![Lam Enseignement de La Grammaire[1]. Ban Sua (2)](https://static.fdocuments.fr/doc/165x107/5572002749795991699ee538/lam-enseignement-de-la-grammaire1-ban-sua-2.jpg)
LAM 4 (myélomonocytaire)
68
LAM 4 (myélomonocytaire) LAM 4 typique Moelle : > 30 % de blastes, 20 à 80 % de cellules monocytaires à différents stades de maturation. Sang : > 5x10 9 /l cellules monocytaires (monoblastes, pro-monocytes, monocytes) M4 éosinophilie : granulations éosinophiles dans le cytoplasme
description
LAM 4 (myélomonocytaire). LAM 4 typique Moelle : > 30 % de blastes, 20 à 80 % de cellules monocytaires à différents stades de maturation. Sang : > 5x10 9 /l cellules monocytaires (monoblastes, pro-monocytes , monocytes) M4 éosinophilie : granulations éosinophiles dans le cytoplasme. - PowerPoint PPT Presentation
Transcript of LAM 4 (myélomonocytaire)

LAM 4(myélomonocytaire)
LAM 4 typique Moelle : > 30 % de blastes, 20 à 80 % de cellules
monocytaires à différents stades de maturation. Sang : > 5x109/l cellules monocytaires (monoblastes, pro-monocytes, monocytes)
M4 éosinophilie : granulations éosinophiles dans le cytoplasme

LAM 5(monoblastique)
LAM5 a : peu différenciée ou monoblastique, plus de 80 % des cellules monocytaires de la moelle sont des monoblastes LAM5b : différenciée, moins de 80 % des cellules de la moelle sont des monoblastes, présence de monocytes et promonocytes.

LAM 6(Erythroleucémies)
Blastes > 30 % des éléments non érythroblastiques Erythroblastes > 50 % des éléments nucléés avec dysérythropoïèse Présence de micromégacaryocytes Corps d ’Auer possibles Evolution possible vers LAM1, LAM2, LAM4

LAM 7(LA mégacaryocytaire)
Mégacaryoblastes Reconnaissance par AC monoclonaux et/ou détection d ’activité péroxydasique en microscopie électronique

2. DEUX RISQUES MAJEURS
I. Syndrome de lyse tumorale (N. LEGUYADER)II. Neutropénie fébrile

Deux risques majeurs
• Syndrome de lyse tumoral– Forme hyperleucocytaire– Forme très tumorale– Risque: troubles ioniques, insuffisance rénale
• Neutropénie fébrile– Chimio-induite– Infection: 1ère cause de mortalité chez le sujet
neutropénique

2. DEUX RISQUES MAJEURS
I. Syndrome de lyse tumorale (N. LEGUYADER)II. Neutropénie fébrile

Facteurs de risque
Masse tumorale élevée ou une hyperleucocytose
Taux de LDH élevé Anomalies préexistantes de la fonction rénale pouvant être liée à la pathologie tumorale :
- infiltration lymphomateuse ou leucémique - uropathie obstructive par compression ou infiltration tumorale

Pathologies et SLT
• LNH de haut grade de malignité (lymphome de Burkitt +++)
• LAL
• LAM
• décrit lors de neuroblastome, hépatoblastome, tératome sacrococcygien, rhabdomyosarcome...

Circonstances d ’apparition• Spontanément (Burkitt ++)• Hyperhydratation ++• Chimiothérapie +++• mais aussi hormonothérapie, radiothérapie,
immunothérapie …
• Dans les 24 - 48 premières heures de prise en charge et jusqu’à 5 jours suivant le début de la chimiothérapie

Physiopathologie
Cellule tumorale
Phosphore Nucléotides
bases puriques Potassium LDH
Anomalies biologiques isolées ou concomitantes
Uricémie

Hyperuricémie
• Anomalie la plus fréquente lors du SLT
• Purines et précurseurs : métabolisme hépatique

