







Langages
Pages
Légal


Syndrome occlusif
néonatal :
Dr R. Dubois – Hôpital Femme Mère Enfant
- Occlusions néonatales
- Maladie de Hischsprung

Diagnostic ante-natal:
• Pour certaines formes seulement
• Signes d ’appel :
- hydramnios
- dilatation digestive
- hyper-échogénicité du grêle
- ascite foetale
- calcifications …
• Si DAN positif : étude du caryotype, étude génétique,
organisation de la prise en charge péri-natale
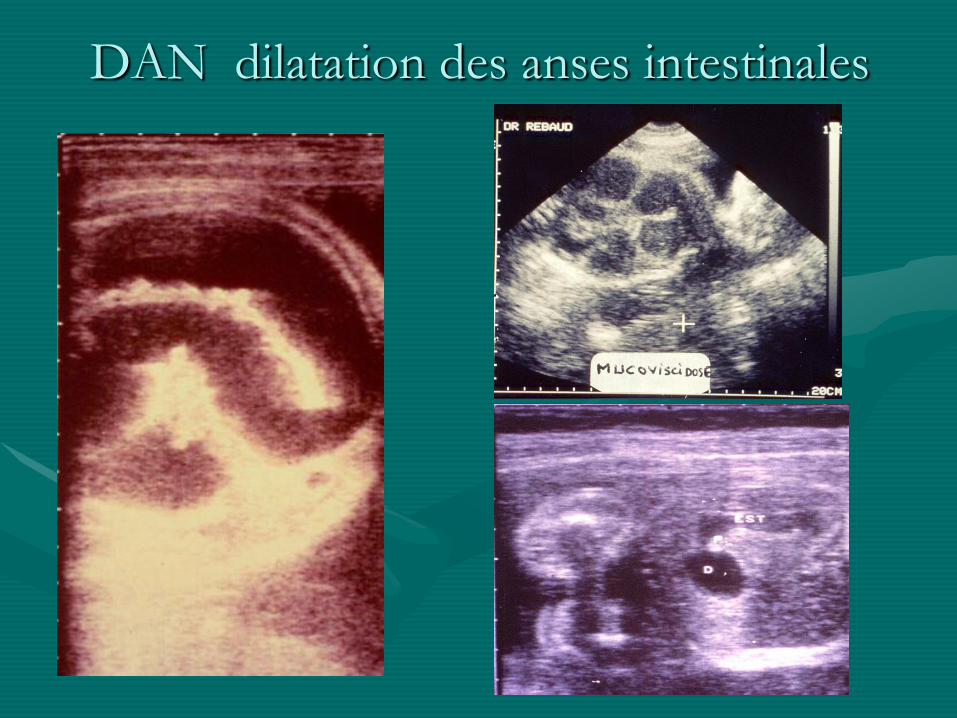
DAN dilatation des anses intestinales

Diagnostic post-natal:
• Examen clinique:
- douleur abdominale +++
- vomissements +++: clairs ou bilieux,
précoces ou tardifs
- émission du méconium +++: avant ou après
24/48 heures
- ballonnement abdominal +++: présent ou
absent, diffus ou localisé

• Examens radiologiques:
- ASP +++ : répartition des gaz, niveau de
l’obsatcle, pneumopéritoine, calcifications, +/-
niveaux hydro-aériques ...
- échographie abdominale +++ : vaisseaux
mésentériques (normalement : artère à gauche), +/-
diagnostic étiologique
- lavement opaque ++ : taille et position du
colon
- TOGD : estomac et duodénum

Diagnostic étiologique:
• Obstructions intrinsèques:
atrésies intestinales +++
iléus méconial ++
• Obstructions extrinsèques:
péritonites et kystes méconiaux
malrotations intestinales +++
• Obstructions fonctionnelles:
maladie de Hirschsprung +++
bouchon méconial
neuro-dysplasies intestinales

Diagnostic topographique:
• Occlusions hautes: duodénum, jéjunum haut:
ballonnement 0 atrésie duodénale, jéjunale haute
vomissements +++ malrotation
• Occlusions intermédiaires: jéjunum, iléon haut:
ballonnement + atrésie grêle, iléus méconial
vomissement ++ péritonite méconiale
• Occlusions basses: iléon terminal, colon:
ballonnement +++ atrésie iléon, colon
vomissement + Hirschsprung, bouchon

Atrésies et sténoses digestives:
• Siège possible sur tous les segments digestifs, surtout œsophage, duodénum, iléon,
• Atrésie duodénale:
- obstacle +/- complet
- trouble précoce de l ’organogénèse
- fréquemment associée à d ’autres malformations et anomalies chromosomiques (trisomie 21 dans 1/3 des cas)
- souvent anomalie du pancréas +/- voies biliaires
- souvent DAN: image de double bulle / hydramnios
- traitement chirurgical: duodéno-duodénostomie +/-modelage
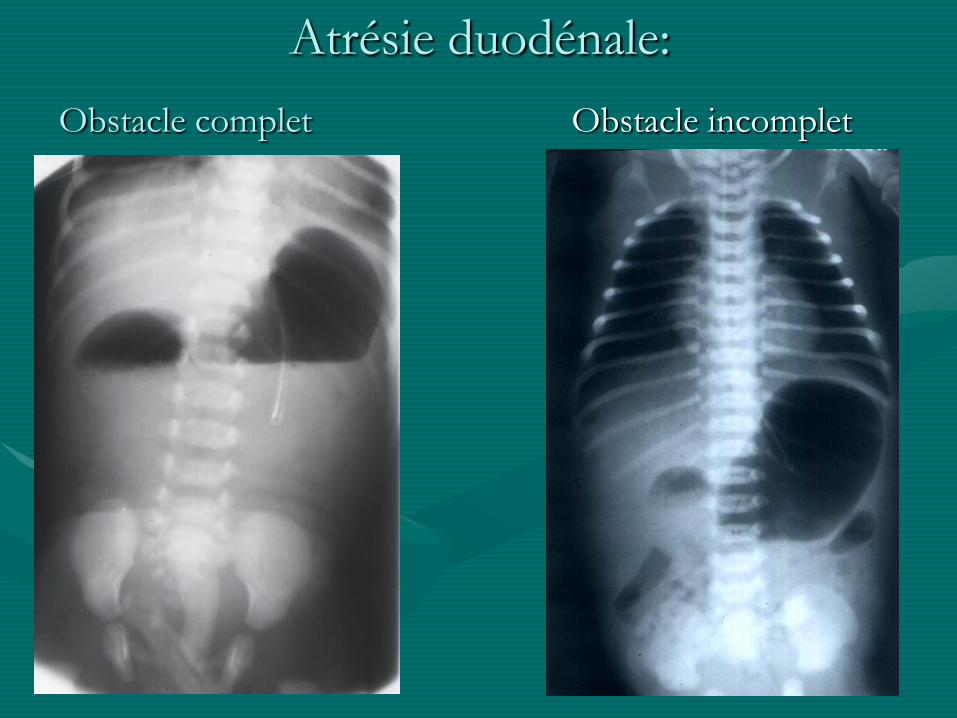
Atrésie duodénale:
Obstacle complet Obstacle incomplet

