livre abstractfinal 2013 paginé
95
1 SOCIETE ALGERIENNE DE PEDIATRIE 34 ème CONGRES NATIONAL DE PEDIATRIE PROGRAMME RECUEIL DES COMMUNICATIONS ORALES ET AFFICHEES Alger : Hôtel Sheraton 11 - 13 décembre 2013
Transcript of livre abstractfinal 2013 paginé

1
SOCIETE ALGERIENNE DE PEDIATRIE
34ème CONGRES NATIONAL DE PEDIATRIE
PROGRAMME
RECUEIL DES COMMUNICATIONS
ORALES ET AFFICHEES
Alger : Hôtel Sheraton 11 - 13 décembre 2013

2
La Société Algérienne de Pédiatrie remercie les laboratoires suivants :
Platinum
SANOFI AVENTIS
Gold Sponsors:
DANONE BABY NUTRITION MILUPA-BLEDINA
PFIZER GSK
NOVONORDISK LFB
LIFESCAN Silver sponsors
BAYER PHARMACEUTICAL HIKMA MSD
ORDESA
Sponsors
BAXTER EREMPHARMA
ROCHE DIAGOSTIC NESTLE
NUTRIBIO ( NACTALIA ) MERCK SERONO
FASSKA CELIA
BIOCODEX BIOMIL
ACCENTIS PHARMA HOSPIMED
STALLERGÈNE MAGPHARM
REMEDE PHARMA MODILAC
PHARMASTRAT (MEDTRONIC) ABBOTT PRODUCTS
ACTELION BIOMARIN

3
Société Algérienne de Pédiatrie
Présidente: Pr. R. Boukari Vice-présidents : Pr. L. Haridi, Pr. L.Smati-Bourtel Secrétaire Générale Pr. S. Chikhi S. Général Adjoint : Pr. K. Berkouk, Pr. Z. Zeroual Trésorier : Pr. C. Kaddache Trésoriers Adjoints : Dr .M. Bennaceur, Pr. H. Maouche
Groupe de travail « Mortalité néonatale» Coordinateur : Pr S. Kermani Pr L. Haridi
M.L Atif, F. Saadaoui, K. Boudhar, Y. Sadi, O. Benrabah, F.A.Saari, M. Yahiaoui, Gadouche, Hamzaoui,T.Fatmi , M ADlaoui, M.HenouneEHS Afflou 0nt collaboré à cette étude les services ou unités de Pédiatrie suivants :
CHU de Blida, HCA Alger, CHU Nafissa Hamoud, Hôpital de Belfort, Pédiatrie Ain Taya, Pédiatrie A CHU Beni Messous, Hôpital d’El Bouni Annnaba, Hôpital de Kouba, Néonatologie CHU Mustapha, Groupe de travail « Mortalité 1 mois- 5ans » Coordinateur : Pr C.Kaddache
D.Arhab CHU TIZI OUZOU O.Ibsaine CHU Alger Est L. Attif CHU Blida Y. INOURI HCA ALGER N.Benmouffok CHU Alger EST L.Kedji CHU BAB EL OUED S.Belamri INSP M.Moumi CHU Alger centre R.Boukari CHU Blida H.Maouche CHU BENI-MESSOUS F.Guerrak EPH Thenia S. Smail CHU Bologhine Dr M.Yahiaoui Z.Zeroual CHU Beni Messous Ont collaboré à cette étude les services de pédiatrie de Pédiatrie et les médecins suivants : N. BOUCHAIR CHU Annaba, S.Taleb Bachtarzi( CHU Constantine) A.DEHIMI CHU Sétif
Groupe de travail : Déficits Immunitaires Primitifs R.Belbouab,KN .Benhalla,N.Boulekhiout,R.Boukari,D.Bouziane,S.Chelah,M.Chaou,A.Dehimi O.Drali, A.Guedouar, L.Kedji,N.Kechout S.Melzi ,Z.Mansouri, FZ.Zemiri,L.Smati,S.Touri, N.Benmouffok, MS Smail
Groupe Maladies Rares
JP.Grangaud, A.Chalabi Benabdelleh, A.Lebied, R.Boukari, M.EKhiari, M.Haridi, S.Chikhi,MT.Hamlaoui,A.Mekki, A.Hadji,L.Benhafssa, N.Benmati, M.Bennaceur.Ma .Keddari
Comité de Lecture : Communications ORALES S.Chikhi, L .Haridi, C.Kaddache, L.Smati
Comité de Lecture : Poster H.Ahmane N.K.N.Benhala, N.Bensadi, K.Berkouk, N.Bouchair, Z.Bouzerar, A .Hadji, O.Ibsaine, L.Kedji, A.Maoudj, H.Maouche, S.Taleb Bachtarzi, Z.Zeroual

4
Sommaire
Programme synoptique…………………………………………………………………………………………………………………… 10 Programme……………………………………………………………………………………………………………………………………….14 CO1 : Suivi et évolution à long terme du Purpura Thrombopénique Immunologique chronique de l’enfant. L. Kedji , ( pédiatrie CHU Bab El Oued, Alger) ………………………………………………………………………….24 CO 2 : La néphropathie lupique à début pédiatrique……………………………………………………………….............24 O.Gacem ,N. Boukhedouma – N. Benmouffok – S. Chabani – A. Lebied CO3 : Expérience clinique d'un traitement prophylactique dans la maladie de von Willebrand. S.Aggoune ………………………………………………………………………………………………………………………………….….25 CO 4: Traitement prophylactique chez l’enfant hémophile A sévère: S.Sokhal-…………………………………….26 CO5 Afibrinogénémie congénitale F. Sadaoui ………………………………………………….…………………………..……27 CO 6: Les déficits immunitaire primitifs selon l’expérience du CHU Béni-Messous. F.Z.Zémiri , ……………………………………………………………………………………………………………………………...………..28 CO7 Alerte pédiatrique : infection du VIH chez l’enfant Y.BELKEBIR………………………………….…………………29 CO 8 : Motif d’hospitalisation des enfants infectés par le VIH à l’hôpital d’enfants d’Oran. A. Radoui ……………………………………………………………………………………………………………………………………………30 CO9 : Evaluation des connaissances des adolescents asthmatiques et leurs parents sur l’asthme et son traitement. Maouche. H,………………………………………………………………………………………..30 CO10 : Prévalence du surpoids et de l’obésité en milieu scolaire dans la wilaya d’Alger : Résultats d’une enquête prospective. A.Hadji-Lehtihet …………………………………………………………………......31 CO11: Tuberculose péritonéale : revue de 10 cas pédiatriques .BOUK'HIL. K.S , …………………………………32 CO12 : Aspects épidémiologiques des intoxications. N.Bensadi, …………………....………………………………….33 CO13- : Place de l’analyse toxicologique en Pédiatrie: L.R Mekacher, …………………………………..…………..34 CO 14 : Les appendicites aigues de l’enfant de moins de 5 ans O.IBSAINE …………………………………………35 CO15 : Syndrome de Guillain - Barré : Expérience d’un service de Pédiatrie H.AHMANE, ………………….35 CO16 Les cholestases intra-hépatiques progressives familiales (syndrome et maladie de Byler) R. Belbouab ………………………………………………………………………………………………………………………………………..36 CO 17 .Prévalence de l’obésité infantile en milieu scolaire à Sétif. Résultats préliminaires. Z. BenaraB , …………………………………………………………………………………………………………………………………………37 CO 18. Impact d’une unité de diabète infantile à l’EPH RelizaneS. kara Mostefa ………………………………38 CO 19 : L’acidose inaugurale dans le diabète de type 1. A. Zennaki ………………………………...……………….38 CO 20 : Utilisation de la pompe à insuline chez l’enfant. A.Zennaki, ………………………………………………….39 CO 21: Le cancer de la thyroïde chez l’enfant (à propos de 39 cas)K………………………………………………….40 CO 22 : Hyperthyroidie de l'enfant Aspects cliniques et evolutifs F.Bouferoua ……………………………….41 CO 23 : prévalence de l'allaitement maternel dans l'ouest algérien .N. Zidane …………………………………42 CO 24 : Méningites purulentes à pneumocoque du nourrisson.F.Z. ZMIT …………………………...…………42 CO 25 Le déficit en vitamine B12 selon l’expérience du CHU Béni-Messous. Zemiri.F.Z …………………..44 CO 26 : Convulsions néonatales révélatrices d’hypovitaminose d maternelle : à propos de 06 cas. Sadaoui..B, ………………………………………………………………………………………………………………………….………..44 CO 27.Prise en charge des cardiopathies congénitales chez les enfants trisomiques (T21) Y.Inouri ,………………………………………………………………………………………………………………………………………….…45 CO 28 .Apport du certificat de décès périnatal et néonatal à la connaissance de la mortalité à Oran Heroual Nabila A.Tadjeddine Semep Ehs Canastel…………………………………………………………………………….46

5
Workshop : Maladies métaboliques
W 1. Mucopolysaccharidose de type1 (MPS1): A propos de trois cas. M. Keddari .Hamdani ,S.Aggoune,S.Mezouari,A.Chergui,A.Mertani,M.Keddari:CMI,Chu Mustapha W 2 La forme infantile de la maladie de Pompe à l’ère de l’enzymothérapie (A propos d’un cas avec revue de la littérature) .L.N.Dris ,Nk .Benhalla ; L. Smati; H.Djenane; A. Yagoubi; D.Douiri ; N.Alim ;O.Redjala ;M. Sari; M. Baghriche Service De Pédiatrie – Eph Bologhine Ibn Ziri – Alger W 3 La maladie de Gaucher chez l’enfant : Expérience de la Clinique Médicale Infantile du CHU Mustapha. A,Hadji-Lehtihet ,S.Sokhal1 ; N.Benali-Khodja1 ; S.Chikhi.1 ; Ghouali2 ; Hallal2 ; B.Imessaoudène 2; A.Berhoune 2 ; A.Mertani1 ; M.Keddari1. 1. Clinique Médicale Infantile CHU Mustapha Alger 2. Laboratoire de Biochimie et de Génétique CHU Mustapha Alger W 4 .Maladies métaboliques : Expérience de 5 ans d’un service de pédiatrie générale M.T. Bencharif, ;S Taleb ; S Benfetima ; F. Sellahi ; A. Bakhouche ; H .Boudbaba ; N. Hezil ; S Benseghir ; A. Mesri ; F.Z .Boulmaiz :Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine W5 . Les hyper ammoniémie s primitives de l’enfant L. Benhafessa, F, Benmati, D .C helighem,K. Boudhar,W.Messek W6 . DEéficit du cycle de l’urée : Discussion de 2 cas cliniques F, Benmati , D .Chelighem, K. Boudhar, W.Messek , L. Benhafessa W7.Maladies héréditaires du métabolisme : mise au point clinique et cas interactifs
A.Mekki Service de Pédiatrie CHU Nafissa Hamoud
Posters 1. Médecine prédictive: le système HLA Rahmoune Hakim, Boutrid Nada, Nedjar Faycal, Belkacem Bioud. 2. Dysplasie ectodermique hypohidrotique: à propos d'un cas. Treki Rima.EPH EL HARROUCH 3.Etude des performances d’une chambre d’inhalation pédiatrique Boukhettala Nabile Porée Thierry, Diot Patrice, Vecellio Laurent CEPR, INSERM U1100 EA 6305, Faculté de Médecine, 10 ter bld tonnellé 37032 Tours FRANCE 4.Syndrome d’Hallermann Streiff François : A propos de deux cas dans une même fratrie. ZerouaL Zoulikha, M.Chaou, S.Tari, F.Bouferoua, L.Atek, N.Bouterfas H.Boucenna, A.Khati, A.Hazzazi, H.Boukhellal, M.Haridi, Z.Bouzerar, M.E.Khiari. Service de pédiatrie « A » Chu Béni-Messous. 5. Histoire naturelle de l’allergie aux protèines du lait de vache Ibsaine Ouardia,A. Benyahia- Narimene Zerrouki - Larbi Rezzag Lebza- Souad.Bellache1- Hassina Berrah.CHU Parnet 6.Une fièvre isolée du nourrisson : pensez à l’appendicite aigue ! BrahmiA Nabila, IbsainE Ouardia, Zerrouki Narrimene, Akkouche Fatima, GhaeouL Ahmes, Berrah Hassine. Service de pédiatrie « B ».C.H.U H.Dey 7. Imperforation choanale bilatérale: que faire chez le nouveau né? Messaoudi Djamel, Afri Yacine. Service ORL CHU Bab El Oued 8.Quel profil de la mortalité infantile au niveau des centres hospitaliers d’Oran ? Heroual Nabila,A.Tadjeddine.Service d’épidémiologie et de médecine préventive, EHS Canastel 9.Education therapeutique d’un enfant malade Rezki Hassiba,Raffa Nassiba,Bettayeb,Rezig Fethia,Ghrieb Samira.Hopital central de la sureté nationale,

6
les glycines, alger 10.Expérience d’un service de pédiatrie a Jiddah (Arabie Saoudite ) avec revue de littérature et dernière mise a jour Sleiman Rola,H.Dey Alger 11.Maladie de Castleman: à propos d'un cas Treki Rima.EPH EL HARROUCH (Wilaya de Skikda) 12.Lymphome Malin Non Hodgkinien à localisation cutanée : à propos d'un cas Chachou khadidja ,Boumeddane amaria, Mecifi raouida, Khelil amina leila, Mahi henni mohamed amine, Mehidi mohammed yacine, Bensid ikram, Oussalah meriem, Maazouzi djamel Service d'oncologie pédiatrique, CAC Emir Abdelkader, ORAN 13.Nanisme thanatophore : à propos d’un cas Mahmoud Abderezak Oumenkhache,Souhila Abed, Fatiha Azi,Ouahiba Benrabeh Unité de néonatologie CHU Nefissa Hamoud ex Parnet; Alger 14.Convulsion fébrile épidémiologie et causes Benallal Khadidja,Benaissa Fatima, Tamert Soumia, Ould said Karima, Machou Nassiba, Mohammedi Abdelatif.Service de pédiatrie CHU de Sidi Belabbes 15.Taux de l’allaitement maternel dans la région de Sidi Belabbes Benallal Khadidja ,Hamdoun Karim, Barket Mnaouer, Larabi Zahira.Service de pédiatrie du CHU de Sidi Belabbes 16.Calcinose pseudo-tumorale compliquant un cas d’osteodystrophie rénale chez l’enfant Oukrif Lamia ,Ouzani Meriem,Boukhil Kais,Bensenouci Abdelatif.Pediatrie B CHU Beni Messous 17.Arthrogrypose: Syndrome et non maladie :A propos de 3 observations Djemane Adel.EPH Gouraya 18.Association Maladie de Crigler Najjar et Syndrome de Gilbert (Rareté et complexité de l’association) Boudhar Kamel ,F.Benmati1, W.Messak, N.Mansouri1, F.Petit2, R. Reding3, S.E.Laalaoui1, L.Benhafessa1. 19. Traitement de l’acidocétose diabetique par perfusion continue d’insuline rapide Ibsaine ouardia, Narimene Zerrouki –Lamri Boukaboub- Samira Medjdoubi- Hassina Berrah.Service de pédiatrie « B ».C.H.U H.Dey 20.Volvulus intestinal sur bride de LADD découverte accidentelle: A propos d’un cas. S. Kerdoudi (1), F. Hallala (1), H. Djabri (1), F. Sallahi (1), A. yousfi (2). 21.Syndrome de hunter forme severe chez un enfant : a propos d’un cas Bennani amal , Bensaadi, Cherifi, Regal, Hamzaoui, Chalah, Iddir, Sifodil,Heddou, Arhab, Ahmane. CHU de tizi-ouzou service de pédiatrie 22.Enquête par questionnaire sur les possibilités et les habitudes des médecins pour le diagnostic de diabète de type 1 Amel Zennaki ,A. Ouzzaa, S. Niar, H. Aichaoui, M. Gharnouti, A. Reguieg, M. Bessahraoui, M. Naceur, K. Bouziane-nedjadi, M. Touhami.Service de pédiatrie C (clinique A. Cabral) CHU d'Oran 23.A propos d’un cas de syndrome de Kasabach-merritt chez un nourrisson traité avec succès Dib saad-eddine,Kandouci-tani fatima, Kaouadji naoual, Benahmed ali, El mezouar shahrazed, Massen Zouhir.Service pédiatrie EHS mère-enfants Tlemcen 24.L’insuffisance renale chronique chez l’enfant experience d’un service de pediatrie generale Widad amel,N Bouchair , J Bounour , A Hamani , F Bouslama , H Sehab ,J Belamri ,L Boustil, M Yaiche.Clinique pediatrique sainte therese chu annaba 25.Cholestase néonatale à cytomégalovirus à propos de deux cas Bendaas N,Manaa M,Guerdouh S .Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine 26.Ostéopétrose à révélation néonatale Laabed Ahlem ,Telhaoui Nabil,EPH Hakim Saadene Biskra 27.Intérêt de l’étude des glycoproteines plaquettaires par la cytométrie en flux dans la thrombasthénie de glanzmann Belhadj Hayat,Fenni Nacéra.Unité de pédiatrie CRMC Blida

7
28.Adrenoleucodystrophie liée à l'X :A propos de deux frères Metidji Bouchra,Hamid Boufenar,Malika Agha,SE Laalaoui.Hôpital central de l'Armée ALGER 29.L’ostéopétrose infantile maligne : une maladie rare mais de diagnostic facile ! Bencharif Madani Tahar, H Boudbaba, NH Salmi, W Nasri, A Rachoui, H Belabed Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine 30.Céphalées mode de révélation de la maladie de behçet : à propos d’un cas Hellal M, Djebri H, Mesri I, Selmi H.Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine 31.Mucoviscidoses difficulté de prise en charge: à propos de 3 cas Helala Fairouz, Jabri Hana, Kerdoudi Sameh.EHS Sidi Mabrouk Constantine 32.La coarctation de l’aorte thoracique.À propos d’un cas diagnostiqué à l’âge de 13 ans Boulmaiz FZ, Bendaas N, Keghouche S.Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine 33.La scoliose secondaire chez l’enfant : à propos de 46 cas. Chadda B, Baaziz H, Albane B.CHU Batna 34.Syndrome de Silver Russell à propos d'un cas Lacete F, Guedouar A, Merzouk A, Lebied A.CHU Parnet 35.Invagination intestinale aiguë révélant une maladie coeliaque:A propos d'un cas. Baghdali N, Khelafi N, Aggoune S, Hadji A, Sokhal S, Mertani A, Keddari M. Clinique médicale infantile,CHU Mustapha,Alger. 36. Rachitisme carentiel a début néonatal :a propos d’un cas Talbi F, Kherra S, Benalioua H, Abba K, Trabelsi F CHU Parnet Bensenouci A .Service de pediatrie B CHU Beni Messous 37.Le syndrome d’hyper IgD: A propos d’un cas Aichaoui H, Zennaki A, Naceur M, Niar S, Bouziane Nadjadi K Service de pédiatrie « C » .Clinique Amilcar Cabral CHU d'Oran 38.Syndrome d’activation macrophagique (SAM) Benmouffok N. Benmoffok N , Boukhedouma N , Boubidi N,Gacem O, Lebied A.Service pédiatrie A Hopital N. Hamoud 39.Histiocytose Langerhansienne :A propos de 7cas Boukhedouma Nabila ,N.Benmouffok , O.Gacem , A.Guedouar , C.Boubidi , F.Lacete , A. Lebied.Service pédiatrie A Hopital N. Hamoud 40.PB/PC et status nutritionnel en pediatrie :" Résultats d'une analyse d'une enquête anthropométrique dirigée" Amireche F,Safi H ,Allas H:Service pédiatrie du Mansourah de Constantine 41.Cystinose infantile :à propos d’une observation Aggoune S , Baghdali N ,Belbouab R, Noumi M, Hadji A, Sokhal S , Keddari M, Mertani A,Keddari M. Clinique médicale infantile CHU Mustapha 42. Exophtalmie bilaterale revelant une leucemie aigue lymphoblastique. a propos d’un cas. AL.Khelil1, A.Boumeddane1, A.Mécifi1, K, Chachou 1, A Meksi1, Z.Taghezout1, K.Mahmoudi 43.La dermatomyosite juvenile (à propos d'un cas) Abdelbaki S,Kendouci-taniF ,Elmezouar C,Kaddour A,Massen Z.Service de pédiatrie EHS mére-enfant Tlemcen 44.Syndrome de Zellweger : à propos d’un cas Boucenna H,Chaou.M, Khati.A, Tari.S, Atek.L,Khiari ME.Service de pédiatrie A, CHU Benimessous, Alger 45.Ecthyma gangénusum a propos d'un cas Boulahlib F, Belkadi S.W. E.P.H Zighout Youcef Ténès chlef 46.Rachitisme vitaminorésistant hypophasphatémique révélateur d’une tyrosinémie héréditaire de type1 Baouia S,.Koumni N , Chikhi S, Benalikhoudja N ,Mertani A, Keddari M CMI, CHU Mustapha Pacha, Alger-centre. 47.Anémie de biermer : cause possible d’anémie normocytaire chez l’adolescent Noumi M,Aggoune S,Berkani R ,Ladj S ,Terrak R,Hassoum,.Mertani A ,.Keddari M Clinique médicale infantile CHU Mustapha 48.Place de la génétique moléculaire dans le diagnostic des SMAs. (Spinal Muscular Atrophy) :(A propos d’un cas SMA1). Delileche H ,Bendjamaa A,Himmi A,.Aissat A ,Semid T , Boukhenfouf N.

8
49.Déficit constitutionnel en facteur VII : à propos d’une observation Amrouni L,Bencheikh F, Kerboua S.EPH Okbi, Guelma 50.Enfants de pierre :Fibrodysplasie Ossifiante Progressive (FOP) : A propos de 2 cas Dris LN,Djenane H1 ,Smail S1, Smati L1 , Douiri D1 ,Baiod S1 , Bendaoud M2, Dahou N3 , Baghriche M1. 51. Erythromelalgie ; difficultés de prise en charge. Hamouda N, Bioud B, Dehimi A., service depédiatrie CHU de Sétif. 52. La leptospirose : une cause rare de défaillance multiviscerale Zerrouki N, Ibsaine O, Drali O, Zeroual S, Boutaba M, Berrah H. CHU H dey 53.Un cas de méningite tuberculeuse chez un enfant vacciné Ait Kaci N ,Meftah A, Mechtoub F-Z, Mohamed Y.EPH Boufarik 54.Hyperthroidie autoimmune de l’enfant (a propos de deux observations) Kerboua ,Amrouni L, Boulares F Z .EHS EL Bouni, Annaba 55.Facteurs influençant la durée de l’allaitement maternel Mazari W,Bouriche.K ,Snouci.D ,Bendeddouche .S .Service de pédiatrie-CHU Tlemcen 56.Aspects épidémiologiques et prise en charge du diabète de type I dans un service de Pédiatrie générale – Tlemcen 13.000 - Algérie Mazari .W, Snouci.D, Bouriche.K, Bendeddouche .S .Service de pédiatrie-CHU Tlemcen 57.Hémophilie A : un mode de révélation exceptionnel Mohand-Oussaid Aïda, Boukhellal-Djemoi Houria, Chaou Mériem, Lamrani Fella, Khiari Mohamed El-Mokhtar.Service Pédiatrie A - CHU Béni Messous 58.Fistule coronaro-camérale (A propos d’01 cas exceptionnel) Boufenar H,Metidji B, Agha M , Ait-idir K , Laalaoui S-E.Service pediatrie HCA 59.Hypoglycémie néonatale persistante...pourquoi? Moussaoui Y, Arrada Z, Oucif Z,Saadaoui,.Rezzaz N,Berrah H .CHU Parnet 60.Le syndrome catastrophique antiphospholipide: a propos d'un cas Rezak R,Bouchetara A,Azzouz S M,Hamiti B. EPH Canastel Oran 61.Coma néonatal révélant une citrullinemie de type I : à propos d’un cas Benmati F,.Messak W ,Boudhar K, Benhafssa L. HCA 62.Maladie de Recklinghausen ou Neurofibromatose de type 1 : A propos de 2 observations et Revue de la littérature Drali O,Boulekhiout N, Zerrouki N,Zebdi N,Ghazoul A,Arrada Z, Berrah H Service Pediatrie B CHU Hussein Dey 63.Nutrition de l'enfant d'âge scolaire Amireche.F, Safi H, Allas H.Service Pédiatrie Mansourah Constantine 64.Maladie de Kawasaki : à propos de 6 cas A L Boustil,H. Sehab1, N. Boukertouta2, W. Boutabia1, N. Bouchair1, M.C. Yaiche1. 1Service de pédiatrie, clinique Sainte Thérèse, CHU Annaba. 2Service de pédiatrie, EHS EL-BOUNI. 65. Syndrome hémolytique et urémique : à propos de 6 observations Boustil Ahmed Lotfi,F. Layachi, Dj. Belamri, H. Sehab, N. Bouchair, M.C. Yaiche. Service de pédiatrie, clinique Sainte Thérèse, CHU Annaba. 66. Gastrite et Allergie aux protéines du lait de vache : à propos de 3 cas. Boudbaba Hanane, T Bencharif Madani ,NH Salmi, W Nasri , A Rachoui Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine 67.Tératome gastrique mature : une tumeur rare Ahmane H,Arhab D, Hamzaoui A, Chellah SA, idir S, Cherifi N, Aidrous L. Bensadi N CHU Nedir Mohamed Tizi-Ouzou 68.Sténose bronchique congénitale: Un autre diagnostic de faux asthme Boulekhiout Nadia,Drali Ourdia,Hadjarab Amina,Arrada Zakia,Berrah hassina CHU Hussein-dey -service pediatrie"B"

9
69.Croissance staturale de l’enfant atteint de Diabète de type 1 associé à la Maladie cœliaque Bouziane-Nedjadi Karim ,Bessahraoui Mimouna,Reguig Ali,Niar Sakina,Naceur Malika Aichaoui Amel,Touhami Mahmoud.Service de Pédiatrie C, 9 Amilcar Cabral 31000 70.'' Primum Non Nocere ''... Boutrid Nada, Rahmoune Hakim, Bouabdallah Saida, Belkacem Bioud, CHU Sétif 71.Les dilatations des sténoses de l'œsophage chez l’enfant : à propos de 93 cas. Benalioua H, Belbouab R, Khiati M. Hopital Belfort 72.Diabète transitoire induit par corticothérapie et L Asparaginase chez 4 enfants atteints de LAL H. Mesbaieh, Guemghar, R. Nemmar, A.Lamraoui, C.Kaddache, R.Boukari Service de Pédiatrie CHU Blida Université Saad dahlab Blida 73.Aplasie médullaire chez l’enfant : étude de 15 cas S.Guemghar, H. Mesbaih, S.Touri, R.Nemmar, A.Lamraoui, L.Haddad, K.Mammeri, C.Kaddache, R.Boukari.Service de Pédiatrie CHU Blida- Université Saad Dahlab 74 .Gastroentérite révélatrice d’une maladie d’Addison. Savoir y penser chez l’enfant ! (A propos d’un cas) 75.Lithiase de la voie biliaire :bile-Plug syndrome : à propos d’un cas Helala Fairouz, Kerdoudi Sameh, Djabri Hana, Allas.H. EHS Constantine 76.La mégacalicose ou maladie de Puigvert. A propos d'un cas Abes H, Benziane N, Bencharif S.EHS Douéra 77. Histiocytose langerhansienne néonatale : Un diagnostic ardu, une prise en charge codifiée.Otmane.A**, Arrada .Z** , Oucif.S**, SadaouI.B** , Berrah.H** , Babaahmed .R* – Djennane .N *. 78. Epidermolyse bulleuse héréditaire simple à révélation néonatale : à propos d’une observation chez un nouveau né âgé de 02 jours Laabed A, Telhaoui N. EPH Hakim Saadene Biskra 79. Sarcome d'Ewing à propos d'un cas rare Bouteldja M1, Boumeddane A2.Clinique Amilcar Cabral CHU Oran . 2Centre Emir Abdelkader Oran– oncologie pédiatrique 80. Dépistage neonatal d’une tyrosinemie asymptomatique Sadaoui B,Arrada Z,Hadjarab A., Rezzaz N, Berrah H. CHU H.Dey 81. Une forme particulière du retard psychomoteur :le syndrome de NEUHAUSER -A propos d'un cas
Mahchouche N.Mekki A,Lebied A,.Baghriche .M .EPH Bologhine Ibn Ziri 82.Une thrombose veineuse révélant une ostéomyélite (A propos d’un cas) Zerrouki NH, Ibsaine O, Drali O, Boutaba K S, Latreche M, Berrah H.CHU Parnet Hussein dey 83.Paraplégie Medicale Sur Malformation Arterio-Veneuse. F.Akli, K.Safi, M.Ammenouche Service : Mpr Ehs Azur Plage

10
Programme Synoptique
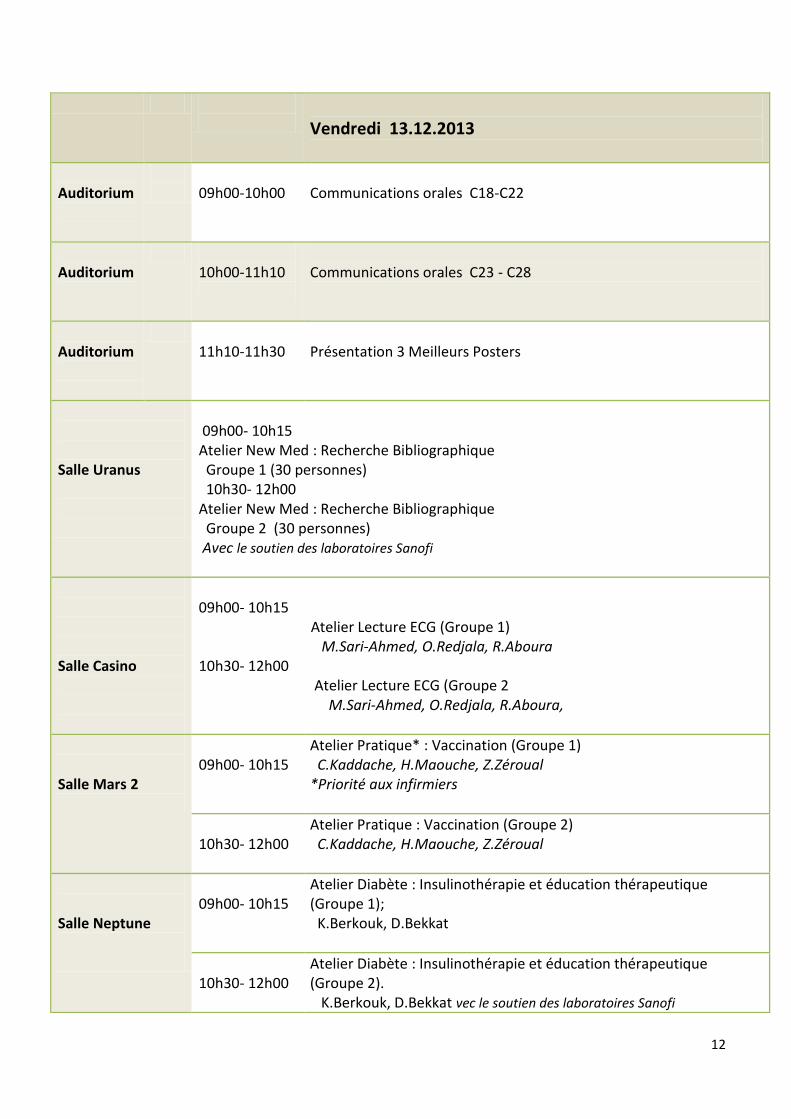
Mercredi 11.12.2013
Auditorium
16h 00
Inauguration
16h10- 16h 30
Conférence. Les objectifs du Millénaire pour le développement en Algérie : Peut-on espérer atteindre les cibles 4 et 5 en 2015 ? D.Lebane
16h 30 18h 30
Table ronde : Problématique Maladies rares
Introduction : A.Laraba
Hémophilie et autres anomalies constitutionnelles de l’hémostase M. Belhani
Déficits Immunitaires primitifs M. Baghriche
Les Maladies rénales A.Bensenouci
Les Maladies Métaboliques M.E Khiari
La Mucoviscidose R.Boukari
Les Maladies rares : Point de vue du MSP/RH Dr Magmoune
Discussion
11
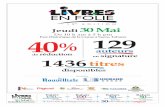
Jeudi 12.12.2013
Auditorium
08h30– 09h45
Table ronde : Mortalité Néo-natale
09h45- 10h 15 Conference : The new approaches and strategies on the enrichment and supplementation of breastfeeding R. Van Elburg Departement de recherche Danone nutrition ELN
10h15- 10h45 Pause café
10h45- 11h 45
Table ronde : Mortalité des enfants de moins de 5 ans (Nné exclu) Mortalité hospitalière : Enquête multicentrique
11h45- 12h 05 Prévention des décès infanto- juvéniles évitables JP Grangaud Avec le soutien des laboratoires Pfizer
12h00 – 12h30
Infections respiratoires basses : Données bactériologiques
H.Tali Mammar
Quelles exigences pour préserver l'efficacité des antibiotiques
L. Smati
Avec le soutien des laboratoires GSK
12h30 - 14h00 Déjeuner
14 h - 15 h 30 Communications orales C1 à C8
15h30 - 17h00 Communications orales C9 - C17
Salle Neptune
Workshop 14h- 16h30
Déficits Immunitaires Primitifs
Salle casino
Workshop 14h- 16h30
Maladies Métaboliques Modérateurs :
Salle Uranus
14h -16h30
Atelier : CAT devant un retard de croissance* : Réflexion autour de cas cliniques (Groupe 1 - Groupe 2) C. Sultan, A. Ladjouze *Avec le soutien des Laboratoires Novonordisk
12
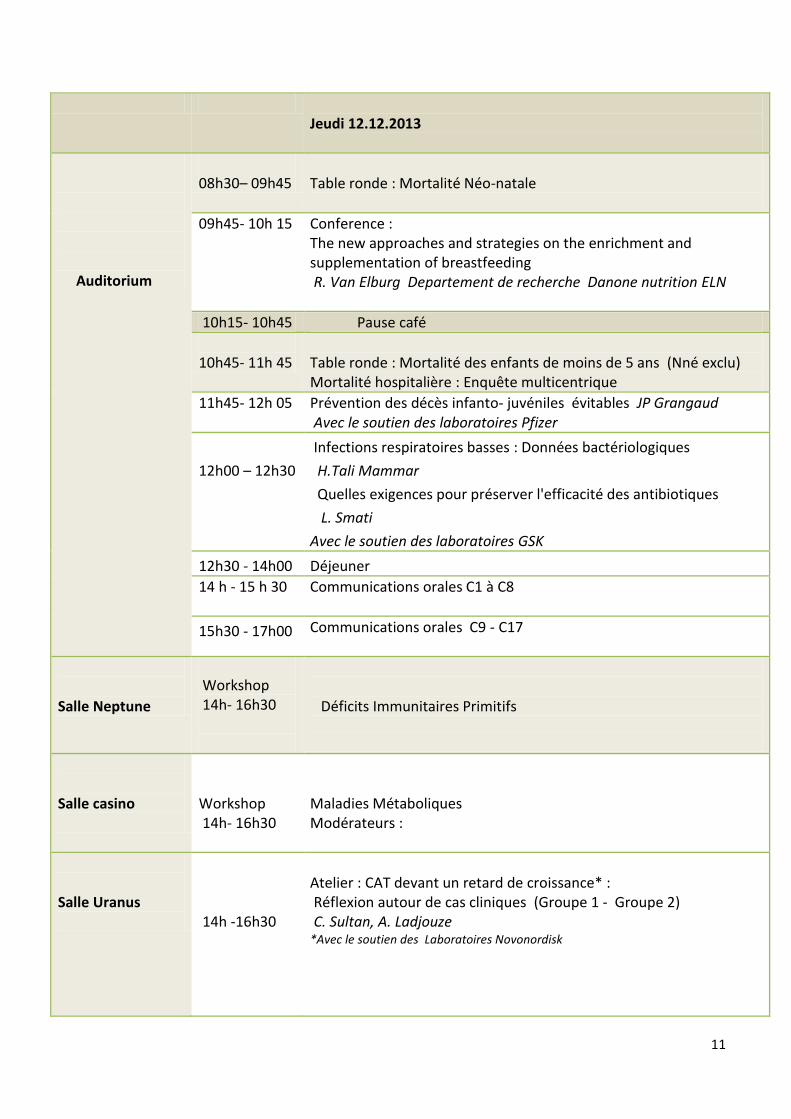
Vendredi 13.12.2013
Auditorium
09h00-10h00
Communications orales C18-C22
Auditorium
10h00-11h10
Communications orales C23 - C28
Auditorium
11h10-11h30
Présentation 3 Meilleurs Posters
Salle Uranus
09h00- 10h15 Atelier New Med : Recherche Bibliographique Groupe 1 (30 personnes) 10h30- 12h00 Atelier New Med : Recherche Bibliographique Groupe 2 (30 personnes) Avec le soutien des laboratoires Sanofi
Salle Casino
09h00- 10h15 Atelier Lecture ECG (Groupe 1) M.Sari-Ahmed, O.Redjala, R.Aboura 10h30- 12h00 Atelier Lecture ECG (Groupe 2 M.Sari-Ahmed, O.Redjala, R.Aboura,
Salle Mars 2
09h00- 10h15
Atelier Pratique* : Vaccination (Groupe 1) C.Kaddache, H.Maouche, Z.Zéroual *Priorité aux infirmiers
10h30- 12h00
Atelier Pratique : Vaccination (Groupe 2) C.Kaddache, H.Maouche, Z.Zéroual
Salle Neptune
09h00- 10h15
Atelier Diabète : Insulinothérapie et éducation thérapeutique (Groupe 1); K.Berkouk, D.Bekkat
10h30- 12h00
Atelier Diabète : Insulinothérapie et éducation thérapeutique (Groupe 2). K.Berkouk, D.Bekkat vec le soutien des laboratoires Sanofi