Hyperuricémie• Sécrétion tubulaire acide urique taux plasmatique • Dans tube distal :
[Urates] + pH + [sels]
Diminution solubilité acide urique
PRECIPITATION acide urique
Néphropathie uratique

Hyperphosphorémie
Hyperphosphaturie : par FG et réabsorption
tubulaire (saturation transport)
Hypocalcémie
Précipitation phosphate de calcium
Néphrocalcinose aiguë
Risque de convulsions +++

Prévention / Traitement = Hyperhydratation (3 l / m2 : G 5%, NaCl et Ca)
sans potassium
+ / - Alcalinisation …
+ Maintien diurèse (furosémide : 0.5 à 1 mg/kg/6h)
+ Traitement hypo-uricémiant :
- Allopurinol (Zyloric)
+ Epuration extra-rénale si troubles ioniques menaçants

Prévention / Traitement
= Hyperhydratation (3 l / m2 : G 5%, NaCl et Ca)
sans potassium
+ / - Alcalinisation
+ Maintien diurèse (furosémide : 0.5 à 1 mg/kg/6h)
+ Traitement hypo-uricémiant ou uricolytique :
- Allopurinol (Zyloric)
- Urate-oxydase

2. DEUX RISQUES MAJEURS
I. Syndrome de lyse tumoraleII. Neutropénie fébrile

Données épidémiologiques• L’infection est la première cause de mortalité chez les
patients neutropéniques– Avant les années 70: septicémie à pyocyanique, 100 %
de mortalité; 1969: 40 % avec carbenicilline et gentamicine
– Aujourd’hui, risque de mortalité par infection dépend du statut de la pathologie sous jacente: < 5% à la phase initiale, mais très élevé en cas de pathologie réfractaire
– Pronostic de certaines infections (aspergillose invasive…) reste très préoccupant

Données épidémiologiques• Facteurs de risques d’infections
– Profondeur (< 500 PNN/mm3) et durée (> 7 jours) de la neutropénie; risque proche de 100 % si < 100 PNN/ mm3 pendant > 3 semaines; étude menée dans le service: chez patients ayant < 500 PNN, RR infection / à ceux ayant > 500 PNN est de 9,2 [3-28]
– Lésions cutanéo-muqueuses chimio-induites– Cathéter central, et autres corps étrangers– Nutrition parentérale pour certains– Corticothérapie et ATB prolongée pour risque
fongique, travaux pour aspergillose

Quels germes ?
• Bactéries digestives– Bacilles gram négatifs
• Bactéries cutanées:– Staphylocoques– Toujours y penser si cathéter central

Infections en aplasie: présentation clinique
• Fièvre ++++– Plusieurs définitions: 1 pic 38,5°C ou 38°C à 3 reprises
en 24 h, actuellement, plutôt 1 pic 38,3°C ou 2 fois 38°C à 1 heure d’intervalle
– Parfois le seul signe d’infection, mais peut manquer, en particulier en cas de corticothérapie +++
– N’est pas d’origine infectieuse dans environ 10 % des cas: néoplasique, chimiothérapie (ara C..), produits sanguins ou Ig, ampho B…
– Documentation microbiologique dans 30% des cas, clinique dans 20% des cas, dite d’origine indéterminée mais possiblement infectieuse dans 40 % des cas

Infections en aplasie: examens de base
• Examen clinique minutieux +++, peau, bouche et siège, signes de gravité (troubles hémodynamiques…)
• Hémocultures (au mieux 2), technique rigoureuse, volume suffisant, anaérobie inutile sauf contexte particulier; frottis-goutte épaisse dans le contexte africain
• CRP et/ou procalcitonine si disponible (Fleischhack et al, 2001, Erten et al 2004)
• Compte de germe dans les selles (ou coproculture)• Radiographie de thorax• Selon contexte ou la disponibilité: prélèvements
parasitologiques, bouche, gorge, peau, ECBU, virémie et Ag CMV, Ag aspergillaire, aspiration rhino-pharyngée pour virologie, PCR mycoplasme…LBA possible?

