................................................................................ 89
Immunomarquage indirect ............................................................................................... 89
Fiche technique N°9 : ............................................................................................................ 91
In situ Proximity Ligation Assay (Duolink) ..................................................................... 91
III. Techniques de physiologie cellulaire ........................................................................... 93
Fiche technique N°10 : .......................................................................................................... 93
Efflux d’iodure ................................................................................................................. 93
Fiche technique N°11 : .......................................................................................................... 95
Etude des variations du potentiel membranaire avec la sonde oxonol ............................. 95
Fiche technique N°12 : .......................................................................................................... 97
Chambre de Ussing .......................................................................................................... 97

Sommaire
3
Résultats ................................................................................................................................ 101
I. Les protéines EDEMs comme cibles thérapeutiques dans la mucoviscidose ........ 101
A. Contexte ........................................................................................................................ 101
B. Résultats ........................................................................................................................ 101
1) Article ....................................................................................................................... 101
2) Résultats complémentaires ....................................................................................... 133
II. Implication de l’α1,2-mannosidase du RE dans la rétention de F508del-CFTR .. 143
A. Contexte ........................................................................................................................ 143
B. Résultats ........................................................................................................................ 144
1) Effet des inhibiteurs des mannosidases sur la fonction de F508del-CFTR ............. 144
2) Effet du siRNA d’ERManI sur F508del-CFTR ....................................................... 144
3) Comprendre l’effet correcteur sur F508del-CFTR de l’extinction d’ERManI ........ 148
Discussion, conclusion et perspectives ................................................................................ 155
A. Synthèse d’une nouvelle famille d’iminosucres : dérivés multivalents du
Deoxymannojirimycin ......................................................................................................... 155
B. La famille des protéines EDEMs : de nouvelles cibles thérapeutique pour la
mucoviscidose ...................................................................................................................... 157
C. Etude du trivalent-DMJ comme correcteur de F508del-CFTR .................................... 159
D. A la recherche d’une autre cible thérapeutique pour la mucoviscidose : α1,2-mannosidase
I du RE ................................................................................................................................. 166
Références bibliographiques ............................................................................................... 171
Annexes ................................................................................................................................. 204
Article scientifique ............................................................................................................... 204

Abréviations
4
Abréviations °C: degré celsius 4PBA: sodium-4-phénylbutyrate
A: alanine AAA+: ATPases associated with diverse cellular activities AAT: α1-antitrypsin
ABC: ATP binding cassette Aha1: Activator of Hsp90 ATPase 1 ADP: adénosine diphosphate AFT: Arginine-Framed Tripeptides AMC: 7-mino-4-methylcoumarin AMPc: adénosine monophosphate cyclique ARN: acide ribonucléide Asn: asparagine ATF6: activating transcription factor-6 ATP: adenosine triphosphate
BAG-1/2: BCL2 associated athanogene 1/2 BCA: bicinchoninique acide BHK: baby hamster kidney BiP: binding immunoglobulin protein BSA: bovine serum albumin
Ca2+: ion calcium CF: cystic fibrosis CFBE: CF branchial epithelial CFTR: CF transmembrane conductance regulator CFTRinh: CFTR inhibiteur CHIP: C-terminus of Hsp70 interacting protein CHO: chinese hamster ovary CHOP: CCAAT/enhancer-binding protein homologous protein Cl-: ion chlorure COPI/II: coat proteins I/II CRD: carbohydrate recognition domain Cu2+: ions cuivriques CypB: cyclophilines B Cys: cystéine
D: aspartate ddp: différence de potentiel del: deletion DMJ: 1-déoxymannojirimycine DNAJB12: DnaJ Hsp40 family member B12 DNJ: deoxynojirimycine DMSO: diméthylsulfoxide
ECL: enhanced chemiluminescence ECLs: extra cellular loops EGF: epidermal growth factor EDEM: ER degradation-enhancing 1,2-manosidase-like protein eIF2α: Eukaryotic Initiation Factor 2α ER: endoplasmic reticulum ERAD: ER associated degradation ERAF: ER associated folding ERdj: ER-localized DnaJ proteins ERES: ER-exit sites ERGIC: ER-Golgi intermediate compartment ERmanI: ER α1,2-mannosidase I ERQC: ER quality control
F: phenylalanine FKBP: FK506-binding proteins Fluo: florescent Fsk: forskoline
GH47: glycoside hydrolase family 47 Glc: glucose GlcNAc: N-Acetylglucosamine Gp78: Glycoprotein 78 Grp: glucose regulated protein Gst: génistéine GTP: guanosine triphosphate
Hdj: human DnaJ HEK293: human embryonic kidney 293
hERG: human ether-a-go-go-related gene
Herp: ER stress-inducible protein Hop: Hsp-organising protein

Abréviations
5
HRD1: HMG-CoA reductase degradation protein 1
HRP: horseradish peroxidase Hsc: heat shock cognate Hsp: heat shock protein HspBP1: Hsp binding protein 1 Htm1: homologous to mannosidase I
I-: ion iodure ICLs: intracellular loops IRE1: inositol requiring 1 Isc: short circuit current IgG: Immunoglobulin G
K+: ion potassium KCl: chlorure de potassium
kDa: kilo Dalton KO: knockout LAMP: lysosomal-associated membranes proteins LC3: microtubules-associated protein-light chain 3
Man: mannose Mns1: α1,2-mannosidase chez la levure Mg: magnésium MSD: membrane-spanning domain
N: asparagine Na+: ion sodium NBD: nucleotide-binding domain NEF: nucleotide exchange factors NHK: null hong kong
OS-9: osteosarcoma amplified 9 OST: oligosaccharyl transférase
PAGE: polyacrylamide gel electrophoresis PAF: paraformaldéhyde PBS: phosphate buffer saline PDI: protein disulfide isomerase PERK: protein kinase RNA-like ER kinase PPIase: peptidyl-prolylisomérases
PKA: protein kinase A PKC: protein kinase C PLA: proximity ligation assay
QCV: quality control vesicles
RD: regulatory domain RE: réticulum endoplasmique RISC: RNA induced silencing complex RMA1: RING-finger protein with membrane anchor-1 RNF5: RING finger protein
SBD: substrate binding domain SDS: sodium dodecyl sulfate SEL1L: suppressor/enhancer of Lin-12-like SERCA: sarco/ER Ca2+-ATPase siRNA: small interfering RNA SPP: signal peptide peptidase SRP: signal recognition particule
SVF: sérum de veau fœtal
T1/2: demi-vie TBS: tris buffer saline TG-SDS: tris-glycin-SDS TMD: transmembrane domain Trx: thiorédoxine
UGGT: UDP-Glucose:glycoprotein glycosyltransferase UPR: unfolded protein response
VIP36: vesicular integral membrane protein of 36 kDa VIPL: VIP36-Like
wt: wild type (sauvage)
XBP-1: x-box binding protein 1 XTP3-B: XTP3-transactivated gene B
protein
Ydj1: yeast dnaJ

Liste des figures
6
Liste des Figures Figure 1 : Reconnaissance du peptide signal par la SRP. ........................................................ 12
Figure 2 : Structure du motif oligosaccharidique N-glycan. .................................................... 13
Figure 3 : Glycosylation d’une protéine naissante. .................................................................. 14
Figure 4 : Formation des liaisons disulfure par les protéines PDI. .......................................... 16
Figure 5 : Réaction d’isomérisation peptidyl-prolyl cis-trans catalysée par des PPIases. ....... 17
Figure 6 : Interaction des glycoprotéines avec la chaperonne calnexine. ................................ 19
Figure 7 : Le Cycle de la calnexine/calréticuline. .................................................................... 20
Figure 8 : Influence des co-chaperonnes du complexe Hsp70 sur le devenir des protéines. ... 24
Figure 9 : Modes de transport des protéines hors du réticulum endoplasmique. ..................... 28
Figure 10 : Transport des glycoprotéines dans la voie sécrétoire (Yamamoto, 2014). ............ 30
Figure 11 : Représentation schématique de la maturation des oligosaccharides N-liés. .......... 31
Figure 12 : Etapes de la dégradation ERAD des glycoprotéines. ............................................ 35
Figure 13 : Structure du motif oligosaccharide N-glycan Glc3Man9GlcNAc2. ........................ 37
Figure 14 : Les trois principales ligases identifiées chez les mammifères. .............................. 38
Figure 15 : Représentation schématique des membres de la famille GH47. ............................ 40
Figure 16 : Modèle de compartimentation du RE dans les cellules de mammifères. .............. 42
Figure 17 : Le processus ERAD tuning. .................................................................................. 46
Figure 18 : Représentation des trois voies de l’UPR. .............................................................. 48
Figure 19 : Du gène à la protéine CFTR. ................................................................................. 50
Figure 20 : Représentation schématique de la structure de CFTR. .......................................... 51
Figure 21 : Régulation des canaux ioniques par la protéine CFTR. ........................................ 52
Figure 22 : Classification des mutations du CFTR et leurs conséquences sur la protéine CFTR.
.......................................................................................................................................... 54
Figure 23 : Localisation de la protéine WT- et F508del-CFTR. .............................................. 55
Figure 24 : Biosynthèse de WT- et F508del-CFTR et assemblage des domaines. .................. 56
Figure 25 : Rôle de Hsp70/90 et leur partenaires dans le contrôle de qualité de CFTR. ......... 59
Figure 26 : Adaptation de la capacité des voies ERAF et ERAD pour corriger les défauts
impliqués dans les pathologies conformationnelles. ........................................................ 65
Figure 27 : Représentation schématique des principales étapes de la biosynthèse de F508del-
CFTR et les sites d’action du Miglustat et des inhibiteurs de pompes Serca................... 71
Figure 28 : Objectifs de notre travail. ...................................................................................... 72

Liste des figures
7
Figure 29 : Principe de la transfection de siRNA. .................................................................... 80
Figure 30 : Principe de la technique du western blot. .............................................................. 82
Figure 31 : Assemblage du sandwich de western blot. ............................................................ 85
Figure 32 : Principe de la biotinylation de surface. .................................................................. 87
Figure 33 : Principe d’immunomarquage indirect. .................................................................. 89
Figure 34 : Principe de la technique de Duolink. ..................................................................... 91
Figure 35 : Exemple de représentation graphique des résultats d’efflux d’iodures. ................ 94
Figure 36 : Structure de la sonde oxonol [DiSBAC2(3)]. ........................................................ 95
Figure 37 : Exemple de représentation graphique de la fluorescence de la sonde oxonol. ...... 96
Figure 38 : Principe de la technique de chambre de Ussing. ................................................... 97
Figure 39 : Chambre de Ussing. ............................................................................................... 99
Figure 40 : Implication de RMA1/RNF5 et CHIP dans la dégradation de F508del-CFTR. .. 133
Figure 41 :Effet du trivalent-DMJ sur l’interaction de F508del-CFTR avec RNF5/RMA1 ou
CHIP. .............................................................................................................................. 134
Figure 42 : Effet du trivalent-DMJ sur l’activité du protéasome 20S intracellulaire. ............ 135
Figure 43 : Effet du trivalent-DMJ sur l’activité du protéasome 20S purifié. ....................... 137
Figure 44 : Effet du DMJ et ses dérivés multivalent sur l’activité du protéasome 20S purifié.
........................................................................................................................................ 138
Figure 45 : Effet de la tunicamycine sur l’expression protéique de BiP/GRP78 et eif2α. ..... 140
Figure 46 : Effet du trivalent-DMJ sur l’expression de la protéine BiP/GRP78 et eIF2α. .... 141
Figure 47 : Efficacité du potentiateur VX-770 sur F508del-CFTR corrigé par le trivalent-DMJ.
........................................................................................................................................ 142
Figure 48 : Effet des inhibiteurs de mannosidase sur l’activité de F508del-CFTR. ............. 144
Figure 49 : Diminution de l’expression protéique d’ERManI ou de Golgi-Man par siRNA. 145
Figure 50 : Effet du siRNA ERManI et Golgi-ManII sur l’activité de F508del-CFTR. ........ 146
Figure 51 : Effet de siRNA ERManI sur la maturation de F508del-CFTR. .......................... 147
Figure 52 : Effet du siRNA ERManI sur l’expression protéique des EDEMs. ...................... 149
Figure 53 : Effet du siRNA ERManI sur l’interaction de F508del-CFTR avec les EDEMs. 150
Figure 54 : Effet du siRNA ERManI sur l’expression protéique de CHIP et RNF5/RMA1. 151
Figure 55 : Effet du siRNA ERManI sur l’interaction de F508del-CFTR avec CHIP ou
RNF5/RMA1. ................................................................................................................. 152
Figure 56 : Effet du siRNA ERManI sur l’activité du protéasome 20S intracellulaire. ........ 153
Figure 57 : Hypothèses de la restauration de F508del-CFTR à la membrane cellulaire sous sa
forme immature par le trivalent-DMJ. ........................................................................... 161

Liste des figures
8
Figure 58 : Principales protéines impliquées dans le repliement et/ou la dégradation de CFTR.
........................................................................................................................................ 163
Figure 59 : Modèle d’ubiquitinylation séquentielle de F508del-CFTR par des complexes
contenant RMA1/RNF5 et Gp78.................................................................................... 164
Figure 60 : Les trois principales ligases identifiées chez les mammifères. ............................ 165
Figure 61 : Devenir des glycoprotéines correctement- ou mal-repliées. ................................ 167
Figure 62 : Récapitulatif des principaux résultats de ce projet de thèse. ............................... 170

Liste des tableaux
9
Liste des tableaux Tableau 1 : Liste non exhaustive des principales protéines du ERQC. .................................... 11
Tableau 2 : Composition du milieu de culture pour les lignées cellulaires Hela WT- et
F508del- CFTR. ............................................................................................................... 74
Tableau 3 : Composition du milieu de culture pour la lignée cellulaire CFBE41o-. ............... 75
Tableau 4 : Composition du milieu de décontamination pour échantillons CF. ...................... 77
Tableau 5 : Composition du milieu de culture primaire CF. .................................................... 78
Tableau 6 : Composition du tampon de lyse cellulaire. ........................................................... 82
Tableau 7 : Composition des gels d’électrophorèse des protéines. .......................................... 84
Tableau 8 : Composition des tampons 4X Lower et Upper. .................................................... 84
Tableau 9 : Composition du tampon de transfert. .................................................................... 84
Tableau 10 : Récapitulatif des anticorps utilisés en western blot. ........................................... 85
Tableau 11 : Récapitulatif des anticorps utilisés en western blot. ........................................... 86
Tableau 12 : Récapitulatif de l’ensemble des anticorps primaires utilisés. ............................. 90
Tableau 13 : Récapitulatif de l’ensemble des anticorps secondaires utilisés. .......................... 90
Tableau 14 : Récapitulatif de l’ensemble des anticorps utilisés en Duolink. ........................... 92
Tableau 15 : Composition du milieu d’efflux utilisé. .............................................................. 93
Tableau 16 : Composition de la solution Krebs sans Cl-.......................................................... 96
Tableau 17 : Composition de la solution de Krebs « normale ». ............................................ 99

Introduction
10
Introduction Dans toutes les cellules vivantes, des programmes génétiques différents contrôlent
toutes les étapes de la naissance, de la vie et de la mort des protéines. Avant de pouvoir exercer
sa fonction biologique, une protéine va subir un ensemble de processus biochimiques
complexes entre sa naissance (au moment de la lecture de l’ARN) et sa localisation définitive
le temps qu’elle soit fonctionnelle.
Nous allons examiner dans cette introduction quelques-uns des mécanismes et déterminants
moléculaires généraux avant d’aborder les particularités de notre protéine d’intérêt, CFTR
(Cystic Fibrosis Transmembrane conductance Regulator).
I. Le contrôle qualité des glycoprotéines
Le contrôle qualité du réticulum endoplasmique (RE), ERQC (Endoplasmic Reticulum
Quality Control) est un mécanisme de surveillance de la biosynthèse et du repliement des
protéines dans le RE. Ce processus est composé de deux voies : la voie ERAF (ER Associated
Folding) qui assiste les protéines au cours de leur repliement et s’assure que seules les protéines
correctement repliées puissent quitter le RE, et la voie ERAD (ER Associated Degradation) qui
séquestre les protéines mal repliées et/ou mutées dans le RE puis les dirige vers la dégradation
(Araki and Nagata, 2011).
Ce système est composé de diverses protéines chaperonnes et enzymes dont les principaux
acteurs ainsi que leur fonction sont présentés dans le tableau 1.

Introduction
11
Familles Principaux membres Fonctions
α-glucosidase I et II
Clivage des résidus glucoses permettant l’entrée des glycoprotéines dans le cycle calnexine ou calréticuline
Enzymes de glycosylation
UGGT (UDP–glucose glycoprotein: glucosyltransferase)
Reconnaissance des glycoprotéines mal repliées et re-glycosylation afin de permettre leur entrée dans un nouveau cycle de repliement calnexin ou calréticuline
ER α-1,2 mannosidase Clivage du résidu mannose permettant la sortie des glycoprotéines du cycle calnexine ou calréticuline
Malectine
Empêche l’agrégation des polypeptides naissants et recrutement de l’α-glucosidases II
Calnexine, Calréticuline Repliement des glycoprotéines
Lectines EDEM1 à 3
Reconnaissance des glycoprotéines mal repliées, clivage des résidus mannoses afin de les diriger vers ERAD
OS-9, XTP3-B
Reconnaissance des glycoprotéines mal repliées et adressage vers le complexe de rétrotranslocation
Hsp70, BiP/grp78, Hsp90, Grp94
Empêche l’agrégation des protéines et aide au repliement
Hsp40 : ERdj1 à 7 Co-chaperonnes qui régulent BiP
Chaperonnes Hsp40 : DNAJB12
Co-chaperonne qui dirige les protéines mal repliées vers ERAD
CHIP (carboxy terminus of Hsc70 interacting protein)
E3 ubiquitine ligase, co-chaperonne qui se lie à Hsc70 ou Hsp70 et dirige les protéines mal repliées vers ERAD
Oxydo- réductases
PDI (protéine disulfure isomérase), ERp57
Formation et isomérisation des ponts disulfures
PPIase Cyclophilines Co-chaperonnes qui facilitent le repliement tardif des substrats de Hsp90
Tableau 1 : Liste non exhaustive des principales protéines du ERQC.
A. La voie ERAF
Chez les eucaryotes, les protéines solubles ou membranaires de la voie de sécrétion sont
synthétisées au niveau du RE. Ces protéines doivent alors être transloquées à travers la
membrane de ce compartiment, et dans le cas des protéines transmembranaires, s’intégrer dans
la bicouche lipidique. Pour cela, les peptides naissants destinés à cette voie de sécrétion
présentent une courte séquence hautement hydrophobe qui assure leur adressage vers la
membrane du RE. Selon le type de la protéine, soit cette séquence est appelée peptide signal,
lorsqu’elle est située dans l’extrémité N-terminal (cas des protéines sécrétées et certaines
protéines transmembranaires de type I), soit elle correspond au premier segment
transmembranaire de la protéine (McCaffrey and Braakman, 2016).

Introduction
12
Dès sa sortie du ribosome, le peptide signal est reconnu par une particule ribonucléoprotéique
SRP (signal-recognition particle) qui après sa fixation provoque un arrêt temporaire de la
traduction en bloquant l’accès aux facteurs d’élongation (Figure 1).
Figure 1 : Reconnaissance du peptide signal par la SRP.
Le peptide signal (rose) de la séquence peptidique (bleu) est reconnu par la SRP. Cette dernière interrompt la traduction et favorise la fixation du ribosome au translocon (Sec61). Cela, permet la dissociation de la SRP de son récepteur, le relâchement du peptide signal, son entré dans le translocon et enfin la reprise de la translocation de manière co-traductionnelle (Modifié d’après http://biochemprotein.blogspot.fr).
La SRP est une GTPase qui possède une faible affinité intrinsèque pour le GTP. Son récepteur
est à la fois ancré dans la membrane du RE et lié au translocon, un complexe hétérotrimérique
de protéines Sec61 (Figure 1).
La fixation de la SRP au complexe « ribosome-chaîne polypeptidique naissante » augmente son
affinité pour le GTP ce qui favorise sa fixation à son récepteur. Le ribosome interagit alors avec
le translocon Sec61 et permet l’hydrolyse du GTP de la SRP et de son récepteur. Cette hydrolyse
provoque la dissociation de la SRP de son récepteur, le relâchement du peptide signal et la
reprise de la translocation qui s’effectue de manière co-traductionnelle.
Le peptide signal est clivé après son intégration dans la membrane du RE par l’enzyme signal
peptidase et est ensuite dégradé par la signal peptide peptidase (SPP). Dans le cas du signal
d’ancrage, la séquence ferait partie de la protéine et permettrait son ancrage dans la bicouche
lipidique (Ng and Walter, 1994; McCaffrey and Braakman, 2016).

Introduction
13
1) Les modifications co- et post-traductionnelles
Dans la lumière du RE, la protéine naissante intègre un processus de repliement lui
permettant d’acquérir une structure tridimensionnelle précise (forme native), indispensable à sa
transition du RE vers l’appareil de Golgi et à sa fonction. Ce processus commence alors que la
chaine polypeptidique est toujours en cours de translocation et de traduction. Il consiste en des
modifications post traductionnelles telles que la N-glycosylation et la formation de ponts
disulfures sur la protéine cliente.
a) N-glycosylation
Dans les cellules eucaryotes, la grande majorité des polypeptides naissants qui entrent
dans la lumière du RE sont glycosylés sur leur résidu asparagine (Asn) contenu dans la séquence
consensus -Asparagine-X-Sérine/Thréonine-, où X correspond à n’importe quel acide aminé
autre que la proline (Caramelo and Parodi, 2015). Elle se caractérise par le transfert en bloc sur
le résidu Asparagine, d’un groupement oligosaccharidique appelé N-glycan. Ce groupement est
constitué de 2 résidus N-acetylglucosamine (GlcNAc), 9 mannoses (Man) et 3 glucoses (Glc)
(Glc3Man9GlcNAc2) réparti en trois branches A, B et C (Figure 2).
Figure 2 : Structure du motif oligosaccharidique N-glycan. L’oligosaccharide préformé est composé de 3 glucoses, 9 mannoses et 2 N-acetylglucosamines. Les branches oligosaccharidiques sont représentées par A, B et C. Les enzymes qui modifient ce motif sont listées sur la figure et leur action est représentée. Le type de liaison glycosidique est représenté en couleur (D'après Tannous et al., 2015).

Introduction
14
Le N-glycan est préalablement assemblé au niveau d’un dolichol-phosphorylé (-PP-
dolichol) ancré dans la membrane du RE (Schwarz and Aebi, 2011; Lannoo and Van Damme,
2015). Il est ensuite transféré sur le résidu Asparagine par l’oligosaccharyl transférase (OST),
un complexe protéique multimérique situé dans la membrane du RE et associé au ribosome et
au translocon sec61 (Harada et al., 2009) (Figure 3).
Figure 3 : Glycosylation d’une protéine naissante. Le motif oligosaccharidique N-glycan Glc3Man9GlcNAc2 est préassemblé sur un dolichol-phosphorylé ancré dans la membrane du RE. Au cours de sa translocation dans le RE via un canal de translocation, le polypeptide néosynthétisé glycosylé par le complexe OST (oligosaccharyl transférase). Ce complexe transfert le motif N-glycan sur le résidu asparagine (Asn) contenu dans la séquence consensus -Asn-X-Sérine/Thréonine-, où X correspond à n’importe quel acide aminé autre que la proline (Modifié d'après Mazola et al., 2011).
Immédiatement après son transfert sur le polypeptide, le N-glycan est modifié par
différentes enzymes glycoside hydrolases (α-glucosidases I et II) et par une glucosyl
transférase, l’UGGT (UDP-Glucose:glycoprotein glycosyltransferase) dans une cascade de
réactions générant différentes structures qui servent de ligand spécifique aux protéines
chaperonnes du RE. Ainsi l’état de glycosylation d’une protéine peut nous renseigner sur son
stade de repliement par le contrôle de qualité.
Les N-glycans jouent un rôle majeur à la fois direct et indirect dans les processus de
repliement et de contrôle qualité des protéines qui les portent. En effet, de part leur nature
encombrante et hydrophile, ces motifs augmentent la solubilité intrinsèque des polypeptides
naissants auxquels ils sont attachés et préviennent leur agrégation. De plus, leur fixation aux
polypeptides pourrait affecter la structure secondaire locale de la protéine en stabilisant la
conformation des résidus proximaux au site de glycosylation. L’analyse de la structure

Introduction
15
secondaire autour de ce dernier a soulevé, en effet, la possibilité que les glycans puissent agir
comme marqueurs des points où des changements se produisent dans la structure secondaire
(Petrescu et al., 2004). De plus, il a été montré que l’interaction du N-glycan avec la chaîne
polypeptidique facilite l’acquisition de structures secondaires particulières telles que les
feuillets β (O’Connor and Imperiali, 1996; Petrescu et al., 2004; Vagin et al., 2008). D’autre
part, il a été observé que des acides aminés aromatiques éloignés dans la séquence, deviennent
proches aux sites de glycosylation dans la structure tertiaire. Ceci suggère que les N-glycans
liés pourraient avoir un rôle encore plus direct dans le repliement, en agissant comme des sites
de nucléation pour des parties éloignées de la chaîne protéique riche en acides aminés
aromatiques. Cette hypothèse est soutenue par des expériences in vitro indiquant que les N-
glycans favorisent le repliement basé sur les interactions hydrophobes glycan-protéine
(Nishimura et al., 1998; Jitsuhara et al., 2002). D’autres études comparant des glycoprotéines
et leur homologues non glycosylés ont montré que la présence du N-glycan augmentait la
stabilité et la résistance à la protéolyse (Beggah et al., 1997; Imperiali and O’Connor, 1999;
Kundra and Kornfeld, 1999; Crothers et al., 2004). Une résistance probablement due à la
restriction d’accès des sites de clivage à la protéase (Vagin et al., 2008). Enfin, les N-glycans
favorisent également le repliement et le contrôle qualité de manière indirecte en recrutant de
manière spécifique les chaperonnes et les enzymes impliquées dans le repliement et la
dégradation des glycoprotéines (Slominska-Wojewodzka and Sandvig, 2015).
b) Formation et réarrangement des ponts disulfures
La formation des ponts disulfures entre deux résidus de cystéine est essentielle pour le
repliement correct et la maturation de beaucoup de protéines de sécrétion. En effet, il s’agit des
liaisons covalentes qui stabilisent les structures des protéines et garantissent le maintien de leur
conformation tridimensionnelle fonctionnelle. Ce processus, appelé repliement oxydatif, prend
place dans le RE et est catalysé par les protéines de la famille PDI (Protein Disulfide-
Isomerase).
Les PDIs sont des protéines localisées principalement dans le RE et sont essentielles
pour le maintien de son environnement oxydatif et son homéostasie. Elles sont dotées d’une
activité oxydoréductase/isomérase et interviennent de manière co- et post-traductionnelle pour
catalyser la formation de ponts disulfures (oxydation des groupements thiols des cystéine), leur
coupure (réduction) et/ou le réarrangement des liaisons incorrectement établies entre deux
cystéines proches (isomérisation) (Wallis and Freedman, 2013) (Figure 4).

Introduction
16
Figure 4 : Formation des liaisons disulfure par les protéines PDI. Mécanismes d’oxydation, de réduction et d’isomérisation des liaisons disulfure catalysées par les
protéines PDI (D'après Tannous et al., 2015).
En plus de leur activité oxydoréductase/isomérase, la majorité des membres de PDI présentent
une fonction chaperonne et jouent un rôle crucial dans l’ERQC. Elles peuvent en effet se lier
sur des protéines nouvellement synthétisées pour contribuer à leur repliement, ou bien sur des
protéines mal repliées pour faciliter leur dégradation (Ushioda et al., 2008; Lee et al., 2010;
Parakh and Atkin, 2015). Ainsi, certains membres de la famille PDI sont des protéines
multifonctionnelles, dont leur fonction comme oxydoréductase ou chaperonne est dépendante
du substrat (Kozlov et al., 2010a). A noter, l’activité chaperonne du PDI semble être
indépendante de son activité enzymatique (Fu and Zhu, 2010).
Plus de 20 membres ont été découverts chez les mammifères. La nécessité d’un si grand
nombre de membres de la famille PDI demeure une question ouverte. Cependant, plusieurs
membres, les plus abondants ont des spécificités de substrat distinctes (Jessop et al., 2009). Bien
que ces membres présentent des séquences en acides aminés et des fonctionnalités diverses, ils
présentent tous au moins un site catalytique qui contient un domaine homologue à la
thiorédoxine (Trx) (Edman et al., 1985; Appenzeller-Herzog and Ellgaard, 2008; Kojima et al.,
2013). PDI est la première oxydoréductase découverte et la plus abondante dans le RE.
Ubiquitaire et très fortement exprimée dans les cellules sécrétoires, PDI a la particularité
d’assurer les diverses fonctions de la famille : oxydase, réductase, isomérase et chaperonne
(Määttänen et al., 2010; Irvine et al., 2014; Okumura et al., 2015).
La protéine ERp57 présente une similarité de séquence significative avec PDI (Oliver
et al., 1997). Cependant, au lieu du domaine assurant l’activité de chaperon pour PDI, ERp57
présente une région de liaison pour les domaines P de calnexine et calréticuline, chaperonnes
impliquées dans le repliement et le contrôle qualité des glycoprotéines (Zapun et al., 1998;
Frickel et al., 2002; Russell et al., 2004; Kozlov et al., 2006). Un rôle spécifique et essentiel
dans l’isomérisation de liaisons disulfure non-natives des glycoprotéines néo-synthétisées lui a
était attribué (Oliver et al., 1997; Jessop et al., 2007) ; cependant, ERp57 ne semble pas avoir

Introduction
17
des propriétés intrinsèques de lectine (Zapun et al., 1998). Ces observations suggèrent
qu’ERp57 fonctionne de concert avec la calnexine et la calréticuline pour moduler
spécifiquement le repliement des glycoprotéines (Oliver et al., 1997; Zapun et al., 1998; Oliver
et al., 1999).
Il a été observé qu’ERp57 peut s’associer à des glycoprotéines qui ne présentent pas de résidu
de cystéines et ne nécessitent donc pas d’isomérisation de disulfure intramoléculaire (Elliott et
al., 1997; Oliver et al., 1997). Le rôle d’ERp57 ne semble donc pas se limiter à la catalyse de
la formation et de l’isomérisation de liaisons disulfure. Mais comme PDI, ERp57 peut avoir un
rôle plus vaste en tant que chaperonne moléculaire. Son extinction in vivo est létal chez des
souris KO (knockout) ERp57 ; en revanche, elle est tolérée in vitro sur des cellules en culture,
suggérant que certaines glycoprotéines peuvent engager d’autres assistants de repliement afin
d’acquérir leur conformation native. Comme par exemple, ERp72 qui semble pouvoir
remplacer ERp57 pour assurer une maturation efficace des glycoprotéines du virus forestier
Semliki (Soldà et al., 2006).
c) Les liaisons peptidiques
Les liaisons peptidiques, reliant n’importe quel acide aminé avec un résidu proline
peuvent adopter une conformation cis ou trans, avec une faible préférence pour la conformation
trans (Fischer and Aumüller, 2003). Les peptidyl-prolylisomérases (PPIases) sont des protéines
ubiquitaires capables de catalyser l’isomérisation des acides aminés prolines des protéines de
conformation trans en cis et inversement (Figure 5). Cela permet le changement d’orientation
de la chaine peptidique dans l’espace.
Figure 5 : Réaction d’isomérisation peptidyl-prolyl cis-trans catalysée par des PPIases. (Tannous et al., 2015)
La correction de l’orientation des liaisons proline a lieu tardivement pendant le
processus de repliement et intervient lorsque la protéine a atteint une conformation quasi-
complète. L’isomérisation de liaisons peptidyl-prolyle mal placées peut-être une étape limitante
pour la vitesse du repliement des protéines (Kiefhaber et al., 1990).

Introduction
18
La famille PPIase des mammifères comprend trois sous-groupes, parvulines, cyclophilines
(Cyps) et protéines de liaison FK506 (FKBPs) (Göthel and Marahiel, 1999; Fischer and
Aumüller, 2003; Stocki et al., 2014). Le RE des mammifères contient 6 membres de FKBP et
2 membres de Cyps. La cyclophiline B est la PPIase la plus caractérisée (Bernasconi et al.,
2010; Bergeron et al., 2011; Jansen et al., 2012; Stocki et al., 2014). Elle a été identifiée dans
un complexe protéique incluant les chaperonnes calnexine et calréticuline. L’absence de site de
fixation chaperonne-like sur cette PPIase suggère son interaction avec ses substrats via d’autres
chaperonnes. Cette hypothèse a été confirmée par des études de cristallographie identifiant un
site d’interaction de CypB à l’extrémité du domaine P de calnexine et calréticuline (Kozlov et
al., 2010b). Ces données supportent le rôle actif de CypB dans le cycle calnexine ou
calréticuline en favorisant l’orientation correcte des liaisons peptidyl-prolyl dans les protéines
en cours de repliement (Kozlov et al., 2010a).
2) Les interactions avec les protéines chaperonnes
a) Les lectines calnexine/calréticuline et les glycosidases
La calnexine et son homologue soluble, la calréticuline, sont deux lectines chaperonnes
responsables du repliement et du contrôle qualité des glycoprotéines dans le RE (Fliegel et al.,
1989; Wada et al., 1991; Michalak et al., 1992; Hebert et al., 1996). La calnexine est une
protéine transmembranaire de type I, de 90 kDa qui interagit avec les glycoprotéines solubles
et transmembranaires ; tandis que la calréticuline interagit préférentiellement avec les
glycoprotéines solubles (Hebert et al., 1997). Ces deux lectines partagent une organisation
générale similaire. Chacune possède dans sa partie N-terminale un domaine globulaire de
liaison aux glucides, permettant la liaison spécifique des glycoprotéines monoglucosylées de
type Glc1Man9GlcNAc2 (Kozlov et al., 2010c; Schrag et al., 2001). À partir de ce domaine
s’étend une longue boucle en forme de bras appelé domaine P pour sa richesse en résidus de
proline. Ce domaine a pour rôle de recruter l’oxydoreductase ERp57 (Frickel et al., 2002;
Kozlov et al., 2006) et la PPIase CypB (Kozlov et al., 2010b). Le domaine globulaire des deux
lectines contient également des sites de liaison pour les ions Ca2+ et l’ATP, deux cofacteurs
régulateurs importants pour la stabilisation des lectines chaperonnes. Le Ca2+ régule également
la liaison des lectines aux substrats (pour revue Lamriben et al., 2016).
Bien que réticulaires, ces deux chaperonnes ont également été localisées hors du RE où elles
joueraient d’autres fonctions. La calréticuline a été localisée dans le cytoplasme, à la surface

Introduction
19
externe de la cellule et la matrice extracellulaire ; et la calnexine à la surface cellulaire de
nombreuses cellules (Michalak et al., 2009; Wang et al., 2012; Lamriben et al., 2016).
i. Cycle de la calnexine/calréticuline
La forme tri-glucosylée Glc3Man9GlcNAc2 du N-glycan des protéines nouvellement
synthétisées a une demi-vie de quelques secondes. Immédiatement après son transfert sur le
résidu asparagine de la chaine polypeptidique naissante, son glucose terminal est clivé avec une
t1/2 < 2 min par l’action de l’α-glucosidase I, une enzyme faisant partie du complexe translocon
avec l’OST (Helenius and Aebi, 2004; Dejgaard et al., 2010; Roth and Zuber, 2017).
Le carbohydrate di-glucosylé Glc2Man9GlcNAc2 ainsi généré recrute et s’associe avec la
malectine, une lectine transmembranaire de type I du RE (Figure 6). Le rôle de cette protéine
induite par le stress du RE serait de prévenir l’agrégation des polypeptides naissants durant la
période de synthèse précoce et de recruter l’α-glucosidase II (Schallus et al., 2008; Galli et al.,
2011).
Figure 6 : Interaction des glycoprotéines avec la chaperonne calnexine. OST : oligosaccharyl transférase, BiP : chaperonne réticulaire, PDI et ERp57 : oxydoréductases, SPC : signal peptide peptidase, GlcI et GlcII : α-glucosidases I et II, Points et traits rouges : liaisons disulfures (D'après Braakman and Hebert, 2013).
L’α-glucosidase II ainsi recrutée clive le deuxième glucose avec une t1/2 ~ 5 min et génère un
carbohydrate mono-glucosylé Glc1Man9GlcNAc2 qui devient ainsi un substrat pour la calnexine
ou la calréticuline (Roth and Zuber, 2017). La calnexine ou calréticuline interagit avec ce N-
glycan au niveau de son domaine globulaire, tandis que sa chaîne polypeptidique forme des
liaisons disulfures mixtes transitoires avec ERp57. Ces interactions aident au repliement de la
glycoprotéine d’une part en réduisant son espace de repliement et en conséquence le nombre de
conformations potentielles, et d’autre part, en prévenant son agrégation et en facilitant
l’isomérisation des ponts disulfures grâce à l’ERp57 (Jessop et al., 2007; Gidalevitz et al., 2013;

Introduction
20
Lamriben et al., 2016). Ce dernier semble jouer un rôle supplémentaire dans la stabilisation de
l’ensemble de ces interactions.
À la fin de son repliement, la glycoprotéine doit être relâchée de la calnexine ou la calréticuline.
L’α-glucosidase II clive alors le dernier glucose du N-glycan et génère le motif Man9GlcNAc2,
avec une t1/2 ~ 20 min (Roth and Zuber, 2017). Le motif généré n’est ainsi plus reconnu par les
deux chaperonnes. Cependant, il pourra être reglucosylé par l’enzyme UGGT afin de restaurer
son affinité pour ces lectines, on parle alors de « cycle calnexine/calréticuline » (Figure 7).
Figure 7 : Le Cycle de la calnexine/calréticuline. (1) Assemblage de l’oligosaccharide Glc3Man9GlcNAc2, (2) Glycosylation de la protéine par l’OST, (3) Clivage de glucose par les α-glucosidases I et II, (4) Liaison de la glycoprotéine à la calnexine ou calréticuline, (5 et 8) Clivage du dernier glucose par l’α-glucosidase II, (6) Re-glucosylation des protéines n’ayant pas atteint leur conformation native par UGGT et (7) leur réentrée dans le cycle calnexine ou calréticuline pour un nouveaux cycle de repliement, (9 et 10) Les protéines correctement repliées sont démannosylées en Man8GlcNAc2 par les mannosidases du RE puis dirigées vers l’appareil de Golgi, (11) Les protéines définitivement mal repliées sont extensivement démannosylées par les mannosidases du RE puis conduites vers la dégradation hors du RE (D'après Lamriben et al., 2016).

Introduction
21
L’UGGT apparait comme étant un senseur de repliement crucial dans le RE extrêmement
sensible, qui peut reglucosyler les glycoprotéines mal repliées pour un nouveau cycle de
repliement et ignorer les glycoprotéines natives et fortement mal conformées. Cependant,
comment cette spécificité est accomplie reste encore mal comprise.
ii. Distinction des protéines correctement repliées des mal repliées
Une des questions importantes dans l’ERQC est de savoir comment ce processus
distingue les glycoprotéines définitivement mal repliées de celles ayant acquis leur
conformation native et surtout des intermédiaires de repliement. Il a été proposé que la présence
de résidus d’acides aminés hydrophobes exposés sur la protéine mal repliée serait le motif de
distinction entre les différentes formes de repliement. Ces motifs seraient reconnus par un
« senseurs de repliement », l’enzyme UGGT (Lamriben et al., 2016) ou BiP, une protéine de la
famille des Hsp70 (Heat Shock Protein at 70 kDa) (Flynn et al., 1991). En effet, tant qu’une
protéine présente des séquences hydrophobes exposées, elle restera retenue dans le RE afin de
subir d’autres cycles de repliement et d’atteindre la bonne conformation. A la fin de chaque
cycle, soit la protéine est correctement repliée et pourra donc quitter le RE pour transiter vers
l’appareil de Golgi, soit elle est reconnue comme définitivement mal repliée et sera alors
envoyée vers le système de dégradation, ou bien, cette protéine présente toujours un repliement
intermédiaire, elle subira dans ce cas-là, une autre tentative de repliement. Le système basé sur
BiP est utilisé principalement pour les protéines non glycosylées ou les glycoprotéines dans
lesquelles le N-glycan se produit relativement tard (Otero et al., 2010). Ainsi, nous détaillerons
ici uniquement le deuxième système, propre aux glycoprotéines.
Le système de contrôle de qualité des glycoprotéines est basé non seulement sur la
reconnaissance des séquences hydrophobes exposées sur la surface des glycoprotéines par
l’enzyme UGGT, mais également sur la structure de leur chaine glycanique, d’où le nom de
« système basé sur le code glycan ». En effet, comme nous l’avons abordé dans la partie
précédente, au cours de son processus de repliement, la glycoprotéine sous sa forme mono
glucosylée Glc1Man9GlcNAc2 interagit avec la chaperonne calnexine/calréticuline et subit un
cycle de repliement « cycle de calnexine/calréticuline ». Suite au clivage de son résidu glucose
par la glucosidaseII, la glycoprotéine sous la forme Man9GlcNAc2 n’est plus reconnue par la
calnexine. A cet instant, elle va être « scannée » par l’enzyme UGGT qui reconnaît et interagit
préférentiellement avec les régions protéiques hydrophobes exposées à la surface des
glycoprotéines n’ayant pas atteint leur conformation native. Ainsi, trois cas de figures sont

Introduction
22
possibles (Braakman and Hebert, 2013) : (1) la glycoprotéine n’expose pas de séquences
hydrophobes, elle est donc correctement repliée et pourra quitter le RE et transiter vers
l’appareil de Golgi (Geva and Schuldiner, 2014a), (2) la glycoprotéine expose des séquences
hydrophobes, elle est donc mal repliée et sera reglucosylée par l’UGGT. Elle reforme alors un
complexe avec calnexine-ERp57 afin de subir un nouveau cycle de repliement (Hebert et al.,
1995), ou (3) la glycoprotéine présente toujours des séquences hydrophobes avec un repliement
anormal, elle sera donc extensivement démannosylée par les mannosidases du RE afin de
fournir un signal pour sa prise en charge par le système de dégradation (Helenius and Aebi,
2004).
b) Les protéines de choc thermique (Hsp)
Le système Hsp (Heat Shock Protein) est un système chaperon majeur dans le RE en
plus du complexe calnexine/calréticuline (Otero et al., 2010; Balchin et al., 2016). Localisées
dans le cytoplasme, le noyau ou même dans le RE, les Hsps sont des chaperonnes inductibles
par le stress (Shastry et al., 2002; McClung et al., 2008). Elles sont recrutées pour aider à la
maturation des protéines non glycosylées ou des domaines sur des protéines glycosylées
(Beckmann et al., 1990). Certains d’entre eux identifient les protéines immatures, aberrantes ou
favorables à l’agrégation de par la présence de segments hydrophobes exposés sur ces protéines
(Glover and Lindquist, 1998; Ben-Zvi et al., 2004; Li and Srivastava, 2004). Ce système est
également impliqué dans la transduction du signal, la régulation du cycle cellulaire, la
différenciation et la mort cellulaire programmée (pour revue Jacob et al., 2017). Leur liaison
est régulée par des cofacteurs adénine-nucléotidiques et cofacteurs spécialisés. Malgré ces
similitudes, leur gamme de substrats et leurs rôles sont divers. Ici, nous allons nous intéresser
principalement aux chaperonnes et co-chaperonnes les plus pertinentes pour notre étude.
i. Hsp70 et Hsp40
La famille des Hsp70 regroupe des protéines d’environ 70 kDa inductibles par un choc
thermique. Les chaperonnes de ce système se lient à des séquences enrichis en résidus
hydrophobes, typiquement exposés par des protéines mal repliées (Rüdiger et al., 1997; Swain
et al., 2007; Qi et al., 2013). Leur rôle dans le repliement des protéines néo-synthétisées se
caractérise par la prévention de l’agrégation, l’aide au repliement et la solubilisation des
protéines agrégées.

Introduction
23
Les membres de cette famille partagent une séquence composée de deux domaines : un
domaine ATPase, nommé NBD (Nucteotide Binding Domain) localisé en N-terminal et un
domaine de liaison au substrat SBD (Substrate Binding Domain) localisé en C-terminal. Ce
dernier domaine contient une région hydrophobe appropriée en forme de poche à laquelle se
fixe la protéine à replier (Mayer and Bukau, 2005). Cette poche est sous le contrôle d’un
couvercle étendu, ATP-dépendant, qui peut s’ouvrir et se fermer en emprisonnant le substrat
dans la poche (Mayer et al., 2000). Suite à la fixation de l’ATP au domaine NBD, le couvercle
s’ouvre, permettant la liaison du substrat au domaine SBD avec une faible affinité. Cette liaison
de substrat stimule l’activité ATPase de la chaperonne et par conséquent l’hydrolyse d’ATP
(Flynn et al., 1989). Hsp70 lié à l’ADP acquiert ainsi une forte affinité pour son substrat et le
couvercle se ferme, isolant la partie de substrat fixé au SBD. Cette liaison avec une haute
affinité protège les substrats d’un repliement prématuré et de l’agrégation. Cependant,
l’échange de l’ADP par un ATP résulte en l’ouverture du couvercle et par conséquent la
libération de la protéine qui peut alors poursuivre son repliement.
Ce cycle de Hsp70 est régulé par des protéines de la famille Hsp40 et des facteurs d’échange
de nucléotides NEF (nucleotide exchange factors). Les co-chaperonnes Hsp40 (DnaJ chez les
bactéries), connue aussi sous le nom de protéines à domaine-J contiennent une région, appelée
domaine J requise pour leur liaison avec les différents membres de Hsp70. Leur liaison stimule
et accélère fortement l’hydrolyse de l’ATP, générant l’état fermé de Hsp70 piégeant le substrat
(Kityk et al., 2012; Nunes et al., 2015). La liaison ultérieure des NEF au NBD quant à elle,
facilite l’échange ADP en ATP, ouvrant le SBD et permettant la libération du substrat pour le
repliement ou le transfert vers les chaperonnes en aval ou vers la machinerie de dégradation
(Sharma et al., 2010; Braakman and Hebert, 2013).
Parmi les membres de la famille Hsp70, on retrouve la protéine GRP78 (Glucose
Regulated Protein), connue sous le nom de BiP pour Binding Immunoglobulin Protein. Il s’agit
d’une protéine soluble réticulaire de 78 kDa qui se lie à la plupart des chaines peptidiques
naissantes qui transitent dans le RE. BiP est l’une des chaperonnes du RE les plus abondantes
et possède de multiples rôles allant du repliement productif à une participation dans le processus
ERAD. Il a été proposé que BiP serait un senseur de repliement qui distingue entre les
différentes formes de repliement des protéines non glycosylées ou des glycoprotéines dans
lesquelles le N-glycan se produit tardivement (Flynn et al., 1991; Otero et al., 2010).

Introduction
24
L’activité ATPase de BiP, mais aussi sa localisation et ses diverses fonctions, nécessitent
l’assistance des co-chaperonnes de la famille Hsp40 connues sous le nom de ERdj 1 à 7 (ER-
localized DnaJ proteins) (Braakman and Hebert, 2013). Certains membres d’ERdj jouent un
rôle dans le processus de repliement (Shen and Hendershot, 2005; Otero et al., 2010), alors que
d’autres s’associent au protéines mal repliées et aident dans l’accélération de leur dégradation
(Dong et al., 2008; Lai et al., 2012). Il a été démontré par exemple qu’ERdj5 interagit avec
EDEM (ER-degradation enhancing alpha-mannosidase like protein), protéine de type α-
mannosidase impliquée dans la dégradation des protéines mal repliées (Ushioda et al., 2008).
De même, ERdj4 et ERdj5 surexprimés interagissent avec p97, une composante de la
machinerie ERAD (Dong et al., 2008).
Les membres de Hsp70 cytoplasmiques participent aussi au bon repliement des
protéines, mais des domaines exposés dans la face cytosolique du RE. Il a été démontré que
Hsp70 en association avec Hsp40 et d’autres co-chaperonnes (telles que Hdj1/2, CHIP (C-
terminus of Hsp70 interacting protein), HspBP1 (Heat Shock Protein Binding 1)) interviennent
en complexe sur des domaines cytoplasmiques des protéines du RE. En fonction du complexe
formé, la protéine sera envoyée soit vers la voie de sécrétion soit vers la voie de dégradation
(Figure 8).
Figure 8 : Influence des co-chaperonnes du complexe Hsp70 sur le devenir des protéines. CHIP : C-terminus of Hsp70 interacting protein. Hdj : Human DnaJ2. Hop : Hsp-organising protein
Hsp : Heat Shock Protein. (Modifié d'après Goldberg, 2003).
Hdj1 et Hdj2 semblent faciliter le repliement des protéines (Meacham, 1999; Farinha et al.,
2002). HspBP1 quant à elle, régule négativement la co-chaperonne CHIP qui augmente
l’activité du système ERAD (Connell et al., 2001; Meacham et al., 2001a; Alberti et al., 2004).
CHIP est une E3 ligase qui interagit avec une famille d’enzymes de conjugaison d’ubiquitine
E2 (E2 ubiquitin conjugating enzymes) et se lie sur Hsp70 ou sa forme constitutivement
exprimée, Hsc70 (heat shock cognate) mais également sur Hsp90 (Cyr et al., 2002). Même si

Introduction
25
les deux chaperonnes lient CHIP de la même affinité, elles présentent des effets opposés sur
l’ubiquitinylation de protéines. En effet, il a été proposé que Hsp70 favorise l’ubiquitinylation
et la dégradation de substrats, tandis que Hsp90 les inhibe (Peng et al., 2009).
ii. Hsp90
La famille des Hsp90 regroupe des protéines d’environ 90 kDa et qui sont présentes
dans la plupart des compartiments cellulaires tels que le cytosol, le RE et les mitochondries.
Elles jouent un rôle dans la prévention de l’agrégation des protéines néo-synthétisées et
facilitent leur repliement ou leur dégradation. Cette famille de protéine joue également un rôle
essentiel dans de nombreux autres processus cellulaires, y compris la réponse de la cellule au
stress, le contrôle du cycle cellulaire, la survie cellulaire et d’autres voies de signalisation (Pour
revue Jackson, 2012; Mayer and Le Breton, 2015; Schopf et al., 2017).
Les membres de HSP90 fonctionnent comme des homodimères et la dimérisation est
essentielle pour leur fonction in vivo (Wayne and Bolon, 2007). Un monomère de HSP90 se
compose de trois domaines hautement conservés : un domaine N-terminal NTD, un domaine
intermédiaire MD et un domaine C-terminal CTD. Le domaine NTD permet la fixation de
l’adénine, du calcium, des co-chaperonnes et régule l’hydrolyse d’ATP (Prodromou et al.,
1997). Le domaine MD possède une large boucle hydrophobe qui contrôle la fixation de l’ATP
et permet la liaison de substrats. Ces 2 domaines NTD et MD sont connectés par un connecteur
long, flexible et chargé qui module les contacts NTD-MD et affecte la fonction de Hsp90
(Hainzl et al., 2009; Tsutsumi et al., 2012; Jahn et al., 2014). Le domaine CTD quant à lui, est
responsable de la dimérisation (Harris et al., 2004; Cunningham et al., 2008). Il contient
également un motif C-terminal important pour l’interaction avec certaines co-chaperonnes
(Buchner, 1999). En l’absence d’ATP, Hsp90 favorise une conformation ouverte. En revanche,
lors de la liaison d’ATP, un segment de couvercle de NTD se ferme sur le nucléotide lié,
conduisant à une dimérisation de NTD. Après l’hydrolyse de l’ATP et la libération de
nucléotides, Hsp90 revient à son état ouvert (Shiau et al., 2006; Hellenkamp et al., 2017).
Hsp90 coopère avec divers co-chaperonnes qui régulent son activité ATPase et recrutent les
substrats (Li et al., 2012; Röhl et al., 2013) . Ces facteurs agissent en séquentiel le long du cycle
Hsp90 et, dans certains cas, forment des complexes mixtes avec la chaperonne (Li et al., 2011).
Parmi ces co-chaperonnes, Hop (Hsp-organising protein) et Cdc37 qui stabilisent la
conformation ouverte du dimère Hsp90, inhibent l’hydrolyse d’ATP et facilitent la liaison avec
le substrat. Hop facilite le transfert de substrat de Hsp70 vers Hsp90, alors que Cdc37

Introduction
26
fonctionne comme un adaptateur pour les substrats kinase. Une autre co-chaperonne, Aha1
(Activator of Hsp90 ATPase protein 1) se lie asymétriquement au MD et NTD du dimère
Hsp90, facilitant la transition vers la conformation fermée et accélérant ainsi l’hydrolyse d’ATP
(Panaretou et al., 2002; Meyer et al., 2004; Koulov et al., 2010; Retzlaff et al., 2010). Quant à
la co-chaperonne P23, elle agit vers la fin du cycle et facilite la maturation du substrat en
stabilisant l’état fermé des domaines N de Hsp90 et en inhibant l’hydrolyse de l’ATP
(Prodromou, 2000; Ali et al., 2006; Li et al., 2012). Hsp90 coopère avec une gamme d’autres
cofacteurs contenant des domaines TPR (Tetratricopeptide). Certains de ces facteurs
contiennent également des domaines peptidyl-prolyl-isomérase (PPIase) et participent au
repliement du substrat sur Hsp90 (Taipale et al., 2012). D’autres facteurs contiennent un
domaine avec une activité ubiquitine ligase comme CHIP, qui se lie aux deux chaperonnes
Hsc70 et Hsp90 à travers son domaine Tetratricopeptide (TPR) (Pratt et al., 2010).
Le mécanisme d’interaction de Hsp90 avec ses substrats et comment son cycle ATPase est
couplé à leur maturation n’est pas encore bien compris. Il a été cependant postulé que
l’hydrolyse de l’ATP de Hsp90 régulerait le transfert du substrat de Hsp70 vers Hsp90,
probablement grâce à un couplage des cycles d’ATP des deux chaperonnes (Kirschke et al.,
2014). Des études avaient démontré en effet que Hsp70 et Hsp90 agiraient ensemble mais en
séquentiel pour prévenir l’agrégation des chaines polypeptidiques en cours de repliement et que
Hsp90 agirait en aval en se liant sur les intermédiaires de repliement tardifs (Kosano et al.,
1998). De plus, le rôle de Hsp40, une co-chaperonne de Hsp70 pour l’activité de Hsp90 a été
démontré in vivo par des études de génétiques dans la levure montrant que des mutations dans
Ydj1 (yeast dnaJ) compromettent la capacité de Hsp90 à chaperonner les récepteurs de stéroïdes
ou la protéine kinase pp60v-src (Caplan et al., 1995; Kimura et al., 1995). Sa nécessité pour
l’activité de Hsp90 a également été démontrée par des études in vitro sur des complexes de
Hsp90 avec le récepteur progestérone (PR) (Kosano et al., 1998; Hernández et al., 2002) ou
glucocorticoïde (GR) (Dittmar et al., 1998; Kirschke et al., 2014). Il a été constaté que cette co-
chaperonne se lie beaucoup plus facilement à PR dans sa forme native que sous sa forme
dénaturée, suggérant que sa liaison serait la première étape pour la voie de chaperon HSP90
pour PR (Hernández et al., 2002).
Parmi les membres de la famille Hsp90, Grp94 (glucose-regulated protein 94),
fortement exprimée dans la lumière du RE (Marzec et al., 2012). Il présente de nombreuses
similitudes avec les Hsp90 cytosoliques. Cependant, Grp94 lie le calcium dans le RE, possède
un ensemble distinct de substrats à plusieurs domaines, tous liées avec des disulfures et ne

Introduction
27
présente pas de motif Tetratricopeptide (TPR) (Soldano et al., 2003; Dollins et al., 2007; Frey
et al., 2007; Immormino et al., 2009; Marzec et al., 2012). Les co-chaperonnes qui pourraient
moduler son activité n’ont pas encore été identifiés. Il a été démontré que GRP94 est souvent
associée à BiP. L’inhibition par voie pharmacologique ou moléculaire de l’une des deux
chaperonnes induit une up-régulation de l’autre chaperonne. GRP94 agirait après BiP en se liant
sur les intermédiaires de repliement tardifs. Cette distinction semble être due à des indices
structurels différents auxquels la GRP94 est sensible (Gidalevitz et al., 2013). Il a été observé
que le knockdown de GRP94 stabilise le substrat classique d’ERAD, α-1-antitrypsin NHK (Null
Hong Kong), suggérant que tout comme BiP, cette chaperonne jouerait un rôle dans l’adressage
de cette protéine mal repliée vers la voie de dégradation ERAD (Christianson et al., 2008). De
plus, une interaction de GRP94 avec OS-9 (OsteoSarcoma amplified 9), une protéine qui
reconnait les glycoprotéines mal repliées et les envoie vers la voie ERAD a été observée
(Christianson et al., 2008; Braakman and Hebert, 2013).
Tout comme Hsp70 cytosolique, Hsp90 cytosolique interagit avec les domaines
cytoplasmiques de différentes protéines insérées dans la membrane du RE, dont la protéine
CFTR (Cystic Fibrosis Transmembrane conductance Regulator), et influence leur stabilité.
c) Les chaperonnes de transport hors le RE
La sortie des protéines correctement repliées du RE se produit au niveau de sites
spécifiques de la membrane du RE, appelés sites de sortie du RE (ERES pour ER-exit sites),
qui sont séparés spatialement des régions où la translocation active a lieu. Au niveau de ces
sites, les protéines correctement repliées sont empaquetées dans des vésicules pour leur
adressage vers un compartiment situé entre le RE et l’appareil de Golgi appelé ERGIC (ER-
Golgi Intermediate Compartment) selon un transport antérograde (Appenzeller-Herzog, 2006;
McCaffrey and Braakman, 2016). Ces vésicules de transport sont appelées vésicules COPII
(COatomer protein II), en référence à leur complexe de revêtement spécifique qui entraîne
l’assemblage et le bourgeonnement de la vésicule. Les vésicules COPII sont composées d’une
enveloppe externe comprenant entre autre les protéines Sec13 et Sec31 qui favorisent la
polymérisation de revêtement (Coat) ainsi que le processus de bourgeonnement. Elles sont
également composées d’une enveloppe interne comprenant les protéines Sec23 et Sec24 qui
trient les protéines néosynthétisées par absorption sélective (Lederkremer et al., 2001; Miller et
al., 2002, 2003; D’Arcangelo et al., 2013). Les protéines correctement repliées sont en effet

Introduction
28
sélectivement concentrées par liaison directe ou indirecte à la sous-unité Sec24 afin de
permettre leur exportation. Les protéines résidentes du RE et les protéines mal/non repliées ne
sont pas reconnues par ce système et ne vont donc pas sortir du RE (Geva and Schuldiner,
2014a).
L’empaquetage des protéines dans les vésicules COPII suivrait deux modèles (Figure 9) :
Figure 9 : Modes de transport des protéines hors du réticulum endoplasmique.
(Geva and Schuldiner, 2014b)
Le transport « bulk flow » : pour ce mode de transport, les protéines sont incorporées
de manière non sélective dans les vésicules COPII. Il est proposé pour ce modèle que les
protéines quittent le RE en suivant le fluide de la membrane des vésicules formées sans
l’intervention de récepteurs. Ce flux de protéines hors du RE est complètement contrôlé par les
chaperonnes et autres composants de l’ERQC qui retiennent les protéines mal/non repliées
telles que les chaperonnes calnexine et calréticuline. Ces interactions réduiraient en effet la
mobilité des protéines dont la mise en conformation n’est pas terminée, leur accès aux vésicules
COPII et ainsi leur sortie du RE. Les protéines ayant atteint leur conformation native, quant à
elles, sont libérées de ces interactions et peuvent ainsi pénétrer dans les vésicules COPII d’une
manière non sélective et quitter le RE (Balch and Gist Farquhar, 1995; Geva and Schuldiner,
2014b).

