8/10/2014 TAMBY Julie L2 CR : Julie Chapon BMCTTM A. …
18
BMCTTM – PARTIE 2 – Mode d'action des médicaments 8/10/2014 TAMBY Julie L2 CR : Julie Chapon BMCTTM A. BOULAMERY 18 pages Mode d'action des médicaments (2) Dans ce cours, on appellera : – M le médicament – L le ligand – R le récepteur – T le transducteur 1/18 Plan : A. Définitions I. Affinité – Kd II. Occupation des récepteurs : Bmax III. Récepteurs de réserve IV. Sélectivité V. Puissance et efficacité B. Notions d'agoniste, antagoniste I. Notion d'agoniste II. Notion d'antagoniste
Transcript of 8/10/2014 TAMBY Julie L2 CR : Julie Chapon BMCTTM A. …

BMCTTM – PARTIE 2 – Mode d'action des médicaments
8/10/2014TAMBY Julie L2 CR : Julie ChaponBMCTTMA. BOULAMERY18 pages
Mode d'action des médicaments (2)
Dans ce cours, on appellera :– M le médicament – L le ligand– R le récepteur– T le transducteur
1/18
Plan :
A. Définitions I. Affinité – Kd II. Occupation des récepteurs : Bmax III. Récepteurs de réserve IV. Sélectivité V. Puissance et efficacité
B. Notions d'agoniste, antagoniste I. Notion d'agoniste II. Notion d'antagoniste

BMCTTM – PARTIE 2 – Mode d'action des médicaments
A. Définitions
Rappel :
• Récepteur pharmacologique : C'est une macromolécule protéique avec laquelle réagit le médicament (ligand) ou un médiateur naturel endogène pour produire une action biologique.
• Action : Cette action peut être modélisée par l'équation suivante :M + R M-R + T → MRT → action pharmacodynamique → effet thérapeutique
M est le médicament.R est le récepteur (ou cible).T est le transducteur : Il permet la transmission de l'information, notamment pour les ligands qui se fixent en dehors de la cellule : ils apportent une information qui sera transmise à l'intérieur de la cellule par le transducteur.
Lorsque le complexe médicament-récepteur-transducteur est formé on a production d'une action pharmacodynamique en aval et au final, on observe, au niveau de l'organisme entier, un effet thérapeutique.
Exemple: Les β-bloquants sont des anti-hypertenseurs qui bloquent les récepteurs par antagonisme compétitif et qui empêchent la contraction de la cellule musculaire lisse. Au niveau de l'organe, du vaisseau, il y a alors une vasodilatation et au niveau de l'organisme entier on observe une chute de la pression artérielle.
I. Affinité – Kd
Affinité : Mesure de la force de liaison du médicament à son récepteur (plus l'affinité est importante plus le médicament se liera fortement à son récepteur).L'affinité est caractérisée par une constante de dissociation appelée Kd. Kd permet de mesurer l'affinité d'un ligand pour son récepteur.La réciproque du Kd est Ka (constante d'association). CR : l'affinité est une notion importante en pré-clinique pour le choix des médicaments à développer.
Notion d'affinité : – La loi d'action de masse → Elle permet de décrire un système en équilibre.
Plus la capacité de fixation, appelée affinité, de M (médicament) sur R (récepteur) est élevée, plus l'équilibre est déplacé vers la droite et l'effet pharmacologique qui en résulte est important. Cette théorie néglige les mécanismes cellulaires d'amplification et le caractère multifactoriel de la réponse.
– Si M active le récepteur c'est un agoniste.– Si M n'active pas le récepteur c'est un antagoniste.
2/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Affinité Activité intrinsèque
K1
[M] + [R] [M+R] effet pharmacodynamique K2
[M] : concentration en médicament.[R] : concentration en récepteur. [M+R] : concentration du complexe médicament-récepteur K1 : constante de vitesse d'association du complexe.K2 : constante de vitesse de dissociation du complexe.
A l'équilibre il y a autant de complexe médicament-récepteur que d’éléments dissociés médicament ET récepteur.A partir du moment ou on modifie un des paramètres du milieu, on modifie la réaction dans un sens ou l'autre.
Affinité et Kd :
• Kd : constante de dissociation à l'équilibre.