13
Vendredi 13.12.2013
Salle Tour
09h00-12h00
Atelier 1 (Médecins Généralistes – Néonatologie) Prématurité, Alimentation PPN M.Haridi, S.E.Smahi, Y.Sadi, K.Boudhar, Benrabah, Boutaghane Avec le soutien des laboratoires Danone NutritionELN
Atelier 2 Puéricultrices : Soins et développement du Nné Techniques d’alimentation du Nné M.Haridi, S.E.Smahi, Y.Sadi, K.Boudhar, Benrabah, Boutaghane Avec le soutien des laboratoires Danone Nutrition ELN
14
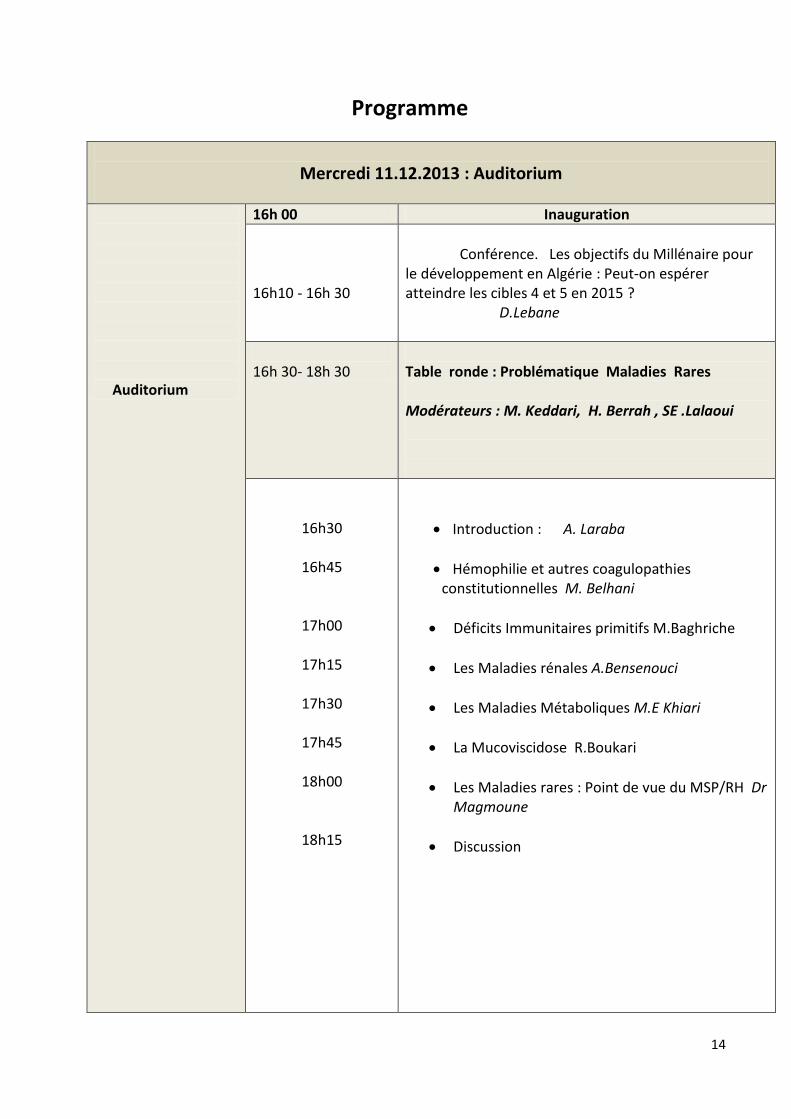
Programme
Mercredi 11.12.2013 : Auditorium
Auditorium
16h 00 Inauguration
16h10 - 16h 30
Conférence. Les objectifs du Millénaire pour le développement en Algérie : Peut-on espérer atteindre les cibles 4 et 5 en 2015 ? D.Lebane
16h 30- 18h 30
Table ronde : Problématique Maladies Rares Modérateurs : M. Keddari, H. Berrah , SE .Lalaoui
16h30
16h45
17h00
17h15
17h30
17h45
18h00
18h15
Introduction : A. Laraba
Hémophilie et autres coagulopathies constitutionnelles M. Belhani
Déficits Immunitaires primitifs M.Baghriche
Les Maladies rénales A.Bensenouci
Les Maladies Métaboliques M.E Khiari
La Mucoviscidose R.Boukari
Les Maladies rares : Point de vue du MSP/RH Dr Magmoune
Discussion
15
Jeudi 12.12.2013 : Auditorium
Auditorium Auditorium Auditorium
08h30 – 09h45 08h30 08h35 08h45 08h55 09h05 09h20 09h30
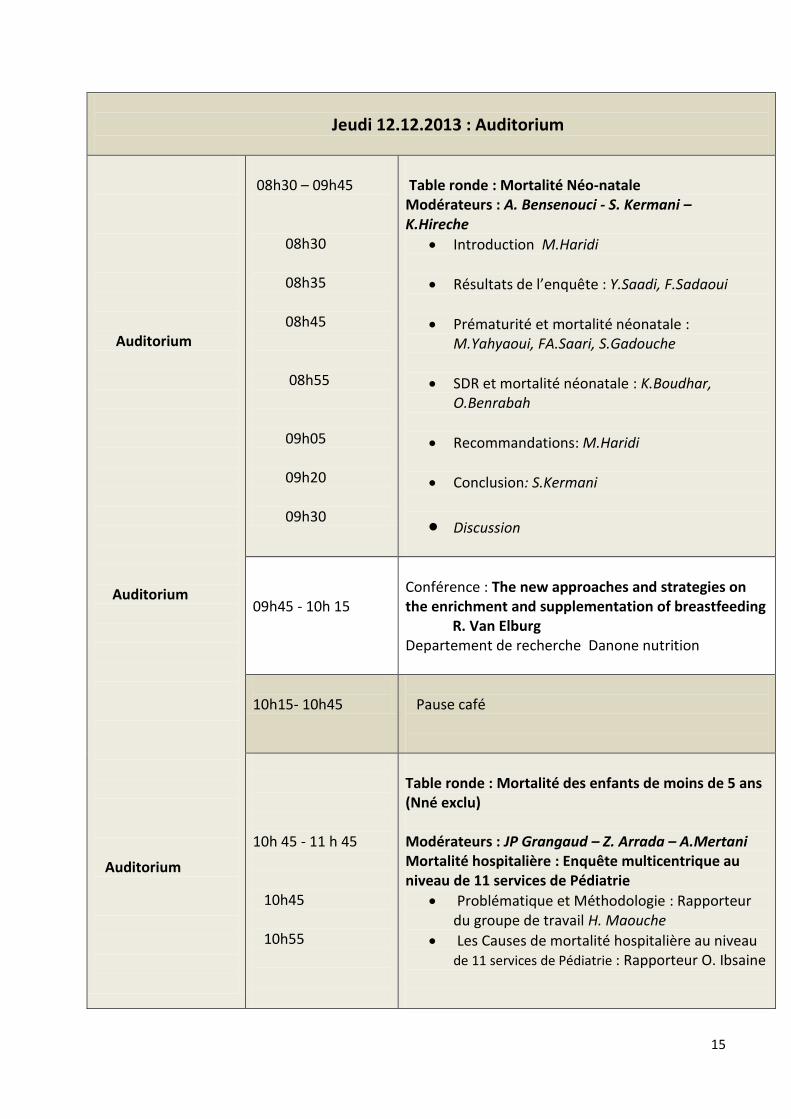
Table ronde : Mortalité Néo-natale Modérateurs : A. Bensenouci - S. Kermani – K.Hireche
Introduction M.Haridi
Résultats de l’enquête : Y.Saadi, F.Sadaoui
Prématurité et mortalité néonatale : M.Yahyaoui, FA.Saari, S.Gadouche
SDR et mortalité néonatale : K.Boudhar, O.Benrabah
Recommandations: M.Haridi
Conclusion: S.Kermani
Discussion
09h45 - 10h 15
Conférence : The new approaches and strategies on the enrichment and supplementation of breastfeeding R. Van Elburg Departement de recherche Danone nutrition
10h15- 10h45
Pause café
10h 45 - 11 h 45 10h45 10h55
Table ronde : Mortalité des enfants de moins de 5 ans (Nné exclu) Modérateurs : JP Grangaud – Z. Arrada – A.Mertani Mortalité hospitalière : Enquête multicentrique au niveau de 11 services de Pédiatrie
Problématique et Méthodologie : Rapporteur du groupe de travail H. Maouche
Les Causes de mortalité hospitalière au niveau de 11 services de Pédiatrie : Rapporteur O. Ibsaine
16
Auditorium Auditorium Auditorium
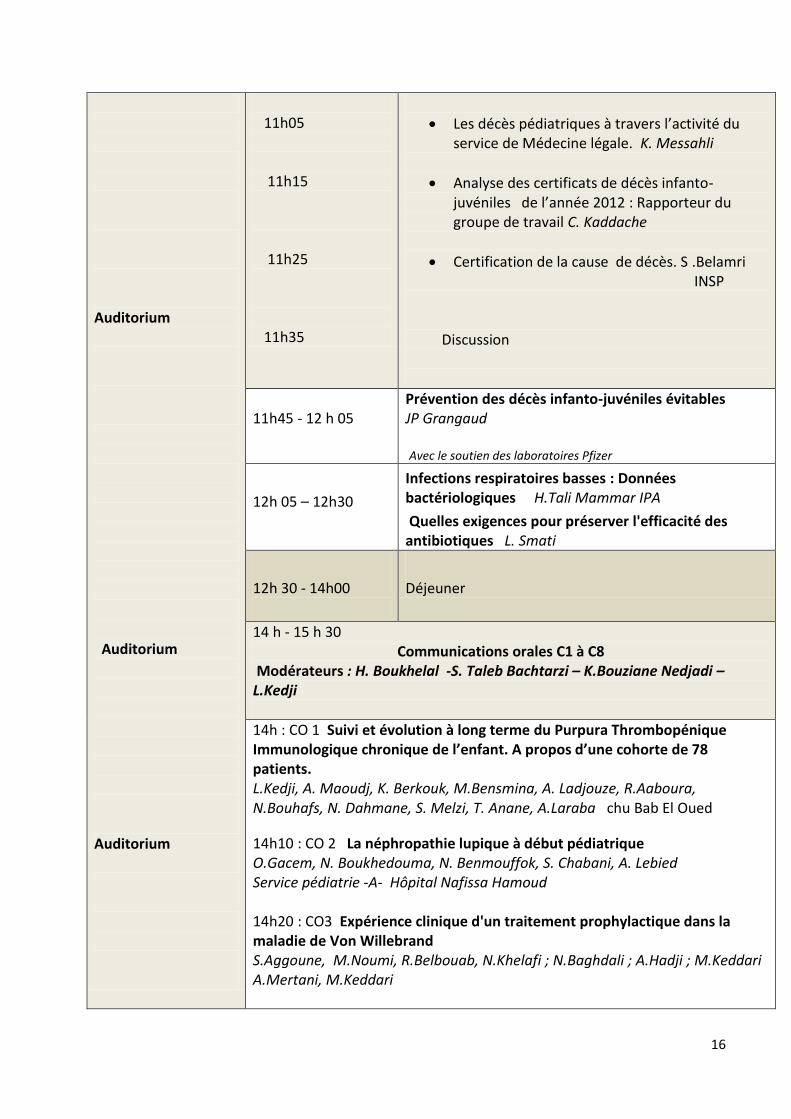
11h05 11h15 11h25 11h35
Les décès pédiatriques à travers l’activité du service de Médecine légale. K. Messahli
Analyse des certificats de décès infanto-juvéniles de l’année 2012 : Rapporteur du groupe de travail C. Kaddache
Certification de la cause de décès. S .Belamri INSP Discussion
11h45 - 12 h 05
Prévention des décès infanto-juvéniles évitables JP Grangaud Avec le soutien des laboratoires Pfizer
12h 05 – 12h30
Infections respiratoires basses : Données bactériologiques H.Tali Mammar IPA
Quelles exigences pour préserver l'efficacité des antibiotiques L. Smati
12h 30 - 14h00
Déjeuner
14 h - 15 h 30 Communications orales C1 à C8 Modérateurs : H. Boukhelal -S. Taleb Bachtarzi – K.Bouziane Nedjadi –L.Kedji
14h : CO 1 Suivi et évolution à long terme du Purpura Thrombopénique Immunologique chronique de l’enfant. A propos d’une cohorte de 78 patients. L.Kedji, A. Maoudj, K. Berkouk, M.Bensmina, A. Ladjouze, R.Aaboura, N.Bouhafs, N. Dahmane, S. Melzi, T. Anane, A.Laraba chu Bab El Oued
14h10 : CO 2 La néphropathie lupique à début pédiatrique O.Gacem, N. Boukhedouma, N. Benmouffok, S. Chabani, A. Lebied Service pédiatrie -A- Hôpital Nafissa Hamoud 14h20 : CO3 Expérience clinique d'un traitement prophylactique dans la maladie de Von Willebrand S.Aggoune, M.Noumi, R.Belbouab, N.Khelafi ; N.Baghdali ; A.Hadji ; M.Keddari A.Mertani, M.Keddari
17
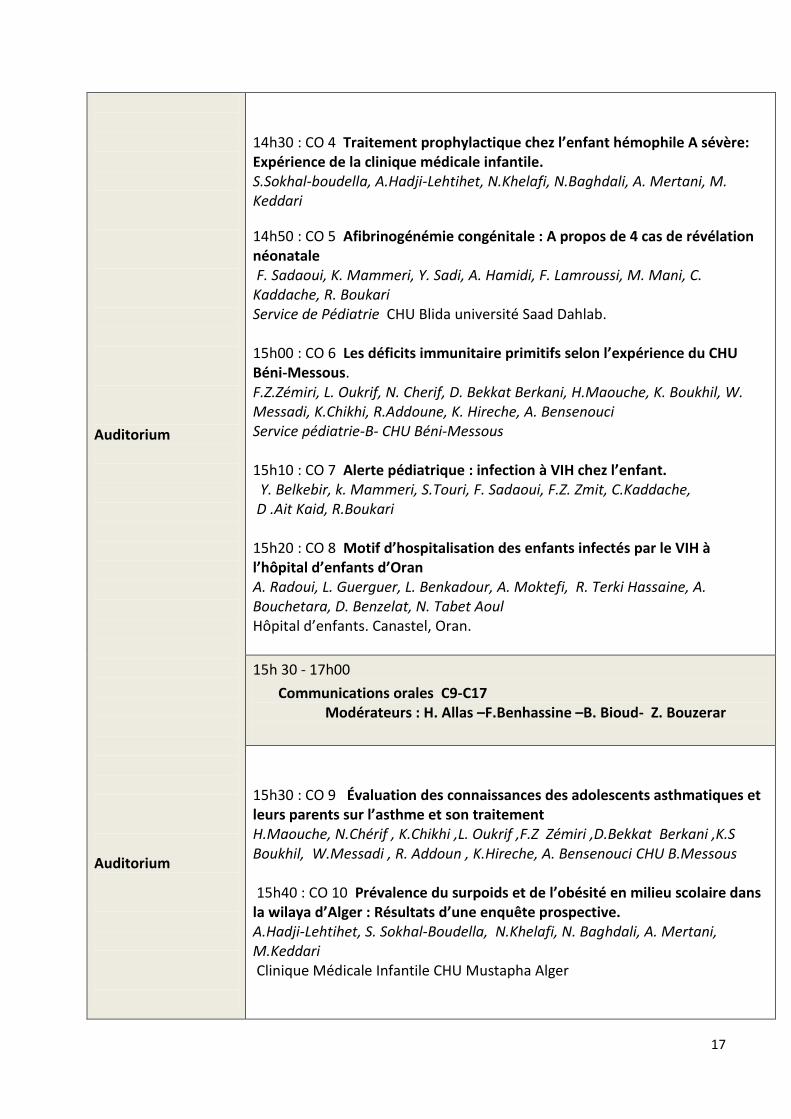
Auditorium Auditorium
14h30 : CO 4 Traitement prophylactique chez l’enfant hémophile A sévère: Expérience de la clinique médicale infantile. S.Sokhal-boudella, A.Hadji-Lehtihet, N.Khelafi, N.Baghdali, A. Mertani, M. Keddari
14h50 : CO 5 Afibrinogénémie congénitale : A propos de 4 cas de révélation néonatale F. Sadaoui, K. Mammeri, Y. Sadi, A. Hamidi, F. Lamroussi, M. Mani, C. Kaddache, R. Boukari Service de Pédiatrie CHU Blida université Saad Dahlab. 15h00 : CO 6 Les déficits immunitaire primitifs selon l’expérience du CHU Béni-Messous. F.Z.Zémiri, L. Oukrif, N. Cherif, D. Bekkat Berkani, H.Maouche, K. Boukhil, W. Messadi, K.Chikhi, R.Addoune, K. Hireche, A. Bensenouci Service pédiatrie-B- CHU Béni-Messous 15h10 : CO 7 Alerte pédiatrique : infection à VIH chez l’enfant. Y. Belkebir, k. Mammeri, S.Touri, F. Sadaoui, F.Z. Zmit, C.Kaddache, D .Ait Kaid, R.Boukari 15h20 : CO 8 Motif d’hospitalisation des enfants infectés par le VIH à l’hôpital d’enfants d’Oran A. Radoui, L. Guerguer, L. Benkadour, A. Moktefi, R. Terki Hassaine, A. Bouchetara, D. Benzelat, N. Tabet Aoul Hôpital d’enfants. Canastel, Oran.
15h 30 - 17h00
Communications orales C9-C17 Modérateurs : H. Allas –F.Benhassine –B. Bioud- Z. Bouzerar
15h30 : CO 9 Évaluation des connaissances des adolescents asthmatiques et leurs parents sur l’asthme et son traitement H.Maouche, N.Chérif , K.Chikhi ,L. Oukrif ,F.Z Zémiri ,D.Bekkat Berkani ,K.S Boukhil, W.Messadi , R. Addoun , K.Hireche, A. Bensenouci CHU B.Messous 15h40 : CO 10 Prévalence du surpoids et de l’obésité en milieu scolaire dans la wilaya d’Alger : Résultats d’une enquête prospective. A.Hadji-Lehtihet, S. Sokhal-Boudella, N.Khelafi, N. Baghdali, A. Mertani, M.Keddari Clinique Médicale Infantile CHU Mustapha Alger

18
Auditorium Auditorium
15h50 : CO 11 Tuberculose péritonéale : revue de 10 cas pédiatriques K.S Bouk’hil, N. Cherif, W.Messadi, L. Oukrif, F.Z Zémiri, H.Maouche, D. Bekkat-Berkani, K. Chikhi, K. Hireche, A. Bensenouci CHU Beni messous
16h00 : CO 12 Aspects épidémiologiques des intoxications par les médicaments et les produits domestiques chez l’enfant en pédiatrie au CHU Nedir Moahmed de Tizi-Ouzou N.Bensaadi, S.Idir, W. Benahmed, N. Benghadid, N. Cherifi, A.Benani, A. Tariket, A.Hamzaoui, D. Arhab, S.A Chalah, H. Haddou, T. Si fodil, K.Kadoun, M.Reghal. CHU Tizi-Ouzou 16h10 : CO 13 Place de l’analyse toxicologique en Pédiatrie: Bilan d’activité au CHU de Tizi-Ouzou: L.R Mekacher, O. Belazougui, W. Sebihi, L. Kaci, M.Mamou, D. Dahmani CHU Tizi-Ouzou 16h20 : CO 14 Les appendicites aigues de l’enfant de moins de 5 ans O.Ibsaine, Y. Kassa, Y. Belhimer, A Hadj- Arab1, Z.Arrada, A. Salem, H.Berrah service de pédiatrie « B ». C.H.U. Hussein.dey 16h30 : CO 15 Syndrome de Guillain - Barré : Expérience d’un service de Pédiatrie H.Ahmane, D. Arhab, A. Hamzaoui, S. Challah, S. Iddir, S. Rami, H.Rebiai, N.Aidrou N. Cherifi, N. Bensaadi CHU Tizi-Ouzou 16h40 : CO 16 Les cholestases intra-hépatiques progressives familiales (syndrome et maladie de Byler) .Présentation clinique et évolution : à propos de 21 malades. R.Belbouab, A. Khelafi, S. Agounne, R. Benalikhoudja, R. Berkani, Ait-Younes*, S.Chikhi, A.Mertani, M.Keddari Service de pédiatrie CMI CHU Mustapha
17h00 : CO 17 Prévalence de l’obésité infantile en milieu scolaire à Sétif. Résultats préliminaires Z.Benarab, S. Hadjit, S. Gabis, K. Dradra, A. Amara korba, B. Bioud Service de pédiatrie hôpital mère et enfant CHU de Sétif.
19
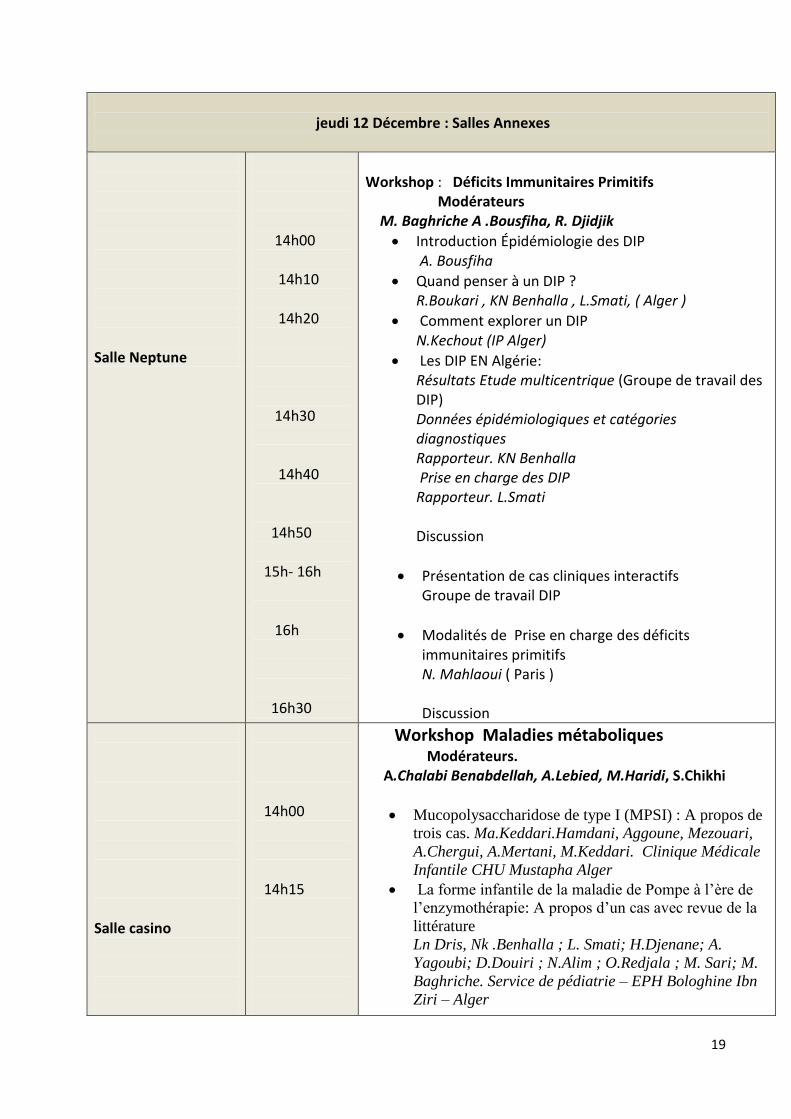
jeudi 12 Décembre : Salles Annexes
Salle Neptune
14h00 14h10 14h20 14h30 14h40 14h50 15h- 16h 16h 16h30
Workshop : Déficits Immunitaires Primitifs Modérateurs M. Baghriche A .Bousfiha, R. Djidjik
Introduction Épidémiologie des DIP A. Bousfiha
Quand penser à un DIP ? R.Boukari , KN Benhalla , L.Smati, ( Alger )
Comment explorer un DIP N.Kechout (IP Alger)
Les DIP EN Algérie: Résultats Etude multicentrique (Groupe de travail des DIP) Données épidémiologiques et catégories diagnostiques Rapporteur. KN Benhalla Prise en charge des DIP Rapporteur. L.Smati Discussion
Présentation de cas cliniques interactifs Groupe de travail DIP
Modalités de Prise en charge des déficits immunitaires primitifs N. Mahlaoui ( Paris ) Discussion
Salle casino
14h00 14h15
Workshop Maladies métaboliques Modérateurs. A.Chalabi Benabdellah, A.Lebied, M.Haridi, S.Chikhi
Mucopolysaccharidose de type I (MPSI) : A propos de
trois cas. Ma.Keddari.Hamdani, Aggoune, Mezouari,
A.Chergui, A.Mertani, M.Keddari. Clinique Médicale
Infantile CHU Mustapha Alger
La forme infantile de la maladie de Pompe à l’ère de
l’enzymothérapie: A propos d’un cas avec revue de la
littérature
Ln Dris, Nk .Benhalla ; L. Smati; H.Djenane; A.
Yagoubi; D.Douiri ; N.Alim ; O.Redjala ; M. Sari; M.
Baghriche. Service de pédiatrie – EPH Bologhine Ibn
Ziri – Alger
20
Salle casino
14h30
14h45
15h00
15h15
15h30
La maladie de Gaucher chez l’enfant :
Expérience de la Clinique Médicale Infantile du CHU
Mustapha
A.Hadji-Lehtihet, S.Sokhal1 ; N.Benali-Khodja1 ;
S.Chikhi.1 ; Ghouali2 ; Hallal2 ; B.Imessaoudène 2;
A.Berhoune 2 ; A.Mertani1 ; M.Keddari1. 1. Clinique Médicale Infantile CHU Mustapha Alger
2. Laboratoire de Biochimie et de Génétique CHU Mustapha Alger
14h45-15h : Maladies métaboliques : Expérience de 5
ans d’un service de pédiatrie générale
T. Bencharif Madani , S Taleb, S Benfetima, F Sellahi,
A Bakhouche, H Boudbaba ; N Hezil, S Benseghir ; A
Mesri ; Fz Boulmaiz
Service de pédiatrie du Mansourah (EHS sidi
mabrouk) de Constantine
Les hyperammoniémies : Démarche diagnostique et
cas interactifs L.Benhafsa, F.Benmati K.Boudhar
Service de Pédiatrie HCA
Déficit du cycle de l’urée : Discussion de 2 cas
cliniques F, Benmati, D .Chelighem, K. Boudhar, W.Messek, L. Benhafessa
Maladies héréditaires du métabolisme : mise au point
clinique et cas interactifs
A.Mekki Service de Pédiatrie CHU Nafissa Hamoud
Discussion
Salle Uranus
14h00-15h30 15h30-16h30
Atelier : CAT devant un retard de croissance : réflexion autour de cas cliniques
Groupe 1
Groupe 2 C. Sultan, A. Ladjouze Avec le soutien des Laboratoires Novonordisk
21
Vendredi 13.12.2013 : Auditorium
Auditorium
09h00-10h00
Communications Orales C18-C22 Modérateurs S.Bendeddouche – N.Bouchair- K.Radoui-K.Benallal
09h00 09h10 09h20 09h30 09h40 09h50
CO 18: Impact d’une unité de diabète infantile à l’EPH Relizane. S.Kara Mostefa, S.Kadari, R. Bendoubaba, R. Mezned, M. Touhami EPH Relizane Mohammed Boudiaf C0 19: L’acidose inaugurale dans le DT1de l’enfant dans la Wilaya d’Oran. Stratégie de réduction de sa fréquence. Zennaki, A. Ouzzaa, S. Niar, H. Aichaoui, M. Gharnouti, A. Reguieg, M. Naceur, K. Bouziane-nedjadi, M. Bessahraoui. Service de pédiatrie « C ». CHU d’Oran. CO20: Utilisation de la pompe à insuline chez l’enfant. Recrutement du service de pédiatrie « C » du CHU d’Oran.A.Zennaki1, K. Benharrats2, M. Gharnouti3, H. Aichaoui1, I. Abdat1, S. Niar1, L. Benhouria4, M. Bessahraoui1, K. Bouziane –Nedjadi1. CO 21: Cancer de la thyroïde chez l’enfant (à propos de 39 cas) K. Bouriche, D.Senouci, W. Mazari, A.S.Bendeddouche Service de pédiatrie. CHU Tlemcen- Algérie CO 22 Hyperthyroïdie de l'enfant Aspects cliniques et évolutifs F. Boufaroua, M.Chaou, S.Tari, Z.Zéroual, A. Atek, H.Boucenna, A. Khati, A Hezazi, ME. Khiari CHU Béni Messous Discussion
22
10h00-11h00
Communications orales C23-C28 Modérateurs L.Benhafsa N.Cherif N.Baghdali
10h00 10h10 10h20 10h30 10h40 10h50 11h00
CO23: Prévalence de l'allaitement maternel dans l'ouest algérien N. Zidane, A. Adel (Chlef) CO24: Méningites purulentes à pneumocoque du nourrisson .Z Zmit, D. Ait Kaid EHS El Kettar C025: Le déficit en vitamine B12 selon l’expérience du CHU Béni-Messous. F.Z Zémiri, L. Oukrif, N. Cherif, D. Bekkat Berkani ,H. Maouche , K. Boukhil , W. Messadi , K. Chikhi , R. Addoun , K. Hireche , A.Bensenouci CO26: Convulsions néonatales révélatrices d’hypovitaminose D maternelle : à propos de 06 cas . B. Sadaoui, Z. Arrada, K. Kebouchi, A. Belache, K. Saheb, M. Latreche S. Oucif H.Berrah Hôpital N. Hamoud, CHU H. Dey, Alger. CO 27: Prise en charge des cardiopathies congénitales chez les enfants trisomiques (T21) Expérience du service de pédiatrie / HCA à propos de 31 cas D. Chelirem , Y. Inouri, H. Boufnar, S.E Lalaoui Service de pédiatrie HCA CO28 Apport du certificat de décès périnatal et néonatal à la connaissance de la mortalité à Oran N.Heroual, A.Tadjeddine Semep Ehs Canastel Discussion
11h10 – 11h30 : Présentation 3 Meilleurs Posters
Auditorium
23
Vendredi 13 Décembre : Salles annexes
Salle Uranus
09h00- 10h15 Atelier New med : Recherche Bibliographique : Groupe 1 (30 personnes) 10h30- 12h00 Atelier New med : Recherche Bibliographique : Groupe 2 (30 personnes) Avec le soutien des laboratoires Sanofi
Salle Casino
09h00- 10h15
Atelier Lecture ECG (Groupe 1 M.Sari-Ahmed, O.Redjala, R.Aboura
10h30- 12h00
Atelier Lecture ECG (Groupe 2 M.Sari-Ahmed, O.Redjala, R.Aboura
Salle Mars 2
09h00- 10h15
Atelier Pratique* : Vaccination (Groupe 1) C.Kaddache, H.Maouche, Z.Zeroual *Priorité aux infirmiers
10h30- 12h00
Atelier Pratique : Vaccination (Groupe 2) C.Kaddache, H.Maouche, Z.Zeroual
Salle Neptune Salle Tour
09h00- 10h15 10h30-11h15 09h- 12h00
Atelier Diabète G 1: Insulinothérapie et ETP ; K.Berkouk, D.Bekkat Atelier Diabète G2 : Insulinothérapie et ETP K.Berkouk, D.Bekkat Avec le soutien des laboratoires Sanofi Atelier « (M G – Néonat) Prématurité, Alimentation PPN M.Haridi, S.E Smahi, N.Boutaghane Avec le soutien des laboratoires Danone Nutrition LEN Atelier Puericultrices : Soins et développement du Nné Techniques d’alimentation du Nné , Y.Sadi, O.Benrabah, K.Boudhar, Avec le soutien des laboratoires Danone Nutrition LEN

24
Abstracts des communications orales
CO1 : Suivi et évolution à long terme du Purpura Thrombopénique Immunologique
chronique de l’enfant. A propos d’une cohorte de 78 patients.
L. Kedji, A.Maoudj ,K. Berkouk, M. Bensmina, A.Ladjouze, R. Aboura, N. Bouhafs, N.Dahmane, S.Melzi , T.Anane, A. Laraba ( pédiatrie CHU Bab El Oued, Alger)
Introduction : lorsque le PTI devient chronique il faut rechercher un déficit immunitaire ou une pathologie auto immune plus large. L’autre point concerne le risque hémorragique et le traitement.
Objectifs : analyser le terrain dysimmunitaire sous-jacent, le saignement, le traitement et l’évolution à long terme des PTIC.
Méthodes : étude observationnelle uni centrique (1991 - 2011).
Paramètres étudiés : les ATCD familiaux, le bilan immunologique, le saignement, le traitement, l’apparition d’autres manifestations d’auto-immunité, et la rémission.
Résultats : 78 patients sont inclus, 14 ont un PTI récurrent : 44 filles et 34 garçons, âge moyen 5,6 ± 3,5 ans (0,5 - 15 ans). On note : un contexte familial dysimmunitaire chez 9 patients (11,5%), des AAN positifs chez 20 patients (26,6%), une dysglobulinémie (71,7%), une hypogammaglobulinémie persistante (11%), un taux bas d’haptoglobine (41%), un test de Coombs Direct positif (7,6%). En excluant les récurrents, le saignement est absent (50,7%), minime (15,8%), modéré (27%), sévère (6,4%). Traitement : 49% n’ont reçu aucun traitement, 44% reçoivent une corticothérapie occasionnelle, 3 patients ont été splénectomisés et 2 ont reçu du Rituximab. Evolution : 3 patients ont développé un syndrome d’Evans, 4 un LED probable. Avec un recul moyen de 67,4 ± 43,7 mois, la rémission complète concerne 44% des patients.
Commentaires : Les anomalies dysimmunitaires sont fréquentes. La majorité des patients sont peu ou pas symptomatiques et ne requièrent aucun traitement. Les traitements de seconde ligne ont des indications restreintes. L’évolution spontanée vers la guérison est toujours possible. Nos résultats rejoignent les données de la littérature.
CO 2 : La néphropathie lupique à début pédiatrique O.Gacem ,N. Boukhedouma – N. Benmouffok – S. Chabani – A. Lebied Introduction Le lupus érythémateux disséminé est une maladie systémique auto-immune d’étiologie inconnue, le début pédiatrique représente 10 % à 16 % des cas. La néphrite est la plus importante cause de morbidité et une cause majeure de mortalité dans cette maladie. Matériel et méthode C’est une étude rétrospective, faite sur une période de 10 ans dans notre service et qui a concerné les enfants dont l’âge est inférieur à 16 ans, répondant tous à au moins 4 critères des 11 critères de (ACR).

25
Résultats Nous avons colligé 25 malades lupiques avec 13 ayant une néphropathie lupique soit 52 % des cas. Parmi ces 13 patients, 11 ont présenté une insuffisance rénale au décours de la maladie soit 84 % des cas. Une ponction biopsie rénale a été effectuée chez la totalité des patients révélant 76 % de cas de néphropathie lupique sévère (stade III et IV). Un traitement d’induction à base de corticoïdes a été préconisé chez les 13 patients soit par voie orale (1,5 à 2 mg/kg/j), soit par des bolus de méthyle Prednisolone par voie intra veineuse suivie d’un traitement d’entretien avec dégression progressive jusqu'à une dose 5mg/j. Le traitement immunosuppresseur a concerné tous nos malades qui ont présente une néphrite lupique (Stade III et IV) soit 76 % des cas à base de cyclophosphamide.
Cette étude a révélé 50 % de rémission partielle et 1/3 de rémission totale. Dans notre série deux décès ont été constatés par insuffisance rénale terminale. Discussion A l’issue de cette étude nous constatons que l’atteinte rénale du lupus est à la fois une des manifestations les plus connues de cette maladie systémique, mais aussi la plus sévère, pesant de façon significative sur le pronostic fonctionnel et vital des patients. Et que la ponction biopsie rénale reste le gold standard pour évaluer l’activité rénale de la maladie. D’où l’intérêt de l’effectuer de façon très précoce, d’autant plus qu’il n’existe pas de corrélation anatomo-clinique et de différence de suivie et de pronostic rénal des classes III et IV versus autres classes. Nous constatons que l’avènement des traitements immunosuppresseurs et immunomodulateurs ont changé le pronostic de la néphropathie lupique et ont réduit de façon signficative le taux de la mortalité. Conclusion L’atteinte rénale dans le lupus érythémateux disséminé chez l’enfant reste une complication redoutable ayant un impact direct sur le pronostic rénal et vital, mais aussi indirecte par les complications additionnelles liées aux traitements. CO3 : Expérience clinique d'un traitement prophylactique dans la maladie de von Willebrand.
S.Aggoune M. Noumi ;R. Belbouab ; N.Khelafi ; Nadia Baghdali ; A.Hadji ; M.Keddari ;A. Mertani ; M. Keddari
INTRODUCTION La maladie de willebrand ( MW) est la plus fréquente maladie constitutionnelle de l’hémostase primaire due à un défaut quantitatif ou qualitatif en facteur Willebrand (FW) qui est une protéine plasmatique indispensable pour l’adhésion des plaquettes aux structures sous endothéliales en cas de lésion de la paroi vasculaire, prédisposant aux hémorragies avec une fréquence voisine de 1 %, elle est de transmission autosomique dominante. Les patients atteints de maladie de von Willebrand peuvent avoir besoin à court ou à long terme

26
de traitement prophylactique. La prophylaxie à court terme est généralement effectuée pour prévenir les saignements excessifs après une chirurgie ou procédures invasives, à long terme celle-ci peut être nécessaire pour contrôler les saignements articulaires et des muqueuses récurrents. MATERIELS ET METHODES Nous rapportons les résultats sur une thérapie secondaire de remplacement VWF prophylactique appliqué chez cinq de nos patients atteints de maladie de willebrand sévère. Chez tous les patients, la décision de lancer la prophylaxie repose sur un score de saignement> 2 avant le diagnostic ; le produit de substitution utilisé a fait appel au wilfactin. Après une période d'observation de 12 mois la fréquence de saignements menstruels chez les filles, et le score de saignement a été significativement réduit par rapport aux valeurs de pré-prophylaxie/pré-diagnostic, les épisodes de saignements récurrents ont été arrêtés chez 4 des 5 patients, aucun patient n’a présenté d’inhibiteur. la durée médiane de la prophylaxie était d’une année et demi. CONCLUSION L'utilisation de la thérapie de remplacement de facteur de von Willebrand prophylactique secondaire est une modalité de traitement efficace tolérée, très bénéfique pour les patients atteints de maladie de von Willebrand, qui présentent des saignements récurrents. CO 4: Traitement prophylactique chez l’enfant hémophile A sévère:Expérience de la Clinique Médicale Infantile. S.Sokhal-Boudella A.Hadji-Lehtihet, N.Khelafi, N.Baghdali, A. Mertani et M. Keddari:clinique medicale infantile CHU Mustapha Alger Abstract : Objectifs: L’hémophilie est une maladie hémorragique liée au chromosome X. L’objectif de ce travail est de rapporter, les résultats de la prophylaxie secondaire réalisée chez 11 enfants hémophiles A sévères suivis au niveau de la clinique médicale infantile. Objectif principal : C’est de réduire les saignements dans les articulations saines et de ralentir la progression de l’arthropathie. Matériels et Méthodes: C’est une étude rétrospective, effectuée de mars 2011 à février 2013. Elle a concerné 11 enfants et adolescents hémophiles A sévères dont l’âge varie de 2,5 à 14 ans avec un âge moyen de 5,3 ans. Les antécédents familiaux sont retrouvés chez 4 enfants. Nous avons décrit les signes cliniques initiaux et l'évolution de la maladie chez les 11 patients traités au préalable à la demande, puis mis sous traitement prophylactique secondaire. Critères d’inclusion: Age > 2ans ; nombre d’hémarthroses > 2 Critères d’exclusion: Age < 2ans ; nombre d’hémarthroses < 2 ; présence d’inhibiteurs Résultats: L'âge au moment du diagnostic était de J7 de vie à 4 ans, l’âge moyen était de 11,9 mois. Trois enfants ont bénéficié d’un diagnostic précoce à 1, 2 et 6 semaines de vie. L’analyse des différents arbres généalogiques a permis de noter: 7 cas sporadiques (64%) et 4 familles (36%) avec des antécédents familiaux de déficit en facteur VIII. Les enfants présentaient une fréquence des saignements allant de 3 à 6 fois par mois. Il s’agissait d’hémarthroses essentiellement majeures : genoux 42%, chevilles 35%, coudes 13% et poignets

