
A coeur vailLant, rien...
190
N° d'ordre : 86-06 THE5E présentée à SYNTHESE L'UNIVERSITE DE LIMOGES POUR OBTENIR LE GRADE DE Soutenue /e 24 1986, devent /B commission d'tlXBmen : Président Examinateurs M. M. DAIRE , Université de Strasbourg MM. M. BILLY Université de Limoges J. MEXMAIN Université de Limoges P. GOURSAT ............ Université de Limoges P. LEFORT E.N.S.C.I. de Limoges - 1986-
Transcript of A coeur vailLant, rien...

N° d'ordre : 86-06
THE5Eprésentée à
SYNTHESE
L'UNIVERSITE DE LIMOGES
POUR OBTENIR LE GRADE DE
Soutenue /e 24 f~vrier 1986, devent /B commission d'tlXBmen :
PrésidentExaminateurs
M. M. DAIRE , Université de StrasbourgMM. M. BILLY Université de Limoges
J. MEXMAIN Université de LimogesP. GOURSAT............ Université de LimogesP. LEFORT E.N.S.C.I. de Limoges
- 1986-

•
A coeur vailLant, rien d'impossible•

A
Monsiewo Nichel BILLY,
ProfesseUl' à la Faculté des Sciences
de t 'Uni.versi.té de Limoges.

A HA FAMILLE
OLIVIER
THIERRY
Landry TAGRO
et
HENRIETTE

Ce travaiL a été réaLisé au Laboratoire de Céramiques NouveLLes
(L.A. C.N.R.S. 320) de L'Université de Limoges.
Monsieur Le Professeur M. BILLY, Directeur de ce Laboratoire a
assuré La direction de cette thèse et a suivi pas à pas L'évoLution de ce
travaiL. Je Le prie de trouver ici L'expression de ma profonde reconnaissance
pour L'aide et Les conseiLs qu'iL n'a cessé de m'apporter tout au Long de
ces recherches.
Je tiens à remercier Monsieur M. DAIRE, Professeur à L'Université
de Strasbourg de L'honneur qu'iL me fait en acceptant de présider ce jury.
Mes pLus vifs remerciements vont aussi à Messieurs Les Professeurs
J. MEXMAIN et P. GOURSAT de L'Université de Limoges qui ont bien vouLu
examiner ce travaiL et accepter de participer à ce jury de thèse.
Je voudrais exprimer toute ma gratitude à Monsieur P. LEFORT,
Maitre Assistant qui a dirigé et suivi au jour Le jour cette thèse, pour Les
conseiLs qu'iL m'a donnés et aussi pour Le caractère amicaL et cordiaL qu'iL
a su donner à nos reLations.
Je tiens à exprimer égaLement toute ma reconnaissance au Ministre
Ange François BARRY BATTESTI, à Monsieur Gervais KOFFI, Secrétaire GénéraL
Adjoint du gouvernement de Côte d'Ivoire et enfin aux Professeurs Jean Jonas
ADOU et HOUSSOU Kouakou pour Leur soutien moraL et financier.
Je remercie aussi MademoiseLLe FLorence MARTY, Messieurs PhiLippe
GUILLO et Gérard GAUTHIER pour Leur coLLaboration précieuse et amicaLe, ainsi
que Messieurs TETARD et LORTHOLARY, Ingénieurs au Laboratoire et en particuLier
MademoiseLLe B. PAROT pour Leur aide efficace dans La réaLisation technique de
ce mémoire.

SOM MAI R F;
INTRODUCTl ON. • . . . . . . . . . . • . . . . . . . . . . . . . . . . . • . . . . . . . . . . . . . . . . . . . . . . 1
Première Partie
APPAREILLAGE ET METHODES F;XPERIMENTALES
A - FRITTAGE SOUS CHARGE......................................... 3
1 - Appareillage2 - Mode opératoire
B 6
C 7
D - ANALYSE 7
E - MESURE DU TAUX DE DENSIFICATION ET DE LA POROSITE............ 8
F - PROPRIETES MECANIQUES....... . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1 Résistance mécanique à l'ambiante2 - Résistance à chaud3 - Résistance aux chocs thermiques4 - Microdureté
G - THERMOGRAVlMETRIE... . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Il

Deuxième Partie
ELABORATION D'OXYNITRURE D'ALUMINIUM
Chapitre l - SYNTHESE DE L'OXYNITRURE PAR REACTION D'ETAT SOLIDE AIN-A1203
A - INTRQDUCTION................................................. 14
B - MATIERES PREMIERES UTILISEES................................. 16
C - LA REACTION DU SYSTEME AIN/A1203.•.....•...............•.....19
D - CONDITIONS OPTIMALES DE SYNTHESE............................. 22
Chapitre II - CARBONITRURATION DE L'ALUMINE - MECANIS~ DE LA REACTION
A - INTRODUCTION................................................. 29
B - CONDITIONS EXPERIMENTALES. . . . . . . . . . . • . . . . . . . . . • . . . . . . . . . . . . . . 30
C - RESULTATS.................................................... 33
D - INTERPRETATION ET DISCUSSION................................. 40
E - CONCLUSION................................................... 46
Chapitre III - PREPARATION DE L'OXYNITRURE D'ALUMINIUM y PAR CARBONITRURATIOND'ALUMINE
A - COMPORTEMENT CINETIQUE...... . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
B - PROCEDE DE SYNTHESE RETENU................................... 48
Chapitre IV - ELABORATION DU MATERIAU TRANSPARENT
A - OPTIMISATION DES CONDITIONS DE FRITTAGE...................... 55
B - CINETIQUE DE GROSSISSEMENT DES GRAINS........................ 59
C - TRANSPARENCE ET TRAITEMENTS THERMIQUES POST-FRITTAGE......... 65

Troisième Partie
FRITTAGE SOUS CHARGE A L'OXYNITRURE
A - MONTAGE ET METHODES EXPERIMENTALES........................... 69
1 - Dispositif de frittage2 - Mode opératoire3 - Les échantillons
B - CINETIQUE DE FRITTAGE........................................ 75
1 - Rappels théoriques? - Etude du premier lot de poudre3 - Second lot de poudre
C - INFLUENCE DE LA G~LOMETRIE................................ 89
Quatrième Partie
RESISTANCE A L'OXYDATION
Chapitre l - OXYDATION DE LA POUDRE
ft - RESULTATS GENERAUX........................................... 92
B - INTERPRETATION................ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
C - CONCLUSION................................................... 102
Chapitre II - OXYDATION DE L'OXYNITRURE MASSIF BRUT DE FRITTAGE
A - RESULTATS GENERAUX........................................... 103
B - INTERPRETATION............................................... 108
C - CONCLUSION................................................... 116
Chapitre III - OXYDATION DE L'OXYNITRURE D'ALUMINIUM TRANSPARENT
A - RESULTATS GENERAUX........................................... 117
B - INTERPRETATION............................................... 122
C - CONCLUSION.................. .. .. .. .. .. .. .. .. • .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. 125

Cinquième Partie
PROPRIETES PHYSIQUES Et MECANIQUES
Chapitre l - CARACTERISTIQUES PHYSIQUES
A - PARAMETRgS DE MAILLE......................................... 128
B - MASSE VOLUMIQUE.............................................. 131
C - COEFFICIENT DE DILATATION THERMIQUE.......................... 132
D - PROPRIETES OPTIQUES.......................................... 132
Chapitre II - CHARGE A LA RUPTURE A LA TEMPERATURE AMBIANTE
A - CHARGE A LA RUPTURE DES FRITTES.............................. 140
B - RESISTANCE A ~A RUPTURE DE L'OXYNITRURE TRANSPARENT.......... 144
Chapitre III - MICRODURETE
1 - Oxynitrure y fritté.............................. 1502 - Oxynitrure y transparent......................... 151
Chapitre IV - EFFET D'ENVIRONNEMENT SUR LES PROPRIETES MECANIQUES
A - RESISTANCE A LA RUPTURE A CHAUD.............................. 154
B - INFLUENCE DE L'OXYDATlON SUR LA CHARGE A LA RUPTURE DESFRITTES. . . . . . . . . . . . . . . . . . • . . . . . . . . . . • . . . . . . . . . . . . . . . . . . . . . . . . 157
C - EFFET D'OXYDATION SUR LA TENUE MECANIQUE DU MATERIAUTRANSPARENT. • . . . . . . . . . . . . . . . . • • • . . • . . . . . . . . . . . . . . . . . . . . . . . . . . 164
D - RESISTANCE AUX CHOCS THERMIQUES.............................. 170
RESUME ET CONCLUSION......... . . . . . . . . . • . . . . . . . . . . . . . . . . . . . . . . . . . . 176
LISTE BIBLXOGRAPHIQUE............................................ 178

INTRODUCTION
Pour certaines applications militaires et industrielles, il est parfois
important de réaliser des pièces céramiques transparentes possédant des
caractéristiques de transmission dans le domaine de l'infrarouge ou du
visible. Il en est ainsi des dômes transparents aux rayonnements infrarouges
destinés aux missiles ou des enveloppes de certaines lampes à vapeur, des
hublots à contraintes sévères d'utilisation ... Pour de telles applications, il
est néçessaire que ces pièces offrent aussi de bonnes propriétés mécaniques et
une excellente résistance à la corrosion par les gaz chauds, l'air notamment.
C'est pourquoi nous avons orienté nos recherches vers des produits
céramiques susceptibles de présenter simultanément des propriétés mécaniques
de qualité et une bonne transparence. A ce titre, l'oxynitrure d'aluminium,
solution solide cubique du diagramme binaire A1N-A1203 signalé en 1964 par
A.M. Lejus (1) para!t répondre à ces impératifs. Son caractère fortement
lacunaire est de nature à favoriser son frittage et donc sa mise en forme par
les méthodes céramiques habituelles. Par ailleurs, l'oxydation de l'oxynitrure
pulvérulent ne se produit qu'au-delà de 650°C (2), ce qui laisse espèrer des
propriétés thermiques convenables pour des pièces massives.
Depuis l'engagement de ce travail (1980), plusieurs publications (3,4)
ou brevets ont confirmé tout l'intérêt de ce matériau et nous avons, pour
notre part, orienté notre étude verS la faisabilité du procédé d'élaboration
de frittage sous charge en l'absence d'ajout, ce qui constitue une voie à la
fois inexplorée à ce jour et a priori optimale quant à la qualité du produit
résultant.
La première partie de ce mémoire présente les techniques expérimentales
utilisées. L'élaboration du produit, les conditions de densification et de
traitement thermique sont développées dans la seconde partie. La cinétique de
frittage sous charge qui fait l'objet de la troisième partie est suivie de
l'étude des caractéristiques physicochimiques tel le comportement en atmos
phère oxydante. D'autres propriétés physiques optiques et mécaniques notamment
sont abordés dans un dernier temps.

APPAREILLAGE ET METHODES EXPERIMENTALES

APPAREILLAGES ET METHODES EXPERIMENTALES
A - FRITTAGE SOUS CHARGE
1 - Appareillage
L'appareil utilisé est un four L.P.A. de type 200 LC. Il comprend:
- un four vertical cylindrique à résistor à graphite (fig. 1),
alimenté en baese pression (22 ou 27 V) pour une puissance de chauffe de la à
15 KW, refroidi par circulation d'eau;
- un système de pressage à simple effet avec pistons en graphite.
La nature du résistor et des pistons nécessite une atmosphère non oxydante
(usuellemnt d'argon ou d'azote). La mise en température du four s'opère en
deux étapes
montée à la vitesse de 2500 0C par heure jusqu'à 1100 0C par
commande manuelle,
- puis pilotage par came jusqu'à la température désirée.
Un pyromètre optique en visée sur le résistor permet la mesure de la
température entre 1100 et 1900 0C. Grâce à un régulateur préalablement
étalonné, on affiche à tout instant la température voulue.
2 - Mode opératoire
Dans la gamme de températures étudiée (1500 à 1900 0C), on utilise une
matrice en graphite qui possède seule la réfractarité et la résistance mécani
que nécessaires. On détermine expérimentalement la pression maximale tolérable
(liée notamment à la morphologie de la matrice et à la qualité du graphite).
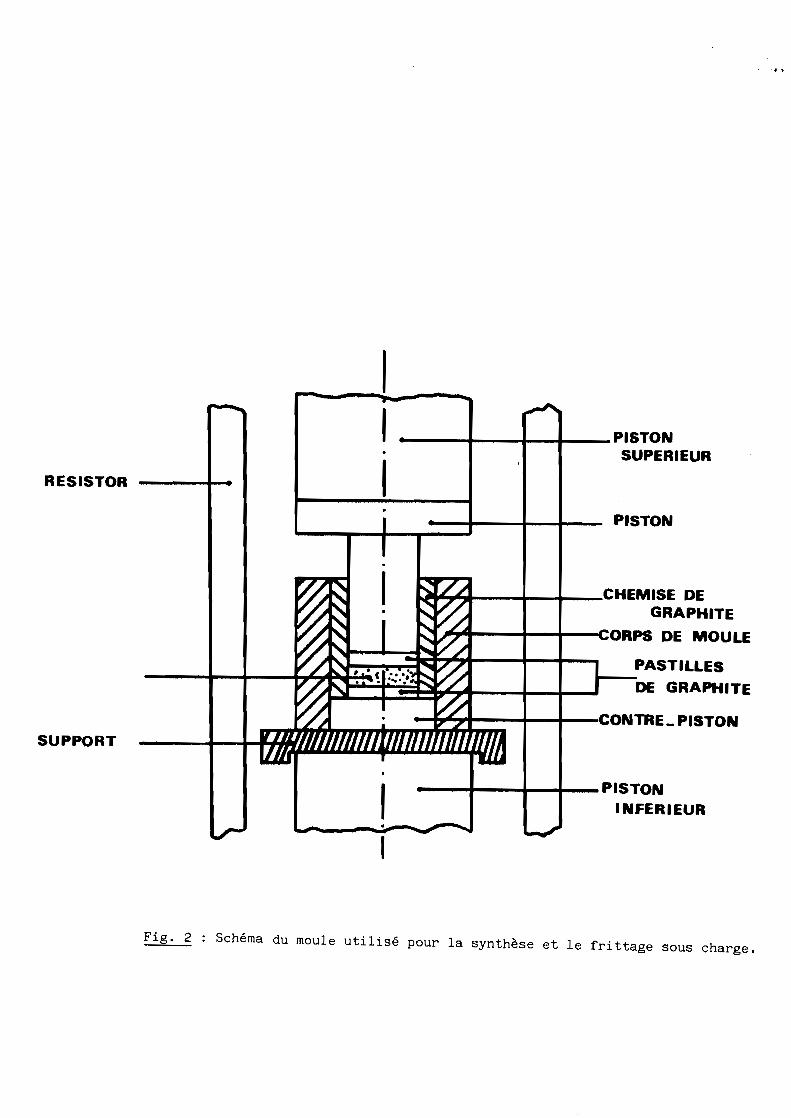
Pour faciliter le démoulage rapide des moules utilisés, une forme géométrique
simple a été choisie avec chemisage (fig. 2). Sa limite d'utilisation en
pression est de 40 MPa.

Fig. 1 Appareillage de frittage sous charge (Equipement LPA).
RESISTOR
---+---+--t--- PISTONSUPERIEUR
PISTON
SUPPORT
UiI~""'---1r---t-__CHEMISE DEGRAPHITE
~"'--+-"""--(.;ORPSDE MOULE
PASTILLES
---r1--t~~:::::t~~~73--+-+-..J"""-DEGRAPHITE
-J.-....--+---II"--CONTRE _ PISTON
1.--......--+-......-_PISTON
INfERIEUR
Fig. 2 Schéma du moule utilisé pour la synthèse et le frittage sous charge.

- 6 -
La poudre est introduite dans le moule, après avoir recouvert au
préalable les parois d'une mince couche de nitrure de bore pulvérulent que
nous avons vérifié être inerte dans nos conditions de travail. Le nitrure de
bore empêche toute diffusion de graphite dans le produit; d'éventuelles traces
en périphérie sont éliminées par grattage et polissage après le démoulage.
On réalise un vide primaire dans l'enceinte du four, puis l'azote
(qualité U) est introduit en légère surpression de 50 Torr pour éviter toute
entrée d'air au cours de l'essai.
En fin de manipulation, le refroidissement s'effectue soit en descente
libre, soit en descente programmée à raison de 2000 K/h. Dans tous les cas, la
charge est arrêtée au-dessous de 1100 oC.
B - ELABORATION DES ECHANTILLONS TRANSPARENTS
Le four sous vide de type convertible 22-12-20 commercialisé par V.A.S.,
est constitué d'un élément vertical cylindrique et d'une armoire d'alimenta
tion délivrant une puissance de 25 kW sous 45 V.
Le four comprend une enceinte de 300 mm de diamètre et 400 mm de hauteur
et un écran formé de 4 couches de feutre en graphite il est refroidi par
circulation d'eau.
La mesure de la température et sa régulation sont effectuées à l'aide
d'un régulateur programmeur pyrométrique à dilatation en graphite. La mise en
température du four s'effectue en deux étapes: après remise sous tension du
système électronique, on vide le corps du four pendant la min à l'aide d'une
pompe primaire, puis on amorce le chauffage jusqu'à atteindre une température
de palier de 250°C, maintenue pendant une demi-heure. Puis on place l'enceinte
sous balayage gazeux d'azote U, la pression étant légérement supérieure à la
pression atmosphérique pour éviter l'oxydation des éléments en graphite. L'ar
rêt du four en fin de manipulation s'effectue après descente programmée de
température à raison de 1900 K/heure.
Les échantillons pulvérulents sont placés dans un récipient de graphite
à parois recouvertes de nitrure de bore afin d'éviter toute contamination par
le carbone; pour les pièces massives, on utilise une bogue constituée d'une
poudre également en nitrure de bore.

y,D :;: KV 2 avec K
- 7 -
C - ANALYSES PHYSICO-CHIMIQUES
1 - Radiocristallographie
On utilise un diffractomètre de type C.G.R. avec générateur SIGMA 2070
qui comporte un ensemble électronique d'enregistrement (Philips) de l'inten
sité du faisceau diffracté.
L'échantillon réduit en poudre est déposé sur une plaque d'aluminium et
exposé aux radiations Ka d'une anticathode de cuivre (un monochromateur en
quartz assure l'élimination de la raie Ka).
2 - Microscopie
L'étude morphologique des échantillons a été effectuée par microscopie
optique (microscope métallographique REICHERT) ou par microscopie électronique
à balayage sur appareil JEOL JSM 35.
D - ANALYSE GRANULOMETRIQUE
Nous avons utilisé un appareil de type Sédigraph 5000 qui permet la
détermination granulométrique d'une poudre sur la base du taux de sédimenta
tion des grains supposés sphériques en suspension dans un milieu dispersif.
Son principe utilise la loi de STOKES qui exprime le diamètre D des
grains en fonction de la vitesse de sédimentation selon l'expression:
Yz18 n
où p et p représentent respectivement les masses volumiques du fluide et deso
grains, n la viscosité du fluide et g l'accélération de la pesanteur.
La cellule de sédimentation se déplace devant un faisceau très fin de
rayons X et l'intensité du rayonnement transmis est reliée au diamètre des
grains. On peut analyser un ensemble de grains de diamètres compris entre 0,1
et 100 ~m. Les résultats sont donnés directement en courbe de fréquence
cumulée en fonction du diamètre.

- 8 -
E - MESURE DU TAUX DE DENSIFICATION ET DE LA POROSITE
La densité apparente des échantillons est déterminée par pesée, par
poussée hydrostatique ou par mesure directe du volume des frittés à l'aide
d'un Palmer. On en déduit le taux de densification 1 (en %) et la porosité
P = 100 - 1 avec :
d1 =_a_ x 100
dt h
da désignant la densité apparente, dt h
la densité théorique déterminée sur la
base des mesures cristallographiques des paramètres de maille.
Comme la porosité totale des échantillons est toujours très faible et
compte tenu des observations morphologiques, on a systématiquement négligé la
porosité ouverte et assimilé la porosité totale à la porosité fermée.
F - PROPRIETES MECANIQUES
1 - Résistance mécanique à l'ambiante
Les essais ont été réalisés à l'aide d'une machine WOLPERT équipée d'un
dispositif de flexion (appuis distants de 19 mm). L'application de la charge
est réalisée à l'aide d'un poussoir solidaire du plateau supérieur avec une
vitesse de déformation de 0,2 mm/min. Les charges appliquées restent infé
rieures à 2000 N. La courbe effort-déformation est enregistrée automatique
ment.
2 - Résistance mécanique à chaud
Nous avons utilisé la même machine d'essai, mais cette fois-ci équipée
d'un four permettant d'opérer jusqu'à 1500 0C.
Le montage comporte deux tubes d'alumine supportant deux plateaux
également en alumine ; le tube inférieur (hauteur 250 mm) est fixé sur le
capteur de force de la machine tandis que le tube supérieur est solidaire de
la traverse du bâti. La distance entre appuis (en alumine) est de 25 mm. Un
thermocouple Pt/Pt Rh 10% placé dans l'axe central du porte-échantillon, sous
l'éprouvette, permet la mesure de la température (fig. 3).

:~~.~3 Appareillage de rupture à froid et à chaud.

- 10 -
La résistance à la rupture est calculée à partir de la relation
où L représente la distance entre appuis, b la dimension de la section de
l'éprouvette perpendiculaire à la direction d'application de la charge, h la
dimension de cette section selon l'autre direction et P la force appliquée.
3 - Résistance aux chocs thermiques
à
y a
à la
la résistance
à la
détermination de l'écart de température c
initiation des fissures.
Au cours des essais, les éprouvettes
température donnée, puis immergées brutalement
ambiante. Après séchage à l'étuve et refroidissement, on mesure leur résistan-
ce à la rupture.
4 - Microdureté
L'appareil utilisé est un microduremètre SHIMADZU type M équipé pour la
mesure de la dureté Vickers (pénétrateur en diamant de forme pyramidale,
d'angle au sommet 8 ~ 136°, appliqué sous une charge P).
Elle s'exprime par un nombre H qui correspond au rapport de la chargev
appliquée P à la surface de l'empreinte S.
p
Hv=-
Savec S
2 sin 0./2
d'où la formule
Hv
P1854,4 2""- .
d

- 11 -
Dans la pratique, les mesures sont effectuées sur des échantillons polis
adhérant à un support fixé par un étau sur la platine du microduremètre.
Après la mise au point sur l'échantillon, le pénétrateur est amené à la
place de l'objectif, puis enfoncé, parcourant ensuite le cycle descente
application-remontée. Lorsque le cycle est terminé, on revient en position de
visée afin de mesurer les dimensions de l'empreinte. On obtient directement Hv
connaissant P et d.
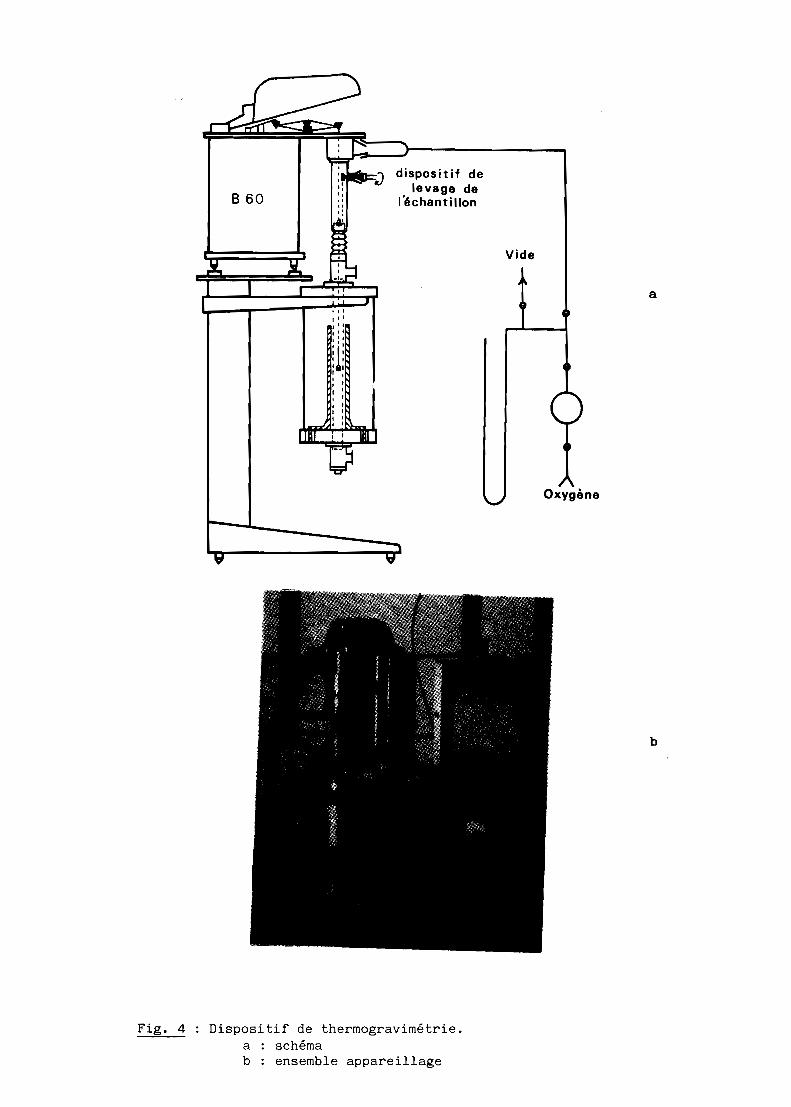
G - THERMOGRAVlMETRIE
L'étude de l'oxydation des échantillons a été réalisée à l'aide d'une
thermobalance enregistreuse de type Ugine Eyraud SETARAM B 60 équipée d'un
four à résistance de chromite de lanthane. Un ensemble programmation régula
tion PRT 3000 assure le cycle thermique, la température étant mesurée par un
thermocouple en platine rhodié 6%-30% (fig. 4).
La masse initiale de l'échantillon est telle que son accroissement reste
de l'ordre de 10 mg. Au cours de la mise en température, la nacelle porte
échantillon (en alumine) est placée sous vide dynamique à l'intérieur de
l'enceinte réactionnelle, reliée au fléau de la balance par l'intermédiaire
d'une chaînette de platine et maintenue hors de la zone chaude du four.
Lorsque la température du palier est atteinte, on introduit l'oxygène à
pression voulue, puis l'échantillon est descendu dans la zone isotherme j
l'instant zéro de la réaction d'oxydation est obtenue avec une faible erreur
vu la petite taille de l'échantillon et de la nacelle.
a
Vide
dispositif delevage de
l'échantillon
.,1"
,, ".,
"
L
B 60
Oxygène
b
Fig. 4 Dispositif de thermogravimétrie.a schémab : ensemble appareillage

ELABORATION DE L'OXYNITRURE D'ALUMINIUM Y.

Chapitre l
SYNTHESE DE L'OXYNITRURE PAR REACTION D'ETAT SOLIDE AIN - AI203
A - INTRODUCTION
Les phases du système binaire A1N/A1203
ont fait l'objet de nombreuses
recherches (1, 5, 6) et en particulier la phase y, de structure spinelle et de
formule Al(64+x)/3 [J (8-x)/3 ° 32-x Nx, présente une grande originalité
puisqu'elle retient l'azote dans sa structure lors de son oxydation (2, 7).
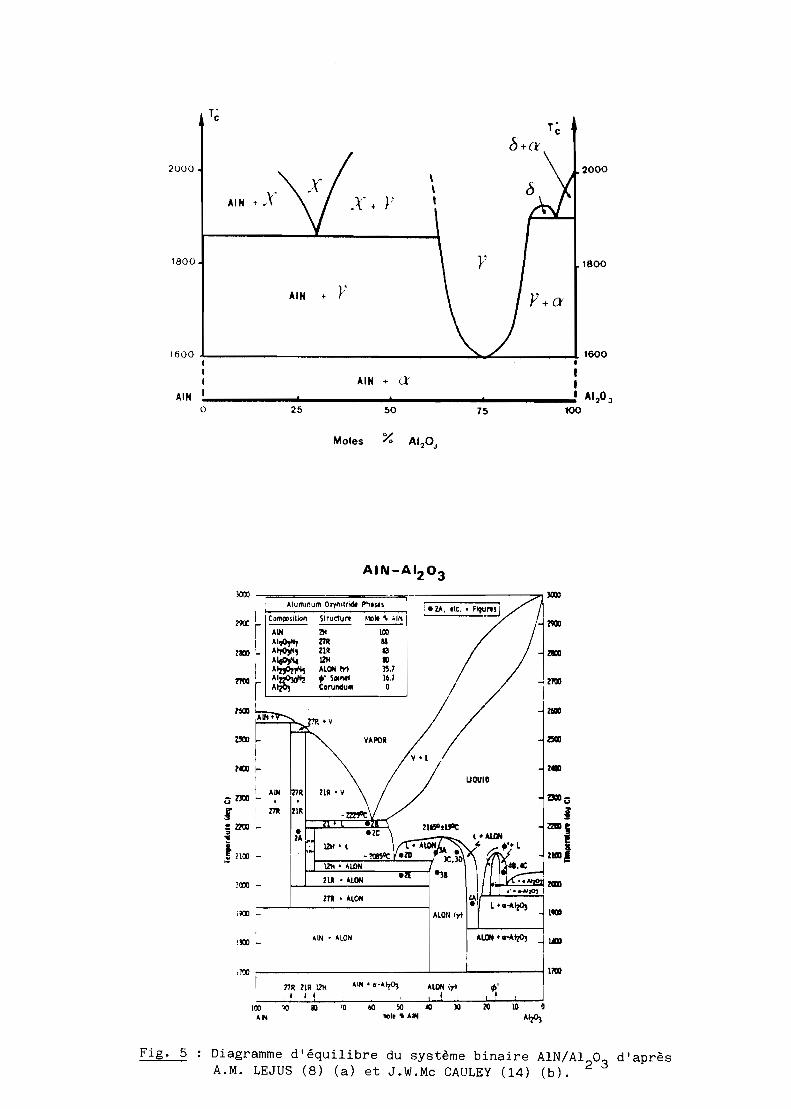
Depuis la publication du premier diagramme de phases par A.M. Lejus (8)
(cf. fig. 5a), les travaux ont particulièrement porté sur la structure de la
phase oxynitrure y (7, 9), sur les composites obtenus par compression à chaud
de mélanges A1N-A1203
(10, 11) et sur la possibilité d'obtenir des produits
massifs transparents avec la phase pure (12, 13).
Sur la base d'une expérimentation menée dans des conditions sensiblement
différentes de celles de A.M. Lejus, Mc Cauley (14) présente une nouvelle
version du diagramme de phases (cf. fig. 5b) mais la comparaison des deux
études montre qu'ils différent très sensiblement. Au surplus, aucune étude de
nature cinétique n'a été entreprise. Il nous a donc paru essentiel pour
l'optimisation du procédé de fabrication de l'oxynitrure à partir des mélanges
A1N/A120 3
de reprendre cette question en définissant
1 - La composition optimale du mélange initial nitrure/oxyde pour
obtenir l'oxynitrure pur.
2 - La température optimale de réaction.
T"cT'c
AIN T.r .r ~ F
AIN ~ F
1800 1800
1600 1600,1
AIN + Cl' 1AIN 1 AI10 J
0 25 50 15 100
Moles % AI20J
2000 2000
171l • "LON
AIN • ALON
Alum.num O'Yn.I'ide P"".. 1
C.mllOlilion SI'vetu,", ~ 1
:~ ~ I~ 1
,,~, ZIR ...."~\Z!4 Il,,~ "LON Ir! 15.7~lzzOld+2 .' SIlOM4 16.1~tzo, Corunclum a
! 1711
2'lCC 1
1"1
t8al-
11
210Q 1rL.-.-.. -----'
2'0011
fCll ~
1
iiŒ) -
1
~ Z200 r'II
f 1100-
1û 1lOO - •
i
1!OO ~----------_---J_--'------1
100AIN
?IR llR 12H,1 1 1
AIN' o'A~
70 60 ~
110'... AIN
"LON (y'1 1 1
Fig. 5 Diagramme d'équilibre du système binaire AlN/A120 3
d'aprèsA.M. LEJUS (8) (a) et J.W.Mc CAULE Y (14) (b).

- 16 -
B - MATIERES PREMIERES UTILISEES
1 - Le nitrure d'aluminium
C'est un produit Koch-Light de pureté affichée 99,9%, d'une densité de
3,24 g/cm3 et de granulométrie supérieure à 50 ~m. L'analyse par activation
neutronique (15) montre que cette poudre peut en fait contenir jusqu'à 6%
d'alumine en poids. Une étude récente a montré que les grains sont en réalité
des agglomérats de grains beaucoup plus fins, submicroniques pour la plupart
(16) •
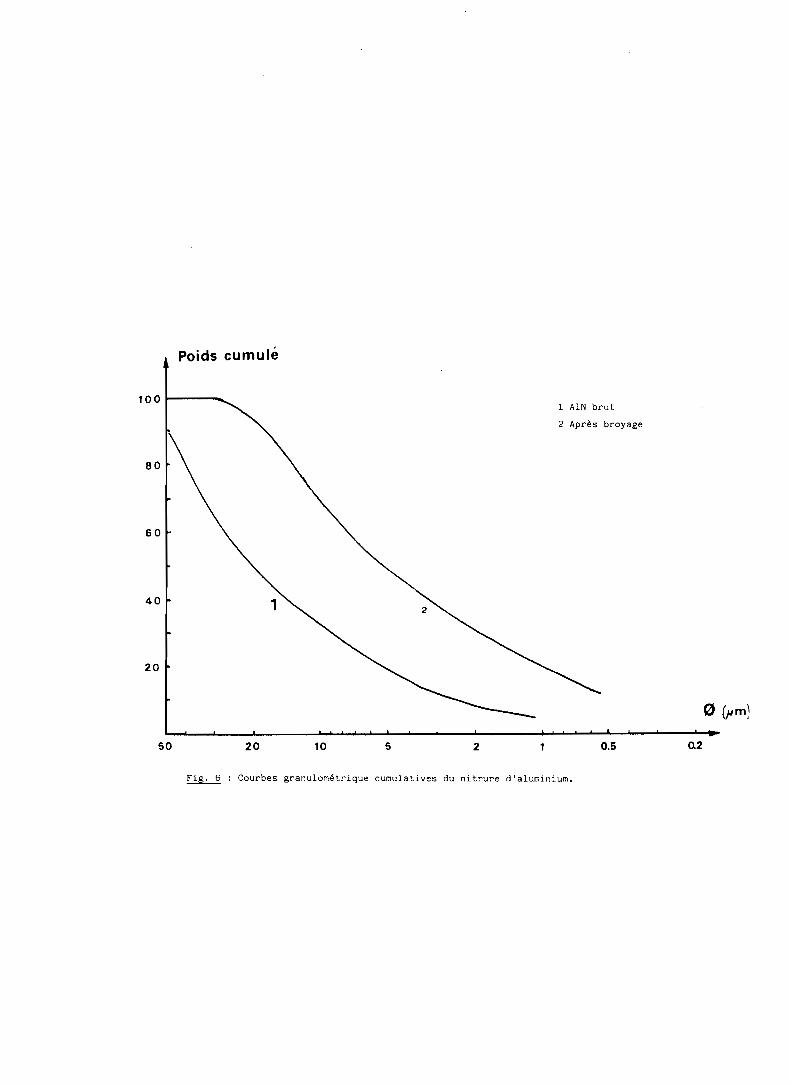
Le nitrure d'aluminium est broyé dans un moule en alumine avec des
boulets de même nature que le moule pendant 4 heures dans un appareil de type
TURBULA. Des prélévements sont effectués après broyage pour déterminer au
sédigraphe la taille des grains. Les résultats présentés en figure 6 montrent
que le broyage ne brise pas complètement les agglomérats. La granulométrie est
toutefois suffisamment réduite pour accroître la réactivité et faciliter le
mélange avec l'alumine dont la taille de grains est sensiblement inférieure.
Un broyage de plus longue durée n'a pas été retenu car il accroît la quantité
d'alumine en provenance du broyage (négligeable pour une durée de 4 heures)
sans pour autant briser les agglomérats.
2 - L'alumine
C'est un produit FLUKA commercialisé par SICCAP EMMOP, de pureté 99,99%;
elle est calcinée à 1300 0C et tamisé à 0,3 ~m. Ses principales impuretés sont
Na ( < 25 pprn ) , Si « 30 ppm) et Fe « 10 pprn ) ,
3 - Le mélange AIN-A1223
Ce mélange est homogénéisé à sec dans l'appareil Turbula pendant 12
heures en utilisant un récipient de matière plastique résistant à l'abrasion
afin de limiter la contamination. La durée retenue a été optimisée en fonction
des résultats observés lors de la réaction.
Poids cumulé
1001---__
BO
60
40
20
l A1N brut
2 Après broyage
50 20 10 5 2 0.5 0.2
Fig. 6 Courbes granulométrique cumulatives du nitrure d'aluminium.

1 Nitrure d aluminium 1
1-------- -------- --------1--------11 h k 1 d A ~ u 1 1/10 1
1-------- -------- --------1--------11 1 0 0 2,70 16,58 1 100 1
1 0 0 2 2,49 18,02 1 60 1
1 1 0 1 2,372 18,95 1 70 1
1 0 2 1,829 24,91 1 20 1
1 1 0 1,557 29,65 1 30 ,1 0 3 1,414 33,01 1 20 1
2 0 0 1,348 34,85 1 6 1
1 1 2 1,320 35,70 1 18 12 0 1 1,301 36,30 1 8 1
202 1,186 40,50 1 6 1
2 0 3 1,047 47,36 1 6 1
2 1 0 1,019 49,10 1 4 1
2 1 1 0,998 50,49 1 6 1
1 0 5 0,934 55,51 1 6 1
2 1 3 0,868 62,50 1 6 1_________, 1
Alumine a 1--------1-------- --------1--------
h k 1 1 d A ~ u 1 1/10
--------1-------- --------1--------o 1 2 1 3,479 12,79 1 751 0 4 1 2,552 17,57 1 901 1 0 1 2,379 18,89 1 40o 0 6 1 2,165 20,84 1 11 1 3 2,085 21,68 1 1002 0 2 1,964 23,09 1 2024 1,740 26,26 1 401 1 6 1,601 28,75 1 802 1 1 1,546 29,87 1 41 2 2 1,514 30,56 1 6o 1 8 1,510 30,65 1 81 2 4 1,404 33,25 1 30030 1,374 34,07 1 501 2 5 1,337 35,15 1 22 0 8 1,276 37,10 1 41 1 1 1 ,239 38,41 1 161 1 9 1,234 38,61 1 82 2 0 1,1898 40,34 1 83 0 6 1,160 41,60 1 1
____________1 -
Oxyni trure d'aluminium y 1
--------1--------1--------1--------1h k 1 1 d A 1 0 u , 1/10 1
--------1--------\-------- --------11 1 1 1 4,5928 9,65 10 1
o 2 2 1 2,8125 15,89 26,51 1 3 1 2,3985 18,73 922 2 2 1 2,2964 19,60 12,5004 1 1,9887 22,79 842 2 4 1 1,6238 28,32 91 1 5 1 1,5309 30,21 35,5044 1 1,4063 33,21 33,21o 2 6 1 1,2578 37,76 353 3 5 1 1,2131 39,41 104 4 4 1 1,1482 42,13 7246 1 1,0630 46,43 51 3 7 1 1,0357 48,05 15o 0 8 1 0,9944 50,77 14o 6 6 1 0,9375 55,24 tf1 5 7 1 0,9186 56,99 Ilo 4 8 1 0,8894 60,00 Il
____ 1 - _
Tableau l - Raies de diffraction des phases AIN, A1203-
a et AION-y(CuK a) •

- 19 -
C - LA REACTION DU SYSTEME AIN/A12Q3
L'examen des diagrammes de phases de la figure 5 montre que l'oxynitrure
d'aluminium y existe selon, A.M. Lejus, au-dessus de 16000 C pour des teneurs
en nitrure AIN variant de 15 à 40% environ. Selon Mc. Cauley, la synthèse
n'est pas possible avant 1700 0 C et le domaine de stabilité est plus réduit
(entre 27% et 40% d'AIN). Au vu de ces chiffres, nous avons testé des mélanges
nitrure/alumine de compositions variables de 10 à 50% molaires en nitrure à
des températures de 1500, 1600, 1700 et 1800 o C, pour une durée de 1 heure.
Chaque mélange a été analysé après réaction par radiocristallographie
quantitative.
1 - Méthode d'analyse
Le produit obtenu est d'abord nettoyé pour enlever le nitrure de bore
qui l'enveloppe. Il est ensuite broyé avec soin dans un mortier d'agate afin
d'éviter les orientations cristallines préférentielles. Compte tenu des trois
phases éventuellement présentes (cf. tableau 1) on a choisi d'observer pour
des raies :
d'intensité moyenne donc mesurables dans une large gamme de composi
tion et suffisamment séparées pour ne pas donner lieu à chevauchements. Il
s'agit des raies
- 024 et 116 pour l'alumine a,
- 002 et 110 pour le nitrure,
- 022 et 115 (ou 004 pour RX2),
pour l'oxynitrure.
On trace ainsi deux courbes d'étalonnage à partir de mélanges-témoin
mettant en relation les rapports massiques des constituants
et
R =1
masse de nitrure dans le mélange
masse d'oxynitrure dans le mélange
masse d'oxynitrure dans le mélange
masse d'alumine dans le mélange

- 20 -
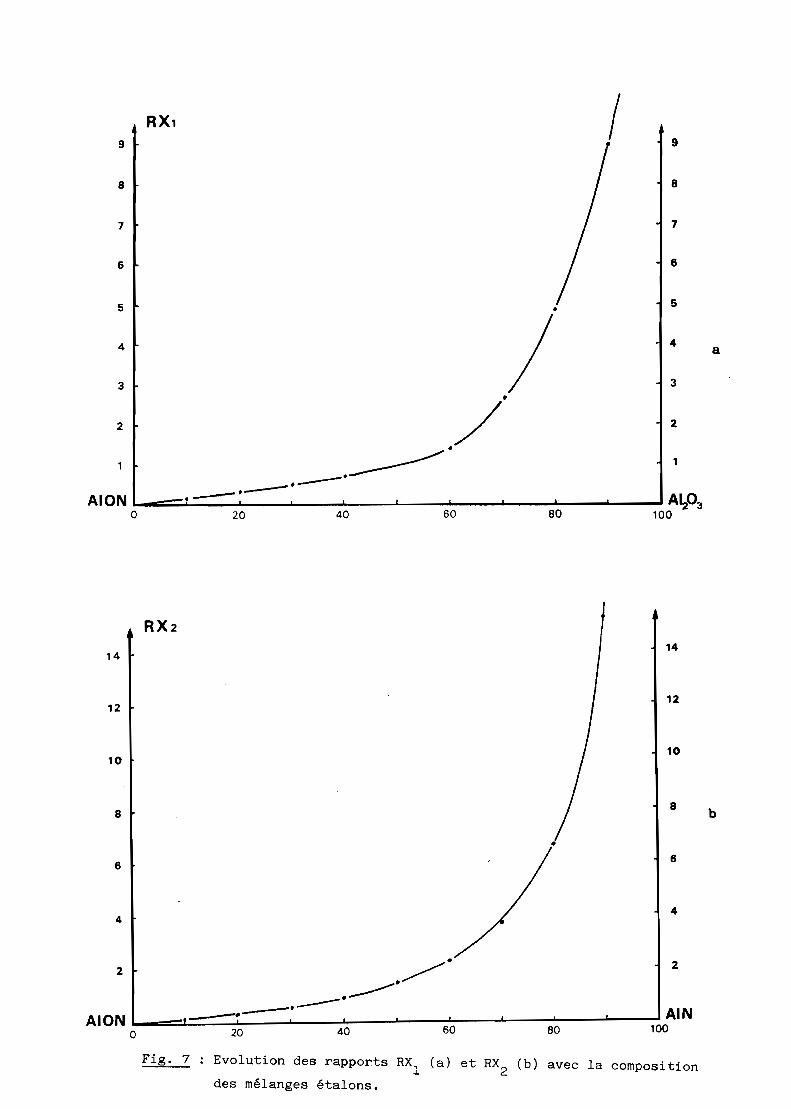
avec les rapports RX1
(fig. 7a) et RX2 (fig. 7b) des intensités et leurs raies
choisies mesurées par une méthode pondérale. A l'inverse, ces courbes
permettent aisément, à partir de mélanges inconnus de ces trois constituants,
de déterminer leurs rapports massiques et leurs proportions respectives
puisque :
% A1 203 + % AION + % AIN = 100
% AIN
% AION
% AION
D'où
100
% AION
% AIN
Il est évidemment possible de transformer ces pourcentages massiques en
pourcentages molaires.
2 - Influence de la température
a) A 1500 oC, aucune réaction ne se produit, quelle que soit la composi
tion des mélanges.
9
8
7
6
5
4
3
2
RX1 1 9
8
7
6
5
4
3
2
a
RX2
1414
1212
1010
8b8
•66
44
.22 .~
.s->_.---AIN~.~
AION40 60 80 1000 20
Fig. 7 Evolution des rapports RX, (a) et RX2
(b) avec la compositions:des mélanges étalons.

