
Emilie ROUTIER thèse de médecinedoxa.u-pec.fr/theses/th0602494.pdf · A tous ceux que j’ai...
91
1 UNIVERSITE PARIS VAL-DE-MARNE FACULTE DE MEDECINE DE CRETEIL ****************** ANNEE 2010 N° THÈSE POUR LE DIPLOME D’ETAT DE DOCTEUR EN MÉDECINE Discipline : DERMATOLOGIE ---------------- Présentée et soutenue publiquement le 21 octobre 2010 A Saint-Antoine ---------------- Par Mlle Emilie ROUTIER Née le 8 janvier 1981 à VILLENEUVE-SAINT-GEORGES ---------------- TITRE : MUTATION DE KIT ET DE PDGFRA DANS LE MELANOME : SERIE DE 107 PATIENTS PRESIDENT DE THESE : LE CONSERVATEUR DE LA Monsieur le Professeur Selim Aractingi BIBLIOTHEQUE UNIVERSITAIRE DIRECTEUR DE THESE : Madame le Docteur Caroline ROBERT Signature du Cachet de la bibliothèque Président de thèse universitaire
Transcript of Emilie ROUTIER thèse de médecinedoxa.u-pec.fr/theses/th0602494.pdf · A tous ceux que j’ai...

1
UNIVERSITE PARIS VAL-DE-MARNE
FACULTE DE MEDECINE DE CRETEIL
******************
ANNEE 2010 N°
THÈSE
POUR LE DIPLOME D’ETAT
DE
DOCTEUR EN MÉDECINE
Discipline :
DERMATOLOGIE
---------------- Présentée et soutenue publiquement le 21 octobre 2010
A Saint-Antoine
---------------- Par Mlle Emilie ROUTIER
Née le 8 janvier 1981 à VILLENEUVE-SAINT-GEORGES
----------------
TITRE : MUTATION DE KIT ET DE PDGFRA DANS LE MELANOME :
SERIE DE 107 PATIENTS
PRESIDENT DE THESE : LE CONSERVATEUR DE LA
Monsieur le Professeur Selim Aractingi BIBLIOTHEQUE UNIVERSITAIRE
DIRECTEUR DE THESE :
Madame le Docteur Caroline ROBERT
Signature du Cachet de la bibliothèque
Président de thèse universitaire

2
REMERCIEMENTS
Au Professeur Sélim ARACTINGI, qui me fait l’honneur de présider ce jury.
Au Docteur Caroline ROBERT, qui m’a proposé ce sujet de travail et m’a fait confiance pour le
mener à bien.
Au Professeur Jean-Charles SORIA qui m’a accordé sa confiance tout au long de cette année en
m’acceptant dans son service. Je suis très honorée que vous ayez accepté de siéger dans le jury de
cette thèse.
Au Docteur Manuelle VIGUIER, qui m’a fait l’honneur de bien vouloir juger ce travail.
Au Docteur Christina MATEUS, dont la disponibilité et l’engagement auprès des patients sont
pour moi des repères dans la pratique de la médecine. Que tu reçoives ici le témoignage de mon
plus grand respect et de ma plus sincère amitié.
Au Docteur Jean BENARD pour son aide précieuse et sa disponibilité, et à toute son équipe du
plateau technique de biologie moléculaire de l’Institut Gustave Roussy.
A tous mes co-internes tout au long de ces années d’internat, pour leur chaleureuse confraternité
et tous les bons moments vécus ensemble.
A Coralie, avec qui j’ai passé un semestre intense (KIT toujours !).
A Anette, co-interne à nos débuts (pas facile tous les jours !) et maintenant co-sportive, cette
année on ira jusqu’au bout ! A ton tour maintenant de soutenir ta thèse...
A Manuel, pour sa relecture attentive, ses conseils et astuces si précieux.
A Hélène et encore Manuel, qui ont su canaliser mes angoisses de dernière minute.
A Emilie, présente tout au long de ces années de médecine pour le meilleur et même le pire,
c’était long, mais c’était cool. Merci pour toute ton aide et tes précieux conseils.

3
A toute l’équipe de Dermatologie, toujours disponible, toujours de bonne humeur qui m’ont mis
la pression jusqu’au bout…et c’est reparti pour deux ans.
A toute l’équipe de SITEP, cette année a été remplie de découvertes, d’échanges, de complicité.
Je vous regrette déjà.
A mes futurs, qui sont un peu aussi mes actuels co-chefs, j’ai hâte de vous rejoindre à temps-
plein (dommage Jean-Phi, que tu ne fasses pas partie de la fête).
A tous ceux que j’ai sollicité pour la relecture de ma thèse et qui ont répondu présent.
A mes amis, présents tout au long de mon parcours et pour longtemps encore je l’espère.
A Léti, qui me supporte depuis tout ce temps, je n’oublierai jamais cette petite séance de mise en
page tardive qui nous a rendu un peu chèvre !!
A Kathia, parmi mes amis, la plus fidèle, et son trio de choc que je considère comme ma famille.
A mes oncles, tantes, cousins et cousines,
A Pierre-Antoine, qui malgré tout et contre tout, a toujours été d’un grand soutien et d’une
disponibilité à toute épreuve. Voilà, je l’ai enfin finie cette thèse !
A mes parents, qui ont toujours cru en moi. Que ce travail soit une marque de mon amour éternel
et de toute ma reconnaissance. Clin d’œil à mon père toujours présent en régie et à ma mère
attentive, à chaque instant, à mes besoins.
A ma sœur, que j’aime tout simplement.

4
A ma grand-mère,
A mon grand-père, j’aurais tellement aimé que tu sois là pour le jour J…
Vous êtes maintenant réunis pour l’éternité.
Je vous dédie cette thèse.

5
INTRODUCTION..................................................................................................................................................... 8 1. GENERALITES................................................................................................................................................. 9 1.1 MELANOME.......................................................................................................................................................................9 1.1.1 Epidémiologie ............................................................................................................................................................9 1.1.2 Traitement du mélanome métastatique ..................................................................................................... 10 1.1.3 Vers une nouvelle classification ...................................................................................................................... 11 1.1.4 Altérations génétiques distinctes dans les différents sous-types de mélanomes ....................... 12 1.1.5 Les différents sous-types de mélanome avec anomalie de KIT.......................................................... 14 1.1.5.1 Les mélanomes acro-‐lentigineux (ALM) ................................................................................................................................. 14 1.1.5.2 Les mélanomes muqueux .............................................................................................................................................................. 15 1.1.5.2.1 Tête et cou ................................................................................................................................................................................... 15 1.1.5.2.2 Tube digestif ............................................................................................................................................................................... 15 1.1.5.2.3 Tractus uro-‐génital .................................................................................................................................................................. 16
1.1.5.3 Mélanome de Dubreuilh ................................................................................................................................................................. 17 1.1.5.4 Mélanome sans primitif retrouvé............................................................................................................................................... 17
1.2 LES THERAPIES CIBLEES.............................................................................................................................................. 18 1.3 ROLE DE KIT ET PDGFRA......................................................................................................................................... 19 1.3.1 Description et activité biologique .................................................................................................................. 19 1.3.2 Les inhibiteurs des tyrosine kinases KIT et PDGFRA.............................................................................. 22 1.3.2.1 Avec une autorisation de mise sur le marché (AMM) (47) ............................................................................................. 22 1.3.2.1.1 Imatinib ........................................................................................................................................................................................ 23 1.3.2.1.2 Sunitinib ....................................................................................................................................................................................... 23 1.3.2.1.3 Sorafenib ...................................................................................................................................................................................... 23 1.3.2.1.4 Dasatinib ...................................................................................................................................................................................... 24 1.3.2.1.5 Nilotinib........................................................................................................................................................................................ 24
1.3.2.2 En cours de développement (49) ............................................................................................................................................... 24 1.3.2.2.1 Masitinib ...................................................................................................................................................................................... 24 1.3.2.2.2 XL820............................................................................................................................................................................................. 24 1.3.2.2.3 Motesanib .................................................................................................................................................................................... 25 1.3.2.2.4 Vatalanib ...................................................................................................................................................................................... 25 1.3.2.2.5 Midostaurin ................................................................................................................................................................................ 25 1.3.2.2.6 Cediranib...................................................................................................................................................................................... 25
1.4 UN MODELE ETABLI : LES TUMEURS STROMALES GASTRO-‐INTESTINALES (GIST).......................................... 26 1.4.1 Prise en charge des GIST avancées : place de l’imatinib ...................................................................... 26 1.4.2 GIST et résistance à l’imatinib......................................................................................................................... 29 1.4.2.1 Résistance primaire.......................................................................................................................................................................... 29 1.4.2.2 Résistance secondaire ..................................................................................................................................................................... 29
1.4.3 Alternatives thérapeutiques ............................................................................................................................. 31 1.4.3.1 Sunitinib ................................................................................................................................................................................................ 31 1.4.3.2 Les autres inhibiteurs de tyrosine kinase............................................................................................................................... 32 1.4.3.2.1 Nilotinib........................................................................................................................................................................................ 32 1.4.3.2.2 Sorafenib ...................................................................................................................................................................................... 32 1.4.3.2.3 Dasatinib ...................................................................................................................................................................................... 32
1.5 MUTATION DE KIT DANS LES AUTRES TUMEURS ................................................................................................... 33 1.5.1 Les tumeurs germinales ..................................................................................................................................... 33 1.5.2 La mastocytose....................................................................................................................................................... 35 1.5.3 La leucémie myéloïde aiguë.............................................................................................................................. 35
2. MATERIEL ET METHODES .......................................................................................................................36 3. RESULTATS...................................................................................................................................................37 3.1 CARACTERISTIQUES DES PATIENTS........................................................................................................................... 37 3.1.1 Mélanome de Dubreuilh ..................................................................................................................................... 37 3.1.2 Mélanomes muqueux ........................................................................................................................................... 38 3.1.3 Mélanomes Acro-lentigineux............................................................................................................................ 42 3.1.4 Mélanome sans primitif retrouvé................................................................................................................... 46

6
3.2 RESULTATS GENERAUX................................................................................................................................................ 47 3.3 OBSERVATIONS DES TROIS PATIENTES TRAITEES PAR INHIBITEUR DE KIT ET DE PDGFRA........................ 49 3.3.1 Observation N°1..................................................................................................................................................... 49 3.3.2 Observation N°2..................................................................................................................................................... 50 3.3.3 Observation N°3..................................................................................................................................................... 51
4. DISCUSSION ..................................................................................................................................................52 4.1 NOS MUTATIONS DANS LA LITTERATURE................................................................................................................. 52 4.1.1 Mutation L576P de l’exon 11............................................................................................................................ 52 4.1.1.1 Dans les GIST....................................................................................................................................................................................... 52 4.1.1.2 Dans le mélanome............................................................................................................................................................................. 52 4.1.1.3 Sensibilité au traitement (Tableau 7)....................................................................................................................................... 52
4.1.2 Mutation Y553C de l’exon 11 ........................................................................................................................... 56 4.1.2.1 Dans le GIST......................................................................................................................................................................................... 56 4.1.2.2 Dans le mélanome............................................................................................................................................................................. 56
4.1.3 Mutation D820V de l’exon 17........................................................................................................................... 56 4.1.3.1 Dans le GIST......................................................................................................................................................................................... 56 4.1.3.2 Dans le mélanome............................................................................................................................................................................. 57 4.1.3.3 Sensibilité au traitement................................................................................................................................................................ 57
4.1.4 Mutation P567S et G569R de l’exon 12........................................................................................................ 58 4.1.4.1 PDGFRA et GIST.................................................................................................................................................................................. 58 4.1.4.2 PDGFRA et mélanome...................................................................................................................................................................... 58 4.1.4.3 Sensibilité au traitement................................................................................................................................................................ 59
4.2 LES MUTATIONS DE KIT DANS LE MELANOME, REVUE DE LA LITTERATURE..................................................... 60 4.2.1 Nos résultats par rapport à la littérature .................................................................................................. 60 4.2.2 Les mélanomes choroïdiens .............................................................................................................................. 64 4.2.3 Correspondance entre immunohistochimie et mutation de KIT ...................................................... 64 4.2.4 Implications thérapeutiques ............................................................................................................................ 65 4.2.4.1 Les essais cliniques décevants..................................................................................................................................................... 65 4.2.4.2 Des cas cliniques encourageants ................................................................................................................................................ 66 4.2.4.3 Les résultats préliminaires des études précoces................................................................................................................. 69 4.2.4.4 Les études en cours (49) ................................................................................................................................................................ 70
5. CONCLUSION ................................................................................................................................................71 ANNEXES................................................................................................................................................................72 ABREVIATIONS ....................................................................................................................................................77 BIBLIOGRAPHIE ..................................................................................................................................................78
Tableau 1 : Principales études cliniques évaluant l’efficacité de l’imatinib dans les GIST avancées...................................................................................................................................27
Tableau 2 : Mutation de KIT dans les tumeurs germinales. ...........................................................34 Tableau 3 : Mélanome de Dubreuilh..............................................................................................37 Tableau 4 : Mélanomes muqueux ..................................................................................................38 Tableau 5 : Mélanomes acro-lentigineux .......................................................................................42 Tableau 6 : Mélanome sans primitif retrouvé ................................................................................46 Tableau 7 : Traitement des patients atteints de mélanome ou de GIST porteur d’une mutation
KITL576P ....................................................................................................................................55 Tableau 8 : Fréquence des mutations de KIT en fonction des sous-types dans le mélanome. .......62 Tableau 9 : Cas clinique mélanome muté en KIT et réponse aux ITK...........................................67

7
Figure 1 : Voie MAP-kinase et PI3 kinase selon Curtin et al. 2005 (13).......................................13 Figure 2 : Structure d’un récepteur tyrosine kinase de classe III ...................................................20 Figure 3 : Voie de signalisation du récepteur KIT (46) .................................................................22 Figure 4 : Localisation et propriétés biochimiques des mutations secondaires de KIT dans les
GIST résistants aux ITK d’après Gramza et al. 2009 (65). .....................................................30 Figure 5 : Distribution et fréquence des mutations publiées dans le mélanome. ...........................60 Annexe 1a : Version finale 2009 AJCC Melanoma Staging and Classification (table 1)…………72 Annexe 1b : Version finale 2009 AJCC Melanoma Staging and Classification (table 2)…………73 Annexe 2 : Code génétique………………………………………………………………………………….......74 Annexe 3 : Liste des 20 acides aminés représentés dans le code génétique ………………………….75 Annexe 4 : Polymorphisme de KIT et de PDGFRA………………………………………………………..76

8
INTRODUCTION
D’immenses progrès ont été réalisés ces dernières années dans la compréhension de la biologie
du mélanome. Les mélanomes sont des tumeurs diverses sur le plan moléculaire et plusieurs
voies de signalisation intracellulaire peuvent être activées selon le type de tumeur. Ainsi, il a été
démontré une corrélation entre les différents sous-types anatomo-cliniques de mélanomes, les
altérations moléculaires somatiques et l’association ou non à une exposition solaire ponctuelle ou
chronique. En effet, les mutations de BRAF sont présentes dans 50 à 60% des SSM (superficial
spreading melanoma), alors qu’elles sont plus rares dans les mélanomes acro-lentigineux (ALM),
muqueux et de Dubreuilh où en revanche les mutations de KIT sont parfois retrouvées.
La mise en évidence de ces anomalies génétiques a aujourd’hui un impact thérapeutique compte-
tenu de la disponibilité d’inhibiteurs des tyrosine kinases spécifiques de KIT et PDGFRA, qui
sont déjà un traitement de référence dans d’autres tumeurs en particulier les tumeurs stromales
gastro-intestinales.
Nous avons étudié la fréquence des mutations de KIT et de PDGFRA dans quatre sous-types
particuliers de mélanomes : les mélanomes acro-lentigineux (ALM), les mélanomes muqueux, les
mélanomes de Dubreuilh et les mélanomes sans primitif retrouvé, sur une série de 107 patients
suivis sur l’Institut Gustave Roussy.

9
1. GENERALITES
1.1 Mélanome
1.1.1 Epidémiologie
Les mélanomes sont des tumeurs malignes développées aux dépens des mélanocytes. L'incidence
croît depuis plusieurs décennies, celle-ci doublant environ tous les 10 ans dans les populations
essentiellement blanches. En France, on estime l'incidence à environ dix nouveaux cas pour 100
000 habitants et par an, proche de l'incidence observée en Europe. A l'échelle planétaire, de
grandes différences s'observent en fonction de la latitude et des caractéristiques ethniques des
populations pouvant atteindre jusqu'à 60 nouveaux cas pour 100 000 habitants et par an chez les
blancs en Australie, alors qu'elle est très faible dans les pays où les sujets sont noirs ou asiatiques.
C’est une tumeur qui touche tous les âges, en dehors de l’enfant chez qui le mélanome est
exceptionnel. La mortalité (1,2 à 1,5 pour 100 000 habitants en France et autour de 5 pour 100
000 en Australie) tend à augmenter moins que l'incidence, ce qui peut être attribué au diagnostic
plus précoce (1-3).
L'exposition solaire est une cause étiologique majeure largement reconnue. Cependant le lien
entre risque et exposition est complexe avec certaines caractéristiques paradoxales : tumeurs plus
fréquentes et plus précoces sur les zones exposées de façon intermittente au soleil (tronc et
membres) que sur les zones chroniquement exposées au soleil (face). Une faible proportion de
mélanomes touche des sites relativement ou totalement protégés du soleil, tels que les paumes,
les plantes ou les muqueuses (4-6). Cette complexité est en partie liée à l'existence de plusieurs
sous-types de mélanomes.

10
1.1.2 Traitement du mélanome métastatique
Le traitement du mélanome au stade métastatique est très décevant, basé sur des mono,
polychimiothérapies ou des biochimiothérapies. Dans le mélanome avancé, on obtient avec une
monochimiothérapie un taux de réponse de 10 à 20% sans franc bénéfice sur la survie globale.
Les réponses complètes sont obtenues dans moins de 2% des cas, la survie médiane est de 9 mois
et seulement 13% des patients sont en vie à 5 ans (7). Les molécules les plus utilisées sont la
dacarbazine (DTIC), le temozolomide (TMZ), la fotemustine, le cisplatine, le carboplatine, le
paclitaxel, le docetaxel et la vinblastine. La monochimiothérapie la plus usitée est la dacarbazine
(8).
La combinaison de plusieurs drogues donne un taux de réponse de 30 à 50% avec un taux de
réponse complète de moins de 5%. Mais il n’y a finalement pas de différence sur la survie ni le
taux de réponse complète comparativement avec le DTIC, avec en plus une toxicité plus
importante non négligeable (9,10).
Le choix des molécules est resté inchangé depuis des décennies et souligne la nécessité d’une
stratégie thérapeutique différente qui ne se contentera plus d’inhiber la prolifération cellulaire
mais qui viserait à atteindre les mécanismes même de l’oncogenèse du mélanome.
Les thérapeutiques ciblées sont désormais des standards thérapeutiques pour plusieurs affections
néoplasiques (leucémie myéloïde chronique (LMC), tumeurs stromales gastro-intestinale (GIST),
adénocarcinome du sein, lymphomes…). De nombreuses avancées dans la biologie du mélanome
ont été faites ces dernières années afin d’identifier de nouvelles cibles thérapeutiques. Les
inhibiteurs de tyrosine kinase et de leur ligand auront de toute évidence une place importante
dans le traitement du mélanome dans les années à venir. Il faut identifier les paramètres
biologiques corrélés à la réponse et à l’efficacité de ces thérapeutiques pour sélectionner les
patients.

11
1.1.3 Vers une nouvelle classification
La classification la plus utilisée est fondée sur l’histogenèse du mélanome avec une classification
anatomo-clinique réunissant quatre grandes variétés (11,12):
- Mélanome de Dubreuilh ou LMM (Lentigo Maligna Melanoma)
- Mélanome acral lentigineux ou ALM (Acral Lentiginous Melanoma)
- Mélanome superficiel extensif ou SSM (Superficial Spreading Melanoma)
- Mélanome Nodulaire ou NM (Nodular Melanoma)
Cependant, cette classification est controversée car elle ne permet pas de classer tous les
mélanomes et n'a pas de valeur pronostique indépendante.
Une hypothèse plus récente suggère que ces tumeurs puissent être classées en fonction de leur
spécificité génétique. Comprendre que l'hétérogénéité du mélanome en fonction de son site, du
degré d'exposition au soleil et des caractéristiques histologiques est due à des types de
mélanomes distincts biologiquement a une grande importance clinique, car cela pourrait nous
orienter vers une approche de thérapie ciblée.

