
REACTIVITE DU NITROMETHANE INDUITE PAR … · d’une pompe primaire (Alcatel 2004A). ... (Alcatel...
42
Université de Provence (Aix-Marseille I) Université de droit d’Economie et des Sciences (Aix-Marseille III) D.E.A. Chimie et Physico-Chimie Moléculaire Organique Option Spectrométrie et Physico-chimie Structurale (Stage effectué de Février à Juin 1999) REACTIVITE DU NITROMETHANE INDUITE PAR IRRADIATIONS SELECTIVES : DETERMINATION DES FORMES ACI ET ACI-ION Carine MANCA Laboratoire de Physique des Interactions Ioniques et Moléculaires UMR 6633 Equipe Spectrométrie et Dynamique Moléculaire Centre Saint Jérôme Maîtres de stage P. ROUBIN C. MARTIN
Transcript of REACTIVITE DU NITROMETHANE INDUITE PAR … · d’une pompe primaire (Alcatel 2004A). ... (Alcatel...

Université de Provence (Aix-Marseille I)
Université de droit d’Economie et des Sciences (Aix-Marseille III)
D.E.A. Chimie et Physico-Chimie Moléculaire Organique Option Spectrométrie et Physico-chimie Structurale
(Stage effectué de Février à Juin 1999)
REACTIVITE DU NITROMETHANE INDUITE
PAR IRRADIATIONS SELECTIVES :
DETERMINATION DES FORMES ACI ET ACI-ION
Carine MANCA
Laboratoire de Physique des Interactions Ioniques et Moléculaires UMR 6633 Equipe Spectrométrie et Dynamique Moléculaire
Centre Saint Jérôme
Maîtres de stage P. ROUBIN C. MARTIN

SOMMAIRE
LISTE DES MOLECULES ETUDIEES 1
1. INTRODUCTION 2
2. TECHNIQUES EXPERIMENTALES 3
2.1. Spectrométrie infrarouge à transformée de Fourier 3 2.1.1. Principe 3 2.1.2. Enregistrement des spectres 3
2.2. Technique cryogénique 4 2.2.1. Intérêt 4 2.2.2. Matériel utilisé 4 2.2.3. Réalisation d’une matrice cryogénique 5
2.3. Irradiation sélective à l’aide d’un laser accordable 1 2.3.1. Principe 1 2.3.2. Matériel utilisé 1 2.3.3. Réalisation d’une irradiation 3
3. ETUDE SPECTROSCOPIQUE DU NITROMETHANE 4
3.1. Etude quantique du nitrométhane 4 3.1.1. Géométrie de la molécule 4 3.1.2. Modes de vibration 5
3.2. Spectres infrarouges du nitrométhane 6 3.2.1. Spectres du nitrométhane en phase condensée 6 3.2.2. Spectres du nitrométhane isolé en matrices 8
3.3. Spectre UV du nitrométhane 12 3.3.1. Réalisation du spectre UV du nitrométhane 12 3.3.2. Interprétation des résultats 12
4. IRRADIATIONS SELECTIVES DU NITROMETHANE EN MATRICE CRYOGENIQUE 14
4.1. Les irradiations réalisées 14 4.1.1. Comparaison des irradiations 14 4.1.2. Identification des produits 15

4.2. Etude cinétique 17 4.2.1. Mécanisme réactionnel 17 4.2.2. Discussion 19
5. IRRADIATIONS SELECTIVES DU NITROMETHANE EN PHASE CONDENSEE 20
5.1. Résultats 20 5.1.1. A l’état liquide 20 5.1.2. A l’état solide 20
5.2. Identification des formes aci et aci ion nitrométhane 23 5.2.1. La forme acide du nitrométhane : l’aci nitrométhane 23 5.2.2. La forme anion du nitrométhane : l’aci ion nitrométhane 24 5.2.3. Discussion 26
6. CONCLUSION 28
ANNEXE I 30
ANNEXE II 32
BIBLIOGRAPHIE 34
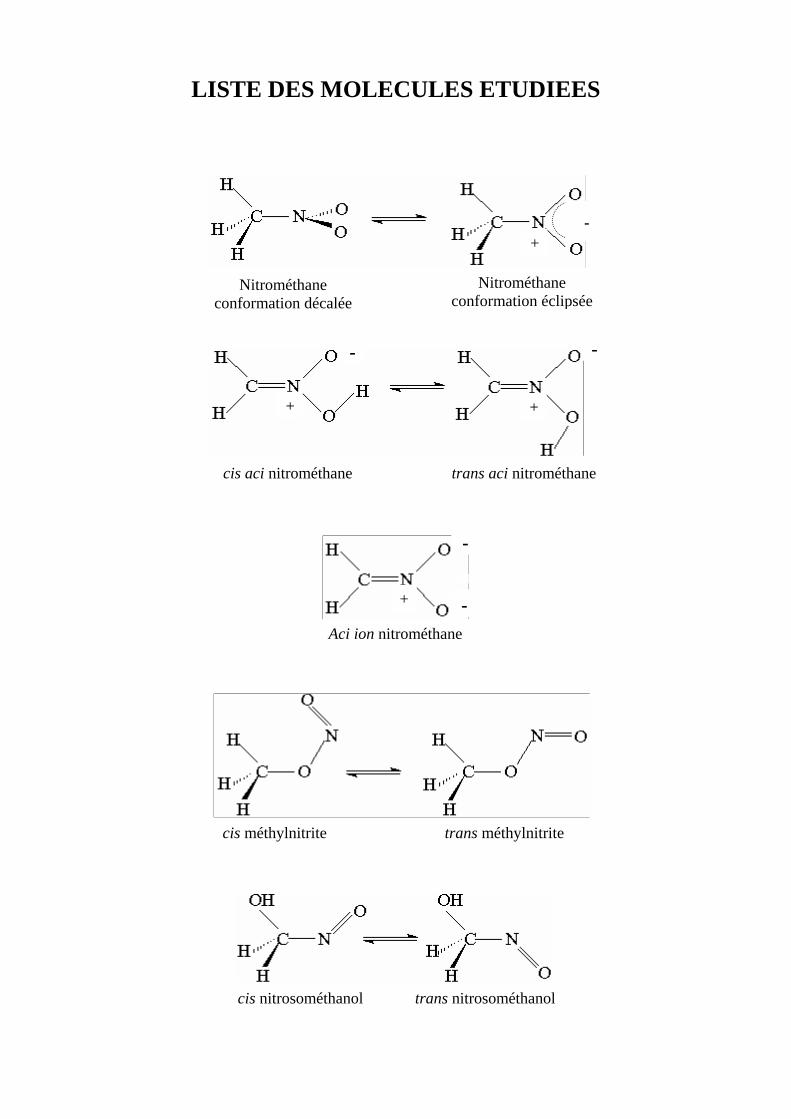
LISTE DES MOLECULES ETUDIEES
Nitromconformati
Nitrométhane conformation décalée
trans aci nicis aci nitrométhane
-
Aci ion nitrométhane
trans méthycis méthylnitrite
trans nitrosométhcis nitrosométhanol
+
éthane on éclips
trométha
lnitrite
anol
-
ée
+
+-
n
-
e
-
+
1. INTRODUCTION Le nitrométhane (CH3NO2) est la plus petite des nitroparaffines. A ce titre, cette molécule a fait l’objet de nombreuses études dans le but de comprendre le mécanisme de détonation1. Il apparaît que la forme anion du nitrométhane appelée aci ion joue un rôle important dans ce mécanisme. Sa formation pourrait s’expliquer par la perte d’un hydrogène pseudo-acide :
CH3NO2 CH2NO2- + H+
nitrométhane aci ion
Le pKa du nitrométhane a été évalué à température ambiante par spectrométrie UV à 10,1 dans une solution aqueuse basique2. Un autre chemin réactionnel a été envisagé en considérant la forme aci nitrométhane, tautomère du nitrométhane :
CH3NO2 CH2NO2H CH2NO2- + H+
nitrométhane aci aci ion A l’état naturel, la forme aci est négligeable par rapport à la forme nitrométhane (la constante d’équilibre a été évaluée à 1,1 10-7)3. La position de cet équilibre et la formation des molécules aci et aci ion peut être une très bonne sonde d’étude des sites basiques de surfaces d’oxyde qui sont jusqu’à ce jour très peu étudiés. Une étude théorique réalisée au laboratoire en vue de modéliser la réactivité du nitrométhane sur une surface de MgO(100)4 montre effectivement la formation des produits aci et aci ion. Si le composé aci ion a été observé par spectrométrie RMN en présence de MgO5 ou lors de l’irradiation UV du nitrométhane1(b), il n’existe actuellement aucun spectre infrarouge expérimental de l’aci nitrométhane. Afin de mieux connaître la réactivité du nitrométhane, nous avons réalisé une série de manipulations sur cette molécule avec, en particulier, l’irradiation en matrice cryogénique et en phases condensées (liquide et solide). Les produits formés ont été observés par spectrométrie infrarouge. Après avoir décrit l’ensemble du dispositif expérimental utilisé au cours du stage, nous rappèlerons l’étude à la fois quantique et spectroscopique menée sur le nitrométhane. Nous présenterons ensuite les résultats obtenus par irradiation du composé en matrice cryogénique que nous comparerons à ceux déjà publiés. Enfin, la dernière partie concernera l’irradiation du nitrométhane réalisée en phase condensée et l’étude théorique effectuée en vue d’identifier les produits obtenus.

2. TECHNIQUES EXPERIMENTALES L’ensemble des techniques utilisées (spectrométrie infrarouge, dispositif cryogénique et laser accordable) est représenté schématiquement sur les figures 1 (page 6) et 2 (page 7).
2.1. Spectrométrie infrarouge à transformée de Fourier
2.1.1. Principe
Les spectres sont enregistrés à l’aide d’un spectromètre infrarouge haute résolution à transformée de Fourier NICOLET 7199 fonctionnant en transmittance. Il est constitué d’une source Globar (un filament de carbure de silicium), d’un interféromètre de Michelson et d’un détecteur (Hg-Cd-Te). L’interféromètre de Michelson est composé d’un miroir fixe M1, d’un miroir mobile M2 et d’une lame séparatrice (S) en KBr-Ge, semi-transparente dans le domaine de l’infrarouge moyen (4000-400 cm-1) qui reçoit le faisceau lumineux issu de la source. La moitié de ce faisceau est réfléchie vers le miroir M1, l’autre est transmise vers le miroir M2 (cf. figure 1). Après réflexion sur les miroirs respectifs, les deux parties du faisceau initial se recombinent au niveau de la séparatrice et traversent l’échantillon contenu dans le cryostat. Elles présentent alors une différence de marche optique liée au déplacement du miroir mobile. Le signal, ainsi obtenu, est recueilli par le détecteur, puis amplifié pour être ensuite utilisé sur l’ordinateur qui pilote le spectromètre. Ce signal est l’interférogramme dont la transformée de Fourier représente la distribution spectrale de luminance de la source ainsi que l’absorption de l’échantillon.
2.1.2. Enregistrement des spectres
Chaque interférogramme enregistré subit une transformée de Fourier, pour obtenir un spectre en émittance I. Au préalable, on a enregistré un spectre du cryostat vide sans échantillon que l’on appellera référence I0. Le rapport de ces deux spectres permet d’obtenir un spectre en transmittance :
T=I/I0
Enfin, pour obtenir le spectre en absorbance A, on effectue la transformation : A=log10 (1/T)
Il faut noter que le nombre de balayages (scans) enregistrés a son importance : plus on enregistre de scans, plus on améliore le rapport signal/bruit sur le spectre après transformations. Chaque spectre est enregistré à une température fixée dans les conditions suivantes :
• Composé à l’état liquide (pur ou solution): Domaine spectral : 900 à 4000 cm-1
Résolution spectrale : 1 cm-1
Nombre de scans : 100

• Composé à l’état solide : Domaine spectral : 400 à 4000 cm-1
Résolution spectrale : 1 cm-1
Nombre de scans : 200
• Composé piégé en matrice : Domaine spectral : 400 à 4000 cm-1
Résolution spectrale : 0,12 cm-1
Nombre de scans : 200 Les limites du domaine spectral sont imposées par les matériaux (le porte-échantillon utilisé pour les matrices cryogéniques ainsi que pour le composé à l’état solide est conçu en KBr : il est transparent dans le domaine 400-4000 cm-1 ; de la même façon, la cellule en fluorure de calcium pour les composés à l’état liquide est transparente dans le domaine 900-4000 cm-1). Nous avons enfin, sur la plupart des spectres, soustrait le spectre correspondant au signal de la source traversant le spectromètre non purgé : il est en effet très difficile d’éliminer toute trace d’eau et de CO2 contenus en phase vapeur dans l’atmosphère au sein du spectromètre.
2.2. Technique cryogénique
2.2.1. Intérêt
L’étude à basse température (de l’ordre de 10 K), présente l’avantage de ralentir les vitesses de réaction. De ce fait, les intermédiaires réactionnels peuvent être observés et identifiés par spectroscopie (infrarouge dans notre cas). Les matrices cryogéniques sont généralement des matrices de gaz inerte tels que l’argon ou le xénon. La molécule piégée dans une telle matrice n’a que peu d’interaction avec les atomes de gaz rare qui l’entourent et donc les fréquences qui lui sont associées varient seulement de quelques cm-1 par rapport à l’état gazeux. De plus, dans une matrice, la rotation de la molécule est bloquée ; d’un point de vue spectroscopique, on observe donc des bandes d’absorption très fines et non un spectre de vibration-rotation. De ce fait, pour les réactions réalisées sur des molécules isolées au sein d’une matrice cryogénique, l’interprétation du spectre est considérablement simplifiée et nous permet d’effectuer un suivi cinétique qui sera confronté aux réactions effectuées en phase condensée (solution et solide).
2.2.2. Matériel utilisé
Le système cryogénique est constitué d’un cryogénérateur, d’un cryostat et d’un système de pompage, comme le montre la figure 1.

