







Langages
Pages
Légal


Génétique de l’obésité humaine
INTRODUCTION : variabilité, monogénique vs. polygénique L'OBÉSITÉ POSSÈDE UN DÉTERMINISME GÉNÉTIQUE • Jumeaux / Familles • Réponse à la suralimentation
OBESITES MONOGENIQUES • Modèles animaux • Obésités monogéniques humaines
OBESITES POLYGENIQUES MULTIFACTORIELLES • Gènes candidats • Interactions avec la nutrition • Etudes pangénomiques (liaison, GWAS) • Valeur prédictive CONCLUSION

VARIABILITE GENETIQUE
• GENOME = 23 paires de chromosomes!→ # 3 milliards paires de bases d'ADN
• VARIATION dans la séquence de l'ADN → 1 base / 300-1000 (⇒ 3-10 millions !) est
différente entre 2 personnes prises au hasard ⇒ INDIVIDUALITE susceptible d'influer sur
l'adaptation à l'environnement...

Maladies monogéniques Maladies multifactorielles et polygéniques
Rares, la plus fréquente = 1/500 (hypercholestérolémie familiale)
Fréquentes, importance en Santé Publique : obésité, diabète, maladies cardiovasculaires…
Variant rare à effet fort Ex.: LDL R
Plusieurs gènes, variants fréquents à effets faibles Ex.: Apo E, CETP
Peu d'interaction avec environnement Interactions avec environnement (nutrition)
Études de familles (liaisons) pour identifier le gène en cause
Familles (±) et/ou populations (association) pour établir les risques individuels ou des populations + identification gènes

L'OBÉSITÉ POSSÈDE UN DÉTERMINISME GÉNÉTIQUE
Corrélations familiales


Comment séparer les effets des gènes des effets de l'environnement ?
Pourcentage de gènes en commun dans les relations familiales : mari - femme : 0 parents - enfants adoptifs : 0 parents - enfants biologiques : 50 membres d'une fratrie : 50 faux jumeaux ( dizygotes ) : 50 vrais jumeaux ( monozygotes ) : 100
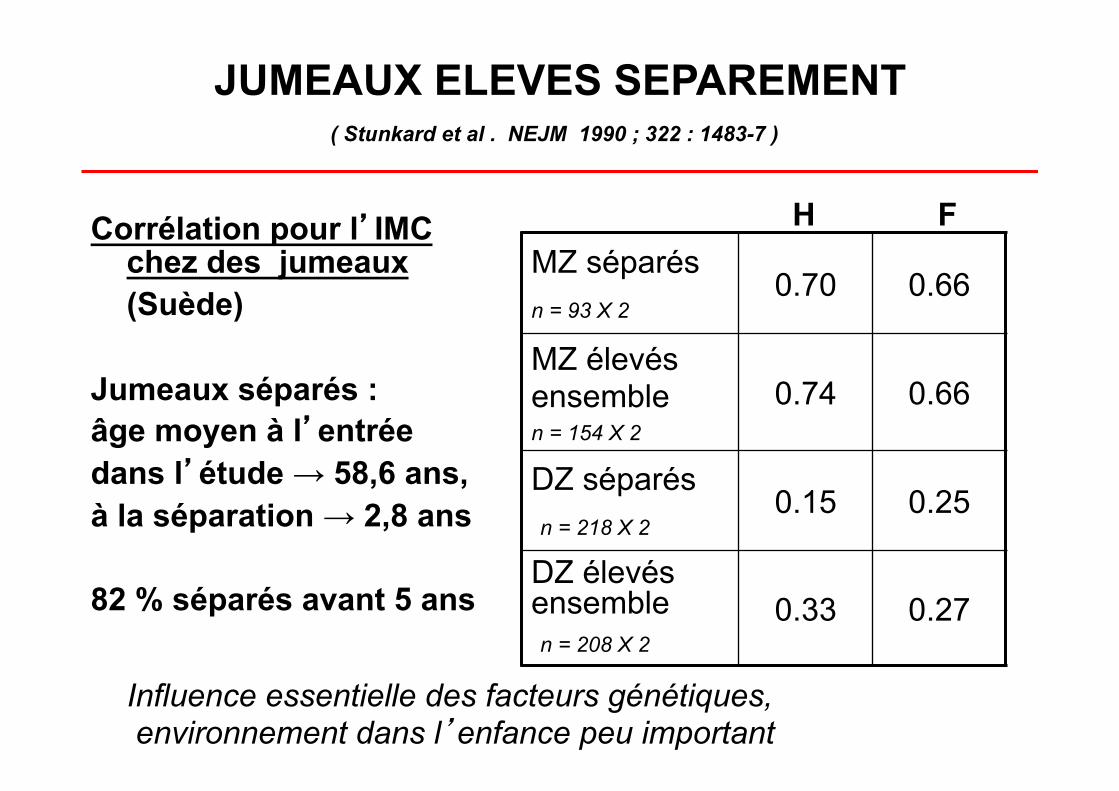
JUMEAUX ELEVES SEPAREMENT
Corrélation pour l’IMC chez des jumeaux (Suède)
Jumeaux séparés : âge moyen à l’entrée dans l’étude → 58,6 ans, à la séparation → 2,8 ans
82 % séparés avant 5 ans
MZ séparés
n = 93 X 2 0.70 0.66
MZ élevés ensemble n = 154 X 2
0.74 0.66
DZ séparés n = 218 X 2
0.15 0.25
DZ élevés ensemble n = 208 X 2
0.33 0.27
H F
Influence essentielle des facteurs génétiques, environnement dans l’enfance peu important
( Stunkard et al . NEJM 1990 ; 322 : 1483-7 )

