CHAPITRE I – Les lymphomes anaplasiques à grandes cellules
237
T T H H È È S S E E En vue de l'obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Cancérologie Présentée et soutenue par Sophie Coronas Le 18 septembre 2008 Titre : Rôle potentiel du phosphatidylinositol 5-phosphate dans l'oncogenèse associée à NPM-ALK: Implication de PIKfyve JURY Pr. Georges Delsol , Président Pr. Banafshe Larijani, Rapporteur Dr. Sylvie Friant, Rapporteur Pr. Gérard Mauco, Examinateur Dr. Hélène Tronchère, Directrice de thèse Ecole doctorale : Biologie, Santé, Biotechnologie de Toulouse Unité de recherche : INSERM U563 Directeur(s) de Thèse : Dr. Hélène Tronchère Rapporteurs : Pr. Banafshe Larijani, Rapporteur; Dr. Sylvie Friant, Rapporteur
Transcript of CHAPITRE I – Les lymphomes anaplasiques à grandes cellules

TTHHÈÈSSEE
En vue de l'obtention du
DDOOCCTTOORRAATT DDEE LL’’UUNNIIVVEERRSSIITTÉÉ DDEE TTOOUULLOOUUSSEE
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Cancérologie
Présentée et soutenue par Sophie Coronas Le 18 septembre 2008
Titre : Rôle potentiel du phosphatidylinositol 5-phosphate dans l'oncogenèse associée à
NPM-ALK: Implication de PIKfyve
JURY
Pr. Georges Delsol , Président Pr. Banafshe Larijani, Rapporteur
Dr. Sylvie Friant, Rapporteur Pr. Gérard Mauco, Examinateur
Dr. Hélène Tronchère, Directrice de thèse
Ecole doctorale : Biologie, Santé, Biotechnologie de Toulouse
Unité de recherche : INSERM U563 Directeur(s) de Thèse : Dr. Hélène Tronchère
Rapporteurs : Pr. Banafshe Larijani, Rapporteur; Dr. Sylvie Friant, Rapporteur

THESE
Présentée devant
L’UNIVERSITE DES SCIENCES PAUL SABATIER – TOULOUSE III
En vue de l’obtention du grade de
DOCTEUR DE L’UNIVERSITE PAUL SABATIER
Spécialité :
Cancérologie
Présentée par
Sophie CORONAS
Rôle potentiel du phosphatidylinositol 5-phosphate dans l’oncogenèse associée à NPM-ALK :
Implication de PIKfyve
Soutenue le 18 septembre 2008 devant la commission d’examen
Pr. Georges Delsol Université Paul Sabatier, Toulouse Président Pr. Banafshe Larijani London Research Institute, Londres Rapporteur Dr. Sylvie Friant UMR7156 CNRS/ULP, Strasbourg Rapporteur Pr. Gérard Mauco INSERM ERM 324, Poitiers Examinateur Dr. Hélène Tronchère INSERM U563, Toulouse Directrice de thèse
« Phosphoinositides et Signalisation dans les cellules hématopoïétiques» INSERM Unité 563
Centre de Physiopathologie de Toulouse Purpan, 31028 Toulouse Cedex 3


Je voudrais tout d'abord remercier les membres du Jury qui m'ont fait l'honneur de lire et
de juger ce travail.
Le Pr. Georges Delsol, pour avoir accepté de présider ce jury de thèse, et pour
m’avoir accueilli au sein du CPTP.
Le Pr. Banafshe Larijani, pour avoir accepté le travail de rapporteur de ce manuscrit
et pour vos conseils scientifiques pertinents.
Le Dr. Sylvie Friant, merci pour votre enthousiasme sur ce sujet de recherche.
Le Pr. Gérard Mauco, merci d’avoir examiner ce travail.
Madame le Dr. Hélène Tronchère, pour son encadrement scientifique depuis mon
DEA, pour son soutien, et ses conseils. Merci pour ta confiance en moi et en ce sujet (pas si
facile). Merci de m’avoir appris à devenir « une grande ». On verra dans les prochains mois si
j’arrive à m’envoler… Merci enfin, pour tout le reste et donc les discussions non scientifiques
et les bons moments que j’espère encore nombreux.

Pour continuer, je voudrais désormais remercier les membres de l’équipe du Pr.
Bernard Payrastre.
Un grand merci au Pr. Bernard Payrastre. Merci à toi, pour ton enthousiasme
scientifique, pour ta passion de la recherche. Merci, de m’avoir soutenu pendant mon DEA et
mes premières années de thèse, de m’avoir donc souvent poussées parfois dans mes
retranchements (j’aurais dû prendre un abonnement chez Kleenex). Merci aussi, pour les
discussions de rugby, de truites et de champignons (je te promets que je t’indiquerai de vrais
coins à champignons). Merci enfin de me faire confiance pour 2 ans de plus.
Je voudrais poursuivre en remerciant les autres membres de l’équipe PI5P et en
particulier: Frédérique. Merci pour ton enthousiasme, tes conseils scientifiques (ou non
scientifiques, d’ailleurs…), et ta bonne humeur contagieuse. Merci aussi aux deux autres
piliers de cette équipe : Damien et Junior. Damien, j’ai beaucoup de choses à dire sur toi, ce
garçon est mon sauveur d’ordinateur (enfin plus maintenant car la bête a rendu l’âme !!), il est
mon garde du corps (il me raccompagne chez moi tous les soirs quand il fait noir en plein
hiver) et il est surtout un ami. Donc un grand merci à toi, un seul slogan « Vive le Gindoul ! ».
Concernant Junior, le lâcheur de PIKfyve, mais je ne t’en veux pas ! Un grand merci à toi
pour ta gentillesse, ton dévouement pour les cellules (tu m’as bien souvent enlevé une épine
du pied…), pour ta bonne humeur et tes qualités humaines. Pour toi aussi le même slogan
que pour Damien. Mais attention, cette équipe ne serait rien sans le Dieu de l’HPLC, j’ai cité
le grand Gaëtan. Cet homme est un roi du VTT, de la commande et des phosphoinositides.
Merci pour tout ça et merci d’être toi. Je voudrais aussi remercier la personne qui m’a appris
le dosage de PI5P, Caro, pour sa gentillesse et sa bonne humeur contagieuse.
Un grand merci aux « Plaquettistes ». Merci à Monique et Marie-Pierre, pour leur
engagement féministe, scientifique et leurs qualités humaines. Merci aussi à leurs disciples,
Marie-Cécile pour ta gentillesse et ton calme (très utile d’ailleurs lors de la préparation des
plaquettes). Merci à la petite dernière, Valérie pour ton humour, tes histoires, j’ai rejoint ton
club « le tabac, c’est tabou, on en viendra tous à bout », en espérant que j’ai autant de volonté
que toi…
Merci aux différents groupes LAM. Ceux qui travaillent sur « le cycle » Stéphane
pour ses conseils lors des réunions d’équipe, Anne V et Anne M, je dois dire que je reste
admirative devant votre organisation. Merci à vous deux pour votre soutien, vos qualités
humaines, vos mots gentils. Passons désormais au groupe LAM mais plutôt côté « cellules
souches ». Merci à Claire pour ta PASSION de la recherche, je ne vois que ce mot pour te
caractériser. A Jessica, merci à ma nouvelle voisine de bureau. Tu verras la route est longue

et semée d’embûches, mais tu t’en sortiras (sur tous les plans, et je sais de quoi je parle !!!).
Merci à Mathieu, mon compagnon de pause clopes, merci à lui pour son oreille attentive et
ses cafés de la machine (qui sont meilleur, soi-disant ??). Tu nous manques, rentre vite. Et
pour finir le groupe LAM et mTOR. Merci donc à Cédric pour nos discussions dans le
bureau, pour ton soutien. A Nathalie, tu seras parfaite dans ta jolie robe, n’oublie pas c’est
celui-là le plus beau jour de ta vie. Annelies, allez courage, bientôt la fin, un dernier effort tu
vas y arriver. Et à la petite dernière Camille.
Un merci aussi à ceux qui ont habité ces murs et qui sont désormais sous d’autres
cieux. Notre anglaise Kelly. Delphine : Vive la Magie de Noël !!
Enfin mes années de thèse n’auraient pas été ce qu’elles ont été sans mes trois
mousquetaires, je parle de Sonia, Audrey et Fabienne. Merci à vous trois, mes épaules, mes
amies… Merci à Sonia, pour être une amie, pour les bons moments mais aussi les moins bons
que l’on a passé ensemble. C’est ça qui crée les liens. Merci d’avoir accepté d’être le témoin
du plus beau jour de ma vie. Merci à Audrey, pour ce que tu es, tes coups de gueule, on ne
sait pas pourquoi, tes coups de blues. Le bureau est bien calme depuis que tu es partie. Enfin,
à ma Fabi, merci pour tes coups de téléphone (bien plus fréquents que les miens), pour ta
présence, pour toutes les rigolades que nous avons eu grâce à toi (il est 11h du matin,
Fabienne est arrivée ????). Un très grand merci à vous trois.
Je voudrais aussi remercier les autres membres du Département et donc de l’étage
jaune, pour leurs mots gentils, leur soutien, leur prêt de réactifs. Et donc un merci particulier à
Christine B, Christine J, Anne et à tous les autres.
Je vais maintenant passer à mon ancien bâtiment, là où tout à commencer : le bâtiment
C, la 326. Un merci donc à Michel, pour m’avoir accueillie dans son équipe. À Safouane, qui
le premier m’a donné le goût de la recherche, et sans qui je ne serais pas Docteur aujourd’hui.
Merci de m’avoir poussé à me présenter en DEA. Merci pour tes qualités humaines.
Je voudrais aussi remercier toutes les personnes de « la 326 » et en particulier les
filles : Stéphanie, Marie, Alex. Un merci particulier à ma copine Monitrice : Camille.
Bientôt le bout du tunnel.
Enfin à vous tous que je viens de citer et à ceux que j’ai sans doute oubliés (désolée),
un grand merci pour votre écoute attentive de toutes mes histoires. Je sais, je parle beaucoup
et de tout. Donc merci pour avoir écouter ma petite vie, en vrac : ma bague de fiançailles
(n’est-ce pas Gaëtan ?), les aventures de Canelle (Coronavirose, ligaments croisées…), mes
divers feux domestiques, mes dégâts des eaux, mes tentatives d’arrêts du tabac, ma mauvaise
humeur parfois, mes histoires avec Laurent et tout le reste. Donc MERCI.

Je voudrais terminer en remerciant ma famille. Je voudrais donc remercier en premier
lieu, mon mari, Laurent. Un grand merci à toi, pour m’avoir soutenu et supporté depuis ces
nombreuses années, pour être toujours là quand j’en ai besoin. Merci à mes parents, pour leur
soutien sans faille depuis toujours. Une dédicace spéciale à Super T, ma Kéké, pour nos
années étudiantes ensemble, pour tous les bons moments et surtout ne change rien. Enfin
merci à ma nouvelle famille, Michel, Anne, Marc et Valérie. Un clin d’œil à la petite
dernière qui fait que je suis pour la première fois tatie : Romane.

RESUME
Les Lymphomes Anaplasiques à Grandes Cellules (ALCL) sont associés dans 75% des cas à
une translocation chromosomique qui juxtapose le gène de la nucléophosmine (NPM) et le
gène de l’Anaplastic Lymphoma Kinase (ALK), qui code pour un récepteur à activité tyrosine
kinase. Cette translocation conduit à l’expression de la d’une protéine chimérique NPM-ALK
constitutivement active, au fort pouvoir transformant via l’activation de voies telles que celles
de la PLC-γ, des facteurs de transcription STAT3 et STAT5, des tyrosines kinases pp60c-scr et
de la PI 3-kinase/Akt. Les phosphoinositides (PIs) sont des régulateurs spatio-temporels de
grandes voies de signalisation qui contrôlent la réorganisation du cytosquelette, la
prolifération et le trafic vésiculaire. La phosphatidylinositol 5-phosphate (PtdIns5P) est l’un
des derniers membres de cette famille à avoir été caractérisé et sa fonction ainsi que son
métabolisme ne sont pas encore totalement connus. Dans ce travail, nous avons étudié le rôle
de ce nouveau second messager lipidique dans l’oncogenèse liée à NPM-ALK.
Nos travaux mettent en évidence la présence de taux élevés de PtdIns5P dans les lignées
surexprimant l’oncogène NPM-ALK. Cette augmentation est dépendante de l’action de la
PtdIns 5-kinase, PIKfyve, une enzyme connue pour son rôle dans l’homéostasie vésiculaire.
Nous montrons que l’oncogène NPM-ALK et PIKfyve s’associent dans un complexe
moléculaire qui fait intervenir un troisième partenaire, la PI 3-kinase. L’utilisation d’un
inhibiteur de NPM-ALK nous permet de mettre en évidence que l’oncogène, via son activité
tyrosine kinase, régule l’activité lipide kinase de PIKfyve. De plus, l’utilisation d’une sonde
spécifique pour ce PtdIns5P nous permet de le localiser dans des extensions membranaires qui
sont des sites actifs du remodelage du cytosquelette d’actine, suggérant ainsi que ce PI
pourrait avoir un rôle dans le contrôle de ce processus. Enfin, nos résultats préliminaires
montrent que la présence de PIKfyve est nécessaire pour permettre la migration et l’invasion
des cellules NPM-ALK positives.
Ce travail met en évidence le rôle important de PIKfyve et du PtdIns5P dans les phénomènes
transformants induits par l’oncogène NPM-ALK, en particulier en ce qui concerne les
mécanismes de migration et d’invasion. Outre la mise à jour de cibles pharmacologiques
originales, cette étude dévoile pour la première fois une interaction entre un processus
oncogénique et le détournement des voies de trafic intracellulaire qui pourrait être généralisé à
d’autres oncogènes.

ABSTRACT Anaplastic large cell lymphomas (ALCL) are associated in 75% of case with a chromosomic
translocation, fusing the gene of nucleophosmin (NPM) to the gene coding for the tyrosine
kinase receptor Anaplastic Lymphoma Kinase (ALK). This translocation leads to the
expression of a constitutive active chimeric NPM-ALK protein in tumoral cells which
strongly activates proliferative and anti-apoptotic pathways such as PLC-γ, STAT3 and
STAT5, pp60c-scr and the PI 3-kinase/Akt pathway. Phosphoinositides (PI) are keys regulators
of signalling pathway involved in cytoskeleton reorganization, proliferation and vesicular
trafficking. Phosphatidylinositol 5-phosphate (PtdIns5P) is one of the last PI characterized
and its metabolism and cellular function are not fully understood. In this work, we focused on
the potential role of PtdIns5P in NPM-ALK-dependant oncogenesis.
We found higher levels of PtdIns5P in ALCL and NPM-ALK-transfected cells. This PtdIns5P
increase is dependant on the PtdIns 5-kinase, PIKfyve, an enzyme know for its role in
membrane trafficking and homeostasis. We show that NPM-ALK and PIKfyve are indirectly
associated within a complex via a third partner, the PI 3-kinase. By using a NPM-ALK
specific inhibitor, we provide evidence that the tyrosine kinase activity of NPM-ALK
regulates the activity of PIKfyve. Moreover, we show that in NPM-ALK-expressing NIH3T3
cells, PtdIns5P is localized in the membrane extensions characteristic of NPM-ALK-
transformed cells, which are dynamic actin remodeling sites, suggesting that the lipid could
play a role in this process. Finally, our preliminary datas show that PIKfyve regulate the
migration and the invasion of NPM-ALK-positive cells.
This work provides evidence for the important role of PIKfyve and PtdIns5P in oncogenesis
induced by NPM-ALK, particularly in migration and invasion.
Overall, in addition to providing a new pharmacological target, this work reveals a link
between vesicular trafficking and oncogenesis.

ABREVIATIONS
ALCL: Anaplastic Large Cell Lymphoma ALK: Anaplastic Lymphoma Kinase ALO17: ALK Lymphoma Ologomerisation partner on chromosome 17 AP180: Adaptor protein 180 Arf: ADP-ribosylation factors ARNO: Arf nucleotide binding site opener ArPIKfyve: Associated regulator of PIKfyve ATP: Adénosine tri-phosphate ATX1: Arabidopsis homologue of trithorax BAR: Bin, Amphiphysin and Rvs Btk: Bruton’s tyrosine kinase CALM: Clathrin Assembly lymphoid Leukemia protein CARS: Cysteinyl-tRNA synthetase enzyme CCT: Chaperonins containing TCP1 CDK: Cyclin Dependent Kinase CHMPs: Charged Multivesicular Body Protein CHO: Chinese Hamster Ovary CHOP: Cyclophosphamide/doxorubicine/vincristine/prednisone CI-MRP: Récepteur au mannose-6-phosphate cation indépendant CLTC1: Clathrin heavy chain-like 1 Cpn60_TCP1: HSP chaperonin_T-complex protein 1 DAG: Diacyglycérol DENN: Différenciellement Exprimé dans les cellules Néoplasiques versus Normales DEP: Dishevelled, Egl10 and Pleckstrin DGK: Diacylglycérol Kinase EGFR: Epidermal Growth Factor Receptor EML4: Echinoderm microtubule-associated protein like 4 ENTH: Epsin NH2-Terminal Homology ERK: Extracellular signal Regulated Kinase ERM: Ezrine/Radixine/Moésine ESCRT: Endosomal Sorting Complex Required for Transport EVH1: Enabled/VASP Homology domain-1 FAK: Focal Adhesion Kinase FAPP: Four Phosphate Adaptator F-MACHOP: 5-fluoroucil/méthothrexate/cytarabine/CHOP FYVE: Fab1p, YOTB, Vps27p et EEA1 Gab1: Grp2-associated protein 1 GEF: Guanine exchange factor Glut4: Glucose Transporter 4 GRAM: Glucosyl transferase, Rab-like GTPase activator and myotubularins Grp1: General receptor for phosphoinositides Hip1: Huntington interacting protein 1 ICAMs: Intracellular Adhesion Molecule IMT: Inflammatory myofibroblastic tumors ING2: Inhibitor of Growth 2 Ins(1,3,4,5)P4: Inositol (1,3,4,5) tetrakisphosphate InsP3 ou IP3: Inositol (1,4,5) trisphosphate

IpgD: Invasion plasmid gene D IRAP: Insulin-responsive aminopeptidase IRS-1: Insulin Receptor Substrate-1 Jak: Janus tyrosine kinase JAK: Just another Kinase LDCV: Large Dense Core Vesicles LPA: Acide lysophosphatidique LTK: Leucocyte tyrosine kinase MACOP-B: Méthothrexate/ doxorubicine/cyclophosphamide/ vincristine/prednisone/bléomycine MAPK: Mitogen activated Protein Kinase MK: Midkine MLF1: Myelodysplasia-Myeloid Leukemia Factor 1 MMAC: Mutated in Multiple Advanced Cancers MMP9: Matrix metalloproteinases 9 MSN: Moesin MTM1: Myutubularin 1 MTMs: Myotubularines MVB: Multivesicular body MYH-9: Non-muscle Myosin Heavy Chain NES: Nuclear Export Signal NIPA: Nuclear Interacting Partner of Anaplastic Lymphoma Kinase NLS: Nuclear Localization Signal NPM: Nucléophosmine OCRL: Oculocerebrorenal Lowe Syndrome OSBP: Oxysterol Binding Protein P130cas: p130 Crk-associated substrate PA: Acide phosphatidique PDE3B: cGMP-inhibited phosphodiesterase 3B PDK: Protein kinase D PDZ-BS: PSD-95/Dlg/ZO-1 binding site PH: Pleckstrin Homology PI 3-kinase: Phosphoinositide 3-kinase PIAS3: Protein Inhibitor of Activated STAT3 PIKfyve: Phosphatidylinositol kinase-Fab1p/YOTB/Vac1p/EEA1 PIs: Phosphoinositides PKC: Protéine kinase C PKC: Protéine Kinase C PLC: Phospholipase C PLC: Phospholipase C PLD: Phospholipase D PLIP: PTEN-like phosphatase PROPPINs: β-Propellers that bind polyphosphoinositides PS: Phosphatidylsérine PTB: Phosphotyrosine Binding Domain PtdIns(3,4)P2: Phosphatidylinositol 3,4-bisphosphate PtdIns(3,4,5)P3 ou PIP3: Phosphatidylinositol 3,4,5-trisphosphate PtdIns(3,5)P2: Phosphatidylinositol 3,5-bisphosphate PtdIns(4,5)P2: Phosphatidylinositol 4,5-bisphosphate PtdIns: Phosphatidylinositol

PtdIns3P: Phosphatidylinositol 3-monophosphate PtdIns4P 5-kinase: Phosphatidylinositol 4-monophosphate 5-kinase PtdIns4P: Phosphatidylinositol 4-monophosphate PtdIns5P: Phosphatidylinositol 5-monophosphate PTEN: Phosphatase and Tensin Homologue deleted on chromosome 10 PTN: Pléiotrophine PTP: Phosphotyrosine Binding PTPMT1: PTP localized to Mitochondrion 1 PX: Phox Homology RAG2: Recombination Activating Gene 2 RARα: Retinoic Acid Receptor α RCPG: Récepteurs couplés aux protéines G hétérotrimériques RID: Rac-Induced recruitement Domain RMN: Resonance magnetique nucléaire SAPK: Stress Activated Protein Kinase SARA: Smad Anchor for Receptor Activation SGK1: Serum and glucocorticoid inducible kinase SH2: Src Homology 2 SID: SET-interacting domain SNXs: Sortin Nexins SPOP: Speckle-type POZ domain protein SRP: Surface Plasma Resonance STATs: Signal Transducers and Activators of Transcription TCR: Récepteur aux lymphocytes T TFIIH: General Transcription factor II H TMP: Tropomyosine TNF: Tumor Necrosis Factor TPR: Translocated Ptomoter Region Vps34: Vesicular protein sorting 34 WSTF: Williams-Beuren syndrome transcription Factor

SOMMAIRE
INTRODUCTION CHAPITRE I .................................................................................................................... 1 Les lymphomes anaplasiques à grandes cellules ................................................................... 1
I-Généralités ........................................................................................................................... 1 1. Définition de l’entité ...................................................................................................... 1 2. Caractéristiques cliniques............................................................................................... 1 3. Caractéristiques histopathologiques............................................................................... 3 4. Les traitements ............................................................................................................... 4
4.1. Les traitements actuels ............................................................................................ 4 4.2. Les nouvelles stratégies thérapeutiques en développement .................................... 4
4.2.1. Les inhibiteurs de l’activité tyrosine kinase..................................................... 4 4.2.2. L’invalidation de l’expression de ALK............................................................ 5 4.2.3. Les stratégies de thérapie génique.................................................................... 6 4.2.4. La vaccination anti-tumorale............................................................................ 6
II. Caractéristiques cytogénétiques ........................................................................................ 7 1. La t(2;5)(p23;q35) .......................................................................................................... 7
1.1. La nucléophosmine ................................................................................................. 7 1.2. Le gène ALK......................................................................................................... 10 1.3. NPM-ALK............................................................................................................. 12
2. Autres translocations impliquant le gène ALK............................................................ 12 2.1 TMP3-ALK ............................................................................................................ 13 2.2. TGF-ALK.............................................................................................................. 13 2.3. ATIC-ALK............................................................................................................ 14 2.4. CLTC1-ALK ......................................................................................................... 15 2.5 MSN-ALK.............................................................................................................. 15 2.6 TPM4-ALK ............................................................................................................ 15 2.7. MYH-9-ALK......................................................................................................... 15 2.8. RanBP-2-ALK....................................................................................................... 16 2.9. ALO17-ALK ......................................................................................................... 16 2.10. CARS-ALK......................................................................................................... 16 2.11. EML4-ALK......................................................................................................... 16 2.12 Sec31L1-ALK ...................................................................................................... 16
III. Autres pathologies impliquant ALK .............................................................................. 17 IV. Mécanismes d’oncogenèse associés à ALK................................................................... 18
1. Effet transformant ........................................................................................................ 18 2. Les modèles animaux ................................................................................................... 19 3. Transduction du signal induit par ALK........................................................................ 20
3.1. NPM-ALK induit la prolifération ......................................................................... 21 3.1.1. La voie des MAP kinases ............................................................................... 21 3.1.2. La phospholipase C-γ (PLC-γ) ....................................................................... 23 3.1.3. La tyrosine kinase pp60c-src ............................................................................ 23 3.1.4. NIPA............................................................................................................... 24
3.2. NPM-ALK induit la survie cellulaire................................................................... 25 3.2.1. La phosphoinositide 3-kinase (PI 3-kinase) ................................................... 25 3.2.2. La voie Jak/STAT .......................................................................................... 27

3.3. NPM-ALK induit la migration cellulaire et le remodelage du cytosquelette ....... 29 3.3.1. La voie p130Cas............................................................................................. 29 3.3.2. Implication des GTPases de la famille Rho ................................................... 29
3.4. NPM-ALK module le phénotype immunitaire via l’expression de CD30........... 30 3.5. Régulation négative de NPM-ALK....................................................................... 31
3. Identification de nouvelles cibles et de nouveaux partenaires de ALK ....................... 32 CHAPITRE II ................................................................................................................ 35 Les Phosphoinositides ............................................................................................................ 35
I- Le métabolisme des phosphoinositides et les enzymes clés............................................. 36 1. Le cycle des phosphoinositides .................................................................................... 36 2. Le métabolisme des phosphoinositides dit « classiques »............................................ 37 3. Le métabolisme des D3-phosphoinositides.................................................................. 38
3.1. Les PI 3-kinases .................................................................................................... 38 3.1.1. Les PI 3-kinases de classe I............................................................................ 39
3.1.1.1. L’activité lipide kinase des PI 3-kinases de classe I ............................... 40 3.1.1.2. L’activité protéine kinase des PI 3-kinases de classe I ........................... 41
3.1.2. Les PI 3-kinases de classe II .......................................................................... 41 3.1.3. La PI 3-kinase de classe III ............................................................................ 42
3.2. Les phosphatases agissant sur les D3-phosphoinositides...................................... 43 3.2.1. La 3-phosphatase PTEN................................................................................. 43 3.2.2. Les 5-phosphatases SHIP1 et SHIP2 ............................................................. 43 3.3.3. Les 3-phosphatases de la famille des myotubularines ................................... 44
4. Les voies permettant la génération de PtdIns5P........................................................... 44 II- Rôles biologiques et localisation des différents phosphoinositides ................................ 44
1. Le PtdIns ...................................................................................................................... 44 2. Le phosphoinositide le mieux caractérisé : le PtdIns(3,4,5)P3 ..................................... 45 3. Les phosphatidylinositol bisphosphates ....................................................................... 46
3.1. Le PtdIns(4,5)P2 .................................................................................................... 46 3.2. Le PtdIns(3,4)P2 .................................................................................................... 47 3.3. Le PtdIns(3,5)P2 .................................................................................................... 48
4. Les phosphatidylinositol monophosphates................................................................... 50 4.1. Le PtdIns4P ........................................................................................................... 50 4.2. Le PtdIns3P ........................................................................................................... 51 4.3. Le PtdIns5P ........................................................................................................... 52
III- Les domaines protéiques d’interaction avec les phosphoinositides............................... 53 1. Les domaines PH.......................................................................................................... 53 2. Les domaines GRAM-PH ............................................................................................ 55 3. Les domaines PX.......................................................................................................... 56 4. Les domaines FYVE .................................................................................................... 57 5. Les domaines FERM.................................................................................................... 59 6. Le domaine PHD.......................................................................................................... 60 7. Les PROPPINs ............................................................................................................. 62 8. Le domaine ENTH ....................................................................................................... 63 9. Le domaine ANTH....................................................................................................... 64 10. Les séquences basiques et hydrophobes..................................................................... 64 11. Les autres domaines de liaison................................................................................... 65
IV- Le PtdIns5P.................................................................................................................... 65 1. Voies de synthèse et de dégradation ............................................................................ 65
1.1. Synthèse à partir du PtdIns(4,5)P2......................................................................... 66 1.1.1. Les enzymes bactériennes .............................................................................. 66

1.1.1.1. IpgD......................................................................................................... 66 1.1.1.2. SigD/SopB............................................................................................... 67
1.1.2. Les enzymes eucaryotes : les PtdIns(4,5)P2 4-phosphatases de type I et II ... 68 1.2. Synthèse à partir du PtdIns(3,5)P2 : rôle des myotubularines (MTMs) ................ 69 1.3. Synthèse à partir du PtdIns : Rôle de PIKfyve...................................................... 72 1.4. Métabolisation du PtdIns5P par les PtdIns5P 4-kinases de type II ....................... 72 1.5. Métabolisation par la phosphatase PLIP ............................................................... 74
2. Fonctions du PtdIns5P.................................................................................................. 75 2.1. Rôle dans l’invasion bactérienne – Potentialisateur de la voie de survie PI 3-kinase/Akt..................................................................................................................... 75 2.2. Fonctions nucléaires.............................................................................................. 76 2.3. Rôle dans le trafic vésiculaire ............................................................................... 78 2.4. Rôle dans la réorganisation du cytosquelette ........................................................ 79
CHAPITRE III .............................................................................................................. 81 La PtdIns3P 5-kinase de type III : PIKfyve......................................................................... 81
I-Structure de PIKfyve ......................................................................................................... 81 II-Localisation de PIKfyve................................................................................................... 84 III-Fonctions......................................................................................................................... 85
1. PIKfyve est une lipide kinase....................................................................................... 85 1.1. PIKfyve synthètise du PtdIns(3,5)P2..................................................................... 86 1.2. PIKfyve synthétise du PtdIns5P............................................................................ 86
2. PIKfyve est une protéine kinase................................................................................... 87 IV-Régulateurs et partenaires de PIKfyve ........................................................................... 88
1. Régulation de son activité enzymatique par des petites molécules.............................. 88 2. Régulation par le proto-oncogène Akt ......................................................................... 89 3. Régulation par le stress osmotique............................................................................... 90 4. Régulation par l’insuline .............................................................................................. 90 5. Le complexe PIKfyve/ArPIKfyve/Sac3....................................................................... 91 6. Rôle de p40 un effecteur de Rab9 ................................................................................ 94 7. PIKfyve s’associe à la PI 3-kinase de classe IA........................................................... 94
V-Rôles biologiques de PIKfyve.......................................................................................... 95 1. PIKfyve et la maintenance de l’homéostasie vésiculaire ............................................. 95 2. PIKfyve et l’exocytose ................................................................................................. 96 3. PIKfyve : contrôle du transport rétrograde des endosomes au TGN ........................... 97 4. Implication dans le métabolisme du glucose en réponse à l’insuline .......................... 97 5. Implication dans le trafic de canaux ioniques .............................................................. 98 6. Rôle dans les voies de dégradation de récepteurs membranaires................................. 99 7. Rôle dans la disparition des fibres de stress............................................................... 100 8. Le récepteur à l’EGF.................................................................................................. 101
VI-Pathologies impliquant PIKfyve ou ses effecteurs ....................................................... 101
RESULTATS Partie I : Les cellules NPM-ALK positives présentent un niveau élevé de PtdIns5P ;
implication de PIKfyve .......................................................................................................... 104 I- Introduction .................................................................................................................... 104 II- Conclusion..................................................................................................................... 106
Partie II : Etude fonctionnelle de l’interaction entre PIKfyve et NPM-ALK ........................ 109 I- Génération d’outils pour l’étude de PIKfyve ................................................................. 109
1. Clonage et expression de PIKfyve sauvage et mutants.............................................. 109 2. Génération d’un anticorps anti-PIKfyve .................................................................... 112

II- Existe-il un complexe PIKfyve/NPM-ALK ? ............................................................... 114 1. Présence d’une activité PtdIns 5-kinase associée à NPM-ALK................................. 114 2. Interaction PIKfyve/NPM-ALK dans un système de transfection ectopique ............ 115 3. Mise en évidence du complexe au niveau endogène.................................................. 116
III- L’oncogène NPM-ALK régule-t-il l’activité de PIKfyve ? ......................................... 118 IV- La relation entre PIKfyve et le proto-oncogène Akt dans les cellules NPM-ALK positives.............................................................................................................................. 119
Partie III : Rôle physiologique de PIKfyve dans l’oncogenèse associée à NPM-ALK ......... 121 I- Effets de PIKfyve sur la prolifération............................................................................. 121 II- Effets sur la migration et l’invasion .............................................................................. 121 III- PIKfyve peut-il réguler NPM-ALK ? .......................................................................... 125
1. Rôle de PIKfyve sur la localisation de NPM-ALK.................................................... 125 2. Rôle de PIKfyve sur le proto-oncogène Akt .............................................................. 127
V- Conclusion-Discussion.................................................................................................. 128
CONCLUSION ET PERSPECTIVES I- Conclusion ...................................................................................................................... 134 II- Perspectives................................................................................................................... 137
1. La relation entre PIKfyve et le proto-oncogène Akt dans les cellules NPM-ALK positives.......................................................................................................................... 137 2. Etude de la balance PtdIns(3,5)P2/PtdIns5P dans les cellules NPM-ALK positives : implication des protéines ArPIKfyve, Sac3 et des 3-phosphatases de la famille des myotubularines ............................................................................................................... 139 3. Rôle de PIKfyve dans les processus invasifs associés à NPM-ALK ......................... 141 4. Rôle de PIKfyve et du PtdIns5P dans les mécanismes de régulation du cytosquelette d’actine dans les cellules NPM-ALK positives.............................................................. 142 5. Rôle de PIKfyve sur NPM-ALK................................................................................ 143 6. Identification de nouveaux effecteurs du PtdIns5P.................................................... 144
BIBLIOGRAPHIE ...............................……………………………………..146
MATERIEL ET METHODES……………………………………..177
ANNEXES

INTRODUCTION

Introduction
CHAPITRE I
Les lymphomes anaplasiques à grandes cellules
I-Généralités
1. Définition de l’entité
Les lymphomes anaplasiques à grandes cellules (ALCL) représentent une entité pathologique
dont les cellules lymphoïdes néoplasiques expriment l’antigène d’activation CD30/Ki-1 et
présentent une morphologie particulière de grandes cellules anaplasiques. La classification de
l’Organisation Mondiale de la Santé définit les ALCL comme étant des lymphomes non
hodgkiniens, dont les cellules malignes sont d’origine T ou de phénotype dit « nul », c’est-à-
dire n’exprimant pas de marqueurs T ou B (Falini, 2001). Les ALCL sont relativement peu
fréquents chez l’adulte où ils ne représentent que 5% de tous les lymphomes non hodgkiniens,
par contre c’est l’une des hémopathies malignes pédiatriques les plus fréquentes puisqu’ils
sont détectés dans 35% des cas de lymphomes non hodgkiniens (Falini, 1998). Les ALCL
sont fréquemment associés à une translocation chromosomique réciproque, la t(2;5)(p23;q35)
(Fiorani, 2001). Cette anomalie chromosomique implique le gène de la nucléophosmine
(NPM) situé en 5q35 et le gène ALK (Anaplastic Lymphoma Kinase) sur le chromosome 2
(Morris, 1994). Bien que formant une entité bien définie, les ALCL peuvent présenter de
nombreuses hétérogénéités tant sur les plans cliniques et morphologiques que cytogénétiques
(Stein, 2000).
2. Caractéristiques cliniques
Les ALCL existent sous 3 formes cliniques : une forme systémique primaire, une forme
cutanée primaire et une forme secondaire (Tilly, 1997). La plupart des ALCL sont des
lymphomes systémiques primaires et affectent préférentiellement les enfants et les jeunes
adultes (Ladanyi, 1997 ; Drexler, 2000). Ces cas sont généralement ALK positifs, affectent
principalement les enfants mâles (distribution homme/femme de 3/1) (Falini, 1999) et sont de
relativement bon pronostic. Ainsi, les différentes études ont montré que la survie à 5 ans de
1

Introduction
ces sujets varie entre 71 et 93% et le pourcentage de rémission complète est de 78%
(Gascoyne, 1999 ; Falini, 1999 ; Kutok, 2002). Les ALCL ALK négatifs surviennent
généralement chez les adultes de plus de 60 ans. Chez ces patients, il ne semble exister
aucune différence en fonction du sexe et le pronostic est plutôt défavorable (Gascoyne, 1999).
Ainsi, selon les études, la survie à 5 ans varie entre 15 et 46% et le taux de rémission
complète est de 56% (Falini, 1999 ; Fiorani, 2001 ; Kutok 2002). Ces désordres
lymphoprolifératifs présentent un haut grade de malignité (Stein, 2000). En effet, la majorité
des patients se présentent au diagnostic à un stade avancé de la maladie (stade III ou IV de la
classification Ann Arbor) (Tableau 1), avec des atteintes extra-ganglionnaires localisées le
plus fréquemment au niveau de la peau, des os, des poumons, de la moelle osseuse et du foie
(Falini, 1999).
Stade
I Atteinte d'une seule aire ganglionnaire ou d'une seule localisation
extra-ganglionnaire
II
Atteinte de deux ou plusieurs aires ganglionnaires situées du même
côté du diaphragme, ou atteinte de plusieurs territoires extra-
ganglionnaires situés du même côté du diaphragme
III Atteintes ganglionnaires situées de part et d'autre du diaphragme
accompagnées éventuellement d'une atteinte extra-ganglionnaire
IV Atteintes disséminées d'une ou plusieurs aires ganglionnaires ou
extra-ganglionnaires
Tableau 1: Classification d’Ann Arbor
La forme cutanée primaire, observée chez les patients âgés (moyenne d’âge 60 ans) et qui
représente 10% des lymphomes cutanés, est ALK négative. Ces lymphomes sont de très bon
pronostic puisque, chez 25% des patients, ils régressent spontanément de façon complète ou
partielle (Ten Berge, 2003). Enfin, quelques rares cas seulement, qualifiés de secondaires,
peuvent apparaître chez certains patients âgés suite à la transformation anaplasique d’un autre
lymphome, comme la maladie de Hodgkin ou les lymphomes T périphériques (Stein, 2000).
Ces lymphomes anaplasiques secondaires sont le plus souvent ALK négatifs et de mauvais
pronostic.
2

Introduction
3. Caractéristiques histopathologiques
Les ALCL présentent de nombreuses morphologies puisque 8 variants morphologiques sont
décrits dans la littérature (Gascoyne, 1999). La forme commune est la plus fréquente : elle est
rencontrée dans plus de 75% des cas (Drexler, 2000) (Figure 1). Elle est composée d’une
population de grandes cellules caractéristiques à cytoplasme abondant, avec un noyau
excentré en forme de fer à cheval et un appareil de Golgi proéminent (Falini, 2001 ; Fiorani,
2001).
Figure 1: Caractéristiques cytologiques du variant de type commun. Coupe histologique d’un cas d’ALCL de type commune. Les cellules tumorales caractéristiques sont indiquées par une flèche. Coloration hémalun/éosine. D’après Benharroch, 1998
Le variant lymphohistiocytaire (environ 10% des cas) est composé de cellules tumorales de
morphologie commune, mêlées à un nombre important d’histiocytes qui peuvent masquer la
population de cellules malignes. Le variant à petites cellules compte pour 8% des cas et se
distingue par une prédominance de petites cellules tumorales et la présence de grandes
cellules de la forme commune, qui restent minoritaires. Ce variant est celui de plus mauvais
pronostic et se caractérise par une évolution clinique particulièrement agressive. En effet, ce
variant est associé à un envahissement de la moelle osseuse par les cellules tumorales et, dans
25% des cas, à une phase leucémique (Villamor, 1999 ; Bayle, 1999 ; Morris, 2001 ; Lamant,
2004). D’autres aspects morphologiques peuvent se rencontrer, mais ils restent marginaux. Il
existe ainsi un variant à cellules géantes, un variant Hodgkin like et un variant sarcomatoïde,
qui comptent chacun pour environ 3% des cas (Falini, 1999).
3

Introduction
4. Les traitements
4.1. Les traitements actuels Il n’existe pas de traitement optimal pour les ALCL. Ainsi, les méthodes de traitements
courantes de ces lymphomes sont adaptées à partir des protocoles de thérapie développés pour
d’autres lymphomes non hodgkiniens B et T (Pulford, 2004b). Les traitements actuels sont
basés sur plusieurs cycles de polychimiothérapies associant différents agents : le CHOP
(cyclophosphamide/doxorubicine/vincristine/prednisone) ou F-MACHOP (5-
fluorouracil/méthotrexate/cytarabine/CHOP) et MACOP-B
(méthotrexate/doxorubicine/cyclophosphamide/vincristine/prednisone/bléomycine) (Bayle,
1999 ; Brugière, 1998). Dans certains cas, le traitement par polychimiothérapies peut être
associé à de la radiothérapie ou à une transplantation de cellules souches autologues (Fiorani,
2001 ; Pulford, 2004b). Bien que ces traitements soient associés à un bon taux de survie, ils
ont des effets limités puisque 30% des patients rechutent rapidement ou y sont réfractaires
(Falini, 2001). De plus, ces traitements présentent une forte toxicité et d’importants effets
secondaires, qui sont d’autant plus délétères dans les cas pédiatriques d’ALCL (Coluccia,
2005). Il est donc indispensable de développer de nouvelles stratégies thérapeutiques plus
efficaces et plus ciblées palliant ces échecs thérapeutiques et garantissant une meilleure
tolérance (Pulford, 2004a).
4.2. Les nouvelles stratégies thérapeutiques en développement
4.2.1. Les inhibiteurs de l’activité tyrosine kinase
Les inhibiteurs de tyrosine kinases ont déjà démontré leur importance dans le traitement des
tumeurs dont l’origine est la dérégulation d’une protéine à activité tyrosine kinase. Par
exemple, le STI-571 ou Gleevec , un inhibiteur compétitif de la poche ATP de la chimère
oncogénique Bcr-Abl, a amélioré considérablement le traitement des leucémies myéloïdes
chroniques (Arora, 2005 ; Tauchi, 2006). L’efficacité du Gleevec a été testée sur des
lignées cellulaires d’ALCL, mais aucune inhibition de l’activité tyrosine kinase de ALK n’a
été observée (Ergin, 2001). D’autres inhibiteurs de tyrosine kinases ont été testés afin
d’évaluer leur efficacité sur l’activité de ALK. C’est le cas de l’herbimycine A, un inhibiteur
des tyrosine kinases non récepteurs qui a été testé sur des lignées cellulaires humaines NPM-
ALK positives. Le traitement par l’herbimycine A, à de faibles doses, entraîne une inhibition
TM
TM
4

Introduction
de l’activité de NPM-ALK et de certaines de ses cibles. De plus, les auteurs montrent que cet
agent est capable de bloquer le cycle cellulaire et permet d’induire une apoptose dans les
cellules exprimant NPM-ALK (Ergin 2001 ; Turturro, 2002a). Malgré une efficacité
démontrée, la faible spécificité de l’herbimycine A qui est un inhibiteur à large spectre rend
son utilisation clinique impossible. D’autres études ont mis en évidence l’inhibition spécifique
de l’activité tyrosine kinase de ALK par d’autres composés. C’est le cas de dérivés des
pyrrolocarbazoles (Wan, 2006) ou des pyridones (Li, 2006). In vitro ces molécules permettent
l’inhibition des propriétés transformantes de NPM-ALK. Lors d’un traitement par un de ces
composés, les cellules présentent une diminution de leur prolifération et entrent en apoptose.
Cependant, ces molécules n’ont pas de bonnes propriétés physico-chimiques, ce qui exclut
également leur utilisation in vivo. De plus, une étude récente de la structure de la poche ATP
de ALK pourrait permettre le développement d’inhibiteurs spécifiques du domaine à activité
tyrosine kinase de ALK comme cela a été fait pour Bcr-Abl avec le Gleevec (Gunby,
2006). Ainsi, les auteurs montre qu’un inhibiteur spécifique de l’activité tyrosine kinase de
ALK, le NVP-TAE684, bloque la prolifération des cellules NPM-ALK positives, prévient le
développement de lymphomes et induit la régression tumorale in vivo (Galkin, 2007). Enfin,
une autre étude montre qu’un inhibiteur de la kinase c-Met, le PF-2341066, est aussi un bon
inhibiteur de la kinase NPM-ALK (Christensen, 2007). Les auteurs montrent que des cellules
NPM-ALK positives traitées par cet agent présentent un arrêt du cycle cellulaire et rentrent en
apoptose. De plus, les auteurs montrent que les voies de signalisation en aval de NPM-ALK
sont inhibées. Enfin, du fait d’une forme administrable oralement, cet inhibiteur pourrait être
un bon candidat pour des traitements cliniques.
TM
4.2.2 L’invalidation de l’expression de ALK
En parallèle à la recherche d’inhibiteurs spécifiques d’ALK, différentes stratégies alternatives
ont été développées pour réguler négativement son activité. L’expression tissulaire restreinte
de la protéine ALK et le fait que les souris invalidées pour le gène ALK soient viables et sans
défaut majeur, suggèrent que les stratégies d’invalidation de ALK par interférence ARN
pourraient être bien tolérées et sans effets secondaires. Ces approches permettraient une plus
grande spécificité en comparaison aux inhibiteurs chimiques, mais la difficulté d’application
en thérapeutique sera de trouver des modes d’administration permettant une persistance de
leurs effets. Ainsi, une étude a montré que l’utilisation de ribozymes ciblant la partie ALK
entraîne la dégradation des transcrits NPM-ALK (Hübinger, 2003). Cependant, probablement
5

Introduction
du fait de la forte expression et de la demi-vie prolongée (48 heures) de la protéine NPM-
ALK, la transfection répétée de ce ribozyme dans la lignée cellulaire d’ALCL Karpas 299, n’a
pas abouti à la réduction significative de son taux d’expression. Enfin, une autre étude montre
que l’utilisation d’shARN (small hairpin) dirigé contre ALK, permet d’invalider
spécifiquement l’expression de NPM-ALK dans des lignées cellulaires NPM-ALK positives
et a pour effet de reverser le phénotype transformé in vitro et in vivo (Piva, 2006a).
4.2.3. Les stratégies de thérapie génique
Des stratégies de thérapie génique ont été suggérées comme approches thérapeutiques
possibles pour le traitement des ALCL ALK positifs. La transduction de p53, grâce à un
vecteur adénoviral, entraîne l’apoptose des cellules de la lignée d’ALCL SUDHL-1 (Turturro,
2000a). In vivo, cette stratégie a permis l’inhibition de la croissance des tumeurs développées
par les souris nude suite à la greffe de cellules SUDHL-1 (Turturro, 2000a). Des approches
similaires, utilisant le même vecteur adénoviral, ont été développées pour transduire les gènes
p27Kip1, p21Waf1 et p16INK4A codant pour des inhibiteurs des CDK (Cyclin Dependent Kinases)
(Turturro, 2002b) et bloquant le cycle cellulaire. Cependant ces thérapies ont leurs limites. En
effet, la transduction ne peut être efficace que si les cellules expriment à leur surface les
récepteurs permettant l’internalisation de l’adénovirus. Au vu des hétérogénéités des ALCL
que nous avons décrites, il est probable que les cellules tumorales présentent aussi des
variations au niveau de l’expression des récepteurs aux adénovirus, compromettant ainsi la
généralisation de telles stratégies.
4.2.4. La vaccination anti-tumorale
Il existe plusieurs raisons pour lesquelles ALK serait une cible idéale pour une telle stratégie.
En effet, les cellules transformées par NPM-ALK sont totalement dépendantes de l’activité
tyrosine kinase de ALK pour promouvoir leur survie et leur prolifération (Piva, 2006). De
plus l’expression de ALK est restreinte à quelques cellules neuronales ce qui minimise les
risques d’une réaction auto-immune après vaccination. Enfin, la protéine ALK est
naturellement immunogène chez les patients présentant un ALCL et entraîne une réponse
cytotoxique médiée par les cellules T suggérant qu’une vaccination dirigée contre ALK
pourrait augmenter cette réponse immunitaire (Ait-Tahar, 2006 ; Passoni, 2006 ; Pulford,
2000). Ainsi, dans des modèles de xénogreffes d’ALCL, l’électroporation, stratégie déjà
6

Introduction
utilisée sur des sujets humains, d’un plasmide codant pour la protéine ALK va abroger
totalement la pousse de la tumeur et augmenter le pourcentage de souris soignées quand ce
traitement est combiné à de la chimiothérapie (Chiarle, 2008). Cette thérapie présenterait
plusieurs avantages. En effet, les lymphomes ALK positifs étant prépondérant chez l’enfant
cette thérapie pourrait être une bonne alternative pour une protection à long terme des
malades et permettre une diminution des rechutes. De plus, comme montré chez les modèles
murins (Chiarle, 2008), il est envisageable que la vaccination en association avec les
inhibiteurs de ALK et/ou la chimiothérapie puisse permettre une survie plus importante des
malades.
II. Caractéristiques cytogénétiques
1. La t(2;5)(p23;q35)
1.1. La nucléophosmine Le gène humain de la nucléophosmine est localisé en position q35 du chromosome 5. Il
possède 11 exons et couvre 25 kbases. Il code pour une phosphoprotéine nucléolaire
ubiquitaire fortement exprimée et conservée au cours de l’évolution (Ladanyi, 1997). NPM,
aussi dénommée B23, NO38 ou numatrine, appartient à la famille des protéines chaperonnes
nucléoplasmiques. Il existe trois isoformes de cette protéine résultant d’un épissage alternatif :
B23.1, isoforme majoritaire de 294 acides aminés et d’un poids moléculaire de 38 kDa ;
B23.2, isoforme tronquée sans les 35 derniers acides aminés C-terminaux, faiblement
exprimée et B23.3, peu étudiée à ce jour. NPM possède un domaine d’oligomérisation en N-
terminal par lequel elle se complexe en homohexamères. Elle comporte aussi deux séquences
de localisation nucléaires (NLS) en C-terminal, ainsi qu’un signal d’export nucléaire (NES)
riche en leucine (Figure 2). Grâce à ces deux domaines, NPM effectue la navette entre le
noyau et le cytoplasme, et est impliquée dans le transport nucléaire de protéines, comme les
protéines ribosomales.
7

Introduction
Figure 2: Représentation schématique de la protéine NPM. Sont figurés ses différents domaines et leur fonction. Le domaine d’oligomérisation en N-terminal couvre les 110 premiers acides aminés, le domaine NLS bipartite est situé en C-terminal. Le domaine de liaison aux acides nucléiques est situé en C-terminal de la protéine. D’après Qi, 2008
Différentes études ont montré que NPM était impliquée dans des processus de cancérogenèse.
En effet, cette protéine a été montrée comme surexprimée dans des cancers solides des
ovaires, du côlon et de la prostate (Grisendi, 2006). Enfin des translocations chromosomiques
autres que celle impliquant le gène ALK ont pu être retrouvées dans des tumeurs
hématopoïétiques (Falini, 2007). C’est le cas par exemple de la translocation t(5;17)(q32;q12)
dans les leucémies aiguës promyélocytaires, NPM est alors réarrangée avec le facteur de
transcription RARα (Retinoic Acid Receptor α) (Yoneda-Kato, 1996). Certains syndromes
myélodysplasiques et leucémies aiguës myéloïdes présentent la translocation t(3;5)(q25;q34)
qui juxtapose le gène MLF1 (Myelodysplasia-Myeloid Leukemia Factor 1) avec NPM
(Redner, 1996). Enfin, le rôle de NPM dans les processus de transformations cellulaires est
renforcé par le fait que cette protéine contrôle de nombreuses fonctions impliquées dans
l’homéostasie cellulaire telles que le transport de protéines impliquées dans la biogenèse des
ribosomes ou bien encore le contrôle de la division cellulaire. Ainsi, la dérégulation de
l’expression de NPM va induire de profondes perturbations dans l’homéostasie cellulaire.
(Figure 3).
8

Introduction
Figure 3: Représentation schématique des différentes fonctions de la protéine NPM. La fonction de navette de NPM, qui transporte les particules pré-ribosomales du cytoplasme au noyau, est essentielle à la biogenèse des ribosomes. Dans le cytoplasme, NPM se lie aux centrosomes non dupliqués et régule leur duplication lors de la division cellulaire. De plus, NPM est capable d’interagir avec le suppresseur de tumeur p53 et ses régulateurs (Hdm2/Mdm2) et influence les fonctions suppresseurs de tumeurs de la voie Hdm2/Mdm2 et p53. D’après Falini, 2007
Ainsi, NPM joue un rôle important dans la progression du cycle cellulaire. Sa forte expression
lors de la transition G1/S est corrélée à son implication dans le contrôle de la duplication des
centrosomes lors de la mitose. En effet, NPM s’associe avec le centrosome non dupliqué et
s’en dissocie au cours de la progression en phase G1, suite à sa phosphorylation par le
complexe CDK2/cycline E (Okuda, 2000 ; Tarapore, 2002). Ce contrôle de la duplication des
centrosomes suggère aussi que NPM jouerait un rôle dans le maintien de la stabilité
génétique. En effet, des travaux ont montré que la mutation du gène NPM chez la souris est
léthale chez cet organisme et que ces mutants présentent une forte instabilité génétique
(Grisendi, 2005). NPM est aussi impliquée dans la biogenèse des ribosomes, d’une part en
assurant le transport des protéines ribosomales du cytoplasme au noyau, et d’autre part par sa
fonction de protéine chaperonne, en empêchant l’aggrégation de protéines lors de
l’assemblage des ribosomes (Szebeni, 1999 ; Hingorani, 2000). D’autres travaux ont montré
que la fonction de protéines chaperonne de NPM s’exerçait aussi sur les histones, impliquant
de ce fait NPM dans la régulation de la transcription (Swaminathan, 2005). Enfin, NPM a été
décrite comme étant un régulateur de l’activité du suppresseur de tumeurs p53. En effet, en
réponse aux dommages à l’ADN, NPM lie Hdm2, un régulateur négatif de p53, empêchant la
formation du complexe Hdm2/p53 et conduisant ainsi à une augmentation de la stabilité et de
l’activité transcriptionnelle de p53 (Colombo, 2002 ; Kurki, 2004). De façon intéressante, des
fonctions inhibitrices de l’activité de p53 ont aussi été attribuées à NPM. Ainsi, NPM est
9

Introduction
capable de se lier à p53, empêchant ainsi sa phosphorylation activatrice par les kinases
ATR/ATM et inhibant l’apoptose (Maiguel, 2004). En conclusion, la surexpression de NPM
observée dans certains types de cancers pourrait conférer aux cellules tumorales une
résistance à l’apoptose (Li, 2004).
1.2. Le gène ALK Le gène ALK est localisé sur le chromosome 2 en position p23. Ce gène code pour un
récepteur transmembranaire à activité tyrosine kinase appartenant à la superfamille du
récepteur à l’insuline. Il comporte 26 exons et l’ADNc humain comporte 6226 paires de
bases. Ce récepteur monomérique compte 1620 acides aminés et le fort taux de glycosylation
au niveau de son domaine extracellulaire N-terminal lui confère un poids moléculaire de 200
kDa (Morris, 1997). Le récepteur ALK présente une forte homologie de séquence avec la
protéine LTK (Leucocyte Tyrosine Kinase), un récepteur de la même famille. Le domaine
extracellulaire de ALK comporte un motif EGF-like, ce qui suggère que ALK lui-même
pourrait agir en tant que son propre ligand. ALK possède aussi un domaine MAM qui joue un
rôle dans les interactions intercellulaires (Iwahara, 1997). Enfin, le domaine extracellulaire de
ALK contient un domaine LDL-A riche en cystéine, participant à la liaison aux lipoprotéines
et retrouvé, entre autres, dans le récepteur aux lipoprotéines de faible densité (Fass, 1997)
(Figure 4).
Figure 4: Représentation schématique du récepteur à activité tyrosine kinase ALK. Sont représentés les différents domaines de ALK. Les sites de liaison au ligand sont en position 391-401. Les sites 1282 et 1283 sont des sites d’autophosphorylation. Le site catalytique à activité tyrosine kinase est porté par le domaine intracellulaire. D’après Pulford 2004b
10

Introduction
Cette protéine est conservée au cours de l’évolution. En effet, la protéine humaine possède
98% d’homologie, au niveau du domaine tyrosine kinase, avec la protéine murine (Morris,
1997) et 52% d’homologie, au niveau de la région intracytoplasmique, avec l’orthologue de
Drosophila melanogaster (Loren, 2001). Même si les fonctions normales de ALK ne sont pas
encore totalement connues, de nombreuses données suggèrent que ce récepteur jouerait un
rôle important dans le développement neuronal. En effet, chez la souris, un fort taux d’ARNm
de ALK est détecté dans le cerveau en développement et une diminution est observée après la
naissance (Morris, 1997). Chez l’homme, l’expression de la protéine ALK est restreinte à des
régions spécifiques du système nerveux central (comme le bulbe olfactif et le thalamus) et
périphérique en développement. Son expression est toutefois maintenue à un faible taux dans
le cerveau adulte, ce qui suggère un rôle de ALK, non seulement dans le développement du
système nerveux et la différenciation neuronale, mais aussi dans la formation et le maintien
des synapses chez l’adulte (Souttou, 2001 ; Pulford, 2001). Cependant, les modèles animaux
invalidés pour le gène ALK sont viables et ne présentent pas de défaut du système neuronal
(Duyster, 2001 ; Morris, 2001). Chez l’homme de faibles taux d’ARNm de ALK ont été
détectés dans les testicules, l’intestin grêle, le côlon et le foie fœtal (Morris, 1994 ; Iwahara,
1997). Des études récentes montrent que chez D. melanogaster, ALK pourrait avoir un rôle
dans la différenciation du mésoderme ventral (Loren, 2001 ; Loren, 2003). Ainsi, différentes
études ont pu mettre en évidence l’existence de protéines impliquées dans des processus
d’embryogenèse et qui sont capables de lier le récepteur ALK. Ainsi, chez D. melanogaster,
un ligand de l’orthologue de ALK, Jelly belly (Jeb) a été identifié (Lee, 2003 ; Englund,
2003). Chez l’homme, deux ligands de ALK ont été caractérisés. Il s’agit de la pléiotrophine
(PTN) et de son homologue midkine (MK) (Stoica, 2001 ; Stoica, 2002). Ces protéines ont des
rôles connus dans l’embryogenèse. Chez l’homme, PTN et MK sont des facteurs de
croissance induisant la prolifération de cellules d’origine variée. Les auteurs ont émis
l’hypothèse que PTN, MK et ALK pouvaient fonctionner comme des couples ligand-
récepteur puisque leurs profils d’expression sont identiques. En effet, les gènes PTN et MK
sont fortement exprimés lors du développement embryonnaire du système nerveux central et
leur expression diminue chez l’adulte. Cette hypothèse a été renforcée par le fait que la
stimulation de cellules exprimant ALK par ces deux facteurs de croissance induit l’activation
de ALK et de ses substrats (Bowden, 2002). Cependant, le rôle de PTN et MK en tant que
ligands de ALK reste controversé (Motegi, 2004 ; Moog-Lutz, 2005). Ainsi, des études
récentes montrent que ALK appartient à la famille des récepteurs à dépendance. Cette famille
comprend des récepteurs impliqués dans le développement et la tumorigenèse qui sont
11

Introduction
caractérisés par une dualité d’action sur les phénomènes apoptotiques. Ainsi, sous sa forme
inactive ALK induit l’apoptose, alors que sous forme active il permet la délivrance d’un
signal anti-apoptotique (Mourali, 2006) suggérant que ce récepteur est capable de contrôler
ces phénomènes sans l’intervention de ligand extérieur.
1.3. NPM-ALK 75% des cas d’ALCL présentent la translocation t(2;5)(p23;q35) qui juxtapose le gène NPM
et le gène ALK (Falini, 2002 ; Pulford, 2004b). Cette translocation est retrouvée plus élevée
dans les cas d’ALCL pédiatriques (Stein, 2000 ; Drexler, 2000). Ce réarrangement
chromosomique va juxtaposer les 4 premiers exons de NPM, codant pour les 116 acides
aminés N-terminaux de la protéine et comportant son domaine d’oligomérisation, avec les
exons codant pour la partie intracytoplasmique de ALK, correspondant aux 563 acides aminés
C-terminaux et contenant son domaine catalytique à activité tyrosine kinase (Morris, 1994).
Ainsi, l’ADNc hybride, code pour une protéine de 680 acides aminés, d’un poids moléculaire
de 80 kDa. Lors de la fusion, les régions extracellulaire et transmembranaire de ALK sont
perdues, ainsi que les domaines NLS de NPM. Bien que les motifs NLS de NPM ne soient
pas maintenus dans la chimère NPM-ALK, la protéine NPM-ALK présente une localisation
cytoplasmique, nucléaire et nucléolaire (Bischof, 1997). Ceci s’explique par la formation de
complexes multimériques NPM natif/NPM-ALK assurée grâce au domaine d’oligomérisation
de NPM (Cordell, 1999). De plus, le signal d’export nucléaire de NPM, conservé dans la
protéine hybride NPM-ALK, contribue au va-et-vient constant de NPM-ALK entre le noyau
et le cytoplasme. L’ARN messager de NPM-ALK (Trumper, 1998) et de la fusion ATIC-
ALK (Maes, 2001) (Cf. Chap. I.II.), ont aussi été retrouvés chez des sujets sains. Ceci suggère
que des évènements secondaires à la translocation chromosomique, encore totalement
inconnus, sont nécessaires pour le développement de la pathologie.
2. Autres translocations impliquant le gène ALK
Plusieurs arguments ont suggéré que des variants de la translocation classique NPM-ALK
pouvaient exister. En effet, des travaux ont montré que lors d’expériences de
immunoempreinte dans les cellules tumorales avec un anticorps dirigé contre ALK, cet
anticorps révélait des protéines de poids moléculaire différent de 80 kDa. De la même façon
lors de marquages immunohistochimiques avec ces mêmes anticorps, les auteurs ont pu
observer un marquage exclusivement cytoplasmique de ALK dans 15% des cas, ce qui était
12

Introduction
en contradiction avec les données déjà publiées. Ces résultats suggéraient qu’il existait des
variants de la translocation classique t(2;5)(p23;q35), dans lesquels un autre gène que NPM
était associé à la kinase ALK (Falini, 1998 ; Pulford, 1999 ; Drexler, 2000). Mis à part pour
les translocations impliquant les gènes MSN et MYH-9, le point de cassure dans le gène ALK
est toujours le même, dans l’intron 16, séparant les exons codant pour les domaines extra- et
juxta-membranaires (Duyster, 2001). Ceci suggère que cet endroit du gène ALK est très
sensible au remaniement chromosomique. A ce jour, plus de 10 variants de translocation
impliquant le gène ALK ont été identifiés (Figure 5). Cependant, il existe encore des
translocations pour lesquelles le partenaire de ALK n’a pas été caractérisé (Pulford, 2004a ;
Pulford, 2004b). Les différents partenaires de fusion de ALK partagent des caractéristiques
communes. Ainsi, ils possèdent un promoteur ubiquitaire fort ce qui leur confère une
expression constitutive et permet l’expression de ALK. De plus, à l’exception de MSN et
MYH-9, ces partenaires possèdent un domaine d’oligomérisation en N-terminal, ce qui
permet l’activation constitutive du domaine tyrosine kinase de ALK par transphosphorylation,
mimant ainsi l’activation du récepteur après sa dimérisation consécutive à l’interaction avec
son ligand.
2.1 TMP3-ALK
La t(1;2)(q25;p23) va impliquer le gène TPM3 de la tropomyosine non musculaire (Lamant,
1999). Après NPM-ALK, ce réarrangement chromosomique est le plus fréquent des variants
de translocation impliquant le gène ALK. La protéine de fusion générée a un poids
moléculaire de 104 kDa et se localise dans le cytoplasme. La protéine TPM3 native,
composant majeur des microfilaments du cytosquelette, possède un domaine coiled-coil
d’oligomérisation en N-terminal. Sa fonction de liaison à l’actine permet la stabilisation des
filaments d’actine.
2.2. TGF-ALK La protéine de fusion TFG (Tyrosine kinase receptor Fused Gene)-ALK est le résultat de la
translocation t(2;3)(p23;q21) (Hernandez, 1999). Suivant le point de cassure dans le gène
TFG, trois protéines chimériques peuvent être générées : TFGS-ALK de 85 kDa, TFGL-ALK
de 97 kDa et TFGXL-ALK de 113 kDa (Hernandez, 2002). La protéine de fusion TFG-ALK
peut former des dimères grâce au domaine coiled-coil de TFG et présente une localisation
13

Introduction
cytoplasmique avec un renforcement au niveau de la région périnucléaire. La fonction de TFG
est encore inconnue.
Figure 5: Représentation schématique du récepteur ALK pleine taille et de différentes protéines de fusion X-ALK. Les nombres font référence au nombre d’acides aminés du partenaire qui sont inclus dans la fusion avec ALK et au nombre total d’acides aminés de la protéine de fusion X-ALK. D’après Pulford, 2004b
2.3. ATIC-ALK Le gène ATIC (5’-aminoimidazole-4-carboxamide ribonucléotide formyltransférase/IMP
cyclohydrolase) est impliqué dans l’inversion inv(2)(p23;q35) (Trinei, 2000). La protéine de
fusion ATIC-ALK a un poids moléculaire de 94 kDa et sa localisation est exclusivement
cytoplasmique. La protéine ATIC joue un rôle dans les deux dernières étapes de la
14

Introduction
biosynthèse des purines et fonctionne sous forme de dimères grâce à son domaine
d’oligomérisation.
2.4. CLTC1-ALK La protéine de fusion CLTC1-ALK (Clathrin heavy chain-like 1) est générée par la
translocation t(2;17)(p23;q23). En immunofluorescence, cette protéine chimérique présente
un marquage cytoplasmique granuleux, cette localisation s’explique du fait de la liaison de
CLTC-ALK avec la clathrine sauvage, qui se situe au niveau des vésicules d’endocytose
(Touriol, 2000). La chimère CLTC-ALK a un poids moléculaire de 250 kDa.
2.5 MSN-ALK
La translocation t(2;X)(p23;q11-12) produit une protéine de fusion MSN-ALK (Moesin) de
125 kDa dont la localisation est restreinte à la membrane plasmique (Tort, 2001). En effet, la
moésine, partenaire de ALK impliqué dans cette fusion, possède un domaine globulaire
permettant son association à la membrane plasmique. MSN appartient à la famille des ERM
(Ezrine/Radixine/Moésine) qui jouent un rôle important dans la motilité et l’adhésion
cellulaire en assurant le lien entre la membrane plasmique et le cytosquelette d’actine.
Cependant, contrairement à l’ezrine, MSN ne possède pas de domaine d’oligomérisation.
Mais sa capacité à se lier à des protéines membranaires, comme CD44, pourrait permettre la
dimérisation de MSN-ALK et conduire ainsi à l’activation du domaine kinase de ALK.
2.6 TPM4-ALK TPM4, la tropomyosine non musculaire, proche de TPM3, est fusionnée avec ALK suite au
réarrangement chromosomique t(2;19)(p23;p13.1) (Lawrence, 2000). Le poids moléculaire de
TPM4-ALK est de 86 kDa.
2.7. MYH-9-ALK La translocation t(2;22)(p23;q11.2) associe ALK avec MYH-9 (non muscle Myosin Heavy
chain), pour former une protéine de 220 kDa. Tout comme MSN, MYH-9 ne possède pas de
domaine d’oligomérisation. C’est par le biais de son interaction avec des protéines
membranaires que MYH-9 pourrait conduire à l’oligomérisation des complexes MYH-9-ALK
et donc à l’activation du domaine kinase de ALK (Lamant, 2003).
15

Introduction
2.8. RanBP-2-ALK RanBP-2 (Ran Binding Protein 2), est une protéine du pore nucléaire liant les GTPases Ran.
Elle est retrouvée fusionnée à ALK suite à l’inversion inv(2)(p23;q11-13) (Ma, 2003). La
chimère RanBP-2-ALK s’oligomérise grâce au domaine N-terminal à motif leucine zipper de
RanBP-2 et se localise au niveau de la membrane nucléaire. La protéine de fusion a un poids
moléculaire de 160 kDa.
2.9. ALO17-ALK
La translocation t(2;17)(p23;q25) juxtapose les gènes ALK et ALO17 (ALK Lymphoma
Oligomerisation partner on chromosome 17), dont la fonction est encore inconnue (Cools,
2002). La protéine de fusion a un poids moléculaire de 190 kDa.
2.10. CARS-ALK CARS (Cysteinyl-tRNA Synthetase enzyme) est fusionnée à ALK, suite à la translocation
t(2;11;2)(p23;p15;q31) qui génère une protéine de fusion de 130 kDa (Cools, 2002) dont la
fonction est encore inconnue.
2.11. EML4-ALK EML4 (echinoderm microtubule-associated protein-like 4) est fusionnée au domaine kinase
de ALK suite à l’inversion sur le chromosome 2, inv(2)(p21;p23). Cette translocation a été
retrouvée dans 75 patients atteints d’une forme de cancer du poumon à cellules non petites
(Soda, 2007). La dimérisation des complexes EML4-ALK semble se faire par un domaine
basique présent dans la partie N-terminale de EML4.
2.12 Sec31L1-ALK Sec31L1 est une protéine appartenant au complexe COPII qui permet la formation de
vésicules de transport à partir du réticulum endoplasmique. La translocation t(2;4)(p23;q21)
impliquant Sec31L1 et ALK a été retrouvée dans des tumeurs myofibroblastiques
inflammatoires (Panagopoulos, 2006) (Cf. Chap.I.III).
Aux vues des différents travaux effectués sur les variants de ALK, il semble que le partenaire
de fusion de ALK ait un rôle plus large que celui de permettre l’activation constitutive du
16

Introduction
domaine tyrosine kinase de ALK. En effet, il semble conditionner la localisation subcellulaire
des protéines chimériques X-ALK, dont l’impact dans la lymphomagenèse reste encore
inconnu. Ainsi, la localisation subcellulaire des différentes protéines X-ALK pourrait moduler
leurs capacités oncogéniques, mettant à jour des propriétés biologiques différentes selon le
variants de translocation, ce qui pourrait avoir des implications cliniques importantes.
III. Autres pathologies impliquant ALK Le récepteur à activité tyrosine kinase ALK est exprimé dans d’autres tumeurs que les
néoplasmes lymphoïdes. En effet, de récentes études rapportent l’expression du gène ALK
dans différentes lignées cellulaires dérivées de carcinomes (mélanome et cancer du sein), ce
qui suggère que ALK pourrait jouer un rôle dans le développement et/ou la progression de
certaines tumeurs solides (Falini, 1998 ; Pulford, 2004b). Ainsi, ALK est un des rares
exemples d’un récepteur à activité tyrosine kinase dont la dérégulation est à la fois impliquée
dans le développement des tumeurs hématopoïétiques et non hématopoïétiques (Tableau 2).
Pathologie Expression de ALK pleine taille ou tronquée associée
ALCL NPM-ALK, TPM3-ALK, CLTC-ALK, TFG-ALK, ATIC-ALK,
ALO17-ALK, MYH9-ALK
IMT TPM3-ALK, TPM4-ALK, CLTC-ALK, CARS-ALK, Ran-BP2-
ALK, SEC31L1-ALK, ATIC-ALK
Neuroblastome/
Glioblastome
ALK pleine taille
Lymphome B ALK pleine taille, CLTC-ALK, NPM-ALK
Tableau 2: Les différentes pathologies associées à l’expression de ALK pleine taille ou sous forme transloquée.
Ainsi, des réarrangements du gène ALK ont pu être retrouvés dans plus de 50% des tumeurs
myofibroblastiques inflammatoires (IMT) (Lawrence, 2000 ; Coffin, 2001). Ces proliférations
néoplasiques mésenchymateuses des tissus mous et des viscères sont relativement rares
(Griffin, 1999). Comme pour les ALCL, les IMT se développent le plus souvent chez les
enfants et les adultes jeunes (Morris, 2001). Ces tumeurs sont caractérisées par la présence de
cellules myofibroblastiques malignes infiltrées par des cellules lymphoïdes saines. Les
17

Introduction
partenaires de fusion de ALK retrouvés dans cette pathologie sont : TPM3 (le plus
fréquemment), TPM4, ATIC, CLTC (Bridge, 2001), RanBP-2 et CARS ou bien encore
SEC31L1-ALK (Panagopoulos, 2006). Récemment, la translocation EML4-ALK a pu être
mise en évidence dans des cancers du poumon à cellules non petites (Soda, 2007). De plus,
l’expression de la protéine ALK pleine taille a aussi été détectée dans des neuroblastomes
primaires (Lamant, 2000) et dans différentes lignées cellulaires établies à partir de tumeurs
solides du neurectoderme, comme les glioblastomes (Dirks, 2002 ; Powers, 2002). Cependant,
la protéine ALK n’a pas été trouvée sous sa forme phosphorylée active, suggérant que son
expression dans ces tumeurs pourrait simplement refléter son expression physiologique dans
les cellules neuronales immatures et n’aurait donc aucun rôle pathologique. Enfin, il a été
rapporté l’existence de quelques rares cas de lymphomes B à grandes cellules exprimant soit
ALK pleine taille (Delsol, 1997 ; Gascoyne, 1999), soit NPM-ALK (Onciu, 2003 ; Adam,
2003) ou CLTC-ALK (Gascoyne, 2003). Contrairement aux ALCL, ces tumeurs sont
observées de façon prédominante chez l’adulte et ont un pronostic défavorable. A ce jour, la
contribution de ALK dans le développement de ces lymphomes reste à déterminer.
IV. Mécanismes d’oncogenèse associés à ALK
1. Effet transformant
La protéine de fusion NPM-ALK étant le plus souvent retrouvée dans les ALCL, la majorité
des études concernant les propriétés oncogéniques de ALK ont été réalisées en utilisant cette
chimère pour modèle. NPM-ALK est un oncogène transformant in vitro de nombreux types
cellulaires, comme les fibroblastes NIH3T3, MEF, HEK293 ou la lignée hématopoïétique
Ba/F3 (Fujimoto, 1996 ; Bischof, 1997 ; Wellmann, 1997 ; Bai, 1998). L’activation du
domaine tyrosine kinase de ALK est indispensable à son pouvoir oncogénique. En effet, des
études ont montré que des mutations au niveau de la poche ATP, rendent son activité tyrosine
kinase inactive, et abrogent ses capacités transformantes (Bischof, 1997). De plus, la
formation de dimères NPM-ALK/NPM-ALK, via le domaine d’oligomérisation de NPM, est
indispensable à l’activation constitutive du domaine tyrosine kinase de ALK. Ainsi, un mutant
de NPM-ALK dont la partie NPM est mutée au niveau de son domaine d’oligomérisation est
incapable d’induire la transformation cellulaire in vitro et in vivo (Fujimoto, 1996 ; Bischof,
1997). De plus, il semble que la localisation de NPM-ALK n’est aucun rôle dans la
transformation cellulaire liée à la présence de cet oncogène. Ainsi, le remplacement de la
18

Introduction
partie NPM N-terminale par TPR (Translocated Promoter Region), va générer une chimère
exclusivement cytoplasmique. Les auteurs montrent que cette nouvelle protéine possède des
capacités transformantes semblables à celles de NPM-ALK, démontrant ainsi que la
localisation nucléaire de NPM-ALK n’est pas indispensable à la transformation cellulaire
(Mason, 1998). D’autres études suggèrent, de la même façon, que le partenaire de fusion de
ALK pourrait avoir une implication dans la transformation cellulaire. Ainsi, il semble que
d’une part, la chimère X-ALK puisse interférer avec les fonctions cellulaires normales du
partenaire, mais d’autre part le partenaire de fusion, qui contrôle la localisation subcellulaire
de la protéine chimère ALK, expose cette dernière à différents substrats, aboutissant ainsi à
l’activation différentielle de voies de signalisation. En effet, la localisation subcellulaire de
ALK est déterminante pour le contrôle et la spécificité des voies de signalisation (Trinei,
2000 ; Gouzi, 2005). Ainsi, une étude réalisée sur différentes protéines chimériques X-ALK a
montré que ces dernières ont des effets transformants différents, notamment en terme de
pouvoir prolifératif et invasif (Armstrong, 2004). Ainsi, le variant TPM3-ALK possède les
capacités de migration et d’invasion les plus importantes. Ces différences de potentiel
transformant in vitro et in vivo semblent dues à l’activation différentielle de diverses voies de
signalisation. Ainsi, la surexpression de TPM3-ALK est corrélée à une plus forte activité de la
PI 3-kinase et induit des changements spécifiques au niveau du cytosquelette d’actine via son
interaction avec la protéine TPM3 native (Armstrong, 2007). En conclusion, il semble que le
rôle du partenaire de fusion de ALK dans les mécanismes d’oncogenèse ne soit pas à négliger.
Il est donc légitime de penser que pour la translocation t(2;5)(p23;q35), la perte de fonction
d’un allèle NPM sauvage et l’interaction de la chimère NPM-ALK avec certains partenaires
de NPM, comme p53, pourraient contribuer à la transformation cellulaire.
2. Les modèles animaux
Actuellement, les modèles animaux développés avec l’oncogène NPM-ALK ne représentent
pas totalement le même phénotype que celui observé chez l’homme (Turner, 2005).
Cependant, ces modèles restent des outils utiles pour l’étude du rôle de NPM-ALK dans la
lymphomagenèse in vivo et la compréhension des mécanismes moléculaires à l’origine de la
transformation tumorale. Ils ont aussi permis de confirmer les résultats établis concernant la
signalisation sur les lignées cellulaires. Ce sont aussi des modèles de choix pour développer
ou tester des moyens alternatifs de thérapie. La tumorigénicité de NPM-ALK in vivo a été
démontrée par la génération d’un modèle murin développant des tumeurs lymphoïdes après
19

Introduction
transplantation de cellules de la moelle osseuse préalablement transduites par un vecteur
rétroviral exprimant NPM-ALK (Kuefer, 1997). Toutefois, dans ce modèle, les souris
chimériques développent des lymphomes B. La période de latence nécessaire au
développement du lymphome se situe entre 4 et 6 mois, ce qui tend à suggérer, comme dit
précédemment que la coopération de différents évènements oncogéniques est nécessaire pour
la lymphomagenèse des ALCL. Ces résultats ont été confirmés par une autre approche de
transfert rétroviral où les souris chimériques NPM-ALK développent de la même façon des
lymphomes B (Miething, 2003). Ainsi, les différents groupes ont développé des modèles de
souris transgéniques dans lesquels l’expression de NPM-ALK est ciblée dans les cellules
lymphoïdes. Une étude a montré que le transgène NPM-ALK mis sous le contrôle du
promoteur de CD4, spécifique des cellules T entraîne un développement de tumeurs de
phénotype T dans 90% des cas (Chiarle, 2003). Dans une autre étude, l’expression du
transgène NPM-ALK est sous le contrôle du promoteur du gène Vav, permettant ainsi son
expression dans le compartiment hématopoïétique. Cependant, dans ce modèle comme dans
les premiers décrits, seul le développement de tumeurs de phénotype B a été observé (Turner,
2003). Bien que ces différents modèles animaux s’avèrent essentiels pour une meilleure
compréhension de la physiopathologie humaine, ils présentent des limites. En effet, parmi
tous ces modèles animaux, seul celui dans lequel NPM-ALK est sous le contrôle du
promoteur de CD4 génère une pathologie dont les cellules tumorales sont de phénotype T,
proche de la pathologie humaine. Il semble donc que le phénotype tumoral induit par NPM-
ALK soit influencé par des facteurs strictement dépendants de l’hôte dans lequel cette
protéine est exprimée (Miething, 2006). En effet, chez la souris, NPM-ALK est capable de
transformer le compartiment de lignage B quel que soit le stade de différenciation des
cellules, alors que la transformation du compartiment T n’a lieu qu’à un stade de
différenciation tardif. Au contraire, chez l’homme, l’expression de NPM-ALK induit presque
exclusivement des lymphomes de type T, suggérant que le compartiment T est plus permissif
à la transformation par NPM-ALK que le compartiment B.
3. Transduction du signal induit par ALK
L’autophosphorylation de NPM-ALK sur de nombreuses tyrosines permet le recrutement de
nombreuses protéines à domaines SH2 (Src Homology 2) ou PTB (PhosphoTyrosine
Binding). Cet ensemble de protéines associées à NPM-ALK est nommé « signalosome ». Ce
20

Introduction
« signalosome » stimule diverses voies de signalisation pro-mitogéniques et anti-apoptotiques
impliquées dans l’oncogenèse dépendant de NPM-ALK (Figure 6).
Figure 6: Le « signalosome » de NPM-ALK. La protéine de fusion NPM-ALK contient 21 sites de phosphorylation qui sont autant de sites potentiels de liaison à des protéines de la signalisation. Sont figurés les résidus tyrosine phosphorylées (Y) dont l’association avec un partenaire de la signalisation a été identifiée. D’après Turner, 2005
Comme nous le verrons au cours de ce paragraphe, il existe une synergie entre les diverses
voies de signalisation initiées par NPM-ALK, ce qui permet d’augmenter les effets
individuels de chacune de ces voies et de potentialiser ainsi l’effet transformant de l’oncogène
(Figure 14).
3.1. NPM-ALK induit la prolifération
3.1.1. La voie des MAP kinases
La voie des MAPK (Mitogen Activated Protein Kinases), est activée en réponse à divers
stimuli et est constituée d’une cascade de kinases agissant les unes sur les autres. Elle se
divise en deux familles : les ERK (Extracellular signal Regulated Kinase) et les SAPK (Stress
Activated Protein Kinases) qui compte les p38 et les JNK (c-Jun N-Terminal Kinase)
(Dhanasekaran, 2007). Ainsi, les MAPK kinase kinases, comme Raf ou MLK1,
phosphorylent et activent les MAPK kinases, comme MEK1 ou MEK4, qui à leur tour
21

Introduction
activent les MAPK, comme ERK ou JNK. Ces dernières activent, entre autres, des facteurs de
transcription contrôlant l’expression de nombreux gènes, impliqués en particulier dans la
survie, la progression du cycle cellulaire et la différenciation (Martin, 2003 ; Platanias, 2003).
Plusieurs études ont démontré une activation de la voie pro-mitogénique des MAPK ERK
sous l’oncogène NPM-ALK. Chez D. melanogaster, l’activation de ALK par son ligand Jeb
induit l’activation de la voie MAPK/ERK (Loren, 2003 ; Englund, 2003 ; Lee, 2003). Cette
activation est retrouvée aussi en aval du récepteur humain ALK pleine taille (Souttou, 2001 ;
Bowden, 2002 ; Motegi, 2004). Concernant les translocations impliquant ALK, il a été
montré, aussi bien dans des lignées cellulaires que chez les animaux transgéniques, un
recrutement des protéines adaptatrices régulatrices de la voie Ras/MAPK, Grb2, IRS-1 et Shc
par NPM-ALK (Fujimoto, 1996 ; Bischof, 1997 ; Miyake, 2002 ; Chiarle, 2003 ; Turner,
2003). Une étude récente vient de confirmer l’activation de la voie MEK/ERK dans les
lignées cellulaires d’ALCL et chez des patients atteints d’ALCL ALK positifs (Marzec,
2007). De façon surprenante, cette activation est indépendante de la kinase Raf, censée activer
les MAPK. Elle contribuerait fortement aux effets pro-mitogéniques et anti-apoptotiques de
l’oncogène. Enfin, l’activation de la voie Ras/MAPK médie certains des effets de NPM-ALK
sur la transcription via l’activation de NFAT/AP-1 (Turner, 2007). Très récemment, une étude
a montré que JNK était un substrat de NPM-ALK et contribuait aux effets pro-mitogéniques
de NPM-ALK par son action sur le facteur de transcription AP-1 en régulant l’expression des
CDK (Leventaki, 2007). La figure 7 représente les différents acteurs de la voie des MAPK
impliqués dans la signalisation an aval de NPM-ALK.
Figure 7: Voies de signalisation impliquant les MAPK en aval de NPM-ALK. D’après Chiarle, 2008.
22