• Kd est la concentration de ligand qui occupe à l'équilibre 50% des récepteurs. → Si [R] = [RL] alors [L] = Kd
Intérêts des tests de liaison et de la détermination du Kd (Ki) : Les tests de liaisons sont des tests pré-cliniques, réalisés in vitro pour calculer notamment la constante de dissociation Kd.
• Kd (Ki) d'un ligand est, pour un type de récepteur, constant quelle que soit sa localisation.
Exemple : Le système adrénergique. Dans le système adrénergique il y a 2 types de récepteurs : α et β et plusieurs sous type de récepteurs : α1, α2, β1, β2...
Localisation des récepteurs :
– α1 : notamment sur les vaisseaux, sur l'utérus– α2 : SNC– β1: essentiellement au niveau du cœur– β2 : au niveau bronchique, utérin et vasculaire
3/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Cas des récepteurs β2 :Ces récepteurs ont plusieurs localisations possibles : lorsque l'on administre un agoniste des récepteurs β2 (exemple du salbutamol = Ventoline®), il se fixe avec une affinité identique sur les différents récepteurs β2 : utérins, bronchiques, vasculaires. Quelle que soit la localisation du récepteur, l'affinité d'un ligand pour un type de récepteur est la même.
• Comparaison des affinités de ligands différents pour un même récepteur et d'un ligand pour des récepteurs différents.Le calcul de Kd permet en fait de comparer les effets de médicaments différents sur un même type de récepteur : on aborde ici la notion de sélectivité.
Exemple :
L'effet thérapeutique recherché pour le salbutamol est : – une bronchodilatation quand on l'administre dans le cas de l'asthme.– une relaxation utérine quand on l'administre dans le cas d'accouchement prématuré.
Le salbutamol est destiné à se fixer sur les récepteurs β2 pour pouvoir obtenir ses effets thérapeutiques mais il se fixe aussi de manière non sélective sur les récepteurs β1 cardiaque avec une affinité qui est décrite par une constante de dissociation particulière, différente de la constante pour les récepteurs β2.
Ce manque de sélectivité est responsable d'une certaine partie des effets indésirables et notamment d'une tachycardie plus ou moins importante (surtout quand le salbutamol est administré par voie intra-veineuse) : effet indésirable dromotrope positif.
Comme le salbutamol exerce un effet agoniste sur les récepteurs β1, on aura aussi un effet chronotrope positif c'est à dire une augmentation de la fréquence cardiaque.
En préclinique, on va comparer le salbutamol avec une autre molécule : on compare leur affinité, non seulement pour les récepteurs β2 mais aussi pour les récepteurs β1. Si l'autre agoniste a une affinité plus importante sur les récepteurs β1 que le salbutamol on choisira préférentiellement le salbutamol.
• Intérêt pour le développement de nouveaux médicaments : la détection de nouveaux ligands a pour but d'augmenter la sélectivité d'action.
Il arrive que par hasard, parce que l'on découvre que tel médicament en cours de développement se fixe sur un autre type récepteur non prévu avec une affinité particulière, on puisse découvrir un autre effet thérapeutique potentiel d'une molécule.
• Étude de la fixation de nouveaux ligands : la prédiction du profil pharmacologique permet la sélection des substances en fonction d'un objectif donné.
4/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
II. Occupation des récepteurs : Bmax
Réponse et théorie d'occupation des récepteurs :
Les conséquences de la formation du complexe LR (ligand-récepteur) sont que l'activité sera l'effet de L et l'efficacité la réponse de R (au niveau du récepteur ou en aval).
Pour qu'une molécule, un médicament exerce son effet maximal théorique, seuls 5 à 20% des récepteurs doivent être occupés.
Exemple : Cas des récepteurs β1Avec un agoniste, pour avoir un effet chronotrope positif considéré comme étant maximal, seulement 5% (au maximum 20%) des récepteurs devront être occupés.
Cela introduit la notion de récepteur de réserve.
A savoir aussi : plus on augmente la dose (ou la concentration) de médicament, plus l'effet sera fort. Et à un moment il y aura saturation. On a une représentation de type hyperbolique.Au niveau fonctionnel on peut donc décrire les relations quantitatives entre les concentrations de ligand et les réponses biologiques par la loi d'action de masse et en considérant que l'effet est proportionnel au pourcentage de récepteurs occupés (avant saturation).
