
Etude par spectroscopie mécanique et microscopie...
146
N° d’ordre 01ISAL00XX Année 2002 Thèse Présentée devant L’Institut National des Sciences Appliquées de Lyon pour obtenir LE GRADE DE DOCTEUR FORMATION DOCTORALE : Génie des Matériaux ECOLE DOCTORALE : Matériaux de Lyon par Bertrand Van de Moortèle ETUDE PAR SPECTROSCOPIE MECANIQUE ET MICROSCOPIE ELECTRONIQUE EN TRANSMISSION DE LA STABILITE THERMIQUE DE VERRES METALLIQUES MASSIFS : EFFETS DE LA DECOMPOSITION ET DE LA NANOCRISTALLISATION Soutenue le 20 décembre 2002 devant la Commission d’examen Jury M. Pierre GUYOT Rapporteur M. Jean-Louis SOUBEYROUX Rapporteur M. Jean-Marc PELLETIER Directeur de thèse M. Thierry EPICIER Directeur de thèse M. Tanguy ROUXEL M. Hervé ARRIBART
Transcript of Etude par spectroscopie mécanique et microscopie...

N° d’ordre 01ISAL00XX Année 2002
Thèse
Présentée devant
L’Institut National des Sciences Appliquées de Lyon
pour obtenir
LE GRADE DE DOCTEUR
FORMATION DOCTORALE : Génie des Matériaux
ECOLE DOCTORALE : Matériaux de Lyon
par
Bertrand Van de Moortèle
ETUDE PAR SPECTROSCOPIE MECANIQUE
ET MICROSCOPIE ELECTRONIQUE EN TRANSMISSION
DE LA STABILITE THERMIQUE DE VERRES METALLIQUES MASSIFS :
EFFETS DE LA DECOMPOSITION ET DE LA NANOCRISTALLISATION
Soutenue le 20 décembre 2002 devant la Commission d’examen
Jury M. Pierre GUYOT Rapporteur M. Jean-Louis SOUBEYROUX Rapporteur M. Jean-Marc PELLETIER Directeur de thèse M. Thierry EPICIER Directeur de thèse M. Tanguy ROUXEL M. Hervé ARRIBART

i
OCTOBRE 2002
INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE LYON Directeur : STORCK A. Professeurs : AUDISIO S. PHYSICOCHIMIE INDUSTRIELLE BABOT D. CONT. NON DESTR. PAR RAYONNEMENT IONISANTS BABOUX J.C. GEMPPM*** BALLAND B. PHYSIQUE DE LA MATIERE BAPTISTE P. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BARBIER D. PHYSIQUE DE LA MATIERE BASTIDE J.P. LAEPSI**** BAYADA G. MATHEMATIQUE APPLIQUEES DE LYON BENADDA B. DEPT GEN BETEMPS M. AUTOMATIQUE INDUSTRIELLE BIENNIER F. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BLANCHARD J.M. LAEPSI**** BOISSON C. VIBRATIONS-ACOUSTIQUE BOIVIN M. (Prof. émérite) MECANIQUE DES SOLIDES BOTTA H. UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOTTA-ZIMMERMANN M. (Mme) UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOULAYE G. (Prof. émérite) INFORMATIQUE BOYER J.C. MECANIQUE DES SOLIDES BRAU J. CENTRE DE THERMIQUE DE LYON - Thermique du bâtiment BREMOND G. PHYSIQUE DE LA MATIERE BRISSAUD M. GENIE ELECTRIQUE ET FERROELECTRICITE BRUNET M. MECANIQUE DES SOLIDES BRUNIE L. INGENIERIE DES SYSTEMES D’INFORMATION BUREAU J.C. CEGELY* CAVAILLE J.Y. GEMPPM*** CHANTE J.P. CEGELY*- Composants de puissance et applications CHOCAT B. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine COMBESCURE A. MECANIQUE DES CONTACTS COUSIN M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures DAUMAS F. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et Thermique DOUTHEAU A. CHIMIE ORGANIQUE DUFOUR R. MECANIQUE DES STRUCTURES DUPUY J.C. PHYSIQUE DE LA MATIERE EMPTOZ H. RECONNAISSANCE DES FORMES ET VISION ESNOUF C. GEMPPM*** EYRAUD L. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE FANTOZZI G. GEMPPM*** FAVREL J. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS FAYARD J.M. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS FAYET M. MECANIQUE DES SOLIDES FERRARIS-BESSO G. MECANIQUE DES STRUCTURES FLAMAND L. MECANIQUE DES CONTACTS FLORY A. INGENIERIE DES SYSTEMES D’INFORMATION FOUGERES R. GEMPPM*** FOUQUET F. GEMPPM*** FRECON L. REGROUPEMENT DES ENSEIGNANTS CHERCHEURS ISOLES GERARD J.F. INGENIERIE DES MATERIAUX POLYMERES GERMAIN P. LAEPSI**** GIMENEZ G. CREATIS** GOBIN P.F. (Prof. émérite) GEMPPM*** GONNARD P. GENIE ELECTRIQUE ET FERROELECTRICITE GONTRAND M. PHYSIQUE DE LA MATIERE GOUTTE R. (Prof. émérite) CREATIS** GOUJON L. GEMPPM*** GOURDON R. LAEPSI****. GRANGE G. GENIE ELECTRIQUE ET FERROELECTRICITE GUENIN G. GEMPPM*** GUICHARDANT M. BIOCHIMIE ET PHARMACOLOGIE GUILLOT G. PHYSIQUE DE LA MATIERE GUINET A. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS GUYADER J.L. VIBRATIONS-ACOUSTIQUE GUYOMAR D. GENIE ELECTRIQUE ET FERROELECTRICITE HEIBIG A. MATHEMATIQUE APPLIQUEES LYON JACQUET RICHARDET G. MECANIQUE DES STRUCTURES JAYET Y. GEMPPM*** JOLION J.M. RECONNAISSANCE DES FORMES ET VISION JULLIEN J.F. UNITE DE RECHERCHE EN GENIE CIVIL - Structures JUTARD A. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE KASTNER R. UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique KOULOUMDJIAN J. INGENIERIE DES SYSTEMES D’INFORMATION LAGARDE M. BIOCHIMIE ET PHARMACOLOGIE LALANNE M. (Prof. émérite) MECANIQUE DES STRUCTURES LALLEMAND A. CENTRE DE THERMIQUE DE LYON - Energétique et thermique LALLEMAND M. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et thermique LAREAL P. UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique LAUGIER A. PHYSIQUE DE LA MATIERE LAUGIER C. BIOCHIMIE ET PHARMACOLOGIE

ii
LEJEUNE P. UNITE MICROBIOLOGIE ET GENETIQUE OCTOBRE 2002 LUBRECHT A. MECANIQUE DES CONTACTS MASSARD N. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE MAZILLE H. PHYSICOCHIMIE INDUSTRIELLE MERLE P. GEMPPM*** MERLIN J. GEMPPM*** MIGNOTTE A. (Mle) INGENIERIE, INFORMATIQUE INDUSTRIELLE MILLET J.P. PHYSICOCHIMIE INDUSTRIELLE MIRAMOND M. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine MOREL R. MECANIQUE DES FLUIDES ET D’ACOUSTIQUES MOSZKOWICZ P. LAEPSI**** MOURA A. GEMPPM*** NARDON P. (Prof. émérite) BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS NIEL E. AUTOMATIQUE INDUSTRIELLE NORTIER P. DREP ODET C. CREATIS** OTTERBEIN M. (Prof. émérite) LAEPSI**** PARIZET E. VIBRATIONS-ACOUSTIQUE PASCAULT J.P. INGENIERIE DES MATERIAUX POLYMERES PAVIC G. VIBRATIONS-ACOUSTIQUE PELLETIER J.M. GEMPPM*** PERA J. UNITE DE RECHERCHE EN GENIE CIVIL - Matériaux PERRIAT P. GEMPPM*** PERRIN J. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PINARD P. (Prof. émérite) PHYSIQUE DE LA MATIERE PINON J.M. INGENIERIE DES SYSTEMES D’INFORMATION PONCET A. PHYSIQUE DE LA MATIERE POUSIN J. MODELISATION MATHEMATIQUE ET CALCUL SCIENTIFIQUE PREVOT P. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PROST R. CREATIS** RAYNAUD M. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux REDARCE H. AUTOMATIQUE INDUSTRIELLE REYNOUARD J.M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures RIGAL J.F. MECANIQUE DES SOLIDES RIEUTORD E. (Prof. émérite) MECANIQUE DES FLUIDES ROBERT-BAUDOUY J. (Mme) (Prof. émérite) GENETIQUE MOLECULAIRE DES MICROORGANISMES ROUBY D. GEMPPM*** ROUX J.J. CENTRE DE THERMIQUE DE LYON – Thermique de l’Habitat RUBEL P. INGENIERIE DES SYSTEMES D’INFORMATION RUMELHART C. MECANIQUE DES SOLIDES SACADURA J.F. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux SAUTEREAU H. INGENIERIE DES MATERIAUX POLYMERES SCAVARDA S. AUTOMATIQUE INDUSTRIELLE SOUIFI A. PHYSIQUE DE LA MATIERE SOUROUILLE J.L. INGENIERIE INFORMATIQUE INDUSTRIELLE THOMASSET D. AUTOMATIQUE INDUSTRIELLE UBEDA S. CENTRE D’INNOV. EN TELECOM ET INTEGRATION DE SERVICES THUDEROZ C. ESCHIL – Equipe Sciences Humaines de l’Insa de Lyon UNTERREINER R. CREATIS** VELEX P. MECANIQUE DES CONTACTS VIGIER G. GEMPPM*** VINCENT A. GEMPPM*** VRAY D. CREATIS** VUILLERMOZ P.L. (Prof. émérite) PHYSIQUE DE LA MATIERE Directeurs de recherche C.N.R.S. : BERTHIER Y. MECANIQUE DES CONTACTS CONDEMINE G. UNITE MICROBIOLOGIE ET GENETIQUE COTTE-PATAT N. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE FRANCIOSI P. GEMPPM*** MANDRAND M.A. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE POUSIN G. BIOLOGIE ET PHARMACOLOGIE ROCHE A. INGENIERIE DES MATERIAUX POLYMERES SEGUELA A. GEMPPM*** Directeurs de recherche I.N.R.A. : FEBVAY G. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS GRENIER S. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS RAHBE Y. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS Directeurs de recherche I.N.S.E.R.M. : PRIGENT A.F. (Mme) BIOLOGIE ET PHARMACOLOGIE MAGNIN I. (Mme) CREATIS** * CEGELY CENTRE DE GENIE ELECTRIQUE DE LYON ** CREATIS CENTRE DE RECHERCHE ET D’APPLICATIONS EN TRAITEMENT DE L’IMAGE ET DU SIGNAL ***GEMPPM GROUPE D'ETUDE METALLURGIE PHYSIQUE ET PHYSIQUE DES MATERIAUX ****LAEPSI LABORATOIRE D’ANALYSE ENVIRONNEMENTALE DES PROCEDES ET SYSTEMES INDUSTRIELS

iii
INSA DE LYON DEPARTEMENT DES ETUDES DOCTORALES ET RELATIONS INTERNATIONALES SCIENTIFIQUES MARS 2002
Ecoles Doctorales et Diplômes d’Etudes Approfondies
habilités pour la période 1999-2003
ECOLES DOCTORALES n° code national
RESPONSABLE
PRINCIPAL
CORRESPONDANT
INSA
DEA INSA
n° code national
RESPONSABLE
DEA INSA
CHIMIE DE LYON
(Chimie, Procédés, Environnement)
EDA206
M. D. SINOU UCBL1 04.72.44.62.63 Sec 04.72.44.62.64 Fax 04.72.44.81.60
M. R. GOURDON 87.53 Sec 84.30 Fax 87.17
Chimie Inorganique 910643
Sciences et Stratégies Analytiques
910634
Sciences et Techniques du Déchet 910675
M. R. GOURDON Tél 87.53 Fax 87.17
ECONOMIE, ESPACE ET
MODELISATION DES COMPORTEMENTS
(E2MC)
EDA417
M.A. BONNAFOUS LYON 2 04.72.72.64.38 Sec 04.72.72.64.03 Fax 04.72.72.64.48
Mme M. ZIMMERMANN 84.71 Fax 87.96
Villes et Sociétés 911218
Dimensions Cognitives et Modélisation
992678
Mme M. ZIMMERMANN Tél 84.71 Fax 87.96 M. L. FRECON Tél 82.39 Fax 85.18
ELECTRONIQUE,
ELECTROTECHNIQUE, AUTOMATIQUE
(E.E.A.)
EDA160
M. G. GIMENEZ INSA DE LYON 83.32 Fax 85.26
Automatique Industrielle 910676
Dispositifs de l’Electronique Intégrée
910696
Génie Electrique de Lyon 910065
Images et Systèmes
992254
M. M. BETEMPS Tél 85.59 Fax 85.35 M. D. BARBIER Tél 85.47 Fax 60.81 M. J.P. CHANTE Tél 87.26 Fax 85.30 Mme I. MAGNIN Tél 85.63 Fax 85.26
EVOLUTION, ECOSYSTEME,
MICROBIOLOGIE , MODELISATION
(E2M2)
EDA403
M. J.P FLANDROIS UCBL1 04.78.86.31.50 Sec 04.78.86.31.52 Fax 04.78.86.31.49
M. S. GRENIER 79.88 Fax 85.34
Analyse et Modélisation des Systèmes Biologiques 910509
M. S. GRENIER Tél 79.88 Fax 85.34
INFORMATIQUE ET INFORMATION
POUR LA SOCIETE
(EDIIS)
EDA 407
M. J.M. JOLION INSA DE LYON 87.59 Fax 80.97
Documents Multimédia, Images et Systèmes d’Information Communicants
992774 Extraction des Connaissances à partir des Données
992099
Informatique et Systèmes Coopératifs pour l’Entreprise 950131
M. A. FLORY Tél 84.66 Fax 85.97 M. J.F. BOULICAUT Tél 89.05 Fax 87.13 M. A. GUINET Tél 85.94 Fax 85.38
INTERDISCIPLINAIRE SCIENCES-
SANTE
(EDISS)
EDA205
M. A.J. COZZONE UCBL1 04.72.72.26.72 Sec 04.72.72.26.75 Fax 04.72.72.26.01
M. M. LAGARDE 82.40 Fax 85.24
Biochimie 930032
M. M. LAGARDE Tél 82.40 Fax 85.24
MATERIAUX DE LYON
UNIVERSITE LYON 1
EDA 034
M. J. JOSEPH ECL 04.72.18.62.44 Sec 04.72.18.62.51 Fax 04.72.18.60.90
M. J.M. PELLETIER 83.18 Fax 84.29
Génie des Matériaux : Microstructure, Comportement Mécanique, Durabilité
910527
Matériaux Polymères et Composites 910607
Matière Condensée, Surfaces et Interfaces
910577
M. J.M.PELLETIER Tél 83.18 Fax 85.28 M. H. SAUTEREAU Tél 81.78 Fax 85.27 M. G. GUILLOT Tél 81.61 Fax 85.31
MATHEMATIQUES ET
INFORMATIQUE FONDAMENTALE
(Math IF)
EDA 409
M. NICOLAS UCBL1 04.72.44.83.11 Fax 04.72.43.00.35
M. J. POUSIN 88.36 Fax 85.29
Analyse Numérique, Equations aux dérivées partielles et Calcul Scientifique
910281
M. G. BAYADA Tél 83.12 Fax 85.29
MECANIQUE, ENERGETIQUE, GENIE
CIVIL, ACOUSTIQUE
(MEGA)
EDA162
M. J. BATAILLE ECL 04.72.18.61.56 Sec 04.72.18.61.60 Fax 04.78.64.71.45
M. G.DALMAZ 83.03 Fax 04.72.89.09.80
Acoustique 910016
Génie Civil
992610 Génie Mécanique
992111
Thermique et Energétique 910018
M. J.L. GUYADER Tél 80.80 Fax 87.12 M. J.J.ROUX Tél 84.60 Fax 85.22 M. G. DALMAZ Tél 83.03 Fax 04.78.89.09.80 M. J. F. SACADURA Tél 81.53 Fax 88.11
En grisé : Les Ecoles doctorales et DEA dont l’INSA est établissement principal

Chapitre 1 : Introduction
Thèse - VAN DE MOORTELE, Bertrand 1
CHAPITRE 1
Introduction
1.1 Contexte de cette étude
Les historiens ont depuis longtemps nommé les périodes historiques par le nom du matériau qui, à
cette époque, a accompagné l'évolution de l'humanité (âge de bronze : 1800-700 avant J.C., âge de fer :
700-52 avant J.C,...). Cette démarche souligne l'importance de la découverte d'un matériau, puisque celle-ci
permet généralement d'ouvrir la voie à de nouvelles applications techniques qu’il n'était pas possible
d'envisager auparavant. La découverte du bronze a permis, par exemple, le développement d'objets comme
des épingles, bracelets, colliers et premières fibules, mais aussi de nouvelles armes telles que des haches,
des poignards, des hallebardes ou des épées dans un premier temps. Les civilisations maîtrisant ce
matériau ont été par la suite dépassées par celles qui ont appris à maîtriser ce qui était à l'époque un
nouveau matériau : le fer. Actuellement, l'enjeu de la science des matériaux ne semble pas aussi important.
Néanmoins, de nombreuses recherches sont menées afin de développer de nouveaux matériaux dans
l'espoir d'applications futures. Généralement, la découverte d'un nouveau matériau se fait au détour d'une
recherche générale exploratoire. Il est alors nécessaire d'entreprendre une caractérisation complète du
matériau pour en connaître toutes les caractéristiques qui détermineront son champ d'application futur.
La découverte des verres métalliques dans les années 60 et leur développement au cours des
années 70-80 ont donné lieu à de nombreuses études portant principalement sur les propriétés physiques
(calorimétrie, résistivité,..). A cette époque les échantillons qui étaient produits l'étaient sous la forme de
rubans d'une épaisseur de 20 à 30 µm ce qui permettait assez peu d’espoir d’application. Depuis la fin des
années 80, de nouvelles compositions ont permis d’aboutir à des échantillons massifs, élargissant ainsi le
champ d’application de tels matériaux. Ainsi par exemple, la caractérisation des propriétés mécaniques a
montré que les verres métalliques possèdent une bonne résistance mécanique alliée à une ténacité élevée,
figure 1.1 qui est un diagramme d’élasticité en fonction de la ténacité d’après Ashby [Ash2001] pour
différentes classes de matériaux.
Une autre propriété intéressante des verres métalliques, et des matériaux amorphes en général, est
de pouvoir être mis en forme à relativement basse température par rapport à la température de fusion. Ainsi,
le verre de silice porté à une température proche de 800°C peut être mis en forme relativement facilement.
Dans le cas des verres métalliques, la mise en forme peut également s’effectuer aisément à une
température appropriée, mais, comme nous le verrons tout au long de ce mémoire, cette montée en
température entraînera une déstabilisation du matériau conduisant à une cristallisation plus ou moins tardive
selon les compositions des alliages, faisant perdre alors à ces matériaux toutes les qualités que lui conférait
l’état amorphe.

Chapitre 1 : Introduction
Thèse - VAN DE MOORTELE, Bertrand 2
Une meilleure compréhension des phénomènes menant à cette cristallisation devrait permettre à
terme l'amélioration de la stabilité à la température des verres métalliques. Mais, pour cela il est nécessaire
de s’intéresser plus particulièrement aux évolutions de la microstructure qui aboutissent à la cristallisation de
la matrice amorphe. Ce travail s'inscrira donc dans une démarche de caractérisation mais également dans
une perspective plus globale de compréhension de l’évolution des propriétés mécaniques au regard des
évolutions microstructurales. Cette thématique, relation microstructure-propriétés mécaniques, est l’axe
principal de recherche du laboratoire Groupe d'Etudes de Métallurgie Physique et de Physique des
Matériaux (GEMPPM) dans lequel s’est effectué ce travail. Plus particulièrement au sein du laboratoire, le
groupe d'étude Polymères, Verres et Matériaux Hétérogènes (PVMH) se consacre depuis de nombreuses
années à l'étude de ces relations propriétés mécaniques-microstructure dans les matériaux amorphes. Des
travaux menés sur des polymères amorphes (PMMA, PET,..), des verres de silice ainsi que d'autres
matériaux amorphes ont permis de dégager des concepts physiques généraux sur la déformation des
matériaux non-cristallins. Ces concepts ont été formalisés dans le modèle de Perez et al.[Per1988] qui décrit
le comportement mécanique dans les domaines linéaires et non-linéaires des matériaux amorphes pour une
large gamme de température autour de la température de transition vitreuse. Dans cette approche, aucune
restriction n’est faite sur la nature du matériau amorphe étudié. Aussi apparaît-il naturel de transférer les
concepts développés par ce modèle dans les polymères amorphes, vers les alliages métalliques amorphes.
Ceci nous permettra de comparer le comportement mécanique expérimental des verres métalliques à celui
prédit par le modèle de Perez et al.
Figure 1.1 : carte représenatnt la ténacité en fonction de la limite élastiquepour différentes familles de matériaux. Les verres métalliques sontreprésentés par le point rouge.

Chapitre 1 : Introduction
Thèse - VAN DE MOORTELE, Bertrand 3
1.2 Objectifs de cette étude
Comme nous venons de l’évoquer brièvement, les verres métalliques sont des matériaux
particulièrement hors d’équilibre et une température trop élevée les mènera à la cristallisation. Aussi cette
étude aura deux objectifs bien distincts.
Dans un premier temps, nous étudierons les propriétés de ces matériaux à l’état amorphe. En
particulier, nous insisterons sur l’évolution des propriétés mécaniques avec la température afin de
déterminer les comportements viscoélastique et viscoplastique de ce type de matériau amorphe.
Dans un second temps, nous aborderons la phase de déstabilisation de l’état amorphe. Comme
dans de nombreuses études, nous mettrons en évidence cet aspect par des méthodes de mesures globales
des propriétés du matériau (calorimétrie différentielle à balayage, pouvoir thermoélectrique). Bien que
l'élaboration d'alliages permettant d'obtenir des alliages métalliques amorphes montre l'importance de la
composition chimique, nous verrons que relativement peu d'études sur la décomposition et la cristallisation
des verres métalliques se préoccupent de cet aspect. Aussi la microscopie électronique en transmission
(imagerie, nanoanalyse, nanodiffraction) nous a semblée un bon moyen technique d'investigation pour
apporter de nouvelles informations. Cette approche sera particulièrement bien adaptée pour mettre en
évidence l’existence d’une décomposition chimique qui s’opère dans certains alliages avant la cristallisation.
Ceci nous permettra également de nous replacer dans l'approche traditionnelle du laboratoire, "relation
propriétés mécaniques-microstructure ", puisque nous tenterons d’interpréter l’évolution des propriétés
mécaniques au regard des évolutions microstructurales.
1.3 Plan de l'exposé
Ce travail s'articulera donc en quatre parties :
Dans une première partie, nous tenterons de situer ce travail dans une double perspective à savoir
matériau amorphe et verre métallique. Pour cela, nous ferons quelques rappels sur la notion de
transition vitreuse et nous présenterons différentes approches qui ont été développées pour modéliser la
déformation des matériaux amorphes. Puis nous présenterons à travers divers exemples choisis dans la
littérature l'évolution des verres métalliques avec la température. En particulier, nous insisterons sur le
fait que peu d'informations sont disponibles sur l'aspect microstructural.
Les techniques expérimentales utilisées ainsi que les différentes compositions d’alliages étudiés seront
détaillées dans une deuxième partie.
La troisième partie sera consacrée à l'étude de l'état amorphe. Après avoir mis en évidence la présence
de phases cristallines dans les échantillons bruts, ainsi que les conséquences lors d'essais de traction à
froid, nous étudierons les effets réversibles qui existent autour de la température de transition vitreuse.
Puis nous aborderons la caractérisation de propriétés mécaniques en présentant les résultats, d'une part
de spectroscopie mécanique qui porte sur la déformation linéaire et d'autre part, des essais de

Chapitre 1 : Introduction
Thèse - VAN DE MOORTELE, Bertrand 4
compression à chaud qui concernent les effets non-linéaires. Les résultats obtenus nous amèneront à
dégager les similitudes et les différences entre le comportement des verres métalliques et celui d'autres
matériaux amorphes. Pour conclure ce chapitre, nous présenterons la modélisation des courbes du
comportement mécanique faites à partir du modèle de Perez et al. Nous montrerons en particulier que
l'accord avec les courbes expérimentales est plus que satisfaisant pour la spectrométrie mécanique.
Dans une dernière partie, nous étudierons le stade de cristallisation des verres métalliques. La
caractérisation par calorimétrie différentielle à balayage et par pouvoir thermoélectrique permettra de
sélectionner une température et des temps de traitement qui nous apparaîtront comme significatifs de
l'évolution de la cristallisation. La microscopie électronique en haute résolution permettra de mettre en
évidence la formation dans les stades les plus précoces de cristaux d'une taille de 2 à 4nm. Les
analyses chimiques par sonde nanométrique montreront que ces premières nano-particules sont
principalement enrichies en titane et appauvries en zirconium. Nous verrons que cette cristallisation est
probablement issue d'une séparation de phase ou démixtion. Dans cette même partie, nous porterons
un regard critique avec l'éclairage des résultats de microscopie électronique sur l'évolution des
propriétés mécaniques des verres métalliques au cours des traitements thermiques.
Enfin nous terminerons en résumant dans une conclusion les points marquants de cette étude. Nous
ferons également quelques propositions sur les extensions de ce sujet qui pourraient faire l'œuvre de futurs
travaux de recherche.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 5
CHAPITRE 2
Bibliographie
CHAPITRE 2 ___________________________________________________________________5 1 La transition vitreuse ________________________________________________________________6
2 Des verres métalliques massifs_________________________________________________________9
3 Des amorphes présentant des défauts __________________________________________________10
4 De l’état vitreux à la cristallisation ____________________________________________________11 4.1-Mode de diffusion dans les verres métalliques _________________________________________________ 12 4-2 Mise en évidence de la cristallisation ________________________________________________________ 14 4-3 Décomposition de la matrice amorphe _______________________________________________________ 16 4-4 Echelle de la cristallisation ________________________________________________________________ 19 4-5 Identification des premières phases cristallographiques __________________________________________ 19 4-6 Des phases quasicristallines _______________________________________________________________ 20 4-7 Devenir des premières phases______________________________________________________________ 23
5-Effet de l’oxygène __________________________________________________________________24
6-Aptitude à la déformation des verres métalliques ________________________________________25 6.1 Description ____________________________________________________________________________ 27 6.2 Approches phénoménologiques de la déformation ______________________________________________ 29 6.3 Modèle des défauts quasi-ponctuels _________________________________________________________ 33

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 6
Bibliographie
Dans un premier temps nous ferons quelques rappels sur le phénomène de transition vitreuse qui
est commun à l’ensemble des matériaux amorphes. Puis après avoir évoqué rapidement l’historique des
verres métalliques, nous verrons à travers plusieurs exemples que le mode de décomposition des verres
métalliques est très sensible à la composition. Nous verrons que de nombreuses questions se posent
encore sur le mode de dévitrification des alliages que nous étudierons (décomposition spinodale ou
nucléation et croissance), en particulier nous insisterons sur le débat qui existe à propos de la formation de
phases quasicristallines dans les verres métalliques. Nous tenterons de mettre en évidence le fait qu’il existe
peu de travaux incluant des analyses chimiques directes qui permettraient de trancher entre les différentes
interprétations. Enfin nous ferons une description des modèles couramment utilisés pour expliquer le mode
de déformation des matériaux amorphes et présenterons le modèle que nous utiliserons par la suite.
1 La transition vitreuse
Un verre est un solide qui se caractérise par l’absence d’ordre à longue distance. La figure 2.1
illustre le cas de la silice cristallisée et amorphe où l’on voit que l’ordre à courte distance n’est pas modifié et
que seul l’ordre à longue distance est différent. Pour comprendre l’origine d’une telle différence, il faut
remonter à la formation d’un amorphe. Lorsque l’on regarde un diagramme du volume spécifique (ou de
l’enthalpie) en fonction de la température, où l’on a tracé les fonctions le volume (ou de l’enthalpie) de l’état
liquide et cristallin, on s’aperçoit qu’en deçà d’une certaine température, l’état le plus stable d’un matériau
est la phase cristalline. Cependant il arrive qu’au passage de la température de cristallisation, la
transformation liquide-cristal ne se produise pas. Ce phénomène se comprend si l’on admet que la
cristallisation nécessite la formation de germes pour lesquels il y a compétition entre l’énergie de surface
luttant contre la croissance de ces germes et l’énergie de volume favorisant leur croissance. Leur
H,V
T
liquide
état vitreux
cristal
Liquide surfondu
T f Tg Figure 2.2 : variation de l’enthalpie ou du volume enfonction de la température.
Figure 2.1 : ordre à courte distance dansla silice cristallisée (a) et amorphe (b).
Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 7
développement ne pourra se faire que s’ils atteignent une taille critique. Dans certaines conditions, il arrive
que ces germes n’atteignent pas cette taille et ceci principalement en raison d’une descente en température
trop rapide au regard de la mobilité atomique. Le système se retrouve dans un état métastable appelé
liquide surfondu. D’un point de vue thermodynamique cela signifie que le système adopte une fonction
d’enthalpie qui est le prolongement vers les plus basses températures de la fonction d’enthalpie décrivant
l’état liquide.
En deçà d’une certaine température, le système est incapable de suivre cet état métastable. Il se produit
alors ce que l’on appelle la transition vitreuse, la courbe d’enthalpie du système s’écarte progressivement de
l’état surfondu pour suivre une droite parallèle à l’enthalpie libre du cristal. Cette transition ne se produit pas
à une température fixe mais à une température d’autant plus haute que la vitesse de refroidissement est
élevée. La transition vitreuse n’est donc pas une transition classique au sens de la thermodynamique
puisque les systèmes étudiés sont hors d’équilibre. Pour s’en persuader, il suffit de rappeler que la
thermodynamique distingue deux types de transition : les transitions du premier et du deuxième ordre. Dans
le cas d’une transition du premier ordre, la fonction d’enthalpie libre est continue mais les dérivées de celle-
ci sont discontinues (exemple de la transition liquide-cristal où le volume subit une discontinuité). Lors d’une
transition du second ordre, l’enthalpie libre et ses dérivées premières sont continues et seules les dérivées
secondes sont discontinues. C’est ce qui est observé dans le cas de la transition vitreuse. Mais de part et
d’autre de la transition, l’état le plus stable est le liquide surfondu, ce qui est contraire à l’idée de transition.
De plus la transition vitreuse est dépendante de l’histoire thermique en ce sens qu’elle est fonction de la
vitesse de trempe ce qui n’est jamais le cas d’une température de fusion par exemple.
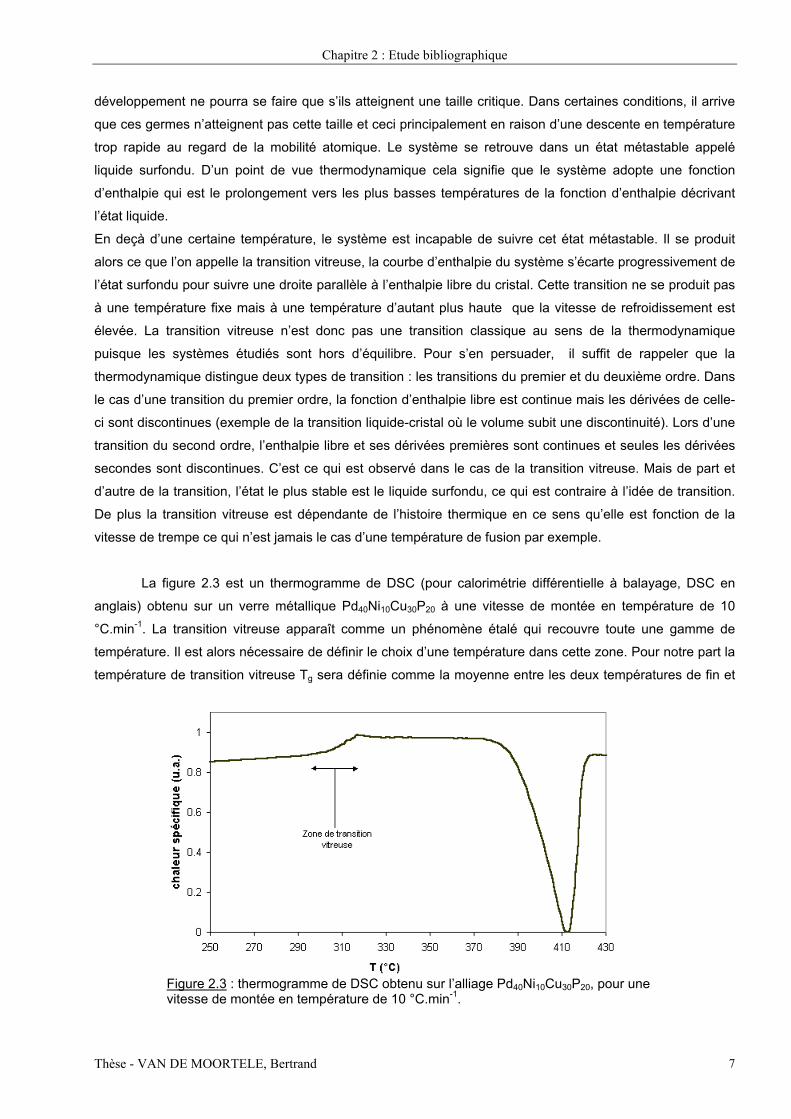
La figure 2.3 est un thermogramme de DSC (pour calorimétrie différentielle à balayage, DSC en
anglais) obtenu sur un verre métallique Pd40Ni10Cu30P20 à une vitesse de montée en température de 10
°C.min-1. La transition vitreuse apparaît comme un phénomène étalé qui recouvre toute une gamme de
température. Il est alors nécessaire de définir le choix d’une température dans cette zone. Pour notre part la
température de transition vitreuse Tg sera définie comme la moyenne entre les deux températures de fin et
Figure 2.3 : thermogramme de DSC obtenu sur l’alliage Pd40Ni10Cu30P20, pour une vitesse de montée en température de 10 °C.min-1.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 8
début de la zone de transition vitreuse comme indiqué sur la figure 2.3. De plus, lorsque l’on donne la valeur
de Tg, il est nécessaire de spécifier les conditions dans lesquelles celle-ci a été mesurée puisque sa valeur
dépend du temps d’observation et des conditions d'évolution de la température.
Pour expliquer la transition vitreuse, plusieurs théories ont été élaborées. La première est celle dite
du volume libre développé par Cohen et Turnbull [Coh1959]. Le concept du volume libre repose sur le fait
qu’un matériau amorphe occupe un volume plus important à l’état amorphe qu’à l’état cristallin. Chaque
atome possède alors en moyenne un volume excédentaire vf. L’hypothèse est que ce volume excédentaire
peut être redistribué entre les atomes sans changement d’énergie du système. A cela s’ajoute le fait que ce
volume libre doit être nul en deçà d’une température T0 et croître continuemenent avec la température au-
dessus de T0. Le temps moyen de relaxation τmol (temps mis par une unité structurale pour se déplacer
d’une distance égale à sa dimension) est donné par la relation de Vogel-Fulcher-Tamman [Tam1926] :
Aussi cette approche permet de décrire correctement l’évolution de la viscosité pour des
températures proches de la transition vitreuse et ce sur plusieurs décades. Pour autant, il faut nuancer cela
en signalant que plusieurs hypothèses de cette théorie sont contredites par l’expérience. Par exemple, il est
prévu que le temps de relaxation doit être constant pour une température inférieure à la température de
transition vitreuse, ce qui est contredit par l’expérience.
Une autre approche est celle d'Adam et Gibbs [Ada1965]. Dans cette théorie, le système est
partagé en N sous-systèmes ou unités structurales. Les mouvements atomiques peuvent s’opérer si sous
l’effet d’une fluctuation thermique, un réarrangement coopératif d’un ensemble d’unités structurales se
produit. On aboutit alors pour le temps de relaxation à l’expression suivante :
avec ∆µ la barrière d’enthalpie libre s’opposant au réarrangement coopératif, ∆Cp la différence de chaleur
spécifique entre état liquide et état vitreux, et enfin T2, température pour laquelle on admet que l’entropie de
configuration s’annulerait si le liquide surfondu pouvait se maintenir en équilibre thermodynamique jusqu'à
cette température. Une telle démarche implique l’existence d’une température critique T2 dont la réalité à
l’heure actuelle est encore discutée, et au sujet de laquelle l’expérience ne peut fournir aucune information.
Plus récemment, Perez et al. [Per1988] ont développé une approche de la transition vitreuse que
nous présenterons dans la section 6 de ce chapitre.
⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜
⎝
⎛
∆µ∆
τ=τ
2
gp0mol
TTlnT
1C
2lnexp
⎟⎠⎞
⎜⎝⎛
−τ=τ
00mol
TTBexp (2.1)
(2.2)

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 9
2 Des verres métalliques massifs
De tous les matériaux amorphes qui nous entourent, c’est le verre minéral qui fut découvert et
maîtrisé le plus tôt, historiquement parlant. Ainsi était-il connu des Egyptiens et des Phéniciens, certaines
traces d’ateliers d’arts du verre remontant à l’Egypte ancienne. Pour comprendre cela, il suffit de considérer
que ces systèmes, comme la silice, sont très facilement amorphisables en raison de leur très forte viscosité
à l’état liquide, expliquant ainsi une maîtrise rapide.
Dans le cas des métaux, les vitesses de trempe nécessaires pour obtenir des amorphes sont élevées voire
impossibles à atteindre dans le cas des métaux purs. Ainsi le premier verre métallique n’a été obtenu qu’en
1960 par Klement et al. [Kle1960], dans le système AuSi. A l’époque, les alliages étudiés étaient
principalement binaires, AuPb ou GdFe par exemple. Ils nécessitaient des vitesses de trempe de l’ordre de
106 °C.s-1. Les techniques de préparations par trempe sur roue ou évaporation permettaient d’atteindre ces
vitesses, mais l’épaisseur des échantillons était faible (environ 25 µm pour la trempe sur roue). Cette forme
d'échantillons ne permettait pas de faire de caractérisation complète des propriétés mécaniques.
Il a fallu attendre près de 15 ans pour que Chen et al. [Che1974] évoquent pour la première fois
l’élaboration d’un verre métallique massif dans le système Pd76Cu6Si18 en atteignant une épaisseur record à
l’époque de 3 mm. Le véritable essor des verres métalliques massifs n’a eu lieu qu’à la fin des années 80
avec la découverte de nombreux systèmes pour lesquels il était possible d’obtenir des amorphes métalliques
massifs : Mg-Ln-TM [Ino1988], Zr-Al-TM [Ino1993] Zr-Ti-TM-Be [Pek1993] (TM étant l’abréviation utilisée en
anglais pour métaux de transition). Les vitesses de trempe limites de ces systèmes, de 100 à 1 °C.s-1 ,
permettent donc d’obtenir des échantillons massifs. Certains de ces alliages, comme le Vit1 et le Vit4, ont
fait l’objet d’une commercialisation. L’idée de pouvoir utiliser ces matériaux pour des applications expliquait
alors l’engouement pour l’étude de ces matériaux. Pour autant, nous évoquerons, dans le chapitre 4, quelles
sont les limites actuelles des applications possibles de ces matériaux.
Dans le même temps des alliages modèles comme le PdCuNiP [Ino1996] permettaient d’atteindre
des épaisseurs de plus de 70 mm.
Figure 2.4 : diagramme ternaire rendant compte de laqualité ‘amorphe’ des alliages en fonction de leurcomposition (d’après [Ino1988]).

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 10
Des études systématiques sur la composition des alliages amorphisables ont été menées afin de
déterminer les diagrammes de composition aboutissant à l'obtention de verres métalliques et d’essayer ainsi
de comprendre ce qui conduisait à ce qu’un alliage devienne amorphe ou non. La figure 2.4 est un
diagramme de composition pour l’alliage LaAlNi dans lequel apparaît l’étendue de la zone de liquide
surfondu en fonction de la composition. Nous voyons que toute une gamme de composition permet d’obtenir
un alliage amorphe avec une zone de liquide surfondu plus ou moins grande. Actuellement, aucune raison
n’a pu être donnée pour expliquer la formation d’un verre pour les différentes compositions. Néanmoins, les
nombreux alliages ayant permis d’obtenir des amorphes métalliques ont conduit à édicter 3 lois empiriques
expliquant qu’un alliage était amorphisable ou ne l’était pas [Ino1995, Ino1997] :
1- le système doit contenir au moins 3 types d’atomes différents,
2- la différence de taille entre les atomes doit être au moins de 15%,
3- l’enthalpie de mélange entre certaines espèces chimiques doit être négative.
Ces trois lois empiriques ont été résumées sous le concept de ‘glass forming ability’ (GFA) que l’on
pourrait traduire par ‘capacité à former des verres’. Actuellement la seule voie d’investigation pour trouver de
nouveaux alliages consiste donc à explorer, en partant de ces lois, les diagrammes de compositions et de
mesurer les propriétés d’amorphicité des alliages obtenus. Dans le cadre de notre collaboration avec le
CRETA de Grenoble nous pouvons citer la thèse de Nicolas CLARET dans laquelle une partie importante
est consacrée à la recherche de nouveaux alliages à base fer ainsi qu’à la caractérisation par méthodes
globales de verres métalliques (diffraction de neutrons par exemple).
3 Des amorphes présentant des défauts
He et al. [He2000] ont montré que dans l’alliage Zr52.5Ni14.6Al10Cu17.9Ti5, il était possible d’observer
des cristaux, que nous appellerons primaires, figure 2.5,
noyés dans une matrice amorphe. La taille de ces
particules, de 0.5 à 1 µm, plaide pour une cristallisation
lors du refroidissement. Les analyses de composition
ont montré que ces cristaux étaient dans ce cas enrichis
en aluminium. Les auteurs ont montré également que
ces particules lors des recuits en température étaient
stables, ne subissaient aucune croissance et ne
perturbaient pas la séquence de cristallisation de la
matrice amorphe environnante.
De la même manière Wanderka et al. ont étudié l’effet de la vitesse de refroidissement sur la
formation de tels cristaux dans l’alliage Zr46.8Ti8.2Cu7.5Ni10Be27.5. Ils ont identifié la phase ZrBe2 contenant un
Figure 2.5 : micrographie en champ clair d’uncristal primaire et diffraction d’un de ces cristauxselon l’azimuth [110] (d’après [He2000]).

