







Langages
Pages
Légal


Antalgiques non opioïdes
A. Muller
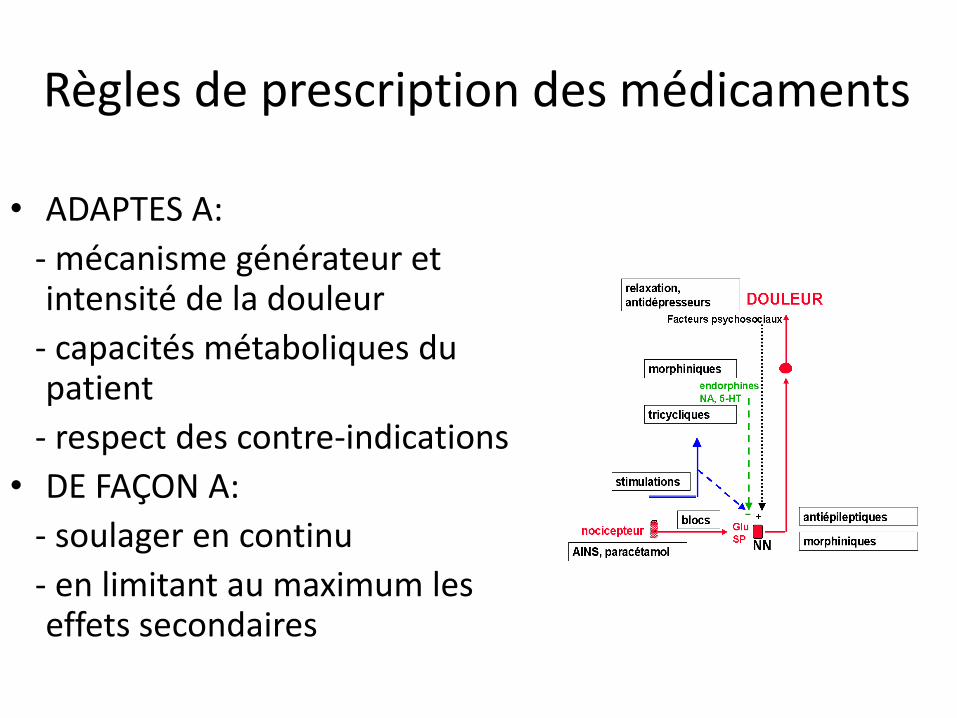
Règles de prescription des médicaments
• ADAPTES A:
- mécanisme générateur et intensité de la douleur
- capacités métaboliques du patient
- respect des contre-indications
• DE FAÇON A:
- soulager en continu
- en limitant au maximum les effets secondaires
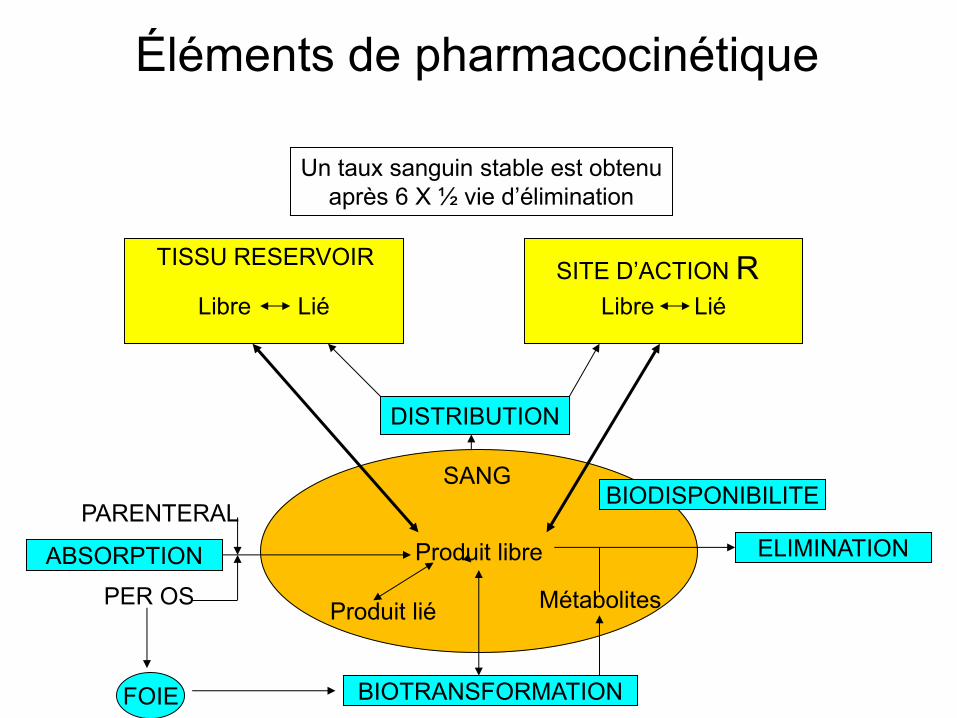
Éléments de pharmacocinétique
Libre Lié
Libre Lié
Produit libre
TISSU RESERVOIR SITE D’ACTION R
SANG
DISTRIBUTION
BIODISPONIBILITE
ABSORPTION ELIMINATION
BIOTRANSFORMATION
Produit lié Métabolites
PARENTERAL
PER OS
FOIE
Un taux sanguin stable est obtenu après 6 X ½ vie d’élimination

Susceptibilité individuelle aux médicaments
• Facteurs génétiques: il y a des différences dans les
récepteurs (µ), les capacités enzymatiques (CYP) de chacun
• Facteurs pharmacocinétiques: selon l’état de nutrition,
d’hydratation, la qualité des fonctions rénales et hépatiques,… la quantité de produit présente au site d’action varie
• Facteurs pharmacodynamiques: la polymédication est
source d’interférences
Notion de titration

EFFET PLACEBO
• Correspond aux effets psycho-physiologiques induits chez un patient par la prescription d’un placebo
• Ces effets peuvent ne pas influencer l’état du patient (sujet placebo non répondeur), l’améliorer (sujet placebo-répondeur), ou l'aggraver (sujet nocebo-répondeur)
• Effet « intervention médicale» = Effet de la thérapeutique + Effet placebo

Mécanismes de l’ analgésie placebo
• Neurobiologiques: implication partielle des opioïdes endogènes; activation des systèmes immunitaires qui influencent DL; importance de la connectivité dans CPFDL
• Psychophysiologiques (ont un support neurochimique!):
- confiance: effet sur tonus muscle et sympathique
- le fait « d’ignorer » la possibilité d’usage du placebo renforce son effet
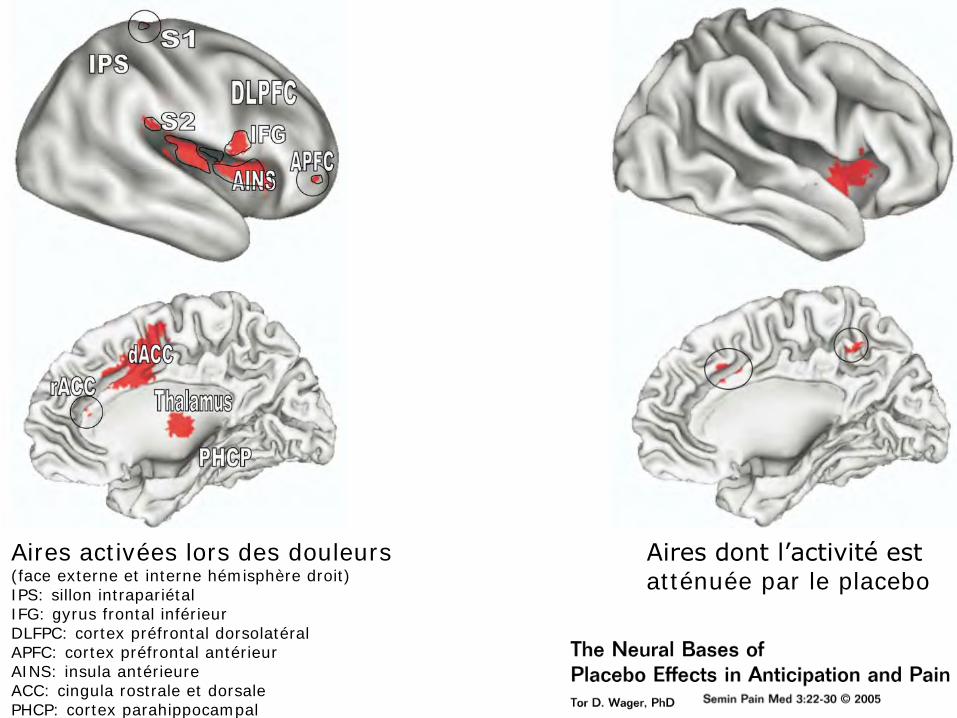
Aires dont l’activité est atténuée par le placebo
Aires activées lors des douleurs (face externe et interne hémisphère droit) IPS: sillon intrapariétal IFG: gyrus frontal inférieur DLFPC: cortex préfrontal dorsolatéral APFC: cortex préfrontal antérieur AINS: insula antérieure ACC: cingula rostrale et dorsale PHCP: cortex parahippocampal
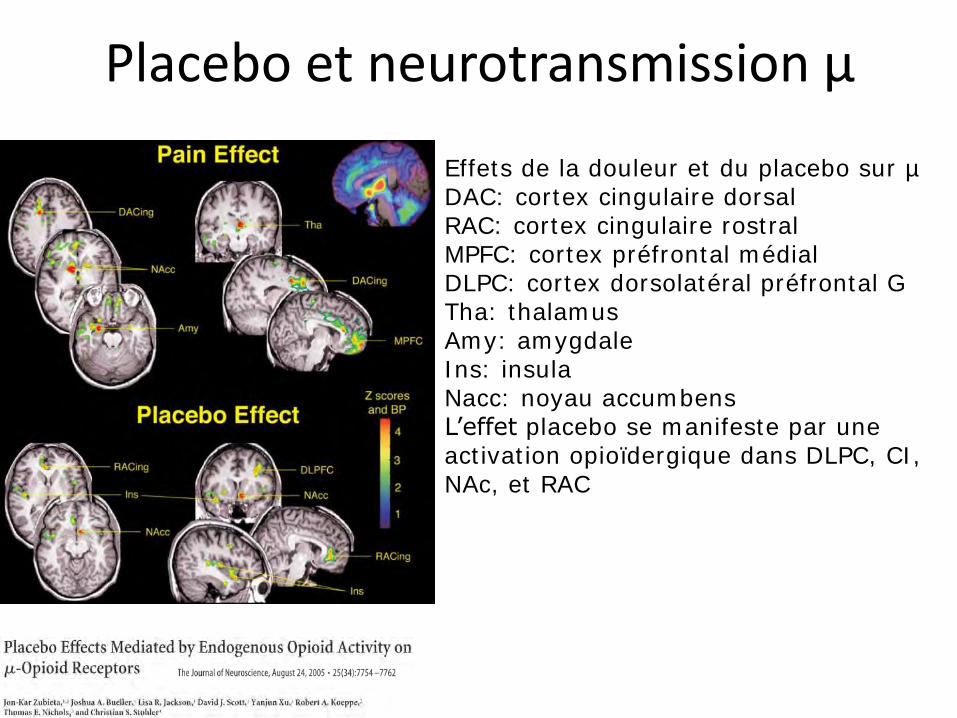
Placebo et neurotransmission µ
Effets de la douleur et du placebo sur µ DAC: cortex cingulaire dorsal RAC: cortex cingulaire rostral MPFC: cortex préfrontal médial DLPC: cortex dorsolatéral préfrontal G Tha: thalamus Amy: amygdale Ins: insula Nacc: noyau accumbens L’effet placebo se manifeste par une activation opioïdergique dans DLPC, CI, NAc, et RAC
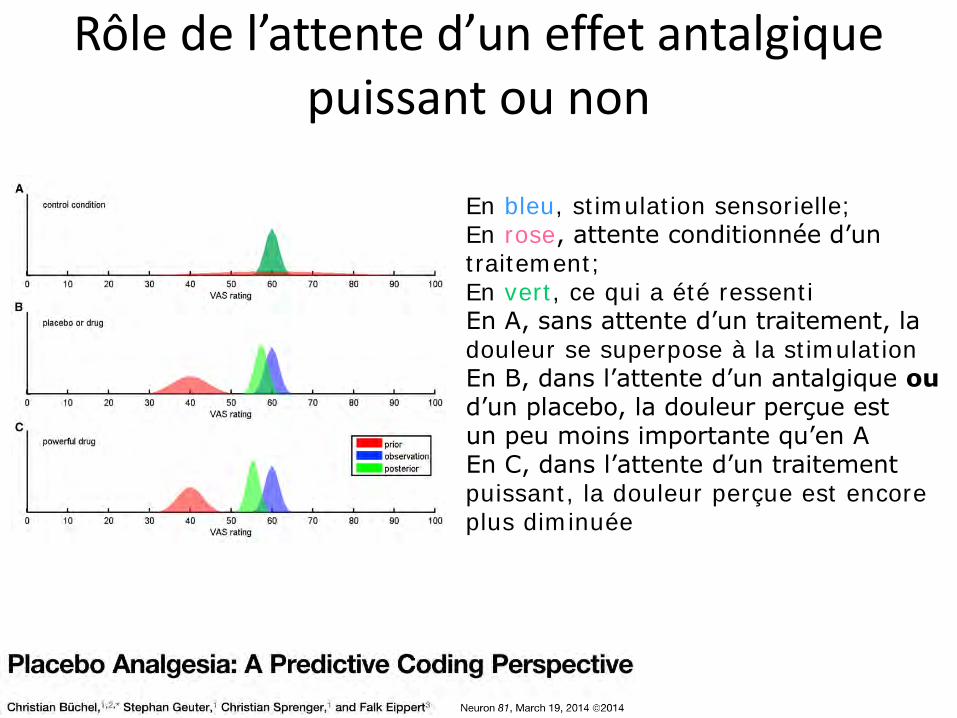
Rôle de l’attente d’un effet antalgique puissant ou non
En bleu, stimulation sensorielle; En rose, attente conditionnée d’un traitement; En vert, ce qui a été ressenti En A, sans attente d’un traitement, la douleur se superpose à la stimulation En B, dans l’attente d’un antalgique ou d’un placebo, la douleur perçue est un peu moins importante qu’en A En C, dans l’attente d’un traitement puissant, la douleur perçue est encore plus diminuée
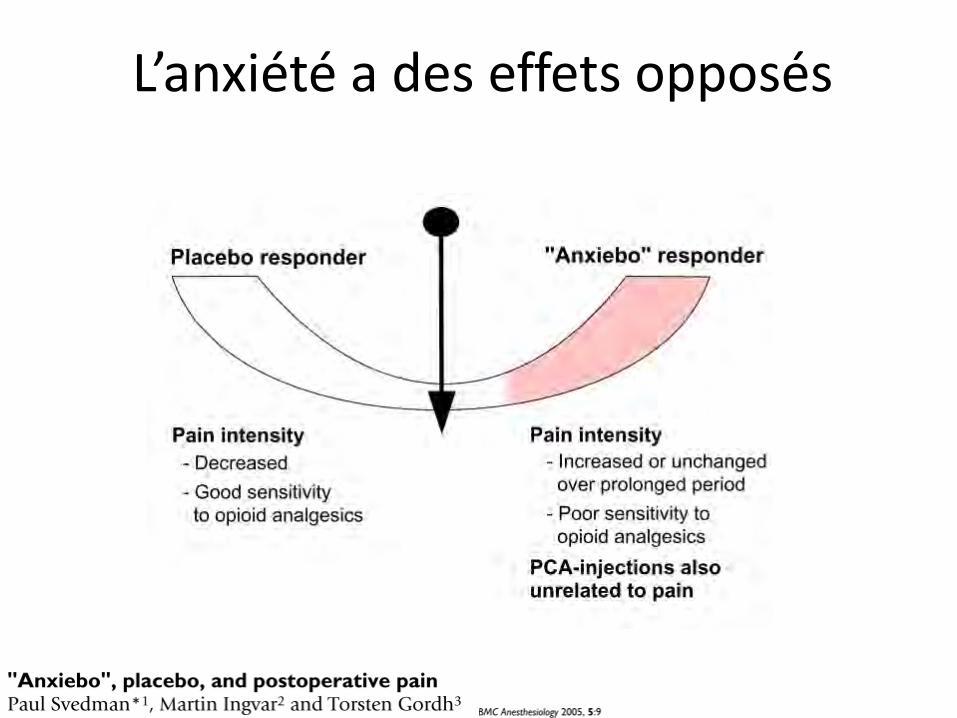
L’anxiété a des effets opposés

