
Electrochimie dans l'acide trifluoroacetique: II. Domaine d'électroactivité—Systèmes de...
11
ELECTROANALYTICAL CHEMISTRY AND INTERFACIAL ELECTROCHEMISTRY 393 Elsevier Sequoia S.A., Lausanne- Printed in The Netherlands ELECTROCHIMIE DANS L'ACIDE TRIF'LUOROACETIQUE II. DOMAINE D'I~LECTROACTIVITI~ SYSTI~MESDE Rt~FI~RENCE--I~TUDEDU SYSTI~ME I~LECTROCHIMIQUE DE L'HYDROGI~NE GI~RARD PETIT ET JACQUES BESSII~RE Laboratoire de Chirnie analytique de la Facult~ des'Sciences associ~ au C.N,R.S., E.P.C.L, 10 rue Yauquelin Paris Ve (France) (Re~u le 23 drcembre 1970) I. INTRODUCTION L'acide trifluoroacrtique, solvant fortement acide et peu basique, permet le dosage de bases qui sont faibles dans reau ou dans l'acide acrtique 1'2. I1 est utilis6 en chimie organique, entant que milieu rractionnel: darts ce sol- vant la cinbtique des rractions de substitution 61ectrophile est grnrralement plus rapide et plus simple que dans l'acide acrtique 3,4. L'acide trifluoroacrtique est en outre difficilement oxydable. De nombreuses esp+ces trrs oxydantes peuvent donc &re 6tudires par 61ectrochimie darts ce solvant. C'est ainsi que sont mis en 6vidence les degrrs d'oxydation (+I) et (+III) de l'iode s. L'acide trifluoroacrtique est un solvant peu dissociant; sa constante dirlectri- que &ant 6gale/l 8.4/~ 20°C 6. On salt que dans de tels milieux les solutions concen- trres de sel ionis6 et partiellement dissocir, sont suffisamment conductrices pour qu'il soit possible d'utiliser les mrthodes 61ectrochimiques d'analyse. Les sels utilisrs corn- me 61ectrolytes doivent donc satisfaire des conditions de solubilit6 et de dissociation. Hara et Cady ont montr6 que les perchlorates et les trtrafluoborates alcalins grnbrale- ment utilisrs en rant que tels dans d'autres solvants, sont peu solubles 7. Par contre la solubilit6 du perchlorate de trtrarthylammonium (Et4NC104), du trifluoroacrtate de sodium et de l'acide perchlorique ainsi que leur constante de dissociation ionique permettent leur utilisation comme 61ectrolyte support s. Ce solvant n'a fait robjet que de quelques &udes 61ectrochimiques 6'9. Conway et Vijh se sont intrressrs ~ la cinrtique 61ectrochimique ~le la rraction de Kolbe, en milieu trifluoroac&ate de sodium,/t des 61ectrodes d'or et de platine poli. Dans ce mrmoire, nous nous sommes limitrs/l la drtermination du domaine d'61ectroactivit6 du solvant h des 61ectrodes de nature diffrrente et/t la description de diff6rents systrmes oxydorrducteurs pouvant servir de systrme de rrfrrence. Tousles systrmes oxydorrducteurs drcrits sont caractrrisrs par des constantes apparentes puisque les 6tudes sont faites en prrsence d'61ectrolyte en grande concen- tration; celui-ci participe aux rractions 61ectrochimiques. Nous montrerons dans un prochain mbmoire qu'il est possible de drterminer des constantes qui ne drpendent pas de l'6lectrolyte support, en tenant compte de la dissociation ionique des paires d'ions. Seule la connaissance de ces constantes permet d'rtablir des corrrlations entre J. Electroanal. Chem., 31 (1971)393-403
-
Upload
gerard-petit -
Category
Documents
-
view
216 -
download
2
Transcript of Electrochimie dans l'acide trifluoroacetique: II. Domaine d'électroactivité—Systèmes de...

ELECTROANALYTICAL CHEMISTRY AND INTERFACIAL ELECTROCHEMISTRY 393 Elsevier Sequoia S.A., Lausanne - Printed in The Netherlands
ELECTROCHIMIE DANS L'ACIDE TRIF'LUOROACETIQUE
II. DOMAINE D'I~LECTROACTIVITI~ SYSTI~MES DE Rt~FI~RENCE--I~TUDE DU SYSTI~ME I~LECTROCHIMIQUE DE L'HYDROGI~NE
GI~RARD PETIT ET JACQUES BESSII~RE Laboratoire de Chirnie analytique de la Facult~ des'Sciences associ~ au C.N,R.S., E.P.C.L, 10 rue Yauquelin Paris Ve (France)
(Re~u le 23 drcembre 1970)
I. INTRODUCTION
L'acide trifluoroacrtique, solvant fortement acide et peu basique, permet le dosage de bases qui sont faibles dans reau ou dans l'acide acrtique 1'2.
