
Protocole de recherche clinique : RTEP5 - oncomip.org · Professeur Bernard DUBRAY Professeur...
46
CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5 page 1/46 Protocole de recherche clinique : R R T T E E P P 5 5 Version 3.0 du 19 septembre 2012 N° ID RCB°: 2011-A00727-34 Autorisation initiale de l’AFSSAPS : 29 décembre 2011 Avis favorable du CPP Nord Ouest I: 06 octobre 2011 PROMOTEUR Centre de lutte contre le cancer Henri Becquerel 1 rue d’Amiens, 76038 Rouen Tél: 02 32 08 25 44 Fax : 02 32 08 25 69 Investigateur coordonnateur Méthodologiste Professeur Pierre VERA Centre Henri Becquerel, Rouen Téléphone : 02 32 08 22 58 Télécopie : 02 32 08 25 50 mail: [email protected] Professeur Philippe Chaumet-Riffaud CHU Bicêtre, Le Kremlin Bicêtre Téléphone : 01 45 21 25 07 Télécopie : 01 45 21 21 12 mail: [email protected] Comité scientifique Professeur Bernard DUBRAY Professeur Françoise MORNEX Professeur Francesco GIAMMARILE Professeur Frédéric COURBON Professeur Jean-Philippe VUILLEZ Monsieur Pierre BOHN Monsieur Sébastien HAPDEY Docteur Isabelle GARDIN Monsieur Maximilien VERMANDEL Etude de phase II pour évaluer l’efficacité et la tolérance d’un complément de dose de radiothérapie (RT) des lésions hypoxi- ques, identifiées par TEP/TDM au F-miso chez les patients atteints d’un cancer broncho-pulmonaire non à petites cellules candidats à une radio-chimiothérapie (RT-CT) à visée curative
Transcript of Protocole de recherche clinique : RTEP5 - oncomip.org · Professeur Bernard DUBRAY Professeur...

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 1/46
Protocole de recherche clinique :
RRTTEEPP55
Version 3.0 du 19 septembre 2012
N° ID RCB°: 2011-A00727-34
Autorisation initiale de l’AFSSAPS : 29 décembre 2011 Avis favorable du CPP Nord Ouest I: 06 octobre 2011
PROMOTEUR
Centre de lutte contre le cancer Henri Becquerel 1 rue d’Amiens, 76038 Rouen
Tél: 02 32 08 25 44 Fax : 02 32 08 25 69
Investigateur coordonnateur
Méthodologiste
Professeur Pierre VERA Centre Henri Becquerel, Rouen Téléphone : 02 32 08 22 58 Télécopie : 02 32 08 25 50 mail: [email protected]
Professeur Philippe Chaumet-Riffaud CHU Bicêtre, Le Kremlin Bicêtre Téléphone : 01 45 21 25 07 Télécopie : 01 45 21 21 12 mail: [email protected]
Comité scientifique Professeur Bernard DUBRAY Professeur Françoise MORNEX Professeur Francesco GIAMMARILE Professeur Frédéric COURBON Professeur Jean-Philippe VUILLEZ Monsieur Pierre BOHN Monsieur Sébastien HAPDEY Docteur Isabelle GARDIN Monsieur Maximilien VERMANDEL
Etude de phase II pour évaluer l’efficacité et la tolérance d’un
complément de dose de radiothérapie (RT) des lésions hypoxi-
ques, identifiées par TEP/TDM au F-miso chez les patients atteints
d’un cancer broncho-pulmonaire non à petites cellules candidats à
une radio-chimiothérapie (RT-CT) à visée curative

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 2/46
Signataires du protocole
Rôle Nom Date Signature
Investigateur-coordonateur
Pr P.Vera 19/09/2012
Méthodologiste Pr P.Chaumet-Riffaud
19/09/2012
Promoteur CLCC Henri Becque-rel représenté par le Pr H.Tilly, Directeur
19/09/2012
Contacts promoteur Radiopharmacien en charge du F-MISO et contrôle qualité du traceur :
Monsieur Pierre Bohn
Tél: 02 32 08 29 23 Fax : 02 32 08 25 50 mail : [email protected]
Unité de Recherche clinique : CLCC Henri Becquerel,1 rue d’Amiens, 76038 Rouen
Monsieur Olivier Rastelli ARC moniteur
Tél: 02 32 08 29 00 Fax : 02 32 08 25 69 mail : [email protected]
Equipe de radiothérapie : Professeur Bernard Dubray
Téléphone : 02 32 08 22 28
Télécopie : 02 32 08 25 04
mail: [email protected] Docteur Sébastien THUREAU
Téléphone : 02 32 08 27 46
mail: [email protected]
Transfert des images et des contours : Monsieur Romain MODZELEWSKI
Télécopie : 02 32 08 25 50
mail: [email protected]
CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 3/46
SYNOPSIS
Titre de l’étude Etude de phase II pour évaluer l’efficacité et la tolérance d’un complément de dose de radiothérapie (RT) des lésions hypoxiques, identifiées par TEP/TDM au F-miso chez les patients atteints d’un cancer broncho-pulmonaire non à petites cellules candidats à une radio-chimiothérapie (RT-CT) à visée curative
Titre abrégé RTEP5 Promoteur CLCC Henri Becquerel Type d’étude Etude phase II, ouverte, multicentrique, non randomisée Coordonnateur Pr P.Vera Méthodologiste Dr P.Chaumet-Riffaud Comité de rédac-tion
P.Vera (Rouen), P.Chaumet-Riffaud (Kremlin-Bicêtre), B.Dubray (Rouen), F.Giammarile (Lyon), F.Mornex (Lyon), F.Courbon (Toulouse), JP.Vuillez (Grenoble), P.Bohn (Rouen), S.Hapdey (Rouen), I.Gardin (Rouen), M.Vermandel (Lille)
Groupe coopéra-teurs
Société Française de Médecine Nucléaire (SFMN), Société Française de Radiothérapie (SFRO)
Objectif principal Estimer le taux de contrôle local après complément de dose des lésions hypoxiques dé-terminées par TEP/TDM au F-miso [dose maximum possible sans que plus de 30% du volume pulmonaire total reçoive une dose supérieure à 20 Gy (V20)]
Objectifs secondai-res
• Evaluation de la tolérance à 3 mois et 1 an • Estimation du pourcentage de sujets pour lesquels la dose de RT a pu être augmen-
tée • Etude physiopathologique de la variation simultanée du métabolisme glucidique et
hypoxie des lésions (tumeur et ganglions) par TEP/TDM en cours de radiothérapie • Valeur prédictive sur la survie à un an, de la variation du métabolisme glucidique et
de l’hypoxie des lésions (tumeur et ganglions) TEP/TDM en cours de radiothérapie Analyse ancillaire Etudier plusieurs scénarii d’optimisation de la radiothérapie en fonction des variations du
métabolisme et de l’hypoxie de la tumeur et des ganglions mesurés en cours de radio-thérapie (étude théorique sur console)
Durée prévue de l’étude
2 ans d’inclusion 1 an de suivi
Analyse statistique Un plan de Gehan en deux étapes est proposé. L’hypothèse sur l’efficacité thérapeutique attendue est p = 0,4; un premier groupe de 6 patients bénéficiera d’une procédure de complément de dose des lésions déterminées comme hypoxiques. Si aucun contrôle local n’est observé sur ces 6 premiers malades, l’essai sera arrêté (puissance 0,95). Dans la deuxième étape, le nombre d’inclusions sera déterminé par le nombre de succès dans l’étape 1 et par la précision souhaitée à ε = 0,10 (soit n=18 pour 1 succès, 19 pour 2 succès, 15 pour 3 succès, 8 pour 4 succès,…).
Nombre de pa-tients prévus
Il est nécessaire de disposer pour l’analyse de 25 sujets évaluables (au maximum) selon le plan décrit ci-dessus. D’après les résultats de l’étude RTEP1 ; • le nombre de patients pré-inclus ayant une TEP/TDM-FDG négative après chimiothé-
rapie d’induction est de 20% ; • 50% des patients participant à l’étude seront éligibles après la réalisation de la TEP
F-miso pré RT-CT ; • Nous posons comme hypothèses supplémentaires que 5 sujets sur 30 éligibles se-
ront non évaluables (soit pour décès précoces avant 3 mois, sortie d’étude,…). Il faudra donc prévoir que : - 75 sujets participeront à l’étude dans la phase de pré-inclusion ; - qu’environ 60 auront une fixation significative sur la TEP/TDM au FDG en pré-RT; - et qu’environ 30 bénéficieront d’une augmentation de dose après identification de lé-sions d’hypoxie lors de la TEP/TDM F-miso en pré-RT.
Centres prévus 22 en France Critères de pré-inclusion
• Homme ou femme • Age supérieur à 18 ans • Bon état général OMS ≤ 1 • Tumeur mesurable selon les critères d’évaluation RECIST • Preuve histologique de cancer bronchique non à petites cellules • Patient candidat à une radio-chimiothérapie thoracique à visée curatrice
CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 4/46
- Absence d’atteinte pleurale, de localisation métastatique pulmonaire ou ex-tra-thoracique
- Absence de co-morbidité contre-indiquant une radio-chimiothérapie • Fonction respiratoire : VEMS ≥ 40% de la valeur théorique et DLCO/VA ≥ 50% de la
valeur théorique et PaO2 ≥ 60 mm Hg. • Fonction hématologique :
- Polynucléaires neutrophiles > 1.5.109/L et plaquettes > 100.109/L - Hémoglobine > 10g/dl
• Une clairance estimée de la créatinine > 60 ml/min • Consentement éclairé signé • Patient affilié à la sécurité sociale ou à une structure assimilée
Critères d’inclusion définitifs
• Fixation tumorale supérieure au bruit de fond médiastinal sur la TEP/TDM au FDG en pré-RT-CT (après la chimiothérapie d’induction)
• L’inclusion définitive est prononcée après réalisation de la dosimétrie prévisionnelle confirmant que les objectifs (dose minimale de 60Gy dans 99% des volumes cible) et les contraintes de dose (poumons, moelle épinière) sont respectés.
Critères de non inclusion
• Histologie autre que cancer bronchique primitif non à petites cellules • Les patients pour lesquels aucune cible tumorale n’est évaluable (rémission complète
après chimiothérapie) • Absence de fixation sur les examens TEP-FDG avant la chimiothérapie d’induction si
cet examen est réalisé • Patients pour lesquels une radiothérapie à visée curative n’est pas indiquée (exten-
sion tumorale, métastases, état général, co-morbidités) • Antécédents de maladie néoplasique de moins de 5 ans ou évolutive • Patient déjà inclus dans un autre essai thérapeutique • Femme enceinte, susceptible de l’être ou en cours d’allaitement • Index de performance OMS ≥2 • Insuffisance rénale contre indiquant un traitement par Cisplatine • Majeurs protégés (sous tutelle ou sous curatelle) • Impossibilité de se soumettre au suivi médical de l’étude pour des raisons géogra-
phiques, sociales ou physiques • Patients diabétiques mal équilibrés avec glycémie ≥10 mmol/L • Hypersensibilité au FDG ou à l’un des excipients du radiopharmaceutique • Hypersensibilité au F-miso ou à l’un des excipients du radiopharmaceutique • Patients en incapacité de comprendre l’étude (problème de langue …)
Critères de non augmentation de dose
• Absence d’hypoxie définie par une évaluation négative de la fixation du F-miso sur les images à 4 heures sur l’image TEP/TDM pré-thérapeutique (TEP/FDG F-miso1)
Evaluation des bénéfices et des risques potentiels attendus
• Augmentation du contrôle local de 17% à 40% • Evaluation du taux de complications • La dosimétrie estimée des 3 examens additionnels TEP/TDM centrés sur le thorax
est de 20 mSv Traitement, pro-duits, matériels utilisés
• Radiothérapie conformationnelle avec correction d’hétérogénéité (radiothérapie par modulation d’intensité (RCMI) et asservissement respiratoire possible)
• Examens TEP/TDM réalisés en position de traitement • TEP/TDM-FDG FDG en pré-RT-CT (injection de produits de contraste (PDC) iodés
possible selon la pratique locale) et TEP/TDM avec injection de F-miso (sans injec-tion de PDC iodés). Délai maximum de 8 jours entre FDG et F-miso pré-RT-CT, et de 72h pendant la RT-CT (à 42 Gy). Ordre à respecter : FDG puis F-miso pour les exa-mens pré-RT-CT, et selon possibilité pour les examens en cours de RT-CT
Critères d’évaluation de l’hypoxie
• Analyse qualitative : Cette analyse servira à la définition de l’hypoxie et décider du complément de dose. Il s’agit d’une analyse visuelle binaire (négative ou positive) qui sera réalisée par 3 relecteurs en insu dans un délai de 8 jours. Les données se-ront centralisées et redistribuées via le réseau SFMN-net (Rischin, JCO 2006)
• Analyse quantitative : Cette analyse permettra de définir le volume hypoxique (HTV) et sera réalisée à postériori pour la réponse aux objectifs secondaires et à l’étude ancillaire. Un GTV sera défini sur l’image TEP/TDM-FDG à partir d’une mé-thode de seuillage relative (40% SUVmax). Les contours de ce volume seront reco-piés sur les images TEP-F-miso 1 après recalage des images. Les pixels dont le rap-
CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 5/46
port signal sur bruit de fond sera supérieur à 1.4 seront considérés comme hypoxi-ques. La réunion de ces pixels constituera le volume hypoxique ou HTV (Nehmeh, IJROBP, 2008)
Complément de dose
• Dose maximum possible sans dépasser comme contrainte de dose aux poumons sains 20 Gy sur plus de 30% du poumon (V20)
Rôle des équipes • Les images TEP/TDM sont acquises localement. Ces images TDM, FDG et F-miso seront transférées à Rouen où seront définis les volumes (GTV et HTV). Ces volumes seront renvoyés au radiothérapeute local. Le plan de traitement définitif sera discuté avec le Pr Dubray. La décision du traitement final appartient au radiothérapeute lo-cal
Critères d’évaluation pour répondre à l’objectif principal
• Pourcentage de contrôle local mesuré par TDM et +/- TEP/TDM-FDG à 3 mois • Analyse en intention de traitement ITT sur tous les sujets inclus et éligibles • Analyse per protocole sur tous les sujets inclus et évaluables
Critères d’évaluation pour répondre aux ob-jectifs secondaires
• Pourcentage de complications à 3 mois et 1 an • Pourcentage de sujets pour lesquels la dose de RT a pu être augmentée • Pourcentage de contrôle local chez les sujets inclus et non éligibles • Mesure de la variation de fixation (SUV) et de volume entre examens TEP/TDM-FDG
et TEP/TDM-F-miso avant et pendant la RT • Taux de survie global à un 1 an • Mesure de la concordance inter-lecteurs pour l’interprétation des images TEP-F-miso
Calendrier prévi-sionnel
75 pré-inclusions sur 2 ans ; suivi pendant 1 an • Début des inclusions : avril 2012 • Fin des inclusions : avril 2014 • Fin de l’étude : avril 2015 • Durée de participation de chaque patient : 1 an
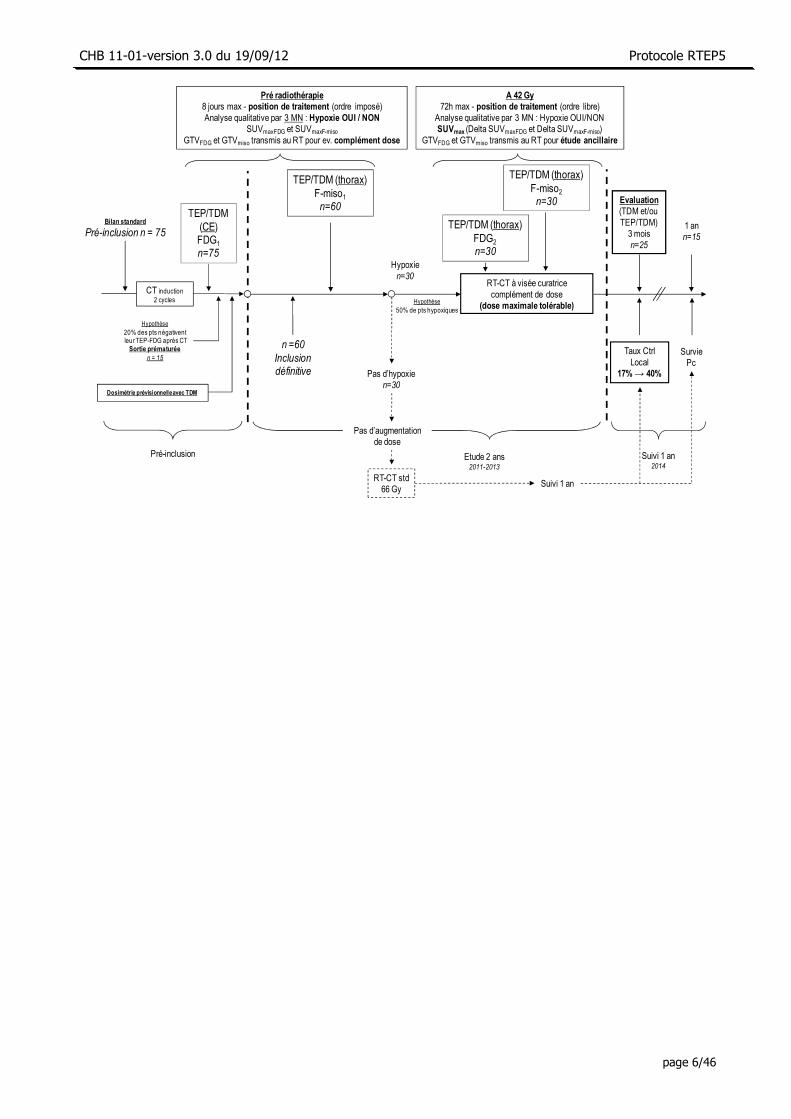
Déroulement et schéma de l’étude
• Pré-inclusion (n=75) - Examen clinique, âge, TNM, OMS, poids, Cr clairance, βHCG, biologie - Fibroscopie bronchique, imagerie cérébrale, scintigraphie osseuse optionnelle - Dosimétrie - TEP/TDM-FDG corps entier
• Etude des patients inclus (n=60) - Avant la RT-CT (n=60): TEP/TDM-F-miso1 centrée sur le thorax - Patients éligibles : A 42 Gy de la RT-CT : TEP/TDM-FDG2 et TEP/TDM-F-miso2
centrée sur le thorax (n=30) • Suivi des patients inclus (n=60)
- 3 mois : Evaluation clinique et TDM (± TEP/TDM-FDG, fibroscopie bronchique) 1 an : Evaluation clinique et TDM + TEP/TDM-FDG (±, fibroscopie bronchique)
CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 6/46
TEP/TDM (thorax)
F-miso1
n=60TEP/TDM
(CE)FDG1
n=75
Pré radiothérapie
8 jours max - position de traitement (ordre imposé)
Analyse qualitative par 3 MN : Hypoxie OUI / NON
SUVmaxFDG et SUVmaxF-miso
GTVFDG et GTVmiso transmis au RT pour ev. complément dose
Etude 2 ans2011-2013
Suivi 1 an2014
TEP/TDM (thorax)
FDG2
n=30
TEP/TDM (thorax)
F-miso2n=30
A 42 Gy
72h max - position de traitement (ordre libre)
Analyse qualitative par 3 MN : Hypoxie OUI/NON
SUVmax (Delta SUVmaxFDG et Delta SUVmaxF-miso)
GTVFDG et GTVmiso transmis au RT pour étude ancillaire
Evaluation
(TDM et/ou
TEP/TDM)
3 mois
n=25
1 an
n=15
Pré-inclusion
Dosimétrie prévisionnelle avec TDM
Hypothèse
50% de pts hypoxiques
Pas d’hypoxie
n=30
RT-CT std
66 Gy
Pas d’augmentation
de dose
Hypoxie
n=30RT-CT à visée curatrice
complément de dose
(dose maximale tolérable)
Bilan standard
Pré-inclusion n = 75
Taux Ctrl
Local
17% → 40%
Survie
Pc
CT induction2 cycles
Hypothèse
20% des pts négativentleur TEP-FDG après CTSortie prématurée
n = 15
Suivi 1 an
n =60
Inclusion
définitive

