
UNIVERSITE FERHAT ABBAS SETIF 1 FACULTE DES … · UNIVERSITE FERHAT ABBAS –SETIF 1 FACULTE DES...
97
UNIVERSITE FERHAT ABBAS –SETIF 1 FACULTE DES SCIENCES DE LA NATURE ET DE LA VIE DEPARTEMENT DU TRONC COMMUN COURS DE BIOCHIMIE STRUCTURALE Polycopié destiné aux étudiants de 2 ème année de Tronc Commun Présenter par : Djarmouni Meriem 2016/2017
Transcript of UNIVERSITE FERHAT ABBAS SETIF 1 FACULTE DES … · UNIVERSITE FERHAT ABBAS –SETIF 1 FACULTE DES...

UNIVERSITE FERHAT ABBAS –SETIF 1
FACULTE DES SCIENCES DE LA NATURE ET DE LA VIE
DEPARTEMENT DU TRONC COMMUN
COURS DE BIOCHIMIE
STRUCTURALE
Polycopié destiné aux étudiants de 2ème
année de Tronc Commun
Présenter par :
Djarmouni Meriem
2016/2017

SOMMAIRE
Introduction………………………………………………..……………...………………..…1
Chapitre I: Les liaisons chimiques…………………...…………..…..……..……….………2
1- Différents types de liaisons ………………………………………..……..…………………2
1.1. Liaisons fortes…………………………………………………...……………………..2
1.2. Liaisons faibles………………………………………….……..……………………….3
Chapitre II: les acides aminés…………………...…….………..……….…...…………....…5
II.1. Définition……………………………………..……………..……………………….……5
II.2. Classification………………………………….…………………………………………..8
II.3. Principales propriétés physiques des aminoacides……...……………………………….10
II.3.1. Stéréochimie ………………………………………………………………………......10
II.3.2. Chiralité…………………………………………………….……………………..…...11
II.3.3. Absorption dans l’ultraviolet……………………………………………………..……11
II.3.4. Ionisation………………………………………….………………………………..….12
II.3.5. Principales propriétés chimiques des acides amines...……………...…………………16
II.3.5.1. Réactions dues à la présence du groupement carboxyle……..………………...……16
II.3.5.2. Réactions dues à la présence du groupement amine……..……..……………...……17
II.3.5.2.Propriétés dues à la présence du groupement carboxyle et amine……………… .…19
Chapitre III: les peptides…………………………………..………………………......……21
III.1. Structure primaire des peptides et des protéines………...………………...................…21
III.1.1. Liaison peptidique…………………………..…………………………….…………..21
III.1.2. Détermination de la structure des chaînes peptidiques ……….…..…………….…....21
II.1.3. Fragmentation des chaînes peptidiques …………..…………………………...…....…23
III.1.4. Séparation de plusieurs chaînes peptidiques…………………......…………...………24
III.1.5. Quelques peptides ayant une importance biologique ……………………...…..……..25
Chapitre IV : les protéines……………………………..…………..…………..……….…..27
IV.1. Introduction …………………………………………………….…….…...………..…..27
IV.2. Structure des protéines…………………………………..………………………….…..28
IV.2.1. Structure primaire………………………………...…………………..………………28
IV.2.2. Structure secondaire……………………………..……………………...…….………29
IV.2.3. Structure tertiaire……………………...……………………………..…………...…..30
IV.2.4. Structure quaternaire…………………………..………………………………..…….31
IV.3. Dénaturation des protéines…………………………….…………………….……….…31
IV.4. Les techniques fondamentales utilisées pour étudier les composés protéiques………..32
IV.4.1. Chromatographie……………………………………………...………….……..……33
IV.4.1.1. Chromatographie par filtration sur gel……………….…………………..….……..33
IV.4.1.2. Chromatographie par échange d’ions…………….…………………..…………….33
IV.4.1.3. Chromatographie par interactions hydrophobes………………..……………....…..34
IV.4.1.4. Chromatographie par affinité…………………….……………………..…………..34
IV.4.2. Electrophorèse...............................................................................................................35
Chapitre V: Enzymes…………………...………………………………..……………….….36
V.1. Définition………………………….………….…………………………………..….…..36
V.2. Classification des enzymes...............................................................................................36
V.3. Site actif des enzymes…………………………..……...……………………….……….37
V.4. Notion de cinétique enzymatique…………………....…………………………………..38
V.5. Influence de différents paramètres sur la vitesse initiale………..…….……………..….40
V.5.1. Influence de la température............................................................................................40
V.5.2. Influence du pH………………………...………………………………………..…......41

LISTE DES TABLEAUX
V.6. Inhibiteurs…………………………………….…………………………..……….….....43
V.6.1. Inhibiteurs compétitifs……………………………………….…….………..………...43
V.6.2. Inhibiteurs non compétitifs............................................................................................45
V.6.3. Inhibiteurs incompétitifs…………………….……………...……….…………..….....47
V.7. L’unité enzymatique……………………………………………………………………..48
V.8. Cofacteurs……………………………………….…….…………………………..….....48
Chapitre VI: les glucides……………………………………...……………..……….…..…50
VI.1. Définition des glucides ………………………………………..……………………….50
VI.2. Importance en Biologie ……………………………….………………...………....…...50
VI.3. Classification des glucides ……………………………..………………………………50
VI.3.1. Les Oses………………………………………………………………...………....….51
VI.3.1.1. Structure linéaire des oses (Modèle de Fischer) …………………………….…..…51
VI.3.1.2. Diversité des oses, Isomérie……………… …………………………….…..……53
VI.3.1.3. Filiation des oses ……………………………………………………….………..…56
VI.3.1.4. Structure cyclique des oses……………………………………………………...….58
VI.3.1.5. propriétés physiques des oses…………………………………………….…...……60
VI.3.1.6. Propriétés chimiques des oses………………………………………………...……61
VI.4. Osides……………………………………………...……………………..……………..67
VI.4.1. Holosides…………………………………………….……………………….…..…..67
VI.4.1.1. La liaison glycosidique……………………….……………………………...…..…67
VI.4.1.2. Nomenclature et convention ………………...……………………….....……...…..67
VI.4.2. Diholosides……………………………………………………………………….…..68
VI.4.3. Polyosides……………………………………………………………………...……..69
VI.4.4. Hétérosides……………………………………….………………………..……….....72
Chapitre VII: Les lipides……………………………...…………………………...………..74
VII.1. Définition………………………..……………………………………………..……...74
VII.2. Les acides gras………………………………………………………………...…….....74
VII.2.1. Propriétés physiques des acides gras ……………………….…………..……….…..79
VII.2.2. Propriétés chimiques……………………………………………..………...………...80
VII.3. Classification des lipides………………………………….……………….…...………82
VII.3.1. Lipides simples ou ternaires………………………………………………….….......82
VII.3.1.1. Les glycérides…………………………………………...…………...……..…..….83
VII.3.1.2. Les cérides…………………………………………………………………..…..…85
VII.3.1.3. Les stérides………………………………………………………………….…….86
VII.3.2. Lipides complexes…………………………………………...……………..….…....86
VII.3.2.1. Les glycérophospholipides……………………………………………………......86
VII.3.2.2. Glycéroglycolipides…………………………………………………………..…...89
VII.3.2.3. Les sphingolipides………………………………………..………………….........89
LISTE DES FIGURES
Figure 1. Formule générale d’un acide aminé ………………………………………...………5
Figure 2 : Aminoacides à chaîne latérale non polaires………………………………..…….…9
Figure 3 : Aminoacides à chaines latérales polaires non chargées……………………...….…9
Figure 4 : Aminoacides acides…………………………………………………….…….…….10
Figure 5 : Aminoacides basiques…………………………………………………………......10
Figure 6 : Spectre d’absorption des aminoacides aromatiques dans l’ultra-violet……...…....12
Figure 7 : Courbe de titration de l’acide aminé Ala………………………………….………14
Figure 8 : Courbe de titration de Glu…………………………………...………………….…15
Figure 9 : courbe de titration de l’acide aminé basique (Lys) ……………………………....16

Figure 10 : Liaison peptidique entre deux aminoacides……………………………...…...…21
Figure 11 : Structure du glutathion réduit…………………………...……………………….25
Figure 12 : Structure de l’ocytocine et de la vasopressine…………………………………...26
Figure 13 : Structure de l’insuline………………………………………………...………....26
Figure 14 : État étiré ou structure en feuillets plissés………………………………..……....29
Figure 15 : État hélicoΪdal ou hélice α…………………………………………….………...30
Figure 16 : Structure tertiaire de la myoglobine………………………………..…………...30
Figure 17. Structure quaternaire de l’hémoglobine …………………………………...…...31
Figure 18 : Différent type de chromatographie………………………………………….….34
Figure 19 : Exemple de l’électrophorèse d’un mélange d’alanine, d’acide aspartique, et de
lysine à pH6…………………………………………………………………………………..35
Figure 20 : Variation de la vitesse initiale en fonction de la concentration en substrat, celle de
l’enzyme étant constante……………………………………………………………...……..39
Figure 21 : Représentation de Lineweaver et Burk et détermination des constantes
cinétiques…………………………………………………………………………………….40
Figure 22 : Influence de la température sur l’activité enzymatique………………………...41
Figure 23 : Influence du pH sur l’activité enzymatique………………………………….…42
Figure 24 : Influence du pH sur l’activité enzymatique de la pepsine…………………….…43
Figure 25 : Analogie structurale entre l’enzyme et l’inhibiteur compétitif……………...….43
Figure 26 : Influence d’un inhibiteur conpétitif sur l’activité enzymatique………….…..…44
Figure 27 : Fixation d’un inhibiteur non conpétitif…………………………………….........45
Figure 28 : Influence d’un inhibiteur non compétitif sur l’activité enzymatique…………...46
Figure 29 : Effets des inhibiteurs en représentation en double inverse……………………...46
Figure 30 : Influence d’un inhibiteur incompétitif sur l’activité enzymatique………………47
Figure 31 : Classification des glucides…………………………………………………...….51
Figure 32 : Filiation des D-aldoses et D-cétoses…………………………………...……..…57
Figure 33. Structure de l’acide palmitoléique………………………………...…….....……..77
Figure 34. Structure de l’acide oléique…………………………………………...…….....…77
Figure 35. Structure de l’acide linoléique……………………...……................................…78
Figure 36. Structure de l’acide alpha-linolénique………………………………...…….....…78
Figure 37. Structure de l’acide arachidonique……………………...…………………......…79
Figure 38. Disposition des micelles……………………...…..............................................…80
Figure 39. Réaction de saponification…………………………………………….....…....…80
Figure 40. Réaction d’esterification……………………...………………………….…....…80
Figure 41. Réaction d’addition d’iode…………………………………………….....…....…81
Figure 42. Oxydation des acides gras insaturés par l’acide performique………………...….81
Figure 43. Structure d’un diol……………………...……………………………………..….81
Figure 44. Oxydation puissante……………………...…....…………………………………82
Figure 45 : Structure des glycérides……………………………………………….….…..…83
Figure 46 :Structure du 1,3 distéaryl-2-palmitylglycérol ……………………….………..…84
Figure 47: Réaction de saponification……………………….………………………………85
Figure 48: Structure des cérides ……………………….………………………………....…85
Figure 49. Molécule de palmitate de cétyle. ……………………………………………..…85
Figure 50. Molécule de palmitate de cholestéryle. ……………………………..………..…86
Figure 51 :Acide phosphorique (=diacylglycérol-P)…………………………………..…….87
Figure 52 : Structure de Sphingosine………………………………………………………...89
Figure 53: Structure de Céramide…………………………………………...…………...…..90
Figure 54: Structure de sphingomyéline………………………….………...….………….....90
Figure 55 : structure de Cérébrogalactosides………………………...………....…………....91
Figure 56 : structure d’un ganglioside.…………………………………………..…………..91

Tableau 1 : Structure des acides aminés………………………………………………...…….5
Tableau 2 : Détermination de l’acide aminé en position N-terminale…………………….…22
Tableau 3 : Détermination de l’acide aminé en position C-terminale………………..……...23
Tableau 4 : Fragmentation des chaines peptidique…………………………………………..23
Tableau 5. Récapitulatif des acides gras naturels……………………………………...…….75
Tableau 6. Récapitulatif des acides gras insaturés………………………………………..….76

Introduction
La biochimie, ou chimie du vivant, a toujours eu et aura toujours une place essentielle dans le
cursus de l’étudiant de biologie, car elle est une base essentielle à la compréhension des
mécanismes de la vie au niveau moléculaire.
La biochimie, science des bases chimiques de la vie, couvre donc un vaste champ
d’investigation, avec de forts recouvrements avec des disciplines comme la biologie
moléculaire et la génétique moléculaire, englobant des domaines de la biologie cellulaire, de la
microbiologie et de la physiologie. Les lois de la physique et de la chimie s’appliquent aux
systèmes vivants : la biochimie s’appuie sur les disciplines physicochimiques (physique,
thermodynamique, chimie physique, minérale et organique).
Les structures des molécules (acides aminés, peptides, protéines, enzymes, glucides et lipides)
sont abordées dans un plusieurs chapitres, ce qui devrait permettre d’intégrer de façon plus
aisée la diversité des molécules du vivant. Les propriétés des acides aminés, de certains
peptides, et des protéines font l’objet de plusieurs chapitres, l’enzymologie est abordée dans un
chapitre à part. Enfin, les propriétés des glucides et des lipides font chacun l’objet d’un chapitre
spécifique.

Chapitre 1 : les liaisons chimiques
La liaison chimique est définie comme une interaction électromagnétique des électrons de
valence des atomes en présence, assurant ainsi une cohésion entre les molécules qui constituent
la matière. D’un point de vue énergétique la molécule formée est d’autant plus stable si les
atomes mettent en commun, donnent ou reçoivent des électrons dans le but d’être entouré de
huit électrons : c’est la règle de l’octet. Cette règle, énonce que les atomes qui rentrent dans la
formation d’un édifice moléculaire ont tendances à acquérir la configuration la plus stable qui
est celle du gaz rare le plus proche (ns2np6).
1- Différents types de liaisons
Généralement on distingue les liaisons fortes et les liaisons faibles. Parmi les premières, on
trouve les liaisons ioniques, covalentes et métalliques. Les liaisons faibles sont principalement
les liaisons par forces de Van der Waals et les liaisons hydrogène.
1.1. Liaisons fortes
Ce type de liaison, dit aussi liaison intramoléculaire, dépend de la différence
d'électronégativité, plus l'électronégativité est importante, plus l'électron est attiré par un atome
particulier et plus la liaison à un caractère ionique. Si l'électronégativité est faible, la liaison est
covalente.
a. Liaison ionique
La liaison ionique se caractérise par le fait que deux atomes ne partagent pas mais échangent
des électrons. Il s’agit au fait d’un type de liaison chimique qui peut être constitué par une paire
d’atomes possédant une grande différence d’électronégativité, typiquement entre un métal et un
non-métal. De sorte que le métal donne un ou plusieurs électrons pour former un cation et le
non-métal capte ces électrons pour former un anion.
Ainsi, les deux ions formés acquièrent souvent la configuration du gaz noble (ils respectent la
règle de l’octet) et la stabilité de la liaison est assurée par l’interaction électrostatique entre le
cation et l’anion.
Exemple : Le chlorure de sodium NaCl (Sel de cuisine) : Na+ + Cl
- NaCl
b. Liaison covalente et de coordinence
b1- Liaison covalente
Une liaison covalente est une liaison dans laquelle chacun des atomes liés met en commun un
électron d’une de ses couches externes (e- de valence) afin de former un doublet d’électrons
liant les atomes. On symbolise ce type de liaison par un tiret
-Les liaisons covalentes sont des liaisons (sigma) les plus fortes, et il ne peut exister qu’une
seule liaison entre deux atomes, si des liaisons supplémentaires sont créées (liaisons
multiples), elles sont faibles et sont de type liaison π (pi).
b2- Liaison covalente de coordinence ou dative

La liaison dative est une liaison dans laquelle le doublet liant mis en commun est apporté par
un seul des atomes donneur de doublet à l’autre qui joue le rôle d’accepteur.
le donneur doit posséder un doublet libre.
l’accepteur doit posséder une case ou orbitale atomique vide (lacune électronique)
c. Liaison métallique
La liaison métallique est une liaison qui permet la cohésion des atomes d’un solide. Ces atomes
mettent en commun un ou plusieurs électrons, dits électrons libres. Ces électrons externes se
délocalisent et se comportent comme s’ils étaient libres, tout en restant dans l’échantillon. C’est
cette libre mobilité des électrons entre les noyaux d’atomes métalliques positifs qui fait que les
métaux sont de bons conducteurs de chaleur et d’électricité. Ainsi, c’est le nombre d’électrons
mis en commun entre les atomes métalliques qui assurera la force de la liaison. Plus un atome
métallique possède d’électrons de valence à mettre en commun avec les autres atomes de métal,
plus la liaison métallique sera forte, le métal sera dur et la température de fusion et d’ébullition
seront élevées.
On peut décrire le métal comme un assemblage d’ions positifs baignant dans un nuage
électronique faible et dont les électrons sont facilement mobiles, d’où la grande conductivité
électronique des métaux.
1.1.1 Polarité des liaisons
La polarisation d’une liaison est étroitement liée à la différence d’électronégativité des atomes
liés entre eux. Plus la différence est importante, plus la liaison est polarisée.
a. Liaison polaire et apolaire
-Une liaison est polaire, si la différence des électronégativités des atomes formant la liaison
n’est pas nulle, créant ainsi une dissymétrie du nuage électronique. En effet, l’atome le plus
électronégatif attire vers lui le doublet de la liaison, acquérant un excédant de charge négative
noté - δ, tandis que l’autre atome, le moins électronégatif, se retrouve avec un déficit de charge
noté + δ, bien entendu l’ensemble reste électriquement neutre.
Exemple : H–O ; C–F ; N–O
-Une liaison est apolaire, si la différence des électronégativités des atomes formant la liaison
est nulle, créant ainsi une symétrie du nuage électronique. Cette liaison est purement covalente.
(voir annexe page 147 : valeurs d’électronégativité)
Exemple : H–H ; Cl–Cl ; C–I ; N–Cl
1-2 Liaisons faibles
La liaison intermoléculaire est une liaison qui unit les molécules. On la définit comme une
force électrostatique résiduelle faible s’établissant entre les dipôles des molécules on distingue
3 types d’interactions ou forces entre les molécules.

