
Utilisation de la spectrométrie de masse pour diverses ... archives/Proteomique/6-rapport... ·...
24
Utilisation de la spectrométrie de masse pour diverses études protéomiques en 2010 Lurlene Akendengue, Cyrielle Arlot, Florence Breil, Gaelle Fontaine, Jérome Funel, Alexandre Gay, Mathieu Hays, Audrey Loiseau, Charlène Perrois, Elodie Regulus, Maya Salhi, Anaïs Vaissiere Professeur encadrant : François Couderc Promotion 2010-2011
Transcript of Utilisation de la spectrométrie de masse pour diverses ... archives/Proteomique/6-rapport... ·...

Utilisation de la spectrométrie de masse pour diverses études protéomiques en
2010
Lurlene Akendengue, Cyrielle Arlot, Florence Breil, Gaelle
Fontaine, Jérome Funel, Alexandre Gay, Mathieu Hays, Audrey Loiseau, Charlène Perrois, Elodie Regulus, Maya
Salhi, Anaïs Vaissiere
Professeur encadrant : François Couderc
Promotion 2010-2011

- 2 -
Sommaire
Introduction 3
I. Applications en protéomique 4
I.1/ Le Laserspray Ionization 4
I.2/ Analyse de protéines membranaire 6
I.3/ Détermination de la partie N-terminale des protéines 7
I.4/ Enrichissement des peptides pour l’analyse en MALDI-TOF 8
I.5/ Autres applications à la taxonomie et à la biominéralisation 8
II. Modifications post traductionnelles 9
II.1/ Phosphorylation 9
II.1.1/ Analyse d’une phosphoprotéine recombinantei 9
II.1.2/ Diverses études de phosphorylations 11
II.2/ Glycosylation 11
II.2.1/ Enrichissement en glycoprotéines sur billes d’hydrazide 11
II.2.2/ Enrichissement par billes couplées à l’acide boronique 12
II.3/ O-Acylation 13
II.4/N-Acétylation et N-méthylation 14
III. Quantification de protéines par spectrométrie de masse 15
III.1/ SILACii : 15
III.2. iTRAQiii
: 17
IV. Applications Médicales 20
IV.1/ Identification par 2D-DIGE et phage display(iv
) 20
IV.2/ Etude des biomarqueurs dans les cancers (v) 21
Conclusion 22

- 3 -
Introduction
La protéomique connaît un essor depuis quelques années et le nombre de publications
ne cesse d’augmenter d’années en années, traduisant un intérêt grandissant pour ce
domaine. L’étude du protéome s’attache à identifier les protéines et à les caractériser.
La spectrométrie de masse dans les études protéomiques combine généralement
l’électrophorèse bidimensionnelle à la détection par MALDI-TOF MS pour réaliser une
cartographie peptidique. Par ailleurs, les techniques nano-chromatographiques liquides
couplées à des spectromètres de masse tandem (MS/MS), sont une approche attractive
par leur capacité à fragmenter des peptides présents en très faible quantité, afin
d’obtenir des informations de séquence en acides aminés. Ce couplage permet dans de
nombreux cas de s’affranchir de l’étape de séparation par électrophorèse
bidimensionnelle et de caractériser ainsi l’ensemble des protéines ainsi que leurs
modifications post-traductionnelles directement à partir de mélanges. Elle permet
également de quantifier les variations du taux d’expression d’une protéine. D’un point
de vue clinique, la détection et la quantification d’un marqueur protéique permet le
diagnostic d’une pathologie.
Cette revue, réalisée par la promotion 11 du Master EGPR, présente une vue d’ensemble
des avancées en spectrométrie de masse qui ne cessent d’émerger dans un souci
d’élaboration de méthodologies performantes, spécifiques et sensibles pour
l’identification et la caractérisation des protéines.

- 4 -
I/ Applications en protéomique
Les progrès des techniques en protéomique permettent de caractériser de plus en plus de
structures et de plus en plus de paramètres physiques et biochimiques.
I.1 Le Laserspray Ionization – une nouvelle méthode pour l’analyse directe sur
un tissu à pression atmosphérique
Le laser spray ionisation permet l’analyse de protéines directement à partir d’un tissu
en utilisant un système d’ionisation à pression atmosphériquevi
Cette alternative pour la production d’ions multichargés est basée sur une découverte récente.
En effet, l’ablation des matrices de MALDI, produit des ions multichargés à pression
atmosphérique de la même manière que l’ESI. Mais cela est effectué directement sur un
échantillon solide (et non pas dans un solvant volatil). Plusieurs ions protéiques de poids
moléculaire inférieur à 20000 Da sont détectés hautement chargés à partir d’un échantillon de
cerveau de souris dépourvue de lipide.
Cette technique a la capacité d’engendrer des ions multichargés ce qui permet
l’analyse de protéines de haut poids moléculaire avec une résolution de masse de 100 000
(FWHH, m/z 200) sur un spectromètre de masse Orbitrap
Figure I.1 A Spectre de masse complet avec « encarts » montrant les ions de protéine et de
peptides multichargés obtenu à partir d’un Orbitrap Exactive à 100,000 (FWHH, m/z 200) de
résolution de masse en rassemblant les acquisitions des 15 premières secondes représentant
les ablations faite par le laser sur la quasi totalité du tissu couvert par une solution de 2,5-
DHAP
B Tissu après les ablations des tirs de laser

- 5 -
Une seule acquisition a été nécessaire pour l’identification d’un fragment basique d’une
protéine du cerveau la myéline. Ceci a été réalisé par la méthode de fragmentation ETD
(Electron Transfert Dissociation) un transfert d’électron sur un cation. Une haute résolution
obtenue ainsi qu’une grande précision sont obtenue en une seule acquisition afin de
déterminer le poids moléculaire qui est confirmé par la base de données des fragments
obtenus par ETD. De plus, la capacité d'acquérir un spectre de masse
à partir d'un seul tir laser utilisant un laser à azote relativement peu coûteux suggère le
potentiel de l’obtention d’une imagerie des tissus rapide.
Figure I.2 Analyse d’un peptide de la myéline en MSMS après un tir de laser sur
l’échantillon. Fragmentation en ions c et z par la méthode de l’ETD
Ions c = H2N-CHR-CO-NH-CHR-CO-NH3+ = M + 1
Ions z = +CHR-CO-NH-CHR-COOH= M -16