Introduction
3.1.2. La phospholipase C-γ (PLC-γ)
La PLC-γ s’associe, via ses domaines SH2, à la majorité des récepteurs à activité tyrosine
kinase ou à des tyrosine kinases non récepteurs. Une fois activée, elle hydrolyse le
phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) membranaire, en inositol 1,4,5-
trisphosphate (InsP3) et 1,2-diacyglycérol (DAG), deux seconds messagers intracellulaires.
L’InsP3 mobilise le calcium intracellulaire et le DAG active la protéine sérine/thréonine
kinase C (PKC) (Patterson, 2005). La PLC-γ est généralement activée en réponse aux facteurs
de croissance et engendre la transmission d’un signal pro-mitogénique. Bai et ses
collaborateurs ont montré que la PLC-γ jouait un rôle important dans la mitogenèse induite
par NPM-ALK (Bai, 1998). Ainsi, dans la lignée lymphoïde murine Ba/F3 transformée par
NPM-ALK et dans la lignée humaine d’ALCL Karpas 299, les auteurs ont observé une
association entre NPM-ALK et la PLC-γ, conduisant à la phosphorylation et à l’activation de
cette dernière. Des expériences de mutagenèse dirigée ont permis d’identifier la tyrosine 664
de NPM-ALK comme étant le site d’interaction entre ces deux protéines.
3.1.3. La tyrosine kinase pp60c-src
La tyrosine kinase c-Src est activée suite à son interaction avec des protéines à activité
tyrosine kinase. Elle mobilise ensuite différentes voies de signalisation impliquées dans la
prolifération (voie Ras/MAP kinases), la survie cellulaire (voie PI 3-kinase/Akt),
l’angiogenèse, la différenciation, l’adhésion et la migration (remodelage du cytosquelette
d’actine via FAK (Focal Adhesion Kinase), par exemple). c-Src active aussi le facteur de
transcription STAT3 (Frame, 2002) (Figure 8). La dérégulation de l’activité tyrosine kinase
de c-Src, observée dans de nombreux cancers, est impliquée dans l’initiation et la progression
tumorales et contribue ainsi au développement de métastases (Irby, 2000). Concernant son
implication dans la signalisation associée à NPM-ALK, notre équipe a identifié le proto-
oncogène pp60c-src comme étant une cible de NPM-ALK (Cussac, 2004). En effet, suite à son
association à la tyrosine 418 de NPM-ALK, pp60c-src est phosphorylée sur sa tyrosine
activatrice 416. Cette activation n’est plus retrouvée dans les cellules exprimant le mutant
inactif de NPM-ALK, ce qui démontre clairement que l’activation de pp60c-src dépend de
l’activité tyrosine kinase de NPM-ALK. L’inhibition de l’activité tyrosine kinase de pp60c-src,
par un inhibiteur spécifique tel que le SU6656 ou par l’abolition de son expression par une
technique d’ARN interférence, diminue fortement la prolifération des cellules NPM-ALK
23
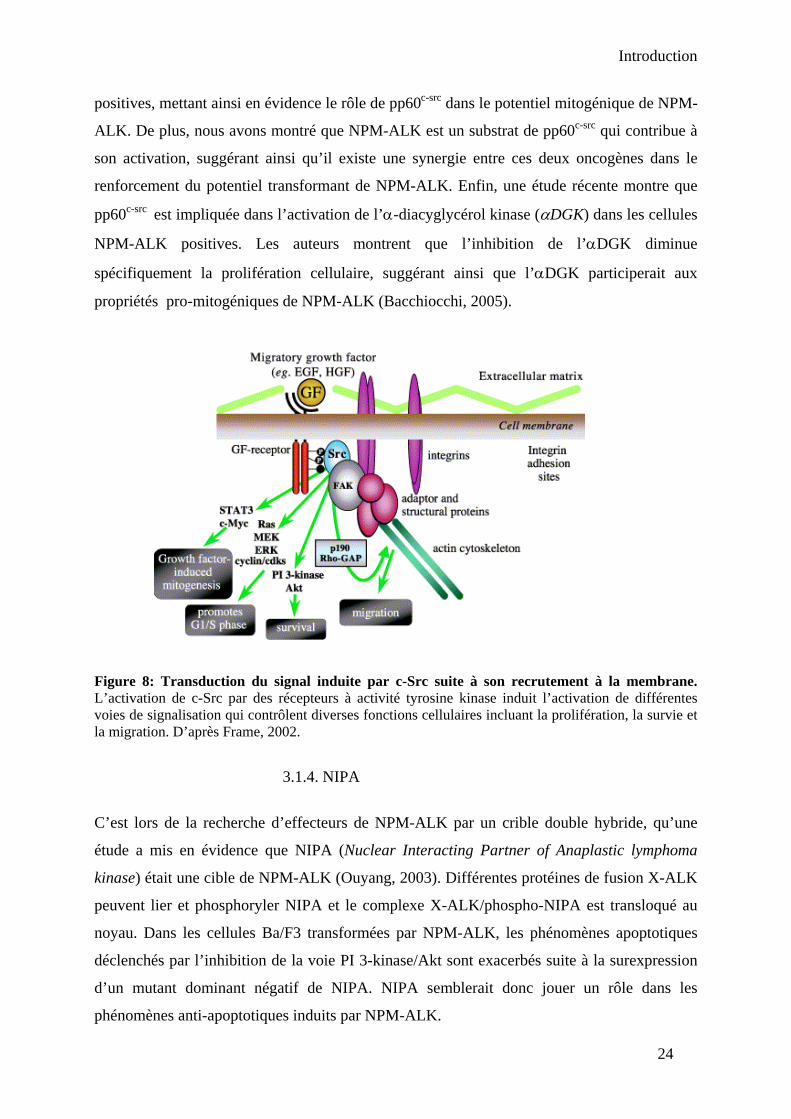
Introduction
positives, mettant ainsi en évidence le rôle de pp60c-src dans le potentiel mitogénique de NPM-
ALK. De plus, nous avons montré que NPM-ALK est un substrat de pp60c-src qui contribue à
son activation, suggérant ainsi qu’il existe une synergie entre ces deux oncogènes dans le
renforcement du potentiel transformant de NPM-ALK. Enfin, une étude récente montre que
pp60c-src est impliquée dans l’activation de l’α-diacyglycérol kinase (αDGK) dans les cellules
NPM-ALK positives. Les auteurs montrent que l’inhibition de l’αDGK diminue
spécifiquement la prolifération cellulaire, suggérant ainsi que l’αDGK participerait aux
propriétés pro-mitogéniques de NPM-ALK (Bacchiocchi, 2005).
Figure 8: Transduction du signal induite par c-Src suite à son recrutement à la membrane. L’activation de c-Src par des récepteurs à activité tyrosine kinase induit l’activation de différentes voies de signalisation qui contrôlent diverses fonctions cellulaires incluant la prolifération, la survie et la migration. D’après Frame, 2002.
3.1.4. NIPA
C’est lors de la recherche d’effecteurs de NPM-ALK par un crible double hybride, qu’une
étude a mis en évidence que NIPA (Nuclear Interacting Partner of Anaplastic lymphoma
kinase) était une cible de NPM-ALK (Ouyang, 2003). Différentes protéines de fusion X-ALK
peuvent lier et phosphoryler NIPA et le complexe X-ALK/phospho-NIPA est transloqué au
noyau. Dans les cellules Ba/F3 transformées par NPM-ALK, les phénomènes apoptotiques
déclenchés par l’inhibition de la voie PI 3-kinase/Akt sont exacerbés suite à la surexpression
d’un mutant dominant négatif de NIPA. NIPA semblerait donc jouer un rôle dans les
phénomènes anti-apoptotiques induits par NPM-ALK.
24

Introduction
3.2. NPM-ALK induit la survie cellulaire
3.2.1. La phosphoinositide 3-kinase (PI 3-kinase)
Les PI 3-kinases sont des lipide kinases qui phosphorylent les phosphoinositides (PIs) en
position 3 du noyau inositol pour générer des D3-phosphoinositides, seconds messagers
intracellulaires impliqués dans l’activation de nombreuses voies de signalisation (Cantley,
2002 ; Engelman, 2006, Payrastre, 2001). La PtdIns3-kinase de classe IA est activée par des
récepteurs à activité tyrosine kinase ou par des protéines à activité tyrosine kinase non
récepteurs. Elle est formée d’une sous-unité régulatrice p85 et d’une sous-unité catalytique
p110. Les domaines SH2 de la sous-unité régulatrice se lient au niveau des tyrosines
phosphorylées, aboutissant ainsi à l’activation de p110 (Bader, 2005). Ainsi activée, la PI 3-
kinase de classe IA génère différents seconds messagers et en particulier le
phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3) (Engelman, 2006). Ce dernier, en
liant le domaine PH (Pleckstrin Homology) de certaines protéines telles que la
sérine/thréonine kinase Akt/PKB peut réguler divers processus biologiques comme la survie
cellulaire, la prolifération, le réarrangement du cytosquelette, la transcription et le
métabolisme (Cantley, 2002). De ce fait, la dérégulation de la voie de la PI 3-kinase contribue
au développement tumoral (Vivanco, 2002). Les différentes classes de PI 3-kinase, leurs
fonctions, localisations et rôles biologiques seront développés au Chapitre II.
Concernant la signalisation en aval de NPM-ALK, plusieurs équipes ont mis en évidence une
association entre NPM-ALK et le domaine SH2 C-terminal de la sous-unité p85, ainsi qu’une
activation de la voie PI 3-kinase/Akt, aussi bien dans des lignées cellulaires transformées par
NPM-ALK ou établies à partir de cas d’ALCL, que dans des biopsies de tumeurs prélevées
chez des patients atteints d’ALCL (Bai, 2000 ; Slupianek, 2001). L’activation de la PI 3-
kinase est aussi retrouvée en aval du récepteur ALK natif (Piccinini, 2002). Cependant, les
tyrosines permettant l’interaction entre ces deux protéines n’ont pas encore été identifiées et il
a été émis l’hypothèse d’une association indirecte entre NPM-ALK et p85, par l’intermédiaire
de protéines adaptatrices comme Gab2 ou Shc. La voie PI 3-kinase/Akt joue un rôle essentiel
dans les effets anti-apoptotiques de NPM-ALK. En effet, Akt inhibe le facteur pro-
apoptotique Bad, empêche ainsi l’activation de la protéine pro-apoptotique pro-caspase-9 et
promeut aussi la transcription de gènes anti-apoptotiques en activant le facteur de
transcription NFκB (Khwaja, 1999 ; Testa, 2001). Mais, cette voie médie aussi des effets pro-
mitogéniques en aval de NPM-ALK en régulant, en particulier, le répresseur du cycle
25

Introduction
cellulaire p27Kip1. En effet, l’activation d’Akt dans les cellules NPM-ALK positives est suivie
de l’inhibition de l’activité transcriptionnelle de FOXO3a, un membre de la famille Forkhead
(FKHD), ce qui a comme conséquence une diminution de l’expression de p27Kip1 (Brunet,
1999 ; Gu, 2004 ; Rassidakis, 2005). De plus, une autre étude a mis en évidence que dans les
cellules transformées par NPM-ALK, la phosphorylation de p27Kip1 par Akt entraînait sa
relocalisation dans le cytoplasme et favorisait ainsi sa dégradation par le protéasome
(Slupianek, 2004). Enfin, Akt, via son activation par la PI 3-kinase, active aussi la voie
mTOR qui permet la synthèse protéique en activant la protéine ribosomale S6 kinase
(p70S6K) et en inhibant 4E-BP1, ce qui permet l’activation du facteur d’initiation de la
traduction des ARNm cap dépendant, eIF4E (Petroulakis, 2006). Il a récemment été mis en
évidence une activation de la voie mTOR et de ses cibles 4E-BP1 et p70S6K en aval de
NPM-ALK, dans des lignées cellulaires d’ALCL et dans des biopsies de tumeurs issues
d’ALCL (Vega, 2006). L’inhibition de cette voie induit l’arrêt du cycle cellulaire et
l’apoptose. Ces effets semblent dépendre de l’activation d’une cible traductionnelle de
mTOR, le facteur de transcription C/EBPβ (Jundt, 2005). De façon intéressante, les auteurs
montrent que dans le cas de l’oncogène NPM-ALK, l’activation de mTOR ne fait pas
intervenir la PI 3-kinase mais la voie Ras/ERK (Marzec, 2007) (Figure 7). ERK va
phosphoryler et donc inactiver le complexe TSC1/TSC2 (Sabatini, 2006). L’ensemble de ces
résultats suggère donc que la voie mTOR contribuerait à l’oncogenèse médiée par NPM-
ALK, ce qui implique que la rapamycine, un inhibiteur de mTOR utilisé en clinique en tant
qu’immunosuppresseur et dans le traitement de certaines pathologies cancéreuses, pourrait
être un complément thérapeutique aux chimiothérapies actuelles pour le traitement des ALCL.
Les différentes données concernant l’activation de la PI 3-kinase par NPM-ALK sont
résumées en figure 9 (Figure 9).
26

Introduction
Figure 9: Voies de signalisation impliquant la PI 3-kinase en aval de NPM-ALK. D’après Chiarle, 2008.
3.2.2. La voie Jak/STAT
La voie des Jak (Janus tyrosine kinases)/ STATs (Signal Transducers and Activators of
Transcription) est activée en aval des récepteurs aux cytokines et aux facteurs de croissance,
dont le domaine intracytoplasmique est associé aux tyrosines kinases non récepteurs Jak. La
dimérisation du récepteur, suite à la fixation de son ligand, provoque l’activation des Jak par
transphosphorylation. Cette transphosphorylation génère ainsi des sites de recrutement pour
d’autres protéines de la signalisation, comme les STATs. Les STATs sont des facteurs de
transcription exprimés ubiquitairement à l’état latent dans le cytoplasme. Leur activation par
les Jak entraine leur dimérisation et leur translocation au noyau (Ward, 2000 ; Imada, 2000).
Les STATs vont contrôler la transcription de nombreux gènes, en particulier ceux codant pour
des protéines anti-apoptotiques ou impliqués dans la progression du cycle cellulaire
(Rawlings, 2004). Les STATs peuvent aussi être activées indépendamment des Jak par la
tyrosine kinase c-Src (Rawlings, 2004). La dérégulation de la voie de signalisation Jak/STAT
a été décrite dans différents cancers (Ward, 2000 ; Blume-Jensen, 2001). Plusieurs études ont
montré que STAT3 est activé de façon constitutive et, est associé à NPM-ALK, aussi bien
dans des lignées cellulaires transformées par l’oncogène, que dans des lignées d’ALCL et sur
des biopsies de tumeurs de patients atteints d’ALCL (Zamo, 2002 ; Zhang, 2002 ; Dalton,
2005). Une autre étude a mis en évidence que l’extinction de STAT3 par une technique
d’ARN interférence, dans un modèle de souris transgéniques NPM-ALK, induit la régression
tumorale, soulignant ainsi le rôle fondamental de STAT3 dans la lymphomagenèse des ALCL
(Chiarle, 2005). En effet, STAT3 participe aux effets anti-apoptotiques et pro-mitogéniques
27

Introduction
de NPM-ALK et sa non activation est corrélée à un meilleur pronostic des ALCL (Khoury,
2003 ; Amin, 2004). L’activation de STAT3 en aval de NPM-ALK semble se faire de façon
indépendante de c-Src. L’expression constitutive de PP2A (phosphatase 2A), un régulateur
positif de l’activation de STAT3 et l’absence d’expression de PIAS3 (Protein Inhibitor of
Activated STAT3), un régulateur négatif de l’activation de STAT3, dans les cellules NPM-
ALK positives, pourraient jouer un rôle important pour le maintien de l’activation de STAT3
en aval de NPM-ALK (Zhang, 2002). Parmi les autres membres de la famille des STAT, seul
STAT5B est constitutivement activé dans les lignées cellulaires NPM-ALK positives
(Nieborowska-Skorska, 2001). Il a été proposé que la tyrosine kinase Jak2 puisse être
l’activateur de STAT5B en aval de NPM-ALK (Ruchatz, 2003). Cependant, cette activation
de STAT5B dépendante de NPM-ALK est controversée, car plusieurs autres équipes ne l’ont
pas mise en évidence (Zhang, 2002 ; Zamo, 2002 ; Chiarle, 2005) et une telle activation reste
sujet à débat. Concernant la protéine Jak3, elle est fréquemment activée dans les ALCL ALK
positifs et co-immunoprécipite avec NPM-ALK (Zamo, 2002 ; Zhang, 2002 ; Lai, 2005). Il
semble que Jak3 joue un rôle dans l’activation de STAT3 dépendante de NPM-ALK (Amin,
2003). Cependant, d’autres études ont pu mettre en évidence que l’expression de mutants de
NPM-ALK ne liant plus Jak3 ou l’inhibition de Jak3 n’affectent pas l’activation de STAT3,
suggérant ainsi que Jak3 n’est pas la seule kinase impliquée dans l’activation de STAT3 par
NPM-ALK (Zamo, 2002 ; Zhang, 2002 ; Marzec, 2005) (Figure 10).
Figure 10: Voies de signalisation impliquant la voie Jak/STAT3 en aval de NPM-ALK. D’après Chiarle, 2008.
28

Introduction
3.3 NPM-ALK induit la migration cellulaire et le remodelage du cytosquelette
3.3.1. La voie p130Cas
Comme dit précédemment, l’oncogène NPM-ALK est capable d’induire des phénomènes tels
que la dépolymérisation des filaments d’actine ou bien encore une perte d’adhésion aux
matrices. Ces processus entraînent des changements morphologiques dans les cellules
transformées par NPM-ALK, et suggèrent que l’oncogène pourrait médier ainsi son pouvoir
transformant et invasif (Ambrogio, 2005). Ainsi, lors de la recherche par spectrométrie de
masse de protéines impliquées dans le réarrangement du cytosquelette associées à NPM-ALK,
les auteurs ont décrit la protéine p130Cas (p130 Crk-associated substrate), une protéine
adaptatrice qui induit des interactions protéine/protéine. Les auteurs montrent que dans des
cellules exprimant NPM-ALK ou dans des lignées issues de patients NPM-ALK positives,
p130Cas s’associe à l’oncogène et entraîne une phosphorylation de NPM-ALK (Ambrogio,
2005). L’utilisation de mutants catalytiquement inactif a permis de mettre en évidence que ces
effets sont dépendants de l’activité kinase de ALK et de Grb2 mais partiellement indépendant
de la kinase pp60c-Src. Enfin, des fibroblastes dépourvus de p130Cas et transfectés par NPM-
ALK présentent un défaut de dépolymérisation des filaments d’actine. Il semble donc que
l’activation de p130Cas soit un acteur important dans les processus de transformation médiés
par NPM-ALK (Ambrogio, 2005).
3.3.2. Implication des GTPases de la famille Rho
Des travaux de notre équipe ont pu mettre en évidence un rôle de la GTPase Rac dans
l’oncogenèse associée à NPM-ALK et plus particulièrement dans les capacités migratoires et
invasives de cet oncogène (Colomba, 2008). Ainsi, ces travaux ont montré que la GTPase
Rac1, un régulateur du cytosquelette est activée aussi bien dans des cellules exprimant NPM-
ALK que dans les lignées d’ALCL issues de patients ou encore des biopsies ganglionnaires de
patients. De plus, cette activation de Rac en aval de NPM-ALK est médiée par le facteur
d’échange guanidique, Vav3. Ainsi, ces résultats montrent que Vav3 est phosphorylée sur sa
tyrosine 173, reflet de son activation, dans les cellules NPM-ALK positives. D’autre part,
notre équipe a pu montrer que l’activation de cette voie Vav3/Rac1 est sous contrôle de la
kinase pp60c-src et que Vav3 était associée en complexe avec NPM-ALK. Enfin, ces résultats
montrent qu’une modulation négative de la voie Vav3/Rac1, par extinction de l’expression de
29

Introduction
Vav3 par une technique de shRNA ou par la surexpression d’une forme dominante négative
de Rac1, affecte les capacités invasives des cellules NPM-ALK positives in vitro (Colomba,
2008). En conclusion, ces données révèle l’existence d’une nouvelle voie de signalisation en
aval de NPM-ALK et démontre un rôle essentiel de la voie Vav3/Rac1 dans les propriétés
transformantes de NPM-ALK, en particulier dans les phénomènes migratoires et invasifs
associés à cet oncogène (Figure 11).
src
Rac
Vav3
PakP
ALKALKNPM
NPM
invasionP P
P P
Figure 11: La voie Vav3/Rac1 : un nouvel acteur dans l’oncogenèse induite par NPM-ALK. NPM-ALK permet, via la tyrosine kinase pp60c-src, l’activation de proto-oncogène Vav3 qui est impliqué dans l’activation de la GTPase Rac1, qui est pour sa part essentielle aux phénomènes migratoires et invasifs dépendant de NPM-ALK. D’après Colomba, 2008.
3.4. NPM-ALK module le phénotype immunitaire via l’expression de CD30 Les ALCL sont caractérisés par l’expression de la protéine CD30. C’est une protéine
membranaire appartenant à la famille des récepteurs aux TNF (Tumor Necrosis Factor)
exprimée dans les lignées lymphocytaires B et T. CD30 est capable de s’associer aux
protéines TRAF 2 et 5 ce qui conduit à l’activation du facteur de transcription NF-κB et
promeut l’apoptose. Dans les ALCL, le rôle exact de l’expression de CD30 reste à déterminer,
mais il semble que la transcription de ce gène soit activée par la présence de NPM-ALK par le
biais des voies de signalisation MAPK/ERK et STAT3 (Hsu, 2006 ; Watanabe, 2005).
Comme pour les lymphomes Hogdkiniens (Horie, 2002), l’engagement de CD30 dans les
cellules NPM-ALK positives va conduire à l’activation des voies canoniques et alternatives
impliquant NF-κB (Wright, 2007 ; Ohno, 2005). En fait, NPM-ALK phosphoryle NPM
sauvage ce qui entraîne la liaison des dimères NPM/NPM-ALK à la protéine TRAF 2 limitant
donc l’activation du récepteur CD30 par TRAF 2 et de la voie canonique d’activation de NF-
κB. Une autre façon de dégrader TRAF2 dans les ALCL et donc d’inhiber le signal
d’activation des voies NF-κB, se fera par la liaison de CD30 à son ligand CD30L. Ces deux
événements permettent donc de diminuer l’activation de la voie canonique d’activationn de
30

Introduction
NF-κB mais l’engagement du récepteur CD30 dans les ALCL va entraîner aussi l’activation
de NF-κB par la voie alterne. Ainsi, cette activation va permettre une augmentation de
l’expression de la sous-unité p100 de NF-κB ce qui conduit à la phosphorylation de Bcl3, et
donc à la transcription de gènes contrôlant l’apoptose et l’arrêt du cycle celulaire (Chiarle,
2008). La dualité de l’activation de NF-κB via les voies canoniques et alternes en aval de
l’engagement de CD30 dans le contexte de NPM-ALK pourrait médier les divers processus
biologiques caractéristiques de cet oncogène à savoir, la prolifération, l’inhibition du cycle
cellulaire, la différenciation cellulaire ou bien encore l’apoptose (Chiarle, 2008) (Figure 12).
Figure 12: Voies de signalisation impliquant la protéine de surface CD30 dans le contexte de l’oncogenèse associée à NPM-ALK. D’après Chiarle, 2008.
3.5. Régulation négative de NPM-ALK Comme détaillé ci-haut, la plupart des travaux réalisées sur la signalisation de NPM-ALK
concerne l’identification des voies de signalisation activées par l’oncogène. Ainsi, les
mécanismes pouvant réguler négativement l’activité tyrosine kinase de NPM-ALK restent
encore largement méconnus. Les protéines tyrosine phosphatases sont des candidats potentiels
pouvant remplir ce rôle. En effet, les mécanismes de tyrosine phosphorylation sont gouvernés
par l’action des tyrosine kinases, mais aussi par les tyrosine phosphatases, qui régulent
d’importantes voies de signalisation impliquées dans le contrôle de la prolifération, de
31

Introduction
l’adhésion et de la migration. De plus, certaines protéines tyrosine phosphatases telles que
PTEN (Phosphatase and Tensin Homologue deleted on chromosome 10) ou SHP-2 ont été
identifiées comme étant des suppresseurs de tumeurs, de par leur action antagoniste sur la
signalisation induite par les tyrosines kinases (Ostman, 2006). Ainsi, des études récentes
suggèrent que la tyrosine phosphatase SHP-1 pourrait être un régulateur négatif de NPM-
ALK. En effet, la perte d’expression de SHP-1, suite à la méthylation de son promoteur, est
fréquemment observée dans des biopsies de tumeurs issues de cas d’ALCL et pourrait
contribuer à l’activation de la voie Jak3/STAT3 dans les ALCL (Khoury, 2004 ; Han, 2006b).
De plus, il existe une corrélation inverse entre l’activité phosphatase de SHP-1 et le taux de
phosphorylation de NPM-ALK, suggérant que NPM-ALK est un substrat de cette
phosphatase. SHP-1 est capable de s’associer en complexe à NPM-ALK et Jak3 et son
invalidation par une technique d’ARN interférence conduit à une augmentation de la
phosphorylation de NPM-ALK et de STAT3, corrélée à une augmentation de la prolifération
cellulaire (Han, 2006a). A l’inverse, la surexpression de SHP-1 est suivie d’une diminution de
la phosphorylation de NPM-ALK et a donc un effet négatif sur la prolifération cellulaire et la
progression tumorale in vivo (Honorat, 2006 ; Han, 2006a). De façon intéressante, une autre
étude récente a pu mettre en évidence un rôle antagoniste de la tyrosine phosphatase SHP-2.
Ainsi, SHP-2 sous sa forme active phosphorylée s’associe à NPM-ALK et est impliquée dans
les propriétés pro-mitogéniques et pro-migratoires de NPM-ALK (Voena, 2007). Ainsi,
malgré leurs fortes homologies de séquence (95%) et de structure, SHP-1 et SHP-2 ont une
fonction opposée, l’une étant anti-tumorale et l’autre participant aux propriétés transformantes
de NPM-ALK (Poole, 2005).
3. Identification de nouvelles cibles et de nouveaux partenaires
de ALK
Bien que les mécanismes moléculaires de la transformation cellulaire induite par NPM-ALK
soient de plus en plus détaillés, les partenaires de la signalisation impliqués dans son pouvoir
transformant restent encore mal connus. Dans le but d’identifier de nouvelles protéines
participant à la lymphomagenèse des ALCL, mais aussi afin de mettre à jour de nouveaux
marqueurs de diagnostic, de pronostic et de nouvelles cibles pour les traitements, des études
d’analyse du transcriptome et du protéome des ALCL ont été entreprises. Les études de
l’analyse comparative du transcriptome entre des lignées cellulaires d’ALCL ALK positives
32

Introduction
et ALK négatives ont été réalisées par puces à ADN ou par hybridation suppressive
soustractive (Villalva, 2002 ; Thompson, 2005 ; Piva, 2006b ; Lamant, 2007). Au niveau
protéomique, différentes stratégies d’analyse par spectrométrie de masse ont étudié divers
aspects du protéome des ALCL. Certaines études sont basées sur l’analyse comparative du
protéome global entre les lignées cellulaires d’ALCL ALK positives et ALK négatives,
d’autres se sont focalisées sur le phospho-protéome et enfin des études ont été consacrées à
l’analyse protéomique des partenaires de ALK (Crockett, 2004 ; Rush, 2005 ; Cussac, 2006).
Ces différentes études ont permis d’identifier un ensemble de gènes et de protéines, dont
seulement un faible pourcentage a déjà été caractérisé comme participant à la transformation
cellulaire induite par ALK. Les nouvelles protéines identifiées ont été regroupées dans
différentes classes suivant leur fonction cellulaire (Figure 13). Ainsi, ces différentes études
ont mis en évidence des protéines impliquées dans : la signalisation (des kinases comme MEK
et PKC, des molécules adaptatrices comme IRS-4), la transcription (comme le facteur de
transcription C/EBPβ) et la stabilité des ARNm (AUF1), le métabolisme, l’organisation du
cytosquelette et la motilité (myosine, tubuline, WASP, Pyk2 p130Cas), le contrôle de la
traduction (eIF4B), l’apoptose (les facteurs anti-apoptotiques Mcl-1 et Bcl-2A2, ou encore la
régulation du cycle cellulaire (cycline D3). Des phosphatases, des protéines chaperonnes
(Hsp60, Hsp70, Hsp90), des petites GTPases (GTPase Ran) et leurs régulateurs (Rho-GDI2,
RhoGAP, GEF de Arf), ont également été identifiés comme étant régulés par et/ou associés à
NPM-ALK (Ambrogio, 2005 ; Jundt, 2005 ; Quintanilla-Martinez, 2006 ; Fawal, 2006 ; Piva,
2006b).
Figure 13: NPM-ALK induit l’expression de nombreuses protéines impliquées dans diverses fonctions cellulaires comme la motilité, la synthèse protéique, la survie, l’invasion et la prolifération. D’après Lim, 2006.
33
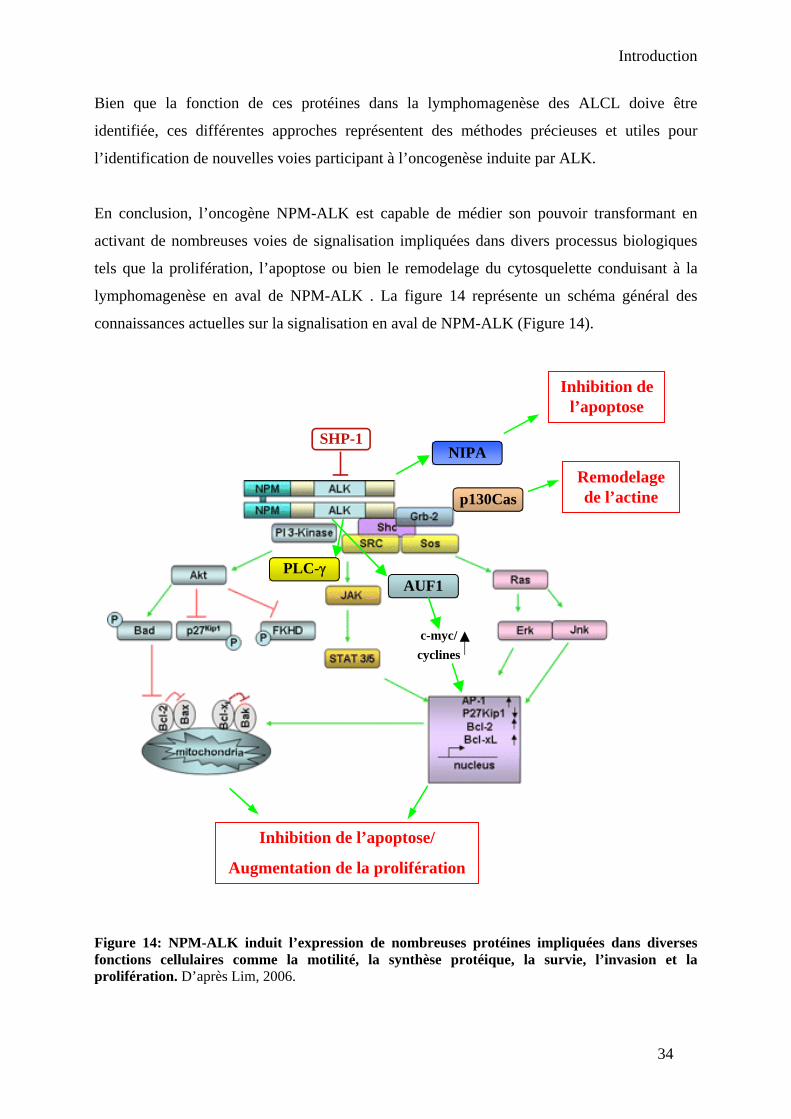
Introduction
Bien que la fonction de ces protéines dans la lymphomagenèse des ALCL doive être
identifiée, ces différentes approches représentent des méthodes précieuses et utiles pour
l’identification de nouvelles voies participant à l’oncogenèse induite par ALK.
En conclusion, l’oncogène NPM-ALK est capable de médier son pouvoir transformant en
activant de nombreuses voies de signalisation impliquées dans divers processus biologiques
tels que la prolifération, l’apoptose ou bien le remodelage du cytosquelette conduisant à la
lymphomagenèse en aval de NPM-ALK . La figure 14 représente un schéma général des
connaissances actuelles sur la signalisation en aval de NPM-ALK (Figure 14).
p130Cas
SHP-1
PLC-γ
NIPARemodelagede l’actine
Inhibition del’apoptose
Inhibition de l’apoptose/
Augmentation de la prolifération
AUF1
c-myc/cyclines
Figure 14: NPM-ALK induit l’expression de nombreuses protéines impliquées dans diverses fonctions cellulaires comme la motilité, la synthèse protéique, la survie, l’invasion et la prolifération. D’après Lim, 2006.
34

Introduction
CHAPITRE II
Les Phosphoinositides
Les phosphoinositides (PIs) sont des glycérophospholipides anioniques membranaires
constitués d’un squelette comprenant un sn-1,2-diacylglycerol associé par une liaison
phosphodiester à un groupement hydroxyl d’un myo-inositol. Les acides gras retrouvés chez
la majorité des PIs sont : l’acide stéarique (C18 :0) et l’acide arachidonique (C20 :4)
respectivement en position 1 et 2 du squelette glycérol (Mauco, 1984) (Figure 15).
Figure 15 : Structure d’un phosphoinositide, le 1-stéaroyl, 2-arachidonoyl, sn glycero-D myo-inositol 4,5-bisphosphate. Seuls les groupements hydroxyl libres en position 3, 4 et 5 peuvent être phosphorylés in vivo.
Les PIs représentent entre 10 et 15% des phospholipides membranaires totaux. Ils sont au
nombre de huit et sont classés selon la position et le nombre de groupements phosphates
présents sur le noyau myo-inositol. Ce noyau myo-inositol contient cinq groupements
hydroxyl libres mais seulement trois de ces positions (3,4 ou 5) peuvent être phosphorylées in
vivo. A côté du : i) phosphatidylinositol (PtdIns) qui est non phosphorylé, on trouve ii) les
phosphatidylinositol monophosphates : le phosphatidylinositol 3-monophosphate (PtdIns3P),
le phosphatidylinositol 4-monophosphate (PtdIns4P) et le phosphatidylinositol 5-
monophosphate (PtdIns5P), iii) les phosphatidylinositol bisphosphates : le
phosphatidylinositol 3,4-bisphosphate (PtdIns(3,4)P2), le phosphatidylinositol 3,5-
bisphosphate (PtdIns(3,5)P2) et le phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) et
iiii) le phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3). En ce qui concerne leur
35

Introduction
répartition, le plus abondant des PIs, le PtdIns, représente 5 à 8% des phospholipides totaux
de la cellule. Le PtdIns4P et le PtdIns(4,5)P2 atteignent au maximum 3% et les autres PIs
représentent moins de 2%. C’est en 1964 que Hokin et Hokin ont mis en évidence pour la
première fois l’hydrolyse des PIs en réponse à une stimulation, posant ainsi les premières
bases du « cycle des PIs » (Hokin, 1964). Des descriptions de plus en plus précises d’une
signalisation des PIs furent alors proposées (Michell, 1975), conduisant à la découverte de la
« voie canonique des PIs » à la fin des années 1970 (Berridge, 1986 ; Berridge, 1987 ;
Berridge, 1984). Plus récemment, la découverte des D3-phosphoinositides, produits de la PI
3-kinase, a suscité l’intérêt de nombreux groupes pour le métabolisme des PIs, et accéléré les
recherches sur les rôles potentiels de ces molécules, dont les taux sont finement régulés et qui
participent à l’organisation de grandes voies de signalisation cellulaires.
I- Le métabolisme des phosphoinositides et les enzymes clés
1. Le cycle des phosphoinositides
Le métabolisme des PIs est complexe et finement régulé par un jeu de kinases, phosphatases
ou phospholipases (Payrastre, 2001 ; Toker, 2002).
Ce cycle peut être découpé en trois voies majeures (Figure 16) :
- la voie « classique » ou « canonique » du PtdIns, PtdIns4P et PtdIns(4,5)P2
conduisant à la génération de seconds messager tels que le diacylglycérol (DAG) et l’inositol-
1,4,5-trisphosphate (IP3), par l’action de phospholipases C (PLCs).
- les voies générant la synthèse des D3-phosphoinositides
- les voies récemment découvertes et permettant la génération de PtdIns5P
36

Introduction
Figure 16 : Voie de synthèse des phosphoinositides et interconversions. En rouge sont représentés les huit phosphoinositides, en bleu les phosphoinositide kinases, en vert les phosphoinositide phosphatases, en noir la phospholipase C et en orange ces deux produits.
2. Le métabolisme des phosphoinositides dit « classiques »
Dans les cellules eucaryotes, il est classiquement admis que les PIs constituant cette « voie
canonique » c’est-à-dire, le PtdIns, le PtdIns4P et le PtdIns(4,5)P2 sont maintenus à des
niveaux relativement constant dans le feuillet interne de la membrane plasmique par des
réactions de phosphorylation/déphosphorylation dépendantes de 4 et 5-kinases et
phosphatases qui ne sont pas toutes identifiées à ce jour (Michell, 1975 ; Berridge, 1984). Le
maintien de cet équilibre représente jusqu’à 7% de la consommation basale d’ATP dans les
cellules.
Le PtdIns est phosphorylé en position 4 de son noyau inositol par des PtdIns 4-kinases, et
génère ainsi du PtdIns4P. A ce jour, seuls deux types de kinases capables de catalyser cette
réaction ont pu être identifiées chez les mammifères, les PtdIns 4-kinases de type II et III
(Balla, 2006). Cette réaction est reversée par des PtdIns 4-phosphatases peu étudiée
actuellement.
37

Introduction
Le PtdIns(4,5)P2 est synthétisé par des PtdInsP-kinases que l’on peut regrouper en deux
classes aux spécificités différentes. La voie de production majeure pour le PtdIns(4,5)P2 va
impliquer les PtdInsP-kinases de type I qui sont capables de phosphoryler le noyau inositol du
PtdIns4P en position 5. Le taux de PtdIns(4,5)P2 membranaire peut encore être augmentée par
l’action d’autres kinases faisant partie des voies nouvellement caractérisées et impliquent le
PtdIns5P. Cette voie fait appel aux PtdInsP-kinases de type II qui sont des PtdIns5P 4-kinases
qui phosphorylent le PtdIns5P en position 4 (Rameh, 1997b). La synthèse de PtdIns(4,5)P2 est
réversée par l’action de 4-phosphatases, comme OCRL (Oculocerebrorenal Lowe syndrome).
La protéine OCRL est responsable d’un syndrome lié à l’X qui se caractérise par des retards
mentaux, des cataractes congénitales et des problèmes rénaux (Lowe, 1952). Les lignées de
cellules rénales issues de patients présentent un taux élevé de PtdIns(4,5)P2 suggérant que
cette enzyme régule bien le taux de ce PI in vivo (Zhang, 1998 ; Wenk, 2003). Cette enzyme
est localisée essentiellement sur l’appareil de Golgi et les endosomes précoces (Ungewickell,
2004) et se relocalise à la membrane plasmique au niveau des ruffles membranaires, en
réponse à des stimuli tels que les facteurs de croissance pour réguler le taux global de
PtdIns(4,5)P2 (Faucherre, 2005).
Le PtdIns(4,5)P2 ainsi produit peut être clivé par des PLCs pour générer du DAG activateur
des protéines kinases C (PKCs), et de l’InsP3 qui permet la libération de calcium depuis le
réticulum endoplasmique (Berridge, 1984 ; Berridge, 1987). Le DAG peut être dégradé par
des lipases ou bien phosphorylé par des DAG-kinases en acide phosphatidique (PA). Le PA
peut alors agir en tant que second messager ou bien, il peut être transporté au réticulum
endoplasmique où il sera métabolisé en PtdIns. Le PtdIns formé sera alors transporté à la
membrane plasmique par des protéines de transfert (PI-TP) afin de compléter le cycle
(Cockcroft, 1998).
3. Le métabolisme des D3-phosphoinositides
3.1. Les PI 3-kinases Les PI 3-kinases appartiennent à une famille d’enzymes très conservées. Il existe huit PI 3-
kinases qui sont regroupées en trois classes (I, II et III) en fonction de leur homologie de
séquence, de leur spécificité de substrat et de leur régulation (Vanhaesebroeck, 2001 ;
Vanhaesebroeck, 2005) (Tableau 3)
38

Introduction
Ces enzymes sont capables de phosphoryler le PtdIns, le PtdIns4P et le PtdIns(4,5)P2 en
position 3 du noyau inositol ce qui va conduire à la génération de PtdIns3P, PtdIns(3,4)P2 et
de PtdIns(3,4,5)P3 que l’on regroupe sous le terme générique de D3-phosphoinositides. Ce
sont des seconds messagers qui jouent un rôle majeur dans de nombreux processus cellulaires
(Toker, 1997 ; Payrastre, 2001).
Classe
Tableau 3: La famille des PI 3-kinases. La figure représente schématiquement la structure des domaines des sous-unités catalytiques et régulatrices des PI 3-kinases de classe I, II et III, et décrit la distribution différentielle tissulaire, la spécificité de substrats et la régulation des différentes isoformes de PI 3-kinases. PtdIns : phosphatidylinositol ; CML : cellules musculaires lisses. D’après Vanhaesebroeck, 2001.
3.1.1. Les PI 3-kinases de classe I
Ce sont les PI 3-kinases les mieux caractérisées à ce jour. In vitro, elles sont capables de
phosphoryler le PtdIns, le PtdIns4P et le PtdIns(4,5)P2 mais in vivo leur substrat préférentiel
est le PtdIns(4,5)P2 génèrant ainsi le PtdIns(3,4,5)P3. Ce sont des hétérodimères composés
d’une sous-unité catalytique de 110 kDa et de différentes sous-unités
adaptatrices/régulatrices. Cette classe est divisée en deux sous-classes, IA et IB qui régulent
respectivement les voies de signalisation en aval de récepteurs à activité tyrosine kinase et en
aval de récepteurs couplés aux protéines G hétérotrimériques (RCPGs) ou Ras.
IA
Classe IB
Classe II
Sous-unitécatalytique
Sous-unitérégulatrice Régulation
SubstratIn vitro In vivo
P110α, β, δ
p110γ p101
p85α, p85βP55γp50α, p55α
Tyr kinasesRasGβγ
pour p110β
GβγTyr kinasesRas
PI3KC2 α,β,γ
Analogue Vps34p p150
Clathrine(α)?
Tyr kinasesChémokinesIntégrines(≠Ras)
Constitutive ?
PtdIns
PtdIns(4)P
PtdIns(4,5)P2
p101
p110γ
SH2SH2P PBHSH3
Domaine catalytiqueRas C2 PIK
p110α,β,δ
p85α
Domaine de liaison à p85
Domaine catalytiqueRas C2 PIK
Domaine catalytiqueRas C2 PIK C2PX
PI3KC2 α,β,γ
Prot kinase domain Heat WD40 p150
Analogue Vps34pDomaine
catalytiqueC2 PIK
PtdIns(4,5)P2
PtdIns(4)P
PtdIns PtdIns
Classe III
PtdIns
PtdIns(4)P
Expression
α,β ubiquitairesδ hématopoïétiqueprincipalement
HématopoïétiqueprincipalementCMLCardiomyocytes
Ubiquitaires
Ubiquitaire
repeats
IAClasse
Classe IB
Classe II
Sous-unitécatalytique
Sous-unitérégulatrice Régulation
SubstratIn vitro In vivo
SubstratIn vitro In vivo
P110α, β, δ
p110γ p101
p85α, p85βP55γp50α, p55α
Tyr kinasesRasGβγ
pour p110β
GβγTyr kinasesRas
PI3KC2 α,β,γ
Analogue Vps34p p150
Clathrine(α)?
Tyr kinasesChémokinesIntégrines(≠Ras)
Constitutive ?
PtdIns
PtdIns(4)P
PtdIns(4,5)P2
p101
p110γ
SH2SH2P PBHSH3
Domaine catalytiqueRas C2 PIK
p110α,β,δ
p85α
Domaine de liaison à p85
SH2SH2SH2P PBHSH3
Domaine catalytiqueRas C2 PIK
p110α,β,δ
p85α
Domaine de liaison à p85
Domaine catalytiqueRas C2 PIK Domaine catalytiqueRas C2 PIK
Domaine catalytiqueRas C2 PIK C2PXDomaine catalytiqueRas C2 PIK C2PX
PI3KC2 α,β,γ
Prot kinase domain Heat WD40 p150
Analogue Vps34pDomaine
catalytiqueC2 PIK Domaine catalytiqueC2 PIK
PtdIns(4,5)P2
PtdIns(4)P
PtdIns PtdIns
PtdIns
PtdIns(4)P
Expression
α,β ubiquitairesδ hématopoïétiqueprincipalement
HématopoïétiqueprincipalementCMLCardiomyocytes
Ubiquitaires
Ubiquitaire
repeats
Classe III
39

Introduction
3.1.1.1. L’activité lipide kinase des PI 3-kinases de classe I
- Les PI 3-kinases de classe IA
Les cellules de mammifères possèdent trois isoformes de la sous-unité catalytique p110 de
classe IA : p110α, p110β et p110δ, codées par trois gènes différents. Ces sous-unités sont
constituées de plusieurs domaines : un domaine N-terminal de fixation à la sous-unité
adaptatrice, un domaine de fixation à Ras, un domaine C2 de fixation aux phospholipides, un
domaine PIK caractéristique des PtdIns-kinases et un domaine catalytique lipide kinase en C-
terminal (Vanhaesebroeck, 1997). Les sous-unités adaptatrices p85α, p55α, p50α (issues de
l’épissage alternatif d’un même gène), p85β et p55γ se lient aux sous-unités catalytiques pour
former des complexes fonctionnels. Elles n’ont pas d’activité catalytique intrinsèque mais
possèdent plusieurs domaines tels que les domaines SH3 qui permet des interactions
protéines-protéines de régions riches en proline et des domaines SH2 séparés par un domaine
inter-SH2 de liaison à la sous-unité catalytique. Lors de l’activation de récepteurs à activité
tyrosine kinase comme le récepteur à l’insuline (Fry, 1994 ; Wymann et Pirola, 1998) ou des
récepteurs tels que famille Src, la kinase FAK ou les kinases JAK (Just Another Kinase)
(Khwaja, 1997 ; Pece, 1999), ces domaines SH2 lient des résidus tyrosine phosphorylés
contenant un motif Y(P)xxM (x représente n’importe quel acides aminés), entraînant
l’activation des PI 3-kinases et leur translocation à la membrane près de leurs substrats
lipidiques.
- Les PI 3-kinases de classe IB
La seule PI 3-kinase de classe IB connue est constituée de la sous-unité catalytique p110γ et
de la protéine adaptatrice p101. Les hétérodimères p110γ/p101 sont activés par les sous-unités
Gβγ des protéines G hétérotrimériques. A ce jour, les données bibliographiques ne permettent
pas de trancher entre un mode d’activation direct de p110γ par les Gβγ ou via une activation
par des adaptateurs tels que la protéine Ras (Krugmann, 1999 ; Suire, 2006). Concernant sa
distribution, la PI 3-kinase de classe IB est présente seulement chez les mammifères et est
retrouvée essentiellement dans les plaquettes sanguines, les globules blancs, les progéniteurs
érythrocytaires, les cellules musculaires lisses ou les cardiomyocytes.
40

Introduction
3.1.1.2. L’activité protéine kinase des PI 3-kinases de classe I
Bien que plus connues pour leur activité lipide kinase, les PI 3-kinases de classe I possèdent
une activité protéine kinase intrinsèque in vitro (Czupalla, 2003a). Cette capacité
d’autophosphorylation abolit l’activité lipide kinase des PI 3-kinases p110 α, δ et β, mais pas
celle de la PI 3-kinase γ (Vanhaesebroeck, 1999b, Czupalla, 2003b). D’autres données ont
montré que les PI 3-kinases peuvent phosphoryler d’autres substrats, c’est le cas de la
protéine adaptatrice IRS-1 (Insulin Receptor Substrate-1) ou de PDE3B (cGMP-inhibited
phosphodiesterase 3B) mais la relevance physiologique de telles phosphorylations reste à ce
jour inconnue (Tanti, 1994 ; Rondinone, 2000 ; Pirola, 2001).
3.1.2. Les PI 3-kinases de classe II
Celles-ci phosphorylent le PtdIns et la PtdIns4P pour générer du PtdIns3P et du PtdIns(3,4)P2.
Cependant, cette conversion n’a pu être démontrée qu’in vitro, cette synthèse reste mal
connue in vivo. Chez les mammifères, il existe trois isoformes de PI 3-kinases de classe II
(C2α, C2β et C2γ) qui sont toutes trois codées par des gènes différents. Les formes C2α et β
sont ubiquitaires, contrairement à l’isoforme C2γ qui est exprimée préférentiellement dans le
foie. Il est aussi intéressant de noter qu’un seul type de PI 3-kinase de classe II a pu être
identifié chez D. melanogaster ou C. elegans et aucune chez la levure ou les plantes. Ces
enzymes sont caractérisées par un domaine C2 en C-terminal, indispensable à leur activité
catalytique. Ce domaine lie les phospholipides de façon calcium indépendante in vitro
(Misawa, 1998 ; Arcaro, 1998). La région N-terminale contient un domaine de liaison à la
protéine Ras mais aucune donnée, à ce jour, n’a pu montrer une liaison des PI 3-kinases de
classe II avec Ras ou bien des protéines adaptatrices. Cette classe de PI 3-kinases contient
aussi un domaine PX (Phox Homology) (Cf. Chap. II.III.) qui lie le PtdIns(4,5)P2 in vitro
(Song, 2001). Le mode d’activation de ces PI 3-kinases par des stimuli extracellulaires n’est
pas clair, cependant une augmentation de l’activité lipide kinase de ces enzymes in vitro a pu
être observée lors de stimulation par l’EGF, l’insuline, l’adhésion des intégrines ou des
chémokines (Brown, 1999 ; Paulhe, 2002 ; Turner, 1998). Des données récentes montrent un
rôle de la PI 3-kinase C2β dans l’adhésion et la mobilité cellulaire en réponse à l’acide
41

Introduction
lysophosphatidique (LPA) et montrent une production atypique de PtdIns3P au niveau de la
membrane plasmique par cette isoforme (Maffucci, 2005).
3.1.3. La PI 3-kinase de classe III
Vps34 (Vesicular protein sorting 34) identifiée en premier lieu chez la levure est la seule PI
3-kinase de classe III connue à ce jour (Odorizzi, 2000 ; pour revue, Backer, 2008). Sa
spécificité de substrat est limitée au PtdIns, ce qui la distingue des autres PI 3-kinases. Elle
semble ainsi être à l’origine de la majorité du PtdIns3P intracellulaire. Il existe des
homologues de cette protéine chez les plantes, C. elegans, D. melanogaster et chez les
mammifères (Volinia, 1995 ; Linassier, 1997 ; Roggo, 2002 ; Zhou, 1995). Chez les
mammifères, hVps34 est exprimée de façon ubiquitaire. Sa structure est composée d’un
domaine C2 comme la PI 3-kinase de classe II, un domaine hélical très peu étudié mais qui a
été décrit comme régulant négativement la capacité d’autophosphorylation des PI 3-kinases
de classe IA (Miled, 2007), et un domaine lipide kinase (Budovskaya, 2002). En plus de son
activité lipide kinase, Vps34 possède une capacité d’autophosphorylation (Stack, 1994) et une
activité protéine kinase in vitro (Panaretou, 1997). A ce jour, aucun substrat protéique de cette
kinase n’a pu être identifié in vivo. Chez les mammifères comme chez la levure, cette enzyme
est retrouvée associée à la sérine/thréonine kinase Vps15. Cette protéine semble être un
régulateur de Vps34, puisqu’un mutant catalytique de Vps15 empêche la production de
PtdIns3P par Vps34. Les mécanismes exacts de cette régulation sont encore sujet à
investigation mais il semble que Vps15 permette l’adressage de Vps34 aux membranes par
myristoylation en N-terminal (Raymond, 1992), ce qui va rapprocher Vps34 de son substrat
lipidique. Chez la levure et les mammifères, Vps34 a pu être impliquée en premier lieu dans
la régulation du trafic vésiculaire entre les endosomes et les lysosomes où le PtdIns3P produit
peut recruter des protéines contenant des domaines de liaison à ce PI. Ainsi, cette kinase peut
recruter une autre enzyme, qui contient un domaine FYVE (Cf. Chap. II.III.), bien connue
pour son rôle dans le trafic vésiculaire, Fab1p (Cf. Chap. III). Chez la levure, la seule voie
connue de production du PtdIns(3,5)P2 fait intervenir ces deux enzymes et permet le contrôle
du trafic vésiculaire. Chez les mammifères, en plus de son rôle de régulateur du trafic
vésiculaire, hVps34 peut intervenir dans la réponse aux nutriments via une activation de
mTOR (mammalian target of Rapamycin) (Byfield, 2005), ou encore dans le contrôle de
l’autophagie (Petiot, 2000).
42

Introduction
3.2. Les phosphatases agissant sur les D3-phosphoinositides
3.2.1. La 3-phosphatase PTEN
PTEN, nommée aussi MMAC (Mutated in Multiple Advanced Cancers), initialement décrite
comme une protéine tyrosine phosphatase, montre une préférence marquée pour les PIs
phosphorylés en position 3 c’est-à-dire le PtdIns3P, le PtdIns(3,4)P2 et le PtdIns(3,4,5)P3,
ainsi que pour l’inositol (1,3,4,5)-tétrakisphosphate (Ins(1,3,4,5)P4) in vitro. In vivo, son
substrat préférentiel reste le PtdIns(3,4,5)P3 (Maehama, 1998). Cette protéine est composée de
deux domaines structuraux : un domaine catalytique contenant une séquence consensus CX5R
spécifique des protéines tyrosine phosphatases qui ont double spécificité de substrat (DSP :
double spécificité de substrat, tyrosine et sérine/thréonine kinase), et un domaine C2 qui va
permettre à PTEN de se localiser à la membrane près de ses substrats (Lee, 1999 ; Das, 2003).
De part sa capacité à déphosphoryler le PtdIns(3,4,5)P3, cette phosphatase va réguler
négativement les effets des PI 3-kinases de classe I. En effet PTEN va diminuer la
translocation à la membrane du proto-oncogène Akt et son activation, engendrant ainsi une
baisse de la prolifération cellulaire et de la survie (Myers, 1998). PTEN est mutée dans de
nombreux cancers humains. Ces mutations, qui affectent son activité lipide phosphatase,
génèrent une suractivation de la voie de survie PI 3-kinase/Akt et perturbent la fonction
suppresseur de tumeurs de cette phosphatase (Myers, 1998 ; Leslie, 2004).
3.2.2. Les 5-phosphatases SHIP1 et SHIP2
Il existe deux membres de cette famille de phosphatases, SHIP1 (Ware, 1996) et SHIP2
(Pesesse, 1997). SHIP1 est exprimée dans les cellules hématopoïétiques (Liu, 1998a ; Liu,
1998b) et SHIP2 est distribuée de façon ubiquitaire (Pesesse, 1997). Le domaine catalytique
des phosphatases SHIP est localisé en position centrale et permet l’hydrolyse du
PtdIns(3,4,5)P3 et du Ins(1,3,4,5)P4. Elles possèdent en N-terminal un domaine SH2 de liaison
avec des résidus tyrosine-phosphorylés, et en C-terminal des séquences consensus NPxY ainsi
que plusieurs séquences riches en proline qui lient les domaines PTB (Phosphotyrosine
Binding Domain) ou SH3. A ce jour, SHIP1 et SHIP2 ont pu être impliquées en tant que
régulateur négatif dans plusieurs voies de signalisation. Ainsi, SHIP1 régule négativement la
prolifération des cellules immunitaires (Lioubin, 1996). Quant à SHIP2, elle inhibe la
translocation d’un transporteur de glucose à la membrane plasmique, GLUT4, via la
déphosphorylation du PtdIns(3,4,5)P3 lors d’une stimulation par l’insuline (Kohn, 1996).
43

Introduction
SHIP2 a pu aussi être impliquée dans la down-régulation du PtdIns(3,4,5)P3 en aval du
récepteur à l’EGF (EGFR), dans les problématiques de diabète (Clement, 2001) et/ou
d’obésité (Sleeman, 2005).
3.3.3. Les 3-phosphatases de la famille des myotubularines
Les myotubularines (MTMs) (Clague, 2005) présentes dans la plupart des organismes
eucaryotes incluant les levures et les plantes, constituent chez l’homme une grande famille de
quatorze membres qui sont capables de déphosphoryler spécifiquement en position 3 le
PtdIns3P et le PtdIns(3,5)P2 (Taylor, 2003 ; Blondeau, 2000 ; Schaletzky, 2003). Cette
métabolisation conduit respectivement à la formation de PtdIns et du PtdIns5P un nouveau
second messager (Tronchère, 2004). L’étude des voies conduisant à la génération du PtdIns5P
ayant fait l’objet de mes travaux de thèse, cette enzyme et ses fonctions seront détaillés au
Chap. II.IV.
4. Les voies permettant la génération de PtdIns5P
Le PtdIns5P, mis en évidence dans les cellules à la fin des années 1990 peut être généré de 3
façons, différentes :
- à partir du PtdIns(3,5)P2 par l’action des MTMs
- à partir du PtdIns(4,5)P2 par les 4-phosphatases telles que IpgD (bactérie)
ou les PtdIns(4,5)P2 4-phosphatases de type I et II (mammifères)
- ou par l’action de la PtdIns 5-kinase, PIKfyve à partir du PtdIns
Ces différentes voies de synthèse seront présentées au Chap. II.IV.
II- Rôles biologiques et localisation des différents
phosphoinositides
1. Le PtdIns
C’est le précurseur majeur des autres PIs via des phosphorylations par des kinases spécifiques
du cycle des PIs. Ce PI ne semble pas avoir de rôle direct dans la régulation de la signalisation
intracellulaire.
44

Introduction
2. Le phosphoinositide le mieux caractérisé : le PtdIns(3,4,5)P3
Le PtdIns(3,4,5)P3 présente un taux de base quasiment indétectable et est produit de façon
rapide et transitoire en réponse à des agonistes (Hinchliffe, 2001) dans la membranes
plasmique où il représentera 1 à 10% du PtdIns(4,5)P2. Le rôle de second messager lipidique
de cette molécule est aujourd’hui bien établi. Le PtdIns(3,4,5)P3 apparaît donc comme un
acteur clé du contrôle de l’organisation spatio-temporelle de grandes voies de signalisation à
la membrane plasmique. Il interagit avec de nombreuses protéines possédant un domaine PH
et conduit à leur localisation membranaire et/ou à leur activation. C’est le cas des facteurs
d’échange des petites protéines G de la famille Arf (ADP-ribosylation factors) comprenant le
récepteur Grp1 (General receptor for phosphoinositides), la cytohesine-1 et ARNO (Arf
nucleotide binding site opener) (Klarlund, 1998), la tyrosine kinase Btk (Bruton’s tyrosine
kinase), les sérine-thréonine kinases PDK1 et Akt, la PLCγ (Bae, 1998), la molécule
adaptatrice Gab1 (Grp2-associated protein 1), et un des facteurs d’échange de la GTPase
Rac1, Vav (Han, 1998). Le PtdIns(3,4,5)P3 est ainsi impliqué dans le contrôle de différentes
fonctions cellulaires comme la survie et la prolifération cellulaire, la réorganisation du
cytosquelette et la mobilité cellulaire, le métabolisme glucidique, la régulation du trafic
vésiculaire ou l’apoptose (Figure 17).
Figure 17: Cibles classiques et fonctions du PtdIns(3,4,5)P3. PH : Pleckstrin Homology ; GAP : GTPase Activating Protein ; GEF : Guanine nucleotide Exchange Factor; PLC: phospholipase C; PDK1: Phosphoinositide Dependent Kinase 1. D’après Astle, 2007.
45

Introduction
3. Les phosphatidylinositol bisphosphates
3.1. Le PtdIns(4,5)P2
Le PtdIns(4,5)P2 est localisé essentiellement à la membrane plasmique et représente environ
10% des PIs totaux. C’est une molécule majeure dans la régulation de différentes voies de
signalisation du fait des variations rapides auxquelles il est soumis (Toker, 1998). En effet, il
sert de précurseurs à la génération de seconds messagers, en tant que substrat de la PLC pour
donner le DAG et l’InsP3 ou bien encore il permet la synthèse de PtdIns(3,4,5)P3 par le jeu
des PI 3-kinases de classe I. Il joue également un rôle majeur dans la régulation du
cytosquelette d’actine (Tall, 2000) (Figure 18) et interagit avec des protéines régulatrices de la
polymérisation de l’actine comme la profiline ou la gelsoline (Hilpela, 2004 ; Yin, 2003). Il
permet la liaison du cytosquelette d’actine à la membrane plasmique, en interagissant avec
des protéines d’ancrage comme la vinculine, la taline ou la filamine. Cette liaison permet
l’interaction de ces protéines avec des glycoprotéines de surface ce qui augmente l’énergie
d’adhésion entre le cytosquelette d’actine et la membrane plasmique (Toker, 1998). Le
PtdIns(4,5)P2 contrôle aussi le trafic vésiculaire, plus particulièrement au niveau de
l’endocytose ou de l’exocytose. Il peut ainsi recruter et séquestrer à la membrane plasmique,
près des sites potentiels d’endocytose, des protéines comme AP2, AP180 ou l’epsine (Ford,
2001 ; Itoh, 2001).
Enfin, il peut servir de substrat aux PtdIns(4,5)P2 4-phosphatases pour générer du PtdIns5P.
Les premières phosphatases ayant cette fonction ont été caractérisées chez les bactéries, IpgD
(Shigella flexneri), SigD/SopB (Salmonella typhimurium) sont des 4-phosphatases qui
synthétisent du PtdIns5P aussi bien in vitro qu’in vivo (Niebuhr, 2002 ; Marcus, 2001, Mason,
2007). Par contre ce n’est que très récemment que l’existence d’un tel type de phosphatase a
été mise en évidence chez les mammifères. Ces PtdIns(4,5)P2 4-phosphatases de type I et II
présentent une homologie avec IpgD uniquement restreinte au site catalytique (Ungewickell,
2005).
46

Introduction
Figure 18 : Rôle du PtdIns(4,5)P2 dans le remodelage de l’actine, la localisation membranaire de ses protéines effectrices et le trafic vésiculaire . PLD : phospholipase D ; GAPs : GTPase Activating Proteins. D’après Astle, 2007.
3.2. Le PtdIns(3,4)P2
Ce PI, provenant de l’hydrolyse du PtdIns(3,4,5)P3 par l’inositol 5-phosphatase SHIP,
représente environ 1 à 10% du PtdIns(4,5)P2 total et est localisé à la membrane plasmique de
façon rapide et transitoire en réponse à des stimulations. Différentes études suggèrent que le
PtdIns(3,4)P2 fonctionne comme un second messager lipidique. Tout comme le
PtdIns(3,4,5)P3, le PtdIns(3,4)P2 est capable de recruter par leur domaine PH les
sérine/thréonine kinases Akt et PDK1, l’impliquant ainsi dans des processus biologiques tels
que la survie, la prolifération ou le métabolisme du glucose. De plus, des membres de la
famille des PKC (PKC ζ/ε/λ) peuvent être activés directement par le PtdIns(3,4)P2 in vitro et
in vivo (Toker, 1994). Ainsi, ce PI augmenté en réponse à des stimuli extracellulaires,
servirait de second messager pour activer les PKC et contrôler les taux de calcium
intracellulaires. Des études complémentaires semblent nécessaires pour mieux comprendre les
rôles de ce lipide.
47

Introduction
3.3. Le PtdIns(3,5)P2
Le PtdIns(3,5)P2 est le plus récent des phosphatidylinositol bisphosphates identifié et
représente moins de 0,1% des PIs totaux. A ce jour, il a pu être essentiellement impliqué dans
le trafic vésiculaire et dans le recyclage des protéines des corps multivésiculaires et des
vésicules des compartiments vacuolaires ou lysosomaux (Dove, 2004 ; Michell, 2006). En
effet, la déplétion de ce PI dans plusieurs types cellulaires engendre une vacuolation anormale
de vésicules cytoplasmiques (Jeffries, 2004). Le métabolisme du PtdIns(3,5)P2 est
relativement bien caractérisé chez la levure, c’est la résultante de l’action consécutive de la PI
3-kinase de type III, ou Vps34 (S. cerevisiae) et d’une PtdIns 5-kinase de type III, Fab1p
(Odorizzi, 2000 ; Gary, 1998). Concernant sa métabolisation la voie majeure de dégradation
fait intervenir, la 5-phosphatase Fig4 qui est capable in vitro de synthétiser du PtdIns3P
(Onishi, 2003 ; Gary, 2002). Aucune étude n’a pu prouver à ce jour l’existence de PtdIns5P
dans la levure ce qui exclut un rôle de 3-phosphatase comme les MTMs. Sac3, l’homologue
de Fig4 chez les mammifères, a été récemment impliqué dans un syndrome neurodégénératif
de type Charcot-Marie-Tooth en régulant, comme Fig4, le taux de PtdIns(3,5)P2 (Chow,
2007) (Cf. Chap. III). (Figure 19). Chez les mammifères, on retrouve également ces voies de
métabolisation du PtdIns(3,5)P2 qui font intervenir hVps34 (Vps34p), PIKfyve (Fab1p) et
Sac3 (Fig4p).
Figure 19: Voie de synthèse et de dégradation du PtdIns(3,5)P2 chez S. cerevisiae. En bleu sont représentés les phosphoinositides conduisant à la synthèse de PtdIns(3,5)P2, en rouge les phosphoinositide kinases, en vert les phosphoinositide phosphatases. D’après Michell, 2006.
De plus, il semble que le taux de PtdIns(3,5)P2 soit aussi contrôlé par les 3-phosphatases de la
famille des MTMs qui vont produire du PtdIns5P. Les MTMs sont une large famille de plus
de quatorze membres capables de déphosphoryler le PtdIns3P et le PtdIns(3,5)P2 pour générer
du PtdIns et du PtdIns5P respectivement (Pendaries, 2003 ; Bertini, 2004 ; Wishart, 2002,
Tronchère, 2003). Ces enzymes ont été impliquées dans différentes maladies génétiques à la
suite de la mise en évidence de mutations sur MTM1 conduisant à la myopathie myotubulaire,
ou encore des mutations de MTMR2 et MTMR13/SBF2 qui entraînent une neuropathie de
48

Introduction
type Charcot-Marie-Tooth 4B1 et 4B2 (Bertini, 2004 ; Wishart, 2002 ; Bolino, 2004). (Détails
Cf. Chap. II.IV.). Un des arguments majeur conduisant à l’hypothèse de régulation du taux de
PtdIns(3,5)P2 par cette famille d’enzyme vient de l’observation par notre groupe de
l’accumulation de PtdIns5P in vivo dans des myotubes qui surexpriment la phosphatase
MTM1 (Tronchère, 2004). Actuellement, aucun groupe n’a pu déterminer quel était le
substrat préférentiel de cette famille d’enzyme in vivo. Plusieurs effecteurs du PtdIns(3,5)P2
ont été identifiés. Chez la levure, c’est le cas des epsines (Ent3p et Ent5p) deux protéines qui
contiennent un domaine ENTH (Epsin NH2-Terminal homology) qui lie le PtdIns(3,5)P2. Ces
protéines sont retrouvées dans les structures vacuolaires et contrôleraient le trafic dépendant
du PtdIns(3,5)P2 des vacuoles vers les corps multivésiculaires (Friant, 2003 ; Eugster, 2004).
Un composant de la machinerie ESCRT-III, Vps24p serait aussi un effecteur du
PtdIns(3,5)P2. Cette protéine appartient à la famille des CHMPs (Charged Multivesicular
Body Protein) et régule le trafic membranaire des corps multivésiculaires vers les vacuoles
(Whitley, 2003). Récemment, une autre famille de protéines liant le PtdIns(3,5)P2 a été
caractérisée, c’est la famille des PROPPINs, qui inclut la protéine Atg18p qui joue un rôle
majeur dans le trafic membranaire des vacuoles vers les corps multivésiculaires (Dove, 2004,
Efe, 2007) et l’autophagie. Cette famille de protéines est détaillée au Chapitre II.III. D’autres
protéines ont été décrites pour lier ce PI mais sans rôle biologique défini, ce sont : la α-
tocopherol-binding protein ATTP (Krugmann, 2002), deux protéines contenant le domaine
Sec14 chez les plantes (Kearns, 1998), la SNX1 (Cozier, 2002) et une protéine du trafic,
Ivy1p (Lazar, 2002) (Figure 20).
Figure 20: Effecteurs et fonctions potentielles du PtdIns(3,5)P2. MVB : Multivesicular Bodies. D’après Michell, 2006.
49

Introduction
4. Les phosphatidylinositol monophosphates
4.1. Le PtdIns4P Ce PI a longtemps été considéré comme un simple précurseur des autres PIs. Il est présent à
un taux relativement constant (10% des PIs totaux) dans la cellule. Cependant, des données
obtenues chez la levure suggèrent un rôle essentiel du PtdIns4P dans la régulation de la
sécrétion constitutive du réseau trans-Golgien (Hama, 1999 ; Walch-Solimena, 1999). Chez
les mammifères, de nombreux travaux ont montré un rôle du PtdIns4P dans la maintenance
du réseau trans-Golgien. En effet, ce PI permet la relocalisation de protéines associées au
Golgi telles que l’EpsinR, l’OSBP (Oxysterol Binding Protein), l’adaptateur de la clathrine
AP-1 et FAPP (Four phosphate adaptator) 1 et 2 au niveau de cet organelle via des domaines
ENTH et PH qui lient le PtdIns4P. De plus, de récents travaux suggèrent que l’interaction
impliquant le PtdIns4P et FAPP2 contrôle la voie de synthèse des glycosphingolipides, un
composant majeur de la membrane plasmique (De Matteis, 2007). Le mode d’action des
protéines du Golgi recrutées par le PtdIns4P reste mal connu, mais des données récentes
suggèrent que ces protéines sont capables de réguler la localisation et la fonction d’autres
protéines dans différents compartiments membranaires du réseau trans-Golgien (De Matteis,
2004 ; Downes, 2005) (Figure 21).
Figure 21: Distribution subcellulaire et implication du PtdIns3P et du PtdIns4P dans le trafic membranaire au niveau du complexe golgien et du système endosomal. D’après Downes, 2005.
50

Introduction
Le PtdIns4P pourrait également jouer un rôle dans la régulation du cytosquelette. En effet, il
est capable d’interagir avec des protéines du cytosquelette telle que la taline par un domaine
ENTH. Il a été montré que cette liaison induit un changement conformationnel de la taline, in
vitro (Martel, 2001). De plus, le PtdIns4P peut réguler la formation du complexe profiline-
actine in vitro mais sa contribution dans ce mécanisme in vivo reste mal connue (Katakami,
1992).
4.2. Le PtdIns3P Le PtdIns3P est constitutivement présent à des taux relativement constants (5 à 15% des
PtdIns monophosphates totaux), dans les cellules de mammifères, et localisé principalement
au niveau des endosomes précoces et au sein des corps multivésiculaires. Chez la levure, S.
cerevisiae, ce PI est le produit de la seule PI 3-kinase connue, Vps34 et joue un rôle critique
dans le recyclage des protéines du Golgi au système vacuolaire (Schu, 1993). En fait, Vps34
est recrutée au Golgi par la sérine/thréonine kinase Vps15p qui stimule alors l’activité lipide
kinase de Vps34. Chez les mammifères, seule la PI 3-kinase de type III est capable de
produire du PtdIns3P in vivo, et comme chez la levure, ce PI est important dans le contrôle du
trafic vésiculaire des organismes supérieurs. Ce PI peut recruter différentes protéines et
déterminer leur localisation via sa capacité à lier des domaines spécifiques (Cullen, 2001,
Downes, 2005). C’est le cas de la protéine EEA1, impliquée dans la fusion des endosomes
précoces qui en interagissant avec le PtdIns3P via son domaine FYVE (Fab1p, YoTB, Vps27p
et EEA1) se localise aux endosomes précoces. En plus de la liaison au PtdIns3P, une
interaction protéine/protéine avec la GTPase Rab5 est nécessaire pour stabiliser la liaison de
EEA1 aux endosomes (Gruenberg, 2004). Le PtdIns3P peut se lier à un autre domaine
protéique comme le domaine PX. Ainsi, il lie le domaine PX de la protéine p40phox et permet
la localisation de cette protéine sur les endosomes. Le taux de PtdIns3P peut être modulé par
différentes kinases et phosphatases. En effet, il est le précurseur du PtdIns(3,5)P2 via l’action
de la kinase PIKfyve (Cf. Chap. III) (Shisheva, 2001) et sert à la localisation de cette enzyme
au niveau des endosomes par sa liaison à son domaine FYVE (Ikonomov, 2001). Il est aussi le
substrat des membres de la famille des MTMs (Tronchère, 2003). L’étude de cette famille
d’enzymes chez plusieurs organismes a permis de mettre en évidence le rôle du PtdIns3P lors
des différentes étapes du trafic vésiculaires. Ainsi, chez la levure, il jouerait un rôle dans
l’homéostasie vésiculaire (Blondeau, 2000) et chez C. elegans, son rôle serait plutôt dans
l’endocytose (Dang, 2004). De plus, des travaux récents suggèrent l’apparition de plusieurs
51

Introduction
pools de PtdIns3P qui seraient néosynthétisés sous activation, en plus du pool constitutif de
PtdIns3P associé aux endosomes. Ainsi, le pathogène humain Mycobacterium tuberculosis
pourrait détourner les voies classiques de maturation des phagosomes en modifiant les taux de
PtdIns3P sur ces corps vésiculaires (Chua, 2004). Des synthèses d’autres pools de PtdIns3P
ont pu être décrites dans des plaquettes sanguines humaines après engagement des intégrines,
ou dans des cellules HeLa et Cos7 après stimulation au LPA (Banfic, 1998 ; Razzini, 2000).
Enfin, la présence de ce PI dans les microdomaines membranaires, « rafts », de cellules
stimulées à l’insuline suggère un rôle du PtdIns3P en tant que second messager (Maffucci,
2003).
4.3. Le PtdIns5P C’est le PtdIns monophosphates découvert le plus tardivement (Rameh, 1997a). Il est présent
dans les cellules de mammifères aussi bien que chez les plantes où il représente une petite
proportion des PIs monophosphates (environ 10%). Il est resté longtemps ignoré du fait de
son faible taux basal dans les cellules (Rameh, 1997b ; Tolias, 1998) et de la difficulté de le
séparer du PtdIns4P. Par des techniques appropriées d’HPLC et une technique de dosage en
masse, il est maintenant possible de quantifier ce lipide (Rameh, 1997a ; Morris, 2000). Le
taux de PtdIns5P peut augmenter sous une stimulation à la thrombine dans des plaquettes
sanguines (Morris, 2000), ou lors d’un choc osmotique dans des cellules de mammifères ou
de plantes (Meijer, 2001, Sbrissa, 2002, Tronchère, 2004, Clarke, 2001). De plus, un pool de
PtdIns5P a été détecté dans le noyau (Clarke, 2001) ou il contrôlerait les réponses au stress
(Bunce, 2007, Jones, 2006). Le premier domaine capable d’interagir avec ce lipide, appelé
PHD (Cf. Chap. II-III.) a été découvert dans une protéine impliquée dans le remodelage de la
chromatine, ING2 (Gozani, 2003). De plus, différents pathogènes bactériens sont capables de
manipuler les niveaux de PtdIns5P dans les cellules qu’ils infectent (Nieburh, 2002) dans le
but de développer des stratégies pour accroitre leur capacité invasive. Enfin, les MTMs, une
famille de 3-phosphatase mutée dans des myopathies myotubulaires sont capables de produire
du PtdIns5P suggérant un rôle pour ce PI dans cette pathologie (Tronchère, 2004). Il semble
donc que ce PI joue différents rôles biologiques dans les cellules et, les connaissances
actuelles seront décrites en Chap.II.IV, ce lipide ayant fait l’objet des recherches lors de ma
thèse.
52

Introduction
III- Les domaines protéiques d’interaction avec les
phosphoinositides
De nombreuses protéines impliquées dans la signalisation possèdent des domaines spécifiques
de reconnaissance des PIs. Le nombre de protéines qui possèdent des domaines de liaisons
aux PIs ne cessent de s’accroitre. L’analyse structurale de ces protéines a permis de
caractériser divers domaines de liaisons présentant des spécificités et des affinités différentes.
Ces différents domaines ainsi que des exemples de protéines qui les possèdent sont
représentés dans la figure 22 (pour revue Lemmon, 2008).
Figure 22: Domaines de liaison des phosphoinositides. Les différents domaines protéiques sont représentés par des symboles de couleurs. Les triangles grisés pointent vers les phosphoinositides liés. Des exemples de protéines contenant des domaines de liaisons sont listés au dessus du domaine correspondant.
1. Les domaines PH
Le domaine PH (Pleckstrin Homology) comprend environ 120 acides aminés, a été décrit
initialement dans la pleckstrine, un substrat de la PKC. Chez l’homme plus de 360 protéines
aux fonctions diverses possèdent un domaine PH telles que des phospholipases (PLCγ, PLCδ,
PLD), des protéines impliquées dans le remodelage du cytosquelette (dynamine, cytohésine),
des protéines adaptatrices (Gab1), des protéines kinases (Akt, PDK1, Btk) et des régulateurs
de petites protéines G (Vav, ARNO, Tiam1) (Lemmon, 2000 ; Lemmon, 2002 ; Cozier,
2004). Sur un plan structural, ces domaines PH présentent une faible homologie de séquence
53

Introduction
mais une structure tertiaire remarquablement conservée (Figure 23). Ils sont constitués de
deux feuillets β orthogonaux antiparallèles composés respectivement de trois et quatre brins
reliés entre eux par des boucles, ainsi qu’une hélice α en position C-terminale. L’ensemble est
organisé de telle sorte que les deux feuillets β forment un « sandwich » fermé par l’hélice α.
Ce type de conformation a pu être retrouvé dans d’autres domaines protéiques, c’est le cas des
domaines PTBs ou des domaines EVH1 (Enabled/VASP Homology domain-1) connus pour
être des domaines d’interaction protéine-protéine. Il est important de noter que les domaines
PH sont aussi des domaines d’interaction protéine-protéine. En effet, le PH domaine de la
protéine β-arrestine, un adaptateur des récepteurs à sept segments transmembranaires (RCPG)
qui permet l’inhibition de la signalisation engendrée par ces RCPG, a été montré pour se lier à
la sous-unité βγ des protéines G hétérotrimériques (Touhara, 1994).
L’interaction des domaines PH avec leur cible lipidique se fait via des acides aminés basiques
(arginine et lysine) qui sont portés par les boucles reliant les feuillets β. Le domaine PH ne
possède pas une spécificité unique pour un PI, ce qui peut s’expliquer par la variabilité et la
longueur selon les protéines (Lemmon, 2001). La plupart des domaines PH vont lier les PIs
de façon spécifiques par des interactions de types électrostatiques entre la face positive du
domaine PH et les phospholipides anioniques (Rameh, 1997a ; Kavran, 1998 ; Dowler, 2000 ;
Lemmon, 2000 ; Singh, 2003 ; Baumeister, 2003, Yu, 2004). In vivo, les différents groupes
proposent qu’il existe une oligomérisation des protéines à domaine PH ce qui permettrait
d’augmenter l’avidité dans l’interaction de ces domaines avec les PIs (Lemmon, 2000). Seul
10% des domaines PH fixent les PIs avec une forte affinité et spécificité. C’est le cas par
exemple du domaine PH de la PLCδ, de la spectrine et de la pleckstrine qui fixent le
PtdIns(4,5)P2 ou bien de ceux des protéines Btk, Tiam-1 qui eux sont spécifiques du
PtdIns(3,4,5)P3 ; et enfin le domaine PH d’Akt lie de façon très spécifique le PtdIns(3,4)P2 et
le PtdIns(3,4,5)P3. De plus, des travaux très récents ont pu montrer que le domaine PH du
facteur nucléaire TFIIH (General transcription factor IIH) serait capable de lier le PtdIns5P
(Di Lello, 2005).
54

Introduction
Figure 23: Structure du domaine PH de la pleckstrine. Les hélices α sont colorées en rouge et en jaune, les feuillets β sont représentés par des flèches bleu ciel. Les séquences de résidus chargés positivement impliquées dans la liaison des phosphoinositides sont en bleu foncé. D’après Itoh, 2002.
2. Les domaines GRAM-PH
Tous les membres de la famille des MTMs (Cf. Chap. II.IV.) contiennent en N-terminal un
domaine GRAM (Glucosyl transferase, Rab-like GTPase activator and myotubularins)
(Doerks, 2000) qui par des études de cristallographie s’est révélé être d’une grande
ressemblance avec les domaines PH, d’où sa dénomination de domaine GRAM-PH (Begley,
2003). Concernant sa spécificité de substrat, des études de liaison sur vésicules et de Fat Blot
ont montré que le domaine GRAM-PH de MTMR2 et MTM1 lie les PIs et plus
particulièrement le PtdIns5P et le PtdIns(3,5)P2 (Tsujita, 2004, Lorenzo, 2005). Ces mêmes
auteurs montrent que ce domaine semble de plus réguler l’activité catalytique des MTMs. En
effet, une mutation de MTMR3 dans le domaine GRAM-PH entraîne une diminution de
l’activité catalytique de la protéine (Lorenzo, 2005). Cette régulation pourrait être un
mécanisme général de régulation des MTMs puisque le même phénomène est observé avec
MTM1, une autre MTMs (Schaletzky, 2003). Enfin, comme beaucoup de domaine de liaison
aux PIs, ce domaine GRAM-PH semble avoir la capacité de lier des protéines. C’est le cas de
MTMR6 qui semble lier les PIs avec une faible affinité mais pourrait lier des protéines qui
influenceraient sa régulation (Choudhury, 2006).
55

Introduction
3. Les domaines PX
Le domaine PX (Phox Homology) est un module protéique de 100 à 140 acides aminés,
identifié initialement dans les sous-unités p40phox et p47phox du complexe NADPH oxydase
des phagocytes. Actuellement, 47 protéines contenant un domaine PX ont pu être identifiées
chez l’homme et 15 chez la levure. Ces diverses protéines ont souvent des fonctions variées et
comprennent par exemple : les PI 3-kinases de classe II (Sato, 2001), des protéines impliquées
dans le trafic membranaire telles que les SNXs (Sorting Nexins) (Simonsen, 2001) ou bien
encore la PLD. Comme pour les domaines PH, la cristallographie du domaine PX a révélé que
leur structure tertiaire est très conservée. Il est constitué de trois brins β et quatre hélices α
(Figure 24) (Hiroaki, 2001). Deux des hélices α sont liées par une boucle riche en proline,
chacune de ces hélices portent un résidu conservé à savoir une phénylalanine et une arginine
qui sont positionnées l’une en face de l’autre. Chez la levure, les 15 protéines contenant ce
domaine PX lient uniquement le PtdIns3P. Lors d’une étude par résonance magnétique
nucléaire (RMN), l’équipe du Dr. Lemmon a pu montrer que seule 4 des protéines contenant
un domaine PX lient le PtdIns3P avec une grande affinité (KD de l’ordre du µM), les autres
lient le PtdIns3P mais avec une moindre affinité (Yu, 2001). De façon intéressante, les auteurs
montrent que ces protéines à domaines PX « de haute affinité » n’ont pas de partenaires
protéiques alors que les protéines à domaines PX « à faible affinité » sont connues pour
s’associer en complexe macromoléculaire ce qui augmenterait leur avidité de substrat pour le
PtdIns3P (Yu, 2001).
Figure 24: Structure du domaine PX de p47phox. Les hélices α sont colorées en rouge et en jaune, les feuillets β sont représentés par des flèches en bleu ciel. Les résidus chargés positivement impliqués dans la liaison des phosphoinositides sont en bleu foncé, les régions hydrophobes impliquées dans l’ancrage à la bicouche lipidique sont en bordeaux. D’après Itoh, 2002.
56