27
10% ; d’ecchymoses et d’hématomes sous-cutanées et musculaires (10 et 15%) superficiels et surtout profonds retrouvés chez 4 enfants : hématomes extra dural cérébelleux et occipito pariétal, au niveau de la cuisse gauche, de l’abdomen, du thorax, de l’avant bras droit et au niveau de la région iliaque gauche. 6 enfants présentaient des plaies de la langue et de la lèvre supérieure. 7 malades ont développé des articulations cibles principalement au niveau des genoux (70%) et les chevilles (43%). 2 malades ont évolué vers l’arthropathie chronique des genoux, un malade a subi une synoviorthèse après évaluation du genou gauche par l’IRM. Tous les patients ont reçu une dose prophylactique du facteur VIII recombinant (octocog alfa) à raison de 25UI/kg une fois par semaine. Sous traitement, nous avons observé une réduction de la fréquence des saignements, 1 à 2 épisodes de saignement par mois chez 6 malades, 1 saignement ? à 3 mois chez 3 enfants et 1épisode ? à 6 mois chez 2. Le score clinique de PedNet a permis de noter une réduction du nombre d’hémarthroses majeures avec tuméfaction mineure : genoux 30%, chevilles 27% et coudes 10% ; la douleur est modérée à légère soulagée par des antalgiques, absence d’atrophie musculaire, absence d’articulations cibles ni de synovite chronique. Le score radiologique de Pettersson n’a pas relevé des modifications notables. L’évaluation biologique : sérologies virales de l’hépatite virale A, B, C et de l’HIV sont négatives pour tous les malades. La recherche des inhibiteurs est négative chez 6 hémophiles et positive chez 4 enfants dont 2 sont fort répondeurs et 2 présentent un taux non titrable. Conclusion: Notre série et les cas pédiatriques rapportés dans la littérature confirment la gravité de l’hémophilie chez l'enfant justifiant un diagnostic et une prise en charge précoces. L’efficacité de la prophylaxie est indiscutable, néanmoins ce traitement induit une augmentation de l'exposition aux concentrés anti-hémophiliques et pourrait majorer les éventuels risques thérapeutiques. Il représente une contrainte et un surcoût immédiat substantiel. L'avenir réside dans la fabrication de facteurs anti-hémophiliques, à longue durée de vie avec des modalités d’administration moins astreignants, et dans la thérapie génique. Mots clés : Hémophilie A; Déficit en Facteur VIII Maladie hémorragique ; prophylaxie secondaire.
CO5 AFIBRINOGENEMIE CONGENITALE : A propos de 4 cas de révélation néonatale
F. Sadaoui K. Mammeri, Y. Sadi, A. Hamidi , F. Lamroussi, M. Mani, C. Kaddache , R. Boukari.Service de Pédiatrie, CHU Blida Université Saad Dahlab.
Introduction : L’afibrinogénémie congénitale (AFC), anomalie constitutionnelle de l’hémostase de transmission autosomique récessive, peut se révéler au stade néonatal par une hémorragie ombilicale à la section du cordon. La gravité du syndrome hémorragique est variable avec mise en jeu du pronostic vital dans les formes graves. Son diagnostic est facile devant un sang incoagulable et un taux de fibrinogène indosable. Objectifs : Description de 4 observations et évaluation de la prise en charge. Matériel et méthodes : Etude rétrospective de 2006 à 2012 Résultats : 4 garçons d’âge moyen actuel 4,5 ans (2 frères) issus d’un mariage consanguin ayant présenté dès la période néonatale une hémorragie ombilicale dont le diagnostic est confirmé par un taux de Fibrinogène < 0,2 g/l. L’évolution est marquée par plusieurs accidents hémorragiques modérés post-traumatiques (ecchymoses, hématomes, gingivorragies, épistaxis) nécessitant la transfusion de plasma frais congelé (PFC) (01 fois / mois en moyenne), seul traitement

28
disponible. Un enfant est décédé à l’âge de 6 ans d’une hémorragie intracrânienne après un traumatisme survenu à l’école. La surveillance virologique est négative, la circoncision a été faite sans incident sous couverture de PFC dans trois cas. L’enquête familiale révèle la présence de cas similaires chez deux enfants. Conclusion : Actuellement le traitement recommandé de l’AFC repose sur le fibrinogène concentré à partir de plasma en prophylaxie ou sur demande. Le diagnostic prénatal est possible pour les familles à risque.
CO 6: Les déficits immunitaire primitifs selon l’expérience du CHU Béni-Messous.
F.Z.Zémiri, L.Oukrif ; N. Chérif ; D.Bekkat ; H.Maouche ; K.Boukhil ; S.Messadi; K.chikhi ; A.Addoune ; K.Hireche ; A.Bensenouci .Service pédiatrie B. CHU Béni-Messous
Les déficits immunitaires primitifs(DIP) sont des maladies individuellement rares, mais dont la fréquence est estimée à 1/5000 dans la population générale. Ils correspondent à environ 300 maladies génétiques actuellement connues, d’expression clinique hétérogène se révélant parfois tardivement chez l’adulte. Toutes ces affections ont en commun un déficit de fonctionnement des défenses de l’organisme. On note une sensibilité accrue aux infections et parfois une auto-immunité. Un diagnostic précoce et une prise en charge sont nécessaires.
Objectifs : Etudier les particularités épidémiologiques, cliniques, paracliniques, thérapeutiques et évolutives de nos malades.
Matériel et méthode : L’étude est rétrospective sur une période de 05ans (2008-2012) selon des fiches préétablies. Nous avons colligés 15cas. Le diagnostic et la classification du déficit immunitaire ont été retenus selon la dernière classification des DIP . Résultats : Deux tiers de nos patients sont des garçons et la moitié des nourrissons. Un malade sur deux a eu au moins une hospitalisation antérieure. Les infections répétées sont surtout respiratoires (75%) digestives (40%) et ORL (35%).Les infections graves (27%) sont des septicémies, abcès ou méningite. La consanguinité est retrouvée dans 75% des cas. Dans 47%, il y’a au moins un décès dans la fratrie. Nous avons colligé 33% DI combiné sévère, 25% DI humoral ,25% DI à lymphocytes T ,17% DI à lymphocytes B avec un déficit du C3 ; 17%DI avec déficit en HLA classe II ,33%syndrome d’hyper IgM avec ataxie-télangiectasies dont un LT-LB-NK-, 33% d’auto-immunité (thrombopénie, anémie, pancytopénie et hépatosplénomégalie),2 cas d’atopie ,un cas syndromique avec hypothyroïdie et malformation cérébrale. Les germes retrouvés sont surtout nosocomiaux .Les infections parasitaires et mycosiques sont aussi notées et même tuberculeuses (2cas) avec poyadénopathies. L’antibiothérapie probabiliste puis selon l’antibiogramme, est instituée chez (54%) des cas ;l’antibioprophylaxie dans (47%),les immunoglobines polyvalentes(47%).L’évolution est favorable dans 75% des cas .Trois décès ont été déplorés.

29
Commentaires : L’âge et le type d’infection à répétition concordent avec la littérature. Au Maghreb, le DI combiné sévère semble le plus fréquent avec le syndrome d’hyper IgM et le défaut d’expression des antigènes HLA de classe II, alors que le déficit en IgA semble être plus rare. En littérature Le DI humoral est le plus fréquent. Dans notre étude, le DI cellulaire est le plus fréquent. L’auto immunité est rare dans la littérature alors que nous l’avons trouvée dans 20%. L’enquête familiale a concerné 3 familles seulement. L’étude génétique aurait été souhaitable car elle est possible ; 170 gènes sont identifiés. Conclusion : Le DI est à suspecter devant toute infection grave ou à répétitions, ou une auto-immunité. Le diagnostic est souvent réalisé par des examens simples mais un immunologiste peut être d’une grande aide. Un traitement adapté et un conseil génétique sont nécessaires.
CO7 Alerte pédiatrique : infection du VIH chez l’enfant.
Y.BELKEBIR OULDHOCINE, K.MAMERI, S.TOURI, F.SADAOUI, CH.KADDACHE, R.BOUKARI
FZ.ZMIT*, Pr AITKAID* Service pédiatrie CHU BLIDA,* EHS ELKettar
INTRODUCTION : Des progrès considérables ont été réalisés dans la prévention de la
transmission mère enfant du VIH. Toutefois ces avancées ne peuvent être appliquées dans notre
pays dés lors que le dépistage chez la mère est pratiquement inexistant.
OBJECTIF : Décrire les caractéristiques cliniques, biologiques et évolutives de l’infection à VIH et
alerter les pédiatres.
MATERIEL : Etude rétrospective sur trois cas de SIDA chez les nourrissons de moins de 12mois
hospitalisés au CHU de Blida au cours de l’année 2012/ 2013.
RESULTATS :Les trois nourrissons (3, 4, 9 mois) sont hospitalisés pour infection pulmonaire avec
détresse respiratoire. Sont tous nés par voie basse, allaités au sein et vaccinés par le BCG.
Les autres manifestations cliniques sont : une hypotrophie, une candidose buccale étendue, et
une diarrhée chronique. L’atteinte respiratoire est associée à une méningite à pneumocoque
dans un cas.
La sérologie VIH positives chez les trois nourrissons et les mères confirme le diagnostic et la
transmission verticale. Deux enfants de la fratrie et deux pères sont infectés par le VIH.
Tous ces cas dépistés sont traités à Hôpital EL-KETTAR.
Un nourrisson est décédé de complication neurologique
Conclusion :Toute pathologie infectieuse chronique et ou récidivante doit faire évoquer une
infection à VIH.Il est impératif de dépister toutes les femmes jeunes en âge de procréer afin
d’instaurer les traitements préventifs de la transmission mère enfant.

30
CO 8 : Motif d’hospitalisation des enfants infectés par le VIH à l’hôpital d’enfants d’Oran. A. Radoui L. Guerguer(1), L. Benkadour(1), A. Moktefi(1), R. Terki Hassaine(2), A. Bouchetara(3), D. Benzelat(4), Tabet Aoul Nabil(5)
Introduction. L’infection à VIH est une cause fréquente de morbi- mortalité de l’enfant dans les pays en voie e de développement comme l’Algérien. Elle se traduit par de nombreuses pathologies qui motivent l’hospitalisation. Méthodologie. Nous avons analysé rétrospectivement sur une période de 5 ans (2009-2013) le motif d’hospitalisation de 19 dossiers d’enfants infectés par le VIH dans le but d’identifier les symptômes les plus caractéristiques qui serviront au diagnostic précoce. Résultats. Le sexe ratio était de 2,1. L’âge moyen à l’hospitalisation était de 2,1 ans (1 mois - 10,4 ans). Le motif principal d’hospitalisation était les pneumopathies aigues ou à répétition (63.1 %), viennent ensuite la candidose buccale (21 %), la malnutrition protéino-énergétique (15,7 %), les diarrhées chroniques (15 ,7 %), la fièvre prolongée (15,7 %), l’adénopathie avec hépato-splénomégalie (10,5 %). Les co -morbidité étaient fréquentes. Le retard staturo-pondéral a été observé dans 63% des cas. L’anémie a été observée chez la majorité des patients. La TDM thoracique pratiquée chez 6 patients a objectivé une pneumopathie interstitielle diffuse avec aspect en verre dépoli. Les diagnostics évoqués avant les résultats de la sérologie VIH étaient principalement le déficit immunitaire, la maladie cœliaque, l’asthme, l’APLV, les broncho-pneumopathies obstructives. La transmission verticale a été évoquée dans la quasi-totalité des cas. Trois enfants sont décédés à un âge moyen de 3 ans [1-7]. Les autres patients étaient orientés au service de maladies infectieuses. Conclusion. Les symptômes de VIH ne sont pas spécifiques et peuvent orienter vers d’autres diagnostics. Devant une symptomatologie chronique ou grave chez l’enfant, ne pas se limiter à rechercher des pathologies classiques mais penser aussi au dépistage du VIH. CO9 : ÉVALUATION DES CONNAISSANCES DES ADOLESCENTS ASTHMATIQUES ET LEURS PARENTS SUR L’ASTHME ET SON TRAITEMENT.
Maouche. H,Chérif N. Chikhi K. Oukrif L. Zémiri F.Z. Bekkat Berkani D. Boukhil K.S. Messadi W. , Addoun R.Hireche K. Bensenouci A.
Introduction : L’asthme est la maladie respiratoire chronique la plus fréquente de l’enfant. Sa prise en charge inclut le traitement des crises et le traitement de fond. Les thérapeutiques utilisées sont sous la dépendance totale du patient et de sa famille. Pour que l’adolescent prenne en charge son traitement quotidien de façon optimale, il est nécessaire que ce dernier ait des connaissances sur sa maladie et sur son traitement. L’acquisition des connaissances s’articule sur un apprentissage simplifié appelé Éducation Thérapeutique. Nos adolescents asthmatiques possèdent ils ces connaissances indispensables pour une meilleure gestion de leur maladie ? Objectif : Évaluer les connaissances des adolescents asthmatiques et leurs parents sur l’asthme et son traitement.

31
Matériels et méthodes : Il s’agit d’une étude prospective sur une période de 1 an. Nous avons recruté 85 patients asthmatiques âgés entre 14 et 17 ans. Nous avons évalué les connaissances sur la maladie : mécanismes, facteurs déclenchants, prodromes, signes de la crise, signes de sévérité de la crise ; sur le traitement : médicaments, technique d’inhalation, utilisation de la chambre d’inhalation, du débitmètre de pointe ; la distinction traitement de la crise/traitement de fond et le niveau de contrôle de la maladie. L’évaluation a été faite à l’aide de questionnaires à choix simple ou multiple et des grilles d’observation avec une échelle de mesure1, 2. La population étudiée étant homogène, un score moyen par malade est calculé pour chaque item évalué.
Résultats : 85 patients ont été évalués. Age moyen 14,92 ans, Sexe ration 1,5. Ancienneté de la maladie : moyenne 74 mois. Niveau de contrôle de la maladie : Contrôlés 0, partiellement contrôlés 84, non contrôlé 01. Suivi antérieur : 48% (pédiatre 26%, pneumologue 11%, Allergologue 04%, généraliste 07%), lieu du suivi :chu ou ehs 22% , centre de santé 05% , cabinet privé 21% ).Les connaissances (scores moyens/malade ) : mécanismes de la maladie : 1,5/10, facteurs déclenchants 3,2/9, les prodromes 1/4, les signes de la crise 4,4/5, les signes de sévérité d’une crise 2,5/4, les médicaments 0,5/6, distinguer le traitement de la crise du traitement de fond 0,4/3.Les habiletés techniques (scores moyens):Technique d’inhalation : 1/15, chambre d’inhalation 2,8/8 (45% n’ont jamais vu une chambre d’inhalation), débitmètre de pointe : 1,4 /9 ( 80% n’ont jamais vu un DEP).
Commentaires : Ces résultats révèlent une carence dans les connaissances des patients sur la maladie, les médicaments et les habiletés techniques. Ce qui est un des facteurs du mauvais contrôle de la maladie. Dans une enquête portant sur 72 enfants asthmatiques de Blic3 trouve que 14% des enfants seulement avaient une technique d’inhalation correcte avec l’aérosol doseur. Chez 364 enfants âgés entre 5 et 18 ans Malot4 rapporte que 75 % d’entre eux ont fait au moins une erreur dans l’utilisation de L’Aérosol Doseur Pressurisé (ADP). Evaluant l’utilisation du système Autohaler chez 379 enfants âgés entre 4 et 17 ans Zureik5 rapporte que 247 ont mal exécuté au moins un item. Chez 103 patients âgés de plus de 12 ans, Agne6 rapporte un score moyen d’utilisation de l’ADP de 5,4/10, seuls 4% avaient une bonne technique de prise de l’ADP et sont tous non contrôlés. Chez 40 enfants asthmatiques âgés entre 8 et 10 ans scolarisés dans 12 écoles de la wilaya de sidi Belabés, Taleb7 rapporte une absence d’informations sur la maladie et son traitement. Conclusion : Des connaissances sur l’asthme et son traitement sont indispensables pour un meilleur contrôle de la maladie. Les praticiens doivent fournir des efforts pour l’apprentissage et l’éducation thérapeutique des patients.

32
CO10 : Prévalence du surpoids et de l’obésité en milieu scolaire dans la wilaya d’Alger : Résultats d’une enquête prospective.
A.Hadji-Lehtihet, S.Sokhal-Boudella ; N.Khelafi ; N.Baghdali ; A.Mertani ; M.Keddari Clinique médicale infantile CHU Mutapha Alger.
L’obésité commune est une véritable épidémie, Peu de chiffres ont été publiés chez l’enfant dans notre pays d’où l’intérêt de cette enquête. Objectifs : Nous avons effectué une enquête prospective de type transversal dans la wilaya d’Alger. L’objectif principal est de déterminer la prévalence du surpoids et de l’obésité en milieu scolaire, prévalence selon l’âge et le sexe. Cette prévalence est évaluée selon les différentes références notamment IOTF2000 et OMS 2007. Résultats : L’enquête a concerné 6180 enfants scolarisés dans les écoles primaires et les collèges des 13 daïras de la wilaya d’Alger et s’est étalée d’avril 2006 à décembre 2007. La prévalence globale du surpoids obésité incluse est de 18% selon les références de l’IOTF 2000 dont 4,1% d’obèses, et de 28,8% selon la référence OMS 2007 dont 8% d’obèses. La prévalence du surpoids est significativement différente selon le sexe, plus faible chez le garçon. L’analyse de la prévalence, selon la commune de résidence, montre des différences significatives, la prévalence du surpoids (obésité incluse) varie de 31,2% à 11%. Conclusion : La prévalence du surpoids et de l’obésité est élevée dans la wilaya d’Alger. Ce chiffre de 18% confirme que l’obésité est un problème majeur de santé publique nécessitant une politique de santé pour le dépistage et la prévention dans les structures de santé et les établissements scolaires. CO11: Tuberculose péritonéale : revue de 10 cas pédiatriques .
BOUK'HIL. K.S , CHERIF .N, MESSADI W, OUKRIF L, ZEMIRI. F.Z, MAOUCHE .H, BEKKAT-BERKANI .D, CHIKHI .K, HIRECHE .K, BENSENOUCI .A Service Pédiatrie B - CHU Béni Messous
Introduction La tuberculose abdominale chez l’enfant est rare, loin derrière l’atteinte ganglionnaire, pleurale, méningée et osseuse. Elle se présente sous 3 formes : intestinale, hépatosplénique ou péritonéale. Cette dernière est insidieuse et peut constituer un problème diagnostique. Objectif Déterminer les particularités épidémiologiques, cliniques, thérapeutiques et évolutives de la tuberculose péritonéale. Méthodes Etude rétrospective des cas colligés au service Pédiatrie B / CHU Béni Messous entre janvier 2011 et octobre 2013. Résultats 10 patients ont été hospitalisés pour tuberculose péritonéale. Ils présentaient une distension abdominale (100%), des douleurs abdominales (70%) et de la fièvre (80%). L’échographie retrouvait une ascite libre (100%) et des lésions hépatospléniques (3 cas). L’ascite était exsudative dans tous les cas avec une prédominance lymphocytaire. Tous nos patients ont eu une exploration cœlioscopique ou chirurgicale, péritonéale, qui retrouvait un aspect nodulaire du péritoine. L’histologie confirmait le diagnostic. Ils ont été traités selon les recommandations du

33
PNLAT Commentaires La tuberculose péritonéale est une maladie subaiguë. L’interrogatoire est contributif, s’il révèle des antécédents personnels ou familiaux de tuberculose. Les symptômes décrits ici sont similaires aux données publiées. Aucun patient ne présentait de tuberculose extra-abdominale contrairement aux autres séries. Il n’existe pas de marqueurs biologiques spécifiques. L’analyse du liquide d’ascite oriente si l’ascite est exsudative avec prédominance lymphocytaire. La recherche à l’examen direct du bK est décevante et l’inconvénient de la culture est le temps nécessaire pour obtenir le résultat. L’imagerie est peu spécifique. La cœlioscopie avec biopsie péritonéale permet d’établir le diagnostic. Conclusion La tuberculose péritonéale, notamment en région d’endémie, doit être suspectée chez tout patient présentant une ascite d’installation progressive, associée à de la fièvre et des douleurs. La cœlioscopie avec biopsie péritonéale est le moyen diagnostique de référence.
CO12 : Aspects épidémiologiques des intoxications par les médicaments et les produits domestiques chez l’enfant en pédiatrie au CHUN.
Nedir Moahmed de Tizi-Ouzou.N.Bensadi, S.Idir, Benahmed,W.Benghadid, N. Cherifi, A.Benani, A.Tariket, A.Hamzaoui, D. Arhab S.A.Chalah, H. Haddou, T.Si Fodil, K.Kadoun, M.Reghal.
Introduction: L’intoxication constitue un des volets de la pathologie accidentelle pédiatrique et une pathologie d’actualité de part le monde, deuxième cause d’accident domestique chez l’enfant en France et en Inde avec respectivement 8% et 22,3%. Elle nécessite une prise en charge rapide et adéquate du fait des déséquilibres rapides qu’elle entraîne. Le pronostic dépend de l’âge, de la nature du toxique et de sa dose, et du délai de la prise en charge. Objectifs : Analyser les caractéristiques épidémiologiques des intoxications de l’enfant, ainsi que les complications constatées. Matériels et méthodes : Une étude rétrospective sur dossiers de cas d'intoxication aiguë enregistrée au Centre Hospitalo-Universitaire (CHU) de Tizi-Ouzou entre Janvier 2011 et septembre l213 Résultats : L’intoxication aigue en dehors de l’intoxication alimentaire a concerné 144 enfants, 77 filles pour 67 garçons. Ceux qui sont le plus à risque d’intoxication sont les petits, âgés de moins de 8ans (83%), 30 % de toutes les intoxications ont été rapportées chez les moins de 2 ans. En ce qui concerne le lieu, la majorité des intoxications sont survenues à domicile et par voie orale. La nature du produit ingérés était médicamenteuse dans 52%, produit ménager dans 45% des cas et cosmétique dans 3%.Quelques intoxications volontaires ont été retrouvées, volontiers polymédicamenteuse avec prédominance féminine. Peu de symptômes inquiétants ont été notés à type de coma, ataxie, dystonie, signes extrapyramidaux, troubles hémodynamiques, lésions buccales et œsophagiennes importantes.

34
L’évolution a été favorable pour presque tous, mis à part 04 œsophagites avec sténose qui sont en cours de dilatation. Discussion : Les intoxications médicamenteuses aigues sont un motif fréquent d’admission au service des urgences de CHU de Tizi_Ouzou. Elles constituent la première cause des intoxications aigues. Vu le grand nombre de familles médicamenteuses mises en vente, ainsi que l’hétérogénéité de la symptomatologie en cas de surdosage, le diagnostic demeure difficile, d’autant plus que le médicament en cause est souvent non précisé. En revanche chez les adolescents, elles sont souvent volontaires pour des raisons sociales. Les produits phytosanitaires particulièrement l’eau de javel utilisée comme désinfectant ménager, viennent en 2eme position dans les intoxications aigues. Conclusion : Les intoxications chez les jeunes enfants sont fréquentes. Même si la plupart d’entre elles sont mineures et n’ont que peu ou pas de conséquences pour la santé de l’enfant, la possibilité que le produit ingéré soit très toxique ne peut être écartée. « Tenir hors de portée des enfants » est une recommandation essentielle en matière de prévention des intoxications mais elle s’avère souvent insuffisante
CO13- : Place de l’analyse toxicologique en Pédiatrie: Bilan d’activité au CHU de Tizi-Ouzou L.R Mekacher, O.Belazougui , Sebihi. W, Kaci.L, Mamou.M, Dahmani.D. Chu De Tizi-Ouzou
Introduction : Les intoxications aiguës et chroniques chez l’enfant sont de plus en plus fréquentes. Le Centre Anti-Poison d’Alger reçoit environ 9000 appels par an dont près de 50% proviennent des services de pédiatrie. Le laboratoire de Toxicologie est confronté aux besoins de ces services et reçoit un nombre de plus en plus important de demande d’analyses. Matériel & Méthodes : Il s’agit d’une étude rétrospective descriptive portant sur le bilan d’activité de Toxicologie pour tous les prélèvements reçus du service de Pédiatrie du CHU de Tizi-Ouzou pour analyses toxicologiques et ceci pour une période allant de décembre 2009 à mars 2013. Résultats et discussions : Pendant cette période nous avons reçu 328 prélèvements du service de Pédiatrie. Ils sont composés de 62.2 % de demande d’analyses après intoxication et 37.8% pour un Suivi Thérapeutique Pharmacologique. On note une augmentation franche du nombre de demande d’analyses sur les années étudiées (2009-2010-2011-2012-2013). Les vacances scolaires et l’échec aux examens expliquent le nombre élevé des intoxications durant la période allant de mars au mois d’août. Pour les intoxications, la proportion des demandes est de 60.4% pour les garçons. Le petit enfant représente la catégorie la plus touchée. Les circonstances des intoxications sont souvent accidentelles (57%). Les médicaments sont en cause dans 98.5% des cas suivis par les pesticides dans 1.5 % des cas.

35
Pour les médicaments, les psychotropes sont les produits les plus souvent incriminés. Il s’agit essentiellement de benzodiazépines et de barbituriques. Le paracétamol occupe une place importante. Le dosage de toxiques en général est dominé par le dosage de l’activité cholinestérasique suivi par le dosage des médicaments. Le Suivi Thérapeutique Pharmacologique concerne essentiellement les antiépileptiques (50.3%), la Digoxine (48.5%) et l’Acide Acétyle Salicylique (1.2%). Le petit enfant représente la catégorie la plus concernée par ce type d’analyse
CO 14 : Les appendicites aigues de l’enfant de moins de 5 ans :
O.IBSAINE ,Y. Kassa1, Y. Belhimer2, A hadj-arab1, Z.Arrada1, A. Salem2, H.Berrah1 L’appendicite aigue (A.A) du jeune enfant est souvent méconnue du personnel médical Objectif Déterminer les caractéristiques cliniques, la place de l’imagerie dans le diagnostic de l’A.A du jeune enfant Matériel et méthodes :C’est une étude transversale, descriptive, prospective (1août au 31 décembre 2012) sur des enfants hospitalisés pour A.A. Tous les enfants ont bénéficié d’une échographie et ont été opérés. Nous avons comparé les enfants présentant une A.A et âgés de moins de 5 ans aux enfants âgés de 5 ans et plus. Le diagnostic de l’A.A était confirmé par une étude anatomo-pathologique. Résultats Sur 107 enfants opérés pour A.A, le diagnostic a été retenu chez 98 (91%) des cas, 25 (25%) enfants étaient âgés de moins de 5 ans. La symptomatologie clinique était atypique chez les enfants de moins de 5 ans (56 % vs 20% p 0,0004). Le diagnostic d’AA était posé au dela de 3 jours (52% vs 20% p 0,0026) et porté après plusieurs consultations chez les jeunes enfants (32% vs 18% p 0,0014). Un traitement antibiotique a été reçu de manière significative dans le 1er groupe (40% vs 4% p < 0,0001). L’échographie était concluante dans les deux groupes (100% vs 89% p 0,08). Une A.A compliquée a été retrouvée chez l’enfant jeune (64% vs 27% p 0,001) avec un taux similaire de perforation (12% vs 8% p 0,4). Conclusion L’A.A du jeune enfant est grave, son diagnostic est tardif car la clinique est souvent atypique d’où l’interêt d’établir un score pour cette tranche d’âge.
CO15 : Syndrome de Guillain - Barré : Expérience d’un service de Pédiatrie
H.AHMANE, D.Arhab, A.HAMZAOUI,S. CHALLAH,S. IDDIR , S.RAMI,H.REBIA ,N.AIDROUS, N.CHERIFI, BENSAADI.NADIA CHU Tizi-Ouzou Introduction : Le Syndrome de Guillain -Barré ( SGB) est considéré comme le résultat d’une réponse immunitaire à une agression le plus souvent viral. Objectif : préciser le profil épidémiologique, les modalités évolutives et la prise en charge par les immunoglobulines. Matériels et Méthodes :13 patients atteint du syndrome de Guillain Barré ont été colligés , au niveau du service de pédiatrie du CHU de Tizi-Ouzou sur une période de 5 années. Résultats : Le sex-ratio G / F est de 0,62 avec un âge moyen de 5,06 années . Une infection des

36
voies aériennes supérieures a précédé la paralysie flasque aréflexique ascendante chez tous les patients. La forme ataxique dans 3/13. Une dissociation albumino-cytologique a été observée dans 5/13. Les formes axonales étaient présentes dans 5/13 dont 2 patients ont nécessité une ventilation assistée. 9 patients avaient bénéficié du traitement par les immunoglobulines . L’évolution était favorable chez tous les enfants. Discussion : Le SGB est la neuropathie acquise la plus fréquente chez l’enfant. L’incidence est estimé entre 0,4 et 1,4/ 100 000 en Europe. Le syndrome de Miller- Fisher représente 5 ? des SGB a été retrouvé chez 3 patients . Des formes axonales sévères sont des formes graves pouvant être mortelles ou laissant des séquelles importantes .
Conclusion : Le traitement repose sur l’administration d’ immunoglobulines , qui réduisent le temps de récupération et la durée d’hospitalisation. Le pronostic du SGB a été considérablement amélioré par les méthodes de réanimation moderne. CO16 Les cholestases intra-hépatiques progressives familiales (syndrome et maladie de Byler) Présentation clinique et évolution : à propos de 21 malades.
R. Belbouab N.Khelafi, S.Agounne,R.Benalikhoudja,R.Berkani,Ait-younes*, S.Chikhi,A.Mertani,M. Keddari.service de pediatrie CMI CHU Mustapha
Introduction : Les cholestases intra hépatiques progressives familiales de types 1 et 2 sont des causes relativement rares des cholestases du nourrisson. Elles évoluent toujours vers l’insuffisance hépatique. Objectifs : Le but de cette étude est de décrire les caractéristiques cliniques, biologiques et histologiques des patients atteints de PFIC 1 et 2 ainsi que leur évolution. Matériels et méthodes : C’est une étude rétrospective sur analyse de dossiers de 21 malades atteints de PFIC 1 et 2 suivis en consultation d’hépatologie de notre service. Le diagnostic a été retenu sur des arguments cliniques, biologiques et histologiques. Ont été étudiés l’âge, le sexe, les antécédents personnels et familiaux, les données de l’examen clinique et des examens biologiques, les résultats de la biopsie du foie ainsi que les modes évolutifs.
Résultats : Age moyen au diagnostic est de 21mois (2mois-36mois), sexe (G/F : 11/10), la notion de consanguinité est retrouvée dans 14 cas/21, deux cas similaires dans la fratrie sont notés, l’âge moyen de début de la maladie est 2 mois et demi (10j-7mois). 12 malades ont eu une cholestase discrète et/ou récurrente. Tous les malades avaient à l’examen une HPM, trois une SPM et cinq un facies particulier. 8 enfants étaient hypotrophes au moment du diagnostic. Le prurit était au premier plan chez 4patients. Une patiente avait des signes extra-hépatiques à type de diarrhée et de surdité, une autre présentait un léger retard mental. La moitie de ces malades avaient une cytolyse importante. Huit malades étaient au stade de cirrhose .une lithiase vésiculaire est retrouvée chez deux patientes. Deux patients sont au stade insuffisance hépatique nécessitant une transplantation hépatique. Un malade a bénéficie d’une dérivation biliaire interne devant un prurit invalidant malgré un traitement intensif.

37
Conclusion : La PFIC est une cause rare des cholestase du nourrisson mais grave vu son évolution vers insuffisance hépatocellulaire d’où la nécessite de développer dans notre pays la transplantation hépatique chez l’enfant.
CO 17 .Prévalence de l’obésité infantile en milieu scolaire à Sétif. Résultats préliminaires. Z. BENARAB , S. Hadjit, S. Gabis, K. Dradra, A. Amara Korba, B. Bioud Service de pédiatrie hôpital mère et enfant CHU de Sétif.
Introduction : L’obésité est un problème sérieux de santé publique. Sa prévalence chez l’enfant comme l’adulte, est en constante augmentation depuis plus de vingt ans dans tous les pays du monde où elle a été évaluée. L’Algérie n’est pas épargnée, représentée dans ce travail par la wilaya des hauts plateaux : Sétif dans laquelle, ce type d’enquête se fait pour la 1ère fois. Objectif : il s’agit d’une étude transversale réalisée auprès de 1158 enfants âgés entre 6 et 12 ans au niveau des écoles du cycle moyen, durant le mois d’avril 2013 à Sétif. A l’aide d’un questionnaire pré établi, avec l’enfant et les parents en direct en plus de la consultation du carnet de santé. Les mesures anthropométriques (P, T, TT, IMC), prises sur place, avec beaucoup de précision. Pour un but primordial de faire un diagnostique sur l’ampleur du problème et les facteurs qui lui sont associés, par le calcul de la prévalence, afin de penser aux voies d’intervention. Résultats : la répartition de l’effectif selon les classes d'âge est comme suit : <= 7 ans : 269, 7<âge<10 :504 et âge sup = 10 ans : 380 élèves. La prévalence de l’obésité est calculé selon les 5 références mondiales : OMS 2007 , référence française , must et al , CDC et IOTF (SP+Ob)% ,Ob%) respectivement comme suit :( 19% 7,6%) (13,9%) (17,8% 9,4%) (15,9% 7,7%) (14,4% 5,1%), la prévalence de l’obésité selon la référence IOTF et selon les classe d’âge est comme suit : 6-7 ans (17,2 %, 7,9 %) ,8-9 ans (16,1 %, 5,4%) , 10-12 ans(11,1% , 3,2%) Avec un P significatif. La prévalence de l’obésité selon la référence IOTF et selon le sexe : -garçon (12,4 %, 5,5 %) -fille : (16,9%, 4,8%) Avec un P significatif: pour le surpoids dans toutes les références. La prévalence selon la référence IOTF et selon la commune est comme suit : -urbaine (15%, 5,4%), -Rurale (10,3%, 2,6) Avec un P significatif: (0,256, 0278). Parmi les facteurs prédisposant, l’obésité parentale est un élément important, la différence est significative entre les surpoids et obèses pour la mère et le père. En effet, l’Odd Ratio varie de 2,36 à 2,91 : le risque retrouvé est de 2,5 à 3. Commentaires : nous avons retrouvé une prévalence du surpoids obésité incluse de 14,4 % (IOTF). Les chiffres sont inférieurs aux résultats nationaux (étude de Mme Hadji 2006/2007 dans la wilaya d’Alger : 18%) et 5,1% d’obèses supérieurs à ces mêmes chiffres (4,1%). Même remarque pour l’enquête effectuée en 2008 à l’EPSP de Bouzareah. Nos résultats sont nettement supérieurs à ceux réalisés dans l’étude Benchihab à Constantine 2008/2009 (SP Obésité incluse :10,16 /Ob :2,92). Nos résultats sont identiques aux résultats dans le monde ; France, USA, quelques pays d’Europe dans les années 80 à 90 donc avec un recul de plus de 20 ans. Ceci pour souligner que l’obésité infantile ne cesse d’augmenter dans notre pays, alors qu’elle se stabilise ailleurs grâce aux programmes de prévention à différents niveaux Conclusion : L’obésité infantile est une épidémie mondiale, commence à s’individualiser comme un problème de santé publique en Algérie, comme en témoigne cette étude à Sétif.