- 6 -
I.2/ Analyse de protéines membranaire
La spectrométrie de masse et l’analyse en gel 2D représentent les méthodes majoritairement
utilisées. L’objectif de nombreuses études est l’identification de protéines selon différentes
conditions d’analyses ou d’expression et les différents profils d’expression des protéines sont
comparés et obtenus à partir de gel 2D.
De façon générale, les protéines sont extraites de tissus ou de cellules, puis séparés par gel
électrophorèse 2D. Les protéines peuvent être visualisées par un traitement du gel à l’argent
ou au Bleu de Coomassie et des logiciels analysent les profils obtenus et quantifient les
différents spots. Le ou les spots d’intérêt sont ensuite récupérés et analysés par spectrométrie
de masse.
De nombreuses applications sont retrouvées dans la littérature. Par exemple, l’analyse de
protéines membranaires représente un challenge. L’étude de ces protéines reste difficile à
cause de leur domaine hydrophobe difficilement dissout, et de nombreux problèmes sont liés
à la solubilité.
Wang et son équipe, propose une stratégie d’étude qui se base sur la dénaturation des
protéines membranaires avec une très forte concentration d’urée, leurs permettant ainsi d’être
ionisées de telle sorte, qu’on puisse les séparer par chromatographie d’échange d’ions (vii
).
Les protéines membranaires préparés à partir de foie de souris sont dissoutes et chargé sur des
appareils de chromatographie en tandem couplés avec Q-Sepharose FF et Sephacryl S-200HR.
Ces colonnes sont capables d’absorber 97,87% des préparations de protéines membranaires.
Les protéines sont éluées par gradient linéaire NaCl (0-1M), et analysés par SDS-PAGE.
Après digestion des bandes obtenues à la trypsine, suivie d’une séparation HPLC en phase
inverse, les protéines sont identifiées par spectrométrie de masse par trappe à ions.
L’approche établissant des colonnes en tandem (ici échangeuses d’ions suivi d’un gel
filtration) a été proposé et largement pratiqué pour l’analyse protéomique. Les résultats
chromatographiques indiquent que la séparation des protéines est significativement améliorée
en utilisant cette technique comparée à l’utilisation d’une seule colonne résine.

- 7 -
I.3/ Détermination de la partie N-terminale des protéines
Xu et al.viii
ont étudiés les extrémités N-terminales des protéines afin de connaître la nature
des séquences reconnues par les différentes aminopeptidases ainsi que les protéines cibles de
ces enzymes. La technique de marquage des peptides N-terminaux des protéines s’appuie sur
le principe de la dégradation d’Edman habituellement utilisée pour le séquençage. La
purification de ces derniers est réalisée grâce à l’affinité de la biotine pour l’avidine (cf
fig I.3).
Les fonctions amines de la protéine (α-amine et ε-amine sur les éventuelles lysines) réagissent
avec le PITC, l’ajout de TFA (acide trifluoroacétique) va induire spécifiquement la
cyclisation intramoléculaire à l’extrémité N-terminale et le clivage de la liaison peptidique
entre le premier et le second acide aminé. La seule fonction amine restée libre en N-terminale
va ensuite réagir avec l’agent biotinylé (NHS-SS-biotine). L’ensemble des protéines est
séparé sur gel polyacrylamide et digéré à la trypsine. Après extraction des peptides du gel, les
peptides N terminaux marqués à la biotine sont purifiés grâce à des billes d’agarose couplées
à de la neutravidine et élués grâce à un agent réducteur le TCEP. Enfin, les peptides purifiés
sont analysés par MALDI-TOF ou par LC/MS/MS.
Fig I.3 : Schéma global et spectres de la stratégie de marquage et de purification des peptides N-terminaux
Ce type d’analyse permet la détermination des séquences N-terminales d’un grand nombre de
protéines et donc de connaître les protéines cibles des différentes peptidases comme les
méthionine aminopeptidases (MetAPs) qui clivent la méthionine en N-terminale. L’analyse
par LC/MS/MS donne également des informations sur la nature de la séquence d’un peptide
signal reconnue par différentes signal peptidases permettant l’adressage des protéines aux
différents compartiments cellulaires. Par exemple les auteurs démontrent que sur la totalité
des protéines étudiées, 57 % des peptides signaux contiennent une région chargée en N-

- 8 -
terminal, 93 % une région centrale hydrophobe et 28 % une région C-terminale polaire non
chargée. De plus uniquement 14 % contiennent les trois régions classiquement décrites pour
caractériser un peptide signal. Un intérêt supplémentaire de ce type d’investigation est montré
par la possibilité de détecter les sites de clivage utilisés lors de l’induction des voies de
signalisation ou bien encore lors de l’induction de l’apoptose de la cellule. La seule restriction
à cette technique commune au séquençage par la dégradation d’Edman est l’impossibilité
d’analyser les protéines possédant un acide aminé N terminal bloqués (ex : acétylés).
I.4/ Enrichissement des peptides pour l’analyse en MALDI-TOF
Un des inconvénients du travail sur un mélange de protéine est qu’un pourcentage des
peptides issus des digestions enzymatiques n’est pas détecté. Cela peut être due à une
mauvaise capacité d’ionisation par rapport à d’autres peptides du mélange ou à une trop faible
concentration dans l’échantillon. Pour remédier à ce problème Rodthongkum et al.ix
ont
proposés une technique de purification de peptides en fonction
de leur pI. Celle-ci est réalisée grâce à des micelles inversés de
l’homopolymère I (voir ci-contre). Après digestion à la
trypsine des protéines d’un mélange, des extractions
liquide/liquide sont réalisées en présence des micelles inversés. Seuls les peptides chargés
négativement ayant un pI inférieur ou égal au pH présent à l’intérieur des micelles seront
purifiés pour être analysés par MALDI-TOF. Cela permet de détecter plus d’ions qui n’était
pas visible par une analyse MALDI-TOF classique.
I.5/ Autres applications à la taxonomie et à la biominéralisation
Lasch et al.x ont mis en place une techniques de détection plus rapide des souches de Yersinia
pathogènes pour l’Homme. Pour cela ils ont étudiés par MALDI-TOF environs 150 souches
de Yersinia des 13 espèces les plus connues et 35 souches d’entérobactéries. L’utilisation en
parallèles de tests statistiques leur a permis de définir des protéines détectables en MALDI-
TOF spécifiques, du genre et des différentes espèces de Yersinia.
Dans un but de recherche plus fondamentale, Mann et al.xi
ont étudié de la biominéralisation
dans un organisme modèle souvent étudié, les spicules de larves d’oursins. Après préparation
des échantillons grâce à une déminéralisation et l’ajout d’acide dilué, les protéines sont
séparées sur un gel SDS et sont digérées directement par la trypsine. L’analyse LC/MS/MS a
permis de déterminer les différentes protéines actrices dans le processus de biominéralisation.

