
Evaluation clinique des dispositifs médicaux - girci-go.org€¦ · › L’essai clinique est de...
58
Evaluation clinique des dispositifs médicaux Particularités méthodologiques Dr P Fabbro-Peray BESPIM - CHU de Nîmes
Transcript of Evaluation clinique des dispositifs médicaux - girci-go.org€¦ · › L’essai clinique est de...

Evaluation clinique des dispositifs médicaux
Particularités méthodologiques
Dr P Fabbro-Peray
BESPIM - CHU de Nîmes

Plan
• Rappels parcours du médicament
• Différences médicament - DM
• Parcours du Dispositif Médical
• Particularités méthodologiques des essais DM
Dr P. Fabbro-Peray - CHU de Nîmes 2

• Très encadré règlementairement : FDA / EMEA / ANSM
– Phase pré-clinique : expérimentation in vitro / animal
– Phase I : 1ère administration à l’Homme = volontaires sains
• tolérance (fixer les limites de la toxicité, pharmacocinétique)
– Phase II : Administration à des malades pour une 1ère évaluation de
l’efficacité pharmacologique
• effet dose, posologie optimale
– Phase III : Evaluation comparative doit apporter la preuve de l’efficacité du
médicament ou de sa supériorité par rapport au médicament de référence (imputation causale)
Parcours du Médicament
Dr P. Fabbro-Peray - CHU de Nîmes 3

Parcours du Médicament
La genèse d’un médicament› Par l’industrie pharmaceutique› 10-15 ans de recherche pour explorer tous les champs d’investigation.› Ces travaux, tests précliniques, essais cliniques et de développement
industriel, sont strictement encadrés par la loi (Loi Huriet 1988 puis loi biomédicale 2004)
Autorisation de mise sur le marché d’un médicament (AMM)› Délivrée par
• l’ANSM (autorité compétente nationale)• l’EMEA (autorité compétente européenne) pour les médicaments innovants
ou destiné à plusieurs états membres de la CEE• Après évaluation du rapport bénéfice-risque qui doit être positif selon 3
critères:– Qualité– Efficacité– Sécurité
• Non comparative
Dr P. Fabbro-Peray - CHU de Nîmes 4

Parcours du Dispositif Médical (DM)
Définition› « On entend par dispositif médical tout instrument, appareil,
équipement, matière, produit, à l’exception des produits d’origine humaine, ou autre article seul ou en association, y compris les accessoires et logiciels intervenant dans son fonctionnement, destinépar le fabricant à être utilisé chez l’homme à des fins médicales et dont l’action principale voulue n’est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. Les DM qui sont conçus pour être implantés en totalité ou en partie dans le corps humain ou placés dans un orifice naturel, et qui dépendent pour leur bon fonctionnement d’une source d’énergie électrique ou de toute source d’énergie autre que celle qui est générée directement par le corps humain ou la pesanteur, sont dénommés dispositifs médicaux implantables actifs. »
Dr P. Fabbro-Peray - CHU de Nîmes 5