12
1.1.4 Altérations génétiques distinctes dans les différents sous-types de
mélanomes
Les mutations de BRAF sont retrouvées plus fréquemment dans les mélanomes se développant
sur peau non exposée chroniquement au soleil que dans les mélanomes se développant sur une
peau protégée du soleil telle que les paumes, les plantes, en sous-unguéal (mélanomes acraux),
les muqueuses (mélanomes muqueux) ou dans les mélanomes survenant sur une peau
chroniquement exposée au soleil (13,14).
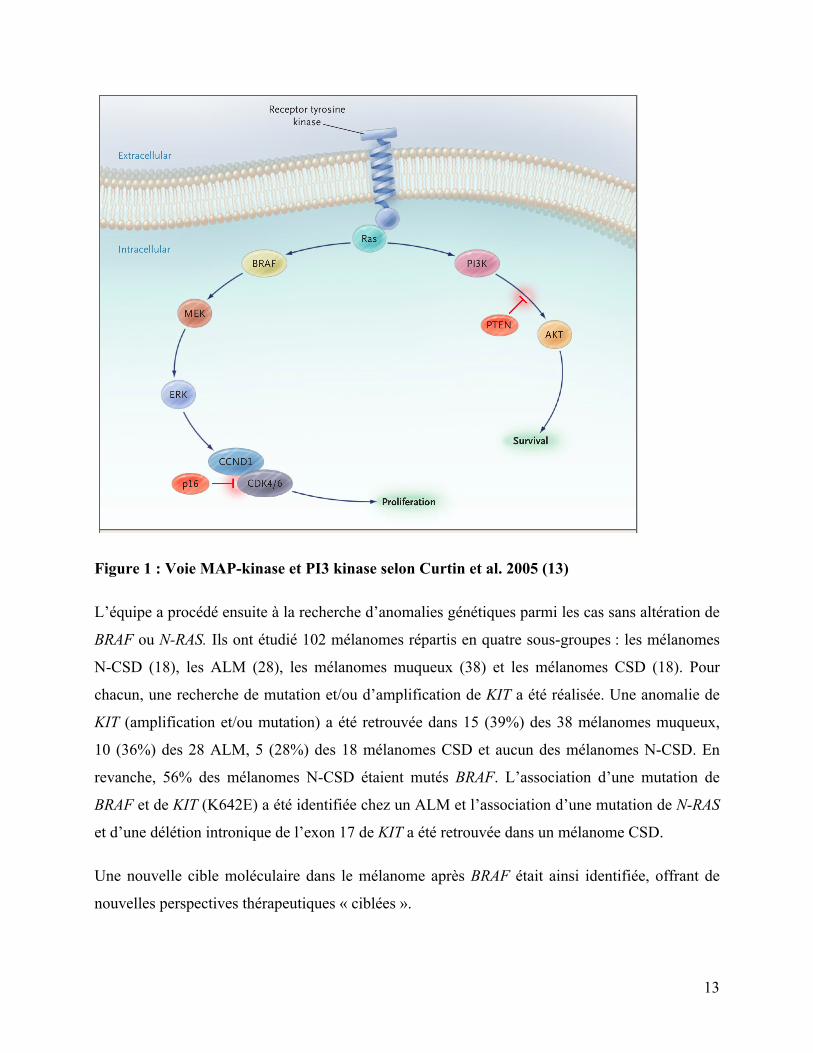
Curtin et al. (13) ont démontré que la voie des MAP-kinases (mitogen-activated protein kinase)
et celle de PI3 kinase (phosphatidylinositol 3 kinase) étaient activées différemment dans quatre
sous-types de mélanomes classés en fonction de la combinaison de l'exposition solaire et du site
anatomique :
- Mélanome acral
- Mélanome muqueux
- Mélanome sur peau chroniquement exposée au soleil ou CSD (chronic sun-induced
damage) équivalent au mélanome de Dubreuilh
- Mélanome sur peau non exposée chroniquement au soleil ou N-CSD (without chronic
sun-induced damage) regroupant les mélanomes de type SSM et nodulaires.
Sur 126 tumeurs, la voie des MAP-kinases et celle de PI3 kinase (figure n°1) étaient
significativement plus activées dans le groupe de mélanomes liés à une exposition intermittente
au soleil par une mutation de BRAF (59% des N-CSD, contre 11% des CSD et des muqueux et
23% des ALM). Dans 5 à 20% des cas, elle était activée par une mutation de N-RAS, sans
différence significative entre les quatre sous-types (22% des N-CSD, 15 % des CSD, 5% des
muqueux et 10% des ALM). Chacune de ces mutations étaient mutuellement exclusives.
13
Figure 1 : Voie MAP-kinase et PI3 kinase selon Curtin et al. 2005 (13)
L’équipe a procédé ensuite à la recherche d’anomalies génétiques parmi les cas sans altération de
BRAF ou N-RAS. Ils ont étudié 102 mélanomes répartis en quatre sous-groupes : les mélanomes
N-CSD (18), les ALM (28), les mélanomes muqueux (38) et les mélanomes CSD (18). Pour
chacun, une recherche de mutation et/ou d’amplification de KIT a été réalisée. Une anomalie de
KIT (amplification et/ou mutation) a été retrouvée dans 15 (39%) des 38 mélanomes muqueux,
10 (36%) des 28 ALM, 5 (28%) des 18 mélanomes CSD et aucun des mélanomes N-CSD. En
revanche, 56% des mélanomes N-CSD étaient mutés BRAF. L’association d’une mutation de
BRAF et de KIT (K642E) a été identifiée chez un ALM et l’association d’une mutation de N-RAS
et d’une délétion intronique de l’exon 17 de KIT a été retrouvée dans un mélanome CSD.
Une nouvelle cible moléculaire dans le mélanome après BRAF était ainsi identifiée, offrant de
nouvelles perspectives thérapeutiques « ciblées ».

14
1.1.5 Les différents sous-types de mélanome avec anomalie de KIT
1.1.5.1 Les mélanomes acro-lentigineux (ALM)
Reed a individualisé en 1976 le sous-type acro-lentigineux, combinant une définition autant
clinique qu'histologique : acral pour sa localisation anatomique sur les extrémités et lentigineux
pour l’histogenèse débutant par une phase de croissance horizontale " en nappe ", au-dessus de la
membrane basale (15). L'atteinte de l'appareil unguéal (mélanome sous-unguéal) est une variante
anatomique qui a été décrite pour la première fois par Boyer en 1854 (cité dans (16)).
La fréquence des ALM est très faible sur peau blanche entre 1 et 7% de tous les mélanomes selon
les séries. Tandis qu'elle atteint 58% des mélanomes dans une série asiatique et est estimée à 60-
70% dans la population noire. La variante sous-unguéale est encore plus rare avec une fréquence
de 1 à 2% de tous les mélanomes dans la population blanche et 10 à 20% dans la population
asiatique (17).
Il est important de noter que tous les mélanomes acraux ne sont pas histologiquement
lentigineux, mais le type histologique acro-lentigineux est le plus fréquent sur ce site anatomique
(la fréquence varie entre 40 à 80%). Le type histologique acro-lentigineux peut aussi survenir sur
le dos des mains ou des pieds et inversement d'autres types histologiques peuvent se voir sur les
paumes et les plantes (SSM ou nodulaire) (18).
Dans notre étude, nous avons tenu compte du site anatomique et inclus des mélanomes acraux de
type SSM et nodulaire.

15
1.1.5.2 Les mélanomes muqueux
Le mélanome muqueux est une entité très rare. Les mélanomes muqueux ont été rapportés dans
1,3 à 1,4% tous mélanomes confondus (19,20). La tête et le cou englobent plus de 55% des
mélanomes muqueux, suivi du mélanome ano-rectal 24%, du mélanome génital féminin 18% et
du tractus urinaire 3%. Chez les blancs, les mélanomes muqueux représentent 1,2% de tous les
mélanomes contre 9,8% dans la population noire (19).
L’incidence des mélanomes muqueux semble stable contrairement à la forme cutanée. Le
pronostic est plus péjoratif, en raison d’un diagnostic souvent réalisé tardivement. Le taux
d’incidence d’envahissement ganglionnaire au diagnostic est de 61% pour la localisation anale,
23% pour le tractus génital féminin, 21% pour la tête et le cou, et 11% pour le tractus urinaire
contre 9% dans le mélanome cutané. La présence de métastases à distance au moment du
diagnostic est de 29% (19). L’exposition solaire n’est pas un facteur de risque. Il n’existe pas de
classification spécifique aux mélanomes muqueux, certains utilisent l’American Joint Committee
on Cancer (AJCC)(annexe 1a et b), d’autres une classification plus simple et pratique selon
Ballantyne : stade I localisé, stade II régional (ganglionnaire), stade III métastatique (21,22). La
prise en charge ne diffère pas du mélanome cutané, en dehors de l’indication de la radiothérapie
adjuvante sur le site opératoire qui est, pour la plupart des auteurs, recommandée (23).
1.1.5.2.1 Tête et cou
Le site le plus fréquent est la cavité naso-sinusale et la cavité buccale. Le palais est le plus
fréquemment atteint dans la cavité buccale, et 80% des mélanomes de la cavité naso-sinusale
débutent dans la cavité nasale contre 20% dans les sinus (21,24). Dans certaines séries, on
retrouve une disparité raciale avec une incidence allant jusqu’à 7,5% parmi les japonais (25) et
jusqu’à 10% sur une série ougandaise (26).
1.1.5.2.2 Tube digestif
Le mélanome peut toucher la muqueuse du tube digestif sur plusieurs sites : l'œsophage, l'intestin
grêle, le rectum et l'anus. Plusieurs études ont confirmées la présence de mélanocytes sur ces
quatre localisations, avec une prédilection pour le canal anal (27,28).

16
Le mélanome anal ou rectal est une tumeur rare, moins de 1% de tous les mélanomes et environ
4% des cancers anaux (29). Le mélanome ano-rectal constitue 16,5% à 24% des mélanomes
muqueux (19,20). Il n'y a pas de différence de fréquence entre la population noire ou blanche
contrairement à la forme primitivement cutanée. Le contrôle local semble amélioré par une
amputation abdomino-pelvienne sans amélioration sur la survie globale (30).
Le mélanome œsophagien est très rare, moins de 0,3% de toutes les tumeurs œsophagiennes. Le
premier cas a été rapporté par Baur en 1907 (cité par (27)) mais c'est Garfinkle en 1952 qui décrit
le premier cas confirmé histologiquement (31).
Le mélanome de l’intestin grêle est extrêmement rare. Distinguer un mélanome primitivement
muqueux d’une localisation secondaire intestinale, d’un mélanome cutané régressif ou d’un
mélanome sans primitif retrouvé peut être difficile, voire impossible (32).
1.1.5.2.3 Tractus uro-génital
Le mélanome du tractus uro-génital chez la femme représente jusqu'à 7% des mélanomes chez la
femme, avec une incidence stable. Le mélanome vulvaire est le plus fréquent, suivi du vaginal (2
à 5%), de l’urétral, du col, de l'ovaire, de la vessie et de l’uretère.
Le mélanome vulvaire est le deuxième cancer le plus fréquent de la vulve après le carcinome
épidermoïde mais représente seulement 5 à 10% des pathologies malignes vulvaires et moins de
0,2 mélanome pour 100 000 femmes par an. Le type histologique le plus commun est lentigineux
suivi du SSM puis du nodulaire. Plus de 25% de ces mélanomes sont achromiques. Des
guidelines ont été proposés par Piura (33).
Le mélanome de la muqueuse urétrale représente seulement 3% de tous les mélanomes muqueux,
c’est une entité extrêmement rare. Chez l’homme, le pénis est le plus souvent atteint parmi les
localisations uro-génitales mais il s’agit pour la majorité des cas de mélanomes cutanés (21).

17
1.1.5.3 Mélanome de Dubreuilh
En 1892, Hutchinson attire l’attention sur le potentiel évolutif de certaines lésions de la face qu’il
appelle « senile freckle » puis « lentigo melanosis » puis en 1894, Dubreuilh le décrit sous le
terme de « lentigo malin des vieillards » puis comme « mélanose circonscrite précancéreuse »
(cité dans (34)).
La moyenne d’âge au moment du diagnostic est souvent élevée. Il siège sur les zones photo-
exposées en particulier la tête et le cou, avec beaucoup plus rarement une autre localisation telle
que les avant-bras, le dos des mains ou les jambes. En effet, le mélanome de Dubreuilh a une
relation directe avec l’exposition solaire chronique (contrairement aux autres formes liées à des
antécédents d’exposition solaire intense et intermittente). Il s’associe souvent aux autres signes
d’héliodermie comme l’élastose, les lentigos et les kératoses actiniques (35).
1.1.5.4 Mélanome sans primitif retrouvé
Cette entité a été définie la première fois par Das Gupta en 1963 (36), par une localisation de
mélanome confirmé histologiquement sous-cutanée, ganglionnaire ou viscérale sans lésion
primitive cutanée, oculaire ou muqueuse retrouvée, ni antécédent de lésion cutanée enlevée ou
brulée sans analyse histologique. A l’heure actuelle, la pathogénie des mélanomes sans lésion
primitive retrouvée n’est pas connue. Les deux grandes hypothèses proposées sont (37) :
- Régression complète de la lésion primitive par un phénomène immunologique,
- Transformation d’un mélanocyte ectopique, c'est-à-dire situé dans un organe extra-cutané.
La localisation principale est ganglionnaire suivie des localisations cutanées ou sous-cutanées
puis des localisations viscérales (37). Une revue de la littérature récente rapporte son incidence à
3,2%, avec un sex ratio homme/femme à 2 : 1 et un âge médian entre la quatrième et cinquième
décennie. Les patients avec une atteinte ganglionnaire ont le même, voire un meilleur pronostic,
que les mélanomes de primitif connu de même stade. Les patients avec une atteinte viscérale
auraient un meilleur pronostic que les mélanomes métastatiques de primitif connu (38).
Nous avons inclus ces patients dans notre étude dans l’hypothèse d’une localisation muqueuse
occulte ou acrale régressive.

18
1.2 Les thérapies ciblées
L’expression « thérapie ciblée » désigne les thérapeutiques dirigées contre des cibles
moléculaires identifiées comme nécessaires à la transformation néoplasique de la cellule et à sa
survie. En ce sens, elle découle de la meilleure connaissance des mécanismes fondamentaux des
processus d’oncogenèse permettant d’identifier les altérations clés inhérentes au phénotype
tumoral. La classification moléculaire a permis de développer des médicaments capables de
bloquer plus ou moins spécifiquement, la fonction de ces protéines activatrices. Ces nouvelles
thérapies ciblées peuvent être classées en plusieurs catégories :
- les thérapeutiques ciblées sur des anomalies moléculaires causales, directement
responsables de la transformation néoplasique. On peut citer comme exemple, le gène de fusion
BCR-ABL des leucémies myéloïdes chroniques (LMC), les mutations activatrices du gène KIT
dans les tumeurs stromales gastro-intestinales (GIST) sur lesquelles on reviendra, les gènes de
fusion impliquant EWS avec les gènes de la famille des Ets (EWS-FLI1 étant le plus fréquent) des
sarcomes d’Ewing et la fusion COL1A1-PDGFB dans les sarcomes de Darrier et Ferrand (DFSP).
- les thérapeutiques ciblées sur des anomalies moléculaires plus tardives, qui contribuent à
la progression tumorale mais qui ne constituent pas l’étape initiale de la transformation, et étant
volontiers un marqueur de mauvais pronostic. L’exemple caractéristique est celui de
l’amplification du gène ERBB2 dans les adénocarcinomes du sein retrouvé dans 15 à 20% des
tumeurs. Autre exemple, les traitements anti-angiogéniques qui agissent aussi sur un événement
moléculaire tardif, la néoangiogénèse, étape indispensable à la croissance tumorale.
- les thérapeutiques ciblées sur des cibles moléculaires qui ne jouent pas un rôle direct dans
la transformation. L’immunothérapie par anticorps monoclonal peut être dirigée contre des
déterminants antigéniques de surface qui ne sont pas nécessairement des molécules de survie
pour la cellule tumorale. Ces anticorps déclenchent la mort cellulaire par différents mécanismes
principalement immunitaire par la voie du complément ou ADCC (antibody-dependent cell
toxicity) (39).

19
1.3 Rôle de KIT et PDGFRA
1.3.1 Description et activité biologique
La protéine KIT ou CD117 est un récepteur transmembranaire à activité tyrosine kinase produit
par le proto-oncogène KIT et dont le ligand naturel est le Stem Cell Factor (SCF). Le proto-
oncogène KIT est localisé sur le bras long du chromosome 4 (4q12) et comprend 21 exons.
PDGFRA est une protéine proche de KIT, dont le gène est également situé sur le bras long du
chromosome 4 et dont le ligand naturel est aussi un facteur de croissance hématopoïétique le
PDGF (Platelet Derived Growth Factor).
Ces récepteurs appartiennent à la grande famille des récepteurs tyrosine kinase (RTK), dont le
rôle dans la physiologie cellulaire est étroitement associé à la régulation de la prolifération et de
la différentiation cellulaire. Les RTK sont classés en sous-famille fondée sur les similitudes de
séquences et les caractéristiques structurales. La classe III des RTK comprend un grand nombre
de proto-oncogènes dont KIT, PDGFR, FLT3 et FMS.
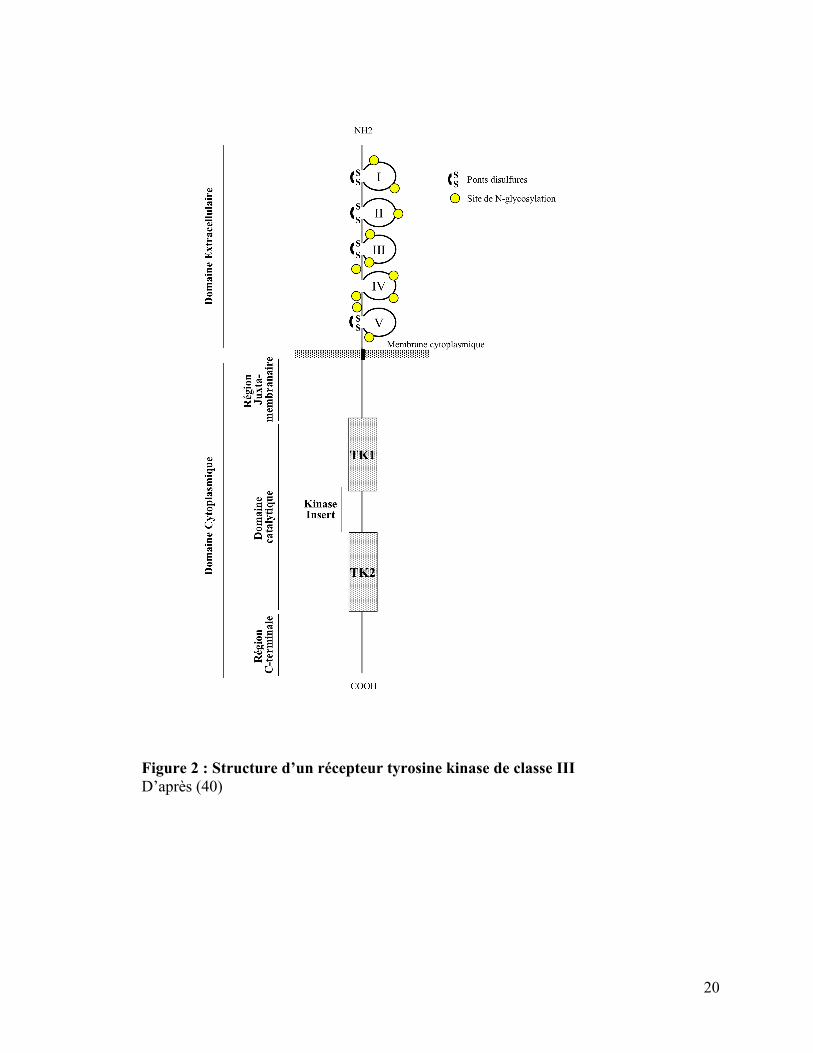
Cette famille de protéines est caractérisée par une structure moléculaire spécifique comprenant un
domaine extracellulaire où se fixe le ligand, composé de cinq boucles immunoglobulin-like, un
domaine cytoplasmique avec une région juxtamembranaire (codée par l’exon 11 dans KIT) et un
domaine tyrosine kinase (TK) divisé en deux partie : TK1 zone de fixation à l’adénosine
triphosphate (ATP), et TK2 le site actif de la phosphotransférase, ces deux parties étant reliées
par une « insert kinase». Les domaines extracellulaire et cytoplasmique sont liés par la région
transmembranaire qui permet l’ancrage du récepteur à la membrane.
20
Figure 2 : Structure d’un récepteur tyrosine kinase de classe III D’après (40)

21
KIT et PDGFR sont activés par la liaison de leur ligand respectif, le SCF et le PDGF. La liaison
avec le ligand sur la portion extracellulaire permet une dimérisation du récepteur qui entraîne
alors des modifications structurelles de la protéine dont la conséquence est l’activation de la
fonction tyrosine kinase. Une autophosphorylation du récepteur signe la mise en œuvre des voies
de transduction d’aval correspondant au recrutement et à la phosphorylation de multiples
effecteurs.
L’activité tyrosine kinase est régulée par son domaine juxtamembranaire qui inhibe son activité
en l’absence de son ligand.
Une altération de la structure de ce domaine notamment en cas de mutation de l’exon 11 de KIT,
codant pour cette région, va entraîner une activation spontanée de KIT, indépendamment de la
liaison à son ligand.
KIT régule de nombreuses fonctions cellulaires incluant la prolifération, l’apoptose, le
chimiotactisme et l’adhésion. Il est un élément essentiel pour le développement et la maintenance
de différents types cellulaires, telles que les cellules hématopoïétiques, les mastocytes, les cellules
germinales, les mélanocytes, les gamètes et les cellules interstitielles de Cajal du système
digestif. PDGFRA est quant à lui impliqué dans la prolifération et la différentiation des cellules
gliales et mésenchymateuses.
L’activité oncogénique de KIT et PDGFRA est ainsi associée à l’activation constitutive, ligand-
indépendante, des récepteurs par le biais d’anomalies moléculaires correspondant dans la plupart
des cas à des mutations activatrices dites « gain de fonction ». Cette activation oncogénique va
entraîner la phosphorylation incontrôlée de protéines impliquées dans la transduction du signal.
Plusieurs voies de signalisation jouant un rôle dans la prolifération et la survie sont concernées,
notamment les voies Ras/Raf/MAP Kinase, STAT, Src kinase, et PI3K/AKT/mTOR (figure 3).
Les cascades de signalisation sont similaires qu’il s’agisse d’altérations touchant KIT ou
PDGFRA (41-43).
Les mutations de KIT et de PDGFRA sont mutuellement exclusives (44,45).

22
Figure 3 : Voie de signalisation du récepteur KIT (46)
1.3.2 Les inhibiteurs des tyrosine kinases KIT et PDGFRA
Les inhibiteurs de tyrosine kinase (ITK) ont initialement été développés pour être actifs contre
une kinase donnée, mais en pratique, les molécules créées ont souvent des cibles multiples. Ces
molécules sont administrées par voie orale.
1.3.2.1 Avec une autorisation de mise sur le marché (AMM) (47)
Ces traitements ont une autorisation sur le marché dans le GIST (imatinib et sunitinib), le cancer
du rein (sorafenib et sunitinib), le dermatofibrosarcome de Darrier et Ferrand (DFSP) (imatinib),
le carcinome hépatocellulaire (sorafenib) et dans certaines pathologies hématologiques en
particulier LMC Philadelphie + (Ph+) (imatinib, nilotinib, dasatinib), leucémie aiguë
lymphoblastique Ph+ (imatinib, dasatinib), les syndromes myélodysplasique et myeloprolifératif
(SMD/SMP) et le syndrome hyperéosinophilique (SHE)(imatinib).

23
1.3.2.1.1 Imatinib
L'imatinib mesylate ou Glivec® (STI571) avait été développé au laboratoire comme inhibiteur
sélectif de l’activité tyrosine kinase du PDGFR. Des études in vitro ont ensuite montré qu’il avait
également une activité inhibitrice sur la protéine c-ABL, la molécule de fusion BCR-ABL et sur
KIT. La dose journalière varie entre 400 et 800 mg/jour. Les principaux effets secondaires sont
nausées/vomissements, diarrhées, douleur abdominale, rash cutané, œdèmes (prédominant en
périorbitaire) et sérites.
1.3.2.1.2 Sunitinib
Le sunitinib ou Sutent® (SU11248) inhibe KIT, PDGFRA et B mais aussi RET, CSF-1, FLT-3 et
les récepteurs VEGFR-1, 2 et 3. Il a été développé pour les cancers du rein métastatique, mais
également pour le traitement des GIST.
Le schéma thérapeutique est de 50 mg/jour 4 semaines sur 6 ou 37,5 mg/jour en continu.
Sa toxicité est marquée par une asthénie, une hypertension artérielle, une protéinurie, une mucite,
un syndrome mains/pieds et une hypothyroïdie.
1.3.2.1.3 Sorafenib
Le sorafenib ou Nexavar® (BAY 43-9006) a été conçu pour inhiber spécifiquement la protéine
RAF. La molécule s’avère également bloquer VEGFR-2 et 3, PDGFRB, FLT-3 et 4, KIT et RET.
La dose recommandée est de 400 mg, deux fois par jour. Les principaux effets secondaires sont le
syndrome mains/pieds, la diarrhée, l’hypertension artérielle.

