
Conf flash Hémato - · LISTE DES ITEMS MODULES N° 143 Agranulocytose médicamenteuse: CAT N°...
50
CONF FLASH HEMATO Martin CARRE, CCA en hématologie clinique Jeudi 16 et Mardi 21 Avril 2015
Transcript of Conf flash Hémato - · LISTE DES ITEMS MODULES N° 143 Agranulocytose médicamenteuse: CAT N°...

CONF FLASH HEMATO
Martin CARRE, CCA en hématologie clinique
Jeudi 16 et Mardi 21 Avril 2015

LISTE DES ITEMS
MODULES
N° 143 Agranulocytose médicamenteuse: CAT
N° 161 Dysmyelopoïèse
N° 162 Leucémies aiguës
N° 163 Leucémie lymphoïde chronique
N° 164 Lymphomes malins
N° 165 Maladie de Vaquez
N° 166 Myélome multiple des os
N° 175 Prescription et surveillance d’un ttt antithrombotique
N° 182 Accident des anticoagulants
N° 178 Transfusions sanguine et produits dérivés du sang
MALADIES ET GRANDS SYNDROMES
N° 222 Anémie par carence martiale
ORIENTATION DIAGNOSTIQUE
N°291 Adénopathie superficielle
N° 297 Anémie
N° 316 Hémogramme
N° 330 Purpura chez l’enfant et l’adulte
N° 332 Splénomégalie
N° 334 Syndrome mononucléosique
N° 335 Thrombopénie
N° 339 Troubles de l’hémostase et de la coag

Mots-clés pour traitements des hémopathies malignes
Consultation d’annonce diagnostiqueDécision thérapeutique (dont surveillance) en RCP
CECOS (+ sérologie syphilis)Contraception efficace
Prothèse capillaireDemande d’ ALD auprès du médecin traitant
PEC psycho-socialeInclusion dans un essai clinique

N° 143 Agranulocytose médicamenteuse: CAT
2 mécanismes physiopathologiques
Périphérique: immunoallergique, Ac anti-granulocytesSeule la lignée granuleuse est touchée
Brutal si 1er contact antérieur ou 8 à 15 jours si primo-introductionMedoc: AINS, anti-thyroïdiens, clozapine , ATB…
Central: aplasie post chimiothérapieBi- voire pancytopénie
8 à 10 jours post J1 de chimioT
Neutrophiles sanguins < 0.5 G/L
MYELOGRAMMEMoelle pauvre
Absence de lignée granuleuse/ Blocage de maturation au stade promyelocytaire
PAS DE MYELOGRAMME
Risque: Choc septique à BGN
CAT diagnostique
Enquête médicamenteuse (imputabilité intrinsèque et extrinsèque)Clinique: Syndrome infectieux +/- signes de choc. Lésions ulcéro-nécrotiques des muqueuses (buccal++)Paraclinique infectieux: Hémocultures + prélèvement orientés par la clinique (penser à VVC/PAC)
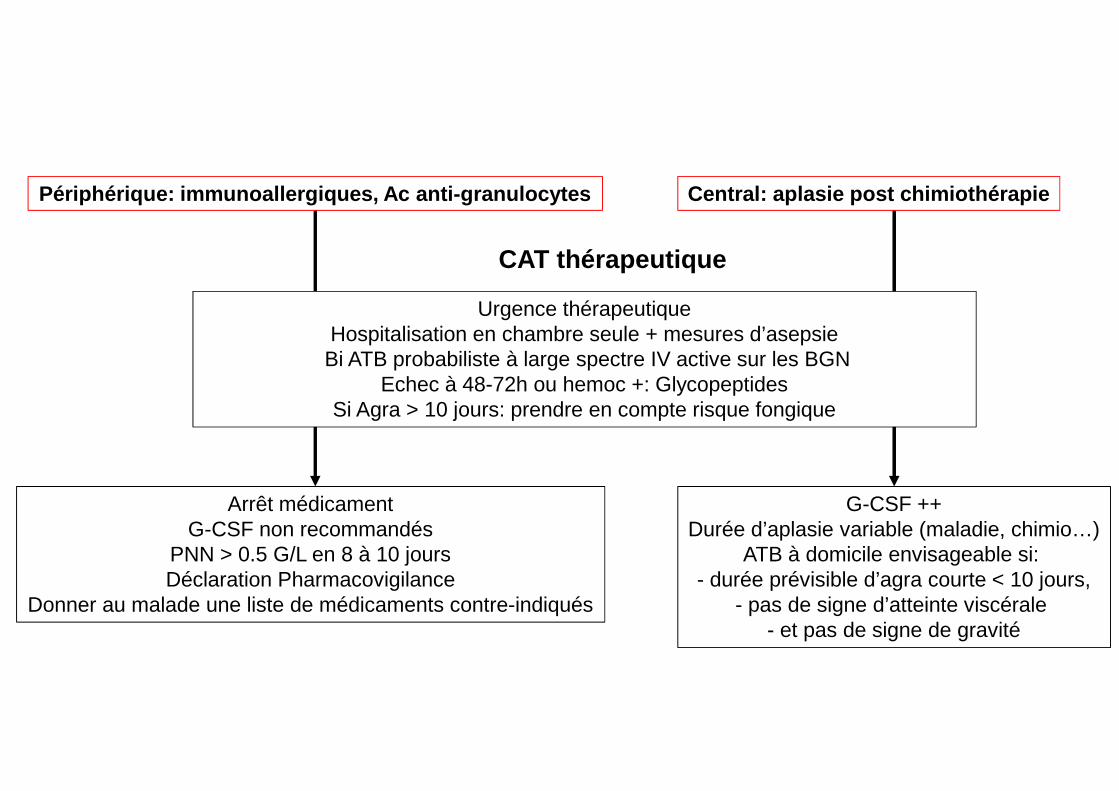
CAT thérapeutique
Arrêt médicamentG-CSF non recommandés
PNN > 0.5 G/L en 8 à 10 joursDéclaration Pharmacovigilance
Donner au malade une liste de médicaments contre-indiqués
Urgence thérapeutiqueHospitalisation en chambre seule + mesures d’asepsie
Bi ATB probabiliste à large spectre IV active sur les BGN Echec à 48-72h ou hemoc +: Glycopeptides
Si Agra > 10 jours: prendre en compte risque fongique
G-CSF ++Durée d’aplasie variable (maladie, chimio…)
ATB à domicile envisageable si: - durée prévisible d’agra courte < 10 jours,
- pas de signe d’atteinte viscérale - et pas de signe de gravité
Périphérique: immunoallergiques, Ac anti-granulocyte s Central: aplasie post chimiothérapie

