
Atelier pratique Incertitudes de mesure - SOMLIT...
47
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach, 1 Estimation de l’incertitude de mesure Etude de cas : Mesure de l’ammonium par colorimétrie Introduction = important dans SOMLIT pour construire une base de donnée fiable, compréhensible et utilisable par les scientifiques d’aujourd’hui et de demain. Cela requière la mise en œuvre d’outils spécifiques comme le système d’assurance qualité et l’estimation des incertitudes en fait partie intégrante. Sachant que les données SOMLIT sont des mesures expérimentales physiques (absorbance, fluorescence, masse), elles répondent pour la plupart à la loi normale et doivent en conséquence être, impérativement, caractérisées par leur moyenne et l’erreur associée. Il n’existe pas de méthode unique pour estimer l’erreur d’une mesure: plusieurs méthodes existent qui peuvent fournir des valeurs différentes et toutes peuvent être justes sous réserve d’être clairement exprimées.
-
Upload
trinhthuan -
Category
Documents
-
view
217 -
download
1
Transcript of Atelier pratique Incertitudes de mesure - SOMLIT...

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
1
Estimation de l’incertitude de mesure
Etude de cas : Mesure de l’ammonium par colorimétrie
Introduction
= important dans SOMLIT pour construire une base de donnée fiable, compréhensible et utilisable
par les scientifiques d’aujourd’hui et de demain.
Cela requière la mise en œuvre d’outils spécifiques comme le système d’assurance qualité et
l’estimation des incertitudes en fait partie intégrante.
Sachant que les données SOMLIT sont des mesures expérimentales physiques (absorbance,
fluorescence, masse), elles répondent pour la plupart à la loi normale et doivent en conséquence
être, impérativement, caractérisées par leur moyenne et l’erreur associée.
Il n’existe pas de méthode unique pour estimer l’erreur d’une mesure: plusieurs méthodes existent
qui peuvent fournir des valeurs différentes et toutes peuvent être justes sous réserve d’être
clairement exprimées.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
2
Comment estimer l’incertitude de mesure sans rien y connaître ?
� Moyen efficace = se baser sur la norme XP-T90-220 : « Qualité de l’eau : Protocole
d’estimation de l’incertitude de mesure associée à un résultat d’analyse pour les méthodes
physico-chimiques » (Août 2003). Cette norme est payante (98 HT), très claire et accessible à
tous.
� Elle décrit très précisément plusieurs méthodes d’estimation.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
3

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
4

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
5
Mise en oeuvre concrète du calcul d’incertitude :
Etude du cas de l’ammonium par colorimétrie
Démarche adoptée : = objet du Stage de DUT 1ière année de Sarah Le Cléach, septembre 2011.
1. mise en œuvre d’une méthode usuelle (« personnelle ») basée sur le GUM : « Guide pour
l’estimation des incertitudes », non normative dans l’objectif de la jauger par comparaison
avec les méthodes normatives
2. mise en œuvre de deux méthodes issues de la norme : méthode GUM et méthode basée sur
les « essais interlaboratoire ».
=> Les résultats offriront matière à débattre en vue d’établir à terme une procédure commune au
réseau SOMLIT pour l’étude des incertitudes des paramètres physico-chimiques mesurés en
laboratoire.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
6
METHODE NON NORMATIVE : approche GUM
Rappel du protocole de dosage de l’NH4 par colorimétrie mis en œuvre à Brest :
=> Remarque : quelques variantes par rapport aux autres station : ex : effet de sel négligeable.
Version : Protocole local ammonium 1.1 Date de création : 7 juin 2011
Date de dernière modification : Date du visa annuel :
Dosage de l'azote ammoniacal
Rédigé par : Thierry Cariou Eric Macé
Visé par : Nicolas Savoye, responsable qualité national Peggy Rimmelin-Maury, responsable qualité-Brest
Le 7 juin 2011

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
7

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
8
I- Introduction : L’azote minéral dissous dans l’eau de mer existe sous forme d’azote gazeux et d’ions ammonium, nitrite et nitrate. Dans le cycle de l’azote, l’ammoniac dissous (sous forme d’ion ammonium) occupe une place particulière. D’une part, le phytoplancton utilise l’ammonium préférentiellement
comme source nutritive azotée, d’autre part, la dégradation de l’azote particulaire et de l’azote dissous par l’activité hétérotrophique donne lieu à la formation d’ammoniac oxydé ensuite en nitrite puis en nitrate. Enfin, le zooplancton contribue à enrichir l’eau de mer en ammoniac par excrétion directe. Les teneurs en ammoniac dissous dans l’eau de mer sont comprises généralement entre 0 et 3 µmoles.l-1. Le dosage est basé sur la réaction de Berthelot (1859). En milieu alcalin (8 < pH < 11.5), l’ammoniac dissous réagit sur l’hypochlorite pour former une monochloramine. Ce composé, en
présence de phénol et en milieu oxydant, donne lieu à la formation d’un bleu d’indophénol. A 20 °C, la réaction catalysée par l’ion nitroprussiate demande 6 heures pour se développer. L’absorption est mesurée par spectrophotométrie à 630 nm. La méthode a été appliquée à l’eau de mer, en particulier par L.Solorzano et F. Koroleff en 1969. L’analyse par fluorimétrie développée par Holmes commence à se répandre dans la communauté des analystes de l’eau de mer. Cette méthode apporte des avantages (par exemple le non-emploi de phénol). Cette méthode ne sera pas décrite dans ce protocole car la majorité des laboratoires utilisent encore la méthode de Koroleff.
Précision et limite de détection : Etant donné l’influence de la turbidité à la longueur d’onde de mesure (630 nm), ainsi que les nombreuses sources de contaminations potentielles de ce paramètre, les écarts-types relatifs obtenus sur des triplicats peuvent être très variables. S’ils se situent généralement entre 1 et 5 % au delà de 1 µmole.l-1, ils peuvent atteindre facilement 20 à 50 % en dessous de 0.5 µmole.l-1.
La limite de détection se situe aux alentours de 0.02 µmole.l-1 (= 3xs0, écart type sur
le blanc « réactif ») pour un trajet optique de 10 cm avec une limite de détection de 0.001 unité DO. La loi de Beer-Lambert est respectée dans la gamme de concentration allant de 0 à 70 µmoles.l-1. Au delà de cette concentration il est possible de diluer l’échantillon. Pour les faibles concentrations (0 à 5 µmoles.l-1) il est conseillé d’utiliser des cuves de 10 cm. Au delà, à moins de diluer, des cuves de 5 cm ou 1 cm seront nécessaires.
II – Précautions particulières
Le risque de contamination est un des paramètres déterminant dans la réussite ou non de la mesure. Les réactifs utilisés sont à conserver à l’obscurité et au réfrigérateur jusqu’à utilisation. Une attention particulière sera apportée lors du prélèvement, mais également au niveau de la manipulation et du stockage du matériel et des échantillons. Le risque majeur est lié à la pollution atmosphérique du laboratoire aussi bien que du lieu de prélèvement (vapeurs azotées, proximité de produits chimiques, présence de fumeurs ou rejets du bateau …) mais également à la manipulation (contact avec la peau de l’échantillon, du
flaconnage, bouchons…) ainsi qu’au matériel de pré-filtration (lorsque celui-ci est indispensable). Le port de gants à usage unique peut être une bonne solution à condition d’être bien ajustés et d’être changés régulièrement. Certains caoutchoucs sont également des sources de

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
9
contaminations notables d’ammonium et il convient donc de les éviter au niveau du bouchage des flacons aussi bien qu’au niveau des tendeurs des bouteilles Niskin. III – Matériel et appareillage utilisé : Prélèvement :
- Bouteilles de prélèvement à clapet Niskin. - Flacons ronds gradués en verre de 100ml (type fisherbrand® grande ouverture, sans bague anti-goutte) : généralement gradués jusqu’à 80 ml, les 100 ml correspondent souvent au rétrécissement du goulot qui assure alors une précision satisfaisante sur le volume d’échantillon analysé. Dans les milieux très turbides (e.g. estuaires de la Gironde) un flacon de 30 ml peut être utilisé. - 1 système de pré-filtration (de type Swinnex Millipore ® en PP) équipé d’une soie de 50
µm décontaminée, par niveau de prélèvement, pour les milieux turbides ou système de filtration (« cocotte » de filtration avec filtre en fibre de verre) dans les systèmes très turbides. - Bac de transport et conservation opaque (pendant la réaction). - 2 dispensettes pour l’addition des réactifs (3ml réactif/100 ml ; 1ml réactif/30 ml).
Préparation des réactifs : - Balance au centième de gramme. - Hotte aspirante. - 1 spatule. - Agitateur magnétique avec barreaux et tige aimantés. - 2 béchers de 500 ml + béchers de 250, 50 ml. - 2 fioles jaugées de 500 ml à bouchon rodé.
- 1 éprouvette graduée. - 1 pissette. - 2 dispensettes à volume fixe de 3 ml. Préparation des standards :
- Balance de précision au centième de milligramme et spatule. - Etuve réglée à 105 °C. - Dessiccateur et silicagel®.
- Petit bécher. - 1 fiole jaugée d’un litre (classe A+) à bouchon rodé. - 4 fioles jaugées de classe A à bouchons rodés de 500 ou 1000ml. - Pipettes automatiques et embouts correspondants. - Pissette.
Mesure : - Spectrophotomètre (630nm) acceptant des cuves de 10 cm de trajet optique, situé sous extracteur. - 1 cuve de 10 cm en verre optique spécialisé (2 cuves appariées si possible en cas de double faisceau). - Papier absorbant et papier optique. Contrôle qualité : Filtre Holmium pour vérification des longueurs d’onde. Référence : filtre Holmium Hellma, 666-F1.
Filtres de vérification des densités optiques. Référence : filtre gris Hellma, 666-F2. Elimination des déchets :

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
10
Recueillir les effluents contenant les réactifs dans un bidon qui sera ensuite retraité avec les déchets chimiques aromatiques. IV – Produits chimiques et réactifs utilisés
Produits chimiques : Eau déminéralisée de très bonne qualité de type Milli-Q fraîchement soutirée. Acide chlorhydrique 32%, d=1.16 (HCl) : Prolabo RP (ref. 20 252 290). Sulfate d’ammonium ((NH4)2SO4) : Merck PA (ref. : 1.01217. xxxx). Citrate trisodique dihydraté (Na3C6H5O7, 2H2O) : Merck PA (ref. 1.06448.xxxx). Hydroxyde de sodium ( NaOH) : Merck PA (ref. 1.06495.xxxx). Solution aqueuse d’hypochlorite de sodium à 3.5% env. de chlore actif (NaClO) : Prolabo GPR
Rectapur (ref. 27896.291). Nitroprussiate de sodium dihydraté (Na2[Fe(CN)5NO],2H2O) : Merck PA (ref. 1.06541.xxxx). Thiosulfate de sodium pentahydraté (Na2S2O3, 5H2O) : Merck PA (ref. 1.06516.xxxx). Phénol (C6H5OH) : Merck PA (ref. 1.00206.xxxx) ou phénol liquide 85 %. Iodure de sodium (NaI) : Merck PA (ref. 1.06523.xxxx). Silicagel. NaCl : Merck PA. Conservation des réactifs : Chacun des réactifs préparés est transvasé dans un flacon brun de 500ml et surmonté d’un distributeur automatique réglé sur 3 ou 1ml (ou de préférence à volume fixe), spécifique et identifié (R1 NH4
+et R2 NH4+). Ils sont conservés au froid (4°C) et à l’obscurité.
Dans ces conditions, le réactif 1 est stable un mois, mais le réactif 2 sera préparé tous les quinze jours. Lors des manipulations sur le terrain, les réactifs R1 et R2 sont transportés, si possible, dans une glacière équipée de blocs de froid et sont replacés au réfrigérateur dès le retour au laboratoire.
V – Préparation du matériel Vérifier que les tendeurs de rappel des clapets des bouteilles « Niskin » n’ont pas été remplacés par un caoutchouc ordinaire (source de contamination importante en ammonium). Flaconnage : Les flacons neufs seront systématiquement remplis d’acide chlorhydrique 1N et laissés à tremper une nuit avant d’être vidés puis parfaitement rincés à l’eau déminéralisée de qualité Milli-Q
(entre 5 et 10 fois). Un blanc de contrôle sera alors effectué. Entretien courant : Entre 2 sorties de prélèvement peu espacées dans le temps, on peut conserver le mélange du dernier prélèvement avec les réactifs. Cela évite les éventuelles contaminations. En cas de blanc trop fort, les flacons seront à nouveau lavés à l’acide puis à l’eau déminéralisée. Un soin tout particulier sera apporté à la manipulation des bouchons et tout contact manuel avec le goulot des flacons sera évité. Le stockage des flacons devra alors se faire dans un endroit exempt de toute contamination
potentielle. Prévoir un bac de transport opaque avec couvercle et correctement équipé pour le transport en toute sécurité de ces flacons. Distributeurs de réactifs :