Atrésies et sténoses digestives:
• Atrésies du grêle +++ et du colon + :
- séquelle d ’une ischémie localisée
- moins souvent DAN
- pas d ’autres malformations sauf mucoviscidose
- divers types, souvent micro-colon
- traitement chirurgical: anastomose ou
dérivation digestive
- pronostic bon sauf dans les formes étendues / apple
peal: possibilité de grêle court
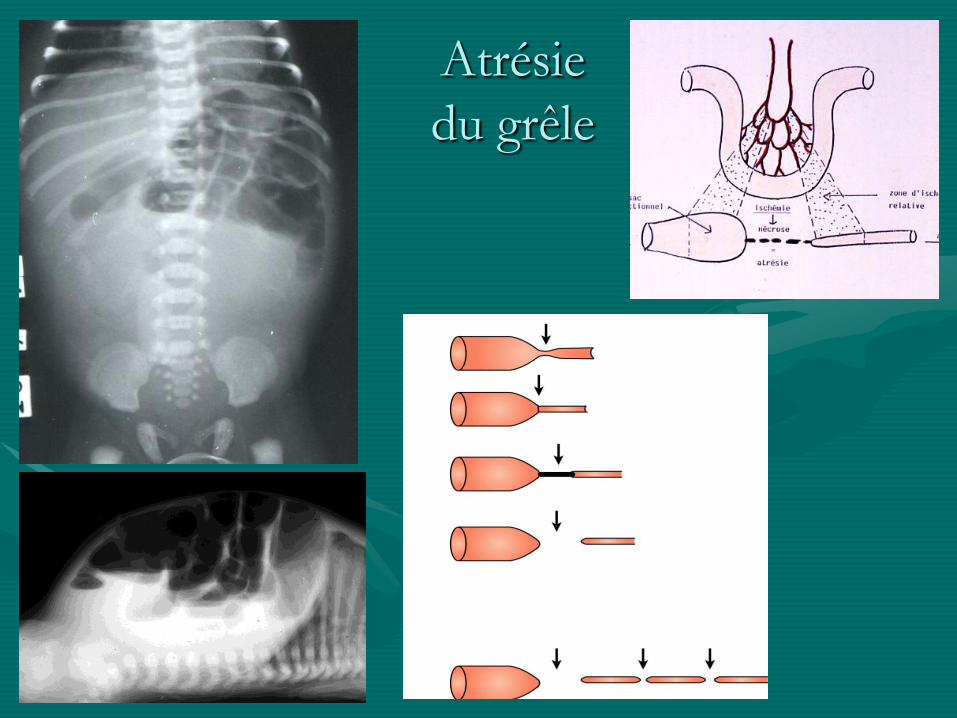
Atrésie
du grêle
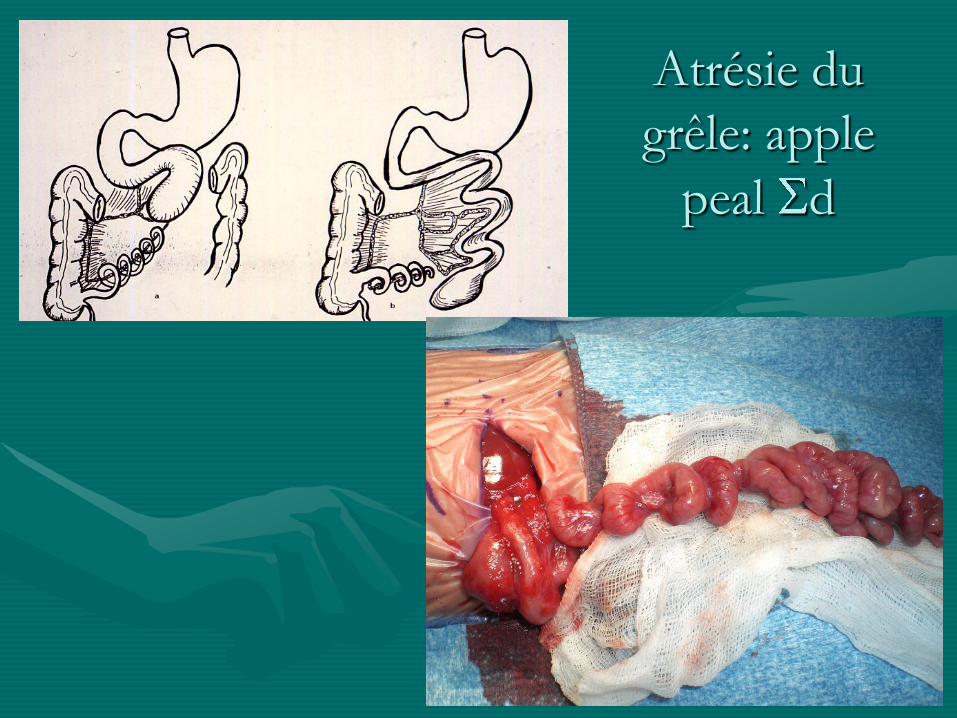
Atrésie du
grêle: apple
peal d