I1 est utilis6 en chimie organique, en tan t que milieu rractionnel: darts ce sol- vant la cinbtique des rractions de substitution 61ectrophile est grnrralement plus rapide et plus simple que dans l'acide acrtique 3,4. L'acide trifluoroacrtique est en outre difficilement oxydable. De nombreuses esp+ces trrs oxydantes peuvent donc &re 6tudires par 61ectrochimie darts ce solvant. C'est ainsi que sont mis en 6vidence les degrrs d'oxydation (+I) et (+II I ) de l'iode s.
L'acide trifluoroacrtique est un solvant peu dissociant; sa constante dirlectri- que &ant 6gale/l 8.4/~ 20°C 6. On salt que dans de tels milieux les solutions concen- trres de sel ionis6 et partiellement dissocir, sont suffisamment conductrices pour qu'il soit possible d'utiliser les mrthodes 61ectrochimiques d'analyse. Les sels utilisrs corn- me 61ectrolytes doivent donc satisfaire des conditions de solubilit6 et de dissociation. Hara et Cady ont montr6 que les perchlorates et les trtrafluoborates alcalins grnbrale- ment utilisrs en rant que tels dans d'autres solvants, sont peu solubles 7. Par contre la solubilit6 du perchlorate de tr trarthylammonium (Et4NC104), du trifluoroacrtate de sodium et de l'acide perchlorique ainsi que leur constante de dissociation ionique permettent leur utilisation comme 61ectrolyte support s.
Ce solvant n'a fait robjet que de quelques &udes 61ectrochimiques 6'9. Conway et Vijh se sont intrressrs ~ la cinrtique 61ectrochimique ~le la rraction de Kolbe, en milieu trifluoroac&ate de sodium,/t des 61ectrodes d'or et de platine poli.
Dans ce mrmoire, nous nous sommes limitrs/l la drtermination du domaine d'61ectroactivit6 du solvant h des 61ectrodes de nature diffrrente et / t la description de diff6rents systrmes oxydorrducteurs pouvant servir de systrme de rrfrrence.
Tousles systrmes oxydorrducteurs drcrits sont caractrrisrs par des constantes apparentes puisque les 6tudes sont faites en prrsence d'61ectrolyte en grande concen- tration; celui-ci participe aux rractions 61ectrochimiques. Nous montrerons dans un prochain mbmoire qu'il est possible de drterminer des constantes qui ne drpendent pas de l'6lectrolyte support, en tenant compte de la dissociation ionique des paires d'ions. Seule la connaissance de ces constantes permet d'rtablir des corrrlations entre
J. Electroanal. Chem., 31 (1971) 393-403

394 G. PETIT. J. BESSI~3~E
<
<
x
e
J. Electroanal. Chem., 31 (1971) 393-403
+ + + + 1 1
d d o d d o I + 1 1 1 1
~ t 7 7 1
I I I I I I

I~LECTROCHIMIE DANS L'ACIDE TRIFLUOROACf~TIQUE. II 395
solvants dissociants et peu dissociants. Nous proposons enfin deux m6thodes de dosage de l'eau dans cet acide.
II. RESULTATS EXPt~RIMENTAUX
Domaine d'dlectroactivitd Le domaine d'61ectroactivit6 du solvant d6pend de son acidit6 et de la nature
des 61ectrodes employ6es. Pour cette 6tude les 61ectrolytes suivants ont 6t6 utilis6s: - -mil ieu neutre non tamponn6: perchlorate de t6tra6thylammonium, solu-
bilit~ voisine de 1.5 M, pKg = 3.7; - -mil ieu acide: acide perchlorique, pKg=4.5; - -mil ieu basique: trifluoroac6tate de sodium, solubilit6 voisine de 1.5 M,
pKg = 4.5. Tout les potentiels sont mesur6s par rapport au potentiel du syst6me de r6-
f6rence Ag(s)IAgC104(s)IEt,NC104 10-1 M (ER) d6crit dans la suite du m6moire. Les limites, donn6es pour une densit6 de courant fix6e arbitrairement A 1 mA
cm- 2, sont d6duites des courbes voltamp6rom6triques. Les r6sultats sont rassembl6s dans le Tableau 1.