Introduction
29
Le transport médié par un récepteur membranaire : ce mode de transport implique
l’interaction des glycoprotéines avec un récepteur membranaire présent aux sites de sorties du
RE et capable d’interagir avec le manteau de la vésicule (Balch et al., 1994). Parmi ces
récepteurs sont retrouvés ERGIC53, VIP36 (36 kDa Vesicular Integral membrane Protein) et
VIPL (VIP36-Like) (Yamamoto, 2014). Ces récepteurs sont classés comme lectines type-L car
ils possèdent un domaine homologue de CRD (Carbohydrate Recognition Domain) (Kamiya et
al., 2008). Des études antérieures ont révélé qu’ERGIC53 et VIP36 présentent des spécificités
et affinités aux substrats différentes malgré des similitudes structurelles dans leur CRD (46%
de similitude), suggérant leurs rôles biologiques distincts dans le trafic intracellulaire.
La lectine ERGIC53 est localisée principalement dans le compartiment ERGIC. Ce
récepteur qui forme des homodimères et des homohexamères, possède un grand domaine
lectine luminale dans la partie N-terminale qui aurait pour rôle de lier les protéines glycosylées,
et un domaine dans sa partie cytosolique qui permet la liaison avec les molécules COPII.
Constitutivement recyclé entre le RE et l’ERGIC, ERGIC53 facilite le transport du RE vers
l’ERGIC de nombreux substrats présentant un motif N-glycan à haute teneur en mannose, de
manière dépendante du Ca2+ et du pH (Kamiya et al., 2008; Satoh et al., 2014a; Yamamoto,
2014) (Figure 10). ERGIC-53 semble lier efficacement le mannose de ses substrats à un pH de
7.4 minimum (Kamiya et al., 2005). Il présente une affinité de liaison plus faible et une
spécificité plus large que VIP36 et VIPL et peut se lier aux glycans avec une branche A
déglucosylée ou monoglucosylée (Kamiya et al., 2008; Satoh et al., 2014b). Il a été révélé que
l’expression d’ERGIC53 avec d’autres chaperonnes du RE est surexprimée dans des conditions
de stress du RE (Qin et al., 2012).

Introduction
30
Figure 10 : Transport des glycoprotéines dans la voie sécrétoire (Yamamoto, 2014). Les protéines ayant acquis leur structure correctement repliée dans le cycle de calnexine/calréticuline sont transportées vers le compartiment ERGIC via le récepteur ERGIC53 dans des vésicule COPII. Une fois dans le compartiment ERGIC, les glycoprotéines sont libérées puis transportées vers l’appareil de Golgi, et ERGIC53 est recyclé vers le RE via des vésicules de type COPI. Dans le cis-Golgi, les glycoprotéines sont soit clivées en Man5 avant la reglucosylation par les glycosyltransférases de Golgi, soit marquées avec le signal lysosomale Man-6-P par l’action séquentielle de phosphotransférase (PT) et phosphodiesterase (PD). Certaines glycoprotéines échappent aux clivages, mais elles peuvent être reconnues par VIP36 dans le trans-Golgi et recyclées au cis-Golgi pour une autre tentative de clivage ou bien recyclées au RE pour les transmettre à la chaperonne BiP. Les protéines transportant des résidus Man-6-P sont reconnues par les MPR (mannose 6-phosphate receptors) dans le trans-Golgi et transportées vers les endosomes via des vésicules revêtues de clathrin. Les glycoprotéines correctement repliées sont transportées vers leur localisation physiologique. Ce processus peut être médiés par des lectines qui sont cependant inconnues (Modifié d'après Hauri et al., 2000; Yamamoto, 2014).

Introduction
31
Une fois dans l’appareil de Golgi, la glycoprotéine est démannosylée par l’α-
mannosidase I de Golgi formant la structure Man5GlcNAc2, qui est un substrat pour N-
acetylglucosaminyltransferases (GnT-I), une enzyme clé dans le développement d’organismes
multicellulaires. L’addition du premier résidu GlcNAc par GnT-I génère
GlcNAcMan5GlcNAc2, substrat pour GnT-III ou α-mannosidase II. La GnT-III oriente la voie
vers des structures hybrides, alors que l’α-mannosidase II clive deux résidus mannose et génère
GlcNAcMan3GlcNAc2. Ce dernier peut être encore modifié par une série de GnTs (GnT-II, IV,
V et VI) qui génèrent différentes branches de complexes N-glycans (Vasconcelos-dos-Santos
et al., 2015) (Figure 11).
Figure 11 : Représentation schématique de la maturation des oligosaccharides N-liés. GNT-I à V : N-acetylglucosaminyltransferases. FucT : Fucosyltransferase. α-Mann I/II : α-Mannosidase I. DMJ : 1-deoxymannojirimycin. LacNac : N-Acetyl-D-lactosamine. Lex : LewisX. SLex : Sialyl LewisX. Ley : Lewisy (D'après Vasconcelos-dos-Santos et al., 2015).
Les protéines mal repliées qui « accidentellement » se retrouvent transportées par les vésicules
COPII jusqu’à l’appareil de Golgi sont recyclées vers le RE selon un transport rétrograde par
des vésicules de type COPI (Hsu et al., 1991; Teasdale and Jackson, 1996). Cela est assuré par
des molécules de capture, tels que les récepteurs de protéines ou le complexe COPI lui-même
qui servent de médiateur à la récupération sélective de protéines mal/non repliées. Ainsi, le
compartiment ERGIC est considéré comme un point de contrôle laissant transiter vers l’appareil
de Golgi uniquement les protéines correctement repliées (Kukushkin et al., 2011).

Introduction
32
VIP36 a été découvert dans un criblage pour les protéines impliquées dans le tri apical
du réseau trans-Golgi (Fiedler et al., 1994). Contrairement à ERGIC53 qui est non glycosylé,
VIP36 a un site de N-glycosylation et acquiert une glycosylation complexe. Cette lectine est
localisée principalement dans l’appareil de Golgi et seulement dans une certaine mesure dans
le compartiment ERGIC (Füllekrug et al., 1999; Reiterer et al., 2010; Hoang et al., 2015).
D’autres études l’ont cependant localisé dans des vésicules sécrétoires post-Golgienne et au
niveau de la membrane plasmique (Hara-Kuge et al., 2002; Shimada et al., 2003). VIP36 se lie
aux oligosaccharides hautement glycosylés ayant la branche A déglucosylée. Elle présente une
dépendance au pH pour lier ses substrats, avec des valeurs de pH optimales d’environ 6.5
(Kamiya et al., 2005, 2008). A noter, les valeurs de pH typiques du réseau RE, cis-Golgi et
trans-Golgi correspondent respectivement aux 7.2, 6.4 et 5.4 (Wu et al., 2001; Weisz, 2003). Il
a été suggéré que VIP36 présente des résidus d’acides aminés « senseurs de pH », responsables
de la détection du pH (Kamiya et al., 2005). Bien que les propriétés de liaison aux
oligosaccharides de VIP36 aient été élucidées de façon assez détaillée, la fonction biologique
de cette lectine reste une énigme. En effet, il a été suggéré d’une part que VIP36 interagit avec
l’α-amylase de rat et la clusterine, et par conséquent, un rôle dans le transport antérograde post-
Golgi lui a été postulé. D’autre part, basé sur l’observation d’une liaison entre VIP36 et l’α1-
antitrypsin mal/non repliée dans l’appareil de Golgi et le RE, il a été suggéré que cette lectine
agirait dans le contrôle de qualité post-ER en se liant à cette protéine qui s’est échappée de
l’ERQC pour la recycler de nouveau dans le RE pour un contrôle de qualité supplémentaire
(Reiterer et al., 2010). De plus, il a été observé que le complexe VIP36/α1-antitrypsine co-
immunoprécipite avec la chaperonne BiP (Reiterer et al., 2010). A noter, VIP36 semble lier un
complexe avec BiP indépendamment de son activité de liaison N-glycan (Nawa et al., 2007).
Enfin, la protéine VIPL a été identifiée lors d’un criblage de structure sur le domaine
CRD (Neve et al., 2003). Comme VIP36, VIPL se lient aux oligosaccharides hautement
glycosylés ayant la branche A déglucosylée. Bien que possédant une séquence génique plus
proche de VIP36 que d’ERGIC53, VIPL est localisée principalement dans le RE (Nufer et al.,
2003; Qin et al., 2012). L’équipe de Pettersson a démontré que l’extinction par siRNA (small
interfering RNA) de VIPL diminue la sécrétion de deux glycoprotéines, proposant ainsi un rôle
de VIPL dans le transport médié par un récepteur membranaire (Neve et al., 2003). Une autre
étude a démontré que VIPL ne transporte pas les protéines du RE et que son rôle serait de
contrôler la fonction d’ERGIC53 en modulant sa sortie du RE (Nufer et al., 2003). Kamiya et
ses collaborateurs quant à eux proposent un autre modèle selon lequel VIPL lie les

Introduction
33
glycoprotéines déglucosylées à haute teneur en mannose immédiatement après leur sortie du
cycle calnexin/calréticuline étant donné sa localisation dans le RE et sa sélectivité élevée pour
lier la branche A complètement déglucosylée. Ainsi, VIPL aurait un rôle pour protéger les
glycoprotéines correctement repliées de la dégradation en empêchant leur démannosylation par
les manosidases du RE et leur interaction avec la famille de protéines EDEM (Kamiya et al.,
2008). Basé sur leur résultats et les études démontrant que la surexpression de VIPL redistribue
ERGIC53 dans le RE (Neve, 2003; Nufer et al., 2003), la même équipe propose que les
glycoprotéines liées au VIPL seraient remises à ERGIC-53 (Kamiya et al., 2008).
B. La voie ERAD
Bien que certaines protéines acquièrent leur conformation native avec une efficacité
proche de 100% telle que la protéine HA de la grippe (Braakman et al., 1991), une grande
partie (environ 75%) de protéines telles que le CFTR, l’apolipoprotéine A ou le récepteur
d’érythropoïétine ont une faible efficacité de repliement avec une dégradation des protéines
n’atteignant pas la bonne conformation (Araki and Nagata, 2011).
Chez les mammifères, il existe plusieurs mécanismes de dégradation des protéines
n’arrivant pas à atteindre leur conformation native. Ces mécanismes peuvent fonctionner d’une
manière parallèle (Fu and Sztul, 2009; Le Fourn et al., 2013) :
La voie de macro-autophagie du RE (réticulophagie) : Il s’agit d’une forme
particulière de l’autophagie qui élimine des protéines en les enveloppant dans des
vésicules à double membrane « les autophagosomes ». Ces dernières fusionnent avec
des lysosomes contenant des hydrolases afin de dégrader les substrats. Ce processus
était attribué à la dégradation des protéines agrégées, afin de remédier aux contraintes
de taille, les empêchant d’être dégradées par la voie ERAD. Cependant, des études ont
démontré que la macro-autophagie n’est pas liée à l’accumulation de protéines dans
l’ERQC. En effet, il a été reporté que la forme mutante de GnRHR (Gonadotropin-
Releasing Hormone Receptor) est dégradée spécifiquement par la voie de l’autophagie
même si elle ne forme pas d’agrégats (Houck et al., 2014). Ceci, suggère que ce
processus de dégradation pourrait être spécifique du substrat. De plus, il a été proposé
que l’autophagie peut être impliquée dans l’élimination des protéines CFTR mal
repliées et qu’elle jouerait un rôle compensatoire lorsque la fonction protéasomique est
surchargée (Fu and Sztul, 2009; Luciani et al., 2011, 2012).

Introduction
34
La voie des vésicules EDEM « EDEMosomes » : cette voie correspond à la
ségrégation des régulateurs d’ERAD dans des vésicules distinctes des autophagosomes
afin de les ségréger hors du RE et de les transporter vers la dégradation en fusionnant
avec des lysosomes. Cependant, des protéines mal repliées ainsi que des complexes
multiprotéiques destinés à la dégradation ont été identifiés dans ces vésicules (Zuber et
al., 2007; Le Fourn et al., 2013). Cette partie sera plus développée en page 46.
La voie de dégradation associée au RE, ERAD:
ERAD est la voie majoritaire de dégradation des protéines mal repliées. Elle consiste en
la rétro-translocation de ses substrats vers le cytosol pour leur dégradation dépendante de
l’ubiquitine-protéasome (Hegde et Ploegh 2010). Un certain nombre de substrats
physiologiques et pathologiques ciblé par ce processus ont été identifiés, dont l’alpha-1
antitrypsine et la protéine CFTR (Vij et al., 2006; Needham and Brodsky, 2013). Ce processus
comprend 4 étapes : la reconnaissance de substrat, son ubiquitinylation, sa rétrotranslocation
vers le cytosol et sa dégradation par le protéasome (Figure 12).

Introduction
35
Figure 12 : Etapes de la dégradation ERAD des glycoprotéines. Reconnaissance : la reconnaissance est médiée par des chaperons réticulaires ou cytoplasmiques, en fonction de l’emplacement de la lésion (étoile rouge). Les lectines sont représentées en rouge. Ubiquitinylation : après la reconnaissance, la machine d’ubiquitinylation est recrutée sur le substrat mal replié, soit directement dans la membrane, soit par des interactions avec des chaperons cytoplasmiques. Une enzyme d’activation de l’ubiquitine (E1) transfère l’ubiquitine (cercle gris) vers un site actif d’une enzyme de conjugaison d’ubiquitine (E2) dans un processus ATP-dépendant. L’ubiquitine est ensuite transférée sur le substrat via une ubiquitine ligase (E3). L’ubiquitinylation à la membrane du RE peut se produire via des E3-ligases cytoplasmiques ou membranaire, toutes deux montrées. Rétro-translocation : la retro-translocation dépend presque toujours du complexe p97/Cdc48, qui comprend Ufd1 et Npl4 et interagit avec l’ubiquitine et les régions mal repliées sur un substrat. P97/Cdc48 fournit la force mécanique par hydrolyse d’ATP pour l’élimination du substrat. Dégradation : suite à leur retro-translocation, les protéines mal repliées sont introduites dans le protéasome 26S et doivent être maintenues solubles pour éviter l’agrégation (Modifié d'après Guerriero and Brodsky, 2012).

Introduction
36
La première étape de la voie ERAD est l’identification des substrats à dégrader. Selon
la localisation du défaut de repliement, ces substrats sont reconnus soit par les chaperonnes
cytosoliques tel que Hsp70 (défaut de repliement dans la partie cytoplasmique) (Grove et al.,
2011) ; ou bien par BiP et des lectines d’ERAD dont EDEM, OS-9 ou XTP3B (XTP3-
transactivated gene B protein) (défaut de repliement dans la partie luminale) (Groisman et al.,
2011).
Malgré les progrès dans la compréhension d’ERAD fait au cours des dernières années, la fin
des cycles de reglucosylation/déglucosylation des glycoprotéines n’est pas à ce jour
complètement comprise. Il est considéré cependant, que chaque glycoprotéine présente un
certain laps de temps pour le repliement correct. Si ce temps de repliement est dépassé et que
la glycoprotéine ne peut pas atteindre sa conformation appropriée, elle est finalement ciblée
pour l’ERAD. Toutefois, certains substrats tels que des polypeptides extensivement mal repliés
ne passent le cycle calnexine/calréticuline qu’une seule fois et ne sont pas reconnus par le
senseur de repliement UGGT.
L’extraction des glycoprotéines identifiées comme définitivement mal repliées du cycle
calnexine/calréticuline semble être effectuée par le/les α1,2-mannosidase(s) dans le RE. Le
clivage de résidus mannose joue en effet un rôle crucial dans la génération de signaux de
dégradation des glycoprotéines mal repliées (Herscovics, 2001; Helenius and Aebi, 2004;
Molinari, 2007). Chez la levure Saccharomyces cerevisiae, deux étapes de clivage de mannose
sont cruciales pour ce processus. La première étape est assurée par l’α1,2-mannosidase Mns1
qui clive un résidu mannose de la branche B du motif Man9GlcNAc2 (M9) et génère ainsi le
motif Man8GlcNAc2 (M8B) ; et la seconde étape est catalysée par l’α1,2-mannosidase Htm1
(homologous to mannosidase I) qui assure la conversion du M8B en un oligosaccharide
exposant le résidu mannose j (Jakob et al., 2001; Clerc et al., 2009) (Figure 13), un ligand pour
la lectine Yos9p qui le dirige vers la dégradation (Quan et al., 2008). Dans les cellules
mammifères, ce processus est cependant beaucoup plus complexe, du fait qu’elles expriment la
protéine ER mannosidase I (ERmanI) comme le seul homologue de MnsI, mais expriment de
multiples homologues pour HtmI, nommés EDEM1, EDEM2 et EDEM3. La contribution
exacte de chaque α1,2-mannosidase au processus de clivage de mannose n’est pas encore
complètement définie (Gonzalez et al., 1999; Hosokawa et al., 2003; Mast et al., 2005; Hirao
et al., 2006; Hosokawa et al., 2010).

Introduction
37
Figure 13 : Structure du motif oligosaccharide N-glycan Glc3Man9GlcNAc2. A, B et C : les branches oligosaccharidiques. Les enzymes qui modifient ce motif sont listées sur la figure et leur action est représentée. Le type de liaison glycosidique est représenté en couleur (D'après Słomińska-Wojewódzka and Sandvig, 2015).
Bien qu’initialement la génération de l’isomère M8B a été supposée comme le signal
pour la dégradation, cet isomère est également porté par les glycoprotéines correctement
repliées qui transitent vers l’appareil de Golgi. Ainsi, il est proposé que d’autres clivages de
résidu mannose, éventuellement trois ou quatre α1,2-mannoses, sont nécessaires pour dériver
les glycoprotéines mal repliées vers la voie ERAD (Frenkel et al., 2003; Clerc et al., 2009).
L’élimination du mannose g (Figure 13) empêche en effet la restauration de l’état
monoglucosylé de la structure glycanique, ce qui extrait de façon irréversible les substrats
ERAD du cycle calnexine/calréticuline. Le clivage du mannose k, quant à lui, expose le
mannose j à liaison α1,6 qui devient une cible pour les régulateurs ERAD cruciaux (Hosokawa
et al., 2009; Yamaguchi et al., 2010; Groisman et al., 2011; Hebert and Molinari, 2012). Le
mannose f peut lui aussi être éliminé. L’accumulation d’évidences indique que les protéines
EDEMs sont les principaux acteurs de ces clivages. Ainsi, ces protéines peuvent contrôler le
statut de repliement/mauvais repliement des glycoprotéines d’une manière très spécifique, ce
qui contribue à éviter une destruction inutile de protéines encore en cours de repliement.
Suite à l’exposition du mannose j, les lectines d’ERAD : OS-9 et XTP3-B (transactivated
protein ou erlectin), deux homologues de Yos9p de la levure, sont recrutées (Christianson et
al., 2008; Hosokawa et al., 2008, 2009; Groisman et al., 2011). Ce sont des protéines solubles
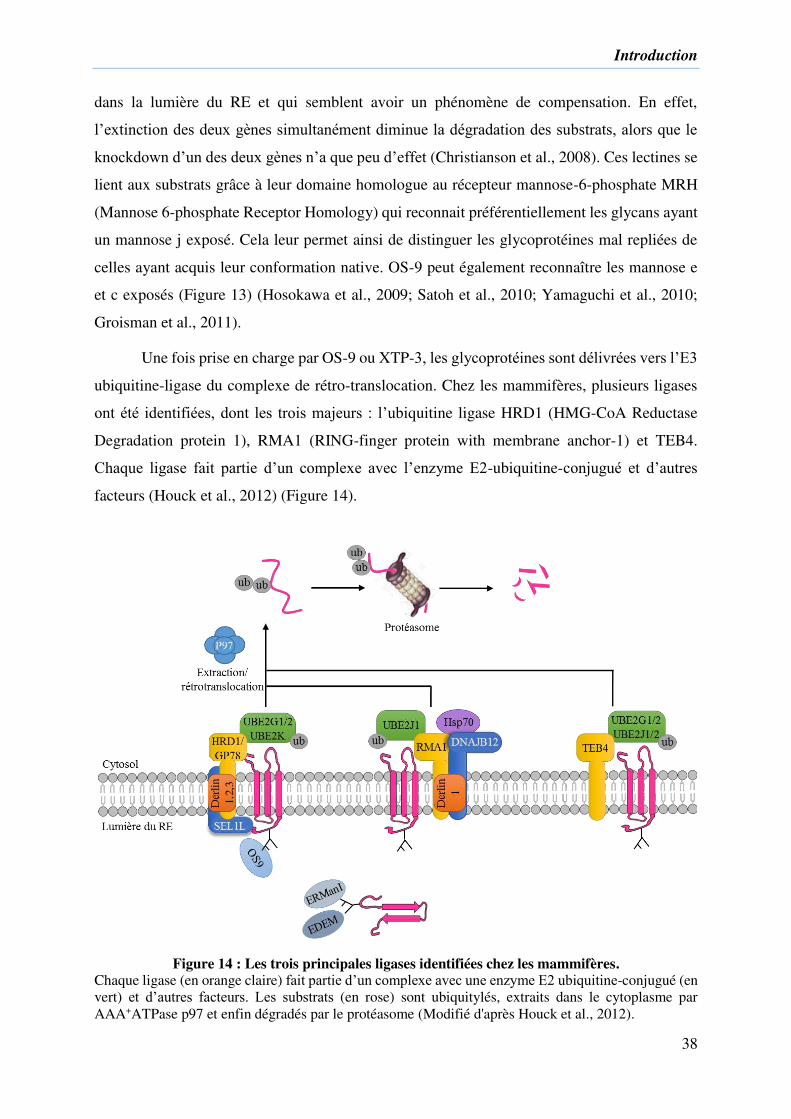
Introduction
38
dans la lumière du RE et qui semblent avoir un phénomène de compensation. En effet,
l’extinction des deux gènes simultanément diminue la dégradation des substrats, alors que le
knockdown d’un des deux gènes n’a que peu d’effet (Christianson et al., 2008). Ces lectines se
lient aux substrats grâce à leur domaine homologue au récepteur mannose-6-phosphate MRH
(Mannose 6-phosphate Receptor Homology) qui reconnait préférentiellement les glycans ayant
un mannose j exposé. Cela leur permet ainsi de distinguer les glycoprotéines mal repliées de
celles ayant acquis leur conformation native. OS-9 peut également reconnaître les mannose e
et c exposés (Figure 13) (Hosokawa et al., 2009; Satoh et al., 2010; Yamaguchi et al., 2010;
Groisman et al., 2011).
Une fois prise en charge par OS-9 ou XTP-3, les glycoprotéines sont délivrées vers l’E3
ubiquitine-ligase du complexe de rétro-translocation. Chez les mammifères, plusieurs ligases
ont été identifiées, dont les trois majeurs : l’ubiquitine ligase HRD1 (HMG-CoA Reductase
Degradation protein 1), RMA1 (RING-finger protein with membrane anchor-1) et TEB4.
Chaque ligase fait partie d’un complexe avec l’enzyme E2-ubiquitine-conjugué et d’autres
facteurs (Houck et al., 2012) (Figure 14).
Figure 14 : Les trois principales ligases identifiées chez les mammifères. Chaque ligase (en orange claire) fait partie d’un complexe avec une enzyme E2 ubiquitine-conjugué (en vert) et d’autres facteurs. Les substrats (en rose) sont ubiquitylés, extraits dans le cytoplasme par AAA+ATPase p97 et enfin dégradés par le protéasome (Modifié d'après Houck et al., 2012).

Introduction
39
Pour la ligase TEB4, les seules protéines adaptatrice connues sont des enzymes de conjugaison
E2 ubiquitine UBE2G1/2 et UBE2J1/2. Bien que peu d’informations sont disponibles pour cette
ligase, son homologue chez la levure, Doa10, ubiquitinyle des substrats ERAD avec des défauts
de repliement dans les domaines cytosoliques (Hirsch et al., 2009; Houck et al., 2012). La ligase
RMA1 forme un complexe avec Derlin-1, l’enzyme de conjugiason E2 ubiquitine UBE2J1 et
DNAJB12 (DnaJ Hsp40 family member B12), une chaperonne de la famille Hsp40 qui coopère
avec le Hsp70 cytosolique pour faciliter l’ubiquitinylation dépendante de RMA1 des substrats
(Yamamoto et al., 2010; Grove et al., 2011). Les protéines de la famille Derlin (Derlin 1-3) sont
des protéines adaptatrices membranaire qui seraient impliquées dans la reconnaissance des
défauts de repliement dans les domaines membranaires de protéines (Houck et al., 2012). Le
complexe de la ligase HRD1 est le complexe le mieux caractérisé. Cette ligase forme un
complexe avec de nombreuses protéines adaptatrices incluant SEL1L (Suppressor/Enhancer of
Lin-12-Like), OS-9, Derlin-1 et les enzymes de conjugaison E2 ubiquitine UBE2G1 et
UBE2G2. Les protéines SEL1L et OS-9 sont impliquées dans la reconnaissance des défauts de
repliement de protéine dans la lumière du RE. SEL1L joue le rôle de récepteur de substrats pour
ce complexe. Il interagit avec d’autres lectines EDEM1, OS-9, XTP3B et BiP. Suite à la
réception du substrat, le complexe HRD1 s’assemble grâce à la protéine Herp (Endoplasmic
Reticulum stress-inducible protein) (Leitman et al., 2014).
Afin d’assurer leur délivrance au protéasome 26S, les substrats sont ubiquitinylés dans
le cytosol, principalement au niveau des résidus lysine (Sadowski and Sarcevic, 2010).
Plusieurs protéines assistent à la rétro-translocation de la protéine dont BiP et des membres de
la famille PDI et PPIase qui déplient la protéine pour faciliter sa rétro-translocation. À la face
cytosolique du complexe ERAD, l’hexamère AAA+ ATPase p97/VC, présent dans tous les
complexes de rétro-translocation, permet l’incorporation des substrats dans le cytosol
(Chapman et al., 2010) (Figure 14). P97 recrute plusieurs modificateurs de chaîne ubiquitine, y
compris les E3 ligases et les facteurs d’allongement de la chaîne (Liang et al., 2006, Mueller et
al., 2008, Schuberth et Buchberger, 2008, Ernst et al., 2009).
Les substrats ubiquitinylés sont transférés au protéasome par des protéines de transport,
connues sous le nom de HR23A/B ou Ubiquilin-1. Ces protéines contiennent des domaines de
type ubiquitine-associated (UBA) et ubiquitine-like (UBL) qui se lient respectivement aux
chaînes poly-ubiquitiques et les sous-unités de protéasome (Rpn10/13, Rpt5) (Deveraux et al.,
1994; Lam et al., 2002; Raasi and Wolf, 2007; Husnjak et al., 2008; Lim et al., 2009).

Introduction
40
1) ERMnI et son rôle dans ERAD
ERManI est une glycoside hydrolase 47 (GH47), une famille qui inclut également les
trois EDEMs et trois α1,2-mannosidases de Golgi (Golgi ManIA, IB et IC). Excepté le domaine
mannosidase, il existe peu ou pas de conservation parmi les membres de cette famille (Figure
15). Ces α-mannosidases de classe I hydrolysent spécifiquement les résidus de mannose liés à
l’α1,2 et nécessitent du calcium pour leur activité enzymatique. Elles sont inhibées par le
1- déoxymannojirimycine (DMJ) et la kifunensine, mais pas par la swainsonine (Tulsiani and
Touster, 1983; Weng and Spiro, 1996; Gonzalez et al., 1999; Tremblay and Herscovics, 1999;
Moremen, 2002).
Figure 15 : Représentation schématique des membres de la famille GH47. La famille GH47 comprend trois sous-familles de protéines, y compris ER α1,2 mannosidase I (ERManI), les Golgi α1,2 mannosidases (GolgiManIA, IB et IC) et les trois EDEMs (EDEM1, EDEM2 et EDEM3). Le domaine de l’homologie de la mannosidase conservé est représenté en rose. EDEM3 contient un domaine associé à la protéase (PA) et le signal de rétention KDEL. Les régions transmembranaires d’ERManI et des GolgiManIA, IB et IC, sont représentées en noir. La région transmembranaire putative d’EDEM1 est indiquée en gris (Modifié d'après Olivari and Molinari, 2007).
ERManI est une protéine transmembranaire de type II qui a initialement été décrite
comme une protéine résidente du RE (Gonzalez et al., 1999; Tremblay and Herscovics, 1999).
L’équipe de Sifers a démontré cependant que cette protéine réside principalement dans
l’appareil de Golgi, où elle est soumise à une O-glycosylation. Il a été suggéré que le ciblage
des substrats, basé sur le code glycan pour ERManI, a lieu dans l’appareil de Golgi (Pan et al.,
2011). Deux ans plus tard, la même équipe a identifié une interaction directe entre ERManI et
γ-COP, la sous-unité gamma du complexe de protéine d’enveloppe I (COPI) (Pan et al., 2013).

Introduction
41
Selon cette étude, les deux protéines assurent une dégradation du substrat d’ERAD, AAT (α-1-
antitrypsin) et son mutant NHK (Null Hong Kong), suggérant qu’ERManI, par son interaction
directe avec γ-COP, contribue à la récupération vers le RE des substrats ERAD capturés,
indépendamment de son activité mannosidase. Cette hypothèse a été appuyée par
l’identification d’une séquence de décapeptide luminale non-enzymatique spécifique aux
vertébrés et hautement conservée, qui contribue à la rétention et à l’élimination du substrat
ERAD (Iannotti et al., 2014). Cependant, cette étude a été controversée par l’équipe de
Lederkremer, qui a démontré que la localisation d’ERManI dans le complexe de Golgi était un
résultat artificiel d’une perturbation de la membrane lors de la fixation cellulaire et de la
perméabilisation (Benyair et al., 2015a). Cette équipe propose que des sous-régions spécialisées
du RE existent, où la concentration de mannosidase atteint des niveaux beaucoup plus élevés
que dans le reste du compartiment (Frenkel et al., 2003; Huyer et al., 2004; Benyair et al.,
2015a). Il semble que la concentration d’ERManI dans ces régions soit similaire aux niveaux
expérimentaux in vitro et provoque une élimination extensive du mannose (Wu et al., 2003;
Hebert and Molinari, 2007). Ainsi, un modèle a été proposé, selon lequel ERManI peut être
séquestré à l’état stationnaire, dans des vésicules dynamiques de contrôle de qualité QCV
(Quality Control Vesicles), et que sous des conditions d’accumulation de substrat (stress du
RE), ces vésicules convergent vers le compartiment ERQC (Benyair et al., 2015b, 2015a).
L’interaction transitoire d’ERManI avec ses substrats semble avoir lieu dans ces vésicules
QCVs (Benyair et al., 2015a) (Figure 16). Ainsi, tout en étant soumises à un contrôle de qualité
dans le but d’obtenir un repliement approprié, les glycoprotéines transitent entre le RE et les
QCV. Ce cycle permet la compartimentation de l’étape de clivage du mannose dans le contrôle
de la qualité, ce qui permet aux glycoprotéines dans le RE d’avoir un repliement approprié sans
subir de coupure de mannose.

Introduction
42
Figure 16 : Modèle de compartimentation du RE dans les cellules de mammifères. Après leur glycosylation (1), les protéines subissent le clivage de 2 résidus glucoses par les α-glucosidases I et II (GlcI et II) afin de rentrer dans le cycle calnexine/calréticuline (CNX/CRT) (2 et 3). Après un cycle de repliement, le dernier glucose est clivé par GlcII (4) permettant la libération des glycoprotéines. Les glycoprotéines correctement repliées (5) subissent un clivage du résidus mannose terminal de la branche B par l’α-mannosidase I (ERManI) (6), permettant leur adressage vers l’appareil de Golgi (7). Les glycoprotéines n’ayant pas atteint leur conformation native (8) seront reglucosylées par l’UGGT (UDP-Glc: glycoprotéine glucosyltransférase) afin de rentrer dans un nouveau cycle de repliement (9). Ces glycoprotéines vont être transportées entre les vésicules de contrôle qualité (QCV) (9) et les compartiments de contrôle qualité, dérivés du RE (ERQC) (10), sous des conditions de stress du RE. Les glycoprotéines identifiées comme définitivement mal repliées subiront un clivage des résidus mannoses à liaison de type α1,2-Man (11), favorisant leur liaison avec les lectines OS-9 (osteosarcoma amplified 9) et XTP3-B (XTP3-transactivated gene B protein) qui les ciblent vers la voie ERAD pour dégradation par le protéasome (12). ERManI résidant dans les QCVs et les composantes d’EDEMosome sont dégradés par un mécanisme autophagique/lysosomal (13 et 14) (adapté de Leitman et al., 2013). ERManI a été désignée comme le « minuteur » qui déclenche la première étape de la
dégradation en clivant le premier résidu mannose de la branche B de l’oligosaccharide N-glycan
(Araki and Nagata, 2011; Caramelo and Parodi, 2015; Zhou et al., 2015). Cependant, d’autres
résultats permettent de proposer un mécanisme entièrement nouveau impliquant un « double
contrôle » des substrats glycosylés d’ERAD dans les cellules de mammifères (Ninagawa et al.,
2014). Selon ce modèle, le clivage du mannose de Man9GlcNAc2 à la forme Man8GlcNAc2 est
effectué principalement par EDEM2, et la transformation de glycan en aval de Man8GlcNAc2
en Man7GlcNAc2 est réalisée principalement par EDEM3 et dans une moindre mesure par
EDEM1.

Introduction
43
2) EDEMs et leurs rôles dans ERAD
Les trois variantes d’EDEM appartiennent à la famille GH47. EDEM1-HA a été
initialement identifiée dans les cellules Cos-7 comme une protéine transmembranaire de type
II qui est insérée dans la membrane sous une forme de 69 kDa. Cette forme est par la suite
convertie en une glycoprotéine de 78 kDa par la fixation d’un motif de glycosylation sur 5
asparagines (Hosokawa et al., 2001). Il a été décrit qu’EDEM1 possède une séquence signal
non clivée qui sert de région transmembranaire liant la région associée à la membrane de la
calnexine et formant un complexe fonctionnel (Oda et al., 2003). Cependant, d’autres
expériences, confirmées aussi par des algorithmes informatiques, ont révélé qu’EDEM1 est une
protéine luminale dans les cellules de rein embryonnaire humain HEK293 (Human Embryonic
Kidney 293) (Olivari et al., 2005; Zuber et al., 2007). Ces résultats contradictoires pourraient
être expliqués par des variations probables dans le clivage du peptide signal dans différentes
lignées cellulaires (Olivari and Molinari, 2007a). Il a été suggéré en effet dans une étude menée
par Tamura et ses collaborateurs que la glycosylation co-traductionnelle et le clivage de la
séquence signal d’EDEM1 sont inefficaces, conduisant à une hypoglycosylation de la protéine
et à la génération de 2 formes de la protéine, soluble et membranaire (Tamura et al., 2011). Les
topologies d’EDEM2 et d’EDEM3 ne sont pas si controversées. Ce sont toutes les deux des
protéines solubles du RE. EDEM2 est une glycoprotéine de 64 kDa avec des oligosaccharides
N-liés au niveau de 4 asparagines (Mast et al., 2005; Olivari et al., 2005) et EDEM3 est une
protéine de 104 kDa glycosylée au niveau de 7 asparagines (Hirao et al., 2006). Comme pour
la séparation spatiale d’ERManI, des travaux ont révélé la présence d’EDEM1 dans des petites
vésicules nommées « EDEMosomes » qui auraient pour rôle la ségrégation de certains acteurs
de la machinerie ERAD hors du RE (détaillé en p.46).
Les membres du groupe des protéines de la famille GH47 partagent un domaine
d’homologie de mannosidase unique et similaire (Figure 15). De plus, tous les résidus
catalytiques nécessaires à l’activité glycolytique et à la liaison de l’inhibiteur spécifique des
α1,2-mannosidases, la kifunensine, sont conservés dans les protéines EDEM (Hosokawa et al.,
2001; Mast et al., 2005; Hirao et al., 2006; Olivari and Molinari, 2007a). Malgré cette
homologie, il y a eu une controverse quant à savoir si les protéines EDEM fonctionnent comme
des enzymes mannosidases ou des lectines. En effet, selon les études initiales sur EDEM1 chez
la levure (Htm1p) (Jakob et al., 2001) et les orthologues chez les mammifères, EDEM1 et
EDEM2, n’ont pas révélé d’activité hydrolytique (Hosokawa et al., 2001, 2003; Molinari et al.,
2003; Oda et al., 2003; Mast et al., 2005; Olivari and Molinari, 2007a). Cependant, d’autres

Introduction
44
études ont indiqué que les orthologues d’EDEM chez les protistes (Banerjee et al., 2007) et les
levures (Frenkel et al., 2003; Quan et al., 2008; Gauss et al., 2011) présentent une activité
mannosidase, et qu’EDEM1 surexprimé dans les cellules humaines accélère la
démannosylation des glycoprotéines mal repliées de la branche A et C des N-glycans. De plus,
il a été démontré qu’EDEM3 possède une activité α1,2-mannosidase in vivo, et qu’EDEM1 et
EDEM3 portant une mutation dans un résidu catalytique conservé parmi les α1,2-mannosidases,
n’accélèrent pas la démannosylation de N-glycan (Hirao et al., 2006; Olivari et al., 2006;
Hosokawa et al., 2010). Cela peut suggérer qu’EDEM1 et EDEM3 sont des mannosidases
actives. L’activité de l’α-mannosidase d’EDEM2 a été la plus controversée, mais les résultats
récemment publiés suggèrent que le clivage du mannose de Man9GlcNAc2 à la forme
Man8GlcNAc2 est effectué principalement par cette protéine et la transformation de glycan en
aval de Man8GlcNAc2 est réalisée principalement par EDEM3 et EDEM1 (Ninagawa et al.,
2014). Il a été observé qu’un traitement avec de la kifunensine diminue l’interaction d’EDEM1
avec SEL1L, suggérant que cette protéine ciblerait ses substrats vers le complexe de dislocation
contenant SEL1L (Cormier et al., 2009). Malgré toutes ces études sur l’activité catalytique des
protéines EDEM, l’activité mannosidase avec les EDEMs purifiées n’a pas encore été
démontrée.
Les protéines EDEMs présentent d’autres fonctions qui n’impliquent pas leur activité
catalytique. En effet, la reconnaissance du substrat à dégrader par EDEM1 et EDEM2 pourrait
être indépendante du glycan et donc non liée à l’activité catalytique des protéines EDEM,
suggérant que ces deux protéines reconnaissent probablement les régions mal repliées ou les
protéines aberrantes (Marin et al., 2012; Slominska-Wojewodzka et al., 2014; Tang et al.,
2014). Une interaction protéine-protéine d’EDEM1 ou EDEM2 a été observée avec certains
substrats tels que : rodopsin (Kosmaoglou et al., 2009), la chaine-A de ricin (Slominska-
Wojewodzka et al., 2006; Sokołowska et al., 2011; Slominska-Wojewodzka et al., 2014), sonic
hedgehog (Tang et al., 2014) et des substrats non-glycosylés de BiP (Shenkman et al., 2013).
D’autre part, il a été démontré qu’EDEM1 inhibe la formation d’agrégats covalents lors de la
libération de protéines mal repliées du cycle calnexine/calréticuline, et cette activité chaperonne
d’EDEM1 est indépendante de son activité catalytique (Hosokawa et al., 2006; Olivari et al.,
2006; Ron et al., 2011; Guiliano et al., 2014). Par mutagenèse dirigée il a également été
démontré qu’EDEM1 régule le taux d’ERManI indépendamment de son activité mannosidase
(Termine et al., 2009).

Introduction
45
Les protéines en cours de repliement sont protégées de la dégradation prématurée par
un processus nommé « ERAD tuning », qui assure la ségrégation des acteurs d’ERAD hors du
RE et d’ERQC. Les protéines mal repliées peuvent s’accumuler dans le RE et engendre ainsi
un stress du réticulum endoplasmique qui déclenche une réponse UPR (Unfolded protein
response).
3) Régulation des régulateurs d’ERAD « ERAD tuning »
Le processus « ERAD tuning » pour régulation d’ERAD consiste en la dégradation
sélective des régulateurs d’ERAD afin de réduire la capacité d’ERAD à des niveaux qui
n’interfèrent pas avec la maturation des protéines nouvellement synthétisées (Calì et al., 2008).
Ce modèle est basé sur des données montrant que de nombreux régulateurs d’ERAD (tels que
ERManI, EDEM1, OS-9, XTP3-B, HERP et SEL1L) sont rapidement éliminés du RE dans des
cellules non stressées, contrairement aux facteurs de repliement (tels que calnexin, calréticuline,
BiP, PDI, ERp57, ERp72 et GRP94). De plus, il a été démontré que la concentration d’EDEM1
dans le RE est strictement régulée en fonction de l’état cellulaire en ce qui concerne le stress
du RE et que cette protéine peut être disloquée dans des compartiments dérivés du RE. Ainsi,
il a été proposé que sous une production extensive et une accumulation de substrats d’ERAD,
la surproduction d’EDEM1 avec PERK (Protein kinase RNA-like ER Kinase) et IRE1 (Inositol
Requiring 1) est dirigée vers le compartiment ERQC (Kondratyev et al., 2007; Groisman et al.,
2011; Benyair et al., 2015b) afin d’améliorer la capacité d’ERAD lorsque les protéines mal
repliées s’accumulent (Ron et al., 2011). En revanche, à l’état d’équilibre, le niveau d’EDEM1
de la même façon que celui d’ERManI doit être réduit. Alors qu’ERManI est concentrée dans
les QCVs (Benyair et al., 2015b), EDEM1 est séparée dans des vésicules couvertes de LC3-I
(LC3 Microtubules-associated protein-light chain 3), nommées EDEMosomes (Calì et al.,
2008; Reggiori et al., 2010). Le fractionnement subcellulaire et les études de microscopie
électronique ont révélé qu’à l’état stationnaire environ 80% d’EDEM1 endogène se localise
dans ces vésicules (Zuber et al., 2007; Calì et al., 2008) (Figure 17).
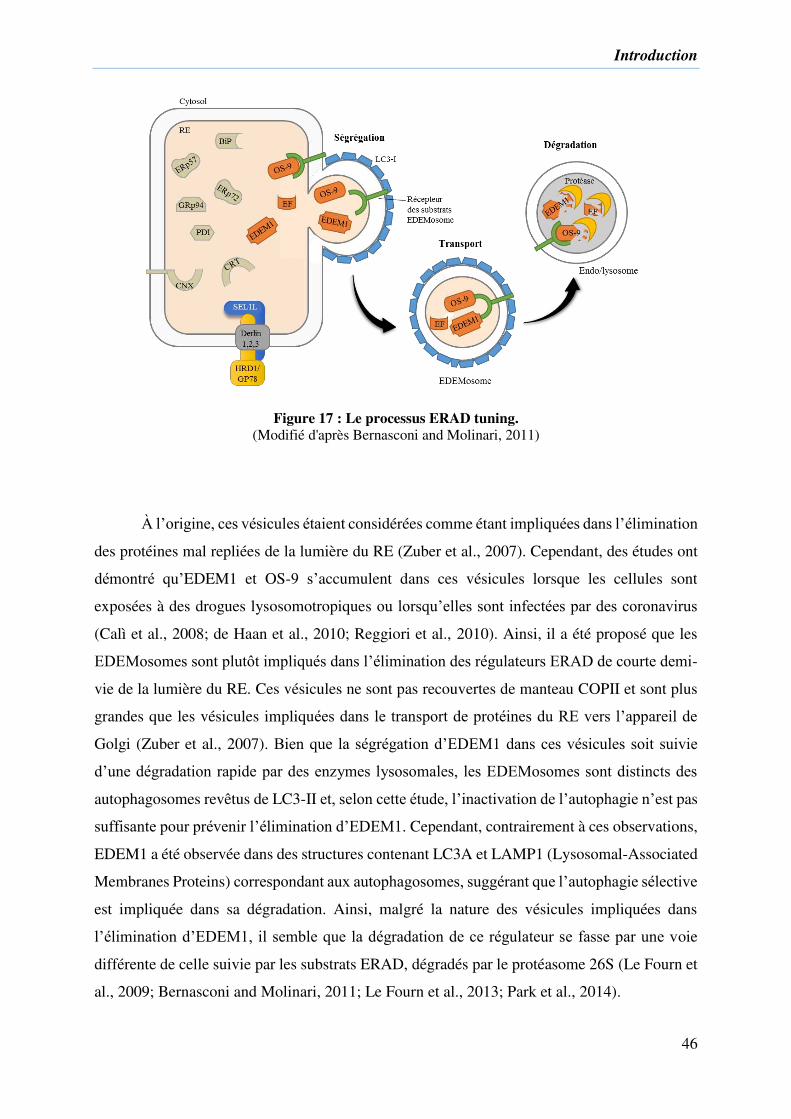
Introduction
46
Figure 17 : Le processus ERAD tuning. (Modifié d'après Bernasconi and Molinari, 2011)
À l’origine, ces vésicules étaient considérées comme étant impliquées dans l’élimination
des protéines mal repliées de la lumière du RE (Zuber et al., 2007). Cependant, des études ont
démontré qu’EDEM1 et OS-9 s’accumulent dans ces vésicules lorsque les cellules sont
exposées à des drogues lysosomotropiques ou lorsqu’elles sont infectées par des coronavirus
(Calì et al., 2008; de Haan et al., 2010; Reggiori et al., 2010). Ainsi, il a été proposé que les
EDEMosomes sont plutôt impliqués dans l’élimination des régulateurs ERAD de courte demi-
vie de la lumière du RE. Ces vésicules ne sont pas recouvertes de manteau COPII et sont plus
grandes que les vésicules impliquées dans le transport de protéines du RE vers l’appareil de
Golgi (Zuber et al., 2007). Bien que la ségrégation d’EDEM1 dans ces vésicules soit suivie
d’une dégradation rapide par des enzymes lysosomales, les EDEMosomes sont distincts des
autophagosomes revêtus de LC3-II et, selon cette étude, l’inactivation de l’autophagie n’est pas
suffisante pour prévenir l’élimination d’EDEM1. Cependant, contrairement à ces observations,
EDEM1 a été observée dans des structures contenant LC3A et LAMP1 (Lysosomal-Associated
Membranes Proteins) correspondant aux autophagosomes, suggérant que l’autophagie sélective
est impliquée dans sa dégradation. Ainsi, malgré la nature des vésicules impliquées dans
l’élimination d’EDEM1, il semble que la dégradation de ce régulateur se fasse par une voie
différente de celle suivie par les substrats ERAD, dégradés par le protéasome 26S (Le Fourn et
al., 2009; Bernasconi and Molinari, 2011; Le Fourn et al., 2013; Park et al., 2014).

Introduction
47
De manière intéressante, les EDEMosomes peuvent aussi contenir des protéines mal
repliées ou des complexes multiprotéiques à plusieurs sous unités à dégrader (Zuber et al.,
2007; Le Fourn et al., 2013). Ainsi, il a été suggéré que les vésicules EDEM constituent une
autre voie de dégradation des protéines du RE, en parallèle de la voie ERAD et de la voie de
réticulophagie. Cependant, à l’heure actuelle, on ne sait pas si cette voie de sortie du RE
fonctionne uniquement comme une voie pour la dégradation d’EDEM1 ou si elle est d’une
importance générale pour un ensemble plus large de mécanismes impliquant la dislocation de
protéines de RE au cytoplasme (Le Fourn et al., 2013).
4) Réponse à l’accumulation des protéines mal repliées UPR
Lorsque la capacité de repliement des protéines dans le RE est dépassée, elles
s’accumulent et peuvent générer un stress du RE qui est toxique pour la cellule. Cette dernière
dispose donc d’un système de secours nommé UPR (Unfolded Protein Response) capable de
déclencher une réponse adaptative permettant de restaurer l’homéostasie du RE : réduction de
la traduction des protéines, diminution de la charge protéique, amélioration de la machinerie
ERQC et augmentation de la capacité du repliement et de dégradation.
Chez les mammifères, l’UPR classique se compose de trois voies de signalisation
initiées par les protéines senseurs de stress : IRE1α, PERK, et ATF6 (Activating Transcription
Factor 6). Il s’agit des facteurs de transcription insérés dans la membrane du RE et maintenus
inactifs dans les conditions standard par leur liaison avec la protéine BiP. Cette liaison se fait
via le domaine luminale des trois facteurs et le site ATPase de BiP. Durant un stress du RE, BiP
se lie aux domaines hydrophobes exposés des protéines mal repliées et se dissocie ainsi des
trois senseurs d’UPR afin de les activer (Figure 18).

Introduction
48
Figure 18 : Représentation des trois voies de l’UPR. (D'après Brambilla Pisoni et al., 2016)
La dissociation de BiP d’ATF6 libère le signal de la localisation golgienne, GLS (Golgi-
Localization Signal) de cette dernière, permettant sa translocation du RE vers l’appareil de
Golgi. Dans ce compartiment, ATF6 subit des protéolyses par les protéases de site 1 et 2 (S1P
et S2P) qui libèrent son domaine cytosolique. Ainsi, ce dernier se dirige vers le noyau où il va
agir comme un facteur de transcription et induire la transcription de plusieurs gènes qui codent
les chaperonnes résidentes du RE et les assistants du repliement, y compris BIP, calnexine,
calréticuline, PDI, CHOP (CCAAT/enhancer-binding protein homologous protein) et XBP1
(X-box-binding protein 1).
La libération d’IRE1α de BiP induit son homodimérisation puis son
autophosphorylation, permettant ainsi d’activer son domaine endoribonucléase. Ce facteur
catalyse l’épissage de l’ARNm de XBP1 qui, sous sa forme épissée XBP1s, se transloque dans
le noyau et régule les gènes impliqués dans l’UPR et ERAD.

Introduction
49
De façon similaire, PERK est activée suite à la dissociation de BiP et subit une
homodimérisation et une autophosphorylation. Cette protéine kinase phosphoryle alors le
facteur d’initiation de traduction eIF2α (Eukaryotic Initiation Factor), provoquant une
diminution de la traduction globale et donc de la charge de protéines dans le RE. eIF2α
augmente spécifiquement la traduction du facteur de transcription ATF4, qui a un rôle à la fois
dans l’adaptation au stress et également dans l’apoptose quand le stress du RE ne peut être
résolu (Brambilla Pisoni et al., 2016).
De part de leur rôle dans la dégradation des glycoprotéines mal repliées, les protéines
EDEM sont des cibles majeures de la voie Ire1/Xbp1 d’UPR (Yoshida et al., 2003; Guiliano et
al., 2014). L’expression d’ERManI quant à elle n’est pas affectée par le stress du RE, ce qui
suggère que son rôle est moins important dans des conditions qui exigent un clivage extensif
du mannose des glycoprotéines mal repliées (Avezov et al., 2008).
II. Exemple du contrôle qualité : la protéine CFTR vs F508del-
CFTR
Le repliement de la protéine CFTR (Cystic fibrosis transmembrane conductance
regulator) est étroitement régulé pour permettre une insertion correcte de la protéine dans la
membrane du RE et une maturation appropriée. Tout comme pour les protéines de la voie de
sécrétion, les protéines CFTR ne parvenant pas à obtenir une conformation native sont retenues
dans le RE. Une rétention favorisée par le mécanisme ERQC, qui reconnaît les protéines mal
repliées et les dirige vers la dégradation par la voie ERAD.
A. Du gène à la protéine CFTR
1) Le gène CFTR
Le locus du gène responsable de la mucoviscidose a été localisé par l’équipe de Tsui sur
le bras long du chromosome 7, grâce à la technique de marqueurs RFLP (Restriction Fragment
Linked Polymorphism) (Tsui et al., 1985). Quatre ans plus tard, la même équipe a déterminé et
caractérisé ce gène, grâce à la technique de clonage positionnel. Ce gène qui fut nommé CFTR
est localisé précisément sur le locus q31 du bras long du chromosome 7 et mesure 250 kb
réparties en 27 exons. Il est transcrit en un ARNm de 6500 pb qui se traduit en une protéine

Introduction
50
membranaire de 1480 acides aminées nommée CFTR (environ 170 kDa lorsqu’entièrement
glycosylée) (Kerem et al., 1989; Riordan et al., 1989; Rommens et al., 1989) (Figure 19).
Figure 19 : Du gène à la protéine CFTR. (D’après Girodon-Boulandet et Costa., 2005)
2) Structure de la protéine CFTR
L’analyse de la séquence polypeptidique primaire de CFTR a permis de prédire sa
structure et là classé parmi les transporteurs ABC (ATP-Binding Cassette) (Riordan J.R. et al.,
1989). Il s’agit d’une famille qui regroupe les protéines transmembranaires dont le rôle est de
transporter de manière active des molécules variables telles que des peptides, des lipides, des
sucres, des métaux ou encore des médicaments. A noter que CFTR est le seul canal ionique de
cette famille et transporte des ions au travers la membrane cytoplasmique (pour revue Dean et
al., 2001; ter Beek et al., 2014; Wilkens, 2015).
Au niveau structural, les transporteurs ABC présentent une architecture commune
composée de 2 domaines transmembranaires MSD1 et MSD2 (Membrane-Spanning Domain)
et 2 domaines cytoplasmiques NBD1 et NBD2 (Nucleotide Binding Domain) (ter Beek et al.,
2014). Cependant, CFTR est un membre à part, car il est composé d’un domaine supplémentaire
appelé le domaine R pour regulatory domain (Riordan et al., 1989) (Figure 20).

Introduction
51
Figure 20 : Représentation schématique de la structure de CFTR. MSD: Membrane Spanning Domain, NBD: Nucleotide Binding Domain, TM: Transmembrane segment, RD: Regulatory Domain, ICLs: IntraCellular Loops, ECLs: ExtraCellular Loops (d'après Farinha and Canato, 2017).
Les domaines MSDs sont des domaines d’ancrage à la membrane et sont composés chacun de
6 segments transmembranaires TMD (TransMembrane domain) : TMD1 à TMD6 pour MSD1
et TMD7 à TMD12 pour MSD2. Organisés en hélice α, ces segments ont pour rôle la formation
du pore hydrophile du canal, laissant passer les anions de manière sélective (Ge et al., 2004;
Linsdell, 2006; Hwang and Kirk, 2013). Ces segments transmembranaires sont reliés deux à
deux par des boucles intra- ou extracellulaires, dont la quatrième boucle extracellulaire reliant
TMD7 à TMD8 présente deux sites de N-glycosylation de la protéine (Patrick et al., 2011). Le
domaine NBD1 est localisé au niveau de la boucle intracellulaire reliant les deux MSDs, et le
domaine NBD2 est localisé dans la partie carboxy-terminale de la protéine (Lewis et al., 2004,
2010). Ces deux domaines ont pour rôle de fixer et hydrolyser des molécules d’ATP afin
d’apporter l’énergie nécessaire pour l’ouverture du canal. Comme pour NBD1, le domaine
régulateur R est présent lui aussi dans la boucle intracellulaire reliant les deux MSDs plus
précisément entre NBD1 et TMD7. Ce domaine présente de multiples sites de phosphorylation
par des protéines kinases, principalement la PKA (protéine kinase dépendante de l’AMPc) et la
PKC (protéine kinase C) (Rich et al., 1991; Csanády et al., 2000). La phosphorylation de ces
sites joue un rôle important dans l’expression du CFTR, son trafic via la voie de biosynthèse,
les interactions intramoléculaires, sa stabilité au niveau de la membrane cellulaire et son
ouverture (pour revue voir Chin et al., 2017).