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
11
Ils seront régulièrement vidés et rincés afin d’éviter une détérioration du piston et d’en maintenir le bon fonctionnement. Une vérification des volumes distribués sera également effectuée par pesée et reportée sur la fiche de contrôle correspondante.(Annexe 1). VI - Préparation des réactifs Ils seront préparés à partir d’eau déminéralisée de haute qualité fraîchement soutirée : eau Milli Q par exemple, et essentiellement de produits MERCK (pro analysis). Les conserver au réfrigérateur et à l’abri de la lumière.
Réactif 1 (R1) : solution de phénol nitroprussiate, à préparer sous hotte aspirante. Dissoudre successivement 17,5g de phénol et 200 mg de nitroprussiate de sodium, dans 400 ml d’eau déminéralisée. Compléter à 500ml en fiole jaugée avec l’eau et homogénéiser.
Dans le cas de l’utilisation de phénol liquide, remplacer les 17.5 g de phénol en cristaux par 20 ml de phénol liquide.
Réactif2 (R2) : solution alcaline complexante au chlore. Dissoudre 11g de soude et 140g d’acide trisodique dans 400ml d’eau. Ajouter la quantité d’hypochlorite nécessaire pour réaliser une solution contenant 0,14% de chlore, soit 22 ml de solution fraîche d’hypochlorite. Compléter à 500 ml par de l’eau déminéralisée. La solution d’hypochlorite peut être remplacée par un autre réactif : le dichloroisocyanurate de sodium dihydraté. La préparation de ce réactif ne sera pas décrite (se reporter au manuel
Aminot-Kerouel pour plus de détails). Quantité de solution d’hypochlorite commerciale à ajouter : Le titre de la solution commerciale est contrôlé périodiquement de la façon suivante : A 1ml de la solution commerciale, on ajoute 50ml de KI à 1% et 0,25ml HCL concentré. L’iode libéré est titré par une solution de thiosulfate de sodium 0,1N. A 1ml de thiosulfate 0,1N correspond 3,54mg de chlore. VII – Prélèvement et conditionnement Le prélèvement des échantillons se fera aussitôt après ceux effectués pour l’analyse de l’oxygène et du pH. Tout contact manuel avec l’eau de prélèvement aussi bien qu’avec toute partie du matériel entrant en contact avec cette eau sera soigneusement évité tout au long de la procédure (voir chapitre II). Après avoir vérifié l’absence de toute contamination potentielle (fumée) sur le lieu de
prélèvement, vider dans la poubelle de déchets 1 à 3 flacons identifiés de leur mélange réactifs-échantillons précédents (ou eau déminéralisée) avant d’en effectuer un triple rinçage avec l’eau de prélèvement. Veiller au rinçage correct des bouchons et utiliser le système de pré-filtration ou de filtration prévu à cet effet, si nécessaire selon le site étudié (le changer pour chaque niveau de prélèvement). Remplir les flacons (+ 1 flacon « turbidité ») sans filtration avec pré-filtration ou filtration (si nécessaire) en ajustant le volume prélevé à (100 +
- 5) ml et ajouter successivement les réactifs 1 puis 2 (3 ml) au moyen des distributeurs correspondants, en prenant soin d’homogénéiser
énergiquement après chaque ajout. Ne pas ajouter de réactif dans le flacon « turbidité ». Réaliser le blanc de réactifs de la même manière au laboratoire avec de l’eau milliQ. VIII – Conservation et stockage

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
12
Aussitôt après le dernier ajout, placer les flacons à l’obscurité dans le bac de transport correspondant. Conserver ce dernier à température ambiante mais à l’abri du soleil et de toute contamination potentielle jusqu’au moment de l’analyse. Le temps de réaction, dépendant de la température, est de 6 heures minimum à environ 20°C. Il est préférable de la laisser se poursuivre une nuit. Le bleu d’indophénol formé étant stable
quelques jours, la lecture des échantillons sera effectuée si possible le lendemain du prélèvement mais peut attendre un week-end lorsque le prélèvement a lieu le vendredi.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
13
IX - Préparation de l’appareil et étalonnage Procéder de façon régulière (tous les 3 mois) à la vérification de votre spectrophotomètre (annexe 3), en utilisant un filtre Holmium qui permettra de contrôler la bonne position des longueurs d'onde du spectre. Vérifier également l'absorbance de filtres étalons. Cela permet de
tester l'optique du spectrophotomètre ou l'état de la lampe ... Etalonnage : Utiliser de l’eau déminéralisée de très bonne qualité et fraîchement soutirée pour préparer la solution mère et utiliser de la verrerie décontaminée. Les solutions étalons seront de préférence préparées dans de l’eau de mer appauvrie et filtrée. En milieu de salinité fortement variable (e.g. estuaires), les solutions peuvent être préparées dans de l’eau Milli-Q légèrement salée (dans l’eau Milli-Q pure, l’ammonium peut s’adsorber sur le
flacon). Il est conseillé de préparer ces solutions dans la même matrice que celle des échantillons mesurés par la suite. Il faut également essayer de travailler avec de l’eau contenant le moins d’ammonium possible. C’est en hiver que l’on aura les eaux marines les moins riches en ammonium. Solution d’eau Milli-Q légèrement salée : Diluer 0.5 g de NaCl dans un litre d’eau Milli-Q ou diluer 100 ml d’eau de mer appauvrie en ammonium dans 900 ml d’eau milli-Q. Solution mère primaire :
Sécher du sulfate d’ammonium ((NH4)2SO4, M = 132.14 g/mol) 1 heure à 105°C et laisser refroidir au dessiccateur. En peser 661mg précisément (au centième de mg) et les dissoudre dans 200ml d’eau. On apportera un soin particulier à la complète récupération des cristaux avant de compléter (avec de l'eau déionisée fraîche) à 1 litre en fiole jaugée de classe A+ : 1ml = 10 µmol d’ammonium. Homogénéiser et transvaser dans un flacon en verre correctement nettoyé et préalablement rincé 3 fois au moyen de cette même solution. Conservée au frais, à l’abri de la lumière, cette solution sera utilisable toute une année sous réserve d’éviter toute évaporation. Il est tout de
même conseillé de la renouveler au bout de 6 mois. Solution mère secondaire : Cette solution sera préparée avec de l'eau déionisée fraîche juste avant usage. Diluer 20 fois la solution primaire (5 ml/100 ml dans une fiole jaugée de classe A+) : 1 ml = 0.5 µmol d’ammonium. Solutions étalons : La gamme étalon dépend des concentrations normalement mesurées dans le milieu étudié et de la
salinité usuellement rencontré : nous à Brest : réalisation de la courbe d’étalonnage dans EMA à S = 35 : aucune correction de l’effet de sel. Cela va également déterminer les volumes utilisés pour préparer les solutions étalons. L’exemple suivant correspond à une mesure dans une gamme de 0 à 1 µmol.l-1 (nous 3 µM) et est simplement indicatif. A partir de la solution secondaire, réaliser au minimum 4 solutions étalons à EDM+0,25; EDM+0.50; EDM+0.75, EDM+1.00 µmole N-NH4
+/1 suivant les concentrations habituellement rencontrées, dans de l’eau de mer filtrée pauvre en ammonium. En milieu estuarien, EDM peut être remplacée par de l’eau Milli-Q légèrement salée.
Traiter ces solutions étalons, sans oublier ni l’eau de mer non dopée, ni le blanc réactif réalisé à partir de l’eau Milli-Q, de la même façon que les échantillons (cf §VII), dans le même type de flacons : Une solution étalon de 1000 ml permet d’effectuer le rinçage des flacons et 3 réplicats de chacune des solutions.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
14
Ces standards préparés avant le départ ou après le retour de la mission seront ainsi mesurés en même temps que les échantillons c'est-à-dire après être restés une nuit à l’obscurité et à température ambiante. La densité optique des solutions est mesurée à 630 nm avec un préchauffage du spectrophotomètre d’une demi-heure minimum avant les mesures.
Vérifier la date du dernier contrôle de bon fonctionnement de l’appareil : longueurs d’ondes (filtre Holmium) et densités optiques mesurées (filtre DO à 630 nm si possible) et effectuer ces vérifications si nécessaire ( cf : dossier métrologie de l’appareil). Commencer par effectuer l’auto-zéro de l’appareil sans présence de cuves afin de vérifier l’absence de problème optique ou électronique. Faire l’auto-zéro avec la (ou les) cuve(s) remplie(s) d’eau déionisée (blanc de cuve), selon le cas d’un simple ou double faisceau optique.
Vider la cuve de mesure, et y transférer, pour rinçage, une fraction du blanc réactif préparé au même moment que les étalons (eau déionisée +les réactifs R1 et R2), bien égoutté puis transférer le contenu pour remplissage de la cuve et lire la mesure d’absorbance (Br = blanc des réactifs) . Consigner la valeur sur les fiches appropriées somlit (cf. annexes 2) Vider de nouveau la cuve de mesure et effectuer les mesures des différentes solutions étalons en veillant à toujours rincer au moins une fois la cuve de mesure avec la solution suivante. Précautions particulières : Spectropohotomètre à double faisceau : Une fois l’auto-zéro effectué, ne plus toucher à la cuve
de référence ; travailler soigneusement avec la même cuve de mesure par rapport à la même cuve de référence. Toutes les mesures seront effectuées par rapport à l’eau dé-ionisée. Positionner la cuve de mesure toujours de la même façon sur le support en veillant à en maintenir les parois propres et sèches sans les rayer (égoutter et tamponner plutôt qu’essuyer et utiliser du papier spécifique) Surveiller la valeur du blanc des réactifs, lorsque les réactifs sont trop âgés, la valeur de ce blanc devient importante. Par exemple un blanc réalisé avec des réactifs « neufs » a une valeur de
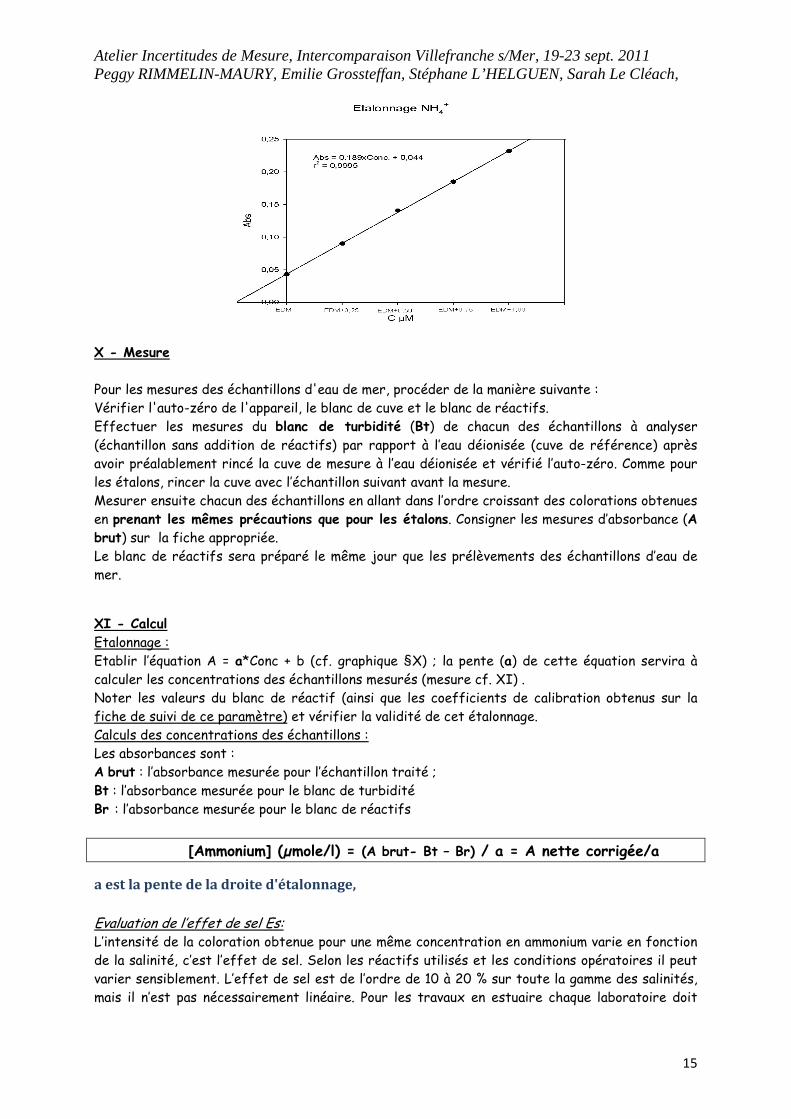
Densité Optique (DO) de 0,005 (cuve de 10 cm). Au delà d’une valeur de 0,015, prévoir de refaire les réactifs. Effectuer les lectures des différents étalons dans l’ordre croissant des concentrations, en prenant soin de rincer préalablement la cuve chaque fois avec l’étalon à mesurer, et consigner les valeurs sur la fiche appropriée. Tracer la droite d’étalonnage : Absorbance = f (concentration). Ne pas omettre de vérifier les valeurs de l’eau déionisée non dopée (blanc de réactifs).
On obtient ce type de tableau permettant le tracé de la courbe d’étalonnage :
R1 R2 R3
blancs réactifs 0,008 0,008 0,007
EDM pauvre 0,052 0,052 0,053
EDM+0,25 0,099 0,099 0,098
EDM+0,50 0,149 0,15 0,15
EDM+0,75 0,198 0,198 0,197
EDM+1,00 0,245 0,246 0,245
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
15
X - Mesure Pour les mesures des échantillons d'eau de mer, procéder de la manière suivante : Vérifier l'auto-zéro de l'appareil, le blanc de cuve et le blanc de réactifs. Effectuer les mesures du blanc de turbidité (Bt) de chacun des échantillons à analyser
(échantillon sans addition de réactifs) par rapport à l’eau déionisée (cuve de référence) après avoir préalablement rincé la cuve de mesure à l’eau déionisée et vérifié l’auto-zéro. Comme pour les étalons, rincer la cuve avec l’échantillon suivant avant la mesure. Mesurer ensuite chacun des échantillons en allant dans l’ordre croissant des colorations obtenues en prenant les mêmes précautions que pour les étalons. Consigner les mesures d’absorbance (A brut) sur la fiche appropriée. Le blanc de réactifs sera préparé le même jour que les prélèvements des échantillons d’eau de mer.
XI - Calcul Etalonnage : Etablir l’équation A = a*Conc + b (cf. graphique §X) ; la pente (a) de cette équation servira à calculer les concentrations des échantillons mesurés (mesure cf. XI) . Noter les valeurs du blanc de réactif (ainsi que les coefficients de calibration obtenus sur la fiche de suivi de ce paramètre) et vérifier la validité de cet étalonnage. Calculs des concentrations des échantillons : Les absorbances sont : A brut : l’absorbance mesurée pour l’échantillon traité ;
Bt : l’absorbance mesurée pour le blanc de turbidité Br : l’absorbance mesurée pour le blanc de réactifs
[Ammonium] (µmole/l) = (A brut- Bt – Br) / a = A nette corrigée/a
a est la pente de la droite d'étalonnage,
Evaluation de l’effet de sel Es: L’intensité de la coloration obtenue pour une même concentration en ammonium varie en fonction de la salinité, c’est l’effet de sel. Selon les réactifs utilisés et les conditions opératoires il peut varier sensiblement. L’effet de sel est de l’ordre de 10 à 20 % sur toute la gamme des salinités, mais il n’est pas nécessairement linéaire. Pour les travaux en estuaire chaque laboratoire doit

