Phylogénie et évolution des Chiroptères : les Phyllostomidae et l ...

Introduction à la
phylogénie moléculaire
Céline Brochier ([email protected]) 2015-2016
(http://www.frangun.org)

Intérêt d’étudier l’évolution moléculaire et la
phylogénie
Prépondérante en biologie
Ecologie
Evolution
Microbiologie
Biologie cellulaire, etc.
Permet d’aborder des questions très variées
Histoire évolutive du matériel génétique
Histoire évolutive des espèces / taxa
Identification / Classification
Etudes comparatives: Analyse de l’évolution des caractères
etc.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Naissance de la phylogénie moléculaire
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
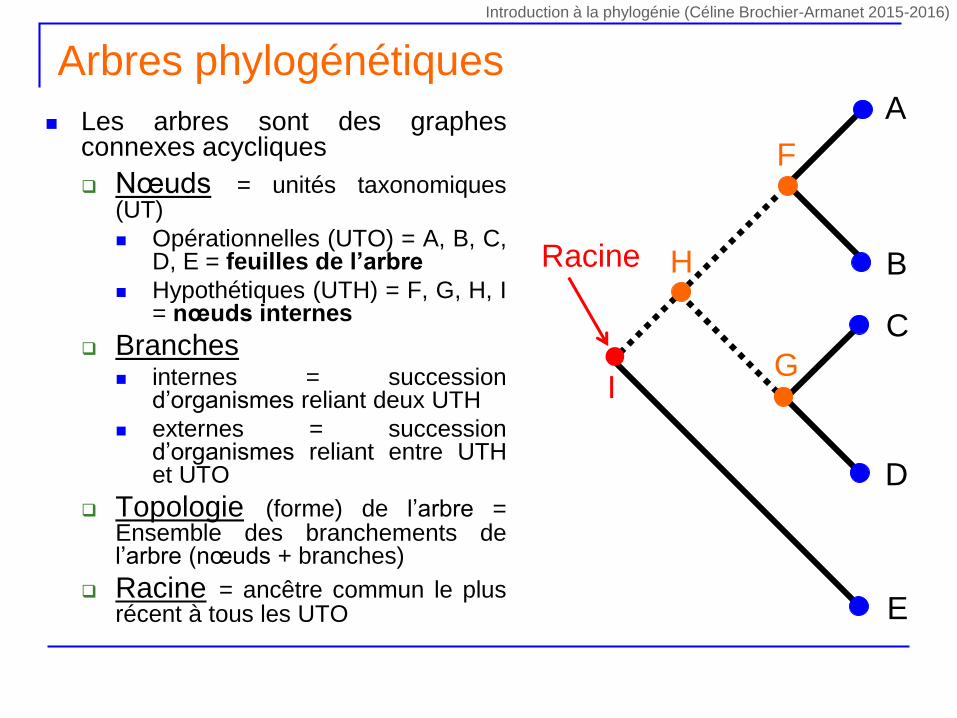
Arbres phylogénétiques
Les arbres sont des graphesconnexes acycliques
Nœuds = unités taxonomiques(UT)
Opérationnelles (UTO) = A, B, C,D, E = feuilles de l’arbre
Hypothétiques (UTH) = F, G, H, I= nœuds internes
Branches internes = succession
d’organismes reliant deux UTH
externes = successiond’organismes reliant entre UTHet UTO
Topologie (forme) de l’arbre =Ensemble des branchements del’arbre (nœuds + branches)
Racine = ancêtre commun le plusrécent à tous les UTO
A
B
C
D
E
F
G
H
I
Racine
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
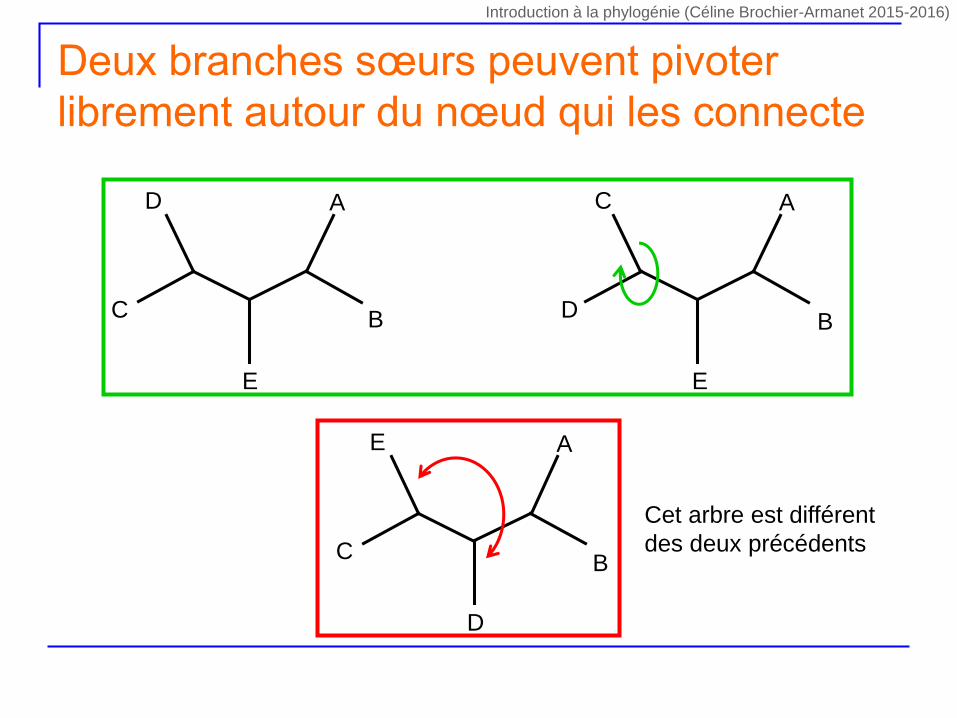
Deux branches sœurs peuvent pivoter
librement autour du nœud qui les connecte
E
C
D A
B
E
D
C A
B
D
C
E A
B
Cet arbre est différent
des deux précédents
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
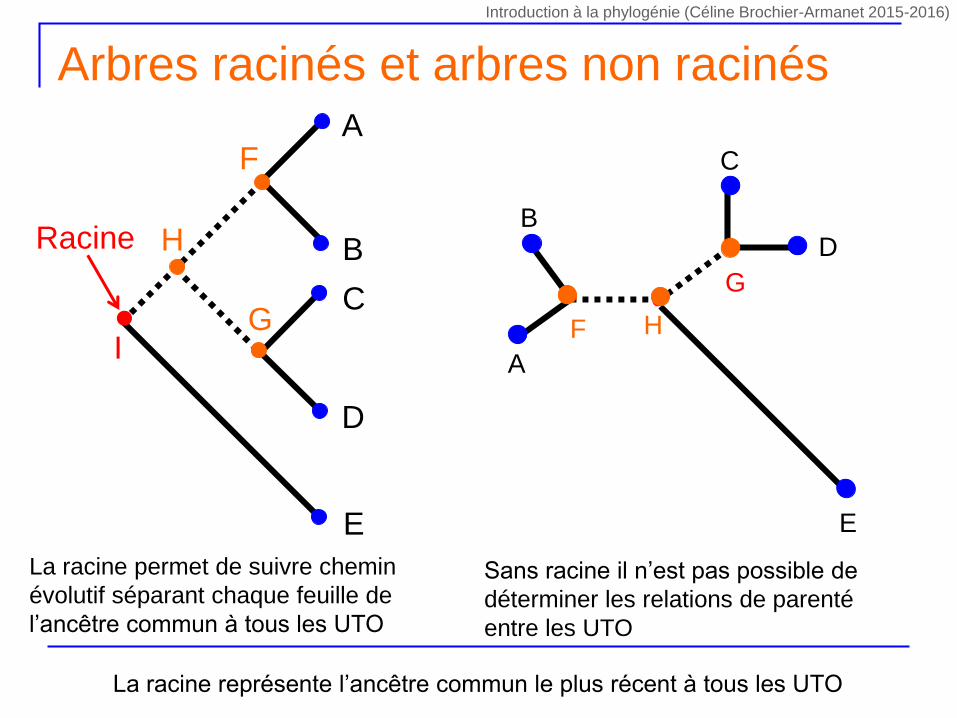
Arbres racinés et arbres non racinés
B
E
H
C
D
G
A
F
A
B
C
D
E
F
G
H
I
Racine
La racine permet de suivre chemin
évolutif séparant chaque feuille de
l’ancêtre commun à tous les UTO
Sans racine il n’est pas possible de
déterminer les relations de parenté
entre les UTO
La racine représente l’ancêtre commun le plus récent à tous les UTO
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
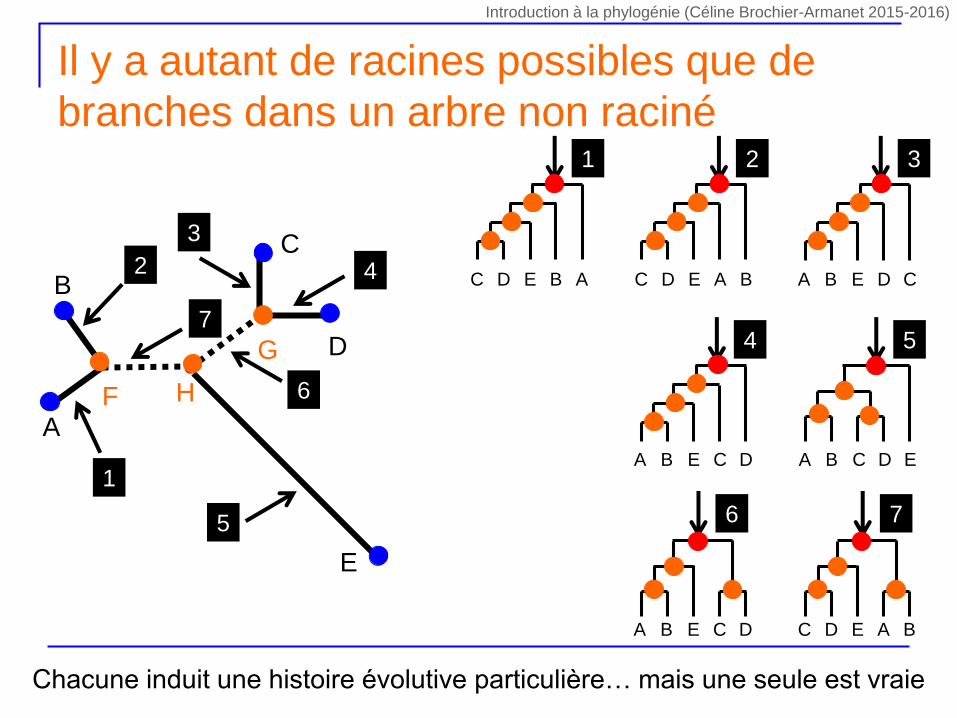
Il y a autant de racines possibles que de
branches dans un arbre non raciné
Chacune induit une histoire évolutive particulière… mais une seule est vraie
B
E
H
C
DG
AF
2
1
7
3
4
5
6
ABC D E
1
BAC D E
2
CDA B E
3
DCA B E
4
EDA B C
5
DCA B E
6
BAC D E
7
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Enraciner un arbre phylogénétique
La majorité des méthodes de reconstruction phylogénétique
produisent des arbres non racinés, car elles n’intègrent pas de
dimension temporelle
L’enracinement se fait donc indépendamment de la méthode choisie
Deux approches:
Enracinement au poids moyen
Enracinement par un groupe extérieur
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
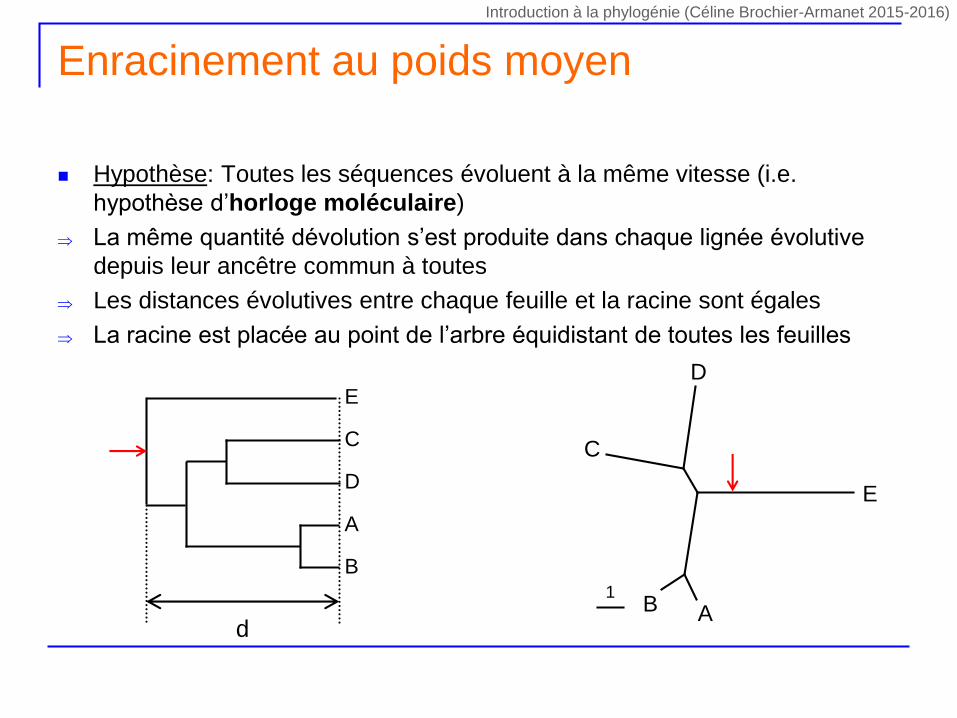
Enracinement au poids moyen
Hypothèse: Toutes les séquences évoluent à la même vitesse (i.e.
hypothèse d’horloge moléculaire)
La même quantité dévolution s’est produite dans chaque lignée évolutive
depuis leur ancêtre commun à toutes
Les distances évolutives entre chaque feuille et la racine sont égales
La racine est placée au point de l’arbre équidistant de toutes les feuilles
E
C
D
A
B
d
1
E
AB
C
D
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Exemple d’enracinement au poids moyen
Fig 5.
Phylogenetic tree showing the relationship of BtubA and BtubB relative to
eukaryotic α and β tubulins. Tree presented is parsimony tree rooted at the
midpoint. Circles indicate bootstrap values. Nodes supported at >75% in the
majority of analyses are indicated by the filled circles. Nodes supported at 50–74%
in most analyses are indicated by the open circles. Unsupported nodes (<50%)
have no circle. (Bar = 0.1 substitutions per site.)
(Jenkins et al. (2002) PNAS)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Cas où l’enracinement au poids moyen
pourrait conduire à une erreur
(van de Peer et al. (2000) gene)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
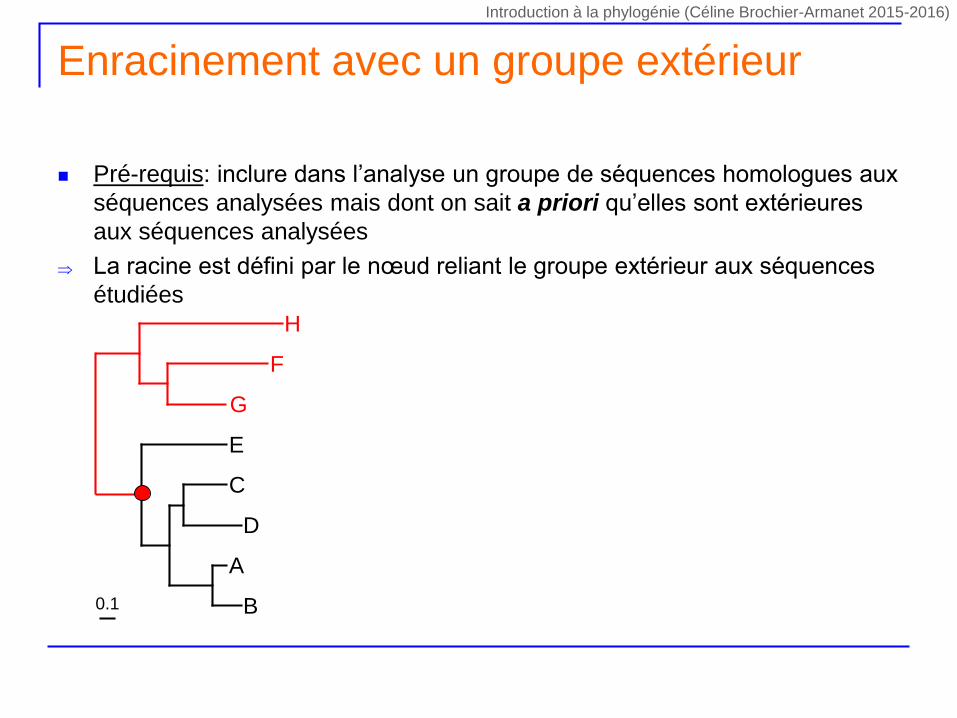
Enracinement avec un groupe extérieur
Pré-requis: inclure dans l’analyse un groupe de séquences homologues aux
séquences analysées mais dont on sait a priori qu’elles sont extérieures
aux séquences analysées
La racine est défini par le nœud reliant le groupe extérieur aux séquences
étudiées
0.1
H
F
G
E
C
D
A
B
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Exemple d’enracinement avec un groupe
extérieur (taxa)
Thermotogae
Autres phyla
bactériens
Fig. S2
Bayesian phylogenetic trees of SSU rRNA. 75 sequences and 1048
unambiguously aligned nucleic acid positions were used. Numbers at
nodes represent posterior probabilities (PP) inferred by MrBayes and
bootstrap values (BV) inferred by TreeFinder. For clarity only PP > 0.50
and BV > 50% are shown. Scale bars represent the average number of
substitutions per site. Thermotogales sequences retrieved from
mesothermic environments are shaded.
(Ben Hania et al (2011) Syst Appl Micro)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
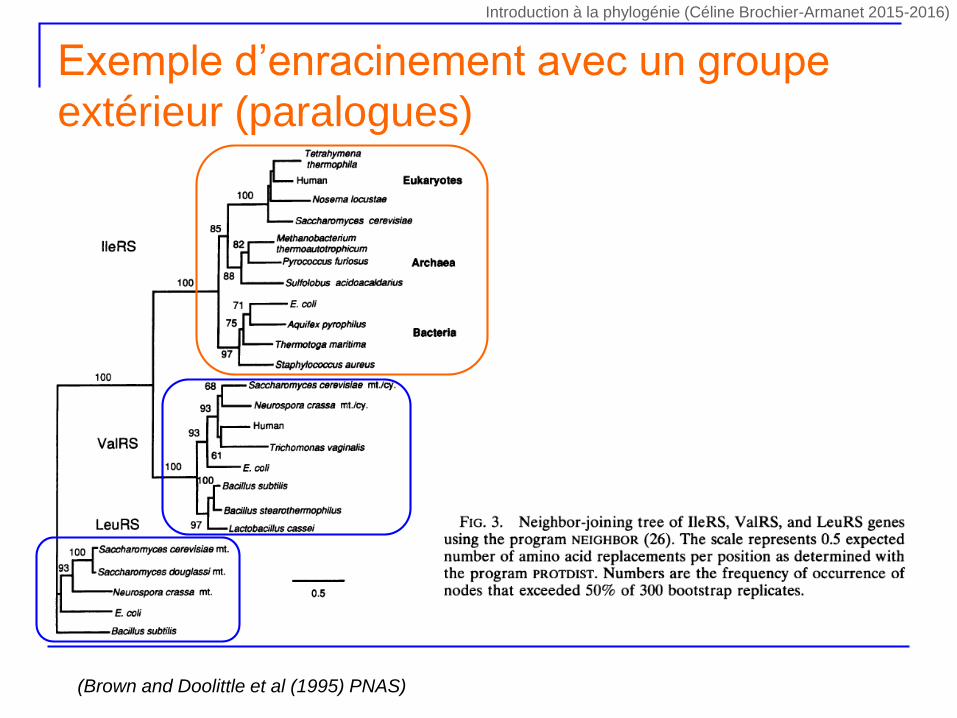
Exemple d’enracinement avec un groupe
extérieur (paralogues)
(Brown and Doolittle et al (1995) PNAS)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
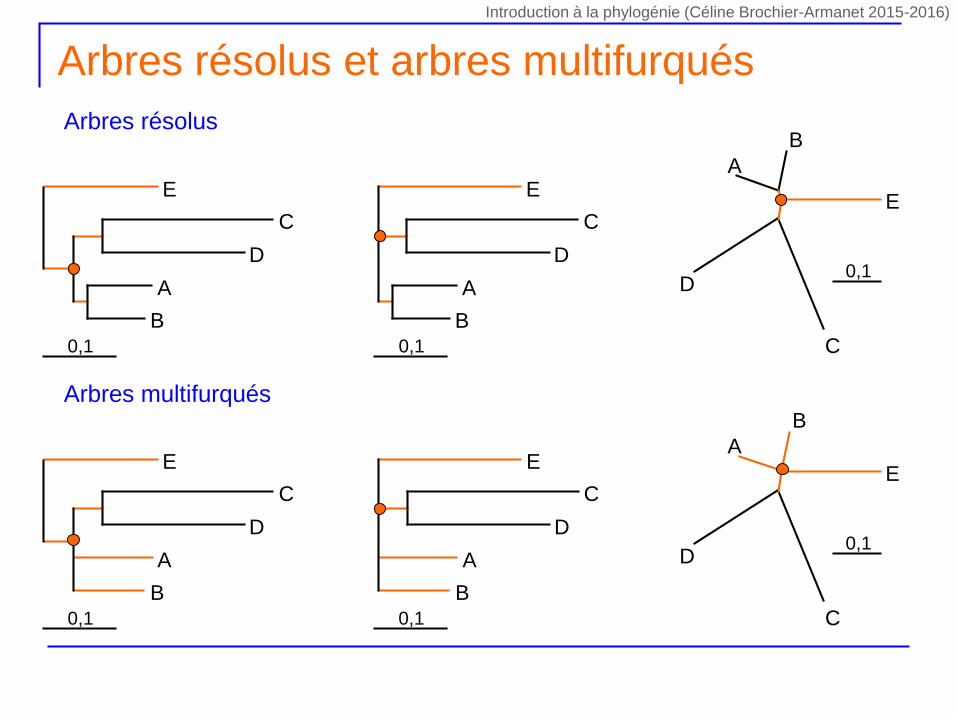
Arbres résolus et arbres multifurqués
Arbres résolus
Arbres multifurqués
0,1
E
C
D
A
B
0,1
E
C
D
A
B
0,1
E
C
D
A
B
0,1
E
C
D
A
B
0,1
E
C
D
A
B
0,1
E
C
D
B
A
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
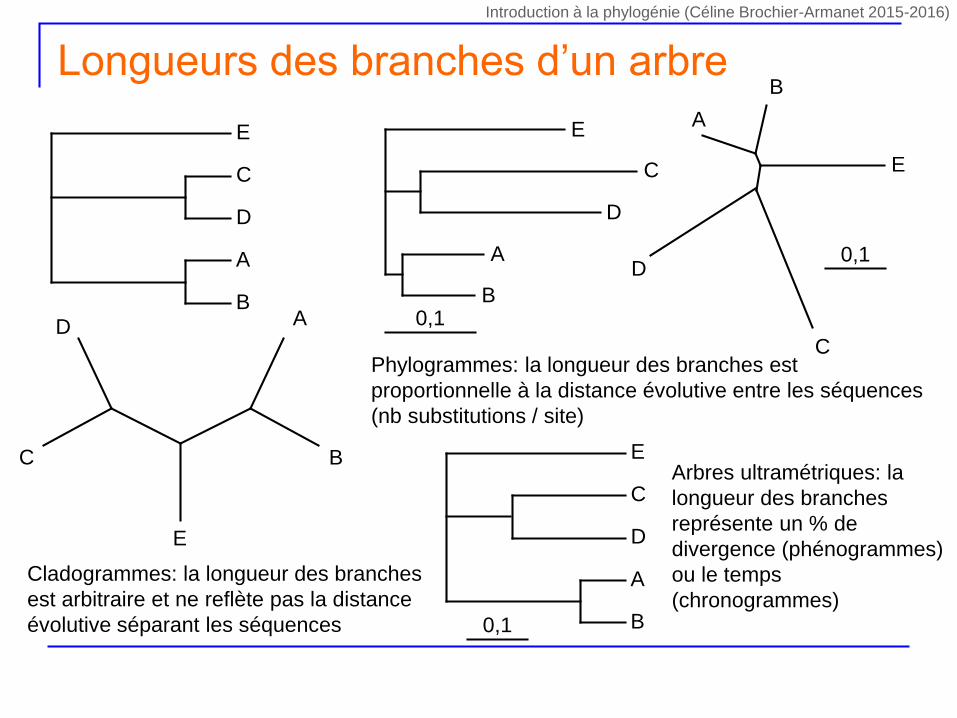
Longueurs des branches d’un arbre
0,1
E
C
D
A
B
E
C
D A
B
Cladogrammes: la longueur des branches
est arbitraire et ne reflète pas la distance
évolutive séparant les séquences
Phylogrammes: la longueur des branches est
proportionnelle à la distance évolutive entre les séquences