Evolution de la théorie d'occupation des récepteurs :
• Un agoniste : molécule qui est capable d'activer un récepteur, entraînant une réponse en aval.
• La réponse maximale obtenue pour une gamme de concentration varie d'un agoniste à l'autre.
Exemple : On a plusieurs molécules agonistes sur les récepteurs β1 : adrénaline, noradrénaline, dopamine et également d'autres médicaments agonistes β1 adrénergique. L'effet maximal théorique chronotrope positif observée avec l'adrénaline sera différent de celui que l'on observe avec la noradrénaline ou avec la dopamine pour une même gamme de concentration.
• On a un facteur de proportionnalité α propre à chaque agoniste appelé activité intrinsèque.
L'équation suivante (qui n'est pas à savoir) traduit la réponse observée lors de l'introduction d'un médicament dans un milieu en fonction de A, son activité et de KA, inverse de la constante de dissociation. On voit aussi apparaître α, qui correspond à l'activité intrinsèque du médicament lui même.
Réponse = αA / (A + KA)
Lorsqu'il s'agit d'un médicament qui est :
➢ Un agoniste entier (complet ou pur) : α= 1→ La réponse est potentiellement maximale lorsque l'on augmente la dose.
➢ Un agoniste partiel : 0<α<1 → A un moment donné, même si on continue à augmenter la dose, on n'atteindra jamais l'effet maximal théorique observé avec un agoniste entier. L'intensité d'effet sera inférieure à celle observée avec l'agoniste entier.
5/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
➢ Un antagoniste neutre : α=0 → Un antagoniste compétitif ou neutre n'a pas d'effet par lui même.Contrairement à l'agoniste qui active le récepteur et entraîne une réponse en aval, l'antagoniste bloque simplement la réponse physiologique.
Exemple des β-bloquants dans le système adrénergique :On parle de β-antagonistes : ce sont des molécules utilisées notamment dans le traitement de l'hyper-tension artérielle pour diminuer la pression artérielle. Quand on utilise un β-bloquant, l'effet thérapeutique recherché se situe surtout sur les récepteurs β1 adrénergique. La stimulation des récepteurs β1 par des agonistes entraîne une stimulation globale du cœur :
– Effet chronotrope positif : augmentation de la fréquence cardiaque. Cet effet vient potentialiser l'effet inotrope.
– Effet inotrope positif : augmentation du volume de l'éjection ventriculaire → augmentation de la force de contraction du cœur.
Au final on observe une augmentation de la pression artérielle, par augmentation de la fréquence cardiaque et augmentation de l'inotropisme.
Maintenant on prend un patient avec une hyper-tension artérielle (stimulation avec antagoniste). On utilise un β-bloquant : Ce médicament vient prendre la place de l'agoniste physiologique (adrénaline, noradrénaline physiologique) sur le récepteur β1 et bloque ainsi la fixation des ligands endogènes.
Effet observé : Empêchement de l'action de l'adrénaline ou de la noradrénaline et au final, diminution de la pression artérielle par un effet qui n'est PAS un effet propre du β-bloquant. C'est pour cela que l'activité intrinsèque est nulle.
➢ Un agoniste inverse : α<0 → Il n'a pas d’intérêt thérapeutique, uniquement expérimental (uniquement à titre indicatif).
III. Récepteurs de réserve
La réponse maximale ne demande qu'une occupation de 5 à 20 % des récepteurs.Cela sous entend 80 à 95 % de récepteurs de réserve libres disponibles potentiellement.
Cette notion de récepteur de réserve est intéressante, notamment dans la situation où l'on est en présence, simultanément, d'un antagoniste et d'un agoniste.
IV. Sélectivité
• La sélectivité définit la fixation d'un médicament sur un type de récepteur préférentiel par rapport à d'autre récepteur.
• Elle conditionne la fiabilité et la pertinence de l'utilisation thérapeutique des ligands. • Il existe une concurrence de liaison du ligand pour le récepteur 1 ou le récepteur 2 :
KDR2 / KDR1
6/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Exemple des β2 agonistes :L'effet thérapeutique recherché est obtenu en passant par la fixation sur les récepteurs β2 mais ils se fixent aussi, par manque de sélectivité, sur les récepteurs β1 entraînant la survenue d'effets indésirables plus ou moins importants.