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
16
l’évaluer en fonction de ses propres conditions de travail. Pour déterminer la correction due à l’effet de sel, opérer de la façon suivante :
- Prélever ou préparer, par dilution d’eau de mer avec de l’eau déionisée, des eaux dont la salinité couvre la gamme de salinité usuelle des échantillons et dont la teneur en ammonium reste faible (0,5 µmole/l). Un minimum de 5 salinités différentes est nécessaire, dont l’une d’elles
identique à celle avec laquelle a été faite la courbe d’étalonnage (eau de mer ou eau douce). - Préparer, pour chacune des salinités, un étalon d’ammonium de même concentration (par
exemple 10 µmol/l). - Effectuer deux analyses de chacun de ces étalons, ainsi que des eaux non dopées
correspondantes, et prendre la moyenne des absorbances. Soustraire l’absorbance des eaux non dopées de celles des eaux dopées.
- Calculer le facteur correctif de l’effet de sel en fonction de la salinité :
AE = Absorbance nette de l’étalon préparé avec de l’eau de salinité identique à celle qui a servi à établir la courbe d’étalonnage,
AS = Absorbance nette d’un étalon à la salinité S. Le facteur correctif de l’effet de sel à la salinité S est : *Es = Ae/As L’absorbance sera multipliée par Es avant conversion en concentration à l’aide de la courbe d’étalonnage. A nette corrigée en tenant compte de l'effet de sel = Es x (A brut- Bt – Br) *Es : facteur correctif de l’effet de sel à la salinité S (il est égal à 1 pour des échantillons de même gamme de salinité que l’eau de mer utilisée pour la préparation des étalons).
XII - Entretien du matériel
Les bouteilles de prélèvement (Niskin) doivent être vidées à la fin des manipulations. Un séjour prolongé d'eau de mer à l’intérieur de celles-ci pourrait les contaminer. Vérifier que les flacons de prélèvement sont propres. Il arrive qu'au bout d'un certains temps d'utilisation un dépôt se fixe sur les parois des flacons. Dans ce cas, procéder à un nettoyage à l'acide. Cf. §V. Vérifier l'état des dispensettes de réactifs. Les réactifs peuvent cristalliser dans le piston. Conserver les flacons à l'abri de toutes contaminations, Lorsque l'on fait des mesures régulières, garder dans les flacons le reliquat de l'échantillon avec ses réactifs à l'abri de la lumière.
XIII - Conservation et entretien de l’appareillage Ne pas oublier de retirer les cuves du porte-cuve de l'appareil. L'atmosphère saline et les réactifs pourraient dégrader l'optique. Faire régulièrement des étalonnages de l'optique à l'aide de filtres adaptés cf. §X. XIV – Evacuation des essais et déchets Recueillir les échantillons et les effluents contenant les réactifs dans un bidon prévu à cet effet. Les déchets recueillis devront être retraités par une société spécialisée. XV - Bibliographie
Aminot A., Kérouel R., 2004. Hydrologie des écosystèmes marins. Paramètres et analyses. Ed. Ifremer, 336 p. Berthelot M., 1859. Répertoire de chimie appliquée. p254.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
17
Koroleff F., 1969. Direct determination of ammonia in natural waters as indophenol blue, ICES/CM/1969/C : 9, Hydrography Commitee, Ref. : L (Plankton C.), 4p. Le Corre P. et Tréguer P. Travaux pratiques Chimie Marine, Université de Bretagne Occidentale, Brest. Solorzano L., 1969. Determination of ammonia in natural waters by the phenol-hypochloite
method. Limnol. Oceanogr., 14, 799-801.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
18
On pose le modèle mathématique appliqué pour le calcul de la grandeur d’intérêt :
a
netteAC =
Équation 1
Sachant que C, (la grandeur qui nous intéresse) est calculée à partir de plusieurs grandeurs (Anette et
a), on combine les Ecart-types de toutes les grandeurs qui ont servi à son calcul conformément à la
loi de propagation des incertitudes :
Sachant que :
« incertitude absolue» = l’écart-type
« incertitude relative» = l’écart-type divisé par la moyenne (équivaut au coefficient de
variation),
lorsque l’on est dans le cas d’une somme (ou soustraction) : Le Carré de l’incertitude
absolue » = Somme des carrés des incertitudes absolues
lorsque l’on est dans le cas d’un produit ou d’un rapport : Le carré de l’incertitude
relative » = Somme des carrés des incertitudes relatives
Remarque : = principe très largement appliqué dans Le Document de référence: le GUM : ou Guide
pour l’expression de l’incertitude de mesure (NF ENV 13005)=> Cf. doc en consultation