Infections en aplasie: conduite à tenir
• URGENCE +++• Antibiothérapie IV dans les 3 heures qui suivent
l’apparition de la fièvre• Large spectre, rapidement bactéricide, association
synergique• Tenir compte
– ATCD infectieux de chaque patient– Signes cliniques (mucite et strepto, douleurs
musculaires ou diarrhée et BGN…)– Évolution dans le temps de l’écologie microbienne chez
les patients d’hémato-oncologie (Aquino et al, 1995)– Écologie de chaque service, et du profil de résistance
des germes rencontrés

Évaluation à 48 H
Apyrexie Syndrome infectieux persistant
Poursuite traitementjusqu’à «sortie d’aplasie »
Ajout glycopeptide
Adaptation selon
bactériologie
AntifongiqueReprise
thermique
ßlactamine + aminoside+/- glycopeptide
ApyrexieÉvaluation à 48-72 H
Syndrome infectieux persistantEt /ou ajout
glycopeptide, changement ATB

Infections en aplasie: conduite à tenir
• Autres mesures– Aciclovir si mucite– Facteur de croissance hématopoïétique, en cas de
syndrome septique non controlé si disponible

Infections en aplasie: conduite à tenir• Durée du traitement anti infectieux
– Si trop court, risque de reprise du processus infectieux, et de choc septique
– Si trop long, risque de sélectionner germe résistant, de favoriser infection fongique, surcoût important
– Classiquement jusqu’à PNN > 500/mm3, voir plus tard si infection documentée, mais arrêt précoce possible (Katz et al 1993, Jones et al 1994…) pour population d’enfants en RC, apyrétique depuis > 24h, bon état clinique, hémocultures négatives et amorce de sortie d’aplasie (plaquettes et/ou monocytes); économie 5000 $ / patient

3. LEUCÉMIES AIGUES LYMPHOBLASTIQUES
I. Données généralesII. Facteurs pronostiquesIII. TraitementsIV. Résultats actuels
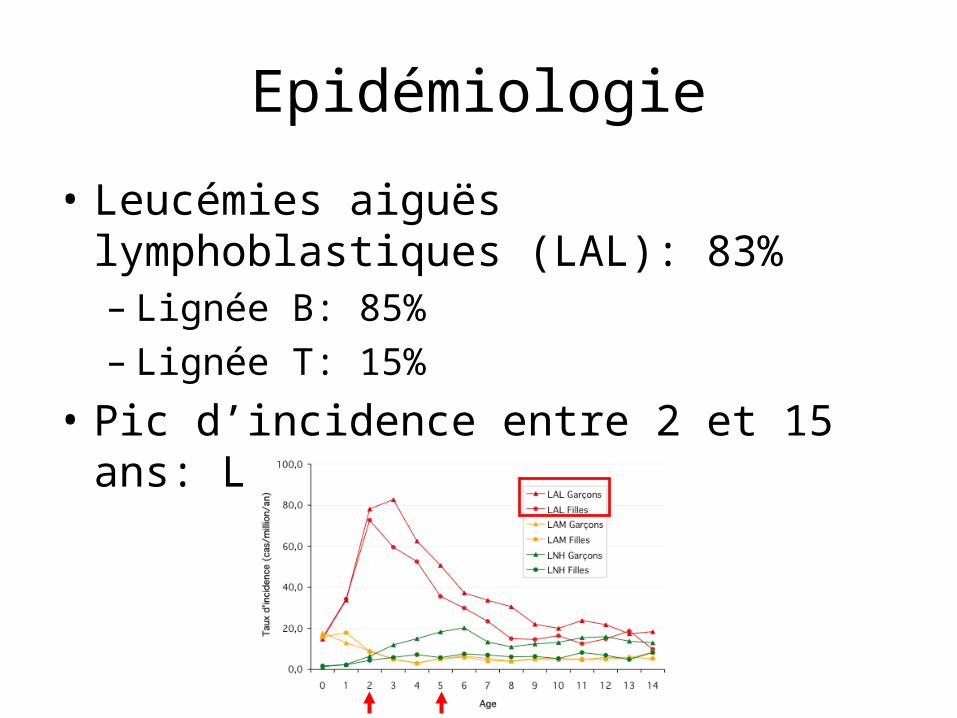
Epidémiologie
• Leucémies aiguës lymphoblastiques (LAL): 83%– Lignée B: 85%– Lignée T: 15%
• Pic d’incidence entre 2 et 15 ans: LAL