- 9 -
Fig.II.1.1: réaction de phosphorylation
II/ Modifications post traductionnelles
II.1/ Phoshorylation
La phosphorylation est une modification post traductionnelle très répandue dans les systèmes
eucaryotes supérieurs, et notamment dans certaines cascades de signalisation (e.g. voie des
MAP kinases). Elle fait intervenir des enzymes (les kinases) qui catalysent la réaction
suivante (fig.II.1.1):
Les résidus concernés par la phosphorylation sont principalement les thréonines et les sérines
et plus marginalement les tryptophanes, les hydroxyprolines et les histidines.
Le moyen le plus rependu pour analyser les phosphorylations reste la spectrométrie de masse
de type MS/MS En effet, lors de l’ajout d’un acide phosphorique sur un des résidus précités,
celui-ci subira un incrément de masse de +80 Da (+ 98 - 18 Da). On observera néanmoins
parfois une perte de 18 Da (+ 80 – 98 Da) du à la perte d’acide phosphorique sur l’ion. Cette
information est directement visible sur un spectre MS/MS en réalisant la différence de masse
entre deux ions consécutifs de même nature (b, y, a, x, c, z).
II.1.1/ Analyse d’une phosphoprotéine recombinantexii
La protéine MAT1 est dans un premier temps purifiée puis analysée par ESI MS : la protéine
est présente sous 2 formes différentes (fig.II.1.1) séparée par un Δ 80 m/z laissant penser à
une forme phosphorylée (P) et une non phosphorylée (NP). Après traitement à la phosphatase
alcaline, on observe la disparition de la forme (P), la phosphosérine devient une
DéHydroAlanine confirmant la présence d’une phosphorylation.
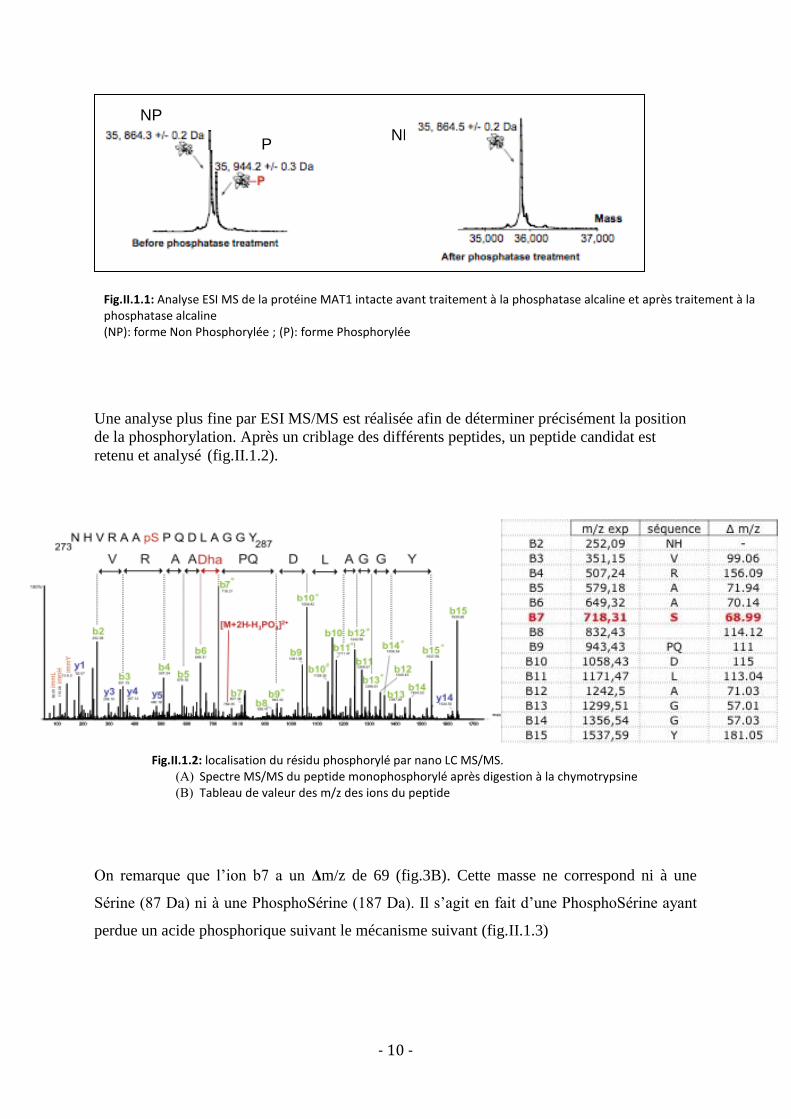
- 10 -
Fig.II.1.1: Analyse ESI MS de la protéine MAT1 intacte avant traitement à la phosphatase alcaline et après traitement à la phosphatase alcaline (NP): forme Non Phosphorylée ; (P): forme Phosphorylée
Fig.II.1.2: localisation du résidu phosphorylé par nano LC MS/MS. (A) Spectre MS/MS du peptide monophosphorylé après digestion à la chymotrypsine
(B) Tableau de valeur des m/z des ions du peptide
Une analyse plus fine par ESI MS/MS est réalisée afin de déterminer précisément la position
de la phosphorylation. Après un criblage des différents peptides, un peptide candidat est
retenu et analysé (fig.II.1.2).
On remarque que l’ion b7 a un Δm/z de 69 (fig.3B). Cette masse ne correspond ni à une
Sérine (87 Da) ni à une PhosphoSérine (187 Da). Il s’agit en fait d’une PhosphoSérine ayant
perdue un acide phosphorique suivant le mécanisme suivant (fig.II.1.3)
NP
NP
P

- 11 -
Fig.II.1.2: Mécanisme de perte d’un ion phosphorique
II.1.2/ Diverses études de phosphorylations
Lorsque 2 sites supposés de phosphorylation sont spatialement proches et inséparables via une
digestion enzymatique, on ne peut pas attribuer la phosphorylation à l’un ou l’autre des
résidus. Les auteurs ont eu recours à la substitution d’un des 2 résidus putatifs par une alanine
pour déterminer le vrai site de phosphorylationxiii
Les auteurs ont mis en évidence qu’un peptide peut porter plusieurs phosphorylations par
étude directe des phosphopeptides en MS/MSxiv
Les auteurs critiquent le logiciel d’analyse de spectres MS/MS. Le logiciel trouve la séquence
d’un peptide qui n’existe pas dans la réalitéxv
II.2/ Glycosylation
II.2.1/ Enrichissement en glycoprotéines sur billes d’hydrazide
Une analyse des N-glycosylations présentes sur les glycoprotéines consiste en la
déglycosylation de ces protéinesxvi
. Pour cela un protocole a été mis en place : les
glycoprotéines sont oxydées au niveau de la partie osidique, il y aura ensuite fixation des
sucres de la protéine sur billes fonctionnalisées à l’hydrazide (Fig II.2.1.a ).