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 7/46
SOMMAIRE
1. Contexte, Problématique, hypothèse(s) .................................................... 10
1.1. Problématique ....................................................................................................... 10 1.2. Etudes cliniques..................................................................................................... 11 1.3. Faisabilité d’une augmentation de la dose totale de radiothérapie .............................. 13 1.4. Conclusion ............................................................................................................ 14
2. Objectifs de la recherche ............................................................................ 14
2.1. Objectif principal ................................................................................................... 14 2.2. Objectifs secondaires ............................................................................................. 14 2.3. Etude ancillaire...................................................................................................... 15
3. Plan de la recherche ................................................................................... 15
3.1. Plan expérimental .................................................................................................. 15 3.2. Calendriers des examens et des bilans realises......................................................... 16
4. Criteres d’inclusion et de non-inclusion ..................................................... 16
4.1. Critères dE PRE-inclusion........................................................................................ 16 4.2. Critères d’inclusion définitive................................................................................... 17 4.3. Critères de non inclusion ........................................................................................ 17 4.4. Critères de non augmentation de dose .................................................................... 17
5. deroulement de l’étude............................................................................... 17
5.1. Phase de pré-sélection ........................................................................................... 17 5.2. Phase d’inclusion ................................................................................................... 18 5.3. Phase de traitement............................................................................................... 18 5.4. Phase de surveillance............................................................................................. 19 5.5. Synthèse des examens TEP à prevoir ...................................................................... 19
6. Toxicité et adaptation de dose ................................................................... 20
6.1. Toxicité liée à la chimiothérapie – Conduite à tenir ................................................... 20 6.2. Toxicités de l’irradiation – Conduite à tenir............................................................... 20
7. sortie, arrêt et fin d’etude ..........................................................................21
7.1. sortie d’étude des patients...................................................................................... 21 7.2. arrêt de l’etude...................................................................................................... 21 7.3. fin de l’etude ......................................................................................................... 21
8. Evaluation de l’efficacité ............................................................................21
8.1. Critère principal ..................................................................................................... 21 8.2. Critères secondaires............................................................................................... 22
9. Evaluation de la sécurité ............................................................................ 22
9.1. Définitions générales.............................................................................................. 22 9.2. Définition d’un événement indésirable grave attendu (EIG-A) .................................... 23 9.3. Définition d’un événement indésirable grave inattendu (EIG-I) .................................. 23 9.4. Notification d’un événement et d’un EFFET INDESIRABLE grave ................................ 23

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 8/46
9.5. Suivi des EIG......................................................................................................... 23 9.6. Paramètres et méthodes d’évaluation de la sécurité.................................................. 24 9.7. Procédures et délais pour la notification et l’enregistrement des effets indésirables ..... 24 9.8. Comité indépendant de surveillance de l’étude ......................................................... 24 9.9. Modalités et durée du suivi des personnes suite à la survenue d’évènements indésirables 25
10. Biométrie et plan d’analyse statistique ................................................... 25
10.1. Les populations DE l’essai....................................................................................... 25 10.2. Nombre de sujets nécessaires................................................................................. 26 10.3. Analyse statistique................................................................................................. 26
11. Calendrier prévisionnel ............................................................................ 28
12. Réalisation des tomodensimométries (TDM ou ScannerS) ..................... 28
13. Réalisation des examens d’imagerie TEP ................................................ 28
13.1. Radiopharmaceutique(s) administré(s) .................................................................... 28 13.2. Procédures d’acquisition ......................................................................................... 29 13.3. Traitement et analyse des images ........................................................................... 30
14. Modalités et procédures de radiothérapie............................................... 31
14.1. Identification et délinéation des volumes cible.......................................................... 31 14.2. Objectifs et contraintes de dose totale..................................................................... 33 14.3. Imagerie de contrôle des faisceaux et du positionnement sous l’appareil .................... 34
15. Role respectif des investigateurs/équipes .............................................. 34
16. Faisabilité ................................................................................................. 34
16.1. Potentiel d’inclusion ............................................................................................... 34 16.2. Travail préparatoire sur le transfert des donnees ...................................................... 35
17. Controle et assurance qualité .................................................................. 35
17.1. Assurance qualité .................................................................................................. 35 17.2. Contrôle qualité ..................................................................................................... 35 17.3. Contrôle de qualité de la radiothérapie .................................................................... 35 17.4. Contrôle qualité des installations TEP-TDM............................................................... 35
18. Justification ALARA, dose efficace totale ................................................ 37
18.1. Justification de la réalisation de deux examens TEP au FDG et au F-miso ................... 37 18.2. Données dosimétriques .......................................................................................... 37
19. Annexe : Justification de l’utilisation du F-miso...................................... 37
20. Annexe : Considérations ethiques et réglementaires.............................. 38
20.1. Comité de Protection des Personnes........................................................................ 39 20.2. Autorité compétente .............................................................................................. 39 20.3. Information et consentement des participants.......................................................... 39 20.4. Responsabilité des investigateurs ............................................................................ 39

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 9/46
21. Annexe : Droit d’accès aux données et documents sources.................... 40
22. Annexe : Regles relative a la publication................................................. 40
23. Annexe : Liste des abréviations ............................................................... 41

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 10/46
1. CONTEXTE, PROBLEMATIQUE, HYPOTHESE(S)
1.1. PROBLEMATIQUE
1.1.1. EPIDEMIOLOGIE ET PRINCIPES DU TRAITEMENT DES CANCERS BRONCHIQUES NON A PETITES CELLULES
La fréquence et la gravité du cancer bronchique primitif non à petites cellules (CBPNPC) justifient la recher-che et la validation d’approches innovantes contribuant à renforcer l’efficacité des traitements. L'incidence annuelle du CBPNPC en France est de 50,5 cas pour 10 000 hommes (12,6 pour 10 000 femmes). En 2005 [www.invs.sante.fr], 30 651 nouveaux cas ont été diagnostiqués et 26 624 patients sont décédés. La sévéri-té du pronostic est due1 :
• au potentiel évolutif de la maladie : relative rareté des formes opérables, fréquence des métastases au diagnostic ou au cours de l’évolution, fréquence des rechutes locales,
• à l’efficacité insuffisante et à la toxicité des traitements,
• aux conséquences des autres affections coexistantes chez le même patient (insuffisance cardio-respiratoire, artériopathie, …), susceptibles de majorer le retentissement toxique des traitements ou d’engager par elles-mêmes le pronostic vital.
1.1.2. PLACE DE LA RADIOTHERAPIE ET DE L’IMAGERIE PRE-THERAPEUTIQUE DANS LE CPNPC
La radiothérapie (RT) est largement utilisée pour le traitement locorégional des CBPNPC, soit à visée curative soit à visée palliative. Selon l’extension tumorale et les facteurs de risque de mauvaise tolérance, la radiothé-rapie est délivrée seule (radiothérapie exclusive) ou associée à une chimiothérapie (CT) de façon séquen-tielle ou concomitante. La RT-CT concomitante est considérée comme un standard de traitement2.
Les volumes cibles de la radiothérapie sont constitués par la tumeur et ses extensions microscopiques de voisinage, et les aires ganglionnaires de drainage lymphatique (cf. annexe selon ICRU 50 et 62). La qualité du bilan d’extension initial est cruciale pour ne pas exclure une zone envahie (éviter la rechute tumorale) et pour ne pas irradier une zone saine (réduire la toxicité notamment pulmonaire). En routine, le scanner tho-racique permet de délinéer les volumes cibles, mais ses performances sont médiocres pour le diagnostic d’extension ganglionnaire. La dose totale de radiothérapie varie classiquement entre 60 et 70 Gy selon les indications (type tumoral et extensions), le volume d’organes sains irradiés (risque de toxicité), et l’association à une chimiothérapie. L’irradiation est administrée au rythme de 9 à 10 Gy par semaine (5 séances hebdomadaires de 1.8 à 2 Gy). Dans les CBPNC stade III, le taux de contrôle histologique local à 1 an est faible, de l’ordre de 17%3.
L’amélioration des résultats thérapeutiques passe par un bilan d’extension tumorale plus performant afin de :
• dépister les métastases présentes au diagnostic (sélection des patients relevant d’un traitement à visée curative).
• identifier et localiser les volumes cible intra-thoraciques accessibles aux traitements locorégionaux (chi-rurgie, radiothérapie).
• évaluer la réponse précoce au traitement pour adapter la stratégie thérapeutique.
La présente étude cible particulièrement sur « l’insuffisance et la toxicité des traitements », la
volonté de « mieux cibler la radiothérapie » et d’augmenter la dose délivrée au volume cible.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 11/46
La TEP au FDG est maintenant incontournable pour délimiter les volumes cibles notamment ganglionnai-res4,5. Elle a aussi montré son intérêt dans le bilan d’extension pré-thérapeutique, le bilan pré-radiothérapie et le suivi post-thérapeutique des patients. En complément du métabolisme énergétique, de nouveaux tra-ceurs TEP permettent d’explorer d’autres aspects de la biologie tumorale : hypoxie (F-miso, Faza, EF3, EF5, Fetnim, ATSM), prolifération cellulaire (FLT, FET), angiogenèse (RGD), apoptose (ML10), métabolisme os-seux (FNa), tumeurs neuroendocrines (f-dopa), récepteurs à la somatostatine (dotatoc) et aux œstrogènes (FES), …
1.1.3. L’HYPOXIE TUMORALE
L’hypoxie tumorale est un phénomène fréquent dans les CBPNPC6 et constitue un facteur important de résis-tance aux traitements cytotoxiques7,8,9,10. In vitro, la dose totale de radiothérapie doit être multipliée par 3 pour obtenir le même effet cytotoxique sur des cellules hypoxiques (pO2 < 5 mmHg) que celui observé sur des cellules normalement oxygénées. Une telle augmentation de dose n’est pas envisageable en clinique, mais plusieurs arguments laissent penser qu’un accroissement de dose plus modeste pourrait avoir un im-pact positif sur le contrôle tumoral, à condition d’être mieux ciblé sur les régions hypoxiques :
• D’un point de vue radio biologique, l’hypoxie tumorale survient soit parce que les cellules sont situées trop loin des vaisseaux sanguins (hypoxie « chronique » par insuffisance de diffusion de l’oxygène), soit parce que les (néo-)vaisseaux tumoraux sont temporairement non fonctionnels (hypoxie « aigüe » par insuffisance de perfusion). Au cours d’une radiothérapie fractionnée, une ré-oxygénation tumorale sur-vient soit parce qu’une partie des cellules situées autour des vaisseaux a été détruite par la(es) frac-tion(s) précédentes, facilitant la diffusion de l’oxygène, soit parce que les (néo-)vaisseaux sont à nou-veau fonctionnels. L’objectif d’une augmentation de dose n’est plus de dépasser la radiorésistance in-duite par l’hypoxie, mais de compenser les quelques séances pendant lesquelles les cellules tumorales étaient hypoxiques. Par ailleurs, une augmentation de dose modérée pourrait être suffisante pour vain-cre la radiorésistance consécutive à l’instabilité génétique observée lorsque des cellules tumorales sont soumises à des stress hypoxiques répétés.
• D’un point de vue clinique, une modification drastique des modalités d’irradiation (accélération de la radiothérapie) a été associée à une augmentation de la survie globale dans un essai randomisé ayant in-clus 500 patients atteints de CBPNPC traités par radiothérapie exclusive11. La démonstration d’un béné-fice consécutif à une modeste augmentation de dose est peu probable sur une population de patients non sélectionnés en fonction du profil biologique de leurs tumeurs, en raison du bruit statistique causé par l’hétérogénéité inter-tumorale, sauf à inclure plusieurs milliers de patients12. L’hypothèse est que l’identification et la localisation de zone(s) hypoxique(s), qui seraient spécifiquement ciblées par une modification des modalités d’irradiation, renforcerait la puissance statistique d’un futur essai thérapeuti-que.
• D’un point de vue technologique, les outils modernes de la radiothérapie en conditions conformationnel-les (imagerie multimodalité en position de traitement, planification dosimétrique avec correction des hé-térogénéités d’absorption, imagerie de contrôle du positionnement sous l’appareil de traitement, irradia-tion avec éventuel asservissement respiratoire ou modulation d’intensité) ont considérablement amélioré la qualité technique de l’irradiation. Nous voulons montrer qu’il est possible d’augmenter modérément la dose totale dans un volume d’intérêt biologique sans compromettre la tolérance de la radiothérapie.
1.2. ETUDES CLINIQUES
Si plusieurs revues de la littérature ont souligné l’intérêt potentiel de la TEP au F-miso pour guider des modi-fications de traitement en radiothérapie13,14,15,16,17, très peu d’études ont évalué l’apport d’un complément de dose guidé par une imagerie fonctionnelle en TEP. Les principales études ont porté sur des cancers ORL et ont débuté en 2006-2007. Celles-ci n’ont inclus que de petits nombres de patients (7 à 20), et essentielle-ment menées par une seule équipe à New York. Pour cette raison, nous présenterons le concept qui a été validé dans les cancers ORL, ainsi que les bases de notre étude sur les CBPNPC.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 12/46
1.2.1. CANCERS ORL
Une revue récente portant sur 397 patients atteints d’un cancer ORL a montré que l’hypoxie tumorale était fortement associée à l’échec du traitement (récidive locale et métastases à distance), indépendamment du stade et de la thérapeutique utilisée18. L’hypoxie tumorale mesurée en TEP permettait d’identifier les patients pouvant recevoir des agents radiosensibilisants des cellules hypoxiques19, des vasodilatateurs ou du carbo-gen comme cela a été utilisé dans l’étude ARCON (Accelerated Radiotherapy with Carbogen and Nicotina-mide)20, ou des cytotoxiques de cellules hypoxiques comme la tirapazamine. L’étude de phase III HEADSTART n’a pas montré d’amélioration de la survie lors de l’addition de tirapazamine à une chimiothéra-pie standard21. Par contre, le contrôle local était meilleur chez les patients qui avaient une hypoxie sur l’image TEP F-miso et qui avaient reçu de la tirapazamine22.
En 2007, Eschmann et al.,23 ont étudié les variations de la fixation du F-miso en TEP chez 14 patients at-teints d’un cancer épidermoïde de la sphère ORL. Des TEP au F-miso ont été réalisées en pré-RT, à 15, 30 et 45 Gy de la RT. Les auteurs ont montré une diminution du SUV chez 12/14 patients. Une récidive est surve-nue chez les 2/14 patients ayant eu une augmentation du SUVmax sur les images F-miso à 45 Gy. Les auteurs ont réalisé des images TEP à 2 et 4 h post-injection et ont évoqué la possibilité d’une hypoxie sévère en cas d’augmentation du SUV entre 2 et 4 h, ou au contraire d’une hypoxie modérée en cas de diminution du SUV entre 2 et 4 h. Les auteurs semblent observer moins de récidive en cas de diminution du SUVmax entre 2 et 4 h sur l’examen réalisé à 30 Gy. Les auteurs ont évoqué le fait que la RT pouvait induire une réoxygénation.
En 2008, Lee et al.24 (New-York) ont cherché à montrer l’intérêt d’une augmentation de dose en RT par mo-dulation d’intensité (IMRT) guidée par la TEP au F-miso. Dix patients ont été inclus. La mesure de l’hypoxie a été réalisée dans ce groupe avec la méthode proposée par Nehmeh et al.25. Les auteurs ont effectué un complément de dose jusqu’à 84 Gy sur le GTV hypoxique. La balistique a permis de respecter les organes à risque dans la mesure où le complément de dose a été effectué sur des zones hypoxiques limitées. En 2009, ce même groupe a étudié l’aspect prédictif de la TEP au F-miso à 4 semaines de la RT-CT26. Dans cette étude, seulement 2/20 patients ont montré une hypoxie persistante à 4 semaines de la RT-CT. Avec une survie médiane de 36 mois, il n’a été montré de récidive locale que chez ces 2 patients. La survie sans réci-dive à 3 ans était de 95% dans cette série.
En 2009, les mêmes auteurs ont réalisé 4 TEP chez 20 patients. Une TEP au FDG à J0, 1 TEP au F-miso à J1 et J4 et 2 TEP au F-miso à 3 jours d’intervalle à 4 semaines du début de la RT-CT (à 70 Gy). 18/20 patients avaient une fixation initiale du F-miso, laquelle a disparu chez 16/18 patients à 4 semaines de la RT. 90% des patients étaient encore vivants à 3 ans. Ces auteurs ont montré qu’il pouvait y avoir des modifications des zones hypoxiques à 72h d’intervalle27.
En 2008, les mêmes auteurs28 ont montré la faisabilité d’un complément de dose de 14 Gy en IMRT sur les zones hypoxiques, mesuré en TEP au F-miso chez 7 patients atteints d’un cancer épidermoïde ORL. Le même groupe a étudié les paramètres cinétiques du F-miso à partir d’un modèle d’analyse compartimen-tale29,30. Sur les mêmes données, les auteurs ont tenté de différencier l’hypoxie chronique de l’hypoxie aigue en étudiant la distribution de l’activité entre les TEP-F-miso réalisées à 72h d’intervalle31.
En 2007, le groupe de M. Schwaiger32 (Allemagne) a étudié 18 patients ayant une tumeur ORL localement avancée (T3-4 ; N0-3) avec une TEP au faza. Les auteurs ont effectué une délinéation manuelle puis auto-matique du GTV à partir des données du scanner seul, puis de la TEP au faza. Les patients ont été irradiés avec modulation d’intensité des faisceaux. Cette étude préliminaire a permis de montrer les rapports du GTV mesurés sur la TEP au faza et sur le scanner, dans la tumeur primitive et les ganglions. Les auteurs ont dis-cuté l’intérêt d’un complément de dose sur les zones hypoxiques, et un essai prospectif est en cours en Al-lemagne (réunion EORTC 28 mai 2010 et congrès MIRO Bruxelles mars 2010).
Une étude de faisabilité a été réalisée pour la comparaison de la TEP à l’EF5 avec la perfusion mesurée en TEP avec de l’H2
150 chez 15 patients ayant une tumeur ORL. Cette étude a montré que l’image précoce re-présentait la perfusion tumorale alors que l’image à 3h post-injection représentait bien l’hypoxie tumorale33.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 13/46
1.2.2. CANCERS BRONCHIQUES NON A PETITES CELLULES
En 2006, Cherk et al.34 ont évalué le degré d’hypoxie par TEP-F-miso chez 17 patients atteints d’un carci-nome pulmonaire non à petites cellules (CBPNPC). Les patients ont également bénéficié d’une TEP-FDG. Les auteurs ont retrouvé une corrélation faible entre la fixation de la TEP au FDG et au F-miso.
En 2006, Gacel et al.35 ont évalué la faisabilité d’une TEP au F-miso (et FDG) chez 7 patients avant et 15 jours après la fin de la RT. Cinq patients ont eu une décroissance du SUVmax en FDG et F-miso, 2 patients ont eu une réponse discordante. Les auteurs ont montré l’absence de corrélation entre le SUVmax du F-miso et la réponse au traitement. Les auteurs ont évoqué que la fixation du F-miso pouvait être une zone de ré-sistance au traitement.
En 2010, nous avons réalisé 3 TEP au FDG, F-miso et FLT chez 5 patients avant et à 46 Gy de la RT pulmo-naire. Cette étude préliminaire a montré la stabilité de l’hypoxie intra-tumorale en cours de radiothérapie (dans la tumeur et les ganglions médiastinaux), une hypoxie plus importante dans la tumeur primitive et une importante corrélation entre la fixation de FDG et du F-miso, pendant la RT (à 42 Gy), mais pas avant la RT.
1.3. FAISABILITE D’UNE AUGMENTATION DE LA DOSE TOTALE DE RADIOTHERAPIE
Les études radiobiologiques classiques ont démontré qu’une augmentation de la dose totale de radiothérapie était associée à une meilleure efficacité anti-tumorale. Les techniques modernes de la radiothérapie en con-ditions conformationnelles autorisent une augmentation de dose dans les volumes cible tumoraux tout en contrôlant précisément l’exposition des organes sains à risque de complication.
Les données cliniques disponibles ont récemment fait l’objet d’une revue extensive36.
1.3.1. RADIOTHERAPIE SANS CHIMIOTHERAPIE CONCOMITANTE
Le groupe de l’université du Michigan37 a mené une étude prospective sur 109 patients traités par radiothé-rapie exclusive, sans chimiothérapie concomitante (19% des patients avaient reçu une chimiothérapie d’induction). L’augmentation de dose était prescrite en fonction des contraintes dosimétriques pulmonaires et 84 (77%) patients ont reçu des doses supérieures à 69 Gy (dose maximale : 103 Gy). Aucune toxicité pulmonaire sévère (grade 4 ou 5) n’a été observée. Une analyse multivariée a montré que le risque de com-plication pulmonaire de grade 2 ou 3 (pneumopathie : 15%, fibrose : 14%) ne dépendait pas de la dose tumorale. La durée médiane de suivi était de 110 mois.
L’essai multicentrique RTOG 931138 a inclus 179 (177 éligibles) patients, dont 14% ont reçu une chimiothé-rapie d’induction. Les doses totales acceptables étaient de 83,8 Gy pour une V20 inférieure à 30% et de 77,4 Gy lorsque la V20 était comprise entre 25 et 36% du volume pulmonaire total.
1.3.2. RADIOTHERAPIE AVEC CHIMIOTHERAPIE CONCOMITANTE
Trois groupes coopérateurs américains ont évalué la faisabilité d’une augmentation de dose lorsque la radio-thérapie était administrée avec une association hebdomadaire de carboplatine et de paclitaxel. L’essai NCCTG 002839 a inclus 15 patients (13 évaluables), l’essai RTOG 011740,41 17 patients et l’essai CALGB 3010542 43 patients. Tous ont conclu en faveur d’une dose totale de 74 Gy. Un essai randomisé rassemblant ces 3 groupes est en cours de préparation.
En Europe, le carboplatine est déconseillé, faute de données cliniques démontrant une efficacité équivalente à celle du cis-platine43. Les associations recommandées sont soit cisplatine – étoposide, soit cisplatine – vino-relbine, administrée toutes les 3 semaines avec une dose totale de radiothérapie de 66 Gy.
Une étude de Cluj (Roumanie)44 a inclus 49 patients, dont 40 remplissaient les critères dosimétriques pulmo-naires et ont reçu jusqu’à 72 Gy en association concomitante avec cis-platine et vinorelbine. Le cisplatine a