24
1.3.2.1.4 Dasatinib
Le dasatinib ou Sprycel® (BMS-354825) est un puissant inhibiteur de Src, mais aussi c-ABL,
protéine de fusion BCR-ABL, KIT et PDGFR. La posologie recommandée est de 100 mg/jour
avec des augmentations pouvant aller jusqu’à 180 mg/jour en hématologie. La phase I dans les
tumeurs solides a déterminé la dose maximale tolérée à 70 mg deux fois par jour en continu ou
120 mg deux fois par jour, 5 jours/7 (48).
Sa toxicité est marquée par une myélosuppression (dans les pathologies hématologiques), des
troubles digestifs (nausée, diarrhée, anorexie), une protéinurie, une rétention hydrique
(épanchement ou œdème), un allongement du QT et une asthénie.
1.3.2.1.5 Nilotinib
Le Nilotinib ou Tasigna® (AMN107) inhibe BCR-ABL, et les récepteurs PDGF, KIT et Ephrine.
La posologie recommandée est de 400 mg deux fois par jour. Sa toxicité est essentiellement
hématologique (thrombopénie, neutropénie et anémie), allongement du QT, céphalée, éruption
cutanée, asthénie, troubles digestifs et œdèmes périphériques.
1.3.2.2 En cours de développement (49)
1.3.2.2.1 Masitinib
Le masitinib mesylate (AB1010) est un nouvel inhibiteur de KIT, PDGFR et FGFR-3.
Actuellement en phase III (NCT00812240) de développement dans les GIST, en comparaison
avec l’imatinib, à la dose de 7,5 mg/kg/jour. Les principaux effets secondaires décrits sont
hématologiques, cutanées et digestifs (50).
1.3.2.2.2 XL820
L’XL820 inhibe les récepteurs de VEGF, KIT et PDGF.
Une phase II (NCT00570635) est en cours dans les GIST après échec d’un traitement par
imatinib et sunitinib dû à une résistance ou à une intolérance.

25
1.3.2.2.3 Motesanib
Le motesanib (AMG706) est une petite molécule orale caractérisée par une activité anti-tyrosine
kinase ciblant KIT, PDGFR et RET ainsi que VEGFR- 1, 2 et 3 (51). Son activité est en cours
d’évaluation pour les GIST après échec de l’imatinib.
1.3.2.2.4 Vatalanib
Le vatalanib (PTK787/ZK 222584) a pour cible VEGFR-1 et 2, KIT et à moindre degré VEGFR-
3, PDGFRB (52,53). Une étude de phase II ainsi qu’une étude de phase I/II en association avec le
DTIC est en cours dans le mélanome (NCT00563823 et NCT00615160).
1.3.2.2.5 Midostaurin
Le midostaurin (PKC412A), la N-benzoyl-staurosorine inhibe la protéine kinase PKCα, VEGFR-
2, KIT, PDGFR et FLT-3. Il a été évalué dans un essai de phase II dans le mélanome
métastatique (non sélectif sur les anomalies moléculaires) et s’est avéré négatif (54). Il est en
cours d’évaluation en hématologie.
1.3.2.2.6 Cediranib
Le cediranib maleate (AZD2171) a pour cible VEGFR, KIT et PDGFR. Une étude de phase II est
en cours dans le mélanome (NCT00243061).

26
1.4 Un modèle établi : les tumeurs stromales gastro-intestinales (GIST)
1.4.1 Prise en charge des GIST avancées : place de l’imatinib
Les tumeurs stromales gastro-intestinales sont des tumeurs rares, pouvant se localiser à tous les
étages du tractus digestif, dont l’incidence est estimée à 15 nouveaux cas par millions d’habitants
par an. Les GIST représentent une entité nosologique particulière depuis la découverte de leur
lien avec les cellules de Cajal, les cellules pacemakers de la motricité digestive. Sur le plan
phénotypique, les cellules tumorales de GIST sont caractérisées par l’expression du marqueur
CD34, commun aux cellules de Cajal et par l’expression du récepteur tyrosine kinase KIT sous
une forme mutée et/ou activée. Ces mutations sont de survenues précoces et constituent même
possiblement l’événement oncogénétique fondateur principal de la maladie.
Les GIST étaient historiquement considérées comme des sarcomes résistants à la chimiothérapie
de pronostic très sombre. Jusqu’à l’introduction de l’imatinib (Glivec®), la médiane de survie
des patients présentant une GIST avancée était comprise entre 18 et 24 mois (55). L’introduction
de l’imatinib a révolutionné le pronostic des GIST évoluées métastatiques. Le contrôle tumoral
est obtenu chez 70 - 90% des patients avec des GIST avancées, et la médiane de survie globale
dépasse 3 ans dans toutes les grandes études cliniques publiées (44,56,57).
Le premier cas clinique rapportant l’efficacité de l’imatinib dans le GIST métastatique fut publié
en 2001 (58). Des études de phase I, II et III européennes et américaines se sont ensuite très vite
succédées. Une synthèse des résultats obtenus en terme de contrôle tumoral est présentée dans le
tableau n° 1.

27
Tableau 1 : Principales études cliniques évaluant l’efficacité de l’imatinib dans les GIST
avancées.
Etudes Phase
Nombre de patients
(dose journalière d’imatinib)
Réponse objective1 Stabilité Contrôle
tumoral2 Références
EORTC I 36 (400-1000 mg) 69,5% 19,5% 89% (59)
EORTC II 27 (800 mg) 71% 18% 89% (60)
US-B2222 II 73 (400 mg) 74 (600 mg)
69% 68%
14% 18%
83% 86%
(61)
Intergroup S0033
III 345 (400 mg) 349 (800 mg)
45% 46%
25% 22%
70% 68%
(56)
EORTC 62005
III 473 (400 mg) 473 (800 mg)
50% 54%
32% 32%
82% 86%
(57)
1 : Réponse partielle + réponse complète 2 : Réponse partielle + réponse complète + stabilité

28
On observe que les patients porteurs de tumeurs mutées pour l’exon 11 de KIT présentent les taux
de réponse les plus élevés au traitement par imatinib. En outre, la médiane de survie globale et
sans progression est également supérieure aux autres GIST (62).
La posologie quotidienne de 400 mg est la règle. Les résultats obtenus sont, en effet, équivalents
aux posologies supérieures pour ce qui est du contrôle tumoral et de la survie globale. Seule une
étude (EORTC 62005) a dégagé une différence significative (p = 0,026) en terme de survie sans
progression : 44% de survie sans progression à 2 ans pour une dose quotidienne de 400 mg contre
52% pour une dose de 800 mg par jour. Cette différence est probablement liée en grande partie au
bénéfice tiré par les patients présentant une mutation de l’exon 9 de KIT recevant 800 mg
d’imatinib par jour (57).
Une posologie initiale plus élevée est désormais proposée d’emblée (800 mg/j) en cas de
mutation connue de l’exon 9 de KIT. L’augmentation de la dose est également discutée pour tout
patient en progression tumorale sous traitement (62). Il n’existe pas d’argument pour interrompre
le traitement chez un patient en rémission sous imatinib. Le traitement doit donc être maintenu
sans interruption aussi longtemps que possible, la tolérance du patient étant le seul facteur
limitant (63,64).

29
1.4.2 GIST et résistance à l’imatinib
L’efficacité n’est pas constante chez l’ensemble des patients, avec notamment près de 10% de
résistance primaire, c’est-à-dire de patients chez lesquels le contrôle tumoral n’a pu être obtenu
après six mois de traitement, mais aussi le développement de résistances acquises dans 40 à 50%
des cas à la suite d’un traitement prolongé (65).
1.4.2.1 Résistance primaire
La résistance primaire concerne tous les sous-types génotypiques des GIST mais elle est
principalement associée à une mutation primaire de l’exon 9 de KIT, de l’exon 18 de PDGFRA
(D842V) et des génotypes sauvages pour KIT et PDGFRA (44,66). Les résultats sont univoques
en ce qui concerne la corrélation manifeste entre le génotype tumoral et la réponse au traitement
par imatinib. L’amplification du gène KIT et/ou PDGFRA est aussi un des mécanismes impliqués
dans la résistance primaire ou secondaire (66-69).
1.4.2.2 Résistance secondaire
Malgré une réponse initiale remarquable aux ITK, une résistance secondaire apparaît
fréquemment durant le traitement. Cette résistance secondaire ou acquise est à distinguer de la
résistance primaire. Elle se définit comme une progression tumorale survenant alors que le
patient a présenté une réponse à l’imatinib pendant plus de six mois. Le développement d’une
résistance secondaire à l’imatinib survient chez 40 à 50% des patients dans les 2 ans. Cette
résistance secondaire serait, la plupart du temps, liée à des mutations acquises de KIT ou de
PDGFRA. Une analyse histologique a été réalisée sur les patients progressifs dans l’étude de
phase II B2222 révélant que 67% des patients, présentant une résistance secondaire, avaient un
clone tumoral avec une ou plusieurs mutations secondaires. Toutes les mutations secondaires
étaient retrouvées dans des GIST présentant une mutation initiale de KIT et la seule mutation
secondaire de PDGFRA identifiée, est apparue chez un patient avec une mutation de PDGFRA
initiale. Les mutations secondaires touchent principalement le TK1 (exon 13 et 14) ou le TK2
(exon 17 et 18) (Figure 4). Aucune mutation secondaire de KIT ou de PDGFRA n’a été identifiée
chez les GIST wild-type (sauvage pour KIT et PDGFRA) (65,67).

30
Figure 4 : Localisation et propriétés biochimiques des mutations secondaires de KIT dans
les GIST résistants aux ITK d’après Gramza et al. 2009 (65).
La fréquence et la localisation des mutations primaires de KIT sont affichées à gauche. Le centre de la
figure est un zoom du domaine kinase (exons 13-18). Les mutations secondaires responsables d’une
résistance sont représentées à droite. L'activité de l'imatinib (IM) ou sunitinib (SU) contre les différentes
mutations secondaires est représentée par des cases colorées. Par exemple, la mutation V654A est
résistante à l'imatinib (rouge), mais reste sensible au sunitinib (vert). Le sunitinib est très efficace contre la
mutation V654A et T670I, mais n'a pas d'activité contre les mutations de la boucle d'activation (exons 17
et 18). Inversement, l'imatinib a une activité minime contre les mutations V654A et T670I, et une activité
variable sur les mutations de la boucle d'activation (inactif contre les mutations des codons 816, 820, ou
823, mais intermédiaire contre les mutations des codons 822 ou 829).

31
1.4.3 Alternatives thérapeutiques
Le développement de nouvelles molécules actives s’est très vite imposé devant d’une part, les
patients présentant une résistance primaire ou ne tolérant pas l’imatinib et d’autre part, la majorité
des patients pour lesquels un contrôle tumoral a été obtenu avec cette molécule, finissant par
développer une résistance secondaire.
1.4.3.1 Sunitinib
La première molécule à disposer d’une autorisation sur le marché (AMM) en Europe depuis
l’avènement de l’imatinib est le sunitinib. L’AMM limite l’indication du traitement aux GIST
non réséquables et/ou métastatiques, après échec d’un traitement par imatinib dû à une résistance
ou une intolérance. Cette molécule a fait preuve de son efficacité, en seconde ligne après l’échec
de l’imatinib, dans des essais de phase I-II et un essai randomisé de phase III contre placebo (70-
72). Les réponses au traitement seraient plus fréquentes et plus durables en cas de mutations sur
l’exon 9 de KIT ou chez les GIST sans mutation de KIT ni de PDGFRA qu’en cas de mutations
de l’exon 11 de KIT. La survie sans progression (PFS) et la survie globale sont significativement
plus longues chez les patients présentant des mutations secondaires de l’exon 13 et 14 que ceux
avec des mutations secondaires de l’exon 17 et 18 (70). La posologie recommandée est de 50 mg
par jour, 4 semaines sur 6. Dans une étude plus récente de phase II, la dose de 37,5 mg par jour
en continu chez les patients résistants ou intolérants à l’imatinib a montré une PFS médiane et
une survie globale de 34 et 107 semaines, respectivement (73).

32
1.4.3.2 Les autres inhibiteurs de tyrosine kinase
Des études sont en cours afin d’identifier de nouvelles molécules pour les patients résistants à
l’imatinib et au sunitinib. Le masitinib, le motesanib, l’XL820, le vatalanib ont été développés
dans le chapitre 1.3.2.2.
1.4.3.2.1 Nilotinib
Un essai de phase II, sur 47 patients en progression après traitement par imatinib et sunitinib, a
évalué l’efficacité et la tolérance du nilotinib. Cinq patients (10%) ont répondu (une réponse
complète et quatre réponses partielles) et 19 patients (37%) étaient stables. La médiane de survie
globale était de 34 semaines (74). Plusieurs études avec le nilotinib dans le GIST sont en cours
dont une étude de phase III (NCT01089595) dans les GIST non réséquables ou métastatiques
comparant le nilotinib et l’imatinib (49).
1.4.3.2.2 Sorafenib
Les résultats préliminaires d’une phase II évaluant le sorafenib ont été présentés à l’ASCO 2009.
Chez 24 patients résistants à l’imatinib et/ou au sunitinib, il y avait un contrôle tumoral dans 71%
des cas, avec une PFS médiane de 5,3 mois, une médiane de survie globale à 13 mois et un taux
de survie à 1 an de 62% (75). Une autre étude présentée à l’ASCO 2009, montrait un taux de
réponse partielle de 19% et une stabilité dans 44% des cas chez des patients traités en quatrième
ligne par sorafenib (après imatinib, sunitinib et nilotinib) (76).
1.4.3.2.3 Dasatinib
Dans l’essai de phase I pour les tumeurs solides, 19 patients atteints de GIST ont été inclus,
aucune réponse objective n’a été retrouvée mais trois patients ont eu une stabilité (48). Une phase
II multicentrique (NCT00568750) est en cours avec le dasatinib en première ligne dans le GIST.

33
1.5 Mutation de KIT dans les autres tumeurs
Les pathologies hématologiques telles que LMC, LAL, SMD et SHE, ainsi que le DFSP ne sont
pas développés dans ce chapitre car ils sont liés à des anomalies de fusion de gènes codant pour
des transcrits de fusion tel que BCR-ABL, FIP1-PDGFRA, COL1A1-PDGFB.
1.5.1 Les tumeurs germinales
Le récepteur membranaire KIT est normalement exprimé par les cellules germinales primitives.
Son expression dans les tumeurs germinales testiculaires est caractéristique des séminomes (77).
Des mutations somatiques de KIT ont été identifiées. Elles portent principalement sur l’exon 17
et beaucoup plus rarement sur l’exon 11 (tableau 2). Cependant, le rôle de KIT dans l’induction
ou dans la progression des tumeurs germinales testiculaires reste à établir. La diminution ou la
perte d’expression de KIT serait liée à une évolution plus agressive ou à une progression vers une
forme non séminomateuse (78). Les inhibiteurs de KIT pourraient donc jouer un rôle dans le
traitement des séminomes. Les résultats cliniques restent quand à eux très modestes. Une étude
de phase II a été effectuée chez 18 patients ayant des tumeurs germinales testiculaires
métastatiques réfractaires à la chimiothérapie. Sur 18 patients, seuls les six qui avaient une
tumeur exprimant KIT en immunohistochimie ont été traités. L’imatinib était administré à une
dose de 600 mg/jour. Cinq patients sur six ont progressé rapidement, un seul a eu une
stabilisation pendant 3 mois. Les patients ont été sélectionné sur l’expression de KIT et non la
mutation ce qui peut expliquer ces résultats décevant (79).

34
Tableau 2 : Mutation de KIT dans les tumeurs germinales.
Auteur Tumeurs Effectif Exon Fréquence Mutation Tian et al. (78) Tumeur
germinale (testiculaire, ovarienne et extra-gonadique)
33 11 17
0% 6%
- D816H
Przygodzki et al. (80) Séminome médiastinal
8 11 17
0% 37,5%
- K818R, D820V1,
N822K Sakuma et al. (81) Tumeur
germinale testiculaire
34 11 17
2,3% 11,8%
W557R D816H, D816V
Kemmer et al. (77) Séminome testiculaire
54 11 17
0% 25,9%
- D816H, D816V, Y823D, Y823C, N822K, T801I
Sakuma et al. (82) Germinome intracrânien
16 11 17
6,25% 18,8%
W557C N822Y, D820V,
D826V Nakai et al. (83) Séminome
testiculaire 22 11
17
9% 31,8%
L576P1, D57 pb (555-573)
D816A, D816H, D816V, D820V
Biermann et al. (84) Tumeur germinale testiculaire
177 11 17
0% 13,5%
- D816H, D816V, S821F, N822K,
Y823D Willmore-Payne et al. (85)
Séminome testiculaire
22 11 17
5% 18%
L576P D816Y, D816V, D816E, D820H,
Y823N 1 : Mutation mise en évidence dans notre série.

35
1.5.2 La mastocytose
Les mastocytoses sont des maladies rares, caractérisées par une prolifération de mastocytes dans
les différents tissus ou organes. Des progrès récents ont été faits dans la compréhension de la
physiopathologie de ce syndrome myeloprolifératif qui est la conséquence d’une mutation
activatrice du récepteur KIT. KIT est décelable dans les mastocytes et leurs progéniteurs.
Plusieurs mutations ont été mises en évidence et la plus fréquente est la D816V sur l’exon 17. En
France, l’AFIRMM a récemment étudié la fréquence de la mutation D816V dans la peau et la
moelle : elle existe dans 85% des cas des formes systémiques et 70% des formes cutanées pures.
D’autres mutations ont été décrites mais sont plus rares, du domaine catalytique sur le codon
816 : (Asp816Tyr, Asp816Phe, Asp816His), du codon 820 : Gly820Val décrit dans un cas de
leucémie à mastocyte, du domaine juxtamembranaire (Val560GLy, Phe522Gly, Phe522Cys,
Asp509Tyr). L’efficacité de l’imatinib a été testée dans les mastocytoses, mais les mutations du
site catalytique de KIT sont connues pour être naturellement résistantes et c’est le cas de la
mutation D816V. D’autres inhibiteurs de tyrosine kinase tels que le nilotinib, le dasatinib, le
masitinib et le midostaurin sont à l’étude (49,86).
1.5.3 La leucémie myéloïde aiguë
Dans la majorité des cas de leucémie aiguë myéloïde (LAM), KIT est surexprimé. Mais les
mutations de KIT restent des anomalies rares (87) où deux sortes de mutations prédominent, l’une
impliquant l’exon 8 sur le codon 419 (88), l’autre touchant le codon 816 de l’exon 17 (D826V et
D826Y) (89).