1. Rdactions ~lectrochimiques en r~duction Quelle que soit la nature de l'61ectrode, le domaine d'61ectroactivit6 est tou-
jours limits par la r6duction des protons de l'acide trifluoroac6tique en milieu neutre et basique:
CF3COOH + Et4N + + e ~ Et,N + CF3COO- +½ H2(g)
et par la r6duction des protons solvat6s en pr6sence d'acide, par exemple:
CF3COOH~FSO 3 +e --* CF3COOH+FSO 3 +½ H2(g )
Ces r6actions sont mises en 6vidence par le trac6 des courbes intensit6--poten- tiel et par coulom6trie. Lors des 61ectrolyses/t intensit6 impos6e, on note un d6gage- ment gazeux fi la cathode.
Aux 61ectrodes de platine platin6 et de mercure, la limite des domaines en milieu neutre non tamponn6 est situ6e A un potentiel plus n6gatif qu'en milieu basique pour la densit6 de courant choisie. L'ordre est invers6 pour une densit6 plus faible. I1 est vraisemblable que le syst6me 61ectrochimique global est plus lent en milieu neutre qu'en milieu basique.
2. Rkaction dlectrochimique en oxydation La limite du domaine d6pend de la nature de l'61eetrode utilis6e. I1 y a soit
oxydation du solvant, soit oxydation du m6tal constituant l'61ectrode. (a) Cas des ~lectrodes inattaquables : platine, or, carbone vitreux. Comme l'ont
montr6 Conway et Vijh 9, le ph6nom6ne global observ6 en milieu trifluoroac6tate est la r6action de Kolbe.
2 NaCF3COO- 2 e -o C2F6(g) + 2 CO2(g) + 2 Na +
I1 est probable que la m6me r6action se produise en milieu neutre. En milieu acide, on observe 6galement un d6gagement gazeux/t l'anode lots des 61eetrolyses ~t intensit6
J. Electroanal. Chem., 31 (1971) 393-403

396 G. PETIT, J. BESSI~E
constante; il est vraisemblablement compos6 de gaz carbonique et d'hexafluoro- 6thane.
La courbe voltamp6rom6trique, trac6e ~t une 6lectrode d'or ou de platine, pr6sente un maximum vers + 1.7 V lorsque le solvant contient des traces d'eau. L'importance du pic est li6e/t l'6tat de surface de l'61ectrode. Conway et Vijh, qui ont observ6 ce ph6nom~ne, l'attribuent /l la formation d'un film mixte d'oxyde et de radicaux CF~ et CFaCOO" ~ la surface du m6tal. Le film aurait selon eux un effet passivant. Leur hypoth6se semble confirm6e par le fait que nous n'ayons pas observ6 de maximum dans la m6me zone de potentiel, lorsque la courbe est trac6e/~ l'aide d'une 61ectrode de carbone vitreux.
(b) Cas des dlectrodes attaquables : argent et mercure. Comme nous le montre- rons ensuite, l'argent est oxyd6 en perchlorate d'argent insoluble en pr6sence de per- chlorate. On pense que dans ce m6me milieu, le mercure est oxyd6 en perchlorate mercureux bgalement insoluble.
En milieu basique, l 'oxydation de l'argent et du mercure conduit aux trifluoro- ac6tates correspondants. Ces sels m6talliques sont solubles.
III. I~TUDE DE QUELQUES SYSTEMES DE Rt~I~RENCE DANS L'ACIDE TRIFLUOROACI3TIQUE
Les syst6mes Ag(s)/AgCIO4(s) en pr6sence de perchlorate de t6tra6thylam- monium, Ag(s)/AgCFaCOO en pr6sence de trifluoroac6tate de sodium et le syst6me de l'hydrog~ne constituent des syst+mes de r6f6rences satisfaisants dans ce solvant.
1. Systbme Ag(s) /AgClO4(s) /Et4NClO 4 10 -~ M La courbe d'oxydation de l'argent en pr6sence de perchlorate de t6tra&hyl-
ammonium 10-1 M est repr6sent6e sur la Fig. 1. Au cours de l'oxydation, un pr6cipit6 blanc de perchlorate d'argent ayant rapidement un effet passivant se d6pose ~t la surface de l'61ectrode. Le potentiel d'6quilibre pris par une 61ectrode d,argent recou- verte de perchlorate d'argent et plongeant dans une solution de perchlorate en con- centration d~termin6e est stable et reproductible. I1 est vraisemblable que la r6action 61ectrochimique soit la suivante:
Ag(s) + E t , N C I O , - e ~ AgC104(s) + Et ,N +
Le potentiel d'6quilibre est donn6 par la relation:
E = E~ + 0.058 log [Et4N+] / [Et ,NCIO,] =
E ; + 0.058 log KD/[CIO2 ]
KD 6tant la constante de dissociation ionique de la paire d'ions Et4NC10 4. Si c o est sa concentration, on montre ais6ment que [CIO2] est peu diff6rent de KDCo.