- 12 -
Fig II.2.1.a : Oxydation et couplage à l’hydrazide.
Le mélange de protéines sera ensuite lavé puis digéré à la trypsine de manière à ne garder que
les peptides glycosylés et liés à l’hydrazine. Les peptides obtenus seront relargués de la résine
couplée à l’hydrazine par la PNGase F (Fig II.2.1.b ). Cette enzyme n’implique pas seulement
la déglycosylation mais elle entraine aussi la transformation de l’asparagine en aspartate ce
qui entraine une différence de masse de 1 qui sera visible par MS/MS.
Fig II.2.1.b : Protocole expérimental d’analyse des protéines N-glycosylées.
Palmisano et al. ont analysé par cette méthode les glycoprotéines présente dans le vin et ils
ont effectivement pu déterminer les 44 sites de N-glycosylation de 28 glycoprotéines.xvii
II.2.2/ Enrichissement par billes couplées à l’acide boronique
Dernièrement, une autre technique d’analyse des glycoprotéines a été mise en place et cette
technique permet de ne pas passer par la déglycosylation des protéines analysées.xviii
La
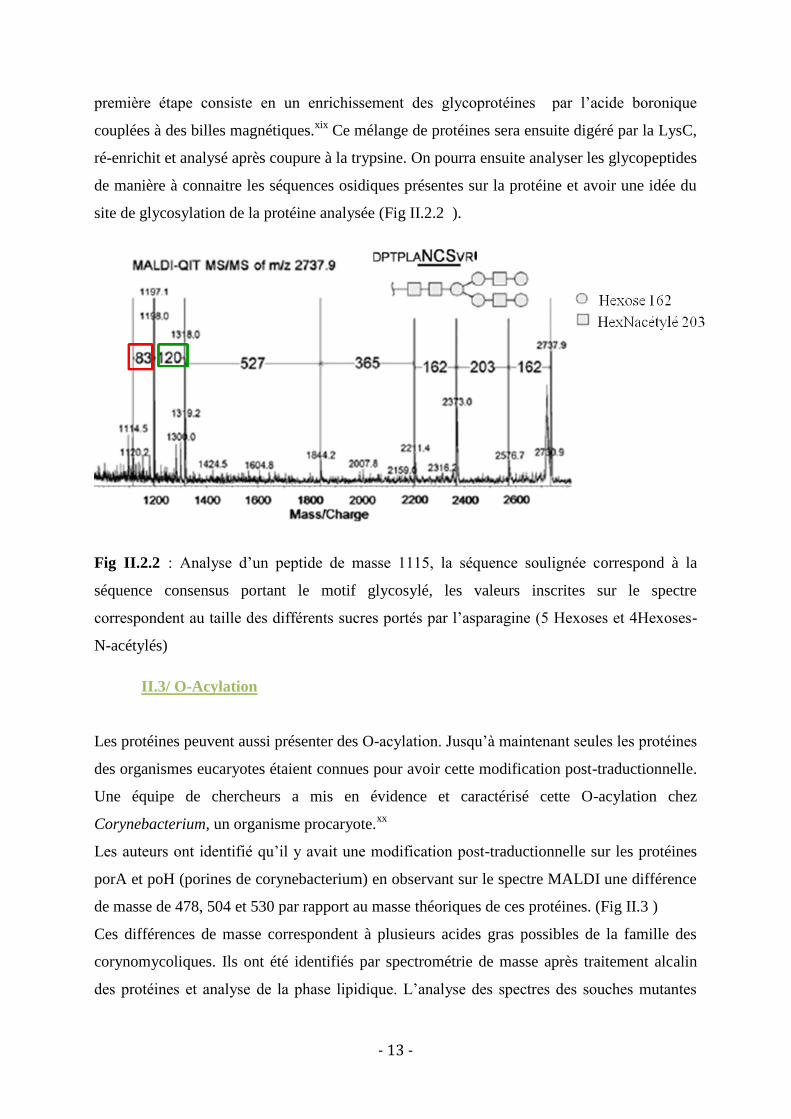
- 13 -
première étape consiste en un enrichissement des glycoprotéines par l’acide boronique
couplées à des billes magnétiques.xix
Ce mélange de protéines sera ensuite digéré par la LysC,
ré-enrichit et analysé après coupure à la trypsine. On pourra ensuite analyser les glycopeptides
de manière à connaitre les séquences osidiques présentes sur la protéine et avoir une idée du
site de glycosylation de la protéine analysée (Fig II.2.2 ).
Fig II.2.2 : Analyse d’un peptide de masse 1115, la séquence soulignée correspond à la
séquence consensus portant le motif glycosylé, les valeurs inscrites sur le spectre
correspondent au taille des différents sucres portés par l’asparagine (5 Hexoses et 4Hexoses-
N-acétylés)
II.3/ O-Acylation
Les protéines peuvent aussi présenter des O-acylation. Jusqu’à maintenant seules les protéines
des organismes eucaryotes étaient connues pour avoir cette modification post-traductionnelle.
Une équipe de chercheurs a mis en évidence et caractérisé cette O-acylation chez
Corynebacterium, un organisme procaryote.xx
Les auteurs ont identifié qu’il y avait une modification post-traductionnelle sur les protéines
porA et poH (porines de corynebacterium) en observant sur le spectre MALDI une différence
de masse de 478, 504 et 530 par rapport au masse théoriques de ces protéines. (Fig II.3 )
Ces différences de masse correspondent à plusieurs acides gras possibles de la famille des
corynomycoliques. Ils ont été identifiés par spectrométrie de masse après traitement alcalin
des protéines et analyse de la phase lipidique. L’analyse des spectres des souches mutantes