• Le cryogénérateur : cryodine 21SC CTI CRYOGENICS Il est composé d’un compresseur relié à une tête froide. Le refroidissement est obtenu par détente d’hélium dans un circuit fermé, ce qui permet une utilisation continue. La tête froide est constituée de deux blocs de refroidissement. Le bloc supérieur est maintenu à 77 K. Le bloc inférieur permet d’atteindre 10 K. Une résistance chauffante située entre le doigt froid et un bloc de cuivre a pour rôle de réguler la température. Une fenêtre en CsBr qui sert de porte-échantillon est fixée sur le bloc de cuivre. Le contrôle de la température est effectué à l’aide d’une diode préalablement étalonnée et située sur la partie supérieure du bloc de cuivre. Un tel système permet d’obtenir des températures variant entre 10 K et 300 K au niveau du porte-échantillon.
• Le cryostat : Il a été construit en fonction des manipulations envisagées dans les ateliers de Physique de l’Université de Provence6. Il se compose de deux parties : la partie supérieure est fixée au cryogénérateur. La partie inférieure est munie de fenêtres en KBr (matériau transparent dans le domaine du moyen infrarouge) et du système d’introduction de gaz : un système de pivot de cette partie du cryostat permet, tantôt de projeter le mélange gazeux à l’aide d’une buse orientée perpendiculairement à la fenêtre en CsBr, tantôt d’enregistrer un spectre infrarouge (le faisceau lumineux de la source arrive perpendiculairement à la fenêtre où a été effectué le dépôt).
• Le système de pompage : Le pompage a pour but d’éviter tout pont thermique qui empêcherait d’atteindre une température de l’ordre de 10 K au niveau du porte-échantillon. Le groupe de pompage est constitué d’un ensemble de deux pompes. Un vide de l’ordre de 10-2 torr est obtenu à l’aide d’une pompe primaire (Alcatel 2004A). La pompe secondaire à diffusion d’huile (Alcatel Cristal 63) permet d’atteindre un vide de l’ordre de 10-5 torr, puis 10-7 torr par cryopompage.
2.2.3. Réalisation d’une matrice cryogénique
Un mélange nitrométhane / gaz inerte (dans notre cas argon ou xénon) dans un rapport 1/500 est projeté lentement (40 mmol/heure) sur une surface préalablement refroidie (20 K pour l’argon, 30 K pour le xénon). Les quantités de gaz sont évaluées par mesure de pression dans une rampe en Inox dont le volume a été préalablement calibré (200 cm3). Cette rampe possède son propre groupe de pompage constitué d’une pompe primaire sèche à membrane (KNF série N813.4 ANE) et d’une pompe turbomoléculaire (Alcatel ATS 100). Une fois le dépôt terminé, un recuit est effectué avec une vitesse de chauffage de 0.5 K/mn (à 30 K pour la matrice d'argon ; à 35 K pour la matrice de xénon). Il permet la réorganisation des composants de la matrice et l’obtention de raies plus fines sur le spectre infrarouge. Pour les deux matrices, tous les spectres sont ensuite enregistrés à 10 K.

Interface spectromètre-ordinateur
Rampe de mélange
Compresseur à hélium
Détecteur
Cryostat
Tête froide
Source (S)
Séparatrice
Miroir fixe M1
Miroir mobile M2
Pompe primaire
Pompe turbomoléculaire
Gaz de mélange Echantill
Irradiation laser
Groupe de pompage relié à la rampe
Groupe de pompage relié
au cryostat
Banc optique
Cryogénérateur Interféromètre de Michelson
Pompe à diffusion
Figure 1 : Schéma du disposit
on
if expérimental

2.3. Irradiation sélective à l’aide d’un laser accordable
2.3.1. Principe
Le fonctionnement d’un laser (Light Amplification by Stimulated Emission of Radiation) repose sur le principe de l’émission stimulée. Des atomes à trois niveaux d’énergie (voire plus) tels que E1 < E2 < E3 constituent le milieu actif. Sous l’effet d’une excitation, les atomes dans l’état fondamental absorbent les photons et accèdent à l’état excité d’énergie E3. Les atomes relaxent spontanément vers le niveau métastable d’énergie E2, à durée de vie relativement longue. Les rapports de population sont alors inversés : c’est le phénomène de pompage. L’émission laser se produit par relaxation induite par un photon du niveau E2 au niveau E1. Les photons réémis ont les mêmes caractéristiques optiques que les photons incidents (même phase, même direction et même polarisation). Dans le cas d’un laser YAG Nd, le milieu actif est constitué d’ions Nd3+ enchâssés dans une matrice de grenat d’yttrium et d’aluminium (Y3Al5O12).
2.3.2. Matériel utilisé
Le laser accordable utilisé au laboratoire est composé d’un laser YAG Nd, d’un dispositif à OPO (Oscillateur Paramétrique Optique) et d’un doubleur (l’ensemble du matériel est schématisé sur la figure 2 et la figure 4 représente une photographie du dispositif).
Oscillateur Paramétrique
Optique VEGA 200
Cristaux doubleurs x2
Vers le cryostat
UVvisible355 nm
Laser YAG Nd 5023 DNS 10
Figure 2 : Schéma d’implémentation des composants du laser accordable
• Le laser YAG Nd : Thomson-CSF laser série 5023 DNS 10 La figure 3 représente le schéma du laser YAG Nd. Il est constitué d’une cavité résonante, d’une chambre d’amplification et d’un ensemble de miroirs dichroïques. Un premier barreau YAG est situé à l’intéreur de la cavité résonante (1) avec des lampes flash qui assurent le pompage. La cellule de Pockels (2) contrôle la durée de l’impulsion (de l’ordre de 10 ns). A la sortie de la cavité, une partie du faisceau est redirigée sans traitement supplémentaire vers un deuxième dispositif (OPO infrarouge). La deuxième partie du faisceau traverse la chambre d’amplification (3). Celle-ci contient un deuxième barreau YAG ainsi que de nouvelles lampes flash. Le faisceau émergeant dont la puissance a augmenté traverse ensuite successivement un doubleur (4), un tripleur (5) et des miroirs dichroïques (6) dont le rôle est de séparer le signal fondamental (1064 nm), le doublé (532 nm) et le triplé (355 nm) du laser YAG. Une puissance moyenne de 3900 mW est disponible à 355 nm.

Figure 3 : Schéma du laser YAG Nd
5
6
4
2 Miroir réfléchissant
3
1
532nm
355nm
1064nm
1064nm
1 – Cavité résonnante contenant le barreau Yag-Nd et les lampes flash ; 2 – Cellule de Pockels ; 3 – chambre d’amplification ; 4 – doubleur ; 5 – tripleur ;
6 - miroir dichroïque séparant 1064 nm et 355 nm ;
3 4
Figure 4 : Photographie du dispositif expérimental : le laser YAG, le laser OPO et le
compartiment de doubleurs

• Le dispositif à OPO : Thomson-CSF laser Véga 202 Le faisceau lumineux de longueur d’onde 355 nm (de pulsation ωP) issu du laser YAG Nd est introduit dans le cristal OPO qui le convertit par effet non linéaire en deux faisceaux : le signal (ωS) et le idler (ωI) tels que :
ωP = ωS + ωI
Le signal est dans le domaine 400 – 800 nm, alors que le idler est dans le domaine du proche infrarouge. L’accordabilité en longueur d’onde est obtenue par rotation du cristal OPO. La puissance disponible est de l’ordre de 200 mW.
• Le doubleur : Le rôle des cristaux du doubleur est de doubler par effet non linéaire la fréquence du rayonnement qui le traverse. On obtient ainsi un rayonnement UV dans la gammes 225-400 nm avec une puissance de l’ordre de 10 mW.
2.3.3. Réalisation d’une irradiation
Les irradiations sont effectuées à une température fixée. Le composé est irradié pendant une durée déterminée, puis on arrête l’irradiation et on enregistre un spectre infrarouge. Cette opération est répétée en faisant varier la durée pendant laquelle le composé est irradié. Ce processus nous permet d’effectuer un suivi cinétique de l’irradiation. Les conditions expérimentales notamment la durée de l’irradiation diffèrent selon l’état physique de l’échantillon : • Composé à l’état liquide : Longueur d’onde : 230 nm et 270 nm Puissance : Environ 20 mW Temps d’irradiation : De 1 à 30 minutes
• Composé à l’état solide : Longueur d’onde : 230 nm Puissance : Environ 10 mW Temps d’irradiation : De 1 à 180 minutes
• Composé piégé en matrices : Longueur d’onde : 230 nm et 270 nm (pour la matrice d’argon) Puissance : Environ 20 mW Temps d’irradiation : De 1 à 105 minutes

3. ETUDE SPECTROSCOPIQUE DU NITROMETHANE
3.1. Etude quantique du nitrométhane
3.1.1. Géométrie de la molécule
A l’état fondamental, le nitrométhane existe sous deux conformations stables. Elles correspondent à deux orientations différentes du groupement méthyl : dans la conformation décalée, un hydrogène du groupement méthyl est perpendiculaire au plan du groupement NO2 ; dans la conformation éclipsée, il est dans le plan du groupement NO2, comme le montre la figure 5.
Conformation éclipsée Conformation décalée
Figure 5 : Représentation des deux conformères du nitrométhane Les géométries des deux conformères ont été déterminées expérimentalement7 et théoriquement8, 9. Il apparaît que la conformation décalée est plus stable, bien que la différence d’énergie entre les deux conformations soit très faible : la barrière de rotation a été évaluée à 6 cal/mol. Nous avons réalisé une optimisation de la géométrie selon deux méthodes de calcul (les principes des calculs ab initio sont rappelés dans l’annexe 1) : la première série de calculs est effectuée avec la base d’orbitales gaussiennes 6-31G** et s’appuie sur une méthode de calcul de la fonctionnelle de la densité LSDA (Local Spin Density Approximation)a. La deuxième série de calculs est effectuée avec le logiciel GAMESS98 dans la base 6-31G**+ et utilise la théorie de perturbation Møller-Plesset au deuxième ordre (MP2). Nous avons trouvé la forme décalée légèrement plus stable de 10-5 Hartree ou 3.10-5 Hartree, soit 6 cal/mol ou 18 cal/mol selon la méthode de calcul utilisée (l’écart d’énergie trouvé par Lammertsma et Prasad10 est de 10 cal/mol). Les paramètres géométriques pour les deux conformations dans les deux méthodes de calculs sont reportés dans le tableau 1. Une comparaison avec les valeurs expérimentales7 nous montre que dans les deux cas, la molécule semble bien modélisée.
a Tous les calculs réalisés selon la méthode LSDA ont été effectués au sein du laboratoire par A. Allouche à l’aide du logiciel GAUSSIAN 94