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 14/46
été remplacé par du carboplatine chez un nombre non précisé de patients. La dose totale limitante MTD n’a pas été atteinte (pneumopathie de grade 3 à 4 : 14%) avec un recul médian de 15,9 mois.
1.3.3. EN RESUME
Le respect strict des contraintes dosimétriques pour les organes à risque (prioritairement les poumons et la moelle épinière) permet une augmentation significative de la dose de radiothérapie, y compris en association concomitante avec la chimiothérapie.
1.4. CONCLUSION
Le mauvais pronostic des cancers bronchiques primitifs tient au potentiel évolutif de la maladie (rechute locale, métastase), à l’efficacité insuffisante et la toxicité des traitements, ainsi qu’à l’existence simultanée d’autres maladies chez le même patient (comorbidités notamment liées au tabac). La TEP au FDG joue déjà un rôle essentiel dans le bilan d’extension initial du cancer et l’identification des volumes cibles intra-thoraciques de la radiothérapie. Parallèlement, sont désormais disponibles des traceurs de l’hypoxie en TEP.
Il existe une justification théorique forte pour envisager un complément de dose chez les patients ayant une tumeur pulmonaire hypoxique. La TEP au F-miso permet in vivo d’identifier et de localiser les zones tumora-les hypoxiques qui constitueraient la cible d’une irradiation à dose totale accrue. Des travaux préliminaires dans les cancers ORL ont démontré la faisabilité et l’intérêt de ce type d’approche. Il n’existe par contre aucune étude concernant le cancer pulmonaire non à petites cellules.
Le groupe porteur de ce projet travaille sur l’imagerie de ciblage pour la radiothérapie des CBPNPC depuis plusieurs années dans le cadre d’un réseau structurant labélisé par l’INCa. Nous proposons de mener une étude de phase II dont l’objectif principal sera de montrer l’augmentation du contrôle local de la maladie grâce à un complément de dose de RT. Ce projet constitue la suite de plusieurs travaux déjà menés et vise à consolider cette thématique au sein des 2 sociétés savantes françaises de médecine nucléaire et radiothéra-pie.
2. OBJECTIFS DE LA RECHERCHE
2.1. OBJECTIF PRINCIPAL
Estimer le taux de contrôle local après complément de dose des lésions hypoxiques déterminées par TEP au F-miso chez les patients éligibles [dose maximum possible sans que plus de 30% du volume pulmonaire total reçoive une dose supérieure à 20 Gy (V20)].
2.2. OBJECTIFS SECONDAIRES
• Evaluation de la tolérance à 3 mois et 1 an.
• Estimation du pourcentage de sujets pour lesquels la dose de RT a pu être augmentée.
• Estimer le taux de contrôle local chez les patients inclus mais non éligibles.
• Etude physiopathologique de la variation simultanée du métabolisme glucidique et de l’hypoxie des lé-sions (tumeur et ganglions) par TEP/TDM en cours de radiothérapie.
• Valeur prédictive sur la survie à un an, de la variation du métabolisme glucidique et de l’hypoxie des lésions (tumeur et ganglions) TEP/TDM en cours de radiothérapie.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 15/46
2.3. ETUDE ANCILLAIRE
Etudier plusieurs scénarii d’optimisation de la radiothérapie en fonction des variations du métabolisme et de l’hypoxie de la tumeur et des ganglions mesurés en cours de la radiothérapie (étude théorique sur console).
3. PLAN DE LA RECHERCHE
3.1. PLAN EXPERIMENTAL
Il s’agit d’une étude de phase II en ouvert, non randomisée, multicentrique nationale, évaluant l’efficacité et la tolérance d’un complément de dose de radiothérapie des lésions hypoxiques, identifiées par TEP/TDM au F-miso chez les patients atteints d’un CBPNPC candidats à une radio-chimiothérapie à visée curative.
• Soixante quinze patients atteints d’un CBPNPC candidats à une radio-chimiothérapie à visée curative seront pré-inclus et bénéficieront d’une chimiothérapie d’induction (2 cycles). Ces 75 patients passeront alors une TEP/TDM-FDG et une dosimétrie sera réalisée. Sur les données de notre étude RTEP1, 20% des patients ont une TEP/TDM-FDG négative au décours de la chimiothérapie d’induction.
• Soixante patients ayant une fixation sur la TEP/TDM-FDG (FDG1) seront donc définitivement inclus. Dans les 8 jours suivants, une TEP/TDM au F-miso sera réalisée (F-miso1). Nous faisons l’hypothèse minima-liste qu’au moins 50% des patients auront une tumeur hypoxique, soit au moins 30 patients (patients dits éligibles; justification en annexe). Ces 30 patients bénéficieront d’un complément de dose de RT et bénéficieront à nouveau d’une TEP/TDM au FDG et au F-miso à 42 Gy de la RT (FDG2 et F-miso2).
• Dans le cadre du suivi (hypothèse de 5 décès et/ou perdus de vue à 3 mois), 25 patients seront encore évaluables à 3 mois (évaluation de l’objectif principal) et 15 seront encore vivants à 1 an (50% de survie à 1 an). Les 30 patients qui n’auront pas bénéficié du complément de dose seront également suivis pen-dant 1 an (évaluation d’un objectif secondaire).
TEP/TDM (thorax)
F-miso1
n=60TEP/TDM
(CE)FDG1
n=75
Pré radiothérapie
8 jours max - position de traitement (ordre imposé)
Analyse qualitative par 3 MN : Hypoxie OUI / NON
SUVmaxFDG et SUVmaxF-miso
GTVFDG et GTVmiso transmis au RT pour ev. complément dose
Etude 2 ans2011-2013
Suivi 1 an2014
TEP/TDM (thorax)
FDG2
n=30
TEP/TDM (thorax)
F-miso2n=30
A 42 Gy
72h max - position de traitement (ordre libre)
Analyse qualitative par 3 MN : Hypoxie OUI/NON
SUVmax (Delta SUVmaxFDG et Delta SUVmaxF-miso)
GTVFDG et GTVmiso transmis au RT pour étude ancillaire
Evaluation
(TDM et/ou
TEP/TDM)
3 mois
n=25
1 an
n=15
Pré-inclusion
Dosimétrie prévisionnelle avec TDM
Hypothèse
50% de pts hypoxiques
Pas d’hypoxie
n=30
RT-CT std
66 Gy
Pas d’augmentation
de dose
Hypoxie
n=30RT-CT à visée curatrice
complément de dose
(dose maximale tolérable)
Bilan standard
Pré-inclusion n = 75
Taux Ctrl
Local
17% → 40%
Survie
Pc
CT induction2 cycles
Hypothèse
20% des pts négativentleur TEP-FDG après CTSortie prématurée
n = 15
Suivi 1 an
n =60
Inclusion
définitive
CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 16/46
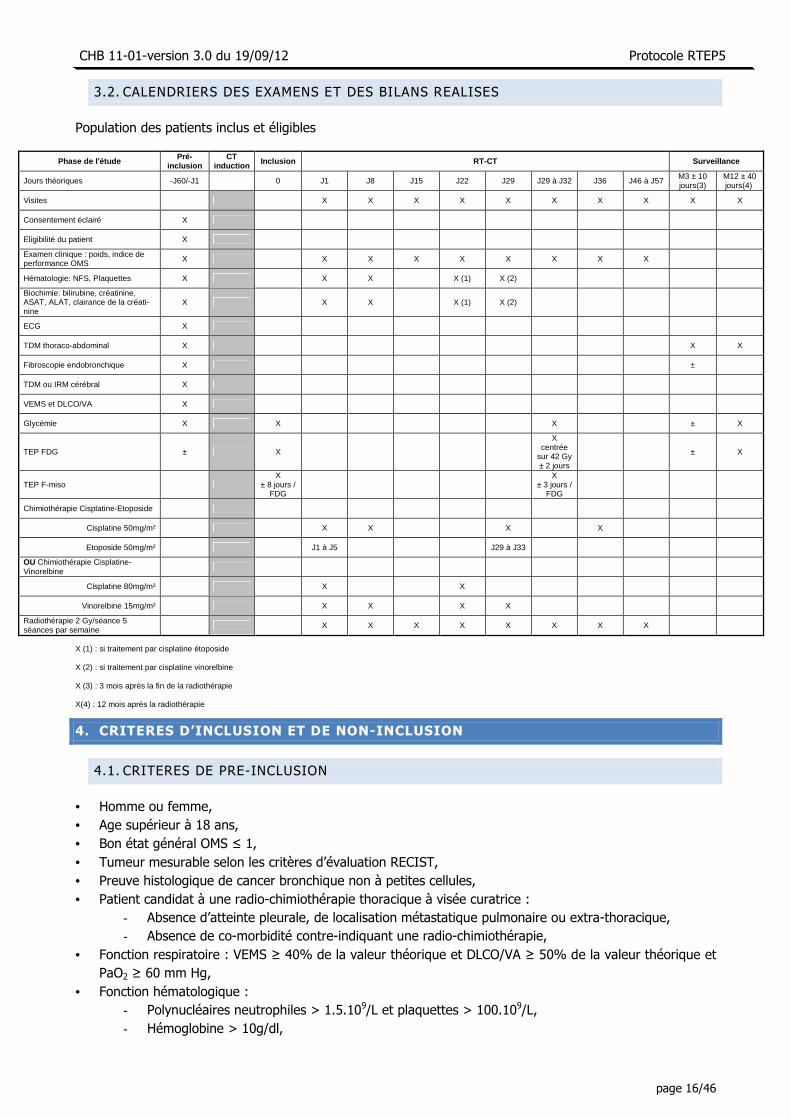
3.2. CALENDRIERS DES EXAMENS ET DES BILANS REALISES
Population des patients inclus et éligibles
Phase de l'étude Pré-
inclusion CT
induction Inclusion RT-CT Surveillance
Jours théoriques -J60/-J1 0 J1 J8 J15 J22 J29 J29 à J32 J36 J46 à J57 M3 ± 10 jours(3)
M12 ± 40 jours(4)
Visites X X X X X X X X X X
Consentement éclairé X
Eligibilité du patient X
Examen clinique : poids, indice de performance OMS X X X X X X X X X
Hématologie: NFS, Plaquettes X X X X (1) X (2)
Biochimie: bilirubine, créatinine, ASAT, ALAT, clairance de la créati-nine
X X X X (1) X (2)
ECG X
TDM thoraco-abdominal X X X
Fibroscopie endobronchique X ±
TDM ou IRM cérébral X
VEMS et DLCO/VA X
Glycémie X X X ± X
TEP FDG ± X
X centrée
sur 42 Gy ± 2 jours
± X
TEP F-miso X
± 8 jours / FDG
X
± 3 jours / FDG
Chimiothérapie Cisplatine-Etoposide
Cisplatine 50mg/m² X X X X
Etoposide 50mg/m² J1 à J5 J29 à J33
OU Chimiothérapie Cisplatine-Vinorelbine
Cisplatine 80mg/m² X X
Vinorelbine 15mg/m² X X X X
Radiothérapie 2 Gy/séance 5 séances par semaine
X X X X X X X X
X (1) : si traitement par cisplatine étoposide
X (2) : si traitement par cisplatine vinorelbine
X (3) : 3 mois après la fin de la radiothérapie
X(4) : 12 mois après la radiothérapie
4. CRITERES D’INCLUSION ET DE NON-INCLUSION
4.1. CRITERES DE PRE-INCLUSION
• Homme ou femme, • Age supérieur à 18 ans, • Bon état général OMS ≤ 1, • Tumeur mesurable selon les critères d’évaluation RECIST, • Preuve histologique de cancer bronchique non à petites cellules, • Patient candidat à une radio-chimiothérapie thoracique à visée curatrice :
- Absence d’atteinte pleurale, de localisation métastatique pulmonaire ou extra-thoracique, - Absence de co-morbidité contre-indiquant une radio-chimiothérapie,
• Fonction respiratoire : VEMS ≥ 40% de la valeur théorique et DLCO/VA ≥ 50% de la valeur théorique et PaO2 ≥ 60 mm Hg,
• Fonction hématologique : - Polynucléaires neutrophiles > 1.5.109/L et plaquettes > 100.109/L, - Hémoglobine > 10g/dl,

