Le rôle du système mélanocortine dans le contrôle de l'activité thermogène du tissu adipeux...
239
Le rôle du système mélanocortine dans le contrôle de l’activité thermogène du tissu adipeux brun. Thèse Boris Monge Roffarello Doctorat en neurobiologie Philosophiae doctor (Ph.D.) Québec, Canada © Boris Monge Roffarello, 2015
-
Upload
boris-monge-roffarello -
Category
Documents
-
view
469 -
download
5
Transcript of Le rôle du système mélanocortine dans le contrôle de l'activité thermogène du tissu adipeux...

I
Le rôle du système mélanocortine dans le contrôle de l’activité thermogène du tissu adipeux brun.
Thèse
Boris Monge Roffarello
Doctorat en neurobiologie Philosophiae doctor (Ph.D.)
Québec, Canada
© Boris Monge Roffarello, 2015


III
Résumé
L’obésité se caractérise par un excès de masse adipeuse corporelle. Elle est due à un
déséquilibre de la balance énergétique qui dépend de l’apport énergétique et les dépenses
énergétiques, notamment via la thermogenèse du tissu adipeux brun (brown adipose tissue —
BAT). Le BAT est sous l’influence de différents systèmes de neurotransmetteurs au sein du
système nerveux central et en particulier au niveau de l’hypothalamus par le système
mélanocortines (SM). Cependant, le rôle du SM dans l’hypothalamus sur le contrôle du BAT
reste à éclaircir. Les travaux présentés dans cette thèse visaient à i) montrer que le noyau
paraventriculaire de l’hypothalamus (PVH) est au centre de la modulation des effets du SM sur la
thermogenèse du BAT par le système endocannabinoïde (SE) et ii) mettre en évidence que le
noyau préoptique médial (MPO) est un site d’activation de la thermogenèse par les
mélanocortines dépendant d’un relais dans le noyau dorsomédian de l’hypothalamus (DMH).
Nous avons observé que l’injection intra-cérébro-ventriculaire de MTII, un agoniste des
récepteurs aux mélanocortines, augmente la dépense énergétique via la thermogenèse du BAT.
D’autre part, les effets du MTII sur le BAT sont inhibés ou partiellement potentialisés par
l’injection respective d’un agoniste (Δ9-THC) ou d’un antagoniste (AM251) du SE. Par la suite,
nous avons mis en évidence que ces observations font intervenir spécifiquement le récepteur aux
mélanocortines 4 et le récepteur aux cannabinoïdes de type 1. Enfin, notre étude a montré que des
neurones du PVH co-expriment ces deux récepteurs et que les effets sur la thermogenèse de
l’injection de MTII dans le PVH, sont totalement inhibés par l’administration de Δ9-THC dans le
4éme ventricule, suggérant donc, que la modulation du SM par le SE a lieu au sein de neurones du
PVH projetant vers la moëlle épinière. D’autre part, l’injection de MTII dans le MPO stimule la
thermogenèse du BAT. Toutefois, des lésions du DMH causées par l’acide kaïnique inhibent les
effets du MTII. De plus, l’expression de gènes impliqués dans le métabolisme du tissu adipeux
blanc inguinal est stimulée par le MTII, mais seulement chez les rats lésés. Ces résultats
permettent donc d’établir que le duo MPO-DMH est une cible importante dans la modulation de
l’homéostasie énergétique par les mélanocortines. L’ensemble de ces résultats montre le rôle
majeur du SM au sein de l’hypothalamus dans le contrôle de l’activité thermogène du BAT.
Cependant, la compréhension des réseaux neuronaux impliquant le SM et ses co-régulateurs reste
à éclaircir avant d’envisager un traitement thérapeutique de l’obésité via le SM.


V
Abstract
Obesity is characterized by an adiposity excess. It is due to an imbalance between energy
intake and energy expenditure, which includes a few components including brown adipose tissue
(BAT) thermogenesis. BAT is controlled by different neuronal systems of the central nervous
system, including the melanocortin system (MS). However, the role of the hypothalamic
melanocortin system in the control of BAT thermogeneis remains unclear. The studies presented
in this thesis were designed to study i) that the paraventricular nucleus of the hypothalamus
(PVH) is central for modulating the effects of MS on BAT thermogenesis by the
endocannabinoid system (ES) and ii) to highlight the influence of the medial preoptic nucleus
(MPO) as a site of the thermogenic action of MS and exploring the role played by the
dorsomedial nucleus of the hypothalamus (DMH) in this action.
We observed that intracerebroventricular (i.c.v) injection of melanocotin II (MTII), a
melanocortin receptor agonist, increased energy expenditure through BAT thermogenesis. The
MTII effects on BAT were inhibited or partially potentiated by an injection of ES agonist (Δ9-
THC) or antagonist (AM251) respectively. Subsequently, we demonstrated that the melanocortin
4 receptor and the cannabinoid receptor type 1 were specifically involved in these observations.
Finally, our study showed that PVH neurons co-expressed both these receptors and the effects on
thermogenesis of the MTII injection into the PVH were completely inhibited by the
administration of Δ9-THC in the 4th ventricle, suggesting that the modulation of MS by the ES
occurred through PVH neurons projecting to the spinal cord. On the other hand, the injection of
MTII in MPO stimulated BAT thermogenesis. Nonetheless, DMH lesions caused by kainic acid
inhibited the effects of MTII. Furthermore, the gene expression involved in inguinal white
adipose tissue metabolism were stimulated by MTII, but only in lesioned rats. These results
indicate that the MPO-DMH duet is an important target in the modulation of energy homeostasis
by the melanocortins. All of these results show the major role of MS within the hypothalamus in
the control of BAT thermogenic activity. However, understanding of neural networks involving
the MS and its co-regulators remains to be clarified before considering therapeutic treatment of
obesity through the activation of MS.


VII
Tables des matières
RESUME ....................................................................................................................................................... III
ABSTRACT .................................................................................................................................................... V
TABLES DES MATIERES ........................................................................................................................ VII
LISTE DES TABLEAUX .............................................................................................................................. XI
LISTE DES FIGURES ............................................................................................................................... XIII
LISTE DES ABREVIATIONS ................................................................................................................... XV
REMERCIEMENTS ................................................................................................................................... XXI
AVANT-‐PROPOS .................................................................................................................................. XXIII
INTRODUCTION .......................................................................................................................................... 1 1 LA REGULATION DE L’HOMEOSTASIE ENERGETIQUE. ...................................................................................... 5
1.1 Les signaux périphériques régulant l’homéostasie énergétique. ............................................. 6 1.1.1 Les signaux sensoriels et la phase céphalique. ......................................................................................................... 8 1.1.2 L’effet des signaux digestifs et des métabolites sur la prise alimentaire. ..................................................... 8
1.1.2.1 Les signaux gastro-‐intestinaux. ............................................................................................................................. 9 1.1.2.2 L’effet des métabolites. .......................................................................................................................................... 11
1.1.3 Les signaux postprandiaux. ............................................................................................................................................ 13 1.1.3.1 L’insuline. ..................................................................................................................................................................... 13 1.1.3.2 La leptine. .................................................................................................................................................................... 14
1.2 La régulation centrale de l’homéostasie énergétique. ............................................................... 15 1.2.1 Le rôle de l’hypothalamus dans l’homéostasie énergétique. ........................................................................... 15
1.2.1.1 Les neurones de premier ordre du noyau arqué. ....................................................................................... 17 1.2.1.2 Les neurones de second ordre. ........................................................................................................................... 21
1.2.2 Le rôle du NTS dans la régulation de l’homéostasie. .......................................................................................... 23 1.2.2.1 Le NTS est le centre de l’activité du SNA. ....................................................................................................... 24 1.2.2.2 Les interactions du NTS et de l’hypothalamus dans la régulation de l’homéostasie. ................. 24
2 LA THERMOGENESE DU TISSU ADIPEUX BRUN. .............................................................................................. 27 2.1 Le tissu adipeux brun. ............................................................................................................................... 28
2.1.1 Les caractéristiques du tissu adipeux brun. ........................................................................................................... 28 2.1.1.1 Origine et développement du tissu adipeux brun. ..................................................................................... 28 2.1.1.2 Localisation du tissu adipeux brun chez le rat et l’homme. ................................................................... 31 2.1.1.3 Le secrétome du tissu adipeux brun. ............................................................................................................... 31 2.1.1.4 Les adipocytes beiges. ............................................................................................................................................ 33 2.1.1.5 La lipogenèse et la lipolyse une compétence commune à tous les adipocytes. ............................. 33

VIII
2.1.2 Les mécanismes cellulaires permettant la production de chaleur. .............................................................. 35 2.1.2.1 L’activation de l’adipocyte brun : la cascade adrénergique. .................................................................. 35 2.1.2.2 Les mécanismes de l’activité thermogène. .................................................................................................... 38 2.1.2.3 Les mécanismes de la capacité thermogène. ................................................................................................ 39 2.1.2.4 Les facteurs endocriniens périphériques influençant l’adipocyte brun. .......................................... 40
2.2 Le contrôle central de l’activité thermogène du tissu adipeux brun. .................................. 42 2.2.1 Le contrôle central du tissu adipeux brun lors de la thermorégulation. .................................................... 42
2.2.1.1 Les afférences de la perception thermique cutanée et centrale. ......................................................... 43 2.2.1.2 Les mécanismes hypothalamiques impliqués dans le contrôle du BAT. .......................................... 45 2.2.1.3 Le système nerveux sympathique. .................................................................................................................... 48
2.2.2 Les autres mécanismes impliqués dans le contrôle central de l’activité du iBAT. ................................. 50 2.2.2.1 Rythme ultradien et stress : rôle de l’orexine. ............................................................................................. 50 2.2.2.2 La fièvre. ....................................................................................................................................................................... 51 2.2.2.3 L’hypoglycémie ......................................................................................................................................................... 52 2.2.2.4 L’hypoxie. ..................................................................................................................................................................... 52
3 IMPLICATION DU SYSTEME MELANOCORTINE DANS LA REGULATION DE L’HOMEOSTASIE
ENERGETIQUE : SON ROLE DANS LA THERMOGENESE. ...................................................................................................... 55 3.1 Le système mélanocortine. ..................................................................................................................... 55
3.1.1 Ses principaux acteurs. .................................................................................................................................................... 55 3.1.1.1 Localisation et synthèse des ligands endogènes. ....................................................................................... 55 3.1.1.2 Localisation et fonction des récepteurs. ......................................................................................................... 56
3.1.2 Ses partenaires dans le contrôle de la prise alimentaire. ................................................................................. 58 3.1.2.1 L’effet de la leptine et de l’insuline sur l’expression de la POMC. ....................................................... 58 3.1.2.2 Les endocannabïnoides. ........................................................................................................................................ 59
3.2 La thermogenèse induite par l’alimentation ................................................................................. 60 3.3 La mise en évidence de l’implication du système mélanocortine dans le contrôle de la
thermogenèse. ...................................................................................................................................................................... 61 3.3.1 Les évidences génétiques ................................................................................................................................................ 62 3.3.2 Les évidences pharmacologiques. ............................................................................................................................... 62 3.3.3 Les évidences anatomo-‐fonctionnelles. .................................................................................................................... 63
PROBLEMATIQUE ET OBJECTIFS GENERAUX DES TRAVAUX. ................................................... 67 Problématique. ........................................................................................................................................................................ 69 Objectifs généraux des travaux. ....................................................................................................................................... 71 Objectifs spécifiques. ............................................................................................................................................................ 71
CHAPITRE 1 ............................................................................................................................................... 73
THE CANNABINOID RECEPTOR 1 IS INVOLVED IN THERMOGENESIS INDUCED BY THE
BRAIN STIMULATION OF THE MELANOCORTIN-‐4 RECEPTOR IN MALE RATS. .............................. 75

IX
Résumé ....................................................................................................................................................................................... 77 Abstract ...................................................................................................................................................................................... 79 Introduction ............................................................................................................................................................................. 81 Research Design and Methods ......................................................................................................................................... 83 Results ......................................................................................................................................................................................... 91 Discussion ................................................................................................................................................................................. 93 Acknowledgements ............................................................................................................................................................... 95 References ................................................................................................................................................................................. 97 Tables ........................................................................................................................................................................................ 103 Figure Legends ...................................................................................................................................................................... 105 Figures ...................................................................................................................................................................................... 109
CHAPITRE 2 ............................................................................................................................................ 117
THE MEDIAL PREOPTIC NUCLEUS AS A SITE OF THE THERMOGENIC AND METABOLIC
ACTIONS OF MELANOTAN II IN MALE RATS. ........................................................................................... 119 Résumé ..................................................................................................................................................................................... 121 Abstract .................................................................................................................................................................................... 123 Introduction ........................................................................................................................................................................... 125 Research Design and Methods ....................................................................................................................................... 127 Results ....................................................................................................................................................................................... 131 Discussion ............................................................................................................................................................................... 133 Acknowledgements ............................................................................................................................................................. 137 References ............................................................................................................................................................................... 139 Tables ........................................................................................................................................................................................ 143 Figure Legends ...................................................................................................................................................................... 145 Figures ...................................................................................................................................................................................... 147
DISCUSSION GENERALE, PERSPECTIVES DE RECHERCHE ET CONCLUSION. ..................... 151 Chapitre 1 ................................................................................................................................................................................ 153 Chapitre 2 ................................................................................................................................................................................ 157 Perceptives de recherche et conclusion ..................................................................................................................... 161
REFERENCES. .......................................................................................................................................... 165


XI
Liste des tableaux
Introduction.
Tableau 1: Principaux facteurs hypothalamiques impliqués dans la régulation de l’homéostasie
énergétique. ............................................................................................................................ 18
Tableau 2: Résumé des principales fonctions, sites d’expression et de l’affinité des ligands pour
les récepteurs aux mélanocortines. ......................................................................................... 57
Chapitre 1.
Table 1: qPCR gene information. ................................................................................................. 103
Table 2: probes characterizations. ................................................................................................ 103
Chapitre 2.
Table 1: qPCR gene information. ................................................................................................. 143
Table 2: Effects of MTII infusion in MPO on blood variation of insulin, NEFA and triglycerides
in DMH-lesioned rats. .......................................................................................................... 144


XIII
Liste des Figures
Introduction.
Figure 1: Régulation de l’homéostasie énergétique. ........................................................................ 6
Figure 2: Représentation spatiotemporelle des différentes phases d’un cycle de prise alimentaire.6
Figure 3: Modèle d’intégration des signaux d'adiposité et de satiété. .............................................. 7
Figure 4: Section sagittale du cerveau de rat montrant l'emplacement de l'hypothalamus. ........... 16
Figure 5: Représentation tridimensionnelle de l'hémisphère droit de l'hypothalamus du rat. ....... 16
Figure 6: La régulation centrale de la balance énergétique. ........................................................... 26
Figure 7: Representation schématique de la différenciation des adipocytes. ................................. 30
Figure 8: Localisation du tissu adipeux brun chez le rat et l’homme. ........................................... 33
Figure 9: L’activation de l’adipocyte brun. .................................................................................... 36
Figure 10: Représentation de la circuiterie neuronale contrôlant l’activité du iBAT. ................... 42
Chapitre 1.
Figure 1: Experimental protocol. ................................................................................................. 109
Figure 2: Effects of Δ9-THC and AM251 on the MTII effects on whole body thermogenesis. . 110
Figure 3: Effects of Δ9-THC and AM251 on the MTII effects on iBAT thermogenesis, whole
body thermogenesis and metabolic activity. ........................................................................ 111
Figure 4: Effects of Δ9-THC and AM251 on the MTII effects on iBAT gene expression. ........ 112
Figure 5: Effect of HS024 or Δ9-THC and AM251 on the MTII effects on iBAT thermogenesis,
whole body thermogenesis and metabolic activity. ............................................................. 113

XIV
Figure 6: Colocalization of MC4R with CB1 and VGLUT2 mRNA in the PVH. ...................... 114
Figure 7: Effect of Δ9-THC on the PVH MTII infusion effect on iBAT thermogenic activity and
capacity. ................................................................................................................................ 115
Chapitre 2.
Figure 1: Histological verification. .............................................................................................. 147
Figure 2: Effects of the MPO infusion of MTII in Sham- and DMH-lesioned rats on iBAT and
whole body thermogenesis. .................................................................................................. 148
Figure 3: Effects of the MPO infusion of MTII on iBAT, iWAT, liver and heart metabolic
activity in Sham- and DMH-lesioned rats. ........................................................................... 149
Figure 4: Effects of the MPO infusion of MTII on iBAT and iWAT gene expression in Sham-
and DMH-lesioned rats. ....................................................................................................... 150

XV
Liste des abréviations
Abréviation Définition
2-AG 2-Arachidonylglycerol
α-MSH Alpha-melanocyte-stimulating hormone
β-MSH Beta-melanocyte-stimulating hormone
γ-MSH Gamma-melanocyte-stimulating hormone
Δ9-THC Delta-9-tetrahydrocannabinol
ACC Acétyl-coenzyme A carboxylase
aCSF Artificial cerebrospinal fluid
ACTH Adrénocorticotrophine
ADRB3 Récepteur β-3 adrénergique
ADP Adénosine diphosphate
AEA Anandamide
AGL Acides gras libres
AGLC- AGL à longue chaine chargée négativement
AGPAT Acylglycérol-phosphate acyltransférase
AgRP Agouti-releated peptide
ALP Acide lysophosphatidique
AMPc Adénosine monophosphate cyclique
ANP Atrial natriuretic peptide
AP Area postrema
APp Acide phosphatidique
ARC Noyau arqué de l’hypothalamus
ARN Acide ribonucléique

XVI
ARNm ARN messager
ATF2 Activating-transcription factor-2
ATGL Lipase des TG du tissu adipeux
ATP Adénosine triphosphate
BAT Tissu adipeux brun
BDNF Brain-derived neurotrophic factor
BMP-7 Bone morphogenetic protein-7
BMP-8b Bone morphogenetic protein-8b
BNP Brain-derived natriuretic peptide
C/EBPβ CCAAT/enhancer-binding protein-β
CART Cocaine- and amphetamine- regulated transcript
CB1 Récepteur aux cannabinoïdes de type 1
CCK Cholécystokinine
CNP C-type natriuretic peptide
CPT1β Carnitine palmitoyl transférase 1β
CRE cAMP response element
CRH Corticotropin-releasing hormone
DG Diglycéride
DGAT Diacylglycérol acyltransférase
DIO2 Deiodinase iodothyronine 2
DMH Noyau dorsomédian de l’hypothalamus
DPP-4 Dipeptidyl-peptidase 4
En1 Engrailed-1
EP3-R Récepteur 3 aux prostaglandines E

XVII
FADH2 Flavine adénine dinucléotide H2
FAS Acides gras synthetase
FATP1 Protéine transporteuse d’acide gras 1
FDG Fluoro-deoxyglucose
FGF-2 Fibroblast growth factor-2
FGF-21 Fibroblast growth factor-21
FOXC2 Forkhead box C2
FOXO1 Forkhead box containing protein 1
G3P Glycerol-3-phosphate
GHS-R Récepteur de la ghréline
GLP-1 Glucagon-like peptide 1
GLUT4 Transporteur au glucose de type 4
GPAT Glycérol-phosphate acyltransférase
H+ Protons
HSL Lipase sensible aux hormones
iBAT BAT interscapulaire
i.c.v. Intra-cérébro-ventriculaire
IGF-1 Insulin-like growth factor-1
IL-6 Interleukine-6
IML Colonne intermediolatérale de la moelle épinière
INS-R Récepteur de l’insuline
Jak2 Janus kinase 2
L-PGDS Lipocalin prostaglandin D synthase
LCR Liquide céphalorachidien

XVIII
LHA Aire latérale de l’hypothalamus
LPS Lipopolysaccharide
MAG Monoacylglycérol
MC3/4R Récepteurs aux mélanocortines 3 et 4
MCH Melanin-concentrating hormone
MCR Récepteurs aux mélanocortines
MGL Lipase du monoacylglycérol
MnPO Noyau préoptique médian
MPO Noyau préoptique médial
MTII Melanotan-II
MVL Médulla ventrolatérale
MVMR Médulla ventromédiale rostrale
Myf5 Facteur myogénique 5
NA Noradrénaline
NADH, H+ Nicotinamide adénine dinucléotide H, H+
NMDA Acide N-méthyl-D-aspartique
NPY Neuropeptide Y
NRF1 Nuclear respiratory factor 1
NRF2 Nuclear respiratory factor 2
NTS Noyau du tractus solitaire
OXM Oxyntomoduline
PAG Substance grise périaqueducale
PBL Noyau parabrachial latéral
PBLd PBL dorsal

XIX
PBLle PBL latéral externe
PC Pro-hormones convertases
PGC1α PPAR-γ co-activator 1α
PGE2 Prostaglandine
PKA Protéine kinase A
PLA2 Phospholipase A2
PI3K Phosphatidylinositol-3-kinase
PNs Peptides natriurétiques
POMC Pro-opiomélanocortine
PP Polypeptide pancréatique
PPAR-γ Peroxisome-proliferator-activated receptor-γ
PPRE PPAR response element
pRB Retinoblastoma protein
PRDM16 PRD1-BF1-RIZ1 homologous domain-containing-16
pSTAT-3 Phosphorylated signal transducer and activator of transcription 3
PVH Noyau paraventriculaire de l’hypothalamus
PYY Peptide YY
PYY (1-36) Peptide YY (1-36)
PYY (3-36) Peptide YY (3-36)
RARE Retinoic acid response elements
RBP-4 Retinol-binding protein-4
RIP40 Receptor-interacting protein 140
RPa Noyau du raphé pallidus
RPar RPa rostral

XX
SNA Système nerveux autonome
SNC Système nerveux central
SNS Système nerveux sympathique
T3 Triiodothyronine
T4 Thyroxine
TEP Tomographie par émission de positrons
TG Triglycéride
TIA Thermogenèse induite par l’alimentation
TRE Thyroid response element
TRH Thyrotropin-releasing hormone
TRP Transient receptor potential family of cation channels
UCP-1 Uncoupling protein 1
VEGF-A Vascular endothelial growth factor-A
VMH Noyau ventromédian de l’hypothalamus
Y1 Récepteurs du NPY de types 1
Y2 Récepteurs du NPY de types 2
Y5 Récepteurs du NPY de types 5

XXI
Remerciements
Je tiens tous d’abord à remercier mon directeur de recherche, le professeur Denis Richard
pour m’avoir accueilli au sein de son équipe de recherche et soutenu lors de mes années d’études
doctorales. J’ai beaucoup apprécié son encadrement bienveillant ainsi que la confiance qu’il m’a
accordée lors de la réalisation de mes travaux. Enfin je le remercie pour ces paroles stimulantes
qui surent m’amener au bout de mon aventure doctorale ; il restera un modèle d’engagement et
d’éthique scientifique.
Je remercie sincèrement le Dr Elena Timofeeva, le Dr David Saint-Pierre et le Dr Yves
Deshaies d’avoir accepté d’examiner cette thèse et de siéger parmi les membres du jury.
J’aimerais remercier particulièrement le Dr Elena Timofeeva pour son soutien et ses conseils lors
de mes travaux et le Dr Yves Deshaies pour ces conseils et son soutien dans mes choix d’avenir
mais aussi pour nos quelques discussions fortement inspirantes sur la place et le rôle d’un
scientifique dans notre société.
Je tiens aussi à remercier chaleureusement et amicalement tous les membres des équipes du
Dr Yves Deshaies, Dr Mathieu Laplante, Dr Fréderic Picard, Dr Elena Timofeeva et Dr Denis
Richard avec qui j’ai eu le grand plaisir de travailler. J’aimerais remercier Julie Plamondon,
Marie Claude Roy, Pierre Samson et Yves Gélinas pour leur soutien et leurs conseils dans mon
travail au laboratoire, pour leur générosité, pour leur accueil et de m’avoir offert leur amitié. Je
remercie aussi Cynthia Bouchard et Audrey Chalifoux pour leur aide à l’animalerie et leurs
sourires. Un grand merci à Annie Moreau pour sa disponibilité et son assistance. J’aimerais par la
suite remercier mes collègues et amis étudiants, à commencer par Alexandre Caron, le Dr
Sébastien Labbé, le Dr Pierre-Gilles Blanchard pour leur aide et leurs précieux conseils. Enfin
pour finir la section collègues de travail, je souhaite remercier plus qu’un collègue, un ami, le Dr
Damien Lanfray, qui m’a apporté son aide et son soutien dans la rédaction de ce manuscrit et
avec qui j’ai partagé en compagnie d’Émilie sa conjointe de nombreux bons moments et bons
repas qui vont me manquer. Merci à vous deux.
Je tiens à remercier mes amis dont certains sont mentionnés ci-dessus et ma famille.
J’exprime toute ma gratitude à mes parents, ma mère et Yann qui m’ont encouragé tout au long
de mes études et de cette aventure. Merci à Estelle et Nicolas qui nous ont permis ces moments

XXII
de détente nécessaire à l’accomplissement d’un doctorat. Enfin, je remercie Agathe ma conjointe,
le mot est faible et je n’aurais pas assez de mots pour lui témoigner toute ma reconnaissance et
mon amour pour être présente à mes cotés. Sans elle rien n’aurait été possible.
Je dédie cette thèse à mon grand-père et ma grand-mère disparus pendant la réalisation de
celle-ci.

XXIII
Avant-propos
Le dépôt de cette thèse à la Faculté des études supérieures et postdoctorales de l’Université
Laval signifie l’évaluation finale en vue de l’obtention du grade de Philosophae doctor ès
Sciences (Ph.D.). Ce manuscrit vise à montrer l’implication du système mélanocortine dans le
contrôle central de l’activité thermogène du tissu adipeux brun. La première section de cet
ouvrage est constituée d’une introduction générale rédigée en français abordant les
caractéristiques du système mélanocortine et des ses partenaires neuronaux impliqués dans la
régulation de la balance énergétique, ainsi que l’innervation sympathique et les caractéristiques
du tissu adipeux brun. Les chapitres 1 et 2 constituent la seconde section, celle-ci s’intéresse aux
travaux dans le domaine sus-cité que j’ai réalisés au cours de ces dernières années dans le
laboratoire de Dr Denis Richard. Étant déjà soumis dans des journaux scientifiques spécialisés
dans le domaine des neurosciences et de la physiologie, ces chapitres prennent la forme d’articles
scientifiques rédigés en langue anglaise, conformément aux exigences éditoriales. La dernière
section de cette thèse discute des résultats, dans le but de démontrer leur nouveauté, leur
pertinence et de proposer de nouvelles orientations de recherche susceptibles de contribuer à
l’avancement des connaissances dans le domaine de la neurobiologie de l’obésité.
Articles présentés
Lors de mon séjour dans le laboratoire du Dr Denis Richard, j’ai eu la chance et le plaisir
d’être sollicité pour contribuer aux travaux de quelques étudiants-chercheurs de talent. Ces
collaborations avec mes collègues ont abouti à la parution de nombreux articles dans des
journaux à fort impact, en plus de diversifier les champs de compétences de chacun d’entre nous.
Cependant, les articles formant le cœur de cette thèse et présentés ici sont l’expression des projets
de recherche pour lesquels j’ai oeuvré à la direction des travaux, de la planification initiale des
hypothèses de travail et des expérimentations à la rédaction, en passant par la mise au point des
protocoles.
La liste des articles qui suit retranscrit mes activités de recherche lors de mes études
doctorales. Les études précédées d'un astérisque et en caractère gras sont celles qui ont été
insérées dans cet ouvrage, elles sont accompagnées d’un avant-propos spécifique aux travaux
qu’elles détaillent.

XXIV
Lanfray, D., Arthaud, S., Ouellet, J., Compère, V., Do Rego, J.-L., Leprince, J.,
Lefranc, B., Castel, H., Bouchard, C., Monge-Roffarello, B., Richard, D., pelletier, G.,
Vaudry, H., Tonon, M-C., Morin, F. (2013). Gliotransmission and Brain Glucose-Sensing:
Critical Role of Endozepines. Diabetes. 62(3):801-10
Lopez, C.A., Guesdon, B., Baraboi, E.-D., Roffarello, B.M., Hétu, M., and Richard,
D. (2011). Involvement of the opioid system in the orexigenic and hedonic effects of
melanin-concentrating hormone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1105–
11.
Richard, D., Monge-Roffarello, B., Chechi, K., Labbé, S.M., and Turcotte, E.E.
(2012). Control and physiological determinants of sympathetically mediated brown adipose
tissue thermogenesis. Front Endocrinol (Lausanne) 3, 36.
* Monge-Roffarello, B.*, Labbe, S.M.*, Roy, M-C., Lemay, M-L., Coneggo, E., Sanson,
P., Lanfray, D., Richard D. (2014). The cannabinoid receptor 1 is involved in thermogenesis
induced by the brain stimulation of the melanocortin-4 receptor in male rats.
Endocrinology. 155(9): 3448-58.
Les travaux présentés dans ce premier chapitre sont le fruit d’une collaboration de
nombreux membres de l’équipe du Dr Richard : Dr Sébastien M. Labbé (stagiaire postdoctoral),
Marie-Claude Roy (M. Sc., Assistante de recherche), Marie-Laurence Lemay (stagiaire d’été,
étudiante en pharmacologie à l’université Laval), Estelle Coneggo (Stage de fin d’étude,
Étudiante à l’école d’ingénieur polytech de l’université de Nice Sophia Antipolis) et le Dr
Damien Lanfray (Stagiaire postdoctoral). Estelle Coneggo a participé aux expériences
d’injections chroniques, ainsi qu’au soin des animaux de laboratoire nécessaires à ces
expériences. Marie-Laurence Lemay a assuré la préparation logistique et le soin des animaux de
laboratoire nécessaires aux expériences de mesure de température du iBAT et injection de traceur
radioactif. Marie-Claude Roy a apporté sa précieuse aide lors du sacrifice des animaux de

XXV
laboratoire et à la lyse des tissus dans le cadre des mesures de captage de C14-bromopalmitate et
de H3-deoxyglucose. Le Dr Damien Lanfray a participé à la lyse des tissus dans le cadre des
mesures de captage de C14-bromopalmitate et de H3-deoxyglucose, mais a surtout partagé son
savoir-faire dans la quantification d’ARNm. Enfin, le Dr Sébastien M. Labbé c’est très
activement impliqué dans les expériences de mesure de température du iBAT ; il a aussi apporté
son expertise lors des injections de traceurs radioactifs et m’a conseillé tout au long des travaux.
Personnellement, ma participation s’est échelonnée tout au long du projet. J’ai participé à
sa conceptualisation avec le Dr Richard, réalisé les chirurgies stéréotaxiques et les injections
centrales. J’ai aussi eu à développer et mettre au point une méthode fiable afin de mesurer la
température du iBAT, ainsi que des procédures d’hybridation in situ couplées à une
immunohistochimie. Enfin, j’ai participé à l’ensemble des mesures effectuées, à la compilation, à
l’analyse des résultats, et finalement à la rédaction du manuscrit. Les Drs Richard et Labbé m’ont
ensuite fait part de leurs corrections qui ont grandement amélioré le manuscrit, qui a été soumis
en décembre 2013 à la revue Endocrinology. La révision de l’article qui a surtout consisté à
retirer les éléments portant sur la colocalisation de MC4R et de CB1 dans le PVH.
* Monge-Roffarello, B., Labbe, S.M., Lenglos, C., Caron, A., Lanfray, D., Samson, P.,
Richard D. (2014). The medial preoptic nucleus as a site of the thermogenic and metabolic
actions of Melanotan II in male rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307(2):
R158-66.
Les travaux présentés dans ce second chapitre sont le fruit d’une collaboration de nombreux
membres de l’équipe du Dr Richard : le Dr Sébastien M. Labbé (stagiaire postdoctoral), Mr
Alexandre Caron (étudiant au doctorat), le Dr Damien Lanfray (Stagiaire postdoctoral) et Mr
Pierre Samson (professionnel de recherche). Mr Pierre Samson a participé à l’étalonnage et à
l’entretien du système permettant d’évaluer la quantité de CO2 exhalée par les animaux lors des
expériences de mesure de température du iBAT et d’injection de traceur radioactif. Le Dr Damien
Lanfray a apporté sa précieuse aide lors de la lyse des tissus dans le cadre des mesures de captage
de 14C-bromopalmitate et de 3H-deoxyglucose, mais a surtout partagé son savoir faire dans la
quantification des ARNm.

XXVI
Personnellement, ma participation s’est échelonnée toute au long du projet. J’ai participé à
sa conceptualisation avec le Dr Richard, réalisé les chirurgies stéréotaxiques et les injections
centrales. J’ai utilisé mes connaissances acquises lors des études réalisées dans le chapitre 1 pour
réaliser l’ensemble des procédures expérimentales chez l’animal de manière autonome. Enfin, j’ai
participé à l’ensemble des mesures effectuées, à la compilation et à l’analyse des résultats, et
finalement à la rédaction du manuscrit dont Mr Alexandre Caron a effectué des relectures qui ont
amélioré la qualité de l’anglais. Par la suite, les Dr Richard et Labbé m’ont fait part de leurs
corrections qui ont grandement amélioré le manuscrit, qui a été soumis en février dernier à la
revue American Journal of Physiology⎯Regulatory, Integrative and Comparative Physiology.
La révision de l’article a consisté à améliorer la figure 1 en ajoutant un gradient de couleur
permettant d’évaluer l’effet de chacun des sites d’injections de Mélanotane II sur l’élévation de la
température du tissu adipeux brun.

1
Introduction


3
L’obésité est définie par un excès de tissu adipeux blanc. Elle a pour origine une
dérégulation à long terme du bilan d’énergie et a des conséquences majeures sur la santé des
sujets atteints. L’obésité a pris une dimension pandémique au cours des 40 dernières années. En
effet, dans les années 70 la prévalence de l’obésité était de 10% au Canada pour atteindre 26% en
2010, une augmentation qui a des conséquences importantes sur la mortalité précoce (environ
20% des cas sont imputables à l’obésité des sujets) et les hospitalisations. Effectivement,
certaines pathologies sont reconnues comme étant une conséquence directe de l’obésité. Parmi
celles-ci, on compte environ 70 % des cas de diabète de type 2, 25 % des cas d’ostéoarthrite, 18
% des cancers colorectaux et 12 % des cas de dépression (Janssen, 2013).
L’augmentation de la prévalence de l’obésité a des conséquences économiques direct et
indirect important. En effet, les patients obèses sont souvent atteints de complications générées
par les facteurs de comorbidités associés aux pathologies citées précédemment, avec pour
conséquence 3,9 milliards de dollars de dépense dans les soins de santé directs en 2006 pour les
hospitalisations, les médicaments, les visites chez le médecin et les services d’urgences. De plus,
Il faut ajouter 3,2 milliards de dollars de coûts indirects reliés à l’incapacité et à la perte de
productivité en raison de la maladie ou de décès prématurés.
Dans ce contexte, l’amélioration du niveau de compréhension des mécanismes qui
aboutissent à la dérégulation du bilan d’énergie apparaît primordiale. La question des
mécanismes contrôlant la prise alimentaire a été largement discutée. Or, celle ayant attrait à la
dépense énergétique fut peu étudiée jusqu'à la mise en évidence de la présence du tissu adipeux
brun (Brown Adipose Tissue — BAT) fonctionnel chez l’adulte, celui-ci participant la
thermogenèse et donc la dépense d’énergie. Il apparaît donc qu’une meilleure compréhension des
mécanismes centraux contrôlant le BAT représente une nouvelle voie de recherche majeure dans
la lutte contre l’obésité.
L’introduction générale de cette thèse se propose donc de s’intéresser à la régulation du bilan
d’énergie, et plus particulièrement au contrôle des deux principaux acteurs qui y participent, la
prise alimentaire et la thermogenèse. Dans les lignes qui vont suivre, nous allons aborder la
régulation du bilan d’énergie, puis nous mettrons l’emphase sur le BAT principal effecteur de la
thermogenèse et son contrôle central lui permettant d’assurer cette fonction. Enfin, nous
détaillerons l’importance du système mélanocortine dans le contrôle de la thermogenèse.


5
1 La régulation de l’homéostasie énergétique.
L’homéostasie énergétique de l’organisme est finement régulée par le contrôle des apports
et de la dépense énergétique. La prise alimentaire constitue l’unique composante de l’apport
exogène tandis que la dépense énergétique correspond à la somme des dépenses liées aux
métabolisme basal, à l’activité physique volontaire et à la thermogenèse (Richard, 2007). Le bilan
énergétique de l’organisme correspond à la différence entre l’apport et la dépense d’énergie.
Ainsi, dans des conditions d’homéostasie énergétique, cette valeur est égale à zéro, on parle alors
de bilan d’énergie nul. Lorsque le bilan d’énergie est positif, c’est-à-dire que l’apport est
supérieur à la dépense énergétique, l’excès calorique est alors stocké sous forme de graisse. Afin
d’éviter l’accumulation excessive de tissu adipeux, des signaux provenant de la périphérie vont
informer le système nerveux central (SNC) du bilan d’énergie et de l’état des réserves de
l’organisme. Par la suite, le SNC et plus particulièrement l’hypothalamus via des neuropeptides
anaboliques et cataboliques ajuste l’apport en énergie. Parallèlement, le système de récompense
réduit la satiété, alors que le système nerveux sympathique (SNS) stimule la thermogenèse, Ainsi,
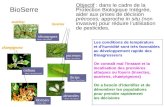
la complémentarité de ces systèmes permet une régulation fine du poids corporel (Figure 1.).
(tractus((gastro+intes/nal)(Ghréline,(GLP+1(
(pancréas,((Tissu(adipeux)((Insuline,(lep/ne((
Réserves d’énergie (graisse)
Prise alimentaire
Anabolique catabolique NPY(AgRP(MCH(
Endocannabïnoides(
α+MSH(CART(
CRH,(TRH(Sérotonine((
Cerveau Hypothalamus
Système de la récompense Complexe dorso-vagal
(
Environnement
Production de chaleur
gas (Gh ( (Gh ( (
R
chaleg (g (
reg (g (
Prode de
SNS(
Thermogénèse(((BAT,(UCP+1)(

6
Figure 1: Régulation de l’homéostasie énergétique.
Abréviations : α-MSH, Alpha-Melanocyte-Stimulating Hormone; AgRP, Agouti-Releated Peptide;
BAT, tissu adipeux brun; CART, Cocaine- and Amphetamine-Regulated Transcript; CRH, Corticotropin-
Releasing Hormone; GLP-1, Glucagon-Like Peptide 1; MCH, Melanin-Concentrating Hormone; NPY,
Neuropeptide Y; SNS, Système Nerveux Sympathique; TRH, Thyrotropin-Releasing Hormone; UCP-1,
Uncoupling Protein 1. Traduit et adapté de Richard and Boisvert., The neurobiology of obesity, Obesity
supp vol. 14, 2006.
1.1 Les signaux périphériques régulant l’homéostasie énergétique.
Les signaux périphériques régulant l’homéostasie énergétique sont libérés de manière
séquentielle par différents organes durant ou consécutivement à l’ingestion de nourriture. Aussi,
selon le moment où ils sont mis en action, on distingue les stimuli de la phase céphalique
(sensoriels), de la phase gastrique (mécaniques, chimiques et hormonaux) et de la phase
postprandiale (nutriments et hormones) (Berthoud, 2002). (Figures 2 et 3.).
Figure 2: Représentation spatiotemporelle des différentes phases d’un cycle de prise
alimentaire.
Le dégradé de noir et blanc représente l’intensité de la sensation de satiété et de faim.
Abréviation : DR, début du repas; FR, fin du repas; PRC, Phase de réponses céphaliques; PSC,
poursuite de stimulation céphalique. Traduit de (Smeets et al., 2010).
PRC$ PSC$Phase$céphalique$
Phase$gastrique$Inges6on$$$$$$Diges6on$$$$$$$$Absorp6on$Sa6a6on$$$$$$$$$$$$$$$$$$$$$$$$$$$$Sa6été$
Faim$
DR$ FR$ DR$FR$ DR$DR$intervalle$entre$les$repas$$
$ striq $
distension$de$l’estomac,$détec6on$des$nutriments$$et$libéra6on$d’hormones$
PRC$ PSC$
La$pensée,$l’an6cipa6on,$la$vision,$l’odorat,$le$gout$et$les$sensa6ons$oropharyngées$$$

7
Figure 3: Modèle d’intégration des signaux d'adiposité et de satiété.
Abréviation : AP, l’area postrema; CCK, la cholécystokinine; GLP-1, Glucagon-Like Peptide 1; NTS, le
noyau du tractus solitaire; OXM, l’oxyntomoduline; PP, le polypeptide pancréatique; PYY (3-36), le
peptide yy (3-36). Traduit de (Wren and Bloom, 2007).
Afférences vagales
Intestin
Pancréas
Tissu adipeux
estomac
étirement
nutriments
Ghréline
insuline
leptine
Adiponectine
Visfatine Resistine

8
1.1.1 Les signaux sensoriels et la phase céphalique.
Avant la phase d’ingestion, les signaux sensoriels visuels, olfactifs, tactiles et gustatifs vont
guider nos choix alimentaires. En effet, au cours de l’apprentissage, ces différents sens font appel
à la partie hédonique du SNC, dans le but d’attribuer une valeur palatable aux aliments. Il a été
montré chez l’humain et d’autres primates que l’information visuelle est intégrée avec les
informations provenant de l’olfaction et du goût pour former des associations polymodales au
niveau du cortex (Berthoud, 2002). De plus, l’activation de ces sens influe sur la physiologie
digestive en permettant d’initier la digestion céphalique, qui prépare le tractus gastro-intestinal,
afin d’optimiser notamment la digestion et l’absorption des nutriments. Enfin, la phase
céphalique permet à l’organisme d’anticiper l’élévation de la glycémie (Smeets et al., 2010), et
intervient également dans l’activation de la thermogenèse (LeBlanc, 2000).
1.1.2 L’effet des signaux digestifs et des métabolites sur la prise alimentaire.
Les signaux digestifs sont secrétés en fonction de la nature physico-chimique du bol
alimentaire. En effet, les caractéristiques physiques du bol alimentaire sont détectées par les
mécanorécepteurs du tube digestif, qui remplissent une fonction très importante dans le contrôle
de la prise alimentaire (Powley and Phillips, 2004). Lors de la distension gastrique, consécutive
au passage du bol alimentaire dans l’estomac, les mécanorécepteurs de la paroi gastrique
stimulent le nerf vague qui communique avec le SNC pour mettre en place une réponse
anorexigène chez le rat (Phillips and Powley, 1996; Berthoud et al., 2001). Ainsi, il a été montré
que l’ingestion d’un bol alimentaire volumineux mais à faible valeur calorique chez l’humain a
un court effet inhibiteur sur la réalimentation (Kral and Rolls, 2004).
Parallèlement, l’intestin est également capable d’intégrer les caractéristiques chimiques du
bol alimentaires. En effet, dans l’intestin les aliments sont dégradés en macro- et
micronutriments, facilement transportables et assimilables par l’organisme. La nature de ces
nutriments peut être détectée grâce à des chémorécepteurs spécifiques présents sur les différents
types de cellules de l’épithélium intestinal (Powley and Phillips, 2004).
L’activation de ces cellules par leur nutriment respectif entraîne une sécrétion d’hormones
anorexigènes (Badman and Flier, 2005) (Figure 3.). D’autre part, les nutriments que sont les

9
métabolites, c’est à dire le glucose, les acides aminés et les acides gras, peuvent agir directement
sur le SNC (Blouet and Schwartz, 2010). De ce fait, l’augmentation de leurs concentrations
plasmatiques, s’accompagne d’une inhibition proportionnelle de la prise alimentaire.
1.1.2.1 Les signaux gastro-intestinaux.
Le tractus gastro-intestinal est l’organe endocrinien le plus volumineux de l’organisme.
Celui-ci synthétise de nombreuses hormones d’environ 10 à 50 acides aminés qui participent au
contrôle de la prise alimentaire et de la dépense énergétique (Lancha et al., 2012). Parmi celles-ci,
la ghréline qui est synthétisée par l’estomac est la seule hormone orexigène circulante connue à
ce jour (Wren et al., 2001). De plus, les signaux anorexigènes comme la cholécystokinine (CCK),
le glucagon-like peptide 1 (GLP-1) et le peptide YY (PYY) sont secrétés par l’intestin (Troke et
al., 2014).
L’estomac.
La ghréline constitue la principale hormone libérée dans la circulation sanguine par
l’estomac. Celle-ci est sécrétée par les cellules X/A du fundus de l’estomac. Son taux
plasmatique augmente consécutivement au jeûne et diminue dès l’absorption de nutriments. Des
études réalisées chez l’Homme et le rongeur indiquent qu’une injection chronique de ghréline par
voie sanguine s’accompagne d’une augmentation de la masse adipeuse (Tschöp et al., 2000;
Wren et al., 2001; Rodríguez et al., 2009) et dont l’augmentation dans le sang chez l’Homme est
corrélé avec une diminution de la dépense énergétique (St-Pierre et al., 2004), indiquant que cette
hormone exerce des effets pro-anaboliques. De plus, des études réalisées chez le rongeur
montrent que les effets de cette hormone sont également observés suite à une injection centrale,
suggérant que les effets de la ghréline sont relayés en partie par le SNC (Tschöp et al., 2000;
Asakawa et al., 2001). Par ailleurs des études réalisées par Chen et ses collaborateurs indiquent
que la ghréline constitue à ce jour l’une des rares hormones orexigènes identifiées, capable de
stimuler l’activité des neurones à neuropeptide Y (NPY) et/ou à agouti-related peptide (AgRP)
situés dans le noyau arqué de l’hypothalamus (ARC) (Chen et al., 2004a), suggérant que les
effets pro-anaboliques sont relayés en partie par l’activation de cette population de neurones.
Enfin, il a été montré chez la souris que l’effet négatif de la ghréline sur la dépense énergétique
est relayé par l’activation du récepteur de la ghréline (GHS-R) présent au niveau du BAT (Lin

10
and Sun, 2012), suggérant que la ghréline exerce ces effets via l’activation simultanée de cibles
centrales et périphérique via le BAT.
L’intestin.
L’intestin est subdivisé en plusieurs parties, incluant le duodénum, situé juste après le
pylore gastrique, puis le jéjunum, l’iléon et le colon qui est relié au rectum.
Au niveau du duodénum et du jéjunum, les cellules de type I libèrent de la CCK en réponse
aux lipides et aux protéines contenus dans le bol alimentaire. La CCK fut la première hormone
gastro-intestinale anorexigène à être identifiée chez le rat (Gibbs et al., 1973). Ainsi, des travaux
réalisé chez l’homme montrent que l’injection périphérique de CCK induit une diminution dose
dépendante de la prise de nourriture (Lieverse et al., 1995). Des études réalisées chez le rongeur
suggèrent de plus que ces effets anorexigènes passent par l’activation des récepteurs CCK 1 et 2
situés sur le nerf vague et dans de nombreuses régions cérébrales (Moran et al., 1997).
Cependant, à long terme les animaux vont compenser l’effet anorexigène aigu observé. Par
ailleurs, des études réalisées par Smith et ses collaborateurs chez le rat, montrent que la CCK ne
modifie pas le comportement alimentaire chez des animaux ayant préalablement subi une
vagotomie, indiquant que les effets anorexigènes de la CCK sont relayés par le nerf vague (Smith
et al., 1981). Enfin, des travaux plus récents indiquent que l’injection périphérique/centrale de la
CCK pourrait également stimuler la dépense énergétique (Lo et al., 2010), toutefois la nature des
voies impliquées dans cette réponse pro-anabolique est actuellement indéterminée.
Plus en aval dans l’intestin, les cellules L de l’iléon secrètent du GLP-1 consécutivement au
passage du bol alimentaire. Celui-ci, en plus d’activer les voies anorexigènes centrales, participe
également à la régulation (i) de la sécrétion d’insuline par les cellules β pancréatiques, (ii) de la
motilité gastrique et (iii) de la concentration plasmatique de glucagon (Drucker, 2005). Le GLP-1
est une hormone essentielle dans la régulation de l’homéostasie énergétique, car en agissant
positivement et rapidement sur la satiété, elle permet de limiter la quantité d’aliments ingérés et
d’augmenter le délai entre les repas (Williams et al., 2009). Enfin, des études réalisées chez le rat
montrent que l’injection intraveineuse de GLP-1 s’accompagne d’une augmentation de la
consommation d’oxygène (Osaka et al., 2005). Toutefois, des études réalisées par la suite chez la
souris montrent que l’invalidation du gène codant pour le récepteur de GLP-1 (GLP-1R) ou bien

11
l’administration périphérique d’un antagoniste de ce récepteur s’accompagnent d’une
augmentation de la dépense énergétique (Hansotia et al., 2007; Knauf et al., 2008), indiquant que
les mécanismes de régulation de ce récepteur nécessite d’être mieux appréhendé.
Enfin, suite au passage du bol alimentaire dans la partie distale de l’intestin, les cellules de
type L localisées au niveau du colon secrètent du PYY dans la circulation sanguine. Le PYY est
une hormone présente chez tous les vertébrés et codée par un gène paralogue au NPY (Tatemoto
and Mutt, 1980). Le PYY est secrété sous deux formes aux caractéristiques pharmacologiques
distinctes, le PYY (1-36) et le PYY (3-36) qui constitue la forme majoritaire (Grandt et al., 1994;
Kirchner et al., 2010). Ainsi, des travaux réalisés chez le rongeur indiquent que le PYY1-36 exerce
des effets orexigènes en se liant spécifiquement aux récepteurs du NPY de types 1 (Y1) et 5 (Y5)
(Kanatani et al., 2000). Toutefois, des études réalisées chez le rongeur et l’homme indiquent que
le PYY (3-36) exerce quant à lui des effets anorexigènes et pro-anaboliques via l’activation du
récepteur du NPY de type 2 (Y2), (Dumont et al., 1995; Batterham et al., 2002; Sloth et al., 2007;
De Silva et al., 2011) dans l’ARC (Teubner and Bartness, 2013). Enfin, la sécrétion de la forme
clivée de PYY est sous le contrôle de la dipeptidyl-peptidase 4 (DPP-4), suggérant que synthèse
de DPP-4 est essentiels dans la régulation de la balance énergétique (Troke et al., 2014).
1.1.2.2 L’effet des métabolites.
Les métabolites, tel que le glucose, les acides aminés et les acides gras, sont issus de la
dégradation des aliments par les processus de digestion et peuvent être stockés puis libérés par le
tissu adipeux et le foie lors d’un jeûne prolongé et même le muscle squelettique dans des cas
extrêmes. Les variations de taux circulants des différents métabolites sont détectées au niveau de
l’hypothalamus afin de déterminer le statut énergétique de l’organisme.
Le glucose.
Les premières études visant à évaluer la capacité du SNC à intégrer les variations de la
glycémie, ont montré qu’une altération centrale de la glycémie s’accompagne d’une diminution
de la dépense énergétique et d’une augmentation de la consommation de nourriture chez le
rongeur (Müller et al., 1973). Des expériences réalisées par la suite chez les rongeurs montrent
que la prise de nourriture consécutive à une période d’hypoglycémie est inhibée par l’injection

12
périphérique de glucose (Campfield et al., 1985), suggérant que le glucose constitue à lui seul un
signal anorexigène. Par ailleurs, une étude réalisée chez le rongeur indique que la prise
alimentaire peut être stimulée par l’injection centrale de 2-deoxyglucose, un analogue non
métabolisable du glucose, chez des animaux nourris ad libitum (Berthoud and Mogenson, 1977).
Enfin, des travaux réalisés en collaboration avec notre laboratoire montrent que l’intégration par
le SNC des variations de la glycémie impliquent la libération d’endozépines par les cellules
astrogliales hypothalamiques (Lanfray et al., 2012).
Les acides aminés.
Au milieu du siècle dernier, Mellinkoff suggéra l’existence d’une relation directe entre les
taux circulants d’acides aminés et le sentiment de satiété (Mellinkoff et al., 1956). Plus tard, des
études ont mis en évidence qu’un régime alimentaire riche en protéines est suivi, dès le premier
jour, par une diminution de la prise alimentaire et une augmentation de la concentration en acides
aminés dans le sang et dans le liquide céphalorachidien (Peters and Harper, 1987; Harper and
Peters, 1989). Par la suite, il a été montré par microdialyse, que les concentrations en acides
aminés essentiels, telles que la leucine, sont augmentées dans certains noyaux hypothalamiques
consécutivement à la prise d’un repas hyper-protéiné (Currie et al., 1995), suggérant que les
variations de leurs concentrations plasmatiques peuvent être intégrées par certaines structures du
SNC. Ainsi, des travaux réalisés par Cota et ses collaborateurs ont montré que l’injection centrale
de leucine induit une diminution de la prise alimentaire chez des rats à jeun (Cota et al., 2006).
Enfin, les travaux de Schwartz et ses collaborateurs ont montré que les effets satiétogènes d’une
injection centrale de leucine sont relayés par l’activation des neurones à pro-opiomélanocortine
(POMC)/ cocaine- and amphetamine- regulated transcript (CART) du ARC et s’accompagnent
d’une stimulation des neurones à ocytocine, du noyau paraventriculaire de l’hypothalamus (PVH)
et des fibres sympathiques du noyau du tractus solitaire (NTS) (Blouet et al., 2009).
Les acides gras.
À la même époque que Mellinkoff, Kennedy suggéra que les acides gras circulants sont
impliqués dans le contrôle de la prise alimentaire (Kennedy, 1953). De nos jours, il est bien établi
que l’intégration centrale des variations de la lipidémie plasmatique participe à la régulation (i)
du comportement alimentaire, (ii) de la sécrétion d’insuline ainsi que (iii) de la production

13
hépatique de glucose. Ainsi, des travaux réalisés chez le rat par Obici et Schwinkendorf ont
montré que l’injection centrale d’acide oléique induit une diminution de la prise alimentaire et de
la production hépatique de glucose (Obici et al., 2002). Ces effets pro-cataboliques
s’accompagnent d’une stimulation des neurones à POMC/CART et d’une inhibition de l’activité
des neurones à NPY/AgRP (Obici et al., 2002; Schwinkendorf et al., 2011), suggérant que ces
populations neuronales participent à l’intégration centrale des variations de la lipidémie.
1.1.3 Les signaux postprandiaux.
Les signaux postprandiaux participant à la régulation de l’homéostasie énergétique sont
libérés par différents tissus en réponse aux variations des taux circulant de métabolites. Les deux
signaux post-prandiaux les mieux caractérisés sont l’insuline et la leptine, qui sont
respectivement produits par le pancréas et le tissu adipeux blanc.
1.1.3.1 L’insuline.
L’insuline est secrétée par les cellules β du pancréas lors de l’augmentation de la glycémie
en condition postprandiale (Granger and Kushner, 2009). L’insuline est une hormone
hypoglycémiante dont les effets sont opposés à ceux du glucagon produit par les cellules α
pancréatiques, et qui permet de maintenir la glycémie proche d’une valeur moyenne de 5 mM
chez le rongeur et l’homme (Jiang and Zhang, 2003). Des études de liaison radioactive, réalisées
au début des années 70, ont permis d’identifier le récepteur de l’insuline (INS-R), un récepteur
ubiquitaire présent chez tous les vertébrés (Freychet et al., 1971). Par la suite, des résultats ont
montré que l’injection centrale d’insuline induit une diminution de la prise alimentaire ainsi
qu’une augmentation de la dépense énergétique chez le singe (Woods et al., 1979), suggérant que
l’insuline est capable d’activer certaines voies neuronales pro-cataboliques. Par ailleurs, des
expériences réalisées chez le rongeur indiquent que l’injection intrapéritonéale d’insuline stimule
l’expression de la POMC dans le ARC, suggérant que cette hormone est capable de traverser la
barrière hémato-encéphalique et exerce ses effets anorexigènes via l’activation de la voie du
système mélanocortine (Kim et al., 1999). Enfin, une étude réalisée chez le rat montre que l’effet
anorexigène d’une injection centrale d’insuline est bloqué par un antagoniste des récepteurs aux
mélanocortines 3 et 4 (MC3/4R) (Benoit et al., 2002), confirmant ainsi que l’effet satiétogène de
l’insuline est relayé par l’activation du système mélanocortinique.