Les paliers de l’OMS L’Organisation Mondiale de la Santé a défini en
1985 trois niveaux d’antalgiques (pour les douleurs cancéreuses nociceptives) :
Antalgiques non opioïdes
Antalgiques opioïdes faibles
Antalgiques opioïdes forts
Palier I
Palier III
Palier II
Utilisation de co-analgésiques pour les douleurs neuropathiques associées
EVA 0
10
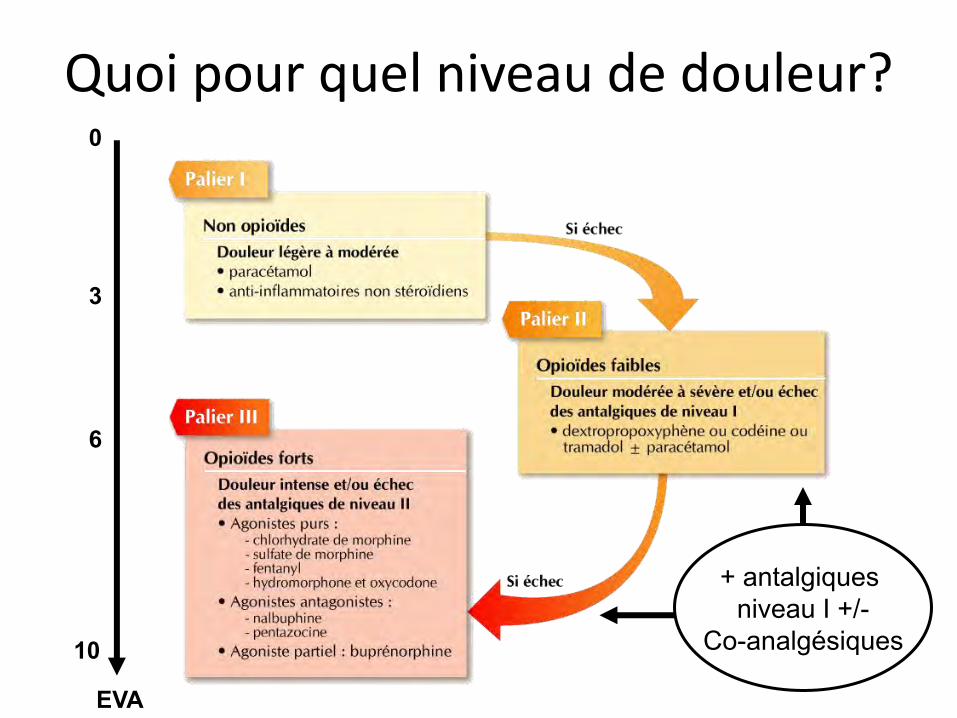
Quoi pour quel niveau de douleur?
+ antalgiques niveau I +/-
Co-analgésiques
EVA
0
10
3
6
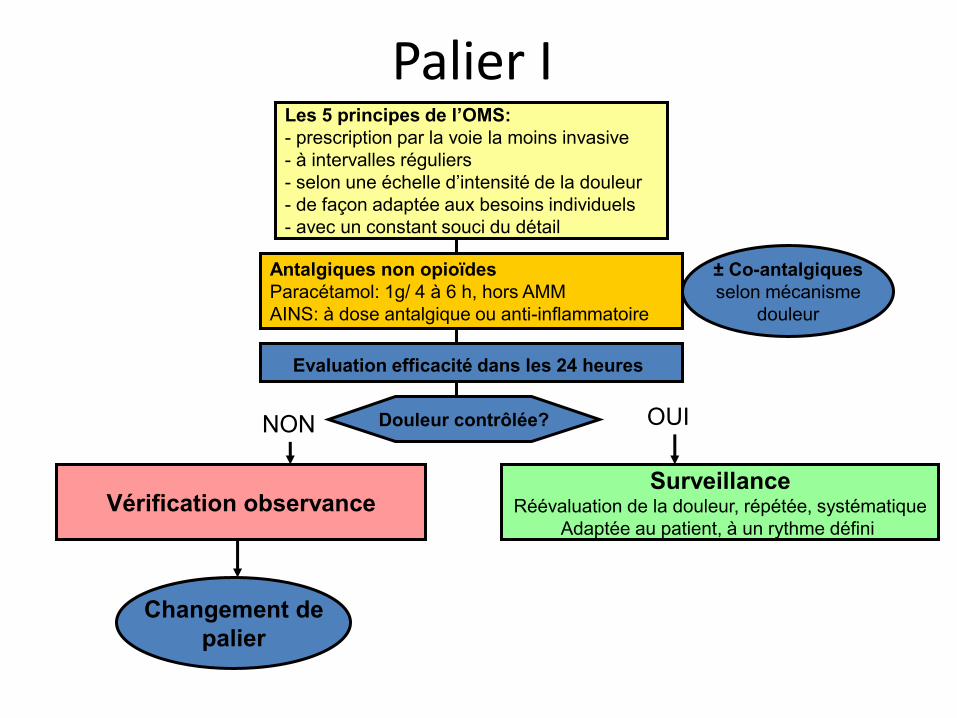
Palier I Les 5 principes de l’OMS: - prescription par la voie la moins invasive - à intervalles réguliers - selon une échelle d’intensité de la douleur - de façon adaptée aux besoins individuels - avec un constant souci du détail
Antalgiques non opioïdes Paracétamol: 1g/ 4 à 6 h, hors AMM AINS: à dose antalgique ou anti-inflammatoire
Evaluation efficacité dans les 24 heures
± Co-antalgiques selon mécanisme
douleur
Douleur contrôlée?
Changement de palier
Vérification observance Surveillance
Réévaluation de la douleur, répétée, systématique Adaptée au patient, à un rythme défini
OUI NON
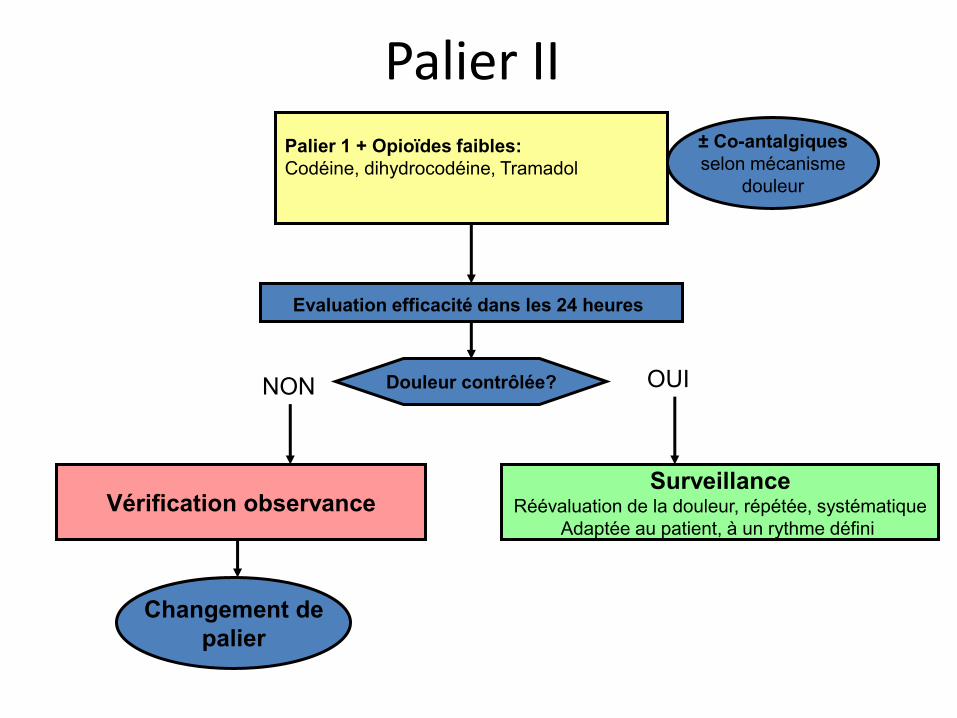
Palier II Palier 1 + Opioïdes faibles: Codéine, dihydrocodéine, Tramadol
Evaluation efficacité dans les 24 heures
± Co-antalgiques selon mécanisme
douleur
Douleur contrôlée?
Changement de palier
Vérification observance Surveillance
Réévaluation de la douleur, répétée, systématique Adaptée au patient, à un rythme défini
OUI NON
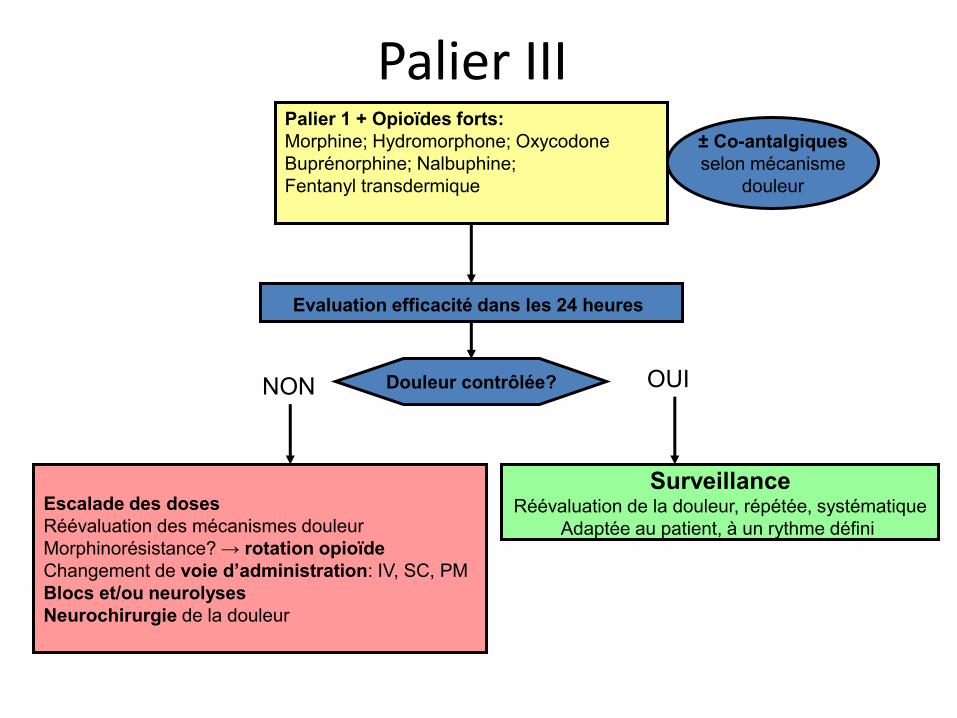
Palier III Palier 1 + Opioïdes forts: Morphine; Hydromorphone; Oxycodone Buprénorphine; Nalbuphine; Fentanyl transdermique
Evaluation efficacité dans les 24 heures
± Co-antalgiques selon mécanisme
douleur
Douleur contrôlée?
Escalade des doses Réévaluation des mécanismes douleur Morphinorésistance? → rotation opioïde Changement de voie d’administration: IV, SC, PM Blocs et/ou neurolyses Neurochirurgie de la douleur
Surveillance Réévaluation de la douleur, répétée, systématique
Adaptée au patient, à un rythme défini
OUI NON

M. ALT 16
ANTALGIQUES MINEURS
• Dérivés du para-amino-phénol (paracétamol et autres) • AINS
– Inhibiteurs sélectifs de la COX1 – Inhibiteurs non sélectifs de la COX correspondant à la majorité
des AINS classiques – Inhibiteurs préférentiels de la COX2 – Inhibiteurs sélectifs de la COX2
• Dérivés de la pyrazolone • Antalgiques purs • Antispasmodiques
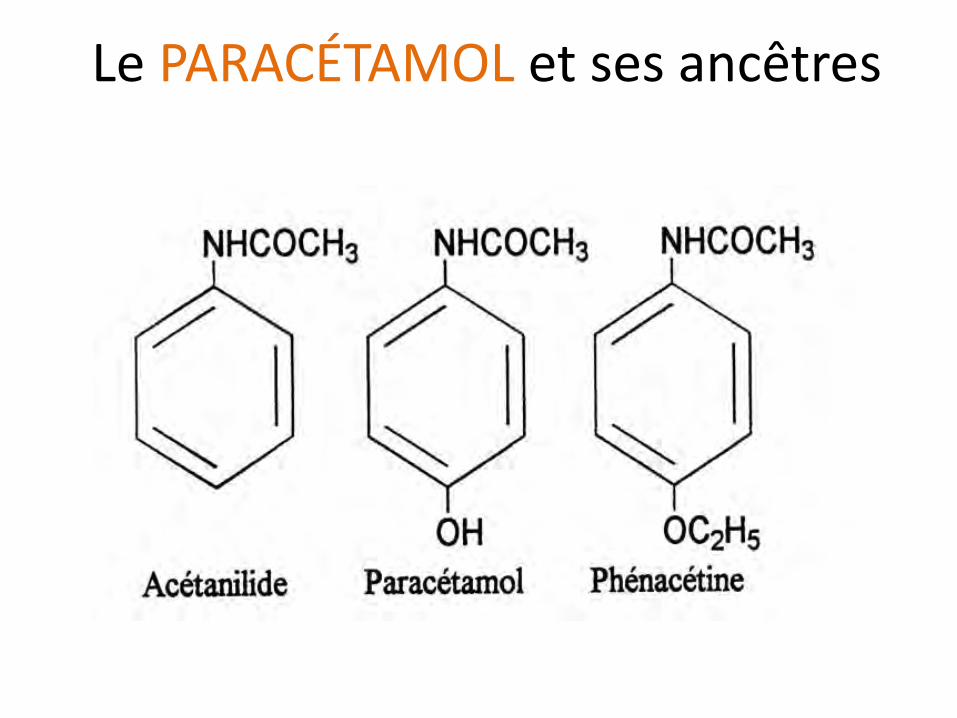
Le PARACÉTAMOL et ses ancêtres
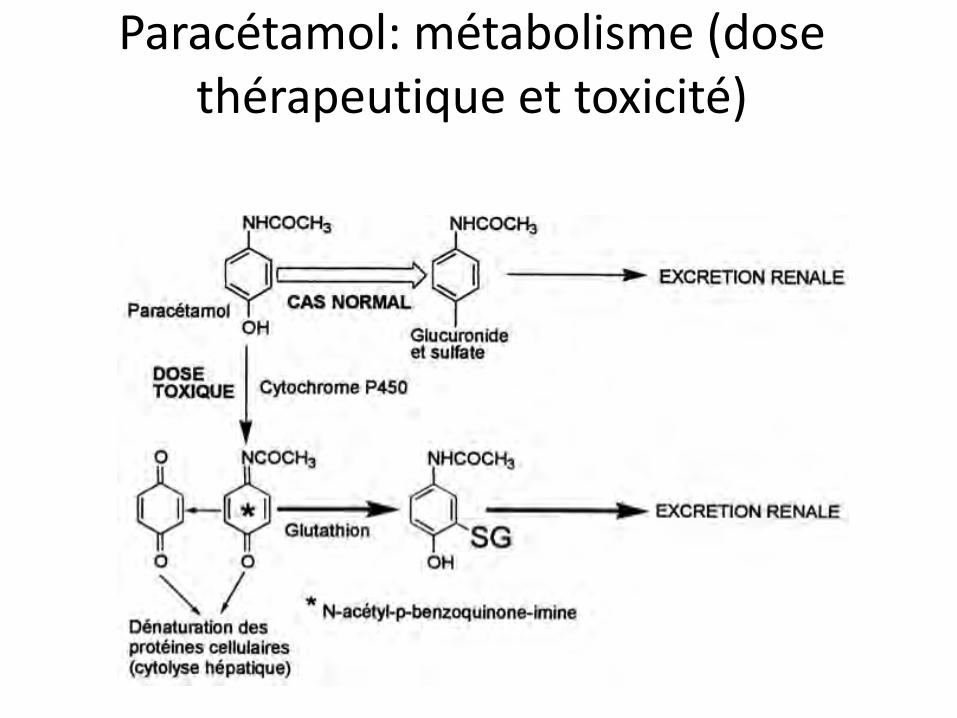
Paracétamol: métabolisme (dose thérapeutique et toxicité)
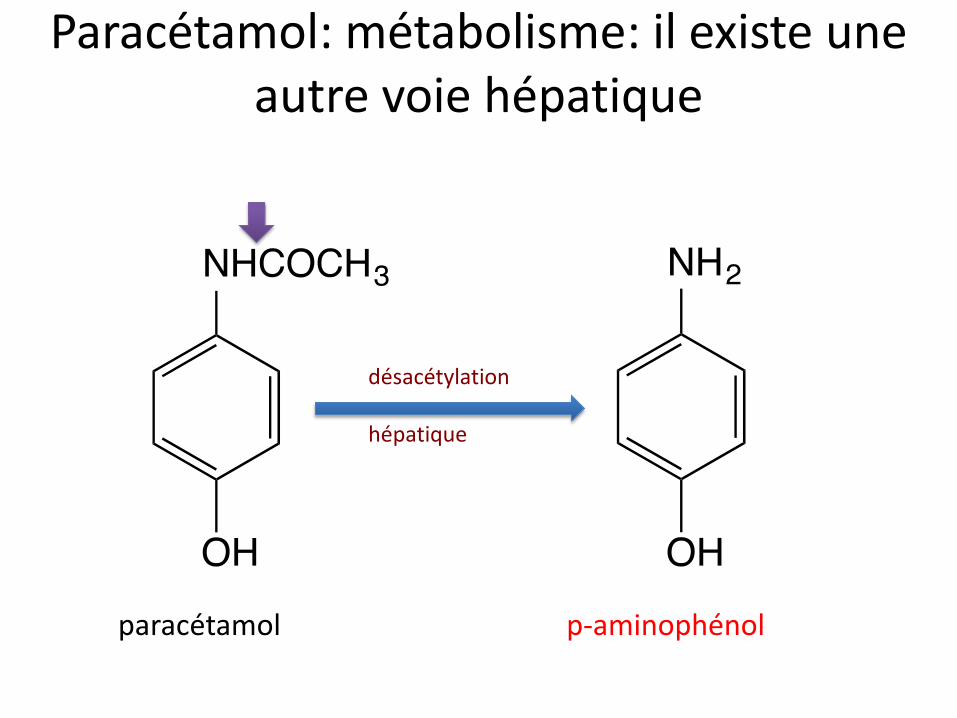
Paracétamol: métabolisme: il existe une autre voie hépatique
paracétamol p-aminophénol
désacétylation hépatique
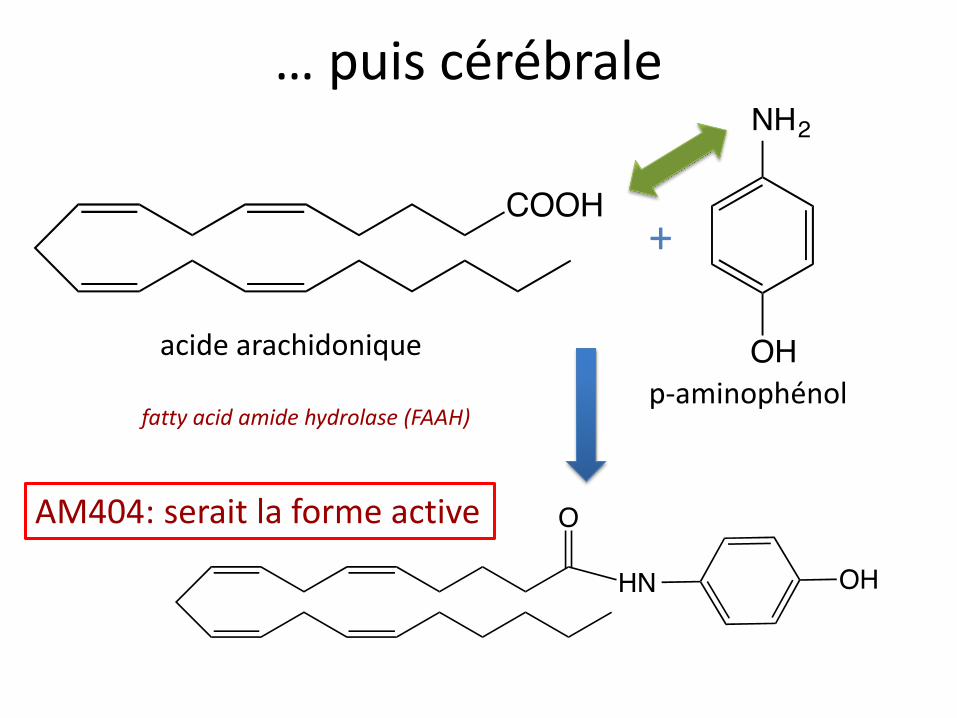
… puis cérébrale
p-aminophénol
acide arachidonique
+
fatty acid amide hydrolase (FAAH)
AM404: serait la forme active

Mécanismes d’action
• Inhibiteur de la COX3 centrale
• Aurait un effet anti-NMDA
• Activateur de la voie sérotoninergique descendante, effet de AM404 via:
- activation des récepteurs TRPV1 centraux
- et/ou inhibition de la dégradation des endocannabinoïdes (activation récepteurs CB1)

Cinétique du paracétamol
• Per os – Taux plasmatique atteint en moins de 2 h
– Demi-vie de 1 à 3 h
– Taux de liaison aux protéines faible (doses thérapeutiques) Si augmentation des doses, taux de liaison augmente
– Élimination urinaire
• Voie – Per os, rectale (paracétamol)
– Parentérale (Perfalgan®)

Prescription
• 4 à 6 g/j selon l’état de la fonction hépatique
• Pour les douleurs nociceptives d’intensité faible
• Peut être associé aux AINS (palier 1) et aux opioïdes faibles (palier 2) ou forts (palier 3)
• Effets secondaires possibles: exceptionnelles réactions allergiques; toxicité hépatique (cytolyse)

AINS
• Les PG périphériques sensibilisent les nocicepteurs; il existe des PG pro-inflammatoires et des PG anti-inflammatoires
• Les PG participent en central à la sensibilisation des voies de la douleur
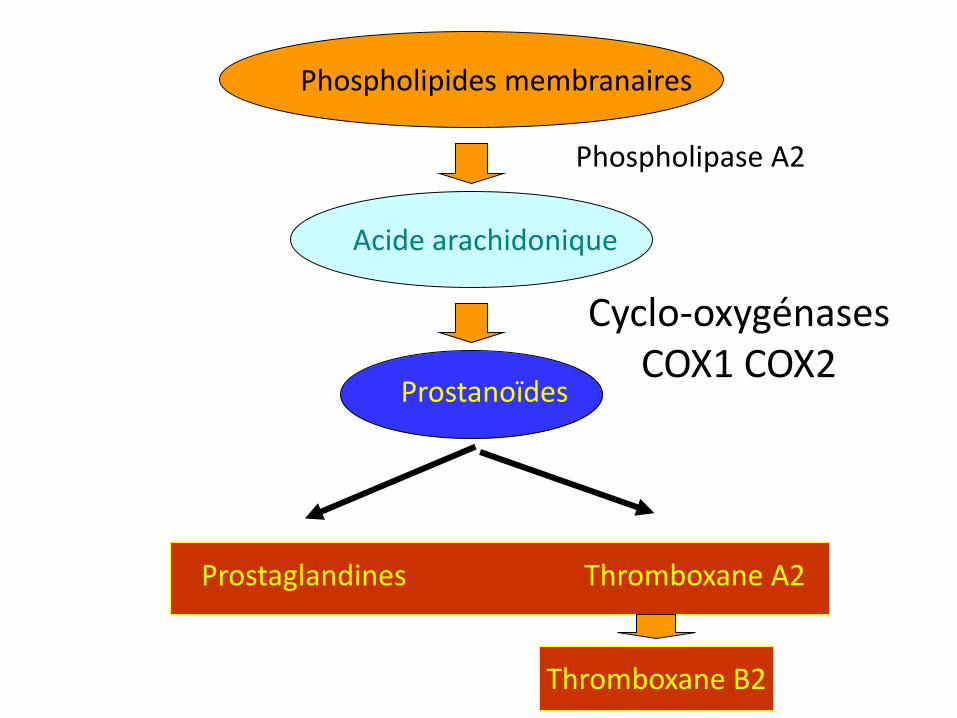
Prostaglandines Thromboxane A2
Phospholipides membranaires
Acide arachidonique
Prostanoïdes
Phospholipase A2
Cyclo-oxygénases COX1 COX2 COX3 (SNC)
Thromboxane B2