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 17/46
• Clairance estimée de la créatinine > 60 ml/min, • Consentement éclairé et signé avant toute procédure spécifique de l’étude. • Patient affilié à la sécurité sociale ou à une structure assimilée.
4.2. CRITERES D’INCLUSION DEFINITIVE
• Fixation tumorale supérieure au bruit de fond médiastinal sur la TEP/TDM au FDG pré-RT-CT réalisée après la CT d’induction et avant la mise en route de la RT-CT combinée.
• L’inclusion définitive est prononcée après réalisation de la dosimétrie prévisionnelle confirmant que les objectifs (dose minimale de 60Gy dans 99% des volumes cible) et les contraintes de dose (poumons, moelle épinière) sont respectés.
4.3. CRITERES DE NON INCLUSION
• Histologie autre que cancer bronchique primitif non à petites cellules, • Les patients pour lesquels aucune cible tumorale n’est évaluable (rémission complète après chimiothéra-
pie d’induction), • Absence de fixation sur un éventuel examen TEP/TDM-FDG avant la chimiothérapie d’induction, • Patients pour lesquels une radiothérapie à visée curative n’est pas indiquée (extension tumorale, métas-
tases, état général, co-morbidités), • Antécédents de maladie néoplasique de moins de 5 ans ou évolutive, • Patient déjà inclus dans un autre essai thérapeutique, • Femme enceinte, susceptible de l’être ou en cours d’allaitement, • Index de performance OMS ≥2, • Insuffisance rénale contre indiquant un traitement par Cisplatine, • Majeurs protégés (sous tutelle ou sous curatelle), • Impossibilité de se soumettre au suivi médical de l’étude pour des raisons géographiques, sociales ou
physiques, • Patients diabétiques mal équilibrés avec glycémie ≥10 mmol/L, • Hypersensibilité au FDG ou à l’un des excipients du radiopharmaceutique, • Hypersensibilité au F-miso ou à l’un des excipients du radiopharmaceutique, • Patients en incapacité de comprendre l’étude (problème de langue …).
4.4. CRITERES DE NON AUGMENTATION DE DOSE
Absence d’hypoxie définie par une évaluation négative de la fixation du F-miso sur les images à 4 heures sur l’image TEP/TDM pré-thérapeutique (F-miso1).
5. DEROULEMENT DE L’ETUDE
5.1. PHASE DE PRE-SELECTION
C’est la période qui se déroule entre le début de la chimiothérapie d’induction et la TEP d’évaluation
• Les patients pourront bénéficier d’une chimiothérapie d’induction à base de Cisplatine.
• Le patient candidat répondant aux critères de pré-inclusion doit avoir signé son consentement éclairé avant de réaliser la TEP
• La TEP est réalisée si possible 3 semaines après le second cycle
• Les données brutes du TEP doivent être enregistrées sur CD. Il s’agit du TEP FDG1

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 18/46
5.2. PHASE D’INCLUSION
Si le résultat de l’examen TEP FDG1 est positif et qu’il montre des lésions évaluables, le patient peut pour-suivre l’étude.
• Les images du TEP FDG1 seront envoyées au promoteur pour qu’il confirme la participation du patient à la deuxième phase du protocole.
• La validation de la participation est faite par un retour de fax autorisant le centre à réaliser la TEP F-miso1
• La TEP F-miso1 doit avoir lieu dans les huit jours qui suivent le PET au FDG
5.3. PHASE DE TRAITEMENT
Chaque patient reçoit une chimiothérapie associée à une radiothérapie +/- un complément de dose selon le résultat du TEP au F-miso.
5.3.1. LA RADIOTHERAPIE
Les modalités de son déroulement sont décrites chapitre 14
Sans complément de dose (patients négatif au F-miso)
• La radiothérapie se déroule dans les conditions standards d’une irradiation conformationnelle :
• La dose totale est de 66 Gy délivrée par fractions quotidiennes de 2 Gy, 5 jours par semaine
Avec complément de dose (patients positifs au F-miso)
La dose dans le PTVmiso est augmentée jusqu’à atteindre l’irradiation maximale tolérable par le poumon.
5.3.2. CHIMIOTHERAPIE
Les centres participants s’engagent à n’utiliser qu’un seul schéma d’association concomitante de chi-miothérapie pour tous les patients inclus dans l’étude. Les deux schémas proposés sont : (J1 étant considéré comme le premier jour de la radiothérapie) :
1-Cisplatine-Etoposide45 :
• Cisplatine 50 mg/m² IV à J1, J8, J29 et J36
• Etoposide 50 mg/m² de J1 à J5 et de J29 et J33
2-Cisplatine-Vinorelbine46 :
• Cisplatine 80 mg/m² IV à J1 et J22
• Vinorelbine 15 mg/m² IV à J1, J8, J22 et J29
Aucun traitement de consolidation n’est prévu durant l’étude compte tenu de l’absence de justification scientifique.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 19/46
5.3.3. TRAITEMENT DE SUPPORT
• Ils seront systématiquement associés à chaque injection de chimiothérapie selon les habitudes de chaque centre. Il est recommandé d’utiliser des sétrons associés à des corticoïdes et l’aprépitant avant et après l’injection de cisplatine.
• La prescription d’érythropoïétine (EPO) et/ou de facteur de croissance granulocytaire (GCSF) de-vra respecter les recommandations de l'EORTC 2010 (Aapro, EJC, 2011).
5.3.4. EXAMENS
Lorsqu’ils auront atteint la dose de 42Gy en radiothérapie les patients devront passer des examens TEP : la TEP FDG2 et la TEP F-miso2.
5.4. PHASE DE SURVEILLANCE
2 périodes de surveillance obligatoires sont prévues par le protocole :
• Surveillance à 3mois +/- 10 jours après la fin de l’irradiation :
- évaluation des lésions par scanner +/- TEP au FDG (critères Recist v 4.0)
- évaluation de la tolérance tardive au traitement
• Surveillance 1 an +/- 20 jours après la fin de l’irradiation
- évaluation des lésions par scanner +/- TEP au FDG (critères Recist v 4.0)
- évaluation de la tolérance tardive au traitement
Les surveillances en dehors de ces périodes sont facultatives, mais leurs données pourront être uti-lisées dans le cadre de l’étude.
Les investigateurs porteront une attention soutenue au recueil des événements indésirables lié à la chimiothérapie et à la radiothérapie. Ceux-ci devront être reportés sur des pages spécifiques du ca-hier d’observation.
5.5. SYNTHESE DES EXAMENS TEP A PREVOIR
Les données brutes de tous les TEP réalisés dans le cadre de l’étude doivent être conservées
• Pré-screening : o 1 TEP-FDG noté TEP-FDG1 réalisé au minimum 15 jours après la dernière cure de chi-
miothrépie
• A l’inclusion : o 1 TEP-FMiso noté TEP-FMiso1 réalisé max 8 jours après TEP-FDG1
• Pendant la radiothérapie : o Pour tous les patients :1 TEP-FDG noté TEP-FDG2 réalisé à 42Gy +/- 4Gy du début de la
radiothérapie o Pour les patients ayant un TEP-FMiso1 positif :1 TEP-FDG noté TEP-FDG2 réalisé à 42Gy
du début de la radiothérapie et 1 TEP-FMiso noté TEP-FMiso2 réalisé +/- 3 jours du TEP-FDG2
• Les examens TEP de surveillance post radiothérapie sont facultatifs.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 20/46
6. TOXICITE ET ADAPTATION DE DOSE
• Les toxicités seront évaluées selon l’échelle du National Cancer Institute Common Criteria NCI-CTC v 4.0 (http://evs.nci.nih.gov/ftp1/CTCAE/About.html)
6.1. TOXICITE LIEE A LA CHIMIOTHERAPIE – CONDUITE A TENIR
Un cycle ne peut débuter que si :
• Hémoglobine ≥ 10 g/dl, • Polynucléaires Neutrophiles (PNN) > 1500 / mm3, • Plaquettes > 100 000/mm3, • Clairance de la créatinine > 45 ml/min, • Neurotoxicité < grade 3, • Etat général compatible, • Absence de fièvre, • Au début de chaque cycle, l’adaptation des doses sera ajustée à la toxicité. L’adaptation des doses se
fera par paliers successifs de 25% pour chacune des molécules prescrites. Aucune augmentation de dose n’est autorisée. Dès récupération, les patients peuvent à nouveau être traités,
• Les toxicités à prendre en compte en vue de réaliser une adaptation des doses sont les suivantes : - En cas de toxicités hématologiques (PNN ≤ 500/mm3 ou plaquettes ≤ 100 000/mm3) - En cas de toxicités digestives grade 3 ou 4 - En cas de neurotoxicité grade 2 :
o pour le protocole cisplatine-étoposide : seule la posologie de cisplatine devra être adaptée avec une diminution de 50%. Aucune adaptation de dose n’est envisagée en cas de toxicité grade 1.
o pour le protocole cisplatine-vinorelbine : diminution du cisplatine de 50% et de 25% de la vi-norelbine.
- En cas de toxicité rénale avec diminution de la clairance de la créatinine : L’investigateur pourra adapter les doses de Cisplatine à la fonction rénale suivant les recommandations ci-dessous ou de remplacer le Cisplatine par du Carboplatine AUC 5.
o Entre 45 et 59ml/min o pour le protocole cisplatine-étoposide : diminution du cisplatine de 50% et de
l’étoposide de 25%, o pour le protocole cisplatine-vinorelbine : diminution du cisplatine de 50% sans adapta-
tion de dose de la vinorelbine. o Inférieure à 45ml/min : report de la cure de 8 jours avec une nouvelle estimation de la clai-
rance de la créatinine (selon la formule MDRD).
6.2. TOXICITES DE L’IRRADIATION – CONDUITE A TENIR
Interruption du traitement de radiothérapie :
• En cas d’interruption due à la défaillance d’une machine, une interruption maximale de 3 jours consé-cutifs du traitement peut être tolérée. L’irradiation devra être effectuée jusqu’aux doses prévues.
• Le nombre total de fractions et de jours doit être soigneusement signalé. • Si une interruption du traitement supérieure à 7 jours consécutifs est due à une maladie intercurrente,
le patient sortira de l’étude et il sera traité à la discrétion de l’investigateur. • Dans le cas d’une œsophagite radique de grade 3 durant le traitement concomitant, la chimiothérapie
sera suspendue et l’irradiation pourra être poursuivie. En cas d’œsophagite de grade 4, la totalité du traitement du protocole sera arrêtée. Dans tous les cas, une prise en charge symptomatique de la

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 21/46
douleur sera entreprise et une alimentation parentérale sera débutée. Le traitement pourra être repris lorsque la toxicité sera revenue à un grade 2 avec adaptation des doses de la chimiothérapie.
• Dans le cas d’une pneumopathie radique ou d’un infiltrat pulmonaire secondaire à la radiothérapie de grade 3, la totalité du traitement sera arrêtée et une corticothérapie sera mise en route.
7. SORTIE, ARRET ET FIN D’ETUDE
7.1. SORTIE D’ETUDE DES PATIENTS
L’arrêt du traitement à l'essai s'impose dans les circonstances suivantes :
• Décision du patient. • Décision de l’investigateur • Évènement grave ou imprévu • Toxicité majeure • Progression de la maladie.
En cas de sortie d’étude prématuré du patient, une visite est prévus au cours de laquelle l’évolution de la pathologie sera précisé et le choix de schéma thérapeutique déterminé par le médecin sera précisé. Une copie des examens ayant conduit à la sortie du patient devra être anonymisée et envoyé par fax au pro-moteur ainsi qu’un formulaire d’arrêt prématuré de l’étude.
Dans la mesure du possible, on s'efforcera d'assurer un suivi des patients après la fin du traitement à l'étude comme spécifié dans le chapitre 5.3
Au regard de la dose de radiothérapie nécessaire au traitement des cancers bronchiques primitifs, la mo-dicité des doses délivrées lors des examens d’imagerie ne justifie pas d’établir une période d’exclusion lors d’essais cliniques ultérieurs.
7.2. ARRET DE L’ETUDE
L'arrêt de l'étude est à la discrétion du promoteur dans l'un ou l'autre de ces cas :
• raisons d'ordre médical ou éthique ayant des conséquences sur la poursuite de l'étude, • difficultés de recrutement des patients.
7.3. FIN DE L’ETUDE
La fin de la période de traitement de l'étude est définie comme le dernier jour de radiothérapie du dernier patient. La fin de la période de suivi de l'étude est définie comme la dernière évaluation par scanner 1an après la fin du traitement.
8. EVALUATION DE L’EFFICACITE
8.1. CRITERE PRINCIPAL
L’objectif principal est d’estimer le taux de contrôle local évalué par TDM (et éventuellement TEP/TDM au FDG) à 3 mois chez les patients ayant reçu un complément de dose de RT
Critères RECIST - Lésion de base évaluable :
• CR (Complete Response : réponse complète) : disparition de toutes les lésions cibles à confirmer à 4 semaines

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 22/46
• PR (Partial Response : rémission partielle) : diminution de plus de 30% du diamètre maximal à confirmer à 4 semaines
• PD (Progression Disease : progression de la maladie) : augmentation de plus de 20% du diamètre maximal à confirmer dans 4 semaines.
• SD (Stable Disease : état stable) : tout le reste
8.2. CRITERES SECONDAIRES
• Estimation du pourcentage de complications à 3 mois et à 1 an, • Estimation du pourcentage de sujets pour lesquels la dose de RT a pu être augmentée, • Taux de contrôle local à 1 an, • Taux de survie à 1 an, • Mesure de la reproductibilité de la lecture des images TEP au F-miso, • Mesure du delta de fixation : corrélations entre les données de la TEP/TDM FDG et F-miso entre les ima-
ges pré- et per-RT-CT.
9. EVALUATION DE LA SECURITE
9.1. DEFINITIONS GENERALES
9.1.1. DEFINITION D’UN EFFET INDESIRABLE
Toute manifestation nocive survenant chez une personne qui se prête à une recherche biomédicale, que cette manifestation soit liée ou non au produit sur lequel porte cette recherche.
9.1.2. DEFINITION D’UN EFFET INDESIRABLE D’UN MEDICAMENT EXPERIMENTAL
Toute réaction nocive et non désirée à un médicament expérimental quelle que soit la dose administrée.
9.1.3. DEFINITION D’UN FAIT NOUVEAU
Toute nouvelle donnée de sécurité, pouvant conduire à une réévaluation du rapport des bénéfices et des risques de la recherche ou du médicament expérimental, ou qui pourrait être suffisant pour envisager des modifications dans l’administration du médicament expérimental, dans la conduite de la recherche.
9.1.4. DEFINITION D’UN EVENEMENT INDESIRABLE GRAVE (EIG)
Est considéré comme un évènement indésirable grave tout évènement:
• Entraînant le décès, • Mettant en jeu le pronostic vital, • Entraînant une hospitalisation ou une prolongation d’hospitalisation, • Provoquant une invalidité permanente ou une incapacité temporaire grave, • Provoquant une anomalie congénitale, une malformation fœtale ou un avortement, • Médicalement significatif. Les termes invalidité et incapacité correspondent à tout handicap physique ou psychique temporaire ou permanent, cliniquement significatif et retentissant sur l’activité physique et/ou la qualité de vie du patient.
Est considéré comme médicalement significatif tout événement clinique ou résultat de laboratoire considéré comme grave par l’investigateur et ne correspondant pas aux critères de gravité définis ci-dessus. Ils peu-vent faire courir un risque au patient et nécessitent une intervention médicale pour prévenir une issue cor-respondant à l’un des critères de gravité mentionnés précédemment (exemples : surdosages, seconds can-cers, grossesses et faits nouveaux peuvent être considérés comme médicalement significatifs).