• Classification des DM en fonction du risque
– Classe I Faible degré de risque
– Classe IIa Degré moyen de risque
– Classe IIb Potentiel élevé de risque
– Classe III Potentiel très sérieux de risque (comprend les DM implantables actifs)
Dr P. Fabbro-Peray - CHU de Nîmes 6
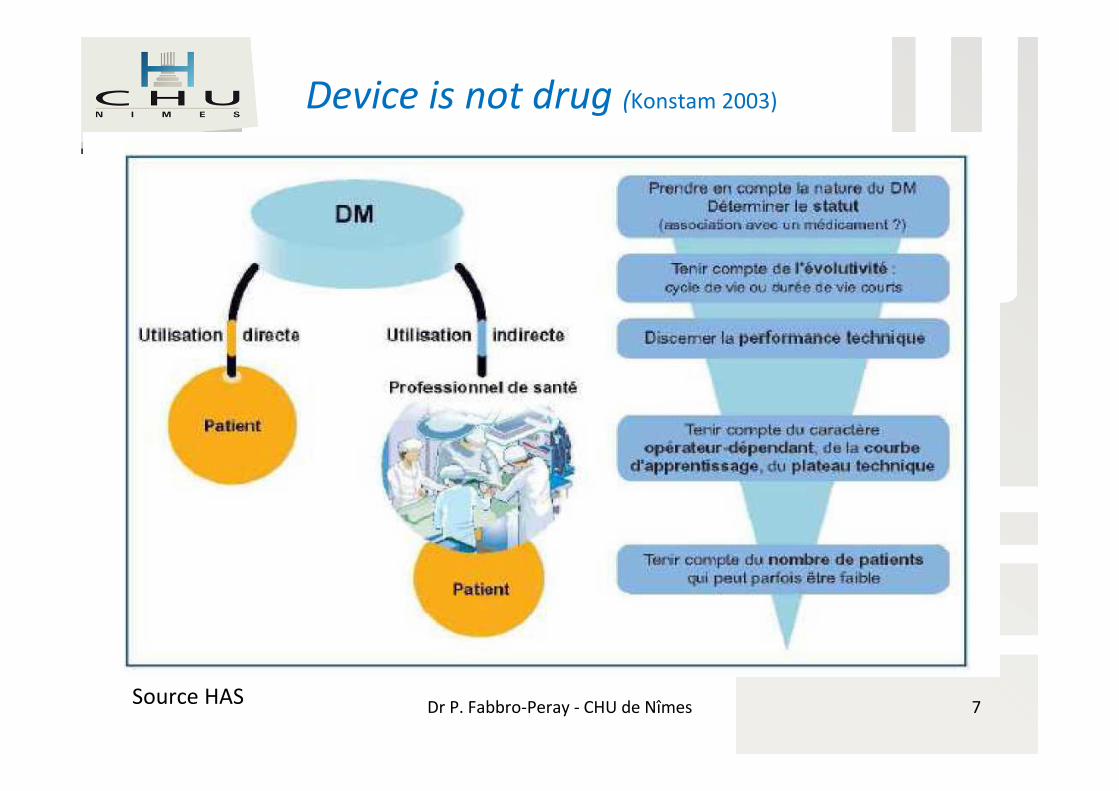
Dr P. Fabbro-Peray - CHU de NîmesSource HAS
Device is not drug (Konstam 2003)
7

Spécificités DM / médicament
Phase préclinique → marquage CEperformance technique ≠ performance clinique
Hétérogénéité +++: tout un mondeDu matelas anti-escarre au robot chirurgical, en
passant par le stent actif!
Statut du produitAssocié ou non à un médicament
Durée de vie Souvent très courte du fait de l’évolution
technologique
Dr P. Fabbro-Peray - CHU de Nîmes 8

Spécificités DM / médicament
Population cible parfois faible
Parfois quelques centaines de patients concernés
Différence essentielle +++
Le bénéfice clinique peut dépendre :
• Du DM lui-même
• Mais aussi des Performances de l’Équipe médicale– courbe d’apprentissage, caractère opérateur dépendant
• Et du Plateau technique – dimension organisationnelle
Dr P. Fabbro-Peray - CHU de Nîmes 9

En matière d’évaluation…Parcours DM
Moins de pré-requis que pour le médicament
Pas de phases précoces standardisées réglementaires mais Le marquage CE a évolué (Directive 2007/47/CE en application depuis le 20 mars 2010)
› Renforcement de la nécessité pour le responsable de la mise sur le marché d’apporter des DONNEES CLINIQUES
› L’essai clinique est de règle pour les dispositifs de classe III
› L’évaluation clinique du marquage CE vise à justifier la revendication d’utilisation médicale en termes de RAPPORT BENEFICE-RISQUE
Les exigences pourront évoluer selon le DM vers un objectif de démonstration de l’impact clinique ou médico-économique› A anticiper dès le début de l’évaluation du DM
→ Développement de partenariats industrie / hôpitaux++
Dr P. Fabbro-Peray - CHU de Nîmes 10

• Dispositifs médicaux
• Chirurgie
• Procédures techniques
• Rééducation
• Psychothérapies
• Interventions comportementales
• Médecines alternatives
• …
Typologie des thérapeutiques non médicamenteuses
Dr P. Fabbro-Peray - CHU de Nîmes
Des points communs méthodologiques
11

Particularités méthodologiques des essais non médicamenteux
• Représentaient en 2000 ≅ ¼ des publications d’essais thérapeutiques
• Interventions souvent complexes à plusieurs composantes– Difficiles à standardiser, à reproduire et à administrer de manière similaire
chez tous les patients
• Aveugle plus difficile à réaliser– Méthodes complexes
– Problèmes éthiques : par exemple simulations d’interventions en chirurgie
• Le même investigateur ou le même centre peut ne pas pouvoir appliquer indifféremment les deux interventions
– Ex: expertise particulière pour une des interventions
– Ex: préférence notoire pour une technique rendant le respect de la randomisation difficile : Cross-over entre les groupes
– Ex: équipement indisponible pour réaliser un des interventions (ex: robot)Dr P. Fabbro-Peray - CHU de Nîmes 12

