
Dr Anthony BEHIN Centre de référence de pathologie...
60
MYOPATHIES MÉTABOLIQUES Dr Anthony BEHIN Centre de référence de pathologie neuromusculaire Paris Est Groupe hospitalier Pitié-Salpêtrière
Transcript of Dr Anthony BEHIN Centre de référence de pathologie...

MYOPATHIESMÉTABOLIQUES
Dr Anthony BEHIN
Centre de référence de pathologie neuromusculaire Paris Est
Groupe hospitalier Pitié-Salpêtrière
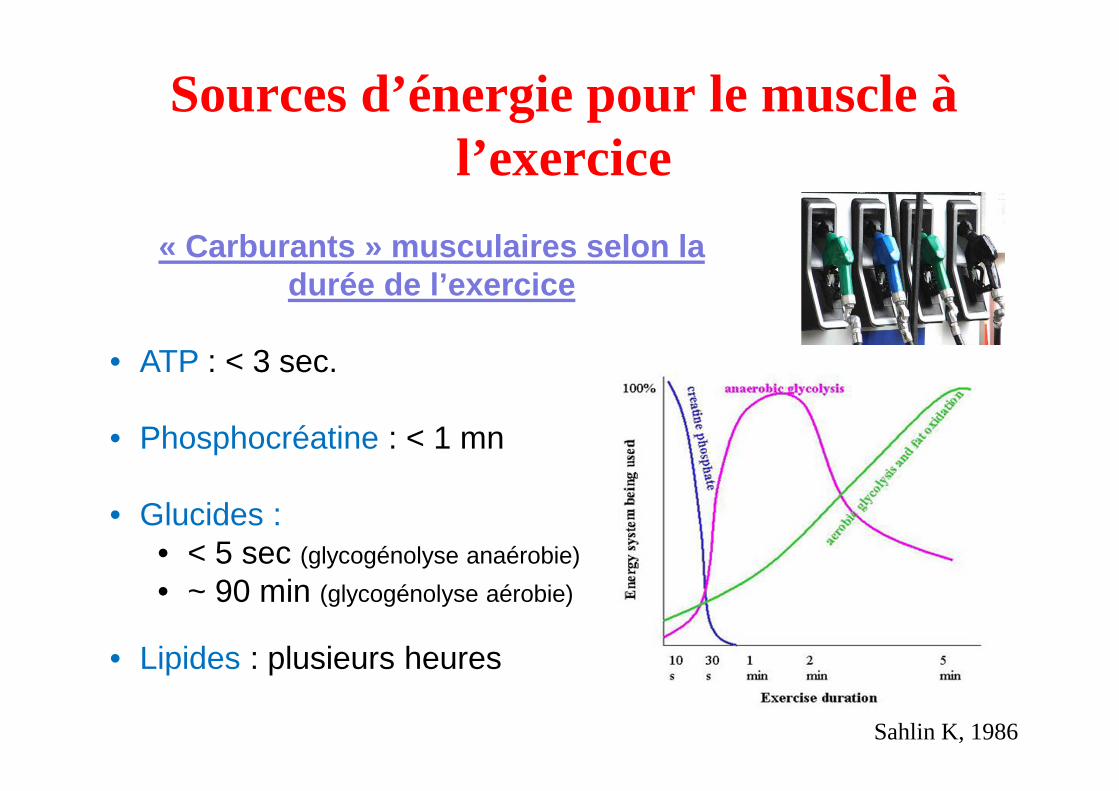
Sources d’énergie pour le muscle à l’exercice
« Carburants » musculaires selon la durée de l’exercice
• ATP : < 3 sec.
• Phosphocréatine : < 1 mn
• Glucides :• < 5 sec (glycogénolyse anaérobie)
• ~ 90 min (glycogénolyse aérobie)
• Lipides : plusieurs heures
Sahlin K, 1986

Où les substrats énergétiques sont-ils stockés ?
• Glucides : glycogène musculaire et hépatique (80% vs 20% des stocks), glucose sanguin
– Source d’énergie primaire pendant l’exercice aérobie intense
• Lipides : tissu adipeux et triglycérides musculaires (97% vs 3% des stocks), acides gras libres du sang
– Source d’énergie pour l’exercice léger à modéré– Les acides gras libres fournissent jusqu’à 80 % de l’énergtie lors de
l’effort prolongé (> 1 heure)

Myopathies métaboliques

Myopathies métaboliques
• Erreurs innées du métabolisme d’origine génétique(la plupart récessives)
• Présentations cliniques chez l’adulte :– Intolérance à l’effort ± épisodes de rhabdomyolyse
– Faiblesse musculaire progressive ± cardiomyopathie (CMH > CMD)
– Manifestations multisystémiques (enfants > adultes)

NAD
NADH
H2O
ADP + Pi
ATP
HgbO2O2
Hgb
Acetyl CoA
FA (n-2)CoA
3 oxyFA CoA
3 hydroxyFA CoA
enoylFA CoA
FA CoA
TCAcycle
B oxidation
ElectronTransport
free fattyacids (FA)
pyruvatelactate
carnitine carnitine
triglyceride
Glycogène
glucose
CPT II VLCAD
LCHAD
ADPATP
Glycogénoses
OAA
Pr Ronald G. Haller
Glycogénose
musculaire
PAS PAS
Déficit en enzyme débranchante
Déficit en
phosphoglucomutase
Maladie de Pompe