- 14 -
Cgl_otsA_treY_treS , pks13::km et fadD32::km connues pour présenter un défaut dans la
chaine de biosynthèse des acides corynomycoliques confirme l’identité de cette modification
post-traductionnelle.
Les auteurs ont ensuite déterminé la localisation de la modification post-traductionnelle par
dégradation d’Edman. En effet lors de la dégradation d’Edman, on obtient un
phénylthiohydantoine déhydroalanine quand la sérine 15 de PorA est dégradée indiquant que
la sérine 15 serait porteuse de la O-mycolation. En analysant le spectre Maldi-Tof d’un
mutant où la protéine PorA dans laquelle la sérine 15 a été mutée en valine, on observe un
seul pic de masse 4715 correspondant à PorA sans mycolation et où une sérine est substituée à
une valine.
Fig II.3 : Mise en évidence de la modification post-traductionnelle de PorA et PorH.
Différence de masse de 478, 504 et 530 par rapport aux masses théoriques de PorA (4680Da
( +H+: 4681Da; +Na
+: 4703Da) et de PorH, 6161Da ( +H+: 6162Da; +Na+: 6184Da).
II.4/N-Acétylation et N-méthylation
Les histones peuvent être modifiées post-traductionnellement. Ces modifications sont
impliquées dans la régulation transcriptionnelle des gènes. En 2010, une équipe de chercheurs
a mis au point une technique pour une caractérisation à haut débit des modifications post-
traductionnelles des histones.xxi
Leur technique consiste à une séparation des histones par
UPLC-MS avec un analyseur de type Orbitrap. L’analyse MS/MS haute résolution de
l’histone H4 révèle grâce à la mesure de la masse exacte une acétylation sur une lysine et non
pas 3 méthylations.

- 15 -
III/ La Quantification de protéines par spectrométrie de masse :
Aucune méthode n’est capable aujourd’hui, en une seule étape simple, d’identifier et
de quantifier les composants d’un échantillon complexe de protéines. Cependant plusieurs
stratégies de marquage ont été développées pour faire de la quantification relative, c'est-à-dire
pour comparer des niveaux de peptides ou protéines dans des conditions différentes, en
établissant des ratios d’intensité des pics de spectrométrie de masse.
Il existe plusieurs types de marquage pour faire de la quantification dont le SILAC,
pour le marquage in vivo, et l’iTRAQ, pour le marquage in vitro.
III.1/ SILACxxii
:
Le marquage SILAC consiste en l’incorporation d’isotopes lourds pendant la synthèse
des protéines. Les protéines que l’on veut quantifier seront marquées sur au moins un acide
aminé (on peut utiliser n’importe quel isotope pour ce marquage : 13
C, 15
N, 2H…) Le principe
repose sur le fait qu’une population de cellules se développe en présence de l’isotope léger, et
la deuxième population se développe en présence de l’isotope lourd. Dans chaque cas les
protéines des différentes conditions sont mélangées, séparées (par chromatographie liquide
généralement) et analysées par MS puis MS/MS. Comme leur ionisation sera identique, elles
produiront le même signal en MS, avec une différence de masse induite par l’incorporation
des isotopes. Un ratio pourra alors être établi entre les protéines marquées à l’isotope léger et
celles marquées par l’isotope lourd, pour en déduire une quantité relative.
Ces techniques de quantification par marquage métabolique sont idéales pour la
quantification à partir de lignées cellulaires, et permettent une analyse précise et assez simple.
En revanche, elles sont limitées à la culture cellulaire et ne sont donc pas adaptées à d’autres
types d’échantillons. De plus, le nombre d’incorporation d’isotope lourd étant variable selon
le temps d’incubation, cela rend les spectres plus difficiles à analyser du fait de la différence
de masse entre les peptides de l’échantillon contrôle et test. Il est également à noter que
l’incorporation d’isotopes varie suivant le compartiment cellulaire.
Un exemple original d’application du marquage SILAC a été réalisé dans le but
d’identifier les interactions spécifiques entre protéines et ARNsxxiii
. Pour cela, ils ont
synthétisé par PCR une population d’ARN contrôle et une population d’ARN dont on veut
identifier les interactants protéine qu’ils ont fusionnés avec un aptamère (obtenu par synthèse