• Sélectivité de l'effet du ligand pour l'effet recherché E1 par rapport à l'effet indésirable E2 :
CE2/CE1
Plus la fixation d'un médicament sur son récepteur sera non sélective et moins on aura de marge entre les concentrations thérapeutiques et celles qui entraîneront des effets indésirables. → Notion de marge thérapeutique qui conditionne la fiabilité et la pertinence de l'utilisation thérapeutique des ligands.
La zone thérapeutique est la zone ou il faut se situer pour être efficace sans être toxique :– Lorsque l'on est en dessous de C1 (concentration minimale efficace) on risque d'être inefficace– Lorsque l'on est au dessus de C2 (concentration maximale non toxique) il y a un risque de toxicité – Entre les deux : zone thérapeutique
Mais attention la zone thérapeutique est variable d'une personne à l'autre.
Différence entre sélectivité et spécificité ? Sélectivité : sélectivité d'effet → apparition de l'effet thérapeutique préférentiellement à l'effet l'indésirable ou inversement. Spécificité : spécificité de la fixation sur un récepteur.(cela est valable pour la pharmacologie, attention cela peut être différent en santé publique par exemple).
V. Puissance et efficacité
Définitions :
• Relation dose (concentration)/effet :
– Hyperbole (ou sigmoïde si semi-log)E = (αEmax x D) / (D+ DE50)Le coefficient α caractérise l'activité intrinsèque d'un ligand, c'est à dire sa capacité à entraîner le couplage du récepteur à l'effecteur.
L'intensité de l'effet peut être représentée par une fonction hyperbolique.
Sur le schéma (voir plus bas) on a l'effet en fonction de la dose ou de la concentration. Plus on augmente la dose ou la concentration du médicament et plus l'effet augmente, et cela pour une certaine gamme de concentration.
– Au bout d'un moment, même si on augmente la dose, l'effet n'augmente plus : on obtient ce qu'on appelle l'effet maximal théorique : Emax
La représentation hyperbolique n'est pas pratique car pour de faibles variations de concentration on a de grandes variation d'effet.Pour faire un « zoom », on prend le logarithme des concentrations sur l'axe des abscisses : cela permet de linéariser la partie de la courbe où il y a de faibles variations d'effet pour de fortes dose ou concentration.
7/18
BMCTTM – PARTIE 2 – Mode d'action des médicaments
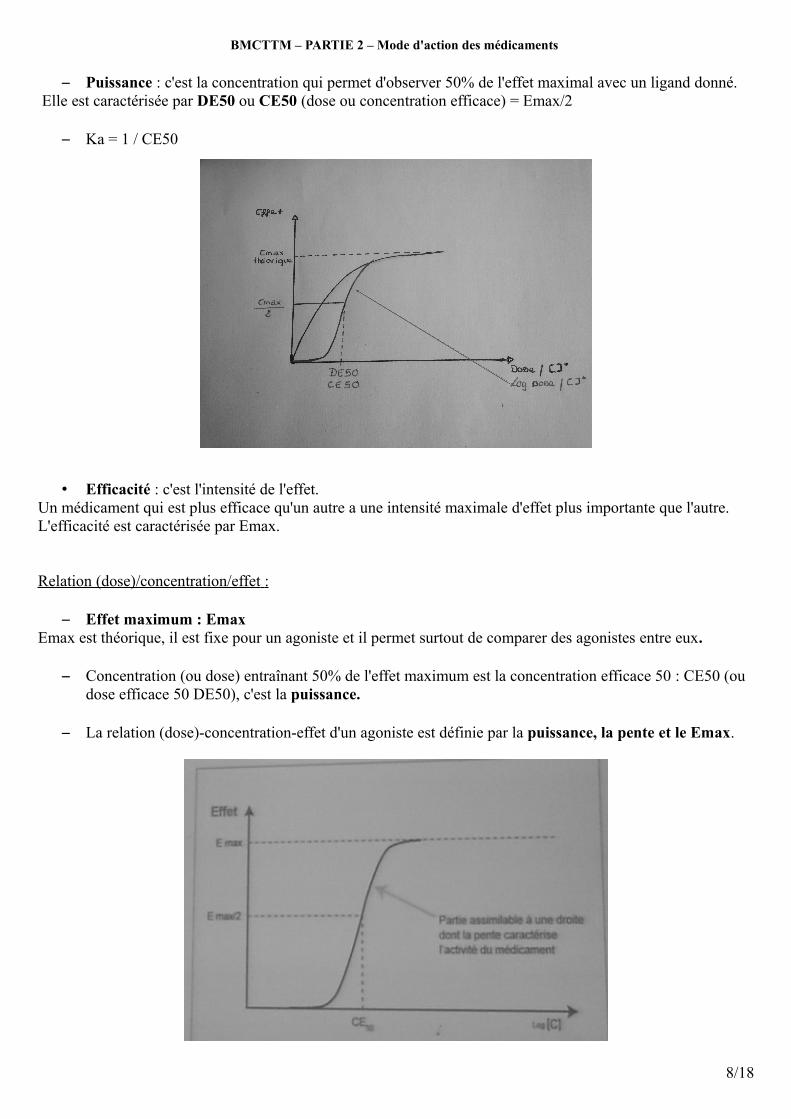
– Puissance : c'est la concentration qui permet d'observer 50% de l'effet maximal avec un ligand donné. Elle est caractérisée par DE50 ou CE50 (dose ou concentration efficace) = Emax/2
– Ka = 1 / CE50
• Efficacité : c'est l'intensité de l'effet.Un médicament qui est plus efficace qu'un autre a une intensité maximale d'effet plus importante que l'autre.L'efficacité est caractérisée par Emax.
Relation (dose)/concentration/effet :
