Corrigé des activités « Alignement de séquences » ACTIVITE ...

Bioinformatique: alignement
de séquences
Céline Brochier-Armanet
Université Claude Bernard, Lyon 1Laboratoire de Biométrie et Biologie évolutive (UMR 5558)

• Problème NP-complet
• Requière l’utilisation d’heuristiques
• > 100 heuristiques disponibles => solutions différentes
• Le choix

Alignement global vs alignement local
Séquence 1
A1 B1 C1 A'1
Séquence 2
A2 C2 B2
A1 B1 C1 A'1
A2 C2 B2
A2
A1
B1
C1
A'1
A2
C2
B2
Smith &
Waterman
BLAST
Needleman
& Wunsch
FASTA
A1 B1 C1 A'1
A2 C2 B2

Alignement de deux séquences vs alignement
multiple
Séquence 1
Séquence 2
Séquence 1
Séquence 2
Séquence 3
Séquence 4

Représentation
• Les résidus (nucléotides, acides aminés) sont superposés de façon à
maximiser la similarité entre les séquences (selon les critères choisis) :
G T T A A G G C G – G G A A A
G T T – – – G C G A G G A C A
* * * * * * * * * *
• Il existe deux sortes de mutations :
– Substitutions (mismatches).
– Insertions et délétions (indels ou gaps).

Quel est le bon alignement ?
• Doit maximiser la « similarité » entre les séquences
Évolution : seront alignés ensemble des résidus homologues,cad descendant d’un même résidu ancestral.
Structure : seront alignés ensembles des résidus occupantune position équivalente des résidus dans des structures 2Dou 3D.
Fonction : seront alignés ensembles des résidus ayant desfonctions similaires.
G T T A C G A
G T T - G G A
* * * * *
G T T A C G A
G T T G - G A
* * * * *
G T T A C - G A
G T T - - G G A
* * * * *
ou
Critères
d’évaluation
/
comparaison
des alignements

Matrices de points (dot-plot)
• Comparaison visuelle de deux séquences :– Une suite de points en
diagonale indique une similarité locale.
– Méthode simple et rapide :
• Algorithme en O(nm).
– Visualisation des répéti-tions directes ou inversées.
– Pas d’alignement global.
– Pas de score associé.
CTGCACGTATTA
C T T G C A C G T A T

Élimination du bruit de fond
• Filtrage en affichant un point uniquement si plusieurs résidus successifs correspondent :– Exemple des hémoglobines et humaines :
Identités = 3/10 Identités = 5/10

Fonction de score de similarité
G T T A A G G C G – G G A A A
G T T – – – G C G A G G A C A
* * * * * * * * * *
Score = Score Identités + Score Différences
Identité = +1
Substitution = 0
Gap = -1Score = 10 - 4 = 6

Modèle d’évolution (ADN)
A C
G T
P(transition) > P(transversion)
G T T A C G A
G T T G - G A
* * * : * *
G T T A C G A
G T T - G G A
* * * * *
>

Matrice de substitution (ADN)
G T T A C G A G T T A C G A
G T T - G G A < G T T G - G A
1 1 1 -1 0 1 1 1 1 1 .5 -1 1 1
Score = 4 Score = 4.5
(A, A) = 1.0
(A, G) = 0.5
(A, –) = -1
A C G T
A
C
G
T
1
1
1
1
0
0 0
0.5
0.5
0

Le cas des acides aminés
• Plus difficile à modéliser que celui des nucléotides :– Un acide aminé peut être remplacé par un autre de différentes
façons (code génétique).
• Asp (GAC) Tyr (UAC, UAU) 1 ou 2 mutations

Le cas des acides aminés
• Plus difficile à modéliser que celui des nucléotides :– Un acide aminé peut être remplacé par un autre de différentes
façons (code génétique).
– Le nombre de substitutions requises pour passer d’un acide aminé à un autre diffère.
Asp (GAC, GAU) Tyr (UAC, UAU) 1 mutation
Asp (GAC, GAU) Cys (UGC, UGU) 2 mutations
Asp (GAC, GAU) Trp (UGG) 3 mutations

Le cas des acides aminés
• Plus difficile à modéliser que celui des nucléotides :– Un acide aminé peut être remplacé par un autre de différentes
façons (code génétique).
– Le nombre de substitutions requises pour passer d’un acide aminé à un autre diffère.
– La probabilité des substitutions au niveau nucléotidique diffère :
P(AAUAsn|GAUAsp) > P(AAUAsn|CAUHis)

Le cas des acides aminés
• Plus difficile à modéliser que celui des nucléotides :– Un acide aminé peut être remplacé par un autre de différentes
façons (code génétique).
– Le nombre de substitutions requises pour passer d’un acide aminé à un autre diffère.
– La probabilité des substitutions au niveau nucléotidique diffère :
– Certaines substitutions peuvent avoir plus ou moins d’effet sur la fonction des protéines.
• Acidité, hydrophobicité, structure des protéines, etc.
Val
H C COOH
C CH3
CH3
NH2
H
Substitutions
conservatrices
Ile
CH2
CH3
H C COOH
C CH3
NH2
H

Modèles d’évolution (prot.)
• Mesure des fréquences de substitution dans des alignements de protéines homologues :– Matrices basées sur des arbres construits en utilisant
le maximum de parcimonie :• PAM (Dayhoff et al., 1978).
• JTT (Jones et al., 1992).
– Matrices basées sur des arbres construits en utilisant le maximum de vraisemblance :
• WAG (Whelan et Goldman, 2001).
– Matrices basées sur des comparaisons par paires utili-sant des alignements locaux :
• BLOSUM (Henikoff et Henikoff, 1992).