- 22 -
b) A 1600 oC, on observe parfois l'apparition des raies de l'oxynitrure
pour des mélanges de teneur initiale 25 ou 30%. Un tel résultat confirme
parfaitement le diagramme de A.M. Lejus quant à la température de formation de
l'oxynitrure, mais il est incompatible avec celui de J.W. Mc Cauley.
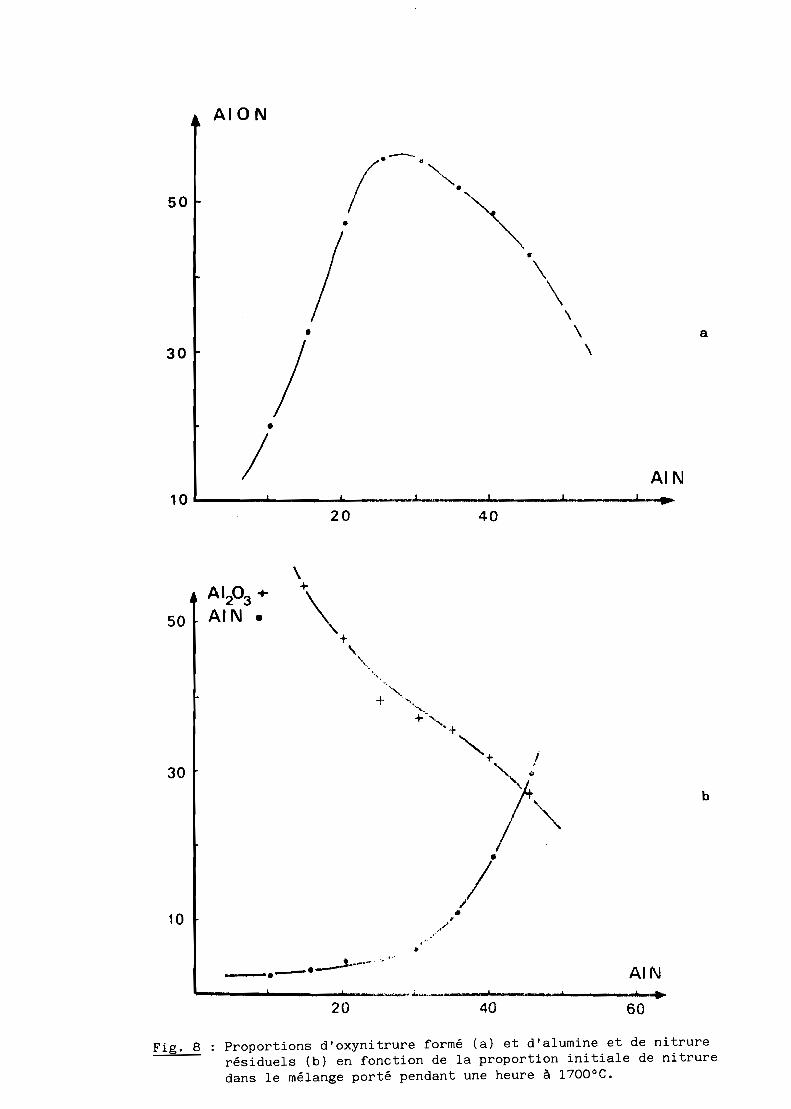
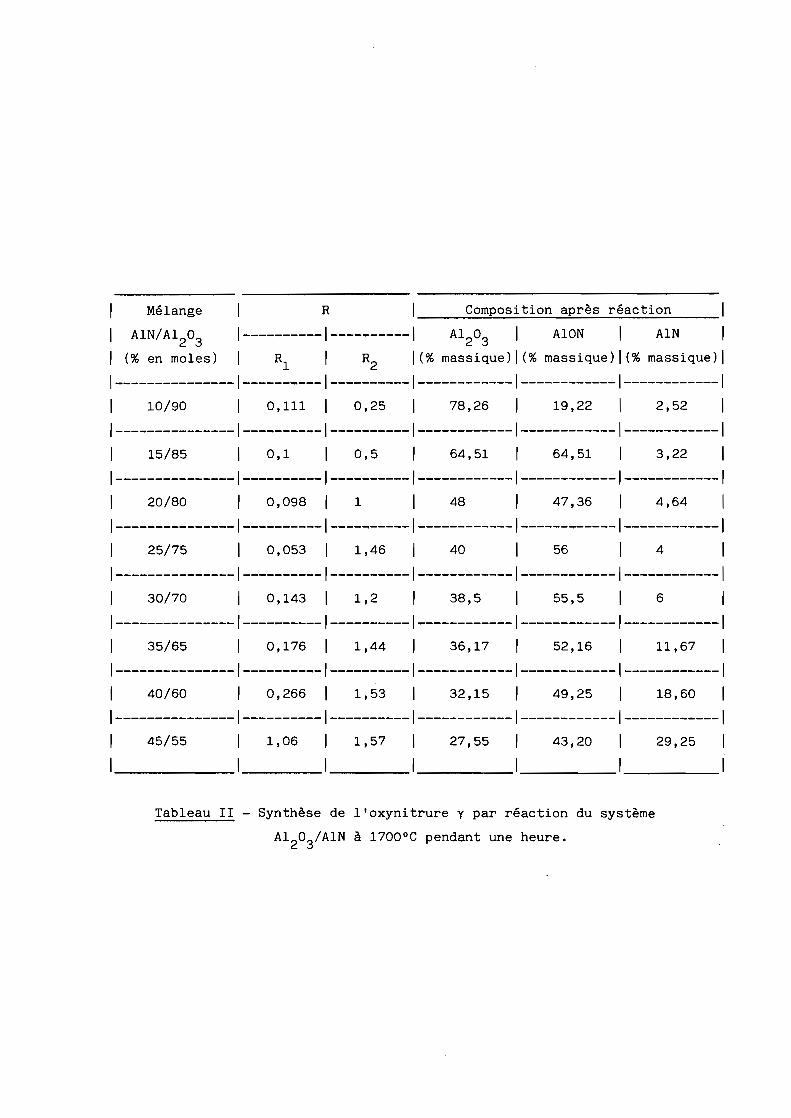
c) Les résultats obtenus à 1700 0C sont donnés au tableau II et illustrés
par les figures 8 a et b. On observe alors une formation appréciable
d'oxynitrure d'aluminium y, avec un maximum pour des teneurs initiales en
nitrure comprises entre 25 et 30%. Mais, la réaction reste incomplète ce qui
implique une augmentation de la température ou de la durée du traitement
thermique.
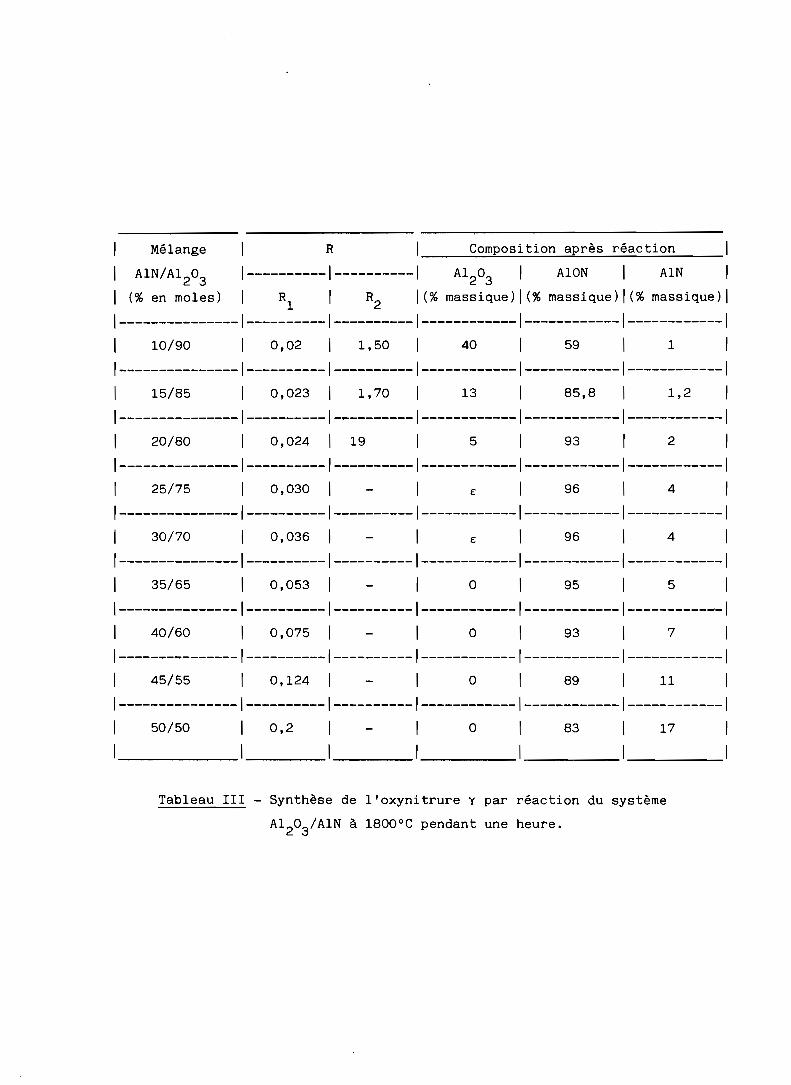
d) A 1800 oC, effectivement, la quantité d'oxynitrure formée est beaucoup
plus importante. Elle passe par un maximum entre 25 et 30% en mole de nitrure
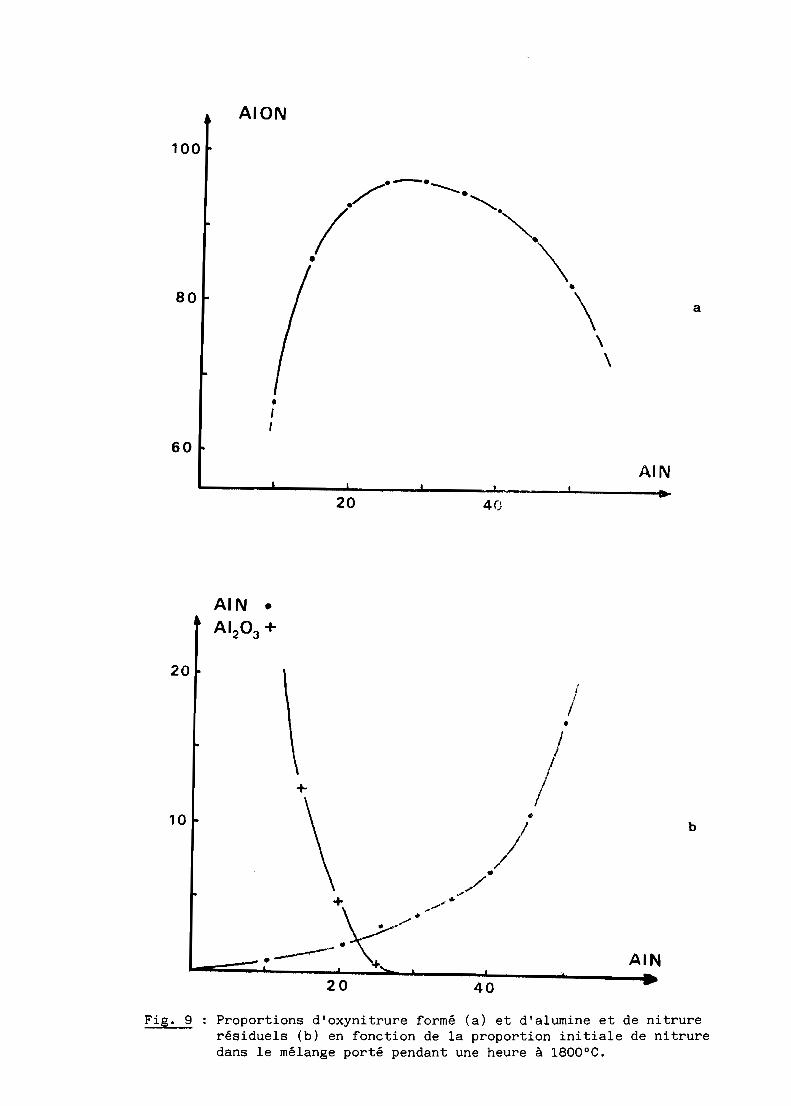
AIN (fig. 9) auquel correspond la formation de 96% (pondéral) d'oxynitrure
d'aluminium
Pour des mélanges riches en nitrure, au-delà de 25% en mole, (cf.
tableau III), la présence d'alumine a dans les produits de réaction se réduit
à des traces non détectables par radiocristallographie.
D - CONDITIONS OPTIMALES DE SYNTHESE
Les précédents résultats montrent que la composition de mélange initial
conduisant au rendement optimal est toujours comprise entre 25 et 30% en mole
de nitrure quelle que soit la température. C'est pourquoi nous l'avons fixée à
27% pour la suite des préparations. Notons que cette composition diffère
sensiblement de celle de Mc Cauley (12) qui admet, sur la base de considéra
tions structurales, que la composition la plus stable doit être observée pour
une teneur en nitrure de 33%.
Il est par ailleurs très important d'éliminer toute trace de réactif
(AIN ou A1 203)dans le produit final, impuretés qui sont préjudiciables à la
transparence du matériau par absorption ou diffusion de la lumière. Il s'agit
donc dans ces conditions optimales de composition préalablement choisies, de
faciliter la réaction soit:
AIO N
.:"'.50,
~•
•\\
\
• \
/a
30 \
•
/ AIN10 .Le ..
20 40
30
10
b
...--"'_.-.-'---20
.,"
40
AIN
Fig. 8 Proportions d'oxynitrure formé (a) et d'alumine et de nitrurerésiduels (b) en fonction de la proportion initiale de nitruredans le mélange porté pendant une heure à 1700 0C.
Mélange
AIN/AI20 3
(% en moles)
R
----------1----------R1 1 R2
Composition après réaction
Al203
AION AIN
(% massique) (% massique) (% massique)
--------------- ---------- ---------- ------------ ------------ ------------10/90 0,111 0,25 78,26 19,22 2,52
--------------- ---_____-1"_ ---------- ------------ ------------ ------------15/85 0,1 0,5 64,51 64,51 3,22
--------------- ---------- ---------- ------------ ------------ ------------20/80 0,098 1 48 47,36 4,64
25/75 0,053 1,46 40 56 4
30/70 0,143 1,2 38,5 55,5 6
35/65 0,176 1,44 36,17 52,16 11,67
18,60
29,251,57
1,53
1,06
0,26640/60
45/55
------------1------------32,15 1 49,25
------------1------------27,55 1 43,20
------- ----- ----- ------,------ ------
Tableau II - Synthèse de l'oxynitrure y par réaction du système
AI20 3/AIN
à 1700 0C pendant une heure.
5
2
7
4
4
1
1,2
11
17
59
96
85,8
93
89
83
96
93
95
E
E
5
Composition après réaction
o
o
o
o
13
40
A1203
A10N A1N
(% massique) (% massique) (% massique)
1,70
1,50
19
R
0,02
0,024
0,030
0,023
0,036
0,053
0,124
0,075
25/75
20/80
Mélange
30/70
50/50
10/90
35/65
15/85
40/60
45/55
A1N/A1203
(% en moles)
1
1---------- ----------1 R
1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ---------- ------------1
---------------1---------- ----------1------------1 0,2 1
______ 1 , _
Tableau III - Synthèse de l'oxynitrure y par réaction du système
A1203/A1N
à 1800 0C pendant une heure.
100
80
60
AJON
•11
/.-.~.
/ <,<,. '\
-\\\
AIN
a
20
20 40
10
AIN
b
20 40
Fig. 9 Proportions d'oxynitrure formé (a) et d'alumine et de nitrurerésiduels (b) en fonction de la proportion initiale de nitruredans le mélange porté pendant une heure à 1800 oC.

- 27 -
1. En augmentant la température à 1850°C pour laquelle la cinétique de
réaction est encore plus rapide.
2. En prolongeant la durée du traitement thermique à 4 heures. Dans ces
conditions, on obtient un produit blanc, pratiquement pur dans la mesure où
des traces de nitrure conduisent à une coloration grise.
3. En améliorant les contacts entre grains de poudre par compactage
préalable, ou pendant le chauffage isotherme. En fait, l'application d'une
charge entraîne un blocage de la réaction par frittage. La composition évolue
alors très lentement et le produit qui renferme encore du nitrure est toujours
de couleur noire. Cette voie qui conduit à la formation de composites (9, la),
doit être abandonnées pour la synthèse de l'oxynitrure d'aluminium pur.
En définitive, le mode opératoire retenu pour la préparation de la phase
est la suivante. Les mélanges d'alumine et de nitrure de composition molaire
73/27% à laquelle correspond le rapport pondéral 87/13%, sont homogènéisés à
sec pendant 12 heures en mélangeur Turbula. Après un chauffage de 4 heures à
1850°C sous atmosphère d'azote, on obtient un produit blanc légèrement fritté
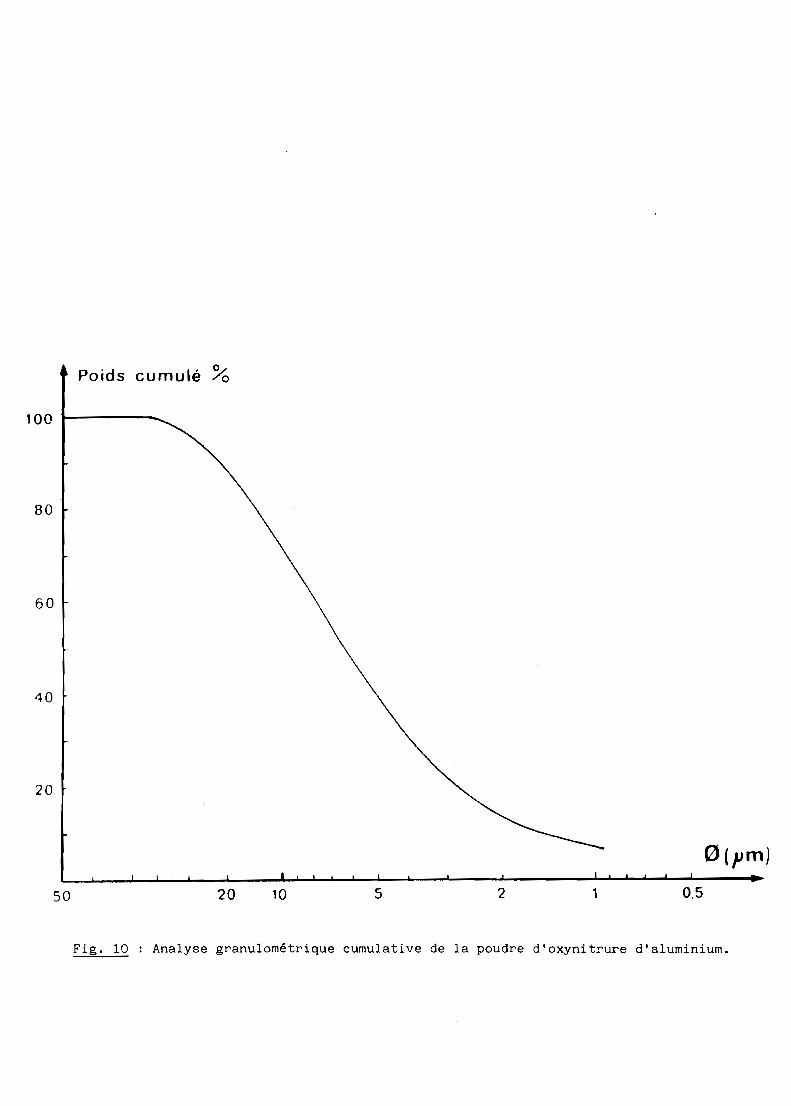
qui, après broyage, conduit à une poudre constituée d'agglomérats de 6 ~m
environ (fig. la).
100
80
60
40
Poids cumulé %
20
50 20 10 5 2
o (,um)
0.5
Fig. 10 Analyse granulométrique cumulative de la poudre d'oxynitrure d'aluminium.

Chapitre II
CARBONITRURATION DE L'ALUMINE
MECANISME DE LA REACTION
Le mode de synthèse de l'oxynitrure d'aluminium par réaction directe du
nitrure avec l'alumine, s'il est le plus pratique a priori pour l'étude des
conditions d'existence et de composition de l'oxynitrure, n'est pas le seul
possible. Il existe en effet d'autres procédés de nitruration de l'alumine,
généralement connus pour donner le nitrure, par réaction directe dans un jet
de plasma d'azote (18, 19) ou dans l'ammoniac (20).
La réduction des oxydes par le carbone en atmosphère nitrurante, appelée
commodément "carbonitruration" est par ailleurs l'un des procédés les plus
anciens de synthèse des nitrures qui revient d'actualité (4, 21). Ainsi,
l'élaboration du nitrure d'aluminium à partir d'alumine est connue depuis fort
longtemps (22). Mais l'application récente à la synthèse de l'oxynitrure (4,
23) n'a fait l'objet d'aucune étude du mécanisme de réaction ou plus simple
ment d'aucune tentative, pourtant essentielle, d'optimisation.
A - INTRODUCTION
On admet ordinairement que la formation du nitrure par carbonitruration
de l'alumine est facilitée lorsque les proportions massiques de carbone dans
le mélange initial dépassent très sensiblement les 26,1% prévus à partir de
la réaction :
[IJ
Le carbone en excès est ensuite éliminé par chauffage en atmosphère
oxydante, ce qui peut conduire à une réoxydation superficielle du nitrure.

- 30 -
B - CONDITIONS EXPERIMENTALES
Les matières premières utilisées dont on trouvera les principales
caractéristiques au tableau IV sont de l'alumine a, du noir du fumée et du
graphite (Carbone-Lorraine) broyé puis tamisé. L'azote employé correspond à la
qualité U fournie par l'Air Liquide.
1 Produits 1 Alumine 1 Noir de fumée 1 Graphite 1
1----------------------------1---------------1---------------1---------------11Granulométrie (~m) 1 0,3 1 0,2 0,3 1 0,2 30 11----------------------------1---------------1---------------1---------------11 Surface spécifique (m
2/g)1 5,7 1 30 1 6,7 1
1----------------------------1---------------1---------------\---------------\1 Granulométrie équivalente 1 1 1 1
1 moyenne (um) 1 0,26 1 0,10 1 0,40 1
1----------------------------1---------------1---------------1---------------11 Pureté % 1 99,99 1 cendres 0,75 1 cendres 0,1 1
1 1 1 1 1
Tableau IV - Caractéristiques des réactifs.
La granulométrie équivalente moyenne est déterminée (24) à partir de
la surface spécifique S et la masse volumique p de la poudre selon la
relation :
6d
S
Le mélange des poudres dans les proportions pondérales définies par la
réaction [IJ, 26,1% et 73,9% respectivement pour le carbone et l'alumine, a
été effectué par voie sèche dans un mélangeur de type "TURBULA" pendant 12
heures. L'homogénéisation est alors satisfaisante malgré la présence d'agglo
mérats dans les mélanges avec le noir de fumée.

- 31 -
Les mélanges sont placés dans un creuset de graphite aux parois recou
vertes d'une couche de nitrure de bore jouant le rôle de barrière de
diffusion. Ils sont ensuite chauffés en montée linéaire de 1600 K/h jusqu'à la
température de l'essai (entre 1500 et 1700 0C). On introduit l'azote sous
pression atmosphérique et assure un balayage gazeux de 0,5 l/min environ tout
au long de la réaction, en général jusqu'à 8 heures. Les produits obtenus sont
analysés par microscopie à balayage.
L'avancement de la réaction est déterminé par mesure de la perte de
masse et/ou par dosage du carbone résiduel par combustion dans l'air. Le
carbone en effet s'élimine à partir de 630°C, et l'oxydation du nitrure AIN
devient prépondérante à température plus élevée comme le montre la figure 11a
obtenue en montée linéaire de 250 oC/h. Dans ces conditions, les analyses ont
été effectuées à 700°C afin d'éviter toute oxydation parasite du nitrure (fig.
llb) •
Des tests d'analyse radiocristallographique quantitative utilisant comme
référence des mélanges étalons d'alumine et de nitrure (cf. chap. précédent)
ont confirmé les résultats obtenus aux températures inférieures à 1600 0C où
l'oxynitrure d'aluminium n'apparaît pas (tableau V).
Degré 1 Perte de masse Dosage du car-
I d'avancement 1 après réaction 1 bone résiduel
Analyse
Iradiocristallographiquel
1--------------1----------------1----------------1-----------------------11 Cl 1 0,66 1 0,66 1 0,675 1
1 1 1 1 1
Tableau V - Degré d'avancement Cl de la réaction d'un mélange (noir de
fumée) après 6 heures à 1500oC, déterminé par 3 méthodes
indépendantes.
La composition des produits de la réaction constitués par des mélanges à
quatre phases A1 203, C, AIN et AION est déterminée par une méthode de dosage
radiocrista11ographique quantitatif analogue à celle présentée plus haut
(chapitre précédent), le carbone étant dosé par combustion dans l'air à 700°C.

-4
-8
-12
200 1000 TOC)
a
6 t J
-5
-10
-15
.àm%'"0 L -========:::1
b
Fig. 11 Oxydation dans l'air d'un mélange carbone/alumine, aprèsréaction partielle, en montée linéaire de température (a)et en isotherme à 700°C (b).

- 33 -
c - RESULTATS
Deux séries d'expérimentations ont été conduites dans la zone de tempé
rature 1500-1700oC, en faisant réagir avec l'alumine a soit le noir de fumée,
soit le graphite. Ces deux expérimentations présentent un ensemble de points
communs qu'il convient d'abord de préciser.
1 - Résultats généraux
a) Le nitrure d'aluminium est le seul produit formé aux températures
inférieures ou égales à 1600 0C. Aux températures supérieures, on observe la
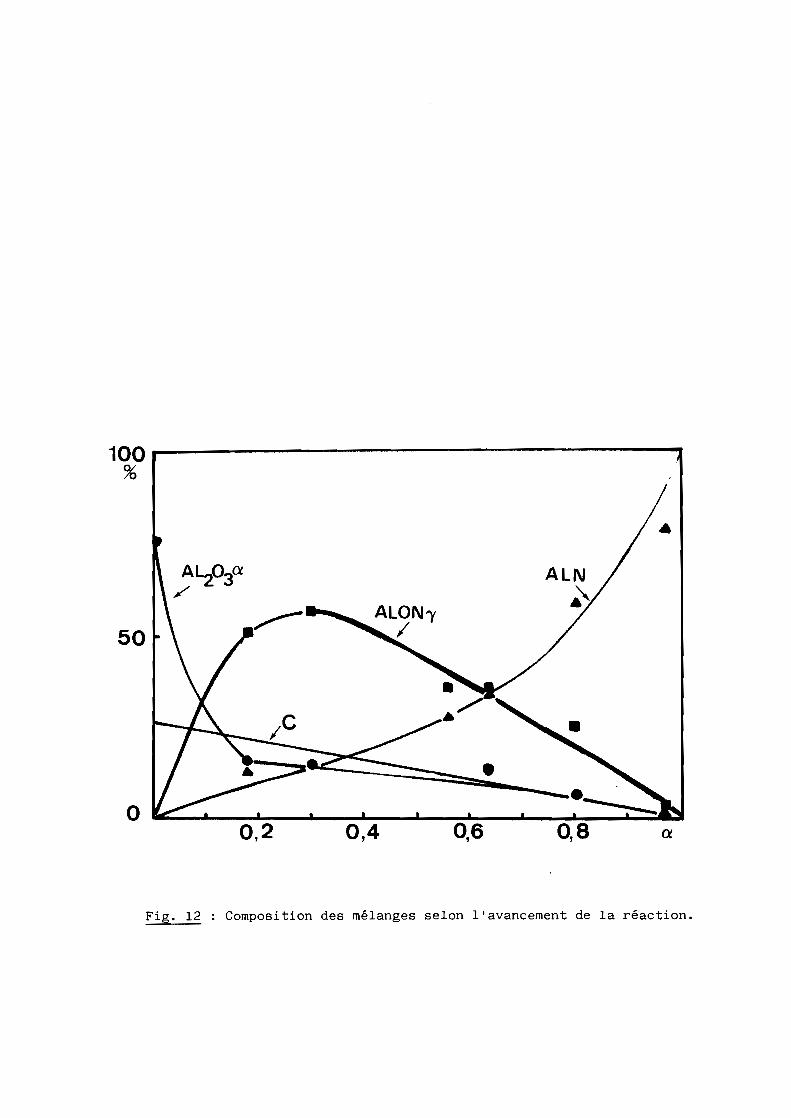
formation de l'AION y (23, 25) en quantité variable avec le degré d'avancement
de la réaction (fig. 12). L'alumine disparaît alors rapidement au profit de
l'AION y, le nitrure lui-même se formant plus tardivement.
b) Il n'y a pas de formation de phase volatile, du moins en quantité
telle qu'elle soit entraînée par le balayage d'azote ou par diffusion en phase
gazeuse. En effet, la comparaison de la pesée des échantillons après réaction
et l'analyse thermogravimétrique du carbone résiduel montre que la totalité de
la perte de masse en cours de réaction est liée à la consommation du carbone.
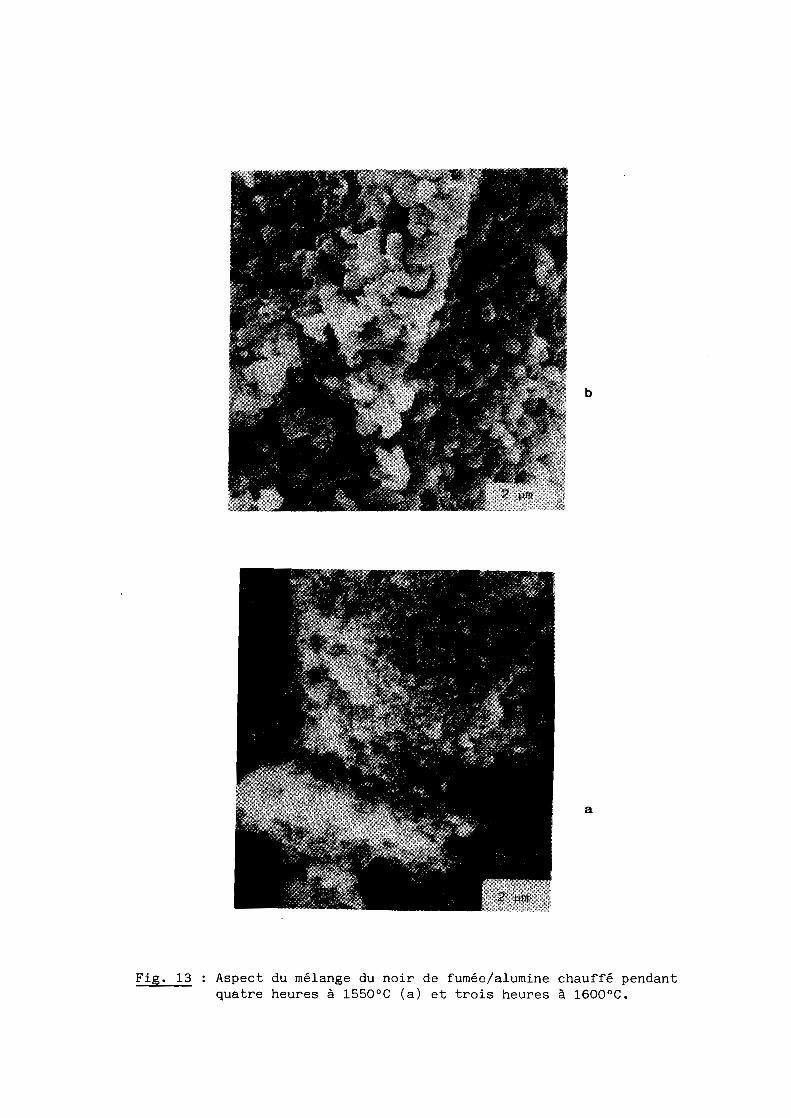
c) On observe un important grossissement des grains qui s'intensifie
avec la température comme l'illustrent les résultats obtenus à 1550 et 1600 0C
pour les mélanges à base de noir de fumée (fig. 13a et 13b respectivement) ou,
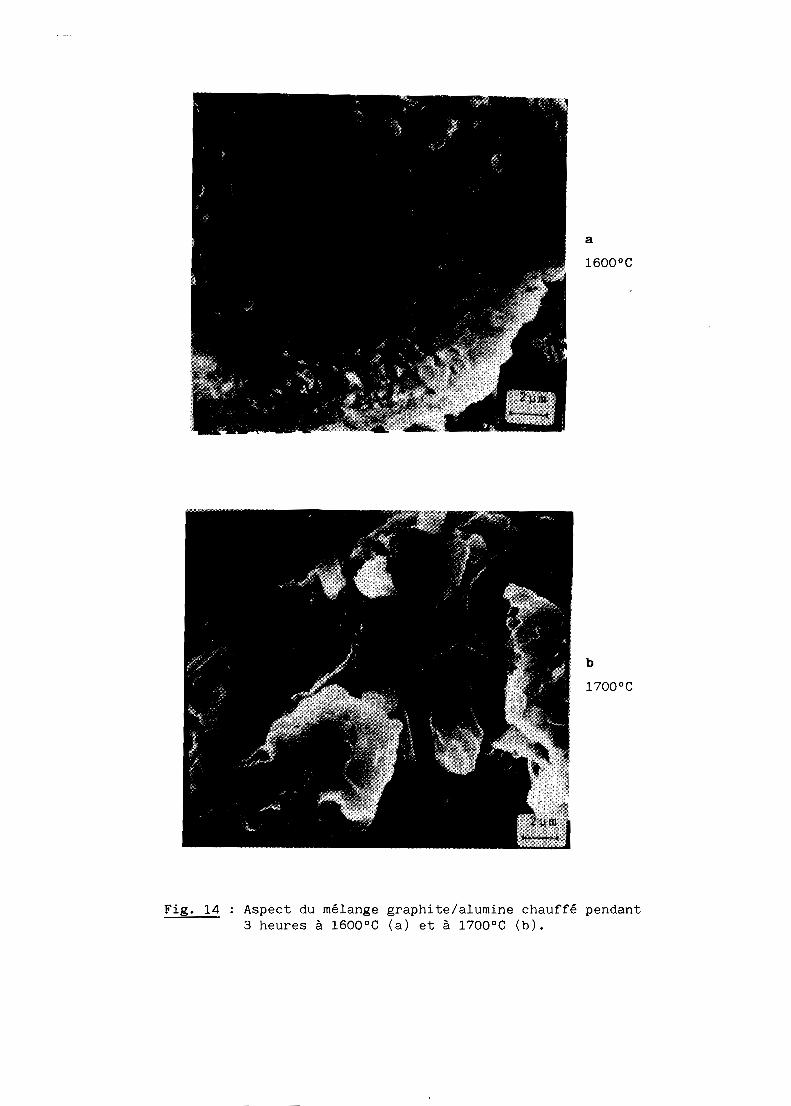
à 1600 et 1700oC, pour ceux utilisant le graphite (fig. 14a et 14b). Le
grossissement des grains est donc indépendant de la présence de la phase
"AION" puisqu'il est sensible dès 1600oC, température pour laquelle cette
phase n'est pas encore observée. Un frittage d'importance relative accompagne
ce grossissement des grains.
2 - Résultats cinétiques
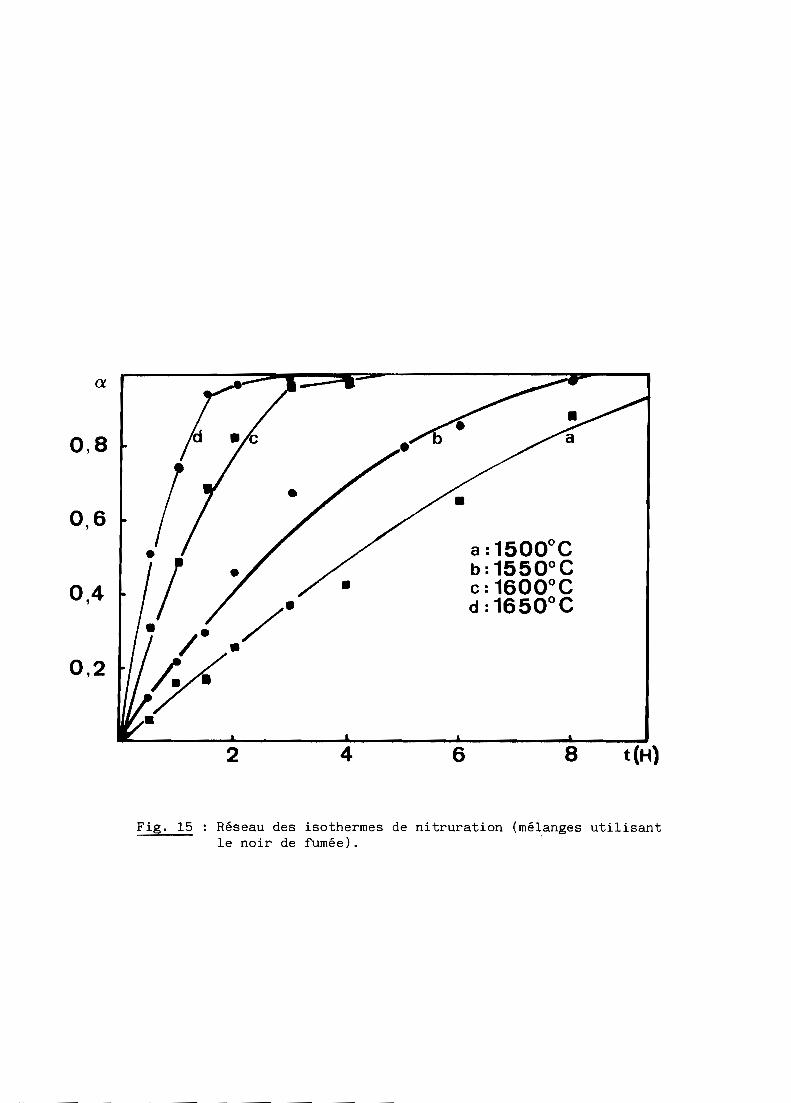
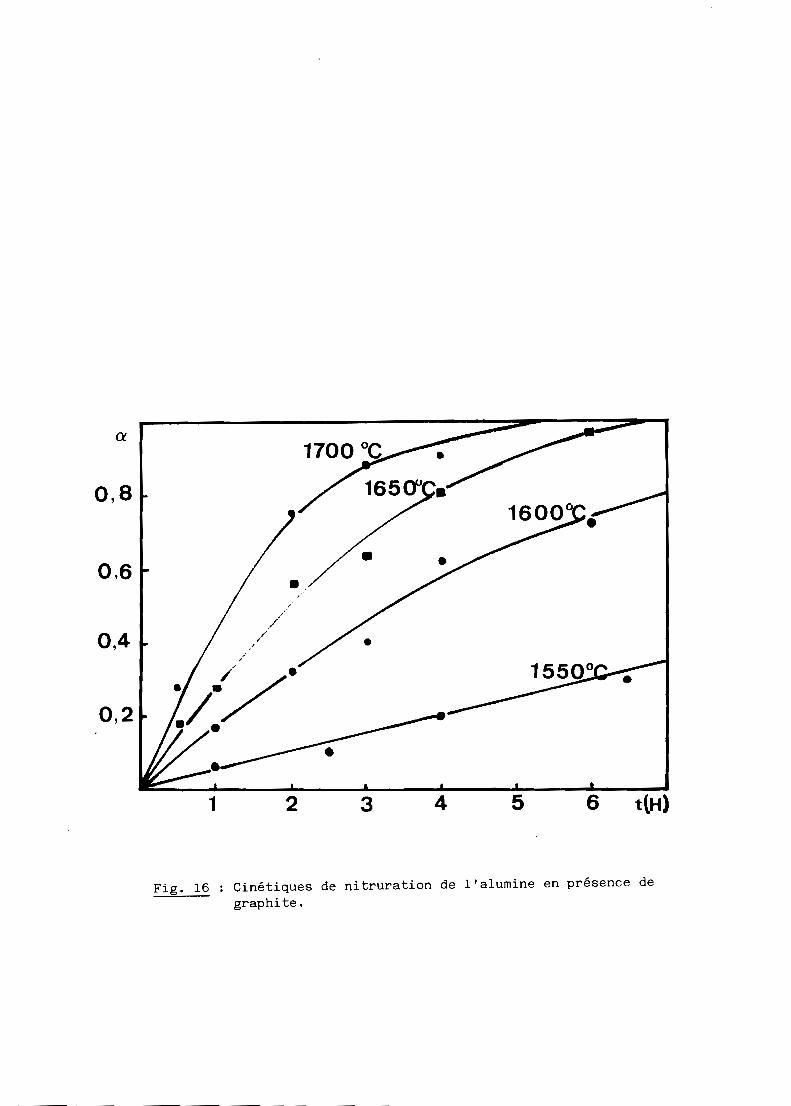
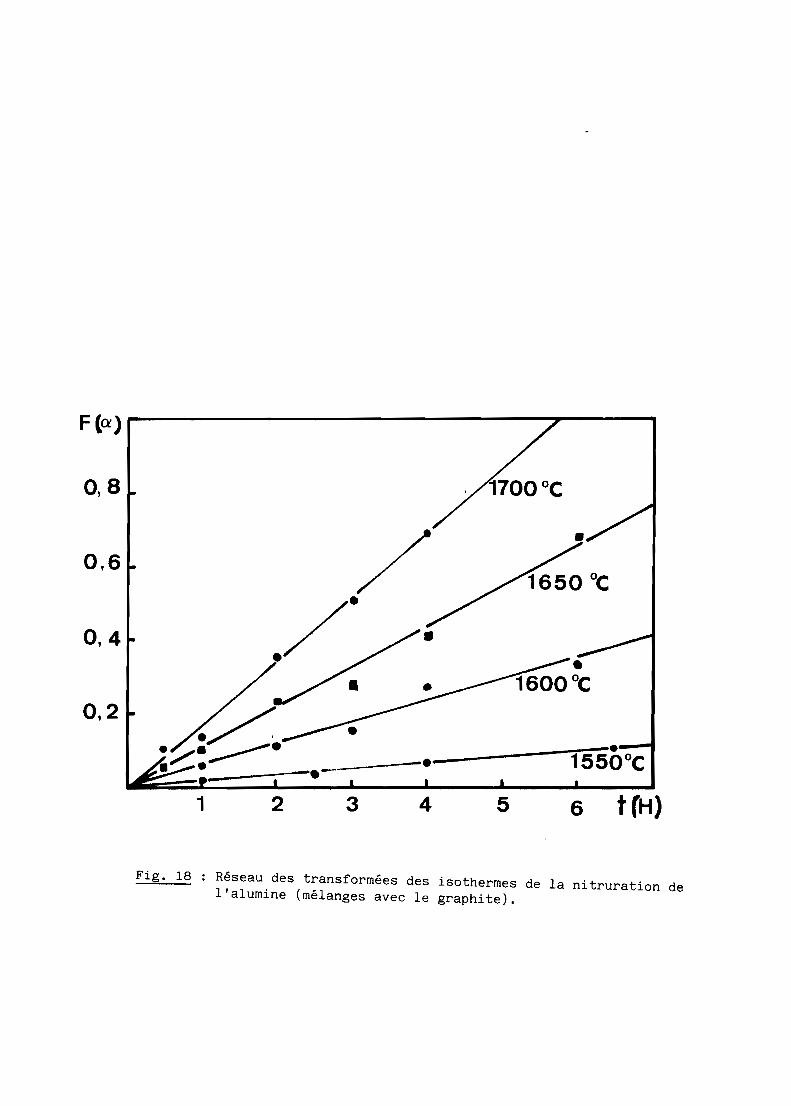
Les figures 15 et 16 représentent les réseaux d'isothermes obtenus par
compilation des analyses d'échantillons portés entre 1500 et 17000C pendant
des durées variables de 0,5 à 8 heures pour les mélanges impliquant respecti
vement le noir de fumée et l'alumine. Chaque point correspond à une expérience
pour laquelle la durée de chauffage est atteinte en une seule fois.
--0,8
•0,60,40,2
o
50
100...-.-------------------,%
Fig. 12 Composition des mélanges selon l'avancement de la réaction.
b
a
Fig. 13 Aspect du mélange du noir de fumée/alumine chauffé pendantquatre heures à 1550°C (a) et trois heures à 1600°C.
a
b
Fig. 14 Aspect du mélange graphite/alumine chauffé pendant3 heures à 1600°C (a) et à 1700°C (b).
0,8
0,6
0,4
0,2
2 4 6 8 t(H)
Fig. 15 Réseau des isothermes de nitruration (mélanges utilisantle noir de fumée).
0,8
0,6
0,4
0,2
1 2 3 4 5 6 tlH)
Fig. 16 Cinétiques de nitruration de l'alumine en présence degraphite.

- 39 -
On constate que les courbes cinétiques sont régulièrement décélérées,
sans incubation initiale, la vitesse étant maximale au début de la réaction.
Cette allure générale est identique pour les deux expérimentations et
l'apparition de phase AION y au-dessus de 1600 0C ne la modifie pas. En
revanche, l'alumine réagit beaucoup moins vite en présence de graphite que de
noir de fumée; à 1600 0C par exemple, le temps de demi-réaction est d'environ
3,5 heures dans le premier cas contre 1 heure seulement dans le second.
Nature du
1 carbone 1 Noir de fumée 1 Graphite broyé 11--------------1-----------------------------\-----------------------------11 Degré 1 0,1 1 0,3 10,5 1 0,7 1 0,9 1 0,1 10,3 10,5 1 0,7 1 0,9 11 d'avancement 1 1 1 1 1 1 1 1 1 1 1
Cl1--------------1-----1-----1-----1-----1-----1-----1-----1-----1-----1-----11 Energie 1 1 1 1 1 1 1 1 1 1 1
1 d'activation 1 397 1 389 1 359 1 364 1 401 1 410 1426 1 347 1 401 1 397 11 en KJ/mole 1 1 1 1 1 1 1 1 1 1 1
1 1_1_1_'_1_1_1_'_1_1_1
Tableau VI - Energie d'activation à différents degrés d'avancement
en fonction de la nature du carbone utilisé.
Sans préjuger du mécanisme réactionnel, ces réseaux d'isothermes se
prêtent au traitement analytique des vitesses instantanées (26). Les résultats
reportés au tableau VI apportent les éléments suivants :
a) L'énergie d'activation est comprise entre les valeurs extrémales de
426 et 347 KJ/mole ; elle fluctue avec l'avancement de la réaction dans les
limites de sensibilité de la méthode.
b) L'énergie d'activation ne dépend pas de la nature du carbone entrant
dans le mélange.
c) La phase intermédiaire AION Y, qui intervient aux températures les
plus élevées, n'introduit aucune modification cinétique.

- 40 -
D - INTERPRETATION ET DISCUSSION
L'allure des courbes cinétiques des deux réseaux, le grossissement des
grains observé à toute température et l'unicité d'énergie d'activation
plaident en faveur d'un régime cinétique unique. En ce cas, il faut rechercher
le ou les processus élémentaires limitants tels qu'ils soient indépendants de
la nature des produits formés (puisque l'apparition de la phase intermédiaire
A10N y ne modifie rien au plan cinétique) et qu'ils justifient par ailleurs la
diminution de vitesse liée à l'augmentation de la granulométrie du carbone
réactif.
La mise en présence de deux réactifs solides, le carbone et l'alumine
pose d'emblée la question de la nature du contact entre ces espèces.
Différents cas sont à envisager: soit le contact direct solide-solide, soit
l'intervention, en plus de l'azote, d'une phase gazeuse issue de l'alumine ou
bien du carbone.
1 - Contact direct entre grains de carbone et d'alumine.
La seule phase gazeuse intervenant dans cette hypothèse serait l'azote.
La formation du nitrure se situerait au niveau des points de contact alumine
carbone-azote. Ensuite, les grains d'alumine se transformeraient progressive
ment en nitrure en passant, en cours de nitruration et pour les températures
les plus hautes, par l'intermédiaire "oxynitrure A10N" respectant en cela les
conditions d'existence de cette phase reportées au diagramme pseudobinaire
A1 203-A1N (fig. 5). Le carbone, quant à lui, serait consommé et disparaîtrait
en phase gazeuse sous forme d'oxyde. Le ralentissement de la réaction avec
l'augmentation de la taille des grains de carbone s'expliquerait par la
diminution corrélative du nombre de points de contact.
Un tel mécanisme impose l'existence d'un transport de l'oxygène et de
l'azote à travers le nitrure formé au voisinage du point de contact. La nature
stoechiométrique et fortement covalente du nitrure d'aluminium ne permet pas
d'envisager ce type de transfert en son sein, ce qui implique un blocage
rapide de la réaction. De plus, la permanence des contacts entre les grains
serait problématique avec la disparition progressive du carbone au fur et à
mesure de sa consommation.