– Effet maximum : EmaxEmax est théorique, il est fixe pour un agoniste et il permet surtout de comparer des agonistes entre eux.
– Concentration (ou dose) entraînant 50% de l'effet maximum est la concentration efficace 50 : CE50 (ou dose efficace 50 DE50), c'est la puissance.
– La relation (dose)-concentration-effet d'un agoniste est définie par la puissance, la pente et le Emax.
8/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
B. Notions d'agoniste, antagoniste
I. Notion d'agoniste
On a deux courbes : – celle de l'agoniste entier (agoniste dit entier par comparaison avec la courbe de l'agoniste partiel)– celle de l'agoniste partiel
La notion d'effet maximal théorique et celle d'agoniste partiel sont relatives à l'existence d'autre molécule qui exerce aussi un effet agoniste sur le même type de récepteur.
Sur la figure : – En ordonné : effet en pourcentage de l'effet maximal observé. L'agoniste entier est pris comme
référence.
– En abscisse : le logarithme des concentrations.
Avec l'agoniste partiel, même si on continue a augmenter les doses ou les concentrations, on ne peut jamais obtenir l'effet maximal que l'on obtient avec l'agoniste entier. L'activité intrinsèque de l'agoniste partiel est moins importante que pour l'agoniste entier, et donc l'effet maximal est moindre.
Exemple : La douleur et la problématique des antalgiques opiacés
Il existe plusieurs classes d'antalgiques dont les antalgiques de classe 3 : ce sont les antalgiques opiacés, dérivés de la morphine puissant. On y retrouve la morphine et aussi d'autres agonistes morphiniques dont certains sont des agonistes partiels.
➢ La morphine est considérée comme étant la référence, c'est donc un agoniste entier des récepteurs μ.
L'effet antalgique de la morphine est considéré comme l'effet de référence, l'effet maximal : plus on augmente la dose avec la morphine, plus l'effet augmente. (La morphine est le seul médicament pour lequel il n'existe pas de dose maximale recommandée et ce qui stoppe l'escalade de dose est donc l'apparition des effets indésirables).
9/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
➢ Il existe d'autres types d'agonistes qui se fixent sur les récepteurs μ, par exemple : la nalbuphine (agoniste des récepteurs μ mais c'est un agoniste partiel)
La nalbuphine a un effet antalgique très intéressant (elle est utilisée notamment en traitement post-opératoire ou dans le cadre des douleurs aiguës). L'effet est rapide mais il est plafonné, malgré l'augmentation des doses l'effet n'augmente plus (en général à partir de 3ampoules de nalbuphine, l'effet n'augmente plus).L'effet maximal est plafonné et est bien inférieur à l'effet observé avec la morphine.
Sur la figure est également représentée la courbe de l'antagoniste neutre : C'est une courbe plate car l'activité intrinsèque est nulle, l'antagoniste neutre n'a pas d'effet qui lui est propre.