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 23/46
N'est pas considéré comme un événement indésirable grave (EIG) :
• Une hospitalisation < à 24 heures, • Une hospitalisation programmée préalablement au début de l’essai et/ou prévue par le protocole (biop-
sie, chimiothérapie...).
9.2. DEFINITION D’UN EVENEMENT INDESIRABLE GRAVE ATTENDU (EIG-A)
Un EIG-A est un événement déjà mentionné dans la version la plus récente de la brochure investigateur ou dans le résumé des caractéristiques du produit (RCP) pour les médicaments ayant déjà une autorisation de mise sur le marché (AMM). Cette définition s’applique également au médicament de l'essai lorsqu’il est ad-ministré pour une même population hors indication de l’AMM.
Les évènements indésirables rapportés dans la littérature ou dans le RCP liés l’utilisation en routine, du FDG et du F-miso sont décrits en annexe.
9.3. DEFINITION D’UN EVENEMENT INDESIRABLE GRAVE INATTENDU (EIG-I)
Un EIG-I est un événement non mentionné ou différent par sa nature, son intensité, son évolution par rap-port à la brochure investigateur ou au résumé des caractéristiques du produit (RCP) pour les médicaments ayant une autorisation de mise sur le marché (AMM).
9.4. NOTIFICATION D’UN EVENEMENT ET D’UN EFFET INDESIRABLE GRAVE
Le promoteur déclare toute suspicion d'effet indésirable grave inattendu lié aux examens TEP/TDM au minis-tre chargé de la santé et au comité de protection des personnes concernées, sans délai et au plus tard dans un délai de sept jours à compter du jour où il en a eu connaissance.
Tous les Événements Indésirables Graves retardés (survenant après cette période de 30 jours) considérés comme raisonnablement liés au(x) traitement(s) protocolaire(s) ou à la recherche doivent être déclarés sans limitation de délai.
Tout EIG doit être communiqué par le formulaire de notification (annexe).
Pour chaque évènement indésirable grave l’investigateur devra émettre un avis sur le lien de causalité de l’évènement avec chaque médicament expérimental ou procédure en cours ou tout autre traitement éven-tuel. L’évolution clinique ainsi que les résultats des éventuels bilans cliniques et des examens cliniques ou des examens diagnostiques et/ou de laboratoire ou toute autre information permettant une analyse adé-quate du lien de causalité seront rapportées soit dans la déclaration initiale d’EIG s’ils sont immédiatement disponibles, soit ultérieurement et le plus rapidement possible pour le suivi de l’EIG.
9.5. SUIVI DES EIG
L’investigateur est responsable du suivi médical approprié des patients jusqu'à la résolution ou la stabilisa-tion de l’effet ou jusqu’au décès du patient. Cela peut impliquer parfois que ce suivi se prolonge après la sortie du patient de l’essai.
Il transmet les informations complémentaires à l’URC du centre Henri Becquerel à l’aide d’un formulaire de déclaration des EIG (en cochant la case Suivi n° X pour préciser qu’il s’agit d’un document de suivi et non d’un rapport initial). Il transmet également le dernier suivi à la résolution ou à la stabilisation de l’EIG.
Il conserve les documents concernant l’effet indésirable présumé afin de permettre, en cas de nécessité de compléter les informations précédemment transmises.
Il répond aux demandes d’informations complémentaires de l’URC pour documenter l’observation initiale.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 24/46
9.6. PARAMETRES ET METHODES D’EVALUATION DE LA SECURITE
La tolérance sera évaluée par le recueil et le suivi des événements indésirables mineurs et graves, les signes vitaux, le poids et la température des patients et le recueil des traitements concomitants.
A la première consultation, l’investigateur enregistrera les maladies actuelles et anciennes et les antécédents chirurgicaux concernant les principaux appareils, les traitements antérieurs et concomitants. Les effets indé-sirables seront recherchés à chaque consultation, tout au long de l’étude.
9.7. PROCEDURES ET DELAIS POUR LA NOTIFICATION ET L’ENREGISTREMENT DES EFFETS INDESIRABLES
Les toxicités aigues seront évaluées pendant toute la durée du traitement. Les toxicités tardives seront re-cherchées et cotées selon la classification CTCAE à chaque bilan de manière exhaustive.
9.7.1. EVENEMENTS INDESIRABLES
Tous les évènements indésirables survenant après que le sujet a signé son consentement éclairé doivent être documentés sur les pages correspondantes du cahier d’observation, ainsi que dans le dossier médical du sujet. L’évènement peut correspondre à un symptôme, un diagnostic ou à un résultat d’examen complé-mentaire jugé significatif. Tous les éléments cliniques ou para-cliniques permettant de décrire au mieux l’évènement correspondant doivent être reportés.
9.7.2. NOTIFICATION DES EFFETS INDESIRABLES GRAVES (INCLUANT LES SUSAR)
Règles pour la notification aux autorités compétentes, aux investigateurs et au CPP des effets indésirables graves inattendus et suspectés d’être associés avec la TEP/TDM.
• Investigateur au promoteur : - notification immédiate sous 48 heures - informations détaillées sur le suivi
• Le promoteur décide du caractère inattendu ou non (SAR ou SUSAR)
• Le promoteur déclare les SUSARs au module EudraVigilance Clinical Trial - dans les 7 jours - informations détaillées de suivi pendant le délai additionnel de 8 jours
Tout effet indésirable grave retardé (survenant après une période de 30 jours) considéré comme raisonna-blement relié aux procédures de l’essai doit être notifié sans limite de temps.
Tous les effets indésirables graves doivent être notifiés selon la feuille spécifique du CRF.
Pour chaque effet indésirable, l’investigateur devra donner son opinion sur la relation de causalité possible avec le traitement ou les procédures de l’essai. L’évolution clinique, les résultats des tests de laboratoire, les données de l’examen Clinique ou des examens paracliniques ou tout autre information pertinente pour ana-lyser une éventuelle relation de causalité seront recueillis dans la fiche initiale de déclaration de l’EIG immé-diatement s’ils sont disponibles ou plus tard mais dès que possible pour le monitoring de l’évolution de l’EIG.
9.8. COMITE INDEPENDANT DE SURVEILLANCE DE L’ETUDE
Il n’est pas prévu de constituer de comité indépendant de surveillance compte tenu de la méthodologie de l’essai basée sur le plan de Gehan.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 25/46
9.9. MODALITES ET DUREE DU SUIVI DES PERSONNES SUITE A LA SURVENUE D’EVENEMENTS INDESIRABLES
Rapports annuels de sécurité : le promoteur rédige les rapports annuels de sécurité et les transmet à l’AFSSAPS, au CPP et à l’investigateur coordonnateur.
Rapport final : il est rédigé par le promoteur et l’investigateur coordonnateur dans un délai d’un an après la fin de l’étude. Tous les investigateurs sont informés des résultats de l’étude. Un résumé est adressé à l’AFSSAPS par le promoteur.
10. BIOMETRIE ET PLAN D’ANALYSE STATISTIQUE
Le cancer du poumon a un pronostic très sombre avec un taux de survie à 5 ans inférieur à 10%. Ceci im-pose de choisir un plan expérimental acceptable d’un point de vue éthique et méthodologique, ce qui oriente notre choix vers la méthode de Gehan. Celle ci permet d’évaluer précocement l’efficacité sur un échantillon de petite taille sans exposer un nombre de patients à un traitement éventuellement inefficace. Il est possible alors d’arrêter rapidement cet essai en cas de non efficacité sans que ce soit préjudiciable pour les patients.
10.1. LES POPULATIONS DE L’ESSAI
10.1.1. PRE INCLUSION
• Hommes ou femmes de plus de 18 ans répondant aux critères d’inclusion et aux critères de non inclu-sion ayant signé un consentement éclairé.
• Ayant un cancer bronchique primitif non à petites cellules, prouvé histologiquement candidat à une as-sociation concomitante de chimiothérapie et de radiothérapie (dose tumorale supérieure à 60 Gy).
• Les patients doivent présenter une tumeur mesurable selon les critères RECIST et leur état général doit être coté inférieur ou égal à 1.
10.1.2. POPULATION NON ELIGIBLE
• Sujets pré-inclus présentant au moins une lésion hypermétabolique sur l’examen TEP/TDM au FDG en pré-RT (FDG1).
• Et ayant une fixation considérée comme non significative lors de l’examen au TEP F-miso en regard de la tumeur bronchique (F-miso1) (sujets considérés comme porteurs de lésions non hypoxiques)
10.1.3. POPULATION ELIGIBLE
• Sujets pré-inclus présentant au moins une lésion hypermétabolique sur l’examen TEP/TDM au FDG en pré-RT (FDG1).
• Et ayant une fixation considérée comme significative lors de l’examen au TEP F-miso en regard de la tumeur bronchique (F-miso1) (sujets considérés comme porteurs de lésions hypoxiques)
10.1.4. POPULATION EVALUABLE
• Patients éligibles pour lesquels on dispose de toutes les données nécessaires à la visite de 3 mois (après avoir éliminé les éventuels inclus à tort, les décès précoces avant 3 mois ou les patients ayant retiré leur consentement).

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 26/46
10.2. NOMBRE DE SUJETS NECESSAIRES
Il est nécessaire de disposer de 25 sujets évaluables. Nous posons comme hypothèses que :
• 20% des sujets inclus ne seront pas éligibles en raison d’une TEP/TDM-FDG négative après chimiothéra-pie (données de l’étude RTEP1)
• 50 % des sujets inclus seront non éligibles après réalisation de la TEP F-miso (justification ci-dessous).
• 5 sujets sur 30 éligibles seront non évaluables : décès précoces, examen non réalisé, retrait de consen-tement.
Nous partons donc sur 75 sujets à inclure, dont 15 ne seront pas éligibles après la TEP/TDM-FDG négative à la suite de la chimiothérapie d’induction. Sur les 60 restants, 50% soit 30 seront éligibles après réalisation de la TEP F-miso. Sur ces 30, 25 sujets seront évaluables.
10.2.1. JUSTIFICATION DE L’HYPOTHESE SUR LE POURCENTAGE DE PATIENTS HYPOXIQUES
Dans notre étude préliminaire portant sur 5 patients, 2 patients étaient hypoxiques de manière visuelle, et un 3ème patient avait des hyperfixations focalisées supérieures à 1.2 fois le bruit de fond. Dans un article sur le cancer non à petites cellules, Cherk et al.22 ont mesuré la fixation intra-tumorale (T) du F-miso chez 17 patients par rapport au bruit de fond calculé dans une région de bruit de fond sur le poumon controlatéral (N). Les auteurs ont montré un rapport T/N supérieur à 1.2 chez 16/17 patients et supérieur à 2 chez 13/17 patients. Gagel et al.47 ont mesuré chez 8 patients atteints d’un cancer non à petites cellules, la fixation du F-miso (T) par rapport à la fixation musculaire ipsilatérale (N). Les auteurs ont trouvé un rapport T/N supé-rieur à 1.2 chez 6/8 patients avant tout traitement. Chez 18 patients atteints d’un cancer pulmonaire, Deh-dashti et al.48 n’ont retrouvé qu’un seul patient qui ne présentait pas de fixation intra-tumorale du 60Cu-ATSM, soit 17/18 patients qui fixaient le traceur de l’hypoxie. Dans une étude de faisabilité du 18F-faza, Postema et al.49, ont montré une fixation du traceur chez 7/13 cancers pulmonaires.
Toutes ces données concernant les cancers bronchiques montrent une fixation des traceurs de l’hypoxie chez plus de 50% des patients. Au vu de ces données de la littérature et de notre étude préliminaire, l’hypothèse consistant à envisager 50% de patients hypoxiques sur l’examen TEP-F-miso pré-thérapeutique nous apparaît donc raisonnable.
10.3. ANALYSE STATISTIQUE
Un plan de Gehan en deux étapes est proposé. L’hypothèse sur l’efficacité thérapeutique attendue est p = 0,4.
• La phase 1 de l’inclusion a pour objectif de décider si la procédure est suffisamment efficace pour néces-siter une étude complémentaire et par la même occasion déterminer le nombre d’inclusion en fonction du taux de succès observé et par la précision souhaitée à ε= 0,10. Si aucun contrôle local n’est observé sur le groupe de 6 patients bénéficiant du complément de dose, l’essai sera arrêté (puissance 0,95).
• La phase 2 de suivi dépendra des résultats observés durant la première phase. On déterminera le nom-bre de sujets selon 4 critères (pourcentage d’efficacité à priori, valeur de risque β retenue, pourcentage de précision (10%), et nombre de succès observés dans la phase préliminaire.
Soit 18 pour un succès, 19 pour 2 succès, 15 pour 3 succès, 8 pour 4 succès.
10.3.1. ANALYSE D’EFFICACITE
L’analyse principale (taux de contrôle local à 3 mois) portera sur la population en intention de traitement, c’est à dire sur la population des sujets éligibles au complément de dose, que celle ci ait été réalisée ou non. Une analyse complémentaire per protocole sera effectuée sur tous les sujets évaluables.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 27/46
10.3.2. ANALYSES SECONDAIRES
Les analyses secondaires porteront sur :
• Le pourcentage de complication à 3 mois et à 1 an, • Le pourcentage de sujets pour lesquels la dose de RT a pu être augmentée, • Le taux de contrôle local dans la population des sujets inclus mais non éligibles • La mesure de la variation de la fixation du TEP/TDM-FDG (SUVmax) et du volume des lésions entre
l’examen de base et l’examen J42, • La mesure de la variation TEP/TDM F-miso, • Le taux de survie global à 1 an, • La mesure de la reproductibilité de la lecture des images TEP-F-miso.
Des analyses descriptives, taille d’échantillon, moyenne, déviation standard, médiane et intervalle de valeurs (minimum et maximum) seront fournies pour toutes les variables quantitatives continues ; les effectifs et pourcentages seront donnés pour les variables catégorielles telles que les données démographiques ou des variables reliées à la tumeur.
Pour chaque lésion, les variations des taux de fixation du [18F]-F-miso entre l’examen F-miso1 baseline TEP/TDM et l’examen F-miso2 TEP/TDM seront analysées selon plusieurs modes de représentation incluant le Standardized uptake values (SUV), et des cartes paramétriques de captation du [18F]-F-miso. Ces modifica-tions seront évaluées pour la recherche d’une association avec la réponse clinique de la lésion sur le groupe de tous les patients avec une approche en variable continue ou en variable dichotomique (répondeurs vs non répondeurs) déterminée à partir de la TDM. Des modèles de régression linéaire seront employés pour examiner l’ajustement de la corrélation entre les mesures en [18F]-FDG TEP, et les mesures de la réponse anatomique.
Les coefficients de corrélation de Pearson seront estimés avec leurs intervalles de confiance à 95% afin de déterminer les bornes inférieures pour le degré d’association entre la fixation en F-miso et la réponse locale au niveau de la tumeur.
10.3.3. ANALYSE DE LA TOLERANCE
Les traitements concomitants à l’inclusion ou démarrés au cours de l’essai seront décrits et listés.
10.3.3.1. EFFETS INDÉSIRABLE
Tous les effets indésirables survenus après le démarrage de l’essai seront notifiés, incluant les événements présents avant l’étude mais s’étant aggravés durant l’essai. Les évènements indésirables seront décrits, listés et cotés selon la classification NCI CTC AE v4.0 (avec classement par organe et terme préférentiel). Des tableaux récapitulatifs donneront la fréquence de survenue de ces effets secondaires (nombre de patients ayant rapporté au moins un épisode de l’effet indésirable), incidence des effets indésirables en fonction de leur intensité, incidence des effets indésirables selon le système de l’organisme, incidence des effets indési-rables ayant conduit à des sorties d’essai et incidence des effets indésirables graves. En ce qui concerne les résumés sur l’intensité et l’attribution, les méthodes adéquates seront employées dans les cas de patients qui auraient souffert à plusieurs reprises du même effet secondaire indésirable (prise en compte de la forme extrême, sévérité la plus forte et liaison la plus élevée avec le produit à l’étude).
Des commentaires et des résumés écrits seront fournis pour tous les effets graves et inattendus et pour tous les autres événements indésirables significatifs jugés comme d’un intérêt spécial en raison de leur impor-tance clinique.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 28/46
10.3.3.2. VALEURS DE LABORATOIRE
Des statistiques descriptives seront présentées avec le nombre de valeurs, les moyennes, déviation standard et intervalles de valeurs pour chacune des visites et pour les variations entre la visite de screening et les visites ultérieures. Des tableaux de changement de classe seront préparés.
10.3.3.3. SIGNES VITAUX
Pour les paramètres vitaux, des statistiques descriptives seront présentées pour les résultats à chacune des visites où ils seront collectés avec le nombre de valeurs recueillies et manquantes, les moyennes, déviation standard et intervalles de valeurs pour chacune des visites.
Toutes les analyses statistiques seront réalisées avec le logiciel SAS® system, version 9.2.
11. CALENDRIER PREVISIONNEL
Il est prévu d’inclure les 75 patients sur deux ans auxquels il faut ajouter 1 an de suivi pour chaque patient ce qui conduit au calendrier prévisionnel suivant :
• Début des inclusions: avril 2012 • Fin des inclusions: avril 2014 • Fin de l’étude: avril 2015
12. REALISATION DES TOMODENSIMOMETRIES (TDM OU SCANNERS)
Plusieurs TDM seront réalisés :
• TDM diagnostiques en début de traitement et d’évaluation à 3 mois. Ces scanners permettront de mesu-rer la taille tumorale,
• TDM de mise en place pour la réalisation du plan de traitement,
• TDM des TEP/TDM FDG (1 et 2) ou F-miso (1 et 2) et TEP/TDM-FDG à 1 an, dont l’intérêt sera la locali-sation anatomique, la correction d’atténuation, le recalage des TEP entres eux et la mesure de la taille tumorale.
Toutes ces TDM auront au moins une reconstruction axiale transverse en coupe jointives de 3 mm pour une comparaison inter-examen. Dans la mesure du possible, ces examens seront réalisés sur le même système. La taille tumorale sera mesurée selon les critères RECIST (plus grande dimension de la tumeur en fenêtre tissulaire : centre 40 HU, largeur 400 HU).
13. REALISATION DES EXAMENS D’IMAGERIE TEP
13.1. RADIOPHARMACEUTIQUE(S) ADMINISTRE(S)
Les patients bénéficieront de 4 examens TEP-TDM (cf. schéma de l’étude) : 2 avant la RT-CT (TEP-TDM FDG1 et TEP-TDM F-miso1) et 2 en cours de RT-CT, autour d’une dose de 42 Gy (TEP-TDM FDG2 et TEP-TDM F-miso2). La TEP/TDM-FDG2 est à réaliser sur l’intervalle de temps 42 Gy à ± 2 jours.
L’ordre est imposé (FDG puis F-miso) uniquement pour la séquence en pré-RT. L’ordre est libre pour la sé-quence à 42 Gy de la RT. Le délai entre les 2 examens est de 8 jours avant la RT-CT et de 72 h pendant la RT-CT. La justification de l’utilisation du F-miso comme traceur de l’hypoxie est donnée en annexe.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 29/46
13.2. PROCEDURES D’ACQUISITION
13.2.1. REALISATION DE L’EXAMEN TEP AU FDG
Les patients se rendront dans le centre effectuant les examens TEP en ambulatoire. Ils doivent être à jeun depuis 6 heures, mais ils peuvent toutefois absorber des boissons non sucrées pendant cette période. Ils doivent être placés au repos environ 1 heure avant l’injection de FDG. Une voie d’abord veineuse sera po-sée.
Une activité de 200 à 400 MBq sera injectée en fonction du poids (4 à 5 MBq/kg de poids). L’activité précise de FDG injectée sera mesurée et consignée dans le cahier d’observation (CRF). La glycémie sera contrôlée avant l’injection de FDG.
Pour l’examen TEP FDG1, le temps t(FDG1) entre l’injection et la réalisation des images doit être de 60 ± 10 min. Cette durée sera consignée dans le CRF. Pour l’examen TEP FDG2, le temps t (FDG2) entre l’injection et la réalisation des images doit être t(FDG1) = ± 5 min.
Les durées et conditions d’acquisition seront identiques pour les examens FDG1 et FDG2. Il est nécessaire de réaliser les examens TEP-FDG en position de traitement (plateau de radiothérapie, matériel de contention et de position des bras, conditions d’acquisition des données TDM à visée de simulation de traitement). A dé-faut, la TEP/TDM devra être impérativement recalée sur le scanner de mise en place. La durée de l’examen est en moyenne de 20 min. Une image corps entier sera réalisée au moyen d’un appareil TEP-TDM, couvrant la région mi-cuisse au vertex du crâne avec généralement 6 à 7 pas de déplacement du lit dans l’appareil nécessaires pour couvrir la zone d’intérêt clinique.
Pour l’examen TEP-TDM FDG2, l’acquisition sera centrée sur le thorax. 1 à 2 positions de lit seront acquises en fonction de la position et la taille de la lésion. Pour ce second examen, les constantes d’acquisitions TDM seront abaissées au maximum afin de réduire notablement l’irradiation du patient.
13.2.2. REALISATION DE L’EXAMEN TEP AU F-MISO
Les auteurs du groupe de New-York qui ont réalisé plusieurs études d’analyse compartimentale n’ont pas montré d’intérêt additionnel à faire une acquisition dynamique pour séparer les différents types d’hypoxie29,30,31. L’étude d’Eschman et al.23 a par contre suggéré de faire une image à 2h et une image à 4h post-injection. Les auteurs ont évoqué la possibilité d’une hypoxie sévère en cas d’augmentation du SUV entre 2 et 4 h, ou au contraire d’une hypoxie modérée en cas de diminution du SUV entre 2 et 4 h. Les au-teurs semblent observer moins de récidive en cas de diminution du SUVmax entre 2 et 4 h sur l’examen réali-sé à 30 Gy. Les hypothèses faites par ce groupe seront vérifiées dans notre série avec un plus grand nombre de patient.
Une activité de 4 à 5 MBq/kg de poids avec un maximum de 450 MBq sera injectée. L’activité précise de F-miso injectée sera mesurée et consignée dans le CRF. Le patient ne devra pas être à jeun.
Comme précédemment, les durées et conditions d’acquisition seront identiques pour les examens F-miso1 et F-miso2. Il est nécessaire de réaliser les examens TEP-F-miso en position de traitement (plateau de radiothé-rapie, matériel de contention et de position des bras, conditions d’acquisition des données TDM à visée de simulation de traitement).
Pour l’examen TEP F-miso1, le temps t1 (F-miso1) entre l’injection et la réalisation des premières images doit être de 120 ± 10 min et le temps t2 (F-miso1) entre l’injection et la réalisation de la seconde série d’images doit être de 240 min ± 10 min. Ces 2 durées seront consignées dans le CRF. Pour l’examen TEP F-miso2, les temps t1,2 (F-miso2) entre l’injection et la réalisation des images doit être de t1,2 (F-miso1) ± 5 min. Entre les deux acquisitions le patient est libre de faire ce qu’il souhaite.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 30/46
Pour les deux examens TEP-TDM F-miso (1 et 2), les acquisitions seront centrées sur la tumeur. 1 à 2 posi-tions de lit seront acquises en fonction de la position et la taille de la lésion. Pour ces deux examens, les constantes d’acquisitions TDM seront abaissées au maximum afin de réduire notablement l’irradiation du patient.
13.3. TRAITEMENT ET ANALYSE DES IMAGES
13.3.1. METHODOLOGIE D’ANALYSE DES IMAGES AU FDG
La reconstruction des données sur un mode itératif fournit des images dans les 3 plans orthogonaux : tran-saxial, coronal et sagittal, ainsi qu’en projection tridimensionnelle et en mode ciné. La reconstruction ano-nymisée, sera relue par 3 médecins nucléaires et comportera une évaluation qualitative (grille d’évaluation) et semi-quantitative (mesure du SUVmax) de tous les sites hypermétaboliques visibles sur l’acquisition TDM.
Les interprétations des images seront réalisées avec le réseau SFMN net. Les résultats de l’analyse visuelle seront transcrits sur une grille pré-codifiée et les informations concernant d’éventuelles sources d’artefacts ou de biais de mesures seront colligées. La fonctionnalité de ce réseau entre les centres investigateurs a été validée entre mars et aout 2011, dans le cadre d’une étude de la reproductibilité de la lecture des images TEP F-miso qui sera utilisé dans cette étude.
L’intensité de fixation du traceur dans la (les) lésions tumorales sera mesurée par la méthode du SUVmax. La valeur maximale de SUVmax sera calculée sur la partie la plus intense de la zone de captation du FDG.
Cette mesure est influencée par plusieurs paramètres (intervalle de temps séparant l’injection et la réalisa-tion des images, glycémie,…). Il faut donc respecter l’intervalle de temps entre l’injection et la réalisation des images le plus proche de 60 min et un contrôle de la glycémie avant la réalisation de l’examen. Ceci est très important pour la répétition des deux examens TEP FDG.
L’image FDG pourra servir pour la détermination du GTV.
13.3.2. METHODOLOGIE D’ANALYSE DE L’HYPOXIE
13.3.2.1. ANALYSE QUALITATIVE DES IMAGES
Les images F-miso (à 2h et 4h) post injection seront analysées visuellement et classées de manière binaire en 2 classes : positives ou négatives. Dès leur acquisition, les images seront transférées sur le réseau SFMN net pour être adressées à 3 relecteurs experts médecin nucléaire sous un délai de 3 jours ouvrés (méthodo-logie validée entre mars et aout 2011). En cas de doute, les images pourront être interprétées en utilisant une analyse quantitative. Nous appliquerons la règle définie par Rischin et al.22. La fixation était définie ini-tialement en 5 classes (0 ; 1 ; 2 ; 3 ; 4), ramenées ensuite en 2 classes (négatif : 0 ; 1) ou positif (2 ; 3 ; 4). C’est l’évaluation de l’image F-miso à 4h qui servira comme critère décisionnel pour le complément de dose de radiothérapie (tumeur hypoxique ou non). La reproductibilité de la décision en 2 classes a été démontrée dans le cadre d’une étude préliminaire. Le critère final de positivité ou négativité sera donné par l’accord des 3 experts ou la majorité en cas de discordance.
• image F-miso positive : fixation focale discrètement ou modérément supérieure au bruit de fond, • image F-miso négative : fixation inférieure ou égal au bruit de fond.
Donc la règle de décision est la suivante : • Images F-miso 2h négative et images F-miso 4h négative : patient non éligible • Images F-miso 2h positive et images F-miso 4h négative : patient non éligible • Images F-miso 2h négative et images F-miso 4h positive : patient éligible • Images F-miso 2h positive et images F-miso 4h positive : patient éligible