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
19
Application :
Le carré de l’incertitude relative d’un rapport de deux grandeurs est égale à la somme du carré des
incertitudes relatives de ces grandeurs :
²²
nette
²
aa
A
A
CC
δδδδ++++
δδδδ====
δδδδ
On en déduit donc l’incertitude de la concentration d’ammonium dans l’eau de mer δδδδC :
²²
nette
nette
aa
A
ACC
∂∂∂∂++++
∂∂∂∂××××====δδδδ
• netteA∂∂∂∂
Même principe : on pose la formule de calcul et on en déduit la formule de calcul de l’écart-
type combiné :
,blancturbiditébrutenetteAAAA −−−−−−−−====
=> loi de propagation des incertitudes : « pour une somme ou une différence : Le carré de
l’incertitude absolue est égal à la somme des carrés des incertitudes absolues de ces grandeurs »,
(((( )))) (((( )))) (((( )))) (((( ))))²,blanc
²
turbidité
²
brute
²
netteAAAA δδδδ++++δδδδ++++δδδδ====δδδδ
Rappel : le coefficient d’étalonnage est réalisé à Brest dans une eau de mer articielle de salinité
proche des échantillons. Ainsi aucun facteur correctif n’est à considérer pour l’Effet de sel. Dans le cas
d’une station qui nécessite la correction de l’effet de sel, une étude des incertitudes sur ce facteur
devra être menée.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
20
On obtient donc l’incertitude sur la concentration en ammonium :
bruteAδδδδ (en ua): ’écart-type obtenu lors du test de réplicats d’échantillonnage
turbiditéAδδδδ (en ua): écart-type obtenu avec les réplicats de turbidité collectés lors du test
de réplicats d’échantillonnage
réactif,blancAδδδδ (en ua): écart-type obtenu avec réplicats de blancs réactifs collectés lors du
test de réplicats d’échantillonnage
aδ (en ua/µM): écart-type de la pente donnée par la droite de régression établie avec la
calibration Absorbance vs Concentration (Cf.« statistiques complémentaires d’Excel») avec l’ensemble des points de calibration obtenus de 5 étalonnages différents.
C (en µM) : valeur moyenne de concentration
netteA : valeur calculée à partir des absorbances moyennes brute,turb et BR
a : valeur de la pente de la droite d’étalonnage (Abs vs Conc) établie avec l’ensemble des points d’au moins 5 étalonnage différents
(((( )))) (((( )))) (((( ))))(((( ))))(((( ))))
²
²
nette
²
réactif,blanc
²
turbidité
²
brute
aa
A
AAACC
δδδδ++++
δδδδ++++δδδδ++++δδδδ××××====δδδδ
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
21
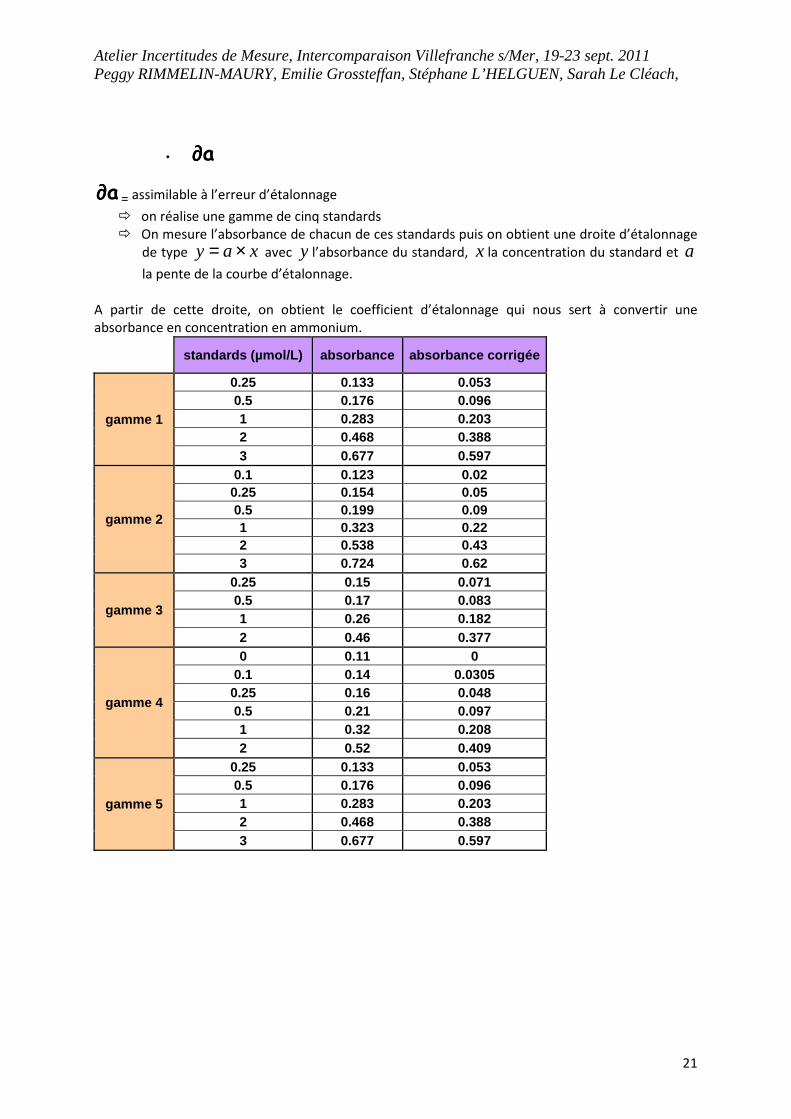
• a∂∂∂∂
a∂∂∂∂ = assimilable à l’erreur d’étalonnage
� on réalise une gamme de cinq standards
� On mesure l’absorbance de chacun de ces standards puis on obtient une droite d’étalonnage
de type xay ×= avec y l’absorbance du standard, x la concentration du standard et a
la pente de la courbe d’étalonnage.
A partir de cette droite, on obtient le coefficient d’étalonnage qui nous sert à convertir une
absorbance en concentration en ammonium.
standards (µmol/L) absorbance absorbance corrigée
gamme 1
0.25 0.133 0.053 0.5 0.176 0.096 1 0.283 0.203 2 0.468 0.388 3 0.677 0.597
gamme 2
0.1 0.123 0.02 0.25 0.154 0.05 0.5 0.199 0.09 1 0.323 0.22 2 0.538 0.43 3 0.724 0.62
gamme 3
0.25 0.15 0.071 0.5 0.17 0.083 1 0.26 0.182 2 0.46 0.377
gamme 4
0 0.11 0 0.1 0.14 0.0305 0.25 0.16 0.048 0.5 0.21 0.097 1 0.32 0.208 2 0.52 0.409
gamme 5
0.25 0.133 0.053 0.5 0.176 0.096 1 0.283 0.203 2 0.468 0.388 3 0.677 0.597
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
22
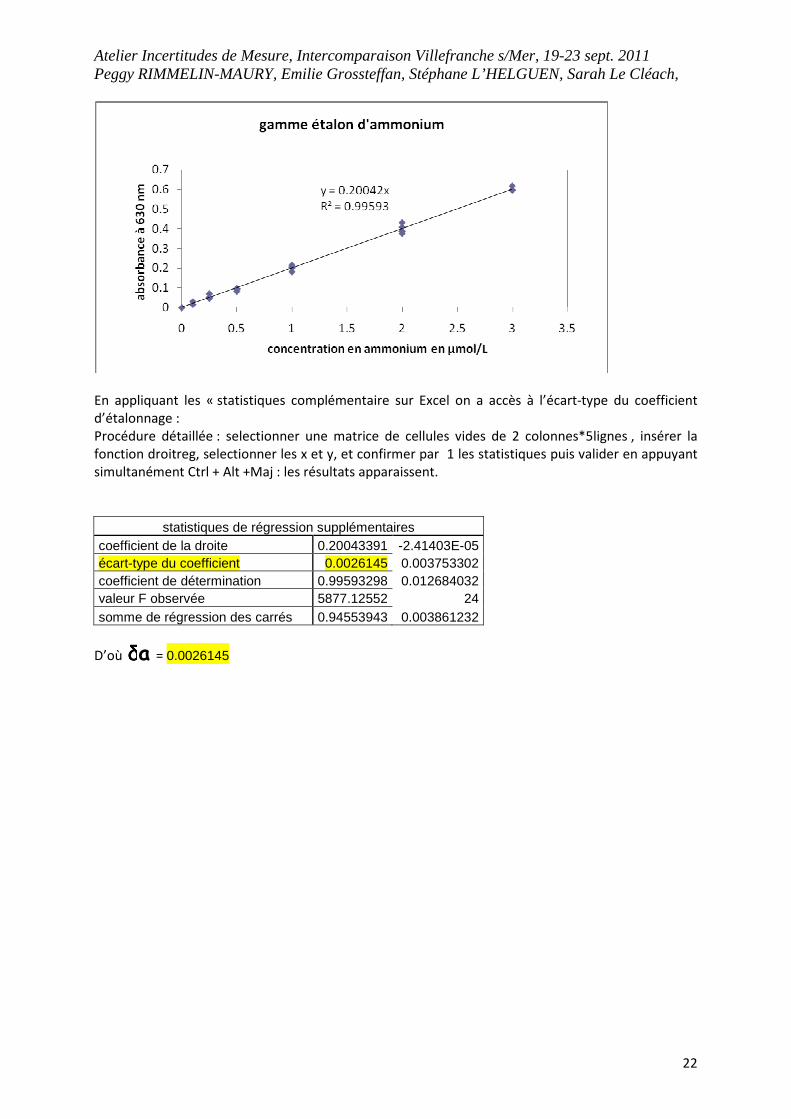
En appliquant les « statistiques complémentaire sur Excel on a accès à l’écart-type du coefficient
d’étalonnage :
Procédure détaillée : selectionner une matrice de cellules vides de 2 colonnes*5lignes , insérer la
fonction droitreg, selectionner les x et y, et confirmer par 1 les statistiques puis valider en appuyant
simultanément Ctrl + Alt +Maj : les résultats apparaissent.
statistiques de régression supplémentaires coefficient de la droite 0.20043391 -2.41403E-05 écart-type du coefficient 0.0026145 0.003753302 coefficient de détermination 0.99593298 0.012684032 valeur F observée 5877.12552 24 somme de régression des carrés 0.94553943 0.003861232
D’où aδδδδ = 0.0026145
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
23
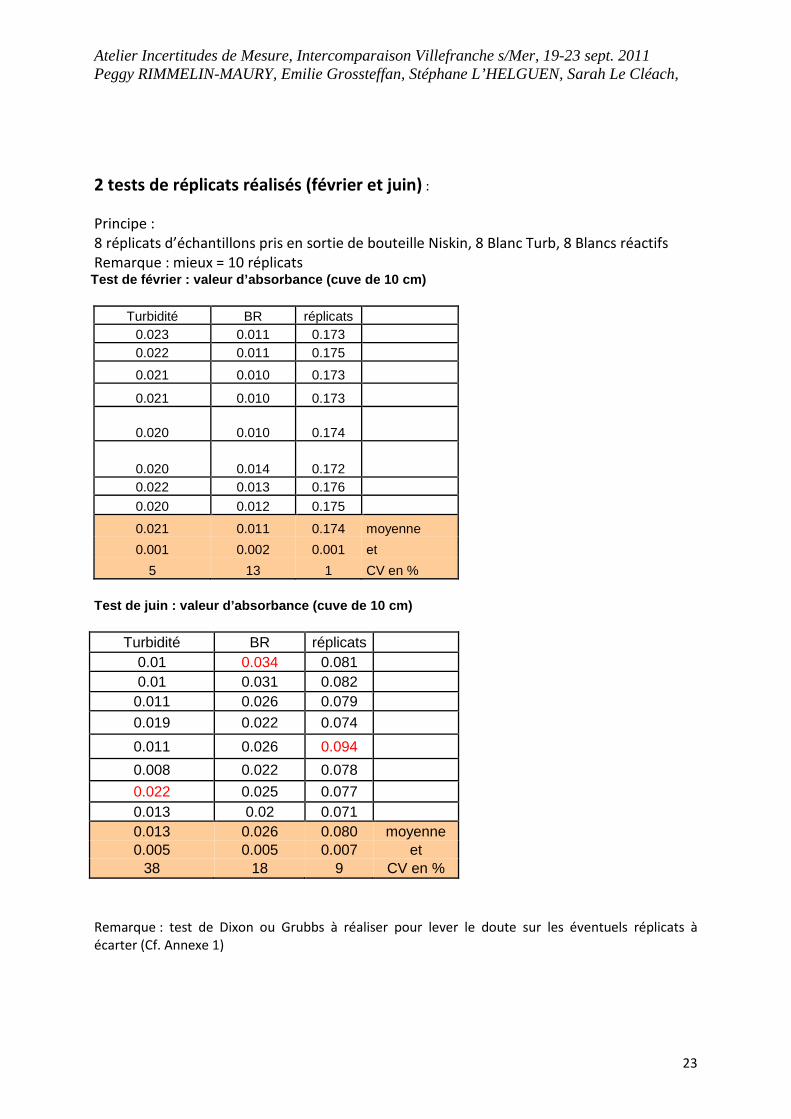
2 tests de réplicats réalisés (février et juin) :
Principe :
8 réplicats d’échantillons pris en sortie de bouteille Niskin, 8 Blanc Turb, 8 Blancs réactifs
Remarque : mieux = 10 réplicats Test de février : valeur d’absorbance (cuve de 10 cm)
Turbidité BR réplicats 0.023 0.011 0.173 0.022 0.011 0.175
0.021 0.010 0.173
0.021 0.010 0.173
0.020 0.010 0.174
0.020 0.014 0.172 0.022 0.013 0.176
0.020 0.012 0.175
0.021 0.011 0.174 moyenne
0.001 0.002 0.001 et
5 13 1 CV en %
Test de juin : valeur d’absorbance (cuve de 10 cm)
Turbidité BR réplicats 0.01 0.034 0.081 0.01 0.031 0.082
0.011 0.026 0.079
0.019 0.022 0.074
0.011 0.026 0.094
0.008 0.022 0.078
0.022 0.025 0.077 0.013 0.02 0.071 0.013 0.026 0.080 moyenne 0.005 0.005 0.007 et
38 18 9 CV en %
Remarque : test de Dixon ou Grubbs à réaliser pour lever le doute sur les éventuels réplicats à
écarter (Cf. Annexe 1)
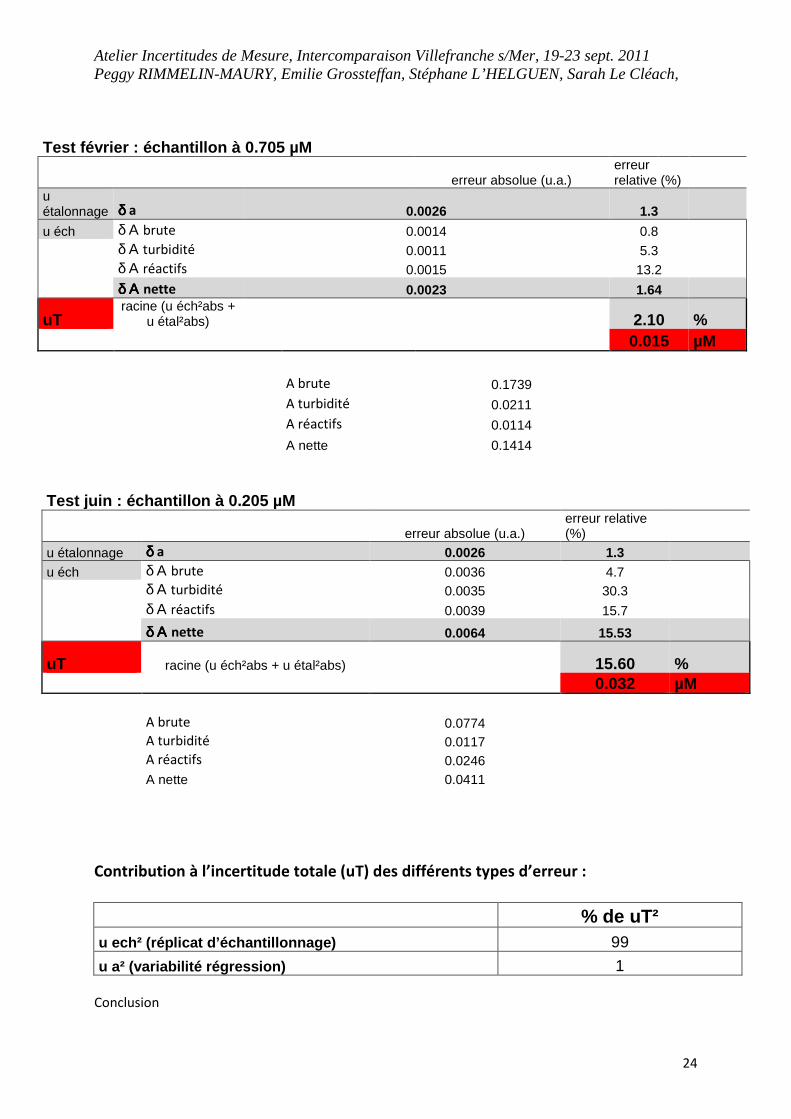
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
24
Test février : échantillon à 0.705 µM
erreur absolue (u.a.) erreur relative (%)
u étalonnage δ δ δ δ a 0.0026 1.3
u éch δ Α brute 0.0014 0.8
δ Α turbidité 0.0011 5.3
δ Α réactifs 0.0015 13.2
δ Α δ Α δ Α δ Α nette 0.0023 1.64
uT racine (u éch²abs +
u étal²abs) 2.10 % 0.015 µM A brute 0.1739
A turbidité 0.0211
A réactifs 0.0114
A nette 0.1414
Contribution à l’incertitude totale (uT) des différents types d’erreur :
% de uT² u ech² (réplicat d’échantillonnage) 99
u a² (variabilité régression) 1
Conclusion
Test juin : échantillon à 0.205 µM
erreur absolue (u.a.) erreur relative (%)
u étalonnage δ δ δ δ a 0.0026 1.3
u éch δ Α brute 0.0036 4.7 δ Α turbidité 0.0035 30.3
δ Α réactifs 0.0039 15.7
δ Α δ Α δ Α δ Α nette 0.0064 15.53
uT racine (u éch²abs + u étal²abs) 15.60 % 0.032 µM A brute 0.0774 A turbidité 0.0117 A réactifs 0.0246 A nette 0.0411
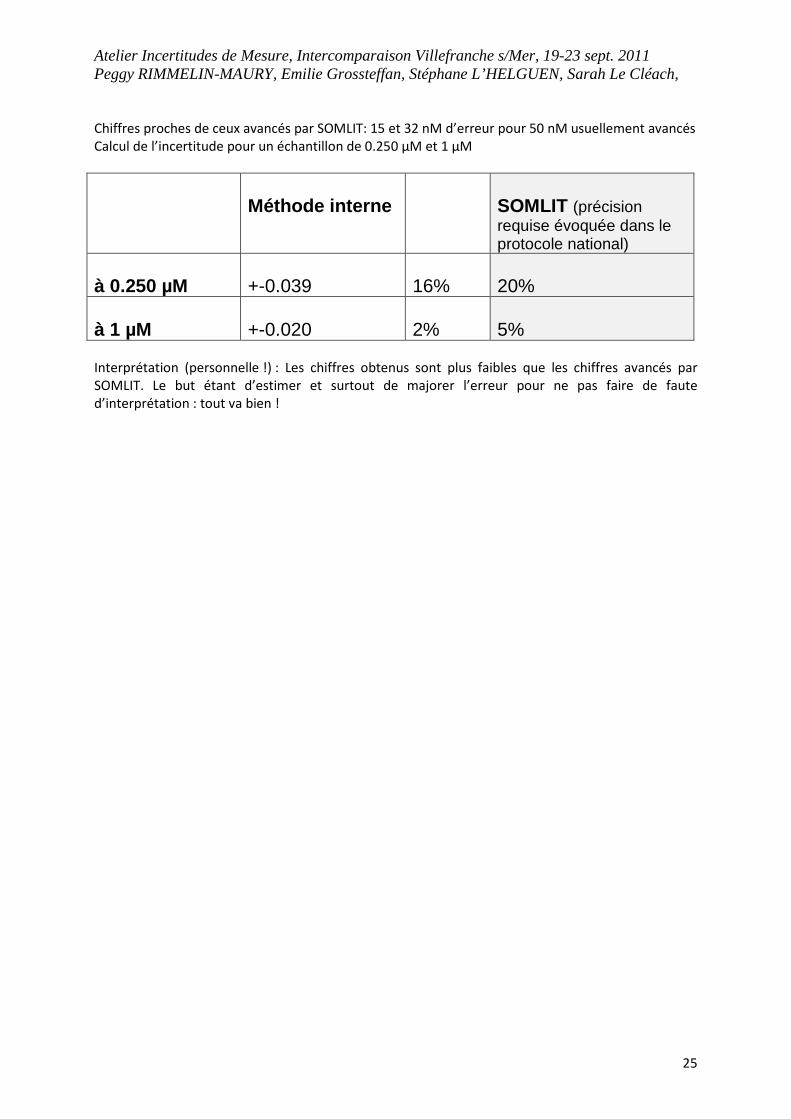
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
25
Chiffres proches de ceux avancés par SOMLIT: 15 et 32 nM d’erreur pour 50 nM usuellement avancés
Calcul de l’incertitude pour un échantillon de 0.250 µM et 1 µM
Méthode interne
SOMLIT (précision requise évoquée dans le protocole national)
à 0.250 µM
+-0.039
16%
20%
à 1 µM
+-0.020
2%
5%
Interprétation (personnelle !) : Les chiffres obtenus sont plus faibles que les chiffres avancés par
SOMLIT. Le but étant d’estimer et surtout de majorer l’erreur pour ne pas faire de faute
d’interprétation : tout va bien !