(nb substitutions / site)
0,1
E
C
D
A
B
E
C
D
A
B
E
C
D
A
B
Arbres ultramétriques: la
longueur des branches
représente un % de
divergence (phénogrammes)
ou le temps
(chronogrammes)
0,1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Apparentement et similarité
B est plus apparenté à A qu’à C, D ou E
B est apparenté de manière égale à C et D
B est plus apparenté à C ou D qu’à E
C est plus apparenté à D qu’à A, B ou E
C est plus apparenté A ou B qu’à E
E est aussi apparenté à A, B, C ou D
A, B, C, D et E sont apparentés de manière égale à leur ancêtre commun
0,1
E
C
D
A
B
H2
H1
H3
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Apparentement et similarité
… mais C est plus similaire à A qu’à D
0,1
E
D
A
B
C
AHHHHHHCACHDHCDCdddddddd
11332222
H2
H1
H3
B est plus apparentés à A qu’à C, D ou E
B est apparenté de manière égale à C et D
B est plus apparenté à C ou D qu’à E
C est plus apparenté à D qu’à A, B ou E
C est plus apparenté A ou B qu’à E
E est aussi apparenté à A, B, C ou D
A, B, C, D et E sont apparenté de manière égale à leur ancêtre commun
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
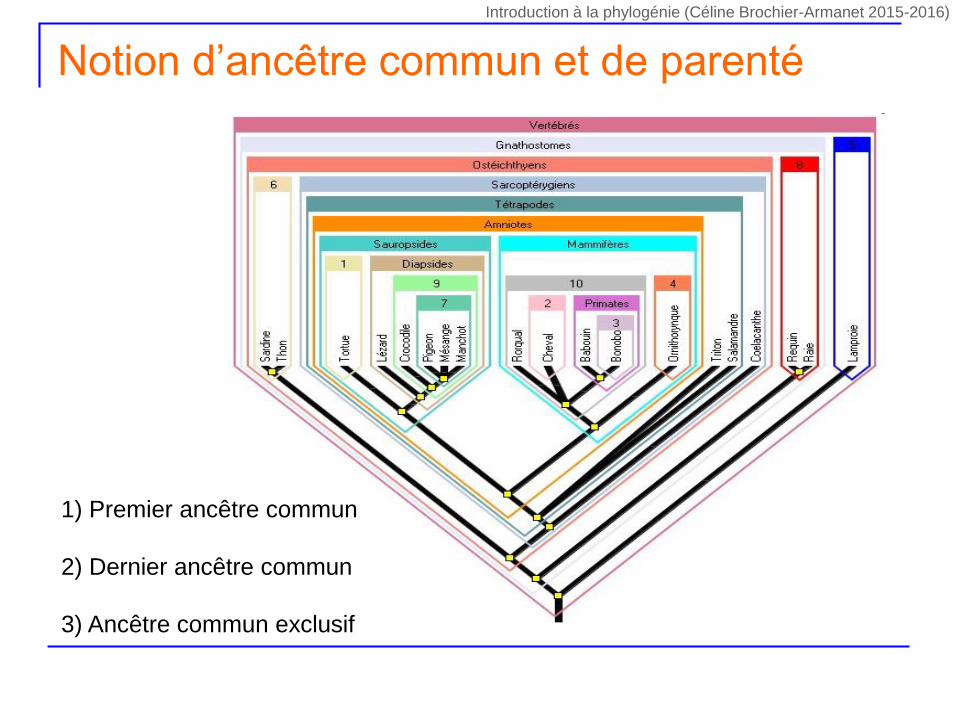
Notion d’ancêtre commun et de parenté
1) Premier ancêtre commun
2) Dernier ancêtre commun
3) Ancêtre commun exclusif
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Divergence ≠ diversification d’un groupe
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
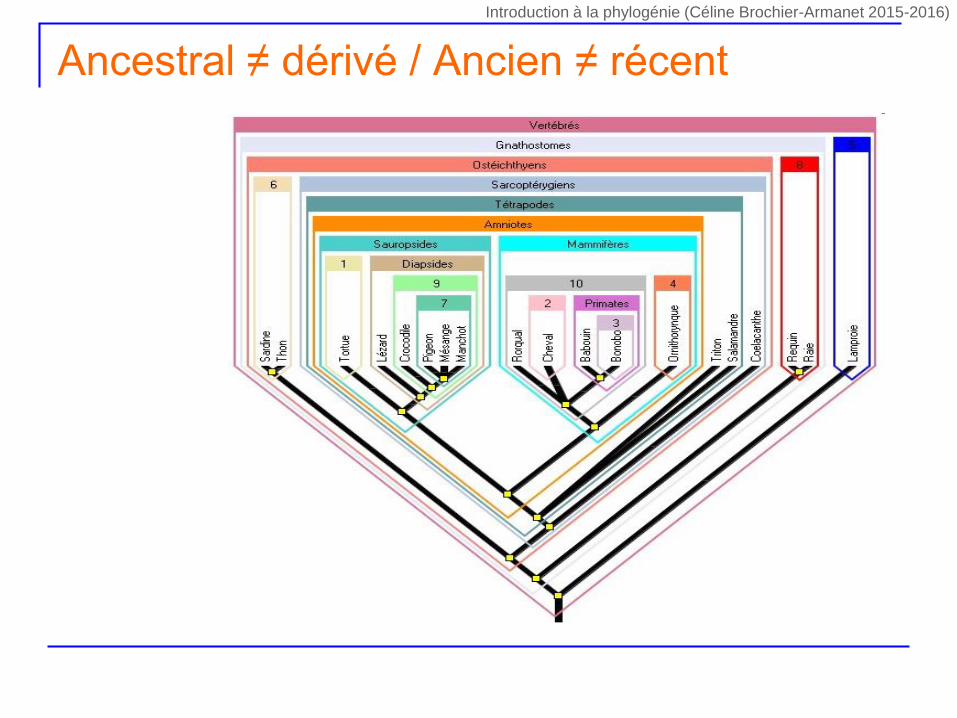
Ancestral ≠ dérivé / Ancien ≠ récent
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
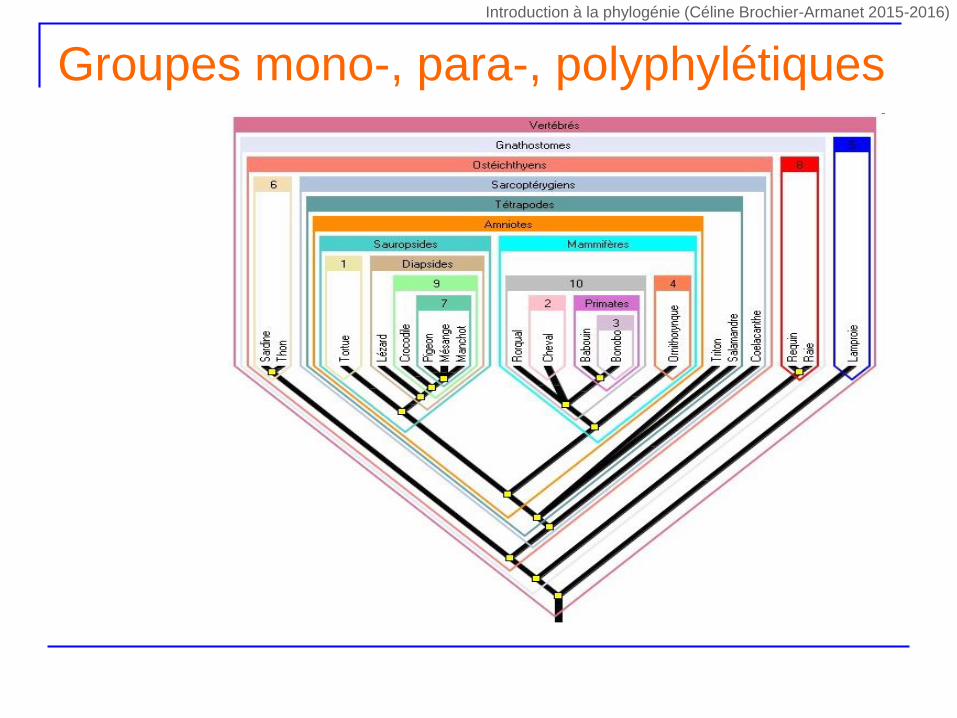
Groupes mono-, para-, polyphylétiques
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
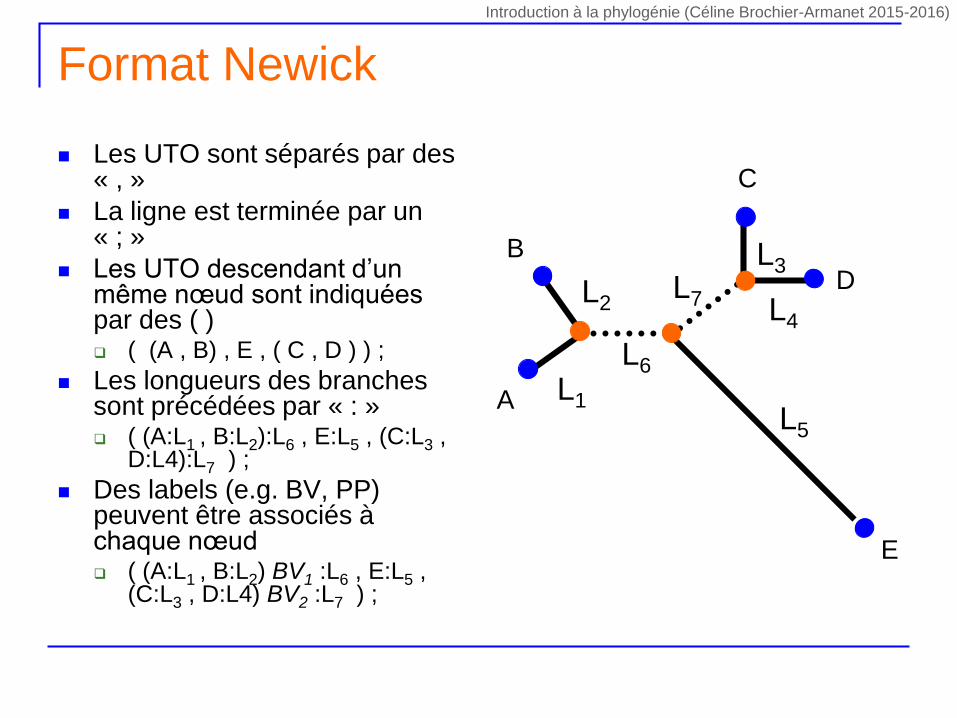
Format Newick
Les UTO sont séparés par des « , »
La ligne est terminée par un « ; »
Les UTO descendant d’un même nœud sont indiquées par des ( ) ( (A , B) , E , ( C , D ) ) ;
Les longueurs des branches sont précédées par « : » ( (A:L1 , B:L2):L6 , E:L5 , (C:L3 ,
D:L4):L7 ) ;
Des labels (e.g. BV, PP) peuvent être associés à chaque nœud ( (A:L1 , B:L2) BV1 :L6 , E:L5 ,
(C:L3 , D:L4) BV2 :L7 ) ;
B
E
C
D
A
L2
L1
L3
L4
L7
L6
L5
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
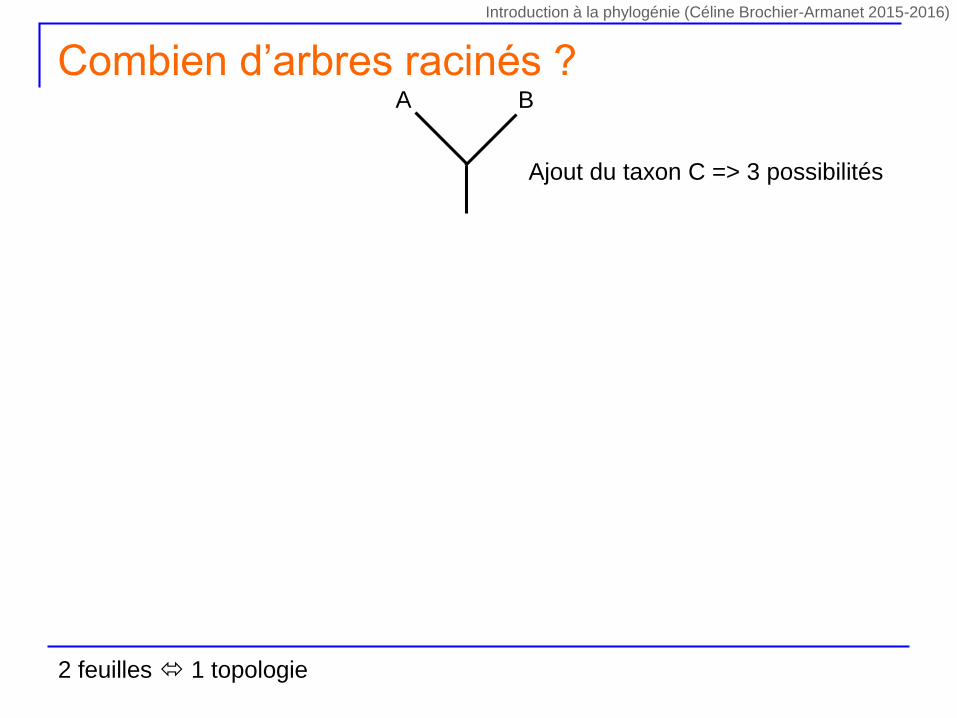
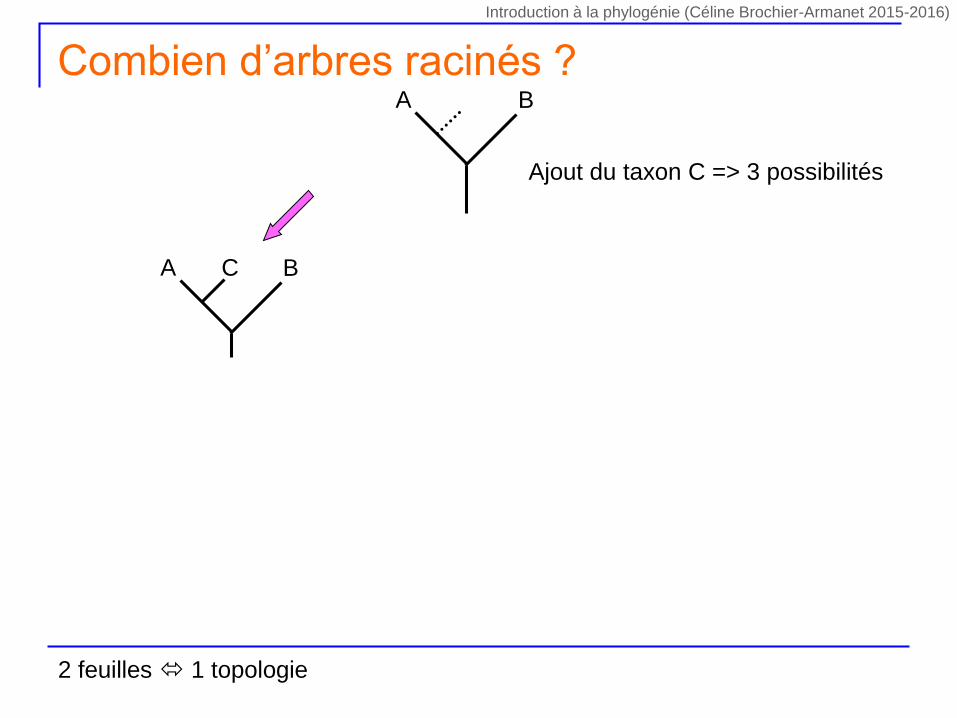
Ajout du taxon C => 3 possibilités
A B
Combien d’arbres racinés ?
2 feuilles 1 topologie
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
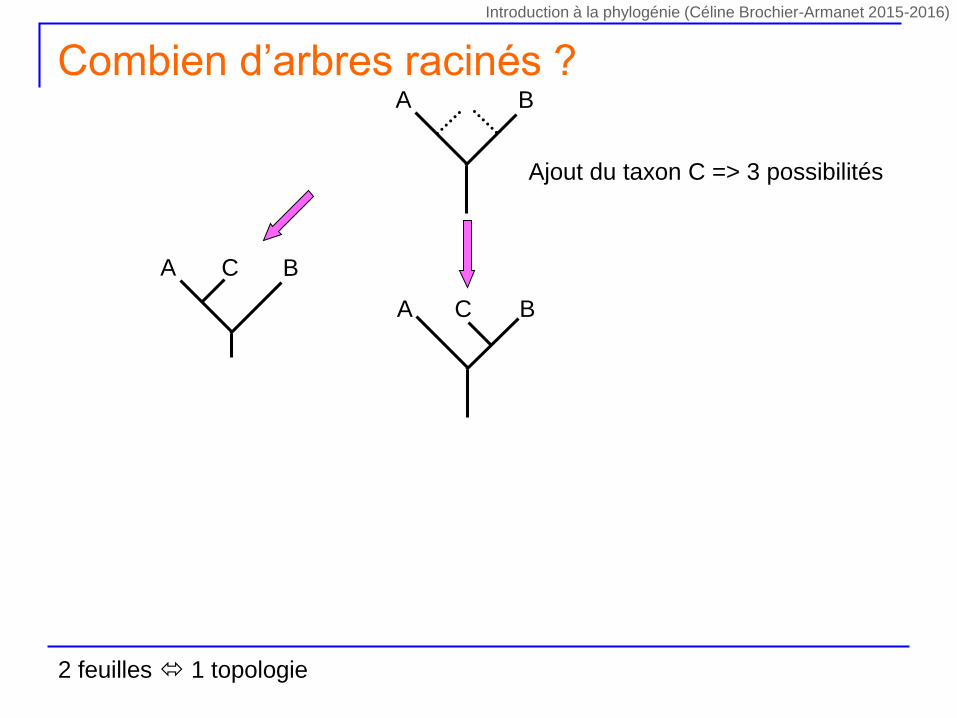
Ajout du taxon C => 3 possibilités
A BC
A B
Combien d’arbres racinés ?
2 feuilles 1 topologie
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
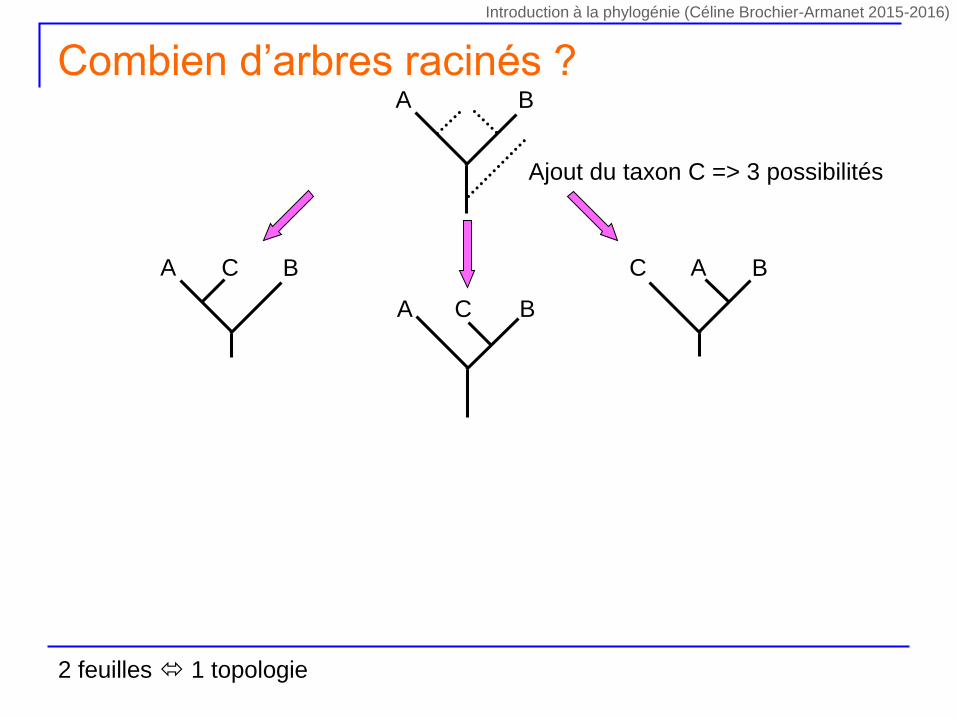
Ajout du taxon C => 3 possibilités
A BC
A B
A BC
Combien d’arbres racinés ?
2 feuilles 1 topologie
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Ajout du taxon C => 3 possibilités
A BC C BA
A B
A BC
Combien d’arbres racinés ?
2 feuilles 1 topologie
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Ajout du taxon C => 3 possibilités
A BC C BA
A B
A BC
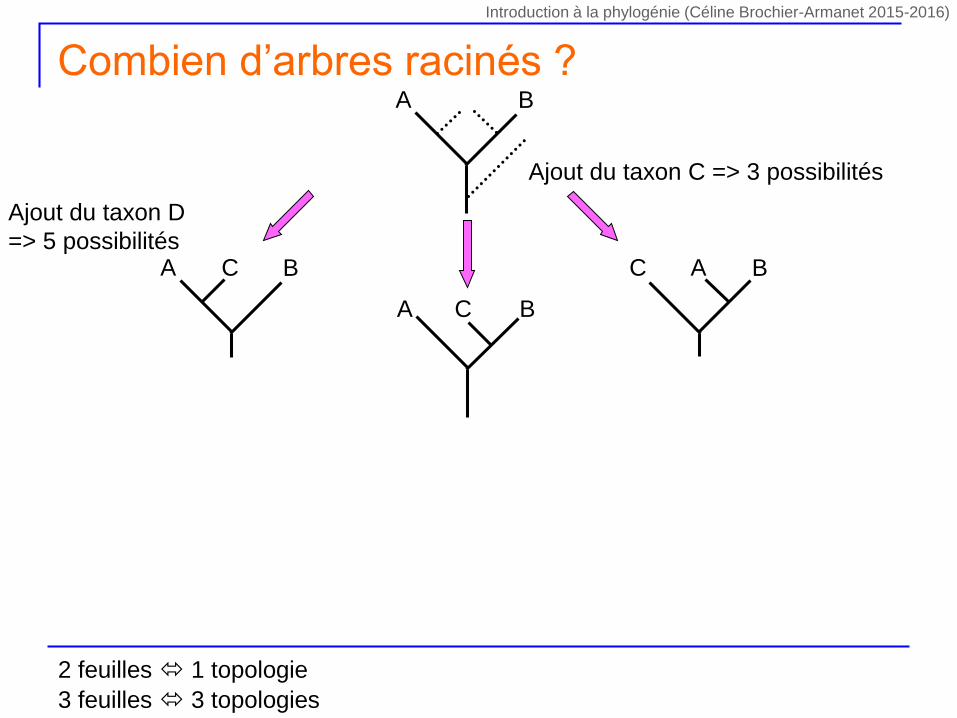
Ajout du taxon D
=> 5 possibilités
Combien d’arbres racinés ?
2 feuilles 1 topologie
3 feuilles 3 topologies
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
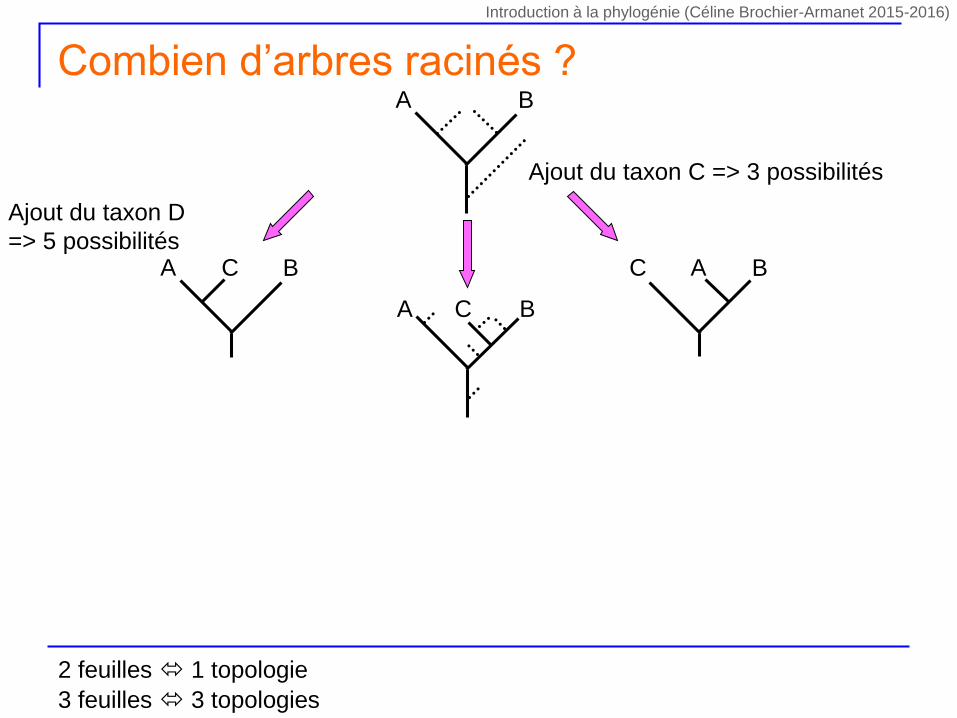
Ajout du taxon C => 3 possibilités
A BC C BA
A B
A BC
Ajout du taxon D
=> 5 possibilités
Combien d’arbres racinés ?
2 feuilles 1 topologie
3 feuilles 3 topologies
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
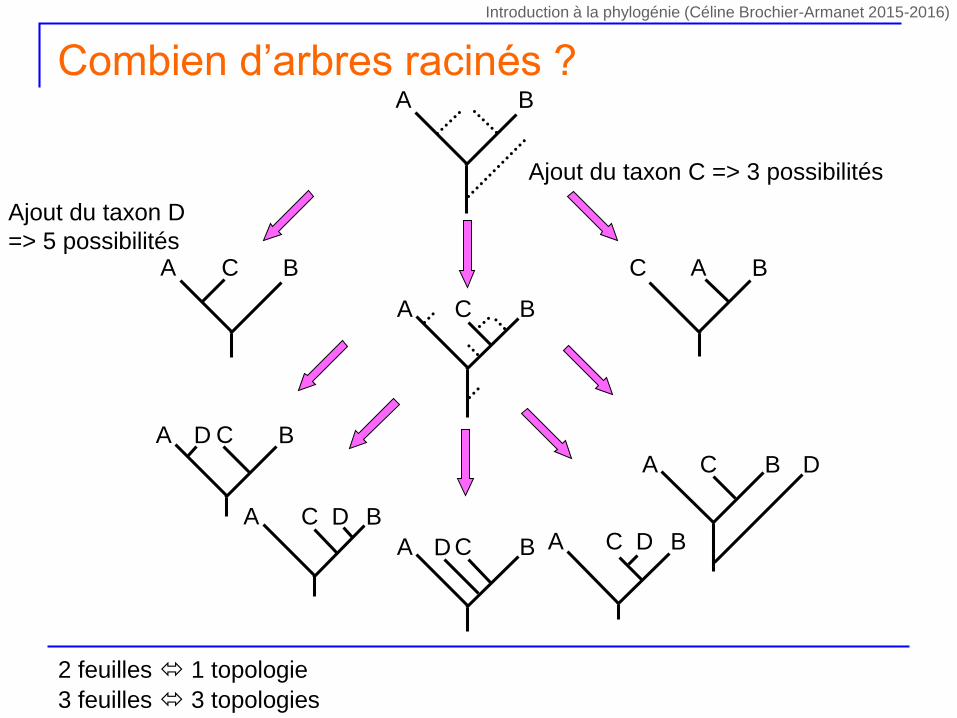
Ajout du taxon C => 3 possibilités
A BC C BA
A B
A BC
A BCD
A BC D
A BC D
A BC DA BCD
Ajout du taxon D
=> 5 possibilités
Combien d’arbres racinés ?
2 feuilles 1 topologie
3 feuilles 3 topologies
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
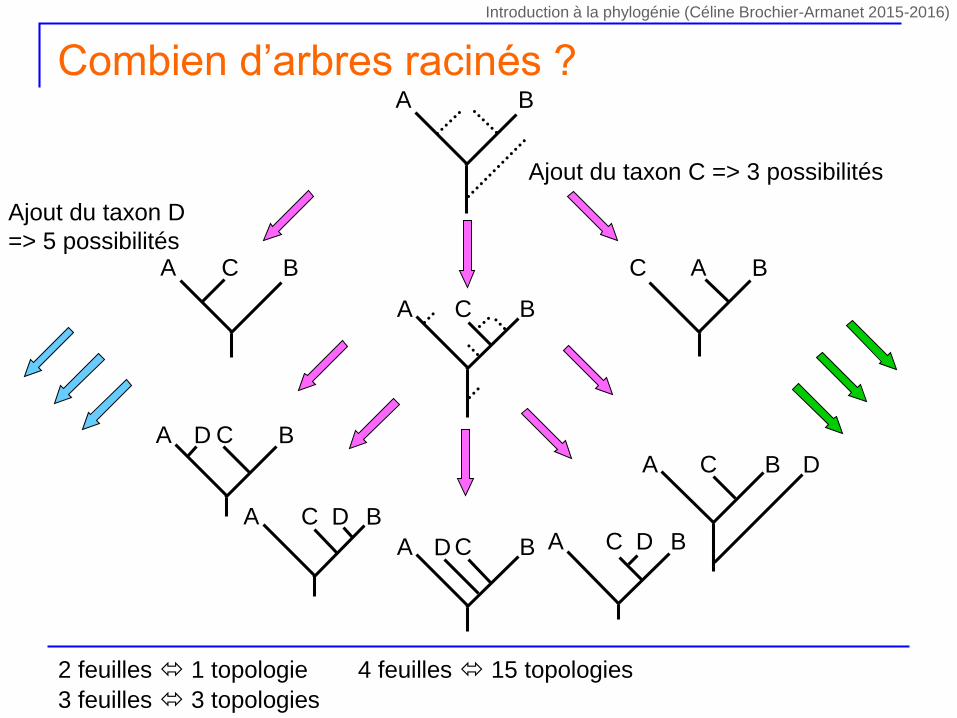
Ajout du taxon C => 3 possibilités
A BC C BA
A B
A BC
A BCD
A BC D
A BC D
A BC DA BCD
Ajout du taxon D
=> 5 possibilités
Combien d’arbres racinés ?
2 feuilles 1 topologie
3 feuilles 3 topologies
4 feuilles 15 topologies
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Combien d’arbres? Arbres racinés
1
3
15
105
945
10 395
135 135
2 027 025
34 459 425
~ 8.2 x 1021
~ 2.75 x 1076
• Nb UTO
2
3
4
5
6
7
8
9
10
20
30
NNR = (2n-5)! / [2n-3 x (n-3)] !
)!2(2
)!32(
2
n
n
n
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
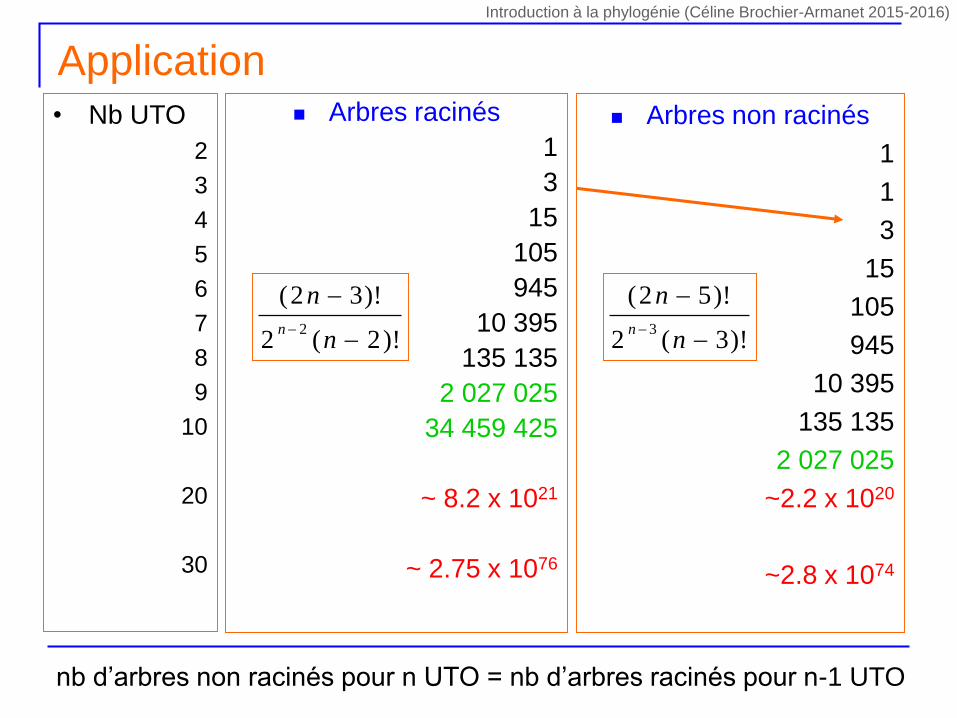
Arbres non racinés
1
1
3
15
105
945
10 395
135 135
2 027 025
~2.2 x 1020
~2.8 x 1074
Application
)!3(2
)!52(
3
n
n
n
NR = (2n-3)! / [2n-2 x (n-2)] ! NNR = (2n-5)! / [2n-3 x (n-3)] !nb d’arbres non racinés pour n UTO = nb d’arbres racinés pour n-1 UTO
Arbres racinés
1
3
15
105
945
10 395
135 135
2 027 025
34 459 425
~ 8.2 x 1021
~ 2.75 x 1076
• Nb UTO
2
3
4
5
6
7
8
9
10
20
30
)!2(2
)!32(
2
n
n
n
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Estimation de la robustesse des arbres :
Bootstrap
Un problème majeur en phylogénie est d’estimer la force des regroupements observés
Idée = estimer la variabilité de l’arbre (ou d’une partie de l’arbre) Étude de la robustesse des arbres
Si un arbre est robuste i.e. fortement soutenu par les données alors sa variabilité sera faible, et les regroupements observés devraient être retrouvés même si on perturbe un peu les données
Si un arbre est peu robuste alors il aura une grande variabilité, et les regroupements observés seront très instables en cas de perturbation des données
Estimation de la force avec laquelle les données (i.e. l’alignement) soutiennent les regroupements observés
Association d’un estimateur de la robustesse à chaque branche de l’arbre
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

