SPEMAI deboeck 12/12/13 11:14 Page1 UE Spécifique ... · - les méthodes d’étude et d’analyse...
54
UE Spécifique Maïeutique Sous la direction de > Rachel Lévy > Michèle Rivière Iconographie > Frédéric Bargy
Transcript of SPEMAI deboeck 12/12/13 11:14 Page1 UE Spécifique ... · - les méthodes d’étude et d’analyse...

UE SpécifiqueMaïeutique
Sous la direction de> Rachel Lévy
> Michèle Rivière
Iconographie> Frédéric Bargy
> R. Lé
vy >
M. R
ivière
UE
Sp
écif
iqu
e M
aïeu
tiq
ue
Cet ouvrage est consacré à l’enseignement de la discipline maïeutique. Ils’adresse aux étudiants de la Première Année Commune des Études deSanté (PACES) qui optent pour cette Unité d’enseignement spécifique. Il constitue un ouvrage de référence rédigé par les praticiens hospitalo-universitaires et les sages-femmes impliqués au quotidien dans les domaines de la maïeutique et dans son enseignement.
Le contenu structuré et richement illustré reflète les qualités pédagogiques et l’expérience de l’enseignement de leurs rédacteurs. Cet ouvrage couvre les 4 modules correspondant au programme de l’UE spécifique maïeutique :- les méthodes d’étude et d’analyse du génome ;- l’anatomie de l’appareil reproducteur féminin et masculin ;- la différenciation sexuelle et l’histologie de l’appareil reproducteur féminin et masculin ;- l’unité foeto-placentaire.
ISBN : 978-2-8041-8284-7
SPEMAI
Pr Rachel Levy est professeur en biologie et médecine du développement et de la reproduction, responsable des enseignements d’embryologie (UE2) et demaïeutique (UE8 spécifique) en PACES à l’Université SMBH, Paris 13 (Bobigny). Michèle Rivière est sage-femme, directrice de l'école de sages-femmes St Antoine/AP-HP-Université Pierre et Marie Curie (Paris VI).
Conc
eptio
n gr
aphi
que
: Prim
o&Pr
imo
DANS LA MÊME COLLECTION
UE3 PACES, Aspects fonctionnels
UE3 PACES, Bases physiques des méthodes d’exploration
www.deboeck.com
• Ouvrage de cours à l’attentiondes étudiants en PACES et enécoles de maïeutique
• Qualité de l’équipe d’auteurs
• Iconographie en couleurs
LES
+
PUBLIC• Étudiants de Première Année
Commune des Études de Santé (PACES).• Étudiants en sciences médicales
(DFGSM 2 et 3 - Diplôme de FormationGénérale en Sciences Médicales).
• Étudiants en sciences maïeutiques(DFGSMa 2 et 3 - Diplôme de Formation Générale en Sciences Maïeutiques )
SPEMAI_deboeck 12/12/13 11:14 Page1


UE spécifique maïeutique
PACMAI-pgeTitre.indd 1 10/12/13 11:56

Dans la même collection
M. Ayadim, QCM de chimie générale
M. Ayadim, QCM de chimie organique
U. Bommas-Erbert, P. Teubner, R. Voss, Cours d’anatomie
P. Chaumet-Riffaud, I. Idy-Peretti, P. Peretti, QCM de physique et de biophysique (UE3),
Bases physiques et aspects fonctionnels des méthodes d’exploration
R. Lüllmann-Rauch, Histologie
C. Moussard, Biochimie et biologie moléculaire
P. Peretti, I. Idy-Peretti, P. Chaumet-Riffaud, Aspects fonctionnels
R. Sutton, B. Rockett, P. Swindells, Chimie pour les étudiants en médecine… et pour tous ceux qui ne seront pas chimistes
PACMAI-pgeTitre.indd 2 10/12/13 11:56

UE spécifique maïeutique> Rachel Rosenzweig-Levy
> Michèle Rivière
PACMAI-pgeTitre.indd 3 10/12/13 11:56

Maquette intérieure : SoftwinRéalisation : Softwin
© De Boeck Supérieur s.a., 2014 Rue des Minimes, 39 B-1000 Bruxelles
Tous droits réservés pour tous pays. Il est interdit, sauf accord préalable et écrit de l’éditeur, de reproduire (notamment par pho-
tocopie) partiellement ou totalement le présent ouvrage, de le stocker dans une banque de données ou de le communiquer au public, sous quelque forme et de quelque manière que ce soit.
Imprimé en Belgique
Dépôt légal : Bibliothèque nationale, Paris: janvier 2014 Bibliothèque royale de Belgique, Bruxelles: 2014/0074/151 ISBN 978-2-8041-8284-7
Pour toute information sur notre fonds et les nouveautés dans votre domaine de spécialisation, consultez notre site web: www.deboeck.com
PACMAI-pgeTitre.indd 4 10/12/13 11:56

V
Avant-propos
La réforme des études médicales a vu la mise en place d’une première année commune aux études de santé (PACES). Celle-ci permet aux étudiants de choi-sir parmi quatre filières : médecine, odontologie, pharmacie et maïeutique. Dès lors, les étudiants peuvent bénéficier d’un enseignement spécifique par discipline, organisé en unités d’enseignement (UE).
Plusieurs manuels leur permettant d’apprendre le plus efficacement possible ont déjà été rédigés. Ils concernent plus volontiers les UE communes, conjuguant des sciences fondamentales (biologie, physique), médicales (anatomie, histolo-gie) ou de sciences humaines. Mais aucun n’est consacré à la maïeutique, disci-pline exercée par les sages-femmes et abordée dans l’UE 8.
Cet ouvrage se propose de compléter l’existant. Il permettra aux futurs étu-diants se destinant à la profession de sage-femme de cibler les connaissances fon-damentales à acquérir et de découvrir certains chapitres propres à cette spécialité, l’objectif étant de se préparer aux épreuves de sélection.
Sa conception et sa rédaction reviennent à des universitaires de différentes universités de médecine (professeurs et maîtres de conférences), ainsi qu’à des enseignants sages-femmes. En effet, certains assurent déjà des cours, tels que la dynamique du liquide amniotique. Leur participation est donc légitime.
Nous espérons que ce nouveau volume correspondra aux attentes des étudiants.


VII
Table des matières
Avant-propos ........................................................................................................... V
Module 1 Méthodes d’étude et d’analyse du génome
Introduction ............................................................................................................. 3
Chapitre 1 Méthodes d’exploration du génome ............................................ 51. Caryotype .......................................................................................................... 5
1.1. Classification des anomalies chromosomiques ................................... 61.2. Mécanique chromosomique anormale ................................................. 71.3. Exemples d’anomalies numériques ....................................................... 81.4. Anomalies de structure .......................................................................... 10
2. Cytogénétique moléculaire ............................................................................. 182.1. Hybridation in situ fluorescente ou FISH
(Fluorescence In Situ Hybridization) ................................................... 182.2. Diagnostic prénatal rapide par la technique
des Bacs-On-Beads (Prenatal BACs-on-BeadsTM) .............................. 322.3. Diagnostic des anomalies chromosomiques par CGH array
en pathologie constitutionnelle post et prénatale .............................. 36
Chapitre 2 Du prélèvement d’ADN à l’étude d’un gène en pathologie humaine : les outils utilisés en biologie moléculaire ..................................... 49
1. Extraction et purification des acides nucléiques .......................................... 491.1. Extraction et purification de l’ADN ..................................................... 491.2. Extraction et purification de l’ARN ...................................................... 511.3. Dosage des acides nucléiques après extraction ................................... 52
2. Les outils enzymatiques utilisés pour l’étude des acides nucléiques ......... 532.1. Les endonucléases de restriction ........................................................... 532.2. Les autres nucléases ................................................................................. 542.3. Les ligases .................................................................................................. 55

VIII
Table des matières
3. Séparation électrophorétique de l’ADN ........................................................ 55
4. Le clonage .......................................................................................................... 574.1. Définition ................................................................................................. 574.2. Les étapes ................................................................................................. 57
5. Les techniques d’hybridation .......................................................................... 595.1. Les facteurs influençant l’hybridation .................................................. 595.2. Le marquage des sondes ......................................................................... 595.3. Les différentes techniques d’hybridation ............................................. 60
6. Amplification génique par la technique de PCR .......................................... 616.1. Principe de la PCR ................................................................................... 616.2. RT-PCR (Reverse Transcription Polymerase Chain Reaction) .......... 636.3. La PCR en temps réel (real time PCR) .................................................. 63
7. Les techniques générales d’étude du génome humain normal et pathologique ................................................................................................. 657.1. Criblage des mutations ........................................................................... 657.2. Séquençage de l’ADN ............................................................................. 677.3. Recherche de mutations connues .......................................................... 72
Chapitre 3 Transgenèse ....................................................................................... 75
1. Introduction ...................................................................................................... 75
2. Les méthodologies ............................................................................................ 75
3. Les outils de thérapie génique ......................................................................... 773.1. Les cassettes d’expression ....................................................................... 783.2. Les vecteurs .............................................................................................. 82
4. Les stratégies de la thérapie génique .............................................................. 884.1. Production d’une protéine thérapeutique............................................ 894.2. Induction de l’apoptose .......................................................................... 894.3. Inhibition de fonction ou « stratégie silencieuse » .............................. 904.4. Stratégie réparatrice ................................................................................ 934.5. Vaccination à ADN ................................................................................. 944.6. Les animaux transgéniques, source thérapeutique ? .......................... 95
5. Les applications de thérapie génique ............................................................. 96

IX
Table des matières
Module 2 Anatomie de l’appareil reproducteur féminin et masculin
Introduction ............................................................................................................. 1011. Le contenant ...................................................................................................... 101
1.1. Le bassin osseux ....................................................................................... 1011.2. La couverture musculaire ....................................................................... 1061.3. Contre la paroi : les vaisseaux et les nerfs ............................................ 111
2. Le contenu ......................................................................................................... 1132.1. Chez la femme ......................................................................................... 1142.2. Chez l’homme ......................................................................................... 1152.3. Crânialement une séreuse : le péritoine ............................................... 115
3. L’appareil ou système génital : développement, croissance et puberté ........................................................... 1173.1. Gonades .................................................................................................... 1173.2. Gonoductes ............................................................................................. 1193.3. Appareil copulatoire ............................................................................... 1203.4. Le cerveau, le gyrus cingulaire et l’hypothalamus .............................. 122
4. Phylogenèse et anatomie comparée du bassin des mammifères (2) ......... 125
Module 3 Histologie de l’appareil reproducteur
Chapitre 1 Tératogenèse : fréquence et principales causes des malformations humaines ....................................................................... 131
1. Définitions ......................................................................................................... 1312. Fréquences ........................................................................................................ 1323. Causes ................................................................................................................ 133
3.1. Les causes génétiques .............................................................................. 1343.2. Les causes extrinsèques (épigenèse : influence du milieu
extérieur sur le développement d’un individu) ................................... 137
Chapitre 2 Différenciation sexuelle normale et pathologique .................... 145Introduction ............................................................................................................. 1451. L’appareil génital indifférencié ....................................................................... 146
1.1. La gonade indifférenciée ou bipotentielle ............................................ 1481.2. Les voies génitales indiférenciées (gonoductes) .................................. 150

X
Table des matières
1.3. Le sinus urogénital .................................................................................. 1521.4. Les organes génitaux externes indifférenciés ...................................... 152
2. La différenciation sexuelle masculine ............................................................ 1542.1. La différenciation testiculaire ................................................................ 1542.2. Les voies génitales masculines ............................................................... 1552.3. Le sinus urogénital .................................................................................. 1562.4. La formation des organes génitaux externes masculins ..................... 1572.5. La migration testiculaire ......................................................................... 159
3. La différenciation sexuelle féminine .............................................................. 1603.1. La différenciation ovarienne .................................................................. 1603.2. Les voies génitales féminines ................................................................. 1613.3. L’évolution du sinus urogénital ............................................................. 1643.4. Les organes génitaux externes féminins ............................................... 164
4. Les malformations de l’appareil génital......................................................... 1654.1. Les malformations en relation avec des anomalies numériques
des chromosomes sexuels ....................................................................... 1654.2. Les malformations isolées de l’appareil génital ................................... 165
Chapitre 3 La différenciation sexuelle du fœtus ............................................. 1671. Détermination de la gonade ............................................................................ 168
1.1. Formation de la gonade bipotentielle ................................................... 1681.2. Formation du testicule ........................................................................... 1691.3. Déterminisme génétique de la différenciation ovarienne [18] ......... 172
2. Différenciation des organes génitaux internes et externes [25, 26] ........... 1732.1. Différenciation des organes génitaux internes (OGI) ........................ 1732.2. Différenciation des organes génitaux externes (OGE) ....................... 175
3. Une bonne connaissance de la différenciation sexuelle permet de comprendre la nouvelle classification (réunion de consensus de Chicago, Novembre 2005) appelée « Anomalies du développement sexuel » « Disorders of Sexual Development » (DSD) ............................................... 176
Chapitre 4 Histologie de l’appareil génital masculin ................................... 1891. Les testicules ...................................................................................................... 189
1.1. Architecture testiculaire ......................................................................... 1891.2. Le tissu interstitiel ou glande endocrine du testicule ......................... 1901.3. Les tubes séminifères .............................................................................. 191