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 11
peu de titane, de nickel et de cuivre [Wan1999a, Wan1999b] et une phase orthorhombique enrichie en
zirconium et appauvrie en nickel et cuivre.
Les deux exemples ont de commun que les matériaux ont été obtenus pour de faibles vitesses de
refroidissement par rapport à ce qui est généralement pratiqué. Les auteurs sous-entendent ainsi que pour
des vitesses plus importantes il n’y a pas de cristallisation primaire. En particulier Wanderka prétend que
pour une vitesse de refroidissement supérieure à 20 °C.s-1, aucun cristal primaire ne se forme dans le Vit4 et
ceci en s’appuyant principalement sur une caractérisation par diffraction des rayons X. Pour autant le même
auteur reconnaît que la diffraction X n’est pas un moyen expérimental suffisant pour mettre en évidence un
taux de cristallisation inférieur à 1 % ou de faible taille [Wan1999a].
Généralement cette cristallisation 'parasite' n’est pas évoquée car elle ne perturbe pas les propriétés du
matériau, sauf dans des cas particuliers que nous aborderons dans le quatrième chapitre. Néanmoins il
semble qu'elle se fasse dans de nombreux alliages. Son origine est parfois attribuée à une cristallisation
hétérogène sur des bords du moule de coulée ou, comme nous le verrons dans la section 5 de ce chapitre,
à la présence de l’oxygène.
4 De l’état vitreux à la cristallisation
L'état amorphe est d'un point de vue thermodynamique un état hors d'équilibre. La représentation
schématique, figure 2.6, de l'énergie du système en fonction d’une coordonnée de configuration du système
permet de mieux comprendre ce que cela signifie et implique sur l'évolution de ces systèmes. Le point A
correspond à un état vitreux. A une température en deçà de Tg, le système bien que hors d'équilibre ne
rejoint pas, à notre échelle de temps, un état métastable ou stable (respectivement C ou D). L'augmentation
de la température jusqu’au voisinage de Tg ou au-delà permet au système de "glisser" dans un premier
temps vers l’état de liquide surfondu qui
correspond au point B. Cette
représentation est valable pour
l'ensemble des systèmes amorphes.
Dans le cas des verres métalliques, un
puits métastable correspond, au cours
d’une montée en température, ou d’un
recuit isotherme, à la formation de
phases cristallines métastables. La
température à laquelle se feront les
traitements isothermes, ou encore la
vitesse de balayage en température,
modifiera les hauteurs respectives des
barrières d’énergie ∆G1 et ∆G2. Comme
nous allons le voir dans la suite de ce
enthalpie libre G
coordonnée de configuration
A
B
C
D
Figure 2.6 : énergie du système en fonction d’une coordonnéede configuration du système.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand
chapitre, l’existence d’une étape intermédiaire est fonction de la composition mais également du type de
traitement thermique (chapitre 5). Dans certains cas, la composition des phases cristallines qui se forment
est fortement différente de celle de l’alliage amorphe. Il est alors nécessaire qu’une diffusion à longue
distance s’opère dans le matériau amorphe. Le phénomène de diffusion, bien connu dans le cas des
métaux, reste sujet à discussion dans le cas des amorphes.
4.1-Mode de diffusion dans les verres métalliques
Dans le cas des alliages métalliques, on distingue deux mécanismes de diffusion différents. Le
premier mécanisme est une diffusion par site interstitiel où un atome vient occuper un espace entre deux
autres atomes. Ceci peut se produire si la taille de l’atome est suffisamment petite au regard des distances
entre les deux autres atomes. C’est le cas du carbone dans l’acier venant se placer entre les atomes de fer.
Le second mécanisme est une diffusion qui se produit grâce aux défauts ponctuels du type lacunaire. Un
atome peut alors passer d’un site à un autre et de proche en proche s’opère une diffusion. On peut
également évoquer la diffusion dans les joints de grains qui dans le cas des verres métalliques n’existera
pas.
Les matériaux amorphes par définition n'ont pas de réseau cristallin et la question du mode de
diffusion peut effectivement se poser. La notion de volume libre, évoquée précédemment, est bien souvent
interprétée comme un partage entre tous les atomes de ce volume supplémentaire. Nous allons voir dans
les quelques exemples qui suivent que la diffusion dans des matériaux amorphes n’est pas interprétée de la
même manière par tous.
Fielitz et al. [Fie1999], ainsi que Macht et al. [Mac2001], ont effectué des mesures expérimentales
du coefficient de diffusion pour différents atomes (Fe,
B, Be) en fonction de la température dans les
alliages Vit1 et Vit4. Afin de décrire les coefficients
de diffusion par une loi du type Arrhénius, il est
nécessaire de distinguer deux régimes de
température pour lesquels il faut définir à chaque fois
un coefficient pré-exponentiel et une énergie
d’activation (figure 2.7). La température à laquelle se
produit ce changement ne semble pas être
directement reliée au rayon atomique de l’atome qui
diffuse. L’interprétation selon ces auteurs est qu’à
haute température la diffusion atomique est
coopérative et ne l’est pas à basse température.
De la même manière, Ehlmer et al.
[Ehm1998], en comparant dans l’alliage Vit4 la
diffusion de deux isotopes du cobalt à longue
Figure 2.7 : coefficients de diffusion en fonctionde la température de différentes espèceschimiques dans les alliages Vit1 et Vit4 (d’après[Fie1999]).12
Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 13
distance, ont montré que la diffusion était gouvernée par un phénomène collectif d’une dizaine d’atomes.
Une autre interprétation a été envisagée par Rehmet et al. [Reh2001]. Pour cela, ils ont étudié la
diffusion du béryllium dans la zone du liquide surfondu du Vit4. Ils en concluent qu’il n’existe qu’un seul
mode de diffusion dans les verres métalliques. Les changements dans les coefficients pré-exponentiels et
les énergies d’activation en fonction de la température observés par Ehlmer et al. [Ehl1998] sont, selon eux,
une conséquence d’un changement structural de l’amorphe (variation du volume libre dans la zone de
liquide surfondu) et non pas d’un nouveau mode de diffusion. Pour ces auteurs la diffusion des atomes se
fait de manière coopérative, et ceci même pour les atomes les plus petits comme le béryllium.
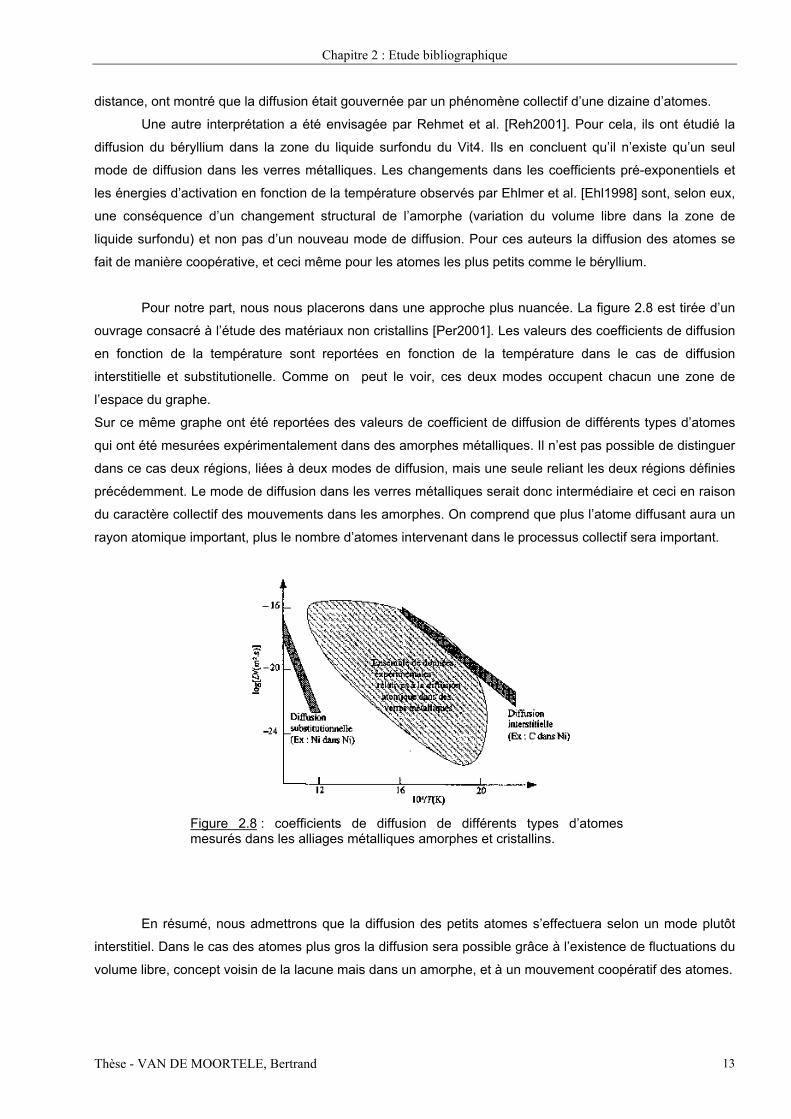
Pour notre part, nous nous placerons dans une approche plus nuancée. La figure 2.8 est tirée d’un
ouvrage consacré à l’étude des matériaux non cristallins [Per2001]. Les valeurs des coefficients de diffusion
en fonction de la température sont reportées en fonction de la température dans le cas de diffusion
interstitielle et substitutionelle. Comme on peut le voir, ces deux modes occupent chacun une zone de
l’espace du graphe.
Sur ce même graphe ont été reportées des valeurs de coefficient de diffusion de différents types d’atomes
qui ont été mesurées expérimentalement dans des amorphes métalliques. Il n’est pas possible de distinguer
dans ce cas deux régions, liées à deux modes de diffusion, mais une seule reliant les deux régions définies
précédemment. Le mode de diffusion dans les verres métalliques serait donc intermédiaire et ceci en raison
du caractère collectif des mouvements dans les amorphes. On comprend que plus l’atome diffusant aura un
rayon atomique important, plus le nombre d’atomes intervenant dans le processus collectif sera important.
En résumé, nous admettrons que la diffusion des petits atomes s’effectuera selon un mode plutôt
interstitiel. Dans le cas des atomes plus gros la diffusion sera possible grâce à l’existence de fluctuations du
volume libre, concept voisin de la lacune mais dans un amorphe, et à un mouvement coopératif des atomes.
Figure 2.8 : coefficients de diffusion de différents types d’atomesmesurés dans les alliages métalliques amorphes et cristallins.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MO
4-2 Mise en évidence de la cristallisation
La figure 2.9 est un thermogramme de DSC obtenu par Xing et al. [Xin1997] sur l’alliage
Zr62Cu20Al10Ni8 lors d’une expérience de montée en température. La partie de la courbe notée [Tg]
correspond à la transition vitreuse décrite précédemment. Elle est suivie d’une zone, notée [ZLS], qui
correspond à la zone de liquide surfondu. Dans le cas des verres métalliques, cette zone est limitée vers les
hautes températures par la cristallisation que l’on reconnaît au pic exothermique noté Tx.
∆Tx est la la
températures de cr
Cette valeur dép
températures de tr
ainsi que de la vites
leur détermination.
L’un des buts des é
d’obtenir un alliage a
possible. La figure
critique en fonct
correspondant à la
pour laquelle il est e
Nous voyons qu’il
valeur ∆Tx et la taille
possible d’obtenir.
Dans de nom
celle de l’alliage Zr6
Figure 2.9 : Thermogramme de DSC dans l'alliage Zr62Cu20Al10Ni8.
[Tg] Tx
[∆Tx]
ZLS
ORTELE, Bertrand 14
rgeur de la zone de liquide surfondu. Elle est définie comme la différence entre les
istallisation et de transition vitreuse.
end donc des définitions des
ansition vitreuse et de cristallisation
se de montée en température lors de
laborateurs de verres métalliques est
vec une valeur de ∆Tx la plus élevée
2.10 est un diagramme de la vitesse
ion de ∆Tx, la vitesse critique
vitesse de descente en température
ncore possible d’obtenir un amorphe.
y a une relation directe entre cette
maximale des échantillons qu’il sera
breux cas, la séquence de cristallisation des verres métalliques est plus complexe que
2Cu20Al10Ni8. Le thermogramme de la figure 2.11 montre que dans le cas de l’alliage
Figure 2.10 : diagramme de la vitesse critique de refoidissement en fonction de ∆Tx pour différents alliages métalliques amorphes.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE
Zr69Cu12Ni10Al9 la cristallisation s’effectue en quatre étapes différentes (d’après [Fan2001a]). La question qui
se pose alors est de savoir à quoi correspondent ces différentes transformations.
Afin de mieux contrô
isothermes à plus basse te
isochrone. C’est le cas cor
Zr58Nb5Cu25Al12 ayant subi u
[Fan2000]. Le temps de tra
puisqu’un second passage
dispersés uniformément dan
remarquer qu'à l'issue du pre
Le taux de cristallisation pe
rapportent qu’à un stade é
l’amorphe a cristallisé. Dans
des rayons X présentent prin
Figu
Figure 2.12 : image en haZr58Nb5Cu25Al12 nanocristallis
re 2.11 : thermogramme de DSC de l’alliage Zr69Cu12Ni10Al9
, Bertrand 15
ler les étapes de la cristallisation, il est fréquent de procéder à des traitements
mpérature que la température de cristallisation déterminée lors d’une montée
respondant à la figure 2.12 qui est une image haute résolution de l’alliage
n traitement thermique pendant 15 min à une température juste au-dessus de Tg
itement correspond à la fin de la réaction relative au premier pic de DSC
montre que celui-ci a disparu. Les cristaux sont de taille nanométrique et
s la matrice amorphe. Le taux de cristallisation est proche de 50 %. On peut
mier pic de DSC, la matrice amorphe n’a pas complètement cristallisé.
ut être extrêmement variable d’un alliage à l’autre. Revesz et al [Rev2001]
quivalent, c'est-à-dire à la fin du premier pic de DSC, seulement 15 % de
le cas d’une faible cristallisation, les diffractogrammes obtenus en diffraction
cipalement un profil amorphe surmonté de très faibles pics.
ute résolution d’un alliageé.
agrandissement d’une zone de la figure 2.12permettant de distinguer correctement lesfranges de haute-résolution.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 16
4-3 Décomposition de la matrice amorphe Dans certains alliages, la cristallisation est précédée d'une décomposition de la matrice amorphe.
Ainsi, Gangopadhyay et al. [Gan2000] ont montré en microscopie électronique que lors d’un recuit isotherme
pour une température comprise dans la région de liquide surfondu l’alliage Al88Gd6La2Ni4 subissait une
décomposition de phases. La figure 2.13 est une micrographie illustrant le propos des auteurs. Le matériau
que ces auteurs ont étudié est un matériau idéal du point de vue outil ‘microscopie électronique’. En effet,
dans un amorphe, la microscopie électronique est principalement sensible à la densité électronique des
atomes. Dans ce cas particulier, les différences de numéro atomique sont telles que l’on obtient un
contraste, ce qui ne sera pas le cas dans des alliages à base zirconium.
Les auteurs concluent, après avoir observé la cristallisation ultérieure du matériau, que la microstructure
conservait des traces de cette décomposition.
Ce genre de décomposition a été également observé dans d'autres alliages. On peut citer l'alliage
Al87Ni10Ce3 dans lequel Tsai et al. [Tsa1997] ont mis en évidence par diffusion des rayons X aux petits
angles un processus identique. De même, Gerold et al. [Ger1997] ont montré que la cristallisation dans
l'alliage Zr11Ti34Cu47Ni8 était précédée d'une décomposition. Dans l’alliage Vit1, des observations similaires
ont été faites comme nous allons le détailler.
Cas du Vit1
Löfller et al. [Lof2000a] ont étudié la décomposition du Vit1 par diffusion de neutrons aux petits
angles. Cette technique permet de mettre en évidence des fluctuations de composition de très faibles
amplitudes et ceci dans les stades les plus précoces. Ils ont montré que pour des températures de
traitement thermique dans la zone de liquide surfondu, il était possible d’observer un maximum
d’interférence qui se développait en fonction du temps comme le montre la figure 2.14. Dans les premiers
instants, l’intensité est une fonction exponentielle du temps et ce d’autant plus longtemps que la température
de traitement est proche de la température de transition vitreuse. Ce comportement a été prédit par Cahn et
Figure 2.13 : image en champ clair montrant la décomposition del’alliage Al88Gd6La2Ni4 en deux phases amorphes.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 17
Hilliard [Cah1958] dans le cadre d‘une décomposition spinodale. Ce mode de décomposition consiste en
des fluctuations régulières de la composition dont l’amplitude augmente avec le temps.
Dans le cas d’une décomposition spinodale, il n’existe pas de barrière d’énergie à franchir et la
décomposition peut se produire dès les premiers instants. Le maximum d’intensité en diffusion de neutrons
se situe à une valeur de vecteur de diffusion qui correspond dans le réseau direct à la distance pour laquelle
se répètent ces fluctuations de composition. Cette distance particulière est appelée longueur d’onde de la
décomposition spinodale. On comprend que plus la température sera basse plus la longueur d’onde
associée à la décomposition sera petite puisque la diffusion ne se fera que sur de petites distances.
En s’appuyant sur la vitesse de croissance des domaines, déduite du rayon de Guinier, ainsi que
des coefficients de diffusion des différents éléments, les auteurs concluent que l’élément qui contrôle le
processus de diffusion est le titane. Dans les stades plus avancés, et ceci quelle que soit la température de
traitement, ils ont observé que ce maximum d’intensité se décalait vers les plus petites valeurs de vecteur
de diffusion. Selon ces auteurs, ce déplacement s’expliquerait par une cristallisation. Or une cristallisation
ne devrait pas entraîner un tel décalage. En effet, la cristallisation qui se produit suite à une décomposition
spinodale s'effectue dans des zones de compositions favorables dont la périodicité est celle de la
décomposition. Il semble donc qu'au moment de la cristallisation s'opèrent des phénomènes plus complexes
qui sont encore inexpliqués.
La figure 2.15 provient d‘un article de Schneider
et al. [Sch1998]. Dans ce travail, ils ont déterminé la
région de la décomposition spinodale pour des alliages
Zr41.2TixCu12.5Ni10Be36.3-x. avec x égal à 8.8, 13.8 ou
16.3 (respectivement 1, 2 et 3 dans le graphe de la
figure 2.15). Selon eux, dans le cas de x=13.8, ce qui
correspond au Vit1, au-delà de 400 °C, aucune
décomposition spinodale ne peut être observée.
Figure 2.15 : courbe de délimitation de ladécomposition spinodale pour l’alliageZr41.2Cu12.5Ni10(Be,Ti)36.3.
Figure 2.14 : évolution en fonction du temps de l’intensité diffusée
Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 18
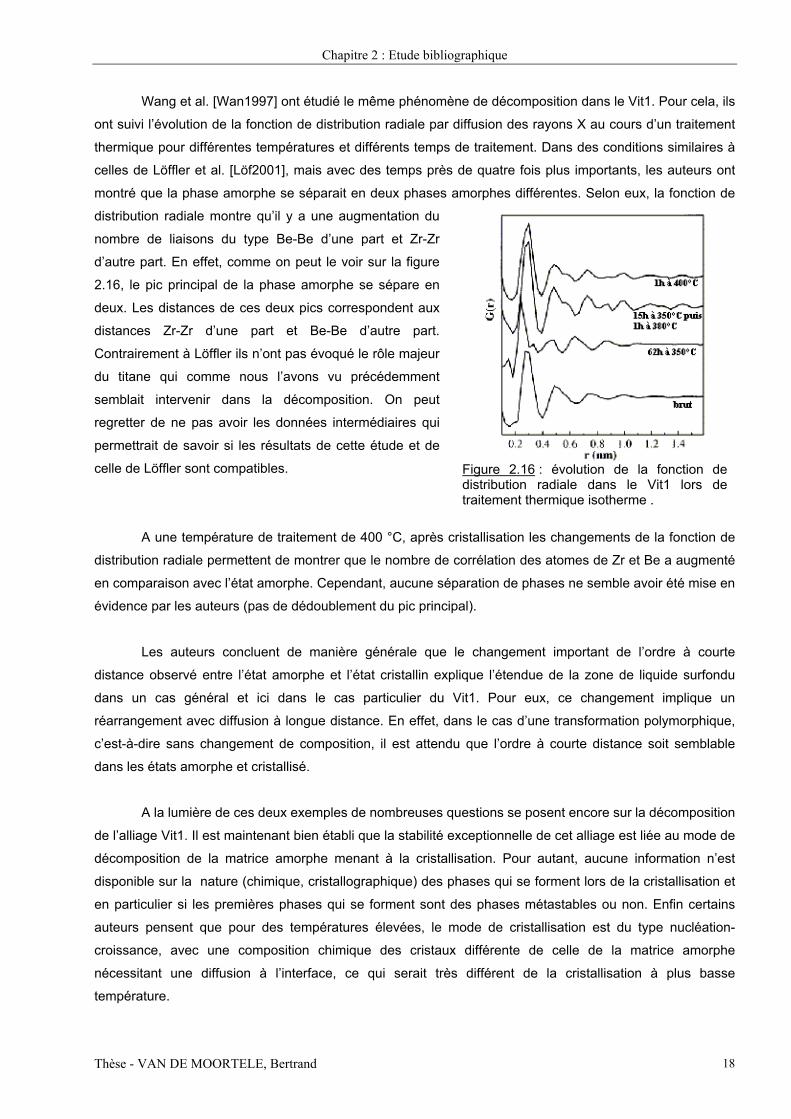
Wang et al. [Wan1997] ont étudié le même phénomène de décomposition dans le Vit1. Pour cela, ils
ont suivi l’évolution de la fonction de distribution radiale par diffusion des rayons X au cours d’un traitement
thermique pour différentes températures et différents temps de traitement. Dans des conditions similaires à
celles de Löffler et al. [Löf2001], mais avec des temps près de quatre fois plus importants, les auteurs ont
montré que la phase amorphe se séparait en deux phases amorphes différentes. Selon eux, la fonction de
distribution radiale montre qu’il y a une augmentation du
nombre de liaisons du type Be-Be d’une part et Zr-Zr
d’autre part. En effet, comme on peut le voir sur la figure
2.16, le pic principal de la phase amorphe se sépare en
deux. Les distances de ces deux pics correspondent aux
distances Zr-Zr d’une part et Be-Be d’autre part.
Contrairement à Löffler ils n’ont pas évoqué le rôle majeur
du titane qui comme nous l’avons vu précédemment
semblait intervenir dans la décomposition. On peut
regretter de ne pas avoir les données intermédiaires qui
permettrait de savoir si les résultats de cette étude et de
celle de Löffler sont compatibles.
A une température de traitement de 400 °C, après cristallisation les changements de la fonction de
distribution radiale permettent de montrer que le nombre de corrélation des atomes de Zr et Be a augmenté
en comparaison avec l’état amorphe. Cependant, aucune séparation de phases ne semble avoir été mise en
évidence par les auteurs (pas de dédoublement du pic principal).
Les auteurs concluent de manière générale que le changement important de l’ordre à courte
distance observé entre l’état amorphe et l’état cristallin explique l’étendue de la zone de liquide surfondu
dans un cas général et ici dans le cas particulier du Vit1. Pour eux, ce changement implique un
réarrangement avec diffusion à longue distance. En effet, dans le cas d’une transformation polymorphique,
c’est-à-dire sans changement de composition, il est attendu que l’ordre à courte distance soit semblable
dans les états amorphe et cristallisé.
A la lumière de ces deux exemples de nombreuses questions se posent encore sur la décomposition
de l’alliage Vit1. Il est maintenant bien établi que la stabilité exceptionnelle de cet alliage est liée au mode de
décomposition de la matrice amorphe menant à la cristallisation. Pour autant, aucune information n’est
disponible sur la nature (chimique, cristallographique) des phases qui se forment lors de la cristallisation et
en particulier si les premières phases qui se forment sont des phases métastables ou non. Enfin certains
auteurs pensent que pour des températures élevées, le mode de cristallisation est du type nucléation-
croissance, avec une composition chimique des cristaux différente de celle de la matrice amorphe
nécessitant une diffusion à l’interface, ce qui serait très différent de la cristallisation à plus basse
température.
Figure 2.16 : évolution de la fonction dedistribution radiale dans le Vit1 lors detraitement thermique isotherme .

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 19
4-4 Echelle de la cristallisation
Le type de microstructure visible sur la micrographie de la figure 2.12 est fréquemment observé dans
les verres métalliques. Selon certains auteurs [Fan2000, Zha1996], le très fort taux de germination pour
aboutir à ce type de microstructure serait lié à l’apparition d’un ordre à courte distance lors du
refroidissement des matériaux. La forte interaction attractive entre certaines espèces chimiques permettrait
de créer des embryons de cristaux dans le liquide surfondu. Ces embryons seraient alors figés et
deviendraient des zones de nucléation facile.
Külik [Kul2001] a montré dans différents alliages à base Fe que seules certaines compositions
permettaient d’obtenir ce genre de microstructure et que cela n’était pas lié à un traitement thermique
particulier.
Une autre approche consiste à considérer que la taille des cristaux observés à la fin de la première
étape de cristallisation est un bon indicateur pour distinguer différents processus de cristallisation existant
dans les verres métalliques. Inoue [Ino1999] a comparé la taille des cristaux dans les alliages Zr67Cu33 (a) et
Zr65Al20Cu15 (b) dont les phases stables après cristallisation sont Zr2Cu et (Zr2Cu, Zr2Al) respectivement.
L’observation en microscopie électronique montre qu’après la fin de la transformation liée au premier pic de
DSC, dans le cas (a), la taille des cristaux est micrométrique et dans le cas (b) nanométrique. Dans le
premier cas, la composition de la matrice amorphe est la même que celle de la phase cristalline stable. Une
réorganisation locale est suffisante pour permettre la cristallisation. Dans le second cas, une diffusion des
éléments est nécessaire. A la température d’observation, la diffusion ne peut pas se faire sur de longues
distances et l'on aboutit alors à une formation de nano-cristaux. C'est précisément cette différence de mode
de dévitrification qui expliquerait les différences de stabilité en température entre certains verres métalliques.
Il est utile de rappeler que les verres métalliques dans lesquels la décomposition de la matrice
amorphe est fortement marquée avant cristallisation ont une taille de cristallisation ou une répartition en
rapport avec cette décomposition.
4-5 Identification des premières phases cristallographiques
Dans certains cas, l’identification des nano-cristaux ne pose pas de problème. La composition de
départ de l’alliage permet de prévoir les phases qui vont se former (composition proche de Zr2Cu et Zr2Al
avec du Nb en substitution pour (a) ) et la diffraction des rayons X permet de le vérifier.
Mais il existe des cas moins triviaux où les premières phases cristallographiques qui se forment ne
sont pas des phases connues et ni identifiables par diffraction des rayons X. Il faut alors faire appel à des
techniques d’analyses chimiques couplées à la microscopie électronique. Cette tâche est d’autant plus
délicate que la taille des cristaux est petite et que les variations de compositions entre les phases
cristallisées et la matrice amorphe ne sont pas forcément très importantes. Aussi il existe très peu de mise
en évidence directe de la nature chimique des premières phases cristallines lorsque celles-ci ne sont pas
déjà connues. Dans le cas du Vit1 très peu d'analyses sont disponibles. Schneider et al. [Sch1996] évoquent

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 20
pour leur part, lors d’un traitement thermique à 350 °C, la formation de nanocristaux riches en cuivre et
titane. La structure cristallographique correspondrait à un cubique à faces centrées, de paramètre
cristallographique voisin de 0.4 nm.
Pour autant, on peut relever quelques points divergents dans les interprétations sur la décomposition du
Vit1. Selon Liu [Liu1997], les phases cristallographiques formées dans le Vit1 sont indépendantes de
l’histoire thermique de l’échantillon, ce qui semble aller à l’encontre de ce qu’ont observé Wang et al.
[Wan1997] lors de leur étude de la fonction de distribution radiale. Les études menées par diffusion (de
neutrons ou de rayons X) ne permettent pas d’avoir des informations directes sur la nature chimique des
fluctuations ou de la cristallisation.
4-6 Des phases quasicristallines
Les premiers quasicristaux ont été obtenus en 1984 par Shechtman et al. [She1984] dans un alliage
métallique à base aluminium-manganèse obtenu par trempe ultra-rapide. Par cette technique, les vitesses
de trempe atteintes ne permettent pas au système de suivre l’équilibre thermodynamique. Il y a alors
formation de phases métastables, les quasi-cristaux étant un cas particulier de celles-ci. Dans le cas des
verres métalliques, les vitesses sont suffisamment élevées pour éviter la cristallisation de la phase
métastable lors du refroidissement. Pour autant, il n’est pas surprenant d’observer des phases
quasicristallines se former au cours de la remontée en température des verres métalliques, ces phases étant
des phases intermédiaires. Si l’on considère les quasicristaux d’un point de vue arrangement atomique, il est
communément admis qu’ils sont une situation intermédiaire entre les cristaux et les amorphes. Un des
arguments avancés pour comprendre la formation de phases quasicristallines est la faible énergie
d’interface entre une phase quasicristalline et la matrice amorphe. Köster et al. [Kos1997] ont estimé dans
l’alliage Zr69.5Cu12Ni11Al7.5 que cette énergie d’interface était dix fois moins grande entre les quasi-cristaux et
la matrice amorphe qu’entre une phase cristalline et la matrice amorphe.
Saida et al. [Sai2000a] ont observé dans l’alliage Zr70Fe20Ni10 (obtenu sous forme de ruban
amorphe) la formation d’une phase quasi-cristalline lors d’un recuit isotherme pendant 120 s à une
température de 397 °C, c’est-à-dire 3 °C en-dessous de la température de cristallisation déterminée en
isochrone pour une vitesse de 40 °C.min-1. Dans ce cas, la taille des quasicristaux est comprise entre 5 et 10
nm et il reste très peu de phase amorphe. La phase quasicristalline formée est une phase métastable, ce qui
est le cas de l’ensemble des phases quasicristallines dans les verres métalliques, puisqu’un traitement à
plus haute température conduit à la disparition de cette phase et la formation de phases plus stables à haute
température telles que Zr2Ni et Zr2Fe. Selon ces auteurs, la formation de la phase quasicristalline serait due,
dans le cas des verres métalliques à base zirconium et métaux de transition, à l’existence d’une forte affinité
entre Zr et Ni d’une part et Zr et les métaux de transition d’autre part, empêchant ainsi une réorganisation à
longue distance. Ceci aurait pour conséquence l’existence dans le matériau amorphe d’un ordre à courte
distance icosahédrique. La cristallisation ne serait alors qu’une réorganisation très locale, ne nécessitant pas
de diffusion d’une espèce chimique expliquant ainsi qu’elle se produise à plus basse température que la

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 21
cristallisation de phase plus stable. Ce même argument a d’ailleurs été repris par Louzguine et al. [Lou2001]
pour expliquer la formation d’une phase quasi-cristalline dans l’alliage Zr55Cu20Ti15Ni10.
Xing et al. [Xin1999] ont également montré l’importance de cet ordre à courte distance préexistant.
Pour ce faire, ils ont étudié le type de cristallisation dans l’alliage Zr57Ti5Cu20Al10Ni8 en fonction de la vitesse
de refroidissement (échantillon massif ou ruban). Ils observent dans les deux cas la formation d’une phase
quasicristalline. Cette phase quasicristalline est associée aux phases Zr2Ni et Zr2Cu pour l’échantillon
massif, c'est-à-dire l'échantillon refroidi plus lentement. Il apparaît que le taux de nucléation de la phase
quasicristalline est plus élevé dans l’alliage refroidi lentement. En considérant que lors du refroidissement, il
y a apparition dans le liquide surfondu d’un ordre à courte distance semblable à l’orientation à celui existant
dans une phase quasicristalline, Xing et al. pensent que plus l’alliage est refroidi lentement plus l’énergie
d’interface est diminuée expliquant ainsi les variations du taux de nucléation.
Cependant l’argument de l’existence d’un ordre icosahédrique dans le liquide surfondu n’en est pas
un selon Mattern et al. [Mat2001, Mat2002]. Pour prouver cela, ils ont comparé, par diffusion des rayons X,
les fonctions de corrélation de paires des différents alliages Zr62-xTixCu20Al10Ni8 avec x compris entre 2 et 7,5
et n’ont constaté aucune différence entre l’alliage dans lequel des quasicristaux ont été observés (x=5) et les
autres. Pour autant, on peut se demander si cette technique, qui est basée sur une information moyennée
sur la taille de la sonde et l’épaisseur de l’échantillon, pourrait déceler des sites d’ordre icosahédrique peu
nombreux.
Une autre approche du débat sur la formation de phase quasicristalline consiste à évaluer les
enthalpies de mélanges des différents espèces chimiques. Pour cela, Saida et al. [Sai2000b] ont observé la
formation de phase quasicristalline dans les alliages Zr70Ni10X20, avec X=Pd, Pt ou Au. Selon eux, l’argument
principal pour expliquer la formation d’une phase quasicristalline réside dans l’enthalpie de mélange qui est
fortement négative pour les paires atomiques Zr-Ni et Zr-Pd, respectivement –49 et –91 kJ.mol-1, alors qu’il
n’y a pas d’affinité chimique entre Ni et Pd. L’idée est donc qu’il y aurait formation d’entités de Zr-Ni et Zr-Pd
suffisamment grandes et liées pour les empêcher d’avoir une mobilité suffisante.
De même, Saida et al. [Sai2001a] ont montré que dans l’alliage Zr70Pd30, la phase quasicristalline
était moins stable que précédemment. Ils pensent que l’absence de nickel facilite légèrement la mobilité en
évitant la formation de paires Zr-Ni, sans pour autant compromettre la formation d’une phase quasicristalline.
De nombreux auteurs s’accordent donc à dire que la formation des quasicristaux est une
transformation polymorphique, sans changement de composition. Il s’agirait juste d’une modification de
l’ordre à courte distance ne nécessitant qu’une faible énergie d’activation. Pour autant, très peu de mesures
directes de la composition chimique des phases quasicristallines ont été effectuées pour appuyer ces
affirmations. Comme nous allons le voir dans les exemples qui suivent, certains auteurs pensent au
contraire que la diffusion joue un rôle non négligeable dans la formation des quasicristaux.
Dans le cas de l’alliage Zr65Al7.5Cu12.5Ni10Ag5 étudié par Lee et al.[Lee2000], lors d’un recuit à 420 °C
pendant 120 min (20 °C en dessous de Tx) toute la matrice amorphe est transformée en quasicristaux dont

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 22
les tailles sont comprises entre 40 et 70 nm. Un des aspects de cette étude est de comparer le taux de
cristallisation dans cet alliage à celui de l’alliage Zr69.5Cu12Ni11Al7.5 étudié par Köster et al. [Kös1997]. Dans
celui-ci pour un traitement thermique similaire, les quasicristaux ne représentent que 10 % du volume et leur
taille est comprise entre 80 et 120 nm. Ceci semble indiquer selon Lee et al. que l’élément argent facilite la
nucléation de quasicristaux. La cinétique de cristallisation, décrite par les équations de Johnson-Mehl-
Avrami, leur permet de conclure dans un premier temps à une transformation polymorphique et donc sans
changement de composition entre la matrice amorphe et la phase quasicristalline. Cette interprétation a été
confirmée par Chen et al. [Che1999] qui sur le même alliage ont effectué des analyses de composition par
EDX. Pour autant, pour un taux de quasicristaux supérieur à 50%, la cinétique de cristallisation décroît et
laisse à penser selon Lee et al. [Lee2000] qu’à ce stade, la cristallisation se fait avec changement de
composition.
Kühn et al. [Küh2000] ont étudié l’influence de la vitesse de trempe sur la formation des quasi-
cristaux dans l’alliage Zr57Ti8Nb2.5Cu13.9Ni11.1Al7.5. Pour ce faire, ils ont préparé deux types : un ruban mince
de 40 µm obtenu par trempe sur roue d’une part et un échantillon cylindrique de 3 mm de diamètre d’autre
part. Les caractérisations par diffraction des rayons X et MET montrent que dans le premier cas, l’échantillon
est amorphe alors que dans le deuxième il est cristallisé sous forme quasicristalline, la taille des
quasicristaux étant de 1 µm. Ils ont montré que le pic exothermique supplémentaire observé dans les
expériences de DSC sur l’échantillon sous forme de ruban correspondait à la formation de la phase
quasicristalline dont la taille des cristaux est comprise entre 5 et 10 nm. Selon ces auteurs, la différence de
taille s’expliquerait par la différence de vitesse de diffusion qui est fonction de la concentration en défauts.
En effet, dans le cas d’un matériau obtenu par trempe sur roue, le système est figé à plus haute température
(vitesses de trempe plus élevées). Le système contient alors, à température ambiante la concentration de
défauts de l’état haute-température. Pour des trempes plus lentes cette concentration est moins élevée. Des
analyses quantitatives par EDX montrent que dans les deux cas les quasi-cristaux ont une composition
différente, et pouvant varier à l’intérieur même des quasi-cristaux.
Köster et al. [Kös1997] dans leur étude sur l’alliage Zr69.5Cu12Ni11Al7.5 ont simulé, en considérant une
nucléation hétérogène, la répartition des tailles des quasi-cristaux. La comparaison des résultats
expérimentaux avec la simulation leur permet de conclure que la cristallisation des quasi-cristaux se fait
selon une transformation primaire, c’est-à-dire par diffusion de certains éléments.
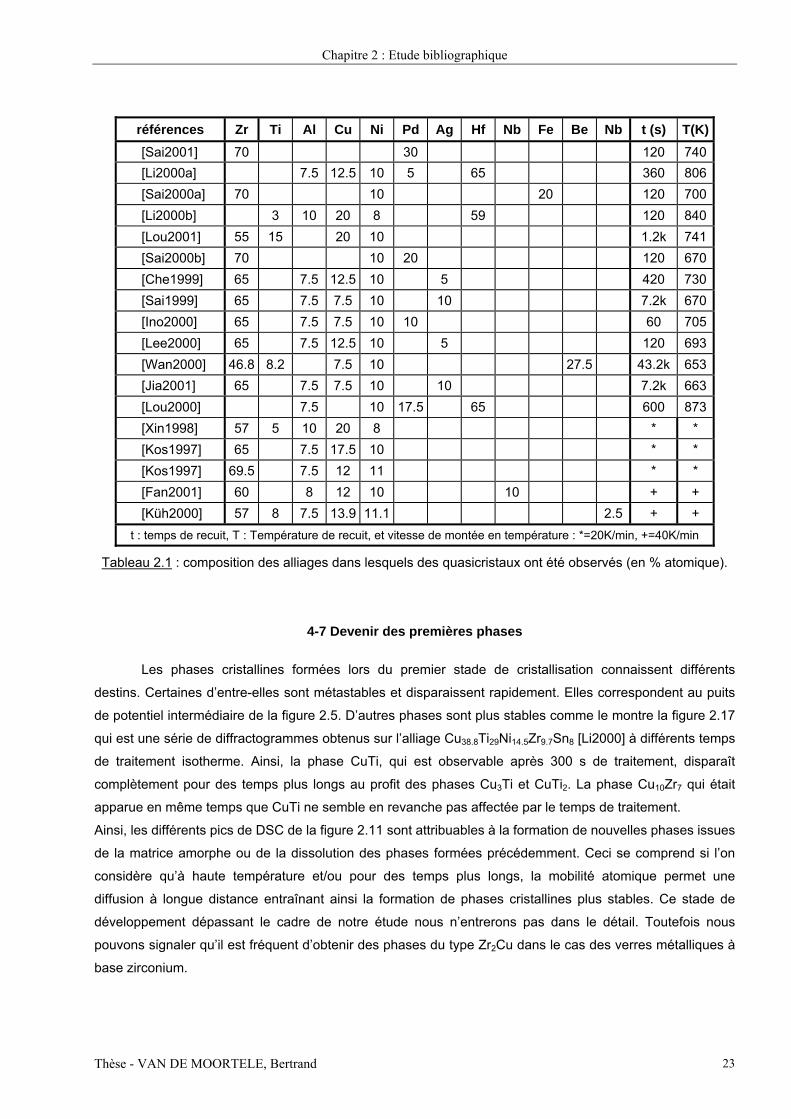
Le tableau 2.1 reprend quelques-unes des compositions des verres métalliques dans lesquels une
quasicristallisation a été observée. Nous avons également indiqué quels étaient les traitements thermiques
effectués pour obtenir ces phases. Dans la majorité des cas, ce sont des traitements à température
constante qui ont été utilisés, ce qui semble être un aspect pratique lié à un meilleur contrôle.
Il est intéressant de remarquer que l’ensemble des alliages du tableau 2.1 ont une proportion de métaux de
transition (Zr, Ti, Hf et Nb) proche de 65-70% alors qu’elle n’est que de 55% dans le Vit4. Nous verrons dans
le chapitre 5 que cette exception n’est qu’apparente et en particulier nous montrerons le rôle majeur que
joue le béryllium dans ce cas.
Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 23
références Zr Ti Al Cu Ni Pd Ag Hf Nb Fe Be Nb t (s) T(K)[Sai2001] 70 30 120 740[Li2000a] 7.5 12.5 10 5 65 360 806[Sai2000a] 70 10 20 120 700[Li2000b] 3 10 20 8 59 120 840[Lou2001] 55 15 20 10 1.2k 741[Sai2000b] 70 10 20 120 670[Che1999] 65 7.5 12.5 10 5 420 730[Sai1999] 65 7.5 7.5 10 10 7.2k 670[Ino2000] 65 7.5 7.5 10 10 60 705[Lee2000] 65 7.5 12.5 10 5 120 693[Wan2000] 46.8 8.2 7.5 10 27.5 43.2k 653[Jia2001] 65 7.5 7.5 10 10 7.2k 663[Lou2000] 7.5 10 17.5 65 600 873[Xin1998] 57 5 10 20 8 * * [Kos1997] 65 7.5 17.5 10 * * [Kos1997] 69.5 7.5 12 11 * * [Fan2001] 60 8 12 10 10 + + [Küh2000] 57 8 7.5 13.9 11.1 2.5 + +
t : temps de recuit, T : Température de recuit, et vitesse de montée en température : *=20K/min, +=40K/min
Tableau 2.1 : composition des alliages dans lesquels des quasicristaux ont été observés (en % atomique).
4-7 Devenir des premières phases
Les phases cristallines formées lors du premier stade de cristallisation connaissent différents
destins. Certaines d’entre-elles sont métastables et disparaissent rapidement. Elles correspondent au puits
de potentiel intermédiaire de la figure 2.5. D’autres phases sont plus stables comme le montre la figure 2.17
qui est une série de diffractogrammes obtenus sur l’alliage Cu38.8Ti29Ni14.5Zr9.7Sn8 [Li2000] à différents temps
de traitement isotherme. Ainsi, la phase CuTi, qui est observable après 300 s de traitement, disparaît
complètement pour des temps plus longs au profit des phases Cu3Ti et CuTi2. La phase Cu10Zr7 qui était
apparue en même temps que CuTi ne semble en revanche pas affectée par le temps de traitement.
Ainsi, les différents pics de DSC de la figure 2.11 sont attribuables à la formation de nouvelles phases issues
de la matrice amorphe ou de la dissolution des phases formées précédemment. Ceci se comprend si l’on
considère qu’à haute température et/ou pour des temps plus longs, la mobilité atomique permet une
diffusion à longue distance entraînant ainsi la formation de phases cristallines plus stables. Ce stade de
développement dépassant le cadre de notre étude nous n’entrerons pas dans le détail. Toutefois nous
pouvons signaler qu’il est fréquent d’obtenir des phases du type Zr2Cu dans le cas des verres métalliques à
base zirconium.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 24
5-Effet de l’oxygène
Bien que les élaborateurs choisissent des matériaux de départ les plus purs possibles, en général
purs à 99.999 %, il est impossible d’obtenir des alliages exempts d’oxygène, et en particulier dans le cas des
alliages à base de zirconium. Dans les deux exemples qui suivent, nous allons voir les conséquences du
point de vue de la facilité de la formation de verres métalliques (GFA : glass forming ability), de la stabilité
des verres métalliques en température ainsi que sur la nature des phases cristallines formées.
Gebert et al. [Geb1998] ont étudié les effets de l’oxygène lors de l’élaboration de l’alliage
Zr65Al7.5Cu17.5Ni10. Ils ont montré que jusqu’à un taux d’oxygène de 0.28%, les échantillons étaient
pleinement amorphes (diffractogramme (a) de la fig.2.18). En revanche, un taux de 0.35% d’oxygène dans
l’alliage ne permettait plus d’obtenir un amorphe comme le montre la présence des pics du diffractogramme
(b).Ces pics sont ceux de la phase métastable Zr2Ni, seules les zones refroidies plus lentement présentent
cette cristallisation. De plus, il apparaît en DSC un second pic de cristallisation à plus basse température
réduisant ainsi la région de liquide surfondu. Ce pic est lié à la formation de la phase Zr2Ni dans les zones
qui n’ont pas cristallisé.
Outre les problèmes d’élaboration que pose la présence d’oxygène, Murty et al. [Mur2000a,
Mur2000b] ont montré que dans l’alliage Zr65Cu27.5Al7.5 la présence d’oxygène modifiait également la
Figure 2.17 : évolution des phases cristallographiques lorsd’un traitement thermique isotherme à 771K.
cristallisation de la phase CuTi
disparition de la phase CuTi

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 25
séquence de cristallisation. Pour des taux d’oxygène de 0.43 % et 0.82 %, ils ont observé lors d’un maintien
à une température de 400 °C la formation d’une phase cristalline métastable se transformant ensuite en
Zr2(Cu, Al) alors que l’échantillon ne contenant que 0.14 % d’O2 cristallise directement sous cette dernière
phase. La figure 2.19 correspond au thermogramme obtenu en DSC pour les trois différents taux
d’oxygène. On voit clairement que l’étendue de la zone de liquide surfondu est diminuée par l'apparition d'un
pic de cristallisation supplémentaire lié à la formation de la phase métastable montrant ainsi les effets de
l’oxygène. Des analyses de composition indiquent que la phase qui se forme à plus basse température est
enrichie en oxygène et appauvrie en aluminium.
6-Aptitude à la déformation des verres métalliques
Lors de la fusion d’un matériau cristallin, la viscosité η subit une chute de plus d’une dizaine d’ordres
de grandeur sur un intervalle de température de quelques degrés. Dans les amorphes, le passage à l’état
liquide n’est pas marqué par une telle variation brutale. Celle-ci est bien plus progressive et l’on observe une
inflexion de la courbe η = f(T) à une valeur autour de 1012 Pa.s-1 pour la température dite de ‘transition
vitreuse’ Tg. Les verriers ont, depuis longtemps, mis à profit cette variation progressive de la viscosité pour la
mise en œuvre de leurs produits. Ils ont remarqué que selon la composition des verres, la variation de la
viscosité au voisinage de Tg est plus ou moins rapide. Ils ont ainsi défini les verres ‘longs’ pour lesquels le
temps disponible pour travailler un objet sortant du four est ‘long’, en raison d’une variation assez lente de la
viscosité lors du refroidissement; à l’inverse, ils parlent de verres ‘courts’. Angel et al. [Ang1988] ont quantifié
(a)
(b)
Figure 2.18 : diffractogramme sur deux échantillonsbruts de Zr65Al7.5Cu17.5Ni10 contenant deux tauxd’oxygène différents.
Figure 2.19 : thermogramme des DSC montrant les effets de la présence de l’oxygène dans l’alliage Zr65Cu27.5Al7.5.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 26
cette notion de ’longueur’ du verre en introduisant un paramètre m appelé fragilité et définit de la façon
suivante :
Un verre long, comme la silice pure, présente une valeur de m voisine de 150 alors que pour un
verre organique comme le o-terphenyl, m a une valeur très faible (2-3). Comme on peut le voir sur la figure
2.20, les verres métalliques sont des matériaux amorphes intermédiaires ; leur fragilité est voisine de celle
de nombreux verres d’oxydes et de polymères amorphes.
La chute de viscosité autour de la température de transition vitreuse suscite toujours de nombreuses
recherches. De nombreux modèles ont été proposés pour comprendre le mode de déformation des
matériaux amorphes dans cette zone de température. Les concepts ont été développés bien souvent pour
des verres de silice ou de polymères mais sont applicables de manière générale à tous les matériaux
amorphes.
La partie bibliographique qui suit doit beaucoup aux articles de Gutzow et al.[Gut1993] et Rekhson
[Rek1991] qui ont accompli un remarquable travail de synthèse sur le sujet et dont nous nous sommes
largement inspirés.
Figure 2.20 : Fragilité de différents matériaux amorphes.
⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜
⎝
⎛
⎟⎠⎞
⎜⎝⎛∂
∂
=
=
TTg
T
TgT
m)(logη
(2.2bis)

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 27
6.1 Description
La figure 2.21 présente des courbes de déformation par compression (obtenue par Inoue [Ino1999])
sur l’alliage Zr65Al10Ni10Cu15 autour de la température de transition vitreuse (653 K à 10 K.min-1) pour une
vitesse de déformation fixe de 5.10-4 s-1. Les courbes de plasticité obtenues présentent de fortes similitudes
avec celles observées sur des verres de polymères ou de silice (figures 2.23 et 2.24). Il est possible de
manière un peu schématique de séparer en plusieurs parties ces courbes de déformations (fig.2.23):
- un stade de déformation élastique (stade 1),
- un stade de déformation plastique et anélastique (stade 2),
- un stade d’écoulement visco-plastique avec un léger adoucissement (stade 3),
- enfin, dans le cas de certains polymères une consolidation peut être observée (stade 4). Elle serait
liée à un alignement des chaînes polymères dans la direction de la contrainte augmentant ainsi la
résistance à la déformation [Qui1995].
Figure 2.21 : courbes de contrainte-déformation dans l’alliage Zr65Al10Ni10Cu15 pour une vitesse dedéformation constante et différentes températures, à gauche, et une température constante etdifférentes vitesses de déformation, à droite.
Figure 2.24 : courbes de contrainte-déformation dans un verre de silice pour différentes vitesses de déformation d’après [Bru1985].
Figure 2.23 : courbe de contrainte déformation dans le
PMMA.
1 2 3 4

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand
E0
elσ
=εE
0el
σ=ε&
&
De façon générale, la réponse d‘un matériau à une contrainte de traction ou de compression
uniaxiale σ0 fait intervenir :
La réponse élastique , d’où
La réponse visqueuse
où ε désigne la déformation, E le module de Young, η0 la viscosité Newtonienne,
dérivées de ε et σ par rapport au temps. D’où :
soit encore :
où E3 0
0η
τ = désigne un temps de relaxation.
Si l’on suppose que τ0 et E sont constants, alors la solution s’écrit très simplement :
Cette équation prévoit une relation linéaire entre les logarithmes de la contrain
déformation. La figure 2.25 est un graphe en échelle log-log de la contrainte en fonc
déformation obtenue dans l’alliage La55Al25Ni20 par Kawamura et al. [Kaw2001]. Comm
(2.5)
(2.3) et (2.4)
.
Figure 2.25 : graphe de la contrainte en fonctionde la vitesse de déformation montrant lecomportement Newtonien à bas niveau decontrainte.
0
0vis 3η
σε =&
.
0
00visel
3E ησ
+σ
=ε+ε=ε&
&&&
ε=τσ
+σ && E0
00
)1exp(ετE(t) t00 τ−×=σ &
ε et σ
te et d
tion d
e le p
28
désignent les
e la vitesse de
e la vitesse de
révoit l’équation
(2.8)
(2.6)
(2.7)

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 29
2.3, pour les faibles contraintes nous observons une droite dont le coefficient directeur est un paramètre
fréquemment utilisé pour caractériser les amorphes métalliques [Ohk1999, Nie2001] : il correspond à la
sensibilité du matériau à la vitesse de déformation.
6.2 Approches phénoménologiques de la déformation
Pour des vitesses de déformation élevées, on observe un écart au comportement purement
Newtonien prédit précédemment. L’équation 2.6 donnant la relation entre la contrainte, la vitesse de
déformation et la viscosité n’est plus valable.
Comme on le voit sur la figure 2.26, la viscosité d’un verre métallique, ici l’alliage Pd40Cu30Ni10P20,
dépend fortement de la vitesse de déformation expliquant ainsi que l’on n’a plus un comportement
Newtonien à forte vitesse de déformation. Cette évolution avait déjà été observée dans les verres de silice.
Aussi Middelman [Mid1962] a introduit la notion de viscosité apparente qui permet de rendre compte de
l’effet de la vitesse de déformation sur la viscosité. On remplace alors dans l’équation 2.5, η0 par la valeur de
la viscosité dite viscosité apparente, ηapp, ce qui donne :
Dans certains cas la dépendance de la viscosité avec la température est décrite par l’intermédiaire
du modèle du volume libre. Kawamura et al. [Kaw2000] utilisent l’équation de Vogel-Fulcher-Tamman (VFT),
basée sur cette notion de volume libre, pour décrire l’évolution de la viscosité en fonction de la température
de l’alliage Pd40Ni40P20.
Dans ce cas, la viscosité en fonction de la température est donnée par :
où D* est le paramètre de fragilité du verre considéré, T0 la température de VFT généralement inférieure à
Tg. Une des limites de cette approche, outre les problèmes intrinsèques à la théorie du volume libre, est son
app
0
3vis
ησ
=ε&
⎟⎟⎠
⎞⎜⎜⎝
⎛−×
×η=ηTTTDexp
0
00equ
*(2.10)
(2.9)
Figure2.26 : dépendance de la viscosité avec la vitesse de déformation et de la contrainte (d’apès Kato [Kat1998]).