36
2. MATERIEL ET METHODES
Nous avons analysé les tumeurs de 107 patients suivis à l’Institut Gustave Roussy, atteints d’un
mélanome localisé ou métastatique :
- 45 mélanomes de localisation primitivement acrale dont
o 37 ALM
o 2 SSM
o 5 nodulaires
o 1 inclassable
- 42 mélanomes de localisation muqueuse dont
o 23 mélanomes de la sphère ORL
o 11 mélanomes vaginaux
o 8 mélanomes anaux
- 6 mélanomes de Dubreuilh
- 14 mélanomes sans primitif retrouvé
La recherche de la mutation était réalisée soit sur la tumeur primitive (57 tumeurs), soit sur une
récidive locale (10 tumeurs), soit sur une localisation métastatique (40 tumeurs dont 15
localisations cutanées, 16 localisations ganglionnaires et 9 localisations à distance) en fonction de
l’accessibilité des prélèvements.
Ces prélèvements ont été analysés entre décembre 2008 et août 2010.
La recherche de mutation du gène KIT a été effectuée sur les exons 8, 9, 11, 13, 17 par
séquençage direct sur ces exons à partir de l’ADN extrait des blocs d’inclusion en paraffine.
Parallèlement, une recherche de mutation du gène PDGFRA sur les exons 12, 14, et 18 a été
réalisée sur 96 des 107 patients.
Les différentes étapes :
- Extraction de l’ADN génomique des blocs fixés en paraffine
- Amplification du matériel génétique par PCR grâce à une ADN polymérase thermostable
(TAQ polymerase), par extension itérative de deux amorces oligonucléotidiques situées
de part et d’autre de la région considérée.
- Les séquences génomiques de référence utilisées sont :
o NM_000222.2 pour KIT et NM_006206.4 pour PDGFRA
37
3. RESULTATS
3.1 Caractéristiques des patients
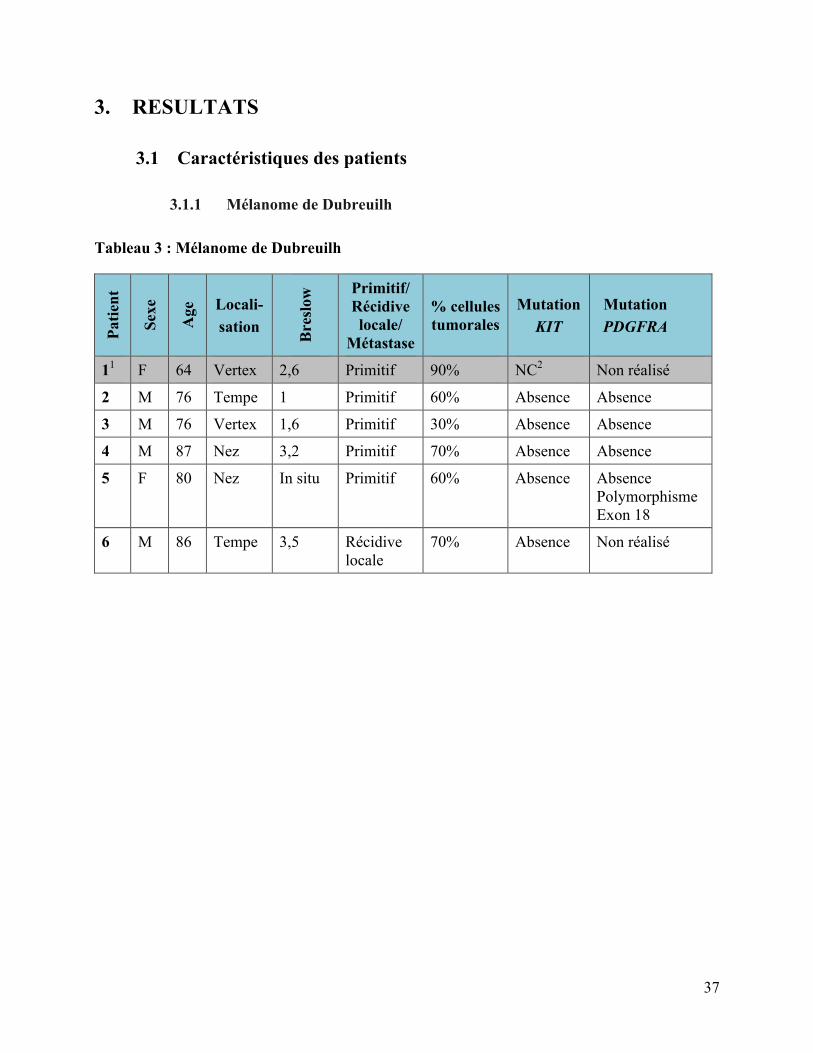
3.1.1 Mélanome de Dubreuilh
Tableau 3 : Mélanome de Dubreuilh
Patie
nt
Sexe
Age
Locali- sation
Bre
slow
Primitif/ Récidive locale/
Métastase
% cellules tumorales
Mutation KIT
Mutation PDGFRA
11 F 64 Vertex 2,6 Primitif 90% NC2 Non réalisé
2 M 76 Tempe 1 Primitif 60% Absence Absence
3 M 76 Vertex 1,6 Primitif 30% Absence Absence
4 M 87 Nez 3,2 Primitif 70% Absence Absence
5 F 80 Nez In situ Primitif 60% Absence Absence Polymorphisme Exon 18
6 M 86 Tempe 3,5 Récidive locale
70% Absence Non réalisé
38
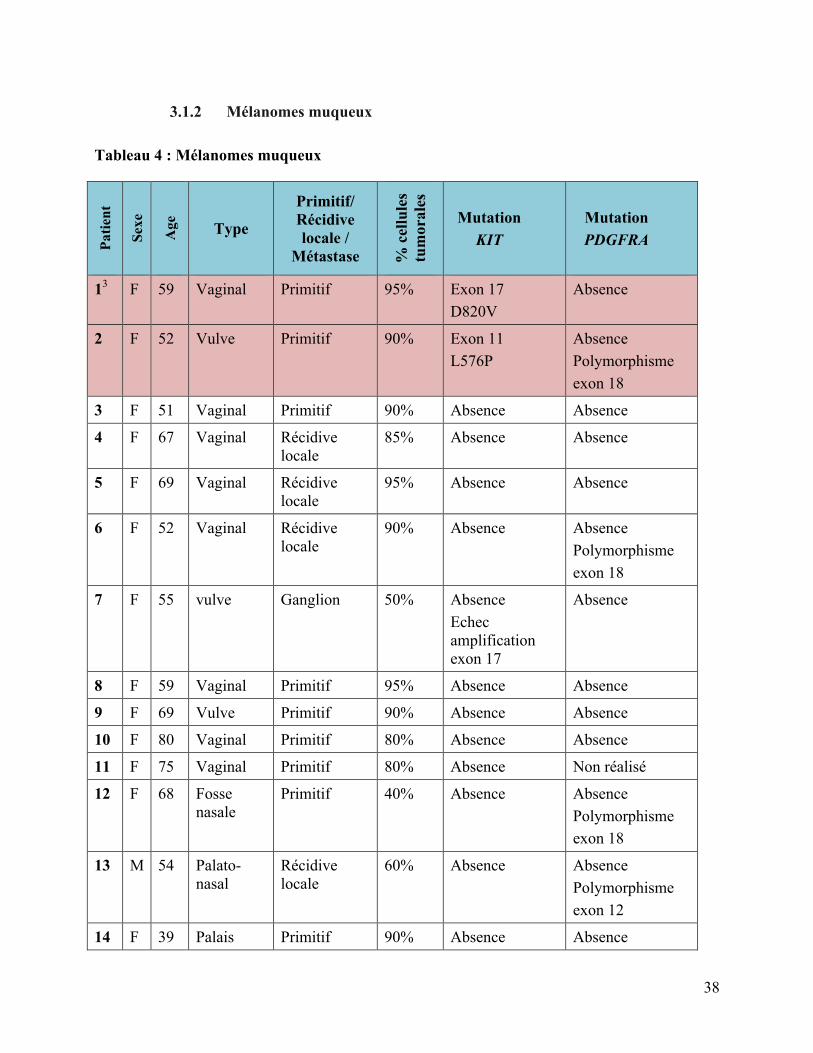
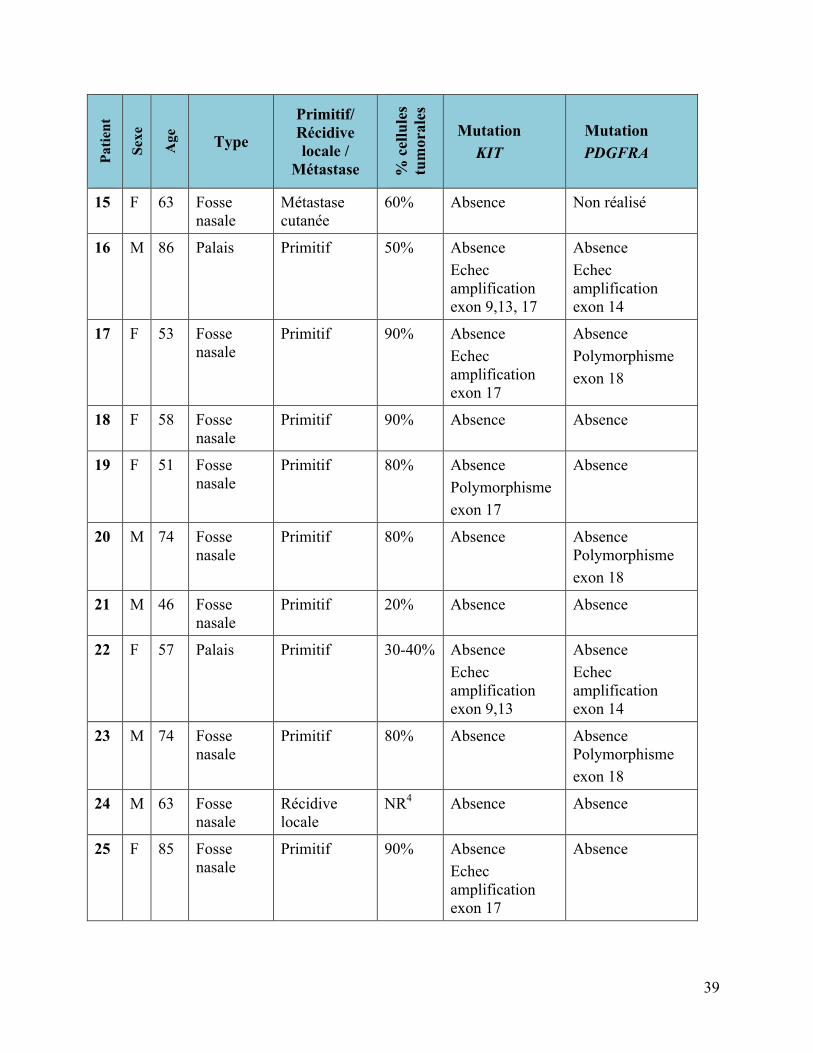
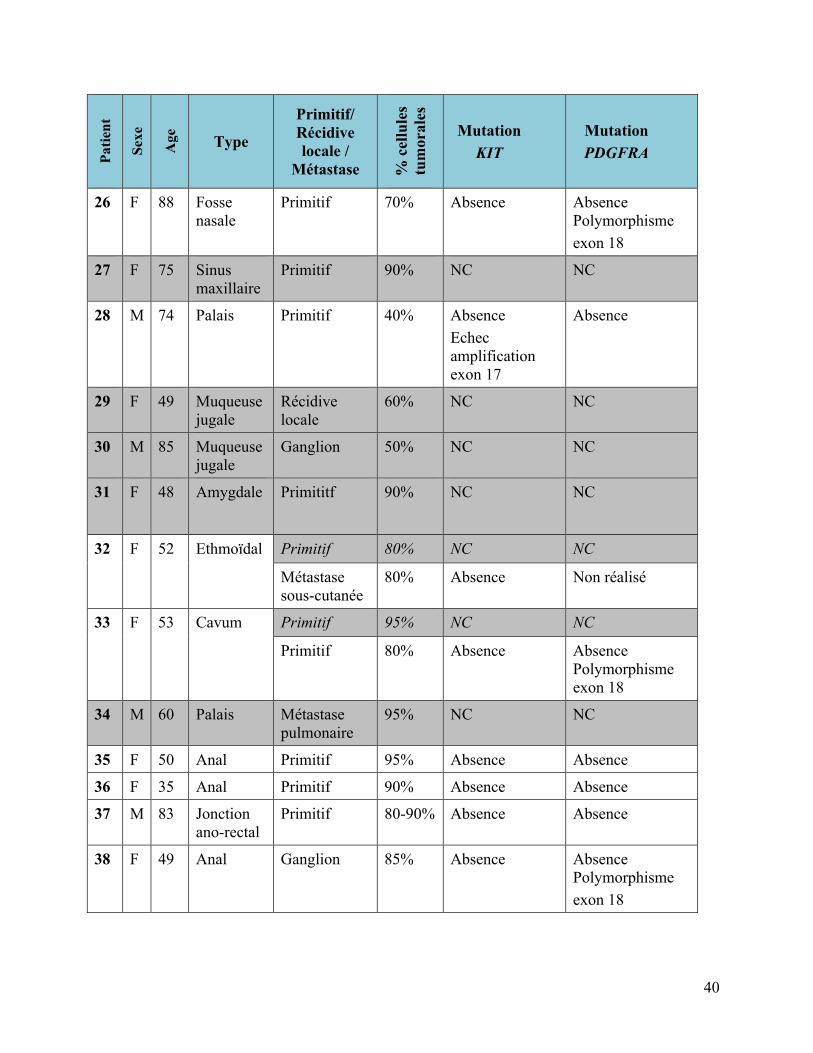
3.1.2 Mélanomes muqueux
Tableau 4 : Mélanomes muqueux
Patie
nt
Sexe
Age
Type
Primitif/ Récidive locale /
Métastase % c
ellu
les
tum
oral
es
Mutation KIT
Mutation PDGFRA
13 F 59 Vaginal Primitif 95% Exon 17 D820V
Absence
2 F 52 Vulve Primitif 90% Exon 11 L576P
Absence Polymorphisme exon 18
3 F 51 Vaginal Primitif 90% Absence Absence
4 F 67 Vaginal Récidive locale
85% Absence Absence
5 F 69 Vaginal Récidive locale
95% Absence Absence
6 F 52 Vaginal Récidive locale
90% Absence Absence Polymorphisme exon 18
7 F 55 vulve Ganglion 50% Absence Echec amplification exon 17
Absence
8 F 59 Vaginal Primitif 95% Absence Absence
9 F 69 Vulve Primitif 90% Absence Absence
10 F 80 Vaginal Primitif 80% Absence Absence
11 F 75 Vaginal Primitif 80% Absence Non réalisé
12 F 68 Fosse nasale
Primitif 40% Absence Absence Polymorphisme exon 18
13 M 54 Palato-nasal
Récidive locale
60% Absence Absence Polymorphisme exon 12
14 F 39 Palais Primitif 90% Absence Absence
39
Patie
nt
Sexe
Age
Type
Primitif/ Récidive locale /
Métastase % c
ellu
les
tum
oral
es
Mutation KIT
Mutation PDGFRA
15 F 63 Fosse nasale
Métastase cutanée
60% Absence Non réalisé
16 M 86 Palais Primitif 50% Absence Echec amplification exon 9,13, 17
Absence Echec amplification exon 14
17 F 53 Fosse nasale
Primitif 90% Absence Echec amplification exon 17
Absence Polymorphisme exon 18
18 F 58 Fosse nasale
Primitif 90% Absence Absence
19 F 51 Fosse nasale
Primitif 80% Absence Polymorphisme exon 17
Absence
20 M 74 Fosse nasale
Primitif 80% Absence Absence Polymorphisme exon 18
21 M 46 Fosse nasale
Primitif 20% Absence Absence
22 F 57 Palais Primitif 30-40% Absence Echec amplification exon 9,13
Absence Echec amplification exon 14
23 M 74 Fosse nasale
Primitif 80% Absence Absence Polymorphisme exon 18
24 M 63 Fosse nasale
Récidive locale
NR4 Absence Absence
25 F 85 Fosse nasale
Primitif 90% Absence Echec amplification exon 17
Absence
40
Patie
nt
Sexe
Age
Type
Primitif/ Récidive locale /
Métastase % c
ellu
les
tum
oral
es
Mutation KIT
Mutation PDGFRA
26 F 88 Fosse nasale
Primitif 70% Absence Absence Polymorphisme exon 18
27 F 75 Sinus maxillaire
Primitif 90% NC NC
28 M 74 Palais Primitif 40% Absence Echec amplification exon 17
Absence
29 F 49 Muqueuse jugale
Récidive locale
60% NC NC
30 M 85 Muqueuse jugale
Ganglion 50% NC NC
31 F 48 Amygdale Primititf 90% NC NC
Primitif 80% NC NC 32 F 52 Ethmoïdal
Métastase sous-cutanée
80% Absence Non réalisé
Primitif 95% NC NC 33 F 53 Cavum
Primitif 80% Absence
Absence Polymorphisme exon 18
34 M 60 Palais Métastase pulmonaire
95% NC NC
35 F 50 Anal Primitif 95% Absence Absence
36 F 35 Anal Primitif 90% Absence Absence
37 M 83 Jonction ano-rectal
Primitif 80-90% Absence Absence
38 F 49 Anal Ganglion 85% Absence Absence Polymorphisme exon 18

41
Patie
nt
Sexe
Age
Type
Primitif/ Récidive locale /
Métastase % c
ellu
les
tum
oral
es
Mutation KIT
Mutation PDGFRA
39 M 67 Rectum Primitif 85% Absence Absence Polymorphisme exon 18
40 F 61 Anal Récidive locale
5% NI5
Absence Echec amplification exon 17
NI Absence
41 F 66 Bas rectum
Primitif 95% NC NC
42 M 60 Anal Récidive locale
90% Absence Absence
42
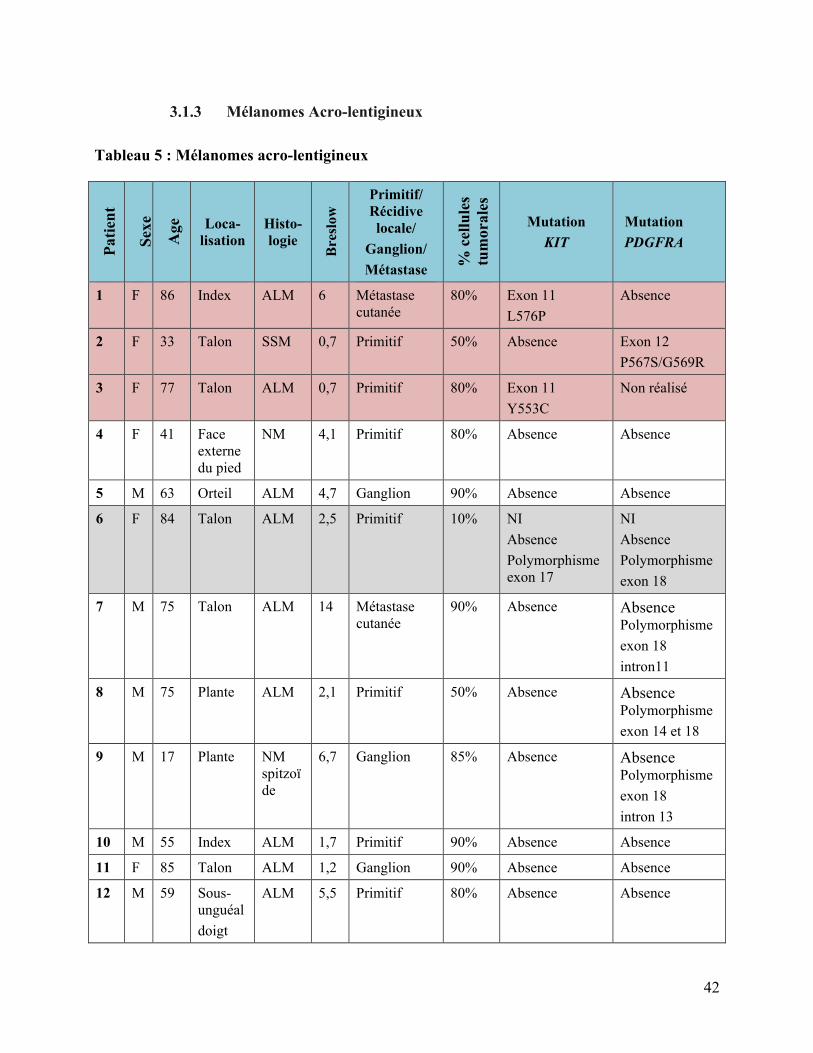
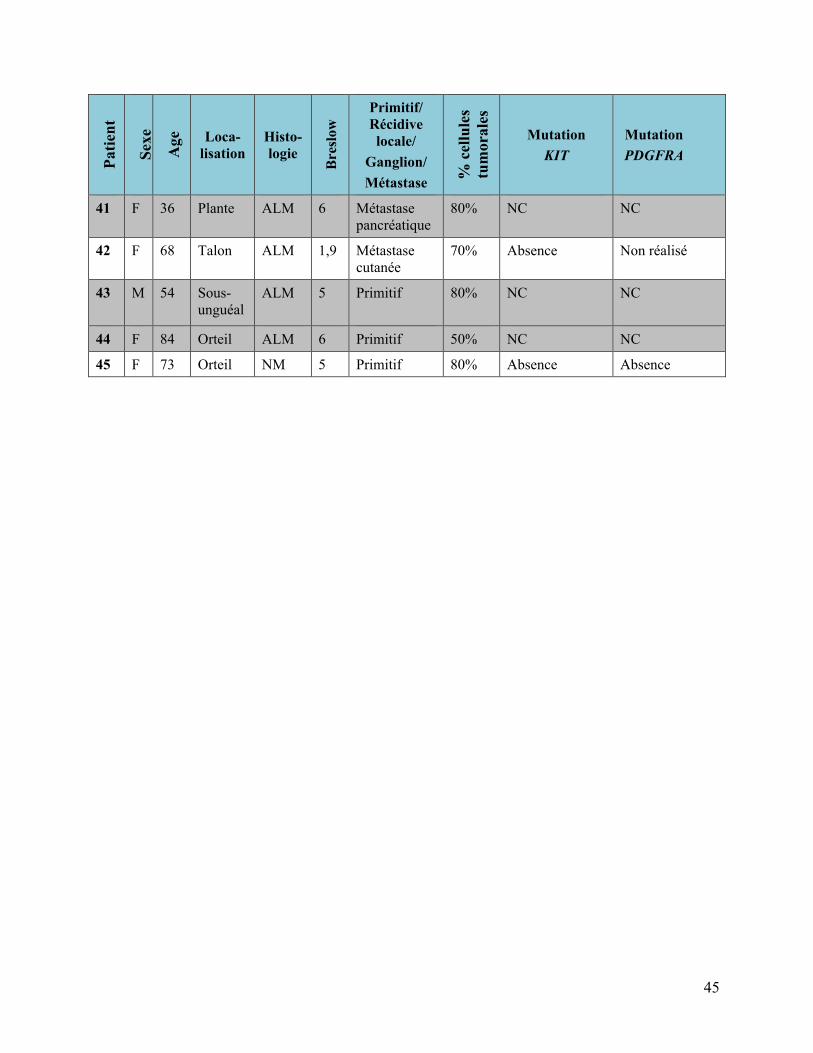
3.1.3 Mélanomes Acro-lentigineux
Tableau 5 : Mélanomes acro-lentigineux
Patie
nt
Se
xe
Age
Loca-lisation
Histo-logie
Bre
slow
Primitif/ Récidive locale/
Ganglion/ Métastase %
cel
lule
s tu
mor
ales
Mutation KIT
Mutation PDGFRA
1 F 86 Index ALM 6 Métastase cutanée
80% Exon 11 L576P
Absence
2 F 33 Talon SSM 0,7 Primitif 50% Absence Exon 12 P567S/G569R
3 F 77 Talon ALM 0,7 Primitif 80% Exon 11 Y553C
Non réalisé
4 F 41 Face externe du pied
NM
4,1 Primitif 80% Absence Absence
5 M 63 Orteil ALM 4,7 Ganglion 90% Absence Absence
6 F 84 Talon ALM 2,5 Primitif 10% NI Absence Polymorphisme exon 17
NI Absence Polymorphisme exon 18
7 M 75 Talon ALM 14 Métastase cutanée
90% Absence Absence Polymorphisme exon 18 intron11
8 M 75 Plante ALM 2,1 Primitif 50% Absence Absence Polymorphisme exon 14 et 18
9 M 17 Plante NM spitzoïde
6,7 Ganglion 85% Absence Absence Polymorphisme exon 18 intron 13
10 M 55 Index ALM 1,7 Primitif 90% Absence Absence
11 F 85 Talon ALM 1,2 Ganglion 90% Absence Absence
12 M 59 Sous-unguéal doigt
ALM 5,5 Primitif 80% Absence Absence

43
Patie
nt
Se
xe
Age
Loca-lisation
Histo-logie
Bre
slow
Primitif/ Récidive locale/
Ganglion/ Métastase %
cel
lule
s tu
mor
ales
Mutation KIT
Mutation PDGFRA
13 F 61 Talon NM 5,4 Primitif 80% Absence Echec amplification exon 17
Absence
14 M 81 Talon ALM 3,2 Primitif 70% Absence Absence
15 M 61 Pouce ALM 1,5 Métastase cutanée
90% Absence Absence
16 M 69 Espace inter-orteil
ALM 1,5 Primitif 30% Absence Echec amplification exon 17
Absence Polymorphisme exon 12
17 F 16 Talon ALM 0,2 Primitif 20% Absence Absence
18 M 42 Talon ALM 5 Ganglion 80% Absence Absence
19 M 41 Talon ALM 3,9 Ganglion 80% Absence Absence
20 F 48 Orteil face plantaire
ALM 1,1 Métastase cutanée
80% Absence Absence
21 M 77 Plante ALM 2,5 Primitif 80% Absence Echec amplification exon 17
Absence
22 M 54 Orteil NM 9 Ganglion 90% Absence Absence
23 F 58 Talon ALM 2,5 Primitif 30% Absence Absence
24 F 39 Espace inter-orteil
ALM 2,6 Primitif 90% Absence Absence Polymorphisme exon 18
25 M 25 Plante ALM 11 Primitif 80% Absence Absence Polymorphisme exon 18
26 F 82 Plante ALM 3,9 Primitif 60% Absence Absence Polymorphisme exon 18