Introduction
L’hétéro- et/ou oligomérisation de ces protéines à domaine PX de « faible affinité » semble se
faire par les domaines coiled-coiled contenus dans ces protéines. C’est le cas chez la levure du
complexe Vps17p-Vps5p (Seaman, 2002). Cette interaction peut aussi se faire par d’autres
domaines, ainsi pour la SNX1 c’est le domaine BAR (Bin, Amphiphysin and Rvs 161/167),
permettant une meilleure liaison aux membranes courbées, qui va permettre la dimérisation
entre deux molécules de SNX1. Cette oligomérisation semble requise pour permettre la
liaison de haute affinité entre la SNX1 et les membranes contenant le PtdIns3P (Zhong, 2005 ;
Zhong, 2002). Chez les mammifères, il existe une diversité de spécificité de liaison pour un
PI. En effet, le domaine PX de p47phox lie le PtdIns(3,4)P2 alors que celui de p40phox interagit
préférentiellement avec le PtdIns3P. Mais ces domaines peuvent lier aussi le PtdIns(4,5)P2,
c’est le cas du domaine PX de la PI 3-kinase de classe II C2α (Song, 2001). Enfin, des
travaux récents ont pu mettre en évidence que le domaine PX de la PLD1 était capable de lier
le PtdIns5P (Du, 2003). De plus, comme le domaine PH, ce domaine est capable de lier des
protéines. En effet, le domaine PX de certaines de ces protéines contient des séquences riches
en proline, PxxP, capables d’interagir avec les domaines de liaison protéiques SH3 (Src
Homology 3). C’est le cas de p47phox qui par une liaison entre les motifs PxxP de son domaine
PX et SH3 qu’elle contient va se replier, changer sa conformation et entrer ainsi dans un état
inactif (Hiroaki, 2001). Des études plus récentes ont montré que ce même domaine est
capable d’interagir avec la moésine, une protéine qui lie les fibres d’actine à la membrane
plasmique, permettant ainsi la localisation de p47phox à la membrane (Wientjes, 2001 ; Zhan,
2004).
4. Les domaines FYVE
Le domaine FYVE comprenant 70 à 80 acides aminés interagit spécifiquement avec le
PtdIns3P (Stenmark, 2002). Son nom est basé sur les initiales des quatre premières protéines
dans lesquelles ce domaine a été identifié : Fab1p, YOPB, Vps27p et EEA1. La structure
tridimensionnelle des domaines FYVE a été établie par RMN à partir de la protéine EEA1
humaine et par cristallographie aux rayons X à partir de Vps27 de levure (Misra, 1999 ;
Kutateladze, 2001) (Figure 25). Les domaines FYVE contiennent plusieurs boucles en N-
terminal, des feuillets β antiparallèles et une hélice α en C-terminal. Cette structure est
stabilisée par deux régions de liaison au zinc formées par huit cystéines, et le site de liaison du
PtdIns3P est formé par des résidus basiques dans la séquence très conservée
57

Introduction
(R/K)(R/K)HHCR. De plus cette structure contient une large boucle hydrophobe qui permet
au domaine de pénétrer dans la bicouche lipidique (Misra, 1999).
Figure 25: Structure du domaine FYVE de EEA1. Les hélices α sont colorées en rouge et en jaune, les feuillets β sont représentés par des flèches en bleu ciel. Les résidus chargés positivement impliqués dans la liaison des phosphoinositides sont en bleu foncé, les régions hydrophobes impliquées dans l’ancrage à la bicouche lipidique sont en bordeaux. Les ions zincs sont indiqués par des sphères bleues. D’après Itoh, 2002.
La liaison du domaine FYVE avec le PtdIns3P se fait en trois étapes. La boucle hydrophobe
va rentrer en contact avec la membrane de façon non spécifique, entraînant une ouverture de
la poche basique qui lie alors la tête polaire du PtdIns3P (Dumas, 2001 ; Kutateladze, 2001).
Du fait de leur forte affinité pour le PtdIns3P, la plupart des protéines contenant un domaine
FYVE sont localisées au niveau des endosomes précoces et des vésicules internes des corps
multivésiculaires et sont impliquées dans le trafic membranaires vésiculaires (Ridley, 2001 ;
Gillooly, 2001). D’autres protéines possédant un domaine FYVE peuvent avoir des rôles dans
d’autres mécanismes biologiques comme la protéine SARA (Smad Anchor for Receptor
Activation), un adaptateur dans la transduction du signal par le récepteur au TGFβ. Ainsi,
SARA en réponse à une stimulation au TGFβ va permettre le recrutement de Smad2 sur le
récepteur au TGFβ et permettre l’activation de différentes voies de signalisation (Tsukazaki,
1998). Dans ce cas, c’est la capacité d’oligomérisation entre deux protéines à domaines
FYVE qui est mise en jeu et non la liaison au PtdIns3P.
58

Introduction
5. Les domaines FERM
Ce sont des domaines d’environ 300 acides aminés qui sont localisés en N-terminal des
protéines de la famille ERMs (Ezrin/Radixin/Moesin) impliquées dans les liens entre le
cytosquelette d’actine et les protéines adhésives de la membrane plasmique telles que CD44
et certaines ICAMs (Intracellular Adhesion Molecule) (Pearson, 2000 ; Bretscher, 2002). Ces
domaines vont lier préférentiellement le PtdIns(4,5)P2. L’étude cristallographique a permis de
mettre en évidence une organisation en trois sous-domaines (A, B et C) (Hamada, 2000 ;
Smith, 2002). Le sous-domaine A se compose d’une longue hélice α et d’un feuillet β à cinq
brins. Le sous-domaine B est composé de quatre longues hélices α et d’une cinquième plus
courte. Le sous-domaine C présente une structure homologue à celle d’un domaine PH avec
deux feuillets β organisés en « sandwich » de quatre et trois brins respectivement. Ainsi, c’est
entre les deux sous-domaines A et C au niveau d’une séquence basique que viendra se fixer la
tête polaire du PtdIns(4,5)P2 (Hamada, 2000) (Figure26). Cette liaison est moins affine que
celle observée entre le PtdIns(4,5)P2 et le domaine PH. A ce jour, la spécificité d’interaction
de ce domaine FERM avec les autres PIs n’a pas été étudiée en détail.
Figure 26: Structure du domaine FERM de la radixine. Les hélices α sont colorées en bleu, les feuillets β sont représentés en rouge. Les « linkers » entre les sous-domaines A-B et B-C sont colorés en vert et marron respectivement. D’après Hamada, 2000.
59

Introduction
6. Le domaine PHD
C’est un domaine en doigt de zinc très conservé qui est présent dans de nombreuses protéines
pour la plupart nucléaires chez les eucaryotes (Aasland, 1995). Ce domaine est retrouvé dans
des facteurs impliqués dans le remodelage de la chromatine comme l’acétyltransférase
CBP/p300, la protéine ACF et dans les protéines suppresseurs de tumeurs de la famille ING
(Feng, 2002 ; Fyodorov, 2002, Kalkhoven, 2002). La résolution de la structure par RMN du
domaine PHD de la protéine WSTF (Williams-Beuren syndrome transcription factor) a révélé
que ce domaine est structuralement proche du domaine FYVE et RING (Misra, 2001 ;
Pascual, 2000) (Figure27). Du fait de leur similitude de structure, il est quelquefois difficile
de différencier les domaines RING des domaines PHD (Aasland, 1995). Cette ressemblance,
a dans un premier temps, suggéré que comme le domaine RING, le domaine PHD était un
domaine d’interaction ADN/protéine ou bien protéine/protéine et donc liait, comme le
domaine RING, la famille des ubiquitine ligase E2 (Aasland, 1995 ; Weissman, 2001).
De façon surprenante, bien que liant des protéines, l’équipe du Dr. Gozani a pu montrer que le
domaine PHD de la protéine ING2 était capable de lier les PIs monophosphates et plus
particulièrement le PtdIns5P (Gozani, 2003). Cependant, tous les domaines PHD ne semblent
pas lier les PIs et le PtdIns5P. Le domaine PHD de la protéine ING2 possède trois régions
distinctes, riches en résidus basiques qui semblent participer à la liaison des PIs en
interagissant avec leurs têtes polaires acides.
Figure 27: Superposition des structures du domaine PHD d’ING2 (en rose) et du domaine FYVE de EEA1 (en bleu ciel) complexés à l’inositol (1,3)-bisphosphate (en gris foncé). Les deux domaines partagent la même structure formée par deux sphères de coordination des ions Zn2+ (ronds) reliés par deux brins β (flèches). Résidus du PHDING2 de droite à gauche : en rouge, résidus lysines K49, K51, K53 ; en vert : résidus K58, R60. Résidus du FYVEEEA1, en bleu foncé, de droite à gauche : arginines R1366, R1370, R1374, R1399. D’après Gozani, 2003.
60

Introduction
L’alignement structural de ce module avec le domaine FYVE a révélé l’existence de trois
lysines K49, K51 et K53 qui forment un groupe de charges positives à la surface du PHD de
ING2 (Gozani, 2003). Ces lysines, comme dans le domaine FYVE, recouvrent de façon
étroite quatre arginines qui forment une partie du site de liaison des PIs (Kutateladze, 2001).
Deux autres séries de résidus basiques (K58 et R60) sont localisées sur une autre face du
domaine PHD de ING2. Un modèle d’association déterminé par modélisation informatique
entre le PtdIns5P et la protéine ING2 est proposé en figure 28. In vitro, le domaine PHDING2
lie le PtdIns5P mais pas seulement puisqu’il présente une certaine affinité pour les autres
phosphatidylinositol monophosphates. Ainsi, par des expériences de Fat Blot ou de RMN, les
auteurs ont pu montrer que le PHDING2 lie le PtdIns3P avec une affinité équivalente à celle du
PtdIns5P et que ce domaine lie aussi le PtdIns4P mais de façon moins affine que le PtdIns3P
et le PtdIns5P (Gozani, 2003 ; Huang, 2007).
Figure 28: Liaison du PtdIns5P et de ses analogues à la surface de la protéine ING2. En rouge liaison avec diC18:1-PtdIns5P, en vert liaison avec l’analogue diC18:1-MP-PtdIns5P et en bleu liaison avec l’analogue diC18:1-PT-PtdIns5P. D’après Huang, 2007.
La même équipe a publié l’existence d’un autre domaine PHD dans la protéine RAG2
(Recombination Activating Gene 2). C’est une protéine responsable de la recombinaison
V(D)J des immunoglobulines et du récepteur des lymphocytes T (TCR) au cours du
développement du système immunitaire (Elkin, 2005). Comme ING2, RAG2 interagit avec la
chromatine et peut lier les monophosphates mais aussi le PtdIns(4,5)P2 contrairement au
domaine PHD d’ING2. Enfin, une autre équipe a montré que chez les plantes la protéine
ATX1 (Arabidopsis homologue of trithorax) possède un domaine PHD qui lie aussi le
PtdIns5P (Alvarez-Venegas, 2006). Les PIs, de part leur liaison à ces protéines, semblent
61

Introduction
donc impliqués dans la régulation de la transcription génique et le remodelage de la
chromatine.
7. Les PROPPINs
Les PROPPINs (β-Propellers that bind polyphosphoinositides) représentent un nouveau
groupe de protéines qui lient le PtdIns(3,5)P2 avec une grande spécificité et affinité. Les
PROPPINS contiennent un domaine β-propeller, c’est une structure en tonneau identifiée
dans la sous-unité β des protéines G. Les chefs de file de ces protéines ont été identifiés chez
S. cerevisiae (Dove, 2004). Par SPR (surface plasma resonance), l’équipe du Dr. Dove a
identifié trois protéines contenant ce domaine, Atg18p, Atg21p et Hsv2p, chez la levure
(Dove, 2004).
Ces protéines contrôlent chez cet organisme la survie cellulaire et différentes étapes du trafic
vésiculaire. Elles sont caractérisées par une partie centrale d’environ 75 résidus conservés qui
contient le motif E/QxRRG. Les alignements de séquence ont pu prédire que ces protéines
contenaient deux à trois domaines WD40 et que ce domaine était constitué de sept hélices
β formant la structure en β-propeller (Figure 29). La position de ces hélices β serait variable
selon les PROPPINs étudiés. Ces protéines lient donc spécifiquement le PtdIns(3,5)P2 et
semblent impliquées dans les mécanismes de transport vésiculaire (Dove, 2004). Par
alignement de séquence, des homologues de mammifères ont pu être identifiés. Des travaux
réalisés sur la protéine humaine WIPI49 semblent indiquer que le domaine PROPPINs qu’elle
contient, lie de façon spécifique aussi bien le PtdIns(3,5)P2 que le PtdIns3P (Jeffries, 2004). Il
semble donc que la sélectivité de ce domaine pour le PtdIns(3,5)P2 n’existe que dans les
protéines issues d’organismes inférieurs.
62

Introduction
Figure 29: Diagramme représentant une hélice β-Propeller. Exemple de C-terminal (en rouge) du domaine WD40 de la protéine la partie Tup1, un corépresseur transcriptionnel chez la levure qui présente 7 feuillets β formant le domaine en hélice.
8. Le domaine ENTH
C’est un motif conservé d’environ 140 acides aminés, identifié en premier lieu dans l’epsine,
une protéine qui fixe l’adaptateur de la clathrine AP-2 (Kay, 1999). Les domaines ENTH sont
constitués de huit hélices α connectées par des boucles variables (De Camilli, 2002) (Figure
30). Des études de cristallographie ont montré que ce domaine est retrouvé essentiellement
dans les protéines impliquées dans l’endocytose à vésicules de clathrine (Itoh, 2002).
Figure 30: Structure du domaine ENTH de l’epsine. Les hélices α sont colorées en rouge et en jaune. Les résidus chargés positivement impliqués dans la liaison des phosphoinositides sont en bleu foncé. D’après Itoh, 2002. Ce domaine ne présente pas de spécificité exclusive pour un PI. En effet, des données
récentes montrent que le domaine ENTH est capable de lier in vivo le PtdIns(4,5)P2 (Itoh,
2001 ; Stahelin, 2003). Par contre, le domaine ENTH des protéines de levure Ent3p et Ent5p
se lient au PtdIns(3,5)P2 (Friant, 2003). Enfin, d’autres études ont montré que le domaine
63

Introduction
ENTH de l’EpsinR, un nouveau membre de la famille des epsines impliqué dans la formation
des vésicules de clathrine via l’adaptateur AP-1, lie le PtdIns4P (Mills, 2003 ; Hirst, 2003).
9. Le domaine ANTH
Ce domaine très conservé présente une grande similitude avec le domaine ENTH. Il a été
identifié en premier lieu chez la protéine AP180 (Adaptor protein 180), une protéine du
cerveau qui permet le recyclage des vésicules synaptiques (Ford, 2002 ; Morgan, 2000), mais
il est présent dans d’autres protéines ayant un rôle dans l’endocytose à vésicules de clathrine
comme CALM (Clathrin Assembly lymphoid Leukemia protein) ou Hip1 (Huntington
interacting protein 1).
Comme le domaine ENTH, ce module est capable de lier les PIs et en particulier le
PtdIns(4,5)P2 (Itoh, 2001). Des études de résonance magnétique nucléaire ont pu mettre en
évidence que la différence majeure entre les domaines ENTH et ANTH vient de leur mode de
liaison au PtdIns(4,5)P2. En effet, le domaine ANTH lie le PtdIns(4,5)P2 par ses hélices 1 et 2
et la boucle qui relie ces deux hélices. Par contre, la liaison du domaine ENTH au
PtdIns(4,5)P2 implique les hélices 1, 3, 4 et la boucle reliant les hélices 1 à 2. De plus, des
études récentes ont montré que le domaine ENTH de l’epsine 1 posséde une neuvième hélice,
nommée helice α0 qui permet la stabilisation de la liaison entre le PtdIns(4,5)P2 et le domaine
ENTH et donc une meilleure insertion dans la bicouche membranaire de l’epsine 1. Les
auteurs proposent que ce mécanisme permettrait de diminuer la quantité d’énergie nécessaire
à la courbure de la membrane lors de la formation de vésicules de clathrine pendant
l’endocytose (Ford, 2002 ; pour revue Legendre-Guillemin, 2004).
10. Les séquences basiques et hydrophobes
Un grand nombre de protéines possèdent des motifs riches en résidus basiques qui sont
capables de lier les PIs. Le motif consensus de liaison est KxxxKxKK où x peut varier de trois
à six acides aminés entre les deux premiers résidus basiques (K). Ce type de motif se retrouve
dans des protéines impliquées dans la régulation et la liaison au cytosquelette d’actine,
régulées par le PtdIns(4,5)P2 telles que la gelsoline, la cofiline, la profiline (Jammey, 1987 ;
Yu, 1992 ; Ojala, 2001). Mais, il peut également être présent dans des phospholipases
spécifiques des PIs comme la PLCβ2 ou PTEN (Simoes, 1995 ; Iijima, 2004). Ces séquences
64

Introduction
sont aussi capables de lier le PtdIns(3,4,5)P3 comme dans le cas de la protéine DOCK180 à
qui un tel motif permettrait l’activation de Rac (Missy, 1998, Kobayashi, 2001).
11. Les autres domaines de liaison
La reconnaissance des PIs par les domaines décrit précédemment semble bien admise. Mais il
existe d’autres motifs dont la liaison aux PIs semble être des exceptions. C’est le cas des
domaines C2 et SH2 qui vont lier les PIs avec une faible affinité. Ces liaisons se feraient par
le biais des surfaces chargées négativement et seraient importantes dans le recrutement de
protéines à la membrane plasmique. Il existe aussi des domaines BAR qui lient
préférentiellement les membranes courbes et régulent la réorganisation du cytosquelette
d’actine et l’endocytose. De façon intéressante, ce domaine est capable de lier la
synaptojanine, une PtdIns phosphatase suggérant ainsi une possible interaction entre ce
domaine et les PIs.
IV- Le PtdIns5P
1. Voies de synthèse et de dégradation
Figure 31 : Voie de synthèse du PtdIns5P. En noir sont représentés les phosphoinositides impliqués dans la synthèse du PtdIns5P, en vert les enzymes impliquées dans la synthèse de ce PIs.
65

Introduction
1.1. Synthèse à partir du PtdIns(4,5)P2
1.1.1. Les enzymes bactériennes
1.1.1.1. IpgD
Certains microbes pathogènes ont développé des stratégies pour pénétrer et survivre dans les
cellules hôtes qu’ils infectent. C’est le cas de la bactérie pathogène intracellulaire Shigella
flexneri responsable de la dysenterie chez l’homme. Cette bactérie a développé des
mécanismes lui permettant d’échapper aux voies classiques de dégradation et d’éviter les
vacuoles d’internalisation, permettant sa multiplication dans l’hôte infecté. Shigella flexneri
utilise un appareil de sécrétion de type III pour injecter dans la cellule hôte plusieurs facteurs
de virulence dont IpgD (invasion plasmid gene D) et promouvoir l’infection par des
mécanismes liés à la macropinocytose (Cossart, 2004). L’analyse de la séquence ne montre
pas d’homologie avec des protéines de mammifères sauf un motif CX5R retrouvé dans le site
actif de plusieurs lipide phosphatases comme MTM et PTEN (Norris, 1998, Niebuhr, 2002).
Divers travaux menés par notre équipe ont pu montrer le rôle d’IpgD en tant que lipide
phosphatase. Des tests d’activité in vitro et in vivo ont montré qu’IpgD est en effet capable
d’hydrolyser plusieurs PIs avec une forte préférence pour le PtdIns(4,5)P2 qu’elle transforme
en PtdIns5P (Niebuhr, 2002). Lors de l’infection bactérienne par S. flexneri, IpgD va entraîner
une hydrolyse massive du PtdIns(4,5)P2 membranaire (environ 35% du PtdIns(4,5)P2 total) et
donc une accumulation de PtdIns5P (Niebuhr, 2002). L’expression d’IpgD entraîne également
dans les cellules de mammifères une forte diminution des tensions membranaires générant
d’importants « ruffling » caractérisés par des invaginations de la membrane plasmique. Ces
remodelages semblent faciliter l’endocytose de la bactérie (Pizzaro-Cerda, 2004). Ces
données suggèrent que l’hydrolyse du PtdIns(4,5)P2, PI bien connu pour son rôle dans le
remodelage du cytosquelette d’actine, engendre une diminution locale de l’adhésion du
cytosquelette à la membrane plasmique. Enfin, nous avons pu montrer que le PtdIns5P
produit lors de l’injection d’IpgD est un puissant activateur de la voie de survie PI 3-
kinase/Akt. En effet, le PtdIns5P active une PI 3-kinase de classe IA, cette activation semble
indirecte et fait intervenir des tyrosines kinases (Pendaries, 2006). L’activation de cette voie
va permettre à la bactérie de maintenir la cellule hôte en vie le temps de se multiplier et de
promouvoir son invasion dans les cellules épithéliales voisines (Pendaries, 2006).
66

Introduction
1.1.1.2. SigD/SopB
Deux homologues d’IpgD portant les motifs présents dans le site actif des inositol
polyphosphate 4-phosphatases de mammifères ont pu être identifiés, chez différentes souches
de salmonelles SopB (Salmonella dublin) et SigD (Salmonella typhimuruim), responsables de
gastroentérites chez l’homme (Marcus, 2001). Tout comme IpgD, ces phosphatases sont
capables de moduler les tensions de la membrane plasmique et affectent le cytosquelette
d’actine afin de pénétrer dans la cellule hôte. SopB a été initialement démontrée comme
capable d’engendrer la déplétion des inositols, InsP6 et InsP5 au profit d’une accumulation
d’InsP4, un métabolite impliqué dans le contrôle des taux d’ions sodium et d’eau lors de
l’infection bactérienne (Norris, 1998). Une seconde étude montre qu’in vitro, SopB est
capable d’hydrolyser plusieurs substrats dont le PtdIns3P, le PtdIns(3,4)P2, le PtdIns(3,5)P2 et
le PtdIns(3,4,5)P3 (Marcus, 2001). In vivo, lors de l’infection par S. dublin, SopB peut induire
une diminution rapide de PtdIns(4,5)P2 au sommet des invaginations membranaires qui
deviennent alors des vésicules contenant les bactéries (Terebiznik, 2002). Le produit de
l’hydrolyse du PtdIns(4,5)P2 n’est, à ce jour, pas encore identifié mais les mécanismes décrits
rappellent fortement l’activité d’IpgD, suggérant que ces deux phosphatases pourraient
fonctionner de la même façon. Parallèlement, d’autres travaux ont pu montrer que SopB
exprimée de façon ectopique dans des cellules de mammifères est localisée sur des
endosomes précoces contenant du PtdIns3P et inhibe le recyclage du récepteur à l’EGF
(EGFR) entre les endosomes et les lysosomes (Dukes, 2006). Les auteurs proposent que le
PtdIns(3,5)P2 serait un des substrat physiologique de cette phosphatase in vivo, la localisation
de SopB sur des endosomes précoces serait le fruit de l’hydrolyse de ce PtdIns(3,5)P2. Enfin,
la perturbation de l’homéostasie vésiculaire classique serait pour la bactérie un mécanisme
d’échappement aux réponses immunitaires (Dukes, 2006). Il semble donc que SopB soit
capable de contrôler de nombreux mécanismes cellulaires du fait de sa capacité d’hydrolyse
de différents PIs et inositol, lui permettant de mettre en place des mécanismes efficaces
d’échappement.
Concernant, la phosphatase SigD, des travaux récents ont pu montrer que cette phosphatase
est capable de transformer le PtdIns(4,5)P2 membranaire en PtdIns5P (Mason, 2007). De la
même façon qu’IpgD, les auteurs montrent que la microinjection de SigD dans des cellules
épithéliales conduit à un profond réarrangement du cytosquelette d’actine, phénomène non
observé lors de l’utilisation d’une forme catalytiquement inactive de SigD (SigDC462S), et que
ce ruffling est bien du à la dégradation massive de PtdIns(4,5)P2 (Mason, 2007). De plus, les
67

Introduction
auteurs démontrent que la déplétion de PtdIns(4,5)P2 et donc probablement l’augmentation de
PtdIns5P induit une inhibition des échanges d’ions sodium et hydrogène dans les cellules
épithéliales. La perte de PtdIns(4,5)P2, et donc sans doute le PtdIns5P, inhibe les échanges
ioniques des cellules épithéliales, un phénomène clé dans les premières étapes de la dysenterie
(Mason, 2007).
1.1.2. Les enzymes eucaryotes : les PtdIns(4,5)P2 4-
phosphatases de type I et II
Récemment, le groupe du Dr. Majerus a pu mettre en évidence l’existence d’inositol 4-
phosphatases chez les mammifères capables de dégrader le PtdIns(4,5)P2 en PtdIns5P comme
le fait IpgD (Ungewickell, 2005). Cette recherche a été basée sur la présence du motif
conservé CX5R retrouvé chez de nombreuses lipides phosphatases (Ungewickell, 2005). Cette
étude à conduit à la découverte de deux phosphatases humaines, les PtdIns(4,5)P2 4-
phosphatases de type I et II. Ces enzymes ne présentent aucune homologie de séquence avec
la phosphatase bactérienne IpgD à part le site catalytique portant le motif conservé CX5R. Ces
enzymes, tout comme IpgD, sont capables de convertir le PtdIns(4,5)P2 en PtdIns5P in vitro
(Ungewickell, 2005). De façon concomitante, le taux de PtdIns(4,5)P2 est diminué d’environ
20% lors de l’expression de la 4-phosphatase de type I. Les auteurs montrent aussi que ces
enzymes sont exprimées de façon ubiquitaire et colocalisent avec LAMP1 et EEA1, deux
marqueurs du système endosomal et lysosomal dans les cellules (Ungewickell, 2005) et
contrôleraient l’homéostasie vésiculaire. Des travaux plus récents montrent que la 4-
phosphatase de type I contrôle l’apoptose induite lors d’un stress dans les cellules de
mammifères (Zou, 2007). Ainsi, les auteurs montrent que dans des cellules HeLa traitées avec
de l’étoposide ou de la doxorubicine, deux agents induisant un stress cellulaire, la fraction
vésiculaire de PtdIns(4,5)P2 4-phosphatase de type I va se relocaliser au noyau et permettre
une augmentation du taux de PtdIns5P nucléaire. L’implication du PtdIns5P dans l’apoptose
dépendante de p53 induite par des agents de stress avait déjà été montrée, et fait appel à la
protéine nucléaire ING2 (Gozani, 2003) (Cf. Chap II.III., Chap.II.IV). Les travaux du Dr. Zou
vont plus loin, puisque cette équipe démontre que l’élévation de PtdIns5P nucléaire par la 4-
phosphatase de type I permet d’augmenter l’acétylation et la stabilisation du suppresseur de
tumeur p53, permettant d’accroitre l’apoptose induite sous un stress génotoxique (Zou, 2007)
(Cf. Chap. II.IV.).
68

Introduction
1.2. Synthèse à partir du PtdIns(3,5)P2 : rôle des myotubularines (MTMs)
La famille des MTMs constitue un grand groupe de la superfamille des tysosine-phosphatases
à double spécificité (PTP/DSP) (Laporte, 2003 ; Tronchère, 2003). Les MTMs sont présentes
dans la plupart des organismes eucaryotes incluant les levures et les plantes et ces 3-
phosphatases ont pour spécificité d’hydrolyser le PtdIns3P et le PtdIns(3,5)P2 en PtdIns et
PtdIns5P respectivement (Tronchère, 2003 ; Tronchère, 2004). Cette famille de phosphatases
contient 14 gènes chez l’homme appelés MTM et MTM Related 1 à 13, plus deux
pseudogènes (Figure 32). MTM1 (myotubularin 1) est mutée dans la myopathie
myotubulaire, une pathologie génétique sévère liée au chromosome X (Laporte, 1996).
MTMR2 et MTMR13, deux autres membres de la famille des MTMs sont impliquées dans
des neuropathies de type Charcot-Marie-Tooth (CMT) 4B1 (CMT4B1) (Berger, 2002 ;
Bolino, 2000) et 4B2 (CMT4B2) (Senderek, 2003) respectivement, des neuropathies de type
démyelinisante autosomale récessive (Wishart et Dixon, 2002). Les membres de la famille
MTM présente un site catalytique conservés (Denu, 1996) mais de façon surprenante la moitié
de ces protéines présentent une mutation dans leur site consensus phosphatase CX5R et sont
donc catalytiquement inactives. Ces deux formes s’associent en dimère, les formes
catalytiquement inactives régulant les sous-unités catalytiquement actives (Mochizuki, 2003 ;
Kim, 2003 ; Robinson, 2005). En ce qui concerne leur organisation, ces protéines sont
constituées de plusieurs domaines et en particulier un domaine GRAM-PH (décrit au Chap.
II.III.), un domaine RID (Rac-Induced recruitement Domain), un domaine catalytique au sein
du motif PTP/DSP, des domaines coiled-coil et un domaine SID (SET-interacting domain).
Le domaine RID a été défini comme nécessaire au recrutement de MTM1 et de ses
homologues (MTMR2, MTMR3 et MTMR9) au niveau de ruffling membranaires induits par
la GTPase Rac (Laporte, 2002a ; Laporte, 2002b). Concernant le domaine coiled-coil, il est
positionné en C-terminal de presque tous les membres de cette famille et est impliqué dans la
dimérisation de deux MTMs (Kim, 2003, Mochizuki, 2003). Le domaine SID de MTMR5
peut interagir avec les protéines SET induisant la transformation oncogénique et la
prolifération des précurseurs des lymphocytes B (De Vivo, 1998). Enfin, certains membres de
la famille des MTMs possèdent des domaines additionnels de liaison à des protéines comme
le domaine DENN (Différenciellement Exprimé dans les cellules Néoplasiques versus
Normales) (MTMR5 et MTMR13), le domaine PDZ-BS (PSD-95/Dlg/ZO-1 binding site)
(MTM1 et MTMR5) (Fabre, 2000) ou des domaines de liaison aux PIs comme le domaine
69

Introduction
FYVE (MTMR3 et MTMR4) et le domaine PH (MTMR5) (Isakoff, 1998). Concernant son
domaine catalytique, notre équipe a pu montrer lors de tests phosphatases in vitro que ces 3-
phosphatases sont capables de métaboliser le PtdIns(3,5)P2 pour former du PtdIns5P
suggérant un rôle des ces enzymes dans le contrôle du taux de PtdIns5P in vivo (Tronchère,
2004). Ainsi, nous avons montré qu’in vivo la surexpression de MTM1 dans des cellules
Jurkat qui présentent un taux basal élévé de PtdIns(3,5)P2, conduit à une augmentation du
taux de PtdIns5P. Le taux de PtdIns(3,5)P2 est modulé par choc osmotique dans les levures et
dans les cellules de mammifères, nous avons analysé l’effet d’un tel traitement sur la
production du PtdIns5P par MTM1. Ainsi, nous avons utilisé des myoblastes L6 différenciés
en myotubes, qui ont été infectés par un adénovirus codant soit pour MTM1 soit pour une
forme catalytiquement inactive (MTM1 D278A). Nous avons observé lors d’un choc
osmotique (traitement au NaCl) une diminution d’environ 50% du taux de PtdIns5P dans les
cellules surexprimant le mutant catalytiquement inactif comparé aux myotubes surexprimant
la forme sauvage de MTM1 (Tronchère, 2004). Ainsi, notre équipe a pu montrer qu’une des
fonctions de MTM1 in vivo est de réguler la production de PtdIns5P à partir de PtdIns(3,5)P2.
Ces résultats suggèrent que dans les myopathies myotubulaires liées à la mutation de MTM1,
cette absence de transformation du PtdIns(3,5)P2 en PtdIns5P pourrait être un composant
important de l’étiologie de ce syndrome. Concernant les fonctions biologiques des MTMs, du
fait de son activité sur le PtdIns3P et le PtdIns(3,5)P2, il a été envisagé qu’elles pourraient
réguler l’homéostasie vacuolaire.
70
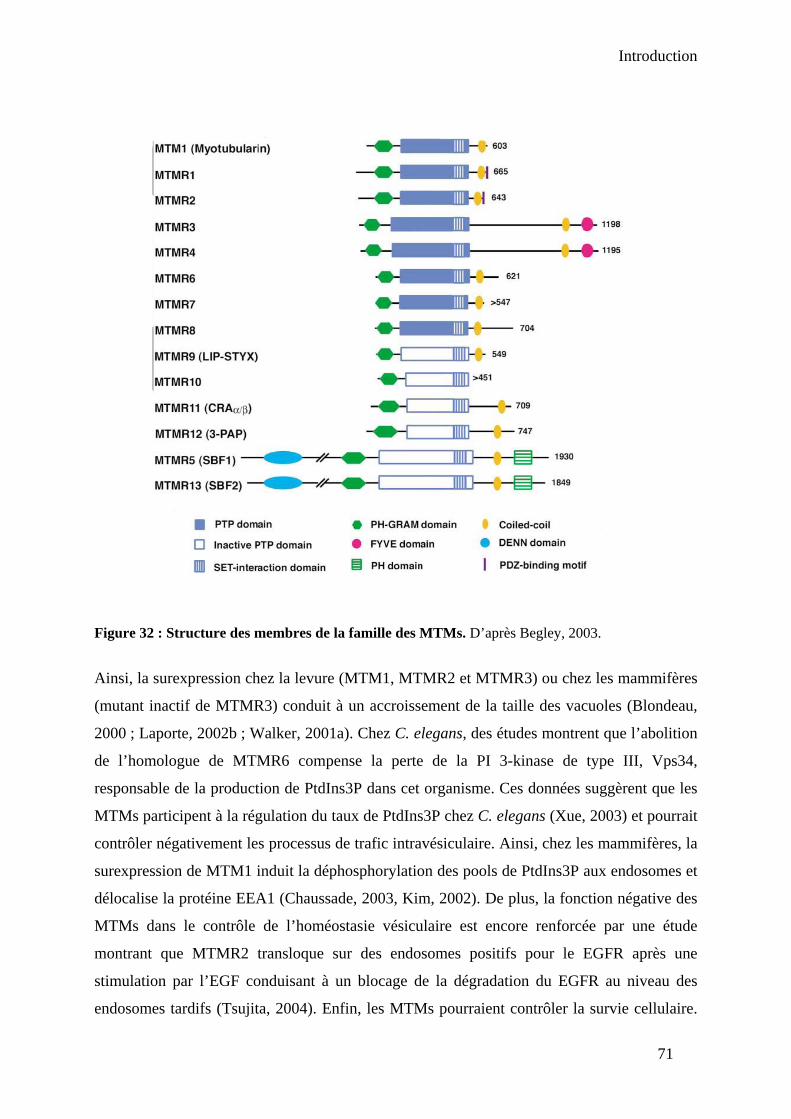
Introduction
Figure 32 : Structure des membres de la famille des MTMs. D’après Begley, 2003.
Ainsi, la surexpression chez la levure (MTM1, MTMR2 et MTMR3) ou chez les mammifères
(mutant inactif de MTMR3) conduit à un accroissement de la taille des vacuoles (Blondeau,
2000 ; Laporte, 2002b ; Walker, 2001a). Chez C. elegans, des études montrent que l’abolition
de l’homologue de MTMR6 compense la perte de la PI 3-kinase de type III, Vps34,
responsable de la production de PtdIns3P dans cet organisme. Ces données suggèrent que les
MTMs participent à la régulation du taux de PtdIns3P chez C. elegans (Xue, 2003) et pourrait
contrôler négativement les processus de trafic intravésiculaire. Ainsi, chez les mammifères, la
surexpression de MTM1 induit la déphosphorylation des pools de PtdIns3P aux endosomes et
délocalise la protéine EEA1 (Chaussade, 2003, Kim, 2002). De plus, la fonction négative des
MTMs dans le contrôle de l’homéostasie vésiculaire est encore renforcée par une étude
montrant que MTMR2 transloque sur des endosomes positifs pour le EGFR après une
stimulation par l’EGF conduisant à un blocage de la dégradation du EGFR au niveau des
endosomes tardifs (Tsujita, 2004). Enfin, les MTMs pourraient contrôler la survie cellulaire.
71

Introduction
En effet, une récente étude, par des techniques d’ARN interférence, qui visait à identifier des
kinases et des phosphatases régulant l’apoptose, a mis en évidence que cinq membres de la
famille des MTMs (MTMR1, MTMR6-8 et MTMR5) étaient des régulateurs potentiels de la
survie cellulaire (MacKeigan, 2005).
Il est intéressant de noter que des travaux récents placent le PtdIns5P en tant qu’activateur
allostérique de MTM1 et MTMR3. Ainsi, in vitro l’ajoût de PtdIns5P augmente l’activité
phosphatase de MTM1 et MTMR3 sur leurs deux substrats, le PtdIns3P et le PtdIns(3,5)P2.
Le PtdIns5P joue un rôle de régulateur allostérique en favorisant la conformation de MTM1
en multimère sous forme d’un anneau. Les auteurs proposent que le PtdIns5P généré par les
MTMs à partir de PtdIns(3,5)P2 pourrait constituer une boucle de rétrocontrôle positif
(Schaletzky, 2003). En conclusion, ces phosphatases semblent réguler de nombreux processus
biologiques incluant l’endocytose ou la survie cellulaire par le contrôle des taux de PtdIns3P
et PtdIns(3,5)P2 et donc de PtdIns5P.
1.3. Synthèse à partir du PtdIns : Rôle de PIKfyve PIKfyve est une PtdIns 5-kinase de type III, homologue de Fab1p chez la levure. Cette
enzyme est décrite pour produire de PtdIns(3,5)P2 in vitro et in vivo (Shisheva, 1999). In
vitro, mPIKfyve est capable de phosphoryler le PtdIns pour générer du PtdIns5P (Sbrissa,
1999). PIKfyve ayant fait l’objet de mes travaux de thèse, elle sera présentée dans le Chapitre
III.
1.4. Métabolisation du PtdIns5P par les PtdIns5P 4-kinases de type II
Le PtdIns5P peut être transformé en PtdIns(4,5)P2 par la famille des PtdIns5P 4-kinases de
type II (PIP4KII). C’est kinases sont présentes dans un grand nombre d’organismes
multicellulaires incluant C. elegans, D. melanogaster, les mammifères mais sont absentes
chez S. cerevisiae. Chez les mammifères, il existe trois isoformes de cette kinase, α, β, et γ
qui présentent des localisations différentes. La forme α est localisée dans le cytosol,
l’isoforme β est essentiellement nucléaire mais semble pouvoir aussi être cytosolique
(Ciruela, 2000 ; Richardson, 2007), et la forme γ a été retrouvée associée au réticulum
endoplasmique (Itoh, 1998). Concernant leur fonction biologique, un rôle du PtdIns5P et des
PIP4KII dans la signalisation nucléaire en réponse au stress et le métabolisme de l’insuline a
été proposé. Concernant son rôle nucléaire, l’équipe du Dr. Majerus a pu montrer que sous un
72

Introduction
stress génotoxique, la PIP4KIIβ va se localiser au noyau où elle sera phosphorylée sur son
résidu S326 par la MAPK impliquée dans les réponses au stress, p38 (Jones, 2006). Cette
phosphorylation va inhiber l’activité kinase de la PIP4KIIβ, qui ne synthétise plus de
PtdIns(4,5)P2, et engendrer une accumulation de PtdIns5P qui contrôlera ainsi l’apoptose p53
dépendante (Gozani, 2003). Très récemment, des travaux ont montré que cette enzyme
pouvait interagir avec la protéine SPOP (speckle-type POZ domaine protéine), une protéine
nucléaire associée aux speckles qui va permettre le recrutement des substrats de la ligase
Cul3. Ces travaux démontrent aussi que Cul3 va ubiquitiner la PIP4KIIβ et donc inhiber son
activité kinase (Bunce, 2008). De plus, d’autres travaux ont pu montrer que les PIP4KII α et
β peuvent être phosphorylées in vivo par la kinase PDK (protein kinase D) (Hinchliffe, 2006),
impliquée dans des fonctions biologiques telles que la division cellulaire ou l’apoptose (pour
revues Rykx, 2003 ; Rozengurt, 2005). Cette phosphorylation régule l’activité de ces kinases
mais le rôle physiologique de ce processus reste à élucider. Enfin, la PIP4KIIβ et le PtdIns5P
ont pu être impliqués dans l’activation d’Akt en réponse à l’insuline. En effet, des souris KO
pour cette kinase sont hypersensibles à l’insuline et présentent une suractivation de la kinase
Akt dans le foie et les muscles squelettiques (Lamia, 2004). De façon concomitante, le même
groupe montre que la surexpression de la PIP4KIIβ, induisant une chute du taux de PtdIns5P,
conduit à la diminution du taux de PtdIns(3,4,5)P3, et donc réduit l’activation d’Akt après une
stimulation à l’insuline (Carricaburu, 2003) suggérant que le PtdIns5P contrôlerait l’activation
d’Akt. Ainsi, le PtdIns5P semble être un régulateur de la voie PI 3-kinase/Akt comme nous
l’avons démontré lors de l’infection bactérienne par IpgD. Concernant les fonctions de la
forme α, une forme recombinante de cette kinase est maintenant utilisée pour mesurer le taux
de PtdIns5P dans des extraits cellulaires lors d’un dosage en masse de ce PI (Morris, 2000).
Des travaux très récents, impliquent cette kinase dans la production de PtdIns5P
extranucléaire en réponse à une stimulation par phosphorylation sur tyrosine (Wilcox, 2008).
En effet, les auteurs montrent que le traitement par du pervanadate, un inhibiteur des tyrosine
phosphatases qui permet donc d’augmenter les phosphorylations sur tyrosines, entraîne une
augmentation du taux de PtdIns5P intracellulaire. De plus, l’invalidation par une technique
d’ARN interférence des différentes formes des PIP4KII a permis de montrer que le PtdIns5P
produit dans ces conditions est du à la forme α (Wilcox, 2008). Enfin, l’implication de la
PIP4KIIγ dans la production de PtdIns5P dans un contexte cellulaire n’est, actuellement, pas
étudiée. En conclusion, la synthèse de PtdIns(4,5)P2 provenant du PtdIns5P par les PIP4KII
est sans doute mineure en comparaison des autres voies de production, c’est-à-dire la synthèse
73

Introduction
provenant du PtdIns4P par des PtdIns4P 5-kinases (Cf. Chap. II.I.), dans le contexte
cellulaire. Mais ces voies semblent finement réguler de nombreux processus biologiques.
1.5. Métabolisation par la phosphatase PLIP PLIP (PTEN-like phosphatase) été considérée un temps comme la seule phosphatase connue
pour hydrolyser spécifiquement le PtdIns5P (Merlot, 2003). C’est lors d’une recherche
génomique d’homologues de PTEN chez Dictyostelium que cette enzyme a été découverte
(Merlot, 2003), et par la suite des homologues procaryotes et eucaryotes ont été caractérisés
(Pagliarini, 2004). A part son site catalytique, PLIP possède peu d’homologie avec PTEN et
arbore un domaine transmembranaire en N-terminal, et son activité 5-phosphatase semble être
dirigée vers le PtdIns5P in vitro (Merlot, 2003). L’orthologue murin de PLIP présente la
même spécificité d’hydrolyse du PtdIns5P, cependant sa capacité à moduler le taux de
PtdIns5P in vivo n’est toujours pas prouvée (Pagliarini, 2004). Chez Dictyostelium, la
surexpression de PLIP conduit à sa localisation sur les membranes golgiennes suggérant la
présence d’un pool de PtdIns5P sur ces organelles. Chez le même organisme, l’abolition de
l’activité catalytique de PLIP conduit à un défaut d’aggrégation cellulaire, néanmoins aucun
lien entre la localisation Golgienne de cette enzyme et le phénotype observé n’a pu être établi
(Merlot, 2003). Jusque récemment, cette enzyme semblait donc être capable de réguler les
taux de PtdIns5P cellulaire mais des données de l’équipe du Dr. Dixon semblent désormais
prouver que PLIP doit être exclue du métabolisme du PtdIns5P. En effet, cette équipe montre
que cette enzyme rebaptisée PTPMT1 (PTP localized to Mitochondrion 1) est une
phosphatase localisée sur les membranes mitochondriales de cellules pancréatiques β
(Pagliarini, 2005). PTPMT1 régulerait la production d’ATP (Adénosine Tri-phosphate) dans
les mitochondries et la sécrétion d’insuline de ces cellules pancréatiques (Pagliarini, 2005).
Enfin, les auteurs montrent que l’abolition de cette phosphatase dans ce type cellulaire ne
conduit pas à un changement du taux de PtdIns5P suggérant donc que cette enzyme ne
contribue pas au métabolisme de ce PI in vivo (Pagliarini, 2005). Ainsi, malgré le fait que
PLIP/PTPMT1 soit capable d’utiliser le PtdIns5P in vitro ; in vivo, cette enzyme ne régule pas
le taux de PtdIns5P intracellulaire.
74

Introduction
2. Fonctions du PtdIns5P
2.1. Rôle dans l’invasion bactérienne – Potentialisateur de la voie de survie PI 3-kinase/Akt
Comme dit au Chapitre II.IV, plusieurs pathogènes microbiens exploitent le métabolisme des
PIs des cellules qu’ils infectent pour promouvoir leur entrée et la synthèse de facteurs de
virulence dans les cellules hôtes (Pizarro-Cerda, 2004). La formation des modules d’entrée de
Salmonella ou S. flexneri est la résultante d’une action concertée entre les protéines qu’injecte
la bactérie et les composants de la cellules hôte. Le facteur de virulence IpgD de S. flexneri est
injecté dans les cellules épithéliales intestinales et donc non phagocytaires par un appareil de
sécrétion de type III. Comme dit précédemment, cette phosphoinositide 4-phosphatase est
capable de convertir le PtdIns(4,5)P2 en PtdIns5P (Niebuhr, 2002). Cette transformation
rapide se situe au niveau des sites d’entrée de la bactérie (Pendaries, 2006). Tandis que cette
disparition rapide de PtdIns(4,5)P2 membranaire, un lipide connu pour son rôle dans la
dynamique du cytosquelette d’actine (Payrastre, 2001) (Cf. Chap. II.II.), conduit à un
réarrangement de la membrane plasmique et du cytosquelette (Niebuhr, 2002), le PtdIns5P
produit va activer des voies de signalisation spécifiques dans la cellule hôte. En effet, notre
équipe a récemment montré que le PtdIns5P joue un rôle clé dans l’activation de la PI 3-
kinase de classe IA et d’Akt dans la cellule hôte. Ce mécanisme est particulièrement
important dans la régulation de la survie de la cellule hôte par le pathogène. Ainsi, la bactérie
maintient la cellule hôte en vie dans le but de réaliser des réplications bactériennes et de
coloniser les cellules voisines (Pendaries, 2006). De plus, nous avons montré que l’expression
ectopique d’IpgD dans de nombreux types cellulaires mais non d’une forme catalytiquement
inactive, ou l’addition de PtdIns5P exogène à courte chaîne, était suffisante pour induire la
phosphorylation de la kinase Akt. Inversement, la baisse du taux de PtdIns5P ou sa
séquestration entraîne une forte diminution de la phosphorylation d’Akt dans des cellules
infectées ou exprimant IpgD. Ainsi, l’infection par S. flexneri met à jour un nouveau
mécanisme d’activation de la voie PI 3-kinase/Akt via la production de PtdIns5P qui va jouer
un important rôle dans la réponse de la cellule hôte comme la survie. Les mécanismes
moléculaires responsables de l’activation des PI 3-kinases de classe IA par le PtdIns5P
semblent impliquer des phosphorylations sur tyrosine et sont en cours de caractérisation par
notre groupe. Comme décrit au Chapitre II.IV. il existe des homologues bactériens d’IpgD
75

Introduction
que sont SopB chez S. dublin et SigD pour S. typhimurium (Marcus, 2001). Des données
récentes montrent que la bactérie S. typhimurium est capable d’induire une production de
PtdIns5P lors de l’infection bactérienne et ceci de façon dépendante de la présence de SigD
(Mason, 2007). Comme dans le cas d’IpgD, les auteurs montrent que le PtdIns5P provient de
l’hydrolyse du PtdIns(4,5)P2 et que les mécanismes de réarrangements membranaires sont
aussi activés lors de l’infection par S. typhimurium (Mason, 2007). La synthèse de PtdIns5P
semblerait donc requise pour l’activation d’Akt dans des cellules HeLa infectées par S.
typhimurium (Steele-Mortimer, 2000 ; Knodler, 2005). Il est donc tentant d’imaginer que
comme dans le cas de l’infection bactérienne par S. flexneri, Salmonella va induire
l’activation de la voie de survie PI 3-kinase/Akt via la production de PtdIns5P. Les
pathogènes bactériens auraient donc mis en place une voie générale d’échappement à la
réponse immunitaire via le détournement du métabolisme du PtdIns5P.
Il est intéressant de noter que malgré des travaux l’impliquant dans le trafic vésiculaire via
son hydrolyse de PtdIns(3,5)P2, la phosphatase SopB de S. dublin, est capable elle aussi
d’hydrolyser le PtdIns(4,5)P2 et de produire les mêmes invaginations membranaires que dans
le cas de l’infection par S. flexneri ; et qu’a ce jour le produit de cette hydrolyse n’est pas
connu (Terebiznik, 2002). Il est donc tout à fait possible d’envisager une activation de la voie
de survie PI 3-kinase/Akt dans le cas de l’infection par S.dublin.
Enfin, en accord avec nos résultats obtenus dans le contexte de l’infection bactérienne
d’autres travaux impliquent le PtdIns5P dans l’activation de cette voie de survie PI 3-
kinase/Akt. Ainsi, la surexpression des PIPKIIβ qui convertissent le PtdIns(4,5)P2 en
PtdIns5P conduit à la chute du taux de PtdIns(3,4,5)P3 et à la réduction de l’activation d’Akt,
dépendante de l’insuline (Carricaburu, 2003). De plus, des souris KO pour cette kinase
présentent une suractivation d’Akt (Lamia, 2004). Tous ces résultats suggèrent un rôle général
du PtdIns5P et des enzymes qui le produisent en tant que régulateur d’Akt.
2.2. Fonctions nucléaires La première preuve d’un rôle possible du PtdIns5P dans le noyau vient de l’observation que
ce PI est retrouvé augmenté de 20 fois dans le noyau de cellules murines d’erythroleucémies
lors de la phase G1 du cycle cellulaire (Clarke, 2001). Les travaux de Gozani et al. ont ensuite
suggéré un rôle du PtdIns5P dans la régulation de la transcription de gènes. Ainsi, le
suppresseur de tumeur nucléaire, ING2 a été montré comme étant un récepteur du PtdIns5P
via son domaine PHD (Gozani, 2003). La liaison du PtdIns5P au domaine PHD d’ING2
régule la capacité d’ING2 à induire une apoptose dépendante de p53 en réponse aux
76

Introduction
dommages à l’ADN causés par des génotoxiques (Gozani, 2003). Comme décrit au Chapitre
II.III, ce domaine est une structure en doigts de zinc ressemblant aux domaines FYVE et
RING. Il est retrouvé dans des protéines nucléaires telles que le régulateur de la chromatine
ACF, l’acétyltransférase CBP/300 et RAG2 (Elkin, 2005) suggérant ainsi un rôle du PtdIns5P
dans le remodelage de la chromatine et la régulation de la transcription. D’autres travaux
impliquent le PtdIns5P dans la régulation génique puisque il est capable d’interagir avec le
domaine PH du facteur de transcription, TFIIH, un composant de l’ARN polymérase de type
II. En fait, le PtdIns5P va rentrer en compétition pour le domaine PH avec le transactivateur
de TFIIH, VP16 (Di Lello, 2005). Des preuves d’un rôle biologique du PtdIns5P dans le
noyau nous sont apportées par des travaux récents. Ceux ci proposent un rôle du PtdIns5P
dans la régulation de la transcription de gènes en réponse au stress et impliquent les voies de
production du PtdIns5P à partir du PtdIns(4,5)P2. Ainsi, des travaux très récents montrent que
dans des cellules HeLa traitées à l’étoposide ou à la doxorubicine, la 4-phosphatase de type I,
nouvellement découverte (Ungewickell, 2005), se relocalise au noyau et permet une
production de PtdIns5P (Zou, 2007). Ce PtdIns5P va alors lier ING2 et augmente l’acétylation
et la stabilisation de p53, induisant alors l’apoptose (Zou, 2007). Parallèlement, l’équipe du
Dr. Divecha prouve le même rôle du PtdIns5P mais en impliquant la PIP4K de type IIβ,
résidente du noyau. Les auteurs montrent que cette kinase est phosphorylée sur son résidu
S326 par la MAP kinase p38 lors d’un stress génotoxique. Cette phosphorylation inhibe la
PIP4KIIβ, ce qui augmente le taux de PtdIns5P nucléaire (Jones, 2006) qui peut alors
contrôler via ING2 l’apoptose p53 dépendante (Bunce, 2006).
Ces résultats conduisent à proposer un modèle dans lequel le PtdIns5P nucléaire produit soit
par la PtdIns(4,5)P2 4-phosphatase de type I ou par l’inhibition de la PIP4KIIβ en réponse à
un stress génotoxique va lier la protéine ING2 qui va alors induire une apoptose p53
dépendante (Figure 33).
Enfin d’autres résultats montrent que le PtdIns5P pourrait réguler l’ubiquitination nucléaire de
certaines protéines. En effet, les travaux récents montrent que la PIP4KIIβ peut lier la
protéine SPOP impliquée dans le recrutement du complexe ubiquitine ligase Cul3 (Bunce,
2008). SPOP et la PIP4KIIβ colocalisent au niveau des speckles nucléaires dans les cellules
de mammifères. De plus, les auteurs montrent que l’activation de p38 et donc l’augmentation
du taux de PtdIns5P nucléaire (Jones, 2006) augmente la capacité d’ubiquitination de Cul3 sur
la PIP4KIIβ. De façon concomitante, l’expression des PtdIns(4,5)P2 4-phosphatases de type I
77

Introduction
et II qui augmentent le taux de PtdIns5P conduit à l’activation de Cul3 et ceci de façon
dépendante de p38 puisqu’un inhibiteur de cette kinase abolit cet effet (Bunce, 2008).
En conclusion, longtemps suggéré comme ayant un rôle dans le noyau, le PtdIns5P semble
désormais y avoir une place importante. En effet, ce lipide semble réguler de façon très
précise les réponses au stress et la transcription de gènes cibles.
Figure 33: Modèle des fonctions nucléaires du PtdIns5P en réponse au stress. En réponse à un stress cellulaire, les 4-phosphatases de type I sont transloquées au noyau où elles hydrolysent le PtdIns(4,5)P2 en PtdIns5P. Les niveaux élevés de PtdIns5P facilitent l’activation de la voie apoptotique p53/ING2 en permettant l’acétylation de p53 dépendante d’ING2. Ainsi, l’activation de p53 et sa stabilité sont augmentées et permettent une augmentation de l’apoptose. D’après Zou, 2007.
2.3. Rôle dans le trafic vésiculaire Plusieurs études suggèrent une implication du PtdIns5P dans le trafic vésiculaire. Ainsi, dans
des cellules CHO (Chinese hamster ovary) exprimant de façon stable le récepteur à l’insuline
et dans des adipocytes 3T3, la synthèse de PtdIns5P après une stimulation à l’insuline conduit
à la translocation de vésicules GLUT4 (Glucose transporter 4) à la membrane plasmique
(Sbrissa, 1999). Le même effet est observé lors de la microinjection de PtdIns5P exogène ou
de la surexpression de PIKfyve (Cf. Chap. III) (Sbrissa, 2004). Inversement, la séquestration
de PtdIns5P intracellulaire par l’expression du domaine PHD de la protéine ING2 inhibe la
78

Introduction
translocation de vésicules GLUT4 à la membrane plasmique (Sbrissa, 2004). De plus, la
surexpression de la 3-phosphatase MTMR2, lors d’une stimulation à l’EGF, conduit à une
inhibition du trafic du EGFR entre les endosomes tardifs et les lysosomes et à une
vacuolisation du système endosomal. Cet effet pourrait impliquer le domaine GRAM-PH de
MTMR2 (Tsujita, 2004). Comme dit précédemment, ce domaine est capable de lier le
PtdIns5P (Lorenzo, 2006) et pourrait donc impliquer ce PI dans le contrôle de la dégradation
du récepteur à l’EGF. D’autres données démontrent un rôle du PtdIns5P dans le contrôle du
trafic de l’EGFR, ainsi la surexpression dans des cellules HeLa des PtdIns(4,5)P2 4-
phosphatases de type I et II, récemment clonées chez l’homme entraîne une activation de la
dégradation du EGFR (Ungewickell, 2005). Dans des cellules infectées par la bactérie S.
dublin, Dukes et al. décrivent que SopB, l’orthologue d’IpgD, peut inhiber la dégradation du
EGFR (Dukes, 2006). L’implication du PtdIns5P n’est pas formellement démontrée mais ce
lipide pourrait être responsable de ces effets. De plus, une étude récente montre qu’un
important régulateur du trafic du EGFR, la SNX5 (sortin nexin 5), qui contrôle la dégradation
de ce récepteur, lie spécifiquement le PtdIns5P via son domaine PX (Liu, 2006). Enfin, de
nombreux travaux ont déjà montré que la kinase PIKfyve, produisant du PtdIns5P, est
impliquée dans le contrôle de l’homéostasie vésiculaire (pour revue, Michell, 2006 ;
Shisheva, 2008) suggérant que le PtdIns5P pourrait lui aussi jouer un rôle dans ce processus.
2.4. Rôle dans la réorganisation du cytosquelette L’étude de l’équipe du Dr. Shisheva suggère que le PtdIns5P pourrait réguler la
réorganisation des fibres de stress en réponse à l’insuline. Ainsi, la microinjection de
PtdIns5P induit une désorganisation des fibres de stress que l’on observe également en
réponse à l’insuline, alors que la séquestration de PtdIns5P par la surexpression du domaine
PHD d’ING2 bloque le désassemblage de l’actine (Sbrissa, 2004). Les données de notre
laboratoire rejoignent ces travaux. En effet, nous avons observé que l’expression ou la
microinjection d’IpgD a un effet sur la réorganisation du cytosquelette d’actine et induit la
perte des fibres d’actine. Dans ce cas, la déplétion du PtdIns(4,5)P2 membranaire qui permet
la synthèse de PtdIns5P par IpgD pourrait coopérer à la réorganisation des fibres d’actine
(Niebuhr, 2002). Cependant, le rôle du PtdIns5P dans l’organisation du cytosquelette d’actine
est encore flou et des recherches supplémentaires sont nécessaires avant que l’on puisse
inclure le PtdIns5P dans la liste des régulateurs du cytosquelette.
79
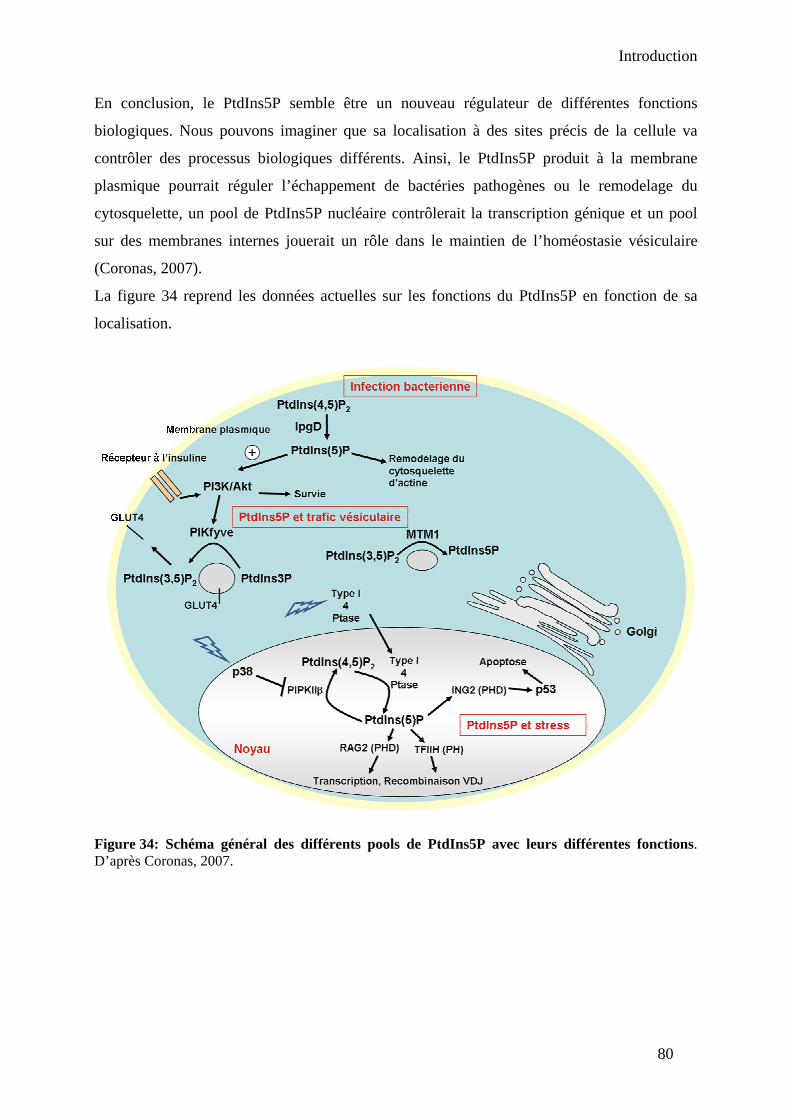
Introduction
En conclusion, le PtdIns5P semble être un nouveau régulateur de différentes fonctions
biologiques. Nous pouvons imaginer que sa localisation à des sites précis de la cellule va
contrôler des processus biologiques différents. Ainsi, le PtdIns5P produit à la membrane
plasmique pourrait réguler l’échappement de bactéries pathogènes ou le remodelage du
cytosquelette, un pool de PtdIns5P nucléaire contrôlerait la transcription génique et un pool
sur des membranes internes jouerait un rôle dans le maintien de l’homéostasie vésiculaire
(Coronas, 2007).
La figure 34 reprend les données actuelles sur les fonctions du PtdIns5P en fonction de sa
localisation.
Figure 34: Schéma général des différents pools de PtdIns5P avec leurs différentes fonctions. D’après Coronas, 2007.
80

Introduction
CHAPITRE III
La PtdIns3P 5-kinase de type III : PIKfyve
C’est en 1999 lors de travaux sur le métabolisme du glucose que l’équipe du Dr. Shisheva a
pu identifier et cloner la kinase de haut poids moléculaire PIKfyve (235 kDa) à partir d’une
banque d’ADNc murine regroupant des transcrits surexprimés dans les muscles et le tissu
adipeux (Shisheva, 1999). Cette kinase, hautement conservée au cours de l’évolution est
l’homologue de Fab1p chez S. cerevisiae, décrite pour produire du PtdIns(3,5)P2 in vitro et in
vivo. Elle appartient à la famille des PtdInsP 5-kinase de type III qui sont capables de
phosphoryler le PtdIns et le PtdIns3P en position 5 produisant respectivement du PtdIns5P et
du PtdIns(3,5)P2. Les travaux réalisés jusqu’à présent suggèrent que PIKfyve contrôle
différentes fonctions cellulaires, en particulier, le trafic vésiculaire et la régulation de
l’homéostasie des vésicules.
I-Structure de PIKfyve Chez la souris, le gène codant pour mPIKfyve est situé sur le chromosome 1 et contient 42
exons. Trois variants protéiques issus d’un épissage alternatif existent, ce sont les formes :
mPIKfyveS (2041 résidus), mPIKfyveL (2052 résidus) et une forme plus longue de 2108
résidus (Shisheva, 1999 ; Shisheva 2008). Ces variants diffèrent par la présence ou l’absence
de l’exon 4 (33 nucléotides) et/ou 12’ (168 nucléotides) et semblent exprimés de façon
équivalente dans les adipocytes murins, suggérant qu’il en est de même dans les autres tissus.
La perte de ces exons ne modifie en aucune façon l’intégrité des domaines de mPIKfyve. A ce
jour, aucune étude ne démontre que les variants de PIKfyve ont des rôles fonctionnels
différents. L’équipe du Dr. Shisheva a pu montrer, par expériences de Northern Blot, que le
transcript de mPIKfyve est augmenté lors de la différenciation des adipocytes (Shisheva,
1999). Cette protéine bien que majoritaire dans le tissu adipeux et les muscles est exprimée de
façon ubiquitaire dans les cellules et les tissus. La forme humaine de PIKfyve code pour une
protéine de 2098 acides aminés, et a été clonée récemment, elle est située en position 2q34 du
chromosome 2, contient 41 exons et correspond au variant mPIKfyveL chez la souris
(Cabezas, 2006). A ce jour, aucune forme issue d’un épissage alternatif n’a pu être identifiée
chez l’homme. PIKfyve est conservée lors de l’évolution, il existe des homologues identifiés
81

Introduction
chez D. melanogaster, C. elegans et S. cerevisiae qui diffèrent de part la présence ou
l’absence de certains domaines structuraux. Il n’existe pas chez ces organismes de formes
issues de l’épissage alternatif.
PIKfyveLM. musculus
85% 1 2052
PIP5KCHK-HomFYVE DEP Cpn60_TCP1
PIKfyveH. sapiens
1 2098
PIP5KCHK-HomFYVE DEP Cpn60_TCP1
PIKfyveD. melanogaster
28% 1 1854
PIP5KCHK-HomFYVE DEP Cpn60_TCP1
pp-k3C.elegans
27% 1 1375PIP5KFYVE Cpn60_TCP1
Fab1pS. cerevisae
20% 1 2278
PIP5KCHK-HomFYVE Cpn60_TCP1
PIKfyveLM. musculus
85% 1 2052
PIP5KCHK-HomFYVEFYVE DEPDEP Cpn60_TCP1
PIKfyveH. sapiens
1 2098
PIP5KCHK-HomFYVE DEP Cpn60_TCP1PIKfyveH. sapiens
1 2098
PIP5KCHK-HomFYVEFYVE DEPDEP Cpn60_TCP1
PIKfyveD. melanogaster
28% 1 1854
PIP5KCHK-HomFYVEFYVE DEPDEP Cpn60_TCP1
pp-k3C.elegans
27% 1 1375PIP5KFYVEFYVE Cpn60_TCP1
Fab1pS. cerevisae
20% 1 2278
PIP5KCHK-HomFYVEFYVE Cpn60_TCP1
Figure 35 : Représentation schématique des différents hortologues de PIKfyve dans plusieurs espèces. Les domaines constituants PIKfyve sont représentés. Sur la droite est indiquée la longueur estimée de la protéine en acides aminés. Le pourcentage d’identité de séquence par rapport à la séquence humaine est indiqué sur la gauche. D’après Cabezas, 2006 ; Shisheva, 2008
PIKfyve contient plusieurs domaines conservés au cours de l’évolution (Figure 35). Le
domaine FYVE situé en N-terminal permet la localisation de PIKfyve sur des membranes
cellulaires contenant du PtdIns3P (Cf. Chap. II.III.) et explique la localisation en partie
vésiculaire de PIKfyve. En effet, la surexpression d’un mutant de PIKfyve délété du domaine
FYVE ou un traitement avec la wortmanine, un inhibiteur des PI 3-kinases qui diminue le
taux de PtdIns3P, engendrent une relocalisation de PIKfyve dans le cytosol (Shisheva, 1999 ;
Sbrissa, 2002 ; Rutherford, 2006). Ce domaine FYVE est situé à côté d’un domaine DEP
(Dishevelled, Egl10 and Pleckstrin), domaine qui n’existe que dans les homologues de
PIKfyve issus des eucaryotes supérieurs (à partir de D. melanogaster) (Shisheva, 2001)
(Figure 35). La fonction de ce domaine reste, à ce jour, encore flou dans le contexte de
PIKfyve, mais il semble contribuer à l’adressage à la membrane et à la stabilité de la protéine
qui le possède. C’est le cas du domaine DEP de la protéine Dishevelled qui par une séquence
basique va localiser cette protéine à la membrane plasmique et permettre ainsi l’activation des
82

Introduction
voies de transduction du signal impliquant la protéine Wnt (Wong, 2000, Chen, 2006). Cette
région basique serait aussi capable d’être engagée dans des liaisons à l’ADN. Enfin, le DEP
domaine est aussi capable de réguler des interactions protéine/protéine par une région appelée
« dipôle électrostatique ». C’est ainsi par ce domaine que se fait l’association entre
Dishevelled et la micro2-adaptine, une sous-unité de l’adaptateur à la clathrine AP-2. Cette
interaction semble médier l’internalisation du récepteur Frizzled engagé dans les voies de
signalisation Wnt (Yu, 2007).
La partie centrale de PIKfyve est occupée par une région « chaperonine-like » dénommée
aussi Cpn60_TCP1 (HSP chaperonin_T-complex protein1) qui semble être impliquée dans
des interactions protéine/protéine. C’est le cas de la Chaperonine de type II, une protéine
CCT (Chaperonins containing TCP1) qui peut lier l’actine et la tubuline (Llorca, 1999 ;
Grantham, 2002) et permettre ainsi de maintenir les conformations originelles de ces
protéines. De façon intéressante, la Chaperonine de type II serait capable de lier des protéines
contenant des domaines β-propellers comme les PROPPINs (Cf., Chap. II.III.), nouveaux
intéracteurs du PtdIns(3,5)P2, un des produit de PIKfyve (Spiess, 2004). Ce domaine semble
être important dans la régulation de PIKfyve puisque sa délétion conduit à l’abolition de
l’activité lipide kinase de mPIKfyve (Sbrissa, 1999). Adjacent à ce domaine « chaperonine-
like » se trouve une région conservée riche en cystéine, histidine et lysine d’environ 300
résidus (domaine CHK-Hom). Cette région contient des homologies de séquence avec les
répétitions spectrine qui sont trouvées dans des protéines impliquées dans le remodelage du
cytosquelette telles que l’α-actinine ou la dystrophine. Ce domaine n’est pas présent chez
l’orthologue de PIKfyve de C. elegans. Le domaine catalytique de PIKfyve est positionné en
C-terminal de la protéine et présente dans sa partie la plus C-terminale une homologie avec la
région catalytique des PtdIns4P 5-kinases de type I et II, qui phosphorylent le noyau inositol
du PtdIns4P en position 5, et avec la PtdIns3P 5-kinase Fab1p de levure (phosphorylation en
position 5 du PtdIns3P) ; la positionnant dans la classe des PtdIns 5-kinases (Shisheva, 1999 ;
Sbrissa, 1999). Mais ce domaine présente aussi, dans sa partie plus N-terminale des séquences
uniques qui le différencient permettant sa classification et celle de son homologue de levure
Fab1p dans une nouvelle classe de PtdIns 5-kinase, la classe III. L’équipe du Dr. Shisheva a
montré que PIKfyve phosphoryle spécifiquement le PtdIns et le PtdIns3P en position 5
(Sbrissa, 1999). A ce jour, c’est la seule kinase qui semble capable de produire du PtdIns5P à
partir de PtdIns.
83

Introduction
II-Localisation de PIKfyve PIKfyve présente une localisation cytosolique et membranaire au niveau de membranes
internes (Ikonomov, 2002 ; Sbrissa, 1999). Par des études de fractionnements subcellulaires
dans des adipocytes 3T3-L1, PIKfyve a pu être localisée à 76% dans le cytosol, à 20% dans
des endomembranes dont le cytosquelette, à 4% à la membrane plasmique et serait exclue du
noyau (Shisheva, 2001). Selon la méthode d’étude et le modèle utilisé, l’identité de ces
membranes est différente. Concernant la localisation de PIKfyve chez les autres organismes,
Fab1p de levure est localisée au niveau du compartiment endosomal et de la
vacuoles/lysosomes. Chez D. melanogaster, PIKfyve est située tout comme chez l’homme au
niveau du système endosomal, d’une part sur les endosomes précoces positifs pour le
marqueur Rab5, et plus particulièrement sue des microdomaines positifs pour le marqueur
Hrs, et d’autre part elle est présente sur des endosomes tardifs où elle colocalise avec la
protéine Rab7 (Rusten, 2006). La localisation de PIKfyveWT chez C.elegans n’a pas été
étudiée mais la délétion de cette protéine dans cet organisme démontre une implication dans
la maturation terminale des lysosomes (Nicot, 2006). Des études de microscopie à
fluorescence montrent un marquage ponctiforme de PIKfyve endogène au niveau des
endosomes dans des cellules 3T3-L1 (Shisheva, 2001). Mais l’absence de moyens fiables de
détection et la faible expression endogène de cette protéine engendre une divergence des
opinions sur la localisation de PIKfyve (Shisheva, 2001 ; Ikonomov, 2006 ; Cabezas, 2006 ;
Rutherford, 2006). D’autres informations concernant la localisation de PIKfyve sont
apportées par l’expression ectopique de cette protéine et sont parfois contradictoires. Ainsi,
dans des cellules HeLa, hPIKfyve exprimée de façon ectopique est localisée en partie sur des
endosomes précoces positifs pour le marqueur EEA1 (Cabezas, 2006). Des études plus
poussées ont pu mettre en évidence que hPIKfyve n’est pas située sur la totalité des
endosomes précoces mais sur des microdomaines distincts de ceux positifs pour EEA1 et Hrs,
un autre marqueur des endosomes précoces (Cabezas, 2006). PIKfyve surexprimée dans des
cellules COS est retrouvée sur les endosomes tardifs où elle colocalise partiellement avec le
récepteur au mannose-6-phosphate cation indépendant (CI-MPR) (Griffiths, 1990 ; Rohn,
2000). Dans ce type cellulaire, mPIKfyve n’est située ni au niveau des endosomes précoces
ni sur les vésicules de recyclages (Shisheva, 2001 ; Ikonomov, 2006). Par contre, des travaux
réalisés par une autre équipe dans des cellules HeLa montrent que mPIKfyve se localiserait
sur les endosomes précoces sur des membranes enrichies en PtdIns3P, par son domaine
FYVE (Rutherford, 2006).
84

Introduction
De plus, des résultats récents de microscopie à fluorescence démontrent que dans des cellules
COS, un dominant négatif de PIKfyve, PIKfyveK1831E, va s’accumuler dans des endosomes
précoces positifs pour EEA1 et Rab5 de façon PtdIns3P dépendante ; contrairement à une
forme sauvage qui se localise dans les endosomes tardifs (Ikonomov, 2006). Ces résultats
suggèrent que PIKfyve pourrait se localiser de façon dynamique en fonction de son activité
enzymatique pour réguler la balance entre le PtdIns3P et le PtdIns(3,5)P2.
En conclusion, PIKfyve semble associée majoritairement au système endosomal où elle
colocalise à différents degrés avec les endosomes précoces, les MVBs, les endosomes tardifs
et le réseau Trans Golgien. Les variations de localisation observées sont vraisemblablement
dus aux types cellulaires étudiés, au niveau d’expression de la protéine et au ratio de
PtdIns3P/ PtdIns(3,5)P2.
III-Fonctions
1. PIKfyve est une lipide kinase
Les premiers résultats in vitro ont montré que PIKfyve phosphoryle le noyau inositol du
PtdIns et du PtdIns3P en position 5 pour synthétiser respectivement du PtdIns5P et du
PtdIns(3,5)P2 (Sbrissa, 1999). Ces mêmes résultats ont pu être reproduits dans un contexte
cellulaire. Premièrement, la surexpression ou l’abolition de PIKfyve dans plusieurs types
cellulaires de mammifères va augmenter ou diminuer, respectivement, le taux de
PtdIns(3,5)P2 (Ikonomov, 2001 ; Sbrissa, 2005 ; Sbrissa, 2007). Deuxièmement, dans des
lignées cellulaires HEK293 exprimant de façon stable soit PIKfyve sauvage soit un dominant
négatif de PIKfyve, PIKfyveK1831E, les auteurs retrouvent respectivement, une augmentation
ou une diminution du taux de PtdIns5P en comparaison à des cellules contrôles HEK293
(Sbrissa, 2002b). Ces résultats indiquent que PIKfyve régulerait la biosynthèse cellulaire du
PtdIns5P et du PtdIns(3,5)P2. Il a été proposé que cette dualité de substrat de l’activité lipide
kinase puisse être contrôlée par des résidus différents du domaine kinase de PIKfyve. Ainsi,
l’équipe du Dr. Shisheva a pu montrer que la mutation ponctuelle dans le domaine kinase sur
la Lys2000 diminue la capacité de PIKfyve à produire du PtdIns5P mais pas du PtdIns(3,5)P2
(Ikonomov, 2002) et inversement une mutation sur le résidu Lys1999 diminue l’activité lipide
kinase de PIKfyve spécifique pour le PtdIns(3,5)P2 (Ikonomov, 2002).
85

Introduction
1.1. PIKfyve synthétise du PtdIns(3,5)P2
Le rôle de PIKfyve dans la synthèse du PtdIns(3,5)P2 est bien documentée in vivo et in vitro.
Les premiers travaux, chez la levure, ont pu mettre en évidence que ce lipide était
essentiellement produit à partir de PtdIns3P par la PtdIns 5-kinase, Fab1p (Whiteford, 1999 ;
Dove, 1997), l’homologue de PIKfyve, et qu’il semblait jouer un rôle majeur dans les
processus de maintien de l’homéostasie vésiculaire (Odorizzi, 2000). Chez les mammifères,
différents travaux ont pu démontrer que PIKfyve est responsable de la production de
PtdIns(3,5)P2. En effet l’expression ectopique de PIKfyve murine dans plusieurs types
cellulaires tels que des adipocytes 3T3-L1, des cellules COS, HEK293 ou CHO entraîne une
augmentation du taux de PtdIns(3,5)P2 de 30 à 55% (Ikonomov, 2001). De plus, le phénotype
observé chez des souches de levures de S. cerevisiae déficientes pour Fab1p, c’est-à-dire une
vacuolation des membranes internes, est observé également lors de l’expression du dominant
négatif de PIKfyveK1831E. Dans ces deux cas, cette dilatation des vésicules intracellulaires est
accompagnée d’une chute concomitante des taux de PtdIns(3,5)P2 (Odorizzi, 2000 ;
Ikonomov, 2002b). Le rôle majeur de PIKfyve dans la production de PtdIns(3,5)P2 est encore
renforcé par sa localisation cellulaire. En effet, son domaine FYVE lui confère la capacité de
se maintenir sur des vésicules contenant du PtdIns3P, lui permettant ainsi une production
efficace de PtdIns(3,5)P2 in vivo malgré le fait que le PtdIns3P soit 400 fois moins abondant
que le PtdIns, l’autre substrat de PIKfyve (Sbrissa, 2002b).
A ce jour, il semble que PIKfyve soit l’enzyme principale permettant la synthèse de
PtdIns(3,5)P2 lui conférant un rôle majeur dans la régulation des processus d’homéostasie
intravésiculaire.
1.2. PIKfyve synthétise du PtdIns5P
Comme dit précédemment, le PtdIns5P, selon les types cellulaires étudiés, représente une
fraction mineure des PIs totaux allant d’un niveau quasi indétectable à 5% des PIs (Shisheva,
2003). A ce jour, chez la levure, aucun groupe n’a été capable de démontrer une production de
PtdIns5P par Fab1p, l’homologue de PIKfyve (Michell, 2006). Ainsi, des expériences menées
sur des souches de levure déficientes pour Fab1p démontrent qu’une complémentation de ses
souches par mPIKfyve ne permet pas de produire du PtdIns5P (McEwen, 1999) suggérant que
chez cet organisme Fab1p produit seulement du PtdIns(3,5)P2 (Sbrissa, 2002b ; Sbrissa,
2004). En ce qui concerne les mammifères, différents travaux du Dr. Shisheva ont pu mettre
86

Introduction
en évidence que la surexpression ectopique de PIKfyve est capable de réguler le taux
intracellulaire de PtdIns5P (Sbrissa, 1999 ; Sbrissa, 2002b). Actuellement, il est bien admis
que PIKfyve est capable de produire du PtdIns5P in vitro. Ainsi, cette équipe a pu démontrer
qu’une surexpression de PIKfyve induit une augmentation du taux de PtdIns5P de 80% dans
les adipocytes 3T3-L1 (Sbrissa, 2002a ; Sbrissa, 1999 ; Shisheva, 2001). Cette augmentation
est retrouvée dans des cellules HEK293 exprimant de façon stable PIKfyveWT par rapport à
une lignée exprimant une forme catalytiquement inactive de PIKfyve (Sbrissa, 2002b).
Cependant, la question d’un rôle direct ou indirect de PIKfyve dans la production du PtdIns5P
in vivo reste posée. En effet, il est tout à fait envisageable que PIKfyve produise
principalement du PtdIns(3,5)P2 qui est alors dégradé en PtdIns5P par des 3-phosphatases. En
effet, plusieurs groupes proposent que le PtdIns5P proviendrait du catabolisme du
PtdIns(3,5)P2, produit par PIKfyve, via sa dégradation par des phosphatases de la famille des
Myotubularines (Walker, 2001 ; Tronchère, 2004 ; Michell, 2006). Les données actuelles de
la littérature ne permettent pas de trancher quant à une implication directe ou indirecte de
PIKfyve dans la production de PtdIns5P, mais il semble que cette protéine contribue au moins
en partie au pool intracellulaire de ce PI.
2. PIKfyve est une protéine kinase
Tout d’abord décrite comme une lipide kinase, PIKfyve possède aussi une activité protéine
sérine/thréonine kinase (comme la PI 3-kinase) et a la capacité de s’autophosphoryler
(Sbrissa, 2000 ; Ikonomov, 2003a). Des travaux récents suggèrent que ce site
d’autophosphorylation serait le résidu S318 montré, dans un premier temps, comme étant le
site de phosphorylation de PIKfyve par le proto-oncogène Akt (Berwick, 2004 ; Ikonomov,
2007). De plus, in vitro, des travaux ont mis en évidence sa capacité à phosphoryler différents
substrats sur résidus sérine dont une protéine de poids électrophorétique de 110 kDa (pp110)
mais dont la séquence et la relevance dans un contexte biologique reste à ce jour inconnues.
Enfin, d’autres travaux ont pu mettre en évidence sa capacité à phosphoryler le facteur de
transport p40, un régulateur du transport de récepteurs agissant entre les endosomes tardifs et
le TGN (Ikonomov, 2003a ; Diaz, 1997). Il semble que les deux activités kinases (protéine et
lipide kinase) de PIKfyve soient indissociables, utilisent le même site de liaison à l’ATP, le
résidu Lys1831 (Sbrissa, 2000). Elles sont sensibles toutes deux au manganèse, insensibles à
la wortmanine et à des concentrations faibles de TritonX100, renforçant l’idée que ces deux
87

Introduction
activités sont portées par le même site. Dans un contexte cellulaire, il semble que ces deux
activités kinases se régulent, et plus particulièrement l’activité lipide kinase de PIKfyve serait
régulée par autophosphorylation, comme cela a été démontré in vitro (Sbrissa, 2000). Ainsi,
ces auteurs ont montré que des immunoprécipités de PIKfyve soumis dans un premier temps à
une autophosphorylation sont incapables de produire du PtdIns5P et/ou du PtdIns(3,5)P2, lors
d’un dosage de l’activité lipide kinase (Sbrissa, 2000). Ces résultats suggèrent donc que
l’activité protéine kinase de PIKfyve inhibe son activité lipide kinase. A ce jour, aucune autre
cible protéique de PIKfyve n’a été identifiée.
IV-Régulateurs et partenaires de PIKfyve
1. Régulation de son activité enzymatique par des petites
molécules
Comme pour les PI 3-kinases, différentes études ont permis de découvrir que des agents
chimiques, des détergents ou encore certains lipides sont capables de réguler l’activité de
PIKfyve. Ainsi, cette enzyme est insensible à la wortmanine, un inhibiteur des PI 3-kinases et
à l’adénosine, un inhibiteur des PtdIns 4-kinases permettant un classement de PIKfyve dans
une autre classe de lipide kinase. Ces mêmes études ont démontré que l’activité enzymatique
de PIKfyve est extrêmement sensible au TritonX-100, puisque sa capacité à phosphoryler le
PtdIns est réduite de 95% en présence de 0,1% de ce détergent. La présence de
phosphatidylsérine (PS) et/ou d’acide phosphatidique (PA) conduit à une abolition totale de
son activité, tandis que la phosphatidylcholine (PC) réduit son activité de 70%. De plus, cette
enzyme présente une préférence pour les ions manganèse par rapport aux ions magnésium
pour phosphoryler ces substrats lipidiques (Sbrissa, 1999). Enfin, l’activité de PIKfyve est
inhibée par la curcumine, un pigment polyphénolique mais qui n’est pas spécifique de
PIKfyve (Ikonomov, 2002). En effet, différentes études ont montré que la curcumine est un
puissant anti-oxydant et un anti-inflammatoire et inhibe l’activité de la lipoxygénase, de la
cycloxygénase 2, du récepteur à l’EGF (EGFR) et du facteur de transcription NFκB ce qui
conduit à l’inhibition de nombreuses cibles incluant les MAPKs, Akt ou la PI 3-kinase (Lin,
2007). Enfin, plus récemment, un inhibiteur spécifique de PIKfyve a été découvert (Jefferies,
2008). C’est lors d’un programme de recherche d’inhibiteurs spécifiques des PI 3-kinases, que
ces auteurs ont pu identifier la molécule YM201636, une pyridofuropyrimidine. Un traitement
de cellules NIH3T3 avec cette molécule conduit à une dilatation des vésicules internes
88