a. Liaison hydrogène : cette liaison est souvent présente dans toute la chimie de la vie. Par
définition une liaison hydrogène se forme lorsqu’un atome d’hydrogène, déjà uni à un premier
atome (A) très électronégatif, peut établir un second lien avec un autre atome (B), également
très électronégatif et porteur d’un ou plusieurs doublets non liants.
L’origine de la liaison hydrogène est essentiellement électrostatique et de type dipôledipôle.
L’hydrogène lié à un atome électronégatif porte une fraction de charge positive localisée qui
interagit fortement avec le dipôle produit par l’autre atome électronégatif fonctionnant comme
accepteur. Les trois atomes A–H et B sont alors alignée.
Généralement les atomes (A) et (B) qui interviennent sont : l’azote, l’oxygène, le fluor et le
chlore. Il faut retenir que le second lien établit, entre H et (B), est souvent représenté par des
pointillés (trait discontinu) pour le distinguer de la liaison covalente établit entre H et (A).
b. Les Forces de Van der Waals :
Les forces de Van der Waals résultent de l’interaction entre les nuages électriques des atomes
ou molécules très proches les uns des autres. La charge négative d’un nuage électronique n’est
pas « figée », à tout moment le nuage électronique fluctue, il n’est que statistique, ce qui permet
les attractions entre un noyau, chargé positivement, et le nuage des électrons d’un atome voisin.
Les interactions de van der Waals comprennent les interactions dipôle- dipôle dont l’énergie
d’interaction diminue. Ces interactions appelées aussi force de London.
Les attractions de van der Waals ne se manifestent donc que dans une zone de distance
interatomique très limitée ; aux températures dites physiologiques l’efficacité des liaisons
dépond du nombre des atomes d’une molécule qui sont en interaction avec les atomes de la
molécule voisine.
c. les interactions hydrophobes: sont la conséquence de la très forte tendance des molécules
d’eau à exclure les groupements et les molécules non polaires. Les interactions hydrophobes ne
résultent pas tant d’une affinité particulière des substances non polaires entre elles, mais du fait
de la très forte interaction entre les molécules d’eau, interaction beaucoup plus forte qu’avec
une molécule non polaire.
Comme la plus forte des interactions possibles entre deux molécules l’emporte sur les autres, la
formation de liaisons hydrogène entre les molécules d’eau polaires exclut les molécules et
groupements non polaires. L’exclusion des substances hydrophobes d’une solution aqueuse et
la tendance des molécules non polaires à s’agglutiner est une conséquence des interactions
préférentielles des molécules d’eau. C’est pour cette raison que les régions non polaires des
macromolécules biologiques sont souvent enfouies à l’intérieur des molécules.

Chapitre II: les acides aminés
II.1. Définition
Les acides aminés (ou amino-acides) sont des molécules qui possèdent une fonction
carboxylique et une fonction amine primaire portée par un même atome de carbone, l’atome
du carbone α : ce sont des acides α-aminés. Ils différent par la nature de la chaine latérale ou
le radical R (figure 1).
Les acides aminés les plus répandus chez l’homme sont définie par la formule suivante :
Figure 1. Formule générale d’un acide aminé
Plus de 300 acides aminés ont été inventoriés. On distingue :
- Les 20 acides aminés constitutifs des protéines naturelles ou acides aminés standards. Ils sont
codés dans l’ADN et incorporés dans la chaîne peptidique lors de la traduction de l’ARNm.
- Et les autres, que l’on trouve soit à l’état libre, soit dans des peptides synthétisés par des
microorganismes ou des végétaux.
On a l’habitude d’utiliser des abréviations à trois lettres ou à une lettre pour cette série de vingt
aminoacides. (tableau1).
Tableau 1. Structure des acides aminés.
A- les acides aminés neutres
A1- Les acides aminés aliphatique
Glycine (Gly, G) Alanine (Ala, A ) Valine (Val , V) pHi= 5,97 ; PM= 75
pHi= 6,02 ; PM= 89
pHi= 5,97 ; PM= 117
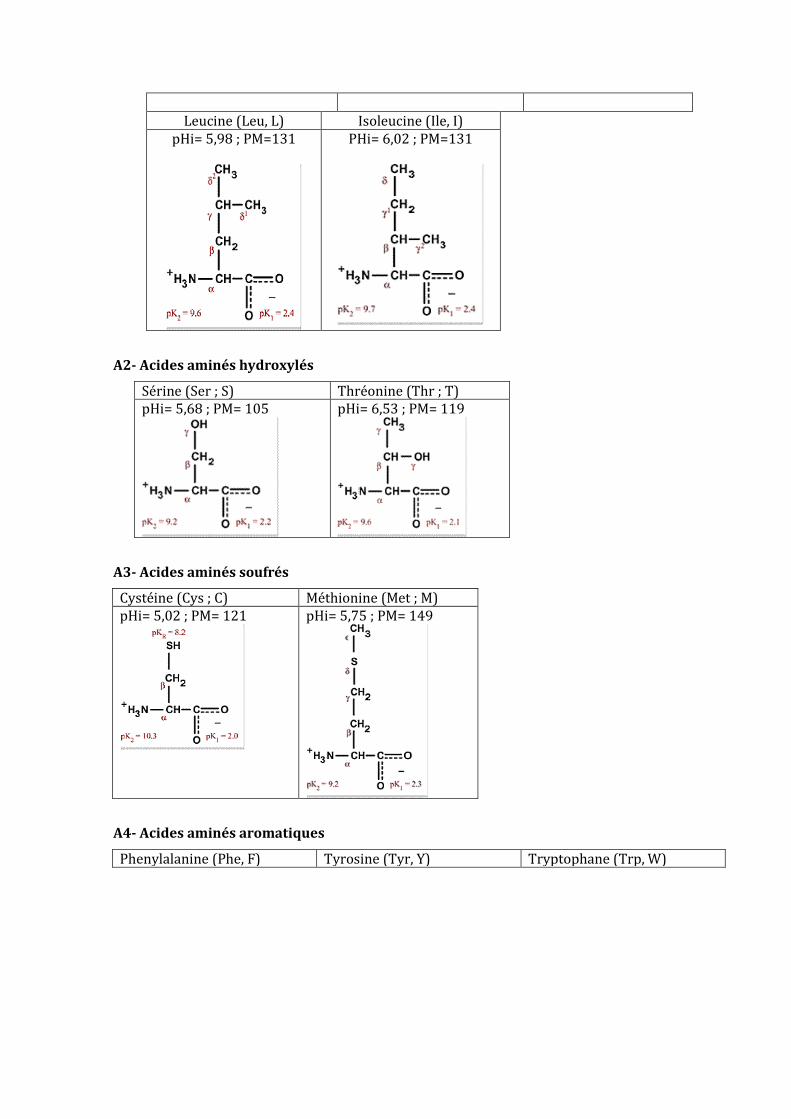
Leucine (Leu, L) Isoleucine (Ile, I) pHi= 5,98 ; PM=131
PHi= 6,02 ; PM=131
A2- Acides aminés hydroxylés
Sérine (Ser ; S) Thréonine (Thr ; T) pHi= 5,68 ; PM= 105
pHi= 6,53 ; PM= 119
A3- Acides aminés soufrés
Cystéine (Cys ; C) Méthionine (Met ; M) pHi= 5,02 ; PM= 121
pHi= 5,75 ; PM= 149
A4- Acides aminés aromatiques
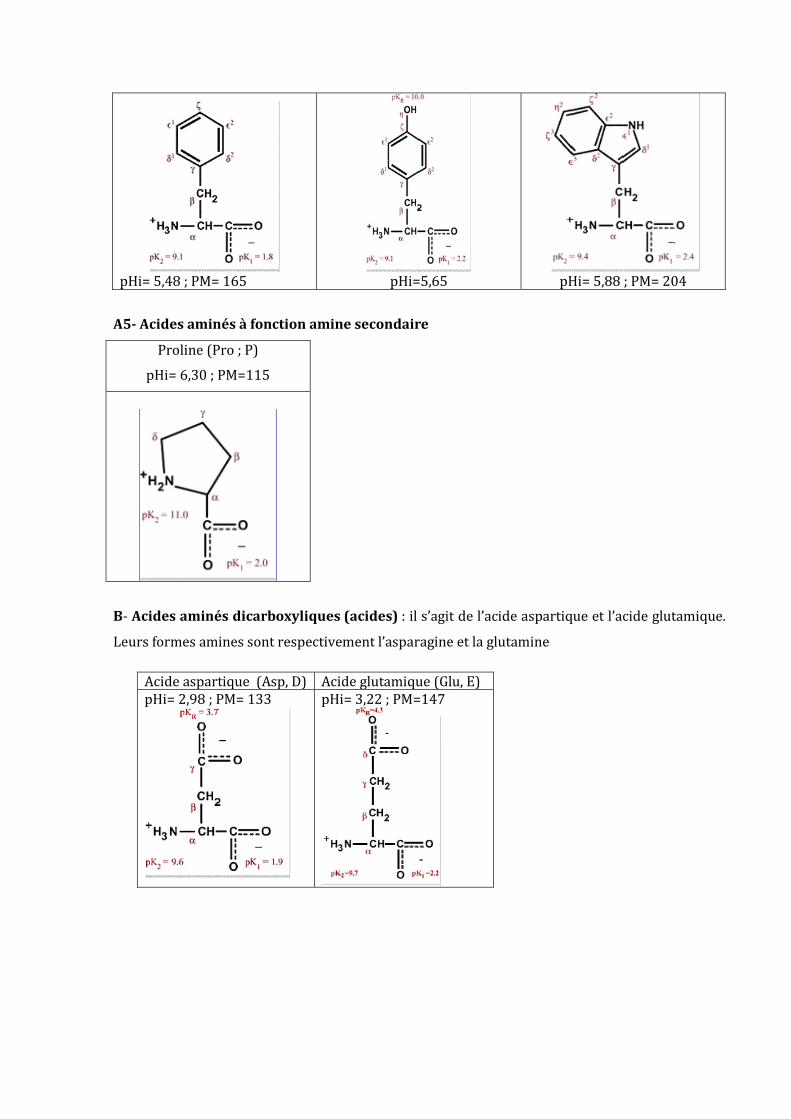
Phenylalanine (Phe, F) Tyrosine (Tyr, Y) Tryptophane (Trp, W)
pHi= 5,48 ; PM= 165
pHi=5,65
pHi= 5,88 ; PM= 204
A5- Acides aminés à fonction amine secondaire
Proline (Pro ; P)
pHi= 6,30 ; PM=115
B- Acides aminés dicarboxyliques (acides) : il s’agit de l’acide aspartique et l’acide glutamique.
Leurs formes amines sont respectivement l’asparagine et la glutamine
Acide aspartique (Asp, D) Acide glutamique (Glu, E) pHi= 2,98 ; PM= 133
pHi= 3,22 ; PM=147

Asparagine (Asn, N) Glutamine (Gln,Q) pHi= 5,41 ; PM= 132
pHi= 5,64 ; PM= 146
C- les acides aminés dibasiques
Lysine (Lys, K) Arginine (Arg, R) Histidine (His, H) pHi= 9,74, PM= 146
pHi=10,76 ; PM= 174
pHi= 7,59, PM= 155
II.2. Classification
Les acides aminés peuvent être classés d’après la structure et la complexité de leur chaine
latérale R. Parmi les différentes classifications possibles, l’une des plus intéressantes repose sur
la polarité et les possibilités d’ionisation de cette chaine.
a. Aminoacides avec une chaîne latérale R non polaire ou hydrophobe
Cette famille contient cinq aminoacides ayant une chaine hydrocarbonée aliphatique : Ala, Leu,
Ile, Val, Gly et Pro, deux possédant des noyaux aromatiques : Phe et Trp et un contenant du
soufre: Met (figure 2).
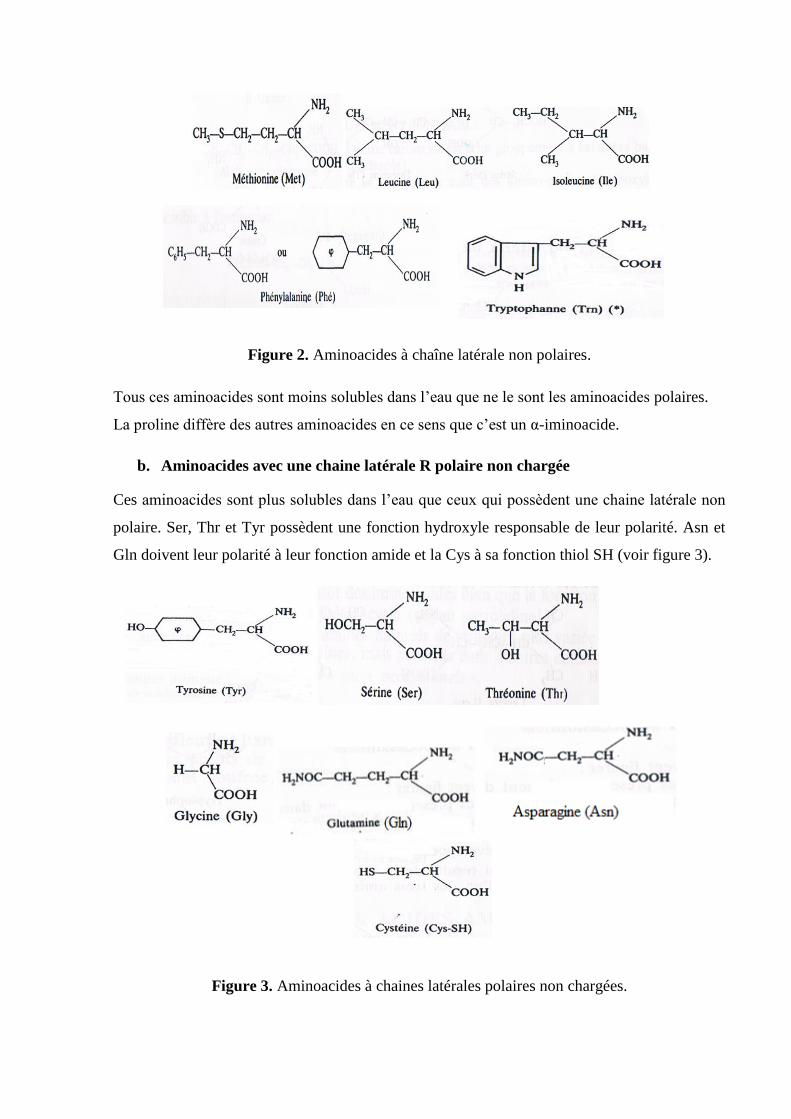
Figure 2. Aminoacides à chaîne latérale non polaires.
Tous ces aminoacides sont moins solubles dans l’eau que ne le sont les aminoacides polaires.
La proline diffère des autres aminoacides en ce sens que c’est un α-iminoacide.
b. Aminoacides avec une chaine latérale R polaire non chargée
Ces aminoacides sont plus solubles dans l’eau que ceux qui possèdent une chaine latérale non
polaire. Ser, Thr et Tyr possèdent une fonction hydroxyle responsable de leur polarité. Asn et
Gln doivent leur polarité à leur fonction amide et la Cys à sa fonction thiol SH (voir figure 3).
Figure 3. Aminoacides à chaines latérales polaires non chargées.
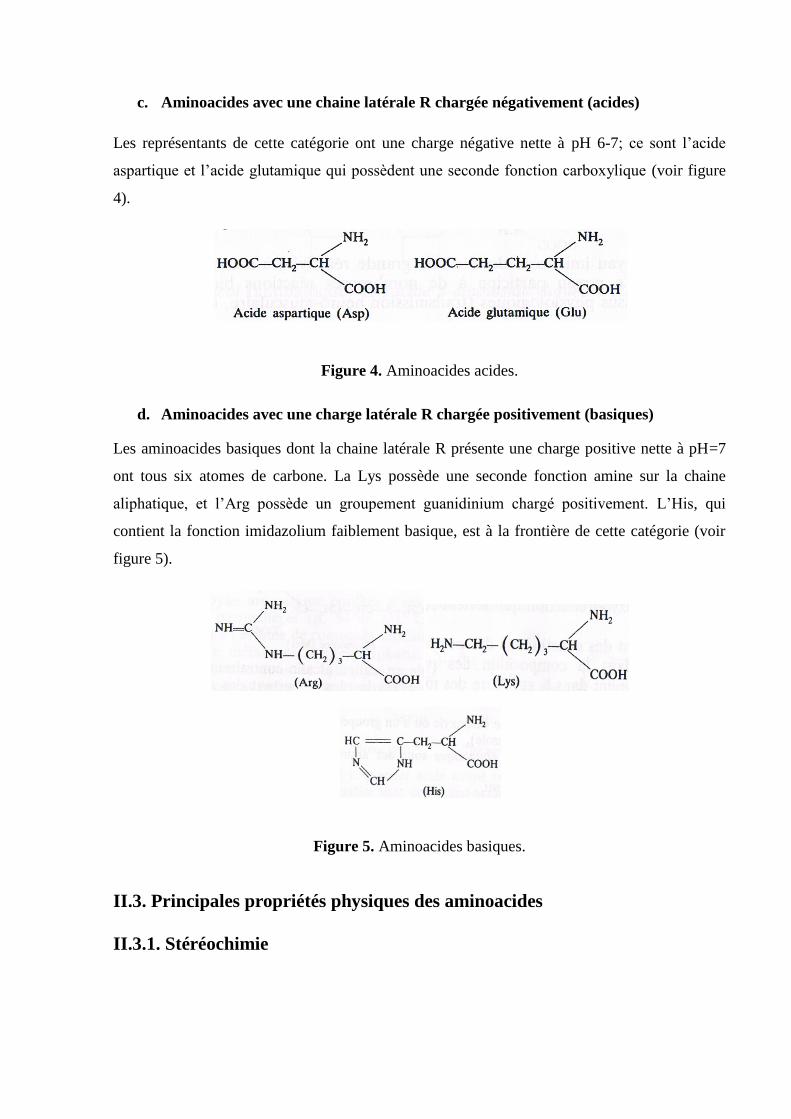
c. Aminoacides avec une chaine latérale R chargée négativement (acides)
Les représentants de cette catégorie ont une charge négative nette à pH 6-7; ce sont l’acide
aspartique et l’acide glutamique qui possèdent une seconde fonction carboxylique (voir figure
4).
Figure 4. Aminoacides acides.
d. Aminoacides avec une charge latérale R chargée positivement (basiques)
Les aminoacides basiques dont la chaine latérale R présente une charge positive nette à pH=7
ont tous six atomes de carbone. La Lys possède une seconde fonction amine sur la chaine
aliphatique, et l’Arg possède un groupement guanidinium chargé positivement. L’His, qui
contient la fonction imidazolium faiblement basique, est à la frontière de cette catégorie (voir
figure 5).
Figure 5. Aminoacides basiques.
II.3. Principales propriétés physiques des aminoacides
II.3.1. Stéréochimie