14
1.1.3.2 La leptine.
La leptine est une adipokine de 16 kDa codée par le gène ob (Zhang et al., 1994),
principalement produite par les adipocytes blancs (Ahima and Osei, 2004). Les taux circulants de
cette adipokine sont proportionnels à la quantité de tissu adipeux et reflètent de ce fait le statut
énergétique de l’organisme. Les obeses présentent par conséquent des taux sanguins de leptine
supérieur à la normale, qui a pour conséquence la mise en place à long terme d’une résistance à la
leptine (Carter et al., 2013). Les modèles transgéniques murins déficients pour le gène de la
leptine (souris ob/ob) ou pour le gène du récepteur de la leptine (souris db/db) présentent un
phénotype obèse caractérisé par une augmentation de la prise alimentaire et une diminution de la
dépense énergétique (Friedman and Halaas, 1998). L’administration chronique de leptine à des
souris ob/ob entraîne une réduction de la prise alimentaire, une augmentation de la dépense
énergétique et une réduction rapide de la masse corporelle (Weigle et al., 1995). Le gène db qui
code pour le récepteur de la leptine permet, par des mécanismes d’épissage alternatif, la synthèse
de plusieurs isoformes (courtes, longues et secrétées) (Moran and Phillip, 2003). Au niveau du
SNC, la leptine se lie à la forme longue Ob-Rb dont l’expression est très élevée dans
l’hypothalamus ventromédian (Funahashi et al., 2003). L’invalidation du gène db, spécifiquement
dans le SNC, induit un phénotype obèse, hyperphage et hypométabolique (Seeley et al., 1996).
Enfin, l’administration de leptine diminue l’expression des acides ribonucléiques messagers
(ARNm) codant pour les gènes NPY et AgRP, tout en augmentant ceux codant pour le gène de la
POMC (Cowley et al., 2001), suggérant que les effets de la leptine sont relayés par la stimulation
et l’inhibition des voies neuronales hypothalamiques respectivement anorexigènes et orexigènes.
Par ailleurs, de nombreuses adipokines tels que l’adiponectine, la resistine et l’acylation
stimulating protein issues elles aussi du tissu adipeux blanc semblent également impliquées dans
la régulation de l’homéostasie énergétique (Lancha et al., 2012), toutefois l’importance de ces
adipokines in vivo reste à être déterminée sachant que leurs secrétions n’est pas synchrone avec la
phase postprandiale.

15
1.2 La régulation centrale de l’homéostasie énergétique.
Le système nerveux central (SNC) est le siège de l’intégration des différents signaux
périphériques relatifs au statut énergétique de l’organisme. Chacun des signaux périphériques
mobilise une ou plusieurs voies neuronales spécifiques afin de générer une multitude de réponses
contrôlant les deux leviers de la régulation de l’homéostasie énergétique. Parmi les différentes
structures du SNC impliquées dans ces réponses, l’hypothalamus est considéré comme le
principal centre intégrateur régulant l’homéostasie énergétique. Néanmoins, d’autres structures,
tels que le noyau du tractus solitaire (NTS), une structure de la région pontique contrôlant
l’activité du système nerveux autonome (SNA) et le métabolisme périphérique (Schwartz et al.,
2000; Myers and Olson, 2012), agissent en synergie avec l’hypothalamus afin de réguler la
balance énergétique.
1.2.1 Le rôle de l’hypothalamus dans l’homéostasie énergétique.
L’hypothalamus est la partie antéro-inférieure du diencéphale. Il est délimité dans sa partie
rostrale par le chiasma optique, la lamina terminalis et la commissure antérieure, alors que dans
sa partie caudale il est circonscrit par un plan vertical postérieur aux corps mamillaires. Au
niveau dorsal, il est bordé par le thalamus, alors que du côté ventral le plancher hypothalamique
est en contacte direct avec le liquide céphalorachidien (LCR) (figure 3).
Le rôle essentiel de l’hypothalamus dans le contrôle de l’homéostasie énergétique fut
suggéré dès les années 40, par des travaux montrant que la lésion du noyau ventromédian
entrainait une hyperphagie et une obésité alors qu’une lésion de l’hypothalamus latéral conduisait
à une réduction de la prise alimentaire et à la perte de poids chez le rat (Hetherington and Ranson,
1940; Anand and Brobeck, 1951). Par la suite, de nombreuses études ont montré l’implication de
l’ARC dans la régulation de l’homéostasie énergétique.

16
Figure 4: Section sagittale du cerveau de rat montrant l'emplacement de l'hypothalamus.
Figure 5: Représentation tridimensionnelle de l'hémisphère droit de l'hypothalamus du rat.
Abréviations : AHA, aire hypothalamique antérieure; ARC, noyau arqué; AV3V, aire
antéroventrale du 3éme ventricule; CI, capsule interne; DP, sous noyau parvocellulaire dorsal du noyau
paraventriculaire (PVH); DMH, noyau dorsomédian; F, fornix; LHA, aire latérale; LM, sous noyau
magnocellulaire latéral du PVH; LPOA, l’aire préoptique latérale; ME, éminence médiane; MP, sous
noyau parvocellulaire médial du PVH; MPO, noyau préoptique médial; OT, tractus optique; SCh, noyau
suprachiasmatique; SON, noyau supraoptique; SI, substantia inomminata; ST, noyau sous-thalamique;
VMH, noyau ventromédian; VP, sous noyau parvocellulaire ventral du PVH. Tirée de (Berthoud, 2002).

17
L’ARC est situé immédiatement au-dessus de l’éminence médiane, un organe circum-
ventriculaire permettant le passage des hormones et des nutriments du sang vers le LCR, et est en
mesure de percevoir les modifications du statut énergétique de l’organisme (Schwartz et al.,
2000; Dhillo, 2007). L’ARC est un noyau composé notamment de deux populations de neurones
distinctes dites de premier ordre, agissant de façon opposée sur la prise alimentaire. L’une
exprime la POMC, le précurseur de l’alpha melanocyte-stimulating hormone (α-MSH) (Poggioli
et al., 1986) et le cocaine and amphetamine-regulated transcript (CART) (Kristensen et al.,
1998; Elias et al., 1998b) qui sont des neuropeptides anorexigènes, tandis que l’autre produit le
NPY (Stanley et al., 1986; Hahn et al., 1998) et l’agouti-related protein (AgRP) (Hahn et al.,
1998; Broberger et al., 1998a), deux neuropeptides orexigènes. Ces deux populations neuronales
projettent vers différents neurones dits de second ordre, situés dans l’ensemble de l’hypothalamus
et notamment dans le noyau paraventriculaire (PVH), l’aire latérale (LHA), le noyau
ventromédian (VMH) et le noyau dorsomédian (DMH) de l’hypothalamus. Ces derniers sont
largement impliqués dans le contrôle de la prise alimentaire et/ou dans la régulation de
l’homéostasie énergétique (DeFalco et al., 2001; Berthoud, 2002; Myers and Olson, 2012; Parker
and Bloom, 2012). Les principaux peptides identifiés comme impliqués dans le contrôle de la
prise alimentaire sont recensés dans le tableau 1.
1.2.1.1 Les neurones de premier ordre du noyau arqué.
Les neurones POMC/CART.
Bien que l’α-MSH ai été découverte à la fin des années 50 (Harris, 1959), son précurseur, la
POMC, ainsi que son principal lieu de synthèse, l’ARC, ne furent identifiés qu’à la fin des années
70 (Mains et al., 1977; Jacobowitz and O'Donohue, 1978). Durant la première moitié des années
90, 5 récepteurs aux mélanocortines (MCR) furent caractérisés (Mountjoy et al., 1992; Chhajlani
et al., 1993; Gantz et al., 1993a; 1993b; Labbé et al., 1994), dont notamment les MCR de types 3
et 4, deux récepteurs présents exclusivement au niveau du SNC (Mountjoy, 2010) (Cf. 3.1.1.2).
Des expériences, réalisées à l’aide de la technique d’hybridation in situ chez le rongeur, indiquent
que le jeûne aigu ou chronique s’accompagne d’une diminution des taux d’ARNm codant la
POMC au niveau de l’ARC, et que ces effets sont renversés suite à la réalimentation des

18
Tableau 1: Principaux facteurs hypothalamiques impliqués dans la régulation de
l’homéostasie énergétique.
peptides Récepteur(s) impliqué(s)
Site(s) d’expression dans l’hypothalamus
références
Facteu
rs orexigène
s
AgRP MC3-‐R, MC4-‐R ARC (Fong et al., 1997) Beacon N.D. ARC, LHA, PVH (Collier et al., 2000)
Dynorphine Kappa PVH (Lambert et al., 1993) GABA GABAa, GABAb ARC, VMH (Patel and Ebenezer, 2004)
Galanine Gal-‐R1, -‐R2, -‐R3 ARC, DMH, LHA (Branchek et al., 2000) Galanin-‐like peptide Gal-‐R2 ARC, PVH (Seth et al., 2004)
Ghréline GHS-‐R ARC (Wren et al., 2001) MCH MCH1-‐R, MCH2-‐R LHA (Qu et al., 1996)
Neuropeptide Y Y1-‐R, Y2-‐R, Y5-‐R ARC, DMH, VMH (Erickson et al., 1996) Nociceptine ORL-‐1 ARC (Polidori et al., 2000) Orexine A Ox1-‐R, Ox2-‐R LHA (Haynes et al., 1999; Shiraishi et al., 2000)
VGF N.D. ARC (Hahm et al., 2002) amandamine CB1R ARC, LHA, VMH, DMH, PVH (Ameri, 1999) 26RFa/43RFa GPR103 LHA (do Rego et al., 2006)
Facteu
rs ano
rexigène
s
α-‐MSH MC3-‐R, MC4-‐R ARC (Fan et al., 1997; Yaswen et al., 1999) Apéline APJ ARC, PVH (Reaux et al., 2002) BDNF TrkB VMH (Xu et al., 2003)
Bombésine/GRP GRP-‐R PVH (Wada et al., 1998) CART N.D. ARC, DMH, LHA (Bannon et al., 2000) CGRP CGRP-‐R ARC, LHA (Dhillo et al., 2003) CRH CRH1-‐R, CRH2-‐R PVH (Richard et al., 2000) ODN RCPG de l’ODN ARC (do Rego et al., 2007)
Dopamine D1, D2 ARC, DMH (Ladurelle et al., 1991)
GLP-‐1 GLP1-‐R ARC, DMH, LHA, PVH, VMH (Tang-‐Christensen et al., 1996; Peters et al., 2001)
GLP-‐2 GLP2-‐R DMH (Tang-‐Christensen et al., 2000) Nesfatine-‐1 N.D. ARC, PVH (Kohno et al., 2008)
Neuromédine S NMU2R PVH (Ida et al., 2005) Neuromédine U NMU2R VMH (Kowalski et al., 2005)
Neuropeptide B et W GPR7 PVH (Ishii et al., 2003) Neurotensine NTR-‐1 ARC, DMH, PVH, VMH (Remaury et al., 2002)
NPFF NPFF-‐1, NPFF-‐2 VMH, DMH (Sunter et al., 2001) Ocytocine OXT-‐R PVH (Arletti et al., 1989; Olson et al., 1991) PACAP N.D. ARC, VMH (Chance et al., 1995; Mizuno et al., 1998) PrRP GPR10 DMH (Gu et al., 2004)
Sérotonine 5HT1, 5HT2 PVH (Dourish, 1995; Currie et al., 2002) Somatostatine N.D. ARC, VMH (Scalera and Tarozzi, 1998)
TRH TRH-‐R1 PVH (Kow and Pfaff, 1991) Urocortine CRH2-‐R VMH, LHA (Ohata et al., 2000; Reyes et al., 2001)
Abreviations: ARC, noyau arqué de l’hypothalamus; DMH, noyau dorsomédian de l’hypothalamus; LHA,
l’aire latérale de l’hypothalamus; PVH, noyau paraventriculaire de l’hypothalamus; VMH, noyau
ventromédian de l’hypothalamus.

19
animaux, indiquant que l’expression de la POMC au niveau de l’ARC est fonction du statut
énergétique de l’organisme (Swart et al., 2002).
Chez la souris, l’invalidation du gène de la POMC induit un phénotype obèse, hyperphage
et hypométabolique, indiquant que la POMC et ses dérivés jouent un rôle essentiel dans le
maintien de l’homéostasie énergétique chez le rongeur (Yaswen et al., 1999; Challis et al., 2004).
De même, plusieurs cas d’obésité sévère diagnostiqués chez l’homme sont associés à la présence
de mutations sur le gène codant pour la POMC (Krude et al., 1998), néanmoins les cas de perte
de fonction complète ont très rarement été répertoriés (Farooqi and O'Rahilly, 2006).
Les premières études pharmacologiques réalisées via l’injection intra-cérébro-ventriculaire
(i.c.v.) d’α-MSH, ou de son analogue cyclique synthétique, le melanotan-II (MTII), montrent une
diminution de la prise alimentaire, qui s’accompagne d’une perte de poids lors d’un traitement
chronique chez le rongeur (Fan et al., 1997), indiquant que l’α-MSH est un puissant facteur
anorexigène. D’autre part, des études réalisées chez le rongeur indiquent que les effets du MTII
sur la prise alimentaire peuvent être abolis par la co-injection d’un antagoniste des MC3/4R
(Rossi et al., 1998). L’utilisation de modèles génétiques a permis de mettre en évidence
l’importance de MC4R dans le contrôle de la prise alimentaire et dans la régulation de la balance
énergétique. En effet, les souris ayant une délétion du gène codant pour MC4R présentent une
hyperphagie accompagnée d’une obésité sévère (Huszar et al., 1997), ainsi qu’une absence
d’inhibition de la prise alimentaire lors de l’administration d’un agoniste aux MC3/4R (Chen et
al., 2000b). D’autre part, la délétion du gène codant MC3R n’entraine par de modification du
comportement alimentaire, cependant ces souris sont obèses, suggérant ainsi un rôle dans la
régulation du métabolisme périphérique (Chen et al., 2000a). Par ailleurs, la présence du MC3R
sur les neurones POMC de l’ARC, suggère que MC3R est un autorécepteur qui module la
synthèse de son propre agoniste, l’α-MSH (Jégou et al., 2000; Lee et al., 2008). Cependant,
l’implication exacte de cette fonction d’autorécepteur dans le métabolisme périphérique reste à
être précisée. Enfin, la double délétion de MC3/4R conduit à une hyperphagie importante et une
obésité extrême chez la souris, démontrant ainsi le rôle primordial du système mélanocortinique
dans la régulation de l’homéostasie énergétique de l’organisme (Atalayer et al., 2010). Chez
l’Homme, les mutations du gène codant pour MC3R sont associées avec de rares cas d’obésités

20
sévères, tandis que des mutations du gène codant pour MC4R sont à l’origine de 5 à 6 % des cas
d’obésité morbide (Yeo et al., 1998; Farooqi and O'Rahilly, 2006).
Le CART est exprimé dans l’ARC au sein des mêmes neurones que la POMC (Elias et al.,
1998a). De façon similaire à la POMC, il voit ses niveaux d’expression diminués chez des rats
soumis à une privation aigue, ainsi que chez plusieurs modèles de souris obèses (Kristensen et al.,
1998), tandis que l’administration de la leptine augmente l’expression de l’ARNm du CART dans
l’ARC (Kristensen et al., 1998). L’administration centrale de CART chez les rongeurs diminue la
prise alimentaire, tandis que l’injection d’anticorps anti-CART l’augmente, démontrant ainsi le
rôle anorexigène de CART (Lambert et al., 1998). De plus, l’injection i.c.v. de CART inhibe
l’effet orexigène du NPY (Kristensen et al., 1998; Lambert et al., 1998). De façon surprenante,
l’injection de CART dans l’ARC augmente la prise de nourriture chez le rongeur (Abbott et al.,
2001). Ces résultats contradictoires suggèrent qu’il existe plusieurs populations neuronales
exprimant le CART et qu’elles exercent des rôles différents dans le contrôle de l’homéostasie
énergétique. Cependant, le récepteur du peptide CART reste à être élucidé (Harrold et al., 2012).
Les neurones NPY/AgRP.
Le NPY est un peptide de 36 acides aminés, dont les extrémités N- et C- terminales sont
constituées de tyrosine (Tatemoto et al., 1982). L’expression de l’ARNm de NPY au niveau de
l’ARC est doublée lors d’un jeûne, aigu ou chronique, puis on observe un retour vers des valeurs
basales après réalimentation des animaux (Sahu et al., 1988; Sanacora et al., 1990; Swart et al.,
2002). L’injection de NPY centralement, ou directement dans le PVH ou le LHA, initie la prise
d’un repas, dont la taille et la durée sont plus importantes que la normale (Stanley et al., 1985).
Par ailleurs, l’injection chronique de NPY chez le rat induit une obésité (Zarjevski et al., 1993).
L’ensemble des effets orexigènes du NPY sont relayés par les récepteurs Y1 et Y5 que l’on
retrouve dans differentes noyaux hypothalamiques et notamment dans le PVH et le LHA (Kopp
et al., 2002; Morin and Gehlert, 2006). Ces résultats montrent l’importance du NPY dans le
contrôle de la prise alimentaire. Cependant, des études sur les modèles de souris transgéniques
n’exprimant pas le NPY montrent que celles-ci ont un poids corporel et une masse adipeuse
normaux, s’ils reçoivent une diète de laboratoire standard (Thorsell and Heilig, 2002), alors que
les animaux nourris avec une diète riche en gras sont résistants à l’obésité (Patel et al., 2006).

21
L’absence d’un phénotype «maigre» chez ces souris transgéniques suggère la présence de
mécanismes orexigènes compensatoires. En effet, une étude réalisée sur ce modèle montre qu’un
jeûne augmente l’expression du gène codant pour l’AgRP dans les neurones de l’ARC et que
l’injection de ce peptide entraine une augmentation importante de la prise alimentaire
comparativement à la souris sauvage (Marsh et al., 1999), indiquant que l’action de ce peptide
pourrait compenser l’absence de NPY chez le rongeur.
L’AgRP fut découverte sur un modèle de souris obèse, hyperphagique et avec un pelage
jaune, dont le phénotype est dû à la surexpression de la protéine agouti, dans l’ensemble de
l’organisme (Lu et al., 1994). Au sein du SNC, l’AgRP est exclusivement synthétisée par des
neurones de l’ARC et 95 % d’entre eux co-expriment le NPY (Broberger et al., 1998b). Lors d’un
jeûne de 24 h l’expression de l’AgRP est augmentée chez la souris suggérant ainsi son
implication dans le contrôle de la prise alimentaire (Swart et al., 2002). En effet, Il a été montré
que comme le NPY, l’AgRP induit des effets orexigènes lorsqu’il est injecté en i.c.v. (Rossi et
al., 1998) ou directement dans le PVH ou le DMH (Kim et al., 2000). L’action de l’AgRP a la
particularité d’être soutenue dans le temps, en effet, l’hyperphagie occasionnée chez le rat peut
durer jusqu'à 7 jours (Hagan et al., 2000). De la même façon que la protéine agouti, l’AgRP agit
comme un antagoniste naturel des récepteurs MC3/4R (Ollmann et al., 1997), le MC4R étant
constitutivement actif en absence de tout ligand, l’AgRP joue conséquemment le rôle d’agoniste
inverse (Nijenhuis et al., 2001).
1.2.1.2 Les neurones de second ordre.
Les principales populations neuronales de second ordre expriment les récepteurs MC3/4R,
Y1 et Y5 et sont présentes dans le LHA, le PVH, le VMH et le DMH.
Dans le LHA, les axones des neurones POMC/CART et NPY/AgRP de l’ARC forment des
contacts synaptiques avec différents neurones secrétant des neuropeptides tels que la melanin-
concentrating hormone (MCH) et l’orexine (Badami et al., 2010). En effet, dès 1998 Broberger et
ses collaborateurs ont montré que les neurones exprimant la MCH et l’orexine dans le LHA sont
les cibles des neurones NPY/AgRP de l’ARC (Broberger et al., 1998a). Par la suite, il a été
montré que l’injection centrale d’AgRP chez des souris nourries ad libitum stimule l’expression
de la MCH, cependant l’AgRP n’a pas d’effet sur l’expression de l’orexine. D’autre part,

22
l’injection i.c.v. d’α-MSH inhibe l’expression de l’orexine sans modifier celle de la MCH dans le
LHA (Hanada et al., 2000). De même, les souris qui présentent une délétion du gène codant pour
la POMC montrent une surexpression de l’orexine dans le LHA (López et al., 2007). L’ensemble
des éléments précédemment cités démontre de façon évidente l’impact des neurones de premier
ordre de l’ARC sur l’expression de la MCH et de l’orexine dans le LHA. Par ailleurs, des études
réalisées à l’aide de certains modèles de souris transgéniques ont permis de mettre en évidence
l’importance de la MCH et de l’orexine dans la régulation de l’homéostasie énergétique. En effet,
la délétion du gène codant pour la MCH entraine une hypophagie chez des souris conduisant à un
phénotype mince (Shimada et al., 1998). De même, l’invalidation du gène codant l’orexine
s’accompagne d’une diminution de la prise de nourriture chez la souris (Hara et al., 2001).
Toutefois, ces souris orexine-/- sont obèses, suggérant une diminution en parallèle de la dépense
énergétique (Cf. 2.2.2.1). Enfin d’un point de vue pharmacologique, il a été montré que ces deux
neuropeptides augmentent la prise de nourriture chez le rongeur, lorsqu’ils sont injectés par voie
i.c.v. (Qu et al., 1996; Székely et al., 2002).
Par ailleurs, la MCH est également secrétée par des neurones du DMH (Badami et al.,
2010). Ces neurones reçoivent également des projections venant des neurones de premier ordre
de l’ARC (Kalra et al., 1999). À leur tour, les neurones à MCH projettent vers différentes
structures du SNC, notamment le PVH et le VMH (Wynne et al., 2005), et ces connexions
pourraient constituer des boucles de rétrocontrôle. Cependant, la population neuronale la mieux
décrite à ce jour dans le DMH est constituée de neurones exprimant le NPY; celle-ci est connue
pour avoir un impact majeur sur la régulation de l’homéostasie énergétique. En effet, l’injection
d’un adenovirus associé avec l’ARNc antisens de NPY dans le DMH inhibe spécifiquement
l’expression de NPY dans le DMH et entraine une diminution de l’hyperphagie et de l’obésité
chez le rat OLETF (Otsuka Long Evans Tokushima Fatty), dont la spécificité est d’être
hyperphage, obèse et diabétique (Yang et al., 2009b; Bi et al., 2012).
Les axones des neurones POMC/CART et NPY/AgRP émettent également des contacts
synaptiques avec d’autres populations neuronales situées dans le PVH. Celles-ci produisent des
neuropeptides anorexigènes tels que la thyréolibérine ou Thyrotropin-Releasing Hormone (TRH)
(Fekete et al., 2000; Harris et al., 2001), la corticolibérine ou Corticotropin-Releasing Hormone
(CRH) (Suda et al., 1993; Valassi et al., 2008) et l’ocytocine (Choy and Watkins, 1977; Parker

23
and Bloom, 2012). Des expériences réalisées chez le rongeur, montrent que l’expression de ces
trois neuropeptides est modifiée suite à une variation du statut énergétique ou à l’injection d’un
agent pharmacologique mimant ce phénomène. En effet, l’expression de l’ARNm de la TRH dans
le PVH est augmentée par l’injection d’α-MSH ou lors d’un jeûne (Fekete et al., 2000), tandis
qu’elle est diminuée après l’administration d’AgRP ou de NPY (Fekete et al., 2004; Valassi et al.,
2008). Par ailleurs, l’infusion de leptine augmente l’expression de l’ARNm de CRH (Masaki et
al., 2003). Ou encore, l’activité des neurones exprimant l’ocytocine est augmentée lors de la prise
d’un repas (Johnstone et al., 2006). De plus, l’injection centrale de TRH ou de CRH chez le rat
diminue sa prise alimentaire et son poids corporel (Valassi et al., 2008). De même, l’injection
centrale d’ocytocine en i.c.v. inhibe la prise de nourriture (Arletti et al., 1989). Il apparaît donc
clairement que le PVH, via la libération de ces neuropeptides dans différentes structures du SNC
est le siège d’une activité neuronale anorexigène.
Au niveau du VMH, les fibres des neurones POMC/CART et NPY/AgRP de l’ARC
forment des contacts synaptiques avec une population de neurones synthétisant un neuropeptide
anorexigène, le brain-derived neurotrophic factor (BDNF) (Rios et al., 2001; Xu et al., 2003;
Sternson et al., 2005). Il a été montré que la délétion du gène codant pour le BDNF chez l’homme
ou le rongeur, s’accompagne d’une hyperphagie et d’une obésité morbide (Lyons et al., 1999;
Yeo et al., 2004). De plus, chez la souris avec une délétion du gène MC4R, l’expression de
BDNF est réduite dans le VMH suggérant ainsi que l’effet satiétogène induit par le système
mélanocortinique passe par des neurones exprimant le BDNF (Xu et al., 2003). Enfin, les
neurones exprimant le BDNF émettent des projections vers plusieurs structures du SNC,
notamment vers le PVH, le LHA, le DMH et vers le NTS, démontrant ainsi le rôle de modulateur
du VMH dans la régulation du bilan d’énergie (McClellan et al., 2006).
1.2.2 Le rôle du NTS dans la régulation de l’homéostasie.
Le NTS se situe dans le tronc cérébral, où il organise l’action du SNA afin de contrôler
l’activité des différents organes périphériques impliqués dans la régulation de l’homéostasie
(Nonogaki, 1999). Il reçoit aussi un grand nombre d’afférences provenant de différents noyaux
hypothalamiques afin de moduler son activité pour réguler le métabolisme énergétique
périphérique (Horst et al., 1989; Schwartz et al., 2000). Cependant, certaines caractéristiques

24
évoquées précédemment lui permettent d’intégrer les signaux périphériques et de participer avec
l’hypothalamus au contrôle la prise alimentaire (Schwartz, 2006).
1.2.2.1 Le NTS est le centre de l’activité du SNA.
Le SNA est contrôlé par le tronc cérébral et plus particulièrement par le NTS, dont les
projections innervent les neurones préganglionnaires des fibres sympathiques (ou
orthosympathiques) et parasympathiques, qui ont un effet opposé sur les organes cibles
(Robertson et al., 2011). Les fibres ortho- et parasympathiques, dont les neuromédiateurs sont
respectivement la noradrénaline et l’acétylcholine, innervent la plupart des organes périphériques
dont notamment le foie, le pancréas, les muscles squelettiques et les adipocytes (Robertson et al.,
2011). Ces organes sont responsables de la mise en réserve, ainsi que de la mise à disposition du
glucose et des acides gras en périphérie. Ainsi, le système parasympathique favorise le stockage
du glucose et des acides gras libres tandis que le SNS favorise leur libération (Scheurink and
Nolan, 1996).
1.2.2.2 Les interactions du NTS et de l’hypothalamus dans la régulation de l’homéostasie.
Le NTS est plus qu’un simple relais pour les entrées viscérales et gustatives. En effet, de
nombreuses études réalisées notamment chez le rongeur indiquent que le NTS, tout comme
l’ARC, présente des neurones à NPY/AgRP et à POMC/CART (Mountjoy et al., 1994; Wynne et
al., 2005; Morin and Gehlert, 2006). Le NTS a une seconde similitude avec l’ARC, car il est
adossé à une petite structure nommée l’area postrema (AP) qui constitue également un organe
circum-ventriculaire (Blouet and Schwartz, 2012). De par ses caractéristiques morphologiques
l’AP est capable de détecter les hormones circulantes et notamment les hormones gastro-
intestinales telles que la CCK (Hill et al., 1987), le GLP1 (Merchenthaler et al., 1999) ou le PYY
(Stanić et al., 2006). Ainsi, l’AP qui possède des connexions directes avec le NTS l’informe de
l’état du système digestif (Berthoud, 2002). De plus, grâce à ses nombreuses interconnexions
avec l’hypothalamus, il intervient dans la régulation de la balance énergétique (Berthoud, 2002).
A titre d’exemple, des expériences réalisées chez le rongeur ont montré que les neurones du
PVH exprimant de l’ocytocine et innervant le NTS (Blevins et al., 2003) sont capables de
moduler la prise alimentaire (Maejima et al., 2009), indiquant l’existence d’une communication

25
bilatérale entre ces deux structures. D’autres études on mis en évidence que l’activation des
neurones situés dans le LHA stimule l’activité du SNA, alors que la modification de l’activité du
SNA par des organes cibles tels que les intestins et le foie, modulerait en retour, via le NTS,
l’activité électrique des populations neuronales du LHA (Kirchgessner, 2002). Enfin, l’activation
des récepteurs MCR4 exprimés sur les neurones de l’ARC qui se projettent vers le NTS diminue
l’appétit via l’activation du nerf vague (Williams et al., 2000).

26
Figure 6: La régulation centrale de la balance énergétique.
Schéma illustrant l’ensemble des connaissances actuelles sur les relations entre les structures du SNC, les
neuropeptides et leurs récepteurs impliqués dans la régulation de l’homéostasie énergétique.
Abréviations : liste des structures cérébrales; Acb, le noyau accumbens; Am, l’amygdale; ARC, le noyau
arqué de l’hypothalamus; DMH, le noyau dorsomédian de l’hypothalamus; DMV, le noyau moteur dorsal du nerf
vague; IML, la colonne intermédiolatérale de la moelle épinière; LHA, l’hypothalamus latéral; NTS, le noyau du
tractus solitaire; PB, le noyau parabrachial; PVH, le noyau paraventriculaire de l’hypothalamus; SCH, le noyau
suprachiasmatique ; VMH, le noyau ventromédian de l’hypothalamus; VTA, l’aire tegmentale ventrale. Les triangles
représentent des récepteurs aux neuropeptides et les losanges représentent des récepteurs aux signaux périphériques
à l’exception du losange vert qui est un transporteur. Liste des récepteurs; CB1, le récepteur aux cannabinoïdes de
type 1; MC3R et MC4R, les récepteurs aux mélanocortines de type 3 et 4; NPY1R et NPY5R, les récepteurs au
neuropeptide Y de type 1 et 5 et le récepteur au Glucagon-Like Peptide-1 (R GLP-1). Les cercles représentent des
neuropeptides. Liste des neuropeptides : AgRP, Agouti-Related Peptide; CART, Cocaine- and Amphetamine-
Regulated Transcript; CRH, Corticotropin-Releasing Hormone; GABA, Acide γ-aminobutyrique; MCH, Melanin-
Concentrating Hormone; NPY, Neuropeptide Y; POMC, Pro-opiomélanocortine; TRH, Thyrotropin-Releasing
Hormone. Les hormones secrétées par la tige pituitaire; TSH, Thyroid-Stimulating Hormone; ACTH,
Adrenocorticotropic hormone. Les hormones secrétées par la thyroïde; T4, la thyroxine; T3, triiodothyronine.
Traduit du Metabolic Syndrome Eposter de Chun-Xia Yi, Lori Zeltser and Matthias Tschöp dans Nature Medecine.
Tige%pituitaire%
Cortex% Hippocampe%
Glande%surrénale%
Bulbe%olfac:f%
Muscle%
Pancréas%
Foie%
Tractus%%gastro>intes:nal%% Tissu%adipeux%
blanc%
Tissu%adipeux%brun%
Senseur%de%l’adiposité%et%de%la%sa:été%
Contrôle%autonome%du%métabolisme%
Contrôle%comportementale%de%l’alimenta:on%
Régulateur%et%senseur%glycémique%
Horloge%et%synchronisa:on%du%métabolisme%
Contrôle%neuroendocrinien%du%métabolisme%
Glande%thyroïdienne%
Transporteurs%du%glucose%dédié%à%sa%percep:on%%
R%lep:ne%
R%insuline%
R%ghréline%
R%GLP>1%
R%estrogène%
MC3R%
MC4R%
NPY1R%
NPY5R%
CB1%
Dopamine%
Ocytocine%
Vasopressine%
TRH%
CRH%
Glutamate%
Acétylcholine%
NPY/AGRP/GABA%
POMC%
CART%
Orexine%
MCH%
Dynorphine%
GABA%
Hormones%%de%croissance% Innerva:on%%
parasympathique%Innerva:on%%
sympathique%
T4/T3%
ACTH%
TSH%
Adrénaline%Cor:sol%
ARC%
DMH%PVH%
VTA%LHA%
Am% VMH%
SCH%
Acb%
PB%
NTS%DMV%
IML%

27
2 La thermogenèse du tissu adipeux brun.
Il existe dans le règne animal deux types de réponses face aux variations de température de
l’environnement. La première, qui consiste à faire varier la température corporelle au gré de
l’environnement, est utilisée par les espèces dites poïkilothermes. La seconde quant à elle,
nécessite de réguler la température corporelle autour d’un point de consigne propre à chaque
espèce, et est adoptée par les espèces homéothermes tels que les oiseaux et les mammifères. Cette
particularité permet aux organismes homéothermes d’assurer l’ensemble de leurs fonctions
physiologiques de façon permanente (Cannon and Nedergaard, 2004).
La thermogenèse est un processus multifactoriel incluant la thermogenèse obligatoire et la
thermogenèse facultative ou adaptative. La thermogenèse obligatoire est la résultante de l’activité
du métabolisme basal, c’est-à-dire une conséquence de l’ensemble des réactions exothermiques.
Cette thermogenèse représente les deux tiers de la chaleur produite par l’organisme (Clapham,
2012). Les termes de thermogenèse facultative ou adaptative regroupent trois effecteurs qui sont,
(i) les muscles dans le cadre de l’activité physique volontaire ou du frissonnement lors d’une
exposition au froid, (ii) le tractus digestif pendant la digestion et (iii) le BAT qui est stimulé
pendant une exposition au froid ou l’ingestion d’aliments (Silva, 2006; Clapham, 2012). Ces trois
sources de thermogenèse sont dites facultatives ou adaptatives, car elles sont activées par le SNC
de façon temporaire en réponse à une modification de l’environnement. Toutefois, elles sont
essentielles à l’adaptation et à la survie de l’organisme aux modifications de l’environnement
(Cannon and Nedergaard, 2004; Silva, 2006).
A la différence du tissu adipeux blanc, dont la fonction est de stocker les lipides sous forme
de triglycérides pour assurer les besoins énergétiques de l’organisme en tout temps, le BAT met
en réserve des lipides pour réaliser la thermogenèse. Le BAT est un acquît évolutif qui s’est
développé très tôt dans l’évolution des mammifères, qui leur a permis de se libérer des
fluctuations environnementales. En effet, lors de l’exposition au froid, le BAT permet à l’animal
de conserver une température corporelle optimale et ainsi d’assurer le bon fonctionnement de son
métabolisme basal et d’empêcher les contractions musculaires réflexes handicapant la mobilité
(Cannon and Nedergaard, 2004).

28
2.1 Le tissu adipeux brun.
Le BAT peut assurer sa fonction thermogène grâce à des caractéristiques particulières en
termes de localisation, d’histologie, mais surtout grâce à la présence d’une protéine découplante
(uncoupling protein-1 UCP-1), qui fut découverte en 1978 (Nicholls et al., 1978; Nicholls and
Locke, 1984).
2.1.1 Les caractéristiques du tissu adipeux brun.
2.1.1.1 Origine et développement du tissu adipeux brun.
Le tissu adipeux brun fut observé pour la première fois par le naturaliste suisse Konrad
Gessner en 1551, qui décrit le BAT comme n’étant “ni graisse ni chair (nec pinguitudo nec
caro)”. Jusqu'à récemment, de nombreux investigateurs assumaient l’origine commune des
adipocytes bruns et blancs. Cette affirmation était basée sur plusieurs observations, telles que la
proximité de morphologie des adipocytes bruns et blancs, la capacité de stocker des triglycérides
dans des gouttelettes lipidiques, ainsi que la similitude des transcriptomes, à l’exception de
l’expression de UCP-1 (Enerbäck, 2010a). Néanmoins, il existe cependant deux différences
histologiques majeures entre ces deux types de tissus. En effet, les adipocytes blancs sont uni-
loculés et comportent peu de mitochondries, alors que les adipocytes bruns sont multi-loculés et
présentent de très nombreuses mitochondries qui expriment le cytochrome C (de couleur cuivre)
leurs conférant une couleur brune (Cinti, 2012; Wu et al., 2013).
Toutefois, avant 2010, l’existence d’une origine embryologique commune entre le derme,
le muscle et la graisse brune fut proposée (Atit et al., 2006; Seale et al., 2007; Timmons et al.,
2007; Seale et al., 2008; Tseng et al., 2008; Kajimura et al., 2009). Depuis, il est admis que les
adipocytes bruns sont issus du mésoderme paraxial contrairement aux adipocytes blancs qui
dérivent du mésoderme latéral. Ainsi, il a été montré chez le rongeur que lors de l’embryogenèse,
certaines cellules du mésoderme paraxial se différentient en dermomyotomes, des cellules
progénitrices qui sont à l’origine de la formation du derme, du muscle et du BAT (Billon and
Dani, 2012). Les premiers indices de ces différences histologiques furent apportés par Atit et ses
collaborateurs en 2006, qui ont montré que l’invalidation du gène engrailed-1 (En1), un gène
exprimé uniquement durant la différenciation des cellules du dermomyotome, perturbe le

29
développement du derme et des muscles mais aussi du BAT interscapulaire (Atit et al., 2006).
Parallèlement, Timmons et ses collaborateurs ont montré à l’aide du micro-array que les cellules
indifférenciées progénitrices des adipocytes bruns ont un profil transcriptionnel comparable à
celui des cellules musculaires (Timmons et al., 2007), (Figure 7A). Ces cellules expriment en
effet différents facteurs myogéniques et notamment le facteur myogénique 5 (Myf5). Les cellules
progénitrices exprimant ce facteur sont nommées Myf5+, tandis que celles ne l’exprimant pas,
sont des cellules qualifiées de Myf5- (Timmons et al., 2007). Par la suite, Tseng et ses
collaborateurs ont mis en évidence le rôle d’un cocktail hormonal contenant du Bone
Morphogenetic Protein-7 (BMP-7), sur la différenciation des dermomyotomes en pré-adipocytes
bruns et la maturation des pré-adipocytes bruns en adipocytes bruns fonctionnels (Tseng et al.,
2008). Toutefois, cette dernière étape est conditionnée à la formation d’une cassette d’expression
constituée par le facteur de transcription PRD1-BF1-RIZ1 homologous domain-containing-16
(PRDM16) et la forme active de CCAAT/enhancer-binding protein-β (C/EBPβ) (Kajimura et
al., 2009). Enfin, certains facteurs sont capables d’orienter le devenir des précurseurs des
adipocytes bruns. Ainsi, la forkhead box C2 (FOXC2) et le peroxisome-proliferator-activated
receptor-γ (PPAR-γ) co-activator 1α (PGC1α) favorisent la différenciation en adipocytes
brun, tandis que la retinoblastoma protein (pRB) ou la receptor-interacting protein 140 (RIP40)
agissent à titre de répresseurs de la différenciation (Kajimura et al., 2010).

30
Figure 7: Representation schématique de la différenciation des adipocytes.
A) L’origine des adipocytes bruns et des cellules musculaires squelettiques. B) L’origine des adipocytes
blancs et beiges. (Traduit et adaptée de (Kajimura et al., 2010)).
Abréviation: BMP-7, Bone Morphogenetic Protein-7; En1+, engrailed-1; Myf5, myogenic factor 5;
PGC1α, PPARγ co-activator 1-α; PPARγ, peroxisome-proliferator-activated receptor-γ ; PRDM16,
PRD1-BF1-RIZ1 homologous domain-containing-16; UCP-1, uncoupling protein-1.
BMP7%
BMP7%
Myoblaste% Muscle%squele1que%
Adipocyte%brun%Pré9adipocyte%brun%
dermomyotome%
4%Précurseur%
d’adipocyte%beige%
Pré9adipocyte%blanc%
BMP4%adipocyte%blanc%
Cellule%souche%mésodermale%
Précurseur%d’adipocyte%brun%(myf59)%du%Dssu%
adipeux%blanc%%%
Adipocyte%brun%«9like»%inducDble%du%Dssu%
adipeux%blanc%%%
pRB;%RIP40%%
PPARγ+;%UCP91+%
PPARγ+%
PPARγ+%
PPARγ+;%PRDM16+;%PGC1α;%UCP91+%
PRDM16+%%
Myf5+;%MyoD+%% MyoD+;%Myogenin+%%
En1+;%Myf5+%%

31
2.1.1.2 Localisation du tissu adipeux brun chez le rat et l’homme.
Les adipocytes bruns sont regroupés en différents dépôts, qui ont été découverts suite à des
nécropsies chez le rat. Ces dépôts sont situés dans les régions interscapulaire (la mieux
caractérisée), cervicale, péri-aortique, péri-rénale, intercostale et médiastinale (Cannon and
Nedergaard, 2004) (Figure 8A).
Chez l’Homme, le BAT fut longtemps considéré comme un organe vestigial régressant
avec l’âge et n’ayant comme seule fonction que le maintien de la température corporelle chez le
nouveau-né (Lean, 1989). Il fallut attendre le développement de l’imagerie médicale et plus
particulièrement de la tomographie par émission de positron (TEP) dans les années 2000, pour
observer des dépôts d’adipocytes bruns actifs chez l’adulte, suggérant que le BAT persiste et
demeure fonctionnel. En effet, en 2002 lors d’un examen de routine en oncologie, l’utilisation du
fluorodésoxyglucose, qui permet de visualiser les tissus métaboliquement actifs en TEP, a révélé
la présence de dépôts symétriques de graisse brune active au niveau supra-claviculaire chez
l’homme (Hany et al., 2002). Par la suite, plusieurs études réalisées chez l’Homme dans des
conditions d’hypothermie ont également permis de révéler l’existence de dépôts cervicaux et
para-spinaux métaboliquement actifs (Figure 8B), (Nedergaard et al., 2007; Cypess et al., 2009;
van Marken Lichtenbelt et al., 2009; Zingaretti et al., 2009; Enerbäck, 2010b; Richard et al.,
2010).
2.1.1.3 Le secrétome du tissu adipeux brun.
Pendant longtemps, le seul rôle connu du tissu adipeux brun était la production de chaleur
dans le but de maintenir la température corporelle. Cependant, des études évaluant l’impact de
l’ablation génétique du BAT et d’UCP1 chez le rongeur ont montré que le BAT a d’autres
fonctions qui vont au-delà de l’oxydation des surplus d’énergie et de la thermorégulation (Lowell
et al., 1993; Enerbäck et al., 1997). Cette découverte a permis de mettre évidence le rôle
autocrine, paracrine mais surtout endocrine du BAT. En effet, le BAT sécrète des facteurs qui
agissent localement, pour favoriser son maintien et son développement. Par exemple,
l’angiogenèse au sein du BAT est favorisée par la libération du vascular endothelial growth
factor-A (VEGF-A) (Xue et al., 2009), la prolifération des précurseurs d’adipocytes bruns est
stimulée par la sécrétion de l’insulin-like growth factor-1 (IGF-1) et du fibroblast growth factor-2

32
(FGF-2) (Yamashita et al., 1994), tandis que la sensibilité des cellules à la noradrénaline (NA) est
maintenue grâce au bone morphogenetic protein-8b (BMP-8b) (Whittle et al., 2012) (figure 9g).
Par ailleurs, certaines études indiquent que le BAT constitue également un organe endocrinien,
capable de libérer des composés de type « Adipokine-like », baptisés batokines (Villarroya et al.,
2013). Plusieurs batokines tels que la triiodothyronine (T3), l’interleukine-6 (IL-6), le fibroblast
growth factor-21 (FGF-21), le BMP-8b, le retinol-binding protein-4 (RBP-4) et la lipocalin
prostaglandin D synthase (L-PGDS), ont ainsi été découvertes chez le rongeur (Villarroya et al.,
2013). Toutefois, seuls l’IL-6 et le FGF-21 ont clairement été identifiés comme intervenant dans
la mise à disponibilité des substrats pour le BAT, la régulation centrale de son activité, ainsi que
sur la sensibilité à l’insuline et l’activité des cellules bêta du pancréas (Villarroya et al., 2013).
A
B
surrénale!!
Para-vertébrale!!
médiastinale!!
supraclaviculaire!!
Le cou!!
intercostale!
Peri-rénale!Peri-aortique!
interscapulaire!