Cardiovasculaire: PGE vasodilatatrices
prostacycline (PGI2) vasodilatatrice
thromboxane (TXA2) vasoconstricteur
Coagulation: PGI2 inhibe l'agrégation plaquettaire
TXA2 proagrégant
Utérus gravide: PGFs, PGE2, PGF2 contraction
Tube digestif: réduction du transit
augmentation production des sécrétions
Rein: augmentation du flux sanguin
augmentation de la diurèse, de la natriurèse
et de la kaliurèse
Effets biologiques des prostanoïdes
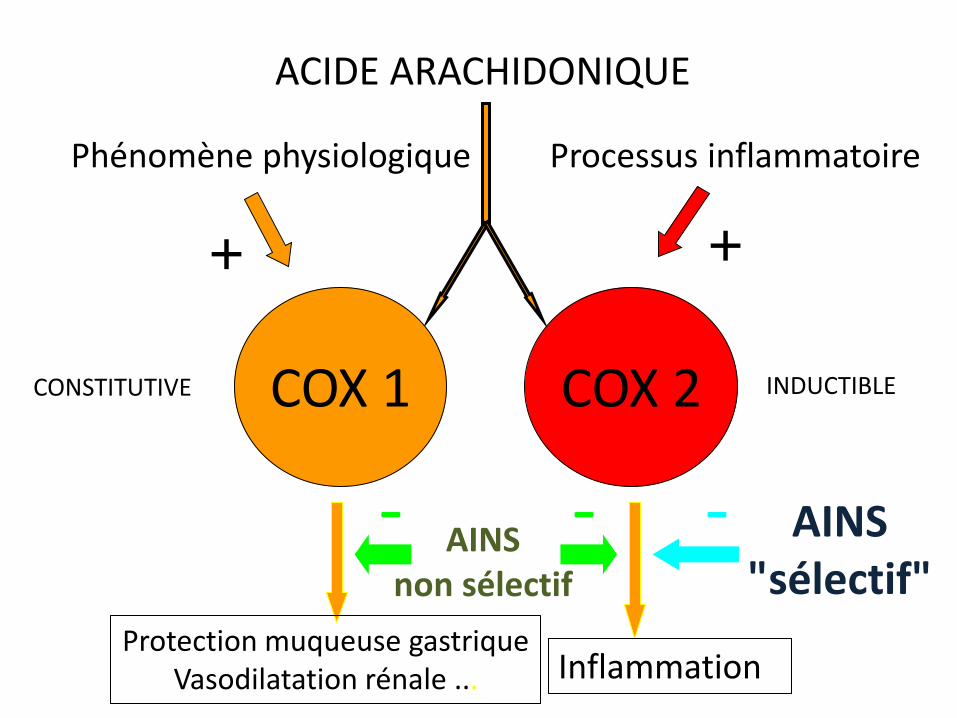
Phénomène physiologique Processus inflammatoire
ACIDE ARACHIDONIQUE
COX 1 COX 2 CONSTITUTIVE INDUCTIBLE
Protection muqueuse gastrique Vasodilatation rénale ... Inflammationn
+ +
AINS non sélectif
- - AINS "sélectif"
-
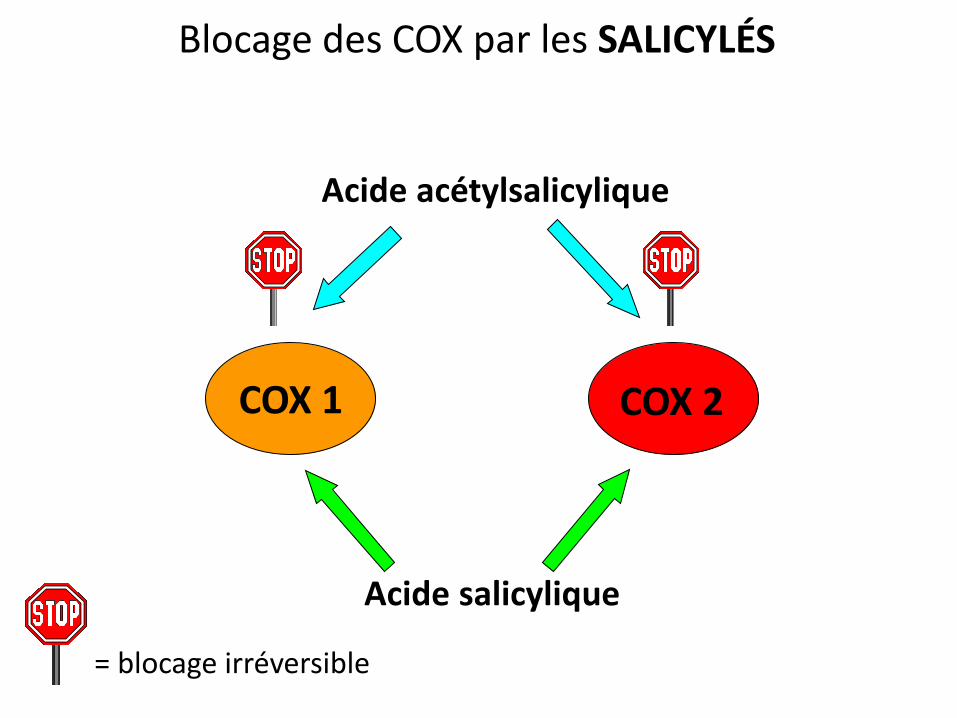
Blocage des COX par les SALICYLÉS
COX 1 COX 2
Acide acétylsalicylique
Acide salicylique
= blocage irréversible
Réversible Réversible

L’aspirine est donc :
• Un très bon antiagrégant plaquettaire (dose faible,
forte sélectivité COX1, effet irréversible)
• Un mauvais AINS (dose forte, réversible, sélectif de la
COX1 et donc mal toléré: estomac et rein ++)

1. Effet anti-inflammatoire
2. Action antalgique
3. Effet antipyrétique
4. Effets hématologiques
5. Effets gastrointestinaux
6. Effets gynéco-obtétricaux
7. Effets observés lors d'une intoxication
8. Le syndrome de Fernand Widal
Effets de l'aspirine
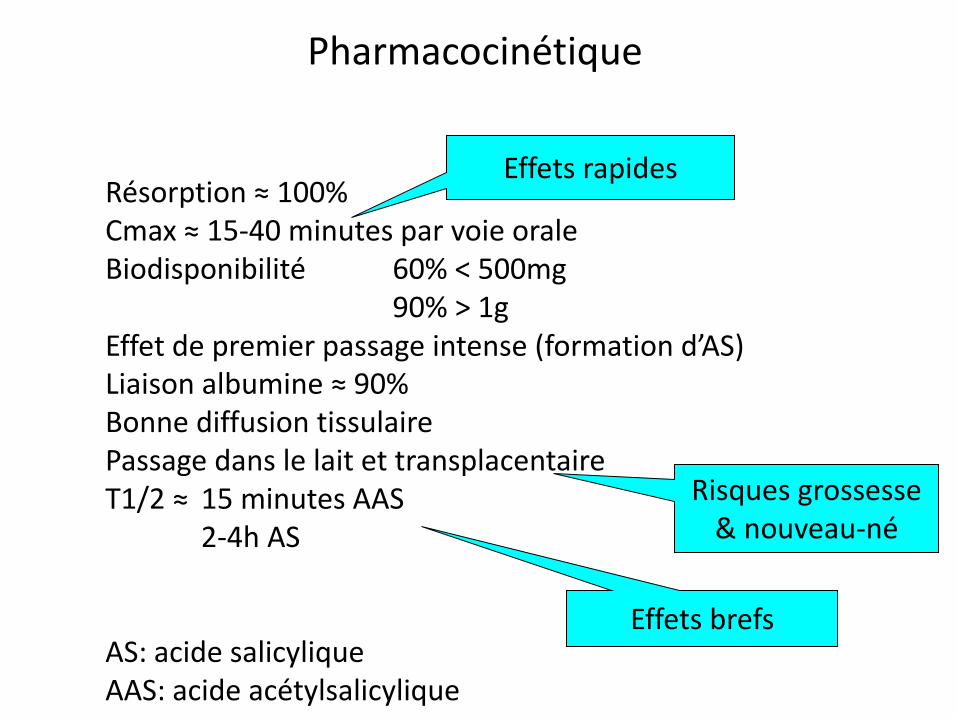
Résorption ≈ 100% Cmax ≈ 15-40 minutes par voie orale Biodisponibilité 60% < 500mg 90% > 1g Effet de premier passage intense (formation d’AS) Liaison albumine ≈ 90% Bonne diffusion tissulaire Passage dans le lait et transplacentaire T1/2 ≈ 15 minutes AAS 2-4h AS AS: acide salicylique AAS: acide acétylsalicylique
Pharmacocinétique
Effets rapides
Risques grossesse & nouveau-né
Effets brefs

Aspirine: effets indésirables
• Certains relèvent du mécanisme d’action • Toxicité digestive
• Inhibition de l’agrégation plaquettaire (risque hémorragique)
• Insuffisance rénale
• Inhibition de la motilité utérine
• Effets sur le fœtus
• Effets broncho-pulmonaires
• Intoxication salicylée chez l’enfant
• D’autres non • Atteintes cutanées
• Atteintes hépatiques
• Néphropathie interstitielle

Aspirine: interactions
• Diurétiques : risque d’insuffisance rénale, réduction de l’effet antihypertenseur
• IEC : risque d’insuffisance rénale
• Potentialise l’activité
• Des anticoagulants oraux (AVK)
• Des sulfamides hypoglycémiants
• Du méthotrexate
– clairance du méthotrexate
– toxicité, notamment hématologique

Autres AINS
• Propriétés communes aux AINS – Anti-inflammatoire (après
quelques jours de traitement) – Antipyrétique – Antiagrégant plaquettaire
(sauf inhibiteurs de la COX2)
• Mécanisme d’action – Inhibition de la synthèse des
prostaglandines par action sur la cyclo-oxygénase
• Plusieurs familles chimiques

Inhibiteurs spécifiques COX2 • Inhibiteurs sélectifs de la COX2
– Célécoxib – Rofécoxib
• Propriétés – Antalgique – Anti-inflammatoire – Pas d’action antiagrégante plaquettaire
• Meilleure tolérance (?) – Deux à trois fois moins d’ulcères gastriques – À court et moyen terme
• Profil de tolérance identiques aux AINS • Effets indésirables rénaux
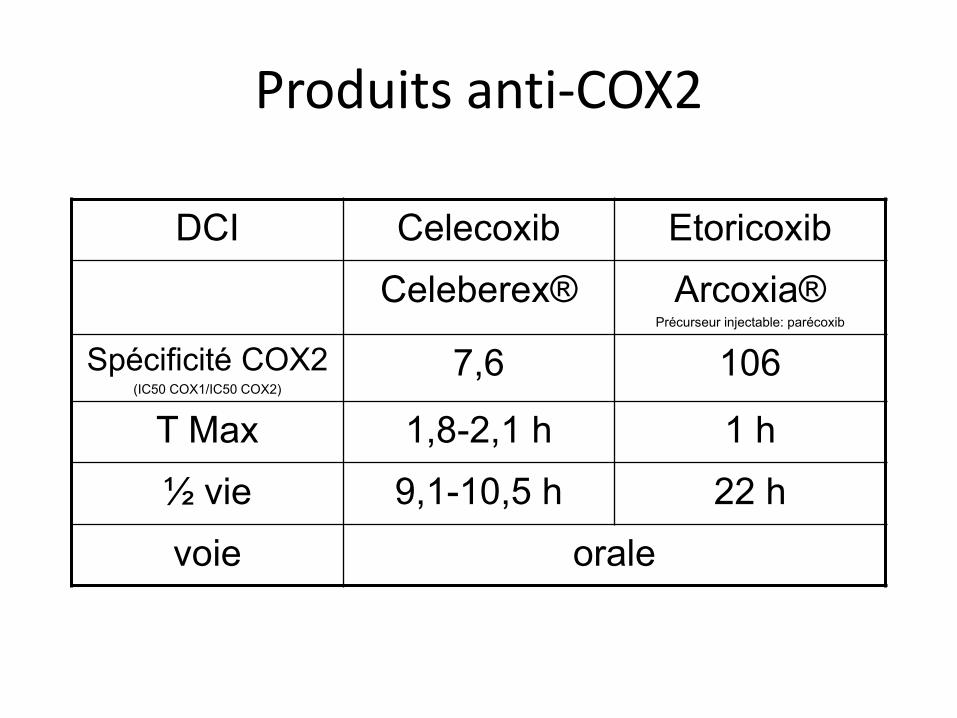
Produits anti-COX2
DCI Celecoxib Etoricoxib Celeberex® Arcoxia®
Précurseur injectable: parécoxib
Spécificité COX2 (IC50 COX1/IC50 COX2)
7,6 106
T Max 1,8-2,1 h 1 h ½ vie 9,1-10,5 h 22 h voie orale

Autres antalgiques mineurs
• Fénines
• Néfopam
• Antispasmodiques: musculotropes ou anticholinergiques

Fénines
• Glafénine : retirée en raison du risque de choc anaphylactique
• Floctafénine (Idarac®)
• Autres propriétés – Peu anti-inflammatoire
– Peu antipyrétique
• Voie orale
• Contre-indication – Allergie à la glafénine, floctafénine

M. ALT 39
Néfopam
• Action centrale complexe (dopamine, noradrénaline, sérotonine)
• Voie IM lente / IV lente
• Effets indésirables – Cardiaques (tachycardie, HTA)
– Oculaires (glaucome)
– Rétention urinaire
– SNC : irritabilité

Musculotropes
• Action directe sur les fibres musculaires lisses (tube digestif, voies urinaires)
• Phloroglucinol (Spasfon®) Trimébutine (Débridat®)
• Bonne tolérance

Anticholinergiques
• Antagonisent les effets muscariniques de l’ACh (muscles lisses du tube digestif, voies urinaires)
• Tiémonium (Viscéralgine®)
• Effets indésirables atropiniques – Sécheresse buccale – Constipation – Tachycardie – Confusion mentale

ANTALGIQUES MAJEURS: OPIOÏDES
• Deux catégories d’après l’OMS: faibles (tramadol, codéine, tapentadol) et forts (morphine, oxycodone, sophidone, fentanyl)
• Agonistes µ préférentiels, agonistes partiels, agonistes-antagonistes, antagonistes
• La méthadone n’a une AMM que pour la substitution des héroïnomanes et en SP
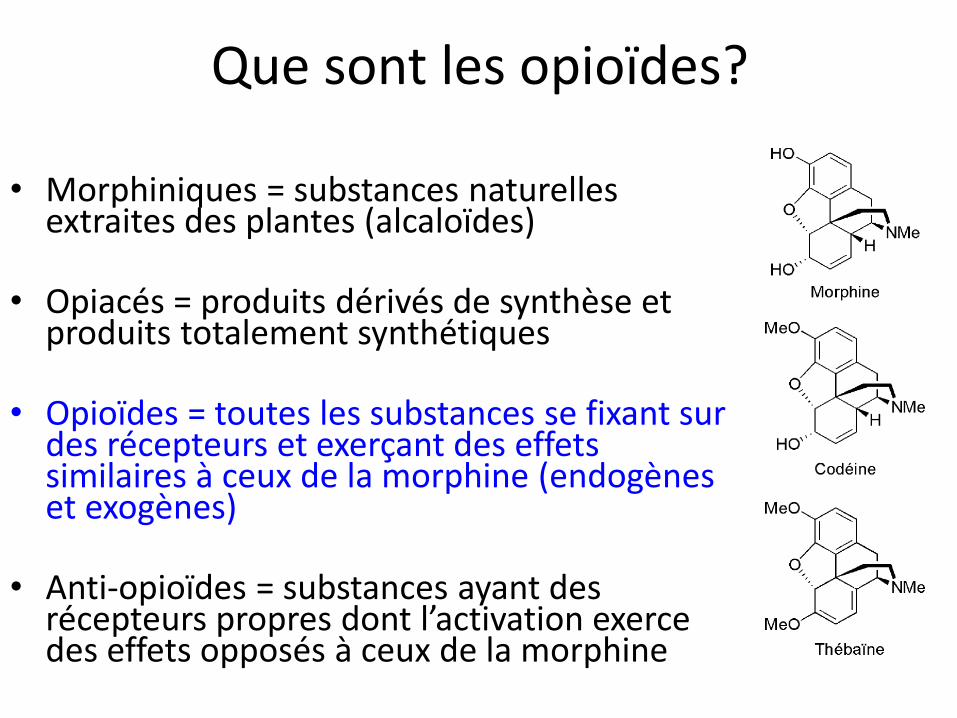
Que sont les opioïdes?
• Morphiniques = substances naturelles
extraites des plantes (alcaloïdes)
• Opiacés = produits dérivés de synthèse et produits totalement synthétiques
• Opioïdes = toutes les substances se fixant sur des récepteurs et exerçant des effets similaires à ceux de la morphine (endogènes et exogènes)
• Anti-opioïdes = substances ayant des récepteurs propres dont l’activation exerce des effets opposés à ceux de la morphine

Où agissent les opioïdes?
• Sur des récepteurs métabotropiques
• Trois variétés: µ, δ, κ, + polymorphisme
• Distribués dans tout l’organisme, en particulier le long des voies nociceptives
• Face à un récepteur, un opioïde a:
- une « force » de fixation = affinité → durée d’action
- une capacité à mobiliser les messagers intracellulaires = efficacité intrinsèque. La puissance (face à un mécanisme donné de douleur) est l’efficacité rapportée à l’unité pondérale
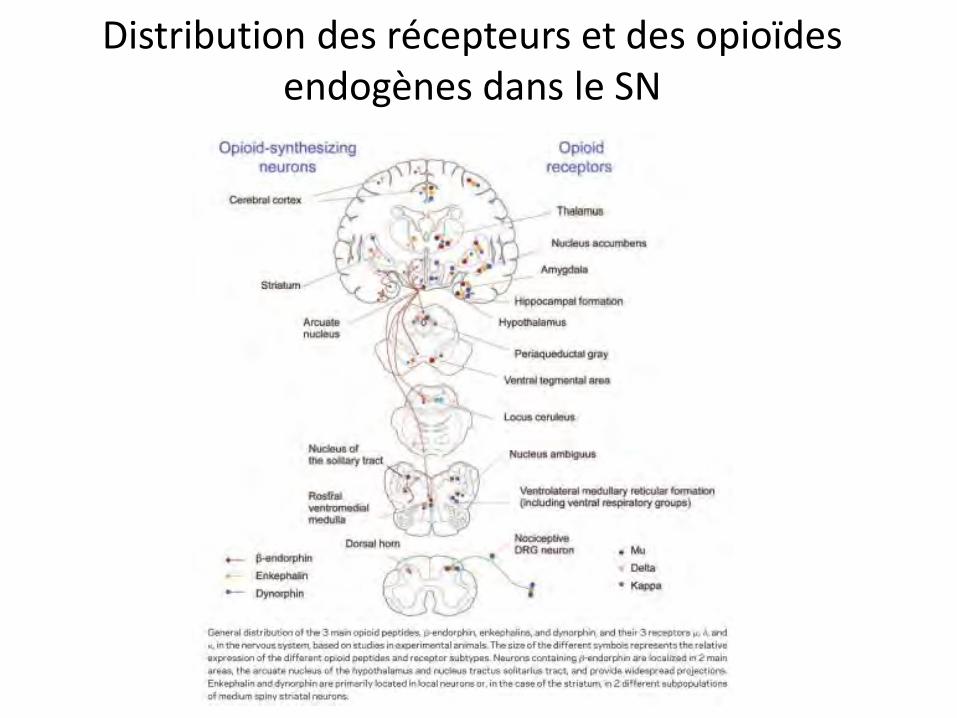
Distribution des récepteurs et des opioïdes endogènes dans le SN
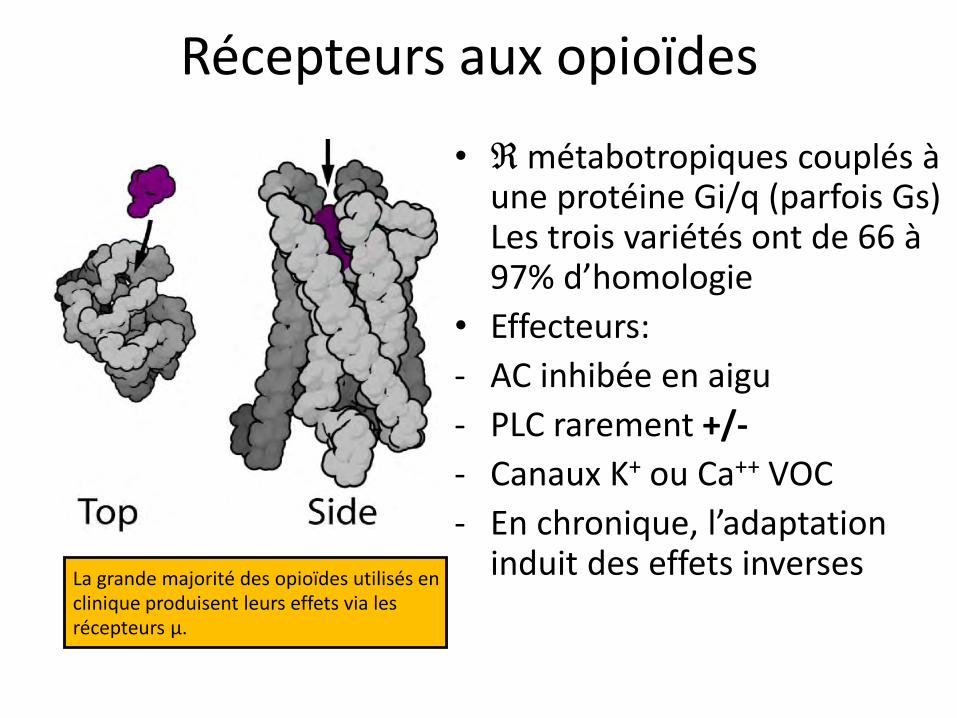
La grande majorité des opioïdes utilisés en clinique produisent leurs effets via les récepteurs µ.
• ℜ métabotropiques couplés à une protéine Gi/q (parfois Gs) Les trois variétés ont de 66 à 97% d’homologie
• Effecteurs:
- AC inhibée en aigu
- PLC rarement +/-
- Canaux K+ ou Ca++ VOC
- En chronique, l’adaptation induit des effets inverses
Récepteurs aux opioïdes