XI
Table des matières
2. Les voies excrétrices ......................................................................................... 1972.1. Les voies intra-testiculaires : tubes droits et rete testis ....................... 1972.2. Les cônes efférents ................................................................................... 1982.3. L’épididyme ou canal épididymaire ..................................................... 1982.4. Le canal déférent ...................................................................................... 1992.5. L’urètre ...................................................................................................... 200
3. Les glandes annexes.......................................................................................... 2003.1. Les vésicules séminales .......................................................................... 2003.2. La prostate ............................................................................................... 2013.3. Les glandes bulbo-urétrales ou glandes de Cowper ............................ 202
4. Le sperme .......................................................................................................... 202
Chapitre 5 Histologie de l’appareil génital féminin ...................................... 2051. Histologie et physiologie de l’ovaire .............................................................. 205
1.1. L’ovogenèse ............................................................................................. 2051.2. La folliculogenèse .................................................................................... 2061.3. L’ovulation ................................................................................................ 2091.4. Le corps jaune .......................................................................................... 210
2. Histologie et physiologie de l’utérus, des trompes utérines (ou trompes de Fallope) et du vagin .............................................................. 2102.1. Les trompes .............................................................................................. 2102.2. L’utérus ..................................................................................................... 2122.3. Le vagin ..................................................................................................... 215
3. Histologie et physiologie du sein .................................................................... 2163.1. Avant la puberté ...................................................................................... 2163.2. À la puberté .............................................................................................. 2163.3. Après la puberté ....................................................................................... 217
Module 4 Unité fœto-placentaire
Chapitre 1 Développement du placenta .......................................................... 2211. Formation du placenta diffus au cours du 1er mois du développement.... 221
1.1. L’implantation : période avilleuse ......................................................... 2211.2. La formation des villosités choriales : période villeuse ...................... 2261.3. La circulation embryonnaire à la fin du premier mois ...................... 229

XII
Table des matières
2. Évolution au cours des 2e et 3e mois : formation du disque placentaire ... 2302.1. Formation de la décidue et des caduques ............................................ 2302.2. Évolution des annexes et formation du cordon ombilical ................. 2312.3. Constitution du disque placentaire ....................................................... 234
3. Évolution du placenta au-delà du 3e mois ..................................................... 2363.1. La plaque choriale.................................................................................... 2363.2. Les villosités .............................................................................................. 2363.3. La plaque basale ....................................................................................... 238
Chapitre 2 La fonction endocrine du placenta ............................................... 239
1. Hormones polypeptidiques placentaires ....................................................... 2391.1. L’hCG ........................................................................................................ 2391.2. L’hCS ou hPL ........................................................................................... 2411.3. GH placentaire (pGH) ............................................................................ 2411.4. Autres hormones peptidiques................................................................ 2411.5. Hormones stéroïdes ................................................................................ 242
2. Autres facteurs .................................................................................................. 243
Chapitre 3 Examen anatomopathologique d’un placenta .......................... 245
1. Examen macroscopique ................................................................................... 2461.1. Examen du sac membranaire................................................................. 2461.2. La configuration du placenta ................................................................. 2491.3. Examen du cordon ombilical ................................................................. 2511.4. Poids du placenta ..................................................................................... 2531.5. Observation de la plaque choriale ......................................................... 2541.6. L’examen de la plaque basale ................................................................. 2551.7. Examen du parenchyme ......................................................................... 2561.8. Cas particulier : les annexes des grossesses multiples ........................ 258
2. Examen histologique ........................................................................................ 260
Chapitre 4 La circulation placentaire ............................................................... 263Introduction ............................................................................................................. 2631. La circulation maternelle ................................................................................. 264
2. La circulation fœtale ......................................................................................... 265
3. La « barrière » placentaire ............................................................................... 266

XIII
Table des matières
Chapitre 5 Les échanges fœto-placentaires .................................................... 2711. La fonction d’échange du placenta ................................................................. 271
1.1. Anatomie fonctionnelle du placenta..................................................... 2721.2. Mécanismes des échanges placentaires ............................................... 273
2. Les échanges gazeux ......................................................................................... 2742.1. Le transfert d’oxygène de la mère au fœtus ......................................... 2742.2. Facteurs influençant les échanges gazeux ............................................ 276
3. Les apports de nutriments au fœtus ............................................................... 2773.1. Glucides .................................................................................................... 2773.2. Protides ..................................................................................................... 2773.3. Lipides ....................................................................................................... 2773.4. Eau, électrolytes, oligo-éléments, vitamines ....................................... 278
4. L’épuration des déchets du métabolisme fœtal ............................................ 2785. Le rôle de barrière protectrice du placenta ................................................... 278
5.1. Médicaments et toxiques ........................................................................ 2785.2. Bactéries et parasites ............................................................................... 2785.3. Virus .......................................................................................................... 279
Chapitre 6 La physiologie du liquide amniotique ......................................... 2811. Définition .......................................................................................................... 2812. Sécrétion ............................................................................................................ 281
2.1. Avant 20 SA (semaines d’aménorrhée) ................................................ 2812.2. Après 20 SA .............................................................................................. 282
3. Résorption ......................................................................................................... 2823.1. Déglutition fœtale .................................................................................... 2823.2. Voie transmembranaire .......................................................................... 2833.3. Voie intra-membranaire......................................................................... 283
4. Volume de liquide amniotique ....................................................................... 2835. Régulation .......................................................................................................... 2846. Composition ...................................................................................................... 284
6.1. Éléments minéraux ................................................................................. 2846.2. Acides aminés .......................................................................................... 2846.3. Éléments biochimiques ........................................................................... 2846.4. Enzymes .................................................................................................... 2856.5. Phospholipides ......................................................................................... 2856.6. Protéines ................................................................................................... 285

XIV
Table des matières
6.7. Hormones ................................................................................................. 2866.8. Cytologie ................................................................................................... 286
7. Études quantitatives ......................................................................................... 2868. Rôles ................................................................................................................... 287
8.1. Antibactérien ........................................................................................... 2878.2. Mécanique ................................................................................................ 2878.3. Environnemental ..................................................................................... 287
Chapitre 7 Grossesse et immunité ..................................................................... 289Introduction ............................................................................................................. 2891. L’interface fœto-maternelle ............................................................................. 290
1.1. Mise en place du placenta : la placentation .......................................... 2901.2. Trophoblaste ............................................................................................ 2911.3. Les membranes fœtales ........................................................................... 2921.4. Cellules déciduales .................................................................................. 2931.5. Cellules fœtales circulantes .................................................................... 293
2. Acteurs du système immunitaire .................................................................. 2942.1. Cellules Natural Killer (NK) .................................................................. 2942.2. Macrophages ............................................................................................ 2952.3. Lymphocytes ........................................................................................... 2952.4. Cellules dendritiques ............................................................................... 2972.5. Système du complément ......................................................................... 297
3. Mécanisme d’adaptation et d’évasion immunitaire .................................... 2983.1. Adaptation de l’immunité innée ........................................................... 2983.2. Impact de l’immunité adaptative : rôle des lymphocytes .................. 2993.3. Rôle de la décidualisation ....................................................................... 3003.4. Mécanismes spécifiques au niveau du placenta .................................. 300
4. Réaction envers les agents pathogènes ......................................................... 3014.1. Cytomégalovirus (CMV) ........................................................................ 3014.2. Listeria monocytogenes (Lm) ................................................................ 301
Chapitre 8 La physiologie du fœtus : développement pulmonaire, surfactant pulmonaire, hémodynamique fœtale, croissance fœtale .... 305
Introduction ............................................................................................................. 3051. Le développement pulmonaire ...................................................................... 305
1.1. Les étapes du développement pulmonaire ........................................... 306

XV
Table des matières
1.2. Facteurs régulant le développement alvéolaire ................................... 3072. Le surfactant pulmonaire ................................................................................ 310
2.1. Composition............................................................................................. 3102.2. Rôle ............................................................................................................ 3102.3. Régulation ................................................................................................. 3112.4. Déficit en surfactant pulmonaire .......................................................... 311
3. L’hémodynamique fœtale ................................................................................ 3113.1. Description de la circulation fœtale ...................................................... 3123.2. Régulation de la circulation fœtale ....................................................... 313
4. La croissance fœtale ......................................................................................... 3134.1. Apports maternels en substrats énergétiques ...................................... 3144.2. Insulinosécrétion fœtale ......................................................................... 3144.3. Facteurs de croissance fœtoplacentaires .............................................. 3154.4. Facteurs génétiques ................................................................................. 316
5. Points clés .......................................................................................................... 3165.1. Développement pulmonaire .................................................................. 3165.2. Surfactant pulmonaire ............................................................................ 3175.3. Hémodynamique fœtale ......................................................................... 3175.4. Croissance fœtale ..................................................................................... 317
Chapitre 9 Grossesses gémellaires ................................................................... 3191. Épidémiologie ................................................................................................... 3192. Mécanismes : jumeaux monozygotes/dizygotes .......................................... 320
2.1. Rappel embryologique ............................................................................ 3202.2. Jumeaux monozygotes (« vrais jumeaux ») ......................................... 3212.3. Jumeaux dizygotes (« faux jumeaux ») ................................................. 3212.4. Détermination de la zygosité à la naissance......................................... 321
3. Placentation ....................................................................................................... 3223.1. Placentation monochoriale. ................................................................... 3223.2. Placentation dichoriale (et diamniotique) ........................................... 3233.3. Le diagnostic de chorionicité ................................................................. 323
4. Physiologie maternelle, complications maternelles ..................................... 3245. Complications périnatales ............................................................................... 325
5.1. Croissance fœtale, restriction de croissance fœtale ........................... 3255.2. Augmentation de la prématurité .......................................................... 325

XVI
Table des matières
5.3. Le développement à plus long terme .................................................... 3265.4. Complications périnatales selon la chorionicité ................................. 326
6. Suivi prénatal et lieu d’accouchement ........................................................... 327
Chapitre 10 Suivi de la grossesse .................................................................... 3291. Les examens médicaux .................................................................................... 329
1.1. Le recueil des données anamnestiques ................................................. 3301.2. L’examen clinique.................................................................................... 3301.3. Les examens paracliniques ..................................................................... 330
2. La préparation à la naissance et à la parentalité ........................................... 331Conclusion ............................................................................................................... 331
Chapitre 11 L’accouchement normal ................................................................ 3331. Définitions ......................................................................................................... 3332. Déroulement de l’accouchement en présentation céphalique .................. 335
2.1. 1re phase : phénomènes dynamiques : effacement et dilatation du col utérin ................................................... 336
2.2. 2e phase : phénomènes mécaniques : parcours du fœtus dans l’excavation pelvienne et expulsion hors des voies génitales ... 337
2.3. 3e phase : la délivrance ........................................................................... 3383. L’accueil du nouveau-né normal à terme ...................................................... 339

Module 1 Méthodes d’étude et d’analyse du génome


3
IntroductionLa cytogénétique est une discipline récente dont l’essor date de 1959 : une
équipe française (Lejeune & col) a établi la corrélation « génotype –phénotype » entre le syndrome de Down (mongolisme) et un chromosome 21 surnuméraire (trisomie 21) [1]. Elle consiste à étudier les anomalies du génome par tous les outils mis à sa disposition. Les anomalies chromosomiques peuvent faire partie de la constitution d’un individu, autorisant un diagnostic pré ou post-natal, ou être acquises au cours d’un processus cancéreux.
À la naissance, les taux observés d’anomalies chromosomiques numériques et de structure chez les enfants vivants sont respectivement de 1/318 et de 1/425. Le plus souvent, les anomalies chromosomiques déséquilibrées ont une expres-sion clinique évidente. Les aneuploïdies (anomalies numériques) en dehors des gonosomes entraînent des malformations graves et caractéristiques. En revanche, les conséquences phénotypiques peuvent être très discrètes voire absentes chez des sujets porteurs d’anomalies des chromosomes sexuels ou de remaniements autosomiques équilibrés. Ce type d’anomalies est le plus souvent rencontré chez les patients adressés en consultation de cytogénétique pour bilan d’infertilité ou bilan de fausses couches spontanées (FCS), [2].
À la naissance, 0,6 à 0,9 % des enfants vivants sont porteurs d’une anomalie chromosomique. Les anomalies de nombre les plus fréquentes à la naissance sont les caryotypes 47, XXX, 47, XXY et 47, XYY (environ 1 pour 1 000 naissances du sexe concerné) pour les gonosomes. Les anomalies de structure les plus fréquentes sont les translocations équilibrées robertsoniennes (0,9 pour 1 000) et réciproques (0,9 à 1,4 pour 1 000) ; les anomalies de structure sont peu fréquentes dans la population générale. Elles sont le plus souvent diagnostiquées lors de l’exploration d’enfants présentant un retard mental isolé ou associé à des malformations.
La fréquence et la répartition des anomalies chromosomiques dans une popu-lation témoin sont rapportées dans le tableau ci-dessous.Fréquence et répartition des anomalies chromosomiques observées à la naissance sur une population normale de 36 950 bébés1.
Anomalie Fréquence dans la population47, XXY 1,05/1000 (dont 0,19/1000 mosaïques)46, XX (phénotype masculin) 0,05/100047, XYY 0,9/100046, X,inv(Y) 0,24/100047, XXX 1/1000Translocations Robertsoniennes 0,74/1000 t(DqDq) et 0,18/1000 t(GqGq)Translocations réciproques équilibrées 0,85 à 1,05/1000Inversions 0,62/1000Marqueurs surnuméraires 0,32/1000
1. D’après (Buckton et al., 1980, 227 ; Hamerton et al., 1975, 223, Jacobs et al., 1974, 359, Lin, 1976, 315 Nielsen, 1975, 1 ; Walzers, 1972, 24)