Glycogénoses : 14 types décrits
• type II : mal. de Pompe
• type III : déficit en enzyme débranchante
• type V : maladie de McArdle

NAD
NADH
H2O
ADP + Pi
ATP
O2
Acetyl CoA
FA (n-2)CoA
3 oxyFA CoA
3 hydroxyFA CoA
enoylFA CoA
FA CoA
TCAcycle
B oxidation
ElectronTransport
Acidesgraslibres
pyruvatelactate
carnitine carnitine
triglyceride
glycogen
glucose
CPT II VLCAD
LCHAD
ADPATP
OAA
Déficit en Multiple acyl-
CoA dehydrogenase
(MADD)
Déficit en VLCAD
TG
Lipidoses

NAD
NADH
H2O
ADP + Pi
ATP
HbO2
Hb
Acetyl CoA
FA (n-2)CoA
3 oxyFA CoA
3 hydroxyFA CoA
enoylFA CoA
FA CoA
B oxidation
ElectronTransport
free fattyacids (FA)
pyruvatelactate
carnitine carnitine
triglyceride
glycogen
glucose
CPT II VLCAD
LCHAD
ADPATP
Myopathies mitochondriales
Pr Ronald G. Haller
SDH COX
TG
O2
Cycle de Krebs

Comment explorer les myopathies métaboliques ?
• Sang: – CK, électrolytes, glucose, TSH– NFS, réticulocytes– lactate, carnitine, profil des acylcarnitines
• Urines: – Chromatographie des acides organiques
(si rhabdomyolyse, ou lipidose sur la biopsie)
autres analyses en fonction du tableau clinique et des résultats de la biopsie musculaire

Étude du métabolismein vivo
Test d’effort de l’avant-bras sans
ischémie(« grip-test »)
Épreuve d’effort sur bicyclette
ergométrique
Spectroscopie RMN du 31P et 13 C

Myopathies métaboliques révélées par une intolérance à l’effort et des
épisodes de rhabdomyolyse
• Glycogénoses: – déficit en myophosphorylase (McArdle)et phosphorylase b kinase – déficits en phosphofructokinase (PFK), PGK, PGM, PGAM, LDH,
aldolase A, β-énolase.
• Anomalies de l’oxydation des acides gras : – déficits en carnitine-palmitoyl–transférase II (CPTII) , – déficit en VLCAD, – déficit en TFP (enzyme trifonctionnelle), – déficit en multiple acyl-CoA déshydrogénases (ETF)
• Maladies mitochondriales : – mutations de l’ADNmt (cytochrome B gene, tRNA), – mutations ISCU mutations (iron-sulfur cluster protein)

Maladie de McArdle• Glycogénose de type V, AR• Début dans l’enfance• Intolérance à l’effort:
– Myalgies et crampes lors des efforts– Phénomène du « second souffle » après ≈ 10
minutes due à une augmentation d’apport des AG et une meilleure consommation du glucose
• Myoglobinurie± ins. rénale dans ≈ 50% cas• CK élevées au repos• Faiblesse musculaire et atrophie après 40 ans
dans ≈≈≈≈ 30 % des casActivité
myophosphorylaseabsente
• Analyse génétique : mutation récurrente : (p.R50X sur un allèle chez 2/3 des patients)
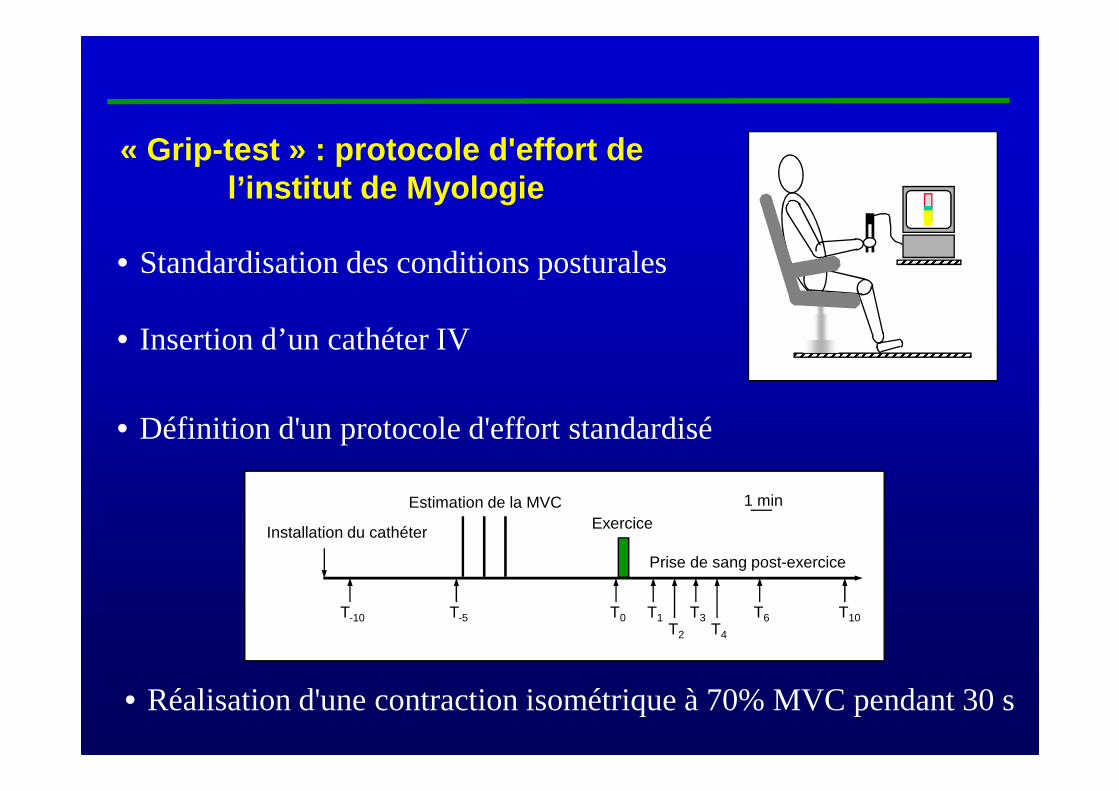
• Définition d'un protocole d'effort standardisé
« Grip-test » : protocole d'effort de l’institut de Myologie
T1T0T-10 T10T3 T6T2 T4
Installation du cathéter
Estimation de la MVC 1 min
Prise de sang post-exercice
Exercice
T-5
• Standardisation des conditions posturales
• Insertion d’un cathéter IV
• Réalisation d'une contraction isométrique à 70% MVC pendant 30 s