Matrice de substitution (prot.)
M R - D W G F
M R W D - G F
* * * * *
M R D W - G F
M R - W D G F
* * * * *>
(D, D) ≠ (W, W)
Certains acides aminés
sont moins facilement
substituables
D E F G
D
E
F
G
W
W
6
2
-3
-1
-4
5
-21-3
-2
-3
6-3
6
11

Matrices de Dayoff ou PAM
– PAM = Percentage of Accepted point Mutation
– Probabilité d'observer la mutation X->Y après un temps évolutif donné. Basé sur alignement de protéines conservées à + de 85%.
Chaque case représente la probabilité de voir ces deux résidus remplacés l'un par l'autre dans un alignement. (matrice lod-score, de "log-odds" ou "log des chances").
•Un exemple de lod-score est: S = log (Fij / (Fi x Fj)) Où Fij est la fréquence de remplacement du résidu i par j, et Fi et Fj sont les fréquences respectives des résidus i et j.
•Dans cette matrice de similitude, plus la valeur est négative, plus la probabilité est faible, plus le remplacement est rare.
•La table est valable pour une certaine distance évolutive.
•La distance est mesurée en PAM: nbre de mutations ponctuelles par 100 aa.
•2 Séquences séparées par une unité PAM: 1 mutation par 100 aa.
•Les valeurs sont déterminées initialement pour des protéines séparées de 6 à 100 PAM, puis extrapolées pour 150, 250 PAM, etc.
•Pour des protéines éloignées, on ne pourrait pas directement extrapoler à partir de valeurs tirées par ex. de PAM 10, car la nature des mutations change avec la distance évolutive. Le code génétique, par exemple, influence les mutations permises sur une courte durée, mais pas sur une longue durée.
Margaret Dayhoff, 1978

A B C D E F G H I K L M N P Q R S T V W Y Z
0.4 0.0 -0.4 0.0 0.0 -0.8 0.2 -0.2 -0.2 -0.2 -0.4 -0.2 0.0 0.2 0.0 -0.4 0.2 0.2 0.0 -1.2 -0.6 0.0 A
0.5 -0.9 0.6 0.4 -1.0 0.1 0.3 -0.4 0.1 -0.7 -0.5 0.4 -0.2 0.3 -0.1 0.1 0.0 -0.4 -1.1 -0.6 0.4 B
2.4 -1.0 -1.0 -0.8 -0.6 -0.6 -0.4 -1.0 -1.2 -1.0 -0.8 -0.6 -1.0 -0.8 0.0 -0.4 -0.4 -1.6 0.0 -1.0 C
0.8 0.6 -1.2 0.2 0.2 -0.4 0.0 -0.8 -0.6 0.4 -0.2 0.4 -0.2 0.0 0.0 -0.4 -1.4 -0.8 0.5 D
0.8 -1.0 0.0 0.2 -0.4 0.0 -0.6 -0.4 0.2 -0.2 0.4 -0.2 0.0 0.0 -0.4 -1.4 -0.8 0.6 E
1.8 -1.0 -0.4 0.2 -1.0 0.4 0.0 -0.8 -1.0 -1.0 -0.8 -0.6 -0.6 -0.2 0.0 1.4 -1.0 F
1.0 -0.4 -0.6 -0.4 -0.8 -0.6 0.0 -0.2 -0.2 -0.6 0.2 0.0 -0.2 -1.4 -1.0 -0.1 G
1.2 -0.4 0.0 -0.4 -0.4 0.4 0.0 0.6 0.4 -0.2 -0.2 -0.4 -0.6 0.0 -0.4 H
1.0 -0.4 0.4 0.4 -0.4 -0.4 -0.4 -0.4 -0.2 0.0 0.8 -1.0 -0.2 -0.4 I
1.0 -0.6 0.0 0.2 -0.2 0.2 0.6 0.0 0.0 -0.4 -0.6 -0.8 0.1 K
1.2 0.8 -0.6 -0.6 -0.4 -0.6 -0.6 -0.4 0.4 -0.4 -0.2 -0.5 L
1.2 -0.4 -0.4 -0.2 0.0 -0.4 -0.2 0.4 -0.8 -0.4 -0.3 M
0.4 -0.2 0.2 0.0 0.2 0.0 -0.4 -0.8 -0.4 0.2 N
1.2 0.0 0.0 0.2 0.0 -0.2 -1.2 -1.0 -0.1 P
0.8 0.2 -0.2 -0.2 -0.4 -1.0 -0.8 0.6 Q
1.2 0.0 -0.2 -0.4 0.4 -0.8 0.6 R
0.4 0.2 -0.2 -0.4 -0.6 -0.1 S
0.6 0.0 -1.0 -0.6 -0.1 T
0.8 -1.2 -0.4 -0.4 V
3.4 0.0 -1.2 W
2.0 -0.8 Y
0.6 Z
Matrice de Dayoff (1979)
University of Nijmegen
W=Tryprophane (Cyclique)
C= Cysteine (Soufre)

Matrices PAM et JTT
• PAM (Point Accepted Mutation) :– 71 familles de gènes nucléaires correspondant à
1300 séquences :• Séquences peu divergentes entre elles (identité ≥ 85 % entre
chaque paire possible dans une famille).
– Alignements globaux.
• JTT (Jones, Taylor and Thornton) :– Construites à partir de 59 190 mutations ponc-tuelles
observées dans 16 300 protéines.
– Alignements globaux.

Seuil pour les matrices PAM%
de
diffé
ren
ce
s
PAM
0 100 200 300 400
5
15
25
35
45
55
65
75
85Twilight Zone

Matrices BLOSUM
• BLOSUM (Blocks Substitution Matrices) :– Utilisation de ~2000 domaines conservés provenant
de 500 familles de protéines.
– Comparaisons effectuées dans les domaines alignés (banque BLOCKS).
– Matrices créées à partir de domaines comprenant des séquences ± divergentes :
• Toutes les paires ayant servi a construire une matrice BLOSUMk ont une identité ≥ à k %.
• Matrices plus adaptées pour des protéines distantes du point de vue évolutif.