- 16 -
chimique). Cet aptamère est un tag biotine qui servira à la purification. A côté de cela, deux
populations cellulaires identiques sont cultivées en présence de l’acide aminé marqué à
l’isotope lourd ou à l’isotope léger. Les conditions de marquage sont ensuite inversées afin de
pouvoir quantifier et établir des ratios qui nous permettront d’établir une zone d’interaction
spécifique. Les protéines et les ARN sont ensuite mélangés et mis en contact par pull down,
qui consiste en l’interaction de l’ARN biotinylé (fixé à des billes de streptavidine) avec les
protéines marquées. Après élution, une analyse MS/MS est réalisée afin de déterminer les
interactions spécifiques. Ces interactions pourront être identifiées grâce à l’intensité des
signaux sur le spectre MS et aux ratios que l’on calculera à partir de ces mêmes spectres.
Figure III.1.a : Stratégie pour l'identification d'interactions ARN/protéines
Le ratio des protéines marquées à l’isotope lourd par rapport aux protéines marquées à
l’isotope léger est établit. Ces ratios permettent de mettre en évidence une zone (représentée
en gris clair sur les graphes) dans laquelle la probabilité de retrouver des interactions
spécifiques est la plus importante. De ce fait, plus le ratio sera important, plus l’interaction
ARN/protéine sera considérée comme la plus spécifique. C’est ce qui est représenté en
exemple sur la figure III.1.a.
Grâce à cette méthode de marquage, les auteurs ont pu identifier de manière très
spécifique et reproductible les interactions protéines/ARN (ARNt, séquence IRES et ARN de
très petite taille), issus d’extraits cellulaires bruts.
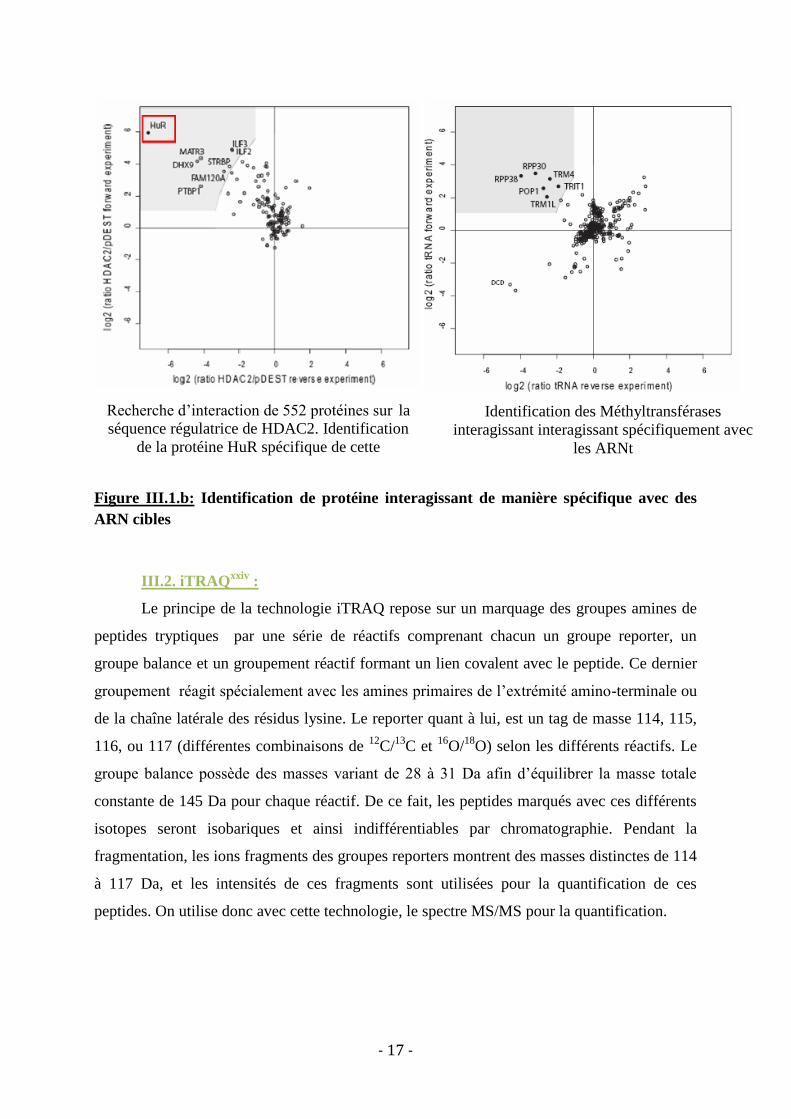
- 17 -
Figure III.1.b: Identification de protéine interagissant de manière spécifique avec des
ARN cibles
III.2. iTRAQxxiv
:
Le principe de la technologie iTRAQ repose sur un marquage des groupes amines de
peptides tryptiques par une série de réactifs comprenant chacun un groupe reporter, un
groupe balance et un groupement réactif formant un lien covalent avec le peptide. Ce dernier
groupement réagit spécialement avec les amines primaires de l’extrémité amino-terminale ou
de la chaîne latérale des résidus lysine. Le reporter quant à lui, est un tag de masse 114, 115,
116, ou 117 (différentes combinaisons de 12
C/13
C et 16
O/18
O) selon les différents réactifs. Le
groupe balance possède des masses variant de 28 à 31 Da afin d’équilibrer la masse totale
constante de 145 Da pour chaque réactif. De ce fait, les peptides marqués avec ces différents
isotopes seront isobariques et ainsi indifférentiables par chromatographie. Pendant la
fragmentation, les ions fragments des groupes reporters montrent des masses distinctes de 114
à 117 Da, et les intensités de ces fragments sont utilisées pour la quantification de ces
peptides. On utilise donc avec cette technologie, le spectre MS/MS pour la quantification.
Recherche d’interaction de 552 protéines sur la
séquence régulatrice de HDAC2. Identification
de la protéine HuR spécifique de cette
séquence.
Identification des Méthyltransférases
interagissant interagissant spécifiquement avec
les ARNt

- 18 -
Figure III.2.a : Représentation schématique des réactifs iTRAQ
Cette technique a pour avantage de travailler simultanément avec quatre échantillons
différents en une seule expérience et permet à la fois identifier et quantifier les éléments
détectables d'un protéome complexe en un court laps de temps (comparé à la méthode
laborieuse des gels 2D). De plus, les peptides marqués sont isobariques et donc induisent une
seule espèce d’ion en MS qui sera utilisée en MS/MS. Ceci augmente l’intensité du signal et
donc la probabilité d’une identification correcte des peptides minoritaires. En marquant un
peptide de quantité connue, on peut alors établir une quantification absolue des autres
peptides. La nouvelle génération de iTRAQ comprend 8 réactifs qui permettent alors de
travailler avec 8 échantillons différents.
Ce type de marquage a, par exemple, été utilisé afin de déterminer les capacités
protectrices de l’haptoglobine sur l’oxydation de l’hémoglobinexxv
. Pour déterminer cela,
quatre échantillons ont été étudiés sous différentes conditions, comme le montre la
figure III.2.b.
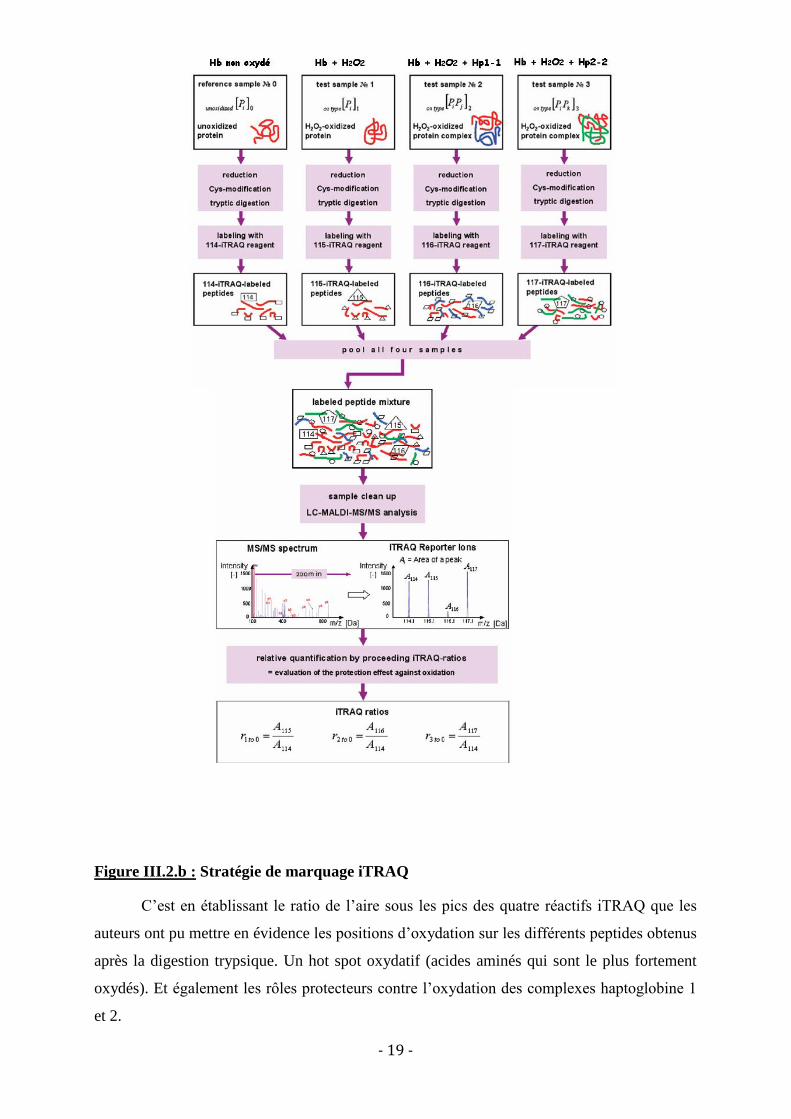
- 19 -
Figure III.2.b : Stratégie de marquage iTRAQ
C’est en établissant le ratio de l’aire sous les pics des quatre réactifs iTRAQ que les
auteurs ont pu mettre en évidence les positions d’oxydation sur les différents peptides obtenus
après la digestion trypsique. Un hot spot oxydatif (acides aminés qui sont le plus fortement
oxydés). Et également les rôles protecteurs contre l’oxydation des complexes haptoglobine 1
et 2.