Valeurs expérimentales LSDA/6-31G** MP2/6-31G**+ Etat gazeux11 Etat solide7(a) Décalée Eclipsée Décalée Eclipsée
C-N 1,489 1,481 1,482 1,482 1,489 1,489 N-O1 1,224 1,223 1,196 1,197 1,243 1,243 N-O2 1,224 1,227 1,196 1,195 1,243 1,244 C-H1 1,089 1,093 1,081 1,076 1,086 1,083 C-H2 1,089 1,092 1,077 1,080 1,083 1,085 C-H3 1,089 1,092 1,077 1,080 1,083 1,085 ONO 125,3 123,7 125,6 125,5 125,4 125,5 CNO1 117,3 118,2 117,1 116,6 117,1 116,6 CNO2 117,3 118,0 117,2 117,8 117,4 117,9 NCH1 107,5 106,4 106,4 108,3 106,9 108,0 NCH2 107,5 108,1 107,8 106,9 107,6 107,1 NCH3 107,5 108,1 107,8 106,9 107,7 107,1
Tableau 1 : Paramètres géométriques des deux conformations du nitrométhane évalués par calculs ab initio
(longueurs en Å ; angles en degrés)
3.1.2. Modes de vibration
Le groupe de symétrie de la molécule en conformation éclipsée ou décalée est le groupe Cs. D’après les règles de sélection, tous les modes sont actifs en spectrométrie infrarouge dans ce groupe de symétrie. La molécule possède 7 atomes, donc 15 modes propres de vibration qui se décomposent en 9 modes de symétrie A’ et 6 modes de symétrie A’’. Les calculs ab initio à partir de la géométrie optimisée de la molécule nous ont permis d’obtenir les fréquences des différents modes de vibration pour les deux conformations envisagées. Ces calculs ont été confrontés aux valeurs expérimentales observées en phase gazeuse par D. Gorse et al.11 dans le tableau 2. Le mode de torsion de la liaison CN dont la fréquence est inférieure à 400 cm-1 n’est pas indiqué dans ce tableau. Les modes propres de vibration sont représentés dans l’annexe 2. On constate que les fréquences des conformations éclipsée et décalée sont très proches (pour une méthode de calcul donnée, l’écart de fréquences ne dépasse pas 6 cm-1). L’utilisation de la théorie de la fonctionnelle de la densité permet de bien modéliser les basses fréquences (en dessous de 1000 cm-1). Le traitement avec des orbitales diffuses permet de mieux reproduire le système délocalisé au niveau du groupement NO2 et ainsi de diminuer l’écart entre la
fréquence théorique et la fréquence expérimentale du mode le plus intense, l’élongation antisymétrique du groupement NO2.
3.2. Spectres infrarouges du nitrométhane
Nous avons réalisé le spectre infrarouge du nitrométhane sous différentes conditions dans le but de caractériser tous ses modes de vibration.
3.2.1. Spectres du nitrométhane en phase condensée
• A l’état liquide :
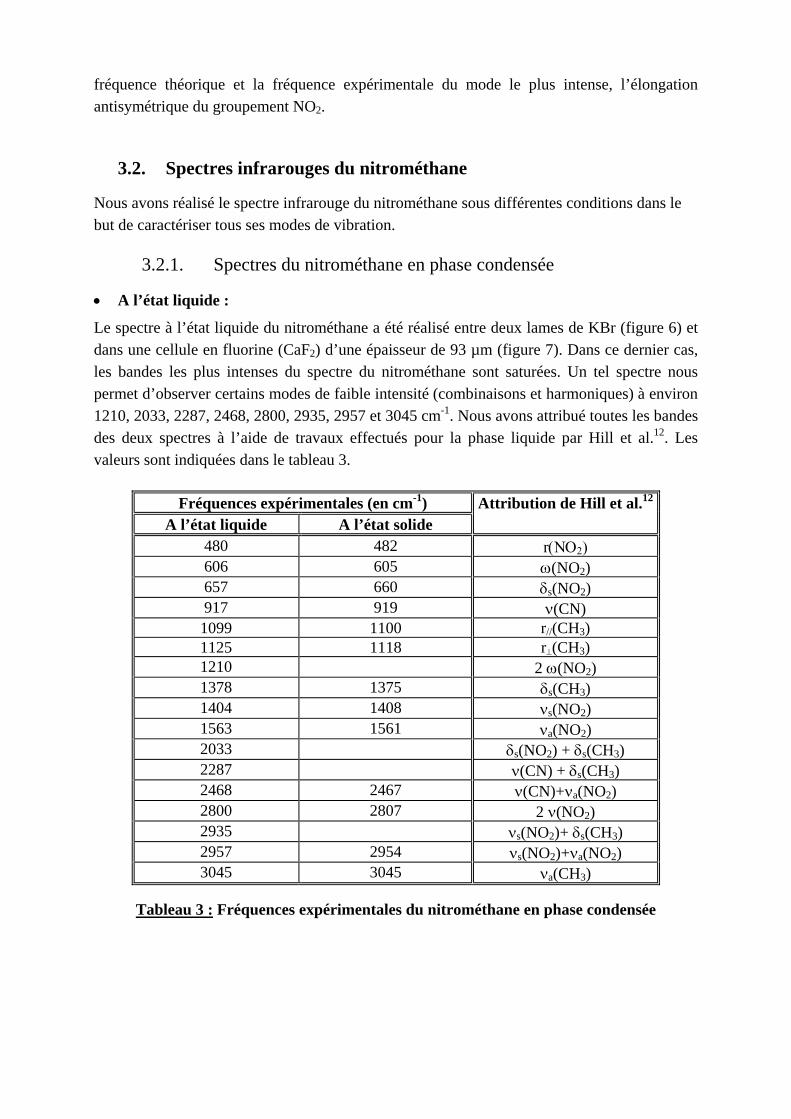
Le spectre à l’état liquide du nitrométhane a été réalisé entre deux lames de KBr (figure 6) et dans une cellule en fluorine (CaF2) d’une épaisseur de 93 µm (figure 7). Dans ce dernier cas, les bandes les plus intenses du spectre du nitrométhane sont saturées. Un tel spectre nous permet d’observer certains modes de faible intensité (combinaisons et harmoniques) à environ 1210, 2033, 2287, 2468, 2800, 2935, 2957 et 3045 cm-1. Nous avons attribué toutes les bandes des deux spectres à l’aide de travaux effectués pour la phase liquide par Hill et al.12. Les valeurs sont indiquées dans le tableau 3.
Fréquences expérimentales (en cm-1) A l’état liquide A l’état solide
Attribution de Hill et al.12
480 482 r(ΝΟ2) 606 605 ω(NO2) 657 660 δs(NO2) 917 919 ν(CN) 1099 1100 r//(CH3) 1125 1118 r⊥(CH3) 1210 2 ω(NO2) 1378 1375 δs(CH3) 1404 1408 νs(NO2) 1563 1561 νa(NO2) 2033 δs(NO2) + δs(CH3) 2287 ν(CN) + δs(CH3) 2468 2467 ν(CN)+νa(NO2) 2800 2807 2 ν(NO2) 2935 νs(NO2)+ δs(CH3) 2957 2954 νs(NO2)+νa(NO2) 3045 3045 νa(CH3)
Tableau 3 : Fréquences expérimentales du nitrométhane en phase condensée

3000 2750 2500 2250 2000 1750 1500 1250 1000 750 500
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Abs
orba
nce
Nombres d'onde (en cm-1)
δs(CH3)
r//(CH3) ν(CN)
δ(NO2)
νa(NO2)
Figure 6: Spectre infrarouge du nitrométhane à l’état liquide enregistré à température ambiante entre deux lames de KBr (Résolution : 1 cm-1)
3400 3200 3000 2800 2600 2400 2200 2000 1800 16000,0
0,5
1,0
1,5
2,0
2,5
3,0
Nombres d'onde (cm-1)
ν(CN) saturé
νs(CH3)
νa(CH3)
2νs(NO2)νa(NO2) + ν(CN)
δs(CH3) + ν(CN)
Absorbance
Figure 7:Figure 7: Spectre infrarouge du nitrométhane à l’état liquide enregistré à température ambiante dans la cellule en CaF (Résolution : 1 cm ) 2
-1

• A l’état solide :
Le solide est obtenu en projetant du nitrométhane gazeux sur le porte-échantillon en CsBr refroidi à 80 K. Il est ensuite lentement réchauffé (0,5 K/mn) jusqu’à 100 K pour favoriser une organisation cristalline du nitrométhane : les bandes du spectre se structurent et s’affinent légèrement. Leur largeur varie de 5 à 25 cm-1 (cf. figure 8) comme dans le cas du spectre à l’état liquide. Tous les modes ont été attribués par comparaison avec les modes observés en phase liquide (les valeurs diffèrent très peu, de l’ordre de 1 à 7 cm-1) et sont référencées dans la colonne 2 du tableau 3.
1600 1400 1200 1000 800 600
0,1
0,2
0,3
0,4
0,5
0,6
0,7
Abs
orba
nce
Nombres d'onde (en cm-1)
ω(NO2) r(NO2)
δ(NO2)
ν (CN)
r(CH3)
δs(CH3)
νs(NO2), δa(CH3)
νa(NO2)
Figure 8 : Spectre du nitrométhane solide enregistré à 100 K (Résolution : 1 cm-1)
3.2.2. Spectres du nitrométhane isolé en matrices
Nous avons réalisé deux matrices différentes de nitrométhane en changeant le gaz matriciel : argon et xénon dans le but d’observer des effets de sites différents. Le spectre de la matrice d’argon est reproduit dans la figure 9. L’argon et le xénon cristallisent selon une maille cubique à faces centrées. Cependant le paramètre cristallin n’est pas le même : Il est plus grand dans le cas de xénon (5,31 Å pour l’argon et 6,12 Å pour le xénon à 10 K)13. L’organisation du nitrométhane au sein de la cage ne sera pas la même pour les deux matrices. Le tableau 4 présente la liste des différentes fréquences observées ainsi que leur attribution. Pour de nombreux modes, on observe une structure de multiplets comme le montrent les figures 10 et 11 (pour un mode donné, la raie la plus intense est indiquée en gras dans le tableau 4).
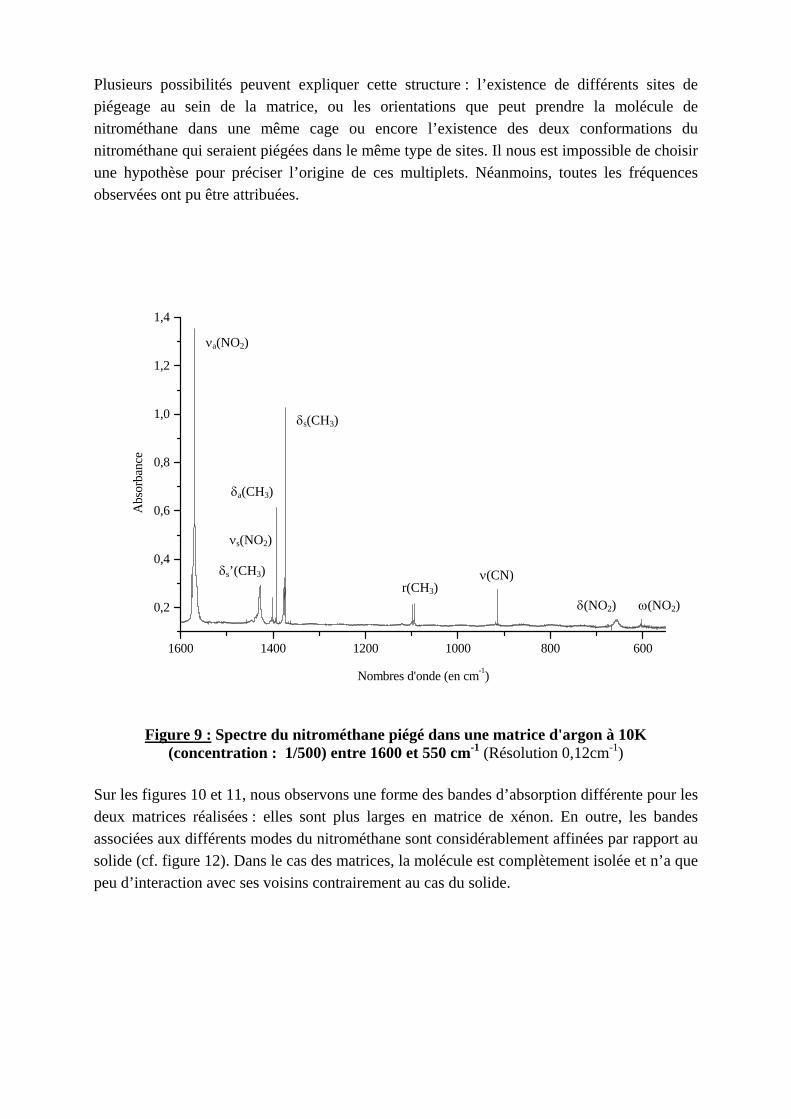
Plusieurs possibilités peuvent expliquer cette structure : l’existence de différents sites de piégeage au sein de la matrice, ou les orientations que peut prendre la molécule de nitrométhane dans une même cage ou encore l’existence des deux conformations du nitrométhane qui seraient piégées dans le même type de sites. Il nous est impossible de choisir une hypothèse pour préciser l’origine de ces multiplets. Néanmoins, toutes les fréquences observées ont pu être attribuées.
1600 1400 1200 1000 800 600
0,2
0,4
0,6
0,8
1,0
1,2
1,4
Abs
orba
nce
Nombres d'onde (en cm-1)
νs(NO2)
ω(NO2) δ(NO2)
ν(CN) r(CH3)
δs’(CH3)
δa(CH3)
δs(CH3)
νa(NO2)
Figure 9 : Spectre du nitrométhane piégé dans une matrice d'argon à 10K (concentration : 1/500) entre 1600 et 550 cm-1 (Résolution 0,12cm-1)
Sur les figures 10 et 11, nous observons une forme des bandes d’absorption différente pour les deux matrices réalisées : elles sont plus larges en matrice de xénon. En outre, les bandes associées aux différents modes du nitrométhane sont considérablement affinées par rapport au solide (cf. figure 12). Dans le cas des matrices, la molécule est complètement isolée et n’a que peu d’interaction avec ses voisins contrairement au cas du solide.
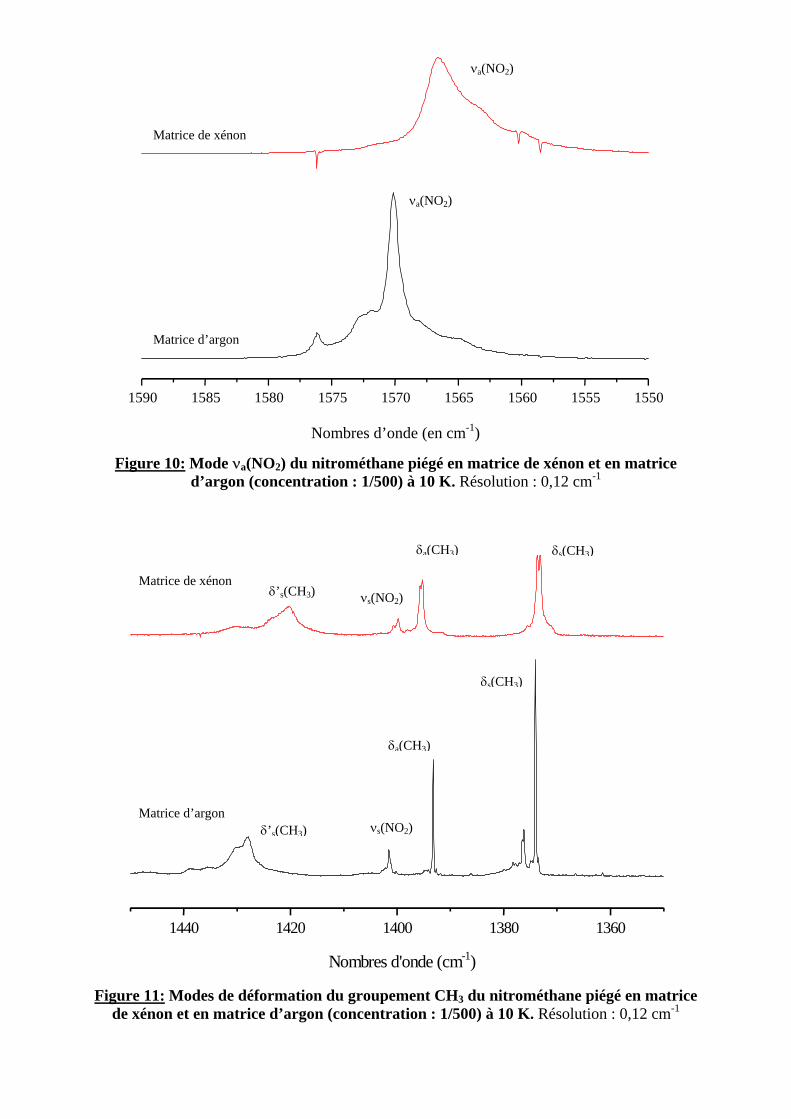
Figure 10: Mode νa(NO2) du nitrométhane piégé en matrice de xénon et en matrice d’argon (concentration : 1/500) à 10 K. Résolution : 0,12 cm-1
νa(NO2)
νa(NO2)
Matrice de xénon
1580 1575 1570 1565 1560 1555 1550
Nombres d’onde (en cm-1)
1585 1590
Matrice d’argon
1440 1420 1400 1380 1360
Nombres d'onde (cm-1)
νs(NO2)
νs(NO2)
δ’s(CH3)
δs(CH3)
δs(CH3)
δa(CH3)
δa(CH3)
δ’s(CH3) Matrice de xénon
Matrice d’argon
Figure 11: Modes de déformation du groupement CH3 du nitrométhane piégé en matrice de xénon et en matrice d’argon (concentration : 1/500) à 10 K. Résolution : 0,12 cm-1