N° 161 Dysmyelopoïèse ( Myélodysplasie)
Affection clonale de cellules souches pluripotentes Cytopénies sanguines à moelle riche ( insuffisance médullaire qualitative avec avortement intramédullaire)Etat préleucémique (risque de transformation en LAM)
Médiane de survenue: 70 ansSMD primitif (pas de cause connue) ou secondaire (ATCD de chimio/radiothérapie/IS, toxiques)Signes dysimmunitaires parfois associés (arthropathie, vascularite…)NFS: Mono/bi/pancytopénie. Anémie arégénérative +/- macrocytaire.
Cytopénie(s) chez la personne plutôt âgée/ ATCD chimio/radioT
CAT diagnostique
SANGNFS + réticulocytes + frottis sanguinVitamine B9, B12, créatinine, TSH
Ferritine, EPO sériqueGroupe Rhésus 2 déterminations + RAI
MOELLE Myelogramme : richesse normale/augmentée,signes de dysérythro/granulo/thrombopoïèseCytochimie : Coloration de Perls (fer)Caryotype+++
Présentation générale
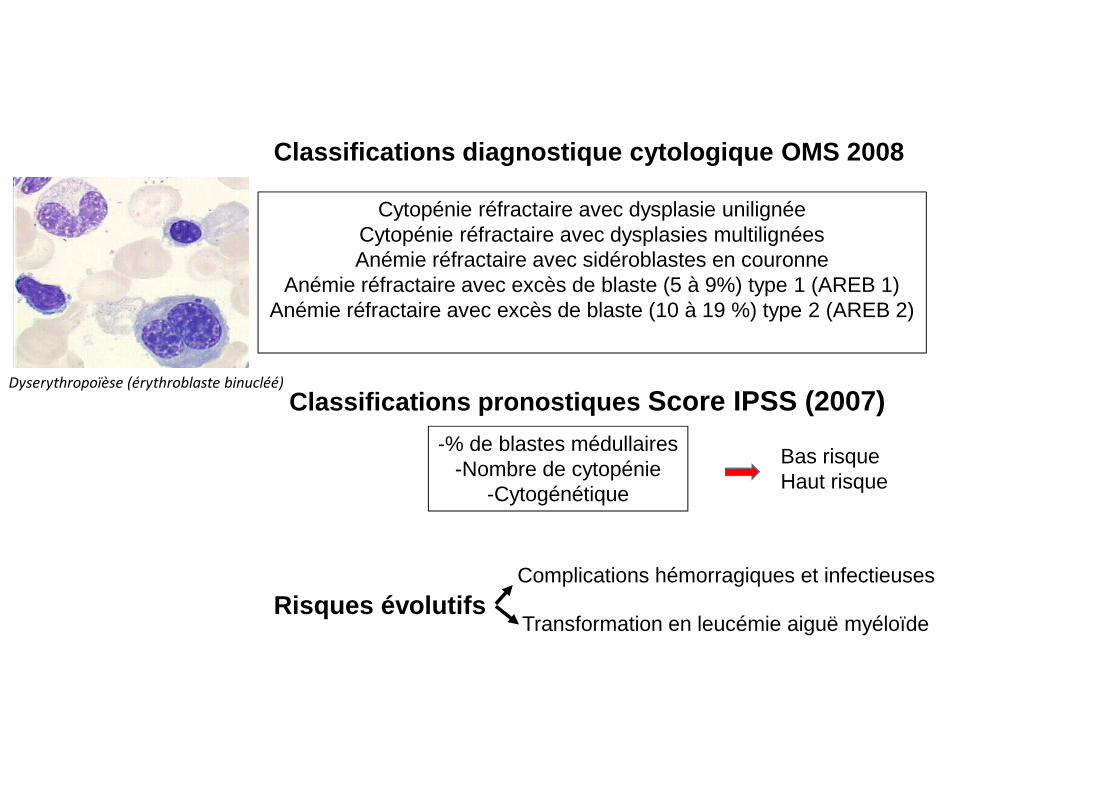
Risques évolutifsComplications hémorragiques et infectieuses
Transformation en leucémie aiguë myéloïde
-% de blastes médullaires-Nombre de cytopénie
-Cytogénétique
Classifications pronostiques Score IPSS (2007)
Classifications diagnostique cytologique OMS 2008
Cytopénie réfractaire avec dysplasie unilignéeCytopénie réfractaire avec dysplasies multilignéesAnémie réfractaire avec sidéroblastes en couronne
Anémie réfractaire avec excès de blaste (5 à 9%) type 1 (AREB 1)Anémie réfractaire avec excès de blaste (10 à 19 %) type 2 (AREB 2)
Bas risqueHaut risque
Dyserythropoïèse (érythroblaste binucléé)

CAT thérapeutique
Catégories de risque Bas
Surveillance simple si cytopénies asympto
Ttt symptomatiqueEPO recombinante
Transfusion en CG et plaquettesChélation martiale si ferritine > 1000 ng/ml
+/- G-CSF si épisode infectieux et blaste < 5%
Catégories de risque Haut
Agent hypométhylant (5 azacytidine)Allogreffe de CSH: seule traitement curatif
(patient < 65 ans sans Co-M importante et donneur disponible)
Toujours penser aux essais cliniques
Cas particulier: le Syndrome del 5q
MDS avec caryotype retrouvant une delétion du 5q isoléePrédominance féminine. NFS: ThrombocytoseMyélogramme: dysmegacaryopoïèse (grands mégacaryocytes hypolobulés)Traitement par Lenalidomid

N° 162 Leucémies aiguësBlastose médullaire ≥ 20 %
Prolifération maligne d’un progéniteur hématopoïétique bloqué à un stade de dif férentiation aboutissant à l’accumulation clonale dans la moelle de cellules immatures de la lignée myéloïde (myéloblastes, LAM ) ou lymphoïde (lymphoblastes, LAL ).
Tableau clinique/biologique aigu ou subaigu
AEG, signes générauxSd infectieux, Sd anémique, Sd hémorragique+/- Sd tumoral
CAT diagnostique
Sang : NFS: Bi- ou pancytopénie +/- blastose périphérique, leucocytose variableréticulocytes + frottis sanguin. TP, TCA, Fibrinogène, PDF, Groupe ABO 2 déterminations + RAIIonogramme sanguin, créat,Ca²+,P+, acide urique, LDH, bilan hépatique
Aspiration médullaire pour examen :- Diagnostique: Myelogramme: moelle riche et blastose ≥ 20 %+ cytochimie (MPO), immunophénotypage- Pronostique: cytogénétique (caryotype +/- FISH), biologie moléculaire- Congélation de cellules (blasthèque)PL si s. neuro, LAL ou LAM hyperleuco > 50 G/L ou à différentiation myelomonocytaire ou monocytaire (LAM 4/5)Préthérapeutique : ECG, échographie cardiaque/ FEVG isotopique
LAMLAL

Leucémies aigues myéloïdes LAM
Epidémiologie
Incidence: 5 nouveaux cas/100.000 habitants/an en FranceAge médian : 65 ansFacteurs favorisants: Génétique constit, Toxique, chimio/radiothérapie, états pré-leucémiques (SMD, NMP)
Classification diagnostique OMS 2008
LAM avec anomalie cytogénétique récurrentet(8;21) (LAM2) t(16;16) ou inv(16) (LAM4Eo) t(15;17) (LAM3) t(9;11), t(6;11) (LAM5)
LAM avec dysplasie multilignée (ATCD ou signe de MDS)
LAM secondaire à un traitement
LAM non classable dans l’un des 3 autres groupes �Classification FAB morphologique (0 à 7)

Leucémies aigues myéloïdes LAM
Classification pronostique ELN 2010
Facteurs pronostiques
Age > 60 ansHyperleucocytaire > 50 G/L
LAM secondaire à une hémopathie ou tttCytogénétique
Biologie moléculaireRéponse au traitement (MRD)
Groupe Cytogénétique + biologie moléculaire
Favorablet(8;21)inv(16) or t(16;16)Caryotype normal et profil moléculaire favorable
Intermédiaire Caryotype normal ou anomalie autre que favorable ou défavorable
Défavorable Caryotype complexe, atteinte du chromosome 3, 5, 7

Cas particuliers
LAM 3 promyélocytaire
CIVD fréquente au diagnostic
Myélogramme : Blocage de maturation au stade promyélocytaire corps d’auer en fagotsCytogénétique : translocation (15;17) dans plus de 90% des cas.Biologie moléculaire : transcrit de fusion des gènes PML - RARA
Traitement : Efficacité de l’ATRAPronostic : Bon une fois passée les complications hémorragiques initiales
LAL Phi +
30% des LAL de l’adulteMauvais pronosticTtt: + Inhibiteur de tyrosine kinase (ITK) et allogreffe

Leucémies aigues lymphoïdes LAL
Epidémiologie
Incidence: 1,5 nouveaux cas/100.000 habitants/an en France80 % des LA chez l’enfant et 20 % chez l’adulte2 pics de fréquence: 2 à 10 ans et après 50 ansFacteurs favorisants: Génétique constitutionnelle (trisomie 21)
Plus fréquent que dans les LAMDouleurs osseuses, Sd tumoral, signes neurologiques, localisation testiculaire
Classification diagnostique OMS 2008
LAL B avec anomalie cytogénétique récurrenteLAL B sans autre spécificationLAL T
Facteurs pronostiques (en plein changement)
Envahissement neuroméningéHyperleucocytose
t(4;11), t(9.22)Cortico résistance, chimiorésistance
Tout gommé par la MRD (Minimal résiduelle minime)??