Atrésie du
grêle
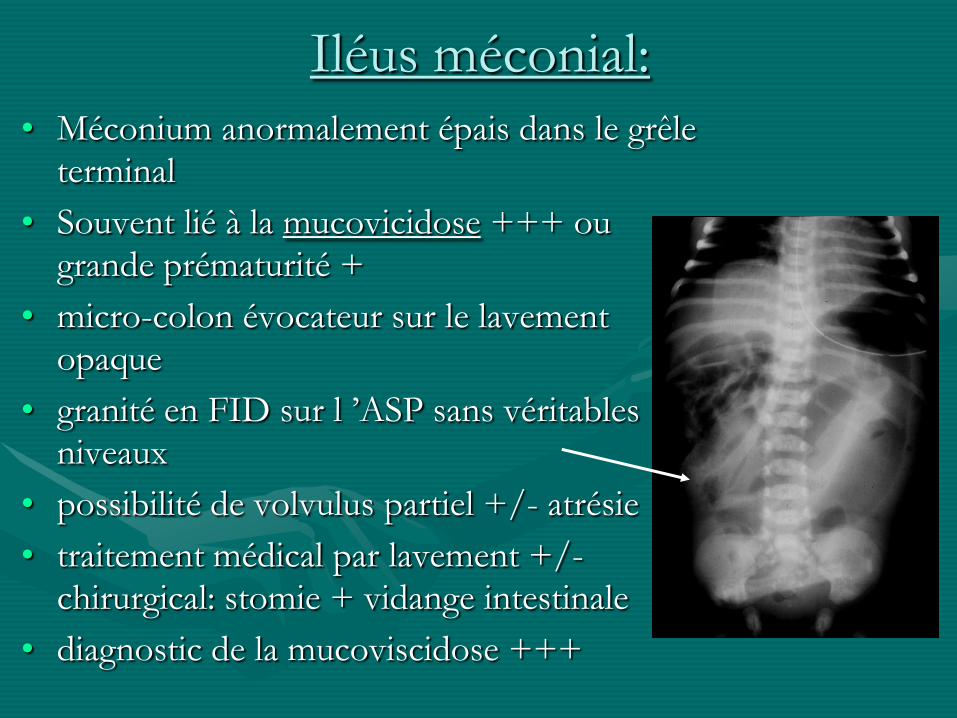
Iléus méconial:
• Méconium anormalement épais dans le grêle terminal
• Souvent lié à la mucovicidose +++ ou grande prématurité +
• micro-colon évocateur sur le lavement opaque
• granité en FID sur l ’ASP sans véritables niveaux
• possibilité de volvulus partiel +/- atrésie
• traitement médical par lavement +/-chirurgical: stomie + vidange intestinale
• diagnostic de la mucoviscidose +++
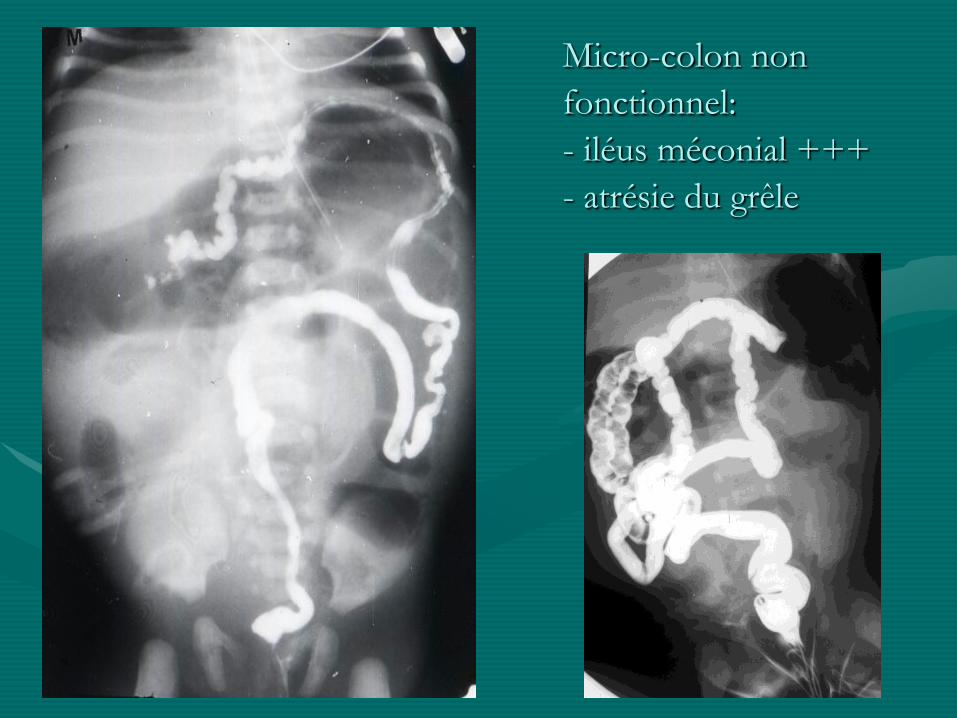
Micro-colon non
fonctionnel:
- iléus méconial +++
- atrésie du grêle