21
22
23
24
25
26
27
ADOPTION DANS L'OBESITE HUMAINE
( Stunkard et al . N Engl J Med 1986 ; 314 : 193-8 )
Mince
IMC
des
Par
ents
(kg/
m2 )
Corpulence des Adoptés (540 Danois adultes)
Pères Mères
Moyen Surpoids Obèse
Parents Biologiques Parents Adoptifs
Mince Moyen Surpoids Obèse

Vrais jumeaux : suralimentation et déficit énergétique!
Entrée dans l’étude âge moyen : 21 ans graisse corporelle: 11% Suralimentation 1000 kcal / jour 100 jours (6j/7) = 84000 kcal suppl.
(Bouchard C et al. NEJM 1990, 1994)
Déficit énergétique 1 000 kcal / jour. exercice physique (ergocycle, deux fois par jour, 9 j/10, 93 jours, à apport calorique constant

LES GENES DE L’OBESITE

Marqueurs génétiques = régions variables de l’ADN, polymorphismes
• Gènes candidats – par leur fonction biologique : implication
physiopathologique possible à métabolismes / syndromes mendéliens d'obésité
– par leur position : régions homologues des modèles de rongeurs
• Répartis sur tout le génome pour identifier des régions du génome comportant des variants de gènes liés (études de familles) ou associés (études cas-témoins) à la maladie

Génétique de l’obésité humaine (Obesity gene map 2005)
• Exceptionnel : monogénique variants génétiques provoquant directement l’obésité, Pas/peu d’interaction avec l’environnement → 11 gènes, < 200 individus
• Cas général : multifactoriel polygénique gènes à variants fréquents, à petits effets, interaction avec l’environnement → > 300 gènes ou régions chromosomiques, majorité des sujets
Rankinen et al. Obesity 2006;14:529-644.

Obésités monogéniques

La souris ob/ob : obèse, diabétique, hyperphage


3 ans : 42 kg 7 ans : 32 kg
Deficit génétique en leptine Avant et après traitement
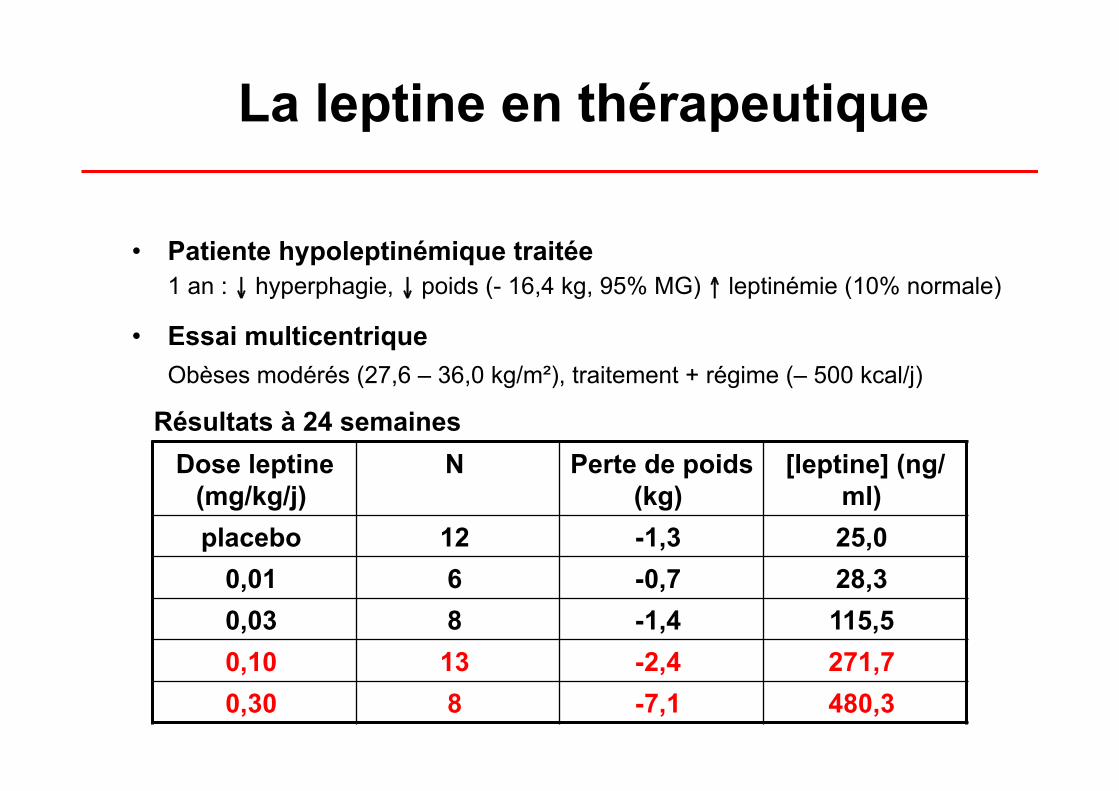
La leptine en thérapeutique
• Patiente hypoleptinémique traitée 1 an : ↓ hyperphagie, ↓ poids (- 16,4 kg, 95% MG) ↑ leptinémie (10% normale)
• Essai multicentrique Obèses modérés (27,6 – 36,0 kg/m²), traitement + régime (– 500 kcal/j)
Dose leptine (mg/kg/j)
N Perte de poids (kg)
[leptine] (ng/ml)
placebo 12 -1,3 25,0 0,01 6 -0,7 28,3 0,03 8 -1,4 115,5 0,10 13 -2,4 271,7 0,30 8 -7,1 480,3
Résultats à 24 semaines