Avant 1950 Durée médiane de survie = 3,4 mois
1950 - 1960 Quelques rémissions < 1 an
1960 2 % de survie à 7 ans
1970 20 % de survie à 7 ans
1980 60 % de survie à 7 ans
1990 75 % de survie à 7 ans
Evolution du pronosticdes LAL de l’enfant

Epidémiologie• Actuellement: 80% de guérison• Survie variable selon les facteurs pronostiques
– Age– Type B ou T– Leucocytose– Localisation: atteinte méningée ?– Réponse au traitement– Cytogénétique
• Risque de rechute: standard / Haut risque La stratégie thérapeutique joue un rôle pronostique Plus un traitement est efficace, moins on retrouve les facteurs
pronostiques.

Principes thérapeutiques des LAL• Adapter le traitement à chaque forme de LAL
– Ne pas sur-traiter les formes à faible risque de rechute– Intensifier les formes à haut risque de rechute
• Centres spécialisés• Optimisation des soins de support
– Transfusion, Nutrition– Prévention des risques métaboliques– Traitement anti-infectieux– Abords veineux
• Etudes multicentriques randomisées

3. LEUCÉMIES AIGUES LYMPHOBLASTIQUES
I. Données généralesII. Facteurs pronostiquesIII. TraitementsIV. Résultats actuels

Facteurs initiaux de pronostic défavorable
• Liés au terrain:– Age: < 1 an, > 10 ans– Sexe: Garçon– Ethnique: race noire
• Liés à la masse tumorale– GB > 50 000/mm3 (LAL B)(LAL B)– Syndrome tumoral important– Elargissement médiastinal– Atteinte du SNC
• Liés au phénotype– Pro-B (CD19+, CD10-)– T
• Liés aux anomalies génétiques:– Hypoploïdie < 45 chromosomes– Translocation: t(9;22), t(4;11)– Réarrangement du gène MLL– Amplification 21q– Transcrit de fusion:
• BCR-ABL• MLL-AF4

Facteurs pronostiques initiaux des LAL
Risque standard
Haut risque
Age 1 à 9 ans 10 ansLeucocytose < 50 000 50 000
EFS à 4 ans (n = 612)
83 % 4
(n=297)
74 % 4
Smith M. et coll J Clin Oncol 1996, 14, 18-24

Anomalies cytogénétiques des LAL
- Anomalies de nombre- Portant sur un seul chromosome:
- perte (monosomie 7, 20, X)- gain (trisomie 21, X)- Bon pronostic: trisomie 4, 10, 17
- Portant sur plusieurs chromosomes- Hyperploïdie > 50, 30 % enfants, bon pronostic- Hypoploïdie < 46, 10 % LAL de l ’enfant, mauvais pronostic- Haploïdie : 26 à 28 chromosomes, 1 % LAL, mauvais pronostic- Pseudoploïdie : 46 chromosomes avec anomalie(s) inhabituelle(s), 35 % LAL, T ou B mature, pronostic intermédiaire
- Anomalies de structures- Bon pronostic : t(12;21)/TEL-AML1- Mauvais pronostic : t (9;22)/BCR-ABL, t(4;11)/MLL-AF4- Autres : t(1;19); t(2;8), t(8;14), t(8;22) des LAL3