Introduction
(Jefferies, 2008) comme observé lors de l’abolition de l’expression de PIKfyve par une
technique d’ARN interférence (Rutheford, 2006). Les auteurs montrent aussi que l’activité
kinase de PIKfyve est engagée dans ce phénomène puisqu’un traitement avec cet inhibiteur
conduit à une diminution du taux de PtdIns(3,5)P2 d’environ 80%. Il est intéressant de noter
que l’YM201636 semble spécifique de la forme mammifère de PIKfyve puisque cette
molécule n’a aucun effet sur Fab1p, l’homologue de levures (Jefferies, 2008).
2. Régulation par le proto-oncogène Akt
PIKfyve a été retrouvée lors d’une étude protéomique visant à identifier les substrats d’Akt
impliqués dans la régulation de la translocation des vésicules GLUT4 à la membrane
plasmique. Dans cette étude, les auteurs ont utilisé un anticorps dirigé contre le site
consensus, RxRxx(pS/pT) phosphorylé par Akt dans grand nombre de ses substrats (Alessi,
1996 ; Berwick, 2004). Les auteurs montrent que PIKfyve est phosphorylée sur son résidu
S318 spécifiquement par le proto-oncogène Akt et ceci de façon dépendante d’une stimulation
à l’insuline (Berwick, 2004). La phosphorylation endogène de PIKfyve par Akt en réponse à
l’insuline serait dépendante des PI 3-kinases puisque la présence de wortmanine induit une
diminution de cette phosphorylation (Berwick, 2004). Les auteurs démontrent aussi que la
phosphorylation de PIKfyve par Akt entraîne une augmentation de son activité lipide kinase
sur le PtdIns3P. L’utilisation d’un mutant ponctuel de PIKfyve sur la S318, PIKfyveS318A,
induit une diminution de la translocation de vésicules GLUT4/IRAP à la surface cellulaire
(Berwick, 2004). Il est intéressant de noter qu’une analyse par spectrométrie de masse
utilisant la protéine recombinante GST-PIKfyve in vitro suggére que ce site S318 ne serait pas
seulement un site de phosphorylation par la kinase Akt (Ikonomov, 2007) mais aussi un site
d’autophosphorylation de PIKfyve. Cette observation apparaît contradictoire, en effet si le site
S318 de PIKfyve est un site d’autophoshorylation, cela signifie que PIKfyve phosphorylée sur
ce site devrait présenter une activité lipide kinase réduite puisque les travaux du Dr.Shisheva
ont montré que l’autophosphorylation de PIKfyve conduit à une diminution de son activité
lipide kinase (Sbrissa, 2000 ; Ikonomov, 2003a). Or, les travaux du Dr. Berwick montrent que
l’activité lipide kinase de PIKfyve est augmentée. Il serait donc intéressant d’évaluer l’activité
lipide kinase du mutant PIKfyveS318A, travaux non réalisés lors de cette étude (Berwick,
2004). Enfin, une autre analyse par spectrométrie de masse utilisant aussi la protéine
89

Introduction
recombinante GST-PIKfyve suggére que PIKfyve pourrait être phosphorylée par Akt sur un
autre site que la S318 (Seebohm, 2007).
3. Régulation par le stress osmotique
Actuellement, l’implication de Fab1p, l’homologue de PIKfyve chez la levure en réponse à un
stress osmotique est bien admis (Michell, 2006). En effet, lors d’un choc osmotique tel qu’un
traitement par du chlorure de sodium ou du sorbitol, le taux de PtdIns(3,5)P2 est augmenté de
20 fois (Dove, 1997 ; Duex, 2006). Fab1p est la seule kinase capable, chez cet organisme, de
produire du PtdIns(3,5)P2, mais la surexpression de cette kinase n’engendre pas une élévation
du taux de ce PI chez la levure, suggérant que l’activité de Fab1p est finement régulée
(Yamamoto, 1995). En effet, plusieurs études ont pu montrer que l’augmentation de
PtdIns(3,5)P2 en réponse à un stress osmotique implique Fab1p et son régulateur Vac14 (Cf.
Chap. III.IV.) (Bonangelino, 2002, Zhang, 2007). Chez, les mammifères une première étude
avait montré une diminution du taux de PtdIns5P, produit par PIKfyve dans des fibroblastes et
des adipocytes en réponse à un choc osmotique (Sbrissa, 2002). Les auteurs ayant observé des
taux élevés de PtdIns(4,5)P2 en réponse à un stress osmotique suggéraient que le PtdIns5P
était métabolisé en ce PI (Sbrissa, 2002). Ce n’est que très récemment qu’une autre étude du
même groupe, dans des adipocytes 3T3-L1 différenciés, a montré que, comme chez la levure,
PIKfyve va produire du PtdIns(3,5)P2 en réponse à un stress osmotique en coopération avec
son régulateur ArPIKfyve (Associated regulator of PIKfyve) (Cf. Chap.III.IV.) (Sbrissa,
2005). Ainsi, les auteurs montrent que lors de l’abolition de mPIKfyve ou de sont régulateur
ArPIKfyve par une technique de siRNA, des adipocytes 3T3-L1 sont incapables de produire
du PtdIns(3,5)P2 en réponse à un traitement au sorbitol (Sbrissa, 2005). Ces résultats montrent
que PIKfyve est activée en réponse à un stress osmotique chez les mammifères et la levure
mais les mécanismes exacts de cette activation restent à élucider. La relevance physiologique
d’une telle réponse dans des cellules de mammifères n’est pas claire, mais les auteurs
proposent que ce mécanisme servirait aux cellules dans le contrôle de leur volume.
4. Régulation par l’insuline
Du fait de sa découverte lors de travaux sur le métabolisme du glucose et d’une augmentation
de ses transcripts dans des cellules sensibles à l’insuline, l’activation de PIKfyve en réponse à
l’insuline a longtemps été supposée. A ce jour, aucune expérience n’a pu montrer une
90

Introduction
augmentation de l’activité lipide kinase de PIKfyve lors d’une stimulation à l’insuline. Seul
des arguments indirects et parfois contradictoires sont en notre possession. L’équipe du Dr.
Shisheva a montré que dans des immunoprécipités de PIKfyve issus d’adipocytes stimulés à
l’insuline et soumis à un test lipide kinase, les taux de PtdIns5P et de PtdIns(3,5)P2 restaient
inchangés par rapport à ces cellules non stimulées (Sbrissa, 1999), mais que par contre le taux
de PtdIns3P était augmenté suggérant un rôle de la PI 3-kinase en synergie avec PIKfyve
(Sbrissa, 1999). Lors d’une stimulation par l’insuline, des résultats récents montrent qu’in
vitro l’activité lipide kinase de PIKfyve est augmentée par la phosphorylation d’Akt et que in
vivo cette phosphorylation promeut le transport de vésicules GLUT4 à la membrane (Cf.
Chap.III.V.) (Berwick, 2004 ; pour revue Welsh, 2005). Ces résultats suggèrent que l’activité
lipide kinase de PIKfyve est augmentée en réponse à l’insuline. De plus, d’autres travaux
montrent qu’un mutant catalytiquement inactif de PIKfyve induit une inhibition de la
translocation de GLUT4 à la membrane plasmique en réponse à l’insuline dans des adipocytes
3T3-L1 (Ikonomov, 2002). Il est intéressant de noter que malgré le manque de preuves
directes (test lipide kinase en condition d’inhibition de la PI 3-kinase), l’insuline reste le seul
facteur de croissance capable d’entraîner une réponse de PIKfyve. En effet, des
immunoprécipités de PIKfyve issus de d’adipocytes 3T3-L1 sont capables de produire du
PtdIns3P en synergie avec la PI 3-kinase avec laquelle elle co-immunoprécipite (Cf. Chap.
III.IV.) seulement après une stimulation à l’insuline et non en réponse à l’EGF ou au PDGF
(Sbrissa, 2001). En conclusion, il n’existe aucune preuve directe d’une activation de PIKfyve
par l’insuline malgré son implication avérée dans le contrôle du métabolisme du glucose en
réponse à une stimulation.
5. Le complexe PIKfyve/ArPIKfyve/Sac3
Comme dit précédemment, PIKfyve chez les mammifères et Fab1p chez la levure ont les
mêmes fonctions biologiques supposant des mécanismes de régulation et des partenaires
protéiques identiques pour les deux orthologues. Un des activateurs connus de Fab1p est la
protéine Vac14 (Bonangelino, 2002 ; Dove, 2002). Cette protéine, conservée au cours de
l’évolution, est requise dans le contrôle du taux basal de PtdIns(3,5)P2. A ce jour, aucune
fonction enzymatique n’a été décrite pour cette protéine qui chez la levure, est associée aux
vacuoles (Bonangelino, 2002). Vac14 est impliquée dans la réponse au choc osmotique, en
effet des mutants de cette protéine présentent une vacuolation des vésicules internes telles que
celles décrites lors de l’abolition de Fab1p. De plus Vac14 contrôle la synthèse de
91

Introduction
PtdIns(3,5)P2 en s’associant et en activant Fab1p (Bonangelino, 1997). A ce jour, le
mécanisme par lequel Vac14 et Fab1p interagissent reste à déterminer (Bonangelino, 2002 ;
Duex, 2006). Ainsi, de part sa capacité d’activation de Fab1p et donc de contrôle des taux
internes de PtdIns(3,5)P2, Vac14 est aujourd’hui considérée comme un osmo-régulateur. Mais
le contrôle du taux de PtdIns(3,5)P2 semble impliquer un autre partenaire. En effet, la protéine
Fig4 a été décrite comme étant une PtdIns 5-phosphatase sur le PtdIns(3,5)P2 (Rudge, 2004).
Fig4 est capable d’interagir directement avec Vac14 chez la levure et contrôle ainsi le
turnover de PtdIns(3,5)P2 (Dove, 2002 ; Gary, 2002 ; Rudge, 2004). De plus, il semble que
Fig4 également soit un régulateur de l’activateur Vac14. En effet, une souche exprimant un
mutant catalytiquement inactif de Fig4 (fig4G519R) ne voit pas son taux de PtdIns(3,5)P2
augmenter lors d’un choc osmotique (Duex, 2006). Les auteurs proposent que Fig4 pourrait
avoir une activité protéine phosphatase comme d’autres phosphoinositides phosphatases à
savoir, PTEN ou les MTMs (Cui, 1998 ; Laporte, 1998) et pourrait contrôler l’activité de
Vac14 par phosphorylation ou déphosphorylation. Le mécanisme par lequel ces trois
protéines contrôlent le taux de PtdIns(3,5)P2 nécessite d’autres recherches, mais des travaux
très récents laissent supposer que la formation du complexe de signalisation impliquant Fab1p
et permettant la synthèse de PtdIns(3,5)P2 nécessite la présence de Fig4 (Botelho, 2008).
Ainsi, par une approche génétique basée sur l’utilisation de mutants des différents domaines
fonctionnels de Fab1p, les auteurs ont pu mettre en évidence que les domaines « Chaperonin-
like » et CHK-Hom de Fab1p sont des régions d’intéraction spécifiques avec les protéines
Vac14 et Fig4. Ces travaux montrent que le complexe Vac14/Fig4 semble préexister dans le
cytosol mais que l’association spécifique de Fab1p avec ces deux protéines ne peut se faire
qu’au niveau des vacuoles internes. Enfin, les auteurs montrent, tout comme dans les travaux
de Duex et al., que Vac14 s’associe à Fab1p mais que cet assemblage nécessite la présence de
Fig4 et n’est pas dépendant, comme initialement suggéré, de l’activité phosphatase de Fig4
(Duex, 2006, Botelho, 2008). Les auteurs proposent donc un modèle dans lequel Fab1p serait
recrutée aux membranes vacuolaires par son domaine FYVE via sa liaison au PtdIns3P. Cette
localisation serait indépendante de la présence des protéines Fig4 et Vac14 qui préexistent en
complexe dans le cytosol. La formation de PtdIns(3,5)P2, lors d’un choc osmotique par
exemple, entraînerait la relocalisation du complexe Vac14/Fig4 aux vacuoles. Vac14 se lierait
à Fab1p par son domaine « Chaperonin-like », cette association permettrait alors la liaison de
Fig4 à Fab1p par le domaine CHK-Hom, et la formation de PtdIns(3,5)P2 (Botelho, 2008).
Ce complexe faisant intervenir Fab1p, Vac14 et Fig4 existe également dans les organismes
supérieurs. Chez les mammifères, ce n’est que très récemment que l’homologue de Vac14 a
92

Introduction
été identifié (Sbrissa, 2004) et comme Vac14, ArPIKfyve s’associe physiquement à PIKfyve
(Sbrissa, 2004a ; Sbrissa, 2005). Les auteurs ont pu montrer que l’abolition de l’expression de
ArPIKfyve s’accompagne d’une déplétion de PtdIns(3,5)P2, alors qu’une surexpression
entraînera une augmentation du taux de PtdIns(3,5)P2 et une diminution concomitante de
PtdIns3P (Sbrissa, 2004a ; Sbrissa, 2005). Ce complexe semble impliquer un troisième
partenaire, la 5-phosphatase Sac3 (Sbrissa, 2007), l’homologue de Fig4. De la même façon,
l’équipe du Dr. Shisheva a pu montrer par des expériences de co-immunoprécipitation, que de
façon endogène, ces trois protéines forment un complexe ternaire (Sbrissa, 2007). Sac3 va
agir sur le PtdIns(3,5)P2, en le métabolisant en PtdIns3P (Sbrissa, 2007). D’autres travaux
supportent l’idée d’une régulation du taux de PtdIns(3,5)P2 et du système endosomal par ce
complexe ternaire impliquant PIKfyve, ArPIKfyve et Sac3. Ainsi, la surexpression de SacWT
(augmentation de PtdIns3P) dans des cellules COS ou bien l’abolition de ArPIKfyve
(diminution de PtdIns(3,5)P2) dans des cellules HEK293 entraîne la formation de larges
vacuoles, phénotype observé lors de la perte de PIKfyve (Sbrissa, 2004 ; Sbrissa, 2007). De
plus, la régulation du taux de PtdIns(3,5)P2 par ce complexe semble d’autant plus importante
que des travaux récents ont pu mettre en évidence que la perte de la phosphatase Sac3/Fig4
est responsable d’un syndrome neurodégénératif de type Charcot-Marie-Tooth (CMT4J) chez
l’homme (Chow, 2007) (Cf. Chap.III.VI) (Figure 36).
Figure 36 : Modèle proposé de l’action de PIKfyve et de ses partenaires dans le contrôle du trafic vésiculaire. Le complexe PIKfyve/ArPIKfyve se localise aux endosomes précoces où il produit du PtdIns(3,5)P2 rapidement métabolisé en PtdIns3P, ceci entraîne le détachement des endosomes. Ces mêmes événements se produisent à plusieurs endroits, en violet l’implication du complexe PIKfyve/ArPIKfyve/Sac 3 dans le contrôle du trafic entre les compartiments. D’après Shisheva, 2008
93

Introduction
6. Rôle de p40 un effecteur de Rab9
C’est au cours de la recherche de partenaires de PIKfyve, par double hybride, en utilisant le
domaine « chaperonine-like » comme hameçon, que la protéine p40 a été trouvée (Ikonomov,
2003a). p40 est un effecteur de la GTPase Rab9, résidente des endosomes tardifs.
L’interaction entre p40 et Rab9 sous forme Rab9-GTP stimule le transport des récepteurs au
mannose (CI-MRP) des endosomes tardifs au TGN in vitro (Diaz, 1997), servant ainsi au
recyclage de ces récepteurs. Le domaine interagissant avec PIKfyve est également impliqué
dans l’interaction de p40 avec Rab9. La séquence de p40 est entièrement composée de six
répétitions internes qui forment des feuillets β à quatre brins qui correspondent à une simple
hélice d’un β-propeller. Cette association p40-PIKfyve a été confirmée in vitro par
expériences de GST-pull-down et fait intervenir la partie C-terminale de p40. Cependant, in
vivo ce complexe pourrait être finement régulé et donc très instable et transitoire (Ikonomov,
2003a). De plus, les auteurs ont pu montrer qu’in vitro, p40 est un substrat de l’activité
protéique kinase de PIKfyve (Ikonomov, 2003a). Enfin, après fractionnement cellulaire de
cellules exprimant un mutant catalytiquement inactif de PIKfyve, p40 est retrouvée largement
cytosolique et non plus associée aux membranes internes. Les auteurs pensent que
l’association p40/PIKfyve permettrait à la protéine p40 de se localiser à la membrane, la
phosphorylation permettrait à p40 de se relarguer de PIKfyve pour interagir avec Rab9 et
réguler le transport rétrograde des récepteurs au mannose (Sbrissa, 2005). L’importance
physiologique de ce complexe est soulignée par des travaux récents suggérant un rôle de
PIKfyve dans l’infection virale. En effet, il semble que la réplication de l’enveloppe de
certains virus et en particulier du VIH serait dépendante d’un transport entre les endosomes
tardifs et le TGN qui impliquerait Rab9, p40, PIKfyve et TIP47 (Murray, 2005). Ainsi,
PIKfyve et ses effecteurs tels que p40 pourraient devenir des cibles importantes dans la
recherche pharmacologique d’inhibiteurs spécifiques contre différents virus.
7. PIKfyve s’associe à la PI 3-kinase de classe IA
L’équipe du Dr. Shisheva, lors d’études sur l’activité lipide kinase de PIKfyve en réponse à
l’insuline a montré que des immunoprécipités de PIKfyve issus d’adipocytes 3T3-L1 étaient
capables de produire du PtdIns5P, du PtdIns(3,5)P2 mais aussi du PtdIns3P (Sbrissa, 1999).
Dans cette étude, les auteurs avait prouvé que cette synthèse de PtdIns3P était sensible à la
94

Introduction
wortmanine suggérant une implication et sans doute une association entre la PI 3-kinase et
PIKfyve (Sbrissa, 1999). Cette association a été confirmée par des expériences
d’immunoprécipitations de PIKfyve par la sous-unité p85 de la PI 3-kinase et inversement
dans le modèle des adipocytes. Il est à noter que 5% des pools cellulaires de chacun des
partenaires est engagé dans cette interaction. Cette interaction entraîne une augmentation du
taux de PtdIns3P en réponse à l’insuline (Sbrissa, 2001) dans des immunoprécipités de
PIKfyve issus de cellules stimulées à l’insuline. L’interaction entre les deux protéines n’est
pas modifiée par l’insuline.
V-Rôles biologiques de PIKfyve
1. PIKfyve et la maintenance de l’homéostasie vésiculaire
De part sa capacité à lier le PtdIns3P et à le métaboliser en PtdIns(3,5)P2, le rôle de PIKfyve
dans le transport de vésicules est largement documenté. Plusieurs études indiquent que
PIKfyve et son produit le PtdIns(3,5)P2 participent à différentes étapes du trafic endosomal en
fonction de l’organisme ou du type cellulaire étudié. Chez S. cerevisiae ou C. elegans, la perte
de PIKfyve affecte les étapes tardives du trafic endocytaire c’est-à-dire endosomes tardifs
vers lysosomes alors que chez des eucaryotes supérieurs (drosophile ou mammifères), les
étapes précoces semblent affectées. Ainsi, l’équipe du Dr. Laporte en collaboration avec notre
groupe a pu montrer que l’orthologue de PIKfyve chez C. elegans, pp-k3 via sa production de
PtdIns(3,5)P2 régule la maturation des lysosomes puisque un défaut de ppk-3 entraîne une
vacuolation dans ces organismes de vésicules positives pour LMP-1, un marqueur des
lysosomes (Nicot, 2006). Chez D. melanogaster, la perte de PIKfyve induit une dilatation des
vésicules d’origine endosomales qui deviennent multivésiculaires de type MVB (multi
vesicular body) (Rusten, 2006). Il semble alors que le recyclage de récepteurs membranaires,
qui empruntent cette voie de façon classique dans les cellules, ne soit perturbé que lors des
étapes tardives au niveau lysosomal lors de leur dégradation. Enfin, il est important de noter
que chez C. elegans et D. melanogaster, la perte totale de PIKfyve induit une létalité au stade
embryonnaire, alors qu’une perte partielle engendrera des retards de croissance chez ces
organismes. Concernant les mammifères, les travaux du Dr. Shisheva impliquent PIKfyve
dans des étapes précoces du trafic vésiculaire au niveau des endosomes précoces (Ikonomov,
2003a). Une autre étude, utilisant un système d’ARN interférence implique PIKfyve dans le
contrôle du transport rétrograde des endosomes précoces au TGN (Rutherford, 2006). Dans
95

Introduction
ces deux études, l’internalisation, le recyclage et la dégradation de récepteurs tel que le
récepteur à l’EGF n’est pas affectés lors de l’inhibition ou de l’invalidation de PIKfyve. Chez
tous les organismes étudiés, la perte de PIKfyve par l’utilisation d’un mutant catalytique ou
par des techniques de siRNA, va conduire à une perturbation profonde du système vésiculaire
avec la formation d’énormes vésicules qui envahissent les cellules. Ces données suggèrent
que PIKfyve aurait un rôle basal dans le maintien de l’homéostasie vésiculaire.
2. PIKfyve et l’exocytose
L’exocytose calcium dépendante est contrôlée par les PIs et en particulier le PtdIns(4,5)P2 et
le PtdIns3P, mais les mécanismes exacts de cette régulation restent à élucider. Ainsi, le
PtdIns3P, un des substrats de PIKfyve est connu pour promouvoir la sécrétion des LDCV
(large dense core vesicles) (Meunier, 2005). Des travaux récents impliquent PIKfyve dans le
contrôle de l’exocytose (Osborne, 2008) des cellules neurosécrétrices PC12 et chromaffines.
Les auteurs montrent, par immunofluorescence, que PIKfyve colocalise partiellement avec
P18 un marqueur des LDVC. De plus, PIKfyve semble réguler la sécrétion de ces cellules,
ainsi sous stimulation nicotinique la fraction de PIKfyve cytosolique va se relocaliser sur ces
structures vésiculaires positives pour le marqueur P18 suggérant que la kinase pourrait y jouer
un rôle. De plus, les auteurs montrent que l’activité lipide kinase de PIKfyve régule
négativement l’exocytose dans ces cellules. En effet, l’inhibition de l’activité de PIKfyve par
un inhibiteur spécifique, l’YM201636 ou l’invalidation par une technique de RNA
interférence, potentialise l’exocytose des LDCV dans des cellules PC12 stimulées par la
nicotine. De façon concomitante, la surexpression de PIKfyve ou de son homologue Fab1p
génère une inhibition de la sécrétion dans ces cellules. Ainsi, les auteurs proposent un rôle
négatif de PIKfyve dans le contrôle de la sécrétion via son action sur le PtdIns3P. La présence
de PIKfyve sur les LDCV permettrait de diminuer le taux de PtdIns3P en le transformant en
PtdIns(3,5)P2 ce qui aurait pour conséquence de diminuer l’exocytose de ses vésicules. Cette
explication semble d’autant plus plausible, que ces mêmes auteurs ont pu montrer que du
PtdIns(3,5)P2 était présent sur les granules sécrétoires LDCV en utilisant la sonde ENTH de
Ent3 (Cf. Chap. II.III.).
96

Introduction
3. PIKfyve : contrôle du transport rétrograde des endosomes au
TGN
Du fait de son association avec p40, l’effecteur de la GTPase Rab9 qui contrôle le transport
rétrograde du CI-MPR des endosomes au TGN, PIKfyve a longtemps été supposée impliquée
dans ce transport (Ikonomov, 2003a). Mais ce n’est que très récemment que des études ont pu
mettre en évidence un tel rôle de PIKfyve (Rutherford, 2006) dans des cellules surexprimant
PIKfyve qui se localise alors sur des endosomes précoces. L’abolition de PIKfyve endogène
par siRNA, induit une vacuolation caractéristique mais a conduit ces auteurs à mettre en
évidence la présence de deux types distincts de vacuoles. Les premières vacuoles sont élargies
et sont positives pour des marqueurs des endosomes tardifs tels que LAMP1 et CD63. Le
second type de vésicules présente un phénotype plus petit, est très difficile à observer en
contraste de phase et semble être originaire des endosomes précoces. De plus, cette étude
montre que, dans ces conditions, le recyclage du EGFR ou du récepteur à la transferrine ne
semblent pas affectés, suggérant que PIKfyve joue un rôle mineur dans le recyclage de ces
récepteurs ou dans les voies de dégradation. Par contre, l’inhibition de l’activité de PIKfyve
conduit à la redistribution sur les endosomes précoces de protéine cargos telles que le CI-
MPR ou la furine, protéines résidentes du TGN. Ainsi, les auteurs suggèrent que PIKfyve
localisé sur les endosomes précoces régulerait le transport rétrograde entre le TGN et ce
compartiment, et que le phénotype classique de vésicules élargies lors de la perte de PIKfyve
serait la résultante d’un manque de fission de ces vésicules. (Rutherford, 2006).
4. Implication dans le métabolisme du glucose en réponse à
l’insuline
GLUT4 est l’un des acteurs qui médie le transport du glucose en réponse à l’insuline dans le
tissu adipeux et les muscles (Bryant, 2002 ; Watson et Pessin, 2006 ; Huang et Czech, 2007).
Au départ, ce transporteur est intracellulaire et cycle lentement entre la membrane plasmique
et les autres compartiments intracellulaires. La stimulation par l’insuline engendre une
translocation accrue d’un facteur 10 dans le but d’augmenter le transport de glucose à la
membrane plasmique. A ce jour, malgré des recherches poussées, aucun groupe n’a pu
déterminer les mécanismes de translocation de ce transporteur à la membrane plasmique. Les
modèles actuels proposent l’existence de deux pools vésiculaires de stockage du glucose. Un
97

Introduction
associé au système endosomal qui serait positif pour des marqueurs endosomals de recyclage
tels que GLUT1 ou le récepteur de la transferrine, et un autre qui serait un compartiment de
stockage de GLUT4, qui contiendrait la protéine IRAP (insulin-responsive aminopeptidase)
mais aucun marqueurs endosomals. Lors d’une stimulation à l’insuline dans des adipocytes, il
semble que ce soit principalement le compartiment de stockage de GLUT4 qui soit mobilisé
ainsi que les voies de recyclage et le trafic inter-endosomal du récepteur GLUT4 à partir des
endosomes précoces (Foster, 2001 ; Karylowski, 2004). Du fait de son implication dans
l’homéostasie vésiculaire, le rôle de PIKfyve dans le transport de GLUT4 en réponse à
l’insuline est hautement probable. Des travaux ont pu montrer que l’expression de
PIKfyveK1831E empêche la translocation du récepteur GLUT4 en réponse à l’insuline dans des
adipocytes 3T3-L1 (Ikonomov, 2002b). Ces résultats sont confirmés par des études d’ARN
interférence dans lesquelles l’invalidation de PIKfyve et/ou d’un de ses effecteurs,
ArPIKfyve, empêche le recyclage de glucose à la membrane plasmique et cela de façon
dépendante de la production de PtdIns(3,5)P2 (Ikonomov, 2007). De façon concomitante,
cette équipe a montré qu’il existe une augmentation de la réponse à l’insuline dans le cas de la
déplétion de Sac3, la 5-phosphatase qui contrecarre l’action de PIKfyve (Sbrissa, 2007).
Ainsi, l’inhibition du complexe PIKfyve/ArPIKfyve et donc une baisse du taux de
PtdIns(3,5)P2 induit un arrêt de la translocation de vésicules GLUT4/IRAP à la membrane
plasmique, mais pas l’inhibition du recyclage de vésicules contenant GLUT1. Ainsi, l’équipe
du Dr. Shisheva propose un modèle dans lequel PIKfyve permettrait l’export des vésicules
GLUT4 à partir des endosomes précoces pour « recharger » les compartiments de stockage de
GLUT4 qui sont rapidement déplétés en glucose après une stimulation à l’insuline via une
augmentation de ses produits le PtdIns(3,5)P2 et le PtdIns5P (Sbrissa, 2004b ; Ikonomov,
2007). En conclusion, il semble que l’activité kinase de PIKfyve entre en jeu dans le
métabolisme du glucose en réponse à l’insuline mais les mécanismes précis et l’implication
des PIs que cette protéine synthétise sont encore à définir.
5. Implication dans le trafic de canaux ioniques
Des travaux récents suggèrent aussi un rôle de PIKfyve dans la régulation de transporteurs
ioniques activés par la kinase SGK1 (serum and glucocorticoid inducible kinase). SGK1
appartient à la famille des protéines kinases de type A, G et C (Lang et Cohen, 2001 ; Loffing,
2006). Son expression est contrôlée par de nombreux stimuli, tels que le sérum, les
98

Introduction
glucocorticoïdes, les stress oxydatifs, les cytokines et le volume cellulaire. Tout comme Akt,
SGK1 est activée par phosphorylation en réponse à une activation de la PI 3-kinase et va
activer des canaux ioniques. Deux études récentes ont pu montrer que PIKfyve est un
intermédiaire dans la phosphorylation de ces canaux par SGK1. Dans une première étude, les
auteurs montrent que le transporteur au glucose SLC5A1 couplé au Na+ est stimulé par
PIKfyve, reproduisant les effets de la kinase SGK1. La régulation de SLC5A1 par SGK1
nécessiterait la phosphorylation de PIKfyve sur le résidu S318 par SGK1 (Shojaiefard, 2007).
SGK1 est aussi impliquée dans l’activation d’autres canaux ioniques tels que des canaux
potassium, KCNQ1/KCNE1. Dans cette seconde étude, les auteurs montrent que SGK1 active
PIKfyve, qui par sa synthèse de PtdIns(3,5)P2, engendre une exocytose de KCNQ1/KCNE1
dépendante de la GTPase Rab11 (Seebohm, 2007). Ces deux études mettent à jour un
nouveau rôle de PIKfyve dans le trafic de canaux ioniques en réponse à des stimuli externes
dans une voie SGK1 dépendante. PIKfyve aurait un rôle positif de part sa production de
PtdIns(3,5)P2 sur l’exocytose de ces deux types de canaux ioniques. Il est important de noter
que dans ces deux études l’activité lipide kinase de PIKfyve via sa production de
PtdIns(3,5)P2 n’a jamais été mesurée.
6. Rôle dans les voies de dégradation de récepteurs
membranaires
Les différents composants de la machinerie de dégradation impliquent trois complexes
protéiques, ESCRT I, II et III (Endosomal Sorting Complex Required for Transport) qui vont
agir de façon séquentielle dans la dégradation de récepteurs membranaires (Babst, 2004 ;
Hurley et Erm, 2006 ; Slagsvold, 2006). Les complexes ESCRT III ; en synergie avec Vps4,
une ATPase de type AAA qui catalyse la dissociation de ces complexes des endosomes
précoces, sont impliqués dans la formation et la transformation de vésicules intraluminales en
endosomes tardifs. PIKfyve n’appartient pas à cette machinerie mais semble cependant y
jouer un rôle. En effet, l’expression ectopique d’une forme mutée de SDK1 l’orthologue de
Vps4 chez les mammifères dans des cellules COS, induit un phénotype vacuolaire comme
observé lors de l’expression du dominant négatif de PIKfyve (Ikonomov, 2002c). Cette
vacuolation nécessite la synthèse de PtdIns(3,5)P2 par PIKfyve puisque ce phénotype est
reversé par l’expression de PIKfyveWT ou la microinjection de PtdIns(3,5)P2 exogène
(Ikonomov, 2002c).
99
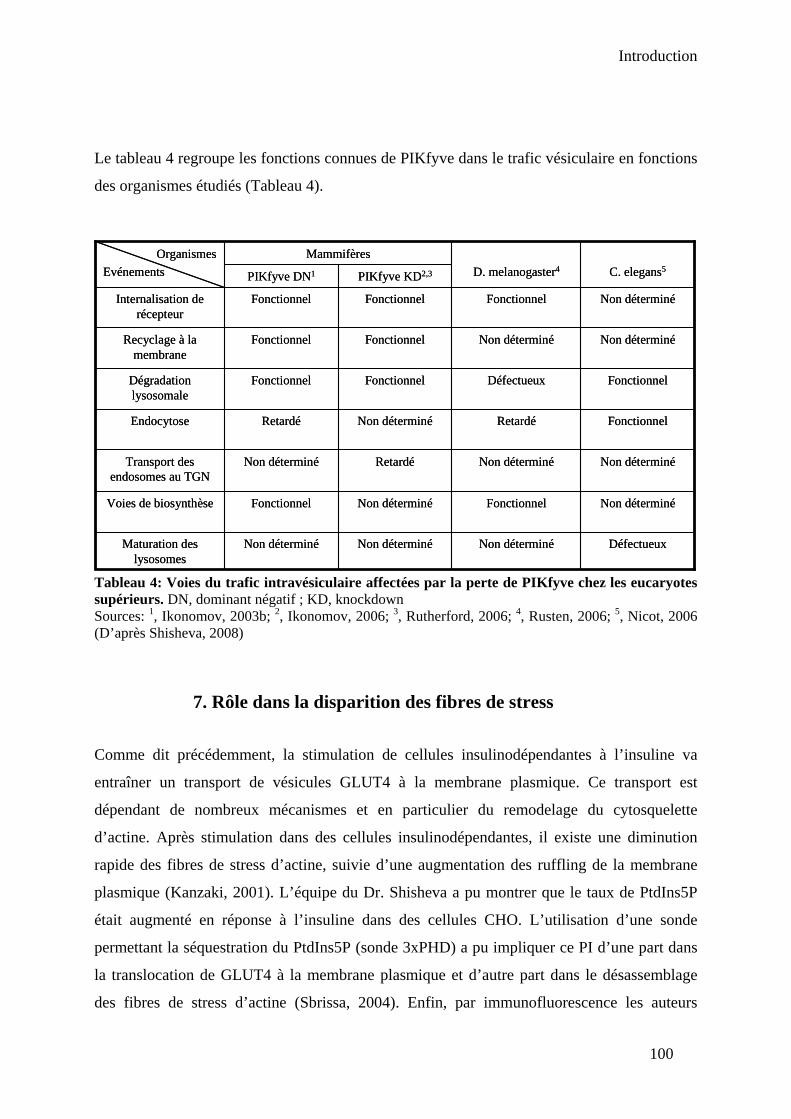
Introduction
Le tableau 4 regroupe les fonctions connues de PIKfyve dans le trafic vésiculaire en fonctions
des organismes étudiés (Tableau 4).
PIKfyve KD2,3PIKfyve DN1
DéfectueuxNon déterminéNon déterminéNon déterminéMaturation des lysosomes
Non déterminéFonctionnelNon déterminéFonctionnelVoies de biosynthèse
Non déterminéNon déterminéRetardéNon déterminéTransport des endosomes au TGN
FonctionnelRetardéNon déterminéRetardéEndocytose
FonctionnelDéfectueuxFonctionnelFonctionnelDégradation lysosomale
Non déterminéNon déterminéFonctionnelFonctionnelRecyclage à la membrane
Non déterminéFonctionnelFonctionnelFonctionnelInternalisation de récepteur
C. elegans5D. melanogaster4MammifèresOrganismes
Evénements PIKfyve KD2,3PIKfyve DN1
DéfectueuxNon déterminéNon déterminéNon déterminéMaturation des lysosomes
Non déterminéFonctionnelNon déterminéFonctionnelVoies de biosynthèse
Non déterminéNon déterminéRetardéNon déterminéTransport des endosomes au TGN
FonctionnelRetardéNon déterminéRetardéEndocytose
FonctionnelDéfectueuxFonctionnelFonctionnelDégradation lysosomale
Non déterminéNon déterminéFonctionnelFonctionnelRecyclage à la membrane
Non déterminéFonctionnelFonctionnelFonctionnelInternalisation de récepteur
C. elegans5D. melanogaster4MammifèresOrganismes
Evénements
Tableau 4: Voies du trafic intravésiculaire affectées par la perte de PIKfyve chez les eucaryotes supérieurs. DN, dominant négatif ; KD, knockdown Sources: 1, Ikonomov, 2003b; 2, Ikonomov, 2006; 3, Rutherford, 2006; 4, Rusten, 2006; 5, Nicot, 2006 (D’après Shisheva, 2008)
7. Rôle dans la disparition des fibres de stress
Comme dit précédemment, la stimulation de cellules insulinodépendantes à l’insuline va
entraîner un transport de vésicules GLUT4 à la membrane plasmique. Ce transport est
dépendant de nombreux mécanismes et en particulier du remodelage du cytosquelette
d’actine. Après stimulation dans des cellules insulinodépendantes, il existe une diminution
rapide des fibres de stress d’actine, suivie d’une augmentation des ruffling de la membrane
plasmique (Kanzaki, 2001). L’équipe du Dr. Shisheva a pu montrer que le taux de PtdIns5P
était augmenté en réponse à l’insuline dans des cellules CHO. L’utilisation d’une sonde
permettant la séquestration du PtdIns5P (sonde 3xPHD) a pu impliquer ce PI d’une part dans
la translocation de GLUT4 à la membrane plasmique et d’autre part dans le désassemblage
des fibres de stress d’actine (Sbrissa, 2004). Enfin, par immunofluorescence les auteurs
100

Introduction
montrent que la microinjection de PtdIns5P dans ces cellules reproduit le désassemblage des
fibres de stress sous insuline (Sbrissa, 2004). Les auteurs proposent donc que le PtdIns5P
produit par PIKfyve soit responsable du remodelage des fibres de stress d’actine en réponse à
l’insuline. C’est pour l’instant le seul rôle proposé pour le PtdIns5P produit par PIKfyve.
8. Le récepteur à l’EGF
Le EGFR a été retrouvé au noyau mais les mécanismes qui régulent la translocation de ce
récepteur vers le compartiment nucléaire sont peu connus. L’étude de Kim et al. montre une
localisation nucléaire du EGFR dans des cellules de lignées humaines de carcinomes de
vessie sous stimulation à l’HB-EGF et une association au promoteur de la cycline D1 (Kim,
2007). L’analyse en spectrométrie de masse de protéines associées au EGFR a permis de
mettre en évidence la présence de PIKfyve dans le complexe suggérant un rôle de PIKfyve
dans le contrôle du trafic du EGFR vers le noyau. La surexpression de PIKfyve entraîne une
augmentation du EGFR dans le noyau, et inversement l’invalidation de PIKfyve inhibe la
translocation vers le noyau et diminue les effets induits par le HB-EGF sur la progression du
cycle cellulaire (Kim, 2007). Les auteurs proposent donc que l’association PIKfyve/EGFR va
faciliter le transport du EGFR de compartiments vésiculaires cytosoliques vers le noyau où ce
récepteur va s’associer rapidement à la chromatine et réguler l’expression génique (Kim,
2007).
VI-Pathologies impliquant PIKfyve ou ses effecteurs
Les travaux récents montrent chez les organismes tels que C. elegans ou D. melanogaster que
PIKfyve est indispensable pendant les stades précoces du développement et de la
différenciation cellulaire (Nicot, 2006 ; Rusten, 2006). La perte complète de PIKfyve chez C.
elegans induit des défauts de développement caractérisés par une létalité embryonnaire, alors
qu’une perte partielle induit un retard de développement (Nicot, 2006). Concernant la
drosophile, l’absence de PIKfyve conduit aussi dans ce cas à une létalité au stade larvaire
(Rusten, 2006). Ainsi, du moins dans des organismes inférieurs, PIKfyve semble
indispensable à un développement normal de l’organisme. Aucun modèle de souris knock-
down (KO) de PIKfyve n’a encore été décrit dans la littérature.
101

Introduction
Chez l’homme, des mutations de PIKfyve ont été impliquées dans la dystrophie cornéenne
mouchetée de François-Neetens, un syndrome autosomal dominant caractérisé par la présence
de nombreuses tâches dispersées dans les différentes couches du stroma (Li, 2005). Ces
mutations sont toutes situées au niveau du domaine « chaperonine-like » et conduisent à
l’expression de codons stop et donc de protéines tronquées pour le domaine catalytique de
PIKfyve (Li, 2005). Une étude histologique a pu montrer que l’aspect moucheté de la cornée
est du à des kératinocytes présentant des vacuoles élargies telles que celles observées lors de
l’inhibition de PIKfyve (Nicholson, 1977). Un défaut de synthèse de PtdIns(3,5)P2 semble
être à l’origine d’autres pathologies retrouvées chez l’homme. Ainsi, des travaux récents
montrent que certains patients présentant une neuropathie motrice et sensorielle ressemblant
aux formes de myopathies de Charcot-Marie-Tooth de type 4J (CMT4J) sont porteurs de
mutations du gène de la phosphatase Sac3/Fig4 (Chow, 2007). Quatre des patients étudiés
portent la mutation I41T. La substitution correspondante dans Fig4 (I59T) chez la levure est
associée à une incapacité de cet organisme d’augmenter son taux de PtdIns(3,5)P2 en réponse
à un choc osmotique (Chow, 2007) et suggère chez l’homme une déficience de l’activité de
PIKfyve à produire du PtdIns(3,5)P2 (Duex, 2006 ; Chow, 2007). Chez la souris, les auteurs
ont pu démontrer de la même façon que chez l’homme, une mutation de Sac3 retrouvée chez
la souris « pale tremor », associée à un syndrome neurodégénératif (Chow, 2007). Ces souris
présentent une létalité juvénile et de nombreux types cellulaires ont de larges vacuoles
indiquant un défaut de PtdIns(3,5)P2. Mais de façon intéressante, cette étude n’a pu mettre en
évidence une baisse significative de PtdIns(3,5)P2 dans ce modèle. Ce résultat surprenant est
vraisemblablement du aux autres régulateurs de PIKfyve, tel que ArPIKfyve dont l’activité
n’a pas été évaluée dans ces travaux, ou bien à une redondance génétique de kinases capables
de produire du PtdIns(3,5)P2 inconnues à ce jour. Ceci est d’autant plus intéressant que chez
des souris KO pour le gène ArPIKfyve, il a été observé le même phénotype que chez des
souris déficientes pour le gène Sac3 (Zhang, 2007). Enfin, chez l’homme, d’autres protéines
contrôlant le taux de PtdIns(3,5)P2 ont aussi été impliquées dans des syndromes
neurodégénératifs. C’est le cas des syndromes neurodégénératifs de type Charcot-Marie-
Tooth (types 4B1 et 4B2) associés à des mutations des myotubularines MTMR2 et MTMR13
ou des myopathies myotubulaires impliquant MTM1. Ce sont des mutations récessives qui
induiraient des taux élevés de PtdIns(3,5)P2 et de PtdIns3P, deux PIs dont ces enzymes
contrôlent la production et qui sont tous deux respectivement substrats et produits de la kinase
PIKfyve (Berger, 2002 ; Robinson and Dixon, 2006).
102

Introduction
En conclusion, PIKfyve, par ses différents effecteurs ou par ses substrats, peut être reliée à
plusieurs syndromes neurodégénératifs ou myopathies.
Le but de mes travaux de thèse à été de mettre à jour un nouveau type de régulation de la
kinase PIKfyve, à savoir, une implication de PIKfyve dans des processus oncogéniques dus à
un récepteur à activité tyrosine kinase, NPM-ALK.
Ces résultats seront développés dans le chapitre « Résultats ».
103

Objectif du travail de thèse
Ainsi que nous l’avons détaillé dans la première partie de l’Introduction, de nombreuses
avancées ont été réalisées quant à l’identification des effecteurs de NPM-ALK. Malgré tout
l’implication de certains de ces partenaires de signalisation dans le pouvoir transformant et la
dissémination métastatique dus à l’oncogène restent encore assez mal connus. Une meilleure
connaissance des mécanismes de transformation par NPM-ALK, ainsi que la caractérisation
du rôle des effecteurs de cet oncogène devrait mettre à jour de nouvelles cibles
pharmacologiques potentielles. Outre la recherche d’inhibiteurs de ALK, il serait en effet
important de cibler les effecteurs clés de l’oncogène notamment dans les cas de résistance aux
inhibiteurs de ALK. Une thérapie ciblée permettrait de remplacer la polychimiothérapie
lourde utilisée actuellement qui comporte de nombreux effets secondaires et n’élimine pas
totalement les risques de rechute.
Parmi les voies de signalisation activées par l’oncogène, la voie pro-mitogène et anti-
apoptotique PI 3-kinase/Akt modulant le métabolisme des PIs a été décrite dans de
nombreuses études. Des travaux menés dans notre équipe ont pu montrer que le PtdIns5P, un
des derniers PIs découvert est un potentialisateur de la PI 3-kinase et donc de la voie de survie
PI 3-kinase/Akt.
Dans ce contexte, notre projet a été d’étudier l’implication de ce second messager lipidique, le
PtdIns5P, dans les mécanismes de transformation cellulaire conduisant au développement des
ALCL exprimant l’oncogène NPM-ALK. Les objectifs de cette étude ont été d’établir les
liens moléculaires et fonctionnels existant entre l’oncogène, le PtdIns5P et les enzymes
régulant la synthèse de ce PIs.

RESULTATS

Résultats
Partie I : Les cellules NPM-ALK positives présentent un niveau élevé de PtdIns5P ;
implication de PIKfyve I- Introduction Comme présenté au cours du chapitre I de l’Introduction, les ALCL sont des proliférations
lymphoïdes malignes de phénotypes T ou nul. Elles représentent 5% des lymphomes non
hodgkiniens chez l’adulte (Fiorani, 2001) mais affectent plus particulièrement les enfants pour
lesquels elles représentent 30% des lymphomes diagnostiqués (Stein, 2000). 75% de ces
tumeurs sont associées à un réarrangement chromosomique, la translocation réciproque
t(2;5)(p23;q35) qui juxtapose la partie N-terminale du gène NPM avec le domaine kinase du
gène ALK (Morris, 1994). Le domaine d’oligomérisation de NPM permet la dimérisation de
la protéine chimérique et l’activation constitutive du domaine kinase de ALK, par
transphosphorylation. NPM-ALK est donc capable d’activer de nombreuses voies de
signalisation pouvant conduire à la prolifération, la migration ou la survie et entraînant des
processus d’oncogenèse (Bai, 2000 ; Chiarle, 2008).
NPM-ALK active de nombreux effecteurs et en particulier des enzymes impliquées dans le
métabolisme des PIs telles que la PLC-γ ou la PI 3-kinase. Les PIs représentent une classe
quantitativement mineure des phospholipides membranaires. Leur métabolisme, finement
régulé, en fait de véritables second messagers. Par leur capacité à lier des domaines protéiques
particuliers tels que le domaine PH, PX ou FYVE, ils sont aujourd’hui considérés comme
essentiels à la régulation spatio-temporelle de grandes voies de signalisation régulant
l’organisation du cytosquelette, la prolifération ou le trafic vésiculaire (Payrastre, 2001). Le
rôle capital de ces PIs est confirmé par la découverte que des mutations sur de nombreuses
enzymes régulant le cycle des PIs peuvent générer des cancers et des maladies génétiques
(Pendaries, 2003). Le PtdIns5P est l’un des derniers PIs découvert et son métabolisme ainsi
que ses fonctions sont encore peu connus (Rameh, 1997). Comme le PtdIns(3,4,5)P3, produit
de la PI 3-kinase, le PtdIns5P pourrait être un second messager. Ainsi, différentes études ont
montré que ce PI voit son niveau augmenter en réponse à une stimulation par un agoniste
(Morris, 2000), à un stress osmotique (Miejer, 2001) ou pendant la phase G1 (Clarke, 2001).
104

Résultats
Différentes voies peuvent conduire à la synthèse de PtdIns5P et impliquent des enzymes
différentes, suggérant l’existence de plusieurs pools intracellulaires de PtdIns5P. Ainsi, notre
équipe a montré, lors de l’infection bactérienne par Shigella flexneri, que la production de
PtdIns5P à la membrane plasmique conduit à l’activation de la voie de survie PI 3-kinase/Akt
(Pendaries, 2006). Une autre étude suggère un rôle du PtdIns5P dans le noyau, où il se lie sur
le domaine PHD du suppresseur de tumeur ING2 et régule l’acétylation de p53 et l’apoptose
(Gozani, 2003). Le taux de PtdIns5P nucléaire pourrait être régulé en réponse à un stress
génotoxique ou aux UV par les PtdIns 4-kinases de type II et la PtdIns(4,5)P2 4-phosphatase
de type I qui convertissent respectivement le PtdIns5P en PtdIns(4,5)P2 et le PtdIns(4,5)P2 en
PtdIns5P (Jones, 2006 ; Zou, 2007). Un autre pool de PtdIns5P généré à partir du
PtdIns(3,5)P2 par les membres de la famille des myotubularines permet de proposer
l’existence d’un pool présent sur des vésicules internes (Tronchère, 2004). Enfin, un dernier
pool peut être issu de l’action de la PtdIns 5-kinase, PIKfyve sur le PtdIns (Sbrissa, 2002).
Du fait de son rôle dans la régulation de la voie de survie PI 3-kinase/Akt (Pendaries, 2006),
et de sa production pendant la phase G1 du cycle cellulaire (Clarke, 2001), nous avons
recherché le rôle possible de ce PI dans les mécanismes d’oncogenèse.
Dans cette première partie de notre travail, nous montrons que le PtdIns5P est augmenté dans
les cellules exprimant la tyrosine kinase NPM-ALK et que sa voie de synthèse implique la
kinase PIKfyve.
Les résultats obtenus sont présentés dans la publication suivante.
105

Biochemical and Biophysical Research Communications 372 (2008) 351–355
Contents lists available at ScienceDirect
Biochemical and Biophysical Research Communications
journal homepage: www.elsevier .com/locate /ybbrc
Elevated levels of PtdIns5P in NPM-ALK transformed cells:Implication of PIKfyve
S. Coronas, F. Lagarrigue, D. Ramel, G. Chicanne, G. Delsol, B. Payrastre, H. Tronchère *
INSERM, U563, Département Oncogénèse, Signalisation et Innovation Thérapeutique, Toulouse, F-31300, FranceUniversité Toulouse III Paul-Sabatier, Centre de Physiopathologie de Toulouse Purpan, Toulouse, F-31300, France
a r t i c l e i n f o
Article history:Received 7 May 2008Available online 22 May 2008
Keywords:Phosphatidylinositol 5-phosphatePIKfyveNPM-ALKAnaplastic large-cell lymphomaOncogenesis
0006-291X/$ - see front matter � 2008 Elsevier Inc. Adoi:10.1016/j.bbrc.2008.05.062
* Corresponding author. Address: INSERM, U563, CPSignalisation et Innovation Thérapeutique, CHU Purpcedex 3, France. Fax: +33 5 62 74 45 58.
E-mail address: [email protected] (H. Tr
a b s t r a c t
Phosphatidylinositol 5-monophosphate (PtdIns5P), one of the latest phosphoinositides discovered, hasbeen suggested to play important cellular functions. Here, we report the presence of higher levels of thislipid in cells expressing the oncogenic tyrosine kinase nucleophosmin anaplastic lymphoma kinase(NPM-ALK), a chimeric protein found in the large majority of anaplastic large cell lymphomas (ALCLs).In addition, we describe that a pool of PtdIns5P is located in the membrane extensions characteristicof NPM-ALK-transformed cells. Finally, we show that the increase of PtdIns5P is controlled by the kinasePIKfyve, which is known for its role in vesicular trafficking. These data suggest for the first time a role ofPtdIns5P and PIKfyve in oncogenesis, potentially linking intracellular trafficking to cancer.
� 2008 Elsevier Inc. All rights reserved.
Nucleophosmin anaplastic lymphoma kinase (NPM-ALK) is anoncogenic tyrosine kinase detected in most anaplastic large celllymphomas (ALCLs) of T or null immunophenotype. This chimeraresults from the t(2;5)(p23;q35) chromosomal translocation whichfuses the N-terminus of nucleophosmin (NPM) with the tyrosinekinase domain of the anaplastic lymphoma kinase (ALK) receptor[1]. The NPM oligomerization domain allows NPM-ALK dimeriza-tion and subsequent autophosphorylation and constitutive activityof the kinase, responsible for the oncogenicity [2] via stimulation ofdownstream signaling pathways leading to cell proliferation,migration and survival [3].
NPM-ALK activates several signaling effectors including phos-phoinositides (PIs)-metabolizing enzymes, such as phospholipaseC and PI 3-kinase. PIs are quantitatively minor phospholipids buthave a highly active and controlled metabolism. Through theircapacity to bind specific protein domains like pleckstrin homology(PH), Fab1p/YOYB/vac1p/EEA1 (FYVE) or Phox homology (PX) do-mains, PIs are now recognized as essential spatio-temporal regula-tors of signaling pathways regulating cytoskeleton reorganization,cell proliferation and vesicular trafficking [4]. Supporting this,mutations in the kinases or phosphatases that specifically regulatetheir metabolism lead to cancer or genetic diseases [5]. Within thePI family, phosphatidylinositol 5-monophosphate (PtdIns5P) wasrelatively recently characterized and its metabolism and function
ll rights reserved.
TP, Département Oncogénèse,an, BP 3028, 31024 Toulouse
onchère).
are yet not fully understood [6]. Like the PI 3-kinase product phos-phatidylinositol-3,4,5-trisphosphate, PtdIns5P may function as a li-pid second messenger and its level was shown to increasefollowing agonist stimulation [7], osmotic shock [8] or during theG1 phase [9].
Several routes for PtdIns5P metabolization have been proposed,suggesting the existence of distinct intracellular PtdIns5P pools.We have shown, in the case of bacterial infection by Shigella flex-neri, that PtdIns5P production at the plasma membrane leads toPI 3-kinase/Akt activation [10]. The virulence factor IpgD, injectedby the bacteria in the host cell, is a phosphatidylinositol-4,5-bis-phosphate (PtdIns(4,5)P2) 4-phosphatase [11] and generatesPtdIns5P at the bacteria entry sites. Other studies suggest a rolefor PtdIns5P in the nucleus, where it can bind the PHD domain ofthe tumor suppressor inhibitor of growth protein-2 (ING2) andregulate p53 acetylation and apoptosis [12]. Another pool ofPtdIns5P is likely to exist at intracellular membranes due to phos-phatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2) hydrolysis viathe myotubularin family members of 3-phosphatases [13].PtdIns5P levels can also be modulated by the type II PIP 4-kinaseand the type I 4-phosphatase, which convert PtdIns5P intoPtdIns(4,5)P2 and PtdIns(4,5)P2 into PtdIns5P, respectively, underUV or genotoxic stress conditions [14,15]. Finally, the 5-kinase PIK-fyve has been proposed to be involved in PtdIns5P synthesis eitherdirectly via PtdIns phosphorylation or indirectly via the productionof PtdIns(3,5)P2 that could be in turn dephosphorylated by 3-phos-phatases like myotubularins [16,17].
The fact that PtdIns5P regulates the PI 3-kinase/Akt pathway[10] and is increased in the nucleus during the G1 phase of the cell
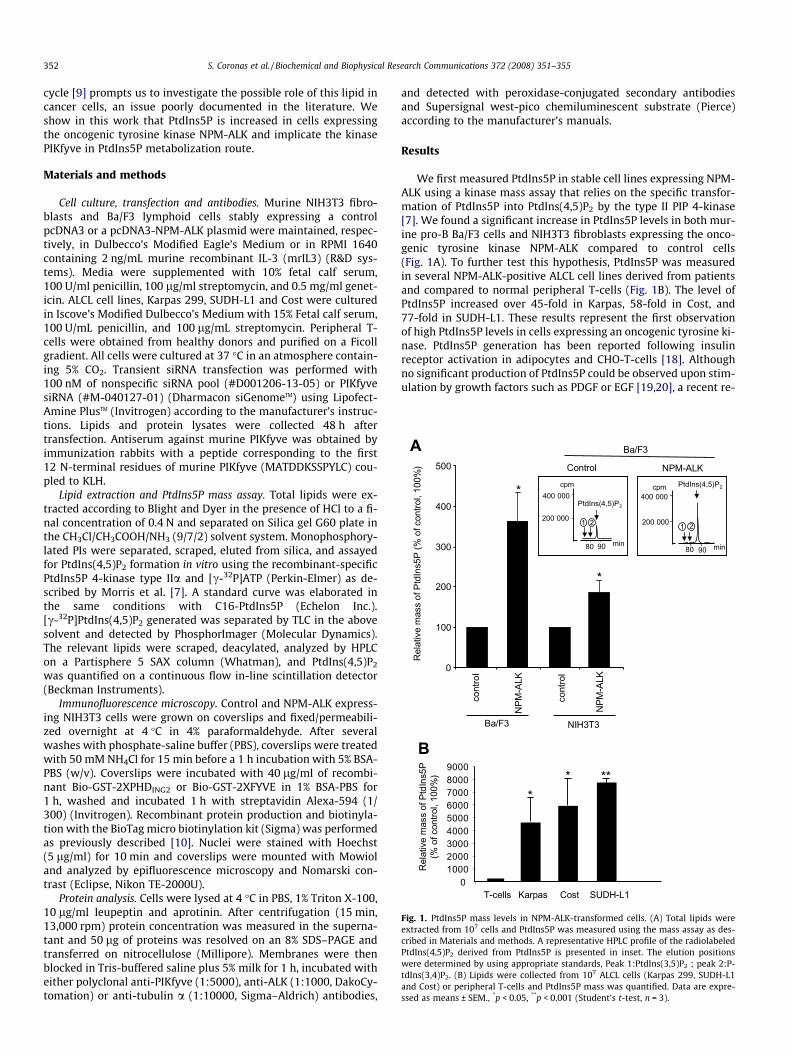
B
Rel
ativ
e m
ass
of P
tdIn
s5P
(% o
f con
trol,
100%
)
0100020003000400050006000700080009000
T-cells Karpas Cost SUDH-L1
** **
NIH3T3Ba/F3
A
PtdIns(4,5)P2cpm
min80 90
400 000
200 000
Ba/F3
Control NPM-ALK
min
PtdIns(4,5)P2
cpm
80 90
400 000
200 000 1 212
*
*
0
100
200
300
400
500
NPM
-ALK
cont
rol
NPM
-ALK
cont
rol
Rel
ativ
e m
ass
of P
tdIn
s5P
(% o
f con
trol,
100%
)
Fig. 1. PtdIns5P mass levels in NPM-ALK-transformed cells. (A) Total lipids wereextracted from 107 cells and PtdIns5P was measured using the mass assay as des-cribed in Materials and methods. A representative HPLC profile of the radiolabeledPtdIns(4,5)P2 derived from PtdIns5P is presented in inset. The elution positionswere determined by using appropriate standards, Peak 1:PtdIns(3,5)P2 ; peak 2:P-tdIns(3,4)P2. (B) Lipids were collected from 107 ALCL cells (Karpas 299, SUDH-L1and Cost) or peripheral T-cells and PtdIns5P mass was quantified. Data are expre-ssed as means ± SEM., *p < 0.05, **p < 0.001 (Student’s t-test, n = 3).
352 S. Coronas et al. / Biochemical and Biophysical Research Communications 372 (2008) 351–355
cycle [9] prompts us to investigate the possible role of this lipid incancer cells, an issue poorly documented in the literature. Weshow in this work that PtdIns5P is increased in cells expressingthe oncogenic tyrosine kinase NPM-ALK and implicate the kinasePIKfyve in PtdIns5P metabolization route.
Materials and methods
Cell culture, transfection and antibodies. Murine NIH3T3 fibro-blasts and Ba/F3 lymphoid cells stably expressing a controlpcDNA3 or a pcDNA3-NPM-ALK plasmid were maintained, respec-tively, in Dulbecco’s Modified Eagle’s Medium or in RPMI 1640containing 2 ng/mL murine recombinant IL-3 (mrIL3) (R&D sys-tems). Media were supplemented with 10% fetal calf serum,100 U/ml penicillin, 100 lg/ml streptomycin, and 0.5 mg/ml genet-icin. ALCL cell lines, Karpas 299, SUDH-L1 and Cost were culturedin Iscove’s Modified Dulbecco’s Medium with 15% Fetal calf serum,100 U/mL penicillin, and 100 lg/mL streptomycin. Peripheral T-cells were obtained from healthy donors and purified on a Ficollgradient. All cells were cultured at 37 �C in an atmosphere contain-ing 5% CO2. Transient siRNA transfection was performed with100 nM of nonspecific siRNA pool (#D001206-13-05) or PIKfyvesiRNA (#M-040127-01) (Dharmacon siGenomeTM) using Lipofect-Amine PlusTM (Invitrogen) according to the manufacturer’s instruc-tions. Lipids and protein lysates were collected 48 h aftertransfection. Antiserum against murine PIKfyve was obtained byimmunization rabbits with a peptide corresponding to the first12 N-terminal residues of murine PIKfyve (MATDDKSSPYLC) cou-pled to KLH.
Lipid extraction and PtdIns5P mass assay. Total lipids were ex-tracted according to Blight and Dyer in the presence of HCl to a fi-nal concentration of 0.4 N and separated on Silica gel G60 plate inthe CH3Cl/CH3COOH/NH3 (9/7/2) solvent system. Monophosphory-lated PIs were separated, scraped, eluted from silica, and assayedfor PtdIns(4,5)P2 formation in vitro using the recombinant-specificPtdIns5P 4-kinase type IIa and [c-32P]ATP (Perkin-Elmer) as de-scribed by Morris et al. [7]. A standard curve was elaborated inthe same conditions with C16-PtdIns5P (Echelon Inc.).[c-32P]PtdIns(4,5)P2 generated was separated by TLC in the abovesolvent and detected by PhosphorImager (Molecular Dynamics).The relevant lipids were scraped, deacylated, analyzed by HPLCon a Partisphere 5 SAX column (Whatman), and PtdIns(4,5)P2
was quantified on a continuous flow in-line scintillation detector(Beckman Instruments).
Immunofluorescence microscopy. Control and NPM-ALK express-ing NIH3T3 cells were grown on coverslips and fixed/permeabili-zed overnight at 4 �C in 4% paraformaldehyde. After severalwashes with phosphate-saline buffer (PBS), coverslips were treatedwith 50 mM NH4Cl for 15 min before a 1 h incubation with 5% BSA-PBS (w/v). Coverslips were incubated with 40 lg/ml of recombi-nant Bio-GST-2XPHDING2 or Bio-GST-2XFYVE in 1% BSA-PBS for1 h, washed and incubated 1 h with streptavidin Alexa-594 (1/300) (Invitrogen). Recombinant protein production and biotinyla-tion with the BioTag micro biotinylation kit (Sigma) was performedas previously described [10]. Nuclei were stained with Hoechst(5 lg/ml) for 10 min and coverslips were mounted with Mowioland analyzed by epifluorescence microscopy and Nomarski con-trast (Eclipse, Nikon TE-2000U).
Protein analysis. Cells were lysed at 4 �C in PBS, 1% Triton X-100,10 lg/ml leupeptin and aprotinin. After centrifugation (15 min,13,000 rpm) protein concentration was measured in the superna-tant and 50 lg of proteins was resolved on an 8% SDS–PAGE andtransferred on nitrocellulose (Millipore). Membranes were thenblocked in Tris-buffered saline plus 5% milk for 1 h, incubated witheither polyclonal anti-PIKfyve (1:5000), anti-ALK (1:1000, DakoCy-tomation) or anti-tubulin a (1:10000, Sigma–Aldrich) antibodies,
and detected with peroxidase-conjugated secondary antibodiesand Supersignal west-pico chemiluminescent substrate (Pierce)according to the manufacturer’s manuals.
Results
We first measured PtdIns5P in stable cell lines expressing NPM-ALK using a kinase mass assay that relies on the specific transfor-mation of PtdIns5P into PtdIns(4,5)P2 by the type II PIP 4-kinase[7]. We found a significant increase in PtdIns5P levels in both mur-ine pro-B Ba/F3 cells and NIH3T3 fibroblasts expressing the onco-genic tyrosine kinase NPM-ALK compared to control cells(Fig. 1A). To further test this hypothesis, PtdIns5P was measuredin several NPM-ALK-positive ALCL cell lines derived from patientsand compared to normal peripheral T-cells (Fig. 1B). The level ofPtdIns5P increased over 45-fold in Karpas, 58-fold in Cost, and77-fold in SUDH-L1. These results represent the first observationof high PtdIns5P levels in cells expressing an oncogenic tyrosine ki-nase. PtdIns5P generation has been reported following insulinreceptor activation in adipocytes and CHO-T-cells [18]. Althoughno significant production of PtdIns5P could be observed upon stim-ulation by growth factors such as PDGF or EGF [19,20], a recent re-
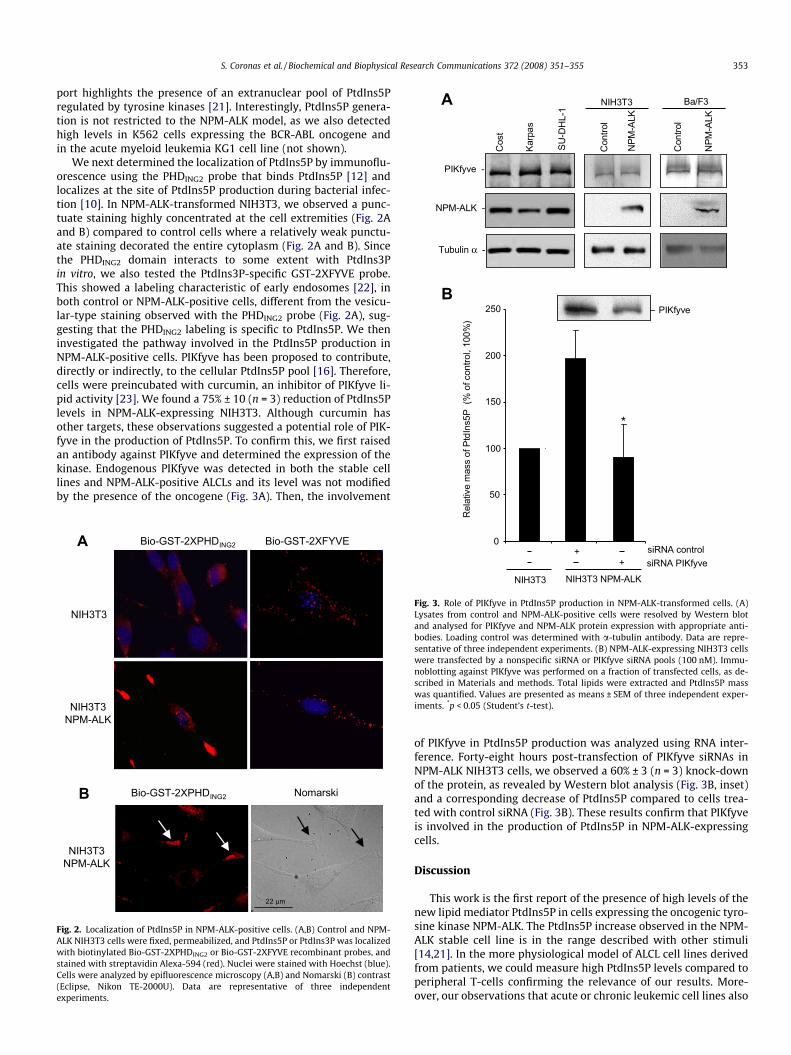
B
A
Tubulin α -
NPM-ALK -
Cos
t
SU-D
HL-
1
PIKfyve -
Karp
as
NPM
-ALK
Con
trol
NIH3T3 Ba/F3
NPM
-ALK
Con
trol
*
PIKfyve
50
100
150
200
250
lativ
em
ass
ofPt
dIns
5P(%
ofco
ntro
l,10
0%)
S. Coronas et al. / Biochemical and Biophysical Research Communications 372 (2008) 351–355 353
port highlights the presence of an extranuclear pool of PtdIns5Pregulated by tyrosine kinases [21]. Interestingly, PtdIns5P genera-tion is not restricted to the NPM-ALK model, as we also detectedhigh levels in K562 cells expressing the BCR-ABL oncogene andin the acute myeloid leukemia KG1 cell line (not shown).
We next determined the localization of PtdIns5P by immunoflu-orescence using the PHDING2 probe that binds PtdIns5P [12] andlocalizes at the site of PtdIns5P production during bacterial infec-tion [10]. In NPM-ALK-transformed NIH3T3, we observed a punc-tuate staining highly concentrated at the cell extremities (Fig. 2Aand B) compared to control cells where a relatively weak punctu-ate staining decorated the entire cytoplasm (Fig. 2A and B). Sincethe PHDING2 domain interacts to some extent with PtdIns3Pin vitro, we also tested the PtdIns3P-specific GST-2XFYVE probe.This showed a labeling characteristic of early endosomes [22], inboth control or NPM-ALK-positive cells, different from the vesicu-lar-type staining observed with the PHDING2 probe (Fig. 2A), sug-gesting that the PHDING2 labeling is specific to PtdIns5P. We theninvestigated the pathway involved in the PtdIns5P production inNPM-ALK-positive cells. PIKfyve has been proposed to contribute,directly or indirectly, to the cellular PtdIns5P pool [16]. Therefore,cells were preincubated with curcumin, an inhibitor of PIKfyve li-pid activity [23]. We found a 75% ± 10 (n = 3) reduction of PtdIns5Plevels in NPM-ALK-expressing NIH3T3. Although curcumin hasother targets, these observations suggested a potential role of PIK-fyve in the production of PtdIns5P. To confirm this, we first raisedan antibody against PIKfyve and determined the expression of thekinase. Endogenous PIKfyve was detected in both the stable celllines and NPM-ALK-positive ALCLs and its level was not modifiedby the presence of the oncogene (Fig. 3A). Then, the involvement
A
B
NIH3T3NPM-ALK
Bio-GST-2XPHDING2 Bio-GST-2XFYVE
NIH3T3
NIH3T3NPM-ALK
Bio-GST-2XPHDING2 Nomarski
22 μm
Fig. 2. Localization of PtdIns5P in NPM-ALK-positive cells. (A,B) Control and NPM-ALK NIH3T3 cells were fixed, permeabilized, and PtdIns5P or PtdIns3P was localizedwith biotinylated Bio-GST-2XPHDING2 or Bio-GST-2XFYVE recombinant probes, andstained with streptavidin Alexa-594 (red). Nuclei were stained with Hoechst (blue).Cells were analyzed by epifluorescence microscopy (A,B) and Nomarski (B) contrast(Eclipse, Nikon TE-2000U). Data are representative of three independentexperiments.
NIH3T3 NPM-ALKsiRNA PIKfyve siRNA control+
+
0
NIH3T3
Re
Fig. 3. Role of PIKfyve in PtdIns5P production in NPM-ALK-transformed cells. (A)Lysates from control and NPM-ALK-positive cells were resolved by Western blotand analysed for PIKfyve and NPM-ALK protein expression with appropriate anti-bodies. Loading control was determined with a-tubulin antibody. Data are repre-sentative of three independent experiments. (B) NPM-ALK-expressing NIH3T3 cellswere transfected by a nonspecific siRNA or PIKfyve siRNA pools (100 nM). Immu-noblotting against PIKfyve was performed on a fraction of transfected cells, as de-scribed in Materials and methods. Total lipids were extracted and PtdIns5P masswas quantified. Values are presented as means ± SEM of three independent exper-iments. *p < 0.05 (Student’s t-test).
of PIKfyve in PtdIns5P production was analyzed using RNA inter-ference. Forty-eight hours post-transfection of PIKfyve siRNAs inNPM-ALK NIH3T3 cells, we observed a 60% ± 3 (n = 3) knock-downof the protein, as revealed by Western blot analysis (Fig. 3B, inset)and a corresponding decrease of PtdIns5P compared to cells trea-ted with control siRNA (Fig. 3B). These results confirm that PIKfyveis involved in the production of PtdIns5P in NPM-ALK-expressingcells.
Discussion
This work is the first report of the presence of high levels of thenew lipid mediator PtdIns5P in cells expressing the oncogenic tyro-sine kinase NPM-ALK. The PtdIns5P increase observed in the NPM-ALK stable cell line is in the range described with other stimuli[14,21]. In the more physiological model of ALCL cell lines derivedfrom patients, we could measure high PtdIns5P levels compared toperipheral T-cells confirming the relevance of our results. More-over, our observations that acute or chronic leukemic cell lines also

354 S. Coronas et al. / Biochemical and Biophysical Research Communications 372 (2008) 351–355
have higher PtdIns5P levels strongly suggest that this feature couldbe a general hallmark of transformed cells expressing deregulatedtyrosine kinase activities. Consistent with the implication of tyro-sine kinases in the regulation of PtdIn5P, a recent report describesa role for type IIa PIP 4-kinase in the control of a tyrosine kinase-dependent pool of PtdIns5P [21]. However we cannot exclude thatPtdIns5P elevation is the consequence of the cellular stress inducedby the expression of the oncogene. Indeed, PtdIns5P was shown toincrease under different stress situations like osmotic, UV or geno-toxic stress [8,12,14,15]. UV or genotoxic stress generated a nucle-ar PtdIns5P pool involved in p53-mediated apoptosis andoriginating from PtdIns(4,5)P2 either by inhibition of the PIP 4-ki-nase or by activation of 4-phosphatases [12,14,15]. However, sofar, the extranuclear pool of PtdIns5P has not been shown to beupregulated by stress [21]. Our results point out to the existenceof a PtdIns5P pool in proliferative cells involving PIKfyve andshowing a punctuate labeling highly concentrated in NPM-ALK-NIH3T3 cell extensions, consistent with an extranuclear pool ofthis lipid. These cytoplasmic extensions characteristic of NPM-ALK-transformed fibroblasts are thought to result from activationof the deregulated tyrosine kinase activity on downstream effec-tors regulating cytoskeleton rearrangements such as Rac1 [24]. Inaddition, we cannot totally exclude the possibility of a nuclear poolof PtdIns5P in these cells as the GST-2XPHDING2 probe does not ac-cess the intranuclear compartment under our experimentalconditions.
In this work, we describe a role for PIKfyve in controlling aPtdIns5P pool in NPM-ALK-positive cells. Downregulation of PIK-fyve by siRNAs in NIH3T3 NPM-ALK cells leads to the reductionof PtdIns5P back to its background level found in NIH3T3 controlcells. As the oncogene does not regulate PIKfyve at the protein orRNA level as observed by q-RT-PCR experiments (not shown), webelieve that PIKfyve activity could be increased in the NPM-ALK-expressing cells. This issue is currently studied in the laboratory.The contribution of PIKfyve to the in vivo PtdIns5P pool is still amatter of debate and PIKfyve could be directly and/or indirectlyimplicated in PtdIns5P production. Like its yeast ortholog Fab1p,PIKfyve, which harbors a FYVE domain able to bind PtdIns3P, phos-phorylates PtdIns3P, and generates PtdIns(3,5)P2 which could be,in turn, dephosphorylated by 3-phosphatases like myotubularinsto generate PtdIns5P. But, PIKfyve could also phosphorylate PtdInsto directly synthetize PtdIns5P, as proposed in vitro [25]. Althoughin our hands recombinant PIKfyve has a marked preference forPtdIns3P over PtdIns as a substrate (not shown), we cannot yet dis-criminate between the two hypotheses and more experiments areneeded to answer that point.
What could be the function of this PIKfyve-dependentPtdIns5P pool in NPM-ALK-transformed cells? PIKfyve has akey role in the regulation of endomembrane homeostasis, affect-ing different steps of the endocytic transport through the syn-thesis of PtdIns(3,5)P2 [17]. The function of PIKfyve throughPtdIns5P production is poorly documented. PtdIns5P has beenimplicated in F-actin stress fiber remodeling in response to insu-lin, however the involvement of PIKfyve in this PtdIns5P synthe-sis remains elusive [18]. Interestingly, transformation by NPM-ALK induces actin depolymerization [26]. Our observations,showing that the main PtdIns5P pool is localized in NPM-ALK-transformed cells extensions, suggest that the lipid could playa role in the regulation of these highly dynamic actin remodelingsites. Moreover, like PtdIns(3,5)P2, whose role in membrane andprotein trafficking is well documented [27], PtdIns5P could alsorepresent a new intermediate in the regulation of intracellulartrafficking. PtdIns5P punctuate localization in NPM-ALK-positivecells could be in agreement with this hypothesis.
The downregulation of growth factor receptor activation by theendocytic pathway underlines a key role of trafficking, however
the issue of its impact in cell transformation mechanisms remainspoorly documented. Such a link was recently proposed for thecytoplasmic tyrosine kinase Src [28]. NPM-ALK-transformed cellsshow enhanced proliferation and migration capacities [3]. There-fore, vesicular trafficking could play a major role through an in-creased exocytosis of molecules like the matrixmetalloproteinases to facilitate degradation of the extracellularmatrix, or through modifications of integrin recycling pathwaysto decrease cell adhesion. PIKfyve, known for its role in vesicularhomeostasis, could be one of the key players in this pathway andits role in cell transformation is currently under investigation inour laboratory.
In conclusion, this study reports the first observation of ele-vated PtdIns5P levels in NPM-ALK-transformed cells and suggestsa role for the lipid and PIKfyve during oncogenesis that could linkvesicle trafficking to cell transformation.
Acknowledgments
We thank O. Gozani for the pGEX-2XPHD and N. Divecha for thetype IIa PIP 4-kinase. This work was supported by grants from IN-SERM, ARC (#4937), ‘‘Cancéropôle Grand Sud-Ouest”, ‘‘Institut Na-tional du Cancer” (INCa), the Région Midi-Pyrénées and the ‘‘Pôlede Compétitivité Cancer-Bio Santé/Fonds uniques interministérielsdes pôles de compétitivité”. SC is supported by grants from Min-istère de la Recherche and ARC. We thank K. Thornber for proof-reading the manuscript.
References
[1] S.W. Morris, M.N. Kirstein, M.B. Valentine, K.G. Dittmer, D.N. Shapiro, D.L.Saltman, A.T. Look, Fusion of a kinase gene, ALK, to a nucleolar protein gene,NPM, in non-Hodgkin’s lymphoma, Science 263 (1994) 1281–1284.
[2] R.Y. Bai, T. Ouyang, C. Miething, S.W. Morris, C. Peschel, J. Duyster,Nucleophosmin-anaplastic lymphoma kinase associated with anaplasticlarge-cell lymphoma activates the phosphatidylinositol 3-kinase/Aktantiapoptotic signaling pathway, Blood 96 (2000) 4319–4327.
[3] R. Chiarle, C. Voena, C. Ambrogio, R. Piva, G. Inghirami, The anaplastic lymphomakinase in the pathogenesis of cancer, Nat. Rev. Cancer 8 (2008) 11–23.
[4] B. Payrastre, K. Missy, S. Giuriato, S. Bodin, M. Plantavid, M. Gratacap,Phosphoinositides: key players in cell signalling, in time and space, Cell.Signal. 13 (2001) 377–387.
[5] C. Pendaries, H. Tronchere, M. Plantavid, B. Payrastre, Phosphoinositidesignaling disorders in human diseases, FEBS Lett. 546 (2003) 25–31.
[6] S. Coronas, D. Ramel, C. Pendaries, F. Gaits-Iacovoni, H. Tronchere, B. Payrastre,PtdIns5P: a little phosphoinositide with big functions?, Biochem Soc. Symp. 74(2007) 117–128.
[7] J.B. Morris, K.A. Hinchliffe, A. Ciruela, A.J. Letcher, R.F. Irvine, Thrombinstimulation of platelets causes an increase in phosphatidylinositol 5-phosphate revealed by mass assay, FEBS Lett. 475 (2000) 57–60.
[8] H.J. Meijer, S.A. Arisz, J.A. Van Himbergen, A. Musgrave, T. Munnik,Hyperosmotic stress rapidly generates lyso-phosphatidic acid inChlamydomonas, Plant J. 25 (2001) 541–548.
[9] J.H. Clarke, A.J. Letcher, C.S. D’Santos, J.R. Halstead, R.F. Irvine, N. Divecha,Inositol lipids are regulated during cell cycle progression in the nuclei ofmurine erythroleukaemia cells, Biochem. J. 357 (2001) 905–910.
[10] C. Pendaries, H. Tronchere, L. Arbibe, J. Mounier, O. Gozani, L. Cantley, M.J. Fry,F. Gaits-Iacovoni, P.J. Sansonetti, B. Payrastre, PtdIns5P activates the host cellPI3-kinase/Akt pathway during Shigella flexneri infection, EMBO J. 25 (2006)1024–1034.
[11] K. Niebuhr, S. Giuriato, T. Pedron, D.J. Philpott, F. Gaits, J. Sable, M.P. Sheetz, C.Parsot, P.J. Sansonetti, B. Payrastre, Conversion of PtdIns(4,5)P(2) intoPtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology,EMBO J. 21 (2002) 5069–5078.
[12] O. Gozani, P. Karuman, D.R. Jones, D. Ivanov, J. Cha, A.A. Lugovskoy, C.L. Baird,H. Zhu, S.J. Field, S.L. Lessnick, J. Villasenor, B. Mehrotra, J. Chen, V.R. Rao, J.S.Brugge, C.G. Ferguson, B. Payrastre, D.G. Myszka, L.C. Cantley, G. Wagner, N.Divecha, G.D. Prestwich, J. Yuan, The PHD finger of the chromatin-associatedprotein ING2 functions as a nuclear phosphoinositide receptor, Cell 114 (2003)99–111.
[13] H. Tronchere, J. Laporte, C. Pendaries, C. Chaussade, L. Liaubet, L. Pirola, J.L.Mandel, B. Payrastre, Production of phosphatidylinositol 5-phosphate by thephosphoinositide 3-phosphatase myotubularin in mammalian cells, J. Biol.Chem. 279 (2004) 7304–7312.
[14] D.R. Jones, Y. Bultsma, W.J. Keune, J.R. Halstead, D. Elouarrat, S. Mohammed,A.J. Heck, C.S. D’Santos, N. Divecha, Nuclear PtdIns5P as a transducer of stresssignaling: an in vivo role for PIP4Kbeta, Mol. Cell 23 (2006) 685–695.

S. Coronas et al. / Biochemical and Biophysical Research Communications 372 (2008) 351–355 355
[15] J. Zou, J. Marjanovic, M.V. Kisseleva, M. Wilson, P.W. Majerus, Type Iphosphatidylinositol-4,5-bisphosphate 4-phosphatase regulates stress-induced apoptosis, Proc. Natl. Acad. Sci. USA 104 (2007) 16834–16839.
[16] D. Sbrissa, O.C. Ikonomov, R. Deeb, A. Shisheva, Phosphatidylinositol 5-phosphate biosynthesis is linked to PIKfyve and is involved in osmoticresponse pathway in mammalian cells, J. Biol. Chem. 277 (2002) 47276–47284.
[17] A. Shisheva, PIKfyve: partners, significance, debates and paradoxes, Cell Biol.Int. (2008), doi:10.1016/j.cellbi.2008.01.006.
[18] D. Sbrissa, O.C. Ikonomov, J. Strakova, A. Shisheva, Role for a novel signalingintermediate, phosphatidylinositol 5-phosphate, in insulin-regulated F-actinstress fiber breakdown and GLUT4 translocation, Endocrinology 145 (2004)4853–4865.
[19] D. Sbrissa, O. Ikonomov, A. Shisheva, Selective insulin-induced activation ofclass I(A) phosphoinositide 3-kinase in PIKfyve immune complexes from 3T3-L1 adipocytes, Mol. Cell. Endocrinol. 181 (2001) 35–46.
[20] H.F. Roberts, J.H. Clarke, A.J. Letcher, R.F. Irvine, K.A. Hinchliffe, Effects of lipidkinase expression and cellular stimuli on phosphatidylinositol 5-phosphatelevels in mammalian cell lines, FEBS Lett. 579 (2005) 2868–2872.
[21] A. Wilcox, K.A. Hinchliffe, Regulation of extranuclear PtdIns5P production byphosphatidylinositol phosphate 4-kinase 2alpha, FEBS Lett. 582 (2008) 1391–1394.
[22] D.J. Gillooly, I.C. Morrow, M. Lindsay, R. Gould, N.J. Bryant, J.M. Gaullier, R.G.Parton, H. Stenmark, Localization of phosphatidylinositol 3-phosphate in yeastand mammalian cells, EMBO J. 19 (2000) 4577–4588.
[23] O.C. Ikonomov, D. Sbrissa, K. Mlak, A. Shisheva, Requirement for PIKfyveenzymatic activity in acute and long-term insulin cellular effects,Endocrinology 143 (2002) 4742–4754.
[24] A. Colomba, D. Courilleau, D. Ramel, D.D. Billadeau, E. Espinos, G. Delsol, B.Payrastre, F. Gaits-Iacovoni, Activation of Rac1 and the exchange factor Vav3are involved in NPM-ALK signaling in anaplastic large cell lymphomas,Oncogene 27 (2008) 2728–2736.
[25] D. Sbrissa, O.C. Ikonomov, A. Shisheva, PIKfyve, a mammalian ortholog of yeastFab1p lipid kinase, synthesizes 5-phosphoinositides. Effect of insulin, J. Biol.Chem. 274 (1999) 21589–21597.
[26] C. Ambrogio, C. Voena, A.D. Manazza, R. Piva, L. Riera, L. Barberis, C. Costa, G.Tarone, P. Defilippi, E. Hirsch, E. Boeri Erba, S. Mohammed, O.N. Jensen, G.Palestro, G. Inghirami, R. Chiarle, p130Cas mediates the transformingproperties of the anaplastic lymphoma kinase, Blood 106 (2005) 3907–3916.
[27] R.H. Michell, V.L. Heath, M.A. Lemmon, S.K. Dove, Phosphatidylinositol 3,5-bisphosphate: metabolism and cellular functions, Trends Biochem. Sci. 31(2006) 52–63.
[28] G. Collin, M. Franco, V. Simon, C. Benistant, S. Roche, The Tom1L1-clathrinheavy chain complex regulates membrane partitioning of the tyrosine kinaseSrc required for mitogenic and transforming activities, Mol. Cell. Biol. 27(2007) 7631–7640.