5
Chapitre 1 Méthodes d’exploration du génome
Pr Brigitte Benzacken1
1. Caryotype Dans l’espèce humaine, l’ensemble du génome se répartit en 46 chromo-
somes, 22 paires de chromosomes numérotés de 1 à 22 appelés autosomes et une paire de chromosomes sexuels appelés gonosomes XX, chez la femme et XY, chez l’homme. Le caryotype consiste à classer les chromosomes par ordre de taille décroissante, en fonction de la position du centromère et grâce aux techniques de marquage mettant en évidence une alternance de bandes claires et sombres (banding). La séquence des bandes observées après les différentes méthodes de marquage [bandes G (Giemsa, par dénaturation enzymatique) ou R (Reverse : inverse des bandes G, par dénaturation thermique] est identique chez deux indi-vidus d’une même espèce en dehors du chromosome Y. La réalisation du caryo-type constitutionnel s’effectue, le plus souvent, par culture lymphocytaire après prélèvement sanguin dans un tube hépariné. La division lymphocytaire in vitro est obtenue par adjonction dans le milieu de culture d’une lectine (la phytohé-magglutinine) qui reproduit de façon non spécifique la réaction antigène-anti-corps. L’introduction de colchicine au bout de 72 heures d’incubation dans le tube permet le blocage des cellules en division au stade métaphasique. La colchi-cine, poison du fuseau mitotique empêche la polymérisation des tubulines dans les microtubules. Un choc hypotonique permet ensuite le gonflement de la cellule et la fragilisation de la membrane plasmique. Après une étape de fixation dans un mélange d’éthanol et d’acide acétique, le culot cellulaire est étalé sur une lame. Les chromosomes dispersés et dépourvus de leur cytoplasme seront ensuite déna-turés soit par la chaleur soit par des enzymes et colorés pour enfin être comptés et analysés. Une nomenclature internationale permet l’établissement de la for-mule chromosomique où figurent le nombre de chromosomes, la formule gono-somique, et la description éventuelle de l’anomalie (ex. : 47, XX, + 21 = formule
1. AP-HP, Laboratoire de Cytogénétique, Hôpital Jean Verdier, Bondy 93140, France, and Université Paris 13, Sorbonne Paris Cité and INSERM, U676.

6
Méthodes d’étude et d’analyse du génome
chromosomique féminine présentant un chromosome 21 surnuméraire). Le bras court du chromosome p et le bras long q sont numérotés en bandes et sous-bandes permettant de localiser les sites de cassures chromosomiques (ex. : 46, XX, del (5) (q13) = formule chromosomique féminine présentant une délétion du bras long d’un chromosome 5 dont le point de cassure est dans la bande 13). Toutes les cel-lules nucléées de l’organisme humain peuvent être caryotypées. Contrairement au caryotype sanguin dont le prélèvement est peu invasif, le caryotype fœtal néces-site un geste invasif : ponction de liquide amniotique, biopsie de trophoblaste (villosités choriales) ou ponction de sang fœtal. Une étape de culture cellulaire est nécessaire pour établir le caryotype.
Quelles que soient les modalités techniques et quels que soient les tissus d’origine, la réalisation du caryotype nécessite l’obtention de cellules en division présentant une dispersion correcte des chromosomes, et l’étalement des prépa-rations afin d’obtenir des images sur lesquelles tous les chromosomes sont bien séparés et dans le même plan. Les analyses chromosomiques réalisées en routine dans les laboratoires de cytogénétique permettent une analyse du caryotype avec une résolution d’environ 400 à 550 bandes, ce qui signifie que les remaniements inférieurs à 5 Mb échappent au diagnostic car la taille d’une bande est d’environ 15 à 20 Mb, d’une sous-bande 5 à 10 Mb, et d’un gène 0,15 Mb. Les techniques en haute résolution (850 à 1 000 bandes) sont beaucoup plus performantes mais restent insuffisantes pour détecter des remaniements inférieurs à 1 Mb.
1.1. Classification des anomalies chromosomiques
On distingue les anomalies chromosomiques constitutionnelles et acquises. Seules les anomalies constitutionnelles seront traitées dans ce chapitre.
Les anomalies chromosomiques constitutionnelles peuvent survenir durant les premiers stades embryonnaires ou être transmises au zygote dès la féconda-tion. Elles résultent, dans ce cas, de malségrégations méiotiques chez des parents sains (de novo) ou de la transmission de façon déséquilibrée d’un remaniement parental (héritées). Les anomalies de nombre sont soit homogènes (retrouvées sur l’ensemble des cellules examinées) ou en mosaïque (l’anomalie ne touchant pas toutes les cellules). Les techniques de banding des chromosomes permettent le diagnostic d’anomalies de structure à condition que celles-ci aient une taille et une colorabilité suffisante et ceci en fonction de la technique utilisée. Cependant, même avec ces méthodes, certains remaniements chromosomiques peuvent pas-ser inaperçus lorsque leur taille est trop fine (anomalies cryptiques) et leur dia-gnostic nécessite alors une approche moléculaire.

7
Méthodes d’exploration du génome
1.2. Mécanique chromosomique anormale
Les anomalies constitutionnelles sont présentes dès la conception ou se for-ment lors des premières divisions du zygote. Le plus souvent, les premières abouti-ront à une anomalie chromosomique homogène, les deuxièmes à un mosaïcisme. On distingue classiquement les anomalies de nombre, qui résultent d’une anoma-lie de la fécondation ou d’une mauvaise répartition des chromosomes lors d’une division cellulaire, et les anomalies de structure qui impliquent une ou plusieurs cassures chromosomiques suivies d’un recollement anormal.
Les anomalies numériques résultent d’une non-disjonction méiotique définie par le fait que deux chromosomes (division I) ou deux chromatides (division II) migrent vers le même pôle lors de l’anaphase et passent ensemble dans la même cellule fille, au lieu de migrer chacun dans une cellule fille. Cette non-disjonction peut se produire lors d’une division méiotique maternelle ou paternelle. Elle peut concerner deux chromosomes homologues, lors de la première division méio-tique, ou deux chromatides-sœurs au cours de la deuxième division méiotique. Dans le premier cas, le gamète reçoit un chromosome de chacun des parents (maternel et paternel) et dans le second, deux exemplaires d’un même chromo-some parental (maternel ou paternel). Ces deux copies ne seront cependant pas génétiquement identiques du fait des recombinaisons qui se produisent en début de méiose (crossing over). Ces accidents déterminés avant la fécondation peuvent aboutir à des aneuploïdies (nombre différent de 46 chromosomes) homogènes.
Toutes les cellules de l’individu ont le même caryotype anormal. L’existence d’une très forte sélection de la conception à la naissance est évidente. Cette sélec-tion porte essentiellement sur les anomalies des autosomes, à l’exception de la monosomie X.
Dans les fausses-couches spontanées du 1er trimestre (15 % des grossesses reconnues), la proportion d’anomalies chromosomiques est de 60 % [2], [4]. Elle n’est plus que de 5 % dans les fausses-couches tardives et chez les enfants mort-nés. La majorité des anomalies ovocytaires résulte d’une non disjonction méiotique de 1re division [5]. Lors de la 1re division méiotique, la non disjonction maternelle conduisant à la formation d’ovocytes aneuploïdes semble être la cause principale de trisomie dans l’espèce humaine [5]. L’âge maternel est le principal facteur étiologique favorisant les non-disjonctions [6].
Les anomalies de structure sont la conséquence de cassures chromosomiques suivies par un ou plusieurs recollements anormaux. Par définition, les trisomies et les monosomies partielles résultent de remaniements de structure.
Les anomalies équilibrées peuvent entraîner, lors de la méiose, la formation de gamètes déséquilibrés donnant des zygotes anormaux, ce qui se traduira par la

8
Méthodes d’étude et d’analyse du génome
survenue de fausses-couches ou par la naissance d’enfants porteurs d’anomalies phénotypiques. Les anomalies non équilibrées peuvent survenir de novo (délé-tions, translocations non équilibrées de novo, etc.) ou être la conséquence d’un remaniement parental équilibré.
1.3. Exemples d’anomalies numériques
Par définition, les anomalies de nombre affectent le nombre des chromo-somes et non leur structure qui demeure normale.
Elles peuvent être homogènes, présentes dans toutes les cellules de l’orga-nisme, ou en mosaïque lorsque au moins deux clones cellulaires présentent la même anomalie. Lorsqu’elles sont homogènes, elles résultent le plus souvent d’une non-disjonction méiotique et peuvent se traduire par une trisomie (pré-sence d’un chromosome excédentaire) ou une monosomie (perte d’un chromo-some). On parle alors d’aneuploïdie.
Les trisomies autosomiques les plus fréquentes à la naissance sont les triso-mies 21, 18 et 13 et la trisomie 8 en mosaïque, (1,4 pour 1 000 dont 1,2 de triso-mie 21). Les fréquences des trisomies 21 et 18 sont directement corrélées à l’âge de la mère.
Figure 1 – Exemples d’anomalies de nombre

9
Méthodes d’exploration du génome
1.3.1. Trisomie 21 (syndrome de Down)
La fréquence est de 1/700 naissances. Le phénotype est caractéristique indé-pendamment du mécanisme chromosomique. La dysmorphie faciale est évocatrice et peut être associée à des malformations viscérales essentiellement cardiaques et rénales. Le retard mental est constant. D’un point de vue cytogénétique, 92,5 % des trisomies 21 sont libres et homogènes et résultent d’une non-disjonction majori-tairement de première division méiotique et le plus souvent maternelle. Les autres mécanismes sont les translocations (4,8 %) et les mosaïques (2,7 %) [8].
1.3.2. Trisomie 18 (syndrome d’Edwards)
La fréquence est de 1/8000 naissances. Le phénotype associe un retard de croissance, une dysmorphie caractéristique, des anomalies des extrémités, che-vauchement des doigts et pieds en piolet, et le plus souvent, des malformations viscérales graves. Dans la majorité des cas, la mort est précoce. Dans 80 % des cas, il s’agit d’une trisomie libre et homogène, dans 10 % des cas d’une mosaïque, et dans 10 % de remaniements plus complexes transmis ou de novo [8].
1.3.3. Trisomie 13 (syndrome de Patau)
La fréquence est de 0,007 % de naissances vivantes. La mort survient le plus souvent in utero. Le phénotype est très malformatif associant une dysmorphie faciale (gueule de loup, microphtalmie) et des malformations viscérales, des membres et des organes génitaux. Dans 75 % des cas, il s’agit d’une trisomie libre et homogène. Dans 20 % des cas, il s’agit d’une translocation transmise ou de novo, enfin dans les 5 % restants, une mosaïque est mise en évidence [8].
1.3.4. Les trisomies des chromosomes sexuels
Elles sont très fréquentes, et portent aussi bien sur le chromosome X que sur le chromosome Y : 47, XXX ; 47, XXY ; 47, XYY. On peut également voir des anomalies de nombre plus importantes : 48, XXXX, etc. Plus le nombre de X aug-mente, plus le risque de retard mental est grand.
Contrairement aux monosomies autosomiques rarement observées à la nais-sance du fait de leur élimination probable dès les premiers stades de la vie embryon-naire, la monosomie X, quand elle est viable, aboutit au syndrome de Turner.
Les anomalies de nombre en mosaïque sont particulièrement fréquentes dans le cas des chromosomes sexuels. Elles sont caractérisées par la présence d’au moins deux clones différents et résultent d’une non-disjonction post-zygotique.