- 41 -
Par ailleurs, la valeur faible du coefficient d'expansion (27) du
nitrure d'aluminium formé sur l'alumine (6 = 0,98) ne justifierait pas le
grossissement des grains par un simple effet volumique lié à la réaction. En
revanche, la tension de vapeur du nitrure d'aluminium aux températures d'étude
est suffisamment élevée (28) pour donner naissance à des processus d'évapora
tion-condensation (24) entraînant des croissances cristallines et des coales
cences de grains.
En l'état, cette hypothèse n'est cependant pas suffisante.
2 - Intervention d'une phase gazeuse issue de l'alumine.
Soulignons tout d'abord qu'aux températures étudiées, de plusieurs
centaines de degrés inférieures à la température de fusion de l'alumine
(2030 0C) et sous atmosphère d'azote, la pression de vapeur considérée ne peut
être que très faible.
La réaction se ramenerait dans ce cas à un modèle de type solide-gaz,
pour lequel la décroissance de la surface réactive du solide consommé, où est
localisée l'étape limitante implique une évolution cinétique classique (23) de
la forme :
F(a)ko
1 - (l_a)1/3 =_e-E/RTx t
oKt
où k représente une constante caractéristique de la réaction, ~ le diamètreo
des grains supposés sphériques, E l'énergie d'activation de la réaction, R la
constante des gaz parfaits et T la température.
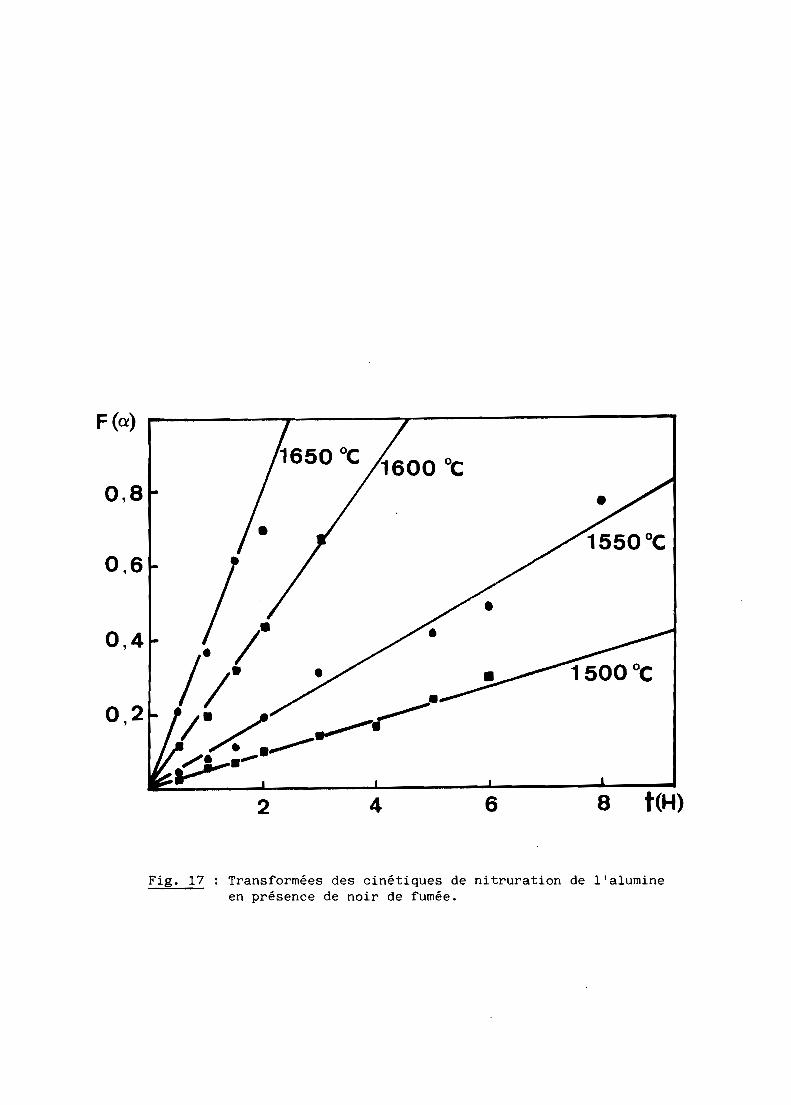
De fait, les isothermes des figures 15 et 16 peuvent être linéarisées
par une transformation F(a) en fonction du temps comme le montrent les figures
17 et 18.
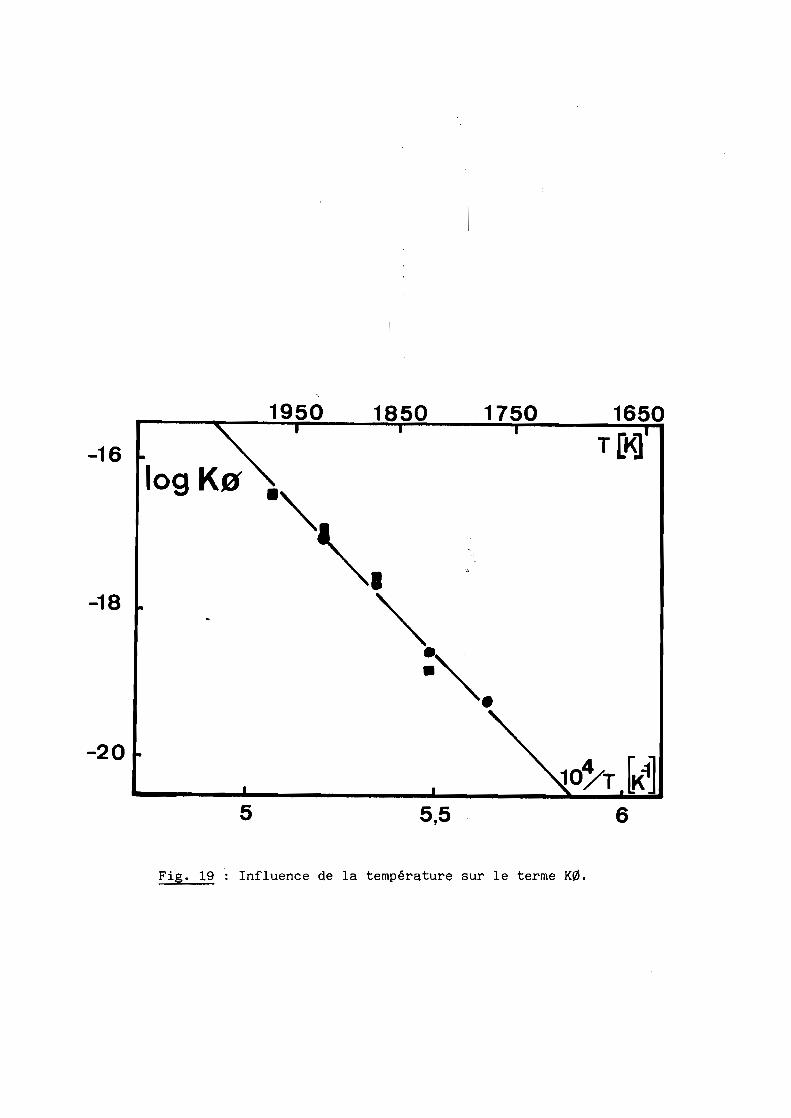
De plus, l'influence du diamètre initial des grains que traduit
l'expression:
ko
-E/RTe
peut être vérifiée sur la base de nos deux séries d'essais obtenues à partir
de graphite (01 ) et de trois de carbone (~2). Les variations de K~ en fonction
0,8
0,6
0,4 . je ./ ja .
0,2/~ .--.--. .~",. ".~.
2 4 6 8 t(H)
f(a)
Fig. 17 Transformées des cinétiques de nitruration de l'alumineen présence de noir de fumée.
t(H)6543
-e----'i55-----.-21
0,8
0,6
0,4
0,2
F(a)
Fig. 18 Réseau des transformées des isothermes de la nitruration del'alumine (mélanges avec le graphite).

- 44 -
de l'inverse de la température sont bien linéaires (fig. 19) et conduisent à-1
une même énergie d'activation: E = 426± 29 KJ.Mol ,valeur en accord avec
celle obtenue directement à partir des vitesses instantanées.
Ainsi donc, la réaction solide-gaz envisagée serait localisée à la
surface des grains de carbone selon un mécanisme unique quelle que soit la~nature ou la granulométrie du carbone.
Mais un tel modèle impliquerait aussi que la formation de la phase soit
relativement plus rapide que sa consommation ; le réacteur étant ouvert, il
serait donc surprenant de ne pas observer de perte de matière par
volatilisation dans le four. En outre, le problème de la barrière de diffusion
que constitue le nitrure d'aluminium fèrmé à la surface du carbone se repose
rait en des termes identiques à ceux mentionnés plus haut. Enfin, la phase
A10N- y et a fortiori sa transformation ultérieure en nitrure trouverait
difficilement sa justification dans un tel modèle.
Cette hypothèse n'est donc pas non plus totalement satisfaisante.
3 - Intervention d'une phase gazeuse issue du carbone
Cette hypothèse est assortie des mêmes réserves que la précédente quant
à ses conditions d'existence et à son importance relative dans l'atmosphère
entourant les grains d'alumine. Le modèle solide-gaz défini plus haut reste là
encore valable.
En inversant simplement les rôles du carbone et de l'alumine par rapport
au cas précédent, on se heurte au même problème d'entraînement de la phase
gazeuse réagissante et l'on ne justifie plus la diminution de la vitesse de
réaction puisque le diamètre des grains à prendre en compte dans l'expression
de la transformée F(a) est celui des grains d'alumine qui demeure constant.
En revanche, si l'on se place dans l'hypothèse o~ l'étape la plus lente
est celle de la formation d'une phase gazeuse issue du carbone, sa consomma
tion étant très rapide sur les grains d'alumine voisins, on comprend qu'il n'y
ait pas de pertes de matière. La vitesse de la réaction est alors gouvernée
par l'évaporation de la phase formée sur le carbone, cas classique en cinéti
que hétérogène qui se résoud analytiquement par la même transformée F(a)
utilisée précédemment. En conséquence, la validité de la représentation
1850 1750 1650
-16 T [t<]
."".-18 -.~•
-2010~T [.<1
5 5,5 6
Fig. 19 : Influence de la température sur le terme K0.

- 46 -
d'Arrhénius de la figure 19 demeure acquise ainsi que ses implications en
terme d'unicité du mécanisme quelle que soit la granulométrie du carbone.
Ce mécanisme expliquerait aussi que la formation du nitrure d'aluminium
sur l'alumine soit sans incidence cinétique puisque non localisé sur l'inter
face où se produit l'étape limitante (carbone/gaz). Ainsi, l'éventuel
processus d'évaporation-condensation et le frittage des grains justifiant leur
grossissement n'auraient-ils aucune influence cinétique, et de même pour la
formation d'oxynitrure par réaction du nitrure avec l'alumine aux températures
les plus élevées.
Ce mécanisme, fondé sur l'existence hypothétique d'une phase volatile
issue du carbone est le seul qui semble assurer une certaine cohérence à
l'ensemble des résultats expérimentaux.
E - CONCLUSION
Dans la perspective de l'application de la carboréduction nitrurante de
l'alumine à la synthèse de l'oxynitrure d'aluminium, l'approche cinétique du
mécanisme de la réaction apporte donc les éléments suivants :
1 - L'oxynitrure se forme préférentiellement au nitrure et apparaît
comme intermédiaire de réaction aux températures supérieures à 16000C. C'est
là un élément très favorable à cette voie de synthèse de la phase A10N, le
risque de subsistance du nitrure après réaction devant être écarté.
2 - Une attention toute particulière doit être apportée au carbone et
spécialement à sa granulométrie qui joue un grand rôle dans la vitesse de la
réaction. Il conviendra donc de retenir le noir de fumée pour la suite de
l'expérimentation.

Chapi tre III
PREPARATION DE L'OXYNITRURE D'ALUMINIUM y PAR
CARBONITRURATION D'ALUMINE
Il s'agit d'obtenir directement un oxynitrure de composition donnée
A12
03 3 N par carboréduction de l'alumine en atmosphère d'azote. Compte-x - x x
tenu des résultats du chapitre II, la suite de réactions conduisant à cet
oxynitrure peut s'écrire
x 3x x 3(1- -) A1
20 3+ - C + - N -+ (1 - x) A1
203 + x A1N + -x CO2 2 2 2 2
3+ - x CO
2
En désignant par C la teneur pondérale du carbone introduit initialement
dans le mélange avec l'alumine, on tire:
18xC =-----
102 ;- 33x
Ainsi, pour un oxynitrure de composition équivalente à celle d'un
mélange A1 203/A1N comportant 27% en mole de nitrure (cf. chap. 1), les
conditions stoechiométriques initiales imposées correspondent à des propor
tions pondérales de 5,22 et 94,78% respectivement pour le carbone et
l'alumine.

- 48 -
Les mélanges répondant à cette composition et utilisant les mêmes
matières premières que précédemment (chapitre II) sont homogénéisées à sec en
mortier d'agate, puis en mélangeur Turbula en récipient de matière plastique
pendant 6 à 12 heures. La poudre est alors transvasée dans un moule en
graphite aux parois recouvertes de nitrure de bore, et l'ensemble porté à
température donnée pour des durées variables.
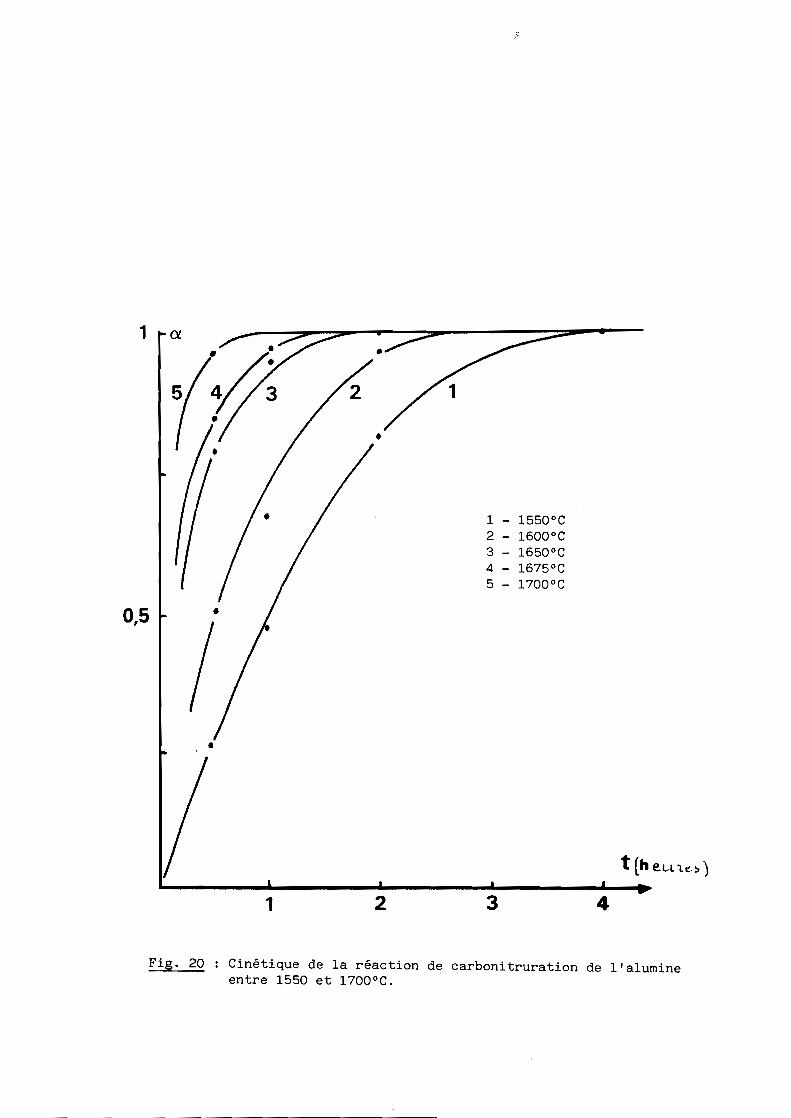
A - COMPORTEMENT CINETIQUE
La marche de la réaction a été suivie dans les mêmes conditions que
précédemment (chapitre II) aux températures 1650, 1600, 1650, 1675 et 1700 0C
pour des temps de palier compris entre 0,5 et 8 heures. L'évolution cinétique
isotherme est reproduite à la figure 20, l'avancement étant encore repéré par
la consommation du carbone.
Comme on pouvait s'y attendre, les résultats obtenus présentent beaucoup
d'analogies avec ceux observés dans les mêmes conditions de température
(chapitre II) mais en présence d'un excès de carbone.
Là encore, il apparaît seulement le nitrure AIN jusqu'à 1600 0C et c'est
au-delà de cette température que se forme l'oxynitrure d'aluminium qui devient
prépondérante en l'absence d'un excès de carbone.
A ce plan cinétique, on vérifie que l'allure des isothermes n'est pas
modifiée au passage à 16000C par la formation de l'oxynitrure, ce qui implique
une étape limitante de réduction de l'alumine par le carbone avec formation de
nitrure suivie d'une étape rapide de dissolution de ce nitrure dans l'alumine
résiduelle, comme le montre d'ailleurs la quasi absence d'AIN dans les
spectres de rayons au cours de la réaction.
On observe par ailleurs un important grossissement des grains à toute
température comme l'attestent les micrographies de la figure 21 obtenues à
1550, 1650 et 1750 0C pour des paliers de 3 heures.
B - PROCEDE DE SYNTHESE RETENU
La qualité du mélange de départ étant importante pour éviter les
hétérogénéités locales génératrices de nitrure difficile à éliminer par la
suite, il faut mélanger soigneusement l'alumine et le noir de fumée d'abord au
mortier puis au mélangeur Turbula pendant au moins quatre heures. Les propor
tions retenues sont 5,22% pour le carbone 94,78% pour l'alumine comme justifiéprécédemment.
1 Œ
0,5
•
•
•
1 - 1550°C2 - 1600°C3 - 1650°C4 - 1675°C5 - 1700°C
1 2 3 4
t (h e.L.l U.:> )
Fig. 20 Cinétique de la réaction de carbonitruration de l'alumineentre 1550 et 1700°C.

a
/ .
b
c
Fig. 21 Observations microscopiques du produit de la réaction de carbonitrurationde l'alumine après 3 heures à 15500C (a) 1650°C (b) et 17500C (c).

- 51 -
En ce qui concerne le traitement thermique, la solution simple consis
tant à opérer à température élevée (1700 0C par exemple) où se forme seul
l'oxynitrure n'a pas été retenue, car on obtient un grossissement et un frit
tage exagérés des grains qui nuisent à la qualité finale du produit, alors
difficile à densifier à 100% (sans doute le grossissement des grains
entraîne-t-il la création d'une porosité intragranulaire qui s'élimine
difficilement) ; le fritté obtenu est par ailleurs gris ou noir, témoin peut
être de la présence de traces de nitrure.
Pour pallier ces inconvénients, on procède en deux temps. D'abord à une
température de 1600 0C pour laquelle les grains conservent une taille raison
nable, et pendant environ trois heures pour s'assurer de l'intégralité de la
réaction de formation du nitrure qui se trouve alors en présence d'alumine
dans les proportions molaires requises AIN/AI203
= 27/73. Puis on élève la
température jusqu'à 18500C afin d'assurer la synthèse de l'oxynitrure. Une
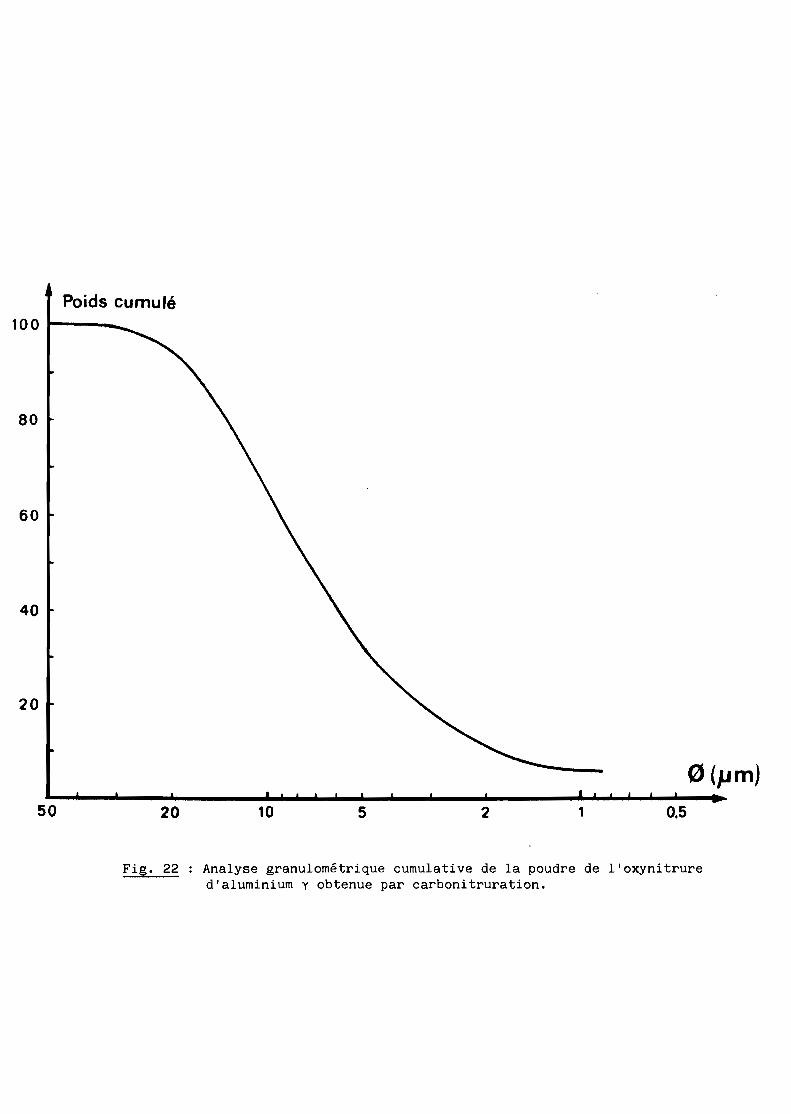
durée de trois heures dans ces conditions permet d'obtenir un produit
homogène, très blanc et très friable qu'un simple broyage rapide au mortier
suffit à réduire en une poudre dont les caractéristiques granulométriques
(valeur moyenne 7 ~m) et morphologiques sont représentées respectivement sur
les figures 22 et 23.
Poids cumulé100 ......---
80
60
40
20
o (,um)50 20 10 5 2 1 0.5
Fig. 22 Analyse granulométrique cumulative de la poudre de l'oxynitrured'aluminium y obtenue par carbonitruration.

Fig. 23 Observation au MEE d'oxynitrure d'aluminium de couleur blancheobtenue par carbonitruration.

Chapitre IV
ELABORATION DU MATERIAU TRANSPARENT
Trois méthodes de densification classiques peuvent être utilisées pour
l'oxynitrure d'aluminium:
- le frittage naturel après synthèse de la poudre,
- le frittage réaction qui permet d'obtenir des pièces de
dimensions désirées mais qui présentent une certaine porosité non négligeable,
- le frittage sous charge qui conduit à des pièces bien densi
fiées, souvent sans porosité.
Divers procédés ont été brevetés (3, 4) mettant en oeuvre les deux
premières méthodes mais, soit ils aboutissent à un produit très poreux donc
seulement translucide (3) par la méthode du frittage-réaction, soit ils
nécessitent l'adjonction d'ajouts le plus souvent néfastes aux propriétés
mécaniques, notamment en frittage naturel (4), qui ne peut d'ailleurs garantir
l'absence totale de porosité. C'est pourquoi nous avons tenté d'utiliser le
frittage sous charge dont l'usage devient de plus en plus fréquent dans
l'industrie et ne constitue plus un obstacle insurmontable au développement
d'un produit.
Nous avons cherché à mettre en évidence le rôle des paramètres de
frittage comme le temps de palier, la température et la pression, en fixant
par exemple deux de ces paramètres et en faisant varier le troisième.

- 55 -
A - OPTIMISATION DES CONDITIONS DE FRITTAGE
Il s'agit de rechercher ici, dans une perspective appliquée, les meil
leures conditions du frittage sous charge visant à éliminer le plus complè
tement possible la porosité.
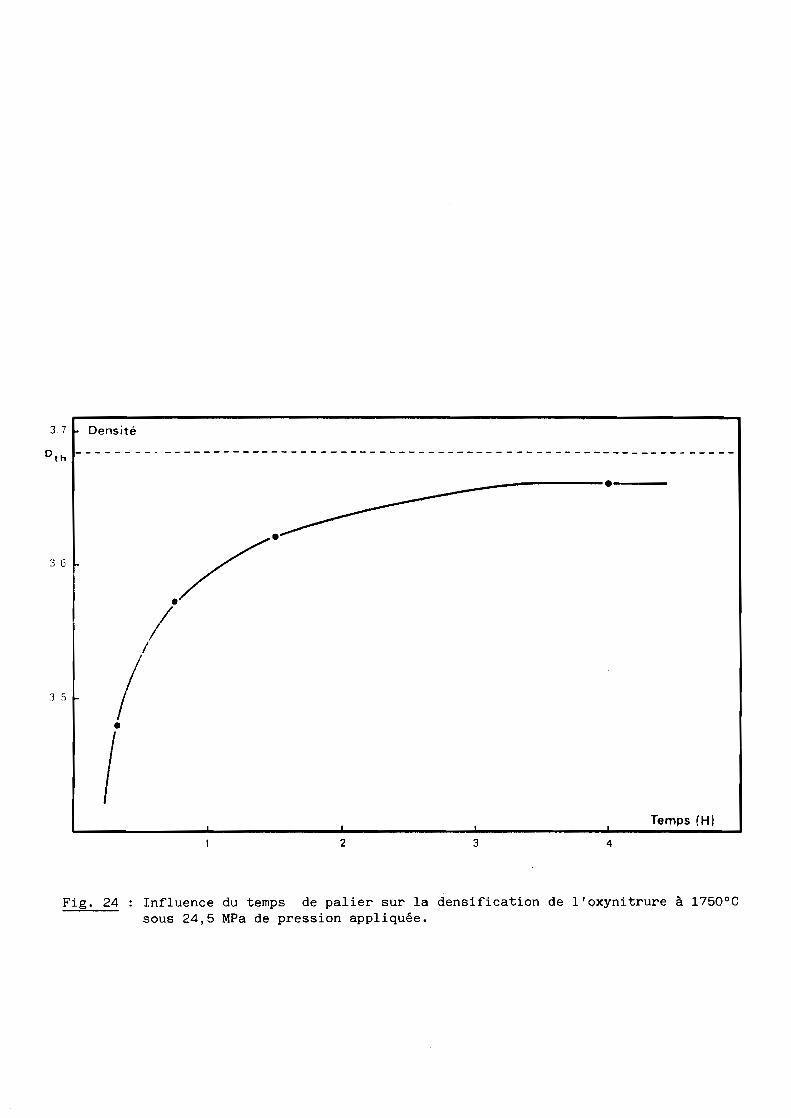
1 - Influence du temps de palier
La température et la charge appliquée restant constantes (l750°C et
24,5 MPa respectivement), nous avons fait varier le temps de palier de 20
minutes à 4 heures.
Les résultats représentés à la figure 24 montrent que le taux de
densification s'accroît avec la durée du palier pour se rapprocher de la
densité théorique, soit 3,68 environ pour la composition retenue (cf. 5ème
Partie).
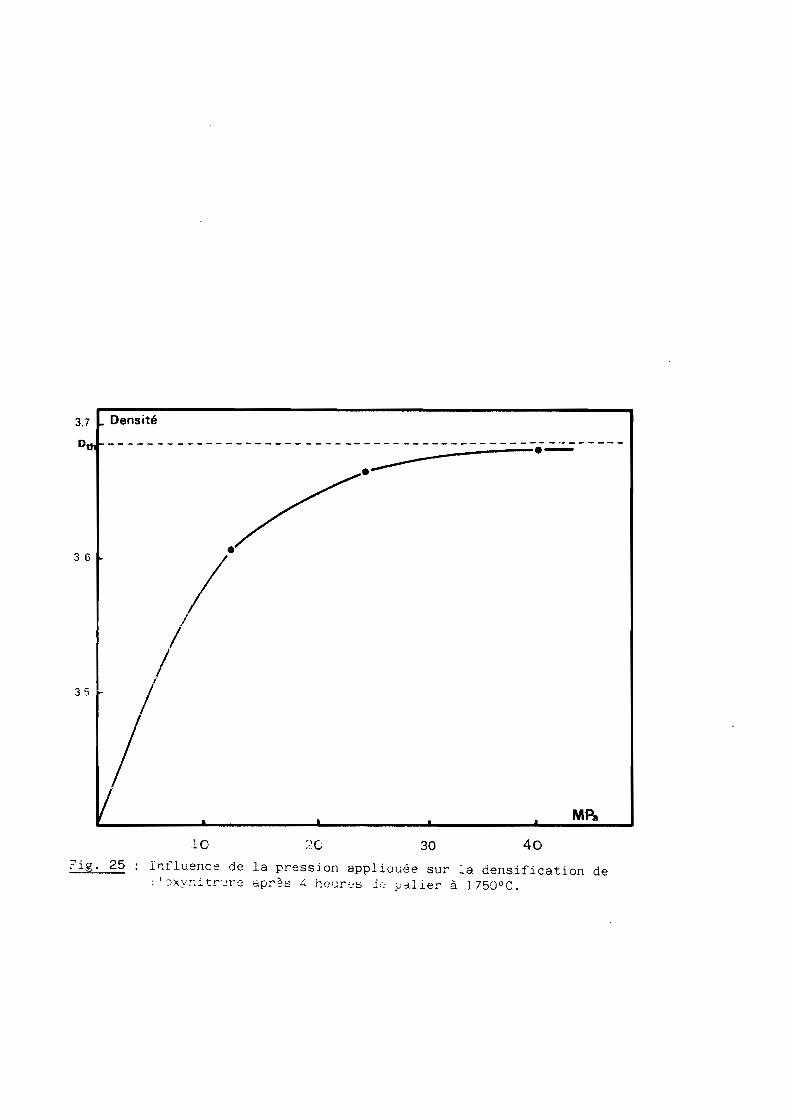
2 - Influence de la charse
Nous l'avons étudiée en fixant cette fois-ci la température et le temps
de palier respectivement à l7500C et 4 heures. Là encore, on observe (cf. fig.
25), une augmentation du taux de densification avec la pression et ce jusqu'à
la limite à ne pas dépasser (40 MPa) compte tenu de la résistance mécanique du
moule de graphite utilisé pour la réalisation des frittés.
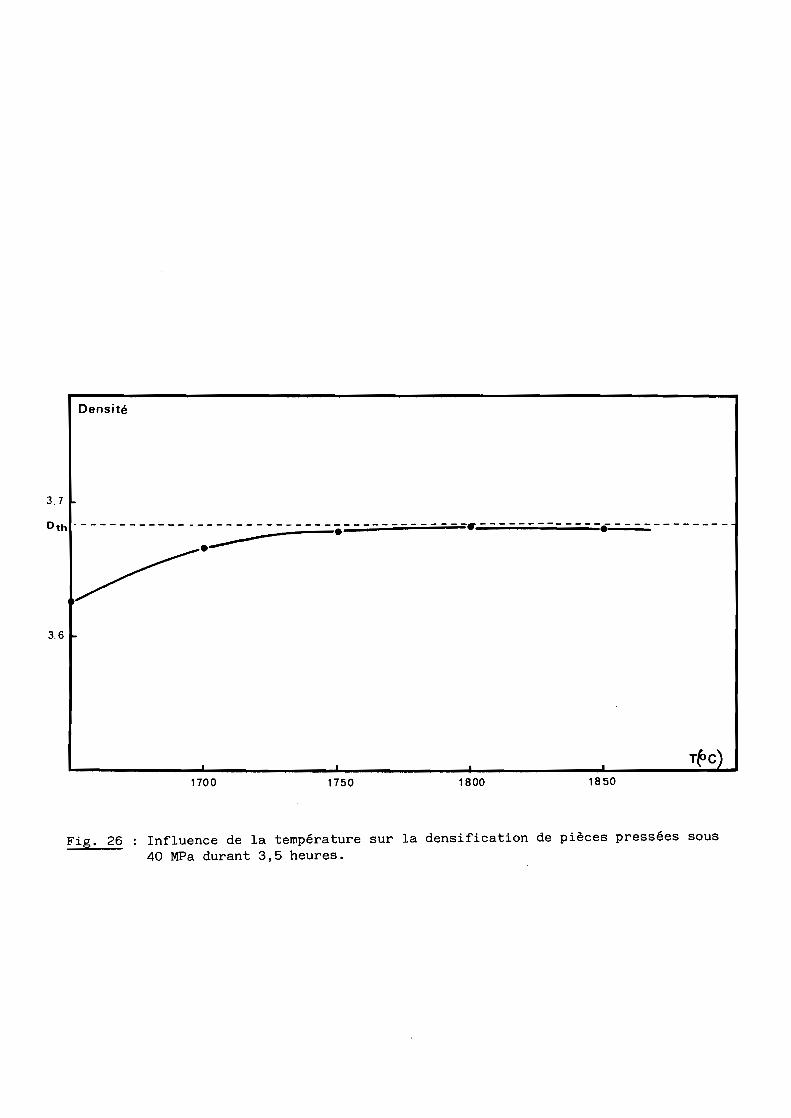
3 - Influence de la température
L'influence de la température pour une durée de traitement thermique de
3,5 heures et une pression de 40 MPa, est illustrée par la figure 26.
On observe une augmentation sensible du taux de densification avec la
température jusqu'à l750oC, suivie d'une très faible variation et même d'une
légère décroissance à l8500C attribuable à un grossissement de grains
accompagné de porosité intergranulaire, comme le confirment les résultats du
tableau VII et l'examen microscopique.
3 7 Densité
__-------e---
] (j /0/
1
/•
12 3 4
Temps (Hl
Fig. 24 Influence du temps de palier sur la densification de l'oxynitrure à 17500C
sous 24,5 MPa de pression appliquée.
3.7 Densité
o -- -~-------------------------------------------------- .-
36
35
MPa
LO 70 30 40Fig. 25 Influence de la pression appliauée sur la densification de
~l~xy~~tr~re après 4 heilr~s J0 palier à l'7500C.
Densité
3. 7
Dth . - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
~.-- .3.6
----.-------------.------------
TfC
1700 1750 1800 1850
Fig. 26 Influence de la température sur la densification de pièces pressées sous40 MPa durant 3,5 heures.

- 59 -
Température Taille moyenne des grains 1
1 (en °C) 1 (en um) 1
1---------------------------------1-----------------------------------11 1750 1 15 1
1---------------------------------1-----------------------------------11 1800 1 50 1
1---------------------------------1-----------------------------------11 1850 1 70 1
1 1 1
Tableau VII - Taille des grains observés en fonction de la température
de frittage sous 40 MPa pendant 3,5 heures.
En définitive, il résulte de cette étude que la température optimale de
frittage de l'oxynitrure d'aluminium est de 1750 0C environ. Par ailleurs, le
temps de palier et la charge doivent être aussi grands que possible, 3,5
heures à 4 heures et 40 MPa respectivement, c'est à dire la pression maximale
tolérable par les moules en graphite utilisés.
De tels résultats sont particulièrement intéressants car ils permettent
d'affirmer que le frittage sous charge de l'oxynitrure donne accès à des
pièces possédant sensiblement la densité théorique, sans ~aire appel pour
autant à des ajouts, et en garantissant une bonne reproductibilité.
B - CINETIQUE DE GROSSISSEMENT DES GRAINS
En dehors des conditions de pureté et de densification optimales du
matériau, la transparence résulte d'un processus de grossissement des grains
par traitement thermique post-frittage à température élevée. On cherche
principalement, dans cette étape, à réduire le nombre des joints de grains qui
constituent autant d'interfaces nuisibles à la propagation des rayons
lumineux.
D'après les expériences préliminaires effectuées, la zone de température
donnant des résultats intéressants se situe entre 1930 et 1980oC, les échan
tillons subissant une volatilisation sensible à partir de 2000 oC.

- 60 -
1 - Rappels théoriques
Il Y a un double intérêt à suivre le grossissement des grains, du point
pratique d'abord pour le choix du traitement thermique le plus adapté, et au
plan théorique aussi pour définir les processus diffusionnels qui gouvernent
la migration des joints de grains.
Certains facteurs comme la pureté du matériau, la densité, la porosité,
la forme et la matière des joints de grains jouent un rôle essentiel dans le
processus de grossissement de ces grains.
Dans le cas de la croissance idéale, on admet généralement (30 à 37) que
la vitesse de migration des joints de grains est directement proportionnelle à
la force motrice F selon l'équation
dGMF
dt
où G est le diamètre moyen des grains et M la mobilité du joint (constante
caractéristique du système).
Plusieurs cas peuvent être dès lors envisagés. Suivant que l'on a
affaire à un système pur monophasé ou à un système monophasé avec impuretés.
a - Système pur monophasé
La force motrice F de migration des atomes correspond au gradient de
potentiel chimique ~ provenant de la différence de pression de chaque côté du
joint de grain. Elle s'exprime par la relation (30) :
3d~, a S 1
F=-=-.-dx k~ W
3 •a représentant le volume atomique, S et Wsont respectivement l'énergie et
l'épaisseur des joints de grains, cr la taille moyenne des grains et k, une
constante dépendant de la température. Dans ces conditions, le flux J des
atomes est de la forme
1J =
a3
dG 1 1-=-MF=dt a3 a3 kT kGW

- 61 -
où D exprime le coefficient de diffusion au niveau de joint de grain. La
vitesse de migration du joint de grain prend alors pour expression:
dG--=dt
3a
D.-
kT
S
kGW
Equation différentielle à variables séparées (G, t) dont l'intégration
conduit à la relation:
soit
-2 -2G - Go
3 D S= 2a • -.-.t
kT kW
-2 -2G - Go = Kt
où G représente la taille initiale des grains.o
b - Système monophasé avec impuretés
Les impuretés supposées en solution solide s'agglomèrent et empêchent le
joint de grain de se déplacer. On montre dans ce cas (30) que le grossissement
des grains suit une loi cinétique du type:
En définitive, la croissance générale des grains peut s'exprimer par une
loi générale de la forme :
n représentant un exposant qui dépend de la nature des phénomènes.

- 62 -
2 - Expérimentation et résultats
Les échantillons frittés dans les conditions optimales précédemment
définies sont portés à température donnée pour des durées variables, dans un
moule en graphite contenant du nitrure de bore pulvérulent.
Après traitement, on procède à un polissage au diamant pendant un quart
d'heure suivi d'un polissage à l'alumine pour mettre en relief les grains par
usure différentielle selon l'orientation cristallographique (figure 27).
La mesure de la taille moyenne G des grains se fait par comptage du
nombre N de joints dans une direction donnée de longueur L suivant la formule:
Lcr = 1,15 ---
N - 1
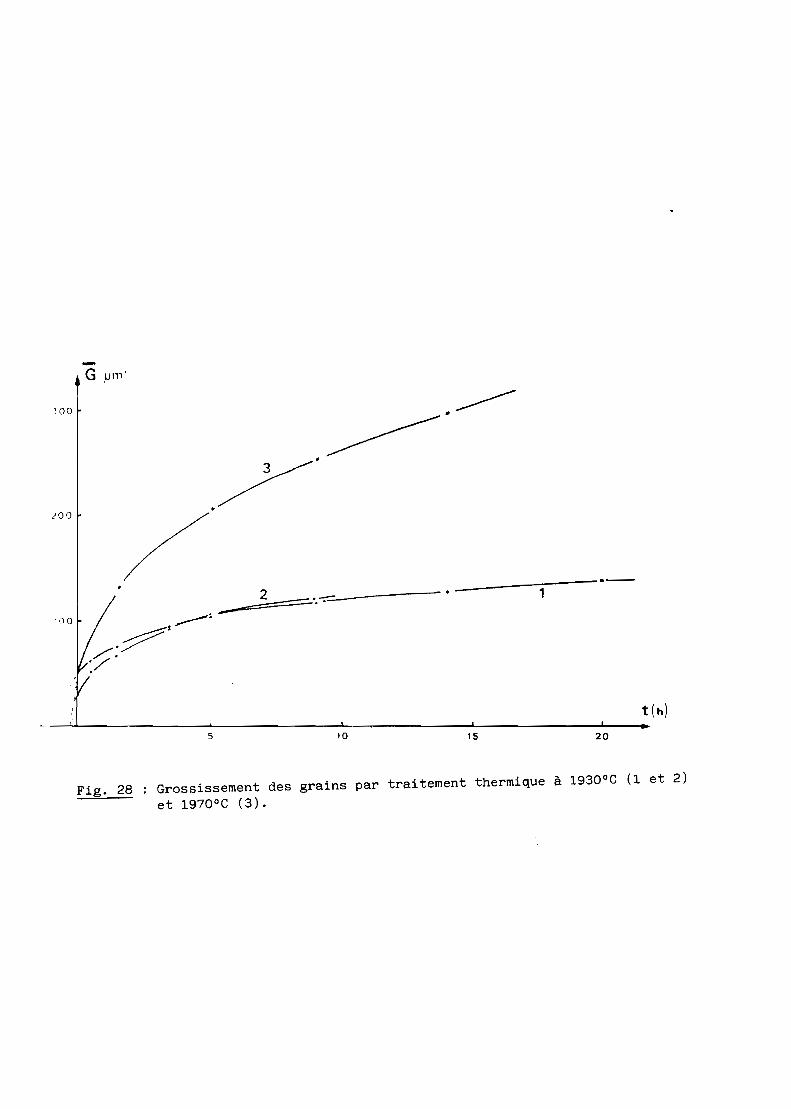
Les résultats obtenus à 1930 puis à 1970°C pour des durées variables de
30 minutes à 20 heures sont consignés au tableau VIII et représentés à la
figure 28.
Temps (en heures) 1
________1
1 Températures (en OC) 1 0 10,5 1 1,5 1 3,5 1 5 1 9 1 14 1 20 11----------------------------1-----\-----1-----1-----1-----1-----1-----1-----11 1930 1 48 1 63 1 77 1 95 1 102 1 117 1 130 1 140 11----------------------------1-----1-----1-----1-----1-----\-----1-----1-----11 1930 1 24 1 56 1 66 1 96 1 107 1 120 1 1 11----------------------------1-----1-----1-----1-----1-----1-----1-----1-----11 1970 1 24 1 1 135 1 1 210 1 255 1 300 1 11 1_1_'_1_1_1_1_1_1
Tableau VIII - Taille des grains (en microns) en fonction de la
température et du temps.
L'analyse de ces résultats selon la loi puissance mentionnée plus haut
conduit aux valeurs de l'exposant n et de la constante K rapportés au tableau
IX à chaque température.

Fig. 27 Aspect des grains observés après polissage sur une pièce transparenteaprès 14 heures de traitement thermique à 1970°C.
300
lOO
G /Jrn
/.o o
1
Fig. 28 Grossissement des grains par traitement thermique à 19300C
(1 et 2)
et 1970 0C (3).

- 65 -
1 Température (OC) 1 n 1 K x 1000 1
1--------------------1-------------1-------------\1 1930 (1) 1 3,2 1 420 1
1--------------------1-------------1-------------11 1930 (2) 1 2,8 1 75 1
1--------------------1-------------1-------------11 1970 (3) 1 2,7 1 330 1
1 1 1 1
Tableau IX - Constantes caractéristiques de la cinétique de grossis
sement des grains obtenues à partir des résultats de la figure 28.
Il s'ensuit une loi cinétique d'évolution des grains de la forme
Ce qui nous amène à conclure au cas d'une phase contenant des impuretés
ou, plus vraisemblablement, en égard à la nature de la phase AIONy, à l'exis
tence de variations locales de composition au niveau des joints de grains.
C - TRANSPARENCE ET TRAITEMENTS THERMIQUES POST-FRITTAGE
Une bonne transparence (cf. 5ème Partie), pour des échantillons de
plusieurs millimètres d'épaisseur, s'obtient lorsque la taille moyenne des
grains est comprise entre 200 et 300 ~m.
D'après les précédents résultats, ceux notamment de la figure 28, la
transparence est donc conditionnée par un traitement thermique de 6 heures à
une température de 1970oC. Mais à cette température, la volatilisation de
l'oxynitrure d'aluminium n'est pas négligeable et, pour, l'éviter, on a intérêt
à opérer à plus basse température, ce qui implique malheureusement un recuit
de plus longue durée. Dans la pratique, nous avons choisi finalement une
température de 19500C qui, maintenue pendant 15 heures, conduit à des
résultats satisfaisants comme l'illustre la figure 29.
En dehors de cette étape indispensable de traitement thermique post
frittage, la transparence est aussi conditionnée par un polissage parfait, de
même que par la planéité et le parallélisme des deux faces de la lame. Un tel
polissage doit être pratiqué à la pâte diamantée car les autres abrasifs
(alumine, carbure de silicium) sont de dureté comparable et, par une usure des
grains suivant des orientations cristallographiques privilégiées, créent des
irrégularités de surface.
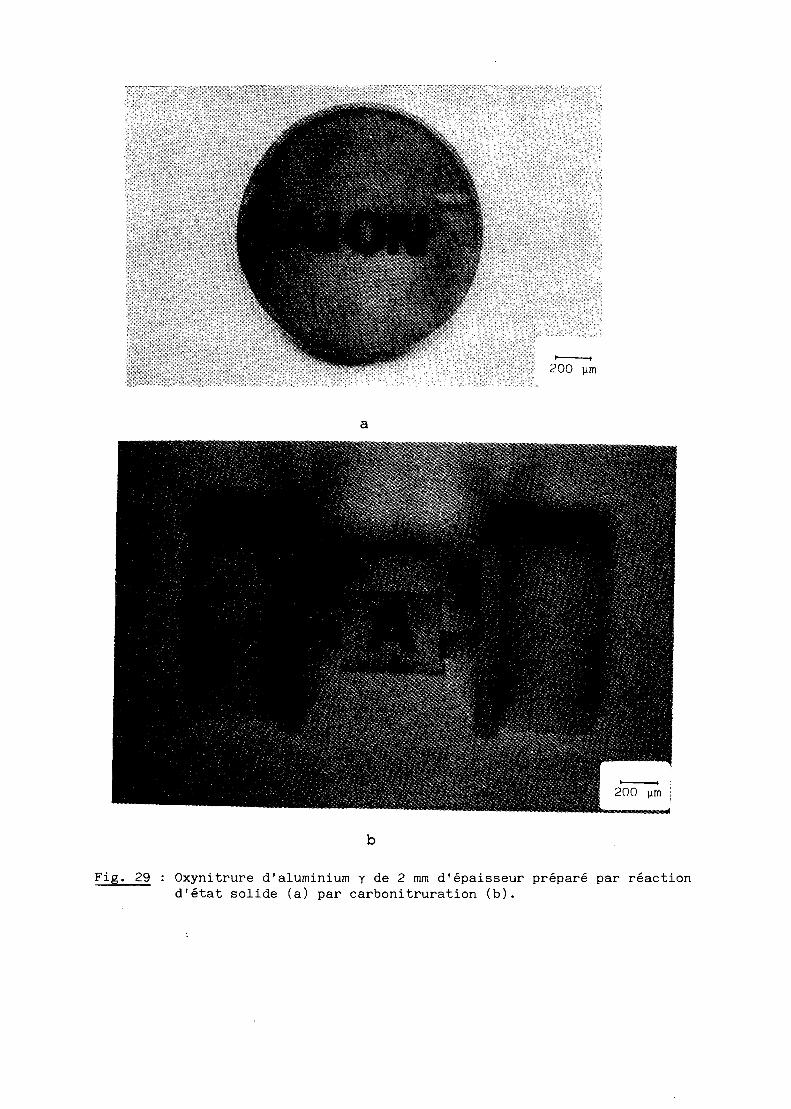
Fig. 29
......--..-.200 Ilm
a
--200 Ilm
b
Oxynitrure d'aluminium Y de 2 mm d'épaisseur préparé par réactiond'état solide (a) par carbonitruration (b).
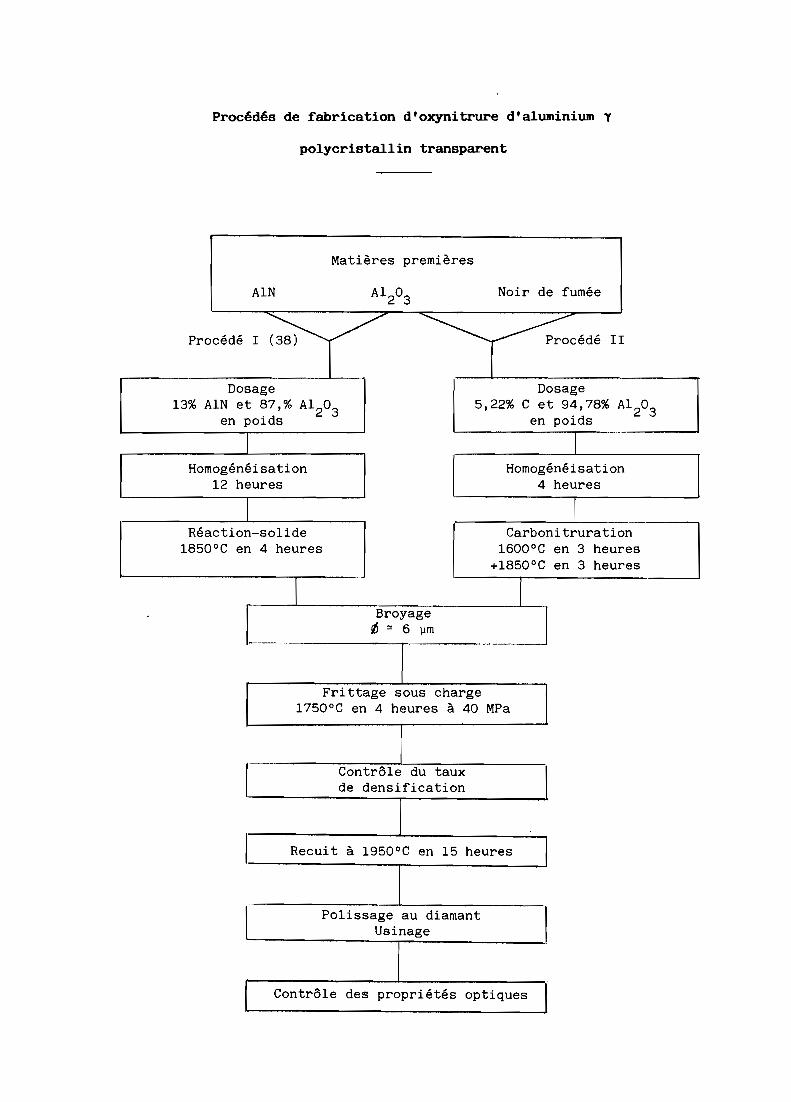
Procédés de fabrication d'oxynitrure d'aluminium Y
polycristallin transparent
Matières premières
AlN Al203
Noir de fumée
Procédé l~/ ~ Procédé II
.Dosage Dosage
13% AlN et 87,% Al203
5,22% C et 94,78% Al203
en poids en poids
1 THomogénéisation Homogénéisation
12 heures 4 heures
1 r
Réaction-solide Carbonitruration1850 0C en 4 heures 1600 0C en 3 heures
+1850 oC en 3 heures
1
_.
1
[ Broyage-1~ '" 6 \lm
Frittage sous charge1750 0C en 4 heures à 40 MPa
Contrôle du tauxde densification
Recuit à 1950 0C en 15 heures
Polissage au diamantUsinage
Contrôle des propriétés optiques

FRITTAGE SOUS CHARGE DE L'OXYNITRURE

FRITTAGE SOUS CHARGE DE L'OXYNITRURE
Nous nous proposons de définir ici les mécanismes du frittage sous
charge de l'oxynitrure d'aluminium qui constitue l'une des étapes de l'élabo
ration du produit transparent.
A - MONTAGE ET METHODES EXPERIMENTALES
1 - Dispositif de frittage
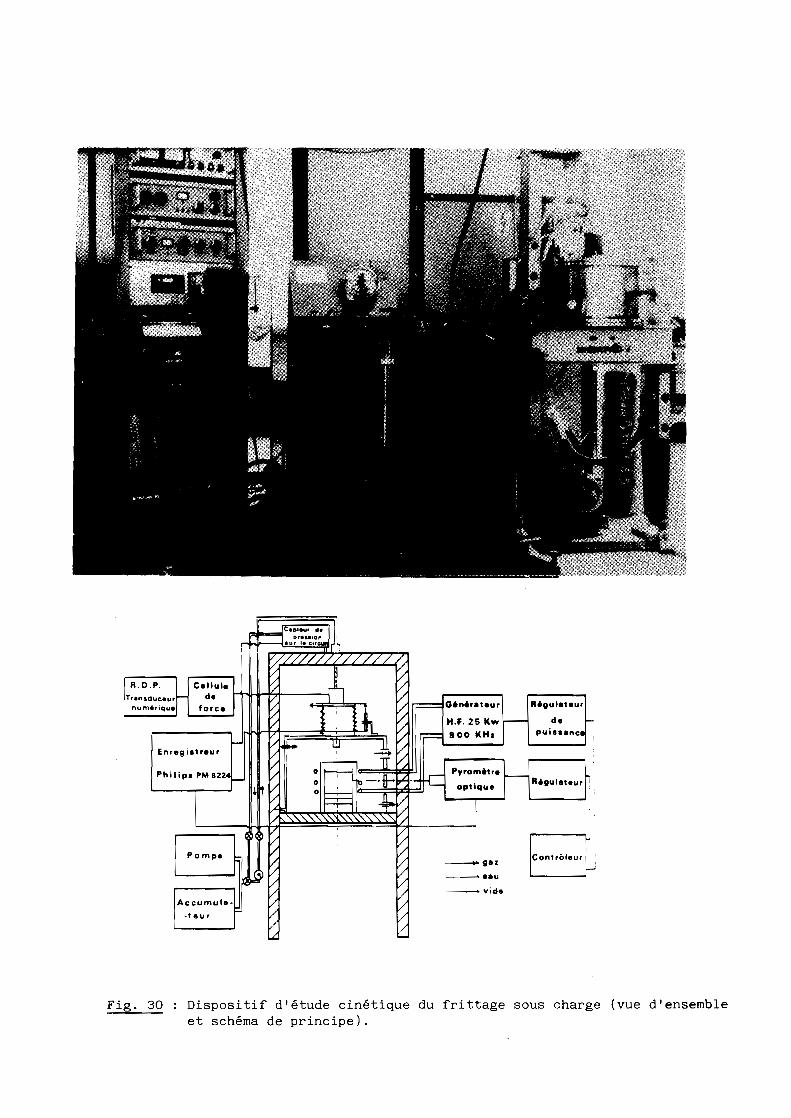
L'équipement utilisé présenté en figure 30 comprend pour l'essentiel un
four haute fréquence opérant sous atmosphère contrôlée, un dispositif de
pressage uniaxial simple effet, et les différents appareils de mesure de
température (pyromètre optique) et de pression.
a - Le four
C'est un four à induction pourvu d'un générateur statique ELPHIAC de
25 KW à fréquence de 900 KHz. Le moule cylindrique en graphite, qui renferme
la poudre à densifier, est placé au centre de la spire parcourue par le
courant haute fréquence ; ses diamètres extérieur et intérieure sont respecti
vement égaux à 70 et 15 mm.
Pour limiter les pertes de chaleur, le moule est entouré d'écrans
thermiques : un feutre de zircone, un tube de silice et, en fermeture, un
chapeau en ciment réfractaire.
La détection et l'enregistrement de la température sont effectués par
l'intermédiaire d'un pyromètre optique IRCON en visée sur le moule et relié à
un enregistreur à quatre voies PHILIPS-PM 8224.
Cet ensemble atteint 1700 0C en 20 minutes environ.
1 1
HC'Pl'U';~Jl' .u~'~:~~~" i1
V////// '//////
~ ~r/
R,D.P. Cellule t- ~T'.nsduce",1- denum'rique force ~ ~
l/ ;:::= G'n'r.'eur R'll u l.,eur,
~f/ H.F. 25 Kw >-- de h~
-;:= 900 KHI pui •••net1-
~ il- ~ -A 1
E.nregistreu r
f/ r-i-- 11' 1/ :Philip. PM 822<4 1/ a~
ID --fi-1,.1 l'-tc Pyrom'''e 1---R'llul.'.ur hf- a ,
~ l/ a r---:- optique
\
l/ .1/R=1l/ ,'\.-, -, ,'\."1.'\ ,'\.",,'\.
~ ~Pompe 1/ Contrôleur
~ ~-------toI> gaz ~.
W 1/ l/-.au
1/ l/ _vÎdeAc cu mu 1.- 1/ 1/
-t e ur V 1/1/~:..::
Fig. 30 Dispositif d'étude cinétique du frittage sous charge (vue d'ensembleet schéma de principe).