Résultats du grip test obtenus dans la maladie de McArdle
0
1
2
3
4
5
6
7
0 2 4 6 8 10Temps (min)
Lact
ate
(mm
ol.l-
1 )
0
50
100
150
200
250
300
0 2 4 6 8 10Temps (min)
Am
mon
iém
ie (
µmol
.l-1 )
Lactate Ammoniémie
• Valeurs de lactates de T1 à T4 inférieures aux normes
• Hyperammoniémie

Oral glucose treatment in McArdle disease
Vissing & Haller. N Engl J Med
2003; 349: 2503-2509.
Constant work
O = placebo
= Energy supplementBPM 160
Above hard
BPM 120
Easy
Effet de la prise orale de glucose dans la maladie
de McArdle
Amélioration du second souffle par le glucose dans la maladie de McArdle

Glycogénoses avec intolérance à l’effort et épisodes de rhabdomyolyse
• Maladie de McArdle : cause la plus fréquente de glycogénose
• Déficit en phosphofructokinase (PFK) ou maladie de Tarui
• Déficit en phosphorylase b kinase
• Maladies rares de la glycolyse : quelques cas connus– Phosphoglucomutase
– Aldolase A
– Phosphoglycérate kinase (PGK)
– Phosphoglycérate mutase (PGAM)
– β-énolase
– Déficit en LDH

Indices cliniques et biologiques utiles au diagnostic
• Début des symptômesaprès quelques minutesd’exercice
• Effet desecond souffle dans la maladie de McArdle• Absence de rhabdomyolyse induite par la fièvre ou le jeûne
(≠ troubles de la bêta-oxydation)
• Pas de faiblesse permanente chez les patients jeunes.
• Faiblesse musculaire permanente possible après 40 ans dans les maladies de McArdle et de Tarui
• Anémie hémolytique (avec augmentation des réticulocytes et de la bilirubine) dans les déficits en PFK et PGK
• Hyperuricémia dans le déficit en PFK
• + crises comitiales et retard mental dans le déficit en PGK

Troubles du métabolisme lipidique
• Défauts de la bêta-oxydation :� Déficit en carnitine Palmitoyl Transferase 2 (CPT 2) � Déficit en very-long-chain acyl-CoAdehydrogenase
(VLCAD) � Déficit en multiple acyl-CoAdehydrogenase (MAD) � Déficit en enzyme trifonctionnelle (LCHAD +++)
• Déficit en lipine
• Déficit primaire en carnitine
• Neutral lipid storage diseases : maladie de Chanarin-Dorfman

• Maladies le plus souvent pédiatriques :– Hypoglycémie récurrente, insuffisance hépatique,
cardiomyopathie, rhabdomyolyse, voire mort subite
• Symptômes musculaires au premier plan chez l’adulte– Intolérance à l’effort +/- épisodes de rhabdomyolyse
• Manifestations cliniques déclenchées par le jeûne, la fièvre et le stress dans les troubles de la bêta-oxydation.
Troubles du métabolisme lipidiqueCaractéristiques cliniques

Déficit de la bêta-oxydation des AG
• Début dans l’enfance ou l’adolescence• Intolérance à l’effort inconstante• Myalgies post-effort et rhabdomyolyses possibles• Facteurs déclenchants en dehors de l’effort :
– fièvre, froid, jeûne, stress• CK et lactates souvent normaux au repos• grip-test et épreuve sur bicyclette le plus souvent normaux• Profil des acylcarnitines essentiel au diagnostic
– À faire en crise ou après 12 heures de jeûne– Peut orienter les études moléculaires vers les gènes
codant pour CPTII, VLCAD, ETF, TFP