Le potentiel varie donc lin6airement avec log Co, la pente th6orique de la droite 6tant de 29 mV par unit6 logarithmique.
E = A-0.029 log c o
L'exp6rience montre que la loi de Nernst est v6rifi6e, la concentration du per- chlorate variant de 10 - 2 ~t 10-1 tool 1 - 1. La pente de la droite a la valeur th6orique. La constante A est 6gale/t -0.029 V par rapport au potentiel de r6f6rence choisi ER (Fig. 2).
J. Electroanal. Chem., 31 (1971) 393-403
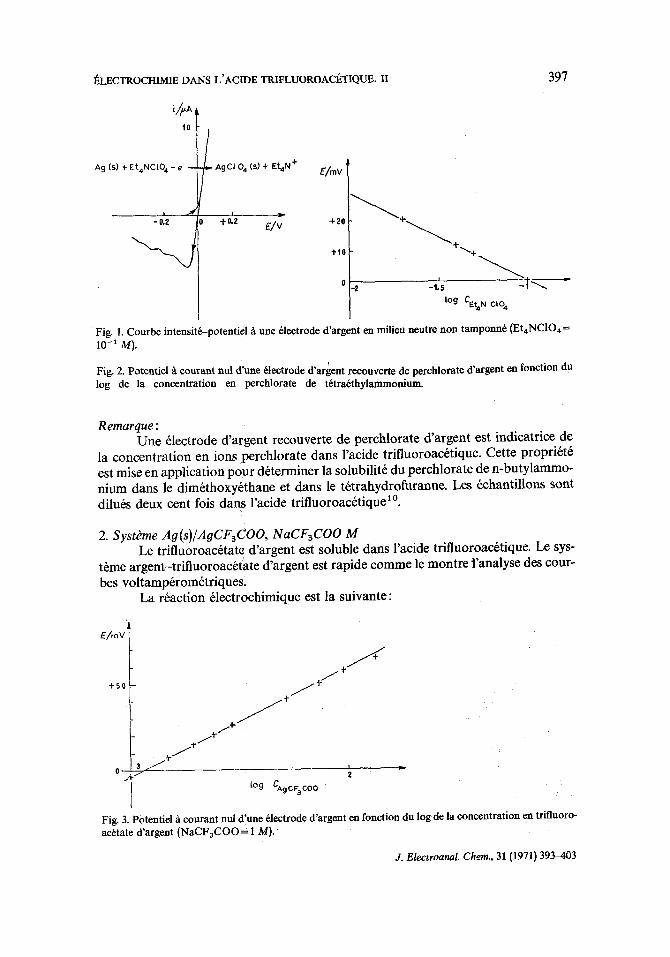
I~LECTROCHIMIE DANS L'ACIDE TRIFLUOROACETIQUE. I I 397
lo
Ag (s) ~- Et4NCIO 4 - e AgC[ 0 4 (s) + Et4N + E/mv l
-,:, log CEt4N Cio4
Fig. 1. Courbe intensit~-potentiel/t une 61ectrode d'argent en milieu neutre non tamponn6 (Et4NCIO4= 10-1 M).
F
Fig. 2. Potentiel A courant nul d'une 61eetrode d'argent recouverte de perchlorate d'argent en fonction du log de la concentration en perchlorate de t6tra6thylammonium.
Remarque : Une 61ectrode d'argent recx)uverte de perchlorate d'argent est indicatrice de
la concentration en iona perchlorate dans l'acide trifluoroac6tique. Cette propri6t6 eat mise en application pour d6terminer la solubilit6 du perchlorate de n-butylammo- nium dana le dim&hoxy~thane et dans le t6trahydrofuranne. Les 6chantillons sont dilu6s deux cent fois dana l'acide trifluoroac6tique 1°.