Situations d’extrême urgence de traitement
Syndrome de leucostase neurologique ou pulmonaire (LAL, LAM 4 ou 5)CIVD avec syndrome hémorragique
Syndrome de lyse tumorale
Principes de traitement
Urgence thérapeutiqueHospitalisation en chambre protégée (flux laminaire), pose d’une VVC ( attention troubles coag)
Chimiothérapie intensive d’induction ( si < 60 ans et « fit ») pour obtention d’une rémission complèteChimiothérapie prophylactique intrathécale dans les LAL, si envahissement neuro dans les LAMTraitement post induction dépend du risque de rechute: chimiothérapie de consolidation +/- allogreffe de CSHsi maladie à haut risque
Prévention du syndrome de lyse tumorale (hyperhydratation, rasburicase, lutte contre hyperkaliémie)Traitement des complications : infectieuses (ATB), transfusion Plq et CG, dénutrition (alimentation entéraleou parentérale), mucite, colite…

N° 163 Leucémie lymphoïde chronique B
Prolifération lymphoïde clonale de lymphocytes matures de morphologie N et de phénotype B.Infiltration médullaire, sanguine, +/- ggaire
Mode de découverteFortuite sur NFSSd tumoral: polyADP superficielles en chapelet, SMGInfections: Zona, PAC à pneumocoqueCytopénies
Lymphocytose isolée persistante > 5 G/L > 3 mois
DiagnosticImmunophénotypage sanguin. PAS DE MYELOGRAMME. Pas de Bx gg
Lymphocytes B CD 5+ 19+ CD 23+Score de Matutes ≥ 4
Bilan d’extensionRéticulocytes, frotti sanguin: petits lymphocytes normaux, ombre de GumprechtEPS, dosage pondéral des Ig, LDH, B2 microglob, Coombs + hémolyse , Séro VHB et VHCFISH interphasique (del 17p) si < 65 ans ou si ttt nécessaireRadio thoracique
Gumprecht
Petits lymphocytes
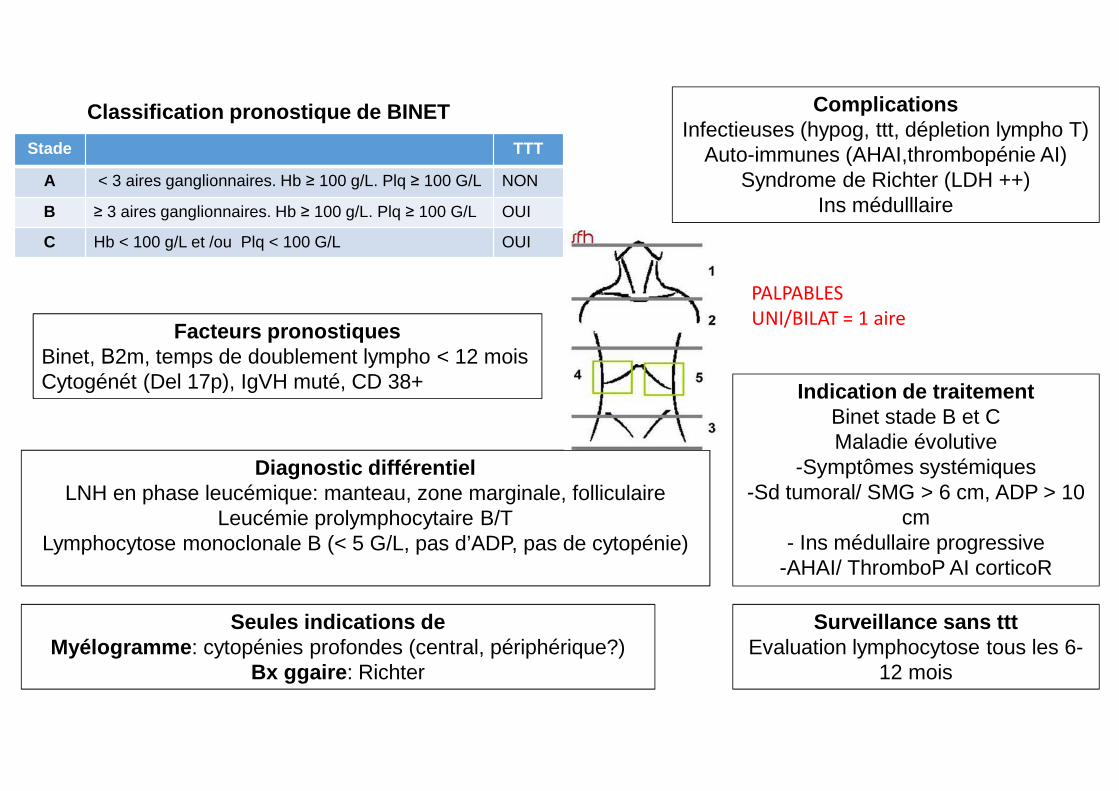
Facteurs pronostiquesBinet, Β2m, temps de doublement lympho < 12 moisCytogénét (Del 17p), IgVH muté, CD 38+
PALPABLES
UNI/BILAT = 1 aire
Stade TTT
A < 3 aires ganglionnaires. Hb ≥ 100 g/L. Plq ≥ 100 G/L NON
B ≥ 3 aires ganglionnaires. Hb ≥ 100 g/L. Plq ≥ 100 G/L OUI
C Hb < 100 g/L et /ou Plq < 100 G/L OUI
ComplicationsInfectieuses (hypog, ttt, dépletion lympho T)
Auto-immunes (AHAI,thrombopénie AI)Syndrome de Richter (LDH ++)
Ins médulllaire
Classification pronostique de BINET
Diagnostic différentielLNH en phase leucémique: manteau, zone marginale, folliculaire
Leucémie prolymphocytaire B/TLymphocytose monoclonale B (< 5 G/L, pas d’ADP, pas de cytopénie)
Seules indications deMyélogramme : cytopénies profondes (central, périphérique?)
Bx ggaire : Richter
Indication de traitementBinet stade B et CMaladie évolutive
-Symptômes systémiques-Sd tumoral/ SMG > 6 cm, ADP > 10
cm- Ins médullaire progressive
-AHAI/ ThromboP AI corticoR
Surveillance sans tttEvaluation lymphocytose tous les 6-
12 mois