Déficit en CPT2 etVLCAD
• Episodes de rhabdomyolyse déclenchés parl’exercice, la fièvre, le froid, le jeûne…
• Absence de déficit musculaire permanent
• Cardiomyopathie rare (VLCAD)
• Absence de lipidose, ou lipidose modérée
• Profil desacylcarnitines typique pour le déficitenVLCAD (pic C14:1)

Déficit en enzyme trifonctionnelle (MTP)
• Episodes de rhabdomyolyse
+ polyneuropathie sensitivo-motrice
+/- rétinopathie +/- cardiomyopathie
• Accumulation d’hydroxyacylcarnitines à longue chaîne+ acidurie 3-OH-dicarboxylique
• Déficit en long-chain 3-hydroxyacyl-CoAdehydroge-nase (LCHAD) en rapport avec une mutation sur la sous-unitéα (gèneHADHA) dans la plupart des cas
• Lipidose modérée et anomalies mitochondrialespossibles à la biopsie musculaire

Déficit en multiple acyl-CoA déshydrogénase (MADD)
• Rhabdomyolyse + douleurs abdominales, vomissements,élévation des enzymes hépatiques +/- encéphalopathie
• Déficit musculaire permanent fréquent (différent des autrestroubles de la bêta-oxydation)
• Lipidose, anomalies mitochondriales, déficit en CoQà laBM
• Elevation combinée de toutes les chaînes d’acylcarnitines(C4 to C18:1)
• Les mutations d’ETFDH sont la cause principales desMADD adultes
• Réponse spectaculaire à CoQ10 et riboflavine possible.

MADD: patient avec lipidose musculaire
MADD: patient avec anomalies mitochondriales

Rhabdomyolyse et myopathies mitochondriales
• Une rhabdomyolyse sévère est une manifestation très raremyopathies mitochondriales
• Se voit avec des mutations de l’ADNmt (gène ducytochrome B, mutations du tRNA) or nucléaire :mutations d’ISCU(iron-sulfur cluster protein)
• Diagnostic à considérer en cas d’acidose lactique associéeà la rhabdomyolyse
• BM : RRF, fibres COX-, ou SDH- (ISCUmutations)
• Toujours envisager un déficit de la bêta-oxydation avecdysfonction mitochondriale secondaire (déficits enVLCAD, MADD, MTP)
•

Myopathies métaboliques avec faiblessemusculaire permanente au premier plan
• Glycogénoses : – maladie de Pompe (GSD II), – déficit en enzyme débranchante (GSD III)
• Lipidoses musculaires :
– déficit multiple en acyl-CoA déshydrogénases (MADD),– déficit primaire en carnitine,
– déficit en triglycéride lipase (Neutral lipid storage disorders)
• Myopathies mitochondriales : – ophtalmoplégies externes progressives, – myopathie isolée (rare)
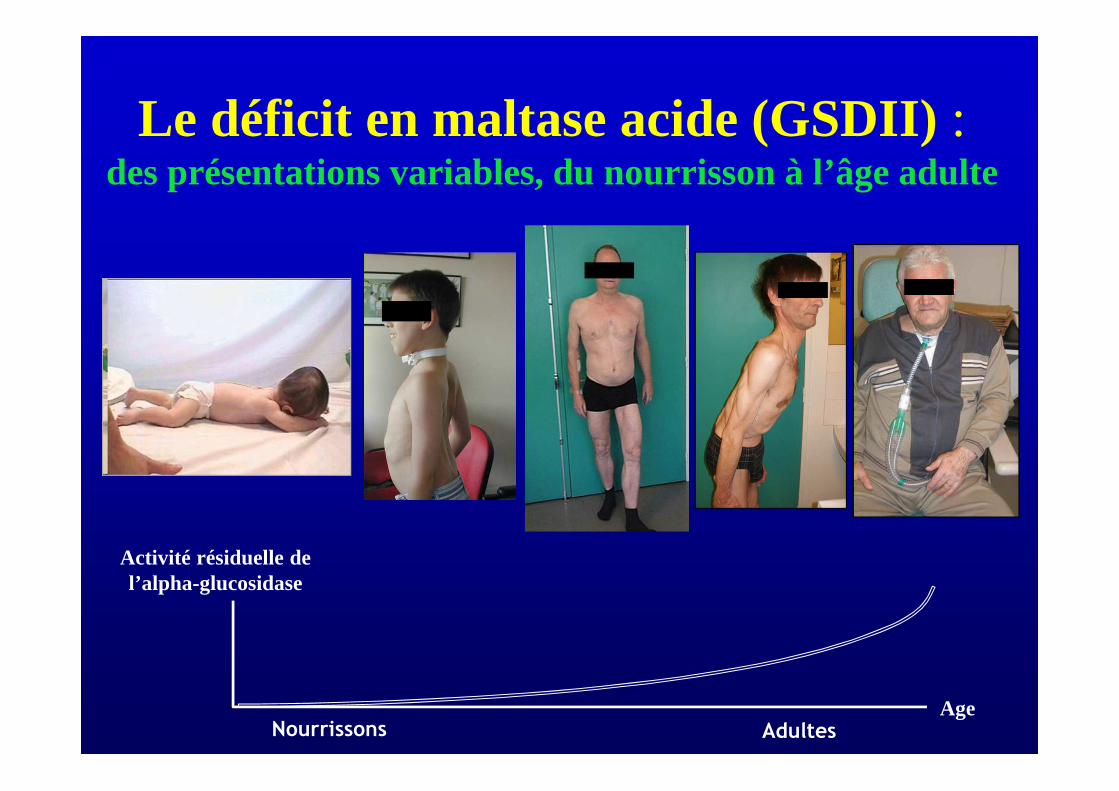
Le déficit en maltase acide (GSDII) : des présentations variables, du nourrisson à l’âge adulte
Nourrissons Adultes
Age
Activité résiduelle de l’alpha-glucosidase