10
Méthodes d’étude et d’analyse du génome
Le zygote d’origine peut être porteur d’une anomalie de nombre. Dans ce cas, la correction d’une monosomie ou d’une trisomie peut être à l’origine d’une disomie uniparentale (les deux chromosomes homologues ont la même origine parentale) [3].
Les polyploïdies correspondent à un nombre anormal de lots haploïdes entiers. La plus fréquente est la triploïdie, caractérisée par la présence de trois lots haploïdes de chromosomes : 69, XXX, ou XXY, ou XYY. Les triploïdies sont dues à des accidents de la fécondation dont le plus fréquent est une dispermie (fécon-dation par deux spermatozoïdes). Quelques grossesses polyploïdes aboutissent à la naissance d’enfants vivants. Ceux-ci meurent précocement au cours des pre-mières heures ou jours de vie.
1.4. Anomalies de structure
Les anomalies de structure peuvent affecter un chromosome ou deux chro-mosomes, homologues ou non homologues, parfois davantage. On parle alors de remaniements complexes.
Elles peuvent être équilibrées ou déséquilibrées. Les anomalies équilibrées n’entraînent pas de déséquilibre du matériel chromosomique et n’ont habituelle-ment pas d’effet phénotypique. Il s’agit des inversions, et des translocations sans perte de matériel génétique.
Sur un caryotype humain les techniques de banding des chromosomes per-mettent le diagnostic d’anomalies de structure à condition que celles-ci aient une taille et une colorabilité suffisante et ceci en fonction de la technique utilisée. Cependant, même avec ces méthodes, certains remaniements chromosomiques peuvent passer inaperçus lorsque leur taille est trop fine (anomalies cryptiques) et leur diagnostic nécessite alors une approche moléculaire.
1.4.1. Anomalies affectant un seul chromosome
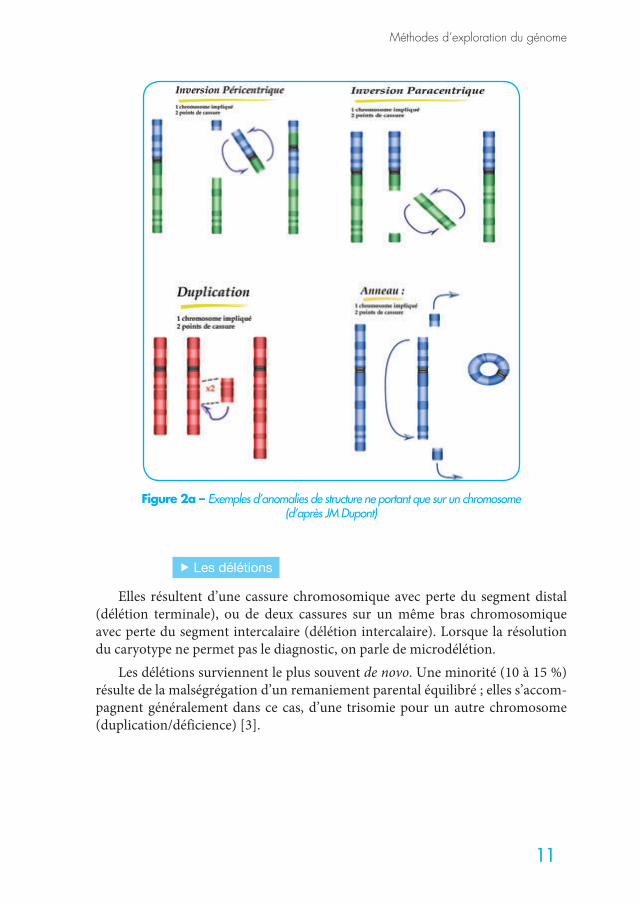
Les principales anomalies de structure suivantes ne portant que sur un seul chromosome sont toujours déséquilibrées (Figure 2a) :
– les délétions terminales ou intercalaires et les microdélétions (del) ; – les isochromosomes (i) ; – les duplications et les microduplications (dup) ; – les chromosomes en anneau r pour « ring » ; – les chromosomes marqueurs (mar).
Certaines anomalies de structure portant sur un seul chromosome sont équilibrées : les inversions péri ou paracentriques (inv).
11
Méthodes d’exploration du génome
f Les délétions
Elles résultent d’une cassure chromosomique avec perte du segment distal (délétion terminale), ou de deux cassures sur un même bras chromosomique avec perte du segment intercalaire (délétion intercalaire). Lorsque la résolution du caryotype ne permet pas le diagnostic, on parle de microdélétion.
Les délétions surviennent le plus souvent de novo. Une minorité (10 à 15 %) résulte de la malségrégation d’un remaniement parental équilibré ; elles s’accom-pagnent généralement dans ce cas, d’une trisomie pour un autre chromosome (duplication/déficience) [3].
Figure 2a – Exemples d’anomalies de structure ne portant que sur un chromosome (d’après JM Dupont)

12
Méthodes d’étude et d’analyse du génome
f Les chromosomes en anneau (r)
Ils résultent d’une cassure à chaque extrémité d’un chromosome suivie par un recollement avec perte des segments distaux. Les structures en anneau sont assimilables à une double délétion.
f Les inversions (inv)
Elles sont dues à deux cassures sur le même chromosome, suivies de recollement après inversion du segment intermédiaire. Elles sont dites péricentriques, si le cen-tromère est compris dans le segment intermédiaire. Elles sont dites paracentriques, si les deux cassures se sont produites sur le même bras chromosomique (p ou q).
Ces inversions sont des remaniements équilibrés mais elles entraînent au moment de la méiose des difficultés d’appariement. Il y a, le plus souvent, for-mation d’une boucle d’appariement. La survenue d’une recombinaison dans le segment inversé entraîne la formation de gamètes anormaux.
f Les isochromosomes (i)
Un isochromosome est un chromosome anormal formé de deux bras longs ou de deux bras courts d’un même chromosome avec perte de l’autre bras. Il résulte d’une séparation des chromatides horizontale et non verticale au moment de la méiose. Il peut être monocentrique ou dicentrique selon le mécanisme de formation.
f Duplications intrachromosomiques (dup)
Ce sont des remaniements rares aboutissant à des trisomies partielles. Les duplications chromosomiques peuvent être :
– directes (en tandem) : le fragment se duplique dans le même sens ; – inverses (en miroir) : le fragment se duplique en sens inverse. Les dupli-
cations en miroir terminales s’accompagnent généralement de la perte de l’extrémité distale du chromosome ; il y a alors co-existence d’une dupli-cation et d’une délétion. Lorsque la résolution du caryotype ne permet pas leur diagnostic, on parle de microduplication.
f Chromosomes marqueurs (mar)
On appelle chromosome marqueur tout fragment chromosomique surnu-méraire dont on ne peut identifier l’origine. Ces chromosomes surnuméraires peuvent être de novo ou hérités. La caractérisation et l’identification de ces mar-queurs nécessitent le plus souvent des investigations complémentaires en cytogé-nétique moléculaire.

13
Méthodes d’exploration du génome
1.4.2. Anomalies affectant deux chromosomes ou plus
Il s’agit : – des translocations réciproques ; – des translocations robertsoniennes ; – des insertions (ins).
Elles peuvent être équilibrées. Il s’agit essentiellement de translocations. Une translocation est caractérisée par deux cassures sur deux chromosomes différents, le plus souvent non-homologues, et recollement après échange des segments dis-taux. On distingue :
– les translocations robertsoniennes (impliquant les chromosomes acrocen-triques 13, 14, 15, 21 et 22) ;
– les translocations réciproques. Ces translocations peuvent être équilibrées ou non équilibrées. Elles peuvent
survenir de novo ou être héritées.
Figure 2b – Exemples de caryotypes avec anomalies de structure impliquant deux chromosomes

14
Méthodes d’étude et d’analyse du génome
f Les insertions
Elles sont plus rares. Elles résultent de deux cassures sur un bras chromoso-mique avec insertion du segment intercalaire au niveau d’un troisième point de cassure en un point quelconque du génome.
f Les translocations robertsoniennes (rob)
Elles se produisent entre chromosomes acrocentriques (13, 14, 15, 21 et 22) par fusion centrique ou, le plus souvent, par cassures dans les régions juxtacen-tromériques (Figure 2b). Les translocations robertsoniennes entraînent la perte apparente d’un centromère, et donc un caryotype à 45 chromosomes dans leur forme équilibrée. La perte du bras court des chromosomes transloqués n’a pas d’effet phénotypique. Il existe lors de la méiose un risque de formation de gamètes déséquilibrés donnant des zygotes trisomiques ou monosomiques pour la totalité d’un chromosome. Les translocations robertsoniennes sont responsables de la majorité des formes familiales de trisomie 21 et 13.
f Les translocations réciproques (t)
Ces translocations sont dues à des échanges de segments chromosomiques entre deux chromosomes le plus souvent non homologues non acrocentriques. Elles peuvent être équilibrées, parfois familiales.
Chez le sujet porteur, la gamétogenèse peut aboutir à la formation de gamètes dont le contenu chromosomique est déséquilibré
Les porteurs d’une translocation réciproque équilibrée peuvent former des gamètes normaux ou équilibrés par ségrégation méiotique de type alterne qui est la plus fréquente (Figure 3). Des gamètes non équilibrés peuvent résulter d’un des trois types de ségrégation suivants.
Le type adjacent-1L’un des deux chromosomes remaniés est transmis avec l’homologue normal
de l’autre chromosome. Il en résulte une duplication de l’un des segments trans-loqués et une monosomie de l’autre segment. Les deux combinaisons réciproques sont bien entendu possibles.
Le type adjacent-2 Il est exceptionnel. L’un des deux chromosomes remaniés est transmis avec
son propre homologue. Il existe alors une duplication/déficience des segments centriques des chromosomes remaniés, sans déséquilibre des parties transloquées.
Ces deux types de ségrégation donnent des zygotes non équilibrés ayant 46 chromosomes.

15
Méthodes d’exploration du génome
Le type 3:1 Il est rare et les zygotes qui en résultent ont 45 ou 47 chromosomes. Les deux
chromosomes remaniés peuvent être transmis avec un homologue normal, ou un seul chromosome remanié est transmis avec les deux homologues normaux.
Les facteurs favorisant les non-disjonctions de type adjacent-2 ou de type 3:1 sont : l’implication d’un acrocentrique au moins dans la translocation ; des segments centriques courts ou porteurs d’une constriction secondaire, celle du 9 en particulier ; le fait que le parent porteur de la translocation équilibrée soit la mère.
Les translocations par échange de bras entiers sont des cas particuliers de translocation réciproque. Ces translocations peuvent donner à la méiose des gamètes disomiques pour un bras chromosomique et nullisomiques pour l’autre. Les zygotes sont rarement viables. Ce type de translocation s’observe plutôt en cas d’avortements à répétition ou de stérilité.
f Les insertions (ins)
Elles se traduisent par le transfert d’un segment intercalaire à l’intérieur d’un autre bras chromosomique. Elles résultent d’un mécanisme à trois cassures, deux sur le chromosome donneur et une sur le chromosome receveur. Les chromo-somes donneur et receveur peuvent être un seul et même chromosome (insertion intrachromosomique). Le segment inséré peut conserver son orientation par rap-port au centromère ou prendre une orientation inverse.
Lors de la méiose on peut observer la formation de gamètes monosomiques ou trisomiques pour le segment inséré [3].
Figure 3 – Schémas représentant les différents modes de ségrégations déséquilibrées (d’après JM Dupont)

16
Méthodes d’étude et d’analyse du génome
1.4.3. Sites fragiles (fra)
On observe chez certains individus une zone de fragilité constitutionnelle localisée sur un autosome donné. Ces zones de fragilité se transmettent comme un caractère dominant et n’ont pas d’effet phénotypique connu. Les autosomes et les sites les plus souvent observés sont : 2q11, 10q23, 10q25, 11q13, 16p12, 16q22, 17p12, 20p11 [3].
Le cas du chromosome X avec le site en Xq27.3 est très particulier du fait de son effet phénotypique. Il est une des causes majeures de retard mental chez le garçon.
L’expression de ces sites fragiles varie avec les conditions de culture, notam-ment la teneur du milieu en acide folique [3].
1.4.4. Les syndromes microdélétionnels ou microduplicationnels
Ils sont définis par des pertes ou des gains chromosomiques de très petite taille (< à 300 Mb). La cytogénétique en haute résolution a permis de rattacher certaines de ces anomalies chromosomiques de petite taille à des entités cliniques décrites de longue date et dont l’origine génétique était suspectée. Ils sont le plus souvent de novo mais peuvent être parfois hérités, en particulier la microdélétion
Figure 4 – Caryotype 46,X(fra)Y

17
Méthodes d’exploration du génome
du chromosome 22. Des répétitions d’un type particulier responsables de recom-binaisons anormales ont été observées au niveau des points de cassure des délé-tions récurrentes. Les manifestations cliniques des microduplications sont liées à l’haploinsuffisance, c’est-à-dire à l’absence d’une des deux copies d’un ou plu-sieurs gènes localisés dans le segment délété. Tous les gènes inclus dans la délé-tion ne sont pas nécessairement sensibles à cet effet de dosage. Lorsque plusieurs gènes interviennent dans le phénotype, on parle de syndrome de gènes conti-gus [3]. Les microduplications ont en général un phénotype moins marqué.
Des sondes commerciales permettant le diagnostic en cytogénétique molé-culaire (FISH pour Fluorescent in situ hybridization) sont disponibles pour la majorité des syndromes microdélétionnels. Elles sont devenues le complément indispensable du caryotype lorsque la clinique évoque l’un de ces syndromes.
Bibliographie
[1] Lejeune J., Gauthier M., Turpin R., Étude des chromosomes somatiques de neuf enfants mongoliens, CR Acad Sci, Paris, 1959, 248:1721-2.
[2] Boué J.G., Boue A., Lazar P., and Gueguen S., Outcome of pregnancies fol-lowing a spontaneous abortion with chromosomal anomalies. Am. J. Obst.Gynec., 1973 ; 161, 806-8.
[3] Turleau C., Types, fréquences et mécanismes de formation des anomalies chromosomiques, Génétique Médicale, Masson, 2004 ; 71-83.
[4] Plachot M., Chromosome analysis of spontaneous abortions after IVF, A euro-pean survey. Hum Reprod, 1989 ; 4, 425-429.
[5] Pellestor F., Frequency and distribution of aneuploidy in human female gametes. Hum Genet.,1991 ; 86, 283-288.
[6] Hassold T.J., Hunt A.P., Sherman S., Trisomy in human : incidence, origin and etiology. Current opinion in genetics and development, 1993 ; 3 : 398-403.
[7] Angell R.R., Xian J., Ledger W. and Baird D.T., First meiotic division abnor-malities in human aneuploidy in human ovocytes : mechanism of trisomy for-mation. Cytogenet Cell Genet., 1994 ; 65, 194-20.
[8] De Grouchy J., Turleau C., Atlas des maladies chromosomiques, Expansion Scientifique Française, 2e édition, Paris, 1984.