Comparaison d'agoniste :
Sur la figure sont représentés 3 médicaments et on observe que les courbes ne sont pas superposées.
– Médicaments A et B : → En terme d'effet, d'efficacité, ils sont aussi efficace l'un que l'autre. On obtient potentiellement le même effet maximal mais les courbes ne sont pas superposables car A est plus puissant que B : il faut plus de médicament B pour obtenir le même effet qu'avec A.
– Médicaments A et C : → A est plus efficace que C → A est un petit peu plus puissant que C
– Médicaments B et C : → B est plus efficace que C → C est plus puissant que B
Tout les cas de figures sont possibles et en fonction de l'objectif thérapeutique que l'on se fixe on choisira plutôt tel ou tel médicament.
10/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Récepteurs de réserve :
Pour obtenir 100 % d'effet maximal il n'est nécessaire d'occuper que 5 à 20 % des récepteurs disponibles.
La figure ci-dessus représente la courbe de l'effet en fonction du logarithme de la dose ou de la concentration. En dessous on peut voir l'occupation des récepteurs :
– Pour l'agoniste entier : Peu de récepteurs sont occupés et pourtant on obtient l'effet maximal considéré ici comme effet maximal de référence.
– Pour l'agoniste partiel : Quand on augmente les concentrations, on va occuper un certain nombre de récepteurs supplémentaires et plus on occupe de récepteurs et plus on a une augmentation de l'effet. Mais au bout d'un moment l'effet plafonne : malgré l'occupation de la totalité des récepteurs on a un effet plafonné qui n'atteint jamais l'effet maximal théorique.
On a une activité intrinsèque de l'agoniste partiel qui est moindre par rapport à celle de l'agoniste entier.
II. Notion d'antagoniste
Antagonisme
• Antagonisme pharmacologique : cf plus loin
• Antagonisme chimique : interaction de type chimique entre deux substances (exemple : avant même l'absorption, deux médicaments qui se retrouvent dans le tube digestif et qui sont chimiquement incompatibles, l'un va précipiter l'autre ou bien les antagonistes chimiques dans les perfusions).
• Antagonisme fonctionnel : On a un effet opposé à celui de l'agoniste mais par une action sur un récepteur différent la plupart du temps.
11/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Exemple d'un patient qui prend des β-bloquants avec une action antagoniste sur les récepteurs β1. Ce patient se retrouve, de manière volontaire, surchargé en β-bloquant (intoxication médicamenteuse volontaire).Cela se traduit par une fréquence cardiaque très diminuée, une baisse de la tension et un inotropisme très diminué.
Ce patient va donc passer en réanimation, il existe différentes manières de le traiter :
– Donner du glucagon– Donner des agonistes des récepteurs adrénergiques pour contrer l'effet antagoniste sur les récepteurs β1.
Quand on administre de manière exogène des médicaments agonistes adrénergiques comme l'adrénaline, on va avoir un antagoniste partiellement fonctionnel car l'adrénaline va exercer son action sur tout les récepteurs y compris sur le récepteur β1.
On aura un antagoniste compétitif, mais aussi un antagoniste fonctionnel par ce que l'adrénaline exerce une action agoniste sur les récepteurs α1 entraînant une vasoconstriction et parallèlement on aura l'effet des β-bloquants sur un autre type de récepteurs: les β1. Les β-bloquants ont une action chronotrope négative et inotrope négative sur les récepteurs β1.
→ on a donc deux actions antagonistes mais sur des récepteurs différents.
Types d'antagonistes pharmacologiques :
• Agoniste / antagoniste partiel
• Antagoniste irréversible : (rare) L'antagoniste est irréversible quand il y a une liaison tellement forte entre le médicament et le récepteur (liaison covalente en général) que l'on ne peut pas dissocier le complexe médicament-récepteur , même avec une molécule agoniste. Cela peut entraîner l'inactivation permanente d'une protéine car l'effet inhibiteur est définitif, mais limité par le taux de renouvellement de la protéine-récepteur.
Exemple : L’inhibition de l'acétylcholine-estérase par les organophosphorés (organophosphorés que l'on retrouve notamment à très faible concentration dans les shampoings anti-poux, à plus forte concentration dans des pesticides, et aussi dans des gaz de combats) A l'heure actuelle, on observe des intoxications aux organophosphorés dues à ces pesticides.