Spectre clinique de la maladie de Pompe
• Maladie de Pompe stricto sensu (forme infantile)– début avant 5 mois– cardiomyopathie, hypotonie, hépatomégalie– décès d’insuffisance cardio-respiratoire < 1 an– activité résiduelle de GAA < 1%
• Forme infantile non classique– age de début < 1 an, décès avant 10 ans– GAA activity < 2%
• Forme juvénile– début dans l’adolescence– myopathie ± cardiomyopathie– activité résiduelle de GAA 2 to 6%
• Forme adulte– début après l’âge de 20 ans– myopathie des ceintures ± ins. respiratoire– activité résiduelle de GAA 7-23 %

Diagnostic de la maladie de Pompe
Biopsie musculaire: myopathie vacuolaire avec surcharge au PAS inconstante (70% des cas)
Doser de l’activité α-glucosidase acide(papier buvard, sang ou fibroblastes) devant une dystrophie des ceintures non étiquetée

Maladie de Pompe :Homme de 45 ans
Déficit pelvien depuis l’âge de 26 ans
Test de marche de 6 mn = 238 m (n > 600)
CPK = 3000 UI/l
Insuffisance respiratoire :
- CVF assis = 2.00 l (41%)- CVF couché = 1.35 l (27%)

Exemples de formes atypiques : ptosis dans une forme évoluée de m. de Pompe

Longue histoire de “myopathie congénitale”
Diminution de l’activité de l’alpha-glucosidase sur leucocytes
Présence de deux mutations : c.525delT et c.517_519delATG
Recherche de mutation dans les gènes SEPN1 et LMNA négative
2 autres cas rapportés (Fadic et al 1997, Kostera-Pruszczyk et al 2006)
(Laforêt P et al.,Neuromuscular disorders 2010)
Exemples de formes
atypiques :
forme avec rigid spine
EFR : CV normale
EMG : décharges pseudo-myotoniques
Biopsie : qqs vacuoles PAS +
Patient de 40 ans

� Années 1960 : échec de la première tentative d’enzymothérapie (enzyme d’Aspergillus Niger et de placenta humain)
� 1996 : GAA recombinante produite à partir de lait de lapine transgénique et cellules CHO
� 1999 : essais de phase I et II avec cette enzyme [alglucosidase alfa, Myozyme™])
� 2000-2001 : publications des essais pilotes (Van den Hout et al.,Lancet 2000; Amalfitano et
al.,Genet Med 2001)
� 2003 : essais cliniques pivots débutés avec Myozyme™
� 2006 : approbation pour la commercialisation par l’EMEA and la FDA de Myozyme™
� 2007 : publication d’un essai de phase III chez 18 enfants de
< 6 mois (Kishnani et al.,Neurology 2007)
Enzymothérapie substitutiveUne longue histoire
CHO = chinese hamster ovary.

Développement de l’enzymothérapiesubstitutive dans la maladie de Pompe
EAP: Expanded Access Protocol
AGLU01602 >6 < 36 mois (n = 21)
AGLU01702 < 6 mois (n = 18)
For
mes
infa
ntile
sF
orm
es d
e dé
but t
ardi
f
LOTS (n = 90)
AGLU 02704
EAP: Expanded Access Protocol
Mini LOTS (n = 5)
AGLU 02804
Histoire naturelle (n = 168)
LOPOS (n = 58)AGLU 02303
French SLOTS (n = 5)
AGLU 03105
Suivi en ouvert (n = 81)
AGLU 3206
International Registry
2006 AMM 2008 2009 AMM adultes
2010

Essais d’enzymothérapie chez l’enfant
Baseline
Week 52
Patient 02.03.308Début de ttt
(4 mois)
Patient 02.03.308Après ttt(19 mois)


-5
0
5
10
15
20
25
30
35
Weeks from Baseline
Ch
an
ge
in M
ean
Dis
tanc
e W
alk
ed (
met
ers)
Myozyme Placebo
Début d’étude Moyenne (DS) 78 semainesMoyenne (SD)Myozyme 332.2 m (126.7) Myozyme 362.7 m (145.3)Placebo 317.9 m (132.3) Placebo 312.7 m (147.2)
Essai LOTS (essai randomisé contreplacebo) Test de marche de 6 minutes
20 40 60 80
(van der Ploeg et al, NEJM 2010)