- 71 -
b - L'enceinte
Le montage comporte une enceinte étanche reliée au vérin par un raccord
souple et constituée par une cuve d'acier double paroi de 340 mm de diamètre
et de 240 mm de hauteur. L'ensemble est relié à une pompe primaire assurant un
vide de 10-2 torr. Une circulation d'eau permet le refroidissement de
l'enceinte. Une fenêtre de quartz est ménagée pour la visée pyrométrique.
c - La presse
Il s'agit d'une presse hydraulique ENERPAC d'environ 1000 MPa alimentée
par un compresseur délivrant au maximum 70 MPa. La pression exercée sur le
vérin hydraulique est transmise au piston en contact avec la poudre par
l'intermédiaire d'un autre piston en graphite. Un ballon tampon gonflé sous
une pression de 20 MPa est ménagé sur le circuit pour assurer la stabilité de
la pression. Un système de vannes assure la mise en pression instantanée et la
remise de celle-ci sur le circuit retour.
La pression est contrôlée directement sur le circuit hydraulique; celle
réellement appliquée sur l'échantillon est mesurée par un capteur de force
(KYOWA, type LC2TF) entre le premier vérin et le premier piston en graphite.
Un capteur de déplacement, solidaire du vérin, permet d'évaluer constamment le
retrait de l'échantillon.
2 - Mode opératoire
On utilise à chaque essai environ trois grammes d'oxynitrure d'aluminium
pulvérulent obtenu dans les conditions précédemment définies (cf. 2ème
Partie). La poudre, dont les caractéristiques sont rapportées au tableau X,
est introduite dans un moule en graphite dont les parois sont là encore
préalablement recouvertes d'une barbotine de nitrure de bore pour éviter une
éventuelle diffusion du carbone dans le produit. L'ensemble (moule et produit)
est placé à l'intérieur du tube de silice, puis, après avoir fait le vide
primaire dans l'enceinte pendant 15 à 20 min, on introduit l'argon dont la
circulation est maintenue pendant toute la manipulation.

- 72 -
La hauteur initiale de l'échantillon est alors déterminée en appliquant
quelques instants la pression, puis l'ensemble amené à la température de
l'essai en 20 minutes environ. Après équilibre thermique, la pression est
introduite à l'aide du ballon-tampon préalablement "gonflé" et maintenue
constante pendant toute la manipulation (45 minutes environ).
Un enregistreur (PHILIPS PM 8224) permet d'enregistrer en continu, la
pression, la température, le temps et le retrait de l'échantillon. La pression
est enfin relâchée au cours du refroidissement. Le cycle de pression et de
température est présenté à la figure 31.
Le produit fritté est décapé pour éliminer le nitrure de bore superfi
ciel, puis sa densité finale df
est mesurée par poussée hydrostatique dans
l'eau j on a :
où Pl et P2 désignent respectivement les poids de la pastille immergée ou non.
On en déduit le taux de densification en rapportant df
à la densité théorique
dt h,
soit
1: = __
Cette donnée, ainsi que le contrôle de la hauteur finale de l'échantil
lon au palmer de précision, permet de traduire la cinétique de retrait en taux
de densification à tout instant.
3 - Les échantillons
Deux lots de granulométries différentes ont été obtenus comme suit
Lot nO 1 : La poudre préalablement tamisée à 100 '~m est broyée pendant 4
heures j ses caractérisitiques sont rapportées au tableau X.
Lot nO 2 : La poudre est directement tamisée entre 50 et 63 ~m. La
répartition granulométrique effectuée au sédigraphe est représentée sur la
figure 32. La dimension moyenne des grains (agglomérats) est de 42 ~m.
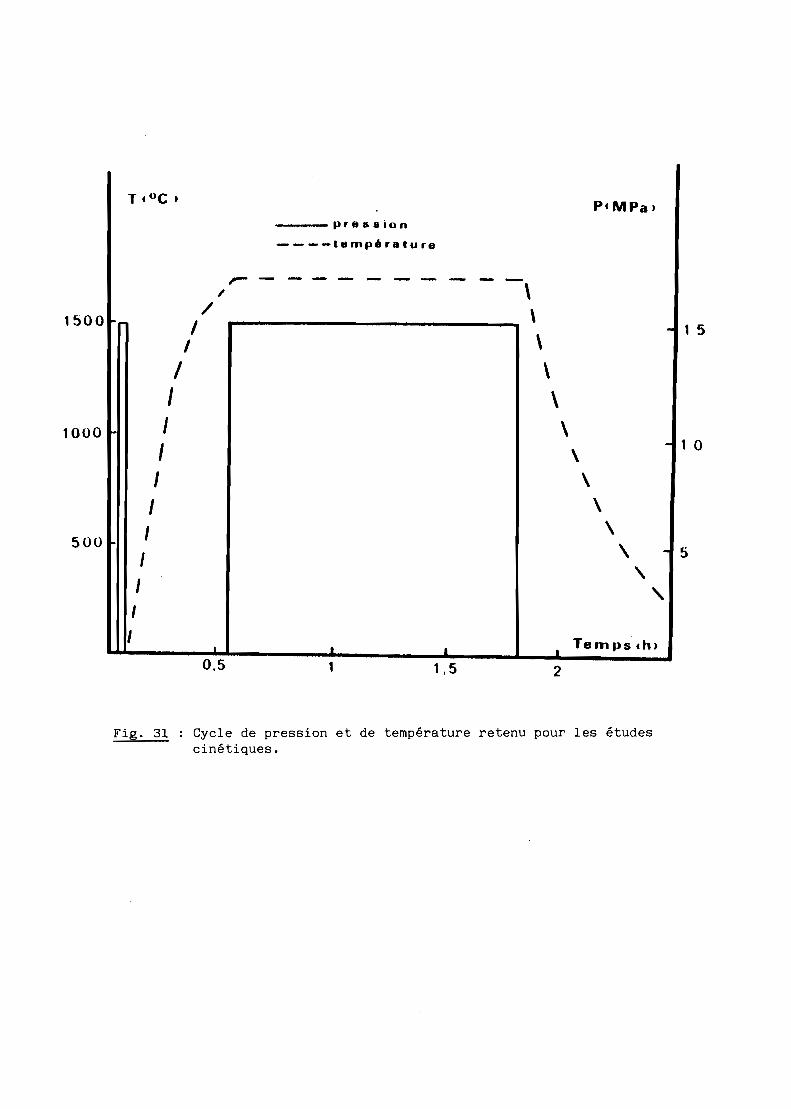
pc MPa»-_-pression
-- --'tt",pérature
------------
- 5
- 1 0
- 1 5
2
. Tem p s ch)
-,\
\\
\\
\\
\\
\ ,'\
t
1.5
t1
0.5
,,/
11
/1/11111111
500
1500
1000
Fig. 31 Cycle de pression et de température retenu pour les étudescinétiques.
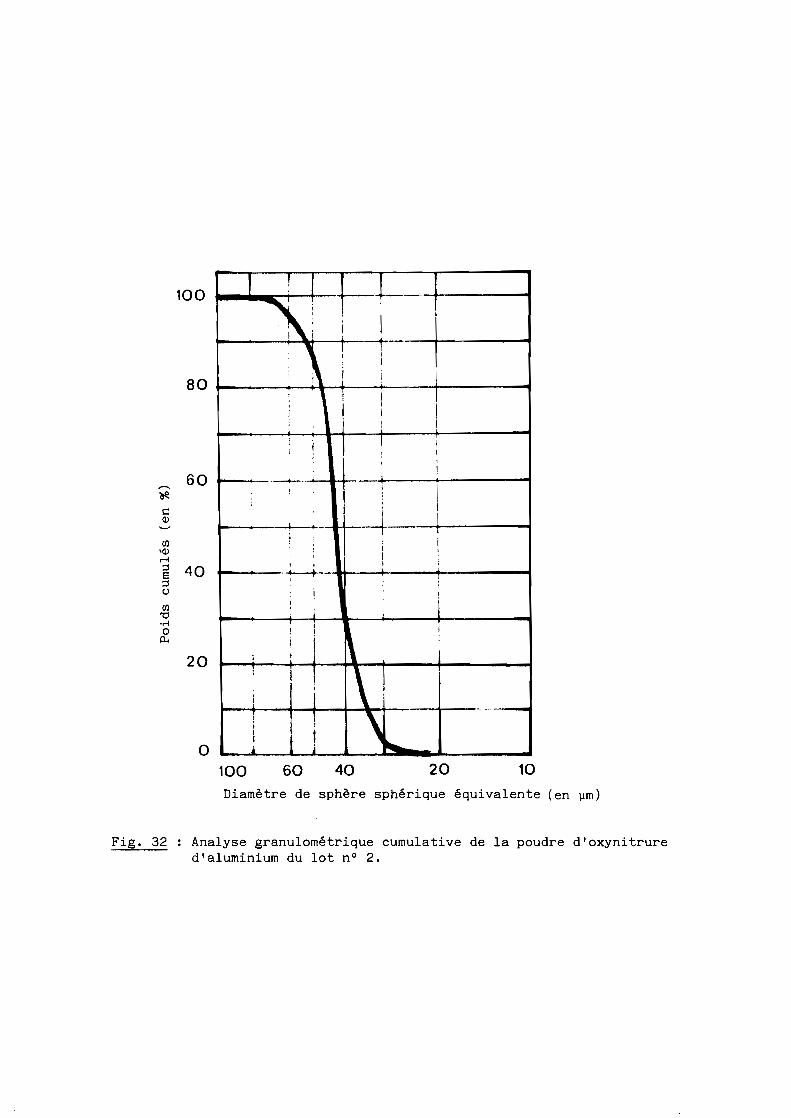
100
80
, '~
. ,
!1
!,i
!1
1
....-----_.+-+---I~._-+---.,....---- .....l ,
t-----.--+------ -i
,rJJ
'Ill.--t::lE::lorJJ"0.,-i
o0..
60
40
20
o100
)111.a
,1
1
60
i
1!1J
1
1:,
H-I
\
40
1
i
1
20 10
Fig. 32
Diamètre de sphère sphérique équivalente (en ~m)
Analyse granulométrique cumulative de la poudre d'oxynitrured'aluminium du lot nO 2.

- 75 -
La surface spécifique de ces deux lots de poudre mesurée par BET (azote)
conduit aux résultats du tableau X.
1 Dimension moyenne Surface spécifique Rayon moyen * 1lot
1 1des agglomérats (u m) 1 (m2/g)
1 (u m) 1
1-----------1--------------------1-------------------------1-----------------11 1 1 6 1 D,58 1 4,21 1
1-----------1--------------------1-------------------------1-----------------11 2 1 42 1 0,685 1 1,19 1
1 1 1 1 1
Tableau X - Caractéristiques des deux lots de poudre d'oxynitrure
d'aluminium (obtenues par réaction nitrure/alumine (cf. 2ème Partie) et
conditionnées pour l'étude cinétique du frittage sous charge.
Le lot 1 est donc constitué de grains de rayon moyen de 4,21 ~m peu
agglomérés et le second d'agglomérats de 42 ~m renfermant des grains de rayon
moyen nettement plus petit (1,19 ~m).
B - CINETIQUE DE FRITTAGE
1 - Rappels théoriques
Au plan cinétique, on distingue trois stades au cours du frittage (fig.
33) •
Le stade initial pour lequel le taux de densification reste inférieur à
0,65 avec formation de ponts entre les grains. Sous l'action combinée de la
tension de surface et des différents rayons de courbures des grains, cette
formation est régie par les lois générales de la thermodynamique : minimisa
tions de l'énergie libre du système par réduction de la surface de contact.
Un stade intermédiaire entre les taux de densification 0,65 et 0,9 au
cours duquel il y a diminution de la porosité ouverte.
Un stade final, au-delà d'un taux de 0,9, assure l'élimination de la
porosité fermée.
* Le rayon moyen est calculé à partir de la surface spécifique en supposantles grains sphériques.
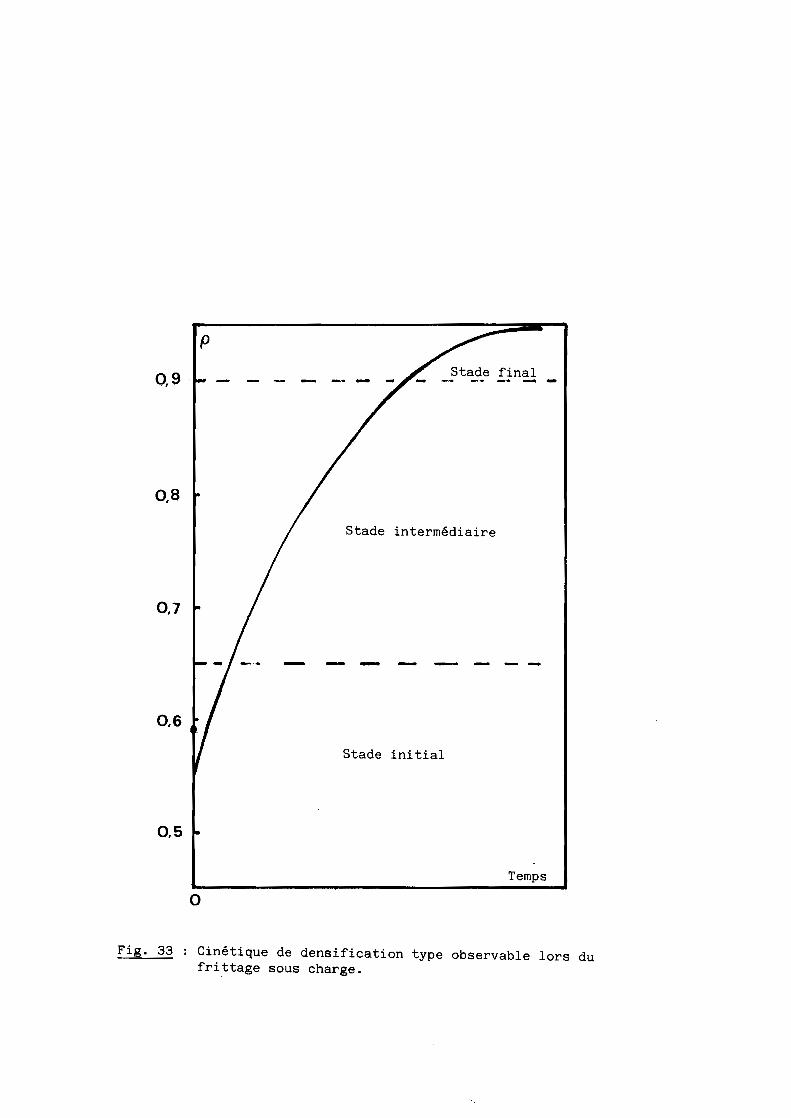
0,9
0,8
0,7
0,6
0,5
p
final
Stade intermédiaire
- -
Stade initial
Fig. 33
Temps
°Cinétique de densification type observable lors dufrittage sous charge.

- 77 -
Il existe des modèles théoriques permettant d'interpréter les cinétiques
de frittage sous charge de matériaux monophasés polycristallins. Mais de tels
modèles restent difficilement transposables aux ensembles polyphasés. Dans le
cas du frittage des céramiques. de nouveaux modèles ont été proposés. Nous en
présenterons une brève description en vue de l'interprétation des courbes
cinétiques de densification.
Parmi tous ces modèles, les plus classiques, car les plus fréquemment
rencontrés, sont ceux où interviennent la diffusion, diffusion en volume selon
NABARRO (39) et HERRING (40), ou diffusion aux joints de grains selon COBLE
(41) •
Dans le premier cas, on considère que la diffusion dans la maille d'un
solide polycristallin intervient par diffusion de matière entre les joints de
grains en compression vers ceux qui sont en tension. Cette diffusion est
possible grâce à l'apparition d'un gradient d'accumulation de lacunes équi
valent à un gradient de potentiel chimique compte tenu de l'application de la
pression. HERRING a contribué largement au développement de cette théorie,
dans le cas particulier de grains sphériques et proposé une équation pour la
diffusion en volume dans l'étape intermédiaire
IIp-= Klit
P
RT d2
où p représente la densité, t le temps, R la constante des gaz parfaits, P la
pression appliquée, T la température, d le diamètre moyen des grains et K une
constante liée au coefficient de diffusion volumique.
Dans le cas de la diffusion aux joints de grains, la diffusion crée des
zones rétrécies entre les particules et entraîne un rapprochement des centres
de gravité des sphères. On aboutit, dans le cas de grains sphériques, à
l'équation suivante de la vitesse de densification
IIp P
-= K' --~lit RT d
3
Ainsi, dans les deux cas, la vitesse de densification évolue linéaire
ment avec la pression appliquée, mais l'influence de la granulométrie initiale
des poudres est différente.

- 78 -
On montre par ailleurs (42) que la vitesse de densification peut encore
s'exprimer par la relation générale:
-=at
K (P _ P )no
avec K
1A(_)m exp
d
E
RT
où P représente la pression appliquée, Po la pression limite au-dessous de
laquelle s'annule la vitesse de densification. n et m sont respectivement les
exposants de la pression et du diamètre des grains. E désigne l'énergie
d'activation et A une constante.
On trouvera au tableau XI les valeurs des exposants m et n selon le
modèle théorique avancé.
2
3
m
oo
1
1
3
1
1
Exposant
granulométrie
1 Exposant
Mécanismes 1 pression
1 n 1
--------------------------------------1----------------SANS PHASE LIQUIDE 1
Déformation plastique du grain 1
• Montée des dislocations 1 3
• Glissement des dislocations 1 4,5
1
- Diffusion en volume 1
(NABARRO-HERRING ) 1 1
Diffusion au joint de grain 1
(COBLE) 1 1
- Réaction d'interface 1
(Montée des dislocations) 1 2
- Glissement grain sur grain 1 2
--------------------------------------1----------------AVEC PHASE LIQUIDE 1
Diffusion au joint de grain 1 1
- Réaction d'interface 1 1
Glissement grain sur grain 1 > 1
----------------,------- -------
Tableau XI - Valeur des exposants n et m de l'équation générale dedes vitesses de densification selon les mécanismes gouvernant lefrittage.

- 79 -
2 - Etude du premier lot
a - Influence de la pression
L'expérimentation a été menée, dans une gamme de pressions comprises
entre 15 et 30 MPa, à la température de 1750 0C pour atteindre commodément un
taux de densification de 0,9. Les courbes de retrait correspondantes sont
représentées sur la figure 34.
A partir de ce réseau, il est possible de calculer les vitesses de
densification pour différentes pressions en fixant un taux de densification
constant. On obtient alors la loi de vitesse-pression. Les résultats dans
l'intervalle P = 0,70 à 0,83 qui correspond au stade intermédiaire de
frittage, sont représentés sur la figure 35.
A chaque taux de densification, on note une régression linéaire très
significative entre l'évolution de la vitesse et celle de la pression comme le
montrent les test t de Student rapportés au tableau XII. La cinétique de
frittage est donc régie par une étape limitante de diffusion comme dans le cas
de la densification du nitrure d'aluminium (43).
On observe, par ailleurs, l'existence d'une pression critique P au-o
dessous de laquelle la vitesse s'annule. Celle-ci varie linéairement avec le
taux de densification comme l'atteste la figure 36.
b - Influence de la température
Différents échantillons pulvérulents du lot 1 ont été densifiés entre
1650 et 17500C sous une pression de 20 MPa (fig. 37). Les vitesses obtenues à
chaque température pour les taux de densification P 0,74, 0,76 et 0,80 sont
représentés en coordonnées Log V, l/T sur la figure 38. Ces représentations
sont toutes linéaires comme l'attestent les valeurs des coefficients de
corrélation obtenus et leur signification statistique dans chaque cas (cf.
tableau XIII).
Les énergies d'activation apparentes tirées des pentes de ces droites
sont 129 ; 182 et 173 KJ/mole respectivement aux taux de densification 0,74
0,76 et 0,80. La valeur moyenne dans cet intervalle, soit E = 162± 29 KJ/mol
est compatible avec celle trouvée lors de la densification du nitrure d'alumi
nium (44) dans la même gamme de température, ce qui implique la présence d'une
même phase A10N aux joints de grains des frittés AIN.
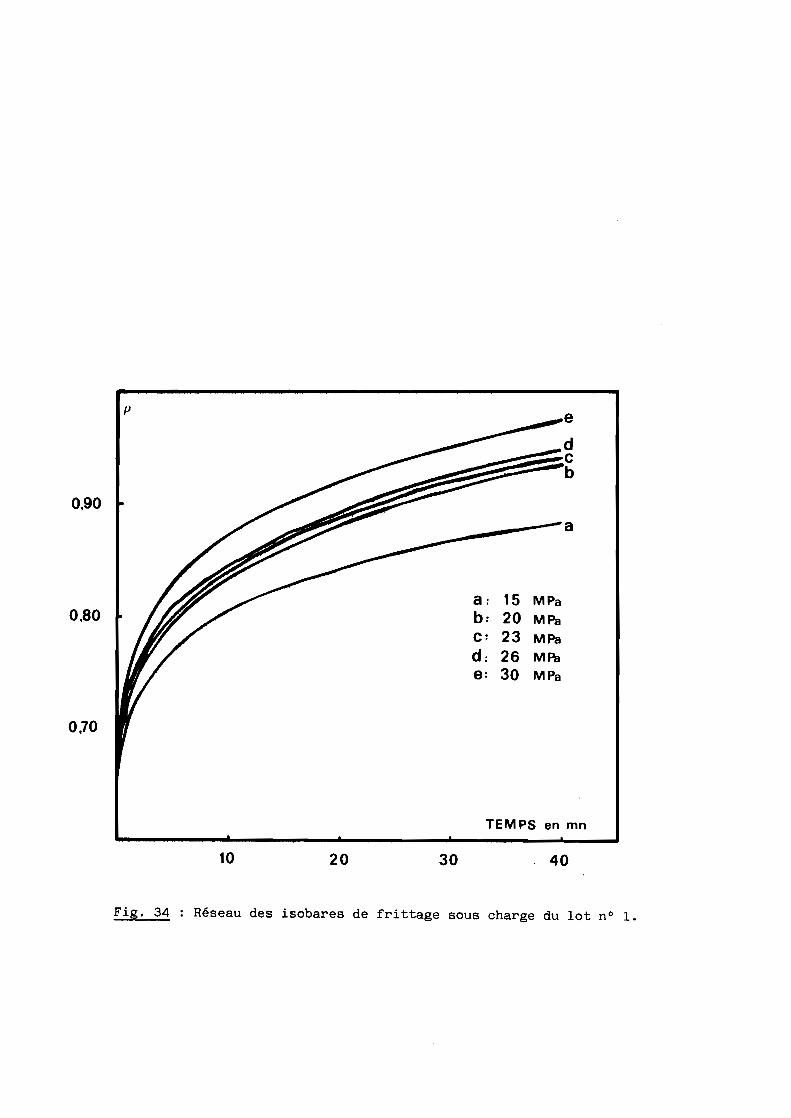
p
0.90
a: 15 MPa0.80 b: 20 MPa
Cl 23 MPad: 26 MPae: 30 MPa
0.70
TEMPS en mn
10 20 30 40
Fig. 34 Réseau des isobares de frittage sous charge du lot nO 1.
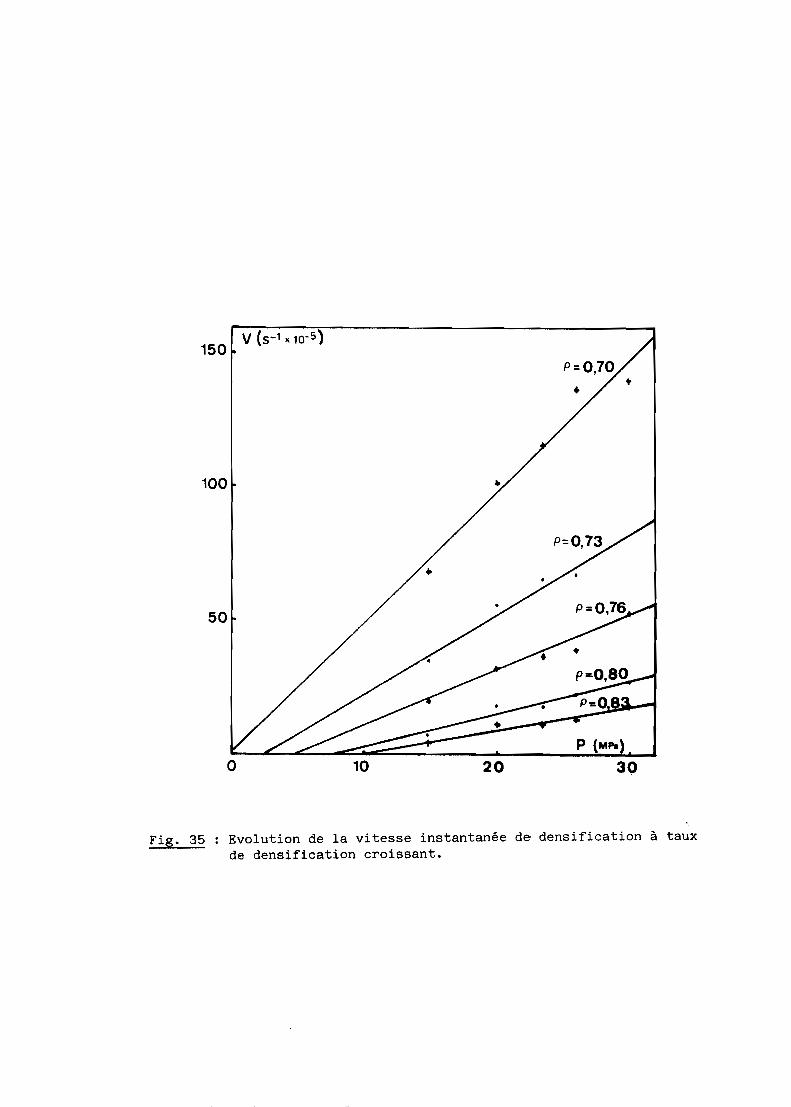
V (5-' x 10-5)150
100
50
o 10 20 30
Fig. 35 Evolution de la vitesse instantanée de densification à tauxde densification croissant.
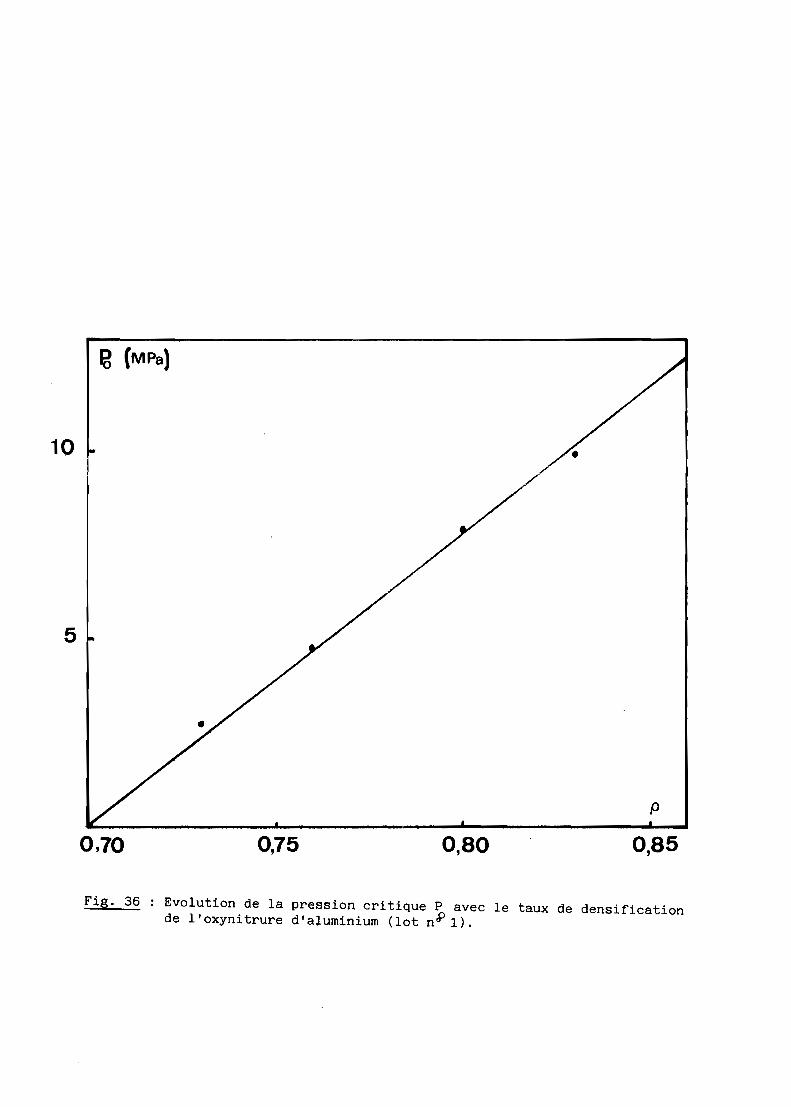
10
5
~ (MPa)
0,70 0,75 0,80
p
Fig. 36 Evolution de la pression critique P avec le taux de densificationde l'oxynitrure d'aluminium (lot n.P 1).

p
0,90
0.80
0.70
10 20 30
al 1650 OCbr 1700 ocCl 1750 0 C
TEMPS en mn
40
Fig. 37 Isothermes de densification du lot nO 1 de poudre d'oxynitrured'al-uminium.
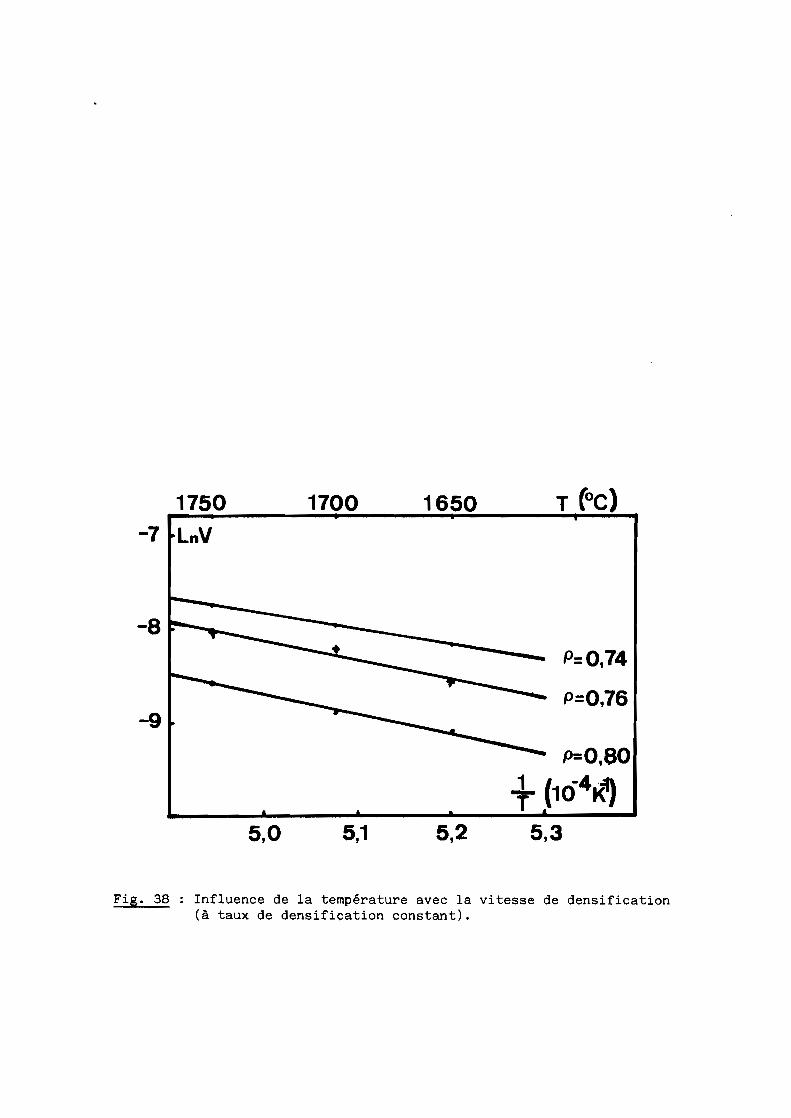
1750
-7 LnV
-9
5,0
1700
5,1
1650
5,2
P=0,74
P=0,76
p=0,80+(10-4K-1)
5,3
Fig. 38 Influence de la température avec la vitesse de densification(à taux de densification constant).
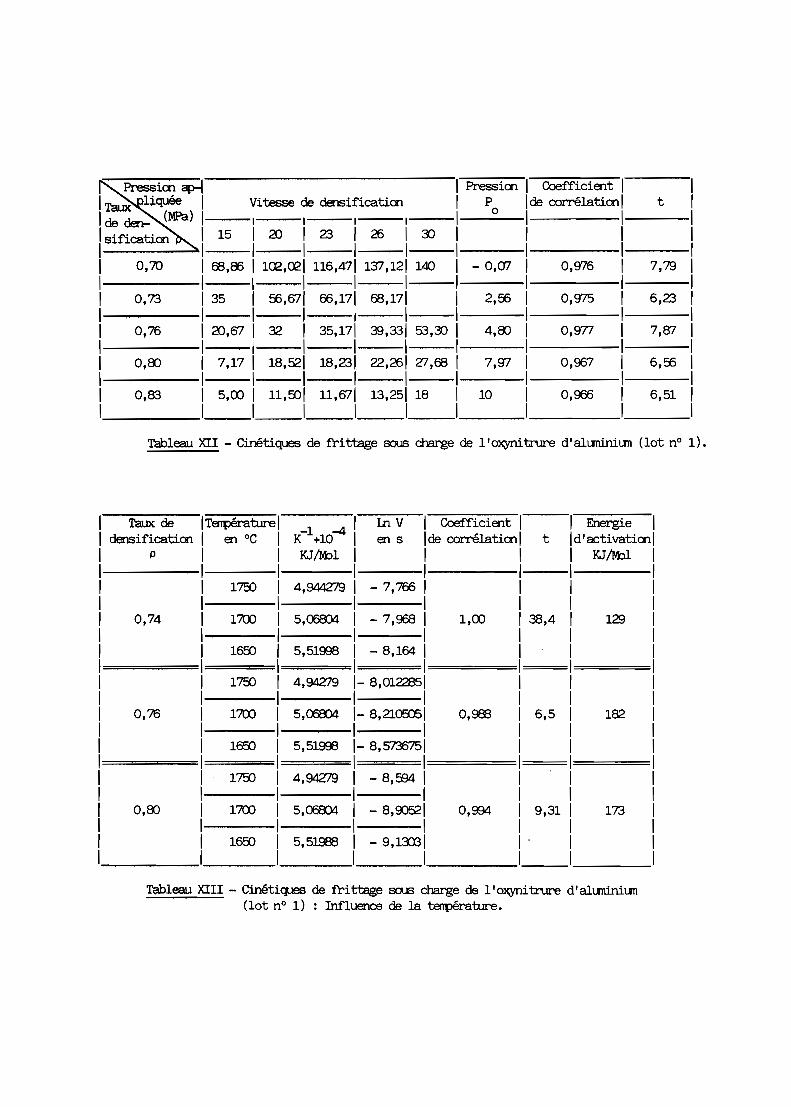
Pressicn ap...j Pressicn 1 Coefficient 1
TauX liquée Vitesse de densificaticn P 1de corrélaticn tde den- (MPa)
011 1 1
sificaticn 15 1 2) 1 23 1 26 3) 1
1 1 1 1 10,70 68,00 1 102,021 116,471 137,12\ MO - 0,07 1 0,976 7,79
1 1 1 1 10,73 35 1 :6,671 66,171 68,171 2,:6 1 0,975 6,23
1 1 1 1 10,76 2),67 1 32 1 35,171 39,331 53,3) 4,00 1 0,977 7,fg
1 1 1 1 10,00 7,17 1 18,521 18,231 22,261 27,68 7,97 1 0,967 6,:6
1 1 1 1 10,83 5,00 1 11,001 11,671 13,251 18 10 1 0,966 6,51
1 1 1 1 1
Tableau XII - Cinétiques de t'rittage sous charge de l'oxyni trure d' aliminnm (lot nO 1).
1 Taux de TaJPéra1J..u"e 1 1 lnV 1 Coefficient 1 1 Energie 11 densificaticn en oC 1 K-1+1O-4 1 ens Ide corrélaticn t Id'activaticn1 p
1 KJ/ltbl 1 1 1 KJ/ltbl1 1 1 1 1
1 17f:O 1 4,944279 1 - 7,766 1 1
1 1 1 1 11 0,74 1700 1 5,068J4 1 - 7,968 1 1,00 38,4 1 1291 1 1 1 1
1 16f:O 1 5,51998 1 - 8,164 1 1
1 1 1 1 117f:O 4,94279 1- 8,0122851 1
1 1 1
0,76 1700 5,068J4 1- 8,21Œ051 0,988 6,5 1 1821 1 1
16f:O 5,51998 1- 8,573675\ 1
1 1 1
17f:O 4,94279 1 - 8,594 1 1
1 1 10,00 1700 5,068J4 1 - 8,00521 0,994 9,31 1 173
1 1 116f:O 5,51988 1 - 9,13)31 1
1 1 1
Tableau XIII - Cinétiques de t'rittage sous charge de l'oxyni trure d' aluniniun(lot n? 1) : Influence de la tenpérature.
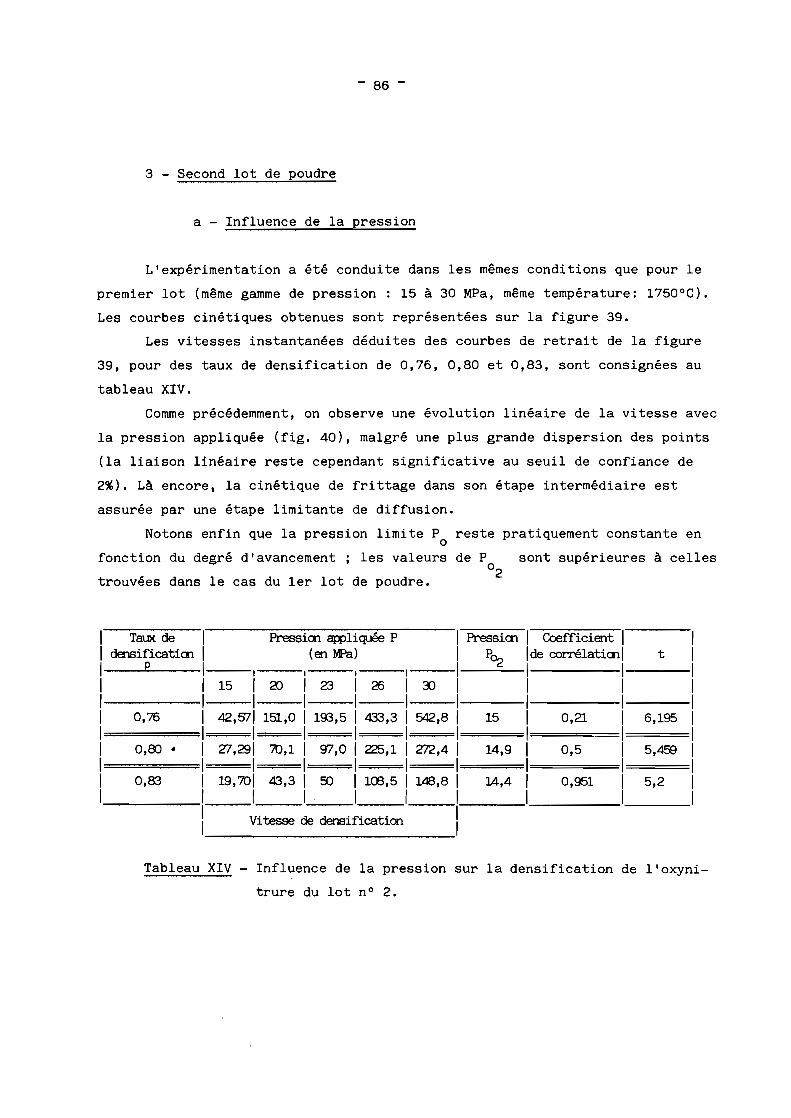
trouvées dans le cas du 1er lot de poudre.
- 86 -
3 - Second lot de poudre
a - Influence de la pression
L'expérimentation a été conduite dans les mêmes conditions que pour le
premier lot (même gamme de pression: 15 à 30 MPa, même température: 1750 oC).
Les courbes cinétiques obtenues sont représentées sur la figure 39.
Les vitesses instantanées déduites des courbes de retrait de la figure
39, pour des taux de densification de 0,76, 0,80 et 0,83, sont consignées au
tableau XIV.
Comme précédemment, on observe une évolution linéaire de la vitesse avec
la pression appliquée (fig. 40), malgré une plus grande dispersion des points
(la liaison linéaire reste cependant significative au seuil de confiance de
2%). Là encore, la cinétique de frittage dans son étape intermédiaire est
assurée par une étape limitante de diffusion.
Notons enfin que la pression limite P reste pratiquement constante eno
fonction du degré d'avancement; les valeurs de P sont supérieures à cellesO2
Taux de Pressicn appli~ P Pressim 1 Coefficient 1
densificatim (en MPa) Pe;z 1de corrêlatacn] tp
1 11
15 1 2) 23 26 3) 1 11 1 1
0,76 42,571 151,0 193,5 433,3 542,8 15 1 0,21 1 6,1951 1 1
0,00 • 27,291 70,1 rrl,O 225,1 272,4 14,9 1 0,5 1 5,4591 1 1
0,83 19,701 43,3 :0 100,5 148,8 14,4 1 0,951 1 5,21 1 1
Vitesse de densification
Tableau XIV Influence de la pression sur la densification de l'oxyni-
trure du lot nO 2.
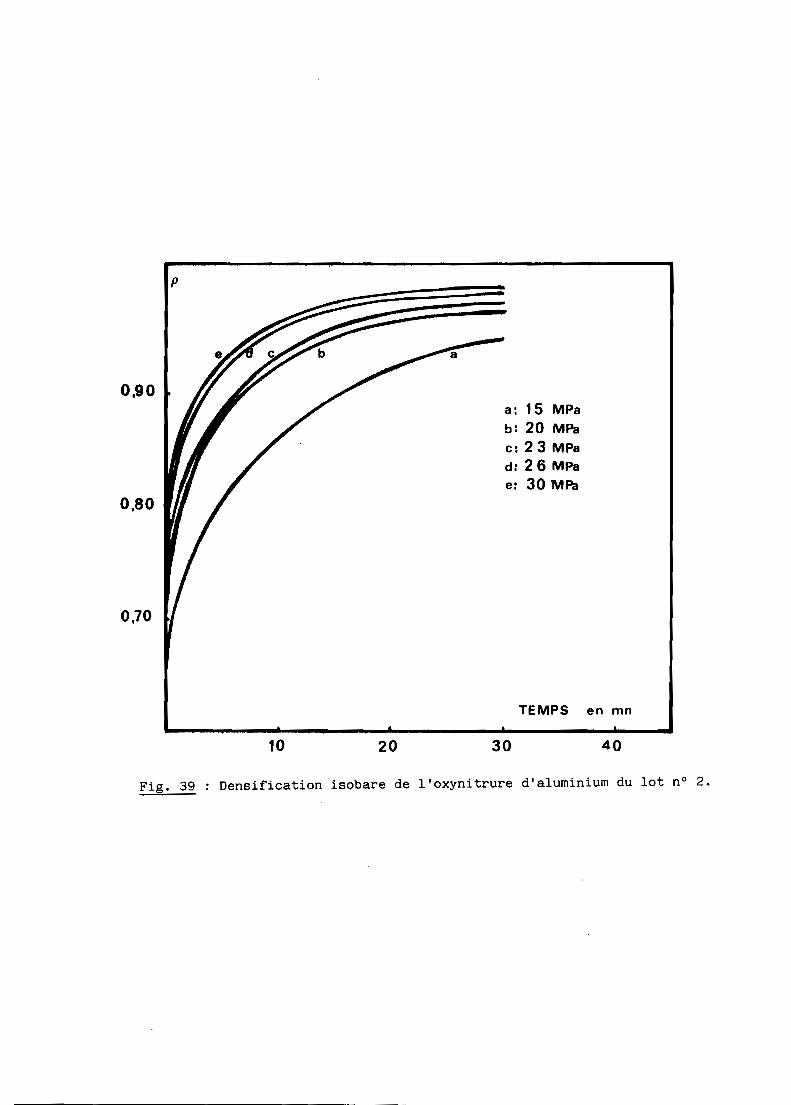
p
0,90
0,80
0,70
a: 15 MPab: 20 MPa
c: 23 MPad: 26 MPae: 30 MPa
TEMPS en mn
10 20 30 40
Fi&. 39 Densification isobare de l'oxynitrure d'aluminium du lot nO 2.
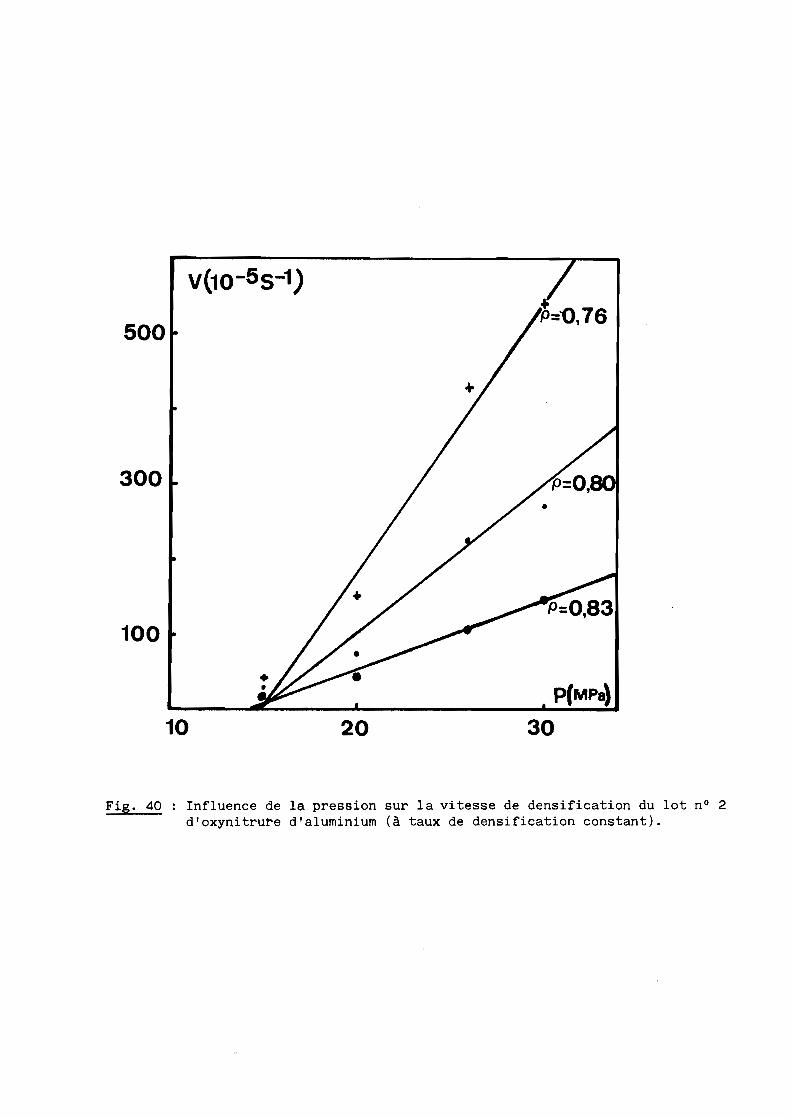
P{MPa)
302010
500
100
300
Fig. 40 Influence de la pression sur la vitesse de densification du lot nO 2d'oxynitru~e d'aluminium (à taux de densification constant).