N° 164 Lymphomes malinsProlifération clonale de cellules lymphoïdes dans organes lymphoïdes secondaires ggaires/extra-ggaire s/non lymphoïde , survenant à différents stades de maturation du lymphocyte mature
MOELLE MOELLE
Lymphocytes B immatures
Lymphocytes matures Plasmocytes
LAL SLP indolentsLLC/L. lymphocytique
L. du manteau
L. folliculaireL. Zone marginale
L. MALT
WaldenströmMyelome
GANGLIONS
Lymphocytes B matures
SLP agressifsL. lymphoblastique
HodgkinL. B à grandes cellules
L. immunoblastique
Lymphomagenèse
Anomalie acquise (survie)
+/- stimulation Agnique chronique (MALT)
Défaut reconnaissance par le Σ immunitaire

Symptômes BFièvre à prédominance vespérale
Sueurs nocturnesAmaigrissement > 10 % en 6 mois ou 5 % en 1 mois
3 tableaux cliniques révélateurs urgentsSCSup
Masse abdominale compressive (Burkitt)Sd compression médullaire
DiagnosticBiopsie tissulaire pour examen:
- Histologique et immunomarquage- Immunophénotypage en cytométrie de flux
- Oncogénétique (caryotype +/- FISH)- Biologie moléculaire (clonalité ou transcrit de
fusion)
Bilan initial
PronostiqueTDM cervico-TAPPET scan (L. agressifs++)BOM+/- PL (L. agressifs++,localisation ORL, sein, testicule)
Immunophénotypage sangLDH et beta2mSéro VIH 1 et 2EPS
Pré-tttECG + ETT ou isotopique(anthracycline)EFR (bléomycine)NFS, rein, foie, coagSéro VHB et VHC
CD 19+ 20+CD 3 +
Cell Reed Sternberg
Schéma daté et signé
Classification pronostique Ann ArborFacteurs pronostiquesregroupés par patho
IPIFLIPI

Maladie de Hodgkin
2 pics: 20-30 ans et > 65 ansPrurit et doul gg avec alcool, Gg sus diaphragmatiquesPas de tropisme méningéHisto: LH classique/ LH à prédominance lymphocytaireTtt: ABVDBon pronostic, but guérison
Principes de traitement des lymphomes
Mots clés communs à toutes les hémopathies malignesPolychimiothrapie systémique: CHOP (LNH), ABVD (LH)+ Immunothérapie dans les Lymphomes B CD 20+: Rituximab (Ac anti-CD 20)Autogreffe de CSH si rechute chimiosensible (chimioT intensive puis réinfusion de CSH)

N° 165 Maladie de VaquezSMP/NMP
95% Mutation JAK2 V617F
Critères diagnostiques OMS 2008: 2 CM + 1 Cm ou le 1er CM + 2 Cm
Critères majeurs (CM) : 1) Hb > 185 g/L chez l’homme et > 165 g/L chez la femme ou augmentation du volume globulaire total
> 25%2) Présence de la mutation JAK2 V617F (ou JAK 2 exon12)
Critères mineurs (Cm) : 1) BOM : hypercellularité avec hyperplasie des 3 lignées. 2) EPO sérique abaissée.3) Pousse spontanée des progéniteurs érythroïdes.
Signes cliniques et biologiquesNFS systématiqueTV/TA, signes d’hyperviscositéErythromélalgie, érythrose faciale, prurit à l‘eau, SMGLeucocytose et thrombocytoseRechercher déshydratation/diurétiques, IRespiC/Tabagisme/SAS

Démarche diagnostique devant une Polyglobulie
Hypoxie: GDSTumeurs rein, fois, utérus: écho
Risques
Thrombose++Myélofibrose
Leucémie aiguë

N° 166 Myélome multiple des os (MM)
MGUS Myélome asymptomatique Myélome symptomatique
Pic monoclonal < 30 G/LPlasmocytose médullaire < 10%Pas de critère CRAB
Pic monoclonal ≥ 30 G/L et/ouPlasmocytose méd ≥ 10%Pas de critères CRAB
Plasmocytose méd ≥ 10%+
≥ 1critère(s) CRAB
1% par an d’évolution en MM sympto
Bilan initialSang : Creat,Alb, Ca, EPS avec quantification du pic,Immunofixation, dosage des Ig, dosage CLL kappa et
lambda + ratioUrines :PU des 24 heures, EPU + BJ. Moelle : myelogramme, cytogénétique++Imagerie : Rx squelette +/- IRM
PU de Bence Jones: chaînes légères libres (CLL) urinaires monoclonales
Urgences diagnostiques IRAHypercalcémieSyndrome d’hyperviscositéCompression médullaire
CRABCa²+ > 2.75
Rein, creat > 173Anémie < 100 g/LBone osteolytic
lesion
Diagnostic différentiel pic monoclonalSLP: LLC et lymphomeWaldenström (Ig M)Infections transitoires
Formes particulièresMM à CL
MM non secrétantLeucémie à plasmocytes (plasmo sg > 2 G/L)
POEMS (lésions ostéocondensantes)

Facteurs pronostiquesSalmon et DurieISS: β2µ et albumineCytogénétique
Rein et MM (60%)Fonctionnelle: hyperCa, AINS, IodeTubulaire: Néphropathie à cylindres myélomateux+++, FanconiGlomérulaire (PU sélective > 85% Alb) : Amylose AL (Rouge congo positif),Maladie de dépôt d’Ig monoclonale (Rouge congo négatif)
Interstitielle : NéphrocalcinoseObstructive: lithiases calciques
Principes de traitementIndication = MM SYMPTOMATIQUE seulement
Polychimiothérapie séquentielle adaptée à l’âge< 65 ans: Chimiothérapie à base de Bortezomib et Corticoïdes+ Intensification par autogreffe>65 ans ou plus jeune mais co-M: Chimiothérapie à base de Bortezomib + corticoïdes
Traitement des complications urgentes:-IRA: Hyperhydratation + Corticoïdes. Nephroprotection-Hypercalcémie : Hyperhydratation, BisP, Corticoïdes-Compression médullaire : IRM pan rachidienne en urgence , Corticoïdes + Avis neurochir ou radiothérapie (antalgique++)- Sd d’hyperviscosité : Echanges plasmatiques
Si lésions osseuses: bisphosphonates
Efficacité: EPS/ CLL (si MM à CL)

Découverte fortuite d’un pic monoclonal Ig G kappa à 24 g/L
EPSImmunofixation
Creat 73 µmol/L, Ca ² 2, 35 mmol/L, Albumine 40 g/L, Hb 135 g/L, Plq 235 G/L , Neutro 4,5 G/L
Rx squellette crâne, os long, gril costal, rachis san s anomalie
Myelogramme: 15% de plasmocytes
Quel traitement?
Aucun, MM asymptomatique. Surveillance simple du pi c monoclonal