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 31/46
13.3.2.2. DETERMINATION DU VOLUME HYPOXIQUE
La mesure du volume hypoxique sera effectuée selon la méthode développée et validée par Nehmeh et al.27. Le volume fonctionnel tumoral en FDG est mesuré par une méthode de seuillage à 40%. Le volume hypoxi-que en F-miso correspond à la réunion de tous les pixels (en 3D) supérieur à 1,4 fois le bruit de fond et si-tués à l’intérieur du volume FDG. La région d’intérêt de bruit de fond sera choisie sur le médiastin à un même niveau de coupe. Ce volume hypoxique sera mesuré sur les images à 2h et 4h post-injection. C’est le volume hypoxique à 4h qui sera transmis au radiothérapeute pour définition du volume hypoxique (HTV). La différence entre le volume hypoxique à 2 et 4h permettra de distinguer les hypoxies sévères et modérées (cf.ci-dessus).
14. MODALITES ET PROCEDURES DE RADIOTHERAPIE
En cas de complément de dose, les images TDM ± TEP/TDM et F-miso et les dosimétries complètes seront transférées à Rouen via le réseau SFMN net. Pour des raisons de reproductibilité, la définition des GTV et HTV sera centralisée et réalisée par une seule équipe avec une définition conjointe par les services de méde-cine nucléaire et de radiothérapie de Rouen (Pr Vera et Pr Dubray). Les GTV et HTV seront ensuite transmis au radiothérapeute local pour finalisation du plan de traitement.
14.1. IDENTIFICATION ET DELINEATION DES VOLUMES CIBLE
14.1.1. IMAGERIE
L’imagerie comporte au minimum
• Soit la TEP-TDM au FDG1 sera réalisé en position de traitement sur lequel la TEP au F-miso1 aura été recalée
L’injection d’un produit de contraste intraveineux radio-opaque est facultative si un scanner diagnostique initial a déjà été réalisé avec injection, et contre-indiquée en cas d’insuffisance rénale ou d’allergie à l’iode en l’absence de prémédication. L’épaisseur maximale des coupes sera de 3 mm.
Les examens TEP pré-RT seront effectués au minimum 15 jours après la dernière cure de chimiothérapie. Aucune chimiothérapie ne sera administrée entre les TEP/TDM-FDG1 et le début de la RT-CT.
Lors des examens en position de traitement, des billes de plomb seront placées sur les points de tatouage du référentiel interne.
Les méthodes de contention et d’asservissement respiratoire (si volumes cible mobiles avec la respiration) sont celles dont l’équipe a l’habitude.
14.1.2. VOLUMES CIBLE ET DES ORGANES A RISQUE
La délinéation des volumes d’intérêt (volumes cible ou organes à risque) peut être manuelle ou automatique selon les habitudes de l’équipe. Chaque contour devra cependant être vérifié par un radiothérapeute senior (paraphe daté dans le dossier technique) avant la réalisation de la dosimétrie. Une délinéation conjointe avec un médecin nucléaire et un radiologue est recommandée.
14.1.2.1. VOLUMES CIBLE MACROSCOPIQUES (« GROSS TUMOR VOLUME » GTV)
Le GTV comprend l’ensemble des lésions tumorales, mesurables, palpables ou visibles avec les moyens ac-tuels d’imagerie50.
Plusieurs GTV peuvent être identifiés chez un même patient :

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 32/46
• Le contour de la tumeur primitive (« GTVt ») est constitué de l’union des volumes délinéés sur le scan-ner (« GTVt-TDM ») et sur le PET-FDG (« GTVt-FDG »). Le GTVt-TDM est dessiné en fenêtre parenchy-mateuse (centre -600 HU; largeur 1200 HU), sauf pour sa partie éventuellement en contact avec le mé-diastin ou la paroi (fenêtre médiastinale : Centre 40 HU; Largeur 400 UH). Le GTVt-FDG est dessiné avec un médecin nucléaire senior (seuillage fixe à 40%). Le GTVt peut être rectifié manuellement par le radiothérapeute lorsque le GTVt-FDG dépasse notoirement le GTVt-TDM. En cas d’atélectasie, le GTVt-TDM peut être rectifié en fonction du GTVt-FDG.
• Le GTV ganglionnaire (« GTVn », indexé selon la numérotation des aires ganglionnaires de Mountain)51 est constitué par tous les ganglions soit le plus petit diamètre au scanner est supérieur ou égal à 10 mm, soit qui sont le siège d’un hypermétabolisme significatif au PET-FDG (quel qu’en soit le diamètre).
• L’ensemble des GTVt-FDG et des GTVn-FDG sera défini à Rouen
Tous les GTV sont délinéés en ROUGE.
14.1.2.2. VOLUMES CIBLE HYPOXIQUES (« HYPOXIC TARGET VOLUME » HTV)
Le HTV correspond au volume considéré comme hypoxique, il sera délinéé par le médecin nucléaire (Pr P.Vera) et le radiothérapeute (Pr Dubray) de Rouen (« HTVmiso », éventuellement indexé selon la nature de la lésion – T/N – et la numérotation des aires ganglionnaires). Plusieurs HTVs peuvent être définis.
Tous les HTV sont délinéés en VIOLET
14.1.2.3. VOLUMES CIBLE ANATOMO-CLINIQUES (« CLINICAL TARGET VOLUME » CTV)
Le CTV correspond à l’ensemble du volume anatomique dans lequel on veut éradiquer la maladie cancéreuse macroscopique et/ou microscopique. La délinéation du CTV implique l’évaluation des risques d’envahissement en fonction des connaissances cliniques, et la prise en compte de risques encourus éven-tuellement par les tissus sains inclus dans ce volume50.
Plusieurs CTV peuvent être définis chez un même patient :
• L’extension microscopique dans le parenchyme pulmonaire autour de la tumeur primitive (« CTVt ») est définie par expansion isotropique de 6 mm pour les carcinomes épidermoïdes et 8 mm pour les adéno-carcinomes52. Le CTVt peut être ensuite corrigé manuellement pour exclure des structures considérées comme non envahies (gros vaisseaux, vertèbres, plèvre, paroi thoracique) et pour tenir compte d’une extension par contiguïté dans le médiastin.
• Le CTV autour des ganglions envahis (« CTVn », indexé selon la numérotation des aires ganglionnai-res51) est constitué par l’ensemble de l’aire ganglionnaire à laquelle appartient le GTVn correspondant, à délinéer selon l’atlas de l’Université du Michigan53.
• L’irradiation prophylactique des aires ganglionnaires adjacentes au(x) CTVn défini(s) au paragraphe pré-cédent et dont le risque d’envahissement microscopique est estimé à plus de 10 % est optionnelle. En pratique, les aires contenant des images ganglionnaires dont le petit diamètre est inférieur à 10mm ou sans hypermétabolisme à la TEP FDG AVANT chimiothérapie ne sont pas irradiées.
• le « CTVmiso » est délinéé avec une marge isotropique de 5 mm (résolution du PET) autour du HTV.
• L’ensemble des CTV sera défini par chaque centre suivant les recommendations précédentes
Tous les CTV sont dessinés en ROSE.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 33/46
14.1.2.4. VOLUMES CIBLE PREVISIONNELS (« PLANNING TARGET VOLUME » PTV)
Le PTV est le volume le plus large, tenant compte de toutes les incertitudes (liées au patient, à la mise en place et aux équipements), et sur lequel s’effectue l’étude dosimétrique.
Idéalement, les marges de sécurité ont été mesurées par chaque équipe tenant compte de ses pratiques et de ses équipements. Les marges isotropiques autour des CTV sont habituellement de 10 mm, parfois éten-dues à 15 mm dans le sens cranio-caudal.
Les PTV sont indexés selon les mêmes règles que les CTV. Pour la clarté de la communication avec les spé-cialistes en dosimétrie et les physiciens, la dose prescrite peut aussi être indiquée (par exemple, « PTVt-66 » pour le volume cible prévisionnel autour de la tumeur primitive et devant recevoir une dose totale de 66 Gy).
Les faisceaux de traitement sont conformés autour du(es) PTV majoré(s) de la pénombre.
• L’ensemble des PTV sera défini par chaque centre suivant les recommandations précédentes
Tous les PTV sont dessinés en BLEU CLAIR.
14.1.2.5. ORGANES A RISQUE
Au minimum, sont délinéés:
• la moelle épinière (ou le canal médullaire), sur toute la hauteur du volume irradié • les 2 poumons en totalité, en excluant le(s) GTV(s) • l’œsophage, de la bouche œsophagienne à la jonction oeso-gastrique • le myocarde
Il est également conseillé de délinéer les gros vaisseaux pour les tumeurs proximales selon les recommanda-tions de Kong FM ; Int J Radiat Oncol Biol Phys; 2011 Dec 1;81(5):1442-57. L’adjonction de marges internes autour des organes à risque est optionnelle.
14.2. OBJECTIFS ET CONTRAINTES DE DOSE TOTALE
Tous les calculs de dose sont faits avec correction des hétérogénéités d’absorption. La dose est prescrite au point ICRU. Le respect des objectifs et contraintes de dose est vérifié par examen visuel des distributions de dose dans les 3 plans de l’espace et par histogramme dose – volume. La dose reçue dans chaque PTV doit idéalement être comprise entre 95% et 107% de la dose prescrite au point ICRU.
14.2.1. OBJECTIFS DE DOSE DANS LES VOLUMES CIBLE
En l’absence d’hypoxie, la dose totale est de 66 Gy en association concomitante avec une chimiothérapie. La dose minimale est de 95% de la dose totale prescrite.
En présence d’hypoxie, la dose dans le PTVmiso est augmentée jusqu’à atteindre l’irradiation maximale tolérable par le poumon (cf. ci-dessous).
L’ensemble de l’irradiation est délivrée par fractions quotidiennes de 2 Gy, 5 jours par semaine.
14.2.2. CONTRAINTES DOSE – VOLUME POUR LES ORGANES A RISQUE
Les contraintes limitantes concernent :
• la moelle épinière (ou canal médullaire) : dose maximale de 45 Gy,

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 34/46
• les poumons : V20 Gy inférieur ou égal à 30% du volume de l’ensemble des 2 poumons (en excluant le GTV).
Pour les autres organes à risque, les contraintes d’optimisation sont :
• œsophage : Le volume recevant 50 Gy devra être inférieur à 30%,
• cœur : le volume recevant 35 Gy devra être inférieur à 30%.
Ces contraintes d’optimisation à l’œsophage et au cœur ne doivent pas faire contre indiquer l’augmentation de dose en cas de respect des contraintes de dose aux poumons.
14.3. IMAGERIE DE CONTROLE DES FAISCEAUX ET DU POSITIONNEMENT SOUS L’APPAREIL
La position et la forme des faisceaux sont contrôlées par imagerie électronique ou film lors de la première séance et lors de chaque modification éventuelle.
La position de l’isocentre est vérifiée par 2 clichés orthogonaux à chaque séance. Une imagerie scannogra-phique sous l’appareil peut être réalisée une fois par semaine (limiter la dose due à l’imagerie).
Les déviations supérieures ou égales à 5 mm sont corrigées avant le traitement. L’imagerie de contrôle de l’isocentre est refaite avant traitement si le(s) décalage(s) est (sont) supérieur(s) ou égal(ux) à 1 cm.
15. ROLE RESPECTIF DES INVESTIGATEURS/EQUIPES
Par centre, il a été défini 6 investigateurs : 1 médecin nucléaire, 1 radiothérapeute, 1 pneumologue, 1 physi-cien médical, 1 radiopharmacien et 1 manipulateur et/ou ARC.
Le médecin nucléaire fera les examens TEP/TDM. Le radiothérapeute fera le plan de traitement et le traite-ment de radiothérapie. Le pneumologue et le radiothérapeute sélectionneront les patients en réunion multi-disciplinaire. Le physicien médical assurera le contrôle de qualité des TEP/TDM et de la radiothérapie. Le radiopharmacien assurera le contrôle de qualité du FDG et du F-miso. Le manipulateur et/ou ARC assurera le transfert des données entre les centres et le remplissage du cahier d’observation (CRF électronique via le logiciel Clinsight, CTD du cancéropôle nord-ouest).
Un informaticien coordinateur (Mr Modzelewski, Rouen) assurera la faisabilité technique du transfert des données en lien avec le réseau SFMN net.
Les images TEP/TDM seront acquises localement. Ces images seront transférées à Rouen ou seront définis les GTV et les HTV par les Pr Vera et Dubray. Ces volumes seront renvoyés au radiothérapeute local. Le plan de traitement définitif sera discuté avec le Pr Dubray. La décision du traitement final appartient au radiothé-rapeute local.
Dans le premier temps du plan de Gehan, seuls les centres de Rouen, Lyon, Lille et Toulouse seront impli-qués. Dans la 2ème phase, l’étude sera progressivement élargie aux autres centres après validation des éta-pes de contrôles qualité (cf.infra).
16. FAISABILITE
16.1. POTENTIEL D’INCLUSION
Le potentiel d’inclusion par les 22 centres français a été évalué à 250 patients par an. Nous ne devrions donc pas avoir de difficulté à recruter 75 patients sur 2 ans.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 35/46
16.2. TRAVAIL PREPARATOIRE SUR LE TRANSFERT DES DONNEES
Le Dr S. Thureau, radiothérapeute à Rouen a évalué durant le 1er semestre 2011, le transfert des images et contours sur les centres participants, ainsi que la validation/portabilité du contourage des volumes cibles métaboliques (GTV) et hypoxiques (HTV) de chaque centre. Ce travail a permis de valider la faisabilité du transfert des images via SFMNnet, leur lecture selon la classification de Risching et al (JCO 2006) ainsi que le transfert de contourage des différents centres participants à Rouen. Ce travail s’est inscrit dans le cadre d’un M2, financé par ailleurs et réalisé au sein du laboratoire QuantIF-LITIS sous la direction des Pr Vera et Du-bray (master 2 Radiothérapie et Radiobiologie, 2010-2011 - Paris XI).
17. CONTROLE ET ASSURANCE QUALITE
17.1. ASSURANCE QUALITE
Le promoteur est responsable de la mise en place et du maintien d’un système d’assurance qualité afin de garantir l’authenticité et la crédibilité des données conformément aux bonnes pratiques cliniques.
Le système comprend:
• la gestion de l’essai selon les procédures de l’URC du centre Henri Becquerel,
• le contrôle qualité des données du site investigateur par l’ARC promoteur.
17.2. CONTROLE QUALITE
Le contrôle qualité des données cliniques et paracliniques (notamment radiothérapie) sera effectué par un attaché de recherche clinique mandaté par le promoteur. La nature et la fréquence des visites de monitoring seront établies selon le risque pour s’assurer du respect du protocole et des exigences liées à la recherche.
Le contrôle portera sur les données recueillies au cours de l’étude et sur le consentement de tous les pa-tients inclus.
Un attaché de recherche clinique du réseau SFMN net prendra en charge spécifiquement tous les aspects reliés à l’assurance et au contrôle qualité des examens d’imagerie.
17.3. CONTROLE DE QUALITE DE LA RADIOTHERAPIE
Avant ouverture du centre, un dossier d’imagerie sera entièrement pris en charge (délinéation des volumes d’intérêt, planification dosimétrique, images de référence pour le contrôle du positionnement).
Une copie électronique (formats DICOM et DICOM-RT) de l’ensemble du dossier d’imagerie et de radiothéra-pie sera adressée pour archivage aux investigateurs principaux.
17.4. CONTROLE QUALITE DES INSTALLATIONS TEP-TDM
La mise en réseau de 22 installations de TEP-TDM nécessite l’harmonisation des protocoles d’acquisitions et de reconstructions. Le contrôle qualité permettra alors d’assurer cette homogénéité tout en garantissant la réalisation d’examens dans des conditions optimales. Le programme d’assurance qualité de l’imagerie FDG et F-MISO respectera plusieurs niveaux de contrôle.
17.4.1. PREMIER NIVEAU : PERFORMANCES INTRINSEQUES DES INSTALLATIONS
Le premier niveau s’adresse en particulier aux tests dits « d’acceptabilité » des installations. Ces tests doi-vent permettre de mesurer les performances intrinsèques du système, de les corréler à la qualité des images