STRATIFICATION INITIALE: 4 grandes entités de LAL chez l ’enfant
Nourrissons LAL-B LAL-B LAL-T < 1an Risque Haut
Standard Risque 1-10 ans > 10 ansGB < 50.000 ou GB > 50.000
ou cytogénétique défavorable
% 2-3% 55-60% 30% 12-15%Survie 30-50% 85-90% 75-80% 70-80%à 5 ans
STRATIFICATION SECONDAIREMENT AFFINEE PAR LA REPONSE PRECOCE AU TRAITEMENT

Facteurs pronostiques sous traitement
- Corticosensibilité - < 1000 blastes / mm3 - à J8 d’un traitement par prednisone et 1 IT de
méthotrexate- BFM 86 :
Patients EFS
Corticosensibles 90% 78 % 2
Corticorésistants 10% 48 % 5

Facteurs pronostiques sous traitement Chimiosensibilité
Myélogramme à J14 (ou J21), M1: < 5 % blastes, M2: 5 à 25 % de blastes,
M3: > 25 % de blastes Résultats Fralle 93 (LAL lignée B, âge > 1 an)
Globale M1 M2 M3
Risque standard patients (n et %) EFS (4 ans)
632 (68 %) 83 4 %
86 % 85 %
9 %
67 %
5 %
35 %
Haut risque patients (n et %) EFS (4 ans)
297 (32 %) 74 6 %
75 % 80 %
13 % 66 %
12 % 48 %

Apport de la génétique moléculaire en cours de traitement:
Etude de la maladie résiduelle

TEMPS
MASSE TUMORALE
DIAGNOSTIC
RC CYTOLOGIQUE
RECHUTE MOLECULAIRE
RC MOLECU LAIRE
SEUIL CYTOLOGIQUE
SEUIL MOLECULAIRE
RECHUTE C LINIQUE
INDUCTION CONSOLIDATION
La Maladie résiduelle

Apport de la génétique moléculaire en cours de traitement
• La persistance d’un niveau élevé de MRD est un facteur indépendant de mauvais pronostic (Cave et al, 1998)

Récapitulatif facteurs mauvais pronostiques
CLINIQUE : Age <1 an ou >10 ans,: Sexe masculin: Race noire: Syndrome tumoral important, élargissement médiastinal (+/-): Atteinte du SNC (clinique et/ou LCR)
HEMOGRAMME : GB > 50.000/mm3 (LAL de la lignée B seulement) IMMUNOPHENOTYPE : Immunophénotype pro-B (CD19+ CD10-)
: Immunophénotype T (+/-) CYTOGENETIQUE : Hypodiploïdie < 45 chromosomes
: Translocations défavorables : t(9;22), t(4;11) FISH : Réarrangement du gène MLL
: Amplification 21q (iAMP21q) BIOLOGIE MOLECULAIRE: Transcrit de fusion BCR-ABL ou MLL-AF4 THERAPEUTIQUE : Chimiorésistance relative
- cortico-résistance à J8 (> 1000 blastes / mm3 dans le sang après 7 jours de stéroïdes + IT MTX)
- blastose médullaire > 5% à J14 ou J21 du traitement d’induction- maladie résiduelle élevée (> 1%) à J35-42 (immunologie ou biologie moléculaire)
: Chimiorésistance avérée
- blastose médullaire persistante à J35-42 (échec d’induction)

3. LEUCÉMIES AIGUES LYMPHOBLASTIQUES
I. Données généralesII. Facteurs pronostiquesIII. TraitementsIV. Résultats actuels

Traitement des LAL : historique- 1948 : aminoptérine - 1950’-60’ : CT: 6-MP, VCR, MTX, - 1960’ : polychimiothérapie- 1960’-70 ’ : prophylaxie du SNC, - 1970’ : stratification, L-ASP, DNR- 1980’ : intensification décalée
progrès du “supportive care”- 1990' : prise en compte de
la réponse précoce - 2000’ : individualisation?
désescalade ?