Fréquences expérimentales (en cm-1) Attribution
Matrice d’Argon Matrice de Xénon 475,6 r(ΝΟ2) 603,5 602,0 ω(NO2) 657,2 658,3 δ(NO2)
913,9 918,1 918,2 ν(CN) 1093,8 1098,8 1097,2 1100,1 r//(CH3)
1122,9 1122,5 r⊥(CH3) 1239,2 2 ω(NO2)
1374,2 1376,2 1373,0 1373,4 1375,4 δs(CH3) 1393,3 1394,7 1395,1 1395,4 δa(CH3) 1401,4 1405,3 1399,7 1400,4 νs(NO2) 1428,0 1435,7 1420,1 1429,6 δs’(CH3) 1570,1 1576,0 1566,5 νa(NO2)
2463,1 2470,9 2477,5 2465,2 ν(CN)+νa(NO2) 2936,3 2932,3 δs(CH3)+νa(NO2) 2956,3 2954,3 νs(NO2)+νa(NO2) 2985,8 νs(CH3)
Tableau 4 : Fréquences expérimentales du nitrométhane piégé en matrice d’argon et de xénon (concentration : 1/500)
1600 1550 1500 1450 1400 1350
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
2,0
2,2
2,4
(a)
(b)
(c)
(a) Matrice d'argon (b) Matrice de xénon (c) Solide
Abs
orba
nce
Nombres d'onde (en cm-1)
Figure 12 : Comparaison des spectres infrarouges du nitrométhane en matrices d’argon et de xénon et à l’état solide entre 1600 et 1350 cm-1Résolution : 0,12 cm-1 en matrice ; 1
cm-1 à l’état solide

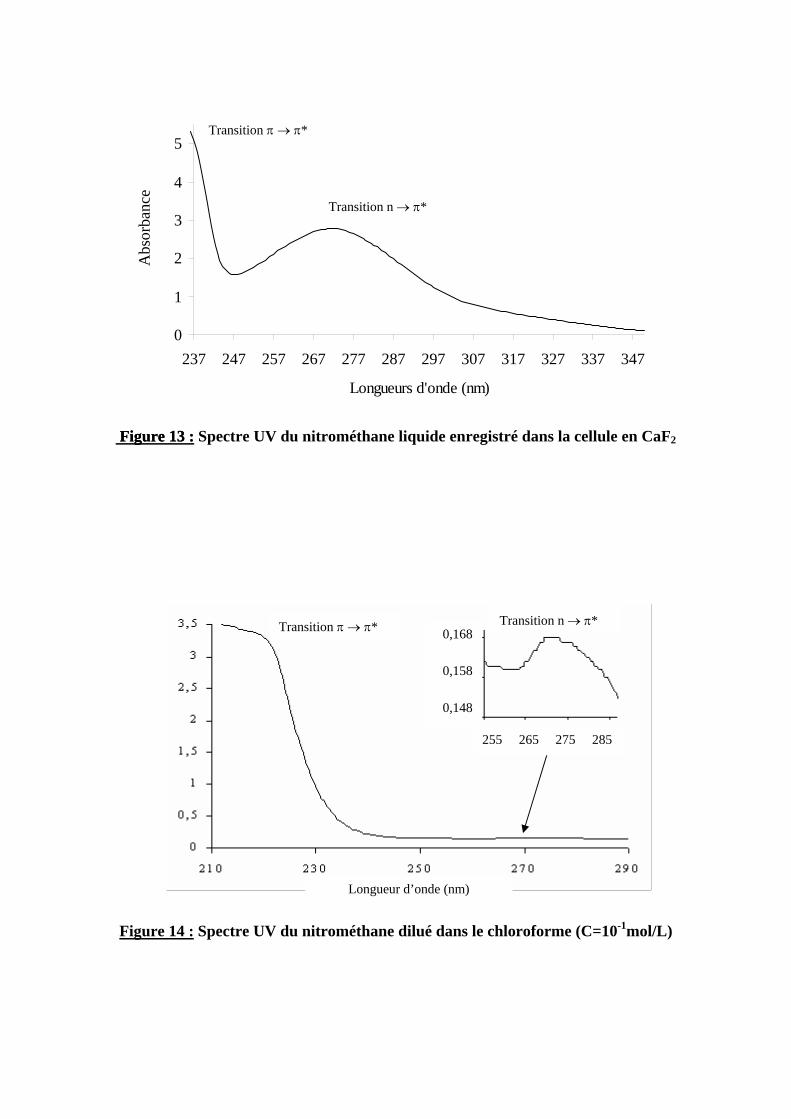
3.3. Spectre UV du nitrométhane
3.3.1. Réalisation du spectre UV du nitrométhane
Nous avons enregistré le spectre UV du nitrométhane à l’état liquide en vue de déterminer les longueurs d’onde à sélectionner pour l’irradiation et pour vérifier que le solvant ne modifie pas ou peu le spectre du nitrométhane. Le choix solvants a été dicté par deux conditions : d’une part, le nitrométhane devait être soluble ; d’autre part, ces solvants doivent perturber le moins possible le spectre infrarouge dans le but d’observer les bandes des produits formés après irradiation. Nous avons utilisé le chloroforme et le tétrachlorure de carbone. Les spectres ont été réalisés avec la cellule en fluorine d’épaisseur 93 µm déjà utilisée pour les spectres infrarouge. Les figures 13 et 14 représentent les spectres UV du nitrométhane pur et dilué dans le chloroforme.
3.3.2. Interprétation des résultats
Dans tous les cas, les différents spectres UV réalisés présentent deux bandes d’absorption d’intensités très différentes. La bande la plus intense aux environs de 200 nm correspond à une transition de type π → π* du groupement NO2. Cette bande a été observée à l’état gazeux à 198 nm et le coefficient d’absorption qui lui est associé a été évalué à 5000 l.cm-1.mol-1 par Nagakura3. La deuxième bande, aux environ de 270 nm correspond à une transition n → π* du groupement NO2. Cette transition étant théoriquement interdite, elle est beaucoup moins intense que la précédente. Nous n’avons pas constaté sur cette bande de modification importante due à un effet de solvant : Ungnade et al.14 ont mesuré la longueur d’onde associée à cette bande à 273 nm, dans le cas du nitrométhane pur ; nous avons observé le maximum de cette bande à 272 nm lorsque le nitrométhane est dilué dans le chloroforme. Ces résultats nous ont permis de choisir des longueurs d’onde précises pour l’irradiation : 230 nm (nous avons choisi d’exciter le « pied » de la bande la plus intense pour éviter de dissocier intégralement le produit dès la première irradiation) et à 270 nm pour comparer.
0
1
2
3
4
5
237 247 257 267 277 287 297 307 317 327 337 347
Longueurs d'onde (nm)
Abs
orba
nce
Transition π → π*
Transition n → π*
Figure 13 : Figure 13 : Spectre UV du nitrométhane liquide enregistré dans la cellule en CaF2
Transition n → π* 0,168
0,158
0,148
255 265 275 285
Transition π → π*
Longueur d’onde (nm)
Figure 14 : Spectre UV du nitrométhane dilué dans le chloroforme (C=10-1mol/L)

4. IRRADIATIONS SELECTIVES DU NITROMETHANE EN MATRICE CRYOGENIQUE
Les matrices réalisées dans la partie 3 dans le but d’identifier les modes de vibration du nitrométhane ont ensuite été irradiées à l’aide du laser accordable.
4.1. Les irradiations réalisées
4.1.1. Comparaison des irradiations
Nous avons réalisé trois expériences différentes d’irradiation du nitrométhane isolé en matrices : deux irradiations pour la matrice d’argon (à 230 nm et 270 nm) et une irradiation pour la matrice de xénon (à 230 nm). Les deux irradiations en matrice d’argon permettent d’étudier la sélectivité de l’irradiation. Les résultats sont tout à fait comparables qualitativement : les mêmes bandes sont observées dans les deux cas. Cependant, il semble que l’irradiation à 230 nm soit plus rapide. Nous avons décidé d’irradier à la même longueur d’onde la matrice de xénon, réalisée dans des conditions similaires. Les résultats de cette troisième irradiation sont comparables à ceux obtenus en matrice d’argon à 230 nm. Néanmoins, l’irradiation du nitrométhane semble plus rapide et plus complète : la figure 15 représente la diminution de l’absorbance de la bande la plus intense du nitrométhane. Nous avons constaté que dans le cas de la matrice de xénon, l’absorbance de cette bande s’annule au bout d’une heure 45 minutes d’irradiation à 230 nm (la puissance du laser étant d’environ 15 mW dans les deux cas), mais pas dans le cas de la matrice d’argon.

