
Unité d’Enseignement LC 202 Atomistique et Liaison ... · Atomistique et Liaison Chimique ......
11
Université Pierre et Marie Curie Année 2006-2007 Unité d’Enseignement LC 202 Atomistique et Liaison Chimique TRAVAUX PRATIQUES: spectroscopie atomique et moléculaire Lab. de Chimie Physique-Matière et Rayonnement 11 rue Pierre et Marie Curie 75231 PARIS cédex 05
Transcript of Unité d’Enseignement LC 202 Atomistique et Liaison ... · Atomistique et Liaison Chimique ......

Université Pierre et Marie Curie Année 2006-2007
Unité d’Enseignement LC 202
Atomistique et Liaison Chimique
TRAVAUX PRATIQUES:
spectroscopie atomique et
moléculaire
Lab. de Chimie Physique-Matière et Rayonnement 11 rue Pierre et Marie Curie
75231 PARIS cédex 05

2
TP n°3: SPECTROSCOPIE D'ABSORPTION DANS L'ULTRAVIOLET
Applications de la spectroscopie d'absorption UV
à l'étude des molécules organiques
A. Rappels théoriques
1. Introduction
Nous définissons le domaine des spectres ultraviolet-visible par les longueurs
d’ondes comprises entre 100 et 800 nm. (Les longueurs d’ondes indiquées ici
sont exprimées en nanomètre, symbole nm,1 nm = 1 mµ =10-9m = 10 Å)
L'absorption d'un photon correspondant à ce domaine peut provoquer une
augmentation (∆E = hc/λ) de l'énergie de la molécule de l'ordre de 1200 à 150
kJ.mol-1 (290 à 40 kcal.mol-1), conduisant à une modification de l'état
énergétique électronique, vibrationnel et rotationnel de la molécule, ainsi :
hν = (∆E) électronique + (∆E) vibrationnelle + (∆E) rotationnelle
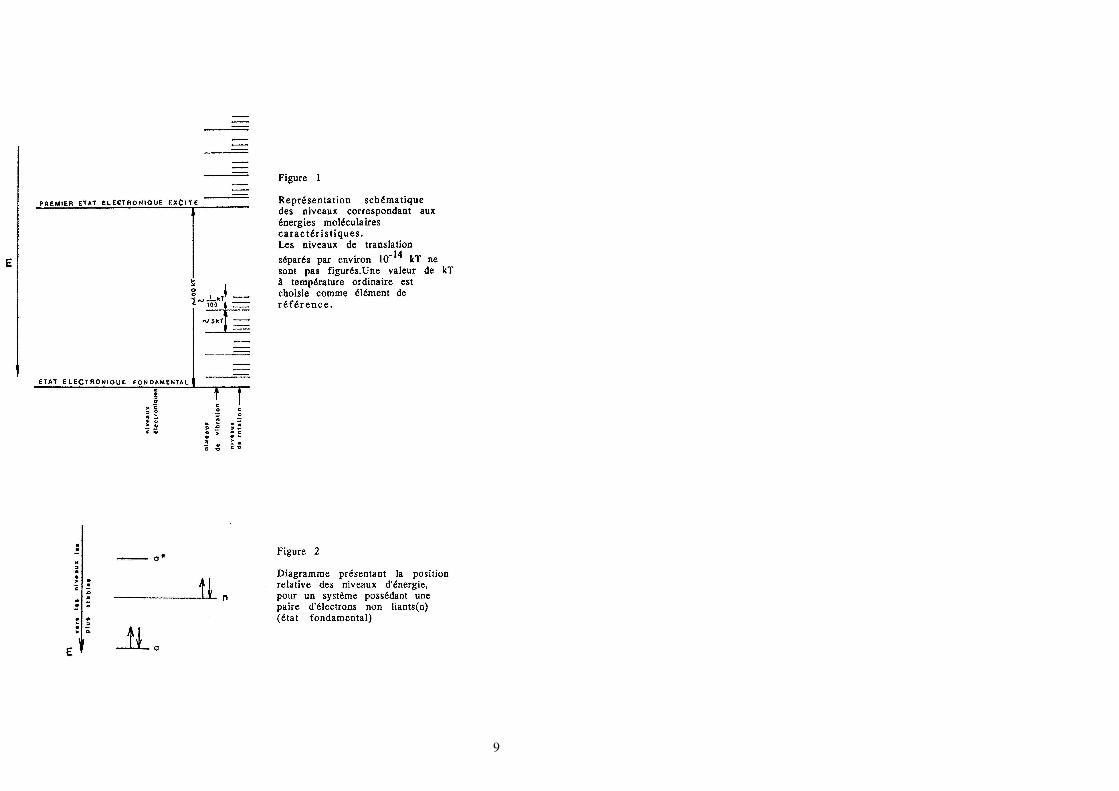
La figure 1 précise l'importance de ces énergies, en prenant kT à température
ordinaire comme élément de référence.