→ Effet observé : inhibition de l'acétylcholine-estérase.
Cette inhibition est tellement puissante que quand l'organophosphoré est fixé il faut attendre le renouvellement de la cible pour avoir un fonctionnement enzymatique adéquat.Or, ce renouvellement n'est pas assez rapide et si on n'intervient pas, on observe le décès du patient :l'objectif est donc de donner l'antidote très rapidement avant que toutes les acétylcholine-estérases ne soient liées aux organophosphorés.
L'acétylcholine-estérase est une enzyme de dégradation de l'acétylcholine et quand il y trop d'acétylcholine, l'activité parasympathique est très fortement marquée. Cela explique les signes observés lors des intoxications aux organophosphorés : signes muscariniques, lipothymiques...
12/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
• Antagoniste réversible :
➢ Type compétitif : liaison à un même site que l'agoniste (exclusion mutuelle), c'est à dire qu'il y a compétition entre l'agoniste et l'antagoniste pour occuper le site de liaison.Exemple des β-bloquants qui empêchent la fixation des ligands endogènes sur les récepteurs β.
➢ Type non compétitif : l'antagoniste se lie à un autre site qui n'empêche pas la liaison de l'agoniste mais qui prévient l'activation du récepteur. Exemple : tout les antagonistes allostériques : modification de la conformation du récepteur empêchant l'action de l'agoniste.
• Agoniste inverse
Théorie conformationnelle des récepteurs (ce n'est pas savoir)
Un récepteur existe sous au moins deux états : – actifs (Ra)– inactif (Ri)
L'équilibre entre ces deux états varie en fonction de l'affinité relative du ligand M pour ces deux conformations.
Le récepteur à l’état de repos, forme inactivée : rond
13/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Le récepteur activé : carré En pratique, un agoniste a une affinité préférentielle pour le récepteur sous la forme activée mais une affinité partielle pour le récepteur sous la forme non activée n'est pas exclue.
Dans le cas de la fixation de l'agoniste sur le récepteur activé on observe un déplacement de la réaction vers la droite : on a une réponse pharmacodynamique en aval.
L'antagoniste a une affinité identique pour le récepteur sous la forme inactivée et celui sous la forme activée.
Donc si on a un antagoniste neutre il bloque le récepteur autant sous sa forme activée que non activée : pas de déplacement vers un effet ou un « non effet ». (Ce n'est pas à savoir!)
Affinité conformationnelle et réponse pharmacologique :
Sur la figure :
- En ordonnée : intensité de l'effet en pourcentage de l'effet basal.Car la théorie sous-entend que le récepteur à l'état basal non activé a quand même une activité en aval (même en l'absence d'agoniste). - En abscisse : logarithme des concentrations.
– L'antagoniste présente une courbe plate mais on a 100% de l'effet basal en absence d'agoniste.– L'agoniste partiel et l'agoniste entier induisent une augmentation de l'effet de base. – L'agoniste inverse entraîne une diminution de l'effet basale du récepteur (intérêt en recherche
uniquement).
14/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
• Une molécule ayant une meilleure affinité pour l'état actif Ra déplace l'équilibre vers M-Ra et entraîne une réponse pharmacologique maximum, c'est un agoniste entier.
• Une molécule ayant une affinité légèrement plus forte pour Ra que pour Ri (récepteur inactivé) déplace peu l'équilibre vers M-Ra, l'amplitude de l'effet est plus faible y compris pour des concentrations saturantes, c'est un agoniste partiel.
• Une molécule ayant la même affinité pour Ra que pour Ri ne modifie pas par elle même l'équilibre mais réduit par compétition la liaison des autres ligands, c'est un antagoniste compétitif.
• Une molécule ayant une affinité plus forte pour Ri que pour Ra, déplace l'équilibre vers M-Ri , réduit la proportion de Ra (qui peut avoir une activité basale) et peut induire un effet opposé à celui de l'agoniste, c'est un agoniste inverse.
Notion d'antagonisme compétitif :
• La puissance d'un antagoniste compétitif réversible est quantifiée par le pA2 : logarithme négatif de la concentration de l'antagoniste qui nécessite le doublement de la concentration de l'agoniste pour maintenir le même effet. Plus le pA2 est élevé, plus l'antagoniste est puissant.