Essai LOTS (essai randomisé contre placebo) capacité vitale assise (FVC)
-5
-4
-3
-2
-1
0
1
2
3
4
5
Weeks from Baseline
Ch
an
ge
in M
ean
% P
red
icte
d
Myozyme Placebo
Début d’étude Moyenne (DS) 78 semainesMoyenne (SD)Myozyme 55.4% (14.4) Myozyme 56.7% (16.4)Placebo 53.0% (15.7) Placebo 51.1% (15.8)
20 40 60 80
(van der Ploeg et al, NEJM 2010)

Déficit en enzyme débranchante(GSD III ou maladie de Cori-Forbes)
• Début dans l’enfance:– hépatomégalie, hypoglycémies de jeûne– Retard de croissance
– Intolérance à l’effort de sévérité variable
• Amélioration spontanée des hypoglycémies à la puberté (régime: maizena, nutrition entérale nocturne)
• Symptômes chez l’adulte: – Faiblesse des membres distale > proximale
(début entre 20 et 30 ans) ± cardiopathie hypertrophique, et rarement insuffisance respiratoire.
• Diagnostis:– Analyse enzymatique sur leucocytes, muscle
ou le foie
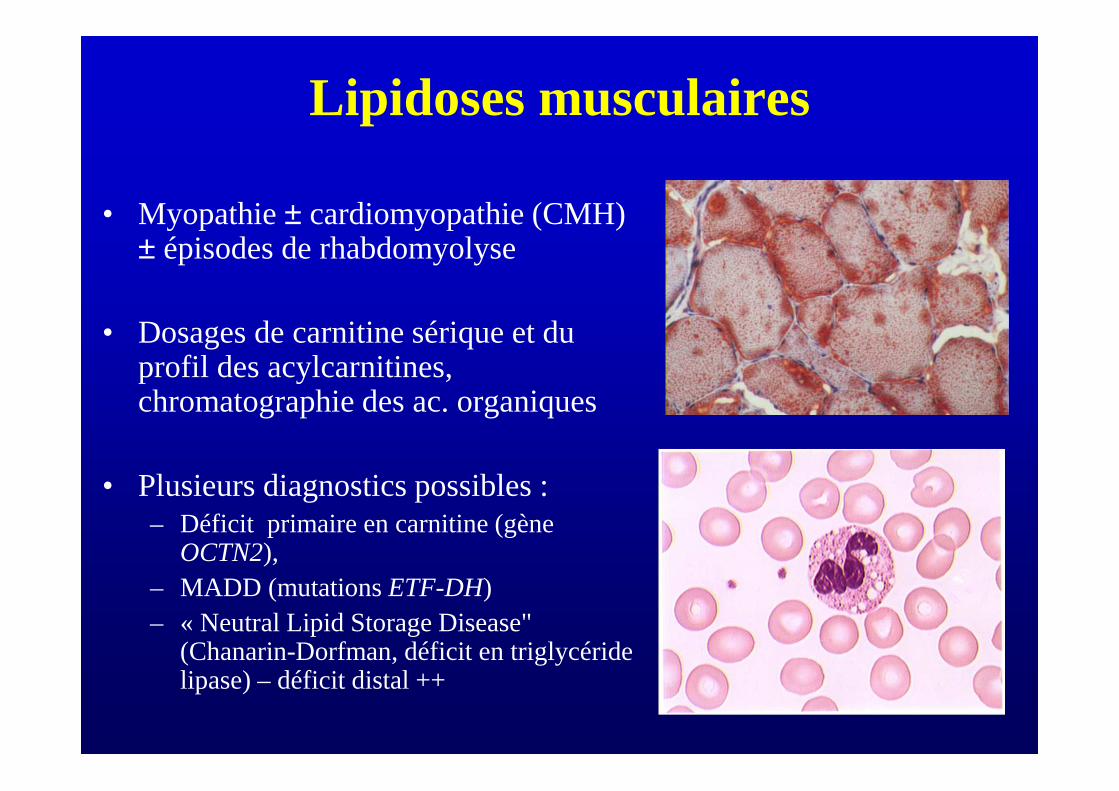
Lipidoses musculaires
• Myopathie ± cardiomyopathie (CMH) ± épisodes de rhabdomyolyse
• Dosages de carnitine sérique et du profil des acylcarnitines, chromatographie des ac. organiques
• Plusieurs diagnostics possibles :– Déficit primaire en carnitine (gène
OCTN2), – MADD (mutations ETF-DH)– « Neutral Lipid Storage Disease"
(Chanarin-Dorfman, déficit en triglycéride lipase) – déficit distal ++