Résultats
II- Conclusion
Dans cette étude, nous avons dosé des taux de PtdIns5P élevés aussi bien dans des cellules
murines pro-B Ba/F3 que dans des fibroblastes NIH3T3 exprimant l’oncogène NPM-ALK en
comparaison avec des cellules contrôles. Ces taux augmentés de PtdIns5P sont également
retrouvés dans des lignées d’ALCL NPM-ALK positives issues de patients. Ces données
suggèrent que l’oncogène peut réguler le taux de PtdIns5P. De plus, nous avons pu montrer
par immunofluorescence que dans les cellules NIH3T3 NPM-ALK, le PtdIns5P présente une
localisation ponctiforme hautement concentrée au niveau des extensions membranaires en
comparaison avec les cellules NIH3T3 contrôles où le marquage du PtdIns5P est plus faible et
plus diffus. Il est intéressant de noter que ces extensions, caractéristiques des cellules NIH3T3
NPM-ALK positives, semblent être des sites actifs du remodelage de l’actine (Colomba,
2007) suggérant que ce lipide pourrait jouer un rôle sur le remodelage du cytosquelette
comme cela a été suggéré dans le contexte d’une stimulation à l’insuline (Sbrissa, 2004).
Enfin, nous montrons par technique de siRNA, que la PtdIns 5-kinase PIKfyve est impliquée
dans cette production de PtdIns5P dans les cellules exprimant NPM-ALK. Cependant, la voie
utilisée par PIKfyve pour produire le PtdIns5P in vivo est encore sujette à débat. En effet,
PIKfyve pourrait produire directement du PtdIns5P par phosphorylation du PtdIns ce qui a été
montré in vitro, mais cette enzyme pourrait produire du PtdIns(3,5)P2 à partir du PtdIns3P qui
pourrait à son tour générer du PtdIns5P via l’action de 3-phosphatases. A ce jour, dans notre
modèle, nous n’avons pas encore tranché entre ces deux hypothèses.
En conclusion, cette étude révèle pour la première fois l’existence d’un pool de PtdIns5P dans
des cellules oncogéniques en prolifération mettant en évidence le rôle de la PtdIns 5-kinase
PIKfyve. Ainsi, nous pouvons nous demander quel est le rôle d’une enzyme largement
documentée pour son rôle clé dans la régulation de l’homéostasie vésiculaire via sa synthèse
de PtdIns(3,5)P2 dans le contexte de l’oncogenèse. Le PtdIns5P produit par PIKfyve aurait
plutôt un rôle dans la réorganisation du cytosquelette d’actine (Sbrissa, 2004), mais un rôle du
PtdIns5P en tant que nouvel intermédiaire dans le contrôle du trafic vésiculaire ne peut être
exclu. Il n’existe à l’heure actuelle que peu d’études reliant le cancer et le trafic
intravésiculaire (Polo, 2004). On peut penser que la régulation du trafic de vésicules est
capital pour de nombreux évènements qui sont exacerbés lors des mécanismes de
transformation cellulaire ; notamment la sécrétion de molécules actives (cytokines,
106
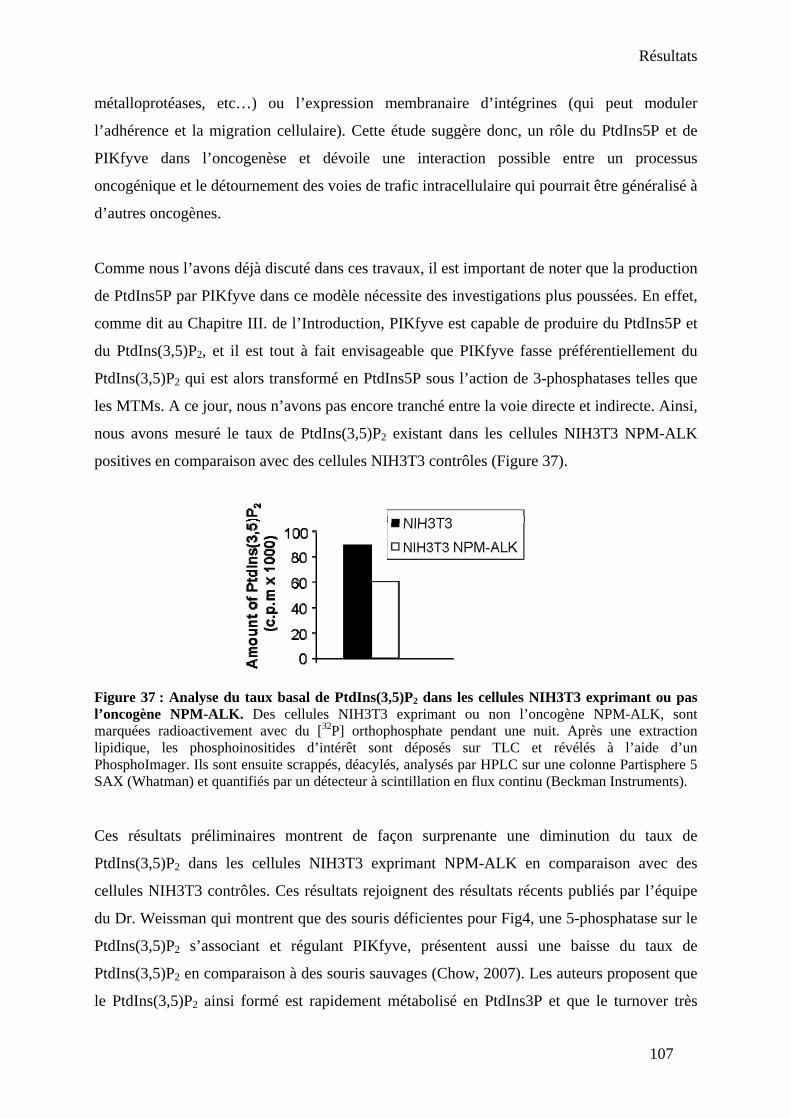
Résultats
métalloprotéases, etc…) ou l’expression membranaire d’intégrines (qui peut moduler
l’adhérence et la migration cellulaire). Cette étude suggère donc, un rôle du PtdIns5P et de
PIKfyve dans l’oncogenèse et dévoile une interaction possible entre un processus
oncogénique et le détournement des voies de trafic intracellulaire qui pourrait être généralisé à
d’autres oncogènes.
Comme nous l’avons déjà discuté dans ces travaux, il est important de noter que la production
de PtdIns5P par PIKfyve dans ce modèle nécessite des investigations plus poussées. En effet,
comme dit au Chapitre III. de l’Introduction, PIKfyve est capable de produire du PtdIns5P et
du PtdIns(3,5)P2, et il est tout à fait envisageable que PIKfyve fasse préférentiellement du
PtdIns(3,5)P2 qui est alors transformé en PtdIns5P sous l’action de 3-phosphatases telles que
les MTMs. A ce jour, nous n’avons pas encore tranché entre la voie directe et indirecte. Ainsi,
nous avons mesuré le taux de PtdIns(3,5)P2 existant dans les cellules NIH3T3 NPM-ALK
positives en comparaison avec des cellules NIH3T3 contrôles (Figure 37).
Figure 37 : Analyse du taux basal de PtdIns(3,5)P2 dans les cellules NIH3T3 exprimant ou pas l’oncogène NPM-ALK. Des cellules NIH3T3 exprimant ou non l’oncogène NPM-ALK, sont marquées radioactivement avec du [32P] orthophosphate pendant une nuit. Après une extraction lipidique, les phosphoinositides d’intérêt sont déposés sur TLC et révélés à l’aide d’un PhosphoImager. Ils sont ensuite scrappés, déacylés, analysés par HPLC sur une colonne Partisphere 5 SAX (Whatman) et quantifiés par un détecteur à scintillation en flux continu (Beckman Instruments). Ces résultats préliminaires montrent de façon surprenante une diminution du taux de
PtdIns(3,5)P2 dans les cellules NIH3T3 exprimant NPM-ALK en comparaison avec des
cellules NIH3T3 contrôles. Ces résultats rejoignent des résultats récents publiés par l’équipe
du Dr. Weissman qui montrent que des souris déficientes pour Fig4, une 5-phosphatase sur le
PtdIns(3,5)P2 s’associant et régulant PIKfyve, présentent aussi une baisse du taux de
PtdIns(3,5)P2 en comparaison à des souris sauvages (Chow, 2007). Les auteurs proposent que
le PtdIns(3,5)P2 ainsi formé est rapidement métabolisé en PtdIns3P et que le turnover très
107

Résultats
rapide du PtdIns(3,5)P2 ne permet pas d’observer l’augmentation attendue de ce PI. Aux vues
de nos résultats et de ceux de Chow et al., nous pouvons envisager que le modèle
d’oncogenèse associée à NPM-ALK puisse impliquer une phosphatase telle que Fig4, ce qui
expliquerait aussi la chute du taux de PtdIns(3,5)P2. Mais, la chute du taux de PtdIns(3,5)P2
pourrait aussi s’expliquer par l’intervention d’une 3-phosphatase de type MTMs, et rejoindrait
notre première observation d’une augmentation du taux de PtdIns5P. Il serait donc important
de doser le taux de PtdIns3P des cellules exprimant l’oncogène NPM-ALK par rapport à des
cellules contrôles pour déterminer l’éventuelle implication d’une phosphatase telle que Fig4,
mais également la présence de 3-phosphatases de la famille des MTMs dans le complexe
NPM-ALK/PIKfyve.
108

Résultats
Partie II : Etude fonctionnelle de l’interaction entre PIKfyve et NPM-ALK
Nos résultats, présentés dans la première partie suggèrent un rôle essentiel de PIKfyve dans la
production de PtdIns5P dépendante de l’oncogène NPM-ALK. La suite de nos travaux,
présentés dans cette deuxième partie, a pour objectif de confirmer la présence de PIKfyve en
aval de l’oncogène et de déterminer sa fonction dans l’oncogenèse médiée par NPM-ALK.
Pour cela nous nous sommes focalisés sur trois questions :
• Existe-il un complexe comprenant NPM-ALK et PIKfyve ?
• PIKfyve est-il un effecteur de NPM-ALK ?
• Quel peut être le rôle d’une enzyme impliquée dans le trafic
vésiculaire et un oncogène ?
Afin de mener à bien ces travaux, j’ai du dans un premier temps générer un certain nombre
d’outils. En effet, aucun outil commercial n’existait pour l’étude de cette enzyme et
l’obtention de ces outils s’est avérée un pré-requis absolument nécessaire. J’ai donc cloné
PIKfyve, généré différents mutants par mutagenèse dirigée et produit un anticorps spécifique
dirigé contre cette kinase.
I- Génération d’outils pour l’étude de PIKfyve
1. Clonage et expression de PIKfyve sauvage et mutants
Du fait de la grande taille de PIKfyve (235 kDa, 6500 pb), le clonage de son ADNc par des
techniques classiques s’est avéré très difficile. Après plusieurs essais infructueux, nous nous
sommes tournés vers la technique du TOPO Cloning. Nous avons utilisé le kit TOPO XL
PCR Cloning (Invitrogen life technologies) qui nous a permis d’insérer directement un
produit de PCR dans un vecteur pcDNA3.1. A partir d’ARN totaux extraits de cellules
NIH3T3 NPM-ALK, nous avons réalisé une reverse transcription puis une PCR en utilisant
des primers qui cadrent le cDNA de PIKfyve de souris (Accession GenBank : AF102777)
(primer sens 5’-ATGGCCACAGATGACAAGAGTTCC-3’ et anti-sens 5’-
TCAGCAATTCAGATCCAACCCTGCCC-3’). Nous avons obtenu un produit majoritaire de
109

Résultats
6,1 kb qui correspond à la taille attendue pour PIKfyve (Figure 38A). Ce produit de PCR a
été purifié et inséré dans un plasmide pcDNA3.1 Topo portant une étiquette GFP en N-
terminal. Plusieurs clones ont été obtenus et vérifiés par digestion enzymatique. Trois clones
ont été choisis et entièrement séquencés. Il est intéressant de noter que les clones obtenus sont
tous de forme PIKfyveS, décrite par l’équipe du Dr. Shisheva comme la forme courte de
PIKfyve qui diffère de la forme PIKfyveL par l’absence de 11 acides aminés entre les résidus
108 et 118 (Shisheva, 1999 ; Shisheva, 2008). L’expression de la protéine a été vérifiée dans
les cellules HEK293. Après 24h de transfection avec des doses croissantes de plasmide GFP-
PIKfyve, l’expression de la protéine est observée par immunoempreinte avec un anticorps
anti-GFP (Figure 38B) et présente une taille d’environ 260 kDa, ce qui correspond à la taille
attendue de la fusion GFP-PIKfyve.
Nous avons également construit plusieurs mutants à partir de l’ADNc codant pour GFP-
PIKfyveWT. Ainsi, à partir des données bibliographiques, nous avons choisi de faire le mutant
ponctuel GFP-PIKfyveK1831E (Shisheva, 1999) dans lequel la lysine du site kinase est
remplacée par une glutamine conduisant à une perte de l’activité kinase, ainsi que le mutant
GFP-PIKfyveS318A, la sérine 318 étant décrite comme un site de phosphorylation de PIKfyve
par Akt (Berwick, 2004). Des données récentes suggèrent également que le résidu 318 serait
un site d’autophosphorylation de PIKfyve qui contrôlerait son état d’activation (Shisheva,
2008). Les mutants (GFP-PIKfyveK1831E et GFP-PIKfyveS318A) ont été générés par mutagène
dirigée avec le kit Quick Change II XL Site-directed Mutagenesis (Stratagene). Les mutations
ont été vérifiées par séquençage.
Figure 38 : Clonage de PIKfyve. (A) Représentation du produit de PCR obtenu lors du clonage de PIKfyve. 1/10ème de la PCR de PIKfyve effectuée après RT à partir d’ARN totaux issus de cellules NIH3T3 NPM-ALK est déposée sur gel TAE/agarose 1% (B) Expression de GFP-PIKfyve dans des cellules HEK293. Les cellules HEK293 sont transfectées avec des doses croissantes de plasmide GFP-PIKfyve. Après 24h de transfection, les cellules sont lavées dans du PBS, lysées dans un tampon Laemli. Les échantillons ainsi obtenus sont soumis à une immunoempreinte révélée par un anticorps anti-GFP (1/1000e).
110
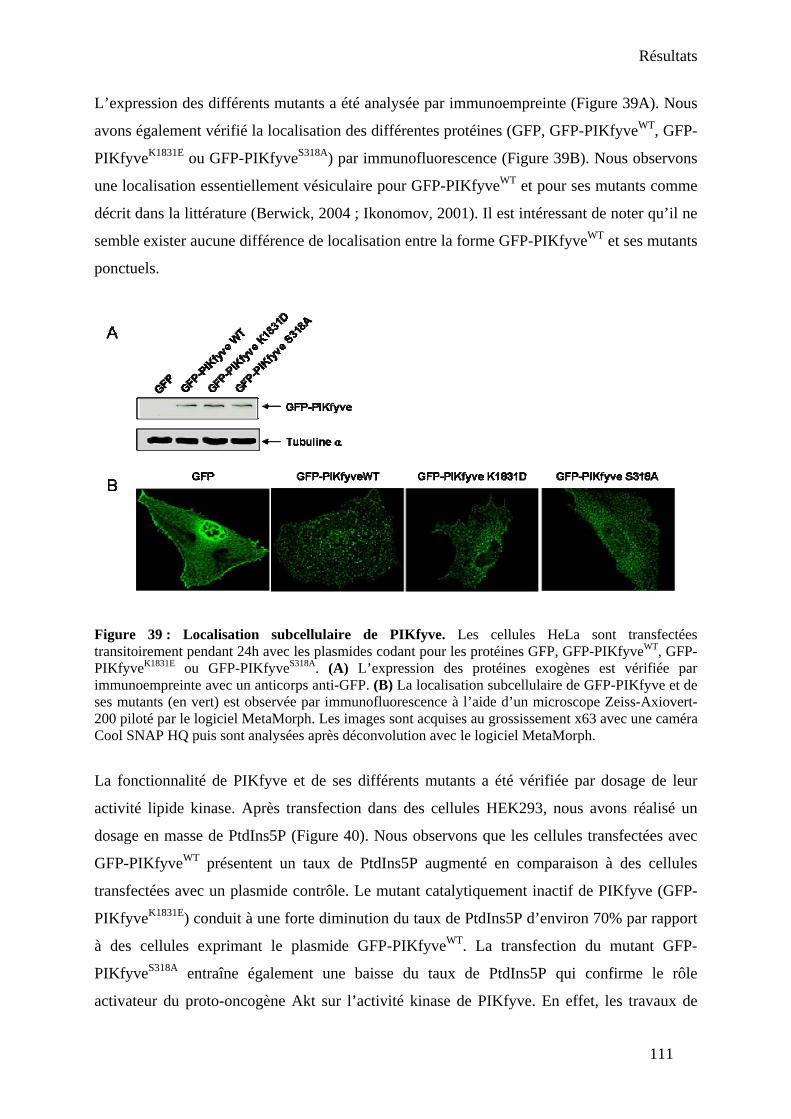
Résultats
L’expression des différents mutants a été analysée par immunoempreinte (Figure 39A). Nous
avons également vérifié la localisation des différentes protéines (GFP, GFP-PIKfyveWT, GFP-
PIKfyveK1831E ou GFP-PIKfyveS318A) par immunofluorescence (Figure 39B). Nous observons
une localisation essentiellement vésiculaire pour GFP-PIKfyveWT et pour ses mutants comme
décrit dans la littérature (Berwick, 2004 ; Ikonomov, 2001). Il est intéressant de noter qu’il ne
semble exister aucune différence de localisation entre la forme GFP-PIKfyveWT et ses mutants
ponctuels.
Figure 39 : Localisation subcellulaire de PIKfyve. Les cellules HeLa sont transfectées transitoirement pendant 24h avec les plasmides codant pour les protéines GFP, GFP-PIKfyveWT, GFP-PIKfyveK1831E ou GFP-PIKfyveS318A. (A) L’expression des protéines exogènes est vérifiée par immunoempreinte avec un anticorps anti-GFP. (B) La localisation subcellulaire de GFP-PIKfyve et de ses mutants (en vert) est observée par immunofluorescence à l’aide d’un microscope Zeiss-Axiovert-200 piloté par le logiciel MetaMorph. Les images sont acquises au grossissement x63 avec une caméra Cool SNAP HQ puis sont analysées après déconvolution avec le logiciel MetaMorph.
La fonctionnalité de PIKfyve et de ses différents mutants a été vérifiée par dosage de leur
activité lipide kinase. Après transfection dans des cellules HEK293, nous avons réalisé un
dosage en masse de PtdIns5P (Figure 40). Nous observons que les cellules transfectées avec
GFP-PIKfyveWT présentent un taux de PtdIns5P augmenté en comparaison à des cellules
transfectées avec un plasmide contrôle. Le mutant catalytiquement inactif de PIKfyve (GFP-
PIKfyveK1831E) conduit à une forte diminution du taux de PtdIns5P d’environ 70% par rapport
à des cellules exprimant le plasmide GFP-PIKfyveWT. La transfection du mutant GFP-
PIKfyveS318A entraîne également une baisse du taux de PtdIns5P qui confirme le rôle
activateur du proto-oncogène Akt sur l’activité kinase de PIKfyve. En effet, les travaux de
111
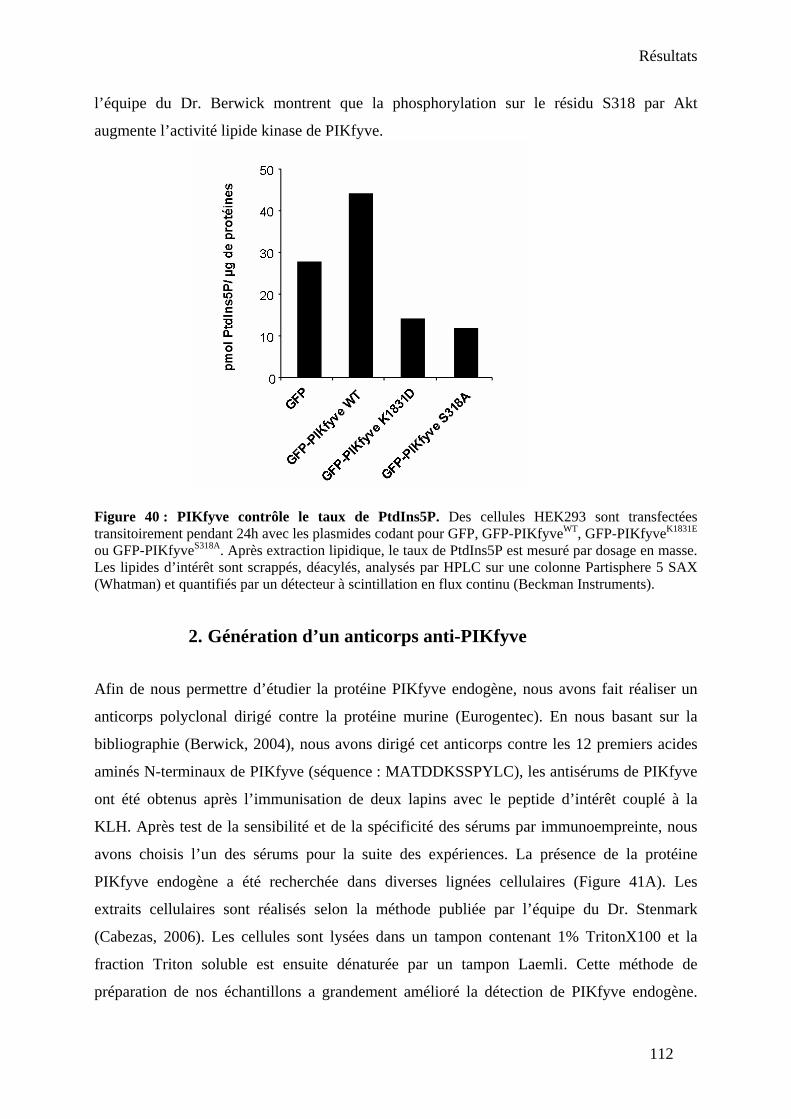
Résultats
l’équipe du Dr. Berwick montrent que la phosphorylation sur le résidu S318 par Akt
augmente l’activité lipide kinase de PIKfyve.
Figure 40 : PIKfyve contrôle le taux de PtdIns5P. Des cellules HEK293 sont transfectées transitoirement pendant 24h avec les plasmides codant pour GFP, GFP-PIKfyveWT, GFP-PIKfyveK1831E ou GFP-PIKfyveS318A. Après extraction lipidique, le taux de PtdIns5P est mesuré par dosage en masse. Les lipides d’intérêt sont scrappés, déacylés, analysés par HPLC sur une colonne Partisphere 5 SAX (Whatman) et quantifiés par un détecteur à scintillation en flux continu (Beckman Instruments).
2. Génération d’un anticorps anti-PIKfyve
Afin de nous permettre d’étudier la protéine PIKfyve endogène, nous avons fait réaliser un
anticorps polyclonal dirigé contre la protéine murine (Eurogentec). En nous basant sur la
bibliographie (Berwick, 2004), nous avons dirigé cet anticorps contre les 12 premiers acides
aminés N-terminaux de PIKfyve (séquence : MATDDKSSPYLC), les antisérums de PIKfyve
ont été obtenus après l’immunisation de deux lapins avec le peptide d’intérêt couplé à la
KLH. Après test de la sensibilité et de la spécificité des sérums par immunoempreinte, nous
avons choisis l’un des sérums pour la suite des expériences. La présence de la protéine
PIKfyve endogène a été recherchée dans diverses lignées cellulaires (Figure 41A). Les
extraits cellulaires sont réalisés selon la méthode publiée par l’équipe du Dr. Stenmark
(Cabezas, 2006). Les cellules sont lysées dans un tampon contenant 1% TritonX100 et la
fraction Triton soluble est ensuite dénaturée par un tampon Laemli. Cette méthode de
préparation de nos échantillons a grandement amélioré la détection de PIKfyve endogène.
112
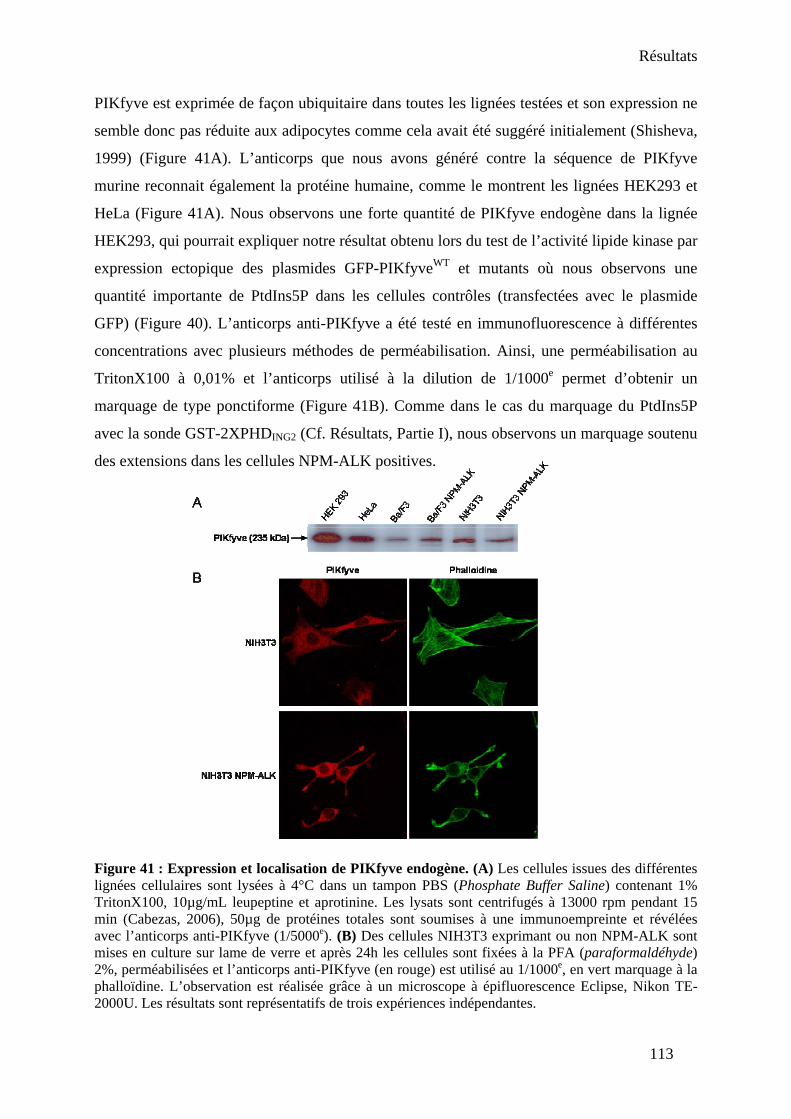
Résultats
PIKfyve est exprimée de façon ubiquitaire dans toutes les lignées testées et son expression ne
semble donc pas réduite aux adipocytes comme cela avait été suggéré initialement (Shisheva,
1999) (Figure 41A). L’anticorps que nous avons généré contre la séquence de PIKfyve
murine reconnait également la protéine humaine, comme le montrent les lignées HEK293 et
HeLa (Figure 41A). Nous observons une forte quantité de PIKfyve endogène dans la lignée
HEK293, qui pourrait expliquer notre résultat obtenu lors du test de l’activité lipide kinase par
expression ectopique des plasmides GFP-PIKfyveWT et mutants où nous observons une
quantité importante de PtdIns5P dans les cellules contrôles (transfectées avec le plasmide
GFP) (Figure 40). L’anticorps anti-PIKfyve a été testé en immunofluorescence à différentes
concentrations avec plusieurs méthodes de perméabilisation. Ainsi, une perméabilisation au
TritonX100 à 0,01% et l’anticorps utilisé à la dilution de 1/1000e permet d’obtenir un
marquage de type ponctiforme (Figure 41B). Comme dans le cas du marquage du PtdIns5P
avec la sonde GST-2XPHDING2 (Cf. Résultats, Partie I), nous observons un marquage soutenu
des extensions dans les cellules NPM-ALK positives.
Figure 41 : Expression et localisation de PIKfyve endogène. (A) Les cellules issues des différentes lignées cellulaires sont lysées à 4°C dans un tampon PBS (Phosphate Buffer Saline) contenant 1% TritonX100, 10µg/mL leupeptine et aprotinine. Les lysats sont centrifugés à 13000 rpm pendant 15 min (Cabezas, 2006), 50µg de protéines totales sont soumises à une immunoempreinte et révélées avec l’anticorps anti-PIKfyve (1/5000e). (B) Des cellules NIH3T3 exprimant ou non NPM-ALK sont mises en culture sur lame de verre et après 24h les cellules sont fixées à la PFA (paraformaldéhyde) 2%, perméabilisées et l’anticorps anti-PIKfyve (en rouge) est utilisé au 1/1000e, en vert marquage à la phalloïdine. L’observation est réalisée grâce à un microscope à épifluorescence Eclipse, Nikon TE-2000U. Les résultats sont représentatifs de trois expériences indépendantes.
113
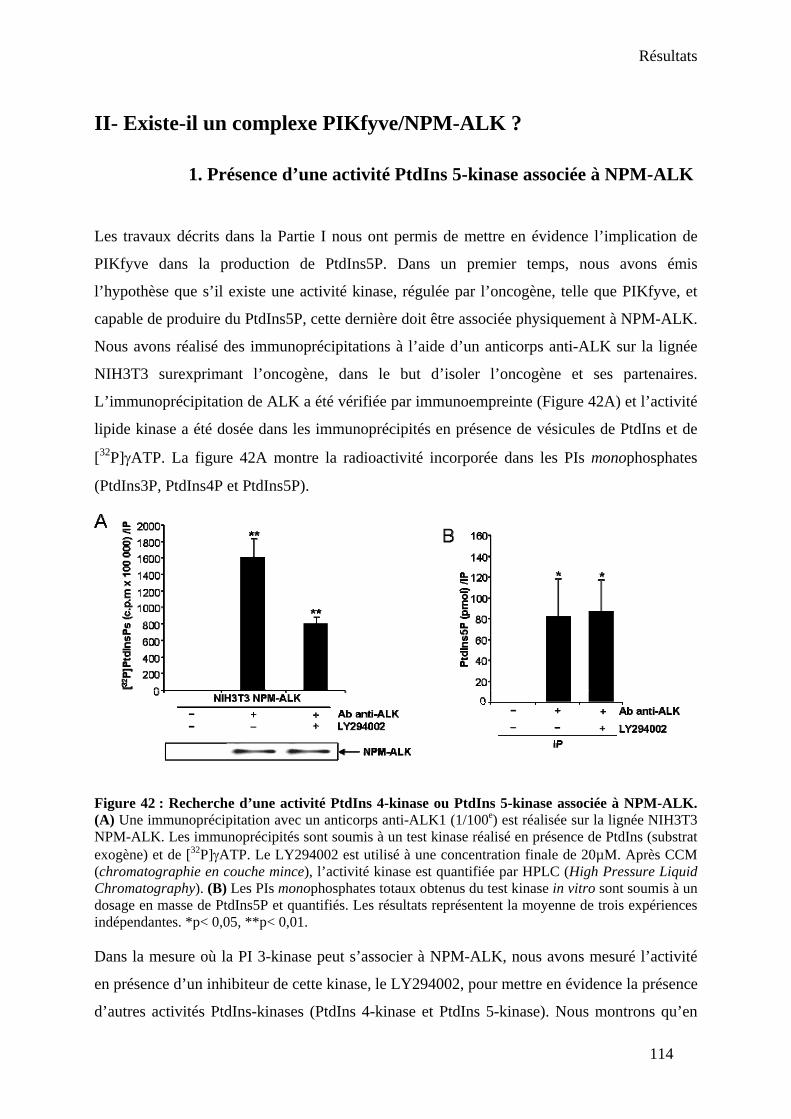
Résultats
II- Existe-il un complexe PIKfyve/NPM-ALK ?
1. Présence d’une activité PtdIns 5-kinase associée à NPM-ALK
Les travaux décrits dans la Partie I nous ont permis de mettre en évidence l’implication de
PIKfyve dans la production de PtdIns5P. Dans un premier temps, nous avons émis
l’hypothèse que s’il existe une activité kinase, régulée par l’oncogène, telle que PIKfyve, et
capable de produire du PtdIns5P, cette dernière doit être associée physiquement à NPM-ALK.
Nous avons réalisé des immunoprécipitations à l’aide d’un anticorps anti-ALK sur la lignée
NIH3T3 surexprimant l’oncogène, dans le but d’isoler l’oncogène et ses partenaires.
L’immunoprécipitation de ALK a été vérifiée par immunoempreinte (Figure 42A) et l’activité
lipide kinase a été dosée dans les immunoprécipités en présence de vésicules de PtdIns et de
[32P]γATP. La figure 42A montre la radioactivité incorporée dans les PIs monophosphates
(PtdIns3P, PtdIns4P et PtdIns5P).
Figure 42 : Recherche d’une activité PtdIns 4-kinase ou PtdIns 5-kinase associée à NPM-ALK. (A) Une immunoprécipitation avec un anticorps anti-ALK1 (1/100e) est réalisée sur la lignée NIH3T3 NPM-ALK. Les immunoprécipités sont soumis à un test kinase réalisé en présence de PtdIns (substrat exogène) et de [32P]γATP. Le LY294002 est utilisé à une concentration finale de 20µM. Après CCM (chromatographie en couche mince), l’activité kinase est quantifiée par HPLC (High Pressure Liquid Chromatography). (B) Les PIs monophosphates totaux obtenus du test kinase in vitro sont soumis à un dosage en masse de PtdIns5P et quantifiés. Les résultats représentent la moyenne de trois expériences indépendantes. *p< 0,05, **p< 0,01. Dans la mesure où la PI 3-kinase peut s’associer à NPM-ALK, nous avons mesuré l’activité
en présence d’un inhibiteur de cette kinase, le LY294002, pour mettre en évidence la présence
d’autres activités PtdIns-kinases (PtdIns 4-kinase et PtdIns 5-kinase). Nous montrons qu’en
114

Résultats
présence de LY294002, il existe une activité PtdIns-kinase résiduelle associée à l’oncogène
NPM-ALK (Figure 42A). Nous avons ensuite déterminé si cette activité pouvait correspondre
à une activité PtdIns 5-kinase (Figure 42B) et donc à PIKfyve. Pour cela, les spots de PIs
monophosphates (PtdInsPs) ont été récupérés de la silice, élués et soumis au dosage en masse
de PtdIns5P (Figure 42A). Nous observons que les immunoprécipités anti-ALK, traités ou
non par le LY294002, contiennent une importante quantité de PtdIns5P (80
picomoles/immunoprécipités). Il existe donc une activité PtdIns 5-kinase associée à
l’oncogène NPM-ALK capable de produire du PtdIns5P à partir de PtdIns. Cette kinase
pourrait être PIKfyve, comme nos résultats avec le siRNA dirigé contre PIKfyve l’ont
suggérés.
2. Interaction PIKfyve/NPM-ALK dans un système de
transfection ectopique
Le fait que PIKfyve soit la seule PtdIns5-kinase caractérisée capable de produire du PtdIns5P
à partir de PtdIns et l’observation de la chute du taux de PtdIns5P dans des cellules NIH3T3
NPM-ALK positives lors de l’abolition de cette protéine, nous ont conduit à vérifier
l’hypothèse d’une association physique entre PIKfyve et NPM-ALK.
Figure 43 : PIKfyve interagit avec NPM-ALK. (A) et (B) Les cellules HEK293 sont transfectées transitoirement avec les plasmides codant pour NPM-ALK et GFP-PIKfyveWT. 24h après transfection, les protéines GFP-PIKfyve et NPM-ALK sont immunoprécipitées et les complexes sont révélés par immunoempreinte. Les résultats dont représentatifs de trois expériences indépendantes.
115

Résultats
L’existence de ce complexe a été recherchée après cotransfection des plasmides codant pour
GFP-PIKfyveWT et pour NPM-ALK dans des cellules HEK293. NPM-ALK a été
immunoprécipité avec l’anticorps anti-ALK1 et la présence de PIKfyve a été révélée par
immunoempreinte avec l’anticorps anti-GFP (Figure 43A). De la même manière, nous
mettons en évidence la présence de NPM-ALK dans un immunoprécipité anti-GFP-PIKfyve
(Figure 43B). Ces résultats confirment l’existence d’un complexe entre PIKfyve et NPM-
ALK.
3. Mise en évidence du complexe au niveau endogène
La recherche du complexe moléculaire contenant PIKfyve et NPM-ALK a été étudiée au
niveau endogène. Malgré nos différents essais, nous avons été incapables de révéler une
interaction entre ces deux protéines de façon endogène. Notre hypothèse est que l’interaction
entre NPM-ALK et PIKfyve n’est pas directe et fait intervenir un ou plusieurs partenaires.
D’après les données bibliographiques il semble que la PI 3-kinase de classe IA puisse être un
bon candidat. En effet, d’une part différentes équipes ont montré que la PI 3-kinase est
capable de s’associer à NPM-ALK (Bai, 2000 ; Slupianek, 2001). L’interaction entre NPM-
ALK et la PI 3-kinase de classe IA n’est probablement pas directe, et ne concerne qu’une
partie du pool de PI 3-kinase de la cellule. D’autre part l’équipe du Dr. Shisheva a identifiée
la PI 3-kinase IA comme partenaire de PIKfyve (Sbrissa, 2001). Dans cette dernière étude les
conséquences de l’action de la PI 3-kinase sur l’activité de PIKfyve n’ont pas été étudiées. La
aussi, seule une partie du pool de PIKfyve s’associe à la PI 3-kinase (Sbrissa, 2001). Nous
pouvons donc imaginer qu’un pool minoritaire de PIKfyve est impliqué dans la signalisation
de l’oncogène NPM-ALK. Il est à noter qu’en plus de la probabilité d’une interaction non
directe, la taille importante de PIKfyve représente une difficulté supplémentaire dans la mise
en évidence du complexe.
116

Résultats
Figure 44 : La PI 3-kinase peut être un intermédiaire de l’interaction entre PIKfyve et NPM-ALK. Une immunoprécipitation avec un anticorps anti-p85 est réalisée sur des cellules NIH3T3 exprimant ou pas NPM-ALK. Après immunoprécipitation, les complexes sont révélés par immunoempreinte avec des anticorps anti-ALK1 (1/1000e) et anti-PIKfyve (1/5000e).
A partir de cellules NIH3T3 exprimant ou pas NPM-ALK, nous avons réalisé des
immunoprécipitations contre la sous-unité régulatrice p85 de la PI 3-kinase de classe IA. Ces
immunoprécipités ont été révélé par immunoempreinte avec des anticorps dirigés contre
PIKfyve et NPM-ALK (Figure 44). Nous avons ainsi pu mettre en évidence qu’il existe dans
les cellules NPM-ALK positives une fraction de la protéine PIKfyve capable de se lier à la PI
3-kinase et qu’une fraction de la protéine NPM-ALK est elle aussi capable de se lier à la PI 3-
kinase (Figure 44). Ces résultats suggèrent fortement l’existence d’un complexe comprenant
la PI 3-kinase, PIKfyve et NPM-ALK où la PI 3-kinase servirait d’intermédiaire entre
PIKfyve et NPM-ALK (Figure 45). Ainsi, la preuve de l’existence de ce complexe ternaire
nécessite d’autres expériences. Il serait intéressant, par exemple, d’abolir l’expression de la PI
3-kinase et de vérifier l’existence de ce complexe.
Figure 45 : Représentation schématique du complexe ternaire faisant intervenir PIKfyve, NPM-ALK et la PI 3-kinase. La PI 3-kinase semble être l’intermédiaire entre PIKfyve et NPM-ALK.
117

Résultats
III- L’oncogène NPM-ALK régule-t-il l’activité de PIKfyve ? Après avoir montré que les protéines NPM-ALK et PIKfyve étaient présentes au sein d’un
complexe multimoléculaire, nous avons voulu savoir si PIKfyve pouvait être régulée par
l’oncogène, et si oui de quelle façon ? Pour cela, PIKfyve a été immunoprécipitée dans des
cellules surexprimant ou pas l’oncogène NPM-ALK et sa capacité lipide kinase a été testée in
vitro. Les immunoprécipités sont soumis à un test lipide kinase in vitro en présence
d’ATPγ[32P] et de vésicules de PtdIns et PtdIns3P (2/1), deux substrats de PIKfyve. L’activité
de l’enzyme a été évaluée par la mesure de l’apparition de PtdIns(3,5)P2 produit. Bien que
lors de ces tests, il y a également production de PtdIns5P, la production de PtdIns(3,5)P2 est
préférentielle suggérant qu’in vitro le PtdIns3P est un meilleur substrat que le PtdIns pour
PIKfyve. Cette observation est confirmée lors de tests similaires in vitro dans lesquels nous
avons incubé la protéine recombinante GST-PIKfyve, produite dans le système du
Bacculovirus, avec de l’ATP radioactif et des vésicules de PtdIns et PtdIns3P où le produit
majoritaire que nous observons est du PtdIns(3,5)P2. Nous observons en figure 46 que la
présence de l’oncogène NPM-ALK induit une augmentation du taux de PtdIns(3,5)P2 par
rapport à des cellules contrôles, mettant en évidence une augmentation de l’activité lipide
kinase de PIKfyve par l’oncogène. Afin de déterminer si l’activation de PIKfyve est
dépendante de l’activité tyrosine kinase de NPM-ALK, nous avons inhibé cette activité
tyrosine kinase par le WHI-154 (Marzec, 2005). Ce composé est capable d’inhiber l’activité
tyrosine kinase de NPM-ALK, d’induire une diminution de la signalisation en aval de
l’oncogène et un arrêt des effets transformants de NPM-ALK. Le test lipide kinase sur les
immunoprécipités de PIKfyve est réalisé après une préincubation des cellules pendant 1h avec
le WHI-154. Nous observons une diminution de l’activité lipide kinase de PIKfyve dans les
cellules NPM-ALK traitées par l’inhibiteur, ramenant cette activité à un niveau basal tel que
celui observé dans les immunoprécipités de PIKfyve issus de cellules NIH3T3 contrôles
(Figure 46).
Ces résultats montrent que PIKfyve est un effecteur de NPM-ALK, et que l’oncogène régule
l’activité lipide kinase de PIKfyve via son activité tyrosine kinase. L’augmentation d’activité
de PIKfyve dans les cellules NPM-ALK positives est dépendante de l’oncogène, et représente
la partie activable de cette enzyme par l’opposition à l’activité basale de PIKfyve dans les
cellules NIH3T3 contrôles.
118
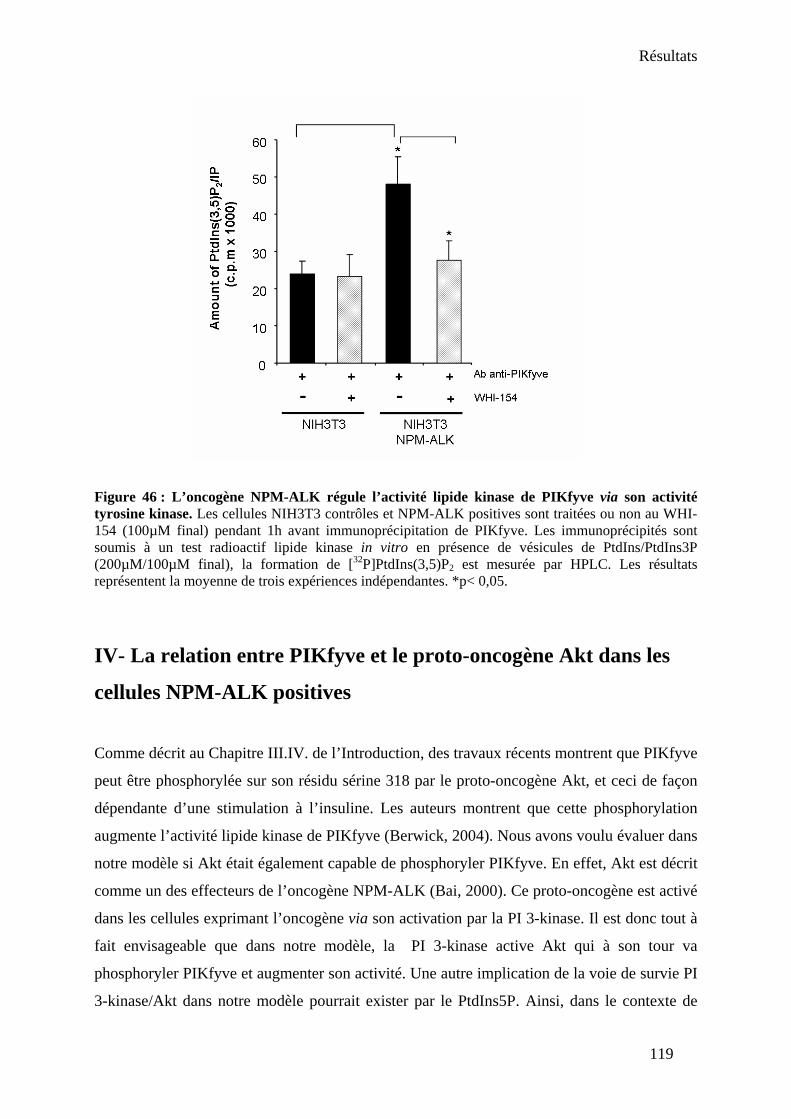
Résultats
Figure 46 : L’oncogène NPM-ALK régule l’activité lipide kinase de PIKfyve via son activité tyrosine kinase. Les cellules NIH3T3 contrôles et NPM-ALK positives sont traitées ou non au WHI-154 (100µM final) pendant 1h avant immunoprécipitation de PIKfyve. Les immunoprécipités sont soumis à un test radioactif lipide kinase in vitro en présence de vésicules de PtdIns/PtdIns3P (200µM/100µM final), la formation de [32P]PtdIns(3,5)P2 est mesurée par HPLC. Les résultats représentent la moyenne de trois expériences indépendantes. *p< 0,05.
IV- La relation entre PIKfyve et le proto-oncogène Akt dans les
cellules NPM-ALK positives
Comme décrit au Chapitre III.IV. de l’Introduction, des travaux récents montrent que PIKfyve
peut être phosphorylée sur son résidu sérine 318 par le proto-oncogène Akt, et ceci de façon
dépendante d’une stimulation à l’insuline. Les auteurs montrent que cette phosphorylation
augmente l’activité lipide kinase de PIKfyve (Berwick, 2004). Nous avons voulu évaluer dans
notre modèle si Akt était également capable de phosphoryler PIKfyve. En effet, Akt est décrit
comme un des effecteurs de l’oncogène NPM-ALK (Bai, 2000). Ce proto-oncogène est activé
dans les cellules exprimant l’oncogène via son activation par la PI 3-kinase. Il est donc tout à
fait envisageable que dans notre modèle, la PI 3-kinase active Akt qui à son tour va
phosphoryler PIKfyve et augmenter son activité. Une autre implication de la voie de survie PI
3-kinase/Akt dans notre modèle pourrait exister par le PtdIns5P. Ainsi, dans le contexte de
119

Résultats
l’infection bactérienne, notre équipe a montré que le PtdIns5P était un potentialisateur de la
voie de survie PI 3-kinase/Akt (Pendaries, 2006). Nous pourrions ainsi imaginer que le
PtdIns5P produit par PIKfyve pourrait alors participer à la voie de survie PI 3-kinase/Akt. A
son tour, Akt pourrait phosphoryler PIKfyve et donc l’activer pour produire du PtdIns5P. Ce
système permettrait la mise en place d’une boucle d’autoactivation du signal via le PtdIns5P
en aval de l’oncogène NPM-ALK.
Nous avons donc tout d’abord recherché le statut de phosphorylation de PIKfyve sur son
résidu 318. L’anticorps dirigé contre le résidu 318 phopshorylé n’étant pas disponible dans le
commerce, nous avons utilisé un anticorps anti-substrats phosphorylés d’Akt dirigés contre
les protéines possédant un site consensus phosphorylable par Akt (RxxRxx(pS/pT)). Les
immunoprécipitations anti-substrats phosphorylés d’Akt ont été révélées avec un anticorps
anti-PIKfyve et inversement. Nos résultats préliminaires montrent que la phosphorylation sur
le site substrat d’Akt est augmentée dans les cellules qui surexpriment l’oncogène NPM-ALK
par rapport aux cellules contrôles, suggérant la participation d’Akt à l’activation de PIKfyve
dans les cellules NPM-ALK positives. Cependant, l’anticorps utilisé lors de ces expériences
n’est pas le meilleur outil, un anticorps dirigé contre le site S318 phosphorylé de PIKfyve, tel
que celui utilisé dans le travail de Berwick et al., donnerait de meilleurs résultats. Nous
envisageons d’une part de tester un anticorps anti-sérine/thréonine phosphorylées et d’autre
part d’évaluer l’activité lipide kinase de PIKfyve dans des cellules exprimant l’oncogène
NPM-ALK lors de tests radioactifs après avoir inhibé l’activité du proto-oncogène Akt par
des techniques de RNA interférence ou au moyen d’un inhibiteur.
120

Résultats
Partie III : Rôle physiologique de PIKfyve dans l’oncogenèse associée à NPM-ALK
Comme décrit dans les partie I et II des Résultats, la production de PtdIns5P en aval de
l’oncogène NPM-ALK fait intervenir PIKfyve. Nous avons donc recherché quel était l’impact
de PIKfyve sur le potentiel oncogénique de la tyrosine kinase.
I- Effets de PIKfyve sur la prolifération Les cellules NIH3T3 transformées par l’oncogène NPM-ALK présentent des capacités
prolifératives augmentées (Marzec, 2007, Armstrong, 2004) qui font intervenir
principalement la voie Ras/Erk (Cf. Chap.I Introduction,). Afin d’appréhender un rôle de
PIKfyve dans la prolifération, nous avons aboli l’expression de PIKfyve par siRNA
interférence. Les cellules NIH3T3 surexprimant ou non NPM-ALK traitées, ont été
ensemencées en plaque 96 puits en 10% sérum. La mesure de la prolifération cellulaire sur
cinq jours a été effectuée par colorimétrie à l’aide du kit Celltiter 96 Aqueous One solution
cell proliferation assay de PromegaTM. Ce test repose sur la détection, par mesure de
l’absorbance à 490 nm, de la conversion d’un dérivé du tétrazolium (MTS) par les cellules
métaboliquement actives.
Nos premiers résultats montrent que PIKfyve ne semble jouer aucun rôle sur la prolifération
en aval de NPM-ALK. Cette étude nécessite des expériences complémentaires car nous ne
pouvons être sûrs que sur les temps longs (cinq jours) le siRNA dirigé contre PIKfyve ait
encore un effet. De plus, dans le contexte de l’oncogenèse associée à NPM-ALK, il semble
que les cellules en suspension telles que les cellules Ba/F3 (en notre possession au
laboratoire) ou les modèles d’ALCL (Cost ou Karpas), soient de meilleurs modèles pour
l’étude de la prolifération en aval de l’oncogène. Ainsi, pour conclure sur l’éventuel rôle de
PIKfyve sur la prolifération induite par NPM-ALK d’autres tests sont nécessaires.
II- Effets sur la migration et l’invasion Dans le Chapitre III de l’Introduction, nous avons détaillé les fonctions de PIKfyve dans
plusieurs processus biologiques. A ce jour, cette protéine semble être largement impliquée
121

Résultats
dans les mécanismes de régulation du trafic vésiculaire et l’homéostasie des vésicules
(Shisheva, 2008). En effet, l’abolition de l’expression de PIKfyve par l’utilisation d’un
inhibiteur spécifique l’YM201636, un mutant catalytiquement inactif ou par une technique de
RNA interférence entraîne dans les cellules une dilatation des vésicules intracellulaires
(Shisheva, 2008 ; Jefferies, 2007). Nous avons pu observer ces résultats dans des cellules
NIH3T3 exprimant ou pas l’oncogène NPM-ALK (Figure 47). Après l’abolition de PIKfyve
par siRNA, nous observons, en contraste de phase, le phénotype caractéristique des cellules
n’exprimant plus cette protéine. La délétion de PIKfyve entraîne aussi bien dans les cellules
contrôles que dans les cellules NPM-ALK positives une apparition de nombreuses vésicules
dans le cytoplasme des cellules. Il est intéressant de noter que les cellules invalidées pour
PIKfyve présentent des capacités adhésives diminuées. Cependant, la présence des vésicules
intracellulaires ne semblent pas affecter la survie de ces cellules.
Figure 47 : La perte de PIKfyve entraîne une perturbation de l’homéostasie vésiculaire dans les cellules exprimant ou pas l’oncogène NPM-ALK. Des cellules NIH3T3 contrôles ou NPM-ALK positives sont ensemencées à une densité de 100000 cellules/puits dans une plaque 6 puits. Les cellules sont transfectées deux fois à 24h et 48h soit par un siRNA contrôle soit un siRNA dirigé contre PIKfyve (100mM) à l’aide de la Lipofectamine (Invitrogen). A 72h d’expression, les cellules sont observées en contraste de phase grâce à un microscope à épifluorescence (Eclipse, Nikon TE-2000U). Les résultats sont représentatifs de trois expériences indépendantes.
Nous nous sommes intéressés au lien fonctionnel qui pouvait exister entre un oncogène et une
protéine connue pour son rôle dans la régulation de l’homéostasie vésiculaire. Le fait que
122

Résultats
l’oncogène NPM-ALK régule l’activité de PIKfyve suggère une relation entre le trafic
vésiculaire et la transformation cellulaire. A ce jour, peu de données bibliographiques
concernant ce sujet existent. Mais le lien entre le trafic vésiculaire et la transformation
cellulaire peut s’envisager à différents niveaux. Tout d’abord, l’altération de la balance entre
le recyclage de récepteurs membranaires ou de molécules adhésives et leur dégradation, peut
amener à une dérégulation de l’expression de surface de ces molécules (Polo, 2004). Ces
perturbations peuvent conduire à une modification des réponses aux facteurs de croissance, à
la perte d’adhésion et à une motilité perturbée que l’on observe dans les cellules transformées.
D’autre part, une modification de l’adressage ou de la maturation des vésicules peut aussi
perturber la sécrétion et par la même participer à la modification du microenvironnement
tumoral, du stade de différenciation ou des propriétés invasives des cellules. De nombreuses
études ont pu mettre en évidence les capacités migratoires accrues pour les cellules qui
surexpriment l’oncogène NPM-ALK (Armstrong, 2004). Ces effets sur la migration sont
associés à des réarrangements du cytosquelette.
Nous avons donc décidé d’évaluer les effets de la perte de PIKfyve sur le pouvoir migratoire
des cellules NPM-ALK positives. Les cellules NIH3T3 contrôles ou NPM-ALK positives ont
été transfectées par un siRNA dirigé contre PIKfyve ou un siRNA contrôle. La migration des
cellules a été évaluée en utilisant le système de chambres d’invasion en transwell constituées
d’une membrane poreuse recouverte de matrigel 3D. Le matrigel 3D va servir à mimer la
matrice extracellulaire et contient des molécules telles que les collagènases. Ainsi, la
migration des cellules ne pourra se faire que si ces dernières sont capables de sécréter des
molécules qui permettent la destruction de cette matrice. Les cellules invasives ayant migré à
travers la membrane sont colorées au crystal violet et comptées. Nous montrons que
l’abolition de l’expression de PIKfyve entraîne une diminution des capacités invasives des
cellules exprimant l’oncogène NPM-ALK. (Figure 48). Bien que ce test soit indicatif quant au
rôle de PIKfyve dans l’oncogenèse médiée par NPM-ALK, cette technique ne nous permet
pas de suivre uniquement les cellules où PIKfyve est invalidée, qui présentent le phénotype
observé en Figure 47. Nous avons donc choisi une autre technique en microscopie nous
permettant de suivre uniquement les cellules invalidées pour PIKfyve. Le déplacement des
cellules ainsi traitées a été mesuré dans du matrigel 3D et suivi en microscopie « live » en
contraste de phase pendant 15h avec une acquisition d’images toutes les 10 mins. L’analyse
du déplacement a été effectuée par le logiciel MétaMorph. Pour les cellules traitées avec le
siRNA PIKfyve, seules les cellules présentant des vésicules ont été analysées. Nous montrons
que la perte de PIKfyve dans les cellules exprimant NPM-ALK entraîne une diminution
123
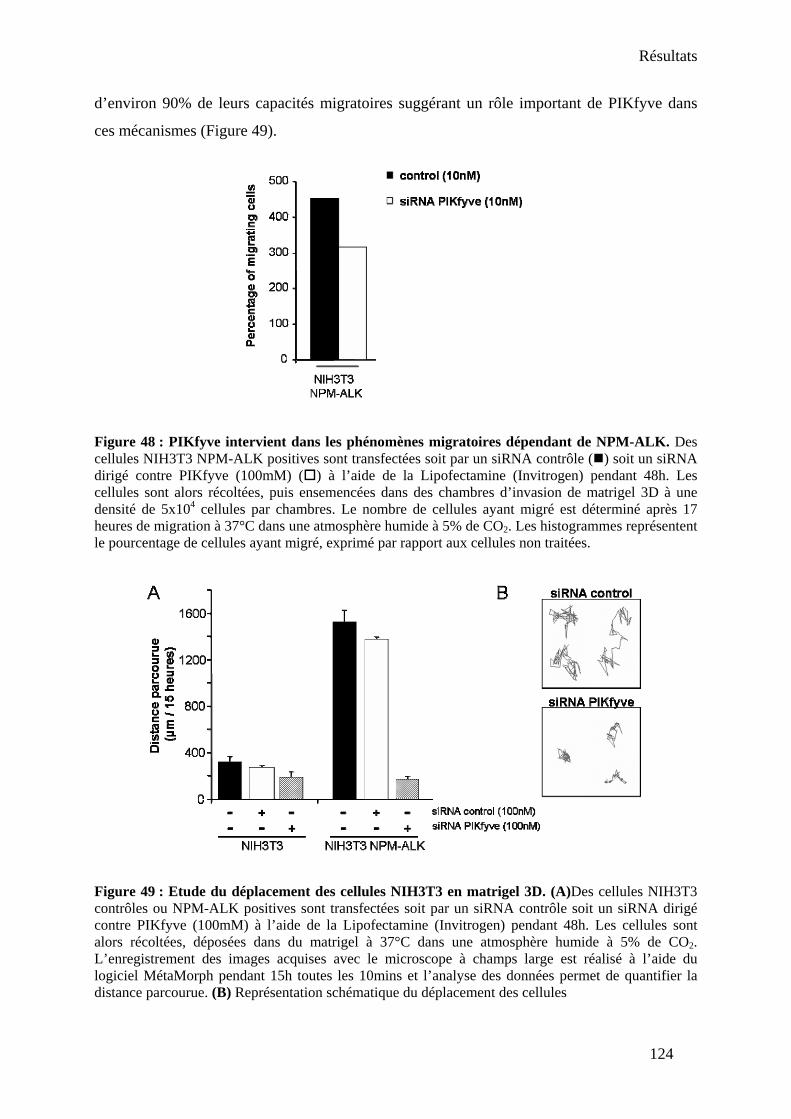
Résultats
d’environ 90% de leurs capacités migratoires suggérant un rôle important de PIKfyve dans
ces mécanismes (Figure 49).
Figure 48 : PIKfyve intervient dans les phénomènes migratoires dépendant de NPM-ALK. Des cellules NIH3T3 NPM-ALK positives sont transfectées soit par un siRNA contrôle ( ) soit un siRNA dirigé contre PIKfyve (100mM) ( ) à l’aide de la Lipofectamine (Invitrogen) pendant 48h. Les cellules sont alors récoltées, puis ensemencées dans des chambres d’invasion de matrigel 3D à une densité de 5x104 cellules par chambres. Le nombre de cellules ayant migré est déterminé après 17 heures de migration à 37°C dans une atmosphère humide à 5% de CO2. Les histogrammes représentent le pourcentage de cellules ayant migré, exprimé par rapport aux cellules non traitées.
Figure 49 : Etude du déplacement des cellules NIH3T3 en matrigel 3D. (A)Des cellules NIH3T3 contrôles ou NPM-ALK positives sont transfectées soit par un siRNA contrôle soit un siRNA dirigé contre PIKfyve (100mM) à l’aide de la Lipofectamine (Invitrogen) pendant 48h. Les cellules sont alors récoltées, déposées dans du matrigel à 37°C dans une atmosphère humide à 5% de CO2. L’enregistrement des images acquises avec le microscope à champs large est réalisé à l’aide du logiciel MétaMorph pendant 15h toutes les 10mins et l’analyse des données permet de quantifier la distance parcourue. (B) Représentation schématique du déplacement des cellules
124

Résultats
De plus, des résultats préliminaires de notre équipe suggéraient que les cellules exprimant
l’oncogène NPM-ALK présentent une sécrétion accrue de molécules de MMP9 (Matrix
metalloproteinases) (F. Lagarrigue, non publiés) et que la neutralisation de la protéine MMP-
9 par un anticorps bloquant induit une diminution des capacités migratoires des cellules
exprimant l’oncogène. Cette protéine participe au développement de cancers en permettant la
digestion de la matrice extracellulaire facilitant ainsi d’une part le grossissement de la tumeur
et d’autre part l’invasion des tissus par les cellules métastatiques. Aux vues de nos résultats, il
est donc tout à fait envisageable que PIKfyve puisse contrôler la sécrétion de MMP-9 et donc
contrôler le pouvoir migratoire des cellules exprimant l’oncogène NPM-ALK. L’étape
suivante sera de rechercher l’impact de PIKfyve sur la sécrétion de MMP-9.
En conclusion, nos résultats suggèrent que l’oncogène NPM-ALK utilise une protéine
régulant le trafic vésiculaire pour réguler la sécrétion de molécules permettant ainsi à
l’oncogène d’accroitre ses capacités invasives et migratoires. L’oncogène détournerait donc le
trafic vésiculaire à son profit afin d’augmenter les processus métastatiques.
III- PIKfyve peut-il réguler NPM-ALK ?
1. Rôle de PIKfyve sur la localisation de NPM-ALK
Comme présenté dans le Chapitre III.V. de l’Introduction, la protéine PIKfyve a été impliquée
dans le contrôle du trafic du EGFR jusqu’au noyau, régulant ainsi l’action de ce récepteur sur
la régulation de gène permettant le contrôle du cycle cellulaire (Kim, 2007). Les auteurs
montrent une association entre le EGFR et PIKfyve, association que nous avons aussi mise en
évidence entre PIKfyve et NPM-ALK, une autre protéine à activité tyrosine kinase. Nous
avons décrit précédemment dans nos travaux le rôle de PIKfyve en tant qu’effecteur de NPM-
ALK. Cependant, on peut également se poser la question d’un rôle de PIKfyve sur
l’oncogène. Nous avons donc décidé d’évaluer l’impact de PIKfyve sur la localisation de
l’oncogène NPM-ALK. NPM-ALK est une tyrosine kinase cytosolique qui présente une
localisation cytosolique, nucléaire et nucléolaire (Figure 50).
125

Résultats
Figure 50 : Localisation de PIKfyve et NPM-ALK dans les cellules NIH3T3 NPM-ALK positives. Des cellules NIH3T3 NPM-ALK positives sont fixées, perméabilisées et incubées avec les anticorps anti- ALK1 et PIKfyve. La révélation est réalisée grâce à des anticorps secondaires couplés au fluorophore Alexa-594 (ALK1) ou 488 (PIKfyve). Les cellules sont observées au grossissement 40 grâce à un microscope à épifluorescence (Eclipse, Nikon TE-2000U).
Au niveau du cytosol, en plus d’une localisation diffuse, on peut observer des granules où la
protéine NPM-ALK phosphorylée active est concentrée. La nature de ces granules n’est pas
encore bien définie mais ils contiennent également la protéine AUF1, un facteur de stabilité
des ARNm (Fawal, 2006). Seuls les dimères NPM/NPM-ALK peuvent aller au noyau. En
effet, NPM possède deux séquences de localisation nucléaire (NLS) lui permettant de faire la
navette entre le cytoplasme et le noyau. Celle-ci va pouvoir former des dimères avec NPM-
ALK et localiser NPM-ALK au noyau sous forme non active.
Afin d’étudier le rôle de PIKfyve sur la localisation de l’oncogène, nous avons aboli
l’expression de PIKfyve par une technique de RNA interférence. La localisation de PIKfyve
et de NPM-ALK a ensuite été déterminée par immunofluorescence. Nous observons sur les
cellules où l’expression de PIKfyve est abolie une perte totale de NPM-ALK dans le noyau.
(Figure 51). Ces résultats suggèrent que, comme dans le cas du EGFR, PIKfyve puisse
contrôler la localisation de l’oncogène. L’interaction entre PIKfyve et NPM-ALK pourrait
soit jouer sur le transport vers le noyau ou sur l’état d’activation de la cellule en favorisant la
formation des dimères NPM-ALK/NPM-ALK.
126
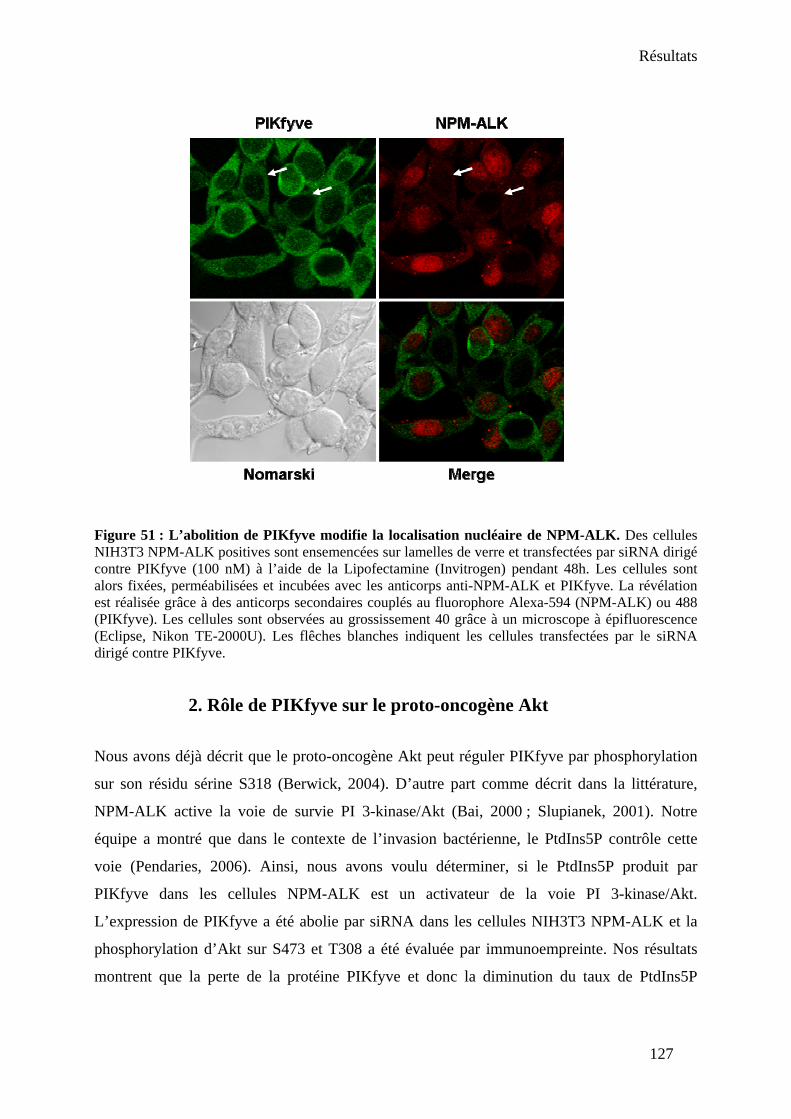
Résultats
Figure 51 : L’abolition de PIKfyve modifie la localisation nucléaire de NPM-ALK. Des cellules NIH3T3 NPM-ALK positives sont ensemencées sur lamelles de verre et transfectées par siRNA dirigé contre PIKfyve (100 nM) à l’aide de la Lipofectamine (Invitrogen) pendant 48h. Les cellules sont alors fixées, perméabilisées et incubées avec les anticorps anti-NPM-ALK et PIKfyve. La révélation est réalisée grâce à des anticorps secondaires couplés au fluorophore Alexa-594 (NPM-ALK) ou 488 (PIKfyve). Les cellules sont observées au grossissement 40 grâce à un microscope à épifluorescence (Eclipse, Nikon TE-2000U). Les flêches blanches indiquent les cellules transfectées par le siRNA dirigé contre PIKfyve.
2. Rôle de PIKfyve sur le proto-oncogène Akt
Nous avons déjà décrit que le proto-oncogène Akt peut réguler PIKfyve par phosphorylation
sur son résidu sérine S318 (Berwick, 2004). D’autre part comme décrit dans la littérature,
NPM-ALK active la voie de survie PI 3-kinase/Akt (Bai, 2000 ; Slupianek, 2001). Notre
équipe a montré que dans le contexte de l’invasion bactérienne, le PtdIns5P contrôle cette
voie (Pendaries, 2006). Ainsi, nous avons voulu déterminer, si le PtdIns5P produit par
PIKfyve dans les cellules NPM-ALK est un activateur de la voie PI 3-kinase/Akt.
L’expression de PIKfyve a été abolie par siRNA dans les cellules NIH3T3 NPM-ALK et la
phosphorylation d’Akt sur S473 et T308 a été évaluée par immunoempreinte. Nos résultats
montrent que la perte de la protéine PIKfyve et donc la diminution du taux de PtdIns5P
127

Résultats
entraîne une baisse de la phosphorylation d’Akt sur ses deux résidus S473 et T308 (Figure
52), reflet d’une perte de son activité.
Figure 52 : PIKfyve contrôle la phosphorylation du proto-oncogène Akt dans les cellules exprimant l’oncogène NPM-ALK. Des cellules NIH3T3 NPM-ALK positives sont ensemencées à une densité de 100000 cellules/puits puis transfectées deux fois à 24h et 48h soit par un siRNA contrôle soit un siRNA dirigé contre PIKfyve (100mM) à l’aide de la Lipofectamine (Invitrogen). A 72h d’expression, les cellules sont privées en sérum pendant une nuit. Les cellules sont lysées à 4°C dans un tampon PBS contenant 1% TritonX100, 10µg/mL leupeptine et aprotinine. Les lysats sont centrifugés à 13000 rpm pendant 15 min (Cabezas, 2006), soumis à une immunoempreinte et révélés avec les anticorps anti-PIKfyve (1/5000e), anti-P-Akt 473 (1/1000e), anti-P-Akt 308 (1/1000e) et anti-Akt total (1/1000e). Les résultats sont représentatifs de trois expériences indépendantes.
Il est intéressant de noter que l’inhibition de PIKfyve par l’utilisation d’un inhibiteur,
l’YM201636, ou par l’abolition de l’expression de la protéine par une technique de siRNA,
n’affecte pas la phosphorylation d’Akt (Jefferies, 2007). Cette différence pourrait être due aux
modèles utilisés. En effet, l’étude de Jefferies et al. utilise un modèle de fibroblastes NIH3T3
contrôles alors que notre modèle de cellules NIH3T3 est transformé par l’oncogène NPM-
ALK connu pour activer plusieurs voies de signalisation qui fonctionnent alors en synergie.
De plus, lors de l’abolition de l’expression de PIKfyve, c’est le taux de PtdIns5P contrôlé par
NPM-ALK qui est inhibé.
En conclusion, nos résultats suggèrent que PIKfyve contrôle la phosphorylation du proto-
oncogène Akt.
V- Conclusion-Discussion Dans l’ensemble de ces travaux, nous montrons que l’oncogène NPM-ALK est capable de
s’associer, dans un complexe multimoléculaire, avec la PtdIns 5-kinase, PIKfyve.
128

Résultats
L’interaction des deux protéines n’est pas directe et nous proposons que la PI 3-kinase est un
intermédiaire. La formation de ce complexe semble contrôler la production de PtdIns5P dans
les cellules NPM-ALK positives. Nous savons que l’oncogène NPM-ALK est capable
d’activer la PI 3-kinase mais rien n’est actuellement connu sur le fait que PIKfyve puisse être
une cible de la PI 3-kinase (Sbrissa, 2001). Nos résultats montrent que l’oncogène NPM-ALK
régule l’activité lipide kinase de PIKfyve via son activité tyrosine kinase ce qui permet de
suggérer un rôle de relai de la PI 3-kinase dans cette activation. Par contre, comme cela a été
décrit dans la littérature, PIKfyve est une cible du proto-oncogène Akt. La phosphorylation
sur le résidu sérine 318 de PIKfyve par Akt (Berwick, 2004) augmente l’activité lipide kinase
de PIKfyve. Nos résultats confirment les travaux de Berwick et al. puisque nous avons pu
mettre en évidence que dans les cellules NPM-ALK positives, la phosphorylation de PIKfyve
sur S318 est plus importante. Ainsi, ces résultats placent la PI 3-kinase et Akt comme
intermédiaire entre NPM-ALK et PIKfyve. Il semble intéressant de vérifier la véracité d’une
telle phosphorylation, en effet nous pouvons imaginer que c’est par ce moyen que l’oncogène
contrôle l’activité de PIKfyve. Nous avons montré que NPM-ALK contrôle l’activité lipide
kinase de PIKfyve et nous savons que dans le contexte de l’oncogenèse associée à NPM-
ALK, la voie de survie PI 3-kinase/Akt est activée (Bai, 2000). Nous pouvons donc proposer
que le proto-oncogène Akt soit capable de phosphoryler PIKfyve sur la S318, ce qui
augmenterait son activité lipide kinase, et serait un moyen pour l’oncogène de contrôler cette
enzyme.
D’autre part, nos résultats suggèrent également un rôle de PIKfyve comme activateur de la
voie PI 3-kinase/Akt. Ainsi, nous avons montré que l’abolition de l’expression de PIKfyve
par une technique de RNA interférence induit une diminution de la phosphorylation d’Akt sur
ses résidus S473 et T308 dans les cellules NPM-ALK positives. Il semble donc que PIKfyve
ou ses produits, le PtdIns5P et le PtdIns(3,5)P2 puissent réguler le proto-oncogène Akt. Dans
le contexte de l’infection bactérienne, notre équipe a montré que la production de PtdIns5P
entraîne une activation de le voie de survie PI 3-kinase/Akt (Pendaries, 2006). Nous pouvons
imaginer que le PtdIns5P produit dans les cellules NPM-ALK positives sert à potentialiser la
voie de survie PI 3-kinase/Akt. Dans un premier temps, il serait intéressant de vérifier
l’implication du PtdIns5P dans l’activation de la voie PI 3-kinase/Akt par la mesure des
produits de la PI 3-kinase dans les cellules NPM-ALK positives après l’abolition de
l’expression de PIKfyve. Cette régulation d’Akt par PIKfyve et le fait qu’Akt puisse contrôler
PIKfyve par phosphorylation suggère un modèle dans lequel il existerait un complexe faisant
intervenir NPM-ALK, PIKfyve, la PI 3-kinase et Akt. La voie de survie PI 3-kinase/Akt
129
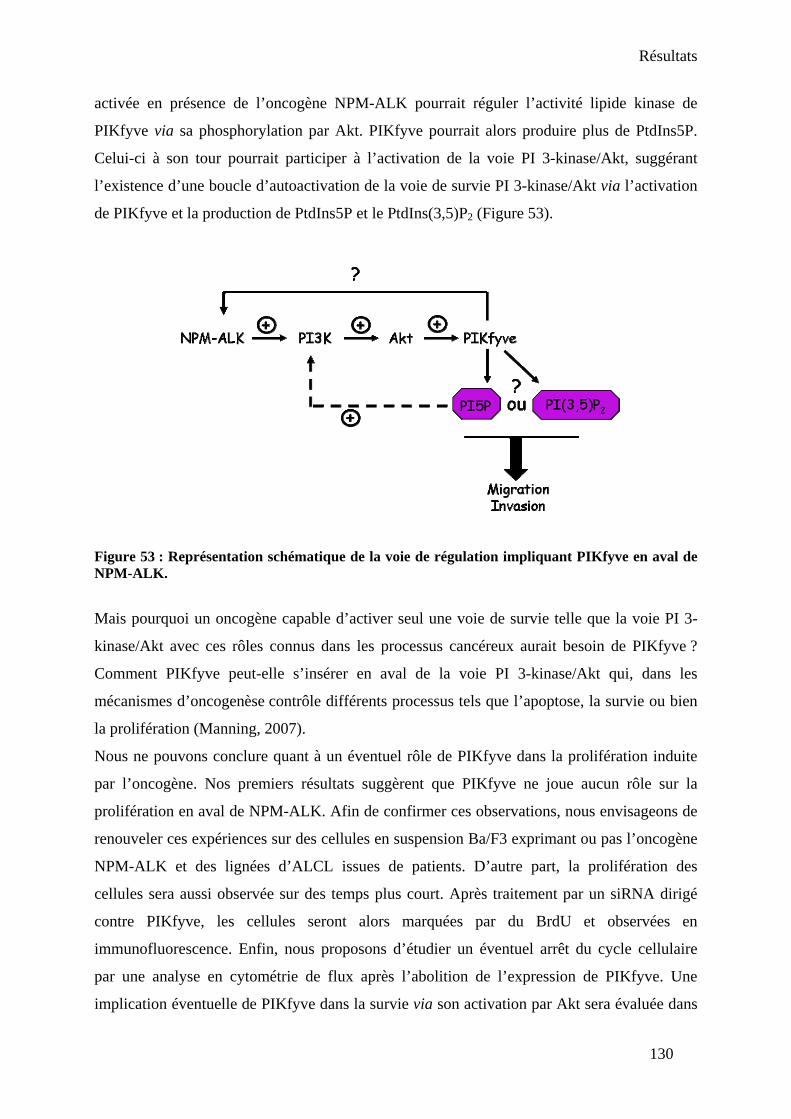
Résultats
activée en présence de l’oncogène NPM-ALK pourrait réguler l’activité lipide kinase de
PIKfyve via sa phosphorylation par Akt. PIKfyve pourrait alors produire plus de PtdIns5P.
Celui-ci à son tour pourrait participer à l’activation de la voie PI 3-kinase/Akt, suggérant
l’existence d’une boucle d’autoactivation de la voie de survie PI 3-kinase/Akt via l’activation
de PIKfyve et la production de PtdIns5P et le PtdIns(3,5)P2 (Figure 53).
Figure 53 : Représentation schématique de la voie de régulation impliquant PIKfyve en aval de NPM-ALK.
Mais pourquoi un oncogène capable d’activer seul une voie de survie telle que la voie PI 3-
kinase/Akt avec ces rôles connus dans les processus cancéreux aurait besoin de PIKfyve ?
Comment PIKfyve peut-elle s’insérer en aval de la voie PI 3-kinase/Akt qui, dans les
mécanismes d’oncogenèse contrôle différents processus tels que l’apoptose, la survie ou bien
la prolifération (Manning, 2007).
Nous ne pouvons conclure quant à un éventuel rôle de PIKfyve dans la prolifération induite
par l’oncogène. Nos premiers résultats suggèrent que PIKfyve ne joue aucun rôle sur la
prolifération en aval de NPM-ALK. Afin de confirmer ces observations, nous envisageons de
renouveler ces expériences sur des cellules en suspension Ba/F3 exprimant ou pas l’oncogène
NPM-ALK et des lignées d’ALCL issues de patients. D’autre part, la prolifération des
cellules sera aussi observée sur des temps plus court. Après traitement par un siRNA dirigé
contre PIKfyve, les cellules seront alors marquées par du BrdU et observées en
immunofluorescence. Enfin, nous proposons d’étudier un éventuel arrêt du cycle cellulaire
par une analyse en cytométrie de flux après l’abolition de l’expression de PIKfyve. Une
implication éventuelle de PIKfyve dans la survie via son activation par Akt sera évaluée dans
130

Résultats
une première approche par l’étude des cibles d’Akt telles que MDM2, GSK3, FOXO, Bad par
immunoempreinte après abolition de l’expression de PIKfyve dans des cellules NPM-ALK
positives.
Un autre rôle de PIKfyve en aval d’Akt pourrait être dans la voie de contrôle du métabolisme
cellulaire. Une des fonctions majeures d’Akt est le contrôle du transport de glucose en
réponse à l’insuline. Ainsi, l’activation d’Akt dans des adipocytes conduit à la stimulation du
transport de vésicules contenant GLUT4, un récepteur au glucose à la membrane plasmique
(Calera, 1998). L’étude de Berwick et al. a montré que la phosphorylation de PIKfyve sur
S318 par Akt entraîne une augmentation de l’activité lipide kinase de PIKfyve et un transport
accru de vésicules contenant GLUT4 à la membrane plasmique en réponse à l’insuline
(Berwick, 2004). Parallèlement, de nombreux travaux ont montré que les cellules
oncogéniques possèdent un métabolisme du glucose augmenté en réponse à l’activation de la
voie PI 3-kinase/Akt (Majumder, 2005 ; Semenza, 2003). On peut donc imaginer que NPM-
ALK utilise PIKfyve en aval d’Akt pour augmenter le métabolisme glucidique des cellules
transformées.
L’activation de PIKfyve par NPM-ALK permet de proposer un rôle fonctionnel de ses
produits lipidiques le PtdIns5P et le PtdIns(3,5)P2 dans les mécanismes d’oncogenèse. Dans la
première partie de nos travaux, nous avons montré une augmentation du taux de PtdIns5P
dépendante de PIKfyve dans les cellules NPM-ALK positives (Cf. Résultats, Partie I).
Concernant le taux de PtdIns(3,5)P2 nos résultats préliminaires montrent que les cellules
surexprimant l’oncogène NPM-ALK ont un taux moins élevé que les cellules contrôles,
suggérant que le PtdIns(3,5)P2 produit par PIKfyve serait métabolisé très rapidement et donc
difficilement détectable. Le rôle des deux produits de PIKfyve comme médiateur de
l’oncogenèse en aval de NPM-ALK reste posée. Si l’on se base sur la littérature, on peut
imaginer que le PtdIns(3,5)P2, même s’il est produit très furtivement joue un rôle dans le
contrôle de mécanismes impliquant le trafic vésiculaire. C’est par ce biais que PIKfyve
pourrait jouer un rôle dans le contrôle du métabolisme du glucose ou dans les mécanismes de
migration. Nos résultats obtenus dans les expériences de migration montrent que la perte
d’expression de PIKfyve entraîne une diminution de la migration des cellules NPM-ALK
positives. Pour migrer, les cellules doivent dégrader une matrice mimant la matrice
extracellulaire, suggérant ainsi l’implication de PIKfyve dans la migration via l’utilisation de
molécules sécrétoires telles que les Matrix Metalloprotéases. De façon intéressante notre
équipe a montré une augmentation de MMP-9 dans des lignées surexprimant l’oncogène
NPM-ALK et dans des biopsies de patients atteints d’ALCL NPM-ALK positifs (en
131