18
Méthodes d’étude et d’analyse du génome
2. Cytogénétique moléculaire
2.1. Hybridation in situ fluorescente ou FISH (Fluorescence In Situ Hybridization)
Pr Jean-Pierre SIFFROI1
2.1.1. Introduction sur la notion d’hybridation
Les molécules d’ADN, qui constituent les chromosomes, sont constituées de deux chaînes polynucléotidiques reliées entre elles par des liaisons de type hydrogènes. La particularité de ces chaînes de nucléotides est d’avoir une orienta-tion chimique inverse et d’être complémentaires, c’est-à-dire que la composition d’une chaîne détermine celle de l’autre.
Les nucléotides renferment des bases dont il existe quatre sortes (Guanine G, Cytosine C, Adénine A, Thymine T) et la loi de complémentarité de ces bases fait qu’une guanine se trouvera toujours face à une cytosine et une adénine face à une thymine. Les paires de bases dans l’ADN sont donc toujours de type G-C ou A-T.
Une autre caractéristique des molécules d’ADN est de pouvoir s’ouvrir, à la manière d’une fermeture éclair, en rompant les liaisons hydrogène entre les bases et en dissociant les deux chaînes polynucléotidiques. Ce phénomène peut avoir lieu in vivo et sur de courts segments, comme lors de la transcription des gènes ou de la réplication de l’ADN, mais également in vitro où le moyen le plus simple pour séparer les chaînes polynucléotidiques l’une de l’autre est de chauffer la molécule d’ADN jusqu’à une température d’environ 90-100 °C. Cette méthode aboutit à ce qu’on appelle la dénaturation de l’ADN et est une des bases des tech-niques de génétique moléculaire.
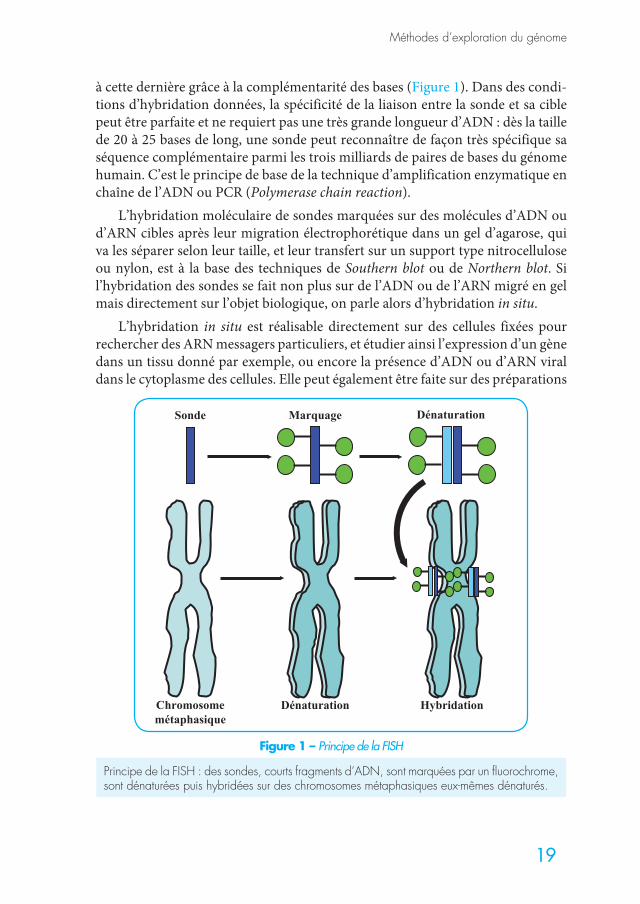
La combinaison de ces deux propriétés de l’ADN (complémentarité des bases et capacité à se dénaturer) a permis l’utilisation d’outils de biologie moléculaire qu’on appelle des sondes. Celles-ci sont des fragments d’ADN de localisation connue dans le génome dont la taille varie de quelques milliers à dizaines ou centaines de milliers de paires de bases (pb). Elles doivent être marquées de façon radioactive, fluorescente ou chemoluminescente pour pouvoir ensuite être repé-rées. Une fois mises en contact avec des molécules d’ADN cibles, et à condition que ces dernières soient dans un état dénaturé, les sondes, elles mêmes dénatu-rées, vont trouver la séquence d’ADN dont elles sont la copie et vont s’hybrider
1. Service de Génétique et d’Embryologie médicales, hôpital Armand Trousseau, APHP, Paris.
19
Méthodes d’exploration du génome
à cette dernière grâce à la complémentarité des bases (Figure 1). Dans des condi-tions d’hybridation données, la spécificité de la liaison entre la sonde et sa cible peut être parfaite et ne requiert pas une très grande longueur d’ADN : dès la taille de 20 à 25 bases de long, une sonde peut reconnaître de façon très spécifique sa séquence complémentaire parmi les trois milliards de paires de bases du génome humain. C’est le principe de base de la technique d’amplification enzymatique en chaîne de l’ADN ou PCR (Polymerase chain reaction).
L’hybridation moléculaire de sondes marquées sur des molécules d’ADN ou d’ARN cibles après leur migration électrophorétique dans un gel d’agarose, qui va les séparer selon leur taille, et leur transfert sur un support type nitrocellulose ou nylon, est à la base des techniques de Southern blot ou de Northern blot. Si l’hybridation des sondes se fait non plus sur de l’ADN ou de l’ARN migré en gel mais directement sur l’objet biologique, on parle alors d’hybridation in situ.
L’hybridation in situ est réalisable directement sur des cellules fixées pour rechercher des ARN messagers particuliers, et étudier ainsi l’expression d’un gène dans un tissu donné par exemple, ou encore la présence d’ADN ou d’ARN viral dans le cytoplasme des cellules. Elle peut également être faite sur des préparations
Chromosomemétaphasique
Dénaturation
Dénaturation
Hybridation
MarquageSonde
Figure 1 – Principe de la FISH
Principe de la FISH : des sondes, courts fragments d’ADN, sont marquées par un fluorochrome, sont dénaturées puis hybridées sur des chromosomes métaphasiques eux-mêmes dénaturés.

20
Méthodes d’étude et d’analyse du génome
chromosomiques classiques sur lesquelles il sera possible d’observer l’accrochage d’une sonde sur les chromosomes métaphasiques mais aussi sur les noyaux en interphase situés tout autour.
2.1.2. De la radioactivité à la fluorescence
La condition nécessaire à l’utilisation des sondes est bien sûr de pouvoir les repérer, que ce soit sur membrane à partir d’un gel d’électrophorèse en cas d’hy-bridation moléculaire ou sur une préparation biologique en cas d’hybridation in situ. Les premières techniques de marquage des sondes utilisaient des nucléo-tides radioactifs qui étaient incorporés dans l’ADN de la sonde par des techniques enzymatiques utilisant les propriétés des ADN polymérases (Nick translation, Random priming ou PCR). En hybridation moléculaire, la révélation de la sonde se faisait en exposant directement la membrane sur laquelle la sonde s’était hybri-dée à l’ADN migré en gel à un film de radiologie. L’exposition durait quelques heures ou jours selon l’intensité du rayonnement radioactif.
En hybridation in situ, le problème était plus compliqué car le signal émis par la sonde devait absolument être localisé sur la structure biologique (cellule par-ticulière dans un tissu, organite intracellulaire particulier, chromosome donné dans une métaphase, etc.). La révélation du signal se faisait en recouvrant la lame de microscope, ou la grille de microscopie électronique, d’un film d’émulsion photographique, sorte de pellicule liquide, sur lequel la radioactivité émise par la sonde réduisait des grains d’argent qui précipitaient sur place. Si le rayonnement radioactif était trop puissant, cette réaction se faisait au niveau de la source, donc de la sonde, mais aussi à distance ce qui interdisait toute localisation précise du signal. Pour pallier cet inconvénient, il était donc nécessaire d’utiliser des radio-éléments de faible activité et à très court trajet, type tritium ou soufre35, ce qui imposait des temps d’exposition très longs qui se comptaient en semaines, voire en mois pour le tritium en microscopie électronique. De tels délais étaient bien sûr incompatibles avec l’utilisation des sondes radioactives en diagnostic courant et leur emploi était limité au domaine de la recherche. De plus, pour les chromo-somes par exemple, la présence de grains d’argent un peu partout sur la prépara-tion nécessitait une étude statistique pour montrer qu’ils étaient plus nombreux sur un chromosome donné par rapport au bruit de fond.
La solution à ce problème fut trouvée à la fin des années 80 par l’emploi de la fluorescence au lieu de la radioactivité. En effet, l’émission d’un signal fluores-cent émis par une sonde pouvait être directement observée après hybridation et résolvait les problèmes de localisation liés à la radioactivité tout en facilitant gran-dement la pratique de la technique en supprimant l’emploi des radio- éléments, potentiellement dangereux. Le principe de l’observation d’un signal fluores-cent est simple : un fluorochrome est excité par une lumière ultraviolette à une