Introduction
52
3) Les fonctions de la protéine CFTR
Le CFTR est un canal ionique dont la fonction principale est de permettre la diffusion
passive des ions chlorures (Cl-) à travers la membrane apicale des cellules épithéliales
(Anderson et al., 1991). Il permet également le passage des petits anions iodures (I-), fluorures
(F-) et bromures (Br-) avec une sélectivité qui s’établie de la manière suivante : Br- ≥ Cl- > I- >
F- (Anderson et al. 1991). De plus, CFTR permet le passage du glutathion, un tripeptide
antioxydant, impliqué dans la protection des cellules des espèces réactives de l’oxygène (ROS)
(Linsdell and Hanrahan, 1998). De nombreuses autres études décrivent le CFTR comme une
protéine régulatrice. Il régule en effet, une large variété de canaux ioniques : ENaC (Epithelial
Na+ Channel) (Stutts et al., 1995; Awayda, 1996; Letz and Korbmacher, 1997), CaCC
(Calcium-activated Cl− Channels) (Kunzelmann, 2001; Wei et al., 2001), ORCC (Outwardly
Rectifying Chloride Channel) (Gabriel et al., 1993; Jovov et al., 1995; Schwiebert et al., 1995,
1998), ROMK1(potassium channel) (Lu et al., 2006; McNicholas et al., 1996) et l’échangeur
Cl/HCO3 (Lee et al., 1999; Shcheynikov et al., 2006; Wang et al., 2006b) (Figure 21). De plus,
il présente un rôle régulateur d’aquaporines (Schreiber et al., 1999; Pietrement et al., 2008) et
certaines connexines, du trafic vésiculaire et fusion des endosomes et du pH intracellulaire. Il
joue également un rôle dans l’infection et l’inflammation, et dans les jonctions cellulaires (pour
revue Saint-Criq and Gray, 2017).
Figure 21 : Régulation des canaux ioniques par la protéine CFTR. (Modifié d’après hopkinscf.org)

Introduction
53
4) Les mutations du CFTR
Des mutations au niveau du gène CFTR sont à l’origine de la mucoviscidose (Kerem et
al., 1989; Riordan et al., 1989; Rommens et al., 1989). Il s’agit d’une maladie génétique létale
à transmission autosomique récessive répandue dans les populations caucasiennes. Elle touche
en moyenne une naissance sur 2500 en Europe et en Amérique du nord. En France, 6585
patients ont été recensés en 2015, selon le registre français de la mucoviscidose. L’absence du
canal CFTR ou l’altération de sa fonction entraine une dérégulation des transports au sein des
épithéliums, provoquant en conséquence, une atteinte fonctionnelle de plusieurs organes :
poumon, intestin, pancréas, l’arbre biliaire, les organes génitaux masculin ainsi que les glandes
sudoripares de la peau. L’atteinte des voies respiratoire est la principale cause de mortalité chez
les malades. L’absence du CFTR à la membrane cellulaire ou l’altération de sa fonction
provoque en effet une déshydratation et un épaississement du mucus favorisant l’accumulation
des micro-organismes opportunistes et des poussières. Il en résulte en conséquence une
infection bactérienne, principalement par Staphylococcus aureus et Pseudomonas aeruginosa,
et provoque une obstruction chronique des bronches associée à une inflammation chronique.
Ces facteurs conduisent à la destruction des tissus pulmonaires provoquant une perte
progressive de la fonction respiratoire chez le patient jusqu’à son décès (Collawn and Matalon,
2014). A l’heure actuelle, il n’existe toujours pas de traitement curatif pour soigner cette
maladie. Seuls les traitements symptomatiques ayant pour but de soulager les patients et une
meilleure prise en charge sont proposés. Aujourd’hui, l’espérance de vie en France est de 47
ans alors qu’elle n’était que de 7 ans en 1965. La transplantation pulmonaire reste le traitement
de dernier recours en cas d’insuffisance respiratoire grave.
La sévérité des manifestations cliniques chez les patients atteints de cette maladie
polyviscérale dépend des conséquences des mutations sur la structure et la fonction de la
protéine CFTR mais également des facteurs génétiques et environnementaux (Wang et al.,
2014; Elborn, 2016). Plus de 2000 mutations ont été identifiées sur le gène CFTR (Cystic
Fibrosis Genetic Analysis Consortium: http://www.genet.sickkids.on.ca./cftr/app). Ces
mutations peuvent affecter la biosynthèse de la protéine CFTR, son trafic, sa fonction ou sa
stabilité à la membrane cellulaire. Ainsi, basées sur les mécanismes moléculaires à l’origine de
la dysfonction du canal CFTR, l’ensemble des mutations identifiées a été répartie en 6 classes
(Welsh and Smith, 1993; Zielenski and Tsui, 1995) (Figure 22).

Introduction
54
Figure 22 : Classification des mutations du CFTR et leurs conséquences sur la protéine CFTR. La classe I regroupe les mutations non-sens et de décalage de cadre de lecture, associées à une absence totale de CFTR dans la cellule. La classe II regroupe les mutations faux sens et des délétions qui perturbent le repliement de la protéine CFTR, qui se trouve séquestrée dans le RE puis dégradée. La protéine est synthétisée, mais absente à la surface cellulaire. La classe III et IV regroupent les mutations faux sens et de substitution qui affectent la régulation du canal Cl- (classe III) ou sa conductance (classe IV). La protéine est présente à la membrane cellulaire mais présente des défauts de régulation ou de conductance. La classe V regroupe les mutations d’épissage faux sens et substitution qui conduisent à la réduction de la synthèse de CFTR et en conséquence la réduction de la protéine au niveau de la membrane cellulaire. La classe VI correspond aux mutations qui touchent la partie C-terminale de CFTR et altèrent sa stabilité à la membrane cellulaire (d'après Lopes-Pacheco, 2016).
Parmi ces mutations, celles appartenant aux classes I et II sont les plus sévères car elles
se caractérisent par une absence du canal CFTR au niveau de la membrane cellulaire.
La délétion d’une phénylalanine en position 508 de la séquence polypeptidique du CFTR
(F508del) est la mutation la plus fréquente chez les malades atteints de mucoviscidose. Elle est
retrouvée chez environ 89% des malades et est à l’origine d’un phénotype sévère. Bien que
cette mutation, située dans le domaine NBD1 ne modifie ni le cadre de lecture du gène, ni la
fixation d’ATP, elle perturbe le repliement post-traductionnelle de F508del-CFTR dans le RE
et provoque sa dégradation et donc son absence à la membrane plasmique (Figure 23). A la
membrane cellulaire, F508del-CFTR est cependant capable d’assurer une conductance chlorure
mais plus faible que celle du CFTR sauvage et aussi moins stable à la membrane (Dalemans et
al., 1991; Lukacs et al., 1993; Moran, 2017). Ainsi, cette mutation de classe II, appartient aussi
à la classe III et à la classe VI.

Introduction
55
Figure 23 : Localisation de la protéine WT- et F508del-CFTR. Localisation de la protéine CFTR (vert) dans des cellules épithéliales primaires hautement différenciées non CF (à gauche) et CF (à droite). Contrairement au CFTR de type sauvage qui présente une localisation au niveau de la membrane apicale, la protéine mutée F508del-CFTR présente une localisation intracellulaire. Les noyaux cellulaire sont marqués en bleu (Riordan, 2008).
B. Biosynthèse et repliement : F508del-CFTR vs WT-CFTR
Comme pour les protéines de la voie de sécrétion, la protéine CFTR ainsi que son mutant
F508del-CFTR sont synthétisées et assemblées dans le RE. Ces deux protéines se traduisent à
raison de 2,7 acide aminés/seconde et par conséquent, la synthèse complète de la protéine se
réalise en 9 minutes environ (Ward and Kopito, 1994). Le repliement de CFTR se fait de
manière co-traductionnelle suivi et/ou complété par un processus post-traductionnel afin
d’acquérir la conformation native (Lu et al., 1998; Kleizen et al., 2005).
Bien que certaines étapes de repliement ne soient pas claires, les données disponibles
soutiennent l’idée que chaque domaine de CFTR se replie indépendamment avec insertion
simultanée de segments transmembranaires dans la membrane du RE. Ce processus nécessite
des interactions domaine-domaine qui commencent dès les premières étapes de repliement de
CFTR et impliquent une interaction spécifique de NBD1 avec MSD2 par ICL4 (intracellular
loops) et NBD2 avec MSD1 par ICL2 (Serohijos et al., 2008). Il est intéressant de noter qu’in
vivo, les domaines CFTR sont reconnus comme mal repliés s’ils sont exprimés seuls ou en
combinaisons incomplètes. Cela suggère que les interactions interdomaines sont essentielles
pour atteindre la conformation native de la protéine. Cette hypothèse est supportée également
par l’observation que les mutations CFTR résidant dans un domaine particulier conduisent à
des défauts conformationnels dans d’autres domaines (Du and Lukacs, 2009), à savoir, les
mutations MSD affectant le repliement de NBD et les mutations NBD affectant l’acquisition
d’une structure MSD compacte (Loo et al., 2008).

Introduction
56
Le déroulement de ce processus complexe se fait dans l’ordre suivant (Figure 24) :
1. Translocation du domaine transmembranaire MSD1 et son positionnement dans la
membrane du RE (Lu et al., 1998),
2. Synthèse du domaine NBD1 et sa liaison transitoire par les deux chaperonnes Hdj2 (Hsp40)
et Hsc70 (Hsp70) qui ont pour rôle de favoriser son repliement et sa stabilisation pendant le
temps nécessaire à la synthèse du domaine R,
3. Synthèse du domaine R puis son interaction avec la moitié de la région N-terminale
synthétisée. Cela conduit en une réduction de la liaison des Hsp40,
4. Synthèse du domaine transmembranaire MSD2 et son insertion dans la membrane du RE.
Cette étape semble stabiliser les interactions NBD1-R, qui conduisent à la libération de la
plupart des Hsp40 de la protéine,
5. La formation d’un complexe MSD1-MSD2 dans la membrane du RE qui pourrait
également stabiliser les domaines NBD1 et R (Ostedgaard et al., 1997). La formation de ce
complexe pourrait elle-même être facilitée par la chaperonne calnexine (Pind et al., 1994),
6. La synthèse du domaine NBD2,
7. Enfin, les modifications post-traductionnelles afin d’acquérir la conformation native.
Figure 24 : Biosynthèse de WT- et F508del-CFTR et assemblage des domaines.
(d'après Farinha and Canato, 2017)

Introduction
57
Au cours de sa translocation et en parallèle des étapes d’assemblage des domaines, la
protéine CFTR est glycosylée sur deux résidus asparagines (N) dans la boucle extracellulaire
ECL4 du domaine MSD2 (N894 et N900) et rentre dans le cycle calnexine (Pind et al., 1994;
Okiyoneda et al., 2004, 2008; Chang et al., 2008). Des études ont démontré que la liaison à la
calnexine est essentielle pour obtenir la conformation appropriée des sous-domaines de CFTR,
y compris les MSDs, qui semblent être stabilisés par le processus de liaison de cette lectine
(Rosser et al., 2008). Cependant, une étude a démontré que la diminution de l’expression
protéique de la calnexine ou bien l’expression d’un CFTR non glycosylé (délétion des N894 et
N900), incapable donc de se lier à cette chaperonne, n’a pas d’effet sur la fonction du WT-
CFTR ni sur son trafic à la membrane plasmique ; néanmoins la protéine peut être moins stable
(Chang et al., 2008). De la même manière, la protéine F508del-CFTR non glycosylée reste
incapable de s’échapper du contrôle de qualité et demeure séquestrée dans le RE (Chang et al.,
2008). D’autres études de susceptibilité aux protéases, agrégation thermique et ubiquitinylation
ont identifié un rôle important de la glycosylation dans la stabilité de CFTR mais pas
nécessairement pour sa fonction ou bien son trafic à la membrane cellulaire (Glozman et al.,
2009). Il a également été constaté que la N-glycosylation est nécessaire pour un recyclage
efficace de CFTR à partir de la membrane apicale et par conséquent, pour le renouvellement du
CFTR de surface (Cholon et al., 2010).
Durant ce cycle calnexine, seules les protéines CFTR ayant atteint leur conformation
native transitent vers l’appareil de Golgi via des vésicules COPII (Wang et al., 2004), où elles
subiront d’autres modifications de glycosylation. La glycoprotéine est ensuite adressée vers la
membrane apicale où elle acquiert sa fonction de canal chlorure et ses autres fonctions de
régulation avant d’être recyclée ou dégradée. Ainsi, au cours de son processus de biosynthèse
et de maturation, CFTR adopte quatre formes de glycosylation distinctes (Cheng et al., 1990).
La forme A d’environ 130 kDa qui correspond au CFTR non glycosylé. La forme B d’environ
140 kDa, nommée « core-glycosylé » ou encore forme immature et qui correspond au CFTR
réticulaire glycosylé. La forme B’ qui correspond à une population de la forme B (environ 30%)
correctement repliée et prête à quitter le RE. En fin, la forme C d’environ 170 kDa qui
correspond au CFTR mature, ayant transité dans l’appareil de Golgi et maturé par glycosylation
(Cheng et al., 1990; Lukacs et al., 1994; Bertrand and Frizzell, 2003). Selon la littérature, de
part de sa grande taille, ses nombreux domaines transmembranaires, et la nécessité de nombreux
contacts intramoléculaires, la biogénèse de CFTR est inefficace et lente, avec 60 à 75% des
protéines séquestrées par l’ERQC puis dégradées (Ward and Kopito, 1994; Abriel and Staub,
2005). De la même manière, la protéine mutée F508del-CFTR, de part sa mutation qui

Introduction
58
l’empêche d’acquérir sa forme native, reste retenue par l’ERQC avant d’être envoyée vers la
voie de dégradation (Cheng et al., 1990).
Plusieurs études tentent de comprendre comment l’absence de la phénylalanine perturbe
le repliement de F508del-CFTR à l’origine de son défaut d’adressage à la membrane cellulaire
(Cheng et al., 1990; Thomas et al., 1992; Lukacs et al., 1993; Welsh and Smith, 1993; Du and
Lukacs, 2009; He et al., 2010; Wang et al., 2014). Il a été suggéré que le défaut de repliement
est dû à un défaut d’interaction du domaine NBD1 avec les autres domaines (défaut
d’assemblage de la protéine) et que la chaîne latérale de l’acide aminée F508 semble être le
point central de ces interactions, en particulier entre NBD1 et les TMDs (Du et al., 2005; Cui
et al., 2007). En effet, un problème de repliement local dans le domaine NBD1 a été observé à
proximité de la mutation. De plus, il a été démontré que l’absence de F508 empêche
l’établissement de contacts entre le domaine NBD1 et la boucle cytoplasmique ICL4 dans le
domaine MSD2 et entraine un mauvais repliement de la partie carboxy-terminale de la protéine,
incluant le domaine NBD2 (Du et al., 2005; Pissarra et al., 2008; Du and Lukacs, 2009; He et
al., 2010; Rabeh et al., 2012; Mendoza et al., 2012; He et al., 2015). Ainsi, au cours de son
processus de repliement dans le RE, F508del-CFTR est reconnue comme une protéine mal
repliée par le système ERQC avant d’être dégradée (Cheng et al., 1990; Chang et al., 1999;
Meacham et al., 2001b; Younger et al., 2006; Riordan, 2008; Määttänen et al., 2010).
C. Le contrôle qualité de CFTR dans le RE et sa dégradation par ERAD
La machinerie ERQC est composée de lectines et de chaperonnes cytosoliques et
réticulaires. Ces protéines interagissent avec le CFTR immature, facilitent son repliement et son
assemblage en structure native et contrôlent son état de repliement. Bien qu’il ne soit pas
complètement compris comment l’ERQC distingue une conformation de protéine native d’une
mal repliée, pour la protéine CFTR, certains acteurs ont été décrits pour être impliqués dans ce
processus.
1) Les principales chaperonnes et lectines
La chaperonne cytosolique Hsp70 et ses co-chaperonnes de la famille Hsp40 ont été
décrites dans le contrôle qualité de CFTR. Comme vu précédemment, Hsc70 et sa
co- chaperonne Hdj-2 interviennent dans les premières étapes de biosynthèse du WT-CFTR au
moment de l’expression du domaine NBD1, afin de faciliter son processus de repliement

Introduction
59
(Figure 25-A). Via son interaction au domaine NBD1, Hsc70 augmente l’efficacité de
repliement du domaine et empêche la formation d’agrégat (Meacham, 1999). Dans le cas de
F508del-CFTR, une association prolongée de Hsc70 avec ce mutant comparée à celle avec le
WT-CFTR a été rapportée (Scott-Ward and Amaral, 2009). Ceci, suggère que l’absence de
F508 augmente l’accessibilité de Hsc70 à un ou plusieurs sites de liaison sur NBD1 provoquant
ainsi sa rétention dans le RE.
Figure 25 : Rôle de Hsp70/90 et leur partenaires dans le contrôle de qualité de CFTR. (A) Lorsque CFTR est synthétisé, de nombreuses chaperonnes et co-chaperonnes (certaines illustrées ici) s’associent au polypeptide naissant sur les côtés luminale et cytosolique de la membrane du RE. Hsp70 et co-chaperonnes interagissent avec NBD1, suivis de l’association de calnexine (CNX) avec TMD2. Hsp90 est recrutée ensuite pour aider à l’assemblage du domaine en conjonction avec la CNX. (B) Le défaut de repliement de la protéine à n’importe quelle étape de la voie de repliement est détecté par la liaison persistante de Hsp70, qui sert à recruter des ligases E3 (RMA1 et CHIP) qui ubiquitinylent CFTR et le ciblent vers le protéasome pour dégradation (Modifié d'après Kim and Skach, 2012).
L’implication du système de chaperon Hsp90 dans la régulation du repliement du CFTR
a également était proposé, bien qu’à un stade ultérieur de sa biogenèse (Loo et al., 1998) (Figure
25-A). Les intermédiaires de repliement de CFTR sont stabilisés en effet en se liant à Hsp90,
ce qui prolonge leur demi-vie et aide à leur trafic et à leur maturation (Loo et al., 1998; Fuller
et Cuthbert, 2000; Wang et al., 2006). Il a été constaté que les protéines WT- et F508del-CFTR

Introduction
60
interagissaient de manière similaire avec Hsp90. Cependant, sa co-chaperonne Aha1 présente
une interaction plus forte avec le F508del-CFTR qu’avec le WT-CFTR (Sun et al., 2008). De
plus, une autre co-chaperonne présentant une activité de peptidylprolyl isomérase, FKBP8
(FK506-binding proteins) semble assister au repliement de CFTR. Cette protéine interagit avec
le CFTR WT ou F508del, et les stabilise via un mécanisme qui requiert une activité isomerase-
prolyl. Hutt et collaborateurs ont démontré que FKBP8 est capable de se lier au CFTR
indépendamment de Hsp90 et présente une préférence pour le pool de CFTR prêt à
l’exportation. Cela suggère que cette protéine est une chaperonne à action tardive et qu’elle
fonctionne en aval de l’étape de repliement dépendante de Hsp90. FKBP8 peut en effet affiner
ou compléter le repliement de la conformation du CFTR à transiter du RE (Hutt et al., 2012).
Les systèmes de chaperonnes Hsp70/40/90 semblent ainsi former un piège chaperonne
(Coppinger et al., 2012) responsable, au moins partiellement, de la délivrance du CFTR mutant
vers la voie de repliement ou de dégradation. En effet, lorsque le CFTR est retenu trop
longtemps dans ce piège, les co-chaperonnes de pro-dégradation remplacent celles qui sont
productives, ciblant ainsi le CFTR vers la dégradation (Matsumura et al., 2013) (Figure 25-B).
L’ubiquitine ligase E3 CHIP, co-chaperonne de Hsc70 a été proposée pour jouer un rôle clé
dans ce processus (Meacham et al., 2001b; Younger et al., 2004). En effet, il semble que Hsp70
maintient les substrats mal repliés dans un état soluble et, lors de l’interaction avec CHIP, le
complexe Hsp70-CHIP détourne le CFTR vers la voie de dégradation. CHIP favorise
l’ubiquitinylation et la dégradation de CFTR en association avec l’enzyme conjuguée
cytosolique E2 ubiquitin UbcH5a. Elle permet la fixation de l’ubiquitine sur le CFTR présenté,
stimulant ainsi sa dégradation par le protéasome (Hohfeld et al., 2001; Meacham et al., 2001a;
Murata et al., 2003; Younger et al., 2004).
En plus du complexe CHIP-Hsp70, qui surveille les domaines cytosoliques de CFTR, il existe
également des protéines transmembranaires qui peuvent éventuellement surveiller l’assemblage
des domaines membranaires de CFTR en séquentiel. Le repliement aberrant de CFTR dans la
bicouche lipidique du RE est proposé pour être identifié par le facteur ERQC Derlin-1 et ses
protéines associées (Sun et al., 2006; Younger et al., 2006) (Figure 25-B). Il a été démontré
qu’un complexe formé par RMA1, appelé aussi RNF5 (RING finger protein), s’associe avec
Derlin-1 et la ligase E2 Ubc6e afin d’ubiquitinyler le CFTR (Younger et al., 2006). Ceci est
suivi par le recrutement d’autres partenaires, tels que Gp78 (Glycoprotein 78), BAP31 (B-cell
receptor-associated protein 31), et p97, qui délivrent le CFTR ubiquitinylé pour le protéasome
(Goldstein et al., 2007; Morito et al., 2008; Wang et al., 2008). Il est intéressant de noter que le

Introduction
61
complexe RMA1 peut détecter l’état d’assemblage des régions N-terminales du substrat CFTR
(Younger et al., 2006).
Les protéines du domaine J, la famille des co-chaperonnes qui potentialisent l’activité ATPase
de Hsp70, jouent également un rôle dans cette voie de dégradation. Prenons comme exemple,
le DNAJB12 qui semble diriger Hsc70 pour agir avec RMA1 dans la dégradation de F508del-
CFTR et du WT-CFTR mal repliée. Une interaction de cette dernière avec Hsc70 et RMA1 a
été en effet observée (Grove et al., 2011). Un autre exemple, les protéines Csp (Cysteine String
Proteins) qui semblent réguler la sortie de CFTR du RE et coordonner la formation du complexe
qui facilite la dégradation du CFTR, à savoir Hsc70/Hsp70 et CHIP (Schmidt et al., 2009). A
noter, une interaction directe entre Csp et CHIP a été observée (Schmidt et al., 2009).
La lectine calnexine a également été proposée pour être impliquée dans le contrôle de
repliement de CFTR. Alors que les deux protéines CFTR, WT et F508del-CFTR interagissent
avec cette lectine, une interaction prolongée avec le CFTR muté a été observée (Pind et al.,
1994; Rosser et al., 2008), suggérant son implication dans la rétention de F508del-CFTR dans
le RE.
2) Motif de rétention dans le RE : AFT (Arginine-Framed Tripeptides)
L’exportation du RE à l’appareil de Golgi de la protéine CFTR dépend de la détection
par la machinerie du trafic, des signaux spécifiques de rétention : tripeptides à arginine AFT
(Arginine-Framed Tripeptides) (Michelsen et al., 2005).
Les AFT sont des motifs de rétention/récupération transitoires avec une séquence consensus :
R-X-R (R correspond à l’arginine et X correspond à n’importe quel acide aminé) (Michelsen et
al., 2005). Le CFTR possède 4 motifs de ce type, répartis sur la queue N-terminale (Arginine29-
Glutamine-Arginine31), le domaine NBD1 (Arginine516-Tyrosine-Arginine518 et Arginine553-
Alanine-Arginine555) et le domaine RD (Arginine764-Arginine-Arginine766). Il a été suggéré que
F508del-CFTR reste séquestrée dans le RE en raison de l’exposition de tels motifs. En effet, la
mutation simultanée des 4 AFT dans la protéine F508del-CFTR par la substitution de l’arginine
par une lysine aux positions 29, 516, 555 et 766 (F508del-4RK-CFTR) restaure un F508del-
CFTR mature et fonctionnel à la membrane cellulaire en favorisant son échappement à l’ERQC
(Chang et al., 1999). Cette restauration d’adressage ne correspond pas à une correction de
repliement de la protéine mais plutôt à la suppression des facteurs de trafic spécifiques qui
peuvent être impliqués dans la rétention (Roxo-Rosa et al., 2006).

Introduction
62
3) Code de sortie du RE : le code diacide
Le compartiment ERGIC constitue un point de contrôle à partir duquel les protéines
correctement repliées progressent vers l’appareil de Golgi tandis que les protéines mal repliées
sont recyclées vers le RE par un transport rétrograde dans des vésicules COPI. Les CFTR WT
et F508del ont été localisées dans l’ERGIC, bien que, seul le WT-CFTR soit transporté jusqu’à
la surface cellulaire (Gilbert et al., 1998). Cette transition repose sur un motif d’exportation
spécifique, le code de sortie diacide, composé de deux acides aspartiques (D) en position 565
et 567 (D565-Alanine-D567), situé dans le domaine NBD1. Cette séquence est en effet nécessaire
pour l’empaquetage, médié par Sec24 des protéines CFTR dans les vésicules de transport
COPII. Autrement dit, pour que CFTR puisse quitter le RE, cette séquence doit être exposé à la
surface de NBD1, afin qu’elle puisse être détectée par Sec24. Il a été démontré que la
substitution du second résidu aspartate de cette séquence par le résidu alanine (A) n’affecte pas
le repliement du CFTR, mais elle réduit son association avec Sec24 et donc sa sortie du RE
(Wang et al., 2004). La mutation simultanée des deux résidus aspartate en alanine (AAA) quant
à elle, inhibe totalement l’exportation de CFTR du RE et en conséquence inhibe sa maturation.
La co-transfection des cellules exprimant DAA-CFTR ou F508del-CFTR, avec Sar1-GTP, une
protéine impliquée dans le trafic du RE vers l’appareil de Golgi ne restaure pas l’exportation ni
la stabilisation du CFTR muté à partir d’ERAD. Cela, suggère que la mutation du motif diacide
dissocie le CFTR de COPII (Wang et al., 2004).
La structure du domaine NBD1 de CFTR a révélé que le motif DAD est situé dans la boucle
extracellulaire contenant l’acide aminé F508. Compte tenu de l’importance probable de la
chaîne latérale aromatique de F508 dans la stabilisation du repliement du domaine NBD1, la
délétion de F508 peut modifier l’orientation de la boucle contenant le motif diacide,
compromettant ainsi son interaction avec Sec24 (Barlowe, 2003; Lewis et al., 2004). Des
expériences de co-immunoprécipitation ont en effet démontré que la liaison de la bande B de
F508del à Sec24 était beaucoup plus faible que celle du WT-CFTR. Ainsi, l’interaction
F508del-CFTR/Sec24 et le transport vers la membrane cellulaire pourrait être restaurés en
favorisant le repliement à 30°C (Wang et al., 2004).
D. F508del-CFTR et la réponse UPR
Les cellules épithéliales CF sont exposées à une inflammation chronique, une rétention
de CFTR mal repliée dans le RE, une surcharge du protéasome et des niveaux élevés de ROS,
facteurs susceptibles d’activer le stress du RE et l’UPR (Blohmke et al., 2012). Cependant, il y

Introduction
63
a encore une certaine controverse quant à savoir si l’UPR est induit dans les cellules CF ou non.
En effet, il a été suggéré que la dégradation rapide de la protéine mutante n’entraine pas une
accumulation importante dans le RE. Toutefois, l’analyse des cellules épithéliales bronchiques
primaires CF n’a pas abouti à des résultats cohérents. En effet, en culture à court terme, ces
cellules présentaient une réponse UPR active ; cependant, les différences observées entre les
cellules témoins et les cellules CF doivent être interprétées avec précaution, car cette réponse
pourrait résulter d’une infection chronique et d’une hyper-inflammation in vivo (Ribeiro and
Boucher, 2010; Ribeiro et al., 2005). En revanche, les cultures à long terme ne présentent pas
de réponse UPR classique dans les conditions basales. Aucune différence dans l’expression de
Grp78/BiP ou la phosphorylation de PERK n’a été observée (Nanua et al., 2006). De plus, une
diminution de la phosphorylation des deux protéines PERK et eIF2α a été rapportée dans des
cellules CF, suggérant une fois de plus l’absence d’activation de l’UPR (Blohmke et al., 2012).
D’autre part, il a été rapporté que la surexpression de F508del-CFTR, mais pas de WT-CFTR
dans diverses cellules, produit le stress du RE et induit l’UPR. En effet, une surexpression de
Grp78/BiP dans des cellules BHK surexprimant F508del-CFTR, mais pas WT-CFTR a été
observée (Gomes-Alves et al., 2010). Kerbiriou et ses collaborateurs ont également observé
l’activation des senseurs de stress du RE, ATF6 et Grp78/BiP dans des cellules épithéliales
humaines transfectées avec la protéine F508del-CFTR. Inversement, la diminution du niveau
d’expression d’ATF6 induit aussi une restauration de F508del-CFTR fonctionnel à la
membrane cellulaire (Kerbiriou et al., 2007). L’augmentation de l’expression de F508del-
CFTR dans des cellules épithéliales séreuses de la glande post-muqueuse humaine non CF,
Calu-3ΔF, exprimant les protéines WT-CFTR endogène et F508del-CFTR recombinant aussi
induit un stress du RE et une activation d’UPR (augmentation des ARNm des marqueurs sXBP1
et BiP). Ce résultat n’a pas été observé lors de l’augmentation d’expression du WT-CFTR
(Bartoszewski et al., 2008a).
L’activation d’UPR, à son tour, inhibe la biogenèse du WT- et F508del-CFTR endogènes en
diminuant les niveaux d’ARNm, et l’efficacité de traduction et de maturation. Le niveau
d’ARNm diminué n’est pas un résultat de l’amélioration de la dégradation, mais de l’inhibition
de la transcription par des mécanismes impliquant la liaison d’ATF6 au promoteur du CFTR,
ainsi que la dés-acétylation des histones et la méthylation du promoteur (Bartoszewski et al.,
2008b; Rab et al., 2007).

Introduction
64
III. Le système de contrôle qualité du RE comme cible
thérapeutique
Le mécanisme ERQC joue un rôle important dans la surveillance du repliement des
protéines et leur trafic soit vers la voie de sécrétion ou de dégradation. Cependant, certaines
protéines, même si elles n’acquièrent pas leur conformation native, conservent leur activité
physiologique si elles gagnent leur destination finale. La destruction « inutile » de ces protéines
est parfois à l’origine de certaines maladies, appelées « maladies conformationnelles », car
résultent d’une conformation anormale des protéines. Ces pathologies peuvent être réparties en
deux groupes distincts (Meusser et al., 2005) :
Le premier groupe correspond aux maladies liées à une perte de fonction des acteurs de
la voie de dégradation ERAD suite à des mutations. Les protéines mutées ou mal
repliées ne sont pas correctement dégradées, elles s’accumulent dans le RE et forment
des agrégats qui peuvent être toxiques pour la cellule. On retrouve dans ce groupe la
maladie de Parkinson, causée par une mutation dans la Parkine, une ubiquitine ligase
impliquée dans la dégradation du récepteur Pael-R. Ce dernier s’accumule dans sa forme
mal repliée et forme des agrégats toxiques pour la cellule (Kahle and Haass, 2004).
Le second groupe de pathologies est causées par une dégradation prématurée des
protéines mal repliées dans le RE, suite à des mutations gain de fonction des acteurs
d’ERAD, ou bien, des mutations dans les substrats, causant leur mauvais repliement.
Les protéines, retenues dans le RE puis envoyées vers la dégradation sont alors
incapables de remplir leur fonction, affectant ainsi l’activité biologique normale de la
cellule. Dans ce groupe on retrouve la mucoviscidose causée par les mutations de CFTR
de la classe II (cf. chapitre II), et une des sarcoglycanopathies, celle causée par la
mutation la plus fréquente chez les patients, R77C. Les sarcoglycanopathies sont des
pathologies génétiques à transmission autosomique récessive. Elles font partie de la
famille des dystrophies de ceintures et sont causées par des mutations d’un des gènes
codant un sarcoglycan (Hack et al., 2000).

Introduction
65
Dans le but de développer des stratégies thérapeutiques pour ces maladies
« conformationnelles », de nombreuses études visant à perturber le système ERQC ont été
menées. En effet, l’augmentation de l’activité de la voie ERAF pourrait favoriser le repliement
des protéines mal repliées/mutées et prévenir leur dégradation inutile par la voie ERAD. De la
même manière, l’augmentation de l’activité d’ERAD pourrait réduire la sécrétion des protéines
déstabilisées et susceptibles de former des agrégats liés aux pathologies dégénératives (Figure
26).
Figure 26 : Adaptation de la capacité des voies ERAF et ERAD pour corriger les défauts impliqués dans les pathologies conformationnelles.
(Modifié d'après Ryno et al., 2013)
Dans le cas d’un ciblage d’ERQC, les chaperonnes Hsp, jouant un rôle crucial dans la
rétention de F508del-CFTR dans le RE. Il a été démontré que la réduction de Hsc70 et
l’induction simultanée de l’expression de Hsp70 par le 4-phénylbutyrate de sodium (4-PBA, un
dérivé non toxique du sodium butyrate) restaure F508del-CFTR à la membrane cellulaire sous
sa forme mature et fonctionnelle (Rubenstein and Zeitlin, 2000; Choo-Kang and Zeitlin, 2001,
2001; Meacham et al., 2001b; Zhang et al., 2001; Singh et al., 2006). En effet, l’interaction avec
Hsc70 est considérée comme une étape clé pour cibler un certain nombre de protéines pour la
dégradation protéasomale. Par conséquent, le 4- PBA corrigerait l’adressage défectueux de

Introduction
66
F508del-CFTR en inhibant sa reconnaissance par la voie de dégradation. Cette molécule est un
des premiers correcteurs à avoir été testé dans un essai clinique pour les patients homozygotes
pour la mutation F508del. Cependant, malgré une amélioration légère de l’activité du CFTR au
niveau de l’épithélium nasal, le 4-PBA n’a pas réduit la concentration en ions chlorure de la
sueur (Rubenstein and Zeitlin, 1998). Le 4-PBA a aussi été testé avec succès sur d’autres
protéines, comme par exemple le récepteur de LDL. Il a été observé en effet, que cette molécule
restaure l’expression membranaire du récepteurs LDL mal conformé, séquestré dans le RE en
ciblant le manteaux COPII (Ma et al., 2017).
Une restauration de l’expression membranaire de F508del-CFTR a été observée dans des
cellules traitées avec la matrine ou l’apoptazole, deux autres inhibiteur de Hsc70. La matrine
régule négativement l’expression de Hsc70 entrainant ainsi une diminution de la dégradation
de F508del-CFTR (Basile et al., 2012). L’apoptazole quant à elle, semble perturber l’activité
ATPase de Hsc70 et en conséquence, diminue l’ubiquitinylation de F508del-CFTR en bloquant
l’interaction de Hsc70 avec sa co-chaperonne, l’ubiquitine ligase CHIP (Cho et al., 2011).
Grove et ses collaborateurs ont démontré que le ciblage d’une autre co-chaperonne de Hsp70,
DNAJB12, entraîne une augmentation significative de l’efficacité de repliement de CFTR et
permet la restauration d’une forme mature de F508del-CFTR. La surexpression de cette co-
chaperonne augmente l’association de CFTR avec Hsc70 et l’ubiquitine ligase RMA1 (Grove
et al., 2011).
D’autre part, il a été démontré que la sous expression de Aha1, la co-chaperonne de Hsp90
restaure l’adressage de F508del-CFTR ainsi que sa fonction de canal chlorure à la membrane
cellulaire ; en revanche sa surexpression diminue sa stabilité (Wang et al., 2006a; Koulov et al.,
2010). De plus, une inhibition pharmacologique spécifique de Hsp90 par la Geldanamycine, un
antibiotique de la famille des ansamycines, perturbe son interaction avec CFTR. Cependant son
effet sur l’adressage de F508del-CFTR est controversé. En effet, la Geldanamycine accélère le
renouvellement du CFTR en augmentant sa dégradation dans des cellules BHK et CHO
(Chinese Hamster Ovary) surexprexprimant stablement la protéine CFTR wt ou F508del (Loo
et al., 1998), Toutefois, Fuller et Cuthbert ont constaté sur des lysats de réticulocytes de lapin
que ce composé interfère avec la dégradation de F508del-CFTR en perturbant son
ubiquitinylation (Fuller and Cuthbert, 2000). Ainsi, d’autres investigations du rôle de Hsp90
dans la restauration de F508del-CFTR à la membrane cellulaire se révèlent nécessaires. A noter,
la Geldanamycine a donné des résultats encourageants sur ErbB2, un récepteur tyrosine kinase
qui est surexprimé dans 20 à 30% des cancers du sein (Slamon et al., 1987; Barr et al., 2008;
Cortese et al., 2013).

Introduction
67
L’inhibition des deux chaperonnes Hsp70 et Hsp90 semble donc rétablir l’expression
membranaire de F508del-CFTR. Il a été observé que la désoxyspergualine, un composé connu
pour perturber l’interaction de CFTR avec Hsc70 et Hsp90, restaure une activité canal chlorure
dans des cellules homozygotes pour la mutation F508del (Nadler et al., 1992; Nadeau et al.,
1994; Jiang et al., 1998; Norez et al., 2008).
Par ailleurs, il a été démontré qu’une protéine calcium-dépendante, la calumenin, agit comme
un modulateur de pompe SERCA (sarco/ER Ca2+-ATPase) dans les cellules épithéliales
bronchiques. La diminution de son taux d’expression restaure l’activité de CFTR à la membrane
cellulaire dans des cellules CF sans modifier la teneur en Ca2+ dans le RE (Philippe et al., 2017).
Cette étude suggère que la calumenin agit comme une chaperonne pour le CFTR régulant son
trafic et sa maturation indépendamment du contenu du calcium réticulaire.
D’autres études se sont intéressées aux complexes de rétrotranslocation et leurs
partenaires impliquées dans la dégradation protéasomale des protéines mal repliées. Par
exemple, dans le cas des sarcoglycanopathies, l’inhibition pharmacologique de l’activité de
HRD1, une E3 ubiquitine ligase, restaure l’expression membranaire du mutant V247M à la fois
dans un modèle cellulaire hétérologue et dans des myotubes de patients portant les mutations
L31P/V247M (Bianchini et al., 2014). Cette restauration a aussi été observée pour plusieurs
mutants suite à l’inhibition du protéasome (Gastaldello et al., 2008). Ainsi, dans le cas de la
mucoviscidose, la surexpression de HspBP1 (inhibiteur de l’activité de CHIP et facteur
d’échange de nucléotides qui favorise la libération de substrats de Hsp70) favorise la maturation
de CFTR (Alberti et al., 2004) ; au contraire, sa surexpression retient le CFTR dans le RE et
facilite son ubiquitinylation et sa dégradation (Meacham et al., 2001a). De même, l’inhibition
de l’activité ubiquitine ligase de CHIP par BAG-2 (BCL2 Associated Athanogene 2), co-
chaperonne de Hsp70 et qui inhibe la coopération CHIP-E2 favorise aussi la maturation de
CFTR (Arndt et al., 2005). De la même manière, il a été suggéré que Bag-1 joue un double rôle
sur la stabilisation de F508del-CFTR : d’une part en contribuant à sa libération de la machinerie
Hsp70 (Meacham et al., 2001a), et d’autre part, en empêchant sa liaison avec l’ubiquitine en
rentrant en compétition avec cette dernière (Mendes et al., 2012). D’autres études sur des
cellules humaines ont montré que le siRNA de la protéine Derlin-1 augmente la stabilité de
F508del-CFTR, tandis que sa surexpression conduit à la rétention de CFTR dans le RE (Sun et
al., 2006; Younger et al., 2006).

Introduction
68
La calnexine a elle aussi fait l’objet d’études dans la voie de biosynthèse de F508del-
CFTR. L’équipe de Kai a démontré que la surexpression de la calnexine augmente à la fois la
stabilité de la bande immature et la bande mature du WT-CFTR, suggérant un rôle protecteur
vis-à-vis de la dégradation (Okiyoneda et al., 2004). Cette surexpression augmente la rétention
de F508del-CFTR et diminue partiellement l’ERAD. Cependant, la même équipe a démontré,
mais cette fois-ci dans un système KO, que l’extinction de la calnexin n’affecte pas la rétention
de F508del-CFTR dans les cellules fibroblastes embryoniques de souris calnexin-KO,
certainement dû à la mise en place par la cellule d’un nouveau contrôle de qualité afin de
compenser l’extinction totale de sa calnexine. Ainsi, une inhibition pharmacologique partielle
de cette lectine semble être un bon compromis pour corriger F508del-CFTR. Dans notre
laboratoire, des études ont montré que l’inhibition de l’entrée de F508del-CFTR dans le cycle
calnexine par un iminosucre inhibiteur de l’α-1,2-glucosidase, le Miglustat (NB-DNJ pour N-
butyldeoxynojirimicyn) restaure partiellement la protéine à la membrane cellulaire dans des
cellules épithéliales mais aussi dans du tissu iléale de souris CF, homozygotes pour la mutation
F508del (Norez et al., 2006a; Noel et al., 2008; Norez et al., 2009). Ces résultats ont été
confirmés par une autre étude montrant que le miglustat appliqué directement sur l’épithélium
respiratoire de souris homozygotes pour la mutation F508del restaure l’activité canal chlorure
dépendante de CFTR (Lubamba et al., 2009). Au regard des résultats prometteurs obtenus avec
ce composé, le Miglustat a été testé lors d’essai clinique de phase 2 afin d’évaluer ses effets sur
la différence du potentiel nasal chez des patients adultes atteints de mucoviscidose et
homozygotes pour la mutation F508del (http://www.clinicaltrials.gov). A noter, ce composé est
déjà indiqué pour les patients atteints de maladie de Gaucher de type I sous le nom de Zavesca®
(Cox et al., 2000; Elstein et al., 2004). Une autre étude réalisée par notre laboratoire a également
démontré que la diminution de l’interaction de F508del-CFTR avec la calnexine par des
inhibiteurs de la pompe Ca2+-ATPase du RE rétabli l’expression membranaire de F508del-
CFTR (Norez et al., 2006b). Ces résultats sont en adéquation avec ceux obtenus précédemment
démontrant que le traitement avec la thapsigargine ou curcumin, inhibiteurs de SERCA restaure
F508del-CFTR fonctionnel à la membrane des cellules épithéliales et chez les souris CF (Egan
et al., 2002, 2004; Berger et al., 2005). Cela a ouvert la porte à l’identification d’autres
modulateurs de l’homéostasie protéique comme par exemple la roscovitine (pour revue Meijer
et al., 2016), qui est en essais cliniques de phase 2 dans le traitement des cancers, de la
polyarthrite rhumatoïde et de la maladie de Cushing. Démontrant de multiples propriétés
thérapeutiques sur les cellules CF, cette molécule est en essai clinique de phase 2 afin d’évaluer
sa tolérance et son efficacité chez les patients atteints de mucoviscidose, homozygotes pour la

Introduction
69
mutation F508del et présentant une infection chronique par Pseudomonas aeruginosa
(http://www.clinicaltrials.gov). En effet, la roscovitine est capable d’améliorer les propriétés
bactéricides des macrophages alvéolaires des patients CF, en abaissant le pH dans les
phagolysosomes, qui présentent un pH anormalement élevé dans les macrophages CF (Di et al.,
2006; Riazanski et al., 2015). De plus, la roscovitine a un effet anti-inflammatoire probablement
par sa capacité à réduire la dégranulation des neutrophiles, de favoriser l’apoptose et la
différenciation des cellules auxiliaires CD4+ T helper en anti-inflammatoires (Rossi et al.,
2006; Koedel et al., 2009; Meijer et al., 2016). Enfin, la roscovitine est capable de rétablir
potentiellement l’expression membranaire de F508del-CFTR dans des cellules CF (Norez et
al., 2014). Bien qu’inhibiteur de protéine kinase, l’effet correcteur de la roscovitine sur
F508del-CFTR est indépendant de ses effets sur les protéines kinases. En effet, il a été démontré
que la roscovitine d’une part empêche l’interaction de F508del-CFTR avec la calnexine en
diminuant la concentration du calcium réticulaire, et d’autre part, inhibe directement l’activité
du protéasome via un mécanisme d’action indépendant du calcium (Norez et al., 2014). Cette
molécule semble corriger l’adressage de F508del-CFTR en modulant à la fois l’ERQC et
l’ERAD.
D’autres études ont révélé que l’inhibition par la Kifunensine d’autres enzymes cette fois-ci
impliquées dans la voie ERAD, α-mannosidases I, corrige l’adressage défectueux de plusieurs
mutants sarcoglycans dans la pathologie des sarcoglycanopathies (Bartoli et al., 2008; Soheili
et al., 2012). Il serait donc intéressant de tester cette stratégie thérapeutique sur l’adressage de
la protéine F508del-CFTR dans des cellules mucoviscidosiques.

Contexte, problématiques et objectifs
70
Contexte, problématiques et objectifs Au cours de ces dernières années, divers correcteurs de CFTR ont été identifiés. Certains
d’entres eux peuvent cibler une ou plusieurs des nombreuses protéines qui orchestrent et
contrôlent son repliement, son adressage et sa stabilité au niveau de la membrane cellulaire (cf.
chapitre III). D’autres correcteurs peuvent interagir avec le CFTR lui-même (pour revue : Cai
et al., 2011; Farinha and Matos, 2016; Schmidt et al., 2016; Fajac and De Boeck, 2017; Zegarra-
Moran and Galietta, 2017), comme par exemple le correcteur pharmacologique VX-809 ou
lumacaftor, qui a fait l’objet d’études cliniques. Cette molécule semble agir tôt dans la
biogenèse de CFTR en interagissant directement avec MSD1 et éviterait ainsi les défauts de
repliement en améliorant les interactions entre les domaines NBD1, MSD1 et MSD2 (Van Goor
et al., 2011; He et al., 2013; Loo et al., 2013; Ren et al., 2013; Okiyoneda et al., 2013; Eckford
et al., 2014; Hudson et al., 2017). Les résultats des essais cliniques de phase II ont révélé que
ce correcteur diminue la concentration d’ions Cl- dans la sueur chez les patients homozygotes
pour la mutation F508del mais n’améliore cependant pas les signes cliniques (Clancy et al.,
2012). Cependant, sa combinaison avec le potentiateur VX-770 (Ivacaftor) connu pour
améliorer l’ouverture du canal muté de classe III, G551D-CFTR a donné de meilleurs résultats
cliniques (Boyle et al., 2014; Ratjen et al., 2017; Rowe et al., 2017; Schneider et al., 2017).
Cette combinaison appelée Orkambi a été approuvée en 2015 pour les patients homozygotes
pour la mutation F508del aux Etats Unis et en France. Une autre combinaison d’Ivacaftor avec
un correcteur de seconde génération, le VX-661 (Tezacaftor) est également en essai clinique.
L’efficacité clinique de cette combinaison est légèrement améliorée par rapport au VX-809
chez des patients âgés d’au moins 12 ans, homozygotes pour la mutation F508del (Pilewski et
al., 2015). Cette combinaison VX-661/Ivacaftor a été à son tour combinée avec VX-152 ou
VX-440, deux autres correcteurs de seconde génération. In vitro, l’activité canal chlorure
obtenu par ces triples combinaison est d’environ trois fois plus élevée que celle obtenue par la
combinaison lumacaftor/ivacaftor dans des cellules HBE. Ces triples combinaison sont en cours
d’evaluation chez les patients homozygotes pour la mutation F508del et les patients avec une
mutation F508del associé à une seconde mutation qui se traduit par une fonction CFTR
minimale (Schmidt et al., 2016).
Bien que les résultats obtenus avec ces correcteurs soient encouragent, la recherche de
correcteurs pour la mutation F508del-CFTR reste toujours primordiale.

Contexte, problématiques et objectifs
71
Des études précédemment menées au sein de notre équipe ont révélé que l’inhibition
des α-1,2-glucosidases du RE par le Miglustat ou le maintien d’une faible concentration en
calcium par des inhibiteurs des pompes SERCA, restaure la protéine F508del-CFTR à la
membrane plasmique (Norez et al., 2006b, 2006a) (Figure 27). Ces études ont également
montré une perturbation de l’interaction de la protéine F508del-CFTR avec la chaperonne
calnexine en concomitance avec cette restauration, amenant à penser que l’inhibition de l’entrée
du F508del-CFTR dans le cycle calnexine empêcherait son envoi vers la voie de dégradation
ERAD au profit de la membrane cellulaire.
Figure 27 : Représentation schématique des principales étapes de la biosynthèse de F508del-
CFTR et les sites d’action du Miglustat et des inhibiteurs de pompes Serca.
Sur la base de ces résultats, nous avons émis l’hypothèse dans ce travail de thèse que l’inhibition
de l’interaction de F508del-CFTR avec d’autres partenaires d’ERQC, et notamment ceux jouant
un rôle majeur dans la voie de dégradation ERAD, pourraient aussi rétablir l’expression
membranaire de F508del-CFTR (Figure 28). Cette étude pourrait contribuer d’une part à une
meilleure compréhension du mécanisme ERQC responsable de la rétention réticulaire de
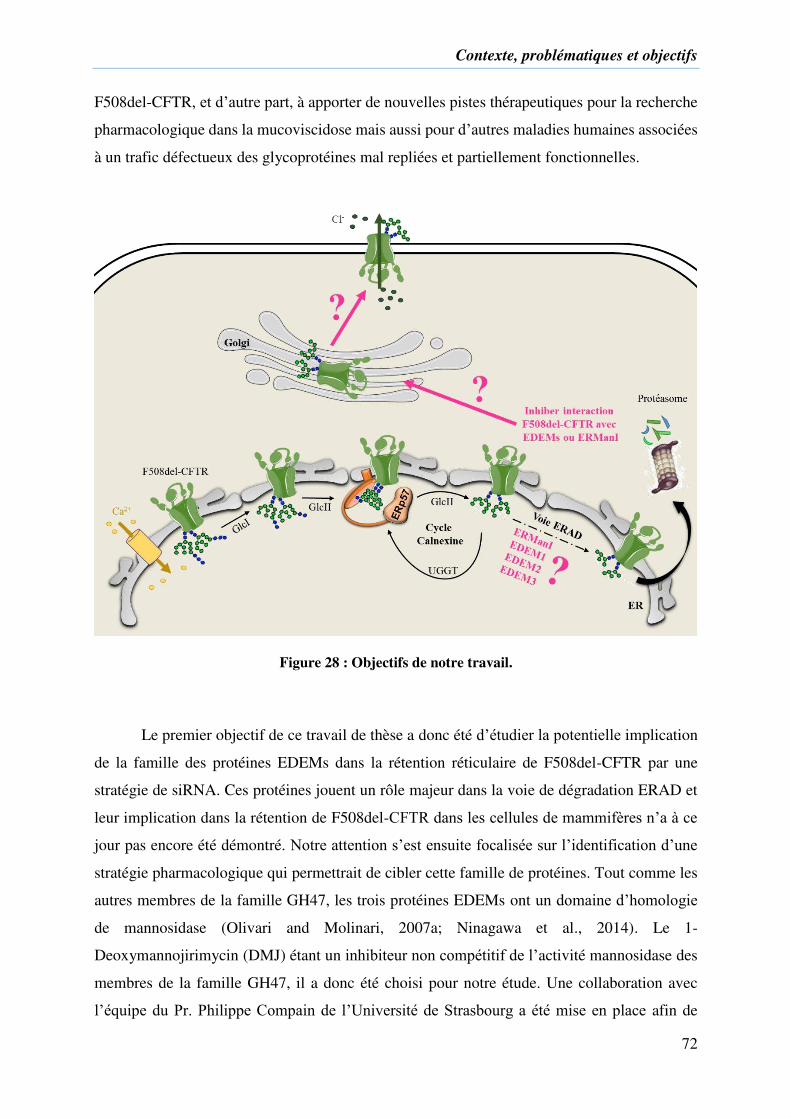
Contexte, problématiques et objectifs
72
F508del-CFTR, et d’autre part, à apporter de nouvelles pistes thérapeutiques pour la recherche
pharmacologique dans la mucoviscidose mais aussi pour d’autres maladies humaines associées
à un trafic défectueux des glycoprotéines mal repliées et partiellement fonctionnelles.
Figure 28 : Objectifs de notre travail.
Le premier objectif de ce travail de thèse a donc été d’étudier la potentielle implication
de la famille des protéines EDEMs dans la rétention réticulaire de F508del-CFTR par une
stratégie de siRNA. Ces protéines jouent un rôle majeur dans la voie de dégradation ERAD et
leur implication dans la rétention de F508del-CFTR dans les cellules de mammifères n’a à ce
jour pas encore été démontré. Notre attention s’est ensuite focalisée sur l’identification d’une
stratégie pharmacologique qui permettrait de cibler cette famille de protéines. Tout comme les
autres membres de la famille GH47, les trois protéines EDEMs ont un domaine d’homologie
de mannosidase (Olivari and Molinari, 2007a; Ninagawa et al., 2014). Le 1-
Deoxymannojirimycin (DMJ) étant un inhibiteur non compétitif de l’activité mannosidase des
membres de la famille GH47, il a donc été choisi pour notre étude. Une collaboration avec
l’équipe du Pr. Philippe Compain de l’Université de Strasbourg a été mise en place afin de

Contexte, problématiques et objectifs
73
synthétiser à partir du DMJ des composés multivalents et de tester leur effet correcteur. En
effet, lors d’une collaboration antérieure, les deux équipes avaient démontré qu’il était possible
d’améliorer l’efficacité du DNJ (un dérivé du miglustat) par une approche de « click-
chemistry », une stratégie qui permet de coupler des composés en une seule molécule (Compain
et al., 2013). Il a ainsi été démontré que les dérivés multivalents du DNJ corrigeaient l’activité
de F508del-CFTR de manière plus efficace que le DNJ. Le composé trivalent-DNJ, qui
correspond à 3 molécules de DNJ associées par un bras chimique, est environ 1000 fois plus
efficace que son monovalent-DNJ, correspondant au DNJ associé au bras chimique (Compain
et al., 2013).
Toujours dans l’optique d’identifier de nouvelles cibles thérapeutiques, nous nous
sommes intéressés par la suite à l’α1,2-mannosidase du RE (ERManI), décrite comme le «
minuteur » qui déclenche la première étape de la dégradation ERAD des glycoprotéines mal
repliées (Araki and Nagata, 2011; Zhou et al., 2015). En effet, on sait que son inhibition
(pharmacologique ou par siRNA) prévient la dégradation des substrats luminaux AAT (α1-
antitrypsin) et NHK (null Hong Kong) (Hosokawa et al., 2003; Wu et al., 2003; Avezov et al.,
2008; Pan et al., 2013). De même, un traitement avec la Kifunensine, un inhibiteur spécifique
de l’α1,2-mannosidase restaure l’expression membranaire de l’α-sarcoglycane R77C in vitro et
in vivo (Bartoli et al., 2008; Soheili et al., 2012). Ainsi, notre deuxième objectif dans cette étude
a été de déterminer si ERManI était impliquée ou non dans la rétention réticulaire de la protéine
mutée F508del-CFTR.
Enfin, lors d’une dernière étude, nous avons tenté de comprendre pourquoi la
multivalence augmentait l’efficacité de nos correcteurs. Ce travail toujours en cours (présentée
en annexe) a nécessité de générer des composés fluorescents par ajout d’une sonde fluorescente,
le BODIPY, abréviation de bore-dipyrométhene (Lepage et al., 2015).

Matériels et méthodes
74
Matériels et méthodes I. Matériel cellulaire
Fiche technique N°1 :
Lignées cellulaires, culture et entretien
Les différentes lignées cellulaires sont manipulées en conditions stériles sous une hotte
à flux laminaire verticale. Elles sont maintenues et cultivées à 37°C sous une atmosphère
humide composée de 5% de CO2 et 95% d’O2.
1- Culture des lignées cellulaires
a) Lignées Hela WT- et F508del-CFTR
La cellule Hela est une lignée cellulaire humaine immortelle issue d’un adénocarcinome
du col utérin de la jeune femme Henrietta Lacks. N’exprimant pas la protéine CFTR, cette
lignée a été stablement transfectée avec un plasmide codant la protéine CFTR sauvage (WT-
CFTR) ou mutée (F508del-CFTR). Le plasmide porte également un gène de résistance à la
zéocin. Les lignées sont mises en culture dans le milieu de culture décrit dans le tableau 2 :
Milieu de culture pour cellules Hela WT- et F508del-CFTR
Milieu DMEM - Dulbecco's Modified Eagle Medium (Gibco) 500 ml
Sérum de veau fœtal (SVF) (Fisher Scientific) 10%
Pénicilline (Sigma Aldrich) 100 U/ml
Streptomycine (Sigma Aldrich) 100 µg/ml
Agent de pression de sélection : Zéocin (Invitrogen) 125 mg/ml
Tableau 2 : Composition du milieu de culture pour les lignées cellulaires Hela WT- et
F508del- CFTR.
b) Lignée CFBE41o-
Il s’agit d’une lignée cellulaire épithéliale bronchique homozygote pour la mutation
F508del-CFTR. Elle a été mise au point par l’équipe du Professeur Gruenert pour étudier le
canal CFTR (Cozens et al., 1994). Cette lignée a la capacité de former une monocouche
polarisée lorsqu’elle est cultivée in vitro en condition air/liquide. Le milieu de culture utilisé
pour cette lignée est décrit dans le tableau 3 :

Matériels et méthodes
75
Milieu de culture pour cellules CFBE41o-
Milieu MEM Eagle with Earle's BSS, without L-glutamine (Lonza) 500 ml
Sérum de veau fœtal (SVF) (Fisher Scientific) 10%
L-Glutamine (Sigma) 2 mM
Plasmocyne (Invitrogen) 5 µg/ml
Tableau 3 : Composition du milieu de culture pour la lignée cellulaire CFBE41o-.
2- Entretien des lignées cellulaires
Lorsque les cellules atteignent environ 80% de confluence, elles sont réensemencées
dans une nouvelle flasque de culture appelée flasque « de routine » et si nécessaire, également
dans des supports adaptés aux manipulations envisagées. Ce repiquage s’effectue une seule fois
par semaine pour tout type cellulaire, selon le protocole suivant :
- Rincer les cellules avec une solution PBS (Phosphate Buffer Saline) dépourvue de
calcium et magnésium (Lonza).
- Répartir sur le tapis cellulaire la trypsine (T4049, Sigma) à raison de 1 ml/25 cm2 et
placer dans l’incubateur (37°C) durant 1 minute pour les lignées Hela WT- ou F508del-
CFTR et 4 minutes pour la lignée CFBE41o-.
- Une fois les cellules décrochées de leur support, stopper l’action de la trypsine en
ajoutant 5 ml de milieu de culture.
- Finir le décollement des cellules en effectuant des aspirations et des refoulements du
milieu puis, transférer la totalité des cellules dans un tube contenant 30 ml de milieu de
culture.
- Centrifuger pendant 7 minutes à 1100 rpm puis éliminer le surnageant.
- Remettre en suspension le culot cellulaire dans 1 ml de milieu de culture.
- Après avoir complété avec 5 ml de milieu de culture supplémentaire, dénombrer les
cellules puis les ensemencer à la dilution souhaitée sur les supports adéquats et selon le
type d’expériences envisagées.
- Tous les 2 jours, aspirer le milieu des cellules et le remplacer par du milieu frais afin de
renouveler les nutriments.

Matériels et méthodes
76
Fiche technique N°2 :
Congélation et décongélation des lignées cellulaires
1- La congélation
Il est possible de constituer un stock des différentes lignées cellulaires pour toute
utilisation ultérieure en les congelant dans de l’azote liquide à -180°C. Le protocole suivi pour
la congélation des cellules utilisées dans cette étude est le suivant :
- Décrocher les cellules de leur support par de la trypsine puis les centrifuger comme
indiqué en fiche technique N°1.
- Resuspendre le culot cellulaire obtenu dans un milieu de congélation qui correspond à
du milieu de culture supplémenté avec 10% de DMSO (DiMéthylSulfOxyde), un agent
cryoprotecteur qui permet d’empêcher la formation de cristaux.
- Transférer de manière très rapide les cellules dans des cryotubes.
- Congeler pendant 24 heures à -80°C.
- Le lendemain, stocker les cellules dans de l’azote liquide (-196°C).
2- La décongélation
La décongélation des cellules doit se faire de manière très rapide afin que les cellules
restent le moins longtemps possible en contact avec le DMSO, toxique à température ambiante.
Le protocole suivi est le suivant :
- Plonger le cryotube dans un bain marie (37°C) le temps de décoller le « glaçon » de
cellules de la paroi du cryotube.
- Transvaser le « glaçon » de cellules dans environ 35 ml de milieu de culture afin de bien
diluer le DMSO.
- Centrifuger l’ensemble à 1100 rpm pendant 7 minutes.
- Remettre en suspension le culot cellulaire obtenu dans 6 ml de milieu de culture.
- Ensemencer la totalité des cellules dans une flasque de culture de 25 cm2 (T25) puis la
placer dans l’incubateur.
- Une fois les cellules adhérentes, aspirer le milieu et le remplacer par du milieu frais.