Précisons que le domaine du visible s'étend de 350 à 800 nm, celui de
l'ultraviolet est donc défini par l'intervalle 100 - 350 nm. Pour des raisons
expérimentales, il faut distinguer la zone de l'UV lointain (100 à 200 nm), plus
difficilement accessible, et celle comprise entre 200 et 350 nm, zone explorée par
la plupart des spectromètres (UV correspondant à la transparence du quartz).
Considérons un échantillon gazeux, comportant de petites molécules, pour une
transition électronique, il existe différentes possibilités pour les transitions
vibrationnelles et rotationnelles, il y a donc absorption dans un assez large
domaine de longueurs d’ondes. (Des règles de sélection, dont nous ne parlerons
pas en détail, gouvernent le passage d'un état initial défini par les niveaux
rotationnel, vibrationnel et électronique, à l'état final).
Si le pouvoir de résolution du spectromètre n'est pas suffisant, nous voyons de
larges bandes d'absorption, et, quelquefois dans des conditions favorables, nous
pouvons résoudre la structure fine de vibration, problème que nous
n'examinerons pas ici. Pour les molécules complexes, (considérons celles non
rigides, formées de plus de cinq atomes, ce qui exclut des entités rigides telles
que le benzène), le nombre de transitions possibles est si grand, que le spectre
obtenu correspond à la superposition des raies très voisines et prend l'apparence
d'un spectre de bandes.
Pour les molécules organiques, nous savons que les électrons des liaisons
covalentes simples, telles que C-C et C-H, exigent des énergies assez importantes
pour qu'il y ait une excitation électronique : passage d'un électron d'une orbitale
moléculaire liante σ vers une orbitale moléculaire antiliante σ*. Les
hydrocarbures saturés absorbent seulement des radiations de grande énergie,
habituellement en-dessous de 160 nm (1600 Å), dans l'ultraviolet lointain. Ainsi
pour le méthane, la bande caractéristique est aux environs de 125 nm, pour
l'éthane à 135 nm.