N° 175 Prescription et surveillance d’un tttantithrombotique
Introduction AVK
J1 AVK + HéparineINR tous les 48-72hArrêt Héparine quand 2 INR cibles consécutifs
Anticoagulants oraux directs AOD
Dabigatran (Pradaxa®): anti IIa. Rivaroxaban (Xarelto®) et Apixaban (Eliquis®): anti XaIndications:Prophylaxie primaire des évènements thromboemboliques au décours des chir prothétiques hanche genouProphylaxie des AVC et embolies systémiques en cas de FA sans facteurs de risque.Rivaroxaban: curatif des TVP et la prophylaxie des récidives
++: PO, peu d’interaction alimentaire, Pas de contrôle bio--: Pas d’antidote spécifique, pas de test bio fiable
AVK- Mesures associées
Education tttiqueInteraction alimentsEviction activités/situation à risqueCarnet de surveillancePort d’une carteInformer tout intervenant médical
Néoplasie active
AVK et AOD non indiqués
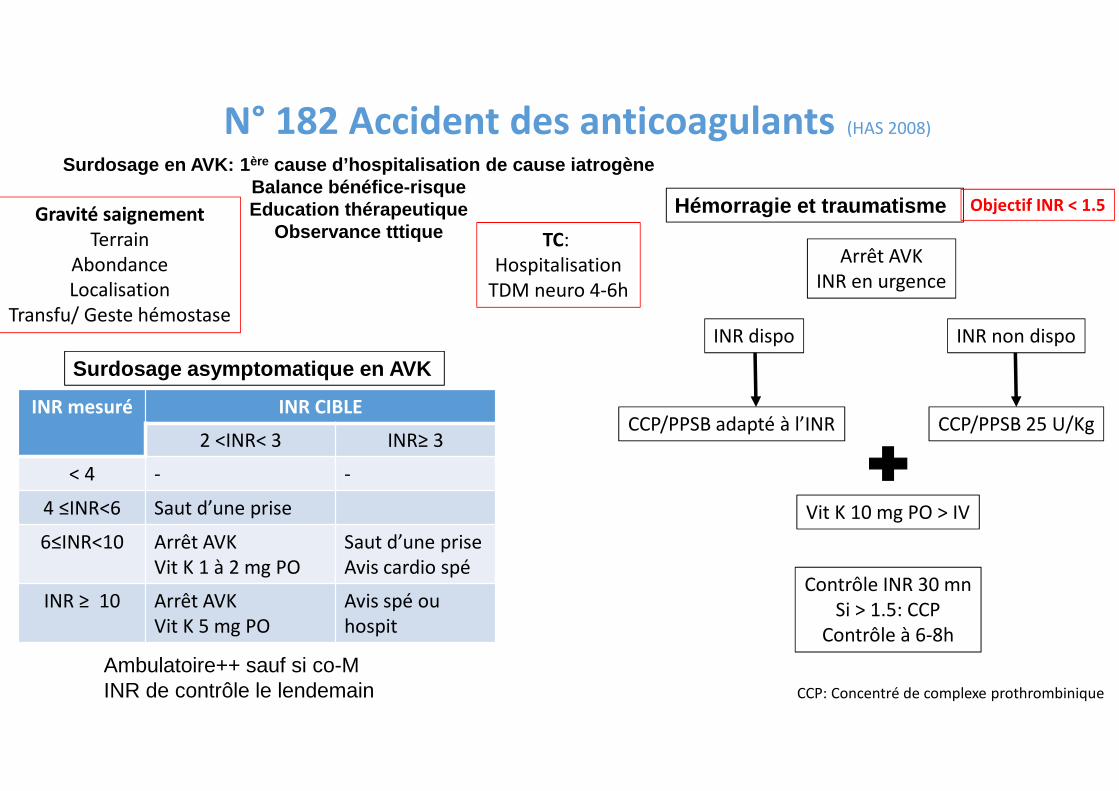
N° 182 Accident des anticoagulants (HAS 2008)
Surdosage asymptomatique en AVK
INR mesuré INR CIBLE
2 <INR< 3 INR≥ 3
< 4 - -
4 ≤INR<6 Saut d’une prise
6≤INR<10 Arrêt AVK
Vit K 1 à 2 mg PO
Saut d’une prise
Avis cardio spé
INR ≥ 10 Arrêt AVK
Vit K 5 mg PO
Avis spé ou
hospit
Ambulatoire++ sauf si co-MINR de contrôle le lendemain
Hémorragie et traumatisme
Arrêt AVK
INR en urgence
INR dispo INR non dispo
CCP/PPSB adapté à l’INR CCP/PPSB 25 U/Kg
Vit K 10 mg PO > IV
Contrôle INR 30 mn
Si > 1.5: CCP
Contrôle à 6-8h
CCP: Concentré de complexe prothrombinique
Gravité saignement
Terrain
Abondance
Localisation
Transfu/ Geste hémostase
TC:
Hospitalisation
TDM neuro 4-6h
Objectif INR < 1.5
Surdosage en AVK: 1 ère cause d’hospitalisation de cause iatrogèneBalance bénéfice-risqueEducation thérapeutique
Observance tttique

Chirurgie et actes invasifs avec interruption AVK n écessaire
Programmé Non programmé
MTEV à risque modéréAC/FA sans ATCD embol
Arrêt AVK Pas de relaisReprise 24-48 h après
Objectif INR
< 1.5
< 1.2 si neurochir
Valve mécaniqueMTEV à haut risque (< 3 mois, récidivante)AC/FA avec ATCD embol
Relais AVK-Héparine1) INR 7j avant en zone ttttique2) Arrêt AVK 5 j avant3) Héparine à dose curative
48h après fluindione/warfarine24h après acenocoumarol
4) INR la veilleSi > 1.5: Vit K 5 mg PO + contôle le matin5) Arrêt pré op de l’héparine
HNF IVSE: 4-6 hHNF SC: 8-12 hHBPM: 24 h
6) Reprise AVK 24-48 h après
INR en urgence + Vit K 5 mg
Si INR > objectif: CPP

Thrombopénie induite par l’héparine TIH type 2J5-J10HNF > HBPMchute des plq > 30% ou < 100 G/LRecherche d’Ac anti PF4 + test fonctionnelRisque thrombotique artériel et veineux +++Arrêt Héparine – Relais Danaporoïde sodique (Orgaran®) ou Argatroban (Arganova®)
Hémorragie sous héparine
Discuter Sulfate de protamine
TIH type 1< J5Non immunologiqueCorrection malgré poursuite héparine

N° 178 Transfusions sanguine et produits dérivés du sang
PSL: CGR, CPA (1 seul donneur), MCP (pool de 5 donneurs isogroupe ABO), PFC → HémovigilancePSS: Albumine, Ig polyvlentes/sp, Facteurs de coagulation → Pharmacovigilance
- PrescriptionRAI < 3 jours (séro. virales pré et post transfu non obligatoires)Groupe ABO Rh D +/-phénotype Rh et kell étendu 2 déterminations- Contrôle ultime au lit du
maladeVérification ID et groupe receveur ainsi que poche (étiquette, intégrité, date et heure péremption)- Distribution dans les 6
heures, max 2 heures par poches
- RAI à 28 jours
Règles de prescription et de transusion en CGRActe médical délégué au personnel infirmier
AB A ABO compatible
A prendre en compteABO (Ac contre Ag non présents) : toujoursRh complet (Ag immunogènes) : femme en âge de procréer, ATCD AIFM

Un patient de groupe A + doit être transfusé en CGR
Quels sont le/les groupe(s) ABO compatibles ?
A et O
Un patient de groupe A + doit être transfusé en Pla sma ?
Quels sont le/les groupe(s) ABO compatibles ?
A et AB

Complications transfusionnelles
AIGUES
TARDIVES
Incompatibilté ABOPrécoce et gravissimeVérification ABO ultimeHémolyse intra vasculaireChoc, CIVD
TACO*FréquentOAP, avec hyperviscosité sanguineTtt: Diurétiques, dérivés nitrés
TRALI*Tableau de SDRA, résolutif en 72 hTtt réanimatoire
InfectieusesATBEnvoi poche en bactério
RFNH*Pdt ou dans les 2 hPeu sévèreCause allo immune Prévention par déleucocytation
Hémochromatose post transfuRisque dès 20 CGRTableau idem HGChélateur de fer
Allo immunisationCompatibilisation ultérieure
InfectieuxVIH, VHB, VHC extrêmement faible
Délai de transfusion en CGRUrgence vitale immédiate: sans délai ( 0- ou 0+ en fonction des stocks)Urgence vitale: 30 mn (ABO)Urgence relative: 2-3 h (ABO et RAI)
Arrêt immédiat de la transfusionExamen cliniqueInformation EFSDéclaration hémovigilance sous 48 heures
*RFNH: Réaction fébrile non hémolytiqueTACO: Transfusion associated circulatory overload
TRALI: Transfusion related acute lung injury