Péritonite méconiale:
• Perforation ante-natale ou péri-natale par :
- accident vasculaire +++ (association possible à une
atrésie intestinale)
- hyper-pression (association au Hirschsprung)
- volvulus intestinal (association à l’iléus méconial)
• ASP caractéristique: micro-calcifications
• parfois asymptomatique
• traitement chirurgical si symptomatique : entérolyse,
résection d ’une atrésie ou d ’un volvulus ...
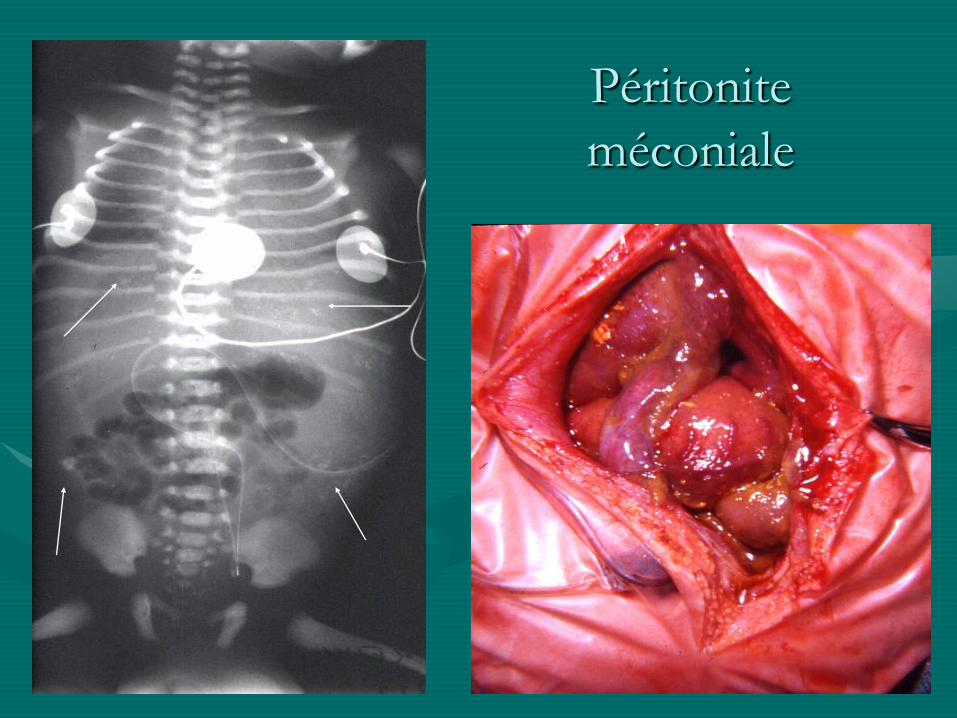
Péritonite
méconiale
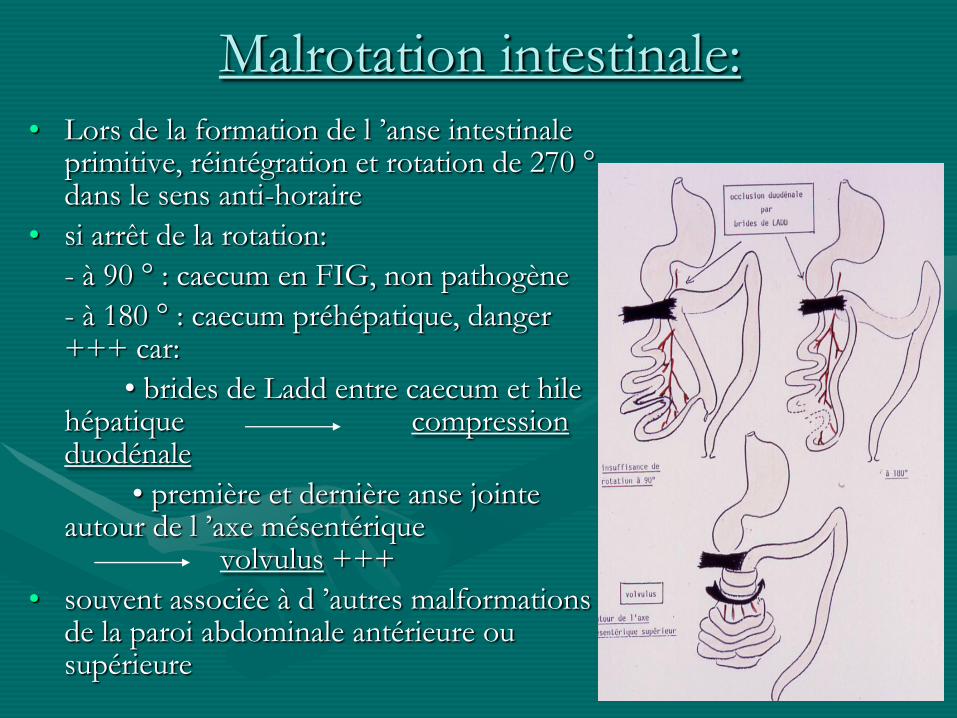
Malrotation intestinale:
• Lors de la formation de l ’anse intestinale primitive, réintégration et rotation de 270 °dans le sens anti-horaire
• si arrêt de la rotation:
- à 90 ° : caecum en FIG, non pathogène
- à 180 ° : caecum préhépatique, danger +++ car:
• brides de Ladd entre caecum et hile hépatique compression duodénale
• première et dernière anse jointe autour de l ’axe mésentérique
volvulus +++
• souvent associée à d ’autres malformations de la paroi abdominale antérieure ou supérieure
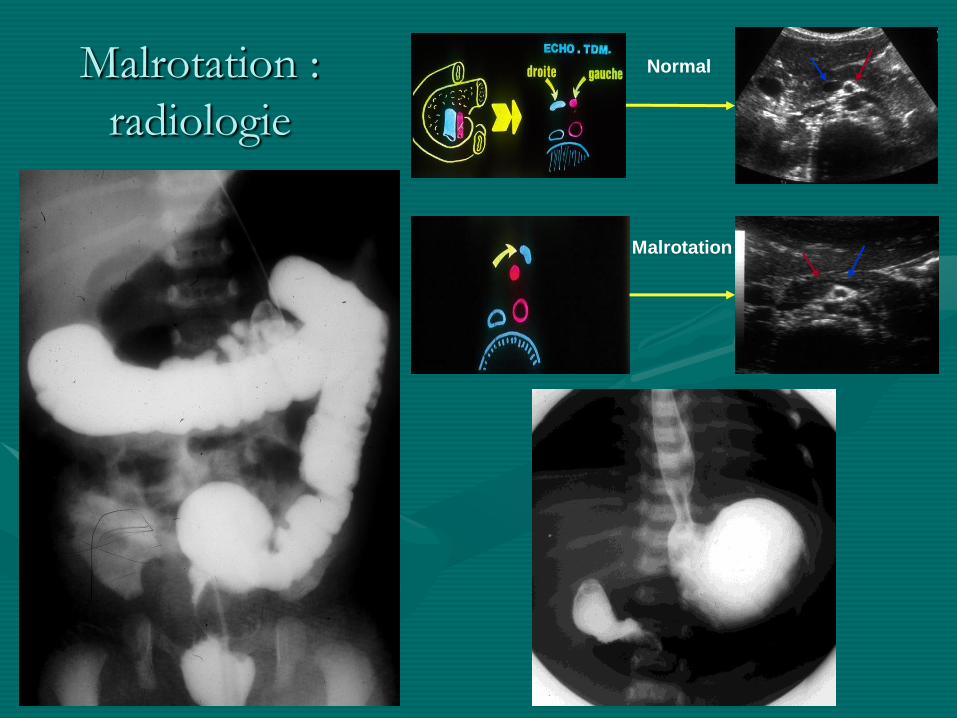
Malrotation :
radiologie
Normal
Malrotation
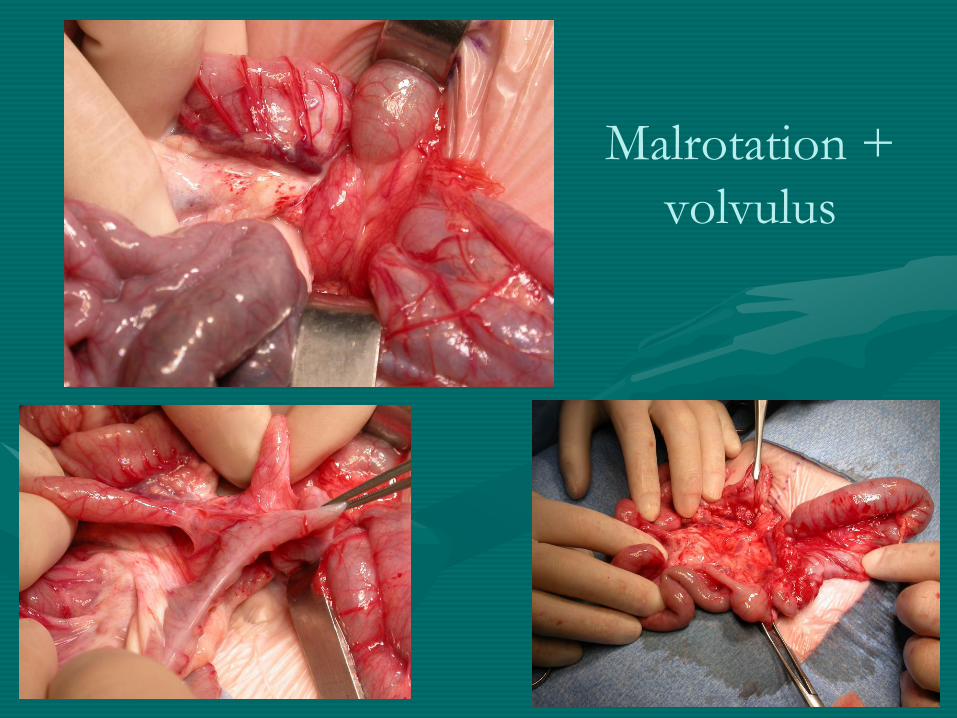
Malrotation +
volvulus
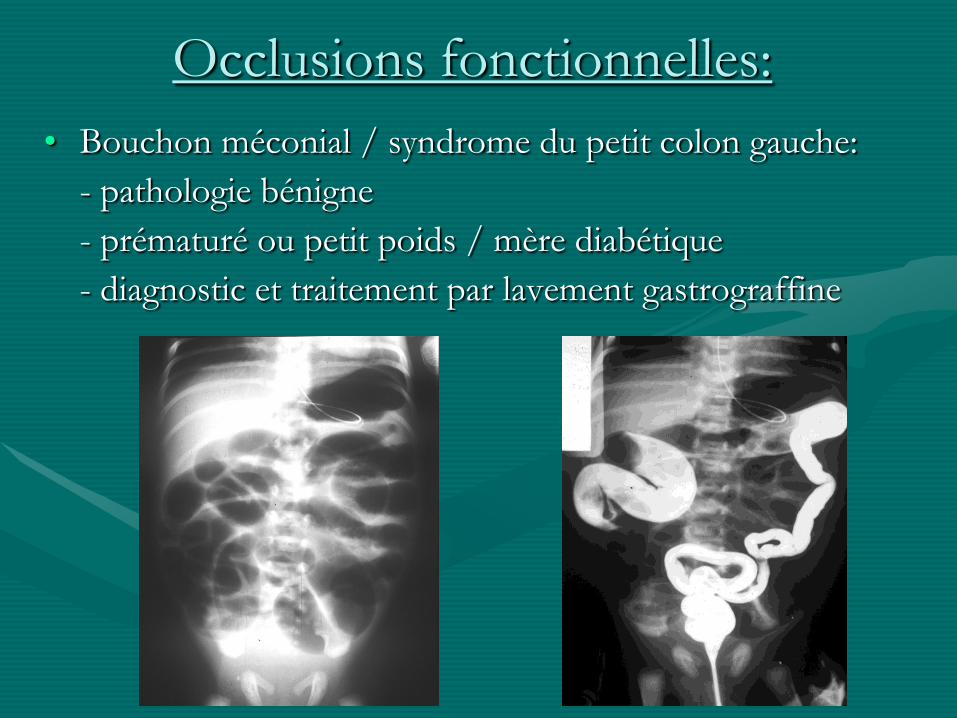
Occlusions fonctionnelles:
• Bouchon méconial / syndrome du petit colon gauche:
- pathologie bénigne
- prématuré ou petit poids / mère diabétique
- diagnostic et traitement par lavement gastrograffine