La première courbe : Milieu dans lequel on a un agoniste seul.
La deuxième courbe : Introduction d'un antagoniste dans le milieu à une certaine concentration. Cette courbe représente la courbe d'effet de l'agoniste en présence d'antagoniste.
En effet, il y a des sites récepteurs qui seront occupés par l'agoniste et des sites récepteurs qui vont être occupés par l'antagoniste β-bloquant. Donc on aura besoin d 'un peu plus d'agoniste adrénaline pour obtenir le même effet que lorsque celle ci est seule : la courbe est déplacée vers la droite.
Les courbes suivantes : elles correspondent aux courbes de l'agoniste en présence de concentrations croissantes d'antagoniste.
15/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Plus on augmente la concentration d'antagoniste dans le milieu (β-bloquants), plus les sites β1 seront occupés, moins l'adrénaline pourra se fixer et plus il faudra d'adrénaline pour obtenir le même effet qu'initialement.
Quand on augmente les concentrations d'adrénaline en présence d'antagoniste, on récupère toujours un effet maximal théorique car il s'agit d'un antagoniste réversible et aussi par ce qu'il y a des récepteurs de réserve.
Notion d'antagonisme non compétitif :
➢ Sans récepteur de réserve :
Exemple : On exclut dans cet exemple l'action de l'adrénaline sur les récepteurs β1.
Première courbe : L'adrénaline seule exerce un effet vasoconstricteur, cela induit une augmentation de la pression artérielle.Plus on augmente la concentration d'adrénaline, plus la pression augmente.
Courbe suivante : Introduction de β-bloquant dans le milieu :cela entraîne un déplacement de la courbe vers la droite. Le β-bloquant va se fixer sur les récepteurs β1 et donc la fréquence cardiaque et l'inotropisme diminuent et s'opposent à l'effet de l'adrénaline sur les récepteurs α1.
Dans le cas ou il n'y a pas de récepteurs de réserve (théorique), on observe un aplatissement de la courbe : malgré l'augmentation de la concentration en adrénaline on ne peut jamais obtenir un effet maximal car les sites sont bloqués par les β-bloquants.
16/18

BMCTTM – PARTIE 2 – Mode d'action des médicaments
➢ Avec récepteur de réserve :
Pour obtenir un effet maximal, l'adrénaline n'est fixée que sur 5 à 20% des récepteurs α1 donc, même en présence de beaucoup de β-bloquant sur les récepteurs β1, quand on augmente la concentration en adrénaline on va occuper les récepteurs de réserve α1 et donc on obtient, pendant un certain temps, un effet maximal de l'adrénaline. Après occupation de tous les récepteurs de réserve, il y a un déplacement des courbes vers le bas et la droite.
Interaction agoniste complet et agoniste partiel :
Exemple : L’interaction entre la morphine, ou un agoniste morphinique puissant et la nalbuphine :
Situation post-opératoire : Les patients opérés bénéficient d'une analgésie per opératoire, c'est à dire que pendant l'intervention on leur administre des antalgiques puissants qui ont un effet beaucoup plus marqué que la morphine.
17/18
Agoniste partiel seul

BMCTTM – PARTIE 2 – Mode d'action des médicaments
Lors de son réveil le patient exprime une douleur, pour traiter cette douleur on peut utiliser un antalgique classe 3 : La nalbuphine. Il faut faire attention à une chose : la nalbuphine est agoniste partiel. Or si le délai entre l'administration de l'agoniste complet (pendant l'opération) et celle de l'agoniste partiel est trop court on observe une recrudescence de la douleur.
Cela s'explique par le fait que l'agoniste partiel va se comporter comme un antagoniste : il vient diminuer l'effet de l'agoniste entier seul. C'est pourquoi on parle parfois d'agoniste/antagoniste.
– Première courbe : agoniste complet seul (agoniste morphinique)
– Deuxième courbe : agoniste partiel seul (nalbuphine)
– La dernière courbe : agoniste complet en présence de l'agoniste partiel.Cette courbe est déplacée vers la droite : pour avoir les mêmes effets antalgiques il faut augmenter la concentration du morphinique car la nalbuphine diminue l'effet de cette agoniste entier.
18/18