Gène muté chromosome Défaut primaire Mutation / individus LEP
Récessif 7q31.3
leptine 2 / 6
LEPR
Récessif 1p31
récepteur de la leptine 1 / 3
POMC Récessif 2p23.3
pro-opio-mélanocortine (ACTH et αMSH)
3 / 8
MC4R
Dominant 18q22
récepteur de l'αMSH 51 / 143
MC3R Dominant 20q13.2-3
récepteur de l'αMSH 1 /2
Proconvertase I (PCSK1)
Récessif 5q15-q21
maturation des hormones (insuline, POMC...)
1 / 1
SIM1
Dominant 6q16
développement de l’hypothalamus 2 / 2
GPR24 22q13.2 récepteur MCH 2 / 2
NTRK2 Dominant 9q22.1
récepteur BDNF (TrkB) 1/1
CRHR1 17q12-q22 récepteur CRH 1 / 1
CRHR2 7p14.3 récepteur CRH 1/ 1
Obésités monogéniques humaines Obesity gene map 2005
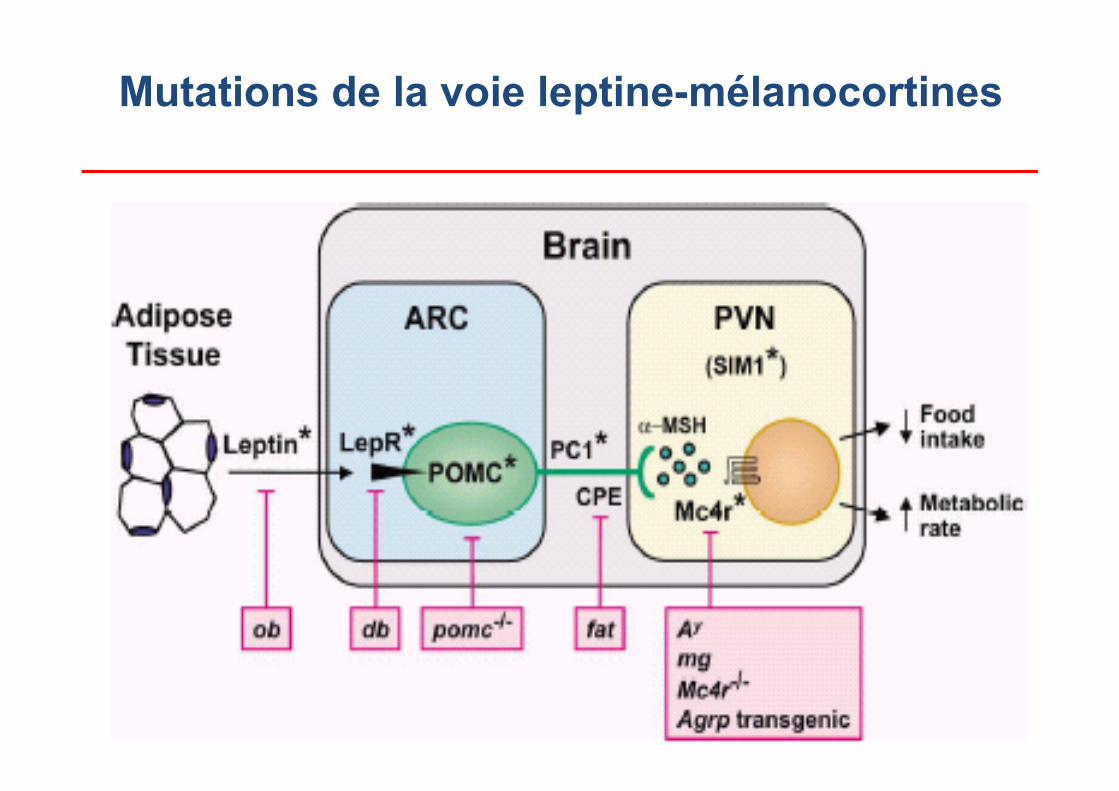
Mutations de la voie leptine-mélanocortines