Matériels et méthodes
77
Fiche technique N°3 :
Isolement des cellules épithéliales bronchiques des patients CF
1- Origine des prélèvements
Les fragments de tissus pulmonaires mucoviscidosiques sont fournis par le service de
chirurgie cardiothoracique de l’hôpital FOCH de Suresnes lors d’une transplantation
pulmonaire sur des patients mucoviscidosiques (collaboration avec le Docteur Bonnette). Une
fois prélevés, les fragments sont conservés à 4°C dans une solution dite de « décontamination
CF » (Tableau 4) jusqu’au moment de la dissection.
Milieu de décontamination CF
Milieu DMEM-HAM F12 500 ml
Pénicilline-streptomycine 1 mg/ml
Amphotéricine B 100 µg/ml
Gentamicine 0.5 mg/ml
Ceftazidine 500 µg/ml
Ticarcilline 500 µg/ml
Tableau 4 : Composition du milieu de décontamination pour échantillons CF.
Durant ces 3 années de thèse, nous n’avons malheureusement reçu que deux
prélèvements provenant de 2 patients hommes (29 et 35 ans), homozygotes pour la mutation
F508del-CFTR. La récupération de ces prélèvements est conforme à la législation française et
approuvée par le comité de protection de personne OUESTIII.
2- Dissection et isolement des cellules épithéliales bronchiques :
Une fois arrivés au laboratoire, les fragments pulmonaires sont rapidement disséqués et
les cellules épithéliales bronchiques sont isolées selon le protocole suivant :
- La manipulation doit être effectuée sous une hôte à flux laminaire.
- Mettre le prélèvement pulmonaire reçu dans une boîte contenant le milieu de
décontamination CF.
- A l’aide des ciseaux de microdissection, dissocier les bronches et bronchioles du
parenchyme puis les transférer dans une boîte propre contenant du PBS (Lonza) froid.
- Ouvrir les bronches et bronchioles dans leur longueur puis effectuer trois bains
successifs dans du PBS.

Matériels et méthodes
78
- A l’aide d’une pince courbe, gratter la surface interne de chaque bronche/bronchiole
afin de décoller l’épithélium puis transférer l’ensemble dans un tube de 50 ml.
- Centrifuger à 2 000 rpm pendant 6 minutes puis remettre le culot cellulaire délicatement
en suspension avec 1 ml de milieu de culture primaire CF (Tableau 5).
Milieu de culture primaire CF
Milieu DMEM-HAM F12 500 ml
Pénicilline-streptomycine 1 mg/ml
Tobramycine 500 µg/ml
Ceftazidine 500 µg/ml
Transferrine 7.5 µg/ml
Insuline 5 µg/ml
EGCS 2 µg/ml
T3 0.03 µg/ml
EGF 2.5 ng/ml
L-Glutamine 1 µM
Hydrocortisone 1 µM
Tableau 5 : Composition du milieu de culture primaire CF.
- Dénombrer les cellules épithéliales sur cellule de Malassez puis les ensemencer sur les
supports adéquats pour les manipulations envisagées.
- Incuber les cellules à 37°C sous une atmosphère humide composée de 5% de CO2 et
95% d’O2 jusqu’au moment de leur manipulation.

Matériels et méthodes
79
II. Techniques de biologie moléculaire et de biochimie
Fiche technique N°4
Mesure de l’activité du protéasome
1- Principe
L’analyse de l’activité enzymatique du protéasome a été effectuée par le kit « 20S
Proteasome Activity Assay kit ». Elle repose sur la détection du fluorophore-7-Amino-4-
methylcoumarin (AMC) libéré après clivage par le protéasome du substrat marqué Leu-Leu-
Val-Tyr-AMC. La fluorescence peut être mesurée par un couple de filtre 380/460 nm d’un
fluoromètre.
2- Protocole
L’activité enzymatique du protéasome est mesurée sur des cellules cultivées dans une
plaque 96 puits jusqu’à atteindre une confluence d’environ 100%, ou bien sur le protéasome
20S purifié du kit. Le test a été réalisé selon les instructions du kit :
- Oter le milieu de culture
- Ajouter 10 µl de 10X Assay Buffer et 80 µl d’H2O.
- Déposer dans l’obscurité, 10 µl de substrat LLVY-AMC.
- Après une incubation de 2h à 37°C, mesurer la fluorescence en utilisant un fluoromètre
(filtre 380/460).
- Pour la mesure de l’activité enzymatique du protéasome purifié 20S, suivre les
instructions du kit.

Matériels et méthodes
80
Fiche technique N°5 :
Transfection de siRNA (small interfering RNA)
1- Principe
Le siRNA est un ARN double brin de petite taille capable de se lier de manière
spécifique sur une séquence d’ARN messager (ARNm) et de le cliver afin d’inhiber sa
traduction en protéine. Il est introduit dans la cellule par lipofection, électroporation ou par des
vecteurs plasmidiques ou viraux. Une fois dans le cytoplasme, le siRNA active le complexe
RISC « RNA Induced Silencing Complex » et le guide jusqu’à l’ARNm cible pour le dégrader.
Ainsi, l’expression protéique de la protéine cible est diminuée dans les cellules transfectées
(Figure 29).
Figure 29 : Principe de la transfection de siRNA.
2- Protocole
Les siRNA et les réactifs de transfection (siRNA Transfection Reagent et siRNA
transfection medium) utilisés dans cette étude proviennent de Santa Cruz Biotechnology. La
transfection a été effectuée sur des cellules cultivées jusqu’à 60% de confluence dans des boites
de culture de 35 mm de diamètre :

Matériels et méthodes
81
- Préparer : Solution A = 100 µl milieu de transfection + 8 µl siRNA
Solution B = 100 µl milieu de transfection + 6 µl agent de transfection
- Mélanger la solution B avec la solution A et incuber pendant 45 minutes à température
ambiante.
- Ajouter 800 µl de milieu de transfection au mélange AB.
- Laver les cellules avec 2 ml de milieu de transfection.
- Déposer le mélange AB sur les cellules.
- Incuber les cellules pendant 7 heures dans l’incubateur (37°C et 5% de CO2).
- Rajouter sur les cellules 1 ml de milieu de culture cellulaire 2 fois supplémenté en milieu
de croissance et en antibiotiques.
- Après 24 heures de transfection, éliminer la solution de siRNA et la remplacer par le
milieu de culture cellulaire standard.
- Effectuer les manipulations envisagées 48 heures après la transfection.

Matériels et méthodes
82
Fiche technique N°6 :
Western blot
1- Principe
Le western blot est une technique qui permet de détecter la présence d’une protéine
particulière dans un extrait cellulaire. Les protéines sont séparées par électrophorèse en fonction
de leur poids moléculaire sur un gel de polyacrylamide dénaturant puis transférées sur une
membrane (nitrocellulose ou PVDF (polyvinylidene difluoride)). Sur cette dernière, la protéine
d’intérêt est reconnue par un anticorps spécifique qui à son tour est reconnu par un second
anticorps couplé à une enzyme, un fluorochrome ou un isotope (Figure 30).
Figure 30 : Principe de la technique du western blot.
2- Protocole
a) Extraction des protéines
L’extraction protéique est effectuée sur des cellules cultivées jusqu’à 100% de
confluence dans des boites de 35 mm de diamètre. Toutes les étapes du protocole sont réalisées
sur glace afin de limiter la dégradation des protéines :
- Rincer les cellules trois fois avec du PBS (Lonza) froid.
- Déposer sur le tapis cellulaire 100 µl de tampon de lyse (Tableau 6) additionné d’un
cocktail d’inhibiteurs de protéase (Roche).
Tampon de lyse
DOC (deoxycholate) 0.5% (w/v) Nonidet P-40 1% (v/v) Tris HCL 10 mM Pefabloc SC 1 mM pH 7,5
Tableau 6 : Composition du tampon de lyse cellulaire.

Matériels et méthodes
83
- À l’aide d’un grattoir, récupérer les cellules puis les transférer dans un tube eppendorf.
- Vortexer pendant 30 secondes puis incuber le lysat cellulaire pendant 30 minutes sur
glace.
- Vortexer de nouveau pendant 30 secondes puis centrifuger le lysat cellulaire pendant 5
minutes à 14 000 rpm et 4°C.
- Récupérer le surnageant (lysat clarifié) puis doser la concentration en protéine totale.
b) Dosage et dénaturation des protéines
La concentration des protéines est déterminée par la méthode BCA (acide
bicinchoninique), un dosage colorimétrique basé sur la capacité des protéines à réduire des ions
cuivriques Cu2+ en Cu+ lorsqu’elles sont dans un milieu alcalin. Les Cu+ libérés forment avec
le BCA un complexe pourpre mesurable à 562 nm. Le protocole utilisé est le suivant :
- Dans un tube, diluer 3 µl de lysat clarifié dans 47 µl d’eau. Pour la solution « blanc »,
mettre uniquement 50 µl d’eau.
- Ajouter dans chaque tube, 1 ml de la solution BCA préalablement préparée (BCA +
sulfate de cuivre dilué au 1/50ème, Thermo Scientific) et incuber pendant 30 minutes à
37°C.
- Mesurer la densité optique à 562 nm.
- La concentration en protéines totales est à déterminer à partir de la densité optique
obtenue grâce à une courbe étalon BSA (Bovine Serum Albumin) préalablement établie.
- Dénaturer les lysats clarifiés :
o Pour la détection de la protéine CFTR : pendant 1h à 37°C dans du Laemmli
2x [125 mM Tris, 4.5% SDS (Sodium Dodecyl Sulfate), 30% glycerol, 0.002%
bleu de bromophenol, 5% α-monothioglycerol].
o Pour les autres protéines : pendant 5 min à 95°C dans du Laemmli 5x (ID1624,
Dgel sciences).
c) Electrophorèse et transfert sur membrane
La séparation des protéines est réalisée par électrophorèse SDS-PAGE (Sodium
DodécylSulfate-PolyAcrylamide Gel Electrophoresis). Il s’agit de séparer les protéines
chargées en fonction de leur poids moléculaire qui migrent au travers d’un gel de
polyacrylamide en présence de SDS, sous l’influence d’un champ électrique. Le protocole
utilisé est le suivant :

Matériels et méthodes
84
- Préparer le gel de migration selon les tableaux 7 et 8.
Gel de séparation Gel de concentration
5% 10% 4%
Eau distillée 2.5 ml 2 ml 1.3 ml
Tampon 4X Lower 1 ml 1 ml -
Tampon 4X Upper - - 500 µl
Acrylamide-Bisacrylamide (40%) 500 µl 1 ml 200 µl
Persulfate d’ammonium (APS) 10% 40 µl 20 µl TEMED (N,N,N',N' tétraméthyléthylènediamine)
4 µl 2 µl
Tableau 7 : Composition des gels d’électrophorèse des protéines. (Le gel de séparation 5% de polyacrylamide est utilisé pour la migration de la protéine CFTR et le gel
de 10% de polyacrylamide pour la migration des autres protéines)
Tris SDS 10% pH
Tampon 4X Lower 1.5 M 0.4% 8,8
Tampon 4X Upper 0.5 M 0.4% 6,8
Tableau 8 : Composition des tampons 4X Lower et Upper.
- Une fois polymérisé, placer le gel dans une cuve à électrophorèse, puis remplir la cuve
avec le tampon de migration TG-SDS 10X (UP91495E, Interchim) dilué au 1/10ème.
- Déposer 50 µg des lysats protéiques dénaturés dans les puits du gel. Afin de faciliter le
suivi de la migration, déposer dans un puits un marqueur de taille coloré RPN800E (GE
Healthcare).
- Appliquer un champ électrique de 180 V et 30 mA pendant 1h40 à l’exception de la
migration de la protéine CFTR (270 V et 30 mA pendant 1 heure).
- Une fois la migration terminée, récupérer le gel puis réaliser le montage dit en «
sandwich » dans le tampon de transfert (Tableau 9), selon la figure 31. La membrane
utilisée est de type nitrocellulose (GE Healthcare).
Tampon de transfert
TG-SDS10X (UP91495E, Interchim) 5%
Ethanol 100% 20%
Tableau 9 : Composition du tampon de transfert.

Matériels et méthodes
85
Figure 31 : Assemblage du sandwich de western blot.
- Mettre le « sandwich » dans la cassette de transfert puis placer l’ensemble dans la cuve
de transfert.
- Après avoir rempli la cuve avec le tampon de transfert froid, appliquer un courant de
170 V et 80 mA pendant 1h30 à l’exception du transfert de la protéine CFTR (400 V et
200 mA pendant 1 heure).
d) Immunodétection
Toutes les étapes de cette partie doivent être réalisées à 4°C et sous agitation :
- A l’issu du transfert, récupérer la membrane et la laver trois fois 5 minutes avec du PBS-
Tween [tampon PBS contenant 0.01% de Tween-20].
- Incuber la membrane pendant 1 heure dans une solution de saturation, PBS-T contenant
0.5% de lait écrémé.
- Après 3 lavages de 5 minutes, incuber la membrane pendant toute une nuit avec
l’anticorps primaire spécifique de la protéine d’intérêt, dilué dans la solution de
saturation (Tableau 10).
Anticorps primaires Fournisseur Dilution
Mouse anti-CFTR monoclonal Merck Millipore 1/1000 Mouse anti- Na+/K+-ATPase α1 monoclonal 1/1000 Goat anti-EDEM1 polyclonal 1/1000 Rabbit anti-EDEM2 polyclonal Santa Cruz 1/2000 Goat anti-EDEM3 polyclonal Biothechnology 1/10 000 Rabbit anti-ERGIC53 polyclonal 1/10 000 Rabbit anti-VIP36 polyclonal 1/1000 Rabbit anti-Hsp70 polyclonal 1/10 000 Rabbit anti-Hsp90 polyclonal Enzo Life Sciences 1/1000 Rabbit anti-Calnexine polyclonal 1/2000 Rabbit anti-Actine monoclonal Sigma-Aldrich 1/10 000
Tableau 10 : Récapitulatif des anticorps utilisés en western blot.

Matériels et méthodes
86
- Le lendemain, laver 3 fois 5 minutes la membrane puis l’incuber pendant 1 heure avec
l’anticorps secondaire dilué dans la solution de saturation (Tableau 11). Cet anticorps
est couplé à la HRP (horseradish peroxidase) et est dirigé contre les IgG de l’organisme
dans lequel l’anticorps primaire utilisé a été produit.
Anticorps secondaires Fournisseur Dilution
Goat anti-Mouse HRP-linked Sigma-Aldrich 1/10 000 Goat anti-Rabbit HRP-linked 1/10 000
Donkey anti-Goat IgG-HRP Santa Cruz
Biothechnology 1/10 000
Tableau 11 : Récapitulatif des anticorps utilisés en western blot.
- Laver la membrane 3 fois 5 minutes.
- La révélation des protéines d’intérêt est réalisée en chambre noire par autoradiographie
à l’aide du kit Enhanced ChemiLuminescence (ECL) (WBKLS0500, Merck Millipore):
o Incuber la membrane pendant 1 minute avec la solution ECL préalablement
préparée (selon les instructions du kit).
o Couvrir la membrane à l’aide d’un plastique (très fin et transparent), puis
appliquer au-dessus un film autoradiographique (GE Healthcare) pendant
quelques minutes.
o Récupérer le film, le plonger dans une solution révélatrice, le rincer avec de
l’eau, le plonger dans une solution fixatrice puis le rincer avec de l’eau distillée.
- Scanner le film et mesurer l’intensité de chaque bande à l’aide du logiciel Photoshop.

Matériels et méthodes
87
Fiche technique N°7 :
Biotinylation de surface
1- Principe
La biotinylation de surface permet de détecter la présence d’une protéine particulière à
la surface cellulaire, de l’isoler et d’estimer sa quantité à un moment donné. Elle est basée,
d’une part, sur la capacité de la biotine, une vitamine soluble à se fixer exclusivement aux
protéines présentes à la surface cellulaire; et d’autre part, sur sa haute affinité avec la
streptavidine, une protéine d’origine bactérienne. La biotine peut être alors utilisée comme
appât pour isoler les protéines membranaires à partir d’un lysat cellulaire. Basée sur ce système,
plusieurs réactifs de biotinylation de propriétés différentes ont été créés. Dans notre étude, le
réactif EZ-Link Sulfo-NHS-SS-Biotin a été choisi pour ses propriétés qui répondent aux critères
pour la détection du canal CFTR. Ce réactif présente en effet, un groupement sulfonate (sulfo-
NHS) qui l’empêche de franchir les membranes cellulaires intactes et aussi permet sa fixation
covalente aux groupements amines primaires des protéines. De plus, il marque de manière
efficace des protéines qui ont une lysine résiduelle accessible et qui sont suffisamment
exposées. Le canal CFTR possède 3 lysines, présentes dans les boucles extracellulaires ECL1,
ECL3 et ECL4. Et enfin, ce réactif présente un pont disulfure intramoléculaire permettant une
élution facile des protéines biotinylées à partir des billes de streptavidine (Figure 32).
Figure 32 : Principe de la biotinylation de surface. A. Structure du EZ-Link Sulfo-NHS-SS-Biotin.
B. Principe de purification des protéines membranaires par la biotinylation.

Matériels et méthodes
88
2- Protocole
Pour notre étude, le kit de réactifs « Pierce™ Cell Surface Protein Isolation Kit »
(Thermo Fisher Scientific) a été utilisé. La technique est réalisée sur des cellules cultivées
jusqu’à 100% de confluence dans des boites de 35 mm de diamètre selon le protocole suivant :
- Rincer les cellules avec du PBS froid.
- Diluer la EZ-Link Sulfo-NHS-SS-Biotin dans du PBS à 0.5 mg/ml.
- Déposer 3 ml de la solution de biotine sur les cellules et incuber sous agitation pendant
30 minutes à 4°C.
- Stopper la réaction par 2 bains successifs avec le tampon Quenching Solution froid.
- Rincer les cellules pendant 5 minutes sous agitation à 4°C avec le TBS (Tris Buffer
Saline) froid.
- Effectuer une lyse cellulaire et doser la concentration des protéines récupérées, comme
indiqué dans la fiche technique N°6.
- Déposer la NeutrAvidin Agarose dans la colonne, et effectuer des lavages avec la
solution Wash Buffer selon les instructions du Kit.
- Déposer le lysat protéique dans la colonne et laisser incuber pendant 1 heure à
température ambiante sous rotation.
- Centrifuger les colonnes pendant 1 minute à 1 000 x g et jeter l’éluât.
- Déposer 500 µl de Wash Buffer additionné d’un cocktail d’inhibiteurs de protéase
(Roche) puis centrifuger pendant 1 minute à 1 000 x g.
- Après avoir jeté l’éluât, répéter cette dernière étape encore 3 fois.
- Eluer les protéines par dénaturation, en incubant les billes avec le tampon Laemmli 2x
[125 mM Tris, 4.5% SDS, 30% glycerol, 0.002% bleu de bromophenol, 5% α-
monothioglycerol] (à raison de 15 µl pour 100 µg de protéines) pendant 1 heure à 37°C.
- Placer les colonnes dans de nouveaux tubes collecteurs et récupérer les protéines
biotinylées par une centrifugation de 2 minutes à 1 000 x g.
- Analyser par western blot (voir la fiche technique N°6) le contenu des échantillons
biotinylés ou stockés à -20°C.

Matériels et méthodes
89
Fiche technique N°8 :
Immunomarquage indirect
1- Principe
L’immunomarquage est une technique qui permet de détecter et/ou localiser un antigène
au sein d’un tissu ou une cellule au moyen d’anticorps. Après fixation et perméabilisation des
membranes cellulaires, un anticorps primaire se fixe par spécificité sur l’antigène étudié. Cet
anticorps est à son tour reconnu par un anticorps secondaire couplé à un fluorochrome (Figure
33). Le complexe ainsi formé « protéine-anticorps primaire-anticorps secondaire » est ensuite
visualisé par microscopie à fluorescence suite à une excitation lumineuse du fluorochrome qui
va émettre une fluorescence.
Figure 33 : Principe d’immunomarquage indirect.
2- Protocole
La technique est réalisée sur des cellules cultivées jusqu'à 80% de confluence sur
lamelles de verre de 12 mm (déposées dans les puits d’une plaque 24 puits). Toutes les étapes
du protocole suivi sont réalisées sous agitation et, sauf contre-indication, à température
ambiante :
- Laver les cellules 3 fois 5 minutes avec le tampon TBS [157 mM NaCl, 20 mM Tris
Base, pH 7.4].
- Fixer les cellules avec du méthanol froid pendant 20 min à -20°C.
- Après 3 lavages de 5 minutes, perméabiliser les membranes cellulaires pendant 15
minutes à température ambiante avec 0.1% de Triton X-100 dilué dans le TBS.
- Laver 3 fois 5 minutes les cellules puis les incuber pendant 1 heure en présence d’une
solution de saturation TBS contenant 1% de BSA (Bovine Serum Albumine) et 0.05%
de saponine (TBS-BSA-Saponine).
Matériels et méthodes
90
- Eliminer la solution et incuber les cellules pendant toute une nuit à 4°C en présence
de/des anticorps primaire(s) approprié(s) dilué(s) dans la solution de saturation (Tableau
12).
Anticorps primaires Fournisseur Dilution
Mouse anti-CFTR monoclonal R&D system 1/100 Goat anti-EDEM1 polyclonal 1/50 Rabbit anti-EDEM2 polyclonal Santa Cruz 1/50 Goat anti-EDEM3 polyclonal Biothechnology 1/50 Mouse anti calnexin polyclonal 1/50 Rabbit anti calnexin polyclonal Enzo Life Sciences 1/200
Tableau 12 : Récapitulatif de l’ensemble des anticorps primaires utilisés.
- Laver les cellules 3 fois 5 minutes puis les incuber pendant 1 heure en présence de/des
anticorps secondaire(s) (dirigé contre les IgG de l’organisme dans lequel l’anticorps
primaire a été produit) dilué(s) dans la solution de saturation (Tableau 13).
Anticorps secondaires Fournisseur Dilution
Donkey anti-Rabbit IgG (H+L), Alexa Fluor 555 1/100 Donkey anti-Mouse IgG (H+L), Alexa Fluor 555 Thermo Fisher 1/50 Donkey anti-Goat IgG (H+L), Alexa Fluor 647 Scientific 1/50 Donkey anti-Rabbit IgG (H+L), Alexa Fluor 647 1/50
Tableau 13 : Récapitulatif de l’ensemble des anticorps secondaires utilisés.
- Après 2 lavages au TBS, monter les lamelles sur des lames de verre à l’aide d’un milieu
de montage avec DAPI (Sigma-Aldrich). Le DAPI, un agent qui s’incorpore dans le
noyau et se lie préférentiellement aux bases A-T de l’ADN (acide désoxyribonucléique)
permet de marquer les noyaux des cellules.
- Conserver les lames à 4°C jusqu’à leur observation au microscope.
Les observations microscopiques ont été réalisées à la plateforme ImageUp (Université de
Poitiers), sur un microscope confocal FV1000 Olympus équipé d’un microscope inversé à
fluorescence (IX-81, Olympus). Les marquages fluorescents ont été obtenus par une excitation
des échantillons à 543 nm (rouge), 633 nm (rouge lointain) et 405 nm (DAPI). La fluorescence
émise a été détectée entre 555-655 nm pour le rouge, 650 nm pour le rouge lointain et entre
425-475 nm pour le DAPI. Les images ont été obtenues à l’aide d’un objectif x60. Le logiciel
Fluoview v1.6 a été utilisé pour l’acquisition des signaux et le traitement des images.

Matériels et méthodes
91
Fiche technique N°9 :
In situ Proximity Ligation Assay (Duolink)
1- Principe
La méthode in situ Proximity Ligation Assay (PLA) est une technique hautement
spécifique et sensible qui permet de détecter, visualiser et quantifier au sein d’un tissu ou d’une
cellule des interactions protéine/protéine, l’expression des protéines seules, ou encore, des
modifications post traductionnelles, telle que la phosphorylation. La détection de l’interaction
entre deux protéines cibles est basée sur l’utilisation de deux anticorps secondaires (PLA-probe
PLUS et PLA-probe MINUS), conjugués chacun avec un fragment d’oligonucléotides. Ces
deux fragments étant complémentaires entre eux, s’hybrident lorsque la distance entre les deux
protéines cibles est inférieure ou égale à 40 nm. Cette hybridation est maintenue grâce à une
ligase puis amplifiée grâce à une polymérase qui incorpore des bases qui fluorescent dans le
rouge (Figure 34).
Figure 34 : Principe de la technique de Duolink.
2- Protocole
La manipulation est effectuée sur des cellules cultivées jusqu’à 80% de confluence sur
lamelles de verre de 12 mm (déposées dans les puits d’une plaque 24 puits). Le kit Duolink in
situ kit (Olink Biosciences) a été utilisé :
- Laver les cellules 3 fois 5 minutes avec le tampon TBS.
- Fixer les cellules pendant 20 minutes à température ambiante avec une solution de
paraformaldéhyde (PAF) diluée à 3% dans le TBS.
- Après 3 lavages de 5 minutes, perméabiliser les membranes cellulaires pendant 10
minutes à température ambiante avec du Triton X-100 dilué à 0.1% dans le TBS.
- Après 3 nouveaux lavages, incuber les cellules pendant 1 heure à température ambiante
avec la solution de saturation TBS contenant 1% de BSA et 0.05% de saponine.

Matériels et méthodes
92
- Ôter la solution de saturation et incuber les cellules à 4°C pendant toute une nuit sous
agitation avec les anticorps primaires dilués dans le tampon de saturation (Tableau 14).
Ces anticorps, dirigés chacun contre une des deux protéines d’intérêt doivent être
produits dans deux espèces différentes.
Anticorps Fournisseur Dilution
Duolink CFTR avec les protéines :
Goat anti-EDEM1 polyclonal 1/200 Rabbit anti-EDEM2 polyclonal Santa Cruz Biotechnology 1/500 Goat anti-EDEM3 polyclonal 1/1000 Rabbit anti-Hsp70 polyclonal 1/300 Rabbit anti-Hsp90 polyclonal Enzo Life Sciences 1/300 Rabbit anti-Calnexin polyclonal 1/300 Mouse anti-CFTR monoclonal Merck Millipore 1/100
Duolink CFTR avec les protéines :
Rabbit anti-VIP36 polyclonal Santa Cruz 1/500 Rabbit anti-ERGIC53 polyclonal Biotechnology 1/1000 Mouse anti-CFTR monoclonal Merck Millipore 1/500
Tableau 14 : Récapitulatif de l’ensemble des anticorps utilisés en Duolink.
- Le lendemain, mélanger les deux sondes PLA PLUS et PLA MINUS diluées à 1/5 dans
la solution de saturation et laisser incuber pendant 20 minutes à température ambiante.
Chaque sonde doit être dirigée contre l’espèce de l’un des anticorps primaires utilisés.
- Laver les cellules 2 fois 5 minutes avec la solution de saturation puis les incuber pendant
1 heure à 37°C en présence du mélange PLA PLUS + PLA MINUS.
- Après 2 lavages avec la solution de lavage A [0.01 M Tris, 0.15 M NaCl, 0.05% Tween
20], incuber les cellules pendant 30 minutes à 37°C en présence d’une solution d’eau
purifiée contenant la solution de ligation et la ligase, à 1/5 et 1/40 respectivement.
- Après deux nouveaux lavages de 2 minutes avec la solution de lavage A, incuber les
cellules pendant 100 minutes à 37°C en présence d’une solution d’eau purifiée
contenant la solution d’amplification et la polymérase, diluées à 1/5 et 1/80
respectivement.
- Laver les cellules 2 fois 10 minutes avec une solution de lavage B 1X [0.2 M Tris, 0.1
M NaCl] puis 1 fois 1 minute avec la solution de lavage B à 0,01X.
- Monter les lamelles sur des lames de verre à l’aide d’un milieu de montage contenant le
DAPI (marqueur des noyaux cellulaires).
- Une fois séchées, les lames sont observées en microscopie comme décrit précédemment
en fiche technique N°8.

Matériels et méthodes
93
III. Techniques de physiologie cellulaire
Fiche technique N°10 :
Efflux d’iodure
1- Principe
Il s’agit d’une technique qui permet d’étudier la fonctionnalité des canaux chlorures
dans une population cellulaire. Elle repose sur la mesure du changement de concentration en
radiotraceur de part et d’autre de la membrane cellulaire. L’iodure radioactif 125I présente de
nombreux avantages qui font de lui le traceur idéal pour l’étude de l’activité des canaux
chlorure, comparé au chlore radioactif 36Cl-. En effet, la plupart de ces canaux présentent une
sélectivité pour l’iodure et pour les ions chlorure. De plus, l’125I présente une durée de vie qui
est beaucoup plus faible que celle du 36Cl- (60 jours contre 300 000 ans) et son utilisation est
beaucoup moins coûteuse (environ 10 fois moins chère).
2- Protocole
La technique est effectuée par le docteur Caroline NOREZ. Elle est réalisée sur des
cellules cultivées jusqu’à 100% de confluence dans une plaque 24 puits selon le protocole
suivant :
- Rincer les cellules avec le milieu d’efflux (Tableau 15) puis déposer un milieu dit « de
charge », composé de milieu d’efflux complémenté par 1 µCi/ml de 125INa (NEN Life
Sciences).
Tableau 15 : Composition du milieu d’efflux utilisé.
- Laisser incuber pendant 1 heure à 37°C, afin de permettre à l’iode radioactif de
s’équilibrer de part et d’autre de la membrane cellulaire.
Milieu d’efflux
NaCl 136.9 mM KCl 5.4 mM CaCl2 1.8 mM MgCl2 0.8 mM NaHCO3 4.2 mM NaH2PO4 0.3 mM KH2PO4 0.3 mM MgSO4 0.4 mM HEPES 10 mM D-glucose 5.6 mM pH 7.5

Matériels et méthodes
94
- A l’issus de cette incubation, les étapes qui suivent sont à réaliser par le robot
MultiPROBE II (Packard).
- Rincer les cellules avec le milieu d’efflux afin d’éliminer l’iodure non incorporé, puis
déposer 500 µl de ce milieu.
- Une minute après, collecter le milieu dans un tube à hémolyse et déposer un volume
équivalent de milieu « propre ».
- Répéter cette dernière étape encore 8 fois :
o Les 3 premiers tubes à collecter correspondent à l’efflux basal de l’iode radioactif
sortit de la cellule de manière passive.
o Les 6 autres prélèvements correspondent aux milieux collecté après l’addition
d’agents pharmacologiques : pour notre étude, il s’agit d’un cocktail activateur de
CFTR composé de 10 µM de forskoline (Fsk), un activateur d’adénylate cyclase et
30 µM de génistéine (Gst), qui active directement le CFTR).
- A la 10ème minute, incuber les cellules pendant 30 minutes en présence d’une solution
de lyse composée de 0,1% SDS et 0,1 M NaOH afin de récupérer et d’évaluer la
radioactivité résiduelle des cellules (restes).
- Mesurer la radioactivité contenue dans les tubes collectés ainsi que dans le lysat (restes)
à l’aide d’un compteur gamma (Cobra II, Packard) en coups par minute (cpm).
3- Analyse des résultats
Les résultats en cpm obtenus par le compteur sont présentés sous la forme de vitesse de
sortit d’iodure radioactif (k) par minute (k (min-1)) (Figure 35) grâce à la formule suivante :
k = [In(125I t1) - In(125I t2))] / (t1-t2). Les 125I t1 et 125I t2 correspondent à la radioactivité
(cpm) mesurée à temps t1 et t2 respectivement. Les résultats présentés en histogrammes sont
représentés en kpeak-kbasal afin de s’affranchir de la sortie passive de l’iodure radioactif.
Figure 35 : Exemple de représentation graphique des résultats d’efflux d’iodures.
kbasal
kpeak Agent pharmacologique

Matériels et méthodes
95
Fiche technique N°11 :
Etude des variations du potentiel membranaire avec la sonde oxonol
1- Principe
La Sonde Oxonol ou bis-(1,3-diethylthiobarbituric acid) trimethine oxonol
(DiSBAC2(3) Molecular Probes, Eugene, OR) est un fluorochrome hydrophobe anionique avec
les longueurs d’ondes d’excitation et d’émission de 546 nm et 580 nm respectivement (Figure
36). L’oxonol est sensible aux variations du potentiel de membrane. En présence des cellules
dépolarisées, la sonde traverse leur membrane et se lie aux protéines membranaires
intracellulaires. Ainsi, l’accumulation d’oxonol dans la cellule au cours de sa dépolarisation se
traduit par une augmentation de la fluorescence émise. A l’inverse, la diminution de la quantité
d’oxonol dans la cellule lors de l’hyperpolarisation provoque une diminution de la fluorescence
émise. Il est donc possible d’explorer l’état d’activation d’un canal ionique en suivant les
changements du potentiel de membrane cellulaire, induits par le mouvement des ions du canal
étudié. Dans le cas du canal CFTR, une augmentation de la fluorescence suite à son activation
sera associée à une dépolarisation de la cellule, induite par la sortie d’ions chlorure par CFTR.
Ainsi, la quantité de fluorescence émise est proportionnelle à l’activité de CFTR.
Figure 36 : Structure de la sonde oxonol [DiSBAC2(3)].
2- Protocole
La mesure de fluorescence est effectuée à température ambiante à l’aide d’une station
de microscope inversé ZEISS Axio observer ZI, et l’acquisition des signaux et le traitement des
images est opéré grâce au module Physiology du logiciel Carl Zeiss AxioVision Release 4.8.2.
La technique est réalisée sur des cellules cultivées dans des boites à fond verre jusqu’à environ
100% de confluence selon le protocole suivant :
- Rincer les cellules 2 fois avec la solution de Krebs dépourvue de Cl- (Tableau 16).

Matériels et méthodes
96
Tableau 16 : Composition de la solution Krebs sans Cl-.
- Incuber les cellules pendant 20 minutes à température ambiante et à l’obscurité avec
250 nM d’oxonol, dilué dans la solution de Krebs dépourvue de Cl-.
- Déposer la boite de cellules sur le microscope et choisir une zone du tapis cellulaire =
zone de travail.
- Prendre une photo de la zone afin d’avoir une référence correspondante à l’intensité de
la fluorescence basale (F0) de chaque pixel de la zone.
- Sélectionner (grâce aux outils du logiciel) 10 à 12 cellules de cette zone afin de suivre
et d’enregistrer les variations de fluorescence de leur sonde avant et après ajout d’agents
pharmacologiques. Ici, il s’agit du cocktail activateur de CFTR (forskoline 10 µM +
génistéine 30 µM) et l’inhibiteur sélectif de CFTR (CFTRinh172).
- Appliquer une excitation par une lampe fluorescente à 546 nm toutes les 30 secondes
(durant 800 ms). À chaque exposition une photo F(t) est prise. Les variations de
fluorescence sont retransmises sous la forme intensité de fluorescence en fonction du
temps.
3- Analyse des résultats
Les résultats obtenus sont présentés en pourcentage de variation de fluorescence (Fx)
selon l’équation suivante : Fx = ([Ft – F0] / F0) x 100. Les Ft et F0 correspondent à l’intensité
de fluorescence au temps t et t0 respectivement (Figure 37).
0 2 4 6 8 10
-2
0
2
4
6
8
10
12
Activateur
Inhibiteur
Temps (min)
[(F
t-F
0)/
F0]*
10
0
Figure 37 : Exemple de représentation graphique de la fluorescence de la sonde oxonol.
Solution de Krebs dépourvue de Cl-
Na-gluconate 101 mM K-acetate 5 mM Ca-gluconate 2 mM Mg-acetate 2 mM Mannitol 50 mM Glucose 5 mM pH 7.4

Matériels et méthodes
97
Fiche technique N°12 :
Chambre de Ussing
1- Principe
La chambre de Ussing est une technique in vitro et ex vivo, qui permet d’étudier le
transport des électrolytes, des éléments nutritifs et de drogues à travers les tissus épithéliaux.
Elle représente donc un bon outil dans les domaines de physiologie et de pharmacologie. Son
principe repose sur la propriété d’étanchéité de l’épithélium, ainsi que sur la distribution
asymétrique des protéines de transport entre le côté apical et basolatéral des cellules. Le
mouvement d’ions se produisant au travers d’une membrane confère à la cellule une polarité
qui se traduit par la ddp (différence de potentiel). L’amplitude de courant électrique annulant
cette ddp (0 mV) appelé courant de court-circuit Isc (short circuit current) traduit donc le
mouvement d’ions se produisant au travers de l’épithélium étudié (Figure 38).
Figure 38 : Principe de la technique de chambre de Ussing.
2- Préparation des cellules
L’expérimentation est réalisée sur des cellules cultivées sur des inserts de type
Transwell 6 puits (Transwell® permeable Supports, Corning). Chaque insert est formé d’une
membrane microporeuse sur laquelle les cellules vont pouvoir se multiplier pour atteindre la
confluence et former une monocouche de cellules, délimitant un pôle apical et un pôle basal «
pseudo-épithélium ». Grâce à la configuration suspendue des inserts, il est possible de cultiver
les cellules en interface air/liquide (coté apical des cellules en contact de l’air environnant, et
coté basal en contact du milieu du puits) permettant ainsi de reproduire l’épithélium bronchique.

Matériels et méthodes
98
L’intégrité de l’épithélium peut être vérifiée en mesurant la résistance électrique transépithéliale
(Rte). En effet, la Rte augmente avec le temps de la culture jusqu’à atteindre un maximum, puis
commence à décroitre.
Dans notre étude, les cellules sont ensemencées à raison de 500 000 cellules/insert et sont
cultivées en condition air/liquide jusqu’à avoir une résistance de 500 Ω/cm² (environ 10 jours
de culture). Les inserts utilisés sont formés d’une membrane en polycarbonate de 12 mm et
constituée de pore de 0.4 μm (Corning Costar Snapwell™, Sigma). La faible taille des pores
permet d’éviter la colonisation de la face basale de la membrane par les cellules.
3- Système expérimental et protocole utilisé dans le laboratoire
Le système utilisé dans notre laboratoire (Easymount, EM-CSYS-6, Physiologic
Instruments) est composé de 6 chambres en plexiglas constituées chacunes de deux hémi-
chambres indépendantes. Chaque insert contenant la culture cellulaire est monté entre deux
hémi-chambres qui peuvent contenir la même solution expérimentale ou deux solutions
différentes. Afin de garder les tissus étudiés en conditions physiologiques tout au long des
expérimentations, le système est relié à un bain-marie, qui permet de maintenir les solutions
des hémi-chambres à 37°C. Il est également relié à une bouteille de gaz contenant un mélange
de 95% d’O2 et 5% de CO2, afin de créer un système de bullage qui sert à l’oxygénation des
tissus et à la circulation des solutions.
Les paramètres électriques du tissu sont déterminés grâce à deux couples d’électrodes
(constituées d’agar 4% et KCl 3M) situées chacun de part et d’autre de l’insert : (i) le couple
d’électrodes de voltage, placé au plus près de l’insert, permet de mesurer la différence de
potentiel (ddp) du tissu en cours d’étude et (ii) le couple d’électrodes de courant, placé plus loin
de l’insert, permet de déterminer le courant de court-circuit Isc. La technique de « voltage clamp
» a été utilisée afin de déterminer ce courant Isc. Le tissu est « court-circuité » par l’injection
d’un courant imposant une ddp de 0 mV au tissu. La valeur de ce courant correspond au courant
de court-circuit (Figure 39).

Matériels et méthodes
99
Figure 39 : Chambre de Ussing.
Les électrodes sont reliées à un préamplificateur, lui-même relié au VCC MC2 (multichannel
voltage-current clamp, Physiologic Instruments), un système composé d’un amplificateur, un
millivoltmètre, un générateur de courant et un microampèremètre. L’amplificateur est relié à
son tour à un ordinateur par le biais d’une interface d'acquisition de données (DI-720-P,
Physiologic Instruments) permettant de transformer les informations électriques ou analogique
reçues de l’amplificateur en données numériques enregistrées grâce au logiciel “Acquire &
Analyse 2.3”.
4- Protocole
- Equilibrer les électrodes :
o Monter un insert vide sans cellules entre les deux hémi-chambres remplies de la
solution apicale et basale (Tableau 17).
Solution de Krebs « normale »
NaCl 107 mM KCl 5 mM CaCl2 1.2 mM MgSO4 1.2 mM NaH2PO4 0.2 mM Na2HPO4 1.8 mM NaHCO3 25 mM Glucose 12 mM pH 7.4
Tableau 17 : Composition de la solution de Krebs « normale ».
o Clamper les cellules pour leur imposer une ddp de 0 mV, puis retirer l’insert.

Matériels et méthodes
100
- Monter l’insert contenant les cellules polarisées et attendre quelques minutes pour que
les échanges et donc le courant de court-circuit basal soit stable.
- Injecter un courant d’une valeur déterminée et non nulle (ici, +2 mV) afin de déterminer
la résistance transépithéliale (Rte) du tissus, grâce à l’équation d’Ohm :
Rte(Ω)=ΔU(mV)/ΔIsc(µA).
- Clamper les cellules afin de maintenir une ddp de 0 mV et commencer l’enregistrement
des courants résultants avant et après ajout d’agents pharmacologiques. Ici, il s’agit du
cocktail activateur de CFTR (forskoline 10 µM + génistéine 30 µM) et l’inhibiteur
sélectif de CFTR (CFTRinh172).

Résultats
101
Résultats I. Les protéines EDEMs comme cibles thérapeutiques dans la
mucoviscidose
A. Contexte
Le contrôle qualité du RE, ERQC, joue un rôle majeur dans l’adressage défectueux de
la protéine mutée F508del-CFTR. Ce processus est devenu la cible de plusieurs études ayant
pour objectif l’identification de correcteurs capables de rétablir l’expression membranaire de
cette protéine mutée. Notre équipe a par exemple démontré que l’inhibition de l’interaction de
F508del-CFTR avec la lectine calnexine restaure son expression membranaire (Norez et al.,
2006b, 2006a, 2009). Suite à ces résultats, nous avons voulu savoir si la modulation d’autres
acteurs de l’ERQC pouvait en faire de même. Dans cette nouvelle étude, nous nous sommes
donc intéressés à la famille des protéines EDEM, jouant un rôle majeur dans la voie de
dégradation ERAD des protéines mal/non-repliées. En effet, EDEM1 a été détectée dans des
complexes protéiques à la fois du WT- et du F508del-CFTR chez les mammifères (Farinha and
Amaral, 2005). Cette protéine a également été identifiée comme étant impliquée dans la
dégradation de la protéine WT-CFTR dans la levure Saccharomyces cerevisiae (Gnann et al.,
2004). Concernant les protéines EDEM2 et EDEM3, leur éventuel lien avec la dégradation de
CFTR, WT ou F508del n’a à ce jour pas été exploré. L’objectif de notre étude était donc de
déterminer si les 3 protéines EDEMs sont impliquées dans la rétention de F508del-CFTR et si
c’est le cas, d’identifier une stratégie pharmacologique qui permettrait de rétablir l’expression
membranaire du canal CFTR en ciblant ces protéines.
B. Résultats
1) Article
Rescue of Cystic Fibrosis and Sarcoglycan Mutations by Inhibition of ER
degradation, enhancing -mannosidase-like proteins EDEM.
K.Sidelarbi, A.Joosten, C.Patissier, J.Bertrand, F.Stauffert, A.Bodlenner, I.Richard, P.Compain,
F.Becq & C.Norez

Résultats
102
Role of the inhibition of ER degradation enhancing -mannosidase-like proteins EDEM in CFTR rescue
K.Sidelarbi1, A.Joosten2, J.Bertrand1, F.Stauffert2, A.Bodlenner2, P.Compain2, F.Becq1 & C.Norez1
1Laboratoire de Signalisation et Transports Ioniques Membranaires, Université de Poitiers, CNRS,
Poitiers, France 2Laboratoire de Synthèse Organique et Molécules Bioactives, CNRS, Université de Strasbourg,
Strasbourg, France

Résultats
103
ABSTRACT
Numerous genetic diseases are directly associated with the recognition of misfolded
proteins by the Endoplasmic Reticulum Quality Control (ERQC), leading to their retention in
the ER and subsequently their retrotranslocation to the cytosol for degradation. In the case of
mutated glycoproteins such as F508del-CFTR (Cystic Fibrosis Transmembrane conductance
Regulator), causing CF pathology, mannose trimming is a key step of this process. It has been
proposed that EDEM family proteins (EDEM1, EDEM2, and EDEM3) play a key role in this
process. Thus, we investigated their involvement in F508del-CFTR trafficking defect using
siRNA strategy. We showed that the knockdown of EDEM1 or EDEM2 restores F508del-
CFTR chloride activity in CF cells, which highlight these proteins as potential pharmacological
targets to correct the F508del-CFTR trafficking defect. Hence, we tested multivalent
derivatives, based mainly on Deoxymannojirimycin (DMJ), an inhibitor of α-mannosidases I
and revealed their better corrective effect on F508del-CFTR. Interestingly, we confirmed the
corrector effect of the most effective derivative on human respiratory epithelial cells freshly
isolated from CF patients and identified its mechanism of action. In conclusion, this study
highlights EDEM proteins as potential pharmacological targets to develop correctors for the
F508del-CFTR trafficking defect. We propose the DMJ series, water soluble and non-toxic as
correctors of F508del-CFTR.
Keywords: F508del-CFTR, endoplasmic reticulum quality control, mannosidase activity,
EDEM.

Résultats
104
INTRODUCTION
The proteins ER degradation, enhancing α mannosidase-like (EDEM), comprising
EDEM1, EDEM2 and EDEM3 are lectins belonging to the glycosyl hydrolase 47 (GH47)
family that also includes ER α 1,2-mannosidase I and the Golgi α 1,2-mannosidases (Olivari
and Molinari, 2007). Overexpression of EDEM1, EDEM2 or EDEM3 in mammalian cells was
shown to accelerate ER-Associated Degradation (ERAD) of misfolded glycoproteins, whereas,
kifunensine and 1-deoxymannojirimycin (DMJ), two inhibitors of α-mannosidase activity
stabilize them (Zhou et al., 2015). In addition, it has been proposed that EDEM1 binds to the
adapter protein SEL1L through its mannosidase-like domain for delivering aberrant proteins to
the ER membrane ubiquitin-ligase complex HRD1-SEL1. Comparing to this interaction,
EDEM2 seems to bind more strongly to SEL1L, whereas EDEM3-SEL1L binding was found
to be very weak. On the other hand, studies revealed that the three variants EDEM1, EDEM2
and EDEM3, initially considered as lectins, can also function as mannosidases and molecular
chaperones (Frenkel et al., 2003; Hirao et al., 2006; Olivari et al., 2006; Banerjee et al., 2007;
Quan et al., 2008; Cormier et al., 2009; Hosokawa et al., 2010; Gauss et al., 2011; Ninagawa et
al., 2014; Jang et al., 2015). EDEMs recognize different classes of substrates (Slominska-
Wojewodzka and Sandvig, 2015). In the case of F508del-CFTR degradation process, Gnanns
and collaborators observed a disturbance in yeast cells deleted from Htm1p lectin, an orthologue
of mammalian EDEM. This defect was complemented by the expression of EDEM, suggesting
that both Htm1p or EDEM are important in the recognition and degradation process of F508del-
CFTR (Gnann et al., 2004). In addition, EDEM was detected in complexes of both WT and
F508del-CFTR in mammalian cells (Farinha and Amaral, 2005). However, the implication of
the three variants of EDEM in F508del-CFTR degradation has still not been described.
Cystic fibrosis (CF) is an autosomal recessive genetic disease caused by mutations in
the gene encoding for the CFTR (CF Transmembrane conductance Regulator), a cAMP-
dependent chloride channel that regulates salt and fluid transports at the apical membrane of
epithelial cells (Riordan, 1993). The most common mutation, found in ~90% of CF patients and
responsible of the severe form of the disease is the deletion of a phenylalanine at position 508
(F508del) (Riordan et al., 1989). This mutation disrupts CFTR folding that remains retained in
the ER resulting in an absence of CFTR channels at the plasma membrane (Du et al., 2005).
Consequently, it results a disruption of ions transport through the cell membrane, depletion of
airway surface liquid, impaired clearance of mucus and bacterial infections in lungs. F508del-
CFTR mutant is recognized as aberrant product by the ER Quality Control (ERQC), which

Résultats
105
delivers it to the proteasomal degradation via a series of event collectively called ER-Associated
Degradation (ERAD) (Ward et al., 1995).
Here, we investigated the role of EDEM in the trafficking defect of F508del-CFTR in
mammalian cells using pharmacological and silencing strategies. We identified by click
chemistry methodology, new iminosugars, multivalent derivative of the DMJ able to rescue
F508del-CFTR mutation.
MATERIALS AND METHODS
Cell culture. Stably transfected Hela cell lines expressing wild-type CFTR (spTCF-wt) or
F508del-CFTR (spTCF-ΔF) were grown in Dulbecco's modified Eagle's medium
(DMEM)+Glutamax supplemented with 10% of fetal bovine serum (FBS), 100 U/ml penicillin,
100 µg/ml streptomycin and 125 mg/ml Zeocine (all from Invitrogen). Human bronchial
epithelial cell lines CFBE41O- were cultured in minimum essential medium Eagle (MEM,
Lonza) containing 10% FBS (Lonza), 2 mM L-glutamine (Gibco) and 5 µg/ml plasmocyne
(Invitrogen). CF human lung tissues were obtained from 2 patients homozygotes for F508del
mutation (the study was approved by our local institutional ethics committee). Following
lobectomy, lung tissues were quickly dissected. Bronchial tubes were removed from connective
tissues, washed three times in PBS (Phosphate Buffered Saline) and then opened along the
length. Epithelium was mechanically dissociated by scraping and then recovered by
centrifugation at 2000 rpm for 5 min. After resuspension in 1 ml of culture medium [Dulbecco’s
modified Eagle medium/HAM-F12, insulin 5 µg/ml, transferrin 7.5 µg/ml, hydrocortisone
10- 6 M, endothelial cell growth supplement 2 µg/ml, epithelial growth factor 25 ng/ml, T3
3.10- 5 M, l-glutamine 200 mM and penicillin + streptomycin 100 µg/ml], dissociated ciliated
epithelial cells were plated onto dishes for 4 hours before experiments. Cells were maintained
at 37°C in a humidified incubator with 5% CO2.
Gene silencing. Small Interfering RNA (siRNA) transfection was performed for 48 hours
according to the manufacturer’s instructions, on cells at 60-70% of confluence. All siRNAs
used in this study were purchased from Santa Cruz Biotechnology: EDEM1 siRNA (sc-43745),
EDEM2 siRNA (sc-77226), EDEM3 siRNA (sc-78683) and control siRNA (sc-37007).
Western blot. Cells were washed with PBS then mechanically harvested in 100 µl of lysis
buffer [10 mM Tris-HCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 1 mM Pefabloc SC,
pH 7.5, supplemented with protease inhibitor cocktail from Roche]. After incubation on ice for

Résultats
106
30 min, lysates were centrifuged for 5 min at 16,500 g and 4°C. Clarified lysates were recovered
and total proteins were denatured at: 37°C for 1 hour in Laemmli sample buffer 2X [125 mM
Tris-HCl, 4,5% SDS, 30% glycerol, 0.002% bromophenol blue and 5% α-monothioglycerol],
for CFTR protein immunoblotting; or 95°C for 5 min in Laemmli 5X (DGel Sciences) for the
detection of the other proteins studied. 50 µg of total protein extract was then separated in 10%
SDS-PAGE gel (5% for CFTR) and then transferred onto nitrocellulose membrane at 4°C for
1 hour. After saturation with PBS containing 0.1% Tween20 and 5% skimmed milk powder,
membranes were incubated overnight at 4°C with primary antibodies: mouse anti-human CFTR
(MAB3480, 1/1000, Merck Millipore), mouse anti-Na+/K+-ATPase α1 subunit (1/1000), goat
anti-EDEM1 (1/1000), rabbit anti-EDEM2 (1/2000), goat anti-EDEM3 (1/10000), rabbit anti-
VIP36 (1/1000), rabbit anti-ERGIC-53 (1/10000) (Santa Cruz Biotechnology) or mouse anti-
β-actin (1/5000, Sigma-Aldrich). The membrane was then incubated at 4°C for 1 hour with
corresponding horseradish peroxidase conjugated secondary antibodies: anti-mouse, anti-rabbit
(1/10000, Sigma-Aldrich) or anti-goat (1/10000, Santa Cruz Biotechnology). After washing
with PBS, bands antibodies were visualized by chemiluminescence using ECL western blotting
detection reagent (GE Healthcare, UK), and then quantified by densitometry using Photoshop
software.
Cell surface biotinylation. Cells were washed with PBS and then incubated for 30 min at
4°C with sulfo-NHS-SS-biotin diluted at 0.5 mg/ml in PBS. Cells were then rinsed two times
with quenching solution and then harvested in 300 µl of lysis buffer. After 30 min of incubation
on ice, lysates were centrifuged at 16,500 g for 5 min at 4°C. Clarified lysate was recovered
and total proteins concentration was determined using the BCA (bicinchoninic acid) assay
(Pierce Biotechnology). Biotinylated proteins were added in the column containing
NeutrAvidin Agarose beads (100 µg of protein for 15 µl of NeutrAvidin Agarose), pre-
equilibrated with the wash buffer as indicated in manufacturer’s instructions. After an
incubation for 1 hour at room temperature on a rotator, column was centrifuged for 1 min at
1000 g to remove the supernatant and to keep only the biotinylated proteins attached to the
beads. Beads were then washed four times with wash buffer containing protease inhibitor
cocktail. Biotinylated proteins were eluted from the beads with an incubation for 1 hour at 37°C
in the presence of Laemmli sample buffer 2X (15 µl for 100 µg of proteins added to column).
Biotinylated CFTR was then analyzed by western blot.
Immunofluorescence staining. Cells cultured on coverslips were washed with TBS (Tris
Buffered Saline) [157 mM NaCl, 20 mM Tris Base, pH 7.4] then fixed for 5 min at -20°C with

Résultats
107
cold methanol. The plasma membrane was permeabilized for 10 min with triton diluted at 0.1%
in TBS, and non-specific binding sites were blocked for 1 hour at 4°C with TBS containing
0.5% of bovine serum albumin (BSA) and 0.05% of saponine. Cells were then incubated
overnight at 4°C with primary antibodies: Goat anti-EDEM1, Rabbit anti-EDEM2, Goat anti-
EDEM3, Mouse anti calnexin (1/50, Santa Cruz Biotechnology), Rabbit anti calnexin (1/50,
Enzo Life Sciences). After washing with TBS, cells were incubated for 1 hour with secondary
antibodies: Donkey anti-Rabbit Alexa Fluor 555, Donkey anti-Mouse Alexa Fluor 555, Donkey
anti-Goat Alexa Fluor 647 or Donkey anti-Rabbit Alexa Fluor 647 (1/50, Thermo Fisher
Scientific). Coverslips were finally mounted on slides using a mounting medium with DAPI to
label nuclei. Fluorescence was examined using a spectral confocal station FV1000 installed on
an inverted microscope CV100 IX-81 (Olympus, Tokyo, Japan) and data acquisition was made
with FV10-ASW 3.0 software.
Proximity Ligation Assay (PLA). The proximity ligation assay was used to detect proteins
interactions: PLA probe anti-mouse PLUS (DUO92001), PLA probe anti-rabbit MINUS
(DUO92005), PLA probe anti-goat MINUS (DUO92006) and detection reagents ORANGE
(DUO92007). Cells cultured on coverslips were fixed for 20 min with TBS-0.3%
paraformaldehyde, permeabilized for 10 min with TBS-0.1% triton and then saturated with
TBS-0.5% BSA-0.05% saponine. Cells were incubated overnight at 4°C with primary specific
antibodies: mouse anti-human CFTR (MAB25031, R&D Systems) at 1/100 with goat anti-
EDEM1 (1/200), rabbit anti-EDEM2 (1/500), goat anti-EDEM3 (1/10000) (Santa Cruz
Biotechnology), Rabbit anti-Hsp70 (1/300), Rabbit anti-Hsp90 (1/300) or Rabbit anti-Calnexin
(1/300) (Enzo Life Sciences); or mouse anti-human CFTR (MAB25031, R&D Systems) at
1/500 with Rabbit anti-VIP36 (1/500) or Rabbit anti-ERGIC53 (1/1000) (Santa Cruz
Biotechnology). After three washes with the blocking solution, cells were incubated for 1 hour
at 37°C in a pre-heated humidity chamber with a PLA mixture (PLA probes PLUS detecting
mouse and PLA probes MINUS detecting rabbit or goat antibodies diluted beforehand at 1/5 in
the blocking solution for 20 min). After two times of washing for 5 min with a wash buffer A
[0.01 M Tris, 0.15 M NaCl and 0.05% Tween 20], samples were incubated for 30 min at 37°C
with a ligation solution consisting of Ligation stock and Ligase diluted in high purity water at
1/5 and 1/40 respectively. Cells were washed again with a wash buffer A and incubated for 100
min with a mixture of Amplification stock (1/5) and Polymerase (1/80) diluted in high purity
water. Samples were then washed two times for 10 min with 1X wash buffer B [0.2 M Tris and
0.1 M NaCl], one time for 1 min in 0.01X wash buffer B, and then, coverslips were mounted

Résultats
108
on slides using a mounting medium with DAPI to label nuclei. Samples were examined on a
spectral confocal station and quantification of fluorescent spots and nuclei number was done
with ImageJ software. Results were done as mean of spots number per nuclei.
Measurements of CFTR transport activity. CFTR-mediated flux was assayed by measuring
the rate of iodide (125I) efflux from cell population using a MultiPROBE IIex robotic liquid
handling system (Perkin Elmer Life Sciences) as previously described (Norez et al., 2004).
Time-dependent rates of 125I efflux of initial intracellular 125I lost during each time point was
calculated from: In(125It1/125It2)/(t1-t2), where 125It is the intracellular 125I at time t, and t1 and t2
successive time points. Curves were constructed by plotting rate of 125I versus time. All
comparisons were based on maximal values for the time-dependent rates (k peak rates, min-1)
excluding the points used to establish the baseline (k peak-k basal, min-1).
CFTR activity was measured by single-cell fluorescence imaging using the potential-sensitive
probe, bis-(1,3-diethylthiobarbituric acid) trimethine oxonol (DiSBAC2[3]; Molecular Probes,
Eugene, OR) as previously described (Norez et al., 2009). Briefly, cells were washed with free
Cl- krebs solution [101 mM Na gluconate, 5 mM K acetate, 2 mM Ca gluconate, 2 mM Mg
acetate, 50 mM Manitol, 5 mM Glucose, 5 mM Hepes-Tris, pH 7.4] and then labeled for 20
min with Bis-oxonol probe diluted at 250 nM in krebs. Fluorescence signal was recorded by
Zeiss Axio observer Z1 inverted microscope. Recordings were performed under Carl Zeiss
AxioVision Release 4.8.2 software, with physiology acquisition Module. Results are presented
as ([Ft−F0]/F0)×100, where Ft and F0 are, respectively, the fluorescent values at the time t and
F0 the fluorescence at the time of addition of CFTR activator.
We used Ussing chambers applied to polarized monolayers of CFBE41O- cells cultured at
air–liquid interface onto 12-mm-diameter Costar Snapwell cell culture inserts (Corning Costar,
Acton, MA; 3801). The apical bath solution contained (in mM) (107 K-gluconate, 4.5 KCl,
25 NaHCO3, 1 MgSO4, 1.8 Na2HPO4, 0.2 NaH2PO4, 5.75 Ca-gluconate, 12 D-glucose, pH 7.4)
and basolateral bath solution (in mM) (111.5 KCl, 25 NaHCO3, 1 MgSO4, 1.8 Na2HPO4, 0.2
NaH2PO4, 1.25 CaCl2, 12 D-glucose pH 7.4). Ussing chambers were connected to the
preamplifier head stage of an amplifier (Physiologic Instruments, USA). Data were collected
with the Acquire and Analyze package software (Physiologic Instruments, USA). CFTR-
dependent current (Isc) was stimulated by addition of the cocktail forskolin (10 µM) + genistein
(30 µM) (to both the apical and basolateral bath solutions for Ussing chamber) and inhibited
with CFTRinh-172 (10 µM) (Ma et al., 2002).