Traitement des LAL: principes
• Stratification: selon les facteurs de risque initiaux et secondaires
• 4 Phases thérapeutiques (2-3 ans) . INDUCTION . CONSOLIDATION . INTENSIFICATION(S) (rarement la greffe)
. ENTRETIEN
• Prophylaxie de l’atteinte du SNC

Traitement d ’induction But : obtenir une rémission complète (RC)
Hémogramme normal (>1000 PNN; >100.000 plaquettes) Myélogramme: < 5% de blastes, moelle de richesse
normale Modalités :
Préphase de corticoïdes Vincristine (VCR) Predniso(lo)ne (PRED) ou Dexamethasone (DXM) Asparaginase (L-ASPA) injections intrathécales (IT) 95% RC Intérêt des anthracyclines : diminuer le risque de rechutes
ultérieur des formes à haut risque.

Traitement de consolidation
• Buts: – Maintenir la RC– Médicaments différents de ceux de l’induction– Pour éviter sélection de clones résistants
• Traitements:– Aracytine, 6-MP, VP16, MTX +/- L-ASPA– Durée: 3 mois

Traitement d’intensification
• Intérêt démontré par les études des groupes BFM et CCSG
• Principe : – Séquence thérapeutique proche du
traitement d'induction – 3 mois après la RC

Traitement d'entretien• But: éradiquer la maladie résiduelle• Modalités:
– Administration continue– 6-MP (Purinethol) administrée tous les jours per
os + Méthotrexate hebdomadaire (per os ou IM)– Adaptation dose: PNN entre 1000 et 1500 /mm3
– Réinductions mensuelles Vincristine + Prednisone + Intra-thécale
• Durée optimale : 2 à 3 ans

Prophylaxie neuroméningée - Systématique- En l’absence de prophylaxie:
Incidence des rechutes méningées 50 % (versus 2-3%)- Pourquoi ?
- SNC: site sanctuaire pour les cellules leucémiques- Chimiothérapie ne passant pas correctement la barrière
méningée- Modalités:
- Injections intrathécales (MTX ± ARAC ± corticoïdes)- Méthotrexate à haute dose (3 à 8 g/m²)- Irradiation encéphale: forme à très haut risque
- Atteinte méningée initiale- Présence d’une t(9;22)- Formes T hyperleucocytaires

FRALLE 2000-APrephase
Prednisone + IT MTXINDUCTION
VCR, DEX, L-Aspa
D21 marrow
M1: group A1 M2: group A2 M3: group A3
Randomization
+ DNR D22/D29
DNR D22 and D29 DNR D22 and D29
CR D35-D42 : evaluation of MRDBefore result : start A1/A2 consolidation
CR D35-D42 :Evluation of MRD
MRD(+)> 10-2
CONSOLIDATION A1/A2
12 weeksVCR, DEX, 6-MP, MTX
If D35 MRD < 10-2
CONSOLIDATION A3
9 weeks / 3 curesVEDA/COPA DM2000/VEDA
INTENSIFICATION N°1 (8 weeks)VDS, DEX, ADRIA, L-Aspa / VP-16, Ara-C, 6-TG
INTERPHASE(8 weeks)
VCR, DEX, 6-MP, MTX
INTERPHASE(8 weeks)
VCR, DEX, 6-MP, MTX+ MTX-HD x 3
INTENSIFICATION N°2(6 weeks)
no anthracyclinVCR, MTX-DI, L-Aspa
INTENSIFICATION N°2(8 weeks)
VCR, PRED, DNR, L-AspaEndoxan, Ara-C, 6-TG
Maintenance 24 months with oral 6-MP/MTXFisrt year :12 pulses (VCR/DEX 5 days)