- 20 -
IV/ Applications Médicales
IV.1/ Identification par 2D-DIGE (DIfferential Gel Electrophoresis) et phage
display(xxvi)
Nous avons choisi de montrer la technique permettant une détection de biomarqueurs.
Cette technique débute par une analyse en gel 2D-DIGE (DIfferential Gel
Electrophoresis) afin de sélectionner les spots de protéines qui présentent une
expression différentielle entre les cellules cancéreuses et les cellules saines. Le gel 2D-
DIGE est une technique d’analyse qui repose sur l’utilisation de marqueurs fluorescents
capables de fixer les chaînes latérales des lysines. 3 marqueurs sont utilisés : le Cy2, Cy3
et Cy5. Les deux échantillons protéiques sont marqués respectivement au Cy3 et Cy5. Le
mélange protéique résultant est séparé par électrophorèse bidimensionnelle sur un gel
unique. Un échantillon contrôle, composé d’un mélange des 2 échantillons précédents
est marqué par un troisième fluorophore Cy2. Puis s’effectue une acquisition des images
correspondant aux protéines marquées respectivement par Cy3, Cy5 et Cy2. Les
résultats obtenus sont présentés en figure 1. Ces spots sont ensuite identifiés par
MALDI-TOF/MS.
B
FigI : le gel 2D-DIGE. A. Principe du gel 2D-DIGE. B. Résultats obtenus
A

- 21 -
Puis on isole des fragments scFv d’anticorps hyper spécifiques des biomarqueurs
identifiés grâce à une technique de gel 2D-DIGE. Le fragment scFv est exprimé à la
surface du phage en fusion avec la protéine membranaire P3 du phage. Entre les deux
protéines, un site de coupure à la thrombine est inséré. Les biomarqueurs d’intérêt sont
fixés sur une membrane de nitrocellulose et mises en présence d’une librairie de phages
présentant à leur surface des fragments scFv. Seront récupérés uniquement les
populations capables de fixer spécifiquement les protéines d’intérêt en répétant les
étapes de sélection. Suite à l’interaction scFv/protéine, le phage contenant l’ADNc du
fragment d’intérêt est récupéré par clivage à la thrombine. On réalise alors des cycles
d’amplification successifs tout en augmentant la stringence (par ajout de détergent) afin
de récupérer les fragments ayant une bonne affinité pour les biomarqueurs étudiés.
Ces fragments scFv sont ensuite utilisés dans des analyses de tissues microarrays ou
TMA afin de vérifier le profil d’expression de ces protéines sur des tissus sains et
malades.
Cette analyse consiste à récupérer différentes coupes de tissus issus de biopsie de
patients sains et malades. Les coupes fines de tissus sont placées sur une plaque de 96
puits, nous pouvons alors faire de l’Immunostaining. Dans un premier temps, tous les
tissus sont mis en présence des fragments scFv sélectionnés auparavant, et qui sont
capables de fixer les biomarqueurs spécifiques du cancer du sein. Puis on fait agir un
anticorps anti-scFv couplé à la peroxydase.
IV.2/ Etude des biomarqueurs dans les cancers (xxvii)
L’expression différentielle de certaines protéines entre des individus sains et souffrants de
cancers, permet de mettre en évidence de potentiels biomarqueurs notamment dans les
cancers oraux. Ces biomarqueurs permettent une détection précoce de la maladie pour une
meilleure prise en charge des patients. A ce jour, deux biomarqueurs ont été identifiés comme
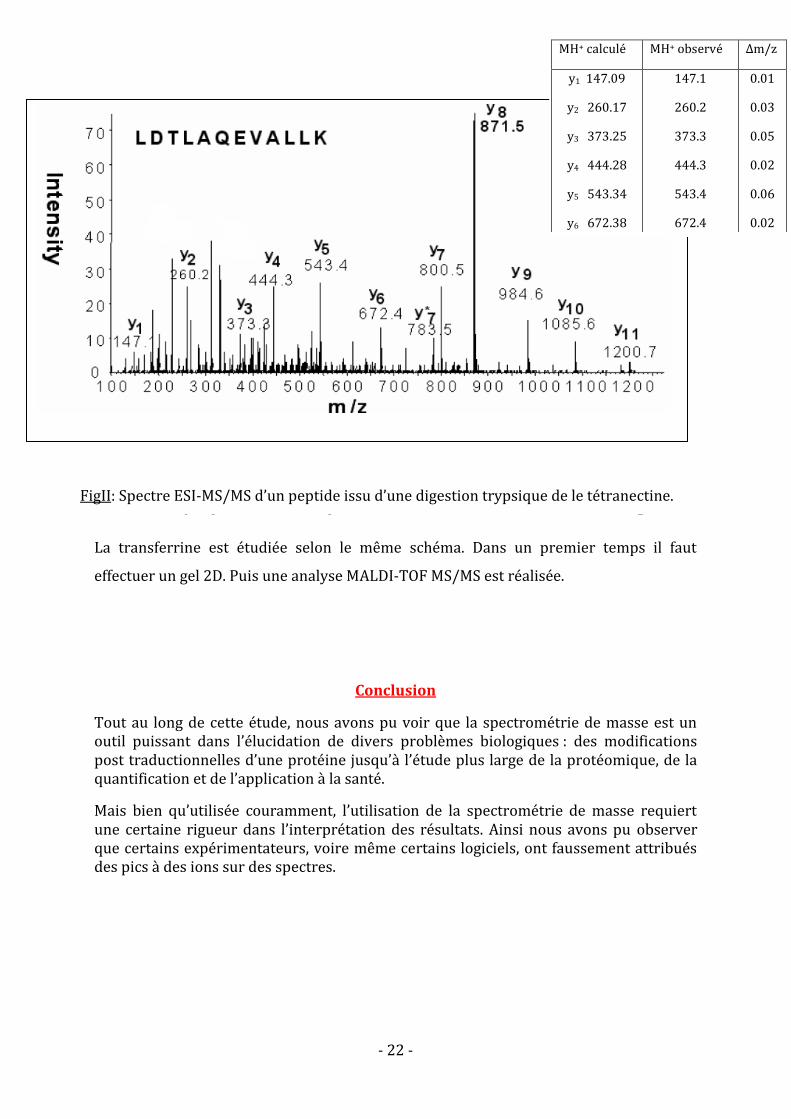
étant des indicateurs des cancers oraux. Il s’agit de la tétranectine et de la transferrine. Il a été
mis en évidence par gel 2D que la tétranectine est sous exprimée chez les patients atteints.
Les spots sont ensuite récupérés pour être identifiés après hydrolyse trypsique par LC/ESI-
MS/MS. Un des spectres obtenu est présenté en figure 2.
- 22 -
Puis une analyse par western blot permet de confirmer les résultats obtenus en gel 2D.
La transferrine est étudiée selon le même schéma. Dans un premier temps il faut
effectuer un gel 2D. Puis une analyse MALDI-TOF MS/MS est réalisée.
Conclusion
Tout au long de cette étude, nous avons pu voir que la spectrométrie de masse est un outil puissant dans l’élucidation de divers problèmes biologiques : des modifications post traductionnelles d’une protéine jusqu’à l’étude plus large de la protéomique, de la quantification et de l’application à la santé.
Mais bien qu’utilisée couramment, l’utilisation de la spectrométrie de masse requiert une certaine rigueur dans l’interprétation des résultats. Ainsi nous avons pu observer que certains expérimentateurs, voire même certains logiciels, ont faussement attribués des pics à des ions sur des spectres.
FigII: Spectre ESI-MS/MS d’un peptide issu d’une digestion trypsique de le tétranectine.
Les masses des différents ions y sont calculés a à partir de la séquence fournit.
MH+ calculé MH+ observé Δm/z
y1 147.09
y2 260.17
y3 373.25
y4 444.28
y5 543.34
y6 672.38
y7 800.43
y8 871.46
y9 984.54
y10 1085.58
y11 1200.6
147.1
260.2
373.3
444.3
543.4
672.4
800.5
871.5
984.6
1085.6
1200.7
0.01
0.03
0.05
0.02
0.06
0.02
0.07
0.04
0.06
0.02
0.1