0 20 40 60 80 100
0,0
0,2
0,4
0,6
0,8
1,0
En matrice d'argon En matrice de xénon
Abs
orba
nce
inté
gré
norm
alisé
e à
1 du
pic
le p
lus i
nten
se
Durée de l'irradiation (en minutes)
Figure 15 : Courbe de décroissance du nitrométhane piégé en matrice d’argon et de xénon en fonction du temps d’irradiation à 230 nm
4.1.2. Identification des produits
Les résultats étant identiques pour les deux matrices, nous ne décrirons en détail qu’une seule des deux expériences, celle de la matrice d’argon. Dès la première minute d'irradiation, on observe sur le spectre infrarouge, une diminution des bandes attribuées au nitrométhane et l'émergence de nouvelles bandes. Les premières observées, à 562, 805-807, 1666-1668 cm-1
sont attribuées au méthyl nitrite en conformation trans15 et cis (625 et 838 cm-1). Ces deux conformations commencent à disparaître au bout de 15 minutes d'irradiation. On observe alors la formation d’un complexe entre le formaldéhyde et HNO associés par liaison hydrogène (bandes à 1174, 1733, 2800, 2812, 2866 cm-1), puis dans une troisième étape, les isomères conformationnels cis et trans du nitrosométhanol16 : les bandes correspondantes sont observées aux environs de 1110, 1126, 1356, 3487, 3640 cm-1. A la fin de l’irradiation, des produits secondaires se forment : l’eau (1596 et 3712 cm-1), le monoxyde de carbone (2145 cm-1) et le monoxyde d’azote (1874 cm-1). Les figures 16 et 17 montrent l’évolution du spectre du nitrométhane en matrice d’argon et de xénon respectivement au cours de l’irradiation.
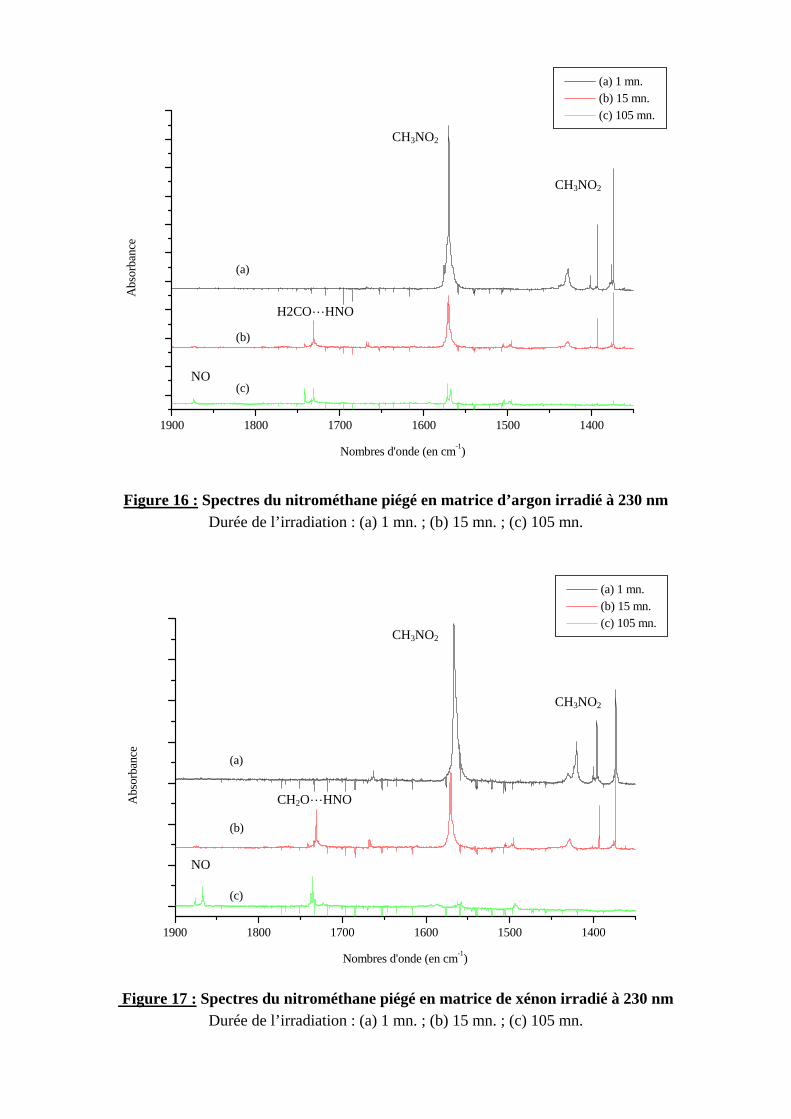
1900 1800 1700 1600 1500 1400
(a)
(b)
(c)
(a) 1 mn. (b) 15 mn. (c) 105 mn.
Abs
orba
nce
Nombres d'onde (en cm-1)
NO
CH3NO2
CH3NO2
H2COLHNO
Figure 16 : Spectres du nitrométhane piégé en matrice d’argon irradié à 230 nm Durée de l’irradiation : (a) 1 mn. ; (b) 15 mn. ; (c) 105 mn.
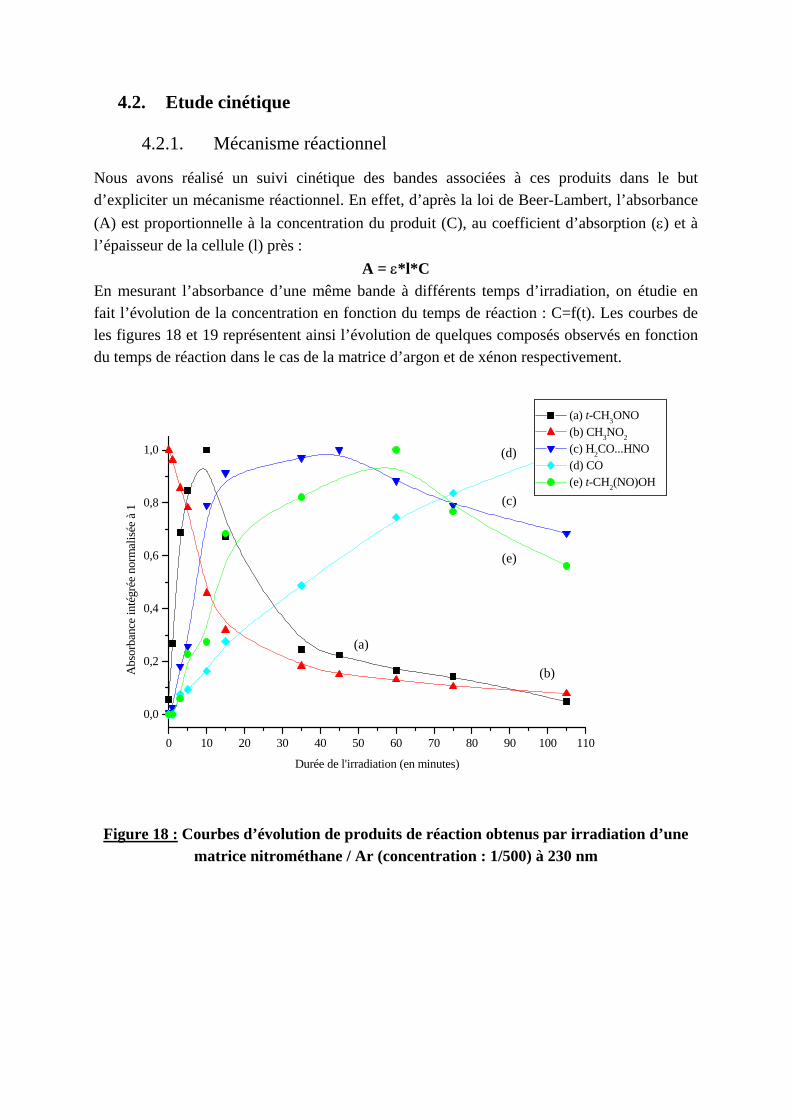
1900 1800 1700 1600 1500 1400
(a)
(b)
(c)
(a) 1 mn. (b) 15 mn. (c) 105 mn.
Abs
orba
nce
Nombres d'onde (en cm-1)
NO
CH2OLHNO
CH3NO2
CH3NO2
Figure 17 : Spectres du nitrométhane piégé en matrice de xénon irradié à 230 nm Durée de l’irradiation : (a) 1 mn. ; (b) 15 mn. ; (c) 105 mn.
4.2. Etude cinétique
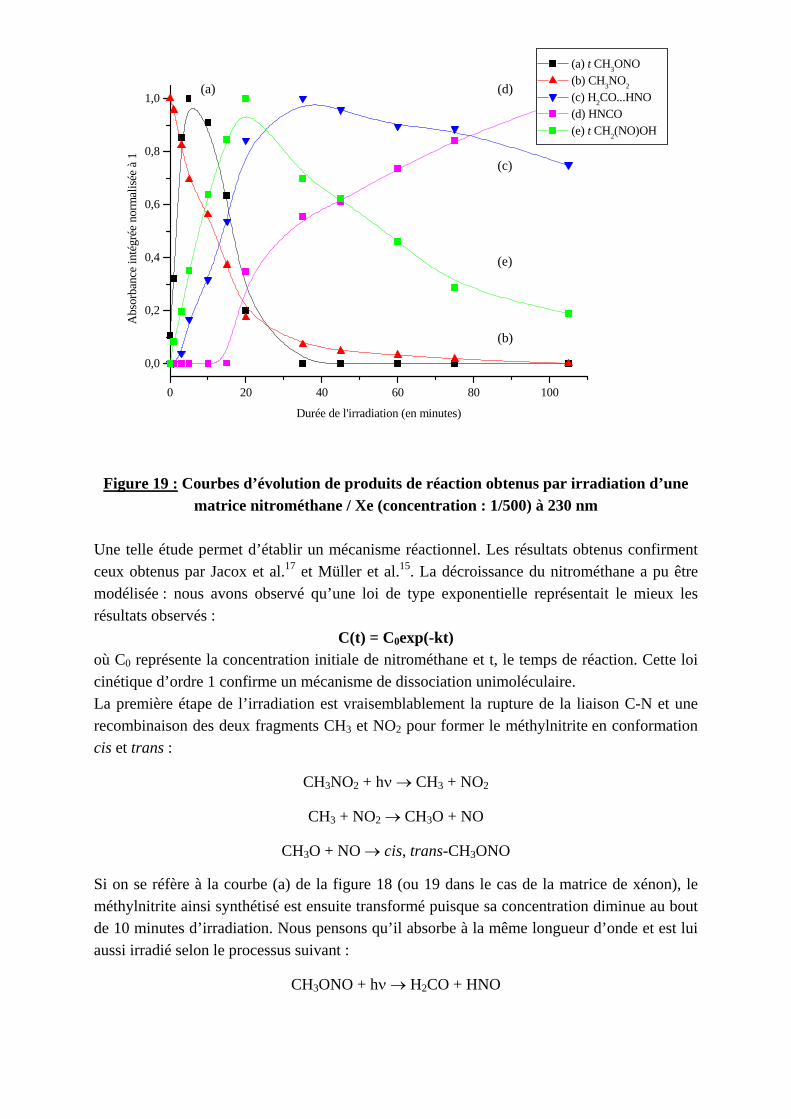
4.2.1. Mécanisme réactionnel
Nous avons réalisé un suivi cinétique des bandes associées à ces produits dans le but d’expliciter un mécanisme réactionnel. En effet, d’après la loi de Beer-Lambert, l’absorbance (A) est proportionnelle à la concentration du produit (C), au coefficient d’absorption (ε) et à l’épaisseur de la cellule (l) près :
A = ε*l*C En mesurant l’absorbance d’une même bande à différents temps d’irradiation, on étudie en fait l’évolution de la concentration en fonction du temps de réaction : C=f(t). Les courbes de les figures 18 et 19 représentent ainsi l’évolution de quelques composés observés en fonction du temps de réaction dans le cas de la matrice d’argon et de xénon respectivement.
0 10 20 30 40 50 60 70 80 90 100 110
0,0
0,2
0,4
0,6
0,8
1,0
(a) t-CH3ONO (b) CH3NO2
(c) H2CO...HNO (d) CO (e) t-CH2(NO)OH
Abs
orba
nce
inté
grée
nor
mal
isée
à 1
Durée de l'irradiation (en minutes)
(c)
(d)
(b)
(e)
(a)
Figure 18 : Courbes d’évolution de produits de réaction obtenus par irradiation d’une
matrice nitrométhane / Ar (concentration : 1/500) à 230 nm
0 20 40 60 80 100
0,0
0,2
0,4
0,6
0,8
1,0
(a) t CH3ONO (b) CH3NO2
(c) H2CO...HNO (d) HNCO (e) t CH2(NO)OH
Abs
orba
nce
inté
grée
nor
mal
isée
à 1
Durée de l'irradiation (en minutes)
(d)
(c)
(e)
(b)
(a)
Figure 19 : Courbes d’évolution de produits de réaction obtenus par irradiation d’une
matrice nitrométhane / Xe (concentration : 1/500) à 230 nm Une telle étude permet d’établir un mécanisme réactionnel. Les résultats obtenus confirment ceux obtenus par Jacox et al.17 et Müller et al.15. La décroissance du nitrométhane a pu être modélisée : nous avons observé qu’une loi de type exponentielle représentait le mieux les résultats observés :
C(t) = C0exp(-kt) où C0 représente la concentration initiale de nitrométhane et t, le temps de réaction. Cette loi cinétique d’ordre 1 confirme un mécanisme de dissociation unimoléculaire. La première étape de l’irradiation est vraisemblablement la rupture de la liaison C-N et une recombinaison des deux fragments CH3 et NO2 pour former le méthylnitrite en conformation cis et trans :
CH3NO2 + hν → CH3 + NO2
CH3 + NO2 → CH3O + NO
CH3O + NO → cis, trans-CH3ONO
Si on se réfère à la courbe (a) de la figure 18 (ou 19 dans le cas de la matrice de xénon), le méthylnitrite ainsi synthétisé est ensuite transformé puisque sa concentration diminue au bout de 10 minutes d’irradiation. Nous pensons qu’il absorbe à la même longueur d’onde et est lui aussi irradié selon le processus suivant :
CH3ONO + hν → H2CO + HNO