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 36/46
acquises et de vérifier les données communiquées par le constructeur. Effectués à la réception de l’installation, ces tests peuvent également être réalisés périodiquement (annuel) et constitueront ici le test initial lors de la mise en place du réseau.
Des protocoles internationalement reconnus permettent déjà d’assurer la réalisation de ces contrôles. Ainsi, ce premier niveau de contrôle se basera sur le protocole NEMA (réf.: NEMA NU 2 2007). Chaque centre par-ticipant fournira alors un rapport de contrôle qualité basé sur le protocole NEMA au début de l’étude. En cas de maintenance importante pendant l’étude, un nouveau rapport de contrôle devra être fourni.
17.4.2. DEUXIEME NIVEAU : SUIVI LONGITUDINAL
Le deuxième niveau consiste au suivi longitudinal de certains paramètres du système par un contrôle interne et périodique afin de détecter et mesurer quantitativement les dérives éventuelles. Ce deuxième niveau est crucial dans la mise en place d’un réseau visant à la réalisation des examens TEP dans des conditions tech-niques reproductibles.
En l’absence de consensus sur le plan national ou d’obligation légale, nous avons retenu une méthode simple et peu couteuse en temps de manipulation afin de minimiser l’impact de ces contrôles sur l’activité clinique. Cette méthode consiste en l’utilisation d’un cylindre homogène rempli d’une activité donnée de FDG. La me-sure de plusieurs zones d’intérêts sur les images reconstruites permettra de vérifier, à la fois, la constance du SUV, liée à la calibration de l’installation, et l’uniformité des images. Afin de minimiser les erreurs de re-productibilités liées à l’analyse des images, les images acquises seront centralisées et analysées par l’attaché de recherche clinique du réseau SFMN net. La périodicité de ce contrôle sera fixée à un mois afin d’avoir les données nécessaires pour observer une éventuelle dérive d’une des installations du réseau.
17.4.3. TROISIEME NIVEAU : QUALITE DE LA SEGMENTATION
Le troisième niveau s’adresse au contrôle des outils de segmentation utilisés afin d’assurer la reproductibilité quelque soit le centre ou l’utilisateur final.
Un fantôme spécifique sera mis à disposition des centres. Une acquisition sera effectuée en respectant les paramètres d’acquisition et reconstructions du protocole. Les images obtenues seront segmentées locale-ment à l’aide de la méthode préconisée et le résultat transmis à l’ARC du réseau SFMN net qui validera les données. Ce processus doit assurer la qualité de l’ensemble de la chaîne : de l’acquisition à la définition du volume hypoxique. Ce test devra être réalisé annuellement ou après une maintenance importante du sys-tème nécessitant une recalibration de l’algorithme de segmentation.
17.4.4. EQUIPEMENTS DES LABORATOIRES
Parallèlement aux contrôles liés à l’imagerie, le contrôle qualité des équipements des laboratoires (chambres à puits…) devra également être assuré puisqu’il s’agit d’un élément important de la chaîne d’acquisition et essentiel dans les aspects quantification. Ces contrôles seront alors effectués conformément à la décision de l’AFSSAPS en respectant les référentiels déjà proposés par le Syndicat des radiopharmaciens.
17.4.5. TRAITEMENT DES NON-CONFORMITES
Dès qu’une non-conformité sera détectée par l’ARC du réseau SFMN net (dérive de la mesure du SUV, pro-blème de segmentation…), le centre sera avisé afin d’isoler et résoudre le problème avec le support des physiciens médicaux associés au réseau si nécessaire.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 37/46
18. JUSTIFICATION ALARA, DOSE EFFICACE TOTALE
18.1. JUSTIFICATION DE LA REALISATION DE DEUX EXAMENS TEP AU FDG ET AU F-MISO
La réalisation d’une TEP-FDG et d’une TEP-F-miso nous apparaît essentielle pour 2 raisons
• Le volume hypoxique déterminé en F-miso sera défini à l’intérieur du volume hypermétabolique (mesuré par une méthode semi-automatique développée dans le laboratoire QuantIF-LITIS défini sur l’image TEP-FDG après recalage des images.
• La corrélation entre métabolisme et hypoxie reste extrêmement débattue dans la littérature avec des résultats discordants54,55,56,57,58,59,60. Dans notre étude préliminaire (5 patients), nous avons montré une absence de corrélation entre le SUVmax du FDG et du F-miso avant la radiothérapie (r = 0.19, p = 0.35), mais une corrélation significative (r = 0.70, p = 0.008) entre ces 2 fonctions à 42 Gy. Ceci tend à mon-trer que la radiothérapie a une influence sur ces 2 fonctions, expliquant possiblement les résultats dis-cordants observés dans la littérature. Nous voudrions confirmer ces résultats sur une plus grande série de patients61.
18.2. DONNEES DOSIMETRIQUES
L’exposition des patients dans le cadre des examens TEP-TDM résulte à la fois de l’irradiation par le radio-pharmaceutique utilisé en TEP, mais aussi par l’imagerie TDM. Pour un patient de 75 kg, la dose effective délivrée au corps entier est estimée à 11,25 mSv pour une TEP au FDG et à 1,95 mSv pour une TEP au F-miso. Dans le cadre de cette étude thérapeutique, 3 examens TEP-TDM dont 1 examen au FDG (FDG2) et 2 examens au F-miso (F-miso1 et F-miso2) seront réalisés spécifiquement pour cette recherche conduisant à un excédent de dose efficace totale d’environ 15 mSv. Afin de minimiser la dose délivrée par les examens TDM associés aux examens TEP, une étude dosimétrique sur fantôme anthropomorphique a été réalisée. Les 3 examens TDM associés aux examens TEP complémentaires induiront une dose effective de 1 mSv par exa-men, soit 3 mSv pour les 3 examens62 (scanner très faible dose pour ces 3 scanners additionnels). Ces 3 scanners additionnels sont apparus indispensables car ils serviront de base au recalage des images avec la TEP-TDM FDG1.
En conclusion, l’excédent de dose dans le cadre de ce protocole est de l’ordre de 20 mSv. Comparativement, la dose délivrée par l’examen TDM corps entier en radiologie est de l’ordre de 30 mSv62.
19. ANNEXE : JUSTIFICATION DE L’UTILISATION DU F-MISO
Concernant le choix du traceur de l’hypoxie tissulaire, une revue exhaustive de la littérature a été effectuée pour trouver le meilleur compromis entre la qualité de ce traceur et la possibilité de distribution sur le terri-toire national (excluant – ou rendant très difficile en dehors d’une étude de Phase I – en France, les traceurs marqués au 60Cu ou 64Cu, et donc l’ATSM). Le tableau suivant résume ces données.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 38/46
P1, P2, P3 : Nombre d’étude de phase I, II, III
Lors de l’analyse de la littérature, il est apparu que le FAZA pouvait présenter un intérêt dans la détection des régions hypoxiques63,64. En effet, il présenterait une élimination rapide des tissus sains tout en conser-vant une bonne captation dans les tissus appauvris en oxygène. Cependant, le F-miso reste la molécule la plus documentée et pourrait prétendre à une AMM (Autorisation de mise sur le marché) simplifiée de type APUCEC (Autorisation Provisoire d’Utilisation et de Commercialisation pour Evaluation Clinique)65. Cette mo-lécule présente une bonne corrélation entre les zones appauvries en oxygène et la captation de la radioacti-vité66. Du fait de sa clairance lente des tissus sains, il est préconisé de réaliser une image plus de deux heu-res après injection. Un ratio tumeur sur bruit de fond de 1,2 à partir de deux heures post-injection est carac-téristique de la captation de ce traceur67. Toutefois, pour garder une densité de coups suffisante, une dose injectée au patient de 4 MBq/Kg avec un maximum à 450 MBq devrait être réalisée pour pouvoir réaliser une image de qualité à 2h et 4h post injection68. Enfin, la disponibilité de cette molécule au niveau national peut être assurée par au moins deux laboratoires différents. Le FETNIM, quant à lui, reste relativement peu do-cumenté et présente un profil pharmacocinétique intermédiaire entre le F-miso et le FAZA. Les deux derniè-res molécules (EF3 et EF5) restent encore peu disponibles du fait de leur statut « universitaire ».
20. ANNEXE : CONSIDERATIONS ETHIQUES ET REGLEMENTAIRES
L'essai clinique doit être conduit conformément :
• aux principes éthiques de la dernière version en vigueur de la déclaration d'Helsinki, • aux Bonnes Pratiques Cliniques de la Conférence Internationale d'Harmonisation (ICH–E6, 17/07/96), • à la Directive Européenne (2001/20/CE) sur la conduite des essais cliniques, • à la loi Huriet (n° 88-1138) du 20 décembre 1988 relative à la Protection des Personnes se prêtant à la
Recherche Biomédicale et modifiée par la loi de Santé Publique (n° 2004-806) du 9 août 2004, • à la loi Informatique et Libertés n° 78-17 du 6 janvier 1978 modifiée par la loi n° 2004- 801 du 6 août
2004 relative à la protection des personnes physiques à l'égard des traitements de données à caractère personnel,

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 39/46
• à la loi bioéthique n° 2004-800 du 6 août 2004.
20.1. COMITE DE PROTECTION DES PERSONNES
Avant de réaliser une recherche biomédicale sur l'être humain, le promoteur est tenu d'en soumettre le pro-jet à l'avis de l'un des comités de protection des personnes compétents pour le lieu où l'investigateur coor-donnateur exerce son activité. La demande d'avis du projet de recherche biomédicale est adressée au comi-té par le promoteur.
Les demandes de modifications substantielles des projets initiaux sont également adressées par le promo-teur au comité pour avis.
20.2. AUTORITE COMPETENTE
Avant de réaliser ou de faire réaliser une recherche biomédicale le promoteur de cette recherche adresse une demande d'autorisation à l’Agence Française de Sécurité Sanitaire des Produits de Santé (AFSSAPS).
20.3. INFORMATION ET CONSENTEMENT DES PARTICIPANTS
Préalablement à la réalisation d'une recherche biomédicale sur une personne, le consentement libre, éclairé et exprès de celle-ci doit être recueilli après qu'elle ait été informée de l’objectif de la recherche, du dérou-lement et de la durée de l’étude, des bénéfices, risques potentiels et contraintes de l’étude et de l'avis donné par le CPP (art. L.1122-1).
Le formulaire de consentement sera daté et signé personnellement par le patient et l’investigateur ou le médecin qui le représente (original archivé par l’investigateur, une copie sera remise au patient ou à son représentant légal).
Les formulaires de fiche d’information et de consentement éclairé (annexe) destinés au patient doivent être associés sur un même document afin d’éviter tout risque de contestation sur le contenu de l’information donnée.
20.4. RESPONSABILITE DES INVESTIGATEURS
L'investigateur principal s'engage à conduire l'essai clinique conformément au protocole qui a été approuvé par le CPP. L'investigateur ne doit apporter aucune modification au protocole sans l'autorisation du promo-teur et sans que le CPP ait donné un avis favorable sur les modifications proposées.
Il est de la responsabilité de l’investigateur coordonnateur :
• de fournir au promoteur son curriculum vitae ainsi que ceux des co-investigateurs ; • d'identifier les membres de son équipe qui participent à l'essai et de définir leurs responsabilités ; • de démarrer le recrutement des patients après autorisation du promoteur ; • de faire le maximum pour inclure le nombre requis de patients dans les limites de la période de recrute-
ment établie. Il est de la responsabilité de chaque investigateur : • de recueillir le consentement éclairé daté et signé personnellement par le patient avant toute procédure
de sélection spécifique à l'essai, • de notifier tout Événement Indésirable Grave (EIG) à la cellule de pharmacovigilance du promoteur dans
les 48 heures ouvrables, • de compléter régulièrement les cahiers d'observation (CRF) pour chacun des patients inclus dans l'essai
et de laisser à l'Assistant de Recherche Clinique (ARC) un accès direct aux documents sources afin que ce dernier puisse valider les données du CRF,
• de dater, corriger et de signer les corrections des CRF pour chacun des patients inclus dans l'essai.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 40/46
Toute la documentation relative à l'essai (protocole, consentements, cahiers d'observation, dossier investiga-teur, etc.…), ainsi que les documents originaux (résultats de laboratoire, radiologies, comptes-rendus de consultations, rapports d'examens cliniques pratiqués, etc.) doivent être détenus dans un lieu sûr et considé-rés comme du matériel confidentiel.
L'archivage des données sera sous la responsabilité de l'investigateur et selon la législation en vigueur.
Ce dernier devra conserver les données, ainsi qu’une liste d'identification des patients pendant une durée minimale de 15 ans après la fin de l’essai.
21. ANNEXE : DROIT D’ACCES AUX DONNEES ET DOCUMENTS SOURCES
Les personnes autorisées à avoir un accès direct conformément aux dispositions législatives et réglementai-res en vigueur, notamment les articles L.1121-3 et R.5121-13 du code de la santé publique (investigateurs, personnes chargées de contrôle qualité, assistants de recherche clinique, auditeurs ainsi que toutes les per-sonnes appelées à collaborer aux essais) doivent prendre toutes les précautions nécessaires en vue d’assurer la confidentialité des informations relatives aux médicaments expérimentaux, aux essais, aux personnes qui s’y prêtent notamment en ce qui concerne leur identité ainsi qu’aux résultats obtenus.
Les données collectées par ces personnes au cours des contrôles qualité ou des audits sont alors rendues anonymes.
22. ANNEXE : REGLES RELATIVE A LA PUBLICATION
Les résultats de cet essai sont la propriété exclusive des responsables du centre investigateur, qui pourront les exploiter librement. Toutes les informations résultant de cette étude sont considérées comme confiden-tielles, au moins jusqu'à ce que l'analyse appropriée et le contrôle par les coordonnateurs soient achevés.
L’ordre des auteurs sera défini de la manière suivante :
- Le premier auteur sera l’investigateur coordonnateur
- Les second et troisième auteurs seront des méthodologistes et/ou des personne ayant participé très activement à l’étude.
- Le dernier auteur sera un radiothérapeute faisant parti du comité de relecture.
- Autrement l’ordre des auteurs sera déterminé en fonction du nombre d’inclusion, à savoir :
� Les centres ayant plus de trois patients inclus dans cette étude auront un médecin nu-cléaire et un radiothérapeute cité comme auteur.
� Les centres ayant moins de trois patients inclus dans l’étude auront un médecin nu-cléaire ou un radiothérapeute cité comme auteur.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 41/46
23. ANNEXE : LISTE DES ABREVIATIONS
• AMM : Autorisation de Mise sur le Marché • APUCEC : Autorisation Provisoire d’Utilisation et de Commercialisation pour Evaluation Clinique • ATSM : Diacetyl-bis-N4-methylthiosemicarbazone • BTV : Biological Target Volume : Volumes cible biologiques • CBPNPC : cancer bronchique primitif non à petites cellules • CR : complete response : réponse complète • CT: Chimiothérapie • CTC AE: Common Terminology Criteria for Adverse Effects • CTV: Clinical Target Volume: Volumes cible anatomo-cliniques • FDG: 18F-Fluoro-deoxyglucose • FET: 18F-fluoroethyl-L-tyrosine • FLT: 18F-fluorothymidine • F-miso: 18F-FluoroMisonidazole • GTV: Gross Tumour Volume: Volume cible macroscopique • HTV : Hypoxic Tumour Volume • ICRU : International Commission on Radiation Units and Measurements • IFCT : Intergroupe Francophone de Cancérologie Thoracique • IMRT: intensity modulated radiation therapy • INCa : Institut national du cancer • IRMd : Imagerie par résonnance magnétique dynamique • PD : Progression Disease • PR : Partial Response • SD : Stable Disease • TDM : tomodensitométrie • TEP : tomographie par émission de positrons • RECIST : Response Criteria in Solid Tumors • RT : radiothérapie • SFMN : Société Française de Médecine Nucléaire • SFRO : Société Française de Radiothérapie Oncologie • SUV : Standardized Uptake Value