• Expertise des « prestataires de soins » et volume d’activitédes centres hétérogènes ++++++– Peuvent influencer l’estimation de l’effet du traitement
– Exemple:
• dans un essai concernant l’Athérosclérose Carotidienne Asymptomatique, 40% des centres possibles sélectionnant seulement ceux ayant des bons résultats en terme de sécurité dans leur file active, cad ceux ayant la plus grosse expérience
� ⇒ taux de mortalité post op 8 fois plus faible que dans d’autres essais avec des critères de sélection des centres moins draconiens
� ⇒ Problème de validité externe ou d’extrapolabilité des résultats
• Si on est moins strict sur les critères de sélection des centres,
– Risque d’effet cluster
Dr P. Fabbro-Peray - CHU de Nîmes
Particularités méthodologiques des essais non médicamenteux
13

Particularités méthodologiques des essais non médicamenteux
• Effet cluster– Dans les essais médicamenteux:
• Hypothèse: les évènements survenant chez les patients inclus par un même médecin et dans le même centre sont indépendants
– Dans les essais non médicamenteux
• Cette hypothèse n’est souvent plus valide : le succès du traitement sera partiellement lié à l’expérience des équipes soignantes et/ou au volume d’activité du centre
• Phénomène aggravé par l’absence d’aveugle
– Possible influence du « prestataire de soins » sur l’estimation de l’effet et dépendant en plus du groupe de traitement
• Dans ces essais les observations des patients traités par le même prestataire de soins ou dans un même centre peuvent être corrélés: EFFET CLUSTER qui entraine une perte de puissance lié au coefficient de corrélation intra cluster
Dr P. Fabbro-Peray - CHU de Nîmes 14

Conséquences sur le protocole• En + des standards habituels
• Définir les critères d’inclusion des « prestataires de soins » et des centres
– Pour les centres (exemple): • volume d’activité
• …
– Pour les prestataires de soins (exemples): • qualification professionnelle,
• années de pratique,
• nombre d’interventions réalisées,
• période de formation avant l’essai,
• courbe d’apprentissage
• …. Dr P. Fabbro-Peray - CHU de Nîmes 15

Conséquences sur le protocole• Décrire précisément les interventions dans les
différents groupes, – Dépend du type d’intervention évaluée
– Chirurgie, procédure technique, dispositif implantable• Détails des procédures préopératoire, intra-opératoire et post-opératoire,
description du dispositif implantable
– Dispositif non implantable• Description & Guide d’utilisation du dispositif
• Décrire les méthodes de standardisation utilisées pour les appliquer– Essais pragmatiques (évaluation en conditions réelles d’utilisation)
• Standardisation = informer les intervenants d’administrer le traitement comme à leur habitude
Dr P. Fabbro-Peray - CHU de Nîmes 16

Conséquences sur le protocole
• Tenir compte de l’effet cluster potentiel
– Dans le calcul du NSN• Prise en compte d’une corrélation intracluster
– Dans l’analyse statistique• Ajustement sur les caractéristiques permettant un contrôle de l’effet
cluster
Dr P. Fabbro-Peray - CHU de Nîmes 17

Conséquences sur le protocole
• Lorsque l’affectation individuelle de l’intervention est impossible ou difficile– Essai en CLUSTERS: Randomisation de « clusters » (groupes)
• Médecins
• Services
• Hôpitaux…
– Pourquoi?• Contraintes logistiques / disponibilité du DM
– Exemples: robot chirurgical
– Impact sur le calcul du NSN et l’analyse statistique• Prise en compte de l’effet cluster
Dr P. Fabbro-Peray - CHU de Nîmes 18

Conséquences sur le protocole
Si Aveugle possible
– Décrire les méthodes permettant de s’assurer du respect de l’aveugle
• Simulation intervention chirurgicale
• Aveugle des hypothèses de l’étude
Si Aveugle impossible ou trop complexe– Importance dans ce contexte d’obtenir l’aveugle des
intervenants dans la chaine de prise en charge et des évaluateurs
Dr P. Fabbro-Peray - CHU de Nîmes 19