Le carbone alpha portant quatre groupements différents, ce carbone est asymétrique. Les acides
aminés sont donc des molécules chirales. On a deux isomères possibles : l'un de la série D
l'autre de la série L. Il existe une exception : la glycine.
II.3.2. Chiralité
L’adjectif « chiral », qualifie une structure dépourvue de plan ou de centre de symétrie et qui,
par voie de conséquence, n’est pas superposable à son image dans un miroir. L’exemple le plus
immédiat est donné par le couple main gauche / main droite. Un atome de carbone relié à
quatre groupements différents reçoit le nom de carbone chiral ou carbone asymétrique.
Il se trouve que tous les acides aminés naturels trouvés dans les molécules du vivant sont de la
série L.
II.3.3. Absorption dans l’ultraviolet
Les aminoacides présentent une absorption importante aux longueurs d’onde inférieures à 230
nm; en outre, certains d’entre eux absorbent entre 250 et 300nm en raison de la présence -dans
leur chaine R- de chromophores tels que le noyau phényle (Tyr) ou le noyau indole (Trp)
permettant ainsi le dosage spectrophotométrique des protéines (figure 6).
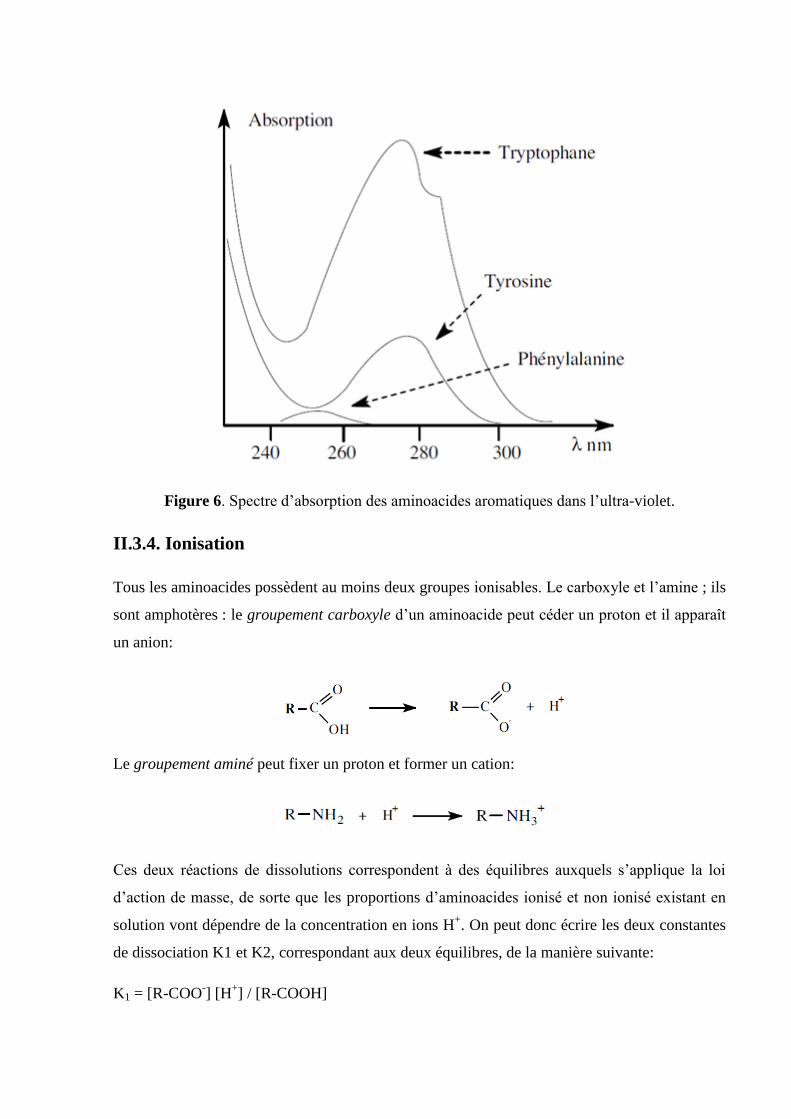
Figure 6. Spectre d’absorption des aminoacides aromatiques dans l’ultra-violet.
II.3.4. Ionisation
Tous les aminoacides possèdent au moins deux groupes ionisables. Le carboxyle et l’amine ; ils
sont amphotères : le groupement carboxyle d’un aminoacide peut céder un proton et il apparaît
un anion:
Le groupement aminé peut fixer un proton et former un cation:
Ces deux réactions de dissolutions correspondent à des équilibres auxquels s’applique la loi
d’action de masse, de sorte que les proportions d’aminoacides ionisé et non ionisé existant en
solution vont dépendre de la concentration en ions H+. On peut donc écrire les deux constantes
de dissociation K1 et K2, correspondant aux deux équilibres, de la manière suivante:
K1 = [R-COO-] [H
+] / [R-COOH]

K2 = [R-NH2] [H+] / [R-NH3
+]
Lorsqu’ on fait passer une solution d’un aminoacide d’un pH bas à un pH élevé, on a les
transformations suivantes:
On veut qu’on passe par un pH où les molécules d’aminoacide sont sous la forme dipolaire
(zwitterion) et où la charge nette de la molécule est nulle, c’est le point isoionique ou
isoélectrique de l’aminoacide. À ce pH, sa solubilité est minimale et il ne migre pas si on le
place dans un champ électrique (contrairement au cation et à l’anion).
pH isoélectrique: défini par [A+] = [A
-]
Nous avons à résoudre le système d’équations suivant :
On peut aisément étudier la dissociation des différentes fonctions polaires d’un aminoacide, en
ajoutant à la solution HCl ou NaOH et en mesurant le pH après chaque addition. On peut ainsi
tracer des courbes de titration, dont l’aspect sera différent selon qu’il s’agit d’un aminoacide
neutre, acide ou basique.
Pour calculer le pKa (ou les concentrations en espèces ioniques à un pH déterminé) on utilise
l’équation de HENDERSON-HASSEBACH:
pKa = pH + log [Accepteur de protons]/ [Donneur de protons]
Connaissant les valeurs des pKa, pHi, il est possible de tracer la courbe de neutralisation d’un
acide aminé.
Exemple : courbe de titration de l’alanine (figure 7):

Elle comporte deux étapes distinctes, chacune correspondant à l’élimination d’un proton de
l’alanine :
→ Au début: (en bas et à gauche du graphique) :
- L’alanine a ses 2 fonctions acido-basique protonée et se trouvent sous une forme cationique
avec une charge positive (NH3+-CH2-COOH),
- Lorsque l’on ajoute de la soude une partie des molécules subissent une dissociation Jusqu’à
arriver à un point où il y a autant de molécules chargées positivement que de molécules neutres,
à ce point le pH est égal au 1er
pK (Pk1=2,3 pour Ala).
pH=Pk1 → 50% sous forme de NH3+-CH2-COOH
50% sous forme de NH3+-CH2-COO
-
- La partie plate autour de ce pK, correspond à une zone tampon, si on continue à ajouter de
la soude, un point d’inflexion est atteint, ce pH correspond au pH isoélectrique ou pHi (pHi=6
pour Ala), toute l’Ala est sous une forme dipolaire avec 2 charges, une (+) et une (-) avec une
charge globale nulle.
pH= pHi →100 % sous forme NH3+-CH2-COO
- (ion Zwitterion)
→ L’ajout de soude provoque une nouvelle dissociation avec perte du proton de l’amine, à
égalité de concentration des 2 espèces, le pH correspond au 2 ème pK (Pk2=9,7 pour Ala).
PH= PK2→ 50% sous forme de NH2-CH2-COO-
50% sous forme de NH3+-CH2-COO
-
- La partie de la courbe relativement plate autour de ce pK correspond à une nouvelle zone
tampon, un dernier ajout de soude va totalement déprotoner l’acide aminé qui va se retrouver
sous une forme chargée négativement NH2-CH2-COO-.
Figure 7 : courbe de titration de l’acide aminé Ala

- Exemple d’un acide aminé à chaîne latérale comportant un groupement acide : (l’acide
glutamique) (figure 8):
Figure 8 : Courbe de titration de Glu
-Exemple d’un acide aminé comportant un groupement à chaîne latérale basique (la
lysine) (figure 9):
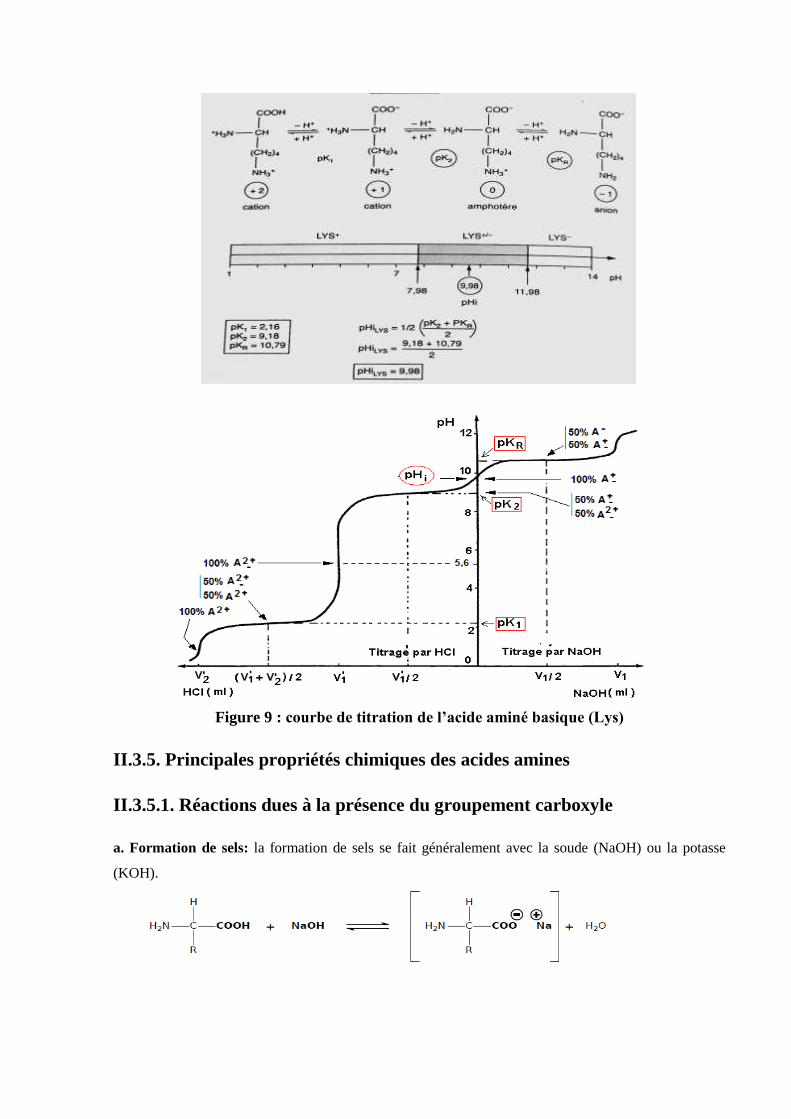
Figure 9 : courbe de titration de l’acide aminé basique (Lys)
II.3.5. Principales propriétés chimiques des acides amines
II.3.5.1. Réactions dues à la présence du groupement carboxyle
a. Formation de sels: la formation de sels se fait généralement avec la soude (NaOH) ou la potasse
(KOH).

b. Estérification: l’estérification se fait grâce à un alcool. On utilise souvent l’alcool n-
butylique (Butan-1-ol) et on obtient des ester n-butyliques. Cette propriété est utilisée lors de
l’analyse par chromatographie en phase gazeuse car ces esters sont volatils. Selon l’ester
obtenu, on pourra connaître l’acide aminé présent dans le mélange.
c.Formation d’un alcool aminé : elle se fait par réduction de la fonction carboxylique en
utilisant le borohydrure de sodium NaBH4 ou le borohydrure de lithium LiBH4, elle aboutit à
la formation d’un alcool α-aminé.
d.Formation d’amide : cette formation est à la base de la liaison peptidique.
e.Décarboxylation: La fonction carboxylique (du carbone α) peut faire l’objet d’une réaction
de décarboxylation conduisant à la formation d’amine, que l’on qualifie de biogène lorsqu’elle
a un rôle biologique.
Certaines de ces amines sont douées d’activité physiologique ou pharmacodynamique :
II.3.5.2. Réaction du NH2 :
a. Addition de carbonyle
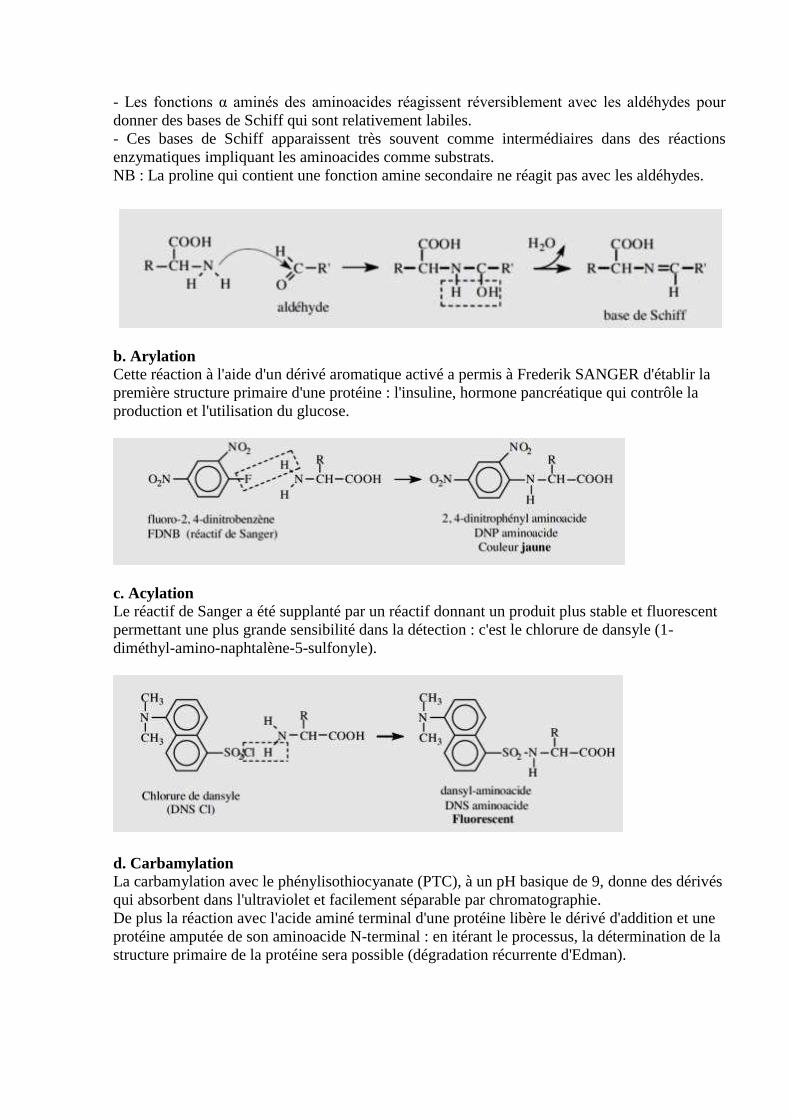
- Les fonctions α aminés des aminoacides réagissent réversiblement avec les aldéhydes pour
donner des bases de Schiff qui sont relativement labiles.
- Ces bases de Schiff apparaissent très souvent comme intermédiaires dans des réactions
enzymatiques impliquant les aminoacides comme substrats.
NB : La proline qui contient une fonction amine secondaire ne réagit pas avec les aldéhydes.
b. Arylation
Cette réaction à l'aide d'un dérivé aromatique activé a permis à Frederik SANGER d'établir la
première structure primaire d'une protéine : l'insuline, hormone pancréatique qui contrôle la
production et l'utilisation du glucose.
c. Acylation
Le réactif de Sanger a été supplanté par un réactif donnant un produit plus stable et fluorescent
permettant une plus grande sensibilité dans la détection : c'est le chlorure de dansyle (1-
diméthyl-amino-naphtalène-5-sulfonyle).
d. Carbamylation
La carbamylation avec le phénylisothiocyanate (PTC), à un pH basique de 9, donne des dérivés
qui absorbent dans l'ultraviolet et facilement séparable par chromatographie.
De plus la réaction avec l'acide aminé terminal d'une protéine libère le dérivé d'addition et une
protéine amputée de son aminoacide N-terminal : en itérant le processus, la détermination de la
structure primaire de la protéine sera possible (dégradation récurrente d'Edman).