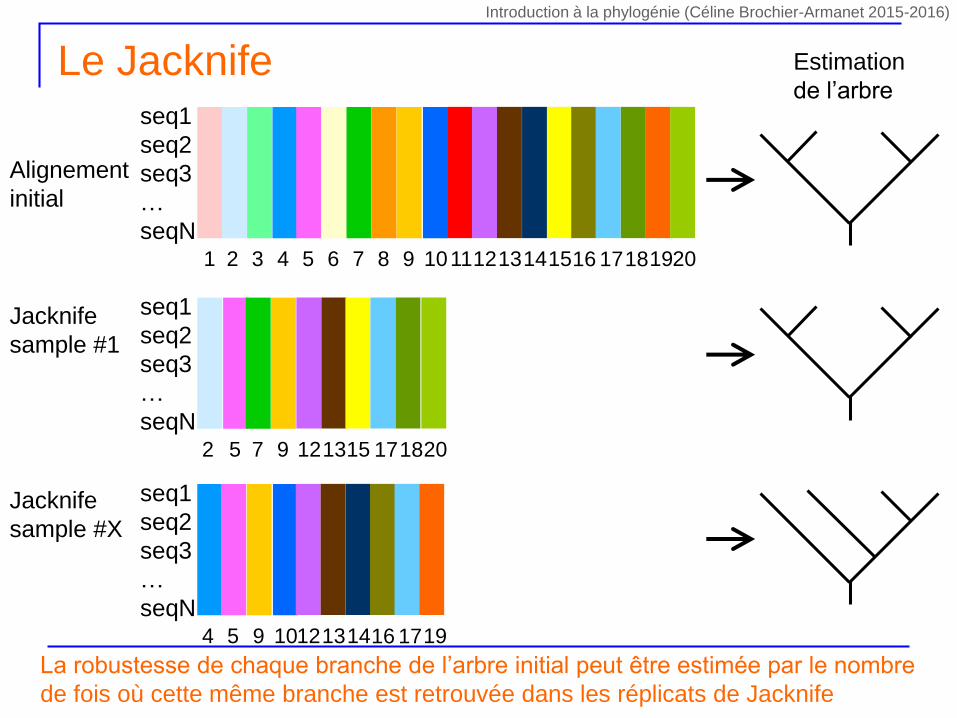
Le Jacknife – Wu 1986 Felsenstein 1985
On réalise X tirages sans remise de n/2 sites au sein du jeu de données initial
Construction d’un nouvel alignement contenant le même nombre de séquences et la moitié des sites de l’alignement initial
Certains sites seront totalement absents du nouvel alignement
pondération des sites par la valeur 0 ou 1
D’un tirage à l’autre les combinaisons de sites absents seront différentes et les combinaisons de sites présents seront différentes
Chaque tirage (i.e. combinaison de sites) est unique, car la pondération des sites change à chaque tirage
Pour chaque tirage on calcule la phylogénie correspondante par la même méthode
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Le Jacknifeseq1
seq2
seq3
…
seqN1 2 3 4 5 6 7 8 9 10111213141516 17181920
Alignement
initial
Jacknife
sample #1
Jacknife
sample #X
seq1
seq2
seq3
…
seqN2 5 7 9 121315 171820
seq1
seq2
seq3
…
seqN4 5 9 1012131416 1719
La robustesse de chaque branche de l’arbre initial peut être estimée par le nombre
de fois où cette même branche est retrouvée dans les réplicats de Jacknife
Estimation
de l’arbre
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

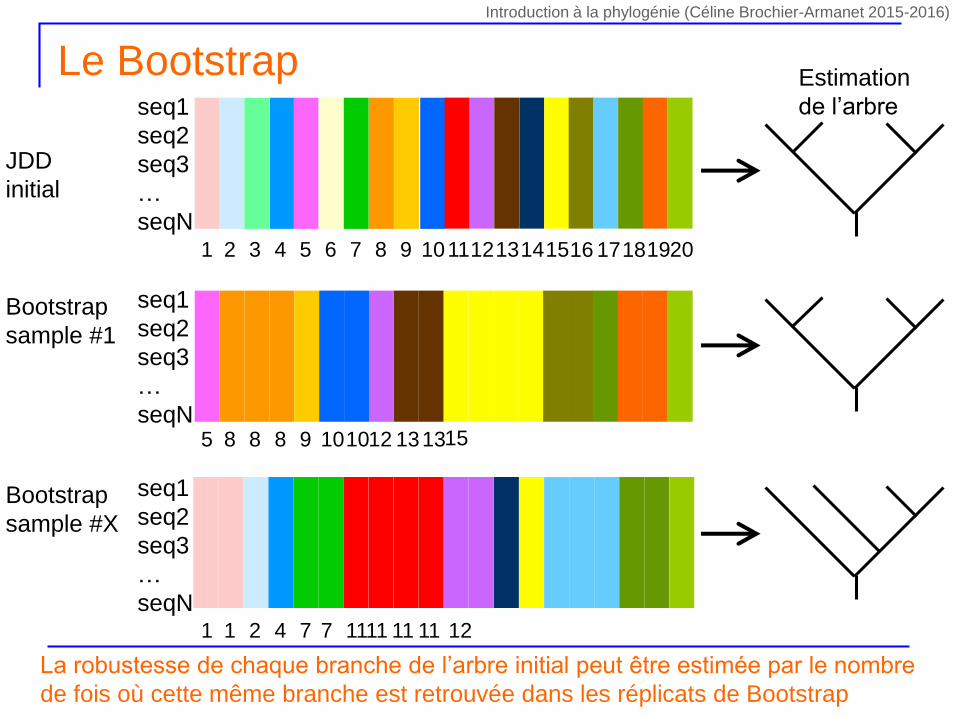
Le Bootstrap
Contrairement au Jacknife, la procédure de bootstrap s’appuie sur des alignements ré-échantillonnés de même taille que l’alignement initial
On réalise X tirages avec remise de n sites parmi les n sites contenus dans l’alignement initial
Certains sites seront présents plusieurs fois dans le nouvel alignement
Certains sites seront absents du nouvel alignement
Pondération des caractères variant entre 0 et n
D’un tirage à l’autre les sites absents ou présents plus d’une fois seront différents
Chaque tirage (i.e. combinaison de sites) est unique car la pondération des sites est aléatoire d’un tirage à l’autre
Pour chaque tirage on calcule la phylogénie correspondante par la même méthode
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
seq1
seq2
seq3
…
seqN1 2 3 4 5 6 7 8 9 10111213141516 17181920
JDD
initial
Bootstrap
sample #1
Bootstrap
sample #X
seq1
seq2
seq3
…
seqN1 1 2 4 7 7 1111 11 11 12
seq1
seq2
seq3
…
seqN5 8 8 8 9 101012 131315
Le Bootstrap Estimation
de l’arbre
La robustesse de chaque branche de l’arbre initial peut être estimée par le nombre
de fois où cette même branche est retrouvée dans les réplicats de Bootstrap
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
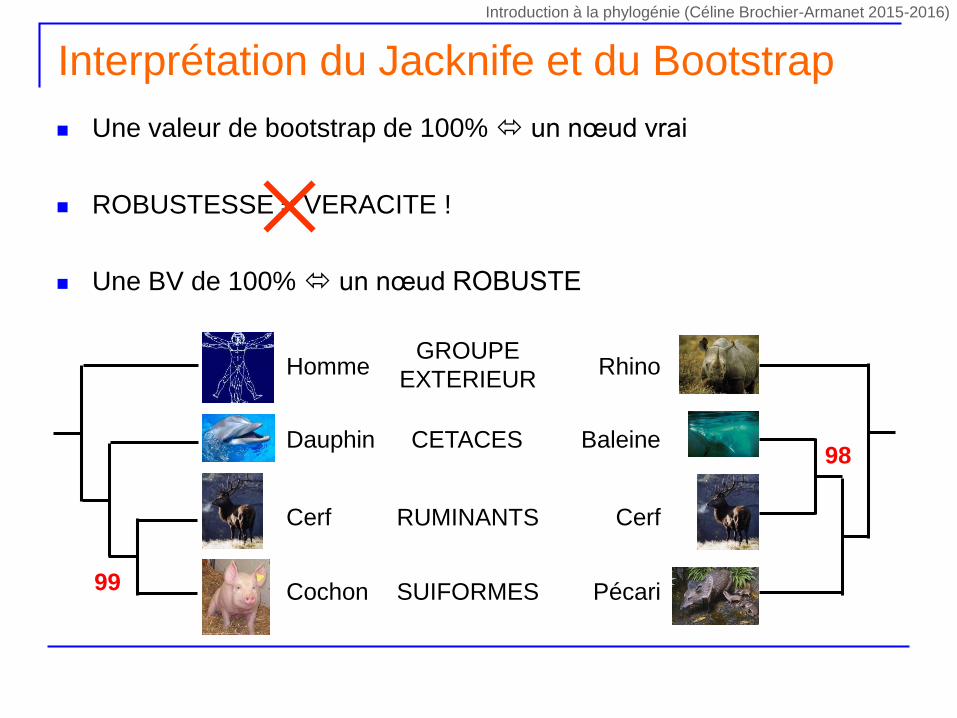
Une valeur de bootstrap de 100% un nœud vrai
ROBUSTESSE VERACITE !
Une BV de 100% un nœud ROBUSTE
Interprétation du Jacknife et du Bootstrap
PécariCochon SUIFORMES
CerfCerf RUMINANTS
BaleineDauphin CETACES
RhinoHommeGROUPE
EXTERIEUR
99
98
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Cause de l’incongruence/problèmes
rencontrés en phylogénie moléculaire
Problèmes d’échantillonnages
Séquences trop courtes => effets stochastiques
Échantillonnage taxonomique trop réduit
Problèmes liés à la divergence des séquences
Séquences pas assez variables
Séquences trop divergentes => saturation
Séquences présentant des taux d’évolution hétérogènes
(Attraction des longues branches)
=> Facteurs non exclusifs !
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Pour vous entrainer
http://www.frangun.org/
Rubrique enseignement
« Tree Thinking Challenge »
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Caractères et états de caractères
Caractère = caractéristique observable d’un organisme
(quantitative ou qualitative)
État de caractère = forme particulière d’un caractère dans une
UTO particulière (variable continue ou discrète)
Exemple
Caractère Taille Pos. 68 CYTB
État de caractères 1,68 cm Alanine
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Arbres vrais et arbres inférés
La succession des événements de spéciation, de duplications et de
transferts qui a conduit aux séquences étudiées est unique !
Parmi tous les arbres possibles un seul représente la véritable
histoire évolutive = ARBRE VRAI
Le (ou les) arbre(s) obtenu(s) à partir d’un gène particulier et une
méthode de reconstruction est appelé ARBRE INFERE
Hypothèse sur la manière dont s’est faite l’évolution
L’ARBRE INFERE ARBRE VRAI
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Reconstruire un arbre
Inférer une phylogénie procédure d’estimation de la véritable
histoire évolutive à partir de données incomplètes
Spécification de critères permettant de sélectionner un ou plusieurs
arbres parmi l’ensemble des arbres possibles
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Reconstruire un arbre
Inférer une phylogénie procédure d’estimation de la véritable
histoire évolutive à partir de données incomplètes
Spécification de critères permettant de sélectionner un ou plusieurs
arbres parmi l’ensemble des arbres possibles
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Meilleur arbre
(maximum global)

Méthodes
Méthodes de distances Recherche l’arbre qui représente au mieux les distances évolutives
entre paires de séquences
Requière l’estimation des distances évolutives entre paires de séquences, sachant un modèle d’évolution
UPGMA, NJ, minimum d’évolution, moindres carrés…
Méthodes cladistiques Recherche l’arbre impliquant le moins de changements évolutifs
permettant d’expliquer les données
Considèrent les sites individuellement
Maximum de parcimonie
Méthodes statistiques Recherche l’arbre ayant la plus forte vraisemblance sous le modèle
d’évolution considéré
Considèrent les sites individuellement
Maximum de vraisemblance, Méthodes bayésiennes
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Données utilisées en phylogénie moléculaire
Point de départ = alignement de séquences homologues
Arrivée = arbre décrivant les liens évolutifs entre les séquences de
l’alignement
0.1
neuroglobin
cytoglobin
myoglobin53
58
beta
delta
98
epsilon
gammaA
gammaG100
80
100
mu
zeta
theta
alpha2
alpha1100
79
76
52
(Alignement des 13 globines humaines réalisé avec clustalW (http://www.frangun.org/HSglobin_A.fasta),
arbre construit avec Seaview (BioNJ, 100 réplicats de bootstrap))
109 / 230 positions
conservées pour l’analyse
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Alignements et gaps Chaque colonne de l’alignement représente une position (ou site)
composée de résidus homologues, cad dérivant d’un même site ancêtre
La qualité des alignements est essentielle
Les régions où l’alignement est ambigu doivent être retirées (automatiquement ou manuellement) avant l’analyse phylogénique
La plupart des méthodes de reconstruction ne prend en compte que les substitutions et non les événements d’insertions/délétions
Les sites contenant des gaps sont ignorés
(ClustalW) (Muscle)
230 218
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Méthodes de distances
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Méthodes de distances - Principe général
Plusieurs méthodes (UMGMA, NJ, ME, etc.)
Deux grandes étapes
Construction de la matrice de distances
Reconstruction d’un arbre phylogénique dont les (di,j) sont les + proches possibles des (di,j) minimise Q
Alignement de séquences
homologues
Modèle d’évo.
4,53,52,51,55
5,43,42,41,44
5,34,32,31,33
5,24,23,21,22
5,14,13,12,11
54321
ddddS
ddddS
ddddS
ddddS
ddddS
SSSSS
n
i
n
j
jijidQ
1
,, )( d
2l
8l
S1S2
S3
S4 S5
1l5l
6l3l
4l
7l
Méthode de dist.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Un estimateur simple: La divergence
observée (p-distance)
Estimateur le + simple
= p-distance
= nb de sub. obs.
= nb de résidus comparés
Variance
Soit 2 séquences ayant des compositions homogènes
(ADN)
(Protéines)
np
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
)1()(
pppVar
p
n
75.00 p
95.00 p
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
Nb sub. obs. = 3 p = 3/14 = 0.214
Nb sub. réel. = 12 /14 d = 0.857
La p-distance d quand la saturation mutationnelle
est faible, cad quand les subs. multiples sont rares
* * * * * *
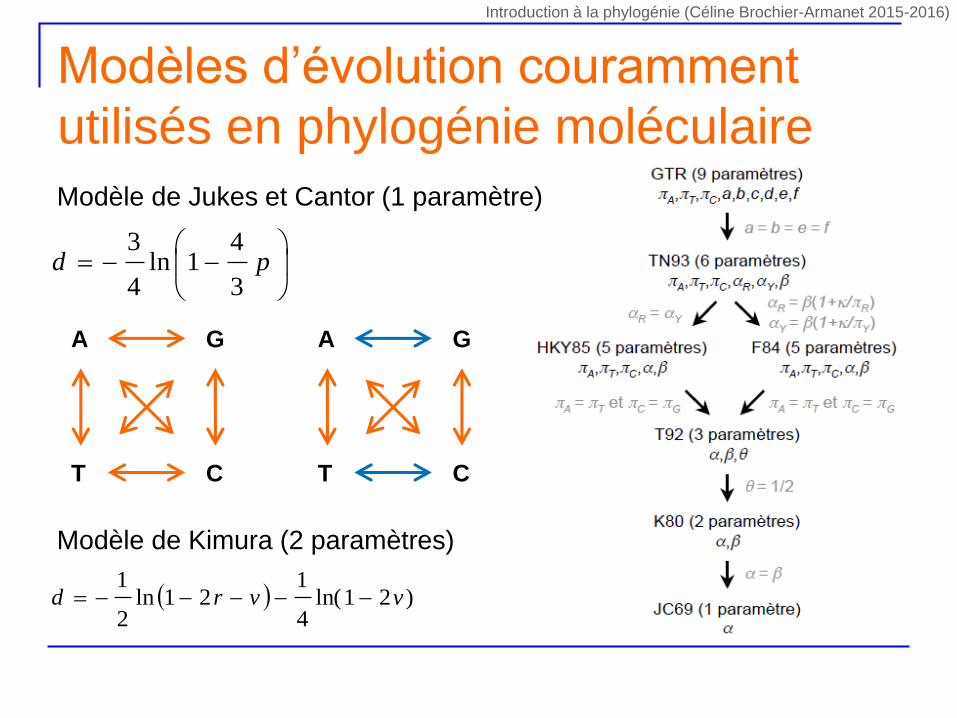
Modèles d’évolution couramment
utilisés en phylogénie moléculaire
pd
3
41ln
4
3
Modèle de Jukes et Cantor (1 paramètre)
Modèle de Kimura (2 paramètres)
)21ln(4
121ln
2
1vvrd
CT
A G
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
CT
A G
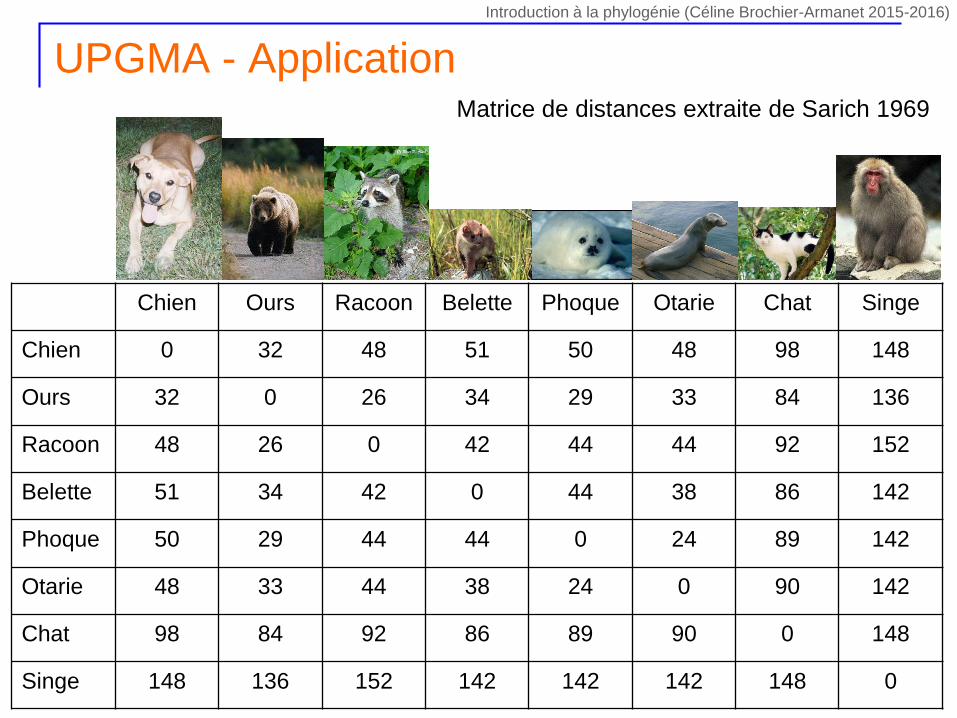
UPGMA - Application
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0 32 48 51 50 48 98 148
Ours 32 0 26 34 29 33 84 136
Racoon 48 26 0 42 44 44 92 152
Belette 51 34 42 0 44 38 86 142
Phoque 50 29 44 44 0 24 89 142
Otarie 48 33 44 38 24 0 90 142
Chat 98 84 92 86 89 90 0 148
Singe 148 136 152 142 142 142 148 0
Matrice de distances extraite de Sarich 1969
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