Il existe plusieurs variants de récepteurs µ
Conséquence clinique: grande variabilité interindividuelle dans la sensibilité à la morphine
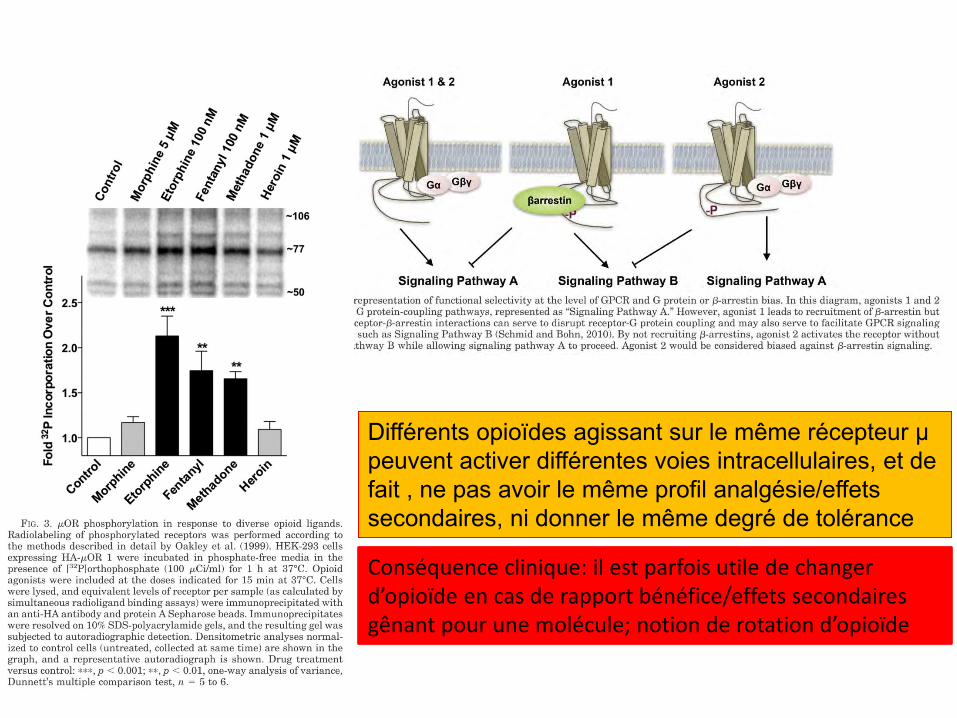
Différents opioïdes agissant sur le même récepteur µ peuvent activer différentes voies intracellulaires, et de fait , ne pas avoir le même profil analgésie/effets secondaires, ni donner le même degré de tolérance
Conséquence clinique: il est parfois utile de changer d’opioïde en cas de rapport bénéfice/effets secondaires gênant pour une molécule; notion de rotation d’opioïde
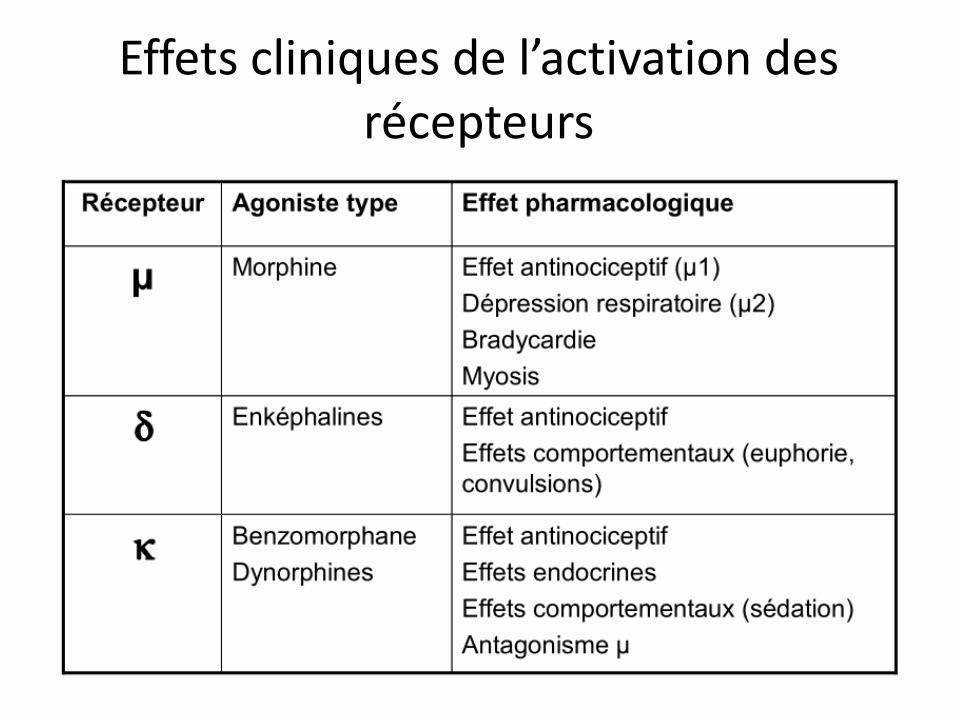
Effets cliniques de l’activation des récepteurs
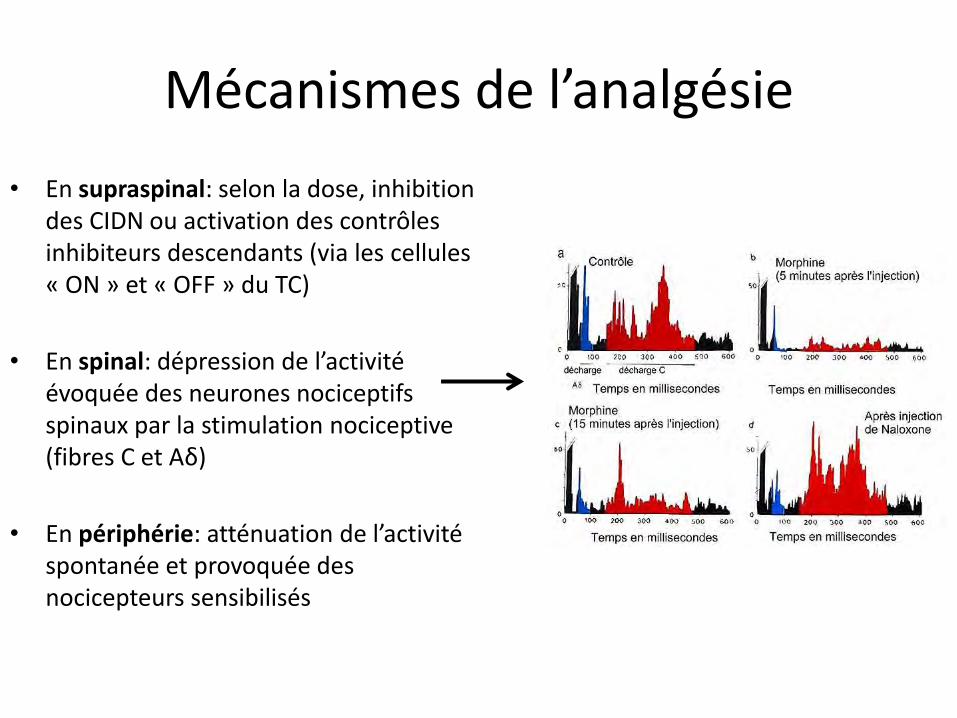
Mécanismes de l’analgésie
• En supraspinal: selon la dose, inhibition des CIDN ou activation des contrôles inhibiteurs descendants (via les cellules « ON » et « OFF » du TC)
• En spinal: dépression de l’activité évoquée des neurones nociceptifs spinaux par la stimulation nociceptive (fibres C et Aδ)
• En périphérie: atténuation de l’activité spontanée et provoquée des nocicepteurs sensibilisés
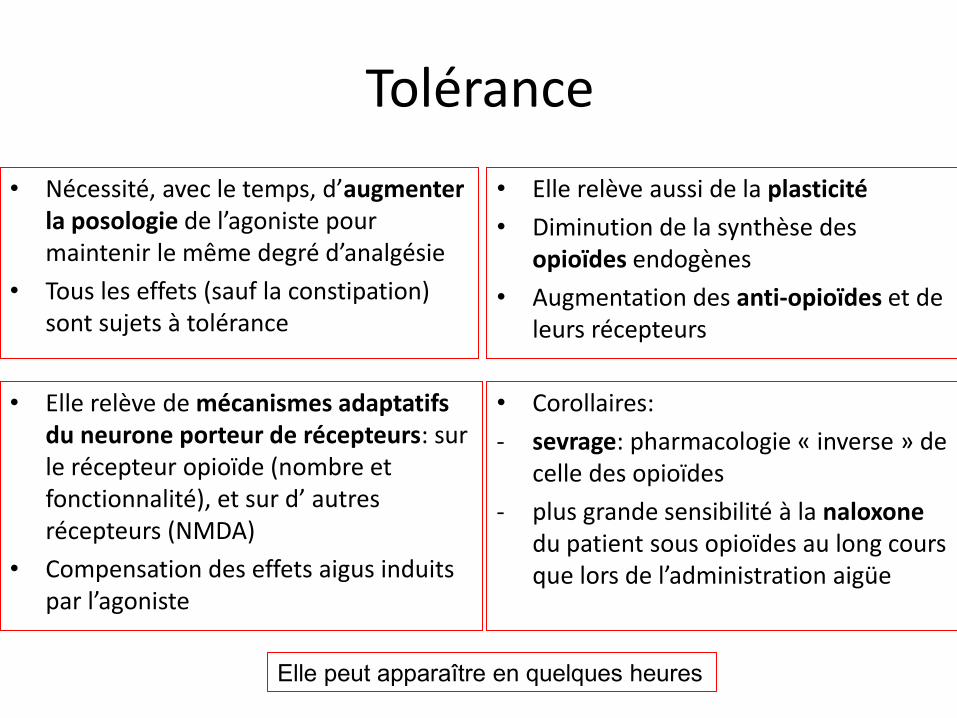
Tolérance
• Nécessité, avec le temps, d’augmenter la posologie de l’agoniste pour maintenir le même degré d’analgésie
• Tous les effets (sauf la constipation) sont sujets à tolérance
• Elle relève aussi de la plasticité
• Diminution de la synthèse des opioïdes endogènes
• Augmentation des anti-opioïdes et de leurs récepteurs
• Elle relève de mécanismes adaptatifs du neurone porteur de récepteurs: sur le récepteur opioïde (nombre et fonctionnalité), et sur d’ autres récepteurs (NMDA)
• Compensation des effets aigus induits par l’agoniste
• Corollaires:
- sevrage: pharmacologie « inverse » de celle des opioïdes
- plus grande sensibilité à la naloxone du patient sous opioïdes au long cours que lors de l’administration aigüe
Elle peut apparaître en quelques heures
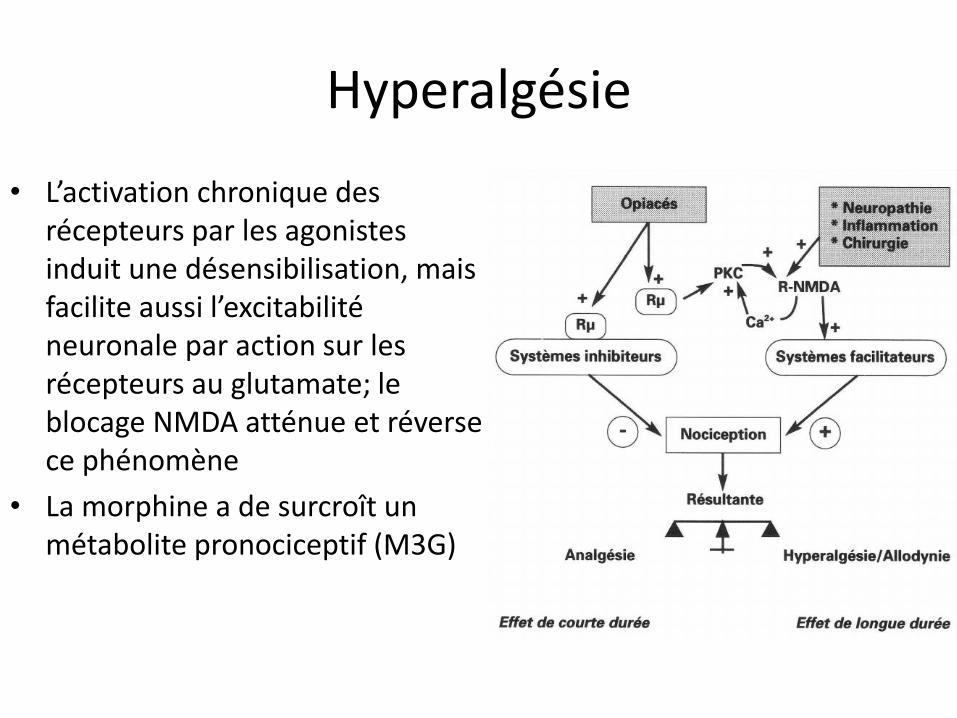
Hyperalgésie
• L’activation chronique des récepteurs par les agonistes induit une désensibilisation, mais facilite aussi l’excitabilité neuronale par action sur les récepteurs au glutamate; le blocage NMDA atténue et réverse ce phénomène
• La morphine a de surcroît un métabolite pronociceptif (M3G)
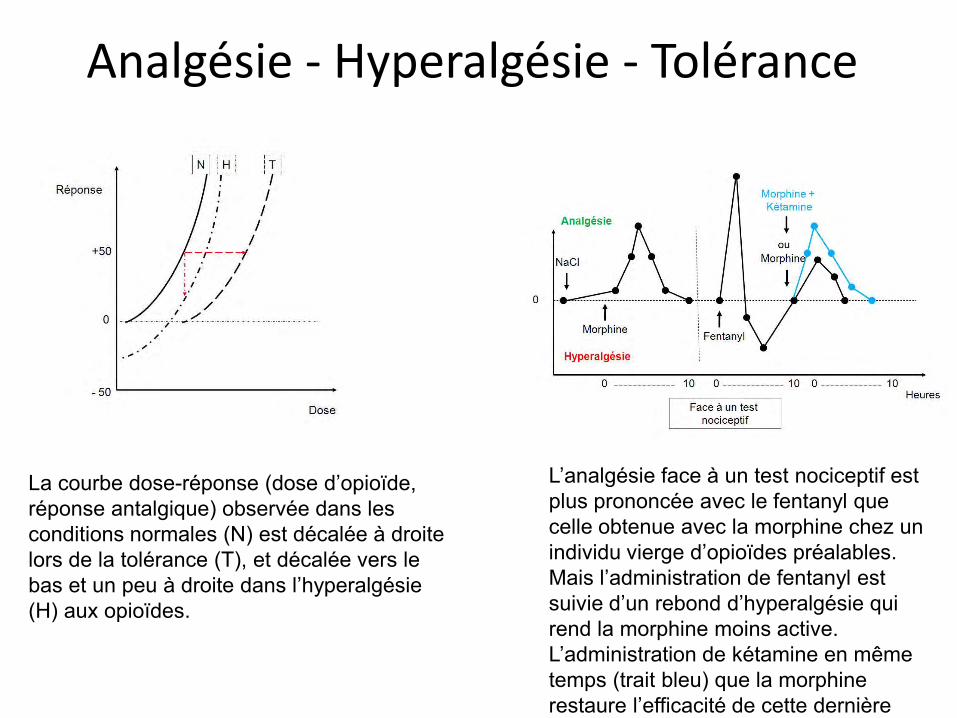
Analgésie - Hyperalgésie - Tolérance
L’analgésie face à un test nociceptif est plus prononcée avec le fentanyl que celle obtenue avec la morphine chez un individu vierge d’opioïdes préalables. Mais l’administration de fentanyl est suivie d’un rebond d’hyperalgésie qui rend la morphine moins active. L’administration de kétamine en même temps (trait bleu) que la morphine restaure l’efficacité de cette dernière
La courbe dose-réponse (dose d’opioïde, réponse antalgique) observée dans les conditions normales (N) est décalée à droite lors de la tolérance (T), et décalée vers le bas et un peu à droite dans l’hyperalgésie (H) aux opioïdes.

Agoniste entier, partiel Antagoniste
• Un agoniste est une substance capable de reconnaitre, de se fixer, et de mimer de façon partielle ou entière l'action du neurotransmetteur endogène.
• Quand tous les récepteurs sont occupés, et que l'effet est total, l'agoniste est dit « entier ».
• Si l'effet est sub-maximal, l'agoniste est dit «partiel ». Emax , c'est-à-dire la « hauteur » du plateau atteinte par le ligand, dépend de l'activité intrinsèque (a) de la molécule
• Cette activité intrinsèque est égale à 1 pour les agonistes entiers, elle est comprise entre 0 et 1 pour les agonistes partiels, et elle est égale à 0 pour les antagonistes.