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
26
APPROCHE 2
METHODE GUM conforme à norme XP-T90-220 :
Soit uT (C), l’incertitude -type total (écar-type combiné) sur la concentration en ammonium C :
)²()²()( CuCuCu grandeursnéchantilloT +=
grandeursu = l’incertitude- type due à l’étalonnage : loi de propagation des incertitudes
)(Cnéchantillou = l’incertitude- type due à l’échantillon : étude de fidélité de lecture du signal de
l’échantillon
Rappel :Fidélité : en géné : erreur de fidélité = composante aléatoire de l’erreur de l’instrument ou
d’un système de mesure : s’exprime par la variance (E-T ou coeff de var.)
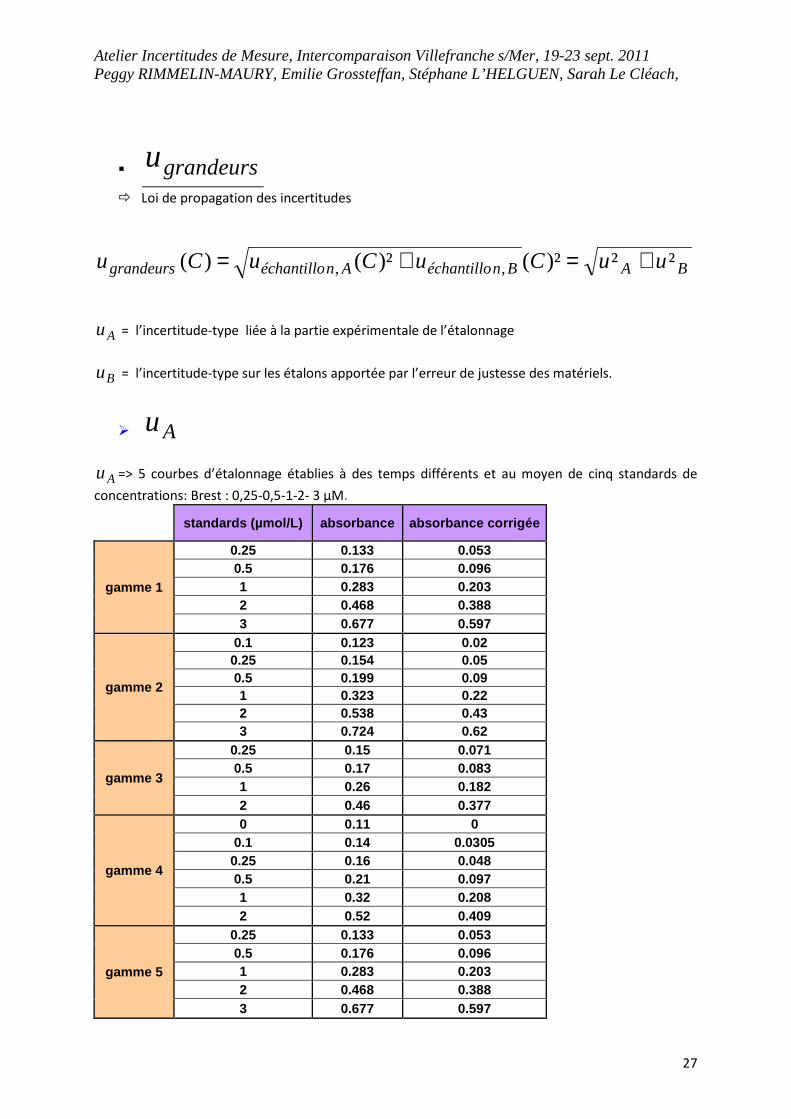
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
27
� grandeursu
� Loi de propagation des incertitudes
BABnéchantilloAnéchantillograndeurs uuCuCuCu ²²)²()²()( ,, +=+=
Au = l’incertitude-type liée à la partie expérimentale de l’étalonnage
Bu = l’incertitude-type sur les étalons apportée par l’erreur de justesse des matériels.
� Au
Au => 5 courbes d’étalonnage établies à des temps différents et au moyen de cinq standards de
concentrations: Brest : 0,25-0,5-1-2- 3 µM.
standards (µmol/L) absorbance absorbance corrigée
gamme 1
0.25 0.133 0.053 0.5 0.176 0.096 1 0.283 0.203 2 0.468 0.388 3 0.677 0.597
gamme 2
0.1 0.123 0.02 0.25 0.154 0.05 0.5 0.199 0.09 1 0.323 0.22 2 0.538 0.43 3 0.724 0.62
gamme 3
0.25 0.15 0.071 0.5 0.17 0.083 1 0.26 0.182 2 0.46 0.377
gamme 4
0 0.11 0 0.1 0.14 0.0305 0.25 0.16 0.048 0.5 0.21 0.097 1 0.32 0.208 2 0.52 0.409
gamme 5
0.25 0.133 0.053 0.5 0.176 0.096 1 0.283 0.203 2 0.468 0.388 3 0.677 0.597
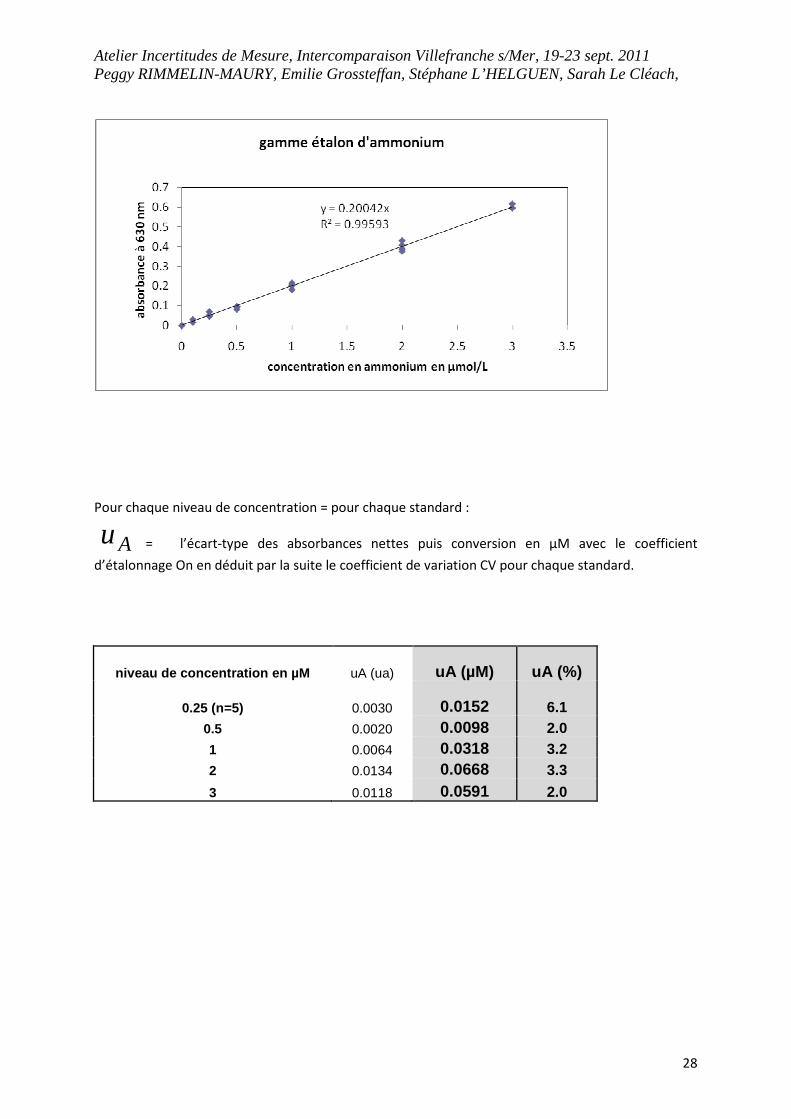
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
28
Pour chaque niveau de concentration = pour chaque standard :
Au
= l’écart-type des absorbances nettes puis conversion en µM avec le coefficient
d’étalonnage On en déduit par la suite le coefficient de variation CV pour chaque standard.
niveau de concentration en µM uA (ua) uA (µM) uA (%)
0.25 (n=5) 0.0030 0.0152 6.1 0.5 0.0020 0.0098 2.0 1 0.0064 0.0318 3.2 2 0.0134 0.0668 3.3
3 0.0118 0.0591 2.0

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
29
� Bu : sur la préparation des standards d’étalonnage
• Incertitudes sur la préparation de la solution mère
La solution mère d’ammonium (sm), est préparée par dissolution d’une masse smm(0.066 g)
dans un volume smV(1L)
:
sm
smsm V
mC =
²
sm
sm
²
sm
sm
²
sm
sm
VV
mm
CC
δδδδ++++
δδδδ====
δδδδ
D’où
²
sm
sm
²
sm
sm
smsm VV
mm
CC
δδδδ++++
δδδδ××××====δδδδ
Équation 1: incertitude-type de la concentration de la solution mère
Erreur-type sur la pesée :
sm
sm
mmδδδδ
- Reproductibilité d’ une pesée : 12.5% sur une pesée de 0.2 g (selon certificat d’étalonnage de
la balance) soit un EMT = 12.5/100*0.066g (masse de sulfate d’ammonium dans 1L) =0.00825
g d’où une erreur associée (telle que décrite dans norme p. 29) : EMT (« écart max toléré »)
u (EMT) = EMT/ =0.0048 g
- Résolution de la balance d= 0.01 g ; incertitude associée (telle que décrite dans norme p. 30) :
ud = =0.0029 g
- Erreur de justesse des masses étalons employées : pour la masse de 0.2 g EMT = pas indiqué
dans le rapport d’étalonnage : on prend 10% d’erreur soit u étal = 0.02g

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
30
Enfin erreur de pesée : uM² = ud² +uEMT²+uétal² = 0.00013 g soit 0.2%
smVδδδδ Erreur sur le volume de la fiole : selon constructeur, l’erreur l’Ecart-type Maximal (ETM)
s’élève à 0.250 mL pour une fiole de 1L soit 0.014%
Enfin erreur globale sur C sm = 0.2%
• Incertitudes sur la préparation des standards
Les standards de concentration stdC sont obtenus par dilution de la solution mère. En effet, on
prélève un volume précis de la solution mère que l’on dilue dans une fiole jaugée d’eau
déminéralisée.
On cherche donc à déterminer les erreurs de justesse de la verrerie utilisée pour la préparation
de nos standards.
Sachant que
fiole
pipeté
smstd V
VCC ××××==== ,
Alors
²
fiole
fiole
²
pipeté
pipeté
²
sm
sm
²
std
std
V
V
V
V
CC
C
C
δδδδ++++
δδδδ++++
δδδδ====
δδδδ
Donc
fiole
fiole
pipeté
pipeté
sm
smstdstd V
V
V
V
C
CCC
δδδδ +
+
×=
²²
Équation 2: incertitude-type de la concentration des standards