Chapitre 2 : Etude bibliographique
Thèse - VAN
incapacité à décrire correctement l’évolution de la viscosité sur une large gamme de température, avec un
seul jeu de paramètres.
Une autre approche pour intégrer l'effet de la vitesse de déformation sur la viscosité, a été
développée par Montrose et al. [Mon1981]. Ils ont proposé une description phénoménologique de la
viscosité qui est la suivante :
où η0 est la viscosité mesurée à une vitesse de déformation très faible, η la viscosité mesurée pour une
vitesse de ε
Re
compte de
comportem
contrainte
vitesses de
déformation
lié à des
déformation
contrainte
d'introduire
courbes ob
silico-sodo-
lim
0
0
0
0
11σηε
+
η=
ατε
+
η=η
&&(2.11)
.
DE MOORTELE, Bertrand 30
, σlim la contrainte limite à la rupture.
khson [Rek1991] a introduit dans cette expression une contrainte seuil permettant de rendre
la transition Newtonien - non Newtonien de la déformation décrivant ainsi correctement le
ent de la viscosité en fonction de la contrainte. Il est ensuite nécessaire d'introduire la notion de
fictive σf pour que ce modèle décrive correctement les pics de contrainte, pour des faibles
déformation, que l'on peut observer dans les courbes de contrainte-déformation à vitesse de
constante. La contrainte fictive tente de rendre compte de la modification de l'état du matériau,
relaxations structurales du fait de l'application d'une contrainte. Le matériau ayant subi une
voit donc une contrainte fictive qui n'est pas la contrainte appliquée mais correspond à une
qui lui serait appliquée s’il n’y avait pas eu de relaxation due à la déformation. Il s'agit ensuite
un jeu de paramètres que l'on ajuste sur des courbes expérimentales. La figure 2.27 montre des
tenues après ajustement des paramètres, comparées aux courbes expérimentales dans un verre
calcique.
Figure 2.27 : courbes de contrainte-déformation expérimentales etsimulées par Rekhson et al.

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 31
Prandtl [Pra1928] puis Eyring [Eyr1936] ont introduit, de manière schématique, l’idée que les atomes
du matériau se situent dans des puits de potentiel symétriques. Pour passer d’un puits à un autre, il est
nécessaire de franchir une barrière de potentiel qui correspond à une énergie d’activation. L’application
d’une contrainte a pour conséquence de modifier la forme des puits de potentiel : dans le sens de la
contrainte, la barrière de potentiel est abaissée alors qu’elle est augmentée dans le sens opposé. La figure
2.28, d’après Gutzow illustre bien ce propos. La mise en équation aboutit alors pour une contrainte en
cisaillement à la formule suivante :
où A et a sont des constantes reliées respectivement au coefficient de diffusion et à un volume d’activation.
Pour les faibles contraintes, on retrouve un comportement à la déformation purement Newtonien
puisque la fonction sinus hyperbolique peut se développer au premier ordre.
Un raffinement supplémentaire de ce modèle proposé par Gutzow [Gut1993] consiste à introduire un
terme rendant compte du désordre et des effets d’interactions lors du franchissement de la barrière
d’énergie. Ceci permet d’aboutir à la relation suivante :
où Γ0 est un paramètre dépendant de la température, de la taille des molécules (dans notre cas, il
correspondrait à des unités structurales de plusieurs atomes), du taux d’enchevêtrement et du temps
caractéristique de saut d’une molécule. Ce modèle a été appliqué avec beaucoup de succès sur des
polymères.
)asinh(A0
σ×η
=γ& (2.12)
(2.13)
Figure 2.28 : schématisation de la modification du puits de potentiel par une contrainte.
)exp()asinh(A0
0
γΓ−σ×η
=γ &&

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand
Un modèle similaire, mais plus spécifiquement développé pour étudier la déformation des verres
métalliques par Spaepen [Spa1977] et Argon [Arg1979] permet d’aboutir à la relation suivante :
où ε0 est la déformation locale induite par la déformation d’un site de volume Ωf, kB la constante de
Boltzmann et T la température.
Ce modèle est actuellement le plus utilisé pour étudier la déformation des verres métalliques. On
peut citer entre autre les travaux récents de Heilmaier [Hei2001] sur la déformation des verres à base
zirconium, de Reger-Lehonard et al. [Reg2000] sur la déformation de l’alliage Zr55Cu30Al10Ni15, dont la figure
2.29 est extraite. Comme on le voit, les paramètres d’ajustement utilisés dans ce modèle permettent de bien
rendre compte de certains faits expérimentaux, en particulier de la courbe log σ en fonction de log
domaines Newtonien et non Newtonien.
Pour autant, les limites de ces différents modèles sont nombreuses. En particulier, aucu
modèles ne permet de décrire le pic de contrainte aux basses températures ou aux vitesses de dé
élevées. Pour l’anecdote, on peut citer Inoue [Ino1999] qui fait référence à un article sur la déform
polymères [Max1979] pour justifier l’existence d’un pic de contrainte tout en restant très évasif su
physique de celui-ci. De plus ces modèles reposent sur une approche qui ne tente pas d’ide
mécanismes physiques à l’origine de la déformation. Ainsi, aucune relation n’est établie entre les p
des différents modèles et la microstructure du matériau.
.
Tksinh
B
f00
Ωσε×ε=ε && (2.14)
Figure 2.29 : courbes de contrainte-déformation expérimentales et simulées de Reger-Lehonard et al.
32
ε dans les
n de ces
formation
ation des
r l’origine
ntifier les
aramètres

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 33
6.3 Modèle des défauts quasi-ponctuels
Aussi nous nous sommes intéressés à une autre approche de la déformation des verres métalliques
proposée par le modèle de Perez et al. [Per1988]. Ce modèle a été développé pour expliquer la déformation
dans les polymères [Cha2002] mais a été appliqué également aux verres minéraux [Abb1987]. Il repose sur
plusieurs hypothèses que nous allons passer en revue.
Dans ce modèle, un amorphe est considéré comme un assemblage désordonné d’unités
structurales dont la cohésion est assurée par des liaisons intra et inter-moléculaire. Dans le cas des verres
métalliques les liaisons sont de nature métallique alors qu’elles sont covalentes dans les verres d’oxydes ou
de nature covalente et Van der Waals dans les polymères (respectivement liaisons intra et inter-
moléculaires). Les unités structurales, qui possèdent un excès d’enthalpie et d’entropie par rapport à la
valeur moyenne de l’ensemble des unités structurales, sont appelées défauts quasi ponctuels (dqp). Au-
dessus de Tg, le système étant à l’équilibre métastable, la concentration de défauts Cd peut être évaluée à
partir d’un raisonnement thermodynamique [Per1988] :
où ∆HF et ∆SF sont respectivement l’enthalpie et l’entropie de formation d’un défaut quasi ponctuel (dqp).
En dessous de Tg, la concentration en défauts est proche de celle que l’on aurait à Tg, elle diminue
lentement par relaxation structurale.
Sous l’application d’une contrainte, des microdomaines cisaillés (mdc) sont nucléés autour des
défauts quasi ponctuels. La croissance des mdc, cf. figure 2.30, est à l’origine de la déformation anélastique.
⎟⎟⎠
⎞⎜⎜⎝
⎛ ∆⎟⎟⎠
⎞⎜⎜⎝
⎛ ∆−+
=
kTH
expkS
exp1
1CdfF (2.15)
Figure 2.30 : Schéma de croissance des « mdc » sous l’effet d’une contrainte.
cisaillement

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand
La croissance des mdc s’effectue selon des mouvements hiérarchiquement corrélés, dépendant de
la concentration de désordre. Plus l’arrangement des unités structurales est désordonné, plus le temps
moyen (τmol) de déplacement sur une distance égale à leur taille est court. Ce temps caractéristique de
mobilité moléculaire s’exprime par la relation suivante :
où τβ est le temps caractéristique associé au m
variant entre 10-12 et 10-7 s suivant les maté
dépendant de la concentration de défauts de la
Dans le domaine linéaire, aux tempéra
(Tg). A des températures supérieures (T > Tg
premier ordre conduit à :
Pour des temps d’application de la cont
dislocations bordant ces mdc s’annihilent e
déformation plastique irréversible (si le matér
déformation est proportionnel à τmol (environ 10
Dans le cas de l’application d’une con
simplement thermiquement activés mais d
caractéristiques diminuent. Ceci conduit à l’expr
où σ0 est la contrainte nécessaire pour franc
élémentaires à la température de 0 K.
τ=τβ 0
)T(χ
(2.17)χ
β
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛ τ=τ1
00mol tt (2.16) ⎟
⎟⎞
⎜⎜⎛
ββ ×τ=τ Uexp0β
T( gχ=χ
avec τ
34
ouvement moléculaire élémentaire, t0 est un facteur d’échelle
riaux. χ est un paramètre d’ordre compris entre 0 et 1,
manière suivante :
tures inférieures à Tg, Cd est constant et donc χ(T) vaut χ
), Cd est fonction de la température. Un développement au
rainte plus longs, il y a coalescence des mdc, les boucles de
t l’énergie élastique emmagasinée s’annule, créant une
iau reste sous Tg). Le temps caractéristique τpl de cette
00 fois plus grand que celui-ci).
trainte élevée, les mouvements élémentaires ne sont plus
eviennent thermomécaniquement activés : les temps
ession suivante :
hir la barrière énergétique Uβ associée aux mouvements
⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜
⎝
⎛⎟⎠⎞
⎜⎝⎛
σσ
−β
β
kT
1Uexp
23
0
(2.18)
)TT(a)T( gg −+χ≅ (2.19)
⎟⎠
⎜⎝ kT
))T(CdCd() g−α+
(2.20)

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 35
De plus, la concentration de défauts (ou le paramètre de corrélation χ) évolue avec la déformation.
La déformation anélastique est responsable de la création de défauts alors que la déformation
viscoplastique engendre leur disparition, ce qui s’écrit de la manière suivante :
vpvpanan AA)T(),T( ε−ε+χ=εχ
où Aan est un paramètre qui rend compte de l’amplitude de l’augmentation du désordre créé par la
composante anélastique, Avp est dans le cas des polymères relié à l’effet du durcissement observé après le
plateau [Cha2002]. Dans le cas des verres métalliques ce durcissement n’a pour l’instant pas pu être mis
clairement en évidence.
Une déformation importante conduit à augmenter la concentration en défauts modifiant ainsi le
paramètre d’ordre χ. Ceci avait déjà été évoqué par Struik [Str1978] pour qui la déformation induisait un
rajeunissement du matériau, c’est-à-dire un procédé inverse au vieillissement physique. On peut d’ailleurs
évoquer à ce sujet un travail récent de Saida et al. [Sai2002] sur la formation d’une phase cristalline dans
l’alliage Zr65Al7.5Ni10Cu12.5Pd5 lors d’un traitement thermique qui disparaît sous l’effet d’une déformation ce
qui semble correspondre à l’augmentation du désordre.
A la suite de cela, il est possible de calculer la courbe de contrainte-déformation par méthode
d’incrémentation. La figure 2.31 est une comparaison entre les données expérimentales obtenues dans un
polymère et la courbe calculée par cette méthode [Cha2002].
En guise de conclusion pour ce chapitre nous pouvons souligner quelques interrogations que suscite
la lecture des travaux qui se font sur les verres métalliques et auxquelles nous tacherons de répondre dans
la suite de ce mémoire :
L’observation par différents auteurs d’une cristallisation primaire de l’ordre de 1 à 2 % indique qu’avant
toute étude portant sur l’évolution des verres métalliques avec la température, il est nécessaire de
0
20
40
60
0 0.1 0.2 0.3ε
σ (
MP
a)
288 K
313 K
295 K
Figure 2.31 : courbes de contrainte-déformationexpérimentales et simulées, d’après [Cha2002].
(2.21)

Chapitre 2 : Etude bibliographique
Thèse - VAN DE MOORTELE, Bertrand 36
caractériser l’état amorphe et en particulier de vérifier ‘l’amorphicité’ des alliages métalliques. Nous
pouvons effectivement nous questionner sur les conséquences de cette cristallisation primaire, dans les
échantillons bruts de trempe, tant sur les propriétés mécaniques que sur la cristallisation ultérieure.
Du point de vue de l’analyse du comportement mécanique de l’état amorphe, nous avons vu que peu de
modèles permettent de faire une description du comportement mécanique sur une large gamme de
température autour de la température de transition vitreuse. De plus, dans bien des cas, aucune relation
n’est faite entre les paramètres des modèles et la microstructure du matériau. Aussi, nous nous
proposons de voir si l’approche basée sur le modèle de Perez et al., permettra de décrire le
comportement mécanique des verres métalliques sur une large gamme de température mais également
de montrer que les concepts développés à l’origine pour des polymères amorphes, et par la suite pour
d’autre matériaux amorphes, peuvent être étendus aux cas des amorphes métalliques.
En ce qui concerne l’évolution microstructurale, il semble que les nombreuses questions qui se posent
sur le mode de décomposition et de cristallisation des verres métalliques pourraient être en grande
partie résolues en utilisant une technique d’analyse adaptée. Par exemple, dans le cas de l’alliage Vit1,
où une décomposition spinodale a été observée à une température de 368 °C, il serait utile d’obtenir des
informations directes sur l’évolution chimique de la matrice amorphe. De la même manière, le débat qui
existe sur la formation des quasicristaux, transformation polymorphique ou non, pourrait être facilement
tranché en ayant l’information sur la chimie des quasicristaux. Aussi, la microscopie électronique en
transmission nous est apparue comme l’outil le mieux adapté pour cela. D’une part, cette technique
permet de faire des observations à une échelle nanométrique et d’autre part, il est possible d‘effectuer
des analyses chimiques locales sur des domaines de taille là aussi nanométrique. Nous verrons que
cette approche nous permettra d’apporter de nouvelles informations sur l’évolution avec la température
des différents alliages que nous étudierons.
Mais avant d’exposer nos résultats expérimentaux, nous allons présenter les différentes techniques
expérimentales qui nous ont permis de mener cette étude ainsi que les matériaux qui ont fait l’objet de notre
étude.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 37
Chapitre 3
Méthodes Expérimentales
Chapitre 3 ____________________________________________________________________37
Méthodes Expérimentales________________________________________________________38 1 Les Matériaux _____________________________________________________________________38
1.1 Elaboration ___________________________________________________________________________ 38 1.2 Compositions __________________________________________________________________________ 39
2 Caractérisation microstructurale _____________________________________________________40 2.1 Diffraction des rayons X_________________________________________________________________ 40 2.2 Microscopie Electronique en Transmission _________________________________________________ 41
2.2.1 Appareillage ________________________________________________________________________ 41 2.2.2 Rappels sur la Microscopie Electronique en Transmission ____________________________________ 41 2.2.3 Techniques d’analyses ________________________________________________________________ 43 2.2.4 Microscopie Electronique en Transmission Filtrée en Energie _________________________________ 48 2.2.5 Préparation des échantillons pour la Microscopie Electronique en Transmission ___________________ 50
3 Mesures Physiques _________________________________________________________________55 3.1 Pouvoir Thermoélectrique (PTE) _________________________________________________________ 55 3.2 Calorimétrie différentielle à balayage (DSC pour Differential Scanning calorimetry) ______________ 56
4 Caractérisation Mécanique __________________________________________________________56 4.1 Spectrométrie mécanique ________________________________________________________________ 56 4.2 Compression __________________________________________________________________________ 59

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 38
Méthodes Expérimentales
Dans ce chapitre, nous allons passer en revue les différentes techniques expérimentales dont nous
avons eu besoin au cours de ce travail. Dans un premier temps, nous présenterons la technique
d’élaboration des verres métalliques ainsi que les différentes compositions que nous avons étudiées. Nous
présenterons également les techniques qui nous ont permis de mettre en évidence l’évolution
microstructurale (diffraction X, Microscopie Electronique en Transmission) observée par ailleurs par des
techniques de caractérisation macroscopique telles que le Pouvoir ThemoElectrique (PTE), la calorimétrie
différentielle (DSC) et les essais mécaniques.
1 Les Matériaux
Comme nous allons le voir, nous avons disposé de plusieurs alliages de compositions différentes
pour effectuer cette étude. Dans un premier temps, nous décrirons le mode d’élaboration le plus utilisé
actuellement pour obtenir des verres métalliques massifs. Puis nous passerons en revue les différents
matériaux que nous avons pu étudier en donnant leurs compositions et leurs provenances.
1.1 Elaboration
Il existe de nombreuses méthodes d’élaboration des verres métalliques. On peut citer la trempe sur
roue qui a permis d’obtenir les premiers verres métalliques dans les années 60, la mécanosynthèse qui part
d’un alliage sous forme de poudre et qui par l’introduction d’un très grand nombre de défauts par broyage
permet d’aboutir à un alliage amorphe sous forme de poudre. Actuellement la technique d’élaboration la plus
utilisée pour obtenir des verres métalliques massifs est la trempe sous vide en moule refroidi.
A titre d’exemple, la technique utilisée par le CRETA (qui nous a fourni de nombreux échantillons à
base de zirconium) est la suivante : les matériaux de départ sont des composants purs à 99.99 % sous
forme de lingot. Ils sont mélangés et fondus à une température de 200 °C au-dessus de la température de
fusion dans un creuset inductif. Ils sont maintenus sous lévitation électromagnétique avec un flux d’argon
pur évitant ainsi au maximum l’oxydation du mélange. Une bonne homogénéisation est assurée grâce au
brassage électromagnétique. Après quelques minutes l’induction magnétique est coupée. Il est alors
possible d’obtenir deux types de verres métalliques. Pour les verres métalliques massifs, le mélange fondu
tombe dans un moule cylindrique refroidi à l’eau. La figure 3.2 est une photographie d’échantillons de
différentes formes et tailles qu’il est possible d’obtenir par cette technique. Les vitesses de refroidissement
que l’on peut obtenir par cette technique sont de l’ordre de 103 °C.s-1.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 39
Dans le cas des amorphes sous forme de ruban, le mélange liquide est éjecté par pression sur une
roue en cuivre non refroidie, pouvant tourner à des vitesses variables (permettant d’obtenir des vitesses de
refroidissement variables également).
1.2 Compositions
Les alliages à base palladium permettent d’obtenir des échantillons de taille record. Ainsi l’alliage
Pd43Cu27Ni10P20 détient le record avec une taille de cylindre de 79 mm de diamètre [Ino993]. Le laboratoire
DLR de Cologne en Allemagne nous a fourni un échantillon de la même composition. Il se présentait sous la
forme d’un cylindre de 9 mm de diamètre et de 60 mm de haut. Nous n’avons disposé que d’un seul cylindre
et n’avons pas pu mener d’études par toutes les techniques de caractérisation que nous avions à
disposition.
Pour autant ces alliages restent des 'curiosités de laboratoire' au regard du coût de fabrication et ne
permettent pas d’entrevoir d’applications de ces matériaux. Aussi des alliages à partir de composants de
base moins chers ont été élaborés. Les alliages à base zirconium ont connu un fort développement à partir
de la fin des années 1980. Elles sont un compromis entre des coûts relativement abordables et la possibilité
d’avoir des échantillons massifs.
Le premier alliage à base zirconium que nous avons étudié nous a été fourni par Jean-Louis Soubeyroux du
CRETA de Grenoble. La composition est la suivante : Zr48.5Ti5.5Al11Cu12.5Ni13. Cet échantillon se présente
sous la forme d’un cylindre de 6 mm de diamètre et de 70 à 80 mm de hauteur.
Le second est un alliage commercialisé sous le nom de Vitrelloy 1 par la société Howmet Corporation de
composition suivante : Zr41.2Ti13.8Cu12.5Ni10Be22.5 La forme de l’échantillon est une plaque de 400 mm de
longueur, 300 mm de large et 3.3 mm de hauteur. Une grande partie de ce mémoire sera basée sur les
résultats obtenus sur cette composition.
Enfin pour les alliages à base zirconium, nous avons étudié l’évolution de l’alliage Zr46.75Ti8.25Cu7.5Ni10Be27.5
également appelé Vit4. Cet échantillon nous a également été fourni par Jean-Louis Soubeyroux. Il se
présente sous la forme d’un cylindre de 9 mm de diamètre et 70 mm de hauteur.
Figure 3.1 : exemples de verres métalliques élaborés par le CRETA de Grenoble.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 40
Pour terminer, le laboratoire de Risöe au Danemark nous a fourni un alliage à base de magnésium de
composition suivante : Mg65Cu25Y10. Ces matériaux ont été développés en raison de leur faible poids. Les
études menées sur cette composition ont porté sur la caractérisation mécanique par spectrométrie, aucune
étude sur la microstructure n’a été faite.
Le tableau 3.1 résume les compositions des alliages étudiés et donne également la dénomination
que nous utiliserons dans la suite de ce manuscrit.
Zr Ti Be Cu Ni Al Pd P Mg Y Provenance Dénomination
41.2 13.8 22.5 12.5 10 Howmet, USA Vit1
46.75 8.25 27.5 7.5 10 CRETA Grenoble Vit4
48.5 5.5 22 13 11 CRETA Grenoble ZrAl
30 10 40 20 Cologne, Allemagne Pd
25 65 10 Risö, Danemark Mg
Tableau 3.1 : compositions et provenances des verres métalliques étudiés.
2 Caractérisation microstructurale
Dans un premier temps nous décrirons assez brièvement la technique de diffraction des rayons X.
Puis après une description des deux microscopes électroniques en transmission dont nous nous sommes
principalement servis, nous ferons quelques rappels sur les principes de base de la microscopie
électronique en transmission et les différentes techniques d’analyses qui lui sont associées. Ensuite nous
aborderons la technique particulière de la microscopie à imagerie filtrée. Enfin, nous évoquerons les
problèmes rencontrés lors de la préparation des échantillons pour la microscopie.
2.1 Diffraction des rayons X
Dans le cadre des études sur les verres métalliques, la diffraction des rayons X est fréquemment
utilisée pour, dans un premier temps, vérifier l’aspect amorphe des échantillons bruts. Les diffractogrammes
obtenus sont constitués de deux bosses amorphes centrées sur des distances qui correspondent aux
premières et secondes sphères de coordination. Lorsque l’échantillon a subi un traitement thermique, il est
possible d’identifier les phases cristallines qui se sont formées, si celles-ci sont connues, et de faire une
estimation de leur taille. Pour autant, dans notre cas, de nombreuses phases cristallines qui se forment
durant les traitements thermiques ne sont pas connues, cette technique s’avère dans ce cas insuffisante.
L’appareil utilisé lors de ce travail est un diffractomètre RIGAKU-DMAXB. Il est doté d’un tube à
anticathode en cuivre (longueur d’onde d’émission λ=0.15406 nm) et d’un monochromateur arrière.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 41
2.2 Microscopie Electronique en Transmission
2.2.1 Appareillage
Durant ce travail, nous avons principalement travaillé sur deux microscopes électroniques. Le
premier est un microscope JEOL 200CX de 200kV dédié à la microscopie dite conventionnelle. La
microscopie à haute-résolution a été réalisée sur un microscope à émission de champ JEOL 2010F1 ayant
lui aussi une tension d’accélération de 200kV. Le tableau 3.2 résume les différentes caractéristiques
techniques de ces deux microscopes ainsi que les moyens d’analyses dont ils sont équipés.
Microscope JEOL 200CX JEOL 2010F résolution ponctuelle 0.4 nm 0.19 nm
information limite 0.4 nm ~0.1 nm
taille minimale de sonde 50 nm (TEM) 0.4 nm
type d’analyse chimique EDX Tracor (fenêtre Be) EDX Oxford (fenêtre mince)
PEELS Gatan - 766
Tableau 3.2 : données techniques sur les microscopes électroniques utilisés au cours de ce travail.
2.2.2 Rappels sur la Microscopie Electronique en Transmission
Microscopie Conventionnelle
La microscopie électronique conventionnelle est basée sur l’interaction entre les électrons du
faisceau incident et l’échantillon. Une partie des électrons est diffusée élastiquement formant ainsi une série
d’ondelettes. Dans des directions particulières, liées au plan atomique concerné, ces ondelettes interférent
entre-elles pour donner naissance au phénomène de diffraction. Ainsi en orientant correctement le cristal
sous le faisceau, il est possible d’obtenir un cliché de diffraction composé d’un faisceau transmis et de
faisceaux diffractés.
Un diaphragme placé au niveau de la lentille objectif permet alors de sélectionner une partie des
faisceaux transmis ou diffractés. Ainsi il sera possible, en centrant le diaphragme sur le faisceau transmis,
d’observer un échantillon avec des électrons n’ayant pas interagi avec l’échantillon, c’est ce que l’on appelle
la technique du champ clair. Cette technique permet de mettre en évidence la présence de défauts cristallins
comme les dislocations mais permet surtout de faire apparaître un contraste. A l’opposé, si l’on utilise un
faisceau diffracté pour former une image, on utilise la technique du champ sombre. Dans le cas d’un
échantillon présentant une fine cristallisation non-orientée, les clichés de diffractions sont constitués de
cercles concentriques assez fins et parfois ponctués. La sélection par le diaphragme d’objectif d’une partie
de ces cercles permet alors de faire des images de cette fine cristallisation.
1 le CLYME, Consortium LYonnais de Microscopie Electronique est remercié pour l'accés à ces différents microscopes.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 42
Microscopie en Haute-Résolution
Le principe de la haute-résolution est d’amener l’ensemble des ondes diffractées à interférer entre-
elles. On obtient alors une figure d’interférences permettant de 'visualiser' les colonnes atomiques des plans
responsables de la diffraction. La résolution maximale que l’on puisse espérer atteindre avec le microscope
2010F est de 0.12 nm. L’obtention d’images de qualité dépend des réglages de la défocalisation ainsi que
de l’astigmatisme.
Figure 3.2 : schéma illustrant le principe de l’imagerie conventionnelle enmicroscopie électronique en transmission : respectivement champ clair, champsombre.
Figure 3.3 : schéma de principe de l’imagerie haute-résolution.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 43
2.2.3 Techniques d’analyses
Les techniques d’analyses chimiques associées à la microscopie électronique en transmission
bénéficient des faibles tailles de sonde qu’il est possible d’obtenir dans cette technique. En effet, la précision
de la détermination de la chimie locale dépend fortement de la taille de sonde qu’il est possible d’obtenir.
Pour comprendre les différentes techniques utilisées il faut rappeler les interactions qui existent entre les
atomes du matériau étudié et les électrons du faisceau, ce qui est résumé sur la figure 3.4.
Spectroscopie par dispersion en énergie des rayons X : EDX
Cette technique développée par Castaing est basée sur l’analyse de l’énergie des rayons émis lors
de la désexcitation d’un atome d’un niveau extérieur vers un niveau profond. Le photon est alors analysé et
comptabilisé en fonction de son énergie. Cette énergie est caractéristique d’une transition électronique c’est-
à-dire d’une espèce chimique. La figure 3.5 montre un exemple de spectre obtenu dans le cas de l’alliage
ZrAl. Les logiciels informatiques qui gèrent l’acquisition permettent alors une très bonne déconvolution des
différentes raies observées liées à la présence de plusieurs éléments. La qualité de la quantification qui en
découle dépend fortement de différents paramètres liés à l’acquisition (temps d’acquisition, temps mort)
mais également de la calibration de la chaîne de mesure. Dans des conditions classiques la résolution est
de 1 à 2 % mais il est possible dans de bonnes conditions d’obtenir des quantifications avec une incertitude
de 0.5 %.
Pour autant cette technique ne permet pas d’obtenir des informations pour des éléments légers tel
que le béryllium. Il est alors nécessaire d’utiliser la spectroscopie de perte d’énergie des électrons (EELS).
Figure 3.4 : différents types d’interaction électrons-échantillon.
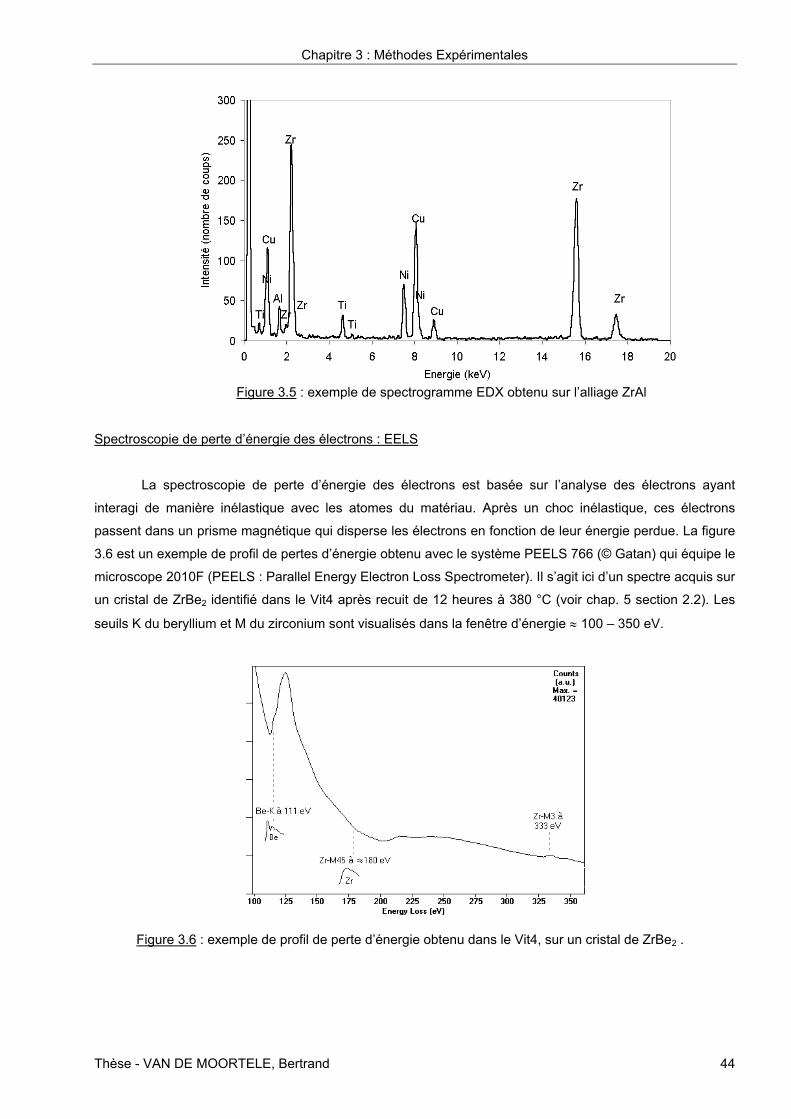
Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 44
Spectroscopie de perte d’énergie des électrons : EELS
La spectroscopie de perte d’énergie des électrons est basée sur l’analyse des électrons ayant
interagi de manière inélastique avec les atomes du matériau. Après un choc inélastique, ces électrons
passent dans un prisme magnétique qui disperse les électrons en fonction de leur énergie perdue. La figure
3.6 est un exemple de profil de pertes d’énergie obtenu avec le système PEELS 766 (© Gatan) qui équipe le
microscope 2010F (PEELS : Parallel Energy Electron Loss Spectrometer). Il s’agit ici d’un spectre acquis sur
un cristal de ZrBe2 identifié dans le Vit4 après recuit de 12 heures à 380 °C (voir chap. 5 section 2.2). Les
seuils K du beryllium et M du zirconium sont visualisés dans la fenêtre d’énergie ≈ 100 – 350 eV.
Figure 3.5 : exemple de spectrogramme EDX obtenu sur l’alliage ZrAl
Figure 3.6 : exemple de profil de perte d’énergie obtenu dans le Vit4, sur un cristal de ZrBe2 .

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 45
Nous avons utilisé la spectroscopie EELS essentiellement pour doser le béryllium, ce qui n’est pas
possible avec l’EDX. Bien entendu, compte-tenu de la résolution énergétique de la technique (proche de 1
eV dans notre cas), il est également possible d’étudier la structure fine des seuils d’ionisation des différents
éléments, et de remonter ainsi aux types de liaison chimique (technique ELNES : Electron Loss Near Edge
Spectroscopy). Dans notre étude, les liaisons chimiques seront peu modifiées lors de la cristallisation,
puisqu’elles restent de type métallique ; nous n’avons pas approfondi de telles études ELNES.
D’une manière générale, le dosage élementaire ‘classique’ en EELS repose sur les différentes étapes
suivantes [Ege1996] :
- correction du spectre brut des réponses des diodes du détecteur,
- déconvolution des pertes proches (diffusion multiple),
- soustraction du fond continu, habituellement à l’aide d’une loi puissance aE-r,
- mesure des aires sous les seuils d’ionisation dans une fenêtre d’énergie de largeur ∆E, et dosage
quantitatif du rapport atomique NA/NB (dans le cas de 2 espèces A et B) à l’aide de la relation :
où SA et SB sont les aires sous les seuils A et B dans la fenêtre ∆E, σA(∆E, βeff) et σB(∆E, βeff) sont les sections efficaces de diffusion inélastiques des 2 seuils intégrées sur la fenêtre ∆E et pour l’angle de collection effectif βeff (dépendant à la fois de l’angle de convergence du faisceau d’électron et de l’angle de collection des électrons inélastiques).
Dans le cas des échantillons de Vit1 et Vit4, notre intérêt s’est porté exclusivement sur les seuils Be-
K et Zr-M, qui présentent l’avantage d’être proches l’un de l’autre, et d’être situés dans une gamme de
relativement faibles pertes d’énergie, où le signal est conséquent. L’analyse chimique ‘complète’ des
matériaux de notre étude sera ainsi effectuée en utilisant les données complémentaires de l’EELS (rapport
Be/Zr) et de l’EDX (rapports atomiques entre les différents éléments métalliques Zr, Ti, Ni et Cu).
Cependant, deux difficultés majeures sont apparues, qui justifient ici de décrire la méthodolgie adoptée pour
déterminer le rapport Be/Zr.
D’une part, la loi-puissance classique pour soustraire le fond continu ‘sous’ les seuils ne nous est
pas apparue adaptée, du fait de la proximité de ces seuils (notamment, seuil K du Be à 110 eV) vis-à-vis des
pics de plasmons à faible perte, vers 20 eV (et 40 eV pour l’harmonique).
Concernant ce dernier point, nous avons donc préféré modéliser le fond par une loi dite de Drude, qui décrit
correctement la décroissance du spectre derrière un pic de plasmon [Ege1996] :
où K est une constante de normalisation liée à l’énergie du maximum du pic de plasmon Ep et à sa largeur à mi-hauteur ∆Ep (K = Ep ∆Ep).
),E(),E(
*SS
NN
eff
eff
B
A
B
A
A
B
β∆σβ∆σ
=
22p
22p
2 E)E()EE(KE)E(g
∆+−=
(3.1)
(3.2)

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 46
La figure 3.7 illustre les soustractions de fond obtenues par la loi puissance - fig. 3.7 a) et b) -et la loi
de Drude- fig. 3.7 c) et d) - ; on constate que, sur la région en énergie servant à extrapoler le fond sous les
seuils Be et Zr (zone bleutée en b) et d)), les 2 approches donnent des résultats satisfaisants, mais que la loi
de Drude reproduit raisonnablement la forme du second plasmon vers 40 eV. L’importance de la méthode
de soustraction du fond sur le résultat final de la quantification est clairement illustré par sa hauteur
différente sous le seuil M du zirconium dans les 2 cas a) et c).
D’autre part, les sections efficaces fournies par le logiciel d’exploitation des spectres EELS (EL/P, ©
Gatan) ne sont pas fiables pour le couple Be-Zr. La section efficace du seuil K du béryllium est en fait
correctement donnée par le modèle hydrogénoïde, ce qui n’est pas le cas pour la raie M de l’espèce ‘lourde’
Zr. Même si le logiciel fournit une section efficace paramétrée à partir de calculs atomistiques de type
Hartree-Slater pour ce dernier seuil, il n’a pas été possible d’obtenir un rapport satisfaisant Be/Zr = 2 pour le
spectre de la figure 3.6 qui correspond pourtant à la phase ZrBe2 (indexée de manière cristallographique - cf.
Chap. V, section3.2). D’ailleurs, la même difficulté est apparue dans le traitement de spectres ‘témoins’
réalisés sur de la zircone pure ZrO2.
Figure 3.7 : soustraction du fond continu sous les seuils Be-K et Zr-M (cristal ZrBe2, VIT4) effectuée avecun programme ‘maison’ de traitement EELS (Pc-EELS, T. Epicier, 2000 - non publié -). a-b) : loi puissance A E-r dans 2 régions d’énergie (i.e. au proche voisinage des seuils et dans la régiondes plasmons respectivement). c-d) : idem a-b) avec une loi de Drude.
a) b) c) d)

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 47
Nous avons donc eu recours à une méthode qui consiste à ajuster les spectres expérimentaux
(après soustraction du fond) à l’aide de 2 spectres ‘de référence’ normalisés pour chacun des seuils Be-K et
Zr-M.
Pour le seuil Be-K, nous avons utilisé le spectre du béryllium pur donné dans l’Atlas Gatan
[Ahn1983]. Comme le montre la figure 3.8, ce seuil présente nénamoins des différences notables, du point
de vue des structures fines, avec le seuil expérimental obtenu dans le ZrBe2 (et d’ailleurs, dans la matrice
Vit4 – cf. chap. 5, section 2.2) .
Figure 3.8 : détail du seuil Be-K (après soustraction du fond continu). a) : comparaison du seuil expérimental dans ZrBe2 (en bas - moyenne de 10 spectres -) avec le seuil du béryllium pur (ATALS Gatan, en haut). b) : seuil de référence construit en extrapolant, aux pertes d’énergie élevées, le seuil expérimental à l’aide du seuil de référence ‘Gatan’.
Pour le seuil Zr-M, nous avons directement utilisé un spectre de référence obtenu sur de la zircone
cubique, les structures fines étant sensiblement identiques pour les 2 matériaux (ZrBe2 et ZrO2, cf. figure
3.9) dans nos conditions expérimentales de résolution en énergie.
Figure 3.9 : détail du seuil Zr-M : en haut, spectre expérimental dans ZrBe2 et soustraction grossière du fond continu pour extraire le seuil Zr-M – en bas - ; au milieu (en grisé) : seuil Zr-M dans la zircone.
Nos spectres de référence Be-Kref et Zr-Mref (fig. 3.8 b) et 3.9 respectivement) ont enfin été
normalisés en intensité, de manière à ce que l’ajustement aux moindres carrés des spectres expérimentaux
du ZrBe2 conduisent à une pondération 2 x Be-Kref et 1 x Zr-Mref (figure 3.10).
a) b)

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 48
Figure 3.10 : ajustement aux moindres carrés des seuils Be-K et Zr-M dans ZrBe2 (après soustraction du fond continu - moyenne de 10 spectres expérimentaux -) avec les références normalisées Be-Kref et Zr-Mref (routine de calcul du logiciel Pc-EELS) : noter le résultat 0.665 Be 0.335 Zr, qui correspond à la stœchiométrie ZrBe1.99.
Dans l’ensemble de notre étude, nous avons donc systématiquement quantifié le rapport atomique
Be/Zr avec cette méthode d’ajustement à l’aide de nos spectres de référence Be-Kref et Zr-Mref. Nous verrons
que la validité de cette approche est renforcée par les résultats très cohérents obtenus sur l’analyse des
matrices brutes dans le Vit1 et le Vit4, puisque des compositions très proches des valeurs nominales ont été
trouvées dans les 2 cas.
2.2.4 Microscopie Electronique en Transmission Filtrée en Energie
Le principe de la microscopie à imagerie filtrée (EFTEM : Energy Filtered Transmission Electron
Microscopy [Rei1995]) utilise un des résultats de la spectroscopie de pertes d’énergie des électrons. En
effet, l’idée est de sélectionner, à l’aide d’une fente physique, les électrons ayant perdu une énergie donnée
pour obtenir une image ou une diffraction. Ainsi en se plaçant au seuil d’un élément, il sera possible
d’obtenir un contraste lié aux fluctuations de sa concentration.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 49
Nous avons utilisé 2 microscopes à filtrage d’énergie dans le cadre de cette étude. Le premier est le
LEO-912 du CLYME installé au laboratoire LTDS de l’Ecole Centrale de Lyon. Cet appareil, d’une tension
d’accélération de 120kV, est un microscope classique avec un filtre Ω inséré dans la colonne (filtre ‘in-
column’) comme illustré sur la figure 3.11. Ce filtre consiste en une série de lentilles, permettant de dévier
sélectivement, de manière réglable et avec une sélectivité également réglable (ce que l’on appellera par la
suite la taille de la fenêtre) les électrons ayant perdu une énergie donnée. Dans cette configuration, il est
alors possible d’observer directement sur l’écran du microscope les images se rapportant à une fenêtre de
perte en énergie, l’acquisition d’image se faisant alors de manière classique sur une caméra numérique de
type Slow-Scan CCD.
Le second est le microscope JEOL 3010, doté d’un filament LaB6, du laboratoire DRFMC/SP2M du
CEA à Grenoble1. Cet appareil, d’une tension d’accélération de 300kV, est équipé pour l’étude des pertes
d’énergie d’un filtre ‘post-column’ GIF (Gatan Imaging Filter). Ce système, outre une résolution en énergie
sur l’image meilleure que le LEO-912 (autour d’1 eV contre 1.5 à 2 eV) a l’avantage d’avoir une tension
d’accélération élevée ce qui, pour des échantillons contenant des atomes relativement lourds, s’avérera
utile.
Dans les 2 cas, nous avons utilisé la technique dite des 3 fenêtres. Le principe est d’acquérir trois
images à trois valeurs de pertes d’énergie, dont deux avant le seuil et une au maximum du seuil. Les deux
1 Pascale Bayle-Guillemaud (CEA Grenoble) est remerciée pour sa collaboration dans la conduite des observations sur ce microscope.
objet
colonne
prisme dispersif
fente de sélection ∆E
image (ou diffraction) filtrée Figure 3.11 : principe de la microscopie à imagerie filtrée en énergie. Ici exemple d'un microscope équipé d'un filtre en Ω in-column.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 50
premières images permettent de calculer la décroissance du fond continu. La troisième, une fois corrigée du
fond, nous donne l’image de la répartition de l’élément responsable du seuil.
Cette méthode est illustrée par la figure 3.12 dans le cas du LEO-912, où des séries d’images filtrées ont été
enregistrées afin de reconstruire l’ensemble du spectre de pertes d’énergie dans la région d’intérêt liée au
seuil.
Figure 3.12: méthode d’imagerie filtrée ‘des 3 fenêtres’ appliqué au seuil Ti-L dans un échantillon VIT1 (logiciel ↓EasyVision © S.I.S. du microscope LEO-912). Le fond continu, déduit d’une loi ajustable pour obtenir le meilleur accord, est approché à l’aide du signal dans les fenêtres W2 et W1 avant le seuil, et l’imagerie se fait à l’aide du signal corrigé du fond dans la fenêtre ‘MAX’. Le spectre expérimental est constitué d’une série de 12 images enregistrées à l’aide d’une fenêtre de largeur environ 3 eV, ce qui explique une ‘résolution’ en énergie médiocre par rapport au spectre de l’Atlas, enregistré lui avec une fenêtre de 1 eV.
2.2.5 Préparation des échantillons pour la Microscopie Electronique en Transmission
Les porte-objets des microscopes électroniques sont conçus pour des objets n’excédant pas 3mm
de diamètre. Dans le cas de l’alliage Vit1 qui se présente sous la forme d’une plaque de dimension
400*300*3.3 mm3, une première découpe par électroérosion était nécessaire. Ensuite, à la scie à fil
diamanté, nous avons découpé des échantillons dont la plus grande dimension ne dépassait pas 3 mm.
Nous avons alors effectué un polissage mécanique pour obtenir une épaisseur comprise entre 80 et 150 µm.
En effet, à l’état brut les amorphes métalliques ne sont pas fragiles et ne craignent pas ce genre d’opération.
spectre expérimental
référence de l’ATLAS LEO
fond continu

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 51
Toutefois, nous avons remarqué que si l’épaisseur à l’issue de cette opération était trop faible, l’échantillon
se déformait.
A ce stade nous procédons à un meulage concave qui nous permet d’obtenir un échantillon dont
l’épaisseur e au centre est comprise entre 5 et 30 µm (cf. figure 3.13). A la suite de cette étape nous
effectuons les traitements thermiques afin de caractériser l’évolution de la microstructure.
Il est apparu nécessaire d’effectuer les traitements thermiques à ce niveau de préparation car les
échantillons recuits deviennent extrêmement fragiles du fait de la nano-cristallisation qui s’opère, et
l’amincissement mécanique d’échantillons massifs devient quasiment impossible (microfissuration).
Il nous a semblé par ailleurs intéressant d’essayer de procéder à des chauffages in situ des lames minces,
pour suivre en temps réel les évolutions microstructurales.
La figure 3.14 est une comparaison de la microstructure de deux échantillons de ZrAl, traités thermiquement
à 459 °C pendant 100 min.
Le premier a été chauffé dans le microscope, le second a été recuit avant amincissement ionique. Il est clair
que les microstructures obtenues n’ont rien à voir entre-elles : dans le cas du second échantillon, on observe
des cristaux d’une taille moyenne de 10 nm (qui fragilisent notablement la lame mince). La diffraction du type
e
Figure 3.13 : stade d’amincissement auquel se feront les traitements thermiques.
100 nm
Figure 3.14 : micrographies d’un échantillon recuit 100 min à 459 °C à gauche, in-situ (champsombre) ; à droite, au stade d’amincissement décrit précédemment.