• POIC: pseudo-obstruction intestinale chronique =
neurodysplasie intestinale :
- groupe très divers: neuropathies, myopathies, ou sans
anomalies histologiques
- âge d ’apparition et étendue variable
- diagnostic difficile: manométrie / histologie / évolution
- syndrome occlusifs à répétition
- +/- atteinte vésicale
- complication parfois d’un Hirschsprung « classique »
- traitement : très souvent dérivation digestive (+/- urinaire)
prolongée
- pronostic parfois (souvent …) mauvais.
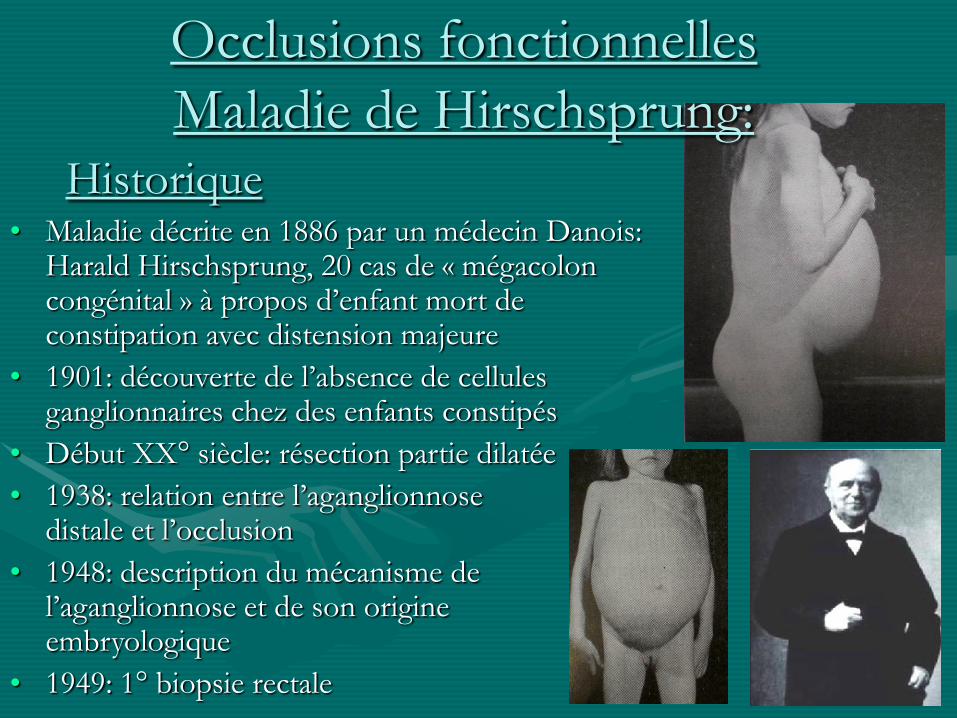
Historique• Maladie décrite en 1886 par un médecin Danois:
Harald Hirschsprung, 20 cas de « mégacolon congénital » à propos d’enfant mort de constipation avec distension majeure
• 1901: découverte de l’absence de cellules ganglionnaires chez des enfants constipés
• Début XX° siècle: résection partie dilatée
• 1938: relation entre l’aganglionnose distale et l’occlusion
• 1948: description du mécanisme de l’aganglionnose et de son origine embryologique
• 1949: 1° biopsie rectale
Occlusions fonctionnelles
Maladie de Hirschsprung:

• Absence de cellules ganglionnaires dans la sous-muqueuse
intestinale
• lieu: débute toujours à l ’extrémité inférieure du rectum et
remonte +/- loin sur le colon:
- forme recto-sigmoïdienne : habituelle : 75 %
- forme colique étendue: 10 %
- forme colique totale : rare : 5 à 10 %
- forme étendue au grêle : exceptionnelle : < 5%
• conséquences: zone aganglionnaire = obstacle fonctionnel,
dilatation en amont (mégacolon)
Physiopathologie:

• Neurochristopathie : arrêt de migration des neuroblastes à
partir des crêtes neurales dans le sens cranio-caudal
• Forme isolée le plus souvent (80 à 80 % des cas):
- forme syndromique parfois : d d’Ondine, Mowat-
Wilson, Waardenburg, Yemenite, Sipple …
- anomalie chromosomique : trisomie 21 +++
- malformations associées : rénale, cardiaque, faciale …
• Forme sproradique le plus souvent :
- forme familiale 5 à 10 % dont 50 % de forme
longue
- gêne RET +++, gênes modificateurs
• Pas de DAN

• Diagnostic clinique le plus souvent en période néonatale,
parfois plus tard dans les formes moins étendues:
- retard d ’émission du méconium +++
- ballonnement abdominal diffus +++
- vomissements ++
- test de la sonde rectale positif +++
• Approche du diagnostic:
- ASP: niveaux coliques, pas d ’air dans le pelvis
- lavement opaque: zone de transition
- rectomanométrie: absence de RRAI (absence de
relâchement du sphincter interne au moment de l’arrivée des
selles)
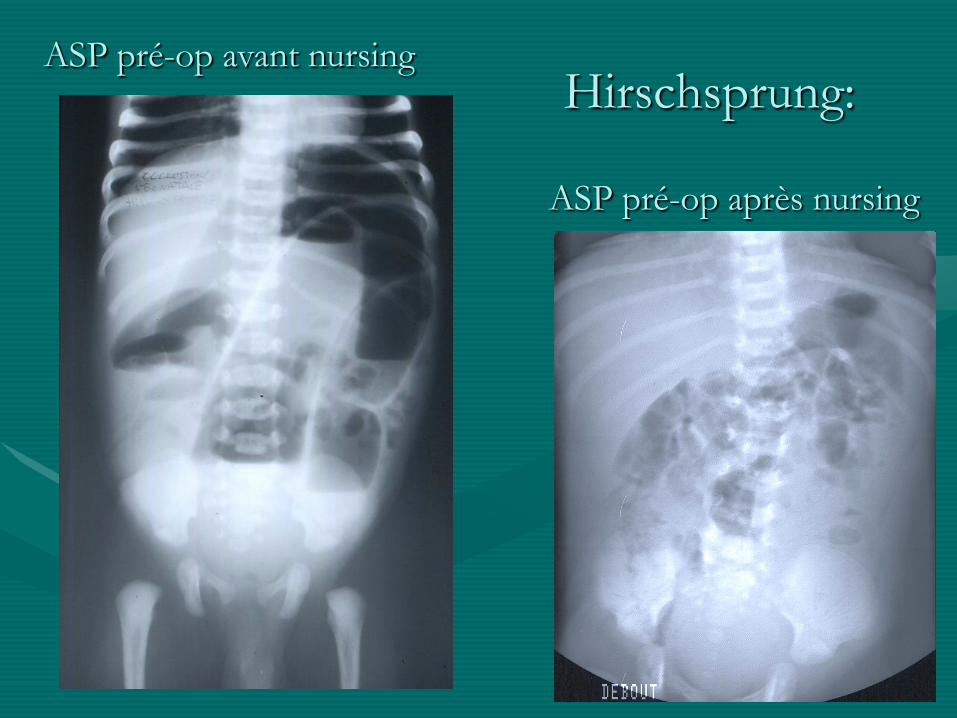
Hirschsprung:ASP pré-op avant nursing
ASP pré-op après nursing
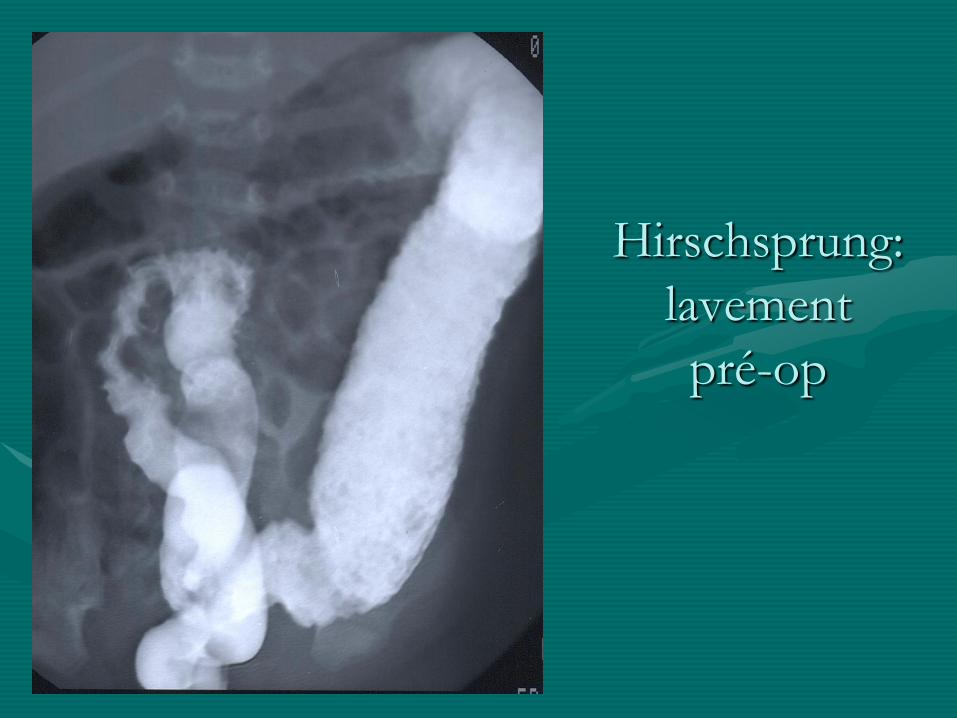
Hirschsprung:
lavement
pré-op

• Diagnostic de certitude par biopsie rectale +++
- sous prémédication
- aiguille de Noblett, aspiration de muqueuse et sous-
muqueuse anapath en 24 h : réponse binaire
• Traitement:
- c ’est une urgence médicale car risque de complications
graves : entérocolite +++, septicémie à point de départ
digestif, perforation caecale ...
* nursing rectal pluriquotidien
* alimentation prudente
* au moindre doute: discuter la colostomie +
antibiotiques

• Traitement (suite):
- traitement « historique »: dérivation digestive en zone saine
dès que le diagnostic était fait, colostomie gauche puis
abaissement du colon sain vers 3 à 4 mois (6 à 7 kg) :
Duhamel, Swenson, Soave ...
- traitement récent: voie trans-anale : nursing rectal pluri-
quotidien pendant quelques jours à quelques semaines, puis
intervention définitive par voie basse exclusive (2.8 à 4 kg)
avantages : intervention précoce, pas de cicatrice abdominale
ni colostomie
inconvénients : nécessité d ’une bonne coopération parentale,
risque de perforation ou entérocolite, sténose anale, peu de
résultats à long terme.