Identité %
100
0
PA
M
BL
OS
UM
30
40
20
90
50
10
80
70
60
90
62
50
30
50
100
120
250
Choix d’une matrice
• Pas de matrice idéale.
• Meilleurs résultats avec les
matrices utilisant des modèles
d’évolution :
– BLOSUM globalement
meilleures que PAM.
• Degré de similarité des
séquences.
• Il est recommandé
d’expérimenter !
0
10
20
30
40
50
pénalit
é
0 5 10 15 20
k
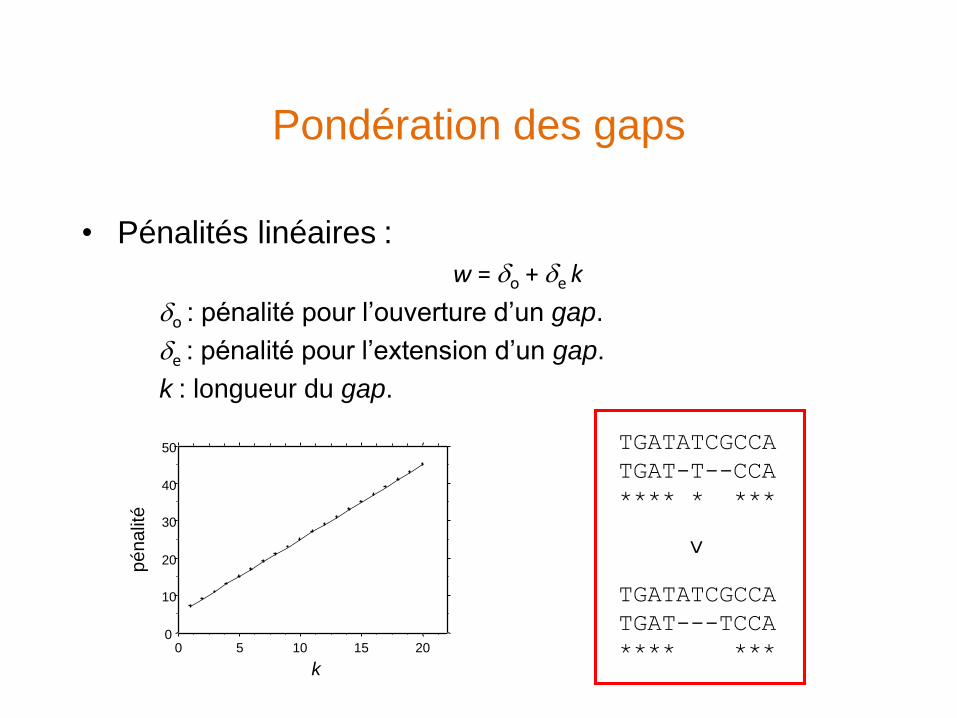
Pondération des gaps
• Pénalités linéaires :
w = o + e k
o : pénalité pour l’ouverture d’un gap.
e : pénalité pour l’extension d’un gap.
k : longueur du gap.
TGATATCGCCA
TGAT---TCCA
**** ***
TGATATCGCCA
TGAT-T--CCA
**** * ***
>

Autres pondérations
• Pénalités
logarithmiques :
w = o + e log(k)
• Pondération par la
distance évolutive :
– e diminue quand la
distance augmente.
• Pondération par la
nature des résidus :
– e diminue dans les
régions hydrophiles.
0
10
20
30
40
pé
na
lité
0 5 10 15 20
k
N
C
Résidus
hydrophiles
Cœur
hydrophobe

Needleman et Wunsch
• Représentation sous la forme d’une trajectoire dans une matrice :
– Détermination de la trajectoire optimisant un score donné.
• Soit deux séquences A et B de longueurs m et n :
– Définition de la matrice de chemin S :
• Dans chaque case de cette matrice on stocke S(i, j), le score optimum de la trajectoire permettant d’arriver à cette case.
a1 … ai …
b1
…
bj
am
…
Séquence A
Sé
qu
en
ce
Bbn
S(i, j)

Construction de la matrice
• Soit S(i, j) la valeur optimum du score dans la case de
coordonnées (i, j) :
– Définition par rapport aux scores dans les trois cases
adjacentes (i – 1, j), (i – 1, j – 1) et (i, j – 1) :
i
j
j – 1
i – 1
S(i, j)
S(i, j) = max
S(i – 1, j) + (ai, –),
S(i – 1, j – 1) + (ai, bj),
S(i, j – 1) + (–, bj)
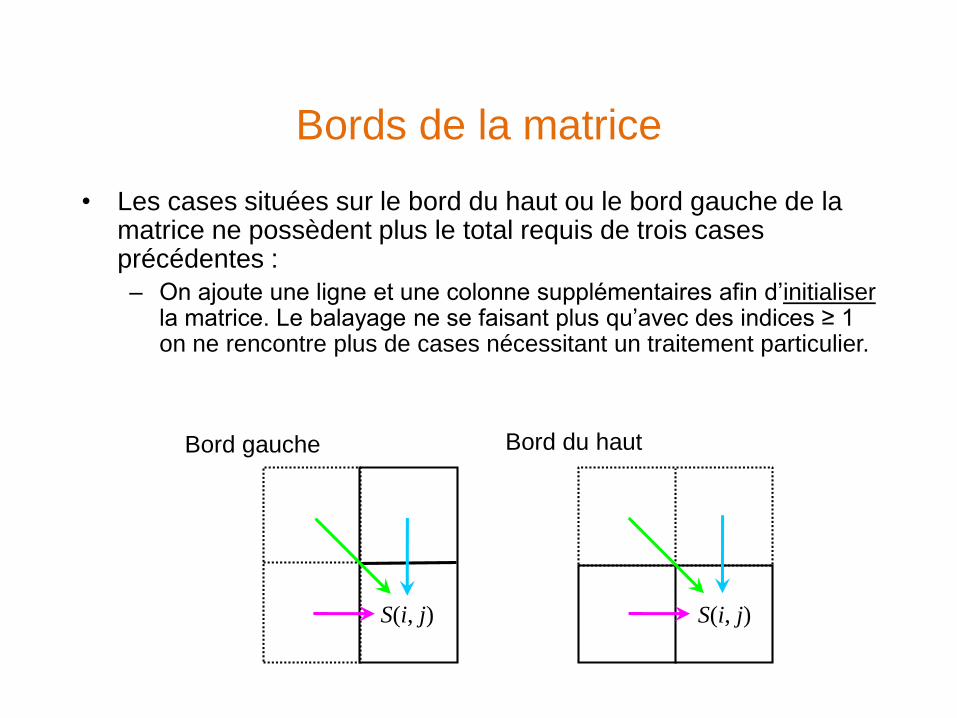
Bords de la matrice
• Les cases situées sur le bord du haut ou le bord gauche de la matrice ne possèdent plus le total requis de trois cases précédentes :
– On ajoute une ligne et une colonne supplémentaires afin d’initialiserla matrice. Le balayage ne se faisant plus qu’avec des indices ≥ 1 on ne rencontre plus de cases nécessitant un traitement particulier.
Bord gauche Bord du haut
S(i, j) S(i, j)