La démarche CONSORT
• Consolidated Standards of Reporting Trials
– 1ère série de recommandations sur la manière de rapporter les essais thérapeutiques
• Groupe d’experts internationaux
– Méthodologistes
– Investigateurs
– Editeurs de journaux
• Consensus
• Remises à jour régulièresDr P. Fabbro-Peray - CHU de Nîmes 20

Dr P. Fabbro-Peray - CHU de Nîmes21

CONSORT
• D’abord pour les essais en groupes parallèles
• ….
• Puis extensions méthodologiques
– Clusters
– Traitements non pharmacologiques
• Check list: Items supplémentaires
• Flow diagram : exigences spécifiques sur le flux de patients concernant les centres, les médecins…
– …
Dr P. Fabbro-Peray - CHU de Nîmes 22

Dr P. Fabbro-Peray - CHU de Nîmes
Source: Ph Ravaud
23

Dr P. Fabbro-Peray - CHU de Nîmes 24

Dr P. Fabbro-Peray - CHU de Nîmes
Check list: 11 items modifiés, 1 nouvel item par rapport au CONSORT essais parallèles
25

Volume 374, Issue 9695, Pages 1105 - 1112, 26 September 2009
No surgical innovation withoutevaluation : the IDEAL
recommendations
Peter McCulloch, Douglas G Altman, W Bruce Campbell , David R Flum, Paul Glasziou, John C Marshall, Jon Nicholl, for the Bal liol Collaboration‡
Dr P. Fabbro-Peray - CHU de Nîmes 26

IDEAL
• Les différentes phases préconisées
– Phase 1 : innovation
– Phase 2a : développement
– Phase 2b : exploration
– Phase 3 : évaluation
– Phase 4: suivi à long terme
Dr P. Fabbro-Peray - CHU de Nîmes
Les recommandations IDEAL peuvent s’appliquer à de nombreux DM associés à des actes de chirurgie ou interventionnels
27

Dr P. Fabbro-Peray - CHU de Nîmes 28
Source: Barkun JS, 2009, et al. Lancet
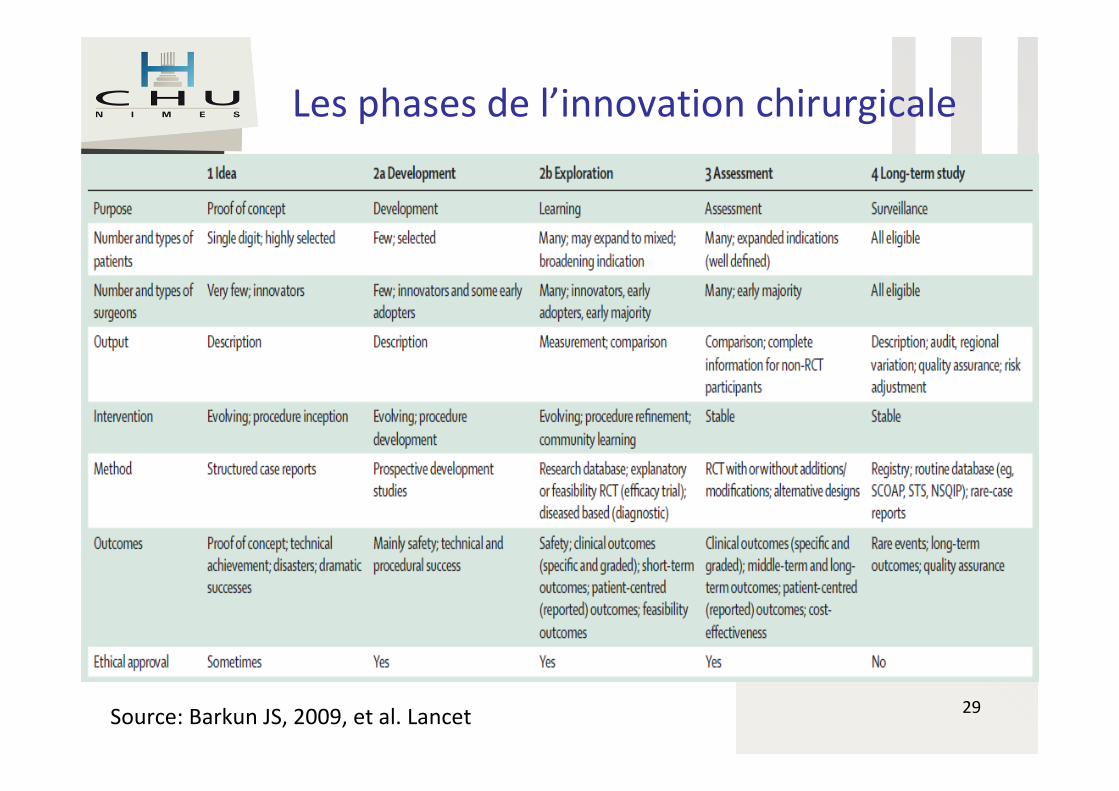
Les phases de l’innovation chirurgicale
29Source: Barkun JS, 2009, et al. Lancet