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 52
Debye-Scherrer (figure 3.15) s’indexe dans la structure de Zr2Ni comme l’avait déjà montré Fan et al. dans
un alliage Zr53Ti5Al12Cu20Ni10 [Fan1999] ainsi que Mattern et al. dans l’alliage Zr54.5Ti7.5Al10Cu20Ni8 [Mat2002].
Dans le cas du premier échantillon, on constate une précipitation plus grossière dont l’analyse en
diffraction électronique et spectroscopie EDX montre qu’il s’agit de 2 populations de précipités : d’une part,
du cuivre métallique (figure 3.16) et d’autre part, de la zircone (figure 3.17). Cette expérience de chauffage
in-situ dans le microscope sur les alliages ZrAl montre à quel point les lames minces sont sensibles à
l’oxydation.
L’origine de l’oxygène menant à cette forme de cristallisation peut être double : soit l’oxygène est
présent en surface de l’échantillon Dans le cas de ces lames minces cette oxydation de surface peut être
suffisamment importante, relativement au volume de l’échantillon, pour aboutir à la précipitation du zirconium
sous forme de zircone, soit l’oxygène provient de la colonne du microscope dans lequel l’expérience de
chauffe in-situ a été réalisée. En effet, selon Külik [Kül2002], il faudrait atteindre des vides de 10-76 atm pour
éviter l’oxydation d’éléments aussi avides d’oxygène que le zirconium ou le titane, ce qui n’est évidemment
pas possible avec une technologie de pompage en vide secondaire.
Pour nous assurer de la possibilité d’un phénomène d’oxydation lié à l’oxygène de surface ou
extérieur à l’échantillon, nous avons déterminé l’ordre de grandeur du parcours moyen de l’oxygène dans
notre matériau. En nous basant sur les travaux de C. Vauglin, la distance de diffusion ((Dt)1/2, avec D
coefficent de diffusion de l’oxygène dans un alliage de zirconium [Vau1984], et t temps du traitement) à
400°C vaut près de 10 nm, ce qui indique qu’une lame mince de 20 nm peut être entièrement oxydée à
cœur du simple fait d’une oxydation de surface !
(211)(220)
(310)
(411)
(420)
(332)
Figure 3.15 : Diffraction en aire sélectionnée du type Debye-Scherrer obtenue sur unéchantillon de ZrAl, après un traitement de 100 min à 459 °C sur une lame pré-amincie. Lesanneaux visibles concordent avec les distances les plus intenses de la phase Zr2Ni (grouped’espace I4/mcm, a = 0.6486 nm, c = 0.5279 nm, fiche JPCDS 38-1170)
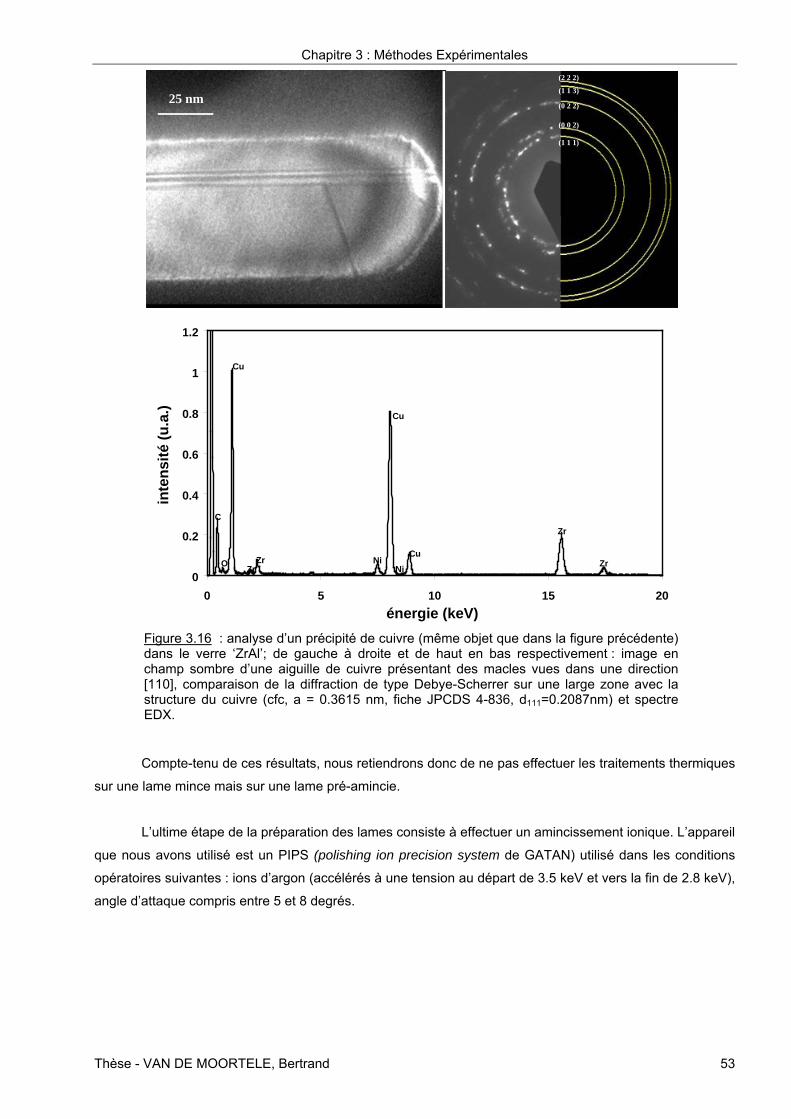
Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 53
Compte-tenu de ces résultats, nous retiendrons donc de ne pas effectuer les traitements thermiques
sur une lame mince mais sur une lame pré-amincie.
L’ultime étape de la préparation des lames consiste à effectuer un amincissement ionique. L’appareil
que nous avons utilisé est un PIPS (polishing ion precision system de GATAN) utilisé dans les conditions
opératoires suivantes : ions d’argon (accélérés à une tension au départ de 3.5 keV et vers la fin de 2.8 keV),
angle d’attaque compris entre 5 et 8 degrés.
25 nm
(1 1 1)
(0 0 2)
(0 2 2)
(1 1 3)
(2 2 2)
0
0.2
0.4
0.6
0.8
1
1.2
0 5 10 15 20énergie (keV)
inte
nsité
(u.a
.) Cu
Zr
Cu
Cu
ZrZr ZrNi
Ni O
C
Figure 3.16 : analyse d’un précipité de cuivre (même objet que dans la figure précédente)dans le verre ‘ZrAl’; de gauche à droite et de haut en bas respectivement : image enchamp sombre d’une aiguille de cuivre présentant des macles vues dans une direction[110], comparaison de la diffraction de type Debye-Scherrer sur une large zone avec lastructure du cuivre (cfc, a = 0.3615 nm, fiche JPCDS 4-836, d111=0.2087nm) et spectreEDX.
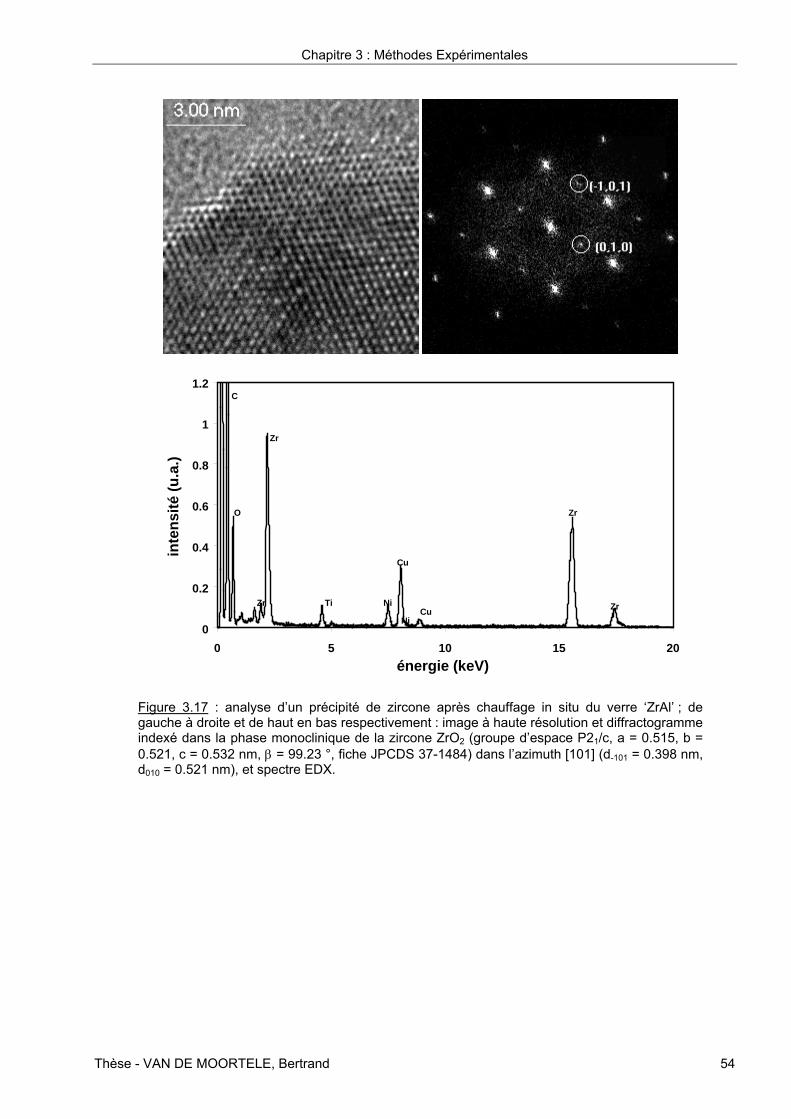
Chapitre 3 : Méthodes Expérimentales
Thèse - VA
Figgauind0.5d01
0
0.2
0.4
0.6
0.8
1
1.2
0 5 10 15 20énergie (keV)
inte
nsité
(u.a
.)
Zr
Cu
TiCu
Zr
Zr
ZrNi
Ni
O
C
ure 3.17 : analyse d’un précipité de zircone après chauffage in situ du verre ‘ZrAl’ ; deche à droite et de haut en bas respectivement : image à haute résolution et diffractogramme
exé dans la phase monoclinique de la zircone ZrO2 (groupe d’espace P21/c, a = 0.515, b =21, c = 0.532 nm, β = 99.23 °, fiche JPCDS 37-1484) dans l’azimuth [101] (d-101 = 0.398 nm,0 = 0.521 nm), et spectre EDX.
N DE MOORTELE, Bertrand 54

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 55
(3.3)
(3.4)
TVS
∆∆
=∆
3 Mesures Physiques
3.1 Pouvoir Thermoélectrique (PTE)
Pour mesurer le pouvoir thermoélectrique on réalise deux jonctions entre l'échantillon et un métal de
référence comme l'indique la figure 3.18. L'une des jonctions est maintenue à une température T, l'autre à
une température légèrement plus élevée T + ∆T. Une différence de potentiel d'origine thermoélectrique ∆V
est ainsi créée par effet Seebeck et peut être mesurée entre les deux extrémités libres du métal de
référence maintenues à une même température T0.
Le pouvoir thermoélectrique ∆S de l'échantillon (PTE) par rapport au métal de référence est donné
par la relation suivante : exprimé en µV/K
Si on appelle SE et SR les pouvoirs thermoélectriques absolus de l'échantillon et du métal de référence :
∆S = SE - SR
Le pouvoir thermoélectrique absolu S varie en fonction de la température et de manière très
différente suivant la nature du matériau considéré. Il est donc nécessaire de définir avec précision les
températures T et T + ∆T choisies. En toute rigueur, la relation (3.3) n'est exacte que pour ∆T tendant vers
zéro; mais elle reste valable pour de faibles différences de température ∆T car on peut considérer alors que
∆V est proportionnel à ∆T (dans la mesure ou SE et SR ne présentent pas de discontinuités dans le domaine
de température <T, T + ∆T>. La température de mesure de ∆S est la moyenne : TM = T + ∆T / 2.
Les valeurs de S peuvent être positives ou négatives et se situent dans l'intervalle <-40 µV/K, +20 µV/K>
pour des métaux ou des alliages métalliques.
L’appareil utilisé a été entièrement conçu au laboratoire.
Figure 3.18 : Schéma de principe de mesure du PTE par effet Seebeck
Différence de potentiel ∆V
Jonction à T Jonction à T + ∆T
Echantillon
Métal de référence To To

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 56
(3.5)
(3.6)
3.2 Calorimétrie différentielle à balayage (DSC pour Differential Scanning Calorimetry)
La calorimétrie différentielle à balayage (DSC) est une technique qui permet de détecter les effets
thermiques liés à des transformations ou évolutions microstructurales. Le principe de la mesure consiste à
déterminer la variation de l’enthalpie lorsque le matériau est soumis à une rampe de montée en température
(isochrone) ou lors d’un recuit à une température donnée (isotherme). Lors d’une expérience de calorimétrie,
le thermogramme obtenu correspond à la dérivée de l’enthalpie par rapport à la température c’est-à-dire à la
chaleur spécifique en fonction de la température.
Dans le cas des matériaux amorphes, il est possible de mettre en évidence par cette technique le
phénomène de transition vitreuse qui s’accompagne d’une variation de la chaleur spécifique, suivie dans
notre cas d’une cristallisation à plus haute température.
Afin d’assurer un bon contact thermique et une température homogène, les échantillons utilisés pour
cette technique expérimentale sont de faible épaisseur. L’appareil utilisé est une DSC commercialisée par
Perkin Elmer sous le nom de DSC7. Les vitesses de montée en température que nous avons utilisées sont
comprises entre 1 et 100 °C.min-1. Toutefois les mesures faites pour des vitesses supérieures à 30 °C.min-1
indiquent plus une tendance qu’un résultat fiable. Le poids des échantillons est compris entre 15 et 40 mg,
au-delà apparaît une saturation de l’appareil.
L'ensemble des traitements thermiques des échantillons de microscopie ont été effectués dans ce
même appareil afin d'assurer un maximum de cohérence entre les observations faites en DSC et en
microscopie.
4 Caractérisation Mécanique
4.1 Spectrométrie mécanique
La spectrométrie mécanique permet l'étude des propriétés viscoélastiques d'un matériau par la
mesure de son module dynamique en fonction de la température ou en fonction de la fréquence de
sollicitation en isotherme. L'échantillon est sollicité par l'application d'une contrainte σ* (ou d'une déformation
ε*) sinusoïdale.
)tiexp(σ0* ω=σ ou )tiexp(0
* ωε=ε
Il en résulte, en régime linéaire, une déformation ε* (ou une contrainte σ*) également sinusoïdale et
déphasée d'un angle φ :
)tiexp(0* φ−ωε=ε ou )tiexp(0
* φ+ωσ=σ

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 57
(3.7)
(3.8)
De manière à rester dans le domaine linéaire, les sollicitations sont de très faible amplitude (entre
10-4 et 10-6) et dans notre cas, il s’agit de contrainte de cisaillement. Le module de cisaillement dynamique
complexe G* s'exprime par le rapport σ*/ε* :
G*=G'+iG"
Ainsi on peut écrire :
φε
σ= cos'G
0
0 et φ
ε
σ= sin''G
0
0
Tan(φ), qui est le rapport de G’’ sur G’, est aussi appelée coefficient de frottement intérieur et représente la
proportion d'énergie dissipée sous forme de chaleur dans un échantillon par rapport à l'énergie élastique
mise en jeu au cours du cycle. Les variations du module complexe G* et de tan(φ), en fonction de la
température ou de la fréquence, correspondent aux différentes relaxations pouvant se produire dans les
matériaux. Dans le cas des polymères, elles sont associées aux divers degrés de liberté des chaînes
moléculaires.
Nous avons utilisé le spectromètre mis au point au laboratoire G.E.M.P.P.M. de l'INSA de Lyon
[Cav1988]. Il s'agit d'un pendule de torsion inversé fonctionnant en régime harmonique forcé, à des
fréquences comprises entre 10-5 et 5 Hz, et sur une gamme de température comprise entre l’ambiante et
900 °C. Il se compose d'une partie mécanique et d'une partie électronique.
Vide
7
10
54
4
3
1
2
5
6
12
8
9
13
14
11
Figure 3.12 : schéma de principe du pendule de torsion.
1 : poulie mécanique et contre-poids 2 : fil 3 : amortisseur 4 : bobines de Helmholtz 5 : aimants 6 : miroir 7 : tige de forte rigidité 8 : four 9 : mors mobile 10 : échantillon 11 : mors fixe 12 : source lumineuse 13 : cellule photovoltaïque différentielle14 : amplificateur

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 58
(3.9)
(3.10)
(3.11)
La partie mécanique est représentée sur la figure 3.12. Le couple de torsion est créé par l’interaction
entre l'aimant et les bobines d'Helmholtz parcourues par un courant sinusoïdal. Il est transmis par
l'intermédiaire d'une tige verticale de forte rigidité à l'extrémité supérieure de l'échantillon. La rotation
angulaire θ de très faible amplitude (≅ 1 °) est mesurée par un système optique. L'échantillon est placé dans
un four thermorégulé. L’ensemble de l’appareil est sous vide de façon à limiter les perturbations (frottement
de l’air, effets de convection dans le four) et les phénomènes d’oxydation auxquels les alliages à base
zirconium sont très sensibles.
La partie électronique interfacée à un ordinateur permet le pilotage de l'expérience. Il est possible de
travailler en contrainte imposée ou en déformation imposée ; dans ce dernier cas, l’électronique asservit la
déformation. Toutes nos mesures ont été réalisées en déformations imposées. L’appareil utilisé donne
également la possibilité de faire des mesures à plusieurs fréquences lors d’une même rampe de montée en
température.
Les échantillons testés sont des parallélépipèdes rectangles. Leurs dimensions sont les suivantes :
30 mm < L < 40 mm
3 mm < l < 4 mm
0.8 mm < e < 1 mm
où e, l, L sont respectivement l’épaisseur, la largeur, et la longueur de l’échantillon.
Le module de cisaillement dynamique G* est calculé de la manière suivante :
*
**
f1G
ΘΓ
=
Γ* est le couple de torsion, Θ* est la rotation angulaire de l’échantillon, f un facteur de forme qui s’écrit pour
des échantillons de forme parallélépipédique :
fe lL
=⋅ ⋅β 3
β varie en fonction du paramètre e/l.
Si l’on compare l’erreur dG faite avec un pendule de torsion et celle dE faite avec un appareil de
traction, on arrive aux résultats suivants :
dGG
dee
dll
dLL
= + +3 et dEE
dee
dll
dLL
= + +
Ainsi, l’erreur faite sur l’épaisseur des échantillons (inévitable avec des échantillons de 1 mm d’épaisseur
tels que les nôtres) a des conséquences trois fois plus importantes sur la valeur du module mesurée en
cisaillement que sur celle mesurée en traction. Aussi nous avons principalement travaillé en mesures

Chapitre 3 : Méthodes Expérimentales
Thèse - VAN DE MOORTELE, Bertrand 59
relatives. Pour cela, les modules mesurés ont été normalisés par le module non relaxé c’est-à-dire le module
mesuré à 20 °C.
4.2 Compression
Seules des expériences en compression sur le Vit1 ont été effectuées. Pour cela, nous avons fait
réaliser par électro-érosion des échantillons cylindriques de 3 mm de diamètre et de 5 mm de haut. Les
essais de compression ont été réalisés au laboratoire GPM2 de Grenoble sur une installation classique
d’essais mécaniques à chaud. Les tests de déformation ont été faits à l’air, l’oxydation, quand elle se produit,
étant un phénomène de surface qui ne modifie pas les propriétés mécaniques. Les vitesses de déformation
utilisées sont comprises entre 10-4 et 10-2 s-1. Lors des expériences isochrones, la vitesse de montée en
température est de 10 °C.min-1, comparable à celles utilisées lors des expériences de DSC.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 60
Chapitre 4
Etude de l’état amorphe
Chapitre 4 ____________________________________________________________________60
Etude de l’état amorphe _________________________________________________________60 1 Caractérisation Microstructurale __________________________________________________61
1.1 Diffraction des rayons X_________________________________________________________________ 61 1.2 Etude et Caractérisation par Microscopie Electronique en Transmission (MET)__________________ 62
1.2.1 ZrAl ______________________________________________________________________________ 62 1.2.2 Vit1 ______________________________________________________________________________ 66 1.2.3 Vit4 ______________________________________________________________________________ 69 1.2.4 Conséquences sur les propriétés mécaniques _________________________________________ 70 1.2.5 Matrice Amorphe __________________________________________________________________ 73
2 Caractérisation Physique par DSC et PTE __________________________________________73 2.1 DSC__________________________________________________________________________________ 73 2.2 PTE__________________________________________________________________________________ 75
3 Réponse Mécanique ______________________________________________________________77 3.1 Elasticité et viscoélasticité : spectrométrie mécanique_________________________________________ 77
3.1.1 Courbes typiques __________________________________________________________________ 77 3.1.2 Effet de la vitesse de montée en température __________________________________________ 79 3.1.3 Effet de la fréquence de sollicitation __________________________________________________ 80 3.1.4 Rôle du vieillissement physique ______________________________________________________ 83
3.2 Viscoplasticité : essais de compression à chaud ______________________________________________ 84 3.3 Analyse de la réponse mécanique _________________________________________________________ 87
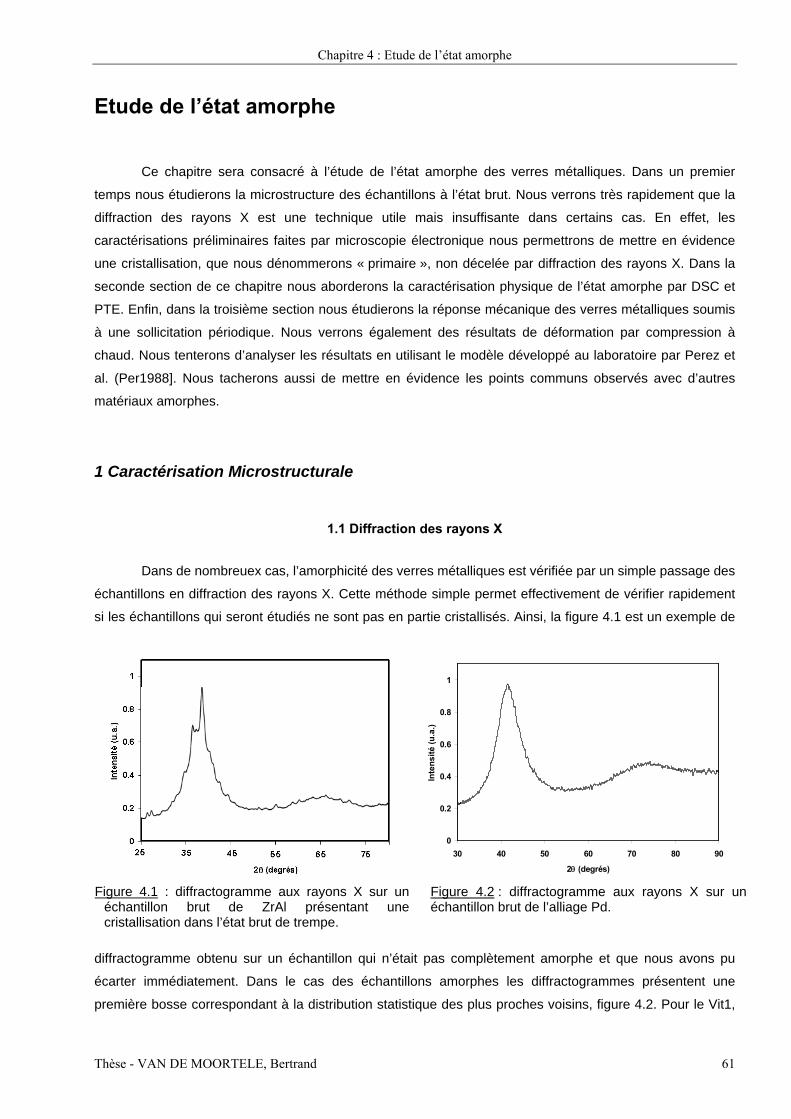
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 61
Etude de l’état amorphe
Ce chapitre sera consacré à l’étude de l’état amorphe des verres métalliques. Dans un premier
temps nous étudierons la microstructure des échantillons à l’état brut. Nous verrons très rapidement que la
diffraction des rayons X est une technique utile mais insuffisante dans certains cas. En effet, les
caractérisations préliminaires faites par microscopie électronique nous permettrons de mettre en évidence
une cristallisation, que nous dénommerons « primaire », non décelée par diffraction des rayons X. Dans la
seconde section de ce chapitre nous aborderons la caractérisation physique de l’état amorphe par DSC et
PTE. Enfin, dans la troisième section nous étudierons la réponse mécanique des verres métalliques soumis
à une sollicitation périodique. Nous verrons également des résultats de déformation par compression à
chaud. Nous tenterons d’analyser les résultats en utilisant le modèle développé au laboratoire par Perez et
al. (Per1988]. Nous tacherons aussi de mettre en évidence les points communs observés avec d’autres
matériaux amorphes.
1 Caractérisation Microstructurale
1.1 Diffraction des rayons X
Dans de nombreuex cas, l’amorphicité des verres métalliques est vérifiée par un simple passage des
échantillons en diffraction des rayons X. Cette méthode simple permet effectivement de vérifier rapidement
si les échantillons qui seront étudiés ne sont pas en partie cristallisés. Ainsi, la figure 4.1 est un exemple de
diffractogramme obtenu sur un échantillon qui n’était pas complètement amorphe et que nous avons pu
écarter immédiatement. Dans le cas des échantillons amorphes les diffractogrammes présentent une
première bosse correspondant à la distribution statistique des plus proches voisins, figure 4.2. Pour le Vit1,
Figure 4.1 : diffractogramme aux rayons X sur unéchantillon brut de ZrAl présentant unecristallisation dans l’état brut de trempe.
0
0.2
0.4
0.6
0.8
1
30 40 50 60 70 80 90
2θ (degrés)
Inte
nsité
(u.a
.)
Figure 4.2 : diffractogramme aux rayons X sur unéchantillon brut de l’alliage Pd.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 62
elle est centrée sur la valeur angulaire 2θ ≈ 35 ° (raie Kα du cuivre) ce qui correspond à une distance de
0.229 nm. A plus grande valeur angulaire on peut distinguer une seconde bosse amorphe qui correspond à
la distance de la seconde sphère de coordination. Comme l’ont évoqué certains auteurs [Wan1999a] la
diffraction des rayons X reste pour autant un moyen insuffisant pour détecter un faible taux de cristallisation.
Aussi avons-nous entrepris sur les échantillons, considérés comme amorphe du point de vue diffraction X,
une étude en microscopie électronique sur l’état brut.
1.2 Etude et Caractérisation par Microscopie Electronique en Transmission (MET)
Spriano et al. [Spr1997] ainsi que Macht et al [Mac2000b] ont déjà évoqué l’existence de cristaux
primaires sans pour autant s’attarder sur ce fait. He et al. [He2000] ont pour leur part fait un travail de
caractérisation plus approfondi. Pour notre part, nous ferons une description générale de la morphologie des
cristaux primaires observés dans les alliages ZrAl, Vit1 et Vit4. Pour certains d’entre eux, nous présenterons
des analyses de compositions qualitatives et dans certains cas nous déterminerons la structure
cristallographique de ces cristaux primaires (cas des alliages ZrAl et Vit1). Nous verrons ensuite les
conséquences de l’existence de cristaux primaires sur les essais mécaniques en traction à froid. Enfin nous
présenterons le cas général de l’étude de la matrice amorphe en prenant un exemple obtenu sur l’alliage
Vit1.
1.2.1 ZrAl
L’image 4.3 est une micrographie en champ clair d’un cristal primaire avec au centre un trou. La
forme générale de ces cristaux est dendritique et semble caractéristique d’une croissance dans un liquide
[Ino1999]. Leur taille est comprise entre 1 et 5 µm et la fraction volumique qu’ils occupent est en-deçà de
1%. La répartition de ces cristaux primaires dans la matrice semble assez uniforme.
1 µm
Figure 4.3 : micrographie en champ clair d’uncristal primaire dans l’alliage ZrAl.
1 µm
Figure 4.4: micrographie en champ clair d’un autre cristal primaire dans l’alliage ZrAl avec là également un trou au centre.
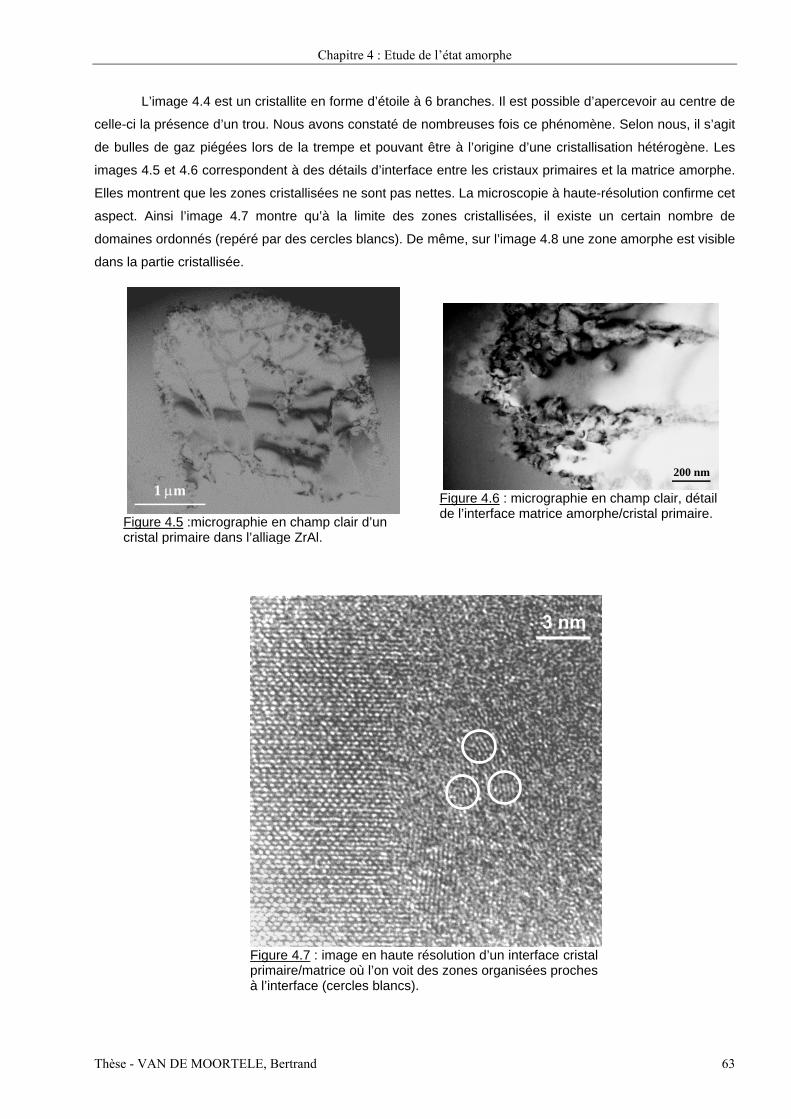
Chapitre 4 : Etude de l’état amorphe
Thèse
L’image 4.4 est un cristallite en forme d’étoile à 6 branches. Il est possible d’apercevoir au centre de
celle-ci la présence d’un trou. Nous avons constaté de nombreuses fois ce phénomène. Selon nous, il s’agit
de bulles de gaz piégées lors de la trempe et pouvant être à l’origine d’une cristallisation hétérogène. Les
images 4.5 et 4.6 correspondent à des détails d’interface entre les cristaux primaires et la matrice amorphe.
Elles montrent que les zones cristallisées ne sont pas nettes. La microscopie à haute-résolution confirme cet
aspect. Ainsi l’image 4.7 montre qu’à la limite des zones cristallisées, il existe un certain nombre de
domaines ordonnés (repéré par des cercles blancs). De même, sur l’image 4.8 une zone amorphe est visible
dans la partie cristallisée.
Fc
200 nm
Figure 4.6 : micrographie en champ clair, détail de l’interface matrice amorphe/cristal primaire.
1 µm
igure 4.5 :micrographie en champ clair d’un ristal primaire dans l’alliage ZrAl.
- VAN DE MOORTELE, Bertrand 63
3 nm
Figure 4.7 : image en haute résolution d’un interface cristal primaire/matrice où l’on voit des zones organisées proches à l’interface (cercles blancs).
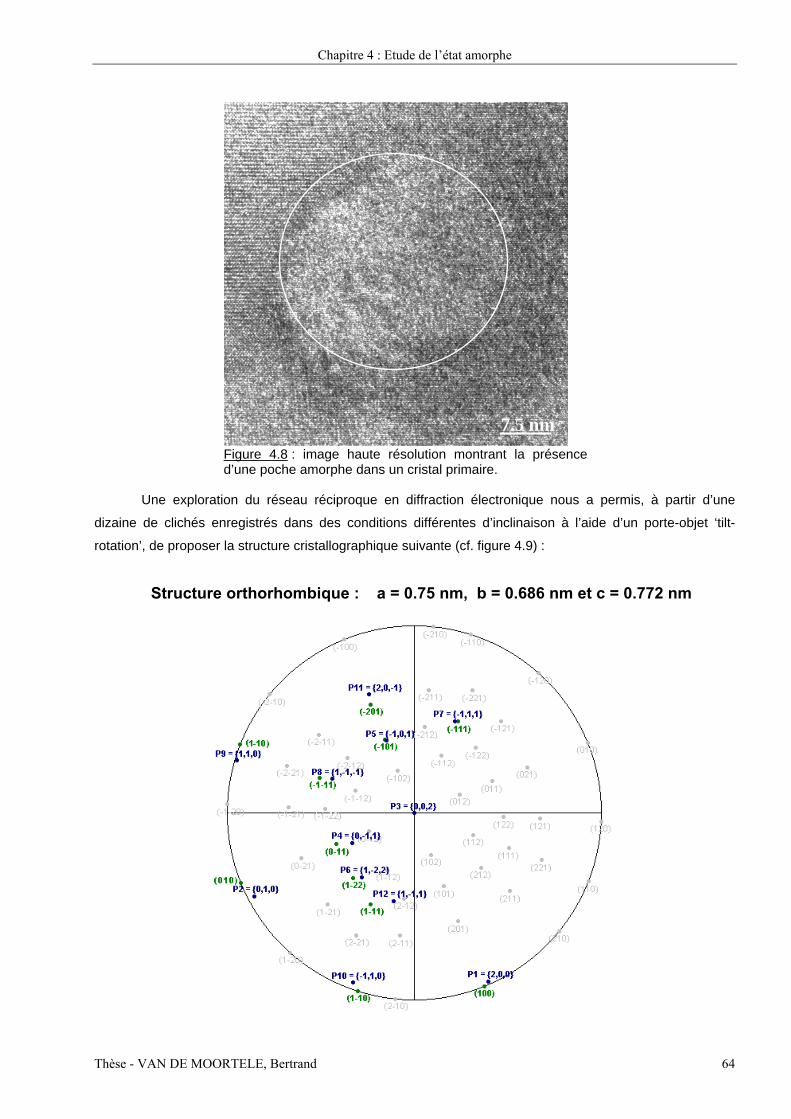
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 64
Une exploration du réseau réciproque en diffraction électronique nous a permis, à partir d’une
dizaine de clichés enregistrés dans des conditions différentes d’inclinaison à l’aide d’un porte-objet ‘tilt-
rotation’, de proposer la structure cristallographique suivante (cf. figure 4.9) :
Structure orthorhombique : a = 0.75 nm, b = 0.686 nm et c = 0.772 nm
7.5 nm
Figure 4.8 : image haute résolution montrant la présence d’une poche amorphe dans un cristal primaire.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 65
Figure 4.9 : indexation de 12 pôles expérimentaux (P1 à P12) dans la projection standard (001) de la phase orthorhombique identifiée, et exemples d’indexation de coupes de bas indices.
Des analyses de compositions chimiques ont été effectuées sur les cristaux primaires et la matrice
amorphe adjacente par EDX en MET, et sur l’échantillon global en MEB. Le taux d’oxygène mesuré par
MEB sur l’échantillon global est proche de 0 en accord avec les analyses effectuées par le CRETA. Les
mêmes mesures effectuées en MET conduisent à des taux d’oxygène proche de 20 %. Cette différence
s’explique sans aucun doute par une contamination superficielle de l’échantillon qui dans le cas du MET
conduit à des résultats complètement faussés. Aussi, il nous a été impossible de nous prononcer sur un
enrichissement des cristaux primaires en oxygène, ce qui pourrait être à l’origine de cette cristallisation

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 66
comme l’ont déjà montré Murty et al. dans l’alliage Zr65Cu27.5Al7.5 [Mur2000b]. Par la suite, nous
considérerons pour les analyses que l’alliage ne contient pas d’oxygène.
Les compositions de la matrice et des cristaux primaires sont assez proches mais on peut
néanmoins dégager une tendance et dire que les cristaux primaires sont enrichis en aluminium et titane,
appauvris en zirconium et nickel et inchangés en cuivre. Ce résultat est assez surprenant puisque l’on
s’attend à former à plus haute température des phases cristallines du type (Zr, Ti)2(Ni, Cu) et donc à
observer un comportement semblable des espèces chimiques Zr et Ti.
1.2.2 Vit1
Dans le cas de l’alliage Vit1, les précipités ont une forme caractéristique assez différente de celle
observée dans les ZrAl, comme le montre la figure 4.10, où l’on peut voir un cristal primaire allongé en forme
de bâtonnet.
La longueur typique de ces cristaux est de 1 µm et la largeur de 0,2 µm. Une image haute-
résolution, figure 4.11, montre l’interface entre un cristal primaire et la matrice amorphe, qui apparaît plus
abrupte que dans le cas des verres ZrAl (notons par ailleurs un petit cristal épitaxié sur le plus gros). Le
tableau 4.2 donne les compositions chimiques obtenues par EDX (rapportées à 100 % hors béryllium) de
% atomique Al Ti Ni Cu Zr Primaire 12.8 7.3 11.1 24.4 44.8
Matrice 9.1 4.6 14 24.7 47.5 Tableau 4.1 : comparaison des compositions chimiques de la matrice amorphe et des précipités primaires dans l’alliage ZrAl.
200 nm
Figure 4.10 : micrographie en champ clair d’un cristalprimaire.
Figure 4.11 : image haute résolution d’uninterface cristal primaire/matrice amorphe.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOO
deux cristaux primaires comparés à l’analyse de la matrice dans les mêmes conditions. On remarque une
forte tendance à l’enrichissement des cristaux primaires en zirconium.
On notera par ailleurs que les résultats d’analyse ‘Matrice 1’ et ‘Matrice 2’ montrent un
appauvrissement en titane corrélé à un enrichissement en nickel par rapport à la composition nominale de
l’alliage donnée par Howmet Corporation. La moyenne des analyses de matrice, obtenue à partir de
plusieurs dizaines de points de mesures, indique la même tendance, même si la différence est moins
marquée. Par la suite, nous utiliserons ce résultat expérimental lorsque nous nous référerons à la
composition ‘brute’ du Vit1.
Parallèlemen
révèlent un net appa
accord avec ce que B
savoir la formation de
Figure 4.12 : analysespectres expérimentaselon la méthode déccarbone nous a condgrisés en gras) condattendue de 0.55) et 0
Tableau 4.
% atomique Zr Ti Ni Cu
Matrice 1 49.3 10.8 20.2 19.7
Matrice 2 53.5 13.4 15.3 17.3
Primaire 1 74.5 3.7 10.6 10.7
Primaire 2 76.7 5. 9.5 8.6
Compo.nom. 53.2 17.8 12.9 16.1
Moyenne matrice 53.6 15.8 14.5 16.1 2 : les analyses sont quantitatives, l’incertitude relative est de l’ordre de 1.0 %.
RTELE, Bertrand 67
t, des analyses par pertes d’énergie confirment un enrichissement en Zr, et surtout
uvrissement en Be dans les cristaux primaires (figure 4.12). Ceci est tout à fait en
usch et al. [Bus1995] ont observé lors d’un refroidissement lent sur un alliage Vit1, à
domaines riches en Zr et pauvres en Be et inversement.
s EELS de la matrice brute du Vit1 (à gauche) et d’un cristal primaire (à droite). Les ux (traits noirs gras) sont ajustés à l’aide des spectres de référence Be-Kref et Zr-Mref rite au chapitre 3. Dans ce cas, la présence d’une contamination non négligeable par le uit à utiliser également un spectre de référence C-Kref . Les spectres reconstruits (traits uisent aux rapports Be/Zr égaux à 0.59 pour la matrice brute (proche de la valeur .15 pour le primaire.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 68
Des images ont été faites en microscopie filtrée en énergie (LEO-912) sur les seuils des éléments Ti,
Zr et Ni. La figure 4.13 montre l’exemple d’un cristal primaire observé dans un échantillon recuit 25 minutes
à 410 °C (ce traitement n’influençant pas la stabilité des cristaux primaires). La zone étudiée est apparue
trop épaisse pour permettre ici une image avec le seuil K du Be à faible perte d’énergie (110 eV), mais les
résultats confirment qualitativement ce qui a été dit précédemment, à savoir un enrichissement en Zr et un
appauvrissement corrélé en Ti (les contrastes autour du précipité primaire proviennent de la nano-
cristallisation s’opérant lors du recuit, et seront ré-analysés en détail au chapitre 5).
Nous avons également effectué une exploration du réseau réciproque de cette phase, comme dans
le cas des cristaux primaires dans le verre ZrAl (§. 1.2.1.), en étayant l’analyse avec un traitement des
distances identifiées en diffraction électronique à l’aide du logiciel TREOR [Wer1990].
Figure 4.13 : analyse d’un cristal primaire dans le Vit1 en EFTEM ; de gauche à droite et de haut en bas : images respectivement élastique et sur les seuils Ti-L, Ni-L et Zr-L (fenêtre d’environ 8 eV). Remarque : le rapport signal-sur-bruit très défavorable sur la dernière image – on discerne des ‘trainées’ horizontales – vient du fait que le seuil L du zirconium est situé à une très grande perte, i.e. vers 2200 eV.
100 nm

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 69
La figure 4.14 résume brièvement cette analyse; l’ensemble des distances mesurées nous ont
permis de proposer une structure cristallographique pour les cristaux primaires présents dans le Vit1 :
structure monoclinique : a = b = 1.5148 nm, c = 0.8076 nm
α = γ = 90° et β = 99.35°
1.2.3 Vit4
Comme le montre la micrographie en champ clair, figure 4.15, les cristaux primaires dans le Vit4
sont de petites tailles (au maximum 0.3*0.3 µm2).
Contrairement aux autres systèmes étudiés, ils sont
groupés et orientés. Cette orientation résulte
semble-t-il d’une cristallisation lors de la coulée dans
le sens de celle-ci. Il faut d’ailleurs se rappeler que
cet alliage se présente sous forme de cylindre de 9
mm de diamètre ce qui induit des vitesses de
refroidissement fortement différentes si l’on se situe
au bord de l’échantillon ou au centre.
L’image 4.16 est une diffraction sur ces
précipités primaires. La comparaison avec la
structure théorique de ZrBe2 montre que certains
des précipités sont identifiables à ZrBe2 (hexagonal,
Figure 4.14 : clichés de diffraction des cristaux primaires du VIT1 indexés dans la structure monoclinique donnée précédemment.
200 nm
Figure 4.15 : micrographie en champ clair decristaux primaires dans le Vit4.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 70
a=0.382 nm, c=0.3239 nm, groupe de symétrie P6/mmm, fiche JPCDS 12-392). D’autres précipités ne
peuvent être indexés dans cette structure confirmant ainsi l’hypothèse de Macht et al. [Mac2001b] de
l’existence d’au moins deux types de cristaux primaires dans le Vit4. Nous avons résumé dans le tableau 4.3
les distances plans inter-réticulaires en séparant celles compatibles avec le ZrBe2 et les autres.
gr
1 gr
2 θ( gr
1, gr
2) (degrés)
nm (hkl) nm (hkl) compatible ZrBe2 (JPCDS 12-392) 0.249 0-11 0.2473 1-11 75.5
gr
1 (nm) gr
2 (nm)
0.466 0.2541 72. non compatible ZrBe2
0.206 0.2566 82.54 Tableau 4.3 : distances et angles obtenus à partir de diffractions en aire sélectionnée de cristaux primaires dans le Vit4, certains cristaux pouvant s’indexer dans la structure de ZrBe2.
1.2.4 Conséquences sur les propriétés mécaniques
Au vu de leur taille, de la fraction volumique et de leur structure non-continue, on comprend que la
diffraction X n’ait pu mettre en évidence ces cristaux primaires. De même, des techniques de caractérisation
comme la DSC sont sensibles aux effets de volume. Pelletier et al. [Pel1980] ont montré que la technique du
PTE pouvait être insensible à la précipitation dans des alliages.
La présence des cristaux primaires ne semble pas affecter les différents stades de cristallisation de
la matrice amorphe puisque l’on obtient les mêmes phases au voisinage ou loin des cristaux primaires (dans
ce cas Zr2Ni). L’image 4.17 est un montage de deux images en champ clair, de même grandissement,
obtenues sur échantillon de ZrAl recuit pendant 100 min à 459°C. Sur l’image de gauche nous pouvons voir
un cristal primaire (partie blanche), il est entouré de matrice amorphe partiellement cristallisée. Sur l’image
de gauche, aucun cristal primaire n’est visible, la matrice amorphe est là également partiellement cristallisée.
La cristallisation étant la même, du point de vue de la taille et de la fraction volumique, nous en avons conclu
Figure 4.16 : diffraction d’un cristal primaire dans le Vit4 pouvant s’indexerdans la structure hexagonale de ZrBe2 selon l'azimuth [121]

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 71
que les cristaux primaires ne favorisent ni ne défavorisent la cristallisation à ce stade de traitement
thermique.
Dans le cadre du DEA de Mourad Arioua réalisé au laboratoire GEMPPM, auquel j’ai été associé
pour la partie microscopie, des essais de traction et de fatigue à froid, également en traction, ont montré que
ces cristaux primaires jouaient un rôle important. La figure 4.18 est un graphe dans lequel est reporté le
nombre de cycles à la rupture en fonction de la contrainte de traction maximale appliquée. Bien que le
nombre de points expérimentaux soit peut important, le comportement observé, c’est-à-dire la diminution du
nombre de cycles à la rupture quand la contrainte augmente, est conforme au comportement des verres
métalliques déjà observé [Ino1999]. Pour autant, la dispersion pour les deux essais effectués pour une
contrainte de 800 MPa est proche de 300 %.
Figure 4.18 : nombre de cycles à la rupture en fonction de la contrainte
Figure 4.17 : micrographie en champ clair après cristallisation du ZrAl montrant lasimilitude entre la précipitation aux abords de précipités primaires et le reste del’échantillon (respectivement gauche et droite).