2. Systbme Ag(s)/AgCF3CO0, NaCF3CO0 M Le trifluoroac6tate d'argent eat soluble dans l'acide trifluoroac6tique. Le sys-
t~me argent-trifluoroac&ate d'argent est rapide comme le montre fanalyse des cour- bes voltamp6rom6triques,
La r6action 61ectrochimique est la suivante:
I E/mV
o 13/~-/*'/~" [
i+ j i
i,,- j
tog CAgc%co o
Fig. 3. Potential A courant nul d'une 61ectrode d'argent en fonction du log de la concentration en trifluoro- ac6tate d'argent (NaCF3COO= 1 M).
J. Electroanal. Chem., 31 (1971) 393-403

398 G. PETIT, J. BESSI~LE
Ag (s) + NaCFaCOO - e --, AgCFaCOO + Na +
Le potentiel d'tquilibre est donn6 par la relation:
E = E' 1 + 0.058 log ([AgCFaCOO] [Na+]/[NaCF3COO])
Le tdfluoroacttate de sodium 6rant en forte concentration, le rapport rNa+]/ [NaCF3COO] est pratiquement constant.
S i c reprtsente la concentration en trifluoroacttate d'argent, le potentiel E varie suivant la relation:
E = B + 0.058 log c
Le tract de la courbe de dilution du trifluoroacttate d'argent, soit E = f(log c) permet de vtrifier la loi de Nernst. La pente de la droite exptrimentalelest ~gale & 62 mV par unit6 de logarithme (Fig. 3). La constante Bes t 6gale & +0.174 V par rapport au potentiel de rtftrence choisi ER.
IV. ~ U D E DU SYSTEME OXYDORE'DUCTEUR DE L'HYDROGF.NE
Comme l'indiquent les courbes intensitt-potentiel, le potentiel & intensit6 nulle est bien dtfini.
E/v 0.500
0,550
0,600
log C B
Fig. 4. Potentiel & courant nul & une 61ectrode & hydrogtne en fonction du log de la concentration en n- butylamine (Et4NC10 4 = 5 x 10-1 M).
1. Milieu basique Si l'on suit le potentiel pris par une 61ectrode & hydrogbne en fonction de l'ad-
dition de n-butylamine (symboliste par B) qui est une base forte, on constate que la loi de Nernst est suivie dans le solvant CFaCOOH, Et4NCIO ~ 5 x 10-1 M.
La pente de la droite E = f(log ca) est de 58 mV par unit6 de logarithme (Fig. 4). Les potentiels d'tquilibre successifs sont stables et reproductibles. La n-butyl- amine rtagit quantitativement sur l'acide trifluoroacttique pour former le trifluoro- acttate de n-butylammonium.
L'tquilibre suivant est totalement dtplac~ vers la droite par suite de la prtsence du perchlorate de tttratthylammonium en grande concentration 11.
J. Electroanal. Chem., 31 (1971) 393-403

I~LECTROCHIMIE DANS L'ACIDE TRIFLUOROACETIQUE. II 399
BH+CF3COO - +Et4N+CIO2 ---, BH+C102 +Et4N+CFaCOO -
On suppose que le d6placement de paire d'ions est rapide et que la r6action 61ectrochimique est la suivante:
CF3C OOH q- Et4N + + e ~ ½ H2 (g) + Et4N + CF3COO-
et non :
C F 3 C O O H + B H + + e ---, ½ H2(g) + B H + C F 3 C O O -
car la droite E=f ( log c8) a une pente de 58 mV, alors que dans l'hypoth6se de la seconde r6action la pente ne serait que de 29 mV.
Le potentiel v6rifie la loi de Nernst:
E = E i + 0.058 log [Et4N + ] / [ C F 3 C O O - Et4N +]
Les ions Et4N + apport6s par la dissociation de l'61ectrolyte Et4NC104 sont en con- centration constante. La concentration du trifluoroac6tate de n-butylammonium est 6gale ~t celle de la n-butylamine ajout6e.
E = D-0.058 log cB
La constante D est 6gale/t -0.631 V. L'61ectrode/t hydrog6ne peut donc servir d'61ectrode de r6f6rence en milieu basique.
2. Milieu acide I1 ne nous a pas 6t6 possible d'6talonner l'61ectrode h hydrog6ne en milieu acide.
La courbe de dilution de l'acide fluorosulfurique diff~re sensiblement de la courbe th6orique, qu'elle soit trac6e err pr6sence ou non de perchlorate de t6tra6thylammo- nium utilis6 comme 61ectrolyte. Les potentiels mesur6s ne sont pas stables, n6anmoins leur valeur moyenne croit avec l'addition d'acide. L'utilisation de l'acide perchlorique d6shydrat6 par l 'anhydrid e trifluoroac6tique ne conduit pas ~ de meilleurs r6sultats.