Résultats
109
Statistics. Results are expressed as means ± SEM. Data sets were compared using one-way
ANOVA or a Student’s t-test (Prism 5.0, GraphPad Software). All experiments were done at
least three times, and differences were considered statistically significant when p<0.05.
Chemicals. Forskolin and genistein were purchased from LC laboratories (Wobum, MA,
USA). VX-809 and VX-770 were obtained from Selleck Chemicals (Houston, TX, USA),
CFTRinh-172 from VWR International (Fontenay/bois, France) and all other chemicals from
Sigma-Aldrich.

Résultats
110
RESULTS
Expression of EDEM variants in cell model and their interaction with F508del-CFTR.
First, we investigated the expression and the localization of the three variants of EDEM
in our cell model, HeLa cell line stably transfected with F508del-CFTR. Using immunoblot
techniques, EDEM1 was detected at 69 kDa and 74 kDa, corresponding respectively to a
soluble- and membrane associated-forms (Fig. 1A up), EDEM2 and EDEM3 were detected
respectively at 70 kDa and 110 kDa (Fig. 1A middle and bottom). We applied then an
immunocytochemistry experiments to observe their subcellular distribution. Confocal
immunofluorescence of EDEM1 (green) showed a localization in punctate structures, which
overlap with the localization of the ER-marker, calnexin (red) (Fig. 1B up). No colocalization
of EDEM1 and calnexin could be observed in our cell model. This result is consistent with
previous studies demonstrating a localization of EDEM1 in vesicles and tubular structures
budding from the ER, called EDEMosomes (Zuber et al., 2007; Calì et al., 2008). Confocal
immunofluorescence of EDEM2 and EDEM3 also showed a localization in structures, which
were distinct from the calnexin and seem to be localized as for EDEM1, in ER-budding vesicles
(Fig. 1B middle and bottom).
EDEM variants play a central role in the recognition and sorting of misfolded proteins to ERAD
pathway (Slominska-Wojewodzka and Sandvig, 2015). In order to determine if these lectins
are involved in the degradation of F508del-CFTR, we examined their interactions using the In
Situ Proximity Ligation Assays. The results from these experiments show the presence of
EDEM1/, EDEM2/ and EDEM3/F508del-CFTR interactions (red spots) (Fig. 1C), suggesting
that these lectins seem to be involved in the trafficking defect of F508del-CFTR.

Résultats
111
FIGURE 1
Figure 1. The three variants EDEM1, EDEM2 and EDEM3 are expressed in Hela F508del-CFTR cell line and interact with F508del-CFTR. (A) The expression of EDEM variants: EDEM1 (up), EDEM2 (middle) and EDEM3 (bottom) in Hela F508del-CFTR cells was assessed by western blot analysis. Actin was used as a loading control. (B) Representative confocal immunofluorescence images of Hela F508del-CFTR cells co-stained with antibodies against an ER marker, the calnexin and EDEM1 (up), EDEM2 (middle) or EDEM3 (bottom) (C) Representative confocal images of proximity ligation assay showing interactions (red spots) between F508del-CFTR and EDEM1 (up), EDEM2 (middle) or EDEM3 (bottom). Nuclei were stained with Dapi (blue). Scale bars: 20 µm. Experiments were repeated more than three times.

Résultats
112
Are EDEM variants implicated in F508del-CFTR retention?
To investigate whether EDEM variants are involved in F508del-CFTR retention, we
used a siRNA strategy to decrease their protein expression level. First, we transfected our cell
model, Hela F508del-CFTR with EDEM1 siRNA and assessed the effect on F508del-CFTR
(Fig. 2). The results show that knockdown of EDEM1 protein level (by about 45%,
supplementary data 1) led to decreased F508del-CFTR/EDEM1 interaction by 34%, compared
to control siRNA condition (Fig. 2A and 2B). To assess the influence of this interaction
decrease on the processing of F508del-CFTR, we analyzed its maturation profile and channel
activity using respectively western-blot and iodide efflux assays. EDEM1 siRNA transfection
did not restore the mature form of CFTR (band C); however, it induced a chloride efflux
response to a cocktail of forskoline+genistein, two well-known activators of CFTR (Fig. 2C
and 2E). Thus, we postulate from our results that decreasing F508del-CFTR/EDEM1
interaction restores an immature but functional F508del-CFTR to the plasma membrane.
The same experiments were performed to investigate the effect of EDEM2 and EDEM3
siRNAs on F508del-CFTR. EDEM2 siRNA transfection decreased F508del-CFTR/EDEM2
interaction by 23%, concomitant with the decrease of EDEM2 protein expression (decrease of
40%, supplementary data 1). Whereas this decrease did not influence the glycosylation state of
F508del-CFTR (Fig 3C and 3D), it restored its chloride activity (Fig.3E and 3F), suggesting
that as for EDEM1, the decrease of EDEM2/F508del-CFTR interaction allows F508del-CFTR
to reach the cell surface in its immature form but functional. In the same way, the decrease of
EDEM3 protein expression level by siRNA (by 50%, supplementary data 1) decreased
F508del- CFTR/EDEM3 interaction by 25% (Fig. 4A and 4B). However, decreasing this
interaction had no effect, neither on F508del-CFTR maturation nor on its function (Fig. 4C and
4E).
All together, these results demonstrate that EDEM1 and EDEM2 are involved in the retention
of F508del-CFTR and highlight these lectins as potential pharmacological targets to develop
new classes of correctors for the F508del-CFTR retention.

Résultats
113
FIGURE 2
Figure 2. Impact of EDEM1 protein Knockdown on F508del-CFTR in HeLa F508del-CFTR cell line. (A) Representative confocal images of proximity ligation assay showing the effect of EDEM1 siRNA on F508del-CFTR/EDEM1 interaction (red dots). Nuclei were stained with Dapi (blue). Scale bars: 20 µm. (B) The histogram reports the quantification of dots/nuclei, representative of the amount of F508del-CFTR/EDEM1 interaction per cell. (C) Immunoblot analysis showing the effect of EDEM1 siRNA on F508del-CFTR protein, compared to control siRNA and untransfected conditions. Treatment with VX809 (10 µM for 24h) was used as a positive control. The α1 NaK-ATPase was used as a loading control. (D) Histogram reporting the relative quantification of [band-C/(band-B+band-C)]. (E) Representative traces of iodide efflux technique showing the effect of EDEM1 siRNA on F508del-CFTR activity. VX809 treatment (10 µM, 24 h) was used as a positive control. The cocktail Forskolin 10 µM + Genistein 30 µM was used to activate CFTR. (F) The histogram summarizes CFTR chloride activity for each condition. All results are presented as normalized of mean ±S.E.M. ns: no significant difference; *P<0.05; ***P<0.001. Experiments were performed 3 times.
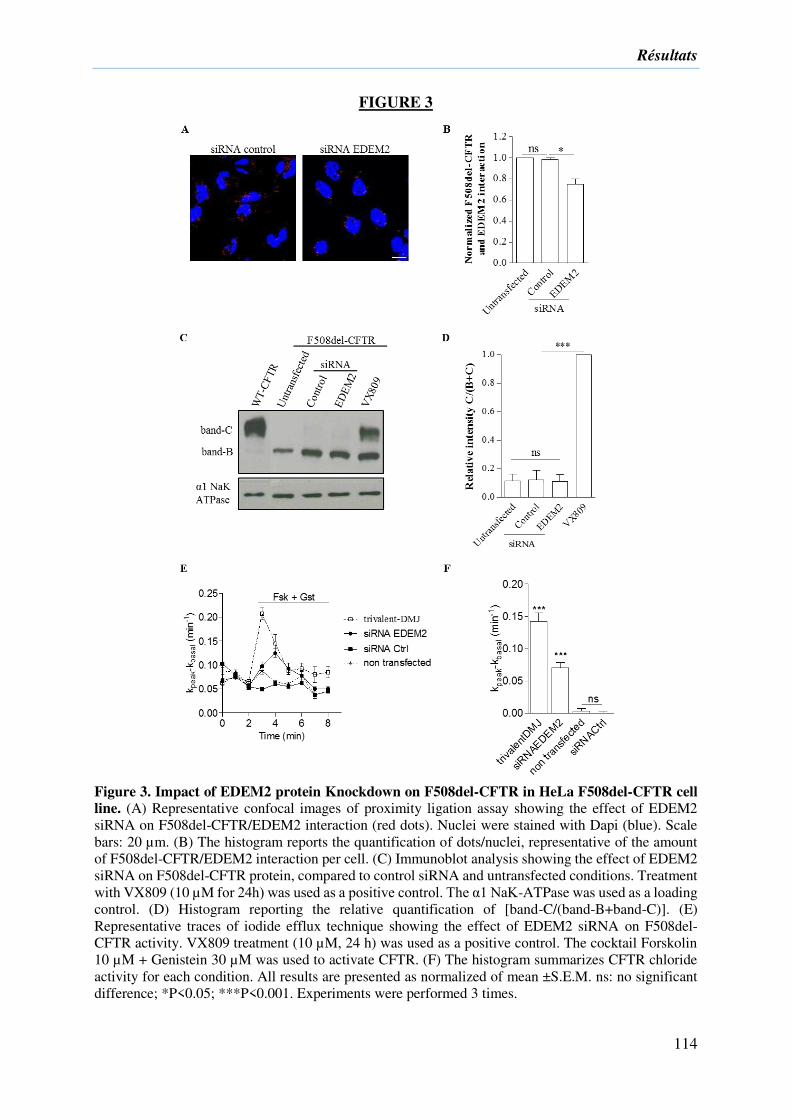
Résultats
114
FIGURE 3
Figure 3. Impact of EDEM2 protein Knockdown on F508del-CFTR in HeLa F508del-CFTR cell line. (A) Representative confocal images of proximity ligation assay showing the effect of EDEM2 siRNA on F508del-CFTR/EDEM2 interaction (red dots). Nuclei were stained with Dapi (blue). Scale bars: 20 µm. (B) The histogram reports the quantification of dots/nuclei, representative of the amount of F508del-CFTR/EDEM2 interaction per cell. (C) Immunoblot analysis showing the effect of EDEM2 siRNA on F508del-CFTR protein, compared to control siRNA and untransfected conditions. Treatment with VX809 (10 µM for 24h) was used as a positive control. The α1 NaK-ATPase was used as a loading control. (D) Histogram reporting the relative quantification of [band-C/(band-B+band-C)]. (E) Representative traces of iodide efflux technique showing the effect of EDEM2 siRNA on F508del-CFTR activity. VX809 treatment (10 µM, 24 h) was used as a positive control. The cocktail Forskolin 10 µM + Genistein 30 µM was used to activate CFTR. (F) The histogram summarizes CFTR chloride activity for each condition. All results are presented as normalized of mean ±S.E.M. ns: no significant difference; *P<0.05; ***P<0.001. Experiments were performed 3 times.
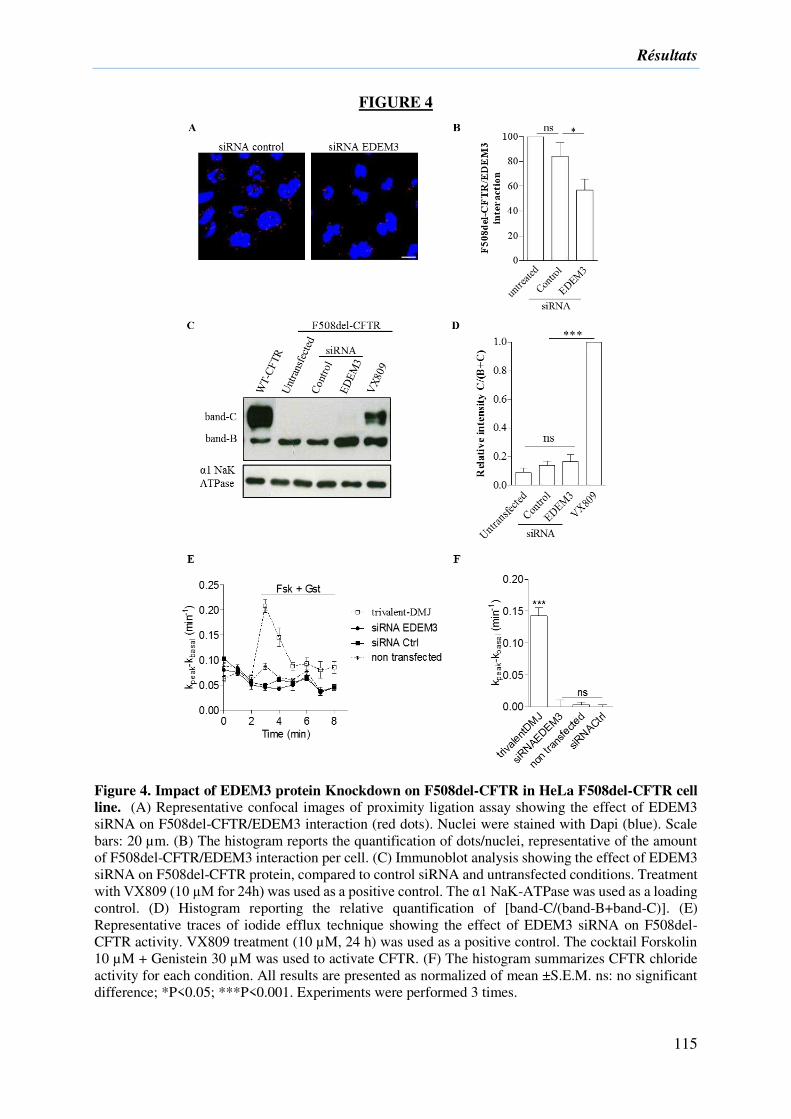
Résultats
115
FIGURE 4
Figure 4. Impact of EDEM3 protein Knockdown on F508del-CFTR in HeLa F508del-CFTR cell line. (A) Representative confocal images of proximity ligation assay showing the effect of EDEM3 siRNA on F508del-CFTR/EDEM3 interaction (red dots). Nuclei were stained with Dapi (blue). Scale bars: 20 µm. (B) The histogram reports the quantification of dots/nuclei, representative of the amount of F508del-CFTR/EDEM3 interaction per cell. (C) Immunoblot analysis showing the effect of EDEM3 siRNA on F508del-CFTR protein, compared to control siRNA and untransfected conditions. Treatment with VX809 (10 µM for 24h) was used as a positive control. The α1 NaK-ATPase was used as a loading control. (D) Histogram reporting the relative quantification of [band-C/(band-B+band-C)]. (E) Representative traces of iodide efflux technique showing the effect of EDEM3 siRNA on F508del-CFTR activity. VX809 treatment (10 µM, 24 h) was used as a positive control. The cocktail Forskolin 10 µM + Genistein 30 µM was used to activate CFTR. (F) The histogram summarizes CFTR chloride activity for each condition. All results are presented as normalized of mean ±S.E.M. ns: no significant difference; *P<0.05; ***P<0.001. Experiments were performed 3 times.

Résultats
116
Effect of Deoxymannojirimycin and its multivalent derivatives on F508del-CFTR.
As decreasing EDEM1 or EDEM2 protein expression readdresses F508del-CFTR to the
plasma membrane, we sought subsequently to identify a pharmacological approach that targets
these proteins. It is known that the Deoxymannojirimycin (DMJ), a cell permeable iminosugar
is a potent noncompetitive inhibitor of mannosidase activity that target members of GH47
family including EDEM1, EDEM2, EDEM3, Golgi α1,2 mannosidases IA, IB, IC and ER α1,2
mannosidase I (Olivari and Molinari, 2007; Ninagawa et al., 2014). In a first step, using the
iodide efflux, we tested the DMJ on F508del-CFTR activity in our cell model. As shown in
Fig. 5B, the DMJ revealed a corrector effect on F508del-CFTR by a restoration of a cAMP-
activated chloride current in response to forskolin+genestein, with a half-maximal effective
concentration (EC50) of 139,7 µM. Next, with the challenge to obtain compounds that restores
F508del-CFTR activity more efficiently than the DMJ, we adapted the concept of multivalency,
reported as a powerful and simple concept to design and synthesis a potent class of molecules
(Compain et al., 2013). Thereby, based on the DMJ cluster (Fig. 5A(1)), four multivalent
iminosugars nontoxic and water soluble were synthesized: the monovalent-DMJ, corresponding
to the DMJ grafted to an alkyl spacer (length C6) (Fig. 5A(2)), the divalent-DMJ, corresponding
to two associated monovalent-DMJ (Fig. 5A(3)) and the trivalent- and tetravalent-DMJ,
corresponding respectively to three or four associated monovalent-DMJ (Fig. 5A(4) and (5)). We
assessed thereafter in our cell model the concentration-response effect of each DMJ-derivative
on F508del-CFTR. Experiments determined an EC50 in a multivalency-increasing order of
112 µM, 86 µM, 0.8 µM and 0.7 µM (Fig. 5B). With its EC50 140-fold more efficient than the
corresponding monovalent, it is evident that the trivalent-DMJ is the best potential corrector of
F508del-CFTR.

Résultats
117
FIGURE 5
Figure 5. Deoxymannojirimycin (DMJ) and its multivalent derivatives as correctors of F508del-CFTR. (A) Structure of multivalent derivatives of the DMJ compound (1) synthesized with a “click’chemistry” methodology: monovalent-DMJ (2), divalent-DMJ (3), trivalent-DMJ (4) and tetravalent-DMJ (5). (B) Concentration-response curves obtained with iodide efflux technique on Hela F508del-CFTR cells showing the half maximal effective concentration (EC50) of each compound. Data points report the normalization of the relative radioactivity collected for each condition.

Résultats
118
The trivalent-DMJ as a corrector of F508del-CFTR.
In the goal to confirm that, the trivalent-DMJ inhibits the interaction of EDEMs with
F508del-CFTR, we studied its effect in our cell model, Hela F508del-CFTR. Confocal images
showed that in cells treated with the trivalent-DMJ, EDEM1/, EDEM2/ and EDEM3/F508del-
CFTR interactions were decreased by 35%, 29% and 16% respectively (Fig. 6A, 6B and 6C).
Moreover, this treatment did not alter the expression level of any EDEM variant (supplementary
data 2), suggesting that the trivalent-DMJ targets the interaction of F508del-CFTR with
EDEMs. Next, we investigated the effect of our compound on the main chaperones of ERQC
and their interaction with F508del-CFTR. Any effect was observed neither on calnexin, Hsp70
or Hsp90 expression level nor on their interaction with F508del-CFTR (supplementary data 3).
Together, these results demonstrates that the trivalent-DMJ has an EDEM dependent
mechanism of action.
In order to confirm its corrector effect, the trivalent-DMJ was tested on F508del-CFTR rescue
using single-cell fluorescence imaging (oxonol probe) and western-blot techniques. In cells
treated with the trivalent-DMJ, the recorded fluorescence signal increased in response to
forskoline+genistein activator and then fully inhibited by CFTRinh-172, which selectively
inhibited the CFTR channel (Ma et al., 2002) (Fig. 6D). However, the trivalent-DMJ did not
restore the maturation (band C) of F508del-CFTR (Fig. 6E), suggesting that as for results
obtained with EDEM1 or EDEM2 siRNA, the trivalent-DMJ rescues F508del-CFTR to the cell
surface in its immature form.

Résultats
119
FIGURE 6
Figure 6. The trivalent-DMJ decreases the interaction of F508del-CFTR with EDEM variants and restores its function. HeLa F508del-CFTR cells were treated or not with the trivalent-DMJ (100 µM, 4h). (A) Representative proximity ligation assay images showing the effect of the trivalent-DMJ on F508del-CFTR/EDEM1 interaction (red dots). The corresponding histogram shows the quantification of dots/nuclei. The same experiment was also performed to investigate the effect of the trivalent-DMJ treatment on the interaction of F508del-CFTR with EDEM2 (B) or with EDEM3 (C). Nucleus were stained with Dapi (blue). Scale bars: 20 µm. (D) Representative time courses of relative CFTR-dependent fluorescence (Fx) of DiSBAC2 after CFTR stimulation with the cocktail forskoline 30 µM + genistein 10 µM on cells treated or not with the trivalent-DMJ. CFTR was inhibited using 10 µM of CFTRinh172. The histogram summarizes mean ± S.E.M of CFTR functionality. (E) Representative immunoblot analysis showing the effect of trivalent-DMJ on F508del-CFTR. Untreated and DMSO (0.1%) conditions were used as negatives controls and VX809 (10 µM, 24 h) was used as a positive control. The α1 Na,K-ATPase was used as a loading control. The histogram reports the quantification of the ratio [band-C/(band-B+band-C)], (3<n<8). All results are presented as mean ± S.E.M. ns: no significant difference; *P<0.05. ***P<0.001.

Résultats
120
The trivalent-DMJ restores F508del-CFTR to the plasma membrane in its immature
form.
In order to confirm that the trivalent-DMJ rescues F508del-CFTR retention, we
observed its effect on the interaction of F508del-CFTR with cargo receptor lectins: ERGIC53
(ER-Golgi Intermediate Compartment protein of 53 kDa) and VIP36 (Vesicular Integral
membrane Protein of 36 kDa). ERGIC-53, the most popular marker of ERGIC, is involved in
the ER export of glycoproteins (Appenzeller et al., 1999). VIP36, distributed to either the pre-
Golgi early secretory pathway or post-Golgi pathway, plays significant roles in vesicular
transport from the ER to the Golgi complex and also from the trans-Golgi network to the plasma
membrane (Satoh et al., 2007; Reiterer et al., 2010). Fig. 7A and 7C show that in cells treated
with the trivalent-DMJ, both F508del-CFTR/ERGIC53 and F508del-CFTR/VIP36 interactions
were significantly increased of about 1.7 times. In order to verify that this increase is not due
to the increase of the expression level of ERGIC53 and VIP36, we investigated the effect of the
trivalent-DMJ on these lectins. Immunoblot analysis showed that ERGIC53 or VIP36 were
equally expressed in untreated and treated cells (Fig.7B and 7D), suggesting that our corrector
did not alter their expression level. From these results, we suggest that the trivalent-DMJ
treatment leads to the exit of F508del-CFTR from the ER and to its transport to the Golgi
apparatus. Moreover, these observations made us to dismiss the hypothesis of F508del-CFTR
rescue via an unconventional secretory proteins pathway.
On the other hand, to further validate our hypothesis, rescue to the cell surface of an immature
F508del-CFTR under the trivalent-DMJ treatment, we performed the cell surface biotinylation
assay. Membrane proteins were biotinylated and then isolated from the cell lysate with
NeutrAvidin beads. F508del-CFTR biotinylated, and hence present at the cell surface was then
detected using immunoblot technique. As expected, no CFTR bands were observed in untreated
Hela F508del-CFTR cells, since this protein considered by the ERQC as misfolded protein
remains retained in the ER. However, once treated with the trivalent-DMJ, the core glycosylated
F508del-CFTR (band B) was detected, suggesting the presence of the immature form of
F508del-CFTR on plasma membranes of these cells. The experiment was also performed on
untreated Hela WT-CFTR cells, and as expected, only the complex glycosylated form of CFTR
(band C) was detected (Fig. 7E).
Based on all these observations, we confirm that the trivalent-DMJ restores an immature but
functional F508del-CFTR to the plasma membrane via a conventional secretory pathway.

Résultats
121
FIGURE 7
Figure 7. The trivalent-DMJ rescue F508del-CFTR in its immature form. HeLa F508del-CFTR cells were treated or not with the trivalent-DMJ (100 µM, 4h). (A,C) Representative proximity ligation assay images showing the effect of the trivalent-DMJ on the interaction of F508del-CFTR with ERGIC53 (A) or VIP36 (C) (red dots). Nucleus were stained with Dapi (blue). Scale bars: 20 µm. Histograms report the quantification of the amount of interactions. (B,D) Representative immunoblots analysis showing the effect of the trivalent DMJ on ERGIC53 (B) or VIP36 (D) protein expression. Actin was used as a loading control. Histograms report the quantification of ratio ERGIC53/actin or VIP36/actin density band. (E) Representative immunoblots analysis showing as indicated, the detection of CFTR and control proteins in total lysates and in biotinylated fractions treated or not with the trivalent-DMJ. Absence of the cytosolic protein ezrin in the biotinylated fraction confirms surface protein-specific labeling in each experiment. Absence of proteins detection in no-biotin condition confirms NeutrAvidin Agarose specificity. The α1 NaK-ATPase was used as a loading control. All experiments were repeated three times, except for (B) and (D) n=5.

Résultats
122
Effect of the Trivalent-DMJ on F508del-CFTR in CFBE cells.
In a second way, we confirmed the corrector effect of the trivalent-DMJ on F508del-
CFTR in more physiological cell models. First, we tested our corrector in polarized monolayers
of CFBE41o− cells, by measuring the short-circuit current on the Ussing chamber. Fig 8.A
shows that in cells treated with the trivalent-DMJ, the addition of forskoline+genistein induced
an increase of Isc current 20 µA.cm2. This signal was fully inhibited following the addition of
CFTRinh-172, reflecting the expression of F508del-CFTR channels on the plasma membrane of
treated cells. Interestingly, we tested the trivalent-DMJ on human bronchial epithelial cells
freshly isolated from CF patients, homozygotes for the mutation F508del, using single-cell
fluorescence imaging (oxonol probe). An increase of the recorded fluorescence signal was
observed following the addition of the cocktail forskoline+genestein only in cells treated with
the trivalent-DMJ. The signal was decreased and then fully inhibited in the presence of
CFTRinh-172 (Fig. 8C and D). These results confirm that the trivalent-DMJ correct the
trafficking defect of F508del-CFTR in CF cells.
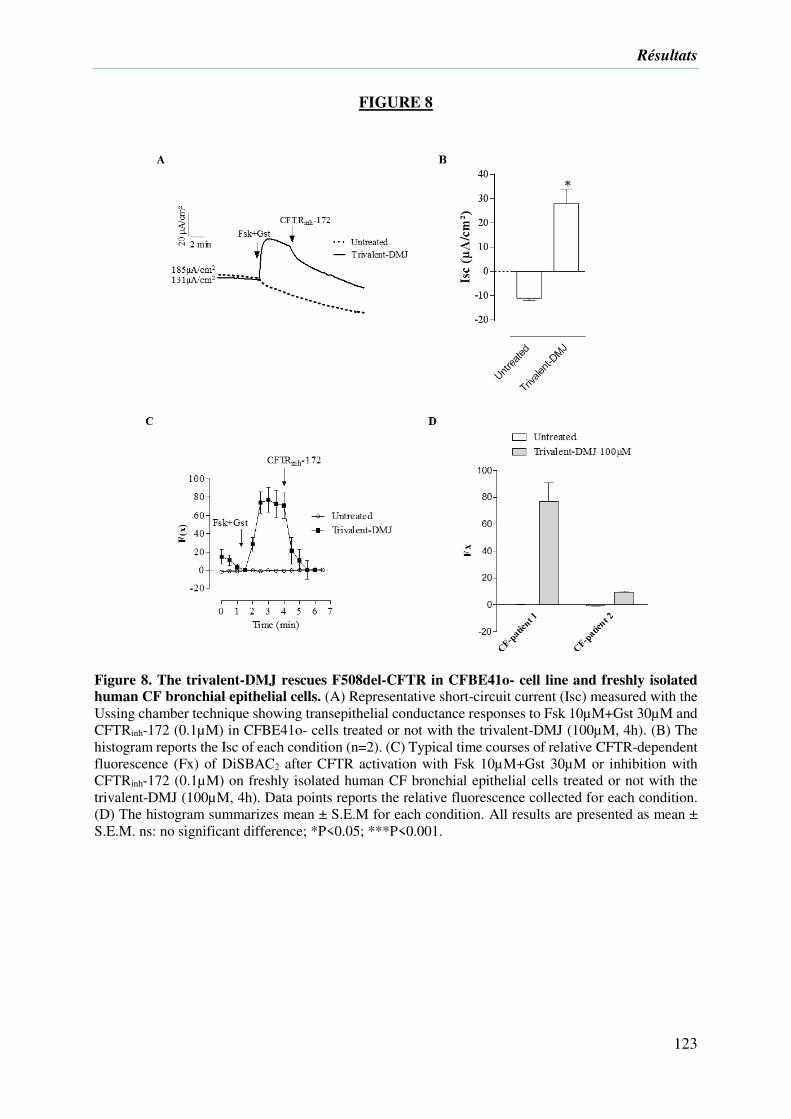
Résultats
123
FIGURE 8
Figure 8. The trivalent-DMJ rescues F508del-CFTR in CFBE41o- cell line and freshly isolated human CF bronchial epithelial cells. (A) Representative short-circuit current (Isc) measured with the Ussing chamber technique showing transepithelial conductance responses to Fsk 10µM+Gst 30µM and CFTRinh-172 (0.1µM) in CFBE41o- cells treated or not with the trivalent-DMJ (100µM, 4h). (B) The histogram reports the Isc of each condition (n=2). (C) Typical time courses of relative CFTR-dependent fluorescence (Fx) of DiSBAC2 after CFTR activation with Fsk 10µM+Gst 30µM or inhibition with CFTRinh-172 (0.1µM) on freshly isolated human CF bronchial epithelial cells treated or not with the trivalent-DMJ (100µM, 4h). Data points reports the relative fluorescence collected for each condition. (D) The histogram summarizes mean ± S.E.M for each condition. All results are presented as mean ± S.E.M. ns: no significant difference; *P<0.05; ***P<0.001.

Résultats
124
DISCUSSION
Our study highlights a role for inhibition of EDEM affecting the trafficking defect of
F508del-CFTR in CF cells and proposes water soluble and nontoxic mannosidase inhibitors as
potential candidates to rescue F508del-CFTR. We showed, using siRNA strategy, that
decreasing the interaction of F508del-CFTR with endogenous EDEM1 or EDEM2 but not with
EDEM3 might restores a functional F508del-CFTR, but as the core glycosylated form
(immature form, band B). In the Yeast model, among the three EDEMs, only EDEM1 was
shown playing a role in the degradation process of F508del-CFTR. Our study is thus the first
showing the involvement of EDEM1 and EDEM2 in F508del-CFTR retention in mammalian
cells. Concerning the EDEM3 siRNA transfection, several hypothesis can be formulated for the
absence of a corrector effect on F508del-CFTR: (i) the loss/decrease of EDEM3 function is
compensated by other EDEMs, (ii) F508del-CFTR has already been engaged in tardive steps
of ERAD pathway, (iii) the decrease level of F508del-CFTR/EDEM3 interaction is not
sufficient to restore F508del-CFTR or (iv) EDEM3 is not involved in the retention of F508del-
CFTR. Further studies on EDEM3 knockout (KO) cells would be needed to answer these
questions. Therefore, our observations suggested EDEM1 and/or EDEM2 as new potential
pharmacological targets to further develop correctors to rescue F508del-CFTR.
In a second part of our study, we focused on the identification of a pharmacological
approach that could targets these lectins to rescue F508del-CFTR to the cell surface. Because
all endogenous EDEMs have a mannosidase activity, we tested DMJ, a general inhibitor of α-
mannosidases and shown its corrector effect on F508del-CFTR. However, this result is not
consistent with that observed by Chang and collaborators, which reported that the inhibition of
EDEM with the DMJ or kifunensine does not rescue F508del-CFTR to the cell surface (Chang
et al., 2008). However, the authors used cells transiently expressing HA-tagged EDEM, and
performed treatments of 20 hours with DMJ, whereas we used endogenous EDEM expressing
cells and 4h treatment with DMJ. Secondly, in order to obtain more efficient correctors of
F508del-CFTR, we synthesized using multivalency (Compain et al., 2013) from the DMJ
clusters, four multivalent derivatives, and showed the best efficiency obtained with the
trivalent-DMJ compound. Trivalent-DMJ decreased the interaction of F508del-CFTR with
EDEM1, EDEM2 and EDEM3 and had no effect on the interaction of F508del-CFTR with the
main chaperones of the ERQC, such as Hsp70, Hsp90 and calnexin, confirming an EDEM-
dependent mechanism of action. Then, we tested our compound on F508del-CFTR to confirm
its corrector effect and showed that treatment of our cell model does not restore the complex
glycosylation form of F508del-CFTR. However, despite the absence of the maturation, using

Résultats
125
oxonol fluorescence, we recorded CFTR-dependent rescue, suggesting a plasma membrane
expression of F508del-CFTR. Therefore, either (i) the trivalent-DMJ is a derivative of the DMJ,
an inhibitor of ER-mannosidases but also of Golgi-mannosidases, which are involved in the
maturation process of glycoproteins (their inhibition block the F508del-CFTR maturation but
not its traffic to the cell surface) or (ii) corrected F508del-CFTR is delivered to the cell surface
via an unconventional secretion pathway. Indeed, it has been reported that F508del-CFTR
surface expression can be rescued in vitro and in vivo by directing it to an unconventional
GRASP-dependent secretion pathway (Gee et al., 2011). Therefore, we investigated the exit of
F508del-CFTR following treatment with the trivalent-DMJ. Interestingly, we found that the
trivalent-DMJ increased the interaction of F508del-CFTR with the cargo receptor lectins
ERGIC53 and VIP36, indicating the exit of F508del-CFTR from the ER, and the rescue of
F508del-CFTR via the conventional pathway. To further confirm that Trivalent-DMJ restores
an immature F508del-CFTR to the cell surface, we observed the glycosylation status of surface
CFTR using a Biotinylation assay. Only the core glycosylated form, corresponding to the
surface-immature F508del-CFTR was observed in cells treated with Trivalent-DMJ.
Interestingly, this result is consistent with that of Morris and collaborators, obtained in both
polarized and unpolarized HT-29 cells showing that treatment of WT-CFTR with the DMJ
reduced the molecular mass of its mature form (170 kDa) by 30 kDa without altering its
transepithelial Cl- current and conductance (Morris et al., 1993). The lack of the complex
glycosylated form (mature form, band C) is without effect on F508del-CFTR. The
glycosylation status of endogenous CFTR does not affect its function (Chang et al., 2008) since
unglycosylated CFTR functions as a chloride channel using membrane vesicles prepared from
stably transfected BHK cells (Chang et al., 2008). However, using similar methods, Chang et
al. also shown that unglycosylated F508del cannot escape ER quality control. Moreover, it was
also shown that the inhibition of EDEM interaction with F508del cannot rescue F508del but
after 20h-treatment with DMJ, kifunensine or tunicamycine (Chang et al., 2008).
Finally, we confirmed the corrector effect of the trivalent-DMJ on human polarized bronchial
epithelial cells (CFBE) using the Ussing Chamber technique and more interestingly, we
confirmed its corrector effect on human respiratory epithelial cells freshly isolated from two
different CF patients using the fluorescent probe oxonol.
In conclusion, we showed that both EDEM1 and EDEM2 are involved in F508del-
CFTR retention and propose them as targets for therapeutically approach in CF. We identified
that the trivalent-DMJ partially restores the trafficking defect of F508del-CFTR as core

Résultats
126
glycosylated immature band B form with an EDEM-dependent mechanism of action and we
propose a new series of water soluble and non-toxic correctors.
ACKNOWLEDGMENTS
We thank association “Vaincre La Mucoviscidose” for their support.

Résultats
127
Supplementary data 1
Supplementary data 1. Knockdown of EDEM1, EDEM2 and EDEM3 proteins with specific siRNAs. (A) Up: Knockdown of EDEM1 by specific siRNA transfection on F508del-CFTR cells. Actin was used as a loading control. Down: Histogram showing the quantification of EDEM1 protein extinction compared to control siRNA. The same experiment was performed to verify EDEM2 (B) or EDEM3 (C) protein extinction by their specific siRNA. All results are presented as mean ± S.E.M. ns: no significant difference; *P<0.05.

Résultats
128
Supplementary data 2
Supplementary data 2. Effect of the trivalent-DMJ on EDEM1, EDEM2 and EDEM3 proteins expression. (A) Representative immunoblot analysis showing EDEM1 protein expression in Hela F508del-CFTR cells treated or not the trivalent-DMJ (100 µM, 4h). Actin was used as a loading control. Corresponding histogram shows the quantification of ratio EDEM1/actin density band. The same experiment was performed to investigate the effect of the trivalent-DMJ treatment on EDEM2 (B) or EDEM3 (C) protein expression. All results are presented as mean ± S.E.M. ns: no significant difference.

Résultats
129
Supplementary data 3
Supplementary data 3. Effect of the trivalent-DMJ on the main chaperones of ERQC. Hela F508del-CFTR cells were treated or not with the trivalent-DMJ (100µM, 4h) (A) Representative proximity ligation assay images showing the effect of the trivalent-DMJ on F508del-CFTR/Calnexin interaction (red dots). Nucleus were stained with Dapi (blue). Scale bars: 20 µm. The histogram reports the quantification of dots/nuclei, representative of F508del-CFTR/calnexin interaction per cell. The same experiment was performed to investigate the effect of the trivalent-DMJ on the interaction of F508del-CFTR with HSP70 (C) or HSP90 (E). (B) Representative immunoblot analysis showing the effect of the trivalent DMJ on calnexin protein expression. Actin was used as a loading control. The corresponding histogram reports the quantification of ratio calnexin/actin density band. The same experiment was performed for the effect of the trivalent-DMJ on HSP70 (D) or HSP90 (F) protein expression. All results are presented as mean ± S.E.M. (n=3). ns: no significant difference.

Résultats
130
REFERENCES
Appenzeller, C., Andersson, H., Kappeler, F., and Hauri, H.P. (1999). The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat. Cell Biol. 1, 330–334.
Banerjee, S., Vishwanath, P., Cui, J., Kelleher, D.J., Gilmore, R., Robbins, P.W., and Samuelson, J. (2007). The evolution of N-glycan-dependent endoplasmic reticulum quality control factors for glycoprotein folding and degradation. Proc. Natl. Acad. Sci. U. S. A. 104, 11676–11681.
Calì, T., Galli, C., Olivari, S., and Molinari, M. (2008). Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 371, 405–410.
Chang, X. -b., Mengos, A., Hou, Y. -x., Cui, L., Jensen, T.J., Aleksandrov, A., Riordan, J.R., and Gentzsch, M. (2008). Role of N-linked oligosaccharides in the biosynthetic processing of the cystic fibrosis membrane conductance regulator. J. Cell Sci. 121, 2814–2823.
Compain, P., Decroocq, C., Joosten, A., de Sousa, J., Rodríguez-Lucena, D., Butters, T.D., Bertrand, J., Clément, R., Boinot, C., Becq, F., et al. (2013). Rescue of Functional CFTR Channels in Cystic Fibrosis: A Dramatic Multivalent Effect Using Iminosugar Cluster-Based Correctors. ChemBioChem 14, 2050–2058.
Cormier, J.H., Tamura, T., Sunryd, J.C., and Hebert, D.N. (2009). EDEM1 Recognition and Delivery of Misfolded Proteins to the SEL1L-Containing ERAD Complex. Mol. Cell 34, 627–633.
Du, K., Sharma, M., and Lukacs, G.L. (2005). The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 12, 17–25.
Farinha, C.M., and Amaral, M.D. (2005). Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Biol. 25, 5242–5252.
Frenkel, Z., Gregory, W., Kornfeld, S., and Lederkremer, G.Z. (2003). Endoplasmic Reticulum-associated Degradation of Mammalian Glycoproteins Involves Sugar Chain Trimming to Man6-5GlcNAc2. J. Biol. Chem. 278, 34119–34124.
Gauss, R., Kanehara, K., Carvalho, P., Ng, D.T.W., and Aebi, M. (2011). A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell 42, 782–793.
Gee, H.Y., Noh, S.H., Tang, B.L., Kim, K.H., and Lee, M.G. (2011). Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760.
Gnann, A., Riordan, J.R., and Wolf, D.H. (2004). Cystic Fibrosis Transmembrane Conductance Regulator Degradation Depends on the Lectins Htm1p/EDEM and the Cdc48 Protein Complex in Yeast. Mol. Biol. Cell 15, 4125–4135.
Hirao, K., Natsuka, Y., Tamura, T., Wada, I., Morito, D., Natsuka, S., Romero, P., Sleno, B., Tremblay, L.O., Herscovics, A., et al. (2006). EDEM3, a Soluble EDEM Homolog, Enhances

Résultats
131
Glycoprotein Endoplasmic Reticulum-associated Degradation and Mannose Trimming. J. Biol. Chem. 281, 9650–9658.
Hosokawa, N., Tremblay, L.O., Sleno, B., Kamiya, Y., Wada, I., Nagata, K., Kato, K., and Herscovics, A. (2010). EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 20, 567–575.
Jang, B.-Y., Ryoo, H., Son, J., Choi, K.-C., Shin, D.-M., Kang, S.-W., and Kang, M.-J. (2015). Role of Drosophila EDEMs in the degradation of the alpha-1-antitrypsin Z variant. Int. J. Mol. Med.
Ma, T., Thiagarajah, J.R., Yang, H., Sonawane, N.D., Folli, C., Galietta, L.J.V., and Verkman, A.S. (2002). Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin–induced intestinal fluid secretion. J. Clin. Invest. 110, 1651–1658.
Morris, A.P., Cunningham, S.A., Benos, D.J., and Frizzell, R.A. (1993). Glycosylation status of endogenous CFTR does not affect cAMP-stimulated Cl- secretion in epithelial cells. Am. J. Physiol. 265, C688-694.
Ninagawa, S., Okada, T., Sumitomo, Y., Kamiya, Y., Kato, K., Horimoto, S., Ishikawa, T., Takeda, S., Sakuma, T., Yamamoto, T., et al. (2014). EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 206, 347–356.
Norez, C., Heda, G.D., Jensen, T., Kogan, I., Hughes, L.K., Auzanneau, C., Dérand, R., Bulteau-Pignoux, L., Li, C., Ramjeesingh, M., et al. (2004). Determination of CFTR chloride channel activity and pharmacology using radiotracer flux methods. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 3 Suppl 2, 119–121.
Norez, C., Antigny, F., Noel, S., Vandebrouck, C., and Becq, F. (2009). A Cystic Fibrosis Respiratory Epithelial Cell Chronically Treated by Miglustat Acquires a Non–Cystic Fibrosis–Like Phenotype. Am. J. Respir. Cell Mol. Biol. 41, 217–225.
Olivari, S., and Molinari, M. (2007). Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 581, 3658–3664.
Olivari, S., Cali, T., Salo, K.E.H., Paganetti, P., Ruddock, L.W., and Molinari, M. (2006). EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 349, 1278–1284.
Quan, E.M., Kamiya, Y., Kamiya, D., Denic, V., Weibezahn, J., Kato, K., and Weissman, J.S. (2008). Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 32, 870–877.
Reiterer, V., Nyfeler, B., and Hauri, H.-P. (2010). Role of the lectin VIP36 in post-ER quality control of human alpha1-antitrypsin. Traffic Cph. Den. 11, 1044–1055.
Riordan, J.R. (1993). The cystic fibrosis transmembrane conductance regulator. Annu. Rev. Physiol. 55, 609–630.

Résultats
132
Riordan, J.R., Rommens, J.M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N., and Chou, J.L. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073.
Satoh, T., Cowieson, N.P., Hakamata, W., Ideo, H., Fukushima, K., Kurihara, M., Kato, R., Yamashita, K., and Wakatsuki, S. (2007). Structural Basis for Recognition of High Mannose Type Glycoproteins by Mammalian Transport Lectin VIP36. J. Biol. Chem. 282, 28246–28255.
Slominska-Wojewodzka, M., and Sandvig, K. (2015). The Role of Lectin-Carbohydrate Interactions in the Regulation of ER-Associated Protein Degradation. Mol. Basel Switz. 20, 9816–9846.
Ward, C.L., Omura, S., and Kopito, R.R. (1995). Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127.
Zhou, T., Frabutt, D.A., Moremen, K.W., and Zheng, Y.-H. (2015). ERManI (Endoplasmic Reticulum Class I α-Mannosidase) Is Required for HIV-1 Envelope Glycoprotein Degradation via Endoplasmic Reticulum-associated Protein Degradation Pathway. J. Biol. Chem. 290, 22184–22192.
Zuber, C., Cormier, J.H., Guhl, B., Santimaria, R., Hebert, D.N., and Roth, J. (2007). EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. 104, 4407–4412.

Résultats
133
2) Résultats complémentaires
Nos résultats présentés dans l’article suggèrent que le trivalent-DMJ restaure F508del-
CFTR à la membrane cellulaire en diminuant son interaction avec les protéines EDEMs. Afin
d’élucider d’avantage le mécanisme d’action de ce correcteur et son effet sur le stress du RE,
des expériences additionnelles ont été conduites.
a) Effet du trivalent-DMJ sur l’interaction de F508del-CFTR avec les principales
ubiquitines ligases RNF5/RMA1 et CHIP
Les protéines CHIP et RNF5 (connue aussi sous le nom de RMA1) sont deux ubiquitines
ligases identifiées comme étant impliquées dans l’ubiquitinylation du F508del-CFTR (Hohfeld
et al., 2001; Meacham et al., 2001b; Murata et al., 2003; Younger et al., 2004, 2006; Morito et
al., 2008) (Figure 40).
Figure 40 : Implication de RMA1/RNF5 et CHIP dans la dégradation de F508del-CFTR. (Modifié de Kim and Skach, 2012)
La protéine RNF5 détecte les défauts de repliement cytosolique de F508del-CFTR pendant la
traduction alors que la protéine CHIP agit de manière post-traductionnelle (Younger et al.,
2006). Afin d’explorer l’effet du trivalent-DMJ sur la dégradation du F508del-CFTR, nous
avons étudié son effet sur l’interaction du F508del-CFTR avec ces protéines. Pour cela, des
expérimentations préliminaires de proximity ligation assay ont été conduites sur des cellules
Hela F508del-CFTR traitées ou non avec le trivalent-DMJ. Les résultats obtenus sont présentés
dans la figure 41.

Résultats
134
A B
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
Inte
racti
on
F5
08
del-
CF
TR
/RN
F5
Figure 41 :Effet du trivalent-DMJ sur l’interaction de F508del-CFTR avec RNF5/RMA1 ou
CHIP. (A) Images représentatives de l’interaction (points rouges) de F508del-CFTR avec RNF5/RMA1 (en bas) ou CHIP (en haut) obtenues par la technique de Proximity Ligation Assay sur des cellules Hela F508del-CFTR traitées ou non avec 100 µM de trivalent-DMJ (4h, 37°C). Echelle : 20 µm. Bleu : noyau cellulaire (B) Histogramme reportant le nombre de points rouges par noyau, représentatif du nombre d’interactions par cellule. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition NT (non traitée) (N = 2, n = 30).
Comme nous pouvons le voir, F508del interagit bien avec les protéines CHIP et RNF5/RMA1.
Une diminution du taux d’interaction entre F508del-CFTR et CHIP est observée après
traitement avec le trivalent-DMJ ; cependant, il apparait que cette diminution n’est pas
significative et des expériences supplémentaires pour augmenter le nombre d’échantillons
(nombre de N) doivent être réalisées pour conclure.
Concernant la protéine RNF5/RMA1, une augmentation d’environ 78% du nombre
d’interactions avec F508del-CFTR a été observée après traitement avec le trivalent-DMJ. Ce
résultat est surprenant car on s’attendait à avoir une diminution du taux d’interaction, dans la
mesure où des études avaient démontré que l’extinction de RNF5/RMA1 diminuait le
phénotype F508del-CFTR (Tomati et al., 2015). Notre composé semblerait donc favoriser
0.0
0.2
0.4
0.6
0.8
1.0
Inte
racti
on
F5
08
del-
CF
TR
/CH
IP
NT Trivalent-DMJ
Interaction F508del-CFTR/CHIP
NT Trivalent-DMJ
Interaction F508del-CFTR/RNF5

Résultats
135
l’interaction de F508del-CFTR avec cette ligase, probablement en bloquant de manière directe
ou indirecte sa rétro-translocation.
En conclusion, le trivalent-DMJ ne semble pas perturber l’interaction de F508del-CFTR
avec CHIP, mais augmenterait son interaction avec RNF5/RMA1.
b) Effet du trivalent-DMJ sur le protéasome 20S
Nous avons étudié l’effet du trivalent-DMJ sur l’activité enzymatique du protéasome
20S, responsable de la dégradation de F508del-CFTR rétro-transloqué dans le cytosol par le
système ERAD.
i. Effet du trivalent-DMJ sur le protéasome 20S intracellulaire
Dans un premier temps, nous avons évalué l’effet du trivalent-DMJ sur l’activité
enzymatique du protéasome intracellulaire sur des cellules Hela F508del-CFTR. Les résultats
obtenus sont présentés dans la figure 42.
NT
Trivale
nt-DM
J
Lactac
ystin
Blanc d
e sub
strat
0.0
0.2
0.4
0.6
0.8
1.0 *****
RF
U d
'AM
C (
46
0 n
m)
Figure 42 : Effet du trivalent-DMJ sur l’activité du protéasome 20S intracellulaire. L’activité enzymatique du protéasome 20S est évaluée à l’aide du kit « 20S Proteasome Activity Assay kit » sur des cellules Hela F508del-CFTR traitées ou non avec le trivalent-DMJ (100 µM, 4h). Le traitement avec la Lactacystin est utilisé comme contrôle positif. La condition « blanc de substrat » correspond aux réactifs dans un puits vide (sans cellules). La fluorescence du substrat AMC libre est quantifiée à l’aide d’un couple de filtre 380/460 nm dans un lecteur de fluorescence, ici Mithras. Les résultats présentés sont les moyennes des triplicats de trois expérimentations indépendantes +/- SEM.

Résultats
136
Les résultats obtenus sont normalisés par rapport à la condition NT « non traité »,
considérée comme égale à 1 et le 0 ou « blanc de substrat » correspondant au réactif sans
cellules.
La Lactacystine, un métabolite de Streptomyces connue entre autres comme un inhibiteur
spécifique et irréversible du protéasome 20S (Fenteany et al., 1995; Tomoda and Omura, 2000)
a été utilisée comme contrôle positif inhibant l’activité du protéasome. Comme attendu, une
diminution d’environ 99% de la fluorescence du substrat AMC a été constatée dans les cellules
traitées avec ce composé, comparée aux cellules non traitées. Cette diminution de fluorescence
est en effet le résultat de la diminution du taux de clivage d’AMC et donc de l’activité du
protéasome intracellulaire.
Concernant les cellules traitées avec le trivalent-DMJ, une diminution de 37% de la
fluorescence d’AMC a été observée, comparée aux cellules non traitées. Cela suggère que notre
correcteur, connu comme un inhibiteur des α 1,2-mannosidases du RE, diminue l’activité
enzymatique intracellulaire du protéasome 20S. Cependant, s’agit-il d’une inhibition directe du
protéasome intracellulaire ou juste d’une conséquence de l’inhibition des α 1,2-mannosidases
du RE ? Un test du trivalent-DMJ sur l’activité enzymatique du protéasome 20S purifié est alors
apparu nécessaire afin de répondre à cette question.
ii. Effet du trivalent-DMJ sur le protéasome 20S purifié
Dans un deuxième temps, nous avons étudié l’effet du trivalent-DMJ sur l’activité
enzymatique du protéasome purifié afin de s’affranchir de l’effet de notre composé sur d’autres
cibles intracellulaires. La fluorescence du substrat AMC a alors été mesurée en présence du
protéasome 20S purifié (Merck millipore) traité ou non avec 100 µM du trivalent-DMJ pendant
4h à 37°C (condition des traitements cellulaires) ou pendant 15 min à température ambiante
(condition de traitement avec l’inhibiteur Lactacystin). Les résultats obtenus à l’aide du lecteur
Mithras (filtre 380/460 nm) sont présentés dans la figure 43.

Résultats
137
NT
Trivale
nt-DM
J
Lactac
ystin
Trivale
nt-DM
J san
s 20S
Blanc d
e sub
strat
0.0
0.2
0.4
0.6
0.8
1.0 *****
ns
*
RF
U d
'AM
C (
DO
460 n
m)
NT
Triv
alent-
DMJ
Lactac
ystin
e
Blanc d
e sub
strat
0.0
0.2
0.4
0.6
0.8
1.0*
***
nsRF
U d
'AM
C (
DO
46
0 n
m)
Figure 43 : Effet du trivalent-DMJ sur l’activité du protéasome 20S purifié.
L’activité enzymatique du protéasome 20S est évaluée à l’aide du kit « 20S Proteasome Activity Assay kit ». Le protéasome 20S purifié est traité avec le trivalent-DMJ pendant 4h à 37°C (à gauche) ou 15 min à température ambiante (à droite). Le traitement avec la Lactacystine est utilisé comme contrôle positif. La condition « blanc de substrat » correspond aux réactifs sans le protéasome 20S. La fluorescence du substrat AMC est quantifiée dans un lecteur de fluorescence (380/460 nm). Les résultats présentés sont les moyennes des triplicats de trois expérimentations indépendantes +/- SEM.
Les résultats sont normalisés par rapport à la condition « non traité » (égal à 1) et « blanc de
substrat », correspondant aux réactifs sans le protéasome 20S (égal à 0).
Comme nous pouvons le voir, nous avons vérifié que le trivalent-DMJ seul ne présente aucun
effet sur la fluorescence du substrat AMC. Aucune différence significative n’a été observée
entre les conditions « blanc de substrat » et « trivalent-DMJ sans protéasome ».
Nous avons constaté une diminution d’environ 65% de la fluorescence d’AMC lorsque le
protéasome 20S est traité pendant 4h à 37°C avec notre composé. Cette diminution de
fluorescence témoignerait d’une inhibition directe du protéasome par le trivalent-DMJ. Ainsi,
afin de vérifier que cette inhibition n’est pas le résultat d’une exposition trop prolongée à 37°C
du protéasome avec notre molécule, nous avons testé le trivalent-DMJ dans les mêmes
conditions de traitement que la Lactacystin, utilisée comme contrôle positif. Comme nous
pouvons le voir, le traitement du protéasome 20S pendant 15 min à température ambiante
diminue aussi le taux de fluorescence d’AMC, confirmant que le trivalent-DMJ inhibe le
protéasome 20S de manière directe.
4 heures à 37°C 15 minutes à température ambiante

Résultats
138
En conclusion, ces résultats mettent en évidence que le trivalent-DMJ inhibe de manière
directe le protéasome 20S. Cependant, l’hypothèse d’une inhibition indirecte du protéasome
intracellulaire en parallèle de l’inhibition directe n’est pas exclue.
Nous nous sommes demandés suite à ces résultats, si l’augmentation de l’effet
correcteur du trivalent-DMJ sur F508del-CFTR était corrélée à l’augmentation de son effet
inhibiteur sur le protéasome. Pour cela, nous avons comparé l’effet inhibiteur du trivalent-DMJ
et de son monovalent sur le protéasome 20S purifié. La fluorescence de son substrat AMC a été
mesurée à l’aide du lecteur Mithras en utilisant un couple de filtre 380/460 nm (Figure 44).
NT
Mon
ovale
nt-DM
J
Trivale
nt-DM
J
Lactac
ystin
e
Blanc d
e sub
strat
Mon
ovale
nt-DM
J san
s 20S
Trivale
nt-DM
J san
s 20S
-0.2
0.0
0.2
0.4
0.6
0.8
1.0
RF
U d
'AM
C (
DO
46
0 n
m)
NT
Mon
ovale
nt-DM
J
Trivale
nt-DM
J
Lactac
ystin
e
Blanc d
e sub
strat
0.0
0.2
0.4
0.6
0.8
1.0
1.2R
FU
d'A
MC
(D
O460 n
m)
Figure 44 : Effet du DMJ et ses dérivés multivalent sur l’activité du protéasome 20S purifié.
L’activité enzymatique du protéasome 20S est évaluée à l’aide du kit « 20S Proteasome Activity Assay kit ». Le protéasome 20S purifié est traité avec le monovalent- ou le trivalent-DMJ pendant 4h à 37°C (à gauche) ou 15 min à température ambiante (à droite). Le traitement avec la Lactacystin est utilisé comme contrôle positif. La condition « blanc de substrat » correspond aux réactifs sans le protéasome 20S. La fluorescence du substrat AMC est quantifiée à l’aide d’un couple de filtre 380/460 nm dans un lecteur de fluorescence, ici Mithras. Les résultats présentés sont les moyennes des triplicats +/- SEM.
Les résultats sont normalisés par rapport aux conditions « non traité » (égal à 1) et « blanc de
substrat », correspondant aux réactifs sans le protéasome 20S (égal à 0).
4 heures à 37°C 15 minutes à température ambiante

Résultats
139
Nous avons vérifié que le monovalent- ou le trivalent-DMJ seul ne présente pas d’effet sur la
fluorescence du substrat AMC. Nous n’avons constaté aucune différence significative entre les
conditions « blanc de substrat » et « monovalent-DMJ sans protéasome » ou « trivalent-DMJ
sans protéasome ».
Nous avons détecté une diminution d’environ 40% de la fluorescence d’AMC lorsque le
protéasome 20S est traité pendant 4 heures à 37°C avec le monovalent- ou le trivalent-DMJ.
Aucune différence significative n’a été observée entre les effets inhibiteurs de ces deux
composés. Nous avons ainsi, testé ces 2 composés dans les mêmes conditions de traitement
que la Lactacystin, utilisée comme contrôle positif. Comme nous pouvons le voir, le traitement
du protéasome 20S pendant 15 min à température ambiante avec le monovalent-DMJ n’a révélé
aucun effet apparent avec la condition non traitée. Au contraire le traitement avec le trivalent-
DMJ diminue l’activité enzymatique du protéasome d’environ 30%. Ces résultats suggèrent
que le trivalent-DMJ pourrait inhiber l’activité enzymatique du protéasome 20S de manière
plus rapide que son monovalent.
En conclusion, ces résultats apporteraient une hypothèse expliquant pourquoi l’effet
correcteur sur F508del-CFTR du trivalent-DMJ est plus efficace que celui de son monovalent.
Il semblerait que la multivalence augmente l’efficacité de l’effet inhibiteur sur le protéasome
20S.
c) Effet du trivalent-DMJ sur la réponse au stress du RE : UPR
Il nous est apparu important de vérifier si le trivalent-DMJ déclenchait la réponse au
stress du RE, l’UPR. Nous avons alors mesuré son effet sur des protéines connues pour être
surexprimées lors d’une réponse UPR, à savoir : BiP/GRP78 et eIF2α phosphorylée (Harding
et al., 1999; Li et al., 2008; Wang et al., 2009; Nishitoh, 2012; Wang and Kaufman, 2014).
i. Effet de la tunicamycine sur l’expression protéique de BiP/GRP78 et eIF2α
Nous avons vérifié dans un premier temps, que le niveau d’expression des protéines
BiP/GP78 et eIF2α est augmenté dans notre modèle cellulaire Hela F508del-CFTR, en réponse
à un stress du RE. La tunicamycine, un antibiotique inhibiteur de la N-glycosylation des
protéines, est utilisée comme contrôle pour l’induction de stress du RE (Samali et al., 2010;
Bartoszewski et al., 2011; Chawla et al., 2011; Wang et al., 2015). Les cellules ont alors été
traitées ou non pendant 6 heures avec des concentrations croissantes de tunicamycine et
l’expression protéique de BiP/GRP78 et d’eIF2α a été testée par western blot (Figure 45).