N°291 Adénopathie superficielle
3 étiologies majeures > 90%:Infectieux/Cancer solide/Hémopathie
Autres: maladie inflammatoire, médicamenteux…
Interrogatoire minutieuxMode de vie, animaux domestiques, sexe, voyages, chasseExamens de dépistage (prostate, colon, seins, utérus), séro VIHNotion de contage, statut vaccinalPE infectieuse: piqûre/morsure/plaie cutanée, rapport sexuel risqueFièvre, AEG, symptômes BMédicaments
Signes fonctionnels en fonction de la localisation de la ou les ADPTroisier: signes digestifs, hématurie…
ADP > 1 cmExamen clinique
Loco-régional : taille, consistance, mobilitéterritoire de drainage, porte d’entrée
Schéma daté et signé+++Général , aires ggaires HSMG
Unique / Multiples
NFS, CRP
Sérologies VIH, EBV ,
CMV , VIH, Toxo
Bartonelle (chat)
Tularémie (chasse)
Syphilis
NFS, CRP, VIH, EBV, CMV , VIH
2eme intention:
Syphilis, Toxo
AAN
TDM TAP
Bx ganglionnaireHisto, bactério, myco
Qq soit localisation:penser MELANOME
Ponction ggaire: Mauvaise SeIntêret si infectieuxSa normalité n’exclue rienNeoplasie: Bx indispensable
1ère intention
2ème intention

N° 297 AnémieDéfinition
H: < 130 g/LF: < 120 g/LNv-né < 140 g/LGrossesse (≥T2) <105 g/L
Toujours éliminer fausse anémie par hémodilution
3 paramètres d’orientation
VGM Réticulocytes
< 80 fl
GrossesseHypersplénsime
Ins cardiaqueHyperproteinémie majeure
80 ≤ ≤ 100 fl > 100 fl
Microcytaire Normocytaire Macrocytaire
< 150 G/L ≥ 150 G/L
Arégénératif Régénératif
Tolérance clinique
Frottis sg

Anémie isolée
Carence martiale Inflammation Thalassémie Régénérative=
Périphérique
Arégénérative=
Central
Hémolyse HémorragieEliminer causes simples
HypothyroïdieAlcool/Foie
IRC
Dosage B12/Folates si macrocytose Effondrées
Supplémentation
NormalesMYELOGRAMME
Mégaloblastose : carence vit B9/B12Moelle riche : envahissement (hémopathie, cancer), MDSMoelle pauvre : aplasie médullaire, Myelofibrose → BOM
Erythroblastopénie : virale, toxique, AI, cong, idioP
Seule anémie µcytaire rég
Microcytaire Normo/macrocytaire= AREGENERATIVE

Anémie hémolytique
Stigmate d’hémolyse: Ictère. ↗Bilirubine libre, LDH. ↘HaptoglobineHémoglobinémie et urie si intravasculaire.
Test de Coombs
positif
AH immunune
Auto-immune (AHAI) primitive ou secondaireAH à Ac froids (MAF,Mycoplasme, EBV)
Immuno-allergique : iatrogèneAllo-immune
négatif
Frottis sanguin
Schizocytes Drépanocytes Sphérocytes Normal
Dosage enzymes érythrocytaires
G6PDPK
AH mécanique
Valve cardiaqueCECMAT

Causes d’anémie hémolytique
Corpusculaire Extra-corpusculaire
Membranaire Enzymatique Hbpathie
Acquise
HPN
Constitutionelles
SphérocytoseElliptocytose
Stomatocytose
Déf G6PDDéf PK
Qualitative Quantitative
Drépanocytose Thalassémie
Immunologique Non -immunologique
MécaniqueInfectieuse
Toxique (Plomb)

Causes carence en B12
ApportMalabsorption (MC, résection iléale)Défaut FI (Biermer, Gastrectomie,Gastrite atrophique, Sd de non dissociationde la vit B12 et de ses prot porteuses)
Causes carence en B9
ApportMalabsorption (MC, coeliaque)Augmentation besoinsIatrogène (MTX, Bactrim)
Maladie de Biermer
Femme d’âge mûr, contexte AITableau clinique hémato et neuroFOGD: atrophie muqueuseAc sérique anti FI et anti cellules pariétales gastr iquesTtt: 1 injection/ j pendant 3 semaines puis entreti en à vie
Folates et B12

N° 222 Anémie par carence martiale
Fer indispensable à la synthèse de l’hème dans les érythroblastes médullaires
Etiologie des carences martialesPertes digestives et gynécologiquesCarence d’apport (nourissons, grossesse, végètariens/taliens)Malabsoprtion (gastrectomie, MC, coeliaque)
Contexte clinique. Signes spécifiques: troubles de phanères, digestifs (perlèche, glossite)Anémie microcytaire (VGM < 80) hypochrome (CCMH < 32) arégénérative. +/- thrombocytose
Suspiçion d’anémie ferriprive
Exploration martiale
Ferritine↘
Bilan étiologique
Femme: explorations gynécologiques. Si négatif, explorations digestivesHomme: explorations digestives: FOGD + Bx puis Coloscopie puis Vidéocapsule

Traitement
EtiologiqueSymptomatique
Sels ferreux 100 à 200 mg par jour PO minimum 3 mois, souvent 6 moisAmélioration clinique, crise réticulocytaire 7-10 jJusqu’à normalisation de la ferritinePrévenir ES (Dyspepsie, nausées, selles noires) pour favoriser l’observance
JAMAIS DE TRANSFUSION SAUF SI MAUVAISE TOLERANCE
Indications de réaliser en 2eme intention:Fer sérique + transferrine: état inflammatoire, IRC, ferritine augmentée de baseRécepteurs solubles de la Tf ( ↗): pas d’indication clairement reconnue.Aide dans les contextes d’affections inflammatoires chroniques
!!Carences multiples !!
Anémie faussement normocytaireIDE(index de distribution érythrocytaire) augmenté > 16

N° 316 Hémogramme
Indications
Sd anémique/infectieux/thrombopéniqueSd tumoralAEGSystématique (grossesse, med travail..)…
NFS sur tube EDTA (anticoagulant). Réticulocytes non inclusPrivilégier valeurs absolus+++ vs %ageChiffres varient avec âge (sauf pour les Plq), sexe, ethnie
PEC URGENTE
Hb < 60 g/LHt > 60%Agra < 0.2 G/LThrombopénie < 10 G/LHyperleuco avec cellules immatures > 20 G/L
Polynucléose neutrophile > 7G/L
PhysiologiqueEffort physiqueStress aiguPost prandialesPost opNv-né, suites de couches
PathologiqueInfection bactérienneInflammation, nécroseCancerHémopathieRégénération médullaireMedoc: cortic, G-CSF
Neutropénie < 1.5 G/LInfectionHémopathieMédicamentsHypersplénismeTroubles de la répartitionConnectivites