Dans le cas d'un peptide de n aminoacides, le PTC-peptide va subir une cyclisation et une
coupure à un pH légèrement acide, libérant un phénylthiohydantoine-aminoacide identifiable
(PTH-aminoacide), qui absorbe dans l'UV, et un peptide de (n-1) aminoacides.
e. Désamination
Pour maintenir la réserve intracellulaire des 20 aminoacides servant à la synthèse protéique, le
métabolisme passera par des désaminations avec oxydation qui produiront des acides α
cétoniques, source principale, sinon la seule, à partir de laquelle les aminoacides sont
synthétisés.
II.3.5.4. Réaction du COOH et NH2 :
- La réaction avec la ninhydrine :
C’est l'une des plus connue et utilisée, elle aboutit à un produit violet pour les amines primaires
et à un autre dérivé de couleur jaune pour les amines secondaires.
L'acide aminé est complètement dégradé par une réaction de désamination et de
décarboxylation.
C’est une réaction qui se déroule en deux étapes :
- La ninhydrine (hydrate de dicéto-hydrindène) est un oxydant puissant qui par désamination
oxydative conduit à l’aldéhyde correspondant avec libération d’ammoniac et de gaz carbonique
et formation de ninhydrine réduite.
- L’ammoniac réagit avec l’hydrindantine et une autre molécule de ninhydrine pour donner un
composé bleu violacé « pourpre de Ruhemann ».
Caractéristiques :
- Réaction colorimétrique sensible.
- Lecture à 570nm.
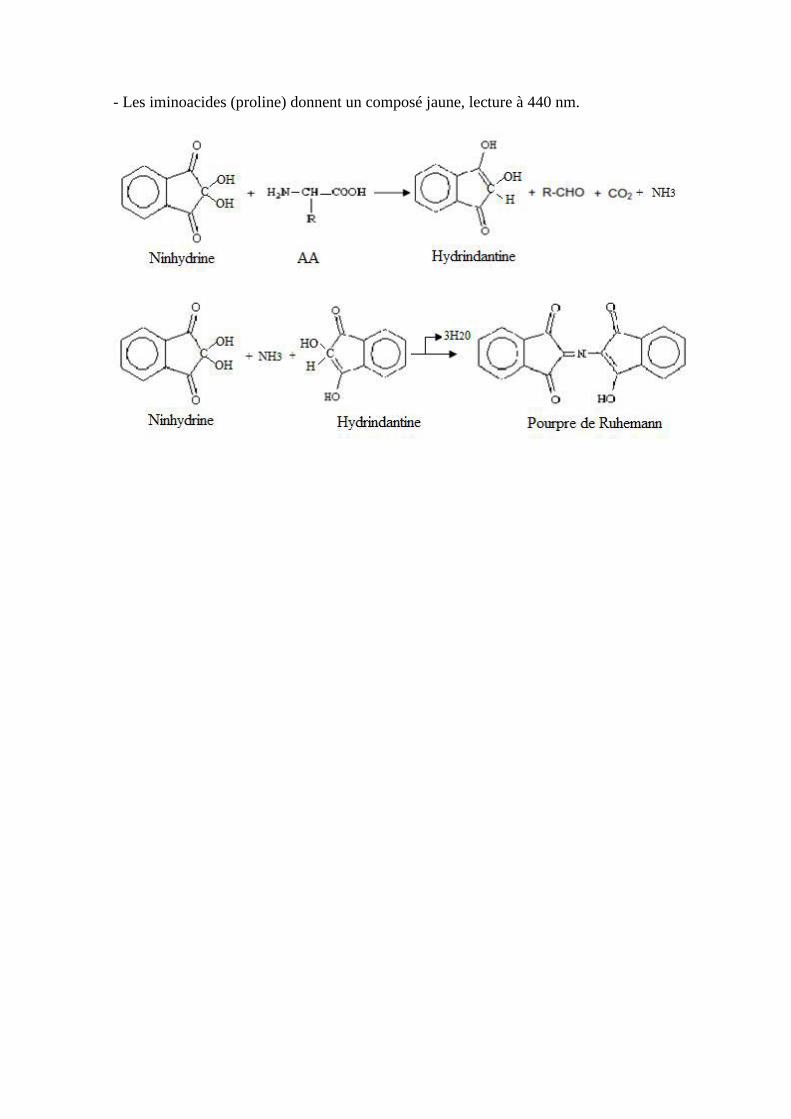
- Les iminoacides (proline) donnent un composé jaune, lecture à 440 nm.

Chapitre III: les peptides
III.1. Structure primaire des peptides et des protéines
III.1.1. Liaison peptidique
Les peptides et les protéines sont le résultat de l’enchainement d’acides aminés reliés entre eux
par une liaison covalente qui est en fait une liaison amide formée par déshydratation entre le
groupement amine d’un acide aminé et le groupement carboxyle d’un autre acide aminé. Cette
liaison est également appelée liaison peptidique, d’où le nom de polypeptides également
donné aux protéines (figure 10).
Figure 10. Liaison peptidique entre deux aminoacides.
On distingue, en générale:
- les oligopeptides: dipeptides (formés par l’union de 2 aminoacides), tripeptides
(3 aminoacides).
- les polypeptides: à partir des tétrapeptides.
- les protéines sont des polypeptides, mais nous verrons un peu plus loin qu’en plus des liaisons
peptidiques unissant les aminoacides, d’autres types d’intéractions interviennent pour conférer
à la protéine une structure tridimensionnelle.
III.1.2. Détermination de la structure des chaînes peptidiques
Pour connaître la séquence des acides aminés il faut d’abord déterminer les acides aminés N- et
C- terminaux, puis l’ordre d’enchaînement des autres acides aminés.
Les tableaux 2 et 3 donnent les principales méthodes utilisées.
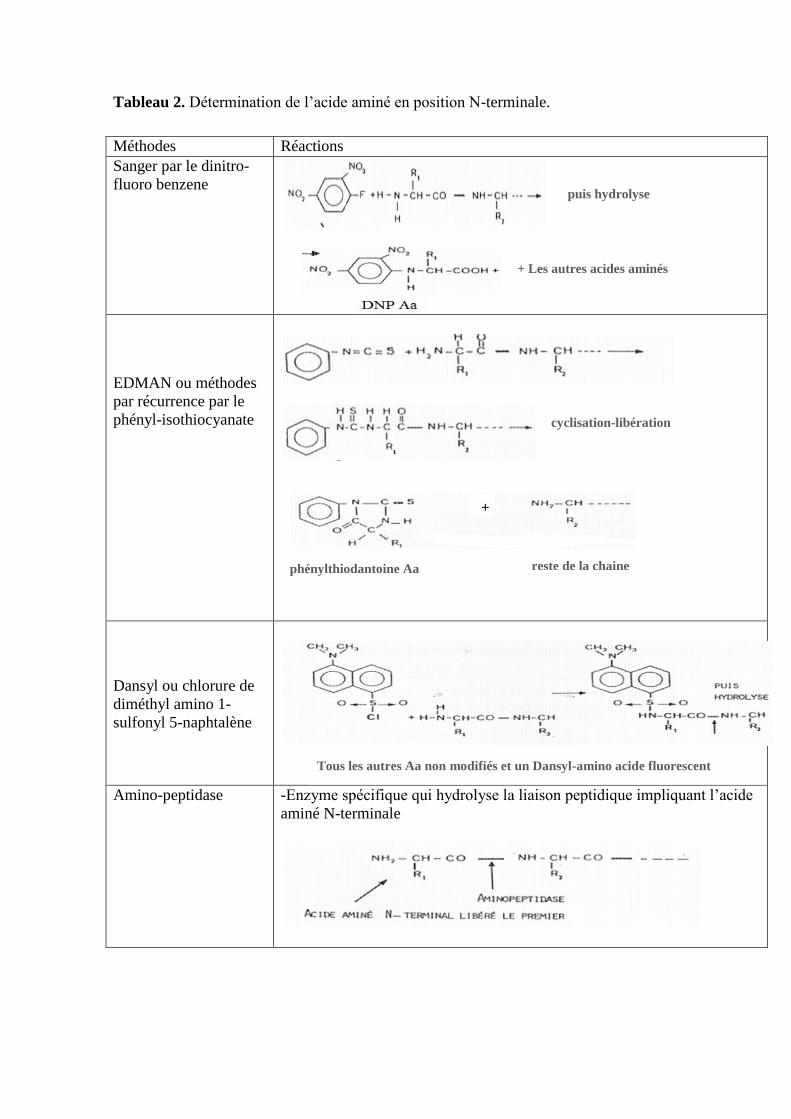
Tableau 2. Détermination de l’acide aminé en position N-terminale.
Méthodes Réactions
Sanger par le dinitro-
fluoro benzene
EDMAN ou méthodes
par récurrence par le
phényl-isothiocyanate
Dansyl ou chlorure de
diméthyl amino 1-
sulfonyl 5-naphtalène
Amino-peptidase -Enzyme spécifique qui hydrolyse la liaison peptidique impliquant l’acide
aminé N-terminale
puis hydrolyse
+ Les autres acides aminés
non modifiés
cyclisation-libération
phénylthiodantoine Aa reste de la chaine
+
Tous les autres Aa non modifiés et un Dansyl-amino acide fluorescent
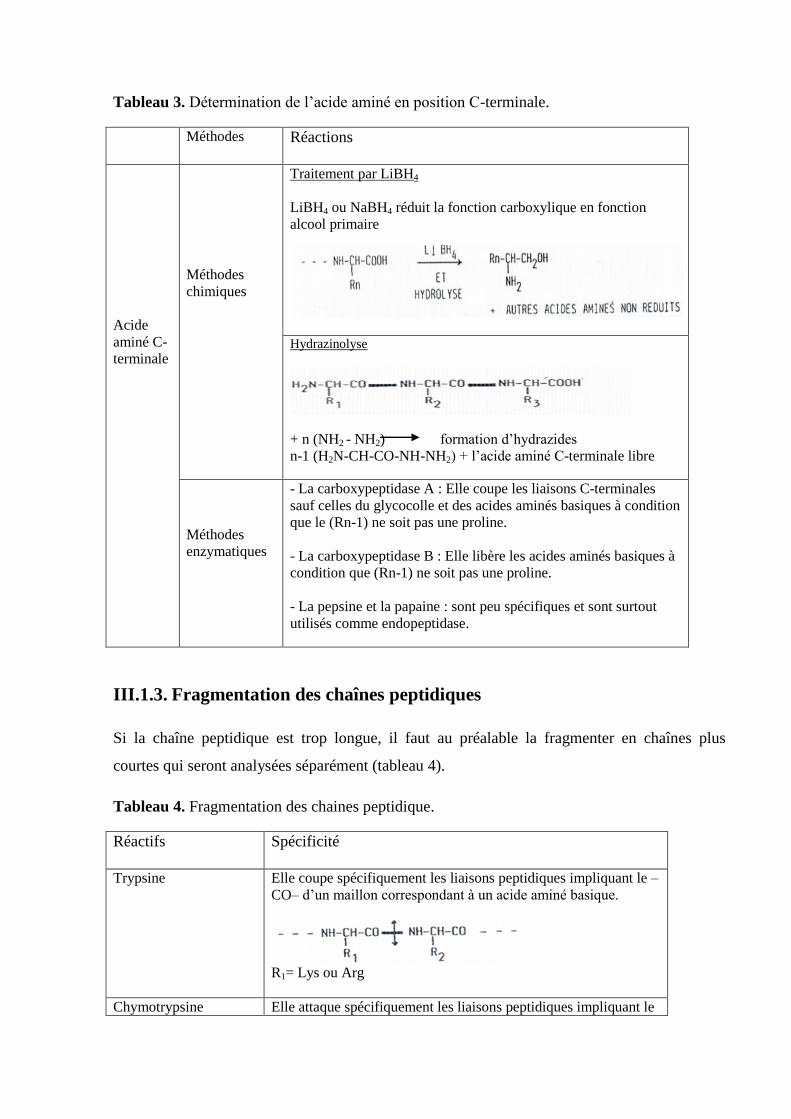
Tableau 3. Détermination de l’acide aminé en position C-terminale.
Méthodes Réactions
Acide
aminé C-
terminale
Méthodes
chimiques
Traitement par LiBH4
LiBH4 ou NaBH4 réduit la fonction carboxylique en fonction
alcool primaire
Hydrazinolyse
+ n (NH2 - NH2) formation d’hydrazides
n-1 (H2N-CH-CO-NH-NH2) + l’acide aminé C-terminale libre
Méthodes
enzymatiques
- La carboxypeptidase A : Elle coupe les liaisons C-terminales
sauf celles du glycocolle et des acides aminés basiques à condition
que le (Rn-1) ne soit pas une proline.
- La carboxypeptidase B : Elle libère les acides aminés basiques à
condition que (Rn-1) ne soit pas une proline.
- La pepsine et la papaine : sont peu spécifiques et sont surtout
utilisés comme endopeptidase.
III.1.3. Fragmentation des chaînes peptidiques
Si la chaîne peptidique est trop longue, il faut au préalable la fragmenter en chaînes plus
courtes qui seront analysées séparément (tableau 4).
Tableau 4. Fragmentation des chaines peptidique.
Réactifs Spécificité
Trypsine Elle coupe spécifiquement les liaisons peptidiques impliquant le –
CO– d’un maillon correspondant à un acide aminé basique.
R1= Lys ou Arg
Chymotrypsine Elle attaque spécifiquement les liaisons peptidiques impliquant le

–CO– d’un maillon correspondant à un acide aminé aromatique.
R1= Phe, Tyr, Trp
Pepsine Spécificité plus faible, elle coupe la liaison peptidique impliquant
le –NH– d’un maillon correspondant à un acide aminé
aromatique.
R2= Phe, Tyr, Trp
Clostripaine
Coupe la liaison peptidique impliquant le –CO– de l’Arg
Protéase
staphylococcique
Coupe la liaison peptidique impliquant le –CO– des résidus
Asp et Glu.
BrCN Coupe la liaison peptidique impliquant le –CO– d’une Met et
transforme le résidu Met en HSL (homosérine lactone).
Hydroxylamine
Coupe la liaison Asn–Gly.
III.1.4. Séparation de plusieurs chaînes peptidiques
Fréquemment les chaînes peptidiques sont unies entre elles par des points disulfures. Les
liaisons disulfures sont scindées par des agents oxydants.
On utilise aussi des agents réducteurs tels que le β mercaptoéthanol.

Pour que le pont disulfure ne se reforme pas, il faut traiter ensuite par un agent d’alkylation
comme l’iodoacétate:
III.2. Quelques peptides ayant une importance biologique
a. Glutathion
C’est un tripeptide (γ-L-glutamyl-L-cystéinyl-glycine) dont on peut voir la structure sur la
figure 11.
Figure 11. Structure du glutathion réduit.
On notera que l’acide glutamique est lié à la cystéine par son γ-COOH (liaison peptidoÏde) et
que son α-COOH et son α-NH2 sont donc libres.
Le glutathion existe sous la forme réduite (ou thiol) et sous la forme oxydée (où deux
molécules sont liées par un pont disulfure, comme dans la cystine), ce qui lui permet de jouer
un rôle dans certaines réactions d’oxydoréduction.
b. Hormones peptidiques
Certaines hormones, notamment celles sécrétées par l’hypophyse et le pancréas, sont des
peptides. Nous allons en voir quelques exemples.
Hormones de la post-hypophyse : ocytocine et vasopressine
Ce sont deux peptides comportant un cycle, en raison d’un pont disulfure, et une chaîne
latérale. Comme on peut le constater sur la figure 12, leurs structures sont très voisines ; la
structure de la vasopressine ne diffère de celle de l’ocytocine que par le remplacement de Ile
par Phe, et de Leu par un aminoacide basique (Lys ou Arg) selon les espèces animales). Les
effets physiologiques sont différents : l’ocytocine stimule la contraction du muscle utérin, alors
que la vasopressine augmente la pression sanguine et a une action antidiurétique.