33
Figure 8: Localisation du tissu adipeux brun chez le rat et l’homme.
A. Distribution des dépôts de tissu adipeux brun chez le rat (tirée de (Cannon and Nedergaard,
2004)). B. Distribution des dépôts de tissu adipeux brun chez l’Humain (tirée de (Nedergaard et al.,
2010)).
2.1.1.4 Les adipocytes beiges.
Certaines études récentes ont permis de mettre en évidence la présence de cellules au
phénotype comparable à celui des adipocytes bruns, au sein des dépôts d’adipocytes blancs.
Ainsi, il a été montré chez le rongeur que ces adipocytes (Wu et al., 2013), baptisés adipocytes
beiges ou brite (brown in white), sont présents notamment dans le dépôt adipeux inguinal et que
leur développement est stimulé par l’injection d’agonistes adrénergiques ou par une exposition au
froid (Cannon and Nedergaard, 2012; Wu et al., 2012). À ce jour, les facteurs et les voies
impliqués dans le développement des adipocytes beiges restent indéterminés. Toutefois,
considérant la nature inductible et la localisation des adipocytes beiges, quatre hypothèses sur
leur origine phylogénique sont actuellement proposées. Selon ces différentes hypothèses, les
adipocytes beiges pourraient dériver (i) de précurseurs d’adipocytes bruns présents au sein du
tissu adipeux blanc (Petrovic et al., 2010), (ii) de précurseurs d’adipocytes blancs (Pisani et al.,
2011), (iii) d’adipocytes blancs matures trans-différenciés (Cinti, 2009; 2012) ou, (iv) de
précurseurs spécifiques aux adipocytes beiges (Wu et al., 2012). En effet, des travaux réalisées
chez le rongeur indiquent que les adipocytes beiges ont un transcriptome unique qui diffère de
ceux des adipocytes blancs et bruns (Wu et al., 2012) (Figure 7B). Toutefois, il a été montré par
Seale et ses collaborateurs que le développement des adipocytes beiges est conditionné à la
présence de PRDM16, un facteur de transcription également impliqué dans la mise en place du
BAT (Seale et al., 2011), indiquant que ce facteur de transcription est essentiel au brunissement
du tissu adipeux.
2.1.1.5 La lipogenèse et la lipolyse une compétence commune à tous les adipocytes.
La lipogenèse et la lipolyse sont deux mécanismes assurant le métabolisme des acides gras
libres (AGL) dans les adipocytes. Les AGL constituent un substrat énergétique essentiel pour
l’organisme. Cependant, l’accumulation excessive d’AGL peut devenir cytotoxique pour
l’adipocyte. Afin de préserver l’intégrité cellulaire les adipocytes stockent les AGL sous forme de

34
triglycéride (TG) dans des gouttelettes lipidiques, grâce à la lipogenèse. Le TG est un composé
hydrophobe et biologiquement inerte formé d’une molécule de glycérol estérifiée par trois acides
gras. Néanmoins, les TG doivent être hydrolysés lors de la lipolyse, pour former de nouveau des
AGL et du glycérol, utilisables par l’adipocyte et l’organisme comme substrats énergétiques.
La lipogenèse de novo.
Durant la phase postprandiale, les niveaux circulants d’insuline augmentent et induisent la
translocation du transporteur du glucose de type 4 (GLUT4, glucose transporter type 4) vers la
membrane plasmique de l’adipocyte, permettant ainsi l’entrée de glucose (James and Pilch,
1988). Une fois dans le cytosol, le glucose est phosphorylé permettant ainsi à ses carbones de
servir de précurseur pour la synthèse d’acide gras suite à l’action séquentielle de l'acétyl-
coenzyme A carboxylase (ACC, acyl-coA carboxylase) et de l'acide gras synthétase (FAS, fatty
acid synthetase) (Shi and Burn, 2004).
D’autre part, les AGL à longue chaine sont en mesure de passer la membrane de façon
passive, toutefois leur transport actif peut être stimulé par l’insuline qui favorise la translocation
de la protéine transporteuse d’acide gras 1 (FATP1, fatty acid transport protein 1) du cytosol vers
la membrane plasmique (Lobo et al., 2007). Les AGL sont alors convertis en acyl-CoA dans le
réticulum endoplasmique par l'acétyl-coenzyme A synthase (ACS, acyl-coA synthase).
Par la suite, cet acyl-CoA est à un glycerol-3-phosphate (G3P) par une réaction d’acylation
réalisée par le glycérol-phosphate acyltransférase (GPAT) pour former un acide
lysophosphatidique (ALP) (Gimeno and Cao, 2008). Puis, l'acylglycérol-phosphate
acyltransférase (AGPAT) lie un second groupement acyl-Coa à ALP pour générer l'acide
phosphatidique (APp) (Shindou et al., 2009). Ensuite, l’APp est converti en diglycéride (DG) par
l’activité phosphatidate phosphatase de la lipine (Reue and Dwyer, 2009). Finalement, l'action de
la diacylglycérol acyltransférase (DGAT) conduit à l'ajout d’un dernier acide gras nécessaire pour
obtenir un TG (Yen et al., 2008). Les TG sont ensuite entreposés dans des gouttelettes lipidiques,
dont la périlipine permet le maintien en inhibant l’action des lipases (Tansey et al., 2001).

35
La lipolyse intracellulaire.
Lors de la lipolyse, les TG contenus dans les gouttelettes lipidiques sont hydrolysés, pour
produire des AGL et du glycérol. Ces produits sont libérés dans la circulation sanguine par les
adipocytes blancs ou utilisés comme source d’énergie pour effectuer la thermogenèse par les
adipocytes bruns (Jaworski et al., 2007) (Cf. 2.1.2.2). Pour réaliser la lipolyse, trois lipases sont
nécessaires, la lipase des TG du tissu adipeux (ATGL, adipose triglyceride lipase), la lipase
sensible aux hormones (HSL, hormone-sensitive lipase) et la lipase du monoacylglycérol (MGL,
monoacylglycerol lipase) (Jaworski et al., 2007). Ces lipases interviennent successivement,
libérant ainsi un acide gras à chaque étape. L’ATGL hydrolyse donc le TG en DG, la HSL
hydrolyse le DG en monoacylglycérol (MAG) et la MGL hydrolyse le MAG en AGL.
Néanmoins, la lipolyse ne serait pas possible sans la phosphorylation de la périlipine par la
protéine kinase A (PKA), qui entraine la fragmentation des gouttelettes lipidiques permettant
ainsi l’accès de l’ATGL au TG. De plus, la PKA phosphoryle les différentes lipases pour
favoriser la production d’AGL (Jaworski et al., 2007).
2.1.2 Les mécanismes cellulaires permettant la production de chaleur.
En réponse au froid ou à l’ingestion de nourriture le SNS libère de la NA au niveau du
BAT. Celle-ci va se lier à un récepteur adrénergique et permet ainsi l’augmentation de l’activité
et de la capacité thermogène du BAT (Richard et al., 2012; Chechi et al., 2013).
2.1.2.1 L’activation de l’adipocyte brun : la cascade adrénergique.
Les adipocytes bruns expriment plusieurs types de récepteurs adrénergiques. Cependant,
seul le récepteur β-3 (ADRB3) stimule la thermogenèse, alors que le récepteur β-1 intervient dans
la maturation du pré-adipocyte brun (Cannon and Nedergaard, 2004). L’ADRB3 est couplé à une
protéine G dont la sous unité alpha est stimulatrice (Gs), celle-ci active l’adénylate cyclase afin
d’augmenter la synthèse d’adénosine monophosphate cyclique (AMPc). L’AMPc permet
l’activation de la PKA (Collins, 2011; Chechi et al., 2013; Harms and Seale, 2013). Le rôle de la
PKA est de phosphoryler différentes protéines situées à la fois dans le cytosol et dans le noyau
(Figure 9a). Ces protéines sont impliquées dans la transformation des triglycérides stockés dans
les gouttelettes lipidiques en AGL et en glycérol (Cf. 2.1.2.2) afin de stimuler la thermogenèse,

36
ainsi que dans les processus transcriptionnels permettant la synthèse des différents acteurs
nécessaires à l‘activité thermogène du BAT (Cf. 2.1.2.3).
Figure 9: L’activation de l’adipocyte brun.
a. La cascade moléculaire du récepteur adrénergique β3 régulant l’activation de PKA. b. La
phosphorylation des protéines impliquées dans la lipolyse et la transformation de l’acide gras en acide
gras-CoA ou acyl-CoA. c. L’acide gras-CoA traverse la membrane de la mitochondrie à l’aide des
carnitine palmitoyl transférases 1 et 2 puis il est pris en charge dans la beta-oxydation et le cycle de
Krebs pour donner des FADH2 (Flavine adénine dinucléotide H2) et des NADH, H+ (Nicotinamide
adénine dinucléotide H, H+) nécessaires au fonctionnement de la chaine respiratoire. d. Le
fonctionnement de la chaine respiratoire. e. Schéma illustrant l’hypothèse actuelle sur le fonctionnement
de UCP-1 (adapté de (Kajimura and Saito, 2013)). f. La phosphorylation de P38 permet la
phosphorylation des acteurs impliqués dans la transcription des gènes mitochondriaux, des co-activateurs
d’UCP-1 et d’UCP-1 (extrait de (Richard et al., 2010)). g. Schéma récapitulatif des molécules libérées
par différents organes et agissant sur l’adipocyte brun (extrait de (Harms and Seale, 2013)).
Abréviations : AC, adénylate cyclase; ACS, acyl-CoA synthétase; AG, acide gras; AGLC, AG à longue
chaine; ADP, Adénosine diphosphate; AMPc, adénosine monophosphate cyclique; ATGL, adipose
triglyceride lipase; ATP, Adénosine triphosphate; β3, récepteur adrénergique β3; β-ox, β-oxidation; CdK,
cycle de krebs; CPT1, carnitine palmitoyl transférase 1; CPT2, carnitine palmitoyl transférase 2; DG,
Diglycéride; Gs, protéine G stimulatrice; HSL, hormone-sensitive lipase; MG, Monoglycéride;
MGL, monoacylglycerol lipase; NA, noradrenaline; P38, P38α mitogen-activated protein kinase; PKA,
protéine kinase A; PLA2, la phospholipase A2; TG, Triglycerides ; UCP-1, uncoupling protein-1.

37
Gs#
β3#
AMPc#ATP#
PKA#inac/ve#
PKA#ac/ve#
+#
AC#
HSL#
lipides#
TG#
SL#
DG#
MG#
AG#ACS#
Acyl<CoA#
Acyl<CoA#
β<ox#
acétyl<CoA#
NADH+,H#FADH2#
CdK#NADH+,H#FADH2#
NADH+,H#
I#II#
III# IV#
H+#H+# H+#
Q#
NAD+#
C#
e<#
FADH2#FAD#
2H+#+#½#O2# H2O#
IV#C#
H+#
V#
2O#H+#
UCP1#
ADP#+#Pi#
ATP#
PLA2#
<#
ATP?#
+
Chaleur#
P38#
? ?
Membrane#externe#mitochondriale#
Membrane#interne#mitochondriale# {#Espace#inter<membranaire#
Matrice#mitochondriale#
NA#N #
a#
b#
c#
e#
NADHd#
? f#
g#
AGLC#

38
2.1.2.2 Les mécanismes de l’activité thermogène.
Suite à l’activation de l’ADRB3, la PKA stimule la lipolyse afin de générer des AGL
(Zechner et al., 2009), (Cf. 2.1.1.5), (Figure 9b). Les AGL à longue chaine carbonée (C16 et plus)
sont alors transformés en acyl-CoA par l’acyl-CoA synthétase. L’acyl-CoA est ensuite pris en
charge par la navette de la carnitine. Dans le détail, la carnitine palmitoyl transférase 1β
(CPT1β) déplace le groupe acylé de l’acyl-CoA sur la carnitine, car le CoA ne passe pas les
membranes de la mitochondrie. L’acyl-carnitine est alors transportée dans la matrice
mitochondriale par une translocase. Une fois dans la matrice mitochondriale, la carnitine acyl
transférase 2, transfère le groupement acyl de l’acyl-carnitine à un CoA présent dans la
mitochondrie, l’acyl-CoA peut alors servir de substrat à la β-oxydation. La prise en charge de
l’acyl-CoA dans la β-oxydation génère un acétyl-CoA, une Flavine adénine dinucléotide H2
(FADH2) et une Nicotinamide adénine dinucléotide H, H+ (NADH, H+) par tour d’hélice de
Lynen (un cycle de β-oxydation). Les acétyl-CoA produits vont être pris en charge par le cycle de
Krebs pour générer des FADH2 et des NADH, H+ supplémentaires. Les protons (H+) ainsi
récupérés par la NAD et la FAD vont être utilisés par la chaine respiratoire (Nicholls and Locke,
1984; Richard and Picard, 2011), (Figure 9c).
La chaine respiratoire est composée de 4 complexes fixes et de 2 transporteurs mobiles
d’électrons. Elle est insérée dans la membrane interne de la mitochondrie et utilise les NADH, H+
et FADH2 pour transporter les H+ de la matrice vers l’espace inter-membranaire de la
mitochondrie. Ce pool de H+ est utilisé par l’ATP synthase afin d’ajouter un groupement
phosphate à l’adénosine diphosphate (ADP) pour obtenir de l’adénosine triphosphate (ATP)
(Nicholls and Locke, 1984; Richard and Picard, 2011). Toutefois, dans les adipocytes bruns
UCP-1 a la capacité de découpler la chaine respiratoire en transportant les protons de l’espace
inter-membranaire vers la matrice mitochondriale. Le découplage de la chaine respiratoire a pour
conséquence de vider la réserve en protons de l’espace inter-membranaire. En réponse, la chaine
respiratoire va s’activer de façon intense et donc générer une quantité très importante de chaleur
en comparaison avec une cellule n’exprimant pas UCP-1 (Nicholls and Locke, 1984; Richard and
Picard, 2011), (Figure 9d). À propos de l’activation et du fonctionnement d’UCP-1 plusieurs
modèles sont envisagés (Divakaruni and Brand, 2011). En effet, la protéine UCP-1 n’est pas
constitutivement active puisque dans des conditions de thermoneutralité et de jeûne, des purines

39
comme l’ATP et l’ADP notamment, sont capables de l’inhiber. Mais très récemment, une étude
utilisant la méthode de patch-clamp réalisée par l’équipe du docteur Kirichok (Fedorenko et al.,
2012) a permis de proposer un mécanisme expliquant l’activation de UCP-1 et le passage des
protons au sein du canal UCP-1. Ce mécanisme implique les AGL à longue chaine chargées
négativement (AGLC-) issus de l’activation de la phospholipase A2 (PLA2), qui est localisée sur
la membrane interne de la mitochondrie. Les AGLC- vont par la suite activer UCP-1 et servir de
substrat pour transporter les protons (Fedorenko et al., 2012; Kajimura and Saito, 2013), (Figure
9e).
2.1.2.3 Les mécanismes de la capacité thermogène.
La protéine UCP-1, est une protéine de 33kDa codée par un gène localisé sur le
chromosome 4 chez l’homme et 8 chez la souris (Puigserver et al., 1992; Picó et al., 1994).
L’expression d’UCP-1 est augmentée par une stimulation adrénergique ou par l’administration
d’un agoniste de PPARγ (Nedergaard et al., 2005). En effet, suite à une stimulation adrénergique,
la PKA est activée et phosphoryle la P38α mitogen-activated protein kinase (Collins et al., 2010).
Celle-ci entraine alors la phosphorylation d’un dimère d’activating-transcription factor-2 (ATF2)
situé sur le cAMP Response Element (CRE) du promoteur de PGC1α induisant de ce fait la
transcription de ce dernier (Puigserver et al., 1998). Ensuite, PGC1α interagit directement avec
les nuclear respiratory factor 1 et 2 (NRF1 et NRF2) pour stimuler la synthèse de novo de gènes
mitochondriaux tels que CPT1β, qui interviennent également dans la thermogenèse (Richard et
al., 2010).
Le promoteur d’UCP-1 contient plusieurs séquences régulatrices appelées éléments de
réponse, permettant la fixation et l’activation de l’ARN polymérase afin de générer des ARNm
d’UCP-1 (Cannon and Nedergaard, 2004). Parmi ces éléments de réponse qui sont le thyroid
response element (TRE), le retinoic acid response element (RARE), le CRE et le PPAR response
elements (PPRE), ces deux dernier sont les plus étudiés, cependant PPRE engendre la plus forte
expression d’UCP-1 à ce jour (Cao et al., 2004a; Chen et al., 2013). Lors d’une activation
adrénergique, un hétérodimère composé de deux récepteurs nucléaires, le retinoid X receptor-α
et le PPARγ va se former et se fixer sur le PPRE. Toutefois, pour que l’expression d’UCP-1 soit
activée, cet hétéromère s’associe à PRDM16 et PGC1α, qui peut alors être phosphorylé et activé

40
par la P38α map kinase. Cette kinase intervient également dans la phosphorylation et l’activation
d’ATF2, qui lie alors le site CRE du promoteur d’UCP-1 et participe ainsi à la mise en place de la
cassette d’expression nécessaire à l’expression d’UCP-1 et à la préparation du BAT aux futurs
modifications environnementales (Cao et al., 2004a; Richard et al., 2010; Collins, 2011). Enfin,
l’expression d’UCP-1 peut être inhibée par un mécanisme impliquant le liver X receptor-α et un
co-répresseur de PPARγ connu sous le nom de RIP140, qui entre en compétition avec le
complexe PGC1α/PRDM16 situé sur le PPRE du promoteur d’UCP-1 (Debevec et al., 2007),
(Figure 9f).
2.1.2.4 Les facteurs endocriniens périphériques influençant l’adipocyte brun.
Outre le SNS, une variété de facteurs circulants identifiés chez le rongeur et chez l’homme
sont également capables de stimuler directement la capacité et l’activité thermogène des
adipocytes bruns classiques, ou encore de favoriser le recrutement des adipocytes beiges
(Kajimura and Saito, 2013; Schulz and Tseng, 2013), (Figure 9g).
Les hormones thyroïdiennes.
La deiodinase iodothyronine 2 (DIO2) permet le passage de la forme inactive, la thyroxine
(T4) en triiodothyronine (T3) active. Des études réalisées chez le rongeur indiquent que la
délétion du gène codant pour la protéine DIO2 entraine une diminution importante de
l’expression d’UCP-1, qui s’accompagne d’un dysfonctionnement de la thermogenèse et a pour
conséquence une hypothermie lors de l’exposition au froid (de Jesus et al., 2001). Cette
expérience démontre que la T3 est indispensable au maintien de l’expression d’UCP-1 et que la
thyroïde et le SNS agissent en synergie dans le maintien de la température corporelle.
Les peptides natriurétiques.
Parmi les peptides natriurétiques (PNs), les plus communément étudiés sont ceux provenant
de l’oreillette du cœur tel que l’atrial natriuretic peptide (ANP), ceux provenant du cerveau tel
que le brain-derived natriuretic peptide (BNP) et ceux de type-C (C-type natriuretic
peptide_CNP) produit par le SNS, les chondrocytes et les cellules de l’endothélium vasculaire
(Lancha et al., 2012; Kajimura and Saito, 2013). Des études récentes ont montré que les PNs
contrôlent la prise alimentaire et la thermogenèse du BAT (Inuzuka et al., 2010; Bordicchia et al.,

41
2012). En effet, l’utilisation d’un modèle de souris déficiente pour le récepteur de la clairance aux
PNs (Bordicchia et al., 2012), récepteur qui a pour fonction de retirer l’ANP et le BNP de la
circulation sanguine (Maack et al., 1987), montre que l’ANP et le BNP stimulent l’expression
d’UCP-1, et le développement des adipocytes beiges au sein du tissu adipeux blanc (Bordicchia
et al., 2012). De plus, l’exposition au froid est associée à une augmentation des niveaux d’ANP et
de BNP circulants, indiquant leurs possibles implications dans la thermorégulation (Bordicchia et
al., 2012). Par ailleurs, les CNP inhibent la thermogenèse en atténuant l’activité du SNS sur le
BAT (Inuzuka et al., 2010). Il apparaît donc clairement que les PNs constituent une nouvelle
cible de recherche dans la compréhension des mécanismes de régulation de la thermogenèse et de
l’homéostasie énergétique.
Le FGF-21.
Le FGF-21 est principalement produit par le foie, il est impliqué dans le métabolisme du
glucose et des lipides. Chez le rongeur nouveau-né, la libération hépatique de FGF-21 qui
survient en réponse aux acides gras du lait, augmente l’expression de plusieurs gènes impliqués
dans la thermogenèse du BAT dont UCP-1 (Hondares et al., 2010). De plus, le FGF-21 est
exprimé et secrété en réponse au froid ou à un traitement avec des agonistes adrénergiques
(Chartoumpekis et al., 2011). Par ailleurs, une étude récente réalisée sur une souris déficiente
pour le gène codant la protéine FGF-21 montre une diminution du nombre d’adipocytes beiges
dans le tissu adipeux blanc sous-cutané. De plus, des études réalisées sur ce modèle indiquent
également qu’une exposition au froid s’accompagne d’une chute plus importante de la
température corporelle (Fisher et al., 2012), suggérant l’implication de FGF-21 dans la
thermorégulation. Enfin, chez l’humain le FGF-21 est augmenté lors d’une exposition modérée
au froid et pourrait donc représenter un bon indicateur de la capacité thermogène (Lee et al.,
2013a).
L’irisine.
L’irisine est une hormone qui a été récemment identifiée chez la souris et chez l’humain
(Boström et al., 2012). C’est la forme clivée et secrétée de la protéine membranaire FNDC5. Elle
est exprimée dans le muscle sous le contrôle de PGC1α en réponse à l’exercice (Boström et al.,
2012). Une augmentation des niveaux circulants d’irisine est associée à une augmentation de 10 à

42
20 fois de l’expression de UCP-1 dans le dépôt adipeux blanc sous-cutané (Boström et al., 2012).
De plus, l’irisine stimule certains gènes impliqués dans la différenciation des pré-adipocytes
beiges (Wu et al., 2012), suggérant ainsi que cette hormone pourrait participer activement à
l’augmentation de la capacité thermogène de l’organisme.
2.2 Le contrôle central de l’activité thermogène du tissu adipeux brun.
Le contrôle central de l’activité thermogène du BAT est avant tout réalisé dans le but de
réguler la température corporelle de l’organisme. Cependant, le SNC est aussi impliqué dans le
contrôle de l’activité thermogène du BAT lors de modifications physiologiques, telles que
l’inflammation ou l’hypoglycémie, cela dans le but de contrôler les dépenses énergétiques.
2.2.1 Le contrôle central du tissu adipeux brun lors de la thermorégulation.
Le contrôle de la température corporelle nécessite la mise en place d’un certain nombre de
mécanismes. En effet, la thermorégulation est réalisée grâce aux afférences sensitives informant
l’hypothalamus, et celui-ci va intégrer les signaux environnementaux afin d’agir adéquatement
sur le SNS, qui stimule l’activité du BAT interscapulaire (iBAT) (Morrison et al., 2012) (Figure
10).
Figure 10: Représentation de la circuiterie neuronale contrôlant l’activité du iBAT.
Abréviation : 5HT, la sérotonine; AC, l’acétylcholine; BAT SC, neurone sensible au chaud; CD, la
corne dorsale; DMH, le noyau dorsomédian de l’hypothalamus; GABA, l’Acide γ-aminobutyrique GLU, le
glutamate; iBAT, tissu adipeux brun interscapulaire; IML, la colonne intermédiolatérale de la moelle
épinière; LHA, l’hypothalamus latéral; MnPO, noyau préoptique médian; MPO, noyau préoptique
médial; MVL, la médulla ventrolatérale; NA, noradrénaline; NTS, le noyau du tractus solitaire; PBLd, le
noyau parabrachial latéral (dorsal); PBLle, le noyau parabrachial latéral (latéral externe); PVH, le
noyau paraventriculaire de l’hypothalamus; RPar, le noyau du raphé pallidus rostral; TRPV1/3 et
TRPM8, transient receptor potentiel V1/3 and M8. Adapté et traduit de (Morrison et al., 2012).

43
2.2.1.1 Les afférences de la perception thermique cutanée et centrale.
La perception thermique est realisée par des cellules nerveuses dont le corps cellulaire se
situe au niveau du derme. Cependant, celles-ci emettent des projetions vers l’épiderme où elles
expriment des récepteurs sensibles à la température externe. Le message nerveux est ensuite
transmis grace à des prolongements axonaux vers la corne dorsale où a lieu la divergence entre la
perception thermique et la nociception. Ensuite, le message codant la température transite par le
noyau parabrachial latéral (PBL) où deux sous régions prennent en charge les stimuli codant pour
le froid et les stimuli codant pour le chaud, tandis que le message nociceptif est conduit vers le
thalamus (Morrison et al., 2012).
CD#
PBLd#
peau#
chaud# froid#
TRPV1/3# TRPM8#
+# +#
+# +#
+# +#
BAT#SC#
_#
DMH#
MPO#
MnPO#
+#
LHA#
iBAT#
RPar#
IML#
PVH#
MVL#NTS#+#+#+#
IML#
MVL#NTS#
GLU#
GLU#+#
GABA#
_#
5HT#_#
GABA#
GABA#
GLU#GABA#
GLU#
GLU#
+#AC#+#
+#+#
+#
NA#
PBLle#

44
Les récepteurs impliqués dans la perception thermique.
Lorsqu’une modification de la température de l’environnement intervient, la température de
la peau change, cette variation de la température est alors détectée par des thermorécepteurs
cutanés situés à la terminaison des nerfs sensitifs primaires, ce qui initie la thermorégulation et
permet ainsi d’éviter une variation de la température corporelle néfaste pour l’organisme. Ces
thermorécepteurs font partie de la famille des récepteurs à potentiels de transition des canaux
cations (transient receptor potentiel (TRP) familly of cation channels) et sont exprimés au niveau
de l’épiderme, du SNC et de l’abdomen pour y permettre le maintien des conditions idéales au
bon fonctionnement des organes vitaux (Morrison et al., 2012). Dans la famille des TRP on
trouve un récepteur sensible au froid, le TRPM8, et trois récepteurs sensible au chaud, les
TRPV1, 3 et 4. Le TRPM8 est réactif à un faible stimulus du froid et la délétion du gène codant
pour ce récepteur s’accompagne d’une réduction de la sensibilité au froid (Dhaka et al., 2007)
pouvant conduire à l’hypothermie. D’autre part, les souris déficientes pour les gènes codant les
protéines TRPV3 et 4 sont incapables de discriminer les températures chaudes (Lee et al., 2005).
Enfin, ces récepteurs présentent un grand intérêt dans la lutte contre l’obésité par le froid, car ils
sont sensibles à certaines molécules. En effet, le menthol entraine une réponse physiologique
comparable à celle produite par le froid (Peier et al., 2002) et pourrait être utilisé comme
molécule anti-obésité tel que le suggère en 2012 Ma et ses collaborateurs (Ma et al., 2012). De
plus, le TRPV1 peut être activé par de la capsaïcine (Tominaga et al., 1998), cette dernière est
aussi considérée comme une drogue anti-obésité (Leung, 2014).
La corne dorsale est le centre de la divergence entre les perceptions thermique et
nociceptive.
Le ganglion nodal, situé en bordure de la corde spinale, contient les neurones de premier
ordre ou fibres somatosensorielles thermales primaires. Ces neurones émettent des projections à
la fois vers les récepteurs thermiques de l’épiderme et vers les neurones de la lamina I situés dans
la corne dorsale de la corde spinale, afin de leur transmettre l’information thermique. Les
neurones de second ordre de la lamina I sont capables de répondre de façon graduelle à des
stimuli froid et chaud (Craig et al., 2001), participant ainsi à la finesse de la réponse centrale. De
plus, c’est au sein des neurones de la corne dorsale que se séparent la voie neuronale impliquée

45
dans la perception consciente de la température (Craig, 2002) et la voie neuronale impliquée dans
la thermorégulation. La première voie fait intervenir le thalamus et le cortex somatosensoriel
primaire et son altération n’a aucun effet sur la réponse sympathique thermogène au froid
(Nakamura and Morrison, 2008a). La seconde voie implique des neurones glutamatergiques qui
émettent des axones vers le PBL, un relais majeur vers les noyaux hypothalamiques contrôlant le
iBAT (Hylden et al., 1989; Li et al., 2006).
Le noyau parabrachial.
Le PBL est subdivisé en deux sous parties. Le PBL latéral externe (PBLle) contient des
neurones glutamatergiques qui se projettent vers le noyau préoptique médian (MnPO), et sont
activés suite à une exposition au froid (Bratincsák and Palkovits, 2004; Nakamura and Morrison,
2008a). Le PBL dorsal (PBLd) contient lui aussi des neurones glutamatergiques qui se projettent
vers le MnPO, mais ceux-ci sont activés suite à une exposition au chaud (Bratincsák and
Palkovits, 2004; Nakamura and Morrison, 2010). Le rôle des neurones du PBL dans la
transmission cutanée et viscérale des signaux thermiques vers l’hypothalamus a été montré par
Kobayashi et Nakamura, et leurs collaborateurs (Kobayashi and Osaka, 2003; Nakamura and
Morrison, 2008a). En effet, lorsque les neurones du PBL sont inactivés par l’injection d’un
antagoniste des récepteurs au glutamate aucune activité du iBAT n’est observée suite aux
variations de température de la peau (Kobayashi and Osaka, 2003; Nakamura and Morrison,
2008a).
2.2.1.2 Les mécanismes hypothalamiques impliqués dans le contrôle du BAT.
Au sein du réseau neuronal régulant la température corporelle, l’hypothalamus et
notamment l’aire préoptique occupent une place centrale entre l’intégration des sensations
cutanées de la température externe et le contrôle de l’activité du iBAT. Grace à ses multiples
interconnexions, l’hypothalamus intègre les signaux sur l’état homéostatique de l’organisme afin
de générer une réponse thermogène adaptée (Morrison et al., 2012).
Le MnPO reçoit les signaux thermiques cutanés et génère la réponse thermogène.
Au sein de l’aire préoptique, les signaux du froid et du chaud sont transmis par des
afférences glutamatergiques provenant du PBL et dirigées vers des neurones respectivement

46
GABA(Acide γ-aminobutyrique)ergiques et glutamatergiques localisés dans le MnPO.
L’importance du MnPO dans la réponse thermogène au froid a été montrée
pharmacologiquement. En effet, l’inhibition de l’activité neuronale du MnPO par l’injection de
glycine ou d’un antagoniste des récepteurs au glutamate bloque la thermogenèse du iBAT induite
par le froid ou par l’activation du PBLle. De plus, la stimulation glutamatergique des neurones du
MnPO entraine une augmentation de la thermogenèse du iBAT similaire à la réponse au froid
(Nakamura and Morrison, 2008b; 2008a). Finalement, l’effet de la stimulation des neurones du
MnPO sur l’activité du BAT peut être renversé par l’administration d’un antagoniste au récepteur
GABAA dans le MPO, suggérant la présence d’interneurones GABAergiques au sein du MnPO
projetant vers le MPO. Dans la littérature plusieurs observations anatomiques supportent cette
hypothèse. En effet, (i) des neurones du MnPO innervent le noyau préoptique médial (MPO)
(Uschakov et al., 2007), (ii) le MnPO contient de nombreux neurones GABAergique (Nakamura
et al., 2002), (iii) il y a beaucoup plus de neurones activés (déterminé par l’expression de la
protéine C-Fos) dans le MnPO que dans le MPO lors d’une exposition au froid ou au chaud
(Bratincsák and Palkovits, 2004), (iv) la concentration extracellulaire de GABA dans l’aire
préoptique augmente lors de l’exposition au froid et diminue lors de l’exposition au chaud
(Ishiwata et al., 2005).
Les neurones sensibles au chaud du MPO, clés de voute de la thermorégulation.
Le MPO contient une population de neurones appelés neurones sensibles à la chaleur. Des
études de patch-clamp, réalisées sur coupe de cerveaux, montrent qu’ils émettent des potentiels
d’action lorsque la température du milieu de survie dépasse les 35 °C (Kobayashi and Osaka,
2003). Cependant, les mécanismes membranaires impliqués dans ce phénomène ne sont pas
encore élucidés. D’autre part, ces neurones libèrent du GABA et sont activés par les afférences
glutamatergiques provenant du MnPO et relayant les stimuli thermiques cutanés chauds. A
l’inverse ils sont inhibés par les afférences GABAergiques provenant du MnPO et relayant les
stimuli thermiques cutanés froids. Ils sont aussi inhibés lors d’une réponse immunitaire grâce à la
présence du récepteur 3 aux prostaglandines E (EP3-R), qui participent à la pyrogénèse (Cf.
2.2.2.2). Ces neurones, qui déchargent de façon spontanée en conditions de thermoneutralité,
voient leur taux de décharge diminuer proportionnelement à la baisse de la temperature, afin de
lever graduellement l’inhibition sur les neurones situé en aval tels que le DMH et le noyau du

47
raphé pallidus (RPa) (Lundius et al., 2010). Enfin, le taux de décharge des neurones sensibles au
chaud du MPO est le principal mécanisme neurophysiologique permettant l’équilibre de la
thermorégulation, lequel est déterminé par les afférences thermosensorielles de la peau et la
température locale du cerveau (Romanovsky, 2004).
Le DMH ou l’origine de l’excitation sympathique.
L’implication du DMH dans le relais de l’information vers le iBAT fut premièrement
suggérée par des expériences de transsection. En effet, une transsection réalisée caudalement à
l’aire préoptique augmente la thermogenèse du iBAT (Chen et al., 1998), alors qu’une
transsection réalisée caudalement à l’hypothalamus n’a aucun effet sur l’activité thermogène du
iBAT (Rothwell et al., 1983). Il en a été conclu qu’il existait une structure au sein de
l’hypothalamus dont l’activité stimule la thermogenèse du iBAT, mais qui est inhibée par les
neurones sensibles au chaud GABAergiques du MPO.
Dans un second temps, il a été montré que l’exposition au froid augmente l’expression de la
protéine C-Fos dans le DMH (Cano et al., 2003) et que la désinhibition des neurones du DMH
augmente à la fois l’activité du SNA du iBAT et la thermogenèse induite par celui-ci (Zaretskaia
et al., 2002; Cao et al., 2004b). Ensuite, Nakumura a montré qu’il y a bien un tonus inhibiteur
GABAergique provenant du MPO qui module l’activité des neurones du DMH (Nakamura et al.,
2005). D’autre part, l’injection d’un antagoniste des récepteurs au glutamate empêche toute
activité thermogène en réponse au froid (Madden and Morrison, 2004). Cependant, l’origine de
cette activation glutamatergique qui pourrait accompagner la levée d’inhibition GABAergique
reste encore obscure. Néanmoins, deux hypothèses sont proposées. La première fait appel aux
neurones qualifiés de sensibles au froid ou de thermiquement insensibles situés dans le MPO
(Boulant, 2006), la seconde pourrait faire intervenir des neurones glutamatergique situés dans la
substance grise périaqueducale (PAG) (de Menezes et al., 2009).
Le DMH ne projette pas directement vers les neurones sympathiques préganglionnaires du
iBAT, mais vers la médulla ventromédiale rostrale (MVMR) (Nakamura et al., 2005), plus
particulièrement vers le RPa rostral (RPar), qui est la région du SNC contenant les neurones pré-
moteurs sympathiques. Une étude de rétrotraçage neuronal réalisée chez le rat a permis de
colocaliser un traceur rétrograde monosynaptique provenant du RPar, avec la protéine C-Fos

48
exprimée dans le DMH suite à une exposition au froid (Sarkar et al., 2007). Enfin, il y a des
évidences qui montrent que le DMH projette aussi vers le PAG pour contrôler l’activité du BAT.
En effet, les neurones du PAG expriment C-Fos en réponse à une exposition au froid (Yoshida et
al., 2005) et sont capables de bloquer l’effet thermogène de la désinhibition du DMH (Rathner
and Morrison, 2006). Cependant, l’implication du PAG comme relais du DMH dans le contrôle
de la thermogenèse du iBAT reste à être plus amplement investiguée.
2.2.1.3 Le système nerveux sympathique.
Le SNS contrôlant le iBAT se situe dans les segments thoraciques 3 à 6 de la colonne
intermediolatérale de la moelle épinière (IML) (Cano et al., 2003; Fan et al., 2005). Celui-ci est
sous l’influence de neurones prémoteurs sympathiques localisés notamment dans le RPa, afin de
libérer la noradrénaline qui active la thermogenèse du iBAT (Morrison et al., 2012).
Les neurones prémoteurs sympathiques du BAT au sein du RPa.
L’inhibition des neurones de la MVMR entraine une chute dramatique de la température
corporelle chez le rat vigile (Zaretsky et al., 2003). De plus, Cano et ses collaborateurs montrent
en 2003 que le RPar est l’une des premières régions infectées par un virus (traceur rétrograde
polysynaptique) injecté préalablement dans le iBAT et que celui-ci est largement colocalisé avec
l’expression de C-Fos induit par l’exposition au froid dans le RPar (Cano et al., 2003). Ces
résultats montrent que le RPar occupe une position fondamentale au sein du réseau neuronal de la
thermorégulation, car il est à l’origine des neurones pré-moteurs sympathiques qui excitent les
ganglions sympathiques du iBAT situés dans la moelle thoraco-lombaire. Cependant, le RPar
n’est pas le seul noyau contenant des neurones pré-moteurs sympathiques du iBAT. En effet,
Cano et ses collaborateurs montrent que le PVH pourrait aussi participer à l’activation des
ganglions sympathiques du iBAT (Cano et al., 2003).
Les neurones pré-moteurs sympathiques du iBAT sont sous l’influence d’afférences
excitatrices glutamatergiques et inhibitrices GABAergiques avec une prédominance inhibitrice
afin de conserver les ressources énergétiques. En effet, l’injection dans le RPar d’un agoniste aux
récepteurs au glutamate ou d’un antagoniste aux récepteurs GABA augmente l’activité du SNA
dans le iBAT ainsi que la thermogenèse (Morrison, 1999; Madden and Morrison, 2003). A

49
l’inverse, l’inhibition de l’activité neuronale du RPar ou le blocage des récepteurs au glutamate
annule toute augmentation de l’activité du SNA du iBAT induit par le froid ou la fièvre
(Nakamura and Morrison, 2007; Ootsuka et al., 2008). De plus, plusieurs autres stimuli
d’origines diverses voient leurs effets sur la thermogenèse du iBAT bloqués par l’inhibition de
l’activité neuronale dans le RPar tel que, (i) le LHA (Cerri and Morrison, 2005), (ii) l’activation
des récepteurs aux opioïdes de type mu (Cao and Morrison, 2005), (iii) l’activation des récepteurs
au CRH dans l’aire préoptique (Cerri and Morrison, 2006), (iv) l’administration systémique de
leptine (Morrison, 2004), (v) l’activation centrale des récepteurs aux mélanocortines (Fan et al.,
2007).
La moelle épinière.
Le relais de l’action du SNC sur le contrôle de l’activité du iBAT dans la moelle épinière se
situe dans l’IML. Parmi les neurones du RPar projetant vers l’IML, la plupart sont
glutamatergiques mais certains expriment ou co-expriment de la sérotonine. En effet, le
transporteur vésiculaire au glutamate de type 3 qui constitue un bon marqueur phénotypique pour
les neurones glutamatergiques se trouve colocalisé dans le RPar, soit avec un virus de la pseudo
rage injecté dans le iBAT, soit avec C-Fos induit par une exposition au froid (Nakamura et al.,
2004). On trouve aussi quelques neurones du RPa qui projettent vers l’IML marqués pour la
glutamate décarboxylase 67, un marqueur phénotypique des neurones GABAergiques (Stornetta
et al., 2005). D’un point de vue fonctionnel, la mise en évidence de projections glutamatergiques
sur l’IML a été réalisée par l’administration de substances pharmacologiques directement dans
l’IML d’un rat anesthésié. En effet, des nano-injections de glutamate ou de l’acide N-méthyl-D-
aspartique (NMDA) active le SNA du iBAT ainsi que sa thermogenèse (Nakamura et al., 2004;
Madden and Morrison, 2006), alors que l’utilisation d’un antagoniste des récepteurs au glutamate
bloque l’augmentation de la thermogenèse suite à l’injection de bicuculine dans le RPa
(Nakamura et al., 2004). Le rôle de la sérotonine dans l’IML pour contrôler l’activité du iBAT fut
rapporté par des expériences pharmacologiques et génétiques. Le blocage des récepteurs à la
sérotonine dans la moelle épinière contrecarre les effets du froid sur le SNA du iBAT (Madden
and Morrison, 2010), tandis que l’injection de sérotonine dans l’IML stimule l’activité du SNA
du iBAT et potentialise même les effets d’une injection de NMDA dans l’IML sur l’activité du
SNA du iBAT ainsi que sa thermogenèse (Madden and Morrison, 2006). Enfin, le rôle significatif

50
des neurones sérotoninergiques dans la réponse normale au froid est supporté par l’absence
d’activité thermogène et la diminution de la température corporelle chez des souris
génétiquement modifiées qui n’ont pas de neurones secrétant la sérotonine (Hodges et al., 2008).
Les mécanismes permettant l’interaction entre les neurones glutamatergiques et sérotoninergiques
au sein de l’IML afin de contrôler l’activité du iBAT restent à être définis. Cependant, quelques
indices sont disponibles. Tout d’abord, Deuchars et ses collaborateurs en 2005 révèlent la
présence d’interneurones GABAergiques capables d’influencer la décharge des neurones
préganglionnaires sympathiques de l’IML (Deuchars et al., 2005). Ensuite, les terminaisons
synaptiques sérotoninergiques de l’IML se trouvent à proximité des neurones GABAergiques
(Morrison et al., 2012). Enfin, l’activation des récepteurs 5-HT1A sur les neurones
GABAergiques de l’IML potentialise l’excitation de neurones préganglionnaires sympathiques
(Madden and Morrison, 2006; 2008).
Par la suite, les neurones préganglionnaires sympathiques de l’IML se projettent vers le
ganglion nodal où ils libèrent de l’acétylcholine, qui active des récepteurs cholinergiques de type
nicotinique N1 situés sur les neurones postganglionaires. Ces derniers se projettent vers le tissu
adipeux brun et libèrent de la noradrénaline pour activer principalement le récepteur β3
adrénergique, qui stimule la thermogenèse.
2.2.2 Les autres mécanismes impliqués dans le contrôle central de l’activité du iBAT.
Au-delà des mécanismes centraux impliqués dans le contrôle de la thermogenèse du iBAT
lors de l’exposition au froid, d’autres systèmes interviennent pour contrôler l’activité du iBAT de
façon chronique comme lors du cycle éveil/sommeil et ultradien ou dans des situations
exceptionnelles telles que le stress, la fièvre, l’hypoglycémie et l’hypoxie.
2.2.2.1 Rythme ultradien et stress : rôle de l’orexine.
La perte des neurones à orexine conduit à différentes altérations, comme des désordres dans
les phases du sommeil, des perturbations métaboliques menant à un risque accru d’obésité et
affecte la thermorégulation associée au rythme ultradien (Hara et al., 2005; Mochizuki et al.,
2006; Plazzi et al., 2011). L’orexine est un neuropeptide synthétisé dans des neurones de
l’hypothalamus latéral, dorsal et périfornical, dont les projections sont dirigées vers le RPar.

51
Récemment, il a été montré que l’injection d’orexine dans le RPar active la thermogenèse
(Tupone et al., 2011). De plus, la toxine du choléra B injectée dans le iBAT migre directement
dans le RPar et est colocalisée avec des neurones orexinergiques (Tupone et al., 2011). D’autre
part, le stress lié à la manipulation des animaux induit une thermogenèse qui implique les
neurones à orexine (Zhang et al., 2010). Enfin, outre son action dans l’activité thermogène du
iBAT, l’orexine joue un rôle important pour la maturation et le maintien fonctionnel du iBAT. En
effet, lors des stades précoces du développement, l’orexine intervient dans la différenciation du
BAT, par la suite, elle est responsable du maintien de la fonctionnalité du iBAT lors du
vieillissement chez la souris (Sellayah et al., 2011; Sellayah and Sikder, 2013). L’ensemble de
ces éléments témoigne de l’impact de l’orexine sur toute la machinerie thermogène.
2.2.2.2 La fièvre.
La fièvre est définie par une température corporelle supérieure à 38°C et elle témoigne de
l’activité du système immunitaire en réponse à une infection ou à une inflammation. Tout
d’abord, la fièvre est considérée comme un mécanisme favorisant la survie de l’hôte à une
infection bactérienne ou virale, car l’élévation de la température corporelle à 39°C diminue la
virulence et le développement de la plupart des organismes pathogènes (Kluger and Vaughn,
1978). Cependant, une fièvre d’une forte intensité peut avoir des effets délétères sur l’organisme
et lorsque la température corporelle approche 43°C la mort de l’hôte peut survenir (Banet, 1986).
La fièvre se décompose en plusieurs phases lors desquelles on observe une élévation de la
température corporelle. Lors d’une induction pharmacologique de la fièvre chez le rat par
l’injection périphérique de lipopolysaccharide (LPS), une molécule présente sur la surface des
bactéries, il est observé une phase précoce à l’intérieur de la première heure et une phase tardive
entre une heure et six heures après l’injection (Elmquist and Scammell, 1996). Néanmoins, ces
deux phases sont dépendantes de la prostaglandine (PGE2) et de son action sur le récepteur EP3-
R situé dans l’aire préoptique et en particulier dans le MnPO et le MPO (Elmquist and Scammell,
1996; Oka et al., 2003). Toutefois, le lieu de synthèse de la PGE2 varie entre ces deux phases. En
effet, 40 minutes après l’injection de LPS, les niveaux d’expression de la cyclooxygenase-2 qui
participe à la biosynthèse de la PGE2 sont très élevés dans les poumons et le foie, alors qu’ils
restent identiques dans l’hypothalamus. De plus, l’injection par voie sanguine d’un anticorps
dirigé contre PGE2 lors de l’injection de LPS inhibe l’augmentation de la température corporelle

52
observée pendant la première phase, suggérant que la PGE2 est synthétisée par les macrophages
hépatiques et pulmonaires (Steiner et al., 2006). Ensuite, il a été montré que la seconde phase fait
intervenir de la PGE2 synthétisée par le réseau vasculaire situé à proximité de l’aire préoptique.
En effet, l’injection d’un inhibiteur de la cyclooxygénase 2 dans l’aire préoptique inhibe
l’élévation de la température corporelle induite par l’administration périphérique de LPS
(Scammell et al., 1998). Enfin, Ootsuka et ses collaborateurs montrent en 2008 qu’une injection
intraveineuse de PGE2 active le iBAT (Ootsuka et al., 2008), suggérant ainsi que l’élévation de la
température corporelle lors d’un épisode de fièvre est la conséquence d’une activation soutenue
du iBAT par l’action de PGE2 sur EP3-R dans l’aire préoptique.
2.2.2.3 L’hypoglycémie
Depuis le début des années 70, il est connu que l’hypoglycémie ou la glucoprivation
entraine une hypothermie chez la souris comme chez l’humain (Freinkel et al., 1972). Dans le cas
d’une hypoglycémie, la baisse de la température corporelle constitue un mécanisme de protection
afin de ralentir le métabolisme et ainsi conserver l’énergie utile au cerveau et au cœur. en effet,
les rats maintenus à leur température d’origine décèdent tous après un hypoglycémie induite par
infusion continue d’insuline (Buchanan et al., 1991). Parmi les centres glucosensibles du SNC, la
médulla ventrolatérale (MVL) est connue pour sa capacité d’inhibition de la thermogenèse du
BAT (Buchanan et al., 1991; Cao et al., 2010). Il a donc été logique pour Madden et ses
collaborateurs en 2012 d’injecter du 5-thio-D-glucose dans la MVL afin de mimer les effets
d’une glucoprivation, avec pour conséquence une inhibition totale de l’activité du SNA du iBAT
induite par le froid (Madden, 2012). Par la suite, celui-ci a réalisé la même expérience, en
effectuant préalablement une transsection ponto-médullaire qui prévient les effets de l’injection,
montrant ainsi la présence d’un relais neuronal situé vers le rostre (Ritter et al., 2011; Madden,
2012).
2.2.2.4 L’hypoxie.
Le tronc cérébral contient des voies neuronales impliquées dans l’inhibition de la
thermogenèse du iBAT qui répondent à l’hypoxie artérielle ; cela participe à un réflexe
permettant de diminuer la consommation d’oxygène par les organes considérés comme non
vitaux afin de permettre une meilleure disponibilité de l’oxygène sanguin pour le SNC. Dans une

53
étude de 2005, l’équipe du Dr Morrison montre que l’hypoxie entraine un arrêt brutal de la
stimulation du iBAT induite par une hypothermie ou par l’administration de PGE2 dans l’aire
préoptique (Madden and Morrison, 2005). Par ailleurs, la forte inhibition de l’activité thermogène
du iBAT provoquée par l’hypoxie est totalement contrecarrée par la section du nerf de Hering
permettant de faire remonter l’information issue de chémorécepteurs situés dans le sinus
carotidien vers le tronc cérébral. De plus, l’inhibition du NTS par une injection de muscimol (un
agoniste du récepteur GABAA) bloque totalement les effets hypothermiques de l’hypoxie
(Madden and Morrison, 2005). Néanmoins, la voie neuronale utilisée entre le NTS et les
neurones préganglionnaires sympathiques activant le iBAT reste à caractériser. Enfin, un étude
canadienne de 2009 indique que la thermosensibilité de l’aire préoptique est diminuée lors d’une
hypoxie entrainant ainsi un déplacement du point de consigne chez le rat de 38°C vers 30°C de
façon concentration dépendante (Tattersall and Milsom, 2009).