3
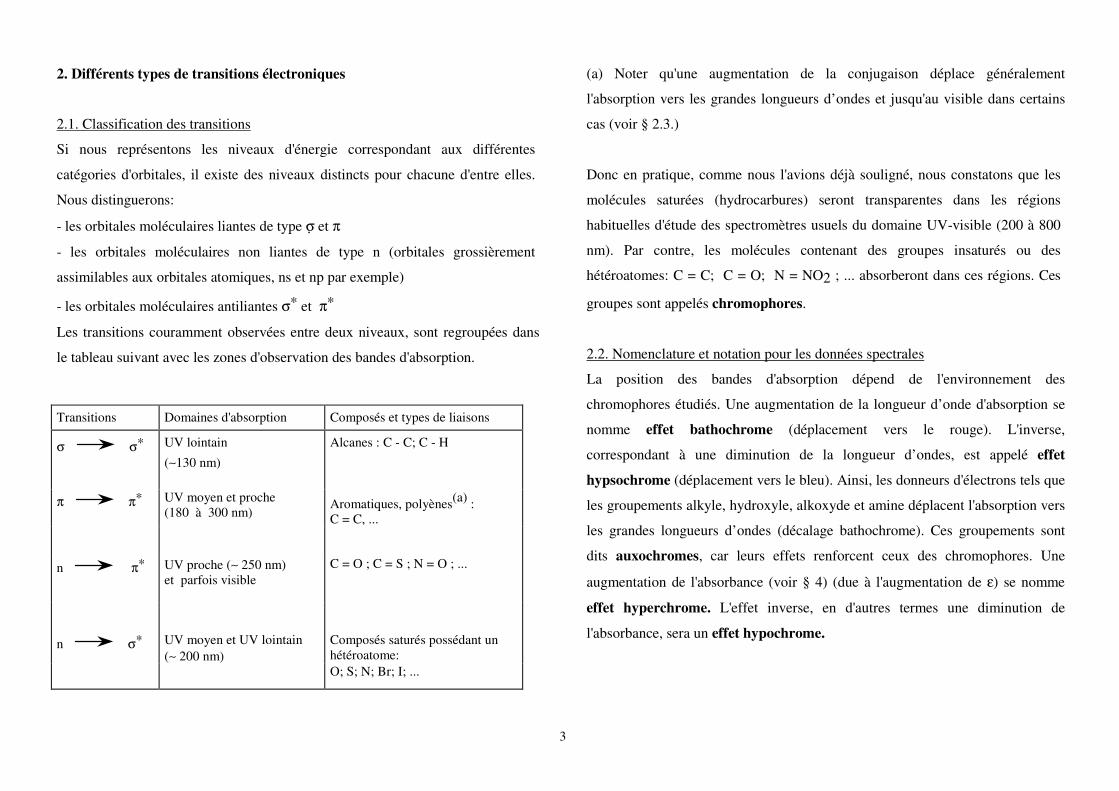
2. Différents types de transitions électroniques
2.1. Classification des transitions
Si nous représentons les niveaux d'énergie correspondant aux différentes
catégories d'orbitales, il existe des niveaux distincts pour chacune d'entre elles.
Nous distinguerons:
- les orbitales moléculaires liantes de type σ et π
- les orbitales moléculaires non liantes de type n (orbitales grossièrement
assimilables aux orbitales atomiques, ns et np par exemple)
- les orbitales moléculaires antiliantes σ* et π*
Les transitions couramment observées entre deux niveaux, sont regroupées dans
le tableau suivant avec les zones d'observation des bandes d'absorption.
(a) Noter qu'une augmentation de la conjugaison déplace généralement
l'absorption vers les grandes longueurs d’ondes et jusqu'au visible dans certains
cas (voir § 2.3.)
Donc en pratique, comme nous l'avions déjà souligné, nous constatons que les
molécules saturées (hydrocarbures) seront transparentes dans les régions
habituelles d'étude des spectromètres usuels du domaine UV-visible (200 à 800
nm). Par contre, les molécules contenant des groupes insaturés ou des
hétéroatomes: C = C; C = O; N = NO2 ; ... absorberont dans ces régions. Ces
groupes sont appelés chromophores.
2.2. Nomenclature et notation pour les données spectrales
La position des bandes d'absorption dépend de l'environnement des
chromophores étudiés. Une augmentation de la longueur d’onde d'absorption se
nomme effet bathochrome (déplacement vers le rouge). L'inverse,
correspondant à une diminution de la longueur d’ondes, est appelé effet
hypsochrome (déplacement vers le bleu). Ainsi, les donneurs d'électrons tels que
les groupements alkyle, hydroxyle, alkoxyde et amine déplacent l'absorption vers
les grandes longueurs d’ondes (décalage bathochrome). Ces groupements sont
dits auxochromes, car leurs effets renforcent ceux des chromophores. Une
augmentation de l'absorbance (voir § 4) (due à l'augmentation de ε) se nomme
effet hyperchrome. L'effet inverse, en d'autres termes une diminution de
l'absorbance, sera un effet hypochrome.