Figure 12. Structure de l’ocytocine et de la vasopressine.
l’insuline, est une hormone hypoglycémiante, sécrétée par le pancréas. Sa masse
moléculaire est de 6 000 environ, qui peut former des polymères par association (12000, 36000,
48000). Elle est considérée à ce titre comme la première protéine dont la structure a été
entièrement déterminée. La molécule est constituée de 51 résidus d’amino-acides groupés en
deux chaînes : la chaîne A de 21 résidus avec un pont disulfure et la chaîne B de 30 résidus. Il
y a trois ponts disulfure : deux ponts interchaînes et un pont intrachaîne (figure 13).
Figure 13. Structure de l’insuline.

Chapitre IV: les protéines
IV.1. Introduction
Constituants fondamentaux des organismes vivants, les protéines sont des polymères formés de
l’enchaînement d’acides aminnés (20 au total, tous de série L) liés par des liaisons covalentes :
les liaisons peptidiques.
Ces protéines sont des molécules de haut poids moléculaire, la plupart sont comprises entre
25 000 D et 150 000 D, certaines possèdent des poids moléculaires plus bas ou beaucoup plus
élevés.
Ces protéines jouent un rôle essentiel dans la cohésion des structures morphologiques et dans le
fonctionnement cellulaire. On citera pour mémoire, quelques grands groupes de protéines :
- Les enzymes (catalyseurs biologiques, responsables de la plupart des réactions chimiques de
la cellule).
- Les anticorps (responsables de la défense des organismes supérieurs, ils forment, dans le sang,
des complexes avec les corps étrangers).
- Les protéines de stockage.
- Les protéines de transport.
- Les hormones (certaines hormones sont de nature protéiques).
- Les histones (liées à l’ADN, elles participent au contrôle de l’expression génétiques).
- Les protéines de structure et de soutien.
En fonction de leur composition, on distingue :
- Les holoprotéines : contenant uniquement des acides aminés.
- Les hétéroprotéines : formées d’une chaîne polypeptidique associée à un groupement
prosthétique, (composé non protéique : lipide, acide nucléique, glucide ect…).
Suivant leur structure on distingue :
- Les protéines fibreuses : de forme allongée, peu solubles, très résistantes, elles entrent dans la
composition des tissus de soutien.
- Les protéines globulaires : de forme compacte, solubles, elles jouent un rôle dynamique dans
la cellule.

Une protéine peut être formée d’une seule chaîne polypeptidique (monomère) ou de plusieurs
chaines polypeptidiques (polymère : dimère, trimère, tétramère,…).
Quelques exemples de protéines :
- Le lysozyme (blanc d’œuf de poule), holoprotéine, globulaire, de PM 14600, monomère,
enzyme responsable de l’hydrolyse des mucopolyssacharides de la paroi cellulaire.
- L’hémoglobine (sang humain), hétéroprotéine (chromoprotéine à groupement héminique
porphyrinique), globulaire, de PM : 500, responsable du transport de l’oxygène dans le sang
des vertébrés.
- La myosine (muscle humain), holoprotéine, fibreuse, de PM : 468000, dimère, la myosine
constitue les filaments épais, stationnaires dans les myofibrilles
- Le collagène : protéine fibreuse, insoluble, très abondante dans tous les organes, chez tous les
mammifères. D’une longueur de 300nm, elle est formée de trois chaînes enroulées en triple
hélice. C’est une holoprotéine riche en glycine (33%) et hydroxyproline.
IV.2. Structure des protéines
On définit quatre niveaux d’organisation : primaire, secondaire, tertiaire et quaternaire (dans le
cas de protéines à plusieurs sous-unités).
IV.2.1. Structure primaire
L’enchainement successif des acides aminés reliés entre eux par une liaison peptidique
constitue la structure primaire ou séquence de la protéine. Dans cette structure chaque acide
aminé prend le nom de résidu. Une séquence en acides aminés est toujours écrite en partant du
résidu N-terminal. Pour décrire la séquence des acides aminés, le suffixe « -yl » est ajouté à
tous les résidus, sauf au C-terminal.

IV.2.2. Structure secondaire
C’est l’organisation de la chaîne polypeptidique dans l’espace par intervention des liaisons
Hydrogène entre éléments constitutifs proches. Cette structure due à la répétition d’un motif
structural de base.
On distingue en générale deux types principaux de structure secondaire : l’état étiré (feuillets
plissés β) et l’état hélicoïdal (hélice α).
a. ETAT étiré ou structure en feuillets plissés β
Les protéines fibreuses possèdent ce type de conformation, schématisé à la figure 14. On voit
deux chaînes polypeptidiques antiparallèles, unies par des liaisons hydrogène interchaînes. Les
atomes de la liaison peptidique sont situés dans un même plan, mais les carbones α
appartiennent simultanément à deux plans différents.
Figure 14. État étiré ou structure en feuillets plissés.
b. Etat hélicoïdal ou hélice α
L’hélice α est représentée à la figure 15. On voit que la chaîne peptidique est maintenue dans
cette configuration hélicoïdale grâce à des liaisons hydrogène intrachaîne. L’hélice comporte
3,7 résidus d’aminoacide par tour de spire. Les liaisons peptidiques forment entre eux un angle
de 80° environ. Les chaines latérales sont dirigées vers l’extérieur et peuvent réagir entre elles
ou avec le milieu.

Figure 15. État hélicoΪdal ou hélice α.
IV.2.3. Structure tertiaire
C’est une structure tridimensionnelle compacte due au repliement et à l’enroulement de la
chaîne polypeptidique sur elle-même par suite d’interactions ioniques, de liaisons hydrogène,
hydrophobes ou covalentes (ponts disulfures). Les chaînes latérales polaires des acides aminés
se trouveront à la surface et hydratées, alors que les chaînes latérales non polaires
(hydrophobes) seront dirigées vers l’intérieur, protégées du contact de l’eau. La figure 16
schématise la structure tridimensionnelle de la myoglobine d’après des études effectuées par
diffraction aux rayons X.
Figure 16. Structure tertiaire de la myoglobine.

IV.2.4. Structure quaternaire (dans le cas de protéines formées de plusieurs sous-
unités):
C’est l’association de plusieurs chaînes polypeptidiques stabilisées par des liaisons de faible
énergie (liaisons électrostatique, hydrogène ou hydrophobes) ou plus rarement des liaisons
covalentes (ponts disulfures).
Figure 17. Structure quaternaire de l’hémoglobine, tétramère formée de 2 sous-unités α et de 2
sous-unités β.
IV.3. Dénaturation des protéines
La dénaturation est une désorganisation de la structure interne (structures secondaire, tertiaire,
quaternaire) des édifices protéique sans rupture de liaison peptidique, ce qui la différencie de
l’hydrolyse.
Des facteurs physiques ou chimiques qui influencent ces interactions peuvent faire évoluer
une protéine d’un état « natif » fonctionnel vers un état « dénaturé » non fonctionnel.
De nombreux paramètres peuvent entrer en jeu, mais trois facteurs sont particulièrement
importants : la température, le pH, et la présence éventuelle d’agents dénaturants.
- la chaleur entraîne une perturbation des liaisons hydrogène et provoque la coagulation des
protéines (c’est le cas de l’ovalbumine lors de la cuisson bu blanc d’œuf). La plupart des
protéines sont dénaturées vers 45°C.
- un pH très acide ou très alcalin dénature les protéines par perturbation des liaisons ioniques.
L’acidité gastrique permet une dénaturation des protéines alimentaires, ce qui facilite leur
digestion par la pepsine.
- Les agents chaotropiques comme l’urée ou le chlorure de guanidinium. Utilisés à des
concentrations élevées (6 à 8 mol.L–1
), ils désorganisent la structure de l’eau, disloquent les
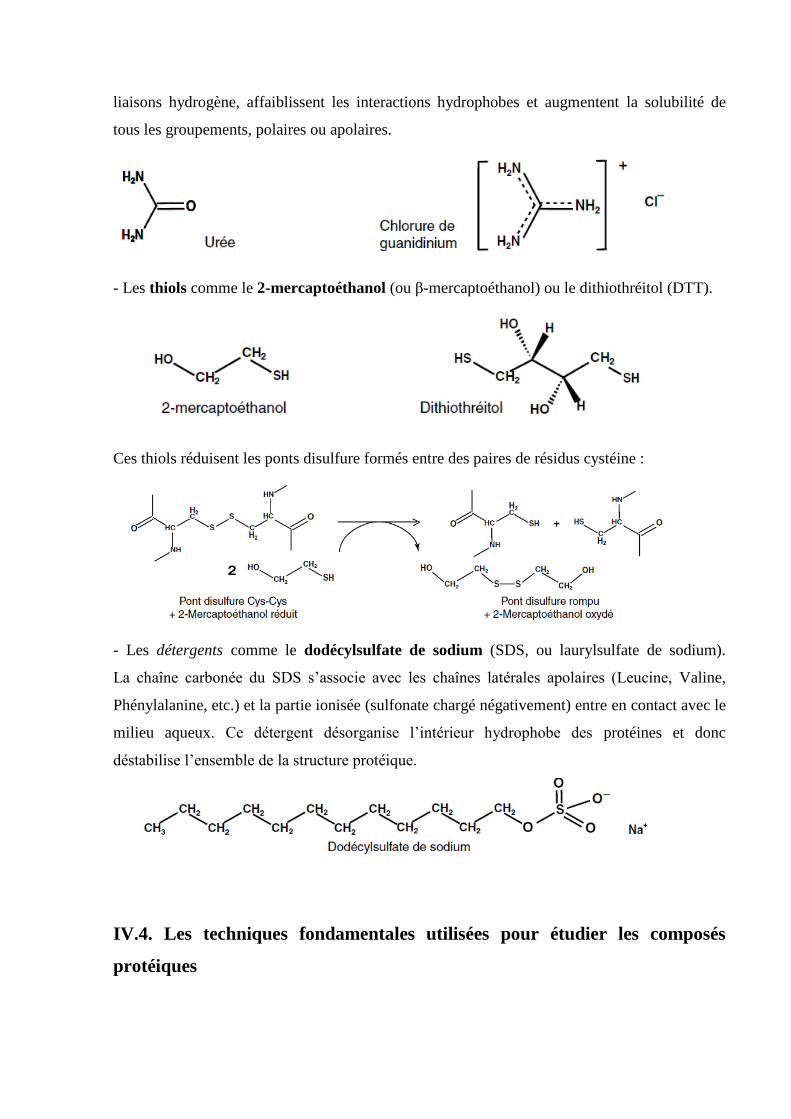
liaisons hydrogène, affaiblissent les interactions hydrophobes et augmentent la solubilité de
tous les groupements, polaires ou apolaires.
- Les thiols comme le 2-mercaptoéthanol (ou β-mercaptoéthanol) ou le dithiothréitol (DTT).
Ces thiols réduisent les ponts disulfure formés entre des paires de résidus cystéine :
- Les détergents comme le dodécylsulfate de sodium (SDS, ou laurylsulfate de sodium).
La chaîne carbonée du SDS s’associe avec les chaînes latérales apolaires (Leucine, Valine,
Phénylalanine, etc.) et la partie ionisée (sulfonate chargé négativement) entre en contact avec le
milieu aqueux. Ce détergent désorganise l’intérieur hydrophobe des protéines et donc
déstabilise l’ensemble de la structure protéique.
IV.4. Les techniques fondamentales utilisées pour étudier les composés
protéiques

IV.4.1. Chromatographie
Le terme chromatographie désigne un ensemble de techniques de séparation qui reposent sur
l’affinité différentielle des molécules d’un mélange pour une phase stationnaire ou matrice,
solide et une phase mobile, liquide ou gazeuse.
Dans le cas des protéines, le mélange est déposé au sommet de la phase stationnaire contenue
dans une colonne. La phase mobile, un éluant, traverse la phase stationnaire et entraîne
progressivement les molécules vers le bas de la colonne où l’effluent est collecté en différentes
fractions qui sont ensuite analysées.
Selon le type de matrice, les molécules de protéines sont retenues et retardées par la colonne en
fonction de paramètres physico-chimiques variés : taille, charge électrique, hydrophobicité de
la surface ou affinité pour des ligands déterminés.
IV.4.1.1. Chromatographie par filtration sur gel
Cette technique chromatographique est également nommée chromatographie par exclusion ou
chromatographie par tamisage moléculaire. Dans cette technique, les molécules sont séparées
selon leur taille et leur forme. L’échantillon est déposé au sommet d’une colonne de gel
constitué de billes de porosité définie. Ces billes sont constituées de dextran (polymère de
glucose), d’agarose (polymère de dérivés du galactose) ou de polyacrylamide.
Les molécules de petite taille (petites protéines et polypeptides) pénètrent dans les billes et sont
donc retardées, alors que les molécules de taille supérieure à celle des pores ne pénètrent pas
dans les billes et traversent donc plus rapidement la colonne (figure 18).
IV.4.1.2. Chromatographie par échange d’ions
Dans cette chromatographie, la matrice (cellulose, polystyrène, agarose, dextran) contient des
groupes ionisés qui fixent des ions de charge opposée (A+, B
–) présents dans la solution ; ces
ions pourront être échangés avec des protéines. On distingue les échangeurs d’anions (matrice
R chargée positivement) et les échangeurs de cations (matrice chargée négativement) :
Échangeurs d’anions : R+B
– + Prot
– R
+Prot
– + B
–
Échangeurs de cations : R–A
+ + Prot
+ R
–Prot
+ + A
+
Les protéines se fixent à la matrice en fonction de leur affinité pour les groupes ionisés.
La colonne est ensuite lavée avec un tampon (tampon d’élution), qui entraine en premier lieu
les protéines les plus faiblement liées à l’échangeur. Les protéines les plus fortement liées

seront libérées en dernier ou resteront fixées sur la matrice. On réussit en général à libérer
l’ensemble des protéines fixées en faisant varier le pH du tampon d’élution ou en augmentant
progressivement sa force ionique (figure 18).
IV.4.1.3. Chromatographie par interactions hydrophobes
Cette méthode met à profit l’existence de zones hydrophobes discrètes à la surface des
protéines. La matrice contenue dans la colonne contient des groupes apolaires (groupes phényl)
qui peuvent retenir les protéines en s’associant à leurs régions hydrophobes.
Cette interaction est d’autant plus forte que la force ionique est élevée. Aussi, et à l’inverse de
la chromatographie d’échange d’ions, la colonne hydrophobe est éluée par un tampon de force
ionique décroissante, ce qui a pour effet d’affaiblir progressivement l’interaction hydrophobe
entre les protéines et la matrice.
En principe, les protéines contenant peu de zones hydrophobes à leur périphérie sont éluées en
premier, les protéines les plus hydrophobes sont éluées en dernier (figure 18).
IV.4.1.4. Chromatographie par affinité
Dans cette technique, les protéines sont séparées en fonction de leur affinité pour un
groupement (analogue de substrat, anticorps spécifique, lectine, etc.) greffé sur la matrice. La
protéine d’intérêt est éluée de la colonne par déplacement avec un analogue de la structure ou
molécule reconnue par le groupement lié (figure 18).
Figure 18. Différent type de chromatographie. (A) Chromatographie par filtration sur gel. Les protéines (disques roses) de taille supérieure au diamètre des
pores ne sont pas retardées par la matrice (disques gris).
(B) Chromatographie par échange d’ions. Les protéines (disques roses) chargées positivement sont retenues par
la matrice (disques gris) chargée négativement.
(C) Chromatographie par interactions hydrophobes. Les protéines (disques roses) les plus hydrophobes sont
retenues par la matrice (disques gris), elle-même hydrophobe.
(D) Chromatographie par affinité. Les protéines (disques roses) les plus affines sont retenues par la matrice
(disques gris) sur laquelle est greffé le ligand spécifique de la protéine d’intérêt.

IV.4.2. Electrophorèse
Les acides aminés en milieu aqueux sont présents sous forme d’espèces ioniques chargées
différemment selon le pH du tampon dans lequel ils sont solubilisés. Si on dépose au milieu
d’une bande de papier un mélange d’acides aminés et si on applique un champ électrique aux
extrémités. Selon leurs charges, au pH du tampon considéré, ils migreront vers le pôle + ou
vers le pôle – (figure 19).
Figure 19. Exemple de l’électrophorèse d’un mélange d’alanine, d’acide aspartique, et de
lysine à pH6.