Ces deux composés forment un complexe associé par liaison hydrogène : H2COLHNO. Ce processus a été observé par Jacox18 et Müller et al.15 en irradiant le méthylnitrite à des longueurs d’onde différentes (respectivement 345 nm et 365 nm). Le complexe disparaît à son tour pour former le nitrosométhanol selon la réaction :
H2COLHNO + hν → CH2(NO)OH
Müller et al.15 ont suggéré que cette réaction se produisait par excitation photochimique du formaldéhyde (la bande d’absorption de ce composé s’étend de 353 à 230 nm). Cette hypothèse semble être confirmée par les résultats que nous avons obtenus. En fin d’irradiation, nous avons observé la formation de petites molécules de type CO (comme le montre la courbe (d) de la figure 18) et NO qui peut s’expliquer par la décomposition de H2CO et HNO respectivement. La molécule HNCO est également détectée (une bande caractérise cette molécule en matrice : 2230 cm-1). Sa formation peut être envisagée par perte d’une molécule d’eau du nitrosométhanol : le composé HCNO formé se réarrange alors en HNCO19 :
CH2(NO)OH → HCNO + H2O → HNCO + H2O
4.2.2. Discussion
Les résultats obtenus par irradiations sélectives du nitrométhane en matrices de gaz rare sont en accord avec ceux déjà publiés. Néanmoins, l’utilisation du laser accordable nous a permis de ne sélectionner qu’une seule longueur d’onde. Les hypothèses soulevées par ces résultats peuvent être confirmées ou infirmées par des travaux supplémentaires en irradiant à une longueur d’onde différente. De plus, le suivi cinétique nous a permis d’effectuer une évaluation plus précise de l’évolution des composés. Il reste cependant des bandes d’intensité beaucoup moins importante ( de l’ordre de 0,01 unité d’absorbance) qui n’ont pas été attribuées jusqu’à présent (820, 1028, 1041, 1180, 1228, 1765 cm-1). Leur intensité trop faible nous a en effet empêchés d’effectuer un suivi de l’absorbance en fonction de la durée de l’irradiation et d’envisager un mécanisme réactionnel.