MyélémiePassage de formes immatures de la lignée granuleuse dans le sang (myélocytes, métamyélocytes…)Infection graveMétastases ostéomédullairesRégénération médullaire: hémorragie, AH, post chimioNMP (LMC)
Lymphocytose (très variable selon l’âge, > 4 G/L ch ez l’adulte)S’assurer qu’il s’agit bien de lymphocytes (centre spécialisé)
Enfant: Réactionelle++ ViroseAdulte: SLP (LLC++), Tabac
Lymphopénie < 1 G/LInfections virales, bactériennesCancer, hémopathieChimioT, radioT, Immunosuppresseurs, corticoTMaladies auto-immunes IRénaleCDéficit immunitaire primitif
Monocytose > 1G/L
RéactionelleCancerInflammationRégénérationInfection: TBC, EI, Brucellose,Palu, leishmaniose
PrimitiveNMP (LMMC)LAM monoblastique
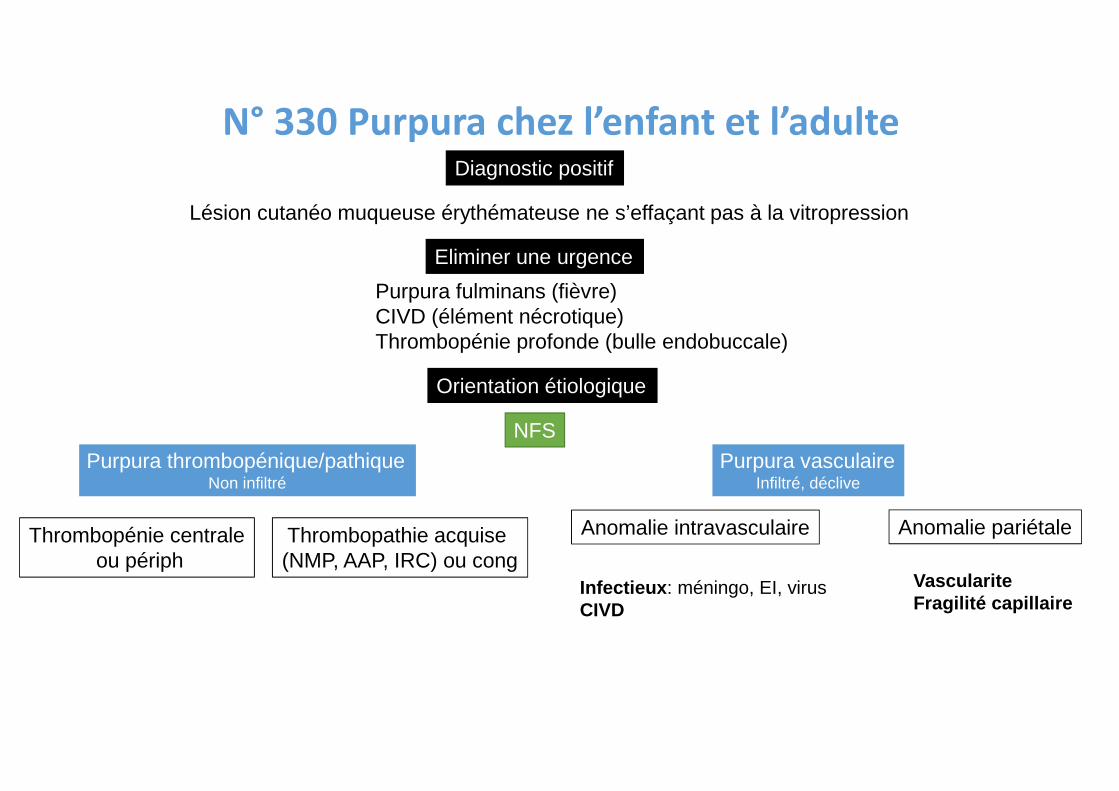
N° 330 Purpura chez l’enfant et l’adulte
Purpura fulminans (fièvre)CIVD (élément nécrotique)Thrombopénie profonde (bulle endobuccale)
Orientation étiologique
Diagnostic positif
Lésion cutanéo muqueuse érythémateuse ne s’effaçant pas à la vitropression
Eliminer une urgence
Purpura thrombopénique/pathiqueNon infiltré
Purpura vasculaireInfiltré, déclive
NFS
Thrombopénie centraleou périph
Thrombopathie acquise (NMP, AAP, IRC) ou cong
Anomalie intravasculaire Anomalie pariétale
Infectieux : méningo, EI, virusCIVD
VasculariteFragilité capillaire

N° 332 SplénomégalieToute rate cliniquement palpable
SMG
Infection
BactérienEI, abcès, septicémie, BKVirus:EBV, CMV, VIHParasitaire:PALU+++, leishmanioseFongémie à Candida
Hépatopathie (HTP)
Obstacle sus hép: Budd chiariObstacle hép: cirrhoseObstacle sous hép: thrombose porte
Hémolyse chronique
Thalassémie, sphérocytose
Hémopathie maligne
SLP: Lymphome, LLCNMP: LMC, PG, TE, MFLA
Causes rares
Maladies inflammatoires: Lupus, PR (Felty), SarcoïdoseMaladie de surcharge: Gaucher, AmyloseMétastases tumeurs solides