FRALLE 2000-BT. Groupe B.
PREPHASE : J1-J7
PREDNISONE + IT MTX
J8-J21 : INDUCTION COMMUNE
VCR, PRED, DNR (J8, J15), L-Aspa x 6 (J8 - J20)
GROUPE B1
GROUPE B2
Suite de l'INDUCTION VCR , PRED + DNR à J22 + L-Aspa x 3 (J22 à J26)
NB : Si MRD à J35-42 > 10-2, passer en B2
Suite de l'INDUCTION
VCR, PRED + DNR à J22 et à J23 + L-ASPA x 3 (J22-J26) + ENDOXAN (J22)
CONSOLIDATION (8 semaines) VP16, ARAC, 6TG puis VCR, PRD, 6MP,MTX
+ 2 cures de MTX-HD (5.000 mg/m2)
CONSOLIDATION (9 semaines) 3 cures : VEDA, COPADM2000, VEDA
INTENSIFICATION n°1 (8 semaines) (1)
VDS / ADRIA / PRED / L-Aspa puis 6-TG / AraC / VP-16
INTERPHASE (8 semaines) VCR, PRED, 6-MP, MTX
+ 2 cures de MTX-HD (5.000 mg/m2)
INTERPHASE (8 semaines) VCR, PRED, 6-MP, MTX
+ 3 cures de MTX-HD (5.000 mg/m2)
IRRADIATION 18 Gy jusqu'à C2 (2)
INTENSIFICATION n°2 (8 semaines) VCR, PRED, DNR, L-ASPA puis 6-TG, Endoxan, AraC
ENTRETIEN 24 mois 6-MP + MTX avec 12 RI : VCR + PRED

FRALLE 2000-BT : Groupe T PREPHASE : J1-J7
PREDNISONE + IT MTX
INDUCTION COMMUNE VCR, PRED, DNR à J8, J9, J10 et J15,
ENDOXAN 1 g/m2 à J8 + L-Aspa x 9
GROUPE T1
NB : Si MRD à J35-J42 > 10-2 , passer en T2
GROUPE T2
CONSOLIDATION (8 semaines) Endoxan, ARAC, 6TG
puis VCR, PRED, 6MP, MTX + 2 cures de MTX-HD (5.000 mg/m2)
CONSOLIDATION (9 semaines) 3 cures : VEDA, COPADM2000, VEDA
INTENSIFICATION n°1 (8 semaines) (1) VDS / ADRIA / PRED / L-Aspa
puis 6-TG, AraC, VP-16
INTERPHASE (8 semaines)
VCR, PRED, 6-MP, MTX + 4 cures de MTX-HD (5.000 mg/m2) (2)
INTERPHASE (8 semaines) VCR, PRED, 6-MP, MTX
+ 3 cures de MTX-HD (5.000 mg/m2)
IRRADIATION 18 Gy jusqu'à C2 (3)
INTENSIFICATION n°2 (8 semaines)
VCR, PRED, DNR, L-ASPA puis 6-TG, Endoxan, AraC
ENTRETIEN 18 mois
6-MP + MTX avec 12 RI : VCR + PRED

INDICATIONS DE GREFFE DE MOELLE EN RC1
Seules des greffes génoidentiques sont indiquées dans ce protocole.
LAL de la lignée B
- La présence d'une t(9;22) ou d'un transcrit BCR-ABL.- La présence d'une t(4;11) ou d'un transcrit MLL-AF4.- Les formes hyperleucocytaires à plus de 100.000 blancs au diagnostic qui sont :
- soit corticorésistantes à J8.- soit chimiorésistantes avec une moelle de type M3 à J21.
-Les patients ayant une MR positive ( > 10-2) à J35-J42 confirmée sur un 2ème prélevement fait 3 à 4 semaines après.
LAL de la lignée T
- une corticorésistance à J8- une chimiorésistance à J21- une MR élevée ( > 10-2) à J35