5. IRRADIATIONS SELECTIVES DU NITROMETHANE EN PHASE CONDENSEE
A titre de comparaison avec l’irradiation de la molécule isolée en matrice cryogénique, nous avons réalisé des manipulations en phase condensée (c’est-à-dire, à l’état liquide et à l’état solide) dans le but d’évaluer l’effet de la proximité d’autres molécules de nitrométhane. Cette étude constitue la partie originale de notre travail. qui n’a pas pu être confronté à d’autres résultats. L’identification des produits s’est donc réalisée par comparaison avec des calculs ab initio.
5.1. Résultats
5.1.1. A l’état liquide
L’irradiation du nitrométhane à l’état liquide constitue une étude préliminaire. Comme dans le cas des matrices, nous avons réalisé deux expériences différentes en irradiant les deux bandes d’absorption caractéristiques du nitrométhane. Nous avons choisi les mêmes longueurs d’onde que celles utilisées pour les matrices (230 nm et 270 nm). Aucune différence remarquable n’a été observée entre ces deux irradiations. Nous avons donc décidé pour la suite d’irradier le composé uniquement à 230 nm. Nous avons également réalisé la même manipulation avec deux solutions de nitrométhane de concentration 10-1 mol/l (les solvants choisis sont ceux utilisés pour les spectres UV : chloroforme et tétrachlorure de carbone). A la fin de l’irradiation, dans les trois cas, on observe une diminution des bandes attribuées au nitrométhane et la croissance de nouvelles bandes (993, 1118, 1160, 1215, 1290, 1725 et 2230 cm-1). Dans le cas du nitrométhane pur, on observe trois autres bandes non détectées dans les solutions aux environs de 978, 1030 et 1640 cm-1 qui apparemment appartiennent au même composé. Nous pouvons alors émettre deux hypothèses : soit le produit formé uniquement dans le nitrométhane n’est pas soluble dans les solvants spectroscopiques utilisés soit il existe un mécanisme bimoléculaire qui permettrait de former ce produit. Pour répondre à cette question, nous avons envisagé d’irradier le nitrométhane pur à l’état solide.
5.1.2. A l’état solide
Une telle manipulation présente deux intérêts : d’une part, nous nous retrouvons dans des conditions très voisines de celles du nitrométhane liquide pur ; d’autre part, la température est plus faible (100 K). Donc la réaction est ralentie et nous pouvons envisager d’effectuer un suivi cinétique comme dans le cas des matrices.
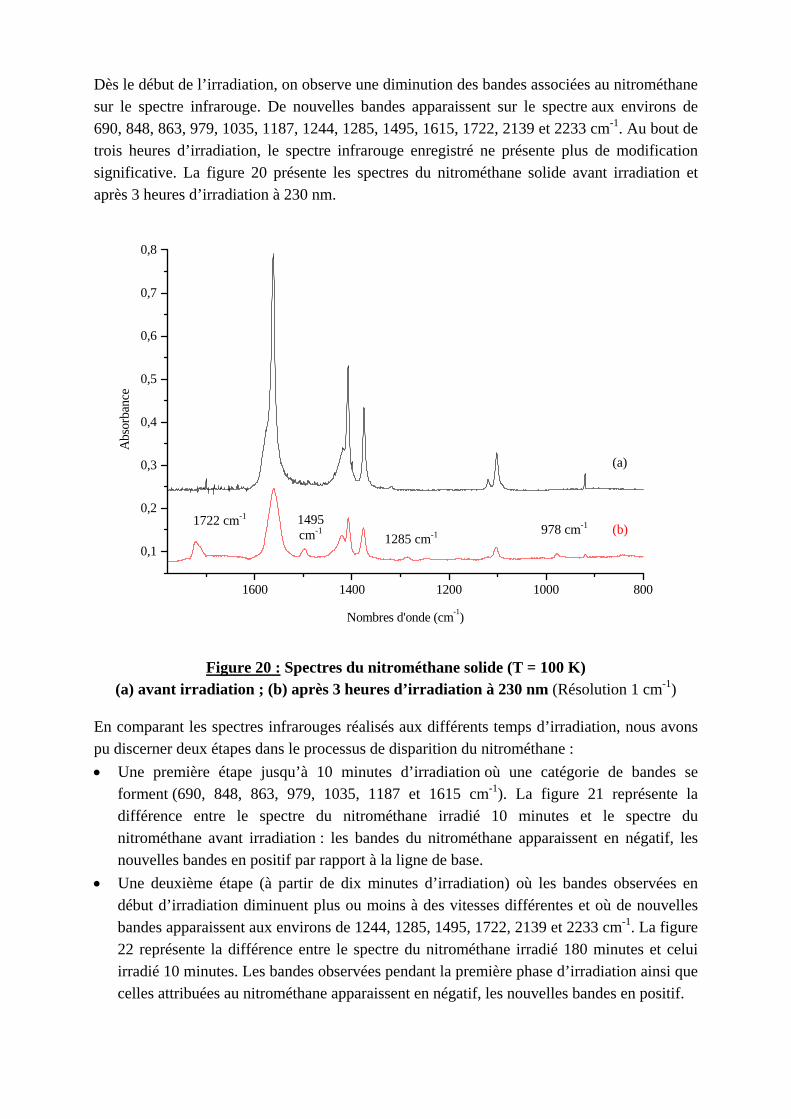
Dès le début de l’irradiation, on observe une diminution des bandes associées au nitrométhane sur le spectre infrarouge. De nouvelles bandes apparaissent sur le spectre aux environs de 690, 848, 863, 979, 1035, 1187, 1244, 1285, 1495, 1615, 1722, 2139 et 2233 cm-1. Au bout de trois heures d’irradiation, le spectre infrarouge enregistré ne présente plus de modification significative. La figure 20 présente les spectres du nitrométhane solide avant irradiation et après 3 heures d’irradiation à 230 nm.
1722 cm-1
1600
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Abs
orba
nce
(a)
(b) 978 cm-1
1285 cm-1
Figure 20 : S(a) avant irradiation ; (b)
En comparant les spectres infrpu discerner deux étapes dans l• Une première étape jusqu
forment (690, 848, 863, 9différence entre le spectnitrométhane avant irradianouvelles bandes en positif
• Une deuxième étape (à padébut d’irradiation diminubandes apparaissent aux en22 représente la différenceirradié 10 minutes. Les bancelles attribuées au nitromé
1495 cm-1
1400 1200 1000 800
Nombres d'onde (cm-1)
pectres du nitrométhane solide (T = 100 K) après 3 heures d’irradiation à 230 nm (Résolution 1 cm-1)
arouges réalisés aux différents temps d’irradiation, nous avons e processus de disparition du nitrométhane : ’à 10 minutes d’irradiation où une catégorie de bandes se 79, 1035, 1187 et 1615 cm-1). La figure 21 représente la re du nitrométhane irradié 10 minutes et le spectre du tion : les bandes du nitrométhane apparaissent en négatif, les par rapport à la ligne de base. rtir de dix minutes d’irradiation) où les bandes observées en
ent plus ou moins à des vitesses différentes et où de nouvelles virons de 1244, 1285, 1495, 1722, 2139 et 2233 cm-1. La figure entre le spectre du nitrométhane irradié 180 minutes et celui des observées pendant la première phase d’irradiation ainsi que thane apparaissent en négatif, les nouvelles bandes en positif.
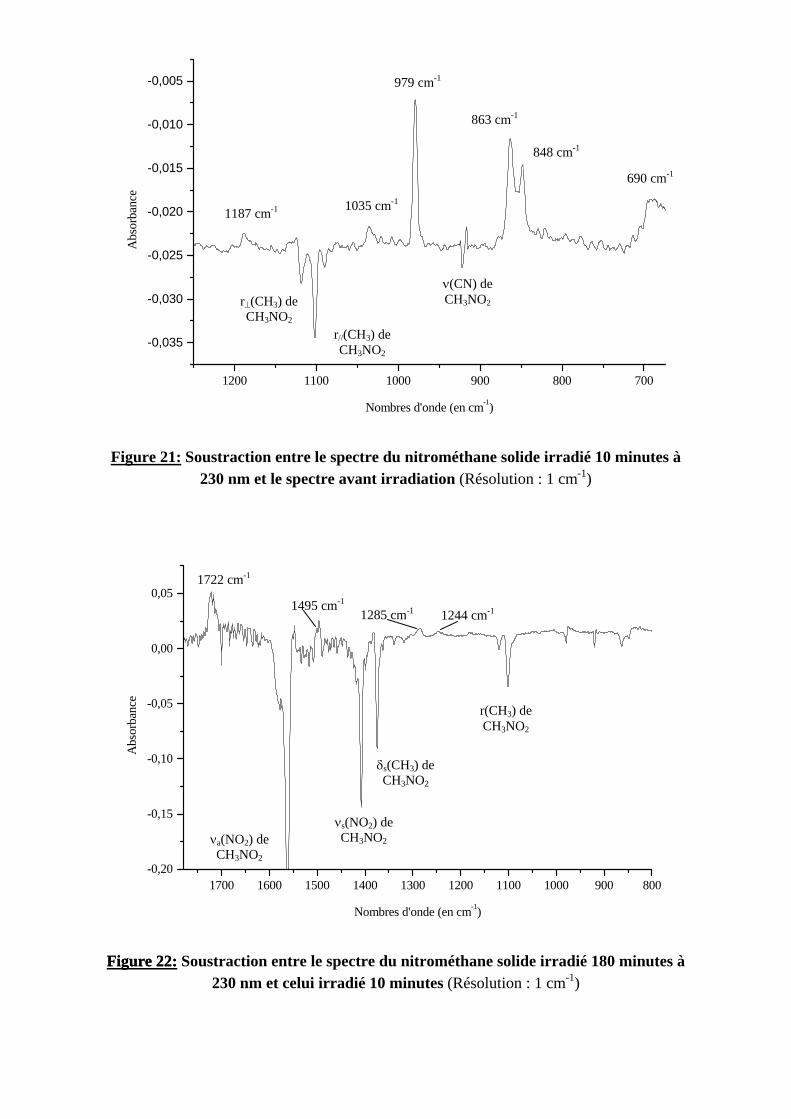
1200 1100 1000 900 800 700
-0,035
-0,030
-0,025
-0,020
-0,015
-0,010
-0,005
Abs
orba
nce
Nombres d'onde (en cm-1)
r⊥(CH3) de CH3NO2
r//(CH3) de CH3NO2
ν(CN) de CH 23NO
1187 cm-1 1035 cm-1
979 cm-1
848 cm-1
863 cm-1
690 cm-1
Figure 21: Soustraction entre le spectre du nitrométhane solide irradié 10 minutes à 230 nm et le spectre avant irradiation (Résolution : 1 cm-1)
1700 1600 1500 1400 1300 1200 1100 1000 900 800-0,20
-0,15
-0,10
-0,05
0,00
0,05
Abs
orba
nce
Nombres d'onde (en cm-1)
1495 cm-1
1285 cm-1 1244 cm-1
1722 cm-1
νa(NO2) de CH3NO2
νs(NO2) de CH3NO2
δs(CH3) de CH3NO2
r(CH3) de CH3NO2
Figure 22:Figure 22: Soustraction entre le spectre du nitrométhane solide irradié 180 minutes à 230 nm et celui irradié 10 minutes (Résolution : 1 cm-1)
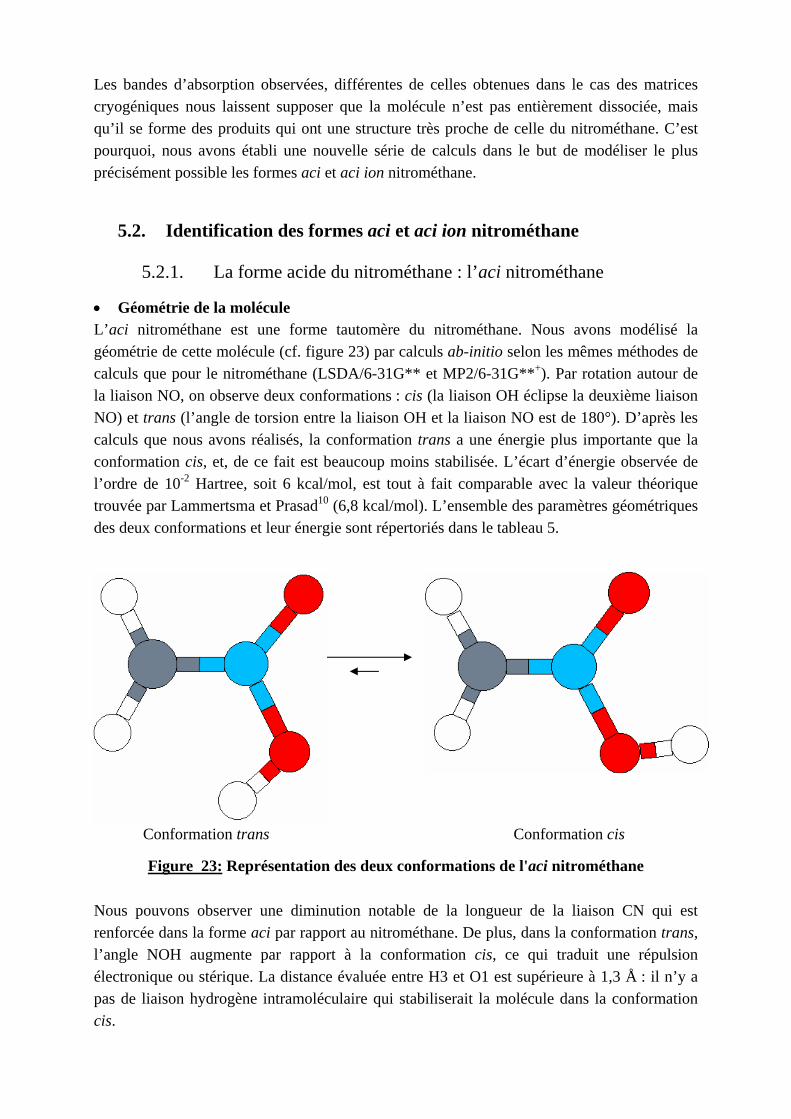
Les bandes d’absorption observées, différentes de celles obtenues dans le cas des matrices cryogéniques nous laissent supposer que la molécule n’est pas entièrement dissociée, mais qu’il se forme des produits qui ont une structure très proche de celle du nitrométhane. C’est pourquoi, nous avons établi une nouvelle série de calculs dans le but de modéliser le plus précisément possible les formes aci et aci ion nitrométhane.
5.2. Identification des formes aci et aci ion nitrométhane
5.2.1. La forme acide du nitrométhane : l’aci nitrométhane
• Géométrie de la molécule L’aci nitrométhane est une forme tautomère du nitrométhane. Nous avons modélisé la géométrie de cette molécule (cf. figure 23) par calculs ab-initio selon les mêmes méthodes de calculs que pour le nitrométhane (LSDA/6-31G** et MP2/6-31G**+). Par rotation autour de la liaison NO, on observe deux conformations : cis (la liaison OH éclipse la deuxième liaison NO) et trans (l’angle de torsion entre la liaison OH et la liaison NO est de 180°). D’après les calculs que nous avons réalisés, la conformation trans a une énergie plus importante que la conformation cis, et, de ce fait est beaucoup moins stabilisée. L’écart d’énergie observée de l’ordre de 10-2 Hartree, soit 6 kcal/mol, est tout à fait comparable avec la valeur théorique trouvée par Lammertsma et Prasad10 (6,8 kcal/mol). L’ensemble des paramètres géométriques des deux conformations et leur énergie sont répertoriés dans le tableau 5.
Conformation trans Conformation cis
Figure 23: Représentation des deux conformations de l'aci nitrométhane Nous pouvons observer une diminution notable de la longueur de la liaison CN qui est renforcée dans la forme aci par rapport au nitrométhane. De plus, dans la conformation trans, l’angle NOH augmente par rapport à la conformation cis, ce qui traduit une répulsion électronique ou stérique. La distance évaluée entre H3 et O1 est supérieure à 1,3 Å : il n’y a pas de liaison hydrogène intramoléculaire qui stabiliserait la molécule dans la conformation cis.

LSDA/6-31G** MP2/6-31G**+
Conformation Cis Trans Cis Trans C-N 1,301 1,309 1,314 1,323 N-O1 1,231 1,215 1,242 1,228 N-O2 1,404 1,419 1,429 1,441 C-H1 1,086 1,087 1,074 1,074 C-H2 1,086 1,088 1,073 1,074 O-H 0,985 0,981 0,975 0,973
CNO1 130,7 129,6 131,2 130,1 CNO2 113,9 117,4 113,0 117,2 ONO 115,4 113,0 115,8 112,7
NO2H3 98,9 105,9 100,2 106,1 NCH1 116,6 116,4 116,1 115,9 NCH2 118,1 120,5 117,7 119,8 HCH 125,2 123,1 126,1 124,3
Tableau 5 : Paramètres géométriques des deux conformations de l'aci nitrométhane
évalués par calculs ab initio (longueurs en Å ; angles en degrés)
• Modes de vibration : La molécule est de symétrie Cs et possède 7 atomes : comme dans le cas du nitrométhane, l’aci-nitrométhane possède 15 modes de vibration qui sont tous actifs en infrarouge (11 modes de symétrie A’ et 4 modes de symétrie A’’). Nous avons déterminé les différents modes de vibration de l’aci nitrométhane par calculs ab initio, selon les deux méthodes précédemment utilisées. Les résultats sont reportés dans le tableau 6. A ce jour, il n’existe aucune valeur expérimentale nous permettant de confronter nos résultats ; cependant, d’autres calculs théoriques de fréquences déjà publiés10 sont en accord avec nos résultats. La comparaison des fréquences expérimentales avec celles prévues par les calculs nous permet d’envisager l’existence de la forme aci dans la deuxième étape de l’irradiation.
5.2.2. La forme anion du nitrométhane : l’aci ion nitrométhane
• Géométrie de la molécule La forme aci ion du nitrométhane (cf. figure 24) est la base conjuguée de la forme aci. Elle peut donc s’obtenir par ajout de base dans une solution de nitrométhane. Cette forme est intimement liée au processus de détonation du nitrométhane1.

Figure 24 : Représentation de la forme aci ion nitrométhane Nous avons modélisé sa géométrie par les deux méthodes de calcul utilisées jusqu’à présent. Les résultats sont donnés dans le tableau 7. Comme pour la molécule aci, la liaison CN est plus courte que dans le cas du nitrométhane. En outre, la molécule modélisée est symétrique (son groupe de symétrie est C2v) : les liaisons et les angles de part et d’autre de cette liaison sont identiques.
LSDA/6-31G** MP2/6-31G**+
C=N 1,316 1,348 N-O1 1,259 1,286 N-O2 1,259 1,286 C-H1 1,070 1,074 C-H2 1,070 1,074 ONO 119,3 120,2 CNO1 120,3 119,9 CNO2 120,3 119,9 HCH 124,5 124,6 NCH1 117,7 117,7 NCH2 117,8 117,7
Tableau 7 : Paramètres géométriques de l'aci ion nitrométhane évalués par calculs ab initio (longueurs en Å ; angles en degrés)
• Modes de vibration Le groupe ponctuel de symétrie de cette molécule est C2v. La molécule possède 6 atomes donc 12 modes de vibration. Ces derniers se décomposent en 5 modes de symétrie A1, un mode de symétrie A2, 2 modes de symétrie B1 et 4 modes de symétrie B2. Une nouvelle fois, des calculs ab initio ont été réalisés dans le but de modéliser les modes de vibration de cette molécule. Les résultats sont présentés dans le tableau 8. Il sont confrontés aux valeurs