38
CO 18. Impact d’une unité de diabète infantile à l’EPH Relizane.
S. kara Mostefa ,S. kadari , R. bendoubaba , R.Mezned , M.Touhami:EPH Relizane Mohammed Boudiaf Introduction : Le diabète de type 1 de l’enfant est un problème de santé publique en Algérie. Sa prise en charge est difficile et différente du diabète de l’adulte (insulinodépendance, éducation individuelle…), cette prise en charge est plus ardue dans les EPH où il n’existe pas de structure d’accueil adéquate, les complications sont retrouvées fréquemment chez les jeunes et le nombre de nouveaux diabétiques augmente d’année en année.
Matériel : En novembre 2012 ouverture de l’unité avec : pédiatre, infirmière ayant bénéficié d’une formation à la clinique A.Cabral CHUOran, psychologue, nutritionniste, partenaires commerciaux.
Résultats (une année d’activité) : 59 cas. La moyenne d’hospitalisation est passée de 8,3j à 3,6j, 54% des malades pris en hôpital du jour, la moyenne l’HbA1c est passée de 11,3% à 9,3% avec 44% de bonne évolution (HbA1c au dessous de 10%), 8% d’échec. 23% malades avaient un diabète de plus de 3 ans, une microalbuminérie systématique a permis de dépister 7 néphropathies, 76% des malade n'avaient aucune connaissance du diabète (pas de support de suivi, jamais entendu parler des complications aiguës ou chroniques). Conclusion : Le diabète augmente chez le jeunes dans le monde entier, les complications à distances son liées non seulement à la qualité de l’équilibre glycémique, mais aussi a l’ancienneté de la maladie, A l’EPH Relizane nous avons agit sur la qualité de prise en charge et nous avons pu modifier le profil de 50% des enfants et stopper les complications déjà installées.
CO19 : L’acidose inaugurale dans le DT1de l’enfant dans la Wilaya d’Oran Stratégie de réduction de sa fréquence
Zennaki, A. Ouzzaa, S. Niar, H. Aichaoui, M. Gharnouti, A. Reguieg, M. Naceur, K. Bouziane-nedjadi, M. Bessahraoui.
Service de pédiatrie « C ». CHU d’Oran.
L'acidocétose diabétique (ACD) inaugurale est une cause non négligeable de mortalité et de co-morbidité dans le diabète de type 1 (DT1). Elle est généralement due à un retard à la reconnaissance des signes d’hyperglycémie. Plusieurs campagnes de sensibilisation et de prévention de l’ACD inaugurale ont été faites à travers le monde et des résultats encourageants ont été obtenus. Objectifs : Faire un état des lieux dans la Wilaya d’Oran pour une campagne de prévention ciblée. Patients et méthode :

39
Les données de 258 nouveaux cas de DT1 originaires de la Wilaya d’Oran, pris en charge au service de pédiatrie « C » ont été analysées. Résultats : Le diagnostic de DT1 est plus fréquent dans la tranche d’âge 5–9 ans : 32 % ont entre 0 et 4 ans, 40 % entre 5 et 9 ans, et 28 % ont entre 10 et 14 ans. L’ACD a été le mode de révélation du DT1 dans 20% des cas. Ce chiffre baisse à 6% quand un apparenté du 1er degré est également atteint de DT1. Elle est plus fréquente dans la tranche d’âge des adolescents : elle représente 12% entre 0 et 4ans, 33% entre 5 et 9ans et 55% entre 10 et 14ans. Conclusion La fréquence de l’acidocétose inaugurale est relativement faible dans le contexte de la Wilaya d’Oran avec un risque élevé pour l’adolescent. Une prochaine campagne ciblée devrait encore améliorer ces chiffres.
CO20 : Utilisation de la pompe à insuline chez l’enfant. Recrutement du service de pédiatrie « C » du
CHU d’Oran. Zennaki1, K. Benharrats2, M. Gharnouti3, H. Aichaoui1, I. Abdat1, S. Niar1, L. Benhouria4, M. Bessahraoui1, K.
Bouziane –Nedjadi1. 1Service de pédiatrie « C ». CHU d’Oran. 2Service de médecine interne EHU d’Oran. 3Service de pédiatrie
HMUR d’Oran. 4Roche Algérie.
L’insulinothérapie par pompe est considérée aujourd’hui comme le gold standard de l’insulinothérapie intensifiée. Un facteur limitant important en est le coût ce qui réduit son utilisation à certaines indications particulières. L’objectif est de rapporter notre expérience, les indications de mise sous pompe à insuline, l’évolution ainsi que les différents problèmes et incidents techniques rencontrés chez l’enfant. Patients et méthode La première pompe à insuline a été mise en place le 23/05/2011. L’indication en était vitale :
résistance à tout autre moyen d’administration de l’insuline. Depuis Août 2012, 9 autres pompes
à insulines ont été placées. Les différentes indications étaient : hypoglycémies sévères chez un
petit nourrisson, hyperglycémies fréquentes, demandes familiale dont une famille multiplex
(deux jeunes sœurs de 3ans et 5 ans) et une grossesse.
Résultats
Trois pompes ont été retirées dont deux pour discorde familiale et la dernière sur conseil médical
intempestif. L’HbA1c moyenne à la mise sous pompe était de 9.15±2.85%, à 1an d’évolution elle
est de 6.97±1.47%. Il n’y a eu aucune hypoglycémie sévère. Le nombre d’hypoglycémies
modérées a été réduit. Il n’y a pas eu de modifications importantes du poids corporel. Divers
incidents techniques sans gravité ont été notés.
Conclusion
L’insulinothérapie par pompe en pédiatrie a des indications restreintes pour le moment.
L’avantage sur la qualité de vie est certain, les conséquences sur l’équilibre glycémique
dépendent de l’utilisation qui en est faite. Sa diffusion reste dépendante du niveau d’éducation

40
et de motivation des familles ainsi que des coûts induits à la charge de ces dernières par défaut
de prise en compte des consommables par l’assurance maladie.
CO 21: Le cancer de la thyroïde chez l’enfant (à propos de 39 cas) K.Bouriche, D.Senuci, W.Mazari, A.S.Bendeddouche Service De Pédiatrie. Chu Tlemcen- Algérie A.Zamalache ; A.Medjahdi, N.Berber Service de médecine nucléaire . CHU Tlemcen- Algérie
Introduction : L’incidence du cancer thyroïdien est en augmentation linéaire depuis plusieurs décennies en raison de l’évolution et la généralisation des moyens de diagnostic, essentiellement de l’échographie et de la cytoponction Le cancer de la thyroïde chez l'enfant représente 0,5 à 1,5 % de tous les cancers pédiatriques. Le but de notre travail est de réaliser une approche épidémiologique des cancers différenciés de la thyroïde chez l’enfant dans notre région. Matériel et méthodes : Notre travail consiste en une analyse rétrospective de cancers différenciés de la thyroïde chez l’enfant, suivis au service de pédiatrie et de médecine nucléaire du CHU Tlemcen sur une période de 20 Ans Nos paramètres d’évaluation concernaient : l’incidence des cancers thyroïdiens, l’influence de l’âge et du sexe sur cette incidence, sur la variété histologique et le pronostic de la maladie. Résultats : Les observations sont colligés sur une période de 20 ans (1993- 2013), 39 cas ont étaient recensés. 30 fille et 9 garçons ;Le sexe ratio est de 3,33 , l’âge moyen à 11,3 ans. Le motif de consultation : adénopathie cervicale dans 20% cas, , un cas d’antécédent familiaux de carcinome thyroïdien et 8 cas de pathologie thyroïdienne bénigne, le reste pour goitre thyroïdien. Le bilan hormonal était normal dans 84% des cas : un cas d’hypothyroïdie congénitale et 5 d’hyperthyroïdie dont 2 cas de maladie de Basedow. La scintigraphie thyroïdienne pratiquée chez 70% des malades a objectivé un nodule froid dans 92% avec seulement dans 2 cas un nodule chaud. Une thyroïdectomie totale a été pratiquée pour tous les malades et complétée par un curage ganglionnaire dans 51% des cas. . L'examen anatomopathologique définitif a conclu à un carcinome papillaire dans 71%, dans 7 cas un carcinome vésiculaire et 4 carcinome moyennement différencié. Nous avons objectivé dans 35% de cas des métastases ganglionnaires dans 2 cas des métastases pulmonaires et dans 1 cas des métastases osseuses et cérébrales. L'irathérapie et l'hormonothérapie frénatrice ont été instituées pour tous les patients, 22 enfants sont en rémission, 10 enfants présentent une maladie résiduelle, 6 perdus de vue. Conclusion Le cancer thyroïdien de l’enfant n’est pas rare, Il y a une nette prédominance féminine, la majorité des cas sont découverts à l’adolescence entre 11 et 15 ans. Le type histologique est dominé par le carcinome papillaire avec nette prédominance d’atteinte ganglionnaires source de

41
récidives et de maladies résiduelles. Les métastases à distance sont très rares, l’évolution à moyen terme a été favorable même en cas de métastases ganglionnaire. Le pronostic est en généralement bon.
CO22 : Hyperthyroidie de l'enfant Aspects cliniques et evolutifs .
F.Bouferoua ,M.chaou, S.Tari, Z.zeroual,A.Atek,H.Boucenna,AKhati,A.Hezazi,M.E.Khiari service de pédiatrie « A » CHU BéniMessous
Introduction L’hyperthyroidie se définit par l’augmentation de synthèse des hormones thyroidiennes. Le diagnostic est aisé lorsque s’associent les éléments majeurs du syndrome, essentiellement tachycardie, amaigrissement, nervosité, tremblement,rétraction de paupière supérieure et thermophobie. Les étiologies sont nombreuses dont la plus fréquente est la maladie de Basedow qui réalise 90% des causes. Objectifs -Mettre en évidence les caractéristiques cliniques et évolutifs de nos patients -Evaluer la prise en charge Matériel et méthodes C’est une étude rétrospective sur une période de 7 ans ( 2006-20013) d’une série de 8 cas. Critères d’inclusion - signes majeurs : Troubles de caractères et comportement , troubles de la croissance staturo pondérale,troubles cardio vasculaires Troubles neuromusculaires ,thermophobie, hypersudation -Manifestations propres à la maladie de Basedow : goitre et ophtalmopathie -Examens complémentaires : bilan thyroidien : TSHus, FT3, FT4, dosage des AC antiTPO et TSI Echographie , ± scintigraphie Résultats Sexe : 6 patients de sexe féminin et 2 de sexe masculin Age : 1 nouveau-né (Nné) de 6 jours de vie et 7 patients dont l’âge moyen de 12,8 ans [10-15] Age au diagnostic : Nné à 2 jours de vie ; et 7 patients avec un délai moyen de 3 mois [1-8] Clinique : troubles du comportement : 6 cas, Troubles cardio vasculaires 7 cas ; troubles neuro musculaires : 5 cas, accélération de la croissance staturale : 1 cas , goitre : 7 cas et ophtalmopathie :4 cas. Bilan hormonal en faveur de l’hyperthyroidie primaire :8 cas ; Ac antiTPO (+) :8 cas, TSI : 5 cas Echograhie thyroidienne : 7 cas Scintigraphie :1 cas Etiologie : Maladie de Basedow :6 cas, Thyroidite de hashimoto : 1 cas, Hypertyroidie néonatale : 1 cas Taitement : Carbimazole :dose d’attaque : 0.5-0.7 mg/kg/j , Propanolpol : 1-2 mg/kg/j Evolution : Recul moyen : Rechute : 2 cas (reprise du TRT), Thyroidectomie : 1 cas, euthyroidie sous traitement : 4 cas, guérison : Nné Commentaires L’âge moyen conforme aux données de la littérature ainsi que la prédominance féminine La maladie de Basedow est l’étiologie la plus fréquente ; diagnostic conforté/ TSI

42
La forme associée est rare mais doit être recherchée systématiquement L’échographie ne permet pas de différencier entre une thyroidite et un Basedow d’où l’intérêt des TSI La scintigraphie a un intérêt très limite dans le diagnostic L’hyperthyroidie néonatale est rare due au passage de TRAK matenels chez le Nné, la guérison est observée avec leur disparition. Le traitement doit être prolongé, seul garant de minimiser le risque de rechute nécessitant pour cela une bonne compliance au traitement. Conclusion L’hyperthyroidie de l’enfant est rare, les étiologies sont nombreuses dominées par la maladie de Basedow. Il est essentiel de faire le diagnostic étiologique sur lequel repose le pronostic qui est différent d’une étiologie à une autre . La prise en charge reste difficile du fait de la fréquence des rechutes auquel cas 2 alternatives seront à discuter , en fonction de lâge de l’enfant : chirurgie ou irathérapie
CO23 : prévalence de l'allaitement maternel dans l'ouest algérien
.N. Zidane ,Adel Ahmed Afin d’évaluer la prévalence de l’allaitement maternel, ainsi que la durée et les principales raisons d’interruptions d’allaitement, nous avons effectué une enquête dans l’ouest Algérien. L’enquête a touché 814 mères d’enfants âgés d’une semaine à deux ans. Un questionnaire, préalablement établi, est soumis aux mères. Notre analyse statistique est fondée sur le test de Chi2 et le calcul des pourcentages. Il ressort de notre travail que la fréquence de l’allaitement maternel est de 96% à la naissance qui chute rapidement à 20% à 6mois. Les facteurs individuels, socioculturels, familiaux et professionnels influencent de manière significative la durée de l’allaitement maternel. Afin d’améliorer ce taux et la durée de l’allaitement, il faut passer obligatoirement par la formation du personnel médical et paramédical à la promotion de l’allaitement maternel, afin qu’ils puissent répondre de façon efficace aux difficultés rencontrées par les mères, susceptibles d’interrompre prématurément l’allaitement. Mots clés : Allaitement maternel, nourrisson, prévalence, ouest algérien.
CO24 : Méningites purulentes à pneumocoque du nourrisson.
F.Z. ZMIT ,D.AITKAID .EHS EL Kettar Introduction-Objectifs Les méningites purulentes du nourrisson sont un véritable problème de santé publique en raison des complications et des séquelles qu’elles entraînent. La virulence des germes en cause impose une surveillance microbiologique vigoureuse. L’étude décrit les caractères évolutifs de ces méningites. Matériels /méthodes Etude rétrospective sur 04 ans (01/01/09 - 31/12/12) durant laquelle 104 cas de méningites

43
purulentes ont été colligées dont 25 méningites à pneumocoque. Recrutement : Nourrissons des 02 sexes Ces derniers ont fait l’objet d’une exploration para-clinique Ponction lombaire Hémoculture EEG, Scanner cérébral à la demande Les méningites sont régulièrement observées avec une recrudescence automno-hivernale. Leur fréquence est maximale pendant la première année de vie. Seulement 45% ont bénéficié de la vaccination anti-haemophilus influenzae b depuis l’introduction de la vaccination. Sur le plan biologique, l’aspect purulent du liquide céphalo-rachidien se voit dans la majorité des cas, une cytologie essentiellement faite de polynucléaires altérés et une albumine supérieure à 01g/l. La confirmation de nos méningites est de l’ordre de 24% à partir de la culture du LCR. Les complications sont fréquentes de l’ordre de 56% des méningites à pneumocoque dominées par les hygromes et les hydrocéphalies.Les séquelles sont de 16% représentées par la surdité. Conclusion Depuis l’introduction de la vaccination anti-Hib nous assistons à une transition bactériologique des méningites purulentes du nourrisson. Le pneumocoque prend une place considérable incitant à une réflexion sur la prise en charge thérapeutique de ces méningites.
CO 25 Le déficit en vitamine B12 selon l’expérience du CHU Béni-Messous. Zemiri.F.Z ;Oukrif.L ; Chérif.N ; Bekkat.D ; Maouche.H ; Boukhil.k ; Messadi.W ;chikhi.K ;Addoune.R ; Hireche.K ; Bensenouci.A . Service pédiatrie B. CHU Béni-Messous
La vitamine B12 ou cobalamine a plusieurs rôles, dont la maturation des globules rouges, la synthèse de l’ADN, la fonction neuronale et la myéline nerveuse. Elle a été isolée par Karl Falker en 1948. Sa synthèse totale, est réalisée en 1972 par Robert Burns Woodward . La carence en vitamine B12 est rare chez l’enfant. Elle est induite par un apport alimentaire insuffisant, un problème métabolique au niveau de son assimilation et de son utilisation, ou une maladie congénitale. Le principal symptôme en est une anémie mégaloblastique. Objectif : Ce travail vise à préciser les caractéristiques épidémiologiques, cliniques et évolutives de l’anémie par carence en vitamine B 12 de nos patients. Matériel et méthode : L’étude est rétrospective de 12 cas recensés sur une période de 5ans (2008-2012) selon des fiches préétablies. Tous les malades présentent une anémie mégaloblastique par déficit en vitamine B12 ou ayant répondu au test thérapeutique. Résultats : 55% de nos patients sont des garçons. 83% sont des nourrissons dont la moitié sont âgés de moins de 1an.75% ont des conditions socioéconomiques mauvaises .Toutes les mères ont allaité leur enfant mais seulement 33% ont introduit la diversification ; et 25% se disent végétariennes. Les signes d’appel les plus fréquents sont la pâleur et l’asthénie. L’état général est conservé chez75% des cas. Le retard staturo-pondéral est retrouvé dans 58% avec des troubles digestifs (50%).Les troubles neurologiques sont rares. L’hémogramme, le frottis sanguin et le médullogramme réconfortent le diagnostic d’anémie mégaloblastique .Une leucopénie et ou thrombopénie ; lui est associée dans 42%des cas. Le dosage vitaminique fait chez la moitié des

44
malades a confirmé le diagnostic. Le taux moyen de vitamine B12 est de 93pg/ml. Le test thérapeutique est positif. La carence d’apport est l’étiologie retenue. Aucune malabsorption n’a été objectivée. Le traitement s’est basé sur les injections de vitamine B12(1000µg) 1jour sur 2 puis espacées ; ainsi que la diversification .L’évolution a été favorable. Commentaires : La carence en vitamine B12 infantile est rare, et n’est guère retrouvée que chez les enfants allaités par une mère végétarienne ou végétalienne, les enfants souffrant d’anémie congénitale, ou souffrant d’un trouble métabolique. Dans notre étude, l’apport insuffisant réalisé par le lait maternel presque exclusif était la cause ;et vue les conditions socioéconomiques mauvaises . Les signes de carence apparaissent au cours de la première année si le stockage in utero et l’apport par le lait maternel ont été faibles. Le dosage de la vitamine B12(sang ,lait) chez les mères aurait été d’une grande utilité. Conclusion : Etant donné le risque potentiel de troubles neurologiques irréversibles lié à la carence en vitamine B12 il serait prudent de recommander une supplémentation en vitamine B12 pour toute femme enceinte ou allaitante. Le végétalisme sans supplémentation lors de ces périodes peut être dangereux pour l’enfant. La diversification est indispensable.
CO26 : CONVULSIONS NEONATALES REVELATRICES D’HYPOVITAMINOSE D MATERNELLE : à propos de 06 cas. Sadaoui .B, Arrada. Z, kebouchi. K, Belache .A, Saheb .K, Latreche .M, Oucif .S, Berrah Hassina
Introduction L’hypovitaminose D maternelle est une étiologie d’hypocalcémie chez le nouveau-né dont le statut vitaminique dépend entièrement de celui de sa mère, devenue fréquente dans notre pratique courante. Objectifs - Attirer l’attention à l’hypovitaminose maternelle comme cause d’hypocalcémie néonatale. - Discuter la prévention par la supplémentation en Vit D durant la grossesse.
Observations Nous rapportons l’analyse de 06 cas d’hypocalcémie néonatale symptomatique (convulsions) en 08 mois, par hypovitaminose D maternelle (sans autre étiologie associée dans 5/6 cas). Nous avons étudié les différents facteurs pouvant être à l’origine de cette hypovitaminose D chez les mères de ces nouveau-nés (à terme et eutrophiques). Résultats-commentaires Les 06 mères - Sont âgées de 23 à 37 ans, indemnes de toute pathologie connue : notamment hépatique ou rénale. - Habitent en milieu urbain, portent des vêtements couvrants, présentent un surpoids et utilisent des crèmes antisolaires : soit une faible Photosynthèse cutanée - Non supplémentées durant la grossesse. - La 25 (OH) D est abaissée en dessous de 10 ng/mL, même cas chez leurs nouveau-nés : soit une carence.

45
- La correction de l’hypocalcémie et la supplémentation en Vit D ont permis une disparition rapide et définitive des symptômes, sauf pour 01cas compliqué d’une ischémie cérébrale localisée. Conclusion Il ne faut pas hésiter à doser la Vit D chez les nouveau-nés et leurs mères en cas d’hypocalcémie néonatale afin de donner une supplémentation, surtout devant ce changement du mode de vie de notre population, et climatique de notre pays. Une prévention par supplémentation au cours de la grossesse doit être discutée.
CO27.Prise en charge des cardiopathies congénitales chez les enfants trisomiques (T21) Expérience du service de pédiatrie / HCA à propos de 31 cas
Y.inouri ,H.boufnar, ,S.E.lalaoui
La trisomie 21 est la plus fréquente des trisomies autosomiques chez l’homme de diagnostic clinique facile, elle associe une hypotonie avec dysmorphie faciale caractéristique, des malformations digestives et cardiaques et un retard mental considérant l’enfant trisomique de mauvais pronostic avec un préjugé que l’investissement dans la correction de ces malformations cardiaques congénitales n’été pas justifié. Depuis une vingtaine d’année la meilleure prise en charge des trisomiques notamment de leurs malformations cardiaques à améliorer leur espérance et qualité de vie. Patients et méthodes : nous présentant ici une analyse de 31 dossiers d’enfant trisomiques 21 entre 2004 – 2012 dont le diagnostic de cardiopathie congénitale a été fait au niveau de notre consultation de cardiologie pédiatrique, puis transféré pour chirurgie au niveau de la clinique saint- Luc en Belgique ,le suivi post opératoire se fait à notre niveau , 54% avait un canal atrioventriculaire complet , 19% des CIV et les 10 autres des malformations variées , tous les enfants ont bénéficié de la correction de leurs malformations Résultats : les CAV complets ont été diagnostiqué à un âge moyen de 54 jours et opéré à un âge moyen de de 11 mois les CIV diagnostiquées à 3.3 mois et opéré à 14 mois. Lors de la chirurgie 37% des CAV complets été en HTAP iso-systémiques non fixés surtout quand le CAV est associé à un PCA, malgré L HTAP tous les CAV ont été opérés en un seul temps avec régression de l’HTAP en post-op avec des complications post-op gérables. Un enfant avait une CIV (pm) +PCA diagnostiqué à 03 ans et transféré à 05 ans avait développé un Eissenmenger et devenu inopérable. Un enfant avait un CAV complet avec PCA décédé en post-op à cause de la présence de malformation vasculaire hépatique associé. 41% des enfants avait des suites opératoires simples sans aucune complications ce sont ceux qui avait un âge entre 4-10 mois avec une HTAP modéré à la chirurgie. Le suivi post opératoire au long cour à montrer une normalisation de la pression pulmonaire chez tous les malades avec bonnes fonctions cardiaques et quelques cas d’IM ou IT minimes. Conclusion : la réparation des cardiopathies congénitales chez le T21 peut avoir les mêmes résultats que les non trisomiques avec amélioration de leur qualité de vie au long court à condition d’être réalisé avant l’installation de L’HTAP imposant l’amélioration de nos capacités diagnostic et de prise en charge en matière de cardiologie pédiatrique

46
CO 28 .Apport du certificat de décès périnatal et néonatal à la connaissance de la mortalité à Oran HEROUAL NABILA A.Tadjeddine SEMEP EHS CANASTEL
Le taux de mortalité périnatale (mortinatalité et mortalité néonatale précoce) est un indicateur remarquable de la qualité des soins obstétricaux et néonatals. Or le système d’information actuel sur les causes de décès est mal adapté pour étudier la mortalité néonatale. L’objectif est d’évaluer les certificats de décès périnatal et néonatal en faisant le point sur son utilisation d’une part et en décrivant les causes de décès d’autre part. Matériels et méthodes : Il s’agit d’une étude rétrospective sur deux ans (2008-2009), qui a porté sur l’ensemble des certificats de décès périnatals et néonatals transmis par les maternités de la wilaya d’Oran à la direction de la santé. Deux types d’informations sont contenus dans le certificat. Résultats : Au total, 235 certificats de décès néonatal ont été transmis à la DSPRH durant la période d’étude. Le taux de remplissage variait de 67,5% pour les maternités des CHU et des hôpitaux, à 49, 38% pour les maternités extra- hospitalières urbaines. 75 ,55% des certificats comprenaient des informations relatives à l’enfant, 51,11% pour l’accouchement, 37% pour les caractéristiques de la mère. Les certificats de décès contenaient une cause de décès dans 80% des cas . Quant aux pathologies obstétricales, sur les 39causes citées, l’hypertension (HTA) et l’iso-immunisation rhésus étaient en causes dans 71% des cas. Conclusion : Il faut relancer le programme en généralisant le certificat de décès périnatal et néonatal à l’échelle national au niveau de l’ensemble des structures concernées pour améliorer les connaissances des causes de décès à cet âge et orienter les actions et les programmes de prévention.

47
Workshop : Maladies métaboliques W 1. Mucopolysaccharidose de type1 ( MPS1): A propos de trois cas.
M. Keddari .Hamdani ,S.Aggoune,S.Mezouari,A.Chergui,A.Mertani,M.Keddari:CMI,CHU Mustapha La mucopolysaccharidose de type1 est une maladie de surcharge lysosomiale multisystémique, de transmission autosomique récessive, due à un déficit en ?-l-iduronidase dont le pronostic a été amélioré par l’enzymothérapie substitutive (ETS ) par la Laronidase Objectif : Intérêt d’un diagnostic et d’une ETS précoces pour optimiser la prise en charge et améliorer le pronostic Matériel : C’est une étude prospective portant sur 4 enfants, une fille et trois garçons dont un perdu de vue, suivis et traités à laCMI depuis une année Résultats : Le diagnostic de MPS1 a été évoqué sur la présence de symptômes cliniques (dysmorphie faciale, raideurs articulaires, opacités cornéennes, otites chroniques,hernies inguinales et ombilicales) , confirmé par les dosages de l’?-l-iduronidase effondrée et des glycosaminoglycanes urinaires (GAG) élevés. L’âge au diagnostic était de 5 ans pour la fille et de 5 ans, 7 ans pour les garçons Le traitement substitutif n’a été institué qu’à l’âge de 9 ans pour la fille et 10ans ,8 ans pour les garçons.
Malgré l’institution tardive de l’ETS, nous avons noté une amélioration de nos patients après une année de traitement portant sur : L’amélioration du test de la marche de 6 minutes L’augmentation de l’amplitude articulaire La diminution de l’encombrement bronchique et la diminution des GAG urinaires Commentaires : La MPSI était une maladie incurable et la plupart des malades décédait à l’âge de 10 ans, par complications cardiaques le plus souvent. Les signes cliniques de la forme sévère de cette maladie apparaissent peu après la naissance et le diagnostic doit être évoqué avant l’âge de un an, devant l’association de : Hernies inguinales ou ombilicales, rhinites et otites chroniques, régression ou modifications morphologiques progressives Hormis les mesures symptomatiques, le traitement actuel de la maladie de hurler est la GCSH et l’ETS, employées seules ou en association Conclusion : Malgré le bénéfice du traitement par la Laronidase sur la qualité de vie, elle ne traverse pas la barrière hémato-encéphalique et reste inefficace sur l’atteinte cognitive osseuse, ophtalmologique et cardiaque. L’idéal est de pouvoir débuter une ETS avant la survenue de signes irréversibles de cette maladie invalidante. Un consensus national pour la prise en charge est souhaitable.

48
W 2 La forme infantile de la maladie de Pompe à l’ère de l’enzymothérapie (A Propos D’un Cas Avec Revue De La Littérature) .L.N.DRIS ,NK .BENHALLA ; L. SMATI; H.DJENANE; A. YAGOUBI; D.DOUIRI ; N.ALIM ;O.REDJALA ;M. SARI; M. BAGHRICHE Service De pédiatrie – EPH Bologhine Ibn Ziri – Alger Introduction : La maladie de Pompe ou glycogénose de type II est une maladie lysosomale de transmission récessive autosomique liée à un déficit en alpha-glucosidase acide. Objectifs : Evoquer les étapes ayant conduit au diagnostic et au traitement de ce patient en mettant l’accent sur la problématique de prise en charge de cette maladie orpheline. Matériel et méthodes : Présentation d’un cas de maladie de Pompe dans sa forme infantile avec revue de la littérature Résultats : Tasnime âgée de 02 mois, issue d’un couple consanguin nous est adressée hypotonieet cardiomyopathie hypertrophique. Les 3 sœurs ainées sont décédées vers l’âge de 05 mois. A l’examen, on constate également chez ce nourrisson, eutrophique, une protrusion de la langue ainsi qu’une hépatomégalie. Cet ensemble symptomatique nous évoque d’emblée une maladie de surcharge avec atteinte musculaire. Parmi ces affections, la maladie de Pompe est l’une des plus probables. Le dosage de l’activité de l’alpha glucosidase intra leucocytaire confirme le diagnostic qui sera ensuite conforté par l’étude génétique : l’enfant est homozygote pour une forme sévère de glycogénose de type 2. Un traitement substitutif a été débuté à l’âge de 13 semaines. On note une stabilisation de la fonction ventriculaire après 4 cures mais l’enfant décède à l’âge de 05mois au cours d’un épisode de détresse respiratoire. Discussion : Dans notre cas, en raison de l’ensemble symptomatique et des antécédents familiaux, nous avons discuté essentiellement les déficits de la chaine respiratoire mitochondriale et la forme infantile de la maladie de Pompe. Celle-ci est caractérisée par une hypotonie globale, une hypo réflexivité, une macroglossie , une hépatomégalie et une myocardiopathie sévère, le plus souvent hypertrophique. Un traitement spécifique par enzymothérapie substitutive est actuellement commercialisé. Il s’agit d’une forme recombinante de l’enzyme. Une amélioration spectaculaire du pronostic vital est parfois observée mais aux dépens de nombreux handicaps. Conclusion : Une cardiomyopathie hypertrophique chez un enfant hypotonique doit faire évoquer une maladie de Pompe

49
W 3 La maladie de Gaucher chez l’enfant : Expérience de la Clinique Médicale Infantile du CHU
Mustapha. A,Hadji-Lehtihet ,S.Sokhal1 ; N.Benali-Khodja1 ; S.Chikhi.1 ; Ghouali2 ; Hallal2 ;
B.Imessaoudène 2; A.Berhoune 2 ; A.Mertani1 ; M.Keddari1.
1. Clinique Médicale Infantile CHU Mustapha Alger
2. Laboratoire de Biochimie et de Génétique CHU Mustapha Alger
La maladie de Gaucher (MG) est l’une des rares maladies génétiques accessible à un traitement
spécifique. C’est une maladie héréditaire de transmission autosomique récessive. C’est un
trouble du métabolisme des sphingolipides due au déficit en ß- Glucosidase acide. Elle se
caractérise par l’accumulation pathologique du substrat non dégradé dans les lysosomes des
cellules du système réticuloendothélial. Elle est responsable d’une maladie multi-systémique:
hépato-splénomégalie anémie, thrombopénie et atteinte osseuse de gravité variable.
Objectifs : Rapporter à travers notre expérience, les résultats et les difficultés diagnostiques et
thérapeutiques de cette maladie.
Matériels et méthodes : L’étude a concerné 10 enfants âgés de 10 mois à 16 ans : 6 filles et 4
garçons. Le diagnostic est évoqué devant la splénomégalie, la bi ou pancytopénie et confirmé par
la mise en évidence du déficit enzymatique de la ß- Glucosidase acide. La recherche des
complications : osseuses par la radiographie standard et l’ostéodensitométrie, hépatospléniques
: clinique, biologique et échographique ; cardiaques et pulmonaires par l’imagerie et la biologie.
La prise en charge a été symptomatique : transfusions sanguines et de plaquettes. Une
splénectomie a été réalisée chez 02 enfants de la même fratrie présentant une énorme
splénomégalie avec hypersplénisme. L’enzymothérapie est débutée en mars 2006 par
l’Imiglucérase (Cérézyme) à la dose de 60UI/kg/15 jours. Un enfant mis sous traitement depuis
un mois ne sera pas évalué dans cette étude.
Résultats : 9 enfants et adolescents âgés de 2 à16 ans. L’âge moyen est de 9,7ans. Prédominance
féminine: 2/1. L’âge au diagnostic varie de 9 mois à 14 ans. Âge moyen de 5,4 ans. Diagnostic
précoce chez 3 enfants (9, 17 et 23 mois). L’origine géographique est variée. Notion de
consanguinité dans 50% des cas. Maladie de Gaucher confirmée par la diminution de l’activité
enzymatique de la ß- Glucosidase acide. 0,10 à 3 nmol/h/mg de protéines. L’étude génétique
effectuée pour la recherche des mutations du gène GBA selon la technique d’amplification –
digestion enzymatique pour les mutations N370S et L444P, et par la technique de séquençage
direct pour la mutation 84GG a permis de confirmer le type de la MG: type I: 4 ; type III: 5. Dans
un cas, il n’y a pas de mutation récurrente.
Conclusion : L’enzymothérapie substitutive a transformé le pronostic des formes sévères de la
maladie de Gaucher,

50
W 4 .Maladies métaboliques : Expérience de 5 ans d’un service de pédiatrie générale
M.T. Bencharif, ;S Taleb ; S Benfetima ; F. Sellahi ; A. Bakhouche ; H .Boudbaba ; N. Hezil ; S Benseghir ; A. Mesri ; F.Z .Boulmaiz :Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine Introduction : Les maladies héréditaires du métabolisme représentent un groupe d’affections génétiques rares ayant en commun une dysfonction d’enzymes ou d’autres protéines impliquées dans le métabolisme cellulaire. Leur diagnostic est souvent difficile à établir du fait surtout de leur présentation clinique très polymorphe et du recours quasi obligatoire à des examens para-cliniques sophistiqués, très coûteux et peu disponibles notamment dans les pays sous-développés. Objectif : Le but de ce travail est de rapporter l’expérience d’un service de pédiatrie générale dans le domaine des maladies métaboliques au cours des 5 dernières années. Méthodes et matériels : Nous avons recensé de manière rétrospective tous les cas confirmés de maladies métaboliques durant la période s’étalant du 1ier octobre 2008 au 1ier octobre 2013. Résultats : 22 cas ont été recensé au total, représentés principalement par les maladies lysosomales, les déficits énergétiques dont les glycogénoses et les cytopathies mitochondriales et enfin les métaboloses par intoxication comme la maladie de Wilson. Conclusion : Le diagnostic des maladies métaboliques demeure essentiellement basé sur la suspicion clinique qui pourra faciliter en 2 ème lieu le choix d’examens complémentaires.

51
Abstracts des communications affichées
1.Médecine prédictive: le système HLA
Rahmoune Hakim,Boutrid Nada, Nedjar Faycal, Belkacem Bioud.
Introduction :
Le système HLA a longtemps été considéré comme vecteur de reconnaissance du soi et non-soi avant qu'un
rôle plus saillant ne lui soit récemment dévolu dans les iassociations morbides auto-immunes et/ou
immunomédiées.
Présentation des cas :
1e cas:
Enfant de 14 ans, suivi et traité pendant 5 ans comme coeliaque, avant qu'un typage HLA n'exclue
définitivement cette pathologie et ne "libère" l'enfant.
2e cas:
Une fille avec arthrite juvénile diagnostiquée comme polyarticulaire séronégative; l'exclusion de la
présence du HLA DR3/DR4 rassure quant à l'éventuel passage à une PR adulte sévère...
Discussion :
Le système HLA (ou Complexe Majeur d’Histocompatibilité, CMH, situé sur le bras court du Chromosome
6 ) est une région polygénique responsable, entre autres, de production de molécules portant le même nom
"HLA" qui sont de plus en plus 'décriées' par les études bio-moléculaires dans des processus aussi divers et
variés que l'auto-immunité, l'infection, l'allergie...
Son rôle peut s'avérer primordial dans certaines pathologies chroniques juvéniles (maladie coeliaque, AJI,
Psoriasis, MICI...) dans l'exclusion - par sa forte valeur prédicitive négative- , dans la confirmation ou dans
la stratification de risque; orientant ainsi grandement le diagnostic, le traitement et le pronostic
Conclusion :
Comme prédit dans les années 1970 par son découvreur et prix Nobel "J.Dausset"; le système HLA est un
pillier -parmi d'autres- d'une médecine dite Prédictive où la susceptibilité génétique préalablement
démasquée permet de guider les choix thérapeutiques.
2.Dysplasie ectodermique hypohidrotique: à propos d'un cas.
Treki Rima.EPH El Harrouch
Dysplasies ectodermiques: groupe hétérogène de maladies génétiques rares caractérisées par l'atteinte d'au
moins 2 dérivés ectodermiques.
DE hypo/anhidrotique:absence totale ou partielle de poils,dents ou glandes,de transmission
autosomique,récessive ou liée à l'X.
dans sa forme moderée, la DEH est la plus fréquente avec une incidence de 17 /100000 naissances
Nous rapportons le cas de CM 8ans ,issue d'un mariage non consanguin;orientée pour une fiévre sans foyer
évident
Notre attention fut attirée par:
-dysmorphie
-hyperpigmentation périorbitaire+hypertélorisme
-eczema péribuccal+hypodontie
-hypotrichose
-hyperkeratose palmoplantaire
l'intérrogatoire révèle une ressemblance avec le père(décédé),une intolérance à la chaleur + hypohidrose et
des bronchopneumopathies à répétition.
tableau qui correspond au syndrome de Christ-Siemens-touraine (sans confirmation génétique)
Prise en charge à base de mesures symptomatiques(perspective d'avenir: ectodysplasine)
Conclusion:Pathologie rare dont le pronostic peut-être grave(30% de décès chez les garçons) d'où l'interet

52
de la connaitre afin de diagnostiquer les sujets symptomatiques,les soumettre aux mesures préventives et de
proposer un conseil génétique.
3.Etude des performances d’une chambre d’inhalation pédiatrique
Boukhettala Nabile Porée Thierry, Diot Patrice, Vecellio Laurent
CEPR, INSERM U1100 EA 6305, Faculté de Médecine, Tours FRANCE
Chez les jeunes enfants asthmatiques, il est recommandé d’utiliser les aérosols-doseurs avec une chambre
d’inhalation. Les objectifs de cette étude sont d’évaluer les performances d’une chambre d’inhalation
pédiatrique avec différents médicaments.
La fluticasone (50µg/dose, GlaxoSmithKline, France) et la béclométhasone (100µg/dose, Medicis, France)
furent délivrés avec un aérosol-doseur seul ou en association avec la chambre d’inhalation Tipshaler
(Protec’Som, France). La distribution de la taille des particules fut mesurée à l'aide d'un impacteur en
cascade NGI (Copley Scientific, Royaume-Uni) selon la méthode de la pharmacopée européenne. Les
concentrations de fluticasone et de béclométhasone furent analysées par spectrophotométrie à 236 nm et
239 nm respectivement.
Dans la trachée, la masse de fluticasone était plus élevée avec un aérosol-doseur seul comparé à Tipshaler
(20 ± 0,6 µg vs 0,9 ± 0,3 µg, p <0,05). La dose de fines particules déposées était similaire en absence ou en
présence de Tipshaler (26 ± 2 µg vs 24 ± 1 µg). Au niveau distal, la masse de fluticasone était plus faible
avec l’aérosol-doseur seul comparé à Tipshaler (3 ± 0,4 µg vs 6 ± 0,2 µg, p <0,05). De plus, la masse de
béclométhasone déposée au niveau de la trachée était plus élevée avec un aérosol-doseur seul par rapport à
Tipshaler (12 ± 0,4 µg vs 1,1 ± 0,2 µg, p <0,05).
En conclusion, la chambre d’inhalation Tipshaler réduit la déposition de médicaments au niveau
oropharyngée et améliore la distribution des corticoides au niveau pulmonaire. Toutefois, chaque
médicament conserve une distribution pulmonaire qui lui est propre.
4.Syndrome d’Hallermann Streiff François : A propos de deux cas dans une même fratrie.
ZerouaL Zoulikha,M.Chaou, S.Tari, F.Bouferoua, L.Atek, N.Bouterfas H.Boucenna, A.Khati, A.Hazzazi,
H.Boukhellal, M.Haridi, Z.Bouzerar, M.E.Khiari. Service de pédiatrie « A » Chu Béni-Messous.
Introduction:
180 cas de Syndrome d’Hallermann Streiff François (HSS) ou syndrome occulo-mandibulo-facial sont
rapportés dans le monde. Observé initialement par Aubry en 1893, puis isolé par Hallermannn en 1948,
Streiff en 1950 et François en 1958.
Objectifs :
Rapporter nos cas cliniques et décrire leurs aspects évolutifs.
Matériels et Méthodes: À propos de deux observations de deux frère et sœur.
Amine le premier enfant d’un couple non consanguin, est hospitalisé à l’âge de 4ans pour exploration d’un
syndrome dysmorphique avec retard statural. Le morphotype a permis de poser le diagnostic clinique de
syndrome occulo-mandibulo-facial, devant une microcéphalie avec un visage petit, un nez mince, un micro
rétrognatisme, une peau fine, une microphtalmie avec strabisme et une dentition mal implantée.
Sur le plan général, il existe un retard de croissance staturo-pondérale avec un nanisme harmonieux. Le
même morphotype est constaté chez la sœur et cela dés la naissance.
Un bilan de malformation est revenu normal pour les deux en dehors du bilan radiologique qui montre des
côtes filiformes. Un retard scolaire marqué est constaté, surtout chez le garçon.
Conclusion :
De rares observations sont rapportée s dans la littérature, la connaissance du SHH permet un diagnostic
rapide et une prise en charge adaptée.