Résultats
comparaison aux lymphocytes sanguins circulants). PIKfyve pourrait donc jouer un rôle dans
le mécanisme de sécrétion des MMPs et il sera important de vérifier cette hypothèse. Mais
PIKfyve pourrait contrôler également d’autres types de molécules en jeu dans les processus
migratoires. En effet, nous pouvons imaginer que PIKfyve puisse être impliquée dans les
mécanismes de recyclage de récepteurs membranaires tels que les récepteurs aux intégrines. Il
sera important de regarder si PIKfyve participe à une accélération du recyclage des récepteurs
aux intégrines ce qui entraînerait une déficience d’adhésion à la matrice extracellulaire et
donc une augmentation des capacités migratoires des cellules qui surexpriment NPM-ALK.
Cependant, nos connaissances sur les fonctions du PtdIns5P ne permettent pas d’exclure un
rôle de ce lipide dans les mécanismes de trafic vésiculaire, et donc de l’impliquer dans le
contrôle du métabolisme glucidique, de la sécrétion de MMPs ou du recyclage des intégrines.
Par contre, le rôle le plus probable pourrait s’inscrire dans le remodelage du cytosquelette
d’actine. En effet, d’après la littérature, le PtdIns5P produit par PIKfyve est capable de
remodeler les fibres de stress d’actine lors d’une stimulation à l’insuline (Sbrissa, 2004). Nous
avons montré que dans les cellules NIH3T3 NPM-ALK positives, le PtdIns5P se localise dans
des prolongements cytoplasmiques qui sont des sites actifs du remodelage du cytosquelette
d’actine. De plus, nous savons que le remodelage du cytosquelette d’actine est un processus
dynamique qui sert à l’invasion des cellules NPM-ALK positives (Colomba, 2008). Nous
pouvons envisager que le PtdIns5P produit par PIKfyve sert à maintenir la cellule
oncogénique dans une dynamique active du cytosquelette d’actine, ce qui lui permettrait de
maintenir ses propriétés invasives actives en permanence. D’autre part, comme déjà décrit ci-
haut, la survie des cellules tumorales nécessitent un métabolisme cellulaire accru, nous
pouvons donc penser que la potentialisation de la voie PI 3-kinase/Akt par PIKfyve et le
PtdIns5P s’inscrive dans un rôle d’une augmentation du métabolisme glucidique contrôlée par
Akt dans la cellule transformée plus qu’un dans le maintien de la survie. Ainsi, le PtdIns5P
tout comme le PtdIns(3,5)P2 pourrait contrôler les mécanismes de trafic intravésiculaire et les
processus invasifs. Ces travaux suggèrent donc un rôle majeur de PIKfyve et de ses produits
dans les processus oncogéniques associés à NPM-ALK. Il nous semble important désormais
de confirmer la voie faisant intervenir PIKfyve, NPM-ALK, la PI 3-kinase et Akt, et de
déterminer quelle part prend chacun des produits de PIKfyve dans les processus qui médient
l’oncogenèse associée à NPM-ALK.
Enfin, un autre contrôle de NPM-ALK par PIKfyve semble exister au niveau de la
localisation de NPM-ALK. Nous avons montré que l’abolition de l’expression de PIKfyve
entraîne une délocalisation de l’oncogène NPM-ALK de sa localisation nucléaire, ce qui
132

Résultats
engendre une relocalisation de l’oncogène dans le cytoplasme et dans des granules
intracytoplasmiques. Comme décrit précédemment, les dimères contenant NPM-ALK
présents dans le noyau sont non phosphorylés et donc inactifs, seul le pool de NPM-ALK
présent dans les granules intracytoplasiques est actif. Nous pouvons envisager que ces
premiers résultats soient une indication quant à un rôle potentiel de PIKfyve dans le contrôle
de la localisation de NPM-ALK. Un tel rôle de PIKfyve a récemment été mis en évidence
avec un autre récepteur à activité tyrosine kinase, le récepteur à l’EGF. En effet, PIKfyve est
capable de s’associer au EGFR et contrôle son transport au noyau où ce récepteur est capable
de réguler la transcription de gènes cibles impliqués dans la prolifération (Kim, 2007). Nous
pouvons imaginer un mécanisme similaire dans le cas de NPM-ALK. En effet, nous pouvons
proposer que PIKfyve puisse réguler la localisation de NPM-ALK nucléaire pour le séquestrer
dans une forme non actif ou bien encore pour permettre à l’oncogène de jouer un rôle sur la
transcription. En effet, à ce jour, nous ne savons pas quel est le rôle de NPM-ALK nucléaire
mais cette localisation nucléaire ne semble pas nécessaire à son pouvoir oncogènique. Nous
pouvons ainsi imaginer que lors de l’abolition de PIKfyve, la délocalisation de NPM-ALK
nucléaire vers une localisation granulaire cytoplasmique permettrait d’augmenter le pool de
NPM-ALK actif.
133

CONCLUSION ET
PERSPECTIVES

Conclusion et perspectives
I- Conclusion
Depuis sa découverte en 1997, le PtdIns5P a fait l’objet de nombreuses études qui ont permis
de mettre à jour différentes fonctions pour ce nouveau second messager lipidique. Plusieurs
pools de PtdIns5P semblent exister et proviennent de différentes voies métaboliques et
interviennent certainement suivant le type cellulaire et l’état d’activation de la cellule. Un
pool de PtdIns5P présent à la membrane plasmique dans le contexte de l’infection bactérienne
par la pathogène Shigella flexneri, serait potentialisateur de la voie de survie PI 3-kinase/Akt
et participerait à la régulation du cytosquelette d’actine (Pendaries, 2006 ; Niebuhr, 2002). Au
noyau, de récentes études montrent qu’il régulerait les processus d’apoptose induite en
réponse à des stress de type UV ou génotoxiques (Gozani, 2003 ; Ungewickell, 2005 ; Zou,
2007 ; Bunce, 2006 ; Bunce, 2008 ; Jones, 2006). Ainsi, selon sa localisation, le PtdIns5P
semble avoir deux rôles antagonistes, un rôle anti-apoptotique à la membrane plasmique et un
rôle pro-apoptotique dans le noyau. Enfin, ce lipide pourrait être impliqué dans la régulation
du trafic vésiculaire du fait de sa production par PIKfyve et les MTMs, des enzymes
largement impliquées dans le maintien de l’homéostasie vésiculaire. L’étude que nous avons
réalisée suggère un nouveau rôle du PtdIns5P dans les processus d’oncogenèse.
Ainsi, dans la première partie nous avons montré que :
- les taux de PtdIns5P sont augmentés dans des cellules exprimant l’oncogène à
activité tyrosine kinase, NPM-ALK. Nous avons également retrouvé des taux
élevés de PtdIns5P dans des lignées de leucémies myéloïdes chroniques exprimant
l’oncogène Bcr-Abl et dans des lignées de leucémies myéloïdes aiguës, suggérant
un rôle plus général dans les processus de transformation. Le PtdIns5P pourrait
donc être un marqueur pronostic de la transformation cellulaire.
- le PtdIns5P se localise dans des prolongements cytoplasmiques dans des
fibroblastes NIH3T3 exprimant NPM-ALK. Ces extensions caractéristiques des
cellules NIH3T3 NPM-ALK positives semblent être des sites actifs du remodelage
de l’actine suggérant que ce lipide pourrait jouer un rôle sur le remodelage du
cytosquelette.
- la production de PtdIns5P dans les cellules NPM-ALK positives implique la PtdIns
5-kinase, PIKfyve.
134

Conclusion et perspectives
Dans la seconde partie de nos travaux nous avons étudié la relation entre PIKfyve et NPM-
ALK au niveau moléculaire et déterminé le rôle physiologique de PIKfyve dans l’oncogenèse
associée à NPM-ALK.
Nous avons pu démontrer que :
- PIKfyve et NPM-ALK sont capables de s’associer dans un complexe. Cette
association ferait intervenir un autre partenaire, la PI 3-kinase.
- l’oncogène NPM-ALK, via son activité tyrosine kinase, régule l’activité lipide
kinase de PIKfyve.
- PIKfyve joue un rôle dans le contrôle de la migration des cellules NPM-ALK.
- PIKfyve pourrait être un intermédiaire dans la régulation de la voie de survie PI 3-
kinase/Akt dans le contexte de l’oncogenèse associée à NPM-ALK et la protéine
peut réguler la localisation de l’oncogène.
Ainsi, l’ensemble de ces travaux montrent un nouveau rôle du PtdIns5P et de PIKfyve, dans
un modèle de cellules transformées. Bien que nous ayons pu démontrer que PIKfyve est
capable de former un complexe avec l’oncogène NPM-ALK où ce dernier régule l’activité
enzymatique de PIKfyve pour promouvoir son pouvoir invasif, de nombreuses questions
demeurent. Si l’on se base sur la littérature, nous pouvons proposer un modèle où NPM-ALK
active la PI 3-kinase qui active le proto-oncogène Akt, ce dernier phosphorylant PIKfyve et
l’activant. Ce modèle place PIKfyve en aval de la voie PI 3-kinase/Akt activée par
l’oncogène. Cette voie contrôlée par l’oncogène n’est apparemment pas impliquée dans une
voie de pro-survie bien que des expériences complémentaires soient nécessaires pour
confirmer ce résultat. Par contre, le fait que PIKfyve soit un substrat d’Akt pourrait placer
PIKfyve en aval d’Akt dans une autre voie contrôlée par Akt qui est la voie du métabolisme
glucidique de la cellule cancéreuse. Cette hypothèse est bien sûr à confirmer. De plus, nos
résultats semblent suggérer que PIKfyve exerce un rétrocontrôle sur Akt permettant une
régulation fine de cette voie. Lors de l’abolition de PIKfyve, nous observons une diminution
des propriétés invasives et migratoires des cellules NPM-ALK. Cette voie pourrait faire
intervenir un autre pool de PIKfyve non contrôlé par la voie PI 3-kinase/Akt. Le rôle de
PIKfyve dans le contrôle de l’homéostasie des membranes que l’on peut visualiser par
l’apparition des vésicules lors de l’abolition de PIKfyve, modifie l’état d’activation de la
cellule et certainement les voies de l’endocytose et de l’exocytose de molécules impliquées
dans l’adhésion des cellules ou la sécrétion de molécules permettant la dégradation de la
135

Conclusion et perspectives
matrice extracellulaire. PIKfyve pourrait agir sur cette voie par l’intermédiaire de son produit
le PtdIns(3,5)P2. On peut également proposer que le PtdIns5P produit en aval de NPM-ALK
par PIKfyve joue un rôle dans le remodelage du cytosquelette. Le rôle de PIKfyve dans le
remodelage des fibres de stress pourrait passer par le PtdIns5P. Ce rôle décrit par les travaux
du Dr. Shisheva dans le modèle des adipocytes (Sbrissa, 2004) pourrait également être
primordial dans le modèle d’oncogenèse associé à NPM-ALK. Les cellules NPM-ALK
présentent un cytosquelette avec une désorganisation des fibres de stress et une augmentation
de l’actine corticale. De plus, nous retrouvons PIKfyve localisé dans les extrémités qui sont
des sites actifs de remodelage permettant de supposer que la lipide kinase PIKfyve joue un
rôle important dans ce phénomène. Un rôle de PIKfyve sur le cytosquelette en aval de NPM-
ALK pourrait faire intervenir une voie PI 3-kinase/petites protéines G. Le rôle du PtdIns5P
produit par PIKfyve sur le cytosquelette pourrait également se répercuter sur le transport de
vésicules. Parallèlement, PIKfyve pourrait intervenir dans la régulation de l’oncogène NPM-
ALK. Nous avons montré que PIKfyve est capable de réguler la localisation de l’oncogène
NPM-ALK. Nous savons que la dégradation de NPM-ALK médiée par des protéines
chaperonnes est une étape importante dans la signalisation en aval de l’oncogène (Bonvini,
2002 ; Bonvini, 2004). Nous pouvons proposer que PIKfyve puisse servir à maintenir NPM-
ALK dans le noyau pour permettre une balance entre les pools de NPM-ALK nucléaires et les
pools de NPM-ALK intracytoplasmiques présents dans les granules donc entre les pools
nucléaires non actifs et les formes actives dans le cytoplasme. Cette régulation pourrait se
faire par le contrôle des protéines chaperonnes, Hsp90 et Hsp70 qui sont associées à
l’oncogène et qui contrôlent sa dégradation (Bonvini, 2002 ; Bonvini, 2004). Ceci pose aussi
une question sur l’activation de la transcription, des travaux récents montrent que PIKfyve
régule le transport du EGFR au noyau et ce transport permet le contrôle de gènes cibles
impliqués dans la prolifération (Kim, 2007). D’autres travaux suggèrent un rôle de NPM et
des dimères NPM/NPM-ALK dans la transcription génique. En effet, d’une part, la protéine
NPM native est capable de s’associer au facteur de transcription NFκB et aux histones et
régule ainsi la condensation de la chromatine (Wang, 1994 ; Okuwaki, 2002). D’autre part, la
régulation de l’expression du récepteur CD30, marqueur spécifique des ALCL, est régulée via
l’activation de la voie NF-κB par les dimères NPM/NPM-ALK (Horie, 2004). PIKfyve
pourrait donc aussi maintenir NPM-ALK dans le noyau dans le but de réguler la transcription
de gènes et plus particulièrement les gènes cibles de la voie NF-κB qui permettent le contrôle
de l’apoptose et de l’arrêt du cycle cellulaire.
136

Conclusion et perspectives
Il semble que le lien qui unit PIKfyve à NPM-ALK soit complexe. Ces deux protéines
semblent être capables de se réguler de façon croisée et donc d’activer de nombreuses voies
de signalisation en aval. Il devient donc indispensable de mieux comprendre la régulation de
cette enzyme, PIKfyve, qui paraît si importante désormais dans les processus d’oncogenèse
pour peut-être mettre à jour de nouvelles cibles thérapeutiques généralisables peut-être à
d’autres oncogènes
II- Perspectives Ce travail ouvre de nombreuses perspectives qui nous permettrons de compléter et intégrer les
données que nous avons déjà acquises et révéler d’autres éléments de la voie de signalisation
impliquant PIKfyve dans l’oncogenèse associée à NPM-ALK.
Les différents axes que nous nous proposons de développer sont :
- la relation entre PIKfyve et le proto-oncogène Akt
- l’étude de la balance PtdIns(3,5)P2/PtdIns5P dans les cellules NPM-ALK
positives : implication des protéines ArPIKfyve, Sac3 et des 3-phosphatases de la
famille des myotubularines
- rôle de PIKfyve dans les processus invasifs associés à NPM-ALK : lien entre
l’oncogenèse et le trafic vésiculaire, implication du cytosquelette
- le rôle de PIKfyve sur NPM- ALK
- détermination de nouveaux partenaires du PtdIns5P
1. La relation entre PIKfyve et le proto-oncogène Akt dans les
cellules NPM-ALK positives
Les résultats que nous avons obtenus suggèrent que PIKfyve est régulé par Akt mais
également régule Akt. Des travaux récents montrent que PIKfyve peut être phosphorylée sur
son résidu sérine 318 par le proto-oncogène Akt, et ceci de façon dépendante d’une
stimulation à l’insuline. Les auteurs ont montré que cette phosphorylation augmente l’activité
lipide kinase de PIKfyve et régule le trafic de vésicules GLUT4/IRAP à la membrane
plasmique (Berwick, 2004). Les résultats préliminaires que nous avons obtenus dans le
modèle NPM-ALK montrent une augmentation de la phosphorylation du résidu sérine 318 de
PIKfyve par Akt. Désormais, nous pensons qu’il serait intéressant de démontrer la véracité de
137

Conclusion et perspectives
la régulation de PIKfyve par Akt dans notre modèle de cellules NPM-ALK positives. Pour
cela, nous proposons d’étudier l’impact du proto-oncogène Akt sur l’activité lipide kinase de
PIKfyve après l’inhibition d’Akt soit à l’aide d’inhibiteurs soit par abolition de l’expression
de la protéine par l’utilisation de RNA interférence. Mais, il serait surtout intéressant de
déterminer le rôle de PIKfyve et d’Akt dans le contrôle du métabolisme. Nous pensons donc
vérifier cette hypothèse par l’étude du transport de vésicules GLUT4 positives à la membrane
plasmique dans les cellules NPM-ALK positives dans un premier temps. Dans un second
temps, cette étude devra être réalisée après l’abolition de PIKfyve par une technique de RNA
interférence ou par l’utilisation de l’inhibiteur spécifique de PIKfyve, l’YM201636 (Jefferies,
2007).
Dans notre modèle d’oncogenèse, nous avons montré que l’abolition de l’expression de
PIKfyve conduit à la diminution de la phosphorylation d’Akt suggérant ainsi que PIKfyve est
capable de réguler Akt. De plus, notre équipe a pu montrer un rôle majeur du PtdIns5P en tant
que potentialisateur de la voie de survie PI 3-kinase/Akt. Il serait donc intéressant d’évaluer,
si comme dans le modèle de l’infection bactérienne, c’est la production de PtdIns5P qui est
responsable de ce phénomène. Ainsi, nous nous proposons d’évaluer l’état de phosphorylation
d’Akt après abolition du taux de PtdIns5P par surexpression du domaine PHDING2 qui va
séquestrer le PtdIns5P ou de la PtdIns5P-4 kinase de type II qui transforme ce dernier en
PtdIns(4,5)P2. Par la suite, afin de vérifier le rôle de PIKfyve dans cette voie pro-survie, il
serait intéressant d’étudier le rôle de PIKfyve et du PtdIns5P sur la signalisation en aval
d’Akt. Nous pensons explorer les effecteurs d’Akt tels que GSK3, le facteur de transcription
Forkhead (FKHD), mTOR, ou bien encore des acteurs de l’apoptose tels Bad, Bax.
Enfin, l’implication de PIKfyve dans la régulation du proto-oncogène, nous amène à nous
demander si PIKfyve ne serait par un intermédiaire dans le voie PI 3-kinase/Akt. De plus, la
phosphorylation de PIKfyve par Akt suppose une interaction entre ces deux protéines. Nous
proposons de vérifier cette hypothèse en réalisant des immunoprécipitations de PIKfyve et
d’Akt suivies d’immunoempreintes de ces deux protéines.
Le modèle que nous proposons où il existerait un complexe protéique impliquant PIKfyve,
NPM-ALK, la PI 3-kinase et Akt dans lequel l’activation d’Akt par phosphorylation
permettrait l’activation de PIKfyve et une production de PtdIns5P activatrice de la PI 3-kinase
ne pourra être vérifié qu’après les expériences que nous proposons ci-dessus (Figure 54). Ces
travaux déterminerait un nouveau partenaire de NPM-ALK et impliquerait PIKfyve dans la
voie de signalisation PI 3-kinase/Akt dans l’oncogenèse associée à NPM-ALK. Cette
138

Conclusion et perspectives
signalisation pourrait conférer à l’oncogène des propriétés de survie accrue ou lui permettre
de contrôler le métabolisme du glucose.
Figure 54 : Représentation schématique du modèle d’activation de PIKfyve dans les cellules NPM-ALK positives.
2. Etude de la balance PtdIns(3,5)P2/PtdIns5P dans les cellules
NPM-ALK positives : implication des protéines ArPIKfyve, Sac3 et
des 3-phosphatases de la famille des myotubularines
La production de PtdIns5P dans les cellules NPM-ALK positives nécessite des investigations
plus poussées. PIKfyve est capable de produire du PtdIns5P et du PtdIns(3,5)P2, les résultats
in vitro suggèrent une préférence de PIKfyve pour le PtdIns3P. Dans notre modèle, à ce jour,
nous n’avons pu déterminer si PIKfyve fait directement du PtdIns5P à partir de PtdIns ou si
cette enzyme phosphoryle le PtdIns3P en PtdIns(3,5)P2 qui est alors métabolisé en PtdIns5P
par des 3-phosphatases. La seule indication que nous avons concerne le taux de PtdIns(3,5)P2
que nous retrouvons diminué dans les cellules exprimant l’oncogène NPM-ALK par rapport à
des cellules contrôles.
Ces premiers résultats seraient plutôt en faveur de la formation première de PtdIns(3,5)P2 qui
serait métabolisé en PtdIns5P par les MTMs. Nous pouvons imaginer, comme cela a été
publié récemment dans des souris déficientes pour Sac3 (une 5-phosphatase sur le
PtdIns(3,5)P2 s’associant et régulant PIKfyve), que le PtdIns(3,5)P2 est très rapidement
transformée en PtdIns3P et que le turnover très rapide du PtdIns(3,5)P2 ne permet pas
d’observer l’augmentation attendue de ce PI (Chow, 2007). Une phénomène identique
139

Conclusion et perspectives
pourrait concerner le taux le transformation de PtdIns(3,5)P2 en PtdIns5P par les MTMs. Pour
déterminer l’implication potentielle d’une 3-phosphatase de type MTMs dans notre modèle,
nous nous proposons d’une part d’étudier l’expression des protéines MTMs par Western blot.
Par la suite, il serait intéressant de déterminer si l’une de ces enzymes appartient au complexe
faisant déjà intervenir PIKfyve, NPM-ALK et la PI 3-kinase. L’implication de ces enzymes
dans la production de PtdIns5P dans le contexte de l’oncogenèse associée à NPM-ALK
pourrait être étudiée par dosage en masse de ce lipide après abolition de l’expression des
MTMs. De la même façon que pour PIKfyve, si l’implication de 3-phosphatases telles que la
famille des MTMs dans l’oncogenèse liée à NPM-ALK est avérée, il serait intéressant
d’étudier la participation de ces enzymes dans les processus d’oncogenèse tels que la
migration, la prolifération, la survie et l’apoptose.
Enfin des données récentes proposent que la régulation du taux de PtdIns(3,5)P2 et donc de
PIKfyve fasse intervenir des protéines partenaires régulatrices. C’est le cas de la protéine
Vac14, un osmo-régulateur (Bonangelino, 2002) décrite en premier lieu chez la levure. Cette
protéine est un activateur de Fab1p, l’homologue de PIKfyve chez la levure. Elle contrôle
ainsi le taux basal de PtdIns(3,5)2 (Duex, 2006). Comme décrit ci-haut, ce complexe fait
intervenir un autre partenaire la 5-phosphatase Fig4, (Rudge, 2004) Chez la levure, Fig4
interagit directement avec Vac14 et contrôle son activité (Dove, 2002 ; Gary, 2002 ; Rudge,
2004). Ce complexe est retrouvé chez les mammifères, ainsi ArPIKfyve, l’homologue de
Vac14, est capable de s’associer à PIKfyve, et Sac3 l’homologue de Fig4 aussi. Ces trois
protéines en s’associant en complexe participent à la régulation du taux de PtdIns(3,5)P2
(Sbrissa, 2004a ; Sbrissa, 2005 ; Sbrissa, 2007). Ainsi, dans notre modèle l’implication de
PIKfyve pose la question de l’éventuelle présence des régulateurs ArPIKfyve et Sac3. Il serait
donc intéressant d’évaluer l’expression de ces deux protéines. Par la suite, nous voudrions
analyser la possible association de ces trois protéines en complexe par des expériences
d’immunoprécipitation. Enfin, nous aimerions étudier la part que prend chacune de ces
protéines dans la synthèse de PtdIns5P, PtdIns(3,5)P2 et PtdIns3P dans le contexte de
l’oncogenèse associée à NPM-ALK. Pour cela, nous proposons de mesurer les niveaux de ces
PIs par dosage en masse et marquage métabolique radioactif après abolition de l’expression
de PIKfyve et/ou Sac3 et/ou ArPIKfyve.
140

Conclusion et perspectives
3. Rôle de PIKfyve dans les processus invasifs associés à NPM-
ALK
Nous avons pu mettre en évidence que PIKfyve joue un rôle dans le contrôle du pouvoir
invasif associé à l’oncogène. En effet, lors d’expériences en matrigel 3D, nous avons montré
que la perte d’expression de PIKfyve entraîne une diminution de la migration des cellules
NPM-ALK positives in vitro. Comme nous l’avons décrit, des expériences préliminaires
obtenues dans notre équipe, suggèrent que l’action de PIKfyve sur ce processus pourrait se
faire via le contrôle de la sécrétion de molécules dans le milieu extracellulaire et suggèrent
l’implication de MMP-9 dans le pouvoir invasif associé à l’oncogène NPM-ALK. Ainsi, il
semble évident que le prochain pas pour compléter cette étude est d’établir un lien entre
PIKfyve et MMP-9. Nous évaluerons l’impact de l’abolition de l’expression de PIKfyve par
l’utilisation de siRNA ou de l’inhibiteur l’YM201636 sur la sécrétion de MMP9 par
immunoempreinte et tests ELISA. Mais, il sera intéressant aussi de déterminer de la même
façon l’impact de PIKfyve sur la sécrétion d’autres molécules connues pour jouer un rôle dans
le maintien et/ou la dégradation de la matrice extracellulaire telles que des cytokines (Il3,
Il10) et des chimioattractants (GM-CSF). Ces molécules pourraient permettre aux cellules
exprimant NPM-ALK, par un phénomène autocrine, d’accroitre leurs capacités prolifératives
ou invasives.
La participation de PIKfyve aux capacités invasives associées à NPM-ALK pourrait aussi être
étudiée in vivo. Nous pensons utiliser des souris Nude auxquelles seront injectées des cellules
NIH3T3 NPM-ALK positives dans lesquelles l’expression de PIKfyve aura été abolie. La
pousse des tumeurs et la dissémination seront alors évaluées. De plus, l’équipe du Pr. G.
Delsol a produit un modèle conditionnel de souris NPM-ALK. Ainsi, nous envisageons de
traiter ces souris avec l’YM201636, l’inhibiteur de PIKfyve et d’évaluer la pousse des
tumeurs et la dissémination métastatiques dans des organes tels que les poumons ou les os par
immunohistochimie.
Comme nous l’avons dit précédemment, lors de l’abolition de l’expression de PIKfyve, les
cellules présentent des capacités adhésives diminuées qui pourrait s’expliquer par le biais de
l’altération de la balance entre le recyclage de récepteurs membranaires impliqués dans
l’adhésion des cellules et leur dégradation. Ceci conduirait à une dérégulation de l’expression
de surface de ces molécules (Polo, 2004) et donc à une perte d’adhésion et à une motilité
perturbée que l’on observe dans les cellules transformées. Ainsi, nous envisageons d’analyser
141

Conclusion et perspectives
l’expression de surface de molécules telles que les intégrines, comme α5β1 présentes
majoritairement sur les fibroblastes, connues pour être impliquées dans les phénomènes
d’adhésion ; dans les cellules NIH3T3 exprimant NPM-ALK, traitées ou non par un siRNA
dirigé contre PIKfyve, par immunofluorescence, immunoempreinte et cytométrie en flux.
Nous étudierons la signalisation en aval des intégrines qui implique des protéines comme
FAK, les kinases Scr, la voie des MAPK ou encore les GTPases Rho.
Ainsi, cette étude pourrait démontrer que l’interaction fonctionnelle entre NPM-ALK et
PIKfyve et donc la production de PtdIns5P et/ou de PtdIns(3,5)P2 pourrait permettre à cet
oncogène de contrôler une partie du trafic vésiculaire à son profit.
4. Rôle de PIKfyve et du PtdIns5P dans les mécanismes de
régulation du cytosquelette d’actine dans les cellules NPM-ALK
positives
Dans la première partie de nos travaux, nous avons montré que dans les cellules NIH3T3
exprimant NPM-ALK, le PtdIns5P produit par PIKfyve, se localise dans des extensions
membranaires qui sont des sites actifs du remodelage du cytosquelette d’actine. Comme nous
l’avons décrit dans la partie Introduction, le PtdIns5P pourrait participer à la réorganisation
des fibres de stress en réponse à l’insuline (Sbrissa, 2004). Les auteurs montrent que la
microinjection de PtdIns5P induit une désorganisation des fibres de stress que l’on observe
également en réponse à l’insuline alors que la séquestration de PtdIns5P par la surexpression
du domaine PHD d’ING2 bloque le désassemblage de l’actine. Ces données rejoignent celles
obtenues dans notre laboratoire (résultats non publiés, D. Ramel). D’une part, D. Ramel a
montré que le PtdIns5P induit la dépolymérisation des fibres de stress d’actine. D’autre part, il
a observé que le PtdIns5P est capable d’activer les GTPases Rac1 et Cdc42, deux GTPases
qui favorisent la formation de structures dynamiques de types lamellipodes et filopodes. De
plus, l’étude du Dr. Colomba, dans notre équipe, a pu mettre en évidence que la voie
Rac1/Vav3 était activée dans les cellules NPM-ALK positives et contrôlait le remodelage du
cytosquelette d’actine (Colomba, 2008). Tous ces résultats suggèrent que, dans le contexte de
l’oncogenèse associée à NPM-ALK, le remodelage du cytosquelette d’actine et les capacités
invasives accrues de l’oncogène pourraient nécessiter l’activation de la voie Vav3/Rac1 par le
PtdIns5P produit par PIKfyve. Ainsi, ce phénomène conférerait ainsi un avantage aux cellules
exprimant l’oncogène NPM-ALK dans les processus migratoires et pourrait permettre aux
142

Conclusion et perspectives
cellules transformées d’accroitre leur pouvoir invasif et de dissémination. Nous proposons
donc de mesurer dans un premier temps les taux de PtdIns5P dans des cellules exprimant
NPM-ALK après l’inhibition par le NSC23766 de la voie Rac1. Le NSC23766 est un
composé soluble bloquant l’interaction entre Rac1 et ses GEF (Guanine exchange factor)
(Goa, 2004), il inhibe l’activation de Rac1 en se fixant au niveau d’un résidu tryptophane
essentiel à l’interaction avec ses GEF. Par la suite, nous pensons abolir l’expression de
PIKfyve par RNA interférence et déterminer l’état d’activation de la voie Rac1/Vav3 par
immunoempreinte. Le statut du cytosquelette d’actine sera étudié par immunofluorescence.
De la même façon, l’état d’activation de la voie Rac1/Vac3 et le cytosquelette d’actine seront
évalués après la diminution du taux de PtdIns5P par l’utilisation soit de la PtdIns5P-4 kinase
de type II qui transforme le PtdIns5P en PtdIns(4,5)P2 soit du domaine PHDING2 qui séquestre
ce PI.
5. Rôle de PIKfyve sur NPM-ALK
De façon intéressante, les résultats que nous avons obtenu suggèrent que PIKfyve pourrait
réguler l’oncogène NPM-ALK. Nous avons montré que l’abolition de l’expression de
PIKfyve entraîne une délocalisation de NPM-ALK nucléaire. Ces résultats suggèrent que
PIKfyve pourrait réguler la localisation initiale de l’oncogène et pourrait servir au maintien de
la balance entre les formes de NPM-ALK, nucléaires non actives et les formes cytosoliques
granulaires actives. Pour aborder ce nouveau champ d’investigation, nous proposons tout
d’abord de vérifier si lors de l’abolition de PIKfyve par une technique de RNA interférence, il
existe une augmentation des formes NPM-ALK actives. La phosphorylation de NPM-ALK
sera étudiée par immunofluorescence et immunoempreinte. Il est intéressant de noter que
NPM-ALK est capable de s’associer à deux protéines chaperonnes, Hsp70 et Hsp90 qui
semblent réguler sa dégradation et la signalisation en aval de l’oncogène. Les auteurs
montrent que l’inhibition de Hsp70 conduit à une diminution de la dégradation de NPM-ALK
et à une relocalisation de la protéine dans des aggrégats qui pourraient représenter des lieux de
stockage de NPM-ALK actifs (Bonvini, 2002 ; Bonvini, 2004). La délocalisation de NPM-
ALK observée lors de l’inhibition de PIKfyve suggère qu’il serait intéressant d’étudier ces
mécanismes de dégradation. Pour cela, nous pourrions évaluer l’expression de Hsp70 et
Hsp90 après l’inhibition de PIKfyve. De plus, nous envisageons d’estimer l’association entre
NPM-ALK, Hsp70 et Hsp90 dans un contexte où l’expression de PIKfyve serait abolie.
143

Conclusion et perspectives
Enfin, il serait intéressant de mesurer l’impact de PIKfyve dans les processus de transcription.
En effet, la délocalisation de NPM-ALK du noyau vers le cytoplasme suggère aussi que
PIKfyve comme dans cas du EGFR (Kim, 2007) pourrait contrôler la transcription de gènes
en aval de NPM-ALK. Ainsi, nous pourrions mesurer les niveaux d’ARNm de facteurs de
transcription tels que STAT3 et NFκB qui ont été montré comme régulant la transcription de
gènes dans le contexte de NPM-ALK, après l’abolition de PIKfyve.
6. Identification de nouveaux effecteurs du PtdIns5P
Afin de mieux comprendre le rôle du PtdIns5P dans l’oncogenèse liée à NPM-ALK et de
façon plus générale dans les cellules, il est nécessaire d’identifier de nouvelles cibles de ce PI.
Nous proposons donc comme première approche l’utilisation de techniques de purification
par affinité. Des cytosols seront incubés avec du PtdIns5P biotinylé et les complexes
protéines/PtdIns5P seront purifiés avec des billes de streptavidine agarose. L’analyse en
spectrométrie de masse permettra l’identification des protéines candidates. Parallèlement, une
seconde approche utilisera le Biacore qui permet de mettre en évidence des interactions
protéines/lipides plus faibles que la technique précédente. Pour cela nous utiliserons des
cytosols préparés à partir de cellules exprimant ou pas l’oncogène. Le couplage du Biacore à
la spectrométrie de masse permettra l’analyse des candidats.
Enfin, le modèle d’oncogenèse associée à NPM-ALK semble désormais impliquer des PIs tels
que le PtdIns(3,5)P2, pour lequel peu d’interacteurs ont été mis en évidence. Nous pensons
donc qu’il serait important de réaliser une étude similaire concernant la recherche des
effecteurs de ce PIs.
144
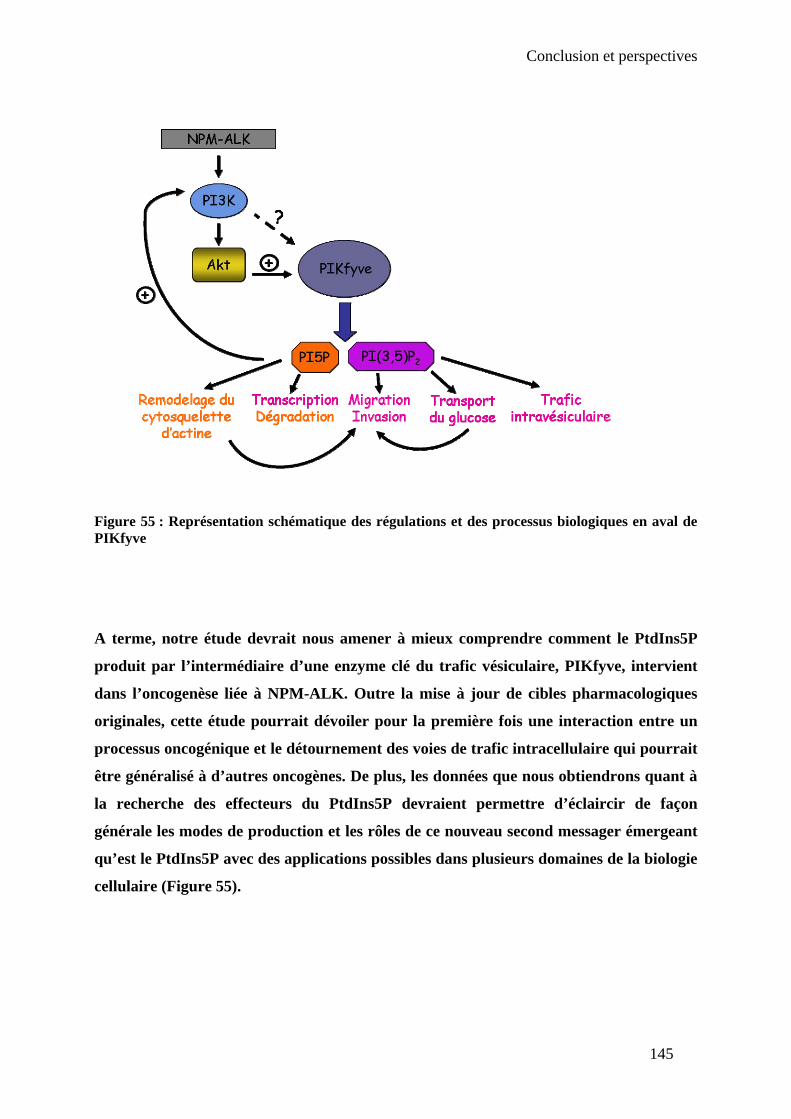
Conclusion et perspectives
Figure 55 : Représentation schématique des régulations et des processus biologiques en aval de PIKfyve
A terme, notre étude devrait nous amener à mieux comprendre comment le PtdIns5P
produit par l’intermédiaire d’une enzyme clé du trafic vésiculaire, PIKfyve, intervient
dans l’oncogenèse liée à NPM-ALK. Outre la mise à jour de cibles pharmacologiques
originales, cette étude pourrait dévoiler pour la première fois une interaction entre un
processus oncogénique et le détournement des voies de trafic intracellulaire qui pourrait
être généralisé à d’autres oncogènes. De plus, les données que nous obtiendrons quant à
la recherche des effecteurs du PtdIns5P devraient permettre d’éclaircir de façon
générale les modes de production et les rôles de ce nouveau second messager émergeant
qu’est le PtdIns5P avec des applications possibles dans plusieurs domaines de la biologie
cellulaire (Figure 55).
145

BIBLIOGRAPHIE

Références bibliographiques
Aasland, R., Gibson, T.J. and Stewart, A.F. (1995) The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci, 20, 56-59.
Adam, P., Katzenberger, T., Seeberger, H., Gattenlohner, S., Wolf, J., Steinlein, C., Schmid, M., Muller-Hermelink, H.K. and Ott, G. (2003) A case of a diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am J Surg Pathol, 27, 1473-1476.
Ait-Tahar, K., Cerundolo, V., Banham, A.H., Hatton, C., Blanchard, T., Kusec, R., Becker, M., Smith, G.L. and Pulford, K. (2006) B and CTL responses to the ALK protein in patients with ALK-positive ALCL. Int J Cancer, 118, 688-695.
Alessi, D.R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P. and Hemmings, B.A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J, 15, 6541-6551.
Alvarez-Venegas, R., Sadder, M., Hlavacka, A., Baluska, F., Xia, Y., Lu, G., Firsov, A., Sarath, G., Moriyama, H., Dubrovsky, J.G. and Avramova, Z. (2006) The Arabidopsis homolog of trithorax, ATX1, binds phosphatidylinositol 5-phosphate, and the two regulate a common set of target genes. Proc Natl Acad Sci U S A, 103, 6049-6054.
Ambrogio, C., Voena, C., Manazza, A.D., Piva, R., Riera, L., Barberis, L., Costa, C., Tarone, G., Defilippi, P., Hirsch, E., Boeri Erba, E., Mohammed, S., Jensen, O.N., Palestro, G., Inghirami, G. and Chiarle, R. (2005) p130Cas mediates the transforming properties of the anaplastic lymphoma kinase. Blood, 106, 3907-3916.
Amin, H.M., McDonnell, T.J., Ma, Y., Lin, Q., Fujio, Y., Kunisada, K., Leventaki, V., Das, P., Rassidakis, G.Z., Cutler, C., Medeiros, L.J. and Lai, R. (2004) Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene, 23, 5426-5434.
Amin, H.M., Medeiros, L.J., Ma, Y., Feretzaki, M., Das, P., Leventaki, V., Rassidakis, G.Z., O'Connor, S.L., McDonnell, T.J. and Lai, R. (2003) Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene, 22, 5399-5407.
Arcaro, A., Volinia, S., Zvelebil, M.J., Stein, R., Watton, S.J., Layton, M.J., Gout, I., Ahmadi, K., Downward, J. and Waterfield, M.D. (1998) Human phosphoinositide 3-kinase C2beta, the role of calcium and the C2 domain in enzyme activity. J Biol Chem, 273, 33082-33090.
Armstrong, F., Duplantier, M.M., Trempat, P., Hieblot, C., Lamant, L., Espinos, E., Racaud-Sultan, C., Allouche, M., Campo, E., Delsol, G. and Touriol, C. (2004) Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene, 23, 6071-6082.
Armstrong, F., Lamant, L., Hieblot, C., Delsol, G. and Touriol, C. (2007) TPM3-ALK expression induces changes in cytoskeleton organisation and confers higher metastatic capacities than other ALK fusion proteins. Eur J Cancer, 43, 640-646.
Arora, A. and Scholar, E.M. (2005) Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther, 315, 971-979.
Babst, M. (2004) GGAing ubiquitin to the endosome. Nat Cell Biol, 6, 175-177. Bacchiocchi, R., Baldanzi, G., Carbonari, D., Capomagi, C., Colombo, E., van Blitterswijk,
W.J., Graziani, A. and Fazioli, F. (2005) Activation of alpha-diacylglycerol kinase is critical for the mitogenic properties of anaplastic lymphoma kinase. Blood, 106, 2175-2182.
Backer, J.M. (2008) The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J, 410, 1-17.
Bader, A.G., Kang, S., Zhao, L. and Vogt, P.K. (2005) Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer, 5, 921-929.
146

Références bibliographiques
Bae, Y.S., Cantley, L.G., Chen, C.S., Kim, S.R., Kwon, K.S. and Rhee, S.G. (1998) Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem, 273, 4465-4469.
Bai, R.Y., Dieter, P., Peschel, C., Morris, S.W. and Duyster, J. (1998) Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol, 18, 6951-6961.
Bai, R.Y., Ouyang, T., Miething, C., Morris, S.W., Peschel, C. and Duyster, J. (2000) Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood, 96, 4319-4327.
Balla, A. and Balla, T. (2006) Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol, 16, 351-361.
Banfic, H., Tang, X., Batty, I.H., Downes, C.P., Chen, C. and Rittenhouse, S.E. (1998) A novel integrin-activated pathway forms PKB/Akt-stimulatory phosphatidylinositol 3,4-bisphosphate via phosphatidylinositol 3-phosphate in platelets. J Biol Chem, 273, 13-16.
Baumeister, M.A., Martinu, L., Rossman, K.L., Sondek, J., Lemmon, M.A. and Chou, M.M. (2003) Loss of phosphatidylinositol 3-phosphate binding by the C-terminal Tiam-1 pleckstrin homology domain prevents in vivo Rac1 activation without affecting membrane targeting. J Biol Chem, 278, 11457-11464.
Bayle, C., Charpentier, A., Duchayne, E., Manel, A.M., Pages, M.P., Robert, A., Lamant, L., Dastugue, N., Bertrand, Y., Dijoud, F., Emile, J.F., Machover, D., Brugieres, L. and Delsol, G. (1999) Leukaemic presentation of small cell variant anaplastic large cell lymphoma: report of four cases. Br J Haematol, 104, 680-688.
Begley, M.J., Taylor, G.S., Kim, S.A., Veine, D.M., Dixon, J.E. and Stuckey, J.A. (2003) Crystal structure of a phosphoinositide phosphatase, MTMR2: insights into myotubular myopathy and Charcot-Marie-Tooth syndrome. Mol Cell, 12, 1391-1402.
Benharroch, D., Meguerian-Bedoyan, Z., Lamant, L., Amin, C., Brugieres, L., Terrier-Lacombe, M.J., Haralambieva, E., Pulford, K., Pileri, S., Morris, S.W., Mason, D.Y. and Delsol, G. (1998) ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood, 91, 2076-2084.
Berger, P., Bonneick, S., Willi, S., Wymann, M. and Suter, U. (2002) Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot-Marie-Tooth disease type 4B1. Hum Mol Genet, 11, 1569-1579.
Berridge, M.J. (1986) Regulation of ion channels by inositol trisphosphate and diacylglycerol. J Exp Biol, 124, 323-335.
Berridge, M.J. (1987) Inositol trisphosphate and diacylglycerol: two interacting second messengers. Annu Rev Biochem, 56, 159-193.
Berridge, M.J., Heslop, J.P., Irvine, R.F. and Brown, K.D. (1984) Inositol trisphosphate formation and calcium mobilization in Swiss 3T3 cells in response to platelet-derived growth factor. Biochem J, 222, 195-201.
Bertini, E., Biancalana, V., Bolino, A., Buj Bello, A., Clague, M., Guicheney, P., Jungbluth, H., Kress, W., Musaro, A., Nandurkar, H., Pirola, L., Romero, N., Senderek, J., Suter, U., Sewry, C., Tronchere, H., Wallgren-Pettersson, C., Wishart, M.J. and Laporte, J. (2004) 118th ENMC International Workshop on Advances in Myotubular Myopathy. 26-28 September 2003, Naarden, The Netherlands. (5th Workshop of the International Consortium on Myotubular Myopathy). Neuromuscul Disord, 14, 387-396.
147

Références bibliographiques
Berwick, D.C., Dell, G.C., Welsh, G.I., Heesom, K.J., Hers, I., Fletcher, L.M., Cooke, F.T. and Tavare, J.M. (2004) Protein kinase B phosphorylation of PIKfyve regulates the trafficking of GLUT4 vesicles. J Cell Sci, 117, 5985-5993.
Bischof, D., Pulford, K., Mason, D.Y. and Morris, S.W. (1997) Role of the nucleophosmin (NPM) portion of the non-Hodgkin's lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol, 17, 2312-2325.
Blondeau, F., Laporte, J., Bodin, S., Superti-Furga, G., Payrastre, B. and Mandel, J.L. (2000) Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet, 9, 2223-2229.
Blume-Jensen, P. and Hunter, T. (2001) Oncogenic kinase signalling. Nature, 411, 355-365. Bolino, A., Bolis, A., Previtali, S.C., Dina, G., Bussini, S., Dati, G., Amadio, S., Del Carro,
U., Mruk, D.D., Feltri, M.L., Cheng, C.Y., Quattrini, A. and Wrabetz, L. (2004) Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol, 167, 711-721.
Bolino, A., Muglia, M., Conforti, F.L., LeGuern, E., Salih, M.A., Georgiou, D.M., Christodoulou, K., Hausmanowa-Petrusewicz, I., Mandich, P., Schenone, A., Gambardella, A., Bono, F., Quattrone, A., Devoto, M. and Monaco, A.P. (2000) Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet, 25, 17-19.
Bonangelino, C.J., Catlett, N.L. and Weisman, L.S. (1997) Vac7p, a novel vacuolar protein, is required for normal vacuole inheritance and morphology. Mol Cell Biol, 17, 6847-6858.
Bonangelino, C.J., Nau, J.J., Duex, J.E., Brinkman, M., Wurmser, A.E., Gary, J.D., Emr, S.D. and Weisman, L.S. (2002) Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol, 156, 1015-1028.
Bonvini, P., Dalla Rosa, H., Vignes, N. and Rosolen, A. (2004) Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res, 64, 3256-3264.
Bonvini, P., Gastaldi, T., Falini, B. and Rosolen, A. (2002) Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res, 62, 1559-1566.
Botelho, R.J., Efe, J.A., Teis, D. and Emr, S.D. (2008) Assembly of a Fab1 Phosphoinositide Kinase Signaling Complex Requires the Fig4 Phosphoinositide Phosphatase. Mol Biol Cell.
Bowden, E.T., Stoica, G.E. and Wellstein, A. (2002) Anti-apoptotic signaling of pleiotrophin through its receptor, anaplastic lymphoma kinase. J Biol Chem, 277, 35862-35868.
Bretscher, A., Edwards, K. and Fehon, R.G. (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol, 3, 586-599.
Bridge, J.A., Kanamori, M., Ma, Z., Pickering, D., Hill, D.A., Lydiatt, W., Lui, M.Y., Colleoni, G.W., Antonescu, C.R., Ladanyi, M. and Morris, S.W. (2001) Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol, 159, 411-415.
Brown, R.A., Domin, J., Arcaro, A., Waterfield, M.D. and Shepherd, P.R. (1999) Insulin activates the alpha isoform of class II phosphoinositide 3-kinase. J Biol Chem, 274, 14529-14532.
148

Références bibliographiques
Brugieres, L., Deley, M.C., Pacquement, H., Meguerian-Bedoyan, Z., Terrier-Lacombe, M.J., Robert, A., Pondarre, C., Leverger, G., Devalck, C., Rodary, C., Delsol, G. and Hartmann, O. (1998) CD30(+) anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood, 92, 3591-3598.
Brunet, A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S., Anderson, M.J., Arden, K.C., Blenis, J. and Greenberg, M.E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857-868.
Bryant, N.J., Govers, R. and James, D.E. (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol, 3, 267-277.
Budovskaya, Y.V., Hama, H., DeWald, D.B. and Herman, P.K. (2002) The C terminus of the Vps34p phosphoinositide 3-kinase is necessary and sufficient for the interaction with the Vps15p protein kinase. J Biol Chem, 277, 287-294.
Buj-Bello, A., Furling, D., Tronchere, H., Laporte, J., Lerouge, T., Butler-Browne, G.S. and Mandel, J.L. (2002) Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet, 11, 2297-2307.
Bunce, M.W., Boronenkov, I.V. and Anderson, R.A. (2008) Coordinated activation of the nuclear ubiquitin ligase Cul3-SPOP by the generation of phosphatidylinositol 5-phosphate. J Biol Chem, 283, 8678-8686.
Bunce, M.W., Gonzales, M.L. and Anderson, R.A. (2006) Stress-ING out: phosphoinositides mediate the cellular stress response. Sci STKE, 2006, pe46.
Byfield, M.P., Murray, J.T. and Backer, J.M. (2005) hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem, 280, 33076-33082.
Cabezas, A., Pattni, K. and Stenmark, H. (2006) Cloning and subcellular localization of a human phosphatidylinositol 3-phosphate 5-kinase, PIKfyve/Fab1. Gene, 371, 34-41.
Calera, M.R., Martinez, C., Liu, H., Jack, A.K., Birnbaum, M.J. and Pilch, P.F. (1998) Insulin increases the association of Akt-2 with Glut4-containing vesicles. J Biol Chem, 273, 7201-7204.
Cantley, L.C. (2002) The phosphoinositide 3-kinase pathway. Science, 296, 1655-1657. Carricaburu, V., Lamia, K.A., Lo, E., Favereaux, L., Payrastre, B., Cantley, L.C. and Rameh,
L.E. (2003) The phosphatidylinositol (PI)-5-phosphate 4-kinase type II enzyme controls insulin signaling by regulating PI-3,4,5-trisphosphate degradation. Proc Natl Acad Sci U S A, 100, 9867-9872.
Chaussade, C., Pirola, L., Bonnafous, S., Blondeau, F., Brenz-Verca, S., Tronchere, H., Portis, F., Rusconi, S., Payrastre, B., Laporte, J. and Van Obberghen, E. (2003) Expression of myotubularin by an adenoviral vector demonstrates its function as a phosphatidylinositol 3-phosphate [PtdIns(3)P] phosphatase in muscle cell lines: involvement of PtdIns(3)P in insulin-stimulated glucose transport. Mol Endocrinol, 17, 2448-2460.
Chen, H.J., Lin, C.M., Lin, C.S., Perez-Olle, R., Leung, C.L. and Liem, R.K. (2006) The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev, 20, 1933-1945.
Chiarle, R., Gong, J.Z., Guasparri, I., Pesci, A., Cai, J., Liu, J., Simmons, W.J., Dhall, G., Howes, J., Piva, R. and Inghirami, G. (2003) NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood, 101, 1919-1927.
Chiarle, R., Simmons, W.J., Cai, H., Dhall, G., Zamo, A., Raz, R., Karras, J.G., Levy, D.E. and Inghirami, G. (2005) Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med, 11, 623-629.
149

Références bibliographiques
Chiarle, R., Voena, C., Ambrogio, C., Piva, R. and Inghirami, G. (2008) The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer, 8, 11-23.
Choudhury, P., Srivastava, S., Li, Z., Ko, K., Albaqumi, M., Narayan, K., Coetzee, W.A., Lemmon, M.A. and Skolnik, E.Y. (2006) Specificity of the myotubularin family of phosphatidylinositol-3-phosphatase is determined by the PH/GRAM domain. J Biol Chem, 281, 31762-31769.
Chow, C.Y., Zhang, Y., Dowling, J.J., Jin, N., Adamska, M., Shiga, K., Szigeti, K., Shy, M.E., Li, J., Zhang, X., Lupski, J.R., Weisman, L.S. and Meisler, M.H. (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature, 448, 68-72.
Christensen, J.G., Zou, H.Y., Arango, M.E., Li, Q., Lee, J.H., McDonnell, S.R., Yamazaki, S., Alton, G.R., Mroczkowski, B. and Los, G. (2007) Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther, 6, 3314-3322.
Chua, J. and Deretic, V. (2004) Mycobacterium tuberculosis reprograms waves of phosphatidylinositol 3-phosphate on phagosomal organelles. J Biol Chem, 279, 36982-36992.
Ciruela, A., Hinchliffe, K.A., Divecha, N. and Irvine, R.F. (2000) Nuclear targeting of the beta isoform of type II phosphatidylinositol phosphate kinase (phosphatidylinositol 5-phosphate 4-kinase) by its alpha-helix 7. Biochem J, 346 Pt 3, 587-591.
Clague, M.J. and Lorenzo, O. (2005) The myotubularin family of lipid phosphatases. Traffic, 6, 1063-1069.
Clarke, J.H., Letcher, A.J., D'Santos C, S., Halstead, J.R., Irvine, R.F. and Divecha, N. (2001) Inositol lipids are regulated during cell cycle progression in the nuclei of murine erythroleukaemia cells. Biochem J, 357, 905-910.
Clement, S., Krause, U., Desmedt, F., Tanti, J.F., Behrends, J., Pesesse, X., Sasaki, T., Penninger, J., Doherty, M., Malaisse, W., Dumont, J.E., Le Marchand-Brustel, Y., Erneux, C., Hue, L. and Schurmans, S. (2001) The lipid phosphatase SHIP2 controls insulin sensitivity. Nature, 409, 92-97.
Cockcroft, S. (1998) Phosphatidylinositol transfer proteins: a requirement in signal transduction and vesicle traffic. Bioessays, 20, 423-432.
Coffin, C.M., Patel, A., Perkins, S., Elenitoba-Johnson, K.S., Perlman, E. and Griffin, C.A. (2001) ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol, 14, 569-576.
Colomba, A., Courilleau, D., Ramel, D., Billadeau, D.D., Espinos, E., Delsol, G., Payrastre, B. and Gaits-Iacovoni, F. (2008) Activation of Rac1 and the exchange factor Vav3 are involved in NPM-ALK signaling in anaplastic large cell lymphomas. Oncogene, 27, 2728-2736.
Colombo, E., Marine, J.C., Danovi, D., Falini, B. and Pelicci, P.G. (2002) Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol, 4, 529-533.
Coluccia, A.M., Gunby, R.H., Tartari, C.J., Scapozza, L., Gambacorti-Passerini, C. and Passoni, L. (2005) Anaplastic lymphoma kinase and its signalling molecules as novel targets in lymphoma therapy. Expert Opin Ther Targets, 9, 515-532.
Cools, J., Wlodarska, I., Somers, R., Mentens, N., Pedeutour, F., Maes, B., De Wolf-Peeters, C., Pauwels, P., Hagemeijer, A. and Marynen, P. (2002) Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer, 34, 354-362.
150

Références bibliographiques
Cordell, J.L., Pulford, K.A., Bigerna, B., Roncador, G., Banham, A., Colombo, E., Pelicci, P.G., Mason, D.Y. and Falini, B. (1999) Detection of normal and chimeric nucleophosmin in human cells. Blood, 93, 632-642.
Coronas, S., Ramel, D., Pendaries, C., Gaits-Iacovoni, F., Tronchere, H. and Payrastre, B. (2007) PtdIns5P: a little phosphoinositide with big functions? Biochem Soc Symp, 117-128.
Cossart, P. and Sansonetti, P.J. (2004) Bacterial invasion: the paradigms of enteroinvasive pathogens. Science, 304, 242-248.
Cozier, G.E., Carlton, J., Bouyoucef, D. and Cullen, P.J. (2004) Membrane targeting by pleckstrin homology domains. Curr Top Microbiol Immunol, 282, 49-88.
Cozier, G.E., Carlton, J., McGregor, A.H., Gleeson, P.A., Teasdale, R.D., Mellor, H. and Cullen, P.J. (2002) The phox homology (PX) domain-dependent, 3-phosphoinositide-mediated association of sorting nexin-1 with an early sorting endosomal compartment is required for its ability to regulate epidermal growth factor receptor degradation. J Biol Chem, 277, 48730-48736.
Crockett, D.K., Lin, Z., Elenitoba-Johnson, K.S. and Lim, M.S. (2004) Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene, 23, 2617-2629.
Cui, X., De Vivo, I., Slany, R., Miyamoto, A., Firestein, R. and Cleary, M.L. (1998) Association of SET domain and myotubularin-related proteins modulates growth control. Nat Genet, 18, 331-337.
Cullen, P.J., Cozier, G.E., Banting, G. and Mellor, H. (2001) Modular phosphoinositide-binding domains--their role in signalling and membrane trafficking. Curr Biol, 11, R882-893.
Cussac, D., Greenland, C., Roche, S., Bai, R.Y., Duyster, J., Morris, S.W., Delsol, G., Allouche, M. and Payrastre, B. (2004) Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell lymphoma recruits, activates, and uses pp60c-src to mediate its mitogenicity. Blood, 103, 1464-1471.
Cussac, D., Pichereaux, C., Colomba, A., Capilla, F., Pont, F., Gaits-Iacovoni, F., Lamant, L., Espinos, E., Burlet-Schiltz, O., Monsarrat, B., Delsol, G. and Payrastre, B. (2006) Proteomic analysis of anaplastic lymphoma cell lines: identification of potential tumour markers. Proteomics, 6, 3210-3222.
Czupalla, C., Culo, M., Muller, E.C., Brock, C., Reusch, H.P., Spicher, K., Krause, E. and Nurnberg, B. (2003a) Identification and characterization of the autophosphorylation sites of phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem, 278, 11536-11545.
Czupalla, C., Nurnberg, B. and Krause, E. (2003b) Analysis of class I phosphoinositide 3-kinase autophosphorylation sites by mass spectrometry. Rapid Commun Mass Spectrom, 17, 690-696.
D'Angelo, G., Polishchuk, E., Di Tullio, G., Santoro, M., Di Campli, A., Godi, A., West, G., Bielawski, J., Chuang, C.C., van der Spoel, A.C., Platt, F.M., Hannun, Y.A., Polishchuk, R., Mattjus, P. and De Matteis, M.A. (2007) Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature, 449, 62-67.
Dalton, R.R., Rassidakis, G.Z., Atwell, C., Wang, S., Oyarzo, M.P. and Medeiros, L.J. (2005) Differential expression of cyclin D3 in ALK+ and ALK- anaplastic large cell lymphoma. Hum Pathol, 36, 806-811.
Dang, H., Li, Z., Skolnik, E.Y. and Fares, H. (2004) Disease-related myotubularins function in endocytic traffic in Caenorhabditis elegans. Mol Biol Cell, 15, 189-196.
Das, S., Dixon, J.E. and Cho, W. (2003) Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A, 100, 7491-7496.
151

Références bibliographiques
De Camilli, P., Chen, H., Hyman, J., Panepucci, E., Bateman, A. and Brunger, A.T. (2002) The ENTH domain. FEBS Lett, 513, 11-18.
De Matteis, M.A. and Godi, A. (2004) Protein-lipid interactions in membrane trafficking at the Golgi complex. Biochim Biophys Acta, 1666, 264-274.
De Vivo, I., Cui, X., Domen, J. and Cleary, M.L. (1998) Growth stimulation of primary B cell precursors by the anti-phosphatase Sbf1. Proc Natl Acad Sci U S A, 95, 9471-9476.
Delsol, G., Lamant, L., Mariame, B., Pulford, K., Dastugue, N., Brousset, P., Rigal-Huguet, F., al Saati, T., Cerretti, D.P., Morris, S.W. and Mason, D.Y. (1997) A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2; 5 translocation. Blood, 89, 1483-1490.
Denu, J.M., Stuckey, J.A., Saper, M.A. and Dixon, J.E. (1996) Form and function in protein dephosphorylation. Cell, 87, 361-364.
Dhanasekaran, D.N. and Johnson, G.L. (2007) MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene, 26, 3097-3099.
Di Lello, P., Nguyen, B.D., Jones, T.N., Potempa, K., Kobor, M.S., Legault, P. and Omichinski, J.G. (2005) NMR structure of the amino-terminal domain from the Tfb1 subunit of TFIIH and characterization of its phosphoinositide and VP16 binding sites. Biochemistry, 44, 7678-7686.
Diaz, E., Schimmoller, F. and Pfeffer, S.R. (1997) A novel Rab9 effector required for endosome-to-TGN transport. J Cell Biol, 138, 283-290.
Dirks, W.G., Fahnrich, S., Lis, Y., Becker, E., MacLeod, R.A. and Drexler, H.G. (2002) Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer, 100, 49-56.
Doerks, T., Strauss, M., Brendel, M. and Bork, P. (2000) GRAM, a novel domain in glucosyltransferases, myotubularins and other putative membrane-associated proteins. Trends Biochem Sci, 25, 483-485.
Dove, S.K., Cooke, F.T., Douglas, M.R., Sayers, L.G., Parker, P.J. and Michell, R.H. (1997) Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature, 390, 187-192.
Dove, S.K., McEwen, R.K., Mayes, A., Hughes, D.C., Beggs, J.D. and Michell, R.H. (2002) Vac14 controls PtdIns(3,5)P(2) synthesis and Fab1-dependent protein trafficking to the multivesicular body. Curr Biol, 12, 885-893.
Dove, S.K., Piper, R.C., McEwen, R.K., Yu, J.W., King, M.C., Hughes, D.C., Thuring, J., Holmes, A.B., Cooke, F.T., Michell, R.H., Parker, P.J. and Lemmon, M.A. (2004) Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. Embo J, 23, 1922-1933.
Dowler, S., Currie, R.A., Campbell, D.G., Deak, M., Kular, G., Downes, C.P. and Alessi, D.R. (2000) Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J, 351, 19-31.
Downes, C.P., Gray, A. and Lucocq, J.M. (2005) Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol, 15, 259-268.
Drexler, H.G., Gignac, S.M., von Wasielewski, R., Werner, M. and Dirks, W.G. (2000) Pathobiology of NPM-ALK and variant fusion genes in anaplastic large cell lymphoma and other lymphomas. Leukemia, 14, 1533-1559.
Du, G., Altshuller, Y.M., Vitale, N., Huang, P., Chasserot-Golaz, S., Morris, A.J., Bader, M.F. and Frohman, M.A. (2003) Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J Cell Biol, 162, 305-315.
152

Références bibliographiques
Duex, J.E., Tang, F. and Weisman, L.S. (2006) The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol, 172, 693-704.
Dukes, J.D., Lee, H., Hagen, R., Reaves, B.J., Layton, A.N., Galyov, E.E. and Whitley, P. (2006) The secreted Salmonella dublin phosphoinositide phosphatase, SopB, localizes to PtdIns(3)P-containing endosomes and perturbs normal endosome to lysosome trafficking. Biochem J, 395, 239-247.
Dumas, J.J., Merithew, E., Sudharshan, E., Rajamani, D., Hayes, S., Lawe, D., Corvera, S. and Lambright, D.G. (2001) Multivalent endosome targeting by homodimeric EEA1. Mol Cell, 8, 947-958.
Duyster, J., Bai, R.Y. and Morris, S.W. (2001) Translocations involving anaplastic lymphoma kinase (ALK). Oncogene, 20, 5623-5637.
Efe, J.A., Botelho, R.J. and Emr, S.D. (2007) Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell, 18, 4232-4244.
Elkin, S.K., Ivanov, D., Ewalt, M., Ferguson, C.G., Hyberts, S.G., Sun, Z.Y., Prestwich, G.D., Yuan, J., Wagner, G., Oettinger, M.A. and Gozani, O.P. (2005) A PHD finger motif in the C terminus of RAG2 modulates recombination activity. J Biol Chem, 280, 28701-28710.
Engelman, J.A., Luo, J. and Cantley, L.C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet, 7, 606-619.
Englund, C., Loren, C.E., Grabbe, C., Varshney, G.K., Deleuil, F., Hallberg, B. and Palmer, R.H. (2003) Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature, 425, 512-516.
Ergin, M., Denning, M.F., Izban, K.F., Amin, H.M., Martinez, R.L., Saeed, S. and Alkan, S. (2001) Inhibition of tyrosine kinase activity induces caspase-dependent apoptosis in anaplastic large cell lymphoma with NPM-ALK (p80) fusion protein. Exp Hematol, 29, 1082-1090.
Eugster, A., Pecheur, E.I., Michel, F., Winsor, B., Letourneur, F. and Friant, S. (2004) Ent5p is required with Ent3p and Vps27p for ubiquitin-dependent protein sorting into the multivesicular body. Mol Biol Cell, 15, 3031-3041.
Fabre, S., Reynaud, C. and Jalinot, P. (2000) Identification of functional PDZ domain binding sites in several human proteins. Mol Biol Rep, 27, 217-224.
Falini, B. (2001) Anaplastic large cell lymphoma: pathological, molecular and clinical features. Br J Haematol, 114, 741-760.
Falini, B., Bigerna, B., Fizzotti, M., Pulford, K., Pileri, S.A., Delsol, G., Carbone, A., Paulli, M., Magrini, U., Menestrina, F., Giardini, R., Pilotti, S., Mezzelani, A., Ugolini, B., Billi, M., Pucciarini, A., Pacini, R., Pelicci, P.G. and Flenghi, L. (1998) ALK expression defines a distinct group of T/null lymphomas ("ALK lymphomas") with a wide morphological spectrum. Am J Pathol, 153, 875-886.
Falini, B. and Mason, D.Y. (2002) Proteins encoded by genes involved in chromosomal alterations in lymphoma and leukemia: clinical value of their detection by immunocytochemistry. Blood, 99, 409-426.
Falini, B., Nicoletti, I., Bolli, N., Martelli, M.P., Liso, A., Gorello, P., Mandelli, F., Mecucci, C. and Martelli, M.F. (2007) Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica, 92, 519-532.
Falini, B., Pileri, S., Zinzani, P.L., Carbone, A., Zagonel, V., Wolf-Peeters, C., Verhoef, G., Menestrina, F., Todeschini, G., Paulli, M., Lazzarino, M., Giardini, R., Aiello, A., Foss, H.D., Araujo, I., Fizzotti, M., Pelicci, P.G., Flenghi, L., Martelli, M.F. and
153

Références bibliographiques
Santucci, A. (1999a) ALK+ lymphoma: clinico-pathological findings and outcome. Blood, 93, 2697-2706.
Falini, B., Pulford, K., Pucciarini, A., Carbone, A., De Wolf-Peeters, C., Cordell, J., Fizzotti, M., Santucci, A., Pelicci, P.G., Pileri, S., Campo, E., Ott, G., Delsol, G. and Mason, D.Y. (1999b) Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood, 94, 3509-3515.
Fass, D., Blacklow, S., Kim, P.S. and Berger, J.M. (1997) Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature, 388, 691-693.
Faucherre, A., Desbois, P., Nagano, F., Satre, V., Lunardi, J., Gacon, G. and Dorseuil, O. (2005) Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum Mol Genet, 14, 1441-1448.
Fawal, M., Armstrong, F., Ollier, S., Dupont, H., Touriol, C., Monsarrat, B., Delsol, G., Payrastre, B. and Morello, D. (2006) A "liaison dangereuse" between AUF1/hnRNPD and the oncogenic tyrosine kinase NPM-ALK. Blood, 108, 2780-2788.
Feng, X., Hara, Y. and Riabowol, K. (2002) Different HATS of the ING1 gene family. Trends Cell Biol, 12, 532-538.
Fiorani, C., Vinci, G., Sacchi, S., Bonaccorsi, G. and Artusi, T. (2001) Primary systemic anaplastic large-cell lymphoma (CD30+): advances in biology and current therapeutic approaches. Clin Lymphoma, 2, 29-37; discussion 38-29.
Ford, M.G., Mills, I.G., Peter, B.J., Vallis, Y., Praefcke, G.J., Evans, P.R. and McMahon, H.T. (2002) Curvature of clathrin-coated pits driven by epsin. Nature, 419, 361-366.
Ford, M.G., Pearse, B.M., Higgins, M.K., Vallis, Y., Owen, D.J., Gibson, A., Hopkins, C.R., Evans, P.R. and McMahon, H.T. (2001) Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science, 291, 1051-1055.
Foster, F.M., Traer, C.J., Abraham, S.M. and Fry, M.J. (2003) The phosphoinositide (PI) 3-kinase family. J Cell Sci, 116, 3037-3040.
Foster, L.J., Li, D., Randhawa, V.K. and Klip, A. (2001) Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem, 276, 44212-44221.
Frame, M.C. (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta, 1602, 114-130.
Friant, S., Pecheur, E.I., Eugster, A., Michel, F., Lefkir, Y., Nourrisson, D. and Letourneur, F. (2003) Ent3p Is a PtdIns(3,5)P2 effector required for protein sorting to the multivesicular body. Dev Cell, 5, 499-511.
Fry, M.J. (1994) Structure, regulation and function of phosphoinositide 3-kinases. Biochim Biophys Acta, 1226, 237-268.
Fujimoto, J., Shiota, M., Iwahara, T., Seki, N., Satoh, H., Mori, S. and Yamamoto, T. (1996) Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc Natl Acad Sci U S A, 93, 4181-4186.
Fyodorov, D.V. and Kadonaga, J.T. (2002) Dynamics of ATP-dependent chromatin assembly by ACF. Nature, 418, 897-900.
Galkin, A.V., Melnick, J.S., Kim, S., Hood, T.L., Li, N., Li, L., Xia, G., Steensma, R., Chopiuk, G., Jiang, J., Wan, Y., Ding, P., Liu, Y., Sun, F., Schultz, P.G., Gray, N.S. and Warmuth, M. (2007) Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A, 104, 270-275.
154

Références bibliographiques
Gao, Y., Dickerson, J.B., Guo, F., Zheng, J. and Zheng, Y. (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A, 101, 7618-7623.
Gary, J.D., Sato, T.K., Stefan, C.J., Bonangelino, C.J., Weisman, L.S. and Emr, S.D. (2002) Regulation of Fab1 phosphatidylinositol 3-phosphate 5-kinase pathway by Vac7 protein and Fig4, a polyphosphoinositide phosphatase family member. Mol Biol Cell, 13, 1238-1251.
Gary, J.D., Wurmser, A.E., Bonangelino, C.J., Weisman, L.S. and Emr, S.D. (1998) Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol, 143, 65-79.
Gascoyne, R.D., Aoun, P., Wu, D., Chhanabhai, M., Skinnider, B.F., Greiner, T.C., Morris, S.W., Connors, J.M., Vose, J.M., Viswanatha, D.S., Coldman, A. and Weisenburger, D.D. (1999) Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood, 93, 3913-3921.
Gascoyne, R.D., Lamant, L., Martin-Subero, J.I., Lestou, V.S., Harris, N.L., Muller-Hermelink, H.K., Seymour, J.F., Campbell, L.J., Horsman, D.E., Auvigne, I., Espinos, E., Siebert, R. and Delsol, G. (2003) ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood, 102, 2568-2573.
Gillooly, D.J., Simonsen, A. and Stenmark, H. (2001) Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem J, 355, 249-258.
Gouzi, J.Y., Moog-Lutz, C., Vigny, M. and Brunet-de Carvalho, N. (2005) Role of the subcellular localization of ALK tyrosine kinase domain in neuronal differentiation of PC12 cells. J Cell Sci, 118, 5811-5823.
Gozani, O., Karuman, P., Jones, D.R., Ivanov, D., Cha, J., Lugovskoy, A.A., Baird, C.L., Zhu, H., Field, S.J., Lessnick, S.L., Villasenor, J., Mehrotra, B., Chen, J., Rao, V.R., Brugge, J.S., Ferguson, C.G., Payrastre, B., Myszka, D.G., Cantley, L.C., Wagner, G., Divecha, N., Prestwich, G.D. and Yuan, J. (2003) The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell, 114, 99-111.
Grantham, J., Ruddock, L.W., Roobol, A. and Carden, M.J. (2002) Eukaryotic chaperonin containing T-complex polypeptide 1 interacts with filamentous actin and reduces the initial rate of actin polymerization in vitro. Cell Stress Chaperones, 7, 235-242.
Griffin, C.A., Hawkins, A.L., Dvorak, C., Henkle, C., Ellingham, T. and Perlman, E.J. (1999) Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res, 59, 2776-2780.
Griffiths, B.B. (1990) Kinetics of hemolysin production in bovine group B streptococci. J Basic Microbiol, 30, 241-250.
Grisendi, S., Bernardi, R., Rossi, M., Cheng, K., Khandker, L., Manova, K. and Pandolfi, P.P. (2005) Role of nucleophosmin in embryonic development and tumorigenesis. Nature, 437, 147-153.
Grisendi, S., Mecucci, C., Falini, B. and Pandolfi, P.P. (2006) Nucleophosmin and cancer. Nat Rev Cancer, 6, 493-505.
Gruenberg, J. and Stenmark, H. (2004) The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol, 5, 317-323.
Gu, T.L., Tothova, Z., Scheijen, B., Griffin, J.D., Gilliland, D.G. and Sternberg, D.W. (2004) NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood, 103, 4622-4629.
155

Références bibliographiques
Gunby, R.H., Ahmed, S., Sottocornola, R., Gasser, M., Redaelli, S., Mologni, L., Tartari, C.J., Belloni, V., Gambacorti-Passerini, C. and Scapozza, L. (2006) Structural insights into the ATP binding pocket of the anaplastic lymphoma kinase by site-directed mutagenesis, inhibitor binding analysis, and homology modeling. J Med Chem, 49, 5759-5768.
Hama, H., Schnieders, E.A., Thorner, J., Takemoto, J.Y. and DeWald, D.B. (1999) Direct involvement of phosphatidylinositol 4-phosphate in secretion in the yeast Saccharomyces cerevisiae. J Biol Chem, 274, 34294-34300.
Hamada, K., Shimizu, T., Matsui, T., Tsukita, S. and Hakoshima, T. (2000) Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. Embo J, 19, 4449-4462.
Han, J., Luby-Phelps, K., Das, B., Shu, X., Xia, Y., Mosteller, R.D., Krishna, U.M., Falck, J.R., White, M.A. and Broek, D. (1998) Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science, 279, 558-560.
Han, Y., Amin, H.M., Franko, B., Frantz, C., Shi, X. and Lai, R. (2006) Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood, 108, 2796-2803.
Hernandez, L., Bea, S., Bellosillo, B., Pinyol, M., Falini, B., Carbone, A., Ott, G., Rosenwald, A., Fernandez, A., Pulford, K., Mason, D., Morris, S.W., Santos, E. and Campo, E. (2002) Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am J Pathol, 160, 1487-1494.
Hernandez, L., Pinyol, M., Hernandez, S., Bea, S., Pulford, K., Rosenwald, A., Lamant, L., Falini, B., Ott, G., Mason, D.Y., Delsol, G. and Campo, E. (1999) TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood, 94, 3265-3268.
Hilpela, P., Vartiainen, M.K. and Lappalainen, P. (2004) Regulation of the actin cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3. Curr Top Microbiol Immunol, 282, 117-163.
Hinchliffe, K.A. (2001) Cellular signalling: stressing the importance of PIP3. Curr Biol, 11, R371-372.
Hinchliffe, K.A. and Irvine, R.F. (2006) Regulation of type II PIP kinase by PKD phosphorylation. Cell Signal, 18, 1906-1913.
Hingorani, K., Szebeni, A. and Olson, M.O. (2000) Mapping the functional domains of nucleolar protein B23. J Biol Chem, 275, 24451-24457.
Hiroaki, H., Ago, T., Ito, T., Sumimoto, H. and Kohda, D. (2001) Solution structure of the PX domain, a target of the SH3 domain. Nat Struct Biol, 8, 526-530.
Hirst, J., Motley, A., Harasaki, K., Peak Chew, S.Y. and Robinson, M.S. (2003) EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol Biol Cell, 14, 625-641.
Hokin, L.E. and Hokin, M.R. (1964) The Incorporation of 32p from Triphosphate into Polyphosphoinositides (Gamma-32p)Adenosine and Phosphatidic Acid in Erythrocyte Membranes. Biochim Biophys Acta, 84, 563-575.
Honorat, J.F., Ragab, A., Lamant, L., Delsol, G. and Ragab-Thomas, J. (2006) SHP1 tyrosine phosphatase negatively regulates NPM-ALK tyrosine kinase signaling. Blood, 107, 4130-4138.
Horie, R., Watanabe, M., Ishida, T., Koiwa, T., Aizawa, S., Itoh, K., Higashihara, M., Kadin, M.E. and Watanabe, T. (2004) The NPM-ALK oncoprotein abrogates CD30 signaling and constitutive NF-kappaB activation in anaplastic large cell lymphoma. Cancer Cell, 5, 353-364.
156

Références bibliographiques
Horie, R., Watanabe, T., Morishita, Y., Ito, K., Ishida, T., Kanegae, Y., Saito, I., Higashihara, M., Mori, S., Kadin, M.E. and Watanabe, T. (2002) Ligand-independent signaling by overexpressed CD30 drives NF-kappaB activation in Hodgkin-Reed-Sternberg cells. Oncogene, 21, 2493-2503.
Hsu, F.Y., Johnston, P.B., Burke, K.A. and Zhao, Y. (2006) The expression of CD30 in anaplastic large cell lymphoma is regulated by nucleophosmin-anaplastic lymphoma kinase-mediated JunB level in a cell type-specific manner. Cancer Res, 66, 9002-9008.
Huang, S. and Czech, M.P. (2007) The GLUT4 glucose transporter. Cell Metab, 5, 237-252. Huang, W., Zhang, H., Davrazou, F., Kutateladze, T.G., Shi, X., Gozani, O. and Prestwich,
G.D. (2007) Stabilized phosphatidylinositol-5-phosphate analogues as ligands for the nuclear protein ING2: chemistry, biology, and molecular modeling. J Am Chem Soc, 129, 6498-6506.
Hubinger, G., Wehnes, E., Xue, L., Morris, S.W. and Maurer, U. (2003) Hammerhead ribozyme-mediated cleavage of the fusion transcript NPM-ALK associated with anaplastic large-cell lymphoma. Exp Hematol, 31, 226-233.
Hurley, J.H. and Emr, S.D. (2006) The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct, 35, 277-298.
Hyun, T.S., Rao, D.S., Saint-Dic, D., Michael, L.E., Kumar, P.D., Bradley, S.V., Mizukami, I.F., Oravecz-Wilson, K.I. and Ross, T.S. (2004) HIP1 and HIP1r stabilize receptor tyrosine kinases and bind 3-phosphoinositides via epsin N-terminal homology domains. J Biol Chem, 279, 14294-14306.
Iijima, M., Huang, Y.E., Luo, H.R., Vazquez, F. and Devreotes, P.N. (2004) Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J Biol Chem, 279, 16606-16613.
Ikonomov, O.C., Sbrissa, D., Dondapati, R. and Shisheva, A. (2007) ArPIKfyve-PIKfyve interaction and role in insulin-regulated GLUT4 translocation and glucose transport in 3T3-L1 adipocytes. Exp Cell Res, 313, 2404-2416.
Ikonomov, O.C., Sbrissa, D., Mlak, K., Deeb, R., Fligger, J., Soans, A., Finley, R.L., Jr. and Shisheva, A. (2003) Active PIKfyve associates with and promotes the membrane attachment of the late endosome-to-trans-Golgi network transport factor Rab9 effector p40. J Biol Chem, 278, 50863-50871.
Ikonomov, O.C., Sbrissa, D., Mlak, K., Kanzaki, M., Pessin, J. and Shisheva, A. (2002a) Functional dissection of lipid and protein kinase signals of PIKfyve reveals the role of PtdIns 3,5-P2 production for endomembrane integrity. J Biol Chem, 277, 9206-9211.
Ikonomov, O.C., Sbrissa, D., Mlak, K. and Shisheva, A. (2002b) Requirement for PIKfyve enzymatic activity in acute and long-term insulin cellular effects. Endocrinology, 143, 4742-4754.
Ikonomov, O.C., Sbrissa, D. and Shisheva, A. (2001) Mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve. J Biol Chem, 276, 26141-26147.
Ikonomov, O.C., Sbrissa, D. and Shisheva, A. (2006) Localized PtdIns 3,5-P2 synthesis to regulate early endosome dynamics and fusion. Am J Physiol Cell Physiol, 291, C393-404.
Ikonomov, O.C., Sbrissa, D., Yoshimori, T., Cover, T.L. and Shisheva, A. (2002c) PIKfyve Kinase and SKD1 AAA ATPase define distinct endocytic compartments. Only PIKfyve expression inhibits the cell-vacoulating activity of Helicobacter pylori VacA toxin. J Biol Chem, 277, 46785-46790.
Imada, K. and Leonard, W.J. (2000) The Jak-STAT pathway. Mol Immunol, 37, 1-11.
157

Références bibliographiques
Irby, R.B. and Yeatman, T.J. (2000) Role of Src expression and activation in human cancer. Oncogene, 19, 5636-5642.
Isakoff, S.J., Cardozo, T., Andreev, J., Li, Z., Ferguson, K.M., Abagyan, R., Lemmon, M.A., Aronheim, A. and Skolnik, E.Y. (1998) Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. Embo J, 17, 5374-5387.
Itoh, T., Ijuin, T. and Takenawa, T. (1998) A novel phosphatidylinositol-5-phosphate 4-kinase (phosphatidylinositol-phosphate kinase IIgamma) is phosphorylated in the endoplasmic reticulum in response to mitogenic signals. J Biol Chem, 273, 20292-20299.
Itoh, T., Koshiba, S., Kigawa, T., Kikuchi, A., Yokoyama, S. and Takenawa, T. (2001) Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science, 291, 1047-1051.
Iwahara, T., Fujimoto, J., Wen, D., Cupples, R., Bucay, N., Arakawa, T., Mori, S., Ratzkin, B. and Yamamoto, T. (1997) Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene, 14, 439-449.
Janmey, P.A. and Stossel, T.P. (1987) Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature, 325, 362-364.
Jefferies, H.B., Cooke, F.T., Jat, P., Boucheron, C., Koizumi, T., Hayakawa, M., Kaizawa, H., Ohishi, T., Workman, P., Waterfield, M.D. and Parker, P.J. (2008) A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep, 9, 164-170.
Jeffries, T.R., Dove, S.K., Michell, R.H. and Parker, P.J. (2004) PtdIns-specific MPR pathway association of a novel WD40 repeat protein, WIPI49. Mol Biol Cell, 15, 2652-2663.
Jones, D.R., Bultsma, Y., Keune, W.J., Halstead, J.R., Elouarrat, D., Mohammed, S., Heck, A.J., D'Santos, C.S. and Divecha, N. (2006) Nuclear PtdIns5P as a transducer of stress signaling: an in vivo role for PIP4Kbeta. Mol Cell, 23, 685-695.
Jundt, F., Raetzel, N., Muller, C., Calkhoven, C.F., Kley, K., Mathas, S., Lietz, A., Leutz, A. and Dorken, B. (2005) A rapamycin derivative (everolimus) controls proliferation through down-regulation of truncated CCAAT enhancer binding protein {beta} and NF-{kappa}B activity in Hodgkin and anaplastic large cell lymphomas. Blood, 106, 1801-1807.
Kalkhoven, E., Teunissen, H., Houweling, A., Verrijzer, C.P. and Zantema, A. (2002) The PHD type zinc finger is an integral part of the CBP acetyltransferase domain. Mol Cell Biol, 22, 1961-1970.
Kanzaki, M. and Pessin, J.E. (2001) Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem, 276, 42436-42444.
Karylowski, O., Zeigerer, A., Cohen, A. and McGraw, T.E. (2004) GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol Biol Cell, 15, 870-882.
Katakami, Y., Katakami, N., Janmey, P.A., Hartwig, J.H. and Stossel, T.P. (1992) Isolation of the phosphatidylinositol 4-monophosphate dissociable high-affinity profilin-actin complex. Biochim Biophys Acta, 1122, 123-135.
Kavran, J.M., Klein, D.E., Lee, A., Falasca, M., Isakoff, S.J., Skolnik, E.Y. and Lemmon, M.A. (1998) Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J Biol Chem, 273, 30497-30508.
Kay, B.K., Yamabhai, M., Wendland, B. and Emr, S.D. (1999) Identification of a novel domain shared by putative components of the endocytic and cytoskeletal machinery. Protein Sci, 8, 435-438.
158