Transitions Domaines d'absorption Composés et types de liaisons
σ σ* UV lointain
(∼130 nm)
Alcanes : C - C; C - H
π π* UV moyen et proche (180 à 300 nm)
Aromatiques, polyènes(a) : C = C, ...
n π* UV proche (∼ 250 nm) et parfois visible
C = O ; C = S ; N = O ; ...
n σ* UV moyen et UV lointain (∼ 200 nm)
Composés saturés possédant un hétéroatome:
O; S; N; Br; I; ...

4
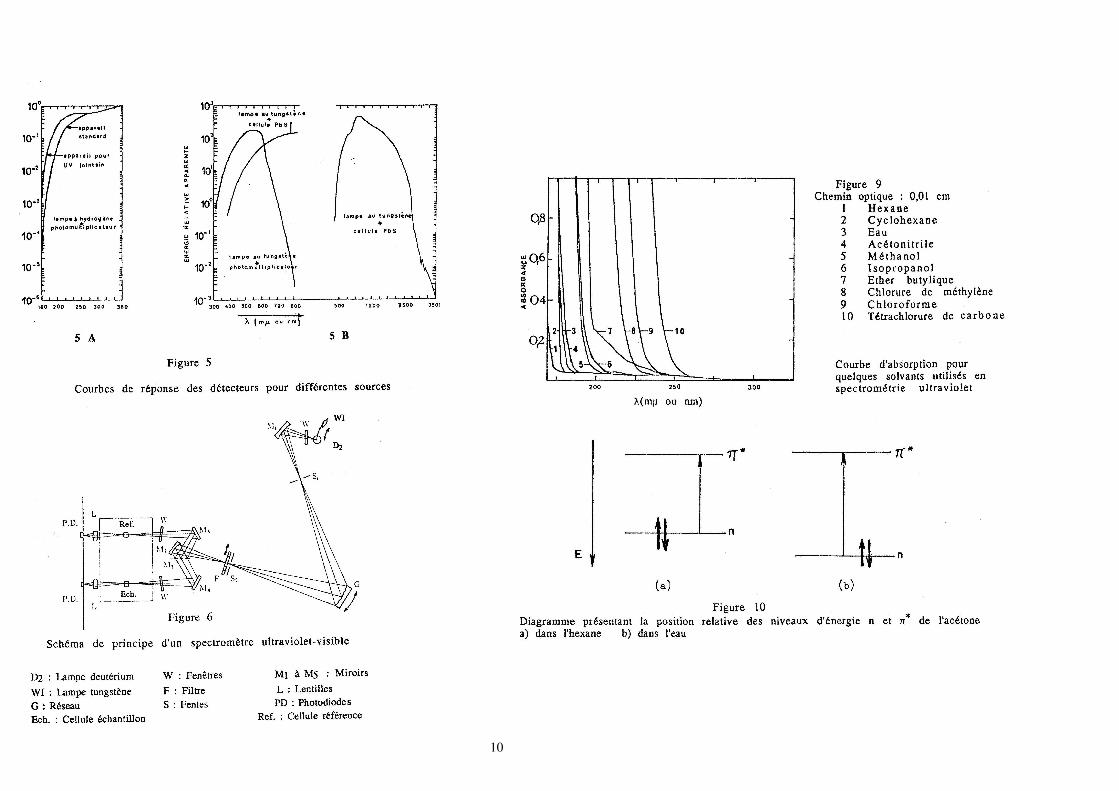
3. Description d'un spectromètre
Les spectromètres du domaine UV-visible fonctionnent sur le même principe que
les spectromètres du domaine infrarouge. Cependant il faut noter quelques
différences dans la réalisation.
Sources: lampe à vapeur de mercure, lampe à tungstène ou lampe à deutérium ou
à hydrogène.
Monochromateur: s'il s'agit d'un prisme, le matériau utilisé doit être transparent
au rayonnement étudié (quartz dans l'UV, LiF dans l'UV lointain ou le visible).