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 72
Les essais de traction ont permis de déterminer l’allongement en fonction de la contrainte et ainsi le
module élastique. Nous avons obtenu un module de Young élastique de 100 GPa à comparer aux 89 GPa
trouvés dans le Zr53Ti5Ni10Cu20Al12 par Fan et al.[Fan1999]. Dans la littérature, on relève des valeurs de la
limite élastique pouvant aller jusque 2 GPa. Ces valeurs sont généralement obtenues lors d’essais de
compression. Lors de nos essais en traction, nous avons obtenu des valeurs plus faibles et surtout très
dispersées, certaines éprouvettes ayant cassé prématurément (vers 0.8 ou 1 Gpa, par exemple)..
L’étude par microscopie électronique en balayage des faciès de rupture a permis de montrer que les
ruptures précoces étaient liées à la présence de cristaux primaires. L’image de la figure 4.19 correspond à
un faciès de rupture typique d’un verre métallique sur une éprouvette de traction. Les cupules que l’on peut
voir sont le résultat de la fusion partielle de l’échantillon lors de la fracture. Les échantillons présentant une
faible durée de vie ont également été observés en microscopie à balayage. La figure 4.20 révèle la présence
d’un cristal primaire à la surface. La forme particulière de ces cristaux primaires ( cf. figure 4.10) privilégie
sans aucun doute les concentrations de contrainte et fragilise le matériau, ce qui expliquerait une rupture
précoce.
Dans certains cas l’analyse EDX nous a permis de mettre en évidence la présence d’impuretés
riches en fer provenant probablement du moule de coulée.
Dans la suite de ce travail nous présenterons des résultats de déformation par compression et de
spectrométrie mécanique qui ne seront pas affectés par la présence de cristaux primaires. En effet, le
comportement des matériaux fragiles n’est absolument pas le même en traction qu’en compression. Dans le
premier cas, la contrainte a tendance à ouvrir d’avantage les fissures qui se forment alors que dans l‘autre il
y a fermeture des fissures. Par ailleurs, les essais de compression seront réalisés à haute température où la
déformation devient homogène et où la viscoplasticité peut se développer.
Figure 4.19 : micrographie d’un faciès de rupture lors d’essais de traction à froid d’un échantillon de Vit1. L’image (a) correspond au faciès typique de la rupture d’un amorphe, l’image (b) montre un cristal primaire semble-t-il à l’origine d’une rupture prématurée.
(a) (b)

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 73
1.2.5 Matrice Amorphe
En dehors de ces zones particulières présentant des cristaux primaires, le matériau est pleinement
amorphe. La figure 4.20 est une diffraction obtenue sur la matrice amorphe de l’alliage Vit1. C’est une
diffraction typique d’un amorphe. On distingue en effet un anneau diffus qui correspond à la première bosse
observée en diffraction des rayons X. Un second anneau est plus difficile à détecter. Une moyenne radiale
de la diffraction permet de retrouver comme le montre la figure 4.21 un profil similaire à celui de la diffraction
des rayons X, mais avec une seconde bosse moins marquée.
2 Caractérisation Physique par DSC et PTE
2.1 DSC
Comme nous l’avons évoqué dans le chapitre 2, la détermination de la température de transition
vitreuse nécessite de préciser non seulement les conditions expérimentales mais également de définir la
convention prise pour déterminer Tg. La figure 4.22 est une partie d’un thermogramme de DSC obtenu sur
l’alliage Vit1 pour une vitesse de montée en température de 10 °C.min-1. La zone de transition vitreuse se
situe entre 345 et 382 °C, Tg1 et Tg3 correspondant respectivement aux températures de début et de fin de
transition vitreuse déterminées en utilisant le point d'intersection des tangentes avant et après la transition
vitreuse. La température que nous appellerons par la suite température de transition vitreuse est définie
comme une température intermédiaire Tg2 qui correspond au point d'inflexion de la zone entre Tg1 et Tg3 et
qui est proche de la moyenne de Tg1 et Tg3.
4 nm-1
Figure 4.20 : diffraction électronique de la matrice amorphe dans le Vit1 brut.
0
200
400
600
800
1000
1200
1400
1600
3 4 5 6 7 8 9 10
distance (nm-1)
inte
nsité
(u.a
.)
Figure 4.21 : profil de diffraction obtenu par moyenne radiale de la diffraction de la figure 4.20.
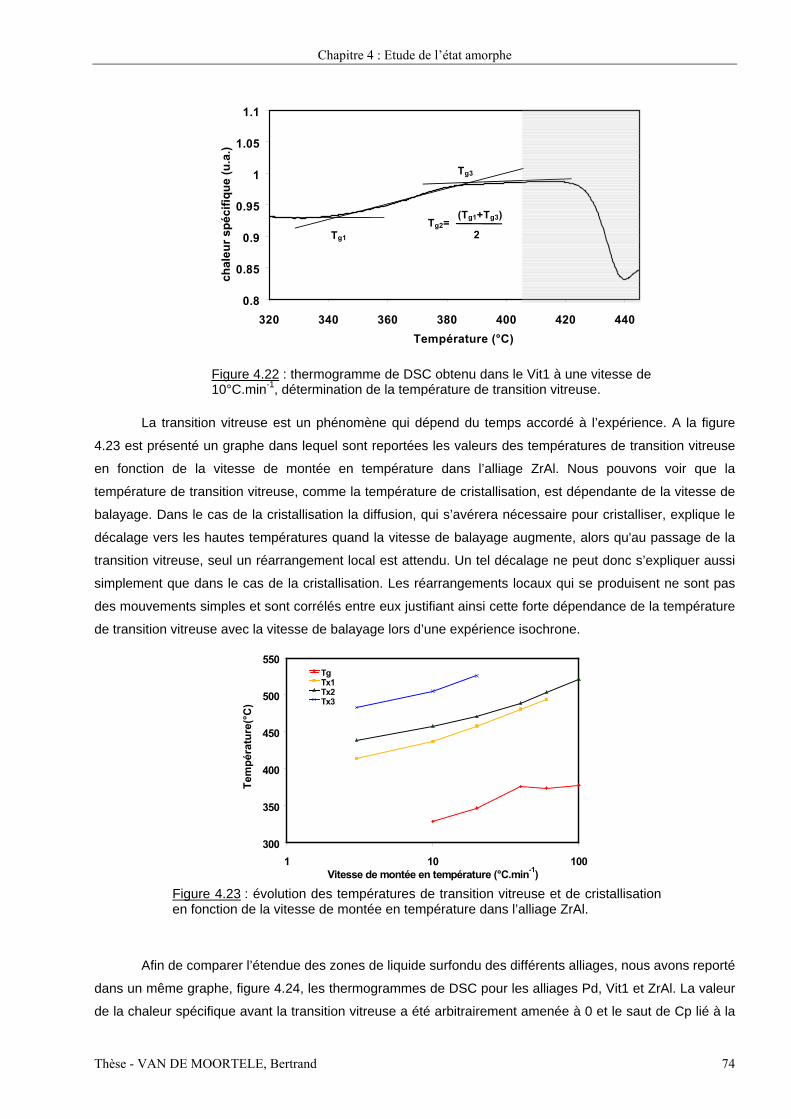
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 74
La transition vitreuse est un phénomène qui dépend du temps accordé à l’expérience. A la figure
4.23 est présenté un graphe dans lequel sont reportées les valeurs des températures de transition vitreuse
en fonction de la vitesse de montée en température dans l’alliage ZrAl. Nous pouvons voir que la
température de transition vitreuse, comme la température de cristallisation, est dépendante de la vitesse de
balayage. Dans le cas de la cristallisation la diffusion, qui s’avérera nécessaire pour cristalliser, explique le
décalage vers les hautes températures quand la vitesse de balayage augmente, alors qu'au passage de la
transition vitreuse, seul un réarrangement local est attendu. Un tel décalage ne peut donc s’expliquer aussi
simplement que dans le cas de la cristallisation. Les réarrangements locaux qui se produisent ne sont pas
des mouvements simples et sont corrélés entre eux justifiant ainsi cette forte dépendance de la température
de transition vitreuse avec la vitesse de balayage lors d’une expérience isochrone.
Afin de comparer l’étendue des zones de liquide surfondu des différents alliages, nous avons reporté
dans un même graphe, figure 4.24, les thermogrammes de DSC pour les alliages Pd, Vit1 et ZrAl. La valeur
de la chaleur spécifique avant la transition vitreuse a été arbitrairement amenée à 0 et le saut de Cp lié à la
0.8
0.85
0.9
0.95
1
1.05
1.1
320 340 360 380 400 420 440Température (°C)
chal
eur s
péci
fique
(u.a
.)Tg1
Tg3
Tg2=(Tg1+Tg3)
2
Figure 4.22 : thermogramme de DSC obtenu dans le Vit1 à une vitesse de10°C.min-1, détermination de la température de transition vitreuse.
300
350
400
450
500
550
1 10 100Vitesse de montée en température (°C.min-1)
Tem
péra
ture
(°C
)
TgTx1Tx2Tx3
Figure 4.23 : évolution des températures de transition vitreuse et de cristallisationen fonction de la vitesse de montée en température dans l’alliage ZrAl.
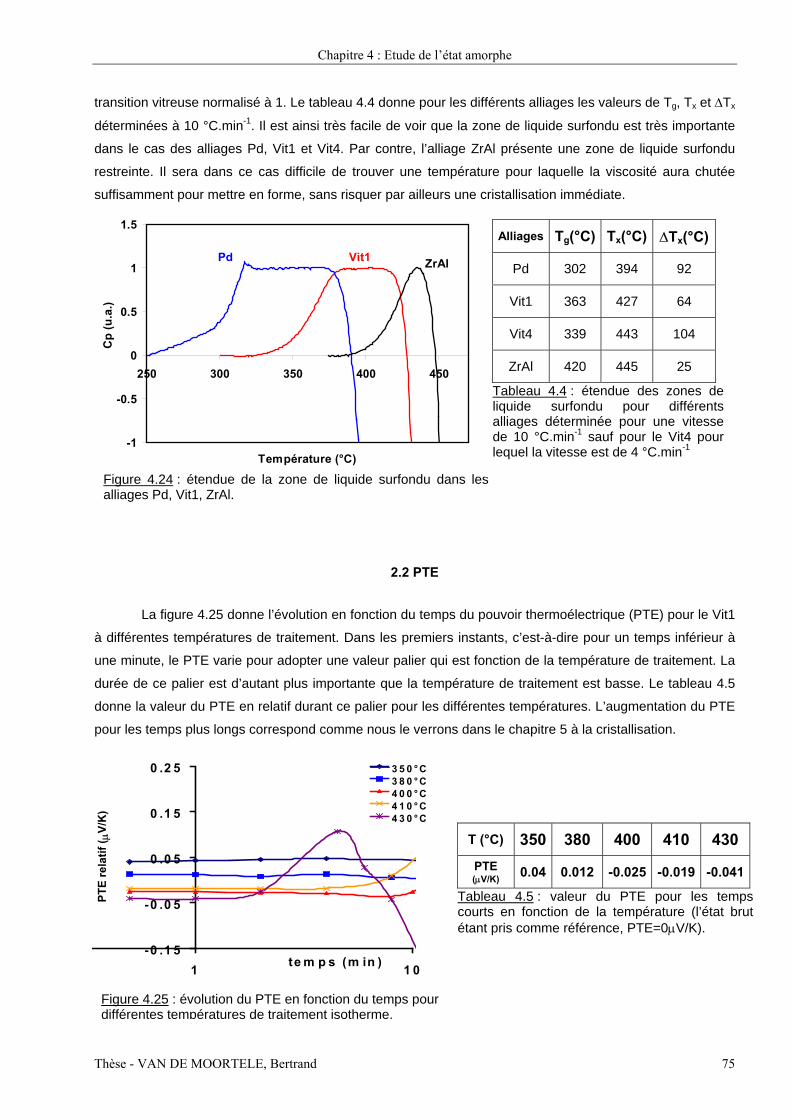
Chapitre 4 : Etude de l’état amorphe
transition vitreuse normalisé à 1. Le tableau 4.4 donne pour les différents alliages les valeurs de Tg, Tx et ∆Tx
déterminées à 10 °C.min-1. Il est ainsi très facile de voir que la zone de liquide surfondu est très importante
dans le cas des alliages Pd, Vit1 et Vit4. Par contre, l’alliage ZrAl présente une zone de liquide surfondu
restreinte. Il sera dans ce cas difficile de trouver une température pour laquelle la viscosité aura chutée
suffisamment pour mettre en forme, sans risquer par ailleurs une cristallisation immédiate.
2.2 PTE
La figure 4.25 donne l’évolution en fonction du temps du pouvoir thermoélectrique (PTE) pour le Vit1
à différentes températures de traitement. Dans les premiers instants, c’est-à-dire pour un temps inférieur à
une minute, le PTE varie pour adopter une valeur palier qui est fonction de la température de traitement. La
durée de ce palier est d’autant plus importante que la température de traitement est basse. Le tableau 4.5
donne la valeur du PTE en relatif durant ce palier pour les différentes températures. L’augmentation du PTE
pour les temps plus longs correspond comme nous le verrons dans le chapitre 5 à la cristallisation.
T (°C) 350 380 400 410 430 PTE (µV/K) 0.04 0.012 -0.025 -0.019 -0.041
Tableau 4.5 : valeur du PTE pour les temps courts en fonction de la température (l’état brut étant pris comme référence, PTE=0µV/K).
-1
-0.5
0
0.5
1
1.5
250 300 350 400 450
Température (°C)
Cp
(u.a
.)
Pd Vit1 ZrAl
Figure 4.24 : étendue de la zone de liquide surfondu dans lesalliages Pd, Vit1, ZrAl.
Alliages Tg(°C) Tx(°C) ∆Tx(°C)
Pd 302 394 92
Vit1 363 427 64
Vit4 339 443 104
ZrAl 420 445 25
Tableau 4.4 : étendue des zones de liquide surfondu pour différents alliages déterminée pour une vitesse de 10 °C.min-1 sauf pour le Vit4 pour lequel la vitesse est de 4 °C.min-1
-0 .1 5
-0 .0 5
0 .0 5
0 .1 5
0 .2 5
1 1 0te m p s (m in )
3 5 0 °C3 8 0 °C4 0 0 °C4 1 0 °C4 3 0 °C
PTE
rela
tif ( µ
V/K
)
Figure 4.25 : évolution du PTE en fonction du temps pourdifférentes températures de traitement isotherme.
Thèse - VAN DE MOORTELE, Bertrand 75

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 76
Avant la cristallisation, des phénomènes de relaxation sont attendus. De précédentes études ont
montré qu’une partie des relaxations avant cristallisation étaient réversibles [Pel1986]. Afin de mettre ceci en
évidence, nous avons procédé à des cycles thermiques entre 410 °C (pendant 1 minute) et 350 °C (5 à 6
minutes). Les valeurs du PTE ont été mesurées à différents temps au cours de l’expérience et sont
reportées au graphe de la figure 4.26. Nous voyons qu’après un cycle entre 350 et 410 °C, le PTE retrouve
les mêmes valeurs, ce qui montre que la relaxation se produisant dans les premiers instants est réversible.
Dans une première approche, la réversibilité d’une part et les valeurs du PTE aux faibles temps
d’autre part, pourraient s’interpréter en revenant sur la courbe d'enthalpie libre en fonction de la température
(figure 4.27). Lorsque l'on porte brutalement l'échantillon à la température de 410 °C, il suit la courbe fictive
d'enthalpie du verre au-delà de la température de transition liquide surfondu-verre. Il faut cependant être
conscient que cet état ne correspond pas à un état
thermodynamique stable, mais cette représentation
schématique permet d’avoir une compréhension de
ce qui se passe. Le maintien à la température de
vieillissement au point A a pour effet de permettre au
système de rejoindre l'équilibre qui correspond au
point B. La trempe à 350 °C met à nouveau le
système hors d'équilibre (point C) en passant par le
point d’inflexion I. A cette température la mobilité est
suffisante pour que le système «glisse» vers des
états métastables sans atteindre d’état stable (cf.
fig2.6) ce qui amène le système au point D. En
revenant sur les mesures faites à différentes températures nous voyons que le signe du PTE indique la
position de la température de traitement par rapport à la température de transition vitreuse (PTE
respectivement négatif et positif pour des températures au-dessus et en-dessous de Tg). De plus, nous
voyons que plus les mesures seront faites loin de la température de transition vitreuse, plus l’amplitude du
phénomène de vieillissement sera importante ce qui se vérifie bien dans les expériences isothermes. La
température d’équilibre, c’est-à-dire celle pour laquelle aucune évolution structurale réversible n’est
observée, est voisine de 390 °C.
Figure 4.26 : mise en évidence des phénomènes réversibles.
H
350 410
A
B
C
Liquide surfondu
Amorphe
T (°C)D
I
Figure 4.27 : schéma de l’enthalpie expliquant la réversibilité du PTE.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 77
3 Réponse Mécanique
Les propriétés mécaniques dans le domaine linéaire (expériences de spectrométrie mécanique) des
verres métalliques ont été mesurées pour l’ensemble des alliages dont nous disposions. Dans cette partie,
nous montrerons principalement les résultats obtenus sur les alliages Vit1 et Vit4, l’ensemble des propriétés
mécaniques étant semblables dans les différents alliages. Pour le Vit1 nous montrerons donc l’effet de la
température, de la vitesse de montée en température et de la fréquence sur les propriétés mécaniques pour
des expériences en isotherme et isochrone. De l’ensemble de ces expériences peuvent être déduites des
courbes maîtresses permettant d'extrapoler le comportement à des fréquences que nous ne pouvons
atteindre expérimentalement. Nous montrerons celles obtenues pour l’alliage Vit4. Puis, nous étudierons les
effets non-linéaires de la déformation en nous intéressant aux expériences de compression effectuées sur
l’alliage Vit1. Enfin, nous aborderons l’analyse de ces résultats en les comparant
3.1 Elasticité et viscoélasticité : spectrométrie mécanique
3.1.1 Courbes typiques
La figure 4.28 montre l’évolution de la partie réelle du module de cisaillement du Vit1 en fonction de
la température, obtenue lors d’une montée en température de 3 °C.min-1 à la fréquence de 0.3 Hz. A basse
température, le module est pratiquement constant. Puis une chute très importante (voisine de trois ordres de
grandeurs) se produit à partir de 380 °C, c’est-à-dire au voisinage de la température de transition vitreuse,
sur un intervalle d’une cinquantaine de degrés. On peut noter dès à présent qu’aucune relaxation sous
vitreuse n’est visible dans ce matériau.
Le plateau observé après la chute du module est communément appelé plateau caoutchoutique.
Dans le cas des polymères, il est lié à l’existence d’une force de rappel entropique entre les chaînes
moléculaires. Dans le cas de verres métalliques aucune interprétation n’a encore été donnée à ce jour.
La remontée du module observée à plus haute température, ici à 480 °C, correspond à la cristallisation du
matériau.
Figure 4.28 : évolution de la partie réelle du module avec la température dans le Vit1 (3 °C.min-1, 0.3 Hz).

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 78
Une fois la température de 600 °C atteinte, les mesures de la valeur du module lors de la descente en
température indiquent clairement que la cristallisation qui s’est opérée à haute température a induit une
augmentation irréversible du module comme on peut le voir sur la figure 4.28 (flêche de droite vers la
gauche).
Comme le montre la figure 4.29, la chute de la partie réelle du module est associée à un pic de la
partie imaginaire du module. Ce pic, centré sur une température Tα, dite température de relaxation
principale, voisine de Tg pour une fréquence de 1Hz, traduit l’existence d’un maximum de dissipation de
l’énergie. Cette relaxation principale est fréquemment associée au début d’une mobilité atomique à longue
distance.
Un comportement similaire a déjà été observé dans d’autres matériaux amorphes. La figure 4.30
montre le module réel de cisaillement d’un verre fluoré en fonction de la température. Le module présente là
également une chute importante suivie très rapidement d’une remontée liée à la cristallisation.
Figure 4.30 : évolution de la partie réelle du module dans un verrefluoré ( a, b et c correspondent aux mesures effectuées à desfréquences de 0.01, 0.1 et 1 Hz respectivement), d'après [Abb1982].
Figure 4.29 : évolution de la partie imaginaire G’’ du module avec la température dans le Vit1 (3 °C.min-1, 0.3 Hz).
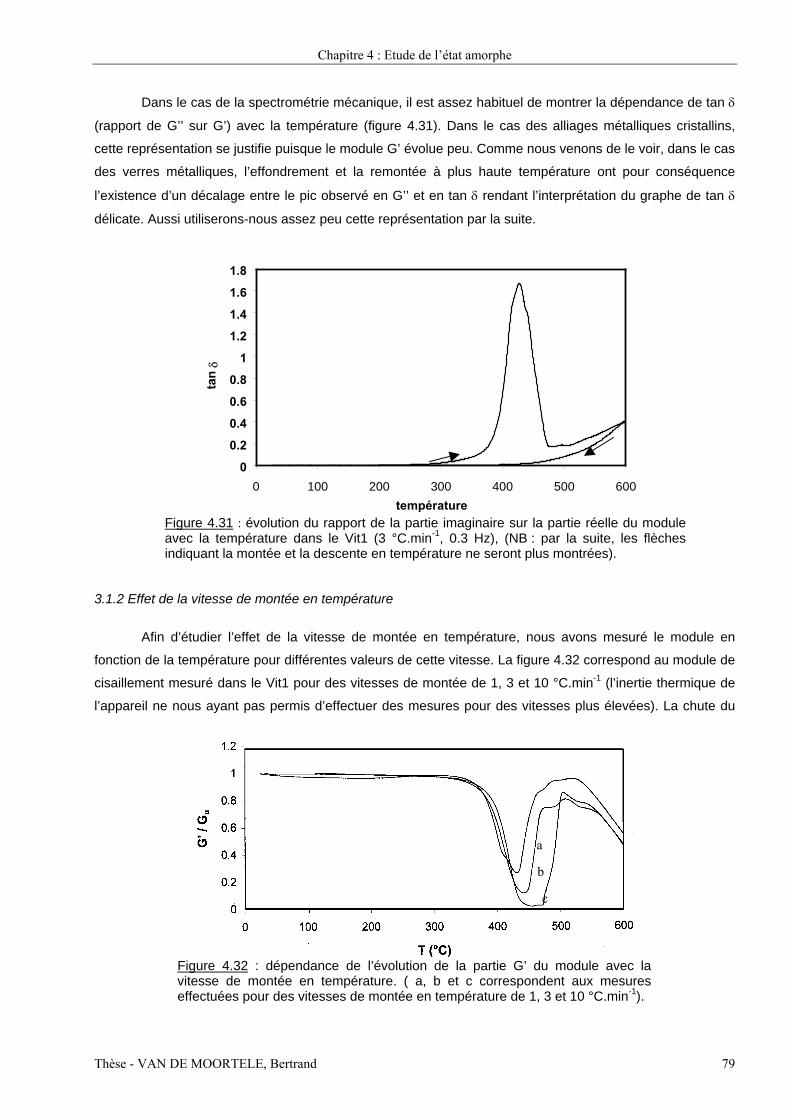
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 79
Dans le cas de la spectrométrie mécanique, il est assez habituel de montrer la dépendance de tan δ
(rapport de G’’ sur G’) avec la température (figure 4.31). Dans le cas des alliages métalliques cristallins,
cette représentation se justifie puisque le module G’ évolue peu. Comme nous venons de le voir, dans le cas
des verres métalliques, l’effondrement et la remontée à plus haute température ont pour conséquence
l’existence d’un décalage entre le pic observé en G’’ et en tan δ rendant l’interprétation du graphe de tan δ
délicate. Aussi utiliserons-nous assez peu cette représentation par la suite.
3.1.2 Effet de la vitesse de montée en température
Afin d’étudier l’effet de la vitesse de montée en température, nous avons mesuré le module en
fonction de la température pour différentes valeurs de cette vitesse. La figure 4.32 correspond au module de
cisaillement mesuré dans le Vit1 pour des vitesses de montée de 1, 3 et 10 °C.min-1 (l’inertie thermique de
l’appareil ne nous ayant pas permis d’effectuer des mesures pour des vitesses plus élevées). La chute du
Figure 4.32 : dépendance de l’évolution de la partie G’ du module avec lavitesse de montée en température. ( a, b et c correspondent aux mesureseffectuées pour des vitesses de montée en température de 1, 3 et 10 °C.min-1).
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0 100 200 300 400 500 600température
tan
δ
Figure 4.31 : évolution du rapport de la partie imaginaire sur la partie réelle du moduleavec la température dans le Vit1 (3 °C.min-1, 0.3 Hz), (NB : par la suite, les flèchesindiquant la montée et la descente en température ne seront plus montrées).
a
b
c
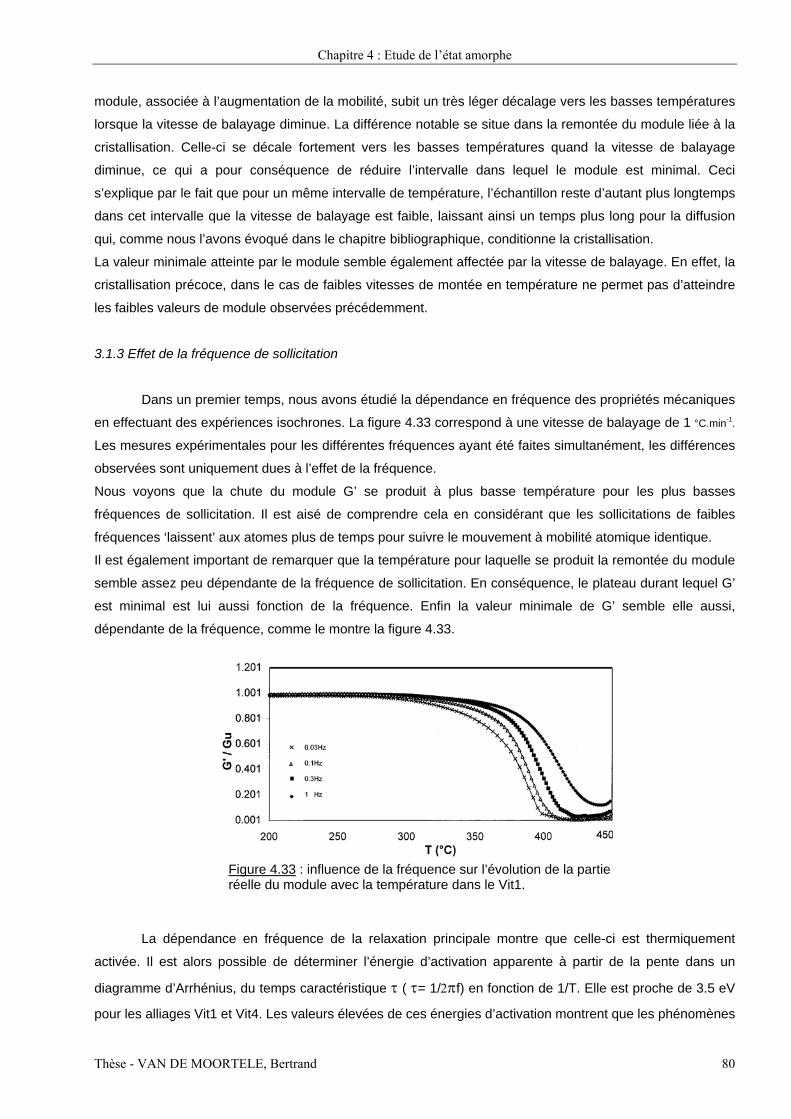
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 80
module, associée à l’augmentation de la mobilité, subit un très léger décalage vers les basses températures
lorsque la vitesse de balayage diminue. La différence notable se situe dans la remontée du module liée à la
cristallisation. Celle-ci se décale fortement vers les basses températures quand la vitesse de balayage
diminue, ce qui a pour conséquence de réduire l’intervalle dans lequel le module est minimal. Ceci
s’explique par le fait que pour un même intervalle de température, l’échantillon reste d’autant plus longtemps
dans cet intervalle que la vitesse de balayage est faible, laissant ainsi un temps plus long pour la diffusion
qui, comme nous l’avons évoqué dans le chapitre bibliographique, conditionne la cristallisation.
La valeur minimale atteinte par le module semble également affectée par la vitesse de balayage. En effet, la
cristallisation précoce, dans le cas de faibles vitesses de montée en température ne permet pas d’atteindre
les faibles valeurs de module observées précédemment.
3.1.3 Effet de la fréquence de sollicitation
Dans un premier temps, nous avons étudié la dépendance en fréquence des propriétés mécaniques
en effectuant des expériences isochrones. La figure 4.33 correspond à une vitesse de balayage de 1 °C.min-1.
Les mesures expérimentales pour les différentes fréquences ayant été faites simultanément, les différences
observées sont uniquement dues à l’effet de la fréquence.
Nous voyons que la chute du module G’ se produit à plus basse température pour les plus basses
fréquences de sollicitation. Il est aisé de comprendre cela en considérant que les sollicitations de faibles
fréquences ‘laissent’ aux atomes plus de temps pour suivre le mouvement à mobilité atomique identique.
Il est également important de remarquer que la température pour laquelle se produit la remontée du module
semble assez peu dépendante de la fréquence de sollicitation. En conséquence, le plateau durant lequel G’
est minimal est lui aussi fonction de la fréquence. Enfin la valeur minimale de G’ semble elle aussi,
dépendante de la fréquence, comme le montre la figure 4.33.
La dépendance en fréquence de la relaxation principale montre que celle-ci est thermiquement
activée. Il est alors possible de déterminer l’énergie d’activation apparente à partir de la pente dans un
diagramme d’Arrhénius, du temps caractéristique τ ( τ= 1/2πf) en fonction de 1/T. Elle est proche de 3.5 eV
pour les alliages Vit1 et Vit4. Les valeurs élevées de ces énergies d’activation montrent que les phénomènes
Figure 4.33 : influence de la fréquence sur l’évolution de la partie réelle du module avec la température dans le Vit1.
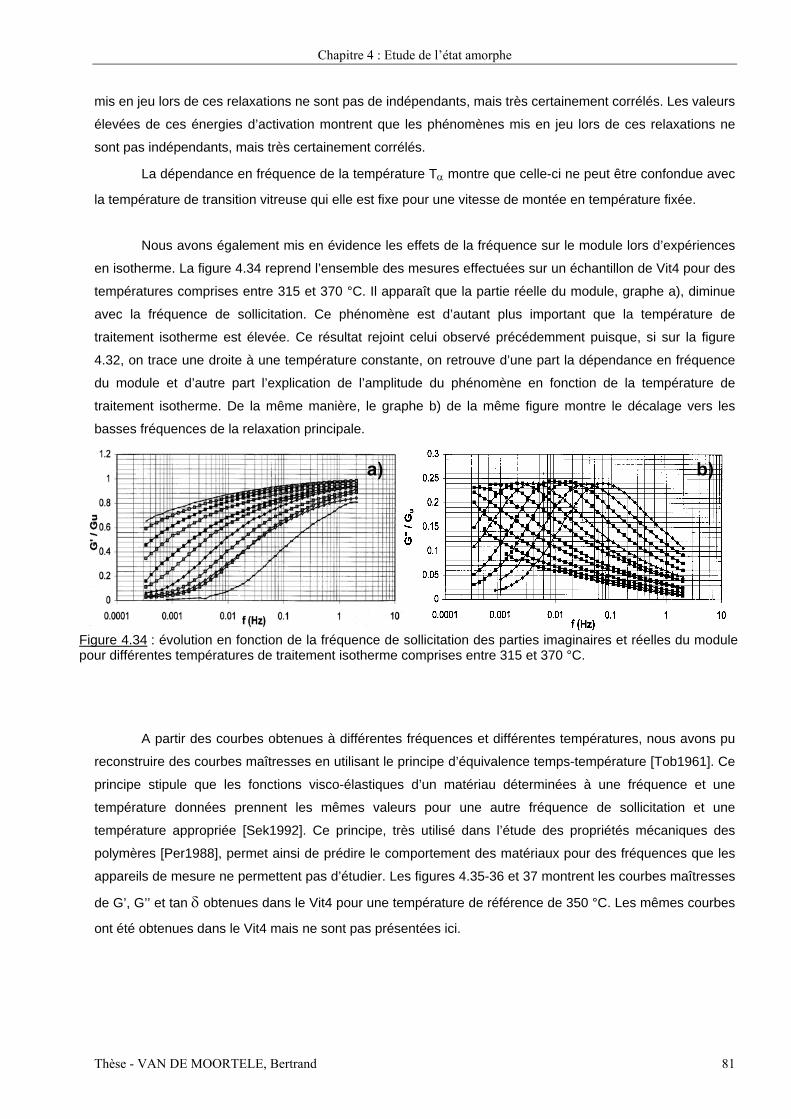
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 81
mis en jeu lors de ces relaxations ne sont pas de indépendants, mais très certainement corrélés. Les valeurs
élevées de ces énergies d’activation montrent que les phénomènes mis en jeu lors de ces relaxations ne
sont pas indépendants, mais très certainement corrélés.
La dépendance en fréquence de la température Tα montre que celle-ci ne peut être confondue avec
la température de transition vitreuse qui elle est fixe pour une vitesse de montée en température fixée.
Nous avons également mis en évidence les effets de la fréquence sur le module lors d’expériences
en isotherme. La figure 4.34 reprend l’ensemble des mesures effectuées sur un échantillon de Vit4 pour des
températures comprises entre 315 et 370 °C. Il apparaît que la partie réelle du module, graphe a), diminue
avec la fréquence de sollicitation. Ce phénomène est d’autant plus important que la température de
traitement isotherme est élevée. Ce résultat rejoint celui observé précédemment puisque, si sur la figure
4.32, on trace une droite à une température constante, on retrouve d’une part la dépendance en fréquence
du module et d’autre part l’explication de l’amplitude du phénomène en fonction de la température de
traitement isotherme. De la même manière, le graphe b) de la même figure montre le décalage vers les
basses fréquences de la relaxation principale.
A partir des courbes obtenues à différentes fréquences et différentes températures, nous avons pu
reconstruire des courbes maîtresses en utilisant le principe d’équivalence temps-température [Tob1961]. Ce
principe stipule que les fonctions visco-élastiques d’un matériau déterminées à une fréquence et une
température données prennent les mêmes valeurs pour une autre fréquence de sollicitation et une
température appropriée [Sek1992]. Ce principe, très utilisé dans l’étude des propriétés mécaniques des
polymères [Per1988], permet ainsi de prédire le comportement des matériaux pour des fréquences que les
appareils de mesure ne permettent pas d’étudier. Les figures 4.35-36 et 37 montrent les courbes maîtresses
de G’, G’’ et tan δ obtenues dans le Vit4 pour une température de référence de 350 °C. Les mêmes courbes
ont été obtenues dans le Vit4 mais ne sont pas présentées ici.
Figure 4.34 : évolution en fonction de la fréquence de sollicitation des parties imaginaires et réelles du modulepour différentes températures de traitement isotherme comprises entre 315 et 370 °C.
a) b)
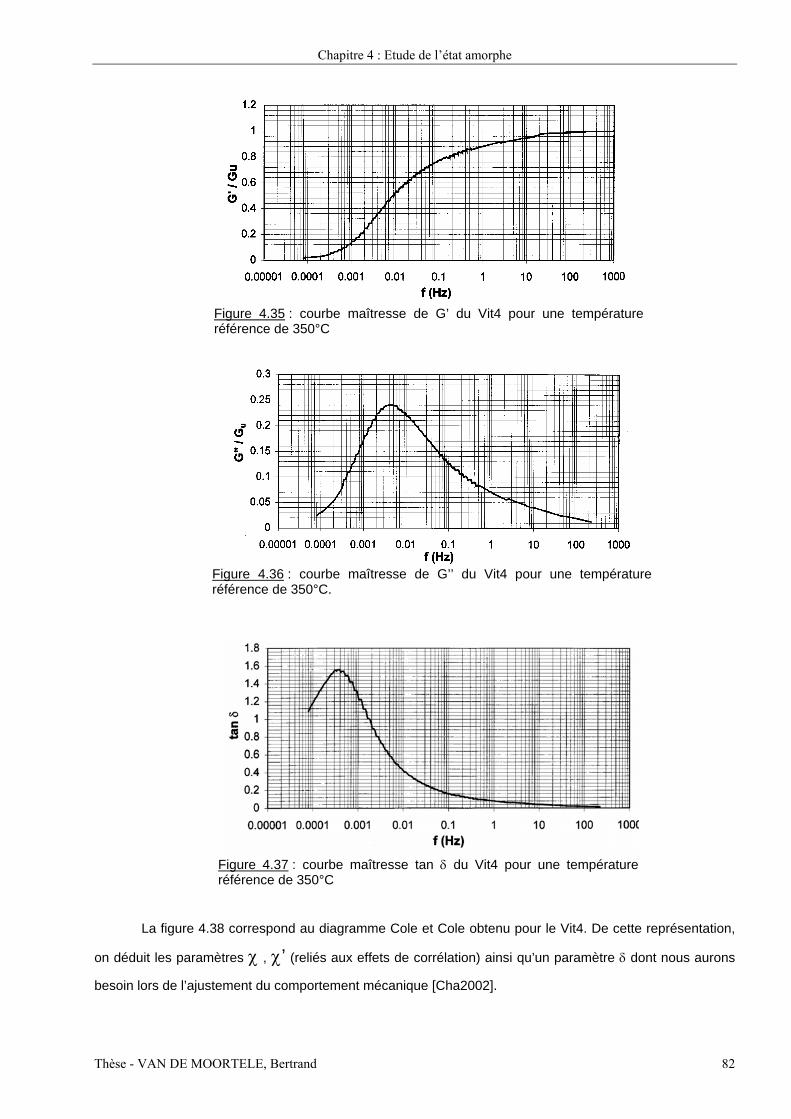
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 82
La figure 4.38 correspond au diagramme Cole et Cole obtenu pour le Vit4. De cette représentation,
on déduit les paramètres χ , χ’ (reliés aux effets de corrélation) ainsi qu’un paramètre δ dont nous aurons
besoin lors de l’ajustement du comportement mécanique [Cha2002].
Figure 4.35 : courbe maîtresse de G’ du Vit4 pour une température référence de 350°C
Figure 4.36 : courbe maîtresse de G’’ du Vit4 pour une température référence de 350°C.
Figure 4.37 : courbe maîtresse tan δ du Vit4 pour une température référence de 350°C

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 83
3.1.4 Rôle du vieillissement physique
De la même manière que lors des mesures de PTE, nous avons étudié les effets réversibles et
irréversibles liés à l’état amorphe du point de vue mécanique. Pour cela, nous avons comparé les courbes
de module obtenues sur un échantillon brut (a) et un échantillon (b) ayant subi un traitement thermique
arrêté à différents stades, refroidi à basse température et à nouveau soumis aux tests mécaniques. Le
traitement thermique est schématisé par la figure 4.39. La vitesse de montée en température a été de 3
°C.min-1 alors que la vitesse de descente a été la plus élevée possible de façon à éviter autant que possible
toute évolution lors de cette descente.
Nous voyons sur la courbe b2 de la figure 4.40 que la valeur du module à basse température a
augmenté de près de 12%. Cette augmentation du module est la conséquence de la modification de la
matrice amorphe, puisque l’échantillon porté à 380 °C n’a pas cristallisé lors du premier passage (courbe
b1). Ensuite, la diminution du module se produit dans le même intervalle de température que lors du
passage b1 pour recouvrer les mêmes valeurs dès 360 °C. Les valeurs du module au-delà de cette
température ne sont pas modifiées par les traitements préalables, puisque l’on retrouve les mêmes résultats
(a)
600Température (°C)
temps
380
430
(b1)
(b2)(b3)
600
temps
Température (°C)
Figure 4.39 : schéma des traitements thermiques pour les échantillons (a) et (b).
Figure 4.38: Diagramme de Cole et Cole pour le Vit4 pour unevitesse de montée en température de 3 °C.min-1 et une fréquencede 1Hz.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 84
que lors du passage a. La courbe b3 montre quant à elle que le module à basse température a légèrement
augmenté et que l’on retrouve par la suite les mêmes valeurs de module.
Comme dans le cas du PTE, nous voyons qu’une partie des modifications induites par la montée en
température est réversible et qu’une autre ne l’est pas. Ainsi, l’augmentation du module après la première
montée jusque 380 °C fait partie des modifications irréversibles. Ensuite, à condition de ne pas aller jusqu’à
la cristallisation, les transformations qui se produisent lors de la montée en température semblent
réversibles.
3.2 Viscoplasticité : essais de compression à chaud (cette partie expérimentale a été réalisée en collaboration avec Qing Wang, Jean-Jacques Blandin et Michel Suery dans le cadre d’un
échange entre la ville chinoise de Shanghai et la région Rhône-Alpes)
La figure 4.41 montre l’évolution de la contrainte en fonction du taux de déformation pour un
échantillon de Vit1 déformé à quatre températures différentes: 346, 355, 364 et 373 °C, à une vitesse de
5.10-4 s-1. Les contraintes d’écoulement sont pour ces quatre températures respectivement de 460, 260, 115
0
100
200
300
400
500
600
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
déformation
cont
rain
te (M
Pa) 346°C
355°C
364°C
373°C
= 5*10-4s-1ε.
Figure 4.41 : courbes contrainte-déformation pour une vitesse de déformation constante et à différentes températures.
Figure 4.40 : mise en évidence des effets réversibles et irréversibles d’un point de vue propriétés mécaniques lors de montées successives en température.
380 °C
430 °C
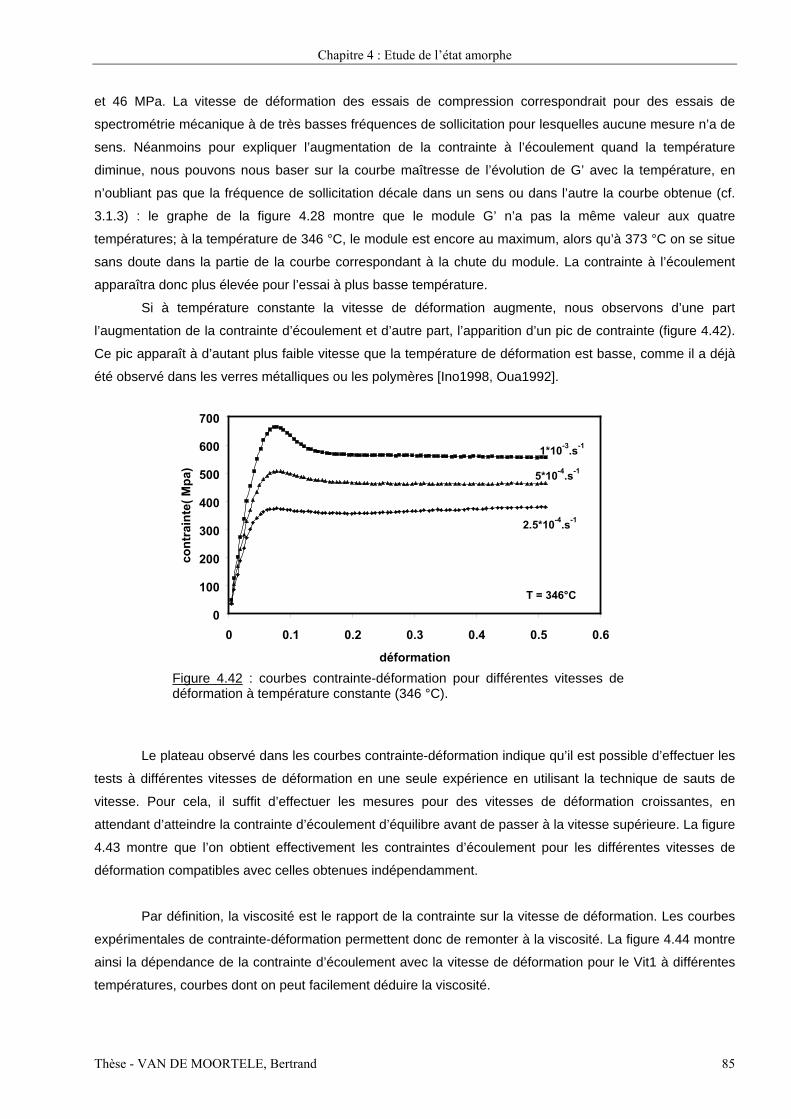
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 85
et 46 MPa. La vitesse de déformation des essais de compression correspondrait pour des essais de
spectrométrie mécanique à de très basses fréquences de sollicitation pour lesquelles aucune mesure n’a de
sens. Néanmoins pour expliquer l’augmentation de la contrainte à l’écoulement quand la température
diminue, nous pouvons nous baser sur la courbe maîtresse de l’évolution de G’ avec la température, en
n’oubliant pas que la fréquence de sollicitation décale dans un sens ou dans l’autre la courbe obtenue (cf.
3.1.3) : le graphe de la figure 4.28 montre que le module G’ n’a pas la même valeur aux quatre
températures; à la température de 346 °C, le module est encore au maximum, alors qu’à 373 °C on se situe
sans doute dans la partie de la courbe correspondant à la chute du module. La contrainte à l’écoulement
apparaîtra donc plus élevée pour l’essai à plus basse température.
Si à température constante la vitesse de déformation augmente, nous observons d’une part
l’augmentation de la contrainte d’écoulement et d’autre part, l’apparition d’un pic de contrainte (figure 4.42).
Ce pic apparaît à d’autant plus faible vitesse que la température de déformation est basse, comme il a déjà
été observé dans les verres métalliques ou les polymères [Ino1998, Oua1992].
Le plateau observé dans les courbes contrainte-déformation indique qu’il est possible d’effectuer les
tests à différentes vitesses de déformation en une seule expérience en utilisant la technique de sauts de
vitesse. Pour cela, il suffit d’effectuer les mesures pour des vitesses de déformation croissantes, en
attendant d’atteindre la contrainte d’écoulement d’équilibre avant de passer à la vitesse supérieure. La figure
4.43 montre que l’on obtient effectivement les contraintes d’écoulement pour les différentes vitesses de
déformation compatibles avec celles obtenues indépendamment.
Par définition, la viscosité est le rapport de la contrainte sur la vitesse de déformation. Les courbes
expérimentales de contrainte-déformation permettent donc de remonter à la viscosité. La figure 4.44 montre
ainsi la dépendance de la contrainte d’écoulement avec la vitesse de déformation pour le Vit1 à différentes
températures, courbes dont on peut facilement déduire la viscosité.
0
100
200
300
400
500
600
700
0 0.1 0.2 0.3 0.4 0.5 0.6
déformation
cont
rain
te( M
pa)
2.5*10-4.s-1
5*10-4.s-1
1*10-3.s-1
T = 346°C
Figure 4.42 : courbes contrainte-déformation pour différentes vitesses dedéformation à température constante (346 °C).