Chapitre V: Enzymes
V.1. Définition
Les enzymes sont des protéines douées d’activité catalytique spécifique. Elles permettent aux
réactions chimiques nécessaires à la vie et à la multiplication cellulaire de s’effectuer à vitesse
élevée et avec une spécificité qui élimine la formation de sous-produits.
V.2. Classification des enzymes
Le nom de la plupart des enzymes est bâti en ajoutant le suffixe «-ase» au terme qualifiant la
réaction ou encore la nature du substrat (par exemple, la lactate déshydrogénase). D’autres sont
désignées par leur nom usuel (par exemple, la pepsine).
Chaque enzyme est désignée par un numéro donné par la Commission des Enzymes de l’Union
Internationale de la Biochimie Moléculaire.
Ce numéro est précédé les lettres EC et comporte quatre chiffres séparés par des points :
EC (W.X.Y.Z) :
Le 1er
chiffre : indique la classe de l’enzyme, il en existe six.
Le second chiffre : la sous classe, la nature du groupement chimique donneur de groupement,
type de fonction du substrat métabolisé.
Le troisième chiffre : la sous-sous-classe, indique la nature chimique de l’accepteur.
Le quatrième chiffre : numéro d’ordre de l’enzyme (dans la sous sous classe), en relation avec
le substrat de l’enzyme
• Classe 1 : Oxydoréductases
Elles catalysent les réactions d’oxydoréduction, c’est-à-dire le transfert de protons et
d’électrons. C’est le cas des déshydrogénases, des réductases, des oxydases … (par exemple, la
lactate déshydrogénase permet la réduction du pyruvate en lactate ou encore l’oxydation du
lactate en pyruvate).
• Classe 2 : Transférases
Elles catalysent les réactions de transfert d’atome ou de groupement d’atomes. C’est le cas des
transaminases qui transfèrent la fonction amine d’un acide aminé sur un acide α-cétonique.
• Classe 3 : Hydrolases
Elles catalysent des réactions de coupure de liaison covalente nécessitant de l’eau. C’est le cas
de toutes les enzymes digestives comme la trypsine (spécialisée dans la coupure des liaisons
peptidiques).

• Classe 4 : Lyases
Elles catalysent les réactions lytiques non hydrolytiques et non oxydantes en créant des doubles
liaisons. Dans la réaction inverse, les lyases catalysent l’addition d’un groupement fonctionnel
sur la double liaison d’un substrat. C’est le cas de l’aldolase (qui transforme le fructose 1,6-
biphosphate en deux triosesphosphate, (glycolyse)).
• Classe 5 : Isomérases
Elles catalysent des réactions d’isomérie, c’est-à-dire des remaniements intramoléculaires.
C’est le cas de l’aconitase qui transforme le citrate en isocitrate (Cycle de Krebs)).
• Classe 6 : Ligases
Elles catalysent les réactions de ligation, de condensation, c’est-à-dire la formation de liaisons
covalentes nécessitant de l’énergie chimique, le plus souvent apportée par l’hydrolase d’ATP.
C’est le cas des synthétases comme la glutamine synthétase, qui permet l’amidification de
l’acide glutamique en glutamine (métabolisme azoté)).
Le système de dénomination internationale attribue à chaque enzyme quatre nombres séparés
par un point.
Le premier nombre correspond à la classe, le deuxième (sous-classe) précise le type de
réaction, le troisième et le quatrième précisent la nature du substrat utilisé.
V.3. Site actif des enzymes
La formation du complexe enzyme-substrat est caractérisée par une spécificité et même une
stéréospécificité, qui est due au fait que la molécule de substrat doit avoir plusieurs
groupements fonctionnels dans une configuration spatiale telle qu’ils puissent réagir avec
groupements fonctionnels correspondants de l’enzyme. Ces groupements sont donc les
repliements de la chaîne peptidique qui les rapprochent pour constituer le site actif.
Les liaisons intervenant dans la formation de ce complexe enzyme-substrat permettent donc
l’union des groupements fonctionnels du substrat et des chaînes latérales des aminoacides du
site actif qui peuvent être divisés en deux groupes :
- Ceux qui interviennent dans la reconnaissance spatiale du substrat en formant avec lui des
liaisons non covalentes.

- Ceux qui participent à la transformation chimique du substrat en produit et qu’on appelle
aminoacide catalytique. Ils sont responsables de la réaction enzymatique.
Les autres aminoacides de l’enzyme sont nécessaires soit au maintien de la conformation
tridimensionnelle active de l’enzyme, soit à d’autres fonctions de l’enzyme : par exemple, ils
peuvent faire partie de sites allostrériques impliqués dans la régulation de l’activité
enzymatique, ou être essentiels au positionnement correct de l’enzyme à l’intérieur de la
cellule.
V.4. Notion de cinétique enzymatique
L’association enzyme-substrat (ES) est stabilisée par des liaisons de faible énergie : liaisons
hydrogène, interactions hydrophobes, liaisons ioniques…
Le complexe ES subit un réarrangement interne qui va permettre la transformation du substrat
en produit (P).
K est la constante de vitesse quantifiant la rapidité de passage d’un état à l’autre.
K1 est la constante de vitesse de formation du complexe ES.
K-1 est la constante de vitesse de dissociation du complexe ES.
Kcat est la constante catalytique.
V0 = (d [P] / dt) t=0 = Kcat [ES]
V = K1 [E] [S] = k-1[ES] + Kcat [ES]
K1 [E] [S] = (k-1 + Kcat) [ES] ou Km [ES] = [E] [S] avec Km = k-1 + Kcat / k1
Km s’appelle constant de Michaelis-Menten. Elle s’exprime en molarité (M).
[E] + [ES] = [E]0
En remplaçant [E] par [E]0 - [ES]
Km [ES] = ([E]0 - [ES]) [S] ou (Km + [S]) [ES] = [E]0 [S]
[ES] = [E]0 [S] / Km + [S]
V0 = Kcat [ES] = Kcat [E]0 [S] / [S] + Km

V0 = Vmax = Kcat [E]0
V0 = Vmax [S] / Km + [S] équation de Michaelis-Menten (figure 20).
Figure 20. Variation de la vitesse initiale en fonction de la concentration en substrat, celle de
l’enzyme étant constante.
- Signification de la vitesse maximale Vmax et constante de Michaélis Km
a- Vmax
Correspond à la vitesse initiale quand l’enzyme est saturée par son substrat
Renseigne sur l’efficacité catalytique de l’enzyme Vmax= k2 [Et] =>Vmax= kcat[Et]
kcat : Turn Over Number « TNO », nombre de molécules de substrat transformé en produit par
unité de temps, c’est l’efficacité catalytique de l’enzyme : la fréquence de l’acte catalytique (s-
1).
b- Km
Affinité de l’enzyme pour le substrat, Km est d’autant plus élevée que l’affinité de
l’enzyme pour le substrat est plus faible (inversement proportionnelle à l’affinité).
Concentration initiale de substrat pour laquelle Vi= 1/2 Vmax, donc la moitié des sites actifs
de l’enzyme est occupée par le substrat.
Quand [S] >>> Km
Vi= Vmax= kcat [Et]
Quand [S] <<< Km
Vi= (kcat/ Km) [Et] [S]
On peut écrire l’équation de Michaelis-Menten de la façon suivante :
1/V0 = Km + [S] / Vmax [S] = Km / Vmax [S] + 1/ Vmax (Equation de Lineweaver et Burk) (figure
21).

Figure 21. Représentation de Lineweaver et Burk et détermination des constantes cinétiques.
Cette représentation est plus commode que la précédente pour déterminer les constantes
cinétiques Km et Vmax puisqu’il suffit de lire sur le graphique les valeurs correspondant aux
intersections de la droite avec les axes de coordonnées. En outre, elle permet de distinguer entre
différent types d’inhibiteurs comme nous le verrons plus loin.
V.5. Influence de différents paramètres sur la vitesse initiale
V.5.1. Influence de la température
Pour qu’une réaction enzymatique se produise, il faut d’une part que les molécules (enzyme et
substrat) se rencontrent et qu’elles aient suffisamment d’énergie pour s’activer.
Dans un premier temps, c’est-à-dire quand la température est basse, la vitesse de la réaction
augmente quand la température augmente (cas le plus fréquent sur une gamme de températures
allant de 0°C à 40°C). L’énergie d’activation nécessaire est fournie au système sous forme
d’énergie thermique. Lorsque l’on atteint la Vmax, on parle alors de température optimale
(figure 22).
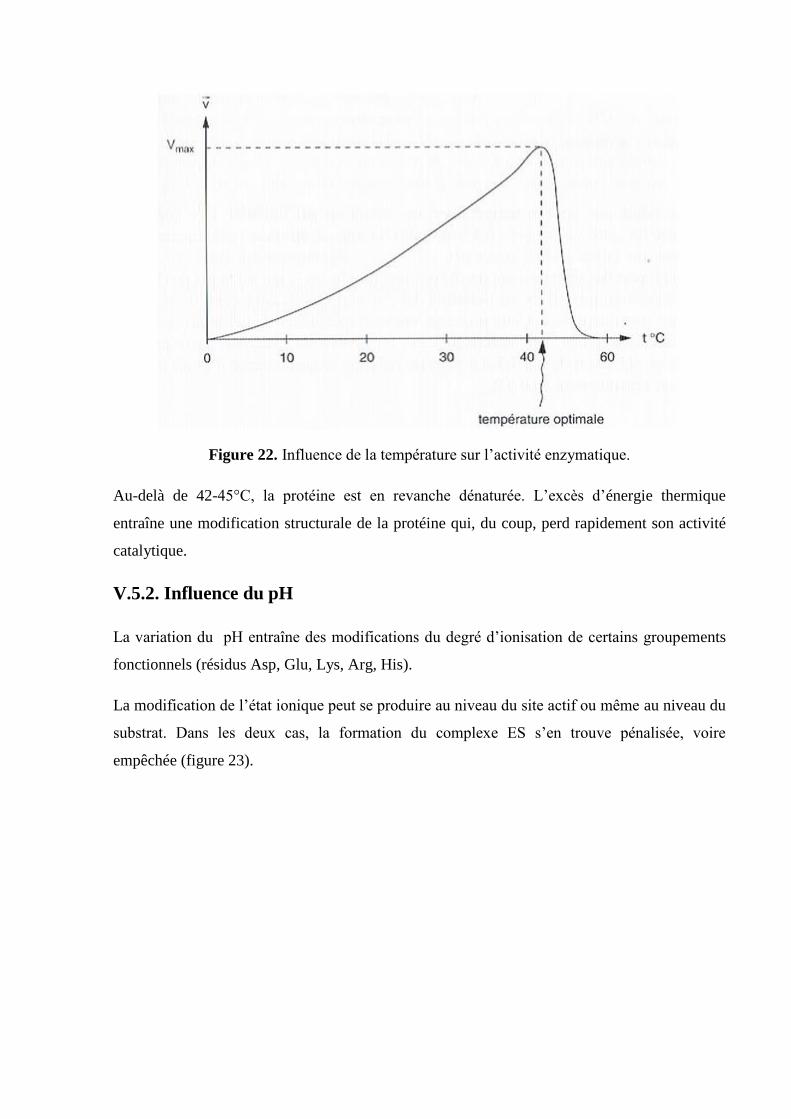
Figure 22. Influence de la température sur l’activité enzymatique.
Au-delà de 42-45°C, la protéine est en revanche dénaturée. L’excès d’énergie thermique
entraîne une modification structurale de la protéine qui, du coup, perd rapidement son activité
catalytique.
V.5.2. Influence du pH
La variation du pH entraîne des modifications du degré d’ionisation de certains groupements
fonctionnels (résidus Asp, Glu, Lys, Arg, His).
La modification de l’état ionique peut se produire au niveau du site actif ou même au niveau du
substrat. Dans les deux cas, la formation du complexe ES s’en trouve pénalisée, voire
empêchée (figure 23).
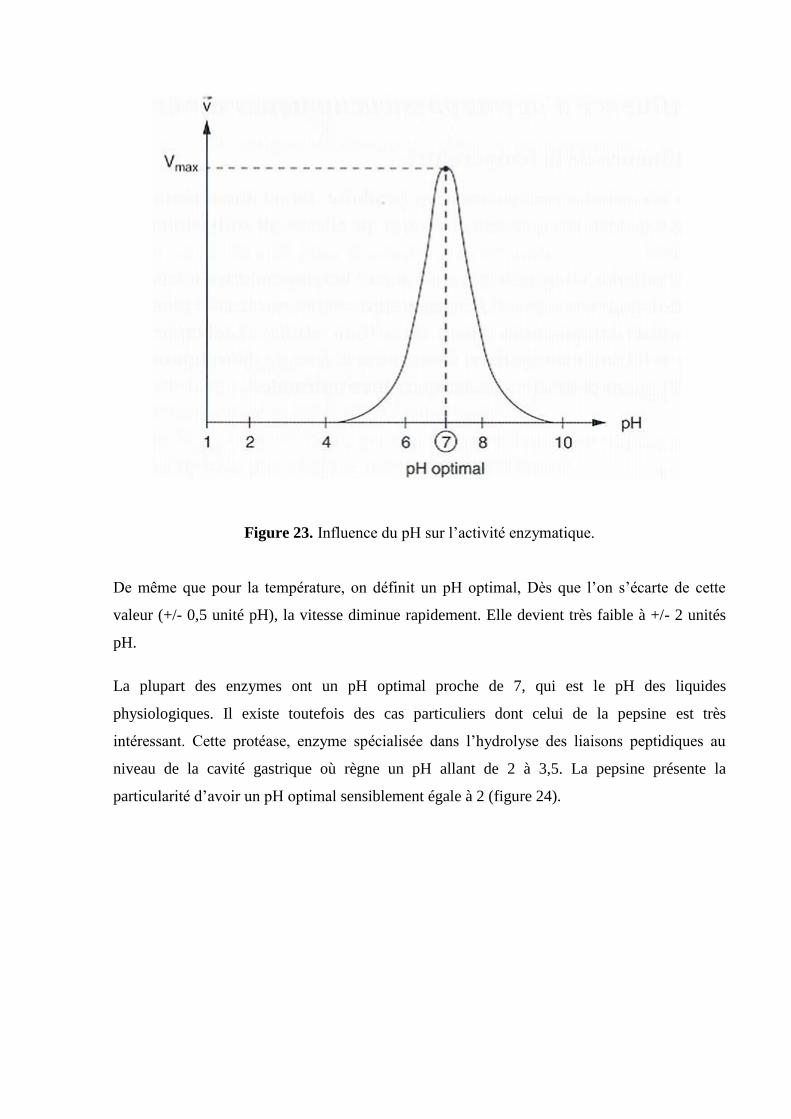
Figure 23. Influence du pH sur l’activité enzymatique.
De même que pour la température, on définit un pH optimal, Dès que l’on s’écarte de cette
valeur (+/- 0,5 unité pH), la vitesse diminue rapidement. Elle devient très faible à +/- 2 unités
pH.
La plupart des enzymes ont un pH optimal proche de 7, qui est le pH des liquides
physiologiques. Il existe toutefois des cas particuliers dont celui de la pepsine est très
intéressant. Cette protéase, enzyme spécialisée dans l’hydrolyse des liaisons peptidiques au
niveau de la cavité gastrique où règne un pH allant de 2 à 3,5. La pepsine présente la
particularité d’avoir un pH optimal sensiblement égale à 2 (figure 24).

Figure 24. Influence du pH sur l’activité enzymatique de la pepsine.
V.6. Inhibiteurs
Un nombre considérable de molécules possédant une action biologique sont des inhibiteurs de
réactions enzymatiques : analogues structuraux de substrats, ou réactifs qui modifient et
inactivent spécifiquement les sites enzymatiques.
V.6.1. Inhibiteurs compétitifs
Ces composés moléculaires présentent une analogie structurale avec le substrat et le site actif
de l’enzyme (figure 25).
Figure 25. Analogie structurale entre l’enzyme et l’inhibiteur compétitif.
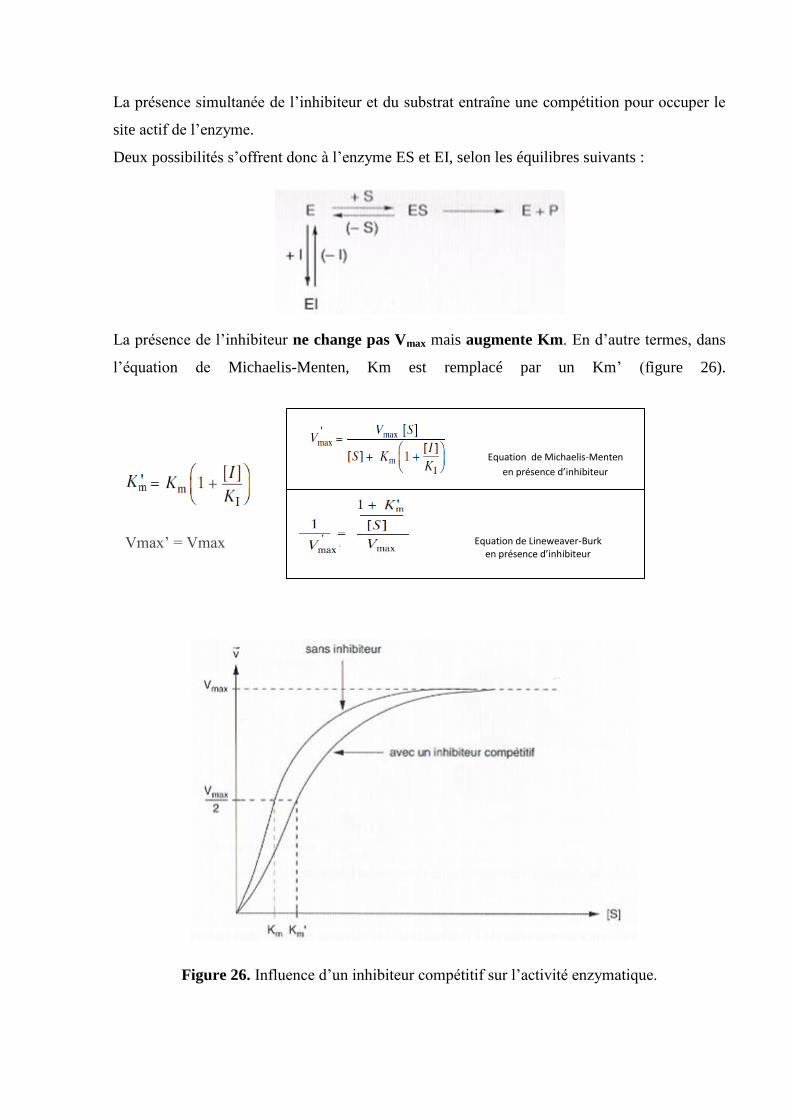
La présence simultanée de l’inhibiteur et du substrat entraîne une compétition pour occuper le
site actif de l’enzyme.
Deux possibilités s’offrent donc à l’enzyme ES et EI, selon les équilibres suivants :
La présence de l’inhibiteur ne change pas Vmax mais augmente Km. En d’autre termes, dans
l’équation de Michaelis-Menten, Km est remplacé par un Km’ (figure 26).
Vmax’ = Vmax
Figure 26. Influence d’un inhibiteur compétitif sur l’activité enzymatique.
Equation de Michaelis-Menten
en présence d’inhibiteur
Equation de Lineweaver-Burk en présence d’inhibiteur