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
31
std
std
C
Cδδδδ
pipetéV Erreur-type sur les volumes pipetés :
Erreur sur les pipettes utilisées pour le pipetage de la solution mère : 0.23 à 0.27% (selon
certification d’étalonnage des pipettes) donc on considère la valeur majorée de 0.27% :
uVpipeté = 0.0027 mL
fioleVδδδδ :Erreur sur fiole de 250 mL : EMT = 0.1 mL soit une erreur de 0.023%
Cf. Tableau de calcul détaillé (ppt)
On obtient finalement une erreur relative sur la concentration des standards
std
std
C
Cδδδδ= 0.208% (très faible : négligeable !!!!)
� néchantillou : fidélité de la mesure de l’échantillon
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
32
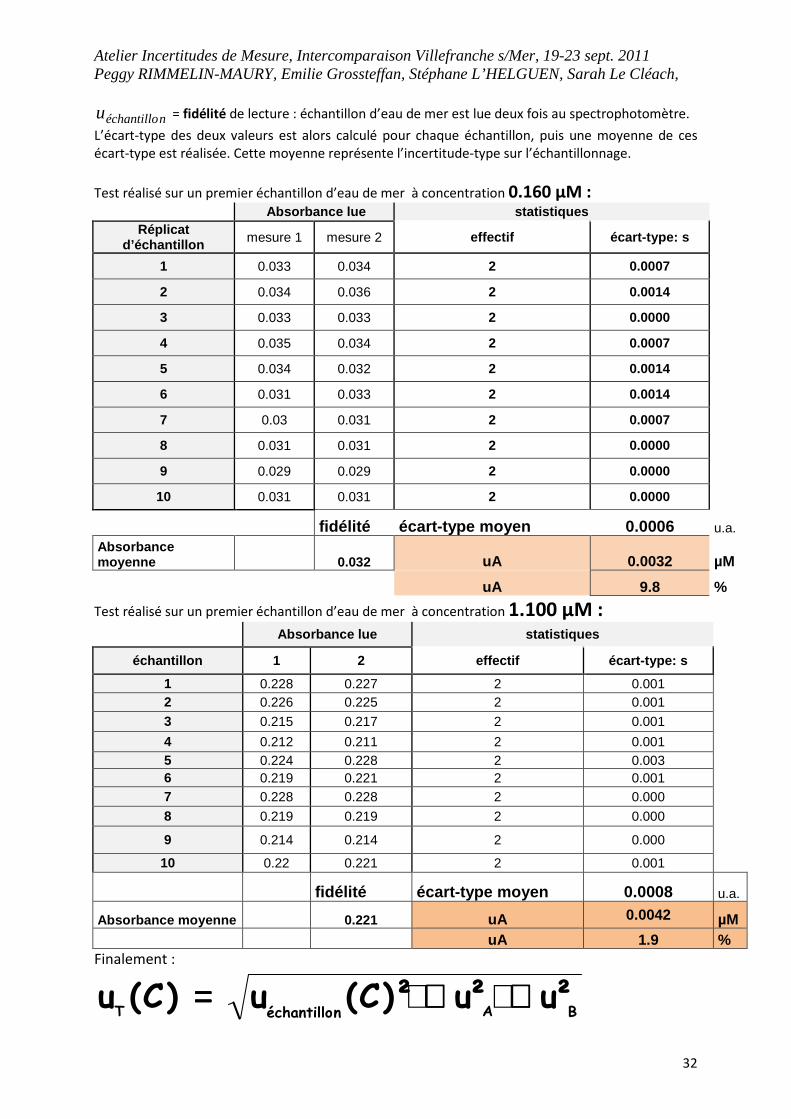
néchantillou = fidélité de lecture : échantillon d’eau de mer est lue deux fois au spectrophotomètre.
L’écart-type des deux valeurs est alors calculé pour chaque échantillon, puis une moyenne de ces
écart-type est réalisée. Cette moyenne représente l’incertitude-type sur l’échantillonnage.
Test réalisé sur un premier échantillon d’eau de mer à concentration 0.160 µM : Absorbance lue statistiques
Réplicat d’échantillon mesure 1 mesure 2 effectif écart-type: s
1 0.033 0.034 2 0.0007
2 0.034 0.036 2 0.0014
3 0.033 0.033 2 0.0000
4 0.035 0.034 2 0.0007
5 0.034 0.032 2 0.0014
6 0.031 0.033 2 0.0014
7 0.03 0.031 2 0.0007
8 0.031 0.031 2 0.0000
9 0.029 0.029 2 0.0000
10 0.031 0.031 2 0.0000
fidélité écart-type moyen 0.0006 u.a. Absorbance moyenne 0.032 uA
0.0032 µM
uA 9.8 %
Test réalisé sur un premier échantillon d’eau de mer à concentration 1.100 µM :
Absorbance lue statistiques
échantillon 1 2 effectif écart-type: s 1 0.228 0.227 2 0.001 2 0.226 0.225 2 0.001 3 0.215 0.217 2 0.001 4 0.212 0.211 2 0.001 5 0.224 0.228 2 0.003 6 0.219 0.221 2 0.001 7 0.228 0.228 2 0.000 8 0.219 0.219 2 0.000
9 0.214 0.214 2 0.000 10 0.22 0.221 2 0.001
fidélité écart-type moyen 0.0008 u.a.
Absorbance moyenne 0.221 uA 0.0042 µM uA 1.9 % Finalement :
BAnéchantilloT²u²u)²C(u)C(u ++++++++====
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
33
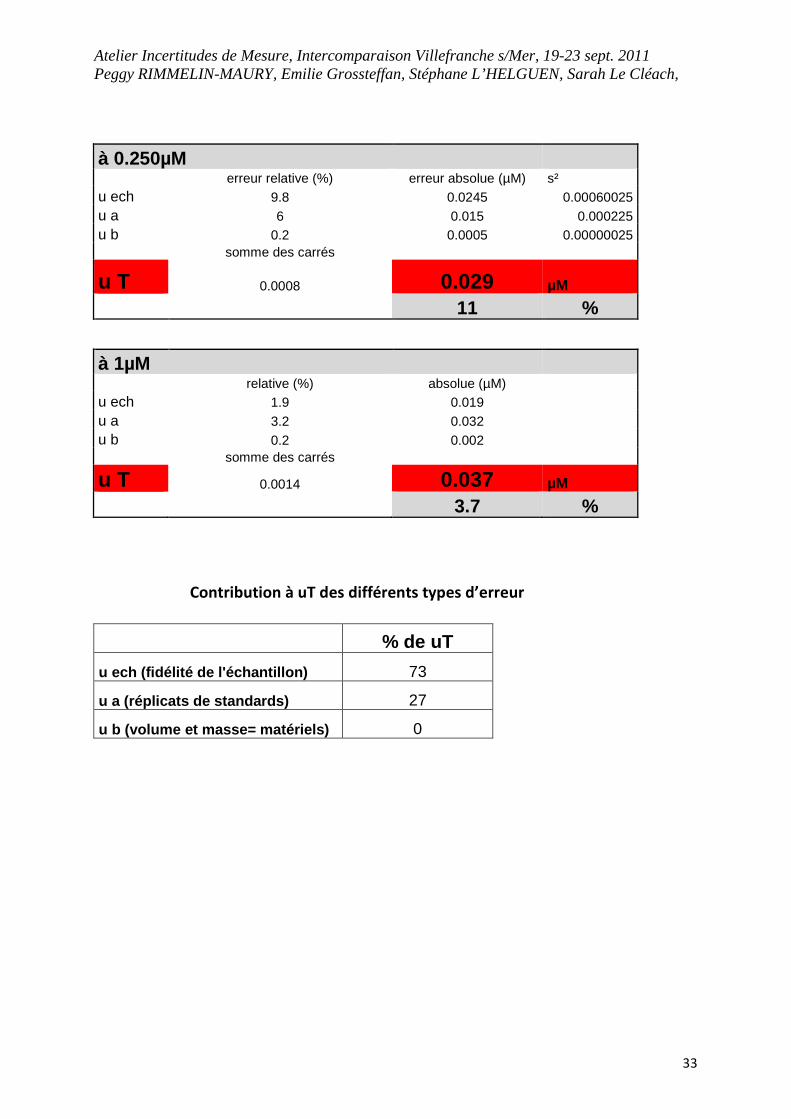
à 0.250µM erreur relative (%) erreur absolue (µM) s² u ech 9.8 0.0245 0.00060025 u a 6 0.015 0.000225 u b 0.2 0.0005 0.00000025 somme des carrés
u T 0.0008 0.029 µM
11 %
à 1µM relative (%) absolue (µM) u ech 1.9 0.019 u a 3.2 0.032 u b 0.2 0.002 somme des carrés
u T 0.0014 0.037 µM
3.7 %
Contribution à uT des différents types d’erreur
% de uT
u ech (fidélité de l'échantillon) 73
u a (réplicats de standards) 27
u b (volume et masse= matériels) 0

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
34
Tableau comparatif des erreurs estimées :
Méthode interne
Méthode norme GUM
SOMLIT
à 0.250 µM
16%
11%
20%
à 1 µM
2%
4%
5%
Chiffres obtenus également plus faibles que chiffres pris en compte dans SOMLIT : confirmation que
chiffre avancés par SOMLIT = OK, sur Brest !
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
35
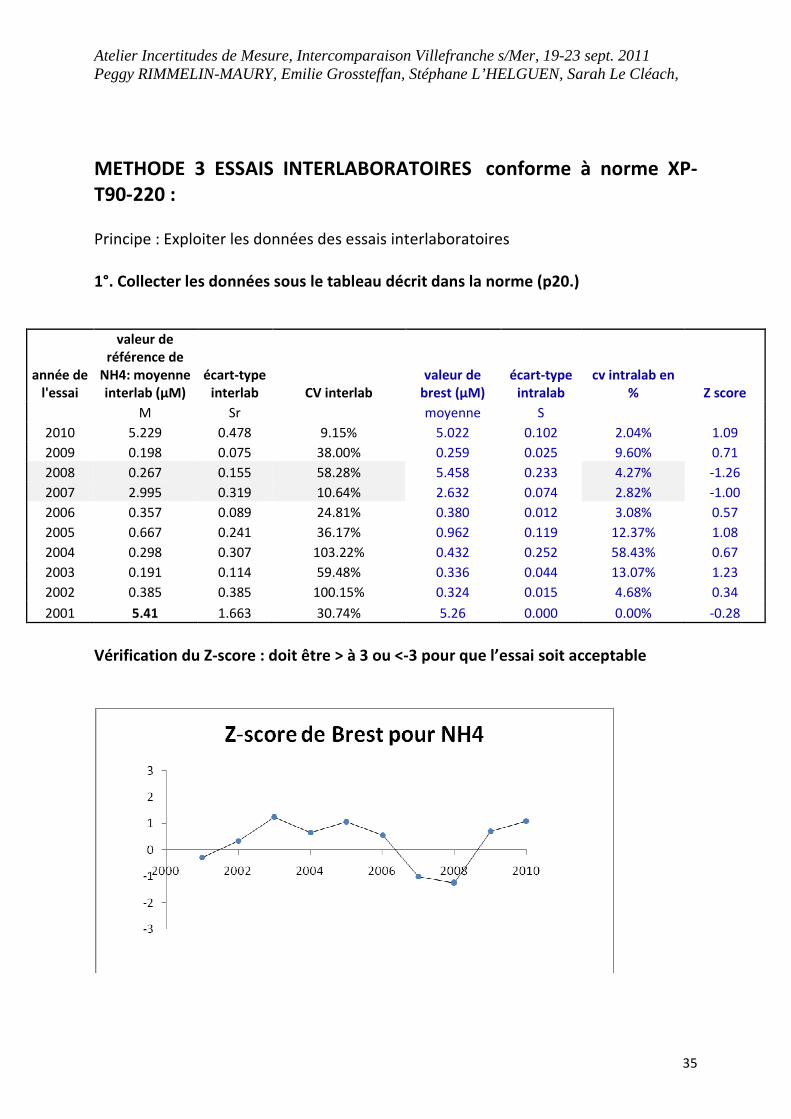
METHODE 3 ESSAIS INTERLABORATOIRES conforme à norme XP-
T90-220 :
Principe : Exploiter les données des essais interlaboratoires
1°. Collecter les données sous le tableau décrit dans la norme (p20.)
Vérification du Z-score : doit être > à 3 ou <-3 pour que l’essai soit acceptable
année de
l'essai
valeur de
référence de
NH4: moyenne
interlab (µM)
écart-type
interlab CV interlab
valeur de
brest (µM)
écart-type
intralab
cv intralab en
% Z score
M Sr moyenne S
2010 5.229 0.478 9.15% 5.022 0.102 2.04% 1.09
2009 0.198 0.075 38.00% 0.259 0.025 9.60% 0.71
2008 0.267 0.155 58.28% 5.458 0.233 4.27% -1.26
2007 2.995 0.319 10.64% 2.632 0.074 2.82% -1.00
2006 0.357 0.089 24.81% 0.380 0.012 3.08% 0.57
2005 0.667 0.241 36.17% 0.962 0.119 12.37% 1.08
2004 0.298 0.307 103.22% 0.432 0.252 58.43% 0.67
2003 0.191 0.114 59.48% 0.336 0.044 13.07% 1.23
2002 0.385 0.385 100.15% 0.324 0.015 4.68% 0.34
2001 5.41 1.663 30.74% 5.26 0.000 0.00% -0.28
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
36
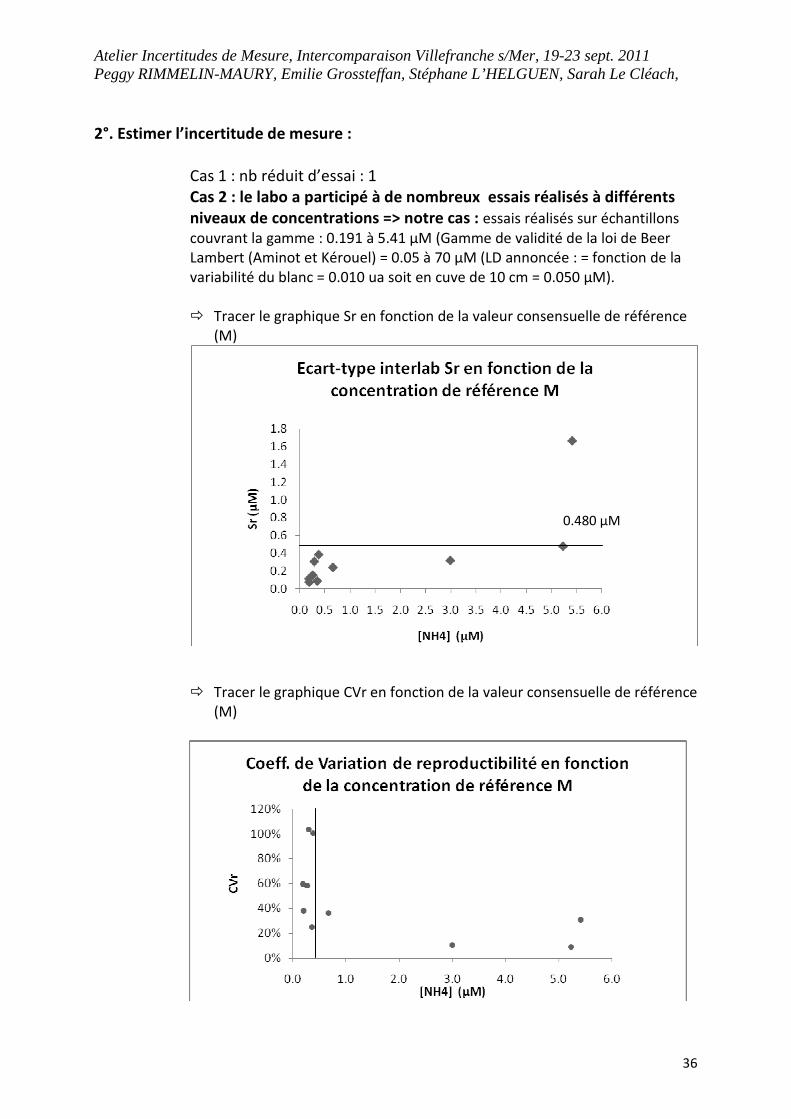
2°. Estimer l’incertitude de mesure :
Cas 1 : nb réduit d’essai : 1
Cas 2 : le labo a participé à de nombreux essais réalisés à différents
niveaux de concentrations => notre cas : essais réalisés sur échantillons
couvrant la gamme : 0.191 à 5.41 µM (Gamme de validité de la loi de Beer
Lambert (Aminot et Kérouel) = 0.05 à 70 µM (LD annoncée : = fonction de la
variabilité du blanc = 0.010 ua soit en cuve de 10 cm = 0.050 µM).
� Tracer le graphique Sr en fonction de la valeur consensuelle de référence
(M)
� Tracer le graphique CVr en fonction de la valeur consensuelle de référence
(M)
0.480 µM