Conclusion
• Innovation et évaluation doivent évoluer de concert
• En tenant compte des spécificités des DM qui rendent les évaluation plus difficiles et plus sujettes à biais
• Communiquer sur les différentes étapes est essentiel– Sur chaque phase de développement d’un DM ou d’une
nouvelle technique
– Enregistrer les données dans des registres
– Rapporter les résultats selon les normes internationales standardisées
Dr P. Fabbro-Peray - CHU de Nîmes 30

Conclusion
• L’industrie pharmaceutique est contrainte par le législateur
• Le DM doit essentiellement montrer qu’il est :
– Techniquement performant
– Sur pour le patient • D’où les moindres contraintes réglementaires ….jusqu’à
récemment
Les acteurs institutionnels se doivent d’être force de proposition et d’innovation dans l’évaluation des nouvelles technologies médicales
Dr P. Fabbro-Peray - CHU de Nîmes 31

Evaluation clinique desdispositifs médicaux
Vigilance et sécurité
Dr. M-P Franceschi
BESPIM CHU de Nîmes

L’environnement réglementaire
� Cadre règlementaire européen : 3 directives dites nouvelle approche: 90/385/CEE(DMIA)*, 93/42/CEE(DM)*, 98/79/CE (DMDIV). *Modifiées par directive 2007/47/CE entrée en application en mars 2010
� Marquage CE: pré-requis à la mise sur le marché des DM et DM-DIV: � Matérialise la conformité du DM/DMDIV aux exigences. � Est apposé par le fabricant sous sa seule responsabilité .
� L’ANSM n’intervient pas dans le processus d’autorisation des DM/DMDIV mais est impliquée dans:� Evalue et autorise les recherches biomédicales.� Evalue le rapport bénéfice/risque et le respect de la règlementation dans le
cadre de la surveillance du marché.
*
Dr. M-P Franceschi BESPIM CHU de Nîmes

Définitions, classification � Dispositif médical:
� Tout instrument, appareil, équipement, matière, produit, à l’exception des produits d’origine humaine, ou autre article seul ou en association, y compris les accessoires et logiciels intervenant dans son fonctionnement, destiné par le fabricant à être utilisé chez l’homme à des fins médicales et dont l’action principale voulue n’est pas obtenue par des moyens pharmacologiques ou immunologiques ni parmétabolisme, mais dont la fonction peut être assistée par de tels moyens.
� Les dispositifs médicaux qui sont conçus pour être implantés en totalité ou en partie dans le corps humain ou placés dans un orifice naturel, et qui dépendent pour leur bon fonctionnement d'une source d'énergie électrique ou de toute source d'énergie autre que celle qui est générée directement par le corps humain ou la pesanteur, sont dénommés dispositifs médicaux implantables actifs.
Art L.5211-1CSP, Art premier point 2.a directive 93/42/CE
Dr. M-P Franceschi BESPIM CHU de Nîmes

Définitions, classification,
� Dispositifs médicaux de diagnostic in vitro :
Produits, réactifs, matériaux, instruments et systèmes, leurs composants et accessoires, ainsi que les récipients pour échantillons, destinés spécifiquement à être utilisés in vitro, seuls ou en combinaison, dans l'examen d'échantillons provenant du corps humain, afin de fournir une information concernant un état physiologique ou pathologique, avéré ou potentiel, ou une anomalie congénitale, pour contrôler des mesures thérapeutiques, ou pour déterminer la sécurité d'un prélèvement d'éléments du corps humain ou sa compatibilité avec des receveurs potentiels. (Art L;5211-1 CSP, ordonnance de transposition n° 2000-198 de la directive 98/79/CE)
Dr. M-P Franceschi BESPIM CHU de Nîmes