• Evolution:
- bon dans la majorité des cas : transit régulier
- constipation modérée parfois : traitement diététique voire médicamenteux
- transit parfois accéléré
- retard acquisition propreté, soiling fréquent avec la voie trans-anale : rééducation > 6 ans
- risque d ’entérocolite +++ pendant 2 ans post-op
- POIC : rare mais mieux connu actuellement, formes +/-précoce, +/- étendues et +/- sévère, nécessité souvent d ’une dérivation digestive +/- colectomie secondaire
- nécessité d ’un suivi à long terme +++ : 10 à 15 ans
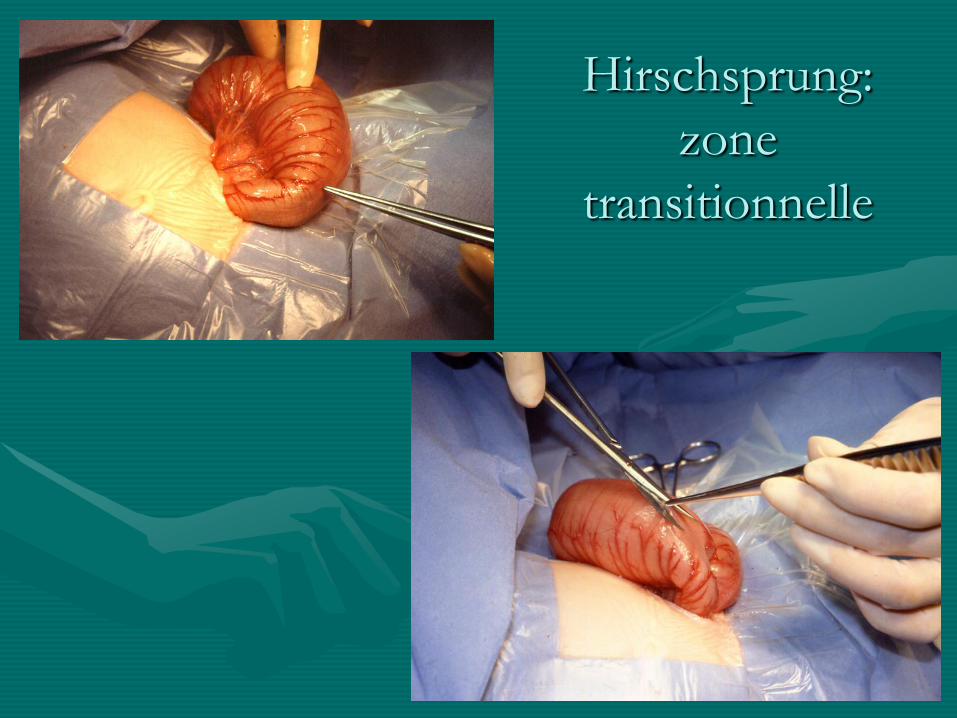
Hirschsprung:
zone
transitionnelle
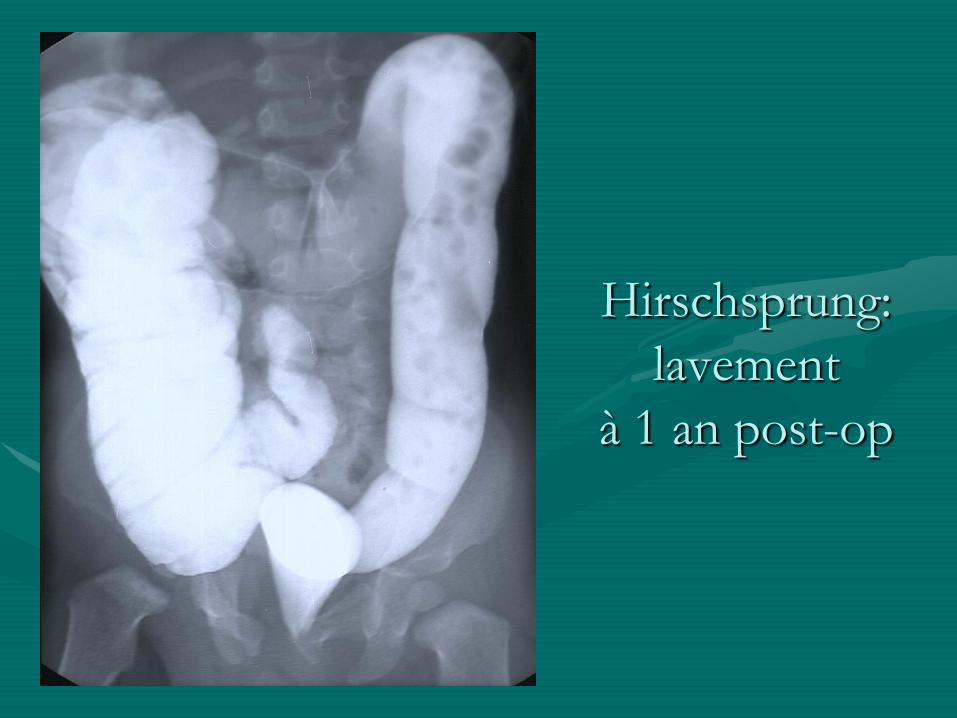
Hirschsprung:
lavement
à 1 an post-op
Top Related