Règle d’or: éviter les « mélanges »
• Opioïde faible + opioïde fort
• Agoniste entier + agoniste partiel
• Agoniste + agoniste/antagoniste
• L’association opioïde + antagoniste par voie orale est utilisée dans la constipation

Rotation des opioïdes
• Stricto sensu: changement de molécule (≠ changement de voie d’administration) pour optimiser le rapport bénéfice / effets secondaires
• En tenant compte des tables d’équianalgésie, et de la cinétique des deux produits
• Repose sur des différences de capacité à activer les récepteurs, selon variabilité individuelle
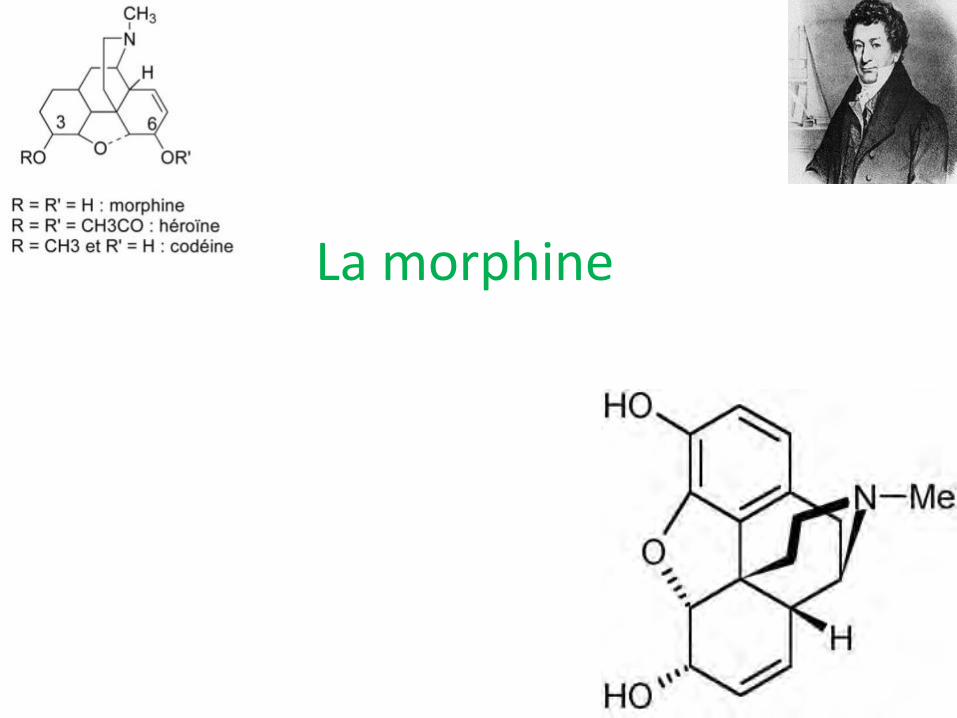
La morphine

Cinétique de la morphine
• Bien absorbée par voie orale; important effet de 1° passage hépatique; biodisponibilité de 15 à 30%
• Ratio per os/sous-cutané/IV: 1/0.5/0.33
• Fixation protéines plasmatiques de 20 à 30%;distribution hépatique, rénale, pulmonaire; VD de 1 à 6 l/kg; passe difficilement BHM
• Glycuroconjugaison hépatique: M3G pronociceptive, M6G 15 à 40 X plus puissante que morphine; sulfonoconjugaison et N-déméthylation sont moins concernées
• ½ vie élimination d’environ 3-4h
• Elimination rénale: morphine libre (<10%); accumulation si insuffisance rénale; clairance d’environ 20 ml/min/kg

Effet recherché de la morphine
• ANALGÉSIE
- DL nociceptive tonique>DL neuropathique
- DL continue plutôt que DL paroxystique
- DL somatique plutôt que viscérale
• Elle s’épuise avec le temps (tolérance), d’autant plus que la DL est peu sensible aux opioïdes et que le patient est « habitué »
Il est des situations douloureuses où les opioïdes NE SONT PAS le traitement de choix: douleurs neuropathiques, douleurs à composante psychogène

Les voies nociceptives sont modifiées par le mécanisme générateur
• Une stimulation nociceptive phasique ne les modifie pas; les opioïdes sont efficaces
• Une stimulation nociceptive tonique produit une hyperalgésie primaire et secondaire; des récepteurs opioïdes sont exprimés sur les afférences au sein du foyer lésionnel; il y a surexpression en central; les opioïdes gagnent en efficacité, au moins initialement
• Les lésions nerveuses conduisent à une baisse des récepteurs au niveau spinal correspondant, et à une surexpression des récepteurs aux anti-opioïdes et des anti-opioïdes; les opioïdes sont peu efficaces, sauf s’il existe une part inflammatoire surajoutée
• Dans la psychogenèse, il existe des modifications de connectivité des structures cérébrales concernées par la nociception
Il est des situations douloureuses où les opioïdes NE SONT PAS le traitement de choix:
douleurs neuropathiques, douleurs à composante psychogène

Effets de la morphine
• Analgésie : elle augmente le seuil de la perception douloureuse quelle que soit son origine.
• Effets psychomoteurs : euphorie, indifférence à la douleur, à la faim, aux sensations désagréables, sédation si la posologie augmente.
• Dépression respiratoire proportionnelle à la dose administrée. Réduction de la réponse des centres respiratoires aux stimuli hypoxémiques et hypercapniques. Rigidité thoracique. Bronchoconstriction. Dépression de la toux
• Nausées et vomissements (20 à 60 % des patients) par action centrale (stimulation de la zone chémoréceptrice) et périphérique (retard à la vidange gastrique)
• Myosis par stimulation centrale du noyau parasympathique du nerf oculaire commun

Effets de la morphine
↓ Libération des catécholamines
Bradycardie sinusale modérée (atropine)
Hypotension orthostatique (personne âgée)
Constipation par inhibition des fibres propulsives, spasme des sphincters, diminution de l’ensemble des sécrétions gastriques (biliaires, gastriques, pancréatiques)
Réduction de la diurèse par augmentation de la libération d’ADH et augmentation du tonus des fibres circulaires du sphincter vésical (rétention urinaire)
Sécrétions endocriniennes : libération d’ADH, diminution de la sécrétion de FSH, LH, TSH, ACTH; aménorrhée et réduction de la sécrétion lactée; réduction de la sécrétion de la testostérone.
Hyperglycémie à des doses supra-thérapeutiques
Hypothermie à fortes doses par altération des centres de la thermorégulation.
Muscle strié : augmentation du tonus et hypertonie à forte dose.
A dose thérapeutique, elle provoque une histaminolibération
Immunosuppression au long cours

Effets secondaires
• Cognitifs: somnolence, sédation
• Respiratoires: ↓FR, ↓ sensibilité au CO2
• Cardio-circulatoires: ↓ FC, TA
• Digestifs: nausées, vomissements, constipation, fermeture sphincters
• Urinaires: rétention
• Histaminolibération
• Prurit
• Myosis
• Au long cours: immunosuppression, troubles cognitifs & hormonaux
• Il y a une tolérance/hyperalgésie, sauf face aux effets digestifs

Indications de la morphine
• DL sensible aux opioïdes
• DL aiguë, DL subaiguë, éventuellement DL chronique
• Opioïdes faibles pour les douleurs d’intensité modérée qu’elle soit ou non liée à un cancer
• Opioïdes forts pour les douleurs intenses, essentiellement cancéreuses, post-traumatiques et post-opératoires aiguës, et, sous réserve de sélection rigoureuse à des DL chroniques nociceptives non cancéreuses
• A éviter dans DLC neuropathiques (même si certains le font) et dans DL psychogènes

Posologie
• Extrêmement variable et à ajuster selon le niveau de douleur observé. Il n’y a pas de posologie maximale, tant que les effets secondaires sont maîtrisables.
• Si possible, pratiquer une titration par injections intraveineuses : 0, 04 mg/kg de morphine toutes les 5 minutes ( 3mg /5min pour un adulte de 70kg) jusqu'à la disparition de la douleur. (Il faut diminuer les doses de 1/3 au delà de 65 ans)
– Cette technique nécessite une surveillance constante
– Puis on commence l'administration S.C. ou orale toutes les 4 heures, ou bien encore on débute la P.C.A.
• Voie sous-cutanée : 5 à 7 mg toutes les 4 à 6 heures pour un adulte de 70 kg, soit environ 0,1 mg/kg toutes les 4 à 6 heures.
• Voie orale :
Forme buvable : 10 à 20 mg toutes les 4 à 6 heures pour un adulte de 70 kg
Comprimés de morphine retard: commencer par 30 mg toutes les 12 heures chez l'adulte sain.
Réévaluer au bout de 36-48h

Suivi du traitement
• En administration aigüe: Vérifier l’effet antalgique; surveiller
paramètres respiratoires et éventuels nausées et vomissements
• En administration chronique: Vérifier l’effet antalgique au bout d’un temps égal à 4 à 6 X la demi-vie du produit. Eventuellement fournir des interdoses de substance à effet immédiat en cas de traitement avec un produit retard. L’apparition d’une hyperalgésie diffuse nécessite de réduire la posologie. Prévenir et surveiller les effets secondaires: troubles digestifs (Laxatifs: émoliants, stimulants, lavements; Relistor ® : methylnaltrexone à utiliser en IV (0.15 mg/kg) chez patients constipés sous opioïdes), somnolence

Règles de prescription en ville • Ordonnance sécurisée
• Identification du prescripteur
• Identification du médicament (posologie, nombre de prises, d’unités)
• Identification du patient (état civil, âge, adresse)
• Identification de la pharmacie qui délivre
• L’ordonnance sécurisée doit dater de moins de 3 mois
• Règle 7 j pour opioïdes injectables (sauf PCA) et de 28 j pour les voies orales
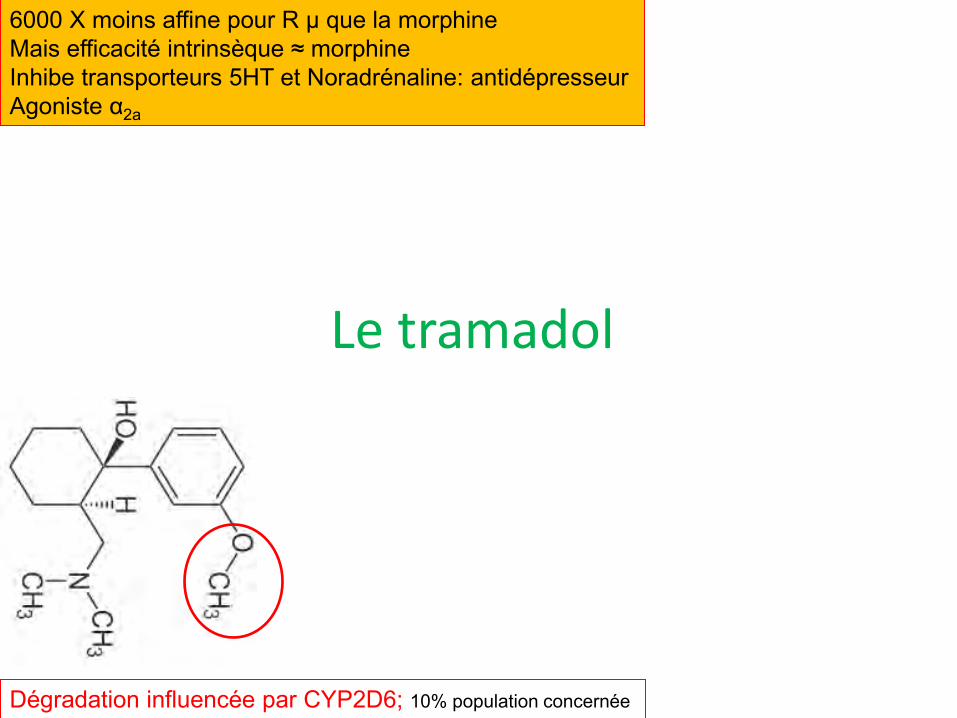
Le tramadol
6000 X moins affine pour R µ que la morphine Mais efficacité intrinsèque ≈ morphine Inhibe transporteurs 5HT et Noradrénaline: antidépresseur Agoniste α2a
Dégradation influencée par CYP2D6; 10% population concernée
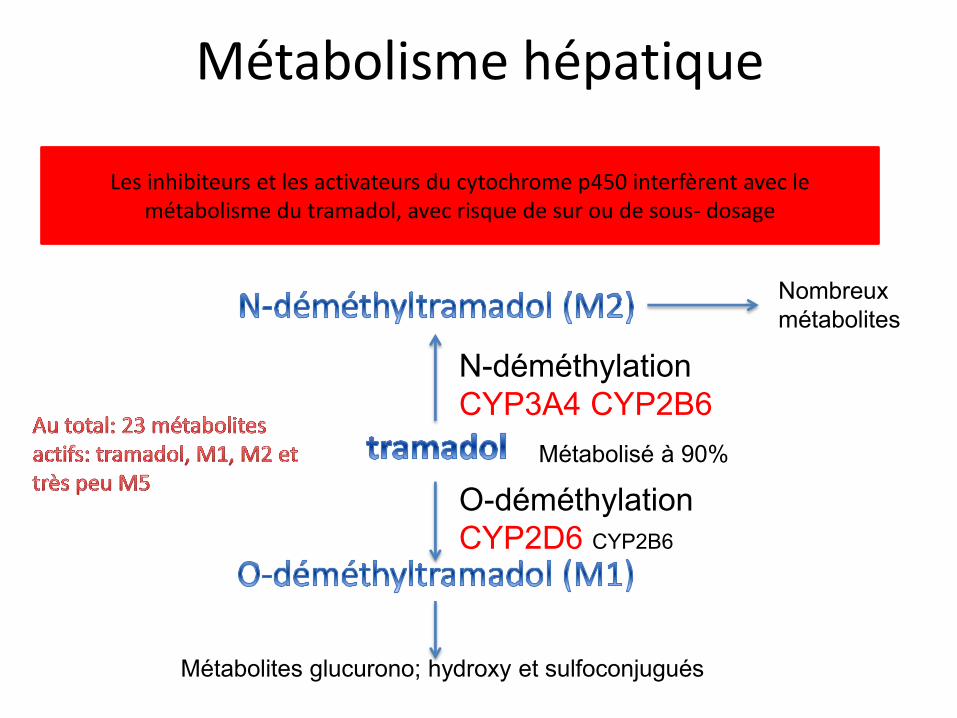
Métabolisme hépatique
O-déméthylation CYP2D6 CYP2B6
Métabolites glucurono; hydroxy et sulfoconjugués
N-déméthylation CYP3A4 CYP2B6
Nombreux métabolites
Métabolisé à 90%
Les inhibiteurs et les activateurs du cytochrome p450 interfèrent avec le métabolisme du tramadol, avec risque de sur ou de sous- dosage

Utilisations
• Douleurs nociceptives peu intenses, mais aussi douleurs neuropathiques
• Existe en cps à 50, 100, 150 mg, les plus dosés étant des formes retard: Topalgic, Contramal, Zamudol,..; Association avec paracétamol: Zaldiar, Ixprim
• Dose maximale 400 mg/j
• Risque sérotoninergique avec ATD 5HT
• Hypoglycémie, troubles du rythme cardiaque
• Atténue éjaculation précoce
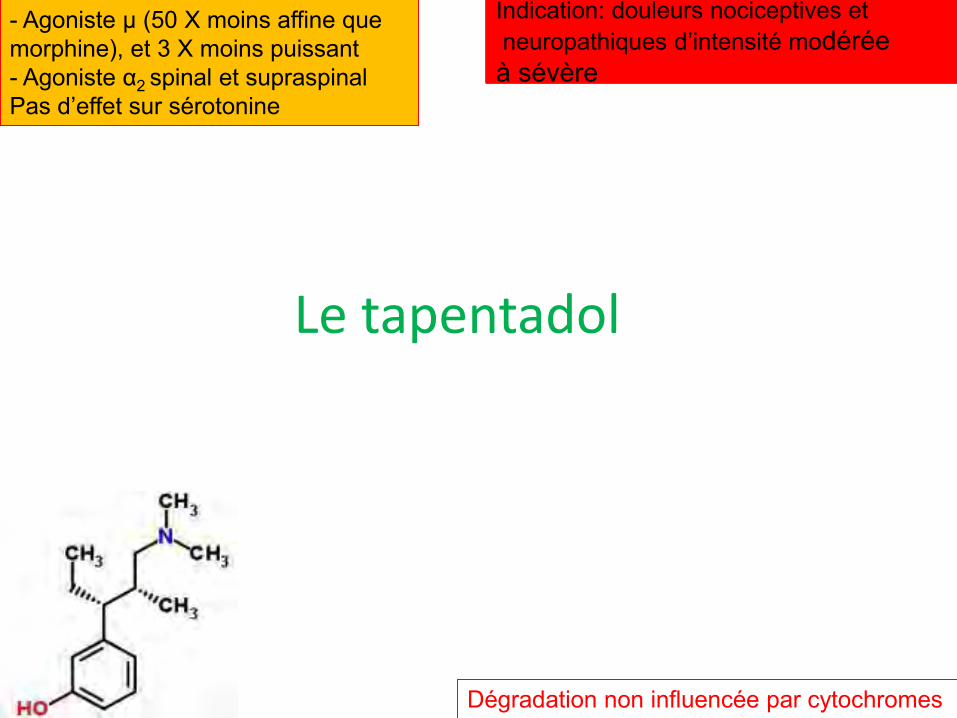
Le tapentadol
- Agoniste µ (50 X moins affine que morphine), et 3 X moins puissant - Agoniste α2 spinal et supraspinal Pas d’effet sur sérotonine
Dégradation non influencée par cytochromes
Indication: douleurs nociceptives et neuropathiques d’intensité modérée à sévère

Cinétique
• Biodisponibilité orale forme LI de 32%
• Fixation protéique 20%
• 50 à 100 mg induisent 4 à 6 h d’analgésie; à donner 4 X/j
• O-glycuronoconjugaison à 99% en métabolite inactif
• Pas d’induction ni d’inhibition des cytochromes
• Elimination rénale de plus de 50% en 4 heures

Efficacité clinique
• Efficace dans douleurs aiguës: chirurgie dentaire, orthopédique,…
• Dans les douleurs chroniques inflammatoires (arthrite, lombosciatalgies) 2 X 200 mg LP/j, considéré comme très efficace par plus de 50% des patients
• Dans les douleurs neuropathiques, chute de 50% de l’intensité douloureuse pour 35% des patients, et de 30% pour 65% des patients
• Sans échappement au bout d’un an; 20% des patients arrêtent pour cause d’effets secondaires
• Effets secondaires: nausées (30%), vomissements (20%), somnolence (15%), vertiges (24%)

La codéine

Cinétique
- La codéine (méthylmorphine) a une meilleure biodisponibilité orale que la morphine car l’effet de premier passage hépatique est moindre. La codéine est transformée partiellement en morphine (10%; il existe des métaboliseurs lents, et des métaboliseurs rapides) et en dihydrocodéine (Dicodin*).
- Efficacité maximum en 1 à 2 heures; durée d'action : 4 à 6 heures; métabolisme hépatique; excrétion urinaire
Conséquence clinique du métabolisme variable:
L’échelle de l’OMS prévoit dans les douleurs cancéreuses nociceptives de passer au palier 3 (morphine, à raison de 60 mg per os /j) en cas d’inefficacité du palier 2 (codéine). Le métaboliseur lent n’aura encore aucune « habitude » de la morphine, contrairement au métaboliseur rapide, et risque plus les effets secondaires
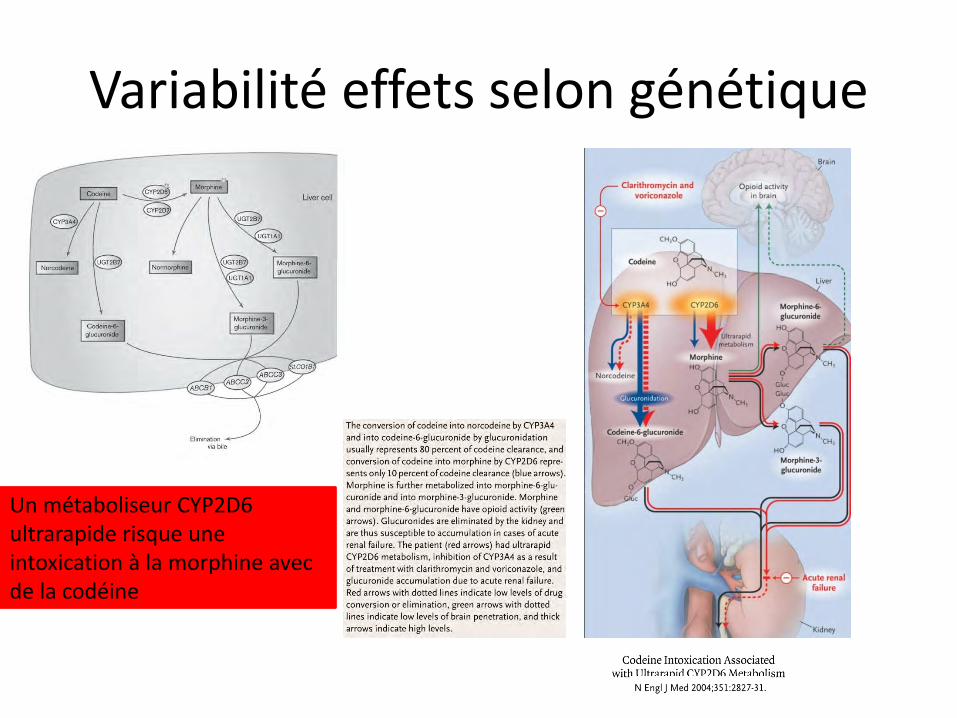
Variabilité effets selon génétique
Un métaboliseur CYP2D6 ultrarapide risque une intoxication à la morphine avec de la codéine

Effets
- Alcaloïde dérivé de la morphine, 6 à 10 fois moins puissant que la
morphine
- Analgésique, antitussif
- Doses équianalgésiques : 10 mg de morphine = 120 mg de codéine
30 mg de morphine = 500 mg d’aspirine.
- De nombreuses spécialités contiennent de la codéine :
• seule : antitussif: Euphon® Netux® Neo-Codion®
• en association avec le paracétamol (synergie d’action sur la douleur de niveau 2) : Codoliprane® 20 mg de codéine et 400 mg de paracétamol; Algisédal®; Efferalgan codéiné®

Indications
• Traitement, dans l’échelle de l’OMS, des douleurs nociceptives d’intensité modérée
• Est toujours utilisée en association avec du paracétamol lors d’une indication d’antalgie
La buprénorphine
Agoniste partiel si donné seul Agoniste-antagoniste si associé

Cinétique
En sublingual
• Important 1° passage hépatique, d’où utilisation sublinguale (analgésie, substitution) ou IV (analgésie)
• Cmax atteinte en 0.5 à 3.5h
• Liaison protéique 96% et important VD
• N-déalkylation via CYP3A4 en norbuprénorphine
• ½ vie d’élimination de 3 à 44h, 15% dans les urines, l’essentiel dans les fèces
En IV 0.15mg/kg
• Cmax atteinte en 30 minutes, second pic de concentration à 1.5h, signant recirculation entérohépatique
• Efficacité antalgique importante pendant 2h
• ½ vie d’élimination de 3h