Définitions, classification,
� Ne sont pas des DMDIV :
� Les instruments, appareils, équipements, matériaux destinés à être utilisés à des fins de recherche.
� Les produits destinés à des usages généraux en laboratoire. � Les dispositifs invasifs destinés à prélever des échantillons en contact
direct avec le corps humain. � Les matériaux de référence utilisés au niveau international et les
matériels utilisés dans les programmes d’évaluation externe de la qualité.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Définitions, classification
� Dispositifs médicaux :� Répartition en 4 classes : I, IIa, IIb; III en fonction du niveau de risque.
� 18 règles de classification définies dans l’annexe IX de la directive 93/42/CEE permettent de déterminer la classe d’un dispositif (si plusieurs classes possibles la plus élevée est retenue).
� Les dispositifs médicaux implantables actifs (DMIA) constituent une classe à part (Directive 90/385/CEE).
� Dispositifs médicaux de diagnostic in vitro:Pas de répartition en différentes classes mais 2 listes* A et B (+ DM autodiagnostic).*Voir annexe II de la directive 98/79/CE
Dr. M-P Franceschi BESPIM CHU de Nîmes

La Global Médical Device NomenclatureGMDN
� Donne la liste des termes, définitions et codes des DM répertoriés par ordre alphabétique suivant leur non de groupe générique.
� Réalisée conjointement par le CEN et l’ISO et publiée en anglais en 2001 est maintenue et développée par la GMDN Agency.
� Outil de coopération et communication entre états membres pour l’enregistrement et de l’analyse des informations réglementaires sur les DM en particulier en cas d’incidents ( rapports de vigilance, et ou suivi de la sécurité des DM).
� Utilisée comme nomenclature dans tous les rapports entre et avec les autorités.
� Utilisée pour la banque de donnée européenne de dispositifs médicaux « Eudamed ».
Dr. M-P Franceschi BESPIM CHU de Nîmes

Quand est-on dans un essai DM?
� Est une recherche biomédicale portant sur un dispositif médical tout essai clinique ou investigation clinique d’un ou plusieurs dispositifs médicaux visant à déterminer ou à confirmer leurs performances ou à mettre en évidence leurs effets indésirables et à évaluer si ceux-ci constituent des risques au regard des performances assignées au dispositif. Art R. 1121-1CSP:
� ANSM: prise en compte de l’objectif principal pour la qualification des essais.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Quand est-on dans un essai DM?
� objectif de l’essai clinique:
� Étude des performances (l’effet), de l’efficacité, de la sécurité d’un ou de plusieurs DM : Essais DM.
� Etude des conséquences de l’utilisation d’un ou de plusieurs DM ,dans une finalité médicale thérapeutique ou diagnostique: Essai DM.
� Etudier la physiologie, la physiopathologie ou l’anatomie en utilisant un ou plusieurs DM, alors le/les DM sont utilisé(s) pour les besoins de la recherche mais ne sont pas l’objet de la recherche: il s’agit d’un essai HPS.
Ne pas confondre performances techniques et performances cliniques
Dr. M-P Franceschi BESPIM CHU de Nîmes