Résultats
140
A
B
NT
1 µg/m
l
2,5 µ
g/ml
5 µg/m
l
7 µg/m
l
10 µ
g/ml
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
Tunicamycine
Rati
o P
ho
sph
o-e
IF2
/Act
ine
Figure 45 : Effet de la tunicamycine sur l’expression protéique de BiP/GRP78 et eif2α. (A) Western blot représentatif de l’expression protéique de BiP/GRP78 (à gauche) et eif2α (à droite) dans les cellules Hela F508del-CFTR traitées ou non avec de la tunicamycine à 1, 2.5, 5, 7 ou 10 µg/ml (6h, 37°C). L’actine est utilisée comme un contrôle de charge. (B) Histogramme reportant la quantification de la protéine BiP/GRP78 et eif2α dans les différentes conditions testées. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition NT (non traitée) (N=2).
La protéine BiP/GRP78 a été détectée à environ 78 kDa. Comme nous pouvons le voir, aucune
différence significative du taux d’expression de la chaperonne BiP/GRP78 n’est observée entre
les cellules non traitées et celles traitées avec de la tunicamycine à 1 µg/ml pendant 6h. Au
contraire, avec des traitements à 2.5 µg/ml, 5 µg/ml, 7 µg/ml et 10 µg/ml, une différence
significative est apparue.
De même, une augmentation du taux d’expression d’eIF2α phosphorylée a été observée à partir
de 1 µg/ml, ce qui est en accord avec la littérature (Brambilla Pisoni et al., 2016).
Ces résultats montrent que les taux d’expression des protéines BiP/GRP78 et d’eIF2α
phosphorylée augmentent bien en réponse à un stress du RE dans les cellules Hela F508del-
CFTR, et que nous pouvons, en conséquence, considérer ces protéines comme des indicateurs
de l’état de nos cellules vis-à-vis du stress du RE.
NT
1 µg/m
l
2,5 µ
g/ml
5 µg/m
l
7 µg/m
l
10 µ
g/ml
0.00.20.4
0.60.81.0
1.21.4
1.6
Tunicamycine
Rati
o B
iP/A
ctin
e
BiP/GRP78 78 kDa
Actine
Tunicamycine
Actine
Tunicamycine
eIF2α-T 36 kDa
eIFα-P 38 kDa

Résultats
141
ii. Effet du trivalent-DMJ sur l’expression protéique de BiP/GRP78 et d’eIF2α
Dans un deuxième temps, nous avons regardé l’effet du trivalent-DMJ sur l’expression
de BiP/GRP78 et eIF2α phosphorylée afin d’examiner son effet sur le stress du RE. Les résultats
obtenus sont présentés dans la figure 46.
0.0
0.2
0.4
0.6
0.8
1.0 ns
Rati
o B
iP/A
cti
ne
0.0
0.2
0.4
0.6
0.8
1.0
Ra
tio
Ph
osp
ho
-eif
2 /A
ctin
e
Figure 46 : Effet du trivalent-DMJ sur l’expression de la protéine BiP/GRP78 et eIF2α. (A) Western blot représentatif de l’expression protéique de BiP/GRP78 (à gauche) et eIF2α (à droite) dans les cellules Hela F508del-CFTR traitées ou non avec le trivalent-DMJ (100 µM, 4h, 37°C). L’actine est utilisée comme un contrôle de charge. (B) Histogramme reportant la quantification de la protéine BiP/GRP78 et eIF2α dans les différentes conditions testées. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition NT (non traitée) (N=5 pour BiP/GRP78 et N=2 pour eIF2α). ns : non significatif.
Aucune différence significative du taux d’expression de la protéine BiP/GRP78 ou d’eIF2α
phosphorylée n’a été détectée entre les cellules non traitées et celles traitées avec le trivalent-
DMJ. Ces résultats montrent que le trivalent-DMJ n’a aucun effet sur l’expression protéique de
BiP/GRP78 ni sur la phosphorylation d’eIF2α.
En conclusion, le trivalent-DMJ ne semble pas induire un stress du RE dans notre
modèle cellulaire.
BiP/GRP78 78 kDa
Actine Actine
eIF2α-P 38 kDa
A
B

Résultats
142
d) Combinaison du trivalent-DMJ avec le potentiateur VX-770
Comme expliqué précédemment, les résultats des essais cliniques du correcteur ciblant
de manière directe la protéine CFTR, VX-809 (Lumacaftor) n’ont pas révélé d’amélioration des
signes cliniques chez les patients homozygotes pour la mutation F508del. En revanche, son
association avec le VX-770 (Ivacaftor) en Orkambi® a apporté une amélioration selon les
études de phase III. Cette combinaison a même obtenu une autorisation de mise sur le marché
pour des patients âgés de 12 ans et plus et homozygotes pour la mutation F508del par l’Agence
Européenne du Médicament (AEM) (d’après https://www.has-sante.fr et http://www.
clinicaltrials.gov).
Comme vu précédemment, le VX-770 est un potentiateur qui augmente la probabilité
d’ouverture du canal CFTR et en conséquence le transport des ions chlorures à la surface
cellulaire. Nous avons évalué l’efficacité de ce composé sur l’activité canal chlorure de
F508del-CFTR restauré à la membrane cellulaire par le trivalent-DMJ. Les résultats obtenus
par la technique des efflux d’iodure sont présentés dans la figure 47.
Figure 47 : Efficacité du potentiateur VX-770 sur F508del-CFTR corrigé par le trivalent-DMJ. Evaluation de l’activité de F508del-CFTR par la technique des efflux d’iodure sur les cellules Hela F508del-CFTR traitées pendant 4h avec 100 µM de trivalent-DMJ. F508del-CFTR est activé en aigu par VX-770 (10 µM), Forskoline (Fsk) (10 µM), Fsk+VX-770, Fsk + Génistéine (Gst) (10 µM) ou Fsk+VX-770+Gst (n=4). ns : non significatif, ***P<0.001.
Comme attendu, l’ajout de la Forskoline seule à une concentration de 10 µM sur les cellules
traitées avec du trivalent-DMJ ne montre pas d’effet activateur sur le canal F508del-CFTR ; au
contraire, son addition en cocktail avec le potentiateur Génistéine induit une activité canal
chlorure. De même, l’addition du potentiateur VX-770 seul ne présente aucun effet sur l’activité
de F508del-CFTR étant donné que cette dernière n’est pas activée.

Résultats
143
L’addition du potentiateur VX-770 en cocktail avec la Fsk induit une activité canal chlorure de
F508del-CFTR similaire à celle induite avec le cocktail Fsk+Gst, suggérant que le VX-770
potentialise l’activité de F508del-CFTR aussi bien que la Gst. Leur association avec la Fsk ne
montre pas de différence significative par rapport aux autres combinaisons.
En conclusion, une combinaison du trivalent-DMJ avec le potentiateur VX-770 pourrait
être envisagée afin d’obtenir un effet thérapeutique plus pertinent in vivo.
II. Implication de l’α1,2-mannosidase du RE dans la rétention de
F508del-CFTR
A. Contexte
Nous avons démontré, dans la première partie de ce travail de thèse, qu’inhiber
l’interaction de F508del-CFTR avec les protéines EDEM permet de rétablir son expression
membranaire. Ces protéines jouent un rôle majeur dans l’envoi des glycoprotéines vers la voie
de dégradation ERAD. Selon la littérature, l’α1,2-mannosidase I du RE (ERManI) serait le
« minuteur » qui déclenche la première étape de cette voie de dégradation en clivant le premier
résidu mannose de la branche B de l’oligosaccharide (Liu et al., 1999; Araki and Nagata, 2011;
Caramelo and Parodi, 2015; Zhou et al., 2015). Il semblerait que l’inhibition de son activité par
siRNA ou par la Kifunensine, un inhibiteur spécifique de l’α1,2-mannosidase, empêche la
dégradation intracellulaire des substrats luminaux d’ERAD, AAT (α1-antitrypsin) et son
mutant NHK (null Hong Kong) (Hosokawa et al., 2003; Wu et al., 2003; Avezov et al., 2008;
Pan et al., 2013). De manière plus intéressante, il a été démontré que le traitement avec de la
Kifunensine restaure in vitro et in vivo l’expression membranaire de l’α-sarcoglycane R77C,
une protéine mutée qui reste séquestrée dans le RE par l’ERQC (Bartoli et al., 2008; Soheili et
al., 2012). D’autre part, le DMJ qui a servi de base à la synthèse du trivalent-DMJ est
commercialisé en tant qu’inhibiteur des mannosidases du RE et de l’appareil de Golgi. Il nous
est alors apparu important de déterminer l’implication d’ERManI dans la rétention réticulaire
de F508del-CFTR. Ceci permettrait de corréler l’effet correcteur du trivalent-DMJ avec un
potentiel effet inhibiteur de mannosidase et aussi une meilleure compréhension du mécanisme
impliqué dans l’adressage défectueux de F508del-CFTR.

Résultats
144
B. Résultats
1) Effet des inhibiteurs des mannosidases sur la fonction de F508del-CFTR
Différents inhibiteurs de mannosidases du RE ont été testés sur l’activité canal chlorure
de F508del-CFTR afin de déterminer l’implication d’ERManI dans sa rétention réticulaire. Les
résultats obtenus par la technique des efflux d’iodure sont présentés dans la figure 48.
Figure 48 : Effet des inhibiteurs de mannosidase sur l’activité de F508del-CFTR. Evaluation de l’activité canal chlorure de F508del-CFTR par la technique des efflux d’iodure sur les cellules Hela F508del-CFTR traitées ou non pendant 4h avec 100 µM de DMJ (Deoxymannojirimicine), NB-DMJ (N-butyl-deoxymannojirimycin), Swainsonine ou Kifunensine. L’activité de CFTR est stimulée par le cocktail Forskoline 10 µM + Génistéine 30 µM. Les résultats sont présentés en moyenne +/- SEM (n=4). ns : non significatif, ***P<0.001.
Comme nous pouvons le voir, les 4 inhibiteurs de l’activité mannosidase induisent une
restauration de la fonction du flux anionique F508del-CFTR. Une différence significative de
cette activité a été observée entre les cellules non traitées et celles traitées avec les différents
composés. Ces résultats suggèrent qu’inhiber l’activité mannosidase rétablit potentiellement la
fonction de F508del-CFTR.
2) Effet du siRNA d’ERManI sur F508del-CFTR
Parce que les inhibiteurs de mannosidase ne sont pas spécifiques de l’enzyme ERManI,
et afin de s’affranchir de leur effet pharmacologique qui pourrait agir sur d’autres cibles dans
la cellule, nous avons opté pour la stratégie de siRNA. Celle-ci nous permettra de réduire de
manière spécifique l’expression protéique d’ERManI, et par conséquent son activité dans les
cellules. Par la même occasion, nous avons étudié l’effet de l’extinction de l’expression
protéique de Golgi-Mannosidases II (Golgi-ManII) sur F508del-CFTR.
untre
ated
DMJ
NB-DM
J
swain
sonin
e
kifun
ensin
0.00
0.05
0.10
0.15
0.20***
ns
*
k pea
k-k b
asal
(m
in-1
)

Résultats
145
a) Efficacité des siRNA ERManI et Golgi-ManII
Afin de vérifier l’extinction de la protéine ERManI et Golgi-ManII après transfection
des cellules Hela F508del-CFTR avec leur siRNA spécifique, nous avons réalisé des
expériences de western blot. La transfection par du siRNA contrôle a servi de contrôle négatif.
Les résultats obtenus sont présentés dans la figure 49.
NT
Contrô
le
ERMan
I0.00
0.25
0.50
0.75
1.00
1.25
1.50 ns **
siRNAR
ati
o E
RM
an
I/A
cti
ne
NT
Contrô
le
Golgi
-Man
0.0
0.2
0.4
0.6
0.8
1.0
siRNA
ns *
Ra
tio
Go
lgiM
an
II/A
cti
ne
Figure 49 : Diminution de l’expression protéique d’ERManI ou de Golgi-Man par siRNA.
(A) Western blot représentatif de l’extinction de la protéine ERManI (en haut) ou Golgi-Man (en bas) par siRNA dans les cellules Hela F508del-CFTR après 48 heures de transfection. Le siRNA contrôle est utilisé comme un contrôle négatif de la transfection. L’actine est utilisée comme un contrôle de charge. (B) Histogramme reportant la quantification de la protéine ERManI (en haut) ou Golgi-Man (en bas) dans les différentes conditions. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition NT (non transfectées) (N=5). ns : non significatif, *P<0.05, **P<0.01.
Les protéines ERManI et Golgi-ManII sont détectées respectivement à environ 80 kDa et 130
kDa. Comme nous pouvons le voir, une extinction d’environ 90% de l’expression protéique
d’ERManI a été constatée dans les cellules transfectées par des siRNAs ERManI, comparée aux
siRNA
Actine
ERManI
siRNA
Actine
Golgi-ManII
A B

Résultats
146
cellules transfectées par des siRNAs contrôle. Ces résultats démontrent que le siRNA utilisé
dans notre étude éteint de manière efficace et quasi-totale l’expression de la protéine ERManI.
De même, une extinction d’environ 40% du taux d’expression de la protéine Golgi-ManII a été
détectée dans les cellules transfectées avec du siRNA Golgi-ManII, suggérant que le siRNA
utilisé éteint de manière partielle l’expression de la protéine Golgi-ManII.
b) Effet de la transfection de siRNA ERManI ou Golgi-ManII sur F508del-CFTR
Dans un premier temps, nous avons exploré l’effet du siRNA ERManI et Golgi-ManII
sur la fonction de transport de F508del-CFTR par la technique des efflux d’iodure. Le cocktail
Forskoline 10 µM + Génistéine 30 µM a été utilisé pour stimuler l’activité du canal CFTR au
niveau de la membrane cellulaire. Les résultats obtenus sont présentés dans la figure 50.
Figure 50 : Effet du siRNA ERManI et Golgi-ManII sur l’activité de F508del-CFTR. Evaluation de l’activité canal chlorure de F508del-CFTR par la technique des efflux d’iodure sur les cellules Hela F508del-CFTR transfectées (48 heures) ou non avec des siRNA ERManI ou siRNA contrôle. L’activité de CFTR est stimulée par le cocktail Forskoline 10 µM+ Génistéine 30 µM. Les résultats sont présentés en moyenne +/- SEM (n=4). ns : non significatif, ***P<0.001.
Comme nous pouvons le voir, aucune activité du transport de F508del-CFTR n’a été détectée
dans les cellules non transfectées ou les cellules transfectées avec du siRNA contrôle, ce qui est
en accord avec la rétention de cette protéine dans le RE. Au contraire, suite à la transfection par
le siRNA ERManI, une augmentation de l’activité de F508del-CFTR est observée, témoignant
de la restauration de cette protéine à la membrane cellulaire. Ces résultats suggèrent ainsi, que
la diminution de l’expression protéique d’ERManI par siRNA restaure l’activité de F508del-
CFTR.
untra
nsfec
ted
siRNA C
trl
siRNA E
R man
.
siRNA G
olgi m
an.
0.00
0.05
0.10
0.15
basalFsk+Gst
nsns
ns
***
k pea
k-k b
asal
(m
in-1
)
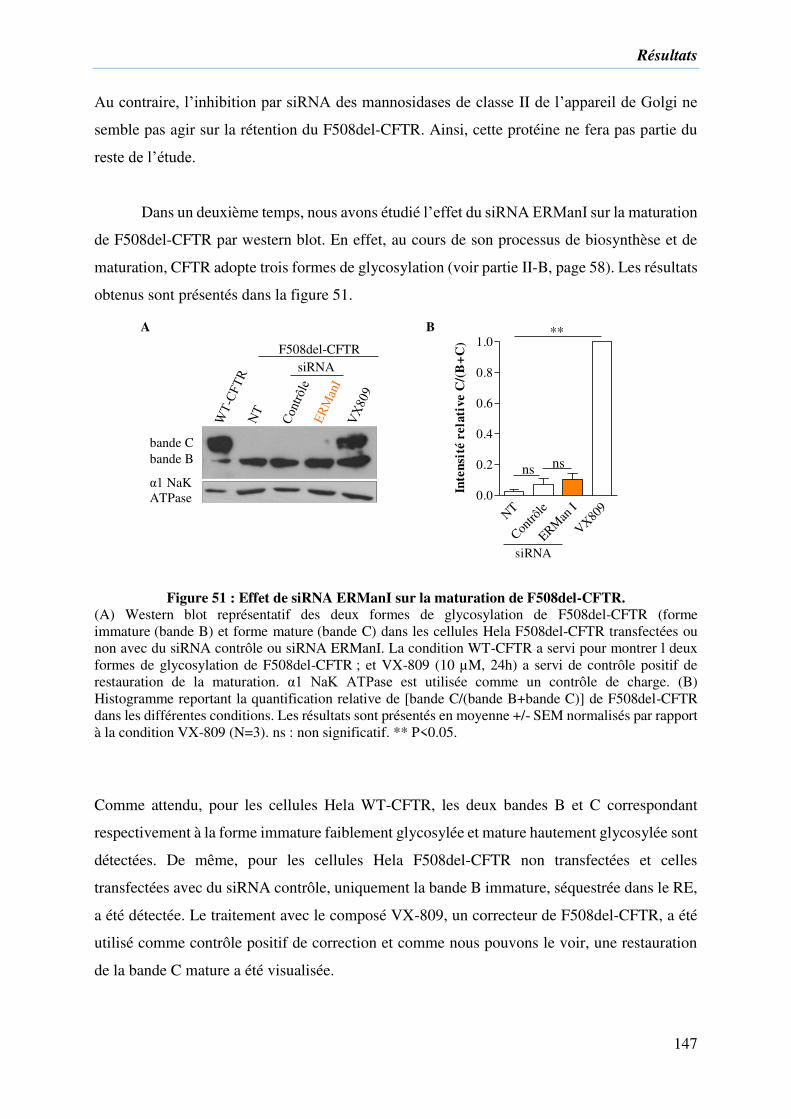
Résultats
147
Au contraire, l’inhibition par siRNA des mannosidases de classe II de l’appareil de Golgi ne
semble pas agir sur la rétention du F508del-CFTR. Ainsi, cette protéine ne fera pas partie du
reste de l’étude.
Dans un deuxième temps, nous avons étudié l’effet du siRNA ERManI sur la maturation
de F508del-CFTR par western blot. En effet, au cours de son processus de biosynthèse et de
maturation, CFTR adopte trois formes de glycosylation (voir partie II-B, page 58). Les résultats
obtenus sont présentés dans la figure 51.
NT
Contrô
le
ERM
an I
VX809
0.0
0.2
0.4
0.6
0.8
1.0**
nsns
siRNA
Inte
nsi
té r
ela
tive C
/(B
+C
)
Figure 51 : Effet de siRNA ERManI sur la maturation de F508del-CFTR. (A) Western blot représentatif des deux formes de glycosylation de F508del-CFTR (forme immature (bande B) et forme mature (bande C) dans les cellules Hela F508del-CFTR transfectées ou non avec du siRNA contrôle ou siRNA ERManI. La condition WT-CFTR a servi pour montrer l deux formes de glycosylation de F508del-CFTR ; et VX-809 (10 µM, 24h) a servi de contrôle positif de restauration de la maturation. α1 NaK ATPase est utilisée comme un contrôle de charge. (B) Histogramme reportant la quantification relative de [bande C/(bande B+bande C)] de F508del-CFTR dans les différentes conditions. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition VX-809 (N=3). ns : non significatif. ** P<0.05.
Comme attendu, pour les cellules Hela WT-CFTR, les deux bandes B et C correspondant
respectivement à la forme immature faiblement glycosylée et mature hautement glycosylée sont
détectées. De même, pour les cellules Hela F508del-CFTR non transfectées et celles
transfectées avec du siRNA contrôle, uniquement la bande B immature, séquestrée dans le RE,
a été détectée. Le traitement avec le composé VX-809, un correcteur de F508del-CFTR, a été
utilisé comme contrôle positif de correction et comme nous pouvons le voir, une restauration
de la bande C mature a été visualisée.
bande C bande B
α1 NaK ATPase
F508del-CFTR siRNA
A B

Résultats
148
Concernant les cellules transfectées avec du siRNA ERManI, seule la bande immature B a été
visualisée. Aucune restauration de la bande mature C n’a été observée. Cependant, d’après les
résultats de fonctionnalité précédemment obtenus, une activité CFTR-dépendante a été
observée dans ces cellules (Figure 50), suggérant une restauration de F508del-CFTR à la
surface cellulaire sous sa forme immature mais fonctionnelle.
En conclusion, la diminution de l’expression de la protéine ERManI par siRNA restaure
un F508del-CFTR immature mais fonctionnel à la surface cellulaire. Ces résultats suggèrent
l’implication de l’enzyme ERManI dans la rétention réticulaire de F508del-CFTR.
3) Comprendre l’effet correcteur sur F508del-CFTR de l’extinction d’ERManI
Afin de comprendre comment et pourquoi la diminution de l’expression protéique
d’ERManI rétablit l’expression membranaire de F508del-CFTR, nous avons étudié l’effet du
siRNA ERManI sur les principaux acteurs connus pour être impliqués dans le contrôle de
qualité de F508del-CFTR.
a) Effet du siRNA ERManI sur l’interaction de F508del-CFTR avec les protéines de
la famille EDEM
Comme nous l’avons démontré dans notre première étude, les protéines de la famille
EDEM, principaux acteurs de l’envoi des protéines mal repliées vers la voie de dégradation,
sont impliquées dans la rétention de F508del-CFTR dans le RE. Nous avons alors étudié l’effet
du siRNA ERManI sur leur niveau d’expression protéique. Les résultats obtenus par western
blot sont présentés dans la figure 52.

Résultats
149
NT
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4 ns
siRNA
Rati
o E
DE
M1
/Acti
ne
NT
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4ns
siRNA
Rati
o E
DE
M2
/Acti
ne
NT
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0ns
siRNA
Rati
o E
DE
M3
/Acti
ne
Figure 52 : Effet du siRNA ERManI sur l’expression protéique des EDEMs. (A) Western blot représentatif du niveaux d’expression protéique d’EDEM1, EDEM2 ou EDEM3 dans les cellules Hela F508del-CFTR transfectées (48 heures) ou non avec du siRNA ERManI ou contrôle. L’actine est utilisée comme un contrôle de charge. (B) Histogramme reportant la quantification du niveau d’expression de la protéine EDEM1, EDEM2 ou EDEM3 dans les différentes conditions. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition NT (non traitée) (N=3). ns : non significatif.
Comme nous pouvons le voir, la protéine EDEM1 est détectée sous deux formes : une forme
soluble à environ 69 kDa, et une forme membranaire à environ 74 kDa. Les protéines EDEM2
et EDEM3 sont quant à elles détectées respectivement à environ 70 kDa et 110 kDa.
Aucune différence significative du niveau d’expression de ces trois protéines n’a été observé
entre les cellules transfectées avec le siRNA ERManI et celles transfectées avec le siRNA
contrôle ou les non transfectées. Le siRNA ERManI ne présente donc aucun effet sur
l’expression protéique totale d’EDEM1, EDEM2 et EDEM3.
EDEM3 EDEM2
siRNA siRNA
EDEM1
siRNA
Actine
A
B

Résultats
150
Dans un deuxième temps, nous avons regardé par la technique de Proximity Ligation
Assay, si le siRNA ERManI perturbait l’interaction de F508del-CFTR avec les 3 protéines
EDEMs (Figure 53).
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0
1.2
siRNA
ns
Inte
racti
on
F5
08
del-
CF
TR
/ E
DE
M1
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0
1.2 ns
siRNA
Inte
racti
on
F5
08
del-
CF
TR
//E
DE
M2
Contrô
le
ERMan
I0.00.20.40.60.81.01.21.41.6
Inte
racti
on
F5
08
del-
CF
TR
//E
DE
M3 ns
siRNA
Figure 53 : Effet du siRNA ERManI sur l’interaction de F508del-CFTR avec les EDEMs. (A) Images représentatives des interactions (points rouges) de F508del-CFTR avec EDEM1 (en haut), EDEM2 (au milieu) ou EDEM3 (en bas) obtenues par la technique de Proximity Ligation Assay sur des cellules Hela F508del-CFTR transfectées (48 heures) ou non avec du siRNA ERManI ou contrôle. Echelle : 20 µm. Bleu : noyau cellulaire (B) Histogramme reportant le nombre de points rouge par noyau, représentatif du nombre d’interaction par cellules. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition siRNA contrôle (N=3). ns : non significatif.
siRNA Contrôle siRNA ERManI
Interaction F508del-CFTR/EDEM1
siRNA Contrôle siRNA ERManI
Interaction F508del-CFTR/EDEM2
siRNA Contrôle siRNA ERManI
Interaction F508del-CFTR/EDEM3
A B

Résultats
151
Les résultats obtenus ne montrent aucune différence significative du taux d’interaction de
F508del-CFTR avec EDEM1, EDEM2 ou EDEM3 entre les cellules transfectées avec du
siRNA ERManI et les cellules transfectées avec du siRNA contrôle. Le siRNA ERManI ne
diminue donc pas l’interaction de F508del-CFTR avec cette famille de protéine.
En conclusion, le siRNA ERManI semble restaurer F508del-CFTR à la membrane
cellulaire via un mécanisme d’action indépendant de l’interaction de F508del-CFTR avec les
protéines EDEMs.
b) Effet du siRNA ERManI sur les ubiquitines ligases CHIP et RNF5/RMA1
Toujours dans le but d’identifier le mécanisme d’action du siRNA ERManI, nous avons
exploré son effet sur l’expression protéique de CHIP et RNF5/RMA1 (voir Figure 40). Les
résultats obtenus par western blot sont présentés dans la figure 54.
NT
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0ns
siRNA
Rati
o C
HIP
/Acti
ne
NT
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0ns
siRNA
Rati
o R
NF
5/A
cti
ne
Figure 54 : Effet du siRNA ERManI sur l’expression protéique de CHIP et RNF5/RMA1. (A) Western blot représentatif du niveau d’expression protéique de CHIP (à gauche) et RNF5/RMA1 (à droite) dans les cellules Hela F508del-CFTR transfectées (48 heures) ou non avec du siRNA ERManI ou contrôle. L’actine est utilisée comme un contrôle de charge. (B) Histogramme reportant la quantification du niveau d’expression de la protéine CHIP ou RNF5/RMA1 dans les différentes conditions. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition NT (non traitée) (N=3). ns : non significatif.
siRNA
Actine
CHIP
siRNA
Actine
RNF5/ RMA1
A
B

Résultats
152
L’immunoblot révèle la protéines CHIP à 35 kDa environ. Une seconde bande plus légère,
correspondant à sa forme glycosylée a également été détectée. La protéine RNF5/RMA1, quant
à elle, a été détectée à environ 18 kDa.
Les résultats obtenus démontrent que le siRNA ERManI ne présente aucun effet sur
l’expression protéique des ubiquitines ligases CHIP et RNF5/RMA1.
Ensuite, nous avons étudié par la technique de Proximity Ligation Assay, si le siRNA
ERManI perturbait l’interaction de F508del-CFTR avec ces protéines (Figure 55).
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0
siRNA
Inte
racti
on
F5
08
del-
CF
TR
/CH
IP
Contrô
le
ERMan
I0.0
0.2
0.4
0.6
0.8
1.0
1.2
Inte
racti
on
F5
08
del-
CF
TR
/RN
F5
siRNA
Figure 55 : Effet du siRNA ERManI sur l’interaction de F508del-CFTR avec CHIP ou RNF5/RMA1.
(A) Images représentatives des interactions (points rouges) de F508del-CFTR avec CHIP (en haut) ou RNF5/RMA1 (en bas) obtenues par la technique de Proximity Ligation Assay sur des cellules Hela F508del-CFTR transfectées (48 heures) ou non avec du siRNA ERManI ou contrôle. Echelle : 20 µm. Bleu : noyau cellulaire (B) Histogramme reportant le nombre de points rouge par noyau, représentatif du nombre d’interaction par cellules. Les résultats sont présentés en moyenne +/- SEM normalisés par rapport à la condition siRNA contrôle (N=2).
siRNA Contrôle siRNA ERManI
Interaction F508del-CFTR/CHIP
siRNA Contrôle siRNA ERManI
Interaction F508del-CFTR/RNF5
A B

Résultats
153
Aucun effet du niveau de l’interaction de F508del-CFTR avec CHIP ou avec RNF5/RMA1 n’a
été observé entre les cellules non transfectées et les cellules transfectées avec du siRNA
ERManI.
Bien que préliminaires, ces résultats suggèrent que le siRNA ERManI rétablit
l’expression membranaire de F508del-CFTR via un mécanisme d’action indépendant des
ubiquitines ligases CHIP et RNF5/RMA1.
c) Effet du siRNA ERManI sur le protéasome 20S
Nous avons exploré l’effet de la diminution du taux d’expression protéique d’ERManI
sur l’activité enzymatique du protéasome intracellulaire sur des cellules Hela F508del-CFTR.
La fluorescence du substrat du protéasome 20S, AMC a été mesurée à l’aide du lecteur Mithras
en utilisant un couple de filtre 380/460 nm (Figure 56).
NT
Lactac
ystin
Blanc d
e sub
strat
0.0
0.2
0.4
0.6
0.8
1.0
RF
U d
'AM
C (
DO
46
0 n
m)
C
ontrô
le
E
RMan
I0.0
0.2
0.4
0.6
0.8
1.0
siRNA
RF
U d
'AM
C (
DO
46
0 n
m)
Figure 56 : Effet du siRNA ERManI sur l’activité du protéasome 20S intracellulaire. (A) L’activité enzymatique du protéasome 20S est évaluée à l’aide du kit « 20S Proteasome Activity Assay kit » sur des cellules Hela F508del-CFTR transfectées ou non pendant 48h avec du siRNA ERmanI. (B) Le traitement avec la Lactacystin est utilisé comme contrôle négatif. La condition « blanc de substrat » correspond aux réactif dans un puits vide (sans cellules). La fluorescence du substrat AMC libre est quantifiée à l’aide d’un couple de filtre 380/460 nm dans un lecteur de fluorescence, ici Mithras. Les résultats présentés sont les moyennes des triplicats de deux expérimentations indépendantes +/- SEM.
Les résultats obtenus sont normalisés par rapport aux conditions « non traité » (égale à
1) et « blanc de substrat » (égale à 0).
A B

Résultats
154
La Lactacystine a été utilisée comme un contrôle positif, inhibant l’activité du protéasome, et
comme attendu, une diminution d’environ 98% de la fluorescence du substrat AMC a été
constatée dans les cellules traitées avec ce composé, comparé aux cellules non traitées.
Concernant les cellules transfectées avec du siRNA ERManI, l’activité enzymatique de leur
protéasome est identique à celle des cellules transfectées avec du siRNA contrôle, suggérant
que le siRNA ERManI ne présente aucun effet sur le protéasome cellulaire.
L’ensemble de ces résultats suggèrent une implication d’ERManI dans la rétention
réticulaire du F508del-CFTR. Cependant, au regard de l’absence d’effet du siRNA ERManI sur
l’interaction de F508del-CFTR avec les protéines EDEMs ou sur l’activité du protéasome, nous
pouvons émettre l’hypothèse que l’inhibition d’ERManI du RE par le trivalent-DMJ n’explique
pas l’effet correcteur de ce composé. Cependant, au regard de l’effet correcteur du siRNA ayant
été observé sur notre modèle, il sera intéressant d’explorer cette voie et de déterminer par quel
mécanisme d’action le siRNA restaure cette protéine à la membrane cellulaire.

Discussion, conclusion et perspectives
155
Discussion, conclusion et perspectives Les études réalisées durant mes trois années de thèse ont permis dans un premier temps,
la conception et la synthèse par nos collaborateurs chimistes de l’équipe du Pr Philippe
Compain, d’une nouvelle classe d’iminosucres multivalents en série DMJ, un inhibiteur de
l’activité enzymatique des α1,2-mannosidases du RE. L’effet multivalent de ces dérivés
observé sur la restauration de l’expression membranaire de F508del-CFTR a permis d’ouvrir
une nouvelle piste thérapeutique pour la mucoviscidose. Nous avons dans un second temps,
apporté de nouveaux éléments concernant le processus de dégradation du F508del-CFTR. Nous
avons montré que la famille des protéines EDEMs ainsi que la mannosidase du RE sont
impliquées dans sa rétention réticulaire. La recherche d’une stratégie pharmacologique ciblant
ces protéines, nous a ensuite conduit à l’identification d’un nouveau correcteur pour F508del-
CFTR, le trivalent-DMJ, et par la même occasion, à la détermination de son mécanisme
d’action. Enfin, au cours d’une étude encore préliminaire, nous avons pu déterminer certains
paramètres impliqués dans l’augmentation d’efficacité des composés avec la multivalence. Pour
plus de facilité, la discussion de l’ensemble de ces études sera présentée en quatre volets. Les
trois premiers volets ayant fait l’objet d’un article scientifique (en cour de soumission), seront
rapidement discutés.
A. Synthèse d’une nouvelle famille d’iminosucres : dérivés multivalents
du Deoxymannojirimycin
Notre première étude, réalisée en collaboration avec l’équipe de chimistes du Pr.
Philippe COMPAIN a permis la synthèse d’une nouvelle classe d’iminosucres multivalents,
solubles dans l’eau et non toxiques. Il s’agit des dérivés du composé DMJ, un inhibiteur de
l’activité mannosidase des membres de la famille GH47 (Olivari and Molinari, 2007b;
Ninagawa et al., 2014). Ces dérivés ont été synthétisés selon la stratégie de « chimie click » qui
consiste à greffer au DMJ un ou plusieurs bras chimiques sur lesquels d’autres composés DMJ
peuvent être fixés.
Les tests de biologie que nous avons réalisés par la suite avec ces composés, nous ont
permis de mettre en évidence leur effet correcteur sur F508del-CFTR. Ceci est en adéquation
avec d’autres études démontrant que les inhibiteurs Kifunensine et DMJ retardent la
dégradation de la protéine NHK (Null Hong Kong), un mutant de la protéine α1-antitrypsin

Discussion, conclusion et perspectives
156
(α1-AT) qui est retenu et dégradé par l’ERQC (Liu et al., 1997, 1999). Ces inhibiteurs
permettent également à la fois la diminution de dégradation de l’α1-ATZ, un autre mutant de
l’α1-AT et l’augmentation de sa sécrétion (Marcus and Perlmutter, 2000).
De manière intéressante, le criblage de ces composés en efflux d’iodure a révélé un effet
croissant sur la restauration de l’activité transporteur de F508del-CFTR avec l’augmentation de
la valence du composé. Le trivalent- et le tétravalent-DMJ, qui correspondent respectivement à
3 molécules et 4 molécules de DMJ associées par un bras chimique, étaient environ 170 fois
plus efficace que le DMJ. Ces résultats apportent de nouveaux arguments quant à l’importance
du concept de multivalence pour la synthèse de correcteurs plus efficaces pour F508del-CFTR.
En effet, lors d’une collaboration antérieure les deux équipes ont démontré que le trivalent-
DNJ, l’inhibiteur des α1,2-glucosidases du RE, était environ 1000 fois plus efficace que son
monovalent (Compain et al., 2013).
En perspectives pour cette étude, et basé sur des études montrant que l’association de
deux ou plusieurs correcteurs agissant à des niveaux différents de la voie de biosynthèse de
F508del-CFTR entraine une augmentation de son activité canal chlorure, il serait intéressant
de combiner des composés permettant de corriger les 3 défauts de F508del-CFTR, à savoir son
adressage, sa fonctionnalité et sa stabilité au niveau de la membrane cellulaire.
Nous avons par la suite cherché à identifier les paramètres impliqués dans
l’amélioration d’efficacité du trivalent-DMJ.
Même si cette étude est en cours de réalisation, certains résultats, encore préléminaires,
obtenus sur les dérivés multivalents fluorescents du composé DNJ (non présentés dans ce
manuscrit), nous ont permis d’identifier quelques paramètres essentiels pour l’augmentation
d’efficacité de composés avec la multivalence. Nous pensons en effet, que l’augmentation
d’efficacité du trivalent pourrait être due à son absorption rapide par les cellules. Le trivalent-
DNJ semble atteindre la saturation dans les cellules plus rapidement que son monovalent.
Cependant, s’agit-il d’une entrée plus rapide dans la cellule ou de sa taille plus importante qui
limiterait la quantité de produit pouvant pénétrer la cellule ?
Nous avons également essayé avec la spectrométrie de masse d’analyser les composés
après leur absorption par les cellules. Notre objectif était de voir si le trivalent-DNJ se fragmente
en plusieurs monovalent-DNJ ou un mélange de monovalent- et divalent-DNJ. Cependant, à ce
jour, nous n’arrivons pas encore à détecter nos composés dans les lysats cellulaires. En effet,

Discussion, conclusion et perspectives
157
après plusieurs tentatives, nous avons remarqué que le tampon de lyse induit l’apparition de
plusieurs pics et masquerait ainsi ceux des composés. Nous avons par la suite opté pour une
lyse mécanique (sonication) dans du H2O et de la trypsine, mais toujours sans succès. Ainsi,
une mise au point de la technique serait nécessaire afin de pouvoir détecter les composés qui
nous intéressent et étudier leur devenir une fois dans la cellule.
Enfin, suite à l’observation de l’effet inhibiteur du trivalent-DMJ sur le protéasome
(Discutée dans la partie C : « Etude du trivalent-DMJ comme correcteur de F508del-CFTR »),
nous nous sommes posé la question à savoir si le trivalent-DMJ inhibait le protéasome de
manière plus importante que son monovalent ? Nous avons observé qu’après 15 minutes de
traitement, le monovalent-DMJ ne présente pas d’effet sur l’activité enzymatique du
protéasome 20S. Ceci ne signifie pas que ce composé ne présente pas d’effet inhibiteur sur le
protéasome, mais que 15 minutes de traitement ne permettent pas son inhibition. Il faut savoir
que nos expérimentations ont été réalisées sur le protéasome 20S purifié, et pour cela, nous
avons opté pour des conditions de traitement identique à celle de la Lactacystine. De manière
intéressante, pour ces mêmes conditions, une diminution d’environ 30% de l’activité du
protéasome a été observée après traitement avec le trivalent-DMJ, suggérant que
l’augmentation d’efficacité de composés multivalents pourrait être due à l’augmentation
d’efficacité sur l’inhibition du protéasome.
B. La famille des protéines EDEMs : de nouvelles cibles thérapeutiques
pour la mucoviscidose
Après avoir souligné l’importance des inhibiteurs de l’activité enzymatique de type
mannosidase dans la correction de l’adressage défectueux de F508del-CFTR, notre attention
s’est focalisée sur les protéines EDEMs et plus particulièrement leur potentielle implication
dans la rétention réticulaire de cette protéine. Nous avons montré la présence de F508del-CFTR
en complexe avec la protéine EDEM1, EDEM2 ou EDEM3 dans des cellules de mammifères,
suggérant d’une potentielle implication de cette famille de protéine dans sa rétention réticulaire.
A notre connaissance, seule l’équipe du Pr. Amaral avait détecté EDEM1 dans des complexes
protéiques à la fois de WT- et de F508del-CFTR (Farinha and Amaral, 2005). Ainsi, c’est la
première fois qu’une interaction de F508del-CFTR avec cette famille de protéine est décrite,
suggérant d’une potentielle implication de cette famille de protéine dans sa rétention réticulaire.

Discussion, conclusion et perspectives
158
Grâce à une stratégie de siRNA, nous avons exploré cette piste, et nous avons montré
que la diminution de l’expression protéique (knockdown) d’EDEM1 ou d’EDEM2 restaure
F508del-CFTR à la membrane cellulaire sous sa forme immature mais fonctionnelle. Ceci n’est
pas le cas du knockdown d’EDEM3. A préciser, l’hypothèse de la restauration de F508del-
CFTR immature mais fonctionnelle à la surface cellulaire est plausible puisqu’il a été décrit
précédemment que la perturbation de l’état de glycosylation de la protéine CFTR-WT endogène
n’impacte pas sa fonction canal chlorure (Chang et al., 2008; Morris et al., 1993). Nos résultats
sont en accord avec d’autres travaux montrant que le knockdown d’EDEM1 inhibe la
dégradation d’un mutant de la protéine hERG (human ether-a-go-go-related gene), Y611H
(Gong et al., 2005). Comme la protéine F508del-CFTR, cette forme mutée du canal hERG
présente un défaut d’adressage, car elle reste séquestrée dans le RE sous sa forme immature
avant d’être dégradée (Zhou et al., 1998). Même si le rôle exact de chacune des trois variantes
d’EDEM n’est pas encore bien établi, il semblerait que le knockdown d’EDEM1 ou d’EDEM2
empêche le clivage extensif des résidus mannoses de F508del-CFTR et par conséquent son
envoi vers le complexe de rétrotranslocation, favorisant sa fuite vers la membrane cellulaire.
Cette hypothèse est soutenue par l’observation que le knockdown d’EDEM1 par siRNA affecte
la voie de dégradation ERAD (Molinari et al., 2003) et réduit la rétro-translocation de la chaîne
A de ricine vers le cytosol (Slominska-Wojewodzka et al., 2006).
Quatre hypothèses ont été émises concernant l’absence d’effet correcteur sur F508del-CFTR
dans les cellules transfectées avec le siRNA d’EDEM3 : (i) EDEM3 n’est pas impliqué dans la
rétention réticulaire de F508del-CFTR, (ii) EDEM3 est impliqué dans cette rétention mais à des
étapes plus tardives, (iii) l’absence d’EDEM3 pourrait être compensée par d’autres protéines,
ou bien encore, (iv) le taux d’extinction d’EDEM3 par siRNA n’était pas suffisant pour rétablir
l’expression membranaire de F508del-CFTR. Il serait alors intéressant d’évaluer l’activité
canal chlorure de F508del-CFTR dans des cellules avec des taux d’expression protéique
d’EDEM3 plus faibles ou sur des cellules EDEM3 KO.
Notre étude propose donc EDEM1 et EDEM2 comme cibles thérapeutiques et ouvre la
porte vers l’identification de stratégies permettant d’inhiber spécifiquement l’interaction de
F508del-CFTR avec ces protéines. En effet, une interaction directe de type protéine-protéine a
été observée entre EDEM1 ou EDEM2 et certains de leurs substrats (Slominska-Wojewodzka
et al., 2006; Kosmaoglou et al., 2009; Sokołowska et al., 2011; Shenkman et al., 2013;
Slominska-Wojewodzka et al., 2014; Tang et al., 2014). Il serait alors intéressant d’étudier si
F508del-CFTR établit ce type d’interactions avec ces protéines. Si c’est le cas, cela nous

Discussion, conclusion et perspectives
159
permettrait d’envisager une stratégie génétique pour inhiber de manière spécifique le site de
fixation de F508del-CFTR avec les protéines EDEMs.
C. Etude du trivalent-DMJ comme correcteur de F508del-CFTR
A la suite de notre étude, nous avons cherché à identifier une stratégie pharmacologique
qui permettrait d’inhiber l’interaction de F508del-CFTR avec les protéines EDEMs. Nous nous
sommes alors intéressé au trivalent-DMJ, celui-ci étant le composé le plus efficace de sa
famille.
Tout d’abord, nous avons vérifié que le trivalent-DMJ diminue l’interaction de F508del-
CFTR avec EDEM1, EDEM2 et EDEM3 dans les cellules Hela F508del-CFTR. Nous avons
également vérifié par western blot que cette diminution n’est pas corrélée à une diminution de
leur niveau d’expression protéique. L’équipe de Hebert a cependant démontré qu’EDEM1 lie
spécifiquement les protéines mal repliées même en présence des inhibiteurs des mannosidases
I (kifunensine et DMJ) et l’inhibiteur de glucosidase (DNJ), et que des mutations sur son site
actif de mannosidase ne semblent pas avoir d’effet sur sa liaison aux glycoprotéines mal repliées
(Cormier et al., 2009). Cependant, il s’agit d’expérimentations réalisées sur les protéines α1AT
et son mutant NHK qui sont toutes les deux des protéines solubles et non membranaires comme
CFTR. EDEM1 est présent dans les cellules sous 2 formes, soluble et membranaire. Il a été
décrit que la forme soluble serait efficace pour accélérer la dégradation des substrats ERAD
solubles, en revanche, la forme membranaire accélèrerait la dégradation de substrats
membranaires (Tamura et al., 2011). Ainsi, il semblerait que c’est la forme soluble d’EDEM1
qui interagit avec son substrat de manière indépendante de son activité catalytique. Une
comparaison de l’effet du trivalent-DMJ sur l’interaction d’EDEM1 avec une protéine soluble
mal repliée telle que l’α1AT et son interaction avec une protéine membranaire mal repliée
confirmerait cela.
Par la suite, nous avons confirmé l’effet correcteur du trivalent-DMJ sur F508del-CFTR
dans différents modèles cellulaires. Sur des cellules Hela F508del-CFTR traitées avec notre
composé, nous avons détecté grâce à la sonde fluorescente Oxonol, une modification de
potentiel de membrane CFTR-dépendant. Nous avons également détecté en chambre de Ussing,
un courant de court-circuit CFTR-dépendant dans un pseudo-épithélium formé par des cellules
CFBE, traitées avec notre composé. De manière plus intéressante, nous avons confirmé l’effet
correcteur du trivalent-DMJ sur des cellules épithéliales bronchiques fraichement isolées de
patients homozygotes pour la mutation F508del.

Discussion, conclusion et perspectives
160
Seule la bande B correspondante à la forme immature de F508del-CFTR a été observée
dans les cellules Hela F508del-CFTR traitées avec le trivalent-DMJ. Nous supposons donc que
notre correcteur adresse le canal F508del-CFTR à la membrane plasmique sous sa forme non
mature mais fonctionnelle. Cette hypothèse a été confirmée par la technique de biotinylation de
surface qui nous a permis de définir le profil de glycosylation de F508del-CFTR, présent au
niveau de la surface cellulaire. Il a déjà été identifié que l’état de glycosylation de CFTR, WT
ou F508del n’affecte pas sa fonction canal chlorure, et que la forme immature de F508del-
CFTR est fonctionnelle si elle est restaurée à la surface cellulaire (Morris et al., 1993; Chang et
al., 2008; Gee et al., 2011). Ainsi, deux hypothèses pourraient expliquer l’absence de
restauration de la forme mature hautement glycosylée (bande C) de cette protéine (Figure 57) :
La première hypothèse est que le F508del-CFTR est restauré à la membrane cellulaire
via la voie non conventionnelle ne passant pas par l’appareil de Golgi. En effet, il a été apporté
que l’expression membranaire de F508del-CFTR pourrait être restaurée in vitro et in vivo en la
dirigeant vers une voie de sécrétion non conventionnelle GRASP-dépendante, activée par le
stress du RE (Gee et al., 2011). Cependant, nous n’avons constaté aucun effet du trivalent-DMJ
sur le niveau d’expression protéique de la chaperonne BiP/GRP78 ou du facteur d’initiation de
la traduction eif2α phosphorylé, connus pour être surexprimés lors d’une réponse UPR
(Harding et al., 1999; Li et al., 2008; Wang et al., 2009; Nishitoh, 2012; Wang and Kaufman,
2014). De plus, nous avons observé que le trivalent-DMJ augmente l’interaction de F508del-
CFTR avec les lectines ERGIC53 et VIP36, deux récepteurs localisés principalement dans le
compartiment ERGIC et l’appareil de Golgi respectivement, suggérant la sortie de F508del-
CFTR du RE et son passage par l’appareil de Golgi. Nous avons également démontré qu’il
s’agissait d’une augmentation du taux d’interaction et non pas du niveau d’expression des
protéines ERGIC53 et VIP36. Ces résultats suggèrent une restauration de F508del-CFTR à la
membrane plasmique via la voie conventionnelle. Néanmoins, il serait intéressant de confirmer
ce point, en vérifiant l’effet du trivalent-DMJ sur F508del-CFTR en inhibant par une stratégie
de siRNA la voie de sécrétion GRASP-dépendent.

Discussion, conclusion et perspectives
161
Figure 57 : Hypothèses de la restauration de F508del-CFTR à la membrane cellulaire sous sa
forme immature par le trivalent-DMJ.
La deuxième hypothèse, la plus plausible, est que le trivalent-DMJ inhibe le processus
de maturation de F508del-CFTR à l’origine de son envoi à la membrane cellulaire sous sa forme
immature. En effet, ce composé est un dérivé du DMJ qui est un inhibiteur de l’activité
mannosidase. Il serait donc possible qu’il puisse inhiber l’activité des mannosidases de
l’appareil de Golgi, impliquées dans le processus de sa maturation. Cette hypothèse est soutenue
par l’étude de Morris et ses collaborateurs montrant que le traitement avec le composé DMJ
d’une ligné cellulaire du côlon humain (HT-29) polarisée ou non, réduit la masse moléculaire
de la forme mature du WT-CFTR par 30 kDa, générant ainsi une forme à 140 kDa,
correspondante à la forme immature de CFTR. De manière intéressante, la localisation
membranaire de CFTR ainsi que le courant chlorure transepithelial n’ont pas été affectés
(Morris et al., 1993). Notre hypothèse est également soutenue par une autre étude montrant que
le DMJ inhibe la conversion du motif oligosaccharidique de type mannose élevé vers un motif
complexe (Fuhrmann et al., 1984). Cette étude a démontré en effet, que les immunoglobulines
IgM et IgD des cellules traitées avec le DMJ, présentaient des niveaux élevés de mannoses et
que cela n’a pas empêché leur sécrétion dans le milieu de culture cellulaire. Les auteurs ont

Discussion, conclusion et perspectives
162
suggéré qu’en présence du DMJ, les modifications terminales ne peuvent pas avoir lieu en
raison de la présence des résidus mannose n’ayant pu être clivés.
Bien que cette hypothèse semble être la plus probable, il serait intéressant de mesurer l’effet
du trivalent-DMJ sur l’activité enzymatique des mannosidases de l’appareil de Golgi
impliquées dans la maturation de CFTR.
Dans un deuxième temps, nous avons cherché à explorer le mécanisme d’action du
trivalent-DMJ mis en place pour la restauration de F508del-CFTR à la membrane cellulaire.
Pour cela, nous avons regardé l’effet du trivalent-DMJ sur les principaux acteurs
impliqués dans le repliement et/ou la dégradation de F508del-CFTR. Les résultats obtenus n’ont
montré aucun effet de notre composé sur le niveau d’expression protéique de la calnexine,
Hsp70 ou Hsp90. De même, aucun effet n’a été observé sur leur interaction avec F508del-
CFTR, suggérant que l’effet correcteur du trivalent-DMJ ne ciblerait pas ces protéines.
Nous avons alors suite à ces résultats cherché à savoir si le trivalent-DMJ diminuait
l’interaction de F508del-CFTR avec l’ubiquitine ligase CHIP, celle-ci, identifiée pour être
impliquée, en complexe avec Hsc70, dans la dégradation de F508del-CFTR (Meacham et al.,
2001a; Alberti et al., 2004; Younger et al., 2006; Grove et al., 2009). En effet, CHIP est une
co-chaperonne de Hsc/Hsp70 et Hsp90 et semble agir de manière post-traductionnelle sur le
CFTR (Younger et al., 2006). Son association avec Hsc/Hsp70 et Hsp90 détourne ces
chaperonnes de la voie de repliement vers la voie de dégradation en liant aux substrats associés
des molécules d’ubiquitine (Figure 58). CHIP semble également jouer un rôle majeur dans la
dégradation de F508del-CFTR restauré à la surface cellulaire ; en revanche elle n’affecte pas la
demi-vie du WT-CFTR (Fu et al., 2012, 2015), suggérant que l’ubiquitinylation médiée par
CHIP permettrait la dégradation sélective du F508del-CFTR mal replié, restauré à la membrane
cellulaire.