Utilisation clinique
• Agoniste partiel µ à effet plafond moins efficace que la morphine (mais plus puissant!)
• Durée d’action de 10 h
• Très forte affinité (peu réversible avec naloxone)
• Cps sublinguaux à 0,2 mg (Temgésic) et à 0.4, 2, 4, 8 mg (Subutex pour la substitution)
• Place « incertaine » dans les paliers de l’OMS
La forte affinité de la buprénorphine pour les récepteurs peut poser en clinique deux problèmes: -Réversibilité difficile par les antagonistes en cas de surdosage -S’il faut ensuite une analgésie plus efficace, il faut de fortes doses de fentanyl; ceci pose aussi le problème du toxicomane substitué qui risque de « forcer » sur un « shoot » complémentaire d’héroïne
*
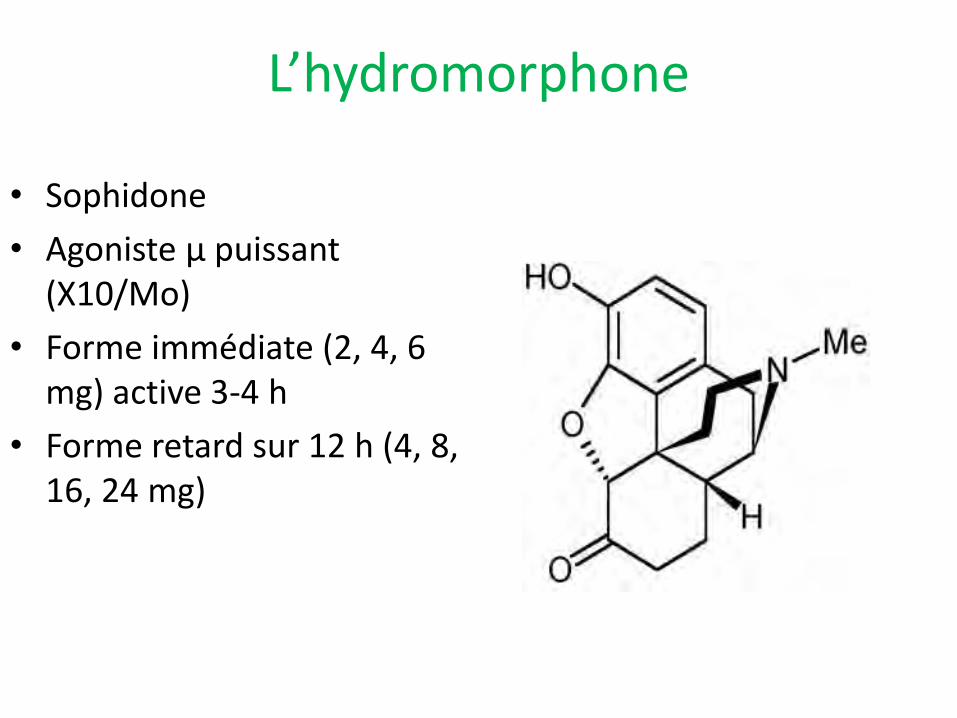
L’hydromorphone
• Sophidone
• Agoniste µ puissant (X10/Mo)
• Forme immédiate (2, 4, 6 mg) active 3-4 h
• Forme retard sur 12 h (4, 8, 16, 24 mg)
L’oxycodone

• 14-hydroxy-7,8-dihydrocodéinone
• Opioïde semi-synthétique
• Métabolites: oxymorphone, noroxycodone et noroxymorphone
• Liposolubilité ≈ morphine
• Agoniste µ1 peu affine (5 à 40 fois moins que morphine), mais facilement transporté dans SNC
• Est aussi agoniste κ2b (et δ)
• Le ratio Mo/OC est de 1,5 : 1 si on passe de Mo à OC; il est de 1,33 : 1 dans l’autre sens
• Il y a moins de tolérance croisée quand on va de Mo vers OC que dans l’autre sens
Recommandation (per os): -Morphine vers oxycodone: 2 : 1 -Oxycodone vers morphine: 1 : 1

Pharmacocinétique
• Chez mouton, équilibre sang/cerveau en 7 min après IV, élimination en 45 min
• Au pic, C cérébrale est 3 X plus élevée
que C plasma du fait d’un transporteur
• Fixation protéines plasma à 45%
• Métabolisée en oxymorphone très active, et en noroxycodone et noroxymorphone peu actives
• Biodisponibilité par voie orale > 60 à 80% (idem en rectal)
• Parentéral, OC 1.5 à 2 fois plus efficace que morphine
• En PM, OC 10 fois moins puissante que morphine, car peu de production d’oxymorphone
• Passe dans le lait maternel: 20% nouveau-nés allaités ont des effets secondaires

Paramètres pharmacocinétiques
• Volume distribution (2-3 l/kg) équivalent à celui de la morphine
• T max 25 min après IV, 80 min après oral LI, 160 min après oral LP
• T1/2 2,5 h après IV, 3 h après oral LI, 8 h après oral LP
• C max 50 ng/ml (après 5 mg IV); 2 X plus élevée après oral LI qu’après la même dose LP
• L’absorption orale est plus élevée chez la femme (âgée) que chez l’homme (jeune)
• Pour une même dose, AUC et C max sont 40% plus élevées chez la femme que chez l’homme
• Pour une clairance de la créatinine < 50 ml/min, C max ↑30%, et T ½ ↑ 50%; les mêmes chiffres sont avancés dans « l’insuffisance hépatique »
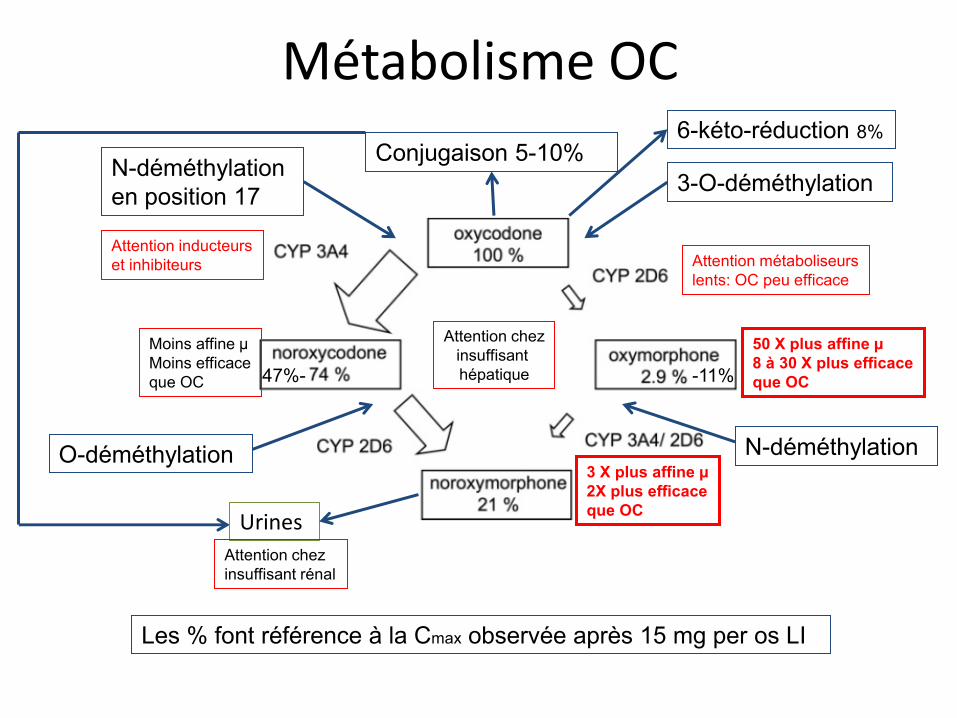
Métabolisme OC
N-déméthylation en position 17 3-O-déméthylation
O-déméthylation N-déméthylation
Conjugaison 5-10%
Urines
Les % font référence à la Cmax observée après 15 mg per os LI
Attention métaboliseurs lents: OC peu efficace
Moins affine µ Moins efficace que OC
50 X plus affine µ 8 à 30 X plus efficace que OC
3 X plus affine µ 2X plus efficace que OC
Attention chez insuffisant rénal
Attention chez insuffisant hépatique
Attention inducteurs et inhibiteurs
-11% 47%-
6-kéto-réduction 8%
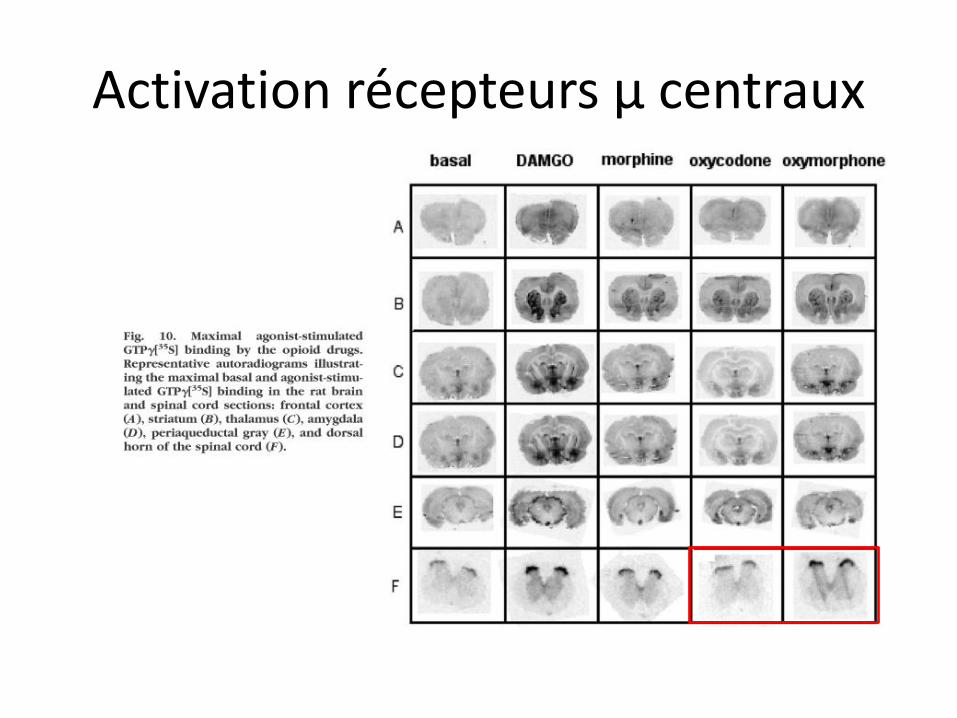
Activation récepteurs µ centraux

Analgésie à OC
• Agoniste µ préférentiel, peu affine, avec peu d’efficacité intrinsèque
• Mais: facilement transporté vers SNC, et oxymorphone très efficace
• Attention aux interférences avec CYP 3A4 (carbamazépine accroît induction, kétoconazole la diminue) et avec CYP 2D6 (blocage, par paroxétine, fluoxétine, quinidine, diminue efficacité OC chez métaboliseurs efficaces)
• DL nociceptive et inflammatoire: 2 à 4 X plus puissante que morphine (sauf par voie périmédullaire où peu voire pas d’effet); efficace en postopératoire, en cancérologie, sur les arthrites chroniques
• DL neuropathique: efficace dans modèles animaux (neuropathie diabétique, ligature sciatique); en clinique, efficace sur DPZ et DL de neuropathies (diabète, chimiothérapie)
• DL viscérale: OC LI plus efficace que morphine LI
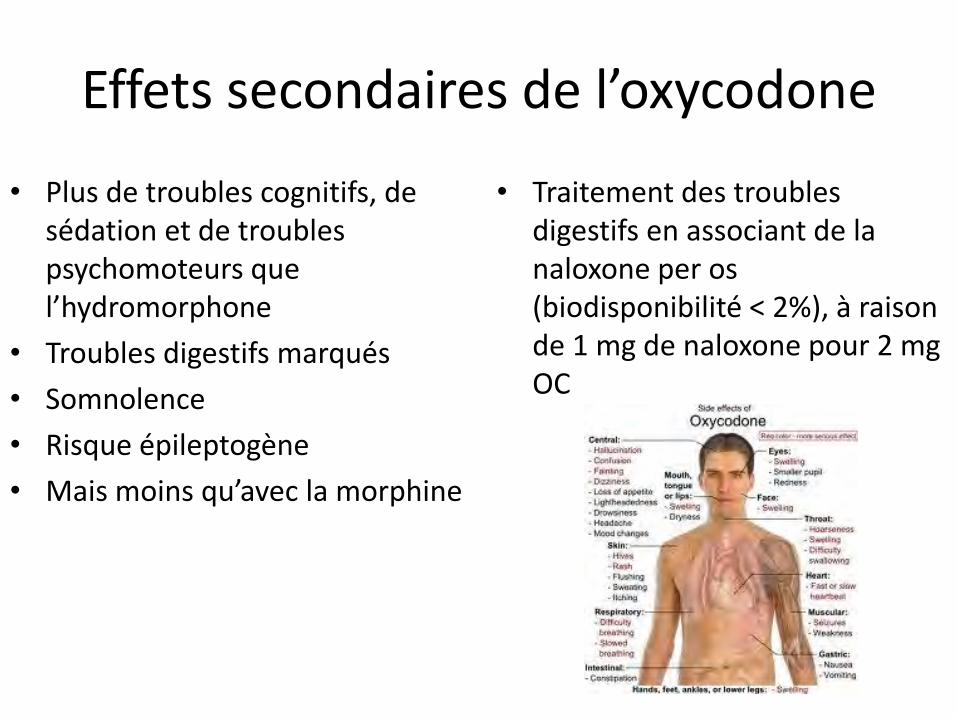
Effets secondaires de l’oxycodone
• Plus de troubles cognitifs, de sédation et de troubles psychomoteurs que l’hydromorphone
• Troubles digestifs marqués
• Somnolence
• Risque épileptogène
• Mais moins qu’avec la morphine
• Traitement des troubles digestifs en associant de la naloxone per os (biodisponibilité < 2%), à raison de 1 mg de naloxone pour 2 mg OC

Effets secondaires au long cours
• C’est pendant les 3 premiers mois que la tolérance est marquée; elle s’atténue ensuite
• Constipation 15%; Nausées 12%; Somnolence 8%; Vomissements 7%; Dépression 2%
• Risque de douleurs thoraciques chez les patients angoreux

La méthadone
• Agoniste µ et δ de puissance équivalente à celle de la morphine
• Antagoniste NMDA; peu d’internalisation des ℛ; d’où peu de tolérance
• Demi-vie de 16 h après une prise unique et de 40 h lors d’une administration chronique
• Cps à 5, 10, 20, 40, 60 mg
• Utilisée pour la substitution
• Cmax atteinte en 2 à 3h
• Fixation protéique 90%
• Métabolisée par CYP3A4
• N-déméthylation en métabolites inactifs
• Pour atteindre un taux sérique de 250 ng/ml, les posologies individuelles vont de 55 à 920 mg/j
• Elimination surtout rénale, accrue par pH urinaire acide

Le fentanyl

Voies d’administration
• Intraveineuse, réservée à l’anesthésie et à la sédation en réanimation
• Périmédullaire: IT en complément d’une rachianesthésie, ED lors de la AECP; par pompe implantée dans les douleurs cancéreuses ou certaines douleurs chroniques
• Transdermique: à délivrance continue (Durogésic®, Matrifène®), ou à la demande (Ionsys®, retiré du commerce), dans les douleurs continues
• Transmuqueuse: orale (Actiq®, Effentora®, Abstral®) ou nasale (Instanyl®, Pecfent), pour les ADP
La liposolubilité élevée fait que les cinétiques sont relativement proches, quelle que soit la voie d’administration
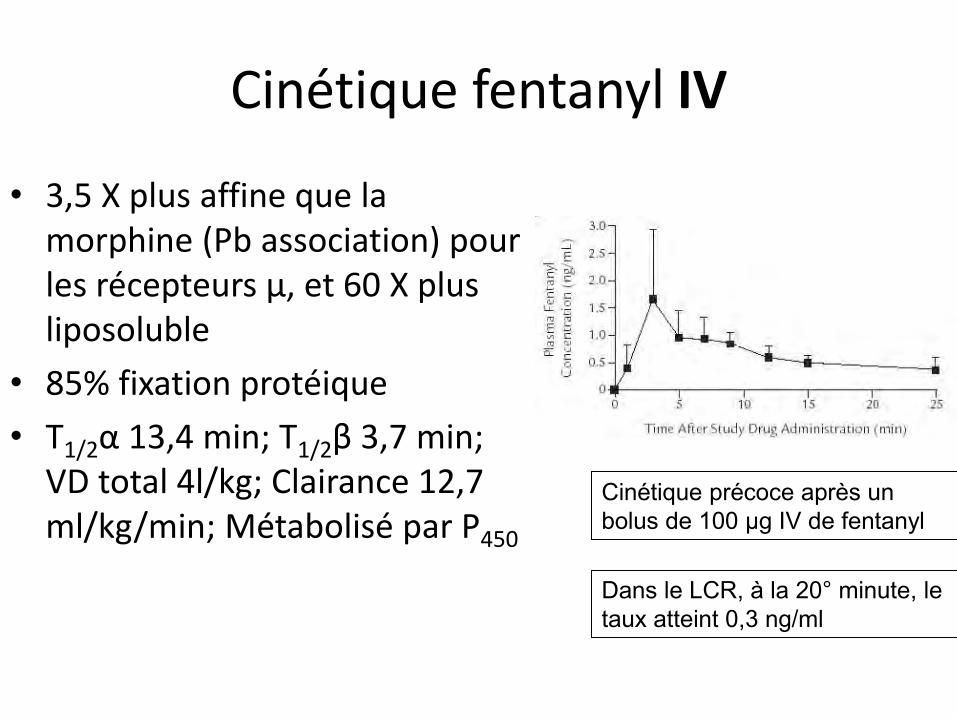
Cinétique fentanyl IV
• 3,5 X plus affine que la morphine (Pb association) pour les récepteurs µ, et 60 X plus liposoluble
• 85% fixation protéique
• T1/2α 13,4 min; T1/2β 3,7 min; VD total 4l/kg; Clairance 12,7 ml/kg/min; Métabolisé par P450
Cinétique précoce après un bolus de 100 µg IV de fentanyl
Dans le LCR, à la 20° minute, le taux atteint 0,3 ng/ml
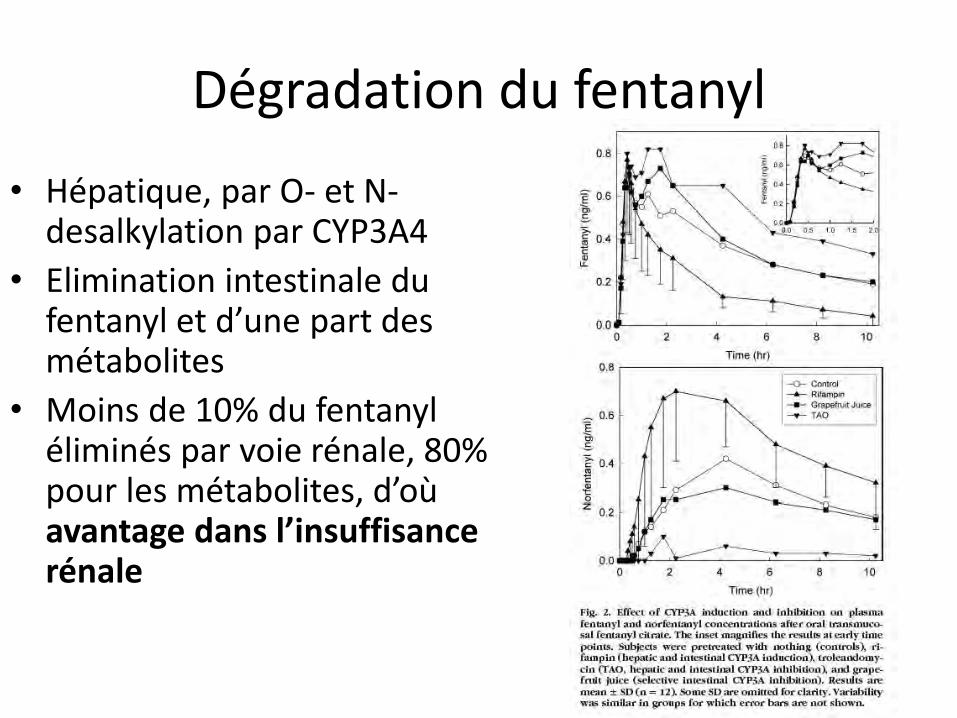
Dégradation du fentanyl
• Hépatique, par O- et N- desalkylation par CYP3A4
• Elimination intestinale du fentanyl et d’une part des métabolites
• Moins de 10% du fentanyl éliminés par voie rénale, 80% pour les métabolites, d’où avantage dans l’insuffisance rénale