- 89 -
C - INFLUENCE DE LA GRANULOMETRIE
Elle permet de préciser si la diffusion, qui est régulatrice du
phénomène d'ensemble, intervient en volume (type NABARRO-HERRING) ou au niveau
du joint de grain (diffusion de type COBLE), en déterminant la valeur de
l'exposant m dans l'expression de la vitesse
KPv =-= aP
mr
Il suffit de comparer les constantes de vitesse al et a2 obtenues pour
un même taux de densification p à partir des lots 1 et 2 dont les granulomé
tries sont dans le rapport r 1/r2 =3,54.
1 p 1 0,76 1 0,80 1 0,83 1
1----------1----------1----------1----------11 a2 1 35,00 1 17,5 1 8,82 1
1----------1----------1----------1----------11 al 1 2,00 1 1,27 1 0,77 1
1----------1----------1----------1----------11 a2/a1 1 17,5 1 13,80 1 11,5 1
1----------1----------/----------1----------11 m 1 2,26 1 2,08 1 1,93 1
1 1 1 1 1
Tableau XV - Influence de la taille des grains sur la vitesse de
densification de l'AION y.
Les chiffres du tableau XV, déduits des représentations linéaires des
figures 35 et 36 montrent que l'exposant m du rayon dans l'équation de la
vitesse est voisin de 2. On en conclut que la résorption de la porosité au
cours de l'étape intermédiaire du frittage se fait par diffusion en volume
selon le modèle de NABARRO-HERRING.

RESISTANCE A L'OXYDATION

RESISTANCE A L'OXYDATION
L'oxynitrure d'aluminium surclassant les verres quant à la température
de fusion, il convient de tester sa résistance à l'oxydation aux températures
relativement élevées que peuvent imposer nombre de ses applications potentiel
les dans l'air. Les études antérieures (2,7) ont révélé que le matériau
pulvérulent s'oxydait à partir de 650°C environ, mais on sait que les produits
massifs possèdent en général une bien meilleure résistance à la corrosion que
les poudres. On a d'ailleurs montré que le comportement à l'oxydation dépen
dait d'une part de la granulométrie, susceptible d'influer sur le mécanisme
lui-même de la rèaction, et d'autre part de la composition, la teneur en azote
gouvernant la nature des produits formés.
Profitant de la recherche des propriétés d'usage en température, nous
avons cherché à préciser le mécanisme d'oxydation d'échantillons massifs
d'oxynitrure d'aluminium y en l'étudiant au travers des trois morphologies
dont nous disposions :
- les poudres
- les pastilles frittées
- les pastilles transparentes

Chapitre l
OXYDATION DE LA POUDRE
La poudre utilisée provient de la synthèse de l'oxynitrure d'aluminium
par le procédé de réaction en phase solide du nitrure avec l'alumine. Ses
caractéristiques sont précisées plus haut (cf. 2ème Partie - Chapitre I). On
utilise principalement comme méthode expérimentale la thermogravimétrie dont
la mise en oeuvre est explicitée en première partie.
A - RESULTATS GENERAUX
1 - Influence de la température
L'oxydation préliminaire d'un lot de poudre sous pression atmosphérique
d'oxygène en montée linéaire de température de 90 K/h, représentée à la figure
41 montre que la prise de masse est sensible à partir de SOooC environ et
qu'elle s'accélère régulièrement jusque vers 1200oC, une perte de masse étant
décelée aux températures supérieures.
En régime isotherme,l'oxydation de la poudre a été suivie entre 1100 0C
et 1225°C sous une pression d'oxygène de 730 Torr. Les cinétiques sont rappor
tées à la figure 42 en coordonnées (a , t) ou a représente le degré d'avance
ment de la réaction au temps t, soit le rapport du gain de masse observé au
temps t au gain de masse maximum obtenu par oxydation totale. Les courbes
présentent une allure globalement décélérée, après une légère accélération
initiale, à relier vraisemblablement à la mise en température de la poudre
pendant les deux premières minutes. Aux températures les plus hautes, on note
une légère dérive témoignant d'une perte de masse qui devient sensible si on
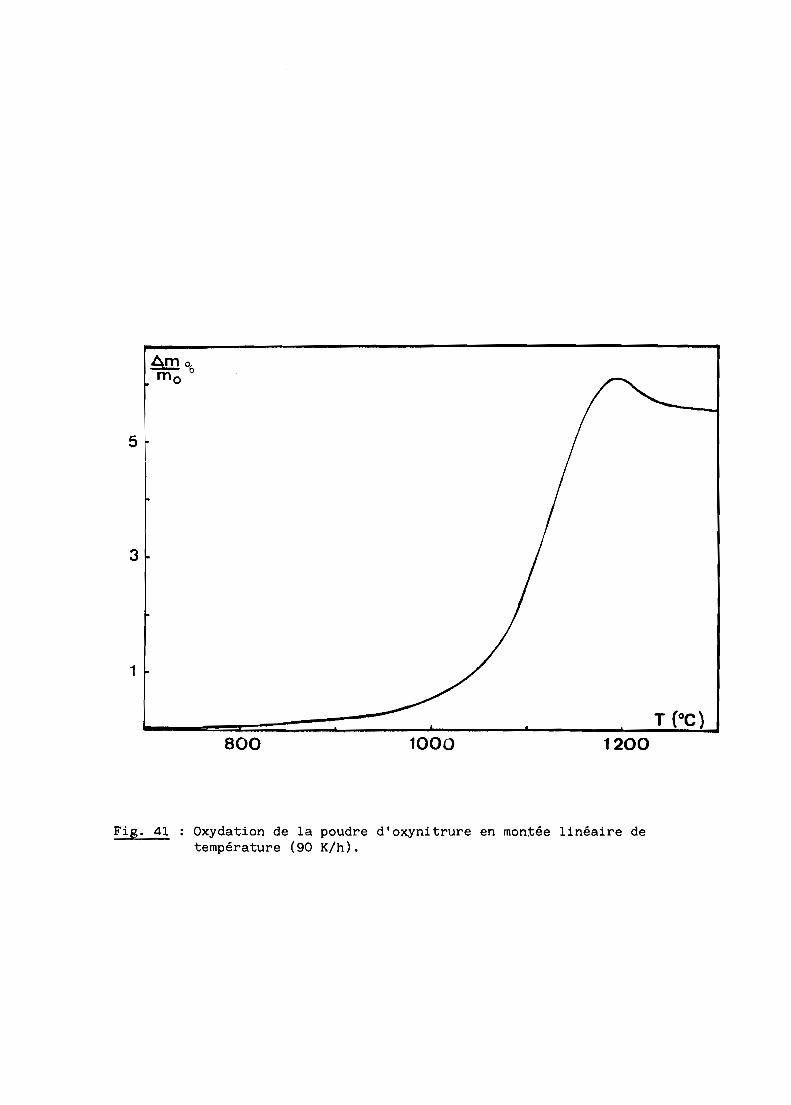
5
3
1
800 1000 1200
Fig. 41 Oxydation de la poudre d'oxynitrure en montée linéaire detempérature (90 K/h).
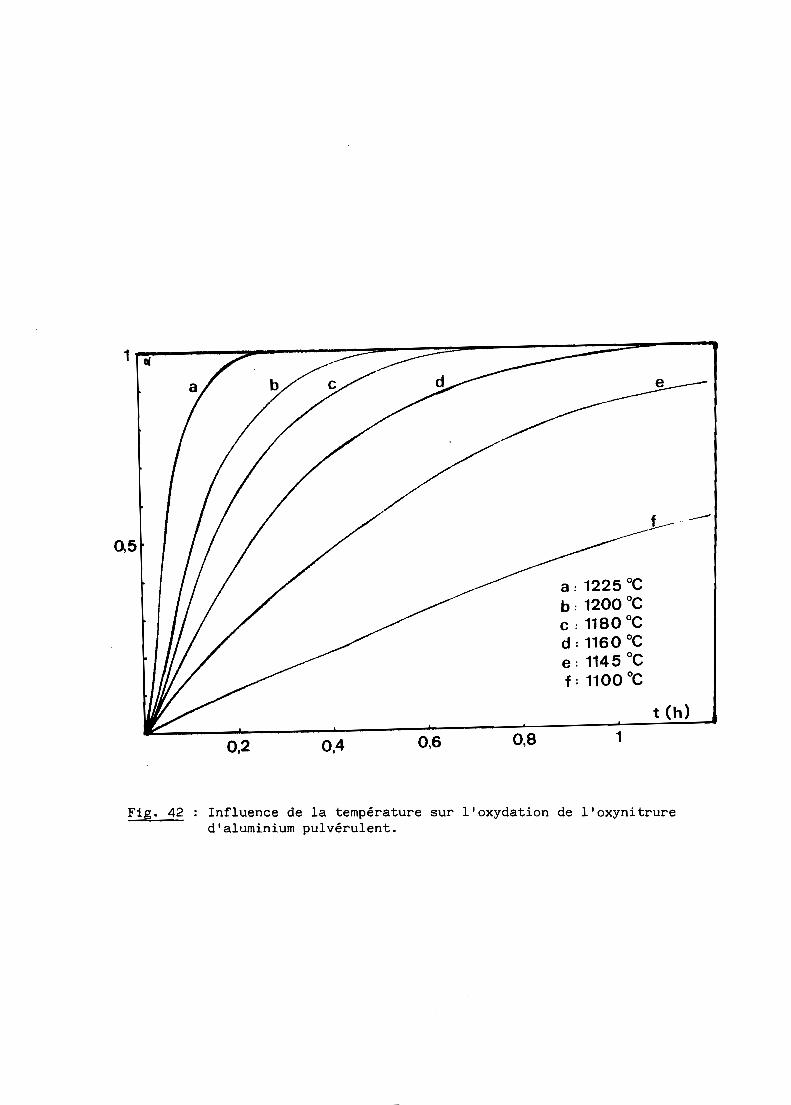
1 0(
0,5
0,2 0,4 0,6 0,8
f_--»->
a: 1225 -cb: 1200 -cc . 1180 -cd: 1160 -ce:1145°Cf: 1100 Oc
t (h)
1
Fig. 42 Influence de la température sur l'oxydation de l'oxynitrure
d'aluminium pulvérulent.

- 95 -
laisse l'échantillon en température pendant plusieurs heures. Par ailleurs, le
gain de masse maximum observé (environ 6,1%) est très nettement supérieur à
celui théoriquement prévisible (3,16%) sur la base de l'équation de la
réaction :
AION + 02 ---. A1 203 + xN2compte tenu de la composition de l'oxynitrure.
L'analyse radiocristallographique du produit de la réaction révèle la
présence des raies de l'alumine y' (17) (de structure spinelle, dont le
spectre est proche de celui de l'oxynitrure de départ) et de l'alumine a en
quantité négligeable en cours de réaction, mais d'autant plus grande que la
température est plus élevée et qu'on maintient la poudre plus longtemps dans
le four après achèvement de l'oxydation.
Les observations de la poudre au microscope électronique à balayage ne
fournissent aucun renseignement supplémentaire, les grains, très fins, parais
sant ne se modifier d'aucune façon en cours de réaction, quel que soit l'avan
cement considéré.
Le traitement analytique direct du réseau des cinétiques isothermes par
la méthode des affinités (26) (qui ne présuppose pas le mécanisme réactionnel)
montre que les courbes sont affines entre elles pour des degrés d'avancement
compris entre 0,05 et 0,90 ; l'énergie d'activation ainsi calculée (cf. fig.
46) ressort à
E = 388 ± 55 kJ.mol-1 (au seuil de probabilité de 95%).
Cette valeur est confirmée, dans les mêmes conditions par la méthode des
vitesses instantanées (26).
2 - Influence de la pression
L'influence de la pression d'oxygène a été testée à 11000C entre 100 et
730 Torr, et la figure 43 montre qu'elle est sensible dans cet intervalle. Les
courbes cinétiques présentent la même allure que celles du réseau d'isother
mes, mais on ne décèle cette fOis par radiocristallographie que les raies de
la phase oxynitrure y' dans le produit formé.
Les observations microscopiques ne font apparaitre aucune modification
d'aspect de la poudre initiale.
Au plan de l'analyse cinétique immédiate, les courbes du réseau isobare,
comme les isothermes, sont affines entre elles. On en déduit (26) que la
vitesse de la réaction (V ) obéit à une loi de pression de la forme:
V = K(a,T) pO,30
vérifiée par la méthode des vitesses instantanées.
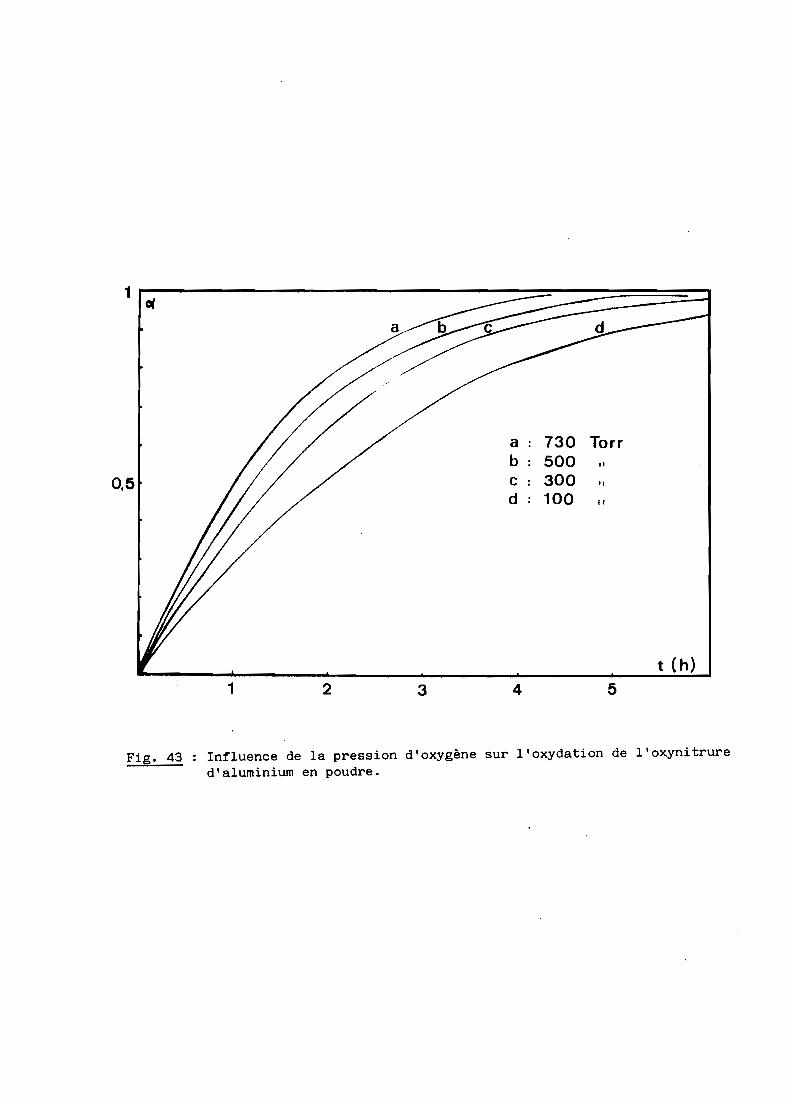
1 01
1 2 3
abcd
4
730500300100
Torr
""1/
5
t (h)
Fig. 43 Influence de la pression d'oxygène sur l'oxydation de l'oxynitrured'aluminium en poudre.

- 97 -
B - INTERPRETATION
1 - Analyse des résultats
Si on néglige l'apparition de très faibles quantités d'alumine aux plus
hautes températures dans le produit formé, et que l'on exclut de l'analyse
dans un premier temps les légères pertes de masses parfois observées après
oxydation, l'ensemble des résultats présentés donnent à penser que l'oxydation
est gouvernée par un régime cinétique simple : les courbes des ~éseaux
isothermes aussi bien qu'isobares sont affines entre elles, donc le régime
cinétique est unique quels que soient l'avancement, la température ou la
pression d'oxygène, et le produit de la réaction est lui-aussi unique, aux
remarques près ci-dessus. Au plan analytique, on en déduit que les variables q)
T et P sont indépendantes et que l'expression de la vitesse de la réaction
répond donc à une équation du type :
daV =-= f(a) g(P) h(T)
dt
L'allure décélérée des cinétiques exclut l'hypothèse d'un régime
d'interface externe et la valeur peu élevée de 1,1 du coefficient d'expansion
volumique (28) (rapport du volume de l'alumine y' formée à celui de l'oxyni
trure disparu) n'est pas a priori favorable (sans toutefois l'interdire) à
l'établissement d'un régime diffusionnel.
En revanche, un régime d'interface interne (sous réserve que l'alumine
soit un semi-conducteur de type p pour justifier de la loi de pression (25,
26» ou, si l'oxyde formé est poreux, un régime de réaction s'accomodent bien
de l'ensemble des éléments présentés. En ce cas, les courbes cinétiques
doivent être linéarisables par la transformée (29) :
F(a) = 1 ~ (1 _ a)1/3
Les réseaux des transformées F(a) des figures 44 (pour les isothermes)
et 45 (pour les isobares) vérifient la validité de l'hypothèse envisagée.
Des valeurs des pentes des droites de ces réseaux, on déduit (cf. fig.
46) la valeur de l'énergie d'activation:-1E = 420 ± 75 kJ. mol
en bon accord avec celle déterminée directement, et on vérifie également la
loi de pression en pO,3.
0,4
0,2
0,2 0,4 0,6 0,8 1
t (H)
Fig. 44 Transformées des isothermes d'oxydation de la poudre d'oxynitrured'aluminium.
0,5 F(U')
0,3
0,1
1 2
a : 730 Torrb : 500 "c : 300 "d : 100 \1
t (H)
Fig. 45 Réseau des transformées des isobares d'oxydation de l'oxynitrured'aluminium pulvérulent.
-0,5
6,7 7 7,3
Fig. 46 Influence de la température sur l'oxydation de l'oxynitrured'aluminium pulvérulent.
+ Méthode des affinités affines
eMéthode des transformées

- 101 -
2 - Comparaison avec les résultats antérieurs
Les travaux antérieurs se rapportant à l'oxydation des poudres d'oxyni
trure d'aluminium ont mis en évidence un phénomène original de rétention
d'azote dans la structure spinelle conservée après réaction (4, 6). La phase
obtenue dite alumine y'. y' est instable à partir de 1100 et 1150oC. Ces
études portaient sur des poudres de différentes granulométries à des tempéra
tUres variant de 900 à 1100oC. Les poudres les plus fines (20<0<30 ~m)
s'oxydaient selon un processus de nature interfacia1e interne tandis que les
plus grossières (40 <0<50 ~m) donnaient lieu à apparition d'une étape diffu
sionne11e limitante.
Comme on le voit, nos résultats qui concernent une poudre sensiblement
plus fine encore que celles étudiées précédemment et un domaine de température
supérieur confirment en tout point ces observations :
1 - quant à la nature du produit formé, l'alumine y' qui évolue vers
l'alumine a pour nos températures d'étude les plus élevées, conformément à ce
que l'on savait de son domaine de stabilité, mais en égard à la rapidité des
cinétiques à ces températures, la transformation y'--.a n'est guère sensible
en cours d'oxydation,
2 - quant à la nature du mécanisme réactionnel qui est identique à celui
observé antérieurement sur des poudres plus fines.
Une différence importante est cependant à souligner: l'énergie d'acti
vation que nous avons calculée est nettement plus importante que celle précé--1 -1demment déterminée: 420 kJ. mol au lieu de 205 ± 17 kJ. mol • On peut
plausiblement en rechercher l'origine d'une part dans la composition de la
phase oxynitrure qui est sensiblement plus pauvre en azote dans la présente
étude (27% de nitrure au lieu de 40%) et dans la perturbation cinétique qui
peut être réduite avec l'apparition du début de la transformation de la phase
y' au-dessus de 1150 oC.

- 102 -
C - CONCLUSION
Conformément aux résultats antérieurs, la poudre fine d'oxynitrure
d'aluminium que nous utilisons s'oxyde dans l'oxygène à température relative
ment peu élevée (800°C environ) pour donner de l'alumine y' de structure
spinelle stabilisée par la présence d'azote résiduel.
Aux températures supérieures à 11000C pour lesquelles l'étude cinétique
n'avait pas été entreprise, la réaction est très rapide et la transformation
de l'alumine y' en a ne se produit pas de façon sensible pendant l'oxydation.
Celle-ci est gouvernée par un régime de nature interfa-ciale interne (ou de
réaction) selon la loi de vitesse:
daV =_= Ct e x (1_a)2/3 x pO,3 x e
dt
420000RT
pour des degrés d'avancement, pressions et températures compris respectivement
dans les intervalles 0,05 à 0,90, 100 et 730 Torr, et 1373 à 1498 K.
L'aspect des grains n'est pas modifié par l'oxydation, mais la phase
alumine y' évolue lentement, après oxydation vers la phase a, avec dégagement
d'azote.

Chapitre II
OXYDATION DE L'OXYNITRURE MASSIF BRUT DE FRITTAGE
Les échantillons employés se présentent sous forme de petits cubes
d'environ 4 mm d'arête découpés dans une pastille de 32 mm de diamètre. Ils
sont polis mécaniquement sur chacune de leurs six faces afin d'obtenir un bon
état de surface avant l'oxydation, pour éviter des perturbations cinétiques au
départ et limiter la microfissuration. Ils proviennent du frittage sous charge
de la poudre d'oxynitrure préparée par réaction à l'état solide entre le
nitrure et l'alumine, dont le comportement à l'oxydation constitue le chapitre
précédent. Une pastille fournit ainsi une trentaine d'échantillons, soit une
quantité suffisante pour l'ensemble de l'étude, ce qui garantit la bonne
homogénéité du lot. Les conditions du frittage sous charge sont celles du
protocole expérimental requis pour l'obtention in fine du produit transparent
; elles ont été précisées en deuxième partie (chapitre II). Les conditions
expérimentales de l'étude cinétique sont identiques à celles mises en oeuvre
pour le suivi de l'oxydation de la poudre (~f. également 1ère partie).
A - IŒSULTATS GENERAUX
1 - Influence de la température
Un échantillon, porté jusqu'à 15500C en montée linéaire de température
de 90 K/h (cf. fig. 47) ne présente de gain de masse par oxydation qu'aux
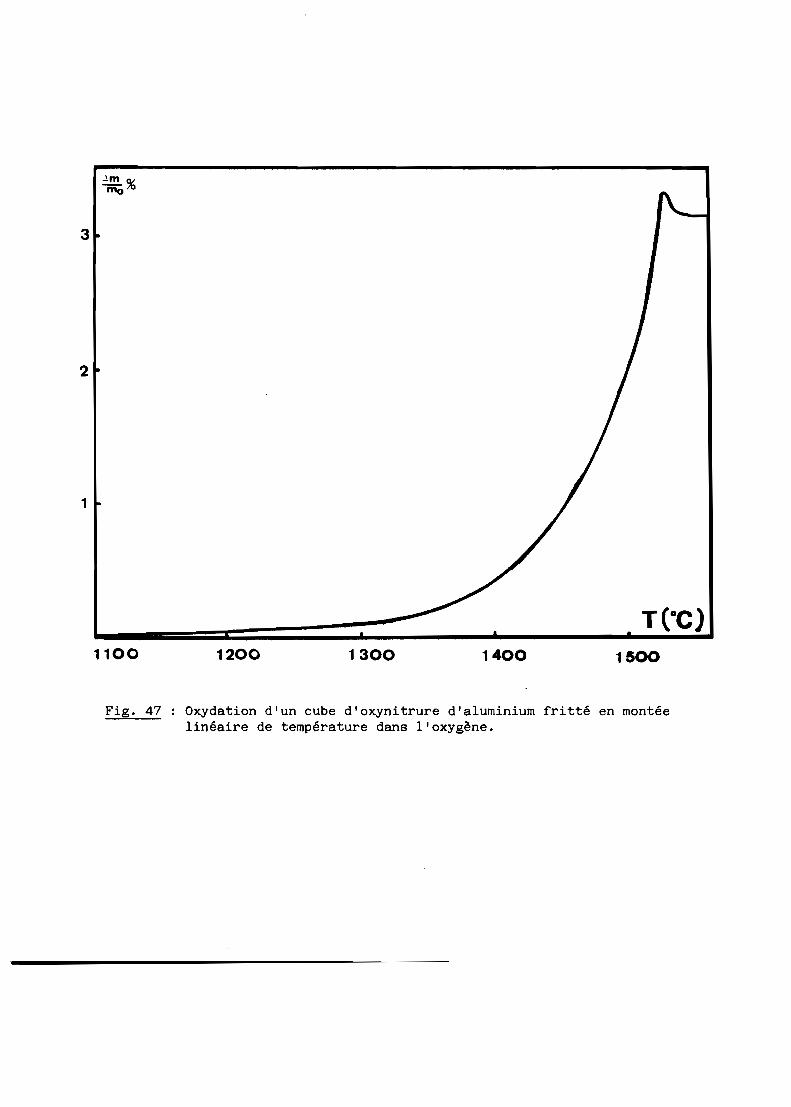
3
2
1
1100 1200 1300 1400 1500
Fig. 47 Oxydation d'un cube d'oxynitrure d'aluminium fritté en montéelinéaire de température dans l'oxygène.

pondéral relatif 6m/m devient vraiment sensible au-delà de 1350oC,o
température à partir de laquelle il s'accélère, l'oxydation qui s'achève vers
1530 oC, étant suivie d'une légère perte de masse (environ 5% du gain de
masse).
Les isothermes d'oxydation de la figure 48 ont été obtenus sous 730 Torr
d'oxygène entre 1420 et 1540oC. L'allure des courbes est régulièrement
décélérée avec vitesse initiale maximale. On a choisi de les tracer en coor-
données (a ,t) où le degré d'avancement expérimental a est déterminé parexp exp
le rapport du gain de masse au temps t à celui maximum observé. Par chauffage
prolongé quelque temps au-delà de la fin de l'oxydation on obtient une perte
de masse allant de 5 à 7% environ analogue à celle observée lors de
l'oxydation en montée linéaire. Dans la pratique, les réactions n'ont pas été
suivies en général jusqu'à l'obtention de cette perte de masse complète, d'où
la représentation choisie, qui présente un avantage analytique.
On vérifie ainsi classiquement que chacune des courbes du réseau se
déduit des autres dans une affinité par rapport au temps pour des degrés
d'avancement compris entre 0,05 et 0,90. L'énergie d'activation de la
réaction, calculée à partir des valeurs des affinités (26), sans faire
d'hypothèse sur le mécanisme de la réaction ressort ainsi (cf. fig. 53) à
E = 441 ± 32 kJ/mol-1
La méthode des vitesses instantanées confirme cette valeur.
2 - Influence de la pression
A 1500 oC, la pression d'oxygène influence la cinétique d'oxydation comme
le montre le réseau d'isobares de la figure 49, tracé en coordonnées (a ,t)exp
dans les mêmes conditions que les isothermes entre 25 et 730 Torr. Les courbes
sont, cette fois encore régulièrement décélérées à partir d'une vitesse
initiale maximale, et on décèle également une légère perte de masse sur les
thermogrammes après obtention du maximum de gain pondéral.
Les isobares constituent, comme les isothermes, un réseau de courbes
affines par rapport au temps pour des valeurs de a comprises entre 0,05 etexp0,90, et les coefficients d'affinité permettent de dégager directement une loi
de pression de type :
v ~ K(a,T) pO,13
qui s'accorde avec celle déterminée par la méthode des vitesses instantanées.
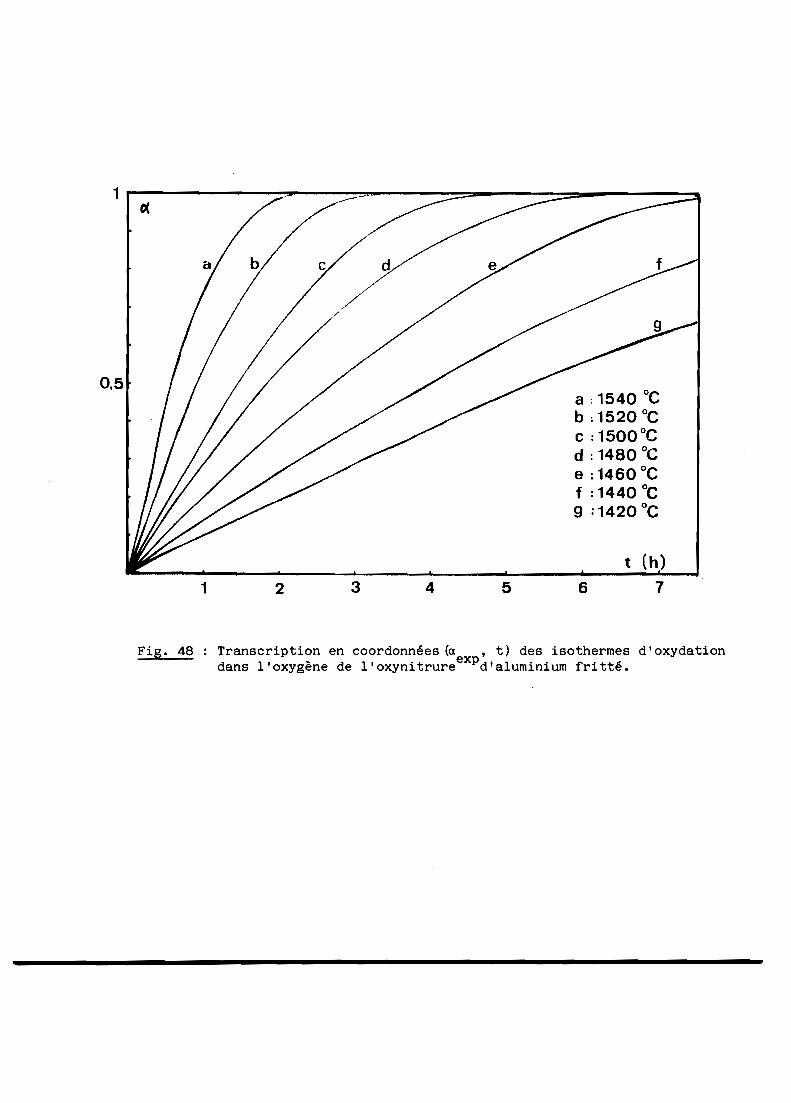
1r-------::::--~~---~=---__==----~
a : 1540 Ocb :1520°Cc : 1500°Cd : 1480 Oce :1460 Ocf : 1440 Oc9 : 1420 Oc
0,5
1 2 3 4 5 6
t (h)
7
Fig. 48 Transcription en coordonnées ~ ,t) des isothermes d'oxydationdans l'oxygène de l'oxynitrureexPd'aluminium fritté.
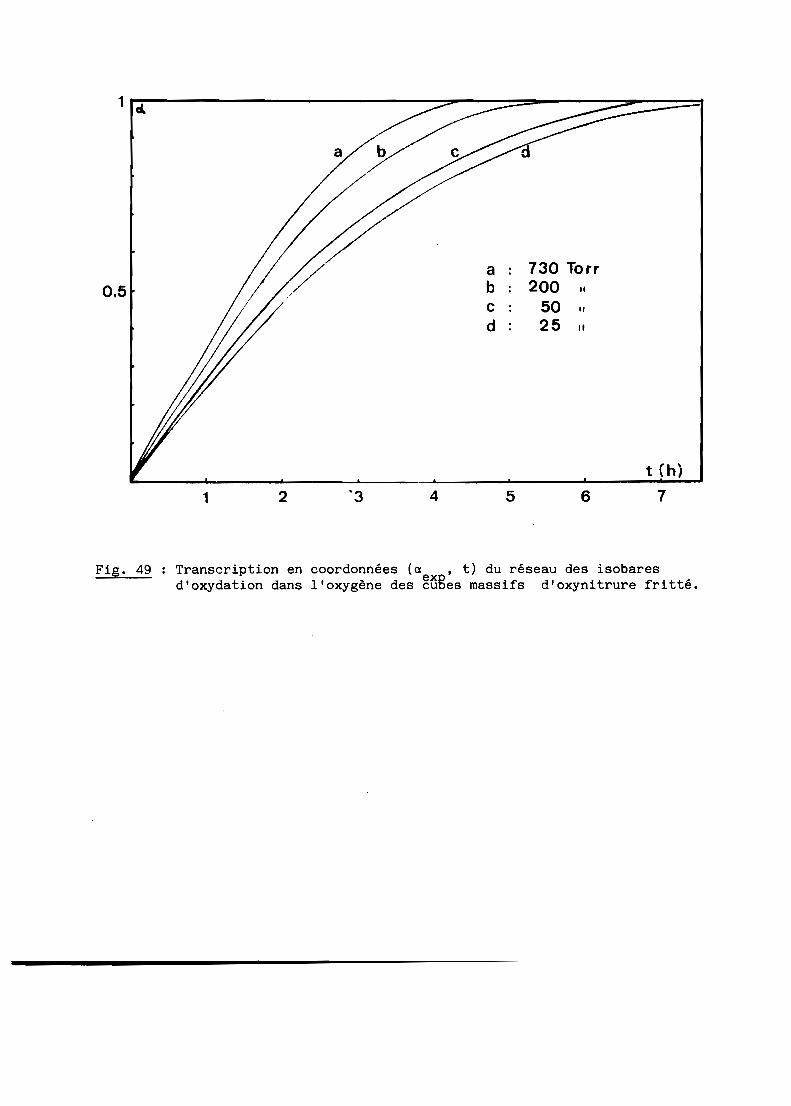
1 Q.
0.5
1 2 '3 4
abcd
5
730 Torr200 "
50 "25 Il
6
t (h)
7
Fig, 49 Transcription en coordonnées (a ,t) du réseau des isobaresd'oxydation dans l'oxygène des ~~ges massifs d'oxynitrure fritté.

L'analyse radiocristallographique du produit formé par oxydation montre
qu'il s'agit d'alumine a. Une analyse fine de la couche au voisinage du subs
trat révèle en plus la présence des raies de l'alumine y' (il est peu probable
qu'il s'agisse de l'oxynitrure qui possède pourtant le même spectre radiocris
tallographique, car la couche d'oxyde et le substrat sont discernables). Le
substrat non encore oxydé ne subit pour sa part aucune modification struc
turale. Il conserve par ailleurs totalement sa cohérence tout au long de
l'oxydation et ne se dilate ni se fissure.
La couche oxydée occupe un volume très nettement supérieur à celui du
nitrure qui l'a généré (contrairement à ce qui était prévisible à priori sur
la base du coefficient d'expansion volumique (27), d'environ 0,98 de
l'alumine formée sur le nitrure). L'oxyde est en effet très poreux et,
partant, fragile et friable (cf. la micrographie de la fig. 50a). Il présente
macroscopiquement un aspect caractéristique dit lien croix de Malte" qui va
en s'accentuant avec l'avancement de la réaction (fig. 50b). L'accrochage
oxyde-substrat est bon à l'endroit où se localise la phase alumine y', mais il
est très difficile à illustrer, l'oxynitrure, l'alumine y' ou a ne se distin
guant pratiquement pas sur les clichés de microscopie électronique. On obtient
ainsi, de l'intérieur vers l'extérieur: le substrat oxynitrure inchangé, puis
vraisemblablement une couche d'alumine y' qui se distingue peu, morphologi
quement du substrat, puis la couche poreuse d'alumine o.
B - INTERPRETATION
L'analyse des cinétiques isothermes et isobares par la méthode des
transformations affines montre que les variables T, P et a sont indépen-exp
dantes (26) ; elle permet d'affirmer ,l'unicité du mécanisme réactionnel. Sa
localisation paraît d'ailleurs relativement aisée puisque la morphologie
générale de la couche d'alumine formée (en particulier sa grande porosité)
implique naturellement un accès plus ou moins libre de l'oxygène jusque vers
l'interface substrat/oxyde. On aurait ici les caractères typiques d'un régime
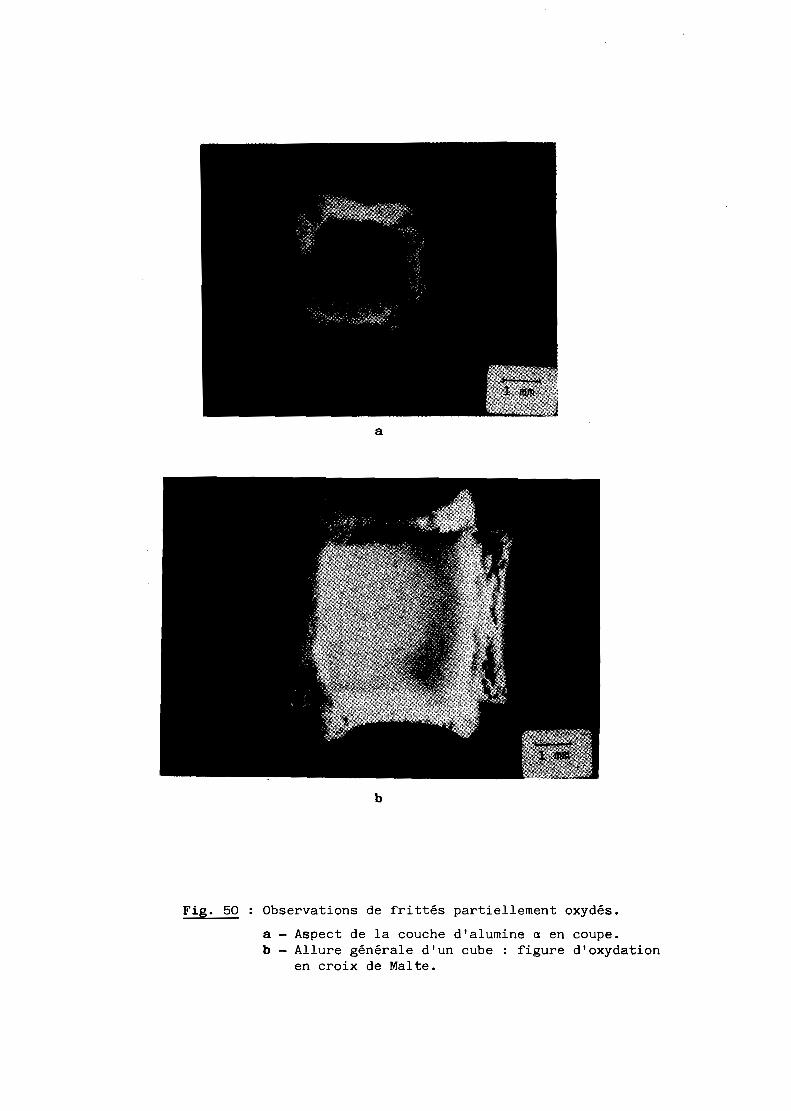
a
b
Fig. 50 Observations de frittés partiellement oxydés.
a Aspect de la couche d'alumine a en coupe.b Allure générale d'un cube: figure d'oxydation
en croix de Malte.

- 110 -
de réaction (absence d'espace de diffusion volumique, influence de la
pression, invariablilité du substrat), mais la présence d'une couche d'alumine
y' complique le mécanisme. Les caractéristiques principales de cette couche
sont sa compacité et sa faible épaisseur (à relier à la faible perte de masse
résiduelle observée à la fin des cinétiques). On a donc affaire à deux
réactions distinctes :
AlaN y ---.
Alumine y' ---.
Alumine y' (compacte)
Alumine a (poreuse)
qui s'accompagnent l'une et l'autre d'un dégagement d'azote.
Le fait que le produit intermédaire (alumine y') existe, fut-ce en
faible quantité, montre que les deux réactions possèdent des vitesses du même
ordre de grandeur, la seconde étant légèrement plus lente que la première.
Au plan cinétique, la réaction qui est responsable de l'oxydation de
l'oxynitrure est à l'évidence la première et c'est bien celle-ci qui est
suivie par thermogravimétrie, mais son effet pondéral se trouve diminué de
près de moitié (accroissement de masse observé de 3,3% environ au lieu
d'environ 6,1%) de par l'effet de la seconde réaction.
Quelle est la nature exacte de chacune de ces réactions ? La thermolyse
de l'alumine y' en a aboutissant à la formation d'un produit poreux est néces
sairement gouvernée par un régime type réaction. Il est très difficile en
revanche d'accéder à la réaction d'oxydation proprement dite. L'oxynitrure se
revêtant d'une couche d'alumine 'compacte dont l'épaisseur est toujours
faible, l'effet pondéral global ne la prend pratiquement pas en compte et la
cinétique doit suivre la loi de vitesse d'un régime de réaction quelle que
soit, au fond, la nature de la réaction elle-même. Seul, l'effet de la
pression doit être à coup sar attribué à la réaction d'oxydation, la
thermolyse n'y étant pas sensible (7). Ce renseignement permet d'affirmer que.
l'étape limitante de l'oxydation ne se situe pas à l'ihterface aluminey'/
alumine a car la loi de pression devrait être alors linéaire, mais en
l'absence d'information sur la nature de la semi-conductivité de l'alumine y',
tous les autres processus élémentaires sont susceptibles d'être limitants.