Quand est-on dans un essai DMDIV?
• Les recherches biomédicales portant sur un dispositif médical de diagnostic in vitro sont entendues comme tout essai clinique visant àévaluer les conséquences cliniques de l'utilisation d'un ou plusieurs dispositifs médicaux de diagnostic in vitro ou tout essai clinique portant sur un dispositif médical de diagnostic in vitro nécessitant pour les seuls besoins de la recherche la pratique d'un acte médical comportant des risques autres que négligeables. Arrêté du 11 mai 2009 relatif aux définitions de certaines catégories de recherches biomédicales, JORF n°0118 du 23 mai 2009 page 8604 texte n° 20
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DM
� Le cadre législatif et règlementaire est totalement calqué sur celui des essais cliniques portant sur des médicaments:
� Mêmes obligations pour les investigateurs.
� Mêmes obligations pour les promoteurs.
� Champ d’application : tous les essais cliniques interventionnels portantsur des DM/DMDIV.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DML’investigateur
� Notifie au promoteur :� Sans délai à compter du jour où il en a connaissance tous les événements
indésirables graves (rapport initial écrit + rapports complémentaires).� Selon les délais et modalités précisés dans le protocole les événements indésirables
et/ou les résultats d’analyse anormaux définis dans le protocole comme déterminants pour la sécurité des personnes participant à l’essai.
� Établit un lien de causalité entre l’EIG et le(s) DM expérimentaux ou le(s) traitement(s) associés.
� S’assure lors de la notification que les éléments suivants sont renseignés :� Identification du patient (n°, âge, sexe), de l’investigateur, de la RBM (n° ID RCB ou
n° du protocole),� dispositif expérimental et/ou un traitement associé,� libellé de l’événement,� relation de causalité et critères de gravité (délai de déclaration).
� Communique au promoteur toute information complémentaire concernant un EIG.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMLe promoteur
� Évalue de façon continue tout au long de la recherche la sécurité d’emploi du dispositif médicalou de tout autre élément expérimental.
� Met en place des structures et procédures écrites afin de garantir le recueil des données, la documentation des cas (événements/effets indésirables, faits nouveaux), leur validation, leur évaluation, leur déclaration et leur archivage.
� Évalue les événements indésirables graves qui lui sont rapportés par les investigateurs (gravité, lien de causalité, caractère inattendu).
� Déclare les suspicions d’effets indésirables graves inattendus (EIGI/SUSAR) à L’ANSM, au CPP et aux investigateurs concernés .
� Déclare les faits nouveaux de sécurité et les mesures urgentes de sécurité à l’ANSM, au CPP et aux investigateurs concernés (si impact sur la conduite de l’essai).
� Prend, le cas échéant des mesures urgentes de sécurité sans attendre l’avis de l’autoritécompétente et/ou du CPP pour protéger les personnes participant à la recherche .
� Transmet un rapport annuel de sécurité à l’autorité compétente et au CPP pendant toute la durée de la recherche ou sur demande.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMLe promoteur
� Évalue les événements indésirables graves :
� Evénement indésirable : manifestation nocive survenant chez une personne participant à la recherche liée ou non à la recherche ou au produit sur lequel porte la recherche (Art R.1123-39 1° CSP).
� Effet indésirable d’un DM ou DMDIV: toute réaction nocive et non désirée à un dispositif médical ou tout incident qui aurait pu entraîner cette réaction si une action appropriée n’avait pas été effectuée, chez une personne qui se prête à la recherche ou chez l’utilisateur du dispositif médical ou tout effet lié à une altération d’un dispositif médical de diagnostic in vitro et néfaste pour la santé d’une personne qui se prête à la recherche. (Art R.1123-39 4° CSP).
� Matériovigilance : signalement obligatoire et sans délai :Le fabricant, les utilisateurs d’un dispositif et les tiers ayant connaissance d’un incident ou d’un risque d’incident mettant en cause un dispositif ayant entrainé ou susceptible d’entrainer la mort ou la dégradation grave de l’état de santé d’un patient, d’un utilisateur ou d’un tiers doivent le signaler sans délai à l’autorité administrative (Art L.5212-12 CSP).
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMLe promoteur
� Évalue les événements indésirables graves:
� Lien de causalité:
� Les symptômes décrits, la localisation, le délai entre la survenue de l’événement et le début d’utilisation du DM sont-ils compatibles?
� A-t-on connaissance d’événement de même typologie avec des DM similaires ?
� Y-a t’il d’autres causes possibles?
� Caractère inattendu, Document de référence:
� Notice d’utilisation/instruction si DM/DMDIV marqué CE et utilisation conforme, BI dans les autres cas.
� Protocole de l’essai.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMLe promoteur
� Déclare
Législation actuelle en France : déclarations à l’ANSM et au CPP :
� suspicions d’effets indésirables graves inattendus lien de causalité DM � événements graves pouvant être liés au geste de mise en œuvre du DM.
Mais
Modification des directives 90/385/CE (DMIA) et 93/42/CE par la directive 2007/47/CE.
Nouvelle législation : «Tous les événements indésirables graves doivent être intégralement enregistrés et communiqués immédiatement à l’ensemble des autorités compétentes des états membres dans lesquels sont réalisées les investigations cliniques ».
Et
Déclaration selon la Phase Pilote mise en place par l’ANSM
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMLe promoteur
� Déclare les faits nouveaux ....
� Fait nouveau : toute donnée de sécurité ou tout fait nouveau qui pourrait: � modifier de manière significative l’évaluation du rapport bénéfices/risques d’un DM/DMDIV ou d’un essai� conduire à envisager des modifications concernant l’utilisation ou la procédure de mise en œuvre du
DM/DMDIV� conduire à envisager des modifications concernant la conduite de l’essai
� Qu’est-ce qu’un fait nouveau dans un essais DM:
Blessure occasionnée à un utilsateur par un DM non marqué CE : fait nouveau
Mais
Blessure occasionnée à un utilsateur par un DM marqué CE utilisé de façon non-conforme: mésusage?
Et
Blessure occasionnée à un utilsateur par un DM marqué CE utilisé de façon conforme : Matériovigilance?
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMLe promoteur
� Envoie des rapports de sécurité :
� Durée de la recherche biomédicale supérieure à 1 an: Envoi du RAS dans les 60 jours suivant la date anniversaire du 1er patient inclus.
� Recherche biomédicale portant sur un DMDMDIV courte:
- Durée de la recherche inférieure à 1 an mais supérieure à 6 mois: idem cas général.
- Durée de la recherche inférieure à 6 mois: dans les 90 jours suivants la fin de recherche.
Dr. M-P Franceschi BESPIM CHU de Nîmes