Le monochromateur est généralement placé avant la traversée de l'échantillon par
le rayonnement, afin d'éviter l'échauffement trop important du produit étudié.
Détecteur: plaque photographique ou cellule photoélectrique ou
photomultiplicateur. Il faut retenir qu'un détecteur choisi n'a pas une courbe de
réponse telle qu'il soit utilisable dans tout un domaine : son emploi est limité à
une région.
L'appareil est du type double faisceau et comme nous l'avons souligné, une des
principales différences entre celui-ci et un spectromètre infrarouge, est liée à
l'emplacement de l'échantillon sur le chemin optique. Sur la figure 6, nous avons
représenté les deux sources: l'une émet dans l'ultraviolet (lampe au deutérium :
200 nm à 340 nm), l'autre dans le visible (lampe au tungstène: 320 nm à 800 nm).
Un système automatique permet d'utiliser l'une ou l'autre selon le domaine
exploré. Après traversée dans la fente d'entrée le faisceau pénètre dans le
monochromateur. Le système dispersif est un réseau holographique concave; le
faisceau provenant de la fente de sortie est monochromatique et va être envoyé
alternativement dans le circuit de mesure et dans le circuit de référence, grâce à
un miroir tournant. Après le passage dans les cellules les faisceaux sont dirigés
sur les détecteurs (2 photodiodes au silicium). Le compartiment de mesure est
maintenu à l'abri de la lumière.
4. Conditions générales relatives aux mesures
L'enregistrement des spectres UV-visible s'effectue à partir de solutions très
diluées. Il faut dissoudre dans le solvant choisi, une quantité de produit
convenable (de l'ordre du milligramme pour 100 cm3 de solvant lorsque la masse
molaire du soluté est voisine de 100 à 200). La cellule est construite de telle
façon que le faisceau incident traverse une épaisseur de solution égale à 1 cm.
Une cellule identique, remplie de solvant pur, est placée sur le faisceau de
référence. La plupart des appareils permettent d'enregistrer l'absorbance A ou
densité optique, en fonction de la longueur d’onde λ.
Dans la relation de BEER-LAMBERT l××ε=== cT1
logI
IlogA 0
Io: intensité de la lumière incidente
I: intensité de la lumière transmise
T: transmission
Les grandeurs intervenant couramment sont:
c: concentration en mol. L-1
λ: épaisseur de la cuve en cm
ε: coefficient d'absorption (ou d'extinction) molaire exprimé en L.cm-1.mol-1
(Ces dernières unités ne sont généralement pas mentionnées.)

5
Les coefficients d'extinction molaire peuvent varier de 10 à 100000 et ainsi c'est
souvent le diagramme log ε en fonction de λ qui se trouve dans les données de la
bibliographie. Par exemple, pour une bande d'absorption donnée (sommet de la
bande), il est d'usage de préciser: λmax = 466 nm; ε = 122000 (pour cette
longueur d’onde) et parfois σ (nombre d’onde) égal ici à 21500 cm-1.
Le solvant employé doit être transparent dans la région à étudier. D'après ce qui a
été vu, sont utilisables en général dans l'ultraviolet lointain les solvants qui ne
comportent que des liaisons σ. Le plus commun est l'éthanol à 95% (l'emploi de
l'éthanol absolu n'est pas indiqué, parce qu'il est obtenu par distillation avec du
benzène (formation d'un azéotrope), or le benzène, même à l'état de traces, se
trouve être une impureté très absorbante). L'éthanol à 95% n'est pas très coûteux,
bon solvant dans la plupart des cas et transparent à partir de 210 nm jusqu'au
domaine du visible compris. Remarquons, que des structures fines peuvent être
obtenues dans le cyclohexane ou d'autres hydrocarbures qui, moins polaires,
présentent peu d'interactions avec des molécules dissoutes.
Nous retiendrons les zones d'emploi des solvants usuels (les longueurs d’ondes
données en nm sont valables avec une cellule de 1 cm).