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 42/46
1 FNCLCC. Standards, Options et Recommandations pour la prise en charge des patients atteints de cancers bronchopulmonaires non à petites cellules. FNCLCC, Coll, Paris, 2000.
2 Aupérin A, Le Péchoux C, Rolland E, Curran WJ, Furuse K, Fournel P, Belderbos J, Clamon G, Ulutin HC, Paulus R, Yamanaka T, Bozonnat MC, Uitterhoeve A, Wang X, Stewart L, Arriagada R, Burdett S, Pignon JP. J Clin Oncol. 2010 28(13):2181-90.
3 Arriagada R, Le Chevalier T, Quoix E, Ruffie P, de Cremoux H, Douillard JY, Tarayre M, Pignon JP, Lap-lanche A. (American Society for Therapeutic Radiology and Oncology) plenary: Effect of chemotherapy on locally advanced non-small cell lung carcinoma: a randomized study of 353 patients. GETCB (Groupe d'Etude et Traitement des Cancers Bronchiques), FNCLCC (Fédération Nationale des Centres de Lutte contre le Can-cer) and the CEBI trialists. Int J Radiat Oncol Biol Phys. 1991 20(6):1183-90.
4 Nestle U, Weber W, Hentschel M, Grosu AL. Biological imaging in radiation therapy: role of positron emis-sion tomography. Phys Med Biol. 2009 54(1):R1-25.
5 Ford EC, Herman J, Yorke E, Wahl RL. 18F-FDG PET/CT for image-guided and intensity-modulated radio-therapy. J Nucl Med. 2009 50(10):1655-65.
6 Cherk MH, Foo SS, Poon AM, Knight SR, Murone C, Papenfuss AT, Sachinidis JI, Saunder TH, O'Keefe GJ, Scott AM. Lack of correlation of hypoxic cell fraction and angiogenesis with glucose metabolic rate in non-small cell lung cancer assessed by 18F-Fluoromisonidazole and 18F-FDG PET. J Nucl Med. 2006 47(12):1921-6.
7 Barendsen GW, Koot CJ, Van Kersen GR, Bewley DK, Field SB, Parnell CJ. The effect of oxygen on impair-ment of the proliferative capacity of human cells in culture by ionizing radiations of different LET. Int J Ra-diat Biol Relat Stud Phys Chem Med. 1966 10:317–327.
8 Hockel M, Schlenger K, Mitze M, Schaffer U, Vaupel P. Hypoxia and radiation response in human tumors. Semin Radiat Oncol. 1996 6:3–9.
9 Gabalski EC, Adam M, Pinto H, Brown JM, Bloch DA, Terris DJ. Pretreatment and midtreatment measure-ment of oxygen tension levels in head and neck cancers. Laryngoscope. 1998 108:1856–1860.
10 Tatum JL, Kelloff GJ, Gillies RJ, et al. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol. 2006 82:699–757.
11 Saunders M, Dische S, Barrett A, Harvey A, Griffith G, Parmar M, (on behalf of the CHART Steering com-mittee Continuous, hyperfractionated, accelerated radiotherapy (CHART) versus conventional radiotherapy in non-small cell lung cancer: mature data from the randomised multicentre trial. Radiother Oncol. 1999 52(2):137-48.
12Thames HD, Schultheiss TE, Hendry JH, Tucker SL, Dubray BM, Brock WA. Can modest escalations of dose be detected as increased tumor control? Int J Radiat Oncol Biol Phys. 1992 22(2):241-6.
13 Bussink J, van Herpen CM, Kaanders JH, Oyen WJ. PET-CT for response assessment and treatment adap-tation in head and neck cancer. Lancet Oncol. 2010 11(7):661-9.
14 Ferrer Albiach C, Conde Moreno A, Rodríguez Cordón M, Morillo Macías V, Bouché, Babiloni A, Beato Tor-tajada I, Sánchez Iglesias A, Francés Muñoz A. Contribution of hypoxia-measuring molecular imaging tech-niques to radiotherapy planning and treatment. Clin Transl Oncol. 2010 12(1):22-6. Erratum in: Clin Transl Oncol. 2010 12(2):156.
15 Troost EG, Schinagl DA, Bussink J, Boerman OC, van der Kogel AJ, Oyen WJ, Kaanders JH. Innovations in radiotherapy planning of head and neck cancers: role of PET. J Nucl Med. 2010 51(1):66-76.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 43/46
16 Schöder H, Fury M, Lee N, Kraus D. PET monitoring of therapy response in head and neck squamous cell carcinoma. J Nucl Med. 2009 50 Suppl 1:74S-88S.
17 Minn H, Grönroos TJ, Komar G, Eskola O, Lehtiö K, Tuomela J, Seppänen M, Solin O. Imaging of tumor hypoxia to predict treatment sensitivity. Curr Pharm Des. 2008 14(28):2932-42.
18 Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, Terris DJ, Overgaard J. Prognostic value of tumor oxygenation in 397 HN tumors after primary radiation therapy: an international multi-center study. Radiother Oncol. 2005 77:18–24.
19 Rosenberg A, Knox S. Radiation sensitization with redox modulators: a promising approach. Int J Radiat Oncol Biol Phys. 2006 64:343–354.
20 Kaanders JH, Pop LA, Marres HA, Bruaset I, van den Hoogen FJ, Merkx MA, van der Kogel AJ. ARCON: experience in 215 patients with advanced HN cancer. Int J Radiat Oncol Biol Phys. 2002 52:769–778.
21 Rischin D, Peters LJ, O'Sullivan B, Giralt J, Fisher R, Yuen K, Trotti A, Bernier J, Bourhis J, Ringash J, Henke M, Kenny L Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J Clin Oncol. 2010 28(18):2989-95.
22 Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, Peters LJ; Trans-Tasman Radiation Oncology Group Study 98.02. Prognostic significance of [18F]-misonidazole PET-detected tumor hypoxia in patients with advanced HN cancer randomly assigned to chemoradiation with or without tirapazamine. J Clin Oncol. 2006 24:2098–2104.
23 Bedeshem C, Machulla HJ, Hehr T, Bamberg M, Bares R. Hypoxia-imaging with (18)F-misonidazole and PET: changes of kinetics during radiotherapy of head-and-neck cancer. Radiother Oncol. 2007 83(3):406-10.
24 Lee NY, Mechalakos JG, Nehmeh S, Lin Z, Squire OD, Cai S, Chan K, Zanzonico PB, Greco C, Ling CC, Humm JL, Schöder H. Fluorine-18-labeled fluoromisonidazole positron emission and computed tomography-guided intensity-modulated radiotherapy for head and neck cancer: a feasibility study. Int J Radiat Oncol Biol Phys. 2008 70(1):2-13.
25 Lee N, Schröder H, Erdi YE, Greco C, Mageras G, Pham HS, Larson SM, Ling CC. Reproducibility of intra-tumor distribution of (18)F-fluoromisonidazole in head and neck cancer. Int J Radiat Oncol Biol Phys. 2008 70(1):235-42.
26 Lee N, Nehmeh S, Schöder H, Fury M, Chan K, Ling CC, Humm J. Prospective trial of incorporating pre-/mid-treatment [18F]-misonidazole PET for HN cancer patients undergoing concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2009 75(1):101-8.
27 Nehmeh S, Lee N, Schröder H, Squire O, Zanzonico P, Erdi Y, Greco C, Mageras G, Pham H, Larson S, Ling C, Humm J. Reproducibility of Intratumor Distribution of 18F-fluoromisonidazole in Head and Neck Cancer. Int J Radiat Oncol Biol Phys. 2008 70(1): 235–242.
28 Zhixiong L, Mechalakos J, Nehmeh S, Schoder H, Nancy L, Humm J, Clifton L. The influence of changes in tumor hypoxia on dose-painting treatment based on 18F-F-miso positron emission tomography. Int. J. Radia-tion Oncology Biol. Phys. 2008 70(4):1219-28.
29 Wang W, Lee N, Georgi JC, Narayanan M, Guillem J, Schöder H, Humm J. Pharmacokinetic Analysis of Hypoxia 18F-Fluoromisonidazole Dynamic PET in Head and Neck Cancer. J Nucl Med. 2010 51(1):37–45.
30 Wang W, Georgi JC, Nehmeh S, Narayanan M, Paulus T, Bal M, O’Donoghue J, Zanzonico P, Schmidtlein R, Lee N, Humm J. Evaluation of a compartmental model for estimating tumor hypoxia via F-MISO dynamic PET imaging. Phys Med Biol. 2009 54(10):3083-99.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 44/46
31 Wang W, Yorke E, Nehmeh S, Humm J, Clifton Ling C. Modeling acute and chronic hypoxia using serial images of 18F-F-MISO PET. Med Phys. 2009 36(10):4400-8.
32 Grosu AL, Souvatzoglou M, Röper B, Dobritz M, Wiedenmann N, Jacob V, Wester HJ, Reischl G, Machulla HJ, Schwaiger M, Molls M, Piert M. Hypoxia imaging with FAZA-PET and theoretical considerations with re-gard to dose painting for individualization of radiotherapy in patients with head and neck cancer. Int J Radiat Oncol Biol Phys. 2007 69(2):541-51.
33 Komar G, Seppänen M, Eskola O, Lindholm P, Grönroos TJ, Forsback S, Sipilä H, Evans SM, Solin O, Minn H. 18F-EF5: a new PET tracer for imaging hypoxia in head and neck cancer. J Nucl Med. 2008 49(12):1944-51.
34 Cherk M, Foo S, Poon A, Knight S, Murone C, Papenfuss A, Sachinidis J, Saunder T, O’Keefe G, Scott A. Lack of Correlation of Hypoxic Cell Fraction and Angiogenesis with Glucose Metabolic Rate in Non–Small Cell Lung Cancer Assessed by 18F-Fluoromisonidazole and 18F-FDG PET. J Nucl Med 2006 47(12):1921–1926.
35 Gagel B, Reinartz P, Demirel C, Kaiser H, Zimny M, Piroth M, Pinkawa M, Stanzel S, Asadpour R, Hamacher K, Coenen K, Buell, Eble M. [18F] fluoromisonidazole and [18F] fluorodeoxyglucose positron emission tomo-graphy in response evaluation after chemo-/radiotherapy of non-small-cell lung cancer: a feasibility study. BMC Cancer. 2006 6:51.
36 Girard N, Mornex F. Radiotherapy for locally advanced non-small cell lung cancer. Eur J Cancer. 2009 45 Suppl 1:113-25.
37 Kong FM, Hayman JA, Griffith KA, Kalemkerian GP, Arenberg D, Lyons S, Turrisi A, Lichter A, Fraass B, Eisbruch A, Lawrence TS, Ten Haken RK. Final toxicity results of a radiation-dose escalation study in patients with non-small-cell lung cancer (NSCLC): predictors for radiation pneumonitis and fibrosis. Int J Radiat Oncol Biol Phys. 2006 15;65(4):1075-86.
38 Bradley J, Graham MV, Winter K, Purdy JA, Komaki R, Roa WH, Ryu JK, Bosch W, Emami B. Toxicity and outcome results of RTOG 9311: a phase I-II dose-escalation study using three-dimensional conformal radio-therapy in patients with inoperable non-small-cell lung carcinoma. Int J Radiat Oncol Biol Phys. 2005 1;61(2):318-28.
39 Schild SE, McGinnis WL, Graham D, Hillman S, Fitch TR, Northfelt D, Garces YI, Shahidi H, Tschetter LK, Schaefer PL, Adjei A, Jett J. Results of a Phase I trial of concurrent chemotherapy and escalating doses of radiation for unresectable non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2006 15;65(4):1106-11.
40 Bradley JD, Moughan J, Graham MV, Byhardt R, Govindan R, Fowler J, Purdy JA, Michalski JM, Gore E, Choy H. A phase I/II radiation dose escalation study with concurrent chemotherapy for patients with inoper-able stages I to III non-small-cell lung cancer: phase I results of RTOG 0117. Int J Radiat Oncol Biol Phys. 2010 1;77(2):367-72.
41 Bradley JD, Bae K, Graham MV, Byhardt R, Govindan R, Fowler J, Purdy JA, Michalski JM, Gore E, Choy H. Primary analysis of the phase II component of a phase I/II dose intensification study using three-dimensional conformal radiation therapy and concurrent chemotherapy for patients with inoperable non-small-cell lung cancer: RTOG 0117. J Clin Oncol. 2010 10;28(14):2475-80.
42 Socinski MA, Blackstock AW, Bogart JA, Wang X, Munley M, Rosenman J, Gu L, Masters GA, Ungaro P, Sleeper A, Green M, Miller AA, et al. Randomized phase II trial of induction chemotherapy followed by con-current chemotherapy and dose-escalated thoracic conformal radiotherapy (74 Gy) in stage III non-small-cell lung cancer: CALGB 30105. J Clin Oncol. 2008 20;26(15):2457-63.
43 Eberhardt WE, Hepp R. Chemotherapy in stage III non-small cell lung cancer. Eur J Cancer. 2009 45 Suppl 1:126-36.
44 Rusu P, Ciuleanu TE, Todor N, Ghilezan N. Dose escalation using three-dimensional conformal radiother-apy (3D-CRT) in concurrent setting with vinorelbine and a platinum compound, preceeded by induction che-

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 45/46
motherapy and followed by consolidation chemotherapy in locally advanced non-small cell lung cancer (NSCLC) - A preliminary report of a modified phase I-II study., Suppl 7[2] ed 2009. p. 536-Abstr.9107.
45 Albain KS, Crowley JJ, Turrisi AT 3rd, Gandara DR, Farrar WB, Clark JI, Beasley KR, Livingston RB. J Clin Oncol. 2002 15;20(16):3454-60.
46 Leopold KA, Perry MC, Miller AA, Green MR. Randomized phase II study of cisplatin with gemcitabine or paclitaxel or vinorelbine as induction chemotherapy followed by concomitant chemoradiotherapy for stage IIIB non-small-cell lung cancer: cancer and leukemia group B study 9431. J Clin Oncol. 2002 15;20(20):4191-8.
47 Gagel B, Reinartz P, Demirel C, Kaiser H, Zimny M, Piroth M, Pinkawa M, Stanzel S, Asadpour R, Hamacher K, Coenen K, Buell, Eble M. [18F] fluoromisonidazole and [18F] fluorodeoxyglucose positron emission tomo-graphy in response evaluation after chemo-/radiotherapy of non-small-cell lung cancer: a feasibility study. BMC Cancer. 2006 6:51
48 Dehdashti F, Mintun MA, Lewis JS, Bradley J, Govindan R, Laforest R, Welch MJ, Siegel BA. In vivo as-sessment of tumor hypoxia in lung cancer with 60Cu-ATSM. Eur J Nucl Med Mol Imaging. 2003 30(6):844-50.
49 Postema EJ, McEwan AJ, Riauka TA, Kumar P, Richmond DA, Abrams DN, Wiebe LI.Initial results of hy-poxia imaging using 1-alpha-D: -(5-deoxy-5-[18F]-fluoroarabinofuranosyl)-2-nitroimidazole ( 18F-FAZA). Eur J Nucl Med Mol Imaging. 2009 36(10):1565-73.
50 Chavaudra J, Bridier A. Cancer Radiother. 2001 ;5(5):472-8.
51 Mountain CF, Dresler CM. Regional lymph node classification for lung cancer staging. Chest. 1997 111(6):1718-23.
52 Giraud P, Antoine M, Larrouy A, Milleron B, Callard P, De Rycke Y, Carette MF, Rosenwald JC, Cosset JM, Housset M, Touboul E. Evaluation of microscopic tumor extension in non-small-cell lung cancer for three-dimensional conformal radiotherapy planning. Int J Radiat Oncol Biol Phys. 2000 1;48(4):1015-24.
53 Chapet O, Kong FM, Quint LE, Chang AC, Ten Haken RK, Eisbruch A, Hayman JA. CT-based definition of thoracic lymph node stations: an atlas from the University of Michigan. Int J Radiat Oncol Biol Phys. 2005 ;63(1):170-8.
54 Busk M, Horsman MR, Jakobsen S, Bussink J, van der Kogel A, Overgaard J. Cellular uptake of PET tracers of glucose metabolism and hypoxia and their linkage. Eur J Nucl Med Mol Imaging. 2008 35(12):2294-303.
55 Dence CS, Ponde DE, Welch MJ, Lewis JS. Autoradiographic and small-animal PET comparisons between (18)F-FMISO, (18)F-FDG, (18)F-FLT and the hypoxic selective (64)Cu-ATSM in a rodent model of cancer. Nucl Med Biol. 2008 35(6):713-20.
56 Pugachev A, Ruan S, Carlin S, Larson SM, Campa J, Ling CC, Humm JL. Dependence of FDG uptake on tumor microenvironment. Int J Radiat Oncol Biol Phys. 2005 62(2):545-53.
57 Wyss MT, Honer M, Schubiger PA, Ametamey SM. NanoPET imaging of (18)F]fluoromisonidazole uptake in experimental mouse tumours. Eur J Nucl Med Mol Imaging. 2006 33(3):311-8.
58 Zanzonico P, Campa J, Polycarpe-Holman D, Forster G, Finn R, Larson S, Humm J, Ling C. Animal-specific positioning molds for registration of repeat imaging studies: comparative microPET imaging of F18-labeled fluoro-deoxyglucose and fluoro-misonidazole in rodent tumors. Nucl Med Biol. 2006 33(1):65-70.
59 Rajendran JG, Mankoff DA, O'Sullivan F, Peterson LM, Schwartz DL, Conrad EU, Spence AM, Muzi M, Far-well DG, Krohn KA. Hypoxia and glucose metabolism in malignant tumors: evaluation by [18F]fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin Cancer Res. 2004 10(7):2245-52.

CHB 11-01-version 3.0 du 19/09/12 Protocole RTEP5
page 46/46
60 Rajendran JG, Wilson DC, Conrad EU, Peterson LM, Bruckner JD, Rasey JS, Chin LK, Hofstrand PD, Grier-son JR, Eary JF, Krohn KA. [(18)F]FMISO and [(18)F]FDG PET imaging in soft tissue sarcomas: correlation of hypoxia, metabolism and VEGF expression. Eur J Nucl Med Mol Imaging. 2003 30(5):695-704.
61 Michael M. Graham, Lanell M. Peterson, Jeanne M. Link, Margaret L. Evans, Janet s. Rasey, Wui-Jin Koh, James H. Caldwell, and Kenneth A. Krohn Fluorine-18-Fluoromisonidazole Radiation Dosimetry in Imaging Studies J Nucl Med. 1997 38: 1631-1636.
62 Logiciel CTDosimetry, http://www.ImpactScan.org
63 Tumour hypoxia Imaging with (18F)FAZA PET in head and neck cancer patients: a pilot study. Souvat-zoglou AL, Grosu AL, Röper B, Krause BJ, Beck R, Reischl G, Picchio M, Machulla HJ, Wester HJ, Piert M. Eur J Nucl Med Mol Imaging. 2007 34:1566-1575.
64 Hypoxia-specific Tumor Imaging with 18F-Fluoroazomycin Arabinoside. Piert M, Machulla HJ, Picchio M, Reischl G,
Ziegler S, Kumar P, Wester HJ, Beck R, McEwan AJB, MBBS, Wiebe LI, Schwaiger M. J Nucl Med. 2005 46:106-113.
65 Vuillez JP, Desruet MD, Mundler O, Karcher G. Le radiopharmaceutique : situation et perspectives depuis la 8e conférence internationale de l’ACOMEN (Bordeaux, 11-12-13 mai 2005). Médecine Nucléaire 2009 33:115-121.
66 [18F]Fluoromisonidazole. The MICAD Research Team. Molecular Imaging and Contrast Agent Database [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004-2010. 2005 Jul 18.
67 Koh W-J, Rasey JS, Evans ML. Imaging of hypoxia in human tumors with [F-18]fluoromisonidazole. Int J Radiat Oncol Biol Phys. 1992 22:199–212.
68 Hypoxia-imaging with (18)F-Misonidazole and PET: changes of kinetics during radiotherapy of head-and-neck cancer. Eschmann SM, Paulsen F, Bedeshem C, Machulla HJ, Hehr T, Bamberg M, Bares R. Radiother Oncol. 2007 Jun;83(3):406-10.


