44
Patie
nt
Se
xe
Age
Loca-lisation
Histo-logie
Bre
slow
Primitif/ Récidive locale/
Ganglion/ Métastase %
cel
lule
s tu
mor
ales
Mutation KIT
Mutation PDGFRA
27 F 68 Plante ALM 20 Récidive locale
80% Absence Absence
28 F 67 Talon SSM 0,9 Métastase cutanée
70% NC NC
29 F 68 Plante ALM 7 Primitif 10% NI Absence
NI Absence
30 M 54 Sous-unguéal doigt
ALM 4,5 Métastase cutanée
NR NC NC
31 M 58 Plante ALM 0,8 Primitif 30% Absence Absence
32 M 60 Talon ALM 4 Métastase sinus
NR NC NC
33 M 80 Talon ALM 2,2 Métastase cutanée
90% NC NC
34 F 48 Plante SSM 5,5 Métastase cutanée
NR Absence Non réalisé
35 F 69 Espace inter-orteil
ALM 0,6 Ganglion 90% NC NC
36 F 45 Sous-unguéal
Inclassable
7 Primitif 90% NC NC
37 M 60 Doigt ALM 15 Ganglion 10% NI Absence
NI Absence Polymorphisme exon 18
38 F 70 Pied ALM NS Ganglion 70% NC Non réalisé
39 F 72 Plante ALM 0,9 Primitif 50% NC NC
40 F 56 Orteil ALM 0,4 Primitif 10% NI Absence Echec amplification exon 13
NI Absence Polymorphisme exon 18
45
Patie
nt
Se
xe
Age
Loca-lisation
Histo-logie
Bre
slow
Primitif/ Récidive locale/
Ganglion/ Métastase %
cel
lule
s tu
mor
ales
Mutation KIT
Mutation PDGFRA
41 F 36 Plante ALM 6 Métastase pancréatique
80% NC NC
42 F 68 Talon ALM 1,9 Métastase cutanée
70% Absence Non réalisé
43 M 54 Sous-unguéal
ALM 5 Primitif
80% NC NC
44 F 84 Orteil ALM 6 Primitif 50% NC NC
45 F 73 Orteil NM 5 Primitif 80% Absence Absence

46
3.1.4 Mélanome sans primitif retrouvé
Tableau 6 : Mélanome sans primitif retrouvé
Patie
nt
Sexe
Age
Stade6 Site recherche
% cellules
tumorales
Mutation KIT
Mutation PDGFRA
1 F 31 Stade III Ganglion 40% Absence Absence
2 F 83 Stade III Cutanée 60% Absence Non réalisé
3 F 21 Stade IV Sous-cutanée
30% Absence Polymorphisme intron 16
Absence Polymorphisme exon 18
4 F 64 Stade IV Osseuse 50% Absence Absence
5 M 59 Stade IV Ganglion 90% Absence Absence
6 F 80 Stade III Sous-cutanée
80% Absence Absence Polymorphisme exon 18
7 F 46 Stade IV Digestive 80% Absence Absence
8 F 36 Stade IV Localisation annexielle
85% Absence Absence
9 M 68 Stade III Ganglion 80% Absence Absence
10 M 44 Stade IV Grêle 95% Absence Echec amplification exon 17
Absence Polymorphisme exon 18
11 F 61 Stade IV Ganglion 90% Absence Absence
12 M 80 Stade III Sous-cutanée
90% Absence Absence
13 F 66 Stade IV Gastrique 30% Absence Absence
14 M 63 Stade IV Pulmonaire 80% Absence Non réalisé 1 : les cases grisées sont les résultats non contributifs 2 : NC : non contributif 3 : les cases rosées sont les résultats positifs 4 : NR : non renseigné 5 : NI : non interprétable 6 : Selon la classification AJCC 2009, en considérant une localisation unique cutanée en stade III
et les localisations cutanées multiples sur différents territoires stade IV en accord avec la classification utilisée dans l’article de Savoia et al. (37)

47
3.2 Résultats généraux
Sur les 107 tumeurs testées, quatre mutations de KIT et deux mutations du même exon de
PDGFRA ont été isolées.
Pour le gène KIT :
Chez deux patientes, il s’agit de la mutation L576P localisée sur l’exon 11. C’est une mutation
ponctuelle liée à une substitution d’une thymidine par une cytosine, remplaçant une leucine par
une proline sur le codon 576 (c.1727T>C) (Annexe 2 et 3) Elle a été mise en évidence :
- pour la première : dans une localisation métastatique sous-cutanée d’un mélanome
primitivement situé sur l’index droit de type acro-lentigineux, Breslow 6 mm, niveau IV
de Clark, ulcéré et d’index mitotique élevé chez une patiente âgée de 84 ans.
- Pour la seconde : sur un mélanome primitif vulvaire para-clitoridien gauche de 7 mm de
Breslow chez une patiente âgée de 52 ans.
Chez la troisième patiente, il s’agit de la mutation D820V localisée sur l’exon 17 liée à la
substitution d’un acide aspartique par une valine sur le codon 820 (c.2459A>T), mise en évidence
sur un mélanome primitif cervico-vaginal chez une patiente âgée de 59 ans.
Chez la quatrième patiente, il s’agit de la mutation Y553C localisée sur l’exon 11 liée à la
substitution d’une tyrosine par une cystéine sur le codon 553 (c.1658A>R), mise en évidence sur
un mélanome primitif du talon de type ALM de 0,7 mm de Breslow, ulcéré avec 3 mitoses/m2
chez une patiente âgée de 77 ans.
Pour le gène PDGFRA :
Nous avons retrouvé deux mutations faux-sens de PDGFRA sur les codons 567 et 569 de l’exon
12 (P567S et G569R), sur un mélanome primitif de type SSM de Breslow 0,7 mm du talon chez
une patiente de 33 ans.

48
Dans 16,8% des cas (18/107) la recherche était non contributive (NC), liée à une mauvaise
qualité de l’ADN après extraction. Pour deux patients, une seconde extraction d’ADN a permis
d’obtenir un ADN utilisable. Dans l’un de ces cas, l’extraction de l’ADN a été faite sur le même
bloc de paraffine, pour l’autre la recherche a été faite sur un nouveau prélèvement tumoral, ces
cas sont intégrés dans les résultats contributifs.
Dans 4,7% des cas (5/107), les résultats ne sont pas interprétables (NI) compte tenu du faible
pourcentage de cellules tumorales sur la zone étudiée (≤10%).
Le pourcentage de cellularité tumorale varie de moins de 10% à 95% selon la répartition
suivante :
- ≤10% : 5
- entre 11 et 49% : 12
- entre 50 et 69% : 15
- ≥ à 70% : 71
- Non renseigné (NR) : 4
Si l’on ne considère que les cas informatifs (84 patients), des mutations de KIT ont été détectées
dans 6,7% (2/30) des ALM, dans 18,2% (2/11) des mélanomes vaginaux, 0% (0/18) des
mélanomes de la sphère ORL, et des mélanomes ano-rectaux (0/6), soit 5,7% (2/35) de tous les
mélanomes muqueux confondus, 0% (0/5) des mélanomes de Dubreuilh et des mélanomes sans
primitif retrouvé ( 0/14 ).
Deux mutations sur le même exon ont été retrouvées sur le gène PDGFRA soit 1/80 (1,3%) des
mélanomes muqueux, de Dubreuilh, ALM et sans primitif retrouvé.
Différents polymorphismes ont été retrouvés sur le gène KIT et PDGFRA, sans conséquence sur
la fonction protéique (voir résultats annexe 4).

49
3.3 Observations des trois patientes traitées par inhibiteur de KIT et de
PDGFRA
Sur nos cinq patients mutés, trois patients ont été traités par inhibiteurs de KIT et de PDGFRA. Les deux autres étant en rémission après chirurgie, aucun traitement complémentaire ne leur a été proposé.
3.3.1 Observation N°1
La première patiente, (N°1 tableau 4) âgée de 60 ans était suivie depuis mai 2009 pour un
mélanome vaginal, traité initialement par chirurgie. En octobre 2009, la patiente a présenté une
récidive métastatique avec des localisations pulmonaires et ganglionnaires sous-
diaphragmatiques. Le séquençage des exons 8, 9, 11, 13, 17 du gène KIT et des exons 12, 14 et
18 du gène PDGFRA a identifié la mutation D820V de l’exon 17 de KIT. Un traitement par
imatinib a débuté le 12 octobre 2009 à la dose de 400 mg deux fois par jour. La première
évaluation à six semaines de traitement montrait une stabilité des lésions pulmonaires. La
tolérance était correcte en dehors d’une éruption maculeuse du tronc peu prurigineuse soulagée
par des anti-histaminiques et des dermocorticoïdes, d’un œdème des paupières prédominant le
matin et d’une asthénie de grade 1. La patiente était revue précocement, fin décembre 2009, pour
des douleurs abdominales non contrôlées. Une échographie abdomino-pelvienne montrait des
collections cloisonnées para-vésicales compatibles avec des adénopathies ou des lymphocèles
postopératoires. En janvier 2010, la patiente a présenté une progression symptomatique avec
aggravation nette des lésions pelviennes. Le traitement par imatinib a été arrêté le 29 janvier
2010. Une seconde ligne de traitement par dacarbazine (DTIC) débutait le 15 février 2010. Le
dasatinib, à la dose de 100 mg par jour, y était associé à partir de la deuxième cure de DTIC le 8
mars 2010. Cette association fait l’objet d’une phase I/II, la dose maximale recommandée serait
DTIC 1000 mg/m2 J1=J21 et dasatinib 70 mg x 2/jour ou 100 mg/j de J2 à J19(90). Le traitement
a été rapidement interrompu (le 26 mars) devant une thrombopénie de grade III et une
neutropénie de grade III. Au cours du traitement par dasatinib, la patiente rapportait une
amélioration clinique avec diminution des douleurs abdominales mais une reprise rapide de ces
dernières à l’arrêt du traitement. Un bilan d’évaluation après 2 cures de DTIC et 20 jours de
dasatinib montrait une progression des lésions pulmonaires, et pelviennes. Le dasatinib était
repris seul à demi-dose soit 50 mg par jour à partir de mi-avril 2010. La patiente décéda, 2
semaines plus tard, d’une occlusion intestinale liée à une progression rapide de la maladie.

50
3.3.2 Observation N°2
La seconde patiente (N°1 tableau ALM), âgée de 86 ans avait été traitée en février 2004 pour un
mélanome acro-lentigineux de l’index de 6 mm de Breslow, niveau IV de Clark, ulcéré, par
amputation trans-P2 de l’index droit. En juillet 2008, une récidive sous la forme d’un nodule
sous-cutané en transit de la face antérieure du bras droit était traitée chirurgicalement. La
mutation L576P sur l’exon 11 de KIT était identifiée sur ce nodule. Le séquençage du gène BRAF
n’a pas révélé de mutation. Une dissémination métastatique était constatée en mars 2010 sous la
forme de localisations pulmonaires bilatérales. Le dasatinib à 100 mg par jour était proposé en
première ligne de traitement le 19 avril 2010 (dose recommandée en hématologie, la dose
maximale tolérée dans les tumeurs solides semble plus élevée (70 mg x2/j) mais compte-tenu de
l’âge du patient, une dose moindre était privilégiée). La tolérance clinique et biologique était
excellente. Le premier bilan d’évaluation retrouvait une réponse partielle (réduction tumorale >
30% selon les critères RECIST) après 2 mois de traitement. La seconde évaluation en septembre
2010 montrait malheureusement, une reprise de sa maladie au plan pulmonaire. Devant
l’excellente tolérance clinique et biologique, le dasatinib a été poursuivi avec une augmentation
des doses à 70 mg x 2 par jour, avec une surveillance rapprochée clinique et une évaluation
précoce.

51
3.3.3 Observation N°3
La troisième patiente (N°2 tableau ALM), âgée de 33 ans avait eu l’exérèse d’un mélanome de
type SSM développé sur naevus pré-existant, de niveau III de Clark, de Breslow 0,7 mm, non
ulcéré, non régressif du talon gauche en décembre 2007. En Janvier 2009, une récidive
ganglionnaire inguinale gauche était traitée par curage inguinal gauche montrant 1N+R-/13N
complété par un curage ilio-obturateur en mars 2009 retrouvant 15N-. La patiente était revue en
urgence en juin 2009 devant l’apparition d’une fièvre et des sueurs associées à une altération de
l’état général. L’examen clinique retrouvait une adénopathie sus-claviculaire droite, la
radiographie de thorax montrait un lâcher de ballon. Un bilan infectieux complet éliminait une
origine infectieuse et une chimiothérapie par dacarbazine et adriamycine était débutée rapidement
le 13 juin 2009 devant la confirmation d’une rechute de son mélanome en stade IV. L’état général
s’améliora dans un premier temps.
Le premier bilan d’évaluation après 3 cures de chimiothérapie montrait une globale stabilité des
lésions pulmonaires, hépatiques mais l’apparition de multiples lésions secondaires cérébrales.
Une recherche de mutation de KIT et de PDGFRA avait été réalisée sur la lésion primitive
compte-tenu de sa localisation acrale, mettant en évidence deux mutations du gène PDGFRA sur
l’exon 12 : P567S et G569R. Malgré une incertitude sur le retentissement de la fonction de la
protéine, un traitement par imatinib à 400 mg associé à la fotemustine était débuté le 19 août
2009. Le traitement d’attaque était réalisé avec une bonne tolérance clinique et biologique, sans
bénéfice clinique. L’état général et neurologique de la patiente se dégrada rapidement, et la
patiente décéda en moins d’un mois.

52
4. DISCUSSION
4.1 Nos mutations dans la littérature
4.1.1 Mutation L576P de l’exon 11
La mutation L576P, mise en évidence chez deux de nos patientes, est une mutation faux-sens
située sur le domaine juxtamembranaire cytoplasmique du récepteur KIT.
Elle a été retrouvée chez l’homme dans les GIST, les séminomes testiculaires, un carcinome
thymique et les mélanomes (41,83,91,92).
4.1.1.1 Dans les GIST
Les mutations de l’exon 11 sont de loin les plus fréquentes comprises entre 65 et 70% des
mutations. Elles sont par ordre de fréquence dues à des délétions, des substitutions simples, des
duplications, des insertions et des mutations complexes (association de plusieurs mécanismes)
(41). Cependant, la mutation L576P est rare dans le GIST, évaluée à 8% des substitutions de
l’exon 11, soit moins de 1% toutes mutations confondues (41,93-95). Elle est corrélée avec un
profil phénotypique plus agressif et peut résulter de mutations secondaires (93).
4.1.1.2 Dans le mélanome
Il s’agit de la première mutation de KIT publiée dans le mélanome en 2004 par Went et al. (91).
Contrairement à ce qui est observé dans le GIST, c’est la mutation la plus fréquente dans le
mélanome estimé à 34,7% dans l’article de Garrido et al. (96).
4.1.1.3 Sensibilité au traitement (Tableau 7)
Le type de mutation de KIT est corrélé au pronostic et à la réponse à l’imatinib.
Les patients atteints de GIST avec mutation de l’exon 11 ont un taux de réponse, une survie sans
progression et une survie globale supérieure à celle des patients avec mutation située dans l’exon
9 ou dans d’autres domaines de la protéine (44).

53
Dans l’essai EORTC de phase III randomisé comparant deux doses d’imatinib (400 mg/jour et
800 mg/jour) chez les patients atteints d’une GIST avancée, il n’y avait pas de différence
significative entre la survie sans progression et la survie globale chez les patients avec une
mutation par substitution de l’exon 11 (29 tumeurs dont huit patients avec une mutation L576P)
et ceux par délétion (137 tumeurs) (62).
Un cas de GIST métastatique muté en L576P traité par imatinib a été publié en 2005. Il a
développé une résistance secondaire en 13 mois de traitement, après avoir répondu partiellement
(67).
L’article de Conca et al. présente deux cas de GIST muté L576P sur l’exon 11 (93).
Le premier a eu une réponse complète pendant 1 an sous imatinib à 400 mg/jour avant une
rechute opérée et suivi d’une augmentation de l’imatinib à 800 mg/jour. Une nouvelle rechute à 6
mois était traitée par sunitinib puis nilotinib sans réponse.
Le deuxième a été traité en néoadjuvant par imatinib à 400 mg/jour pendant 10 mois avec
une réponse radiologique permettant un geste chirurgical limité.
Antonescu et al. ont étudié la sensibilité aux ITK de lignées cellulaires de souris exprimant la
mutation L576P. Ils ont testés in vitro le dasatinib (Sprycel®), le nilotinib (Tasigna®) et
l’imatinib (Glivec®) sur la lignée cellulaire Ba/F3 KITL576P et la lignée cellulaire Ba/F3 KITV559D
qui est sensible à l’imatinib. Le dasatinib inhibe la prolifération cellulaire (IC50 = 54 nM) et la
phosphorylation dans les cellules de la lignée Ba/F3 KITL576P. L’imatinib et le nilotinib inhibent
également, dans une moindre mesure, la prolifération cellulaire (IC50 = 253 nM et 185 nM
respectivement). La lignée Ba/F3 KITV559D est bien plus sensible aux ITK avec des IC50 pour le
dasatinib, l’imatinib et le nilotinib à 22 nM, 27 nM et 34 nM respectivement (94).
L’activité in vitro du dasatinib, de l’imatinib, du nilotinib et du sorafenib a aussi été étudiée par
l’équipe de Woodman, sur une lignée cellulaire de mélanome humain WM3211 muté L576P et
comparée avec deux lignées cellulaires sans mutation de KIT : A375 (BRAF V600E muté) et
Mewo (BRAF wild-type) (97). Le dasatinib mais non l’imatinib, ni le nilotinib ni le sorafenib
inhibe les cellules WM3211. S’appuyant sur ces données, deux patients ont été traités :

54
- Une patiente de 55 ans avec un mélanome vulvaire porteur d’une mutation KITL576P,
rechuta après un traitement adjuvant par imatinib à 400 mg par jour pendant 3 mois puis 300 mg
pendant 8 mois. Elle reçut ensuite de multiples lignes de chimiothérapie standard ou dans le cadre
de protocole. Après l’échec de ces traitements, le dasatinib fut instauré à 70 mg, 2 fois par jour.
En 1 mois, l’état clinique de la patiente s’améliora avec une réponse radiologique de 53,5% mais
une progression survint après 4 mois de traitement. Des nouveaux prélèvements tumoraux ne
mettaient pas en évidence de mutations secondaires de KIT.
- Une patiente de 61 ans, avec un mélanome anal porteur d’une mutation KITL576P traitée
par radiothérapie adjuvante après l’exérèse chirurgicale, qui présenta une récidive hépatique 4
mois après l’intervention. Après 1 mois de traitement par dasatinib, l’évaluation au TEP-scanner
montrait une disparition d’une des lésions et une réduction de 54% de l’autre lésion. Le
traitement fut interrompu après 3 mois de traitement pour une reprise de la maladie sur les mêmes
sites.
Ces deux patientes ont donc présenté une réponse franche clinique et métabolique initiale mais
une reprise de l’activité métabolique et de la croissance tumorale en 3-4 mois.
Handolias et al. présentent le cas d’une patiente de 38 ans suivie pour un mélanome vulvaire
métastatique présentant une mutation L576P de l’exon 11. Un traitement par imatinib à 400 mg
par jour était débuté après échec de la chimiothérapie standard. Après une réponse clinique,
biologique (diminution des LDH) et radiologique (-42% des lésions cibles mais apparition d’une
nouvelle lésion vésicale), et malgré l’augmentation du traitement à 600 mg par jour, le traitement
fut arrêté au bout de 16 semaines sur progression et la patiente décéda rapidement de métastases
cérébrales (98).
Notre observation N°2 s’accorde avec les résultats in vitro et les quelques cas cliniques publiés
du dasatinib sur les mélanomes mutés en L576P, avec une réponse partielle à 2 mois de
traitement mais une reprise évolutive à 5 mois.
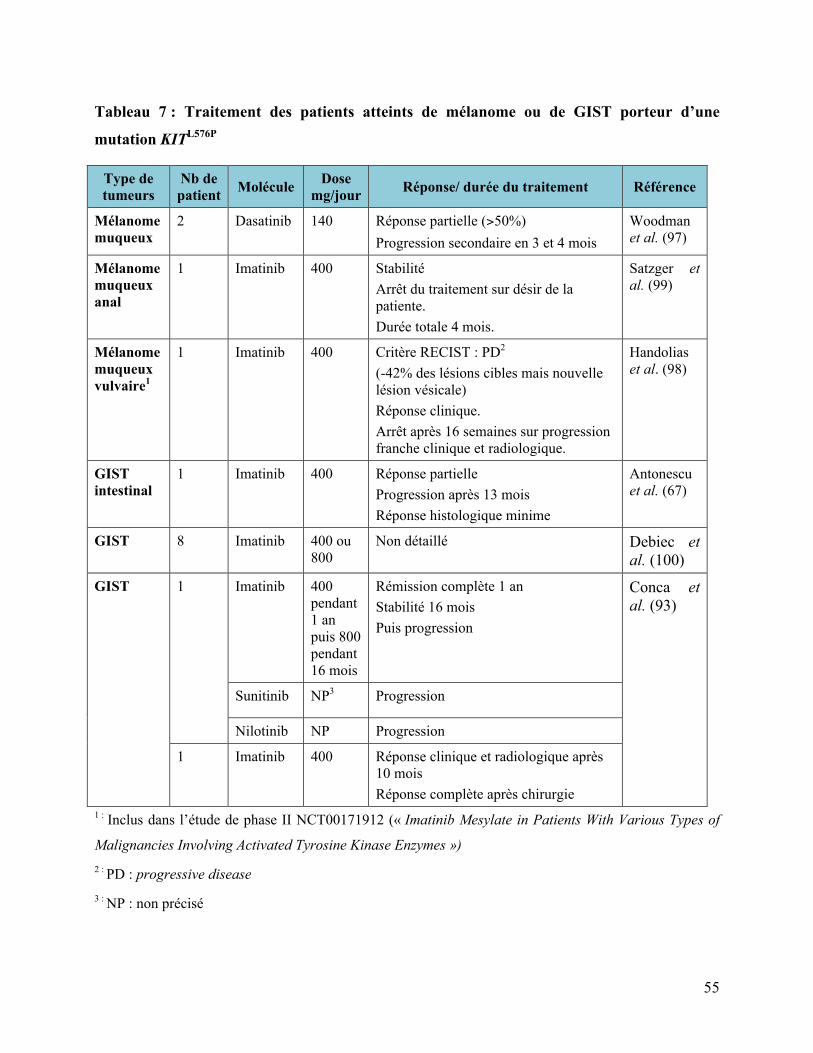
55
Tableau 7 : Traitement des patients atteints de mélanome ou de GIST porteur d’une
mutation KITL576P
Type de tumeurs
Nb de patient Molécule Dose
mg/jour Réponse/ durée du traitement Référence
Mélanome muqueux
2 Dasatinib 140 Réponse partielle (>50%) Progression secondaire en 3 et 4 mois
Woodman et al. (97)
Mélanome muqueux anal
1 Imatinib 400 Stabilité Arrêt du traitement sur désir de la patiente. Durée totale 4 mois.
Satzger et al. (99)
Mélanome muqueux vulvaire1
1 Imatinib 400 Critère RECIST : PD2
(-42% des lésions cibles mais nouvelle lésion vésicale) Réponse clinique. Arrêt après 16 semaines sur progression franche clinique et radiologique.
Handolias et al. (98)
GIST intestinal
1 Imatinib 400 Réponse partielle Progression après 13 mois Réponse histologique minime
Antonescu et al. (67)
GIST 8 Imatinib 400 ou 800
Non détaillé Debiec et al. (100)
Imatinib 400 pendant 1 an puis 800 pendant 16 mois
Rémission complète 1 an Stabilité 16 mois Puis progression
Sunitinib NP3 Progression
1
Nilotinib NP Progression
GIST
1 Imatinib 400 Réponse clinique et radiologique après 10 mois Réponse complète après chirurgie
Conca et al. (93)
1 : Inclus dans l’étude de phase II NCT00171912 (« Imatinib Mesylate in Patients With Various Types of
Malignancies Involving Activated Tyrosine Kinase Enzymes ») 2 : PD : progressive disease 3 : NP : non précisé

56
4.1.2 Mutation Y553C de l’exon 11
4.1.2.1 Dans le GIST
Cette mutation par substitution n’a été citée qu’une fois dans le GIST, chez une femme de 53 ans
avec une localisation gastrique (101) (recherche réalisée dans la base de donnée COSMIC du site
Trust Sanger Institute (102)). Le codon 553 est impliqué majoritairement dans des mutations
dues à des délétions (41).
4.1.2.2 Dans le mélanome
Cette mutation n’avait jamais été décrite jusqu’à présent. Une mutation du codon 553 a cependant
déjà été décrite avec une substitution de tyrosine en asparagine (Y553N) (103).
4.1.3 Mutation D820V de l’exon 17
Cette mutation se situe sur la boucle activation du domaine tyrosine kinase 2 (TK2).
Elle a déjà été isolée dans le GIST, un séminome médiastinal, un séminome testiculaire et un
germinome intra-cranien (41,80,82,83).
4.1.3.1 Dans le GIST
Les mutations de l’exon 17 (qui code pour le domaine tyrosine kinase 2) sont extrêmement rares
dans le GIST, décrites sur une série de 322 GIST à 0,6% par Corless et al. (104). Le profil
clinico-pathologique des GIST avec une mutation de l’exon de 17 a été spécifiquement étudié.
Dans 71,4% il s’agit d’une substitution Asn822Lys, la médiane d’âge et le sex ratio sont
équivalents aux GIST mutées sur d’autres exons, l’atteinte du grêle est deux fois plus fréquente
que l’estomac et le potentiel agressif ne semble pas varier que ce soit dans sa localisation
digestive ou gastrique (105).
La mutation D820V a déjà été isolée dans le GIST à deux reprises : un cas sur 21 d’une étude
multicentrique, il s’agissait d’une femme de 73 ans avec une localisation gastrique de 15 cm, et
dans un contexte de mutations secondaires multiples touchant les codons 816, 820 et 822. Les
mutations primaires ou secondaires de l’exon 17 se situent sur les mêmes « hot spot » (41).