Cas général : exemples de gènes candidats dans l’obésité commune, interactions avec
la nutrition

Les influences génétiques s’expriment en interaction avec la nutrition
!

0
10
20
30
40
50
Gene-Environment Interaction in the Pathogenesis of Obesity
Bod
y M
ass
Inde
x (k
g/m
2 )
Ravussin E et al. Diabetes Care 1994;17:1067-1074.
Pima Indians
Maycoba, Mexico Arizona
P <0.0001

Gènes candidats !
Gènes à rôle potentiel : leur produit est impliqué dans les métabolismes !
Susceptibilité à l’obésité • Prise alimentaire
Leptine et récepteur, 5-HT2AR, 5-HTT, NPY • Dépense énergétique
UCP(s), leptine et récepteur, récepteurs catécholamines • Biologie adipocytaire
PPARγ, adiponectine, CEBP, SREBP • Métabolisme lipidique
Lipoprotéine lipase (LPL), Apos E, B, A-I/C-III/A-IV, CETP
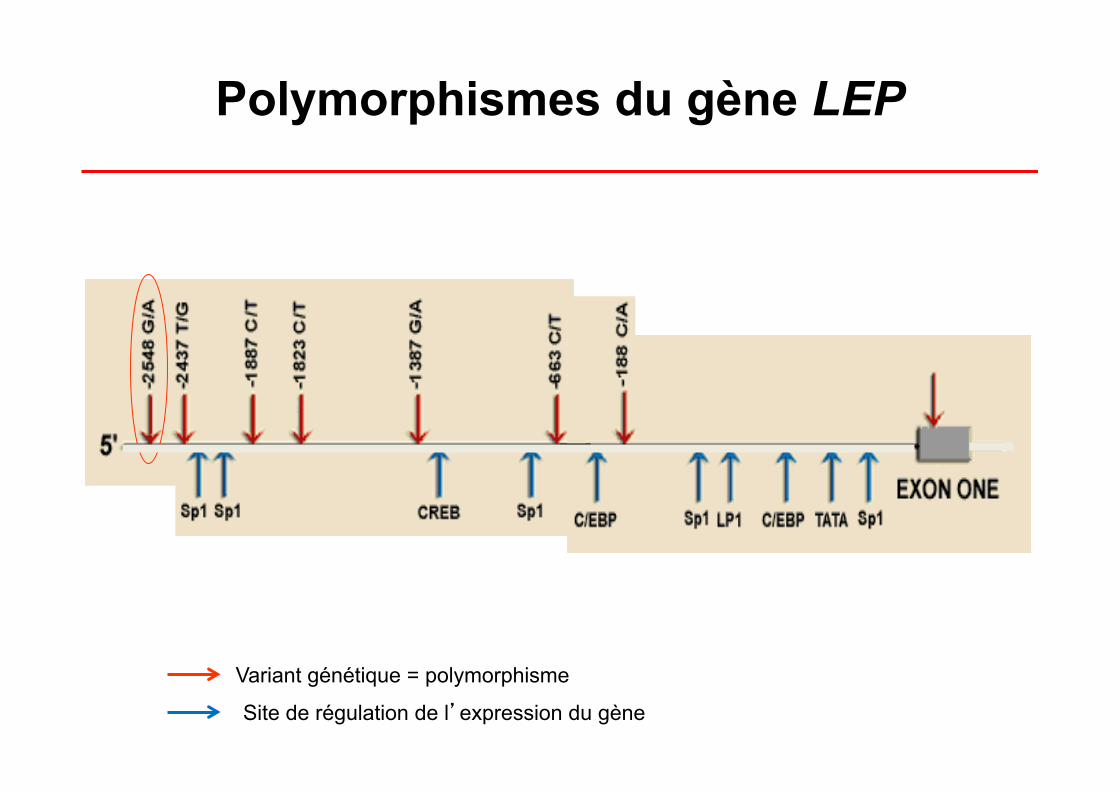
Polymorphismes du gène LEP
Variant génétique = polymorphisme
Site de régulation de l’expression du gène