Solvant Zones utilisables pour les solvants usuels (λλλλ en nm)
eau 200 - 800
cyclohexane 210 - 800
hexane 200 - 800
méthanol 210 - 800
éthanol 210 - 800
éther 220 - 800
B. Etude de quelques exemples
1. Mise en évidence des différentes transitions. Influence des substituants.
Quelques molécules possèdent, à l'état fondamental, des configurations électroniques
qui peuvent subir différentes modifications, pour aboutir à un état électronique excité
d'énergie élevée. Nous avons vu que le cas se présente par exemple avec des
composés contenant le groupe:
Pour un tel groupe, il y a la possibilité de porter les électrons π vers l'état excité π*,
comme pour une oléfine, pour donner lieu à une transition π π*.
Dans l'autre éventualité, les électrons non liants de l'oxygène peuvent être excités
vers l'état électronique de plus haute énergie π*, et l'absorption serait alors
caractérisée par une transition n π*, où n symbolise un état initial non liant
pour l'électron. Par exemple, pour l'acétone dans l'alcool éthylique, il existe 3
bandes à 150 nm, 190 nm et 270 nm, correspondant respectivement aux transitions
notées habituellement n σ*, π π* et n π*.
La substitution par des radicaux alkyle, hydroxyle ou des halogènes, déplace la
bande n π*. Les effets inductifs et de résonance agissent souvent
simultanément :
°°°
°C O
R
R' R'
R
C O°°°°°°
+ -
C O°°°°

6
2. Influence du solvant
La position d'une bande d'absorption qui met en jeu des électrons non liants
(n π* entre autres) est particulièrement sensible à la polarité du solvant
utilisé, ce qui est logique puisque ces électrons sont privilégiés pour subir l'effet
du solvant polaire. Si le groupement en question est plus polaire dans l'état
fondamental que dans l'état excité, les électrons non liants dans l'état fondamental
sont stabilisés (par rapport à l'état excité). Ceci est réalisé, s'il y a formation d'une
liaison par pont hydrogène ou s'il existe une interaction électrostatique avec le
solvant. Par exemple, en présence de solvants hydroxylés, une paire libre
d'électrons du groupe carbonyle peut agir comme un donneur d'électrons vis à vis
de l'atome d'hydrogène du solvant pour former une liaison par pont hydrogène.
La formation de cette liaison abaisse l'énergie de l'orbitale n d'une quantité
approximativement égale à l'énergie de la liaison par pont hydrogène. L'énergie
d'association par liaison par pont hydrogène, devient nulle dans l'état excité, car
un électron quitte l'orbitale non liante et l'électron restant n'est plus suffisant pour
maintenir à lui seul l'association, l'état excité est donc le même dans les deux cas.
Donc le déplacement hypsochrome (en passant de l'hexane à l'alcool) est en
rapport avec l'énergie de la liaison hydrogène qui existait dans l'état fondamental.
3. Etude du phénomène de tautomérie (desmotropie)
Les composés possédant deux groupements carbonyles non conjugués,
présentent, à côté d'une bande d'absorption n π*, un spectre compliqué
par le phénomène de tautomérie cétone-énol.
Exemple: l'acétylacétone
Forme cétonique Forme énolique
CH 3C
O
CH 2C
O
CH 3 CH 3C
O
CHC
OH
CH 3 CH 3C
CH
O
C
O
CH 3
H
Les deux formes, cétonique et énolique, absorbent toutes deux aux alentours de
270 nm (cf. Tableau précédent), mais l'intensité de la bande n π* de la
forme cétonique est beaucoup plus faible que celle de la bande π π* de
l'énol (ε = 100 pour la première, 12000 pour la seconde). L'expérience montre
que c'est le composé donnant la plus grande résonance qui présente la plus
grande valeur de ε. Nous pouvons donc déterminer le degré de transformation par
une simple mesure de l'absorbance. La constante d'équilibre cétone-énol varie
avec la nature du solvant. L'énol (en configuration cis) est stabilisé par une
liaison par pont hydrogène interne. En présence de solvant polaire, cette liaison
est brisée et remplacée par une liaison par pont hydrogène entre le solvant et le
groupe carbonyle, et la concentration d'énol diminue. Donc, plus le solvant est
polaire et protique, plus le pourcentage d'énol est faible. Cette variation peut être
étudiée par spectrométrie UV.