Exemple de calcul
A
T
T
A
A G C T A
0 Identité : +1
Mismatch : +0
Gap : -2
-2 -4 -6 -8 -10
-2
-4
-6
-8
-4
-4
+1
-6
-1
-2
-8
-4
-3
-10
-6
-5
-12
-7
-7
-1
-6
-2
-3
-8
-4
-5
-10
-5
-3
-3
+1
-1
-5
-1
-3
-7
-3
-5
-1
-1
-3
-3
+1
-7
-3
-2
-9
-4
-5
-4
-1
0
-6
-2
-2
-1
-5
-1
-2
-3
+1
-4
-1
+1
A G C T A
A – T T A
+1 -2 +0 +1 +1
S = +1
A G C T A
A T – T A
+1 +0 -2 +1 +1
S = +1

Smith et Waterman
• Algorithme dérivé de Needleman et Wunsch :
– Initialisation des bords à 0.
– N’importe quelle case de la matrice peut être
considérée comme point de départ pour le calcul
du score. i
j
j – 1
i – 1
S(i, j)
S(i, j) = max
S(i – 1, j) + (ai, –),
S(i – 1, j – 1) + (ai, bj),
S(i, j – 1) + (–, bj)
0
S(i, j) < 0 S(i, j) = 0

L’alignement de n séquences
• Application possible du Needleman & Wunch à plus de deux séquences (en théorie)
• Le nombre de possibilité pour aligner n séquences est proportionnel à 2n – 1.
• Le besoin en mémoire et ressources de calcul augmentent de manière exponentielle avec le nombre de séquences
Application d’heuristiques
Pairwise Alignment:three possibilities
Alignment of threesequences : seven possibilities

Outils pour la recherche par similarité
• Utilisation d’outils comme BLASTP/TBLASTN pour la recherche par
similarité dans différentes banques de données de séquences
protéiques/nucléiques
• Banques de séquences protéiques types
– SWISS-PROT : banque non redondante de séquences protéiques
confirmées (Août 2010: 519 348 entrées (158,316 en 2004), incluant de
nombreuses annotations et références croisées avec d’autres banques
de séquences, de structures, de familles protéiques, de références
bibliographiques, de descriptions de la fonction et du rôle biologique des
protéines…
– TrEMBL : banque non redondante de traduction des CDS soumis à
EMBL (Août 2010: 11 636 205 entrées (1 400 820 en 2004)

Séquence banque
Séquence requête
Longueur du mot = w
Score ≥ T
Mot
Extension du
segment similaire
Séquence banque
Séquence requête
HSP : High Scoring Pair
Score
Extension du segment
Extension stoppée quand :
- la fin d’une des deux séquences est atteinte
- score ≤ 0
- score ≤ score_max - xT
Score max.
x
BLAST : principe général
©Guy Perrière

S L A A L L N K C K T P Q G Q R L V N Q W
P Q G 18
P E G 15
P R G 14
P K G 14
P N G 13
P D G 13
P H G 13
P M G 13
P S G 13
P Q A 12
P Q N 12
...
Liste
de mots
voisins
Score seuil T = 13
Query : 325 S L A A L L N K C K T P Q G Q R L V N Q W 345
+ L A + + L + T P G R + + + W
Sbjct : 290 T L A S V L D C T V T P M G S R M L K R W 310
(P, P) = 7
(Q, R) = 1
(G, G) = 6x
T
Exemple
©Guy Perrière

Versions de BLAST
• blastp : protéine vs.protéine.
• blastn : utile pour le non-codant.
• blastx : séquences co-dantes non identifiées.
• tblastn : homologues dans un génome non complètement annoté.
Nucléique
Protéique
Nucléique
Protéiqueblastp
blastnT
Banque
tblastxT
T
Séquence
©Guy Perrière

Évaluation statistique
©Guy Perrière
• Similarités détectées :
– Relations significatives.
– Similarités dues au hasard.
• Fonction de score :
– Mesure sous la forme :
• D’une espérance mathématique (E-value).
• Valeur en bits.
– Basée sur une distribution calculée à partir séquences non homologues.
– Les scores dépendent de la taille de la banque.

E-value, bits et similarité
• Soit E, l’espérance mathématique d’avoir une similarité ≥ au score S observé :
E = Kmn e–S
Avec m et n les longueurs des deux séquences considérées, et K et deux paramètres dérivés de la distribution précédente.
• Le score en bits S' est donné par :
S' = [S – log(K)] / log(2)
• La relation entre E et S' est donc donnée par :
E = mn 2–S'
©Guy Perrière

Recherche par Blast au NCBI

Choix des paramètres
Choix des paramètres

Choix des paramètres avancés

Résultats du BLAST : Entête
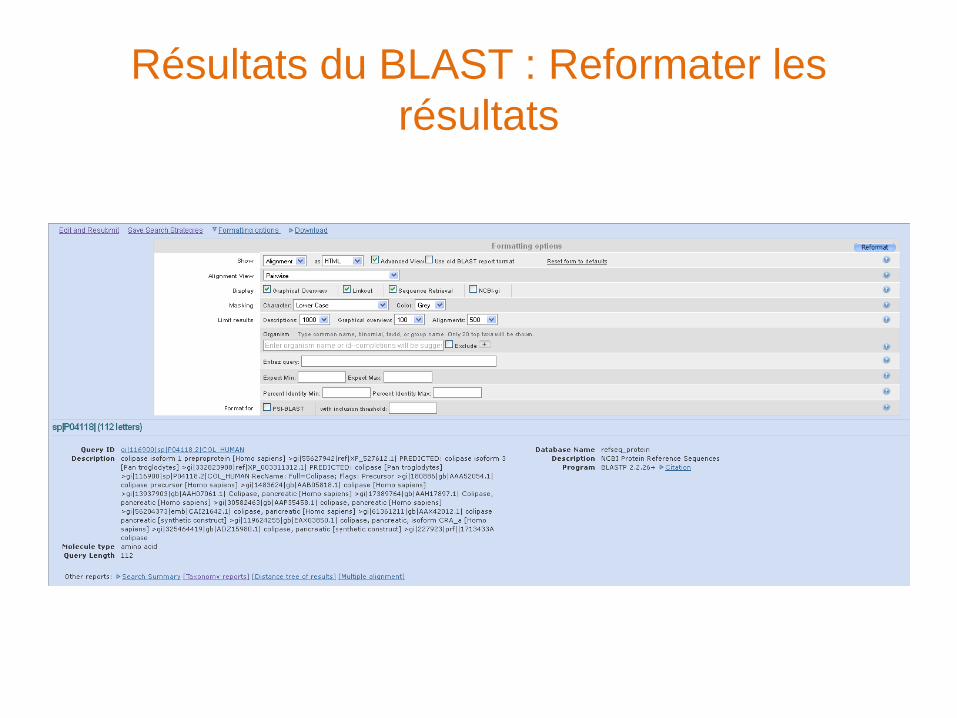
Résultats du BLAST : Reformater les
résultats