Polymorphisme LEP G-2548A Fréquences dans la Cohorte STANISLAS
Génotypes (%) Allèles (%)
GG GA AA G A
Normal (IMC < 27) 27.4 52.5 20.1 53.7 46.4
Surpoids (IMC ≥ 27) 38.5 51.4 10.1 64.2 35.8
P (χ2)= 0.019; P (tendance) = 0.005
P (χ2)= 0.007
(Mammès O et al., Ann Hum Genet 2000)

Polymorphisme LEP G-2548A Réponse au régime hypocalorique
(Mammès O et al. Diabetes 1998)

• Récepteur de la leptine
• UCP1 àdépense énergétique
• Lipoprotéine-lipase àmétabolisme lipidique, risque CV
• etc…
Résultats similaires avec :
Autres gènes

PPARγ : régulation du métabolisme des lipides.
PPARγ
Stockage des lipides
Importation des acides gras Lipogénèse
Différenciation adipocytaire
Tissu adipeux blanc

Interaction entre consommation lipidique et Pro12Ala de PPARγ : étude D.E.S.I.R.
Chez les grands consommateurs de lipides, les Ala/Ala ont un BMI plus élevé que les sujets porteurs d’un allèle Pro, p interaction = 0,02
Lamri et al. IJO, 2012

Prédisposition aux complications de l’obésité
Exemple : LPL et HTG

Polymorphisme LPL/Hind III et interactions
0,9
2,26
0,88
2,08
1,02
2,46
0,98
1,56
0
0,5
1
1,5
2
2,5
3
Normo TG Hyper TG
Trig
lycé
rides
(m
mol
/l)
Triglycéridémie en fonction du régime et du polymorphisme HindIII de la LPL
H1H1 + H1H2 avantH1H1 + H1H2 aprèsH2H2 avantH2H2 après
Interaction P < 0,001
(Jemaa et al. IJO 1997)

Etudes pangénomiques
études de liaison familiale
= transmission conjointe des marqueurs et des traits phénotypiques
dans les familles (clonage positionnel)

INDIENS PIMA OBÉSITÉ ET MÉTABOLISME ÉNERGÉTIQUE
ETUDES DE LIAISON PAR MARQUEURS ANONYMES
Phénotypes % graisse (densitométrie), répartition (taille / cuisse),
dépense énergétique / 24h, métabolisme de sommeil, quotient respiratoire / 24h
Sujets
Composition corporelle : 127 familles, 362 sibs, 451 sib-pairs Métabolisme énergétique :
82 familles, 220 sibs, 236 sib-pairs

INDIENS PIMA OBÉSITÉ ET MÉTABOLISME ÉNERGÉTIQUE
Marqueurs 516 microsatellites, distance # 6,4 cM
Résultats (LOD > 2) % graisse : chr. 11q21-22, 18q21 énergie 24h : 11q23-24 QR 24h : 1p31-p21, 20q11.2
Gènes candidats LEPR (1p31), ASIP (20q)

Clonages positionnels
USA (Philadelphie)
1) 92 familles (n = 513), (BMI > 40 + BMI >30 + BMI < 27) 354 marqueurs (# 10 cM) → 4 marqueurs positifs en 20q 2) 124 familles (n = 713)
25 marqueurs en 20q → 20q13 (idem Pima) candidats de la région : ASIP, ADA, MC3R, MODY1, PCK1, CEBPB

Clonages positionnels
FRANCE 158 familles nucléaires (n = 514), super obèses (BMI > 40 + BMI > 27), 380 microsatellites (9,1 ± 2,5 cM) → obésité : chr 10 (D10S197 : LOD = 4,85) → [leptine] : chr. 2p (LOD = 2,68, région POMC), Chr. 5 (LOD = 2,93)
MEXICAINS-AMÉRICAINS 10 familles (n = 459), 20 cM → [leptine] et masse grasse : chr 2p21 (LOD = 4,95) candidats : POMC, GCKR (glucokinase regulatory protein)

NHLBIFHS GENOME SCAN
National Heart, Lung and Blood Institute Family Heart Study = Framingham Heart Study + Utah Health Family Tree Study + Atherosclerosis Risk in
Communities Study
Phénotype : BMI 2 populations
1) recrutement sur la base de CHD ou risque CHD élevé 1184 sujets, 317 fratries 243 marqueurs, distance # 20 cM
2) recrutement sur la taille des familles 3027 sujets, 401 familles sur 3 générations 404 marqueurs, distance # 8,5 cM
Résultats (LOD > 2) 7q32.3 LOD = 4.9 (P < 0,00001) ⇒ 28% BMI expliqués 13q14 LOD = 3.2 (P = 0,00006)
Gènes candidats : LEP (7q31.33), 5-HTR2A, esterase D (13q14)