Références bibliographiques
Kearns, M.A., Monks, D.E., Fang, M., Rivas, M.P., Courtney, P.D., Chen, J., Prestwich, G.D., Theibert, A.B., Dewey, R.E. and Bankaitis, V.A. (1998) Novel developmentally regulated phosphoinositide binding proteins from soybean whose expression bypasses the requirement for an essential phosphatidylinositol transfer protein in yeast. Embo J, 17, 4004-4017.
Khoury, J.D., Medeiros, L.J., Rassidakis, G.Z., Yared, M.A., Tsioli, P., Leventaki, V., Schmitt-Graeff, A., Herling, M., Amin, H.M. and Lai, R. (2003) Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK+ and ALK- anaplastic large cell lymphoma. Clin Cancer Res, 9, 3692-3699.
Khoury, J.D., Rassidakis, G.Z., Medeiros, L.J., Amin, H.M. and Lai, R. (2004) Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood, 104, 1580-1581.
Khwaja, A. (1999) Akt is more than just a Bad kinase. Nature, 401, 33-34. Khwaja, A., Rodriguez-Viciana, P., Wennstrom, S., Warne, P.H. and Downward, J. (1997)
Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. Embo J, 16, 2783-2793.
Kim, J., Jahng, W.J., Di Vizio, D., Lee, J.S., Jhaveri, R., Rubin, M.A., Shisheva, A. and Freeman, M.R. (2007) The phosphoinositide kinase PIKfyve mediates epidermal growth factor receptor trafficking to the nucleus. Cancer Res, 67, 9229-9237.
Kim, S.A., Taylor, G.S., Torgersen, K.M. and Dixon, J.E. (2002) Myotubularin and MTMR2, phosphatidylinositol 3-phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. J Biol Chem, 277, 4526-4531.
Kim, S.A., Vacratsis, P.O., Firestein, R., Cleary, M.L. and Dixon, J.E. (2003) Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci U S A, 100, 4492-4497.
Klarlund, J.K., Rameh, L.E., Cantley, L.C., Buxton, J.M., Holik, J.J., Sakelis, C., Patki, V., Corvera, S. and Czech, M.P. (1998) Regulation of GRP1-catalyzed ADP ribosylation factor guanine nucleotide exchange by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem, 273, 1859-1862.
Knodler, L.A., Finlay, B.B. and Steele-Mortimer, O. (2005) The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem, 280, 9058-9064.
Kobayashi, S., Shirai, T., Kiyokawa, E., Mochizuki, N., Matsuda, M. and Fukui, Y. (2001) Membrane recruitment of DOCK180 by binding to PtdIns(3,4,5)P3. Biochem J, 354, 73-78.
Kohn, A.D., Summers, S.A., Birnbaum, M.J. and Roth, R.A. (1996) Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem, 271, 31372-31378.
Krugmann, S., Anderson, K.E., Ridley, S.H., Risso, N., McGregor, A., Coadwell, J., Davidson, K., Eguinoa, A., Ellson, C.D., Lipp, P., Manifava, M., Ktistakis, N., Painter, G., Thuring, J.W., Cooper, M.A., Lim, Z.Y., Holmes, A.B., Dove, S.K., Michell, R.H., Grewal, A., Nazarian, A., Erdjument-Bromage, H., Tempst, P., Stephens, L.R. and Hawkins, P.T. (2002) Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol Cell, 9, 95-108.
Krugmann, S., Hawkins, P.T., Pryer, N. and Braselmann, S. (1999) Characterizing the interactions between the two subunits of the p101/p110gamma phosphoinositide 3-kinase and their role in the activation of this enzyme by G beta gamma subunits. J Biol Chem, 274, 17152-17158.
159

Références bibliographiques
Kuefer, M.U., Look, A.T., Pulford, K., Behm, F.G., Pattengale, P.K., Mason, D.Y. and Morris, S.W. (1997) Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood, 90, 2901-2910.
Kurki, S., Peltonen, K., Latonen, L., Kiviharju, T.M., Ojala, P.M., Meek, D. and Laiho, M. (2004) Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell, 5, 465-475.
Kutateladze, T. and Overduin, M. (2001) Structural mechanism of endosome docking by the FYVE domain. Science, 291, 1793-1796.
Kutok, J.L. and Aster, J.C. (2002) Molecular biology of anaplastic lymphoma kinase-positive anaplastic large-cell lymphoma. J Clin Oncol, 20, 3691-3702.
Ladanyi, M. (1997) The NPM/ALK gene fusion in the pathogenesis of anaplastic large cell lymphoma. Cancer Surv, 30, 59-75.
Lai, R., Rassidakis, G.Z., Lin, Q., Atwell, C., Medeiros, L.J. and Amin, H.M. (2005) Jak3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum Pathol, 36, 939-944.
Lamant, L., Dastugue, N., Pulford, K., Delsol, G. and Mariame, B. (1999) A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood, 93, 3088-3095.
Lamant, L., de Reynies, A., Duplantier, M.M., Rickman, D.S., Sabourdy, F., Giuriato, S., Brugieres, L., Gaulard, P., Espinos, E. and Delsol, G. (2007) Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood, 109, 2156-2164.
Lamant, L., Espinos, E., Duplantier, M., Dastugue, N., Robert, A., Allouche, M., Ragab, J., Brousset, P., Villalva, C., Gascoyne, R.D., Al Saati, T. and Delsol, G. (2004) Establishment of a novel anaplastic large-cell lymphoma-cell line (COST) from a 'small-cell variant' of ALCL. Leukemia, 18, 1693-1698.
Lamant, L., Gascoyne, R.D., Duplantier, M.M., Armstrong, F., Raghab, A., Chhanabhai, M., Rajcan-Separovic, E., Raghab, J., Delsol, G. and Espinos, E. (2003) Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer, 37, 427-432.
Lamant, L., Pulford, K., Bischof, D., Morris, S.W., Mason, D.Y., Delsol, G. and Mariame, B. (2000) Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol, 156, 1711-1721.
Lamia, K.A., Peroni, O.D., Kim, Y.B., Rameh, L.E., Kahn, B.B. and Cantley, L.C. (2004) Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta-/- mice. Mol Cell Biol, 24, 5080-5087.
Lang, F. and Cohen, P. (2001) Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci STKE, 2001, RE17.
Laporte, J., Bedez, F., Bolino, A. and Mandel, J.L. (2003) Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Hum Mol Genet, 12 Spec No 2, R285-292.
Laporte, J., Blondeau, F., Buj-Bello, A., Tentler, D., Kretz, C., Dahl, N. and Mandel, J.L. (1998) Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum Mol Genet, 7, 1703-1712.
Laporte, J., Blondeau, F., Gansmuller, A., Lutz, Y., Vonesch, J.L. and Mandel, J.L. (2002a) The PtdIns3P phosphatase myotubularin is a cytoplasmic protein that also localizes to Rac1-inducible plasma membrane ruffles. J Cell Sci, 115, 3105-3117.
Laporte, J., Hu, L.J., Kretz, C., Mandel, J.L., Kioschis, P., Coy, J.F., Klauck, S.M., Poustka, A. and Dahl, N. (1996) A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet, 13, 175-182.
160

Références bibliographiques
Laporte, J., Liaubet, L., Blondeau, F., Tronchere, H., Mandel, J.L. and Payrastre, B. (2002b) Functional redundancy in the myotubularin family. Biochem Biophys Res Commun, 291, 305-312.
Lawrence, B., Perez-Atayde, A., Hibbard, M.K., Rubin, B.P., Dal Cin, P., Pinkus, J.L., Pinkus, G.S., Xiao, S., Yi, E.S., Fletcher, C.D. and Fletcher, J.A. (2000) TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol, 157, 377-384.
Lazar, T., Scheglmann, D. and Gallwitz, D. (2002) A novel phospholipid-binding protein from the yeast Saccharomyces cerevisiae with dual binding specificities for the transport GTPase Ypt7p and the Sec1-related Vps33p. Eur J Cell Biol, 81, 635-646.
Lee, H.H., Norris, A., Weiss, J.B. and Frasch, M. (2003) Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature, 425, 507-512.
Lee, J.O., Yang, H., Georgescu, M.M., Di Cristofano, A., Maehama, T., Shi, Y., Dixon, J.E., Pandolfi, P. and Pavletich, N.P. (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell, 99, 323-334.
Legendre-Guillemin, V., Wasiak, S., Hussain, N.K., Angers, A. and McPherson, P.S. (2004) ENTH/ANTH proteins and clathrin-mediated membrane budding. J Cell Sci, 117, 9-18.
Lemmon, M.A. (2008) Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol, 9, 99-111.
Lemmon, M.A. and Ferguson, K.M. (2000) Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J, 350 Pt 1, 1-18.
Lemmon, M.A. and Ferguson, K.M. (2001) Molecular determinants in pleckstrin homology domains that allow specific recognition of phosphoinositides. Biochem Soc Trans, 29, 377-384.
Lemmon, M.A., Ferguson, K.M. and Abrams, C.S. (2002) Pleckstrin homology domains and the cytoskeleton. FEBS Lett, 513, 71-76.
Leslie, N.R. and Downes, C.P. (2004) PTEN function: how normal cells control it and tumour cells lose it. Biochem J, 382, 1-11.
Leventaki, V., Drakos, E., Medeiros, L.J., Lim, M.S., Elenitoba-Johnson, K.S., Claret, F.X. and Rassidakis, G.Z. (2007) NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood, 110, 1621-1630.
Li, J., Zhang, X., Sejas, D.P., Bagby, G.C. and Pang, Q. (2004) Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J Biol Chem, 279, 41275-41279.
Li, R., Xue, L., Zhu, T., Jiang, Q., Cui, X., Yan, Z., McGee, D., Wang, J., Gantla, V.R., Pickens, J.C., McGrath, D., Chucholowski, A., Morris, S.W. and Webb, T.R. (2006) Design and synthesis of 5-aryl-pyridone-carboxamides as inhibitors of anaplastic lymphoma kinase. J Med Chem, 49, 1006-1015.
Li, S., Tiab, L., Jiao, X., Munier, F.L., Zografos, L., Frueh, B.E., Sergeev, Y., Smith, J., Rubin, B., Meallet, M.A., Forster, R.K., Hejtmancik, J.F. and Schorderet, D.F. (2005) Mutations in PIP5K3 are associated with Francois-Neetens mouchetee fleck corneal dystrophy. Am J Hum Genet, 77, 54-63.
Lin, J.K. (2007) Molecular targets of curcumin. Adv Exp Med Biol, 595, 227-243. Linassier, C., MacDougall, L.K., Domin, J. and Waterfield, M.D. (1997) Molecular cloning
and biochemical characterization of a Drosophila phosphatidylinositol-specific phosphoinositide 3-kinase. Biochem J, 321 ( Pt 3), 849-856.
161

Références bibliographiques
Lioubin, M.N., Algate, P.A., Tsai, S., Carlberg, K., Aebersold, A. and Rohrschneider, L.R. (1996) p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev, 10, 1084-1095.
Liu, H., Liu, Z.Q., Chen, C.X., Magill, S., Jiang, Y. and Liu, Y.J. (2006) Inhibitory regulation of EGF receptor degradation by sorting nexin 5. Biochem Biophys Res Commun, 342, 537-546.
Liu, Q., Oliveira-Dos-Santos, A.J., Mariathasan, S., Bouchard, D., Jones, J., Sarao, R., Kozieradzki, I., Ohashi, P.S., Penninger, J.M. and Dumont, D.J. (1998a) The inositol polyphosphate 5-phosphatase ship is a crucial negative regulator of B cell antigen receptor signaling. J Exp Med, 188, 1333-1342.
Liu, Q., Shalaby, F., Jones, J., Bouchard, D. and Dumont, D.J. (1998b) The SH2-containing inositol polyphosphate 5-phosphatase, ship, is expressed during hematopoiesis and spermatogenesis. Blood, 91, 2753-2759.
Llorca, O., McCormack, E.A., Hynes, G., Grantham, J., Cordell, J., Carrascosa, J.L., Willison, K.R., Fernandez, J.J. and Valpuesta, J.M. (1999) Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature, 402, 693-696.
Loffing, J., Flores, S.Y. and Staub, O. (2006) Sgk kinases and their role in epithelial transport. Annu Rev Physiol, 68, 461-490.
Loren, C.E., Englund, C., Grabbe, C., Hallberg, B., Hunter, T. and Palmer, R.H. (2003) A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep, 4, 781-786.
Loren, C.E., Scully, A., Grabbe, C., Edeen, P.T., Thomas, J., McKeown, M., Hunter, T. and Palmer, R.H. (2001) Identification and characterization of DAlk: a novel Drosophila melanogaster RTK which drives ERK activation in vivo. Genes Cells, 6, 531-544.
Lorenzo, O., Urbe, S. and Clague, M.J. (2005) Analysis of phosphoinositide binding domain properties within the myotubularin-related protein MTMR3. J Cell Sci, 118, 2005-2012.
Lorenzo, O., Urbe, S. and Clague, M.J. (2006) Systematic analysis of myotubularins: heteromeric interactions, subcellular localisation and endosome related functions. J Cell Sci, 119, 2953-2959.
Lowe, C.U., Terrey, M. and Mac, L.E. (1952) Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child, 83, 164-184.
Ma, Z., Hill, D.A., Collins, M.H., Morris, S.W., Sumegi, J., Zhou, M., Zuppan, C. and Bridge, J.A. (2003) Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer, 37, 98-105.
MacKeigan, J.P., Murphy, L.O. and Blenis, J. (2005) Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol, 7, 591-600.
Maehama, T. and Dixon, J.E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem, 273, 13375-13378.
Maes, B., Vanhentenrijk, V., Wlodarska, I., Cools, J., Peeters, B., Marynen, P. and de Wolf-Peeters, C. (2001) The NPM-ALK and the ATIC-ALK fusion genes can be detected in non-neoplastic cells. Am J Pathol, 158, 2185-2193.
Maffucci, T., Brancaccio, A., Piccolo, E., Stein, R.C. and Falasca, M. (2003) Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. Embo J, 22, 4178-4189.
162

Références bibliographiques
Maffucci, T., Cooke, F.T., Foster, F.M., Traer, C.J., Fry, M.J. and Falasca, M. (2005) Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol, 169, 789-799.
Maiguel, D.A., Jones, L., Chakravarty, D., Yang, C. and Carrier, F. (2004) Nucleophosmin sets a threshold for p53 response to UV radiation. Mol Cell Biol, 24, 3703-3711.
Majumder, P.K. and Sellers, W.R. (2005) Akt-regulated pathways in prostate cancer. Oncogene, 24, 7465-7474.
Manning, B.D. and Cantley, L.C. (2007) AKT/PKB signaling: navigating downstream. Cell, 129, 1261-1274.
Marcus, S.L., Wenk, M.R., Steele-Mortimer, O. and Finlay, B.B. (2001) A synaptojanin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett, 494, 201-207.
Martel, V., Racaud-Sultan, C., Dupe, S., Marie, C., Paulhe, F., Galmiche, A., Block, M.R. and Albiges-Rizo, C. (2001) Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem, 276, 21217-21227.
Martin, G.S. (2003) Cell signaling and cancer. Cancer Cell, 4, 167-174. Marzec, M., Kasprzycka, M., Liu, X., El-Salem, M., Halasa, K., Raghunath, P.N., Bucki, R.,
Wlodarski, P. and Wasik, M.A. (2007a) Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene, 26, 5606-5614.
Marzec, M., Kasprzycka, M., Liu, X., Raghunath, P.N., Wlodarski, P. and Wasik, M.A. (2007b) Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene, 26, 813-821.
Marzec, M., Kasprzycka, M., Ptasznik, A., Wlodarski, P., Zhang, Q., Odum, N. and Wasik, M.A. (2005) Inhibition of ALK enzymatic activity in T-cell lymphoma cells induces apoptosis and suppresses proliferation and STAT3 phosphorylation independently of Jak3. Lab Invest, 85, 1544-1554.
Mason, D., Mallo, G.V., Terebiznik, M.R., Payrastre, B., Finlay, B.B., Brumell, J.H., Rameh, L. and Grinstein, S. (2007) Alteration of epithelial structure and function associated with PtdIns(4,5)P2 degradation by a bacterial phosphatase. J Gen Physiol, 129, 267-283.
Mason, D.Y., Pulford, K.A., Bischof, D., Kuefer, M.U., Butler, L.H., Lamant, L., Delsol, G. and Morris, S.W. (1998) Nucleolar localization of the nucleophosmin-anaplastic lymphoma kinase is not required for malignant transformation. Cancer Res, 58, 1057-1062.
Mauco, G., Dangelmaier, C.A. and Smith, J.B. (1984) Inositol lipids, phosphatidate and diacylglycerol share stearoylarachidonoylglycerol as a common backbone in thrombin-stimulated human platelets. Biochem J, 224, 933-940.
McEwen, R.K., Dove, S.K., Cooke, F.T., Painter, G.F., Holmes, A.B., Shisheva, A., Ohya, Y., Parker, P.J. and Michell, R.H. (1999) Complementation analysis in PtdInsP kinase-deficient yeast mutants demonstrates that Schizosaccharomyces pombe and murine Fab1p homologues are phosphatidylinositol 3-phosphate 5-kinases. J Biol Chem, 274, 33905-33912.
Meijer, H.J., Berrie, C.P., Iurisci, C., Divecha, N., Musgrave, A. and Munnik, T. (2001) Identification of a new polyphosphoinositide in plants, phosphatidylinositol 5-monophosphate (PtdIns5P), and its accumulation upon osmotic stress. Biochem J, 360, 491-498.
Merlot, S., Meili, R., Pagliarini, D.J., Maehama, T., Dixon, J.E. and Firtel, R.A. (2003) A PTEN-related 5-phosphatidylinositol phosphatase localized in the Golgi. J Biol Chem, 278, 39866-39873.
163

Références bibliographiques
Meunier, F.A., Osborne, S.L., Hammond, G.R., Cooke, F.T., Parker, P.J., Domin, J. and Schiavo, G. (2005) Phosphatidylinositol 3-kinase C2alpha is essential for ATP-dependent priming of neurosecretory granule exocytosis. Mol Biol Cell, 16, 4841-4851.
Michell, R.H. (1975) Inositol phospholipids and cell surface receptor function. Biochim Biophys Acta, 415, 81-47.
Michell, R.H., Heath, V.L., Lemmon, M.A. and Dove, S.K. (2006) Phosphatidylinositol 3,5-bisphosphate: metabolism and cellular functions. Trends Biochem Sci, 31, 52-63.
Miething, C., Grundler, R., Fend, F., Hoepfl, J., Mugler, C., von Schilling, C., Morris, S.W., Peschel, C. and Duyster, J. (2003) The oncogenic fusion protein nucleophosmin-anaplastic lymphoma kinase (NPM-ALK) induces two distinct malignant phenotypes in a murine retroviral transplantation model. Oncogene, 22, 4642-4647.
Miething, C., Peschel, C. and Duyster, J. (2006) Targeting the oncogenic tyrosine kinase NPM-ALK in lymphoma: the role of murine models in defining pathogenesis and treatment options. Curr Drug Targets, 7, 1329-1334.
Miled, N., Yan, Y., Hon, W.C., Perisic, O., Zvelebil, M., Inbar, Y., Schneidman-Duhovny, D., Wolfson, H.J., Backer, J.M. and Williams, R.L. (2007) Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science, 317, 239-242.
Mills, I.G., Praefcke, G.J., Vallis, Y., Peter, B.J., Olesen, L.E., Gallop, J.L., Butler, P.J., Evans, P.R. and McMahon, H.T. (2003) EpsinR: an AP1/clathrin interacting protein involved in vesicle trafficking. J Cell Biol, 160, 213-222.
Misawa, H., Ohtsubo, M., Copeland, N.G., Gilbert, D.J., Jenkins, N.A. and Yoshimura, A. (1998) Cloning and characterization of a novel class II phosphoinositide 3-kinase containing C2 domain. Biochem Biophys Res Commun, 244, 531-539.
Misra, S. and Hurley, J.H. (1999) Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting motif, the FYVE domain of Vps27p. Cell, 97, 657-666.
Misra, S., Miller, G.J. and Hurley, J.H. (2001) Recognizing phosphatidylinositol 3-phosphate. Cell, 107, 559-562.
Missy, K., Van Poucke, V., Raynal, P., Viala, C., Mauco, G., Plantavid, M., Chap, H. and Payrastre, B. (1998) Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J Biol Chem, 273, 30279-30286.
Miyake, I., Hakomori, Y., Shinohara, A., Gamou, T., Saito, M., Iwamatsu, A. and Sakai, R. (2002) Activation of anaplastic lymphoma kinase is responsible for hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene, 21, 5823-5834.
Mochizuki, Y. and Majerus, P.W. (2003) Characterization of myotubularin-related protein 7 and its binding partner, myotubularin-related protein 9. Proc Natl Acad Sci U S A, 100, 9768-9773.
Moog-Lutz, C., Degoutin, J., Gouzi, J.Y., Frobert, Y., Brunet-de Carvalho, N., Bureau, J., Creminon, C. and Vigny, M. (2005) Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem, 280, 26039-26048.
Morgan, J.R., Prasad, K., Hao, W., Augustine, G.J. and Lafer, E.M. (2000) A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J Neurosci, 20, 8667-8676.
Morris, J.B., Hinchliffe, K.A., Ciruela, A., Letcher, A.J. and Irvine, R.F. (2000) Thrombin stimulation of platelets causes an increase in phosphatidylinositol 5-phosphate revealed by mass assay. FEBS Lett, 475, 57-60.
164

Références bibliographiques
Morris, S.W., Kirstein, M.N., Valentine, M.B., Dittmer, K.G., Shapiro, D.N., Saltman, D.L. and Look, A.T. (1994) Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science, 263, 1281-1284.
Morris, S.W., Naeve, C., Mathew, P., James, P.L., Kirstein, M.N., Cui, X. and Witte, D.P. (1997) ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene, 14, 2175-2188.
Morris, S.W., Xue, L., Ma, Z. and Kinney, M.C. (2001) Alk+ CD30+ lymphomas: a distinct molecular genetic subtype of non-Hodgkin's lymphoma. Br J Haematol, 113, 275-295.
Motegi, A., Fujimoto, J., Kotani, M., Sakuraba, H. and Yamamoto, T. (2004) ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci, 117, 3319-3329.
Mourali, J., Benard, A., Lourenco, F.C., Monnet, C., Greenland, C., Moog-Lutz, C., Racaud-Sultan, C., Gonzalez-Dunia, D., Vigny, M., Mehlen, P., Delsol, G. and Allouche, M. (2006) Anaplastic lymphoma kinase is a dependence receptor whose proapoptotic functions are activated by caspase cleavage. Mol Cell Biol, 26, 6209-6222.
Murray, J.L., Mavrakis, M., McDonald, N.J., Yilla, M., Sheng, J., Bellini, W.J., Zhao, L., Le Doux, J.M., Shaw, M.W., Luo, C.C., Lippincott-Schwartz, J., Sanchez, A., Rubin, D.H. and Hodge, T.W. (2005) Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J Virol, 79, 11742-11751.
Myers, M.P., Pass, I., Batty, I.H., Van der Kaay, J., Stolarov, J.P., Hemmings, B.A., Wigler, M.H., Downes, C.P. and Tonks, N.K. (1998) The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A, 95, 13513-13518.
Nicholson, D.H., Green, W.R., Cross, H.E., Kenyon, K.R. and Massof, D. (1977) A clinical and histopathological study of Francois-Neetens speckled corneal dystrophy. Am J Ophthalmol, 83, 554-560.
Nicot, A.S., Fares, H., Payrastre, B., Chisholm, A.D., Labouesse, M. and Laporte, J. (2006) The phosphoinositide kinase PIKfyve/Fab1p regulates terminal lysosome maturation in Caenorhabditis elegans. Mol Biol Cell, 17, 3062-3074.
Nieborowska-Skorska, M., Slupianek, A., Xue, L., Zhang, Q., Raghunath, P.N., Hoser, G., Wasik, M.A., Morris, S.W. and Skorski, T. (2001) Role of signal transducer and activator of transcription 5 in nucleophosmin/ anaplastic lymphoma kinase-mediated malignant transformation of lymphoid cells. Cancer Res, 61, 6517-6523.
Niebuhr, K., Giuriato, S., Pedron, T., Philpott, D.J., Gaits, F., Sable, J., Sheetz, M.P., Parsot, C., Sansonetti, P.J. and Payrastre, B. (2002) Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. Embo J, 21, 5069-5078.
Norris, F.A., Wilson, M.P., Wallis, T.S., Galyov, E.E. and Majerus, P.W. (1998) SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc Natl Acad Sci U S A, 95, 14057-14059.
Odorizzi, G., Babst, M. and Emr, S.D. (2000) Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem Sci, 25, 229-235.
Ohno, H., Nishikori, M., Maesako, Y. and Haga, H. (2005) Reappraisal of BCL3 as a molecular marker of anaplastic large cell lymphoma. Int J Hematol, 82, 397-405.
Ojala, P.J., Paavilainen, V. and Lappalainen, P. (2001) Identification of yeast cofilin residues specific for actin monomer and PIP2 binding. Biochemistry, 40, 15562-15569.
Okuda, M., Horn, H.F., Tarapore, P., Tokuyama, Y., Smulian, A.G., Chan, P.K., Knudsen, E.S., Hofmann, I.A., Snyder, J.D., Bove, K.E. and Fukasawa, K. (2000)
165

Références bibliographiques
Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell, 103, 127-140.
Okuwaki, M., Tsujimoto, M. and Nagata, K. (2002) The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol Biol Cell, 13, 2016-2030.
Onciu, M., Behm, F.G., Downing, J.R., Shurtleff, S.A., Raimondi, S.C., Ma, Z., Morris, S.W., Kennedy, W., Jones, S.C. and Sandlund, J.T. (2003) ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood, 102, 2642-2644.
Onishi, M., Nakamura, Y., Koga, T., Hirata, A. and Fukui, Y. (2003) Inportance of phosphatidylinositol 3-phosphate in sporulation of Schizosaccharomyces pombe. Biosci Biotechnol Biochem, 67, 1191-1193.
Osborne, S.L., Wen, P.J., Boucheron, C., Nguyen, H.N., Hayakawa, M., Kaizawa, H., Parker, P.J., Vitale, N. and Meunier, F.A. (2008) PIKfyve negatively regulates exocytosis in neurosecretory cells. J Biol Chem, 283, 2804-2813.
Ostman, A., Hellberg, C. and Bohmer, F.D. (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer, 6, 307-320.
Ouyang, T., Bai, R.Y., Bassermann, F., von Klitzing, C., Klumpen, S., Miething, C., Morris, S.W., Peschel, C. and Duyster, J. (2003) Identification and characterization of a nuclear interacting partner of anaplastic lymphoma kinase (NIPA). J Biol Chem, 278, 30028-30036.
Pagliarini, D.J., Wiley, S.E., Kimple, M.E., Dixon, J.R., Kelly, P., Worby, C.A., Casey, P.J. and Dixon, J.E. (2005) Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol Cell, 19, 197-207.
Pagliarini, D.J., Worby, C.A. and Dixon, J.E. (2004) A PTEN-like phosphatase with a novel substrate specificity. J Biol Chem, 279, 38590-38596.
Panagopoulos, I., Nilsson, T., Domanski, H.A., Isaksson, M., Lindblom, P., Mertens, F. and Mandahl, N. (2006) Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int J Cancer, 118, 1181-1186.
Panaretou, C., Domin, J., Cockcroft, S. and Waterfield, M.D. (1997) Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 3-kinase. Substrate presentation by phosphatidylinositol transfer protein to the p150.Ptdins 3-kinase complex. J Biol Chem, 272, 2477-2485.
Pascual, J., Martinez-Yamout, M., Dyson, H.J. and Wright, P.E. (2000) Structure of the PHD zinc finger from human Williams-Beuren syndrome transcription factor. J Mol Biol, 304, 723-729.
Passoni, L., Gallo, B., Biganzoli, E., Stefanoni, R., Massimino, M., Di Nicola, M., Gianni, A.M. and Gambacorti-Passerini, C. (2006) In vivo T-cell immune response against anaplastic lymphoma kinase in patients with anaplastic large cell lymphomas. Haematologica, 91, 48-55.
Patterson, R.L., van Rossum, D.B., Nikolaidis, N., Gill, D.L. and Snyder, S.H. (2005) Phospholipase C-gamma: diverse roles in receptor-mediated calcium signaling. Trends Biochem Sci, 30, 688-697.
Paulhe, F., Perret, B., Chap, H., Iberg, N., Morand, O. and Racaud-Sultan, C. (2002) Phosphoinositide 3-kinase C2alpha is activated upon smooth muscle cell migration and regulated by alpha(v)beta(3) integrin engagement. Biochem Biophys Res Commun, 297, 261-266.
166

Références bibliographiques
Payrastre, B., Missy, K., Giuriato, S., Bodin, S., Plantavid, M. and Gratacap, M. (2001) Phosphoinositides: key players in cell signalling, in time and space. Cell Signal, 13, 377-387.
Pearson, M.A., Reczek, D., Bretscher, A. and Karplus, P.A. (2000) Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell, 101, 259-270.
Pece, S., Chiariello, M., Murga, C. and Gutkind, J.S. (1999) Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J Biol Chem, 274, 19347-19351.
Pendaries, C., Tronchere, H., Arbibe, L., Mounier, J., Gozani, O., Cantley, L., Fry, M.J., Gaits-Iacovoni, F., Sansonetti, P.J. and Payrastre, B. (2006) PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. Embo J, 25, 1024-1034.
Pendaries, C., Tronchere, H., Plantavid, M. and Payrastre, B. (2003) Phosphoinositide signaling disorders in human diseases. FEBS Lett, 546, 25-31.
Pesesse, X., Deleu, S., De Smedt, F., Drayer, L. and Erneux, C. (1997) Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem Biophys Res Commun, 239, 697-700.
Petiot, A., Ogier-Denis, E., Blommaart, E.F., Meijer, A.J. and Codogno, P. (2000) Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem, 275, 992-998.
Petroulakis, E., Mamane, Y., Le Bacquer, O., Shahbazian, D. and Sonenberg, N. (2006) mTOR signaling: implications for cancer and anticancer therapy. Br J Cancer, 94, 195-199.
Piccinini, G., Bacchiocchi, R., Serresi, M., Vivani, C., Rossetti, S., Gennaretti, C., Carbonari, D. and Fazioli, F. (2002) A ligand-inducible epidermal growth factor receptor/anaplastic lymphoma kinase chimera promotes mitogenesis and transforming properties in 3T3 cells. J Biol Chem, 277, 22231-22239.
Pirola, L., Zvelebil, M.J., Bulgarelli-Leva, G., Van Obberghen, E., Waterfield, M.D. and Wymann, M.P. (2001) Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase alpha (PI3Kalpha ). Functions of lipid kinase-deficient PI3Kalpha in signaling. J Biol Chem, 276, 21544-21554.
Piva, R., Chiarle, R., Manazza, A.D., Taulli, R., Simmons, W., Ambrogio, C., D'Escamard, V., Pellegrino, E., Ponzetto, C., Palestro, G. and Inghirami, G. (2006a) Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood, 107, 689-697.
Piva, R., Pellegrino, E., Mattioli, M., Agnelli, L., Lombardi, L., Boccalatte, F., Costa, G., Ruggeri, B.A., Cheng, M., Chiarle, R., Palestro, G., Neri, A. and Inghirami, G. (2006b) Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest, 116, 3171-3182.
Pizarro-Cerda, J. and Cossart, P. (2004) Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat Cell Biol, 6, 1026-1033.
Platanias, L.C. (2003) Map kinase signaling pathways and hematologic malignancies. Blood, 101, 4667-4679.
Polo, S., Pece, S. and Di Fiore, P.P. (2004) Endocytosis and cancer. Curr Opin Cell Biol, 16, 156-161.
Poole, A.W. and Jones, M.L. (2005) A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal, 17, 1323-1332.
167

Références bibliographiques
Powers, C., Aigner, A., Stoica, G.E., McDonnell, K. and Wellstein, A. (2002) Pleiotrophin signaling through anaplastic lymphoma kinase is rate-limiting for glioblastoma growth. J Biol Chem, 277, 14153-14158.
Pulford, K., Falini, B., Banham, A.H., Codrington, D., Roberton, H., Hatton, C. and Mason, D.Y. (2000) Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood, 96, 1605-1607.
Pulford, K., Falini, B., Cordell, J., Rosenwald, A., Ott, G., Muller-Hermelink, H.K., MacLennan, K.A., Lamant, L., Carbone, A., Campo, E. and Mason, D.Y. (1999) Biochemical detection of novel anaplastic lymphoma kinase proteins in tissue sections of anaplastic large cell lymphoma. Am J Pathol, 154, 1657-1663.
Pulford, K., Lamant, L., Espinos, E., Jiang, Q., Xue, L., Turturro, F., Delsol, G. and Morris, S.W. (2004a) The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci, 61, 2939-2953.
Pulford, K., Morris, S.W. and Mason, D.Y. (2001) Anaplastic lymphoma kinase proteins and malignancy. Curr Opin Hematol, 8, 231-236.
Pulford, K., Morris, S.W. and Turturro, F. (2004b) Anaplastic lymphoma kinase proteins in growth control and cancer. J Cell Physiol, 199, 330-358.
Quintanilla-Martinez, L., Pittaluga, S., Miething, C., Klier, M., Rudelius, M., Davies-Hill, T., Anastasov, N., Martinez, A., Vivero, A., Duyster, J., Jaffe, E.S., Fend, F. and Raffeld, M. (2006) NPM-ALK-dependent expression of the transcription factor CCAAT/enhancer binding protein beta in ALK-positive anaplastic large cell lymphoma. Blood, 108, 2029-2036.
Rameh, L.E., Arvidsson, A., Carraway, K.L., 3rd, Couvillon, A.D., Rathbun, G., Crompton, A., VanRenterghem, B., Czech, M.P., Ravichandran, K.S., Burakoff, S.J., Wang, D.S., Chen, C.S. and Cantley, L.C. (1997a) A comparative analysis of the phosphoinositide binding specificity of pleckstrin homology domains. J Biol Chem, 272, 22059-22066.
Rameh, L.E., Tolias, K.F., Duckworth, B.C. and Cantley, L.C. (1997b) A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature, 390, 192-196.
Rassidakis, G.Z., Feretzaki, M., Atwell, C., Grammatikakis, I., Lin, Q., Lai, R., Claret, F.X., Medeiros, L.J. and Amin, H.M. (2005) Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood, 105, 827-829.
Rawlings, J.S., Rosler, K.M. and Harrison, D.A. (2004) The JAK/STAT signaling pathway. J Cell Sci, 117, 1281-1283.
Raymond, C.K., Howald-Stevenson, I., Vater, C.A. and Stevens, T.H. (1992) Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol Biol Cell, 3, 1389-1402.
Razzini, G., Brancaccio, A., Lemmon, M.A., Guarnieri, S. and Falasca, M. (2000) The role of the pleckstrin homology domain in membrane targeting and activation of phospholipase Cbeta(1). J Biol Chem, 275, 14873-14881.
Redner, R.L., Rush, E.A., Faas, S., Rudert, W.A. and Corey, S.J. (1996) The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood, 87, 882-886.
Richardson, J.P., Wang, M., Clarke, J.H., Patel, K.J. and Irvine, R.F. (2007) Genomic tagging of endogenous type IIbeta phosphatidylinositol 5-phosphate 4-kinase in DT40 cells reveals a nuclear localisation. Cell Signal, 19, 1309-1314.
Ridley, S.H., Ktistakis, N., Davidson, K., Anderson, K.E., Manifava, M., Ellson, C.D., Lipp, P., Bootman, M., Coadwell, J., Nazarian, A., Erdjument-Bromage, H., Tempst, P., Cooper, M.A., Thuring, J.W., Lim, Z.Y., Holmes, A.B., Stephens, L.R. and Hawkins, P.T. (2001) FENS-1 and DFCP1 are FYVE domain-containing proteins with distinct functions in the endosomal and Golgi compartments. J Cell Sci, 114, 3991-4000.
168

Références bibliographiques
Robinson, F.L. and Dixon, J.E. (2005) The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-Tooth disease. J Biol Chem, 280, 31699-31707.
Robinson, F.L. and Dixon, J.E. (2006) Myotubularin phosphatases: policing 3-phosphoinositides. Trends Cell Biol, 16, 403-412.
Roggo, L., Bernard, V., Kovacs, A.L., Rose, A.M., Savoy, F., Zetka, M., Wymann, M.P. and Muller, F. (2002) Membrane transport in Caenorhabditis elegans: an essential role for VPS34 at the nuclear membrane. Embo J, 21, 1673-1683.
Rohn, W.M., Rouille, Y., Waguri, S. and Hoflack, B. (2000) Bi-directional trafficking between the trans-Golgi network and the endosomal/lysosomal system. J Cell Sci, 113 ( Pt 12), 2093-2101.
Rondinone, C.M., Carvalho, E., Rahn, T., Manganiello, V.C., Degerman, E. and Smith, U.P. (2000) Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J Biol Chem, 275, 10093-10098.
Rozengurt, E., Rey, O. and Waldron, R.T. (2005) Protein kinase D signaling. J Biol Chem, 280, 13205-13208.
Ruchatz, H., Coluccia, A.M., Stano, P., Marchesi, E. and Gambacorti-Passerini, C. (2003) Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp Hematol, 31, 309-315.
Rudge, S.A., Anderson, D.M. and Emr, S.D. (2004) Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell, 15, 24-36.
Rush, J., Moritz, A., Lee, K.A., Guo, A., Goss, V.L., Spek, E.J., Zhang, H., Zha, X.M., Polakiewicz, R.D. and Comb, M.J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol, 23, 94-101.
Rusten, T.E., Rodahl, L.M., Pattni, K., Englund, C., Samakovlis, C., Dove, S., Brech, A. and Stenmark, H. (2006) Fab1 phosphatidylinositol 3-phosphate 5-kinase controls trafficking but not silencing of endocytosed receptors. Mol Biol Cell, 17, 3989-4001.
Rutherford, A.C., Traer, C., Wassmer, T., Pattni, K., Bujny, M.V., Carlton, J.G., Stenmark, H. and Cullen, P.J. (2006) The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci, 119, 3944-3957.
Rykx, A., De Kimpe, L., Mikhalap, S., Vantus, T., Seufferlein, T., Vandenheede, J.R. and Van Lint, J. (2003) Protein kinase D: a family affair. FEBS Lett, 546, 81-86.
Sabatini, D.M. (2006) mTOR and cancer: insights into a complex relationship. Nat Rev Cancer, 6, 729-734.
Sato, T.K., Overduin, M. and Emr, S.D. (2001) Location, location, location: membrane targeting directed by PX domains. Science, 294, 1881-1885.
Sbrissa, D., Ikonomov, O. and Shisheva, A. (2001) Selective insulin-induced activation of class I(A) phosphoinositide 3-kinase in PIKfyve immune complexes from 3T3-L1 adipocytes. Mol Cell Endocrinol, 181, 35-46.
Sbrissa, D., Ikonomov, O.C., Deeb, R. and Shisheva, A. (2002a) Phosphatidylinositol 5-phosphate biosynthesis is linked to PIKfyve and is involved in osmotic response pathway in mammalian cells. J Biol Chem, 277, 47276-47284.
Sbrissa, D., Ikonomov, O.C., Fu, Z., Ijuin, T., Gruenberg, J., Takenawa, T. and Shisheva, A. (2007) Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve-PIKfyve complex. J Biol Chem, 282, 23878-23891.
169

Références bibliographiques
Sbrissa, D., Ikonomov, O.C. and Shisheva, A. (1999) PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. Effect of insulin. J Biol Chem, 274, 21589-21597.
Sbrissa, D., Ikonomov, O.C. and Shisheva, A. (2000) PIKfyve lipid kinase is a protein kinase: downregulation of 5'-phosphoinositide product formation by autophosphorylation. Biochemistry, 39, 15980-15989.
Sbrissa, D., Ikonomov, O.C. and Shisheva, A. (2002b) Phosphatidylinositol 3-phosphate-interacting domains in PIKfyve. Binding specificity and role in PIKfyve. Endomenbrane localization. J Biol Chem, 277, 6073-6079.
Sbrissa, D., Ikonomov, O.C., Strakova, J., Dondapati, R., Mlak, K., Deeb, R., Silver, R. and Shisheva, A. (2004a) A mammalian ortholog of Saccharomyces cerevisiae Vac14 that associates with and up-regulates PIKfyve phosphoinositide 5-kinase activity. Mol Cell Biol, 24, 10437-10447.
Sbrissa, D., Ikonomov, O.C., Strakova, J. and Shisheva, A. (2004b) Role for a novel signaling intermediate, phosphatidylinositol 5-phosphate, in insulin-regulated F-actin stress fiber breakdown and GLUT4 translocation. Endocrinology, 145, 4853-4865.
Sbrissa, D. and Shisheva, A. (2005) Acquisition of unprecedented phosphatidylinositol 3,5-bisphosphate rise in hyperosmotically stressed 3T3-L1 adipocytes, mediated by ArPIKfyve-PIKfyve pathway. J Biol Chem, 280, 7883-7889.
Schaletzky, J., Dove, S.K., Short, B., Lorenzo, O., Clague, M.J. and Barr, F.A. (2003) Phosphatidylinositol-5-phosphate activation and conserved substrate specificity of the myotubularin phosphatidylinositol 3-phosphatases. Curr Biol, 13, 504-509.
Schu, P.V., Takegawa, K., Fry, M.J., Stack, J.H., Waterfield, M.D. and Emr, S.D. (1993) Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science, 260, 88-91.
Seaman, M.N. and Williams, H.P. (2002) Identification of the functional domains of yeast sorting nexins Vps5p and Vps17p. Mol Biol Cell, 13, 2826-2840.
Seebohm, G., Strutz-Seebohm, N., Birkin, R., Dell, G., Bucci, C., Spinosa, M.R., Baltaev, R., Mack, A.F., Korniychuk, G., Choudhury, A., Marks, D., Pagano, R.E., Attali, B., Pfeufer, A., Kass, R.S., Sanguinetti, M.C., Tavare, J.M. and Lang, F. (2007) Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ Res, 100, 686-692.
Semenza, G.L. (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer, 3, 721-732. Senderek, J., Bergmann, C., Weber, S., Ketelsen, U.P., Schorle, H., Rudnik-Schoneborn, S.,
Buttner, R., Buchheim, E. and Zerres, K. (2003) Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet, 12, 349-356.
Shisheva, A. (2001) PIKfyve: the road to PtdIns 5-P and PtdIns 3,5-P(2). Cell Biol Int, 25, 1201-1206.
Shisheva, A. (2003) Regulating Glut4 vesicle dynamics by phosphoinositide kinases and phosphoinositide phosphatases. Front Biosci, 8, s945-946.
Shisheva, A. (2008) PIKfyve: Partners, significance, debates and paradoxes. Cell Biol Int. Shisheva, A., Sbrissa, D. and Ikonomov, O. (1999) Cloning, characterization, and expression
of a novel Zn2+-binding FYVE finger-containing phosphoinositide kinase in insulin-sensitive cells. Mol Cell Biol, 19, 623-634.
Shojaiefard, M., Strutz-Seebohm, N., Tavare, J.M., Seebohm, G. and Lang, F. (2007) Regulation of the Na(+), glucose cotransporter by PIKfyve and the serum and glucocorticoid inducible kinase SGK1. Biochem Biophys Res Commun, 359, 843-847.
170

Références bibliographiques
Simoes, A.P., Reed, J., Schnabel, P., Camps, M. and Gierschik, P. (1995) Characterization of putative polyphosphoinositide binding motifs from phospholipase C beta 2. Biochemistry, 34, 5113-5119.
Simonsen, A. and Stenmark, H. (2001) PX domains: attracted by phosphoinositides. Nat Cell Biol, 3, E179-182.
Singh, S.M. and Murray, D. (2003) Molecular modeling of the membrane targeting of phospholipase C pleckstrin homology domains. Protein Sci, 12, 1934-1953.
Slagsvold, T., Pattni, K., Malerod, L. and Stenmark, H. (2006) Endosomal and non-endosomal functions of ESCRT proteins. Trends Cell Biol, 16, 317-326.
Sleeman, M.W., Wortley, K.E., Lai, K.M., Gowen, L.C., Kintner, J., Kline, W.O., Garcia, K., Stitt, T.N., Yancopoulos, G.D., Wiegand, S.J. and Glass, D.J. (2005) Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat Med, 11, 199-205.
Slupianek, A., Nieborowska-Skorska, M., Hoser, G., Morrione, A., Majewski, M., Xue, L., Morris, S.W., Wasik, M.A. and Skorski, T. (2001) Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res, 61, 2194-2199.
Slupianek, A. and Skorski, T. (2004) NPM/ALK downregulates p27Kip1 in a PI-3K-dependent manner. Exp Hematol, 32, 1265-1271.
Smith, W.J. and Cerione, R.A. (2002) Crystallization and preliminary crystallographic analysis of the ezrin FERM domain. Acta Crystallogr D Biol Crystallogr, 58, 1359-1361.
Soda, M., Choi, Y.L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., Fujiwara, S., Watanabe, H., Kurashina, K., Hatanaka, H., Bando, M., Ohno, S., Ishikawa, Y., Aburatani, H., Niki, T., Sohara, Y., Sugiyama, Y. and Mano, H. (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 448, 561-566.
Song, X., Xu, W., Zhang, A., Huang, G., Liang, X., Virbasius, J.V., Czech, M.P. and Zhou, G.W. (2001) Phox homology domains specifically bind phosphatidylinositol phosphates. Biochemistry, 40, 8940-8944.
Souttou, B., Carvalho, N.B., Raulais, D. and Vigny, M. (2001) Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J Biol Chem, 276, 9526-9531.
Spiess, C., Meyer, A.S., Reissmann, S. and Frydman, J. (2004) Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol, 14, 598-604.
Stack, J.H. and Emr, S.D. (1994) Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J Biol Chem, 269, 31552-31562.
Stahelin, R.V., Long, F., Peter, B.J., Murray, D., De Camilli, P., McMahon, H.T. and Cho, W. (2003) Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J Biol Chem, 278, 28993-28999.
Steele-Mortimer, O., Knodler, L.A., Marcus, S.L., Scheid, M.P., Goh, B., Pfeifer, C.G., Duronio, V. and Finlay, B.B. (2000) Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector sigD. J Biol Chem, 275, 37718-37724.
Stein, H., Foss, H.D., Durkop, H., Marafioti, T., Delsol, G., Pulford, K., Pileri, S. and Falini, B. (2000) CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood, 96, 3681-3695.
Stenmark, H., Aasland, R. and Driscoll, P.C. (2002) The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett, 513, 77-84.
171

Références bibliographiques
Stoica, G.E., Kuo, A., Aigner, A., Sunitha, I., Souttou, B., Malerczyk, C., Caughey, D.J., Wen, D., Karavanov, A., Riegel, A.T. and Wellstein, A. (2001) Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem, 276, 16772-16779.
Stoica, G.E., Kuo, A., Powers, C., Bowden, E.T., Sale, E.B., Riegel, A.T. and Wellstein, A. (2002) Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem, 277, 35990-35998.
Suire, S., Condliffe, A.M., Ferguson, G.J., Ellson, C.D., Guillou, H., Davidson, K., Welch, H., Coadwell, J., Turner, M., Chilvers, E.R., Hawkins, P.T. and Stephens, L. (2006) Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol, 8, 1303-1309.
Swaminathan, V., Kishore, A.H., Febitha, K.K. and Kundu, T.K. (2005) Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol Cell Biol, 25, 7534-7545.
Szebeni, A. and Olson, M.O. (1999) Nucleolar protein B23 has molecular chaperone activities. Protein Sci, 8, 905-912.
Tall, E.G., Spector, I., Pentyala, S.N., Bitter, I. and Rebecchi, M.J. (2000) Dynamics of phosphatidylinositol 4,5-bisphosphate in actin-rich structures. Curr Biol, 10, 743-746.
Tanti, J.F., Gremeaux, T., Van Obberghen, E. and Le Marchand-Brustel, Y. (1994) Insulin receptor substrate 1 is phosphorylated by the serine kinase activity of phosphatidylinositol 3-kinase. Biochem J, 304 ( Pt 1), 17-21.
Tarapore, P., Okuda, M. and Fukasawa, K. (2002) A mammalian in vitro centriole duplication system: evidence for involvement of CDK2/cyclin E and nucleophosmin/B23 in centrosome duplication. Cell Cycle, 1, 75-81.
Tauchi, T. and Ohyashiki, K. (2006) The second generation of BCR-ABL tyrosine kinase inhibitors. Int J Hematol, 83, 294-300.
Taylor, G.S. and Dixon, J.E. (2003) PTEN and myotubularins: families of phosphoinositide phosphatases. Methods Enzymol, 366, 43-56.
ten Berge, R.L., de Bruin, P.C., Oudejans, J.J., Ossenkoppele, G.J., van der Valk, P. and Meijer, C.J. (2003) ALK-negative anaplastic large-cell lymphoma demonstrates similar poor prognosis to peripheral T-cell lymphoma, unspecified. Histopathology, 43, 462-469.
Terebiznik, M.R., Vieira, O.V., Marcus, S.L., Slade, A., Yip, C.M., Trimble, W.S., Meyer, T., Finlay, B.B. and Grinstein, S. (2002) Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol, 4, 766-773.
Testa, J.R. and Bellacosa, A. (2001) AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A, 98, 10983-10985.
Thompson, M.A., Stumph, J., Henrickson, S.E., Rosenwald, A., Wang, Q., Olson, S., Brandt, S.J., Roberts, J., Zhang, X., Shyr, Y. and Kinney, M.C. (2005) Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol, 36, 494-504.
Tilly, H., Gaulard, P., Lepage, E., Dumontet, C., Diebold, J., Plantier, I., Berger, F., Symann, M., Petrella, T., Lederlin, P. and Briere, J. (1997) Primary anaplastic large-cell lymphoma in adults: clinical presentation, immunophenotype, and outcome. Blood, 90, 3727-3734.
Toker, A. (1998) The synthesis and cellular roles of phosphatidylinositol 4,5-bisphosphate. Curr Opin Cell Biol, 10, 254-261.
Toker, A. (2002) Phosphoinositides and signal transduction. Cell Mol Life Sci, 59, 761-779.
172

Références bibliographiques
Toker, A. and Cantley, L.C. (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature, 387, 673-676.
Toker, A., Meyer, M., Reddy, K.K., Falck, J.R., Aneja, R., Aneja, S., Parra, A., Burns, D.J., Ballas, L.M. and Cantley, L.C. (1994) Activation of protein kinase C family members by the novel polyphosphoinositides PtdIns-3,4-P2 and PtdIns-3,4,5-P3. J Biol Chem, 269, 32358-32367.
Tolias, K.F., Rameh, L.E., Ishihara, H., Shibasaki, Y., Chen, J., Prestwich, G.D., Cantley, L.C. and Carpenter, C.L. (1998) Type I phosphatidylinositol-4-phosphate 5-kinases synthesize the novel lipids phosphatidylinositol 3,5-bisphosphate and phosphatidylinositol 5-phosphate. J Biol Chem, 273, 18040-18046.
Tort, F., Pinyol, M., Pulford, K., Roncador, G., Hernandez, L., Nayach, I., Kluin-Nelemans, H.C., Kluin, P., Touriol, C., Delsol, G., Mason, D. and Campo, E. (2001) Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab Invest, 81, 419-426.
Touhara, K., Inglese, J., Pitcher, J.A., Shaw, G. and Lefkowitz, R.J. (1994) Binding of G protein beta gamma-subunits to pleckstrin homology domains. J Biol Chem, 269, 10217-10220.
Touriol, C., Greenland, C., Lamant, L., Pulford, K., Bernard, F., Rousset, T., Mason, D.Y. and Delsol, G. (2000) Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood, 95, 3204-3207.
Trinei, M., Lanfrancone, L., Campo, E., Pulford, K., Mason, D.Y., Pelicci, P.G. and Falini, B. (2000) A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res, 60, 793-798.
Tronchere, H., Buj-Bello, A., Mandel, J.L. and Payrastre, B. (2003) Implication of phosphoinositide phosphatases in genetic diseases: the case of myotubularin. Cell Mol Life Sci, 60, 2084-2099.
Tronchere, H., Laporte, J., Pendaries, C., Chaussade, C., Liaubet, L., Pirola, L., Mandel, J.L. and Payrastre, B. (2004) Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin in mammalian cells. J Biol Chem, 279, 7304-7312.
Trumper, L., Pfreundschuh, M., Bonin, F.V. and Daus, H. (1998) Detection of the t(2;5)-associated NPM/ALK fusion cDNA in peripheral blood cells of healthy individuals. Br J Haematol, 103, 1138-1144.
Tsujita, K., Itoh, T., Ijuin, T., Yamamoto, A., Shisheva, A., Laporte, J. and Takenawa, T. (2004) Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem, 279, 13817-13824.
Tsukazaki, T., Chiang, T.A., Davison, A.F., Attisano, L. and Wrana, J.L. (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell, 95, 779-791.
Turner, S.D. and Alexander, D.R. (2005) What have we learnt from mouse models of NPM-ALK-induced lymphomagenesis? Leukemia, 19, 1128-1134.
Turner, S.D., Tooze, R., Maclennan, K. and Alexander, D.R. (2003) Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes B-cell lymphomas with hyperactive Jun kinase. Oncogene, 22, 7750-7761.
Turner, S.D., Yeung, D., Hadfield, K., Cook, S.J. and Alexander, D.R. (2007) The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell Signal, 19, 740-747.
Turner, S.J., Domin, J., Waterfield, M.D., Ward, S.G. and Westwick, J. (1998) The CC chemokine monocyte chemotactic peptide-1 activates both the class I p85/p110
173

Références bibliographiques
phosphatidylinositol 3-kinase and the class II PI3K-C2alpha. J Biol Chem, 273, 25987-25995.
Turturro, F., Arnold, M.D., Frist, A.Y. and Pulford, K. (2002a) Model of inhibition of the NPM-ALK kinase activity by herbimycin A. Clin Cancer Res, 8, 240-245.
Turturro, F., Arnold, M.D., Frist, A.Y. and Seth, P. (2002b) Effects of adenovirus-mediated expression of p27Kip1, p21Waf1 and p16INK4A in cell lines derived from t(2;5) anaplastic large cell lymphoma and Hodgkin's disease. Leuk Lymphoma, 43, 1323-1328.
Turturro, F., Heineke, H.L., Drevyanko, T.F., Link, C.J., Jr. and Seth, P. (2000) Adenovirus-p53-mediated gene therapy of anaplastic large cell lymphoma with t(2;5) in a nude mouse model. Gene Ther, 7, 930-933.
Ungewickell, A., Hugge, C., Kisseleva, M., Chang, S.C., Zou, J., Feng, Y., Galyov, E.E., Wilson, M. and Majerus, P.W. (2005) The identification and characterization of two phosphatidylinositol-4,5-bisphosphate 4-phosphatases. Proc Natl Acad Sci U S A, 102, 18854-18859.
Ungewickell, A., Ward, M.E., Ungewickell, E. and Majerus, P.W. (2004) The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci U S A, 101, 13501-13506.
Vanhaesebroeck, B., Ali, K., Bilancio, A., Geering, B. and Foukas, L.C. (2005) Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci, 30, 194-204.
Vanhaesebroeck, B., Higashi, K., Raven, C., Welham, M., Anderson, S., Brennan, P., Ward, S.G. and Waterfield, M.D. (1999) Autophosphorylation of p110delta phosphoinositide 3-kinase: a new paradigm for the regulation of lipid kinases in vitro and in vivo. Embo J, 18, 1292-1302.
Vanhaesebroeck, B., Leevers, S.J., Ahmadi, K., Timms, J., Katso, R., Driscoll, P.C., Woscholski, R., Parker, P.J. and Waterfield, M.D. (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem, 70, 535-602.
Vanhaesebroeck, B., Welham, M.J., Kotani, K., Stein, R., Warne, P.H., Zvelebil, M.J., Higashi, K., Volinia, S., Downward, J. and Waterfield, M.D. (1997) P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A, 94, 4330-4335.
Vega, F., Medeiros, L.J., Leventaki, V., Atwell, C., Cho-Vega, J.H., Tian, L., Claret, F.X. and Rassidakis, G.Z. (2006) Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res, 66, 6589-6597.
Villalva, C., Trempat, P., Greenland, C., Thomas, C., Girard, J.P., Moebius, F., Delsol, G. and Brousset, P. (2002) Isolation of differentially expressed genes in NPM-ALK-positive anaplastic large cell lymphoma. Br J Haematol, 118, 791-798.
Villamor, N., Rozman, M., Esteve, J., Aymerich, M., Colomer, D., Aguilar, J.L., Campo, E. and Montserrat, E. (1999) Anaplastic large-cell lymphoma with rapid evolution to leukemic phase. Ann Hematol, 78, 478-482.
Vivanco, I. and Sawyers, C.L. (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer, 2, 489-501.
Voena, C., Conte, C., Ambrogio, C., Boeri Erba, E., Boccalatte, F., Mohammed, S., Jensen, O.N., Palestro, G., Inghirami, G. and Chiarle, R. (2007) The tyrosine phosphatase Shp2 interacts with NPM-ALK and regulates anaplastic lymphoma cell growth and migration. Cancer Res, 67, 4278-4286.
Volinia, S., Dhand, R., Vanhaesebroeck, B., MacDougall, L.K., Stein, R., Zvelebil, M.J., Domin, J., Panaretou, C. and Waterfield, M.D. (1995) A human phosphatidylinositol
174

Références bibliographiques
3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. Embo J, 14, 3339-3348.
Walch-Solimena, C. and Novick, P. (1999) The yeast phosphatidylinositol-4-OH kinase pik1 regulates secretion at the Golgi. Nat Cell Biol, 1, 523-525.
Walker, D.M., Urbe, S., Dove, S.K., Tenza, D., Raposo, G. and Clague, M.J. (2001) Characterization of MTMR3. an inositol lipid 3-phosphatase with novel substrate specificity. Curr Biol, 11, 1600-1605.
Wan, W., Albom, M.S., Lu, L., Quail, M.R., Becknell, N.C., Weinberg, L.R., Reddy, D.R., Holskin, B.P., Angeles, T.S., Underiner, T.L., Meyer, S.L., Hudkins, R.L., Dorsey, B.D., Ator, M.A., Ruggeri, B.A. and Cheng, M. (2006) Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood, 107, 1617-1623.
Wang, D., Baumann, A., Szebeni, A. and Olson, M.O. (1994) The nucleic acid binding activity of nucleolar protein B23.1 resides in its carboxyl-terminal end. J Biol Chem, 269, 30994-30998.
Ward, A.C., Touw, I. and Yoshimura, A. (2000) The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood, 95, 19-29.
Ware, M.D., Rosten, P., Damen, J.E., Liu, L., Humphries, R.K. and Krystal, G. (1996) Cloning and characterization of human SHIP, the 145-kD inositol 5-phosphatase that associates with SHC after cytokine stimulation. Blood, 88, 2833-2840.
Watanabe, M., Sasaki, M., Itoh, K., Higashihara, M., Umezawa, K., Kadin, M.E., Abraham, L.J., Watanabe, T. and Horie, R. (2005) JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and reed-sternberg cells of Hodgkin lymphoma. Cancer Res, 65, 7628-7634.
Watson, R.T. and Pessin, J.E. (2006) Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci, 31, 215-222.
Weissman, A.M. (2001) Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol, 2, 169-178.
Wellmann, A., Doseeva, V., Butscher, W., Raffeld, M., Fukushima, P., Stetler-Stevenson, M. and Gardner, K. (1997) The activated anaplastic lymphoma kinase increases cellular proliferation and oncogene up-regulation in rat 1a fibroblasts. Faseb J, 11, 965-972.
Welsh, G.I., Hers, I., Berwick, D.C., Dell, G., Wherlock, M., Birkin, R., Leney, S. and Tavare, J.M. (2005) Role of protein kinase B in insulin-regulated glucose uptake. Biochem Soc Trans, 33, 346-349.
Whiteford, C.C., Brearley, C.A. and Ulug, E.T. (1997) Phosphatidylinositol 3,5-bisphosphate defines a novel PI 3-kinase pathway in resting mouse fibroblasts. Biochem J, 323 ( Pt 3), 597-601.
Whitley, P., Reaves, B.J., Hashimoto, M., Riley, A.M., Potter, B.V. and Holman, G.D. (2003) Identification of mammalian Vps24p as an effector of phosphatidylinositol 3,5-bisphosphate-dependent endosome compartmentalization. J Biol Chem, 278, 38786-38795.
Wientjes, F.B., Reeves, E.P., Soskic, V., Furthmayr, H. and Segal, A.W. (2001) The NADPH oxidase components p47(phox) and p40(phox) bind to moesin through their PX domain. Biochem Biophys Res Commun, 289, 382-388.
Wilcox, A. and Hinchliffe, K.A. (2008) Regulation of extranuclear PtdIns5P production by phosphatidylinositol phosphate 4-kinase 2alpha. FEBS Lett, 582, 1391-1394.
Wishart, M.J. and Dixon, J.E. (2002) PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol, 12, 579-585.
175

Références bibliographiques
Wong, H.C., Mao, J., Nguyen, J.T., Srinivas, S., Zhang, W., Liu, B., Li, L., Wu, D. and Zheng, J. (2000) Structural basis of the recognition of the dishevelled DEP domain in the Wnt signaling pathway. Nat Struct Biol, 7, 1178-1184.
Wright, C.W., Rumble, J.M. and Duckett, C.S. (2007) CD30 activates both the canonical and alternative NF-kappaB pathways in anaplastic large cell lymphoma cells. J Biol Chem, 282, 10252-10262.
Wymann, M.P. and Pirola, L. (1998) Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta, 1436, 127-150.
Xue, Y., Fares, H., Grant, B., Li, Z., Rose, A.M., Clark, S.G. and Skolnik, E.Y. (2003) Genetic analysis of the myotubularin family of phosphatases in Caenorhabditis elegans. J Biol Chem, 278, 34380-34386.
Yamamoto, A., DeWald, D.B., Boronenkov, I.V., Anderson, R.A., Emr, S.D. and Koshland, D. (1995) Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol Biol Cell, 6, 525-539.
Yin, H.L. and Janmey, P.A. (2003) Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol, 65, 761-789.
Yoneda-Kato, N., Look, A.T., Kirstein, M.N., Valentine, M.B., Raimondi, S.C., Cohen, K.J., Carroll, A.J. and Morris, S.W. (1996) The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene, 12, 265-275.
Yu, A., Rual, J.F., Tamai, K., Harada, Y., Vidal, M., He, X. and Kirchhausen, T. (2007) Association of Dishevelled with the clathrin AP-2 adaptor is required for Frizzled endocytosis and planar cell polarity signaling. Dev Cell, 12, 129-141.
Yu, F.X., Sun, H.Q., Janmey, P.A. and Yin, H.L. (1992) Identification of a polyphosphoinositide-binding sequence in an actin monomer-binding domain of gelsolin. J Biol Chem, 267, 14616-14621.
Yu, J.W. and Lemmon, M.A. (2001) All phox homology (PX) domains from Saccharomyces cerevisiae specifically recognize phosphatidylinositol 3-phosphate. J Biol Chem, 276, 44179-44184.
Yu, J.W., Mendrola, J.M., Audhya, A., Singh, S., Keleti, D., DeWald, D.B., Murray, D., Emr, S.D. and Lemmon, M.A. (2004) Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell, 13, 677-688.
Zamo, A., Chiarle, R., Piva, R., Howes, J., Fan, Y., Chilosi, M., Levy, D.E. and Inghirami, G. (2002) Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene, 21, 1038-1047.
Zhan, Y., He, D., Newburger, P.E. and Zhou, G.W. (2004) p47(phox) PX domain of NADPH oxidase targets cell membrane via moesin-mediated association with the actin cytoskeleton. J Cell Biochem, 92, 795-809.
Zhang, Q., Raghunath, P.N., Xue, L., Majewski, M., Carpentieri, D.F., Odum, N., Morris, S., Skorski, T. and Wasik, M.A. (2002) Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol, 168, 466-474.
Zhang, X., Hartz, P.A., Philip, E., Racusen, L.C. and Majerus, P.W. (1998) Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J Biol Chem, 273, 1574-1582.
Zhang, Y., Zolov, S.N., Chow, C.Y., Slutsky, S.G., Richardson, S.C., Piper, R.C., Yang, B., Nau, J.J., Westrick, R.J., Morrison, S.J., Meisler, M.H. and Weisman, L.S. (2007) Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci U S A, 104, 17518-17523.
176

Références bibliographiques
Zhong, Q., Lazar, C.S., Tronchere, H., Sato, T., Meerloo, T., Yeo, M., Songyang, Z., Emr, S.D. and Gill, G.N. (2002) Endosomal localization and function of sorting nexin 1. Proc Natl Acad Sci U S A, 99, 6767-6772.
Zhong, Q., Watson, M.J., Lazar, C.S., Hounslow, A.M., Waltho, J.P. and Gill, G.N. (2005) Determinants of the endosomal localization of sorting nexin 1. Mol Biol Cell, 16, 2049-2057.
Zhou, K., Takegawa, K., Emr, S.D. and Firtel, R.A. (1995) A phosphatidylinositol (PI) kinase gene family in Dictyostelium discoideum: biological roles of putative mammalian p110 and yeast Vps34p PI 3-kinase homologs during growth and development. Mol Cell Biol, 15, 5645-5656.
Zou, J., Marjanovic, J., Kisseleva, M.V., Wilson, M. and Majerus, P.W. (2007) Type I phosphatidylinositol-4,5-bisphosphate 4-phosphatase regulates stress-induced apoptosis. Proc Natl Acad Sci U S A, 104, 16834-16839.
177

MATERIELS ET
METHODES

Matériels et méthodes
Cultures cellulaires
Les lignées de lymphomes anaplasiques humains (Fepd, Cost, Karpas, SUDHL-1) sont
cultivées dans du milieu Iscove (GibcoBRL) supplementé de 15% de sérum de veau fœtal
(SVF) et de 20μg/ml de pénicilline/streptomycine (P/S). Les cellules Baf/3 transfectées de
façon stable soit avec le vecteur pcDNA 3.1 vide soit le vecteur pcDNA 3.1 NPM-ALK sont
cultivées dans du milieu RPMI (GibcoBRL) supplémenté de 10% SVF, de P/S et de
généticine (G418, GibcoBRL). Les cellules adhérentes NIH 3T3 exprimant ou non de façon
stable l’oncogène NPM-ALK sont cultivées dans du milieu DMEM (GibcoBRL) supplémenté
de 10% SVF, de P/S et de généticine (G418, GibcoBRL). Les cellules HEK293 et Hela sont
cultivées dans du milieu DMEM (GibcoBRL) supplémenté de 10% SVF et de P/S.Les
cultures cellulaires sont maintenues à 37°C dans une atmosphère saturée en humidité
contenant 5% de CO2.
Clonage Le clonage de PIKfyve en fusion avec la GFP en N-terminal a été réalisé grâce au kit TOPO
XL PCR Cloning (Invitrogen) qui permet l’insertion d’un fragement de PCR dans un vecteur
pcDNA3.1 selon les recommandations du fabriquant. Des ARN totaux ont été extraits de
cellules NIH3T3 exprimant l’oncogène NPM-ALK (Kit Qiagen) et soumis à une réverse
transcription puis une PCR en utilisant des primers qui cadrent le cDNA de PIKfyve murin
(Accession GenBank : AF102777) (primer sens, 5’-ATGGCCACAGATGACAAGAGTTCC-
3’ et anti-sens 5’-TCAGCAATTCAGATCCAACCCTGCCC-3’). Deux mutants ponctuels
PIKfyve[S318A] et PIKfyve[K1831E] ont été construits à partir du cDNA GFP-PIKfyve
sauvage en utilisant le kit Quick Change II XL Site-directed Mutagenesis (Stratagene) selon
les recommandations du fabriquant.
Transfections Le plasmide codant pour la protéine NPM-ALK a été généreusement fourni par le Pr. Georges
Delsol. Les transfections des cellules HEK293 et Hela sont réalisées avec le kit de
transfection Effecten (Qiagen) selon les recommandations du fabriquant. 100nM de siRNA
contrôle (#D001206-13-05) ou anti-PIKfyve « smartpool » (#M-040127-01) (Dharmacon)
sont transfectés dans les cellules NIH3T3 grâce au kit LipofectAmine PlusTM (Invitrogen)
selon les recommandations du fabriquant.
178

Matériels et méthodes
Génération d’un anticorps dirigé contre PIKfyve Les antisérums de PIKfyve ont été obtenus après l’immunisation de deux lapins avec un
peptide dirigé contre les 12 premiers acides aminés N-terminaux de PIKfyve
(MATDDKSSPYLC) couplé KLH (Eurogentec). La spécificité des sérums a été évaluée par
immunoempreinte.
Purification de la kinase recombinante GST-PIP4-Kinase IIα Des bactéries Escherichia coli de type BL21-RIL+ (Stratagene) transformées par le vecteur
pGEX-PIP4-Kinase IIα (AmpR) sont mises en préculture dans du milieu LB (Luria-Broth)
contenant 100μg/ml d’ampicilline et 50μg/ml de chloramphénicol, pendant une nuit à 37°C.
La préculture saturée est diluée dans 500ml de milieu LB sans antibiotiques et remise à
pousser jusqu’à une DO de 0.6-0.8. La production de la protéine recombinante est induite par
addition d’IPTG (Bioprobe) à 0.2mM, 3h à 37°C. Les bactéries sont centrifugée
(6400rpm,20min, 4°C) et le culot bactérien repris dans 18ml de PBS froid. Les bactéries sont
lysées par sonication (6 x 30sec) et incubées en présence de TritonX100 1% final (30min,
4°C). Après centrifugation, le surnageant est incubé avec 500μl de billes de glutathione-
sépharose (Amersham), 3h à 4°C. Les billes sont ensuite sédimentées par centrifugation
(1000rpm, 5min à 4°C) et lavées trois fois dans un tampon PBS, NaCl 0,3M, 1% Tween final.
Les billes sont mises à sec et reprises dans 500μl dans un tampon Tris HCl pH 7.4, 20%
glycérol final. Les aliquots sont conservés à –80°C.
Mesure de la prolifération cellulaire
Les cellules sont ensemencées à raison de 2.000 cellules par puits dans 100μl de milieu de
culture sur une plaque de 96 puits. Après 24, 48, 72 ou 96h, sont ajoutés 20µl d'une solution
de Méthylthiazoltétrazolium (MTS-Promega). Le MTS est métabolisé par les mitochondries
en un produit coloré détectable par spectrophotomètrie. Après 3 h à 37°C sous 5% CO2, la
lecture de la densité optique, corrélée au nombre de cellules vivantes, est effectué à 490 nm.
Les essais sont effectués en triplicate.
Immunoprécipitations
Les cellules NIH3T3 exprimant ou pas l’oncogène NPM-ALK sont lysées dans 800μl de
tampon contenant 50mM de Tris HCl pH 7.4, 100mM NaCl, 5mM EDTA, 1% TritonX100,
179

Matériels et méthodes
NaF 25mM, Orthovanadate 1mM et 10μg/mL d'aprotinine et de leupeptine. Les lysats sont
agités 30min à 4°C puis centrifugés 10min à 13200rpm à 4°C afin d'éliminer les débris
cellulaires. Une « pré-clairance » à l’aide de billes de protéine A-Sépharose (20μl/point,
Amersham) est réalisée puis les homogénats sont incubés avec l’anticorps monoclonal ALK1
(10μl/point, Dako) ou l’anticorps polyclonal PIKfyve (10µL/point, Eurogentec) et la protéine
A-Sépharose (40μl/point) pendant 3h à 4°C sous agitation puis sédimentée par centrifugation.
Les culots sont lavés 3 fois avec 1mL de tampon contenant 50mM Tris-HCl pH 7.4, 100mM
NaCl, 0.1% de TritonX100, NaF 25mM, Orthovanadate 1mM et 5μg/mL d'aprotinine et de
leupeptine. Les immunoprécipitats sont alors soumis à un test lipide kinase et/ou à une
immunoempreinte.
Immunoempreintes Les cellules sont lysées à 4°C dans ne solution de PBS1X contenant 1%TritonX100, 10µg/mL
de leupeptine et d’aprotinine pendant 15min. Après une centrifugation (13200rpm, 4°C,
15min), la concentration en protéines est mesurée dans le surnageant et les lysats sont repris
dans un tampon SDS-PAGE (0.625M Tris HCl pH 6,8, 10% SDS, 50% glycérol, 0.1% bleu
de bromophénol, 8% β-mercaptoéthanol, 5mM EDTA). Les protéines sont ensuite dénaturées
5min à 100°C puis soumises à une électrophorèse sur gel de polyacrylamide SDS-PAGE
avant transfert sur membrane de nitrocellulose. La membrane est saturée dans du TBST
(500mM NaCl ; 20mM Tris base ; 0.05% Tween 20) contenant 5% lait, puis incubée
consécutivement pendant 1 heure avec une solution d'anticorps primaire (ALK1, 1:1000,
Dako ; GFP, 1:1000, Santa Cruz ; PIKfyve, 1:5000, Eurogentec ; P-Akt 473, 1:1000, Cell
Signaling ; P-Akt 308, 1:1000, Cell Signaling ; Tubuline 1:10000, Sigma-Aldrich), puis une
solution d’anticorps secondaire anti-immunoglobulines de souris ou de lapin couplé HRP
( 1:5000, Promega W402B). L'immunoempreinte est révélée par chimiluminescence (ECL,
Amersham).
Immunofluorescence Les cellules sont ensemencées dans des boites de 24 puits sur des lamelles de verre.
Lorsqu’elles ont atteints 70% de confluence, elles sont lavées dans du PBS 1X puis fixées et
perméabilisées avec du paraformaldéhyde (Sigma) (4%, 4°C, overnight) lors de l’utilisation
des sondes Bio-GST-2XPHDING2 et Bio-GST-2XFYVE. La biotinylation est effectuée à l’aide
du kit Biotintag Micro de biotinylation (SIGMA B-TAG), technique validée par l’équipe. 6
180

Matériels et méthodes
lavages de 5mins dans du PBS 1X sont réalisés puis les cellules sont incubées avec 50mM
NH4Cl pendant 15min. Pour les marquages de PIKfyve et de NPM-ALK, à 70% de
confluence, les cellules sont lavées en PBS1X, fixées pendant 1h avec du paraformaldéhyde à
2%, lavées 3 fois 10min dans du PBS1X et perméabilisées dans une solution de PBS-1%
BSA-0,01%Triton. Dans les deux cas, la saturation des sites aspécifiques est effectuée dans
du PBS-5% BSA pendant 1h. Les cellules sont incubées soit avec les protéines recombinantes
biotinylées Bio-GST-2XPHDING2 et Bio-GST-2XFYVE (40μg/ml dans PBS-BSA 1%, 1h)
soit avec les anticorps anti-PIKfyve (1:1000, Eurogentec), anti-ALK1 (1:100, Dako). Elles
sont lavées 6 fois 5mins dans une solution de PBS-BSA1% puis incubées avec de la
streptavidine Alexa-594 (Molecular probe) (1μl/300μl, 1h) ou avec des anticorps secondaires
couplés à un fluorophore Alexa-594 ou Alexa-488 (1:300, Molecular Probe) à l’obscurité. Les
cellules sont alors lavées 6 fois 5mins dans une solution de PBS-BSA 1%. Une dernière étape
de lavage est effectuée dans du PBS 1X et les lamelles sont montées sur lames avec du
mowiol. L’observation des lames se fait par microscopie à fluorescence classique et confocale
(Zeiss).
Dosage en masse de PtdIns5P Les lipides cellulaires totaux sont extraits selon la technique de Bligh et Dyer en présence
d’un mélange CHCL3/CH3OH/H2O (v/v/v) contenant de l’HCl 0.4N final. La phase organique
est récupérée puis évaporée sous azote, reprise par CHCL3/CH3OH (v/v) et déposée sur une
plaque de silice préalablement pré-migrée dans un tampon oxalate de potassium pendant 3h30
et activée à 70°C pendant 10min. Les lipides sont séparés par chromatographie en couche
mince (CCM) dans le solvant CHCl3/CH3OH/NH4 2.4N (9/7/2). L’utilisation d’un standard de
PtdIns4P (Echelon) permet de repérer la zone de migration des phosphoinositides
monophosphates qui sont récupérés par grattage de silice. Une gamme étalon de PtdIns5P pur
(Echelon) (0, 50, 100 pmoles) traitée comme les échantillons à analyser est réalisée, et servira
à la quantification du [32P]PtdIns(4,5)P2 produit. Les phosphoinositides monophosphates
purifiés par CCM sont alors repris en présence de 20nmoles de PtdIns (Sigma) dans 60µl de
tampon kinase (50mM Tris-HCl pH 7.4, 1.5mM DTT, 100mM NaCl, 0.5mM EDTA, 5mM
MgCl2) contenant 10µM final d’ATP ajouté extemporanément, puis soniqués brièvement pour
former des vésicules. La kinase recombinante GST-PI4P-Kinase IIα (0.6µg) et de l’
[32P]γATP (10µCi) (Perkin Elmer) sont ajoutés à chaque point. Après 1h d’incubation à 30°C,
la réaction est arrêtée par addition de 100µl d’H2O, 30µl d’HCl 10N, 400µl de CHCl3/CH3OH
181

Matériels et méthodes
(v/v), et 5µl de mélange de Folch (carrieur des lipides). Les lipides sont à nouveau extraits par
la technique de Bligh et Dyer et séparés par CCM dans le solvant CHCl3/CH3OH/NH4 2.4N
(9/7/2). Le [32P]PtdIns(4,5)P2 produit est visualisé à l’aide d’un phosphorImager (445Si
Molecular Dynamics) et quantifié par HPLC.
Quantification des divers phosphoinositides par HPLC Les lipides séparés sur CCM et révélés au phosphorImageur sont récupérés par grattage de la
silice et sont ensuite incubés pendant 50min à 53°C dans 700µl d’une solution de déacylation
CH3NH2/H2O/CH3OH/C3H7OH (26.8/16.1/45.7/11.4, v/v/v/v). Après évaporation à sec sous
azote, resuspension dans l’eau des glycérophosphoinositides et filtrage, les échantillons sont
analysés et quantifiés par chromatographie HPLC sur colonne échangeuse d’ions (Parisphère
SAX).
Test lipide kinase Les immunoprécipités NPM-ALK et/ou PIKfyve sont lavés 2 fois dans un tampon de réaction
contenant 25mM Hépès, 120mM NaCl, 2.5mM MgCl2, 2.5mM MnCl2 et mis en présence de
vésicules de PtdIns à 200µM (Sigma) ou de vésicules de PtdIns/PtdIns3P (200/100µM)
(Sigma, Echelon), 50µM d’ATP, 20µCi d’ [32P]γATP et selon le cas d’inhibiteurs tels que le
LY294002 (Sigma), ou le WHI-154 (Calbiochem). Après 30min d’incubation à 37°C, la
réaction est arrêtée par addition de 100µl d’H2O, 30µl d’HCl 10N, 400µl de CHCl3/CH3OH
(v/v), et 5µl de mélange de Folch. Les lipides extraits par la technique de Bligh et Dyer sont
séparés par CCM dans le solvant CHCl3/CH3OH/NH4 2.4N (9/7/2). Les [32P]PtdInsP générés
sont visualisés à l’aide d’un phosphorImager (445Si Molecular Dynamics) et quantifiés par
HPLC.
Etude du pouvoir invasif des cellules en matrigel 3D 2x104 cellules NIH3T3 transfectées ou pas avec les siRNA contrôle, ou dirigés contre
PIKfyve (Dharmacon) sont récoltées par ajout de PBS contenant 5mM d’EDTA, lavées dans
du PBS puis sédimentées par centrifugation à 90g pendant 5min. Les cellules sont reprises
dans 175µL de milieu de culture complet. Après ajoût de 25µL de marigel (BD Biosciences),
les cellules sont déposées au fond d’un puit d’une LAB-TEK à 8 chambres incubée à 37°C
pendanr 40min pour permettre au matrigel de se solidifier puis placée sur la platine du
microscope au sein d’une étuve à atmosphère humide régulée à 37°C et 5% CO2. L’analyse
182

Matériels et méthodes
des déplacements cellulaires est réalisée à l’aide du microscope Zeiss-Axiovert-200 piloté par
le logiciel MetaMorph avec une caméra Cool SNAP HQ permettant l’enregistrement des
images acquises à intervalles de 10min au grossissement x10. La distane parcourue par les
cellules durant 15h est mesurée à l’aide du logiciel MétaMorph.
183

ANNEXES

ARTICLE IN PRESS
Advan. Enzyme Regul. 45 (2005) 201–214
0065-2571/$ -
doi:10.1016/j
�CorrespoE-mail ad
www.elsevier.com/locate/advenzreg
Emerging roles of phosphatidylinositolmonophosphates in cellular signaling
and trafficking
Caroline Pendaries, Helene Tronchere, Claire Racaud-Sultan,Frederique Gaits-Iacovoni, Sophie Coronas, Stephane Manenti,Marie-Pierre Gratacap, Monique Plantavid, Bernard Payrastre�
Inserm U563-CPTP, IFR 30, Department of Oncogenesis and signaling in haematopoıetic cells,
CHU Purpan, 31024 Toulouse, France
Introduction
Phosphoinositides are a quantitatively minor family of membrane glyceropho-spholipids composed of eight members; their metabolism is highly active. Theirstructural backbone corresponds to a sn-glycerol esterified in position 1 and 2 byfatty acids (essentially stearic and arachidonic acids, respectively), and bearing inposition 3 a myo-inositol head group connected to the diacylglycerol by aphosphodiester bound. The myo-inositol of phosphoinositides contains five freehydroxyls, only three of them (positions D-3, D-4 or D-5) being phosphorylated invivo probably because of steric hindrance (Fig. 1). Phosphatidylinositol (PtdIns), themost abundant member of the family, can be sequentially phosphorylated by specifickinases to generate seven polyphosphoinositides implicated in the regulation ofcritical cellular functions (Rameh and Cantley, 1999; Payrastre et al., 2001; Toker,2002 for reviews). Phosphoinositides exert their role either as precursors of theimportant phospholipase C-generated second messengers, inositol 1,4,5-trispho-sphate (Ins(1,4,5)P3) and diacylglycerol (DAG), or directly by interacting with
see front matter r 2005 Elsevier Ltd. All rights reserved.
.advenzreg.2005.02.006
nding author. Tel.: +335 62 74 45 25; fax: +33 5 62 74 45 58.
dress: [email protected] (B. Payrastre).