Résultats du BLAST : Domaines conservés

Résultats du BLAST : Vue graphique

Résultats du BLAST : Descriptions

Résultats du BLAST : Alignements locaux

Résultats du BLAST : Alignements locaux

Taxonomy report

Caractéristiques des principaux logiciels
d’alignement multiple
Composants principaux
des algorithmes
Principaux logiciels
(Chatzou et al. 2015)

L’alignement progressif
• Principe = procédure itérative basée sur le regroupement d’alignements deux à deux pour construire un alignement multiple
• Trois étapes :
– Alignement de paires de séquences
– Construction d’un arbre guide
– Alignement de groupes de séquences déjà alignées (alignement progressif).
• CLUSTAL (Higgins, Sharp 1988, Thompson et al., 1994), le programme d’alignement multiple le plus cité.
• MULTALIN, PILEUP, T-Coffee, Muscle

L’homologie, base théorique de l’alignement
multiple
• Les séquences homologues sont reliées d’un point de vue évolutif
• Idée = construire progressivement un alignement, à partir de séries de séquences (ou de groupes de séquences) alignées deux à deux, suivant un ordre de branchement donné par un arbre phylogénétique– Alignement des séquences les plus proches d’un point de vue
phylogénétique
– Intégration progressive des séquences un peu plus éloignées
• Approche suffisamment rapide pour permettre la construction d’alignements contenant un grand nombre de séquences

Alignement progressif
• Construction itérative par groupement des alignements de paires de séquences :– Alignement de toutes les paires possibles :
• Établissement d’une matrice de distances basée sur les scores des alignements.
– Groupement des paires et / ou des séquences.
– Groupement des alignements (alignement progressif proprement dit).
• Différentes implémentations disponibles :– CLUSTAL, MULTALIN, MUSCLE.

Algorithme de CLUSTAL W
• Alignement de toutes les paires de séquences deux à deux par l’algorithme de Needleman et Wunsh
• Construction d’une matrice de distances d’après la divergence mesurée entre chaque paire de séquences
• Calcul d’un arbre guide à partir de la matrice de distances
• Alignement progressif des séquences suivant l’ordre de branchement donné par l’arbre

Exemple
• Alignement de 7 séquences de globines:
– Hémoglobine b Humaine (Hbb_H)
– Hémoglobine a Humaine (Hba_H)
– Hémoglobine b Cheval (Hbb_C)
– Hémoglobine a Cheval (Hba_C)
– Myoglobine de cétacé Physeter catodon (Myo)
– Hémoglobine V de lamproie Petromyzon marinus (Glb5)
– Leghémoglobine II de Lupin (Lgb)

Alignement des séquences 2 à 2 et
construction de la matrice de distances
• Alignement des séquences 2 à 2 par programmation dynamique
(algorithme de Needleman et Wunsh) connaissant une matrice de
similarité et les pénalité dues aux gaps (ouverture et extension)
• Score = nombre d’identités / nb de résidus comparés (excluant les
gaps)
• % de divergence = 1 - score
• Remarque : le calcul du score ne tient pas compte des substitutions
multiples, mais on peut utiliser des modèles d’évolution comme
Kimura ou JC pour en tenir compte

Alignement des séquences 2 à 2 et
construction de la matrice de distances
Hbb_H Hbb_C Hba_H Hba_C Myo Glb5 Lgb
Hbb_H -
Hbb_C 0.17 -
Hba_H 0.59 0.60 -
Hba_C 0.59 0.59 0.13 -
Myo 0.77 0.77 0.75 0.75 -
Glb5 0.81 0.82 0.73 0.74 0.80 -
Lgb 0.87 0.86 0.86 0.88 0.93 0.90 -

Construction de l’arbre guide
• Arbre phylogénétique non raciné construit par la
méthode du Neighbor-Joining à partir de la matrice de
distances calculée précédemment
– Longueur des branches <=> proportionnelle à la
divergence estimée
– Racine placée au « poids moyen » <=> Longueur des
branches d’un côté de la racine = longueur des
branches de l’autre côté

Construction de l’arbre guide
Hba_H
Hba_C
Hbb_H
Hbb_C
Myo
Glb5
Leg
0.081
0.084
0.226
0.055
0.065
0.219
0.0
61
0.398
0.389
0.504
Positionnement de la racine au poids moyen (point à partir duquel les
longueurs moyennes des branches de chaque côté du nœud sont égales)

Placement de la racine
Hba_H
Hba_C
Hbb_H
Hbb_C
Myo
Glb5
Leg
0.081
0.084
0.226
0.055
0.065
0.219
0.0
61
0.398
0.389
0.504
Positionnement de la racine au poids moyen (point à partir duquel les
longueurs moyennes des branches de chaque côté du nœud sont égales)
ROOT

Arbre guide raciné
Hba_H
Hba_C
Hbb_H
Hbb_C
Myo
Glb5
Leg
0.081
0.084
0.226
0.055
0.065
0.219
0.398
0.389
0.442
0.015
0.061
0.062
ROOT

Pondération des séquences
• Principe : attribuer un poids à chaque branche de l’arbre
=> Dépend de la taille de la branche et du nombre de taxa partageant cette branche (redondance de l’information)
=> longueur de la branche / nombre de taxa partageant cette branche
• Poids d’une séquence = des longueurs des branches pondérées de la racine au taxon considéré

Pondération des séquences
Hba_H
Hba_C
Hbb_H
Hbb_C
Myo
Glb5
Leg
0.081
0.084
0.226
0.055
0.065
0.219
0.398
0.389
0.442
0.015
0.061
0.062
ROOT
W1 = 0.062/6 + 0.015/5 + 0.061/4 +
0.226/2 + 0.081 = 0.221
W2 = 0.062/6 + 0.015/5 + 0.061/4 +
0.226/2 + 0.084 = 0.225
W3 = 0.194
W4 = 0.203
W5 = 0.411
W6 = 0.398
W7 = 0.442