7
C. Manipulation
ATTENTION: prendre soin de rincer, soigneusement et plusieurs fois, les cuves
avec de l'acétone (technique) avant chaque mesure. Il est alors indispensable de
les sécher à l'aide d'un jet d'air comprimé en opérant avec le plus grand soin.
- Influence du solvant sur les transitions n → → → → ππππ* de l’acétone
1) Représenter schématiquement la molécule d’acétone.
2) Calculer le volume d’acétone pure à prélever pour préparer 25 mL d’une
solution d’acétone à 5.10-2 mol.L-1 dans les deux solvants fournis: eau distillée et
cyclohexane.
3) Calculer la concentration exacte de la solution d’acétone diluée en tenant
compte du volume d’acétone pure effectivement prélevé.
4) À partir des spectres d’absorption enregistrés sur le domaine 220–320 nm pour
chacune des deux solutions d’acétone diluées:
a) Déterminer la valeur de la longueur d’onde λmax correspondant au maximum
d’absorption.
b) Représenter sur un diagramme énergétique la position des niveaux
électroniques n et π* de l’acétone en fonction du solvant utilisé.
c) Calculer l’écart énergétique ∆E (kJ.mol-1) de ces niveaux électroniques.
d) En déduire la valeur de l’énergie ∆EH (kJ.mol-1) de la liaison hydrogène.
e) Indiquer les valeurs respectives de l’absorbance A.
f) En déduire la valeur du coefficient d’absorption molaire εsol.
g) Conclure en commentant l’effet du solvant sur:
i) la valeur de λmax
ii) la valeur de εsol
Données:
2- Influence du solvant sur l’équilibre des formes cétonique et énolique de
l’acétylacétone
1) Représenter schématiquement les formes cétonique et énolique de la molécule
d’acétylacétone.
2) Calculer les volumes de solution-mère à prélever pour préparer 10 mL de
solution-fille de concentrations respectives regroupées dans le tableau suivant:
Concentration (mol.L-1) Solution-mère Solution-fille
Acétylacétone + eau 1,02.10-2 3.10-4
Acétylacétone + cyclohexane 9,95.10-4 5.10-5
Masse volumique (g.cm-3) Nombre d’Avogadro (mol-1)
ρacétone = 0,79 NA = 6,022.1023

8
3) Calculer la concentration exacte de chacune des solutions-filles en tenant
compte du volume de solution-mère effectivement prélevé.
4) À partir des spectres d’absorption mesurés sur le domaine 220–320 nm pour un
échantillon de chacune de ces deux solutions-filles:
a) Déterminer la valeur de la longueur d’onde λmax correspondant au maximum
d’absorption.
b) Indiquer les transitions responsables du maximum d’absorption dans la gamme
de longueurs d’onde [220–320] nm.
c) Indiquer les valeurs respectives de l’absorbance A.
d) En déduire la valeur du coefficient d’absorption molaire εsol. Commenter son
sens de variations.
5) En raisonnant à partir du fait que chacune des solutions-filles est constituée
d’un mélange des formes cétonique et énolique, dont la constante d’équilibre
varie sensiblement en fonction du solvant utilisé:
a) Exprimer le coefficient d’absorption molaire εsol de la solution en fonction du
coefficient d’absorption molaire des formes énolique et cétonique pures.
b) En déduire l’expression du % de forme énolique présente en solution en
fonction de εsol, εénol pur et εcétone pur.
c) Calculer le % de forme énolique présente dans chacune des deux solutions-
filles. Conclure.
Données:
ε ( mol-1.L.cm-1)
Forme énolique εénol pur = 12000
Forme cétonique εcétone pur = 100
9
10

11


