UPGMA (Unweighted pair-group method
with arithmetic means)
Algorithme itératif de clustering: création à chaque étape d’un nouveau cluster regroupant deux clusters proches (Sokal et Michener, 1958)
L'arbre est construit "de bas en haut" : on part des feuilles et à chaque étape on rajoute un nœud au-dessus des précédents
Condition d’application
Hypothèse d’horloge moléculaire constance des taux d’évolution le long des lignées
Caractéristiques des arbres obtenus
Ils sont racinés
Les longueurs des branches allant de la racine à n’importe quelle feuille sont égales
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

UPGMA - Algorithme
1. Trouver les deux UTO i et j pour lesquels la distance dij est la plus petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij = ni + nj (ni et nj nombre d’UTO dans les groupes i et j)
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui correspond au nouveau groupe (ij). Attribuer aux branches Li
et Lj connectant i à (ij) et j à (ij) la longueur dij /24. Calculer la distance entre le nouveau groupe (ij) et tous les
autres groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk / (ni
+ nj)5. Éliminer les colonnes et les lignes correspondant aux groupes
i et j et ajouter celles correspondant au nouveau groupe (ij)6. Si il reste un seul élément dans la matrice, arrêter, sinon
retourner en 1.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
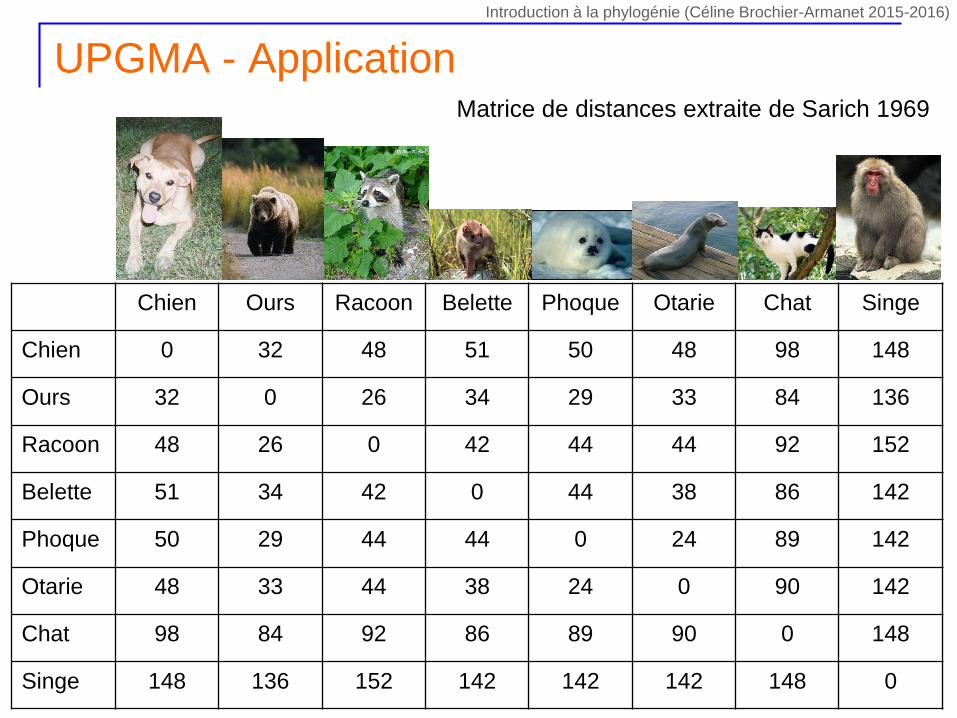
UPGMA - Application
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0 32 48 51 50 48 98 148
Ours 32 0 26 34 29 33 84 136
Racoon 48 26 0 42 44 44 92 152
Belette 51 34 42 0 44 38 86 142
Phoque 50 29 44 44 0 24 89 142
Otarie 48 33 44 38 24 0 90 142
Chat 98 84 92 86 89 90 0 148
Singe 148 136 152 142 142 142 148 0
Matrice de distances extraite de Sarich 1969
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

1. Trouver les deux UTO i et j pour lesquels la distance dij est la plus
petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij = ni +
nj (ni et nj nombre d’UTO dans les groupes i et j)
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui
correspond au nouveau groupe (ij). Attribuer aux branches Li et Lj
connectant i à (ij) et j à (ij) la longueur dij /2
4. Calculer la distance entre le nouveau groupe (ij) et tous les autres
groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk / (ni + nj)
5. Éliminer les colonnes et les lignes correspondant aux groupes i et j
et ajouter celles correspondant au nouveau groupe (ij)
6. Si il reste un seul élément dans la matrice, arrêter, sinon retourner
en 1.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
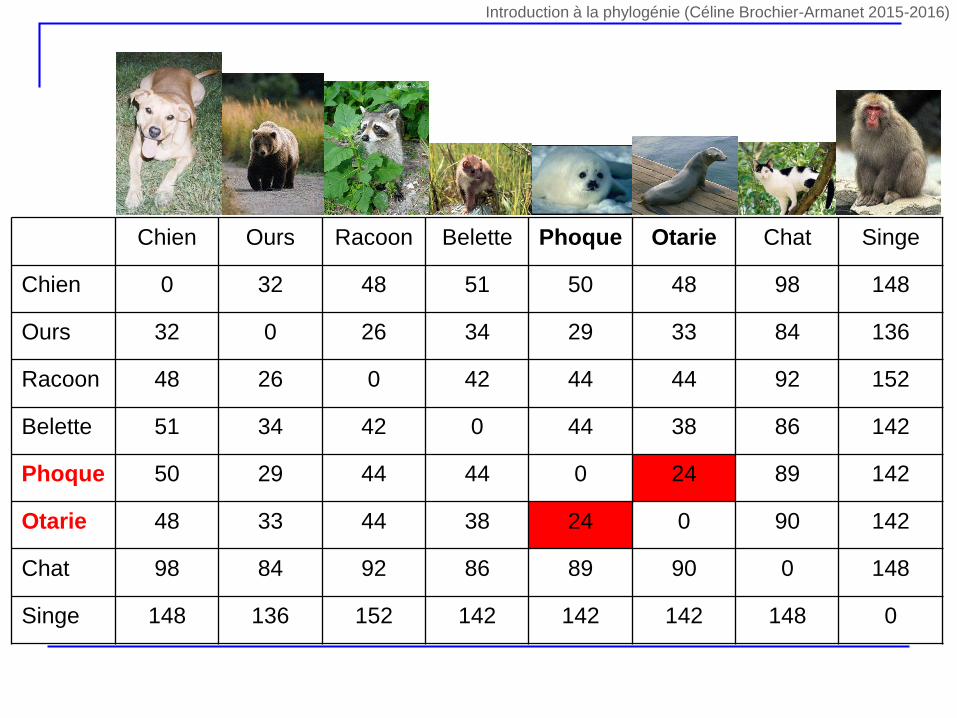
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0 32 48 51 50 48 98 148
Ours 32 0 26 34 29 33 84 136
Racoon 48 26 0 42 44 44 92 152
Belette 51 34 42 0 44 38 86 142
Phoque 50 29 44 44 0 24 89 142
Otarie 48 33 44 38 24 0 90 142
Chat 98 84 92 86 89 90 0 148
Singe 148 136 152 142 142 142 148 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

1. Trouver les deux UTO i et j pour lesquels la distance dij est la plus
petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij = ni +
nj (ni et nj nombre d’UTO dans les groupes i et j) nij = ni + nj
nij = 1 + 1 = 2
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui
correspond au nouveau groupe (ij). Attribuer aux branches Li et Lj
connectant i à (ij) et j à (ij) la longueur dij /2
4. Calculer la distance entre le nouveau groupe (ij) et tous les autres
groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk / (ni + nj)
5. Éliminer les colonnes et les lignes correspondant aux groupes i et
j et ajouter celles correspondant au nouveau groupe (ij)
6. Si il reste un seul élément dans la matrice, arrêter, sinon retourner
en 1.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

1. Trouver les deux UTO i et j pour lesquels la distance dij est
la plus petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij
= ni + nj (ni et nj nombre d’UTO dans les groupes i et j)
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui
correspond au nouveau groupe (ij).
Attribuer aux branches Li et Lj connectant i à (ij) et j à (ij) la
longueur dij /2 soit Li = 24/2 = 12 et Lj = 24/2 =12
4. Calculer la distance entre le nouveau groupe (ij) et tous les
autres groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk /
(ni + nj)
5. Éliminer les colonnes et les lignes correspondant aux
groupes i et j et ajouter celles correspondant au nouveau
groupe (ij)
6. Si il reste un seul élément dans la matrice, arrêter, sinon
retourner en 1.12 12
Ph
oq
ue
Ota
rie
(ij)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

1. Trouver les deux UTO i et j pour lesquels la distance dij est la plus
petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij = ni +
nj (ni et nj nombre d’UTO dans les groupes i et j)
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui
correspond au nouveau groupe (ij). Attribuer aux branches Li et Lj
connectant i à (ij) et j à (ij) la longueur dij /2
4. Calculer la distance entre le nouveau groupe (ij) et tous les autres
groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk / (ni + nj) dphoque-
otarie,chien = nphoque x dphoque,chien / (nphoque + notarie) + notarie x dotarie,chien /
(nphoque + notarie) = (50 + 48)/2 = 49
5. Éliminer les colonnes et les lignes correspondant aux groupes i et j
et ajouter celles correspondant au nouveau groupe (ij)
6. Si il reste un seul élément dans la matrice, arrêter, sinon retourner
en 1.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
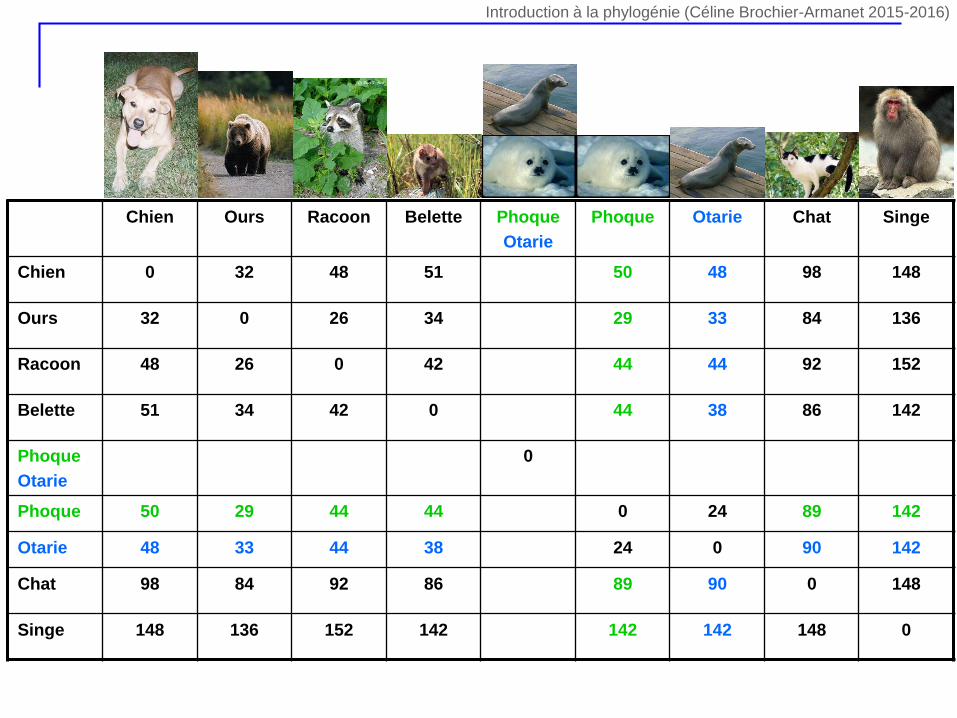
Chien Ours Racoon Belette Phoque
Otarie
Phoque Otarie Chat Singe
Chien 0 32 48 51 50 48 98 148
Ours 32 0 26 34 29 33 84 136
Racoon 48 26 0 42 44 44 92 152
Belette 51 34 42 0 44 38 86 142
Phoque
Otarie
0
Phoque 50 29 44 44 0 24 89 142
Otarie 48 33 44 38 24 0 90 142
Chat 98 84 92 86 89 90 0 148
Singe 148 136 152 142 142 142 148 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
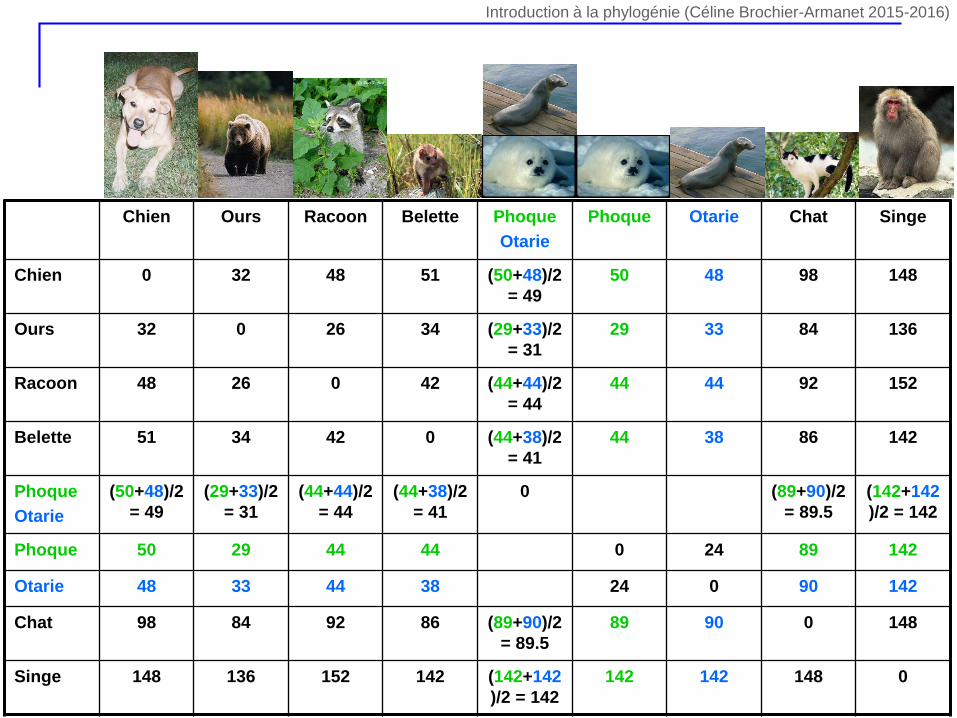
Chien Ours Racoon Belette Phoque
Otarie
Phoque Otarie Chat Singe
Chien 0 32 48 51 (50+48)/2
= 49
50 48 98 148
Ours 32 0 26 34 (29+33)/2
= 31
29 33 84 136
Racoon 48 26 0 42 (44+44)/2
= 44
44 44 92 152
Belette 51 34 42 0 (44+38)/2
= 41
44 38 86 142
Phoque
Otarie
(50+48)/2
= 49
(29+33)/2
= 31
(44+44)/2
= 44
(44+38)/2
= 41
0 (89+90)/2
= 89.5
(142+142
)/2 = 142
Phoque 50 29 44 44 0 24 89 142
Otarie 48 33 44 38 24 0 90 142
Chat 98 84 92 86 (89+90)/2
= 89.5
89 90 0 148
Singe 148 136 152 142 (142+142
)/2 = 142
142 142 148 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

1. Trouver les deux UTO i et j pour lesquels la distance dij est la
plus petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij =
ni + nj (ni et nj nombre d’UTO dans les groupes i et j)
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui
correspond au nouveau groupe (ij). Attribuer aux branches Li
et Lj connectant i à (ij) et j à (ij) la longueur dij /2
4. Calculer la distance entre le nouveau groupe (ij) et tous les
autres groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk / (ni
+ nj)
5. Éliminer les colonnes et les lignes correspondant aux groupes
i et j et ajouter celles correspondant au nouveau groupe (ij)
6. Si il reste un seul élément dans la matrice, arrêter, sinon
retourner en 1.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
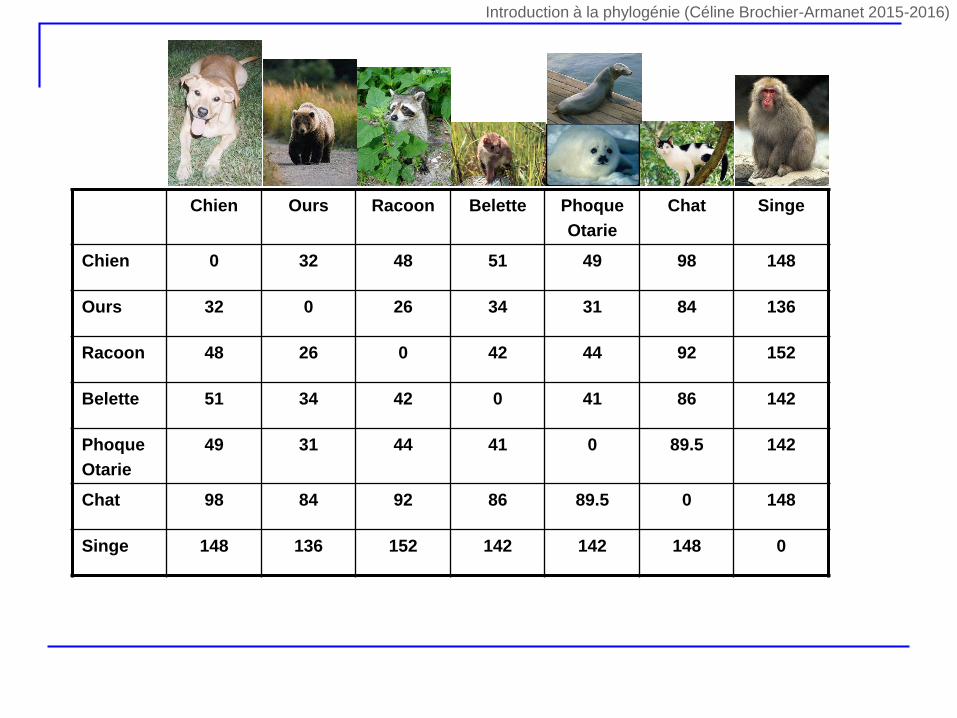
Chien Ours Racoon Belette Phoque
Otarie
Chat Singe
Chien 0 32 48 51 49 98 148
Ours 32 0 26 34 31 84 136
Racoon 48 26 0 42 44 92 152
Belette 51 34 42 0 41 86 142
Phoque
Otarie
49 31 44 41 0 89.5 142
Chat 98 84 92 86 89.5 0 148
Singe 148 136 152 142 142 148 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