- 111 -
En résumé, l'ensemble du mécanisme qui vient d'être décrit est celui
d'un régime mixte oxydation/thermolyse complexe qui répond approximativement
aux lois cinétiques d'un régime de réaction.
Dans
réaction V
la surface
le cas de la symétrie cubique, la vitesse instantanée de lada
= dt est, pour un régime de réaction directement proportionnelle à
du cube d'arête a du substrat non encore attaqué; elle-même
fonction de l'avancement S(a)
daA(P)S(a)
dt
Dans ce cas, S(a) 2= 6 a ,
a = 1
a étant lié à l'avancement a. En effet
Vr
Vo
si Vr est le volume du substrat restant au temps t et Vo le volume du cube
initial d'arête a • On obtient alorso
3a
Soit encore
a = 1 ---,a 3
o
a = a (1 _ a )1/3 .o
L'équation de vitesse devient alors
da--:;:
dt6A(P)a 2 (1 _ a)2/3 •
o
Equation qui s'intègre en
da3 (1_a)1/3 = -6 A(P)a 2 t + ete
o

- 112 -
Les conditions initiales (a= 0, t = 0) imposent à la constante la
valeur 3, et l'on peut alors exprimer la transformée F(a) par la relation
F(a) 1 - (1 - a)1/3 = Kt
avec K 2a 2 A (P).o
Il est bien évident que, dans le cas de l'oxydation de l'oxynitrure
d'aluminium, le degré d'avancement a de la réaction, tel qu'il a été définiexpexpérimentalement au paragraphe précédent pour l'établisssement des figures 48
et 49, ne correspond à rien en toute rigueur, puisqu'il prend en compte les
deux réactions d'oxydation et de thermolyse, sa valeur maximum étant atteinte
lorsque les deux vitesses sont égales. Cependant, comme l'ensemble des résul
tats expérimentaux permet d'affirmer que l'erreur commise en le confondant au
degré d'avancement de la réaction d'oxydation a correspond à une majoration de
ce dernier qui avoisine 5% seulement, on se propose de tester la transformée
empirique suivante des cinétiques des réseaux des figures 48 et 49.
F(a ) = 1 - (1 - 0,95 a )1/3exp exp
Les figures 51 et 52 montrent la validité de celle-ci qui assure une
linéarisation satisfaisante des courbes.
Le réseau des transformées des isothermes permet de calculer l'énergie
d'activation expérimentale de l'ensemble de la réaction (cf. fig. 53).
-1E = 452 ± 39 kJ.mol
compatible avec la valeur déterminée directement.
Le réseau des transformées des isobares confirme pour sa part la loi de
pression déterminée directement, aux incertitudes près
A(P) = pO, 16
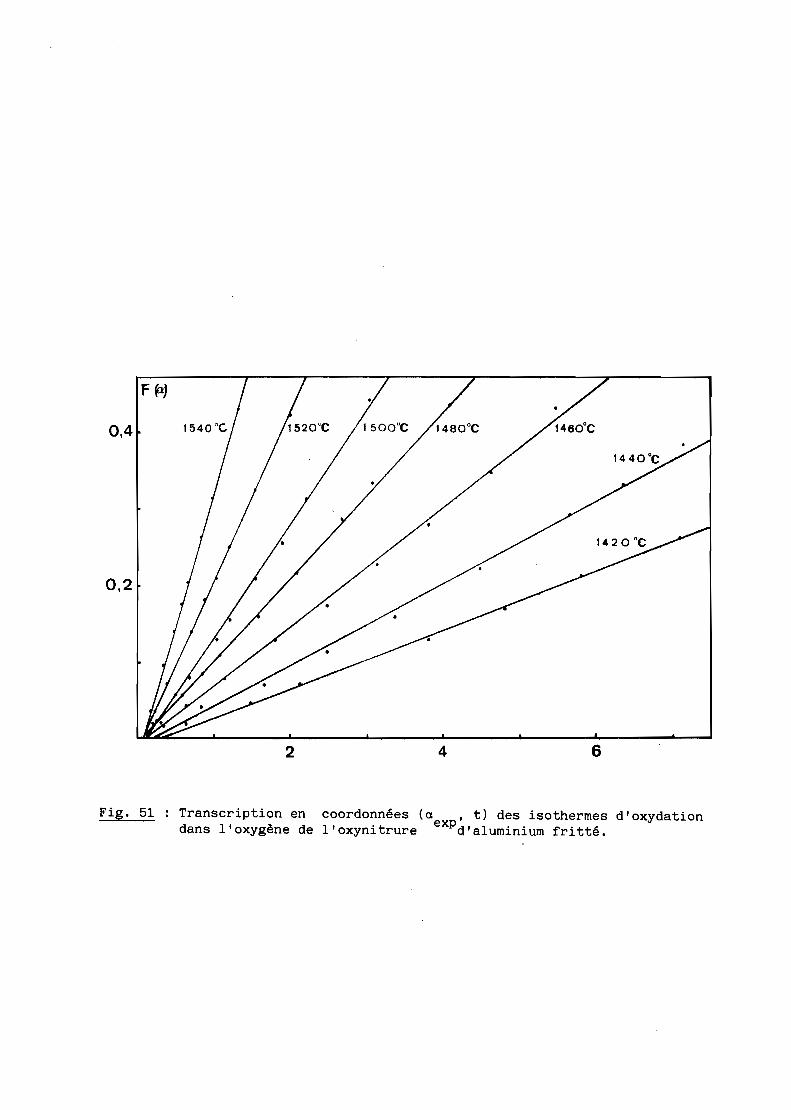
0,4
0,2
2 4
Fig. 51 Transcription en coordonnées (a • t) des isothermes d'oxydationdans l'oxygène de l'oxynitrure eXPd'aluminium fritté.
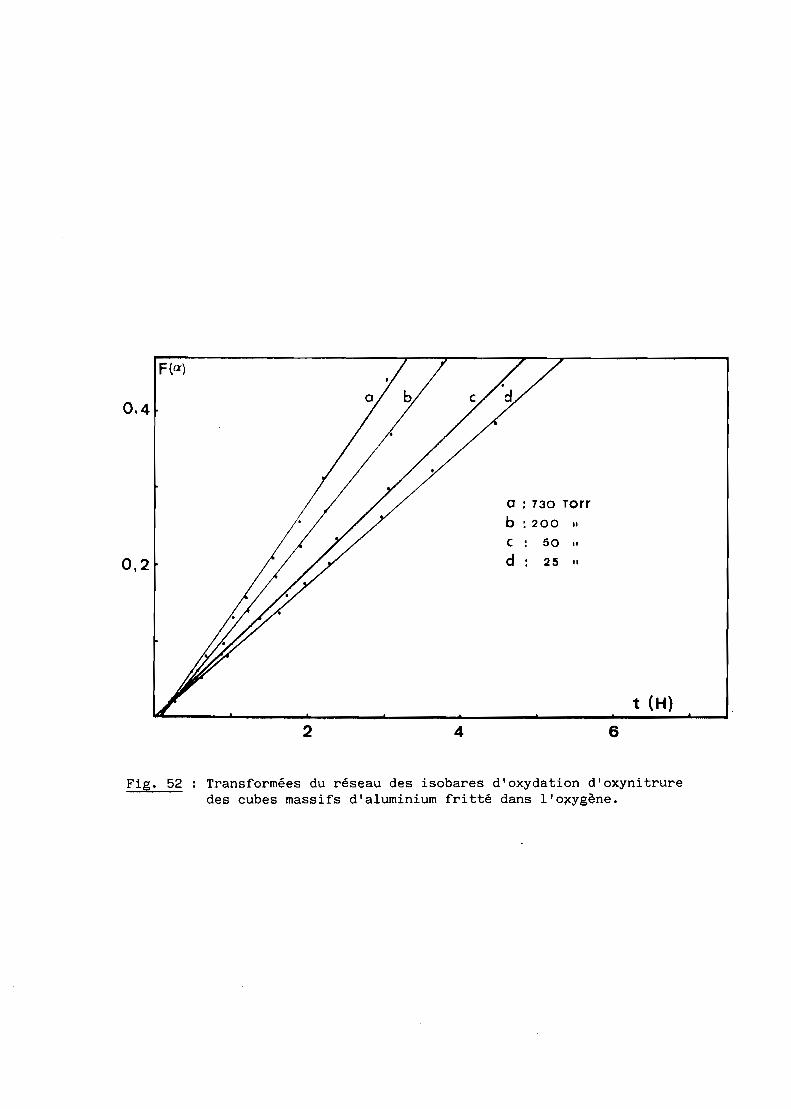
a ; 730 Torr
b :200 "C 50 "
0,2 d 25 "
t (H)
2 4 6
Fig. 52 Transformées du réseau des isobares d'oxydation d'oxynitruredes cubes massifs d'aluminium fritté dans l'o~ygène.
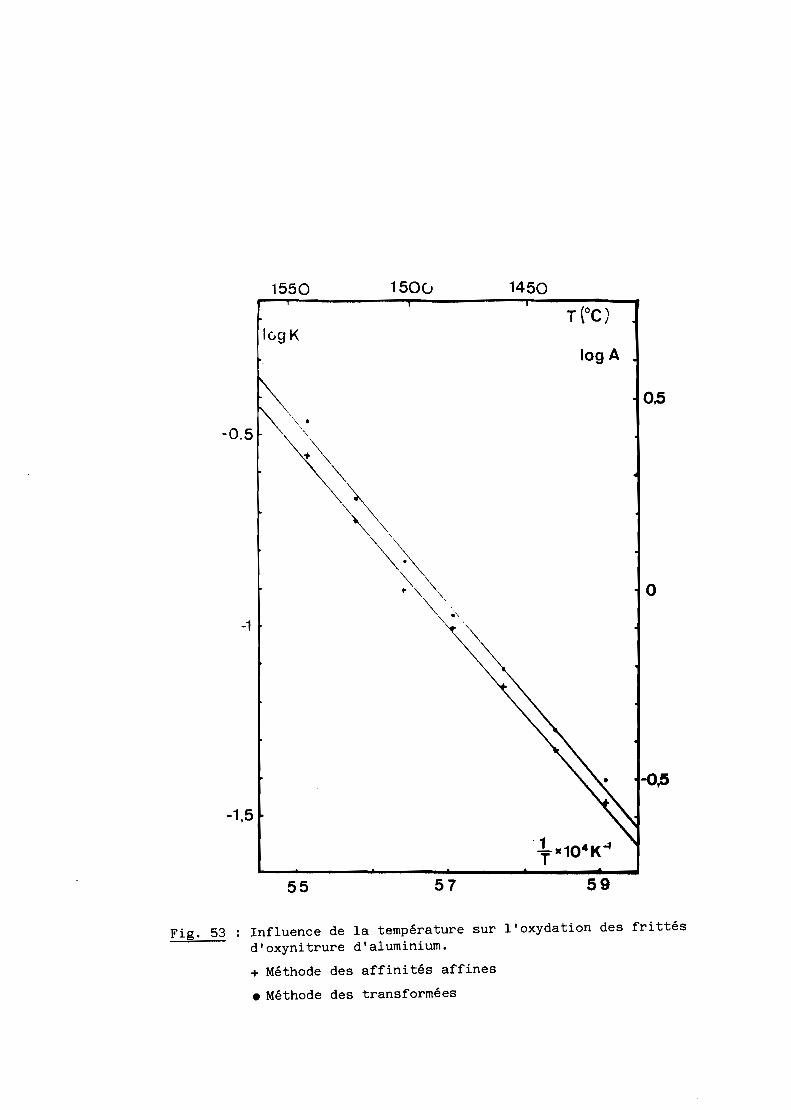
-0,5
-1
-1,5
1550
55
1500
57
1450
lagA
59
0.5
o
-0,5
Fig. 53 Influence de la température sur l'oxydation des frittésdtoxynitrure d'aluminium.
+ Méthode des affinités affines
• Méthode des transformées

- 116 -
C - CONCLUSION
dt
v =--__ = ete x (l _ 0,95 a )2/3 x pO,16x e- 452000/RTexp
L'oxydation des frittés d'oxynitrure d'aluminium est gouvernée par un
mécanisme complexe qui fait intervenir deux réactions, la première oxydation
proprement dite formant l'alumine y', l'autre de thermolyse en alumine a du
produit de la première, et légèrement plus lente. En fin de réaction, on
n'observe plus que la seconde pendant quelque temps, ce qui se traduit par une
perte de masse égale à 5 à 6% du maximum de prise de masse observé.
Analytiquement, si l'on définit pour la partie des thermogrammes corres
pondant à l'augmentation pondérale, un degré d'avancement expérimental aexp
rapport du gain de masse au temps t à celui maximum observé, les courbes
cinétiques répondent à la loi de vitesse :
da exp
pour des avancements, pression et températures compris respectivement entre
0,05 et 0,90, 25 et 730 Torr, et 1693 et 1813 K, qui décrit l'ensemble du
mécanisme mixte mis en évidence. La réaction d'oxydation est gouvernée par un
processus influencé par la pression qui est soit de nature diffusionnelle à
travers la mince couche d'alumine y' formée, soit de nature interfaciale
interne (ou de réaction). La thermolyse qui forme l'alumine a poreuse suit un
régime de réaction.
Dans l'ensemble, ces résultats sont cohérents avec ceux relatifs à
l'oxydation de la poudre puisque la nature des produits formés est la même
(même si, en raison des températures d'étude différentes, leurs proportions
sont inversées, l'alumine y' étant beaucoup plus instable dans le domaine de
température d'oxydation des cubes) et que la loi de vitesse est analogue. On
notera également que les valeurs expérimentales des énergies d'activation
sont proches.

Chapi t.r-e 11...
OXYDATION DE L'OXYNITRURE
D'ALUMINIUM TRANSPARENT
Les échantillons, issus du procédé de fabrication présenté en seconde
partie, se présentent sous forme de petits cubes de 4 mm d'arête obtenus par
découpe à la scie lente dans des disques de 45 mm de diamètre et de 5 mm
d'épaisseur. Chaque échantillon subit sur toutes ses faces, le même polissage
mécanique qui assure un état de surface initial satisfaisant et uniforme. Les
cinétiques d'oxydation sont obtenues dans des conditions expérimentales ana
logues à celles retenues pour l'étude de la poudre ou des frittés.
A - RESULTATS GENERAUX
1 - Influence de la température
L'oxydation en montée linéaire de température de 90 K/h sous pression
atmosphèrique d'oxygène, transcrite sur la figure 54 en gain de masse relatif
6m/m , montre que le début de la réaction se produit vers 1200 0C ; elle neo
s'accélérent vraiment qu'à partir de 1400 0C environ. L'on remarque aussi sur
la courbe plusieurs lègères ondulations. Vers 1520oC, ~'échantillon cesse de
prendre du poids et, au contraire, une perte se manifeste aux températures
supérieures.
Le gain de masse maximum observé (3,4 % environ) est supérieur à celui
attendu pour la réaction donnant l'alumine a, soit 3,16% ; et l'on retrouve
donc certainement là le phénomène de rétention d'azote dans l'alumine y'
signalé dans les chapitres précédents lors de l'oxydation de la poudre ou des
frittés (2).
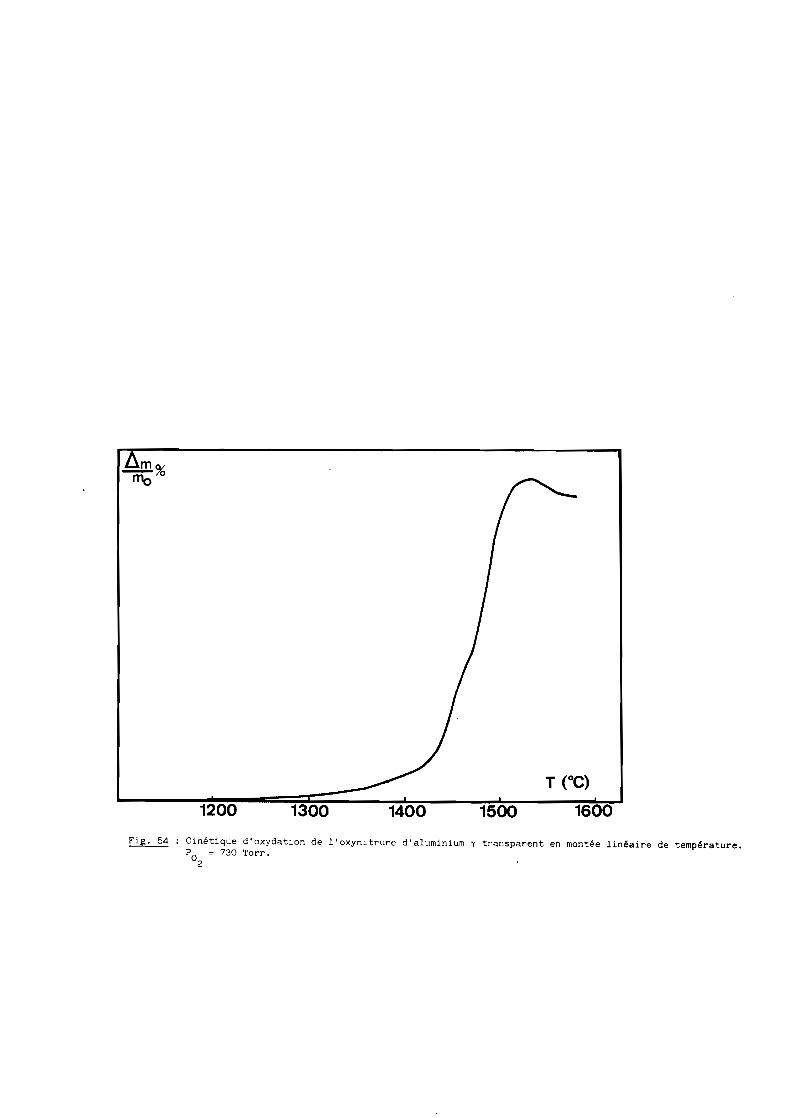
1200 1300 1400 1500 1600
~ Cinétique d'oxydation de l'oxynitrure d'aluminium y transparent en montée linéaire de température.Po = 730 Torr.
2

- 119 -
Le réseau des isothermes présente en figure 55 les thermogrammes obtenus
entre 1420 et 1520°C sous pression atmosphérique d'oxygène. Leur allure est
globalement accélérée jusque vers leur achèvement, sans incubation et avec
vitesse initiale non nulle, mais ils présentent de nombreuses irrégularités
qui prennent la forme d'accélérations exagérées parfois suivies de ralentis
sements. Les cinétiques ont été arrêtés au voisinage du gain de masse maximum.
Au delà, on observe une très lente perte de masse qui s'estompe progressive
ment, prenant pour asymptote la valeur calculée de 3,16%, en gain de masse
relatif global, de la réaction fournissant l'alumine a. Des tests de reproduc
tibilité montrent le caractère aléatoire des irrégularités cinétiques. Il
n'existe évidemment pas d'affinité entre ces différentes courbes et l'on ne
peut donc en faire l'analyse cinétique directe.
2 - Influence de la pression
Les thermogrammes rapportés à la figure 56, obtenus pour des pressions
d'oxygène dans l'enceinte réactionnelle de 12,5 à 730 Torr ont la même allure
que les isothermes et leurs irrégularités aléatoires rendent également impos
sible la description analytique de la loi de pression, les courbes n'étant pas
affines entre elles.
3 - Observations morphologiques
L'observation macroscopique de petits cubes incomplétement oxydés montre
qu'ils s'entourent, au début de la réaction, d'une couche d'oxydation blanche
et opaque qui se fissure rapidement au niveau des arêtes en donnant une figure
d'oxydation en croix de Malte (cf. le cliché de la fig. 57a). Aux stades plus
avancés de la réaction, on peut faire les observations suivantes :
a - la surface des faces oxydées demeure bien cohérente et
conserve la mémoire des joints de grains de l'oxynitrure de départ;
b - au-dessous apparaît une couche d'oxyde très friable et poreuse
constituée uniquement d'alumine a et en tout point semblable à celle observée
sur les frittés non transparents partiellement oxydés ;
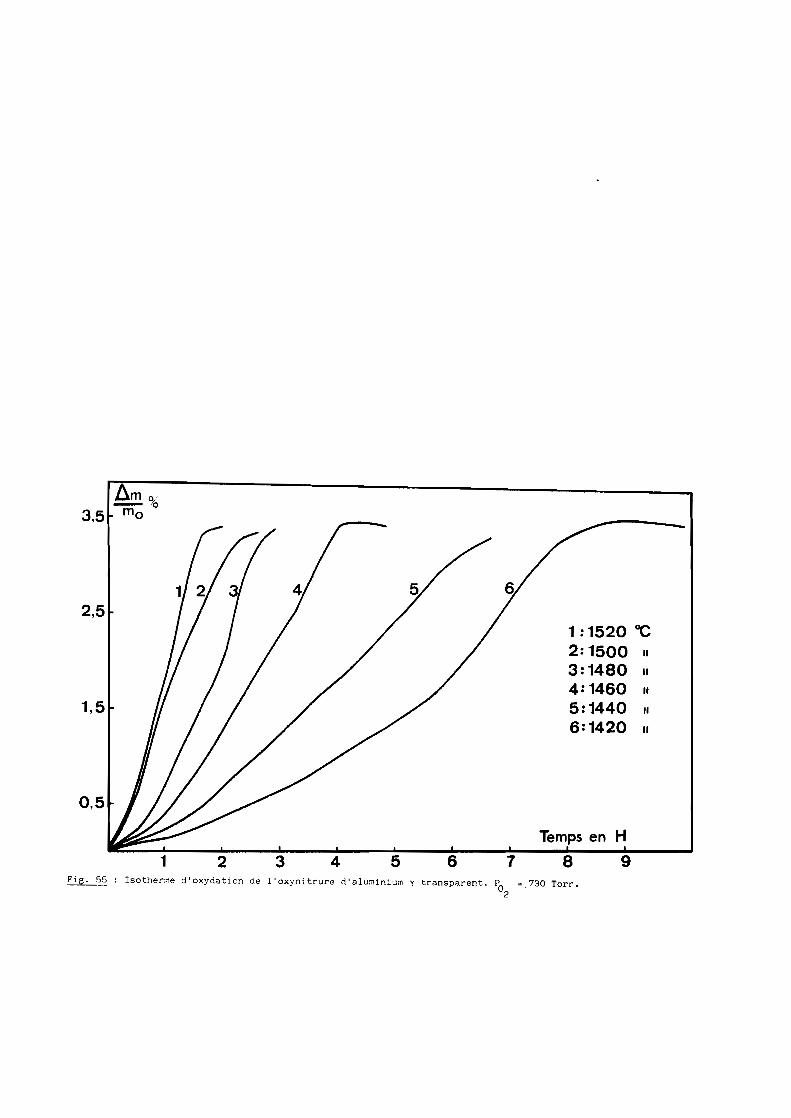
8 9Temps en H
7654321
am 0,_70
3,5 mo
2,51 :1520 oc2: 1500 "3:1480 Il
4: 1460 Il
1,5 5:1440 "6:1420 "
0,5
~ : Isotherme d'oxydation de l'oxynitrure d'aluminium y transparent. Po ;,730 Torr.
2
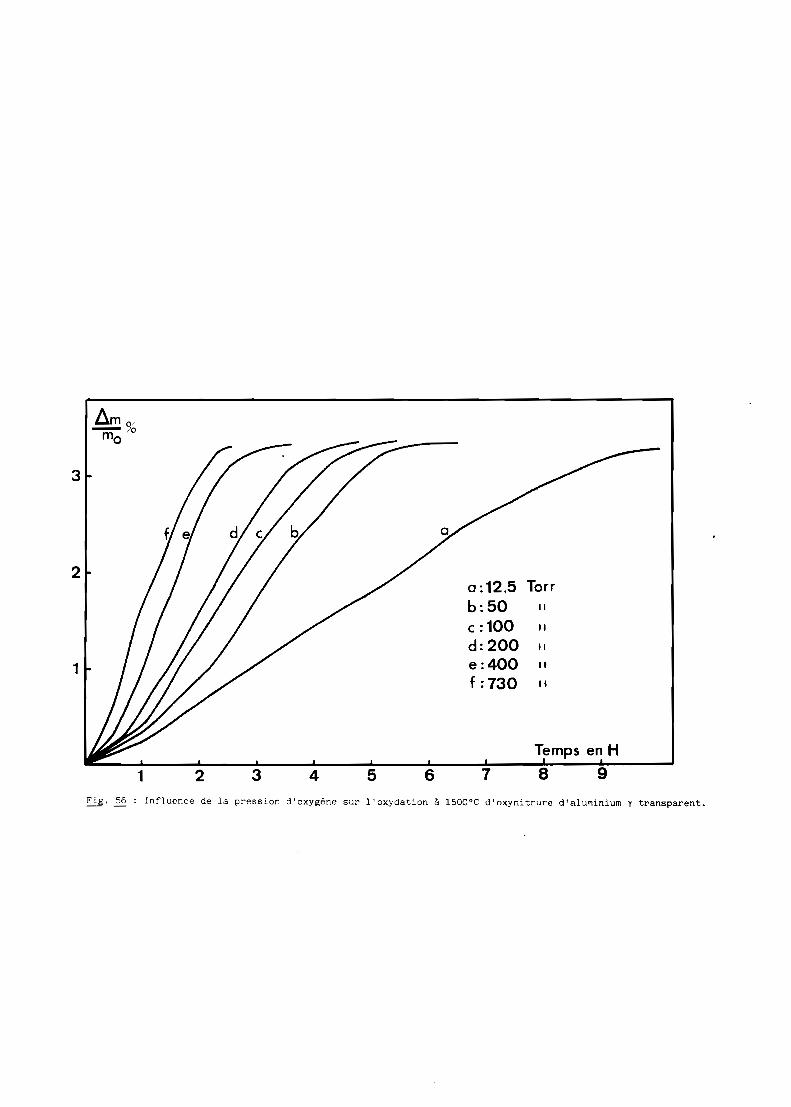
8 9Temps en H
7654321
/im 0'-70mo
3
~ Influence de la pression d'oxygène sur l'oxydation à l5000C d'oxynitrure d'aluminium y transparent.

- 122 -
c - au voisinage du noyau central d'oxynitrure, il existe une
couche également poreuse, mais formée de grains pour partie assez bien
individualisés (cf. fig. 57b) dont le spectre radiocristallographique fournit
les raies de l'alumine 0 et de l'oxynitrure Y (ou/et de l'alumine y' qui ne
s'en distingue spectralement que très difficilement). La preuve indirecte de
la présence de l'alumine y' à cet endroit demeurant l'excessive prise de masse
observée en thermogravimétrie
d - Le coeur de l'échantillon, de forme cubique au début, tend à
devenir progressivement sphérique. Son analyse radiocristallographique ne fait
apparaître aucune modification structurale par rapport au matériau initial. En
fin de réaction, la nacelle porte-échantillon contient une poudre blanche avec
quelques blocs d'allure parfois pyramidale et très fragiles, vestiges de la
structure en croix de Malte.
B - INTERPRETATION
L'allure irrégulière des thermogrammes et la complexité de la texture et
de la composition de la couche d'oxydation prouve de façon évidente que le
mécanisme réactionnel n'est pas simple. On répertorie cependant quelques
caractères principaux.
1 - Il existe deux réactions distinctes
Conformément aux résultats observés sur la poudre, puis sur les frittés
bruts (cf. supra) on vérifie la coexistence de deux réactions, l'une d'oxyda
tion de l'oxynitrure produisant un dégagement d'azote et l'alumine y', l'autre
de thermolyse de l'alumine y' en alumine 0, accompagnée d'un nouveau dégage
ment d'azote. A tout instant, ces réactions se produisent avec des effets
pondéraux opposés, à des vitesses légèrement différentes (seule subsiste la
seconde en fin de réaction) et donc en des localisations sensiblement dis
tinctes au sein de l'échantillon.
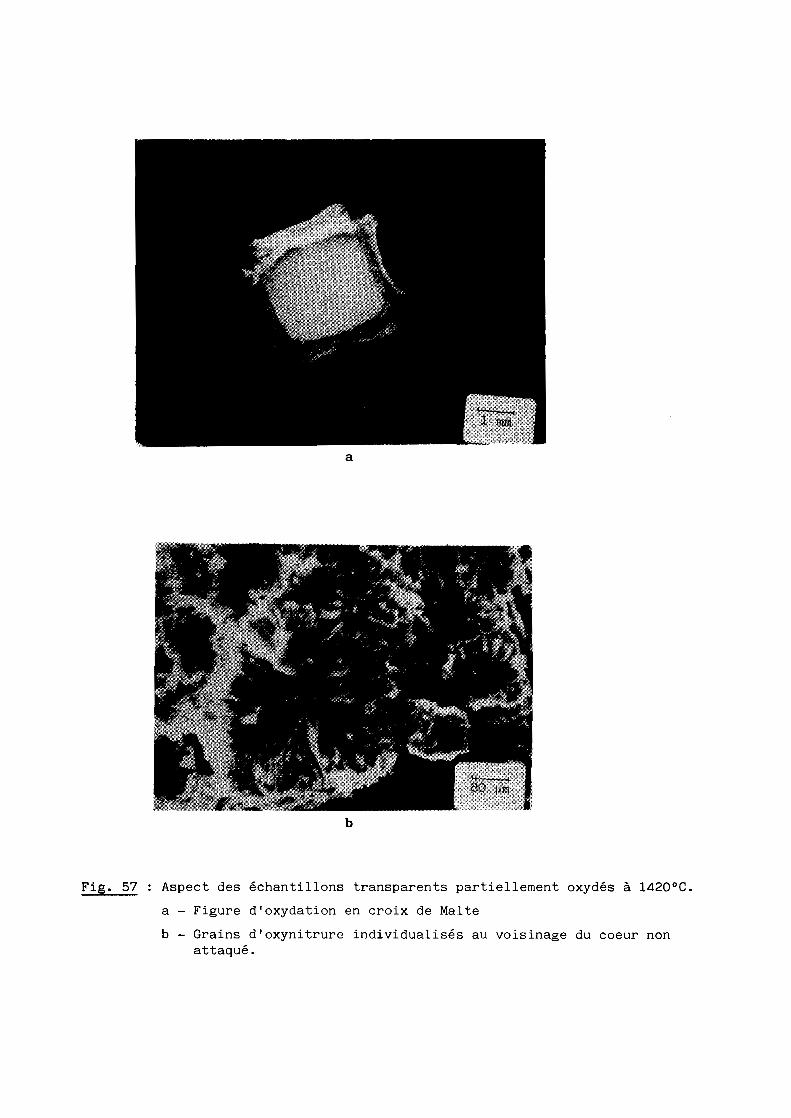
a
b
Fig. 57 Aspect des échantillons transparents partiellement oxydés à 1420 oC.
a - Figure d'oxydation en croix de Malte
b - Grains d'oxynitrure individualisés au voisinage du coeur nonattaqué.

- 124 -
2 - La localisation des réactions est difficile
La couche oxydée étant poreuse et délitée, on ne peut pas plus que dans
le cas des cubes massifs bruts de frittage définir d'interface interne et
externe séparés par un espace de diffusion volumique. Mais on ne peut conclure
non plus à l'établissement d'un régime de réaction localisé à la surface du
noyau central d'oxynitrure, car les observations morphologiques montrent que
l'oxydation provoque un déchaussement des grains qui continuent de réagir
après avoir été individualisés, alors même que le front de réaction a pénétré
plus avant dans l'oxynitrure.
Ces résultats impliquent qu'il est très difficile d'accéder à la locali
sation exacte des réactions, en particulier pour celles qui se produisent au
niveau des grains où coexistent les phases oxynitrure, et alumines a et r'.
Ainsi l'hypothèse de participation d'une diffusion volumique n'est pas à
exclure puisque les grains fraîchement décollés du substrat sont entourés
d'une couche d'oxyde qui paraît assez compacte (cf. fig. 57b) ce qui serait
compatible avec la valeur de 1,1 du coefficient d'expansion volumique pour la
transformation de l'oxynitrure en alumine r'.
Par ailleurs, l'instabilité de l'alumine r' aux températures d'étude
entraîne corrélativement un dégagement d'azote et une modification structurale
à effet volumique négatif (justifiant la grande fragilité de l'alumine a formée
in fine) ; cette réaction est également impossible à localiser précisément.
3 - L'influence de la pression n'est pas quantifiable
La texture de la couche contenant les produits d'oxydation permet à
l'oxygène de pénétrer par diffusion gazeuse jusqu'à l'oxynitrure, et l'azote
dégagé s'évacue également de cette façon, en contre-courant de l'oxygène, dont
il est dès lors impossible de connaître la pression partielle régnant réelle
ment au niveau de la (ou des) surface(s) réactive(s) (46). L'influence de la
pression dans l'enceinte réactionnelle est donc bien un paramètre à prendre en
compte (puisque sa variation joue nécessairement pour partie sur celle qui
existe à l'intérieur de l'échantillon) mais l'ampleur de cette influence sur
la marche des réactions ne peut être déterminée.

- 125 -
4 - L'allure des cinétiques est justifiée
L'étendue du terrain réactionnel quelle que soit son exacte localisation
dépend de trois facteurs différents :
a - la réduction de la surface du front de réaction au fur et à
mesure de la diminution des dimensions du noyau d'oxynitrure non encore
attaqué, qui joue dans le sens d'une décélération des cinétiques (29) ;
b - la croissance constante du nombre de grains d'oxynitrure
individualisés par suite de la progression du front réactionnel, qui accélère
d'autant la vitesse globale de l'oxydation. L'augmentation du nombre de grains
détachés dépend beaucoup de la structure de l'oxynitrure qui est d'autant plus
fragile que les grains sont plus gros. Ainsi, vu les différences locales de
taille des grains, selon que le front réactionnel rencontre une zone à gros
grains ou à grains plus fins, la vitesse de décollement de ceux-ci est corré
lativement plus ou moins rapide : on comprend ainsi les ondulations des
thermogrammes
c - la consommation progressive des grains isolés qui tend à
limiter l'influence du facteur précédent.
Ainsi, l'allure globalement accélérée des cinétiques s'explique-t-elle
par la part de plus en plus importante prise par les grains individualisés qui
s'oxydent, tandis que les ondulations des thermogrammes doivent être reliées à
la plus ou moins grande résistance mécanique locale de l'oxynitrure. Pour
achever la justification de l'allure des cinétiques, il suffit de prendre en
compte l'effet pondéral de thermolyse de l'alumine y' (perte de masse) qui
devient prépondérante en fin de réaction.
C - CONCLUSION
L'oxydation de l'oxynitrure d'aluminium transparent met encore en
évidence (cf. les études sur la poudre et le fritté brut) la coexistence de
deux réactions distinctes, l'une d'oxydation proprement dite fournissant
l'alumine Y', l'autre de thermolyse de celle-ci.

- 126 -
On observe ici un ensemble de phénomènes encore plus complexes que dans
les cas précédents puisque, à l'imbrication des processus chimiques mise en
évidence sur les frittés bruts se surajoute l'effet du paramètre mécanique
qui, par les perturbations cinétiques qu'il génère, rend absolument impossible
toute approche du mécanisme réactionnel.
Il apparaît cependant que c'est l'effet de la taille des grains (et plus
généralement de la résistance mécanique, cf. infra) que différencie principa
lement les deux dernières études. En fait, aucune donnée expérimentale ne les
oppose et il y a tout lieu d'affirmer que les oxydations des matériaux bruts
de frittage et transparent sont gouvernées par des processus chimiques
analogues et que les mécanismes réactionnels se trouvent modifiés par les
caractéristiques mécaniques différentes des deux produits, de façon désormais
classique en particulier sur les céramiques de type nitrure (47).

PROPRIETES PHYSIQUES ET MECANIQUES

Chapitre l
CARACTERISTIQUES PHYSIQUES
A - PARAMETRE DE MAILLE
D'après les travaux antérieurs (1, 12), le paramètre de maille de la
phase spinelle AlaN y présente une variation linéaire en fonction de la
quantité d'azote substituée aux atomes d'oxygène dans les sites anioniques.
Compte tenu de la divergence constatée (fig. 58) entre les résultats, il nous
a paru utile de reprendre ces expériences.
1 - Mode opératoire
La détermination des paramètres de maille a été effectuée par diffracto
métrie de RX aux grands angles à l'aide d'un diffractomètre Hubert à antica
thode de cuivre muni d'un monochromateur et alimenté par un générateur Philips
avec une vitesse de défilement de 0,025°/min. Les échantillons ont été
analysés en présence d'un étalon de silicium. Etant donnée la finesse des
raies obtenues, le paramètre de maille est déterminé à partir de raies 115,
044 et 026 de l'oxynitrure. Ce choix permet d'obtenir des résultats
reproductibles et exclut la prise en compte des imprécisions observées au
niveau des petits angles.
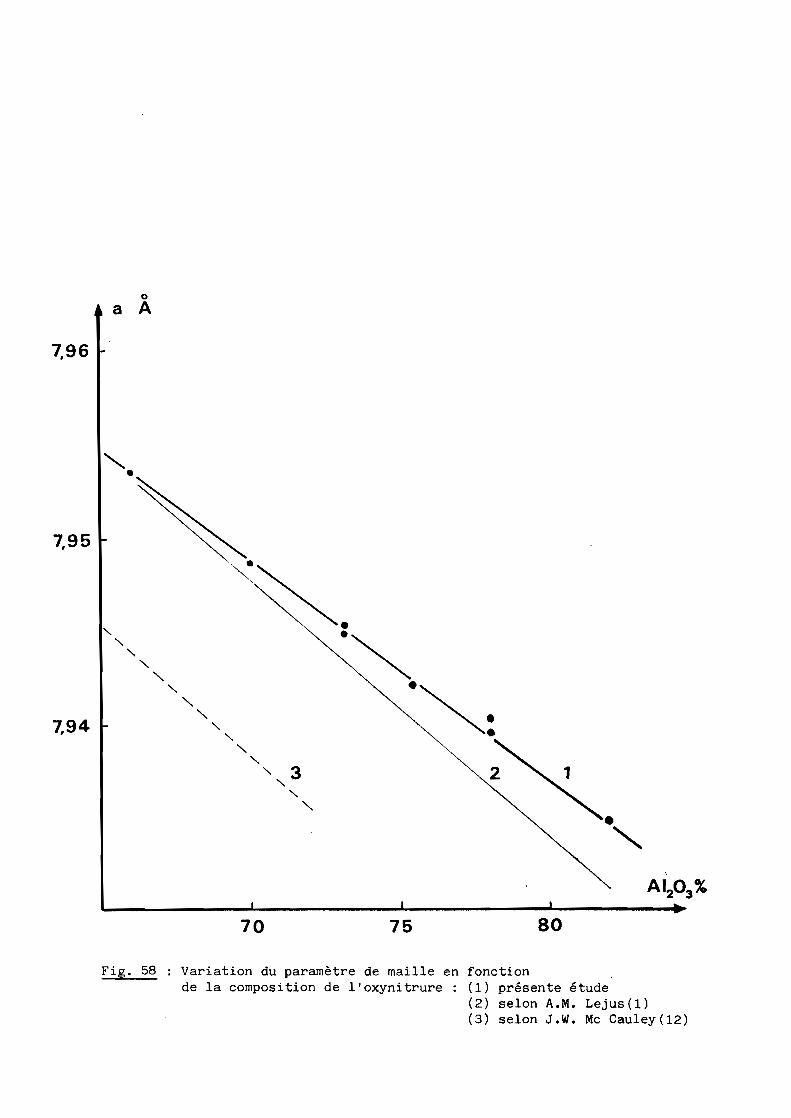
7,96
7,95
7,94
oa A
<,•
<,<,
"- -,-,
-,-,
-,-,
-,-,
-,"- 3-,
-,-,
70 75 80
•-,
Fig. 58 Variation du paramètre de maille en fonctionde la composition de l'oxynitrure : (1) présente étude
(2) selon A.M. Lejus(l)(3) selon J.W. Mc Cauley(12)

- 130 -
2 - Paramètre de l'oxynitrure d'aluminium aux différents stades de
fabrication
Les échantillons résultant de la réaction entre A1 203et AIN ont été
analysés. Les résultats présentés au tableau XVI sont très proches de ceux
obtenus par A.M. LEJUS (l) ; et ils témoignent de la stabilité du produit du
point de vue de sa composition, de la poudre au fritté, ou au matériau trans
parent.
1 Températures de traitement Valeur du paramètre a 1Nature du produitlien 0 C 1 en A 1
1--------------------1----------------------------1-------------------------11 Poudre 1 1850 1 7,9431 ± 0,0005 1
1--------------------\----------------------------1-------------------------\1 Matériau fritté 1 1750 1 7,9428 ± 0,0005 1
1--------------------1----------------------------1-------------------------1IMatériau transparent 1 1950 1 7,9432 ± 0,0005 1_________~__----_I 1
Tableau XVI Valeurs du paramètre a en fonction de la température de traite
ment et de la nature du produit.
La valeur centrale du paramètre de maille de l'oxynitrure fabriqué par
réaction d'état solide ressort ainsi: a = 7,9430 ± 0,0005, l'incertitude
retenue prenant en compte l'ensemble des mesures.
Dans le cas de l'AION obtenu par carbonitruration en faisant varier la
proportion de carbone, on obtient des produits de compositions molaires
équivalentes en nitrure comprises entre 18 et 34%. Ces résultats sont présen
tés à la figure 58. Ils vérifient également l'évolution linéaire du paramètre
de maille en fonction de la quantité d'a~ote fixée dans la maille spinelle
(par substitution à l'oxygène) avec:

~ 131 -
Ces résultats sont très comparables à ceux de A.M. LEJUS (1). Le
décalage de 1 à 2% const~té en composition provient vraisemblablement des
différences observées dans les méthodes de fabrication et se situe dans un
domaine d'incertitude raisonnable.
Quant aux résultats de Mc CAULEY, la différence est très sensible et ne
peut se justifier que par des glissements de composition par rapport à la
teneur nominale en a~ote, peut-être en liaison avec la volatilisation du
nitrure qui intervient au cours du traitement thermique des poudres à tempéra
ture élevée.
Au Vu de cette étude et de celles relatives à la synthèse (cf. 2ème
partie Chapitre 1), le diagramme de phase présenté par A.M. LEJUS est en tout
point conforme à nos résultats, contrairement à celui de J.W.Mc CAULEY qui
paraît plus éloigné de la réalité.
B - MASSE VOLUMIQUE
La densité théorique de la phase AION y a été déterminée à partir du3- 2-paramètre de maille en supposant que les ions N et 0 sont répartis dans
les 32 sites anioniques de la maille spinelle. La synthèse de l'oxynitrure
étant en accord avec la formule :
A1 2 03 3 Nx ,-x - x
la maille renferme, dans le modèle dit "à anion constant" (12).
2o3 - 2x
32(3-3x)32x
3 - 2x
3---- ions N et----- ions. A13 +lons ,
3 - 2x
32(2-x)
pour une masse de :
3264 - 1952 x
M :; ------- X ----g .3 - 2x 6,023
La connaissance du paramètre de maille permet donc celle de la masse
volumique, soit:
3264 - 1952 x 10-23
(3-2x) (7,914+0,117x)3 6,023

~ 132 ~
3La variation de la masse volumique, comprise entre 3,66 et 3,70 glcm et
correspondant au domaine étudié, est représentée sur la figure 59. Pour une3
composition molaire de 27% A1N et 73% A12
03
, elle est égale à 3,683 glcm .
C - COEFFICIENT DE DI~ATATION THERMIQUE
L'analyse dilatométrique a été réalisée à l'aide d'un dilatomètre élec
tronique ADAMEL jusqu'à 100Q oe à Partir d'éprouvettes d'oxynitrure à porosité
inférieure à 1%. Le diagramme de la figure 60 représente la courbe dilatomé
trique en fonction de la température. L'évolution linéaire entre 20 et 700 0C
est suivie d'une légère variation de la pente aux températures supérieures.
Les moyennes obtenues
sont très voisines de celles mentionnées par KIEFFER et al. (10). Le comporte
ment dilatométrique de l'AION se rapproche de celui de l'alumine-6
(~=8,3.10 loc de 0 à 100QPC), mais il est isotrope compte tenu de la struc-
ture cubique.
D - PROPRIETES OPTIQUES
L'étude des propriétés optiques du matériau transparent porte sur des
mesures de transmission d'absorption et de réflexion, ainsi que sur la mesure
de l'indice de réfraction. Elles ont été réalisées dans le visible, l'ultra
violet et l'infrarouge à l'aide des spectrophotomètres PERKIN-ELMER 551 S et
225.
1 - Transmission, réflexion et absorption
Le spectre représenté sur la figure 64 indique une zone de transmission
dans le domaine de longueurs d'onde de 0,2 à 5,5 ~m, couvrant ainsi toute la
zone du visible et une partie importante de l'infrarouge. La pente ascendante
du spectre vers les grandes longueurs d'ondes s'interprète par les défauts du
polissage des échantillons et par l'influence de leur porosité résiduellefigure 62 (48, 49).
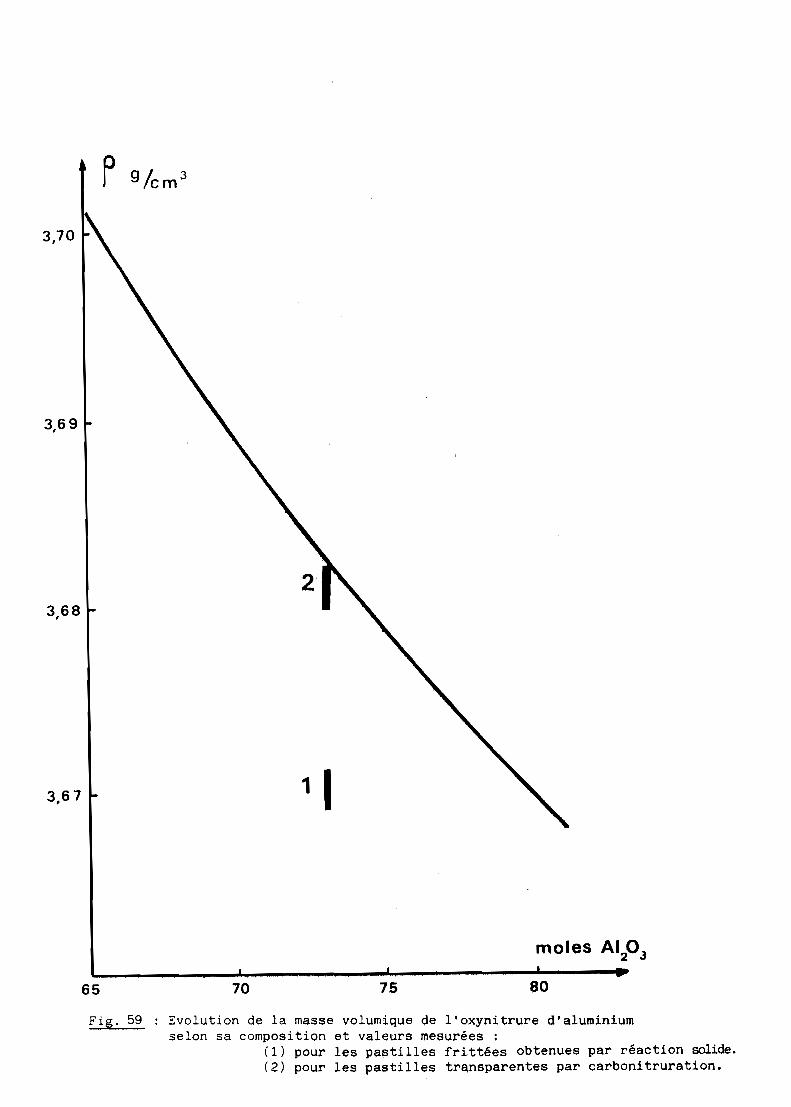
3,70
3,69
3,68
3,67
65 70 75
Fig. 59 Evolution de la masse volumique de l'oxynitrure d'aluminiumselon sa compositiQn et valeurs mesurées:
(1) pour les pa~tilles frittées obtenues par réaction solide.(2) pour les pastilles transparentes par carbonitruration.
8
6
4
2
200 400 600 800 1000
Fig. 60 Courbe dilatométrique d'un échantillon dense d'oxynitrure d'aluminium(porosité inférieure à 1%).
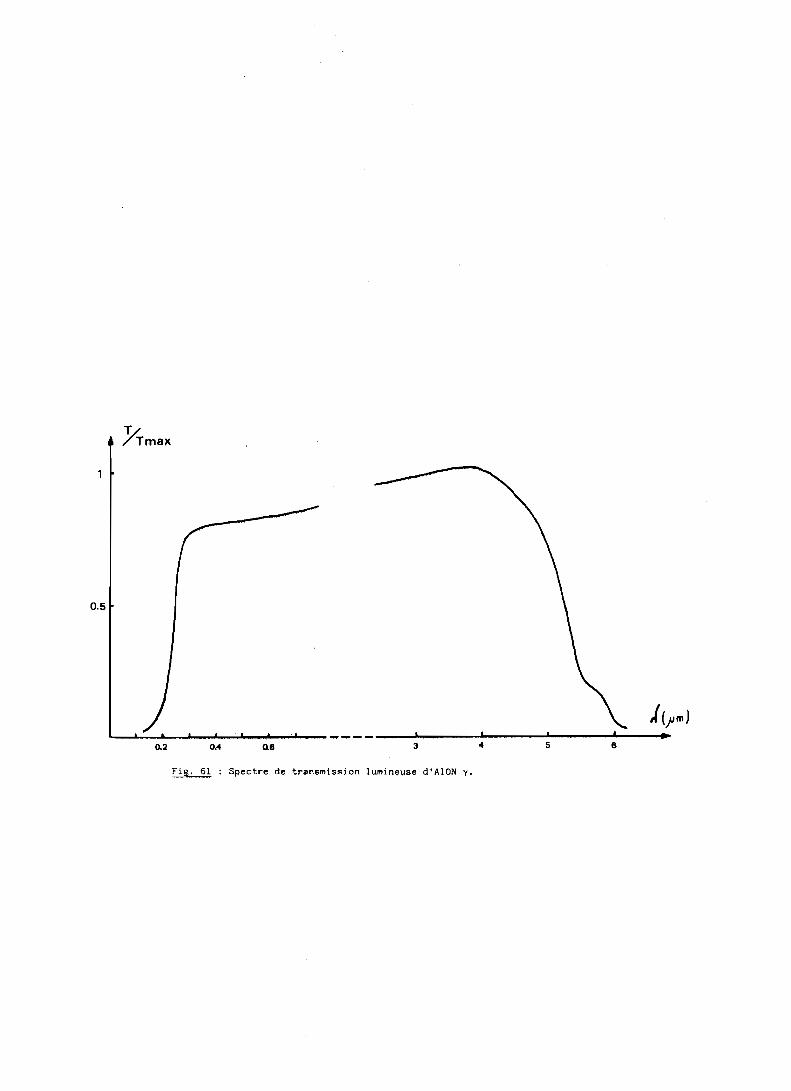
0.5
JiTmax
....-"-_..&.._...._ ...._..,;,,0 "-_..... .......__---....-----......----.....---.....
0.4 0.0 3 4 5 6
~ : Spectre de transmission lumineuse d'AlDN y.
T%100
10
1
0.1
0.01
p%
0.01 0.02 0.03
Fig. 62 Transmission optique en fonction de la porosité pour différentestailles de pores pour une alumine polycristalline (49).
- 137 -
On notera que la bande passante ne dépend ni du procédé d'élaboration, ni de
la composition de ~a phase oxynitrure.
Dans le cas d'un échantillon obtenu par réaction entre le nitrure et
l'alumine, d'épaisseur 2 mm, on obtient une transmission directe (fig. 63) de
40 à 50% dans le visible et dans l'infrarouge pour des longueurs d'onde allant
de 0,6 à 4 ~m. Mais lorsqu'on adapte une sphère intégrante au spectrophoto
mètre (PERKIN ELMER 551 S), cet échantillon donne une transmission globale
supérieure à 60% pour un angle de résolution de 10 degrés. Les mesures de
réflexion effectuées en utilisant une sphère intégrante fournissent un taux de
réflexion de 15% dans une zone de longueur comprise entre 0,3 et 0,8 ~m. Ce
taux de réflexion dans le visible est un peu plus élevé que dans le cas des
verres ordinaires, ce qui implique un indice de réfraction supérieur.
Des échantillons de même épaisseur obtenus par carbonitruration
affichent une transmission directe de 65 à 80% dans le visible et l'infrarouge
pour des longueurs d'ondes de 0,6 à 4 ~m. L'absorption dans ce cas est infé
rieure à 5%.
Ces résultats qui sont rapportés au tableau XVII montrent que l'oxyni
trure d'aluminium y est une céramique réellement transparente. Les meilleurs
résultats sont observés pour le matériau obtenu par carbonitruration de
l'alumine.
Echantillcn Réflexicn Transmissicn directe Transmissicn Absoqlticn Bande passanteobtenu par maxirrale maxi.n'ele totale rraxi , maxirrale en um
(en %) (en %) (en %) (en %)
Réacticnsolide 15 50 65 25 0,2 à 5,5
All'i + Al203
Carbanitru-raticn de 15 85 5 0,2 à 5,5l'alunine
Tableau XVII Différentes valeurs de la transmission en fonction du
procédé de synthèse.
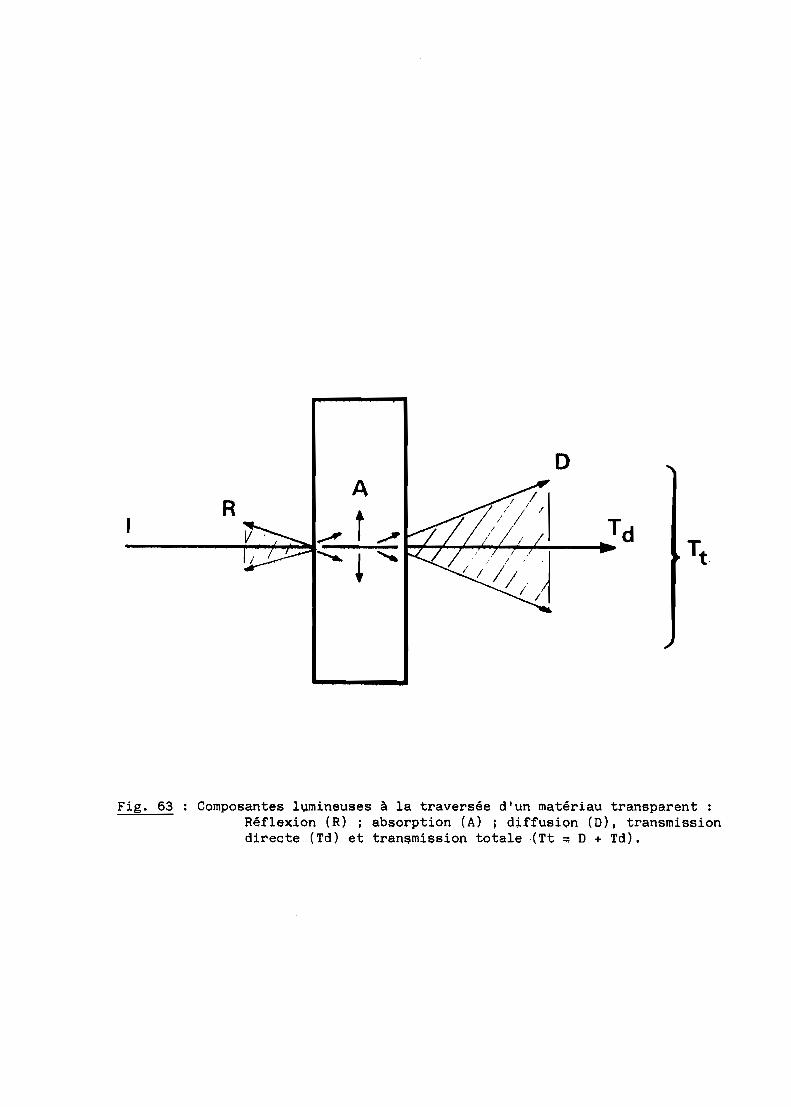
o
R
Fig. 63 Composantes lumineuses à la traversée d'un matériau transparent:Réflexion (R) ; absorption (A) ; diffusion (D), transmissiondirecte (rd) et transmission totale ·(Tt ~ D + Td).