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MO
De la même
température permet
déformation. Nous v
de déformation, ce q
déformation, la visco
Figde de
Ft
0
50
100
150
200
250
300
0 0.05 0.1 0.15 0.2 0.25 0.3 0.35
ε
cont
rain
te(M
Pa)
2.5*10-4s-15*10-4s-1
1*10-3s-1
2.5*10-3s-1
5*10-3s-1
T=373°C
ure 4.43 : courbe contrainte-déformation pour différentes vitessesdéformation expériences de sauts de vitesse à une température373 °Cdans le Vit1.
ORTELE, Bertrand 86
manière qu’en spectrométrie mécanique, l’application du principe d’équivalence temps-
d’obtenir une courbe maîtresse de la viscosité pour une large gamme de vitesse de
oyons, sur la figure 4.45, qu’à faible vitesse la viscosité est indépendante de la vitesse
ui par définition est un comportement Newtonien. En revanche, à plus forte vitesse de
sité devient dépendante de cette vitesse ce qui est caractéristique d’un comportement
igure 4.45 : courbe maîtresse de la viscosité dans le Vit1 pour une
empérature de référence de 373°C (355, 364 et 373°C).
domaine linéaire
domaine non linéaire
10
100
1000
1.E-04 1.E-03 1.E-02vitesse de déformation ( s-1)
cont
rain
te(M
Pa)
355°C
364°C
373°C
Figure 4.44 : dépendance de la contrainte d’écoulement avec lavitesse de déformation à différentes températures.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 87
(4.1)
non-Newtonien. La figure 4.46 est un exemple de l’évolution de la viscosité en fonction de la vitesse de
déformation dans une fibre de verre, autre matériau amorphe. La similitude est frappante.
3.3 Analyse de la réponse mécanique
Comme nous l’avons évoqué dans la section 6 du chapitre 2, de nombreux modèles ont été utilisés
pour décrire le comportement mécanique des verres métalliques. Pour autant aucun d’entre eux ne permet
une description complète du comportement mécanique des verres métalliques pour une large gamme de
températures inférieures ou proches de Tg.
Le modèle de défauts quasi ponctuels développé au sein du laboratoire GEMPPM pour décrire dans un
premier temps le comportement mécanique des polymères a été étendu par la suite aux verres d’oxydes. La
similitude du comportement mécanique des verres métalliques avec les autres matériaux amorphes a
conduit à étendre de nouveau ce modèle à ce type de matériau pour tester ainsi l’universalité des concepts
qui y sont développés. Rappelons brièvement que ce modèle est basé sur trois idées essentielles qui ont été
plus largement évoquées dans le second chapitre et dont on retrouvera le détail dans le chapitre 5 :
il existe dans le matériau des zones de plus forte enthalpie, ce sont les défauts quasi-ponctuels,
la mobilité atomique s’effectue de manière hiérarchiquement corrélée,
l’application d’une contrainte conduit à produire, dans ces zones de plus grande mobilité, un
cisaillement localisé. Ces zones sont appelées des microdomaines cisaillés.
La déformation d’un matériau soumis à une contrainte est la somme de trois contributions : élastique,
viscoélastique et viscoplastique. L’écriture de ces différentes contributions permet d’aboutir à l’expression du
module dynamique sous la forme dite bi-parabolique :
où Gu est le module non relaxé, Gr le module résiduel et δ un paramètre d’ajustement proche de l’unité mais
dépendant de la concentration en défaut Cd. Le paramètre χ est un facteur de corrélation qui vaut 0 lorsque
( ) resres G
)iω)iωδ1G-Giω*G 1
molχ
mol
u+
τ(+τ(+= −−
Figure 4.46 : évolution de la viscosité en fonction de lavitesse de déformation dans une fibre de verre.

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOO
les mouvements sont tous corrélés et 1 lorsqu’il n’existe aucune corrélation entre eux. τmol est la mobilité
moléculaire (équation 2.15).
L’ajustement numérique se fait sur les courbes maîtresses de G’ et G’’ en fonction de la fréquence
de sollicitation pour une température de référence. Les figures 4.47 et 4.48 correspondent à la comparaison
des courbes maîtresses de G’ et G’’ respectivement avec les courbes obtenues par modélisation dans le cas
du Vit4. Outre un bon accord entre la modélisation et les points expérimentaux pour G’ et G’’, nous voyons
que la courbe maîtresse du G’’ ne présente qu’un maximum qui correspond à la relaxation principale.
L’intensité de ce pic, 0.3, indique que les mouvements atomiques lui donnant naissance ne sont pas des
phénomènes simples puisque dans ce cas l’intensité du pic devrait être de 0.5 (dans le cas du modèle
simple de Debye). De plus, la forme dissymétrique du pic montre un étalement important des temps de
relaxation. L’étalement des temps de relaxation peut s’interpréter de manière qualitative d’une part par les
tailles différentes des atomes et d’autre part par l’existence possible d’agrégats de plusieurs atomes liés par
affinité chimique.
Figure 4expérim
Figure 4.47 : comparaison dans le Vit 4 des courbes maîtresses de G’expérimentale et théorique pour une température de référence de 350°C.
0
0.05
0.1
0.15
0.2
0.25
0.3
1.E-05 1.E-04 1.E-03 1.E-02 1.E-01 1.E+00 1.E+01 1.E+02 1.E+03
f (Hz)
G"
/ Gu
points expérimentauxmodélisation
.48 : comparaison dans le Vit 4 des courbes maîtresses de G’’
entale et théorique pour une température de référence de 350°C.
RTELE, Bertrand 88

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 89
Le tableau 4.5 résume les différentes valeurs pour le paramètre de corrélation et l’énergie d’activation des
mouvements élémentaires déduits des courbes expérimentales et de la modélisation pour les différents
verres métalliques. La comparaison avec un verre non fragile, la silice, et un verre organique fragile, le o-
terphenyl, montre le caractère intermédiaire des verres métalliques.
alliage Vit1 Vit4 Pd SiO2 o-terphenyl χ 0.41 0.4 0.4 0.5 0.2
Uβ(eV) 0.68 0.7 1.1 2 0.4 Tableau 4.5 : ensemble de paramètres déduits des courbes expérimentales et de la modélisation pour les différents alliages (d’après [Per2001] ).
Les figures 4.49 et 4.50 sont des courbes maîtresses obtenues pour d’autres matériaux amorphes : le
PMMA et un verre silico-sodo-calcique. Nous voyons avec ces deux exemples que la relaxation principale
ou alpha est commune à l’ensemble des matériaux amorphes.
Par contre, nous remarquons qu’alors que dans le PMMA, la relaxation principale est précédée
d’une relaxation secondaire ou béta, dans le Vit1 et le verre silico-sodo-calcique aucune relaxation de ce
type n’est visible. Néanmoins, la figure 4.51 montre que cette relaxation secondaire existe dans l’alliage
Figure 4.50 : courbe maîtresse de la partie réelle du module dans un verre silico-sodo-calcique [Se1992].
Figure 4.49 : courbes maîtresses expérimentaleet simulée de G’’ dans le PMMA [Cha2002].
0.0E+00
2.0E+09
4.0E+09
6.0E+09
8.0E+09
1.0E+10
1.2E+10
1.4E+10
50 100 150 200 250 300 350T (°C)
Figure 4.51 : évolution de G’’ dans l'alliage Pd lors d'une montée en température à 1 °C.min-1 ; pour différentes fréquences comprises entre 0,01 et 1 Hz
relaxation α
relaxation β

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 90
(4.3)
(4.3)
(4.5)
amorphe PdNiCuP. Inoue et al., par l’étude de l’évolution de la fonction de distribution radiale par diffusion
des rayons X, attribue cette relaxation à un réarrangement de l’ordre local autour des atomes de phosphore
(seul élément non métallique de cet alliage Pd-Ni-Cu-P). Le volume concerné par cette relaxation est bien
plus faible que dans le cas de la relaxation principale ce qui explique qu’elle se produit à plus basse
température.
Enfin dans le cas des verres métalliques, il n’est pas possible de se prononcer sur l’existence d’un
module résiduel du fait de la cristallisation qui se traduit par la remontée du module G’.
A présent, nous allons analyser les courbes expérimentales de compression et en particulier de
l’écoulement. Il est généralement admis que la contrainte d’écoulement est associée à la propagation de
défauts de cisaillement. Si l’on admet que cette propagation peut être décrite par un mécanisme unique,
thermiquement activé, un simple modèle à deux puits, proposé par Eyring en 1936 [Eyr1936] conduit à
l’expression suivante :
où σa est la contrainte au seuil d'écoulement. Les dérivées partielles de cette expression permettent
d'aboutir au volume d'activation apparent (Va) et à l'énergie d'activation apparente (∆Ha) qui sont :
Les valeurs déterminées expérimentalement par ce modèle à deux puits sont : Va=50Å3 et ∆Ha=4.9eV.
Le modèle de Perez et al. introduit, comme nous l’avons vu, la notion de microdomaines cisaillés; la
contrainte d’écoulement est alors associée à un processus d’équilibre dynamique entre la création et la
propagation de ces domaines et leur coalescence. Un modèle a été développé pour rendre compte de ces
phénomènes [Cha2002]. Retenons simplement que la contrainte réduit le temps caractéristique de la
mobilité moléculaire. Elle augmente également le paramètre de corrélation χ. On aboutit à l’expression du
volume d’activation qui est la suivante :
où Uβ est l'énergie d'activation de la relaxation secondaire, χ est le paramètre de corrélation et σ0 est la
contrainte qu'il serait nécessaire d'appliquer pour que les mouvements moléculaires ou atomiques
élémentaires se produisent sans apport d'énergie thermique, à température nulle.
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛ σ∆−σε=ε
kT)T,(Gexp)T,( a
ap 0&&
structure,Ta
pa
lnkTV ⎟⎠⎞
⎜⎝⎛
σ∂ε∂
=&
structure,pa
aT
TH ε⎟⎠⎞
⎜⎝⎛
∂σ∂
−=∆ &
⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜
⎝
⎛
χσ
⎟⎠⎞
⎜⎝⎛
σσ
−= β
0
21
0a
2
1U3V
et (4.4)

Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE M
Par ce modèle, nous obtenons une valeur de volume d'activation de : Va=80 Å3, c'est-à-dire en assez bon
accord avec le résultat obtenu par le modèle de Eyring (50 Å3).
Alors que, comme le montre le tableau 4.6, le volume d'activation dans les polymères varie de 300 à
1000 Å 3, il n'est que de 80 Å3 dans le Vit1. Cette différence s'explique principalement par la taille des
structures mises en jeu lors de la déformation (chaînes de polymères dans un cas et structure de quelques
atomes dans le cas des verres métalliques).
Matériaux PMMA S1M3 S2M2 S3M1 PS
Va exp(Å3) 220 330 410 530 940
Tableau 4.6 : volume d’activation déterminé pour différents polymères (d’après [Per2001] ).
Pour décrire le comportement non-linéaire (cas de déformation en compression,), contrairement à la
spectrométrie mécanique, où n’interviennent que des contraintes faibles (régime linéaire), il n’est plus
possible d’établir des expressions analytiques simples et seul un modèle numérique est envisageable. Dans
le cas des polymères, cette modélisation a pu être menée à terme grâce aux nombreuses études
entreprises notamment du point de vue expérimental. Dans notre cas, seules des tendances ont été
dégagées et nous n’avons réalisé qu’une première approche de description qualitative des courbes à partir
de ce modèle.
Les figures 4.52 et 4.53 sont des courbes contrainte-déformation du Vit1 simulées. Nous voyons que
l'allure générale est proche des courbes obtenues expérimentalement (4.42 et 4.43). La déformation par
compression induit des phénomènes non linéaires.
Comme n
semblable aux au
l’ensemble des a
ce pic de relaxa
associés à cette
Figu 364°
re 4.52 : courbe simulée de contrainte-déformation pour une température fixe deC et à différentes vitesses de déformation comprises entre 0.25 et 5.10-3s-1.
OORTELE, Bertrand 91
ous venons de le voir, les verres métalliques ont un comportement à la déformation
tres matériaux amorphes. Une relaxation principale, α, a pu être mise en évidence dans
lliages étudiés. L’énergie d’activation apparente de cette relaxation, la valeur maximale de
tion et sa forme dissymétrique tendent à démontrer que les mouvements atomiques
relaxation d’une part sont hiérarchiquement corrélés et d’autre part mettent probablement
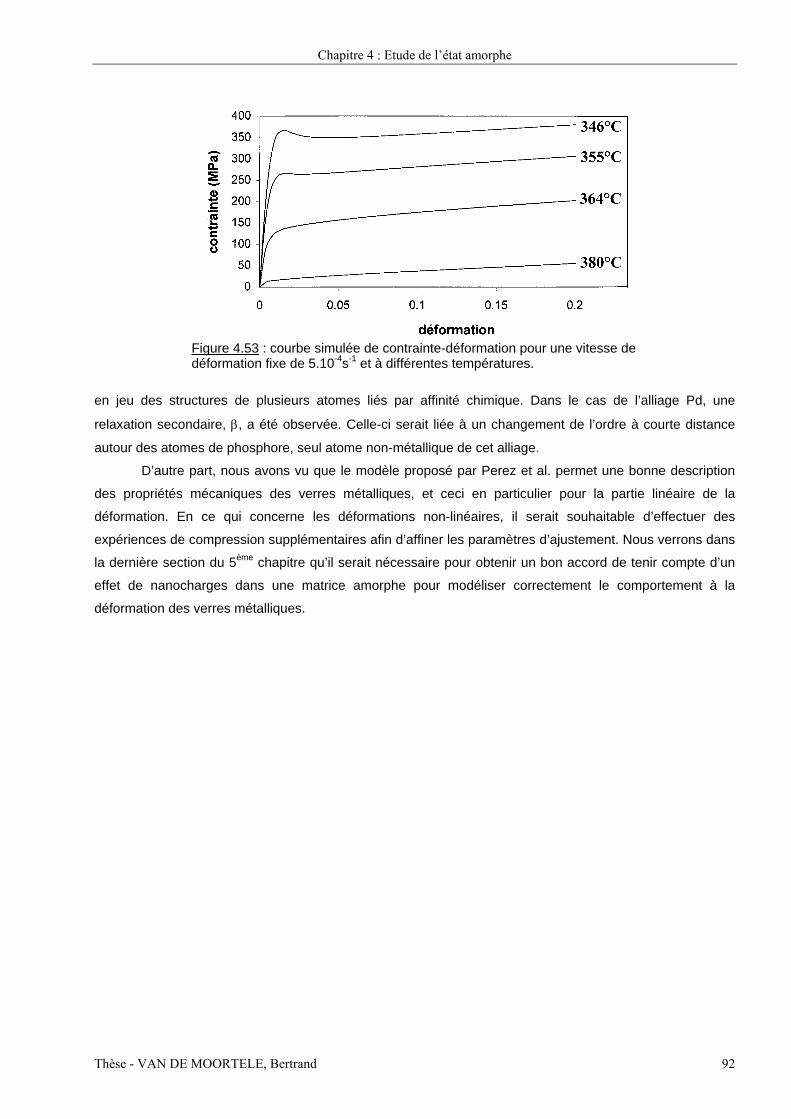
Chapitre 4 : Etude de l’état amorphe
Thèse - VAN DE MOORTELE, Bertrand 92
en jeu des structures de plusieurs atomes liés par affinité chimique. Dans le cas de l’alliage Pd, une
relaxation secondaire, β, a été observée. Celle-ci serait liée à un changement de l’ordre à courte distance
autour des atomes de phosphore, seul atome non-métallique de cet alliage.
D’autre part, nous avons vu que le modèle proposé par Perez et al. permet une bonne description
des propriétés mécaniques des verres métalliques, et ceci en particulier pour la partie linéaire de la
déformation. En ce qui concerne les déformations non-linéaires, il serait souhaitable d’effectuer des
expériences de compression supplémentaires afin d’affiner les paramètres d’ajustement. Nous verrons dans
la dernière section du 5ème chapitre qu’il serait nécessaire pour obtenir un bon accord de tenir compte d’un
effet de nanocharges dans une matrice amorphe pour modéliser correctement le comportement à la
déformation des verres métalliques.
Figure 4.53 : courbe simulée de contrainte-déformation pour une vitesse de déformation fixe de 5.10-4s-1 et à différentes températures.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 93
Chapitre 5
Décomposition et Cristallisation
Chapitre 5 ____________________________________________________________________93
Décomposition et Cristallisation __________________________________________________94 1 Mise en évidence par DSC et PTE_____________________________________________________94
2 Analyse de la microstructure_________________________________________________________99 2.1 Vit1 : Isotherme à 410°C _______________________________________________________________ 100
2.1.1. Traitement 5 minutes________________________________________________________________ 100 2.1.2. Traitement 7 minutes________________________________________________________________ 100 2.1.3. Traitement ‘intermédiaire’ à 9 minutes __________________________________________________ 104 2.1.4. Traitements ‘longs’ de 25 et 60 minutes _________________________________________________ 105 2.1.5. Bilan et Interprétation _______________________________________________________________ 110
2.2 Vit4 _________________________________________________________________________________ 114 2..2.1. Traitement isochrone _______________________________________________________________ 114 2.2.2. Traitement isotherme________________________________________________________________ 115
2.2.2.1 observation de la microstructure ____________________________________________________ 115 2.2.2.2 analyse cristallographique_________________________________________________________ 116
précipités A ________________________________________________________________________ 116 précipités B ________________________________________________________________________ 117
2.2.3. Observation in-situ de la cristallisation du Vit4. ___________________________________________ 121 2.2.4. Interprétation______________________________________________________________________ 123
4 Conséquences sur les propriétés mécaniques___________________________________________126 4.1 cas du Vit1 ___________________________________________________________________________ 126 4.2 Cas du Vit4 __________________________________________________________________________ 128

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN
Décomposition et Cristallisation
Dans ce chapitre nous allons aborder la décomposition et la cristallisation des alliages Vit1 et Vit4.
Dans un premier temps, nous mettrons en évidence les évolutions de la matrice amorphe par des mesures
des propriétés physiques (DSC et PTE). Ceci nous permettra de choisir une température de traitement
thermique et des temps caractéristiques de l’évolution des matériaux. Une large partie de ce chapitre sera
consacrée à l’étude de l’évolution microstructurale des alliages Vit1 et Vit4. La microscopie électronique
couplée aux techniques d’analyses chimiques nous permettra de proposer le scénario d’un processus de
décomposition de la matrice amorphe suivie d’une cristallisation dans le cas du Vit1. En ce qui concerne le
Vit4, nous nous attacherons plus particulièrement à l’évolution du matériau lors d’un traitement thermique
proche de la température de transition vitreuse. Enfin, nous reviendrons sur l’évolution des propriétés
mécaniques dans les alliages Vit1 et Vit4 en reliant les observations microstructurales aux modifications et
évolutions du comportement mécanique étudiées dans le chapitre précédent.
1 Mise en évidence par DSC et PTE
1.1 DSC
Nous avons vu dans le chapitre 4, que la transition vitreuse était suivie d’une zone de liquide
surfondu (correspond à la région grisée de la figure 5.1). La partie claire de la figure 5.1 correspond au
thermogramme obtenu sur un échantillon de Vit1 pour une montée isochrone de 10 °C.min-1 présentant la fin
de la zone de liquide surfondue, suivie de la cristallisation. Dans le cas du Vit1 la cristallisation s’effectue en
deux étapes distinctes, bien qu’à cette vitesse les pics soient proches. Nous avons déterminé les débuts de
cristallisation des deux pics à 427 °C et 458 °C. Le premier pic de cristallisation est assez large et semble
Figmosu
0
0.2
0.4
0.6
0.8
1
400 420 440 460 480T°C
Cp
(u.a
.)
ure 5.1 : thermogramme de DSC du Vit1 pour une vitesse de balayage de 10 °C.min-1,ntrant les deux pics de cristallisation. La zone grisée correspond à la zone de liquide
rfondu étudiée dans le chapitre 4.
DE MOORTELE, Bertrand 94
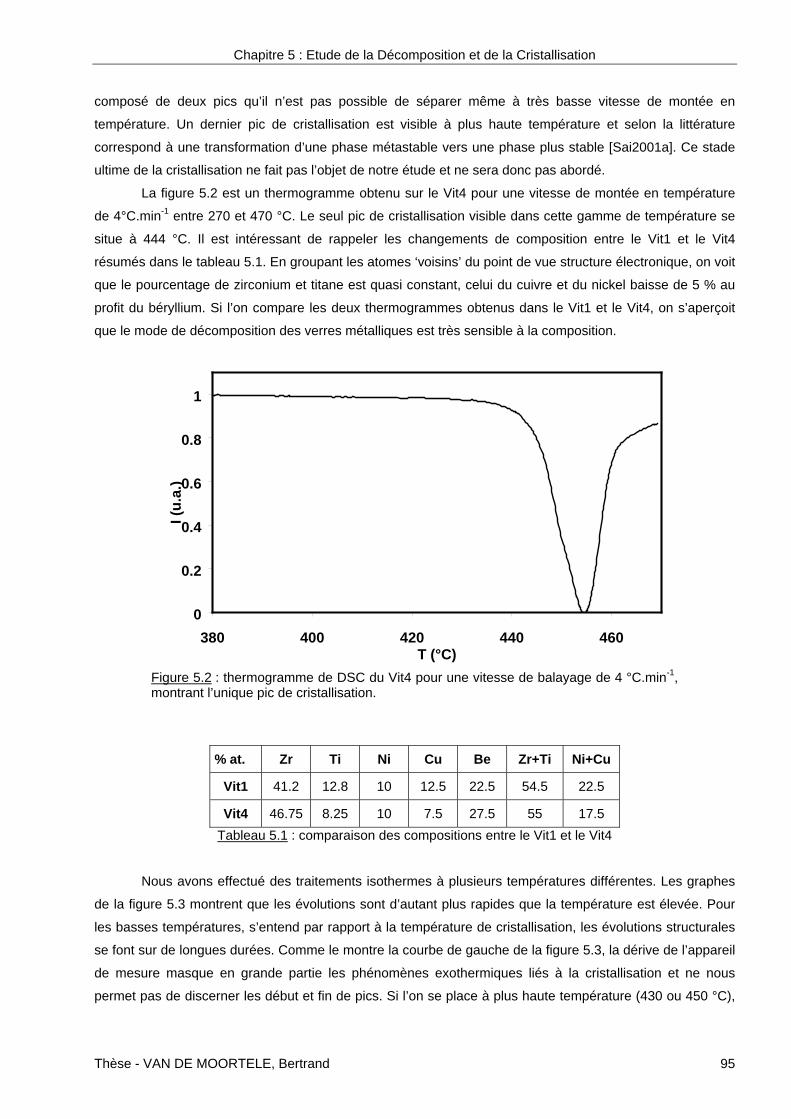
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 95
composé de deux pics qu’il n’est pas possible de séparer même à très basse vitesse de montée en
température. Un dernier pic de cristallisation est visible à plus haute température et selon la littérature
correspond à une transformation d’une phase métastable vers une phase plus stable [Sai2001a]. Ce stade
ultime de la cristallisation ne fait pas l’objet de notre étude et ne sera donc pas abordé.
La figure 5.2 est un thermogramme obtenu sur le Vit4 pour une vitesse de montée en température
de 4°C.min-1 entre 270 et 470 °C. Le seul pic de cristallisation visible dans cette gamme de température se
situe à 444 °C. Il est intéressant de rappeler les changements de composition entre le Vit1 et le Vit4
résumés dans le tableau 5.1. En groupant les atomes ‘voisins’ du point de vue structure électronique, on voit
que le pourcentage de zirconium et titane est quasi constant, celui du cuivre et du nickel baisse de 5 % au
profit du béryllium. Si l’on compare les deux thermogrammes obtenus dans le Vit1 et le Vit4, on s’aperçoit
que le mode de décomposition des verres métalliques est très sensible à la composition.
% at. Zr Ti Ni Cu Be Zr+Ti Ni+Cu
Vit1 41.2 12.8 10 12.5 22.5 54.5 22.5
Vit4 46.75 8.25 10 7.5 27.5 55 17.5 Tableau 5.1 : comparaison des compositions entre le Vit1 et le Vit4
Nous avons effectué des traitements isothermes à plusieurs températures différentes. Les graphes
de la figure 5.3 montrent que les évolutions sont d’autant plus rapides que la température est élevée. Pour
les basses températures, s’entend par rapport à la température de cristallisation, les évolutions structurales
se font sur de longues durées. Comme le montre la courbe de gauche de la figure 5.3, la dérive de l’appareil
de mesure masque en grande partie les phénomènes exothermiques liés à la cristallisation et ne nous
permet pas de discerner les début et fin de pics. Si l’on se place à plus haute température (430 ou 450 °C),
0
0.2
0.4
0.6
0.8
1
380 400 420 440 460T (°C)
I (u.
a.)
Figure 5.2 : thermogramme de DSC du Vit4 pour une vitesse de balayage de 4 °C.min-1,montrant l’unique pic de cristallisation.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN D
les deux stades de cristallisation qui avaient déjà été vus en isochrone, sont bien visibles mais très
rapprochés dans le temps.
Aussi la température de 410 °C, pour effectuer les traitements thermiques et suivre ainsi l’évolution
du Vit1 en fonction du temps, nous a semblé être un bon compromis entre des phénomènes suffisamment
marqués en DSC pour dégager des points particuliers de l’évolution et des phénomènes suffisamment lents
pour piloter correctement les temps de recuit. La figure 5.4 correspond au thermogramme obtenu en
isotherme à 410 °C. Nous avons indiqué les points pour lesquels nous avons caractérisé la microstructure
par MET et diffraction des rayons X que nous verrons dans la troisième section de ce chapitre.
Figureéchanmicro
1 10 100 1000temps (min)
Cp
(u.a
.)
380°C
1 10 100temps (min)
Cp
(u.a
.)
450°
Figure 5.3 : thermogramme de DSC en isotherme sur des échantillons de Vit1, montrant l’influence de la température sur la cinétique de cristallisation. Le choix de la température de traitement isotherme quenous effectuerons par la suite sera un compromis entre ces deux situations à savoir une températured’une part suffisamment élevée pour avoir des phénomènes marqués en DSC et d’autre partsuffisamment basse pour pouvoir contrôler correctement les temps de traitements.
0123456789
10
1 10 100temps (min)
Cp
(u.a
.)
5 min
12 min9 min
7 min
25 min
5.4 : thermogramme des DSC obtenu durant une isotherme à 410 °C sur untillon brut de Vit1. Les temps indiqués sur ce graphe correspondent aux étatsstructuraux que nous caractériserons en microscopie électronique.
E MOORTELE, Bertrand 96
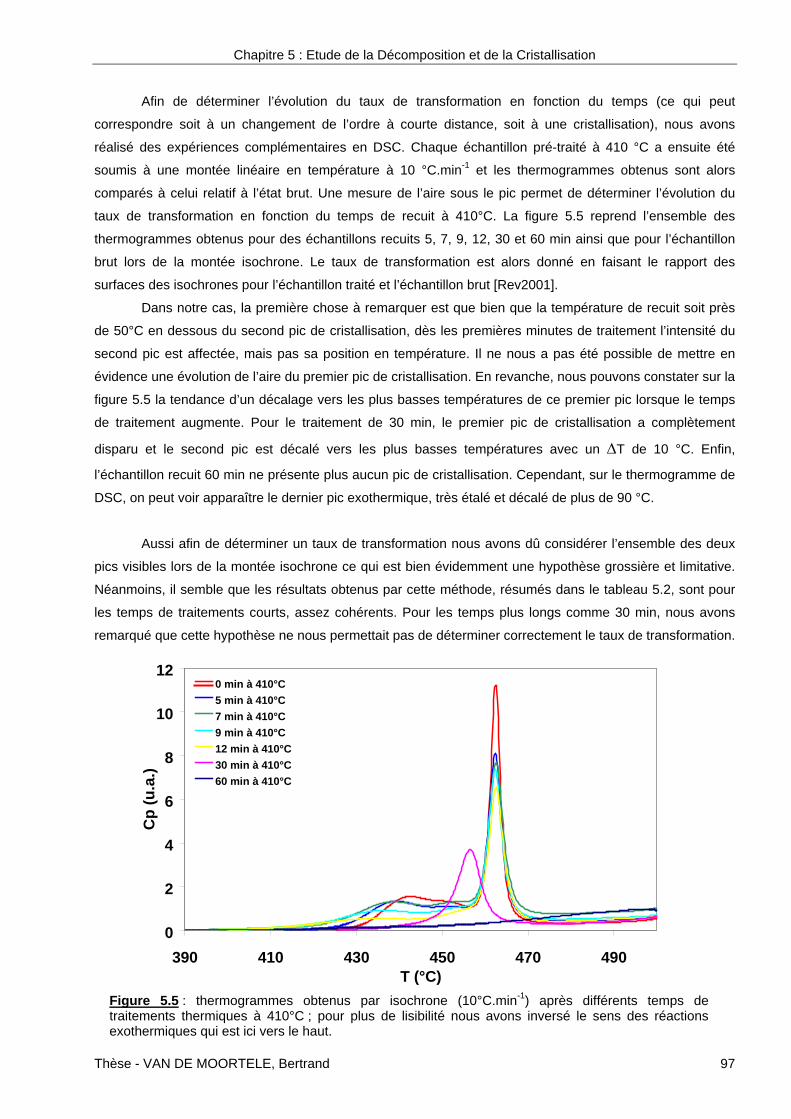
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse
Afin de déterminer l’évolution du taux de transformation en fonction du temps (ce qui peut
correspondre soit à un changement de l’ordre à courte distance, soit à une cristallisation), nous avons
réalisé des expériences complémentaires en DSC. Chaque échantillon pré-traité à 410 °C a ensuite été
soumis à une montée linéaire en température à 10 °C.min-1 et les thermogrammes obtenus sont alors
comparés à celui relatif à l’état brut. Une mesure de l’aire sous le pic permet de déterminer l’évolution du
taux de transformation en fonction du temps de recuit à 410°C. La figure 5.5 reprend l’ensemble des
thermogrammes obtenus pour des échantillons recuits 5, 7, 9, 12, 30 et 60 min ainsi que pour l’échantillon
brut lors de la montée isochrone. Le taux de transformation est alors donné en faisant le rapport des
surfaces des isochrones pour l’échantillon traité et l’échantillon brut [Rev2001].
Dans notre cas, la première chose à remarquer est que bien que la température de recuit soit près
de 50°C en dessous du second pic de cristallisation, dès les premières minutes de traitement l’intensité du
second pic est affectée, mais pas sa position en température. Il ne nous a pas été possible de mettre en
évidence une évolution de l’aire du premier pic de cristallisation. En revanche, nous pouvons constater sur la
figure 5.5 la tendance d’un décalage vers les plus basses températures de ce premier pic lorsque le temps
de traitement augmente. Pour le traitement de 30 min, le premier pic de cristallisation a complètement
disparu et le second pic est décalé vers les plus basses températures avec un ∆T de 10 °C. Enfin,
l’échantillon recuit 60 min ne présente plus aucun pic de cristallisation. Cependant, sur le thermogramme de
DSC, on peut voir apparaître le dernier pic exothermique, très étalé et décalé de plus de 90 °C.
Aussi afin de déterminer un taux de transformation nous avons dû considérer l’ensemble des deux
pics visibles lors de la montée isochrone ce qui est bien évidemment une hypothèse grossière et limitative.
Néanmoins, il semble que les résultats obtenus par cette méthode, résumés dans le tableau 5.2, sont pour
les temps de traitements courts, assez cohérents. Pour les temps plus longs comme 30 min, nous avons
remarqué que cette hypothèse ne nous permettait pas de déterminer correctement le taux de transformation.
Figutraiteexot
0
2
4
6
8
10
12
390 410 430 450 470 490T (°C)
Cp
(u.a
.)
0 min à 410°C5 min à 410°C7 min à 410°C9 min à 410°C12 min à 410°C30 min à 410°C60 min à 410°C
re 5.5 : thermogrammes obtenus par isochrone (10°C.min-1) après différents temps dements thermiques à 410°C ; pour plus de lisibilité nous avons inversé le sens des réactions
hermiques qui est ici vers le haut.
- VAN DE MOORTELE, Bertrand 97
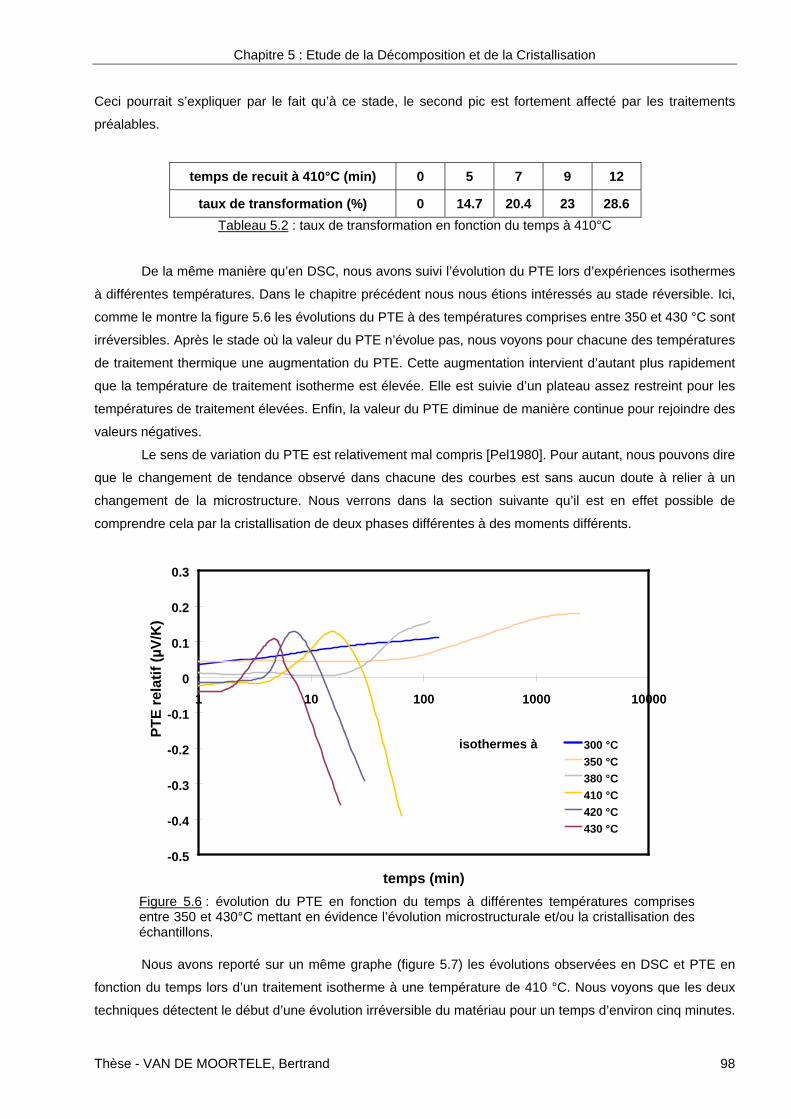
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse -
Ceci pourrait s’expliquer par le fait qu’à ce stade, le second pic est fortement affecté par les traitements
préalables.
temps de recuit à 410°C (min) 0 5 7 9 12
taux de transformation (%) 0 14.7 20.4 23 28.6 Tableau 5.2 : taux de transformation en fonction du temps à 410°C
De la même manière qu’en DSC, nous avons suivi l’évolution du PTE lors d’expériences isothermes
à différentes températures. Dans le chapitre précédent nous nous étions intéressés au stade réversible. Ici,
comme le montre la figure 5.6 les évolutions du PTE à des températures comprises entre 350 et 430 °C sont
irréversibles. Après le stade où la valeur du PTE n’évolue pas, nous voyons pour chacune des températures
de traitement thermique une augmentation du PTE. Cette augmentation intervient d’autant plus rapidement
que la température de traitement isotherme est élevée. Elle est suivie d’un plateau assez restreint pour les
températures de traitement élevées. Enfin, la valeur du PTE diminue de manière continue pour rejoindre des
valeurs négatives.
Le sens de variation du PTE est relativement mal compris [Pel1980]. Pour autant, nous pouvons dire
que le changement de tendance observé dans chacune des courbes est sans aucun doute à relier à un
changement de la microstructure. Nous verrons dans la section suivante qu’il est en effet possible de
comprendre cela par la cristallisation de deux phases différentes à des moments différents.
fonction
techniqu
Feé
-0.5
-0.4
-0.3
-0.2
-0.1
0
0.1
0.2
0.3
1 10 100 1000 10000
temps (min)
PTE
rela
tif (µ
V/K
)
300 °C350 °C380 °C410 °C420 °C430 °C
isothermes à
igure 5.6 : évolution du PTE en fonction du temps à différentes températures comprisesntre 350 et 430°C mettant en évidence l’évolution microstructurale et/ou la cristallisation deschantillons.
VAN DE MOORTELE, Bertrand 98
Nous avons reporté sur un même graphe (figure 5.7) les évolutions observées en DSC et PTE en
du temps lors d’un traitement isotherme à une température de 410 °C. Nous voyons que les deux
es détectent le début d’une évolution irréversible du matériau pour un temps d’environ cinq minutes.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 99
Par contre, le début d’une forte diminution du PTE intervient alors que la fin du premier pic de DSC n’est pas
encore atteinte. Nous verrons dans la section suivante que ce premier pic de DSC pourrait correspondre à la
cristallisation de différentes phases cristallines que la DSC, contrairement au PTE, ne pourrait discriminer.
Les mesures effectuées en DSC et PTE nous ont permis de sélectionner une température de
traitement et des temps qui correspondent à des évolutions importantes. Nous allons voir dans la section
suivante à quoi correspondent d’un point de vue microstructural ces évolutions.
2 Analyse de la microstructure
Cette section sera séparée en deux sous sections bien distinctes dans lesquelles interviendront les
caractérisations faites aussi bien par diffraction X que par microscopie électronique, une technique
complétant l’autre. Dans un premier temps nous nous intéresserons au cas du Vit1. L’évolution du PTE très
marquée aux temps longs (i.e. au-delà de 20 minutes) nous a naturellement incité à étudier les états
microstructuraux correspondants à ces temps longs. Néanmoins, il nous est apparu nécessaire de procéder
chronologiquement en entamant logiquement l’étude microstructurale par les temps les plus courts.
Dans un second temps nous verrons que la microstructure observée dans le Vit4 est fortement
dépendante des traitements thermiques appliqués à cet alliage. Pour cela nous étudierons assez rapidement
un échantillon ayant subi une montée isochrone puis nous nous consacrerons bien plus longuement à
l’étude des phases cristallines présentes dans un échantillon ayant subi un traitement à température
constante.
500
700
900
1100
1300
1500
1 10 100time (min)
Cp
(u.a
.)
-0.5
-0.4
-0.3
-0.2
-0.1
0
0.1
0.2
PTE
(µV/
K)
410°C
1
4
3
2DSC
PTE
Figure 5.7 : évolution comparée par DSC et PTE d’un échantillon de Vit1 lors d’un traitementthermique isotherme à 410°C

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 100
2.1 Vit1 : Isotherme à 410°C
2.1.1. Traitement 5 minutes
La DSC et le PTE indiquent qu’après cinq minutes à 410°C, les propriétés physiques commencent à
changer. Pour autant, à ce stade, la diffraction X ne permet pas de voir d’évolution du matériau. Aussi
avons-nous effectué des observations sur la microstructure par MET. La figure 5.10 est une diffraction
obtenue sur cet état de traitement thermique. Seuls les anneaux amorphes liés à la diffusion moyenne sont
visibles et aucun affinement de ceux-ci n’a pu être mis en évidence. La figure 5.11 est une image en champ
clair sur le même échantillon où aucun contraste de diffraction n’est visible. De la même manière l’imagerie
haute résolution ne nous a pas permis d’observer de cristallisation.
2.1.2. Traitement 7 minutes
La durée de traitement de sept minutes correspond à l’amorçage du premier pic de cristallisation lors
d’une isotherme à 410°C. Les observations effectuées à ce stade de développement révélent la présence
d’une très fine cristallisation, c’est-à-dire inférieure à 5 nm. Une diffraction électronique faite sur une large
zone nano-cristallisée se présente sous la forme d’un cliché de Debye-Scherrer fortement ponctué (figure
5.12).
De manière surprenante, chacun de ces anneaux a pu être indexé dans une structure simple de type
c.f.c., dont le paramètre est a ≈ 0.404 nm très proche de la structure identifiée par Schneider et al. [Sch1996]
lors d’un traitement isotherme à 350 °C.
L’imagerie à Haute Résolution complète cette observation : la figure 5.13 montre une première zone
où plusieurs nano-cristaux d’une taille d’environ 3 à 4 nm sont visibles. Le diffractogramme numérique
indique que la structure de ces particules est compatible avec l’hypothèse ‘c.f.c.’ précédente, puisque les
taches de diffraction sont réparties sur 2 anneaux correspondant aux distances d111 (0.23 nm) et d200 (0.2
nm) respectivement.
Figure 5.11 : image en champ clair sur unéchantillon de Vit1 traité 5min à 410 °C.Aucun contraste de diffraction n'est visible.
0.5 µm
Figure 5.10 : diffraction en aire sélectionnéesur un échantillon de Vit1 recuit 5 min à410°C.
4 nm-1
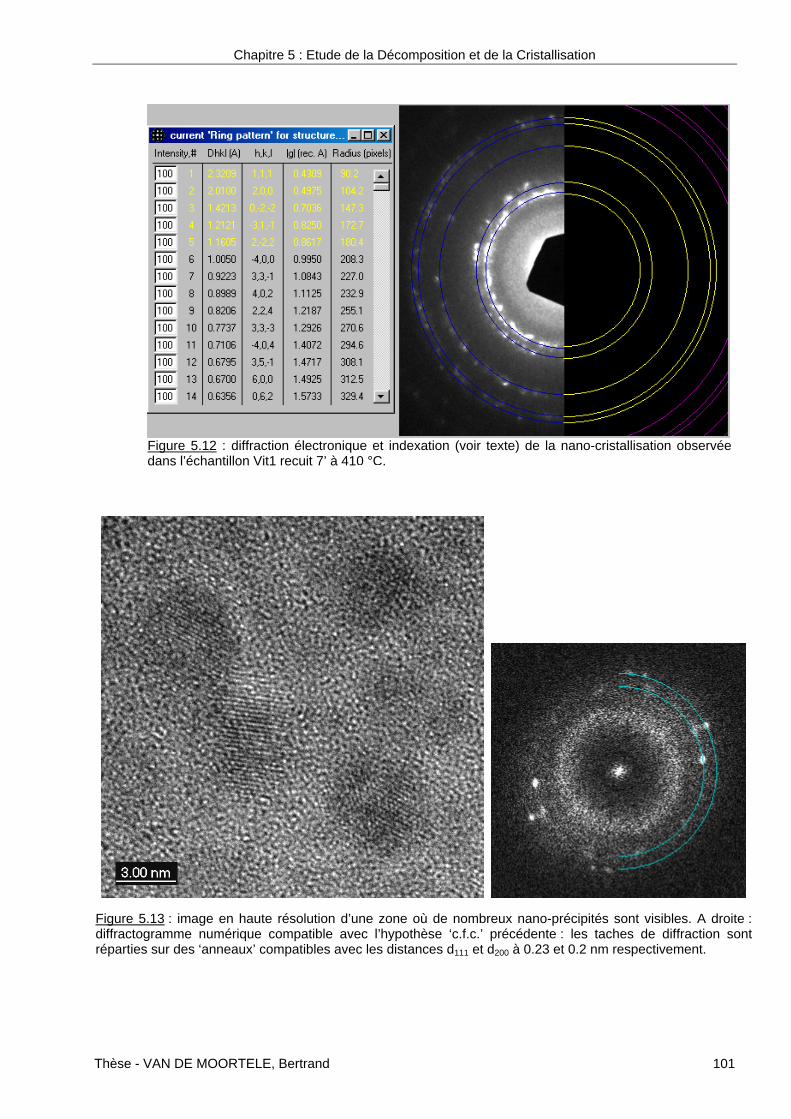
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - V
Fd
Figure 5.diffractogréparties
igure 5.12 : diffraction électronique et indexation (voir texte) de la nano-cristallisation observéeans l’échantillon Vit1 recuit 7’ à 410 °C.
AN DE MOORTELE, Bertrand 101
13 : image en haute résolution d’une zone où de nombreux nano-précipités sont visibles. A droite :ramme numérique compatible avec l’hypothèse ‘c.f.c.’ précédente : les taches de diffraction sontsur des ‘anneaux’ compatibles avec les distances d111 et d200 à 0.23 et 0.2 nm respectivement.
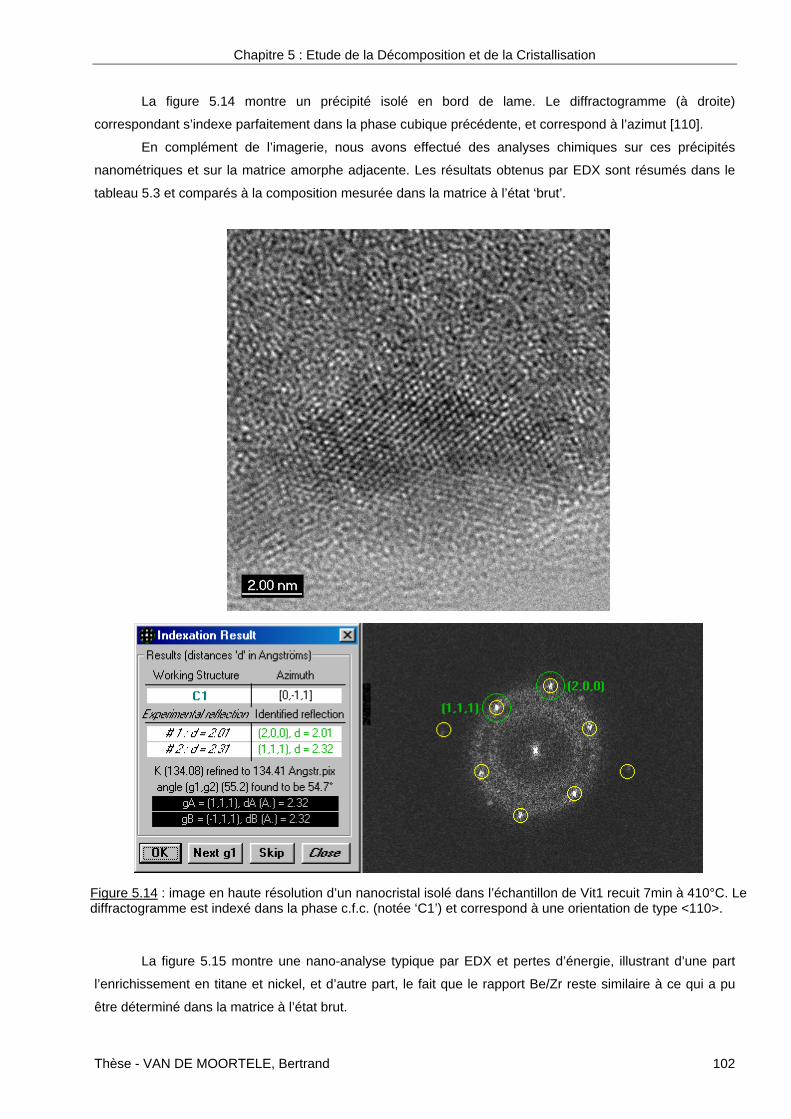
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 102
La figure 5.14 montre un précipité isolé en bord de lame. Le diffractogramme (à droite)
correspondant s’indexe parfaitement dans la phase cubique précédente, et correspond à l’azimut [110].
En complément de l’imagerie, nous avons effectué des analyses chimiques sur ces précipités
nanométriques et sur la matrice amorphe adjacente. Les résultats obtenus par EDX sont résumés dans le
tableau 5.3 et comparés à la composition mesurée dans la matrice à l’état ‘brut’.
La figure 5.15 montre une nano-analyse typique par EDX et pertes d’énergie, illustrant d’une part
l’enrichissement en titane et nickel, et d’autre part, le fait que le rapport Be/Zr reste similaire à ce qui a pu
être déterminé dans la matrice à l’état brut.
Figure 5.14 : image en haute résolution d’un nanocristal isolé dans l’échantillon de Vit1 recuit 7min à 410°C. Lediffractogramme est indexé dans la phase c.f.c. (notée ‘C1’) et correspond à une orientation de type <110>.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 103
Les analyses de composition de la matrice amorphe sont troublantes dans le sens où elles sous-
évaluent systématiquement le titane au profit du zirconium (et du nickel) par rapport à la composition de
départ. Notons ici que les zones d’analyse ‘de matrice’ étaient assez larges (environ 100 nm) pour admettre
que s’il existait des fluctuations, celles-ci seraient moyennées. Par la suite, nous donnerons des tendances
d’enrichissement relatif sans être quantitatif dans l’absolu.
% atomique Zr Ti Ni Cu
Particule 1 52.3 18.0 13.2 16.5
Particule 2 53.4 16.5 14.6 15.5
Particule 3 55.6 15.4 13.7 15.3
Particule 4 55.8 16.8 12.5 14.9
Moyenne 54.3 16.65 13.5 15.55
Matrice 1 58.0 12.6 12.9 16.5
Matrice 2 58.4 13.1 12.8 15.7
Matrice 3 58.5 13.0 12.1 16.4
Matrice 4 60.3 12.7 11.8 15.2
Moyenne Matrice 58.8 12.85 12.4 15.95
Compo. ‘brute’ 53.6 15.8 14.5 16.1
Tableau 5.3 : composition par analyse EDX de différentes zones cristallisées après 7’ à 410 °C.
Figure 5. 15 : analyse chimique typique d’un nano-précipité de phase c.f.c. après recuit de 7’ à 410 °C(sonde 2 nm). A gauche ; spectre EDX (aplat clair) comparé à un spectre typique de la matrice à l’état brut,montrant l’enrichissement en Ti et Ni. A droite : analyse EELS du rapport Be/Zr ; notez que le rapport Be/Zr(0.51), déduit d’un ajustement du spectre expérimental avec les spectres de référence normalisés Be-Kref etZr-Mref, est proche de celui de la matrice à l’état brut (0.59, cf. fig. 4.12).
Be/Zr = 0.51