Alignement progressif
• Principe : utiliser une série de paires d’alignements pour
aligner des groupes de séquences de plus en plus
larges, en respectant l’ordre de branchement dans
l’arbre guide (des feuilles vers la racine)

Alignement progressif
• Dans l’exemple des globines, on aligne dans
l’ordre:
– Les b globines humaines et de cheval
– Les a globines humaines et de cheval
– Les a et b hémoglobines
– Les a, b hémoglobines et la myoglobine
– Les hémoglobines, myoglobine et l’hémoglobine de
lamproie
– La leghémoglobine avec toutes les autres

Alignement progressif
• Calcul du score à une position = moyenne des scores
obtenus par toutes les comparaisons 2 à 2 des
séquences de chaque groupe pondérés par le poids de
chaque séquence

Alignement progressif
• Exemple: on cherche à aligner un groupe de 4 séquences (déjà alignées) avec un groupe de 2 séquences (déjà alignées)
Calcul du score:
1 PEEKSAVTAL M(T,V) x w1 x w5 +
2 GEEKAAVLAL M(T,I) x w1 x w6 +
3 PADKTNVKAA M(L,V) x w2 x w5 +
4 AADKTNVKAA M(L,I) x w2 x w6 +
M(K,V) x w3 x w5 +
5 EGEWQLVLHV M(K,I) x w3 x w6 +
6 AAEKTKIRSA M(K,V) x w4 x w5 +
M(K,I) x w4 x w6 / 8
Score associé à la comparaison d’un gap = 0 plus mauvais score possible

gi|122615|sp|P02023|HBB_HUMAN -------MVHLTPEEKSAVTALWGKVN--VDEVGGEALGRLLVVYPWTQR
gi|70401|pir||HBHO --------VQLSGEEKAAVLALWDKVN--EEEVGGEALGRLLVVYPWTQR
gi|122412|sp|P01922|HBA_HUMAN --------MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKT
gi|2144717|pir||HAHO --------MVLSAADKTNVKAAWSKVGGHAGEYGAEALERMFLGFPTTKT
gi|127687|sp|P02185|MYG_PHYCA ---------VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLE
gi|121233|sp|P02208|GLB5_PETMA PIVDTGSVAPLSAAEKTKIRSAWAPVYSTYETSGVDILVKFFTSTPAAQE
gi|126238|sp|P02240|LGB2_LUPLU --------GALTESQAALVKSSWEEFNANIPKHTHRFFILVLEIAPAAKD
*: : : * . : .: * :
gi|122615|sp|P02023|HBB_HUMAN FFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDN-----LKGTF
gi|70401|pir||HBHO FFDSFGDLSNPGAVMGNPKVKAHGKKVLHSFGEGVHHLDN-----LKGTF
gi|122412|sp|P01922|HBA_HUMAN YFPHF-DLS-----HGSAQVKGHGKKVADALTNAVAHVDD-----MPNAL
gi|2144717|pir||HAHO YFPHF-DLS-----HGSAQVKAHGKKVGDALTLAVGHLDD-----LPGAL
gi|127687|sp|P02185|MYG_PHYCA KFDRFKHLKTEAEMKASEDLKKHGVTVLTALGAILKKKGH-----HEAEL
gi|121233|sp|P02208|GLB5_PETMA FFPKFKGLTTADQLKKSADVRWHAERIINAVNDAVASMDDT--EKMSMKL
gi|126238|sp|P02240|LGB2_LUPLU LFSFLKGTSEVP--QNNPELQAHAGKVFKLVYEAAIQLQVTGVVVTDATL
* : . . .:: *. : . :
gi|122615|sp|P02023|HBB_HUMAN ATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVA
gi|70401|pir||HBHO AALSELHCDKLHVDPENFRLLGNVLVVVLARHFGKDFTPELQASYQKVVA
gi|122412|sp|P01922|HBA_HUMAN SALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLA
gi|2144717|pir||HAHO SNLSDLHAHKLRVDPVNFKLLSHCLLSTLAVHLPNDFTPAVHASLDKFLS
gi|127687|sp|P02185|MYG_PHYCA KPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGDFGADAQGAMNKALE
gi|121233|sp|P02208|GLB5_PETMA RDLSGKHAKSFQVDPQYFKVLAAVIADTVAAG---------DAGFEKLMS
gi|126238|sp|P02240|LGB2_LUPLU KNLGSVHVSKGVAD-AHFPVVKEAILKTIKEVVGAKWSEELNSAWTIAYD
*. * . : .: : .: ...
gi|122615|sp|P02023|HBB_HUMAN GVANALAHKYH------
gi|70401|pir||HBHO GVANALAHKYH------
gi|122412|sp|P01922|HBA_HUMAN SVSTVLTSKYR------
gi|2144717|pir||HAHO SVSTVLTSKYR------
gi|127687|sp|P02185|MYG_PHYCA LFRKDIAAKYKELGYQG
gi|121233|sp|P02208|GLB5_PETMA MICILLRSAY-------
gi|126238|sp|P02240|LGB2_LUPLU ELAIVIKKEMNDAA---
. :

L‘alignement multiple n’est pas toujours
optimal
• Seul l’un de ces alignements est optimal

MuscleEdgar (2004) Nucleic Acids Res. 32:1792
http://www.drive5.com/muscle/

Global Alignments, Block alignments

DialignMorgenstern et al. 1996 PNAS 93:12098
• Search for similar blocks without gap
• Select the best combination of consistent similar blocks (uniforms or not) : heuristic (Abdeddaim 1997)
• Alignment anchored on blocks
• Slower than progressive alignment, but better when sequences contain large indels
• Do not try to align non-conserved regions