57
4.1.3.2 Dans le mélanome
Contrairement au GIST, les mutations de l’exon 17 semblent plus fréquentes et s’élèveraient à
14,4% des mutations de KIT dans le mélanome (96,98,106-109)(figure 5).
Une mutation sur le codon 820 a déjà été retrouvée à quatre reprises soit dans 4,4% des
mutations, avec une substitution de l’acide aspartique pour une tyrosine (D820Y) dans deux
ALM et un mélanome anal (41,44,62) mais n’a jamais été décrite avec une substitution pour la
valine. Nous avons donc mis en évidence une nouvelle mutation dans le mélanome.
4.1.3.3 Sensibilité au traitement
In vitro, plusieurs lignées cellulaires avec mutations secondaires ont été testées et en particulier
sur une lignée mutée en D820Y (Ba/F3 KITV559D/D820Y), le nilotinib semble plus efficace par
rapport au dasatinib, au sorafenib et à l’imatinib avec des IC50 respectivement à 297 nmol/l, 432
nmol/l, 944 nmol/l et 3202 nmo/l (110).
Les données cliniques sur la sensibilité à l’imatinib des mutations touchant l’exon 17 sont
limitées du fait de leur rareté. Chez des patients atteints de GIST mutées sur l’exon 17, quatre
réponses partielles, une stabilité et une résistance primaire ont été décrites, mais il ne s’agissait
pas de la mutation D820V (44,62,95).
L’émergence d’une résistance secondaire à l’imatinib est en lien avec l’apparition de mutations
secondaires qui prédominent sur les exons 13, 14, 17 et 18 avec un profil de résistance à
l’imatinib et au sunitinib pour les mutations de l’exon 17 touchant les codons 816, 820 ou 823.
Cela laisse penser que les mutations primaires de l’exon 17 devraient entraîner une résistance à
l’imatinib et au sunitinib (65).
Un cas de réponse partielle prolongée (plus de 15 mois) sous sorafenib (400 mg/jour) a été publié
chez un patient atteint d’un carcinome thymique muté en D820E (111).
Dans notre observation, la patiente a eu probablement un bénéfice des deux inhibiteurs de
tyrosine kinase donnés (imatinib et dasatinib). En effet, l’imatinib a maintenu une stabilité
pendant deux mois et le dasatinib a eu un effet rapide sur les symptômes mais a été interrompu
suite à une toxicité cumulée avec le DTIC.

58
4.1.4 Mutation P567S et G569R de l’exon 12
4.1.4.1 PDGFRA et GIST
Parmi les GIST sans mutation de KIT, on retrouve dans un tiers des cas une mutation du gène
PDGFRA (soit 5 à 7 % de l’ensemble des GIST) (44,45,112). C’est l’exon 18 codant pour la
boucle d’activation du domaine kinase 2 qui est le plus souvent concerné (89,6% des GIST
mutées en PDGFRA). La mutation la plus souvent retrouvée est la D842V (62,6% des cas). Les
autres exons sont beaucoup plus rarement mutés, l’exon 12 est concerné dans 9,3% des cas et
l’exon 14 dans 1,2% des cas (112). La présence d’une double mutation sur le même exon a déjà
été retrouvée sur l’exon 18 (112).
Les deux mutations ponctuelles faux-sens de notre série sont uniques, jamais décrites. Leur
conséquence fonctionnelle n’est pas connue.
4.1.4.2 PDGFRA et mélanome
Une seule mutation primaire de PDGFRA a été publiée à ce jour, il s’agit de la mutation V824L
de l’exon 18, le type histologique du mélanome n’est pas précisé (113).
A l’ASCO 2010, une mutation secondaire de l’exon 12 de PDGFRA (W559stop) a été publiée
chez un patient avec un mélanome métastatique en progression après une réponse partielle à
l’imatinib (114).
Cependant, il n’existe que peu d’articles publiés sur les mutations de PDGFRA et le mélanome.
L’équipe de Curtin a étudié 26 tumeurs : dix tumeurs avec une amplification du locus 4q12 et
seize tumeurs sans amplification et sans mutation de KIT ou de BRAF. L’histologie des 26
tumeurs comprenait sept ALM, treize mélanomes muqueux, trois mélanomes CSD et trois N-
CSD. Les exons 10, 12, 14 et 18 de PDGFRA étaient étudiés. Aucune mutation n’a été mise en
évidence (115).
Antonescu et al. n’ont pas mis en évidence de mutation de PDGFRA sur 20 mélanomes anaux
(94).

59
Une autre étude sur 14 patients atteints de mélanome dont le type histologique n’est pas précisé
ne retrouve pas de mutation de PDGFRA (116).
Terada étudia le gène PDGFRA (exon 12 et 18) de deux mélanomes muqueux achromiques de
l’œsophage, les deux résultats étaient négatifs (117).
Et enfin, dans un article récent, Terada explora le gène PDGFRA de douze patients d’origine
japonaise, atteints de mélanome dont deux ALM, trois CSD et sept N-CSD. Aucune mutation de
PDGFRA n’a été retrouvée (118).
4.1.4.3 Sensibilité au traitement
La mutation D842V de l’exon 18 de PDGFRA, la plus fréquente dans le GIST engendre
fréquemment une résistance primaire à l’imatinib et au sunitinib (104). Cependant, d’autres
substitutions du même codon ou sur d’autres codons ont montré des réponses in vitro et cliniques
(112).
Le patient avec la mutation PDGFRAV824L a été traité dans le cadre d’un protocole de phase 1
évaluant le sorafenib en association avec le tipifarnib, et a été stable pendant 14 mois (113).
Notre troisième observation ne nous permet pas de conclure sur l’efficacité ou non du traitement
par imatinib qui a été débuté très tardivement, sur une maladie déjà très évolutive au plan
cérébral.

60
4.2 Les mutations de KIT dans le mélanome, revue de la littérature
4.2.1 Nos résultats par rapport à la littérature
Nous avons donc isolé à deux reprises la mutation la plus fréquemment retrouvée dans le
mélanome et mis en évidence deux mutations jamais décrites jusqu’alors. La figure 5 résume les
mutations de KIT publiées à ce jour et leur fréquence dans le mélanome.
Figure 5 : Distribution et fréquence des mutations publiées dans le mélanome.
(85,91,94,97-99,103,106-109,119-129)

61
Nous avons retrouvé un faible pourcentage de mutations de KIT (4,8%) sur les 84 résultats
contributifs de notre série de 107 patients :
- 6,7% (2/30) des ALM,
- 18,2% (2/11) des mélanomes vaginaux,
- 0% (0/18) des mélanomes de la sphère ORL,
- 0% (0/6) des mélanomes ano-rectaux,
o soit 5,7% (2/35) de tous les mélanomes muqueux confondus,
- 0% (0/5) des mélanomes de Dubreuilh,
- 0% (0/14) des mélanomes sans primitif retrouvé
Ces résultats sont inférieurs à ceux de la littérature pour les mélanomes muqueux et à la limite
inférieure pour les ALM.
En effet, la fréquence des mutations de KIT dans les mélanomes acro-lentigineux varie entre 6 et
23%, celle des mélanomes muqueux entre 15 et 37,5%.
Il est difficile de comparer nos résultats dans les mélanomes de Dubreuilh avec la littérature
compte-tenu de notre faible échantillon. Seules deux études ont mises en évidence des mutations
sur les mélanomes CSD à 16,7 et 34,6%, une autre étude sur treize patients est négative
(103,114,130).
Les mélanomes sans primitif retrouvé n’ont pas été beaucoup étudiés jusqu’à présent, notre série
fait partie des plus grandes. En effet, le taux de mutation de KIT dans ce type de mélanome n’a
été recherché que sur deux séries : la plus grande a été présenté à l’ASCO 2010 dans une étude de
phase II testant l’imatinib dans les mélanomes avec aberrations de KIT. Sur 42 mélanomes sans
primitif retrouvé, cinq patients ont une mutation ou une amplification de KIT soit 11,9% (114).
L’autre série sur neuf patients est négative (108).
Le taux d’analyse non contributive pour échec de PCR s’élève à 16,8% des cas (18/107) et est
attribuable en grande partie à la mélanine qui entre en compétition avec la Taq Polymérase. Ces
taux sont comparables aux taux d’échec des autres études compris entre 5,1 et 32%.
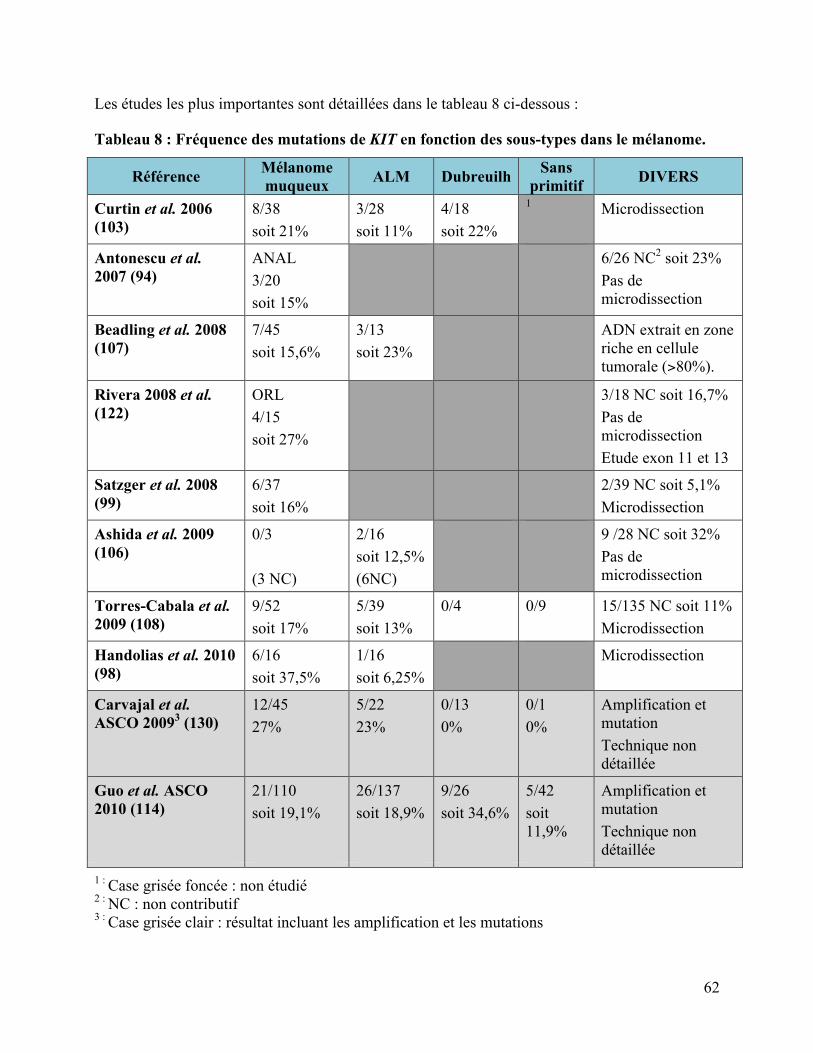
62
Les études les plus importantes sont détaillées dans le tableau 8 ci-dessous :
Tableau 8 : Fréquence des mutations de KIT en fonction des sous-types dans le mélanome.
Référence Mélanome muqueux ALM Dubreuilh Sans
primitif DIVERS
Curtin et al. 2006 (103)
8/38 soit 21%
3/28 soit 11%
4/18 soit 22%
1 Microdissection
Antonescu et al. 2007 (94)
ANAL 3/20 soit 15%
6/26 NC2 soit 23% Pas de microdissection
Beadling et al. 2008 (107)
7/45 soit 15,6%
3/13 soit 23%
ADN extrait en zone riche en cellule tumorale (>80%).
Rivera 2008 et al. (122)
ORL 4/15 soit 27%
3/18 NC soit 16,7% Pas de microdissection Etude exon 11 et 13
Satzger et al. 2008 (99)
6/37 soit 16%
2/39 NC soit 5,1% Microdissection
Ashida et al. 2009 (106)
0/3 (3 NC)
2/16 soit 12,5% (6NC)
9 /28 NC soit 32% Pas de microdissection
Torres-Cabala et al. 2009 (108)
9/52 soit 17%
5/39 soit 13%
0/4 0/9 15/135 NC soit 11% Microdissection
Handolias et al. 2010 (98)
6/16 soit 37,5%
1/16 soit 6,25%
Microdissection
Carvajal et al. ASCO 20093 (130)
12/45 27%
5/22 23%
0/13 0%
0/1 0%
Amplification et mutation Technique non détaillée
Guo et al. ASCO 2010 (114)
21/110 soit 19,1%
26/137 soit 18,9%
9/26 soit 34,6%
5/42 soit 11,9%
Amplification et mutation Technique non détaillée
1 : Case grisée foncée : non étudié 2 : NC : non contributif 3 : Case grisée clair : résultat incluant les amplification et les mutations

63
Toutes ces études sont assez hétérogènes. Certaines études n’étudiant qu’un sous-type, d’autres
mélangeant les différents mélanomes muqueux. Elles sont aussi hétérogènes dans la technique de
recherche de mutation et en particulier, pour l’extraction de l’ADN, qui est réalisé pour certains
par microdissection, augmentant ainsi le pourcentage de cellularité tumorale. Notre laboratoire ne
réalise pas dans le cadre de la production (technique de routine) l’extraction par microdissection,
et nos échantillons de tumeurs sont très variables dans la cellularité tumorale.
Il est possible que cela engendre des faux négatifs liés au fait que nous avons tenu compte des
résultats obtenus sur des échantillons tumoraux contenant parfois un faible pourcentage de
cellules tumorales.
Certaines équipes se fixent un seuil minimum de cellularité tumorale beaucoup plus élevé, de
l’ordre de 70% pour Blokx et al. voire même 80% pour Beadling et al. (107,131). Ces difficultés
liées à la cellularité tumorale n’interviennent peu ou pas dans les GIST qui sont des tumeurs
extrêmement riches et homogènes en cellules tumorales contrairement au mélanome.
Nos cinq mutations ont effectivement été retrouvées au sein de mélanome à forte densité de
cellules tumorales (50, 80, 80, 90 et 95% respectivement).
Par ailleurs, nous n’avons pas recherché de mutation de l’exon 18 (car non recherchée
habituellement dans les GIST ni dans les pathologies hématologiques). Cependant, ces mutations
sont rarement retrouvées dans les mélanomes, ne représentant que 3,3% des cas dans la littérature
(99,103,132). Ceci n’explique donc probablement pas la différence d’incidence des mutations
entre nos cas et ceux rapportés dans la littérature.
Une autre hypothèse que l’on peut évoquer est celle d’une différence de fréquence des mutations
de KIT en fonction de l’origine géographique des patients. Les séries de patients étudiées
proviennent des Etats-Unis d’Amérique, du Japon, de la Chine, de l’Australie et de l’Allemagne.
Il s’agit de la première série française.

64
4.2.2 Les mélanomes choroïdiens
Nous n’avons pas étudié les mélanomes choroïdiens pour plusieurs raisons.
Notre service n’est pas un centre de référence pour ce sous-type de mélanome compte-tenu de
l’absence de service d’ophtalmologie sur l’Institut Gustave Roussy, donc peu de patients avec ce
type de mélanome sont suivis dans notre service.
Par ailleurs, même si les mélanomes choroïdiens surexpriment dans plus de 50% des cas le
récepteur KIT, aucune mutation n’a été mise en évidence à ce jour (107,133,134). Au plan
ophtalmique, seules deux mutations de KIT ont été décrites : K558N et V559G de l’exon 11.
Dans les deux cas, il s’agissait de mélanomes conjonctivaux et non choroïdiens (107,108).
4.2.3 Correspondance entre immunohistochimie et mutation de KIT
Nous n’avons pas analysé l’expression de KIT en immunohistochimie, car il n’y a pas de
correspondance entre l’existence d’une mutation de KIT et l’hyperexpression de la protéine.
Effectivement, des mutations de KIT ont déjà été retrouvées sur des tumeurs n’exprimant pas ou
peu le récepteur KIT (103,122).
Cependant, une étude de Torres-Cabala et al. a montré que l’expression de KIT dans moins de
10% des cellules de la composante invasive des mélanomes acraux et muqueux semblait être un
facteur prédictif négatif pour les mutations de KIT (108). Inversement, Satzger et al. ont observé
une expression significativement plus élevée de KIT dans les mélanomes muqueux arborant une
mutation de KIT comparativement aux tumeurs non mutées (99).
Par ailleurs, il semble que la simple surexpression de la protéine, telle qu’on peut la mettre en
évidence en immunohistochimie ne semble pas être un facteur prédictif puissant de l’efficacité du
traitement par inhibiteurs de KIT et les cas de patients répondeurs sont le plus souvent ceux dont
les mélanomes sont mutés sur KIT (cf. 4.2.4.2) (135,134).

65
4.2.4 Implications thérapeutiques
Sur nos cinq patients mutés, trois patients ont été traités par inhibiteurs de KIT, les deux autres
étant en rémission après chirurgie, aucun traitement complémentaire ne leur a été proposé (cf.
3.2).
4.2.4.1 Les essais cliniques décevants
L'importance de la sélection des patients a été confirmée par l'expérience d’essais cliniques
négatifs avec l'imatinib chez des patients atteints de mélanome non sélectionnés sur leurs
anomalies moléculaires.
Trois essais de phase II, évaluant l'imatinib dans des mélanomes métastatiques, ont été publiés et
se sont avérés très décevants.
Dans le premier, sur 16 patients évaluables, un patient a eu une stabilité pendant 2,6 mois tandis
que tous les autres patients ont progressé après 2 à 8 semaines de traitement. Sur les 16 patients,
il y avait un mélanome acral et un mélanome muqueux, le statut KIT et PDGFRA n’a pas été
étudié (136,137).
Dans le deuxième essai, 26 patients ont été inclus dont 23 mélanomes cutanés, deux mélanomes
oculaires et un mélanome muqueux. Dix-huit patients étaient évaluables, et l’expression de KIT
et PDGFRA et B était étudiée chez 15 patients. Seules deux stabilités ont été documentées avec
une durée de 3 et 3,5 mois (les caractéristiques des patients n’ont pas été détaillées) (138).
Dans le troisième essai, sur les 21 patients traités, quatre ont eu une stabilité ne dépassant pas 12
semaines, et un patient a répondu partiellement pendant 12,8 mois et progressa ensuite sur ses
métastases ganglionnaires et cérébrales. Chez les cinq patients, répondeurs ou stables, aucune
mutation de KIT n’a été mise en évidence, le statut PDGFRA n’a pas été étudié. Deux patients
avaient une expression de KIT dans plus de 75% des cellules d’intensité modérée, un patient
avait une expression de KIT dans plus de 75% des cellules d’intensité forte (la réponse partielle),
un patient avait une expression de KIT entre 26 et 75% des cellules d’intensité faible et un patient
n’exprimait pas KIT. Il est important de noter que le patient répondeur avait un mélanome de
type acro-lentigineux (139).