53
5.Histoire naturelle de l’allergie aux protèines du lait de vache
Ibsaine Ouardia,A. Benyahia- Narimene Zerrouki - Larbi Rezzag Lebza- Souad.Bellache1- Hassina
Berrah.CHU Parnet
L’allergie aux protèines du lait de vache (APLV) peut parfois persister (20 à 30%) au dela de 3 ans
Objectif :
Déterminer les caractéristiques évolutives de l’ APLV et identifier les facteurs prédictifs de la tolérance
Matériel et méthodes : Nous avons réalisé une étude prospective, descriptive des cas d’ APLV, diagnostiqués sur une période de
10 ans (2000-2009) et suivis jusqu’à l’âge de la tolérance. Le diagnostic de l’APLV était retenu devant
l’association de symptômes évocateurs, un bilan immunologique positif et/ou un test de provocation positif
Résultats :
150 enfants ont présenté une APLV dont 85 cas (57%) à IgE médiées. L’âge moyen au moment du
diagnostic est de 102,7 jours ±5,31. La durée moyenne de suivie était de 33,24 mois. La tolérance a été
obtenue dans 47% à 2 ans, 75% à 3 ans et 92% à 6 ans.
La tolérance est acquise plus rapidement dans le groupe non IgE médiée que dans le groupe IgE médié
(89% à 2 ans et 100% à 5 ans vs 31% à 2 ans et 80% à 5 ans, p=0,001). Les facteurs prédictifs d’acquisition
de la tolérance aux PLV sont : l’âge de diagnostic inférieur à 3 mois (OR : 2,6 ; p 0,0006), le type retardé
des manifestations cliniques (OR : 3,5 ; p 0,004), l’absence de servenue d’une allegie alimentaire associée
(OR :2,93 ; p 0,01) et d’asthme (OR :5,17 ; p 0,00001)
Conclusion :
L’APLV n’est plus une allergie transitoire surtout dans les Formes à IgE médiées et à début tardif
6.Une fièvre isolée du nourrisson : pensez à l’appendicite aigue !
BrahmiA Nabila,IbsainE Ouardia, Zerrouki Narrimene, Akkouche Fatima, GhaeouL Ahmes, Berrah
Hassine. Service de pédiatrie « B ».C.H.U H.Dey
L’Appendicite aiguë touche l’enfant de plus de 6 ans (90% des cas). Elle est rare avant 3 ans et
exceptionnelle avant l’âge de 1 an.
Observation : Nous rapportons le cas de Laeticia, âgée de 15 mois, suivis depuis l’âge de 9mois pour infection urinaire,
hospitalisée pour fièvre isolée et hématémèse de petite abondance survenant après prise de trois fois la dose
journalière du Celestene.
A l’admission l’enfant était obnubilée, fébrile à 39°c, tachycarde (FC 140 bat/mn), avec un mauvais état
général et une légère pâleur cutanéo-muqueuse. L’examen clinique était normal en particulier l’abdomen
était souple. L’exploration a révélé sur le plan biologique une hyperleucocytose à 22 500 elmts/mm3 et une
CRP positive. Le bilan d’hemostase était normal. Le bilan infectieux était négatif (PL, hémocultures,
ECBU, Radiographie du thorax). Le scanner cérébral était normal. Malgré un traitement antibiotique à
doses septicémiques, l’enfant est restée fébrile avec apparition vers le 3ème jour de vomissements et d’une
distension abdominale. L’abdomen était devenu sensible avec des images hydro-aériques à l’ASP.
L’ionogramme sanguin ne notait pas d’hypokalièmie. L’échographie abdominale avait retrouvé du liquide
épais au niveau de la FID, distension des anses digestives sans boudin d’invagination. La laparotomie a
retrouvé une appendicite retro-coecale, phlegmoneuse, perforée à sa base et compliquée de péritonite .Une
appendicectomie a été réalisée. Les suites opératoires étaient bonnes sous double antibiothérapie (C3G et
metronidazole).
Conclusion : Le diagnostic de l’appendicite aigue est souvent difficile et tardif lié à un tableau clinique souvent atypique,
avec des conséquences qui peuvent être

54
7. Imperforation choanale bilatérale: que faire chez le nouveau né?
Messaoudi Djamel ,Afri Yacine. Service ORL CHU Bab El Oued
L'imperforation choanale est une malformation congénitale rare mais non exceptionnelle. Elle consiste en
une obstruction totale ou subtotale des orifices postérieurs des fosses nasales.
Quand elle est bilatérale elle s'exprime dès les premières minutes de la vie et peut compromettre le
pronostic vital chez le nouveau-né, du fait de la détresse respiratoire qu'elle engendre.
La gravité réside aussi dans les malformations majeures qui peuvent y être associées, et qui sont des
facteurs de comorbidité.
Le diagnostic étant facilité par l’avènement de l'endoscopie nasale, le dépistage systématique par le passage
d'une sonde du plus petit calibre reste indispensable.
La conduite chez le nouveau-né est d'abord d'assurer la liberté des voies aériennes supérieures, ensuite de
rétablir la continuité des fosses nasales chirurgicalement.
La voie endoscopique a révolutionné l'attitude chirurgicale, du fait de sa facilité et son innocuité relative
mais les autres voies d'abord conservent bien leurs indications.
En fin l'imperforation choanale bilatérale est une affection grave du nouveau-né, qui nécessite une gestion
urgente et immédiate du fait du pronostic vital qui peut être compromis.
8.Quel profil de la mortalité infantile au niveau des centres hospitaliers d’Oran ?
Heroual Nabila,A.Tadjeddine.Service d’épidémiologie et de médecine préventive, EHS Canastel
Les objectifs du Millénaire pour le développement ciblent la réduction de mortalité infantile et maternelle
d’ici l’an 2015. En Algérie, des programmes de santé ont été mis en place pour réduire ces taux et
améliorer l’état de santé des mères et des enfants. L’objectif de ce travail est d’identifier les causes de
mortalité infantile dans la wilaya d’Oran.
Matériel et méthodes :
Les données analysées sont issues d’une base de données sur les causes médicalisées de décès survenus
chez les enfants âgés de moins de 15 ans au niveau de l’ensemble des structures hospitalières de la wilaya
d’Oran entre 2003-2007.
Les causes de décès sont codées selon les recommandations de la classification Internationale des maladies
CIM-10.
Résultats : Sur les cinq ans d’étude, le nombre de décès des moins d’un an représentait plus de 79% des décès. La
mortalité néonatale précoce a représenté 77,4% de l’ensemble des décès néonataux. Les décès survenus en
période post natale ont représentés près de 27% de l’ensemble des décès. Parmi les causes de décès, en
période néonatale, 77,5% des décès étaient dû aux affections périnatales dont les affections respiratoires
(39,8%) et la prématurité (11, 4%) suivies par le groupe des malformations congénitales (15, 3%) qui
portaient sur l’appareil digestif (07%). En période postnatale, les groupes de pathologies en causes étaient
maladies endocriniennes, nutritionnelles et métaboliques, les malformations congénitales surtout de
l’appareil circulatoire et maladies infectieuses avec respectivement 22,7%, 19,9% et 14,6%.
Conclusion : L’étude a permis de dresser un profil global de la mortalité pendant la période infantile. Cependant,
plusieurs causes de décès sont évitables d’où la nécessité de réorienter et renforcer les programmes de lutte
contre la mortalité materno-infantile pour atteindre le 4ème objectif du Millénaire.

55
9.Education therapeutique d’un enfant malade
Rezki Hassiba,Raffa Nassiba,Bettayeb,Rezig Fethia,Ghrieb Samira.Hopital central de la sureté nationale,
les glycines, alger
Introduction: Selon l’OMS: « L’éducation thérapeutique du patient vise à aider les patients à acquérir ou maintenir les
compétences dont ils ont besoin pour gérer au mieux leur vie avec une maladie chronique. Elle fait partie
intégrante et de façon permanente de la prise en charge du patient.
Elle comprend des activités organisées, y compris un soutien psychosocial, conçues pour rendre les patients
conscients et informés de leur maladie, des soins, de l’organisation et des procédures hospitalières, et des
comportements liés à la santé et à la maladie. »
Objectif: Attirer l’attention des pédiatres sur l’intérêt d’une éducation thérapeutique de qualité qui doit :
• être centrée sur l’enfant et son entourage
• être scientifiquement fondée (recommandations professionnelles, littérature scientifique pertinente,
consensus professionnel)
• faire partie intégrante du traitement et de la prise en charge
• être un processus permanent, qui est adapté à l’évolution de la maladie et au mode de vie de l’enfant
• être réalisée par des professionnels de santé formés à la démarche d’éducation thérapeutique du patient et
aux techniques pédagogiques
• s’appuyer sur une évaluation des besoins et de l’environnement de l’enfant : (diagnostic éducatif)
• se construire avec le patient, et impliquer autant que possible les proches du patient
• s’adapter au profil éducatif et culturel du patient
Conclusion: L’éducation thérapeutique du patient est une pratique et un domaine scientifique jeune, évolutif, qui trouve
un ancrage à la fois dans la médecine, la pédagogie de la santé et les sciences humaines et sociales.
10.Maldie de Kawazaki :Expérience d’un service de pédiatrie a Jiddah (Arabie Saoudite ) avec revue
de littérature et dernière mise a jour Sleiman Rola,H.Dey Alger
Introduction : La maladie de kawazaki est une vascularité systémique fébrile , le diagnostique Clinique peut être difficile
en absence d’un examen complémentaire de choix .
Un traitement précoce peut prévenir les complications cardiaques à type d’anévrismes coronariennes qui
peuvent survenir dans 30 % des cas non traités .
Méthodes :
revue des dossiers électroniques des malades atteints de la maladie de kawazaki admis au service de
pédiatrie à l’hôpital Soliman Fakeeh Jiddah( hôpital à 350 lits de capacité ) Durant 5 ans(2008-2013)
Résultats : 21 cas de malades de kawazaki , 10 garçons et 11 filles dont 13 d’ origine saoudienne , 8 non saoudienne ,
La moyenne d’ âge des malades est de 28 mois (5mois-9 ans ),
la fièvre et la desquamation cutanée sont des signes Cliniques quasi constants (100%).
D’autres signes Clinique sont moins fréquents:
éruption cutanée 80%(17 cas),
conjonctivite non purulente 66% (14cas)
érythème et sécheresse des lèvres 66% (14 cas),
lymphe adénopathie cervical (5 cas) 25%,
irritabilité 25%,arthrite 19% (4 cas) œdème des extrémités 4 cas (19%)
toux, diarrhée , vomissement 3 cas ,
méningiomes 2cas .
Les anévrismes coronariens sont retrouvés chez 2 cas (9%), le traitement au immunoglobuline était fait
immédiatement pour 10 malades , 7 avec retard de 3 jours en moyenne ,3 cas l’ont eu deux fois

56
Discussion : comparaison de ces résultats à la littérature .
Conclusion : la maladie de kawazaki dans la population arabe peut être sous estimée et mal diagnostiquée
d'ou la nécessite d'y penser .
11.Maladie de Castleman: à propos d'un cas
Treki Rima.EPH EL HARROUCH (Wilaya de Skikda)
La maladie de Castleman est une hyperplasie lymphoïde angiofolliculaire
Exceptionnelle chez l'enfant, de l'ordre de 1-9/1000000 cas
Nous rapportons le cas de A.M âgé de 3ans ,adressé au service pur une fièvre au long cours et chez qui
l'exploration radiologique a objectivé :
- une pneumopathie micronodulaire
- des adénopathies mésentériques d'allure réactionnelle
Devant la persistance de la fièvre, des images radiologiques et l'apparition de nodules hépato-spleniques,
une TDM abd-pelvienne a été réalisée et le malade a bénéficié d'une biopsie ganglionnaire par voie
coelioscopique,
L' étude anatomo-pathologique a conclu à une maladie de Castleman dans sa forme vasculaire hyaline
Le patient a été confié au service d'oncologie pédiatrique du CHU de Constantine
L'objectif de cette observation est de mettre en lumière une pathologie extrêmement rare chez l'enfant,
souvent méconnue.
fréquemment asymptomatique dans sa forme localisée, elle peut mimer différents tableaux (TBC
intestinale, lymphome) quand elle est multicentrique; d'où l'intérêt de l'étude anapath dans le redressement
diagnostic.
Bien qu'il s'agisse d'un syndrome lymphoprolifératif bénin, le traitement et le pronostic dépendent de la
forme de la maladie.
Forme localisée: exérèse chirurgicale,
Forme multicentrique: polychimiothérapie ou Anticorps monoclonaux.
12.Lymphome Malin Non Hodgkinien à localisation cutanée : à propos d'un cas
Chachou khadidja ,Boumeddane amaria, Mecifi raouida, Khelil amina leila, Mahi henni mohamed amine,
Mehidi mohammed yacine, Bensid ikram, Oussalah meriem, Maazouzi djamel
Service d'oncologie pédiatrique, CAC Emir Abdelkader, ORAN
Introduction : Le lymphome malin non hodgkinien est une prolifération cellulaire maligne d’origine lymphoïde
caractérisée par une croissance tumorale rapide et précoce, c’est une urgence diagnostique et thérapeutique.
Il représente après les leucémies la 2ème hémopathie maligne de l’enfant
Les localisations habituellement retrouvées :
Abdominales 40%, thoraciques 25%, ORL 15%, ganglionnaires périphériques 10%, Autres 10%.
Objectif : A partir d’un cas de notre pratique médicale et des données de la littérature, les auteurs se proposent de
décrire un cas de lymphome T sous-cutané chez une fille âgée de 5ans découvert suite à une panniculite
récidivante
Matériel et méthode : Une fille âgée de 5 ans et demi présentait depuis l’âge de 3ans des lésions nodulaires sous cutanées chaudes
et douloureuses de taille variable1 à 3 cm, siégeant au niveau du visage, l’abdomen, le dos et la face latéro-
externe de la jambe à évolution récidivante dans un tableau général fait de fièvre prolongée, asthénie,
anorexie ; ne panniculite fébrile récidivante type Weber-Christian était initialement évoquée.
L’évolution était favorable (apyrexie et régression des nodules cutanés) sous corticothérapie par voie
générale à raison de 2mg/kg/j maintenue pendant 4mois après échec des AINS.
A l’arrêt des corticoïdes, une reprise de la symptomatologie antérieure était constatée et l’examen
histologique d’un nodule biopsié associé aux immunomarquages était évocateur d’un lymphome T sous–
cutané type panniculite de phénotype CD3+, CD8+
Résultat : Après un bilan d’extension (ponction lombaire, medullogramme et scanner thoraco-abdomino-pelvien)

57
revenu sans anomalie; la malade a été classée stade 3 selon la classification de Murphy et mise sous
chimiothérapie selon le protocole LMT 89 le 12/12/2012
L’évolution a été marqué par la disparition totale des nodules sous cutanée après la 2ème cure de
chimiothérapie laissant place à des taches hyper chromiques.
Actuellement elle est en rémission complète et reçoit sa chimiothérapie d’entretien.
Conclusion : Le lymphome T cutané reste une affection rare chez l’enfant, la forme type panniculite d’évolution
indolente est exceptionnellement rapportée et doit inciter à réaliser des biopsies cutanées devant toute
panniculite récidivante
Bibliographie : _ Cancer de l’enfant Flammarion 2008.O.Hartmann, Jean Lemerle.
_ Hématologie de l’enfant : G.Schaison.Flammarion.
13.Nanisme thanatophore : à propos d’un cas
Mahmoud Abderezak Oumenkhache,Souhila Abed, Fatiha Azi,Ouahiba Benrabeh
Unité de néonatologie CHU Nefissa Hamoud ex Parnet; Alger
Introduction : Le nanisme thanatophore est une ostéochondrodysplasie rare classée en deux types I et II.
Elle est due à une mutation du gène FGFR3 (fibroblast growth factor receptor 3) localisé sur le bras court
du chromosome 4. C’est une anomalie morphologique toujours létale.
Observation : Nous rapportons le cas de B.A nouveau-né de sexe masculin issu d’un couple jeune non
consanguin, grossesse suivie (Echographie obstétricale de la 27e semaine d’aménorrhée objective un fœtus
de sexe masculin en présentation du siège avec thorax étroit, cotes courtes, vertèbres plates en H et
hydramnios) et menée à terme avec accouchement par voie haute (présentation du siège et macrocéphalie),
bonne adaptation à la vie extra utérine.
Le nouveau-né à présentais un poids et une taille dans les normes avec macrocéphalie (PC=41 cm), un
facies particulier (front bombé, cou court et dépression de la racine du nez), un thorax étroit contrastant
avec un abdomen proéminents, des membres très courts avec doigts courts et boudinés. Par ailleurs
apparition d’une détresse respiratoire précoce modérée mis sous oxygène et double antibiothérapie
L’examen radiologique retrouva des os long très court avec fémur incurvé et vertèbres en H
Le nouveau-né est décédé à 31 heures de vie dans un tableau de détresse respiratoire aiguë.
Conclusion : le nanisme thanatophore de diagnostic anténatale est rapidement mortel et représente
l’exemple de maladie génétique sporadique et le conseil génétique doit être donc rassurant
14.Convulsion fébrile épidémiologie et causes Benallal Khadidja,Benaissa Fatima, Tamert Soumia, Ould said Karima, Machou Nassiba, Mohammedi
Abdelatif.Service de pédiatrie CHU de Sidi Belabbes
Introduction : Les convulsions fébriles constituent un problème pédiatrique fréquent : 2 à 5 %
des enfants présentent une convulsion fébrile dans leur vie.
Objectif : décrire le profil des malades admis dans notre service pour convulsion fébrile et faire le point sur
l’indication de la ponction lombaire dans notre contexte.
Patients et méthodes : il s’agit d’une étude rétrospective faite sur dossiers de malades admis pour
convulsions fébrile sur une période de cinq ans.
Résultats : on a colligé 442 malades dont 275 garçons et 167 filles (sex ratio= 1,6), âgés de 1mois à 12 ans
, 72,8% sont âgés de 6 à 24 mois, la crise était simple chez 411 malades et complexe chez 31 malades, des
antécédents d’épilepsie sont retrouvés chez 41% des cas, une ponction lombaire a été indiquée chez 311
malades, elle est revenu normale sans réaction cellulaire chez 259 des cas (83,3%) et pathologique chez 52
malades (16,7%) dont 44 méningite lymphocytaire (84,4%) et seulement 8 malades présentant une
méningite bactérienne. Le diagnostic étiologique final est représenté essentiellement par les infections extra
crânienne dominé dans 87% des par les infections ORL, et seulement 5,1% des malades présentaient une
méningite dont 1,8% étaient bactériennes.

58
Conclusion : la ponction lombaire ne devrait pas être effectuée de routine si l’état général de l’enfant est
bon et l’examen neurologique est normal. Elle devrait être fortement envisagée dans les cas de déficit
neurologique résiduel ou de convulsions prolongées. Donc examen clinique minutieux permet d’orienter la
conduite pratique.
15.Taux de l’allaitement maternel dans la région de Sidi Belabbes
Benallal Khadidja ,Hamdoun Karim, Barket Mnaouer, Larabi Zahira.Service de pédiatrie du CHU de Sidi
Belabbes
Introduction : l’évolution des sociétés et le progrès de l’industrie laitière sont responsables du recul de la
pratique de l’allaitement maternel qui demeure préoccupant dans les pays en développement.
Objectif : évaluer le taux de l’allaitement maternel dans la région de Sidi Belabbes.
Matériels et méthode : on a étudié deux groupes d’enfants, le premier groupe a colligé 99 nourrissons et
enfants âgés de 1 à 36 mois hospitalisés dans notre service de pédiatrie pour divers problèmes dont les plus
fréquents sont infectieux, on a étudié certains paramètres liés aux nourrissons principalement le type
d’allaitement et la durée de l’allaitement maternel exclusif, et des paramètres liés à la mère, le deuxième
groupe a concerné 300 nourrissons sains âgés de 1 à 24 mois qui se sont présentés pour vaccination dans
deux centres de la ville de Sidi Belabbes, on a recueillis surtout les information concernant le type
d’allaitement et la durée de l’allaitement maternel exclusif.
Résultats : le premier groupe est représenté par 54 fille et 45 garçon (sex ratio=0,88), l’âge moyen des
mères est de 30 ans, 89,9% sont des multipares et 12% seulement avait un emploi, le deuxième groupe est
représenté par 157 filles et 143 garçons (sex ratio=0,91), l’âge moyen est de 6,5 mois, l’allaitement était
artificiel chez 29,6%, mixte chez 39,6% et maternel chez 30,6% des nourrissons. Le taux moyen de
l’allaitement maternel exclusif était de 82,5 jours dans le premier groupe vs 75,4 jours dans le deuxième
groupe.
Conclusion : nos taux sont faibles par rapport aux chiffres des pays développés, il est primordial de faire
des actions de sensibilisation.
16.Calcinose pseudo-tumorale compliquant un cas d’osteodystrophie rénale chez l’enfant
Oukrif Lamia ,Ouzani Meriem,Boukhil Kais,Bensenouci Abdelatif.Pediatrie B CHU Beni Messous
Introduction : La calcinose pseudotumorale est dans la majorité des cas, idiopathique avec une histoire familiale retrouvée
dans 30% des cas. C’est une cause rare de calcifications intra tissulaires chez les insuffisants rénaux
chroniques hémodialysés. Elle est due à l’ hyperparathyroïdie secondaire. Et sa fréquence est estimée entre
0,5 et 7%
Objectif : Rapporter un cas exceptionnel de calcinose chez un enfant atteint d’insuffisance rénale
chronique
Materiel et méthodes :
Nous rapportons le cas d’un garçon âgé de 3ans, suivi depuis la naissance pour insuffisance rénale
chronique sur RVU bilatéral grade V, sous traitement conservateur mal suivi, admis pour cervicalgies
importantes évoluant depuis 2 mois rebelles aux antalgiques.
L’examen clinique retrouve un déficit staturo pondéral important, des déformations, en varus des membres
inferieurs avec un aspect des tibias en « lame de sabre » responsables d’un arrêt de la marche. L’examen du
rachis cervical révèle une douleur vive à sa mobilisation, sans tuméfaction, sans déficit neurologique.
Résultats : Le diagnostic d’ostéodystrophie rénale a été posé devant les déformations osseuses, la déminéralisation
osseuse diffuse, les fractures, l’hypercalcémie, l’hyperphosphorémie, le taux de PTH très élevé et confirmé
par l’histologie
Les radiographies du rachis cervical montrent une opacité des parties molles avec des calcifications. La
TDM cervicale retrouve une masse des parties molles de C1 à C3, de densité renforcée en périphérie.
L’IRM confirme la calcinose pseudo-tumorale cervicale.

59
Nous avons réajusté les doses de la calcithérapie, malheureusement l’état général de l’enfant s’est
rapidement détérioré avec aggravation de son insuffisance rénale puis décès.
Commentaires : La Calcinose des insuffisants rénaux siège habituellement au niveau des hanches, des épaules, des genoux,
des coudes, poignets et doigts. Ses principaux facteurs de risque sont l’hyperparathyroïdie, l’augmentation
du produit phosphocalcique et probablement des facteurs locaux traumatiques. La particularité de notre
patient demeure dans le siège cervical exceptionnel avec infiltration et ostéolyse des vertèbres. Son
traitement demeure controversé. Pour certains, le traitement de l’hyperparathyroïdie permet une
amélioration spectaculaire avec une disparition de la masse calcifiée en quelques semaines, pour d’autres,
l’exérèse chirurgicale et même la para thyroïdectomie subtotale sont recommandées.
Conclusion : L’hyperparathyroïdie secondaire à l’insuffisance rénale chronique peut être pourvoyeuse d’une calcinose
pseudo-tumorale. Le siège vertébral cervical est exceptionnel, et seul un traitement conservateur bien
conduit permet de l’éviter.
17.Arthrogrypose: Syndrome et non maladie :A propos de 3 observations
Djemane Adel.EPH Gouraya
Introduction: l’arthrogrypose ne constitue qu’un signe clinique et non une entité pathologique. De
nombreuses affections.Nous allons rapporté ici l’observation de 3 type différents.
Observation 1 : B.A garçon né a terme sans incident qui est admis en néonatologie pour trouble
alimentaire, a l’examen on retrouve un nouveau né eutrophique qui présente des contractures articulaires :
Camptodactylie , déviation cubitale des mains , un pied bot varus équin combiné a un philtrum allongé
narines rétrécies et une microstomie accompagné d’un microglossie donnant une expression du visage
particulière en museau de carpe ou aspect de bébé siffleur concordant avec le diagnostic de syndrome de
Freeman-Sheldon ou Arthrogrypose distale de type 2A.
Observation 2 : S.A fille de 18mois est adressée en consultation pour exploration d’un syndrome
malformatif . A l’examen l’enfant présente des raideurs articulaire multiples : main-coudes-poignets , pieds
bots varus équins de façon symétrique ainsi qu’une scoliose sévère et une luxation de hanche gauche
négligée le tout sans trouble neurologique notable. Ce groupement de signes nous permet de retenir le
diagnostic d’Arthrogrypose multiple congénitale.
Observation 3 : G.Z garçon de 4 ans est adresse pour infections répétées sur syndrome malformatif
.l’examen clinique retrouve des raideurs articulaire avec présence de palmures cutanées au niveau des
zones de flexion limitant la mobilité au niveau des membres. un hypertélorisme, une micrognathie avec
petite ouverture de bouche, fente palatine, Camptodactylie et Cyphoscoliose .ce qui nous permet de retenir
le diagnostic de Syndrome du Pterygium multiple type Escobar forme non létale.
Discussion : L'arthrogrypose est un syndrome et non une maladie : elle est caractérisée par un ensemble de
signes et non par une cause univoque. On estime sa fréquence à 1/3000 naissances. Les arthrogryposes
entrent aujourd’hui dans le vaste cadre de ce qu’il est convenu d’appeler les syndromes d’immobilité fœtale
.Devant une telle variété de tableaux cliniques, le choix du traitement est des plus difficiles. Le but est
avant tout d’essayer d’améliorer la fonction et l’autonomie de ces sujets.
Conclusion : l’arthrogrypose est une affection présente dès la naissance, nécessitant un diagnostic précoce
et précis afin d’instaurer une prise en charge adaptée et garantir l’autonomie de ces enfants .

60
18.Association Maladie de Crigler Najjar et Syndrome de Gilbert
(Rareté et complexité de l’association)
Boudhar Kamel ,F.Benmati1, W.Messak, N.Mansouri1, F.Petit2, R. Reding3, S.E.Laalaoui1,
L.Benhafessa1.
1 Service de réanimation néonatale HCA Alger
2 Laboratoire de génétique moléculaire, Hôpital Antoine Béclère France
3 Unité de chirurgie pédiatrique abdominale et générale, Cliniques Universitaires Saint Luc Bruxelles
Introduction : La maladie de Crigler Najjar et le syndrome de Gilbert sont deux affections héréditaires à transmission
autosomique récessive liées à l’atteinte de la glycuronyl transférase. Leur diagnostic repose sur la mise en
évidence d’une mutation du gène UGT1A1. Isolé, la première est une maladie grave et le second est bénin.
Observation : Oussaid est originaire de Tebessa, né d’un mariage consanguin 2eme degré, 3eme d’une fratrie dont les
deux frères sont décédés suite à un ictère nucléaire. Il présentait un ictère constatait au 7éme jour de vie à
bilirubine libre non hémolytique grave et prolongé. Le diagnostic de la maladie de Crigler Najjar était
suspecté et confirmé par étude génétique qui a révélé en plus la présence du syndrome de Gilbert. Le
traitement consistait à une photothérapie jusqu’à l’âge de 1 ans ou l’enfant a bénéficié d’une greffe
hépatique.
Discussion : L'anomalie du promoteur seule est responsable du syndrome de Gilbert. Les mutations dans la séquence
codante du gène UGT1A1 sont responsables soit du syndrome de Gilbert soit de la maladie de Crigler-
Najjar. D'un point de vue moléculaire, la présence de l'anomalie du promoteur avec une ou deux mutations
dans les exons d'UGT1A1 est un facteur aggravant. Dans le cas de notre patient, l'anomalie du promoteur
TA7 a toujours été retrouvée associée à la mutation c.1070A>G. Au niveau cellulaire, il est difficile de dire
si la mutation isolée donnerait une maladie de Crigler-Najjar de type 1 mais l'association TA7/c.1070A>G
à l'état homozygote est responsable d'une forme sévère de maladie de Crigler-Najjar.
Conclusion : L’association maladie de Crigler Najjar et Syndrome de Gilbert est exceptionnelle, elle est observée surtout
chez la population tunisienne, responsable de la forme grave de la maladie de Crigler Najjar dont le seul
traitement est la greffe hépatique.
19. Traitement de l’acidocétose diabetique par perfusion continue d’insuline rapide
Ibsaine ouardia ,Narimene Zerrouki –Lamri Boukaboub- Samira Medjdoubi- Hassina Berrah.Service de
pédiatrie « B ».C.H.U H.Dey
Le traitement de l’acidocétose diabétique (ACD) associe une réanimation hydrolélectrolytique et une
insulinithérapie.
Objectif : Evaluer l’éfficacité et les incidents liés au traitement de l’ACD par perfusion continue d’insuline rapide.
Matériel et méthodes :
C’est une étude rétrospective, descriptive, portant sur notre population d’enfants hospitalisée pour ACD sur
une période de 02 ans (2008-2009). Tous les enfants ont bénéficié d’une perfusion continue d’insuline
rapide et une rehydratation (Groupe A). Ce groupe a été comparé à un groupe de malades reçevant de
l’insuline rapide dans les solutés de rehydratation (Groupe B). Le diagnostic de l’ACD était confirmé par
une glycémie et une gazométrie.
Résultats : 21 enfants ont reçu le protocole A ont été comparés à 22 qui ont bénéficié du protocole B. L’âge médian
était de 8 ans (groupe A) et de 10 ans (groupe B). Le taux d’ACD sévère était similaire dans les 2 groupes
(24% vs 23%). Chez les enfants ayant une ACD sévère, le temps moyen de résolution de l’hyperglycémie
et le nombre d’hypoglycémies était similaire dans les 2 groupes (p : 0,2). En cas d’ACD légère le temps
moyen de résolution de l’hyperglycémie était significativement plus court dans le groupe A (p 0,0004) et le
nombre d’hypoglycémie était plus important (61% vs 4,5% p 0,0002). La durée de séjour était réduite dans

61
le premier groupe ( p 0,008)
Conclusion : La perfusion continue d’insuline rapide est le traitement approprié pour l’acidocétose sévère. . Une autre
thérapeutique doit être proposée dans les formes légères.
20.Volvulus intestinal sur bride de LADD découverte accidentelle: A propos d’un cas.
S. Kerdoudi (1), F. Hallala (1), H. Djabri (1), F. Sallahi (1), A. yousfi (2).
1 : service de pédiatrie, EHS Sidi Mabrouk, Constantine
2 : service de chirurgie pédiatrique, EHS Sidi Mabrouk, Constantine
Introduction : Les anomalies de rotation du mésentère correspondent à un arrêt du processus rotatoire du tube
duodénojéjunal.Le mésentère commun est dit incomplet si l’interruption survient à 180° suspendant ainsi le
grêle de part et d’autre du mésentère et accolant le caecum à droite du duodénum à la région hépato
vésiculaire par des bandes péritonéales malformatives « Bride de LADD »
Le diagnostic est porté le plus souvent par un tableau d’une urgence chirurgicale, mais des manifestations
atypiques peuvent être révélatrices.
Objectif : Connaitre le volvulus chronique du grêle dans sa forme atypique comme mode de révélation fréquent chez
le grand enfant
Matériel : Nous rapportons le cas d’une fille de 5ans, qui présente des vomissements chroniques depuis la période
néonatale, à l’origine de plusieurs hospitalisations. A son admission les signes de déshydratation sont
patents, avec un retard pondéral marqué sans retentissement statural .Biologiquement on note une
hypokaliémie et une insuffisance rénale fonctionnelle. l’échographie montre un estomac distendu avec une
stase , le mode doppler visualise le signale vasculaire mésentérique avec image en tour de spire, la FDH
met en évidence une dilatation du D1 ,D2 infranchissable par la sonde, Le TOGD révèle une dilatation
pylorique , le scanner abdominal visualise une image typique d’un volvulus .Opérée selon l’intervention de
LADD , les suites opératoires sont favorables, et la prise en charge nutritionnelle est en cours.
Résultat : Notre cas illustre la possibilité de diagnostiquer des malformations embryonnaires digestives se manifestant
habituellement en période néonatale, mais des signes atypiques en dehors de toute urgence chirurgicale
sont révélateurs chez le grand enfant.
Commentaire et Conclusion : Le volvulus chronique du grêle chez l’enfant sur bride de LADD est une entité clinique rare, qu’il faut
savoir évoquer et confirmer par des examens non invasifs même en présence de manifestations atypiques
21.Syndrome de Hunter forme severe chez un enfant : a propos d’un cas
Bennani amal , Bensaadi, Cherifi, Regal, Hamzaoui, Chalah, Iddir, Sifodil,Heddou, Arhab, Ahmane.
CHU de tizi-ouzou service de pediatrie
Introduction : Le syndrome de Hunter (mucopolysaccharidose de type II) est une maladie génétique rare,
la seule mucopolysaccharidose transmise sur le mode récessif lié à l'X.
Observation :B.Abdelmadjid, 2ans, unique enfant d’un couple non consanguin, aux antécédents familiaux
chargés de décès de tous les oncles maternels et des cousins avant l’age de 7 ans, vue en consultation pour
exploration d’un retard psychomoteur progressif (marche et langage non acquis) associant un faciès
grossier particulier, une hépatosplénomégalie, une cyphoscoliose avec enraidissement. Le diagnostic de
mucopolysaccharidose de type II est évoqué confirmé rapidement par la biologie et l’étude génétique.
L’enquête familiale est en cour.
Discussion : Le syndrome de Hunter est une maladie très grave dont les symptômes apparaissent progressivement:

62
L'enfant est normal à la naissance ; Le tableau clinique des formes sévères associe hernies, dysmorphie
faciale (macroglossie, bouche entrouverte, traits épais), hépatosplénomégalie, limitations articulaires,
syndrome du canal carpien, dysostose multiple, petite taille, troubles du comportement et dégradation
psychomotrice, surdité, atteinte cardiaque et respiratoire, signes cutanés. L’espérance de vie est très
diminuée.
L'excrétion urinaire accrue de dermatane-sulfate et d'héparane sulfate oriente le diagnostic biologique
confirmé par la mise en évidence du déficit enzymatique en iduronate-2-sulfatase.
Une thérapeutique enzymatique substitutive est proposée mais à défaut de traitements complètement
efficaces, les soins palliatifs restent d'une importance capitale.
Conclusion : La mucopolysaccharidose de type II est une maladie de surcharge lysosomale considérée
comme une maladie orpheline. La rareté de la maladie, la lenteur de l'apparition et la diversité des
symptômes mènent souvent à un diagnostic tardif.
Enquête sur les moyens et les comportements des médecins de première ligne en cas de suspicion de
diabète de type 1 chez l’enfant
A. Zennaki, A. Ouzzaa, S. Niar, H. Aichaoui, I. Abdat, M. Gharnouti, A. Reguieg, M. Naceur,
M. Bessahraoui, K. Bouziane-nedjadi.
Service de pédiatrie « C », CHU d’Oran.
Introduction
L'acidocétose diabétique (ACD) inaugurale est une cause de mortalité et de morbidité supplémentaire chez
les enfants atteints de diabète de type 1 (DT1). On estime sa fréquence à 20% dans notre contexte de
travail. Il a été démontré qu’il était possible, par des moyens simples et peu coûteux, d’en réduire encore la
fréquence.
Objectifs
Connaitre les habitudes et les possibilités des médecins recevant des enfants à leur consultation, en vue
d’une campagne de prévention de l’ACD inaugurale dans le DT1
Matériel et méthodes
Un questionnaire concernant les moyens diagnostics et la conduite en cas de suspicion de DT1 a été
distribué aux médecins présents lors de deux journées de formation médicale continue en février et mai
2013 à Oran. Cent vingt cinq médecins, dont 12 du secteur libéral et 5 spécialistes, ont répondu aux
questions concernant la disponibilité de lecteurs de glycémie, de bandelettes d’analyse de glycémie
capillaire, de bandelettes d’analyse d’urine, de laboratoire d’analyse et leur conduite en cas de suspicion de
DT1.
Résultats
Quatre-vingt et un pour cent des médecins disposent de lecteurs de glycémie à demeure et 52% d’entre eux
de bandelettes en permanence.
Soixante-sept pour cent des médecins exercent dans une structure disposant d’un laboratoire, avec la
possibilité pour 63% d’entre eux d’obtenir une glycémie veineuse à tout moment.