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
37
� Si Sr ou CVr constant : prise en compte de cette valeur sur tout le
domaine
� Sinon, exprimer incertitude variable pour des domaines séparés : Pas
simple !
Selon Sr, on peut interpréter : Erreur de ± 0.480 µM sur tout le domaine
Selon CVr, on peut interpréter :
Pour concentration < à 0.3 µM, l’incertitude max s’élève à 103%
Pour concentration > 0.3 µM, l’incertitude max s’élève à 36 %
Finalement, pour estimer une incertitude selon cette méthode pour des concentrations
comparables aux deux études précédentes, choix personnel de prendre le CVr pour des
concentrations proches (discutable):
CVr à 0.267 µM (2008) = 58.28% soit pour la concentration de référence 0.250 ±0.146 µM
CVr à 2.995 µM (2007) = 10.64% soit pour 1.000 ± 0.106 µM
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
38
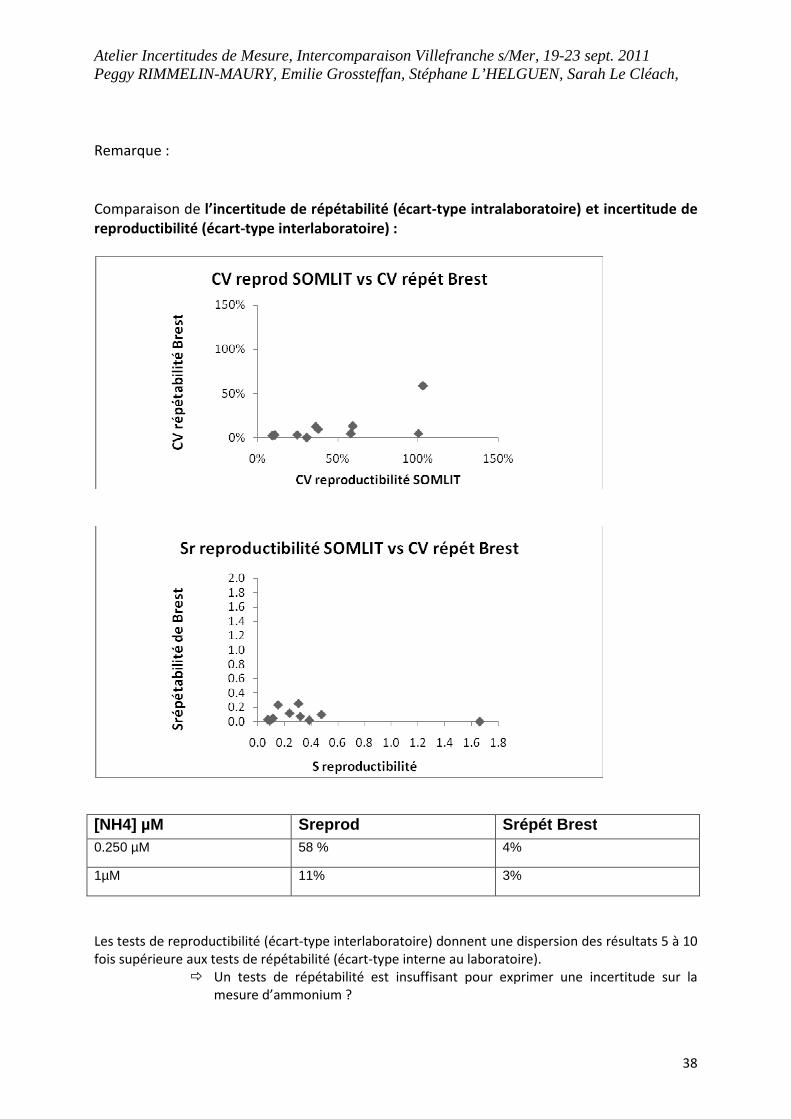
Remarque :
Comparaison de l’incertitude de répétabilité (écart-type intralaboratoire) et incertitude de
reproductibilité (écart-type interlaboratoire) :
[NH4] µM Sreprod Srépét Brest 0.250 µM 58 % 4%
1µM 11% 3%
Les tests de reproductibilité (écart-type interlaboratoire) donnent une dispersion des résultats 5 à 10
fois supérieure aux tests de répétabilité (écart-type interne au laboratoire).
� Un tests de répétabilité est insuffisant pour exprimer une incertitude sur la
mesure d’ammonium ?
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
39
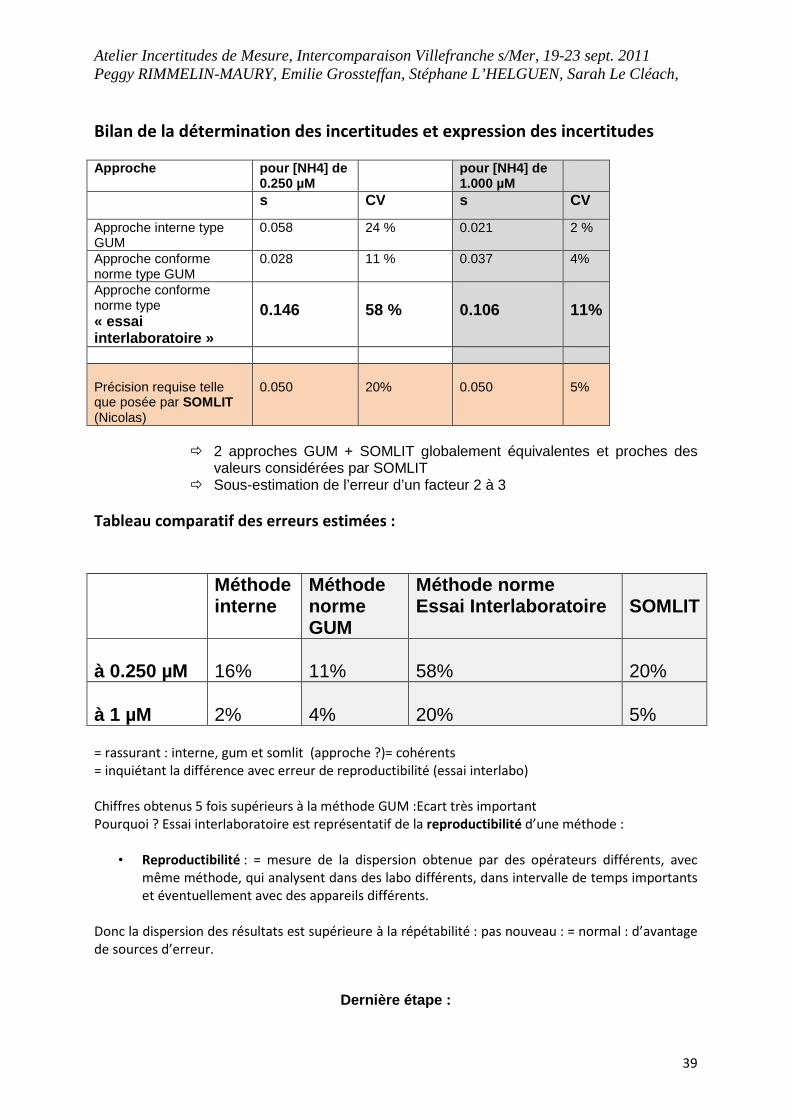
Bilan de la détermination des incertitudes et expression des incertitudes
Approche pour [NH4] de 0.250 µM
pour [NH4] de 1.000 µM
s CV s CV
Approche interne type GUM
0.058 24 % 0.021 2 %
Approche conforme norme type GUM
0.028 11 % 0.037 4%
Approche conforme norme type « essai interlaboratoire »
0.146
58 %
0.106
11%
Précision requise telle que posée par SOMLIT (Nicolas)
0.050
20%
0.050
5%
� 2 approches GUM + SOMLIT globalement équivalentes et proches des
valeurs considérées par SOMLIT � Sous-estimation de l’erreur d’un facteur 2 à 3
Tableau comparatif des erreurs estimées :
Méthode interne
Méthode norme GUM
Méthode norme Essai Interlaboratoire
SOMLIT
à 0.250 µM
16%
11%
58%
20%
à 1 µM
2%
4%
20%
5%
= rassurant : interne, gum et somlit (approche ?)= cohérents
= inquiétant la différence avec erreur de reproductibilité (essai interlabo)
Chiffres obtenus 5 fois supérieurs à la méthode GUM :Ecart très important
Pourquoi ? Essai interlaboratoire est représentatif de la reproductibilité d’une méthode :
• Reproductibilité : = mesure de la dispersion obtenue par des opérateurs différents, avec
même méthode, qui analysent dans des labo différents, dans intervalle de temps importants
et éventuellement avec des appareils différents.
Donc la dispersion des résultats est supérieure à la répétabilité : pas nouveau : = normal : d’avantage
de sources d’erreur.
Dernière étape :
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
40
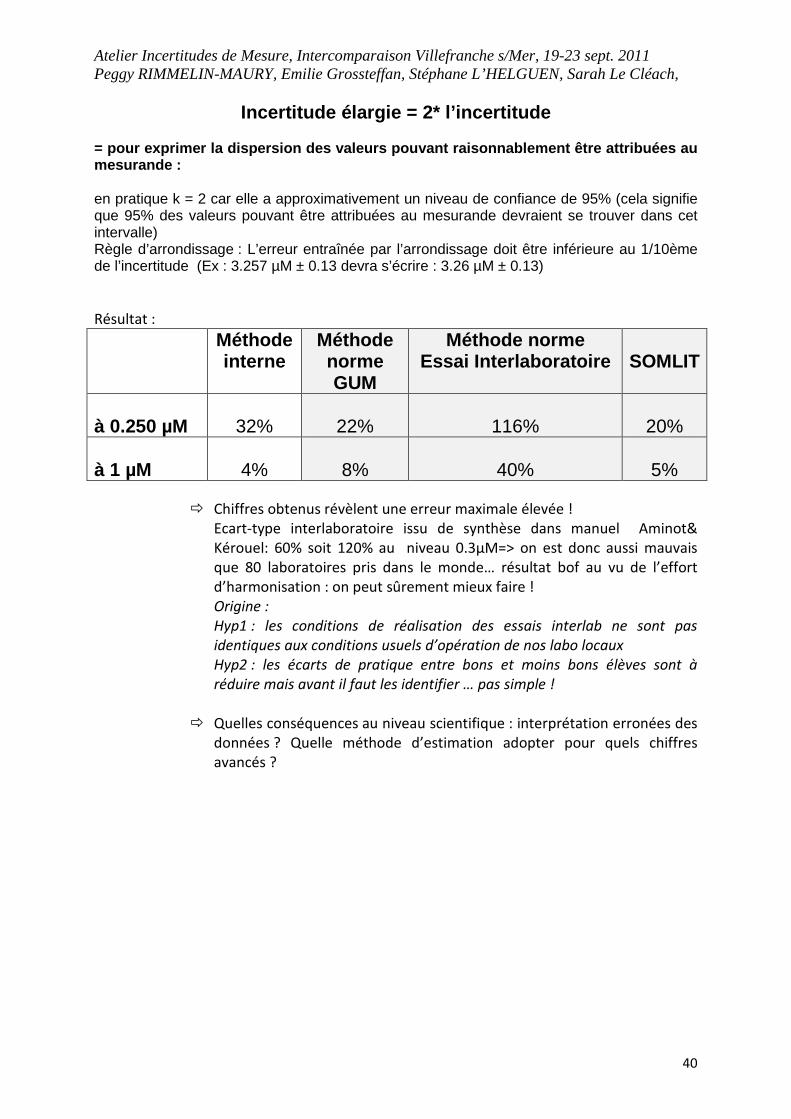
Incertitude élargie = 2* l’incertitude = pour exprimer la dispersion des valeurs pouvant r aisonnablement être attribuées au mesurande : en pratique k = 2 car elle a approximativement un niveau de confiance de 95% (cela signifie que 95% des valeurs pouvant être attribuées au mesurande devraient se trouver dans cet intervalle) Règle d’arrondissage : L’erreur entraînée par l’arrondissage doit être inférieure au 1/10ème de l’incertitude (Ex : 3.257 µM ± 0.13 devra s’écrire : 3.26 µM ± 0.13)
Résultat :
Méthode interne
Méthode norme GUM
Méthode norme Essai Interlaboratoire
SOMLIT
à 0.250 µM
32%
22%
116%
20%
à 1 µM
4%
8%
40%
5%
� Chiffres obtenus révèlent une erreur maximale élevée !
Ecart-type interlaboratoire issu de synthèse dans manuel Aminot&
Kérouel: 60% soit 120% au niveau 0.3µM=> on est donc aussi mauvais
que 80 laboratoires pris dans le monde… résultat bof au vu de l’effort
d’harmonisation : on peut sûrement mieux faire !
Origine :
Hyp1 : les conditions de réalisation des essais interlab ne sont pas
identiques aux conditions usuels d’opération de nos labo locaux
Hyp2 : les écarts de pratique entre bons et moins bons élèves sont à
réduire mais avant il faut les identifier … pas simple !
� Quelles conséquences au niveau scientifique : interprétation erronées des
données ? Quelle méthode d’estimation adopter pour quels chiffres
avancés ?