Myopathies mitochondriales
• Principales manifestations: ophtalmoplégie externe progressive (OEP)
• ± BAV, rétinite pigmentaire, rétinopathie, surdité, ataxie, neuropathie axonale ou sensitive (“OEP +” et syndrome de Kearns-Sayre)
• + Pseudo-obstruction intestinale (POIC) = MNGIE syndrome
• Modes d’hérédité variables:– Délétion mtDNA sur le muscle– Mutations ponctuelles mtDNA – Gènes nucléaires (POLG, Twinkle,
ANT1, TYMP)

Maladies mitochondriales• Âge de début variable: enfance ou
adulte• Intolérance à l’effort:
– Myalgies, fatigabilité, essoufflement à l’effort
• Myoglobinurie exceptionnelle• ± atteinte multisystémique• Le taux de CK peut être normal au
repos
Taux de lactate normaux chez un patient avec mutation CytB
Lactate (mmol/l)
0,0
2,0
4,0
6,0
8,0
-5 0 5 10t (min)
• Élévation de la lactactémieau repos ou après effort– Le grip-test peut être normal– Test sur bicyclette plus sensible

Maladies mitochondriales• Importance de la biopsie musculaire:
- aspects morphologiques: fibres RRF et COX - le plus souvent
– étude des complexes de la chaîne respiratoire– recherche de délétion(s) / déplétion de l’ADNmt– dosage du taux de Coenzyme Q10– génétique : screening des mutations de l’ADNmt

Myopathie des ceintures révélatrice de maladie mitochondriale
• Faiblesse musculaire proximale isolée rarement observée
• Myopathie + lipomesassocié à la mutation MERRF(A8344G mutation lysine)
• Atteinte des ceintures sévère, ophtalmoplégie et ins. respiratoire
• Déplétion de l’ ADNmtsur le muscle
• Mutation sur le gène de la thymidine Kinase (TK2)

Pièges et diagnostics différentiels des myopathies
métaboliques

• Patient âgé de 33 ans• Suivi depuis 8 ans pour des douleurs des membres
inférieurs survenant pour des efforts intenses• Plusieurs épisodes de myoglobinurie• Présence d’un phénomène de « second souffle »• Examen clinique: hypertrophie des mollets• Taux de CPK au repos > 5000 UI/l• Parents cousins germains, et une cousine présente une
faiblesse musculaire avec cardiopathie
Cas clinique


Bilan métabolique
• Épreuve d’effort sur bicyclette : élévation de la lactacidémie sans élévation de l’ammoniémie: déficit en myoadénylate déaminase
• Spectroscopie RMN : anomalie du métabolisme mitochondrial (lenteur de la resynthèse de la PCr)
• Dosage des enzymes de la glycolyse érythrocytaire, de la CPTII normal
• 1ère biopsie à 26 ans : confirmation du déficit biochimique en myoadénylate déaminase, étude de la chaîne respiratoire mitochondriale normale
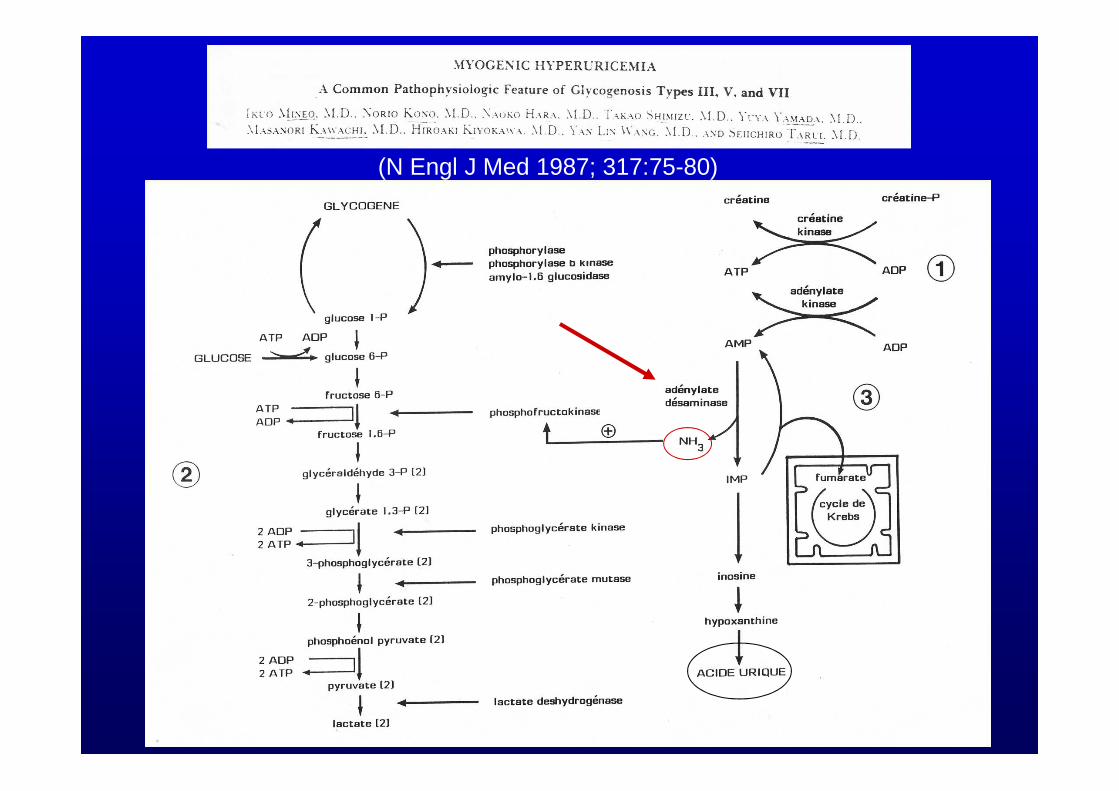
(N Engl J Med 1987; 317:75-80)