Types de patch transcutanés
Indiqués dans les douleurs continues
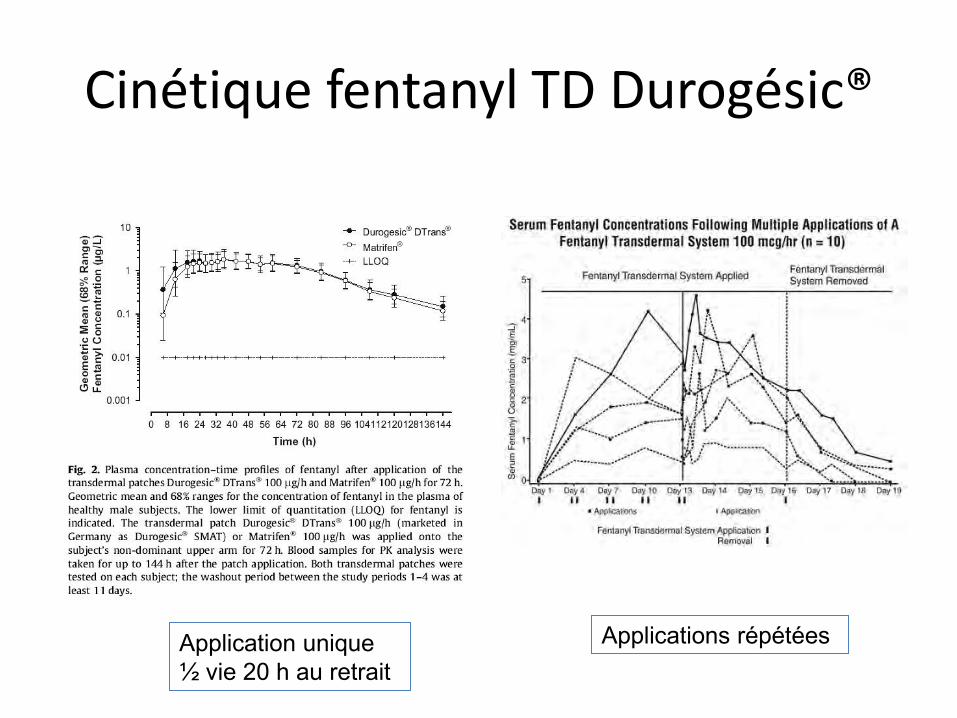
Cinétique fentanyl TD Durogésic®
Application unique ½ vie 20 h au retrait
Applications répétées

Avantages / Inconvénients
• Application facile
• Délivrance contrôlée et fiable sur les patchs matriciels
• Pas d’effet de premier passage hépatique
• Traitement des douleurs continues, de fond
• Décollement par sudation
• Surdosage si application de plusieurs patchs
• Risque de détournement

Formes à libération immédiate
• Réservées aux accès douloureux paroxystiques survenant chez les cancéreux recevant par ailleurs un traitement opioïde de fond
• Même si cela se fait, éviter de mettre du fentanyl LI alors que le traitement de fond est à base de morphine
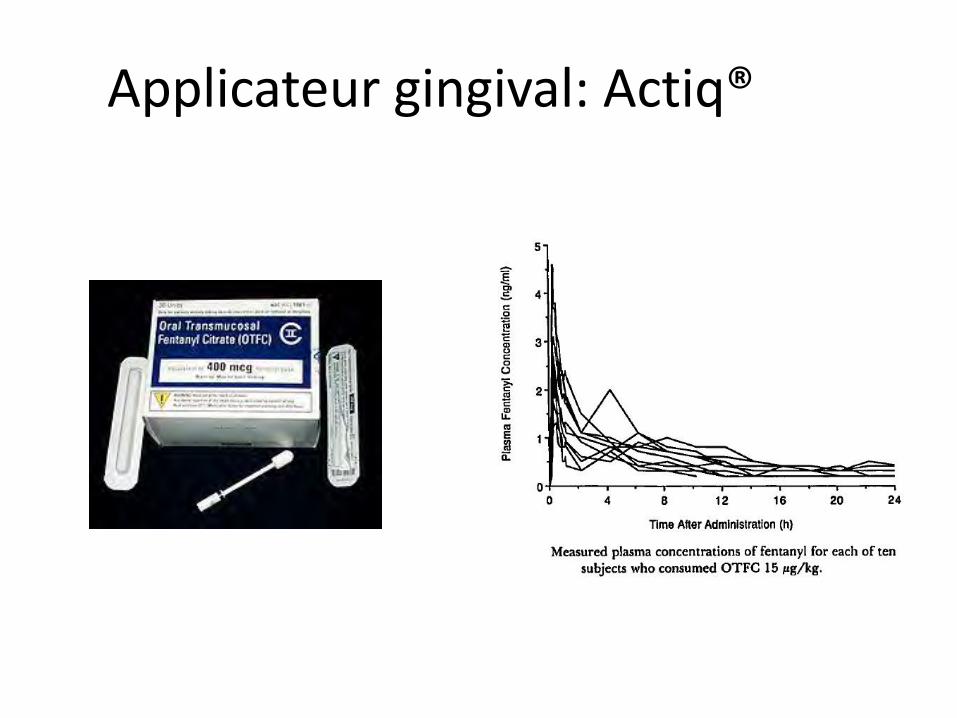
Applicateur gingival: Actiq®

Comprimé sublingual: Abstral® • Cp sublingual à dissolution
rapide (100, 200, 300, 400, 600, 800 µg/cp)
• Absorption rapide en 30 minutes
• Biodisponibilité estimée à environ 70 %
• Concentrations plasmatiques maximales moyennes de fentanyl comprises entre 0,2 et 1,3 ng/ml (après administration de 100 à 800 µg). Elles sont obtenues respectivement en 22,5 et 240 minutes.

Comprimé gingival: Effentora®
Comprimés à 100, 200, 400, 600, 800 µg
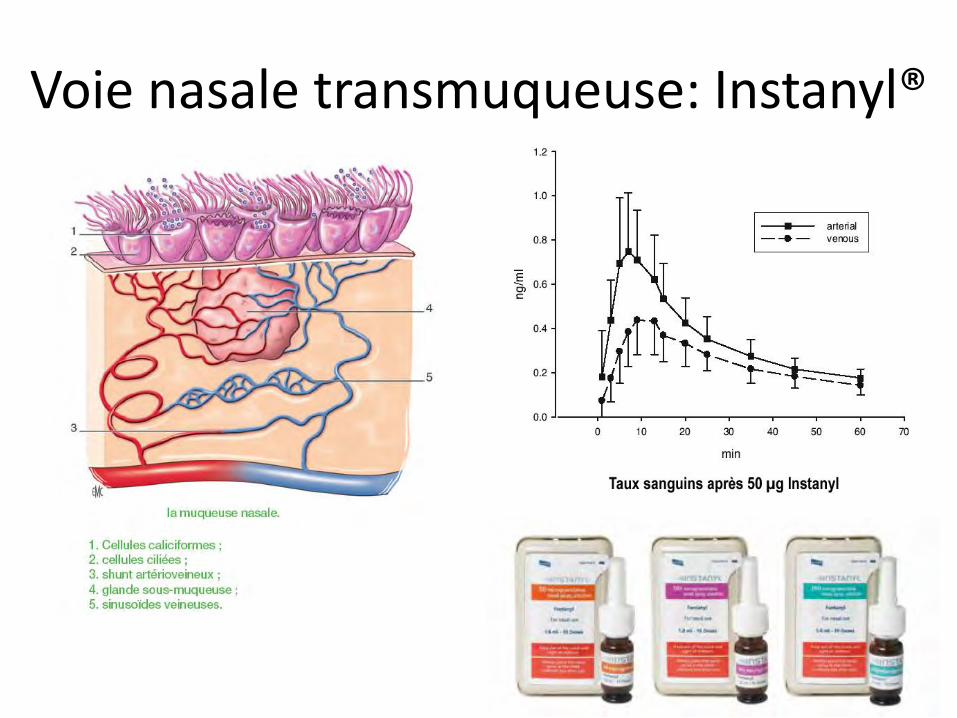
Voie nasale transmuqueuse: Instanyl®
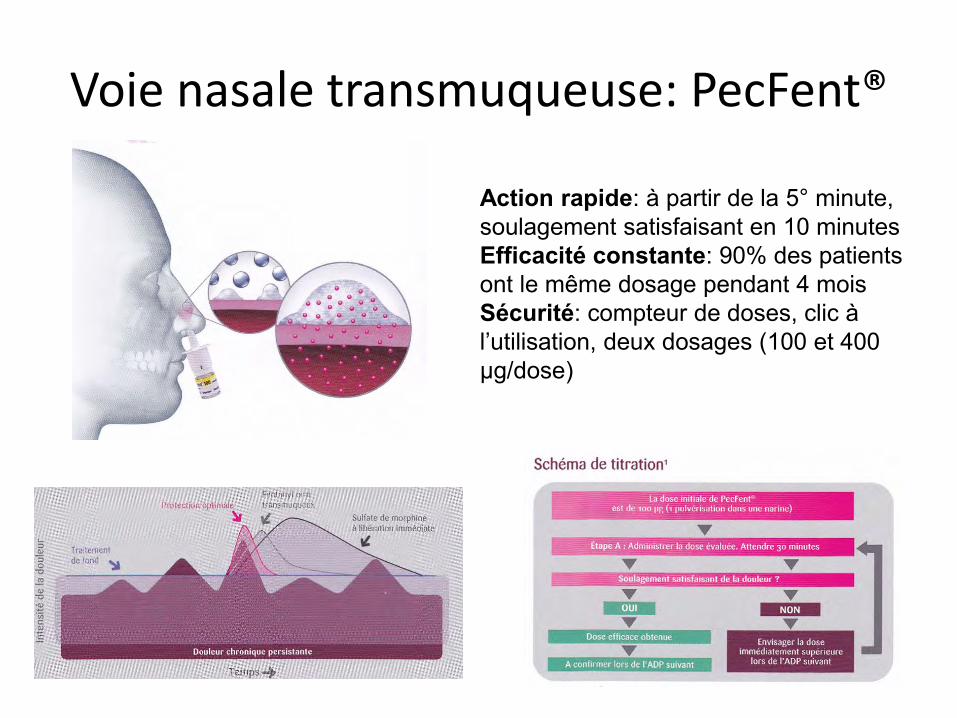
Voie nasale transmuqueuse: PecFent®
Action rapide: à partir de la 5° minute, soulagement satisfaisant en 10 minutes Efficacité constante: 90% des patients ont le même dosage pendant 4 mois Sécurité: compteur de doses, clic à l’utilisation, deux dosages (100 et 400 µg/dose)
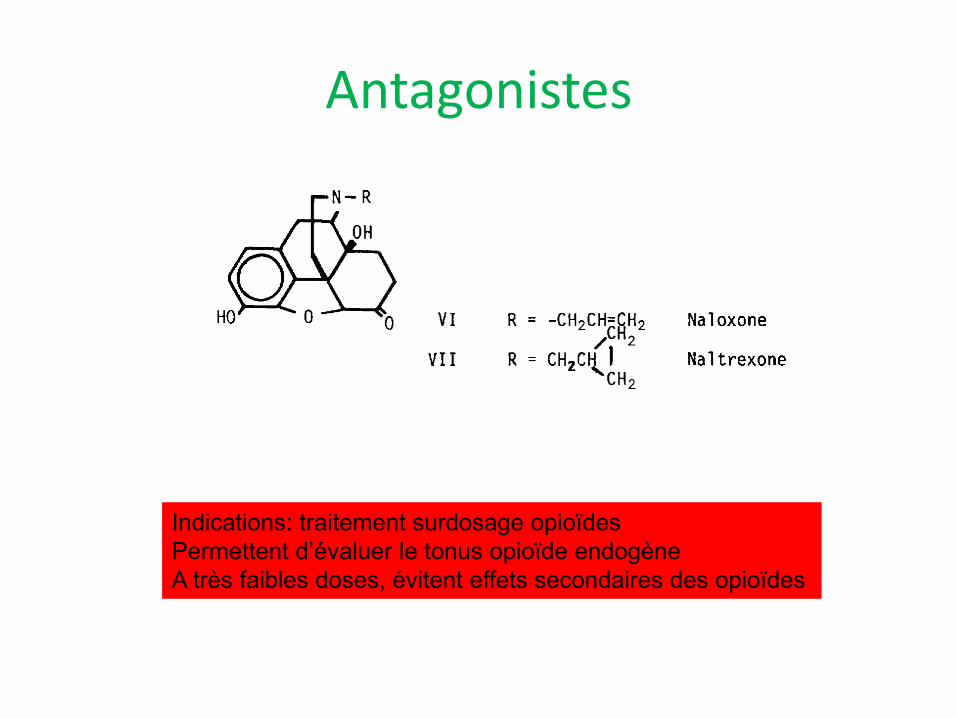
Antagonistes
Indications: traitement surdosage opioïdes Permettent d’évaluer le tonus opioïde endogène A très faibles doses, évitent effets secondaires des opioïdes

Naloxone
• Dérivé de noroxymorphone; Agoniste µ, δ, κ
• IV, IM, ou SC: agit dès la 2° minute; ampoule à 0.4 mg
• Absorbé à 75% par TGI, mais effet de 1° passage hépatique
• Distribution rapide
• Métabolisée par le foie; 30% élimination urinaire sous forme inchangée
• ½ vie de 30 à 80 minutes
• Réversion surdosage opioïdes
• Risque sevrage brutal avec OAP, AC
• A très faible dose (0.05 mg), peut annuler tolérance à la morphine; aurait effet anti-inflammatoire
• Utilisée per os dans constipation aux opioïdes
• Associée à buprénorphine dans substitution
Réversion opioïdes doit être progressive D’autant plus que le patient est depuis longtemps sous opioïdes

Naltrexone
• Antagoniste 3 X plus puissant que la naloxone
• Absorption digestive complète
• Conjugaison et dégradation en 6β-naltrexol (surtout)
• ½ vie plasmatique de 4 à 12h
• Elimination urinaire 60% en 24 h
• Réversion surdosage opioïdes en injectable (50 à 200 mg PO, 1 à 50 mg SC)
• Maintenance sevrage opioïdes ou alcool en formes LP (implants de 1 à 3g qui assurent pour 3 à 4 mois un taux de 2 ng/ml; injectable à 380 mg)
• Prévention effets secondaires dans association fixe avec opioïde (morphine, oxycodone)
• A très faible dose, potentialisation de l’effet des opioïdes dans douleurs inflammatoires
Volontaires. IV 1mg. SC 5 mg. PO 50 mg. % dose dans sang

CO-ANTALGIQUES
Antidépresseurs
• Pour la composante continue des douleurs neuropathiques, et parce que douleur et dépression sont liés
• Plusieurs produits: amitryptiline, venlafaxine, le plus utilisé étant la duloxétine
Antiépileptiques
• Pour les accès fulgurants des douleurs neuropathiques
• Plusieurs produits: CBZ, OCBZ, GBP, PGB, le plus récent étant le lacosamide
• Les neuroleptiques n’ont pas d’effet antalgique, sauf dans ténesmes en
cancérologie (chlorpromazine, métotriméprazine)
• Les anxiolytiques sont utiles dans les douleurs neuropathiques (clonazépam), et
au stade avancé chez les cancéreux
Efficacité, dans les essais cliniques, correspond à une diminution de 50% de l’intensité des douleurs chez au moins 50% des patients; certains ne sont pas soulagés mais ont des effets secondaires qui conduisent à l’arrêt du traitement

Antidépresseurs
• Les plus actifs sur les douleurs neuropathiques sont ceux qui agissent sur les récepteurs noradrénergiques du SNC, et à un moindre degré sur les récepteurs sérotoninergiques
• Modes d’action: Renforcement des voies inhibitrices descendantes (NA et 5HT); effet sur le ganglion rachidien du corps lés (effet nécessitant la présence de récepteurs δ aux opioïdes et de récepteurs β2-adrénergiques)
• Tricycliques: agissent sur recapture NA, 5HT, et D; bloquent R HT2A; bloquent aussi R muscariniques, R H1, R Aα2A, et canaux Na+; d’où les effets secondaires et les contre-indications; efficaces plus rapidement (8 jours) et à doses moindres (1/4) que dans la dépression
• Les ISRS (fluoxétine, sertraline, citalopram) sont les mieux tolérés (sauf hyponatrémies, et syndrome sérotoninergique) mais les moins efficaces
• Les ISRNA (venlafaxine, duloxétine) sont les plus efficaces, ont des effets secondaires (sédation ou agitation, HTA, troubles du rythme)
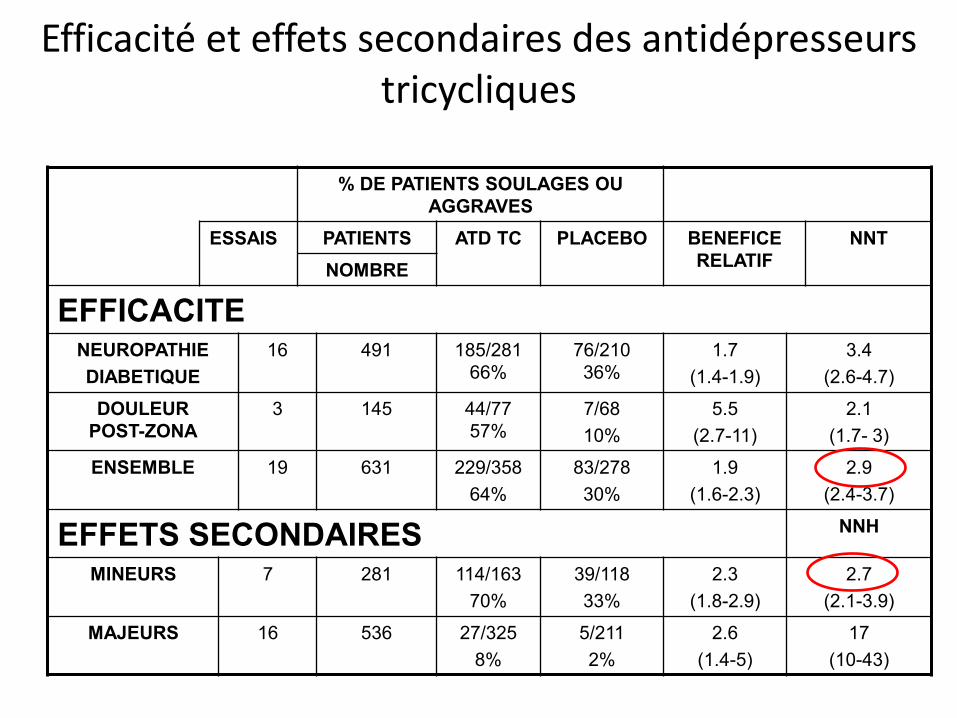
Efficacité et effets secondaires des antidépresseurs tricycliques
% DE PATIENTS SOULAGES OU AGGRAVES
ESSAIS PATIENTS ATD TC PLACEBO BENEFICE RELATIF
NNT NOMBRE
EFFICACITE NEUROPATHIE DIABETIQUE
16 491 185/281 66%
76/210 36%
1.7 (1.4-1.9)
3.4 (2.6-4.7)
DOULEUR POST-ZONA
3 145 44/77 57%
7/68 10%
5.5 (2.7-11)
2.1 (1.7- 3)
ENSEMBLE 19 631 229/358 64%
83/278 30%
1.9 (1.6-2.3)
2.9 (2.4-3.7)
EFFETS SECONDAIRES NNH
MINEURS 7 281 114/163 70%
39/118 33%
2.3 (1.8-2.9)
2.7 (2.1-3.9)
MAJEURS 16 536 27/325 8%
5/211 2%
2.6 (1.4-5)
17 (10-43)

Duloxétine, les effets II fréquents (>1%)
• Céphalées (13,8 %), somnolence (10,7 %) ; Sensations vertigineuses, tremblements, léthargie, paresthésies ; Vision floue ; Acouphènes ; Spasmes musculaires, douleurs musculosquelettiques, tension musculaire; Fatigue, douleur abdominale; Insomnie, rêves anormaux, anxiété, agitation.
• Bâillements.
• Nausées (21,7 %), sécheresse de la bouche (13,2 %) ; Diarrhée, constipation, vomissements, dyspepsie, flatulence.
• Rash, hypersudation, sueurs nocturnes.
• Baisse de l'appétit; Perte de poids.
• Bouffées de chaleur.
• Dysfonction érectile; Baisse de la libido, orgasmes anormaux.
• Palpitations.