Etudes pangénomiques
grandes études d’association: GWAS
2007 : Genome Wide Association Studies Puces de 100 000 à 1 M de polymorphismes (SNPs) testés dans des études cas-témoins



Un variant fréquent du gène FTO est associé à l’IMC et prédispose à l’obésité infantile et adulte
• Effet sur l’IMC (variant rs9939609 [A]), répliqué dans 13 cohortes incluant 38 759 participants.*
• Les 16% d’adultes homozygotes AA pèsent environ 3 kg de plus et ont un risque d’obésité X 1,67 comparés aux sujets TT.*
• Variation génétique de FTO rendrait compte de 22% des individus obèses communs.**
* Frayling et al. Science 2007 ** Dina et al. Nature Genetics 2007
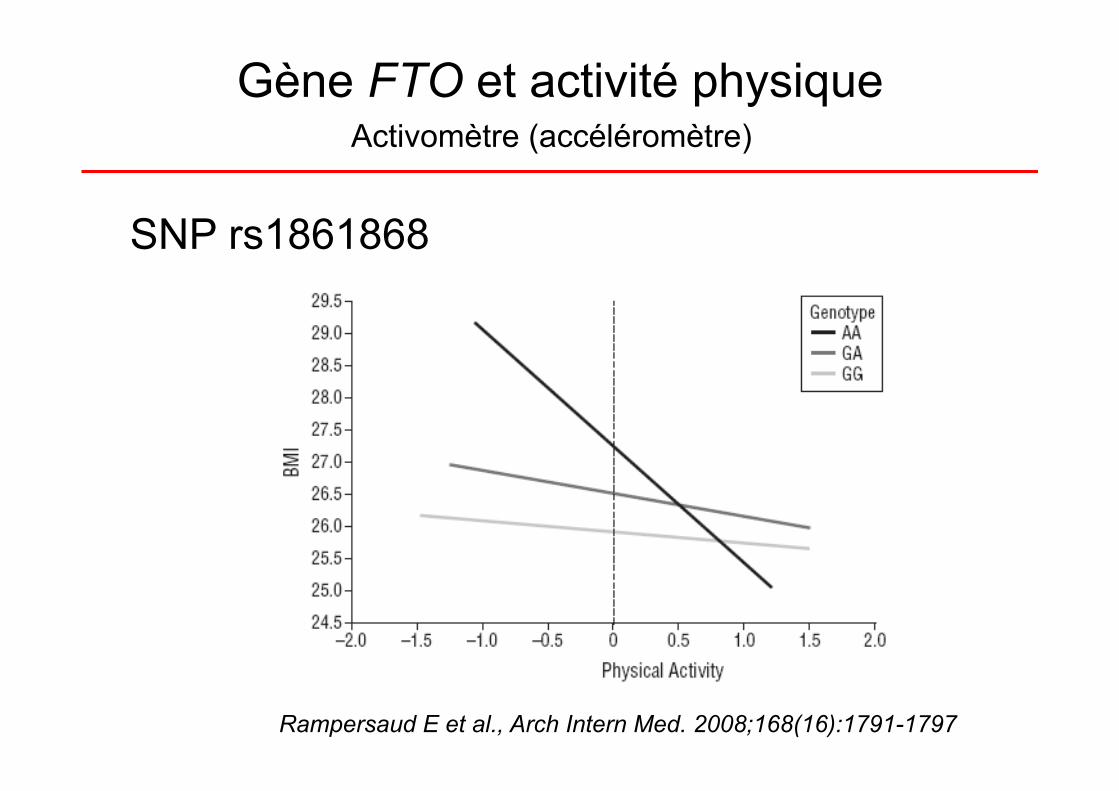
Gène FTO et activité physique
Rampersaud E et al., Arch Intern Med. 2008;168(16):1791-1797
Activomètre (accéléromètre)
SNP rs1861868

Gène FTO et comportement alimentaire (SNP rs9939609)
Chez des enfants, A augmente la consommation calorique indépendamment du poids corporel, avec préférence pour les aliments denses en énergie (repas tests) Cecil et al., N Engl J Med 2008;359:2558-66
A diminue la sensation de satiété (questionnaire-
test). Cette réponse explique en partie l’adiposité augmentée. Wardle et al., J Clin Endocrino Metab 2008;93:3640-3643

MC4R 108 auteurs, dont certains sont des consortiums!!!

MC4R et obésité précoce
a) Évolution du BMI avec l’âge en fonction du génotype b) % de masse grasse en fonction du génotype chez les enfants de 9 ans c) méta-analyse: effet sur le risque d’obésité sévère par allèle chez 10 583 enfants/ados de 3 études

GWAS 2009
NATURE GENETICS 2009; 41(1) • Thorleifsson et al.
– Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity pp 18-24
• Willer et al. – Six new loci associated with body mass index highlight a
neuronal influence on body weight regulation pp 25-34
FTO et MC4R répliqués, + nouveaux variants impliqués dans la signalisation et le développement nerveux, impliqués dans le développement des centres cérébraux de la prise alimentaire ?