- 139 -
2 - Indice de réfraction
L'indice de réfraction n est mesuré par une méthode de visée sur les
faces à partir d'échantillons transparents d'épaisseur e = 2 mm. La mise au
point est réalisée successivement sur les deux faces et le mouvement e' de la
platine du microscope déterminé à l'aide d'un comparateur de précision. Le
rapport n =~, donne la valeur de l'indice de réfraction qui est dee'
1,76 ± 0,01, l'incertitude étant définie par la précision de la mesure.
Pour vérifier ce résultat, d'autres mesures ont été faites sur un
réfractomètre à angle limite, puis par utilisation de la méthode de Brewter
(mesure de l'angle d'iB tel que la réflexion d'un faisceau polarisé soit
minimale tg i B = n). Ces méthodes confirment la valeur déterminée précédem
ment.

Chapi tre II
CHARGE A LA RUPTURE A LA TEMPERATURE AMBIANTE
A - CHARGE A LA RUPTURE DES FRITTES
1 - Echantillons
La charge à la rupture à l'ambiance a été déterminée en flexion 3 points
sur des éprouvettes 5 x 5 x 25 mm environ reposant sur deux appuis distants de
19 mm, avec une contrainte imposée au centre de l'éprouvette.
Sa valeur exprimée en MPa est calculée à partir de l'expression déjà
mentionnée dans la première partie
et la moyenne déterminée sur la base de quatre essais au moins avec un inter
valle de confiance au seuil de probabilité de 90% (50).
Les essais ont été effectués perpendiculairement (1) et parallèlement
(II) à la direction du frittage sous charge. Les résultats sont rapportés au
tableau XVIII pour des frittés obtenus avec des poudres issues de la réaction
directe entre le nitrure et l'alumine et présentant une porosité résiduelle
inférieure à 0,4%.

- 141 -
1 Direction de rupture 1 Nbre d'éprouvettes 1 of (MPa) 1 Ecart type 1I--~-------------------I--------------------I----------1------------1
1 l 1 4 1 32~ 1 29 1
1----------------------1--------------------1----------1------------11 / / 1 8 1 285 1 20 1
1 1 1 1 1
Tableau XVIII Résistanae à la rupture en flexion 3 points.
Ils font apparaître une meilleure r~sistance à la rupture perpendicu
lairement à la direct~on de frittage, mais la différence d'environ 15%, n'est
pas statistiquement significative. La tenue mécanique de l'oxynitrure est par
ailleurs, intermédiaire entre celles de l'alumine (51) et du nitrure d'alumi
nium (52), respectivment de 250 et 450 MPa dans les mêmes conditions opératoi-
res.
2 - Influence de la porosité
Les résultats obtenus po~r des échantillons de porosité comprise entre
0,05 et 5,6 % sont consignés au tableau XIX et illustrés sur la figure 64. On
constate que la résistance à la rupture reste sensiblement constante pour des
porosités comprises entre 1 et 5%.
Aux porosités inférieures à 0,3%, on note cependant une nette diminu
tion de of' en corrélation secondaire avec la taille des grains qui justifie
la rupture plus facile des éprouvettes.
Le maximum de résistance à la rupture est obtenu pour des barrettes de
porosité voisine de 0,4% qui proviennent de pastilles réalisées à 17500C sous
une charge de 40 MPa pendant 4 heures, conditions retenues pour l'élaboration
du produit transparent (38) (cf. 2ème Partie - Chapitre III). Les éprouvettes
présentent alors une densification maximale sans présenter pour autant un
grossissement excessif des grains. C'est au voisinage de ce maximum qu'appa
raît cependant une légère anisotropie entre la résistance à la rupture selon
les directions de l'essai par rapport à celle d'application de la charge.
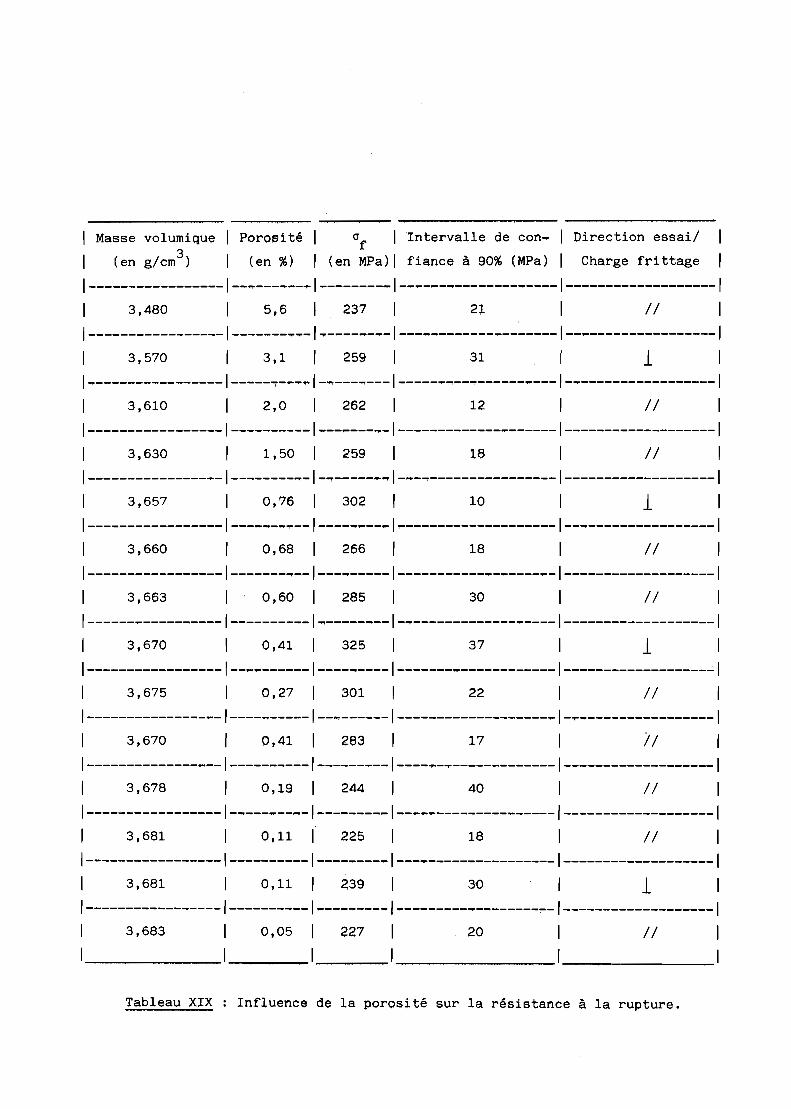
Masse volumique 1 Porosité 1 crf 1Intervalle de con~ 1Direction essail 1(en g/cm3
) 1 (en %) 1 (en MPa) 1 fiance à 90% (MPa) 1 Charge frittage 1
-----------------�----------�--------~I---------------~----I-------------------I
3,480 1 5,6 1 237 1 21 1 Il 1
----------------- ----------1--------- --------------------\-------------------13,570 3,1 1 259 31 1 l ,
~---------------- -----~---~I-~---~--- ------~--~------~---I-------------------I
3,610 2,0 1 262 12 1 Il 1
----------------- ----------1--------- --------------------1-------------------13,630 1,50 1 259 18 1 Il 1
---------------~- -~--------I-~------~ -~-~----------------I-------------------
3,657 0,76 1 302 10 1 l----------------- ----------1--------- --------------------1-------------------
3,660 0,68 1 266 18 1 Il
----------------- ----------1--------- --------------------1-------------------3,663 0,60 1 285 30 1 Il
----------------- ----------1--------- --------------------1-------------------3,670 0,41 1 325 37 1 I
----------------- ----------1--------- --------------------I------------------~
3,675 0,27 1 301 22 1 Il----------------- ----------1--------- --------------------1-------------------
3,670 0,41 1 283 17 , il----------------- ----------1--------- --------------------1-------------------
3,678 0,19 1 244 40 1 Il----------------- -~--------I--------- --------------------1-------------------
3,681 0,11 1 225 18 1 Il----------------- ----------1--------- --------------------1-------------------
3,681 0,11 1 ~39 30 1 1----------------- ----------1--------- ---------~----------I-------------------
3,683 0,05 1 227 20 1 Il____________ 1 , _
Tableau XIX Influence de la porosité sur la résistance à la rupture.
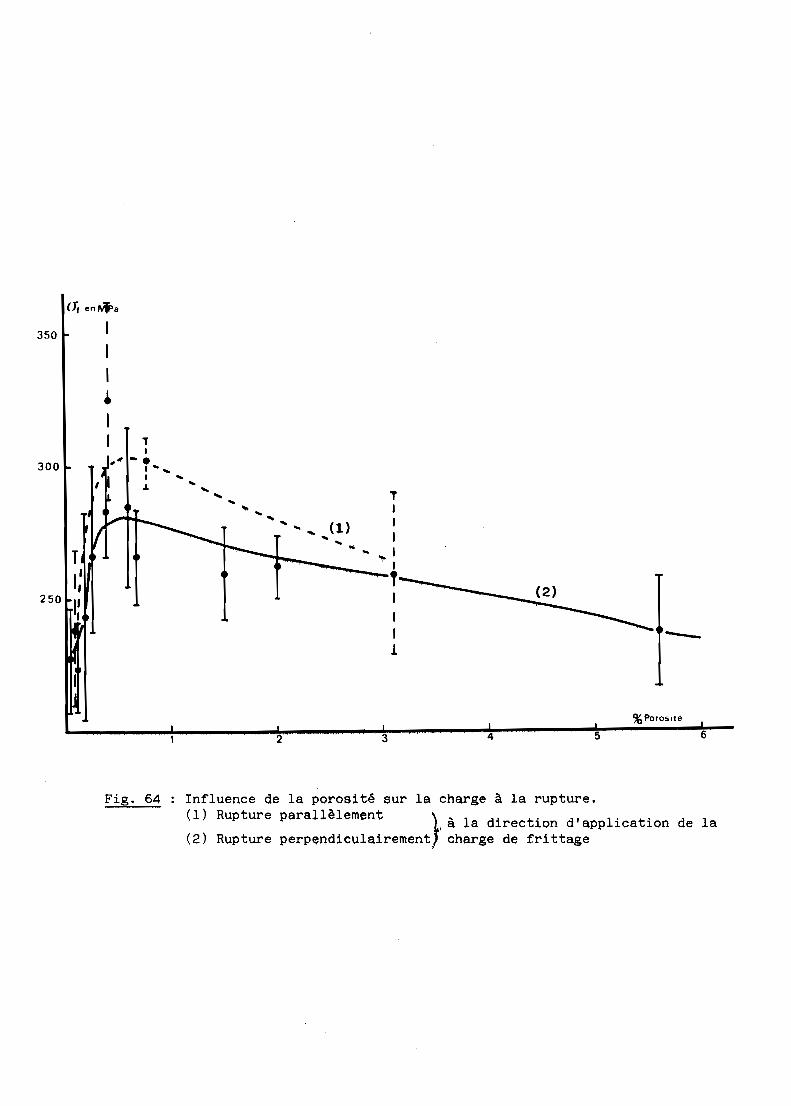
T1
(1) 1... , 1, .. ... 1"1
T(2)1
11 ---l
(J, en rvf>a
350 1
1
1
•11 T
1
300 #' ....... ,1 ,Jo
, ... ... , .. ,
T1:
250l',
%Poros,té
2 3 4 5 6
Fig. 64 Influence de la porosité sur la charge à la rupture.(1) Rupture p~rallèlement } '1 d· t· d' 1· t· de la,a a ar-ec 10n app r ca 10n(2) Rupture perpendiculairement, charge de f.rittage

- 144 -
B - RESISTANCE A LA RUPTUR! DE L'OXYNITRURE TaANSPARENT
On sait (cf. 2ème Partie - Chapitre IV) que le critère de transparence
est relié à la taille des grains qui augmente fortement au cours du traitement
thermique, et il est également bien connu que cette augmentation de taille
s'accompagne d'une diminution corrélative de la résistance à la rupture. Il
nous a donc semblé intéressant de suivre l'évolution de la charge à la rupture
des pièces d'oxynitrure lorsqu'elles deviennent transparentes après traitement
thermique à 1950°C.
1 - Influence de la taille des irains.
Les échantillons frittés sont traités à 1950 QC pendant des durées varia
bles de 1 à 24 heures. Les éprouvettes après polissage ont des dimensions
voisines de 5 x 5 x 25 mm. On détermine la taille des grains comme dans le cas
de l'étude cinétique (2ème Partie) et la charge à la rupture est mesurée dans
les mêmes conditions que précédemment.
La figure 65, qui présente les résultats obtenus avec les barres d'in
certitude pour un intervalle de confiance à 90%, montre que l'on obtient effec
tivement la décroissance attendue de la charge à la rupture lorsque s'élève la
taille des grains.
La théorie de la rupture fragile précise que la résistance of varie comme
l'inverse de la racine carrée de la taille des grains G
lorsque le solide comporte des défauts uniformément répartis (30) et que l'on
observe généralement une modification en liaison avec l'existence de contrain
tes résiduelles aux joints de grains (empilement de dislocations) lorsque leur
nombre devient grand, ç'est-à-dire lorsque la taille des grains devient faible.
Le diagramme de la figure 66 présente ainsi l'allure habituellement observée de
la courbe of ~ f (G-U) pour un solide réel (53).
Les résultats obtenus pour l'oxynitrure d'aluminium sont rapportés au
tableau XX et représentés en coordonnées of' G-Uà la figure 67.

100
200
(Tf en MPa
300
'I", ~l1'--!-t-!-1
100 2 0 300 400 G en /lm
Fig. 65 Charge à la rupture en fonction de la taille des grains.
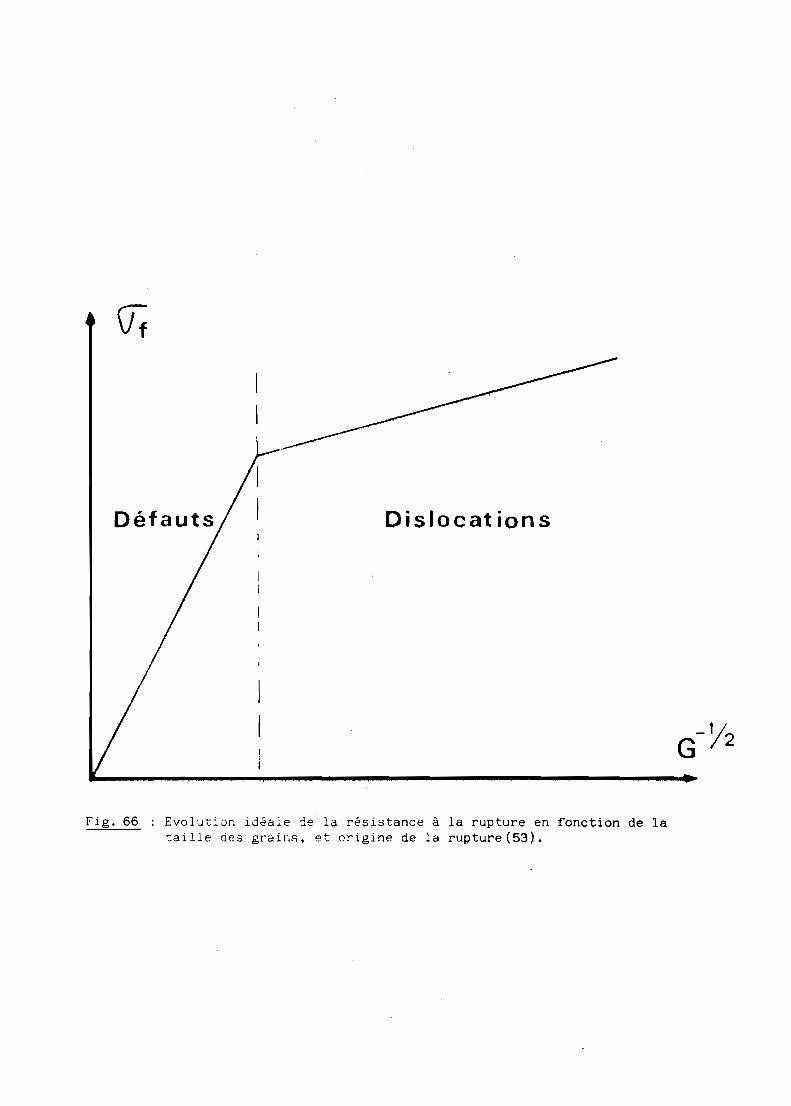
Défauts Dislocations
Fig. 66 Evolu~ion id~ale de la rés~stance à la rupture en fonction de lataille des grains, et origine de la rupture (53).
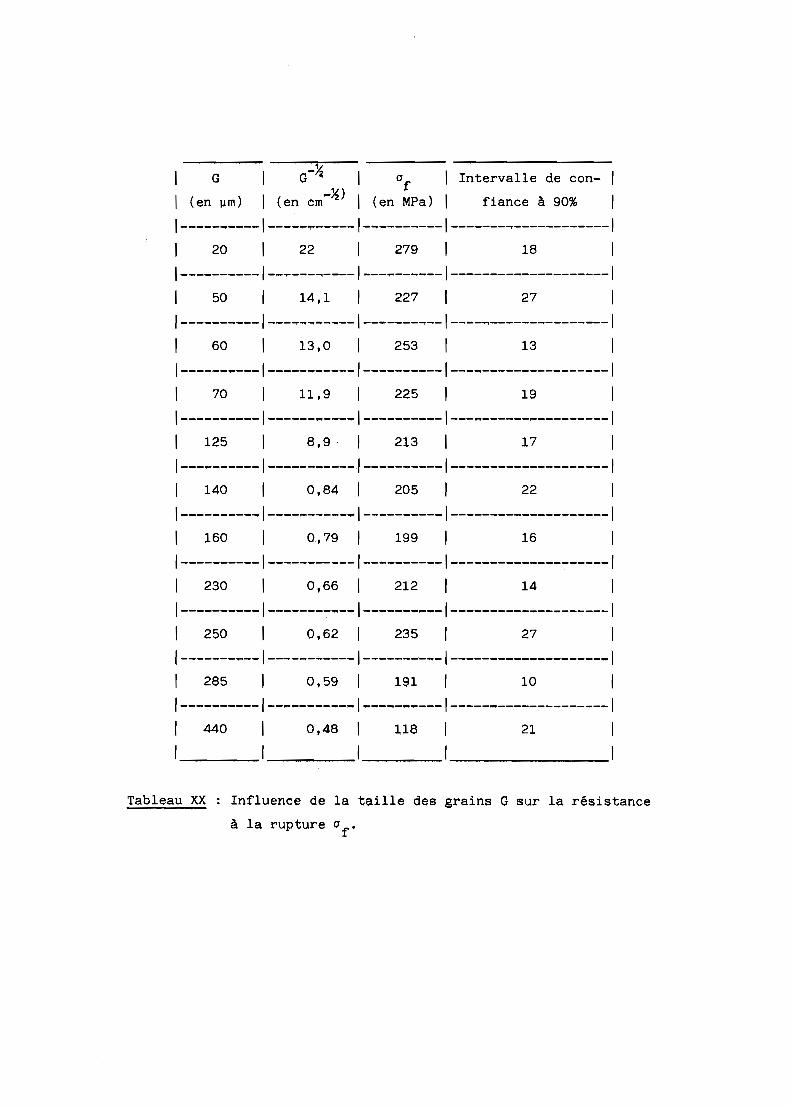
G G-~ 1 or 1 Intervalle de con-
1 (en ~m) (en cm-~) 1 (en MPa) 1 fiance à 90%
I---~------ ----~~-----I----------I--------------------
1 20 22 1 279 1 18
1---------- -----------1----------1--------------------1 50 14,1 1 227 1 27
---~-------I-------~-- ----~---------------
60 13,0 1 253
---------- -----------1----------70 11,9 1 225
---------- -----------1----------125 8,9 1 213
----------1----------- ----------
13
19
17
140 0,84 205 22
160 0,79 199 16
---------- ----------- --~------- --------------------230 0,66 212 14
---------- ----------~ ---------- --------------------250 0,62 235 27
285 0,59 191 10
440 0,48 118 21
Tableau XX Influence de la taille des grains G sur la résistance
à la rupture of'
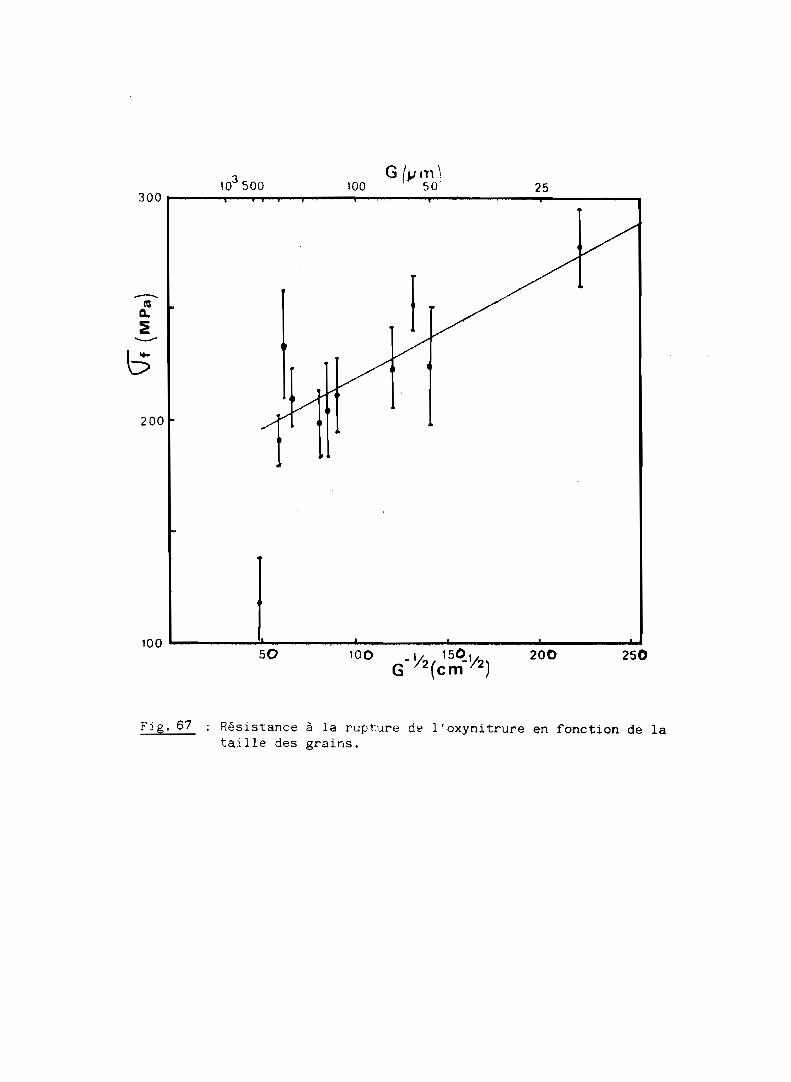
25G (".Jln \
501003 00 ...---T--",.....,..-...-...,....--T----T------".-.-----,
200
50 100 200 250
Fig. 67 Résistance à la rupture de l'oxynitrure en fonction de lataille des grains.

- 149 -
L'allure linéaire observée, qui se traduit par l'expression
-~or = 0,415 G + 183
-6 6pour 20.10 < G < 400.10- m s'inscrit donc logiquement dans l'interprétation
théorique envisagée.
2 - Transparence et charge à la rupture
La transparence s'obtient pour des tailles de grains de 200 à 300 ~m. On
voit donc que la charge à la rupture est très sensiblement inférieure à celle
du produit fritté ; elle dépasse à peine 200 MPa. Un grossissement des grains
plus poussé n'améliore pas la transparence mais aboutit à un produit plus
fragile.
L'ensemble de ces résultat a été obtenu à partir d'échantillons préparés
en utilisant la méthode de synthèse directe à partir de mélanges aluminel
nitrure. Un essai effectué sur des échantillons transparents obtenus par
carbonitruration montre que la charge à la rupture observée est beaucoup plus
élevée :
Of = 360 ± 20 MPa
(l'incertitude représentant toujours l'intervalle de confiance à 90%). Cette
amélioration n'a par reçu de justification expérimentale, mais on peut penser
qu'elle résulte d'une différence de microstructure, d 1une plus grande homogé
néité du fritté et, peut-8tre, de sa plvs grande pureté.

Chapitre II l
MlCRODURETE
Nous avons déterminé la dureté Vicker~ des frittés et du matériau trans
parent en fonction de la porosité. On sait en effet que la dureté varie avec la
porosité selon une loi exponentielle (54) du type :
Hp :: Ha
(-aP)e
H et Hp désignant les densités respectives des échantillons denses et dea
porosité P.
Les éprouvettes sont d'abord métallisées par dépôt sous vide, pour
accentuer la netteté des contours de l'empreinte.
1 - Oxynitrure fritté
Un matériau densifié à 1750 0C sous 40 MPa pendant 3 heures 30 minutes
présente une dureté moyenne (sous une contrainte de 300 g) définie à partir de
25 essais :
H :: 1285v
-2kg mm

- 151 -
L'influence de la porosité a été étudiée de façon identique sur des
échantillons métallisés et les résultats reproduits sur la figure 68. On
observe une décroissance de la microdureté avec la porosité P que traduit
l'expression (fig. 69) :
Hv
-19P= 1370 ~
La microdureté de l'oxynitrure fritté est donc sensiblement inférieure à
celle (1560 Kg mm-2 ) du nitrure d'aluminium parfaitement dense obtenue dans les
mêmes conditions (15).
2 - Oxynitrure transparent
La moyenne obtenue à partir de 50 mesures prend pour valeur
H = 1820 kgv
-2mm
Compte tenu de la grosseur des grains qui varie entre 200 et 300 ~m, les
mesures affectent en général un seul grain. Ainsi, la valeur trouvée correspond
elle à celle d'un monocristal, ce qui explique la différence avec la dureté de
l'échantillon fritté.
La dispersion importante des normes que traduit l'écart type a vraisem
blablement pour origine l'anisotropie cristalline due aux orientations diffé
rentes des cristaux.
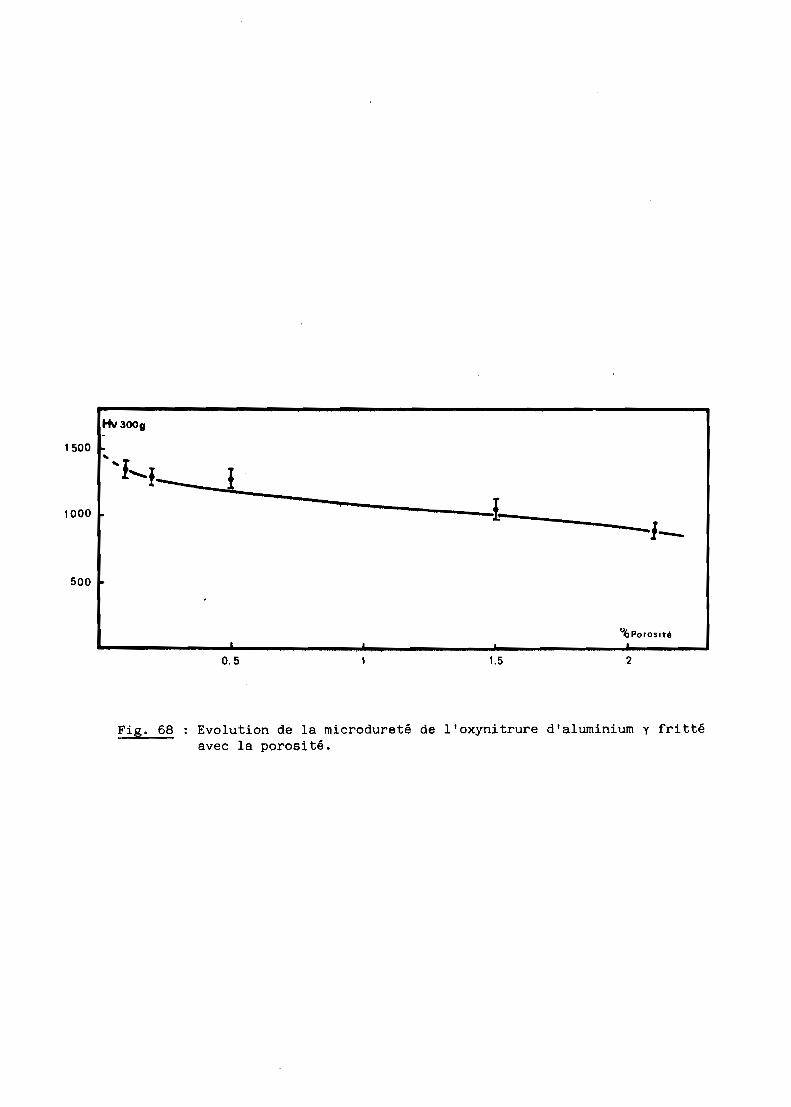
1500
1000
500
Hv3OO9
1
°oPorosité
0.5 1.5 2
Fig. 68 Evolution de la microdureté de l'oxynitrure d'aluminium y frittéavec la porosité.
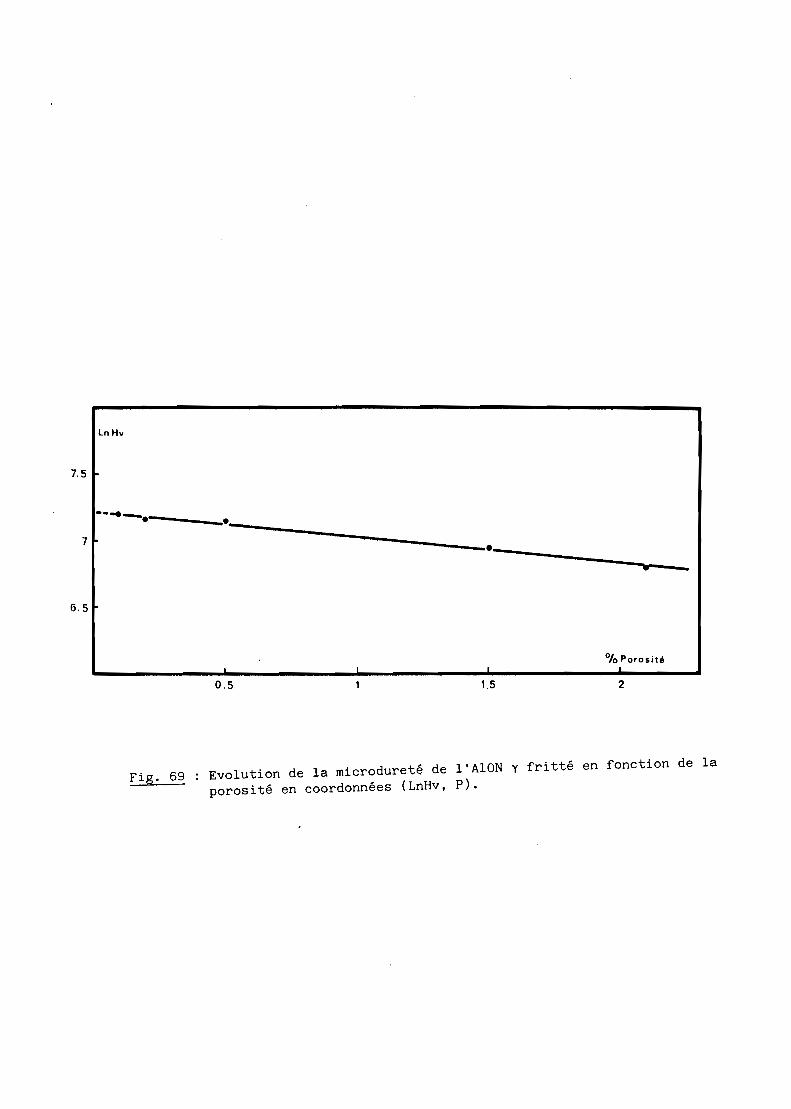
lnHv
7.5
-•
..---..-------_..-------------_.---------~
7
6.5
0/0 Porosité
0.5 1.5 2
Fig. 69Evolution de la microdureté de l'AlüN y fritté en fonction de laporosité en coordonnées (LnHv, P).

Chapitre IV
EFFETS D'ENVIRONNEMENT SUR LES PROPRIETES MECANIQUES
Compte tenu des possibilités d'utilisation de l'oxynitrure d'aluminium à
température élevée et de sa dégradation en atmosphère oxydante, il nous a
semblé intéressant d'étudier l'effet d'environnement sur les propriétés méca
niques.
Le présent chapitre rapporte les résultats obtenus en ce qui concerne la
résistance à la rupture à chaud, l'influence de l'oxydation sur la tenue méca
nique et la résistance aux chocs thermiques. L'appareillage utilisé et les
conditions opératoires sont identiques à ceux définis précédemment.
A - RESISTANCE A LA RUPTURE A CHAUD
1 - Oxynitrure d'aluminium fritté
Les essais de rupture à chaud ont été effectués perpendiculairement à la
direction d'application de la charge de frittage sur un support en alumine
entre 1000 et 1450oC. La.figure 70 représente les résultats (les barres d'in
certitude indiquent l'intervalle de confiance à 90%).
A 1000oC, la valeur obtenue (326 ± 50 MPa) (cf. le tableau XXI) est
analogue à celle (328 ± 35 MPa) observée à l'ambiante. Aux températures plus
élevées, on remarque une décroissance sensible de la résistance à la rupture
qui tombe à 132 ± 15 MPa, et qui est acquise, pour l'essentiel vers 1200oC.
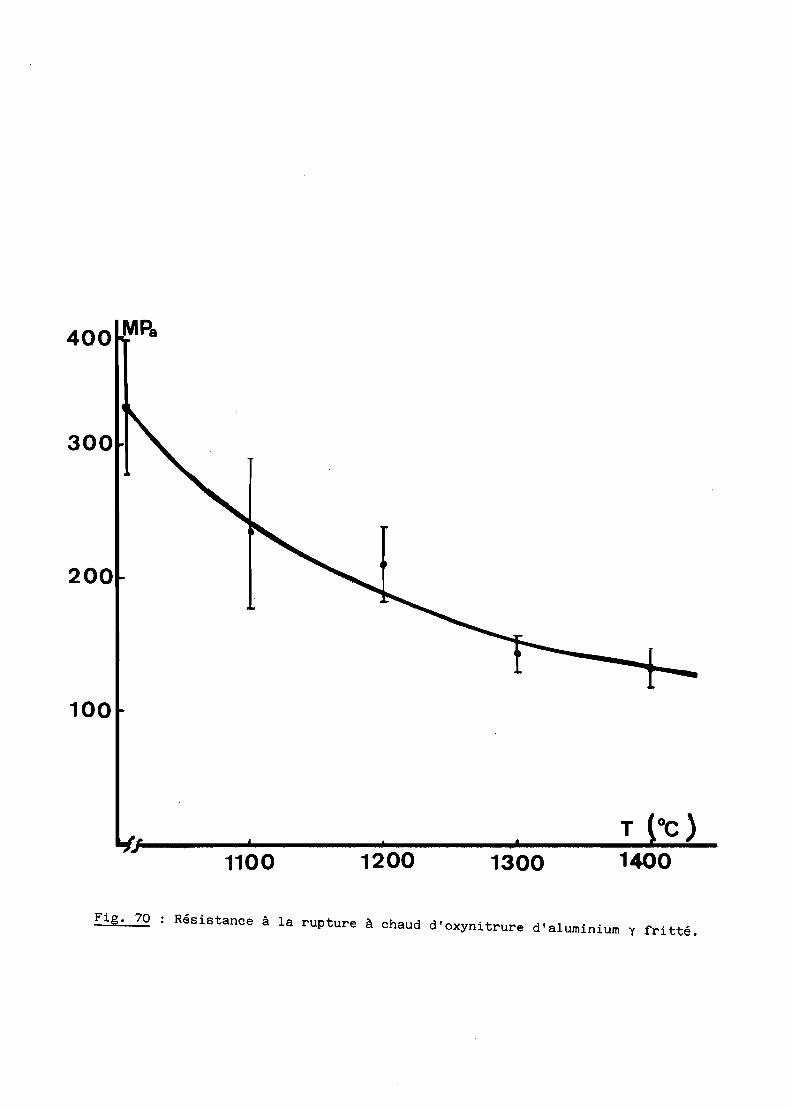
400 MPa
300
200
100
1100,
1200 1300
Fig. 70 Résistance à la rupture à chaud d'oxynitrure d'aluminium y fritté.
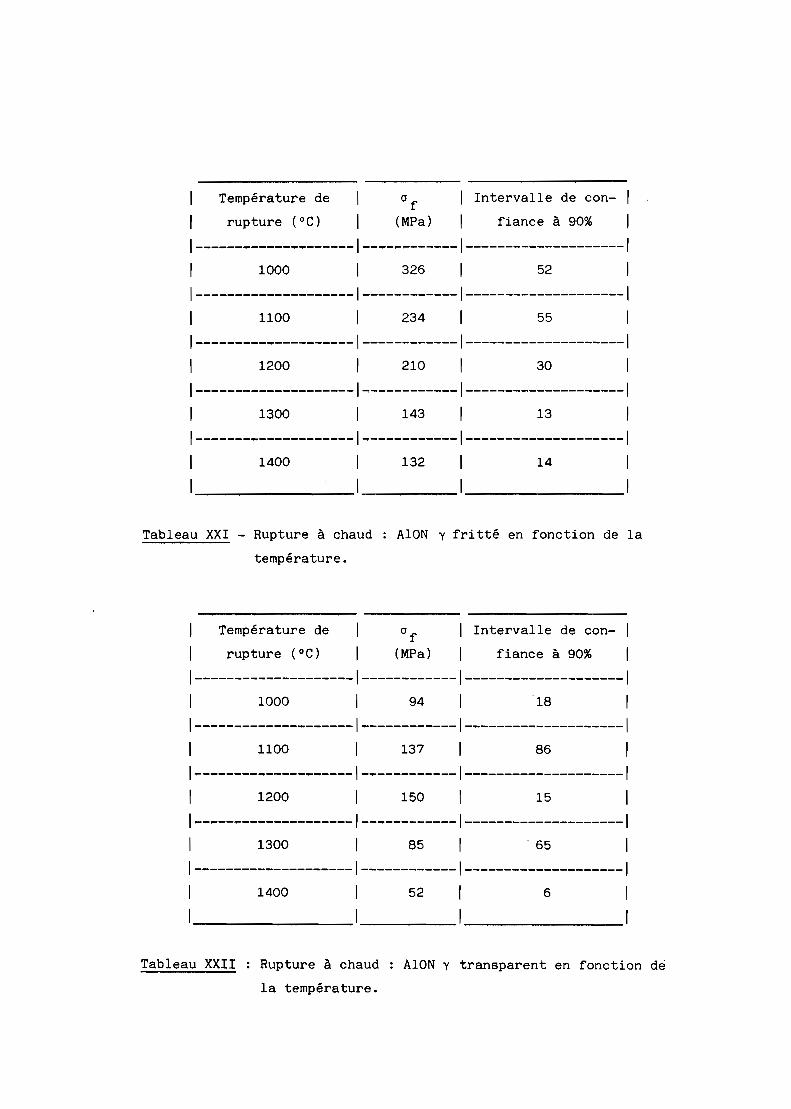
Température de of Intervalle de con-
rupture (OC) (MPa) fiance à 90%
-----~------~~------ --~--------- --------------------1000 326 52
1100 234 55
-------------------- -------~---- ---~-----------~----
1200 210 30
1300 143 13
1400 132 14
Tableau XXI - Rupture à chaud
température.
AlON y fritté en fonction de la
6
18
15
86
. 65
Intervalle de con
fiance à 90%
52
85
94
137
150
1 Température de
1 rupture (OC)
1--------------------1 1000
1--------------------1 1100
1--------------------1 1200
1--------------------1 1300
I---------------~----
1 1400
1 --- _
Tableau XXII Rupture à chaud AlON y transparent en fonction de
la température.