55
3 Implication du système mélanocortine dans la régulation de
l’homéostasie énergétique : son rôle dans la thermogenèse.
3.1 Le système mélanocortine.
Le système mélanocortine est phylogénétiquement bien conservé, il est présent chez les
invertébrés, les poissons, les amphibiens et bien sûr les mammifères. Chez ces derniers, ce
système est présent dans l’ensemble de l’organisme et est impliqué dans de nombreuses fonctions
physiologiques, tels que la pigmentation, la reproduction, l’anxiété ou la régulation de
l’homéostasie énergétique.
3.1.1 Ses principaux acteurs.
Les fonctions physiologiques du système mélanocortine sont définies par l’action opposée
des agonistes (les peptides dérivant de POMC) et des antagonistes (l’agouti et l’AgRP) sur les 5
récepteurs aux mélanocortines connus à ce jour (Pritchard et al., 2002; Mountjoy, 2010;
Biebermann et al., 2012).
3.1.1.1 Localisation et synthèse des ligands endogènes.
La POMC est une pro-hormone de 31-kDa dont la synthèse a lieu dans de nombreux
organes. En effet la POMC est exprimée dans la tige pituitaire et le SNC (uniquement dans
l’ARC et le NTS) (Joseph et al., 1983), mais également au niveau de la peau, de l’intestin, du
placenta et du pancréas (Smith and Funder, 1988).
La clivage de la POMC dépend du tissu (Pritchard et al., 2002). En effet, chaque tissu
synthétisant la POMC exprime aussi des pro-hormones convertases (PC) et d’autres protéines
nécessaires au clivage de celle-ci en neuropeptides actifs. Dans le SNC la présence des PC
(PC1/3, PC2 et PACE4), de la carboxypeptidase E et de la n-acetyltransférase permet la synthèse
de l’α-MSH, de la β-MSH et de la γ-MSH, à l’exception des rongeurs qui ne possèdent pas le site
de clivage nécessaire à la synthèse de la β-MSH (Pritchard et al., 2002). Tandis que l’absence de
PC2 dans la glande pituitaire arrête la clivage de POMC à l’étape de l’adrénocorticotrophine
(ACTH) (Shimizu et al., 2007; Biebermann et al., 2012). Enfin, la dernière étape de la régulation
de POMC a été mise en évidence en 2009 dans une étude de Wallingford et ses collaborateurs,

56
décrivant le rôle de la prolyl carboxypeptidase, dans la dégradation de l’α-MSH (Wallingford et
al., 2009). Par ailleurs, ces processus sont reconnus comme étant essentiels au maintien de la
spécificité de fonction des tissus, car les récepteurs aux mélanocortines n’ont pas la même affinité
pour les dérivés de la POMC, et notamment le MC2R qui ne lie que l’ACTH (Pritchard et al.,
2002; Biebermann et al., 2012).
Le système mélanocortine est unique parmi les systèmes de neuropeptides connus, car il
est aussi régulé par deux antagonistes endogènes (Ellacott and Cone, 2006), l’Agouti (Bultman et
al., 1992; Woychik and Young, 1992) et l’AgRP (Ollmann et al., 1997; Shutter et al., 1997).
L’Agouti est exprimé en périphérie et particulièrement dans la peau et les cheveux où il intervient
dans la couleur (Bultman et al., 1992), tandis que l’AgRP est uniquement produit par des
neurones de l’ARC (Ollmann et al., 1997; Shutter et al., 1997). Tout comme la POMC, l’AgRP
est une pro-hormone. Cependant, la forme clivée et non clivée sont actifs et son clivage, réalisé
en une seule étape par la PC1/3, permet la formation de 3 neuropeptides dont seul l’AgRP 83-132
semble activer les MC3/4R (Biebermann et al., 2012).
3.1.1.2 Localisation et fonction des récepteurs.
Dans les années 90, les cinq récepteurs aux mélanocortines MC1-5R connus actuellement
ont été caractérisés (Mountjoy et al., 1992; Chhajlani et al., 1993; Gantz et al., 1993a; 1993b;
Labbé et al., 1994). Ces récepteurs sont des récepteurs à sept domaines transmembranaires
couplés aux protéines G stimulatrices dont les fonctions sont nommées ci-dessous (Ellacott and
Cone, 2006; Mountjoy, 2010). Parmi ces récepteurs, on distingue ceux dont l’expression a lieu en
périphérie, le MC1R, le MC2R et le MC5R, et ceux dont l’expression a lieu dans le SNC, le
MC3R et le MC4R. Leur lieu de synthèse, leurs affinités et leurs principales fonctions sont
recensés dans le tableau 2.
Le MC3R est exprimé dans le SNC et particulièrement dans l’hypothalamus (Roselli-
Rehfuss et al., 1993). À l’instar du MC3R, le MC4R est exprimé dans le SNC mais de façon
beaucoup plus importante. En effet, des études d’immunohistochimie et d’hybridation in situ ont
relevé la présence de MC4R dans de très nombreuses structures cérébrales telles que
l’hypothalamus et le tronc cérébral (Kishi:2003jy Gelez et al., 2010). Le MC4R est essentiel pour
la régulation de l’homéostasie énergétique (Cf. 1.2 et 3.3), mais participe également à la

57
régulation du stress et de la reproduction notamment (Mountjoy, 2010). Enfin, dans le système
mélanocortine, l’expression des récepteurs ainsi que sa liaison avec son ligand sont contrôlés par
des protéines accessoires. Pour exemple, les melanocortin receptor accessory protein 1 et 2
diminuent l’expression de MC4R (pour revue (Cerdá-Reverter et al., 2013)) alors que syndecan-3
régule le site de liaison de l’AgRP sur MC4R, afin de stimuler la prise alimentaire (Reizes et al.,
2006).
Tableau 2: Résumé des principales fonctions, sites d’expression et de l’affinité des ligands
pour les récepteurs aux mélanocortines.
Les récepteurs
L’affinité pour les ligands
Les sites d’expressions
La principale fonction références
MC1R α-MSH = ACTH > β-MSH > γ-MSH, Agouti Mélanocytes Pigmentation (Rees, 2003)
MC2R ACTH, Agouti Glandes surrénales Stéroïdeogenèse
(Mountjoy et al., 1992; Ellacott and
Cone, 2006)
MC3R α-MSH = ACTH = β-MSH = γ-MSH, AgRP SNC Homéostasie
énergétique
(Roselli-Rehfuss et al., 1993; Butler et
al., 2000)
MC4R α-MSH = ACTH > β-MSH > γ-MSH, AgRP SNC Homéostasie
énergétique (Gelez et al., 2010; Mountjoy, 2010)
MC5R α-MSH > ACTH > β-MSH > γ-MSH, Agouti
Glandes exocrines
Régulation de la sécrétion des
glandes exocrines
(Chen et al., 1997)

58
3.1.2 Ses partenaires dans le contrôle de la prise alimentaire.
Le système mélanocortine est très impliqué dans le contrôle de la prise alimentaire. Par
conséquent, l’expression de la POMC est largement influencée par d’autres acteurs importants
tels que la leptine, l’insuline ou les endocannabinoïdes.
3.1.2.1 L’effet de la leptine et de l’insuline sur l’expression de la POMC.
Comme nous l’avons vu, la leptine est produite par les adipocytes blancs, elle est ensuite
libérée dans la circulation sanguine et agit centralement sur les neurones POMC de l’ARC et du
NTS, grâce à la présence de son récepteur au sein de ces deux populations neuronales (Cheung et
al., 1997). D’autre part, l’effet anorexigène de la leptine est partiellement bloqué par
l’administration d’un antagoniste des MCR, confirmant ainsi que le système mélanocortine est
l’un des principaux médiateurs de la leptine dans ces effets pro-cataboliques (Seeley et al., 1997).
Par la suite, Munzberg et ses collaborateurs ont montré que l’administration périphérique de
leptine chez le rongeur phosphoryle la Janus Kinase 2 (Jak2) qui active une cascade de
signalisation impliquant la phosphorylated signal transducer and activator of transcription 3
(pSTAT-3) (Münzberg et al., 2003), Parallèlement, l’expression de la POMC est augmentée dans
l’ARC. De plus, la délétion du gène codant pour STAT-3 chez la souris entraine une diminution
de l’expression de la POMC qui s’accompagne d’une obésité sévère (Gao et al., 2004), suggérant
de ce fait que pSTAT-3 est le relais de l’action de la leptine sur la POMC en agissant comme
facteur de transcription (Xu et al., 2007).
L’action de la pSTAT-3 comme facteur de transcription nécessite la dimérisation de deux
pSTAT-3 puis sa liaison avec le complexe SP1-POMC sur le promoteur de la POMC (Morton et
al., 2006). Parallèlement, la Jak2 phosphoryle l’insulin receptor substrate proteins 1 and 2, qui
activent la voie phosphatidylinositol-3-kinase (PI3K), qui aboutit à la phosphorylation et à
l’exclusion du noyau de la Forkhead box containing protien 1 (FOXO1) (Yang et al., 2009a).
L’exclusion nucléaire de FOXO1 qui inhibe la liaison du dimère de pSTAT3 sur le complexe
SP1-POMC favorise l’expression de la POMC. De plus, la voie PI3K est également utilisée par
l’insuline pour agir sur FOXO1 et ainsi favoriser l’action de pSTAT3, suggérant que l’insuline et
la leptine agissent en synergie sur l’activité du promoteur de la POMC (Varela and Horvath,
2012). À l’inverse, dans les neurones AgRP, pSTAT-3 inhibe le promoteur de l’AgRP alors que

59
FOXO1 le stimule (Kitamura et al., 2006). Enfin, pSTAT-3 agit simultanément comme facteur de
transcription du gène suppressor of cytokine signaling 3, lequel inhibe la phosphorylation du
récepteur à la leptine (Elias et al., 1999; Bjorbak et al., 2000) dans le but de réduire la
transcription de la POMC.
3.1.2.2 Les endocannabïnoides.
Le système endocannabïnoide est constitué de deux principaux récepteurs aux
cannabinoïdes : le récepteur aux cannabinoïdes de type 1 (CB1) est exprimé dans le SNC alors
que le récepteur aux cannabinoïdes de type 2 est exprimé en périphérie (Matsuda et al., 1990;
Munro et al., 1993). De plus, il possède deux principaux ligands endogènes dérivant de
phospholipides contenant de l’acide arachidonique, l’anandamide (AEA) (Devane et al., 1992) et
le 2-arachidonylglycerol (2-AG) (Mechoulam et al., 1995), mais son ligand le plus connu est le
delta-9-tetrahydrocannabinol (Δ9-THC) (Gaoni and Mechoulam, 1964). Le système
endocannabïnoide a quelques particularités. En effet, le récepteur CB1 sous sa forme protéique
est exprimé au niveau pré-synaptique, alors que ses ligands sont libérés à la demande du coté
post-synaptique (Wilson and Nicoll, 2001). L’activation de CB1 qui est un récepteur à sept
domaines transmembranaires couplé à une protéine Gi/o (Pertwee, 1997) inhibe la libération de
neurotransmetteur dans la fente synaptique par un mécanisme désinhibant les canaux potassiques
de type A, lesquels vont faire sortir du K+ du neurone conduisant à son hyperpolarisation (Ameri,
1999).
L’injection d’un agoniste CB1 dans l’hypothalamus ou le noyau accumbens augmente la
prise alimentaire chez le rongeur (Jamshidi and Taylor, 2001; Kirkham et al., 2002), alors que le
jeûne augmente la concentration d’AEA et de 2-AG dans ces structures cérébrales (Kirkham et
al., 2002), montrant ainsi que le système endocannabïnoide est étroitement lié au comportement
alimentaire. Enfin, les souris ayant une délétion du gène codant pour CB1 montrent une
augmentation de la thermogenèse (Quarta et al., 2010) suggérant que le système
endocannabïnoide est impliqué dans la régulation de la balance énergétique.
Le système endocannabïnoide influence le système mélanocortine. En effet, l’injection
chronique de Δ9-THC en périphérie augmente l’expression de la POMC dans l’ARC et le NTS,
suggérant que les neurones exprimant les récepteurs aux mélanocortines ont besoin de plus de

60
ligands pour réaliser leurs actions après l’inhibition opérée par le Δ9-THC (Corchero et al., 1997;
1999). De plus, Verty et ses collaborateurs ont observé que la co-injection centrale d’un agoniste
des récepteurs aux mélanocortines et d’un antagoniste CB1, inefficace seul, inhibent en synergie
la prise alimentaire chez le rat (Verty et al., 2004). Enfin, l’expression de l’ARNm de CB1 est
très fortement exprimée dans le PVH, un noyau hypothalamique très impliqué dans le contrôle de
la balance énergétique et qui exprime également le MC4R (Richard et al., 2009).
3.2 La thermogenèse induite par l’alimentation
La thermogenèse réalisée par le BAT a pour principale fonction de réguler la température
corporelle, et les mécanismes relatifs à cette fonction ont été développés dans le chapitre 2. Par
ailleurs, le BAT participe activement à la régulation de l’homéostasie énergétique (Richard et al.,
2012; Carey and Kingwell, 2013). En effet, il permet de dissiper le surplus d’énergie ingérée lors
d’un repas ou en réserve dans le tissu adipeux blanc, par un phénomène appelé la thermogenèse
induite par l’alimentation (TIA) (Rothwell and Stock, 1979). Celle-ci survient en concomitance
avec l’ingestion d’aliments. Néanmoins, elle ne doit pas être confondue avec la thermogenèse
issue directement des processus de la digestion des aliments. En effet, la TIA résulte de l’action
du SNC sur le BAT, suite à l’intégration des signaux périphériques, traduisant à la fois la quantité
d’énergie procurée par le repas et l’état des réserves énergétique de l’organisme (Rothwell and
Stock, 1979; 1981; Golay et al., 1982). Bien que la TIA soit largement argumentée, le débat sur
sa pertinence physiologique reste ouvert. En effet, dès les années 70-80, il a été mis en évidence
que la masse du BAT, l’expression protéique de gènes impliqués dans la thermogenèse comme
UCP-1 et la calorimétrie indirecte sont augmentées suite à un régime riche en graisse, en sucre ou
en protéine (Rothwell and Stock, 1979). Cependant, l’équipe du docteur Kozak remet en question
ce phénomène ; la dissipation d’énergie sans nécessité physiologique apparait pour eux comme
un non sens évolutif (Kozak, 2010). En montrant que la perte de fonction thermogène du BAT,
par la délétion du gène codant pour la protéine UCP-1 chez des souris hébergées à
thermoneutralité et soumises un régime riche en graisse, n’entraine pas de diminution de la
consommation d’oxygène, ni d’augmentation de la masse corporelle, le docteur Kozak et ses
collaborateurs ont suscité le débat (Anunciado-Koza et al., 2008; Kozak, 2010). Enfin, une étude
récente d’imagerie chez l’homme vient appuyer leur hypothèse. En effet, une expérience de
captage de fluoro-deoxyglucose (FDG) en tomographie par émission de positrons (TEP), indique

61
que le BAT n’est pas activé lors de la suralimentation chez l’humain (Schlogl et al., 2013).
Cependant, d’autres équipes de recherche ont observé un développement accru de l’obésité chez
des souris hébergées à thermoneutralité nourries avec une diète riche en graisse dont le gène
codant pour la protéine UCP-1 a été invalidé (Feldmann et al., 2009), ainsi qu’une augmentation
du captage de FDG dans le BAT chez l’humain après un repas, traduisant ainsi une activation
postprandiale du BAT (Vosselman et al., 2013). Malgré cette contradiction dans la littérature, la
grande majorité des auteurs s’accordent sur le rôle majeur de l’activité thermogène du BAT dans
la survenue de l’obésité ainsi que sur ca capacité à dissiper des calories. D’ailleurs, une
dysfonction du BAT est corrélée avec l’apparition de l’obésité chez l’humain (Cypess et al.,
2009; Ouellet et al., 2011), alors que lorsque le BAT est pleinement activé pendant 1 an, il
pourrait consommer plus de 4 kilogrammes de lipides selon une projection menée par Virtanen et
ses collaborateurs (Virtanen et al., 2009). Enfin, d’après Butler et ses collaborateurs, la TIA est
sous le contrôle du système mélanocortine, car la délétion du gène codant pour MC4R chez la
souris inhibe l’augmentation de la consommation d’oxygène induite par le changement de régime
alimentaire (Butler et al., 2001; Butler, 2006). La mesure de la consommation d’oxygène permet
d’évaluer la dépense énergétique, mais constitue aussi une mesure indirecte reconnue de la
thermogenèse (Simonson and DeFronzo, 1990).
3.3 La mise en évidence de l’implication du système mélanocortine dans le contrôle de
la thermogenèse.
L’utilisation de modèles génétiques du système mélanocortine, mais aussi l’injection
d’agonistes et antagonistes du système mélanocortine déjà connus pour affecter la balance
énergétique chez l’humain et les rongeurs (Cone, 1999; Butler and Cone, 2002; Farooqi and
O'Rahilly, 2006; Myers and Olson, 2012) ou encore, des techniques de traçage neuronal associées
à de l’immunohistochimie et d’hybridation in situ sur tranche de cerveau de rongeur (Voss-
Andreae et al., 2007; Song et al., 2008), ont permis d’améliorer notre compréhension du role du
système mélanocortine, et ainsi mis en évidence son implication dans le contrôle de la dépense
énergétique.

62
3.3.1 Les évidences génétiques
La diminution de la consommation d’oxygène constatée chez des souris dont le gène
POMC est invalidé, suggère que certains neuropeptides dérivés de celui-ci ont un rôle essentiel
dans la dépense énergétique (Challis et al., 2004; Tung et al., 2007). Parallèlement, l’utilisation
d’un modèle de souris présentant une délétion pour le gène de l’AgRP présente une augmentation
de la consommation d’oxygène (Wortley et al., 2005), suggérant ainsi que les neurones exprimant
la POMC et l’AgRP participent au contrôle de la dépense énergétique.
D’autre part, des études sur MC3R et MC4R, les deux seuls récepteurs aux mélanocortines
présents au sein du SNC, montrent que seul le MC4R est impliqué dans le contrôle de la dépense
énergétique (Garfield et al., 2009). En effet, les souris présentant une délétion pour le gène
MC4R consomment 20% d’oxygène de moins que les souris sauvages (Ste Marie et al., 2000).
De plus, lorsque ces souris sont nourries avec une diète riche en gras, elles sont incapables de
diminuer leur prise alimentaire ainsi que d’augmenter leur consommation d’oxygène, tel que le
font les souris sauvages (Butler et al., 2001). Par ailleurs, l’effet de la leptine sur l’expression
d’UCP-1 est absent chez ces souris transgéniques (Ste Marie et al., 2000), alors que celles-ci
conservent une température corporelle similaire à celle des souris sauvages lors d’une exposition
de 120h à 4°C (Butler et al., 2001). Il apparaît donc évident que le système mélanocortine joue un
rôle capital dans le contrôle de la dépense énergétique et conséquemment sur la thermogenèse via
son récepteur MC4R, dans le but de réguler la balance énergétique.
3.3.2 Les évidences pharmacologiques.
Des études utilisant des ligands endogènes et synthétiques ont été réalisées chez le rat afin
de compléter et d’affiner les connaissances sur le rôle des acteurs révélés par des études
génétiques chez la souris. En effet, l’injection i.c.v. de MTII, un agoniste des récepteurs aux
mélanocortines, augmente la consommation d’oxygène lors d’un traitement aigu (Hwa et al.,
2001) ou chronique (Jonsson et al., 2001). De plus, l’administration répétée de MTII sur une
journée augmente significativement l’expression d’UCP-1 dans le iBAT (Williams et al., 2003).
Enfin, l’injection centrale de MTII augmente le renouvellement de la noradrénaline dans le iBAT,
ainsi que sa température (Brito et al., 2007; Skibicka and Grill, 2008), suggérant de ce fait que la

63
stimulation des récepteurs aux mélanocortines permet l’augmentation de la dépense énergétique
via l’activité du iBAT.
À l’inverse, la consommation d’oxygène et la température corporelle diminuent lors de
l’injection centrale d’un antagoniste MC3/4R chez le rat (Adage et al., 2001; Small et al., 2003).
De plus, l’administration centrale et chronique de SHU9119 (un antagoniste MC3/4R) chez le rat
diminue fortement l’expression d’UCP-1 dans le iBAT (Nogueiras et al., 2007). De même, l’effet
de la leptine sur l’expression d’UCP-1 est inhibé par l’injection de SHU9119 (Satoh et al., 1998).
Enfin, il a été constaté que la température du iBAT diminue chez le hamster sibérien après
l’injection i.c.v. de l’antagoniste endogène (AgRP) des MC3/4R (Brito et al., 2007), suggérant
donc que les agonistes du système mélanocortine exercent un tonus positif sur la dépense
énergétique et ses acteurs.
3.3.3 Les évidences anatomo-fonctionnelles.
Les effets centraux du système mélanocortine sur la thermogenèse du iBAT sont
principalement relayés par le SNS. En effet, l’injection de MTII augmente le renouvellement de
la noradrénaline dans le iBAT (Brito et al., 2007) et la dénervation du iBAT conduit à un blocage
de l’augmentation de l’expression d’UCP-1 dans le iBAT suite à une injection de MTII dans le
3ème ventricule chez le rat (Williams et al., 2003).
L’activité du SNS contrôlant le iBAT est gouvernée par plusieurs structures du SNC (Cf.
2.2.1.3). Cependant, l’injection d’un virus de la pseudo-rage (traceur rétrograde polysynaptique)
dans le iBAT chez des rats sacrifiés à différents temps, suivie d’une hybridation in situ ou d’une
immunohistochimie dirigées contre MC4R, a permis d’établir une chronologie d’infection des
structures du SNC innervant le iBAT. Cette méthode a aussi permis de quantifier les neurones
exprimant le MC4R et le virus de la pseudo-rage au sein des même structures (Voss-Andreae et
al., 2007; Song et al., 2008). L’étude menée par Song, en collaboration avec notre laboratoire, a
révélé que parmi les differentes structures du SNC connectées au iBAT, le PVH, le PAG et l’aire
préoptique présentent le plus grand nombre de neurones marqués par le virus de la pseudo-rage
(Song et al., 2008). Enfin, 84, 83 et 77% des neurones exprimant MC4R dans le PVN, le PAG et
l’aire préoptique, respectivement, sont marqués aussi par le virus de la pseudo-rage, traduisant

64
ainsi les connexions indirectes de ces neurones avec le iBAT (Song et al., 2008) et le rôle
potentiel de MC4R au sein de ces structures, dans le contrôle de l’activité thermogène du iBAT.
Ces trois structures cérébrales sont connues pour leurs actions sur la thermogenèse du
iBAT. En effet, Le PVH est connecté au iBAT (Bamshad et al., 1999; Oldfield et al., 2002).
Cependant, le rôle du PVH sur le iBAT est controversé ; les premières études ont montré que le
PVH stimule le iBAT via une connexion directe sur l’IML (Zhang et al., 2000) et que sa lésion
n’affecte pas la réponse au froid de l’organisme (Lu et al., 2001). À l’inverse, l’activation du
PVH par l’injection de bicuculline inhibe la thermogenèse du iBAT et bloque la réponse au froid
(Madden and Morrison, 2009). Enfin, l’injection de NPY dans le PVH diminue l’activité du SNS
du iBAT (Egawa et al., 1991), suggérant existence d’au moins deux sous populations de neurones
dans le PVH dont les effets sont opposés (Morrison et al., 2012). La controverse est aussi
présente quant au rôle des neurones du PVH exprimant MC4R. En effet, Balthasar et ses
collaborateurs ont rapporté que la restauration de l’expression de MC4R dans le PVH chez des
souris transgéniques ne l’exprimant pas, permet un retour à l’état normal du contrôle de la prise
alimentaire, mais pas de l’augmentation de la consommation d’oxygène postprandiale, suggérant
ainsi que l’action de MC4R sur la dépense énergétique est relayé par une autre structure du SNC
(Balthasar et al., 2005). D’autre part, deux groupes de recherche ont montré que l’injection de
MTII dans le PVN augmente la température du corps ou du iBAT (Song et al., 2008; Skibicka
and Grill, 2009), suggérant ainsi que les neurones du PVH exprimant MC4R jouent un rôle
essentiel dans le contrôle de la thermogenèse du iBAT. Enfin, les rôles de l’aire préoptique et du
PAG dans le contrôle de la thermogenèse ont été abordés dans la section 2.2. Quant au rôle du
système mélanocortine dans le contrôle de la thermogenèse au sein de ces structures cérébrales, il
n’y a rien de décrit à ce jour.
Cependant, le système mélanocortine peut aussi influencer la libération d’hormones
thyroïdiennes, via son action sur l’axe hypothalamus-pituitaire-thyroïdien, permettant ainsi la
synthèse de la T3, dont les effets sur l’augmentation de l’expression d’UCP-1 dans le iBAT sont
connus (de Jesus et al., 2001). En effet, chez le rat les neurones du PVH exprimant de la TRH
sont innervés par des fibres qui libèrent de l’α-MSH et de l’AgRP (Fekete et al., 2000). De plus,
certains neurones du PVH expriment conjointement l’ARNm de MC4R et de la pro-TRH (Harris

65
et al., 2001). Enfin, l’injection centrale d’α-MSH chez des animaux à jeun augmente de 2,5 fois
la concentration de la T4, le précurseur de la T3 (Fekete et al., 2000).


67
Problématique et Objectifs généraux des travaux.


69
Problématique.
L’obésité est la conséquence d’une dérégulation de l’homéostasie énergétique et se définit
par un excès de masse adipeuse. Tout d’abord, il est connu que les signaux périphériques, tels que
la leptine, informent le SNC de l’état des réserves énergétiques de l’organisme afin de réguler
l’homéostasie énergétique. Ensuite, il a été montré que le système mélanocortine est l’un des
relais majeurs du SNC dans la régulation de l’homéostasie énergétique en contrôlant l’apport
énergétique, mais également la dépense énergétique via son action sur le BAT, qui réalise la
thermogenèse. Enfin, la découverte récente de BAT physiologiquement actif chez l’homme
adulte offre de nouvelles perceptives thérapeutiques dans la lutte contre l’obésité.
Actuellement le BAT est connu pour assurer deux grandes fonctions. Tout d’abord, il
participe au maintien de la température corporelle des animaux homéothermes, et à un
phénomène appelé la TIA. Il est établi que la TIA participe à la régulation de l’homéostasie
énergétique et nécessite l’implication du système mélanocortine. En effet, la délétion du gène
codant pour la protéine MC4R chez la souris a pour conséquence de bloquer la TIA. De plus,
differentes études ont mis en évidence le rôle du MTII, un agoniste des récepteurs aux
mélanocortines dans la thermogenèse du iBAT. Enfin, il a été mis en évidence que le iBAT est
indirectement connecté à différents noyaux hypothalamiques, parmi lesquels le PVH et le MPO
présentent un nombre important de neurones marqués avec le rétrotraceur préalablement injecté
dans le iBAT et dont environ 80% de ces neurones sont colocalisés avec le MC4R, suggérant
ainsi que ces deux noyaux hypothalamiques sont impliqués dans le contrôle du iBAT via leurs
activités mélanocortiniques.
Actuellement, il est connu que l’injection de MTII dans le PVH entraine une augmentation
de la température du iBAT. De plus, de nombreux neurones du PVH expriment CB1, un
récepteur du système endocannabïnoide connu pour être impliqué dans la thermogenèse et dont
l’interaction avec le système mélanocortine participe déjà au contrôle de la prise alimentaire chez
le rat. Par ailleurs, le MPO est un relais majeur au niveau de l’aire préoptique dans le contrôle de
la thermorégulation, et l’action du MPO sur le iBAT passe par le DMH. Il apparaît donc que les
systèmes mélanocortines et endocannabinoïdes pourraient interagir dans le PVH pour contrôler
l’activité du iBAT, mais aussi que les neurones exprimant le MC4R dans le MPO sont
susceptibles de contrôler le iBAT via leurs actions sur le DMH.


71
Objectifs généraux des travaux.
1) Mettre en évidence l’interaction entre les systèmes mélanocortine et endocannabinoïde
dans le contrôle de la thermogenèse du tissu adipeux brun.
2) Montrer l’implication des récepteurs MC4R exprimés par des neurones du noyau
préoptique médial dans la thermogenèse.
Objectifs spécifiques.
1.a) Mesurer différents paramètres de la thermogenèse du iBAT, lors de l’injection par voie
i.c.v. d’un agoniste des récepteurs aux mélanocortines et la modulation de cette réponse,
lors de l’injection par voie i.c.v. d’un agoniste ou d’un antagoniste des récepteurs aux
endocannabïnoides.
1.b) Montrer que les effets observés sur la thermogenèse du iBAT lors de l’injection par voie
i.c.v. d’un agoniste des récepteurs aux mélanocortines, sont imputable à l’activation de
MC4R.
1.c) Déterminer si la modulation de l’action du système mélanocortine sur la thermogenèse du
iBAT par le système endocannabïnoide passe par le récepteur CB1.
1.d) Mettre en évidence que l’interaction entre les systèmes mélanocortine et
endocannabïnoide dans le contrôle de la thermogenèse du iBAT a lieu dans les neurones
du PVH exprimant MC4R et CB1.
2.a) Déterminer le rôle des neurones du MPO exprimant MC4R dans le contrôle de la
thermogenèse du iBAT.
2.b) Évaluer le rôle des neurones du DMH répondant au glutamate dans la transmission de
l’activation MC4R du MPO sur la thermogenèse du iBAT.


73
Chapitre 1


75
The cannabinoid receptor 1 is involved in thermogenesis induced by the brain stimulation of the melanocortin-4 receptor in male rats.
Boris Monge-Roffarello*, Sebastien M. Labbé*, Marie-Claude Roy, Marie-Laurence
Lemay, Estelle Coneggo, Damien Lanfray et Denis Richard
Centre de Recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de
Québec, Université Laval, Québec, Canada.
Article soumis à Endocrinology


77
Résumé
Cette étude a été conçue dans le but d’investiguer l’implication du récepteur aux
cannabinoïdes de type 1 (CB1) sur la stimulation de la dépense énergétique et de la thermogenèse
du tissu adipeux brun (BAT) induite par un agoniste du récepteur aux mélanocortines 4 (MC4R).
Dans la première série d’expérimentation, la dépense énergétique et la thermogenèse du BAT
sont évaluées chez des rats injectés dans le 3éme ventricule avec un agoniste MC4R, le Melanotan-
II (MTII) et un agoniste CB1, le delta-9-tetrahydrocannabinol (Δ9-THC) ou un antagoniste CB1,
l’AM251. La dépense énergétique a été mesurée par calorimétrie indirecte, alors que la
thermogenèse du BAT a été mesurée par l’enregistrement de la température du BAT
interscapulaire (iBAT). L’effet du MTII sur la dépense énergétique et la thermogenèse du iBAT
est inhibé par l’administration de Δ9-THC alors qu’il a tendance à être potentialisé par l’injection
d’AM251. Le Δ9-THC bloque aussi les effets du MTII sur le captage de C14-bromopalmitate et de
H3-désoxyglucose par le iBAT. En plus, le Δ9-THC attenue les effets du MTII sur l’expression de
l’ARNm du peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α), de
la désiodinase iodothyronine 2 (Dio2), la carnitine palmitoyltransférase 1B (Cpt1b) et la protéine
découplante 1 (Ucp1). Dans la seconde série d’expérimentation, nous avons traité de
l’implication du noyau paraventriculaire de l’hypothalamus (PVH) dans les effets du CB1 sur
l’activation de la thermogenèse du iBAT induite par le MTII, lesquels ont été évalués par
l’injection de MTII dans le PVH et de Δ9-THC dans le 4éme ventricule du cerveau. Nous avons
confirmé la présence de l’ARNm de CB1 dans les neurones MC4R du PVH et démontré la
capacité du Δ9-THC à bloquer l’augmentation de la température du iBAT induite par le MTII. Le
Δ9-THC bloque aussi l’effet du MTII dans le PVH sur le captage de C14-bromopalmitate aussi
bien que sur l’expression de Pgc1α et de Dio2 dans le iBAT. Tout compte fait, ces résultats
démontrent l’implication du CB1 dans la stimulation centrale de la thermogenèse induite par le
MTII et mettent l’emphase sur le rôle potentiel du PVH dans cette stimulation.


79
Abstract
The present study was designed to investigate the involvement of the cannabinoid receptor
1 (CB1) in the stimulating effects of melanocortin-4 receptor (MC4R) agonism on whole body
and brown adipose tissue (BAT) thermogenesis. In a first series of experiments, whole body and
BAT thermogeneses were assessed in rats infused in the third ventricle of the brain with the
MC4R agonist melanotan II (MTII) and the CB1 agonist delta-9-tetrahydrocannabinol (Δ9-THC)
or the CB1 antagonist AM251. Whole body thermogenesis was measured by indirect calorimetry
and BAT thermogenesis from interscapular BAT (iBAT) temperature. Δ9-THC blunted the
effects of MTII on whole body and iBAT thermogenesis, whereas AM251 tended to potentiate
the MTII effects. Δ9-THC also blocked the stimulating effect of MTII on 14C-bromopalmitate and 3H-deoxyglucose uptakes in iBAT. Additionally, Δ9-THC attenuated the effect of MTII on
peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α), type II
iodothyronine deiodinase (Dio2), carnitine palmitoyltransferase 1B (Cpt1b) and uncoupling
protein 1 (Ucp1). In a second series of experiments, we addressed the involvement of the
paraventricular hypothalamic nucleus (PVH) in the CB1-mediated effects of MTII on iBAT
thermogenesis, which was assessed by injecting MTII in the PVH and Δ9-THC in the fourth
ventricle of the brain. We confirmed the presence of CB1 mRNA in PVH MC4R neurons and
demonstrated the ability of Δ9-THC to blunt MTII-induced increase in iBAT temperature. Δ9-
THC also blocked the PVH effect of MTII on 14C-bromopalmitate uptake as well as on Pgc1α
and Dio2 expression in iBAT. Altogether the results demonstrate the involvement of the CB1
receptor in the brain stimulation of thermogenesis by MTII and emphasize the potential
involvement of the PVH in such stimulation.


81
Introduction
The melanocortin system plays a key role in energy homeostasis (1,2) and there is strong
evidence that this system is involved not only in energy intake but also in energy expenditure
(1,3-6). Consistently, activation of the brain melanocortin-4 receptor (MC4R) with melanotan II
(MTII), a peptide analogue of alpha-melanocyte stimulating hormone (α-MSH), decreases food
intake (7) and body weight (8). Moreover, loss-of-function mutations of proopiomelanocortin
(POMC) (9,10) and the MC4R (11-13) lead to massive obesity in laboratory animals as well as in
humans, providing evidence for the catabolic role of the stimulated brain melanocortin system.
MC4R agonists exert their catabolic effects not only by reducing food intake but also by
inducing energy expenditure likely through brown adipose tissue (BAT) thermogenesis (14-16).
The role of the melanocortin system in the control of BAT thermogenesis is further supported
anatomically. In that regard, we (14) and others (17) have identified numerous brain populations
of MC4R mRNA-expressing neurons that are (poly)synaptically connected to interscapular BAT
(iBAT) (14,17). The brown adipocyte represents a thermogenesis effector, whose role in energy
homeostasis has been postulated (18-20). The interest for the study of BAT thermogenesis has
recently been rejuvenated by the demonstration of active BAT in adult humans (21-23).
The mechanisms whereby the melanocortin system influences energy homeostasis remain
to be fully delineated. Recently, the suggestion was made that the endocannabinoid system could
modulate MC4R-mediated catabolic activity (24-26). Activation of the endocannabinoid system
is anabolic in that it stimulates food intake while reducing energy expenditure (24,27-30). Recent
experiments (25) demonstrated that the orexigenic effect of Δ9-tetrahydrocannabinol (Δ9-THC)
was blocked by the concomitant administration of α-MSH and that rimonabant, a cannabinoid
receptor 1 (CB1) antagonist, impaired the hyperphagia led to by the MC4R antagonist JKC-363.
The neuronal circuit behind the metabolic interaction of the melanocortin and endocannabinoid
systems remains to be determined. CB1 is known to exert its action at the presynaptic levels (31)
and it could involve the PVH, where both the CB1 and MC4R are expressed (24). Furthermore,
the PVH hosts neurons projecting to the intermediolateral column (IML) (32) and a large
proportion of these neurons, which likely control the SNS activity in BAT, express the MC4R
(14).

82
The aim of the present study was to further investigate the interaction of the
endocannabinoid and melanocortin systems on energy expenditure and whole-body and iBAT
thermogenesis in rats. Two series of experiments were carried out (i) to test the ability of
intracerebroventricular (icv) infusions of Δ9-THC (CB1 agonist) and AM251 (CB1 antagonist) to
modulate the stimulating effect of the MC4R agonist MTII on whole body and iBAT
thermogenesis and (ii) to assess the implication of the PVH as a potential site of the CB1-
mediated effect of MTII on iBAT thermogenesis.

83
Research Design and Methods
Animals
Male Wistar rats (Charles River Laboratories, St. Constant, QC, Canada), initially
weighing ~300 g, were individually housed, one week before the surgery, at 23 ± 1°C under a
12:12-h light-dark cycle (light on at 06:00). The rats had ad libitum access to water and pelleted
standard chow (Charles River Rodent Diet no. 5075). All animal treatments and procedures were
approved by Université Laval Animal Ethics Committee and in agreement with the Canadian
Guide for the Care and Use of Laboratory Animals.
Drugs
Δ9-THC (Tocris Bioscience, Bristol, UK) was first mixed in ethanol containing a few
drops of Tween 80. The suspension was stirred continuously under a steady stream of nitrogen
gas until all ethanol was evaporated. Artificial cerebrospinal fluid (aCSF) was then added and the
solution was stirred until the Tween 80 / Δ9-THC suspension was well dispersed. The final
vehicle solution contained 15 µl Tween 80 / 2 ml aCSF. Δ9-THC was administered at doses of 0
(vehicle only) or 3 µg in a volume of 3 µl. The CB1 antagonist AM251 (Tocris Bioscience,
Bristol, UK) was mixed with ethanol solution and was administered at doses of 0 or 3 µg in a
volume of 3 µl. The MC4R agonist MTII (Phoenix Pharmaceutical Inc, Burlingame CA, USA)
was dissolved in aCSF and administered at doses of 0, 30 or 300 ng in a volume of 3 µl in the
third ventricle or at doses of 0 or 100 ng in a volume of 200 nl (100 nl in each side of the PVH).
The MC4R antagonist HS024 (Tocris Bioscience, Bristol, UK) was dissolved in aCSF and
administered at doses of 0 or 100 ng in a volume of 3 µl. All doses and volumes of MTII and Δ9-
THC are in accordance with previous studies (25,33). Doses of HS024 and AM251 are in
accordance with previous studies, and volumes are in accordance with Δ9-THC volume.
Brain icv cannulation and drug infusions
Rats were chronically implanted with guide-cannulas (Plastics One, Roanoke, VA, USA),
aimed at the third ventricle (22-gauge), PVH (26-gauge bilateral guide-cannula) or caudal fourth

84
ventricle (22-gauge). The stereotaxic coordinates were as follows: third ventricle, 0.8 mm
posterior to bregma on the midline and 5.0 mm ventral to the brain surface; PVH, 1.8 mm
posterior to bregma on the midline with 1.0 mm center to center bilateral cannula and 7.0 mm
ventral to the brain surface; the caudal fourth ventricle, 2.5 mm posterior to interaural on the
midline and 6.0 mm ventral to the brain surface (34). The guide-cannulas were secured with
screws and cranioplastic cement (Lang Dental Manufacturing co., Inc, Wheeling, IL, USA). To
prevent clogging, sterile obturators were inserted into the guide-cannulas. The viability of the icv
cannulations was tested by evaluating the dipsogenic response to angiotensin II (Sigma-Aldrich;
50 ng). Only rats drinking more than 10 ml in a 30 minute period were included in the
experiments. The placement of the cannulas was further confirmed by the observation of the
infusion site on the brain slices at the end of the study.
Prior to every brain infusion, rats were familiarized with the testing procedures for at least
three days (only for free moving rats experiments). While animals were gently handled an
infusion cannula [26 gauge (third and fourth ventricle) or bilateral 30-gauge (PVH)] was inserted
into the guide-cannula. Brain infusions were carried out using a 50 µl Hamilton syringe
connected to a syringe pump (Harvard Apparatus 55-2226). In each protocol, the solutions were
carefully infused during 1 minute (3 µl/min) or 30 sec (200 nl/min) for PVH infusion.
Effects of third ventricle infusions of MTII, ΔΔ9-THC and AM251 on thermogenesis
Whole body thermogenesis
Twelve (12) groups of rats were used to test the effects of Δ9-THC and AM251 on the
stimulating effect of MTII on whole body thermogenesis, which was assessed by indirect
calorimetry. Figure 1A illustrates the experimental schedule for whole body thermogenesis
measurements. Indirect calorimetry was used as previously described (35,36). Rats were placed
in metabolic cages 48-h prior to monitoring oxygen consumption (VO2) and carbon dioxide
production (VCO2). This enabled animals to become accustomed to their new surroundings
before drug infusion. Measurements were made continuously using an open-circuit system. Air
was circulated through the metabolic cages at the rate of 1800-ml/min. VO2 and VCO2 were
measured using gas analyzers (model S-3A, Applied Electrochemistry Incorporated, Sunnyvale,

85
CA, USA). The animals were fasted for 8 hours, to completely blunt diet-induced thermogenesis,
before the infusion of MTII, which was preceded by the infusion of Δ9-THC or AM251. MTII
was infused at either 0, 30 or 300 ng per rat.
IBAT thermogenesis and metabolic activity
Eight (8) groups of rats were used to assess the effects of HS024, Δ9-THC and AM251 on
the stimulating effect of MTII on iBAT thermogenesis, which was assessed by measuring iBAT
temperature in isoflurane-anesthetized animals. iBAT temperature was monitored using a copper-
constantan thermocouple (Physitemp Instruments, Inc. Clifton, NJ, USA) inserted into the intact,
right iBAT pad. During the measurement, core temperature was monitored with a second
thermocouple inserted 6 cm into the rectum (37). Skin temperature was also assessed with a third
copper-constantan thermocouple being placed on the left-rear leg. The last two measurements
were used to control the animal state before and during the beginning of the experiment. The first
injection was done when the core body temperature was between 37.0 and 37.5°C. Measurement
of CO2 production was performed during the acute treatment and was used as a surrogate of VO2
to assess whole body thermogenesis. For technical reasons inherent to the use of the isoflurane
mask, VO2 could not be measured.
In addition to measuring iBAT temperatures we also measured iBAT 14C-bromopalmitate
([1-14C]-2-bromopalmitic acid) and 3H-deoxyglucose ([1,2-3H(N)]-Deoxy-D-glucose) uptakes to
further validate changes in iBAT thermogenic/metabolic activity. Both tracers (Moravek
Biochemicals, Inc. Brea, CA, USA) were dissolved in normal saline supplemented with 4%
bovine serum albumin and administered at 20-µCi doses. 14C-bromopalmitate is routinely used to
determine fatty acid uptake (38,39), oxidation (39-41) and esterification (39) and 3H-
deoxyglucose for the determination of glucose uptake (42). Figure 1B shows the timeline for the
determination of 14C-bromopalmitate and 3H-deoxyglucose uptakes in iBAT depots after icv
infusions. Thirty minutes after the third ventricle infusion of MTII, an intravenous bolus of 20
µCi of each tracer was injected in the tail vein, in a volume of 350 ul. Arterial blood sampling
from the carotid was done at 31, 32, 33, 35, 40, 50 and 60 minutes post MTII infusion for
clearance determination according to a procedure previously described (38). Sixty minutes after
MTII infusion, animals were euthanized by an overdose of anaesthetic and iBAT, liver, heart and

86
skeletal muscle (gastrocnemius) were removed on ice, weighed and stored at -80°C pending
future analyses. Tissue-specific uptakes of 14C-bromopalmitate and 3H-deoxyglucose were
determined on samples of tissues (~250 mg), which were homogenized (Polytron PT10-35,
Kinematica, inc. Bohemia, NY, USA) in ice-cold Chappell-Perry buffer (0.075 M sucrose, 0.225
M sorbitol, 1 mM EGTA, 0.1% fatty-acid-free BSA, and 10 mM Tris–HCl, pH 7.4). An aliquot
was kept to determine total [14C and 3H] activity in the tissue as previously described (39).
Fractional uptakes of 14C-bromopalmitate and 3H-deoxyglucose were determined by measuring
[14C] and [3H] activity in tissue extracts using a scintillation counter (liquid scintillation analyzer
Tri-Carb 2900TR, PerkinElmer, Montreal, QC, Canada). They were expressed as the percentage
of injected dose (%ID) of [14C] and [3H] recovered per gram of tissues.
IBAT gene expression
Eight (8) groups of rats were used to assess the effects of MTII on iBAT gene expression
in the presence or absence of HS024, Δ9-THC or AM251. Rats were killed either 60 minutes after
MTII and the CB1 ligands or 24 hours after drug treatments, which were administered 3 times at
12-hour intervals.
The expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha
(Pgc1α), Type II iodothyronine deiodinase (Dio2), carnitine palmitoyltransferase 1B (Cpt1b),
uncoupling protein 1 (Ucp1) was assessed as previously described (43). Briefly, RNA was
isolated from the iBAT depot with QIAzol and the RNeasy Lipid Tissue Kit (QIAGEN,
Mississauga, ON, Canada). For cDNA synthesis, expand reverse transcriptase (Invitrogen,
Burlington, ON, Canada) was used following manufacturer's instructions, and cDNA was diluted
in DNase-free water (1:25) before quantification by real-time PCR. mRNA transcript levels were
measured in duplicate samples using a Rotor Gene 3000 system (Montreal Biotech, Montreal,
QC, Canada). The primers used for the PCR reactions are shown in Table 1. At the end of each
run, melt curve analyses were performed, and a few samples representative of each experimental
group were run on agarose gel to ensure the specificity of the amplification. Results were
expressed as the ratio of the expression of the target gene to that of the housekeeping gene Arbp,
which was selected as no significant variation in its expression was observed after treatments.

87
Involvement of the PVH in the CB1-mediated effects of MTII on thermogenesis
MC4R colocalisation with CB1 and vesicular glutamate transporter 2 (VGLUT2) mRNA
in the PVH
Untreated rats were used to determine whether MC4R neurons expressed CB1 mRNA.
We hypothesized that MCR4-containing neurons were stimulatory and therefore verified if they
were glutamatergic by colocalizing MCR4 and VGLUT2.
Colocalisations were performed by histochemistry as performed previously (44). Rats
were anesthetized with 1.5 ml mixture containing 20 mg/ml ketamine and 2.5 mg/ml xylazine.
Once the animals showed no more reflexes, they were intracardially perfused with 50 ml of ice-
cold isotonic saline followed by 500 ml of paraformaldehyde (4%) solution. Brains were
removed and kept in paraformaldehyde for an additional period of 7 days. They were then
transferred overnight to a solution containing paraformaldehyde (4%) and sucrose (10%) and
sliced using a sliding microtome (Histoslide 2000, Reichert-Jung, Heidelberg, Germany). Thirty-
micrometer thick sections of the PVH were collected and stored at −30°C in a cold
cryoprotecting solution containing sodium phosphate buffer (50-mM), ethylene glycol (30%),
and glycerol (20%).
In situ hybridization histochemistry was used to reveal CB1 mRNA and VGLUT2 mRNA
on the brain sections taken from the whole brain sections in the antero-posterior level around the
hypothalamus. The protocol used was largely adapted from the technique described by Simmons
et al. (45).
Briefly, one out of every six brains sections were mounted onto poly-L-lysine coated slides.
The sections were then successively fixed in paraformaldehyde (4%), digested with proteinase K
(10-µg/ml in 100-mM Tris–HCl containing 50-mM EDTA (pH 8.0)), acetylated with acetic
anhydride (0.25% in 0.1 M triethanolamine (pH 8.0)), and dehydrated through graded
concentrations (50, 70, 95, and 100%) of alcohol. Thereafter, 100 µl hybridization mixture, which
contains an antisense 35S-labeled cRNA probe, were spotted on each slide. The slides were sealed
with coverslips and incubated overnight at 70°C in a slide warmer. The next day, the coverslips

88
were removed and the slides were rinsed four times with SSC (0.6 M NaCl, 60 mM trisodium
citrate buffer (pH 7.0)), digested with RNAse A (20 µg/ml in 10 mM Tris–500-mM NaCl
containing 1 mM EDTA), washed in descending concentrations of SSC (2×, 10 min; 1×, 5 min;
0.5×, 5 min; 0.1×, 30 min at 60°C) and dehydrated through graded concentrations of alcohol. An
X-ray film (Kodak) was exposed to the slides. Once removed from the autoradiography cassettes,
the slides were defatted in toluene, dipped in NTB2 nuclear emulsion (Eastman Kodak), and
exposed for 7 days, before being developed in D 19 developer and fixed in rapid fixer (Kodak).
The cRNA probes (Table 2) were generated from cDNA fragments subcloned into pGem-
T plasmid (Promega, Madison, WI, USA), and linearized with restriction enzymes (New England
Biolabs, Whitby, ON, Canada) and transcribed in vitro using RNA polymerases (Stratagene, La
Jolla, CA) in the presence of 35S-labeled UTP (PerkinElmer, Boston, MA) to generate sense and
antisense probes. To measure the specificity of each probe, a confirmation by the absence of
positive signal in sections hybridized with the sense probe was done.
MC4R was immunostained (46-48) after the in situ hydridization. Briefly, brain slices
were incubated overnight at 4°C with the MC4R antibody (1/400) (#ab24233; Abcam Inc.,
Toronto, ON, Canada) in KPBS (50-mM) with Triton X-100 (0.3%) goat serum (1%) and BSA
(5%). Following incubation at 4°C with the first antibody, the brain slices were rinsed in sterile
KPBS and incubated with the same previous mixture of KPBS in which we added a biotinylated
goat anti-rabbit second antibody (1/150) (Vector Laboratories Inc., Burlington, ON, Canada) for
2 hours. Sections were then rinsed with KPBS and incubated at room temperature for 60 min
with an avidin–biotin–peroxidase complex (Vectasain ABC Elite Kit; Vector labs, Burlingame,
CA). After several rinses in sterile KPBS, the brain slices were allowed to react in a mixture
containing sterile KPBS, the chromagen 3,3′ -diaminbenzidine tetrahydrochloride (DAB,
0.05%) and 0.3% hydrogen peroxide. Thereafter, tissues were rinsed in sterile KPBS and covered
with a coverslip.
All slides were examined using an Olympus BX61 microscope (Olympus Canada,
Richmond Hill, ON, Canada). Images were acquired with a DVC-2000C digital camera (DVC
Company Inc., Austin, TX, USA).