Discussion, conclusion et perspectives
163
Figure 58 : Principales protéines impliquées dans le repliement et/ou la dégradation de CFTR. (Modifié d'après Kim and Skach, 2012)
Nous avons constaté dans notre étude encore préliminaire que le trivalent-DMJ n’aurait
pas d’effet sur l’interaction de F508del-CFTR avec cette ligase. Ceci n’est pas étonnant, dans
la mesure où une étude menée par Morito et ses collègues a suggéré que la dégradation de
F508del-CFTR se fasse par au moins deux voie indépendantes (Morito et al., 2008) : une voie
médiée par CHIP et une voie médiée par la coopération de RMA1-Gp78, d’autres ligases
impliquées dans l’ubiquitinylation de F508del-CFTR. RMA1/RNF5 est une E3 ubiquitine
ligase ancrée à l’extérieur de la membrane du RE et semble reconnaitre les défauts de
repliement de CFTR de manière co-traductionnelle ; GP78 quant à elle présente à la fois une
activité E3 et E4 ubiquitine ligase (Vij et al., 2006; Younger et al., 2006; Morito et al., 2008).
Sur des cellules HEK293 sur-exprimant transitoirement F508del-CFTR, cette équipe avait
constaté que le Gp78 favorise l’ubiquitinylation de F508del-CFTR dans les cellules CHIP-
knockdown, mais pas dans les cellules RMA1-knockdown. De plus, le knockdown de Gp78
altère l’ubiquitinylation de F508del-CFTR et provoque un retard dans sa dégradation. Ce délai
de dégradation est plus prononcé suite à la diminution de l’expression protéique de RMA1. De
plus, le knockdown simultané de Gp78 et de RMA1 n’a pas montré d’effet additif sur le retard
de la dégradation de F508del-CFTR. Ainsi, cette équipe suggère que RMA1 est spécifiquement
requis en tant qu’enzyme E3 en amont pour l’activité E4-like de Gp78 et propose un transfert

Discussion, conclusion et perspectives
164
direct de F508del-CFTR d’E3 à E4 dans le même complexe (Figure 59). Les auteurs ont
démontré que la diminution du taux d’expression de la protéine CHIP a provoqué un retard dans
la dégradation de F508del-CFTR, comparable à celui provoqué par Gp78-knockdown, et
suggèrent que la voie de dégradation de F508del-CFTR médiée par CHIP serait indépendante
de celle médiée par RMA1-Gp78.
Figure 59 : Modèle d’ubiquitinylation séquentielle de F508del-CFTR par des complexes contenant RMA1/RNF5 et Gp78.
(Modifié d'après Morito et al., 2008)
Afin de vérifier que le trivalent-DMJ n’a pas d’effet sur l’ubiquitinylation médiée par la ligase
CHIP, il serait alors intéressant de regarder son effet sur l’interaction de Hsc/Hsp70 avec les
co-chaperonnes productives à savoir, Hdj-2 et plus particulièrement HspB1, connue pour
inhiber l’activité ubiquitine ligase de CHIP, et BAG-1, connue pour coopérer avec CHIP afin
de déplacer l’activité de Hsc/Hsp70 du processus de repliement vers la dégradation (Demand
et al., 2001; Alberti et al., 2004). Il serait aussi intéressant de comparer par co-
immunoprécipitation, l’état du complexe protéique formé avec F508del-CFTR avant et après
le traitement cellulaire par le trivalent-DMJ. Cela nous permettrait de caractériser les
protéines qui s’associent ou qui se dissocient du CFTR après traitement avec notre composé.
Une autre étude a démontré que CHIP et RMA1/RNF5 agissent en séquentiel dans la
membrane du RE et le cytosol respectivement, pour détecter et contrôler l’état de repliement de
CFTR (Younger et al., 2006). Nous avons alors regardé l’effet du trivalent-DMJ sur
l’interaction de F508del-CFTR avec RMA1/RNF5, et de manière surprenante, une
augmentation importante (~ 80%) est apparue dans les cellules traitées avec notre correcteur.
Deux hypothèses peuvent être envisagées pour expliquer cette augmentation :

Discussion, conclusion et perspectives
165
La première hypothèse est que le trivalent-DMJ en diminuant l’interaction de F508del-
CFTR avec les protéines EDEMs, empêche l’envoi de F508del-CFTR vers le récepteur SEL1L,
un récepteur des substrats mal repliés au niveau du complexe de retrotranslocation HRD1/Gp78
et par conséquent favorise son envoie vers le complexe RMA1 (Figure 60). Cette hypothèse
repose d’une part, sur le fait que les deux ligases HRD1/Gp78 et RMA1/RNF5 ont été
identifiées pour être impliquées dans la dégradation de F508del-CFTR, et d’autre part, sur la
nécessité du domaine mannosidase d’EDEM1 pour son interaction avec SEL1L (Cormier et al.,
2009). Afin de vérifier cette hypothèse, il serait intéressant de vérifier l’effet du trivalent-DMJ
sur l’interaction de F508del-CFTR avec les acteurs du complexe HRD1/Gp78. Une co-
immunoprécipitation permettrait également de distinguer quels acteurs du complexe de rétro-
translocation précipiteraient avec F508del-CFTR.
Figure 60 : Les trois principales ligases identifiées chez les mammifères. Chaque ligase (en orange claire) fait partie d’un complexe avec une enzyme E2 ubiquitine-conjugué (en vert) et d’autres facteurs. Les substrats (en rose) sont ubiquitylés, extraits dans le cytoplasme par AAA+ATPase p97 et enfin dégradés par le protéasome (Modifié d'après Houck et al., 2012).
La deuxième hypothèse est que le trivalent-DMJ inhibe la voie de dégradation de
F508del-CFTR en aval de RMA1/RNF5, bloquant ainsi sa rétrotranslocation vers le cytosol et
induisant son accumulation au niveau du complexe de rétro-translocation RMA1/RNF5.
Afin de vérifier cette hypothèse, nous avons regardé l’effet du trivalent-DMJ sur le protéasome,
la voie dominante pour l’élimination du CFTR mal replié dans les cellules de mammifères
(Gelman et al., 2002). Contre toute attente, une diminution de l’activité enzymatique du

Discussion, conclusion et perspectives
166
protéasome intracellulaire et du protéasome purifié a été observée suite au traitement avec le
trivalent-DMJ. Cela suggère que notre correcteur, connu comme un inhibiteur des α 1,2-
mannosidases du RE inhibe de manière directe l’activité enzymatique du protéasome 20S. A
notre connaissance, nos résultats seraient les premiers à démontrer un effet inhibiteur de la série
DMJ sur l’activité enzymatique du protéasome. Cependant, l’origine de cet effet inhibiteur reste
inconnu : s’agit-il de la présence d’un site catalytique dans le composé DMJ, ou bien, d’une
conséquence des modifications chimiques, nécessaires pour la synthèse des composés
multivalents ? Là encore, c’est un tout nouveau domaine à explorer.
L’ensemble de ces résultats nous laisse penser que le mécanisme d’action du trivalent-
DMJ serait dépendant de la famille des protéines EDEM mais également du protéasome
cellulaire.
D. A la recherche d’une autre cible thérapeutique pour la mucoviscidose :
α1,2-mannosidase I du RE
Nous avons mis en évidence dans cette partie de l’étude, l’implication de l’α1,2-
mannosidase I du RE (ERManI) dans la rétention réticulaire de F508del-CFTR. Son extinction
partielle par son siRNA spécifique a été associée avec une restauration de la fonction CFTR-
dépendante. Comme pour le traitement au trivalent-DMJ, ERManI knockdown restaure
F508del-CFTR à la membrane cellulaire sous sa forme immature (bande B). Plusieurs études
décrivent ERManI comme le « minuteur » qui déclenche la première étape de la voie de
dégradation en clivant le premier résidu mannose de la branche B de l’oligosaccharide (Araki
and Nagata, 2011; Caramelo and Parodi, 2015; Zhou et al., 2015). Ainsi, l’absence de
restauration de la forme mature de F508del-CFTR pourrait être expliquée par le fait que
l’extinction d’ERManI par siRNA empêcherait la génération du motif oligosaccharidique de
forme M8GlcNAc2 de F508del-CFTR et favoriserait ainsi son transport vers l’appareil de Golgi
avec le motif M9GlcNAc2, gênant le processus de maturation (Figure 61). En effet, après le
cycle de calnexine/calréticuline, les glycoprotéines natives ou mal repliées subissent par
l’enzyme ERManI un clivage du résidu mannose de la branche B de leur motif M9GlcNAc2
pour générer le motif M8GlcNAc2. Les glycoprotéines mal repliées subissent d’autres clivages
de mannose qui les envoient vers la dégradation ; au contraire, les glycoprotéines natives
transitent vers l’appareil de Golgi (Figure 61).

Discussion, conclusion et perspectives
167
Figure 61 : Devenir des glycoprotéines correctement- ou mal-repliées. L’oligosaccharide Glc3Man9GlcNAc2 : carrés bleu : N-acétylglucosamine, cercles vert : mannoses, cercle bleu glucose. GlcI : α-glucosidases I. UGGT : UDP-glucose: glycoprotein glucosyltransferase. Au regard du rôle d’ERManI dans la dégradation des glycoprotéines mal/non repliées,
nous avons émis l’hypothèse, que son knockdown inhiberait l’envoi de F508del-CFTR vers la
voie de dégradation. Cette hypothèse repose sur des études démontrant que l’inhibition de
l’activité enzymatique de cette protéine, que ce soit par siRNA ou par Kifunensine, stabilise les
substrats luminaux d’ERAD AAT et son mutant NHK (Avezov et al., 2008; Hosokawa et al.,
2003; Pan et al., 2013; Wu et al., 2003). Afin d’apporter la preuve qui confirmerait notre
hypothèse, il serait alors important de suivre le niveau d’expression de F508del-CFTR au cours
du temps par la technique de pulse-chase (marquage métabolique au 35S). Elle repose sur un
marquage des protéines nouvellement synthétisées par incorporation de méthionine et de
cystéine marquées pendant une période déterminée (pulse), puis un suivi au cours du temps
(chase), mais cette fois-ci dans un environnement non marqué, du devenir de ces protéines
marquées.
Nous avons par la suite, entrepris la caractérisation du mécanisme d’action impliqué
dans cette correction. Partant du fait qu’ERManI est le minuteur qui déclenche la première étape
de dégradation en clivant le premier résidu de mannose, nous avions émis l’hypothèse que
l’inhibition de l’interaction de F508del-CFTR avec cette enzyme diminuerait son interaction
avec les protéines EDEMs. Cependant, nous n’avons détecté aucun effet de la diminution du
taux d’expression d’ERManI sur l’expression protéique d’EDEM1, EDEM2 ou EDEM3, ni sur
leur interaction avec F508del-CFTR, suggérant que le mécanisme d’action impliqué dans la
correction de F508del-CFTR n’est pas dépendant des protéines EDEMs. Trois hypothèses ont

Discussion, conclusion et perspectives
168
été émises pour expliquer ces résultats surprenants : (1) ERManI agirait dans une voie de
dégradation indépendante de celle des EDEMs, (2) ERManI n’affecte pas l’interaction des
protéines EDEMs avec F508del-CFTR mais l’inhibition du clivage du premier résidu mannose
inhiberait leur activité mannosidase. Cette hypothèse est basée sur des études montrant que les
protéines EDEMs peuvent interagir avec leur substrat de manière indépendante de leur activité
mannosidase (Slominska-Wojewodzka et al., 2006; Kosmaoglou et al., 2009; Sokołowska et
al., 2011; Shenkman et al., 2013; Slominska-Wojewodzka et al., 2014; Tang et al., 2014), ou
bien, (3) ERManI agirait en aval des protéines EDEMs. En effet, une étude réalisée en 2014 par
l’équipe de Mori sur la lignée de poule DT40 et la lignée tumorale de colon HCT116, a
démontré par un système d’extinction de gènes, que le premier clivage de résidus mannose
serait effectué par EDEM2 et non pas, par ERManI (Ninagawa et al., 2014).
A ce stade de notre étude, il est impossible de trancher entre ces trois hypothèses. En effet, bien
que l’implication des trois EDEMs et d’ERManI dans la démannosylation des glycoprotéines
mal/non repliées soit bien établie, l’ordre de leur action n’est à ce jour pas encore déterminé.
Ainsi, face à la divergence sur le mode d’action d’ERManI, il serait envisageable d’éteindre
les protéines EDEMs ou ERManI séparément par un système KO, et définir ensuite le profil en
résidus mannose du N-glycane de F508del-CFTR comme décrit dans l’étude réalisée par
l’équipe de Mori (Ninagawa et al., 2014). Cela nous permettrait d’identifier dans quel type de
cellules les protéines avec le profil M9 s’accumulent et par conséquent quel type de
mannosidase serait responsable de la catalyse du clivage de premier résidu mannose.
Dans la poursuite de notre étude, mais cette fois-ci du côté cytosolique, une diminution
de l’interaction de F508del-CFTR avec l’ubiquitine ligase CHIP, la co-chaperonne de
Hsp/Hsc70 et Hsp90 a été observée dans les cellules transfectées par le siRNA ERManI. Même
s’il s’agit de résultats encore préliminaires, la diminution observée semble être significative.
Nous nous sommes assurés par western blot que cette diminution n’est pas corrélée à une
diminution du niveau d’expression protéique de CHIP, ce qui suggère qu’ERManI knockdown
restaure F508del-CFTR à la membrane cellulaire en prévenant l’interaction de CHIP au
complexe Hsc/Hsp70. F508del-CFTR n’étant alors pas ubiquitinylé, ne subira pas de
rétrotranslocation vers le cytosol pour dégradation, favorisant sa progression dans la voie de
repliement. Une étude de l’effet du siRNA ERManI sur les co-chaperonne productives tell que
Hdj-2, Hop et Bag1, montrant une augmentation de leur interaction avec F508del-CFTR
confirmerait cela.

Discussion, conclusion et perspectives
169
Suite à ces résultats, nous nous sommes intéressés à l’ubiquitine ligase RMA1/RNF5,
agissant en séquentiel avec CHIP pour la détection et le contrôle du repliement de CFTR. Le
knockdown d’ERManI n’a montré aucun effet sur l’interaction de RMA1/RNF5 avec F508del-
CFTR (résultat préliminaire) ni sur leur niveau d’expression. Au regard de son rôle de détection
des défauts de repliement de CFTR, nous pensons que RMA1/RNF5 reste en permanence
attaché au F508del-CFTR et que c’est sa coopération avec d’autres partenaires tel que
l’ubiquitine ligase Gp78, Derlin-1 ou la co-chaperonne de Hsc/Hsp70, DNAJB12, qui
déclenche le processus de dégradation de F508del-CFTR (Morito et al., 2008; Grove et al.,
2011). Enfin, nous avons démontré que le knockdown d’ERManI ne présente aucun effet sur
l’activité enzymatique du protéasome. Il serait intéressant d’explorer l’effet de knockdown
d’ERManI sur les autres acteurs d’ERAD et les partenaires des complexes de
rétrotranslocation par des techniques de Duolink et/ou de co-immunoprécipitation.
En conclusion, notre travail propose la protéine ERManI et la famille de protéines
EDEM, plus particulièrement EDEM1 et EDEM2 comme cibles pour la recherche de
correcteurs de F508del-CFTR (Figure 62-A). Dans cette optique, nous avons révélé l’effet
correcteur du trivalent-DMJ et avons tenté de comprendre son mécanisme d’action (Figure 62-
B). Il semblerait que ce composé connu comme un inhibiteur de l’activité mannosidase
rétablissent l’expression membranaire de F508del-CFTR sous sa forme immature mais
fonctionnelle en diminuant son interaction avec les protéines EDEMs, mais également en
inhibant l’activité enzymatique du protéasome. Ainsi, la suite logique de ce travail sera de
continuer à élucider le mécanisme d’action de ce correcteur et de le tester dans d’autres cas de
pathologies.

Discussion, conclusion et perspectives
170
A
B
Figure 62 : Récapitulatif des principaux résultats de ce projet de thèse. (A) Implication des protéines ERManI, EDEM1 et EDEM2 dans la rétention réticulaire de F508del-CFTR. (B) L’effet correcteur sur F508del-CFTR du trivalent-DMJ et son mécanisme d’action.

Références bibliographique
171
Références bibliographiques Abriel, H., and Staub, O. (2005). Ubiquitylation of Ion Channels. Physiology 20, 398–407.
Alberti, S., Böhse, K., Arndt, V., Schmitz, A., and Höhfeld, J. (2004). The Cochaperone HspBP1 Inhibits the CHIP Ubiquitin Ligase and Stimulates the Maturation of the Cystic Fibrosis Transmembrane Conductance Regulator. Mol. Biol. Cell 15, 4003–4010.
Ali, M.M.U., Roe, S.M., Vaughan, C.K., Meyer, P., Panaretou, B., Piper, P.W., Prodromou, C., and Pearl, L.H. (2006). Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature 440, 1013–1017.
Anderson, M.P., Gregory, R.J., Thompson, S., Souza, D.W., Paul, S., Mulligan, R.C., Smith, A.E., and Welsh, M.J. (1991). Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253, 202–205.
Appenzeller-Herzog, C. (2006). The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J. Cell Sci. 119, 2173–2183.
Appenzeller-Herzog, C., and Ellgaard, L. (2008). The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1783, 535–548.
Araki, K., and Nagata, K. (2011). Protein Folding and Quality Control in the ER. Cold Spring Harb. Perspect. Biol. 3, a007526–a007526.
Arndt, V., Daniel, C., Nastainczyk, W., Alberti, S., and Höhfeld, J. (2005). BAG-2 Acts as an Inhibitor of the Chaperone-associated Ubiquitin Ligase CHIP. Mol. Biol. Cell 16, 5891–5900.
Avezov, E., Frenkel, Z., Ehrlich, M., Herscovics, A., and Lederkremer, G.Z. (2008). Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-glycan trimming to Man5-6GlcNAc2 in glycoprotein ER-associated degradation. Mol. Biol. Cell 19, 216–225.
Awayda, M.S. (1996). Regulation of Epithelial Sodium Channels by the Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 271, 4725–4732.
Balch, W.E., and Gist Farquhar, M. (1995). Beyond bulk flow. Trends Cell Biol. 5, 16–19.
Balch, W.E., McCaffery, J.M., Plutner, H., and Farquhar, M.G. (1994). Vesicular stomatitis virus glycoprotein is sorted and concentrated during export from the endoplasmic reticulum. Cell 76, 841–852.
Balchin, D., Hayer-Hartl, M., and Hartl, F.U. (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354-aac4354.
Banerjee, S., Vishwanath, P., Cui, J., Kelleher, D.J., Gilmore, R., Robbins, P.W., and Samuelson, J. (2007). The evolution of N-glycan-dependent endoplasmic reticulum quality control factors for glycoprotein folding and degradation. Proc. Natl. Acad. Sci. U. S. A. 104, 11676–11681.

Références bibliographique
172
Barlowe, C. (2003). Signals for COPII-dependent export from the ER: what’s the ticket out? Trends Cell Biol. 13, 295–300.
Barr, D.J., Ostermeyer-Fay, A.G., Matundan, R.A., and Brown, D.A. (2008). Clathrin-independent endocytosis of ErbB2 in geldanamycin-treated human breast cancer cells. J. Cell Sci. 121, 3155–3166.
Bartoli, M., Gicquel, E., Barrault, L., Soheili, T., Malissen, M., Malissen, B., Vincent-Lacaze, N., Perez, N., Udd, B., Danos, O., et al. (2008). Mannosidase I inhibition rescues the human alpha-sarcoglycan R77C recurrent mutation. Hum. Mol. Genet. 17, 1214–1221.
Bartoszewski, R., Rab, A., Jurkuvenaite, A., Mazur, M., Wakefield, J., Collawn, J.F., and Bebők, Z. (2008a). Activation of the Unfolded Protein Response by ΔF508 CFTR. Am. J. Respir. Cell Mol. Biol. 39, 448–457.
Bartoszewski, R., Rab, A., Twitty, G., Stevenson, L., Fortenberry, J., Piotrowski, A., Dumanski, J.P., and Bebök, Z. (2008b). The Mechanism of Cystic Fibrosis Transmembrane Conductance Regulator Transcriptional Repression during the Unfolded Protein Response. J. Biol. Chem. 283, 12154–12165.
Bartoszewski, R., Brewer, J.W., Rab, A., Crossman, D.K., Bartoszewska, S., Kapoor, N., Fuller, C., Collawn, J.F., and Bebok, Z. (2011). The Unfolded Protein Response (UPR)-activated Transcription Factor X-box-binding Protein 1 (XBP1) Induces MicroRNA-346 Expression That Targets the Human Antigen Peptide Transporter 1 (TAP1) mRNA and Governs Immune Regulatory Genes. J. Biol. Chem. 286, 41862–41870.
Beckmann, R.P., Mizzen, L.E., and Welch, W.J. (1990). Interaction of Hsp 70 with newly synthesized proteins: implications for protein folding and assembly. Science 248, 850–854.
ter Beek, J., Guskov, A., and Slotboom, D.J. (2014). Structural diversity of ABC transporters. J. Gen. Physiol. 143, 419–435.
Beggah, A.T., Jaunin, P., and Geering, K. (1997). Role of glycosylation and disulfide bond formation in the beta subunit in the folding and functional expression of Na,K-ATPase. J. Biol. Chem. 272, 10318–10326.
Benyair, R., Ogen-Shtern, N., Mazkereth, N., Shai, B., Ehrlich, M., and Lederkremer, G.Z. (2015a). Mammalian ER mannosidase I resides in quality control vesicles, where it encounters its glycoprotein substrates. Mol. Biol. Cell 26, 172–184.
Benyair, R., Ogen-Shtern, N., and Lederkremer, G.Z. (2015b). Glycan regulation of ER-associated degradation through compartmentalization. Semin. Cell Dev. Biol. 41, 99–109.
Ben-Zvi, A., De Los Rios, P., Dietler, G., and Goloubinoff, P. (2004). Active solubilization and refolding of stable protein aggregates by cooperative unfolding action of individual hsp70 chaperones. J. Biol. Chem. 279, 37298–37303.
Berger, A.L., Randak, C.O., Ostedgaard, L.S., Karp, P.H., Vermeer, D.W., and Welsh, M.J. (2005). Curcumin Stimulates Cystic Fibrosis Transmembrane Conductance Regulator Cl – Channel Activity. J. Biol. Chem. 280, 5221–5226.

Références bibliographique
173
Bergeron, M.J., Burzle, M., Kovacs, G., Simonin, A., and Hediger, M.A. (2011). Synthesis, Maturation, and Trafficking of Human Na+-Dicarboxylate Cotransporter NaDC1 Requires the Chaperone Activity of Cyclophilin B. J. Biol. Chem. 286, 11242–11253.
Bernasconi, R., and Molinari, M. (2011). ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 23, 176–183.
Bernasconi, R., Soldà, T., Galli, C., Pertel, T., Luban, J., and Molinari, M. (2010). Cyclosporine A-Sensitive, Cyclophilin B-Dependent Endoplasmic Reticulum-Associated Degradation. PLoS ONE 5, e13008.
Bertrand, C.A., and Frizzell, R.A. (2003). The role of regulated CFTR trafficking in epithelial secretion. AJP Cell Physiol. 285, C1–C18.
Blohmke, C.J., Mayer, M.L., Tang, A.C., Hirschfeld, A.F., Fjell, C.D., Sze, M.A., Falsafi, R., Wang, S., Hsu, K., Chilvers, M.A., et al. (2012). Atypical Activation of the Unfolded Protein Response in Cystic Fibrosis Airway Cells Contributes to p38 MAPK-Mediated Innate Immune Responses. J. Immunol. 189, 5467–5475.
Boyle, M.P., Bell, S.C., Konstan, M.W., McColley, S.A., Rowe, S.M., Rietschel, E., Huang, X., Waltz, D., Patel, N.R., and Rodman, D. (2014). A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir. Med. 2, 527–538.
Braakman, I., and Hebert, D.N. (2013). Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 5, a013201–a013201.
Braakman, I., Hoover-Litty, H., Wagner, K.R., and Helenius, A. (1991). Folding of influenza hemagglutinin in the endoplasmic reticulum. J. Cell Biol. 114, 401–411.
Brambilla Pisoni, G., Molinari, M., Jan Bergmann, T., and 1 Institute for Research in Biomedicine (IRB), Bellinzona, Switzerland (2016). Quality control mechanisms of protein biogenesis: proteostasis dies hard. AIMS Biophys. 3, 456–478.
Buchner, J. (1999). Hsp90 & Co. - a holding for folding. Trends Biochem. Sci. 24, 136–141.
Cai, Z., Liu, J., Li, H., and Sheppard, D.N. (2011). Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis. Acta Pharmacol. Sin. 32, 693–701.
Calì, T., Galli, C., Olivari, S., and Molinari, M. (2008). Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 371, 405–410.
Caplan, A.J., Langley, E., Wilson, E.M., and Vidal, J. (1995). Hormone-dependent Transactivation by the Human Androgen Receptor Is Regulated by a dnaJ Protein. J. Biol. Chem. 270, 5251–5257.
Caramelo, J.J., and Parodi, A.J. (2015). A sweet code for glycoprotein folding. FEBS Lett. 589, 3379–3387.

Références bibliographique
174
Chang, X. -b., Mengos, A., Hou, Y. -x., Cui, L., Jensen, T.J., Aleksandrov, A., Riordan, J.R., and Gentzsch, M. (2008). Role of N-linked oligosaccharides in the biosynthetic processing of the cystic fibrosis membrane conductance regulator. J. Cell Sci. 121, 2814–2823.
Chang, X.B., Cui, L., Hou, Y.X., Jensen, T.J., Aleksandrov, A.A., Mengos, A., and Riordan, J.R. (1999). Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol. Cell 4, 137–142.
Chawla, A., Chakrabarti, S., Ghosh, G., and Niwa, M. (2011). Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J. Cell Biol. 193, 41–50.
Cheng, S.H., Gregory, R.J., Marshall, J., Paul, S., Souza, D.W., White, G.A., O’Riordan, C.R., and Smith, A.E. (1990). Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834.
Chin, S., Hung, M., and Bear, C.E. (2017). Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants. Cell. Mol. Life Sci. CMLS 74, 57–66.
Cho, H.J., Gee, H.Y., Baek, K.-H., Ko, S.-K., Park, J.-M., Lee, H., Kim, N.-D., Lee, M.G., and Shin, I. (2011). A small molecule that binds to an ATPase domain of Hsc70 promotes membrane trafficking of mutant cystic fibrosis transmembrane conductance regulator. J. Am. Chem. Soc. 133, 20267–20276.
Cholon, D.M., O’Neal, W.K., Randell, S.H., Riordan, J.R., and Gentzsch, M. (2010). Modulation of endocytic trafficking and apical stability of CFTR in primary human airway epithelial cultures. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L304-314.
Choo-Kang, L.R., and Zeitlin, P.L. (2001). Induction of HSP70 promotes DeltaF508 CFTR trafficking. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L58-68.
Christianson, J.C., Shaler, T.A., Tyler, R.E., and Kopito, R.R. (2008). OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 10, 272–282.
Clancy, J.P., Rowe, S.M., Accurso, F.J., Aitken, M.L., Amin, R.S., Ashlock, M.A., Ballmann, M., Boyle, M.P., Bronsveld, I., Campbell, P.W., et al. (2012). Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67, 12–18.
Clerc, S., Hirsch, C., Oggier, D.M., Deprez, P., Jakob, C., Sommer, T., and Aebi, M. (2009). Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 184, 159–172.
Collawn, J.F., and Matalon, S. (2014). CFTR and lung homeostasis. AJP Lung Cell. Mol. Physiol. 307, L917–L923.
Compain, P., Decroocq, C., Joosten, A., de Sousa, J., Rodríguez-Lucena, D., Butters, T.D., Bertrand, J., Clément, R., Boinot, C., Becq, F., et al. (2013). Rescue of Functional CFTR Channels in Cystic Fibrosis: A Dramatic Multivalent Effect Using Iminosugar Cluster-Based Correctors. ChemBioChem 14, 2050–2058.

Références bibliographique
175
Connell, P., Ballinger, C.A., Jiang, J., Wu, Y., Thompson, L.J., Höhfeld, J., and Patterson, C. (2001). The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96.
Coppinger, J.A., Hutt, D.M., Razvi, A., Koulov, A.V., Pankow, S., Yates, J.R., and Balch, W.E. (2012). A Chaperone Trap Contributes to the Onset of Cystic Fibrosis. PLoS ONE 7, e37682.
Cormier, J.H., Tamura, T., Sunryd, J.C., and Hebert, D.N. (2009). EDEM1 Recognition and Delivery of Misfolded Proteins to the SEL1L-Containing ERAD Complex. Mol. Cell 34, 627–633.
Cortese, K., Howes, M.T., Lundmark, R., Tagliatti, E., Bagnato, P., Petrelli, A., Bono, M., McMahon, H.T., Parton, R.G., and Tacchetti, C. (2013). The HSP90 inhibitor geldanamycin perturbs endosomal structure and drives recycling ErbB2 and transferrin to modified MVBs/lysosomal compartments. Mol. Biol. Cell 24, 129–144.
Cox, T., Lachmann, R., Hollak, C., Aerts, J., van Weely, S., Hrebícek, M., Platt, F., Butters, T., Dwek, R., Moyses, C., et al. (2000). Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. The Lancet 355, 1481–1485.
Crothers, J.M., Asano, S., Kimura, T., Yoshida, A., Wong, A., Kang, J.W., and Forte, J.G. (2004). Contribution of oligosaccharides to protection of the H,K-ATPase beta-subunit against trypsinolysis. Electrophoresis 25, 2586–2592.
Csanády, L., Chan, K.W., Seto-Young, D., Kopsco, D.C., Nairn, A.C., and Gadsby, D.C. (2000). Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116, 477–500.
Cui, L., Aleksandrov, L., Chang, X.-B., Hou, Y.-X., He, L., Hegedus, T., Gentzsch, M., Aleksandrov, A., Balch, W.E., and Riordan, J.R. (2007). Domain Interdependence in the Biosynthetic Assembly of CFTR. J. Mol. Biol. 365, 981–994.
Cunningham, C.N., Krukenberg, K.A., and Agard, D.A. (2008). Intra- and Intermonomer Interactions Are Required to Synergistically Facilitate ATP Hydrolysis in Hsp90. J. Biol. Chem. 283, 21170–21178.
Cyr, D.M., Höhfeld, J., and Patterson, C. (2002). Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 27, 368–375.
Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Crystal, R.G., Pavirani, A., Lecocq, J.P., and Lazdunski, M. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528.
D’Arcangelo, J.G., Stahmer, K.R., and Miller, E.A. (2013). Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1833, 2464–2472.
Dean, M., Rzhetsky, A., and Allikmets, R. (2001). The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166.

Références bibliographique
176
Dejgaard, K., Theberge, J.-F., Heath-Engel, H., Chevet, E., Tremblay, M.L., and Thomas, D.Y. (2010). Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J. Proteome Res. 9, 1763–1771.
Demand, J., Alberti, S., Patterson, C., and Höhfeld, J. (2001). Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. CB 11, 1569–1577.
Deveraux, Q., Ustrell, V., Pickart, C., and Rechsteiner, M. (1994). A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem. 269, 7059–7061.
Di, A., Brown, M.E., Deriy, L.V., Li, C., Szeto, F.L., Chen, Y., Huang, P., Tong, J., Naren, A.P., Bindokas, V., et al. (2006). CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 8, 933–944.
Dittmar, K.D., Banach, M., Galigniana, M.D., and Pratt, W.B. (1998). The Role of DnaJ-like Proteins in Glucocorticoid Receptor·hsp90 Heterocomplex Assembly by the Reconstituted hsp90·p60·hsp70 Foldosome Complex. J. Biol. Chem. 273, 7358–7366.
Dollins, D.E., Warren, J.J., Immormino, R.M., and Gewirth, D.T. (2007). Structures of GRP94-Nucleotide Complexes Reveal Mechanistic Differences between the hsp90 Chaperones. Mol. Cell 28, 41–56.
Dong, M., Bridges, J.P., Apsley, K., Xu, Y., and Weaver, T.E. (2008). ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 19, 2620–2630.
Du, K., and Lukacs, G.L. (2009). Cooperative Assembly and Misfolding of CFTR Domains In Vivo. Mol. Biol. Cell 20, 1903–1915.
Du, K., Sharma, M., and Lukacs, G.L. (2005). The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 12, 17–25.
Eckford, P.D.W., Ramjeesingh, M., Molinski, S., Pasyk, S., Dekkers, J.F., Li, C., Ahmadi, S., Ip, W., Chung, T.E., Du, K., et al. (2014). VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 21, 666–678.
Edman, J.C., Ellis, L., Blacher, R.W., Roth, R.A., and Rutter, W.J. (1985). Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin. Nature 317, 267–270.
Egan, M.E., Glöckner-Pagel, J., Ambrose, C.A., Cahill, P.A., Pappoe, L., Balamuth, N., Cho, E., Canny, S., Wagner, C.A., Geibel, J., et al. (2002). Calcium-pump inhibitors induce functional surface expression of ΔF508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med. 8, 485–492.
Egan, M.E., Pearson, M., Weiner, S.A., Rajendran, V., Rubin, D., Glöckner-Pagel, J., Canny, S., Du, K., Lukacs, G.L., and Caplan, M.J. (2004). Curcumin, a Major Constituent of Turmeric, Corrects Cystic Fibrosis Defects. Science 304, 600–602.
Elborn, J.S. (2016). Cystic fibrosis. The Lancet 388, 2519–2531.

Références bibliographique
177
Elliott, J.G., Oliver, J.D., and High, S. (1997). The Thiol-dependent Reductase ERp57 Interacts Specifically with N -Glycosylated Integral Membrane Proteins. J. Biol. Chem. 272, 13849–13855.
Elstein, D., Hollak, C., Aerts, J.M.F.G., van Weely, S., Maas, M., Cox, T.M., Lachmann, R.H., Hrebicek, M., Platt, F.M., Butters, T.D., et al. (2004). Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 27, 757–766.
Fajac, I., and De Boeck, K. (2017). New horizons for cystic fibrosis treatment. Pharmacol. Ther. 170, 205–211.
Farinha, C.M., and Amaral, M.D. (2005). Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Biol. 25, 5242–5252.
Farinha, C.M., and Canato, S. (2017). From the endoplasmic reticulum to the plasma membrane: mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 74, 39–55.
Farinha, C.M., and Matos, P. (2016). Repairing the basic defect in cystic fibrosis - one approach is not enough. FEBS J. 283, 246–264.
Farinha, C.M., Nogueira, P., Mendes, F., Penque, D., and Amaral, M.D. (2002). The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 366, 797–806.
Fenteany, G., Standaert, R., Lane, W., Choi, S., Corey, E., and Schreiber, S. (1995). Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268, 726–731.
Fiedler, K., Parton, R.G., Kellner, R., Etzold, T., and Simons, K. (1994). VIP36, a novel component of glycolipid rafts and exocytic carrier vesicles in epithelial cells. EMBO J. 13, 1729–1740.
Fischer, G., and Aumüller, T. (2003). Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 148, 105–150.
Fliegel, L., Burns, K., MacLennan, D.H., Reithmeier, R.A., and Michalak, M. (1989). Molecular cloning of the high affinity calcium-binding protein (calreticulin) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 264, 21522–21528.
Flynn, G., Chappell, T., and Rothman, J. (1989). Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245, 385–390.
Flynn, G.C., Pohl, J., Flocco, M.T., and Rothman, J.E. (1991). Peptide-binding specificity of the molecular chaperone BiP. Nature 353, 726–730.
Frenkel, Z., Gregory, W., Kornfeld, S., and Lederkremer, G.Z. (2003). Endoplasmic Reticulum-associated Degradation of Mammalian Glycoproteins Involves Sugar Chain Trimming to Man6-5GlcNAc2. J. Biol. Chem. 278, 34119–34124.

Références bibliographique
178
Frey, S., Leskovar, A., Reinstein, J., and Buchner, J. (2007). The ATPase cycle of the endoplasmic chaperone Grp94. J. Biol. Chem. 282, 35612–35620.
Frickel, E.-M., Riek, R., Jelesarov, I., Helenius, A., Wuthrich, K., and Ellgaard, L. (2002). TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proc. Natl. Acad. Sci. U. S. A. 99, 1954–1959.
Fu, L., and Sztul, E. (2009). ER-associated complexes (ERACs) containing aggregated cystic fibrosis transmembrane conductance regulator (CFTR) are degraded by autophagy. Eur. J. Cell Biol. 88, 215–226.
Fu, X.-M., and Zhu, B.T. (2010). Human pancreas-specific protein disulfide-isomerase (PDIp) can function as a chaperone independently of its enzymatic activity by forming stable complexes with denatured substrate proteins. Biochem. J. 429, 157–169.
Fu, L., Rab, A., Tang, L.P., Rowe, S.M., Bebok, Z., and Collawn, J.F. (2012). Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 441, 633–643.
Fu, L., Rab, A., Tang, L. ping, Bebok, Z., Rowe, S.M., Bartoszewski, R., and Collawn, J.F. (2015). ΔF508 CFTR Surface Stability Is Regulated by DAB2 and CHIP-Mediated Ubiquitination in Post-Endocytic Compartments. PLOS ONE 10, e0123131.
Fuhrmann, U., Bause, E., Legler, G., and Ploegh, H. (1984). Novel mannosidase inhibitor blocking conversion of high mannose to complex oligosaccharides. Nature 307, 755–758.
Füllekrug, J., Scheiffele, P., and Simons, K. (1999). VIP36 localisation to the early secretory pathway. J. Cell Sci. 112 ( Pt 17), 2813–2821.
Fuller, W., and Cuthbert, A.W. (2000). Post-translational disruption of the delta F508 cystic fibrosis transmembrane conductance regulator (CFTR)-molecular chaperone complex with geldanamycin stabilizes delta F508 CFTR in the rabbit reticulocyte lysate. J. Biol. Chem. 275, 37462–37468.
Gabriel, S.E., Clarke, L.L., Boucher, R.C., and Stutts, M.J. (1993). CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship. Nature 363, 263–266.
Galli, C., Bernasconi, R., Soldà, T., Calanca, V., and Molinari, M. (2011). Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PloS One 6, e16304.
Gauss, R., Kanehara, K., Carvalho, P., Ng, D.T.W., and Aebi, M. (2011). A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell 42, 782–793.
Ge, N., Muise, C.N., Gong, X., and Linsdell, P. (2004). Direct Comparison of the Functional Roles Played by Different Transmembrane Regions in the Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channel Pore. J. Biol. Chem. 279, 55283–55289.
Gee, H.Y., Noh, S.H., Tang, B.L., Kim, K.H., and Lee, M.G. (2011). Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760.

Références bibliographique
179
Gelman, M.S., Kannegaard, E.S., and Kopito, R.R. (2002). A Principal Role for the Proteasome in Endoplasmic Reticulum-associated Degradation of Misfolded Intracellular Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 277, 11709–11714.
Geva, Y., and Schuldiner, M. (2014a). The back and forth of cargo exit from the endoplasmic reticulum. Curr. Biol. CB 24, R130-136.
Geva, Y., and Schuldiner, M. (2014b). The Back and Forth of Cargo Exit from the Endoplasmic Reticulum. Curr. Biol. 24, R130–R136.
Gidalevitz, T., Stevens, F., and Argon, Y. (2013). Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1833, 2410–2424.
Gilbert, A., Jadot, M., Leontieva, E., Wattiaux-De Coninck, S., and Wattiaux, R. (1998). Delta F508 CFTR localizes in the endoplasmic reticulum-Golgi intermediate compartment in cystic fibrosis cells. Exp. Cell Res. 242, 144–152.
Glover, J.R., and Lindquist, S. (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82.
Glozman, R., Okiyoneda, T., Mulvihill, C.M., Rini, J.M., Barriere, H., and Lukacs, G.L. (2009). N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 184, 847–862.
Gnann, A., Riordan, J.R., and Wolf, D.H. (2004). Cystic Fibrosis Transmembrane Conductance Regulator Degradation Depends on the Lectins Htm1p/EDEM and the Cdc48 Protein Complex in Yeast. Mol. Biol. Cell 15, 4125–4135.
Goldberg, A.L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899.
Goldstein, R.F., Niraj, A., Sanderson, T.P., Wilson, L.S., Rab, A., Kim, H., Bebok, Z., and Collawn, J.F. (2007). VCP/p97 AAA-ATPase does not interact with the endogenous wild-type cystic fibrosis transmembrane conductance regulator. Am. J. Respir. Cell Mol. Biol. 36, 706–714.
Gomes-Alves, P., Couto, F., Pesquita, C., Coelho, A.V., and Penque, D. (2010). Rescue of F508del-CFTR by RXR motif inactivation triggers proteome modulation associated with the unfolded protein response. Biochim. Biophys. Acta BBA - Proteins Proteomics 1804, 856–865.
Gong, Q., Keeney, D.R., Molinari, M., and Zhou, Z. (2005). Degradation of Trafficking-defective Long QT Syndrome Type II Mutant Channels by the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 280, 19419–19425.
Gonzalez, D.S., Karaveg, K., Vandersall-Nairn, A.S., Lal, A., and Moremen, K.W. (1999). Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 274, 21375–21386.
Göthel, S.F., and Marahiel, M.A. (1999). Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell. Mol. Life Sci. CMLS 55, 423–436.

Références bibliographique
180
Groisman, B., Shenkman, M., Ron, E., and Lederkremer, G.Z. (2011). Mannose trimming is required for delivery of a glycoprotein from EDEM1 to XTP3-B and to late endoplasmic reticulum-associated degradation steps. J. Biol. Chem. 286, 1292–1300.
Grove, D.E., Rosser, M.F.N., Ren, H.Y., Naren, A.P., and Cyr, D.M. (2009). Mechanisms for Rescue of Correctable Folding Defects in CFTR F508. Mol. Biol. Cell 20, 4059–4069.
Grove, D.E., Fan, C.-Y., Ren, H.Y., and Cyr, D.M. (2011). The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol. Biol. Cell 22, 301–314.
Guerriero, C.J., and Brodsky, J.L. (2012). The Delicate Balance Between Secreted Protein Folding and Endoplasmic Reticulum-Associated Degradation in Human Physiology. Physiol. Rev. 92, 537–576.
Guiliano, D.B., Fussell, H., Lenart, I., Tsao, E., Nesbeth, D., Fletcher, A.J., Campbell, E.C., Yousaf, N., Williams, S., Santos, S., et al. (2014). Endoplasmic reticulum degradation-enhancing α-mannosidase-like protein 1 targets misfolded HLA-B27 dimers for endoplasmic reticulum-associated degradation. Arthritis Rheumatol. Hoboken NJ 66, 2976–2988.
de Haan, C.A.M., Molinari, M., and Reggiori, F. (2010). Autophagy-independent LC3 function in vesicular traffic. Autophagy 6, 994–996.
Hack, A.A., Lam, M.Y., Cordier, L., Shoturma, D.I., Ly, C.T., Hadhazy, M.A., Hadhazy, M.R., Sweeney, H.L., and McNally, E.M. (2000). Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci. 113 ( Pt 14), 2535–2544.
Hainzl, O., Lapina, M.C., Buchner, J., and Richter, K. (2009). The Charged Linker Region Is an Important Regulator of Hsp90 Function. J. Biol. Chem. 284, 22559–22567.
Harada, Y., Li, H., Li, H., and Lennarz, W.J. (2009). Oligosaccharyltransferase directly binds to ribosome at a location near the translocon-binding site. Proc. Natl. Acad. Sci. U. S. A. 106, 6945–6949.
Hara-Kuge, S., Ohkura, T., Ideo, H., Shimada, O., Atsumi, S., and Yamashita, K. (2002). Involvement of VIP36 in intracellular transport and secretion of glycoproteins in polarized Madin-Darby canine kidney (MDCK) cells. J. Biol. Chem. 277, 16332–16339.
Harding, H.P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274.
Harris, S.F., Shiau, A.K., and Agard, D.A. (2004). The Crystal Structure of the Carboxy-Terminal Dimerization Domain of htpG, the Escherichia coli Hsp90, Reveals a Potential Substrate Binding Site. Structure 12, 1087–1097.
Hauri, H., Appenzeller, C., Kuhn, F., and Nufer, O. (2000). Lectins and traffic in the secretory pathway. FEBS Lett. 476, 32–37.
He, L., Aleksandrov, L.A., Cui, L., Jensen, T.J., Nesbitt, K.L., and Riordan, J.R. (2010). Restoration of domain folding and interdomain assembly by second-site suppressors of the DeltaF508 mutation in CFTR. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 24, 3103–3112.

Références bibliographique
181
He, L., Kota, P., Aleksandrov, A.A., Cui, L., Jensen, T., Dokholyan, N.V., and Riordan, J.R. (2013). Correctors of F508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 27, 536–545.
He, L., Aleksandrov, A.A., An, J., Cui, L., Yang, Z., Brouillette, C.G., and Riordan, J.R. (2015). Restoration of NBD1 Thermal Stability Is Necessary and Sufficient to Correct ∆F508 CFTR Folding and Assembly. J. Mol. Biol. 427, 106–120.
Hebert, D.N., and Molinari, M. (2007). In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377–1408.
Hebert, D.N., and Molinari, M. (2012). Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci. 37, 404–410.
Hebert, D.N., Foellmer, B., and Helenius, A. (1995). Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81, 425–433.
Hebert, D.N., Foellmer, B., and Helenius, A. (1996). Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of influenza hemagglutinin in microsomes. EMBO J. 15, 2961–2968.
Hebert, D.N., Zhang, J.-X., Chen, W., Foellmer, B., and Helenius, A. (1997). The Number and Location of Glycans on Influenza Hemagglutinin Determine Folding and Association with Calnexin and Calreticulin. J. Cell Biol. 139, 613–623.
Helenius, A., and Aebi, M. (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049.
Hellenkamp, B., Wortmann, P., Kandzia, F., Zacharias, M., and Hugel, T. (2017). Multidomain structure and correlated dynamics determined by self-consistent FRET networks. Nat. Methods 14, 174–180.
Hernández, M.P., Chadli, A., and Toft, D.O. (2002). HSP40 binding is the first step in the HSP90 chaperoning pathway for the progesterone receptor. J. Biol. Chem. 277, 11873–11881.
Herscovics, A. (2001). Structure and function of Class I alpha 1,2-mannosidases involved in glycoprotein synthesis and endoplasmic reticulum quality control. Biochimie 83, 757–762.
Hirao, K., Natsuka, Y., Tamura, T., Wada, I., Morito, D., Natsuka, S., Romero, P., Sleno, B., Tremblay, L.O., Herscovics, A., et al. (2006). EDEM3, a Soluble EDEM Homolog, Enhances Glycoprotein Endoplasmic Reticulum-associated Degradation and Mannose Trimming. J. Biol. Chem. 281, 9650–9658.
Hirsch, C., Gauss, R., Horn, S.C., Neuber, O., and Sommer, T. (2009). The ubiquitylation machinery of the endoplasmic reticulum. Nature 458, 453–460.
Hoang, H.-D., Maruyama, J., and Kitamoto, K. (2015). Modulating Endoplasmic Reticulum-Golgi Cargo Receptors for Improving Secretion of Carrier-Fused Heterologous Proteins in the Filamentous Fungus Aspergillus oryzae. Appl. Environ. Microbiol. 81, 533–543.

Références bibliographique
182
Hohfeld, J., Cyr, D.M., and Patterson, C. (2001). From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2, 885–890.
Hosokawa, N., Wada, I., Hasegawa, K., Yorihuzi, T., Tremblay, L.O., Herscovics, A., and Nagata, K. (2001). A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2, 415–422.
Hosokawa, N., Tremblay, L.O., You, Z., Herscovics, A., Wada, I., and Nagata, K. (2003). Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 278, 26287–26294.
Hosokawa, N., Wada, I., Natsuka, Y., and Nagata, K. (2006). EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded alpha1-antitrypsin. Genes Cells Devoted Mol. Cell. Mech. 11, 465–476.
Hosokawa, N., Wada, I., Nagasawa, K., Moriyama, T., Okawa, K., and Nagata, K. (2008). Human XTP3-B Forms an Endoplasmic Reticulum Quality Control Scaffold with the HRD1-SEL1L Ubiquitin Ligase Complex and BiP. J. Biol. Chem. 283, 20914–20924.
Hosokawa, N., Kamiya, Y., Kamiya, D., Kato, K., and Nagata, K. (2009). Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 284, 17061–17068.
Hosokawa, N., Tremblay, L.O., Sleno, B., Kamiya, Y., Wada, I., Nagata, K., Kato, K., and Herscovics, A. (2010). EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 20, 567–575.
Houck, S.A., Singh, S., and Cyr, D.M. (2012). Cellular Responses to Misfolded Proteins and Protein Aggregates. In Ubiquitin Family Modifiers and the Proteasome, R.J. Dohmen, and M. Scheffner, eds. (Totowa, NJ: Humana Press), pp. 455–461.
Houck, S.A., Ren, H.Y., Madden, V.J., Bonner, J.N., Conlin, M.P., Janovick, J.A., Conn, P.M., and Cyr, D.M. (2014). Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol. Cell 54, 166–179.
Hsu, V.W., Yuan, L.C., Nuchtern, J.G., Lippincott-Schwartz, J., Hammerling, G.J., and Klausner, R.D. (1991). A recycling pathway between the endoplasmic reticulum and the Golgi apparatus for retention of unassembled MHC class I molecules. Nature 352, 441–444.
Hudson, R.P., Dawson, J.E., Chong, P.A., Yang, Z., Millen, L., Thomas, P.J., Brouillette, C.G., and Forman-Kay, J.D. (2017). Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 92, 124–135.
Husnjak, K., Elsasser, S., Zhang, N., Chen, X., Randles, L., Shi, Y., Hofmann, K., Walters, K.J., Finley, D., and Dikic, I. (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488.
Hutt, D.M., Roth, D.M., Chalfant, M.A., Youker, R.T., Matteson, J., Brodsky, J.L., and Balch, W.E. (2012). FK506 Binding Protein 8 Peptidylprolyl Isomerase Activity Manages a Late Stage of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Folding and Stability. J. Biol. Chem. 287, 21914–21925.

Références bibliographique
183
Huyer, G., Longsworth, G.L., Mason, D.L., Mallampalli, M.P., McCaffery, J.M., Wright, R.L., and Michaelis, S. (2004). A striking quality control subcompartment in Saccharomyces cerevisiae: the endoplasmic reticulum-associated compartment. Mol. Biol. Cell 15, 908–921.
Hwang, T.-C., and Kirk, K.L. (2013). The CFTR Ion Channel: Gating, Regulation, and Anion Permeation. Cold Spring Harb. Perspect. Med. 3, a009498–a009498.
Iannotti, M.J., Figard, L., Sokac, A.M., and Sifers, R.N. (2014). A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J. Biol. Chem. 289, 11844–11858.
Immormino, R.M., Metzger, L.E., Reardon, P.N., Dollins, D.E., Blagg, B.S.J., and Gewirth, D.T. (2009). Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J. Mol. Biol. 388, 1033–1042.
Imperiali, B., and O’Connor, S.E. (1999). Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 3, 643–649.
Irvine, A.G., Wallis, A.K., Sanghera, N., Rowe, M.L., Ruddock, L.W., Howard, M.J., Williamson, R.A., Blindauer, C.A., and Freedman, R.B. (2014). Protein Disulfide-Isomerase Interacts with a Substrate Protein at All Stages along Its Folding Pathway. PLoS ONE 9, e82511.
Jackson, S.E. (2012). Hsp90: Structure and Function. In Molecular Chaperones, S. Jackson, ed. (Berlin, Heidelberg: Springer Berlin Heidelberg), pp. 155–240.
Jacob, P., Hirt, H., and Bendahmane, A. (2017). The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol. J. 15, 405–414.
Jahn, M., Rehn, A., Pelz, B., Hellenkamp, B., Richter, K., Rief, M., Buchner, J., and Hugel, T. (2014). The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. 111, 17881–17886.
Jakob, C.A., Bodmer, D., Spirig, U., Battig, P., Marcil, A., Dignard, D., Bergeron, J.J., Thomas, D.Y., and Aebi, M. (2001). Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2, 423–430.
Jansen, G., Maattanen, P., Denisov, A.Y., Scarffe, L., Schade, B., Balghi, H., Dejgaard, K., Chen, L.Y., Muller, W.J., Gehring, K., et al. (2012). An Interaction Map of Endoplasmic Reticulum Chaperones and Foldases. Mol. Cell. Proteomics 11, 710–723.
Jessop, C.E., Chakravarthi, S., Garbi, N., Hämmerling, G.J., Lovell, S., and Bulleid, N.J. (2007). ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 26, 28–40.
Jessop, C.E., Watkins, R.H., Simmons, J.J., Tasab, M., and Bulleid, N.J. (2009). Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J. Cell Sci. 122, 4287–4295.
Jiang, C., Fang, S.L., Xiao, Y.F., O’Connor, S.P., Nadler, S.G., Lee, D.W., Jefferson, D.M., Kaplan, J.M., Smith, A.E., and Cheng, S.H. (1998). Partial restoration of cAMP-stimulated

Références bibliographique
184
CFTR chloride channel activity in DeltaF508 cells by deoxyspergualin. Am. J. Physiol. 275, C171-178.
Jitsuhara, Y., Toyoda, T., Itai, T., and Yamaguchi, H. (2002). Chaperone-like functions of high-mannose type and complex-type N-glycans and their molecular basis. J. Biochem. (Tokyo) 132, 803–811.
Jovov, B., Ismailov, I.I., Berdiev, B.K., Fuller, C.M., Sorscher, E.J., Dedman, J.R., Kaetzel, M.A., and Benos, D.J. (1995). Interaction between cystic fibrosis transmembrane conductance regulator and outwardly rectified chloride channels. J. Biol. Chem. 270, 29194–29200.
Kahle, P.J., and Haass, C. (2004). How does parkin ligate ubiquitin to Parkinson’s disease? EMBO Rep. 5, 681–685.
Kamiya, Y., Yamaguchi, Y., Takahashi, N., Arata, Y., Kasai, K., Ihara, Y., Matsuo, I., Ito, Y., Yamamoto, K., and Kato, K. (2005). Sugar-binding Properties of VIP36, an Intracellular Animal Lectin Operating as a Cargo Receptor. J. Biol. Chem. 280, 37178–37182.
Kamiya, Y., Kamiya, D., Yamamoto, K., Nyfeler, B., Hauri, H.-P., and Kato, K. (2008). Molecular Basis of Sugar Recognition by the Human L-type Lectins ERGIC-53, VIPL, and VIP36. J. Biol. Chem. 283, 1857–1861.
Kerbiriou, M., Le Drévo, M.-A., Férec, C., and Trouvé, P. (2007). Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 1772, 1236–1249.
Kerem, B., Rommens, J.M., Buchanan, J.A., Markiewicz, D., Cox, T.K., Chakravarti, A., Buchwald, M., and Tsui, L.C. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080.
Kiefhaber, T., Quaas, R., Hahn, U., and Schmid, F.X. (1990). Folding of ribonuclease T1. 2. Kinetic models for the folding and unfolding reactions. Biochemistry (Mosc.) 29, 3061–3070.
Kim, S.J., and Skach, W.R. (2012). Mechanisms of CFTR Folding at the Endoplasmic Reticulum. Front. Pharmacol. 3.
Kimura, Y., Yahara, I., and Lindquist, S. (1995). Role of the protein chaperone YDJ1 in establishing Hsp90-mediated signal transduction pathways. Science 268, 1362–1365.
Kirschke, E., Goswami, D., Southworth, D., Griffin, P.R., and Agard, D.A. (2014). Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 157, 1685–1697.
Kityk, R., Kopp, J., Sinning, I., and Mayer, M.P. (2012). Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 48, 863–874.
Kleizen, B., van Vlijmen, T., de Jonge, H.R., and Braakman, I. (2005). Folding of CFTR Is Predominantly Cotranslational. Mol. Cell 20, 277–287.
Koedel, U., Frankenberg, T., Kirschnek, S., Obermaier, B., Häcker, H., Paul, R., and Häcker, G. (2009). Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 5, e1000461.

Références bibliographique
185
Kojima, R., Okumura, M., and Inaba, K. (2013). Structural Basis of Disulfide Bond Formation in the Bacterial Periplasm and Mammalian ER. In eLS, John Wiley & Sons Ltd, ed. (Chichester, UK: John Wiley & Sons, Ltd), p.
Kondratyev, M., Avezov, E., Shenkman, M., Groisman, B., and Lederkremer, G.Z. (2007). PERK-dependent compartmentalization of ERAD and unfolded protein response machineries during ER stress. Exp. Cell Res. 313, 3395–3407.
Kosano, H., Stensgard, B., Charlesworth, M.C., McMahon, N., and Toft, D. (1998). The Assembly of Progesterone Receptor-hsp90 Complexes Using Purified Proteins. J. Biol. Chem. 273, 32973–32979.
Kosmaoglou, M., Kanuga, N., Aguilà, M., Garriga, P., and Cheetham, M.E. (2009). A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 122, 4465–4472.
Koulov, A.V., LaPointe, P., Lu, B., Razvi, A., Coppinger, J., Dong, M.-Q., Matteson, J., Laister, R., Arrowsmith, C., Yates, J.R., et al. (2010). Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 21, 871–884.
Kozlov, G., Maattanen, P., Schrag, J.D., Pollock, S., Cygler, M., Nagar, B., Thomas, D.Y., and Gehring, K. (2006). Crystal structure of the bb’ domains of the protein disulfide isomerase ERp57. Struct. Lond. Engl. 1993 14, 1331–1339.
Kozlov, G., Määttänen, P., Thomas, D.Y., and Gehring, K. (2010a). A structural overview of the PDI family of proteins: PDI family of proteins. FEBS J. 277, 3924–3936.
Kozlov, G., Bastos-Aristizabal, S., Maattanen, P., Rosenauer, A., Zheng, F., Killikelly, A., Trempe, J.-F., Thomas, D.Y., and Gehring, K. (2010b). Structural Basis of Cyclophilin B Binding by the Calnexin/Calreticulin P-domain. J. Biol. Chem. 285, 35551–35557.
Kozlov, G., Pocanschi, C.L., Rosenauer, A., Bastos-Aristizabal, S., Gorelik, A., Williams, D.B., and Gehring, K. (2010c). Structural basis of carbohydrate recognition by calreticulin. J. Biol. Chem. 285, 38612–38620.
Kukushkin, N.V., Alonzi, D.S., Dwek, R.A., and Butters, T.D. (2011). Demonstration that endoplasmic reticulum-associated degradation of glycoproteins can occur downstream of processing by endomannosidase. Biochem. J. 438, 133–142.
Kundra, R., and Kornfeld, S. (1999). Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J. Biol. Chem. 274, 31039–31046.
Kunzelmann, K. (2001). CFTR: interacting with everything? News Physiol. Sci. Int. J. Physiol. Prod. Jointly Int. Union Physiol. Sci. Am. Physiol. Soc. 16, 167–170.
Lai, C.W., Otero, J.H., Hendershot, L.M., and Snapp, E. (2012). ERdj4 Protein Is a Soluble Endoplasmic Reticulum (ER) DnaJ Family Protein That Interacts with ER-associated Degradation Machinery. J. Biol. Chem. 287, 7969–7978.
Lam, Y.A., Lawson, T.G., Velayutham, M., Zweier, J.L., and Pickart, C.M. (2002). A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 416, 763–767.

Références bibliographique
186
Lamriben, L., Graham, J.B., Adams, B.M., and Hebert, D.N. (2016). N -Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle: The Calnexin Binding Cycle. Traffic 17, 308–326.
Lannoo, N., and Van Damme, E.J.M. (2015). Review/N-glycans: The making of a varied toolbox. Plant Sci. Int. J. Exp. Plant Biol. 239, 67–83.
Le Fourn, V., Gaplovska-Kysela, K., Guhl, B., Santimaria, R., Zuber, C., and Roth, J. (2009). Basal autophagy is involved in the degradation of the ERAD component EDEM1. Cell. Mol. Life Sci. CMLS 66, 1434–1445.
Le Fourn, V., Park, S., Jang, I., Gaplovska-Kysela, K., Guhl, B., Lee, Y., Cho, J.W., Zuber, C., and Roth, J. (2013). Large protein complexes retained in the ER are dislocated by non-COPII vesicles and degraded by selective autophagy. Cell. Mol. Life Sci. CMLS 70, 1985–2002.
Lederkremer, G.Z., Cheng, Y., Petre, B.M., Vogan, E., Springer, S., Schekman, R., Walz, T., and Kirchhausen, T. (2001). Structure of the Sec23p/24p and Sec13p/31p complexes of COPII. Proc. Natl. Acad. Sci. 98, 10704–10709.
Lee, M.G., Wigley, W.C., Zeng, W., Noel, L.E., Marino, C.R., Thomas, P.J., and Muallem, S. (1999). Regulation of Cl-/ HCO3- exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J. Biol. Chem. 274, 3414–3421.
Lee, S.-O., Cho, K., Cho, S., Kim, I., Oh, C., and Ahn, K. (2010). Protein disulphide isomerase is required for signal peptide peptidase-mediated protein degradation. EMBO J. 29, 363–375.
Leitman, J., Ron, E., Ogen-Shtern, N., and Lederkremer, G.Z. (2013). Compartmentalization of endoplasmic reticulum quality control and ER-associated degradation factors. DNA Cell Biol. 32, 2–7.
Leitman, J., Shenkman, M., Gofman, Y., Shtern, N.O., Ben-Tal, N., Hendershot, L.M., and Lederkremer, G.Z. (2014). Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 25, 1050–1060.
Lepage, M.L., Mirloup, A., Ripoll, M., Stauffert, F., Bodlenner, A., Ziessel, R., and Compain, P. (2015). Design, synthesis and photochemical properties of the first examples of iminosugar clusters based on fluorescent cores. Beilstein J. Org. Chem. 11, 659–667.
Letz, B., and Korbmacher, C. (1997). cAMP stimulates CFTR-like Cl- channels and inhibits amiloride-sensitive Na+ channels in mouse CCD cells. Am. J. Physiol. 272, C657-666.
Lewis, H.A., Buchanan, S.G., Burley, S.K., Conners, K., Dickey, M., Dorwart, M., Fowler, R., Gao, X., Guggino, W.B., Hendrickson, W.A., et al. (2004). Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23, 282–293.
Lewis, H.A., Wang, C., Zhao, X., Hamuro, Y., Conners, K., Kearins, M.C., Lu, F., Sauder, J.M., Molnar, K.S., Coales, S.J., et al. (2010). Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 396, 406–430.