21
Méthodes d’exploration du génome
longueur d’onde spécifique et émet une fluorescence dans une autre longueur d’onde, elle-même spécifique du fluorochrome utilisé. Cette technique ne peut donc pas être employée en microscopie électronique et nécessite l’utilisation de microscopes équipés d’une source de lumière ultraviolette et de filtres d’excita-tion et d’émission répondant au fluorochrome qui a été choisi pour le marquage de la sonde.
Il fallut cependant attendre quelques années pour pouvoir disposer de sondes marquées par des fluorochromes directement utilisables sur des préparations biologiques. Une étape intermédiaire consista à utiliser des sondes marquées par des protéines (Biotine, Digoxigénine) et à révéler celles-ci par des anticorps spécifiques couplés à des fluorochromes. Cette méthode d’immunofluorescence permettait d’amplifier le signal grâce à l’utilisation d’anticorps se reconnaissant les uns les autres (technique dite en sandwich). Aujourd’hui, la qualité des fluo-rochromes, et celle des microscopes maintenant équipés de systèmes d’analyse d’images performants, autorisent le marquage direct des sondes. Le dernier avan-tage de la fluorescence, et non des moindres, est de pouvoir disposer de fluoro-chromes de couleurs différentes les unes des autres et donc de pouvoir hybrider plusieurs sondes sur une même préparation biologique, chacune reconnaissant une cible donnée.
2.1.3. Types de sondes utilisées en FISH sur chromosomes
Les sondes utilisées en FISH sur chromosomes sont le plus souvent des sondes ADN, ayant donc une structure classique bicaténaire. D’autres types de sondes existent, comme les sondes ARN constituées d’une seule chaîne polynu-cléotidique mais celles-ci sont surtout utilisées en hybridation in situ sur coupes de tissus à la recherche d’ARN messagers spécifiques et pour étudier l’expression d’un gène particulier.
Des essais sur chromosomes furent également effectués avec des oligonucléo-tides, courtes chaînes monocaténaires de 20-25 nucléotides de long qui s’hybri-daient de façon spécifique mais nécessitaient un marquage par élongation une fois hybridés pour pouvoir être repérés (technique PRINS). Si les résultats étaient globalement satisfaisants, cette dernière technique ne fut jamais généralisée et ce furent les sondes ADN qui s’imposèrent.
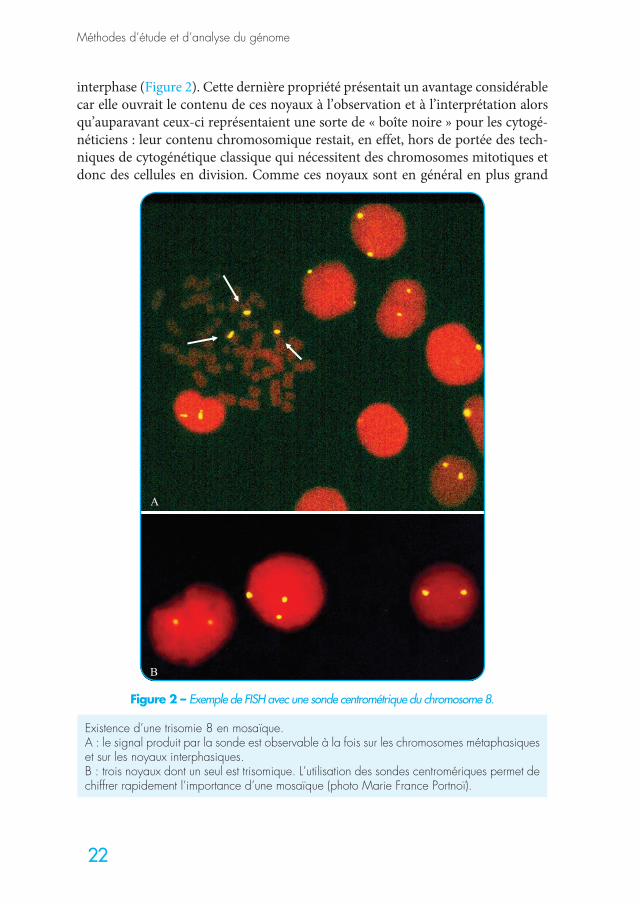
Pour des raisons d’intensité de signal, les premières sondes utilisables en FISH furent celles reconnaissant des séquences répétées dans le génome comme celles constituant les régions centromériques des chromosomes ou l’hétérochro-matine. Elles généraient un signal puissant, facilement observable, mais suffi-samment ponctuel pour permettre l’identification du chromosome reconnu par la sonde, à la fois dans les métaphases mais également dans les noyaux en
22
Méthodes d’étude et d’analyse du génome
interphase (Figure 2). Cette dernière propriété présentait un avantage considérable car elle ouvrait le contenu de ces noyaux à l’observation et à l’interprétation alors qu’auparavant ceux-ci représentaient une sorte de « boîte noire » pour les cytogé-néticiens : leur contenu chromosomique restait, en effet, hors de portée des tech-niques de cytogénétique classique qui nécessitent des chromosomes mitotiques et donc des cellules en division. Comme ces noyaux sont en général en plus grand
A
B
Figure 2 – Exemple de FISH avec une sonde centrométrique du chromosome 8.
Existence d’une trisomie 8 en mosaïque.A : le signal produit par la sonde est observable à la fois sur les chromosomes métaphasiques et sur les noyaux interphasiques.B : trois noyaux dont un seul est trisomique. L’utilisation des sondes centromériques permet de chiffrer rapidement l’importance d’une mosaïque (photo Marie France Portnoï).
23
Méthodes d’exploration du génome
nombre que les métaphases sur les préparations chromosomiques, la possibilité de les analyser représentait un gain de temps considérable pour certaines applications.
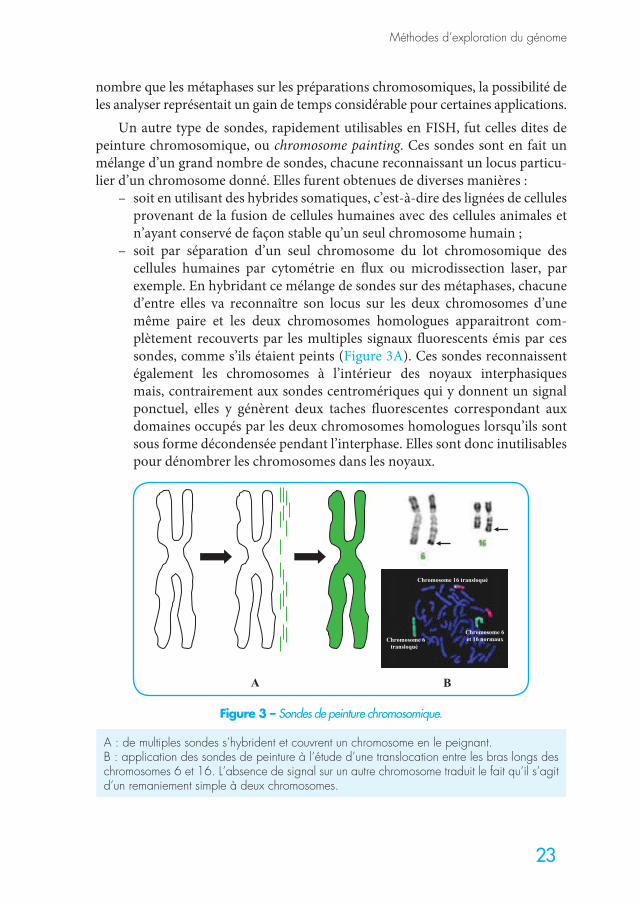
Un autre type de sondes, rapidement utilisables en FISH, fut celles dites de peinture chromosomique, ou chromosome painting. Ces sondes sont en fait un mélange d’un grand nombre de sondes, chacune reconnaissant un locus particu-lier d’un chromosome donné. Elles furent obtenues de diverses manières :
– soit en utilisant des hybrides somatiques, c’est-à-dire des lignées de cellules provenant de la fusion de cellules humaines avec des cellules animales et n’ayant conservé de façon stable qu’un seul chromosome humain ;
– soit par séparation d’un seul chromosome du lot chromosomique des cellules humaines par cytométrie en flux ou microdissection laser, par exemple. En hybridant ce mélange de sondes sur des métaphases, chacune d’entre elles va reconnaître son locus sur les deux chromosomes d’une même paire et les deux chromosomes homologues apparaitront com-plètement recouverts par les multiples signaux fluorescents émis par ces sondes, comme s’ils étaient peints (Figure 3A). Ces sondes reconnaissent également les chromosomes à l’intérieur des noyaux interphasiques mais, contrairement aux sondes centromériques qui y donnent un signal ponctuel, elles y génèrent deux taches fluorescentes correspondant aux domaines occupés par les deux chromosomes homologues lorsqu’ils sont sous forme décondensée pendant l’interphase. Elles sont donc inutilisables pour dénombrer les chromosomes dans les noyaux.
A B
Chromosome 6transloqué
Chromosome 16 transloqué
Chromosome 6et 16 normaux
Figure 3 – Sondes de peinture chromosomique.
A : de multiples sondes s’hybrident et couvrent un chromosome en le peignant.B : application des sondes de peinture à l’étude d’une translocation entre les bras longs des chromosomes 6 et 16. L’absence de signal sur un autre chromosome traduit le fait qu’il s’agit d’un remaniement simple à deux chromosomes.

24
Méthodes d’étude et d’analyse du génome
Une variante des peintures chromosomiques est représentée par les sondes multi-FISH ou par la cytogénétique spectrale (Spectral Karyotyping ou SKY). Dans ce cas, un ou plusieurs mélanges de sondes de peinture sont constitués et, pour une paire chromosomique donnée, les sondes ont été marquées en utili-sant des fluorochromes différents et en proportions bien définies. Chaque paire de chromosomes est alors reconnue par des sondes qui ont un spectre de fluo-rescence bien spécifique. Si les différences ne se voient pas à l’œil, l’utilisation d’un analyseur d’images dédié à cette technique permet de mesurer ce mélange de fluorescence de façon très précise et, associé à la taille du chromosome analysé, d’assigner un numéro de paire chromosomique et une fausse couleur à ce chro-mosome. Le résultat est un caryotype tout en couleur et assez impressionnant pour les non spécialistes.
Cependant, l’intérêt de ces deux techniques est principalement l’étude des caryotypes très remaniés qu’on observe dans certains cancers ou leucémies et pas du tout les pathologies chromosomiques constitutionnelles.
Le dernier type de sondes utilisé en FISH correspond à des sondes qui recon-naissent un locus unique sur un chromosome donné. Appelées « locus spéci-fique », ces sondes furent les dernières à apparaître et à être utilisées en pratique courante du fait de leur petite taille et du faible signal qu’elles émettaient. Les pro-grès en matière de marquage par les fluorochromes et d’imagerie ont fait qu’elles sont maintenant couramment utilisées.
En diagnostic de routine, la plupart des sondes sont des sondes commerciales répondant à des critères de qualité stricts quant à leur spécificité et à l’intensité du signal qu’elles émettent. Néanmoins, la majorité des laboratoires de cytogénétique sont maintenant capables de fabriquer leurs propres sondes pour des applications particulières. C’est surtout vrai pour les sondes « locus spécifique » qui peuvent être obtenues à partir des milliers de clones bactériens recombinés disponibles dans des banques, c’est-à-dire de clones d’E. coli contenant un fragment d’ADN humain porté par un vecteur qui peut être un plasmide, un cosmide, un fosmide ou plus généralement un BAC (Bacterial Artificial Chromosome). Ces derniers sont le produit direct des techniques qui ont permis le séquençage du génome humain et ils sont maintenant disponibles après consultation des banques de données qui les répertorient. Une fois le clone bactérien amplifié par culture, l’ADN est extrait et marqué puis hybridé sur les chromosomes. Seule la partie spécifiquement humaine de l’ADN extrait s’hybridera et génèrera un signal au locus correspondant. Ces sondes offrent donc maintenant la possibilité d’étudier en FISH n’importe quelle région des chromosomes humains, notamment pour la caractérisation des anomalies chromosomiques repérées au caryotype (Figure 4) ou pour vérifier les résultats obtenus avec les méthodes les plus modernes d’étude du génome humain comme la CGH-Array.
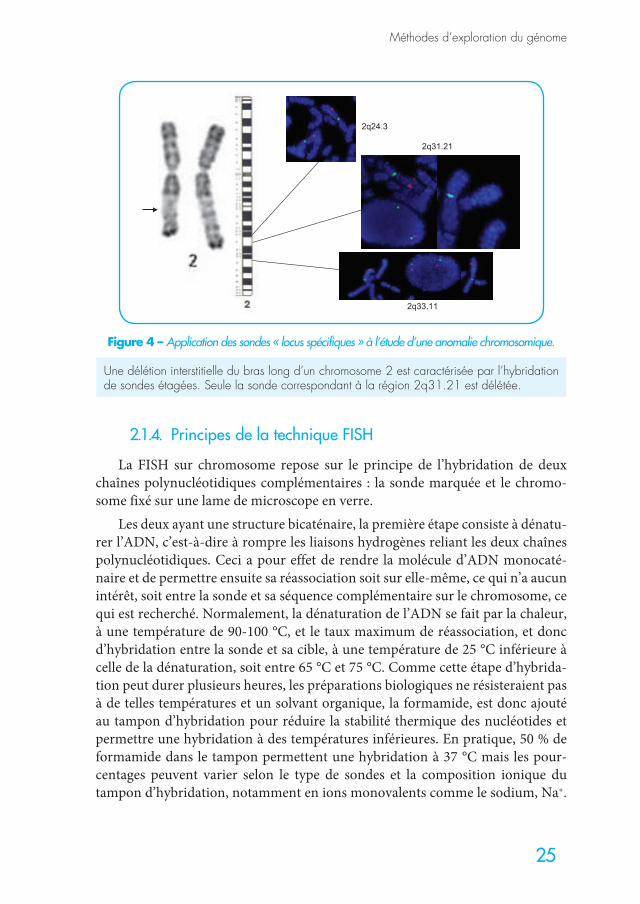
25
Méthodes d’exploration du génome
2.1.4. Principes de la technique FISH
La FISH sur chromosome repose sur le principe de l’hybridation de deux chaînes polynucléotidiques complémentaires : la sonde marquée et le chromo-some fixé sur une lame de microscope en verre.
Les deux ayant une structure bicaténaire, la première étape consiste à dénatu-rer l’ADN, c’est-à-dire à rompre les liaisons hydrogènes reliant les deux chaînes polynucléotidiques. Ceci a pour effet de rendre la molécule d’ADN monocaté-naire et de permettre ensuite sa réassociation soit sur elle-même, ce qui n’a aucun intérêt, soit entre la sonde et sa séquence complémentaire sur le chromosome, ce qui est recherché. Normalement, la dénaturation de l’ADN se fait par la chaleur, à une température de 90-100 °C, et le taux maximum de réassociation, et donc d’hybridation entre la sonde et sa cible, à une température de 25 °C inférieure à celle de la dénaturation, soit entre 65 °C et 75 °C. Comme cette étape d’hybrida-tion peut durer plusieurs heures, les préparations biologiques ne résisteraient pas à de telles températures et un solvant organique, la formamide, est donc ajouté au tampon d’hybridation pour réduire la stabilité thermique des nucléotides et permettre une hybridation à des températures inférieures. En pratique, 50 % de formamide dans le tampon permettent une hybridation à 37 °C mais les pour-centages peuvent varier selon le type de sondes et la composition ionique du tampon d’hybridation, notamment en ions monovalents comme le sodium, Na+.
2q33.11
2q24.3
2q31.21
Figure 4 – Application des sondes « locus spécifiques » à l’étude d’une anomalie chromosomique.
Une délétion interstitielle du bras long d’un chromosome 2 est caractérisée par l’hybridation de sondes étagées. Seule la sonde correspondant à la région 2q31.21 est délétée.