89
BAT thermogenesis
Four (4) groups of rats were used to determine BAT thermogenesis and metabolic
activity. Anesthetized rats were infused in the PVH (bilaterally) with MTII at the dose of 50-ng
administered in each side and with Δ9-THC at the dose of 3-µg in the fourth ventricle. The doses
were selected based on the observations made in the previous series of experiments. In this series
of experiments, we did not use AM251 but rather focused on Δ9-THC. BAT thermogenesis and
metabolic activity were measured as described above. We also examined the expression of
Pgc1α, Dio2, Cpt1b and Ucp1 in rats killed 60 minutes after the administration of the drugs.
Statistical analyses
Values obtained from rats with improper placement of cannula were excluded from the
statistical analyses. Results are expressed as means ± SEM. Comparisons were done on normally
distributed data using Mann-Whitney or two-way ANOVAs followed by Bonferroni post hoc
tests (when appropriate) to assess the differences between various conditions with Graph pad
Prism Software version 6.0 for Mac (San Diego, CA, USA). Differences were considered
statistically significant when P values were less than 0.05.


91
Results
Effects of third ventricle infusions of MTII, ΔΔ9-THC and AM251 on thermogenesis.
Figure 2 illustrates the effects of third ventricle infusions of Δ9-THC and AM251 on
MTII-induced whole body thermogenesis. Acute icv infusion of MTII dose-dependently
increased VO2 and energy expenditure. Neither Δ9-THC nor AM251, given alone, influenced
those variables. However, Δ9-THC blunted the stimulating effects of MTII (Fig. 2, A-D); rats
treated with MTII and Δ9-THC had VO2 and energy expenditure values lower than those of
animals treated with MTII alone. On the other hand, AM251 tended to potentiate the MTII effects
on VO2 and energy expenditure (Fig. 2, E-H) since rats treated with 30 ng of MTII and AM251
had higher VO2 and energy expenditure than rats treated with MTII. The latter effects
disappeared at the MTII dose of 300 ng, likely because that dose led to a near maximal effect.
None of the treatments used altered the respiratory quotient (Fig. 2, C and G).
The effects of third ventricle infusions of Δ9-THC and AM251 on MTII-induced iBAT
thermogenesis, whole body CO2 production and 14C-bromopalmitate and 3H-deoxyglucose
uptakes are shown in Figure 3. Δ9-THC and AM251 blunted and potentiated, respectively, the
effects of MTII on iBAT temperature (Fig. 3, A-B). Δ9-THC also blocked the stimulating effects
of MTII on whole body CO2 production and 14C-bromopalmitate and 3H-deoxyglucose uptakes
whereas AM251 was without effect on those variables (Fig. 3, C-F). There were significant
correlations between iBAT temperature and VCO2 and between iBAT temperature and 14C-
bromopalmitate uptake (Fig. 3, G-H).
Figure 4 demonstrates the effects of MTII, Δ9-THC and AM251 on the expression of
mRNAs coding for proteins involved in BAT thermogenesis. We looked at the expression of
Pgc1α, Dio2, Cpt1b and Ucp1 in rats euthanized one hour after the infusion of the drugs (Fig. 4,
A-B). Δ9-THC blocked the stimulating effect of MTII on Pgc1α and Dio2 (Fig. 4A) whereas
AM251 was without effect (Fig. 4B). Δ9-THC and AM251 alone did not affect the expression of
the genes. MTII did not change the expression of Cpt1b and Ucp1 when rats were killed 60
minutes after infusion. However, significant effects of MTII were observable after 3 infusions of
the drugs given at 12-hour intervals over a period of 24 hours. In that context, Δ9-THC again

92
attenuated the inducing effect of MTII on Cpt1b and Ucp1 (Fig. 4C) whereas AM251 was
without effect (Fig. 4D).
To conclude the series of experiments on third ventricle infusions, we confirmed the role
of the MC4R and CB1 on the respective effects of MTII and Δ9-THC on iBAT thermogenesis.
The selective MC4R antagonist HS024 blocked the stimulating effects of MTII on iBAT
thermogenesis (Fig. 5, A-B), whole body CO2 production (Fig. 5, C-D), expression of Pgc1α and
Dio2 (Fig. 5E) and 14C-bromopalmitate and 3H-deoxyglucose uptakes (Fig. 5F). On the other
hand, AM251 prevented the inhibitory effects of Δ9-THC on MTII-induced BAT thermogenesis
(Fig. 5, G-H).
Involvement of the PVH in the CB1-mediated effects of MTII on thermogenesis.
To test the hypothesis that PVH was involved in the CB1-mediated effects of MTII on
thermogenesis, we conducted experiments to first demonstrate the neuronal colocalisation of
MC4R with CB1 mRNA in the PVH and then to verify whether Δ9-THC could prevent the BAT
thermogenic response induced by the PVH infusion of MTII.
Figure 6 illustrates the co-localization of the MC4R with CB1 mRNA (Fig. 6, A-B). The
MC4R also appeared to be present in glutamate stimulatory neurons as it co-localized with
VGLUT2 mRNA (Fig. 6, C-D). When infused in the PVH, MTII activated iBAT and whole body
thermogenesis and those effects were blocked by Δ9-THC when injected into the fourth ventricle
(Fig. 7, A-D). We injected Δ9-THC into the fourth ventricle to irrigate the brainstem and the
intermediolateral column (IML), possible sites of CB1 projections. It is noteworthy that the rise
in iBAT temperature following PVH infusion of MTII was higher than that observed following
the third ventricle infusion. Δ9-THC also blocked MTII-induced Pgc1α and Dio2 expression in
iBAT (Fig. 7E). Finally, MTII raised iBAT uptake of 14C-bromopalmitate and this effect was
blocked with Δ9-THC (Fig. 7F). None of the treatments altered the uptake of 3H-deoxyglucose
and the expression of Cpt1b and Ucp1 in rats killed 60 minutes after infusion (Fig. 7G-H).

93
Discussion
The present results confirm the modulating role of the CB1 on the stimulating effects of
MC4R agonism on thermogenesis. Icv administration of Δ9-THC and AM251 blunted and
potentiated, respectively, the stimulation by MTII of whole body and BAT thermogenesis. Δ9-
THC blunted iBAT expression of genes involved in thermogenesis and BAT uptakes of the
glucose and NEFA tracers 3H-deoxyglucose and 14C-bromopalmitate respectively. The present
results also demonstrate the implication of the PVH in the CB1-modulated effects of the MC4R
stimulation of iBAT thermogenesis and metabolic activity. Indeed, icv administration of Δ9-THC
blocked the stimulating effect of intra-PVH administration of MTII on BAT thermogenesis,
iBAT expression of genes involved in thermogenesis and uptake of 14C-bromopalmitate. We also
confirmed the presence of CB1 mRNA in the PVH MC4R neurons and demonstrated that the
latter were in large part glutamatergic neurons (expressing VGLUT2), therefore, stimulatory
neurons.
The present study further emphasizes the role of MC4R agonism in stimulating
thermogenesis. We confirmed that the ability of MTII to stimulate thermogenesis (49) could be
blocked by the specific MC4R antagonist HS024. MTII not only elevated whole body
thermogenesis and iBAT temperature but also the uptake 3H-deoxyglucose and 14C-
bromopalmitate, which are used as markers of glucose (42) and NEFA uptakes (38,39). The
importance of the MC4R in thermogenesis has been claimed before from investigations carried
out in MC4R KO mice (50) and is supported anatomically by retrograde transneuronal viral tract
tracing studies, which have demonstrated, in mice (17) and hamsters (14), the link between the
brain MCR4-expressing neurons and iBAT. In fact, in regions such as the PVH, preoptic area and
raphe, it has been estimated that more than 80% of MC4R neurons govern the SNS outflow to
iBAT (14).
This study emphasizes the role played by the CB1 in modulating the stimulating effect of
MC4R agonism on thermogenesis. The CB1 agonist Δ9-THC blocked the stimulating effects of
the MC4R agonist MTII on whole body VO2 and VCO2, iBAT temperature, iBAT uptakes of
glucose and NEFA and iBAT expression of genes involved in thermogenesis. Consistently, the
CB1 antagonist AM251 proved capable of potentiating the stimulating effects of MTII at the dose

94
of 30 ng. This effect was not seen when the 300 ng dose of MTII was used as that dose possibly
produced a ceiling effect on most variables studied. The modulating effect of CB1 ligands on the
stimulating effect of MC4R agonism on whole body thermogenesis had not been extensively
investigated but was predictable from the work of Verty et al (25), who demonstrated that CB1
signaling could prevent the melanocortin system from acting on food intake. The melanocortin
and endocannabinoid systems are known to exert opposite actions on energy balance due to
effects exerted on both food intake and energy expenditure (24,51). In this study, Δ9-THC and
AM251 given alone did not affect thermogenesis at the doses that were selected and acutely
administered. There is, however, evidence that subchronic administration of rimonabant (52) or
Δ9-THC (53) can enhance or prevent thermogenesis, respectively.
Our results demonstrate the role of the PVH as a potential site for the CB1-modulated
thermogenic effect of MTII. The PVH plays a major role in energy balance and has been
demonstrated to be a site for the melanocortin action on energy homeostasis (14,54). MTII
injection in the PVH has been reported to increase iBAT thermogenesis (14). Additionally, the
PVH has been reported to host a large number of MC4R-expressing neurons, which are
connected to BAT (14) and which express the CB1 (Fig. 6, A-B). In this study, we were able to
block the thermogenic action of the PVH infusion of MTII with Δ9-THC. We infused Δ9-THC in
the fourth ventricle as we expected the action of the drug on presynaptic CB1 synthesized in PVH
neurons projecting down in the IML. The CB1 is in fact known to exert its action at the
presynaptic levels (31). The CB1 would control the activity of PVH MC4R neurons harbouring
CB1 receptors on their terminals and projecting to the spinal cord. Those neurons could be
stimulatory as suggested by the neuronal colocalization in the PVH of MC4R and VGLUT2. This
circuitry however needs to be fully delineated. Similar to the CB1 modulation of the MC4R-
mediated hypophagic effect (25), that of MC4R-mediated thermogenic effect appears located
downstream from the melanocortin receptors.
In conclusion, the present results confirm the thermogenic role of the melanocortin
system. They emphasize the involvement of the CB1 in modulating the brain stimulation of
thermogenesis by MC4R agonism and they demonstrate a potential involvement of the PVH in
that stimulation.

95
Acknowledgements
This work was supported by a grant from the Canadian Institutes of Health Research
(CIHR — MOP-119436) to D.R. and was performed at the Quebec Heart and Lung Institute
Research Center, which is funded by the Fonds de la recherche du Québec — Santé (FRQ-S). B.
Monge-Roffarello holds a fellowship from the CIHR Training Program in Obesity/Healthy Body
Weight Research. SM Labbé is the recipient of a CIHR postdoctoral fellowship. We are grateful
to Julie Plamondon and Pierre Samson for their helpful laboratory assistance.


97
References
1. Butler AA 2006 The melanocortin system and energy balance. Peptides 27:281–290
2. Pandit R, de Jong JW, Vanderschuren LJMJ, Adan RAH 2011 Neurobiology of overeating and obesity: The role of melanocortins and beyond. Eur. J. Pharmacol. 660:28–42
3. Xu Y, Elmquist JK, Fukuda M 2011 Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann. N. Y. Acad. Sci. 1243:1–14
4. Mountjoy KG 2010 Distribution and function of melanocortin receptors within the brain. Advances in experimental medicine and biology [Internet] 681:29–48
5. Ellacott KLJ, Cone RD 2006 The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 361:1265–1274
6. Garfield AS, Lam DD, Marston OJ, Przydzial MJ, Heisler LK 2009 Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol. Metab. 20:203–215
7. Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD 1997 Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385:165–168
8. Thiele TE, van Dijk G, Yagaloff KA, Fisher SL, Schwartz M, Burn P, et al. 1998 Central infusion of melanocortin agonist MTII in rats: assessment of c-Fos expression and taste aversion. Am. J. Physiol. [Internet] 274:R248–R254
9. Yaswen L, Diehl N, Brennan MB, Hochgeschwender U 1999 Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med. [Internet] 5:1066–1070
10. Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A 1998 Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. [Internet] 19:155–157
11. Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, et al. 1997 Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141
12. Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S 1998 A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet. [Internet] 20:111–112
13. Vaisse C, Clement K, Guy-Grand B, Froguel P 1998 A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 20:113–114

98
14. Song CK, Vaughan CH, Keen-Rhinehart E, Harris RBS, Richard D, Bartness TJ 2008 Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown adipose tissue: neuroanatomical and functional evidence. AJP: Regulatory, Integrative and Comparative Physiology 295:R417–R428
15. Brito MN, Brito NA, Baro DJ, Song CK, Bartness TJ 2007 Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology 148:5339–5347
16. Ste Marie L, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD 2000 A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. U.S.A. 97:12339–12344
17. Voss-Andreae A, Murphy JG, Ellacott KLJ, Stuart RC, Nillni EA, Cone RD, et al. 2006 Role of the Central Melanocortin Circuitry in Adaptive Thermogenesis of Brown Adipose Tissue. Endocrinology 148:1550–1560
18. Rothwell NJ, Stock MJ 1997 A role for brown adipose tissue in diet-induced thermogenesis. Obes. Res. 5:650–656
19. Cannon B, Nedergaard J 2004 Brown adipose tissue: function and physiological significance. Physiol. Rev. 84:277–359
20. Richard D, Monge-Roffarello B, Chechi K, Labbé SM, Turcotte EE 2012 Control and physiological determinants of sympathetically mediated brown adipose tissue thermogenesis. Front Endocrinol (Lausanne) 3:36
21. Ouellet V, Labbé SM, Blondin DP, Phoenix S, Guerin B, Haman F, et al. 2012 Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Invest. 122:545–552
22. Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, et al. 2013 Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 19:635–639
23. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JMAFL, Kemerink GJ, Bouvy ND, et al. 2009 Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360:1500–1508
24. Richard D, Guesdon B, Timofeeva E 2009 The brain endocannabinoid system in the regulation of energy balance. Best Pract. Res. Clin. Endocrinol. Metab. 23:17–32
25. Verty ANA, McFarlane JR, McGregor IS, Mallet PE 2004 Evidence for an interaction between CB1 cannabinoid and melanocortin MCR-4 receptors in regulating food intake. Endocrinology 145:3224–3231
26. Sinnayah P, Jobst EE, Rathner JA, Caldera-Siu AD, Tonelli-Lemos L, Eusterbrock AJ, et al. 2008 Feeding induced by cannabinoids is mediated independently of the

99
melanocortin system. PLoS ONE Public Library of Science; 3:e2202
27. Kunos G, Osei-Hyiaman D, Liu J, Godlewski G, Bátkai S 2008 Endocannabinoids and the control of energy homeostasis. J. Biol. Chem. 283:33021–33025
28. Cota D 2008 Role of the Endocannabinoid System in Energy Balance Regulation and Obesity. Obesity and Metabolism [Internet] Basel: Karger Publishers; 36:135–145
29. Woods SC 2007 The Endocannabinoid System: Mechanisms Behind Metabolic Homeostasis and Imbalance. The American Journal of Medicine 120:S9–S17
30. Matias I, Di Marzo V 2007 Endocannabinoids and the control of energy balance. Trends in Endocrinology & Metabolism 18:27–37
31. Di Marzo V, Matias I 2005 Endocannabinoid control of food intake and energy balance. Nat. Neurosci. 8:585–589
32. Pyner S, Coote JH 2000 Identification of branching paraventricular neurons of the hypothalamus that project to the rostroventrolateral medulla and spinal cord. Neuroscience [Internet] 100:549–556
33. Hwa JJ, Ghibaudi L, Gao J, Parker EM 2001 Central melanocortin system modulates energy intake and expenditure of obese and lean Zucker rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281:R444–R451
34. Paxinos G, Watson C 2006 The rat brain in stereotaxic coordinates: hard cover edition.
35. Guesdon B, Paradis E, Samson P, Richard D 2009 Effects of intracerebroventricular and intra-accumbens melanin-concentrating hormone agonism on food intake and energy expenditure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296:R469–R475
36. Sell H, Berger JP, Samson P, Castriota G, Lalonde J, Deshaies Y, et al. 2004 Peroxisome proliferator-activated receptor gamma agonism increases the capacity for sympathetically mediated thermogenesis in lean and ob/ob mice. Endocrinology 145:3925–3934
37. Gordon CJ 1990 Thermal biology of the laboratory rat. Physiol. Behav. 47:963–991
38. Ménard SL, Ci X, Frisch F, Normand-Lauzière F, Cadorette J, Ouellet R, et al. 2008 Mechanism of Reduced Myocardial Glucose Utilization During Acute Hypertriglyceridemia in Rats. Mol Imaging Biol 11:6–14
39. Menard SL, Croteau E, Sarrhini O, Gelinas R, Brassard P, Ouellet R, et al. 2010 Abnormal in vivo myocardial energy substrate uptake in diet-induced type 2 diabetic cardiomyopathy in rats. Am. J. Physiol. Endocrinol. Metab. 298:E1049–E1057
40. Oakes ND, Thalén P, Aasum E, Edgley A, Larsen T, Furler SM, et al. 2006 Cardiac metabolism in mice: tracer method developments and in vivo application revealing

100
profound metabolic inflexibility in diabetes. Am. J. Physiol. Endocrinol. Metab. 290:E870–E881
41. Ci X, Frisch F, Lavoie F, Germain P, Lecomte R, van Lier JE, et al. 2006 The effect of insulin on the intracellular distribution of 14(R,S)-[18F]Fluoro-6-thia-heptadecanoic acid in rats. Mol Imaging Biol 8:237–244
42. Ferré P, Leturque A, Burnol AF, Pénicaud L, Girard J 1985 A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaesthetized rat. Biochem. J. 228:103–110
43. Chechi K, Blanchard P-G, Mathieu P, Deshaies Y, Richard D 2013 Brown fat like gene expression in the epicardial fat depot correlates with circulating HDL-cholesterol and triglycerides in patients with coronary artery disease. Int J Cardiol 167:2264–2270
44. Timofeeva E, Picard F, Duclos M, Deshaies Y, Richard D 2002 Neuronal activation and corticotropin‐releasing hormone expression in the brain of obese (fa/fa) and lean (fa/?) Zucker rats in response to refeeding. Eur. J. Neurosci. [Internet] Blackwell Science, Ltd; 15:1013–1029
45. Simmons DM, Arriza JL, Swanson LW 1989 A Complete Protocol for In Situ Hybridization of Messenger RNAs in Brain and Other Tissues With Radio-labeled Single-Stranded RNA Probes. Maney Publishing; :169–181
46. Chitravanshi VC, Bhatt S, Sapru HN 2009 Microinjections of alpha-melanocyte stimulating hormone into the nucleus ambiguus of the rat elicit vagally mediated bradycardia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296:R1402–R1411
47. Peter J-C, Bekel A, Lecourt A-C, Zipfel G, Eftekhari P, Nesslinger M, et al. 2009 Anti-melanocortin-4 receptor autoantibodies in obesity. J. Clin. Endocrinol. Metab. 94:793–800
48. Spencer JD, Schallreuter KU 2009 Regulation of pigmentation in human epidermal melanocytes by functional high-affinity beta-melanocyte-stimulating hormone/melanocortin-4 receptor signaling. Endocrinology 150:1250–1258
49. Vaughan CH, Shrestha YB, Bartness TJ 2011 Characterization of a novel melanocortin receptor-containing node in the SNS outflow circuitry to brown adipose tissue involved in thermogenesis. Brain Res.
50. Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M, Cone RD 2001 Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat. Neurosci. 4:605–611
51. Butler AA, Cone RD 2002 The melanocortin receptors: Lessons from knockout models. Neuropeptides [Internet] 36:77–84
52. Verty ANA, Allen AM, Oldfield BJ 2009 The effects of rimonabant on brown adipose

101
tissue in rat: implications for energy expenditure. Obesity (Silver Spring) 17:254–261
53. Verty AN, Evetts MJ, Crouch GJ, McGregor IS, Stefanidis A, Oldfield BJ 2011 The Cannabinoid Receptor Agonist THC Attenuates Weight Loss in a Rodent Model of Activity-Based Anorexia. Neuropsychopharmacology 36:1349–1358
54. Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD 1999
Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular
nucleus: evidence of a cellular basis for the adipostat. Neuron 24:155–163


103
Tables
Table 1: qPCR gene information.
Gene Forward sequence Reverse sequence Annealing temperature °C
Arbp TAAAGACTGGAGACAAGGTG GTGTAGTCAGTCTCCACAGA 60
Cpt1b CGGAAGCACACCAGGCAGTA GCAGCTTCAGGGTTTGTCGGAATA 64
Dio2 ATGGGACTCCTCAGCGTAGA GCACAGGCAAAGTCAAGAAG 60
Pgc1α TCCTGTTACTATTATGAATCAAGCC AAACCATAGCTGTCTCCATCATCC 64
Ucp1 TGGTGAGTTCGACAACTTCC GTGGGCTGCCCAATGAATAC 64
Table 2: probes characterizations.
Probes Accenssion number Bp number Sequence Restriction
enzyme S/AS RNA polymerase
S/AS
CB1 NM_012784 400 294-693 Not-Nco1 T7-SP6
VGLUT2 NM_053427 829 2889-3717 Nco1-Nde1 SP6-T7


105
Figure Legends
Figure 1. Experimental protocol. A. Experimental protocol for the measurement of whole
body thermogenesis by indirect calorimetry. B. Experimental protocol for the measurement of
iBAT thermogenesis, metabolic activity and gene expression with 3rd ventricle injections. C.
Experimental protocol for the measurement of iBAT thermogenesis, metabolic activity and gene
expression with PVH and 4th ventricle injections.
Figure 2. Effects of Δ9-THC and AM251 on the MTII effects on whole body
thermogenesis. Twelve distinct groups of 7-10 rats were used to test the effects of MTII (0, 30
and 300 ng) with Δ9-THC or AM251. Drugs were infused in the third ventricle of the brain (3V).
Δ9-THC and AM251 were infused 15 minutes before MTII. The measure of oxygen
consumption: curves (A, F) and the area under the curve (AUC — from baseline) (B, G). The
AUC of energy expenditure (C, H). The AUC was measured over 180 min following the MTII
infusion. The measures of respiratory quotient: curves (D, I) and bar graphs during the last hour
(E, J). * P < 0.05 vs. Vehicle-Vehicle, # P < 0.05 vs. Vehicle-MTII (30 ng) and † P < 0.05 vs.
Vehicle-MTII (300 ng).
Figure 3. Effects of Δ9-THC and AM251 on the MTII effects on iBAT thermogenesis,
whole body thermogenesis and metabolic activity. Six distinct groups of 6-8 rats were used to test
the effect of MTII (0 or 300 ng) with Δ9-THC (3 µg) or AM251 (3 µg). Drugs were infused in the
third ventricle (3V). Δ9-THC and AM251 were infused 15 minutes before MTII. The area under
the curve (AUC — from baseline) for Δ-iBAT temperature (A, B), Δ-CO2 exhaled (indirect
assessment of whole body thermogenesis) (C, D) was measured over 60 min following the MTII
infusion. The 14C-bromopalmitate (E) and 3H-deoxyglucose (F) uptakes in iBAT (over 30
minutes prior to euthanasia) are expressed as % of the injected dose per gram of tissue. The
Pearson correlations between the AUCs of Δ-iBAT temperature and Δ-CO2, and the AUCs of Δ-
iBAT temperature and the % bromopalmitate uptake are illustrated in G and H, respectively. * P
< 0.05 vs. Vehicle-Vehicle, # P < 0.05 vs. Vehicle-MTII 300 ng.
Figure 4. Effects of Δ9-THC and AM251 on the MTII effects on iBAT gene expression. A
and B. Six distinct groups of 6-8 rats were used to test the effect of MTII (0 or 300 ng)
administered with Δ9-THC (A) or AM251 (B). The drugs were infused in the third ventricle (3V).

106
Δ9-THC and AM251 were infused 15 minutes before MTII. Pgc1α, Dio2, Cpt1b and Ucp1
mRNA expressions in iBAT were measured in rats sacrificed 1 hour after the last infusion. C and
D. Twelve distinct groups of 6-7 rats were used to test the effect of three doses of MTII (0, 30
and 300 ng) given with Δ9-THC (3 µg) (C) or AM251 (3 µg) (D). Drugs were infused in the third
ventricle (3V) over a period of 24 hours with 12 hours between each treatment. Δ9-THC and
AM251 were infused 15 minutes before MTII. Rats were killed 2 hours after the last infusion. *
P < 0.05 vs. Vehicle-Vehicle, # P < 0.05 vs Vehicle-MTII (30 ng), † P < 0.05 vs. Vehicle-MTII
(300 ng).
Figure 5. Effect of HS024 or Δ9-THC and AM251 on the MTII effects on iBAT
thermogenesis, whole body thermogenesis and metabolic activity. Ten distinct groups of 6-8 rats
were used to test the effect of MTII (0 or 300 ng) given with HS024 (1 µg) (A-F) and the effect
of MTII (0 or 300 ng) given with Δ9-THC (3 µg) and AM251 (3 µg) (G-H). Drugs were infused
in the third ventricle (3V). HS024 or Δ9-THC and AM251 were infused 15 minutes before MTII.
The area under the curve (AUC — from baseline) for Δ-iBAT temperature (B), Δ-CO2 exhaled
(D) were measured over 60 min following the last infusion. The Pgc1α and Dio2 mRNA
expression in iBAT, 1 hour after the MTII infusion is illustrated in E. The 14C-bromopalmitate
and 3H-deoxyglucose uptakes in iBAT (over 30 minutes prior to euthanasia) are expressed as %
of injected dose per gram of tissue in F. The AUC — from baseline) for iBAT temperature was
measured over 60 min following the MTII infusion (H). * P < 0.05 vs. Vehicle-Vehicle, # P <
0.05 vs. Vehicle-MTII.
Figure 6. Colocalization of MC4R with CB1 and VGLUT2 mRNA in the PVH. The
MC4R was revealed in brown by immunohistochemistry staining. CB1 mRNA (A-B) and
VGLUT2 mRNA (C-D) were revealed as clusters of silver gains following in situ hybridization.
Neurons presenting a colocalization are highlighted by red arrows.
Figure 7. Effect of Δ9-THC on the PVH MTII infusion effect on iBAT thermogenic
activity and capacity. Four distinct groups of 6-7 rats were used to test the effect of MTII (0 or
100 ng) given with Δ9-THC (3 µg). MTII was infused in the PVH, bilaterally, and Δ9-THC was
administered in caudal fourth ventricle (4V). Δ9-THC was infused 15 minutes before MTII. The
area under the curve (AUC — from baseline) for Δ-iBAT temperature (B), Δ-CO2 exhaled (D)
was measured over 60 min following the last infusion. The Pgc1α and Dio2 mRNA expression

107
(E) or Cpt1b and Ucp1 (G) mRNA expression in iBAT was measured in rats killed 1 hour after
the MTII infusion. The 14C-bromopalmitate (F) and 3H-deoxyglucose (H) uptakes in iBAT (over
30 minutes prior to euthanasia) are expressed as % of injected dose per gram of tissue. * P < 0.05
vs. Vehicle-Vehicle, # P < 0.05 vs. Vehicle-MTII.


109
Figures
Figure 1: Experimental protocol.

110
Figure 2: Effects of Δ9-THC and AM251 on the MTII effects on whole body
thermogenesis.

111
Figure 3: Effects of Δ9-THC and AM251 on the MTII effects on iBAT thermogenesis,
whole body thermogenesis and metabolic activity.

112
Figure 4: Effects of Δ9-THC and AM251 on the MTII effects on iBAT gene expression.

113
Figure 5: Effect of HS024 or Δ9-THC and AM251 on the MTII effects on iBAT
thermogenesis, whole body thermogenesis and metabolic activity.

114
Figure 6: Colocalization of MC4R with CB1 and VGLUT2 mRNA in the PVH.

115
Figure 7: Effect of Δ9-THC on the PVH MTII infusion effect on iBAT thermogenic activity
and capacity.


117
Chapitre 2


119
The medial preoptic nucleus as a site of the thermogenic and metabolic actions of
Melanotan II in male rats.
Boris Monge-Roffarello, Sebastien M. Labbe, Alexandre Caron, Damien Lanfray,
Pierre Samson, Denis Richard
Centre de Recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de
Québec, Université Laval, Québec, Canada.
Article soumis à American Journal of Physiology, Endocrinology and Metabolism


121
Résumé
Cette étude a été élaborée afin d’investiguer le rôle du noyau préoptique médial (MPO)
comme site d’action des effets thermogènes et métaboliques d’un analogue de l’α−MSH, le
Melanotan II (MTII). Nous avons aussi évalué l’implication du noyau hypothalamique
dorsomédian (DMH), grâce à l’étude des effets de l’infusion de MTII dans le MPO chez des rats
donc le DMH a subi des lésions par l’injection d’acide kaïnique. L’injection de MTII dans le
MPO conduit à l’augmentation de la température et du captage de C14-bromopalmitate dans le
tissu adipeux brun interscapulaire (iBAT). La température et le captage de C14-bromopalmitate
sont deux variable corrélées (r=0.63, p<0.001). Ces deux augmentations sont bloquées par des
lésions du DMH. Les lésions du DMH bloquent aussi l’effet du MTII sur l’expression des ARNm
codant pour des protéines impliquées dans (i) la thermogenèse [déiodinase iodothyronine de type
II (Dio2) et le peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1α)],
(ii) la lipolyse [hormone sensitive lipase (Hsl)] et (iii) la lipogenèse [diacylglycérol O-
acyltransférase 2 (Dgat2) et la fatty acid synthase (Fas)], dans le iBAT de rats sacrifiés une heure
après l’infusion de MTII dans le MPO. L’expression de gènes dans le tissu adipeux blanc
inguinal est aussi stimulée par le MTII, mais seulement chez les rats avec des lésions du DMH.
Ces gènes incluent le glucose transporter member 4 (Glut4), la glycerol-3-phosphate
acyltransférase 3 (Gpat3), la Dgat1, la Dgat2, la triglyceride lipase (Atgl), la Hsl et le Cpt1β.
L’ensemble de ces résultats révèle que le MPO est un site majeur des effets thermogènes et
métaboliques du MTII. En outre, ils contribuent aussi à établir que le duo MPO-DMH est une
cible importante dans la modulation de l’homéostasie énergétique par les mélanocortines.


123
Abstract
The present study was designed to investigate the role of the medial preoptic nucleus
(MPO) as a site of the thermogenic and metabolic effects of the α−MSH analog Melanotan II
(MTII). We also assessed the involvement of the dorsomedial hypothalamic nucleus (DMH) by
investigating the effects of the MPO infusion of MTII in rats with DMH lesions produced by
kainic acid. Injection of MTII in the MPO led to increases in interscapular BAT (iBAT)
temperature and iBAT uptake of 14C-bromopalmitate. Both increases were blocked by DMH
lesions. iBAT temperature increase (area under curve) and 14C-bromopalmitate uptake emerged
as two correlated variables (r=0.63, p<0.001). DMH lesions also blocked MTII-induced
expression of mRNAs coding for proteins involved in (i) thermogenesis [type II iodothyronine
deiodinase (Dio2) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha
(Pgc1α)], (ii) lipolysis [hormone sensitive lipase (Hsl)] and (iii) lipogenesis [diacylglycerol O-
acyltransferase 2 (Dgat2), fatty acid synthase (Fas)], in iBAT of rats killed one hour after MPO
infusion of MTII. MTII also stimulated expressions of genes in iWAT but only in rats with DMH
lesions. These genes included glucose transporter member 4 (Glut4), glycerol-3-phosphate
acyltransferase 3 (Gpat3), Dgat1, Dgat2, triglyceride lipase (Atgl), Hsl and Cpt1β. Altogether,
the present results reveal the MPO as a site of the thermogenic and metabolic actions of MTII.
They also contribute to establish the MPO-DMH duet as a significant target for melanocortins to
modulate energy homeostasis.


125
Introduction
The role of the brain melanocortin system in energy balance regulation is acknowledged (7,
13, 18, 32, 43). This system comprises the proopiomelanocortin (POMC) neurons of the
hypothalamic arcuate nucleus (ARC) and the widely distributed melanocortin-4 receptors
(MC4R), which are important partners in controlling food intake and energy expenditure. POMC
and Mc4r knockout or loss-of-function mutations are associated with severe obesity in laboratory
rodents (20) as well as in humans (16). Consistent with this, the POMC product
alpha-melanocyte-stimulating hormone (α−MSH) and the α−MSH synthetic analog Melanotan
II, which represent MC4R/MC3R agonists, suppress food intake and stimulate energy
expenditure (14, 21).
In addition to reducing food intake, MC4R agonism has been reported to increase energy
expenditure through stimulating brown adipose tissue (BAT) thermogenesis (6, 15, 38, 39, 41).
Increases in BAT temperature (38, 39), BAT sympathetic nervous system (SNS) activity (15) and
BAT expression of uncoupling protein 1 (UCP1) and accessory thermogenic genes (42) have all
been observed after brain ventricular and parenchymal injections of MC4R agonists. BAT
involvement in MC4R-mediated thermogenic effects is also supported by observations obtained
in Mc4r knockout or SHU9119 (MC4R antagonist)-treated animals, which exhibit diminished
BAT thermogenic capacity/activity (21, 41). Additionally, the role of the MC4R in controlling
BAT thermogenesis is anatomically supported by retrograde viral transneuronal tracing studies,
which have demonstrated the strong connection between MC4R mRNA-expressing neurons and
the SNS outflow to iBAT (39).
There is good evidence that the paraventricular hypothalamus (PVH) (39), ARC (12), and
brainstem (38) are sites of the thermogenic action of MC4R agonists. However, the brain circuits
involved in the MC4R-mediated control of BAT thermogenesis remain to be fully unraveled.
Several brain regions expressing the MC4R have not been systematically investigated as sites of
the MC4R-mediated action on BAT thermogenesis. Among those regions, the preoptic area
(POA) is certainly a worthy candidate. The POA represents the second hypothalamic region, next
to the PVH, with the highest proportion of neurons connected to BAT (via the SNS) that express
the MC4R (39). Within the POA, MC4R mRNA is widely expressed in the medial preoptic

126
nucleus (MPO) (23), which is involved in thermoregulatory thermogenesis (29, 30). The MPO
hosts inhibitory neurons that modulate the activity of the excitatory neurons of the dorsomedial
hypothalamic nucleus (DMH) that control interscapular BAT (iBAT) activity via the raphe
pallidus (RPa) (26, 34). The observation that inhibition of neurons found in the rostral RPa
blocks the thermogenic effect of ventricular injection of MT II (15) supports the involvement of a
MPO-DMH-RPa-SNS circuit in the MC4R-mediated effect on iBAT activity.
The objective of the present study was to verify whether the MPO could be a site where
MTII could stimulate brown fat thermogenesis and metabolism with the complicity of the DMH.
The effects of the MPO infusion of MTII on iBAT temperature as well as on iBAT 14C-
bromopalmitate and 3H-deoxyglucose uptake were assessed in male Wistar rats with or without
DMH lesions. The uptake of the radiolabelled energy substrate was also measured in inguinal
white adipose tissue (iWAT), liver and heart. Furthermore, iBAT and iWAT expression of
thermogenic and metabolic genes was measured in animals killed one hour following MTII with
or without DMH lesions. iWAT was studied since it represents a WAT depot where brown
adipocytes can develop under appropriate adrenergic stimulation (3) and since it is, similar to
iBAT, connected to the MPO-MC4R neurons via the SNS (40).

127
Research Design and Methods
Animals
Male Wistar rats (Charles River Laboratories, St. Constant, QC, Canada), initially
weighting ~300 g, were individually housed at 23 ± 1°C under a 12:12-h light-dark cycle (light
on at 06:00). The rats had ad libitum access to water and pelleted standard chow (Charles River
Rodent Diet no. 5075). All animal treatments and procedures were approved by the Université
Laval Animal Ethics Committee and were in agreement with the Canadian Guide for the Care
and Use of Laboratory Animals.
Drugs
The MC4R/MC3R agonist MTII (Phoenix Pharmaceutical Inc, Burlingame CA, USA)
was dissolved in aCSF and administered at dose of 200 ng in a volume 200 nl (100 nl in each side
of the MPO). Kainic acid (1 mg/ml, Sigma) was dissolved in sterile saline and adjusted to pH
7.2–7.4 by using 0.1 M NaOH. The dose of MTII (21) and kainic acid (25) were selected from
previous studies (25).
MPO MTII infusions
Rats were chronically implanted with bilateral (1.0 mm between tubes) guide-cannulas
(26-gauge — Plastics One, Roanoke, VA, USA), aimed at the MPO, using the following
coordinates relative to bregma: 0.5 mm on each side of the midline, 0.6 mm posterior and 6.5 mm
ventral to the brain surface (36). The cannula was fixed in place using dental cement (Lang
Dental Manufacturing co., Inc, Wheeling, IL, USA) and anchoring screws. Sterile obturators
were inserted into the guide-cannulas to avoid blockage. The placement of the cannulas was
further confirmed under microscope the end of the study and the injection sited were estimated.
(Figure 1, A). Each surgery was done 1-week before the experiment to allow complete recovery
at the time of infusion. Brain MTII infusions were carried out using a 50 µl Hamilton syringe
connected to a syringe pump (Harvard Apparatus 55-2226). In each protocol, the solutions were
carefully infused in the brain during 30 sec (200 nl/min).

128
DMH lesions
DMH lesions were performed prior to the MPO guide-cannula implantation. Briefly, a 26-
gauge bilateral (1.0 mm between tubes) injection cannula (Plastics One, Roanoke, VA, USA) was
stereotaxically aimed at the DMH. The coordinates were as follows: 0.5 mm on each side of the
midline, 3.2 mm posterior to bregma and 8.5 mm ventral to the brain surface (36). Bilateral
infusions of 200 nl of kainic acid or sterile saline (control animals) were performed over a 10-
minute period. The cannula was left in place for another 10 minutes to allow the complete
diffusion of the kainic acid. Kainic acid damages perikarya and dendrites of neurons harboring
kainate receptors while preserving the fibers of passage of other neurons having their bodies
outside the targeted nuclei (4). DMH-lesions are illustrated in figure 1B. DMH-lesioned rats were
subjected to the MT II infusions 10 days after the injections of kainic acid. Their averaged weight
at that time was significantly lower (p<0.05) than that of sham-lesioned animals (331.9 ± 3.9 vs
347.8 ± 4.2). Such an effect had been reporter before (4).
iBAT thermogenesis and iBAT and iWAT energy substrate uptakes
Four (4) groups of rats with of without DMH lesions were used to assess the effects of
aCSF or MPO-infused MTII on iBAT thermogenesis. iBAT temperature was assessed under
isoflurane anesthesia, using a copper-constantan thermocouple (Physitemp Instruments, Inc.
Clifton, NJ, USA) inserted into the right iBAT pad. During the measurement, core temperature
was monitored with a second thermocouple inserted 6 cm into the rectum (19). Skin temperature
was also assessed with a third copper-constantan thermocouple placed on the left-rear leg. Core
and skin measurements were performed before the MPO infusions in order to avoid body
temperature variations. Measurement of CO2 production was assessed using a CO2 analyser
(model S-3A, Applied Electrochemistry Incorporated, Sunnyvale, CA, USA). CO2 production
was used to gauge whole-body thermogenesis. VO2 could not be measured for technical reasons
inherent to the use of the isoflurane mask during the MPO infusions of MTII.
In addition to the iBAT temperature and CO2 production measurements, we also
measured iBAT, iWAT, liver and heart 14C-bromopalmitate ([1-14C]-2-bromopalmitic acid) and 3H-deoxyglucose ([1,2-3H(N)]-Deoxy-D-glucose) uptakes. 14C-bromopalmitate was used to
assess non-esterified fatty acid (NEFA) uptake (27, 28) and to estimate lipid oxidation (10, 27,

129
35) and esterification (27), whereas 3H-deoxyglucose was used to trace glucose uptake (17). Both
radioactive tracers (Moravek Biochemicals, Inc. Brea, CA, USA) were dissolved in normal saline
supplemented with 4% bovine serum albumin (BSA). Thirty minutes after the MPO infusion of
MTII, an intravenous bolus of 20 µCi of each tracer were injected in the tail vein, in a total
volume of 350 µl. Arterial blood sampling was done from carotid prior to the MTII infusion at
time -30, -15, 0 minutes for the measurement of plasma levels of insulin, NEFA and triglycerides
(TG) as previously described (28). Additionally, blood was sampled at time 31, 32, 33, 35, 40, 50
and 60 minutes post MTII infusion to determine the radioactivity clearance. Sixty minutes after
MTII infusion, animals were euthanized with an overdose of anesthetic and tissues were collected
on ice, weighed and stored at -80°C pending analyses. iBAT, iWAT, liver and heart-specific
uptakes of 14C-bromopalmitate and 3H-deoxyglucose were determined on samples of tissues
(~250 mg), which were homogenized (Polytron PT10-35, Kinematica, inc. Bohemia, NY, USA)
in ice-cold Chappell-Perry buffer (0.075 M sucrose, 0.225 M sorbitol, 1 mM EGTA, 0.1% fatty-
acid-free BSA, and 10 mM Tris–HCl, pH 7.4). An aliquot was kept to determine total [14C and 3H] activity in the tissue as previously described (27). Fractional uptakes of 14C-bromopalmitate
and 3H-deoxyglucose were determined by measuring [14C] and [3H] activity in tissue extracts
using a scintillation counter (liquid scintillation analyzer Tri-Carb 2900TR, PerkinElmer,
Montreal, QC, Canada). Uptakes data were expressed as the percentage of injected dose (% of
ID) of [14C] and [3H] recovered per gram of tissues.
iBAT and iWAT expression of metabolic genes
We determined the expression of genes involved in (i) energy substrate uptake [fatty acid
binding protein 1 (Fabp1) and glucose transporter member 4 (Glut4)], (ii) lipid synthesis
[diacylglycerol O-acyltransferase 1 and 2 (Dgat1 and Dgat2), fatty acid synthase (Fas), glycerol-
3-phosphate acyltransferase 3 (Gpat3)] (iii) lipolysis [triglyceride lipase (Atgl), hormone
sensitive lipase (Hsl)] and (iv) thermogenesis-related genes [carnitine palmitoyltransferase 1β
(Cpt1β), Type II iodothyronine deiodinase (Dio2), peroxisome proliferator-activated receptor-γ
coactivator 1-alpha (Pgc1α), Ucp1]. Briefly, RNA was isolated from iBAT and iWAT depots
with QIAzol and the RNeasy Lipid Tissue Kit (QIAGEN, Mississauga, ON, Canada). Expand
reverse transcriptase (Invitrogen, Burlington, ON, Canada) was used for cDNA synthesis,
accpording to manufacturer's instructions. cDNAs were diluted in DNase-free water (1:25) before

130
quantification by real-time PCR. mRNA transcript levels were measured in duplicate samples
using a Rotor Gene 3000 system (Montreal Biotech, Montreal, QC, Canada). The primers used
for the PCR reactions are shown in Table 1. At the end of each run, melt curve analyses were
performed, and a few samples, representative of each experimental group, were run on agarose
gel. Results were expressed as the ratio of the expression of the target gene to that of the
housekeeping gene acidic ribosomal phosphoprotein (Arbp), which was selected as no significant
variation in its expression was observed after our treatment.
Statistical analyses
Values obtained from rats with improper placement of cannula were excluded from the
statistical analyses. Results are expressed as means ± SEMs. Comparisons were done on normally
distributed data using two-way ANOVAs followed by Bonferroni post hoc tests (when
appropriate) to assess the differences between various conditions with Graph pad Prism Software
version 6.0 for Mac (San Diego, CA, USA). Differences were considered statistically significant
when P values were less than 0.05.

131
Results
MTII infusion in the MPO led to an increase (above aCSF) in iBAT temperature, which
was abolished by DMH lesions (Figure 2, A-B). The infusion of MTII also produced an elevation
in CO2 production (Figure 2, C-D), which did not reach statistical significance in rats with DMH
lesions. The correlation between iBAT temperature elevation and whole body CO2 production
(Figure 2, E) proved to be highly significant (r = 0.79, P < 0.001).
Together with measuring iBAT thermogenesis and VCO2 production, we also measured 14C-bromopalmitate and 3H-deoxyglucose uptake in iBAT, iWAT, liver and heart (Figure 3).
MPO infusion of MTII led to an increase (above aCSF) in 14C-bromopalmitate uptake in iBAT,
which was prevented by DMH lesions (Figure 3, A). However, we did not observe any increase
in iBAT 3H-deoxyglucose uptake following MTII (Figure 3, B), revealing NEFA uptake as a
better predictor of BAT thermogenesis than glucose uptake in the context of the present study.
Correlation analyses, which showed a significant Pearson's r (0.63, p < 0.001) solely between
iBAT temperature elevation and 14C-bromopalmitate uptake (Figure 3, C-D), support this
deduction. MTII infusion in MPO increased (above aCSF) 14C-bromopalmitate and 3H-
deoxyglucose uptakes in iWAT (Figure 3, E-F). The effect was however statistically significant
(p < 0.05) only for the 14C-bromopalmitate uptake. MPO infusion of MTII also stimulated liver 14C-bromopalmitate uptake, which was prevented by DMH lesions (Figure 3, G). MTII and DMH
lesions did not influence liver 3H-deoxyglucose uptake (Figure 3, H). Finally, MTII did not
increase heart 14C-bromopalmitate uptake (Figure 3, I), whereas DMH lesions significantly
(p<0.05) reduced heart 3H-deoxyglucose uptake, regardless of whether aCSF or MTII was
injected in the MPO (Figure 3, H).
The effects of MTII and DMH lesions on the expression of genes involved in iBAT
thermogenesis and metabolism are illustrated in figure 4A. We observed that MTII increased
expression of genes involved in lipogenesis (Fas and Dgat2), lipolysis (Hsl) and thermogenesis
(Pgc1α and Dio2) in sham-lesioned rats. The effects of MTII were however abolished following
DMH lesions. As revealed by the correlations presented in Figure 4, B-C, 14C-bromopalmitate
uptake in iBAT correlated well with the expression of Hsl (r = 0.62, P < 0.001), which is
implicated in lipolysis, and Dgat2 (r = 0.51, P < 0.01), which is implicated in the synthesis of
TG. Figure 4, D illustrates the effects of the various experimental treatments on the iWAT

132
expression of the genes reported for BAT in Figure 4, A. In sham-lesioned animals, MTII did not
lead to any increase in gene expression. In fact, it reduced Glut4 and Gpat3 expressions.
However, in DMH-lesioned rats, MTII induced Glut4, Gpat3, Dgat1, Dgat2, Hsl, Atgl and Cpt1
β. As illustrated in Figure 4, E-F, the mRNA coding for Fas, which is implicated in the
synthesis of TG correlated well with 14C-bromopalmitate (r = 0.46, P = 0.02) and 3H-
deoxyglucose (r = 0.59, P < 0.01) uptake in iWAT.
Table 2 presents the differences (Δ) between post- and pre-infusion levels of circulating
NEFA, TG, and insulin. DMH-lesioned rats showed a Δ insulin that was significantly lower than
that of aCSF-sham-lesioned animals.

133
Discussion
The present results point to the MPO as a site of the thermogenic and metabolic actions of
MTII, the synthetic analog of α-MSH and a MC4R/MC3R agonist. The MPO injection of MTII,
led to an increase in iBAT temperature, which was accompanied by increases in iBAT 14C-
bromopalmitate uptake and iBAT expression of genes involved in the thermogenesis and energy
metabolism. This study also demonstrated the role of DMH in the effect of the MPO infusion of
MTII. DMH lesions caused by a neurotoxic dose of kainic acid blunted the MTII-induced
increases in iBAT temperature, 14C-bromopalmitate uptake and iBAT expression of
metabolic/thermogenic genes. Finally, the present results also suggest that the MPO melanocortin
receptors could, through the MPO-DMH duet, be involved in energy substrate metabolism in
other tissues such as iWAT and liver.
The injection of MTII into the MPO led to an increase in iBAT temperature, which was
abolished by DMH lesions. Together with revealing a new site of the thermogenic action of
MTII, these findings further support the role of the POA (which comprises the MPO) and DMH
in the control of iBAT thermogenesis. These two structures have indeed been flagged to for their
participation in cold-induced thermogenesis (29). The POA has also been reported to be the site
of thermogenic action of corticotropin-releasing factor (8, 11) and prostaglandin E2 (2, 26),
which seemingly act by releasing the inhibitory effect of POA GABA neurons on the DMH to
stimulate the RPa neurons governing the SNS outflow to iBAT (8, 29). Within the POA, both the
MC4R and MC3R are expressed in relative abundance in the MPO (32). However, the MC4R is
most probably the melanocortin receptor mediating the thermogenic effect of MTII since it is
expressed in neurons that are polysynaptically connected to iBAT (39) and iWAT (40) via the
SNS. Further supporting this assumption are recent results from our group demonstrating the
ability of HS024, a specific MC4R agonist, to block the iBAT-stimulating effect of the third
ventricle infusion of MTII in rats (Monge-Roffarello, B; Labbé, S and Richard, D, unpublished
results). Finally, the observation that the MTII infusion in the MPO caused an increase in BAT
thermogenesis further supports the integrative role of the POA in the effects of the melanocortins
in energy balance regulation. This role was suggested in a previous investigation pointing to the
MPO as one of the major sites of the hypophagic action of α-MSH (22).