66
Un essai clinique de phase II présenté à l’ASCO 2009, évaluant le dasatinib en première ligne de
traitement chez des patients atteints de mélanome métastatique non sélectionné sur leur anomalie
moléculaire a retrouvé une modeste activité. Sur les 30 patients, il y avait cinq ALM, quatre
mélanomes oculaires et trois mélanomes muqueux. Trois patients ont eu une réponse partielle
confirmée de 16, 6 et 5 mois et un patient une réponse partielle non confirmée avec progression
après 4 mois. Un des patients répondeurs avait une mutation de l’exon 13, un patient avec une
délétion de l’exon 11 n’a pas répondu (140).
4.2.4.2 Des cas cliniques encourageants
Au moins une quinzaine de cas cliniques ont été publiés, sur des patients atteints de mélanomes
métastatiques, répondant aux ITK.
Les cas cliniques des patients avec une mutation de l’exon 11 en L576P sous imatinib ou
dasatinib ont déjà été résumés dans le tableau 7.
D’autres patients atteints de mélanome muté sur d’autres exons ont était traités avec succès par
des ITK telles que l’imatinib, le sunitinib et le sorafenib. Ils sont résumés dans le tableau 9.
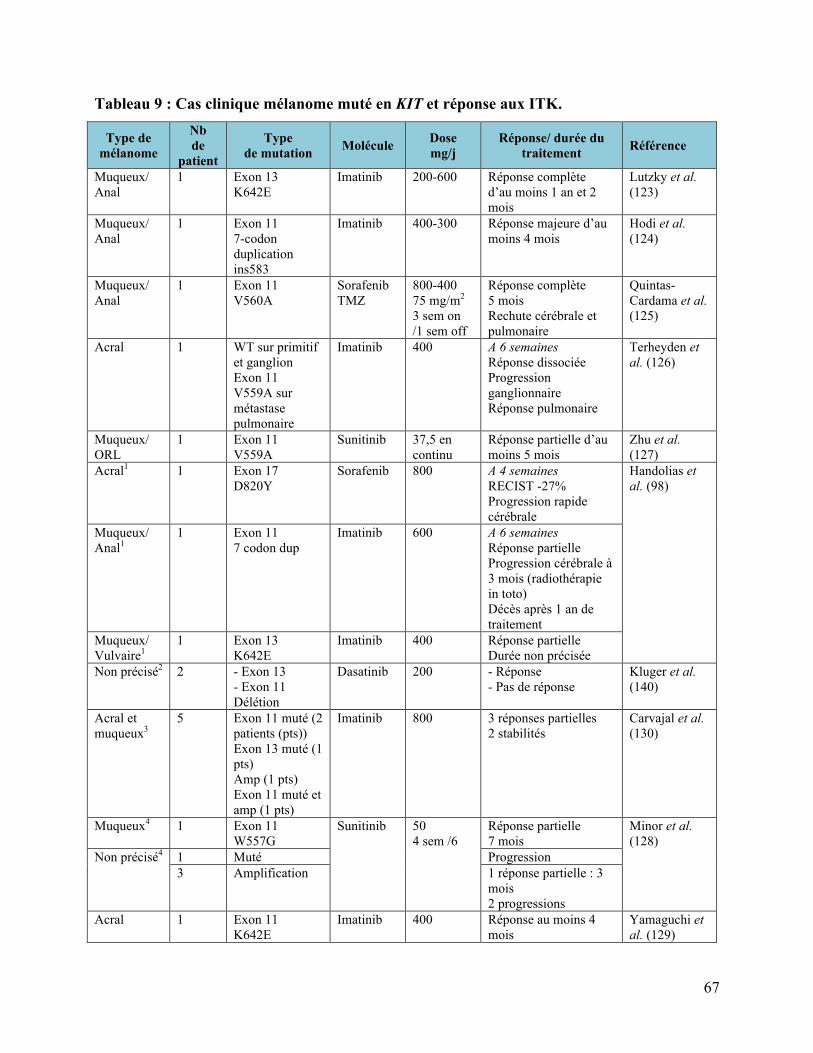
67
Tableau 9 : Cas clinique mélanome muté en KIT et réponse aux ITK.
Type de mélanome
Nb de
patient
Type de mutation Molécule Dose
mg/j Réponse/ durée du
traitement Référence
Muqueux/ Anal
1 Exon 13 K642E
Imatinib 200-600 Réponse complète d’au moins 1 an et 2 mois
Lutzky et al. (123)
Muqueux/ Anal
1 Exon 11 7-codon duplication ins583
Imatinib
400-300 Réponse majeure d’au moins 4 mois
Hodi et al. (124)
Muqueux/ Anal
1 Exon 11 V560A
Sorafenib TMZ
800-400 75 mg/m2
3 sem on /1 sem off
Réponse complète 5 mois Rechute cérébrale et pulmonaire
Quintas-Cardama et al. (125)
Acral 1 WT sur primitif et ganglion Exon 11 V559A sur métastase pulmonaire
Imatinib 400 A 6 semaines Réponse dissociée Progression ganglionnaire Réponse pulmonaire
Terheyden et al. (126)
Muqueux/ ORL
1 Exon 11 V559A
Sunitinib 37,5 en continu
Réponse partielle d’au moins 5 mois
Zhu et al. (127)
Acral1 1 Exon 17 D820Y
Sorafenib 800 A 4 semaines RECIST -27% Progression rapide cérébrale
Muqueux/ Anal1
1 Exon 11 7 codon dup
Imatinib 600 A 6 semaines Réponse partielle Progression cérébrale à 3 mois (radiothérapie in toto) Décès après 1 an de traitement
Muqueux/ Vulvaire1
1 Exon 13 K642E
Imatinib 400 Réponse partielle Durée non précisée
Handolias et al. (98)
Non précisé2 2 - Exon 13 - Exon 11 Délétion
Dasatinib 200 - Réponse - Pas de réponse
Kluger et al. (140)
Acral et muqueux3
5 Exon 11 muté (2 patients (pts)) Exon 13 muté (1 pts) Amp (1 pts) Exon 11 muté et amp (1 pts)
Imatinib 800 3 réponses partielles 2 stabilités
Carvajal et al. (130)
Muqueux4 1 Exon 11 W557G
Réponse partielle 7 mois
1 Muté Progression Non précisé4
3 Amplification
Sunitinib 50 4 sem /6
1 réponse partielle : 3 mois 2 progressions
Minor et al. (128)
Acral 1 Exon 11 K642E
Imatinib 400 Réponse au moins 4 mois
Yamaguchi et al. (129)

68
Légendes tableau n°9 1 : Inclus dans l’étude NCT00171912 : « Imatinib Mesylate in Patients With Various Types of Malignancies Involving Activated Tyrosine Kinase Enzymes » 2 : Inclus dans l’étude NCT00436605 : « A Phase 2 Study of Dasatinib in Advanced Melanoma » 3 : Inclus dans l’étude NCT00470470 : « A Phase II Study of Imatinib Mesylate in Patients With Inoperable AJCC Stage III or IV « Melanoma Harboring Somatic Alterations of C-KIT » 4 : Inclus dans l’étude NCT00631618 : « A Phase II Trial of Sutent in Metastatic Melanoma Patients With KIT Aberrations »
Le cas de Lutzky et al. est très intéressant car il démontre un effet dose-dépendant, avec une
rechute à la décroissance de l’imatinib à 200 mg et de nouveau une réponse complète à
l’augmentation à 600 mg. Cela nous interroge sur la dose initiale à recommander d’imatinib (400
mg par jour ou 800 mg par jour). La recherche d’une mutation de KIT a été réalisée sur cinq
échantillons tumoraux différents (lésion primitive, métastases cutanées, adénopathie) et était
présente sur chaque prélèvement (123).
Le cas clinique de Terheyden et al. est remarquable sur la corrélation de la réponse en présence
ou non d’une mutation de KIT sur les différentes localisations métastatiques (pulmonaire et
ganglionnaire), qui fait craindre le risque de perte de mutation ou d’apparition de mutations
secondaire entraînant une résistance secondaire au traitement, comme dans les GIST et dans la
présentation de Guo et al. à l’ASCO 2010 (cf. infra) (114,126).
Dans certains cas publiés et notre première observation, on peut remarquer que la réponse
clinique est très précoce sur les symptômes, et l’état général des patients. En effet, en trois jours
les saignements anaux se sont arrêtés pour la patiente de Hodi et al., en quelques jours la patiente
de Handolias et al était sevrée en oxygène et ambulatoire, en deux semaines l’état général et la
sensation de corps étranger anal se sont est améliorés pour la patiente de Satzger et al., en moins
de quatre semaines, les nodules de perméation ont totalement disparu pour la patiente de Lutzky
et al., en un mois l’état général et les douleurs abdominales se sont améliorées pour la patiente de
Woodman et al.. Cette amélioration clinique était associée à une réponse biologique
(décroissance des LDH) et métabolique (97,98,123,124).
Ce type de réponse franche et rapide ressemble aux réponses observées chez les patients mutés
BRAFV600E répondeurs aux PLX4032 (141).

69
Il est important de noter que quatre patients répondeurs ont progressé sur leurs métastases
cérébrales, entraînant le décès de ces derniers (98,125). Ceci pourrait être expliqué par la
pénétration limitée de la barrière hémato-encéphalique par les petites molécules ITK, comme déjà
documenté dans d’autres pathologies telles que les leucémies aigues Ph+ ainsi que dans les GIST
où des rechutes et/ou des progressions dans le système nerveux ont été rapportées (142,143).
Cependant, les ITK de seconde génération tel que le dasatinib semblerait plus efficace dans ce
contexte (144,145). Une étude de phase II est en cours, évaluant l’efficacité du sunitinib dans les
métastases cérébrales de mélanome métastatique et de cancer du rein (NCT00462982) (49). Les
études à grande échelle nous permettront de mieux statuer sur ce risque qui pourrait être une
véritable limite, car les métastases cérébrales sont fréquentes dans le mélanome.
4.2.4.3 Les résultats préliminaires des études précoces
Les résultats préliminaires de certaines études de phase II, évaluant l’imatinib et le sunitinib sur
les patients sélectionnés sur leur anomalie moléculaire ont été présentés à l’ASCO 2009 et 2010
(114,128,130).
Dans l’étude de Carvajal et al. évaluant l’imatinib dans les mélanomes stade III inopérables ou
stade IV, cinq patients arborant une anomalie moléculaire (mutation ou amplification) étaient
évaluables. Trois réponses partielles et deux stabilités ont permis l’expansion de la cohorte,
l’étude est toujours en cours de recrutement (NCT00470470 : « A Phase II Study of Imatinib
Mesylate in Patients With Inoperable AJCC Stage III or IV Melanoma Harboring Somatic
Alterations of C-KIT ») (49,130).
Dans l’étude de Guo et al. sur 26 patients évaluables, six patients ont eu une réponse partielle et
neuf une stabilité. Les patients ne répondant pas à 400 mg, n’ont pas répondu à l’augmentation de
l’imatinib à 600 mg et n’ont pas toléré l’imatinib à 800 mg.
Sur cinq patients qui ont progressé après réponse partielle, des prélèvements tumoraux ont été
réalisés sur les nouvelles lésions ou les métastases évolutives. Chez un patient, avec de nouvelles
lésions hépatiques, une mutation de BRAF (V600E) et de PDGFRA (W559stop) ont été isolées,
(alors qu’elles n’existaient pas sur la lésion biopsiée initialement), associées à une disparition de
la mutation de KIT (114).

70
L’apparition de mutations secondaires responsable d’une résistance secondaire au traitement est
l’un des facteurs principaux d’échappement à l’imatinib dans le GIST. Dans la phase II B2222,
l’apparition d’une mutation secondaire de PDGFRA ne fut retrouvée que chez un patient déjà
muté PDGFRA en baseline (65).
Dans l’étude de Minor et al., le sunitinib a été prescrit à 50 mg/jour quatre semaines sur six. Sur
sept patients traités et évaluables, il y a eu trois réponses partielles : sept mois pour un mélanome
muté W557G, trois mois pour un mélanome avec amplification de KIT et une réponse chez un
mélanome non muté non amplifié mais avec une surexpression de KIT en immunohistochimie
(128).
4.2.4.4 Les études en cours (49)
De multiples études de phase I, II sont en cours dans le mélanome pour évaluer la place de ces
ITK.
On dénombre au moins trois essais de phase II avec l’imatinib dans les mélanomes avec anomalie
moléculaire de KIT ainsi que des études de phase I évaluant l’association de l’imatinib avec le
temozolomide, le bevacizumab et le cyclophosphamide à dose métronomique. Une étude en
association avec l’everolimus a été interrompue pour toxicité.
Le sunitinib fait aussi l’objet d’essai de phase II dans le mélanome avec ou sans aberration
moléculaire de KIT, dont une évaluant l’efficacité sur les métastases cérébrales. Un essai de
phase I/II associant DTIC et sunitinib a été interrompu précocement pour toxicité. Trois essais de
phase I/II évaluent la tolérance de l’association temozolomide-sunitinib, une a été arrêtée
précocement.
Il existe aussi des essais avec le dasatinib dans le mélanome avec ou sans aberration moléculaire
de KIT dont une phase I/II en association avec le DTIC.
Trois essais de phase II évaluent le nilotinib chez les mélanomes métastatiques ou inopérables
avec anomalie de KIT dont une se déroule en France depuis septembre 2010 (Hôpital Saint-
Louis). Un essai de phase III multicentrique international en ouvert comparant le nilotinib et le
DTIC dans le mélanome inopérable ou métastatique, avec anomalie de KIT de l’exon 11, 13 ou
Y822D et Y823D de l’exon 17 est en cours depuis avril 2010.

71
5. CONCLUSION
L’existence de mutations de KIT dans certains sous-types de mélanomes fait de ce récepteur une
cible thérapeutique majeure du fait de la disponibilité de plusieurs puissants inhibiteurs de cette
molécule en clinique.
Nous avons recherché des mutations de KIT et de PDGFRA dans une sous-population de
mélanomes acro-lentigineux, muqueux, de Dubreuilh et sans primitif retrouvé provenant d’une
cohorte de 107 patients. Nous avons mis en évidence quatre mutations de KIT (4,8%) sur les 84
résultats contributifs et deux mutation du même exon de PDGFRA (1,3%) sur les 80 contributifs :
trois mutations sur des mélanomes acraux (KITL576P, KITY553C et PDGFRAP567S/G569R) et deux
mutations sur des mélanomes vaginaux (KITL576P et KITD820V).
Ce pourcentage de mutation de KIT est inférieur à ce qui a été antérieurement rapporté dans ces
sous-groupes de mélanomes, peut-être du fait d’une faible proportion de cellules tumorales dans
certains de nos échantillons. Cette série est singulière car nous avons découvert trois nouvelles
mutations dans le mélanome KITY553C, KITD820V et PDGFRAP567S/G569R. Actuellement, il n’existe
pas de données in vitro ou in vivo sur le retentissement fonctionnel de ces mutations dans les
cellules, ni sur leur sensibilité aux inhibiteurs de tyrosine kinase.
Sur trois patients traités par ITK, nous avons eu une réponse partielle pendant cinq mois sous
dasatinib, une stabilité de deux mois sous imatinib et un patient progressif rapidement sur
métastases cérébrales préexistantes sous imatinib.
Des explorations complémentaires en immunohistochimie et en biologie moléculaire sont en
cours afin de caractériser plus précisément ces sous-types particuliers de mélanomes.
Aujourd’hui, même si nous disposons encore de peu de données sur l’efficacité des inhibiteurs de
KIT dans le mélanome, plusieurs cas cliniques anecdotiques et résultats préliminaires d’essais de
phases précoces sont très encourageants, faisant espérer que le succès thérapeutique de ces
thérapies dans les GIST puisse se transposer dans les mélanomes avec anomalie moléculaire de
KIT ou PDGFRA. Ces traitements sont actuellement évalués dans le cadre d’essais de phase II et
III, nous avons tous l’espoir de pouvoir confirmer bientôt notre entrée dans l’ère du traitement du
mélanome guidé par la biologie.

72
ANNEXES
Annexe 1 a : Version finale 2009 AJCC Melanoma Staging and Classification (146)
73
Annexe 1b : Version finale 2009 AJCC Melanoma Staging and Classification (146)

74
Annexe 2 : Code génétique
Le code génétique définit la correspondance entre la séquence nucléotidique de l'ARN messager
(et donc celle de l'ADN) et la séquence en acides aminés de la protéine. La séquence des acides
nucléiques est une combinaison de 4 lettres (A, C, G, T dans l'ADN ; A, C, G, U dans l'ARN), la
combinaison de ces 4 bases va permettre de "coder" pour les 20 acides aminés.
La rangée indique la première lettre du codon, la colonne indique la seconde et la dernière
colonne indique la dernière lettre du codon.
U C A G UUU UCU UAU UGU U UUC
phénylalanine UCC UAC
tyrosine UGC
cystéine C
UUA UCA UAA UGA stop/sélénocystéine A U
UUG UCG
sérine
UAG stop
UGG tryptophane G CUU CCU CAU CGU U CUC CCC CAC
histidine CGC C
CUA CCA CAA CGA A C
CUG
leucine
CCG
proline
CAG glutamine
CGG
arginine
G AUU ACU AAU AGU U AUC ACC AAC
asparagine AGC
sérine C
AUA isoleucine
ACA AAA AGA A A
AUG méthionine/start ACG
thréonine
AAG lysine
AGG arginine
G GUU GCU GAU GGU U GUC GCC GAC
acide aspartique GGC C
GUA GCA GAA GGA A G
GUG
valine
GCG
alanine
GAG acide glutamique GGG
glycine
G

75
Annexe 3 : Liste des 20 acides aminés représentés dans le code génétique :
Nom Code à 3 lettres Code à une lettre Alanine Ala A
Arginine Arg R
asparagine Asn N
aspartate ou acide aspartique Asp D
cystéine Cys C
glutamate ou acide glutamique Glu E
glutamine Gln Q
glycine Gly G
histidine His H
isoleucine Ile I
leucine Leu L
lysine Lys K
méthionine Met M
phénylalanine Phe F
proline Pro P
sérine Ser S
thréonine Thr T
tryptophane Trp W
tyrosine Tyr Y
valine Val V

76
Annexe 4 : Polymorphisme de KIT et de PDGFRA
Polymorphisme de PDGFRA :
- Exon 18 : c.2472 C>T ; p.Val824Val ; rs2228230
- Exon 12 : c.1731 G>R ; p.Pro577Pro ; rs55830582
- Exon 14 : c.1896G>A ; p.Trp588Stop
- Intron 11 : c.1654-24G>S
- Intron 13 : c. 1892-45G
- Intron 16 : c. 2362-77G>R; rs 4864921 et c. 2362-107T>W ; rs 11935331
Polymorphisme de KIT :
- Exon 17 : c.2394C>T ; p.Ile798Ile ; rs55789615

77
ABREVIATIONS
AFIRMM : Association Française pour les Initiatives de Recherche sur le Mastocyte et
les Mastocytoses
AJCC : American Joint Committee on Cancer
ALM : Mélanome acral lentigineux ou Acral Lentiginous Melanoma
ASCO : American Society of Clinical Oncology
COSMIC : Catalogue Of Somatic Mutations In Cancer
CSD : chronic sun-induced damage ou mélanome sur peau chroniquement exposée au soleil
DFSP : Dermatofibrosarcome de Darrier et Ferrand ou dermatofibrosarcoma protuberans
DTIC : Dacarbazine, Déticène®
EORTC : European Organisation for Research and Treatment of Cancer
GIST : Tumeur stromale gastro-intestinale
IHC : Immunohistochimie
ITK : Inhibiteur de tyrosine kinase
LMC : Leucémie myéloïde chronique
LMM : Mélanome de Dubreuilh ou Lentigo Maligna Melanoma
MAP-kinase : Mitogen-activated protein kinase
N-CSD : Without chronic sun-induced damage ou mélanome sur peau non exposée
chroniquement au soleil
NM : Mélanome nodulaire ou Nodular Melanoma
ORL : Oto-rhyno-laryngé
PDGF : Platelet Derived Growth Factor
PI3 kinase : Phosphatidylinositol 3 kinase
PFS : Survie sans progression ou progression free survival
RECIST : Response Evaluation Criteria In Solid Tumors
RTK : Récepteur tyrosine kinase
SCF : Stem Cell Factor
SSM : Mélanome superficiel extensif ou Superficial Spreading Melanoma
TK : Domaine tyrosine kinase
TMZ : Temozolomide, Temodal®

78
BIBLIOGRAPHIE
1. Marks R. Epidemiology of melanoma. Clinical dermatology. Review article. Clin Exp Dermatol. 2000 : 25 : 459-463. 2. Grange F. Epidémiologie du mélanome cutanée : données descriptives en France et en Europe. Ann Dermatol Venereol. 2005 : 132 : 975-982. 3. Lipsker D, Engel F, Cribier B, Velten M, Hedelin G. Trends in melanoma epidemiology suggest three different types of melanoma. Br J Dermatol. 2007 : 157 : 338-343. 4. Holman CD, Armstrong BK, Heenan PJ. Relationship of cutaneous malignant melanoma to individual sunlight-exposure habits. J Natl Cancer Inst. 1986 : 76 : 403-414. 5. Elwood JM. Melanoma and sun exposure. Semin Oncol. 1996 : 23 : 650-666. 6. Elwood JM, Gallagher RP. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. Int J Cancer. 1998 : 78 : 276-280. 7. Lee ML, Tomsu K, Von Eschen KB. Duration of survival for disseminated malignant melanoma: results of a meta-analysis. Melanoma Res. 2000 : 10 : 81-92. 8. Hersey P, Bastholt L, Chiarion-Sileni V, Cinat G, Dummer R, Eggermont AMM, et al. Small molecules and targeted therapies in distant metastatic disease. Ann Oncol. 2009 : 20 : 35-40. 9. Chapman PB, Einhorn LH, Meyers ML, Saxman S, Destro AN, Panageas KS, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999 : 17 : 2745-2751. 10. Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004 : 351 : 998-1012. 11. Clark WH, From L, Bernardino EA, Mihm MC. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969 : 29 : 705-727. 12. McGovern VJ, Mihm MC, Bailly C, Booth JC, Clark WH, Cochran AJ, et al. The classification of malignant melanoma and its histologic reporting. Cancer. 1973 : 32 : 1446-1457. 13. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005 : 353 : 2135-2147. 14. Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003 : 95 : 1878-1890.

79
15. Reed R. Acral lentiginous melanoma. Dans : New concepts in surgical pathology of the skin. New York, Wiley : 1976 : p.89-90. 16. Patterson RH, Helwig EB. Subungual malignant melanoma: a clinical-pathologic study. Cancer. 1980 : 46 : 2074-2087. 17. Phan A, Touzet S, Dalle S, Ronger-Savle S, Balme B, Thomas L. Acral lentiginous melanoma: a clinicoprognostic study of 126 cases. Br J Dermatol. 2006 : 155 : 561-569. 18. Kuchelmeister C, Schaumburg-Lever G, Garbe C. Acral cutaneous melanoma in caucasians: clinical features, histopathology and prognosis in 112 patients. Br J Dermatol. 2000 : 143 : 275-280. 19. Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1998 : 83 : 1664-1678. 20. McLaughlin CC, Wu X, Jemal A, Martin HJ, Roche LM, Chen VW. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005 : 103 : 1000-1007. 21. Patrick RJ, Fenske NA, Messina JL. Primary mucosal melanoma. J Am Acad Dermatol. 2007 : 56 : 828-834. 22. Ballantyne AJ. Malignant melanoma of the skin of the head and neck. An analysis of 405 cases. Am J Surg. 1970 : 120 : 425-431. 23. Wagner M, Morris CG, Werning JW, Mendenhall WM. Mucosal melanoma of the head and neck. Am. J Clin Oncol. 2008 : 31: 43-48. 24. McLean N, Tighiouart M, Muller S. Primary mucosal melanoma of the head and neck. Comparison of clinical presentation and histopathologic features of oral and sinonasal melanoma. Oral Oncol. 2008 : 44 : 1039-1046. 25. Takagi M, Ishikawa G, Mori W. Primary malignant melanoma of the oral cavity in Japan. With special reference to mucosal melanosis. Cancer. 1974 : 34 : 358-370. 26. Broomhall C. Malignant melanoma of the oral cavity in Ugandan Africans. Br J Surg. 1967 : 54 : 581-584. 27. Schuchter LM, Green R, Fraker D. Primary and metastatic diseases in malignant melanoma of the gastrointestinal tract. Curr Opin Oncol. 2000 : 12 : 181-185. 28. Clemmensen OJ, Fenger C. Melanocytes in the anal canal epithelium. Histopathology. 1991 : 18 : 237-241.