Effets cumulatifs des polymorphismes
• 12 SNPs (de 12 gènes, dont FTO et MC4R) issus des GWAS ont été génotypés chez 20 431 individus (age: 39–79 ans) provenant de la cohorte EPIC (European Prospective Investigation into Cancer and Nutrition) –Norfolk (GB). .
Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association Studies. (Li et al. Am J Clin Nutr 2010;91:184–90)
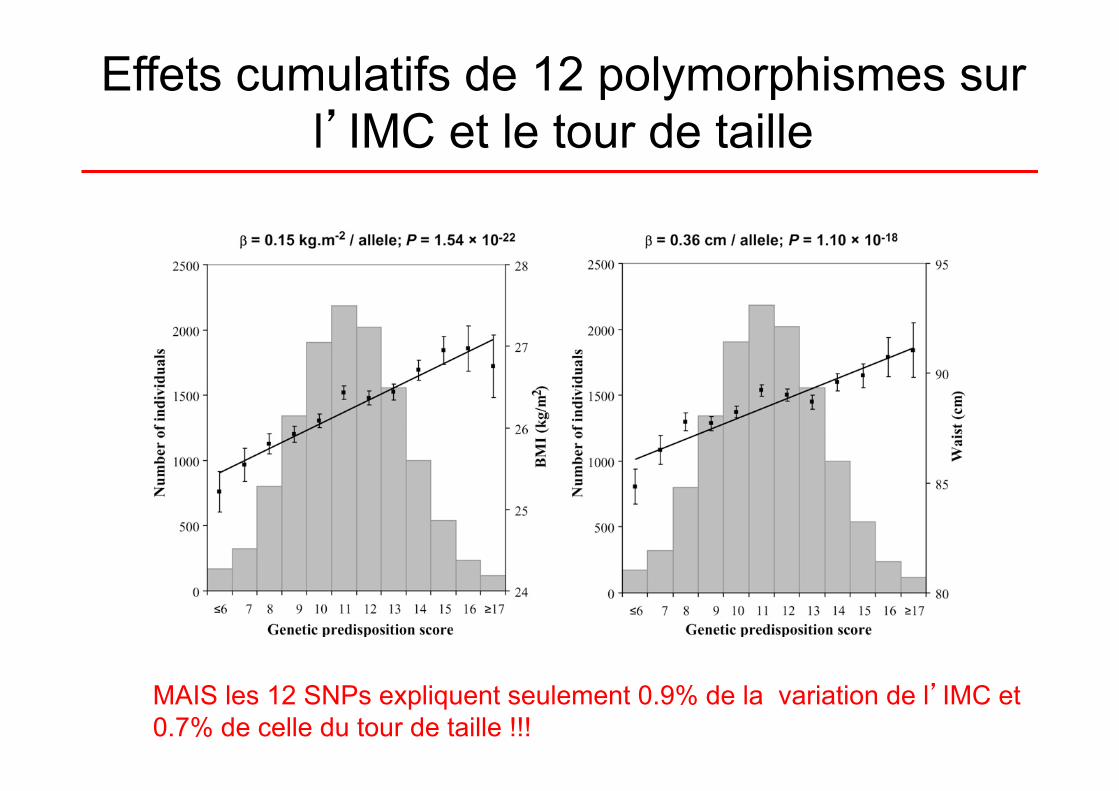
Effets cumulatifs de 12 polymorphismes sur l’IMC et le tour de taille
MAIS les 12 SNPs expliquent seulement 0.9% de la variation de l’IMC et 0.7% de celle du tour de taille !!!