134
The DMH-dependent stimulating effect of the MPO infusion of MTII on iBAT
temperature was accompanied by an increase in iBAT 14C-bromopalmitate uptake. This tracer is
taken up by cells as native NEFA and is trapped in mitochondria (27). 14C-bromopalmitate
uptake correlated well with the change in iBAT temperature. This was not the case with 3H-
deoxyglucose uptake, which appeared, in this study, as a poor predictor of the MTII effects on
BAT thermogenesis, both in sham- and DMH-lesioned groups. 14C-bromopalmitate uptake also
significantly correlated with iBAT Hsl and Dgat2 mRNA expression further indicating that 14C-
bromopalmitate uptake can be used to predict lipid oxidation (10, 27, 35) and esterification (27).
Noteworthily, Hsl (lipolysis) and Dgat2 (lipogenesis) were among those genes, which also
included Fatp1 (fatty acid uptake), Fas (de novo lipogenesis) and Pgc1α and Dio2
(thermogenesis), whose expression underwent a DMH-dependent stimulating effect of MTII. We
did not observe any induction of Ucp1 following MTII, this likely because of the short period
time (one hour) elapsing between the MTII treatment and the killing of the rats.
We also observed DMH-dependent metabolic effects of the MPO infusion of MTII in
tissues other than BAT. Indeed, MPO infusion of MTII increased 14C-bromopalmitate uptake in
the liver and iWAT and DMH lesions blunted those effects. Such findings further support the
extensive role for the MPO melanocortin receptors and POA/DMH duet in energy metabolism
(29, 37). Given the recognition of iWAT as a WAT depot capable of developing brown
adipocytes (beige depot) (3) and the neuroanatomical link between iWAT and the POA via the
SNS (40), we choose to examine the same panel of genes in this fat depot as the one we
scrutinized in iBAT. The stimulating effects of MTII were however only seen in iWAT of rats
with DMH lesions. Indeed, in those rats, MTII induced genes involved in the glucose transport
(Glut4), lipogenesis (Gpat3, Dgat1 and Dgat2), lipolysis (Hsl, Atgl) and
thermogenesis/metabolism (Cpt1β). The different effects seen in iBAT and iWAT cannot be
easily explained. Nonetheless, these findings demonstrate the importance of the DMH in
modulating and potentially fine-tuning energy homeostasis. DMH comprises excitatory
glutamatergic neurons that were reported to be key players in the control of SNS-mediated iBAT
thermogenesis (30, 31, 33). It seems reasonable to assume that those glutamatergic neurons were
affected by kainic acid (1, 4), which prevented the temperature rise in iBAT induced by the MPO
infusion of MTII. On the other hand, the increased expression of genes such as Hsl, Atgl and
Cpt1β, which we saw in iWAT after MTII only in rats with damaged DMH, suggests an

135
enhanced iWAT metabolic activity, reminiscent of the effects seen after DMH NPY knockdown
(5). Knockdown of DMH NPY has indeed been reported to stimulate metabolism and the
browning of iWAT (due to the development of brown adipocytes) (5). The DMH neurons can
produce NPY in neurons that also expressed CART (24) and enhanced expression of NPY in the
DMH has been associated with excess fat deposition (44), high fat feeding (24) and lactation (9).
The possibility that the NPY-expressing neurons could have been damaged by kainic acid cannot
be excluded. Further studies are however needed to verify this possibility and further clarify the
complex interplay the POA and DMH in iWAT metabolism.
In conclusion, the present results revealed the MPO as a site of the thermogenic and
metabolic actions of MTII. They also established the MPO-DMH duet as a target for the
melanocortin effects on energy homeostasis.


137
Acknowledgements
This work was supported by a grant from the Canadian Institutes of Health Research
(CIHR — MOP-119436) and was performed at the Quebec Heart and Lung Institute Research
Center, which is funded by the Fonds de la recherche du Québec — Santé (FRQ-S). B. Monge-
Roffarello holds a fellowship from the CIHR Training Program in Obesity/Healthy Body Weight
Research. SM Labbé is the recipient of a CIHR postdoctoral fellowship. A. Caron holds a
fellowship from the CIHR Training Program in Obesity/Healthy Body Weight Research. We are
grateful to Julie Plamondon for its helpful laboratory assistance.


139
References
1. Acosta-Galvan G, Yi C-X, van der Vliet J, Jhamandas JH, Panula P, Angeles-Castellanos M, Del Carmen Basualdo M, Escobar C, Buijs RM. Interaction between hypothalamic dorsomedial nucleus and the suprachiasmatic nucleus determines intensity of food anticipatory behavior. Proceedings of the National Academy of Sciences 108: 5813–5818, 2011.
2. Amir S, Schiavetto A. Injection of prostaglandin E2 into the anterior hypothalamic preoptic area activates brown adipose tissue thermogenesis in the rat. Brain Res. 528: 138–142, 1990.
3. Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol (October 22, 2013). doi: 10.1038/nrendo.2013.204.
4. Bellinger LL, Williams FE. Aphagia and adipsia after kainic acid lesioning of the dorsomedial hypothalamic area. Am. J. Physiol. 244: R389–99, 1983.
5. Bi S, Li L. Browning of white adipose tissue: role of hypothalamic signaling. Ann. N. Y. Acad. Sci. 1302: 30–34, 2013.
6. Brito MN, Brito NA, Baro DJ, Song CK, Bartness TJ. Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology 148: 5339–5347, 2007.
7. Butler AA. The melanocortin system and energy balance. Peptides 27: 281–290, 2006.
8. Cerri M, Morrison SF. Corticotropin releasing factor increases in brown adipose tissue thermogenesis and heart rate through dorsomedial hypothalamus and medullary raphe pallidus. Neuroscience 140: 711–721, 2006.
9. Chen P. Melanocortin 4 Receptor-Mediated Hyperphagia and Activation of Neuropeptide Y Expression in the Dorsomedial Hypothalamus during Lactation. Journal of Neuroscience 24: 5091–5100, 2004.
10. Ci X, Frisch F, Lavoie F, Germain P, Lecomte R, van Lier JE, Bénard F, Carpentier AC. The effect of insulin on the intracellular distribution of 14(R,S)-[18F]Fluoro-6-thia-heptadecanoic acid in rats. Mol Imaging Biol 8: 237–244, 2006.
11. Dagnault A, Richard D. Involvement of the medial preoptic area in the anorectic action of estrogens. [Online]. Am. J. Physiol. 272: R311–7, 1997.
12. do Carmo JM, da Silva AA, Rushing JS, Pace BR, Hall JE. Differential control of metabolic and cardiovascular functions by melanocortin-4 receptors in proopiomelanocortin neurons. AJP: Regulatory, Integrative and Comparative Physiology (July 10, 2013). doi: 10.1152/ajpregu.00518.2012.
13. Ellacott KLJ, Cone RD. The role of the central melanocortin system in the regulation of

140
food intake and energy homeostasis: lessons from mouse models. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 361: 1265–1274, 2006.
14. Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385: 165–168, 1997.
15. Fan W, Morrison SF, Cao W-H, Yu P. Thermogenesis activated by central melanocortin signaling is dependent on neurons in the rostral raphe pallidus (rRPa) area. Brain Res. 1179: 61–69, 2007.
16. Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348: 1085–1095, 2003.
17. Ferré P, Leturque A, Burnol AF, Pénicaud L, Girard J. A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaesthetized rat. Biochem. J. 228: 103–110, 1985.
18. Garfield AS, Lam DD, Marston OJ, Przydzial MJ, Heisler LK. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol. Metab. 20: 203–215, 2009.
19. Gordon CJ. Thermal biology of the laboratory rat. Physiol. Behav. 47: 963–991, 1990.
20. Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88: 131–141, 1997.
21. Hwa JJ, Ghibaudi L, Gao J, Parker EM. Central melanocortin system modulates energy intake and expenditure of obese and lean Zucker rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281: R444–51, 2001.
22. Kim MS, Rossi M, Abusnana S, Sunter D, Morgan DG, Small CJ, Edwards CM, Heath MM, Stanley SA, Seal LJ, Bhatti JR, Smith DM, Ghatei MA, Bloom SR. Hypothalamic localization of the feeding effect of agouti-related peptide and alpha-melanocyte-stimulating hormone. Diabetes 49: 177–182, 2000.
23. Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J. Comp. Neurol. 457: 213–235, 2003.
24. Lee SJ, Verma S, Simonds SE, Kirigiti MA, Kievit P, Lindsley SR, Loche A, Smith MS, Cowley MA, Grove KL. Leptin stimulates neuropeptide Y and cocaine amphetamine-regulated transcript coexpressing neuronal activity in the dorsomedial hypothalamus in diet-induced obese mice. Journal of Neuroscience 33: 15306–15317, 2013.

141
25. Li F, Li M, Cao W, Xu Y, Luo Y, Zhong X, Zhang J, Dai R, Zhou X-F, Li Z, Li C. Anterior cingulate cortical lesion attenuates food foraging in rats. Brain Res. Bull. 88: 602–608, 2012.
26. Madden CJ, Morrison SF. Excitatory amino acid receptors in the dorsomedial hypothalamus mediate prostaglandin-evoked thermogenesis in brown adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286: R320–5, 2004.
27. Menard SL, Croteau E, Sarrhini O, Gelinas R, Brassard P, Ouellet R, Bentourkia M, van Lier JE, Rosiers CD, Lecomte R, Carpentier AC. Abnormal in vivo myocardial energy substrate uptake in diet-induced type 2 diabetic cardiomyopathy in rats. Am. J. Physiol. Endocrinol. Metab. 298: E1049–E1057, 2010.
28. Ménard SL, Ci X, Frisch F, Normand-Lauzière F, Cadorette J, Ouellet R, Lier JE, Bénard F, Bentourkia M, Lecomte R, Carpentier AC. Mechanism of Reduced Myocardial Glucose Utilization During Acute Hypertriglyceridemia in Rats. Mol Imaging Biol 11: 6–14, 2008.
29. Morrison SF, Madden CJ, Tupone D. Central Control of Brown Adipose Tissue Thermogenesis. Front Endocrinol (Lausanne) 3, 2012.
30. Morrison SF, Nakamura K, Madden CJ. Central control of thermogenesis in mammals. Exp. Physiol. 93: 773–797, 2008.
31. Morrison SF. 2010 Carl Ludwig Distinguished Lectureship of the APS Neural Control and Autonomic Regulation Section: Central neural pathways for thermoregulatory cold defense. Journal of Applied Physiology 110: 1137–1149, 2011.
32. Mountjoy KG. Distribution and function of melanocortin receptors within the brain. Advances in experimental medicine and biology 681: 29–48, 2010.
33. Nakamura K, Morrison SF. A thermosensory pathway that controls body temperature. Nat. Neurosci. 11: 62–71, 2007.
34. Nakamura K, Nakamura K, Morrison SF, Morrison SF. Central efferent pathways mediating skin cooling-evoked sympathetic thermogenesis in brown adipose tissue. AJP: Regulatory, Integrative and Comparative Physiology 292: R127–R136, 2006.
35. Oakes ND, Thalén P, Aasum E, Edgley A, Larsen T, Furler SM, Ljung B, Severson D. Cardiac metabolism in mice: tracer method developments and in vivo application revealing profound metabolic inflexibility in diabetes. Am. J. Physiol. Endocrinol. Metab. 290: E870–81, 2006.
36. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates [Online]. academic press.
37. Richard D, Monge-Roffarello B, Chechi K, Labbé SM, Turcotte EE. Control and physiological determinants of sympathetically mediated brown adipose tissue

142
thermogenesis. Front Endocrinol (Lausanne) 3: 36, 2012.
38. Skibicka KP, Grill HJ. Energetic Responses Are Triggered by Caudal Brainstem Melanocortin Receptor Stimulation and Mediated by Local Sympathetic Effector Circuits. Endocrinology 149: 3605–3616, 2008.
39. Song CK, Vaughan CH, Keen-Rhinehart E, Harris RBS, Richard D, Bartness TJ. Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown adipose tissue: neuroanatomical and functional evidence. AJP: Regulatory, Integrative and Comparative Physiology 295: R417–R428, 2008.
40. Song CK. Melanocortin-4 receptor mRNA is expressed in sympathetic nervous system outflow neurons to white adipose tissue. AJP: Regulatory, Integrative and Comparative Physiology 289: R1467–R1476, 2005.
41. Ste Marie L, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. U.S.A. 97: 12339–12344, 2000.
42. Williams DL, Bowers RR, Bartness TJ, Kaplan JM, Grill HJ. Brainstem melanocortin 3/4 receptor stimulation increases uncoupling protein gene expression in brown fat. Endocrinology 144: 4692–4697, 2003.
43. Xu Y, Elmquist JK, Fukuda M. Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann. N. Y. Acad. Sci. 1243: 1–14, 2011.
44. Zheng F, Kim YJ, Chao P-T, Bi S. Overexpression of neuropeptide y in the dorsomedial hypothalamus causes hyperphagia and obesity in rats. Obesity 21: 1086–1092, 2013.

143
Tables
Table 1: qPCR gene information.
Gene Forward sequence Reverse sequence Annealing temperature °C
Arbp TAAAGACTGGAGACAAGGTG GTGTAGTCAGTCTCCACAGA 60
Atgl CACTTTAGCTCCAAGGATGA TGGTTCAGTAGGCCATTCCT 60
Cpt1b CGGAAGCACACCAGGCAGTA GCAGCTTCAGGGTTTGTCGGAATA 64
Dio2 ATGGGACTCCTCAGCGTAGA GCACAGGCAAAGTCAAGAAG 60
Dgat1 TATTACTTCATCTTTGCTCC AAAGTAGGTGACAGACTCAG 60
Dgat2 TGCCCGCAGCGAGAACAAGAATAAA GGCGTGTTCCAGTCAAATGCCA 64
Fabp1 GCCAAGAGAACTTTGAGCCC CTTGTAGACGATGTCACCCAGT 60
Fas GAGTCCGAGTCTGTCTCCCGCTTGA GCCGTGAGGTTGCTGTTGTCTGTAG 64
Glut4 AGTGACTGGGACACTGGTCCTT ACATTGTTGGCCAGCATAGC 64
Gpat3 TGGACTGATGGGGATCATTCAGAGA GCCCAGCTTGTCATTATCCGAA 64
Hsl CCTGCTGACCATCAACCGAC CCTCGATCTCCGTGATATTCCAGA 60
Pgc1α TCCTGTTACTATTATGAATCAAGCC AAACCATAGCTGTCTCCATCATCC 64
Ucp1 TGGTGAGTTCGACAACTTCC GTGGGCTGCCCAATGAATAC 64

144
Table 2: Effects of MTII infusion in MPO on blood variation of insulin, NEFA and
triglycerides in DMH-lesioned rats.
Groups Δ-Insulin (pmol/L) Δ-NEFA (mmol/L) Δ-TG (mmol/L)
aCSF-Sham-lesioned -28.29±5.99 -0.127±0.028 -0.168±0.248
MTII-Sham-lesioned -25.37±10.62 -0.011±0.050 -0.104±0.071
aCSF-DMH-lesioned -29.18±8.76 0.020±0.073 0.048±0.117
MTII-DMH-lesioned -71.08±10.50* -0.035±0.029 -0.002±0.150

145
Figure Legends
Figure 1. Histological verification. A) Schematic representation of the MPO infusions of
MTII in the MPO. The different groups are presented as follows: white circles, aCSF-Sham-
lesioned; black circles, MTII-Sham-lesioned; white squares, aCSF-DMH-lesioned; black squares,
MTII-DMH-lesioned). B) Pictures of brain coronal sections taken at the level of DMH in Sham-
and DMH-lesioned rats.
Figure 2. Effects of the MPO infusion of MTII in Sham- and DMH-lesioned rats on iBAT
and whole body thermogenesis. Four distinct groups of 6-7 rats were used to test the effect of
MTII (0 or 100 ng). The area under the curve (AUC — from baseline) for Δ-iBAT temperature
(A, B), Δ-CO2 exhaled (indirect assessment of whole body thermogenesis) (C, D) were measured
over 60 minutes following the MTII infusion. (E) Correlation between the AUC of Δ-iBAT
temperature and Δ-CO2 exhaled. * P < 0.05 vs. aCSF-Sham-lesioned, # P < 0.05 vs MTII-Sham-
lesioned.
Figure 3. Effects of the MPO infusion of MTII on iBAT, iWAT, liver and heart metabolic
activity in Sham- and DMH-lesioned rats. Four distinct groups of 6-7 rats were used to test the
effect of MTII (0 or 100 ng) on 14C-bromopalmitate (A) and 3H-deoxyglucose (B) uptake in
iBAT, on 14C-bromopalmitate (E) and 3H-deoxyglucose (F) uptake in iWAT, on 14C-
bromopalmitate (G) and 3H-deoxyglucose (H) uptake in liver and on 14C-bromopalmitate (I) and 3H-deoxyglucose (J) uptake in heart. (C) Correlation between 14C-bromopalmitate uptake and
AUC of Δ-iBAT temperature. (D) Correlation between 3H-deoxyglucose uptake and AUC of Δ-
iBAT temperature. * P < 0.05 vs. aCSF-Sham-lesioned, # P < 0.05 vs MTII-Sham-lesioned.
Figure 4. Effects of the MPO infusion of MTII on iBAT and iWAT gene expression in
Sham- and DMH-lesioned rats. Four distinct groups of 6-7 rats were used to test the effects of
MTII (0 or 100 ng) on iBAT (A) and iWAT (D) mRNA expressions, which were measured in

146
rats sacrificed 1 hour after MTII infusion. (C) Correlation between 14C-bromopalmitate uptake
and Hsl mRNA expression in iBAT. (D) Correlation between 14C-bromopalmitate uptake and
Dgat2 mRNA expression in iBAT. (E) Correlation between 14C-bromopalmitate uptake and Fas
mRNA expression in iWAT. (F) Correlation between between 3H-deoxyglucose uptake and Fas
mRNA expression in iWAT. * P < 0.05 vs. aCSF-Sham-lesioned, # P < 0.05 vs MTII-aham-
lesioned, † P < 0.05 vs aCSF-DMH-lesioned.

147
Figures
Figure 1: Histological verification.
A"
"B"
"
Bregma'(0.48mm'
Bregma'(0.60mm'
Bregma'(0.72mm'
Bregma'(0.84mm'
DMH$Sham) DMH$lesioned)

148
Figure 2: Effects of the MPO infusion of MTII in Sham- and DMH-lesioned rats on iBAT
and whole body thermogenesis.
0 15 30 45 60
-1
0
1
2
Time (min)
Δ-iB
AT
Tem
pera
ture
°`C
inj.
Sham-lesioned-MTIIDMH-lesioned-aCSFDMH-lesioned-MTII
Sham-lesioned-aCSF
-20
0
20
40
60
80
AU
C Δ
-iBA
T te
mpe
ratu
re
*
#
Sham-lesioned DMH-lesioned
0 15 30 45 60
90
100
110
120
Time (min)
Δ-V
CO
2 %
inj.
-200
0
200
400
600
AU
C Δ
-VC
O2
Sham-lesioned DMH-lesioned
*
-50 0 50 100 150 200
-500
0
500
1000
1500
2000
AUC Δ-iBAT temperature
AU
C Δ-V
CO
2
P < 0.001 r = 0.788
A B
C D
E
aCSF MTII

149
Figure 3: Effects of the MPO infusion of MTII on iBAT, iWAT, liver and heart metabolic
activity in Sham- and DMH-lesioned rats.
A B
C D
E F
G H
I J
Brown adipose tissue
0.0
0.2
0.4
0.6
0.8
% In
jecte
d do
se o
f [14
C]-b
rom
opal
mita
te / g
ram
Sham-lesioned DMH-lesioned
*
#
-50 0 50 100 150
0.0
0.2
0.4
0.6
0.8
AUC Δ-iBAT temperature
% In
ject
ed d
ose
of
[14C]
-bro
mop
alm
itate
/ gr
am
P < 0.001r = 0.632
Inguinal white adipose tissue
0.0
0.1
0.2
0.3
% In
jecte
d do
se o
f [14
C]-b
rom
opal
mita
te / g
ram
Sham-lesioned DMH-lesioned
*#
Liver
0
1
2
3
4
% In
jecte
d do
se o
f [14
C]-b
rom
opal
mita
te / g
ram
Sham-lesioned DMH-lesioned
*
Heart
0.0
0.2
0.4
0.6
0.8
% In
jecte
d do
se o
f [14
C]-b
rom
opal
mita
te / g
ram
Sham-lesioned DMH-lesioned
Brown adipose tissue
0.0
0.1
0.2
0.3
% In
jecte
d do
se o
f [3 H
]-deo
xygl
ucos
e / g
ram
Sham-lesioned DMH-lesioned
-50 0 50 100 150
0.00
0.05
0.10
0.15
AUC Δ-iBAT temperature%
Inje
cted
dos
e of
[3 H
]-deo
xygl
ucos
e / g
ram
P = 0.11 r = 0.31
Inguinal white adipose tissue
0.0
0.1
0.2
0.3
% In
jecte
d do
se o
f [3 H
]-deo
xygl
ucos
e / g
ram
Sham-lesioned DMH-lesioned
#
Liver
0.0
0.1
0.2
0.3
% In
jecte
d do
se o
f [3 H
]-deo
xygl
ucos
e / g
ram
Sham-lesioned DMH-lesioned
Heart
0.0
0.1
0.2
0.3
% In
jecte
d do
se o
f [3 H
]-deo
xygl
ucos
e / g
ram
Sham-lesioned DMH-lesioned
aCSFMTII
aCSFMTII
aCSFMTII
aCSFMTII
aCSFMTII
aCSFMTII
aCSFMTII
aCSFMTII

150
Figure 4: Effects of the MPO infusion of MTII on iBAT and iWAT gene expression in
Sham- and DMH-lesioned rats.
Fatp1Glut4 Fas
Gpat3Dgat1
Dgat2 HslAtgl
Pgc1α Dio2Cpt1βUcp1
0
1
2
3
4
5
iBAT g
ene e
xpres
sion /
ARBP
mRN
A
aCSF-Sham-lesionedMTII-Sham-lesionedaCSF-DMH-lesionedMTII-DMH-lesioned
**
* *
*
#
Fatp1Glut4 Fas
Gpat3Dgat1
Dgat2 HslAtgl
Pgc1αDio2Cpt1βUcp1
0
2
4
6
8
10
iWAT
gene
expre
ssion
/ ARB
P mRN
A
aCSF-Sham-lesionedMTII-Sham-lesionedaCSF-DMH-lesionnedMTII-DMH-lesionned
†
*
*
*
*
* *#
#
#
#
#
# #
†
†
†
ND
A
B C
D
0.0 0.2 0.4 0.6 0.80
1
2
3
% Injected dose of [14C]-bromopalmitate . gram-1
Dgat2
mRN
A ex
pres
sion
Brown adipose tissue
r = 0.51P < 0.01
0.0 0.2 0.4 0.6 0.80
1
2
3
% Injected dose of [14C]-bromopalmitate . gram-1
Hsl m
RNA
expr
essio
n
Brown adipose tissue
r = 0.62P < 0.001
0.00 0.05 0.10 0.15 0.200
1
2
3
% Injected dose of [14C]-bromopalmitate . gram-1
Fas m
RNA
expr
essio
n
Inguinal white adipose tissue
r = 0.46P = 0.02
0.00 0.05 0.10 0.15 0.20 0.250
1
2
3
% Injected dose of [3H]-deoxyglucose . gram-1
Fas m
RNA e
xpres
sion
Inguinal white adipose tissue
r = 0.59P < 0.01
E F

151
Discussion générale, perspectives de recherche et conclusion.


153
Chapitre 1
Le système mélanocortine est un acteur majeur de la régulation de l’homéostasie
énergétique (Butler, 2006; Pandit et al., 2011), il agit aussi bien sur la prise alimentaire que sur la
dépense énergétique et en particulier sur le BAT, qui comme il a été vu dans l’introduction est le
tissu effecteur de la thermogenèse sans frisson (Cannon and Nedergaard, 2004; Clapham, 2012).
En effet, la délétion du gène codant pour MC4R, le principal récepteur aux mélanocortines
impliqué dans la régulation de la balance énergétique a pour conséquence une réduction de
l’activité du BAT et notamment en réponse à une suralimentation (Butler et al., 2001). De plus,
l’injection de MTII, un agoniste du système mélanocortine, en i.c.v. ou dans le PVH entraine une
augmentation de la thermogenèse du iBAT (Song et al., 2008). Par ailleurs, le PVH est la
structure centrale qui compte le plus de neurones innervant indirectement le iBAT et dont 80% de
ceux-ci expriment MC4R (Song et al., 2008). Enfin, il a été montré que le système
endocannabinoïde interagit avec le système mélanocortine dans le contrôle de la prise alimentaire
(Verty et al., 2004) et que son récepteur central, CB1, est très exprimé au niveau du PVH
(Richard et al., 2009), suggérant ainsi que ces deux systèmes pourraient interagir dans le contrôle
de la thermogenèse du iBAT.
Dans un premier temps, nos résultats confirment les observations décrites dans la littérature
à propos de l’action des mélanocortines sur la dépense énergétique. En effet, l’injection centrale
de MTII entraine une augmentation de la dépense énergétique qui se traduit par une élévation de
la consommation d’oxygène sans modification significative du quotient respiratoire (Cowley et
al., 1999). De plus, la mesure directe de la température du iBAT (Fan et al., 2007; Song et al.,
2008; Vaughan et al., 2011) associée à l’injection de C14-bromopalmitate et de H3-désoxyglucose
qui sont des marqueurs respectivement des NEFAs (Ménard et al., 2009; 2010) et du glucose
(Ferré et al., 1985), permet d’établir avec force, l’effet stimulateur du MTII sur l’activité
thermogène du iBAT. Ensuite, l’augmentation de l’expression des gènes Pgc1α, Dio2, Cpt1β et
Ucp1, connus pour être impliqués dans la thermogenèse lors d’un traitement aigu ou sub-
chronique confirme le rôle du système mélanocortine dans la mise en place de la capacité
thermogène du iBAT (Williams et al., 2003; Richard et al., 2010).

154
Cette étude apporte des éléments nouveaux concernant le rôle du système
endocannabinoïde dans la modulation de la dépense énergétique induite par l’activité du système
mélanocortine. En effet, il est déjà connu que le système endocannabinoïde interagit avec le
système mélanocortine dans le contrôle de la prise alimentaire (Verty et al., 2004) et qu’il a des
effets opposés au système mélanocortine sur la thermogenèse. Néanmoins, notre étude va plus
loin en montrant que le système endocannabinoïde agit comme régulateur de l’activité du
système mélanocortine sur le contrôle de la thermogenèse et pas comme un compétiteur. En effet,
l’injection préalable dans le 3eme ventricule de Δ9-THC, un agoniste des récepteurs aux
cannabinoïdes à une dose qui n’a pas d’effet propre observé, bloque l’augmentation induite par
l’administration de MTII dans le 3eme ventricule sur de nombreuses composantes de la
thermogenèse telles que la consommation d’oxygène, la température du iBAT, le captage du C14-
bromopalmitate et l’expression génique de Pgc1α, Dio2, Cpt1β et Ucp1. De plus, l’utilisation de
l’AM251, un antagoniste sélectif de CB1, potentialise une partie des effets du MTII sur la
dépense énergétique, comme l’augmentation de la consommation d’oxygène lors de l’injection de
la faible dose de MTII (30ng) et étonnant de la température du iBAT avec la forte dose de MTII
(300ng), alors que la production de CO2 ne varie pas, laissant suggérer que le iBAT dissipe plus
de chaleur sans pour autant oxyder plus de substrats énergétiques. L’absence de potentialisation
du captage de C14-bromopalmitate et de H3-deoxyglucose vient supporter cette hypothèse. Enfin,
les expériences de mesure de la température et de captage de substrat du iBAT n’ont été réalisées
qu’avec la forte dose de MTII, celle-ci occasionnant un effet plateau, il ne nous est pas possible
de démontrer une potentialisation de l’utilisation des substrats énergétiques par un antagoniste du
système endocannabinoïde.
Notre étude s’est par la suite intéressée aux récepteurs impliqués dans la modulation du
système mélanocortine par le système endocannabinoïde. L’injection d’un antagoniste sélectif du
récepteur MC4R (HS024) a permis de mettre en évidence l’implication unique de MC4R dans les
effets positifs du MTII sur les différentes composantes de la thermogenèse citées précédemment.
En effet, l’administration de HS024 15 minutes avant le MTII bloque l’intégralité des effets de
celui-ci sur l’élévation, (i) de la température du iBAT, (ii) de la production de CO2 systémique et
(iii) du captage de substrats énergétiques par le iBAT, démontrant ainsi le rôle primordial de
MC4R au sein du système mélanocortine dans la régulation de l’homéostasie énergétique.
Parallèlement, on observe que l’administration concomitante de l’AM251 puis de Δ9-THC avant

155
le MTII n’entrave pas la potentialisation de la température du iBAT induite par l’AM251,
suggérant que les effets obtenus lors de l’injection du Δ9-THC sont relayés par le récepteur CB1.
Par ailleurs, la particularité du système endocannabinoïde qui consiste à exprimer ses récepteurs
au niveau pré-synaptique nous à permis d’envisager que la modulation opérée par CB1 ait lieu
en aval de l’activation MC4R, soit sur l’un des neurones relayant le message nerveux soit sur le
même neurone.
La seconde partie de nos résultats s’intéresse au rôle du PVH dans la modulation du
système mélanocortine par le système endocannabinoïde et s’appuie sur deux études menées en
collaboration avec notre laboratoire (Song et al., 2008; Richard et al., 2009). Celles-ci avaient
déjà mis l’emphase sur le PVH en démontrant que 80% neurones exprimant MC4R le constituant
sont connectées au iBAT (Song et al., 2008) et que l’expression de l’ARNm de CB1 y est très
largement retrouvé (Richard et al., 2009). Dans un premier temps, nos travaux
d’immunohistochimie dirigés contre la protéine MC4R, couplés à une hybridation in situ dirigée
contre l’ARNm de CB1 pour tenir compte de la spécificité du système endocannabinoïde, nous
ont permis de montrer que certains neurones du PVH expriment les deux récepteurs et donc
d’envisager que la modulation de l’activité du système mélanocortine par le système
endocannabinoïde pouvait avoir lieu dans ces neurones du PVH. De plus, la colocalisation dans
certains neurones du PVH de la protéine MC4R avec l’ARNm de Vglut2, un marqueur
phénotypique des neurones glutamatergiques, associé à la précocité de l’infection du PVH par un
virus rétrograde injecté dans le iBAT (Voss-Andreae et al., 2007) laissent supposer une
projection stimulatrice directe du PVH vers l’IML. Ensuite, d’un point de vue fonctionnel,
l’injection de MTII dans le PVH entraine une élévation de la température du iBAT comme cela
avait été observé lors d’une étude menée en collaboration avec notre laboratoire, mais aussi à une
augmentation (i) de la production de CO2, (ii) du captage de C14-bromopalmitate et (iii) de
l’expression de l’ARNm de Pgc1α et de Dio2, confirmant ainsi le rôle du PVH dans le contrôle
de la thermogenèse du iBAT. Enfin, l’injection de Δ9-THC dans le 4éme ventricule au plus près
de l’IML conduit à une inhibition totale de l’action du MTII dans le PVH, confirnant ainsi que
l’action des endocannabinoïdes sur l’activité des neurones MC4R du PVH a lieu en aval du PVH.

156
En conclusion, nos résultats affirment le rôle du système mélanocortine dans le contrôle de
la thermogenèse, dans lequel le récepteur MC4R a le rôle principal. Ensuite, ils mettent en
lumière la modulation du système mélanocortine par le système endocannabinoïde, avec le
récepteur CB1 comme acteur de premier plan. Enfin, ils mettent l’emphase sur la place du PVH
dans cette modulation.

157
Chapitre 2
La thermorégulation est contrôlée par un réseau neuronal complexe qui a été décrit dans
l’introduction. Au sein de celui-ci, le MPO occupe une place centrale en recevant les messages
thermiques en provenance de la peau, puis après les avoir intégrés grâce à des neurones
spécialisés dans la perception de la température, il émet des projections vers le DMH afin de
contrôler l’activité thermogène du iBAT (Morrison et al., 2014). D’autre part, une étude conduite
en collaboration avec notre laboratoire a montré que des neurones du MPO expriment des
récepteurs MC4R, et parmi ceux-ci plus de 75% sont connectés indirectement avec le iBAT
(Song et al., 2008). Enfin, il a été montré que l’injection d’un agoniste ou d’un antagoniste du
système mélanocortine dans le MPO, conduit respectivement à une hypophagie ou une
hyperphagie chez le rat (Kim et al., 2000). Il apparaissait donc évident que notre étude s’intéresse
aux rôles des neurones exprimant MC4R au sein du MPO dans le contrôle de la thermogenèse du
iBAT.
Notre étude est la première à notre connaissance à montrer que l’injection de MTII, un
agoniste MC4/3R, dans le MPO augmente la température du iBAT et la production de CO2
systémique. Néanmoins, plusieurs études avaient déjà suggéré que le POA dont fait partie le
MPO est impliqué dans le contrôle de la thermogenèse du iBAT. En effet, l’injection de CRH
(Cerri and Morrison, 2006) et de PGE2 (Madden and Morrison, 2005) dans le POA augmente la
température du iBAT, alors que l’injection d’un antagoniste GABAa inhibe la stimulation du
BAT évoquée par une baisse de la température de la peau (Nakamura and Morrison, 2007). Nos
travaux viennent aussi confirmer que le système mélanocortine joue un rôle déterminant dans la
régulation de la balance énergétique. En effet, il avait déjà été mis en évidence que l’injection
d’α-MSH et d’AgRP ont respectivement des effets hypophagiques et hyperphagiques (Kim et al.,
2000). Enfin, l’action du MTII dans le MPO sur la thermogenèse du iBAT se fait probablement à
travers l’activation de neurones exprimant le MC4R du MPO. Cette hypothèse est soutenue par la
colocalisation importante de MC4R avec le traceur rétrograde provenant du iBAT dans le MPO
(Song et al., 2008) et par l’inhibition des effets du MTII sur la thermogenèse lors de l’injection
i.c.v. de HS024, un antagoniste sélectif du MC4R (résultats observés dans le premier chapitre)

158
Les résultats décrits dans cet article montrent que lors de lésions du DMH obtenues par
l’injection d’acide kaïnique, le MPO perd sa capacité à stimuler la thermogenèse du iBAT suite à
l’injection de MTII, suggérant ainsi que le DMH est le relais majeur de l’action mélanocortinique
sur la thermogenèse dans le MPO. Nos résultats montrent que l’augmentation de la température
du iBAT suite à l’injection de MTII dans le MPO est dépendante du DMH mais que c’est aussi le
cas pour le captage du C14-bromopalmitate, un traceur permettant de mesurer l’entrée de NEFA
dans les cellules. Dans le cas de cette étude le captage du C14-bromopalmitate est corrélé
significativement avec la température du iBAT, ce qui en fait un bon indicateur de l’activité
thermogène du iBAT pour l’ensemble des groupes. De plus, le C14-bromopalmitate est
significativement corrélé avec l’expression génique de Hsl and Dgat2 dans le iBAT, faisant aussi
du C14-bromopalmitate un bon indicateur de l’oxydation des lipides (Ci et al., 2006; Oakes et al.,
2006; Ménard et al., 2010) et de leur estérification (Ménard et al., 2010). Fait intéressant,
l’administration de MTII augmente l’expression d’un certain nombre de gènes impliqués dans le
métabolisme des lipides et la thermogenèse, tels que Hsl (la lipolyse) et Dgat2 (la lipogenèse),
mais aussi Fatp1 (le captage des acides gras), Fas (la lipogenèse de novo) ou encore Pgc1α et
Dio2 (la thermogenèse) de manière dépendante du DMH. Enfin, nous n’observons aucun effet de
l’injection du MTII sur les niveaux d’expression de l’ARNm de Ucp1. Cependant, le délai entre
l’injection et le sacrifice de l’animal est trop court pour observer une augmentation. En effet,
l’étude précédente et Williams et ses collaborateurs montrent qu’il faut une stimulation de 24h
pour entrainer des variations de l’expression d’Ucp1 (Williams et al., 2003) (résultats décrits dans
le premier chapitre).
En parallèle, nous avons aussi observé que de façon dépendante du DMH, l’injection de
MTII dans le MPO avait des conséquences métaboliques sur d’autres tissus que le iBAT. En
effet, l’injection de MTII dans le MPO augmente le captage de C14-bromopalmitate dans le foie
et le tissu adipeux blanc inguinal, alors que chez les animaux ayant des lésions du DMH cet effet
est complètement aboli. Ces données confortent l’idée selon laquelle les récepteurs aux
mélanocortines du MPO et le duo MPO-DMH ont un rôle très large dans le métabolisme
énergétique (Morrison et al., 2012; Richard et al., 2012; Morrison et al., 2014). À la vue des
connaissances sur le iWAT, lequel est capable de développer des adipocytes bruns au sein du
tissu adipeux blanc (Bartelt and Heeren, 2014) et de sa forte connexion avec le POA à travers le
SNS (Song et al., 2009), nous avons choisi d’étudier les mêmes gènes que ceux analysés dans le

159
iBAT. Cependant, fait surprenant, les effets de stimulation du MTII ne sont observés que chez les
rats avec des lésions du DMH. En effet, chez ces rats, le MTII induit des gènes impliqués dans le
transport du glucose (Glut4), dans la lipogenèses (Gpat3, Dgat1 et Dgat2), dans la lipolyse (Hsl
et Atgl) et dans la thermogenèse et le métabolisme (Cpt1β). La différence dans les résultats entre
le iBAT et le iWAT sur l’expression génique n’est pas évidente à expliquer. Cependant, ces
résultats montrent l’importance du DMH dans la modulation de l’homéostasie énergétique.
Néanmoins, le DMH contient de nombreux neurones glutamatergiques excitateurs, qui sont l’un
des acteurs clés du contrôle de la thermogenèse du iBAT par le SNS (Morrison et al., 2008;
Nakamura and Morrison, 2008a; Morrison, 2011). Il semble donc raisonnable d’assumer que ces
neurones glutamatergiques sont affectés par l’injection de l’acide kaïnique (Bellinger and
Williams, 1983; Acosta-Galvan et al., 2011), ce qui pourrait donc inhiber l’élévation de la
température du iBAT induite par l’injection de MTII dans le MPO. D’un autre coté,
l’augmentation de l’expression des gènes comme Hsl, Atgl et Cpt1β observée dans le iWAT après
l’injection de MTII, seulement chez des rats ayant subi des lésions du DMH, suggère un
renforcement de l’activité métabolique du iWAT qui rappelle les effets de la délétion du gène
NPY dans le DMH (Bi and Li, 2013). En effet, il a été rapporté que la délétion du gène codant
pour NPY dans le DMH stimule le métabolisme et le brunissement du iWAT grâce au
développement d’adipocytes brun/beige (Chao et al., 2011). Tandis que la surexpression de NPY
dans les neurones du DMH est associée avec un excès d’accumulation de gras (Zheng et al.,
2013), de consommation de diète riche en gras (Lee et al., 2013b) et de lactation (Chen et al.,
2004b). La possibilité que les neurones exprimant le NPY soient altérés par l’acide kaïnique ne
peut être exclue. Cependant, d’autres études seront nécessaires pour vérifier cette hypothèse et
approfondir nos connaissances sur l’interaction complexe entre le MPO et le DMH qui gouverne
le métabolisme du iWAT.
En conclusion, ces résultats révèlent que le MPO est un site d’activation thermogène et
métabolique du MTII. Ils permettent aussi d’établir que le duo MPO-DMH est une cible
privilégiée pour l’action des mélanocortines sur l’homéostasie énergétique.


161
Perceptives de recherche et conclusion
Nos travaux ont mis en lumière le rôle du système mélanocortine hypothalamique dans le
contrôle de l’activité thermogène du iBAT et permettent d’envisager de nouvelles interactions
pharmacologiques et anatomiques dans la régulation de l’homéostasie énergétique. Cependant,
des études complémentaires pourraient être réalisées afin d’élaborer une stratégie
pharmacologique impliquant les systèmes aux mélanocortines et endocannabinoïde, et d’affiner
nos connaissances sur les connexions neuroanatomiques impliquées dans le contrôle du iBAT.
Dans le chapitre 1 nous avons montré que l’action du système mélanocortine sur l’activité
thermogène du iBAT est modulée par le système endocannabinoïde. Cependant, notre étude met
plus l’emphase sur l’inhibition des effets du système mélanocortine et n’approfondit pas la
potentialisation des effets du système mélanocortine par un antagoniste CB1. Il serait donc
intéressant de poursuivre cette étude en raffinant les doses de MTII et d’AM251 utilisées afin
d’obtenir un cocktail pharmacologique efficace sur la stimulation du iBAT et/ou la diminution de
la prise alimentaire comme l’ont montré Verty et ses collaborateurs en 2004 (Verty et al., 2004).
Néanmoins, l’utilisation d’un antagoniste du système endocannabinoïde comme piste de
recherche pour lutter contre l’obésité a déjà subi un revers important avec le Rimonabant à la fin
des années 2000 (Christensen et al., 2007; Buggy et al., 2011), et cela pourrait expliquer
l’absence de poursuite de recherche dans cette voie. Toutefois, le retour sur expérience découlant
de l’utilisation du Rimonabant à grande échelle nous permettrait d’élaborer des études
investiguant les effets de notre cocktail pharmacologique sur la dépression et ainsi prévenir un
nouvel échec de la thérapie pharmacologique dans la lutte contre l’obésité.
Toujours dans le chapitre 1, nous avons mis en avant le rôle du PVH dans la modulation de
l’action du système mélanocortine sur l’activité thermogène du iBAT par le système
endocannabinoïde. Nous avons aussi émis l’hypothèse que les endocannabinoïdes agissent au
niveau pré-synaptique des neurones du PVH projetant dans la moelle épinière. Cette hypothèse se
base sur nos travaux qui montrent que l’injection de Δ9-THC dans le 4éme ventricule inhibe les
effets induits par l’injection de MTII sur l’activité thermogène du iBAT et sur des travaux
réalisés par Voss-Andreae et ses collaborateurs qui montrent que lors de l’injection d’un traceur
rétrograde dans le iBAT le PVH est l’une des premières structures cérébrales à être marquées

162
(Voss-Andreae et al., 2007). Nous proposons donc une étude permettant de vérifier cette
hypothèse. Il nous semble que l’injection d’un traceur antérograde dans le PVH suivie d’une
colocalisation du traceur antérograde avec la protéine CB1 dans l’IML de la moelle épinière
permettrait d’éclaircir ce point.
Dans le chapitre 2, nous avons mis en évidence le rôle du système mélanocortine au sein du
MPO dans le contrôle de l’activité thermogène du iBAT ainsi que l’importance du relais DMH
dans cette stimulation du iBAT. Cependant, il serait intéressant de déterminer la nature des
neurones impliqués. Morisson et ses collaborateurs ont montré que les neurones qui participent à
la thermorégulation au sein du MPO et qui innervent le DMH sont de nature GABAergique
(Morrison et al., 2014). Néanmoins, dans notre contexte d’étude, il paraît plus probable qu’ils
soient glutamatergiques dans le cas d’une connexion directe entre le MPO et le DMH. Toutefois,
la présence d’interneurones GABAergiques ne peut être exclue. Donc, dans le but d’identifier la
nature des neurones impliqués, nous pourrions réaliser une colocalisation entre MC4R et un
marqueur phénotypique des neurones GABAergiques ou glutamatergiques, puis valider nos
observations histologiques par la mesure de la température du iBAT après injection de MTII dans
le MPO et d’un antagoniste des récepteurs GABA ou glutamate dans le DMH. Enfin, concernant
l’hypothèse émise dans la discussion du chapitre 2 sur l’implication de neurones exprimant NPY
dans les effets observés sur l’expression génique du iWAT, nous pourrions quantifier
l’expression de l’ARNm de NPY dans le DMH chez des animaux avec des lésions du DMH
avant d’aller plus loin. Nous pourrions aussi injecter un antagoniste des recepteur Y1/Y5 dans le
DMH afin de regarder le brunissement du iWAT.
Par ailleurs, il reste encore d’autres centres pouvant participer au contrôle mélanocortinique
du iBAT, tel que le MnPO ou le PAG ventrolatéral que l’on pourrait investiguer avec la méthode
de mesure de la température du iBAT développée au sein du laboratoire (Song et al., 2008).
En conclusion, la compréhension des réseaux neuronaux impliqués dans le contrôle du
BAT et de la prise alimentaire va nous permettre d’élaborer des thérapies pharmacologiques
ciblant plus efficacement les acteurs impliqués dans la régulation de l’homéostasie énergétique
tout en minimisant les effets secondaires. Cependant, la complexité des réseaux neuronaux
impliqués, la multipotence des systèmes neuropeptidiques et les échecs successifs des traitements
pharmacologiques de l’obésité (Adan, 2013) nous obligent à avoir un discours prudent sur la

163
possibilité à court ou moyen terme d’une issue pharmacologique à l’obésité vis à vis de la
société, et par conséquent encourager en tant que scientifique et citoyen la prévention et
l’éducation des saines habitudes de vie.


165
Références.
Abbott, C.R., Rossi, M., Wren, A.M., Murphy, K.G., Kennedy, A.R., Stanley, S.A.,
Zollner, A.N., Morgan, D.G., Morgan, I., Ghatei, M.A., et al. (2001). Evidence of an orexigenic
role for cocaine- and amphetamine-regulated transcript after administration into discrete
hypothalamic nuclei. Endocrinology 142, 3457–3463.
Acosta-Galvan, G., Yi, C.-X., van der Vliet, J., Jhamandas, J.H., Panula, P., Angeles-
Castellanos, M., Del Carmen Basualdo, M., Escobar, C., and Buijs, R.M. (2011). Interaction
between hypothalamic dorsomedial nucleus and the suprachiasmatic nucleus determines intensity
of food anticipatory behavior. Proc. Natl. Acad. Sci. U.S.a. 108, 5813–5818.
Adage, T., Scheurink, A.J., de Boer, S.F., de Vries, K., Konsman, J.P., Kuipers, F., Adan,
R.A., Baskin, D.G., Schwartz, M.W., and van Dijk, G. (2001). Hypothalamic, metabolic, and
behavioral responses to pharmacological inhibition of CNS melanocortin signaling in rats. J.
Neurosci. 21, 3639–3645.
Adan, R.A.H. (2013). Mechanisms underlying current and future anti-obesity drugs. Trends
Neurosci. 36, 133–140.
Ahima, R.S., and Osei, S.Y. (2004). Leptin signaling. Physiol. Behav. 81, 223–241.
Ameri, A. (1999). The effects of cannabinoids on the brain. Prog. Neurobiol. 58, 315–348.
ANAND, B.K., and BROBECK, J.R. (1951). Hypothalamic control of food intake in rats
and cats. Yale J Biol Med 24, 123–140.
Anunciado-Koza, R., Ukropec, J., Koza, R.A., and Kozak, L.P. (2008). Inactivation of
UCP1 and the glycerol phosphate cycle synergistically increases energy expenditure to resist
diet-induced obesity. J. Biol. Chem. 283, 27688–27697.
Arletti, R., Benelli, A., and Bertolini, A. (1989). Influence of oxytocin on feeding behavior
in the rat. Peptides 10, 89–93.

166
Asakawa, A., Inui, A., Kaga, T., Yuzuriha, H., Nagata, T., Ueno, N., Makino, S., Fujimiya,
M., Niijima, A., Fujino, M.A., et al. (2001). Ghrelin is an appetite-stimulatory signal from
stomach with structural resemblance to motilin. Gastroenterology 120, 337–345.
Atalayer, D., Robertson, K.L., Haskell-Luevano, C., Andreasen, A., and Rowland, N.E.
(2010). Food demand and meal size in mice with single or combined disruption of melanocortin
type 3 and 4 receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1667–74.
Atit, R., Sgaier, S.K., Mohamed, O.A., Taketo, M.M., Dufort, D., Joyner, A.L., Niswander,
L., and Conlon, R.A. (2006). Beta-catenin activation is necessary and sufficient to specify the
dorsal dermal fate in the mouse. Dev. Biol. 296, 164–176.
Badami, V.M., Rice, C.D., Lois, J.H., Madrecha, J., and Yates, B.J. (2010). Distribution of
hypothalamic neurons with orexin (hypocretin) or melanin concentrating hormone (MCH)
immunoreactivity and multisynaptic connections with diaphragm motoneurons. Brain Res. 1323,
119–126.
Badman, M.K., and Flier, J.S. (2005). The gut and energy balance: visceral allies in the
obesity wars. Science 307, 1909–1914.
Balthasar, N., Dalgaard, L., Lee, C., Yu, J., and Funahashi, H. (2005). Divergence of
melanocortin pathways in the control of food intake and energy expenditure. Cell.
Bamshad, M., Song, C.K., and Bartness, T.J. (1999). CNS origins of the sympathetic
nervous system outflow to brown adipose tissue. Am. J. Physiol. 276, R1569–78.
Banet, M. (1986). Fever in mammals: is it beneficial? Yale J Biol Med 59, 117.
Bannon, A.W., Seda, J., Carmouche, M., Francis, J.M., Norman, M.H., Karbon, B., and
McCaleb, M.L. (2000). Behavioral characterization of neuropeptide Y knockout mice. Brain Res.
868, 79–87.
Bartelt, A., and Heeren, J. (2014). Adipose tissue browning and metabolic health. Nat Rev
Endocrinol 10, 24–36.

167
Batterham, R.L., Cowley, M.A., Small, C.J., Herzog, H., Cohen, M.A., Dakin, C.L., Wren,
A.M., Brynes, A.E., Low, M.J., Ghatei, M.A., et al. (2002). Gut hormone PYY(3-36)
physiologically inhibits food intake. Nature 418, 650–654.
Bellinger, L.L., and Williams, F.E. (1983). Aphagia and adipsia after kainic acid lesioning
of the dorsomedial hypothalamic area. Am. J. Physiol. 244, R389–99.
Benoit, S.C., Air, E.L., Coolen, L.M., Strauss, R., Jackman, A., Clegg, D.J., Seeley, R.J.,
and Woods, S.C. (2002). The catabolic action of insulin in the brain is mediated by
melanocortins. J. Neurosci. 22, 9048–9052.
Berthoud, H.-R. (2002). Multiple neural systems controlling food intake and body weight.
Neurosci Biobehav Rev 26, 393–428.
Berthoud, H.R., and Mogenson, G.J. (1977). Ingestive behavior after intracerebral and
intracerebroventricular infusions of glucose and 2-deoxy-D-glucose. Am. J. Physiol. 233, R127–
33.
Berthoud, H.R., Lynn, P.A., and Blackshaw, L.A. (2001). Vagal and spinal
mechanosensors in the rat stomach and colon have multiple receptive fields. Am. J. Physiol.
Regul. Integr. Comp. Physiol. 280, R1371–81.
Bi, S., and Li, L. (2013). Browning of white adipose tissue: role of hypothalamic signaling.
Ann. N. Y. Acad. Sci. 1302, 30–34.
Bi, S., Kim, Y.J., and Zheng, F. (2012). Dorsomedial hypothalamic NPY and energy
balance control. Neuropeptides 46, 309–314.
Biebermann, H., Kühnen, P., Kleinau, G., and Krude, H. (2012). The neuroendocrine
circuitry controlled by POMC, MSH, and AGRP. Handb Exp Pharmacol 47–75.
Billon, N., and Dani, C. (2012). Developmental origins of the adipocyte lineage: new
insights from genetics and genomics studies. Stem Cell Rev 8, 55–66.