expérimentales de Jonathan20. La majorité des fréquences observées par Jonathan se retrouvent après irradiation du nitrométhane solide. Mais, les rapports d’intensité des différents modes sont pour la plupart inversés.
5.2.3. Discussion
Concernant la première étape des irradiations du nitrométhane, les fréquences observées sont en bon accord avec les résultats obtenus par N. Jonathan : il semble que l’aci ion soit formé. Cependant les attributions des résultats expérimentaux ne correspondent pas à celles des calculs théoriques (le mode r(CH2) est décalé de 600 cm-1 et les modes νa(NO2) et δ(CH2) sont inversés). Il reste également des bandes qui n’ont pas pu être reconnues comme mode propre de vibration de la forme aci ion, notamment celle à 1615 cm-1. De plus, dans notre travail, deux des bandes observées n’ont pas été attribuées jusqu’à présent (848 cm-1 et 863 cm-1) et disparaissent plus rapidement pendant l’irradiation que celles attribuées à l’aci ion. Ces résultats montrent qu’il existe au moins une deuxième molécule formée pendant la première étape. Comme dans le cas des matrices, nous avons modélisé la décroissance du nitrométhane. La loi obtenue :
C(t) = C0 / (1+C0kt) traduit une cinétique d’ordre 2 et donc un mécanisme bimoléculaire. Dans l’hypothèse d’un tel mécanisme, nous avons envisagé de modéliser la forme protonée du nitrométhane (CH3NO2H+) ; l’étude est en cours actuellement. Dans la deuxième étape (à partir de 10 minutes d’irradiation), de nouvelles bandes apparaissent : les principaux modes de la forme aci ont pu être caractérisés à l’aide des calculs quantiques effectués. La formation de l’aci peut s’expliquer par la réaction entre une molécule aci ion et une molécule de nitrométhane qui conduirait à la formation d’une molécule aci et d’une molécule aci ion par transfert de proton :
CH3NO2 + CH2NO2- → CH2NO2
- + CH2NO2H
La structure cristalline du nitrométhane montre que des atomes d’hydrogène et d’oxygène de deux molécules différentes sont proches dans l’espace (cf. figure 25). Cette position faciliterait le transfert de proton. De plus, ce mécanisme confirmerait aussi l’hypothèse de Lammertsma et Prasad selon laquelle l’équilibre tautomérique s’établirait via l’aci-ion10. Peu de temps après l’observation des bandes attribués à la forme aci, on observe une nouvelle bande à 2233 cm-1. Cette bande peut être attribuée à la molécule HNCO qui se formerait par perte d’une molécule d’eau au niveau de la forme aci :
CH2NO2H → HNCO + H2O
Ces résultats, tout à fait nouveaux, sont en accord avec l’étude théorique menée par McKee sur les réarrangements possibles de la molécule de nitrométhane21. Il existerait deux chemins possibles : le premier conduirait au méthylnitrite (l’énergie d’activation est évaluée à 73,5 kcal/mol) qui se décomposerait selon le processus que nous avons observé ; le deuxième avec une énergie d’activation légèrement plus importante (75 kcal/mol) amènerait à la formation de l’aci. Nous pouvons penser qu’à l’état solide, ce chemin réactionnel est favorisé.

Figure 25 : Représentation du plan (110) du cristal de nitrométhane à 78 K
Paramètres de la maille7(a) : a = 5,2 Å ; b = 6,2 Å ; c0 = 8,5 Å

6. CONCLUSION Nous avons étudié la réactivité du nitrométhane irradié à l’aide d’un laser accordable à 230 nm essentiellement. Une étude quantique et spectroscopique du nitrométhane a été réalisée au préalable ; elle nous a permis de reconnaître l’ensemble des modes de vibration de la molécule piégée en matrice de gaz rare (argon et xénon), à l’état liquide et à l’état solide. Les irradiations réalisées en matrice sont en accord avec celles déjà publiées, bien que nous ayons utilisé un laser donc d’un faisceau quasi-monochromatique. Nous avons établi un suivi de la plupart des bandes d’absorption des différents composés au cours du temps. Elles conduisent à la formation et à la dégradation bien connue du méthylnitrite. Mais, les résultats en phase condensée sont complètement différents. En effet, nous n’observons aucun des produits formés lors de l’irradiation en matrice. Les premières interprétations montrent qu’il existe deux étapes distinctes. Des calculs quantiques ont été réalisés dans le but de caractériser les produits formés. Dans la première étape, l’aci ion serait formé bien que certaines attributions ne correspondent pas à celles réalisées dans les travaux antérieurs et à l’étude théorique. Dans la deuxième étape, cette molécule se protonnerait pour former l’aci dont l’attribution est confirmée par les calculs. Ce travail est nouveau en ce qui concerne les résultats expérimentaux de l’aci. A notre connaissance, aucun spectre infrarouge de la forme aci nitrométhane n’a été réalisé à ce jour. C’est pourquoi, des manipulations complémentaires, telles que l’utilisation d’isotopes, sont à réaliser dans le but de confirmer ces deux attributions. Dans le cas des matrices où la molécule est entièrement entourée de gaz inerte, le groupement NO2 est excité et l’énergie est transférée au niveau de la liaison C-N qui se rompt. Il semble que le transfert d’énergie ne se produise pas de la même façon en phase condensée puisque l’observation de la forme aci ion puis aci en phase condensée suppose la rupture d’une liaison C-H (dans la première étape) et la formation d’une liaison O-H (dans la deuxième étape). L’environnement semble donc jouer un rôle primordial dans le processus d’irradiation du nitrométhane.

ANNEXE I : PRINCIPES DU CALCUL AB INITIO
Les approximations simplificatrices Une approche théorique d’un système polyatomique se traduit par la résolution de l’équation de Schrödinger :
HΨ = EΨ
E représente l’énergie du système dans l’état Ψ appelé fonction d’onde. L’opérateur H contient l’énergie cinétique constituant le système (noyaux et électrons) ainsi que leurs interactions électrostatiques. Une résolution analytique complète est possible uniquement dans les cas monoélectroniques simples ; dans le cas général, on effectue une série de simplifications et d’approximations. La première d’entre elles est l’approximation de Born-Oppenheimer selon laquelle, les noyaux, plus lourds que les électrons, sont considérés immobiles. L’expression de l’hamiltonien se réduit alors au terme électronique :
∑ ∑ ∑∑∑ −+∇−=>
électrons
i
électrons
i
noyaux
AiA
A
ijij
électrons
i
2i
élec
rZ
r1
21H
rij représente la distance entre les électrons i et j, ZA, la charge nucléaire et riA, la distance entre le noyau A et l’électron i. En considérant que la fonction d’onde du système peut se décomposer en un produit d’une fonction nucléaire et d’une fonction électronique, l’équation de Schrödinger devient alors :
HélecΨélec = EelecΨélec
La deuxième approximation utilisée est l’approximation orbitalaire : on considère que la fonction d’onde électronique qui décrit le système de n électrons peut être décomposée en un produit antisymétrique de n fonctions monoélectroniques Φi, appelées orbitales moléculaires. Dans la théorie Hartree-Fock, l’écriture de l’hamiltonien électronique est alors modifiée : le terme représentant l’interaction entre électrons est remplacé par un potentiel moyen (c’est la théorie du champ autocohérent : Self Consistent Field). De ce fait, l’écriture de l’équation de Schrödinger se transforme en un problème de n équations indépendantes du type :
FiΦi = εiΦi
où Fi est l’opérateur de Fock associé à l’électron i. Choix de bases et méthodes de calculs Les fonctions Φi sont des combinaisons d’orbitales atomiques. Pour décrire ces orbitales atomiques, l’utilisation d’orbitales de Slater (Slater Type Orbital) implique des calculs très complexes et coûteux. On leur substitue donc des orbitales de type gaussiennes (exp (-αr2)) qui donnent des expressions mathématiques très simples au niveau des intégrales multicentriques dans l’expression de l’opérateur de Fock. La base la plus simple est la base

STO-3G : chaque orbitale de Slater est représentée par une combinaison de trois orbitales gaussiennes. Dans notre cas, la molécule de nitrométhane présentant une délocalisation importante au niveau du groupement NO2, nous avons voulu minimiser les écarts entre les valeurs théoriques et expérimentales, notamment au niveau du calcul des fréquences que nous n’avons volontairement pas pondérées. C’est pourquoi, nous avons utilisé une base de calcul plus conséquente : la base 6-31G**+. Les orbitales sont définies de la façon suivante : pour chaque noyau, les orbitales de cœur sont décrites par 6 orbitales gaussiennes. Les orbitales de valence (c’est-à-dire les orbitales 2s et 2p pour les atomes C, N et O) sont développées sur un ensemble de 3 gaussiennes et d’une gaussienne isolée. Le premier astérisque signifie que l’on a rajouté des orbitales de polarisation sur les atomes « lourds » (soit des orbitales de type 3d sur les atomes C, N et O), le deuxième astérisque concerne les atomes d’hydrogène auxquels on ajoute des orbitales de type 2s. Enfin le « + » précise qu’on utilise également des orbitales diffuses sur les atomes lourds (ce sont des gaussiennes dont le coefficient α est très petit, qui par conséquent s’annule très loin du noyau ; elle reproduisent assez bien les délocalisations au sein des molécules). Les calculs effectués avec de telles orbitales ne nécessitent pas d’approximation supplémentaire pour la détermination de toutes les intégrales biélectroniques. Ils portent le nom de calculs ab initio.
Grandeurs modélisées par les logiciels Gaussian94 et Gamess98 Les logiciels de calculs ab initio utilisés permettent d’évaluer de nombreuses propriétés : • La géométrie de la molécule après optimisation (c’est-à-dire celle pour laquelle l’énergie
est minimale) • L’énergie de la molécule ainsi que les vecteurs propres obtenus après résolution de
l’équation d’Hartree-Fock • La correction Møller-Plesset au deuxième ordre au niveau de l’énergie • L’analyse des populations des orbitales moléculaires et les charges de Mülliken • La matrice F des constantes de forces en coordonnées cartésiennes utilisé pour l’analyse
vibrationnelle • L’analyse vibrationnelle en coordonnées normales dans l’approximation harmonique : les
fréquences (en cm-1), leurs intensités et les modes normaux qui leur sont associés.
ANNEXE II : REPRESENTATION SCHEMATIQUE DES PRINCIPAUX MODES DE VIBRATION DU
NITROMETHANE Nous avons représenté ici les modes de vibration du nitrométhane uniquement pour la conformation éclipsée ; ces modes sont tout à fait équivalents pour la conformation décalée. Les élongations :
ν(CN) νa(NO2) νs(NO2)
νa(CH3) νs’(CH3) νs(CH3)

Les déformations :
δs(CH3) δa(CH3) δ(NO2)
r(NO2) r//(CH3) δs’(CH3)
ω(NO2)
-
+
+
+
+
+
r⊥(CH3)

BIBLIOGRAPHIE
1 (a) R. Engelke, W. L. Earl, C. McMichael Rohlfing, J. Chem. Phys., 84-1, 142 (1986)
(b) R. Engelke, W. L. Earl, C. McMichael Rohlfing, J. Chem. Phys., 90, 545 (1986)
(c) R. Engelke, D. Schiferl, C. B. Storm, W. L. Earl, J. Phys. Chem., 92, 6815 (1988) 2 S. Nagakura, Mol. Phys., 3, 152 (1960) 3 R. G. Pearson, R. L. Dillon, J. Am. Chem. Soc., 75, 2439 (1943) 4 A. Allouche, J. Phys. Chem., 100, 1820 (1996) 5 A. Kheir, J. F. Haw, J. Am. Chem. Soc., 116, 817 (1994) 6 M. Monnier, Thèse d’Etat de l’Université de Provence (1991) 7 (a) S.F. Trevino, E. Prince, C. R. Hubard, J. Chem. Phys., 73, 2996 (1980)
(b) D. T. Cromer, R. R. Ryan, D. J. Schifferl, J. Phys. Chem., 89, 2315 (1985) 8 M. L. McKee, J. Am. Chem. Soc., 107,1900 (1985) 9 R. Engelke, D. Schifferl, W. L. Earl, J. Mol. Struct. (Theochem), 180, 141 (1988) 10 K. Lammertsma, B. V. Prasad, J. Am. Chem. Soc., 115, 2348 (1993) 11 D. Gorse, D. Cavagnat, M. Pesquier, C. Lapouge, J. Phys. Chem., 97, 4262 (1993) 12 J. R. Hill, D. S. Moore, S. C. Schmidt, C. B. Storm, J. Phys. Chem., 95, 3037 (1991) 13 B. Meyer, Low Temperature Spectroscopy, American Elsevier publishing Company, Inc.,
New York (1971) 14 H. E. Ugnade, E. D. Loughran, L. W. Kissinger, J. Phys. Chem., 54, 1410 (1960) 15 M. Bodenbinder, S. E. Ulic, H. Willner, J. Phys. Chem., 98, 6441 (1994) 16 R. P. Müller, J. Robert Huber, J. Mol. Spec., 104, 209 (1984) 17 M. Jacox, J. Phys. Chem., 88, 3373 (1984) 18 M. E. Jacox et F. L. Rook, J. Phys. Chem., 86, 2899-2904, (1982) 19 V. E. Bondybey, J. H. English, C. W. Mathews, R. J. Contolini, J. Mol. Spect., 92, 431
(1982) 20 N. Jonathan, J. Mol. Spec., 7, 105 (1961) 21 M. L. McKee, J. Am. Chem. Soc., 108, 5784 (1986)


