63
Devant une glycémie anormalement élevée, 73% des médecins recherchent sucre et acétone à la bandelette
urinaire, 66% demandent une glycémie veineuse, 86% orientent le patient vers un service spécialisé dans la
prise en charge du diabète et 30% seraient prêts à entamer l’insulinothérapie sur place.
Conclusion
Il convient de continuer de doter d’avantage de médecins de bandelettes d’analyse de glycémie capillaire
et/ou d’urine, moyen simple et rapide pour le diagnostic de DT1, et d’habituer les médecins à orienter
rapidement les patients vers un service spécialisé sans demander de confirmation veineuse en raison du
risque d’évolution rapide vers l’acidocétose
23.A propos d’un cas de syndrome de Kasabach-merritt chez un nourrisson traité avec succès
Dib saad-eddine,Kandouci-tani fatima, Kaouadji naoual, Benahmed ali, El mezouar shahrazed, Massen
Zouhir.Service pédiatrie EHS mère-enfants Tlemcen
Introduction : Le syndrome ou phénomène de Kasabach-Merritt (SKM) se définit par l'association d'une
tumeur vasculaire, d'une thrombopénie majeure et d'une coagulopathie de consommation plus ou moins
marquée (fibrinogène bas, présence de complexes solubles à des taux élevés, D-dimères très élevés le plus
souvent). Ce syndrome est très rare, mais sa prévalence est inconnue. Le déclenchement est soudain avec
constitution d'une masse ecchymotique et inflammatoire. Les tumeurs vasculaires du SKM sont
congénitales ou acquises en, général avant 6 mois. Le SKM, tel qu'il fut décrit en 1940, concerne par
définition toujours des nourrissons, et il a pour substratum anatomique une tumeur vasculaire.
Matériel et méthodes : il s’agit d’un nourrisson de sexe féminin âgé de 2 mois admis dans notre service
pour prise en charge d’un hémangiome géant au niveau de l’épaule gauche associé à une thrombopénie.
Résultats : La conduite à tenir a été de la mettre d’abord sous corticoides puis d’associer un béta-bloqueur
(avlocardyl) malgré cela la tumeur a continué à augmenter de volume. On a alors eu recours à la vincristine
à raison d’une cure par semaine associée à la ticlopidine et l’aspirine, ce qui a entrainé une réduction
notable du volume de la tumeur et normalisation du taux de plaquettes.
Commentaires : le syndrome de Kasabach et merritt a longtemps été considéré comme un hémangiome
infantile. Pourtant, ni les aspects cliniques, ni les lésions résiduelles, ni les aspects histologiques ne sont
typiques d'un hémangiome. On sait aujourd'hui que la tumeur sur laquelle se greffe le syndrome de
Kasabach-Merritt est soit un angiome en touffes soit un hémangio-endothéliome kaposiforme. Un élément
est venu confirmer le fait que la tumeur qui fait le lit de ce grave phénomène hématologique diffère de
l'hémangiome infantile : l'immunophénotype GLUT1 est constamment négatif dans les tumeurs associées
au SKM alors qu'il est positif dans 100% des hémangiomes. Malgré une fréquente confusion dans la
littérature, il faut distinguer le SKM de la coagulopathie intravasculaire chronique, associée aux
malformations veineuses ou lymphatiques (malformations à flux lent) ou encore observée dans un contexte
de tumeur maligne (angiosarcome, fibrosarcome). Cette distinction est importante car les traitements sont
totalement différents. L'hétérogénéité des réponses thérapeutiques est une particularité du SKM : la réponse
est imprévisible et le choix empirique. Actuellement, les quatre traitements les plus régulièrement efficaces
sont la corticothérapie générale, l'interféron alpha 2a ou 2b, la vincristine, et une association anti-agrégante:
ticlopidine et aspirine. Lorsqu'elle est possible, l'exérèse chirurgicale est suivie d'une guérison immédiate
du phénomène biologique. L'embolisation thérapeutique ou la radiothérapie sont plus rarement proposées
en complément du traitement pharmacologique. Les transfusions de plaquettes sont à éviter, car elles
exacerbent la thrombopénie et le risque hémorragique viscéral.
24.L’insuffisance renale chronique chez l’enfant experience d’un service de pediatrie generale
Widad amel,N Bouchair , J Bounour , A Hamani , F Bouslama , H Sehab ,J Belamri ,L Boustil, M
Yaiche.Clinique pediatrique sainte therese chu annaba
Introduction :IRC chez l’enfant est une pathologie posant de réels problèmes de prise en charge dans notre
pays quelque soit son étiologie
Objectif :.notre étude a pour but de mettre le doigt sur nos difficultés et essayer d’améliorer le sort de ces
enfants devant l’absence de greffe rénale.
Matériels et méthodes:Il s’agit d’une étude rétrospective portant sur 20 malades suivis pour IRC au seins du

64
CHU d’ Annaba au service de pédiatrie durant la période:janvier 2008 à mars 2013.
Résultats :
L’âge moyen de nos patients est de 8,3 ans avec un sexe ratio F/M 1,22
Cliniquement le retard staturo-pondéral associé a une anémie sont presents dans 70% des cas suivi par une
HTA dans 40%.
75% de nos malades lors du diagnostic étaient au stade d’IRC terminale
Les uropathies malformatives retrouvées dans 40%des cas fut l’étiologie la plus retrouvé suivie des
glomerulopathies dans 30%.
Le traitement adjuvant a été administré a tous nos malades et 06 malades ont été traités par dialyse
péritonéale et 09 patients par hémodialyse.
Nous déplorons 05 décès dues essentiellement par un œdème aigu du poumon
Conclusion :il ya beaucoup a faire afin d’améliorer la prise en charge de ces malades tans par le dépistage
précoce des uropathies malformatives, la disponibilité des cathéters pour dialyse péritonéale que par le
développement des centres de dialyse pédiatrique et surtout par l lancement de la greffe rénale chez nos
enfants
25.Cholestase néonatale à cytomégalovirus à propos de deux cas
Bendaas N,Manaa M,Guerdouh S .Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine
Introduction : L’infection néonatale à cytomégalovirus (CMV) est une cause classique des cholestases du nouveau né et
du nourrisson dont le diagnostique n’est retenu qu’après avoir éliminé les causes fréquentes et urgentes
comme l’atrésie des voies biliaires. La mise en route du traitement obéie à des indications précises.
Presentation des cas : Il s’agit de 2 nourrissons âgés successivement 2 mois, 50 jours, hospitalisés à notre niveau pour d’un
syndrome cholestatique partiel et intermittent ; dont l’imagerie et la biologie avaient éliminé préalablement
des anomalies des voies biliaires et les autres causes les plus fréquentes. Le diagnostic de l’hépatite à CMV
avait été retenu sur des bases cliniques, biologiques et anatomopathologiques typiques.
Le nourrisson de 2 mois a bénéficié en plus du traitement adjuvant d’un traitement par le GANCICLOVIR
vu qu’il présentait une forme sévère d’infection: atteinte hépatique, hématologique, auriculaire ; alors que
le nourrisson de 50jours n’a bénéficié que du traitement adjuvant et d’une surveillance.
Les 2 nourrissons ont très bien évolués.
A travers ces 2 observations et une revus de la littérature, nous rappelons la présentation clinique et para
clinique ainsi que les indications du traitement par le GANCICLOVIR.
Conclusion : L’infection à CMV reste encore une cause fréquente de cholestase néonatale dont les indications de
traitement sont très limités.
26.Ostéopétrose à révélation néonatale
Laabed Ahlem ,Telhaoui Nabil,EPH Hakim Saadene Biskra
Introduction : L'ostéopétrose est une maladie osseuse rare, due principalement à un dysfonctionnement des ostéoclastes,
qui sont des cellules d'origine hématopoïétique, de la lignée macrophagique. Il existe différentes formes
d'ostéopétrose qui ont une évolution variable allant du décès in utero à la forme restant totalement
asymptomatique.
L’ostéopetrose affection génotypique autosomique,offre deux aspects essentiels : la forme néonatale,très
sévère, et la forme à révélation plus tardive,parfois latente.
Observation : Nous rapportons le cas d’une fille agée de 1 mois, premier enfant d’un couple consanguin, née à terme,
présentant une anémie à la naissance,hospitalisée en pédiatrie pour une pancytopénie avec hépato-
splénomégalie.
La clinique ainsi que le bilan radiologique ont permis de poser le diagnostic qui a été confirmé par la
biopsie osseuse .

65
Conclusion : L'ostéopétrose est une maladie congénitale rare de la résorption osseuse caractérisée par une densification
généralisée du squelette, de diagnostic facile mais dont la prise en charge est lourde , son diagnostic
prénatal peut être réalisé à la fin du 2e trimestre de grossesse par échographie, mais le diagnostic
moléculaire, s'il est disponible, est recommandé car il peut être effectué plus tôt, à 11-13 semaines de
gestation sur prélèvement de villosités choriales
27.Intérêt de l’étude des glycoproteines plaquettaires par la cytométrie en flux dans la
thrombasthénie de glanzmann
Belhadj Hayat,Fenni Nacéra.Unité de pédiatrie CRMC Blida
Introduction : La thrombasthénie de Glanzmann est une maladie hémorragique caractérisée par un défaut d’agrégation
plaquettaire en rapport avec un déficit quantitatif ou qualitatif des glycoprotéines plaquettaires.
Observation : K. Aya âgée de 03 ans est suivie depuis l’âge de 10 mois pour thrombasthénie de Glanzmann.
Elle est issue d’un mariage consanguin du 2eme degré, il n’ya pas d’antécédents familiaux connus, en
dehors d’une tante paternelle polytransfusée (?).
Le diagnostic de thrombasthénie de Glanzmann a été évoqué à l’âge de 10 mois devant un hématome
fessier secondaire à une injection intra-musculaire.
Le diagnostic de thrombasthénie de Glanzmann a été posé sur un temps de saignement allongé et une étude
des fonctions plaquettaires par agrégométrie objectivant une agrégation nulle à l’ADP , à l’acide
arachidonique et au collagène. Aya a présenté plusieurs hémorragies cutanéo-muqueuses spontanées et
provoquées (ecchymoses, hématomes, epistaxis….)
L’étude de l’expression des glycoprotéines plaquettaires par cytométrie en flux effectuée à l’age de 03 ans
a montré une diminution importante de l’expression du CD41b (GPIIb) et du CD61 (GPIIa) en faveur
d’une thrombasthénie de Glanzmann de type I (sévère) selon la classification de Nurden. Les tests effectués
chez les parents retrouvent le même profil.
Discussion : La thrombasthénie de Glanzmann est un syndrome rare de prévalence inconnue. Sa sévérité est variable
fonction de l’ampleur de déficience des plaquettes des protéines en glycoprotéines plaquettaires.
Trois types de sévérité croissante sont rapportés selon la classification de Nurden et all :
Type I : taux < 5% de la normale
Type II : taux entre 5 et 20%
Type III : anomalies qualitatives avec taux sub-normal.
Conclusion : L’étude de l’expression des glycoprotéines par cytométrie en flux est indispensable pour confirmer le
diagnostic de thrombasthénie de Glanzmann et évaluer son pronostic et son mode de transmission.
28.Adrenoleucodystrophie liée à l'X :A propos de deux frères
Metidji Bouchra,Hamid Boufenar,Malika Agha,SE Laalaoui.Hôpital central de l'Armée ALGER
L'adrenoleucodystrophie liée à l'X (ALD)est une maladie neurodégénérative sévère liée à l'X,elle associe
une démyélinisation progressive du SNC et périphérique,une insuffisance surrénale et une accumulation
d'acides gras à très longue chaîne(AGTLC) dans le plasma,les fibroblaste et les tissus.il existe une forme
cérébrale qui touche les garçons entre 5 et 12 ans ,parfois les hommes adultes et une forme limitée à une
atteinte de la moelle épinière (adrenomyéloneuropathie) qui atteint les adultes entre 20et50 ans.L'ALD est
due à une mutation du gène ABCD1 codant pour une protéine peroxysomiale de transport.
Le diagnostic repose sur le dosage des AGTLC dans le plasma.
Nous rapportons les cas de deux frères issus d'un mariage consanguin ,la fratrie est composée de 3 frères,il
n'y a pas d'antécédents familiaux pathologiques.
Le premier cas concerne le plus jeune, chakib, 7ans,qui présente depuis une année des troubles visuels,une
diminution de son rendement scolaire associés à des troubles du comportement et lors d'une crise
convulsive,une IRM cérébrale est réalisée objectivant des lésions de la substance blanche,l'association

66
d'une insuffisance surrénalienne biologique nous fait évoquer l'ADL et l'augmentation des AGTLC
confirmait le diagnostic.Le dépistage systématique de ses frères ,zakaria 16 ans et mohamed 20 ans ,alors
asymptomatiques
objectivait une atteinte cérébrale sans insuffisance surrénalienne chez le premier et un bilan normal chez le
deuxième. Chakib est décédé 3 ans aprés et zakaria présente actuellement des signes neurologiques .La
greffe de moelle osseuse est un moyen thérapeutique intéressant à condition qu'elle soit réalisée avant le
premier signal de la substance blanche.
Tout l’intérêt du dépistage et de la surveillance de toute insuffisance surrénalienne isolée.
29.L’ostéopétrose infantile maligne : une maladie rare mais de diagnostic facile !
Bencharif Madani Tahar, H Boudbaba , NH Salmi, W Nasri,A Rachoui , H Belabed
Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine
Introduction : L’Ostéopétrose ou « maladie des os de marbre » est un groupe hétérogène de maladies héréditaires rares lié
à un défaut de résorption osseuse par anomalies de différenciation ou de fonctionnement des ostéoclastes et
ayant en commun une densification osseuse radiologique. L’ostéopétrose infantile maligne de transmission
autosomique récessive étant la forme la plus sévère et la plus précoce de cette maladie.
Objectif : A travers cette observation descriptive et une revue de la littérature, nous rappelons la présentation clinique
et surtout radiologique de l’ostéopétrose infantile maligne.
Observation : Nous rapportons le cas d’un nourrisson âgé de 7 mois admis dans un tableau d’anémie et de thrombopénie
sévère et présentant une dysmorphie faciale, un retard staturo-pondéral et une hépato-splénomégalie, le
diagnostic d’ostéopétrose a été facilement porté devant la constatation d’une densité osseuse exagérée sur
les radiographies standards. Le début précoce de la symptomatologie et la notion de consanguinité
orientaient vers la forme sévère de la maladie.
Conclusion : De présentation clinique sévère et précoce, l’ostéopétrose infantile maligne est facilement diagnostiquée
par une simple analyse de radiographies osseuses.
30.Céphalées mode de révélation de la maladie de behçet : à propos d’un cas Hellal M,Djebri H,Mesri I,Selmi H.Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine
Introduction :
La maladie de Behçet est une vascularite primitive non nécrosante multi systémique, Décrite comme “une
aphtose buccale, génitale et une uvéite à hypopion”, d’étiologies inconnues. Son diagnostic est purement
clinique, reposant sur la constatation en l’absence d’autres explications, d’aphtes buccaux, récidivant plus
de 3 fois par an, associés à 2 autres manifestations parmi les aphtes génitaux ou des lésions oculaires ou
cutanées. Il n’existe aucun critère biologique d’appoint. Le groupage du système HLA n’a qu’un intérêt
épidémiologique. Elle évolue par poussées, capricieuses et imprévisibles, sans parallélisme entre les lésions
cutanéomuqueuses et viscérales.
Observation : Nous rapportons le cas d’un garçon âgé de 12ans, aux antécédents de convulsion fébrile à l’âge de 9 mois,
admis dans un tableau de céphalées frontales intenses rebelles évoluant depuis plus de 03 mois, associé à
des aphtes endo-buccaux multiples et récidivants, une hyper sensibilité au test cutané ( pseudofoliculite aux
points de piqure ) , une Vs accélérée, typage HLA B5( 51) avec absence d’Anticorps anti nucléaires et anti
phospholipides, un œdème papillaire stade 2 bilatéral au FO et une thrombophlébite cérébrale diffuse à
l’IRM, devant ces résultats le diagnostic de la maladie de Behçet a été retenu.
Conclusion : La maladie de Behçet est une affection multi systémique, ses complications neurologiques sont
polymorphes, dominées par la méningo-encéphalo-myélite et les thrombophlébites cérébrales superficielles
et profondes qui sont classiquement de mauvais pronostic tant sur le plan vital que fonctionnel, Elles
doivent être prise en charge précocement afin d’éviter les séquelles motrices et cognitives.

67
31.Mucoviscidoses difficulté de prise en charge: à propos de 3 cas Helala Fairouz ,Jabri Hana,Kerdoudi Sameh.EHS Sidi Mabrouk Constantine
Introduction : La mucoviscidose est une maladie génétique autosomique récessive, liée à des mutations du gène CFTR
situé sur le chromosome 7 ; elle est caractérisée par la production de sécrétions muqueuses trop épaisses.
Observations : Nous rapportons 3 cas de mucoviscidoses, dont les signes révélateurs sont typiques mais les évolutions
cliniques sont différentes.
1èr cas : un garçon de 3 mois présentant une détresse respiratoire sifflante depuis l’âge de 2 mois, une
insuffisance pondérale sévère, et des troubles de la ventilation à la radiographie du thorax;
2ème cas : une fille de 8 mois, présentant une insuffisance pondérale sévère, une diarrhée chronique, et une
détresse respiratoire fébrile.
3ème cas : une fille de 4mois, présentant une détresse respiratoire sur une bronchiolite sévère récidivante.
Ils sont issus de couples non consanguins, leurs tests de sueur sont fortement positifs, et la recherche de la
mutation génétique est en cours.
L’évolution du 1er nourrisson est marquée par des infections pulmonaires récidivantes et un gain pondéral
difficile. Celle du 2éme nourrisson est favorable tant sur le plan respiratoire que sur le plan pondéral. Par
contre celle du 3éme nourrisson a été rapidement fatale.
Discussion : Nos cas illustrent les difficultés thérapeutiques et le retard diagnostique de cette maladie qui devait être
dépistée en période néonatale
Conclusion : La mucoviscidose est une maladie fréquente dont on ignore l’incidence exacte dans notre pays. Son
pronostic est lié à la précocité et la sévérité de l’atteinte respiratoire.
32.La coarctation de l’aorte thoracique.À propos d’un cas diagnostiqué à l’âge de 13 ans
Boulmaiz FZ,Bendaas N,Keghouche S.Service de pédiatrie du Mansourah (EHS sidi mabrouk) de
Constantine
Introduction : La coarctation de l’aorte représente 6% des cardiopathies congénitales dont le diagnostic est facile et
repose sur l’asymétrie pulsatile et tensionelle des pouls des membres inférieurs et supérieurs, toutefois le
diagnostic anténatal est possible et les cas découvert chez le grand enfant ne sont pas rares.
Objectif : Rapporter un cas de coarctation de l’aorte thoracique dans sa forme inhabituelle, révélé par une
hypertension artérielle diagnostiquée à l’âge de 13 ans.
Observation : Garçon âgé de 13ans admis pour des lipothymies dont l’examen clinique a objectivé un souffle systolique
4/6 ème au foyer tricuspide irradiant en sous Clavière gauche, aux vaisseaux du cou et au dos en para
vertébrale gauche sous scapulaire, pouls radiaux bondissants, pouls fémoraux faibles et pédieux absents et
une HTA menaçante.
Devant ce tableau clinique la coarctation de l’aorte est fortement suspectée, confirmé par une radiographie
du thorax qui montre des encoches costales multiples, un amortissement du flux au niveau des artères
rénales à écho doppler rénal et une échocardiographie qui confirme la coarctation isthmique serrée, dont
l’importance est estimé à l’angioscanne à plus de 95% avec important réseaux de suppléance mammaire
interne et intercostale associe à une hypertrophie du ventricule gauche.
L’enfant a bénéficié d’une réparation chirurgicale de la coarctation avec une bonne évolution post
opératoire
Conclusion : La coarctation de l’aorte est généralement une cardiopathie simple de diagnostic, de traitement facile et de
pronostic excellent après réparation. Ce ci n’est toutefois pas la règle.

68
33.La scoliose secondaire chez l’enfant : à propos de 46 cas.
Chadda B,Baaziz H, Albane B.CHU Batna
Introduction : La scoliose est une déformation tridimensionnelle du rachis qui touche 3 à 5% des enfants. Elle est souvent
idiopathique mais elle peut être secondaire dans 20 à 30% des cas. La scoliose secondaire est due,
essentiellement, à des pathologies neuromusculaires, congénitales et dysplasiques. Un diagnostic précoce
permet d'éviter les complications d'ordre esthétiques, fonctionnelles et psychologiques.
Objectif : Nous rapportons une série de cas de scoliose secondaire pour montrer les pathologies qui donnent
cette déformation et l’intérêt de la rechercher précocement.
Patients et méthodes : Nous avons, rétrospectivement, colligé les cas de scoliose secondaire chez les
enfants adressés à la consultation de médecine physique et réadaptation entre le 01 janvier 2010 et le
31décembre 2012.
Tous les enfants ont bénéficié d’un examen clinique et d'une radiographie du rachis en totalité face et profil
en position debout, une IRM rachidienne a été pratiqué uniquement pour les enfants ayant des formes
atypiques ou malformatives.
Résultats : Sur un total de 110 cas de scoliose, 46 sont secondaires (41,81%). L’âge moyen est de 6ans et 9 mois
(04mois-16ans. Les causes dysplasiques sont les plus fréquentes, retrouvées dans 45.65% des cas (21 /46),
les causes congénitales sont incriminées dans 36.96% (17 /46) et les pathologies neuromusculaires dans
17.39% des cas (08/46).la scoliose est isolée dans 39 cas et combinée à une cyphose dans 07 cas.
La kinésithérapie vertébrale et respiratoire a été pratiquée chez 27 cas, la natation a été indiquée pour 30
cas et le corset pour 14 cas. Tous les malades ont bénéficié d’une surveillance clinique et radiologique
périodique. Le traitement chirurgical est indiqué chez un seul cas après échec du traitement orthopédique et
déféré chez 05 cas vue la stabilité de la courbure, ce qui permet le maximum de croissance.
Conclusion : L'examen du rachis doit se faire systématiquement chez tous les malades qui présentent des
pathologies génératrices de la scoliose.
34.Syndrome de Silver Russell à propos d'un cas
Lacete F,Guedouar A,Merzouk A,Lebied A.CHU Parnet
Introduction : Le syndrome de Silver Russell est une maladie génétique rare sa prévalence dans le monde est de
1/100.000 caractérisée, par une grande diversité phénotypique les signes cliniques majeurs sont retard de
croissance intra utérin, retard staturo-pondéral postnatal, dysmorphie crânio-faciale et une hémi-
hypertrophie corporelle.
Objectifs : Discuter à travers ce cas les aspects cliniques du syndrome de Silver Russell pour pouvoir le reconnaitre.
Matériels et méthodes : Nous rapportons le cas d’un petit garçon issu d’un mariage non consanguin aux antécédent de retard de
croissance intra-utérin, un retard staturo-pondéral et dysmorphie crânio-faciale caractéristique, une hémi-
hypertrophie corporelle et des taches cutanées café au lait.
Résultats et discussions :
Le diagnostic chez le malade à été retenu par la coexistence des trois signes majeurs sus cités
Le diagnostic différentiel se pose devant les causes environnementales et génétiques du retard staturo-
pondéral éliminées par les signes caractéristiques existants chez notre malade.
Un interrogatoire détaillé, examen morphologique et clinique complet permettent généralement d’en faire
le diagnostic.
Sur le plan génétique c’est une pathologie de l’empreinte parentale
Conclusion : Le syndrome de Silver Russell est une maladie génétique rare, son diagnostic est essentiellement clinique.

69
35.Invagination intestinale aiguë révélant une maladie coeliaque:A propos d'un cas.
Baghdali N ,Khelafi N, Aggoune S, Hadji A, Sokhal S,Mertani A, Keddari M.
Clinique médicale infantile,CHU Mustapha,Alger.
Introduction : L’invagination intestinale aigue (IIA) de l’enfant est le plus souvent idiopathique. La maladie cœliaque
(MC) en est rarement la cause, mais représente l’un des facteurs favorisants.
Objectif : Nous rapportons le cas d’un nourrisson dont le diagnostic de MC a été porté au décours d’une IIA
spontanément régressive .
Observation : Soundous, 2 ans, est hospitalisée pour diarrhée chronique glairo-sanglante , associée à une cassure de la
courbe pondérale. Puis apparaissent vomissements et douleurs abdominales.
L’examen clinique révèle une pâleur cutanée, un pli de déshydratation avec amaigrissement de 2 kg.
L’abdomen est ballonné, douloureux.
Le bilan biologique objective une anémie microcytaire hypochrome, une hypoalbumi-némie avec
hypogammaglobulinémie.
Les anticorps de la MC sont positifs. Le dosage des immunoglobulines E spécifiques du lait de vache est
positif .L’échographie abdominale révèle un aspect en cocarde caractéristique.
Le diagnostic de MC est confirmé par la biopsie jéjunale objectivant une atrophie villositaire totale.
Après réhydratation ,antibiothérapie, l’enfant est mis sous régime sans gluten (RSG).
L’évolution ultérieure a été favorable. l’IIA a été résolutive sans récidive avec prise pondérale .
Commentaires : L’IIA dans le cadre de la MC a été décrite chez l’adulte en 1967.Sa description est plus récente chez
l’enfant.
Dans notre observation, les éléments en faveur du diagnostic étaient l’aspect échographique typique avec
résolution spontanée.
La particularité des invaginations dans le cadre de la MC est leur caractère récidivant.
Conclusion : Le caractère atypique voire récidivant d’une IIA peut participer au diagnostic précoce de MC, le RSG évite
les récidives d’invagination.
36.Rachitisme carentiel a début néonatal :a propos d’un cas Talbi F, Kherra S, Benalioua H,Abba K,Trabelsi F CHU Parnet
Bensenouci A .Service de pediatrie B CHU Beni Messous
Le rachitisme carentiel est un syndrome résultant d’un déficit de minéralisation du tissu pré-osseux
nouvellement formé de l’organisme en croissance et du a une carence en vitamine D, mais fort
heureusement la situation s’est améliorée depuis ces dernières années et la fréquence du rachitisme a
nettement diminué grâce a la prévention par l’administration systématique de vitamine D à tous les
nourrissons à 1 mois et a 6 mois
Nous vous rapportons le cas d’un nouveau né de sexe masculin âgé de 21 jours de vie admis pour prise en
charge d’un état de mal convulsif.
Il est issu d’un mariage non consanguin, grossesse mal suivie menée à terme, la mère rapporte la notion de
paresthésies au niveau des membres supérieurs non explorées (vit D et calcium non reçu durant la
grossesse)
Condition sociaux économiques médiocres.
A son admission le nouveau né présentait des convulsions à type de ramage aux membres supérieurs et
pédalage aux membres inferieures, clignement palpébral le tout dans un contexte apyrétique , n’ayant pas
répondu aux anticonvulsivants.
Un bilan métabolique fait retrouvant une hypocalcémie sévère a 41 mg/l confirmée par une deuxième

70
calcémie 45 mg/l, (arrêt des convulsions après correction de la calcémie par voie intra veineuse),
phosphatase alcaline élevée et le dosage de 25(OH) D effondré a 10 nmol/l.
Radiographie du poignet gauche met en évidence une discrète déminéralisation épiphysaire
Calcémie et dosage de vit D chez la mère sont diminués.
Le diagnostic de rachitisme néonatal (début précoce) retenu sur
Les éléments anamnestiques : Condition sociaux économiques médiocres
Symptomatologie maternelle
Calcium et vit D non reçu chez la mère
Les éléments cliniques et biologiques: Etat de mal convulsif
Calcémie effondrée
Dosage de 25(OH) D diminué
Radiologie
Dosage de la calcémie maternelle et de la vit D diminués
Conclusion:
Le rachitisme a début néonatal du a une carence maternelle en vit D est une forme rare , il en découle que
l'apport vitaminique D doit être adéquat chez la femme enceinte, non seulement pour le fœtus mais aussi
pour elle, idéalement 1 000 UI par jour durant le 3e trimestre, ou à défaut une dose de 100 000 UI au 6e et
8e mois ou une dose unique de 2 à 300 000 UI au 6e mois (100 000 UI est une posologie insuffisante).
37.Le syndrome d’hyper IgD: A propos d’un cas
Aichaoui H,Zennaki A, Naceur M, Niar S, Bouziane Nadjadi K
Service de pédiatrie « C » .Clinique Amilcar Cabral CHU d'Oran
Introduction : Le syndrome d’hyper IgD de cause probablement génétique et de transmission autosomique récessive, se
caractérise par des manifestations paroxystiques périodiques débutant dès le jeune âge et évoluant
jusqu’après l’âge de 50 ans, associant une fièvre constante avec des manifestations digestives, articulaires,
cutanéo-muqueuses et des adénopathies.
Le but de notre travail :
Faire connaitre cette maladie à travers notre observation
Observation : Il s’agit d’un enfant de sexe masculin, dont le diagnostic de l’hyper IgD a été posé à l’âge de 7 ans.
Le tableau clinique se résumait à une fièvre périodique depuis l’âge de 2 mois, qui dure 3 à 7 jours associée
à des adénopathies cervicales, douleurs abdominales et arthralgies, parfois une éruption cutanée et
splénomégalie. Ce tableau sans diagnostic précis, régresse spontanément avant même le début d’un
traitement antibiotique. Le malade est asymptomatique en dehors de ces poussées.
Ce sont les complications post opératoire inhabituelles d’une appendicectomie avec l’échec aux traitements
antibiotiques qui ont fait pousser l’investigation; le dosage sérique et urinaire de l’acide mévalonique a
posé le diagnostic positif.
Le malade a bénéficié d’un traitement spécifique.
Conclusion : Devant un tableau de fièvre périodique, le syndrome de l’hyper IgD est envisageable. Le diagnostic positif
est difficile, le traitement est lourd, mais le pronostic est bon puisque les crises ne laissent aucune séquelle
ou complication à long terme, et ne modifient pas l’espérance de vie
38.Syndrome d’activation macrophagique (SAM)
Benmouffok N . Benmoffok N , Boukhedouma N , Boubidi N,Gacem O, Lebied A.Service pédiatrie A
Hopital N. Hamoud
Introduction : Le syndrome d’activation macrophagique (SAM) ou (SALH) hémophagocytose lymphohistiocytaire est
une entité clinico-biologique qui constitue un ensemble de signes cliniques et biologiques qui sont le
conséquence d’une production non régulière de cytokines (orage cycokinique) aboutissant à une activation

71
et à une prolifération lymphocytaire et histiocytaire bénigne. Maladie rare mais probablement sous estimée
car non de diagnostiquée. Il n’ya pas d’âge de prédilection (étiologie change avec l’âge). Au japon 51 cas
de SAM par an (adultes, et enfants). En Algérie aucune étude épidémiologique n’a été faite à ce jour. Le
tableau clinico-biologique est peu spécifique d’où le retard diagnostique et la mortalité élevée. La prise en
charge est jusqu’à ce jour mal codifiée, aucune étude thérapeutique n’a été réalisée en dehors du cadre des
hémophagocytoses héréditaires de l’enfant.
Objectif : Notre étude a pour objectif d’évaluer les signes cliniques et biologiques les plus fréquemment rencontrés et
de les comparer à la littérature. De faire ressortir les étiologies les plus fréquentes.
D’évaluer la prise en charge ainsi que le délai diagnostic.
Matériel et méthodes : - Etude rétrospective : du 01 Janvier 2011 au 31 Décembre 2012 ou 09 cas ont été colligés au niveau du
service de pédiatrie « A » CHU Pr. N. HAMOUD (ex. Parnet). 07 SAM secondaires et 2 primitifs. L’âge de
nos patients varie de 3 mois à 12 ans. Diagnostic est retenu sur les critères révisés de Henter 2004.
Commentaires et résultats de l’étude : Le SAM est une entité clinique et biologique qui n’est pas rare puisque nous avons colligé 09 cas, en une
année, sa gravité est lié à la pathologie sous jacente et le délai diagnostic. Le début est souvent brutal
dominé par une altération fébrile de l’état général quasi constante (100%) dans notre série.
L’organomégalie (hépato splénomégalie) est retrouvé dans 90% des cas. Les signes neurologiques 3 fois 9.
La biologie est peut spécifique mais très contributive. L’Hémogramme a montré une bicytopénie dans
100% des cas. Le fibrinogène est diminué 3 fois 9 associe à TP bas, la CIVD est retrouvé dans chez 3
patients. La ferritinémie est bon marqueur de l’activité SAM 8 fois 9. Quant à l’épreuve histologique est
confirmée chez tous les patients. Le pronostic est meilleur dans la série des SAM secondaires cependant il
est sombre dans les SAM primitifs (100% de décès).
Conclusion : Le syndrome d’activation macrophagique est une complication rare potentiellement mortelle. Le traitement
est mal codifié, bien qu’il repose en grande partie sur le traitement étiologique. Il faut y penser devant toute
enfant qui présente une altération de l’état général fébrile avec organomégalie. Entamer rapidement une
enquête étiologique exhaustive et réunir les autres critères diagnostiques.
39.Histiocytose Langerhansienne :A propos de 7cas
Boukhedouma Nabila ,N.Benmouffok , O.Gacem , A.Guedouar , C.Boubidi , F.Lacete , A. Lebied.Service
pédiatrie A Hopital N. Hamoud
Introduction :
L’histiocytose langerhansienne est une maladie rare, multi systémique, liée à l’accumulation de cellules de
Langerhans, au sein de différents organes. Son incidence annuelle est estimée à 4,6 cas/1000.000 ce qui
représente 55 nouveaux cas / an.
Sa physio pathogénie reste encore mal connue.
Elle se caractérise par un polymorphisme clinique ce qui rend le pronostic très varié.
Le diagnostic est histologique et immunohitochimique (CD1a)
Matériel et méthode : Il s’agit d’une étude rétrospective dans notre service sur une période de 2 ans (octobre 2011 – Octobre
2013) qui a intéressé des enfants moins de 15 ans.
Résultats : Nous avons colligé 7 cas d’histiocytose Langerhansienne. On a noté une légère prédominance masculine 4
garçons et 3 filles avec un sexe ratio de 1,33.
L’âge au diagnostic s’étend de 2 mois à 13 ans avec un âge moyen de 2,8 ans.
Les formes cliniques sont variées allant de la forme localisée à des lésions généralisées, respectivement :
- 1 granulome éosinophile
- 2 cas d’atteinte osseuse multifocale
- 4 cas d’atteinte multi viscérale (litterer siwe)

72
Le diagnostic est posé par immunohistochimie dans 5 sur 7 cas.
Le traitement proposé chirurgie dans un cas, protocole LCH III dans les autres cas.
L’évolution est émaillée de survenue de syndrome d’activation macrophagique chez 5 sur 7 cas dont un
décès.
Commentaires :
Actuellement le diagnostic pose peu de problèmes, vu la disponibilité de l’immuno-marquage.
Le pourcentage élevé des formes multi viscérales chez le nourrisson mettant en jeu le pronostic vital pose
un problème thérapeutique, d’autant plus qu’on ne dispose pas de biomarqueurs de suivi de l’activité de la
maladie.
Conclusion :
La sévérité de la maladie tient à l’agressivité des formes hématologiques chez le très jeune enfant.
Bien que des progrès aient été réalisés dans la compréhension de sa pathogénie, la méconnaissance de
celle-ci limite les capacités d’optimiser l’approche thérapeutique.
40.PB/PC et status nutritionnel en pediatrie :" Résultats d'une analyse d'une enquête
anthropométrique dirigée"
Amireche F,Safi H ,Allas H:Service pédiatrie du Mansourah de Constantine
L’évaluation de l’état nutritionnel et son evolution sous traitement constitue une preoccupation essentielle
dans la prise des enfants denutris, notamment dans les pays en voie de developpement ou la malnutrition
demeure un probleme crutial de sante publique.
Les mesures anthropometriques restent, de notre temps, parmi les elements essentiels dans l’ observation d’
un statut nutritionnel. une place toute particuliere a ete reservee a l’ appreciation du perimetre brachial ( p.b
) et partant de l’ evaluation du pb / pc. cet indicateur propose en 1970 par mac-laren et kanawati a le merite
d’ etre un critere anthropometrique simple, applicable aux premieres annes de la vie ( 3 mois – 4 ans ),
independant de l’age et du sexe et ne necessitant aucun appareillage sophistique ni recours a des tables
anthropometriques.
Cependant la determination de seuils pour cet indice anthropometrique destines a orienter les enfants vers
des programmes adaptes est une operation tres difficile necessitant encore de nombreuses recherches. il
faut aussi souligner qu’aucune explication physiopathologique plausible n’a ete fournie pour la validation
des differents seuils preconises.
Pour repondre a cette attente pediatrique, il nous a paru utile d’analyser ce precieux parametre lors d’une
enquete nutritionnelle menee a constantine aupres d’un echantillon de 2397 enfants sains entre 0 et 60
mois. dans cette etude nous avons evaluer ce nouveau critere en fonction de l’age et du sexe puis le
comparer aux indicateurs biometriques classiques.
41.Cystinose infantile :à propos d’une observation
Aggoune S , Baghdali N ,Belbouab R, Noumi M, Hadji A, Sokhal S , Keddari M, Mertani A,Keddari M.
Clinique médicale infantile CHU Mustapha
Introduction La cystinose est une maladie métabolique familiale rare récessive autosomique, caractérisée par
l'accumulation de cystine dans les lysosomes des cellules de la plupart des organes. Elle réalise, dans sa
forme infantile de loin la plus fréquente, le type le plus complet et le plus grave d'insuffisance tubulaire
proximale. Son incidence a été récemment estimée à 1/200 000 naissances, en France à 1/167 000
naissances récemment.
Observation : Nous rapportons le cas clinique d’un petit nourrisson âgé actuellement de 6ans, qui a été admis à l’âge de
3ans pour prise en charge d’un retard staturo pondéral compliquée d’un rachitisme sévère développé en
dépit d'une prévention correcte.
L’examen clinique retrouve un nanisme harmonieux chez un enfant blanc aux cheveux clair cendré, il

73
existait une glycosurie massive avec normo glycémie, une protéinurie de type tubulaire rentrant dans le
cadre d’un syndrome de Toni Debrè Fanconi.
L’examen ophtalmologique était normal au début, ce n’est que vers l’âge de 5ans qu’on retrouvait des
dépôts cornéens de cystine avec début de photophobie.
Le tableau clinique s’est compliqué d’une hypothyroïdie, ainsi que d’une insuffisance rénale sévère malgré
une supplémentation en eau et électrolytes et l’usage d’indométacine au tout début de la maladie.
Le dosage de cystine intra leucocytaire était positif, et le malade est mis depuis sous cystéamine sous forme
de bitartrate (Cystagon®) à raison de 50mg/kg/j en 4 prises journalières.
Conclusion : Le diagnostic de cystinose doit être porté précocement devant une hypotrophie associée à des signes
urinaires malgré l’absence d’atteinte oculaire au début, bien que ce diagnostic se trouve heurté a la
réalisation du dosage lui-même chez nous d’où le retard à l’instauration du traitement.
Cette observation soulève aussi la gravité de cette maladie et l’évolution rapide vers l’insuffisance rénale
d’où l’importance du diagnostic et du conseil génétique.
42.Exophtalmie bilaterale revelant une leucemie aigue lymphoblastique. a propos d’un cas.
AL.Khelil1, A.Boumeddane1, A.Mécifi1, K, Chachou 1, A Meksi1, Z.Taghezout1, K.Mahmoudi 2.
1 - Service d’oncologie pédiatrique, Etablissement Spécialisé en Oncologie
EHS Emir Abdelkader Oran ; 2- Service d’ophtalmologie EHS pédiatrique Boukhrofa Aek Canastel Oran.
Introduction :
Dans le cadre des localisations extra-médullaires des leucémies aigues, l’atteinte oculo-orbitaire se situe au
troisième rang après les localisations méningées et testiculaires. Il s’agit de complications oculaires en
rapport avec l’insuffisance médullaire et l’hyperviscosité sanguine, elle pose un problème diagnostique et
pronostique.
Matériels et Méthodes : À travers cette observation, nous présentons le cas d’une exophtalmie bilatérale ayant révélé une leucémie
aiguë lymphoblastique chez un nourrisson.
Observation : Il s’agit d’un nourrisson de sexe masculin âgé de 13 mois, hospitalisé pour une exophtalmie bilatérale non
inflammatoire, associée à une paleur cutanéo muqueuse.
La tomodensitométrie oculo-orbitaire confirme l’exophtalmie, et montre une infiltration musculaire
orbitaire. L’hémogramme a retrouvé une anémie, une thrombopénie, une hyperleucocytose avec présence
de blastes, et le myélogramme a confirmé le diagnostic de leucémie aiguë lymphoblastique de type L2 avec
envahissement à 75% par des blastes. Le patient a bénéficié d’une chimiothérapie avec une bonne réponse
hématologique et une régression de l’exophtalmie
Discussion : L’atteinte orbitaire inaugurale dans les leucémies aigues peut prêter à confusion avec d’autres affections
orbitaires notamment les autres étiologies d’exophtalmie douloureuse et/ou inflammatoire. Le diagnostic
histologique est difficile, d’où l’intérêt de la réalisation de l’hémogramme et du myélogramme qui
permettent un diagnostic et un traitement précoce : chimiothérapie L’atteinte orbitaire constitue souvent un
facteur de mauvais pronostic.
Conclusion : L’atteinte orbitaire reste une localisation peu commune de la leucémie aiguë, qui doit inciter à la réalisation
d’un bilan hématologique devant toute exophtalmie en vue d’un diagnostic et d’une prise en charge précoce
afin d’améliorer le pronostic.