26
Méthodes d’étude et d’analyse du génome
La réassociation de la sonde sur elle-même dépend du rapport entre la concentra-tion de celle-ci dans le tampon d’hybridation et le nombre de molécules d’ADN cibles, donc de chromosomes ou de noyaux interphasiques sur la lame de micro-scope. Augmenter la concentration de la sonde dans le tampon n’est donc pas forcément bénéfique et ne fait que renchérir le coût de l’examen.
L’existence dans le génome de séquences répétées, type Alu ou Line1, fait que les sondes qui ont une origine génomique en contiennent également. Pour éviter une hybridation non spécifique qui viendrait noyer le signal d’hybridation dans un bruit de fond trop important, les sondes marquées sont co-hybridées avec de l’ADN humain non marqué enrichi en de telles séquences, type ADN Cot1, qui va servir de compétiteur.
Les étapes de lavage post-hybridation conditionnent également la réussite de la technique soit en ne supprimant pas tous les accrochages non spécifiques de la sonde, soit, au contraire, en décrochant la sonde de sa séquence cible. Des fac-teurs importants de concentration ionique et de température sont en jeu dans ces lavages et définissent ce qu’on appelle la « stringence » de la réaction : un signal fluorescent obtenu dans des conditions très stringentes a donc toutes les chances d’être spécifique.
L’observation au microscope nécessite un appareil équipé d’une source de lumière ultraviolette et de filtres adaptés au fluorochrome qui a été utilisé pour le marquage de la sonde. Les chromosomes sont tout d’abord contre-colorés par un agent fluorescent, en général le DAPI (4’,6’-diamilino-2-phénylindole) qui donne une coloration bleue, pour que les mitoses soient repérables au micros-cope à faible grossissement. Une fois les chromosomes localisés, un fort gros-sissement est effectué sur la mitose pour observer le signal généré par la sonde. Normalement, un filtre correspond à une fluorescence donnée mais il en existe certains qui sont à double voire triple bandes et qui permettent donc de visualiser simultanément plusieurs signaux de couleurs différentes.
2.1.5. La FISH en pratique courante
Depuis la fin des années 90, la FISH est devenue un outil incontournable pour le diagnostic d’anomalies chromosomiques, à tel point que sa maîtrise et sa pra-tique courante conditionnent l’obtention des autorisations nécessaires pour faire des diagnostics en cytogénétique. Sa mise en route peut se faire d’emblée, dès l’arrivée d’un prélèvement au laboratoire et en dehors de toute anomalie chromo-somique connue, ou bien dans un deuxième temps, pour vérifier ou préciser une anomalie du caryotype repérée grâce aux techniques de marquage en bandes des chromosomes. Selon l’anomalie chromosomique suspectée ou à vérifier, tel ou tel type de sonde sera employé.

27
Méthodes d’exploration du génome
f Anomalies de nombre des chromosomes ou aneuploïdies
Le diagnostic des aneuploïdies a beaucoup bénéficié du développement des techniques de FISH car la rapidité de lecture autorise l’analyse d’un grand nombre de cellules. En effet, ces anomalies peuvent être recherchées à la fois sur des mitoses mais également sur des noyaux interphasiques. La condition tech-nique est de disposer d’une sonde spécifique générant un signal ponctuel (sondes centromériques ou locus spécifique).
L’intérêt de l’utilisation de la FISH pour ce type de diagnostic est de savoir si l’anomalie est homogène, c’est-à-dire présente dans toutes les cellules d’un indi-vidu, ou en mosaïque c’est-à-dire uniquement dans un pourcentage variable de cellules, les autres ayant un caryotype normal. Le travail long et fastidieux qui consistait auparavant à parcourir au faible grossissement des lames de micros-cope à la recherche de mitoses, à mettre une goutte d’huile pour passer au fort grossissement puis à compter les chromosomes et repérer celui qui était éven-tuellement en trop ou en moins, a été remplacé par un comptage rapide en FISH du nombre de signaux correspondant à ce chromosome à la fois dans les mitoses mais surtout dans les noyaux interphasiques qui sont en grand nombre sur les lames (Figure 2). Ainsi, un comptage portant sur plusieurs centaines de noyaux peut-il être maintenant réalisé en quelques dizaines de minutes.
Ce qu’on appelle désormais la FISH interphasique (pour laquelle il existe une formulation précise dans la nomenclature internationale) peut être mise en œuvre immédiatement après l’obtention des cellules et sans culture préalable. C’est ce qui est fait, par exemple, pour le diagnostic rapide en FISH des aneuploï-dies fœtales sur des cellules amniotiques prélevées par amniocentèse ou encore sur des cellules sanguines pour la confirmation rapide d’une trisomie 21 chez un nouveau-né. Elle peut être également réalisée pour confirmer et chiffrer une ano-malie de nombre repérées sur des mitoses par des techniques de cytogénétique classique et après culture.
f Anomalies de structure des chromosomes
Les anomalies de structure les plus fréquentes sont les translocations qui comportent à la fois les translocations Robertsoniennes, ou fusions centriques, et les translocations réciproques qui sont un échange mutuel de matériel chromo-somique entre deux chromosomes de paires différentes. Dans de rares cas, il peut s’agir de remaniements chromosomiques plus complexes impliquant trois voire quatre paires de chromosomes.
Le plus souvent, les techniques de marquage en bandes des chromosomes suf-fisent à caractériser les translocations mais ces techniques peuvent quand même

28
Méthodes d’étude et d’analyse du génome
passer à côté d’un remaniement complexe qui, du fait de la très petite taille d’un des fragments échangés, ne serait pas ou difficilement repérable. L’étude en FISH des translocations, même celles qui paraissent à l’évidence des remaniements à deux chromosomes, est maintenant systématiquement réalisée avec des sondes de pein-tures pour vérifier qu’un troisième chromosome n’est pas impliqué (Figure 3B).
Une telle erreur diagnostique aurait en effet des conséquences dramatiques pour la pratique d’un diagnostic prénatal.
Le deuxième exemple où la FISH est nécessaire à la caractérisation d’une translocation est lorsque les fragments échangés sont de trop petites tailles pour être vus après simple marquage en bandes. Ces translocations sont dites cryp-tiques et concernent les segments les plus terminaux des chromosomes qu’on appelle les télomères. Elles sont d’autant plus redoutables qu’elles sont impos-sibles à repérer sur le caryotype classique et que, lorsqu’elles sont transmises à l’état déséquilibré, elles ne donnent pas obligatoirement de fausses couches. La probabilité qu’un enfant atteint naisse est alors importante. À titre d’exemple, on estime à environ 5 % le pourcentage d’enfants présentant un retard mental sans diagnostic connu dont le handicap serait lié à la transmission déséquilibrée d’une translocation parentale cryptique. Le diagnostic de ces remaniements chromoso-miques cryptiques repose sur l’utilisation de sondes qui s’hybrident sur les régions sub-télomériques. Comme il existe 23 paires de chromosomes, donc 46 régions sub-télomériques, si on considère les deux extrémités des chromosomes, cela fait autant de techniques de FISH à réaliser pour balayer tous les télomères. Comme les chromosomes X et Y ont les mêmes télomères et que les cinq paires de chro-mosomes acrocentriques ne sont étudiés que sur le télomère de leur bras long (13, 14, 15, 21, 22), il reste 41 techniques de FISH différentes à faire, ce qui représente toujours un travail considérable pour analyser tous les télomères chez un patient.
En pratique, il existe des dispositifs commercialisés (mélanges de sondes cor-respondant à des chromosomes de tailles très différentes donc facilement dis-cernables les uns des autres ou lames de microscope quadrillées présentant les sondes télomériques d’une seule paire chromosomique par case) qui permettent maintenant de faire cette étude très rapidement et en une fois.
f Caractérisation de points de cassures chromosomiques et identification de gènes responsables de maladies héréditaires
Dans la grande majorité des cas, les anomalies de structure équilibrées des chromosomes n’ont pas d’effet phénotypique, en dehors d’une possible infertilité chez les hommes. Il arrive cependant qu’un tel remaniement soit associé à une maladie cliniquement connue, et même bien répertoriée quant à son mode de transmission, mais dont le gène responsable n’est pas identifié. Il s’agit là d’une

29
Méthodes d’exploration du génome
situation rare mais particulièrement intéressante puisque le remaniement chro-mosomique indique deux endroits (puisqu’il y a deux points de cassure) dans le génome où pourrait se situer ce gène. Le problème est que la résolution du mar-quage en bandes est, au mieux, de 5 à 10 millions de paires de bases et que, dans un tel intervalle, peuvent se situer des centaines de gènes.
Il convient alors de réduire au maximum cet intervalle en réalisant ce qu’on appelle une « marche sur le chromosome » avec des sondes de FISH spécifiques des deux régions concernées pour voir à partir de quel endroit une sonde norma-lement située à un locus donné change de position, c’est-à-dire passe sur un autre chromosome en cas de translocation ou change d’emplacement sur le même chromosome en cas d’inversion. Cette démarche peut être longue mais aboutit à caractériser un espace minimum dans lequel, avec un peu de chance, se situent uniquement quelques gènes, l’hypothèse étant que l’un d’entre eux soit inactivé par le point de cassure. En regardant dans les bases de données le rôle de chacun de ces gènes, il est alors possible d’en choisir un comme étant un bon candidat et de chercher des mutations ponctuelles de ce dernier dans des familles de patients atteints par la maladie en question. L’identification de telles mutations signe alors la responsabilité de ce gène dans la maladie.
fMicrodélétions et microduplications
Il a pu être montré qu’un certain nombre de syndromes cliniques connus et bien étiquetés (syndromes de DiGeorge, Williams-Beuren, Smith Magenis, Prader Willi, Angelman, etc.) étaient dus en fait à l’existence de microdélétions interstitielles, le plus souvent invisibles au caryotype classique, et détectables uni-quement en FISH grâce à l’utilisation de sondes codant pour les régions spécifi-quement impliquées dans ces syndromes (Figure 5). La fréquence et la relative similarité des points de cassure chromosomique chez les sujets atteints est le reflet du mode de formation de ces microdélétions, à savoir des recombinaisons anor-males intra chromosomique ou entre chromosomes homologues à la méiose chez un des parents. Le résultat de ce type d’accident chromosomique est la formation d’un chromosome délété pour une région et d’un autre qui est dupliqué pour la même région. Il existe donc des syndromes dus à des micro-duplications dont la clinique peut être voisine de celle observée dans les microdélétions, comme dans le syndrome de DiGeorge, ou bien différente voire en miroir comme dans le syn-drome de Williams-Beuren.
D’une façon générale, le diagnostic des duplications en FISH est plus difficile que celui des délétions. En effet, les signaux générés par deux sondes qui s’hybrident l’une à côté de l’autre sur un même chromosome fusionnent, le plus souvent, en un seul signal pour lequel il est parfois possible de dire qu’il est plus intense que celui
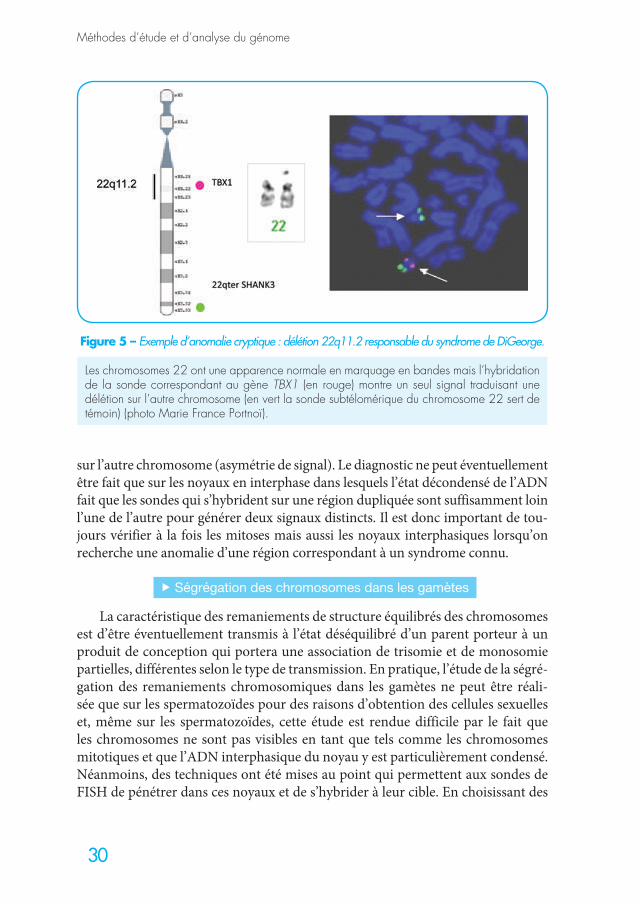
30
Méthodes d’étude et d’analyse du génome
sur l’autre chromosome (asymétrie de signal). Le diagnostic ne peut éventuellement être fait que sur les noyaux en interphase dans lesquels l’état décondensé de l’ADN fait que les sondes qui s’hybrident sur une région dupliquée sont suffisamment loin l’une de l’autre pour générer deux signaux distincts. Il est donc important de tou-jours vérifier à la fois les mitoses mais aussi les noyaux interphasiques lorsqu’on recherche une anomalie d’une région correspondant à un syndrome connu.
f Ségrégation des chromosomes dans les gamètes
La caractéristique des remaniements de structure équilibrés des chromosomes est d’être éventuellement transmis à l’état déséquilibré d’un parent porteur à un produit de conception qui portera une association de trisomie et de monosomie partielles, différentes selon le type de transmission. En pratique, l’étude de la ségré-gation des remaniements chromosomiques dans les gamètes ne peut être réali-sée que sur les spermatozoïdes pour des raisons d’obtention des cellules sexuelles et, même sur les spermatozoïdes, cette étude est rendue difficile par le fait que les chromosomes ne sont pas visibles en tant que tels comme les chromosomes mitotiques et que l’ADN interphasique du noyau y est particulièrement condensé. Néanmoins, des techniques ont été mises au point qui permettent aux sondes de FISH de pénétrer dans ces noyaux et de s’hybrider à leur cible. En choisissant des
Figure 5 – Exemple d’anomalie cryptique : délétion 22q11.2 responsable du syndrome de DiGeorge.
Les chromosomes 22 ont une apparence normale en marquage en bandes mais l’hybridation de la sonde correspondant au gène TBX1 (en rouge) montre un seul signal traduisant une délétion sur l’autre chromosome (en vert la sonde subtélomérique du chromosome 22 sert de témoin) (photo Marie France Portnoï).