Bilan diagnostique
Orienté par interrogatoire et examen cliniqueImagerie: penser au doppler/injectionJAMAIS DE BIOPSIE SPLENIQUE+++Splénectomie diagnostique en dernier recours
Complications
DouleursInfarctus spléniqueRupture sléniqueAsplénie fonctionnelleHémodilution HypersplénismeDiminution rendement transfu
Splénectomie
Risque immédiat: thrombose portePrévention des infections par germes encapsulés+++Vaccins Anti pneumocoque/méningocoque/HibProphylaxie ATB par Oracilline pendant 2 ans chez l’adulte
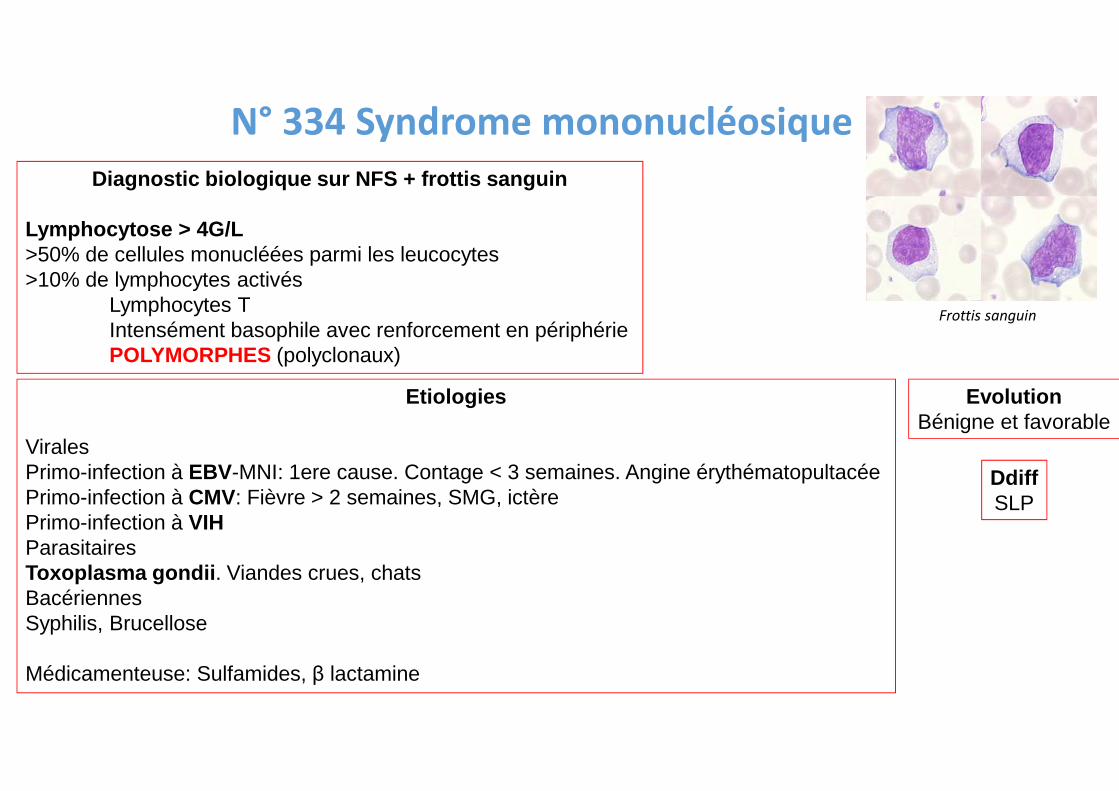
N° 334 Syndrome mononucléosique
Diagnostic biologique sur NFS + frottis sanguin
Lymphocytose > 4G/L>50% de cellules monucléées parmi les leucocytes>10% de lymphocytes activés
Lymphocytes TIntensément basophile avec renforcement en périphériePOLYMORPHES (polyclonaux)
Etiologies
ViralesPrimo-infection à EBV-MNI: 1ere cause. Contage < 3 semaines. Angine érythématopultacéePrimo-infection à CMV: Fièvre > 2 semaines, SMG, ictèrePrimo-infection à VIHParasitairesToxoplasma gondii . Viandes crues, chatsBacériennesSyphilis, Brucellose
Médicamenteuse: Sulfamides, β lactamine
EvolutionBénigne et favorable
DdiffSLP
Frottis sanguin
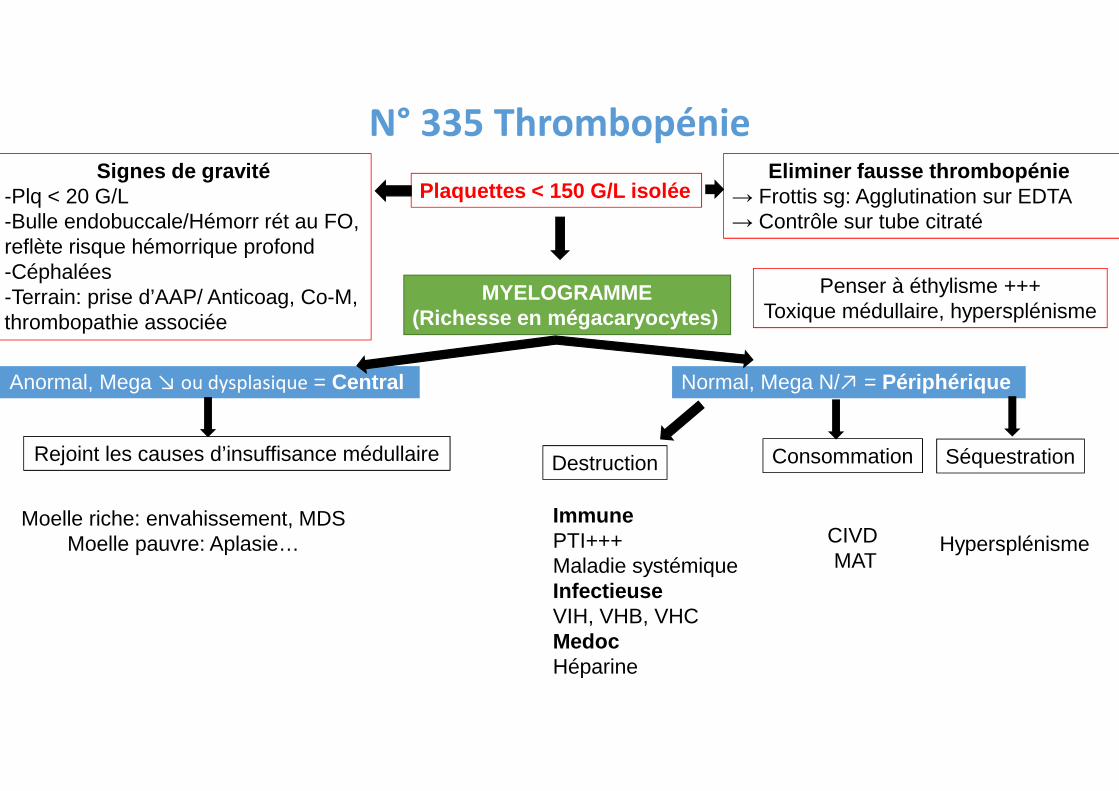
N° 335 Thrombopénie
Plaquettes < 150 G/L isoléeEliminer fausse thrombopénie
→ Frottis sg: Agglutination sur EDTA→ Contrôle sur tube citraté
Signes de gravité-Plq < 20 G/L-Bulle endobuccale/Hémorr rét au FO,reflète risque hémorrique profond-Céphalées-Terrain: prise d’AAP/ Anticoag, Co-M,thrombopathie associée
MYELOGRAMME(Richesse en mégacaryocytes)
Anormal, Mega ↘ ou dysplasique = Central Normal, Mega N/↗ = Périphérique
Destruction Consommation Séquestration
ImmunePTI+++Maladie systémiqueInfectieuseVIH, VHB, VHCMedocHéparine
CIVD MAT
Hypersplénisme
Rejoint les causes d’insuffisance médullaire
Moelle riche: envahissement, MDSMoelle pauvre: Aplasie…
Penser à éthylisme +++Toxique médullaire, hypersplénisme

PTI / PTAI
Destruction périphérique des plaquettesDiagnostic d’élimination (BH, echo abdo, VIH, VHB, VHC, AI, Breath test, EPS)Indications formelles de myelogramme:> 60 ans, ano qualitatives des autres lignées, frottis anormal, organomégalie, réfractaire
Ttt: si Plq < 30 G/L ou signe hémorragique1ere I: CorticoT 1 mg/Kg 3 semaines puis décroissance. IgIV ssi Sd hémorragique sévère (enfant: score de Buchannan)JAMAIS DE TRANSFUSION PLAQUETTAIRE sauf si hémorragie grave
PTI chronique si persistance après 12 mois: Splénectomie, Rituximab, Agoniste de la TPO, Immunosupp,

N° 339 Troubles de l’hémostase et de la coag
Tps de saignement TS: Hémostase primaire
Indication: Sd hémorragique non expliqué par thrombopénie et coag NMéthode: standardisée IVY, N < 10 mnAllongement: Plq < 80 G/L, AAP, Ht < 35%, Willebrand typique, thrombopathies acquises/constit
TP: Voie exogène VII, X, V, II, I
TCA: Voie endogène XII, XI, IX, VIII, X, V, II, I
IHC, Déficit en Vit K (1972), Déficit isolé en facteur VII
Héparine, ACC de type lupique, Déficit isolé en facteur de la voie endogène (Hémophilie)
Willebrand ou Hémophile
Pas d’IM. Carte. Avis spécialiste

Circonstance déclenchante de MTEV
Majeures : Immobilisation plâtrée/Fracture MIChir sous AG > 30 mnAlitement > 3 jCancer actif < 2 anModérées ou mineuresGrossesse/ Post partumContraception OP, THS < 1 an, Voyage > 6 hIdiopathique si rien de tout ça
Indication bilan de thrombophilie
< 50 ans avec MTEV idiopathiqueHistoire familiale de MTEVHistoire perso suggestive de SAPLMTEV x nFemme en âge de procréer avec histoire perso/fam de MTEVMTEV sites inhabituels (cerebral, hépatique, mésentérique, porte)
Bilan de thrombophilie
Déficit en AT (thrombogène++), PC ,PSMutation Facteur V LeidenMutation Facteur 2 G20210AaPL
Intêret
Modification thérapeutiqueChoix d’une contraception OPDurée d’une anticoagulation
Maladie thrombo-embolique veineuse

Encore du courage pour des questions?
Bon courage à tous


