Antiépileptiques
• Selon la famille, on distingue plusieurs mécanismes d’action: gabaergique (BDZ, topiramate); blocage de canaux sodium (CBZ, valproate, phénytoïne, lamictal, lacosamide, topiramate); sous-unité α2δ de canaux calciques neuronaux (GBP, PGB)
• Tous abaissent le seuil d’excitabilité neuronale
• Les gabapentinoïdes ont aussi un effet antihyperalgésiant qui peut être préventif (utilisation en prémédication avant opération)
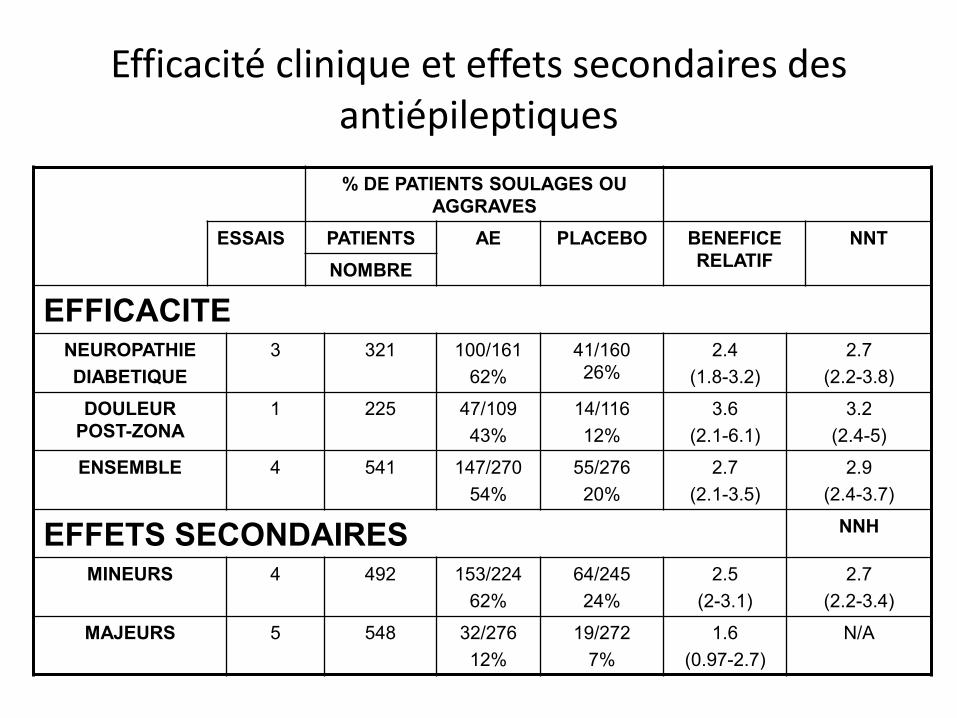
Efficacité clinique et effets secondaires des antiépileptiques
% DE PATIENTS SOULAGES OU AGGRAVES
ESSAIS PATIENTS AE PLACEBO BENEFICE RELATIF
NNT NOMBRE
EFFICACITE NEUROPATHIE DIABETIQUE
3 321 100/161 62%
41/160 26%
2.4 (1.8-3.2)
2.7 (2.2-3.8)
DOULEUR POST-ZONA
1 225 47/109 43%
14/116 12%
3.6 (2.1-6.1)
3.2 (2.4-5)
ENSEMBLE 4 541 147/270 54%
55/276 20%
2.7 (2.1-3.5)
2.9 (2.4-3.7)
EFFETS SECONDAIRES NNH
MINEURS 4 492 153/224 62%
64/245 24%
2.5 (2-3.1)
2.7 (2.2-3.4)
MAJEURS 5 548 32/276 12%
19/272 7%
1.6 (0.97-2.7)
N/A

Prégabalin, les effets II fréquents (>1%)
• Augmentation de l'appétit; Prise de poids
• Humeur euphorique, confusion, irritabilité, diminution de la libido
• Étourdissements, somnolence; Ataxie, troubles de la coordination, tremblements, dysarthrie, troubles de la mémoire, troubles de l'attention, paresthésies; Troubles de la marche, sensation d'ébriété, fatigue, œdème périphérique; Vision trouble, diplopie; Vertiges
• Dyspnée, sécheresse nasale
• Vomissements, bouche sèche, constipation, flatulences
• Troubles de l'érection

Précautions
• Avant de prescrire: examen neurologique, cutané; NFS, ionogramme,
créatinine, bilan hépatique; recherche d’interférences avec les médicaments déjà en cours
• Contre-indications: - CBZ: BAV, hypoplasie médullaire, porphyrie, hypersensibilité, insuffisance
hépatique ou cardiaque, glaucome, grossesse, antiviraux
- Valproate: hépatite, porphyrie, méfloquine, hypersensibilité, grossesse, association salicylés
- GBP, Lamotrigine: hypersensibilité connue, grossesse, insuffisance rénale ou hépatique
- Topiramate: lithiase rénale, contraceptifs oraux

Lithium
• Indication: Algie vasculaire chronique de la face, avec 2/3 de bons résultats à 6 mois
• Agit par action sur noradrénaline et 5HT centrales; baise de synthèse des PG; baisse de l’activité des PDE; effet AL-like
• Lithémie à obtenir 0,5 mEq/l
• Théralite 250 LI ou 400 LP; Neurolithium
• Avant prescription: bilan cardiaque, rénal, thyroïdien et digestif; ECG; NFS, ionogramme
• CI: salidiurétiques, hyponatrémie, insuffisance rénale, troubles du rythme, grossesse; AINS, CBZ, hypoglycémiants

Précautions
• Avant: bilan cardiaque, rénal, thyroïdien et digestif; ECG; NFS, ionogramme
• CI: salidiurétiques, hyponatrémie, insuffisance rénale, troubles du rythme, grossesse; AINS, CBZ, hypoglycémiants
• Surveillance:
- Effet sur douleur
- Effets II: tremblement, prise de poids, agitation, excitation, polyurie
- Dosage sanguin

AUTRES MEDICAMENTS
• Corticoïdes • Anesthésiques locaux • Clonidine • Kétamine • Antibiotiques • MEOPA

Corticoïdes
• Action anti-inflammatoire et stabilisante de membrane sur les lésions nerveuses
• Compression médullaire • Céphalées par HTIC • Hépatomégalies douloureuses • Carcinoses péritonéales • Synacthène® efficace (endomorphines)

Anesthésiques locaux
• Inhibition des canaux sodiques • Voie orale : mexilétine 200 mg x 2 • Voie parentérale : lidocaïne 0,5 mg/kg/h • Voie locorégionale : blocs • Les topiques locaux : EMLA

Clonidine
• Potentialise l’action des morphiniques • Agoniste alpha 2 adrénergique • Patch, voie parentérale, voie péridurale • Petite doses • Mécanisme d’action: inhibition de l’activité des neurones
spinaux, sédation

Kétamine
• Anesthésique général, antagoniste canalaire des récepteurs NMDA, impliqués dans la sensibilisation des voies nociceptives, d’où une action antihyperalgésiante, et antidépressive
• Agit sur les douleurs nociceptives, neuropathiques et sur l’HAO, avec restauration de l’efficacité antalgique des opioïdes
• Administration IV continue (1 mg/kg/j) ou per os (1 mg/kg/j en 3 prises), avec dans plus de la moitié des cas un post(effet de 2 à 8 semaines à l’arrêt du traitement
• L’ifenprodil est sélectif des R NMDA ayant la sous-unité NR2B (récepteurs des voies nociceptives en spinal)

Cyclines
• La minocycline est la cycline qui franchit le mieux la BHM
• Outre ses effets antibiotiques, elle est capable d’inhiber l’activation des cellules microgliales qui participent à la sensibilisation des voies nociceptives
• 2 X 100 mg/j per os permet d’atténuer tous les types de douleurs

MEOPA Mélange équimoléculaire d’oxygène et
de protoxyde d’azote (N2O)

N2O
• Gaz hilarant
• Propriétés analgésiques (élévation du seuil
de douleur et modification de la perception) connues depuis 1800 (impliquant:
récepteurs opioïdes κ, NMDA, voies
adrénergiques), et anesthésiques (MAC 104%, potentialisation GABA, interaction avec
membranes)
• Très diffusible (35X plus que N2)
Comburant à T°> 50°C ou en présence de corps gras

N2O: Implications de sa diffusibilité
Avantages
• Induction d’analgésie extrêmement rapide (en moins de 5 minutes)
• « Réveil » très rapide (en moins de 5 minutes)
• Peu soluble dans sang: équilibration rapide pression alvéolaire et cérébrale
• Permet en anesthésie « d’économiser » des morphiniques
Inconvénients
• Diffuse dans toutes les cavités aériques normales (sinus) ou pathologiques (pneumothorax, intestin occlus), cavités dont il peut augmenter le volume
• En fin d’administration, sa concentration augmente dans l’alvéole et peut entraîner une hypoxie si le patient est à l’air (effet Finck)

Effets anesthésiques & analgésiques
Anesthésie
• Peu soluble
• MAC de 104%
• Effet anesthésique peu puissant
• Réduit MAC des gaz anesthésiques
• Réflexes conservés
• Inotrope négatif
• Stimulation orthosympathique
Analgésie
• Fi 25% # 15 mg morphine IM
• Pic d’analgésie en 3-5 minutes
• Atténuation de toutes les perceptions
• Euphorisant

Une nouvelle indication?
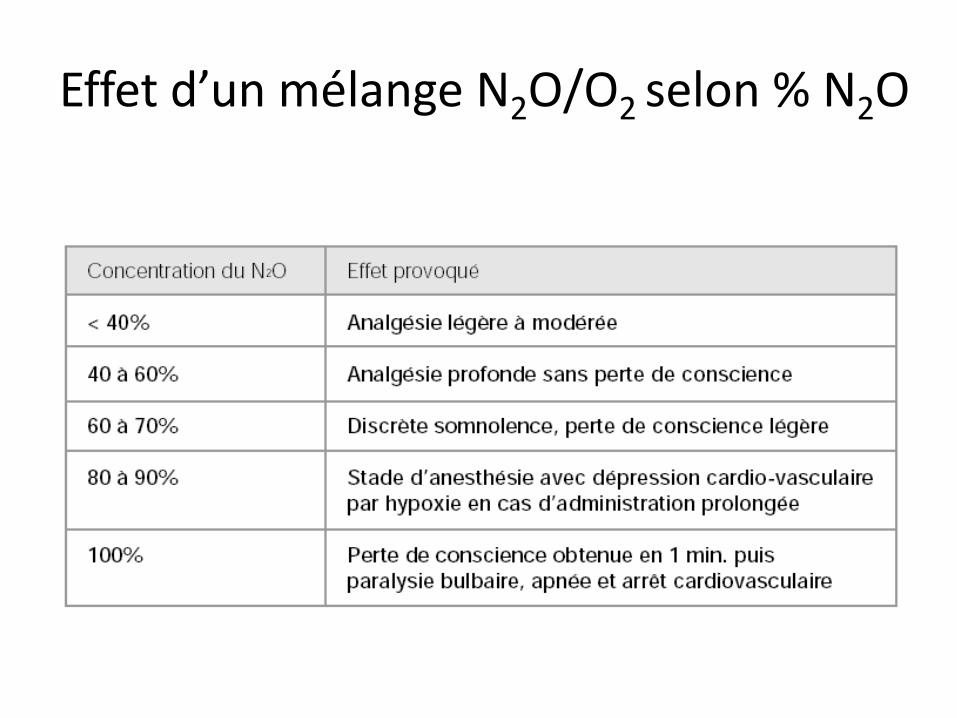
Effet d’un mélange N2O/O2 selon % N2O

MEOPA = O2/N20 à 50/50
• Kalinox®, Oxynox®, Entonox®, Antasol®
• Mélange N2O 50%, O2 50%
• Bouteilles de 5, 15, ou 20 l
• Conditionnées à 70 bars (minimum > à 10 avant utilisation)
• Instable si T°< -5° (liquéfaction N2O)
• Inodore, incolore, D 1,5
• Métabolisme quasi nul (0,004%)
• Retentissement cardiocirculatoire limité, vasodilatateur cérébral
• Analgésique, amnésiant
Utilisable par personnel infirmier formé

Bouteilles blanches avec bandes bleues
Boulland et al., AFAR 2005

Manodétendeur
Ballon, valve anti-retour, masque
Dispositif type Bain
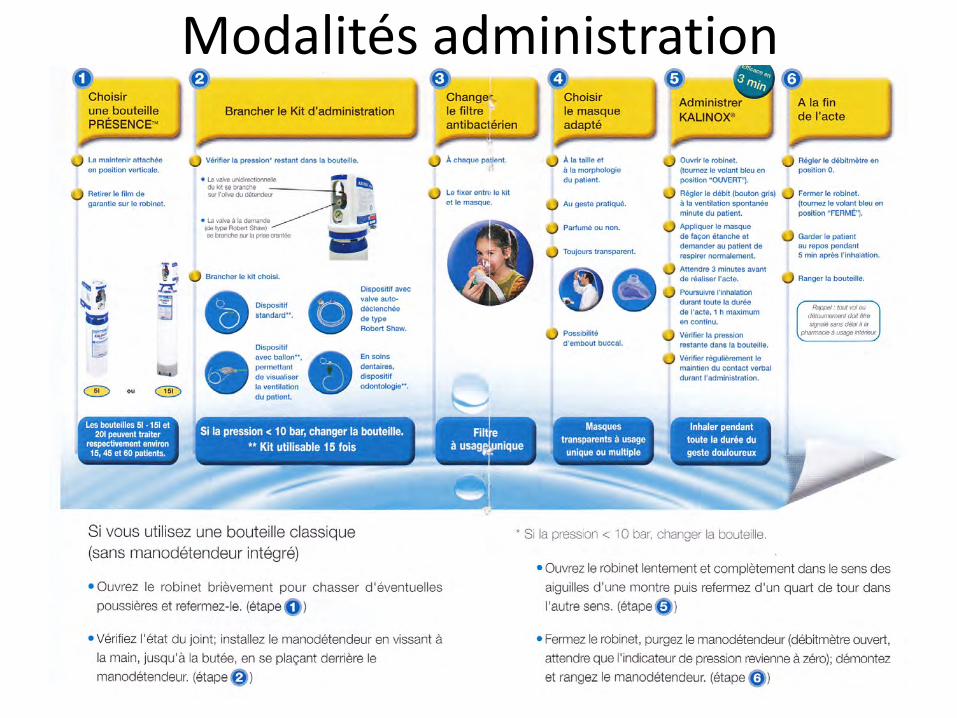
Modalités administration

Principales indications
• Analgésie en aide médicale urgente: brûlés, traumatisés, lors des mobilisations
• Analgésie lors de la douleur provoquée par les soins: ponctions veineuses ou artérielles, PL, BOM, petite chirurgie (exploration plaie, ablation agrafes,…), pansements de brûlés, de plaies, d’escarre, réduction fractures ou luxations, endoscopies, biopsies (hépatique, osseuse, prostatique,…), laser, soins dentaires, obstétrique,…

Contre indications
• Nécessité d’une oxygénation > 50%
• Altération de la conscience (coopération indispensable du patient)
• HTIC
• Cavités gazeuses: PNO, sinusite, emphysème, occlusion; embolie gazeuse, accident plongée
• Traumatisme maxillo-facial (masque à appliquer)
• T°< 5°C

Qualité de l’analgésie
• Atténuation de l’anxiété liée au geste algique
• Bonne acceptation du MEOPA
• Diminution de la douleur d’au moins 60% lors des soins douloureux
• Inefficace chez 10 à 20% des patients
• Modifications des perceptions sensorielles
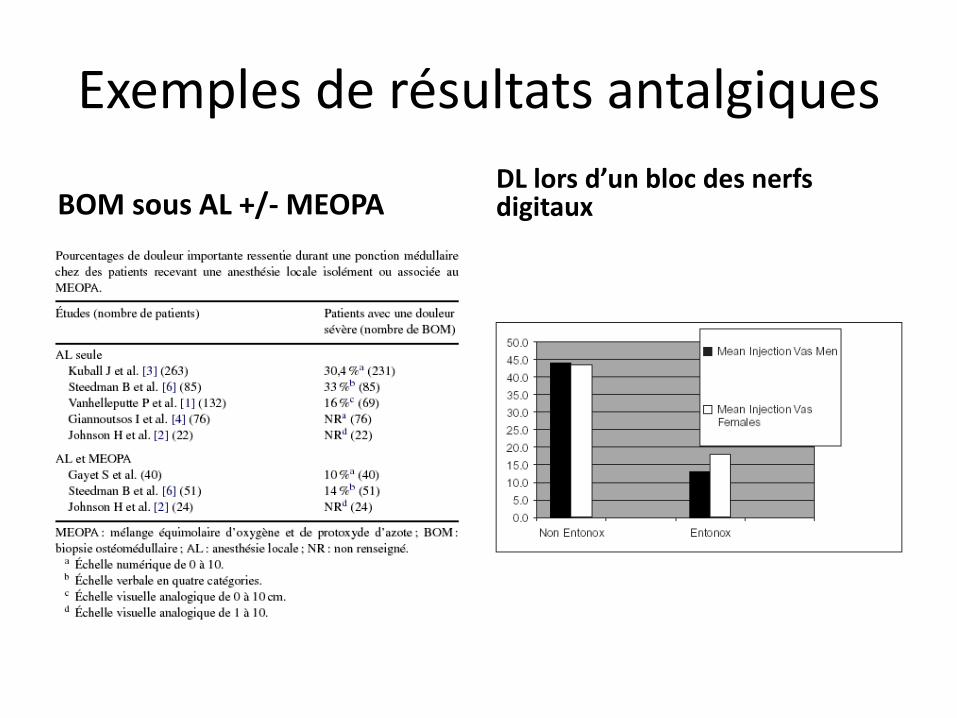
Exemples de résultats antalgiques
BOM sous AL +/- MEOPA DL lors d’un bloc des nerfs digitaux

Exemples de résultats antalgiques
MEOPA vs N2/O2 dans gestes en cancérologie (drains, ponctions,…
Procédure urologique
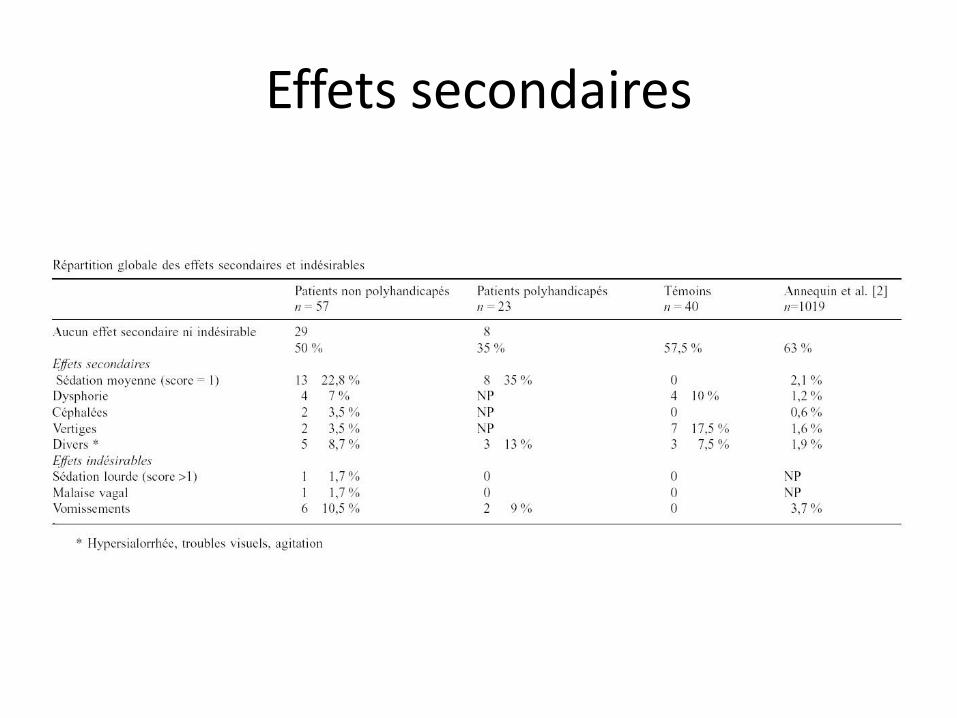
Effets secondaires

Risques
• En administration répétée ou prolongée, vitamine B12 inactivée
• D’où toxicité hématologique et neurologique
• Tératogène chez le rat, non confirmé chez l’homme

Sans MEOPA
Top Related