168
Bjorbak, C., Lavery, H.J., Bates, S.H., Olson, R.K., Davis, S.M., Flier, J.S., and Myers,
M.G. (2000). SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol.
Chem. 275, 40649–40657.
Blevins, J.E., Eakin, T.J., Murphy, J.A., Schwartz, M.W., and Baskin, D.G. (2003).
Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin. Brain Res. 993,
30–41.
Blouet, C., and Schwartz, G.J. (2010). Hypothalamic nutrient sensing in the control of
energy homeostasis. Behav. Brain Res. 209, 1–12.
Blouet, C., and Schwartz, G.J. (2012). Brainstem Nutrient Sensing in the Nucleus of the
Solitary Tract Inhibits Feeding. Cell Metab. 16, 579–587.
Blouet, C., Jo, Y.-H., Li, X., and Schwartz, G.J. (2009). Mediobasal hypothalamic leucine
sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J. Neurosci.
29, 8302–8311.
Bordicchia, M., Liu, D., Amri, E.-Z., Ailhaud, G., Dessì-Fulgheri, P., Zhang, C.,
Takahashi, N., Sarzani, R., and Collins, S. (2012). Cardiac natriuretic peptides act via p38 MAPK
to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest.
122, 1022–1036.
Boström, P., Wu, J., Jedrychowski, M.P., Korde, A., Ye, L., Lo, J.C., Rasbach, K.A.,
Boström, E.A., Choi, J.H., Long, J.Z., et al. (2012). A PGC1-α-dependent myokine that drives
brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468.
Boulant, J.A. (2006). Counterpoint: Heat-induced membrane depolarization of
hypothalamic neurons: an unlikely mechanism of central thermosensitivity. Am. J. Physiol.
Regul. Integr. Comp. Physiol. 290, R1481–4; discussion R1484.
Branchek, T.A., Smith, K.E., Gerald, C., and Walker, M.W. (2000). Galanin receptor
subtypes. Trends Pharmacol. Sci. 21, 109–117.

169
Bratincsák, A., and Palkovits, M. (2004). Activation of brain areas in rat following warm
and cold ambient exposure. Neuroscience 127, 385–397.
Brito, M.N., Brito, N.A., Baro, D.J., Song, C.K., and Bartness, T.J. (2007). Differential
activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation.
Endocrinology 148, 5339–5347.
Broberger, C., De Lecea, L., Sutcliffe, J.G., and Hökfelt, T. (1998a). Hypocretin/orexin-
and melanin-concentrating hormone-expressing cells form distinct populations in the rodent
lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems.
J. Comp. Neurol. 402, 460–474.
Broberger, C., Johansen, J., Johansson, C., Schalling, M., and Hökfelt, T. (1998b). The
neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and
monosodium glutamate-treated mice. Proc. Natl. Acad. Sci. U.S.a. 95, 15043–15048.
Buchanan, T.A., Cane, P., Eng, C.C., Sipos, G.F., and Lee, C. (1991). Hypothermia is
critical for survival during prolonged insulin-induced hypoglycemia in rats. Metab. Clin. Exp. 40,
330–334.
Buggy, Y., Cornelius, V., Wilton, L., and Shakir, S. (2011). Risk of Depressive Episodes
with Rimonabant - Springer. Drug Safety.
Bultman, S.J., Michaud, E.J., and Woychik, R.P. (1992). Molecular characterization of the
mouse agouti locus. Cell 71, 1195–1204.
Butler, A.A. (2006). The melanocortin system and energy balance. Peptides 27, 281–290.
Butler, A.A., and Cone, R.D. (2002). The melanocortin receptors: lessons from knockout
models. Neuropeptides 36, 77–84.
Butler, A.A., Kesterson, R.A., Khong, K., Cullen, M.J., Pelleymounter, M.A., Dekoning, J.,
Baetscher, M., and Cone, R.D. (2000). A unique metabolic syndrome causes obesity in the
melanocortin-3 receptor-deficient mouse. Endocrinology 141, 3518–3521.

170
Butler, A.A., Marks, D.L., Fan, W., Kuhn, C.M., Bartolome, M., and Cone, R.D. (2001).
Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat.
Neurosci. 4, 605–611.
Campfield, L.A., Brandon, P., and Smith, F.J. (1985). On-line continuous measurement of
blood glucose and meal pattern in free-feeding rats: the role of glucose in meal initiation. Brain
Res. Bull. 14, 605–616.
Cannon, B., and Nedergaard, J. (2004). Brown adipose tissue: function and physiological
significance. Physiol. Rev. 84, 277–359.
Cannon, B., and Nedergaard, J. (2012). Cell biology: Neither brown nor white. Nature 488,
286–287.
Cano, G., Passerin, A.M., Schiltz, J.C., Card, J.P., Morrison, S.F., and Sved, A.F. (2003).
Anatomical substrates for the central control of sympathetic outflow to interscapular adipose
tissue during cold exposure. J. Comp. Neurol. 460, 303–326.
Cao, W., Daniel, K.W., Robidoux, J., Puigserver, P., Medvedev, A.V., Bai, X., Floering,
L.M., Spiegelman, B.M., and Collins, S. (2004a). p38 mitogen-activated protein kinase is the
central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1
gene. Mol. Cell. Biol. 24, 3057–3067.
Cao, W.-H., and Morrison, S.F. (2005). Brown adipose tissue thermogenesis contributes to
fentanyl-evoked hyperthermia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R723–32.
Cao, W.-H., Fan, W., and Morrison, S.F. (2004b). Medullary pathways mediating specific
sympathetic responses to activation of dorsomedial hypothalamus. Neuroscience 126, 229–240.
Cao, W.-H., Madden, C.J., and Morrison, S.F. (2010). Inhibition of brown adipose tissue
thermogenesis by neurons in the ventrolateral medulla and in the nucleus tractus solitarius. Am.
J. Physiol. Regul. Integr. Comp. Physiol. 299, R277–90.
Carey, A.L., and Kingwell, B.A. (2013). Brown adipose tissue in humans: Therapeutic
potential to combat obesity. Pharmacol. Ther. 140, 26–33.

171
Carter, S., Caron, A., Richard, D., and Picard, F. (2013). Role of letin resistance in the
development of obesity in older patients. Clin. Interv. Aging. 8, 829–44.
Cerdá-Reverter, J.M., Agulleiro, M.J., Cortés, R., Sánchez, E., Guillot, R., Leal, E.,
Fernández-Durán, B., Puchol, S., and Eley, M. (2013). Involvement of melanocortin receptor
accessory proteins (MRAPs) in the function of melanocortin receptors. Gen. Comp. Endocrinol.
188, 133–136.
Cerri, M., and Morrison, S.F. (2005). Activation of lateral hypothalamic neurons stimulates
brown adipose tissue thermogenesis. Neuroscience 135, 627–638.
Cerri, M., and Morrison, S.F. (2006). Corticotropin releasing factor increases in brown
adipose tissue thermogenesis and heart rate through dorsomedial hypothalamus and medullary
raphe pallidus. Neuroscience 140, 711–721.
Challis, B.G., Coll, A.P., Yeo, G.S.H., Pinnock, S.B., Dickson, S.L., Thresher, R.R., Dixon,
J., Zahn, D., Rochford, J.J., White, A., et al. (2004). Mice lacking pro-opiomelanocortin are
sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-
36). Proc. Natl. Acad. Sci. U.S.a. 101, 4695–4700.
Chance, W.T., Thompson, H., Thomas, I., and Fischer, J.E. (1995). Anorectic and
neurochemical effects of pituitary adenylate cyclase activating polypeptide in rats. Peptides 16,
1511–1516.
Chao, P.-T., Yang, L., Aja, S., Moran, T.H., and Bi, S. (2011). Knockdown of NPY
expression in the dorsomedial hypothalamus promotes development of brown adipocytes and
prevents diet-induced obesity. Cell Metab. 13, 573–583.
Chartoumpekis, D.V., Habeos, I.G., Ziros, P.G., Psyrogiannis, A.I., Kyriazopoulou, V.E.,
and Papavassiliou, A.G. (2011). Brown adipose tissue responds to cold and adrenergic
stimulation by induction of FGF21. Mol. Med. 17, 736–740.
Chechi, K., Carpentier, A.C., and Richard, D. (2013). Understanding the brown adipocyte
as a contributor to energy homeostasis. Trends in Endocrinology & Metabolism.

172
Chen, A.S., Marsh, D.J., Trumbauer, M.E., Frazier, E.G., Guan, X.-M., Yu, H., Rosenblum,
C.I., Vongs, A., Feng, Y., Cao, L., et al. (2000a). Inactivation of the mouse melanocortin-3
receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 26, 97–102.
Chen, A.S., Metzger, J.M., Trumbauer, M.E., Guan, X.-M., Yu, H., Frazier, E.G., Marsh,
D.J., Forrest, M.J., Gopal-Truter, S., Fisher, J., et al. (2000b). Role of the melanocortin-4 receptor
in metabolic rate and food intake in mice. Transgenic Res. 9, 145–154.
Chen, H.Y., Liu, Q., Salter, A.M., and Lomax, M.A. (2013). Synergism between cAMP
and PPARγ Signalling in the Initiation of UCP1 Gene Expression in HIB1B Brown Adipocytes.
PPAR Res 2013, 476049.
Chen, H.Y., Trumbauer, M.E., Chen, A.S., Weingarth, D.T., Adams, J.R., Frazier, E.G.,
Shen, Z., Marsh, D.J., Feighner, S.D., Guan, X.-M., et al. (2004a). Orexigenic action of
peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology 145,
2607–2612.
Chen, P., Williams, S.M., Grove, K.L., and Smith, M.S. (2004b). Melanocortin 4 receptor-
mediated hyperphagia and activation of neuropeptide Y expression in the dorsomedial
hypothalamus during lactation. J. Neurosci. 24, 5091–5100.
Chen, W., Kelly, M.A., Opitz-Araya, X., Thomas, R.E., Low, M.J., and Cone, R.D. (1997).
Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of
exocrine gland function by melanocortin peptides. Cell 91, 789–798.
Chen, X.M., Hosono, T., Yoda, T., Fukuda, Y., and Kanosue, K. (1998). Efferent
projection from the preoptic area for the control of non-shivering thermogenesis in rats. J.
Physiol. (Lond.) 512 ( Pt 3), 883–892.
Cheung, C.C., Clifton, D.K., and Steiner, R.A. (1997). Proopiomelanocortin neurons are
direct targets for leptin in the hypothalamus. Endocrinology 138, 4489–4492.
Chhajlani, V., Muceniece, R., and Wikberg, J.E. (1993). Molecular cloning of a novel
human melanocortin receptor. Biochem. Biophys. Res. Commun. 195, 866–873.

173
Choy, V.J., and Watkins, W.B. (1977). Immunocytochemical study of the hypothalamo-
neurohypophysial system. II. Distribution of neurophysin, vasopressin and oxytocin in the normal
and osmotically stimulated rat. Cell Tissue Res. 180, 467–490.
Christensen, R., Kristensen, P., Bartels, E., and Bliddal, H. (2007). Efficacy and safety of
the weight-loss drug rimonabant: a meta-analysis of randomised trials. The Lancet.
Ci, X., Frisch, F., Lavoie, F., Germain, P., Lecomte, R., van Lier, J.E., Bénard, F., and
Carpentier, A.C. (2006). The effect of insulin on the intracellular distribution of 14(R,S)-
[18F]Fluoro-6-thia-heptadecanoic acid in rats. Mol Imaging Biol 8, 237–244.
Cinti, S. (2009). Transdifferentiation properties of adipocytes in the adipose organ. Am. J.
Physiol. Endocrinol. Metab. 297, E977–86.
Cinti, S. (2012). The adipose organ at a glance. Dis Model Mech 5, 588–594.
Clapham, J.C. (2012). Central control of thermogenesis. Neuropharmacology 63, 111–123.
Collier, G.R., McMillan, J.S., Windmill, K., Walder, K., Tenne-Brown, J., de Silva, A.,
Trevaskis, J., Jones, S., Morton, G.J., Lee, S., et al. (2000). Beacon: a novel gene involved in the
regulation of energy balance. Diabetes 49, 1766–1771.
Collins, S. (2011). β-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored
Fat and Energy Expenditure. Front Endocrinol (Lausanne) 2, 102.
Collins, S., Yehuda-Shnaidman, E., and Wang, H. (2010). Positive and negative control of
Ucp1 gene transcription and the role of β-adrenergic signaling networks. Int J Obes (Lond) 34
Suppl 1, S28–33.
Cone, R. (1999). The Central Melanocortin System and Energy Homeostasis. Trends
Endocrinol. Metab. 10, 211–216.
Corchero, J., Fuentes, J.A., and Manzanares, J. (1997). delta 9-Tetrahydrocannabinol
increases proopiomelanocortin gene expression in the arcuate nucleus of the rat hypothalamus.
Eur. J. Pharmacol. 323, 193–195.

174
Corchero, J., Manzanares, J., and Fuentes, J.A. (1999). Repeated administration of delta9-
tetrahydrocannabinol produces a differential time related responsiveness on proenkephalin,
proopiomelanocortin and corticotropin releasing factor gene expression in the hypothalamus and
pituitary gland of the rat. Neuropharmacology 38, 433–439.
Cota, D., Proulx, K., Smith, K.A.B., Kozma, S.C., Thomas, G., Woods, S.C., and Seeley,
R.J. (2006). Hypothalamic mTOR signaling regulates food intake. Science 312, 927–930.
Cowley, M.A., Pronchuk, N., Fan, W., Dinulescu, D.M., Colmers, W.F., and Cone, R.D.
(1999). Integration of NPY, AGRP, and Melanocortin Signals in the Hypothalamic
Paraventricular Nucleus. Neuron 24, 155–163.
Cowley, M.A., Smart, J.L., Rubinstein, M., Cerdán, M.G., Diano, S., Horvath, T.L., Cone,
R.D., and Low, M.J. (2001). Leptin activates anorexigenic POMC neurons through a neural
network in the arcuate nucleus. Nature 411, 480–484.
Craig, A.D. (2002). How do you feel? Interoception: the sense of the physiological
condition of the body. Nat. Rev. Neurosci. 3, 655–666.
Craig, A.D., Krout, K., and Andrew, D. (2001). Quantitative response characteristics of
thermoreceptive and nociceptive lamina I spinothalamic neurons in the cat. J. Neurophysiol. 86,
1459–1480.
Currie, P.J., Chang, N., Luo, S., and Anderson, G.H. (1995). Microdialysis as a tool to
measure dietary and regional effects on the complete profile of extracellular amino acids in the
hypothalamus of rats. Life Sci. 57, 1911–1923.
Currie, P.J., Coiro, C.D., Niyomchai, T., Lira, A., and Farahmand, F. (2002). Hypothalamic
paraventricular 5-hydroxytryptamine: receptor-specific inhibition of NPY-stimulated eating and
energy metabolism. Pharmacol. Biochem. Behav. 71, 709–716.
Cypess, A.M., Lehman, S., Williams, G., Tal, I., Rodman, D., Goldfine, A.B., Kuo, F.C.,
Palmer, E.L., Tseng, Y.-H., Doria, A., et al. (2009). Identification and importance of brown
adipose tissue in adult humans. N Engl J Med 360, 1509–1517.

175
de Jesus, L.A., Carvalho, S.D., Ribeiro, M.O., Schneider, M., Kim, S.-W., Harney, J.W.,
Larsen, P.R., and Bianco, A.C. (2001). The type 2 iodothyronine deiodinase is essential for
adaptive thermogenesis in brown adipose tissue. J. Clin. Invest. 108, 1379–1385.
de Menezes, R.C.A., Zaretsky, D.V., Fontes, M.A.P., and Dimicco, J.A. (2009).
Cardiovascular and thermal responses evoked from the periaqueductal grey require neuronal
activity in the hypothalamus. J. Physiol. (Lond.) 587, 1201–1215.
De Silva, A., Salem, V., Long, C.J., Makwana, A., Newbould, R.D., Rabiner, E.A., Ghatei,
M.A., Bloom, S.R., Matthews, P.M., Beaver, J.D., et al. (2011). The gut hormones PYY 3-36 and
GLP-1 7-36 amide reduce food intake and modulate brain activity in appetite centers in humans.
Cell Metab. 14, 700–706.
Debevec, D., Christian, M., Morganstein, D., Seth, A., Herzog, B., Parker, M., and White,
R. (2007). Receptor interacting protein 140 regulates expression of uncoupling protein 1 in
adipocytes through specific peroxisome proliferator activated receptor isoforms and estrogen-
related receptor alpha. Mol. Endocrinol. 21, 1581–1592.
DeFalco, J., Tomishima, M., Liu, H., Zhao, C., Cai, X., Marth, J.D., Enquist, L., and
Friedman, J.M. (2001). Virus-assisted mapping of neural inputs to a feeding center in the
hypothalamus. Science 291, 2608–2613.
Deuchars, S.A., Milligan, C.J., Stornetta, R.L., and Deuchars, J. (2005). GABAergic
neurons in the central region of the spinal cord: a novel substrate for sympathetic inhibition. J.
Neurosci. 25, 1063–1070.
Devane, W., Hanus, L., Breuer, A., Pertwee, R., Stevenson, L., Griffin, G., Gibson, D.,
Mandelbaum, A., Etinger, A., and Mechoulam, R. (1992). Isolation and structure of a brain
constituent that binds to the cannabinoid receptor. Science 258, 1946–1949.
Dhaka, A., Murray, A.N., Mathur, J., Earley, T.J., Petrus, M.J., and Patapoutian, A. (2007).
TRPM8 is required for cold sensation in mice. Neuron 54, 371–378.
Dhillo, W.S. (2007). Appetite regulation: an overview. Thyroid 17, 433–445.

176
Dhillo, W.S., Small, C.J., Jethwa, P.H., Russell, S.H., Gardiner, J.V., Bewick, G.A., Seth,
A., Murphy, K.G., Ghatei, M.A., and Bloom, S.R. (2003). Paraventricular nucleus administration
of calcitonin gene-related peptide inhibits food intake and stimulates the hypothalamo-pituitary-
adrenal axis. Endocrinology 144, 1420–1425.
Divakaruni, A.S., and Brand, M.D. (2011). The regulation and physiology of mitochondrial
proton leak. Physiology (Bethesda) 26, 192–205.
do Rego, J.-C., Leprince, J., Chartrel, N., Vaudry, H., and Costentin, J. (2006). Behavioral
effects of 26RFamide and related peptides. Peptides 27, 2715–2721.
do Rego, J.-C., Orta, M.-H., Leprince, J., Tonon, M.-C., Vaudry, H., and Costentin, J.
(2007). Pharmacological characterization of the receptor mediating the anorexigenic action of the
octadecaneuropeptide: evidence for an endozepinergic tone regulating food intake.
Neuropsychopharmacology 32, 1641–1648.
Dourish, C.T. (1995). Multiple serotonin receptors: opportunities for new treatments for
obesity? Obes. Res. 3 Suppl 4, 449S–462S.
Drucker, D.J. (2005). Biologic actions and therapeutic potential of the proglucagon-derived
peptides. Nat Clin Pract Endocrinol Metab 1, 22–31.
Dumont, Y., Fournier, A., St-Pierre, S., and Quirion, R. (1995). Characterization of
neuropeptide Y binding sites in rat brain membrane preparations using
[125I][Leu31,Pro34]peptide YY and [125I]peptide YY3-36 as selective Y1 and Y2 radioligands.
J. Pharmacol. Exp. Ther. 272, 673–680.
Egawa, M., Yoshimatsu, H., and Bray, G.A. (1991). Neuropeptide Y suppresses
sympathetic activity to interscapular brown adipose tissue in rats. Am. J. Physiol. 260, R328–34.
Elias, C.F., Aschkenasi, C., Lee, C., Kelly, J., Ahima, R.S., Bjørbaek, C., Flier, J.S., Saper,
C.B., and Elmquist, J.K. (1999). Leptin differentially regulates NPY and POMC neurons
projecting to the lateral hypothalamic area. Neuron 23, 775–786.

177
Elias, C.F., Lee, C., Kelly, J., Aschkenasi, C., Ahima, R.S., Couceyro, P.R., Kuhar, M.J.,
Saper, C.B., and Elmquist, J.K. (1998a). Leptin activates hypothalamic CART neurons projecting
to the spinal cord. Neuron 21, 1375–1385.
Elias, C.F., Saper, C.B., Maratos-Flier, E., Tritos, N.A., Lee, C., Kelly, J., Tatro, J.B.,
Hoffman, G.E., Ollmann, M.M., Barsh, G.S., et al. (1998b). Chemically defined projections
linking the mediobasal hypothalamus and the lateral hypothalamic area. J. Comp. Neurol. 402,
442–459.
Ellacott, K.L.J., and Cone, R.D. (2006). The role of the central melanocortin system in the
regulation of food intake and energy homeostasis: lessons from mouse models. Philos. Trans. R.
Soc. Lond., B, Biol. Sci. 361, 1265–1274.
Elmquist, J., and Scammell, T. (1996). Distribution of fos-like immunoreactivity in the rat
brain following intravenous lipopolysaccharide administration - Elmquist - 1998 - Journal of
Comparative Neurology - Wiley Online Library. Journal of ….
Enerbäck, S. (2010a). Brown adipose tissue in humans. Int J Obes (Lond) 34, S43–S46.
Enerbäck, S. (2010b). Human brown adipose tissue. Cell Metab. 11, 248–252.
Enerbäck, S., Jacobsson, A., Simpson, E.M., Guerra, C., Yamashita, H., Harper, M.E., and
Kozak, L.P. (1997). Mice lacking mitochondrial uncoupling protein are cold-sensitive but not
obese. Nature 387, 90–94.
Erickson, J.C., Clegg, K.E., and Palmiter, R.D. (1996). Sensitivity to leptin and
susceptibility to seizures of mice lacking neuropeptide Y. Nature 381, 415–421.
Fan, W., Boston, B.A., Kesterson, R.A., Hruby, V.J., and Cone, R.D. (1997). Role of
melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168.
Fan, W., Morrison, S.F., Cao, W.-H., and Yu, P. (2007). Thermogenesis activated by
central melanocortin signaling is dependent on neurons in the rostral raphe pallidus (rRPa) area.
Brain Res. 1179, 61–69.

178
Fan, W., Voss-Andreae, A., Cao, W.-H., and Morrison, S.F. (2005). Regulation of
thermogenesis by the central melanocortin system. Peptides 26, 1800–1813.
Farooqi, S., and O'Rahilly, S. (2006). Genetics of obesity in humans. Endocr. Rev. 27, 710–
718.
Fedorenko, A., Lishko, P.V., and Kirichok, Y. (2012). Mechanism of fatty-acid-dependent
UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413.
Fekete, C., Légrádi, G., Mihály, E., Huang, Q.H., Tatro, J.B., Rand, W.M., Emerson, C.H.,
and Lechan, R.M. (2000). alpha-Melanocyte-stimulating hormone is contained in nerve terminals
innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic
paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing
hormone gene expression. J. Neurosci. 20, 1550–1558.
Fekete, C., Marks, D.L., Sarkar, S., Emerson, C.H., Rand, W.M., Cone, R.D., and Lechan,
R.M. (2004). Effect of Agouti-related protein in regulation of the hypothalamic-pituitary-thyroid
axis in the melanocortin 4 receptor knockout mouse. Endocrinology 145, 4816–4821.
Feldmann, H.M., Golozoubova, V., Cannon, B., and Nedergaard, J. (2009). UCP1 ablation
induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by
living at thermoneutrality. Cell Metab. 9, 203–209.
Ferré, P., Leturque, A., Burnol, A.F., Pénicaud, L., and Girard, J. (1985). A method to
quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the
anaesthetized rat. Biochem. J. 228, 103–110.
Fisher, F.M., Kleiner, S., Douris, N., Fox, E.C., Mepani, R.J., Verdeguer, F., Wu, J.,
Kharitonenkov, A., Flier, J.S., Maratos-Flier, E., et al. (2012). FGF21 regulates PGC-1α and
browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281.
Fong, T.M., Mao, C., MacNeil, T., Kalyani, R., Smith, T., Weinberg, D., Tota, M.R., and
Van der Ploeg, L.H. (1997). ART (protein product of agouti-related transcript) as an antagonist of
MC-3 and MC-4 receptors. Biochem. Biophys. Res. Commun. 237, 629–631.

179
Freinkel, N., Metzger, B.E., Harris, E., Robinson, S., and Mager, M. (1972). The
hypothermia of hypoglycemia. Studies with 2-deoxy-D-glucose in normal human subjects and
mice. N Engl J Med 287, 841–845.
Freychet, P., Roth, J., and Neville, D.M. (1971). Insulin receptors in the liver: specific
binding of ( 125 I)insulin to the plasma membrane and its relation to insulin bioactivity. Proc.
Natl. Acad. Sci. U.S.a. 68, 1833–1837.
Friedman, J.M., and Halaas, J.L. (1998). Leptin and the regulation of body weight in
mammals. Nature 395, 763–770.
Funahashi, H., Yamada, S., Kageyama, H., Takenoya, F., Guan, J.L., and Shioda, S.
(2003). Co-existence of leptin- and orexin-receptors in feeding-regulating neurons in the
hypothalamic arcuate nucleus-a triple labeling study. Peptides 24, 687–694.
Gantz, I., Konda, Y., Tashiro, T., Shimoto, Y., Miwa, H., Munzert, G., Watson, S.J.,
DelValle, J., and Yamada, T. (1993a). Molecular cloning of a novel melanocortin receptor. J.
Biol. Chem. 268, 8246–8250.
Gantz, I., Miwa, H., Konda, Y., Shimoto, Y., Tashiro, T., Watson, S.J., DelValle, J., and
Yamada, T. (1993b). Molecular cloning, expression, and gene localization of a fourth
melanocortin receptor. J. Biol. Chem. 268, 15174–15179.
Gao, Q., Wolfgang, M.J., Neschen, S., Morino, K., Horvath, T.L., Shulman, G.I., and Fu,
X.-Y. (2004). Disruption of neural signal transducer and activator of transcription 3 causes
obesity, diabetes, infertility, and thermal dysregulation. Proc. Natl. Acad. Sci. U.S.a. 101, 4661–
4666.
Gaoni, Y., and Mechoulam, R. (1964). Isolation, Structure, and Partial Synthesis of an
Active Constituent of Hashish. J. Am. Chem. Soc. 86, 1646–1647.
Garfield, A.S., Lam, D.D., Marston, O.J., Przydzial, M.J., and Heisler, L.K. (2009). Role of
central melanocortin pathways in energy homeostasis. Trends Endocrinol. Metab. 20, 203–215.

180
Gelez, H., Poirier, S., Facchinetti, P., Allers, K.A., Wayman, C., Bernabé, J., Alexandre, L.,
and Giuliano, F. (2010). Neuroanatomical distribution of the melanocortin-4 receptors in male
and female rodent brain. J. Chem. Neuroanat. 40, 310–324.
Gibbs, J., Young, R.C., and Smith, G.P. (1973). Cholecystokinin elicits satiety in rats with
open gastric fistulas. Nature 245, 323–325.
Gimeno, R.E., and Cao, J. (2008). Thematic review series: glycerolipids. Mammalian
glycerol-3-phosphate acyltransferases: new genes for an old activity. J. Lipid Res. 49, 2079–
2088.
Golay, A., Schutz, Y., Meyer, H.U., Thiébaud, D., Curchod, B., Maeder, E., Felber, J.P.,
and Jéquier, E. (1982). Glucose-induced thermogenesis in nondiabetic and diabetic obese
subjects. Diabetes 31, 1023–1028.
Grandt, D., Schimiczek, M., Beglinger, C., Layer, P., Goebell, H., Eysselein, V.E., and
Reeve, J.R. (1994). Two molecular forms of peptide YY (PYY) are abundant in human blood:
characterization of a radioimmunoassay recognizing PYY 1-36 and PYY 3-36. Regul. Pept. 51,
151–159.
Granger, A., and Kushner, J.A. (2009). Cellular origins of beta-cell regeneration: a legacy
view of historical controversies. J. Intern. Med. 266, 325–338.
Gu, W., Geddes, B.J., Zhang, C., Foley, K.P., and Stricker-Krongrad, A. (2004). The
prolactin-releasing peptide receptor (GPR10) regulates body weight homeostasis in mice. J. Mol.
Neurosci. 22, 93–103.
Hagan, M.M., Rushing, P.A., Pritchard, L.M., Schwartz, M.W., Strack, A.M., Van der
Ploeg, L.H., Woods, S.C., and Seeley, R.J. (2000). Long-term orexigenic effects of AgRP-(83---
132) involve mechanisms other than melanocortin receptor blockade. Am. J. Physiol. Regul.
Integr. Comp. Physiol. 279, R47–52.
Hahm, S., Fekete, C., Mizuno, T.M., Windsor, J., Yan, H., Boozer, C.N., Lee, C., Elmquist,
J.K., Lechan, R.M., Mobbs, C.V., et al. (2002). VGF is required for obesity induced by diet, gold

181
thioglucose treatment, and agouti and is differentially regulated in pro-opiomelanocortin- and
neuropeptide Y-containing arcuate neurons in response to fasting. J. Neurosci. 22, 6929–6938.
Hahn, T.M., Breininger, J.F., Baskin, D.G., and Schwartz, M.W. (1998). Coexpression of
Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1, 271–272.
Hanada, R., Nakazato, M., Matsukura, S., Murakami, N., Yoshimatsu, H., and Sakata, T.
(2000). Differential regulation of melanin-concentrating hormone and orexin genes in the agouti-
related protein/melanocortin-4 receptor system. Biochem. Biophys. Res. Commun. 268, 88–91.
Hansotia, T., Maida, A., Flock, G., Yamada, Y., Tsukiyama, K., Seino, Y., and Drucker,
D.J. (2007). Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and
energy expenditure. J. Clin. Invest. 117, 143–152.
Hany, T.F., Gharehpapagh, E., Kamel, E.M., Buck, A., Himms-Hagen, J., and Schulthess,
von, G.K. (2002). Brown adipose tissue: a factor to consider in symmetrical tracer uptake in the
neck and upper chest region. Eur. J. Nucl. Med. Mol. Imaging 29, 1393–1398.
Hara, J., Beuckmann, C.T., Nambu, T., Willie, J.T., Chemelli, R.M., Sinton, C.M.,
Sugiyama, F., Yagami, K., Goto, K., Yanagisawa, M., et al. (2001). Genetic ablation of orexin
neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30, 345–354.
Hara, J., Yanagisawa, M., and Sakurai, T. (2005). Difference in obesity phenotype between
orexin-knockout mice and orexin neuron-deficient mice with same genetic background and
environmental conditions. Neurosci. Lett. 380, 239–242.
Harms, M., and Seale, P. (2013). Brown and beige fat: development, function and
therapeutic potential. Nat. Med. 19, 1252–1263.
Harper, A.E., and Peters, J.C. (1989). Protein intake, brain amino acid and serotonin
concentrations and protein self-selection. J. Nutr. 119, 677–689.
Harris, J.I. (1959). Studies on pituitary polypeptide hormones. 3. The structure of α-
melanocyte-stimulating hormone from pig pituitary glands. Biochemical Journal 71, 451.

182
Harris, M., Aschkenasi, C., Elias, C.F., Chandrankunnel, A., Nillni, E.A., Bjøorbaek, C.,
Elmquist, J.K., Flier, J.S., and Hollenberg, A.N. (2001). Transcriptional regulation of the
thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J. Clin. Invest. 107,
111–120.
Harrold, J.A., Dovey, T.M., Blundell, J.E., and Halford, J.C.G. (2012). CNS regulation of
appetite. Neuropharmacology 63, 3–17.
Haynes, W.G., Morgan, D.A., Djalali, A., Sivitz, W.I., and Mark, A.L. (1999). Interactions
between the melanocortin system and leptin in control of sympathetic nerve traffic. 33, 542–547.
Hetherington, A., and Ranson, S. (1940). Hypothalamic lesions and adiposity in the rat -
Hetherington - 2005 - The Anatomical Record - Wiley Online Library. The Anatomical Record.
Hill, D.R., Campbell, N.J., Shaw, T.M., and Woodruff, G.N. (1987). Autoradiographic
localization and biochemical characterization of peripheral type CCK receptors in rat CNS using
highly selective nonpeptide CCK antagonists. J. Neurosci. 7, 2967–2976.
Hodges, M.R., Tattersall, G.J., Harris, M.B., McEvoy, S.D., Richerson, D.N., Deneris, E.S.,
Johnson, R.L., Chen, Z.-F., and Richerson, G.B. (2008). Defects in breathing and
thermoregulation in mice with near-complete absence of central serotonin neurons. J. Neurosci.
28, 2495–2505.
Hondares, E., Rosell, M., Gonzalez, F.J., Giralt, M., Iglesias, R., and Villarroya, F. (2010).
Hepatic FGF21 expression is induced at birth via PPARalpha in response to milk intake and
contributes to thermogenic activation of neonatal brown fat. Cell Metab. 11, 206–212.
Horst, ter, G.J., de Boer, P., Luiten, P.G., and van Willigen, J.D. (1989). Ascending
projections from the solitary tract nucleus to the hypothalamus. A Phaseolus vulgaris lectin
tracing study in the rat. Neuroscience 31, 785–797.
Huszar, D., Lynch, C.A., Fairchild-Huntress, V., Dunmore, J.H., Fang, Q., Berkemeier,
L.R., Gu, W., Kesterson, R.A., Boston, B.A., Cone, R.D., et al. (1997). Targeted Disruption of
the Melanocortin-4 Receptor Results in Obesity in Mice. Cell 88, 131–141.

183
Hwa, J.J., Ghibaudi, L., Gao, J., and Parker, E.M. (2001). Central melanocortin system
modulates energy intake and expenditure of obese and lean Zucker rats. Am. J. Physiol. Regul.
Integr. Comp. Physiol. 281, R444–51.
Hylden, J.L., Anton, F., and Nahin, R.L. (1989). Spinal lamina I projection neurons in the
rat: collateral innervation of parabrachial area and thalamus. Neuroscience 28, 27–37.
Ida, T., Mori, K., Miyazato, M., Egi, Y., Abe, S., Nakahara, K., Nishihara, M., Kangawa,
K., and Murakami, N. (2005). Neuromedin s is a novel anorexigenic hormone. Endocrinology
146, 4217–4223.
Inuzuka, M., Tamura, N., Yamada, N., Katsuura, G., Oyamada, N., Taura, D., Sonoyama,
T., Fukunaga, Y., Ohinata, K., Sone, M., et al. (2010). C-type natriuretic peptide as a new
regulator of food intake and energy expenditure. Endocrinology 151, 3633–3642.
Ishii, M., Fei, H., and Friedman, J.M. (2003). Targeted disruption of GPR7, the endogenous
receptor for neuropeptides B and W, leads to metabolic defects and adult-onset obesity. Proc.
Natl. Acad. Sci. U.S.a. 100, 10540–10545.
Ishiwata, T., Saito, T., Hasegawa, H., Yazawa, T., Kotani, Y., Otokawa, M., and Aihara, Y.
(2005). Changes of body temperature and thermoregulatory responses of freely moving rats
during GABAergic pharmacological stimulation to the preoptic area and anterior hypothalamus
in several ambient temperatures. Brain Res. 1048, 32–40.
Jacobowitz, D.M., and O'Donohue, T.L. (1978). alpha-Melanocyte stimulating hormone:
immunohistochemical identification and mapping in neurons of rat brain. Proc. Natl. Acad. Sci.
U.S.a. 75, 6300–6304.
James, D.E., and Pilch, P.F. (1988). Fractionation of endocytic vesicles and glucose-
transporter-containing vesicles in rat adipocytes. Biochem. J. 256, 725–732.
Jamshidi, N., and Taylor, D.A. (2001). Anandamide administration into the ventromedial
hypothalamus stimulates appetite in rats. Br. J. Pharmacol. 134, 1151–1154.

184
Janssen, I. (2013). The public health burden of obesity in Canada. Can J Diabetes 37, 90–
96.
Jaworski, K., Sarkadi-Nagy, E., Duncan, R.E., Ahmadian, M., and Sul, H.S. (2007).
Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue.
Am. J. Physiol. Gastrointest. Liver Physiol. 293, G1–4.
Jégou, S., Boutelet, I., and Vaudry, H. (2000). Melanocortin-3 receptor mRNA expression
in pro-opiomelanocortin neurones of the rat arcuate nucleus. J. Neuroendocrinol. 12, 501–505.
Jiang, G., and Zhang, B.B. (2003). Glucagon and regulation of glucose metabolism. Am. J.
Physiol. Endocrinol. Metab. 284, E671–8.
Johnstone, L.E., Fong, T.M., and Leng, G. (2006). Neuronal activation in the hypothalamus
and brainstem during feeding in rats. Cell Metab. 4, 313–321.
Jonsson, L., Skarphedinsson, J.O., Skuladottir, G.V., Atlason, P.T., Eiriksdottir, V.H.,
Franzson, L., and Schiöth, H.B. (2001). Melanocortin receptor agonist transiently increases
oxygen consumption in rats. Neuroreport 12, 3703–3708.
Joseph, S.A., Pilcher, W.H., and Bennett-Clarke, C. (1983). Immunocytochemical
localization of ACTH perikarya in nucleus tractus solitarius: evidence for a second opiocortin
neuronal system. Neurosci. Lett. 38, 221–225.
Kajimura, S., and Saito, M. (2013). A New Era in Brown Adipose Tissue Biology:
Molecular Control of Brown Fat Development and Energy Homeostasis. Annu. Rev. Physiol.
Kajimura, S., Seale, P., and Spiegelman, B.M. (2010). Transcriptional control of brown fat
development. Cell Metab. 11, 257–262.
Kajimura, S., Seale, P., Kubota, K., Lunsford, E., Frangioni, J.V., Gygi, S.P., and
Spiegelman, B.M. (2009). Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta
transcriptional complex. Nature 460, 1154–1158.

185
Kalra, S.P., Dube, M.G., Pu, S., Xu, B., Horvath, T.L., and Kalra, P.S. (1999). Interacting
appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 20,
68–100.
Kanatani, A., Mashiko, S., Murai, N., Sugimoto, N., Ito, J., Fukuroda, T., Fukami, T.,
Morin, N., MacNeil, D.J., Van der Ploeg, L.H., et al. (2000). Role of the Y1 receptor in the
regulation of neuropeptide Y-mediated feeding: comparison of wild-type, Y1 receptor-deficient,
and Y5 receptor-deficient mice. Endocrinology 141, 1011–1016.
KENNEDY, G.C. (1953). The role of depot fat in the hypothalamic control of food intake
in the rat. Proc. R. Soc. Lond., B, Biol. Sci. 140, 578–596.
Kim, E.M., Grace, M.K., Welch, C.C., Billington, C.J., and Levine, A.S. (1999). STZ-
induced diabetes decreases and insulin normalizes POMC mRNA in arcuate nucleus and pituitary
in rats. Am. J. Physiol. 276, R1320–6.
Kim, M.S., Rossi, M., Abusnana, S., Sunter, D., Morgan, D.G., Small, C.J., Edwards, C.M.,
Heath, M.M., Stanley, S.A., Seal, L.J., et al. (2000). Hypothalamic localization of the feeding
effect of agouti-related peptide and alpha-melanocyte-stimulating hormone. Diabetes 49, 177–
182.
Kirchgessner, A.L. (2002). Orexins in the brain-gut axis. Endocr. Rev. 23, 1–15.
Kirchner, H., Tong, J., Tschöp, M.H., and Pfluger, P.T. (2010). Ghrelin and PYY in the
regulation of energy balance and metabolism: lessons from mouse mutants. Am. J. Physiol.
Endocrinol. Metab. 298, E909–19.
Kirkham, T.C., Williams, C.M., Fezza, F., and Marzo, V.D. (2002). Endocannabinoid
levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation:
stimulation of eating by 2-arachidonoyl glycerol. Br. J. Pharmacol. 136, 550–557.
Kitamura, T., Feng, Y., Kitamura, Y.I., Chua, S.C., Xu, A.W., Barsh, G.S., Rossetti, L., and
Accili, D. (2006). Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food
intake. Nat. Med. 12, 534–540.

186
Kluger, M.J., and Vaughn, L.K. (1978). Fever and survival in rabbits infected with
Pasteurella multocida. J. Physiol. (Lond.) 282, 243–251.
Knauf, C., Cani, P.D., Ait-Belgnaoui, A., Benani, A., Dray, C., Cabou, C., Colom, A.,
Uldry, M., Rastrelli, S., Sabatier, E., et al. (2008). Brain glucagon-like peptide 1 signaling
controls the onset of high-fat diet-induced insulin resistance and reduces energy expenditure.
Endocrinology 149, 4768–4777.
Kobayashi, A., and Osaka, T. (2003). Involvement of the parabrachial nucleus in
thermogenesis induced by environmental cooling in the rat. Pflugers Arch. 446, 760–765.
Kohno, D., Nakata, M., Maejima, Y., Shimizu, H., Sedbazar, U., Yoshida, N., Dezaki, K.,
Onaka, T., Mori, M., and Yada, T. (2008). Nesfatin-1 neurons in paraventricular and supraoptic
nuclei of the rat hypothalamus coexpress oxytocin and vasopressin and are activated by
refeeding. Endocrinology 149, 1295–1301.
Kopp, J., Xu, Z.-Q., Zhang, X., Pedrazzini, T., Herzog, H., Kresse, A., Wong, H., Walsh,
J.H., and Hökfelt, T. (2002). Expression of the neuropeptide Y Y1 receptor in the CNS of rat and
of wild-type and Y1 receptor knock-out mice. Focus on immunohistochemical localization.
Neuroscience 111, 443–532.
Kow, L.M., and Pfaff, D.W. (1991). The effects of the TRH metabolite cyclo(His-Pro) and
its analogs on feeding. Pharmacol. Biochem. Behav. 38, 359–364.
Kowalski, T.J., Spar, B.D., Markowitz, L., Maguire, M., Golovko, A., Yang, S., Farley, C.,
Cook, J.A., Tetzloff, G., Hoos, L., et al. (2005). Transgenic overexpression of neuromedin U
promotes leanness and hypophagia in mice. J. Endocrinol. 185, 151–164.
Kozak, L.P. (2010). Brown fat and the myth of diet-induced thermogenesis. Cell Metab. 11,
263–267.
Kral, T.V.E., and Rolls, B.J. (2004). Energy density and portion size: their independent and
combined effects on energy intake. Physiol. Behav. 82, 131–138.

187
Kristensen, P., Judge, M.E., Thim, L., Ribel, U., Christjansen, K.N., Wulff, B.S., Clausen,
J.T., Jensen, P.B., Madsen, O.D., Vrang, N., et al. (1998). Hypothalamic CART is a new
anorectic peptide regulated by leptin. Nature 393, 72–76.
Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., and Grüters, A. (1998).
Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC
mutations in humans. Nat. Genet. 19, 155–157.
Labbé, O., Desarnaud, F., Eggerickx, D., Vassart, G., and Parmentier, M. (1994).
Molecular cloning of a mouse melanocortin 5 receptor gene widely expressed in peripheral
tissues. Biochemistry 33, 4543–4549.
Ladurelle, N., Duterte-Boucher, D., and Costentin, J. (1991). Stimulation of D1 and D2
dopamine receptors produces additive anorectic effects. Fundam Clin Pharmacol 5, 481–490.
Lambert, P.D., Couceyro, P.R., McGirr, K.M., Dall Vechia, S.E., Smith, Y., and Kuhar,
M.J. (1998). CART peptides in the central control of feeding and interactions with neuropeptide
Y. Synapse 29, 293–298.
Lambert, P.D., Wilding, J.P., al-Dokhayel, A.A., Bohuon, C., Comoy, E., Gilbey, S.G., and
Bloom, S.R. (1993). A role for neuropeptide-Y, dynorphin, and noradrenaline in the central
control of food intake after food deprivation. Endocrinology 133, 29–32.
Lancha, A., Frühbeck, G., and Gómez-Ambrosi, J. (2012). Peripheral signalling involved in
energy homeostasis control. Nutr Res Rev 25, 223–248.
Lanfray, D., Arthaud, S., Ouellet, J., Compère, V., Do Rego, J.-L., Leprince, J., Lefranc,
B., Castel, H., Bouchard, C., Monge-Roffarello, B., et al. (2012). Gliotransmission and Brain
Glucose-Sensing: Critical Role of Endozepines. Diabetes.
Lean, M.E. (1989). Brown adipose tissue in humans. Proc Nutr Soc 48, 243–256.
LeBlanc, J. (2000). Nutritional implications of cephalic phase thermogenic responses.
Appetite 34, 214–216.

188
Lee, H., Iida, T., Mizuno, A., Suzuki, M., and Caterina, M.J. (2005). Altered thermal
selection behavior in mice lacking transient receptor potential vanilloid 4. J. Neurosci. 25, 1304–
1310.
Lee, M., Kim, A., Conwell, I.M., Hruby, V., Mayorov, A., Cai, M., and Wardlaw, S.L.
(2008). Effects of selective modulation of the central melanocortin-3-receptor on food intake and
hypothalamic POMC expression. Peptides 29, 440–447.
Lee, P., Brychta, R.J., Linderman, J., Smith, S., Chen, K.Y., and Celi, F.S. (2013a). Mild
cold exposure modulates fibroblast growth factor 21 (FGF21) diurnal rhythm in humans:
relationship between FGF21 levels, lipolysis, and cold-induced thermogenesis. J. Clin.
Endocrinol. Metab. 98, E98–102.
Lee, S.J., Verma, S., Simonds, S.E., Kirigiti, M.A., Kievit, P., Lindsley, S.R., Loche, A.,
Smith, M.S., Cowley, M.A., and Grove, K.L. (2013b). Leptin stimulates neuropeptide Y and
cocaine amphetamine-regulated transcript coexpressing neuronal activity in the dorsomedial
hypothalamus in diet-induced obese mice. J. Neurosci. 33, 15306–15317.
Leung, F.W. (2014) Capsaicin as an anti-obesity drug. Prog. Drug. Res. 68, 171–9.
Li, J., Xiong, K., Pang, Y., Dong, Y., Kaneko, T., and Mizuno, N. (2006). Medullary dorsal
horn neurons providing axons to both the parabrachial nucleus and thalamus. J. Comp. Neurol.
498, 539–551.
Lieverse, R.J., Jansen, J.B., Masclee, A.A., and Lamers, C.B. (1995). Satiety effects of a
physiological dose of cholecystokinin in humans. Gut 36, 176–179.
Lin, L., and Sun, Y. (2012). Thermogenic characterization of ghrelin receptor null mice.
Meth. Enzymol. 514, 355–370.
Lo, C.-M., King, A., Samuelson, L.C., Kindel, T.L., Rider, T., Jandacek, R.J., Raybould,
H.E., Woods, S.C., and Tso, P. (2010). Cholecystokinin knockout mice are resistant to high-fat
diet-induced obesity. Gastroenterology 138, 1997–2005.

189
Lobo, S., Wiczer, B.M., Smith, A.J., Hall, A.M., and Bernlohr, D.A. (2007). Fatty acid
metabolism in adipocytes: functional analysis of fatty acid transport proteins 1 and 4. J. Lipid
Res. 48, 609–620.
Lowell, B.B., S-Susulic, V., Hamann, A., Lawitts, J.A., Himms-Hagen, J., Boyer, B.B.,
Kozak, L.P., and Flier, J.S. (1993). Development of obesity in transgenic mice after genetic
ablation of brown adipose tissue. Nature 366, 740–742.
López, M., Lage, R., Tung, Y.C.L., Challis, B.G., Varela, L., Virtue, S., O'Rahilly, S.,
Vidal-Puig, A., Diéguez, C., and Coll, A.P. (2007). Orexin expression is regulated by alpha-
melanocyte-stimulating hormone. J. Neuroendocrinol. 19, 703–707.
Lu, D., Willard, D., Patel, I.R., Kadwell, S., Overton, L., Kost, T., Luther, M., Chen, W.,
Woychik, R.P., and Wilkison, W.O. (1994). Agouti protein is an antagonist of the melanocyte-
stimulating-hormone receptor. Nature 371, 799–802.
Lu, J., Zhang, Y.H., Chou, T.C., Gaus, S.E., Elmquist, J.K., Shiromani, P., and Saper, C.B.
(2001). Contrasting effects of ibotenate lesions of the paraventricular nucleus and
subparaventricular zone on sleep-wake cycle and temperature regulation. J. Neurosci. 21, 4864–
4874.
Lundius, E.G., Sanchez-Alavez, M., Ghochani, Y., Klaus, J., and Tabarean, I.V. (2010).
Histamine influences body temperature by acting at H1 and H3 receptors on distinct populations
of preoptic neurons. J. Neurosci. 30, 4369–4381.
Lyons, W.E., Mamounas, L.A., Ricaurte, G.A., Coppola, V., Reid, S.W., Bora, S.H.,
Wihler, C., Koliatsos, V.E., and Tessarollo, L. (1999). Brain-derived neurotrophic factor-
deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic
abnormalities. Proc. Natl. Acad. Sci. U.S.a. 96, 15239–15244.
Ma, S., Yu, H., Zhao, Z., Luo, Z., Chen, J., Ni, Y., Jin, R., Ma, L., Wang, P., Zhu, Z., Li,
L., Zhong, J., Liu, D., Nilius, B., and Zhu, Z. (2012). Activation of the cold-sensing TRPM8
channel triggers UCP1-dependent thermogenesis and prevents obesity. J. mol. Cell. Biol. 4, 88–
96.