ARTICLE IN PRESS
PIK-FYVE
PtdIns 4-Kinase II α,βPtdIns 4-Kinase III α,β
PI 3-Kinase (Class IIIand possibly IIand I)
CO
O CH2
CH2
CHOC
O
O P O
O
O-
HOOH OH
OH
OHArachidonic acid
Stearic acid
1 23
6 54
Fig. 1. Structure of phosphatidylinositol and its phosphorylation to phosphatidyinositol monopho-
sphates. PtdIns is the precursor of the other phosphoinositides through sequential phosphorylations by
specific kinases. The kinases able to phosphorylate its inositol head group at the positions (D-3, D-4 and
D-5) to produce PtdIns(3)P, PtdIns(4)P and PtdIns(5)P are indicated.
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214202
proteins and modulating their localization, conformation, and activity. Indeed, overthe last decade, a number of protein domains able to bind phosphoinositides, with awide range of affinities and specificities, have been identified. These includepleckstrin homology (PH), phox homology (PX), Fab1p/YOTB/Vac1p/EEA1(FYVE) domains or lysine/arginine rich sequences (Itoh and Takenawa, 2002).The interaction of phosphoinositides with these domains is the key to theorganization of many signaling complexes involved in the regulation of criticalcellular functions such as proliferation and survival, trafficking or even glucosemetabolism. The discovery of these domains sheds light on the importance ofphosphoinositides to give spatial and temporal cues mandatory to the correctintegration of cellular processes. Some phosphoinositide binding domains specificfor a given lipid have been fused to the green fluorescent protein (GFP) or to thegluthatione S-transferase (GST) and used to visualize the location of these lipids incells (Balla et al., 2000). These approaches together with biochemical data suggestthat pools of phosphoinositides can be rapidly synthesized and degraded throughdifferent metabolic pathways in discrete membrane domains, in organelles, or evenin subnuclear areas. In fact, specific lipid kinases, phosphatases and phospholipasesthat are often cytosolic and targeted to specific subdomains tightly control the levelof these versatile lipids. Upon extracellular signals, they change the phosphoinositideprofile, which in turn regulates downstream targets involved in the control of propercellular responses. The critical role of these lipids is emphasized by recent discoveriesindicating that mutations in several phosphoinositide phosphatases take part in thedevelopment of human diseases including cancer, myotubular myopathy or Lowesyndrome (Pendaries et al., 2003 for review).
Until recently, only some members of the family, the phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and the phosphoinositide 3-kinase (PI 3-kinase)products, mainly phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P2) andphosphatidylinositol 3,4-bisphosphate (PtdIns(3,4)P2), were thought to be importantin terms of cellular functions. Phosphatidylinositol monophosphates (PtdInsP)were considered as intermediate metabolites of the synthesis pathways of these

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214 203
polyphosphoinositides. However, a few years ago, it was shown that yeasts usephosphatidylinositol 3-monophosphate (PtdIns(3)P) to regulate vesicular traffickingby interacting with a set of FYVE domain-containing proteins involved in vacuolarsorting (Gaullier et al., 1998). Evidence is now accumulating that this phosphoinosi-tide is also implicated in vesicular trafficking in mammals (Wurmser et al., 1999).More recently, phosphatidylinositol 4-monophosphate (PtdIns(4)P), which was longonly considered as a precursor for PtdIns(4,5)P2, has been shown to act as aregulator on its own, by recruiting PtdIns(4)P-binding proteins such as FAPPs (Godiet al., 2004) or the clathrin adaptor AP-1 to the trans-Golgi network (Wang et al.,2003). Finally, phosphatidylinositol 5-monophosphate (PtdIns(5)P), the leastcharacterized phosphoinositide, is also emerging as a potentially important signalingmolecule.
In this review, we will focus on recent data showing that phosphatidylinositolmonophosphates (PtdIns(3)P, PtdIns(4)P and PtdIns(5)P) are not relegated to asecondary role but are critical lipid messengers able to interact with protein domains(Table 1) and required for the regulation of important cell functions.
A Role of PtdIns(3)P in Intracellular Trafficking
PI 3-kinases are lipid kinases that phosphorylate the D3 position of the myo-inositol ring of phosphoinositides to generate directly or indirectly the different 3-phosphorylated phosphoinositides: PtdIns(3)P, PtdIns(3,4)P2, PtdIns(3,5)P2 andPtdIns(3,4,5)P3. By opposition to PtdIns(3,4,5)P3, which levels transitory increaseunder cell stimulation by a variety of agonists, PtdIns(3)P is constitutively present ata rather constant level (5–15% of total PtdInsP) in mammalian cells and mainlylocated on intravesicular membranes (endosome and multivesicular bodies). In theyeast S. cerevisiae, the only PI 3-kinase, vps34 (vacuolar protein sorting 34), isresponsible for the production of PtdIns(3)P and plays a critical role in the sorting ofproteins from the Golgi to the vacuole (Schu et al., 1993). The cytosolic vps34 isrecruited from the cytosol to the Golgi by the serine/threonine kinase vps15p, whichalso stimulates its lipid kinase activity. In mammalian cells, three different classes ofPI 3-kinases have been found, all of them being able to phosphorylate PtdIns toPtdIns(3)P in vitro (Foster et al., 2003, for review). The human homologue of vps34,hVps34 (or class III PI 3-kinase), however, shows a specificity over PtdIns and isunable to phosphorylate PtdIns(4)P or PtdIns(4,5)P2. Like in yeast, hVps34 isregulated by its interaction with p150, the human homologue of vps15p, and playsan important role in the membrane trafficking pathway to the lysosome.
PtdIns(3)P levels in cells can be modulated by several ways implicating differentlipid kinases or phosphatases (Table 1). First, PtdIns(3)P is a potential substrate fora putative 4-kinase generating PtdIns(3,4)P2, however, this way needs to be clearlydocumented. Second, PtdIns(3)P is the precursor of PtdIns(3,5)P2 by the action ofthe 5-kinase PIKfyve (Shisheva, 2001, for review). PIKfyve can also use PtdIns as asubstrate and generates PtdIns(5)P, a recently characterized PI (see chapter onPtdIns(5)P below). Like its yeast homologue fab1p, which was shown to be essentialto maintain vacuole morphology (Odorizzi et al., 1998), PIKfyve localizes to late
ARTICLE IN PRESS
Table
1
Bindingdomainsandmetabolizingenzymes
ofmonophosphoinositides
PtdInsP
Bindingdomains
Metabolizingenzymes
Localizationto
mem
branes
EC
number
PtdIns(3)P
FYVE
Product
of:
-Class
IIIPI3-kinase
GC,E
2.7.1.137
PX
-Class
IIPI3-kinase
PM,GC,E,N
2.7.1.154
PH
-Class
IPI3-kinase
PM,N
2.7.1.153
-typeI,II
4-phosphatases
ND
Substrate
of:
-5-kinase
PIK
fyve
LE
2.7.1.68
-3-phosphatasesMTM
family(M
TM1,MTMR1-8)
PM
3.1.3.48;3.1.3.-
-3-phosphatase
PTEN
PM,GC,N
3.1.3.67
PtdIns(4)P
PH
Product
of:
-PtdIns4-K
inasesII
a;b
GC,E(a),PM
(b)
A/ENTH
-PtdIns4-K
inasesIIIa;b
GC,ER
(a),N
(b)
-5-phosphatase
IIND
-5-phosphatase
OCRL
GC,E,Ly
3.1.3.36
-5-phosphatase
Synaptojanin
SV,Mi
3.1.3.36
-5-phosphatase
PIPP
PM
3.1.3.56
-5-phosphatase
SKIP
ER,PM
3.1.3.56
-3-phosphatase
PTEN
PM,GC,N
3.1.3.67
Substrate
of:
-Class
I,II
PI3-K
inases
PM,N,GC,E
-typeIPtdInsP
kinases(PIP5K
a;b;g)
PM
-4-phosphataseshSac1,2,3
GC,ER
-4-phosphatasesSynaptojanin
1,2
CCV,Mi
3.1.3.36
PtdIns(5)P
PHD
Product
of:
-5-kinase
PIK
fyve
LE
2.7.1.68
PX
-3-phosphatase
MTM1,MTMR2-3
PM
3.1.3.48;3.1.3.-
Substrate
of:
-typeII
PtdInsP
kinases(PIP4K
a;b;g)
PM
(a;b;),ER
(g)
-5-phosphatase
PLIP
GC
TheTable
liststhebindingdomainsofPtdInsP,thekinasesandthephosphatasesfrom
higher
eukaryotes,
whichmetabolize
theselipids,
alongwiththeir
intracellularlocalizationsandtheirEnzymeCommissionnumber.Abbreviations:CCV,clathrincoatedvesicles;E,endosomes;ER,endoplasm
icreticulum;
GC,Golgicomplex;LE,late
endosomes;Ly,Lysosomes;Mi,mitochondria;N,nucleus;ND,NotDetermined;PM,plasm
amem
brane;SV,synapticvesicles.
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214204

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214 205
endosomes, through binding of its FYVE domain to PtdIns(3)P (see below) and isnecessary for endosome integrity (Ikonomov et al., 2001). The role of PIKfyve inregulating the balance of the PtdIns(3)P and PtdIns(3,5)P2 pools in vesiculartrafficking needs to be precise. Finally, PtdIns(3)P levels can be decreased by theaction of the 3-phosphatases from the myotubularin family (Tronchere et al., 2003for review). MTM1 is mutated in the X-linked recessive myotubular myopathy(XLMTM), a muscular genetic disease, and was shown to specifically depho-sphorylate PtdIns(3)P and PtdIns(3,5)P2 (Blondeau et al., 2000; Taylor et al., 2000;Tronchere et al., 2004). MTMR2, mutated in the demyelinating neuropathyCharcot-Marie-Tooth disease 4B (CMT4B1), and MTMR3, also show this strictsubstrate specificity. Results obtained in yeast strongly suggest a role for MTM1 invesicular trafficking, through regulation of PtdIns(3)P and PtdIns(3,5)P2 levels(Blondeau et al., 2000). Similarly, two myotubularin homologues in C. elegans havebeen shown to play an essential role in the endocytosis pathway involving PtdIns(3)Pand the Arf6 GTPase (Dang et al., 2004). However, a role for myotubularin familymembers in membrane traffic or maintenance in eukaryotes is not yet clearlydemonstrated. In mammalian cells, overexpression of MTM1, but not MTMR2, wasable to partly dephosphorylate the endosomal pool of PtdIns(3)P (Kim et al., 2002).However, studies of PtdIns(3)P endosomal pool on normal or XLMTM patientmuscular cell lines revealed identical staining patterns, pointing to a more discreterole of MTM1 in vivo (Tronchere et al., 2004). This observation is reinforced by thefact that in most of the cell lines studied PtdIns(3)P hydrolysis by MTM1 does notaccount for more than 10% of the total PtdIns(3)P cellular pool (Kim et al., 2002;Tronchere et al., unpublished results). MTM1 activity could be directed to specificminor pools of PtdIns(3)P, located on membranes others than vesicular, like theplasma membrane, as shown in C. elegans (Dang et al., 2004) and suggested by thefact that MTM1 localize in part to Rac1-induced membrane ruffles in mammaliancells (Laporte et al., 2002).
The importance of PtdIns(3)P as a spatial regulator came from the finding thatPtdIns(3)P binding domains are present in a number of proteins involved in vesiculartrafficking. The FYVE domain was the first PtdIns(3)P binding domain character-ized and FYVE domain proteins are mainly implicated in endosomal/vacuolartrafficking. The FYVE domain is a small domain (80 a.a.) of Ring-zinc finger typewith two b-hairpin and a C-terminal a-helix. The basic motif (R/K)(R/K) HHCR inthe b strand is critical for the PtdIns(3)P binding (Misra and Hurley, 1999). SeveralFYVE domains have been crystallized and help understand the mechanism and thespecificity of the PtdIns(3)P binding (Misra et al., 2001; Stenmark et al., 2002;Lemmon, 2003 for reviews). Hydrophobic interactions of the non-polar side chainsof the FYVE domains which insert into the membranes help for the targeting.Moreover, it has been shown that dimerization of certain FYVE domains, such asthe FYVE domain of EEA1, is necessary for proper localization to endosomes. Atandem of FYVE domain fused to GFP or GST can be used to visualize endosomalpools of PtdIns(3)P (Gillooly et al., 2000). However, the binding affinity of theFYVE domains for PtdIns(3)P is rather weak, and recent data suggest that theassociation of FYVE domain with membranes in vivo requires other interactions to

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214206
be stable. In the case of EEA1, a protein–protein interaction between EEA1 and theGTPase Rab5 also stabilizes the binding to the endosomal membrane. Inmammalian cells, some FYVE domain-containing proteins function in cellularprocesses other than membrane trafficking. This is the case of the signal transductionprotein SARA, implicated in the TGF-b signaling (Seet and Hong, 2001) or the Fgd1family of proteins, which are exchange factors for the small GTPase cdc42, andtherefore regulators of the actin cytoskeleton (Nagata et al., 1998).
Besides FYVE domains, PX domains have been shown to be PtdIns(3)P bindingmodules. The PX domain (120 a.a.) was originally described in proteins of diversefunction in cells, some of them implicated in trafficking (Ponting, 1996). The crystalstructure of the PX domain containing protein p40phox bound to PtdIns(3)P wasresolved and shows a N-terminal 3 b-sheet structure linked to a four a-helicalstructure. The binding site for PtdIns(3)P lays between the b-sheet and the helicaldomain (Bravo et al., 2001). In yeast S. cerevisiae, all the PX protein found in thegenome (15) show a specificity for PtdIns(3)P (Yu and Lemmon, 2001). PX domainsaffinity for PtdIns(3)P ranks from low to high and many of the low affinity PXproteins are part of multimolecular complexes, in contrast to high affinity PXdomains that would be sufficient to target a protein to its endosomal location. Ineukaryotes, the number of PX proteins is around 54 with varying specificity towardsPI. The majority of PX proteins binds PtdIns(3)P, with the exception of preferentialbinding of the p47phox to PtdIns(3,4)P2 and the PI 3-kinase class II to PtdIns(4,5)P2.The increasing number of proteins bearing PtdIns(3)P binding domain certainlyfavors a key role for this PtdIns in the spatial regulation of vesicular trafficking.However, recent studies point to a stimulatory control of certain pools of PtdIns(3)P,besides the constitutively endosome-associated PtdIns(3)P pool. The humanpathogen Mycobacterium tuberculosis has been shown to interfere with thephagosome maturation pathway by modifying oscillatory waves of PtdIns(3)P onthe phagosomes (Chua and Deretic, 2004). Other regulated PtdIns(3)P pools havebeen described in stimulated cells, as in human platelets upon integrin engagement(Banfic et al., 1998), or in Hela and Cos7 cells after lysophosphatidic stimulation(Razzini et al., 2000). Finally, the presence of PtdIns(3)P in plama membranemicrodomains (rafts) in insulin stimulated cells suggests a role of lipid secondmessenger for PtdIns(3)P (Maffucci et al., 2003).
PtdIns(4)P: a Regulator of Golgi Functions
PtdIns (�10% of total cell glycerophospholipids in mammalian cells) undergoescontinuous sequential and reversible phosphorylations at position D-4 and D-5 byspecific kinases (Fig. 1). Two families of PtdIns 4-kinases (type II and III) are knownto phosphorylate PtdIns to PtdIns(4)P. PtdIns(4)P can then be phosphorylated bytype I PtdInsP-kinases to yield PtdIns(4,5)P2 (Table 1). PtdIns and its phosphoryla-tion products, PtdIns(4)P and PtdIns(4,5)P2 (both representing about 10% of totalphosphoinositides), are constitutive components of cells. It is widely acceptedthat these three phosphoinositides are kept in a steady state in the membranesthrough phosphorylation/dephosphorylation reactions by specific 4 and 5-kinases

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214 207
and -phosphatases. The so called ‘‘canonical pathway’’ describes PtdIns(4)P as ametabolic precursor for PtdIns(4,5)P2, permanently providing a pool of substrate forphospholipase C. By an alternative pathway that remains elusive, PtdIns(4)P canalso be phosphorylated by type II (and possibly type I) PI 3-kinases to producePtdIns(3,4)P2.
A few years ago, data obtained in yeast suggested that PtdIns(4)P was essential forthe regulation of constitutive secretion from the late Golgi (Hama et al., 1999;Walch-Solimena and Novick, 1999). Recent data demonstrate that, in mammaliancells, the Golgi complex is rich in PtdIns(4)P but poor in PtdIns(4,5)P2 at steady state(De Matteis et al., 2004; Wang et al., 2003). This composition strikingly differs fromthat of the plasma membrane where PtdIns(4,5)P2 is mainly found. The enzymaticmachinery involved in the maintenance of the specific lipid composition of the Golgiis still poorly characterized but the type-III Arf1-associated PtdIns 4-kinase b hasbeen proposed to play an important role (Godi et al., 1999). Moreover, it wasrecently shown by RNA interference that the Golgi resident type II PtdIns 4-kinase aalso accounts for the maintenance of the PtdIns(4)P pool in this organelle (Wanget al., 2003). PtdIns-transfer proteins that are expected to drive the formation ofseparated phosphoinositide pools by delivering PtdIns to specific places within cells,may provide PtdIns to the Golgi complex (Simon et al., 1998).
Interestingly, PtdIns(4)P appears to target Golgi-associated proteins such asepsinR, oxysterol binding protein (OSBP), the clathrin adaptor AP-1 and the four-phosphate-adaptor protein 1 and 2 (FAPP 1 and 2) to this organelle (Hirst et al.,2003; Levine and Munro, 2002; Wang et al., 2003). Recently, the role of PtdIns(4)Pas a signature establishing the Golgi’s unique organelle identity that specifies thedocking of the AP-1 coat machinery has been proposed (Wang et al., 2003). It ismajor move to generate vesicles transporting cargo proteins to endosomes. AP-1directly binds PtdIns(4)P probably through a positively charged amino-acid richpatch. In contrast to the plasma-membrane-associated AP-2 adaptor, AP-1 does notinteract with PtdIns(4,5)P2 nor with PtdIns(3,4,5)P3. AP-1 also interacts withepsinR, another PtdIns(4)P (and possibly PtdIns(5)P) binding protein via itsENTH domain (Mills et al., 2003; Hirst et al., 2003). AP-1, predominantly found inthe trans-Golgi network and endosomes, can also localize to phagocytic cups,suggesting that local production of PtdIns(4)P may play a role in regulatingearly steps of phagocytosis as well. A recent study (Godi et al., 2004) showsthat PtdIns(4)P and Arf target FAPP 1 and 2 to the trans-Golgi network viainteraction with their PH-domains. These proteins may then ensure coordination ofbudding and fission reactions required for generation of carriers that are competentfor fusion with the plasma membrane. Another protein whose role is still poorlycharacterized, OSBP, translocates to the Golgi in the presence of oxysterols viainteraction of its PH-domain with PtdIns(4)P (Ridgway et al., 1992; Levine andMunro, 1998).
The ultimate mode of action of Golgi proteins recruited by PtdIns(4)P is notalways well defined but recent data suggest that they may sort various types of cargoproteins by directing them to different post-Golgi membrane compartments (DeMatteis and Godi, 2004, for review).

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214208
In addition to its important role in Golgi complex, PtdIns(4)P has also been shownto bind to cytoskeletal proteins including talin. Interestingly, PtdIns(4)P was foundto interact with talin in cells in suspension, whereas at early stages of adhesion, it wasrather PtdIns(4,5)P2 that associated with this protein (Heraud et al., 1998; Martel etal., 2001). The functional consequences of the interaction of PtdIns(4)P with talin arestill unknown but, as shown for PtdIns(4,5)P2 binding, it can induce conformationalchanges of the protein in vitro (Martel et al., 2001). Via the recruitment and theactivation of type I PtdInsP 5-kinase g to the FERM domain of talin, PtdIns(4)Pmay be transformed into PtdIns(4,5)P2 leading to enhanced interaction of talin withintegrins (Martel et al., 2001), a final step in integrin activation (Calderwood, 2004).Moreover, like PtdIns(4,5)P2, PtdIns(4)P can modulate the profilin-actin complex
in vitro (Katakami et al., 1992), but its exact contribution to this mechanism in vivoremains unclear. A recent report indicates that in yeast both the GTPase Cdc42 anda pool of PtdIns(4)P specifically recruit the p21-activated protein kinase-relatedkinase Cla 4 to sites of polarized growth (Wild et al., 2004). Since Cla 4, where thePH domain interacts with PtdIns(4)P, is involved in mitotic exit, this observationmay account for the PtdIns(4)P regulation of cell division previously reported(Muhua et al., 1998).
Finally, PtdIns(4)P has been proposed to act at other levels of cell biology,but those functions remain rather uncharacterized. They include the controlof stomatal movements in plant (Jung et al., 2002), and the regulation of neuronalCl�-ATPaseXpump (Wu et al., 2002).
PtdIns(5)P: a New Lipid Second Messenger
PtdIns(5)P has long been ignored because it is generally present in small amounts(�1–5% of total PtdInsP) and is difficult to separate from PtdIns(4)P (Rameh et al.,1997; Tolias et al., 1998). The metabolic pathway and the roles of this newlydiscovered phosphoinositide are still poorly understood but recent data definePtdIns(5)P as a potential new lipid second messenger.
So far, only a few enzymes have been shown to potentially participate in themetabolism of this lipid (Table 1). PtdIns(5)P can be synthesized by phosphorylationof PtdIns via the 5-kinase PIKfyve (Sbrissa et al., 2002; Shisheva, 2001). It can alsobe generated by dephosphosphorylation of PtdIns(3,5)P2 via 3-phosphatases such asmembers of the myotubularin family (Tronchere et al., 2003, 2004; Walker et al.,2001). PtdIns(5)P is the preferred substrate of type II PtdInsP-kinase (a; b and g)which transforms it into PtdIns(4,5)P2 (Rameh et al., 1997). The level of PtdIns(5)Pmay also be decreased by the PTEN-like phosphatase (PLIP) which exhibits a uniquepreference for PtdIns(5)P in vitro (Pagliarini et al., 2004). However, PLIP is mainlylocalized in the Golgi, with its phosphatase domain facing the cytoplasmiccompartment (Merlot et al., 2003) and should thus hydrolyze specific local poolsof PtdIns(5)P.
By using recombinant type II PtdInsP-kinase a; Morris et al. (2000) developed amass assay to monitor the level of cellular PtdIns(5)P. This sensitive testdemonstrated that the level of PtdIns(5)P is very low in resting cells, but rises upon

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214 209
thrombin stimulation in human platelets (Morris et al., 2000) or during osmotic-stress in mammalian and in plant cells (Meijer et al., 2001; Sbrissa et al., 2002;Tronchere et al., 2004). Moreover, a pool of PtdIns(5)P has been found in thenucleus of murine erythroleukemia cells, with a 20-fold increase during the G1phase unraveling a potential role for PtdIns(5)P in cell-cycle progression (Clarkeet al., 2001).
So far, the most important production of PtdIns(5)P in mammalian cells wasobserved in cells infected by the human intestinal pathogen Shigella flexneri, aGram-negative bacillus known to cause bacillary dysentery (Niebuhr et al., 2002).Among the proteins injected into the host cell by S. flexneri through its type IIIsecretion system, IpgD and its chaperone IpgE are implicated in entry focusformation. Interestingly, IpgD has two motifs related to the active site ofmammalian inositol polyphosphate 4-phosphatase (Norris et al., 1998). Wedemonstrated that IpgD is a phosphoinositide phosphatase that specificallyhydrolyzes the host cell PtdIns(4,5)P2 into PtdIns(5)P during infection of epithelialcells (Niebuhr et al., 2002). Transfection of IpgD in various mammalian cells inducesthe formation of PtdIns(5)P. The transformation of PtdIns(4,5)P2 into PtdIns(5)P byIpgD during S. flexneri invasion promotes a local decrease in cytoskeletal-membraneadhesion, allowing the formation of membrane ruffling at the entry site (Niebuhr etal., 2002, 2000; Pendaries et al., 2003). In agreement, expression of IpgD infibroblasts facilitates the development of Cdc42 and Rac-mediated cytoskeletonrearrangement, such as filopodia and lamellipodia, upon agonist stimulation. Thus,transformation of PtdIns(4,5)P2 into PtdIns(5)P by IpgD, in cooperation with Cdc42and Rac (two GTPases activated by/and involved in the entry process of S. flexneri),allows the formation of membrane ruffles at the entry site to improve the bacterialentry and virulence.
Like S. flexneri, Salmonella typhimurium uses a type III secretory system to injectinto the host cell, SigD (also known as SopB), the homolog of IpgD which is also aneffective inositol phosphatase able to decrease the amount of PtdIns(4,5)P2 at thebase of the ruffles (Terebiznik et al., 2002). The product of PtdIns(4,5)P2 hydrolysisby SigD has not been identified yet but based on its homology with IpgD, one canpropose that it is PtdIns(5)P. A reduced rigidity of the membrane was also observedafter SigD action during S. typhimurium invasion. As in the case of IpgD, this waslikely due to a release of membrane–cytoskeleton interactions, thereby facilitatingplasmalemmal deformation at the entry foci. This focal disappearance ofPtdIns(4,5)P2 facilitates sealing of the invaginations by contributing to the removalof F-actin and its associated proteins (Terebiznik et al., 2002). This event appears tobe required for an efficient formation of Salmonella-containing vacuoles. Accord-ingly, the invasion of SigD-deficient Salmonella is significantly delayed.
Those data show a role for PtdIns(4,5)P2/PtdIns(5)P during cell invasion, but it isstill not clear whether PtdIns(4,5)P2 degradation or PtdIns(5)P synthesis isresponsible for membrane remodeling at the entry site. Clearly the local decreasein PtdIns(4,5)P2 is directly implicated in this mechanism as this lipid interacts withseveral proteins involved in the actin cytoskeleton remodeling (Takenawa and Itoh,2001). However, PtdIns(5)P may also play an important role in the control of cell

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214210
morphology and actin assembly (Sbrissa et al., 2004) or in the initiation ofintracellular mechanisms used by the pathogen to increase its virulence.
We and others have investigated the functions of PtdIns(5)P by manipulation ofits intracellular level. Recently, it was shown that loss of PtdIns(5)P, via conversionto PtdIns(4,5)P2 by the type II PtdInsP-kinase b; resulted in a decrease in Aktactivity in response to insulin (Carricaburu et al., 2003). Conversely, cells expressingIpgD had higher levels of basal and insulin-stimulated Akt phosphorylation and typeII PtdInsP-kinase b expression partially reversed this effect. Consistent with thosedata, increased insulin sensitivity and reduced adiposity were observed in type IIPtdInsP-kinase b knockout mice (Lamia et al., 2004). One interpretation of thesedata is that PtdIns(5)P pathway could negatively regulate a PtdIns(3,4,5)P3
phosphatase and in turn increase the level of this phosphoinositide and thesubsequent activation of Akt. However, the role of PtdIns(5)P in regulatingphosphoinositide phosphatases remains unclear. Indeed, two in vitro studies reportthat this phosphoinositide rather increases the activity of PTEN (Campbell et al.,2003) and of various myotubularin family members (Schaletzky et al., 2003),presumably through allosteric regulation.
Interestingly, a novel phosphoinositide binding domain, the plant homeodomain(PHD) of inhibitor of growth protein 2 (ING2), a conserved Cys4-HisCys3 zincfinger, was recently identified as a nuclear PtdIns(5)P receptor (Gozani et al., 2003).ING2 is a candidate tumor suppressor that induces growth arrest and apoptosis in ap53-dependent manner. Mutations resulting in loss of its interaction with PtdIns(5)Paffect the localization and the activity of ING2, leading to a decrease in ING2-mediated apoptosis and p53 acetylation. The identification of nuclear-specificphosphoinositide binding domains establishes potential novel functions forphosphoinositides, particularly PtdIns(5)P, within the nucleus (Jones and Divecha,2004 for review). Signals that induce cell responses such as differentiation, cellproliferation and stress adaptation/apoptosis lead to a remodeling of nuclearinositol-lipid and -phosphate profiles (Irvine, 2003). Nuclear phosphoinositides,including PtdIns(5)P, may play an important role in regulating changes in chromatinstructure to modulate gene expression, replication or DNA repair. However, howPtdIns(5)P is produced in the nucleus remains an important open question.
Altogether, recent data indicate that different pools of PtdIns(5)P may exist indifferent localization of the cell including the plasma membrane, where it could acton the PI 3-kinase/Akt pathway and membrane/cytoskeleton remodeling; the Golginetwork, where two PtdIns(5)P metabolizing enzymes, PIKfyve and PLIP, arelocated; and the nucleus, where it interacts with chromatin remodeling proteins.Ongoing work will put further light on the whereabouts of PtdIns(5)P as a secondmessenger.
Summary
The phosphoinositide metabolism that is highly controlled by a set of kinases,phosphatases and phospholipases leads to the production of several second

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214 211
messengers playing critical roles in intracellular signal transduction mechanisms.Recent discoveries have unraveled unexpected roles for the three phosphatidylino-sitol monophosphates, PtdIns(3)P, PtdIns(4)P and PtdIns(5)P, that appear now asimportant lipid messengers able to specifically interact with proteins. The formationof functionally distinct and independently regulated pools of phosphatidylinositolmonophosphates probably contributes to the specificity of the interactions with theirtargets. The relative enrichment of organelles in a particular species of phosphoi-nositides (i.e. PtdIns(3)P in endosomes, PtdIns(4)P in Golgi and PtdIns(4,5)P2 inplasma membrane) suggests the notion of lipid-defined organelle identity. PtdIns(3)Pis now clearly involved in vesicular trafficking by interaction with a set of FYVEdomain-containing proteins both in yeast and in mammals. PtdIns(4)P, which untilnow was only considered as a precursor for PtdIns(4,5)P2, appears as a regulator onits own, by recruiting a set of proteins to the trans-Golgi network. PtdIns(5)P, themost recently discovered inositol lipid, is also emerging as a potentially importantsignaling molecule.
Acknowledgements
We wish to thank Pr. P. Sansonetti and Dr. C. Erneux for many helpful dis-cussions and collaboration. This work was supported by grants from ‘‘Associationpour la Recherche Contre le Cancer’’ (ARECA-Toulouse and contract no. 4794) and‘‘Association Franc-aise contre les Myopathies’’.
References
Balla T, Bondeva T, Varnai P. How accurately can we image inositol lipids in living cells? TiPS
2000;21:238–41.
Banfic H, Tang X, Batty IH, Downes CP, Chen C, Rittenhouse SE. A novel integrin-activated pathway
forms PKB/akt-stimulatory phosphatidylinositol 3,4-bisphosphate via phosphatidylinositol 3-phos-
phate in platelets. J Biol Chem 1998;273:13–6.
Blondeau F, Laporte J, Bodin S, Superti-Furga G, Payrastre B, Mandel J-L. Myotubularin, a phosphatase
deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol
3-phosphate pathway. Hum Mol Genet 2000;9:2223–9.
Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, butler PJG, Lavenir I,
Perisic O, Hawkins PT, Stephens L, Williams RL. The crystal structure of the PX domain from
p40(phox) bound to phosphatidylinositol 3-phosphate. Mol Cell 2001;8:829–39.
Calderwood DA. Talin controls integrin activation. Biochem Soc Trans 2004;32:434–7.
Campbell RB, Liu F, Ross AH. Allosteric activation of PTEN phosphatase by phosphatidylinositol
4,5-bisphosphate. J Biol Chem 2003;278:33617–20.
Carricaburu V, Lamia KA, Lo E, Favereaux L, Payrastre B, Cantley LC, Rameh LE. The
phosphatidylinositol (PI)-5-phosphate 4-kinase type II enzyme controls insulin signaling by regulating
PI-3,4,5-trisphosphate degradation. Proc Natl Acad Sci USA 2003;100:9867–72.
Chua J, Deretic V. Mycobacterium tuberculosis reprograms waves of phosphatidylinositol 3-phosphate on
phagosomal organelles. J Biol Chem 2004; in press.
Clarke JH, Letcher AJ, D’Santos C S, Halstead JR, Irvine RF, Divecha N. Inositol lipids are regulated
during cell cycle progression in the nuclei of murine erythroleukemia cells. Biochem J 2001;357:905–10.

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214212
Dang H, Li Z, Skolnik EY, Fares H. Disease-related myotubularins function in endocytic traffic in
Caenorhabditis elegans. Mol Biol Cell 2004;15:189–96.
De Matteis MA, Godi A. PI-loting membrane traffic Nature. Cell Biol 2004;6:487–92.
Foster FM, Traer CJ, Abraham SM, Fry MJ. The phosphoinositide (PI) 3-kinase family. J Cell Sci
2003;116:3037–40.
Gaullier JM, Simonsen A, D’Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind
PtdIns(3)P. Nature 1998;394:432–3.
Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant N, Gaullier JM, Parton RG, Stenmark H.
Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J 2000;
19:4577–88.
Godi A, Di Campli A, Konstantokopoulos A, Di Tullio G, Alessi AR, Kular GS, Daniele T, Marra P,
Lucocq JM, De Matteis A. FAPPs control Golgi-to-cell-membrane traffic by binding to ARF and
PtdIns(4)P. Nat Cell Biol 2004;6:393–404.
Godi A, Santone I, Pertile P, Marra P, Di Tullio G, Luini A, Corda D, De Matteis MA. ADP-ribosylation
factor regulates spectrin skeleton assembly on the Golgi complex by stimulating phosphatidylinositol
4,5-bisphosphate synthesis. Biochem Soc Trans 1999;27:638–42.
Gozani O, Karuman P, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL,
Villasenor J, Mehrotra B, Chen J, Rao VR, Brugge JS, Ferguson CG, Payrastre B, Myszka DG,
Cantley LC, Wagner G, Divecha N, Prestwich GD, Yuan J. The PHD finger of the chromatin-
associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell 2003;114:99–111.
Hama H, Schnieders EA, Thorner J, Takemoto JY, DeWald DB. Direct involvement of phosphatidy-
linositol 4-phosphate in secretion in the yeast Saccharomyces cerevisiae. J Biol Chem 1999;274:
34294–300.
Heraud JM, Racaud-Sultan C, Gironcel D, Albiges-Rizo C, Giacomini T, Roques S, Martel V, Breton-
Douillon M, Perret B, Chap H. Lipid products of phosphoinositide 3-kinase and phosphatidylinositol
40,50-bisphosphate are both required for ADP-dependent platelet spreading. J Biol Chem 1998;273:
17817–23.
Hirst J, Motley A, Harasaki K, Peak Chew SY, Robinson MS. EpsinR: an ENTH domain-containing
protein that interacts with AP-1. Mol Biol Cell 2003;14:625–41.
Ikonomov OC, Sbrissa D, Shisheva A. Mammalian cell morphology and endocytic membrane
homeostasis require enzymatically active phosphoinositide 50-kinase PIKfyve. J Biol Chem 2001;276:
26141–7.
Irvine RF. Nuclear lipid signalling. Nat Rev Mol Cell Biol 2003;4:349–60.
Itoh T, Takenawa T. Phosphoinositide-binding domains functional units for temporal and spatial
regulation of intracellular signalling. Cell Signal 2002;14:733–43.
Jones DR, Divecha N. Linking lipids to chromatin. Curr Opin Genet Dev 2004;14:196–202.
Jung JY, Kim YW, Kwak JM, Hwang JU, Young J, Schroeder JI, Hwang I, Lee Y. Phosphatidylinositol
3- and 4-phosphate are required for normal stomatal movements. Plant Cell 2002;14:2399–412.
Katakami Y, Katakami N, Janmay PA, Hartwigh JH, Stossel TP. Isolation of the phosphatidylinositol 4-
monophosphate dissociable high-affinity profilin-actin complex. Biochim Biophys Acta
1992;1122:123–35.
Kim SA, Taylor GS, Torgersen KM, Dixon JE. Myotubularin and MTMR2, phosphatidylinositol 3-
phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. J Biol
Chem 2002;277:4526–31.
Lamia KA, Peroni OD, Kim YB, Rameh LE, Kahn BB, Cantley LC. Increased insulin sensitivity and
reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta-/- mice. Mol Cell Biol 2004;24:
5080–7.
Laporte J, Blondeau F, Gansmuller A, Lutz Y, Vonesh JL, Mandel J-L. The PtdIns3P phosphatase
myotubularin is a cytoplasmic protein that also localizes to Rac1-inducible plasma membrane ruffles.
J Cell Sci 2002;115:3105–17.
Lemmon MA. Phosphoinositide recognition domains. Traffic 2003;4:201–13.
Levine TP, Munro S. Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns
4-kinase-dependent and -independent components. Curr Biol 2002;12:695–704.

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214 213
Levine TP, Munro S. The pleckstrin homology domain of oxysterol-binding protein recognises a
determinant specific to Golgi membranes. Curr Biol 1998;8:729–39.
Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M. Insulin induces phosphatidylinositol-3-
phosphate formation through TC10 activation. EMBO J 2003;22:4178–89.
Martel V, Racaud-Sultan C, Dupe S, Marie C, Paulhe F, Galmiche A, Block MR, Albiges-Rizo C.
Conformation, localization, and integrin binding of talin depend on its interaction with
phosphoinositides. J Biol Chem 2001;276:21217–27.
Meijer HJ, Berrie CP, Iurisci C, Divecha N, Musgrave A, Munnik T. Identification of a new
polyphosphoinositide in plants, phosphatidylinositol 5-monophosphate (PtdIns5P), and its accumula-
tion upon osmotic stress. Biochem J 2001;360:491–8.
Merlot S, Meili R, Pagliarini DJ, Maehama T, Dixon JE, Firtel RA. A PTEN-related 5-
phosphatidylinositol phosphatase localized in the Golgi. J Biol Chem 2003;278:39866–73.
Mills IG, Praefcke GJK, Vallis Y, Peter BJ, Olesen LE, Gallop JL, Butler PJG, Evans PR, McMahon HT.
EpsinR: an AP1/clathrin interacting protein involved in vesicle trafficking. J Cell Biol 2003;160:213–22.
Misra S, Hurley JH. Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting
motif, the FYVE domain of Vps27p. Cell 1999;97:657–66.
Misra S, Miller GJ, Hurley JH. Recognizing phosphatidylinositol 3-phosphate. Cell 2001;107:559–62.
Morris JB, Hinchliffe KA, Ciruela A, Letcher AJ, Irvine RF. Thrombin stimulation of platelets causes an
increase in phosphatidylinositol 5-phosphate revealed by mass assay. FEBS Lett 2000;475:57–60.
Muhua L, Adames NR, Murphy MD, Schields CR, Cooper JA. A cytokinesis checkpoint requiring the
yeast homologue of an APC-binding protein. Nature 1998;393:487–91.
Nagata K, Driessens M, Lamarche N, Gorski JL, Hall A. Activation of G1 progression, JNK mitogen-
activated protein kinase, and actin filament assembly by the exchange factor FGD1. J Biol Chem
1998;273:15453–7.
Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ,
Payrastre B. Conversion of PtdIns(4,5)P2 into PtdIns(5)P by the Shigella flexneri effector IpgD
reorganizes host cell morphology. Embo J 2002;21:5069–78.
Niebuhr K, Jouihri N, Allaoui A, Gounon P, Sansonetti PJ, Parsot C. IpgD, a protein secreted by the type
III secretion machinery of Shigella flexneri, is chaperoned by IpgE and implicated in entry focus
formation. Mol Microbiol 2000;38:8–19.
Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. SopB, a protein required for virulence of
Salmonella dublin, is an inositol phosphate phosphatase. Proc Natl Acad Sci USA 1998;95:14057–9.
Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the
multivesicular body. Cell 1998;95:847–58.
Pagliarini DJ, Worby CA, Dixon JE. A PTEN-like phosphatase with a novel substrate specificity. J Biol
Chem 2004;279:38590–6.
Payrastre B, Missy K, Giuriato S, Bodin S, Plantavid M, Gratacap MP. Phosphoinositides key players in
cell signalling, in time and space. Cell Signal 2001;13:377–87.
Pendaries C, Tronchere H, Plantavid M, Payrastre B. Phosphoinositide signaling disorders in human
diseases. FEBS Lett 2003;546:25–31.
Ponting CP. Novel domains in NADPH oxidase subunits, sorting nexins, and PtdIns 3-kinases: binding
partners of SH3 domains? Protein Sci 1996;11:2353–7.
Rameh LE, Tolias KF, Duckworth B, Cantley LC. A new pathway for synthesis of phosphatidylinositol-
4,5-bisphosphate. Nature 1997;390:192–6.
Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem
1999;274:8347–50.
Ridgway ND, Dawson PA, Ho YK, Brown MS, Goldstein JL. Translocation of oxysterol binding protein
to Golgi apparatus triggered by ligand binding. J Cell Biol 1992;116:307–19.
Razzini G, Brancaccio A, Lemmon MA, Guarnieri S, Falasca M. The role of pleckstrin homology domain
in membrane targeting and activation of phospholipase Cb 1. J Biol Chem 2000;275:14873–81.
Sbrissa D, Ikonomov OC, Deeb R, Shisheva A. Phosphatidylinositol 5-phosphate biosynthesis is linked to
PIKfyve and is involved in osmotic response pathway in mammalian cells. J Biol Chem 2002;277:
47276–84.

ARTICLE IN PRESS
C. Pendaries et al. / Advan. Enzyme Regul. 45 (2005) 201–214214
Sbrissa D, Ikonomov OC, Strakova J, Shisheva A. Role for a novel signalling intermediate,
phosphatidylinositol 5-phosphate in insulin-regulated F-actin stress fiber breakdown and GLUT4
translocation. Endocrinol 2004;145:4853–65.
Schaletzky J, Dove SK, Short B, Lorenzo O, Clague MJ, Barr FA. Phosphatidylinositol-5-phosphate
activation and conserved substrate specificity of the Myotubularin phosphatidylinositol 3-phospha-
tases. Curr Biol 2003;13:504–9.
Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase
encoded by yeast VPS34 gene essential for protein sorting. Science 1993;260:88–91.
Seet LF, Hong W. Endofin, an endosomal FYVE domain protein. J Biol Chem 2001;276:42445–54.
Shisheva A. PIKfyve: the road to PtdIns 5-P and PtdIns 3,5-P(2). Cell Biol Int 2001;25:1201–6.
Simon JP, Morimoto T, Bankaitis VA, Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. An essential
role for the phosphatidylinositol transfer protein in the scission of coatomer-coated vesicles from the
trans-Golgi network. Proc Natl Acad Sci USA 1998;95:11181–6.
Stenmark H, Aasland R, Driscoll PC. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS
Lett 2002;513:77–84.
Takenawa T, Itoh T. Phosphoinositides, key molecules for regulation of actin cytoskeletal organization
and membrane traffic from the plasma membrane. Biochim Biophys Acta 2001;1533:190–206.
Taylor GS, Maehama T, Dixon JE. Myotubularin, a protein tyrosine phosphatase mutated in myotubular
myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl
Acad Sci USA 2000;97:8910–5.
Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM, Trimble WS, Meyer T, Finlay BB, Grinstein S.
Elimination of host cell PtdIns(4,5)P2 by bacterial SigD promotes membrane fission during invasion
by salmonella. Nat Cell Biol 2002;4:766–73.
Toker A. Phosphoinositides and signal transduction. Cell Mol Life Sci 2002;59:761–79.
Tolias KF, Rameh LE, Ishihara H, Shibasaki Y, Chen J, Prestwich GD, Cantley LC, Carpenter CL.
Type I phosphatidylinositol-4-phosphate 5-kinases synthesize the novel lipids phosphatidylinositol
3,5-bisphosphate and phosphatidylinositol 5-phosphate. J Biol Chem 1998;273:18040–6.
Tronchere H, Buj-Bello A, Mandel JL, Payrastre B. Implication of phosphoinositide phosphatases in
genetic diseases: the case of myotubularin. Cell Mol Life Sci 2003;60:2084–99.
Tronchere H, Laporte J, Pendaries C, Chaussade C, Liaubet L, Pirola L, Mandel JL, Payrastre B.
Production of phosphatidylinositol 5-phosphate by the phosphoinositide 3-phosphatase myotubularin
in mammalian cells. J Biol Chem 2004;279:7304–12.
Walch-Solimena C, Novick P. The yeast phosphatidylinositol 4-OH kinase pik1 regulates secretion at the
Golgi. Nat Cell Biol 1999;1:523–5.
Walker DM, Urbe S, Dove SK, Tenza D, Raposo G, Clague MJ. Characterization of MTMR3, an
inositol lipid 3-phosphatase with novel substrate specificity. Curr Biol 2001;11:1600–5.
Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin
HL. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the
Golgi. Cell 2003;114:299–310.
Wild AC, Yu JW, Lemmon MA, Blumer KJ. The p21-activated protein kinase-related kinase Cla4 is a
coincidence detector of signaling by Cdc42 and phosphatidylinositol 4-phosphate. J Biol Chem
2004;279:17101–10.
Wu B, Kitagawa K, Yagyu K, Zhang NY, Hattori N, Inagaki C. Phosphatidylinositol and PI-4-
monophosphate recover amyloid b protein-induced inhibition of Cl- -ATPase activity. Life Sci
2002;72:455–63.
Wurmser AE, Gary JD, Emr SD. Phophoinositide 3-kinases and their FYVE domain-containing effectors
as regulators of vacuolar/Lysosomal membrane trafficking pathway. J Biol Chem 1999;269:31552–62.
Yu JW, Lemmon MA. All phox homology (PX) domains from Saccharomyces cerevisiae specifically
recognize phosphatidylinositol 3-phosphate. J Biol Chem 2001;276:44179–84.

185
1To whom correspondence should be addressed: (email [email protected]).
16Biochem. Soc. Symp. 74, 185–196(Printed in Great Britain)© 2007 The Biochemical Society
PtdIns5P: a little phosphoinositide with big functions?S. Coronas, D. Ramel, C. Pendaries, F. Gaits-Iacovoni, H. Tronchère and B. Payrastre1
Inserm Unité 563, CPTP, Département d’Oncogenèse et Signalisation dans les cellules hématopoïétiques, IFR30, Hôpital Purpan, 31059 Toulouse, France
Abstract
Phosphoinositides are minor constituents of cell membranes playing a critical role in the regulation of many cellular functions. Recent discoveries indicate that mutations in several phosphoinositide kinases and phosphatases generate imbalances in the levels of phosphoinositides, thereby leading to the development of human diseases. Although the roles of phosphoinositide 3-kinase products and of PtdIns(4,5)P
2 were largely studied these last years,
the potential role of phosphatidylinositol monophosphates as direct signalling molecules is just emerging. PtdIns5P, the least characterized phosphoinositide, appears to be a new player in cell regulation. This review will summarize the current knowledge on the mechanisms of synthesis and degradation of PtdIns5P as well as its potential roles.
Introduction
Phosphoinositides are relatively low abundant lipids (approx. 10% of total phospholipids), their myo-inositol moiety contains fi ve free hydroxy groups and three of them (positions D-3, D-4 and D-5) can be phosphorylated by specifi c kinases. Thus PtdIns, the most abundant member of the family, can be sequentially phosphorylated to generate the seven polyphosphoinositides. These bioactive lipids exert their role either as precursors of second messengers (such as Ins(1,4,5)P
3 and diacylglycerol) or directly by interacting with proteins
through a set of well defi ned phosphoinositide binding domains [including PH (pleckstrin homology), FYVE, PX (phox homology) or FERM] and modulating their localization, conformation or activity [1].
BY0074-0016.indd 185BY0074-0016.indd 185 10/12/06 7:16:30 PM10/12/06 7:16:30 PM

186 S. Coronas et al.
© 2007 The Biochemical Society
Phosphoinositides can be rapidly synthesized and degraded via different metabolic pathways involving specifi c lipid kinases, phosphatases and phos-pholipases in discrete membrane domains, in organelles or in subnuclear areas. These phosphoinositide kinases and phosphatases regulate very dynamically the interconversions between the different polyphosphoinositides [2]. The turno-ver rate of the monoester phosphates of polyphosphoinositides is rapid and a change in kinase or phosphatase activity can result in quick, and often local, modifi cation of the concentration of these versatile lipids. Based on the use of phosphoinositide binding domains as reporters for a given phosphoinositide [3], recent data suggest that some phosphoinositides are specifi cally enriched in different organelles. For instance PtdIns(4,5)P
2 appears mainly present in the
inner leafl et of the plasma membrane, PtdIns3P in the early endosomes and PtdIns4P in the Golgi. However, these phosphoinositide binding domains have often other determinants for their localization and it might be that these probes preferentially report peculiar pools of a phosphoinositide and do not refl ect the whole picture of its localization [4]. Biochemical evidence indeed suggest that PtdIns(4,5)P
2 is also present in the Golgi and in the nucleus whereas PtdIns3P
and mainly PtdIns4P are also present in the plasma membrane.The critical role of phosphoinositides is emphasized by recent discoveries
indicating that mutations in several phosphoinositide kinases and phosphatases take part in the development of human diseases including cancer, X-linked myotubular myopathy, fl eck corneal dystrophy or Lowe syndrome [5].
Until recently, phosphatidylinositol monophosphates (PtdIns3P, PtdIns4P and PtdIns5P) were considered as intermediate metabolites of the synthesis pathways of polyphosphoinositides. Among the phosphatidylinositol mono-phosphates, PtdIns4P is from far the most abundant isomer (approx. 70%). It was long only considered as a precursor for PtdIns(4,5)P
2, however recent
data suggest that it can contribute to the recruitment of certain proteins such as FAPPs (four-phosphate adaptor proteins) [6] or the clathrin adaptor AP-1 (activator protein 1) to the trans-Golgi network [7]. PtdIns3P, which represents about 15–20% of total phosphatidylinositol monophosphates, is implicated in the regulation of vesicular traffi cking by interacting with a set of FYVE domain-containing proteins involved in vacuolar sorting in yeast [8] and in intracellular traffi cking in mammals [9]. PtdIns5P, the least characterized phos-phoinositide [10], is just now emerging as a potentially important signalling molecule. This lipid was discovered in 1997 [10,11]. It is present in mammalian cells as well as in plant cells where it represents a small proportion of total phos-phatidylinositol monophosphates, commonly less than 10% in resting cells. The fact that it is diffi cult to biochemically separate PtdIns5P from PtdIns4P explains why it has long been ignored. Using an appropriate HPLC technique [10] and a mass assay [12] it is now possible to monitor the level of this phosph-oinositide. Interestingly, the amount of PtdIns5P, classically very low in resting cells, rises upon stimulation of blood platelets [12] or during osmotic-stress in mammalian and plant cells [13–15]. Moreover, a pool of PtdIns5P has been de-tected in the nucleus [16] and the fi rst PtdIns5P interacting domain, called PHD (plant homeodomain) was recently discovered in ING2, a protein involved in chromatin remodelling [17]. Moreover, some bacterial pathogens can also
BY0074-0016.indd 186BY0074-0016.indd 186 10/12/06 7:16:31 PM10/12/06 7:16:31 PM

Functions of PtdIns5P 187
© 2007 The Biochemical Society
manipulate the level of PtdIns5P in the host cell [18] as part of the strategy developed to increase their virulence. Finally, myotubularin, a mammalian phos-phatase mutated in X-linked myotubular myopathy, can transform PtdIns(3,5)P
2
into PtdIns5P suggesting a potential role of these lipids in the aetiology of the disease. Thus evidence is accumulating to suggest that PtdIns5P is playing an important role in the regulation of different cell functions.(Figure 1)
PtdIns(5)P synthesis and degradation
Bacterial enzymes
IpgDShigella fl exneri is a facultative intracellular pathogen responsible for
bacillary dysentery. This bacterial pathogen uses a type III secretion system to inject virulence factors into the host cell to promote its uptake by a mechanism related to macropinocytosis [19]. Among injected proteins, the virulence factor IpgD (invasion plasmid gene D) has two motifs related to the mammalian inositol 4-phosphatase active site. In vitro as well as in vivo, IpgD has a preference for PtdIns(4,5)P
2 and transforms this lipid into PtdIns5P [18]. During infection with
S. fl exneri, we observed a massive hydrolysis of PtdIns(4,5)P2 (approx. 35% of
its amount) and an accumulation of PtdIns5P reaching up to 280 pmol/mg of proteins in HeLa cells [18].
Figure 1 Multiple pools of PtdIns5P for different functions?
The different localizations of PtdIns5P and the associated roles of this lipid are indicated in the Figure.
BY0074-0016.indd 187BY0074-0016.indd 187 10/12/06 7:16:31 PM10/12/06 7:16:31 PM

© 2007 The Biochemical Society
SigD/SopBTwo homologues of IpgD, SopB in Salmonella dublin and SigD in
Salmonella typhimurium (responsible for gastroenteritis) also have two motifs related to the mammalian inositol 4-phosphatase active site [20]. SopB was fi rst demonstrated to induce the depletion of Ins6P and Ins5P and the accumulation of Ins4P, a metabolite involve in chloride ion and water secretion during infection [21]. In vitro, recombinant SopB can hydrolyse PtdIns3P, PtdIns(3,4)P
2 and
PtdIns(3,4,5)P3 [21]. Previously, it was shown that upon infection with S. dublin,
SopB induces a rapid disappearance of host cell PtdIns(4,5)P2 at the bottom of
the invagination which will become the bacteria containing vacuole [22].Whether SopB/SigD are capable of transforming PtdIns(4,5)P
2 into
PtdIns5P is unknown but our recent data suggest that Salmonella induces PtdIns5P production during invasion in a SigD-dependent manner (Masson et al., manuscript submitted).
Thus IpgD, SopB and SigD are able to hydrolyse PtdIns(4,5)P2 which may
explain the fact that they all contribute to actin cytoskeleton and membrane rearrangement during bacterial entry. Whether SopB and SigD, like IpgD, control the PtdIns(4,5)P
2/PtdIns5P ratio remains to be fi rmly established. However,
some of these phosphatases may also have other phosphoinositides and inositol phosphates as substrate, a diversity that could contribute to the different cellular responses against S. fl exneri and Salmonella which have different intracellular lifestyles.
Eukaryotic enzymes
Type I and II PtdIns(4,5)P2 4-phosphatasesRecently, a database search based on the conserved CX
5R phosphatase
motif led to the discovery of two mammalian orthologues of IpgD [23]. Actually, apart from the phosphatase active site, there is no marked homology with IpgD but the characterization of these two novel human phosphatases has revealed that they are PtdIns(4,5)P
2 4-phosphatases (type I and II). They
are able to convert PdtIns(4,5)P2 into PtdIns5P in vitro. Accordingly, the level
of PdtIns(4,5)P2 was reduced by approx. 20% in an inducible stable cell line
expressing the type I enzyme. However, it remains to be established whether these phosphatases transform PdtIns(4,5)P
2 into PtdIns5P in physiological or
pathological in vivo situations. Both enzymes are ubiquitously expressed and colocalize with LAMP1 and EEA1, two late endosomal/lysosomal specifi c markers.
MyotubularinsMTM1 (myotubularin 1) is the prototype of the large myotubularin family
of 3-phosphatases, with a substrate specifi city for PtdIns3P and PtdIns(3,5)P2
[15,24]. MTM1 is mutated in the myotubular myopathy, a severe genetic disease linked to chromosome X, leading to a defect in myotube maturation [25]. MTMR2 and MTMR13 are mutated in two forms of Charcot-Marie-Tooth Disease (CMT4B1 and CMT4B2), a neuropathy with defective myelination and myelin outfolding [26,27]. MTM1 knockout mice reproduce the human
188 S. Coronas et al.
BY0074-0016.indd 188BY0074-0016.indd 188 10/12/06 7:16:31 PM10/12/06 7:16:31 PM

© 2007 The Biochemical Society
phenotype and show a default in maintaining the mature myotubes [28]. In MTMR2 knockout mice, the myelin defect phenotype was also associated with impaired spermatogenesis and azoospermia [29]. About half of the members of the myotubularin family presents a mutation in the consensus CX
5R phosphatase site and are catalytically inactive phosphatases. Several
studies have now described the heterodimerization of active and inactive members of the myotubularin family. The interaction involves the coiled-coil domains of the partners and enhance the enzymatic activity of the phosphatase active myotubularin and/or changes in its subcellular localization [30–33]. Since myotubularins substrates are PtdIns3P and PtdIns(3,5)P
2, it was envisioned
that these phosphatases could regulate the endocytic pathway. Several studies in mammalian cells or with the yeast and Caenorhabditis elegans myotubularin orthologues propose a role for myotubularins as negative regulators of the endocytic traffi cking [34–37]. It is now clear that myotubularins can transform PtdIns(3,5)P
2 into PtdIns5P [15,24,38]. Interestingly, PtdIns5P was found to be
an allosteric activator of MTM1 and MTMR3 lipid phosphatase activity and a component of a positive feedback loop for myotubularin activity [38]. In the same study, it was described that MTM1 assembles into heptameric ring structures. The PH-GRAM domain of myotubularins binds phosphoinositides [35,38] but there are still discussions about its specifi city for PtdIns(3,5)P
2 and/or
PtdIns5P. A convincing recent work of Lorenzo et al. [39], describes PtdIns5P as the prefered binder of the PH-GRAM domain that could be the allosteric binding site for this lipid. A recent large scale RNAi (RNA interference) screen to identify kinases and phosphatases regulating apoptosis has pointed out fi ve members of the myotubularin family (MTMR1, MTMR6–8 and MTMR5/SBF1) as phosphatases regulating cell survival [40]. It is interesting to note that MTMR5 was originally found to interact with SET proteins and induce oncogenic transformation and growth stimulation of B cell precursors [41]. Thus myotubularin could control several intracellular functions like endocytosis or cell survival either by controlling the level of PtdIns3P and PtdIns(3,5)P
2 or by
generating PtdIns5P.
PIKfyvePIKfyve is the mammalian type III PtdIns 5-kinase orthologue of the
yeast Fab1p fi rst described to produce PtdIns(3,5)P2 in vitro and in vivo [42].
It is a dual-specifi city enzyme with a lipid and protein kinase activity [43]. The structure of PIKfyve is highly conserved from prokaryotes to eukary-otes. PIKfyve is ubiquitously expressed and harbours different conserved domains including a Zn2+/PtdIns3P binding FYVE domain and a catalytic ‘phosphoinositide phosphate kinase’ domain [44]. The endogenous protein localizes on intracellular membrane structures of the late endocytic pathway via its FYVE domain interacting with PdtIns3P [14]. Because of its localization and its ability to produce PtdIns(3,5)P
2, several studies involve PIKfyve in
vesicular transport (for an excellent review on PIKfyve and PtdIns(3,5)P2
see [45]). Recently, the human PIKfyve orthologue was cloned and shown to localize in microdomains in early endosomes containing EEA1 and Hrs markers [46]. Mutations in human PIKfyve appear to be responsible for a rare
Functions of PtdIns5P 189
BY0074-0016.indd 189BY0074-0016.indd 189 10/12/06 7:16:31 PM10/12/06 7:16:31 PM

190 S. Coronas et al.
© 2007 The Biochemical Society
autosomal dominant corneal dystrophy, the François-Neetens Mouchetée Fleck Corneal Dystrophy [47]. It has been shown that in vitro the mouse PIKfyve can phosphorylate PtdIns to produce PtdIns5P [48] and that expression of PIKfyve in mammalian cells increases the level of PtdIns5P. There is still some controversy as to whether PIKfyve is able to directly produce PtdIns5P in vitro and in vivo [45]. In vivo, the effect of this kinase may be indirect since one cannot exclude that the PtdIns(3,5)P
2 produced by PIKfyve could be transformed into PtdIns5P
by myotubularins. For a better understanding of the role of PIKfyve, it will be important to determine whether or not this kinase is able to directly produce PtdIns5P in vivo.
Type II PtdInsP kinasesIn 1997, Cantley’s laboratory demonstrated that the type II PtdInsP kinase
is a 4-kinase synthesizing PtdIns(4,5)P2 from PtdIns5P [10]. This study provided
the fi rst evidence of the existence of PtdIns5P in vivo. Three isoforms of type II PtdInsP kinase (α, β and γ) exist in mammalian cells. The recombinant type II PtdInsP kinase α is now used to monitor the amount of PtdIns5P in cell extracts by a sensitive mass assay [12]. Although a recent study suggests that the type II PtdInsP kinase β is not very effi cient in transforming basal PtdIns5P into PtdIns(4,5)P
2 [49], it seems to perform the conversion when the level of
PtdIns5P has increased. In normal cells, the amount of PtdIns(4,5)P2 synthesized
from PtdIns5P is probably minor compared to the amount formed by other pathways (i.e. via PtdIns4P 5-kinase). As discussed below, these kinases appears to play an important function in cell regulation [10].
PLIPPLIP, or PTEN (phosphatase and tensin homologue deleted on chromosome
10)-like phosphatase, is so far the only mammalian lipid phosphatase described to specifi cally hydrolyze PtdIns5P. The enzyme was originally found in a Dictyostelium database genomic search for additional PTEN homologues [50] and it defi nes a new family with orthologues in eukaryotes and prokaryotes [51]. Apart from the phosphatase signature, PLIP has only a weak overall similarity to PTEN and harbours a transmembrane domain in its N-terminus. Interestingly, in contrast to the 3-phosphatase activity of PTEN on PtdIns(3,4)P
2 and PtdIns(3,4,5)P
3, PLIP exhibits a highly specifi c
5-phosphatase activity against PtdIns5P in vitro [50]. The murine orthologue of PLIP shows the same high specifi city towards PtdIns5P, however its capacity to modulate PtdIns5P in vivo is still not proven [51]. In Dictyostelium, overexpressed PLIP localizes in the Golgi, suggesting the presence of a co-localized PtdIns5P pool. The knockout of PLIP in Dictyostelium indicates that this phosphatase is required for cell aggregation, however the link between a defect in the Golgi system and the aggregation phenotype was not established [50]. Thus whether this PTEN related phosphatase is regulating specifi c mechanisms via PtdIns5P degradation remains to be demonstrated. Table 1.
BY0074-0016.indd 190BY0074-0016.indd 190 10/12/06 7:16:32 PM10/12/06 7:16:32 PM
Functions of PtdIns5P 191
© 2007 The Biochemical Society
Tab
le 1
En
zym
es
invo
lved
in
Ptd
Ins5
P m
eta
bo
lism
.
Enzy
me
Act
ivity
Su
bstr
ate
Org
anis
m
Kno
ckou
t ph
enot
ype
Ass
ocia
ted
dise
ases
R
efer
ence
s
Type
II P
tdIn
sP k
inas
es
4-K
inas
e
PtdI
ns5P
Ca
enor
habd
itis
M
ice:
incr
ease
d in
sulin
no
t de
scri
bed
[56,
57]
( α,β
,γ)
eleg
ans,
Dro
soph
ila
sens
itivi
ty, r
educ
edPI
5P4K
II
m
elan
ogas
ter,
gr
owth
rat
es a
nd lo
wer
fat
m
amm
als
PIK
fyve
(PIP
KIII
) 5-
Kin
ase
PtdI
ns
Con
serv
ed fr
om
not
desc
ribe
d
Fran
çois
-Nee
tens
Mou
chet
ée
[44,
47]
PtdI
ns3P
ye
ast
to m
amm
als
Fl
eck
Cor
neal
Dys
trop
hy
MT
M1
3-Ph
osph
atas
e Pt
dIns
(3,5
)P2
Con
serv
ed fr
om
Mic
e: g
ener
aliz
ed a
nd
X-li
nked
myo
tubu
lar
myo
path
y [2
4,28
]
Pt
dIns
3P
yeas
t to
mam
mal
s pr
ogre
ssiv
e m
yopa
thy
star
ting
arou
nd 4
wee
ks o
f
ag
e, w
ith a
myo
trop
hy a
nd
accu
mul
atio
n of
cen
tral
nu
clei
in s
kele
tal m
uscl
e
fi b
res,
lead
ing
to d
eath
aft
er
6 –1
4 w
eeks
PtdI
ns(4
,5)P
2
4-Ph
osph
atas
e P
tdIn
s(4,
5)P 2
Hum
an
not
desc
ribe
d
not
desc
ribe
d [2
3]4-
Phos
phat
ase
Si
gD/S
opB
4-Ph
osph
atas
e Pt
dIns
(4,5
)P2
Salm
onel
la d
ublin
gast
roen
teri
tis
[20,
21,2
2]
& ty
phim
uriu
m
Ip
gD
4-Ph
osph
atas
e Pt
dIns
(4,5
)P2
Shig
ella
fl ex
neri
ba
cilla
ry d
ysen
tery
[1
8,53
]PL
IP
5-Ph
osph
atas
e Pt
dIns
5P
Hig
hly
cons
erve
d
Dict
yost
eliu
m: d
efec
t in
no
t de
scri
bed
[50,
51]
fr
om P
rotis
ta t
o
aggr
egat
ion
of c
ells
m
amm
als
BY0074-0016.indd 191BY0074-0016.indd 191 10/12/06 7:16:32 PM10/12/06 7:16:32 PM

192 S. Coronas et al.
© 2007 The Biochemical Society
Functions of PtdIns5P
Role in bacterial invasion
Interestingly, several microbial pathogens exploit the phosphoinositide metabolism of host cells to promote their entry and develop their virulence [52]. The formation of Salmonella or S. fl exneri entry structures results from the concerted action of injected bacterial proteins and components of the host cell. The virulence factor IpgD, delivered into nonphagocytic cells by the type III secretion system of the pathogen S. fl exneri is a phosphoinositide 4-phosphatase transforming a signifi cant part of PtdIns(4,5)P
2 into PtdIns5P
[18]. This transformation is rapid and occurs primarily at the entry foci of bacteria [53]. While this local breakdown of PtdIns(4,5)P
2, a lipid known to
regulate actin cytoskeleton dynamics [2], results in dramatic plasma membrane and cytoskeleton rearrangements [18], PtdIns5P production activates specifi c host cell signalling pathways. Indeed, we recently showed that PtdIns5P plays a key role in class IA PI3K (phosphoinositide 3-kinase)/Akt (protein kinase B) activation in the host cell, a mechanism particularly important to regulate the survival of infected cells in order to maintain effi cient bacterial replication and colonization [53]. Ectopic expression of IpgD in various cell types, but not of its inactive mutant, or addition of short chain penetrating PtdIns5P are suffi cient to induce Akt phosphorylation. Conversely, sequestration of PtdIns5P or reduction of its level strongly decreases Akt phosphorylation in infected cells or in IpgD expressing cells. Thus, S. fl exneri parasitism is shedding light on a new mechanism of PI3K/Akt activation via PtdIns5P production that play an important role in host cell responses such as survival. The molecular mechanism responsible for PtdIns5P-induced class IA PI3K activation involves a tyrosine phosphorylation process and is currently under characterization.
Two homologues of IpgD, SopB in S. dublin and SigD in Salmonella typhimurium are also inositol polyphosphate-phosphatases [20]. Recent data suggest that Salmonella induces PtdIns5P production during invasion in a SigD-dependent manner (Masson et al, manuscript submitted) and is required for Akt activation in Hela cells infected with Salmonella [54,55]. Thus it is tempting to propose that, as in the case of S. fl exneri infection, Salmonella induces the PI3K/Akt survival pathway via PtdIns5P production.
In agreement with the results obtained in the S. fl exneri infection model, overexpression of the type II PtdInsP kinase β [which transforms PtdIns5P into PtdIns(4,5)P
2] reduces the level of PtdIns(3,4,5)P
3 and, in turn, decreases Akt
activation under insulin stimulation [56]. Accordingly, type II PtdInsP kinase β knockout mice show a hypersensitivity to insulin [57]. Akt activation induced by insulin was greatly increased in skeletal muscle and liver from the knockout mice. These results suggest a general regulatory role of PtdIns5P upstream of Akt. In this context, type II PtdInsP kinase β may be a sensor modulating the level of PtdIns5P and in turn the PI3K/Akt signalling pathway.
Nuclear functions
The fi rst biochemical evidence for a potential role of PtdIns5P in the nucleus came from the observation that the amount of this phosphoinositide (as well
BY0074-0016.indd 192BY0074-0016.indd 192 10/12/06 7:16:32 PM10/12/06 7:16:32 PM

Functions of PtdIns5P 193
© 2007 The Biochemical Society
as others) increased by 20-fold in the nucleus of murine erythroleukaemia cells during the G
1-phase of the cell cycle [16]. Moreover, the type II PtdInsP kinase
β has also been found in the nucleus [58]. More recently, the nuclear tumor suppressor ING2 (inhibitor of growth 2) has been shown to be a PtdIns5P receptor via its PHD domain [17]. PtdIns5P binding to the PHD of ING2 regulates the ability of ING2 to induce p53-dependent apoptotic pathways as part of the nuclear response to DNA damage [17]. The PHD domain is organized in a zinc fi nger structure and is related to the FYVE and RING domains. It is mainly found in nuclear proteins including chromatin regulators as ACF, acetyltransferase like CBP/300 and RAG2 [59] suggesting a role for PtdIns5P in chromatin remodelling and transcription regulation. The potential role of PtdIns5P in transcription is also suggested by its interaction with the PH domain of a general transcription factor, TFIIH, a RNA polymerase II component. PtdIns5P may compete with the transactivator VP16 binding site, also located on the PH domain of TFIIH [60]. Finally, by imaging PtdIns5P with a recombinant biotinylated PHD probe we observed that, after 1 h of infection by S. fl exneri, a signifi cant pool of PtdIns5P is present in the nucleus of infected cells [53]. Altogether these results suggest a role for PtdIns5P in the nucleus, however its exact function in this cell compartment remains to be established and the next few years should bring very exciting information in this area.
Vesicular transport
Although a direct implication of PtdIns5P in vesicular traffi cking has not been clearly demonstrated so far, several studies suggest the involvement of this lipid.
In CHO (Chinese hamster ovary) cells stably expressing the insulin receptor and in 3T3-L1 adipocytes, PdtIns5P production under insulin stimulation results in GLUT4 (glucose transporter 4) vesicle translocation to the cell surface. Interestingly, the same effect was observed when PtdIns5P was microinjected or when PIKfyve was overexpressed. Conversely, sequestration of intracellular PtdIns5P by expression of the ING2 PHD domain abrogates GLUT4 vesicle translocation [61]. This effect seems to be independent of PI3K but remains ill-defi ned.
As mentioned above, the 5-phosphatase PLIP, known to hydrolyse PtdIns5P, localizes at the Golgi membrane [50] suggesting that a pool of PtdIns5P could exist in this organelle. However, in Dictyostelium, the knockout of PLIP does not lead to an evident implication of Golgi membrane traffi cking [50]. Interestingly, overexpression of MTMR2, leads to an inhibition of the EGF (epidermal growth factor) receptor traffi cking from late endosomes to lysosomes under EGF stimulation and induces a large endosomal vacuolization. It has been suggested that this effect implicates an interaction between the GRAM domain of MTMR2 and PdtIns(3,5)P
2 [35]. However, the GRAM domain has recently
been shown to bind PtdIns5P which could play a role in this process.Overexpression of the recently cloned human PtdIns(4,5)P
2 4-phosphatases
in HeLa cells enhanced the degradation rate of EGF receptors [23]. In cells infected with S. dublin, Dukes et al. also describe that SopB/SigD, the orthologue of IpgD, can inhibit EGF receptor degradation [62], but it is not clear which
BY0074-0016.indd 193BY0074-0016.indd 193 10/12/06 7:16:33 PM10/12/06 7:16:33 PM

194 S. Coronas et al.
© 2007 The Biochemical Society
lipid is involved in this effect. Finally, a recent study shows that an important regulator of the traffi cking of the EGF receptor, SNX5, whose overexpression leads to EGF receptor degradation, specifi cally binds PtdIns5P via its PX domain [63]. The identifi cation of PtdIns5P interacting proteins may help to better understand its role in vesicular traffi cking.
Cytoskeleton organization
A recent study suggests that PtdIns5P is involved in insulin-induced actin stress fi bre disassembly. Microinjection of PtdIns5P mimics the disassembly of actin stress fi bres evoked by insulin in cells, whereas sequestration of PtdIns5P, by overexpression of a tandem of PHD domain, blocked this actin disassembly. This effect of PtdIns5P appears independent of PI3K [61]. Accordingly, we also observed that ectopic expression of IpgD or micro-injection of this phosphatase has a dramatic effect on actin cytoskeleton organization with loss of actin fi bres. In this case, in addition to PtdIns5P production, the decrease in PtdIns(4,5)P
2
level may strongly cooperate to effi ciently reorganize the actin fi lament system [18]. The role of PtdIns5P in actin cytoskeleton organization is still unclear and additional investigation of the molecular mechanisms is required before this lipid might increment the list of cytoskeleton regulators.
Conclusion
Evidence reviewed in the present chapter strongly suggests that PtdIns5P is a new important player in cell regulation. The strategy developed by a microbial pathogen to precisely manipulate the level of this lipid has provided interesting insights into the role of PtdIns5P. Moreover, several exiting ongoing studies concerning the nuclear pool of PtdIns5P and its receptors as well as the potential roles for this phosphoinositide in vesicular traffi cking and cytoskeleton organization should provide new lipid-mediated mechanisms of cell regulation.
The authors thank Dr M.P. Plantavid, Dr C. Racaud-Sultan, Dr M.P. Gratacap and Dr
S. Manenti for critical reading of the manuscript. This work was supported by grants
from Inserm, Association pour la Recherche contre le Cancer, la Ligue Nationale
Contre le Cancer and Association Française contre les Myopathies.
References1. Itoh, T. and Takenawa, T.(2002) Cell Signalling 14, 733–7432. Payrastre, B., Missy, K., Giuriato, S., Bodin, S., Plantavid, M. and Gratacap, M.(2001)
Cell Signalling 13, 377–3873. Balla, T., Bondeva, T. and Varnai, P.(2000) Trends Pharmacol. Sci. 21, 238–2414. Carlton, J.G. and Cullen, P.J.(2005) Trends Cell. Biol. 15, 540–5475. Pendaries, C., Tronchere, H., Plantavid, M. and Payrastre, B.(2003) FEBS Lett. 546, 25–316. Godi, A., Di Campli, A., Konstantakopoulos, A., Di Tullio, G., Alessi, D.R., Kular, G.S.,
Daniele, T., Marra, P., Lucocq, J.M. and De Matteis, M.A.(2004) Nat. Cell Biol. 6, 393–4047. Wang, Y.J., Wang, J., Sun, H.Q., Martinez, M., Sun, Y.X., Macia, E., Kirchhausen,
T., Albanesi, J.P., Roth, M.G. and Yin, H.L.(2003) Cell 114, 299–3108. Gaullier, J.M., Simonsen, A., D’Arrigo, A., Bremnes, B., Stenmark, H. and Aasland,
R.(1998) Nature 394, 432–433
BY0074-0016.indd 194BY0074-0016.indd 194 10/12/06 7:16:33 PM10/12/06 7:16:33 PM

Functions of PtdIns5P 195
© 2007 The Biochemical Society
9. Wurmser, A.E., Gary, J.D. and Emr, S.D.(1999) J. Biol. Chem. 274, 9129–913210. Rameh, L.E., Tolias, K.F., Duckworth, B.C. and Cantley, L.C.(1997) Nature 390, 192–19611. Tolias, K.F., Rameh, L.E., Ishihara, H., Shibasaki, Y., Chen, J., Prestwich, G.D., Cantley,
L.C. and Carpenter, C.L.(1998) J. Biol. Chem. 273, 18040–1804612. Morris, J.B., Hinchliffe, K.A., Ciruela, A., Letcher, A.J. and Irvine, R.F.(2000) FEBS Lett.
475, 57–6013. Meijer, H.J., Berrie, C.P., Iurisci, C., Divecha, N., Musgrave, A. and Munnik, T.(2001)
Biochem. J. 360, 491–49814. Sbrissa, D., Ikonomov, O.C., Deeb, R. and Shisheva, A.(2002) J. Biol. Chem. 277,
47276–4728415. Tronchere, H., Laporte, J., Pendaries, C., Chaussade, C., Liaubet, L., Pirola, L., Mandel,
J.L. and Payrastre, B.(2004) J. Biol. Chem. 279, 7304–731216. Clarke, J.H., Letcher, A.J., D’Santos C, S., Halstead, J.R., Irvine, R.F. and Divecha,
N.(2001) Biochem. J. 357, 905–91017. Gozani, O., Karuman, P., Jones, D.R., Ivanov, D., Cha, J., Lugovskoy, A.A., Baird, C.L.,
Zhu, H., Field, S.J., Lessnick, S.L. et al. (2003) Cell 114, 99–11118. Niebuhr, K., Giuriato, S., Pedron, T., Philpott, D.J., Gaits, F., Sable, J., Sheetz, M.P., Parsot,
C., Sansonetti, P.J. and Payrastre, B.(2002) EMBO J. 21, 5069–507819. Cossart, P. and Sansonetti, P.J.(2004) Science 304, 242–24820. Marcus, S.L., Wenk, M.R., Steele-Mortimer, O. and Finlay, B.B.(2001) FEBS Lett. 494,
201–20721. Norris, F.A., Wilson, M.P., Wallis, T.S., Galyov, E.E. and Majerus, P.W.(1998) Proc. Natl.
Acad. Sci. U.S.A. 95, 14057–1405922. Terebiznik, M.R., Vieira, O.V., Marcus, S.L., Slade, A., Yip, C.M., Trimble, W.S., Meyer,
T., Finlay, B.B. and Grinstein, S.(2002) Nat. Cell Biol. 4, 766–77323. Ungewickell, A., Hugge, C., Kisseleva, M., Chang, S.C., Zou, J., Feng, Y., Galyov, E.E.,
Wilson, M. and Majerus, P.W.(2005) Proc. Natl. Acad. Sci. U.S.A. 102, 18854–1885924. Tronchere, H., Buj-Bello, A., Mandel, J.L. and Payrastre, B.(2003) Cell Mol. Life Sci. 60,
2084–209925. Laporte, J., Hu, L.J., Kretz, C., Mandel, J.L., Kioschis, P., Coy, J.F., Klauck, S.M., Poustka,
A. and Dahl, N.(1996) Nat. Genet. 13, 175–18226. Bolino, A., Muglia, M., Conforti, F.L., LeGuern, E., Salih, M.A., Georgiou, D.M.,
Christodoulou, K., Hausmanowa-Petrusewicz, I., Mandich, P., Schenone, A. et al. (2000) Nat. Genet. 25, 17–19
27. Senderek, J., Bergmann, C., Weber, S., Ketelsen, U.P., Schorle, H., Rudnik-Schoneborn, S., Buttner, R., Buchheim, E. and Zerres, K.(2003) Hum. Mol. Genet. 12, 349–356
28. Buj-Bello, A., Laugel, V., Messaddeq, N., Zahreddine, H., Laporte, J., Pellissier, J.F. and Mandel, J.L.(2002) Proc. Natl. Acad. Sci. U.S.A. 99, 15060–15065
29. Bolino, A., Bolis, A., Previtali, S.C., Dina, G., Bussini, S., Dati, G., Amadio, S., Del Carro, U., Mruk, D.D., Feltri, M.L., Cheng, C.Y., Quattrini, A. and Wrabetz, L.(2004) J. Cell. Biol. 167, 711–721
30. Mochizuki, Y. and Majerus, P.W.(2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9768–977331. Kim, S.A., Vacratsis, P.O., Firestein, R., Cleary, M.L. and Dixon, J.E.(2003) Proc. Natl.
Acad. Sci. U.S.A. 100, 4492–449732. Nandurkar, H.H., Layton, M., Laporte, J., Selan, C., Corcoran, L., Caldwell, K.K.,
Mochizuki, Y., Majerus, P.W. and Mitchell, C.A.(2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8660–8665
33. Robinson, F.L. and Dixon, J.E.(2005) J. Biol. Chem. 280, 31699–3170734. Chaussade, C., Pirola, L., Bonnafous, S., Blondeau, F., Brenz-Verca, S., Tronchere, H.,
Portis, F., Rusconi, S., Payrastre, B., Laporte, J. and Van Obberghen, E.(2003) Mol. Endocrinol. 17, 2448–2460
35. Tsujita, K., Itoh, T., Ijuin, T., Yamamoto, A., Shisheva, A., Laporte, J. and Takenawa, T.(2004) J. Biol. Chem. 279, 13817–13824
36. Parrish, W.R., Stefan, C.J. and Emr, S.D.(2004) Mol. Biol. Cell 15, 3567–357937. Xue, Y., Fares, H., Grant, B., Li, Z., Rose, A.M., Clark, S.G. and Skolnik, E.Y.(2003) J. Biol.
Chem. 278, 34380–3438638. Schaletzky, J., Dove, S.K., Short, B., Lorenzo, O., Clague, M.J. and Barr, F.A.(2003) Curr.
Biol. 13, 504–50939. Lorenzo, O., Urbe, S. and Clague, M.J.(2005) J. Cell Sci. 118, 2005–2012
BY0074-0016.indd 195BY0074-0016.indd 195 10/12/06 7:16:33 PM10/12/06 7:16:33 PM

196 S. Coronas et al.
© 2007 The Biochemical Society
40. MacKeigan, J.P., Murphy, L.O. and Blenis, J.(2005) Nat. Cell Biol. 7, 591–60041. De Vivo, I., Cui, X., Domen, J. and Cleary, M.L.(1998) Proc. Natl. Acad. Sci. U.S.A. 95,
9471–947642. Shisheva, A., Sbrissa, D. and Ikonomov, O.(1999) Mol. Cell Biol. 19, 623–63443. Sbrissa, D., Ikonomov, O.C. and Shisheva, A.(2000) Biochemistry 39, 15980–1598944. Shisheva, A.(2001) Cell Biol. Int. 25, 1201–120645. Michell, R.H., Heath, V.L., Lemmon, M.A. and Dove, S.K.(2006) Trends Biochem. Sci. 31,
52–6346. Cabezas, A., Pattni, K. and Stenmark, H.(2006) Gene 371, 34–4147. Li, S., Tiab, L., Jiao, X., Munier, F.L., Zografos, L., Frueh, B.E., Sergeev, Y., Smith, J.,
Rubin, B., Meallet, M.A., Forster, R.K., Hejtmancik, J.F. and Schorderet, D.F.(2005) Am. J. Hum. Genet. 77, 54–63
48. Sbrissa, D., Ikonomov, O.C. and Shisheva, A.(1999) J. Biol. Chem. 274, 21589–2159749. Roberts, H.F., Clarke, J.H., Letcher, A.J., Irvine, R.F. and Hinchliffe, K.A.(2005) FEBS
Lett. 579, 2868–287250. Merlot, S., Meili, R., Pagliarini, D.J., Maehama, T., Dixon, J.E. and Firtel, R.A.(2003) J. Biol.
Chem. 278, 39866–3987351. Pagliarini, D.J., Worby, C.A. and Dixon, J.E.(2004) J. Biol. Chem. 279, 38590–3859652. Pizarro-Cerda, J. and Cossart, P.(2004) Nat. Cell Biol. 6, 1026–103353. Pendaries, C., Tronchere, H., Arbibe, L., Mounier, J., Gozani, O., Cantley, L., Fry, M.J.,
Gaits-Iacovoni, F., Sansonetti, P.J. and Payrastre, B.(2006) EMBO J.54. Steele-Mortimer, O., Knodler, L.A., Marcus, S.L., Scheid, M.P., Goh, B., Pfeifer, C.G.,
Duronio, V. and Finlay, B.B.(2000) J. Biol. Chem. 275, 37718–3772455. Knodler, L.A., Finlay, B.B. and Steele-Mortimer, O.(2005) J. Biol. Chem. 280, 9058–906456. Carricaburu, V., Lamia, K.A., Lo, E., Favereaux, L., Payrastre, B., Cantley, L.C. and
Rameh, L.E.(2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9867–987257. Lamia, K.A., Peroni, O.D., Kim, Y.B., Rameh, L.E., Kahn, B.B. and Cantley, L.C.(2004)
Mol. Cell Biol. 24, 5080–508758. Ciruela, A., Hinchliffe, K.A., Divecha, N. and Irvine, R.F.(2000) Biochem. J. 346, 587–59159. Elkin, S.K., Ivanov, D., Ewalt, M., Ferguson, C.G., Hyberts, S.G., Sun, Z.Y., Prestwich,
G.D., Yuan, J., Wagner, G., Oettinger, M.A. and Gozani, O.P.(2005) J. Biol. Chem. 280, 28701–28710
60. Di Lello, P., Nguyen, B.D., Jones, T.N., Potempa, K., Kobor, M.S., Legault, P. and Omichinski, J.G.(2005) Biochemistry 44, 7678–7686
61. Sbrissa, D., Ikonomov, O.C., Strakova, J. and Shisheva, A.(2004) Endocrinology 145, 4853–4865
62. Dukes, J.D., Lee, H., Hagen, R., Reaves, B.J., Layton, A.N., Galyov, E.E. and Whitley, P.(2006) Biochem. J. 395, 239–247
63. Liu, H., Liu, Z.Q., Chen, C.X., Magill, S., Jiang, Y. and Liu, Y.J.(2006) Biochem. Biophys. Res. Commun. 342, 537–546
BY0074-0016.indd 196BY0074-0016.indd 196 10/12/06 7:16:34 PM10/12/06 7:16:34 PM