1. Trouver les deux UTO i et j pour lesquels la distance dij est la
plus petite
2. Créer le nouveau groupe (ij) contenant nij membres avec nij =
ni + nj (ni et nj nombre d’UTO dans les groupes i et j)
3. Connecter i et j dans l’arbre à un nouveau nœud (ij) qui
correspond au nouveau groupe (ij). Attribuer aux branches Li
et Lj connectant i à (ij) et j à (ij) la longueur dij /2
4. Calculer la distance entre le nouveau groupe (ij) et tous les
autres groupes en utilisant dij,k = ni x dik / (ni + nj) + nj x djk / (ni
+ nj)
5. Éliminer les colonnes et les lignes correspondant aux groupes
i et j et ajouter celles correspondant au nouveau groupe (ij)
6. Si il reste un seul élément dans la matrice, arrêter, sinon
retourner en 1.
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
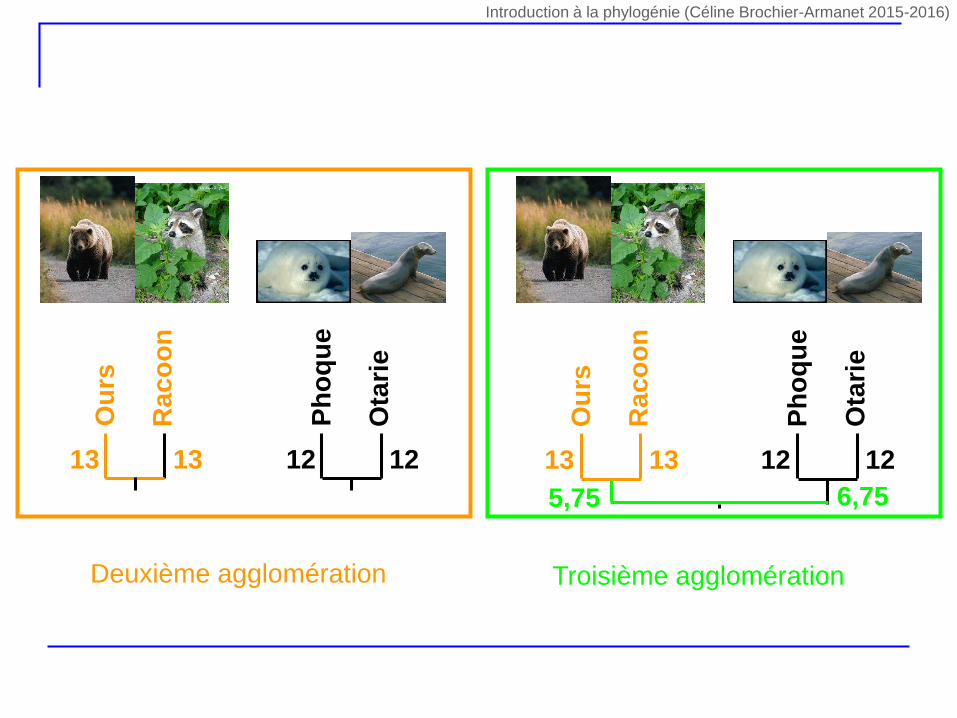
Ph
oq
ue
Ota
rie
12 12
Ou
rs
Raco
on
13 13
Ph
oq
ue
Ota
rie
12 12
Ou
rs
Ra
co
on
13 13
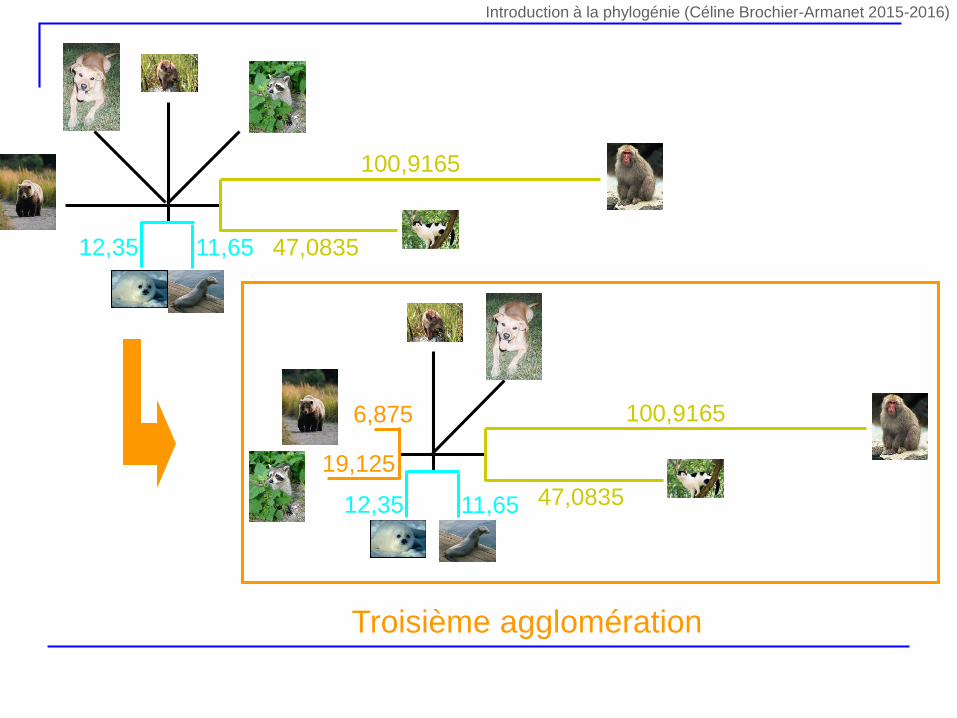
5,75 6,75
Deuxième agglomération Troisième agglomération
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
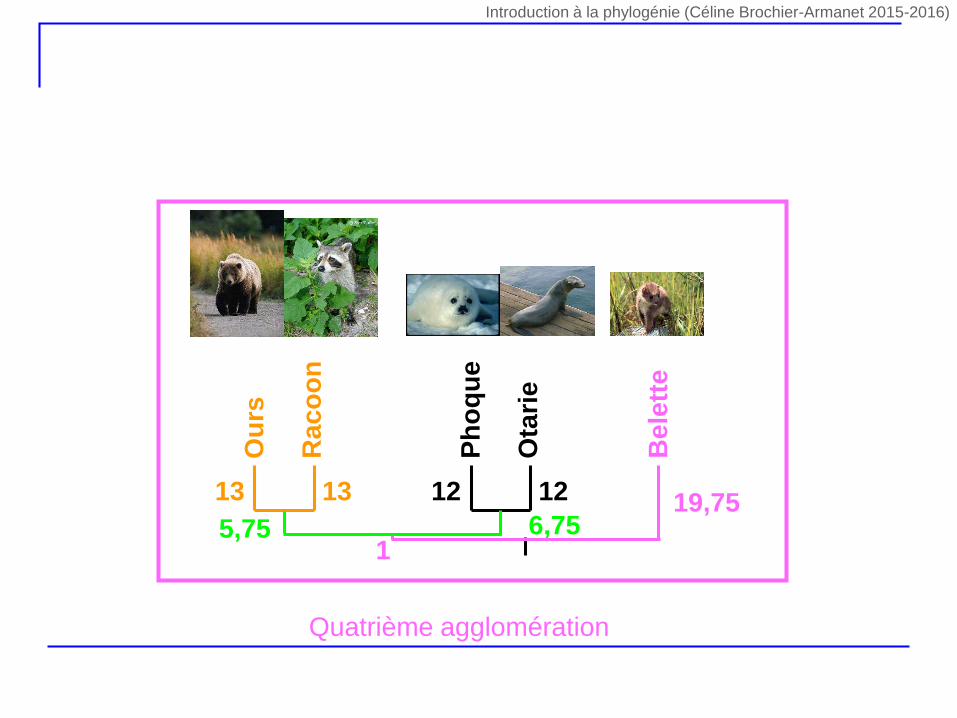
Ph
oq
ue
Ota
rie
12 12
Ou
rs
Ra
co
on
13 13
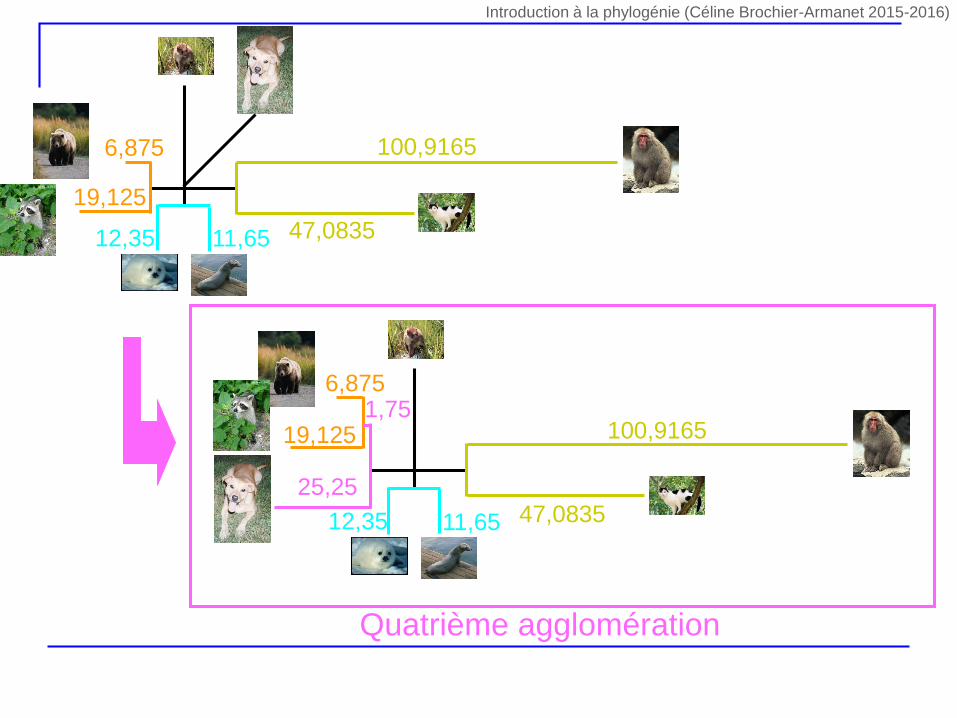
5,75 6,7519,75
Be
lett
e
1
Quatrième agglomération
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
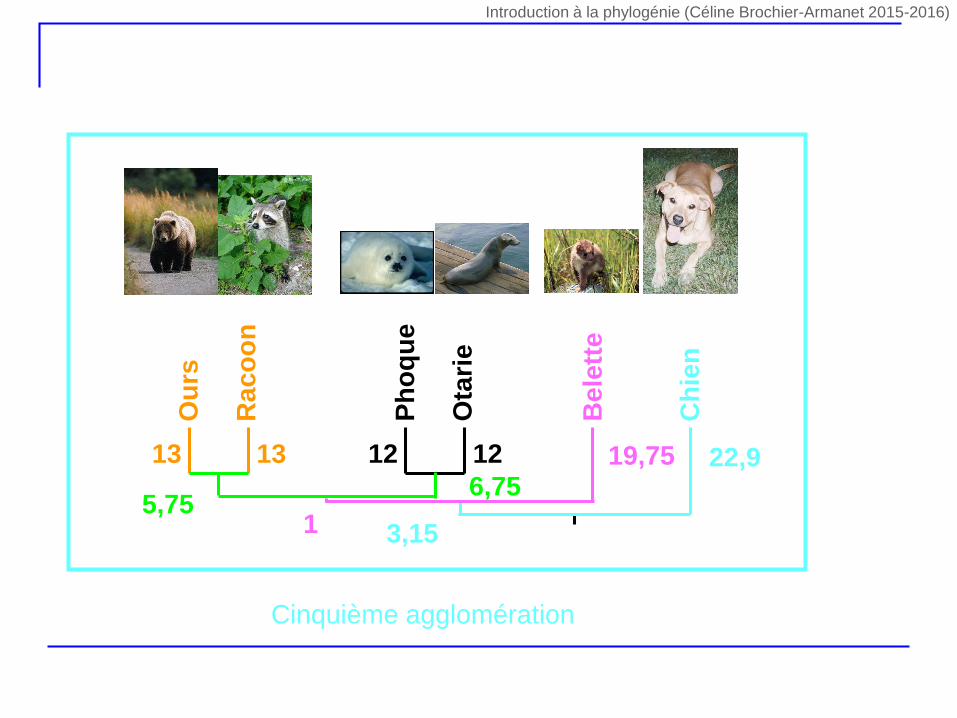
Ph
oq
ue
Ota
rie12 12
Ou
rs
Ra
co
on
13 13
5,756,75
19,75
Be
lett
e
1
Ch
ien
22,9
3,15
Cinquième agglomération
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
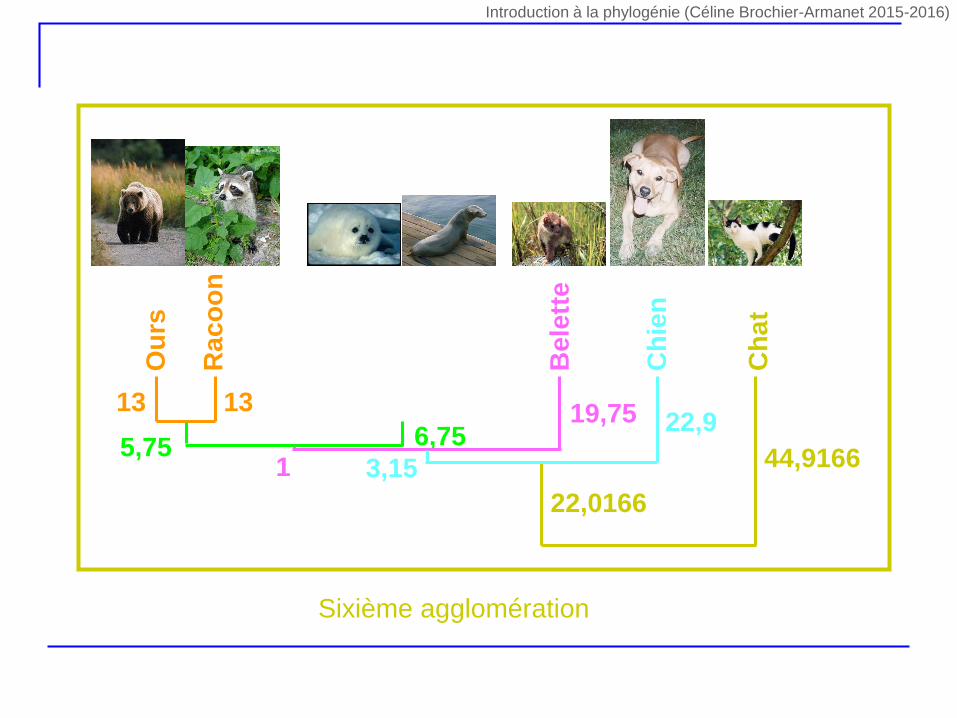
Ph
oq
ue
Ota
rie
Ou
rs
Raco
on
Be
lett
e
Ch
ien
12 1213 13
5,75 6,7519,75
1
22,9
3,15 44,9166
Ch
at
22,0166
Sixième agglomération
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
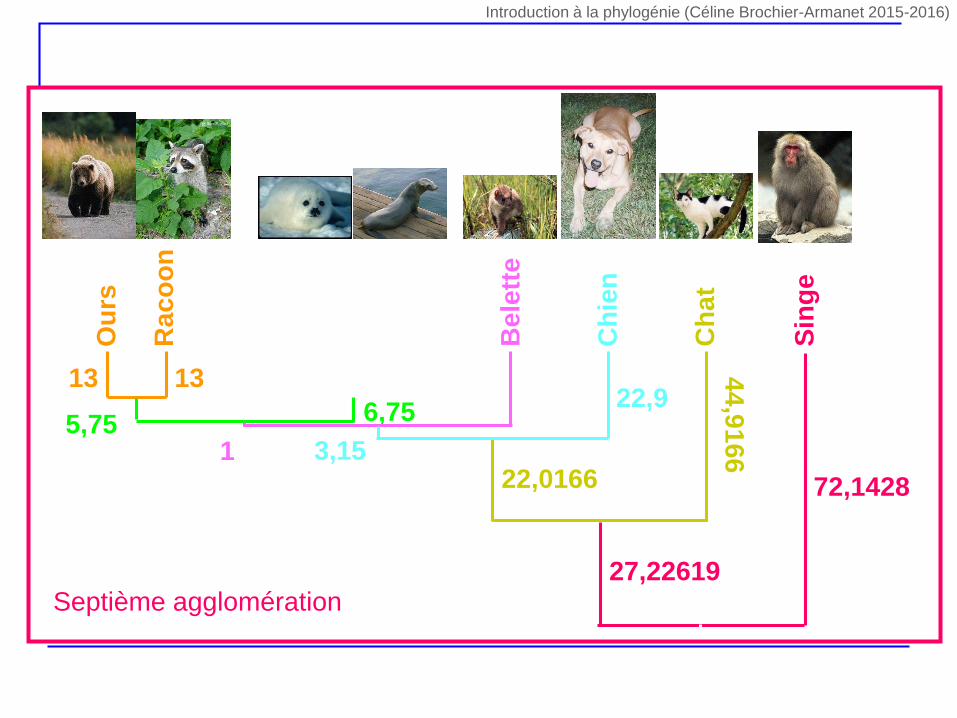
Ph
oq
ue
Ota
rie
Ou
rs
Ra
co
on
Be
lett
e
Ch
ien
12 1213 13
5,75 6,7519,75
1
22,9
3,15
44
,91
66
Ch
at
22,0166
Sin
ge
72,1428
27,22619Septième agglomération
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Conclusions sur l’UPGMA
Avantages de l’algorithme:
Rapidité & simplicité
Critiques:
Hypothèse de l’égalité des taux d’évolution entre les lignées.
Résultats faux si les distances de la matrice n’obéissent pas au critère
d’horloge moléculaire
N’est presque plus utilisée
Peut être réaliste si on étudie des espèces très proches
A
B C
D
13 4 4 112 2
A B C D
A 0 17 21 28
B 17 0 12 19
C 21 12 0 15
D 28 19 15 0
B C AD
6 6 8,5 11
2,52,5
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Le neighbor-joining (NJ)
Développé par Saitou et Nei (1987) est une approximation de l’algorithme pour trouver l’arbre le plus court (minimum évolution)
Avantages
Rapidité => permet de travailler avec un très grand nombre de taxons (plusieurs centaines)
Bonne approximation de la méthode du minimum d’évolution
Retrouve l’arbre vrai si la matrice de distances est un reflet exact d’un arbre
Conditions d’application
Les taux d’évolution ne sont pas les mêmes dans toutes les lignées
Caractéristiques des arbres obtenus
Ils sont non racinés
Principe:
A chaque étape, rechercher le couple d’UTO qui minimise la longueur totale de l’arbre
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
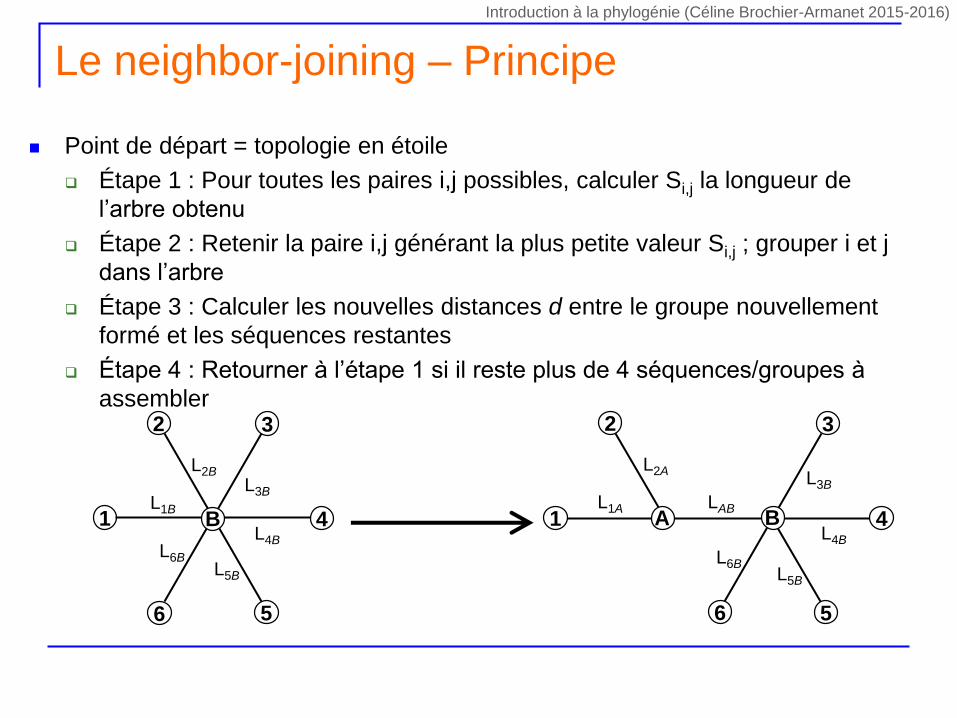
Le neighbor-joining – Principe
Point de départ = topologie en étoile
Étape 1 : Pour toutes les paires i,j possibles, calculer Si,j la longueur de
l’arbre obtenu
Étape 2 : Retenir la paire i,j générant la plus petite valeur Si,j ; grouper i et j
dans l’arbre
Étape 3 : Calculer les nouvelles distances d entre le groupe nouvellement
formé et les séquences restantes
Étape 4 : Retourner à l’étape 1 si il reste plus de 4 séquences/groupes à
assembler
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
L1B
L2B
L3B
L4B
L5B
L6B
1
2 3
4
56
BL1A
L2AL3B
L4B
L5B
L6B
LAB1
2 3
4
56
A B

Le neighbor-joining – Algorithme simplifié
(Studier et Keppler 1988)(1) Pour chaque feuille i calculer ui = Ri / (m-2) = m
k=1 dik / (m-2)
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2
Lj = dij/2 + (uj-ui)/2
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
(5) Éliminer les colonnes et les lignes correspondant aux groupes i et j et ajouter celles correspondant au nouveau groupe (ij)
(6) Si il reste un seul élément dans la matrice connecter les deux derniers nœuds, sinon retourner en 1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Le neighbor-joining – Application
(1) Pour chaque feuille i calculer ui = Ri / (m-2) = mk=1 dik /(m-2)
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2
Lj = dij/2 + (uj-ui)/2
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
(5) Éliminer les colonnes et les lignes correspondant aux groupes i et j et ajouter celles correspondant au nouveau groupe (ij)
(6) Si il reste un seul élément dans la matrice connecter les deux derniers nœuds, sinon retourner en 1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
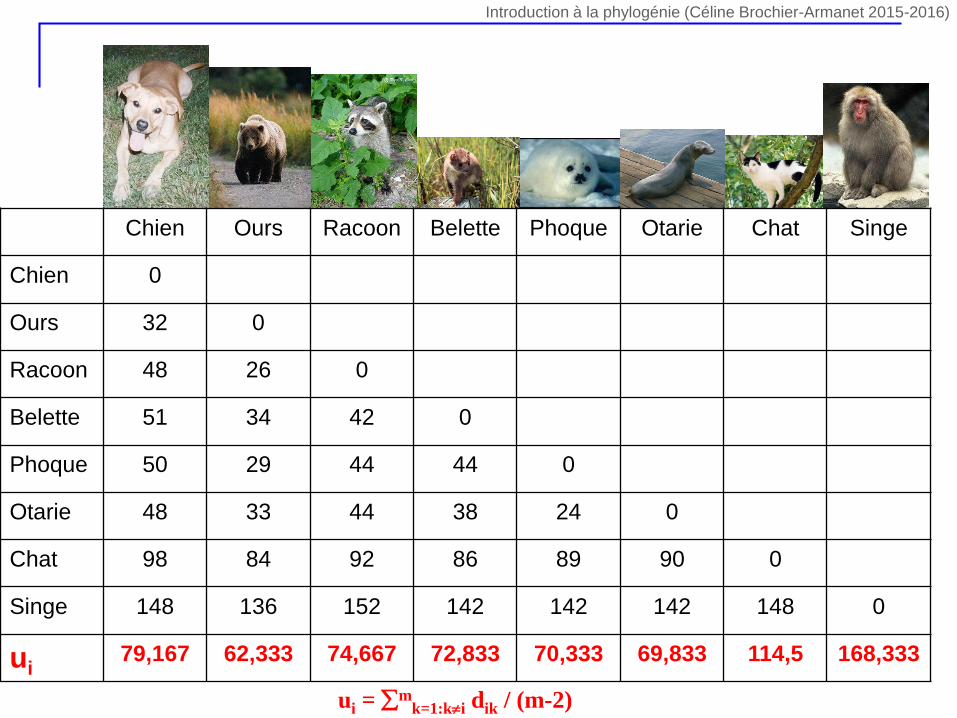
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0
Ours 32 0
Racoon 48 26 0
Belette 51 34 42 0
Phoque 50 29 44 44 0
Otarie 48 33 44 38 24 0
Chat 98 84 92 86 89 90 0
Singe 148 136 152 142 142 142 148 0
ui79,167 62,333 74,667 72,833 70,333 69,833 114,5 168,333
ui = mk=1:ki dik / (m-2)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

(1) Pour chaque feuille i calculer ui = Ri / (m-2) = mk=1 dik / (m-2)
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2
Lj = dij/2 + (uj-ui)/2
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
(5) Éliminer les colonnes et les lignes correspondant aux groupes i et j et ajouter celles correspondant au nouveau groupe (ij)
(6) Si il reste un seul élément dans la matrice connecter les deux derniers nœuds, sinon retourner en 1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
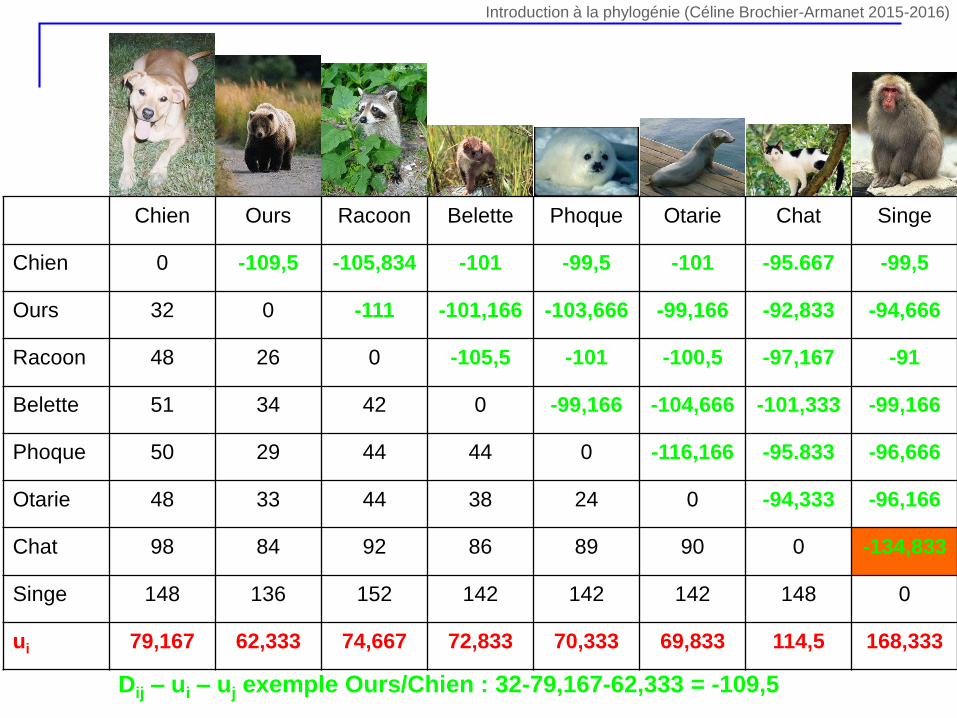
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0 -109,5 -105,834 -101 -99,5 -101 -95.667 -99,5
Ours 32 0 -111 -101,166 -103,666 -99,166 -92,833 -94,666
Racoon 48 26 0 -105,5 -101 -100,5 -97,167 -91
Belette 51 34 42 0 -99,166 -104,666 -101,333 -99,166
Phoque 50 29 44 44 0 -116,166 -95.833 -96,666
Otarie 48 33 44 38 24 0 -94,333 -96,166
Chat 98 84 92 86 89 90 0 -134,833
Singe 148 136 152 142 142 142 148 0
ui 79,167 62,333 74,667 72,833 70,333 69,833 114,5 168,333
Dij – ui – uj exemple Ours/Chien : 32-79,167-62,333 = -109,5
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
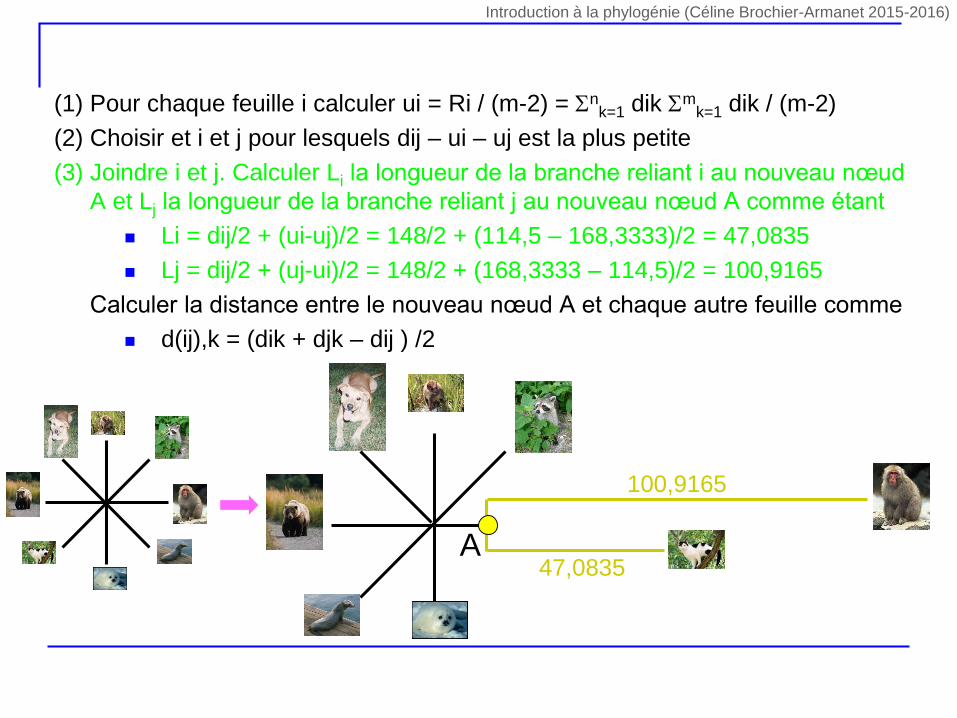
(1) Pour chaque feuille i calculer ui = Ri / (m-2) = nk=1 dik m
k=1 dik / (m-2)
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud
A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2 = 148/2 + (114,5 – 168,3333)/2 = 47,0835
Lj = dij/2 + (uj-ui)/2 = 148/2 + (168,3333 – 114,5)/2 = 100,9165
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
47,0835
100,9165
A
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