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 104
On peut donc voir que les premiers cristaux qui se forment dans le Vit1 lors d’un traitement
thermique à 410°C semblent enrichis en titane et appauvris en zirconium, et ceci à l’encontre de ce qui a été
rapporté dans la littérature. En effet, la première cristallisation attendue dans les verres métalliques à base
zirconium est du type (Zr,Ti)2(Ni,Cu) [Jia2000, Rev2001]. Nous nous attendions donc à observer les mêmes
tendances de variation de composition de Zr et Ti, mais nous observons exactement l’inverse. Par contre,
nous pouvons mettre ceci en relation avec les travaux de Löffler et al. [Löf2000b] relatant des expériences
de diffusion de neutrons qui concluent au rôle majeur du titane dans les premiers stades de décomposition.
2.1.3. Traitement ‘intermédiaire’ à 9 minutes
Pour ce temps de traitement, la nano-cristallisation précédente s’est généralisée, et la taille des
précipités est de l’ordre de 15 à 20 nm, comme en témoigne la figure 5.16 (images en champ clair et champ
sombre respectivement).
% atomique Zr Ti Ni Cu
Particule 1 58.5 18.8 11.6 11.1
Particule 2 57.0 19.8 12.6 10.6
Particule 3 57.6 19.4 10.7 12.3
Moyenne 57.7 19.3 11.6 11.4
Matrice 1 62.2 14.1 11.4 12.3
Matrice 2 63.6 13.2 10.6 12.6
Matrice 3 61.5 14.3 10.2 14.0
Matrice 4 64.9 13.3 10.0 11.8
Moyenne 63.1 13.7 10.5 12.7
Compo. ‘brute’ 53.6 15.8 14.5 16.1 Tableau 5.4 : composition par analyse EDX de différentes zones cristallisées
200 nm
200 nm
figure 5.16 : microstructure de l’échantillon Vit1 recuit 9 min à 410 °C.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 105
Sur le plan de la composition chimique des cristaux, le tableau 5.4 résume quelques analyses
représentatives ; il apparait que les tendances observées après 7 minutes (tableau 5.3) se retrouvent, avec
apparemment un enrichissement plus marqué en titane.
2.1.4. Traitements ‘longs’ de 25 et 60 minutes
Après 25 min à 410°C, la diffraction X, figure 5.17, montre clairement que les échantillons ont
cristallisé. De larges pics de cristallisation surmontent une bosse amorphe résiduelle. Les observations faites
par microscopie électronique en transmission confirment une cristallisation généralisée de taille
nanométrique, figure 5.18. La taille des cristaux observés varie de 10 à 50 nm, la moyenne étant de 22 nm.
Afin de déterminer la chimie de ces nanocristaux, nous avons effectué des analyses EDX qui sont
résumées dans le tableau 5.5. Nous avons pu classer en deux catégories les précipités observés : les uns
sont enrichis en zirconium, les autres en titane, et dans une moindre proportion en nickel et cuivre.
Comme on peut le voir au travers de ces diverses analyses, les résultats montrent une tendance
d’enrichissement en zirconium ou en titane, et la précision qu’on peut espérer est au mieux de l’ordre du
pourcent atomique.
Les analyses effectuées dans la matrice amorphe présentent également de fortes variations de
composition. Afin de voir si celles-ci étaient régulières, nous avons effectué un profil de composition dans la
matrice amorphe par analyse EDX. Avant tout , nous nous sommes assurés qu’aucune cristallisation ne
perturberait nos mesures. Pour cela nous nous sommes placés dans une zone où aucun contraste de
diffraction n’était visible et seules les variations de composition liées à la matrice ont été mises en évidence
Figure 5.18 : image en champ clair d’un échantillon de Vit1 recuit 25min à 410 °C
0.0
0.2
0.4
0.6
0.8
1.0
1.2
30 40 50 60 70 80 902θ (degrés)
I (u.
a.)
Figure 5.17 : diffractogramme de rayons X sur unéchantillon de Vit1 après 25 min à 410 °C.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 106
par ces analyses. Bien que le Vit1 contienne 22.5% de béryllium, il a été très difficile d’obtenir des spectres
de perte d’énergie sur le seuil du béryllium qui se situe à 111 eV. En effet, soit l’échantillon était trop épais,
soit la pollution liée au carbone augmentait artificiellement l’épaisseur, ce qui modifiait la décroissance des
plasmons en masquant ce seuil. Nous avons essayé de limiter au maximum les sources de pollution en
effectuant l’amincissement ionique le plus tardivement possible mais aucune de ces précautions n’a permis
de s’affranchir de ces problèmes.
cristal Zr Ti Ni Cu 1 60.7 11.61 12.85 14.84
2 56.03 9.71 13.54 20.72
3 67.05 10.5 8.41 14.04
4 60.95 9.24 14.38 15.43
moyenne 61.2 10.3 12.3 16.2
cristal Zr Ti Ni Cu 5 47.0 18.4 13.4 21.2
6 45.3 15.6 16.5 22.6
7 46.2 18.3 15.7 19.8
8 46.2 18.4 16.0 19.4
9 46.0 15.7 15.9 22.4
moyenne 46.1 17.3 15.5 21.1
Comp. nominale 53.2 17.8 12.9 16.1 Tableau 5.5 : composition par analyse EDX de différentes zones cristallisées
Nous nous sommes aperçus tardivement qu’en utilisant un porte-objet froid nous limitions le
phénomène de pollution lié au carbone et améliorions ainsi la qualité des analyses au détriment de la
résolution. Nous ne présenterons donc pour le Vit1 que les analyses chimiques obtenues par EDX. Ce sont
les images de cartographie chimique, qui seront présentées un peu plus loin, qui nous permettront d’avoir
une lecture a posteriori des évolutions de la chimie, en particulier concernant le béryllium, de notre
échantillon à ce stade et aux stades antérieurs.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 107
La figure 5.20 représente les variations de composition que nous avons observées pour les quatre
éléments détectables par EDX (Zr, Ni, Ti et Cu). Sur une distance de 200 nm, nous avons effectué des
analyses avec un pas de 17 nm et une taille de sonde de 2 nm. Les conditions expérimentales ne
permettent pas de quantifier proprement ces analyses. Néanmoins, elles permettent de dégager des
tendances significatives du comportement des différents éléments. A ce stade avancé de la cristallisation, la
matrice amorphe n’est plus uniforme ainsi que l’avaient déjà évoqué Wang et al. [Wan1997] en étudiant
l’évolution de la fonction de distribution radiale. Il est intéressant de voir que les variations du titane et du
zirconium sont opposées, comme le confirment la composition des cristaux qui se forment. Il semble que les
variations de composition en nickel suivent celles du titane. Les variations du cuivre bien qu’importantes ne
peuvent être reliées à l’un ou l’autre des éléments.
Pour confirmer les résultats des analyses EDX, nous avons effectué de l’imagerie chimique en
EFTEM (microscope 300 kV du CEA Grenoble). Les images de la figure 5.21 sont réalisées sur les seuils
respectifs des éléments suivants : béryllium, zirconium, titane, nickel et cuivre.
On peut dès à présent remarquer que l’image du cuivre ne présente aucun contraste particulier,
contrairement à ce qui avait été vu précédemment par EDX.
Ceci peut avoir deux explications : soit les variations de composition en cuivre, si elles existent, sont
en-deçà de la sensibilité de cette technique, soit les variations observées en EDX sont liées à des erreurs de
statistique. Les images du zirconium et du béryllium présentent les mêmes contrastes, ce qui indique que
ces deux espèces chimiques migrent vers les mêmes zones. De même les images du titane et du nickel ont
0
10
20
30
40
50
60
0 50 100 150 200distance (nm)
% a
tom
ique
ZrCuNiTi
maxima de Ni et Ti correspondant aux minima de Zr
maxima de Zr correspondant aux minima de Ni et Ti
Figure 5.20 : profil de composition obtenu dans la matrice amorphe résiduelle. Nous pouvons voir que les fluctuations de compositions du titane et du nickel sont en phase alors que les fluctuations du zirconium semblent opposées.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 108
un contraste semblable et opposé au contraste du zirconium et du béryllium, ce qui confirme les résultats de
l’EDX.
Figure 5.21 : images filtrées en énergie sur les seuils de Be, Zr, Ti, Ni et Cu respectivement de gauche àdroite et de haut en bas. La dernière image correspond à une image en champ clair de la matrice amorphe(sur la partie inférieure aucun contraste de diffraction n’est visible). On peut distinguer une nette corrélationentre Zr et Be d’une part et Ti et Ni d’autre part. De plus, il existe une anti-corrélation entre Zr/Be et Ti/Ni. NB : ce que l’on aperçoit en bas à gauche de l’image en champ clair ainsi que dans les images filtrées enénergie ne correspond pas à un cristal primaire mais à un bord de la lame mince qui s’est retourné.
120 nm
Be Zr
Ti Ni
Cu

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 109
A ce stade de transformation, comme l’avaient déjà montré les analyses EDX et comme l’atteste
l’imagerie filtrée, la matrice amorphe n’est plus uniforme et connaît donc les mêmes tendances de
fluctuations de compositions que les parties cristallines. D’un point de vue processus de décomposition, nos
observations sont en accord avec les travaux de la littérature, à savoir que la cristallisation s’opère après
une décomposition de la matrice amorphe [Liu1997, Wan1996]. En ce qui concerne la nature chimique des
atomes responsables de cette décomposition, nos résultats indiquent que seul le cuivre ne subit pas de
variation importante.
Si nous admettons que la cristallisation observée dans le Vit1 est issue de la décomposition de la
matrice amorphe, nous pouvons conclure que les nanocristaux riches en zirconium sont également riches en
béryllium et inversement les nanocristaux riches en nickel et titane seront appauvris en béryllium.
Malheureusement nous n’avons pas eu le temps de vérifier cela par des analyses directes de pertes
d’énergie sur les deux types de nanocristaux. Cet aspect pourrait donc faire l’objet d’un travail
complémentaire pour confirmer l’hypothèse qui a été faite. Il pourrait être également intéressant de mener
une étude comparative de la taille des cristaux en fonction de leur chimie respective.
Pour compléter ces observations sur des traitements ‘longs’, un échantillon a été recuit 60 minutes à
410 °C.
La figure 5.22 montre la microstructure obtenue, avec des nano-cristaux encore plus grossiers.
Pour améliorer la lisibilité du cliché de diffraction, nous présentons dans la figure 5.23 une
comparaison du profil moyenné en rotation avec celui obtenu à partir de clichés sur les états 5, 7 min (où la
phase c.f.c. de paramètre 0.4 nm a été identifiée - fig. 5.12 -) et 25 minutes.
Figure 5.22 : microstructure d’un échantillon de Vit1 recuit 60 min à 410 °C. A gauche : champ clair, à droite :diffraction électronique en aire sélectionnée filtrée en énergie sur le pic élastique.
100 nm

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 110
Sur cette figure, on constate que dès 25 minutes, l’épaulement perceptible à gauche (i.e., aux faibles
valeurs de ‘q’) de la raie 111 de la phase c.f.c. à 7 minutes est devenu un pic marqué, qui est évidemment
encore plus visible à 60 minutes (réflexion à d = 0.24 nm). En outre, une autre réflexion est apparue à d =
0.255 nm.
A la lumière de nos résultats d’analyse chimique, qui montrent clairement l’existence de 2
populations de précipités aux temps de recuit longs, nous pouvons émettre l’hypothèse probable qu’une
seconde phase, autre que la phase c.f.c. visible à 7 minutes, a précipité dès 25 minutes. Cette seconde
phase correspondrait donc aux cristaux enrichis en zirconium (numérotés 1 à 4 dans le Tableau 5.5).
Nous avons cherché à imager cette seconde phase en MET à Haute Résolution, mais nous n’avons
pu produire d’images dans des azimuts de bas indices ; toutefois, des cristaux où la distance prépondérante
à 0.24 nm est présente, ont clairement été imagés (cf. figure 5.24).
2.1.5. Bilan et Interprétation
Avant de passer à l’étude de la microstructure dans le Vit4, il nous semble nécessaire de résumer en
quelques lignes les principaux résultats que nous avons obtenus au cours de cette étude sur le Vit1 lors de
traitements isothermes à 410 °C.
1 : durant les 5 à 6 premières minutes, aucune cristallisation n’est visible. Pour autant la diffusion modifie
localement la composition de la matrice, et induit une décomposition qui mènera à la cristallisation. Il ne
q (Å-1)
Figure 5.23 : profils de diffraction électronique sur des échantillons de Vit1 recuits 5, 7, 25 et 60 minutes à410 °C.
d = 0.240 nm d = 0.255 nm d111 = 0.232 nm d220 = 0.142 nm d200 = 0.201 nm

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 111
nous a pas été possible de montrer que cette décomposition était une décomposition spinodale comme cela
a été rapporté dans la littérature pour cet alliage à des températures plus basses (368 °C). Néanmoins, les
résultats d’imagerie filtrée effectuée après un traitement de 25 minutes à 410 °C (figure 5.21) et, dans une
moindre mesure, d’EDX (fig. 5.20) indiquent clairement la présence de nano-domaines de chimie différente
dans la matrice amorphe : d’une part, un enrichissement en Ti et Ni, et d’autre part, un enrichissement en Be
et Zr. Bien que nous n’ayons pu, faute de temps, confirmer cette ‘microstructure’ dans les recuits les plus
courts, nous pouvons raisonnablement supposer qu’une telle organisation est la preuve d’une décomposition
de la matrice lors d’un traitement thermique.
2 : après 7 minutes, les premiers nano-cristaux sont visibles (3 à 4 nm). Leur cristallographie est compatible
avec une structure c.f.c. de paramètre très proche de 0.4 nm. Ce résultat est très comparable à celui de
Schneider et al. [Sch1996] qui ont identifié une même structure avec le même paramètre. L’analyse EDX
révèle que les précipités sont enrichis en titane et appauvris en zirconium. Il y a par contre là désaccord
avec les spéculations de Schneider et al. qui, sans analyse en microscopie, avaient émis l’hypothèse d’une
phase métastable enrichie en titane mais surtout en cuivre. Nos résultats d’analyse chimique de ces
nanocristaux formés lors d’un recuit isotherme sont à notre connaissance les premiers concernant ce
matériau. Il est donc difficile de les comparer à la littérature. Néanmoins, rappelons que la plupart des
auteurs insistent sur le fait que le titane est l’élément important, du point de vue de la diffusion, du début de
la transformation. On peut supposer qu’à ce stade, la cristallisation a eu lieu dans des zones de la matrice
où la composition permettait de former un germe cristallin par simple transformation polymorphique : à la
lumière des résultats en EFTEM, ces zones amorphes sont des domaines enrichis en Ti et Ni.
Figure 5.24 : image à Haute Résolution d’un cristal présentantune famille de plans réticulaires de distance 0.24 nm(échantillon de Vit1 recuit 25 min à 410 °C).
dd ≈≈ 00..2244 nnmm

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 112
3 : Entre 7 et 12 minutes, il n’y a pas de grand changement. Les cristaux formés sont un peu plus gros et
toujours plus riches en titane.
4 : Après 25 minutes : les analyses EDX montrent qu’il existe alors deux types de cristaux. Ceux présents
depuis la 7ème minute, riches en titane (et qui semblent à ce stade s’être de nouveau enrichis en nickel), et
d’autres riches en zirconium. Il apparaît donc qu’entre 12 et 25 minutes à 410°C, les zones de la matrice
amorphe enrichies en zirconium ont pu être le siège d’une seconde nano-cristallisation. L’analyse
cristallographique de ces précipités n’a pu être complétée, du fait de la difficulté à orienter les cristaux dans
des axes de zones simples ; de plus, les diagrammes d’anneaux en diffraction électronique (cf. fig. 5.12 et
profils à 25 et 60 minutes de la fig. 5.22) ne nous ont pas permis de proposer d’hypothèse sur le type de
structure cristalline, du fait notamment de la présence persistante de la première phase c.f.c. .
A titre de complément, nous pouvons noter que les résultats ‘cristallographiques’ obtenus en
diffraction électronique (fig. 5.23) sont tout à fait cohérents avec ceux obtenus en diffraction des rayons X,
puisque les mêmes pics de diffraction sont identifiés pour des temps de recuit comparables (cf. figure 5.25).
Ces résultats en diffraction X sont d’ailleurs eux-mêmes très similaires avec ceux publiés dans la littérature
[Löffler et Johnson 2001].
q (Å-1)
Figure 5.25 : diagrammes de diffraction X pour le Vit1 recuit à différents temps à410 °C (à comparer aux résultats de diffraction électronique - fig. 5.23 -).

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 113
Compte-tenu de ces différents éléments, nous pouvons proposer un scénario cohérent de la
séquence de nano-précipitation au sein du Vit1 à 410 °C, illustré à la figure 5.26.
On notera que nos résultats d’analyse chimique concernant le béryllium sont parcellaires ;
néanmoins, il a toujours été vu que le béryllium suivait la même tendance que le zirconium (dans les nano-
domaines non cristallins – fig. 5.21 -, et dans la première phase cristalline c.f.c. - fig. 5.15, montrant un
rapport Be/Zr très semblable à ce qu’il est dans la matrice à l’état brut -). Nous pouvons donc supposer que
pour la seconde phase cristalline (fig. 5.24) où la teneur en béryllium n’a pas été déterminée, il y a, comme
pour le zirconium, enrichissement en Be.
Figure 5.26 : évolution du Vit1 lors d’un traitement isotherme à 410 °C : a) : matrice amorphe à l’état brut(chimiquement homogène) ; b) : décomposition (spinodale ?) en ‘nano-domaines’ amorphes riches en Ti, Ni(zones sombres) et riches en Be, Zr (zones claires) après un recuit court (inférieur à 7 min) ; c) : nano-précipitation d’une phase c.f.c. (a environ 0.4 nm) riche en Ti,Ni dans les domaines amorphes de mêmetendance chimique dès 7 min de recuit ; d) : seconde nano-précipitation (phase de cristallographie nondéterminée) riche en Be, Zr dans les domaines amorphes de même tendance chimique aux temps detraitement plus longs.
a) b)
≈≈ 110000 nnmm
c) d)

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE
2.2 Vit4
2..2.1. Traitement isochrone
Dans un premier temps nous avons étudié par diffraction X (figure 5.26) l’état microstructural d’un
échantillon ayant subi une montée en température de 270 à 470 °C, suivie d’une trempe rapide. L’indexation
des pics permet d’identifier les phases cristallines ZrBe2, Zr2Cu et Zr2Ni. Certains pics ne sont attribuables à
aucune de ces phases et n’ont pour l’instant pas été identifiés. Le fond continu du spectre est plat ce qui
signifie qu’il n’y a plus ou très peu de phase amorphe. Il n’a pas été fait de microscopie électronique sur ces
échantillons mais à partir du spectre de diffraction X, il est possible de retrouver la taille des particules par la
formule de Scherrer :
0cosL)2(
θ×λ
=θ∆ (5.1)
avec ∆ (2θ) largeur angulaire à mi-hauteur qui est la somme de l’élargissement lié à la taille des cristaux
et de la largeur instrumentale ( déterminée par L. Gremillard [Gre2002] et valant 0.13° dans cette gamme de
valeur angulaire),
L taille des cristaux diffractants,
λ longueur d’onde,
θ0 valeur de l’angle de diffraction.
En utilis
graphe 5.26), o
Fi
0
0.2
0.4
0.6
0.8
1
25 35 45 55 65 75 852θ (degrés)
Inte
nsité
(u.a
.)
+ *°°
° ° °
°
+ ++ *
*
**+
Zr2NiZr2Cu
ZrBe2
igure 5.26 : diffractogramme d’un échantillon de Vit4 après un traitement
sochrone entre 270 et 470 °C à une vitesse de balayage de 4 °C.min-1.
0
0.2
0.4
0.6
0.8
1
34.6 35.1 35.6
2θ (degrés)
I nte
nsité
(u.a
.)
∆θ = 0.41°
MOORTELE, Bertrand
ant le pic le plus intense, centré à 35.34 ° et de 0.41
n obtient une taille de cristaux de 38 nm. Ce type de na
114
° de largeur à mi-hauteur (détail du
no-cristallisation a fréquemment été

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse
observé dans les verres métalliques et fait l’étude d’une recherche pour contrôler la taille des cristaux afin de
faire des matériaux composites.
2.2.2. Traitement isotherme
Sur le même échantillon brut nous avons effectué des traitements thermiques isothermes à 380 °C,
ce qui correspond à 40 °C au-dessus de Tg et 60 °C en-dessous de Tx. Le graphe 5.27 est un spectre de
diffraction X obtenu pour un recuit de 12 h. Ce spectre diffère fortement de celui observé précédemment. Il
présente une bosse amorphe importante, indiquant l’existence d’une phase amorphe résiduelle, surmontée
de pics intenses qui comme nous le verrons par la suit correspondent à une phase quasicristalline.
L’emplacement des pics principaux ne correspond pas à ceux déjà observés. L’évolution des pics (intensité,
largeur à mi-hauteur) principaux en fonction du temps de recuit, pour un domaine angulaire plus restreint,
est visible en insertion dans le même graphe.
On note également des pics très faibles, qui après examen précis s’indexent formellement dans la
phase hexagonale ZrBe2.
2.2.2.1
microsc
microg
précipit
l’image
0
0.2
0.4
0.6
0.8
1
25 35 45 55 65 752θ (degrés)
Inte
nsi
té (
a.u
.)
110000100000
200000
311111111101
101000
210000 110010
111100
210001
100ZrBe2
101 ZrBe2 110ZrBe2211000-211001
- *
**
** **
0
0.2
0.4
0.6
0.8
1
25 35 45 55 65 752θ (degrés)
Inte
nsi
té (
a.u
.)
110000100000
200000
311111111101
101000
210000 110010
111100
210001
100ZrBe2
101 ZrBe2 110ZrBe2211000-211001
- *
**
** **
110000100000
200000
311111111101
101000
210000 110010
111100
210001
100ZrBe2
101 ZrBe2 110ZrBe2211000-
211000-211001
-211001- *
**
** **
Figure 5.27 : diffractogramme d’un échantillon de Vit4 après un traitement isotherme de12 heures à 380 C. Les pics de cristallisation correspondent à ceux d’une phasequasicristalline de paramètre 0.4 nm.
0.3
0.5
0.7
0.9
1.1
1.3
34 35 36 37 38
angle (2θ)
I rel
ativ
e
1 h à 380 °C3 h à 380 °C12 hà 380 °C
- VAN DE MOORTELE, Bertrand 115
observation de la microstructure
Afin d’effectuer une observation directe de la microstructure, nous avons préparé une lame de
opie à partir d’un échantillon recuit pendant 12 heures à 380 °C. La figure 5.28 est un montage de
raphies en champ clair et champ sombre. On distingue clairement la présence de deux types de
és englobés dans la matrice amorphe résiduelle. Le premier type de précipité, noté (A) sur
, est de forme hexagonale aux contours nets, présentant des plans cristallins automorphes ce qui

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 116
laisse penser à une phase cristalline hexagonale. Le second type de précipité noté (B) a une forme plus
sphérique. La taille de ces cristaux est comprise entre 50 et 200 nm. Leur répartition est uniforme dans le
matériau.
2.2.2.2 analyse cristallographique
précipités A
Nous nous sommes attachés à déterminer la nature cristallographique de ces phases en
commençant par la phase (A). La forme particulière des précipités et l’existence dans ce matériau à des
températures plus élevées de la phase hexagonale ZrBe2 nous a orienté pour l’indexation de cette phase.
L’image 5.29 est une micrographie en haute-résolution d’une particule A avec sa transformée de Fourier en
insertion. Nous avons également effectué des diffractions électroniques en aire sélectionnée sur ces phases
Figure 5.29 : image en Haute Résolution d’une particule cristalline de type (A) et sa transformée de Fourrier en insertion.
A B
50 nm
Figure 5.28 : images en champ clair/champ sombre de deux types departicules cristallisées présentes dans le Vit4 après un traitementthermique de 12 heures à 380 °C.
B A

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 117
(image 5.30). Dans les deux cas, nous pouvons indexer les clichés dans la phase ZrBe2 sans ambiguité,
comme reporté par exemple sur la figure 5.30. Ce résultat est cohérent avec les résultats de diffraction X
(faibles pics de ZrBe2, cf. fig. 5.28).
précipités B
L’étude de la phase B montre qu’il s’agit de quasicristaux, comme l’avaient déjà observé Wanderka
et al [Wan2001] dans le Vit4. Les images 5.31-a, b, et c correspondent à des diffractions révélant
respectivement les symétries d’ordre 5, 3 et 2 d’une phase icosahédrique. L’image 5.32 est une image en
haute résolution où l’on peut voir la symétrie d’ordre 5.
.
(1010)_
(0110)_
(1100)_
(0001)
Figure 5.30 : diffraction électronique sur une particule de type A indexée dans la structure hexagonale de ZrBe2.
Figure 5.31 : diffraction électronique sur la particule quasicristalline selon les axes de symétrie d’ordre cinq,trois et deux respectivement.
55 nnmm--11
a) b) c)

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 118
2.2.2.3 analyses chimiques
Dans le cas des matériaux formant des phases quasi-cristallines, il existe très peu d’analyses de
composition, mis à part quelques travaux effectués en sonde atomique (APFIM : Atom Probe Field Ion
Microscopy – cf. [Wan1999] -). De nombreux auteurs pensent, sans pour autant en apporter la preuve
directe, que la transformation de matrice amorphe en phase quasi-cristalline est une transformation
polymorphique, c’est-à-dire consistant en une réorganisation locale sans modification de la composition
chimique. L’argument principal pour cette interprétation est que l’état quasi-cristallin est un état métastable
du matériau et de ce fait contradictoire avec l’idée d’une réorganisation à longue distance. Il nous est apparu
nécessaire de compléter cette étude par des analyses de composition chimique des différentes phases.
Les analyses de composition par EDX effectuées sur les quasi-cristaux et la matrice amorphe sont
résumées dans le tableau 5.6. Les compositions relatives (sans le béryllium) sont comparées à celles
données par l’élaborateur, dite nominales. Il est à noter que nous avons obtenu un très bon accord entre la
matrice amorphe et la composition nominale en ce qui concerne le zirconium et le cuivre. Par contre nous
détectons moins de titane (7.2% at. au lieu de 11.45) et plus de nickel que ce que nous devrions. La
composition relative des quasi-cristaux semble très proche de celle de la matrice avec juste un
enrichissement en titane au détriment du nickel.
Figure 5.32 : image en Haute Résolution sur une particule quasicristalline selon l’axe desymétrie d’ordre cinq (après filtrage de Fourier).
22..55 nnmm

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 119
% atomique Ti Ni Cu Zr
quasi-cristaux 14,4±1,3 12,4±0,4 10,2±0,9 63,0±1,9
matrice amorphe 7,2±1,1 16,6±0,8 11,7±0,9 64,4±2,5
composition nominale 11,5 13,8 10,3 64,4 Tableau 5.6 : composition chimique partielle de la phase quasi-cristalline et de la matrice amorphe
Afin de vérifier que la phase (A) correspond bien à une structure du type ZrBe2, nous avons effectué
sur des précipités de ce type des analyses EDX quantitatives. Le spectre de la figure 5.35 est un exemple
de ce que l’on a pu observer. De manière qualitative, le zirconium semble être le composant ultra-majoritaire
détecté par l’analyse EDX. On remarque également la présence d’autres éléments que l’on peut attribuer
soit à la matrice amorphe environnante, soit à la présence de ces espèces dans la phase ZrBe2. Le tableau
5.7 donne la composition moyenne de la phase (A) déterminée par EDX. La présence d’éléments
minoritaires dans une phase de
type ZrBe2 avait déjà été évoquée
par Wanderka et al. [Wan1999].
Après un refroidissement lent sur le
Vit4, ils ont observé la formation de
cristaux primaires. Une des phases
observées dans ces conditions était
du ZrBe2 contenant du titane et
dans une moindre mesure du nickel
et du cuivre (composition
également donnée dans le
tableau).
% atomique Zr Ti Ni Cu
Zone A 92.8±0,9 3.4±0,1 1.4±0.1 2.4±0.1
ZrBe2 dans Vit4 d’après Wanderka et al. 89.0 4.2 3.9 2.8
Tableau 5.7 : composition partielle de la phase de type A et de ZrBe2 d’après Wanderka et al. (pour ces derniers résultats, la composition hors béryllium est déduite de la composition suivante : Zr31.6 ± 3.5 Be64.5 ± 3.6 Ti1.5±0.9 Ni1.4±0.9 Cu1±0.8).
La faible proportion de titane, atome voisin du zirconium de par sa taille et sa structure électronique
externe ([Kr].4d2.5s2 pour le zirconium et [Ar].3d2.4s2 pour le titane) ainsi que la faible concentration de Ni et
Cu explique est que les paramètres du réseau cristallin de ZrBe2 sont peu ou pas perturbés.
0
100
200
300
400
500
600
700
800
0 2 4 6 8 10 12 14 16 18
energie (keV)
Inte
nsité
Zr
Zr
ZrZr ZrZr
Ti Ni
TiNi
Ni
NiCu
C Cu CuTi
Figure 5.35 : spectre EDX obtenu dans une particule (A)
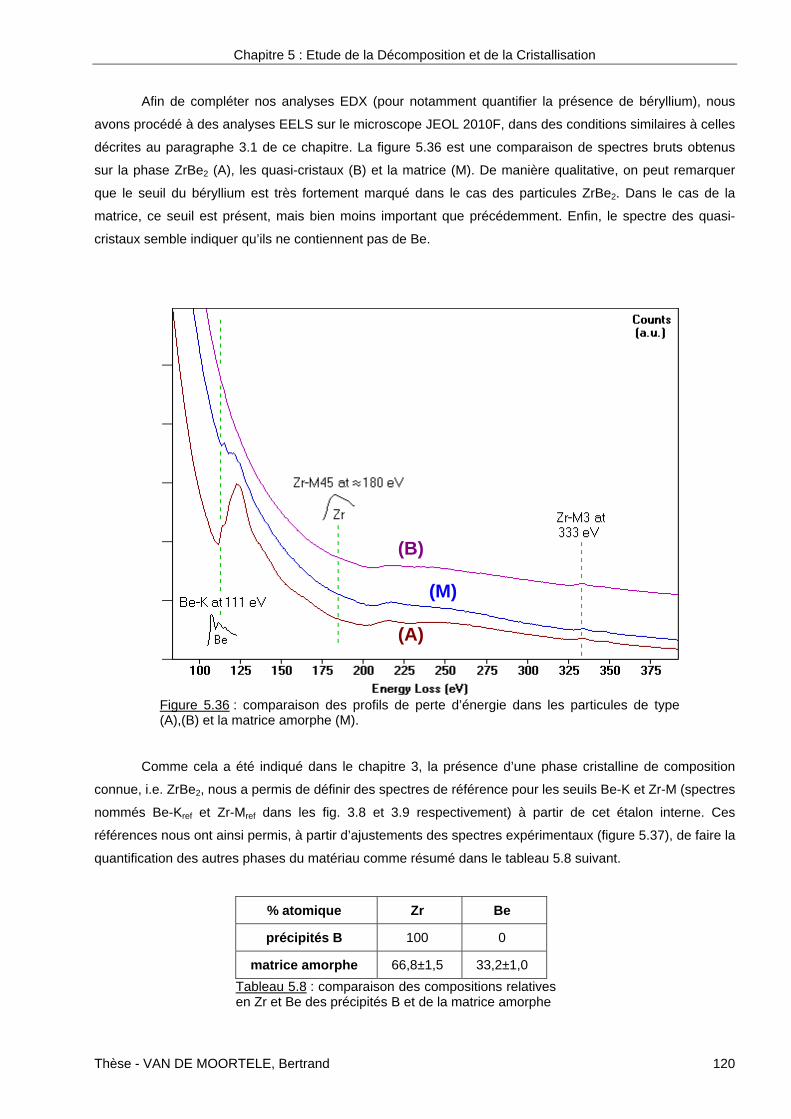
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 120
Afin de compléter nos analyses EDX (pour notamment quantifier la présence de béryllium), nous
avons procédé à des analyses EELS sur le microscope JEOL 2010F, dans des conditions similaires à celles
décrites au paragraphe 3.1 de ce chapitre. La figure 5.36 est une comparaison de spectres bruts obtenus
sur la phase ZrBe2 (A), les quasi-cristaux (B) et la matrice (M). De manière qualitative, on peut remarquer
que le seuil du béryllium est très fortement marqué dans le cas des particules ZrBe2. Dans le cas de la
matrice, ce seuil est présent, mais bien moins important que précédemment. Enfin, le spectre des quasi-
cristaux semble indiquer qu’ils ne contiennent pas de Be.
Comme cela a été indiqué dans le chapitre 3, la présence d’une phase cristalline de composition
connue, i.e. ZrBe2, nous a permis de définir des spectres de référence pour les seuils Be-K et Zr-M (spectres
nommés Be-Kref et Zr-Mref dans les fig. 3.8 et 3.9 respectivement) à partir de cet étalon interne. Ces
références nous ont ainsi permis, à partir d’ajustements des spectres expérimentaux (figure 5.37), de faire la
quantification des autres phases du matériau comme résumé dans le tableau 5.8 suivant.
% atomique Zr Be
précipités B 100 0
matrice amorphe 66,8±1,5 33,2±1,0 Tableau 5.8 : comparaison des compositions relatives en Zr et Be des précipités B et de la matrice amorphe
Figure 5.36 : comparaison des profils de perte d’énergie dans les particules de type(A),(B) et la matrice amorphe (M).
(B) (M) (A)

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 121
En combinant les résultats EDX et EELS, nous avons recalculé les pondérations de chaque
élément ; le tableau 5.9 résume les résultats de la quantification chimique des précipités de type (A) et (B) et
de la matrice amorphe (M). Ces résultats confirment que la composition de la matrice amorphe a peu
évolué, puisque l’on retrouve la composition de départ du Vit4.
% atomique Zr Ti Ni Cu Be
Matrice amorphe 46,9± 2,5 5,3±1,1 8,5±0,8 12,1±0,9 27,2
Précipités A 32,4 0,9 _ _ 66,7
Précipités B 63,0±1,9 14,4±1,3 12,4±0,4 10,2±0,9 0
Composition Nominale 46.7 8.3 10 7.5 27.5
Tableau 5.9 : compositions des différentes phases et de la matrice amorphe comparées à la composition
nominale.
2.2.3. Observation in-situ de la cristallisation du Vit4.
Afin de suivre l’évolution de la phase quasi-cristalline, il a été effectué une expérience de diffraction
de neutrons in situ à l’Institut Laue-Langevin (ILL), en collaboration avec J.-L. Soubeyroux. La figure 5.38 est
un graphique à 3 dimensions où l’on peut suivre l’évolution de l’intensité diffractée en fonction du temps et
de l’angle de diffraction.
La figure 5.39 donne l'évolution de la température lors de cette expérience in-situ. Une première
montée linéaire en température s'effectue à une vitesse de 2 °C.min-1 jusqu'à une température d'environ
380°C. En effet, dans ce genre d'expérience, la position du thermocouple ne permet pas d'être en contact
avec l'échantillon ce qui induit un certain décalage en température entre les indications du thermocouple et
la température de l'échantillon (ce qui nécessite ensuite de recaler les températures). Les pics de la phase
quasicristalline apparaissent alors après environ 1h de traitement isotherme (ce qui comme nous le verrons
Figure 5.37 : ajustement de spectres EELS expérimentaux (moyenne de 10 mesures après soustraction dufond continu et déconvolution des pertes proches) de la matrice (M) (à gauche) et de la phasequasicristalline (B) (à droite) par les spectres de référence; le rapport Be/Zr vaut respectivement 0.5 et 0pour ces 2 phases.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 122
semble indiquer que la température de traitement isotherme était légèrement supérieure à 380°C) et leur
position angulaire ne change pas pendant l’expérience de recuit isotherme.
temps
Température
380°C1 2
3
1 : montée linéaire en température
2 : palier isotherme vers 380°C
3 : montée linéaire en température
isotherme
Figure 5.39. : schéma de traitement thermique sur les échantillonsde Vit4 lors de l'expérience de diffraction in-situ à l'ILL.
Figure 5.38 : diffraction de neutrons lors d’une expérience in-situ, dont le traitement thermique est explicitépar la figure 5.39, sur un échantillon de Vit4. Les pics de la phase quasicristalline (indiqués par les flèchesvertes) apparaissent après une heure. Un fois que la phase quasicristalline s’est bien développée, uneisochrone a permis de suivre l’apparition de phases cristallines (indiquées par les flèches marrons) audétriment de la phase quasicristalline.
pics de la phase quasicristallineidentiques à ceux observés endiffraction des rayons x.
pics de phases cristallinesse développant à plushaute température.
bosse amorphe

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 123
Seule l’intensité et la largeur à mi-hauteur évoluent. Bien que la proportion de phase amorphe soit
encore très importante, aucune évolution des pics n’est par la suite détectable. Enfin, une montée linéaire
vers les plus hautes températures permet d’observer alors la disparition de la phase quasicristalline au profit
d’autres phases cristallines comme le montre la partie supérieure (sombre) de la figure 5.38.
L’observation en microscopie conventionnelle d’un échantillon ayant subit un traitement de 3 heures
révèle une faible cristallisation alors qu’après 4h la cristallisation est générale (figure 5.40 a et b). On peut
noter ici que des observations faites en microscopie électronique après une heure à 380 °C ne révèlent
aucune cristallisation, ce qui indique que la température lors de l'expérience in-situ était légèrement
supérieure à 380 °C.
2.2.4. Interprétation
L’ensemble de ces résultats expérimentaux nous a permis d’éclaircir l’interprétation de la formation
de cette phase quasi-cristalline. Wanderka et al. [Wan2000] ont déjà proposé une séquence de cristallisation
qui est la suivante :
Cette hypothèse implique dans une première étape, une transformation polymorphique de la phase
amorphe en une phase quasi-cristalline, suivie d’une modification de la chimie des quasi-cristaux, cette
Matrice Amorphe
Quasi-Cristaux + Matrice Amorphe
Quasi-Cristaux + ZrBe2 + phases non-identifiées + Matrice Amorphe
ZrBe2 + Zr2Cu + phases non-identifiées
200 nm 200 nmFigure 5.40 : évolution de la cristallisation de la phase quasicristalline dans le Vit4 lors d’un recuitisotherme à 380 °C. Les figures a) et b) sont des images en champ clair pour des échantillons traités 3et 4 heures respectivement.
a) b)

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 124
interprétation étant basée sur l’idée qu’une phase quasi-cristalline ne peut se former à la suite d’une
diffusion à longue distance. Ceci est contredit par les résultats combinés de l’analyse chimique et de la
diffraction de neutrons in-situ : la figure 5.38 montre en effet que les pics attribuables à la phase
quasicristalline n’évoluent pas en position lors de la montée en température, ce qui indique qu’il n’y a pas de
modification cristallographique. De surcroît, nos analyses EELS montrent que la phase quasicristalline ne
contient pas de béryllium, donc la phase ZrBe2 qui se forme ne peut s’alimenter en béryllium à partir des
quasicristaux.
Néanmoins, nos résultats montrent que ce scénario de transformation est qualitativement correct. La
diffusion joue un rôle important, en particulier celle du béryllium; à partir des résultats expérimentaux de
Rehmet et al. [Reh2001] sur la diffusion du Be dans le Vit1, on peut estimer la valeur du coefficient de
diffusion du Be à 380°C à 6.10-17 m2.s-1. Pour des temps de 3 à 4 h, le parcours moyen du Be est ainsi de
l’ordre du demi-micron, ce qui est compatible avec l’absence de Be dans les quasi-cristaux puisqu’après 4 h,
leur taille est comprise entre 50 et 100 nm. On doit donc conclure qu’il se produit un phénomène de rejet
solutal menant d’une part à la formation d’une phase quasi-cristalline stable, et d’autre part à la formation de
zones riches en Be, permettant à terme la précipitation de cristaux de ZrBe2. On comprend alors que des
cristaux de ZrBe2 puissent être englobés dans des quasi-cristaux. En effet, un cristal de ce type peut se
former dans une zone entourée de quasicristaux qui ont continué leur développement et donner ainsi
l’impression d’une structure ‘cœur-écorce’ que nous avons pu fréquemment observer (cf. fig. 5.28).
Le fait que la phase quasicristalline n’ait jamais été observée lors d’une montée isochrone en
température dans le Vit4 tend également à montrer que la formation de celle-ci n’est pas issue d’une
transformation polymorphique. Dans ce cas, la diffusion du béryllium ne peut s’effectuer suffisamment à
longue distance avant que la diffusion des autres éléments ne devienne efficace, permettant ainsi la
cristallisation de phases plus stables, comme Zr2Cu.
On peut remarquer que la composition chimique en (zirconium et titane) de la phase quasicristalline
est très proche de celles des amorphes métalliques dans lesquels des quasi-cristaux ont été observés par
divers auteurs (cf. tableau 2.1). Il est étonnant que personne jusqu’alors ne se soit aperçu que le Vit4 faisait
figure d’intrus dans cette famille des verres métalliques. En admettant le scénario de cristallisation que nous
proposons, nous voyons que ce paradoxe n’est qu’apparent.
Cette interprétation de la formation de quasi-cristaux, par diffusion du béryllium dans le Vit4 implique
également, comme le proposaient Mattern et al. [Mat1999, Mat2002], que la formation d’une phase quasi-
cristalline n’est pas nécessairement corrélée à l’existence d’un ordre à courte distance préexistant dans le
liquide surfondu.
A la suite des analyses de composition de la phase quasi-cristalline et afin de vérifier notre propos,
Jean-Louis Soubeyroux (CRETA) a réalisé un échantillon sous forme de ruban, de composition similaire à
celle déterminée pour les quasi-cristaux : Zr63Ti14.4Ni12.4Cu10.2. L’expérience in-situ faite sur celui-ci montre
que lors de la montée en température isochrone, une phase quasicristalline se forme pour une température

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 125
proche de 335 °C, qui présente les mêmes positions de raies que celles observées dans le Vit4 (cf. fig.5.38).
Ces quasicristaux disparaissent très rapidement vers 385 °C au profit de la phase cristalline Zr2Cu. Ceci
confirme que dans le cas du Vit4, le béryllium n’entre pas dans la composition des quasi-cristaux qui se
forment dans cet alliage, mais intervient comme un élément déstabilisateur de la matrice amorphe.
De nombreuses questions restent encore en suspens au sujet de la formation de ces quasi-cristaux.
Un des points sur lesquels nous n’avons pas encore de réponse est la limite de croissance des quasi-
cristaux alors que toute la phase amorphe n’a pas été transformée. L’explication la plus simple est de
supposer que cette croissance est limitée par un équilibre des potentiels chimiques empêchant le béryllium
de diffuser vers des zones déjà riches. Ainsi les quasi-cristaux ne pourraient plus se développer puisque le
béryllium les en empêcherait. On peut d’ailleurs s’étonner que cet aspect n’ait jamais été évoqué dans la
littérature. En effet, dans le cas d’une transformation polymorphique, il est difficile d’imaginer une explication
de l’arrêt de croissance des quasi-cristaux.
Un autre problème est de comprendre la faible intensité des pics de ZrBe2 dans les spectres de
diffraction X au regard de ceux des quasi-cristaux, bien que ceux-ci soient indubitablement détectés (cf.
fig.5.28). On peut également s’étonner qu’il n’ait jamais été observé de quasi-cristaux dans l’alliage Vit1 où
la concentration de béryllium est de 22,5% atomique, et la concentration relative en Zr et Ti identique à celle
du Vit4, comme nous l’avons montré dans la partie de ce chapitre consacrée au Vit1.
Figure 5.41 : diffraction de neutrons lors d’une expérience in-situ en isochrone sur un échantillon rubande composition Zr63Ti14.4Ni12.4Cu10.2. Les pics de la phase quasicristalline qui apparaissent (partie vertede la courbe) correspondent aux mêmes que ceux observés dans le Vit4.
pics de la phase quasicristallineidentiques à ceux observés endiffraction de neutrons lors del'expérience in situ sur le Vit4.
pics de phases cristallines sedéveloppant à plus hautetempérature (Zr2Cu entre autre).
bosse amorphe

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 126
4 Conséquences sur les propriétés mécaniques
Nous avons vu que lors d’une montée linéaire en température, la cristallisation dans le cas du Vit1
s’effectuait en deux étapes, à la fin de la première étape l’échantillon n’étant que partiellement cristallisé,
alors qu’une seule étape de cristallisation est visible pour le Vit4. Dans cette section nous allons aborder les
conséquences d’un point de vue mécanique de cette différence de séquence de cristallisation.
4.1 cas du Vit1
Dans le cas du Vit1, nous avons étudié l’effet des traitements thermiques à 410 °C, c’est-à-dire la
même température que lors de l’étude microstructurale faite en microscopie. Ainsi, la figure 5.42 présente
l’évolution des modules de cisaillement avec la température pour des échantillons pré-traités à 410°C pour
des temps compris entre 0 min et 1 h. Afin de pouvoir comparer les essais mécaniques, chacune des
courbes obtenues a été recalée sur les autres en admettant que le module mesuré à basse température
après complète cristallisation est indépendant de l’histoire thermique. Le module mesuré à basse
température sur l’échantillon brut a été, quant à lui, normalisé à l’unité. Le premier effet visible du traitement
thermique isotherme est d’augmenter la valeur du module élastique à basse température. Ceci est
semblable à ce qui a été observé dans les échantillons montés en température en plusieurs étapes (cf.
figure 40 du chapitre 4). L’augmentation de module, proche de 10 % pour les échantillons recuits entre 15 et
60 min, est d’environ 20 % pour un temps de traitement de 4 h. Elle est probablement liée à un effet de
renfort de nanoparticules, c’est-à-dire les nanocristaux observés en microscopie, dans une matrice amorphe.
Cet effet de renfort par des nanocharges est fréquemment observé dans les polymères [Cha2002]. La
persistance d’une relaxation principale, même pour les échantillons prétraités pour les temps les plus longs,
indique la présence d’une phase amorphe encore importante. Il apparaît que la chute du module élastique
est d’autant plus décalée vers les hautes températures que le traitement thermique préalable a été long.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
300 350 400 450 500 550 600
T (°C)
G' /
Gu
brut
4h
15 min
cristallin
1 h
Figure 5.42 : Courbes de frottement intérieur pour des échantillons de Vit1pré-traités à 410°C pendant les temps indiqués.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 127
Ainsi en se plaçant à une température donnée, le module élastique mesuré paraîtra d’autant plus élevé que
le traitement thermique préalable a été long.
De la même manière qu’en spectrométrie mécanique, les courbes de compression sur des
échantillons pré-traités présentent des changements notables. En particulier, nous voyons sur la figure 5.43
que la contrainte d’écoulement est bien plus importante dans le cas des échantillons recuits. Là également,
ceci peut être attribué à l’effet de la présence des nanocristaux, mais également aux modifications de la
phase amorphe. Ainsi, la figure 5.44 montre que les échantillons ayant subi un traitement isotherme à 373°C
présentent un décalage de la température de transition vitreuse vers les plus hautes températures. Ceci est
à rapprocher de la séparation de la matrice amorphe en deux phases amorphes l’une riche en Ti et Ni,
l’autre en Zr et Be, observée en microscopie électronique. Ainsi les fluctuations de composition observées
dans la matrice amorphe du Vit1 modifient notablement les propriétés mécaniques.
6 0 0 6 5 0 7 0 0 7 5 0T (K )
Cp
(u.a
.)
1 0 K /m in
a m o rp h e
p a r t ie lle m e n tc r is ta l l is é
Figure 5.44 : thermogramme montrant le décalage vers les plushautes températures de la transition vitreuse des échantillons deVit1 pré-traités à 373 °C pendant 2h.
0100200300400500600700800900
1000
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7déformation
cont
rain
te (M
Pa)
brut
ε = 5*10-4s-1
373°C
364°C
373°C
364°C
.
Figure 5.43 : comparaison des courbes de compression à 364 et 373 °Cpour des échantillons bruts et pré-traités 2 h à 373 °C.