3. LEUCÉMIES AIGUES LYMPHOBLASTIQUES
I. Données généralesII. Facteurs pronostiquesIII. TraitementsIV. Résultats actuels

INTERFANT 99: 481 pts
LAL du Nourrisson < 1 an

Fralle 2000 A: « risque standard »
• 524 patients inclus• Recul médian: 28 mois
– Survie globale à 3 ans : 97.6% (96.0-99.2) – Survie sans évènement à 3 ans : 95.4% (93.2-
97.7)– Survie sans maladie à 3 ans : 95.5% (93.0-98.0)
• 495 patients randomisés– 247 DNR+– 248 DNR-

SURVIE GLOBALE (whole cohort)
0 10 20 30 40 50
0.0
0.2
0.4
0.6
0.8
1.0
Months
P(s
urvi
val)

Survie sans évènement (whole cohort)
0 10 20 30 40 50
0.0
0.2
0.4
0.6
0.8
1.0
Months
P(n
o ev
ent)

Survie sans maladie (whole cohort)
0 10 20 30 40 50
0.0
0.2
0.4
0.6
0.8
1.0
Months
P(n
o ev
ent)

FRALLE 2000 BT: « haut risque »Devenir à 3 ans
- Survie globale : 85 ± 6%- Survie sans évènement : 77 ± 5%- Survie sans maladie : 81 ± 4%

LAL chez l’adolescent: 15 à 19 ans

0
,2
,4
,6
,8
1
0 1 2 3 4 5 6
FRALLE 93
LALA 94
P<0.0001
41 % (± 14)
67 % (± 13)
5-year EFS
EFS
Time (years)
EFS (N=177)

RESULTATS PRELIMINAIRES FRALLE 2000
• RC: 73/76 (96%)• Groupe après la RC - BR ( B1/T1): 50 - MR (B2/T2): 233y EFS: 86.5 + 9 % (73 + 10 % F 93)3y DFS: 90 + 9 %3y OS: 97 + 5 %

4. LEUCÉMIES AIGUES MYELOBLASTIQUES
I. Données généralesII. Facteurs pronostiquesIII. TraitementsIV. Résultats actuelsV. Cas particulier: LAM3

Epidémiologie: 0 à 14 ansRegistre national, hémopathies malignes, 1990-1999
J. Clavel, European Journal of Cancer Prevention, 2004
- Population:- Age : 0 à 14 ans- Enfants français métropolitains- Leucémies aiguës : 4 479 cas
- LAM : 770 cas soit 17 %- (LAL : 3 642, autres LA : 67)
- Répartition - Enfants < 1 an- Puis homogène dans toutes les tranches d'âge

Epidémiologie chez l’adolescent
Registre national des tumeurs solides de l'enfant B. Lacour, E. Desandes
- Age : 15 à 19 ans- Cancers : 700/an (1/1000 adolescents)- Leucémies : 12 %- LAM : 3,5 % ( 25/an en France)

Leucémies aiguës myéloblastiques de l’enfantType FAB Terminologie commune Corps d'Auer Réactions
cytochimiques Fréquence %
M0 Myéloblastique avec différenciation myéloïde
minime
- MP – (< 3 %) 3
M1 Myéloblastique sans maturation
MP + 13
M2 Myéloblastique avec maturation
+ MP + 32
M3 Promyélocytaire +++ (en fagots) MP + 8
M4 Myélomonocytaire MP + NSE + 12
M4 éos Myélomonocytaire avec éosinophilie
MP + NSE + 5
M5 Monoblastique sans (a) ou avec (b) différenciation
NSE + 20
M6 Erythroleucémie MP + PAS + 2
M7 Mégacaryoblastique - MP plaquettaire + 6
MP : myélopéroxydase, NSE : estérases non spécifiques, PAS : periodic acid schiff


