190
Maack, T., Suzuki, M., Almeida, F.A., Nussenzveig, D., Scarborough, R.M., McEnroe,
G.A., and Lewicki, J.A. (1987). Physiological role of silent receptors of atrial natriuretic factor.
Science 238, 675–678.
Madden, C.J. (2012). Glucoprivation in the ventrolateral medulla decreases brown adipose
tissue sympathetic nerve activity by decreasing the activity of neurons in raphe pallidus. Am. J.
Physiol. Regul. Integr. Comp. Physiol. 302, R224–32.
Madden, C.J., and Morrison, S.F. (2003). Excitatory amino acid receptor activation in the
raphe pallidus area mediates prostaglandin-evoked thermogenesis. Neuroscience 122, 5–15.
Madden, C.J., and Morrison, S.F. (2004). Excitatory amino acid receptors in the
dorsomedial hypothalamus mediate prostaglandin-evoked thermogenesis in brown adipose tissue.
Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R320–5.
Madden, C.J., and Morrison, S.F. (2005). Hypoxic activation of arterial chemoreceptors
inhibits sympathetic outflow to brown adipose tissue in rats. J. Physiol. (Lond.) 566, 559–573.
Madden, C.J., and Morrison, S.F. (2006). Serotonin potentiates sympathetic responses
evoked by spinal NMDA. J. Physiol. (Lond.) 577, 525–537.
Madden, C.J., and Morrison, S.F. (2008). Brown adipose tissue sympathetic nerve activity
is potentiated by activation of 5-hydroxytryptamine (5-HT)1A/5-HT7 receptors in the rat spinal
cord. Neuropharmacology 54, 487–496.
Madden, C.J., and Morrison, S.F. (2009). Neurons in the paraventricular nucleus of the
hypothalamus inhibit sympathetic outflow to brown adipose tissue. Am. J. Physiol. Regul. Integr.
Comp. Physiol. 296, R831–43.
Madden, C.J., and Morrison, S.F. (2010). Endogenous activation of spinal 5-
hydroxytryptamine (5-HT) receptors contributes to the thermoregulatory activation of brown
adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R776–83.
Maejima, Y., Sedbazar, U., Suyama, S., Kohno, D., Onaka, T., Takano, E., Yoshida, N.,
Koike, M., Uchiyama, Y., Fujiwara, K., et al. (2009). Nesfatin-1-regulated oxytocinergic

191
signaling in the paraventricular nucleus causes anorexia through a leptin-independent
melanocortin pathway. Cell Metab. 10, 355–365.
Mains, R.E., Eipper, B.A., and Ling, N. (1977). Common precursor to corticotropins and
endorphins.
Marsh, D.J., Miura, G.I., Yagaloff, K.A., Schwartz, M.W., Barsh, G.S., and Palmiter, R.D.
(1999). Effects of neuropeptide Y deficiency on hypothalamic agouti-related protein expression
and responsiveness to melanocortin analogues. Brain Res. 848, 66–77.
Masaki, T., Yoshimichi, G., Chiba, S., Yasuda, T., Noguchi, H., Kakuma, T., Sakata, T.,
and Yoshimatsu, H. (2003). Corticotropin-releasing hormone-mediated pathway of leptin to
regulate feeding, adiposity, and uncoupling protein expression in mice. Endocrinology 144,
3547–3554.
Matsuda, L.A., Lolait, S.J., Brownstein, M.J., Young, A.C., and Bonner, T.I. (1990).
Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346,
561–564.
McClellan, K.M., Parker, K.L., and Tobet, S. (2006). Development of the ventromedial
nucleus of the hypothalamus. Front Neuroendocrinol 27, 193–209.
Mechoulam, R., Ben-Shabat, S., Hanus, L., Ligumsky, M., Kaminski, N.E., Schatz, A.R.,
Gopher, A., Almog, S., Martin, B.R., and Compton, D.R. (1995). Identification of an endogenous
2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical
Pharmacology 50, 83–90.
MELLINKOFF, S.M., FRANKLAND, M., BOYLE, D., and GREIPEL, M. (1956).
Relationship between serum amino acid concentration and fluctuations in appetite. J. Appl.
Physiol. 8, 535–538.
Merchenthaler, I., Lane, M., and Shughrue, P. (1999). Distribution of pre-pro-glucagon and
glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J. Comp.
Neurol. 403, 261–280.

192
Ménard, S.L., Ci, X., Frisch, F., Normand-Lauzière, F., Cadorette, J., Ouellet, R., van Lier,
J.E., Bénard, F., Bentourkia, M., Lecomte, R., et al. (2009). Mechanism of reduced myocardial
glucose utilization during acute hypertriglyceridemia in rats. Mol Imaging Biol 11, 6–14.
Ménard, S.L., Croteau, E., Sarrhini, O., Gélinas, R., Brassard, P., Ouellet, R., Bentourkia,
M., van Lier, J.E., Rosiers, Des, C., Lecomte, R., et al. (2010). Abnormal in vivo myocardial
energy substrate uptake in diet-induced type 2 diabetic cardiomyopathy in rats. 298, E1049–
E1057.
Mizuno, Y., Kondo, K., Terashima, Y., Arima, H., Murase, T., and Oiso, Y. (1998).
Anorectic effect of pituitary adenylate cyclase activating polypeptide (PACAP) in rats: lack of
evidence for involvement of hypothalamic neuropeptide gene expression. J. Neuroendocrinol. 10,
611–616.
Mochizuki, T., Klerman, E.B., Sakurai, T., and Scammell, T.E. (2006). Elevated body
temperature during sleep in orexin knockout mice. Am. J. Physiol. Regul. Integr. Comp. Physiol.
291, R533–40.
Moran, O., and Phillip, M. (2003). Leptin: obesity, diabetes and other peripheral effects--a
review. Pediatr Diabetes 4, 101–109.
Moran, T.H., Baldessarini, A.R., Salorio, C.F., Lowery, T., and Schwartz, G.J. (1997).
Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. Am.
J. Physiol. 272, R1245–51.
Morin, S.M., and Gehlert, D.R. (2006). Distribution of NPY Y5-like immunoreactivity in
the rat brain. J. Mol. Neurosci. 29, 109–114.
Morrison, S.F. (1999). RVLM and raphe differentially regulate sympathetic outflows to
splanchnic and brown adipose tissue. Am. J. Physiol. 276, R962–73.
Morrison, S.F. (2004). Activation of 5-HT1A receptors in raphe pallidus inhibits leptin-
evoked increases in brown adipose tissue thermogenesis. Am. J. Physiol. Regul. Integr. Comp.
Physiol. 286, R832–7.

193
Morrison, S.F. (2011). 2010 Carl Ludwig Distinguished Lectureship of the APS Neural
Control and Autonomic Regulation Section: Central neural pathways for thermoregulatory cold
defense. J. Appl. Physiol. 110, 1137–1149.
Morrison, S.F., Madden, C.J., and Tupone, D. (2012). Central control of brown adipose
tissue thermogenesis. Front Endocrinol (Lausanne) 3.
Morrison, S.F., Madden, C.J., and Tupone, D. (2014). Central Neural Regulation of Brown
Adipose Tissue Thermogenesis and Energy Expenditure. Cell Metab.
Morrison, S.F., Nakamura, K., and Madden, C.J. (2008). Central control of thermogenesis
in mammals. Exp. Physiol. 93, 773–797.
Morton, G.J., Cummings, D.E., Baskin, D.G., Barsh, G.S., and Schwartz, M.W. (2006).
Central nervous system control of food intake and body weight. Nature 443, 289–295.
Mountjoy, K.G. (2010). Distribution and function of melanocortin receptors within the
brain. Adv. Exp. Med. Biol. 681, 29–48.
Mountjoy, K.G., Mortrud, M.T., Low, M.J., Simerly, R.B., and Cone, R.D. (1994).
Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control
circuits in the brain. Mol. Endocrinol. 8, 1298–1308.
Mountjoy, K.G., Robbins, L.S., Mortrud, M.T., and Cone, R.D. (1992). The cloning of a
family of genes that encode the melanocortin receptors. Science 257, 1248–1251.
Munro, S., Thomas, K.L., and Abu-Shaar, M. (1993). Molecular characterization of a
peripheral receptor for cannabinoids. Nature 365, 61–65.
Müller, E.E., Panerai, A., Cocchi, D., Frohman, L.A., and Mantegazza, P. (1973). Central
glucoprivation: some physiological effects induced by the intraventricular administration of 2-
deoxy-d-glucose. Experientia 29, 874–876.

194
Münzberg, H., Huo, L., Nillni, E.A., Hollenberg, A.N., and Bjørbaek, C. (2003). Role of
signal transducer and activator of transcription 3 in regulation of hypothalamic
proopiomelanocortin gene expression by leptin. Endocrinology 144, 2121–2131.
Myers, M.G., and Olson, D.P. (2012). Central nervous system control of metabolism.
Nature 491, 357–363.
Nakamura, K., and Morrison, S.F. (2007). Central efferent pathways mediating skin
cooling-evoked sympathetic thermogenesis in brown adipose tissue. Am. J. Physiol. Regul.
Integr. Comp. Physiol. 292, R127–36.
Nakamura, K., and Morrison, S.F. (2008a). A thermosensory pathway that controls body
temperature. Nat. Neurosci. 11, 62–71.
Nakamura, K., and Morrison, S.F. (2008b). Preoptic mechanism for cold-defensive
responses to skin cooling. J. Physiol. (Lond.) 586, 2611–2620.
Nakamura, K., and Morrison, S.F. (2010). A thermosensory pathway mediating heat-
defense responses. Proc. Natl. Acad. Sci. U.S.a. 107, 8848–8853.
Nakamura, K., Matsumura, K., Hübschle, T., Nakamura, Y., Hioki, H., Fujiyama, F.,
Boldogköi, Z., König, M., Thiel, H.-J., Gerstberger, R., et al. (2004). Identification of
sympathetic premotor neurons in medullary raphe regions mediating fever and other
thermoregulatory functions. J. Neurosci. 24, 5370–5380.
Nakamura, K., Matsumura, K., Kaneko, T., Kobayashi, S., Katoh, H., and Negishi, M.
(2002). The rostral raphe pallidus nucleus mediates pyrogenic transmission from the preoptic
area. J. Neurosci. 22, 4600–4610.
Nakamura, Y., Nakamura, K., Matsumura, K., Kobayashi, S., Kaneko, T., and Morrison,
S.F. (2005). Direct pyrogenic input from prostaglandin EP3 receptor-expressing preoptic neurons
to the dorsomedial hypothalamus. Eur. J. Neurosci. 22, 3137–3146.
Nedergaard, J., Bengtsson, T., and Cannon, B. (2007). Unexpected evidence for active
brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 293, E444–52.

195
Nedergaard, J., Bengtsson, T., and Cannon, B. (2010). Three years with adult human brown
adipose tissue. Ann. N. Y. Acad. Sci. 1212, E20–36.
Nedergaard, J., Petrovic, N., Lindgren, E.M., Jacobsson, A., and Cannon, B. (2005).
PPARgamma in the control of brown adipocyte differentiation. Biochim. Biophys. Acta 1740,
293–304.
Nicholls, D.G., and Locke, R.M. (1984). Thermogenic mechanisms in brown fat. Physiol.
Rev. 64, 1–64.
Nicholls, D.G., Bernson, V.S., and Heaton, G.M. (1978). The identification of the
component in the inner membrane of brown adipose tissue mitochondria responsible for
regulating energy dissipation. Experientia Suppl. 32, 89–93.
Nijenhuis, W.A., Oosterom, J., and Adan, R.A. (2001). AgRP(83-132) acts as an inverse
agonist on the human-melanocortin-4 receptor. Mol. Endocrinol. 15, 164–171.
Nogueiras, R., Wiedmer, P., Perez-Tilve, D., Veyrat-Durebex, C., Keogh, J.M., Sutton,
G.M., Pfluger, P.T., Castaneda, T.R., Neschen, S., Hofmann, S.M., et al. (2007). The central
melanocortin system directly controls peripheral lipid metabolism. J. Clin. Invest. 117, 3475–
3488.
Nonogaki, K. (1999). Obesity: autonomic circuits versus feeding. Nat. Med. 5, 742–743.
Oakes, N.D., Thalén, P., Aasum, E., Edgley, A., Larsen, T., Furler, S.M., Ljung, B., and
Severson, D. (2006). Cardiac metabolism in mice: tracer method developments and in vivo
application revealing profound metabolic inflexibility in diabetes. Am. J. Physiol. Endocrinol.
Metab. 290, E870–81.
Obici, S., Feng, Z., Morgan, K., Stein, D., Karkanias, G., and Rossetti, L. (2002). Central
administration of oleic acid inhibits glucose production and food intake. Diabetes 51, 271–275.
Ohata, H., Suzuki, K., Oki, Y., and Shibasaki, T. (2000). Urocortin in the ventromedial
hypothalamic nucleus acts as an inhibitor of feeding behavior in rats. Brain Res. 861, 1–7.

196
Oka, T., Oka, K., Kobayashi, T., Sugimoto, Y., Ichikawa, A., Ushikubi, F., Narumiya, S.,
and Saper, C.B. (2003). Characteristics of thermoregulatory and febrile responses in mice
deficient in prostaglandin EP1 and EP3 receptors. J. Physiol. (Lond.) 551, 945–954.
Oldfield, B.J., Giles, M.E., Watson, A., Anderson, C., Colvill, L.M., and McKinley, M.J.
(2002). The neurochemical characterisation of hypothalamic pathways projecting
polysynaptically to brown adipose tissue in the rat. Neuroscience 110, 515–526.
Ollmann, M.M., Wilson, B.D., Yang, Y.K., Kerns, J.A., Chen, Y., Gantz, I., and Barsh,
G.S. (1997). Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related
protein. Science 278, 135–138.
Olson, B.R., Drutarosky, M.D., Stricker, E.M., and Verbalis, J.G. (1991). Brain oxytocin
receptor antagonism blunts the effects of anorexigenic treatments in rats: evidence for central
oxytocin inhibition of food intake. Endocrinology 129, 785–791.
Ootsuka, Y., Blessing, W.W., Steiner, A.A., and Romanovsky, A.A. (2008). Fever response
to intravenous prostaglandin E2 is mediated by the brain but does not require afferent vagal
signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1294–303.
Osaka, T., Endo, M., Yamakawa, M., and Inoue, S. (2005). Energy expenditure by
intravenous administration of glucagon-like peptide-1 mediated by the lower brainstem and
sympathoadrenal system. Peptides 26, 1623–1631.
Ouellet, V., Routhier-Labadie, A., Bellemare, W., Lakhal-Chaieb, L., Turcotte, E.,
Carpentier, A.C., and Richard, D. (2011). Outdoor Temperature, Age, Sex, Body Mass Index,
and Diabetic Status Determine the Prevalence, Mass, and Glucose-Uptake Activity of 18F-FDG-
Detected BAT in Humans. Journal of Clinical Endocrinology & Metabolism 96, 192–199.
Pandit, R., de Jong, J.W., Vanderschuren, L.J.M.J., and Adan, R.A.H. (2011).
Neurobiology of overeating and obesity: The role of melanocortins and beyond. Eur. J.
Pharmacol. 660, 28–42.
Parker, J.A., and Bloom, S.R. (2012). Hypothalamic neuropeptides and the regulation of
appetite. Neuropharmacology 63, 18–30.

197
Patel, H.R., Qi, Y., Hawkins, E.J., Hileman, S.M., Elmquist, J.K., Imai, Y., and Ahima,
R.S. (2006). Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in
obesity-prone mice. Diabetes 55, 3091–3098.
Patel, S.M., and Ebenezer, I.S. (2004). The effects of intraperitoneal and
intracerebroventricular administration of the GABAB receptor antagonist CGP 35348 on food
intake in rats. Eur. J. Pharmacol. 503, 89–93.
Peier, A.M., Moqrich, A., Hergarden, A.C., Reeve, A.J., Andersson, D.A., Story, G.M.,
Earley, T.J., Dragoni, I., McIntyre, P., Bevan, S., et al. (2002). A TRP channel that senses cold
stimuli and menthol. Cell 108, 705–715.
Pertwee, R.G. (1997). Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol.
Ther. 74, 129–180.
Peters, C.T., Choi, Y.H., Brubaker, P.L., and Anderson, G.H. (2001). A glucagon-like
peptide-1 receptor agonist and an antagonist modify macronutrient selection by rats. J. Nutr. 131,
2164–2170.
Peters, J.C., and Harper, A.E. (1987). Acute effects of dietary protein on food intake, tissue
amino acids, and brain serotonin. Am. J. Physiol. 252, R902–14.
Petrovic, N., Walden, T.B., Shabalina, I.G., Timmons, J.A., Cannon, B., and Nedergaard, J.
(2010). Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of
epididymally derived white adipocyte cultures reveals a population of thermogenically
competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J.
Biol. Chem. 285, 7153–7164.
Phillips, R.J., and Powley, T.L. (1996). Gastric volume rather than nutrient content inhibits
food intake. Am. J. Physiol. 271, R766–9.
Picó, C., Herron, D., Palou, A., Jacobsson, A., Cannon, B., and Nedergaard, J. (1994).
Stabilization of the mRNA for the uncoupling protein thermogenin by
transcriptional/translational blockade and by noradrenaline in brown adipocytes differentiated in
culture: a degradation factor induced by cessation of stimulation? Biochem. J. 302 ( Pt 1), 81–86.

198
Pisani, D.F., Djedaini, M., Beranger, G.E., Elabd, C., Scheideler, M., Ailhaud, G., and
Amri, E.-Z. (2011). Differentiation of Human Adipose-Derived Stem Cells into “Brite” (Brown-
in-White) Adipocytes. Front Endocrinol (Lausanne) 2, 87.
Plazzi, G., Moghadam, K.K., Maggi, L.S., Donadio, V., Vetrugno, R., Liguori, R., Zoccoli,
G., Poli, F., Pizza, F., Pagotto, U., et al. (2011). Autonomic disturbances in narcolepsy. Sleep
Med Rev 15, 187–196.
Poggioli, R., Vergoni, A.V., and Bertolini, A. (1986). ACTH-(1-24) and alpha-MSH
antagonize feeding behavior stimulated by kappa opiate agonists. Peptides 7, 843–848.
Polidori, C., de Caro, G., and Massi, M. (2000). The hyperphagic effect of
nociceptin/orphanin FQ in rats. Peptides 21, 1051–1062.
Powley, T.L., and Phillips, R.J. (2004). Gastric satiation is volumetric, intestinal satiation is
nutritive. Physiol. Behav. 82, 69–74.
Pritchard, L.E., Turnbull, A.V., and White, A. (2002). Pro-opiomelanocortin processing in
the hypothalamus: impact on melanocortin signalling and obesity. J. Endocrinol. 172, 411–421.
Puigserver, P., Herron, D., Gianotti, M., Palou, A., Cannon, B., and Nedergaard, J. (1992).
Induction and degradation of the uncoupling protein thermogenin in brown adipocytes in vitro
and in vivo. Evidence for a rapidly degradable pool. Biochem. J. 284 ( Pt 2), 393–398.
Puigserver, P., Wu, Z., Park, C.W., Graves, R., Wright, M., and Spiegelman, B.M. (1998).
A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–
839.
Qu, D., Ludwig, D.S., Gammeltoft, S., Piper, M., Pelleymounter, M.A., Cullen, M.J.,
Mathes, W.F., Przypek, R., Kanarek, R., and Maratos-Flier, E. (1996). A role for melanin-
concentrating hormone in the central regulation of feeding behaviour. Nature 380, 243–247.
Quarta, C., Bellocchio, L., Mancini, G., Mazza, R., Cervino, C., Braulke, L.J., Fekete, C.,
Latorre, R., Nanni, C., Bucci, M., et al. (2010). CB(1) signaling in forebrain and sympathetic

199
neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. 11,
273–285.
Rathner, J.A., and Morrison, S.F. (2006). Rostral ventromedial periaqueductal gray: a
source of inhibition of the sympathetic outflow to brown adipose tissue. Brain Res. 1077, 99–
107.
Reaux, A., Gallatz, K., Palkovits, M., and Llorens-Cortes, C. (2002). Distribution of apelin-
synthesizing neurons in the adult rat brain. Neuroscience 113, 653–662.
Rees, J.L. (2003). Genetics of hair and skin color. Annu. Rev. Genet. 37, 67–90.
Reizes, O., Clegg, D.J., Strader, A.D., and Benoit, S.C. (2006). A role for syndecan-3 in the
melanocortin regulation of energy balance. Peptides 27, 274–280.
Remaury, A., Vita, N., Gendreau, S., Jung, M., Arnone, M., Poncelet, M., Culouscou, J.-
M., Le Fur, G., Soubrié, P., Caput, D., et al. (2002). Targeted inactivation of the neurotensin type
1 receptor reveals its role in body temperature control and feeding behavior but not in analgesia.
Brain Res. 953, 63–72.
Reue, K., and Dwyer, J.R. (2009). Lipin proteins and metabolic homeostasis. J. Lipid Res.
50 Suppl, S109–14.
Reyes, T.M., Lewis, K., Perrin, M.H., Kunitake, K.S., Vaughan, J., Arias, C.A.,
Hogenesch, J.B., Gulyas, J., Rivier, J., Vale, W.W., et al. (2001). Urocortin II: a member of the
corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF
receptors. Proc. Natl. Acad. Sci. U.S.a. 98, 2843–2848.
Richard, D. (2007). Energy expenditure: a critical determinant of energy balance with key
hypothalamic controls. Minerva Endocrinol. 32, 173–183.
Richard, D., and Picard, F. (2011). Brown fat biology and thermogenesis. Front Biosci
(Landmark Ed) 16, 1233–1260.

200
Richard, D., Carpentier, A.C., Doré, G., Ouellet, V., and Picard, F. (2010). Determinants of
brown adipocyte development and thermogenesis. Int J Obes (Lond) 34 Suppl 2, S59–66.
Richard, D., Guesdon, B., and Timofeeva, E. (2009). The brain endocannabinoid system in
the regulation of energy balance. Best Pract. Res. Clin. Endocrinol. Metab. 23, 17–32.
Richard, D., Huang, Q., and Timofeeva, E. (2000). The corticotropin-releasing hormone
system in the regulation of energy balance in obesity. Int J Obes (Lond) 24 Suppl 2, S36–9.
Richard, D., Monge-Roffarello, B., Chechi, K., Labbé, S.M., and Turcotte, E.E. (2012).
Control and physiological determinants of sympathetically mediated brown adipose tissue
thermogenesis. Front Endocrinol (Lausanne) 3, 36.
Rios, M., Fan, G., Fekete, C., Kelly, J., Bates, B., Kuehn, R., Lechan, R.M., and Jaenisch,
R. (2001). Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads
to obesity and hyperactivity. Mol. Endocrinol. 15, 1748–1757.
Ritter, S., Li, A.-J., Wang, Q., and Dinh, T.T. (2011). Minireview: The value of looking
backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit.
Endocrinology 152, 4019–4032.
Robertson, D., Biaggioni, I., Burnstock, G., and Low, P. (2011). Primer on the autonomic
nervous system (3rd Ed.) BIAGGIONI Italo, BURNSTOCK Geoffrey, LOW Phillip A., PATON
Julian F.R., ROBERTSON Da: Librairie Lavoisier.
Rodríguez, A., Gómez-Ambrosi, J., Catalán, V., Gil, M.J., Becerril, S., Sáinz, N., Silva, C.,
Salvador, J., Colina, I., and Frühbeck, G. (2009). Acylated and desacyl ghrelin stimulate lipid
accumulation in human visceral adipocytes. Int J Obes (Lond) 33, 541–552.
Romanovsky, A.A. (2004). Do fever and anapyrexia exist? Analysis of set point-based
definitions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R992–5.
Roselli-Rehfuss, L., Mountjoy, K.G., Robbins, L.S., Mortrud, M.T., Low, M.J., Tatro, J.B.,
Entwistle, M.L., Simerly, R.B., and Cone, R.D. (1993). Identification of a receptor for gamma

201
melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system.
Proc. Natl. Acad. Sci. U.S.a. 90, 8856–8860.
Rossi, M., Kim, M.S., Morgan, D.G., Small, C.J., Edwards, C.M., Sunter, D., Abusnana, S.,
Goldstone, A.P., Russell, S.H., Stanley, S.A., et al. (1998). A C-terminal fragment of Agouti-
related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating
hormone in vivo. Endocrinology 139, 4428–4431.
Rothwell, N.J., and Stock, M.J. (1979). A role for brown adipose tissue in diet-induced
thermogenesis. Nature 281, 31–35.
Rothwell, N.J., and Stock, M.J. (1981). A role for insulin in the diet-induced thermogenesis
of cafeteria-fed rats. Metab. Clin. Exp. 30, 673–678.
Rothwell, N.J., Stock, M.J., and Thexton, A.J. (1983). Decerebration activates
thermogenesis in the rat. J. Physiol. (Lond.) 342, 15–22.
Sahu, A., Kalra, P.S., and Kalra, S.P. (1988). Food deprivation and ingestion induce
reciprocal changes in neuropeptide Y concentrations in the paraventricular nucleus. Peptides 9,
83–86.
Sanacora, G., Kershaw, M., Finkelstein, J.A., and White, J.D. (1990). Increased
hypothalamic content of preproneuropeptide Y messenger ribonucleic acid in genetically obese
Zucker rats and its regulation by food deprivation. Endocrinology 127, 730–737.
Sarkar, S., Zaretskaia, M.V., Zaretsky, D.V., Moreno, M., and Dimicco, J.A. (2007).
Stress- and lipopolysaccharide-induced c-fos expression and nNOS in hypothalamic neurons
projecting to medullary raphe in rats: a triple immunofluorescent labeling study. Eur. J. Neurosci.
26, 2228–2238.
Satoh, N., Ogawa, Y., Katsuura, G., Numata, Y., Masuzaki, H., Yoshimasa, Y., and Nakao,
K. (1998). Satiety effect and sympathetic activation of leptin are mediated by hypothalamic
melanocortin system. Neurosci. Lett. 249, 107–110.

202
Scalera, G., and Tarozzi, G. (1998). Somatostatin administration modifies food intake,
body weight, and gut motility in rat. Peptides 19, 991–997.
Scammell, T., Griffin, J., and Elmquist, J. (1998). Microinjection of a cyclooxygenase
inhibitor into the anteroventral preoptic region attenuates LPS fever | Regulatory, Integrative and
Comparative Physiology. American Journal of ….
Scheurink, A.J., and Nolan, L.J. (1996). Food intake, fuel homeostasis, and the autonomic
nervous system. Appetite 26, 304.
Schlogl, M., Piaggi, P., Thiyyagura, P., Reiman, E.M., Chen, K., Lutrin, C., Krakoff, J.,
and Thearle, M.S. (2013). Overfeeding over 24 hours does not activate brown adipose tissue in
humans. Journal of Clinical Endocrinology & Metabolism.
Schulz, T.J., and Tseng, Y.-H. (2013). Brown adipose tissue: development, metabolism and
beyond. Biochem. J. 453, 167–178.
Schwartz, G.J. (2006). Integrative capacity of the caudal brainstem in the control of food
intake. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 361, 1275–1280.
Schwartz, M.W., Woods, S.C., Porte, D., Seeley, R.J., and Baskin, D.G. (2000). Central
nervous system control of food intake. Nature 404, 661–671.
Schwinkendorf, D.R., Tsatsos, N.G., Gosnell, B.A., and Mashek, D.G. (2011). Effects of
central administration of distinct fatty acids on hypothalamic neuropeptide expression and energy
metabolism. Int J Obes (Lond) 35, 336–344.
Seale, P., Bjork, B., Yang, W., Kajimura, S., Chin, S., Kuang, S., Scimè, A., Devarakonda,
S., Conroe, H.M., Erdjument-Bromage, H., et al. (2008). PRDM16 controls a brown fat/skeletal
muscle switch. Nature 454, 961–967.
Seale, P., Conroe, H.M., Estall, J., Kajimura, S., Frontini, A., Ishibashi, J., Cohen, P., Cinti,
S., and Spiegelman, B.M. (2011). Prdm16 determines the thermogenic program of subcutaneous
white adipose tissue in mice. J. Clin. Invest. 121, 96–105.

203
Seale, P., Kajimura, S., Yang, W., Chin, S., Rohas, L.M., Uldry, M., Tavernier, G., Langin,
D., and Spiegelman, B.M. (2007). Transcriptional control of brown fat determination by
PRDM16. Cell Metab. 6, 38–54.
Seeley, R.J., van Dijk, G., Campfield, L.A., Smith, F.J., Burn, P., Nelligan, J.A., Bell, S.M.,
Baskin, D.G., Woods, S.C., and Schwartz, M.W. (1996). Intraventricular leptin reduces food
intake and body weight of lean rats but not obese Zucker rats. Horm. Metab. Res. 28, 664–668.
Seeley, R.J., Yagaloff, K.A., Fisher, S.L., Burn, P., Thiele, T.E., van Dijk, G., Baskin,
D.G., and Schwartz, M.W. (1997). Melanocortin receptors in leptin effects. Nature 390, 349.
Sellayah, D., and Sikder, D. (2013). Orexin restores aging-related brown adipose tissue
dysfunction in male mice. Endocrinology.
Sellayah, D., Bharaj, P., and Sikder, D. (2011). Orexin is required for brown adipose tissue
development, differentiation, and function. Cell Metab. 14, 478–490.
Seth, A., Stanley, S., Jethwa, P., Gardiner, J., Ghatei, M., and Bloom, S. (2004). Galanin-
like peptide stimulates the release of gonadotropin-releasing hormone in vitro and may mediate
the effects of leptin on the hypothalamo-pituitary-gonadal axis. Endocrinology 145, 743–750.
Shi, Y., and Burn, P. (2004). Lipid metabolic enzymes: emerging drug targets for the
treatment of obesity. Nat Rev Drug Discov 3, 695–710.
Shimada, M., Tritos, N.A., Lowell, B.B., Flier, J.S., and Maratos-Flier, E. (1998). Mice
lacking melanin-concentrating hormone are hypophagic and lean. Nature 396, 670–674.
Shimizu, H., Inoue, K., and Mori, M. (2007). The leptin-dependent and -independent
melanocortin signaling system: regulation of feeding and energy expenditure. J. Endocrinol. 193,
1–9.
Shindou, H., Hishikawa, D., Harayama, T., Yuki, K., and Shimizu, T. (2009). Recent
progress on acyl CoA: lysophospholipid acyltransferase research. J. Lipid Res. 50 Suppl, S46–51.

204
Shiraishi, T., Oomura, Y., Sasaki, K., and Wayner, M.J. (2000). Effects of leptin and
orexin-A on food intake and feeding related hypothalamic neurons. Physiol. Behav. 71, 251–261.
Shutter, J.R., Graham, M., Kinsey, A.C., Scully, S., Lüthy, R., and Stark, K.L. (1997).
Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and
diabetic mutant mice. Genes Dev. 11, 593–602.
Silva, J.E. (2006). Thermogenic mechanisms and their hormonal regulation. Physiol. Rev.
86, 435–464.
Simonson, D.C., and DeFronzo, R.A. (1990). Indirect calorimetry: methodological and
interpretative problems. Am. J. Physiol. 258, E399–412.
Skibicka, K.P., and Grill, H.J. (2008). Energetic responses are triggered by caudal
brainstem melanocortin receptor stimulation and mediated by local sympathetic effector circuits.
Endocrinology 149, 3605–3616.
Skibicka, K.P., and Grill, H.J. (2009). Hypothalamic and hindbrain melanocortin receptors
contribute to the feeding, thermogenic, and cardiovascular action of melanocortins.
Endocrinology 150, 5351–5361.
Sloth, B., Holst, J.J., Flint, A., Gregersen, N.T., and Astrup, A. (2007). Effects of PYY1-36
and PYY3-36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in
obese and lean subjects. Am. J. Physiol. Endocrinol. Metab. 292, E1062–8.
Small, C.J., Liu, Y.L., Stanley, S.A., Connoley, I.P., Kennedy, A., Stock, M.J., and Bloom,
S.R. (2003). Chronic CNS administration of Agouti-related protein (Agrp) reduces energy
expenditure. Int. J. Obes. Relat. Metab. Disord. 27, 530–533.
Smeets, P.A.M., Erkner, A., and de Graaf, C. (2010). Cephalic phase responses and
appetite. Nut. Rev. 68, 643–655.
Smith, A.I., and Funder, J.W. (1988). Proopiomelanocortin processing in the pituitary,
central nervous system, and peripheral tissues. Endocr. Rev. 9, 159–179.

205
Smith, G.P., Jerome, C., Cushin, B.J., Eterno, R., and Simansky, K.J. (1981). Abdominal
vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 213, 1036–1037.
Song, C.K., Schwartz, G.J., and Bartness, T.J. (2009). Anterograde transneuronal viral tract
tracing reveals central sensory circuits from white adipose tissue. Am. J. Physiol. Regul. Integr.
Comp. Physiol. 296, R501–11.
Song, C.K., Vaughan, C.H., Keen-Rhinehart, E., Harris, R.B.S., Richard, D., and Bartness,
T.J. (2008). Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown
adipose tissue: neuroanatomical and functional evidence. Am. J. Physiol. Regul. Integr. Comp.
Physiol. 295, R417–28.
St-Pierre, D.H., Karelis, A.D., Cianflone, K., Conus, F., Mignault, D., Rabasa-Lhoret, R.,
St-Onge, M., Tremblay-Lebeau, A., and Poehlman, E.T. (2004). Relationship between ghrelin
and energy expenditure in healthy young women. Journal of Clinical Endocrinology &
Metabolism 89, 5993–5997.
Stanić, D., Brumovsky, P., Fetissov, S., Shuster, S., Herzog, H., and Hökfelt, T. (2006).
Characterization of neuropeptide Y2 receptor protein expression in the mouse brain. I.
Distribution in cell bodies and nerve terminals. J. Comp. Neurol. 499, 357–390.
Stanley, B.G., Chin, A.S., and Leibowitz, S.F. (1985). Feeding and drinking elicited by
central injection of neuropeptide Y: evidence for a hypothalamic site(s) of action. Brain Res.
Bull. 14, 521–524.
Stanley, B.G., Kyrkouli, S.E., Lampert, S., and Leibowitz, S.F. (1986). Neuropeptide Y
chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia
and obesity. Peptides 7, 1189–1192.
Ste Marie, L., Ste Marie, L., Miura, G.I., Miura, G.I., Marsh, D.J., Marsh, D.J., Yagaloff,
K., Yagaloff, K., Palmiter, R.D., and Palmiter, R.D. (2000). A metabolic defect promotes obesity
in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. U.S.a. 97, 12339–12344.

206
Steiner, A.A., Ivanov, A.I., Serrats, J., Hosokawa, H., Phayre, A.N., Robbins, J.R., Roberts,
J.L., Kobayashi, S., Matsumura, K., Sawchenko, P.E., et al. (2006). Cellular and molecular bases
of the initiation of fever. PLoS Biol. 4, e284.
Sternson, S.M., Shepherd, G.M.G., and Friedman, J.M. (2005). Topographic mapping of
VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 8,
1356–1363.
Stornetta, R.L., Rosin, D.L., Simmons, J.R., McQuiston, T.J., Vujovic, N., Weston, M.C.,
and Guyenet, P.G. (2005). Coexpression of vesicular glutamate transporter-3 and gamma-
aminobutyric acidergic markers in rat rostral medullary raphe and intermediolateral cell column.
J. Comp. Neurol. 492, 477–494.
Suda, T., Tozawa, F., Iwai, I., Sato, Y., Sumitomo, T., Nakano, Y., Yamada, M., and
Demura, H. (1993). Neuropeptide Y increases the corticotropin-releasing factor messenger
ribonucleic acid level in the rat hypothalamus. Brain Res. Mol. Brain Res. 18, 311–315.
Sunter, D., Hewson, A.K., Lynam, S., and Dickson, S.L. (2001). Intracerebroventricular
injection of neuropeptide FF, an opioid modulating neuropeptide, acutely reduces food intake and
stimulates water intake in the rat. Neurosci. Lett. 313, 145–148.
Swart, I., Jahng, J.W., Overton, J.M., and Houpt, T.A. (2002). Hypothalamic NPY, AGRP,
and POMC mRNA responses to leptin and refeeding in mice. Am. J. Physiol. Regul. Integr.
Comp. Physiol. 283, R1020–6.
Székely, M., Pétervári, E., Balaskó, M., Hernádi, I., and Uzsoki, B. (2002). Effects of
orexins on energy balance and thermoregulation. Regul. Pept. 104, 47–53.
Tang-Christensen, M., Larsen, P.J., Göke, R., Fink-Jensen, A., Jessop, D.S., Møller, M.,
and Sheikh, S.P. (1996). Central administration of GLP-1-(7-36) amide inhibits food and water
intake in rats. Am. J. Physiol. 271, R848–56.
Tang-Christensen, M., Larsen, P.J., Thulesen, J., Rømer, J., and Vrang, N. (2000). The
proglucagon-derived peptide, glucagon-like peptide-2, is a neurotransmitter involved in the
regulation of food intake. Nat. Med. 6, 802–807.

207
Tansey, J.T., Sztalryd, C., Gruia-Gray, J., Roush, D.L., Zee, J.V., Gavrilova, O., Reitman,
M.L., Deng, C.X., Li, C., Kimmel, A.R., et al. (2001). Perilipin ablation results in a lean mouse
with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced
obesity. Proc. Natl. Acad. Sci. U.S.a. 98, 6494–6499.
Tatemoto, K., and Mutt, V. (1980). Isolation of two novel candidate hormones using a
chemical method for finding naturally occurring polypeptides. Nature 285, 417–418.
Tatemoto, K., Carlquist, M., and Mutt, V. (1982). Neuropeptide Y--a novel brain peptide
with structural similarities to peptide YY and pancreatic polypeptide. Nature 296, 659–660.
Tattersall, G.J., and Milsom, W.K. (2009). Hypoxia reduces the hypothalamic thermogenic
threshold and thermosensitivity. J. Physiol. (Lond.) 587, 5259–5274.
Teubner, B.J.W., and Bartness, T.J. (2013). PYY(3-36) into the arcuate nucleus inhibits
food deprivation-induced increases in food hoarding and intake. Peptides 47, 20–28.
Thorsell, A., and Heilig, M. (2002). Diverse functions of neuropeptide Y revealed using
genetically modified animals. Neuropeptides 36, 182–193.
Timmons, J.A., Wennmalm, K., Larsson, O., Walden, T.B., Lassmann, T., Petrovic, N.,
Hamilton, D.L., Gimeno, R.E., Wahlestedt, C., Baar, K., et al. (2007). Myogenic gene expression
signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc.
Natl. Acad. Sci. U.S.a. 104, 4401–4406.
Tominaga, M., Caterina, M.J., Malmberg, A.B., Rosen, T.A., Gilbert, H., Skinner, K.,
Raumann, B.E., Basbaum, A.I., and Julius, D. (1998). The cloned capsaicin receptor integrates
multiple pain-producing stimuli. Neuron 21, 531–543.
Troke, R.C., Tan, T.M., and Bloom, S.R. (2014). The future role of gut hormones in the
treatment of obesity. Ther Adv Chronic Dis 5, 4–14.
Tschöp, M., Smiley, D.L., and Heiman, M.L. (2000). Ghrelin induces adiposity in rodents.
Nature 407, 908–913.

208
Tseng, Y.-H., Kokkotou, E., Schulz, T.J., Huang, T.L., Winnay, J.N., Taniguchi, C.M.,
Tran, T.T., Suzuki, R., Espinoza, D.O., Yamamoto, Y., et al. (2008). New role of bone
morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004.
Tung, Y.C.L., Rimmington, D., O'Rahilly, S., and Coll, A.P. (2007). Pro-opiomelanocortin
modulates the thermogenic and physical activity responses to high-fat feeding and markedly
influences dietary fat preference. Endocrinology 148, 5331–5338.
Tupone, D., Madden, C.J., Cano, G., and Morrison, S.F. (2011). An orexinergic projection
from perifornical hypothalamus to raphe pallidus increases rat brown adipose tissue
thermogenesis. J. Neurosci. 31, 15944–15955.
Uschakov, A., Gong, H., McGinty, D., and Szymusiak, R. (2007). Efferent projections
from the median preoptic nucleus to sleep- and arousal-regulatory nuclei in the rat brain.
Neuroscience 150, 104–120.
Valassi, E., Scacchi, M., and Cavagnini, F. (2008). Neuroendocrine control of food intake.
Nutr Metab Cardiovasc Dis 18, 158–168.
van Marken Lichtenbelt, W.D., Vanhommerig, J.W., Smulders, N.M., Drossaerts,
J.M.A.F.L., Kemerink, G.J., Bouvy, N.D., Schrauwen, P., and Teule, G.J.J. (2009). Cold-
activated brown adipose tissue in healthy men. N Engl J Med 360, 1500–1508.
Varela, L., and Horvath, T.L. (2012). Leptin and insulin pathways in POMC and AgRP
neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 13, 1079–1086.
Vaughan, C.H., Shrestha, Y.B., and Bartness, T.J. (2011). Characterization of a novel
melanocortin receptor-containing node in the SNS outflow circuitry to brown adipose tissue
involved in thermogenesis. Brain Res. 1411, 17–27.
Verty, A.N.A., McFarlane, J.R., McGregor, I.S., and Mallet, P.E. (2004). Evidence for an
interaction between CB1 cannabinoid and melanocortin MCR-4 receptors in regulating food
intake. Endocrinology 145, 3224–3231.

209
Villarroya, J., Cereijo, R., and Villarroya, F. (2013). An endocrine role for brown adipose
tissue? Am. J. Physiol. Endocrinol. Metab. 305, E567–72.
Virtanen, K.A., Lidell, M.E., Orava, J., Heglind, M., Westergren, R., Niemi, T., Taittonen,
M., Laine, J., Savisto, N.-J., Enerbäck, S., et al. (2009). Functional brown adipose tissue in
healthy adults. N Engl J Med 360, 1518–1525.
Voss-Andreae, A., Murphy, J.G., Ellacott, K.L.J., Stuart, R.C., Nillni, E.A., Cone, R.D.,
and Fan, W. (2007). Role of the central melanocortin circuitry in adaptive thermogenesis of
brown adipose tissue. Endocrinology 148, 1550–1560.
Vosselman, M.J., Brans, B., van der Lans, A.A., Wierts, R., van Baak, M.A., Mottaghy,
F.M., Schrauwen, P., and van Marken Lichtenbelt, W.D. (2013). Brown adipose tissue activity
after a high-calorie meal in humans. Am. J. Clin. Nutr. 98, 57–64.
Wada, K., Wada, E., Watase, K., Yamada, K., and Ohki-Hamazaki, H. (1998). Bombesin,
obesity, and social behavior. Mol. Psychiatry 3, 204–206.
Wallingford, N., Perroud, B., Gao, Q., Coppola, A., Gyengesi, E., Liu, Z.-W., Gao, X.-B.,
Diament, A., Haus, K.A., Shariat-Madar, Z., et al. (2009). Prolylcarboxypeptidase regulates food
intake by inactivating alpha-MSH in rodents. J. Clin. Invest. 119, 2291–2303.
Weigle, D.S., Bukowski, T.R., Foster, D.C., Holderman, S., Kramer, J.M., Lasser, G.,
Lofton-Day, C.E., Prunkard, D.E., Raymond, C., and Kuijper, J.L. (1995). Recombinant ob
protein reduces feeding and body weight in the ob/ob mouse. J. Clin. Invest. 96, 2065–2070.
Whittle, A.J., Carobbio, S., Martins, L., Slawik, M., Hondares, E., Vázquez, M.J., Morgan,
D., Csikasz, R.I., Gallego, R., Rodriguez-Cuenca, S., et al. (2012). BMP8B increases brown
adipose tissue thermogenesis through both central and peripheral actions. Cell 149, 871–885.
Williams, D.L., Baskin, D.G., and Schwartz, M.W. (2009). Evidence that intestinal
glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 150, 1680–1687.

210
Williams, D.L., Bowers, R.R., Bartness, T.J., Kaplan, J.M., and Grill, H.J. (2003).
Brainstem melanocortin 3/4 receptor stimulation increases uncoupling protein gene expression in
brown fat. Endocrinology 144, 4692–4697.
Williams, D.L., Kaplan, J.M., and Grill, H.J. (2000). The role of the dorsal vagal complex
and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology
141, 1332–1337.
Wilson, R.I., and Nicoll, R.A. (2001). Endogenous cannabinoids mediate retrograde
signalling at hippocampal synapses. Nature 410, 588–592.
Woods, S.C., Lotter, E.C., McKay, L.D., and Porte, D. (1979). Chronic
intracerebroventricular infusion of insulin reduces food intake and body weight of baboons.
Nature 282, 503–505.
Wortley, K.E., Anderson, K.D., Yasenchak, J., Murphy, A., Valenzuela, D., Diano, S.,
Yancopoulos, G.D., Wiegand, S.J., and Sleeman, M.W. (2005). Agouti-related protein-deficient
mice display an age-related lean phenotype. Cell Metab. 2, 421–427.
Woychik, N.A., and Young, R.A. (1992). Genes encoding transcription factor IIIA and the
RNA polymerase common subunit RPB6 are divergently transcribed in Saccharomyces
cerevisiae. Proc. Natl. Acad. Sci. U.S.a. 89, 3999–4003.
Wren, A.M., and Bloom, S.R. (2007). Gut hormones and appetite control. Gastroenterology
132, 2116–2130.
Wren, A.M., Seal, L.J., Cohen, M.A., Brynes, A.E., Frost, G.S., Murphy, K.G., Dhillo,
W.S., Ghatei, M.A., and Bloom, S.R. (2001). Ghrelin enhances appetite and increases food intake
in humans. Journal of Clinical Endocrinology & Metabolism 86, 5992.
Wu, J., Boström, P., Sparks, L.M., Ye, L., Choi, J.H., Giang, A.-H., Khandekar, M.,
Virtanen, K.A., Nuutila, P., Schaart, G., et al. (2012). Beige adipocytes are a distinct type of
thermogenic fat cell in mouse and human. Cell 150, 366–376.

211
Wu, J., Cohen, P., and Spiegelman, B.M. (2013). Adaptive thermogenesis in adipocytes: is
beige the new brown? Genes Dev. 27, 234–250.
Wynne, K., Stanley, S., McGowan, B., and Bloom, S. (2005). Appetite control. J.
Endocrinol. 184, 291–318.
Xu, A.W., Ste-Marie, L., Kaelin, C.B., and Barsh, G.S. (2007). Inactivation of signal
transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes
decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology
148, 72–80.
Xu, B., Goulding, E.H., Zang, K., Cepoi, D., Cone, R.D., Jones, K.R., Tecott, L.H., and
Reichardt, L.F. (2003). Brain-derived neurotrophic factor regulates energy balance downstream
of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742.
Xue, Y., Petrovic, N., Cao, R., Larsson, O., Lim, S., Chen, S., Feldmann, H.M., Liang, Z.,
Zhu, Z., Nedergaard, J., et al. (2009). Hypoxia-independent angiogenesis in adipose tissues
during cold acclimation. Cell Metab. 9, 99–109.
Yamashita, H., Sato, Y., Kizaki, T., Oh, S., Nagasawa, J., and Ohno, H. (1994). Basic
fibroblast growth factor (bFGF) contributes to the enlargement of brown adipose tissue during
cold acclimation. Pflugers Arch. 428, 352–356.
Yang, G., Lim, C.-Y., Li, C., Xiao, X., Radda, G.K., Li, C., Cao, X., and Han, W. (2009a).
FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3
interaction with specificity protein 1. J. Biol. Chem. 284, 3719–3727.
Yang, L., Scott, K.A., Hyun, J., Tamashiro, K.L., Tray, N., Moran, T.H., and Bi, S.
(2009b). Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and
energy balance. J. Neurosci. 29, 179–190.
Yaswen, L., Diehl, N., Brennan, M.B., and Hochgeschwender, U. (1999). Obesity in the
mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med.
5, 1066–1070.

212
Yen, C.-L.E., Stone, S.J., Koliwad, S., Harris, C., and Farese, R.V. (2008). Thematic
review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49,
2283–2301.
Yeo, G.S., Farooqi, I.S., Aminian, S., Halsall, D.J., Stanhope, R.G., and O'Rahilly, S.
(1998). A frameshift mutation in MC4R associated with dominantly inherited human obesity.
Nat. Genet. 20, 111–112.
Yeo, G.S.H., Connie Hung, C.-C., Rochford, J., Keogh, J., Gray, J., Sivaramakrishnan, S.,
O'Rahilly, S., and Farooqi, I.S. (2004). A de novo mutation affecting human TrkB associated
with severe obesity and developmental delay. Nat. Neurosci. 7, 1187–1189.
Yoshida, K., Konishi, M., Nagashima, K., Saper, C.B., and Kanosue, K. (2005). Fos
activation in hypothalamic neurons during cold or warm exposure: projections to periaqueductal
gray matter. Neuroscience 133, 1039–1046.
Zaretskaia, M.V., Zaretsky, D.V., Shekhar, A., and Dimicco, J.A. (2002). Chemical
stimulation of the dorsomedial hypothalamus evokes non-shivering thermogenesis in
anesthetized rats. Brain Res. 928, 113–125.
Zaretsky, D.V., Zaretskaia, M.V., and Dimicco, J.A. (2003). Stimulation and blockade of
GABA(A) receptors in the raphe pallidus: effects on body temperature, heart rate, and blood
pressure in conscious rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R110–6.
Zarjevski, N., Cusin, I., Vettor, R., Rohner-Jeanrenaud, F., and Jeanrenaud, B. (1993).
Chronic intracerebroventricular neuropeptide-Y administration to normal rats mimics hormonal
and metabolic changes of obesity. Endocrinology 133, 1753–1758.
Zechner, R., Kienesberger, P.C., Haemmerle, G., Zimmermann, R., and Lass, A. (2009).
Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 50, 3–
21.
Zhang, W., Sunanaga, J., Takahashi, Y., Mori, T., Sakurai, T., Kanmura, Y., and Kuwaki,
T. (2010). Orexin neurons are indispensable for stress-induced thermogenesis in mice. J. Physiol.
(Lond.) 588, 4117–4129.

213
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J.M. (1994).
Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432.
Zhang, Y.H., Lu, J., Elmquist, J.K., and Saper, C.B. (2000). Lipopolysaccharide activates
specific populations of hypothalamic and brainstem neurons that project to the spinal cord. J.
Neurosci. 20, 6578–6586.
Zheng, F., Kim, Y.J., Chao, P.-T., and Bi, S. (2013). Overexpression of neuropeptide Y in
the dorsomedial hypothalamus causes hyperphagia and obesity in rats. Obesity (Silver Spring)
21, 1086–1092.
Zingaretti, M.C., Crosta, F., Vitali, A., Guerrieri, M., Frontini, A., Cannon, B., Nedergaard,
J., and Cinti, S. (2009). The presence of UCP1 demonstrates that metabolically active adipose
tissue in the neck of adult humans truly represents brown adipose tissue. Faseb J. 23, 3113–3120.