(1) Pour chaque feuille i calculer ui = Ri / (m-2) = nk=1 dik /m
k=1 dik / (m-2)
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2
Lj = dij/2 + (uj-ui)/2
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
(5) Éliminer les colonnes et les lignes correspondant aux groupes i et j et ajouter celles correspondant au nouveau groupe (ij)
(6) Si il reste un seul élément dans la matrice connecter les deux derniers nœuds, sinon retourner en 1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
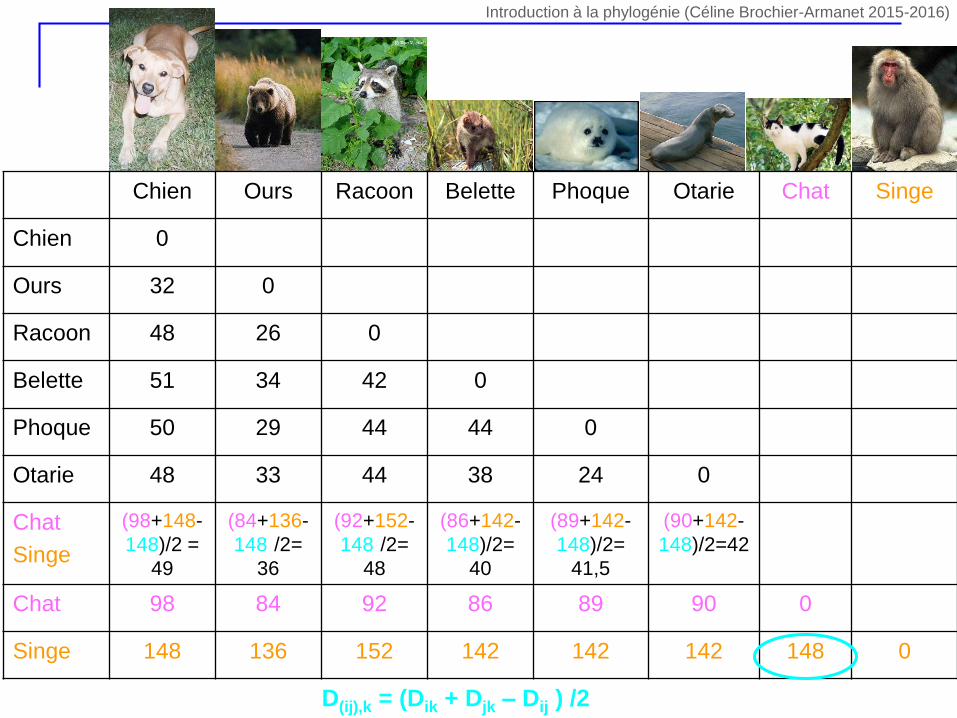
D(ij),k = (Dik + Djk – Dij ) /2
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0
Ours 32 0
Racoon 48 26 0
Belette 51 34 42 0
Phoque 50 29 44 44 0
Otarie 48 33 44 38 24 0
Chat
Singe
(98+148-
148)/2 =
49
(84+136-
148)/2=
36
(92+152-
148)/2=
48
(86+142-
148)/2=
40
(89+142-
148)/2=
41,5
(90+142-
148)/2=42
Chat 98 84 92 86 89 90 0
Singe 148 136 152 142 142 142 148 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

(1) Pour chaque feuille i calculer ui = Ri / (m-2) = mk=1 dik
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2
Lj = dij/2 + (uj-ui)/2
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
(5) Éliminer les colonnes et les lignes correspondant aux groupes i et j et ajouter celles correspondant au nouveau groupe (ij)
(6) Si il reste un seul élément dans la matrice connecter les deux derniers nœuds, sinon retourner en 1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
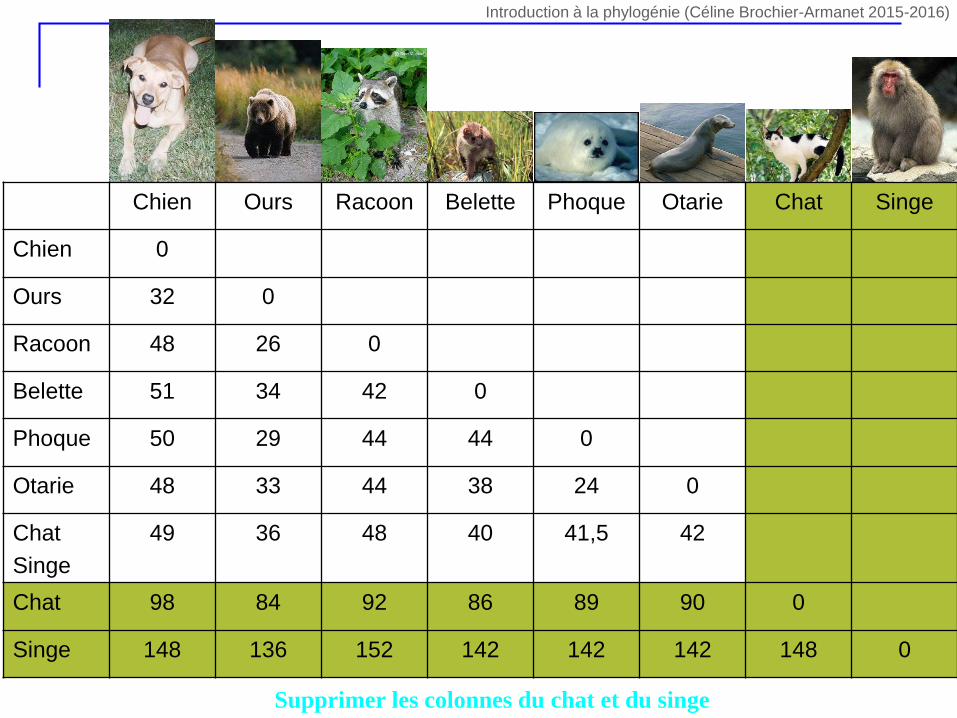
Supprimer les colonnes du chat et du singe
Chien Ours Racoon Belette Phoque Otarie Chat Singe
Chien 0
Ours 32 0
Racoon 48 26 0
Belette 51 34 42 0
Phoque 50 29 44 44 0
Otarie 48 33 44 38 24 0
Chat
Singe
49 36 48 40 41,5 42
Chat 98 84 92 86 89 90 0
Singe 148 136 152 142 142 142 148 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
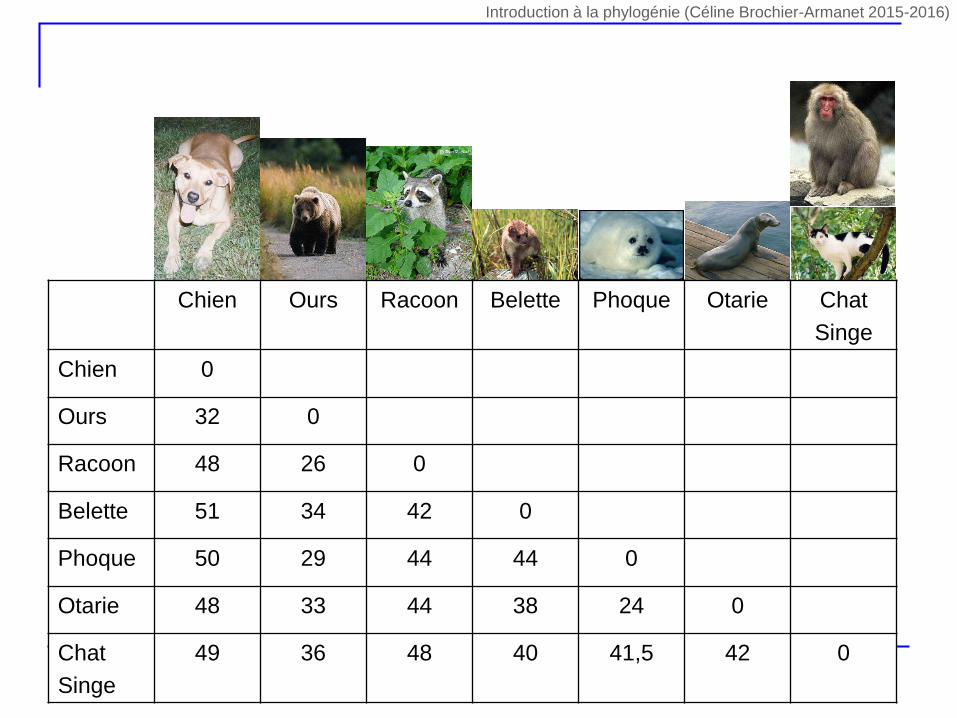
Chien Ours Racoon Belette Phoque Otarie Chat
Singe
Chien 0
Ours 32 0
Racoon 48 26 0
Belette 51 34 42 0
Phoque 50 29 44 44 0
Otarie 48 33 44 38 24 0
Chat
Singe
49 36 48 40 41,5 42 0
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

(1) Pour chaque feuille i calculer ui = Ri / (m-2) = mk=1 dik
(2) Choisir et i et j pour lesquels dij – ui – uj est la plus petite
(3) Joindre i et j. Calculer Li la longueur de la branche reliant i au nouveau nœud A et Lj la longueur de la branche reliant j au nouveau nœud A comme étant
Li = dij/2 + (ui-uj)/2
Lj = dij/2 + (uj-ui)/2
(4) Calculer la distance entre le nouveau nœud A et chaque autre feuille comme
d(ij),k = (dik + djk – dij ) /2
(5) Éliminer les colonnes et les lignes correspondant aux groupes i et j et ajouter celles correspondant au nouveau groupe (ij)
(6) Si il reste un seul élément dans la matrice connecter les deux derniers nœuds, sinon retourner en 1
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
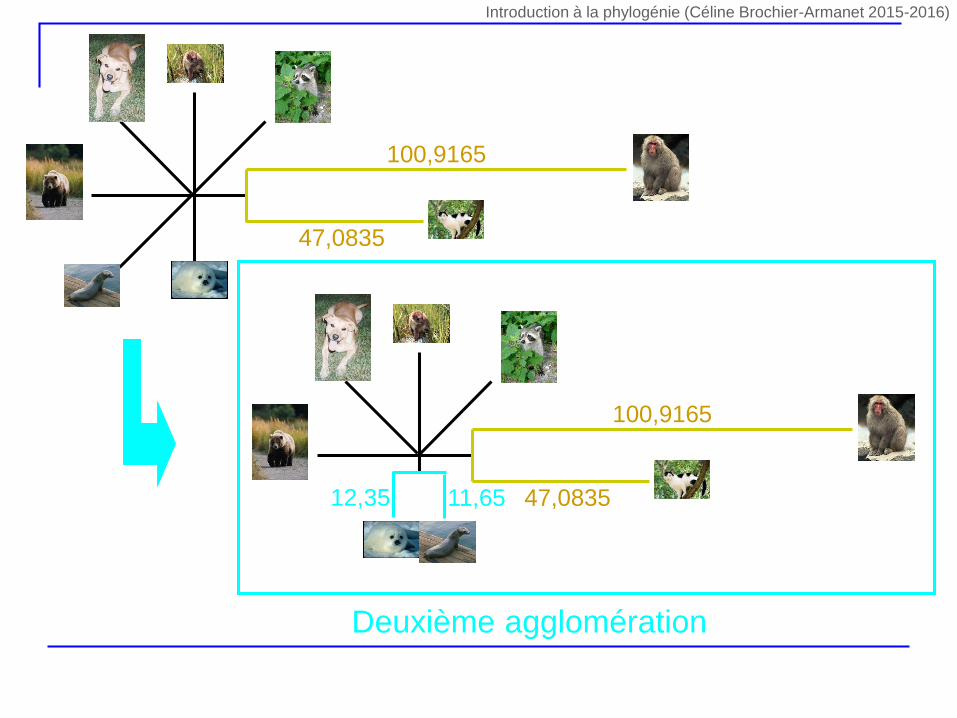
47,0835
100,9165
47,0835
100,9165
12,35 11,65
Deuxième agglomération
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
47,0835
100,9165
12,35 11,65
Troisième agglomération
47,0835
100,9165
12,35 11,65
6,875
19,125
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
Quatrième agglomération
47,0835
100,9165
12,35 11,65
6,875
19,125
47,0835
100,9165
11,6512,35
6,875
19,1251,75
25,25
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
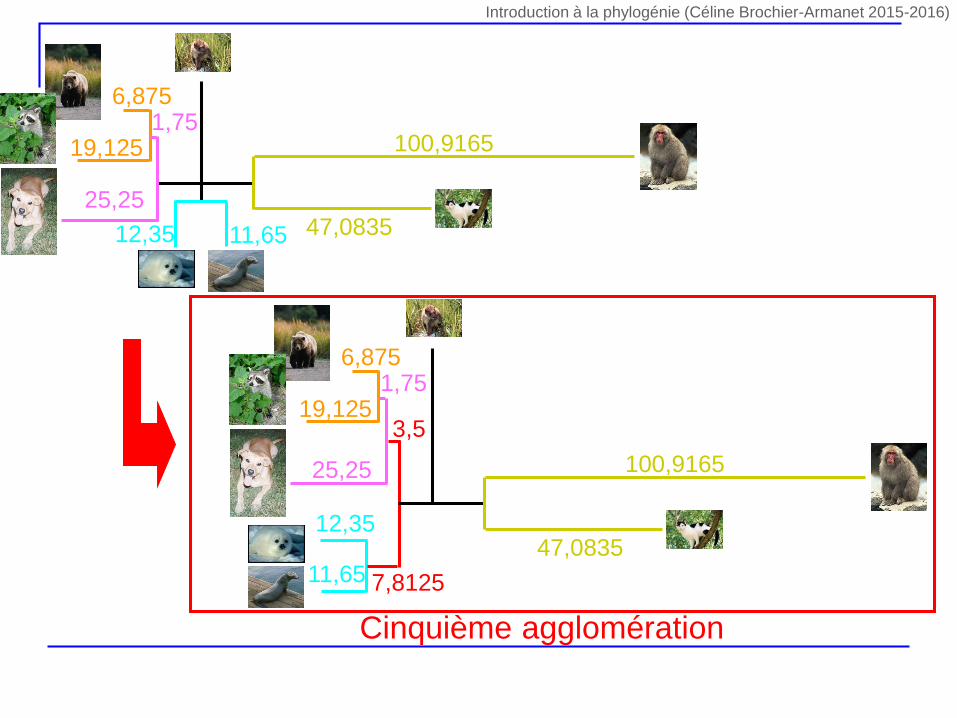
Cinquième agglomération
47,0835
100,9165
11,6512,35
6,875
19,1251,75
25,25
47,0835
100,9165
11,65
12,35
6,875
19,1251,75
25,25
3,5
7,8125
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
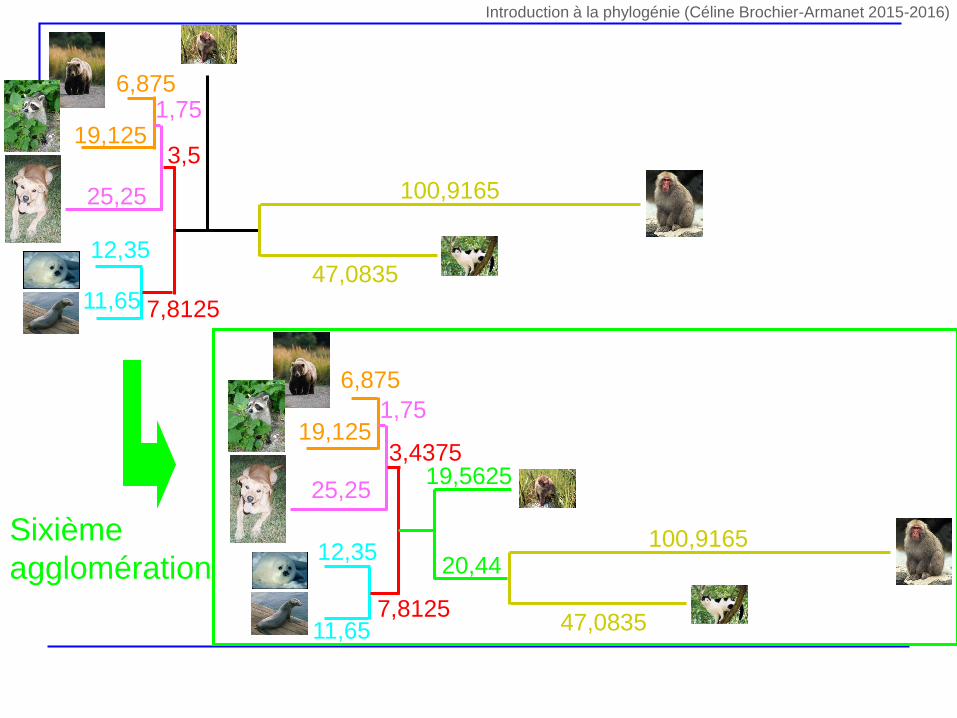
Sixième
agglomération
47,0835
100,9165
11,65
12,35
6,875
19,1251,75
25,25
3,5
7,8125
47,083511,65
12,35
6,875
19,125
25,25
1,75
100,9165
19,56253,4375
7,8125
20,44
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
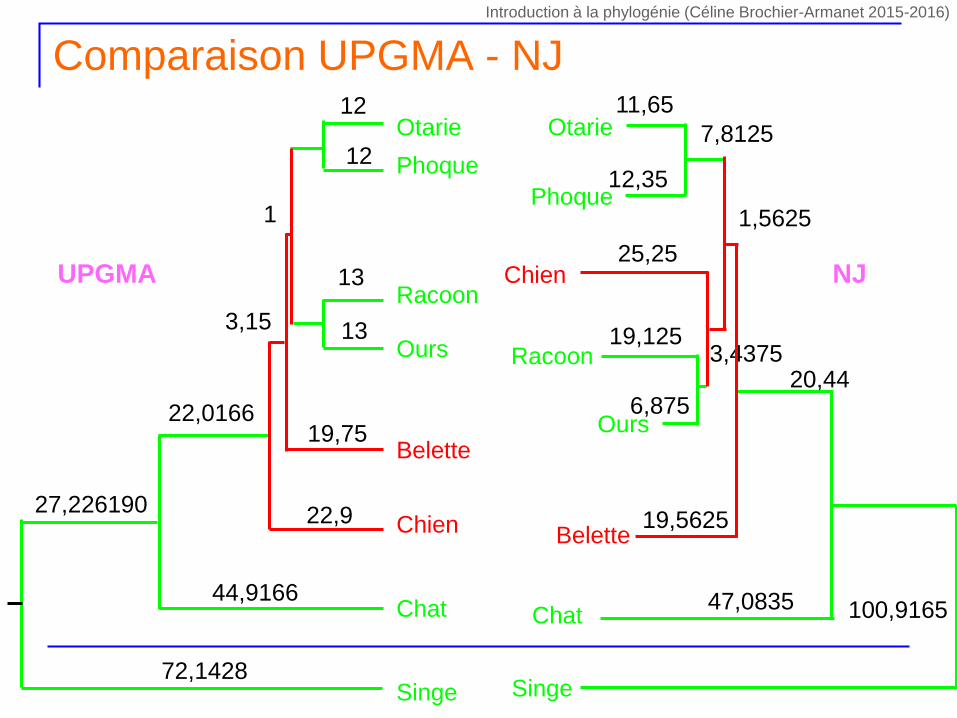
Comparaison UPGMA - NJ
47,0835 100,9165
3,437520,44
11,65
12,35
6,875
19,125
25,25
1,75
19,5625
7,8125
1,5625
13
Phoque
Otarie
Ours
Racoon
Belette
Chien
12
12
13
5,75
6,75
19,75
1
22,9
3,15
44,9166Chat
22,0166
Singe72,1428
27,226190
Phoque
Otarie
Ours
Racoon
Chien
Chat
Singe
Belette
UPGMA NJ
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Maximum de Parcimonie
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