74
43.La dermatomyosite juvenile (à propos d'un cas)
Abdelbaki S,Kendouci-taniF ,Elmezouar C,Kaddour A,Massen Z.Service de pédiatrie EHS mére-enfant
Tlemcen
Introduction : La DMJ maladie orpheline ,associe cliniquement un syndrome cutané ,musculaire et général . nous
apportant un cas de DMJ colligé dans le service de pédiatrie EHS Tlemcen (MARS 2013).
Objectif : souligner l’importance de l’examen clinique dans l’orientation diagnostique et le caractère
orphelin de la maladie .
Patient et méthodes : Garçon de 12 ans présentait depuis 02 mois avant son hospitalisation ,un érythème facial des deux joues et
du nez ,des nodules rouges violacés en regard de la face d’extension des IPP et MCP, une faiblaisse
musculaire proximale des deux ceitures : pelvienne et scapulaire avec œdème des quatre membres plus
accentué au niveau du membre supérieur gauche .
CPK :2050 UI/L ,LDH :814 UI/L , ALDOLASE :24 UI/L
Bilan auto-immun :négatif
L’EMG : atteinte axonale motrice avec traçés myogènes diffus
L’EFR :légères anomalies restrictives
Une corticothérapie à 2 mg /kg/j peros donnée pendant un mois permettait une régression des signes
cliniques et biologiques
La kinésithérapie passive et douce à la phase aiguë,puis active réhabilite le malade par des exercices
modérés.
La photoprotection est recommandée.
44.Syndrome de Zellweger : à propos d’un cas
Boucenna H,Chaou.M, Khati.A, Tari.S, Atek.L,Khiari ME.Service de pédiatrie A, CHU Benimessous, Alger
Le syndrome de Zellweger (syndrome cérébro-hépato-rénal) est une maladie rare du métabolisme
peroxysomal liée une mutation au niveau du gène PEX1. Il en résulte une accumulation de métabolites
toxiques et des altérations dans le développement des cellules neurales.
Observation : Nous rapportons le cas de N.B, âgée de 5 ans, suivie depuis la période néonatale pour hydrocéphalie
biventriculaire, retard psychomoteur, hospitalisée pour pris en charge d’une insuffisance rénale chronique.
Le diagnostic du syndrome de Zellweger est évoqué devant le faciès caractéristique, l’atteinte neurologique
et rénale, confirmé par la présence de la mutation PEX1 à l’étude génétique.
Conclusion : Le diagnostic du syndrome de Zellweger est essentiellement clinique (un bon indice) : un faciès aplati, une
fontanelle antérieure large, des sutures écartées, un front proéminent et haut placé, un occiput plat, des
fentes palpébrales orientées en haut et en dehors, une arrête nasale large, un épicanthus et des rebords
supra-orbitaux hypoplasiques, une macrocéphalie ou une microcéphalie, un palais ogival, une protrusion
linguale ou une micrognathie.
L’étude génétique à la recherche de la mutation PEX1 permet de confirmer le diagnostic.
45.Ecthyma gangénusum a propos d'un cas
Boulahlib F, Belkadi S.W. E.P.H Zighout Youcef Ténès chlef
L’ecthyma gangénusum est une infection cutanée rare et invasive habituellement associé à une septicémie,
causée dans la majorité des cas par pseudomonas aérugénosa. Les lésions caractéristiques de l’ecthyma
Gangrenosum sont des pustules ou des vésicules hémorragiques qui évoluent vers des ulcérations
nécrotiques avec des bords érythémateux.
Nous aborderons le cas d’un nourrisson atteint d’un ecthyma Gangrenosum sans septicémies avec
seulement des lésions ulcéro-nécrotique, la culture et l’examen direct des prélèvements de ces lésions
révèle la présence de pseudomonas aérugénosa, la patiente à très bien répondue à une double
antibiothérapie associant amikacine et imipénème et facteur de croissance épidermique humain.

75
46.Rachitisme vitaminorésistant hypophasphatémique révélateur d’une tyrosinémie héréditaire de
type1
Baouia S,.Koumni N , Chikhi S, Benalikhoudja N ,Mertani A, Keddari M
CMI, CHU Mustapha Pacha, Alger-centre.
Introduction : Le rachitisme vitaminorésistant (RVR) hypophosphatémique se caractérise par une hypophosphatémie avec
troublemajeur de la réabsorption tubulaire de phosphate,il peut être associé ou non à une tubulopathie.
Observation :
Nous rapportons le cas de la fillette H.Z âgée de 4 ans issue d’un mariage consanguin, aux antécédents
d’une tyrosinémie héréditaire de type 1 (HT1) chez la sœur.L’enfant s’est présentée à la consultation avec
signes cliniques de rachitisme :genu valgum bilatéral ;pas d’hépato-splénomégalie ni retard staturo-
pondéral. La radiographie standard retrouvait des signes nets de rachitisme.La biologie objectivait une
hypophosphatémie à 32[N:40-72mg/L],une tubulopathie :PH
urinaire=7,glucosurie(+),proteinurie(+)=0.14g/L,une réabsorption tubulaire des phosphsphates(RTP) très
diminuée à 15.18[N:85-90%];La fonction hépatique ainsi que la crase sanguine étaient normales .Sur les
antécédents familiaux de HT1,le dosage du succinylacétone urinaire était fait avec un taux à 47[N:0-
10µmol/l],la tyrosinémie à 130[N:10-30mg/L]et un taux d’?-foetoproteine était élevé à 500 U/ml. le
diagnostic de HT1 est retenu.
Discussion : La HT1 est une affection rare liée à un déficit en fumarylacétoacétate hydrolase(FAH),de transmission
autosomique récessive.Dans sa forme chronique et non traitée on peut trouver une tubulopathie avec fuite
continue rénale de phosphate laquelle est responsable du RVR hypophosphastémique comme nous avons
constaté chez notre patiente chez qui la HT1 n’est découverte que tardivement par RVR.
Conclusion : La HT1 reste une maladie rare,à évoquer devant un RVR hypophosphatémique avec tubulopathie,d’où
l’intérêt du dépistage précoce chez les familles à risque afin d’ éviter les complications invalidantes de la
maladie.
47.Anémie de biermer : cause possible d’anémie normocytaire chez l’adolescent
Noumi M,Aggoune S,Berkani R ,Ladj S ,Terrak R,Hassoum,.Mertani A ,.Keddari M
Clinique médicale infantile CHU Mustapha
La maladie de biermer est une maladie auto-immune rarement décrite en pédiatrie, sa présentation
hématologique est classiquement macrocytaire avec un volume globulaire moyen (VGM) supérieur a 100
fl.
Chez l’enfant, une carence martiale peu être associe, responsable alors d’une errance diagnostique, Nous
rapportons l’observation d’une adolescente de 14 ans ,aux antécédent d’anémie ferriprive a âge de 02 ans
,de syndrome néphrotique a âge de 9ans et de pyélonéphrite a âge de 13 ans ,admise pour l’exploration
d’anémie profonde normocytaire normo chrome a régénérative ,avec un syndrome neurologique fait de
fourmillement des membres supérieur et paraplégie flasque évoluant depuis 09 mois .
L’HB a 06 g/dl, fer sérique et ferritinémie abaissées, le taux de vitamine B12 <100ng/l(160-970ng/l),la
recherche d’anticorps antifacteurs intrinsèques et d’anticorps anticellules pariétales est positive ,taux de
folates est normale.
La FOGD avec biopsie révèle une muqueuse gastrique fundique siège de lésion de gastrite chronique avec
helicobacter pylori(++) confirmant ainsi le diagnostic de maladie de biermer.
La supplémentation martiale orale et vitamine B12 parentérale a permet une correction en quelque mois le
taux HB et du VGM, par contre on note une amélioration que partiel des signes neurologiques.
Le bilan thyroïdien et la glycémie sont normaux et l’examen clinique ne retrouve pas de signes orientant
vers une maladie auto-immune associée.

76
48.Place de la génétique moléculaire dans le diagnostic des SMAs.
(Spinal Muscular Atrophy) :(A propos d’un cas SMA1).
Delileche H ,Bendjamaa A,Himmi A,.Aissat A ,Semid T , Boukhenfouf N.
La SMA (Spinal Muscular Atrophy) appelée aussi amyotrophie spinale proximale est une maladie rare
neurodégénérative caractérisée par une paralysie musculaire progressive due à une dégénérescence des
motoneurones du cortex moteur primaire.
On distingue 3 types chez l’enfant selon l’âge et le mode de début à gravité décroissante.
(Le type 4 concerne l’adulte)
Observation : Nous rapportons le cas d’un nourrisson âgé de 4 mois et demi adressé de l’hôpital de Reggane pour
complément diagnostic d’une hypotonie.
La mère constate une hypotonie avec diminution des mouvements des quatre membres surtout inferieurs et
une constipation chronique. Elle consulte chez un pédiatre qui après examen clinique, neurologique, bilan
biologique complet, sérologie rubéole et toxoplasmose, bilan thyroïdien et scanner cérébral, conclue à une
hypotonie par atteinte de la corne antérieure de la moelle épinière, pour cela un EMG et Biopsie musculaire
sont demandés d’où son admission.
L’anamnèse objective :
- Des Parents consanguins 1er degrés
-Frère décédé dans un tableau neurologique par détresse respiratoire.
-Cas similaire dans la fratrie.
L’examen clinique objective un enfant eutrophique avec distension abdominale importante.
A l’examen neurologique on note :
- Nourrisson éveillé, cris faible, mimique normale
- Hypotonie axiale et segmentaire (images affichées)
- Mouvements spontanés faibles présents au niveau des membres supérieurs, absents au niveau des
membres inferieurs avec abolition des Reflexes ostéo-tendineux
-Pas d’amyotrophie, ni de déformation, sensibilité superficielle et profonde conservée.
CPK normal : 135 UI/L (N < 190 UI/L).
EMG : Atteinte neurogéne mais surtout myogène intéressant les muscles proximaux et distaux.
Diagnostic suspecté devant : le début avant l’âge de 6 mois, l’amyotrophie, l’aréflexie, le déficit musculaire
à prédominance proximale, enzymes musculaires non élevées et l’atteinte myogène à l’EMG, confirmé par
l’étude génétique qui met en évidence une délétion homozygote de l’exon 7 du chromosome 5 (5q-SMA).
Prise en charge : Orthopédique, nutritionnelle, support psychologique et conseil génétique.
Conclusion -LA SMA type 1est une maladie à transmission autosomique récessive par délétion homozygote ou
conversion de l’exon 7 du gène SMN1.
-l’examen clinique, l’EMG et l’étude génétique suffisent à poser le diagnostic.
-La biopsie n’a aucun intérêt dans le diagnostic des SMAs.
-Le Diagnostic prénatal est possible ainsi que l’étude génétique de la parenté.
-Aucune thérapeutique curative n’est disponible à ce jour. Seule une Prise en charge Pluridisciplinaire est
possible (Orthopédique, Respiratoire, Cardiaque, Nutritionnelle)

77
49.Déficit constitutionnel en facteur VII : à propos d’une observation
Amrouni L,Bencheikh F, Kerboua S.EPH Okbi, Guelma
Introduction : Le déficit constitutionnel en facteur VII (FVII) est une maladie hémorragique héréditaire autosomique
récessive due à la diminution ou l'absence du facteur VII. C'est une pathologie rare et d'expression clinique
est très variable. L’objectif de notre travail est de déterminer l’intérêt d’un dépistage systématique des
troubles de la coagulation avant toute intervention chirurgical afin d’éviter des complications
hémorragiques pouvant être grave.
Observation : Nous rapportons l’Observation d’un enfant de cinq ans, sexe masculin, sans antécédents particuliers, lors
d’un bilan préopératoire pour une circoncision, nous avons noté un taux de prothrombine bas alors que
Temps de céphaline kaolin était normal. Le dosage de facteurs de coagulation a montré un taux bas de
FVII.
L’enfant a été opéré sous couverture du facteur VII, sans complication hémorragique .
Discussion.
Le déficit en facteur VII est une pathologie rare, avec une présentation clinique très polymorphe,mais la
sévérité du syndrome hémorragique n’est pas corrélée aux taux résiduels en facteur VI. La prévention des
complications hémorragiques pouvant survenir en cours d'intervention chirurgicale repose d’abord sur le
dépistage de la pathologie par le bilan de la coagulation, et surtout la perfusion de facteur VII.
Conclusion. Le déficit en facteur VII peut, dans sa forme sévère, engager le pronostic fonctionnel et même
vital. Il importe de dépister les troubles de la coagulation avant toute chirurgie ; Nous insistons sur
l’importance du dépistage des troubles de la coagulation, ainsi que l’enquête familiale et sur le conseil
génétique chez les parents d’un enfant atteint.
50.Enfants de pierre :Fibrodysplasie Ossifiante Progressive (FOP) : A propos de 2 cas
Dris LN,Djenane H1 ,Smail S1, Smati L1 , Douiri D1 ,Baiod S1 , Bendaoud M2, Dahou N3 , Baghriche
M1.
1Service de pédiatrie – EPH Bologhine Ibn Ziri – Alger
2Service de radiologie – EPH Bologhine Ibn Ziri – Alger
3Service de rhumatologie – CHU BEO – Alger
La FOP est une affection rare du tissu conjonctif avec une fréquence de 1 cas pour 2 millions.
1ère observation Chahd, 03 ans, 5 /6 enfants consulte pour hématomes multiples des membres inferieurs après des
traumatismes minimes.
L’examen outre les hématomes retrouve de multiples masses solides non mobiles au niveau des jambes et
des cuisses sans retentissement fonctionnel, à noter un hallux valgus des pieds.
Après élimination d'un trouble de l'hémostase, l'imagerie des masses retrouve au niveau des membres
inferieurs des calcifications de type lamellaire d’allure symétrique au niveau des parties molles des cuisses
et en amont du tibia, sans atteinte articulaire ou osseuse réalisant l’aspect de bandes d’os corticalisé.
2ème observation Akram, 09 ans 3/3 consulte à l’âge de 05 ans pour masse dorsale douloureuse. On retrouve une tuméfaction
douloureuse de la face postérieure du cou et du de l’épaule gauche, avec limitation des mouvements du
membre supérieur. La radiographie retrouve des ossifications ectopiques aux niveaux axillaire et dorsal ;
des mobilisations des articulations douloureuses ainsi qu'un un hallux valgus des pieds.
En raison des ossifications hétérotopiques ,le diagnostic de FOT est évoqué.
Discussion La FOP qui résulte d’une métaplasie osseuse du tissu interstitiel des muscles est une maladie soit génétique
autosomique dominante soit cas sporadiques (les plus fréquent).
Le diagnostic est aisé car aspect de squelette ectopique, sans nécessité de recourir à la biopsie pouvant
aggraver les lésions. La progression des ossifications va entraîner une perte de mobilité, prédisposant si
atteinte temporo-mandibulaire à des troubles de la déglutition.

78
L'iconographie disponible dans ces cas est caractéristique de l'affection.
Le mécanisme d’ossification enchondrale qui caractérise la FOP est secondaire à une dysrégulation de
BMP 4 (Bone Morphogenic Proteine) et des axes de recherche thérapeutique sont en cours.
Conclusion Le diagnostic de la FOP affection rare mais grave est radiologique. La prise en charge reste symptomatique
(rééducation douce) et préventive (éviter les traumatismes des tissus mous ) d’où l’intérêt du diagnostic
précoce et la lutte contre les facteurs aggravants.
51.Erythromelalgie ; difficultés de prise en charge .
Hamouda N,Bioud B, Dehimi A. , service depédiatrie CHU de Sétif.
L’érythromelalgie est une affection dont la prévalence est estimée à 0,36 / 100 000 habitants , elle est
caractérisée par une triade permettant de poser le diagnostic, cette triade est faite de sensations de brulures
des extrémités avec un érythème et une chaleur , primaire ou secondaire dont la prise en charge reste
complexe .
Nous rapportons le cas d’une fille K.Salsabil âgée de six ans pour souligner la difficulté de prise en charge
de cette pathologie , il s’agit de la patiente K.Salsabil sans antécédents familiaux ni personnels particuliers
admise à notre niveau pour des douleurs invalidantes aux extrémités chez qui le diagnostic fut porté vu la
présence de la triade sus citée ,notre recherche étiologique menée dans ce sens ne retrouve aucune étiologie
concluant ainsi à une érythromelalgie primitive après un traitement inefficace à base d’aspirine puis de B
bloquant et vu la détérioration clinique faite de douleurs invalidantes et macération , une instauration d’un
traitement à base de carbamazepine et anti dépresseur imipraminique s’est révélée très bénéfique avec une
réponse spectaculaire . l’érythromelalgie reste une pathologie rare dont la prise en charge reste mal
codifiée.
52.La leptospirose : une cause rare de défaillance multiviscerale
(A propos d’un cas)
Zerrouki N, Ibsaine O, Drali O, Zeroual S, Boutaba M , Berrah H. CHU H dey
La leptospirose est une maladie infectieuse grave et rare provoquée par une bactérie spiralée, du genre
Leptospira qui vit essentiellement parmi les rongeurs. L’atteinte multiviscérale (rénale et pulmonaire) peut
engager le pronostic vital.
Observation : Nous vous rapportons le cas d’un garçon âgé de 14 ans admis pour une fièvre évoluant depuis 5 jours
associé à un ictère avec urine foncées, selles décolorées et hémoptysie. A l’admission, l’enfant était fébrile
à 39,5°, se plaignant de myalgies diffuses. L’examen clinique retrouvait un ictère cutanéo-muqueux franc,
un escare noiratre au niveau de la jambe droite et une hepathomégalie. L’exploration a revélé sur le plan
biologique, une hyperleucocytose à 11 500 elts/mm3 associée à une thrombopénie à 36 000 elts/mm3, un
bilan hépatique perturbé (syndrome de cytolyse modéré SGOT 128 UI/L SGPT 62 UI/L, un syndrome de
cholestase Bilirubine totale 111 mg/L Bil libre 27 mg/l directe 24 mg/l) sans insuffisance hepathocellulaire,
une protèinurie au labstix et une insuffisance rénale (urée 2,50 g/L créatinémie 84 mg/L). Les hémocultures
ne retrouvaient pas de germes. La radiographie du thorax notait une pneumonie interstitielle disséminée. La
sérologie de l’hépatite virale A et l’Ag HBs était négatifs. Le diagnostic de leptospirose a été évoqué
devant la notion de baignade en eaux douce 4 jours avant le tableau clinique, confirmé que 3 semaines plus
tard par une sérologie. L’évolution était favorable sous traitement antibiotique.
Conclusion : La leptospirose est une maladie de diagnostic difficile en raison du polymorphisme clinique et l’absence de
tests biologiques rapides et fiables.

79
53.Un cas de méningite tuberculeuse chez un enfant vacciné Ait Kaci N ,Meftah A, Mechtoub F-Z, Mohamed Y.EPH Boufarik
Introduction
La survenue d’une méningite tuberculeuse chez un enfant vacciné par le BCG ne remet pas en cause le
bien-fondé de cette vaccination, offre une efficacité plus de 50%. La présentation clinique peut être très
variable, revêt parfois des tableaux trompeurs, pronostic réservé par sa mortalité et séquelles invalidantes.
Observation
Nous rapportons un cas de méningite tuberculeuse confirmée sur la culture de LCR chez un enfant de 5
ans, sans antécédent médical. Correctement vacciné avec présence d’une cicatrice de BCG, pas de notion
de contage tuberculeux. Début des manifestations une semaine avant son hospitalisation par de la fièvre,
céphalées, vomissements, douleurs abdominales, sans amélioration sous antipyrétique. A l’examen état
général moyen, fébrile à 39,1 °c, bonnes constantes hémodynamiques, pas de syndrome méningé ni
troubles neurologiques. PL: LCR clair 464 éléments /? 100%lymphocytaire glucorrachie 0,37g /l, pas
d’anomalie à l’hémogramme VS 23/50, téléthorax et scanner cérébrale sans anomalie, sous céftriaxone
enfant reste fébrile céphalalgique, état général altéré, délirant, agité, confus, devant l’aggravation
neurologique une 2ème TDM et IRM cérébrale montrant une hydrocéphalie tétraventriculaire, 2ème PL :
LCR clair 117 éléments /? 80%lymphocytaire glucorrachie 0,15g albuminorrachie 0,68g /l ; devant ces
éléments clinico-biologues et radiologique un traitement antituberculeux associés à une corticothérapie est
entamé avec pose d’une sonde ventriculo-péritonéale à 3semaines des antituberculeux.
Conclusion La méningite tuberculeuse n'a pas disparu, même chez les enfants vaccinés par le BCG, reste une forme
sévère de la maladie. Des techniques comme la PCR ou l'IRM contribuent à un diagnostic précoce.
54.Hyperthroidie autoimmune de l’enfant (a propos de deux observations) Kerboua ,Amrouni L, Boulares F Z .EHS EL Bouni, Annaba
Introduction : L'hyperthyroïdie auto-immune est une maladie rare et sévère chez l’enfant qui résulte de la stimulation du
récepteur thyréotrope par des autoanticorps. L’objectif de notre travail est de préciser la difficulté de la
prise en charge en âge pédiatrique, en rapportant deux observations d'hyperthyroïdie auto-immune.
Observations: Deux filles âgées respectivement de 7 et 8 ans, présentent un amaigrissement avec hyperexcitabilité et
troubles du sommeil, associés à des palpitations et une exophtalmie bilatérale symétrique chez l’une d’entre
elle. Un goitre diffus est retrouvé dans les deux cas. Les examens biologiques retrouvent une anémie
hémolytique dans un cas, avec une hyperthyroïdie et positivité des anticorps anti thyroglobulines.
Les antithyroïdiens de synthèse et les bétabloquants ont été prescrits chez les deux patientes, des
corticoïdes et une transfusion sanguine ont été pratiqués dans un cas.
Commentaire: L'hyperthyroïdie auto-immune est 08 fois plus fréquente chez le sexe féminin. Elle débute rarement avant
l’âge de 10 ans. Elle peut être associée à d'autres pathologies auto-immunes telles que L’anémie
hémolytique auto-immune dans 50 % des cas, sa prise en charge reste un sujet de controverse et la durée
optimale du traitement médical pour induire une rémission de la maladie, reste à définir.
Conclusion:
L’hyperthyroïdie de l’enfant peut s’associer à d'autres pathologies auto-immunes, sa prise en charge est
améliorée par l’utilisation d’un score pronostique avec une adaptation de la durée du traitement en fonction
des caractéristiques des patients au diagnostic.
Mots clés :
Hyperthyroïdie, Anémie auto-immune, Diagnostic, Thérapie.

80
55.Facteurs influençant la durée de l’allaitement maternel
Mazari W,Bouriche.K ,Snouci.D ,Bendeddouche .S .Service de pédiatrie-CHU Tlemcen
Tandis que divers actions de santé publique nationale ou locale sont entreprises pour améliorer le taux et la
durée d’allaitement maternel en Algérie nous avons conduit une étude afin de décrire les facteurs associés à
sa pratique à la maternité de l’hôpital « Mère –Enfant » de Tlemcen, Algérie.
Méthodes : Cette étude prospective descriptive portant sur les nouveau-nés de maternité de l’hôpital « Mère –Enfant »
de Tlemcen, Algérie (exclusion des grands prématurés).
Résultats: femmes incluses dans l’étude : 470,césarienne : 9%, poids normal : 70%,Multiparité:60%, niveau
universitaire :15% , préparation à l’accouchement 5% ,préparation à l’allaitement maternnel : 45% . Le
taux d’allaitement à 1mois : 60% , 6 mois : 50% femmes sans niveau d’instruction (70 et 61 %), femmes
habitant en zone rurale (78 et 67 %), la cause de l’arrêt est le tarissement du lait dans 35% , et la prise de
contraceptifs dans 12%.
Discussion: Le taux d’allaitement au sein à 1mois est comparable pour les primi et pour les multipares ( OR :0,87 [
0,50- 1,48],Il n’y a pas une différence statistiquement significative entre celles les femmes de niveau
l’instruction supérieur ( OR : 0,88 [ 0,62-1,24] par rapport à un niveau plus bas),l’âge ? à 30 ans de la mère
(OR :1,25 [ 0,28- 5,2 ] et le milieu rural ( OR : 1, 3 [ 0,18- 9,10 ] sont liés à la pratique de l’allaitement
maternel.
A 6 mois la pratique de l’allaitement maternel est trois fois plus élevée chez les femmes ayant un niveau
d’études primaire (OR : 3,11 [8 10-6 – 11,21 5] par rapport à un niveau supérieur),la différence est
nettement significative en ce qui concerne la multiparité ( OR : 1,40 [ 0,13 – 14,31] et pour les femmes de
plus de 30ans (OR : 1,25 [ 0,42- 3,66]
La pratique de l’allaitement en Algérie est voisine de celle des autres pays arabe avec un taux de 93% en
Algérie, 90% en Soudan, 94% en Libye, et 91% au Yémen .
56.Aspects épidémiologiques et prise en charge du diabète de type I dans un service de Pédiatrie
générale – Tlemcen 13.000 - Algérie Mazari .W,Snouci.D ,Bouriche.K ,Bendeddouche .S .Service de pédiatrie-CHU Tlemcen
Introduction : Nous assistant actuellement une véritable épidemie du diabète type 1 notamment dans les pays en voie de
développement ,justifiant l’étude de nouvelles approches thérapeutiques.
Materiels et methodes :
Etude épidémiologique rétrospective sur une période de trois ans du 01/01/2008 au 31/12/2010 sur des
enfants diabétiques hospitalisés au service de Pédiatrie de l’hôpital « mère- enfant » -Tlemcen- Algérie et
suivis en consultation spécialisée,
Schéma utilisés :
- Schéma 1 : mélange d’insuline rapide et intermédiaire ou une d’une insuline analogue rapide + analogue
intermédiaire : une injection le matin et soir
- Schéma 2 : schéma 1 + une injection d’analogue rapide à midi
- Schéma 3 : avec 4 injections : trois injections d’analogues rapides aux moments des repas + une injection
d’analogue basal le soir ou le jour
Résultats : 142 dossiers d’enfants diabétiques entre 1 et 15 ans ont pu être exploités sur une durée de 3 ans :
Sexe ratio : garçon / fille = 1.27
Age moyen de découverte 6,5 ans
Antécédents familiaux de diabète présents dans 71% des cas
Circonstances de découvertes : 74% acidocétose, 24% syndrome polyuro-polydyspsique
Insulinothérapie :
- Schéma 1: 18%,- Schéma 2 : 6% ;- Schéma 3 : 76%
Complications :
- Accident hypoglycémique dans 43 % ;- Rétinopathies 2.3 % ;- Néphropathies 0.7 %

81
Maladies associés : thyroïdite auto-immune : 6.2 %, maladie cœliaque : 1.4%
Équilibre glycémique en fonction de l’Hb glyquée :- Schéma 1 : 9.05%- Schéma 2 : 8.55 %
- Schéma 3 : 9.43 %
Discussion : - L’âge moyen de survenu, le sexe ratio et les circonstances de découverte sont comparables à toute les
études retrouvées dans la littérature.
- Notre étude démontre que le schéma 3 est le plus utilisé avec un équilibre ( HbA1C moyenne : 9.81%),
alors que le schéma 2 donne un meilleur équilibre glycémique avec une: HbA1C moyenne de 8.55 %.
Prédominances d’un IMC chez la plus part des petits diabétiques
Proportion non négligeable des enfants ayant une diététique inadéquate 70% ;
Les sites d’injections préférentiels restent les avant bras et la face externe des cuisses
Importance des complications aigues notamment les hypoglycémies majeurs.
Presque la moitié des enfants ont un suivi médical anarchique.
57.Hémophilie A : un mode de révélation exceptionnel Mohand-Oussaid Aïda,Boukhellal-Djemoi Houria, Chaou Mériem, Lamrani Fella, Khiari Mohamed El-
Mokhtar.Service Pédiatrie A - CHU Béni Messous
Introduction : L'hémophilie A est une coagulopathie congénitale secondaire à un déficit en facteur VIII plasmatique,
transmise sur le mode récessif lié à l'X. Toutefois, dans un tiers des cas, elle peut être diagnostiquée chez
des patients sans aucun antécédent. De plus, bien que rare, l'hémophilie peut être révélée à tout âge par une
hémorragie grave pouvant mettre en jeu le pronostic vital et donc nécessiter un traitement approprié en
urgence.
Objectif : Illustrer par une observation clinique un mode de révélation exceptionnel d'une hémophilie A chez un
nourrisson jusque-là bien portant.
Observation
Nous rapportons le cas d'un nourrisson, âgé de 04 mois, sans antécédents familiaux ou personnels
d'hémophilie, qui est amené en urgence, suite à la constatation par ses parents d'une pâleur. Lors de
l'examen en salle de consultation, les médecins notent la présence d'un volumineux hématome en regard de
la région scapulaire droite, associé à une pâleur intense mal tolérée qui a nécessité une réanimation
hématologique avant réalisation d'un bilan d'hémostase puis de coagulopathie, qui ont confirmé le
diagnostic d'hémophilie A.
Conclusion : Les patients présentant une hémophilie jusque-là méconnue, consultent le plus souvent à l'occasion
d'accidents hémorragiques graves qui mettent en jeu leur pronostic vital. Un retard diagnostique va dès lors
entraîner un retard dans la mise en route d'un traitement efficace et ciblé avec des conséquences
potentiellement catastrophiques.
58.Fistule coronaro-camérale (A propos d’01 cas exceptionnel)
Boufenar H,Metidji B, Agha M , Ait-idir K , Laalaoui S-E.Service pediatrie HCA
La fistule coronaro-cardiaque est une communication anormale entre une artère coronaire (droite+++) et
une cavité cardiaque ou avec la circulation systémique ou pulmonaire, la coronaire gauche est rarement en
cause. Elle représente 0,4% des malformations cardiaques. La clinique est orientée par un souffle méso
cardiaque continu, elle peut se compliquer d’insuffisance cardiaque congestive, de rupture ou de
thrombose, d’HTAP (shunt GD) et d’endocardite infectieuse. Le diagnostic est fait par échographie-doppler
cardiaque, le coroscanner et la coronographie sélective sont nécessaires à l’orientation diagnostique et
thérapeutique. La fermeture spontanée est rare, le traitement est chirurgical (ligature simple sous CEC) ou
embolisation par cathétérisme cardiaque.
Nous rapportons le cas d’une forme anatomique exceptionnelle de communication entre la coronaire
gauche et le ventricule droit chez un nourrisson de sexe masculin, âgé de 02 mois, bien portant, ou la
découverte fortuite d’un souffle cardiaque continu a motivé une exploration. Radiographie thoracique et
ECG sans particularités. L’echocardio-doppler a montré une coronaire gauche très dilatée, (16mm à son

82
origine et 09mm de diamètre ensuite), se drainant à l’apex du ventricule droit sans anomalies de naissance
des coronaires. Le scanner cardiaque a confirmé l’anomalie, son diamètre, son caractère non compliqué et
son trajet tortueux. La stratégie thérapeutique était basée sur une surveillance régulière avec abstention
médicale. Une embolisation percutanée à un âge adéquat (06mois ou plus) est de règle en absence
d’indication chirurgicale urgente.
59.Hypoglycémie néonatale persistante...pourquoi?
Moussaoui Y, Arrada Z, Oucif Z,Saadaoui,.Rezzaz N,Berrah H .CHU Parnet
Introduction : L’hypoglycémie constitue une urgence métabolique néonatale le plus souvent transitoire.
Dans certains cas rares, elle est persistante et réfractaire aux traitements classiques, orientant vers d’autres
étiologies plus complexes, dont l’hyperinsulinisme reste la cause la plus fréquente.
Le diagnostic doit toujours être évoqué précocement du fait de sa gravité, et le traitement doit être instauré
rapidement.
Observation : Farah nouveau né de sexe féminin, issue d’un couple consanguin, aux antécédents familiaux d’une sœur
unique suivie pour hyperinsulinisme primaire congénital diagnostiqué tardivement après séquelles
neurologiques dues aux différents états de mal convulsif par hypoglycémie en périodes néonatale,
hospitalisée à l’âge de 48 heures pour hypoglycémies précoces, sévères, réfractaires d’horaire anarchique
mais asymptomatique.
Le diagnostic d’hyperinsulinisme congénital est alors évoqué devant la consanguinité, l’antécédent
familial, les caractères de l’hypoglycémie, la macrosomie (poids de naissance à 5kg500) ainsi qu’une
épreuve positive au glucagon. confirmé par plusieurs insulinémies normales qui étaient comprises entre 63
et 74pmol/l , dont une concomitante à une hypoglycémie à 0,25g/l.
Le traitement par Diazoxide (25 mg/kg) a été sans résultats, la somatostatine (40µg/kg) à été alors indiqué
avec stabilisation des chiffres glycémiques.
Conclusion : L’hyperinsulinisme est une cause rare d’hypoglycémie néonatale, méconnu de pronostic péjoratif, avec
possibilités de séquelles neurologiques graves.
Farah, actuellement âgée de 3 mois, contrairement à sa sœur a bénéficié d’un diagnostic et d’une prise en
charge précoce, ayant bien évoluée sous traitement médical.
Le PET scan, étape cruciale pour préciser la forme topographique (localisée ou diffuse) qui aura un impact
thérapeutique majeur dans la prise en charge ultérieure (éventualité d’un acte chirurgical dans la forme
localisée).
L’étude génétique permis dans certaines situations de préciser la forme topographique et des formes
familiales sont décrites.
60.Le syndrome catastrophique antiphospholipide: a propos d'un cas
Rezak R,Bouchetara A,Azzouz S M,Hamiti B. EPH Canastel Oran
Introduction :
Le syndrome des antiphospholipide (SAPL) est une maladie auto-immune définit par l’association de
thromboses et la présence d’auto-anticorps antiphospholipes ; sa forme rare et gravissime qui est le
»syndrome catastrophique « appelé syndrome d’asherson., conduit à une défaillance multiviscérale,
compromettant le pronostic vital ; c est le cas de notre observation
Observation :
la patiente A Nawel âgée de 12 ans suivie a notre niveau pour maladie cœliaque ,est hospitalisée dans un
tableau de malnutrition severe avec infection grave :malade opérée d’un plastron appendiculaire compliqué
d’un sepsie de la paroi , et en arrêt de régime sans gluten depuis l’intervention !!
Le diagnostic du syndrome des antiphospholipes était retenu sur :la présence du thrombose veineuse
cliniquement et biologiquement par la positivité des auto-anticorps antiphospholipides type AC
anticardiolipine a deux reprises et a 6semaines d'intervalle.
La forme catastrophique de ce syndrome retenu sur la présence des thromboses étagées étendues cavo-ilio-

83
fémorales et femoro-poplitées et une atteinte cérébrale type AVC ischémique.
Malgres la réanimation hématologique , nutritionnelle ; métabolique et la thérapie spécifique qui a fait
appel a la corticotherapie et les anti-coagulants; la malade est décédée suite sa complication neurologique
Resultat :
le syndrome antiphospholipidique peut etre primaire mais il est souvent secondaire a une maladie auto-
immune type LED ( AC antinucléaire négatif dans notre cas), chez notre patiente le syndrome était
secondaire a une infection grave sur suite post-operatoire défaillante.Malgres le traitement etait défavorable
par l'apparition du syndrome catastrophique.
Conclusion :
le syndrome catastrophique des antiphospholipides est exceptionnel chez l’enfant, ; il complique moins de
1% du syndrome antiphospholipide, et il peut etre inaugural et revelateur du SAP dans environs 50%. ,
malgres les progres thérapeutique le deces survient dans plus de 30% des cas .
Savoir penser a ce syndrome devant toute thrombose chez un enfant avec ou sans terrain auto-immun est
l'objectif de notre présentation.
61.Coma néonatal révélant une citrullinemie de type I : à propos d’un cas
Benmati F,.Messak W ,Boudhar K, Benhafssa L. HCA
Introduction: La citrullinemie de type I est une erreur innée du métabolisme par déficit en une enzyme du cycle de l’urée.
Son risque principal est l’évolution vers une encéphalopathie hyperammoniémique pouvant conduire au
décès. Une fois le diagnostic posé, une stratégie thérapeutique doit être rapidement mise en œuvre afin d’en
améliorer le pronostic.
Matériel et méthode : K .Nadir 1er enfant d’un couple non apparenté, né à terme, admis à J7 de vie pour exploration d’un coma
d’étiologie inexpliquée .A son admission Nadir est somnolent, hypotone, ses réflexes archaïques sont
absents. Aucune convulsion n’est constatée. Il est polypneique mais n’est pas en détresse respiratoire. Sa
glycémie est dans les normes ainsi que sa natrémie et sa calcémie. L’exploration neuroradiologique est sans
anomalie .L’étude des gaz du sang met en évidence une alcalose respiratoire. On retrouve une
Ammoniémie > 700 ?mol/l.
Un traitement médicamenteux épurateur et vitaminique est instauré avec arrêt transitoire des apports
protidiques. A J7 d’hospitalisation nadir sort de son coma. L’Ammoniémie se normalise progressivement
en 9 jours .Nadir sort de son coma à J10 de vie.
Le diagnostic de déficit en ASS sera porté par la chromatographie des acides aminés.
Discussion :
La citrullinemie de type I est une maladie rare à transmission récessive autosomique. Elle est liée à un
déficit en ASS. La maladie s'exprime le plus souvent en période néonatale par un coma
hyperammoniémique. Parfois son mode de révélation est tardif avec anorexie, vomissements, hypotonie,
retard de croissance, retard psychomoteur et convulsions.Le diagnostic repose sur l'hyperammoniémie et la
chromatographie des acides aminés plasmatiques et urinaires qui montre une élévation majeure de la
citrulline, de la glutamine et de l'alanine, et un abaissement de l'arginine avec une acidurie orotique. Le
traitement consiste en un régime hypoprotidique, associé à une supplémentation en arginine, en benzoate et
phénylbutyrate de sodium. Le diagnostic anténatal est possible. Une enquête familiale doit être réalisée
étant donné le risque encouru en cas de transmission de la mutation.
Conclusion : Les déficits en enzymes du cycle de l’urée tel le déficit en ASS sont rares, mal connus et donc peu évoqués.
Le principal progrès à réaliser dans cette pathologie réversible en cas de prise en charge adaptée, est
d’évoquer le diagnostic dès que se présente une encéphalopathie d’étiologie indéterminée.

84
62.Maladie de Recklinghausen ou Neurofibromatose de type 1 : A propos de 2 observations et Revue
de la littérature
Drali O,Boulekhiout N, Zerrouki N,Zebdi N,Ghazoul A,Arrada Z, Berrah H
Service Pediatrie B CHU Hussein Dey
Introduction : La neurofibromatose de type 1 (NF1) est une maladie neurocutanée héréditaire multi systémique,
prédisposant au développement de tumeurs bénignes et malignes. L'expression clinique est très variable
Objectif :
Nous rapportant 02 cas de neurofibromatose de type 1 et en rappelons les éléments diagnostiques ,
thérapeutiques et pronostiques
Matériels et Méthodes :
Le premier cas est une fille âgée de 6 ans qui a consultée pour une toux chronique et des céphalées évoluant
depuis 2 mois .Le deuxième cas est une fille de 4 ans qui a consultée pour exploration de broncho-
pneumopathies a répétition
Résultats :l’examen clinique retrouve dans le premier cas 03 tuméfactions sous cutanée au niveau du dos et
de nombreuses taches café au lait au niveau des bras de dimension variable. L’échographie des parties
molles a objectivée 3 neurofibromes sous cutané.L’IRM cérébrale a révélée un hamartome du putamen
gauche.
Le deuxième cas, la patiente présentait des taches café au lait disséminées sur tout le corps et une
déformation du rachis dorsal a type de scoliose .La TDM thoracique a révélée la présence d’une hyper
clarté parenchymateuse au niveau du segment para cardiaque lobaire inférieur gauche associée a une
scoliose et un pectus excavatum. L’examen ophtalmologique a objectivé deux taches de lish spécifique de
la NF1 au niveau de l’iris
Conclusion : Le diagnostic de la neurofibromatose de type 1 de l’enfant est clinique ; le début est marquée par des taches
café au lait qui constituent la circonstance de découverte. Le traitement requiert une équipe
multidisciplinaire .
63.Nutrition de l'enfant d'âge scolaire
Amireche.F,Safi H,Allas H.Service Pédiatrie Mansourah Constantine
Les préoccupations sanitaires recentes relatives aux enfants scolarises nous interpellent et font l’objet
aujourd’hui d’une prise de conscience nouvelle de la part des educateurs et des professionnels de la sante.
Ces jeunes constituent sur le plan nutritionnel un groupe a risque en raison des modifications liees a leur
croissance et leur maturation notamment pubertaire chez les eleves de plus de 11 ans. En effet, a cet age de
la vie, le developpement staturo-ponderal peri-pubertaire est rapide et impressionnant, necessitant un apport
anabolique et energetique considerable, rendant l’organisme tres vulnerable a tout desequilibre alimentaire.
Il est a noter, par aileurs que cette tranche d’age represente une periode critique ou peuvent se developper
des fluctuations comportementales pouvant aboutir a des troubles de la conduite alimentaire,
particulierement chez les filles.
Dans la presente communication nous effectuerons une revue des methodes classiques et modernes
d’evaluation des etats nutritionnels, puis nous tenterons de preciser les apports nutritionnels et faire-part
des recommandations dietetiques utiles a cet age de la vie.