Alignement multiples locaux
• MEME
• MATCH-BOX
• PIMA
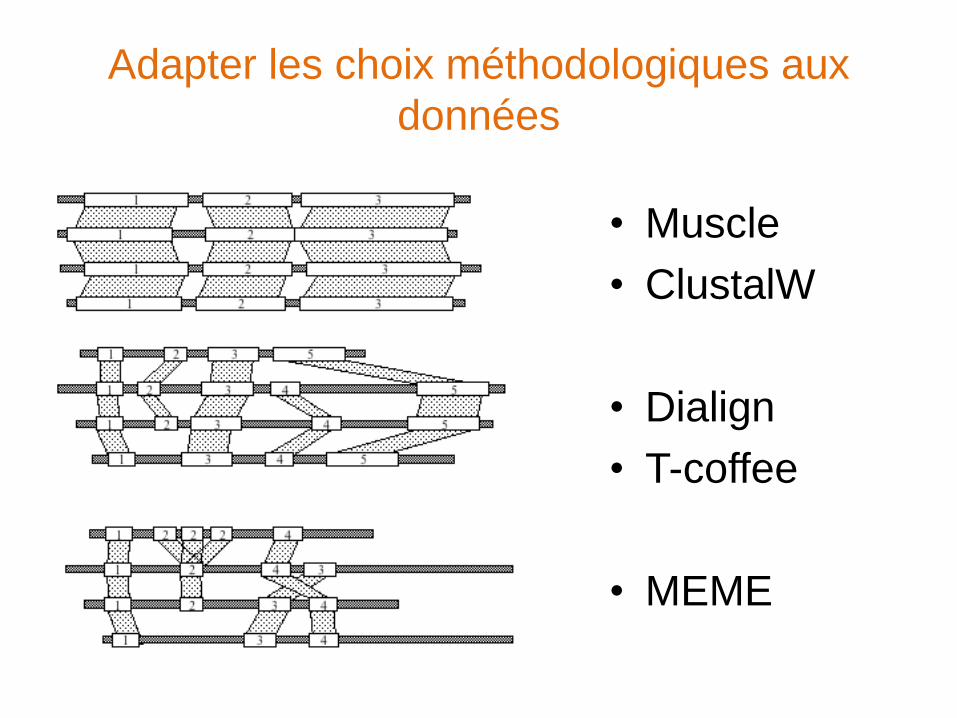
Adapter les choix méthodologiques aux
données
• Muscle
• ClustalW
• Dialign
• T-coffee
• MEME

Multiple alignment editor

Cas spéciaux

Alignement de séquences d’ADN codantes
(1) Alignement des séquences protéiques
(2) Utilisation de l’alignement obtenu comme guide pour aligner les séquences d’ADN
protal2dna: http://bioweb.pasteur.fr/seqanal/interfaces/protal2dna.html
L F L
F
CTT TTC CTT TTC
CTC --- --- CTC
L - - L
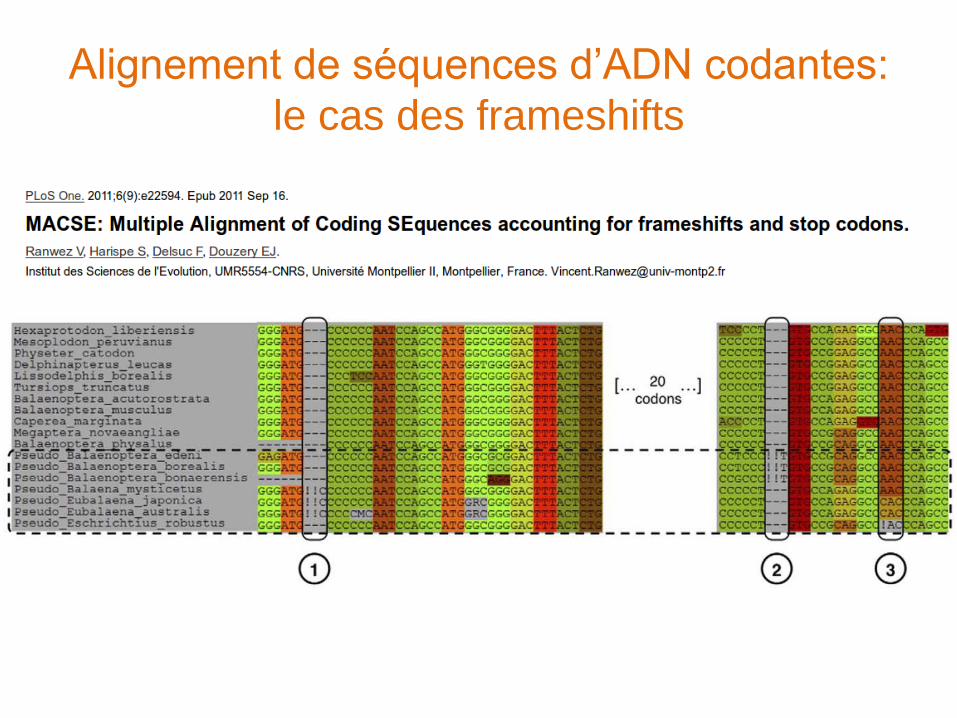
Alignement de séquences d’ADN codantes:
le cas des frameshifts
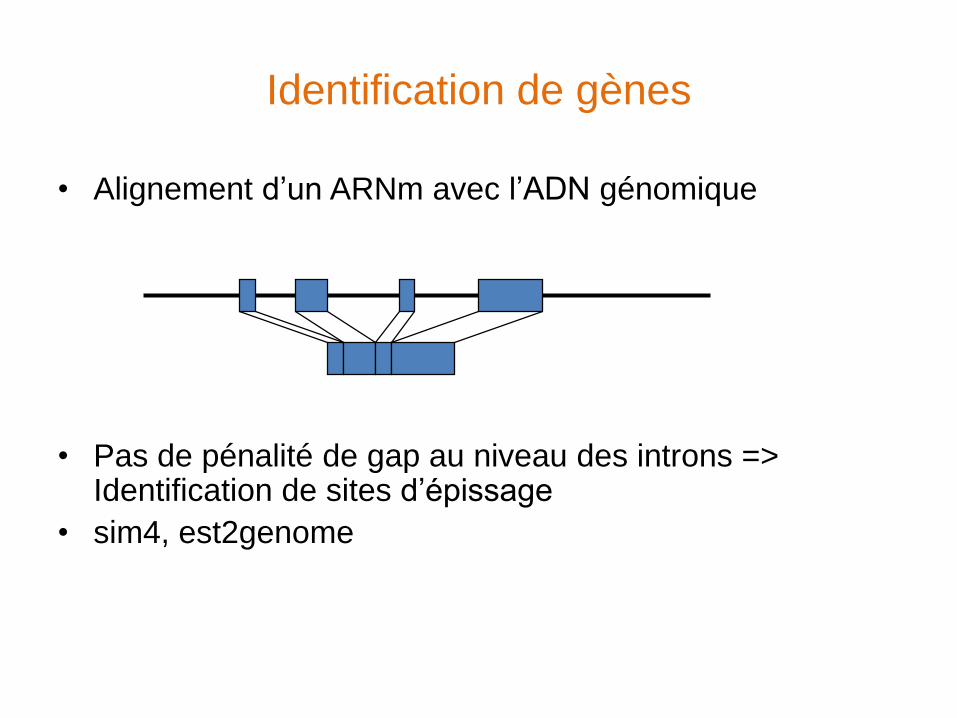
Identification de gènes
• Alignement d’un ARNm avec l’ADN génomique
• Pas de pénalité de gap au niveau des introns => Identification de sites d’épissage
• sim4, est2genome