3. Application Utilisation de l'61ectrode h hydrog6ne pour le dosage de reau: l'eau manifeste
des propri6t6s basiques dans l'acide trifluoroac6tique ~. Elle peut &re dos6e par potentiom6trie ~ l'aide Aune 61ectrode/t hydrog~ne. Les r6sultats obtenus sont con- firm6s par spectrophotom6trie.
(a) M~thode potentiomdtrique L'eau est dosOe par l'acide fluorosulfurique HFSO3. La force de cet acide est
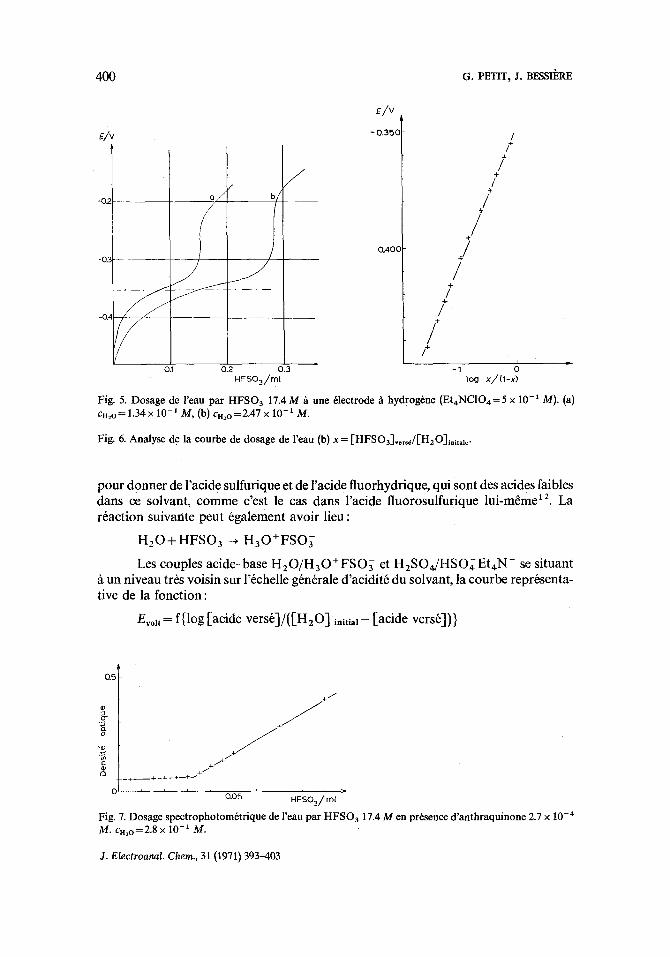
voisine de celle de l'acide perchlorique. L'61ectrode/t hydrog~ne ayant un comporte- ment d6fectueux en milieu acide, la courbe de titrage de l'eau apr~s la neutralisation ne pr6sente pas la forme th6orique. Cependant, le saut de potentiel au point 6quiva- lent est suffisamment important pour que la pr6cision du dosage soit satisfaisante.
Deux courbes de neutralisation de l'eau sont repr6sent6es sur la Fig. 5. La premi6re est relative ~t urt 6chantillon de solvant contenant 1.34 × 10-1 mole d'eau par litre (a); la seconde, h u n 6chantillon indentique auquel on a ajout6 la m~me quantit6 d'eau (b). :
Dans l'acide trifluoroac6tique, l'eau peut r6agir sur l'acide fluorosulfurique
J. Electroanal. Chem., 31 (1971) 393-403
400 G. PETIT, J. BESSIERE
~v
-0,~ - -
-0..:
-0.4
0.1
a/
/
E/V i
- 0.350
0.400
0.2 0.3 H F S O 3 / m I log x / ( l - x )
/ /+
/ 2 +
/ +
+/ / /
+ /
+ /
/ /+
-~ ~>
Fig. 5. Dosage de l'eau par HFSO 3 17.4M h une 61ectrode h hydrog6ne (Et4NCIO4=5 x 10 -1 M). (a) Cn2o = 1.34 x 10- t M, (b) cn2 o =2.47 x 10- t M.