85
64.Maladie de Kawasaki : à propos de 6 cas
A L Boustil,H. Sehab1, N. Boukertouta2, W. Boutabia1, N. Bouchair1, M.C. Yaiche1.
1Service de pédiatrie, clinique Sainte Thérèse, CHU Annaba.
2Service de pédiatrie, EHS EL-BOUNI.
Introduction : La maladie de Kawasaki (MK) est une vascularite systémique des moyens vaisseaux qui touche le
nourrisson et le jeune enfant. D’étiologie inconnue, son diagnostic est clinique, et sa gravité réside dans le
risque d’anévrismes coronaires potentiellement mortels.
Objectifs :
Évaluer les caractéristiques épidémiologique, clinique, et évolutive de la MK.
Matériel et méthodes :
Étude rétrospective descriptive bicentrique (clinique Sainte-Thérèse et EHS EL-BOUNI) d’une série de 6
cas de MK hospitalisés entre le 1er janvier 2008 et le 30 septembre 2013.
Résultats : Six cas de MK ont été colligés (4 garçons et 2 filles), âgés en moyenne de 25 mois. Le délai moyen de
consultation était de 8 jours. Le motif de consultation était représenté majoritairement par la fièvre (5 cas).
Parmi les critères classiques de la MK, l’énanthème et l’exanthème étaient présent dans 5 cas, la
conjonctivite et la modification des extrémités dans la moitié des cas, l’adénopathie cervicale dans un cas.
La NFS avait retrouvé une hyperleucocytose, une thrombocytose et une anémie inflammatoire dans 5 cas.
Une élévation de la VS et de la CRP était constatée dans 4 cas. Un cas s’est révélé par une méningite
lymphocytaire aseptique. Deux cas se sont compliqués de dilatation anévrysmale coronarienne dont un
ayant évolué vers l’ischémie myocardique. Tous les cas avaient reçu 2g/kg d’immunoglobulines IV et 80
mg/kg/24h d’aspirine.
Conclusion :
La MK est probablement sous diagnostiquée en Algérie. Il importe de la reconnaitre, de la prendre en
charge précocement, et de la répertorier dans un registre national.
65.Syndrome hémolytique et urémique : à propos de 6 observations
Boustil Ahmed Lotfi,F. Layachi, Dj. Belamri, H. Sehab, N. Bouchair, M.C. Yaiche.
Service de pédiatrie, clinique Sainte Thérèse, CHU Annaba.
Introduction : Le syndrome hémolytique et urémique (SHU) associe une anémie hémolytique avec schizocytes, une
thrombopénie et une insuffisance rénale aigue.
Objectifs : Préciser la fréquence et les caractéristiques des formes de SHU observées dans notre service.
Matériel et méthodes :
Étude rétrospective descriptive ayant concerné les hospitalisations pour SHU entre le 1er juillet 2008 et le
31 juillet 2013.
Résultats : Six cas de SHU ont été colligés. La majorité d’entre eux (5 cas) étaient de sexe féminin et âgé de moins de
3 ans. Dans 5 cas le SHU a été précédé de prodromes digestifs associant diarrhée sanglante et
vomissements avec un intervalle moyen de 8 jours. A la phase aigue, la pâleur était omniprésente.
L’oligurie, l’hématurie et l’HTA étaient présente dans la moitié des cas. La NFS avait retrouvé une anémie
dans tous les cas avec un taux moyen d’hémoglobine de 6,4 g/dl, une thrombopénie dans 5 cas avec un taux
moyen de plaquettes de 39500 /mm3, et une hyperleucocytose dans 4 cas avec un taux moyen de globules
blanc de 17050 /mm3. La valeur moyenne de créatininémie était de 401 µmol/L. Quatre cas ont nécessité
une transfusion de culot globulaire, trois cas un traitement antihypertenseur, et deux cas une dialyse
péritonéale. L’évolution était favorable dans 4 cas. Un cas a évolué vers une IRC, et un autre est décédé.
Conclusion : Les formes de SHU typique (post-diarrhéique) sont les plus fréquentes. Elles sont possiblement secondaires
à une infection à E. Coli d’origine alimentaire ou environnementale.

86
66.Gastrite et Allergie aux protéines du lait de vache : à propos de 3 cas.
Boudbaba Hanane, T Bencharif Madani ,NH Salmi, W Nasri , A Rachoui
Service de pédiatrie du Mansourah (EHS sidi mabrouk) de Constantine
Introduction : L’allergie aux protéines de lait de vaches (APLV) dans sa forme non IgE-médiée est à l’origine de
nombreuses manifestations cliniques le plus souvent digestives. En dehors des formes classiques comme
l’entérocolite allergique et l’entéropathie chronique, d’autres symptômes digestifs non spécifiques peuvent
être secondaire à l’APLV. Dans ces cas, l’absence d’autres causes identifiées et surtout la mise en route
d’un régime d’épreuve permettent de les rattacher à cette pathologie assez fréquente.
Objectif :
Le but de ce travail est de démontrer la place de l’APLV dans les gastrites du nourrisson.
Matériels et méthodes : Observation descriptive de 3 cas et revue de la littérature.
Observation :
Nous rapportons 3 cas de gastrite liée à une APLV, le premier est celui d’une fille âgée de 4 mois
présentant une hématémèse et une fissure anale sans constipation associée, le deuxième est un tableau
d’hématémèse chez un garçon du même âge présentant par ailleurs une diarrhée chronique à début néonatal
et le 3ème cas est celui d’un nourrisson de 3 mois hospitalisé à plusieurs reprises pour hématémèse
récidivante et résistante au TRT anti-sécrétoire. Les 3 enfants ont bénéficié d’une fibroscopie digestive
haute revenant en faveur d’une gastrite. Un régime adapté est entrepris en association à un traitement anti-
sécrétoire. L’évolution était bonne pour les 3 enfants tant sur le plan clinique qu’endoscopique.
Conclusion : L’APLV doit être évoqué en premier lieu devant toute gastrite du nourrisson d’autant plus que cette
dernière est associée à d’autres symptômes digestifs inexpliqués.
67.Tératome gastrique mature : une tumeur rare
Ahmane H,Arhab D, Hamzaoui A, Chellah SA, idir S, Cherifi N, Aidrous L. Bensadi N
CHU Nedir Mohamed Tizi-Ouzou
Introduction :
Le tératome gastrique est une tumeur extrêmement rare en pédiatrie, ce qui représente moins de 1 % de
tous les tératomes diagnostiqués chez l’enfant. Patients et méthodes un cas de tératome gastrique est
rapporté chez un garçon de 6 mois.
Résultat :
nourrisson de 6 mois sans antécédents particuliers qui a présenté une hémorragie digestive faite de méléna
et d’hématémèse. Une pâleur cutanéo-muqueuse franche et une masse de l’hypochondre gauche sont
retrouvées à l’examen clinique. Sur le plan biologique, une anémie ferriprive profonde et un bilan
inflammatoire positif. Calcification de l’aire gastrique à l’ASP et volumineuse masse intra-péritonéale
inter-spléno-gastrique contenant des calcifications à l’échographie et la tomodensitométrie abdominale. Le
diagnostic de tératome gastrique mature a été retenu sur l’étude histologique de la masse gastrique de 10
cm de diamètre pesant 200grammes. Discussion : Actuellement seulement moins de 100 cas de tératomes
ont été rapporté dans la littérature. Le premier cas a été décrit en 1922 par Eustermann et al. La majorité
des tératomes gastrique sont exo-gastriques (60%des cas), des croissances tumorales endo-gastriques sont
présents dans 30% des cas,ceux à croissance mixte sont rares. Environ 85% sont des lésions matures bien
différenciés et environ 15% sont des tumeurs immatures ayant le potentiel de transformation maligne.
Conclusion :
Le tératome gastrique est une tumeur extrêmement rare de l'enfance et dans presque tous les cas c’est
bénin. l’exploration radiologique précise l'origine gastrique de la masse et permet ainsi d’exclure les autres
causes de masses abdominales rencontrées chez l’enfant.

87
68.Sténose bronchique congénitale: Un autre diagnostic de faux asthme
Boulekhiout Nadia,Drali Ourdia,Hadjarab Amina,Arrada Zakia,Berrah hassina
CHU Hussein-dey -service pediatrie"B"
Introduction: les malformations tracheo-bronchiques sont rares et peuvent simuler les manifestations de l’asthme
lorsqu’elles sont révélées par des sifflements récurrents ou encore une toux chronique.
Observation: Hadil, 8 ans ,née à terme d'une grossesse gémellaire,poids de naissance 2.9kg,aux antécédents de plusieurs
épisodes++ d'infection respiratoire avec une toux chronique productive et sifflements récurrents,mise sous
Budesonid et B2mimétiques puis arrêtée âpres une année car inefficace.A l'occasion d'un épisode de toux
(3ans) avec fièvre à 39° ,l'auscultation pulmonaire retrouve des râles bronchiques seulement à gauche ,à
droite l'examen est normal .
radiographie du thorax de face est normale.
L'endoscopie bronchique trouve une diminution de calibre de la bronche souche gauche sans fistule.
La TDM thoracique objective une sténose serrée de la bronche souche gauche.
L'angio-scanner a éliminé une anomalie vasculaire compressive.
Hadil a été opérée il ya 3 ans, le chirurgien a réséqué la zone sténosée avec anastomose termino-terminale.
Évolution:6 mois après reprise des symptômes,le scanner thoracique confirme la sténose au niveau de
l'anastomose.
Conclusion: les sténoses bronchiques congénitales sont des malformations rares,leur diagnostic doit être guidé par une
anamnèse et un examen clinique soigneux pour éviter les faux diagnostics.
Le traitement par voie endoscopique donne de bon résultats ,peut-être que Hadil pourra en bénéficier.
69.Croissance staturale de l’enfant atteint de Diabète de type 1 associé à la Maladie cœliaque
Bouziane-Nedjadi Karim ,Bessahraoui Mimouna,Reguig Ali,Niar Sakina,Naceur Malika
Aichaoui Amel,Touhami Mahmoud.Service de Pédiatrie C, 9 Amilcar Cabral 31000
Objectif :
Etudier les répercussions de l’association diabète de type 1 (DT1)-maladie coeliaque (MC) sur la
croissance staturale.
Patients et méthodes : Etude longitudinale de la croissance staturale des DT1-MC. Le diabète avait été
diagnostiqué avant l’âge de 15 ans. Les patients avaient été suivis du début du DT1 jusqu’à l’âge de 18 ans
au moins. La courbe de croissance staturale des DT1-MC avait été comparée à celle de sujets appariés MC
isolés, DT1 isolés, et témoins provenant des courbes de Sempé et Pédron.
Résultats :
163 DT1-MC ont été étudiés (78 garçons et 85 filles), avec un âge moyen de 8,77± 3,9 ans au diabète, et de
12,82±4,65 ans à la MC.
La taille moyenne au diabète était de -1,10 ±1,47 DS chez les garçons, et de - 0,98 ±1,37 DS chez les filles.
La taille moyenne à la MC était de -2,58 ±1,54 DS chez les garçons, et de - 2,15 ±1,31 DS chez les filles.
L’étude de la vitesse moyenne de croissance staturale avait montré chez les DT1-MC, l’absence
d’accélération en période pubertaire.
À 18 ans, la taille des garçons DT1-MC étaient de - 2,92 ± 1,85 DS (DT1 isolé : -0,86±1,09 DS, p<0,001),
( MC isolée : -0,99 ± 1,21 DS p<0,001).
À 18 ans, la taille des filles DT1-MC étaient de - 2,22± 1,54 DS (DT1 isolé : -0,68±1,10 DS p<0,001), (MC
isolée : -1,25±1,02 DS, p<0,001)
Conclusion :
Par rapport aux patients DT1 et MC isolés, les sujets DT1-MC avaient présenté un retard statural
significatif.

88
70.'' Primum Non Nocere ''...
Boutrid Nada,Rahmoune Hakim, Bouabdallah Saida, Belkacem Bioud
CHU Sétif
Introduction : Le valproate de sodium, le plus fréquemment utilisé des anti-épileptiques, peut s’avérer être un redoutable
hémorrhéique…comme le dénote notre cas.
Présentation du cas : Yacine, nouveau-né diagnostiqué précocément comme déficit sévère en facteur VII, présente, lors de la
dégression des doses de Novoseven® vers l’âge de 40 jours, une hémorragie cérbroméningée révélée par
des convulsions ;suite à quoi, il est mis sous Valproate (VPA) à raison de 20 mg/kg/j .
Dès lors, il présente deux autres épisodes suivis de deux saignement intra-péritonéaux malgré une
prophylaxie intermittente au Facteur VII.
La normalité de l’EEG demandé alors au 4e mois (conjuguée au bon développement psychomoteur) fait
interrompre le valrpoate.
Et ainsi,après un recul de 5 mois, il n’a plus refait pareille hémorragie grave.
Discussion : Les toxicités hématologiques du VPA sont bien reconnues : des coagulopathies induites fréquentes et
variables mais le plus souvent infra-cliniques.
Cette myriade va des troubles plaquettaires (thrombopathies-thrombopénies ) au déficit acquis du facteur
de von Willebrand,en passant par d'autres atteintes :protéines C et S, Anti-Thrombine III; et notamment le
Facteur VII...!!!
Heureusement, la majorité des enfants présentent une atteinte mineure et transitoire, et notre patient a bien
répondu à la suspension du VPA
Conclusion : Le valproate, communément utilisé chez l’enfant, n’est pas dénué de risque hématologique.
Son usage chez des sujets présentant déjà des coagulopathies doit être minutieusement pesé et
régulièrement revu.
71.Les dilatations des sténoses de l'œsophage chez l’enfant : à propos de 93 cas.
Benalioua H, Belbouab R, Khiati M . Hopital Belfort
Introduction :
Les sténoses de l'œsophage notamment celles secondaires aux lésions caustiques
sont fréquentes dans notre pays, leur traitement repose essentiellement sur les dilatations instrumentales
aux bougies de Savary-Gilliard.
L’objectif de ce travail est d’évaluer l’efficacité, les limites et les complications de ce type traitement dans
la prise en charge des sténoses de l’œsophage.
Matériels et méthodes : C’est une étude rétrospective qui concerne 93 enfants atteints de sténoses de l'œsophage, pris
en charge dans notre service (unité d’endoscopie digestive) entre février 2008 au février 2013 ayant
nécessité des dilatations endoscopiques .Age moyen est de 18 mois, les garçons sont plus touchés G/F : 1,7.
Les dilatations ont été réalisé en ambulatoire en salle d’endoscopie, après prémédication à hypnovel ® (
midazolame chlorhydrate ) et gardé en observation deux heures après la dilatation, le matériel utilisé était
dans tous les cas les bougies de Savary-Gilliard.
Nous avons analysé : les caractéristiques de la sténose, la cause, le nombre de séance de dilatations, les
incidents ainsi que l'évolution sous dilatations
L’évolution était jugée à la fin du programme de dilatations : l’enfant était considéré comme guéri lorsqu’il
arrive à s’alimenter librement sans dysphagie.
Résultats : 9 3 cas de sténose de l’œsophage ayant nécessité des dilatations endoscopiques ont été enregistrés dont la
nature était la suivante. :
64 sténoses caustiques, 13 sténoses peptiques, 12 sténoses anastomotiques (dont 8 atrésies de l’œsophage

89
opérées et 4 caustiques opérées) 02 sténoses congénitales, 02 sténoses secondaires à une épidermolyse
bulleuse.
79 malades ont terminés leur programme de dilatation et 14 sont toujours en cours de dilatation.
Les dilatations ont été réalisés avec succès chez 68 malades soit 86 %, les malades en échec de dilatation
(14 %) étaient tous de nature caustique et ont bénéficiés d’un remplacement œsophagien.
L évolution selon la cause de la sténose était la suivante :
- 07 sténoses anastomotiques sur atrésie de l’œsophage tous guéris (une moyenne 8 séances par malade)
- 0 4 sténose anastomotique sur remplacement œsophagien tous guéris (une moyenne de 10.5 séances par
malade)
- 12 sténoses peptiques opérées ont tous évolué favorablement (une moyenne de 5.5 séances par malade)
- 01 Sténose due à l’épidermolyse après 6 séances de dilatation.
- 02 Sténose congénitale, guéris avec une moyenne 04 séance par malade
- 53 cas de sténoses caustiques : 42 cas de succès soit 79 %, et 11 échecs soit 20 %, ces sténoses ont
nécessité une moyenne de 12 séances par malade
Les sténoses caustiques sont celles qui nécessitent le plus grande nombre de séances de dilatations (10 à 34
séances)
Les complications : on a enregistré 2 cas de perforation ayant concerné des sténoses caustiques, qui ont été
traité médicalement et ayant bien évolué , 2 malades ont eu une coudure du fil- guide en sous- sténotique
ayant nécessité un extrait chirurgical.
Conclusion : La dilatation instrumentale des sténoses de l’œsophage par les bougies de savary est une méthode efficace
chez l’enfant, de réalisation simple, et de cout faible.
.
72.Diabète transitoire induit par corticothérapie et L Asparaginase chez 4 enfants atteints de LAL H.
Mesbaieh, Guemghar, R. Nemmar, A.Lamraoui, C.Kaddache, R.Boukari
Service de Pédiatrie CHU Blida Université Saad dahlab Blida
Introduction : A côté du diabète de type 1 d’origine auto-immune de l’enfant, d’autres étiologies sont possibles
notamment le « diabète induit » par la chimiothérapie (Asparaginase) ou la corticothérapie prolongée.
Objectifs : Description de 4 cas de diabète chimio-induit chez des enfants traités pour leucémie aigue lymphoblastique
(LAL) dans notre unité d’oncologie pédiatrique par le protocole EORTC 2002.
Méthodes :
Le protocole EORTC comporte à la 1ère phase (induction ) et à la 4ème Phase ( réinduction) une
corticothérapie haute dose prolongée (Prednisolone 60 mg/m2/ j/1 mois /induction et dexaméthasone 6
mg/m2/ j/36j / réinduction) associée à L Asparaginase ( 20 000 UI /m2).
Résultats :
Les 4 enfants (5, 10,12 ,14 ans) sans antécédents particuliers traités pour LAL type T , LAL type B ( 2) ,
LAL type 2 ont présenté au décours des phases d’induction et/ou de réinduction des signes cliniques et
biologiques de diabète avec hyperglycémie ( 2,77 à 4,5 g/l) et glycosurie objectivés à plusieurs reprises et
une HbA1c variant de 4,5 % à 6,25 %.
Tous ont reçu une insulinothérapie conventionnelle (1UI/Kg/j). Son arrêt a été progressif après une durée
de traitement de 6 mois, 3 mois, 2 mois, 1 mois.
Avec un recul de 5 mois, 6 mois, 4 ans et 5 ans, bilan glucidique et HbA1c restent normaux
Conclusion : Le diabète insulino dépendant est une complication inhabituelle mais possible de la chimiothérapie anti
cancéreuse. Sa survenue complique la prise en charge des hémopathies malignes et nécessite des mesures
préventives (diététique - monitoring glycémie).

90
73.Aplasie médullaire chez l’enfant : étude de 15 cas S.Guemghar,H. Mesbaih, S.Touri, R.Nemmar, A.Lamraoui, L.Haddad, K.Mammeri , C.Kaddache,
R.Boukari.Service de Pédiatrie CHU Blida- Université Saad Dahlab
Introduction : L’aplasie médullaire est une maladie de pronostic sévère dont le traitement repose sur la greffe de moelle
osseuse (GMO) ou sur le traitement immunosuppresseur en l’absence de donneur HLA identique.
Objectif : Décrire les caractéristiques de la maladie et souligner les difficultés de la prise en charge.
Matériels et méthodes : Etude rétrospective sur dossiers recueillis de janvier 2005 à décembre 2012.
Résultat : 15 enfants (08 G/07F) dont l’âge moyen est de 9,3 ans (2 - 15 ans) sont colligés. Les symptômes à
l’admission sont : Pâleur (100 %), Fièvre (40 %), Syndrome hémorragique (86%).
Les signes biologiques sont un taux moyen d’ Hb 5,6 g/dl, une leucopénie (200 – 3190 leucocytes /mm3) et
une thrombopénie (1000 et 48000 plaquettes/mm3)
Au myélogramme la moelle est désertique (40 %) ou pauvre (60%). La BOM a confirmé le diagnostic dans
tous les cas.
L’aplasie médullaire a été classée sévère (86%) ou modérée ( 14% ) ( selon les critères de Camitta)
L’aplasie médullaire est idiopathique dans 60% des cas. Une anémie de Fanconi est retenue dans 33% des
cas.
Le traitement a fait appel à la corticothérapie (73%), aux androgènes (46%), aux immunosuppresseurs (20
%). 2 enfants ont bénéficié d’une GMO.
10 décès (67 %) sont survenus dans un délai moyen de 04 mois, 2 transformations malignes ( lymphome de
Burkitt , leucémie aigue) ont été observées.
Conclusion
La prise en charge de l’aplasie médullaire reste insuffisante en l’absence d’accès à la GMO.
74 .Gastroentérite révélatrice d’une maladie d’Addison. Savoir y penser chez l’enfant !
(A propos d’un cas)
Delileche Hana ,A.Bendjamaa, R.Aissat, A.Himmi , N.Boukhenfouf.
Introduction : La maladie d’Addison est une endocrinopathie chronique rare parfois sous diagnostiquée due à un
dysfonctionnement des glandes surrénales, potentiellement grave en raison du risque d’insuffisance
surrénalienne aigue d’où l’intérêt d’un traitement adapté et rapide.
Observation : Nous rapportons le cas d’un garçon âgé de 13 ans adressé pour prise en charge d’une gastroentérite avec
retentissement clinique important.
L’anamnèse précise que l’enfant présente depuis 25 jours des vomissements, nausées, douleurs
abdominales et diarrhées sans fièvre.
L’examen clinique objective :- une Asthénie chronique, amaigrissement de 8 kg et une déshydratation à
7%.
-Hyperpigmentation cutanée = mélanodermie (images affichées)
-Hypotension artérielle : 80 /40 mm Hg
-Glycémie capillaire : 0,7 gr /dl.
Le bilan sanguin retrouve :
- Hyponatrémie : 126 mmol /l
- Hyperkaliémie : 5.6 mmol/l
- Hypercalcémie 120 mg/l et des signes d’hémoconcentration avec hyperéosinophilie.
Devant ce syndrome de perte de sel, un bilan hormonal a été fait :
-Cortisolémie du matin : 162 nmol /l (N : 101,2 - 535,7)
-ACTH élevée : 2000 pg/ml (Il s’agit d’une atteinte primitive).

91
- Test de stimulation au tetracoside (synacténe®) négatif (cortisolémie: 180 nmol/l).
-S DHEA : 35ug /dl (N : 149 +/- 8).
-Testostérone <0.0025ng/ml.
-Bilan d’étiologie négatif (Tuberculose, hémorragie, carcinome surrénaliens…).
-Bilan de maladies auto-immunes associées négatif (Diabète, hypothyroïdie, candidose…).
Le diagnostic de maladie d’Addison auto-immune primitive sans autres atteintes auto-immunes
a été retenu.
L’enfant a été émis sous traitement substitutif (Hémisuccinate d’Hydrocortisone) avant les résultats du
bilan et après correction de l’hypoglycémie et des troubles hydro électrolytique.
Par voie parentérale à raison de 2 mg/Kg chaque 4 à 6 heures puis relais per os à raison de 25 mg/m2/j
reparties en deux prises. Du sel à raison de 2gr/j. (minéralocorticoïdes non disponibles).
L’évolution sous traitement, éducation thérapeutique et support psychologique a été bonne.
Conclusion : - La maladie d’Addison est un état pathologique complexe, aux causes variées.
- L’insuffisance surrénalienne aigue peut se présenter comme une gastroentérite sévère ou un
sépsis grave, Savoir y penser chez l’enfant.
- Un traitement journalier bien équilibré et à vie permet à la plupart des enfants atteints de continuer à
mener une vie aussi bien qu’avant la maladie ou presque.
75.Lithiase de la voie biliaire :bile-Plug syndrome : à propos d’un cas Helala Fairouz,Kerdoudi Sameh,Djabri Hana,Allas.H.
EHS Constantine
Introduction :
bile-Plug syndrome est une particularité de lithiase de la voie biliaire du nourrisson et du nouveau né, la
lithiase est toujours de nature pigmentaire et dans 40% des cas idiopathique.
Observation : nous rapportons le cas du nourrisson Mohamed Amine âgé de 3 mois, issu d’un mariage non
consanguin aux antécédents d’un diabète gestationnel et d’une macrosomie qui consulte pur un ictère
cholestatique évoluant depuis une semaine avec chlestase totale et permanente avec des coliques
abdominales. Examen clinique : eutrophique, bon développement psychomoteur, pas d’hépatomégalie ni de
splénomégalie.
Echographie abdominale : dilatation de la voie biliaire principale à 6,5 mm et des voies biliaire intra
hépatique sans obstacle écho décelable .Bili-IRM : dilatation de la voie biliaire principale et des voies
biliaires intra hépatique sans obstacle individualisé.
Evolution : clinique disparition de la cholestase, à l’écho cholédoque et voies biliaires intra hépatiques
deviennent normaux.
Notre cas illustre la difficulté diagnostique d’une dilatation des voies biliaires sans visualisation de calculs
à l’écho.
Conclusion :
le diagnostic d’une lithiase biliaire éliminée est retenu après l’évolution spontanée favorable clinique et
radiologique.
76.La mégacalicose ou maladie de Puigvert. A propos d'un cas Abes H,Benziane N, Bencharif S.EHS Douéra
Introduction : La mégacalicose ou maladie de Puigvert est une uropathie malformative rare (50 cas publiés seulement),
caractérisée par une dilatation non obstructive des calices avec hypoplasie de la médullaire rénale.
Restant longtemps asymptomatique, elle ne fait parler d’elle qu’au stade de complications.
Le but de notre travail est de présenter les particularités cliniques, thérapeutiques et évolutives de cette
pathologie.
Cas clinique :
Nous raportons le cas d’un nourrisson agé de 18mois présentant des douleurs abdominales chroniques sans
manifestations urinaires. Sur le plan biologique, sa fonction rénale était correcte et l’examen

92
Cytobactériologique des urines (ECBU) négatif.
L’échographie rénale montrait au niveau du rein droit un aspect hypotonique et dilaté des cavités calicielle,
un uretère non dilaté et absence d’image lithiasique intra rénale.
On a complété le bilan radiologique par une cystographie rétrograde,uiv et une scintigraphie rénale .
Discussion : Le principal diagnostic différentiel de la mégacalicose est
L’hydronéphrose secondaire a un syndrome de jonction pyélo-urétérale
Les critères pour le diagnostic sont : l’aspect caractéristique des calices ; les dimensions normales du
bassinet et de l’uretère, l’absence d’un reflux vésico-urétéral et l’absence d’obstruction aux examens
isotopiques.
Conclusion : Cette malformation devrait être connue et distinguée de l’hydronéphrose car si l’hydronéphrose nécessite
une intervention réparatrice, le traitement de la mégacalicose est souvent conservateur, on n’intervient
chirurgicalement qu’en cas de complications.
77 . Histiocytose langerhansienne néonatale :Un diagnostic ardu ,une prise en charge codifiée.
Otmane.A**,Arrada .Z** , Oucif.S**, SadaouI.B** , Berrah.H** , Babaahmed .R* – Djennane .N *.
** Service de pédiatrie B Hopital Nafissa Hamouda ex Parnet H .Dey- Alger.* Service anatomo-pathologie
Hopital BEO.
Introduction :
L’histiocytose langerhansienne « HL » ou Histiocytose X est une maladie mystérieuse et polymorphe, varie
de la forme locale à la forme grave
multisystémique.
C’est une prolifération non maligne, de cellules langerhansiennes ; elle touche souvent l’enfant et très
rarement le nouveau né.
Nous rapportons l’observation d’un nouveau né de sexe masculin âgé de 17jours de vie, qui présente une
fièvre , hépatomégalie , splénomégalie , atteinte cutanée nodulaires siégeant aux niveaux des membres
supérieurs et au cuir chevelu , atteinte médullaire avec un syndrome d’activation macrophagique (SAM).
Apres avoir éliminé les autres étiologies du SAM, l’étude anatomo histochimique par immuno marquage
du CD1a et de la protéine S100 sur biopsie cutané abouti à une HL.
Une réponse partiellement favorable est constatée sous chimiothérapie à base de corticoïdes et de
VP16.Malgré cette chimiothérapie, l’enfant décède 2 mois après.
L'HL est une hemopathie non maligne,
elle regroupe 4 formes cliniquement hétérogènes : le granulome éosinophile, maladie de Hand- Schuller-
Christian, maladie d’Hashimoto- Pritzker, maladie de Letterer- Siwe, comme c’est le cas de notre malade.
Le diagnostic de certitude et l’étude anatomo histochimique.
Le traitement est codifié par une chimiothérapie.
Conclusion : L’évolution capricieuse, la variété du tableau clinique et la non connaissance des mécanismes
étiopathogéniques, font toute la difficulté de la prise en charge.
Malgré sa rareté chez le nouveau né cette forme ne doit pas être méconnu, car bien que sévère, la guérison
peut être obtenu si le diagnostic est posé précocement et le traitement administré à temps.
78. Epidermolyse bulleuse héréditaire simple à révélation néonatale : à propos d’une observation
chez un nouveau né âgé de 02 jours
Laabed A,Telhaoui N. EPH Hakim Saadene Biskra
Introduction : Les épidermolyses bulleuses héréditaires sont des maladies génétiques, caractérisées par une fragilité de la
peau et des muqueuses. Cette fragilité se traduit par l’apparition de bulles ou d’érosions cutanées lors des
traumatismes minimes de la peau, ou survenant même de manière spontanée.
Les épidermolyses bulleuses sont des maladies génétiques secondaires à des mutations de gènes intervenant
dans la fabrication de protéines permettant la cohésion des différents composants cellulaires de la
peau.Dans l’épidermolyse bulleuse simple le défaut de cohésion se situe entre les cellules de l’épiderme

93
(couche la plus superficielle de la peau).
Observation : Nous rapportons le cas d’un nouveau né ,qui a présenté à l’âge de 48 heures une éruption bulleuse débutant
au niveau des paumes des mains et des plantes des pieds, puis rapidement les lésions se sont étendues à
tous le corps et notamment au niveau des zones de frottement et de la muqueuse buccale.les érosions
cutanées sont variables dans leurs profondeur et étendue, se répètent au moindre traumatisme et même
spontanément.
Sous traitement symptomatique l’évolution était lentement favorable.
Conclusion : L’épidermolyse bulleuse simple est la forme la plus fréquente des épidermolyses bulleuses héréditaires, elle
est de bon pronostic.
79. Sarcome d'Ewing à propos d'un cas rare
Bouteldja M1,Boumeddane A2.Clinique Amilcar Cabral CHU Oran . 2Centre Emir Abdelkader Oran–
oncologie pédiatrique
Introduction : Le sarcome d’Ewing est une tumeur osseuse maligne correspondant à la forme indifférenciée du
neuroépithéliome (variété rare de tumeurs primitives du système nerveux périphérique d’aspect épithélial,
localisée dans n’importe quelle partie molle). C’est une tumeur qui touche principalement les adolescents et
les jeunes adultes (la moyenne d'âge est de 13 ans). Le sarcome d’Ewing est souvent localisé au niveau des
os plats (60% des cas), en particulier au niveau du bassin et des côtes, mais peut intéresser les os longs
(40% des cas). Nous rapportons un cas rare de sarcome d’Ewing diagnostiqué chez un enfant de 2ans et
demi présentant une tumeur située à l’extrémité supérieure du fémur.
Observation : B.I, garçon de 2 ans et demi, originaire et demeurant à Mostaghanem, sans antécédents personnels et
familiaux particuliers, consulte le 18-09-2013 pour une fracture de l’extrémité supérieure de la diaphyse
fémorale droite lors d’un traumatisme minime (chute de sa hauteur sur le siège) l’examen clinique retrouve
un enfant apyrétique en bon état général présentant une tuméfaction de la cuisse droite ferme, douloureuse
avec peau tendue et luisante. La radiographie standard complétée par une TDM montre un trait de fracture,
un processus tissulaire ostéolytique de l’extrémité supérieure du fémur avec rupture des corticales,
envahissement des partie molles et une réaction périostée en feu d’herbe. L’anapath de la pièce de biopsie
chirurgicale est revenue en faveur d’un sarcome d’Ewing. Après un bilan d’extension qui n’a pas montré
une localisation secondaire locorégionale ou à distance, on a entamé une chimiothérapie néo-adjuvante le
14/10/2013 dont le but est de réduire la taille de la tumeur avant le traitement chirurgical.
Discussion : Une tumeur osseuse survenue chez un enfant de moins de 5ans et à l’extrémité supérieure du fémur doit
faire discuter à priori les tumeurs osseuses malignes secondaires :
• Neuroblastome stade IV : surrénales d’aspect TDM normal, pas de tumeur dans les chaines sympathiques
paravertébrales, taux normal des catécholamines urinaires.
• Hémopathies malinges: pas de syndrome d’insuffisance médullaire associé au syndrome tumoral,
médullogramme objectivant une moelle cytologiquement normale.
• Histiocytose X : aspect radiologique et anapath ne sont pas en faveur.
Conclusion : La survenue d’une tumeur osseuse primitive chez un nourrisson ou un jeune enfant est rare et souligne
l’intérêt d’une enquête étiologique rigoureuse avant de retenir ce diagnostic.

94
80. Dépistage neonatal d’une tyrosinemie asymptomatique Sadaoui B,Arrada Z,Hadjarab A., Rezzaz N, Berrah H.
Unité « Nid » Pr Arrada Z., Sce de pédiatrie « B » Pr Berrah H., hôpital N. Hamoud,CHU H.Dey , Alger.
La tyrosinémie est une maladie métabolique orpheline (quelques dizaines de cas en Algérie), grave et mal
connue, souvent diagnostiquée tardivement, alors qu’aujourd’hui elle est une des seules maladies
héréditaires traitées avec succès, et ce, à trois niveaux:
- Test diagnostic
- Traitement efficace
- Test de détection des porteurs
Nous rapportons le premier cas dépisté en période néonatale et pris en charge avant l’installation d’une
symptomatologie clinque.
Ce dépistage précoce, motivé par la présence de la maladie dans sa forme grave chez le frère, nous a permis
d’éviter les complications hépato rénales et neurologiques grâce à une prise en charge précoce.
et d’identifier une autre forme clinique dans la même famille.
La prise en charge précoce d’une tyrosinémie, une des seules maladies orpheline traitées aujourd’hui avec
succès est indiscutable.
81. Une forme particulière du retard psychomoteur :le syndrome de NEUHAUSER -A propos d'un
cas
Mahchouche N.Mekki A,Lebied A,.Baghriche .M .EPH Bologhine Ibn Ziri
Il s’agit d’un syndrome malformatif extrêmement rare, à transmission autosomique récessive, associant un
retard mental , une hypotonie musculaire, une mégalocornée congénitale non progressive et des traits
dysmorphiques variables(MIM 249310).
Très peu cité dans la littérature ; puisque, depuis sa première description en 1975 seulement 36 cas ont été
rapportés (2010).Ce petit nombre de cas publiés peut être dû au fait que l’association des deux signes
cardinaux ; retard psychomoteur (retrouvé dans 100% des cas) et mégalocornée (88%) jusque-là méconnue,
échappe à notre attention.
Le propositus est un nourrisson de 17mois, 3eme et denier enfant d’un couple non consanguin, examiné à
la consultation de neurologie pédiatrique du CHU HUSSEIN DEY pour retard des acquisitions
psychomotrices.
La recherche de la cause des retards mentaux congénitaux, en règle délicate, parce que d’origine génétique
dans prés de la moitié des cas, a été vite orientée chez notre malade par l’aspect de la dysmorphie faciale et
les signes associés.
Le risque de récurrence n’est pas établi dans ce syndrome pour procéder à un conseil génétique. Les
éléments cliniques retrouvés dans ce premier cas décrit en Algérie complètent la classification de Vérloes
et al (Am.J.Med. Genet.1993 ; 46 :132-7) lesquels proposent 5 sous-groupes, ce qui laisse suggérer des
génotypes différents. Des recherches sont en cours pour clarifier le mode de transmission, l’étiopathogénie
et le pronostic de ce syndrome afin d’affiner une prise en charge adaptée.
82.Une thrombose veineuse révélant une ostéomyélite (A propos d’un cas)
Zerrouki NH, Ibsaine O, Drali O, Boutaba K S, Latreche M, Berrah H.CHU Parnet Hussein dey
Les thromboses veineuses de l’enfant sont très rares. Elles peuvent compliquer des ostéomyélites aiguës
(OMA) hématogènes.
Observation
Nous vous rapportons le cas de Fares âgé de 4ans, hospitalisé pour tuméfaction douleureuse de la jambe
droite évoluant depuis 5 jours dans un context fébrile, traité par immobilisation platrée et anti-inflamatoire
par les chirurgiens pédiatres. A l’admission, l’enfant était fébrile à 38,5°, présentant une jambe droite
douloureuse, tuméfiée, rouge et un homans positif. Le doppler avait objectivé une thrombose de la saphène
interne. L’exploration a révélé sur le plan biologique une hyperleucocytose à 17 100 elmts/mm3, une CRP

95
positive et uneVS accélérée à 85 mm la première heure. Le bilan de la thrombophilie était normal (protèine
S, C, antithrombine II et les antiphopholipides normaux). La radio avait objectivé un abcés sous périosté du
tibia droit ayant nécéssité un drainage chirurgical. L’analyse bactériologique du pus avait objectivé un
staphylocoque aureus alors que les hémocultures étaient négatives. L’évolution était favorable sous
traitement anti-coagulant pendant 3 semaines entrainant une reperméabilisation complete de la saphène
interne et une antibiothérapie anti staphylococcique pendant 6 mois.
Conclusion
Les OMA compliquées de thromboses veineuses sont rares. L’existence d’une thrombose constitue un
facteur d’essaimage bactérien par l’intermédiaire d’emboles septiques, notamment aux poumons.
83 : Paraplégie Medicale Sur Malformation Arterio-Veneuse.
F.Akli, K.Safi, M.Ammenouche Service : Mpr Ehs Azur Plage La paraplégie est une paralysie des deux membres inferieurs, résultant d’une atteinte de la moelle épinière d’origine traumatique (accidentelle) le plus souvent, mais parfois médicale (maladie), c’est le cas de notre malade. Nous rapportons le cas d’une malformation artério-veineuse médullaire dorsale, chez un adolescent de 14 ans, révélée par une paraplégie totale, avec évolution favorable sous corticothérapie, embolisation suivie d’une rééducation. -la fréquence de l’évolution spontanément favorable d’une malformation artério-veineuse est mal connue, mais ne doit pas être sous-estimée, elle pourrait être influencée par la corticothérapie embolisation suivie d’une rééducation.


