Identification de gènes
• Alignement d’une protéine avec l’ADN génomique
• Pas de pénalité de gap au niveau des introns => Identification de sites d’épissage
• genewise
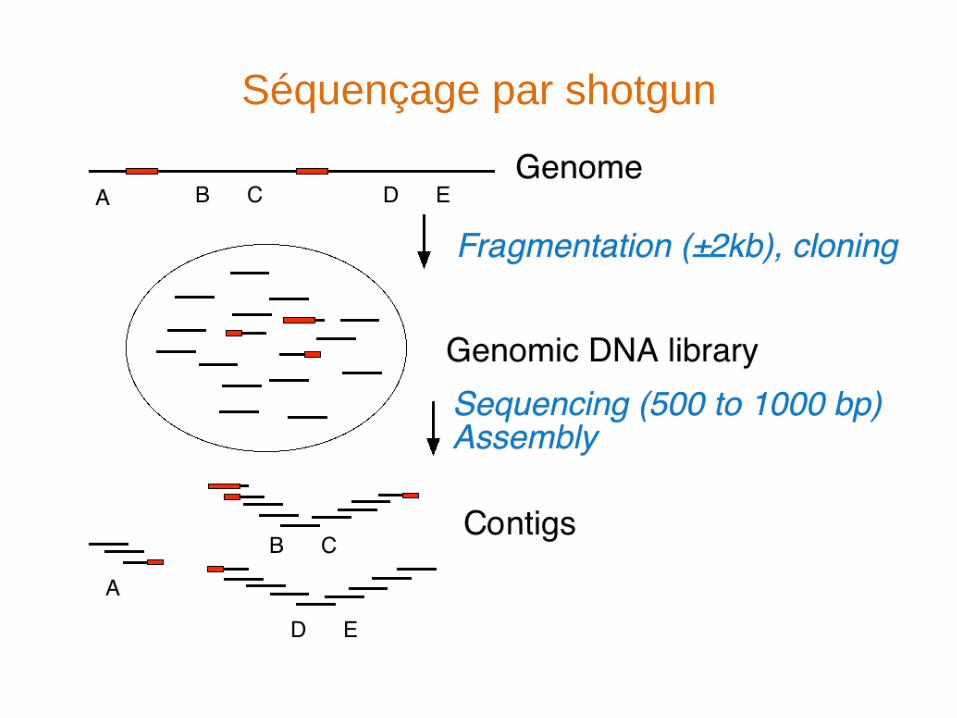
Séquençage par shotgun

Assemblage
• Recherche de séquences chevauchantes entre les reads
• Autoriser / prise en compte des erreurs de séquençage
et/ou du polymorphisme
• Prise en compte de la qualité des séquences
• cap3, phred/phrap (il existe des outils plus sophistiqués
pour l’assemblage de génomes)

A C D E F G H I K L M N P Q R S T V W Y -
1 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 16
2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 16
3 1 0 0 1 0 0 0 0 0 0 1 0 0 3 0 1 0 0 0 0 10
4 2 0 0 0 0 2 0 0 0 0 0 0 1 0 2 1 0 0 0 0 9
5 1 0 0 1 0 0 0 0 0 0 0 0 0 0 2 0 3 0 0 1 9
6 0 0 0 0 0 0 1 0 0 0 0 0 2 0 0 0 4 1 0 0 9
7 1 0 0 0 0 9 0 0 0 0 0 4 0 0 1 2 0 0 0 0 0
8 0 0 0 0 1 0 0 8 0 0 5 0 1 0 0 0 0 2 0 0 0
9 2 0 1 0 2 0 0 0 0 0 0 0 0 0 0 0 0 12 0 0 0
10 0 5 6 4 0 0 0 0 0 0 0 0 1 0 1 0 0 0 0 0 0
11 0 0 1 12 1 0 0 0 1 0 0 0 0 1 0 0 0 0 0 1 0
12 0 17 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
13 0 17 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
14 0 0 0 1 5 0 0 0 0 4 1 0 0 0 1 0 1 0 0 4 0
15 0 0 0 0 0 0 0 1 2 0 0 5 0 1 5 2 0 1 0 0 0
16 0 0 0 1 0 2 0 2 0 0 0 0 5 1 0 3 0 2 0 1 0
17 0 17 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
18 0 0 4 0 0 0 0 0 0 0 0 0 0 0 0 4 9 0 0 0 0
19 0 0 1 0 0 0 0 1 1 5 0 1 1 1 0 0 1 4 0 1 0
20 1 0 6 0 0 1 0 0 0 0 0 0 0 2 3 3 0 0 0 1 0
21 0 0 1 3 0 0 0 0 1 1 0 0 0 2 1 1 1 6 0 0 0
22 0 0 0 0 1 0 0 0 0 14 0 0 0 0 0 1 0 1 0 0 0
23 2 0 0 4 0 0 0 0 1 5 0 0 0 1 2 0 0 1 0 1 0
24 1 0 0 1 0 0 0 0 3 1 1 1 0 1 0 4 4 0 0 0 0
25 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 15 0
26 0 17 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
27 2 0 1 0 0 2 0 0 0 0 0 2 2 0 0 0 1 0 0 2 5
28 0 0 0 0 0 0 0 0 1 0 0 0 2 0 1 0 1 0 0 1 11
29 0 0 0 0 0 0 0 0 0 1 0 0 1 1 1 0 0 0 0 1 12
30 1 0 0 0 0 0 0 1 1 0 0 0 0 0 0 1 0 0 0 0 13
Matrix of position-specific amino-acid frequency (A-chain of insulin)

Alignment of SeqA with the matrix of position-specific amino-acid frequency

Alignment of SeqB with the matrix of position-specific amino-acid frequency