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
41
Détermination de la limite de détection et de quant ification
Tout comme pour l’incertitude associée à la grandeur, il est important de préciser la méthode employée dans la détermination de la limite de détection que l’on avance. On peut distinguer notamment limite de détection de l’appareil et limite de détection de la méthode. La limite de détection de l’appareil est une notion claire. Si l’on consulte la littérature, on trouvera les définitions suivantes :
• visuellement : conc mini à laquelle le signal est détectée de manière fiable
• déviation standard de la réponse et de la pente
DL = 3.3 σ/S où σ est la déviation standard (écart-type) de la réponse, S est la pente de la courbe de calibration. σ estimé = déterminé de différentes manière : - l’’écart-type du blanc : analyse d’un nombre approprié de blanc et en calculant l’écart- type de leur réponse - sur la courbe d’étalonnage : une courbe d’étalonnage spécifique, dans le domaine de la DL doit être réalisée. L’écart-type résiduel de la régression linéaire ou de l’y-intercept des courbes d’étalonnage peut être utilisée comme SD (statistiques complémentaires sur excel). Recommandation concernant les données : la LD et la méthode utilisée doivent être explicitées : graphe fourni ou par un nombre de points suffisants.
• rapport signal sur bruit : uniquement applicable pour les techniques qui émettent des lignes de base : le rapport est réalisé en comparant le signal d’échantillons de concentration faible connue et le signal du blanc : un rapport entre 3 et 2 est généralement considéré comme acceptable pour la limite de détection
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
42
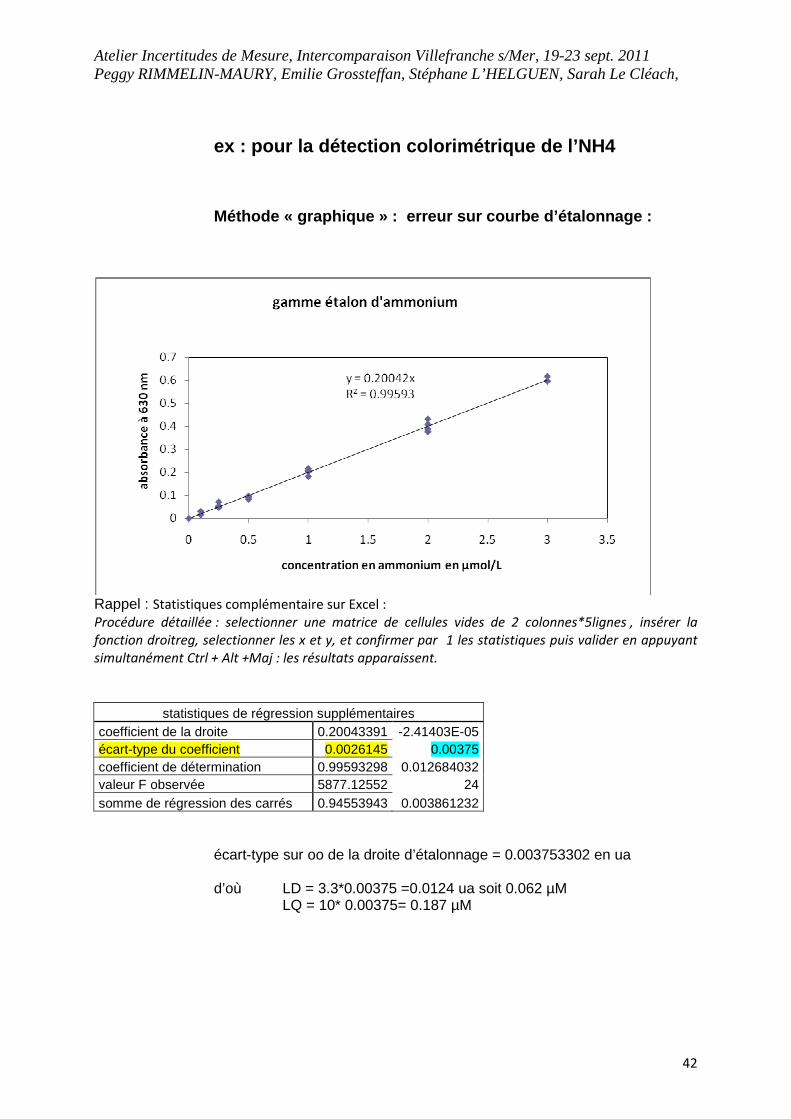
ex : pour la détection colorimétrique de l’NH4 Méthode « graphique » : erreur sur courbe d’étalon nage :
Rappel : Statistiques complémentaire sur Excel :
Procédure détaillée : selectionner une matrice de cellules vides de 2 colonnes*5lignes , insérer la
fonction droitreg, selectionner les x et y, et confirmer par 1 les statistiques puis valider en appuyant
simultanément Ctrl + Alt +Maj : les résultats apparaissent.
statistiques de régression supplémentaires coefficient de la droite 0.20043391 -2.41403E-05 écart-type du coefficient 0.0026145 0.00375 coefficient de détermination 0.99593298 0.012684032 valeur F observée 5877.12552 24 somme de régression des carrés 0.94553943 0.003861232
écart-type sur oo de la droite d’étalonnage = 0.003753302 en ua d’où LD = 3.3*0.00375 =0.0124 ua soit 0.062 µM LQ = 10* 0.00375= 0.187 µM
Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
43
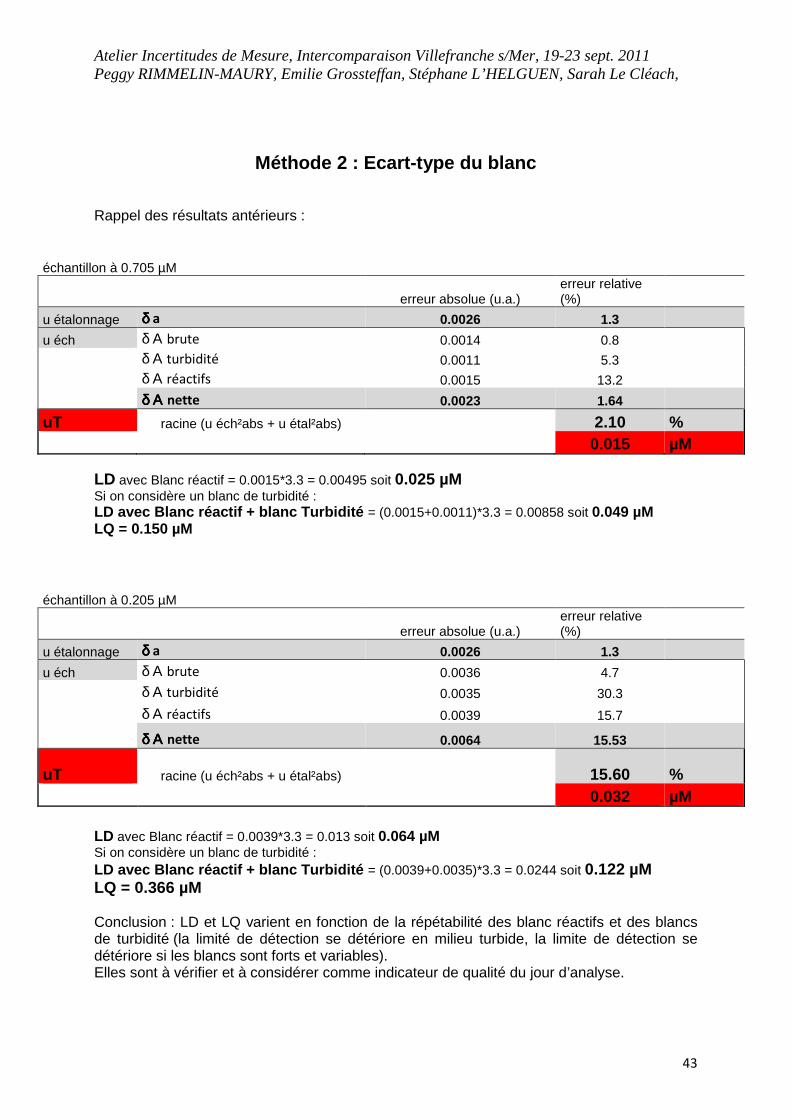
Méthode 2 : Ecart-type du blanc Rappel des résultats antérieurs :
échantillon à 0.705 µM
erreur absolue (u.a.) erreur relative (%)
u étalonnage δ δ δ δ a 0.0026 1.3
u éch δ Α brute 0.0014 0.8
δ Α turbidité 0.0011 5.3
δ Α réactifs 0.0015 13.2
δ Α δ Α δ Α δ Α nette 0.0023 1.64
uT racine (u éch²abs + u étal²abs) 2.10 % 0.015 µM
LD avec Blanc réactif = 0.0015*3.3 = 0.00495 soit 0.025 µM Si on considère un blanc de turbidité : LD avec Blanc réactif + blanc Turbidité = (0.0015+0.0011)*3.3 = 0.00858 soit 0.049 µM LQ = 0.150 µM
échantillon à 0.205 µM
erreur absolue (u.a.) erreur relative (%)
u étalonnage δ δ δ δ a 0.0026 1.3
u éch δ Α brute 0.0036 4.7
δ Α turbidité 0.0035 30.3
δ Α réactifs 0.0039 15.7
δ Α δ Α δ Α δ Α nette 0.0064 15.53
uT racine (u éch²abs + u étal²abs) 15.60 % 0.032 µM
LD avec Blanc réactif = 0.0039*3.3 = 0.013 soit 0.064 µM Si on considère un blanc de turbidité : LD avec Blanc réactif + blanc Turbidité = (0.0039+0.0035)*3.3 = 0.0244 soit 0.122 µM LQ = 0.366 µM Conclusion : LD et LQ varient en fonction de la répétabilité des blanc réactifs et des blancs de turbidité (la limité de détection se détériore en milieu turbide, la limite de détection se détériore si les blancs sont forts et variables). Elles sont à vérifier et à considérer comme indicateur de qualité du jour d’analyse.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
44
Méthode 3 : basée sur le plus petit standard préparé : 0.250 µM à Brest
niveau de concentration en µM uA (ua) uA (µM) uA (%)
0.25 0.0030 0.0152 6.1
LD = 0.0152 * 3.3 = 0.050 ua soit 0.250 µM
LQ = 0.152 soit 0.760 µM
Bilan de l’estimation de la limite de détection
Selon directive, la limite de détection doit être d’au moins 30% de la valeur seuil (NQE) : « norme de
qualité environnement » : existe pour les eaux marines ?
Méthode graphique
Méthode Blancs
Méthode norme Plus petit standard
SOMLIT
LD (µM)
0.049
0.122
0.250
0.050
LQ (µM)
0.150
0.366
0.760
0.150
Conclusion : Méthode de dosage du NH4 à Brest :
La plus petite quantité d’ammonium détectée de manière fiable sur une
eau de mer naturelle à Brest est au mieux de 0.122 µM ± 40%
d’incertitude étendue de répétabilité et ± 116% d’incertitude étendue de
reproductibilité interlaboratoire.

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
45
Annexe 1. Traitement des points aberrants
Avant d’éliminer définitivement un point qui nous semble aberrant, il est nécessaire d’avoir recours à
un test statistique. Le test de Dixon et le test de Grubbs sont ici adaptés.
L’absorbance de turbidité de l’eau de mer nous donne deux valeurs aberrantes à première vue (0.019
et 0.022).
Il est donc indispensable de vérifier si ces valeurs sont statistiquement considérées comme
aberrantes.
Appliquons d’abord le test de Dixon.
� Test de Dixon
Ce test consiste à comparer la distance entre les points les plus éloignés du modèle et les points
immédiatement plus voisins à l’étendue totale des résidus.
Tout d’abord on classe nos résultats dans l’ordre croissant, ici l’absorbance de turbidité mesurée de
nos 8 échantillons d’eau de mer.
absorbance turbidité
0,01 0,01 0,011 0,019 0,011 0,008 0,022 0,013
Tableau 1: résultats Excel: absorbance turbidité de l'eau de mer
Dans notre cas, 8 ≤n ≤12, avec n le nombre d’échantillons d’eau de mer analysés.
Soit R1
ou Rn les points aberrants à la suite du classement.
Hypothèses à tester :
H0 : les valeurs 0.019 et 0.022 ne sont pas aberrantes.
H1 : les valeurs 0.019 et 0.022 sont aberrantes.
Critère de décision :
11
121 RR
RRQ
n −−=
−
Équation 3
� Pour la valeur d’absorbance de 0,019 : 182,0008,0019,0
008,001,01 =
−−=Q
absorbance turbidité 0,008 0,01 0,01
0,011 0,011 0,013 0,019 0,022
Tri dans l’ordre croissant

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
46
� Pour la valeur d’absorbance de 0,022 : 40,0008,0013,0
008,001,01 =
−−=Q
Interprétation du test :
Q1
ou Q2
> Q (1%) alors R1
ou Rn
sont aberrants � on décide H1
Q (5 %) < Q1
ou Q2
< Q (1 %) alors R1
ou Rn
sont douteux
Q1
ou Q2
< Q (5 %) alors R1 ou R
n ne sont pas aberrants � on décide H0
Dans la table de Dixon, on lit la valeur Q de référence avec α le risque d’erreur égal à 5% :
Q α =5% = 0.554
Donc : Q1< Q α =5% on décide donc H0 .
Conclusion:
Les valeurs 0.019 et 0.022 ne sont pas aberrantes. On doit donc les prendre en compte dans la suite
de nos calculs.
Afin de confirmer le test de Dixon, on peut appliquer le test de Grubbs dans les mêmes conditions et
en conservant les mêmes hypothèses.
Le test de Grubbs a un intérêt considérable puisqu’il permet le rejet de deux points aberrants dans
une série de mesures, dans le cas de petits échantillons.
Critère de décision :
1005,0
008,0013,011 =−=−=
s
xmG
Équation 4
8,1005,0
013,0022,01 =−=−=
s
mxG n
Équation 5
G1
ou Gn
> G (α = 1 %) x1
ou xn
est aberrant � on décide H1
G (α = 5 %) < G1
ou Gn
< G (α = 1 %) x1
ou xn
est douteux
G1
ou Gn
< G (α = 5 %) x1
ou xn
est non aberrant � on décide H0
Dans la table de Grubbs, on lit la valeur G de référence avec α le risque d’erreur égal à 5% :
G (α = 5 %)= 2,032

Atelier Incertitudes de Mesure, Intercomparaison Villefranche s/Mer, 19-23 sept. 2011 Peggy RIMMELIN-MAURY, Emilie Grossteffan, Stéphane L’HELGUEN, Sarah Le Cléach,
47
Donc G1
et Gn < G (α = 5 %) on décide H0
Conclusion:
Les valeurs 0.019 et 0.022 ne sont pas aberrantes. On doit donc les prendre en compte dans la suite
de nos calculs. Ce test confirme donc en plus le test de Dixon.


