Références bibliographique
187
Li, Z., and Srivastava, P. (2004). Heat-Shock Proteins. In Current Protocols in Immunology, J.E. Coligan, B.E. Bierer, D.H. Margulies, E.M. Shevach, and W. Strober, eds. (Hoboken, NJ, USA: John Wiley & Sons, Inc.), p.
Li, J., Ni, M., Lee, B., Barron, E., Hinton, D.R., and Lee, A.S. (2008). The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 15, 1460–1471.
Li, J., Richter, K., and Buchner, J. (2011). Mixed Hsp90–cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Biol. 18, 61–66.
Li, J., Soroka, J., and Buchner, J. (2012). The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1823, 624–635.
Lim, P.J., Danner, R., Liang, J., Doong, H., Harman, C., Srinivasan, D., Rothenberg, C., Wang, H., Ye, Y., Fang, S., et al. (2009). Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J. Cell Biol. 187, 201–217.
Linsdell, P., and Hanrahan, J.W. (1998). Glutathione permeability of CFTR. Am. J. Physiol. 275, C323-326.
Linsdell, P. (2006). Mechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. Exp. Physiol. 91, 123–129.
Liu, Y., Choudhury, P., Cabral, C.M., and Sifers, R.N. (1997). Intracellular Disposal of Incompletely Folded Human α 1 -Antitrypsin Involves Release from Calnexin and Post-translational Trimming of Asparagine-linked Oligosaccharides. J. Biol. Chem. 272, 7946–7951.
Liu, Y., Choudhury, P., Cabral, C.M., and Sifers, R.N. (1999). Oligosaccharide Modification in the Early Secretory Pathway Directs the Selection of a Misfolded Glycoprotein for Degradation by the Proteasome. J. Biol. Chem. 274, 5861–5867.
Loo, M.A., Jensen, T.J., Cui, L., Hou, Y., Chang, X.-B., and Riordan, J.R. (1998). Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 17, 6879–6887.
Loo, T.W., Bartlett, M.C., and Clarke, D.M. (2008). Processing Mutations Disrupt Interactions between the Nucleotide Binding and Transmembrane Domains of P-glycoprotein and the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). J. Biol. Chem. 283, 28190–28197.
Loo, T.W., Bartlett, M.C., and Clarke, D.M. (2013). Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 86, 612–619.
Lopes-Pacheco, M. (2016). CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 7.
Lu, M., Leng, M., Egan, M., Caplan, M., Boulpaep, E.L., Giebisch, G.H., and Steven, C.H. (2006). CFTR is required for PKA-regulated ATP sensitivity of Kir1.1 potassium channels in mouse kidney. J. Clin. Invest. 116, 797–807.

Références bibliographique
188
Lu, Y., Xiong, X., Helm, A., Kimani, K., Bragin, A., and Skach, W.R. (1998). Co- and Posttranslational Translocation Mechanisms Direct Cystic Fibrosis Transmembrane Conductance Regulator N Terminus Transmembrane Assembly. J. Biol. Chem. 273, 568–576.
Lubamba, B., Lebacq, J., Lebecque, P., Vanbever, R., Leonard, A., Wallemacq, P., and Leal, T. (2009). Airway Delivery of Low-Dose Miglustat Normalizes Nasal Potential Difference in F508del Cystic Fibrosis Mice. Am. J. Respir. Crit. Care Med. 179, 1022–1028.
Lukacs, G.L., Chang, X.B., Bear, C., Kartner, N., Mohamed, A., Riordan, J.R., and Grinstein, S. (1993). The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 268, 21592–21598.
Lukacs, G.L., Mohamed, A., Kartner, N., Chang, X.B., Riordan, J.R., and Grinstein, S. (1994). Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 13, 6076–6086.
Ma, W., Goldberg, E., and Goldberg, J. (2017). ER retention is imposed by COPII protein sorting and attenuated by 4-phenylbutyrate. eLife 6.
Määttänen, P., Gehring, K., Bergeron, J.J.M., and Thomas, D.Y. (2010). Protein quality control in the ER: the recognition of misfolded proteins. Semin. Cell Dev. Biol. 21, 500–511.
Marcus, N.Y., and Perlmutter, D.H. (2000). Glucosidase and Mannosidase Inhibitors Mediate Increased Secretion of Mutant α1 Antitrypsin Z. J. Biol. Chem. 275, 1987–1992.
Marin, M.B., Ghenea, S., Spiridon, L.N., Chiritoiu, G.N., Petrescu, A.-J., and Petrescu, S.-M. (2012). Tyrosinase degradation is prevented when EDEM1 lacks the intrinsically disordered region. PloS One 7, e42998.
Marzec, M., Eletto, D., and Argon, Y. (2012). GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1823, 774–787.
Mast, S.W., Diekman, K., Karaveg, K., Davis, A., Sifers, R.N., and Moremen, K.W. (2005). Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 15, 421–436.
Matsumura, Y., Sakai, J., and Skach, W.R. (2013). Endoplasmic Reticulum Protein Quality Control Is Determined by Cooperative Interactions between Hsp/c70 Protein and the CHIP E3 Ligase. J. Biol. Chem. 288, 31069–31079.
Mayer, M.P., and Bukau, B. (2005). Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684.
Mayer, M.P., and Le Breton, L. (2015). Hsp90: Breaking the Symmetry. Mol. Cell 58, 8–20.
Mayer, M.P., Schröder, H., Rüdiger, S., Paal, K., Laufen, T., and Bukau, B. (2000). Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 7, 586–593.

Références bibliographique
189
Mazola, Y., Chinea, G., and Musacchio, A. (2011). Integrating Bioinformatics Tools to Handle Glycosylation. PLoS Comput. Biol. 7, e1002285.
McCaffrey, K., and Braakman, I. (2016). Protein quality control at the endoplasmic reticulum. Essays Biochem. 60, 227–235.
McClung, J.P., Hasday, J.D., He, J.-R., Montain, S.J., Cheuvront, S.N., Sawka, M.N., and Singh, I.S. (2008). Exercise-heat acclimation in humans alters baseline levels and ex vivo heat inducibility of HSP72 and HSP90 in peripheral blood mononuclear cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R185-191.
McNicholas, C.M., Guggino, W.B., Schwiebert, E.M., Hebert, S.C., Giebisch, G., and Egan, M.E. (1996). Sensitivity of a renal K+ channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cystic fibrosis transmembrane regulator. Proc. Natl. Acad. Sci. U. S. A. 93, 8083–8088.
Meacham, G.C. (1999). The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 18, 1492–1505.
Meacham, G.C., Patterson, C., Zhang, W., Younger, J.M., and Cyr, D.M. (2001a). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105.
Meacham, G.C., Patterson, C., Zhang, W., Younger, J.M., and Cyr, D.M. (2001b). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105.
Meijer, L., Nelson, D.J., Riazanski, V., Gabdoulkhakova, A.G., Hery-Arnaud, G., Le Berre, R., Loaëc, N., Oumata, N., Galons, H., Nowak, E., et al. (2016). Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 8, 330–349.
Mendes, F., Farinha, C.M., Felício, V., Alves, P.C., Vieira, I., and Amaral, M.D. (2012). BAG-1 Stabilizes Mutant F508del-CFTR in a Ubiquitin-Like-Domain-Dependent Manner. Cell. Physiol. Biochem. 30, 1120–1133.
Mendoza, J.L., Schmidt, A., Li, Q., Nuvaga, E., Barrett, T., Bridges, R.J., Feranchak, A.P., Brautigam, C.A., and Thomas, P.J. (2012). Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174.
Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005). ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772.
Meyer, P., Prodromou, C., Liao, C., Hu, B., Mark Roe, S., Vaughan, C.K., Vlasic, I., Panaretou, B., Piper, P.W., and Pearl, L.H. (2004). Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 511–519.
Michalak, M., Milner, R.E., Burns, K., and Opas, M. (1992). Calreticulin. Biochem. J. 285 ( Pt
3), 681–692.

Références bibliographique
190
Michalak, M., Groenendyk, J., Szabo, E., Gold, L.I., and Opas, M. (2009). Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666.
Michelsen, K., Yuan, H., and Schwappach, B. (2005). Hide and run. Arginine‐based endoplasmic‐reticulum‐sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 6, 717–722.
Miller, E., Antonny, B., Hamamoto, S., and Schekman, R. (2002). Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 21, 6105–6113.
Miller, E.A., Beilharz, T.H., Malkus, P.N., Lee, M.C.S., Hamamoto, S., Orci, L., and Schekman, R. (2003). Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114, 497–509.
Molinari, M. (2007). N-glycan structure dictates extension of protein folding or onset of disposal. Nat. Chem. Biol. 3, 313–320.
Molinari, M., Calanca, V., Galli, C., Lucca, P., and Paganetti, P. (2003). Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400.
Moran, O. (2017). The gating of the CFTR channel. Cell. Mol. Life Sci. 74, 85–92.
Moremen, K.W. (2002). Golgi α-mannosidase II deficiency in vertebrate systems: implications for asparagine-linked oligosaccharide processing in mammals. Biochim. Biophys. Acta BBA - Gen. Subj. 1573, 225–235.
Morito, D., Hirao, K., Oda, Y., Hosokawa, N., Tokunaga, F., Cyr, D.M., Tanaka, K., Iwai, K., and Nagata, K. (2008). Gp78 Cooperates with RMA1 in Endoplasmic Reticulum-associated Degradation of CFTR F508. Mol. Biol. Cell 19, 1328–1336.
Morris, A.P., Cunningham, S.A., Benos, D.J., and Frizzell, R.A. (1993). Glycosylation status of endogenous CFTR does not affect cAMP-stimulated Cl- secretion in epithelial cells. Am. J. Physiol. 265, C688-694.
Murata, S., Chiba, T., and Tanaka, K. (2003). CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int. J. Biochem. Cell Biol. 35, 572–578.
Nadeau, K., Nadler, S.G., Saulnier, M., Tepper, M.A., and Walsh, C.T. (1994). Quantitation of the interaction of the immunosuppressant deoxyspergualin and analogs with Hsc70 and Hsp90. Biochemistry (Mosc.) 33, 2561–2567.
Nadler, S.G., Tepper, M.A., Schacter, B., and Mazzucco, C.E. (1992). Interaction of the immunosuppressant deoxyspergualin with a member of the Hsp70 family of heat shock proteins. Science 258, 484–486.
Nanua, S., Sajjan, U., Keshavjee, S., and Hershenson, M.B. (2006). Absence of typical unfolded protein response in primary cultured cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 343, 135–143.
Nawa, D., Shimada, O., Kawasaki, N., Matsumoto, N., and Yamamoto, K. (2007). Stable interaction of the cargo receptor VIP36 with molecular chaperone BiP. Glycobiology 17, 913–921.

Références bibliographique
191
Needham, P.G., and Brodsky, J.L. (2013). How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: The early history of ERAD. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1833, 2447–2457.
Neve, E. (2003). VIPL, a VIP36-like membrane protein with a putative function in the export of glycoproteins from the endoplasmic reticulum⋆. Exp. Cell Res. 288, 70–83.
Neve, E.P.A., Svensson, K., Fuxe, J., and Pettersson, R.F. (2003). VIPL, a VIP36-like membrane protein with a putative function in the export of glycoproteins from the endoplasmic reticulum. Exp. Cell Res. 288, 70–83.
Ng, D.T., and Walter, P. (1994). Protein translocation across the endoplasmic reticulum. Curr. Opin. Cell Biol. 6, 510–516.
Ninagawa, S., Okada, T., Sumitomo, Y., Kamiya, Y., Kato, K., Horimoto, S., Ishikawa, T., Takeda, S., Sakuma, T., Yamamoto, T., et al. (2014). EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 206, 347–356.
Nishimura, I., Uchida, M., Inohana, Y., Setoh, K., Daba, K., Nishimura, S., and Yamaguchi, H. (1998). Oxidative refolding of bovine pancreatic RNases A and B promoted by Asn-glycans. J. Biochem. (Tokyo) 123, 516–520.
Nishitoh, H. (2012). CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. (Tokyo) 151, 217–219.
Noel, S., Wilke, M., Bot, A.G.M., De Jonge, H.R., and Becq, F. (2008). Parallel Improvement of Sodium and Chloride Transport Defects by Miglustat (n-Butyldeoxynojyrimicin) in Cystic Fibrosis Epithelial Cells. J. Pharmacol. Exp. Ther. 325, 1016–1023.
Norez, C., Noel, S., Wilke, M., Bijvelds, M., Jorna, H., Melin, P., DeJonge, H., and Becq, F. (2006a). Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the α-glucosidase inhibitor miglustat. FEBS Lett. 580, 2081–2086.
Norez, C., Antigny, F., Becq, F., and Vandebrouck, C. (2006b). Maintaining Low Ca2+ Level in the Endoplasmic Reticulum Restores Abnormal Endogenous F508del-CFTR Trafficking in Airway Epithelial Cells: Low Ca2+ Levels Restore Endogenous F508del-CFTR Trafficking. Traffic 7, 562–573.
Norez, C., Pasetto, M., Dechecchi, M.C., Barison, E., Anselmi, C., Tamanini, A., Quiri, F., Cattel, L., Rizzotti, P., Dosio, F., et al. (2008). Chemical conjugation of DeltaF508-CFTR corrector deoxyspergualin to transporter human serum albumin enhances its ability to rescue Cl- channel functions. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L336-347.
Norez, C., Antigny, F., Noel, S., Vandebrouck, C., and Becq, F. (2009). A Cystic Fibrosis Respiratory Epithelial Cell Chronically Treated by Miglustat Acquires a Non–Cystic Fibrosis–Like Phenotype. Am. J. Respir. Cell Mol. Biol. 41, 217–225.
Norez, C., Vandebrouck, C., Bertrand, J., Noel, S., Durieu, E., Oumata, N., Galons, H., Antigny, F., Chatelier, A., Bois, P., et al. (2014). Roscovitine is a proteostasis regulator that corrects the trafficking defect of F508del-CFTR by a CDK-independent mechanism: Roscovitine as a novel corrector of F508del-CFTR. Br. J. Pharmacol. 171, 4831–4849.

Références bibliographique
192
Nufer, O., Mitrovic, S., and Hauri, H.-P. (2003). Profile-based data base scanning for animal L-type lectins and characterization of VIPL, a novel VIP36-like endoplasmic reticulum protein. J. Biol. Chem. 278, 15886–15896.
Nunes, J.M., Mayer-Hartl, M., Hartl, F.U., and Müller, D.J. (2015). Action of the Hsp70 chaperone system observed with single proteins. Nat. Commun. 6, 6307.
O’Connor, S.E., and Imperiali, B. (1996). Modulation of protein structure and function by asparagine-linked glycosylation. Chem. Biol. 3, 803–812.
Oda, Y., Hosokawa, N., Wada, I., and Nagata, K. (2003). EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397.
Okiyoneda, T., Harada, K., Takeya, M., Yamahira, K., Wada, I., Shuto, T., Suico, M.A., Hashimoto, Y., and Kai, H. (2004). ΔF508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol. Biol. Cell 15, 563–574.
Okiyoneda, T., Niibori, A., Harada, K., Kohno, T., Michalak, M., Duszyk, M., Wada, I., Ikawa, M., Shuto, T., Suico, M.A., et al. (2008). Role of calnexin in the ER quality control and productive folding of CFTR; differential effect of calnexin knockout on wild-type and ΔF508 CFTR. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1783, 1585–1594.
Okiyoneda, T., Veit, G., Dekkers, J.F., Bagdany, M., Soya, N., Xu, H., Roldan, A., Verkman, A.S., Kurth, M., Simon, A., et al. (2013). Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 9, 444–454.
Okumura, M., Kadokura, H., and Inaba, K. (2015). Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 83, 314–322.
Olivari, S., and Molinari, M. (2007a). Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 581, 3658–3664.
Olivari, S., and Molinari, M. (2007b). Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 581, 3658–3664.
Olivari, S., Galli, C., Alanen, H., Ruddock, L., and Molinari, M. (2005). A Novel Stress-induced EDEM Variant Regulating Endoplasmic Reticulum-associated Glycoprotein Degradation. J. Biol. Chem. 280, 2424–2428.
Olivari, S., Cali, T., Salo, K.E.H., Paganetti, P., Ruddock, L.W., and Molinari, M. (2006). EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 349, 1278–1284.
Oliver, J.D., van der Wal, F.J., Bulleid, N.J., and High, S. (1997). Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 275, 86–88.
Oliver, J.D., Roderick, H.L., Llewellyn, D.H., and High, S. (1999). ERp57 Functions as a Subunit of Specific Complexes Formed with the ER Lectins Calreticulin and Calnexin. Mol. Biol. Cell 10, 2573–2582.

Références bibliographique
193
Ostedgaard, L.S., Rich, D.P., DeBerg, L.G., and Welsh, M.J. (1997). Association of Domains within the Cystic Fibrosis Transmembrane Conductance Regulator †. Biochemistry (Mosc.) 36, 1287–1294.
Otero, J.H., Lizák, B., and Hendershot, L.M. (2010). Life and death of a BiP substrate. Semin. Cell Dev. Biol. 21, 472–478.
Pan, S., Wang, S., Utama, B., Huang, L., Blok, N., Estes, M.K., Moremen, K.W., and Sifers, R.N. (2011). Golgi localization of ERManI defines spatial separation of the mammalian glycoprotein quality control system. Mol. Biol. Cell 22, 2810–2822.
Pan, S., Cheng, X., and Sifers, R.N. (2013). Golgi-situated endoplasmic reticulum α-1, 2-mannosidase contributes to the retrieval of ERAD substrates through a direct interaction with γ-COP. Mol. Biol. Cell 24, 1111–1121.
Panaretou, B., Siligardi, G., Meyer, P., Maloney, A., Sullivan, J.K., Singh, S., Millson, S.H., Clarke, P.A., Naaby-Hansen, S., Stein, R., et al. (2002). Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 10, 1307–1318.
Parakh, S., and Atkin, J.D. (2015). Novel roles for protein disulphide isomerase in disease states: a double edged sword? Front. Cell Dev. Biol. 3.
Park, S., Jang, I., Zuber, C., Lee, Y., Cho, J.W., Matsuo, I., Ito, Y., and Roth, J. (2014). ERADication of EDEM1 occurs by selective autophagy and requires deglycosylation by cytoplasmic peptide N-glycanase. Histochem. Cell Biol. 142, 153–169.
Patrick, A.E., Karamyshev, A.L., Millen, L., and Thomas, P.J. (2011). Alteration of CFTR transmembrane span integration by disease-causing mutations. Mol. Biol. Cell 22, 4461–4471.
Peng, H.-M., Morishima, Y., Clapp, K.M., Lau, M., Pratt, W.B., and Osawa, Y. (2009). Dynamic Cycling with Hsp90 Stabilizes Neuronal Nitric Oxide Synthase Through Calmodulin-Dependent Inhibition of Ubiquitination. Biochemistry (Mosc.) 48, 8483–8490.
Petrescu, A.-J., Milac, A.-L., Petrescu, S.M., Dwek, R.A., and Wormald, M.R. (2004). Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology 14, 103–114.
Philippe, R., Antigny, F., Buscaglia, P., Norez, C., Huguet, F., Castelbou, C., Trouvé, P., Becq, F., Frieden, M., Férec, C., et al. (2017). Calumenin contributes to ER-Ca2+ homeostasis in bronchial epithelial cells expressing WT and F508del mutated CFTR and to F508del-CFTR retention. Cell Calcium 62, 47–59.
Pietrement, C., Da Silva, N., Silberstein, C., James, M., Marsolais, M., Van Hoek, A., Brown, D., Pastor-Soler, N., Ameen, N., Laprade, R., et al. (2008). Role of NHERF1, cystic fibrosis transmembrane conductance regulator, and cAMP in the regulation of aquaporin 9. J. Biol. Chem. 283, 2986–2996.
Pilewski, J.M., Cooke, J., Lekstrom-Himes, J., and Donaldson, S. (2015). WS01.4 VX-661 in combination with ivacaftor in patients with cystic fibrosis and the F508del-CFTR mutation. J. Cyst. Fibros. 14, S1.

Références bibliographique
194
Pind, S., Riordan, J.R., and Williams, D.B. (1994). Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 269, 12784–12788.
Pissarra, L.S., Farinha, C.M., Xu, Z., Schmidt, A., Thibodeau, P.H., Cai, Z., Thomas, P.J., Sheppard, D.N., and Amaral, M.D. (2008). Solubilizing Mutations Used to Crystallize One CFTR Domain Attenuate the Trafficking and Channel Defects Caused by the Major Cystic Fibrosis Mutation. Chem. Biol. 15, 62–69.
Pratt, W.B., Morishima, Y., Peng, H.-M., and Osawa, Y. (2010). Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp. Biol. Med. Maywood NJ 235, 278–289.
Prodromou, C. (2000). The ATPase cycle of Hsp90 drives a molecular clamp’ via transient dimerization of the N-terminal domains. EMBO J. 19, 4383–4392.
Prodromou, C., Roe, S.M., O’Brien, R., Ladbury, J.E., Piper, P.W., and Pearl, L.H. (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75.
Qi, R., Sarbeng, E.B., Liu, Q., Le, K.Q., Xu, X., Xu, H., Yang, J., Wong, J.L., Vorvis, C., Hendrickson, W.A., et al. (2013). Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 20, 900–907.
Qin, S.-Y., Kawasaki, N., Hu, D., Tozawa, H., Matsumoto, N., and Yamamoto, K. (2012). Subcellular localization of ERGIC-53 under endoplasmic reticulum stress condition. Glycobiology 22, 1709–1720.
Quan, E.M., Kamiya, Y., Kamiya, D., Denic, V., Weibezahn, J., Kato, K., and Weissman, J.S. (2008). Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 32, 870–877.
Raasi, S., and Wolf, D.H. (2007). Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 18, 780–791.
Rab, A., Bartoszewski, R., Jurkuvenaite, A., Wakefield, J., Collawn, J.F., and Bebok, Z. (2007). Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am. J. Physiol. Cell Physiol. 292, C756-766.
Rabeh, W.M., Bossard, F., Xu, H., Okiyoneda, T., Bagdany, M., Mulvihill, C.M., Du, K., di Bernardo, S., Liu, Y., Konermann, L., et al. (2012). Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148, 150–163.
Ratjen, F., Hug, C., Marigowda, G., Tian, S., Huang, X., Stanojevic, S., Milla, C.E., Robinson, P.D., Waltz, D., Davies, J.C., et al. (2017). Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 5, 557–567.
Reggiori, F., Monastyrska, I., Verheije, M.H., Calì, T., Ulasli, M., Bianchi, S., Bernasconi, R., de Haan, C.A.M., and Molinari, M. (2010). Coronaviruses Hijack the LC3-I-positive

Références bibliographique
195
EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7, 500–508.
Reisin, I.L., Prat, A.G., Abraham, E.H., Amara, J.F., Gregory, R.J., Ausiello, D.A., and Cantiello, H.F. (1994). The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 269, 20584–20591.
Reiterer, V., Nyfeler, B., and Hauri, H.-P. (2010). Role of the lectin VIP36 in post-ER quality control of human alpha1-antitrypsin. Traffic Cph. Den. 11, 1044–1055.
Ren, H.Y., Grove, D.E., De La Rosa, O., Houck, S.A., Sopha, P., Van Goor, F., Hoffman, B.J., and Cyr, D.M. (2013). VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 24, 3016–3024.
Retzlaff, M., Hagn, F., Mitschke, L., Hessling, M., Gugel, F., Kessler, H., Richter, K., and Buchner, J. (2010). Asymmetric Activation of the Hsp90 Dimer by Its Cochaperone Aha1. Mol. Cell 37, 344–354.
Riazanski, V., Gabdoulkhakova, A.G., Boynton, L.S., Eguchi, R.R., Deriy, L.V., Hogarth, D.K., Loaëc, N., Oumata, N., Galons, H., Brown, M.E., et al. (2015). TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc. Natl. Acad. Sci. U. S. A. 112, E6486-6495.
Ribeiro, C.M.P., and Boucher, R.C. (2010). Role of Endoplasmic Reticulum Stress in Cystic Fibrosis-Related Airway Inflammatory Responses. Proc. Am. Thorac. Soc. 7, 387–394.
Ribeiro, C.M.P., Paradiso, A.M., Schwab, U., Perez-Vilar, J., Jones, L., O’Neal, W., and Boucher, R.C. (2005). Chronic Airway Infection/Inflammation Induces a Ca2+i-dependent Hyperinflammatory Response in Human Cystic Fibrosis Airway Epithelia. J. Biol. Chem. 280, 17798–17806.
Rich, D.P., Gregory, R.J., Anderson, M.P., Manavalan, P., Smith, A.E., and Welsh, M.J. (1991). Effect of deleting the R domain on CFTR-generated chloride channels. Science 253, 205–207.
Riordan, J.R. (2008). CFTR function and prospects for therapy. Annu. Rev. Biochem. 77, 701–726.
Riordan, J.R., Rommens, J.M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N., and Chou, J.L. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073.
Röhl, A., Rohrberg, J., and Buchner, J. (2013). The chaperone Hsp90: changing partners for demanding clients. Trends Biochem. Sci. 38, 253–262.
Rommens, J.M., Iannuzzi, M.C., Kerem, B., Drumm, M.L., Melmer, G., Dean, M., Rozmahel, R., Cole, J.L., Kennedy, D., and Hidaka, N. (1989). Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059–1065.
Ron, E., Shenkman, M., Groisman, B., Izenshtein, Y., Leitman, J., and Lederkremer, G.Z. (2011). Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol. Biol. Cell 22, 3945–3954.

Références bibliographique
196
Rosser, M.F., Grove, D.E., Chen, L., and Cyr, D.M. (2008). Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Mol. Biol. Cell 19, 4570–4579.
Rossi, A.G., Sawatzky, D.A., Walker, A., Ward, C., Sheldrake, T.A., Riley, N.A., Caldicott, A., Martinez-Losa, M., Walker, T.R., Duffin, R., et al. (2006). Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064.
Roth, J., and Zuber, C. (2017). Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochem. Cell Biol. 147, 269–284.
Rowe, S.M., McColley, S.A., Rietschel, E., Li, X., Bell, S.C., Konstan, M.W., Marigowda, G., Waltz, D., Boyle, M.P., and VX09-809-102 Study Group (2017). Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann. Am. Thorac. Soc. 14, 213–219.
Roxo-Rosa, M., Xu, Z., Schmidt, A., Neto, M., Cai, Z., Soares, C.M., Sheppard, D.N., and Amaral, M.D. (2006). Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc. Natl. Acad. Sci. 103, 17891–17896.
Rubenstein, R.C., and Zeitlin, P.L. (1998). A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am. J. Respir. Crit. Care Med. 157, 484–490.
Rubenstein, R.C., and Zeitlin, P.L. (2000). Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am. J. Physiol. Cell Physiol. 278, C259-267.
Rüdiger, S., Buchberger, A., and Bukau, B. (1997). Interaction of Hsp70 chaperones with substrates. Nat. Struct. Mol. Biol. 4, 342–349.
Russell, S.J., Ruddock, L.W., Salo, K.E.H., Oliver, J.D., Roebuck, Q.P., Llewellyn, D.H., Roderick, H.L., Koivunen, P., Myllyharju, J., and High, S. (2004). The Primary Substrate Binding Site in the b′ Domain of ERp57 Is Adapted for Endoplasmic Reticulum Lectin Association. J. Biol. Chem. 279, 18861–18869.
Ryno, L.M., Wiseman, R.L., and Kelly, J.W. (2013). Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr. Opin. Chem. Biol. 17, 346–352.
Sadowski, M., and Sarcevic, B. (2010). Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 5, 19.
Saint-Criq, V., and Gray, M.A. (2017). Role of CFTR in epithelial physiology. Cell. Mol. Life
Sci. 74, 93–115.

Références bibliographique
197
Samali, A., FitzGerald, U., Deegan, S., and Gupta, S. (2010). Methods for Monitoring Endoplasmic Reticulum Stress and the Unfolded Protein Response. Int. J. Cell Biol. 2010, 1–11.
Satoh, T., Chen, Y., Hu, D., Hanashima, S., Yamamoto, K., and Yamaguchi, Y. (2010). Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol. Cell 40, 905–916.
Satoh, T., Suzuki, K., Yamaguchi, T., and Kato, K. (2014a). Structural Basis for Disparate Sugar-Binding Specificities in the Homologous Cargo Receptors ERGIC-53 and VIP36. PLoS ONE 9, e87963.
Satoh, T., Suzuki, K., Yamaguchi, T., and Kato, K. (2014b). Structural basis for disparate sugar-binding specificities in the homologous cargo receptors ERGIC-53 and VIP36. PloS One 9, e87963.
Schallus, T., Jaeckh, C., Fehér, K., Palma, A.S., Liu, Y., Simpson, J.C., Mackeen, M., Stier, G., Gibson, T.J., Feizi, T., et al. (2008). Malectin: a novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol. Biol. Cell 19, 3404–3414.
Schmidt, B.Z., Watts, R.J., Aridor, M., and Frizzell, R.A. (2009). Cysteine String Protein Promotes Proteasomal Degradation of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) by Increasing Its Interaction with the C Terminus of Hsp70-interacting Protein and Promoting CFTR Ubiquitylation. J. Biol. Chem. 284, 4168–4178.
Schmidt, B.Z., Haaf, J.B., Leal, T., and Noel, S. (2016). Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: current perspectives. Clin. Pharmacol. Adv. Appl. 8, 127–140.
Schneider, E., Reyes-Ortega, F., Li, J., and Velkov, T. (2017). Can Cystic Fibrosis Patients Finally Catch a Breath With Lumacaftor/Ivacaftor? Clin. Pharmacol. Ther. 101, 130–141.
Schopf, F.H., Biebl, M.M., and Buchner, J. (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360.
Schrag, J.D., Bergeron, J.J., Li, Y., Borisova, S., Hahn, M., Thomas, D.Y., and Cygler, M. (2001). The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol. Cell 8, 633–644.
Schreiber, R., Nitschke, R., Greger, R., and Kunzelmann, K. (1999). The Cystic Fibrosis Transmembrane Conductance Regulator Activates Aquaporin 3 in Airway Epithelial Cells. J. Biol. Chem. 274, 11811–11816.
Schwarz, F., and Aebi, M. (2011). Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 21, 576–582.
Schwiebert, E.M., Egan, M.E., Hwang, T.H., Fulmer, S.B., Allen, S.S., Cutting, G.R., and Guggino, W.B. (1995). CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell 81, 1063–1073.

Références bibliographique
198
Schwiebert, E.M., Morales, M.M., Devidas, S., Egan, M.E., and Guggino, W.B. (1998). Chloride channel and chloride conductance regulator domains of CFTR, the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. U. S. A. 95, 2674–2679.
Scott-Ward, T.S., and Amaral, M.D. (2009). Deletion of Phe508 in the first nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator increases its affinity for the heat shock cognate 70 chaperone: Interaction of Hsc70 with F508del-NBD1. FEBS J. 276, 7097–7109.
Serohijos, A.W.R., Hegedus, T., Aleksandrov, A.A., He, L., Cui, L., Dokholyan, N.V., and Riordan, J.R. (2008). Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. 105, 3256–3261.
Sharma, S.K., De Los Rios, P., Christen, P., Lustig, A., and Goloubinoff, P. (2010). The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 6, 914–920.
Shastry, S., Toft, D.O., and Joyner, M.J. (2002). HSP70 and HSP90 expression in leucocytes after exercise in moderately trained humans. Acta Physiol. Scand. 175, 139–146.
Shcheynikov, N., Ko, S.B.H., Zeng, W., Choi, J.Y., Dorwart, M.R., Thomas, P.J., and Muallem, S. (2006). Regulatory Interaction between CFTR and the SLC26 Transporters. In Novartis Foundation Symposia, D.J. Chadwick, and J. Goode, eds. (Chichester, UK: John Wiley & Sons, Ltd), pp. 177–192.
Shen, Y., and Hendershot, L.M. (2005). ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol. Biol. Cell 16, 40–50.
Shenkman, M., Groisman, B., Ron, E., Avezov, E., Hendershot, L.M., and Lederkremer, G.Z. (2013). A Shared Endoplasmic Reticulum-associated Degradation Pathway Involving the EDEM1 Protein for Glycosylated and Nonglycosylated Proteins. J. Biol. Chem. 288, 2167–2178.
Shiau, A.K., Harris, S.F., Southworth, D.R., and Agard, D.A. (2006). Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 127, 329–340.
Shimada, O., Hara-Kuge, S., Yamashita, K., Tosaka-Shimada, H., Yanchao, L., Einan, L., Atsumi, S., and Ishikawa, H. (2003). Localization of VIP36 in the post-Golgi secretory pathway also of rat parotid acinar cells. J. Histochem. Cytochem. Off. J. Histochem. Soc. 51, 1057–1063.
Singh, O.V., Vij, N., Mogayzel, P.J., Jozwik, C., Pollard, H.B., and Zeitlin, P.L. (2006). Pharmacoproteomics of 4-Phenylbutyrate-Treated IB3-1 Cystic Fibrosis Bronchial Epithelial Cells. J. Proteome Res. 5, 562–571.
Slamon, D.J., Clark, G.M., Wong, S.G., Levin, W.J., Ullrich, A., and McGuire, W.L. (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182.

Références bibliographique
199
Slominska-Wojewodzka, M., and Sandvig, K. (2015). The Role of Lectin-Carbohydrate Interactions in the Regulation of ER-Associated Protein Degradation. Mol. Basel Switz. 20, 9816–9846.
Slominska-Wojewodzka, M., Gregers, T.F., Wälchli, S., and Sandvig, K. (2006). EDEM is involved in retrotranslocation of ricin from the endoplasmic reticulum to the cytosol. Mol. Biol. Cell 17, 1664–1675.
Slominska-Wojewodzka, M., Pawlik, A., Sokołowska, I., Antoniewicz, J., Węgrzyn, G., and Sandvig, K. (2014). The role of EDEM2 compared with EDEM1 in ricin transport from the endoplasmic reticulum to the cytosol. Biochem. J. 457, 485–496.
Soheili, T., Gicquel, E., Poupiot, J., N’Guyen, L., Le Roy, F., Bartoli, M., and Richard, I. (2012). Rescue of sarcoglycan mutations by inhibition of endoplasmic reticulum quality control is associated with minimal structural modifications. Hum. Mutat. 33, 429–439.
Sokołowska, I., Wälchli, S., Węgrzyn, G., Sandvig, K., and Słomińska-Wojewódzka, M. (2011). A single point mutation in ricin A-chain increases toxin degradation and inhibits EDEM1-dependent ER retrotranslocation. Biochem. J. 436, 371–385.
Soldà, T., Garbi, N., Hämmerling, G.J., and Molinari, M. (2006). Consequences of ERp57 Deletion on Oxidative Folding of Obligate and Facultative Clients of the Calnexin Cycle. J. Biol. Chem. 281, 6219–6226.
Soldano, K.L., Jivan, A., Nicchitta, C.V., and Gewirth, D.T. (2003). Structure of the N-terminal Domain of GRP94: BASIS FOR LIGAND SPECIFICITY AND REGULATION. J. Biol. Chem. 278, 48330–48338.
Stocki, P., Chapman, D.C., Beach, L.A., and Williams, D.B. (2014). Depletion of Cyclophilins B and C Leads to Dysregulation of Endoplasmic Reticulum Redox Homeostasis. J. Biol. Chem. 289, 23086–23096.
Stutts, M.J., Canessa, C.M., Olsen, J.C., Hamrick, M., Cohn, J.A., Rossier, B.C., and Boucher, R.C. (1995). CFTR as a cAMP-dependent regulator of sodium channels. Science 269, 847–850.
Sugita, M., Yue, Y., and Foskett, J.K. (1998). CFTR Cl- channel and CFTR-associated ATP channel: distinct pores regulated by common gates. EMBO J. 17, 898–908.
Sun, F., Zhang, R., Gong, X., Geng, X., Drain, P.F., and Frizzell, R.A. (2006). Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 281, 36856–36863.
Swain, J.F., Dinler, G., Sivendran, R., Montgomery, D.L., Stotz, M., and Gierasch, L.M. (2007). Hsp70 Chaperone Ligands Control Domain Association via an Allosteric Mechanism Mediated by the Interdomain Linker. Mol. Cell 26, 27–39.
Taipale, M., Krykbaeva, I., Koeva, M., Kayatekin, C., Westover, K.D., Karras, G.I., and Lindquist, S. (2012). Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 150, 987–1001.
Tamura, T., Cormier, J.H., and Hebert, D.N. (2011). Characterization of Early EDEM1 Protein Maturation Events and Their Functional Implications. J. Biol. Chem. 286, 24906–24915.

Références bibliographique
200
Tang, H.-Y., Huang, C.-H., Zhuang, Y.-H., Christianson, J.C., and Chen, X. (2014). EDEM2 and OS-9 are required for ER-associated degradation of non-glycosylated sonic hedgehog. PloS One 9, e92164.
Tannous, A., Pisoni, G.B., Hebert, D.N., and Molinari, M. (2015). N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 41, 79–89.
Teasdale, R.D., and Jackson, M.R. (1996). Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu. Rev. Cell Dev. Biol. 12, 27–54.
Termine, D.J., Moremen, K.W., and Sifers, R.N. (2009). The mammalian UPR boosts glycoprotein ERAD by suppressing the proteolytic downregulation of ER mannosidase I. J. Cell Sci. 122, 976–984.
Thomas, P.J., Shenbagamurthi, P., Sondek, J., Hullihen, J.M., and Pedersen, P.L. (1992). The cystic fibrosis transmembrane conductance regulator. Effects of the most common cystic fibrosis-causing mutation on the secondary structure and stability of a synthetic peptide. J. Biol. Chem. 267, 5727–5730.
Tomati, V., Sondo, E., Armirotti, A., Caci, E., Pesce, E., Marini, M., Gianotti, A., Ju Jeon, Y., Cilli, M., Pistorio, A., et al. (2015). Genetic Inhibition Of The Ubiquitin Ligase Rnf5 Attenuates Phenotypes Associated To F508del Cystic Fibrosis Mutation. Sci. Rep. 5, 12138.
Tomoda, H., and Omura, S. (2000). Lactacystin, a proteasome inhibitor: discovery and its application in cell biology. Yakugaku Zasshi 120, 935–949.
Tremblay, L.O., and Herscovics, A. (1999). Cloning and expression of a specific human alpha 1,2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan biosynthesis. Glycobiology 9, 1073–1078.
Tsutsumi, S., Mollapour, M., Prodromou, C., Lee, C.-T., Panaretou, B., Yoshida, S., Mayer, M.P., and Neckers, L.M. (2012). Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. 109, 2937–2942.
Tulsiani, D.R., and Touster, O. (1983). Swainsonine causes the production of hybrid glycoproteins by human skin fibroblasts and rat liver Golgi preparations. J. Biol. Chem. 258, 7578–7585.
Ushioda, R., Hoseki, J., Araki, K., Jansen, G., Thomas, D.Y., and Nagata, K. (2008). ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572.
Vagin, O., Kraut, J.A., and Sachs, G. (2008). Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. AJP Ren. Physiol. 296, F459–F469.
Van Goor, F., Hadida, S., Grootenhuis, P.D.J., Burton, B., Stack, J.H., Straley, K.S., Decker, C.J., Miller, M., McCartney, J., Olson, E.R., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. 108, 18843–18848.

Références bibliographique
201
Vasconcelos-dos-Santos, A., Oliveira, I.A., Lucena, M.C., Mantuano, N.R., Whelan, S.A., Dias, W.B., and Todeschini, A.R. (2015). Biosynthetic Machinery Involved in Aberrant Glycosylation: Promising Targets for Developing of Drugs Against Cancer. Front. Oncol. 5.
Vij, N., Fang, S., and Zeitlin, P.L. (2006). Selective Inhibition of Endoplasmic Reticulum-associated Degradation Rescues F508-Cystic Fibrosis Transmembrane Regulator and Suppresses Interleukin-8 Levels: THERAPEUTIC IMPLICATIONS. J. Biol. Chem. 281, 17369–17378.
Wada, I., Rindress, D., Cameron, P.H., Ou, W.J., Doherty, J.J., Louvard, D., Bell, A.W., Dignard, D., Thomas, D.Y., and Bergeron, J.J. (1991). SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J. Biol. Chem. 266, 19599–19610.
Wallis, A.K., and Freedman, R.B. (2013). Assisting oxidative protein folding: how do protein disulphide-isomerases couple conformational and chemical processes in protein folding? Top. Curr. Chem. 328, 1–34.
Wang, M., and Kaufman, R.J. (2014). The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597.
Wang, B., Heath-Engel, H., Zhang, D., Nguyen, N., Thomas, D.Y., Hanrahan, J.W., and Shore, G.C. (2008). BAP31 Interacts with Sec61 Translocons and Promotes Retrotranslocation of CFTRΔF508 via the Derlin-1 Complex. Cell 133, 1080–1092.
Wang, H., Wang, X., Ke, Z.-J., Comer, A.L., Xu, M., Frank, J.A., Zhang, Z., Shi, X., and Luo, J. (2015). Tunicamycin-induced unfolded protein response in the developing mouse brain. Toxicol. Appl. Pharmacol. 283, 157–167.
Wang, M., Wey, S., Zhang, Y., Ye, R., and Lee, A.S. (2009). Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 11, 2307–2316.
Wang, W.-A., Groenendyk, J., and Michalak, M. (2012). Calreticulin signaling in health and disease. Int. J. Biochem. Cell Biol. 44, 842–846.
Wang, X., Matteson, J., An, Y., Moyer, B., Yoo, J.-S., Bannykh, S., Wilson, I.A., Riordan, J.R., and Balch, W.E. (2004). COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 167, 65–74.
Wang, X., Venable, J., LaPointe, P., Hutt, D.M., Koulov, A.V., Coppinger, J., Gurkan, C., Kellner, W., Matteson, J., Plutner, H., et al. (2006a). Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 127, 803–815.
Wang, Y., Soyombo, A.A., Shcheynikov, N., Zeng, W., Dorwart, M., Marino, C.R., Thomas, P.J., and Muallem, S. (2006b). Slc26a6 regulates CFTR activity in vivo to determine pancreatic duct HCO3- secretion: relevance to cystic fibrosis. EMBO J. 25, 5049–5057.
Wang, Y., Wrennall, J.A., Cai, Z., Li, H., and Sheppard, D.N. (2014). Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int. J. Biochem. Cell Biol. 52, 47–57.

Références bibliographique
202
Ward, C.L., and Kopito, R.R. (1994). Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 269, 25710–25718.
Wayne, N., and Bolon, D.N. (2007). Dimerization of Hsp90 Is Required for in Vivo Function: DESIGN AND ANALYSIS OF MONOMERS AND DIMERS. J. Biol. Chem. 282, 35386–35395.
Wei, L., Vankeerberghen, A., Cuppens, H., Cassiman, J.J., Droogmans, G., and Nilius, B. (2001). The C-terminal part of the R-domain, but not the PDZ binding motif, of CFTR is involved in interaction with Ca(2+)-activated Cl- channels. Pflugers Arch. 442, 280–285.
Weisz, O.A. (2003). Acidification and protein traffic. Int. Rev. Cytol. 226, 259–319.
Welsh, M.J., and Smith, A.E. (1993). Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73, 1251–1254.
Weng, S., and Spiro, R.G. (1996). Endoplasmic Reticulum Kifunensine-Resistant α-Mannosidase Is Enzymatically and Immunologically Related to the Cytosolic α-Mannosidase. Arch. Biochem. Biophys. 325, 113–123.
Wilkens, S. (2015). Structure and mechanism of ABC transporters. F1000Prime Rep. 7.
Wu, M.M., Grabe, M., Adams, S., Tsien, R.Y., Moore, H.-P.H., and Machen, T.E. (2001). Mechanisms of pH Regulation in the Regulated Secretory Pathway. J. Biol. Chem. 276, 33027–33035.
Wu, Y., Swulius, M.T., Moremen, K.W., and Sifers, R.N. (2003). Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc. Natl. Acad. Sci. U. S. A. 100, 8229–8234.
Yamaguchi, D., Hu, D., Matsumoto, N., and Yamamoto, K. (2010). Human XTP3-B binds to alpha1-antitrypsin variant null(Hong Kong) via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology 20, 348–355.
Yamamoto, K. (2014). Intracellular lectins are involved in quality control of glycoproteins. Proc. Jpn. Acad. Ser. B 90, 67–82.
Yamamoto, Y., Kimura, T., Momohara, S., Takeuchi, M., Tani, T., Kimata, Y., Kadokura, H., and Kohno, K. (2010). A novel ER J-protein DNAJB12 accelerates ER-associated degradation of membrane proteins including CFTR. Cell Struct. Funct. 35, 107–116.
Yoshida, H., Matsui, T., Hosokawa, N., Kaufman, R.J., Nagata, K., and Mori, K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271.
Younger, J.M., Ren, H.-Y., Chen, L., Fan, C.-Y., Fields, A., Patterson, C., and Cyr, D.M. (2004). A foldable CFTRΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70–CHIP E3 ubiquitin ligase. J. Cell Biol. 167, 1075–1085.
Younger, J.M., Chen, L., Ren, H.-Y., Rosser, M.F.N., Turnbull, E.L., Fan, C.-Y., Patterson, C., and Cyr, D.M. (2006). Sequential Quality-Control Checkpoints Triage Misfolded Cystic Fibrosis Transmembrane Conductance Regulator. Cell 126, 571–582.

Références bibliographique
203
Zapun, A., Darby, N.J., Tessier, D.C., Michalak, M., Bergeron, J.J., and Thomas, D.Y. (1998). Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J. Biol. Chem. 273, 6009–6012.
Zegarra-Moran, O., and Galietta, L.J.V. (2017). CFTR pharmacology. Cell. Mol. Life Sci. 74, 117–128.
Zhang, Y., Nijbroek, G., Sullivan, M.L., McCracken, A.A., Watkins, S.C., Michaelis, S., and Brodsky, J.L. (2001). Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12, 1303–1314.
Zhou, T., Frabutt, D.A., Moremen, K.W., and Zheng, Y.-H. (2015). ERManI (Endoplasmic Reticulum Class I α-Mannosidase) Is Required for HIV-1 Envelope Glycoprotein Degradation via Endoplasmic Reticulum-associated Protein Degradation Pathway. J. Biol. Chem. 290, 22184–22192.
Zhou, Z., Gong, Q., Epstein, M.L., and January, C.T. (1998). HERG Channel Dysfunction in Human Long QT Syndrome: INTRACELLULAR TRANSPORT AND FUNCTIONAL DEFECTS. J. Biol. Chem. 273, 21061–21066.
Zielenski, J., and Tsui, L.C. (1995). Cystic fibrosis: genotypic and phenotypic variations. Annu. Rev. Genet. 29, 777–807.
Zuber, C., Cormier, J.H., Guhl, B., Santimaria, R., Hebert, D.N., and Roth, J. (2007). EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. 104, 4407–4412.

Annexes
204
Annexes Article scientifique
Pushing the limits of catalytic C-H amination in polyoxygenated cyclobutanes
Pierre-Antoine Nocqueta, Raphael Hensiennea, Joanna Wencel-Delord‡a, Eugenie Laigrea, Khadidja Sidelarbib, Frederic Becqb, Caroline Norezb, Damien Hazelarda and Philippe
Compaina
a. Laboratoire de Synthese Organique et Molecules Bioactives (SYBIO), Universite de
Strasbourg/CNRS (UMR 7509), Ecole Europeenne de Chimie, Polymeres et Materiaux
(ECPM), 25 rue Becquerel, 67087 Strasbourg, France. E-mail:[email protected] b. Laboratoire Signalisation et Transports Ioniques Membranaire (STIM), Universite de Poitiers
et CNRS (ERL7368), 1 rue Georges Bonnet, 86000 Poitiers, France.
‡ Present address: SynCat, Universite de Strasbourg/CNRS (UMR 7509), Ecole Europeenne de
Chimie, Polymeres et Materiaux (ECPM), 25 rue Becquerel, 67087 Strasbourg, France

Annexes
205

Annexes
206

Annexes
207

Annexes
208

Annexes
209

Annexes
210

Annexes
211

Annexes
212

Annexes
213

Annexes
214

Annexes
215

Annexes
216

Annexes
217

Annexes
218

Annexes
219

Annexes
220

221
Khadidja SIDELARBI 32 ans, Célibataire
94 rue de la cathédrale, N° 59 Poitiers 86000
06 95 65 71 86
Formations et diplômes obtenus
2017 : Doctorat en cours ; Biologie Santé, Secteur de recherche Biomolécules, pharmacologie, thérapeutique. Université de Poitiers, France.
2014 : Master 2 en Biologie Santé Science du médicament - Recherche et Ingénierie en Biosanté ; parcours : Biologie Cellulaire et Moléculaire ; Université de Poitiers, France.
2012 : Licence 3 en Biochimie Biologie Moléculaire Cellulaire et Génétique; Université de Poitiers, France.
2006 : Diplôme des études supérieures (Bac+4) en Biologie, spécialité Génétique; Université Es- senia d’Oran, Algérie.
Expériences professionnelles
Emploi
Depuis septembre 2017 : Attaché temporaire d’enseignement et de recherche, section Biologie cellulaire (65ème section) > Université de Poitiers.
Janvier à Juillet 2010 (6 mois) : Ingénieur d’application > Laboratoire d’analyses de biologie médicale; Oran, Algérie. Postes occupés : analyses de sérologie virale, biochimie, hématologie, immunologie, prélèvements sanguins, secrétariat.
Décembre 2006 à Décembre 2008 (2 ans) : Ingénieur d’application > Centre de Transfusion Sanguine (CTS) ; Ain Témouchent, Algérie. Postes occupés : analyses de sérologie virale avec le test ELISA (HIV, hépatite B et C, brucellose Wright), test du groupe sanguin, banque du sang et distribution des produits sanguins, collectes sanguines, prélèvements sanguins, secrétariat.
Stage
2014 – 2017 (3 ans) : Doctorante sur le projet « Implication du chaperome de la protéine F508del-CFTR dans son transport intracellulaire et/ou sa dégradation : Rôle de la lectine EDEM et de la mannosidase du RE ». Laboratoire : Signalisation et Transports Ioniques Membranaires (STIM). Equipe Transports Ioniques et Mucoviscidose (TIM); CNRS ERL7368, Université de Poitiers, France. Sous la direction du Dr Caroline NOREZ et Pr. Frédéric BECQ.
Janvier à Juin 2014 (6 mois) : Stage de master 2 sur le projet « Étude des inhibiteurs de mannosidase comme correcteurs de la protéine F508del-CFTR ». Laboratoire STIM, équipe TIM ; Université de Poitiers, France. Sous la direction du Dr Caroline NOREZ.
Avril à Juin 2013 (2 mois) : Stage de master 1 sur le projet « Étude des gouttelettes lipidiques et leurs implications dans le mécanisme de l’autophagie ». Institut de Physiologie et de Biologie Cellulaires, laboratoire « Génétique des levures », CNRS FRE3511, Université de Poitiers, France.
Avril à Mai 2011 (1 mois) : Stage de licence 3 sur le projet « Étude du mode d'implantation des légionelles dans les biofilms pour leur maîtrise par lutte biologique ». Laboratoire de chimie et microbiologie de l'eau (LCME) ; Université de Poitiers, France.

222
Domaines de compétences
Techniques - Biologie cellulaire et physiologie : Transfection siRNA, immunomarquage, proximity ligation
assay (duolink), transformation des bactéries et des levures, culture de lignées cellulaire, dissection de bronches issues de biopsies pulmonaires humaines, mesure de potentiel membranaire (sonde Oxonol), mesure de courants transépitheliaux sur cellules polarisées en chambre de Ussing.
- Biochimie et biologie moléculaire : Extraction des protéines, western blot, biotinylation de surface, extraction et purification d’ADN et d’ARN, test ELISA, électrophorèse, mini-préparation.
- Microbiologie : Ensemencement et repiquage de bactéries/levures, préparation des milieux de culture.
Autres - Encadrement des stagiaires : Licence 3 CMI (Master en Ingénierie BioSanté) en stage pratique,
Licence 3 BBMCG (Biochimie Biologie Moléculaire Cellulaire et Génétique) et des élèves de 3ème en stage d’observation.
- Membre élu représentante des doctorants dans le conseil de laboratoire.
Informatique : GraphPad prism, Photoshop, ImageJ, AxioVision, logiciels bibliographiques Zotero et Référence Manager, logiciels courants sous Windows.
Langues : Français : bilingue Anglais : Moyen Arabe : langue maternelle
Publications et communications
Publications
Article en cours de soumission : K.Sidelarbi, A.Joosten, J.Bertrand, F.Stauffert, A.Bodlenner, P.Compain, F.Becq & C.Norez. Role of the inhibition of ER degradation enhancing -mannosidase-like proteins EDEM in CFTR rescue.
Article publié : PA.Nocquet, R.Hensienne, JW.Delord, E.Laigre, K.Sidelarbi, F.Becq, C.Norez, D.Hazelard, P.Compain. Pushing the limits of catalytic C-H amination in polyoxygenated cyclobutanes. Organic & Biomolecular Chemistry. 2016. DOI: 10.1039/c5ob02602d
Abstract publié : K.Sidelarbi, J.Bertrand, A.Joosten, F.Stauffert, A.Bodlenner, F.Becq, P.Compain & C.Norez. Involvement of EDEM1 and EDEM2 in F508del-CFTR trafficking defect and identification of a new series of correctors with an EDEM-dependent mechanism of action. Pediatric Pulmonology. 2016, Vol 51, S45, p210.
Communications orales
Mars 2016 : 13th European Cystic Fibrosis Society “Basic Science Conference” : « EDEM1 and EDEM2 lectins are involved in F508del-CFTR trafficking defect in cystic fibrosis ». K.Sidelarbi, F.Becq, & C.Norez; Pise, Italie.
Février 2016 : 10th European CF Young Investigator Meeting : « ERManI/EDEM, a tandem ushering in new insights on F508del-CFTR correction ». K.Sidelarbi, F.Becq, P.Compain & C.Norez; Paris, France.
Janvier 2015 : 2ème Journée du laboratoire STIM : « Les inhibiteurs de l’α1,2-mannosidase I comme correcteurs du défaut d’adressage de la protéine F508del-CFTR » K.Sidelarbi, F.Becq, P.Compain & C.Norez ; Poitiers, France.
Communications posters
Mars 2017 : 14th European Cystic Fibrosis Society “Basic Science Conference” : « Inhibition of F508del-CFTR/EDEMs interaction restores a functional but immature F508del CFTR to the plasma membrane ». K.Sidelarbi, F.Becq, P.Compain & C.Norez ; Faro, Portugal.

223
Février 2016 : 17ème colloque Français des jeunes chercheurs en mucoviscidose : « ER α1,2-mannosidase et EDEM comme cibles thérapeutique pour F508del-CFTR dans la mucoviscidose ». K.Sidelarbi, F.Becq, P.Compain & C.Norez ; Paris, France.
Mars 2015 : 12th European Cystic Fibrosis Society “Basic Science Conference” : « Iminosugars acting as mannosidase inhibitors in Cystic Fibrosis : Design of multivalent correctors and identification of new therapeutically targets». K.Sidelarbi, J.Bertrand, F.Becq, P.Compain & C.Norez; Faro, Portugal.
Février 2015 : 16ème colloque Français des jeunes chercheurs en mucoviscidose : « Inhibiting the interaction of F508del-CFTR with the lectin EDEM restores its trafficking defect in cystic fibrosis ». K.Sidelarbi, J.Bertrand, A.Joosten, F.Stauffert, A.Bodlenner, F.Becq, P.Compain & C.Norez ; Paris, France.
Références
Pr. Frédéric Becq > Tél : +33 5 49 45 37 29 / Email : frédé[email protected]
Dr. Caroline Norez > Tél : +33 5 49 45 39 33/ Email : [email protected]

Résumé : De nombreuses maladies génétiques sont directement liées à la reconnaissance de protéines mal repliées par le contrôle qualité du réticulum endoplasmique (RE), conduisant à leur rétrotranslocation vers le cytosol puis leur dégradation. Dans le cas des glycoprotéines mutées, comme la protéine F508del-CFTR (Cystic Fibrosis Transmembrane conductance Regulator), la démannosylation extensive de leur résidus mannoses constitue une étape clé dans ce processus. L’objectif de ce travail a été d’identifier et de caractériser des molécules capables de rétablir l’expression membranaire de F508del-CFTR en ciblant cette activité enzymatique.
Dans un premier temps, nous avons testé des dérivés multivalents basés sur le Deoxymannojirimycin (DMJ), un inhibiteur spécifique de la classe I des α-mannosidases et révélé leur effet correcteur puissant sur F508del-CFTR. Nous avons par la suite mis en évidence leur mécanisme d’action et tenté d’expliquer l’augmentation d’efficacité observée entre les monovalents et les multivalents. Nous nous sommes enfin focalisés sur le rôle des principales mannosidases dans le RE, l’α1,2-mannosidase I du RE (ERManI) et la famille des lectines EDEM (ER degradation-enhancing α-mannosidase-like protein). Nous avons montré par une stratégie de siRNA qu’ERManI, EDEM1 et EDEM2 sont impliquées dans la rétention réticulaire du F508del-CFTR. Notre étude ouvre ainsi de nouvelles perspectives quant à l’identification de nouveaux agents pharmacologiques ciblant ces protéines. Mots clés : F508del-CFTR, contrôle qualité du réticulum endoplasmique, activité mannosidase, EDEM. Summary: Numerous genetic diseases are directly associated to the recognition of misfolded proteins by the endoplasmic reticulum (ER) quality control, leading to their retention and subsequently their retrotranslocation to the cytosol for degradation. In the case of mutated glycoproteins such as F508del-CFTR (Cystic Fibrosis Transmembrane conductance Regulator), causing CF pathology, mannose trimming is a key step of this process. Our objective was to identify and characterize molecules targeting ER-mannosidases activity, with the goal to restore F508del-CFTR to the plasma membrane.
First, we tested multivalent derivatives, based mainly on Deoxymannojirimycin (DMJ), an inhibitor of α-mannosidases I and revealed their better corrective effect on F508del-CFTR and their mechanism of action. Then we explored the mechanism explaining the higher efficiency of the multivalents compared to the monovalent. Finally, we focused on the role of key players of mannose trimming, ER-α1,2-mannosidase I (ERManI) and EDEMs (ER degradation-enhancing α-mannosidase-like protein) proteins on F508del-CFTR trafficking defect. We showed the implication of ERManI, EDEM1 and EDEM2 in F508del-CFTR retention, using a siRNA strategy. In conclusion, our study highlights these proteins as potential pharmacological targets to develop correctors for the F508del-CFTR trafficking defect. Keywords: F508del-CFTR, endoplasmic reticulum quality control, mannosidase activity, EDEM.