La parcimonie
Fondement: rasoir d’Occam
« Les multiples ne doivent pas être utilisés sans nécessité. »
(pluralitas non est ponenda sine necessitate) ou sous une forme
plus moderne « les hypothèses les plus simples sont les plus
vraisemblables »
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
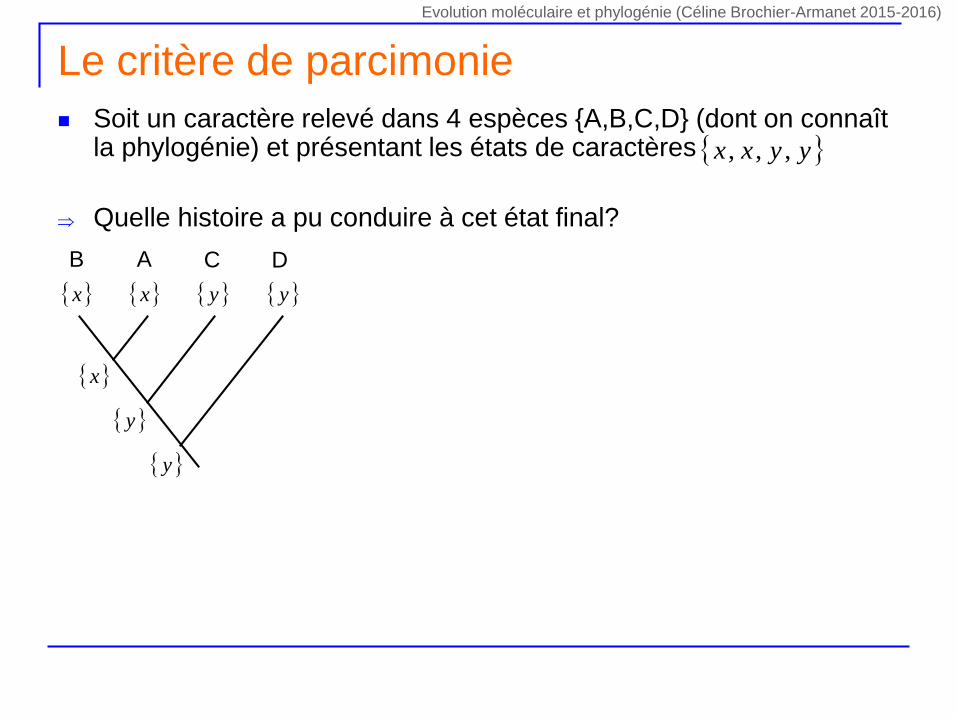
Le critère de parcimonie
Soit un caractère relevé dans 4 espèces {A,B,C,D} (dont on connaît la phylogénie) et présentant les états de caractères
Quelle histoire a pu conduire à cet état final?
yyxx ,,,
DCAB
x y x y
y
x
y
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
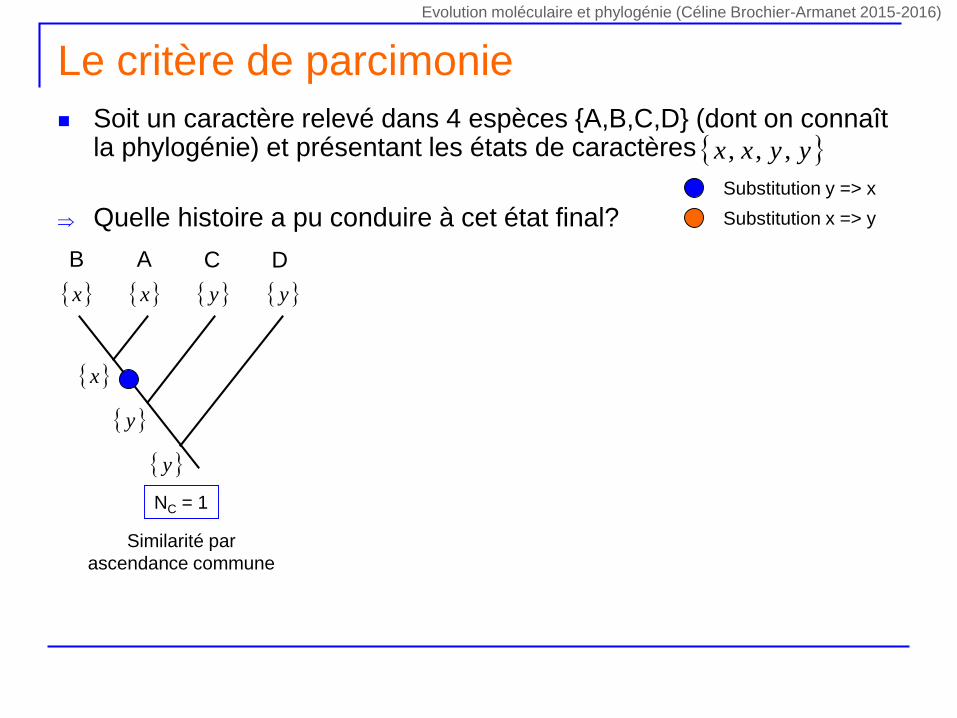
Le critère de parcimonie
Soit un caractère relevé dans 4 espèces {A,B,C,D} (dont on connaît la phylogénie) et présentant les états de caractères
Quelle histoire a pu conduire à cet état final?
yyxx ,,,
NC = 1
DCAB
x y x y
y
x
y
Similarité par
ascendance commune
Substitution y => x
Substitution x => y
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
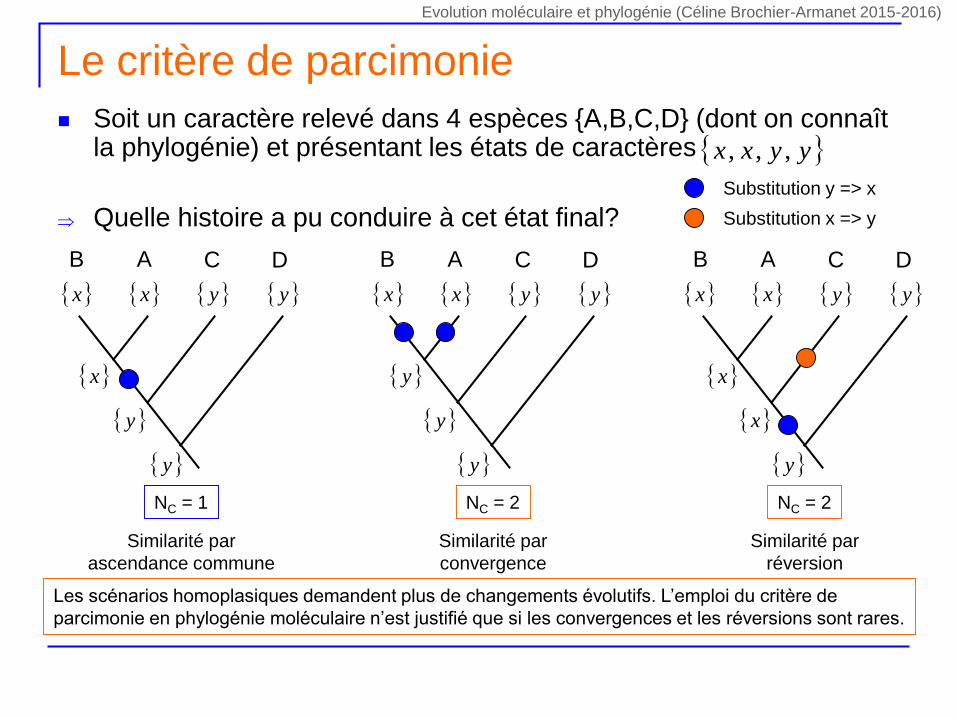
Le critère de parcimonie
Soit un caractère relevé dans 4 espèces {A,B,C,D} (dont on connaît la phylogénie) et présentant les états de caractères
Quelle histoire a pu conduire à cet état final?
yyxx ,,,
NC = 1
DCAB
x y x y
y
x
y
Similarité par
ascendance commune
NC = 2
DCAB
x y x y
y
y
y
Similarité par
convergence
NC = 2
DCAB
x y x y
x
x
y
Similarité par
réversion
Substitution y => x
Substitution x => y
Les scénarios homoplasiques demandent plus de changements évolutifs. L’emploi du critère de
parcimonie en phylogénie moléculaire n’est justifié que si les convergences et les réversions sont rares.
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Maximum de parcimonie - Généralités
Identifier la topologie T qui implique le plus petit nombre de changements évolutifs suffisant pour rendre compte des différences observées entre les séquences étudiées.
L’arbre le plus parcimonieux plus court chemin conduisant aux états de caractères observés
Caractéristique des arbres obtenus
Solutions multiples => plusieurs arbres impliquant un même nombre minimal de changements peuvent être obtenus
Ne possèdent pas de longueur de branche
Arbres non racinés
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Le maximum de parcimonie
Principe: rechercher parmi l’espace des arbres définissant les liens entre n séquences la topologie qui minimise le nombre de changements évolutifs
Quelle est la topologie qui implique le moins de changements d’état de caractères pour rendre compte des différences observées entre les UTO étudiées
Procédure:
1) pour une topologie fixée et pour un site donné de l’alignement, calculer (NC) le nombre de changements évolutifs nécessaires pour expliquer les états de caractères observés
2) calculer (NC) pour chaque site de l’alignement => , la longueur de l’arbre
3) calculer pour toutes les topologies possibles => retenir l’arbre le plus parcimonieux (cad l’arbre le plus court)
T
TL
L
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Parcimonie: Etape 1
Pour une topologie fixée et pour un site
donné de l’alignement, calculer (NC) le
nombre de changements évolutifs
nécessaires pour expliquer les états de
caractères observés
T
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Algorithme de Fitch: calcul du nombre
minimal de changements évolutifs Soit une topologie fixée et racinée de manière arbitraire, soit
l’ensemble de ses nœuds
Pour tout on définit:
, le nombre minimal de changements dans le sous-arbre dont
est la racine
, l’état de , cad l’ensemble des résidus en compatibles
avec changements évolutifs dans le sous-arbre raciné par .
Soit et les deux nœuds fils de
T V
Vp
pC
p
pS pp
p
pC
q r p
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Algorithme de Fitch: Application
La racine est placée de manière
arbitraire et n’a aucune influence
sur le nombre de changements
évolutifs inférés
Les états de caractères inférés
aux nœuds ne représentent pas
des caractères ancestraux, ni tous
les états de caractères possibles !
Initialisation du calcul récursif aux
feuilles de l’arbre
-P = {x} = résidu présent à cette feuille
-Cp = 0
NC = 4
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
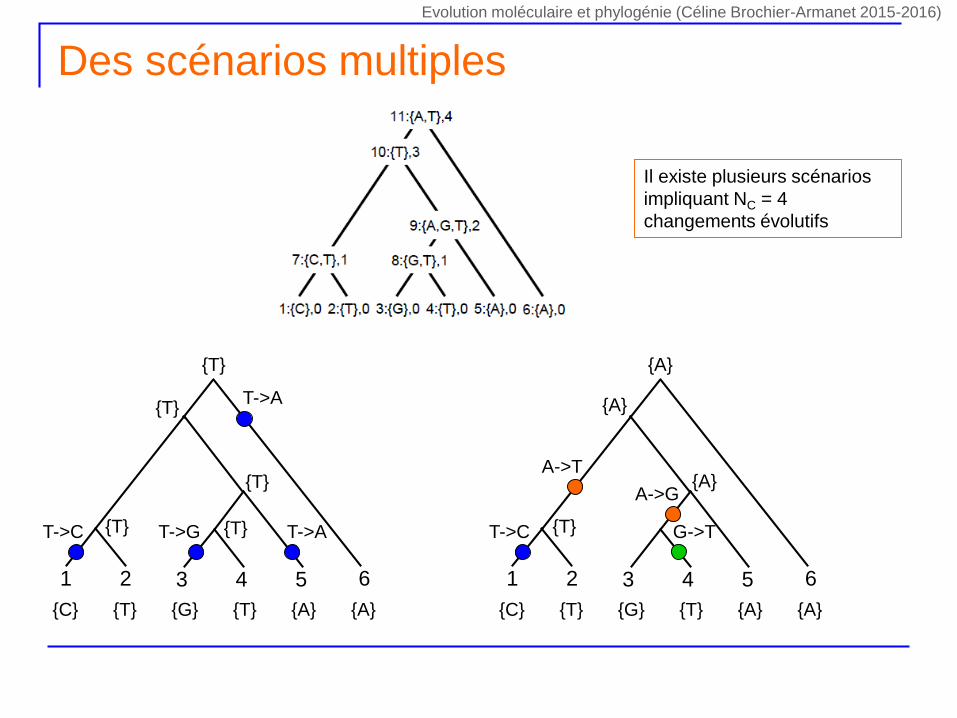
Des scénarios multiples
61 2 543
{C} {T} {G} {T} {A} {A}
61 2 543
{C} {T} {G} {T} {A} {A}
{T} {A}
T->C
T->A
T->G
{T}
{T}
{T} {T} T->A
{A}
{A}
{T}T->C
A->T
A->G
G->T
Il existe plusieurs scénarios
impliquant NC = 4
changements évolutifs
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Parcimonie: Etapes 2 et 3
Etape 2:
Calculer NC pour chaque site de l’alignement
Sommer tous les valeurs de NC pour l’ensemble des sites
Calculer , la longueur totale de l’arbre
Etape 3:
Répéter l’étape 2 pour chaque topologie composant
l’espace des arbres possibles à n feuilles
Retenir l’arbre de longueur minimale arbre le plus
parcimonieux
T
L
L
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Tous les sites ne sont pas équivalents
Tous les sites ne contiennent pas une information
permettant de discriminer les topologies
Les sites constants (1 seul état de caractère)
Ne sont pas informatifs
Sites variables (au moins 2 états de caractères)
Informatifs: présentent au moins deux états de caractères
chacun partagés par au moins deux séquences
Non informatifs: tous les autres
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Tous les sites ne sont pas équivalents
Soit A, B, C et D quatre séquences
d’ADN homologues alignées
Il existe 3 topologies non racinées
possibles
Il existe 4 états de caractères
{A,T,C,G}
Il existe 44 = 256 motifs différents
observables à une position
Seuls 36 sont informatifs, et sont
tous du type {x,x,y,y}, {x,y,x,y} ou
{x,y,y,x} (avec x ≠ y et x,y E
{A,T,C,G})
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Parcimonie: Récapitulatif & propriétés
Produit des arbres non racinés
Le positionnement des changements dans un arbre n’est pas unique ne permet pas d’inférer des longueurs de branches de manière unique
Plusieurs arbres équiparcimonieux peuvent être trouvés Inférence de consensus
Le nombre d’arbre croissant de manière rapide avec le nombre de séquences, seul un sous-ensemble des topologies est testé pour identifier l’arbre le plus parcimonieux Utilisation d’heuristiques pour explorer l’espace des arbres de manière rationnelle
Aucune certitude d’identifier l’arbre le plus parcimonieux à la fin de l’analyse
Absence de critères pour discriminer le(les) arbre(s) le(s) plus parcimonieux des arbres légèrement moins parcimonieux ex. est-ce qu’un arbre comptant 2504 pas est significativement meilleur que les
20 arbres comptant 2506 pas ?
La parcimonie classique (algorithme de Fitch) considère toutes les substitutions comme équivalentes Parcimonie pondérée (algorithme de Sankoff) permet de pondérer les types de
changements
Pour approfondir ces notions: Concepts et méthodes en phylogénie moléculaire (2010) Perrière &
Brochier-Armanet (Springer)
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
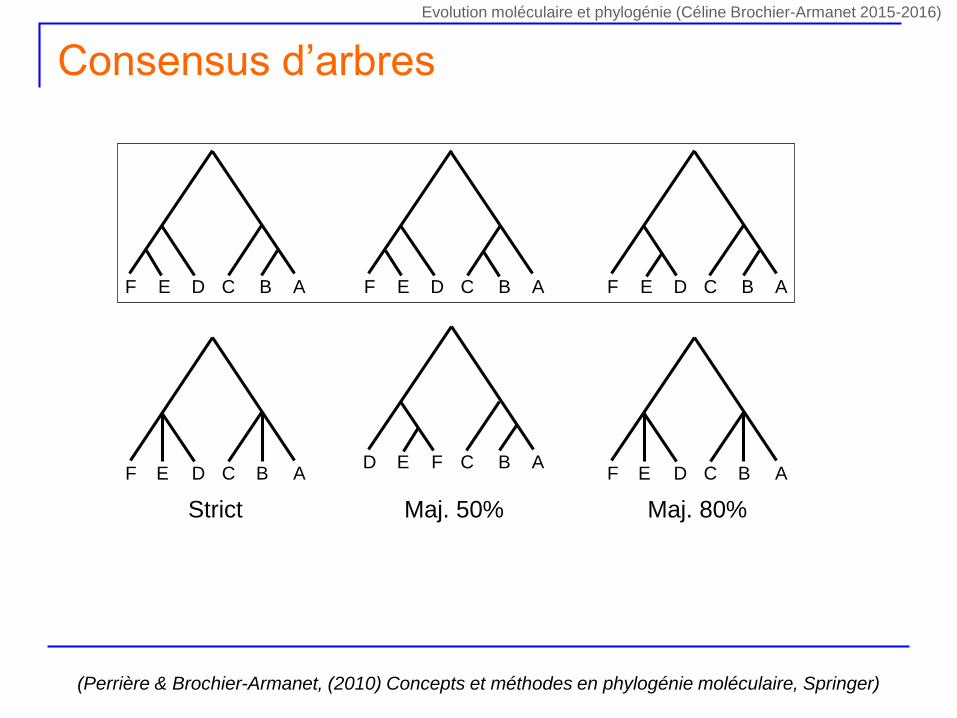
Consensus d’arbres
F E D C B A F E D C B A F E D C B A
F E D C B A F E D C B A
Strict
D E F C B A
Maj. 50% Maj. 80%
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)