Valeur prédictive de 12 polymorphismes pour l’obésité
Modèle 1: âge, âge², sexe (n = 10 871); AUC:0,572 modèle 2: tous les SNPs (n = 6452); AUC: 0,574 modèle 3: âge, âge², sexe , tous les SNPs (n = 6452); AUC: 0,597
àprise en compte des facteurs génétiques : amélioration de 2% / modèle 1
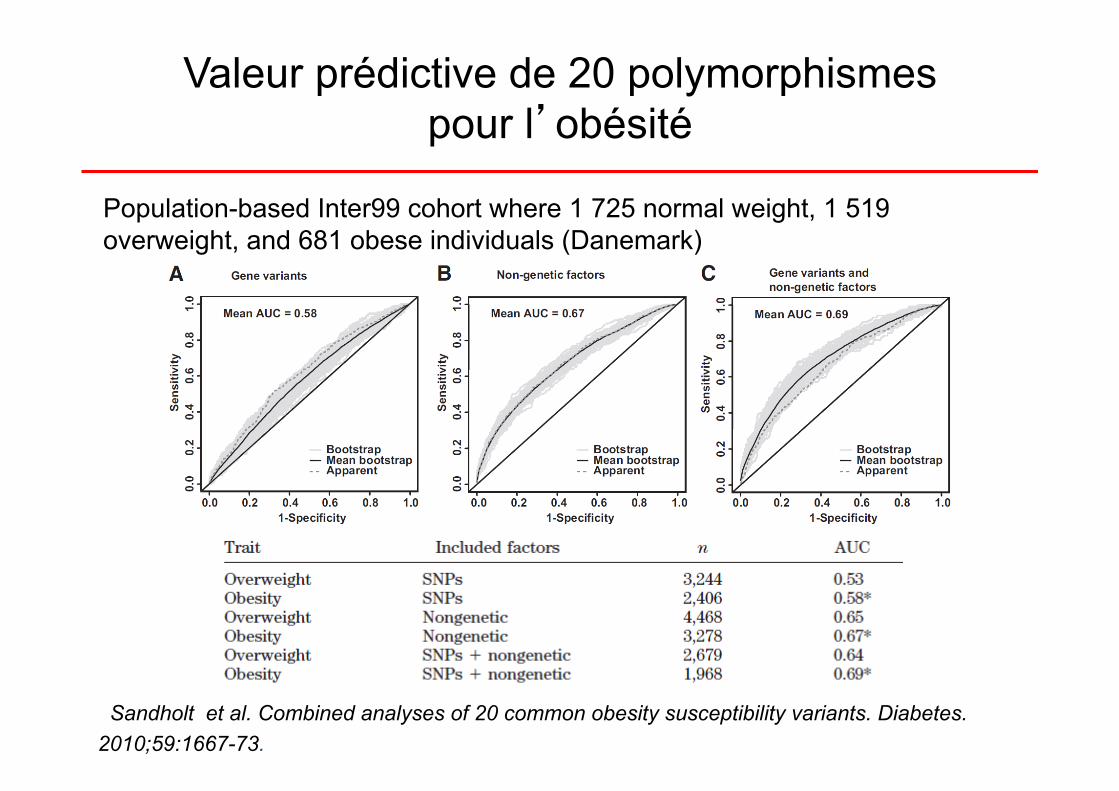
Valeur prédictive de 20 polymorphismes pour l’obésité
Sandholt et al. Combined analyses of 20 common obesity susceptibility variants. Diabetes. 2010;59:1667-73.
Population-based Inter99 cohort where 1 725 normal weight, 1 519 overweight, and 681 obese individuals (Danemark)

MAIS…
SNPs expliquent quelques % de l’héritabilité (études familiales) à Problème de l’héritabilité manquante
Les GWAS • ne contiennent pas tous les polymorphismes • manquent les effets des variants rares, et des
variants de structure chromosomique (insertions, délétions, répétitions)

Séquençage des gènes candidats
Ahituv et al. Am J Hum Genet 2007;80:779-791
Variants observés uniquement soit chez les obèses, soit chez les minces. (A) 21 gènes impliqués dans les obésités monogéniques (B) 37 gènes candidats des voies potentiellement impliquées dans l’obésité
58 gènes (96kb) séquencés (exons codants, jonctions avec introns) chez 379 individus obèses et 378 minces à1074 variants dont 852 rares (< 1%)

Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Duplication dans la même région 16p11 (n= 138 /95000) : Recrutement : cohortes de sujets avec retards mentaux, développement, pb psychiatriques, mais aussi pop. générales) Chez les enfants, ä poids de naissance, cachexie, retard de croissance Adultes: RR maigreur (IMC < 18,5) = 8,3 Symptômes accompagnés de comportement alimentaire sélectif/restrictif + ä circonférence cranienne “Each of the observed phenotypes is the converse of one reported in carriers of deletions at this locus.”
Jacquemont et al. NATURE. 2011 Aug 31;478(7367):97-102

Conclusion générale
• Les obésités purement génétiques sont rares, mais leur étude a permis de découvrir des nouvelles voies métaboliques, impliquées principalement dans la régulation nerveuse de la prise alimentaire
• La majorité des obésités comporte une composante héréditaire due à des variations génétiques dont les effets sont faibles, mais qui agissent…
• en interaction avec l’environnement nutritionnel / activité physique. • Puisque les gènes agissent en interaction avec l’environnement, il faut
agir sur l’environnement !!!
Génétique ≠ Fatalité !

Epigénétique Rôle dans l’obésité
• Empreinte: région 15q11 (si père : Prader-Willi), mutation locus GNAS (ostéodystrophie d’Albright, obésité si mère)
• Environnement in utero à risque d’obésité et diabète à la 1ère génération + effets transgénérationnels
• Régime riche en graisses : profils méthylations MC4R, LEP, POMC
• FTO = déméthylase ADN • PPARγ : interactions avec acétylations histones pdt
adipogénèse
Top Related