- 157 -
2 - Oxynitrure d'aluminium transparent
Les résultats sont représentés sur la figure 71 et repris au tableau
XXII. On note essentiellement leur très grande dispersion, les domaines
d'incertitude prenant parfois des proportions considérables. Ceci ne constitue
pas a priori un élément surprenant puisque les échantillons sont ici composés
de très gros grains, ce qui maximalise le rôle des défauts aux joints de grains
et des imperfections superficielles. On note cependant, comme pour l'oxynitrure
fritté, que le transparent perd beaucoup de ses qualités de résistance mécani
que à haute température, tout en conservant jusqu'à 12000C une valeur de la
charge à la rupture de l'ordre de 150 MPa compatible avec la plupart des
impératifs liés à son usage en tant que produit transparent.
B - INFLUENCE DE L'OXYDATION SUR LA CHARGE A LA RUPTURE DES FRITTES
Après avoir testé le comportement du matériau en fonction de la porosité,
de la densité et de la taille des grains, nous reprenons cette étude pour
rechercher l'influence de l'oxydation sur la charge à la rupture en fonction de
la température et du temps d'oxydation.
Les pastilles sont d'abord découpées et polies sous forme parallépipédi
que (5 x 5 x 30 mm environ), puis oxydées dans l'air à température donnée.
Quatre éprouvettes sont utilisées pour chaque essai • Les mesures des
charges à la rupture ont lieu après oxydation et retour à la température
ambiante. Dans tous les cas, la charge est appliquée dans la direction perpen
diculaire à la pression du frittage.
1 - Influence de la température
Les résultats obtenus après oxydation des éprouvettes pendant 48 heures à
des températures comprises entre 1100 et 1350°C, sont consignées au tableau
XXII et représentés sur la figure 72. Sur cette même figure sont également
représentées en regard les épaisseurs du revêtement d'alumine formée par oxyda
tion telles qu'elles se déduisent des variations de poids et de l'observation
directe au microscope.
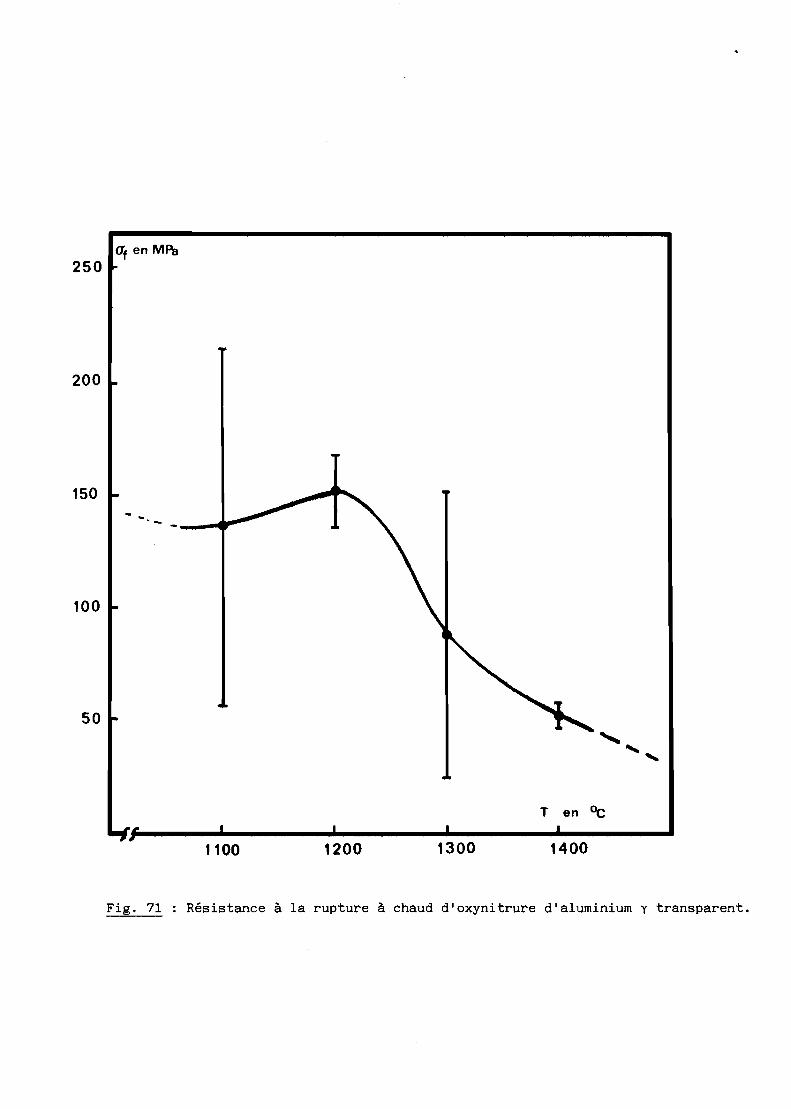
250
200
150
100
50
Of en MPa
T en OC
1100 1200 1300 1400
Fig. 71 Résistance à la rupture à chaud d'oxynitrure d'aluminium y transparent.
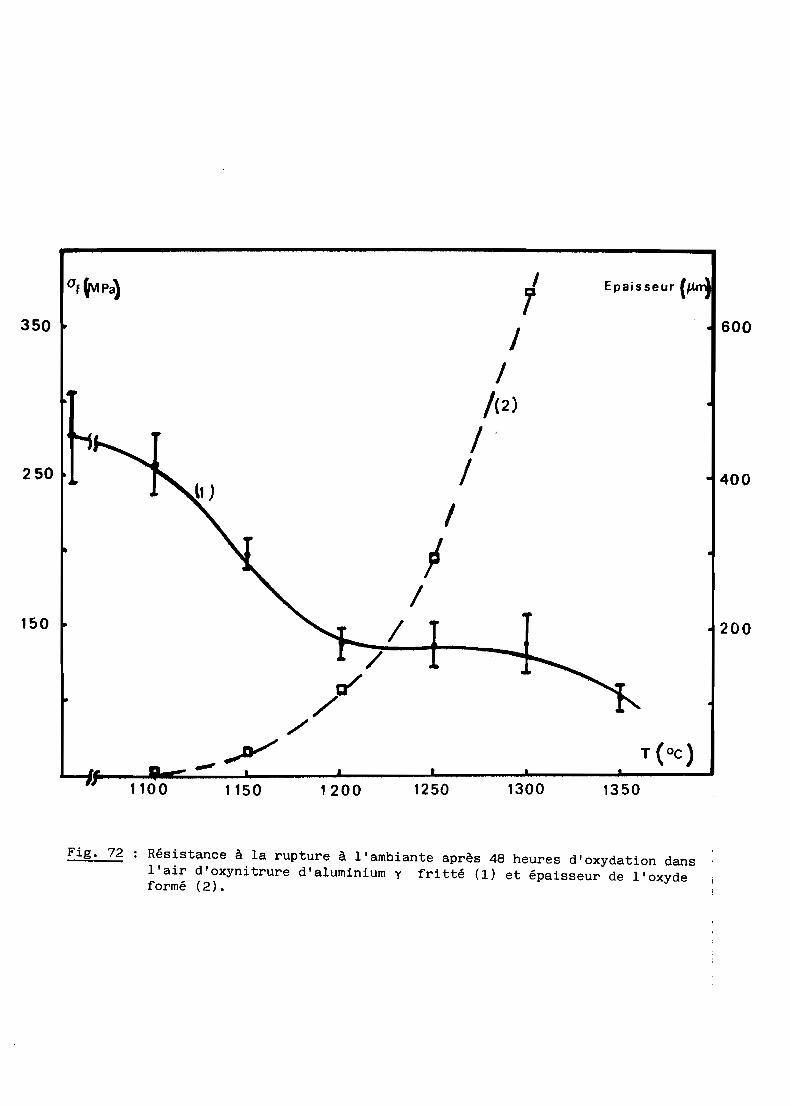
af~Pa) 1 Epaisseur (r350
1 600
//(2)
/250 / 400
1
f/
150 / 200
/
/./'
.",~ T (OC)1150 1200 1250 1300 1350
Fig. 72 Résistance à la rupture à l'ambiante après 48 heures d'oxydation dansl'air d'oxynitrure d'aluminium y fritté (1) et épaisseur de l'oxydeformé (2).
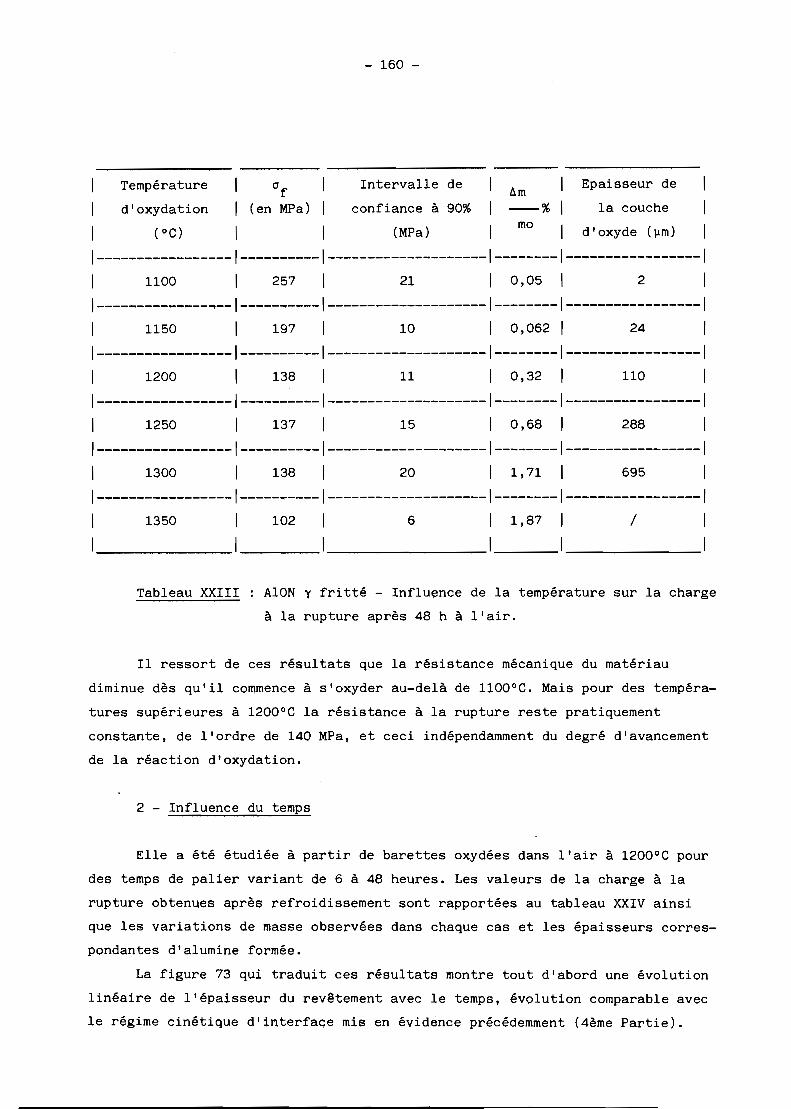
- 160 -
2
/
24
288
110
695
d'oxyde (~m)
Epaisseur de
la couchemo
1,87
1,71
0,32
0,062
0,05
0,68
tlm
-%
6
21
20
15
la
11
(MPa)
Intervalle de
confiance à 90%
138
137
257
138
197
102
Of(en MPa)
1250
1350
1300
1200
1100
1150
Température
d'oxydation
1
1
1
-~--------I--------------------
1
----------I-~------------------
1
----------1--------------------
r
----------1--------------------1
----------1--------------------
1
----------1--------------------
1
_____________ 1 --- _
Tableau XXIII AlaN y fritté - Influence de la température sur la charge
à la rupture après 48 h à l'air.
Il ressort de ces résultats que la résistance mécanique du matériau
diminue dès qu'il commence à s'oxyder au-delà de 1100oC. Mais pour des tempéra
tures supérieures à 1200 0C la résistance à la rupture reste pratiquement
constante, de l'ordre de 140 MPa, et ceci indépendamment du degré d'avancement
de la réaction d'oxydation.
2 - Influence du temps
Elle a été étudiée à partir de barettes oxydées dans l'air à 12000C pour
des temps de palier variant de 6 à 48 heures. Les valeurs de la charge à la
rupture obtenues après refroidissement sont rapportées au tableau XXIV ainsi
que les variations de masse observées dans chaque cas et les épaisseurs corres
pondantes d'alumine formée.
La figure 73 qui traduit ces résultats montre tout d'abord une évolution
linéaire de l'épaisseur du revêtement avec le temps, évolution comparable avec
le régime cinétique d'interfaGe mis en évidence précédemment (4ème Partie).
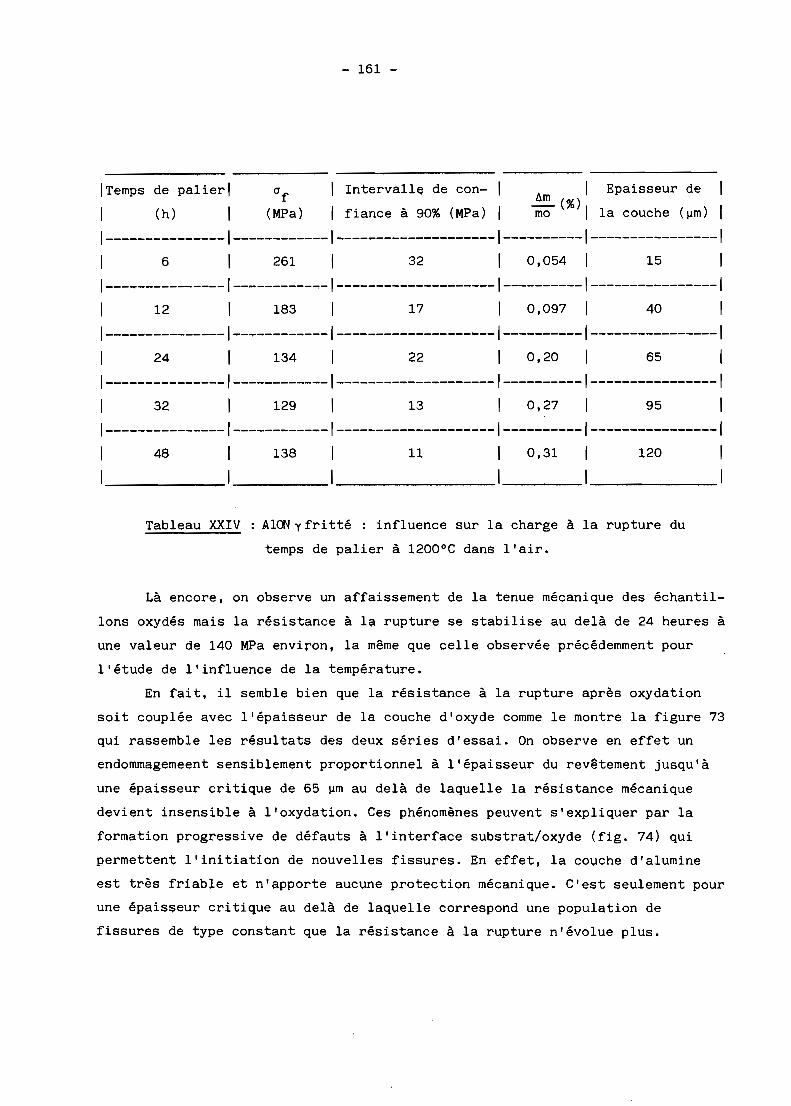
- 161 -
Temps de palier of Intervalle de con-~(%)
Epaisseur de
(h) (MPa) fiance à 90% (MPa) mo la couche (~m)
-----------,...--- ------------ -------------------- ---------- ----------------6 261 32 0,054 15
--------------- ----------~- ------------------~-----------12 183 17 0,097
--------------- ------------ -------------------- ----------24 134 22 0,20
32 129 13 0,27
48 138 Il 0,31
40
65
95
120
Tableau XXIV Alm y fri tté : influence sur la charge à la rupture du
temps de palier à l2000C dans l'air.
Là encore, on observe un affaissement de la tenue mécanique des échantil
lons oxydés mais la résistance à la rupture se stabilise au delà de 24 heures à
une valeur de 140 MPa environ, la même que celle observée précédemment pour
l'étude de l'influençe de la température.
En fait, il semble bien que la résistance à la rupture après oxydation
soit couplée avec l'épaisseur de la couche d'oxyde comme le montre la figure 73
qui rassemble les résultats des deux séries d'essai. On observe en effet un
endommagemeent sensiblement proportionnel à l'épaisseur du revêtement jusqu'à
une épaisseur critique de 65 ~m au delà de laquelle la résistance mécanique
devient insensible à l'oxydation. Ces phénomènes peuvent s'expliquer par la
formation progressive de défauts à l'interface substrat/oxyde (fig. 74) qui
permettent l'initiation de nouvelles fissures. En effet, la couche d'alumine
est très friable et n'apporte aucune protection mécanique. C'est seulement pour
une épaisseur critique au delà de laquelle correspond une population de
fissures de type constant que la résistance à la rupture n'évolue plus.
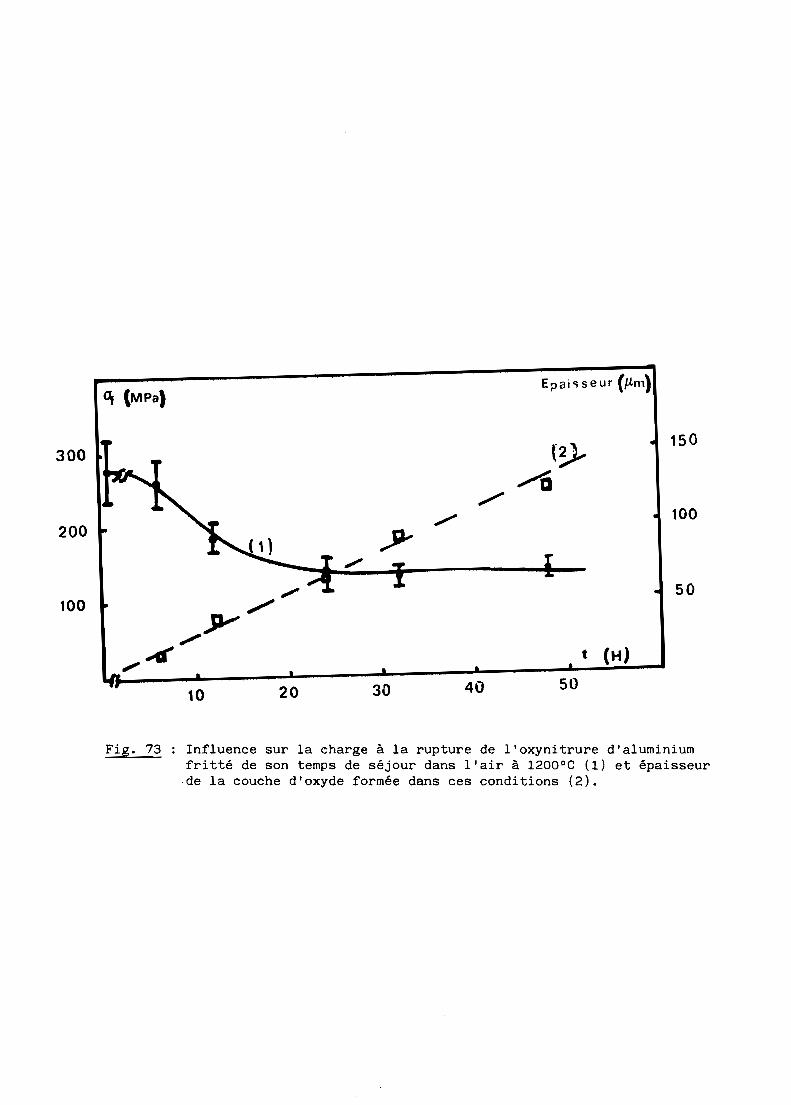
300
200
100
~ (MPa)
10 20 30 40
Epai<;seur (JLm)
t CH)
50
150
100
50
Fig. 73 Influence sur la charge à la rupture de l'oxynitrure d'aluminium
fritté de son temps de séjour dans l'air à 12000C (1) et épaisseur
de la couche d'oxyde formée dans ces conditions (2).

Fig. 74
Substrat
Micrographie en coupe d'une éprouvette d'oxynitrure d'aluminium y
fritté oxydé dans l'air à 1200 0C en 48 heures.
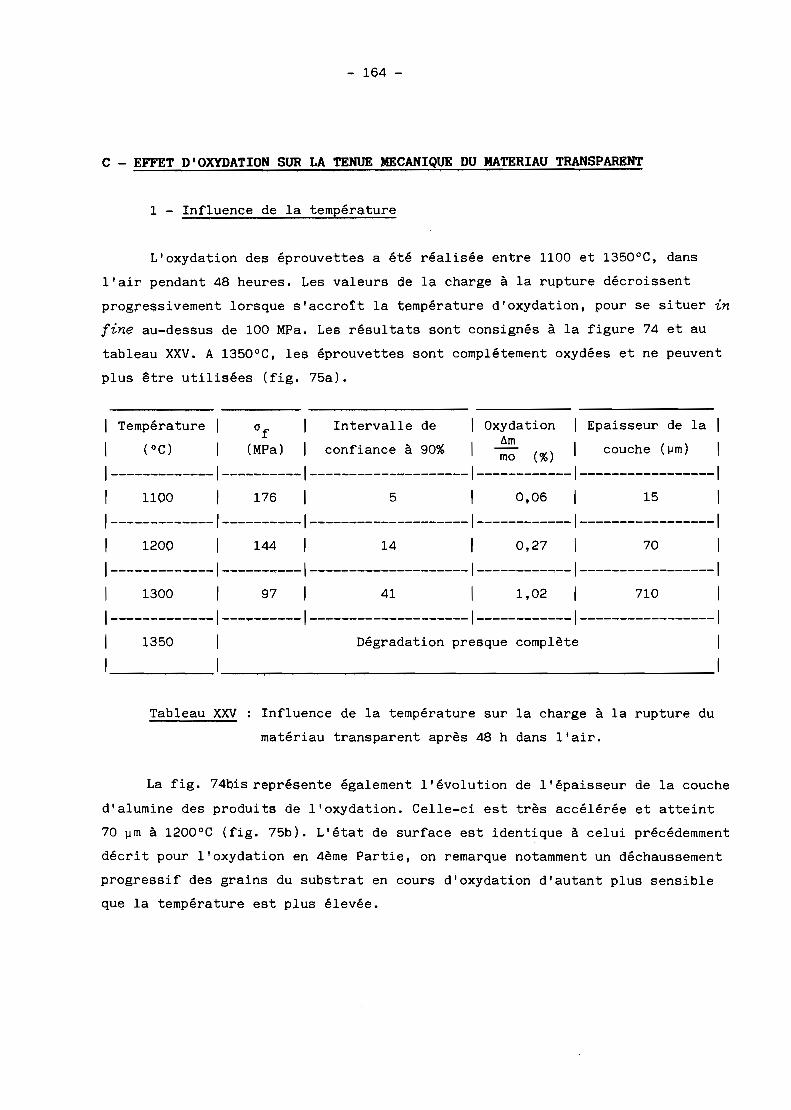
- 164 -
C - EFFET DIOXYDATION SUR LA TENUE MECANIQUE DU MATERIAU TRANSPARENT
1 - Influence de la température
L'oxydation des éprouvettes a été réalisée entre 1100 et 1350 oC, dans
l'air pendant 48 heures. Les valeurs de la charge à la rupture décroissent
progre~sivement lorsque s'accroît la température d'oxydation, pour se situer in
fine au-dessus de 100 MPa. Les résultats sont consignés à la figure 74 et au
tableau XXV. A 1350°C, les éprouvettes sont complétement oxydées et ne peuvent
plus être utilisées (fig. 75a).
Température Of Intervalle de Oxydation Epaisseur de la 1llm
1 (OC) 1 (MPa) 1 confiance à 90% 1 mo (%) 1 couche (~m) 1
1-------------1----------1--------------------1------------1-----------------11 1100 1 176 1 5 1 0,06 1 15 1
I-------------I----------I--------~-----------I------------1-----------------1
1 1200 1 144 1 14 1 0,27 1 70 1
1-------------1----------\--------------------1------------1-----------------11 1300 1 97 1 41 1 1,02 1 710 1
1-------------1----------1--------------------1------------1-----------------11 1350 1 Dégradation presque complète 1
1 1 1
Tableau XXV Influence de la température sur la charge à la rupture du
matériau transparent après 48 h dans l'air.
La fig. 74bis représente également l'évolution de l'épaisseur de la couche
d'alumine des produits de l'oxydation. Celle-ci est très accélérée et atteint
70 ~m à 1200 0C (fig. 75b). L'état de surface est identique à celui précédemment
décrit pour l'oxydation en 4ème Partie, on remarque notamment un déchaussement
progressif des grains du substrat en cours d'oxydation d'autant plus sensible
que la température est plus élevée.
Of (MPa) Epaisseur (p.m)400 800
(/ 600
2/
200 / 400
/100 / 200/
,,/T (oc)- --~
1100 1200 1300
Fig. 74bis Influence de la température d'oxydation sur la charge à la ruptured'oxynitrure d'aluminium y transparent et le gain de masse correspondant.
a
Couche d'alumine
Substrat ---__
b
Fig. 75 Eprouvettes d'oxynitrure d'aluminium partiellement oxydéesen 48 heures dans l'air à 1350°C (a) et 1200 0C (b) (en coupe).
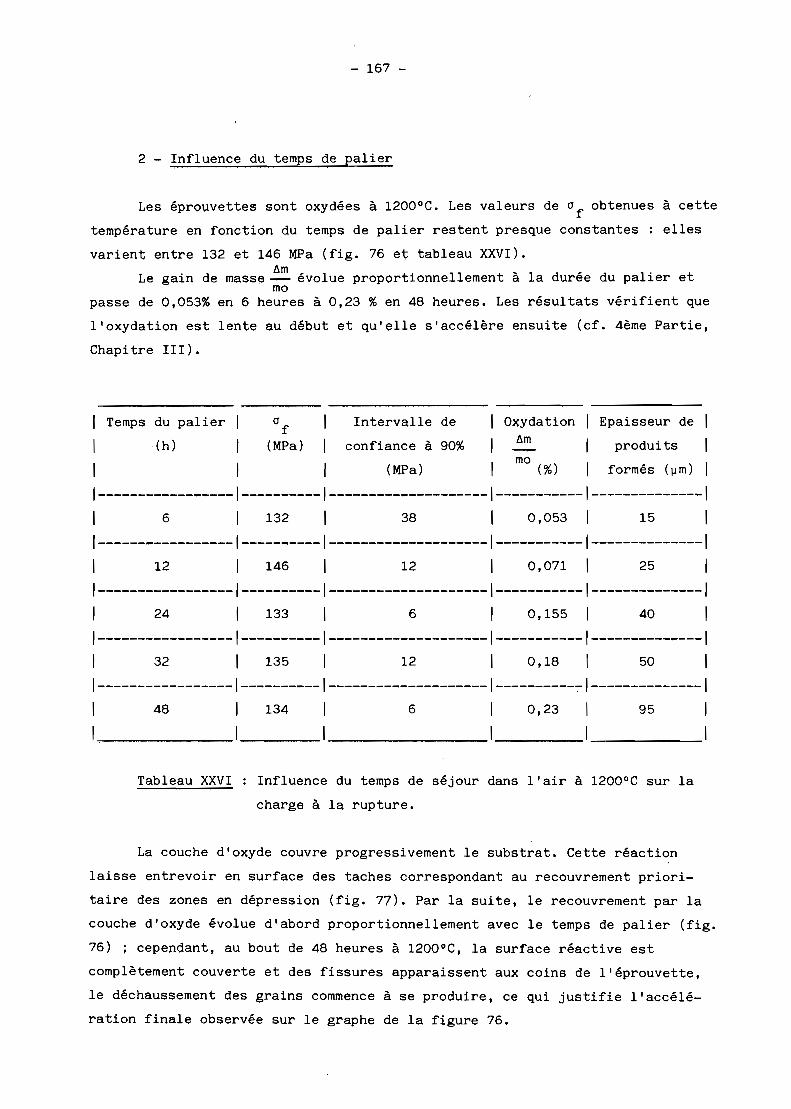
- 167 -
2 ~ Influence du temps de palier
Les éprouvettes sont oxydées à 1200°C. Les valeurs de of obtenues à cette
température en fonction du temps de palier restent presque constantes : elles
varient entre 132 et 146 MPa (fig. 76 et tableau XXVI).
Le gain de masse~ évolue proportionnellement à la durée du palier etmo
passe de 0,053% en 6 heures à 0,23 % en 48 heures. Les résultats vérifient que
l'oxydation est lente au début et qu'elle s'accélère ensuite (cf. 4ème Partie,
Chapitre III).
Temps du palier ° Intervalle def
(h ) (MPa) confiance à 90%
(MPa)
----------------- ---------- --------------------6 132 38
6
6
12
12
24 133
12 146
32 135
48 134
Oxydation 1 Epaisseur deflm 1 .produ1.ts
mo (%) 1 ( )formés ]lm
-----------1--------------0,053 1 15
-----------1--------------0,071 1 25
-----------1--------------0,155 1 40
-----------1--------------0,18 1 50
-----------1--------------0,23 1 95
~ 1 -
Tableau XXVI Influence du temps de séjour dans l'air à 12000C sur la
charge à la rupture.
La couche d'oxyde couvre progressivement le substrat. Cette réaction
laisse entrevoir en surface des taches correspondant au recouvrement priori
taire des zones en dépression (fig. 77). Par la suite, le recouvrement par la
couche d'oxyde évolue d'abord proportionnellement avec le temps de palier (fig.
76) ; cependant, au bout de 48 heures à 1200oC, la surface réactive est
complètement couverte et des fissures apparaissent aux coins de l'éprouvette,
le déchaussement des grains commence à se produire, ce qui justifie l'accélé
ration finale observée sur le graphe de la figure 76.
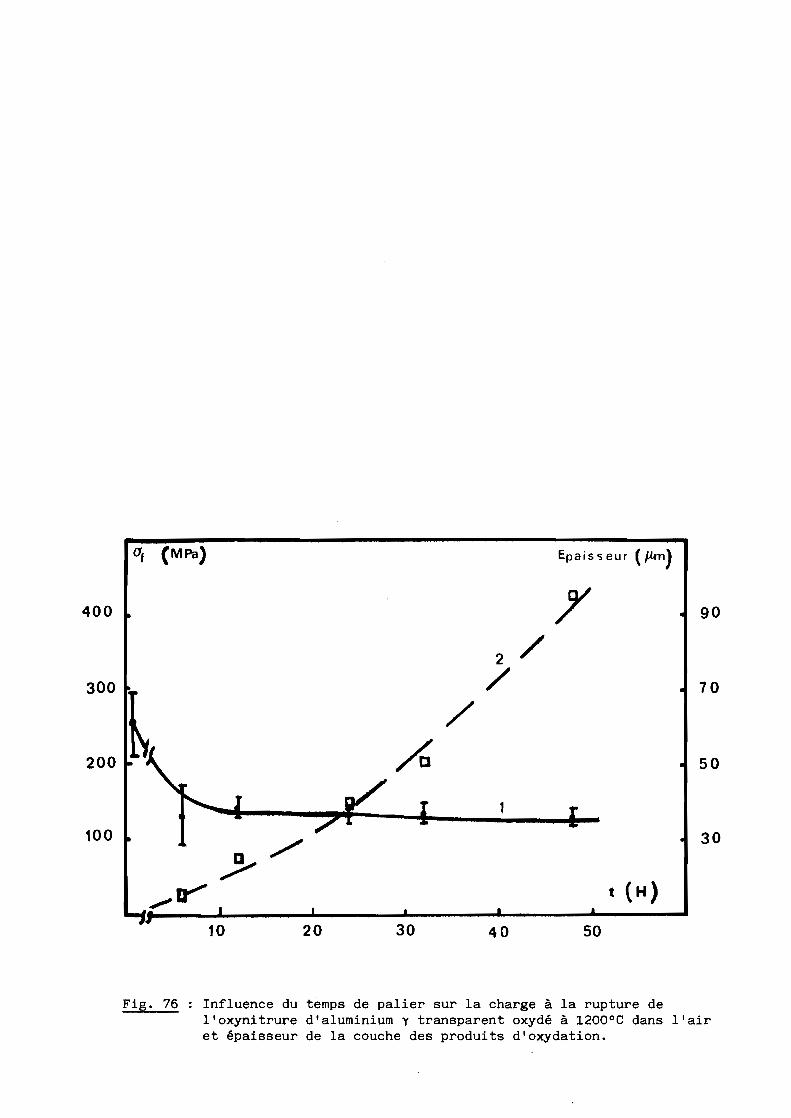
Ut (MPa) Epaisseur (JLm)
400 Y 90
2 // 70
/200 A: 50
7(': • i-100
/' 30
>t (H)
10 20 30 40 50
Fig. 76 Influence du temps de palier sur la charge à la rupture del'oxynitrure d'aluminium y transparent oxydé à 12000C dans l'airet épaisseur de la couche des produits d'oxydation.

Fig. 77
-...,..-- Couche d'alumine
----Substrat
Irrégularités d'épaisseur de la couche des produits formés paroxydation d'une éprouvette partiellement oxydée pendant 32 heuresà 1200°C.

- 170 -
3 - Conclusion
Placé dans des conditions très sévères d'utilisation pour un produit
transparent, l'oxynitrure d'aluminium y se recouvre après quelques dizaines
d'heures à 1100 0C d'une couche d'alumine qui le rend opaque et lui fait perdre
sa transparence. La résistance à la rupture chute elle aussi mais de façon plus
régulière et moins sensible que pour l'oxynitrure fritté non transparent. A
1200°C, à partir d'un séjour d'une quinzaine d'heures, dans l'air, les deux
produits présentent à peu près la même valeur de la résistance à la rupture,
alors que le fritté est très sensiblement plus résistant à l'ambiante
(328 ± 35 MPa contre 215 ± 17 MPa).
D ~ RESISTANCE AUX CHOCS THERMIQUES
Caractéristique importante des céramiques destinées à être utilisées à
température élevée, la résistance aux chocs thermiques se traduit en terme de
propriété d'usage par la différence de température critique ~Tc, valeur au delà
de laquelle le choc thermique se traduit par une dégradation importante de la
résistance mécanique de la pièce.
La méthode classique de détermination de la différence de température
critique est celle d'essais destructifs sur des éprouvettes portées à une
température T donnée, puis trempées brutalement dans l'eau (qui se trouve alors
portée à ébullition au voisinage de l'éprouvette) c'est-à-dire subissant une
chute extrêmement rapide de température :
AT T - 100 (T en OC).
Les échantillons ainsi traités sont alors casssés selon le protocole
expérimental défini plus haut (cf. 5ème Partie - Chapitre II) pour déterminer
l'évolution de leur résistance à la rupture après choc thermique.
Les éprouvettes de dimension 4 x 4 x 30 mm sont polies puis portées par
groupe de trois à des températures de 100 à 450°C par un séjour d'une heure
dans un four. Elles sont alors trempées dans l'eau initialement à 20°C, dès
leur sortie du four. Leur résistance à la rupture est ensuite mesurée après
séchage de 24 heures à l'étuve à 120°C.

- 171 -
Les résultats reportés au tableau XXVII et à la figure 78 représentent
pour chaque point la moyenne des trois mesures effectuées sur l'oxynitrure
fritté. On constate que, portés jusqu'à une température de 200°C, les échantil
lons sont peu altérés par leur trempe. En revanche, au-dessus, ils perdent
rapidement l'essentiel de leur résistance à la rupture qui chute de plus de 250
à moins de 50 MPa. Ces résultats conduisent à déterminer l'écart de température
critique comme voisin de :
Cette valeur peut être comparée à celle obtenue par le calcul à partir
des constantes mécaniques et thermiques du matériau selon la formule :
0'(1 - v)
où O'f est la résistance à la rupture à froid, v le coefficient de Poisson, E le
module de Young et a le coefficient d'expansion thermique. Sur la base des
mesures précédentes (cf. supra) et d'autres faites antérieurement au
laboratoire (55) on dispose des valeurs suivantes pour ces constantes
cr ; 328 MPa (direction perpendiculaire à la charge de frittage)
v ; 0,26
E ::;: 325 GPa-6 -1a ; 7,5.10 K
qui conduisent à la valeur suivante de âTC
âTC ; 99,6 K
En égard à l'incertitude relativement importante sur la détermination
expérimentale de ATC' l'accord entre la valeur nominale mesurée et la valeur
calculée est tout à fait remarquable.
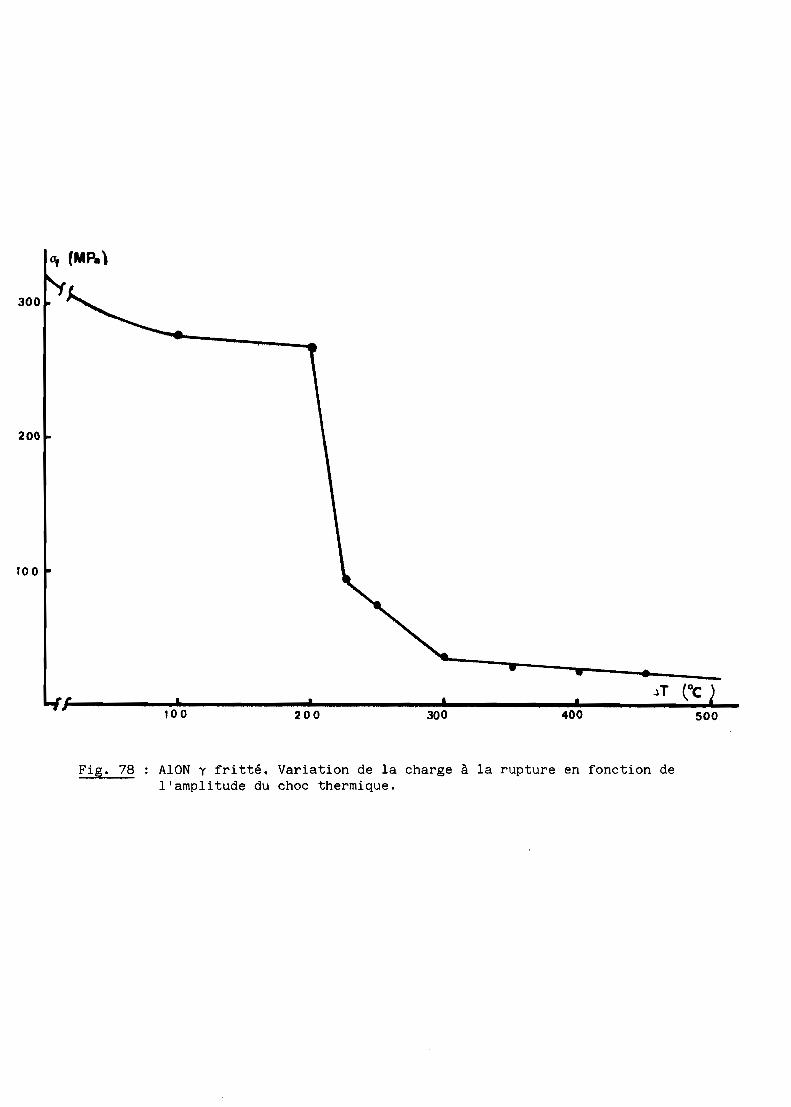
300
200
io 0
ut (MF\.)
,.,100
,
2001
300
,400 500
Fig. 78 AlaN y fritté. Variation de la charge à la rupture en fonction del'amplitude du choc thermique.
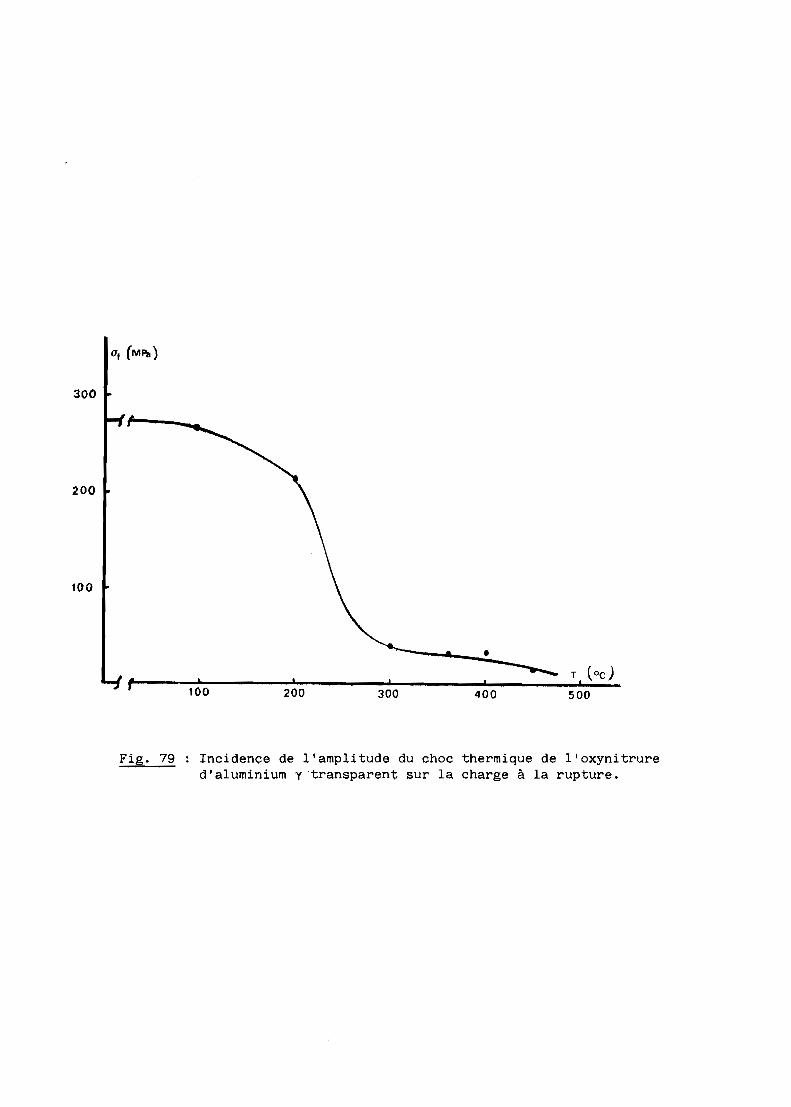
300
200
100
1
J 100 200 300 500
Fig. 79 Incidence de l'amplitude du ohoc thermique de l'oxynitrured'aluminium y 'transparent sur la charge à la rupture.

Fig. 80Micrographie des fissurations aux joints de grains observéessur l'oxynitrure d'aluminium Y transparent.
- 175 -
En ce qui concerne l'oxynitrure transparent, le tableau XXVII et la
figure 79 traduisent les résultats expérimentaux obtenus dans les mêmes
conditions. La résistance à la rupture chute moins brutalement que pour le
fritté, mais l'écart de température critique peut également être estimé comme
voisin de 6TC
~ 100 K.
Les observations des échantillons ayant subi les chocs thermiques les
plus rigoureux montrent, à l'instar du cliché de la figure 80que des fissures
importantes ont pris naissance aux joints de grains, et il est bien évident que
c'est donc cette fissuration intergranulaire qui est à l'origine de la chute
des qualités de résistance à la rupture des barettes d'oxynitrure d'aluminium.
-------------- ---~ -- -- --- --- --- ---
Tableau XXVII Résistance à la rupture après choc thermique de l'oxyni
trure d'aluminium porté à la température T.

RESUME ET CONCLUSION
L'oxynitrure d'aluminium y, phase spinelle non stoechiométrique du
diagramme binaire AIN-A1203,
a été préparé par réaction d'état solide entre
l'alumine a et le nitrure d'aluminium ou par réduction carbothermique de
l'alumine en atmosphère d'azote pour un mélange AIN/A1203
en proportions
molaires 27/73.
La cinétique de la carbonitruration qui conduit soit au nitrure, soit à
l'oxynitrure à température élevée (supérieure à 1600 oC) a montré l'existence
d'une phase gazeuse non identifiée, issue du carbone dont la réaction avec
l'alumine régie ~a vitesse globale.
Par frittage sous charge, la poudre d'oxynitrure d'aluminium conduit à
un produit dense dont la porosité est inférieure à 0,5%. L'étude cinétique
indique une linéarité entre la vitesse de densification et la pression qui
d'après l'influence de la granulométrie s'interpréte par un régime de diffu
sion en volume de type Nabarro-Herring dû à la structure fortement lacunaire
de la phase spinelle AION y. Ce mécanisme, original dans le contexte des
nitrures céramiques, rend compte du grossissement des grains et de la possibi
lité de formation d'un produit dense transparent en absence d'impureté. Cette
transparence (jusqu'à 5,5 ~m) est obtenue après traitement thermique à 1950 0C
pendant quelques heures.
L'oxynitrure d'aluminium y résiste bien à l'oxydation dans l'oxygène ou
dans l'air jusqu'à 1000oC. Le mécanisme d'oxydation a été précisé par trois
séries d'expérimentations sur des échantillons pulvérulents, bruts de frittage
puis transparents. Il y a cohexistence de deux réactions difficiles à séparer
que l'on peut schématiser comme suit:
~ Alumine a + N2

- 177 -
L'oxydation dans un premier temps s'accompagne de la formation d'une
autre phase spinelle de type aluminer mais retenant dans sa structure l'azote
initial (d'où le non d'alumine y'). Cette phase n'est stable qu'au dessous de
1000-1100oC et, en se décomposant, conduit à l'alumine a avec perte de
l'azote. Le chevauchement de ces deux étapes explique la complexité des
phénomènes observés, surtant dans le cas du matériau transparent où l'oxyda
tion préférentielle aux joints de grains se double d'un processus de décohé
sion aux températures élevées.
L'oxynitrure d'aluminium Y présente une masse volumique de 3,683 g/cm3
et des coefficients de dilatation thermique.
aT
7,5 10-6/ oC de 20 à 700°C
et aT
~ 9,0 10-6/ oC de 700 à 10000C
Sa transparence par transmission directe dans le domaine de longueur
d'onde 0,2 - 5,5 ~m dépasse 80%.
Le matériau présente par ailleurs une bonne dureté 1820 Hv et une
résistance à la rupture en flexion trois points supérieures à 300 MPa ou 200
MPa pour le produit transparent après grossissement des grains. Les effets
d'oxydation sur les propriétés mécaniques, de même que la résistance aux chocs
thermiques ont également été étudiés.
C'est la première fois qu'une céramique polycristalline transparente de
type non oxyde a pu être préparée. Ses possibilités d'application sont
multiples aussi bien dans le domaine militaire (blindage transparent, dômes de
missiles à guidage infrarouge) qu'industriel où le matériau peut être utilisé
comme envo1oppes de lampes ou tubes de décharge, hublots de four, etc •••

B l B LOG R A PHI E
1 - A.M. LEJUS - Rev. Int. Htes Temp. Refract. Fr., 1, 1964, 53.
2 - P. GOURSAT, p. GOEURIOT, M. BILLY - Mater. Chem., 1, 1976, 97.
3 - J.W. Mc CAULEY, WAKEFIELD, N.D. CORBIN - Brevet américain nO 4 241 000,
1980.
4 - RAYTHEON Co - Brevets Français nO 2 512 003 et 2 512 012, 1982.
5 - G. LONG, L.M. FOSTER - J. Amer Ceram. Soc., 44, 1961, 255.
6 - J. ADAMS, R.T. AUCOIN, G.A. WOLFF - J. Elect. Soc., 109, 1962, 1050.
7 - P. GOURSAT, M. BILLY, P. GOEURIOT, J.C. LABBE, J.M. VILLECHENOUX,
G. ROULT, J. BARDOLLE - Materials Chemistry, 6, 1981, 81.
8 - R. COLLONGES, J.C. GILLES, A.M. LEJUS - Bull. Soc. Chim. Fr., 11, 1962,
2113.
9 - J.W. CAULE Y - J. Amer. Ceram. Soc., 61, 1978, 372.
10 - R. KIEFFER, W. WRUSS, B. WILLER - Rev. Int. Htes Temp. et Refract.,
1976, 98.
11 - D. TURPIN-LAUNAY, F. THEVENNOT, F. DELVOYE, P. BOCH - 5ème Cimtech
Faenza, 1982.
12 - J.W. Mc CAULEY, N.D. CORBIN, J. Am. Ceram. Soc., 62, 1979, 476.
13 - T.M. HARTNETT, E.A. MAGUIRE, R.L. GENTILMAN, N.D. CORBIN, J.W. Mc CAULEY
Ceramic Proceeding, 3, 1982, 67.
14 - J.W.Mc CAULLEY and N.D. CORBIN in F. RILEY Ed "Progres in Nitrogen
Ceramics Martinius Nijhoff Publisher", Boston, 1983, p. 111.
15 - J.P. LECOMPTE - Thèse, Université de Limoges, 1982.
16 - K.S. HAN - Thèse, Université de Limoges, 1984.
17 - P. GOEURIOT - Thèse, Université de Limoges, 1975.
18 - R. COLLONGES, F. LEPRINCE-RINGUET - Bull. Soc. Chim. Fr., 1962, 522.
19 - G.A. SLACK, T.T. Mc NELLY - J. Cristal Growth, 34, 1976, 263.
20 - M. HOCH, K.M. NAIR - Am. Ceram. Soc. Bull., 58, 1979, 187.
21 - S. IWANA, K. JAYAKAWA, T. ARIZUMI - J. Cristal Growth, 56, 1982, 265.
22 - W.L. LI, L.P. HUANG, X.Z. HUANG, G. KUANG, S.H. TAN, S.R. FWU, T.S. YEN
P. VINCENZINI - Ed. Ceramic Powders, Elsevier, Amsterdam 1983, p. 403.

- 179 -
23 - M. ISH-SHALOM - J. Mater. Sei. Lett., 1, 1982, 147.
24 - C.A. JOUENNE - Traité de céramique et matériaux minéraux, Septima, Paris,
1975.
25 - W. RAFANIELLO, I.B. CUTLER - Corn. Am. Ceram. Soc., 64, 1981, C 128.
26 - P. BARRET - Cinétique Hétérogène, Gauthier-Villars, Paris, 1973.
27 - M.B. PILLING, R.E. BEDWORTH - J. Inst. Metal., 29, 1923, 529.
28 - L.H. DREGER, V.V. DADAPE, J.L. MARGRAVE - J. Pnys. Chim., 66, 1962,
1556.
29 - M. BILLY, G. VALENSI - Proceedings of the 6th C.I.T.C.E. Butterworths,
London, 1955, p. 371.
30 - R.J. BROOK - Treatrise on Materials Science and Technology Edts. Franklin,
F.Y. Wang, Academie Press New York, 9, 1976, 331.
31 - J.E. BURKE - Proceeoings of the third International Materials Symposium
"Ceramic Microstructures" at University of California, Berkeley Edits
Willy New York, 1966, 681.
32 - M. HILLERT - Acta. Met., 13, 1965, 227.
33 - G.W. GREENWOOD - Acta. Met., 4, 1956, 243.
34 - I.M. LIFSHITZ, V.V. SLYOZOV - J. Phys. Chem. Solids, 19, 1961, 35.
35 - I.M. STEPHENSON, J. WHITE - Trans. But. Ceram. Soc., 66,1967, 443.
36 - T. IKEGAMI, S. MATSUDA, Y. MORIYOSHI, H. SUZUKI - J. Mater. Sei., 17,
1982, 2855.
37 - S.J.L. KANG, DUK N. YOON - J. Mater. Sei., Letters 6, 1983, 291.
38 - G. ADO, M. BILLY, P. GUILLO, P. LEFORT - Brevet Fr. na 8320037.
39 - F.R.N. NABARRO - Phys. Soc., London, 1948, 75.
40 - G. HERRING - J. Appl. Phys., 21, 1950, 437.
41 - R.L. COB LE - J. Appl. Phys., 21, 1963, 1679.
42 - R.J. BROOK - J. Amer. Ceram. Soc., 65, 1980, 648.
43 - KYONG-SOP HAN - Thèse, Université de Limoges, 1984.
44 - J.P. LECOMPTE - Thèse, Université de Limoges, 1982.
45 - J. BESSON, P. SARAZIN - J. Chim. Phys., 64, 1967, 852 et 65, 1968, 1957.
46 - A. PELOUX - Thèse, Université de Grenoble, 1969.
47 - P. LEFORT - Thèse, Université de Limoges, 1981.
48 - W.D. KINGERY, DOWEN, UHLMANN - Introduction to ceramics, Ed. A. Willey,
p. 676.
49 - D.W. LEE, W.D. KINGERY - J. Am. Ceram. Soc., 43, 1960,
50 - B. WILLIAM, L. RAY, M. WAYNE PARKER - Ceram. Bull., 55, 1976, 1004.

- 180 -
,~~./«"r>.1' j<.'~'" »e "'~~'
51 - J. C. GLANDUS - Universi té,/tt,;;~e~~~!:52 - C. DEVIN - Thèse, Univer~i:t ~s} }983.
53 - S.C. CARNIGLIA - J. Am. C~\f.~" 55'.1.9.,~~.. 243.
54 - 1. SOROKA, P.J. SEREDA - J. ,m. ~,",?bC., 51, 1968, 337.! C ment .'~;,,!4:,
55 - P. GUI LLO - Thèse, Université de Lïmoges, 1985.



