80
29. Klas JV, Rothenberger DA, Wong WD, Madoff RD. Malignant tumors of the anal canal: the spectrum of disease, treatment, and outcomes. Cancer. 1999 : 85 : 1686-1693. 30. Zhang S, Gao F, Wan D. Abdominoperineal resection or local excision? a survival analysis of anorectal malignant melanoma with surgical management. Melanoma Res. 2010 : 20 : 338-341. 31. Garfinkle JM, Cahan WG. Primary melanocarcinoma of the esophagus; fist histologically proved case. Cancer. 1952 : 5 : 921-926. 32. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009 : 10 : 516-521. 33. Piura B. Management of primary melanoma of the female urogenital tract. Lancet Oncol. 2008 : 9 : 973-981. 34. Wayte DM, Helwig EB. Melanotic freckle of Hutchinson. Cancer. 1968 : 21 : 893-911. 35. Ollivaud L, Basset-Seguin N, Archimbaud A. Mélanome de Dubreuilh. Ann Dermatol Venereol. 2001 : 128 : 172-176. 36. Dasgupta T, Bowden L, Berg JW. Malignant melanoma of unknown primary origin. Surg Gynecol Obstet. 1963 : 117 : 341-345. 37. Savoia P, Fava P, Osella-Abate S, Nardò T, Comessatti A, Quaglino P, et al. Melanoma of unknown primary site: a 33-year experience at the Turin Melanoma Centre. Melanoma Res. 2010 : 20 : 227-232. 38. Kamposioras K, Pentheroudakis G, Pectasides D, Pavlidis N. Malignant melanoma of unknown primary site. To make the long story short. A systematic review of the literature. Critical Reviews in Oncology/Hematology. doi:10.1016/j.critrevonc.2010.04.007 39. Blay J, Le Cesne A, Alberti L, Ray-Coquart I. Targeted cancer therapies. Bull Cancer. 2005 : 92 : 13-18. 40. Bourgin, Céline. Caractérisation des évènements de signalisation induits par le M-CSF au cours de la différenciation monocytaire/macrophagique. 171f. Thèse d'exercice : Médecine : Lyon : 2003. 41. Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008 : 53 : 245-266. 42. Longley BJ, Reguera MJ, Ma Y. Classes of c-KIT activating mutations: proposed mechanisms of action and implications for disease classification and therapy. Leuk Res. 2001 : 25 : 571-576.

81
43. Heinrich MC, Rubin BP, Longley BJ, Fletcher JA. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol. 2002 : 33 : 484-495. 44. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003 : 21 : 4342-4349. 45. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen C, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003 : 299 : 708-710. 46. Heinrich MC. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Human Pathology. 2002 : 33 : 484-495. 47. VIDAL le dictionnaire. 2010. 48. Demetri GD, Lo Russo P, MacPherson IR, Wang D, Morgan JA, Brunton VG, et al. Phase I Dose-Escalation and Pharmacokinetic Study of Dasatinib in Patients with Advanced Solid Tumors. Clin Cancer Res. 2009 : 15 : 6232-6240. 49. ClinicalTrials.gov [Internet]. Available from: http://clinicaltrials.gov 50. Le Cesne A, Blay J, Bui BN, Bouché O, Adenis A, Domont J, et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur J Cancer. 2010 : 46 : 1344-1351. 51. Caenepeel S, Renshaw-Gegg L, Baher A, Bush TL, Baron W, Juan T, et al. Motesanib inhibits Kit mutations associated with gastrointestinal stromal tumors. J Exp Clin Cancer Res. 2010 : 29 : 96. 52. Joensuu H, De Braud F, Coco P, De Pas T, Putzu C, Spreafico C, et al. Phase II, open-label study of PTK787/ZK222584 for the treatment of metastatic gastrointestinal stromal tumors resistant to imatinib mesylate. Ann Oncol. 2008 : 19 : 173-177. 53. Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000 : 60 : 2178-2189. 54. Millward MJ, House C, Bowtell D, Webster L, Olver IN, Gore M, et al. The multikinase inhibitor midostaurin (PKC412A) lacks activity in metastatic melanoma: a phase IIA clinical and biologic study. Br J Cancer. 2006 : 95 : 829-834. 55. Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007 : 369 : 1731-1741.

82
56. Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008 : 26 : 626-632. 57. Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004 : 364 : 1127-1134. 58. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001 : 344 : 1052-1056. 59. Van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001 : 358 : 1421-1423. 60. Verweij J, Van Oosterom AT, Blay J, Judson I, Rodenhuis S, van der Graaf W, et al. Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003 : 39 : 2006-2011. 61. Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008 : 26 : 620-625. 62. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, Van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006 : 42 : 1093-1103. 63. Blay J, Adenis A, Ray-Coquard I, Cassier PA, Le Cesne A. Is there a role for discontinuing imatinib in patients with advanced gastrointestinal stromal tumour? Curr Opin Oncol. 2009 : 21 : 360-366. 64. Blay J, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol. 2007 : 25 : 1107-1113. 65. Gramza AW, Corless CL, Heinrich MC. Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin Cancer Res. 2009 : 15 : 7510-7518. 66. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular Correlates of Imatinib Resistance in Gastrointestinal Stromal Tumors. J Clin Oncol. 2006 : 24 : 4764-4774.

83
67. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired Resistance to Imatinib in Gastrointestinal Stromal Tumor Occurs Through Secondary Gene Mutation. Clin Cancer Res. 2005 : 11 : 4182-4190. 68. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008 : 216 : 64-74. 69. Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005 : 128 : 270-279. 70. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J Clin Oncol. 2008 : 26 : 5352-5359. 71. Faivre S. Safety, Pharmacokinetic, and Antitumor Activity of SU11248, a Novel Oral Multitarget Tyrosine Kinase Inhibitor, in Patients With Cancer. J Clin Oncol. 2006 : 24 : 25-35. 72. Demetri GD, Vanoosterom A, Garrett C, Blackstein M, Shah M, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006 : 368 : 1329-1338. 73. George S, Blay J, Casali P, Le Cesne A, Stephenson P, DePrimo S, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009 : 45 : 1959-1968. 74. Montemurro M, Schöffski P, Reichardt P, Gelderblom H, Schütte J, Hartmann J, et al. Nilotinib in the treatment of advanced gastrointestinal stromal tumours resistant to both imatinib and sunitinib. Eur J Cancer. 2009 : 45 : 2293-2297. 75. Wiebe L, Kasza KE, Maki RG, D'Adamo DR, Chow WA, Wade III JL, et al. Activity of sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): A phase II trial of the University of Chicago Phase II Consortium. J Clin Oncol (Meeting Abstracts). 2008 : 26 : 10502. 76. Reichardt P, Montemurro M, Gelderblom H, Blay J, Rutkowski P, Bui B, et al. Sorafenib fourth-line treatment in imatinib-, sunitinib-, and nilotinib-resistant metastatic GIST: A retrospective analysis. J Clin Oncol (Meeting Abstracts). 2009 : 27 : 10564. 77. Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, et al. KIT mutations are common in testicular seminomas. Am J Pathol. 2004 : 164 : 305-313. 78. Tian Q, Frierson HF, Krystal GW, Moskaluk CA. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999 : 154 : 1643-1647.

84
79. Einhorn LH, Brames MJ, Heinrich MC, Corless CL, Madani A. Phase II study of imatinib mesylate in chemotherapy refractory germ cell tumors expressing KIT. Am J Clin Oncol. 2006 : 29 : 12-13. 80. Przygodzki RM, Hubbs AE, Zhao F, O'Leary TJ. Primary mediastinal seminomas: evidence of single and multiple KIT mutations. Lab Invest. 2002 : 82 : 1369-1375. 81. Sakuma Y, Sakurai S, Oguni S, Hironaka M, Salto K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003 : 94 : 486-491. 82. Sakuma Y, Sakurai S, Oguni S, Satoh M, Hironaka M, Saito K. c-kit gene mutations in intracranial germinomas. Cancer Sci. 2004 : 95 : 716-720. 83. Nakai Y, Nonomura N, Oka D, Shiba M, Arai Y, Nakayama M, et al. KIT (c-kit oncogene product) pathway is constitutively activated in human testicular germ cell tumors. Biochem Biophys Res Commun. 2005 : 337 : 289-296. 84. Biermann K, Göke F, Nettersheim D, Eckert D, Zhou H, Kahl P, et al. c-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J Pathol. 2007 : 213 : 311-318. 85. Willmore-Payne C, Holden JA, Chadwick BE, Layfield LJ. Detection of c-kit exons 11- and 17-activating mutations in testicular seminomas by high-resolution melting amplicon analysis. Mod Pathol. 2006 : 19 : 1164-1169. 86. Georgin-Lavialle S, Barete S, Suarez F, Lepelletier Y, Bodemer C, Dubreuil P, et al. Actualités sur la compréhension et le traitement des mastocytoses systémiques. Rev Med Interne. 2009 : 30 : 25-34. 87. Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002 : 20 : 1692-1703. 88. Gari M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, et al. c-kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol. 1999 : 105 : 894-900. 89. Beghini A, Peterlongo P, Ripamonti CB, Larizza L, Cairoli R, Morra E, et al. C-kit mutations in core binding factor leukemias. Blood. 2000 : 95 : 726-727. 90. Algazi AP, Weber JS, Andrews S, Urbas P, Arimura E, Hwang J, et al. A phase I/II trial of DTIC and dasatinib in metastatic melanoma. J Clin Oncol. (Meeting Abstracts). 2010 : 28 : 8532. 91. Went PT, Dirnhofer S, Bundi M, Mirlacher M, Schraml P, Mangialaio S, et al. Prevalence of KIT expression in human tumors. J Clin Oncol. 2004 : 22 : 4514-4522.

85
92. Yoh K, Nishiwaki Y, Ishii G, Goto K, Kubota K, Ohmatsu H, et al. Mutational status of EGFR and KIT in thymoma and thymic carcinoma. Lung Cancer. 2008 : 62 : 316-320. 93. Conca E, Negri T, Gronchi A, Fumagalli E, Tamborini E, Pavan GM, et al. Activate and resist: L576P-KIT in GIST. Mol Cancer Ther. 2009 : 8 : 2491-2495. 94. Antonescu CR, Busam KJ, Francone TD, Wong GC, Guo T, Agaram NP, et al. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. Int J Cancer. 2007 : 121 : 257-264. 95. Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol. 2006 : 23 : 91-102. 96. Garrido MC, Bastian BC. KIT as a therapeutic target in melanoma. J Invest Dermatol. 2010 : 130 : 20-27. 97. Woodman SE, Trent JC, Stemke-Hale K, Lazar AJ, Pricl S, Pavan GM, et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009 : 8 : 2079-2085. 98. Handolias D, Hamilton AL, Salemi R, Tan A, Moodie K, Kerr L, et al. Clinical responses observed with imatinib or sorafenib in melanoma patients expressing mutations in KIT. Br J Cancer. 2010 : 102 : 1219-1223. 99. Satzger I, Schaefer T, Kuettler U, Broecker V, Voelker B, Ostertag H, et al. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br J Cancer. 2008 : 99 : 2065-2069. 100. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, Van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006 : 42 : 1093-103. 101. Sihto H, Franssila K, Tanner M, Vasama-Nolvi C, Sarlomo-Rikala M, Nupponen NN, et al. Platelet-derived growth factor receptor family mutations in gastrointestinal stromal tumours. Scand J Gastroenterol. 2006 : 41 : 805-811. 102. COSMIC : Catalogue Of Somatic Mutations In Cancer. [Internet]. Available from : http://www.sanger.ac.uk/genetics/CGP/cosmic/ 103. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006 : 24 : 4340-4346. 104. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004 : 22 : 3813-3825.

86
105. Lasota J, Corless CL, Heinrich MC, Debiec-Rychter M, Sciot R, Wardelmann E, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008 : 21 : 476-484. 106. Ashida A, Takata M, Murata H, Kido K, Saida T. Pathological activation of KIT in metastatic tumors of acral and mucosal melanomas. Int J Cancer. 2009 : 124 : 862-868. 107. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008 : 14 : 6821-6828. 108. Torres-Cabala CA, Wang W, Trent J, Yang D, Chen S, Galbincea J, et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol. 2009 : 22 : 1446-1456. 109. Handolias D, Salemi R, Murray W, Tan A, Liu W, Viros A, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res. 2010 : 23 : 210-215. 110. Guo T, Agaram NP, Wong GC, Hom G, D'Adamo D, Maki RG, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007 : 13 : 4874-4881. 111. Bisagni G, Rossi G, Cavazza A, Sartori G, Gardini G, Boni C. Long lasting response to the multikinase inhibitor bay 43-9006 (Sorafenib) in a heavily pretreated metastatic thymic carcinoma. J Thorac Oncol. 2009 : 4 : 773-775. 112. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005 : 23 : 5357-5364. 113. Hong DS, Sebti SM, Newman RA, Blaskovich MA, Ye L, Gagel RF, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res. 2009 : 15 : 7061-7068. 114. Guo J, Si L, Kong Y, Xu XW, Flaherty KT, Corless CL, et al. A phase II study of imatinib for advanced melanoma patients with KIT aberrations. J Clin Oncol (Meeting Abstracts). 2010 : 28 : 8527. 115. Curtin JA, Pinkel D, Bastian BC. Absence of PDGFRA mutations in primary melanoma. J Invest Dermatol. 2008 : 128 : 488-489. 116. Sihto H, Sarlomo-Rikala M, Tynninen O, Tanner M, Andersson LC, Franssila K, et al. KIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumors. J Clin Oncol. 2005 : 23 : 49-57.

87
117. Terada T. Amelanotic malignant melanoma of the esophagus: report of two cases with immunohistochemical and molecular genetic study of KIT and PDGFRA. World J Gastroenterol. 2009 : 15 : 2679-2683. 118. Terada T. Low incidence of KIT gene mutations and no PDGFRA gene mutations in primary cutaneous melanoma: an immunohistochemical and molecular genetic study of Japanese cases. Int J Clin Oncol. DOI: 10.1007/s10147-010-0087-0 119. Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005 : 36 : 486-493. 120. Wu JM, Alvarez H, García P, Rojas PL, Wong G, Maitra A, et al. Melanoma hyperpigmentation is strongly associated with KIT alterations. Am J Dermatopathol. 2009 : 31 : 619-625. 121. Monsel G, Ortonne N, Bagot M, Bensussan A, Dumaz N. c-Kit mutants require hypoxia-inducible factor 1alpha to transform melanocytes. Oncogene. 2010 : 29 : 227-236. 122. Rivera RS, Nagatsuka H, Gunduz M, Cengiz B, Gunduz E, Siar CH, et al. C-kit protein expression correlated with activating mutations in KIT gene in oral mucosal melanoma. Virchows Arch. 2008 : 452 : 27-32. 123. Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008 : 21 : 492-493. 124. Hodi FS, Friedlander P, Corless CL, Heinrich MC, Mac Rae S, Kruse A, et al. Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol. 2008 : 26 : 2046-2051. 125. Quintás-Cardama A, Lazar AJ, Woodman SE, Kim K, Ross M, Hwu P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008 : 5 : 737-740. 126. Terheyden P, Houben R, Pajouh P, Thorns C, Zillikens D, Becker JC. Response to imatinib mesylate depends on the presence of the V559A-mutated KIT oncogene. J Invest. Dermatol. 2010 : 130 : 314-316. 127. Zhu Y, Si L, Kong Y, Chi Z, Yuan X, Cui C, et al. Response to sunitinib in Chinese KIT-mutated metastatic mucosal melanoma. J Clin Oncol (Meeting Abstracts). 2009 : 27 : 20017. 128. Minor DR, O'Day S, Kashani-Sabet M, Daud A, Salman Z, Bastian B. Sunitinib therapy for metastatic melanomas with KIT aberrations. J Clin Oncol (Meeting Abstracts). 2010 Mai : 28 : 8545.

88
129. Yamaguchi M, Harada K, Ando N, Kawamura T, Shibagaki N, Shimada S. Marked response to imatinib mesylate in metastatic acral lentiginous melanoma on the thumb. Clin Exp Dermatol. DOI: 10.1111/j.1365-2230.2010.03885. 130. Carvajal RD, Chapman PB, Wolchok JD, Cane L, Teitcher JB, Lutzky J, et al. A phase II study of imatinib mesylate (IM) for patients with advanced melanoma harboring somatic alterations of KIT. J Clin Oncol (Meeting Abstracts). 2009 : 27 : 9001. 131. Blokx WAM, van Dijk MCRF, Ruiter DJ. Molecular cytogenetics of cutaneous melanocytic lesions - diagnostic, prognostic and therapeutic aspects. Histopathology. 2010 : 56 : 121-132. 132. Daniotti M, Ferrari A, Frigerio S, Casieri P, Miselli F, Zucca E, et al. Cutaneous melanoma in childhood and adolescence shows frequent loss of INK4A and gain of KIT. J Invest Dermatol. 2009 : 129 : 1759-1768. 133. Pache M, Glatz K, Bösch D, Dirnhofer S, Mirlacher M, Simon R, et al. Sequence analysis and high-throughput immunohistochemical profiling of KIT (CD 117) expression in uveal melanoma using tissue microarrays. Virchows Arch. 2003 : 443 : 741-744. 134. Hofmann UB, Kauczok-Vetter CS, Houben R, Becker JC. Overexpression of the KIT/SCF in uveal melanoma does not translate into clinical efficacy of imatinib mesylate. Clin Cancer Res. 2009 : 15 : 324-329. 135. Alexis JB, Martinez AE, Lutzky J. An immunohistochemical evaluation of c-kit (CD-117) expression in malignant melanoma, and results of imatinib mesylate (Gleevec) therapy in three patients. Melanoma Res. 2005 : 15 : 283-285. 136. Ugurel S, Hildenbrand R, Zimpfer A, La Rosée P, Paschka P, Sucker A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005 : 92 : 1398-1405. 137. Becker JC, Bröcker EB, Schadendorf D, Ugurel S. Imatinib in melanoma: a selective treatment option based on KIT mutation status? J Clin Oncol. 2007 : 25 : 9. 138. Wyman K, Atkins MB, Prieto V, Eton O, McDermott DF, Hubbard F, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006 : 106 : 2005-2011. 139. Kim KB, Eton O, Davis DW, Frazier ML, McConkey DJ, Diwan AH, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008 : 99 : 734-740. 140. Kluger HM, Dudek A, McCann C, Rink L, Ritacco J, Adrada C, et al. A phase II trial of dasatinib in advanced melanoma. J Clin Oncol (Meeting Abstracts). 2009 : 27 : 9010. 141. McArthur GA, Puzanov I, Ribas A, Chapman PB, Kim KB, Sosman JA, et al. Early FDG-PET responses to PLX4032 in BRAF-mutant advanced melanoma. J Clin Oncol (Meeting Abstracts). 2010 : 28 : 8529.

89
142. Takayama N, Sato N, O'Brien SG, Ikeda Y, Okamoto S. Imatinib mesylate has limited activity against the central nervous system involvement of Philadelphia chromosome-positive acute lymphoblastic leukaemia due to poor penetration into cerebrospinal fluid. Br J Haematol. 2002 : 119 : 106-108. 143. Hughes B, Yip D, Goldstein D, Waring P, Beshay V, Chong G. Cerebral relapse of metastatic gastrointestinal stromal tumor during treatment with imatinib mesylate: Case report. BMC Cancer. 2004 : 4 : 74. 144. Porkka K, Koskenvesa P, Lundan T, Rimpilainen J, Mustjoki S, Smykla R, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008 : 112 : 1005-1012. 145. Papageorgiou SG, Pappa V, Economopoulou C, Tsirigotis P, Konsioti F, Ionnidou E, et al. Dasatinib induces long-term remission in imatinib-resistant Philadelphia chromosome-positive acute megakaryoblastic leukemia but fails to prevent development of central nervous system progression. Leuk Res. 2010 : 34 : 254-256. 146. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009 : 27 : 6199-206.

90
ANNEE : 2010
NOM ET PRENOM DE L’AUTEUR : ROUTIER Emilie
PRESIDENT DE THESE : Professeur Selim ARACTINGI
DIRECTEUR DE THESE : Docteur Caroline ROBERT
TITRE DE LA THESE : Mutation de KIT et de PDGFRA dans le mélanome : série de 107 patients
Introduction : Les mélanomes sont des tumeurs diverses sur le plan moléculaire. Il a été démontré une corrélation entre les différents sous-types anatomo-cliniques de mélanome, les altérations moléculaires somatiques et l’association à une exposition solaire ponctuelle ou chronique. En effet, les mutations de BRAF sont présentes dans 50 à 60% des SSM, alors qu’elles sont plus rares dans les mélanomes de types acro-lentigineux, muqueux et les Dubreuilh où en revanche les mutations de KIT sont parfois retrouvées. Méthode : Nous avons recherché les mutations de KIT et de PDGFRA sur les tumeurs de 107 patients atteint d’un mélanome : 45 acro-lentigineux, 42 muqueux dont 23 de la sphère ORL, 11 vaginaux et 8 anaux, 6 Dubreuilh et 14 sans primitif retrouvé. Résultats : Nous avons mis en évidence quatre mutations de KIT (4,8%) sur les 84 résultats contributifs et deux mutations du même exon de PDGFRA (1,3%) sur les 80 contributifs. Trois mutations sur des mélanomes acraux (KITL576P, KITY553C et PDGFRAP567S/G569R) et deux mutations sur des mélanomes vaginaux (KITL576P et KITD820V). Sur trois patients traités par thérapie ciblée anti-KIT, nous avons eu une réponse partielle pendant cinq mois sous dasatinib, une stabilité de deux mois sous imatinib et un patient dont les métastases cérébrales ont rapidement progressé sous imatinib. Conclusion : Notre travail confirme l’existence de mutations de KIT et de PDGFRA dans certains sous-types de mélanomes. Même si nous avons trouvé un pourcentage de mutations plus faible que dans des publications antérieures, notre série est particulière car nous avons identifié trois nouvelles mutations dans le mélanome : KITY553C, KITD820V et PDGFRAP567S/G569R. Aujourd’hui, même si nous disposons encore de peu de données sur l’efficacité des inhibiteurs de KIT dans le mélanome, plusieurs cas cliniques anecdotiques et résultats préliminaires d’essais de phases précoces sont très encourageants, faisant espérer que le succès thérapeutique de ces thérapies dans les GIST puisse se transposer dans les mélanomes avec anomalie moléculaire de KIT ou PDGFRA.
MOTS-CLES :
- Mélanome - KIT - PDGFRA - Thérapie ciblée
ADRESSE DE L’U.F.R. : 8, Rue du Général Sarrail
94010 CRETEIL

91


