31
Méthodes d’exploration du génome
sondes de couleurs différentes et correctement positionnées sur les chromosomes impliqués dans le remaniement de structure, il est dès lors possible de déduire le type de ségrégation chromosomique de la combinaison de couleurs observée.
Ce type d’étude n’a qu’une valeur pronostique, en permettant de consta-ter qu’un homme porteur d’une translocation produit tant de pourcentage de gamètes déséquilibrés, mais en aucun cas, elle ne peut autoriser l’utilisation des gamètes hybridés pour une fécondation ultérieure.
2.1.6. Conclusion
Il est légitime de dire que la FISH a révolutionné la pratique de l’analyse chro-mosomique en autorisant une résolution très fine et en permettant l’accès au contenu des noyaux interphasiques. Cependant, contrairement au caryotype clas-sique qui donne une vue d’ensemble du génome, les sondes de FISH n’apportent des renseignements que sur la région chromosomique dont elles sont la copie.
Leur utilisation ne peut donc être que ciblée soit par des signes cliniques observés chez un patient soit par un remaniement chromosomique visible au caryotype, soit encore par le fait qu’une anomalie va être recherchée en FISH parce qu’elle est particulièrement fréquente dans une population de patients. Malgré ces limites, la FISH reste un outil incontournable de l’analyse chromo-somique car elle fournit des données topographiques sur des anomalies qui sont, de plus en plus souvent, diagnostiquées par des méthodes purement moléculaires (CGH-Array, par exemple). Ces données sont absolument nécessaires à l’établis-sement d’un conseil génétique fiable.
Bibliographie
Landegent J.E., Jansen in de Wal N., Ommen G.J.B. et al. Chromosomal moca-lization of a unique gene by non-autoradiographic in situ hybridization. Nature 1985;317:175-77.
Lichter P., Cremer T., Chang Tang CJ. Et al. Rapid detection of human chro-mosome 21 aberrations by in situ hybridization. Proc Natl Acad Sci USA 1988;85:9664-68.
Pinkel D., Straume T., Gray J.W. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA 1986;83:2934-38.
Ried T., Landes G., Dackowski W. et al. Multicolor fluorescence in situ hybridiza-tion for the simultaneous detection of probe sets for chromosomes 13, 18, 21, X and Y in uncultured amniotic fluid cells. Hum Mol Genet 1992;1:307-313.

32
Méthodes d’étude et d’analyse du génome
Schröck E., du Manoir S., Veldman T. et al. Multicolor spectral karyotyping of human chromosomes. Science 1996;273:494-97.
Siffroi J.P., Chantot-Bastaraud S. L’avenir de la cytogénétique après le séquençage du génome humain. Morphologie 2004;88:19-23.
Trask B. Fluorescence in situ hybridization: applications in cytogenetics and gene mapping. Trends Genet 1991;7:149-54.
Trask B.J. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat Rev Genet 2002;3:769-778.
2.2. Diagnostic prénatal rapide par la technique des Bacs-On-Beads (Prenatal BACs-on-BeadsTM)
Dr Céline Dupont1, Pr Brigitte Benzacken2
Comme il a été décrit précédemment, l’examen de référence en matière de diagnostic prénatal chromosomique est l’établissement du caryotype conven-tionnel sur liquide amniotique ou villosités choriales. Les principales anoma-lies chromosomiques retrouvées sont des aneuploïdies (anomalie de nombre des chromosomes) principalement la trisomie 21. L’établissement du caryotype conventionnel nécessite une étape de culture cellulaire qui peut prendre 15 jours (liquide amniotique) ou plus, 3 semaines à 1 mois dans le cas des biopsies de trophoblastes. Cette étape de plusieurs semaines est difficile à supporter pour les femmes enceintes, particulièrement en cas de terme avancé ou de signes d’appel échographiques. Des techniques de diagnostic rapide existent permettant de don-ner un résultat en 24 à 48 h au moins concernant les principales aneuploïdies et le sexe du fœtus.
La FISH (Fluorescence In Situ Hybridization) est la technique la plus com-munément utilisée à l’heure actuelle pour ce diagnostic rapide.
Parallèlement, de nouvelles techniques de cytogénétique moléculaire (micro-puces à ADN), permettant l’étude du génome dans sa globalité à la recherche de perte ou de gain de matériel chromosomique, sont apparues. La difficulté de leur utilisation en prénatal réside principalement tout d’abord dans le fait qu’il existe des régions polymorphes dans le génome dont le gain ou la perte sont de significati-vité mal connue ou inconnue et ensuite que ces techniques permettent de faire des diagnostics présymptomatiques de maladie dont la pénétrance et l’expressivité sont variables. Des problèmes d’ordre éthique sont alors posés et le conseil génétique est
1. PH Cytogénétique Robert Debré.2. Service d’Histologie Embryologie Cytogénétique, Biologie de la Reproduction, CECOS Hôpital
Jean Verdier.

33
Méthodes d’exploration du génome
extrêmement difficile. Des études tentent à l’heure actuelle de proposer des lignes directrices concernant l’utilisation de ces micropuces à ADN en diagnostic prénatal.
La nouvelle technique des BACs-on-BeadsTM (Kit Prenatal BoBsTM) est un outil de cytogénétique moléculaire intermédiaire, de dépistage ciblé. Il utilise la technique des CGH-array (micropuces à ADN par hybridation génomique comparative) (Shaffer et al., 2011) mais en suspension sur des microsphères ou microbilles. En ce qui concerne les BoBsTM, l’ADN du fœtus est extrait à partir de liquide amniotique natif (ou cultivé le cas échéant) ou à partir de villosités cho-riales ou même de sang fœtal.
L’ADN fœtal à tester, marqué par un fluorochrome, est ensuite mis en compé-tition avec deux ADNs de référence (un mâle et un femelle) également marqués. Ces trois ADNs sont alors cohybridés tous les trois sur des microbilles (les Beads) qui portent chacune un oligonucléotide particulier (les BACs) correspondant aux principales aneuploïdies (13, 18 et 21), aux chromosomes sexuels (X et Y), et à 9 syndromes (Sd) microdélétionnels dont le pronostic sévère est connu (syn-drome de DiGeorge 1-22q11.2 et 2-10p14, syndrome de Williams-Beuren-7q11.2, syndrome de Prader-Willi-15q11, syndrome d’Angelman-15q11, syndrome de Smith-Magenis-17p11, syndrome de Wolf Hirschhorn-4p16, syndrome du Cri du Chat-5p15, syndrome de Langer-Giedion-8q23, et syndrome de Miller Dieker-17p13). Après hybridation sur la nuit, les billes hybridées sont lues par un appareil de cytométrie (Luminex®) capable de reconnaître chaque identité de chaque bille (elles sont chacune codées par une couleur spécifique). Une fois la bille reconnue par le Luminex®, cet appareil calcule le ratio de fluorescence émis par l’ADN fœtal et l’ADN des références. Ce calcul permet d’établir un profil avec deux courbes (une contre la référence mâle-bleue et une contre la référence femelle-rouge) (Figure 6) et permet ainsi de diagnostiquer des pertes et des gains du génome correspondant à ces régions ciblées particulièrement importantes en diagnostic prénatal.
Les pertes, monosomies ou délétions chromosomiques ont un profil décalé vers la gauche (Figure 7) et les gains, trisomies, ou duplications ont un profil décalé vers la droite (Figure 8). Lorsque la région est disomique (normale, en deux copies), le profil est au centre sur la ligne du chiffre 1 (ratio de fluorescence test/références).
La technique des BACs-on-BeadsTM est une technique fiable, rapide (rendu des résultats en 24-48 h), reproductible, permettant l’analyse de nombreux patients en même temps (environ 80) et ne nécessitant pas de culture cellulaire. Cependant, elle possède également quelques inconvénients, communs aux micropuces à ADN. En effet, elle ne permet pas de détecter les anomalies équilibrées de la structure des chromosomes, les polyploïdies (ex. : triploïdies) et les faibles taux de mosaïcisme. On sait également qu’à partir d’une contamination par au moins 70 % de cellules maternelles, on peut ignorer une anomalie par cette technique (Vialard et al., 2011).
34
Méthodes d’étude et d’analyse du génome
Figu
re 6
– P
rofil
BoBs
fém
inin
nor
mal


UE SpécifiqueMaïeutique
Sous la direction de> Rachel Lévy
> Michèle Rivière
Iconographie> Frédéric Bargy
> R. Lé
vy >
M. R
ivière
UE
Sp
écif
iqu
e M
aïeu
tiq
ue
Cet ouvrage est consacré à l’enseignement de la discipline maïeutique. Ils’adresse aux étudiants de la Première Année Commune des Études deSanté (PACES) qui optent pour cette Unité d’enseignement spécifique. Il constitue un ouvrage de référence rédigé par les praticiens hospitalo-universitaires et les sages-femmes impliqués au quotidien dans les domaines de la maïeutique et dans son enseignement.
Le contenu structuré et richement illustré reflète les qualités pédagogiques et l’expérience de l’enseignement de leurs rédacteurs. Cet ouvrage couvre les 4 modules correspondant au programme de l’UE spécifique maïeutique :- les méthodes d’étude et d’analyse du génome ;- l’anatomie de l’appareil reproducteur féminin et masculin ;- la différenciation sexuelle et l’histologie de l’appareil reproducteur féminin et masculin ;- l’unité foeto-placentaire.
ISBN : 978-2-8041-8284-7
SPEMAI
Pr Rachel Levy est professeur en biologie et médecine du développement et de la reproduction, responsable des enseignements d’embryologie (UE2) et demaïeutique (UE8 spécifique) en PACES à l’Université SMBH, Paris 13 (Bobigny). Michèle Rivière est sage-femme, directrice de l'école de sages-femmes St Antoine/AP-HP-Université Pierre et Marie Curie (Paris VI).
Conc
eptio
n gr
aphi
que
: Prim
o&Pr
imo
DANS LA MÊME COLLECTION
UE3 PACES, Aspects fonctionnels
UE3 PACES, Bases physiques des méthodes d’exploration
www.deboeck.com
• Ouvrage de cours à l’attentiondes étudiants en PACES et enécoles de maïeutique
• Qualité de l’équipe d’auteurs
• Iconographie en couleurs
LES
+
PUBLIC• Étudiants de Première Année
Commune des Études de Santé (PACES).• Étudiants en sciences médicales
(DFGSM 2 et 3 - Diplôme de FormationGénérale en Sciences Médicales).
• Étudiants en sciences maïeutiques(DFGSMa 2 et 3 - Diplôme de Formation Générale en Sciences Maïeutiques )
SPEMAI_deboeck 12/12/13 11:14 Page1