Fig. 6. Analyse de la courbe de dosage de l'eau (b) x = [HFSOa]versJ[H20]initale.
pour donner de l'acide sulfurique et de l'acide fluorhydrique, qui sont des acides faibles dans ce solvant, c o m m e c'est le cas dans l'acide f luorosulfurique lui -m~me 12. La r6action suivante peut egalement avoir l ieu:
H 2 0 + H F S O 3 ~ H a O + F S O 3
Les couples ac ide-base H 2 0 / H 3 0 ÷ F S O { et H 2 S O U H S O ~ Et4N ÷ se situant fi un niveau trbs vois in sur l'6chelle g6n6rale d'acidit6 du solvant, la courbe repr6senta- tive de la fonct ion:
Evolt = f{log [acide vers6]/([HzO] in i t ia l - - [acide vers6])}
i
O.5
0
+ /
_ . _ _ 4 _ _ . ~ _ ÷ - - + J q
, , >
' ' o . ~ H F ~ o ~ / n ~
Fig. 7. Dosage spectrophotom6trique de l'eau par HFSO 3 17.4 M en prgsence d'anthraquinone 2.7 x 10 -4 M. Ca2o=2.8 x 10 -1 M.
J. Electroanal. Chem., 31 (1971) 393-403

I~.LECTROCHIMIE DANS L'ACIDE TRIFLUOROACI~TIQUE. II 401
ne nous autorise pas h pr6ciser quelle r6action se produit lors du dosage (voir Fig. 6); des mesures conductim6triques nous permettraient peut-~tre de conclure. La droite a une pente de 0.058 V par unit6 logarithmique; le potentiel de demi-neutralisation, tr6s reproductible, est 6gal ~ -0.350 V.
(b) Mkthode spectrophotom&rique L'eau est dos6e par l'acide HSO3F en pr6sence d'un indicateur color6: l'an-
thraquinone de pKn2 o 6gal 13/t -8.2. La densit6 optique est suivie/l 400 nm au maximum d'absorption de la forme
acide de l'indicateur. La densit6 optique demeure constante tant qu'il reste de l'eau en solution; elle augmente apr6s la neutralisation (Fig. 7).
Le pK de la r6action
An+HFSO3 ~--- AnH+,FSO3
est voisin de 1 unit6. Nous avons dos6 l'eau r6siduelle contenue dans le solvant ainsi que des 6chan-
tiUons auxquels des concentrations d&ermin6es d'eau avaient 6t6 ajout6es. Les r6sultats des dosages d'eau dans l'acide trifluoroac6tique par la m6thode
de Karl Fischer (r6actif en formamide) sont en accord avec ceux obtenus par potentio- m6trie et par spectrophotom6trie.
PARTIE EXPI~RIMENTALE
1. Le solvant L'acide et l'anhydride trifluoroac6tique sont des produits Eastman Kodak.
L'acide commercial contient g6n6ralement plus de 10-1 mole d'eau par litre. On le d6shydrate en le chauffant ~ reflux pendant douze heures en pr6sence d'anhydride. On le distille ensuite; l'acide bout h 72. 4 ° C sous la pression atmosph6rique. La teneur en eau est inf6rieure/i 24 p.p.m, apr6s distillation. L'acide est conserv6 h l'abri de l'hu- midit6. U est d6plac6 de la r6serve dans la cellule de mesure par pression d'azote.
2. Les r~actifs Le trifluoroac6tate de sodium est pr6par6 selon la m6thode pr6conis6e par
Conway et Vijh 9. L'acide perchlorique est le produit commercial Prolabo. I1 est d6shydrat6 en solution dilu6e dans le solvant par de l'anhydride trifluoroac6tique en quantite egale/~ celle de l'eau pr6sente initialement. Les autres r6actifs : perchlorate de t6tra6thylammonium, n-butylamine, trifluoroac6tate d'argent, anthraquinone sont des produits Eastman Kodak. L'acide fluorosulfurique est pr6par6 selon une m6thode pr6c6demment d6crite 12,1s.
3. Techniques ~lectrochimiques Les courbes intensit6-potentiel sont trac6es/t l'aide d'un montage potentio-
statique classique ~t trois 61ectrodes. Les 61ectrodes indicatrices de platine, d'or, de carbone et d'argent sont des micro61ectrodes ~t disque de 1 mm de diam~tre. Leur vitesse de rotation est fix6e/t 660 tours par min.
L'61ectrode/t hydrog6ne est constitu6e par une rnicro61ectrode de platine poli plongeant dans une solution d'acidit6 donn6e sous une pression d'hydrog6ne de une
J. Electroanal. Chem., 31 (1971) 393-403

402 G. PETIT, J. BESSIERE
atmosph6re. Le platinage a lieu dans l'eau. L'61ectrode est ensuite s6ch6e ~t l'azote puis maintenue une demi-heure dans une solution d'acide trifluoroac6tique contenant un: 16ger exc6s d'anhydride.