Vigilance et sécurité des essais DMQuelques réflexions
� Veiller à la qualité des données recueillies: un formulaire d’EIG renseignéde façon précise permet d’optimiser l’évaluation de l’événement.
� Sécuriser le circuit des DM expérimentaux dans les centres investigateurs:
� Les essais de DM se déroulent la plus part du temps au bloc opératoire ou au niveau de plateaux techniques (radio/cardio interventionnelles) où le personnel est nombreux à intervenir : tous ne sont pas formés à la recherche clinique!
� Sensibiliser les intervenants: un DM expérimental est « réservé » à un essai particulier et ne doit être utilisé que pour les patients inclus da ns l’essai .
� Bien identifier les DM expérimentaux et leur lieu de stockage afin d’éviter l’utilisation hors essais (et vice-versa)!
� Attention les DM implantables sont soumis à traçabilité : penser à récupérer une étiquette de traçabilité pour le cahier d’observation (il y en a plusieurs par conditionnement).
Dr. M-P Franceschi BESPIM CHU de Nîmes

Le formulaire de déclaration d’EIG
Identifier la recherche
Dr. M-P Franceschi BESPIM CHU de Nîmes

Le formulaire de déclaration d’EIG
Identifier le patient
Dr. M-P Franceschi BESPIM CHU de Nîmes

Le formulaire de déclaration d’EIG
Identifier l’événement
Dr. M-P Franceschi BESPIM CHU de Nîmes

Le formulaire de déclaration d’EIG
Identifier le dispositif médical
Il s’agit de l’utilisation du DM, dans le cas d’un DM implanté, si il n’est explanté suite à l’EIG l’utilisation continue : ne pas confondre avec la date de fin d’EIG
Dr. M-P Franceschi BESPIM CHU de Nîmes

Le formulaire de déclaration d’EIG
Identifier le dispositif médicalEt non pas Prolène qui
est un nom de marque
Dans ce cas précis la réf est importante car à une référence correspond un fil ayant des caractéristiques d’aiguille etun diamètre précis.
Peut être un lot de fabrication le plus souvent mais aussi un lot de stérilisation
Dans cet exemple il s’agit un fil de suture non résorbable indiquéen chirurgie cardiaque, vasculaire
Dr. M-P Franceschi BESPIM CHU de Nîmes

Le formulaire de déclaration d’EIG
Identifier le dispositif médical
Autre exemple: les ballons d’angioplastie périphérique qui existent en différents diamètres et longueur
BALLON WANDA H965SCH505050
04MMX10CM cathéter 075CM
BALLON WANDA H965SCH505040
04MMX04CM cathéter 075CM
BALLON WANDA H965SCH505030
04MMX02CM cathéter 075CM
BALLON WANDA H965SCH505260
03MMX8CM 120CM
BALLON WANDA H965SCH505010
03MMX4CM 135CM
BALLON WANDA H965SCH505240
03MMX4CM 080CM
BALLON WANDAH965SCH505240
03MMX2CM 135CMBallon coaxial sur guide 0.035
Haute pression
BALLON ANGIOPLASTIE PERIPHERIQUE
Libellé fournisseur Gamme produit (tailles)Descriptif Dénomination commune
Non CommercialFournisseur: BOSTON SCIENTIFIC
Référence
Dr. M-P Franceschi BESPIM CHU de Nîmes

En guise de conclusion: La vigilance des essais cliniques est un
travail d’équipe
Dr. M-P Franceschi BESPIM CHU de Nîmes

Merci pour votre attention
Dr. M-P Franceschi BESPIM CHU de Nîmes


