- 23 -
i Fouillen L, Abdulrahman W, Moras D, Dorsselaer AV, Poterszman A, Sanglier-Cianférani
S. Anal Biochem. 2010, 407, 34-43.
ii Ong SE, Foster LJ, Mann M. Methods. 2003, 29, 124-30.
iii Latterich M, Abramovitz M, Leyland-Jones B. Eur J Cancer. 2008, 44, 2737-41.
iv Imai S, Nagano K, Yoshida Y, Okamura T, Yamashita T, Abe Y, Yoshikawa T, Yoshioka
Y, Kamada H, Mukai Y, Nakagawa S, Tsutsumi Y, Tsunoda S. Biomaterials. 2011, 32, 162-9. v Arellano-Garcia ME, Li R, Liu X, Xie Y, Yan X, Loo JA, Hu S. Int J Mol Sci. 2010, 11,
3106-21. vi
Inutan ED, Richards AL, Wager-Miller J, Mackie K, McEwen CN, Trimpin S. Mol Cell
Proteomics. 2011, 10, M110.
vii Wang et al 2010
viii Xu G, Shin SB, Jaffrey SR. Global profiling of protease cleavage sites by chemoselective
labeling of protein N-termini. (2009)
ix Nadnudda Rodthongkum, Yangbin Chen, S. Thayumanavan, and Richard W. Vachet.
Selective Enrichment and Analysis of Acidic Peptides and Proteins Using Polymeric Reverse
Micelles and MALDI-MS. (2010)
x Peter Lasch,, Michal Drevinek, Herbert Nattermann, Roland Grunow, Maren Stammler,
Ralf Dieckmann, Torsten Schwecke, and Dieter Naumann. Characterization of Yersinia Using
MALDI-TOF :Mass Spectrometry and Chemometrics. (2010)
xi
Mann K, Wilt FH, Poustka AJ. Proteomic analysis of sea urchin spicule matrix. (2009)
xii Laetitia Fouillen et al., 2009
xiii Kamikawaji et al., 2009
xiv Fujii et al., 2010
xv Gant-Branum, 2009
xvi Zhou et al. 2007, Isolation of N-Linked Glycopeptides from Plasma
xvii
G. Palmisano et al. 2010, Glycoproteomic profile in wine: a ‘sweet’ molecular renaissance
xviii
Chen et al, 2010, One-pipeline approach achieving glycoprotein identification and
obtaining
intact glycopeptide information by tandem mass spectrometry
xix Sparbier et al, 2006, Exploring the binding profiles of ConA, boronic acid and WGA by
MALDI-TOF/TOF MS and magnetic particles.

- 24 -
xx
Huc et al, 2010, O-Mycoloylated Proteins from Corynebacterium ANUNPRECEDENTED
POST-TRANSLATIONAL MODIFICATION IN BACTERIA
xxi
Contrepois et al, 2010, Ultra-High Performance Liquid Chromatography-Mass
Spectrometry
for the Fast Profiling of Histone Post-Translational Modifications
xxii
Ong, et al. (2003), Mass spectrometric-based approaches in quantitative proteomics
xxiii
Butter et al. (2010), Unbiased RNA–protein interaction screen by quantitative proteomics
xxiv
Latterich et al. (2008), Proteomics: New technologies and clinical applications
xxv Pimenova et al. (2010), Quantitative mass spectrometry defines an oxydative hotspot in
Hemoglobin that is Specifically protected by haptoglobin
xxvi Imai S, Nagano K, Yoshida Y, Okamura T, Yamashita T, Abe Y, Yoshikawa T, Yoshioka
Y, Kamada H, Mukai Y, Nakagawa S, Tsutsumi Y, Tsunoda S. Development of an antibody
proteomics system using a phage antibody library for efficient screening of biomarker
proteins. Biomaterials. 2011 Jan;32(1):162-9. Epub 2010 Oct 8.
xxvii
Arellano-Garcia ME, Li R, Liu X, Xie Y, Yan X, Loo JA, Hu S. Identification of
tetranectin as a potential biomarker for metastatic oral cancer. Int J Mol Sci. 2010 Sep
2;11(9):3106-21.


