V.6.2. Inhibiteurs non compétitifs
Dans ce cas, la fixation de l’inhibiteur se fait sur l’enzyme au niveau d’un site différent du site
de fixation du substrat. Il n’ya donc aucune compétition entre le substrat et l’inhibiteur pour
l’occupation du site actif (figure 27).
Figure 27. Fixation d’un inhibiteur non compétitif.
La fixation de l’inhibiteur sur l’enzyme ne gêne pas la fixation du substrat sur le site
catalytique. L’affinité du complexe ES n’est donc pas modifiée, Km ne change pas, la Vmax est
alors diminuée (figure 28, 29).
L’équation devient :
Km’ = Km
Equation de Michaelis-Menten
en présence d’inhibiteur
Equation de Lineweaver-Burk en présence d’inhibiteur
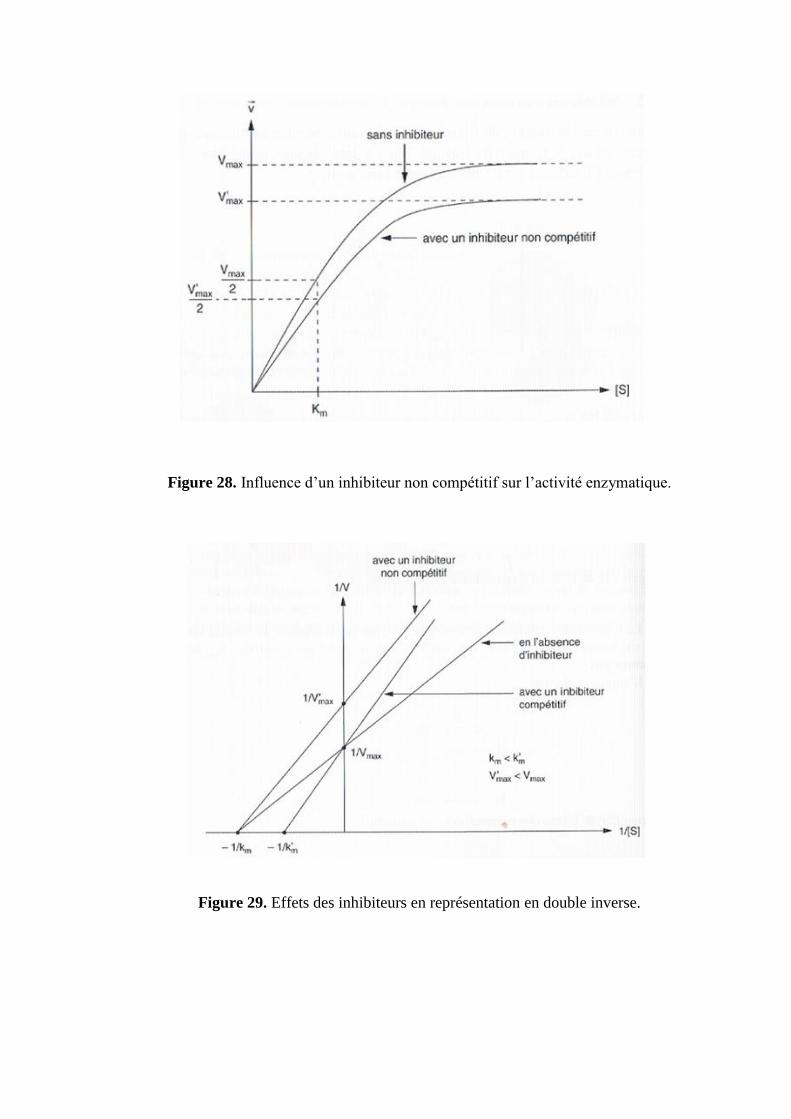
Figure 28. Influence d’un inhibiteur non compétitif sur l’activité enzymatique.
Figure 29. Effets des inhibiteurs en représentation en double inverse.
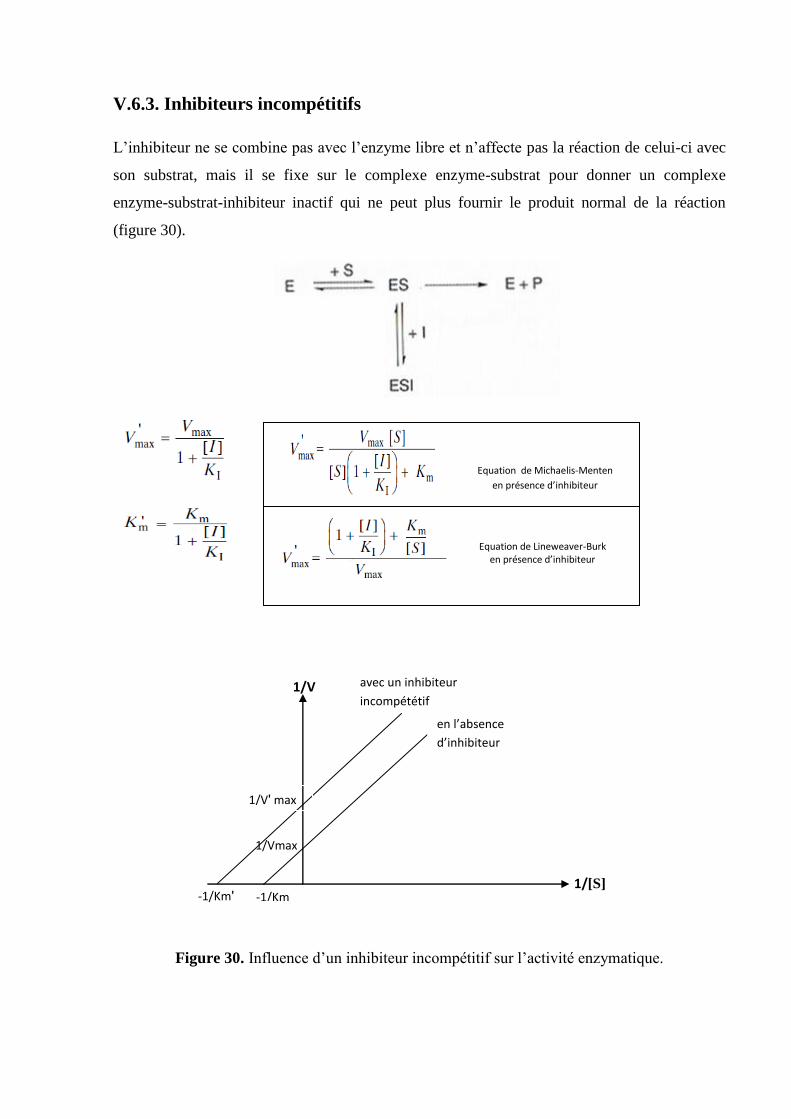
V.6.3. Inhibiteurs incompétitifs
L’inhibiteur ne se combine pas avec l’enzyme libre et n’affecte pas la réaction de celui-ci avec
son substrat, mais il se fixe sur le complexe enzyme-substrat pour donner un complexe
enzyme-substrat-inhibiteur inactif qui ne peut plus fournir le produit normal de la réaction
(figure 30).
Figure 30. Influence d’un inhibiteur incompétitif sur l’activité enzymatique.
Equation de Michaelis-Menten
en présence d’inhibiteur
Equation de Lineweaver-Burk en présence d’inhibiteur
1/[S]
1/V
1/V' max
1/Vmax
-1/Km -1/Km'
en l’absence
d’inhibiteur
avec un inhibiteur
incompététif

V.7. L’unité enzymatique
C’est la quantité d’enzyme qui catalyse la transformation d’une certaine quantité de substrat par
unité de temps. Actuellement il en existe deux :
- L’unité internationale « UI » : la quantité d’enzyme qui catalyse la transformation d’une
micromole de substrat par minute.
- Le katal « kat » : la quantité d’enzyme qui catalyse la transformation d’une mole de substrat
par seconde.
L’activité enzymatique spécifique
Nombre de molécules de substrat transformées par min et par milligramme d’enzyme
Elle permet de vérifier la pureté d’une préparation enzymatique.
L’activité enzymatique moléculaire
Nombre de molécules de substrat transformées par min et par molécule d’enzyme
V.8. Cofacteurs
De nombreuses enzymes ont besoin pour exercer leur activité catalytique d’un cofacteur.
Il existe plusieurs sortes de cofacteurs.
a. Ion métalliques :
Le cation métallique est un oligoélément fourni par l’alimentation à la cellule où fonctionne la
métallo-enzyme.
Un exemple est le cation Zn2+
présent dans le site actif de nombreuses enzymes :
carboxypeptidase, phosphatase alcaline, par exemple. Cet ion est fortement lié à la protéine.
Il participe à la fois à la reconnaissance du substrat et à la catalyse, mais il joue aussi un rôle de

structuration en stabilisant la conformation spatiale efficace du site actif. Il existe un très grand
nombre d’autre métallo-enzymes utilisant divers cations tels que Mg2+
, Mn2+
, Ca2+
, Cu2+
, Fe2+
.
b. Groupement prosthétique ou coenzyme vrais :
Ce sont des molécules organiques de petite taille et de nature non protéique, fortement liées au
site actif de l’enzyme, par des liaisons covalentes. Leur présence est indispensable à
l’expression de l’activité catalytique. Un bon exemple est la porphyrine liée aux cytochromes+.
Coenzymes mobiles ou cosubstrat :
Cette catégorie ne mérite pas vraiment le nom de coenzyme, mais plutôt de cosubstrat, capable
de se fixer réversiblement au site actif de l’enzyme. Un bon exemple est fourni par les dérivés
du nicotinamide, NAD et NADP. Ils permettent le transfert d’hydrogène et d’électrons d’un
substrat, qui sera donc oxydé, à un autre qui sera réduit :
AH2 + NAD+ A + NADH + H
+
NADH + H+
+ B NAD+ + BH2
Les vitamines :
Les vitamines sont des composés organiques que certains organismes sont incapables de
synthétiser et qui doivent donc leur être fournis par l’alimentation, régulièrement mais en
faibles quantités.
Les principaux vitamines hydrosolubles : vitamines du groupe B et vitamines C.

Chapitre VI: les glucides
VI.1. Définition des glucides
Les glucides sont des composés essentiels pour tous les organismes vivants, et sont les
molécules biologiques les plus abondantes.
Le terme hydrate de carbone s’explique par leur formule brute générale Cn(H2O)n, où n ≥3.
Les unités de base des glucides sont appelés oses ou monosaccharides.
VI.2. Importance en Biologie
1. Rôle énergétique : 40 à 50 % des calories apportées par l’alimentation humaine sont des
glucides. Ils ont un rôle de réserve énergétique dans le foie et les muscles (glycogène).
2. Rôle structural : Les glucides interviennent comme éléments de soutien (cellulose), de
protection et de reconnaissance dans la cellule. Ce sont les constituants de molécules
fondamentales : acides nucléiques, coenzymes, vitamines, …
VI.3. Classification des glucides
On distingue les oses et les osides.
Les Oses : Monoses, sucre simple ou encore monosaccharides non hydrolysables.
Les Osides : Hydrolysables, formés par la condensation de deux ou plusieurs molécules d’oses
ou dérivé d’ose et pouvant à leurs tour être divisé en deux grandes classes :
Holosides : Liaison de n molécules d’oses par des liaisons glycosidiques. Elle se deviser on :
Oligosides : sont des polymères de 2 à 10 résidus d'oses, les plus communs étant les
disaccharides.
Polyosides (Polysaccharides) : Ce sont des polymères, formés d’oses liés en longues
chaines linéaires ou ramifiées (plus de 10 molécules d’oses).On distingue :
Hétérosides :
• Ils donnent par hydrolyse: oses + aglycone (partie non sucrée).
• Liaison à des Protéines (glycoprotéines), à des Lipides (glycolipides), à des bases (figure 31).

Figure 31. Classification des glucides
VI.3.1. Les oses
VI.3.1.1. Structure linéaire des oses (Modèle de Fischer)
A. Définition
Les oses simples sont des molécules constituées d’une chaîne carbonée, de 3 à 9 éléments
carbones (les monoses). Les oses principalement impliqués dans les voies métaboliques sont
des oses constitués de 3 à 6 éléments carbones.
Chaque molécule à n éléments carbone contient un (1) groupement carbonyle et (n-1)
groupements hydroxyles. Suivant l’emplacement du groupement carbonyle sur la chaîne
carbonée, on observera une fonction aldéhyde ou une fonction cétone. Dans le premier cas les
molécules seront appelées des aldoses, dans le second cas, des cétoses. Le carbone portant le
groupement carbonyle a toujours le numéro le plus petit, à savoir : (numéro1 pour les Aldose et
numéro 2 pour les cétoses).
Les atomes de carbone d'un ose sont numérotés à partir du carbone le plus oxydé.

B. Classification des Oses
Elle repose à la fois sur deux critères :
Nombre d’atomes de carbone de l’ose (le premier élément ayant 3C)
Nature de la fonction carbonylique ou réductrice.
La combinaison de ces deux critères permet de caractériser un ose (voir tableau ci-après).
C. Configuration absolue : Appartenance à la série D ou L
L’appartenance à la série D ou L pour un ose à n C est déterminé par la configuration du Cn-1
Série D OH du Cn-1 est à droite
Série L OH du Cn-1 est à gauche
Les glucides naturels sont de la série D

VI.3.1.2. Diversité des oses, Isomérie
a. Configuration absolue et isomérie
Tout carbone asymétrique (C*) est définit par sa configuration absolue qui décrit
l'arrangement dans l
'espace des atomes ou groupes fonctionnels auxquels il est lié (ses
substituants).
Pour le glycéraldéhyde, deux configurations absolues sont possibles (1C*). On a deux
molécules différentes de glycéraldéhyde non superposables l'une à l
'autre.
Ce sont deux formes stéréoisomères du glycéraldéhyde. Cette stéréoisomérie est appelée
énantiomérie.
Les deux molécules ont des activités optiques contraires, déviant le plan de polarisation de
la lumière d'une même valeur d
'angle, mais dans les deux directions opposées.
Un mélange équimoléculaire des deux énantiomères d'une même molécule est appelé :
mélange racémique (n'a pas d
'activité optique).
b. Pouvoir rotatoire ou activité optique:
Toute molécule chirale possède la particularité d’être optiquement active ou douée de pouvoir
rotatoire : Traversée par un faisceau de lumière polarisée plan, elle provoque la rotation du
plan de polarisation de la lumière.
La mesure de l'angle de rotation α permet de définir le pouvoir rotatoire spécifique,
caractéristique d'une substance optiquement active, à une température donnée et pour une
longueur d'onde donnée. α = [α]20
D C l

[α] : est le pouvoir rotatoire spécifique de la substance étudiée,
l: est la longueur de la cuve polarimétrique en dm
C : la concentration de la solution étudiée en g/ ml
α ou R: l’angle de rotation
Lorsque la rotation est vers la droite le composé est dit dextrogyre et son pouvoir
rotatoire est positif.
Lorsque la rotation est vers la gauche le composé est dit lévogyre et son pouvoir
rotatoire est négatif.
- Le pouvoir rotatoire d’un mélange de substances est la somme des pouvoirs rotatoires de
chaque substance.
NB :
La série D ou L de Fischer ne préjuge en rien du caractère dextrogyre (+) ou lévogyre
(-) de la molécule.
Ainsi, le D(+) glucose est bien dextrogyre (= +52°), mais le D(-) fructose, lui, est
fortement lévogyre (= -92,4°).
E/ Les stéréo-isomères: sont des composés ayant la même formule brute et développée mais
diffèrent par l’arrangement spatial des groupements OH.
D’une façon générale pour n C* on a 2n stéréo-isomères :
Pour des aldoses a n atomes de carbone on a n-2 C* et donc 2n-2
stéréo-isomères.
Pour les cétoses on a un C* de moins que leurs aldoses isomères, donc pour des cétoses
a n atomes de carbone on a n-3 C* et donc 2n-3
stéréo-isomères.
Exp: aldose n=6 C*=6-2=4 I= 2 4 =16 (8 série D+ 8 série L)
cétose n =6 C*=6-3=3 I= 2 3 =8 (4 série D+ 4série L)
c. Formes d’isomérie :
Les énantiomères : deux isomères qui différant par la configuration absolue de tous
leurs carbones asymétriques et sont images l'un de l'autre dans un miroir.