H.E. T.G.
α- DGβ- DG
Déficit en alpha-dystroglycan Mutations homozygote FKRP L276I

β
dystroG.
α dystroglycane
α β γ δ
Calpaïne
Dysferline
sarcoglycanes
25 sarcospan
Laminine
Sarcolemme
Lame basale
Dystrophine
Actine
Espace extracellulaire
CytoplasmeSyntrophines
Dystrobrevine
Caveoline
Dystrophies musculaires:Anomalies des protéines membranaires
Western-blot :dystrophine, sarcoglycanes, dysferline, calpaïne, AlphadystroglycaneCaveoline
Immunomarquage :idem moins calpaïne
Dysferlinopathies DystrophinopathiesSarcoglycanopathiesCalpaïnopathies Alpha-dystroglycanopathiesCaveolinopathies,...

Examen clinique
• Faiblesse musculaire diffuse à prédominance proximale
• Face et oculomotricité normales (léger ptosis constaté à 43 ans)
• Absence de troubles bulbaires• Flessum des genoux• CV normale• Bilan cardiaque normal

Biopsie deltoidienne: surcharge lipidique, prédominance des fibres de type I.
Diagnostic: myopathie métabolique

Examens complémentaires
• Taux de CPK normal• Étude de la chaîne respiratoire: déficit combiné en
complexes I et III• Déficit de l’oxydation du palmitate sur myoblastes
en culture accentué par le stress métabolique( - 26 à – 44% après culture dans milieu sans glucose)
• Traitement : carnitine, Vit B2, puis bezafibrate• Consulte plusieurs spécialistes de maladies
métaboliques: hésitation entre une anomalie de la chaîne respiratoire et de la bêta-oxydation

• Étude de la transmission neuromusculaire (Dr E Fournier) : décrément de – 30 à – 50% des réponses motrices à la stimulation répétitive
• Analyse moléculaire: mutations du gène DoK7
• Diagnostic:syndrome myasthénique congénital
• Anomalies mitochondriales secondaires ?

Myopathies métabolique: diagnostics différentiels
• Présentation “pseudo-métabolique” des dyst. musculaires :– Toutes les dystrophies peuvent se révéler par des myalgies à
l’effort + élévation du taux de CK au repos (> 1000)– immunomarquages + western blot sur la biopsie musculaire
• Syndromes myasthéniques congénitaux:– EMG pour recherche de BNM– Possibilité de lipidose sur la biopsie musculaire
• Channelopathies:– Faiblesse épisodiques et fluctuantes des 4 membres ± douleur– EMG pour recherche de myotonie et tests d’effort
• “Fibromyalgie”– Douleurs et fatigabilité au moindre effort, taux de CPK
normaux, examen clinique et EMG normaux

Arguments en faveur d’une dystrophie musculaire devant une intolérance à l’effort
• antécédents familiaux de dystrophie• hypertrophie ou atrophie des mollets• cardiopathie (dilatée)• élévation marquée des CPK au repos (> 1000 UI/l)• anomalies de l’imagerie musculaire• Biopsie musculaire systématique si CPK > 1000 UI/l
au repos: formule de nécrose/régénération, immunomarquages et Western Blot +++

Arguments en faveur de l’origine métabolique d’une myopathie
• Antécédents familiaux (atteintes pédiatriques)• Manifestations précoces parfois spontanément régressives: *
syndrome de Reye, hépatopathie, hypoglycémies, vomissements, encéphalopathie…
• Fluctuation des symptômes • Décompensations aiguës après effort, stress, fièvre ou jeûne ** • Atteinte multisystémique • Cardiomyopathie hypertrophique ***
* déficit en en enzyme débranchante, en VLCAD, en acyl-CoA déshydrogénases,
** anomalies de la bêta-oxydation*** glycogénoses, maladies mitochondriales et bêta-oxydation

Traitement des myopathies métaboliques
• Éviter le jeûne et les efforts intenses• Maladie de McArdle: réentrainement à l’effort, prise de
sucre avant les efforts• Maladie de Pompe:
– enzymothérapie substitutive (alglucosidase alfa = Myozyme®) – perspectives: molécules chaperonnes, inhibition de substrat,
réentrainement à l’effort, thérapie génique…
• Déficit primaire en carnitine: supplémentation en carnitine• Déficit multiple en acyl CoA-déshydrogénases:
Riboflavine, Carnitine• Déficit en CPTII et VLCAD:
– régimes enrichis en triglycérides à chaînes moyennes– acide triheptanoïque (TG à nombre impair de carbones)– bezafibrate (étude pilote encourageante, essai en cross-over
contre placebo en cours)


