Explorer l’espace des topologies
n < 12: Exploration exhaustive
n < 20: branch-and-bound
n > 20: heuristiques
Utilisé pour la parcimonie, mais aussi les moindres carrés, le
maximum de vraisemblance, etc.
Topologie de départ?
Topologie aléatoire
Meilleure topologie issue d’une recherche séquentielle
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
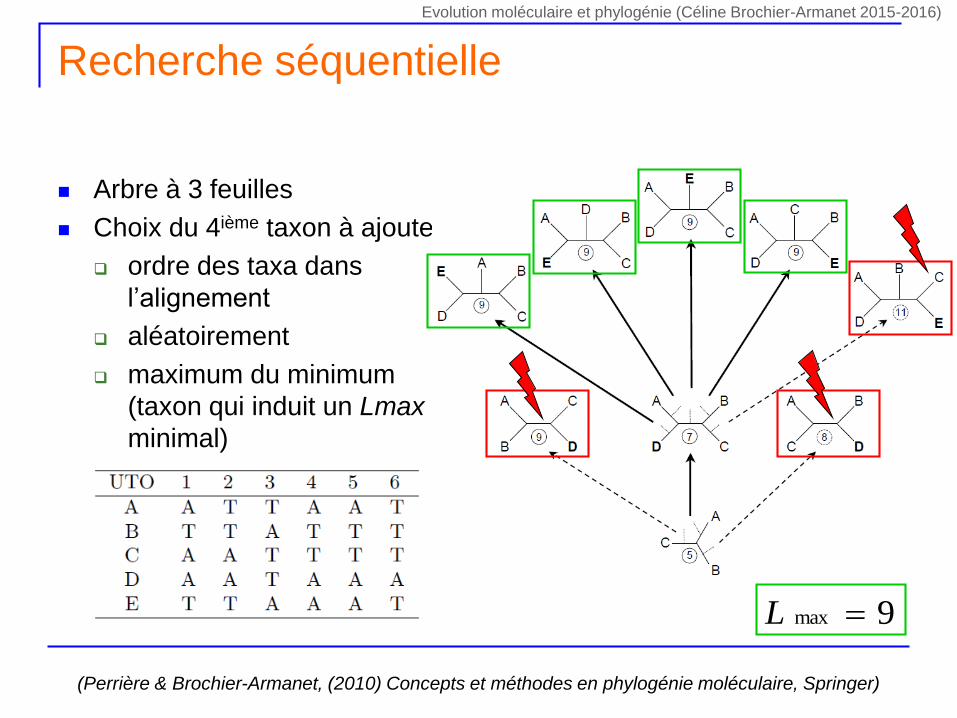
Recherche séquentielle
Arbre à 3 feuilles
Choix du 4ième taxon à ajouter
ordre des taxa dans
l’alignement
aléatoirement
maximum du minimum
(taxon qui induit un Lmax
minimal)
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
9max L
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
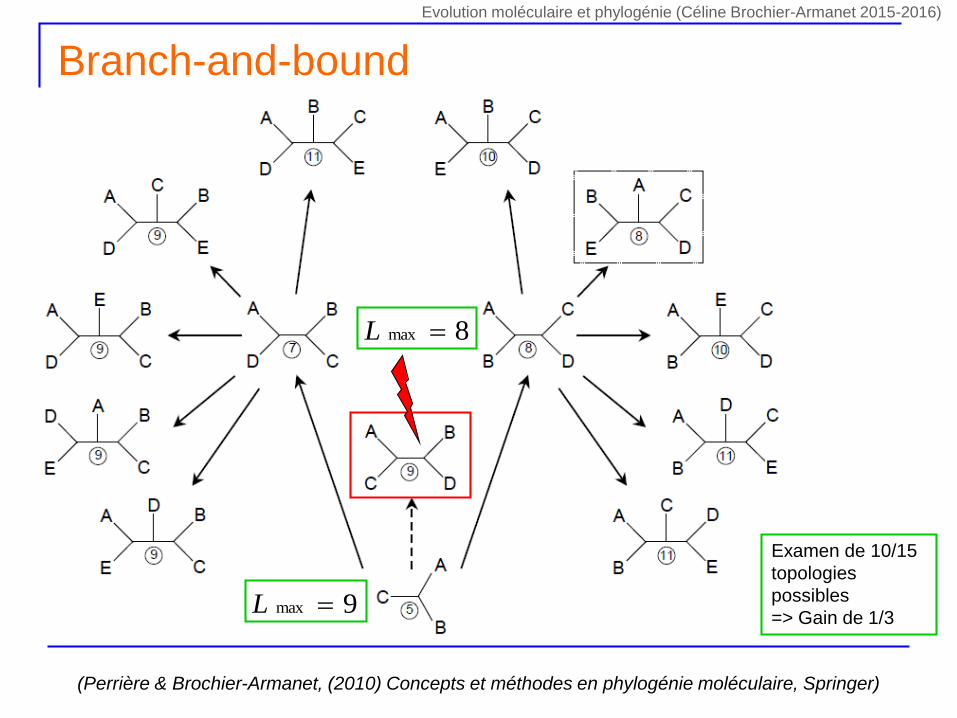
Branch-and-bound
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
9max L
Examen de 10/15
topologies
possibles
=> Gain de 1/3
8max L
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
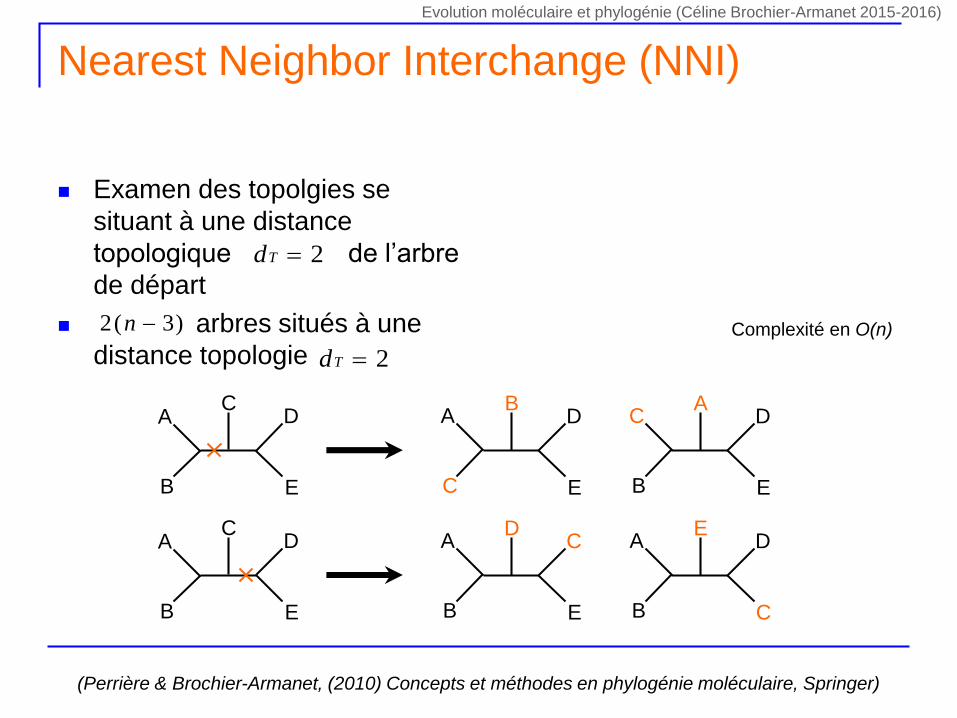
Nearest Neighbor Interchange (NNI)
Examen des topolgies se
situant à une distance
topologique de l’arbre
de départ
arbres situés à une
distance topologie
2Td
2Td
)3(2 n
A D
EB
CA D
EC
BC D
EB
A
A D
EB
CA C
EB
DA D
CB
E
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
Complexité en O(n)
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
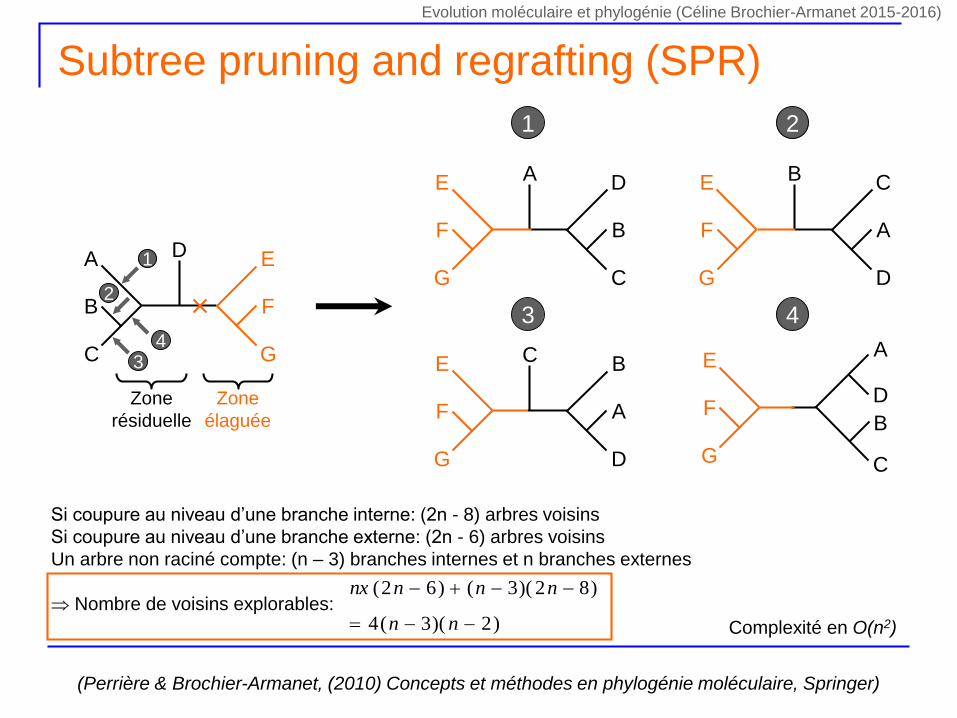
Subtree pruning and regrafting (SPR)
CE B
F A
G D
AE D
F B
G C
BE C
F A
G D
B
A
D
C
E
F
G
DA E
B F
C G
Zone
élaguée
Zone
résiduelle
1
2
34
1 2
3 4
Complexité en O(n2)
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
Si coupure au niveau d’une branche interne: (2n - 8) arbres voisins
Si coupure au niveau d’une branche externe: (2n - 6) arbres voisins
Un arbre non raciné compte: (n – 3) branches internes et n branches externes
Nombre de voisins explorables:)2)(3(4
)82)(3()62(
nn
nnnnx
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
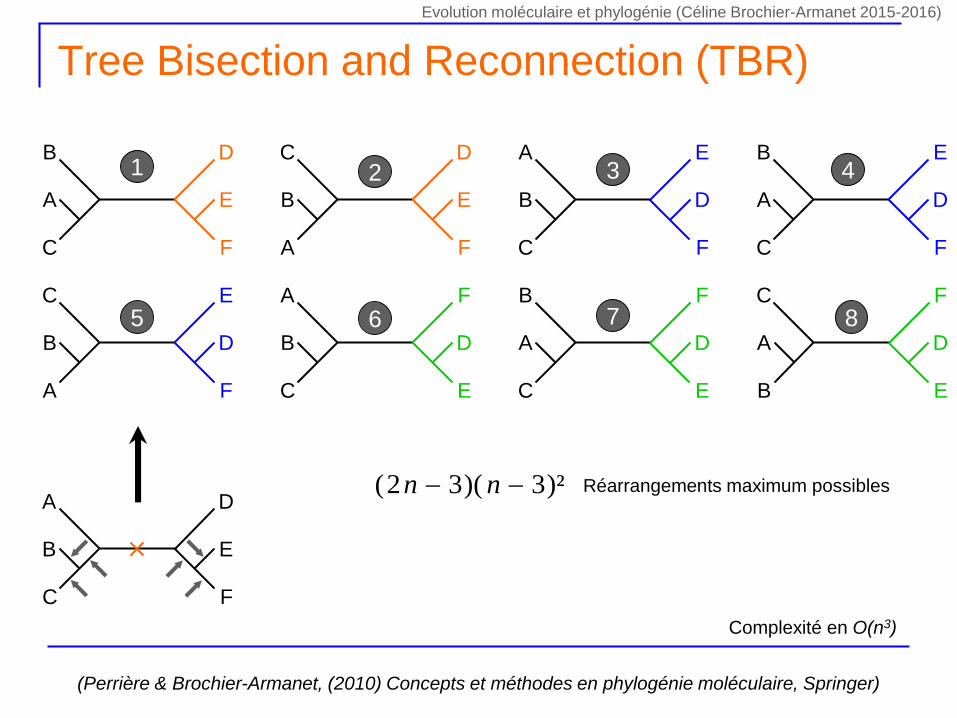
Tree Bisection and Reconnection (TBR)
A D
B E
C F
(Perrière & Brochier-Armanet, (2010) Concepts et méthodes en phylogénie moléculaire, Springer)
D
E
F
C D
B E
A F
A E
B D
C F
B E
A D
C F
E
D
B
A
C
C
B
A F
A F
B D
C E
B F
A D
C E
C F
A D
B E
1 2 3 4
5 6 7 8
)²3)(32( nn Réarrangements maximum possibles
Complexité en O(n3)
Evolution moléculaire et phylogénie (Céline Brochier-Armanet 2015-2016)
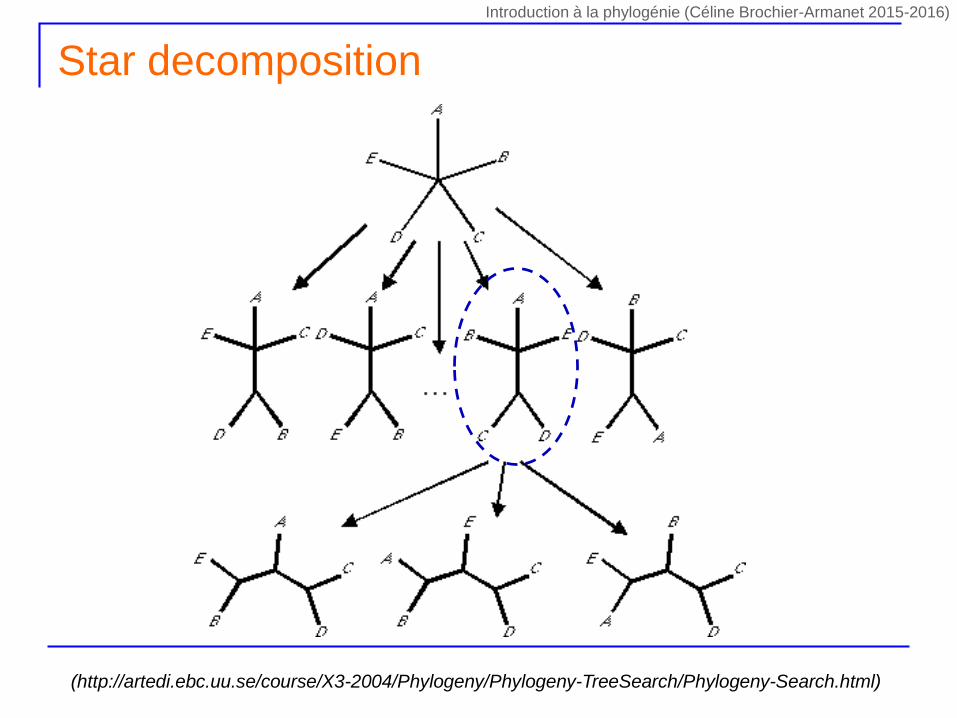
Star decomposition
(http://artedi.ebc.uu.se/course/X3-2004/Phylogeny/Phylogeny-TreeSearch/Phylogeny-Search.html)
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Maximum de vraisemblance
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Généralité
Introduit par Edwards et Cavalli-Sforza (1964) pour l’étude de données de type fréquences de gènes
Appliquée à la phylogénie moléculaire par Neyman (1971)
Élargissement par Kashyap et Subas (1974) et Felsenstein (1981)
IDEE DE BASE
Étant donné un modèle d’évolution, on peut estimer une phylogénie avec des méthodes statistiques comme le maximum de vraisemblance
PROPRIETES des estimations par Maximum de vraisemblance
Bonne consistance convergent vers la valeur correcte du paramètre
Bonne efficience variance faible autour de la vraie valeur du paramètre
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Principe général
Basé sur des lois de probabilité conditionnelles
La vraisemblance de l’hypothèse H connaissant les données D est définie par:
probabilité d’observer les données D sous l’hypothèse H
: probabilité de l’hypothèse H sachant les données
Si on dispose de n observations indépendantes
)( HDPL
)( DHPL
)(...)()()()()3()2()1(
HDPHDPHDPHDPLn
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Un exemple simple
Estimation de la probabilité p d’obtenir face d’une pièce lancée 11 fois
Hypothèses :
Indépendance des lancés
Tous les lancés on la même probabilité p (inconnue) d’obtenir face
Données :
Résultats observés : FFPPFPFFPPP
Définition de la fonction de vraisemblance
Soit
)( pDPL
65)1(
)1()1()1()1()1()1()(
pp
ppppppppppppDPL
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
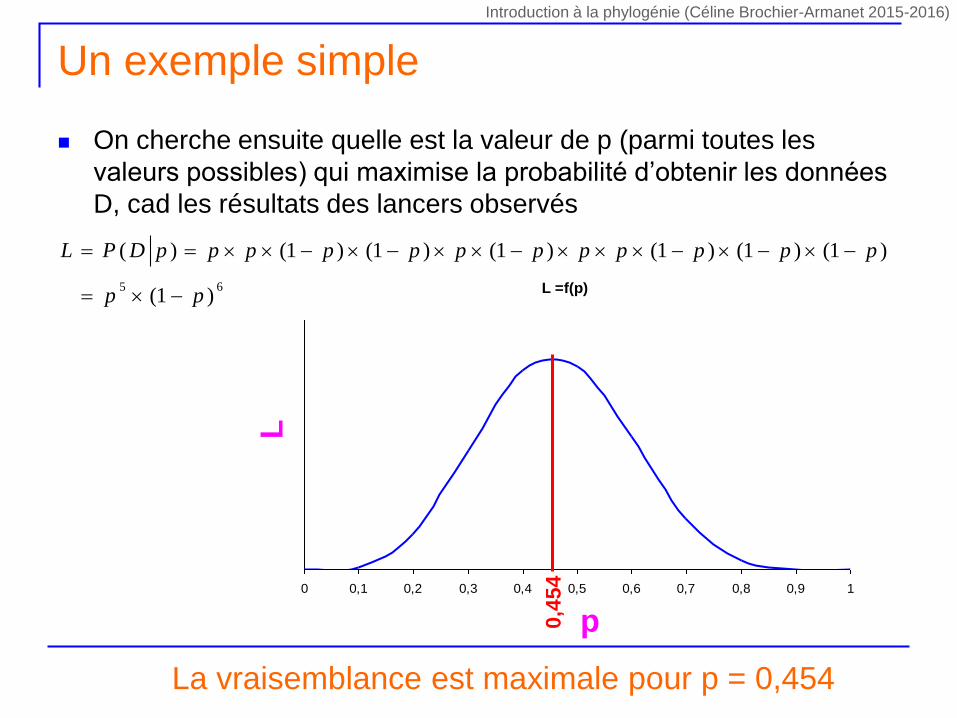
Un exemple simple
On cherche ensuite quelle est la valeur de p (parmi toutes les
valeurs possibles) qui maximise la probabilité d’obtenir les données
D, cad les résultats des lancers observés
65)1(
)1()1()1()1()1()1()(
pp
ppppppppppppDPL
L =f(p)
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1
p
L
0,4
54
p
L
La vraisemblance est maximale pour p = 0,454
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Application aux phylogénies moléculaires
HYPOTHESES Le processus de substitution suit un modèle probabiliste dont
l’expression mathématique est connue a priori, mais dont les paramètres ne sont pas connus (modèle connu mais paramètres inconnus => calcul des probabilité de passage d’un état i à un état j le long d’une branche de longueur t)
Les sites évoluent indépendamment les uns des autres
Les probabilités de substitution ne changent pas au cours du temps
Tous les sites obéissent au même processus de substitution
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Définition de la vraisemblance
Données Séquences d’ADN lignées (n sites)
Modèle d’évolution M (JK, K2P, HKY…)
Hypothèses Paramètres du modèle , topologie , longueurs de
branches
Décomposition de la vraisemblance
T
),,(
)(
)(...)()(
)()(
)(
1
)(
1
)()2()1(
TDP
HDPL
HDPHDPHDPL
HDPHDPL
in
i
in
i
n
La vraisemblance est
calculée de manière
indépendante à
chaque site
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Application
Sites
Séquences 1 2 3 4 5 6 7 8 n
A A A G A G T T C N
B A G C C G T T C N
C A G A T A T C C N
D A G A G A T C C N
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
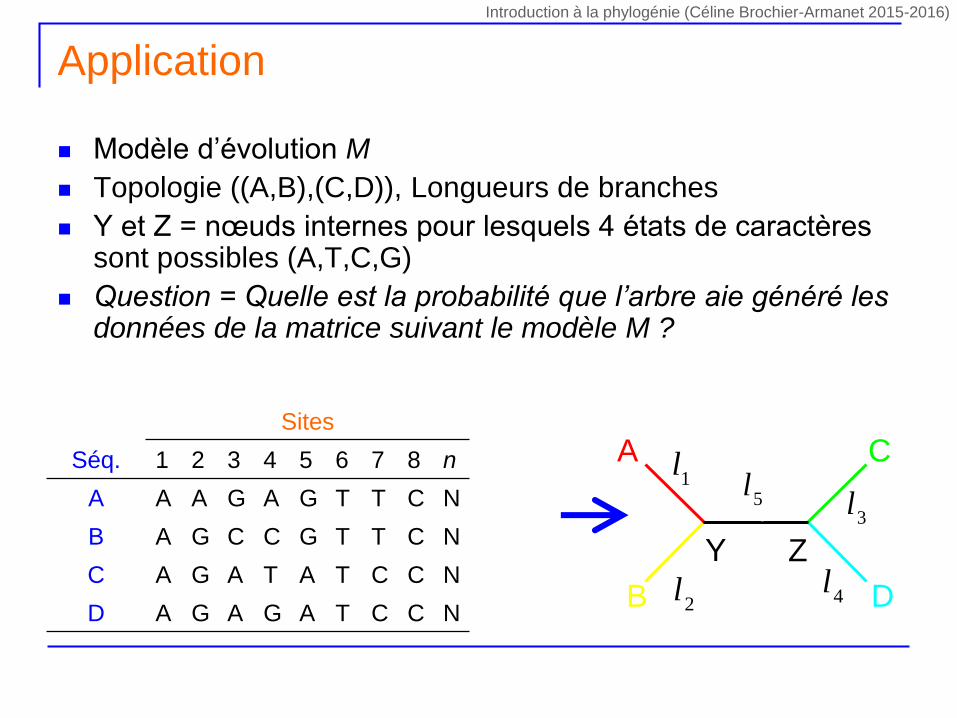
Modèle d’évolution M
Topologie ((A,B),(C,D)), Longueurs de branches
Y et Z = nœuds internes pour lesquels 4 états de caractères sont possibles (A,T,C,G)
Question = Quelle est la probabilité que l’arbre aie généré les données de la matrice suivant le modèle M ?
Application
Sites
Séq. 1 2 3 4 5 6 7 8 n
A A A G A G T T C N
B A G C C G T T C N
C A G A T A T C C N
D A G A G A T C C N
1lA
B
C
D
Y Z
l55
l
4l
3l
2l
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
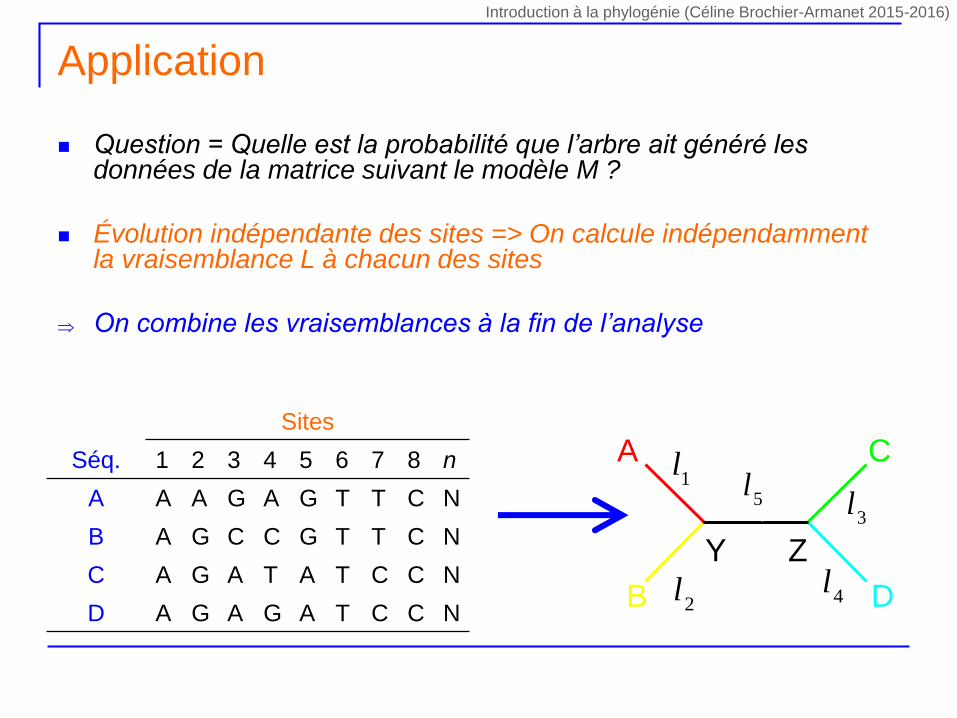
Question = Quelle est la probabilité que l’arbre ait généré les données de la matrice suivant le modèle M ?
Évolution indépendante des sites => On calcule indépendamment la vraisemblance L à chacun des sites
On combine les vraisemblances à la fin de l’analyse
Application
Sites
Séq. 1 2 3 4 5 6 7 8 n
A A A G A G T T C N
B A G C C G T T C N
C A G A T A T C C N
D A G A G A T C C N
1lA
B
C
D
Y Z
l55
l
4l
3l
2l
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
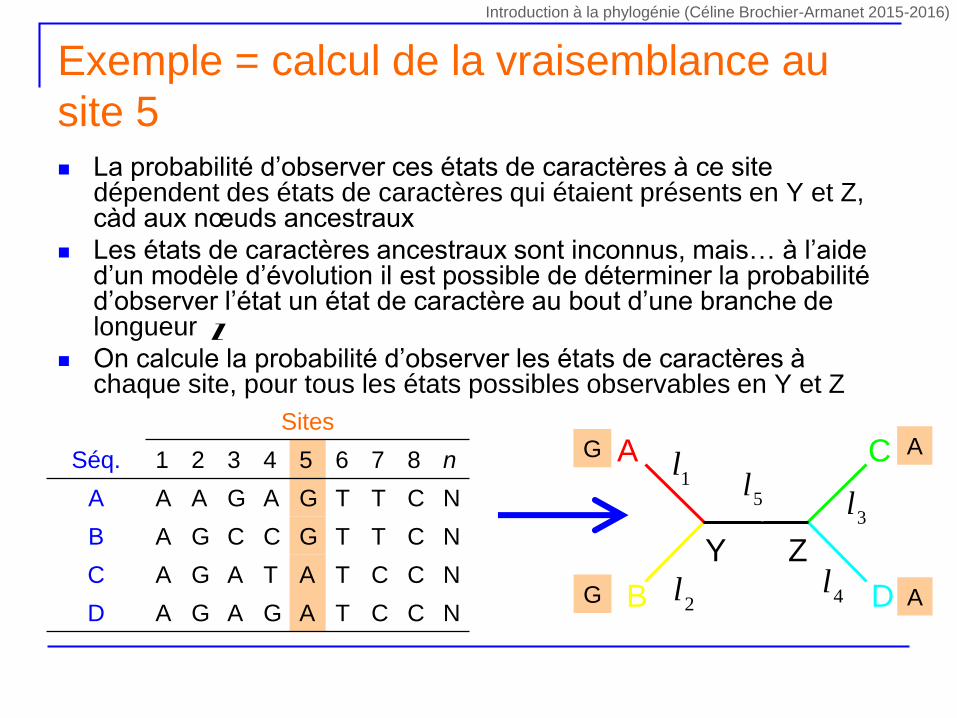
La probabilité d’observer ces états de caractères à ce site dépendent des états de caractères qui étaient présents en Y et Z, càd aux nœuds ancestraux
Les états de caractères ancestraux sont inconnus, mais… à l’aide d’un modèle d’évolution il est possible de déterminer la probabilité d’observer l’état un état de caractère au bout d’une branche de longueur
On calcule la probabilité d’observer les états de caractères à chaque site, pour tous les états possibles observables en Y et Z
Exemple = calcul de la vraisemblance au
site 5
Sites
Séq. 1 2 3 4 5 6 7 8 n
A A A G A G T T C N
B A G C C G T T C N
C A G A T A T C C N
D A G A G A T C C N
1lA
B
C
D
Y Z
l55
l
4l
3l
2l
G
G
A
A
l
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
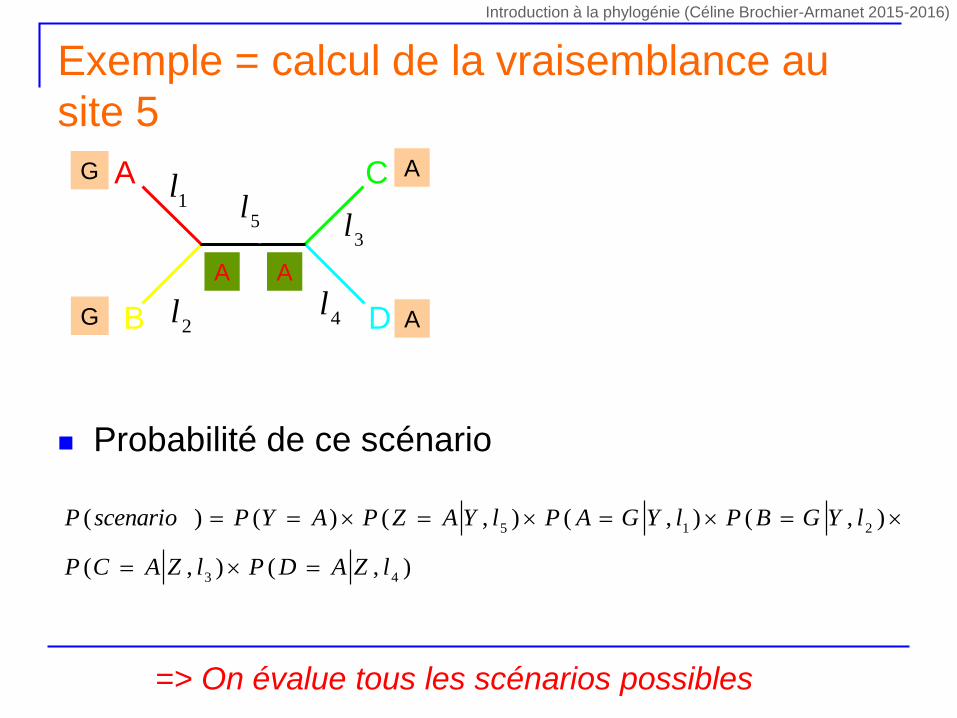
Exemple = calcul de la vraisemblance au
site 5
Probabilité de ce scénario
=> On évalue tous les scénarios possibles
),(),(
),(),(),()()(
43
215
lZADPlZACP
lYGBPlYGAPlYAZPAYPscenarioP
1lA
B
C
D
l55
l
4l
3l
2l
G
G
A
A
A A
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)
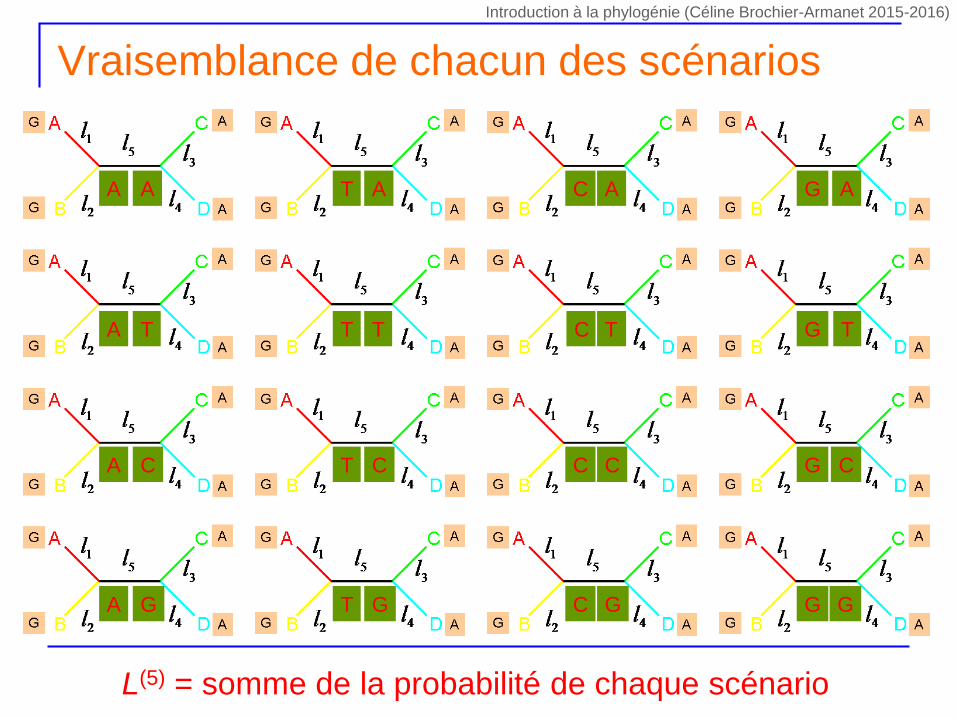
Vraisemblance de chacun des scénarios
A A
A T
A C
A G
T A
T T
T C
T G
C A G A
C T G T
C C
C G
G C
G G
L(5) = somme de la probabilité de chaque scénario
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Calcul de la vraisemblance d’un arbre
L(5) = somme des probabilités individuelles de chaque scénario
Vraisemblance de l’arbre
n
i
i
n
n
i
i
n
LL
LLLL
LL
LLLL
1
)(
)()2()1(
1
)(
)()2()1(
ln
ln...lnln
...
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Maximisation de la vraisemblance
(1) On considère une topologie , un site et un ensemble de longueurs de branches
(2) On calcule la vraisemblance des paramètres = probabilité d’observer les états de caractères au site en fonction des paramètres
(3) On fait le calcul pour tous les caractères
(4) On calcule les longueurs de branches et les paramètres du modèle qui maximisent la vraisemblance
(5) On calcule la vraisemblance pour toutes les topologies possibles
(6) On retient la topologie qui a la plus grande grande vraisemblance
T
),,( lT
l
l
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)

Propriétés du maximum de vraisemblance
C’est une des méthodes les plus justifiées d’un point de vue théorique
Les simulations montrent que cette méthode est supérieure aux autres dans
beaucoup de cas. En particulier elle est moins sensible aux artefacts
d’attraction des longues branches
Coûteuse en temps de calcul
Impossible d’évaluer tous les arbres utilisation d’heuristiques n’est
plus certain d’obtenir l’arbre le plus vraisemblable
Des tests statistiques dérivés du maximum de vraisemblance permettent
d’évaluer si des topologies ayant une vraisemblance moins bonne que la
topologie la plus vraisemblable sont significativement différentes
Introduction à la phylogénie (Céline Brochier-Armanet 2015-2016)