Le syst6me de r6f6rence Ag(s)/AgC104(s)/Et4NCIO4 10-1 M est d6crit pr6c6- demment. L'61ectrode est pr6par6e selon la m6thode propos6e par Billon ~6.
Les 61ectrolyses sont effectu6es h l'aide d'un montage coulombtrique Tacussel.
4. Spectrophotom~trie Les mesures spectrophotom6triques ont 6t6 r6alis6es h l'aide d'un spectro-
photom6tre Graphispectral Jouan.
REMERCIEMENTS
Nous tenons ~ remercier Madame J. Badoz-Lambling, Directeur de Recher- che au C.N.R.S. pour l'int6r~t qu'eUe a bien voulu porter ~t cette 6tude, ainsi que le Commissariat ~ l'Energie Atomique qui a subventionn6 ces recherches.
RESUMI~
Le domaine d'61ectroactivit6 de l'acide trifluoroac6tique a 6t6 d&ermin6 ~ des 61ectrodes de platine platin6, de platine poli, d'or, de carbone vitreux, d'argent et de mercure, ~ diff6rentes acidit6s. Le domaine le plus 6tendu est obtenu ~t l'61ectrode de platine (,,~ 3 V en milieu NaCF3COO M).
Les couples oxydo-r6ducteurs: Ag(s)/AgC104(s)/Et4NCIO 4 et Ag(s)/AgCF3- COO,NaCF3COO constituent des syst6mes de r6f6rence satisfaisants dans ce solvant.
Le syst6me 61ectrochimique de l'hydrog6ne peut 6galement servir de syst6me de r6f6rence en milieu basique.
L'eau est dos6e par potentiom6trie; les r6sultats concordent avec ceux que l'on obtient par~ spectrophotom6trie.
SUMMARY ¢
The electroactivity range of trifluoroacetic acid has been determined with platinized platinum and bright platinum, gold, silver, mercury and vitreous carbon electrodes at different acidities. The widest domain is obtained with the platinum electrode (,-~ 3 V in NaCFaCOO (M) medium).
The redox systems: Ag(s)/AgCIO4(s)/Et4NC10 4 and Ag(s)/AgCFaCOO,Na- C F a C O O c a n be used as satisfactory reference systems in this solvent. The hydrogen redox system can be used as reference system in basic media. Water is titrated poten- tiometrically; the results obtained agree with those obtained spectrophotometrically.
BIBLIOGRAPHIE
1 G. CHARLOT ET B. TRI~MILLON, Les R(actions Chimiques dans les Solvants et les Sels Fondus, Gauthier- Villars, Paris, 1963, p. 239.
2 J. BESSI~RE, Bull: Soc. Chim. France, 9 (1969) 3356. 3 H: C. BROWN ET R. A. WIRKKALA, J. Amer. Chem. Soc., 88 (1966) 1447, 1453.
J. Electroanal. Chem., 31 (1971) 393-403

I~LECTROCHIMIE DANS L'ACIDE TRIFLUOROACt~TIQUE. II 4(~3
4 P. ALCAIS, F. ROTHENBERG ET J. E. DUaOlS, J. Chim. Phys., 64 (1967) 1818. 5 G. PETIT ET J. BESSII/RE, J. Electroanal. Chem., 25 (1970) 817. 6 J. H. SXMMONS ET K. E. LORENTZEN, J. Amer. Chem. Soc., 74 0952) 4746. 7 R. HARA ET G. H. CADY, J. Amer. Chem. Soc., 76 (1954) 4285. 8 J. BESSli~RE, Th~se, Paris, 1969. 9 B. E. CONWAY ETA. K. VUH, J. Phys. Chem., 71 (1967) 3637, 3655.
10 A. CAILLET, Th~se, Paris, 1970. 11 J. BESSI[/RE, Bull. Soc. Chim, France, 7 (1968) 3074. 12 W. LANGE, Z. Anorg. Allg. Chem., 215 (1933) 321. 13 L. P. HAMMETT ET M. A. PAUL, J. Amer. Chem. Soc., 56 (1934) 827. 14 L. A. FLEXER, L. P. HAMMETT ETA. DINGWALL, J/Amer. Chem. Soc., 57 0935) 2103. 15 G. BRAUER, Handbuch der Priiparativen Anorganischen Chemie, Stuttgart, 1951, p. 146. 16 J. P. BILLON, J. Electroanal. Chem., 1 (1960) 486.
J. Electroanal. Chem., 31 (1971) 393--403


