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 128
Malheureusement nous n’avons pas encore eu le temps d’étudier par microscopie électronique ni la
microstructure des échantillons avant déformation mais ayant subi un pré-traitement thermique à ces
températures, ni celle des échantillons déformés. L’analyse de la composition de la matrice résiduelle
pourrait confirmer les changements que nous avons observés à la température de 410 °C.
Dans le cas des matériaux nano-composites à base polymères, une modélisation du comportement
mécanique a été proposée. Elle prend en compte les propriétés de la matrice et des renforts, ainsi que des
effets de couplage mécanique entre matrice et renforts [Cha2002]. Dans le cas des verres métalliques, nous
pourrions envisager le même type d’approche. Cependant, nous nous heurtons à une difficulté majeure : la
décomposition de la matrice évolue de façon notable lors de la cristallisation comme l’ont prouvé les
analyses par microscopie électronique ou le décalage de la température de transition vitreuse. De ce fait,
nous ne disposons pas d’informations fiables sur les propriétés de la matrice. De la même façon, les
nanocristaux évoluent non seulement en taille mais aussi en composition lors des traitements isothermes ce
qui pose un problème pour connaître les caractéristiques mécaniques de ces nano-particules. Une telle
modélisation semble donc impossible à réaliser dans notre cas. En revanche, il serait particulièrement
intéressant de réaliser une étude dans le cas des matrices amorphes de compositions connues, stables,
renforcées par des nano-charges stables, par exemple des carbures. Ceci serait à poursuivre.
4.2 Cas du Vit4
Dans le cas du Vit4, nous avons étudié les conséquences de la cristallisation et du changement de
la composition de la matrice amorphe. La figure 5.45 présente l’évolution de la partie imaginaire du module
avec le temps, à une température de 380 °C, c’est-à-dire à la température à laquelle se forme la phase
quasicristalline observée et décrite précédemment. Nous voyons que durant les trois premières heures,
l’évolution du module est faible. Puis une chute se produit dans l’intervalle de temps de 3 à 4 h. D’un point
de vue microstructural, cela correspond à l’intervalle de temps durant lequel se forment les quasi-cristaux.
0
0.2
0.4
0.6
0.8
1
1.2
1 10 100 1000 10000temps (min)
G"
/ G"0
-0.6
-0.5
-0.4
-0.3
-0.2
-0.1
0
0.1
∆ (P
TE)
µV/K
Figure 5.45 : évolutions comparées de la partie imaginaire du module et des valeurs duPTE (courbes avec les marques) lors d’une isotherme à 380°C dans le Vit4.
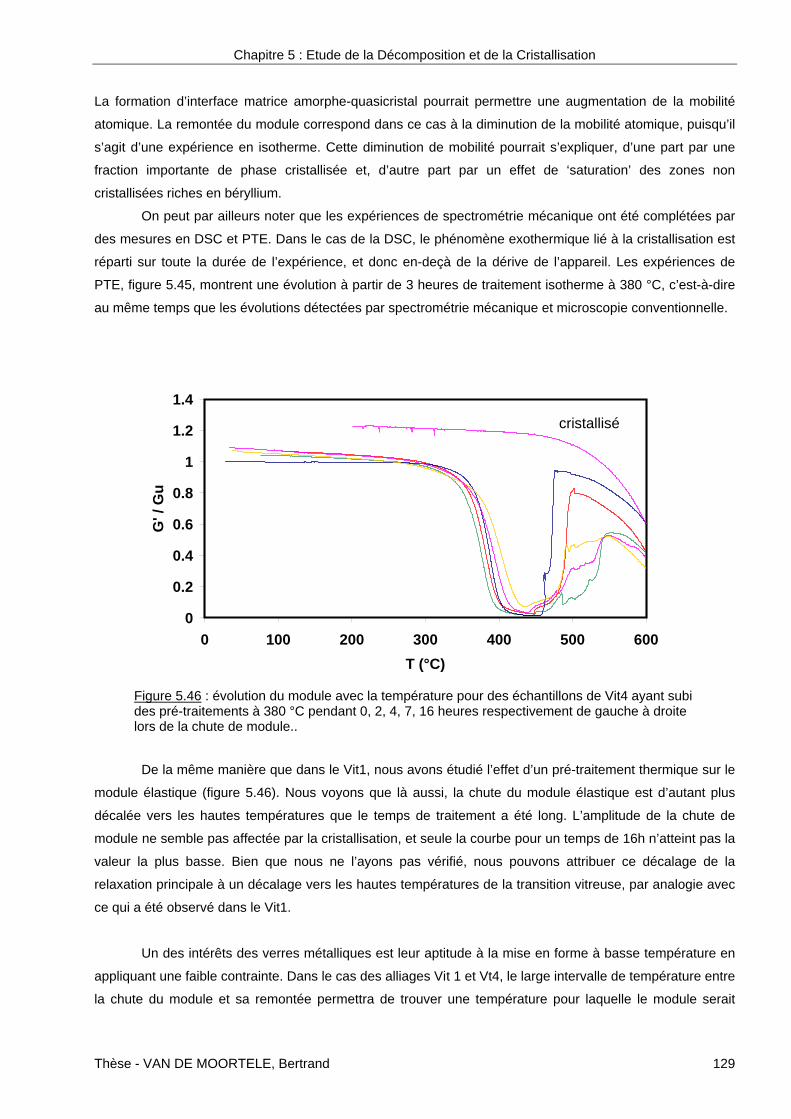
Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 129
La formation d’interface matrice amorphe-quasicristal pourrait permettre une augmentation de la mobilité
atomique. La remontée du module correspond dans ce cas à la diminution de la mobilité atomique, puisqu’il
s’agit d’une expérience en isotherme. Cette diminution de mobilité pourrait s’expliquer, d’une part par une
fraction importante de phase cristallisée et, d’autre part par un effet de ‘saturation’ des zones non
cristallisées riches en béryllium.
On peut par ailleurs noter que les expériences de spectrométrie mécanique ont été complétées par
des mesures en DSC et PTE. Dans le cas de la DSC, le phénomène exothermique lié à la cristallisation est
réparti sur toute la durée de l’expérience, et donc en-deçà de la dérive de l’appareil. Les expériences de
PTE, figure 5.45, montrent une évolution à partir de 3 heures de traitement isotherme à 380 °C, c’est-à-dire
au même temps que les évolutions détectées par spectrométrie mécanique et microscopie conventionnelle.
De la même manière que dans le Vit1, nous avons étudié l’effet d’un pré-traitement thermique sur le
module élastique (figure 5.46). Nous voyons que là aussi, la chute du module élastique est d’autant plus
décalée vers les hautes températures que le temps de traitement a été long. L’amplitude de la chute de
module ne semble pas affectée par la cristallisation, et seule la courbe pour un temps de 16h n’atteint pas la
valeur la plus basse. Bien que nous ne l’ayons pas vérifié, nous pouvons attribuer ce décalage de la
relaxation principale à un décalage vers les hautes températures de la transition vitreuse, par analogie avec
ce qui a été observé dans le Vit1.
Un des intérêts des verres métalliques est leur aptitude à la mise en forme à basse température en
appliquant une faible contrainte. Dans le cas des alliages Vit 1 et Vt4, le large intervalle de température entre
la chute du module et sa remontée permettra de trouver une température pour laquelle le module serait
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 100 200 300 400 500 600T (°C)
G' /
Gu
cristallisé
Figure 5.46 : évolution du module avec la température pour des échantillons de Vit4 ayant subi des pré-traitements à 380 °C pendant 0, 2, 4, 7, 16 heures respectivement de gauche à droite lors de la chute de module..

Chapitre 5 : Etude de la Décomposition et de la Cristallisation
Thèse - VAN DE MOORTELE, Bertrand 130
assez faible et sans risque de cristallisation. Nous avions évoqué dans le chapitre bibliographique que la
qualité des verres est généralement ramenée à l’étendue de la zone de liquide surfondu. Ainsi l’alliage à
base palladium qui présente une très large région de liquide surfondu subit une cristallisation
‘catastrophique’. Il semble donc qu’il faudrait nuancer l’étendue de la ZLS en évaluant également la largeur
de la zone de ‘travail’.

Chapitre 6 : Conclusion
Thèse - VAN DE MOORTELE, Bertrand 131
CHAPITRE 6
Conclusions et Perspectives
Conclusions
Ce travail s’inscrit dans une longue lignée de travaux portant sur les matériaux amorphes au sein du
laboratoire GEMPPM. On peut citer des études précédentes sur le comportement mécanique au voisinage
de la transition vitreuse des verres fluorés, des polymères, ainsi que des verres de silice. La possibilité
offerte par les verres métalliques massifs d’étudier le comportement mécanique d’un matériau amorphe
d’une autre catégorie et de le comparer au comportement mécanique d’autres matériaux amorphes ne
pouvait donc laisser indifférent. Comme nous l'avons vu dans ce mémoire, l'instabilité en température des
verres métalliques fait qu'à haute température ils cristallisent partiellement ou complètement modifiant ainsi
les propriétés mécaniques. Aussi avons-nous été amenés dans ce mémoire à séparer l'étude du matériau
amorphe à proprement parler de celle du matériau décomposé et/ou cristallisé.
L'étude par microscopie électronique en transmission sur des échantillons n’ayant subit aucun
traitement thermique a mis en évidence une cristallisation 'primaire' dans l’ensemble des verres métalliques
observés (excepté pour l’alliage Pd). Cette cristallisation qui n'avait pu être mise en évidence par la
diffraction des rayons X est à l'origine de la rupture prématurée lors d’essais de traction à froid comme nous
avons pu le constater dans le cas de l’alliage ZrAl.
L’analyse des propriétés mécaniques par spectrométrie mécanique ainsi que par compression à
chaud a permis de mettre en évidence l'existence d'une relaxation principale, thermiquement activée, dans
l'ensemble des verres métalliques étudiés. Ceci replace donc les verres métalliques parmi les autres
matériaux amorphes dans lesquels une telle relaxation est bien connue déjà. L'intensité du pic de relaxation
et sa forme dissymétrique ainsi que les paramètres physiques déduits des différentes expériences de
spectrométrie mécanique (énergie d'activation, temps de mobilité) permettent d'avancer que les
phénomènes de relaxation qui se produisent sont d'une part hiérarchiquement corrélés et d'autre part
distribués en temps. Par contre, hormis pour l'alliage Pd, aucune relaxation secondaire n'a pu être observée.
Le modèle de déformation des matériaux amorphes proposé par Perez et al., développé dans un
premier temps pour décrire la déformation des polymères amorphes, pour une large gamme de température
autour de la température de transition vitreuse Tg, a été rapidement étendu à d'autres matériaux amorphes.
Dans notre cas, nous avons vu que les résultats expérimentaux de déformation dans le domaine linéaire des

Chapitre 6 : Conclusion
Thèse - VAN DE MOORTELE, Bertrand 132
verres métalliques, correspondant à la spectrométrie mécanique, présentent un bon accord avec les
prédictions de ce modèle, mettant ainsi en évidence l'unicité des concepts utilisés dans celui-ci.
Une première approche qualitative de la modélisation de l'aspect non-linéaire de la déformation,
correspondant aux expériences de compression, laisse entrevoir là également un accord satisfaisant mais
moins bon. Des expériences complémentaires de compression à chaud seraient nécessaires dans ce
domaine pour affiner les paramètres d'ajustement.
La déstabilisation et/ou la cristallisation du matériau brut a dans un premier temps été mise en
évidence par l'étude des propriétés physiques de chaleur spécifique et de pouvoir thermoélectrique. Ceci
nous a permis de choisir les états microstructuraux du matériau que nous voulions étudier par microscopie
électronique en transmission.
Dans le cas du Vit1, nous avons montré par microscopie électronique en imagerie filtrée qu'à une
température de 410°C, une décomposition, peut-être spinodale, se développait, la matrice amorphe se
séparant en deux matrices amorphes : l'une riche en Ti et Ni d'une part et l'autre en Zr et Be d’autre part. Le
cuivre ne semble pas ou peu intervenir dans le cadre de cette décomposition.
La première phase cristalline qui se forme par la suite dans ces conditions expérimentales semble
identique d'un point de vue cristallographique à celle observée par Wang et al. à plus basse température.
Ceci indique que, contrairement à ce qu’il est fréquent de lire dans la littérature, le processus de
décomposition et cristallisation du Vit1 est le même à 350 et 410 °C. Les analyses chimiques par EDX ont
prouvé un enrichissement en Ti et Ni de cette première phase cristalline qui se formerait dans des zones de
fluctuations chimiques maximales de la décomposition (spinodale ?) de la matrice amorphe. La seconde
phase se formerait quant à elle dans les extrema inverses de la décomposition.
Les images de microscopie en haute résolution et les diffractions électroniques faites sur les phases
cristallines qui se développent dans le Vit4 pendant un traitement isotherme une température proche de la
transition vitreuse ont confirmé la présence de quasicristaux et de la phase cristalline ZrBe2. Les analyses
chimiques par EDX et EELS ont dans un premier temps permis de révéler l’absence de béryllium dans la
phase quasicristalline. Une quantification plus précise de cette phase a abouti à la composition suivante de
la phase quasicristalline : Zr64.4Ti7.2Cu11.75Ni16.6. L’élaboration par le CRETA, d’un ruban amorphe d’un alliage
de même composition cristallisant sous la forme d'un quasicristal de même paramètre cristallographique a
confirmé le résultat des analyses chimiques, c'est-à-dire l’absence de béryllium dans les quasicristaux. Du
point de vue des élaborateurs ce résultat semble surprenant puisque l’augmentation du taux de béryllium
entre le Vit4 et le Vit1 (ainsi que dans d’autres alliages) facilite la formation des quasi-cristaux. Grâce à
l’analyse chimique nous avons montré que dans ce genre d’alliage, le béryllium de part sa taille sans doute,
est un élément que diffuse à plus basse température permettant ainsi d’atteindre localement des
compositions voisines de la composition de la phase quasicristalline.
Enfin, la microscopie électronique a apporté un éclairage sur les modifications des propriétés
mécaniques induites par la cristallisation. L’augmentation du module à basse température est probablement

Chapitre 6 : Conclusion
Thèse - VAN DE MOORTELE, Bertrand 133
due à une nanocristallisation qui fait office de renfort, cet effet de renfort ayant déjà été observé dans de
nombreux matériaux polymères (PVC ou PET par exemple). D’autre part, la séparation de la phase amorphe
en ‘deux’ phases amorphes à une température de 410°C et le décalage de la température de transition
vitreuse explique le décalage de la chute de module vers les plus hautes températures.
Perspectives
En ce qui concerne la modélisation de la déformation mécanique se pose le problème de la
présence de nanoparticules. En effet, dans notre cas, la nanocristallisation s'accompagne d'une modification
des propriétés de la matrice amorphe par rapport à l'état brut. Il est alors difficile de distinguer les parts
respectives du changement dues d'une part aux modifications de la matrice amorphe et d'autre part à la
présence de nanoparticules de plus haut module élastique. Comme nous l'avons vu, la décomposition de la
matrice amorphe en deux phases amorphes a pour conséquence de décaler vers les plus hautes
températures la relaxation principale et par-là même la chute du module. Ainsi à une température donnée,
en dessous de la température de cristallisation, lors d'un essai de compression, le module, et donc la
viscosité, apparaît plus élevé dans le cas de la matrice décomposée. Pour s'affranchir de cet aspect, il serait
nécessaire de mener une étude de déformation à chaud sur les deux matériaux suivants :
une matrice amorphe de type (M),
un matériau nanocomposite de matrice (M) contenant des nanoparticules de compositions et de
propriétés biens connues.
Du point de vue de l'élaboration, l'ajout de particules dans un tel alliage pourrait mener lors de
l'élaboration à une cristallisation de type hétérogène. C'est pourquoi le carbure de zirconium pourrait s'avérer
intéressant en raison de l'affinité chimique importante entre ces particules et la matrice participant ainsi au
principe de confusion menant à l'obtention de verres métalliques. Une collaboration entre le laboratoire
GEMPPM et l'université de Shanghai pourrait porter sur ce sujet dans le cadre de la thèse en co-tutelle de
Qing Wang.
On peut également s'interroger sur les conséquences de la déformation sur le processus de
cristallisation. Nous avons vu que dans les verres métalliques possédant la zone de liquide surfondu la plus
large, la cristallisation nécessitait une diffusion atomique à longue échelle. Dans le cas d'une déformation, le
nombre de défauts sera augmenté ce qui aura pour conséquence de faciliter la diffusion. D'un autre côté, la
déformation pourra avoir pour conséquence de détruire les germes pouvant conduire à la cristallisation.
Nous voyons donc que l'influence de la déformation sur la cristallisation sera le résultat d'une compétition
entre phénomènes favorable et défavorable. Un début de réponse pourrait être apporté par les travaux de
microscopie actuellement en cours sur des échantillons déformés et non déformés.

Chapitre 6 : Conclusion
Thèse - VAN DE MOORTELE, Bertrand 134
L'apport de la microscopie électronique dans ce genre d'étude est incontestable. On peut d'ailleurs
s'étonner qu'assez peu de travaux fassent appel à cette approche expérimentale. Aussi, la méthodologie
développée dans le cadre de notre étude pourrait être étendue à toute étude à l'échelle nanoscopique de
phénomène de démixtion dans les verres, par exemple dans les verres minéraux, ou même les alliages
dans lesquels une mise en ordre chimique ou une précipitation a lieu.
Nous pouvons dégager principalement deux axes intéressants vers lesquels des travaux en
microscopie électronique pourraient êtres développés :
ainsi, l'étude de l'évolution de l'ordre à courte distance par diffusion des rayons X dans le cas d'une
décomposition spinodale ne semble pas la plus pertinente. D'une part, la taille des faisceaux qu'il est
possible d'atteindre est de l'ordre de 0.1*0.1 mm2, c'est-à-dire bien plus grande que la taille des
domaines de décomposition, d'autre part il n'est pas possible de savoir où l'on se situe par rapport à ces
domaines de composition. Dans le cas de la microscopie, les zones étudiées peuvent être de 10*10 nm2
mais surtout les analyses chimiques permettent immédiatement de distinguer les zones analysées.
un travail de cristallographie pourrait être mené sur les phases qui se forment dans le Vit1. Il apparaît
clairement que pour pouvoir mieux détailler la chronologie de la formation de ces phases, il serait
nécessaire d'opérer des traitements thermiques à plus basse température et des temps plus longs au
détriment de la caractérisation des propriétés physiques. Ce travail devrait être complété par des
analyses chimiques plus fines sur les nano-précipités, en particulier en ce qui concerne le béryllium, afin
de confirmer ce que nous avons pu observer.
Enfin, de manière plus éloignée mais tout aussi intéressante, l'absence du béryllium dans les
quasicristaux observés dans le Vit4 pose un problème plus large sur la formation des quasicristaux dans les
verres métalliques contenant du béryllium. La formation d'une phase quasicristalline dans les alliages à base
zirconium pourrait ne se faire que dans une certaine gamme de composition et sans béryllium. Pour
confirmer cela, des échantillons nous ont été envoyés par le Pr. Kim du Laboratoire des Sciences
Appliquées de l'université de Chongju en Corée du Sud (Divison of Applied Science). Des analyses
chimiques devraient être effectuées sur ces matériaux de composition Ti40Zr25Ni8Cu9Be18 dans lesquels une
phase quasicristalline a été observée [Kim2002]. Elles devraient permettre de savoir si dans ce cas
également les quasicristaux ne contiennent pas de béryllium et d'apporter dans ce cas une réponse à cette
question.
Dans le même esprit, l'observation de quasicristaux dans le Vit1 devrait être confirmée et des
analyses chimiques de compositions devraient être menées.

135
[Abb1987] Abbes K. Etude des propriétés micromécaniques des verres fluorés à base de ZrF4. Thèse INSA
de Lyon, 1987.120p.
[Ada1965] Adam G., Gibbs J.H.,On the temperature dependence of cooperative relaxation in glass-forming
liquids. Journal of chemical physics, 1965, p.139-146.
[Ahn1983] Ahn C.C., Krivanek O.L., EELS Atlas. Publié en 1983 par ASU HREM facilty© et GATAN©, 169p.
[Ang1988] Angell A., Perspective on the glass transition. Journal of chemical physics, 1988, vol.49, p.863-
871.
[Arg1979] Argon A.S., Plastic deformation in metallic glasses. Acta Metallurgica, vol.27, p.47-58.
[Ash2001] Ashby M., Bréchet Y. et Salvo L., Sélection des matériaux et des procédés de mise en œuvre,
Traité des matériaux, 20ème volume, chapitre 1. Presses polytechniques et Universitaires Romandes, 478p.
[Bru1985] Bruckner R., Response and structure of glass melts under extreme forming process. Journal of
Non-Crystalline Solids, 1985, vol.73, p.421-449.
[Bus1995] Busch R., Schneider S., Peker A. et Johnson W.L., Decomposition and primary crystallization in
undercooled Zr41.2Ti13.8Cu12.5Ni10Be22.5 melts. Applied Physics Letters, 1995, vol.67, p.1544-1546.
[Cah1958] Cahn J.W. et Hilliard J.E., Free Energy of a Nonuniform system. I. Interfacial free energy. Journal
of chemical physics, 1958, vol. 28, p.258-267.
[Cav1988] Cavaillé J. Y., Salvia M., Merzeau P. Un nouvel outil d'analyse de spectrométrie mécanique : le
micromécanalyseur. Spectra 2000., 1988, vol.16, p.37-45.
[Cha2002] Chabert E. Propriétés mécaniques de nanocomposites à matrice polymère : approche
expérimentale et modélisation. Thèse INSA de Lyon, 2002. 218p.
[Che1974] Chen H.S. Acta Metallurgica, 1974, vol.22, p.1505-1511.
[Che1999] Chen M.W., Zhang T., Inoue A. et Sakuraï T., Quasicrystals in a partially devitrified
Zr65Al7.5Ni10Cu12.5Ag5. Appl. Phys. Lett., 1999, vol.75, p.1697-1699.
[Che2000] Chen M.W., Inoue A., Zhang T., Sakai A. et Sakuraï T., Formation of icosahedral in an annealed
Zr65Al7.5Ni10Cu12.5Ag5 metallic glass. Phil. Mag. Letters, 2000, vol.80, p.263-269.
[Coh1959] Cohen M.H. et Turnbull D.J., Molecular transport in liquid and glasses. Journal of Chem.Phys.,
1959, vol.31, p.1164-1169.
[Ege1996] Egerton R.F., Electron Energy-Loss Spectroscopy in the Electron Microscope. , 2nd Edition,
Plenum Publishing Co., New York, 1996, 495p.

136
[Ehm1998] Ehmler H., Heesemann A., Rätzke K. et Faupel F., Mass dependence of diffusion in a
supercooled Metallic melt. Physical Review Letters, 1998 , vol.80, p.4919-4922.
[Eyr1936] Eyring M.J., Viscosity, plasticity, and diffusion as examples of absolute reactions rates. Journal of
Chemical Physics, 1936, vol.4, p.283-291.
[Fan1999] Fan C., Louzguine D.V., Li C. et Inoue A., Nanocrystalline composites with high strength obtained
in Zr-Ti-Ni-Cu-Al bulk amorphous alloys. Appl. Phys. Lett., 1999, vol.75, p.340-342.
[Fan2000] Fan C., Li C. et Inoue A.. Nanocrystal composites in Zr-Nb-Cu-Al metallic glasses. Journal of Non-
Crystalline Solids, 2000, vol.270, p.28-33.
[Fan2001a] Fan C., Li C. et Inoue A., Effects of Nb addition on icosahedral phase formation and glass-
forming ability of Zr-Nb-Ni-Cu-Al metallic glasses. Appl. Phys. Lett., 2001, vol.79, p.1024-1026.
[Fan2001b] Fan C. et Inoue A., Formation of nanoscale icosahedral quasicrystals and glass-forming ability in
Zr-Nb-Ni-Cu-Al metallic glasses. Scripta Mater., 2001, vol.115, p.115-120.
[Fie1999] Fielitz P., Macht M.P., Naundorf V. et Frohberg G. Diffusion in ZrTiCuNiBe bulk glasses at
temperatures around the glass transition. Journal of Non-Crystalline Solids, 1999, vol.250-252, p.674-678.
[Gan2000] Gangopadhyay A.-K., Croat T.-K. et Kelton K.-F., The effect of phase separation on subsequent
crystallization in Al88Gd6La2Ni4. Acta Mater., 2000, vol.48, p.4035-4043.
[Geb1998] Gebert A., Eckert J. et Schultz L., Effect of oxygen on phase formation et thermal stability of
slowly cooled Zr65Al7.5Cu17.5Ni10 metallic glass. Acta mater., 1998, vol.46, p.5475-5482.
[Ger1997] Gerold U., Wiedenmann A., Keiderling U. et Fecht H.J., Decomposition and crystallization of the
bulk amorphous Zr11Ti34Cu47Ni8 alloy studied by SANS. Physica B, 1997, vol.234-236, p.995-996.
[Gut1993] Gutzow I., Dobreva A., et Schmelzer J., Rheology of non-Newtonian glass-forming melts Part I :
Flow stress relations. Journal of Materials Science, 1993, vol.28, p.890-900.
[He2000] He G., Bian Z. et Chen G.-L., Investigation of phases on a Zr-based bulk glass alloy. Materials
Science and Engineering, 2000, vol. A279, p.237-243.
[Hei2001] Heilmaier M., Deformation behavior of Zr-based metallic glasses. Journal of Materials Processing
Technology, 2001, vol.117, p.374-380.
[Ino1988] Inoue A., Kohinata M., Tsai A.P. et Masumoto T., Mg-Ni-La Amorphous Alloys with a Wide
Supercooled Liquid Region. Materials Transactions, JIM, 1988, vol.30, p.378-381.
[Ino1993] Inoue A., Zhang T. et Masumoto T., Glass-forming ability of alloys. Journal of non-Crystalline
Solids, 1993, vol.156-158, p.473-480.

137
[Ino1995] Inoue A., High Strength Bulk Amorphous Alloys with Low Critical Cooling Rates. Materials
Transactions,1995, JIM, vol.36, p.866-875.
[Ino1996] Inoue A., Nishiyama N et Matsuda T., Preparation of Bulk Glassy Pd40Ni10Cu30P20 Alloy of 40
mm in Diameter by Water Quenching. Materials Transactions, JIM, 1996, vol.37, p.181-184.
[Ino1997] Inoue A. et Zhang T., Thermal stability and glass-forming ability of amorphous Nd-Al-TM (TM =
Fe, Co, Ni or Cu) alloys . Materials Science and Engineering A, 1997, vol. 226-228, p.393-396.
[Ino1999] Inoue A., Bulk Amorphous Alloys : practical characteristics and applications. Materials Science
Foundations, 1999, vol.6, 148p.
[Ino1999] Inoue A., Stabilization and high strain-rate superplasticity of metallic supercooled liquid, Materials
Science and Engineering A, 1999, vol. 267, p.171-183.
[Ino2000] Inoue A., Zhang T, Chen M.W., Sakurai T., Saida J. et Matsushita M., Ductile quasicrystalline
alloys. Appl. Phys. Lett., 2000, vol.76, p.967-969.
[Jia2001] Jiang J.Z., Saskl K., Rasmussen H., Watanuki T., Ishimatsu, N., et Shimomara, O., High-pressure
x-ray diffraction of icosahedral Zr-Al-Ni-Cu-Ag quasicrystals. Appl. Phys. Lett., 2001, vol.7, p.1112-1114.
[Kat1998] Kato H., Kawamura Y., Inoue A. et Chen H.S., Newtonian to non-Newtonian master curves of a
bulk glass alloy Pd40Ni10Cu30P20. Appl. Phys. Lett., 1998, vol. 73, p.3665-3667.
[Kaw2000] Kawamura Y. et Inoue A., Newtonoian viscosity of supercooled liquid in a Pd40Ni40P20 metallic
glass. Appl. Phys. Lett., 2001, vol. 76, p.1114-1116.
[Kaw2001] Kawamura Y., Nakamura T., Kato H., Mano H et Inoue A., Newtonoian and non-Newtonian
viscosity of supercooled liquid in metallic glasses. Materials Science and Engineering, 2001, vol.A304-306,
p.674-678.
[Kim2002] Kim Y.C., Park J.M., Lee J.K., Bae D.H., Kim W.T. et Kim D.H., Amorphous and icosahedral
phases in Ti-Zr-Ni-Cu-Be alloys. Conférence orale, Congrès RQ11, Oxford, 2002, à paraître dans Scripta
Materialia et conversation privée.
[Kle1960] Klement W., Willens R.H., et Duwez P., Non-crystalline structure in solidified Gold-Silicon alloys.
Nature, 1960, vol.187, p.869-870.
[Kös1997] Koster, U., Meinhardt, J., Roos, S. et Busch, R. Formation of quasicrystals in bulk glass forming
Zr-Cu-Ni-Al alloys. Mat. Sci. Eng. A, 1997, vol.226-228, p.995-998.
[Küh2000] Kühn U., Eckert J., Mattern N., et Schults L., As-cast quasicrystalline phase in a Zr-based
multicomponent bulk alloy. Appl. Phys. Lett., 2000, vol. 77, n°20, p.3176-3178.

138
[Kul2001] Kulik T., Nanocrystallization of metallic glasses. Journal of Non-Crystalline Solids, 2001, vol.287,
p.145-161.
[Kül2002] Külik T., Magnetically soft nanocrystalline materials obtained by devitrification of metallic glasses.
Conférence orale, Congrès RQ11, Oxford, 2002, à paraître dans Scripta Materialia.
[Lee2000] Lee J.K., Choi G., Ki, D.H., et Kim W.T., Formation of icosahedral phase formation from
amorphous Zr65Al7.5Cu12.5Ni10Ag5 alloys. Appl. Phys. Lett. 2000, vol.77, p.978-980.
[Li2000a] Li C., Saida J., Matsushita M., et Inoue A., Precipitation of icosahedral quasicrystalline phase in
Hf65Al7.5Ni10Cu12.5Pd5 metallic glass. Appl. Phys. Lett., 2000, vol.77, p.528-530.
[Li2000b] Li C., Saida J., Matsushita M., et Inoue A., Precipitation of icosahedral quasicrystalline phase in
Hf59Ni8Cu20Al10Ti3 metallic glass. Phil. Mag. Letters, 2000, vol.80, p.621-626.
[Li2000c] Li C., Saida J., Matsushita M., et Inoue A., Effect of Sn addition on the glass forming ability in
(Cu40Ti30Ni15Zr10)(100—x)/95Snx (x=0, 2, 4, 6 and 8) alloys. Scripta Mat., 2000, vol.42, p.923-927.
[Liu1997] Liu J.M., Decomposition of amorphous Zr41Ti14Cu12.5Ni10Be22.5 alloy : as investigated by small angle
neutron scattering. Appl. Phys. Lett., vol.70, p.1968-1970
[Lof2000a] Löffler J.L. et Johnson W.L., Model for decomposition and nanocrystallization of deeply
undercolled Zr41.2Ti13.8Cu12.5Ni10Be22.5. Appl. Phys. Lett., vol.76, n°23, p.3394-3396.
[Lof2000b] Löffler J.L., Bossuyt S., Glade S.C., Johnson W.L., Wagner W. et Thiyagarajan, Crystallization of
bulk amorphous Zr-Ti(Nb)-Cu-Ni-Al. Appl. Phys. Lett., 2000, vol.77, p.525-527.
[Lof2001a] Löffler J.L. et Johnson W.L., Crystallization pathways of deeply undercooled Zr-Ti-Cu-Ni-Be
melts. Scripta Materialia, 2001, vol.44, p.1251-1255.
[Löf2001b] Löffler J.L. et Johnson W.L., Model for decomposition et crystallization of Zr-based bulk
amorphous alloys near the glass transition. Materials Science et Engineering, 2001, vol. A304-306, p.670-
673.
[Lou2000] Louzguine, D.V., Ko, M.S. et Inoue A., Nanoquasicrystalline phase produced by devitrification of
Hf-Pd-Ni-Al metallic glass. Appl. Phys. Lett., 2000, vol.76, p.3424-3426.
[Lou2001] Louzguine, D.V. et Inoue A., Formation of a nanoquasicrystalline phase in Zr-Cu-Ti-Ni metallic
glass. Appl. Phys. Lett., 2001, vol.78, p.1841-1843.
[Mac2001a] Macht M.P., Naundorf V., Fielitz P., Rüsing J., Zumkley Th. et Frohberg G., Dependence of
diffusion on the alloy composition in ZrTiCuNiBe bulk glasses. Materials Science and Engineering, 2001, vol.
A304-306, p.646-649.

139
[Mac2001b] Macht M.P., Wanderka N., Wei Q., Sieber I. et Deyneka N., Tendancy of primary crystal
formation in ZrTiCuNiBe metallic glasses. Materials Science and Engineering A, 2001, vol. A304-306,
p.701-705.
[Mat2001] Mattern N., Kühn U. et Eckert J., Structure formation and crystallisation of Zr62-xTixAl10Cu20Ni8 bulk
metallic glasses. Proceeding of the 22nd Riso International Symposium on Materials science, 2001, p.323-
328.
[Mat2002] Mattern N., Kühn U., Hermann H., Ehrenberg H., Neuefeind J. et Eckert J., Short-range order of
Zr62-xTixAl10Cu20Ni8 bulk metallic glasses. Acta Materialia, 2002, vol.50, p.305-314.
[Max1979] Maxwell B.et Nguyen M., Measurement of the elastic properties of polymers melts. Polymer
Engeneering Science, 1979, vol.19, p.1140-1150.
[Mid1962] Middleman S., The Flow of High Polymers, chapitre 4. Academic press. New York, 1962.
[Mon1981] Montrose C.J., Litovitz T.A., Heyes D.M., Simmons J.H., Mohr R.K., Kim J.J., Grant S.D.,
Rekhson S.M., Dynamical nonlinear material response and failure. Catholic University of America, Ofice of
Naval research, Technical Report CUA/VSL, 1981.
[Mur2000a] Murty B.S., Ping D.H., Hono K. et Inoue A., Direct evidence for oxygen stabilization of
icosahedral phase during crystallization of Zr65Cu27.5Al7.5 metallic galsses. Appl. Phys. Lett., 2000, vol.76,
p.55-57.
[Mur2000b ] Murty B.S., Ping D.H., Hono K. et Inoue A., Influence of oxygen on the crystallization behavior
of Zr65Cu27.5Al7.5 and Zr66.7Cu33.3 metallic galsses. Acta mater., 2000, vol.48, p.3985-3996.
[Muz1991] Muzeau E., Perez J. et Johari G.P., Mechanical spectrometry of the β relaxation in Poly(methyl-
methacrylate). Macromolecules, 1991, vol.24, p.4713-4723.
[Nie2001] Nieh T.G., Wadsworth J., Liu C.T., Ohkubo T. et Hirotsu Y., Plasticity and structural instability in a
bulk metallic glass deformed in the supercooled liquid region. Acta Materialia, 2001, vol.49, p.2887-2896.
[Oua1992] Ouali N., Etude de la déformation non élastique (faible et forte contrainte) de polymères
amorphes monophasés et polyphasés base PMMA. Thèse INSA de Lyon, 1992, 237p.
[Pek1993] Peker A.L. et Johnson W.L., A highly processable metallic glass: Zr41.2Ti13.8Cu12.5Ni10.0Be22.5.
Applied physics Letters, 1993, vol.63, n°17, p.2342-2344.
[Pel1980] Pelletier J.M., Pouvoir thermoélectrique d’alliages à base de cuivre et d’aluminium ; étude des
solutions solides et rôle de la précipitation d’une seconde phase. Thèse Insa de Lyon, 185p., 1980.

140
[Pel1986] Pelletier J.M., Fouquet F. et Bouras S, Reversible and Irreversible changes in amorphous alloys :
detection by termoelectric power measurements. Materials Science and Engineering, 1986, vol.77 p.175-
179.
[Per1988] Perez J., Study of polymer materials by mechanical spectroscopy methods. Polymer Science,
ser.B, 1988, vol.40, p.17-46.
[Per1992] Perez J., Physique et mécanique des polmères amorphes. Paris : Lavoisier, 1992, 384p.
[Per2001] Perez J., Matériaux non cristallins et science du désordre. Presse Polytechnique et Universitaires
Romandes, 2001, 557p.
[Pra1928] Prandtl L., Ein Gedankenmodell zu kinetischen Theorie der festen Körper, Seminar on the theory
of elasticity. ZAMM, 1928, p.85-106.
[Qui1995] Quinson R., Caractérisation et modélisation de la déformation non élastique des polymères
amorphes à l’état solide. Thèse INSA de Lyon, 1995, 252p.
[Reg2000] Reger-Leonhard A., Heilmaier M. et Eckert J., Newtonian flow of Zr55Cu30Al10Ni5 bulk metallic
glassy alloys. Scripta mater., 2000, vol. 43, p.459-464.
[Reh2001] Rehmet A., Rätzke K., Faupel F., Eversheim P.D., Freitag K., Geyer U. et Schneider S., Be tracer
diffusion in a deeply supercooled Zr46.7Ti8.3Cu7.5Ni10Be27.5 melt. Applied Physics Letters, 2001, vol. 78,
p.2892-2894.
[Rei1995] Reimer L., Energy-Filtering Transmission Electron Microscopy, Springer Series in Optical
Sciences, vol. 71, Springer-Verlag, Berlin, 342p.
[Rek1991] Rekhson S., A model for non-linear viscoelastic relaxation. Journal of Non-Crystalline Solids,
1991 , vol. 131-133, p.467-475.
[Rev2001] Revesz A., Donnadieu P., Simon J.-P., Guyot P. et Ochin P. Nanocrystallization in a
Zr57Ti5Cu20Al10Ni8 metallic glass. Phil.Mag.Letters. 2001, vol.81, p.767-775.
[Sai1999] Saida, J., Matsushita, M., Zhang, T., Inoue, A., Chen, M.W. et Sakurai T., Precipitation of
icosahedral phase from a supercooled liquid region in Zr65Cu7.5Al7.5Ni10Ag10 metallic glass. Appl. Phys. Lett.,
1999, vol.75, p.3497-3499.
[Sai2000a] Saida, J., Matsushita, M., et Inoue, A., Nano icosahedral phase formation by crystallization of Zr-
based ternary glassy alloys. Scripta Mat., 2000, vol.44, p.1245-1249.
[Sai2000b] Saida J., Li C., Matsushita M., et Inoue A., Nano-icosahedral quasicrystalline phase formation
from a supercooled liquid state in Zr-Fe-Ni ternary metallic glass. Appl. Phys. Lett., 2000, vol.76, p.3037-
3039.

141
[Sai2001a] Saida J., Matsushita M., Li C. et Inoue A., Formation of the icosahedral quasicrystalline phase
un Zr70Pd30 binary glassy alloy. Phil. Mag. Letters, 2001, vol.81, p.39-44.
[Sai2001b] Saida J., Matsushita M. et Inoue A., Nano icosahedral phase formation by crystallization of Zr-
based ternary glassy alloys. Scripta Materialia, 2001, vol.44, p.1245-1249.
[Sai2002] Saida J., Ishihara S., Kato H., Inoue A. et Chen H.S., Supression of quasicrystallization by
nonlinear viscous flow in Zr-Al-Ni-Cu-Pd glassy alloys. Applied Physics Letters, 2002, vol.80, n°25, p.4708-
4710.
[She1984] Schechtman D., Blech I., Gratias D. et Cahn J.W., Metallic phase with long-range orientational
order and no translational symmetry. Physical Rewiev Letters, 1984, vol.53, n°20, p.1951-1954.
[Sch1996] Schneider S., Thiyagarajan P. et Johnson W.L., Formation of nanocrystals on decomposition in
the amorphous Zr41Ti14Cu12.5Ni10Be22.5 alloy. Appl. Phys. Lett., 1996, vol. 68,n°4, p.493-495.
[Sch1998] Schneider S., Thiyagarajan P., Geyer U. et Johnson W.L., SANS of bulk metallic ZrTiCuNiBe
glasses. Physica B, 1998, vol.241-243, p.918-920.
[Sek1992] Seekat A., Comportement micromécanique de système vitreux de nature différente autour de la
température de transition vitreuse : approche expérimentale et modélisation. Thèse INSA de Lyon, 1992,
231p.
[Spa1977] Spaepen F., A microscopic mechanism for steady state in homogeneous flow in metallic glasses,"
Acta Metallurgica, 1977, vol.25, p.407-415.
[Spr1997] Spriano S, Antonione C., Doglione R., Battezati L., Cardoso S., Soares J.C. et Da Silva M.F.,
Crystallization and mechanical behaviour of bulk Zr-Ti-Ni-Cu-Be metallic glasses. Phil. Mag. Letters B,
1997,vol.76, p.529-540.
[Str1978] Struik L.C.E., Physical ageing in amorphous polymers and other materials. Elseviers Scientific
Publishing Company, 1978, Amsterdam 1978, 230p.
[Tam1926] Tamman G. et Hesse G., Die abhängigkeit der viscosität von der temperatur bei unterkühlten
flüssigkeiten Z. Amorg. Allem. Chem., 1926, vol.156 , p.245-251.
[Tob1961] Tobolsky, A.V. Viscoelastics properties of polymers, New York : John Wiley and sons, 1961,
431p.
[Tsa1997] Tsai A.P., Kamiyama T., Kawamura Y., Inoue A. et Masumoto T., Formation et precipitation
mechanism of nanoscale Al particles in Al-Ni base amorphous alloys. Acta Mater., 1997, vol.45, p.1477-
1487.
[Vau1984] Vauglin C., Etude par mesure de PTE de la restauration et de la recristallisation des tôles de
zircaloy-4 laminées. Thèse INSA de Lyon, 1984, 160p.

142
[Wan1997] Wang W.H., Wei Q., Friedrich S., Macht M.P., Wanderka N. et Wollenberger H., Microstructure
studies of Zr41Ti14Cu12.5Ni10Be22.5 bulk amorphous alloy by electron diffraction intensity analysis. Appl. Phys.
Lett., 1997, vol.71, p.1053-1055.
[Wan1999a] Wanderka N., Czubayko U., Schubert-Bischoff P. et Macht M.P., Primary crystals in
ZrTiCUNiBe bulk glasses. Materials Science and Engineering, 1999, vol.A270, p.44-47.
[Wan1999b] Wanderka N., Wei Q., Sieber I., Czubayko U. et Macht M.P., Indentification of primary crystals
in ZrTiCuNiBe metallic glasses. Materials Science Forum, 1999, vol.312-314, p.369-374.
[Wan2000] Wanderka N., Macht M.P., Seidel M., Mechler S., Stahl K. et Jiang J.Z., Formation of
quasicrystals in Zr46.8Ti8.2Cu7.5Ni10Be27.5. Appl. Phys. Lett., 2000, vol 77, p.3935-3937.
[Wer1990] Werner P.E., logiciel treor, 1990.
[Xin1997] Xing L.Q., Herlach D.M., Cornet M., Bertrand. C., Dallas J.P., Trichet M.F. et Chevalier J.P.,
Mechanical properties of Zr57Ti5Al10Cu20Ni8 amorphous and partially nanocrystallized alloys. Materials
Science and Engineering, 1997, vol.226-228, p.874-877.
[Xin1998] Xing., L.Q., Eckert, J., Loser, W. et Schultz, L. Effect of cooling rate on the precipitation of
quasicrystals from the Zr-Cu-Al-Ni-Ti amorphous alloy. Appl. Phys. Lett., 1998, vol.73, p.2110-2112.


