Les diastéréo-isomères : représentent le cas des isomères qui ont au moins 2 carbones
asymétriques différents.
Les épimères: sont des stéréo-isomères qui diffèrent par la position de leur groupe
hydroxyle au niveau d’un seul carbone asymétrique.
Les isomères de fonctions: deux isomères de fonction, on la même configuration,
même nombre d’atomes de C, ils différent par la fonction carbonyle.

Ex : le D-Glucose et le D-fructose ont la même formule C6H12O6 mais pas la même formule
développée (car ils diffèrent par leur fonction).
VI.3.1.3. Filiation des oses
a. Synthèse cyanhydrique de Killiani-Fisher (HCN)
L’acide cyanhydrique s’additionne sur la fonction aldéhyde pour former un nitrile alcool ou
cyanhydrine. Par hydrolyse, il est possible de passer à l’amide, puis à l’acide et de là par
réduction à l’aldéhyde, c’est- à-dire à un nouvel aldose possédant un atome de carbone de plus,
on obtient 2 stéréoisomère épimère en C2.
Quand on passe d’un ose à l’ose supérieur, un groupe H-C-OH est ajouté après le carbone de la
fonction carbonyle.

b. La dégradation de WOHL-ZEMPLEN
Par condensation avec l’hydroxylamine, la fonction aldéhyde passe à l’état d’oxime de laquelle
on peut faire dériver la cyanhydrine puis, en présence d’oxyde d’argent, un aldose à (n-1)
carbone.
La grande majorité des oses naturels appartient à la série D de Fischer, mais des oses de série L
existent également.
Tout aldose dérive théoriquement d'un glycéraldéhyde par une ou plusieurs étapes d'insertion
d'un chaînon asymétrique H-C-OH, selon le principe dit de la filiation des oses (Figure 32).
Figure 32. Filiation des D-aldoses et D-cétoses.

VI.3.1.4. Structure cyclique des oses
Les oses ne sont pas des structures rigides et rectilignes. La forme linéaire des oses est une représentation simple mais incomplète, elle ne permet pas d'expliquer les propriétés des oses.
a. Diverses objections à la formule linéaire des oses :
- Si le glucose se comporte comme un aldéhyde vrai avec certains réactifs, il n'en est pas de même dans toutes les réactions caractéristiques de la fonction. - Il ne réagit pas non plus de la même manière que les autres aldéhydes avec le méthanol en milieu acide : Les aldéhydes et les cétones sous forme hydratée, réagissent avec 2 molécules d'alcool pour donner des Acétals alors que les oses se combinent seulement avec 1 seule molécule d'alcool pour donner un Hémi acétal.
- Le pouvoir rotatoire d'une solution de D glucose fraîchement préparée diminue pour se stabiliser au bout d'environ 1 heure. - Ce changement (mutarotation) traduit une modification de structure qui ne peut pas être expliquée par la forme linéaire.
- On voit qu’à temps égale à zéro (t=0) la solution a un [α] 20°D = +113°. On déduit qu’elle est
formée: α-D-glucose mais cette valeur démunie et devient égale à [α] 20°D = +18°,7. On déduit
qu’elle est formée que de Β-D glucose, pour arriver avec le temps à une valeur stable qu’est de
+53°.
35% d’α-Dglucopyranose +112°
65% de β-Dglucopyranose +18,7°
0,005% de D-glucose linéaire
En conclusion on sait juste qu’une solution de glucose contient 1/3 de forme α et 2/3 de forme
β plus une très faible quantité de la forme ouverte (0,005 %)
b. Représentation cyclique du glucose
Pour expliquer ces anomalies, Tollens, en 1883 va émettre une hypothèse pour les expliquer et arriver a une représentation cyclique du glucose :
• Un pont oxydique s'établit par formation d'une liaison hémi-acetalique (Hémi- =moitié, vient du grec) interne entre la fonction aldéhyde et une des fonctions alcool du même ose, formant ainsi un cycle.
• Le Cl devient alors un nouveau centre d'asymétrie.
c. Mécanisme de la cyclisation
} +52,7°
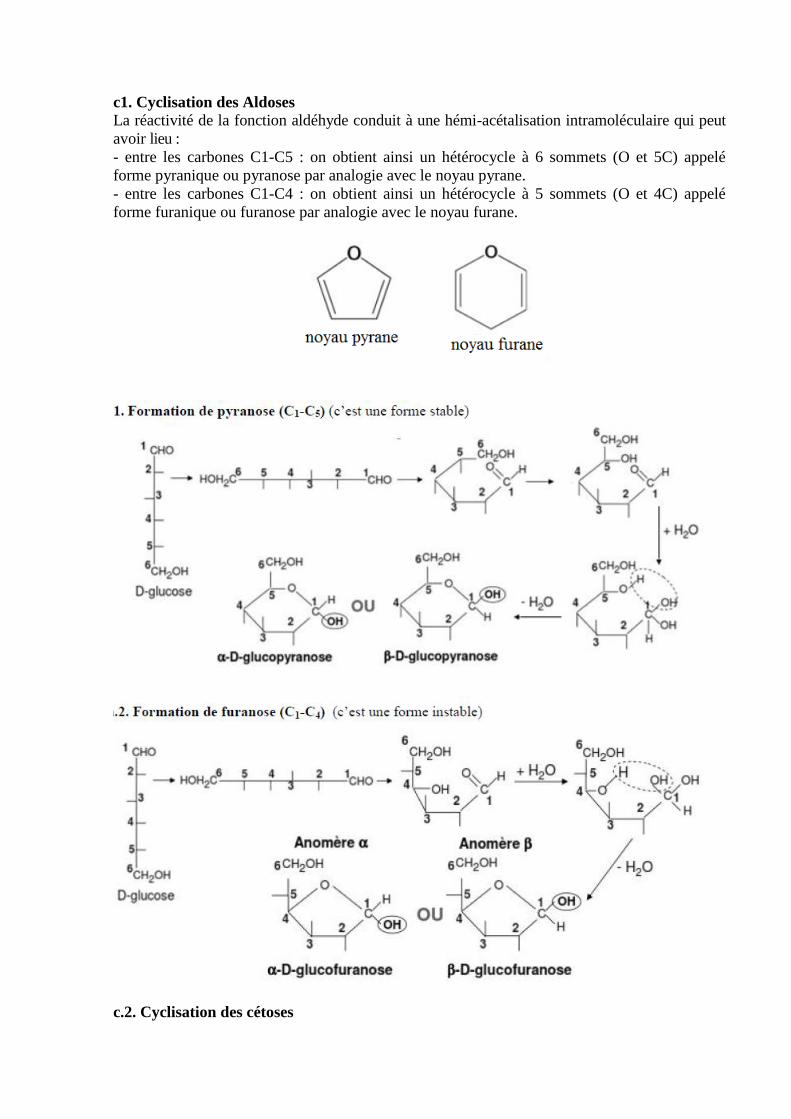
c1. Cyclisation des Aldoses La réactivité de la fonction aldéhyde conduit à une hémi-acétalisation intramoléculaire qui peut
avoir lieu :
- entre les carbones C1-C5 : on obtient ainsi un hétérocycle à 6 sommets (O et 5C) appelé
forme pyranique ou pyranose par analogie avec le noyau pyrane.
- entre les carbones C1-C4 : on obtient ainsi un hétérocycle à 5 sommets (O et 4C) appelé
forme furanique ou furanose par analogie avec le noyau furane.
c.2. Cyclisation des cétoses
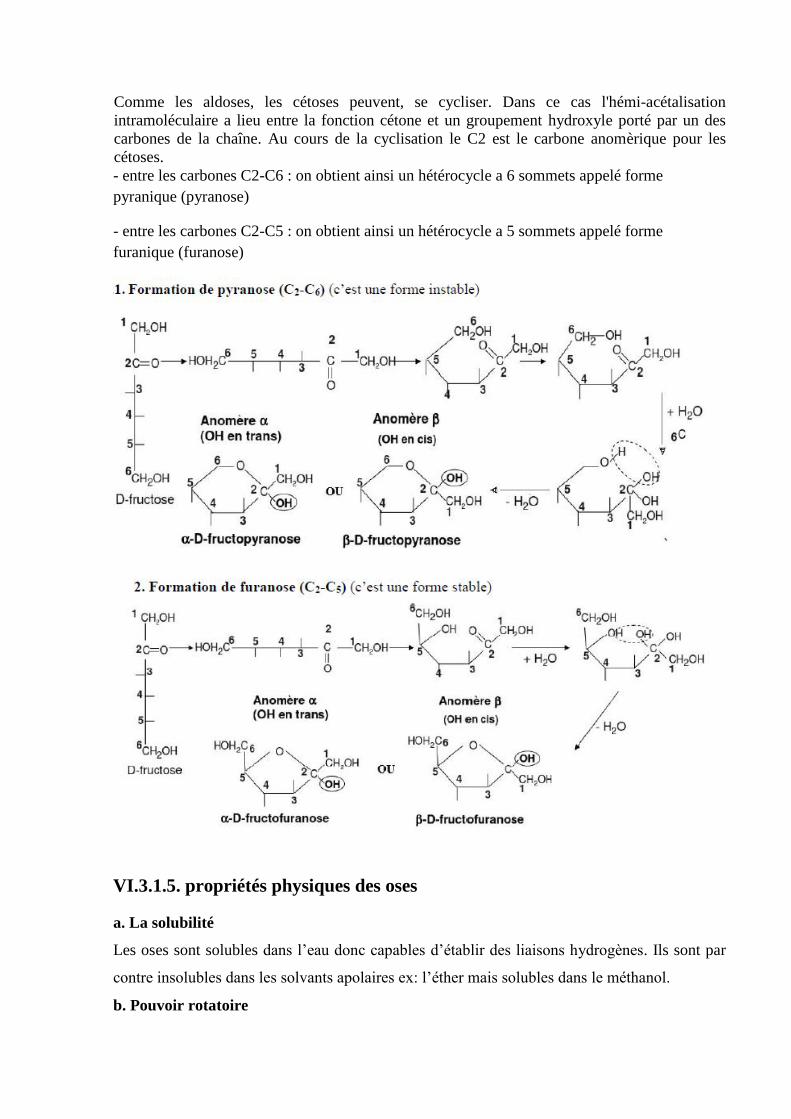
Comme les aldoses, les cétoses peuvent, se cycliser. Dans ce cas l'hémi-acétalisation
intramoléculaire a lieu entre la fonction cétone et un groupement hydroxyle porté par un des
carbones de la chaîne. Au cours de la cyclisation le C2 est le carbone anomèrique pour les
cétoses.
- entre les carbones C2-C6 : on obtient ainsi un hétérocycle a 6 sommets appelé forme
pyranique (pyranose)
- entre les carbones C2-C5 : on obtient ainsi un hétérocycle a 5 sommets appelé forme
furanique (furanose)
VI.3.1.5. propriétés physiques des oses
a. La solubilité
Les oses sont solubles dans l’eau donc capables d’établir des liaisons hydrogènes. Ils sont par
contre insolubles dans les solvants apolaires ex: l’éther mais solubles dans le méthanol.
b. Pouvoir rotatoire

Les molécules qui ont des carbones asymétriques dévient le plan de polarisation d'une lumière
polarisée. Tous les oses (sauf dihydroxy-acétone) ont une activité optique.
c. Spectre d’absorption
Les glucides absorbent peu dans le visible et l’ultraviolet. Ils possèdent par contre un spectre
Infra-Rouge caractéristique.
VI.3.1.6. Propriétés chimiques des oses
a. Propriétés dues à la fonction carbonyle
a.1. Réduction des oses : obtention d’alditols (ositols)
Les aldoses et les cétoses sont irréversiblement réduits en alditols par addition des agents
alcalins : borohydrures alcalins (NaBH4, LiBH4)
Les noms des alditols s'obtiennent en remplaçant le suffixe -ose par le suffixe -itol.
Par exemple le D-glucose donne le D-glucitol (D-sorbitol) et le D-mannose donne le D-
mannitol, etc...
La réduction du D-fructose par NaBH4 donne un mélange équimoléculaire de D-glucitol et de
D-mannitol, alditols épimères en C2.

a.2. Oxydation des oses :
Oxydation douce en milieu alcalin :
Les oxydants doux, comme le brome ou l’iode (Br2, I2) en milieu alcalin, l’acide nitrique très
dilué, oxydent la fonction aldéhydique des aldoses en groupement caboxylique, conduisant à la
formation d’un acide aldonique (R-COOH). Le D-glucose donne ainsi l’acide D-gluconique.
Oxydation par les sels de métaux lourds (Le pouvoir réducteur des aldoses)
Réaction d'oxydation des aldoses par la liqueur de Fehling : à chaud en milieu alcalin, l'oxyde
cuivrique (bleu) est réduit en oxyde cuivreux (rouge brique) insoluble, tandis que l'aldose
s'oxyde en acide aldonique.
Exp : Action de la liqueur de fehling avec les sels cuivriques (à chaud en présence d’un ose
réducteur)
Oxydation forte = oxydation nitrique :
L'oxydation forte d'un aldose conduit à l'attaque simultanée de l'alcool primaire terminal et de
l'aldéhyde. On obtient un di-acide carboxylique appelé acide aldarique.
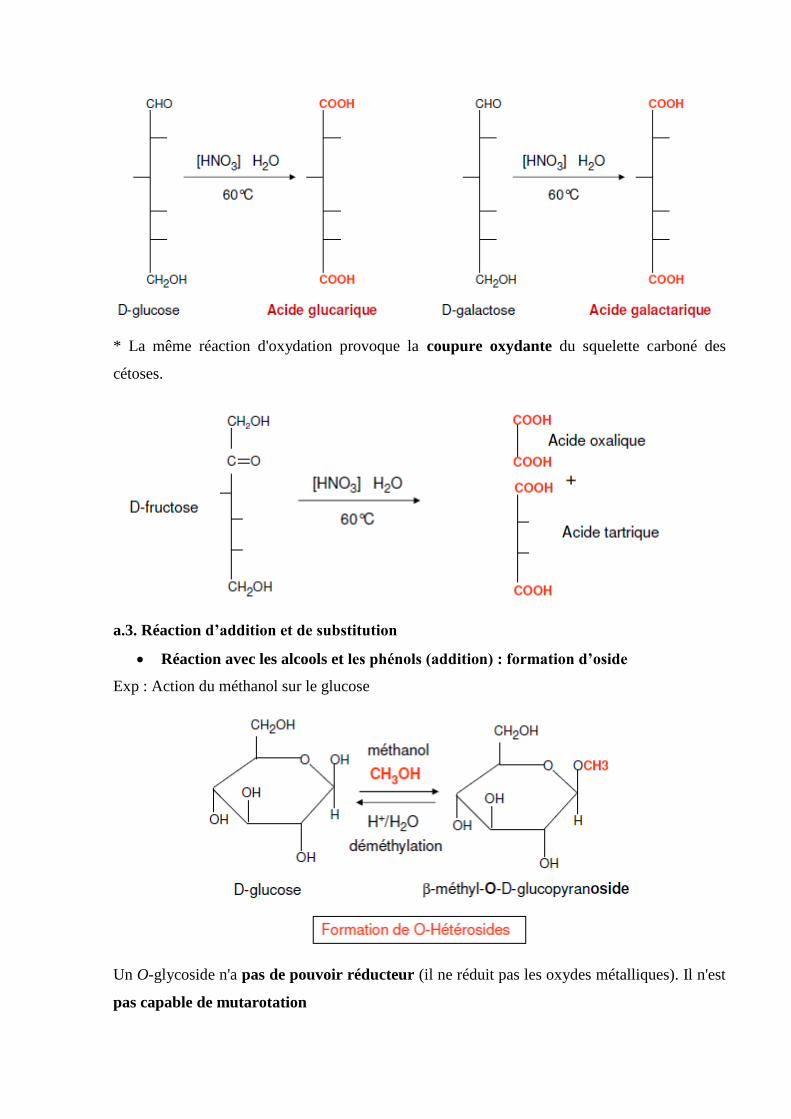
* La même réaction d'oxydation provoque la coupure oxydante du squelette carboné des
cétoses.
a.3. Réaction d’addition et de substitution
Réaction avec les alcools et les phénols (addition) : formation d’oside
Exp : Action du méthanol sur le glucose
Un O-glycoside n'a pas de pouvoir réducteur (il ne réduit pas les oxydes métalliques). Il n'est
pas capable de mutarotation

Action des amines (substitution)
Les aldoses et les cétoses se condensent avec les amines primaires pour donner des imines
cycliques.
Les imines cycliques ou glycosylamines N-substituées, ou encore N-glycosides. Comme les
O-glycosides, les N-glycosides entrent dans la composition de nombreuses molécules
biologiques, dont les plus connues sont les nucléosides et les nucléotides, constitutifs des
acides nucléiques.
Action des thiols (substitution) : Le groupement aldéhydique des aldoses se combine
avec des thiols (R-SH) pour donner naissance à des S-Hétérosides. Le groupement cétonique
des cétoses par contre ne se combinent pas.
b. Propriétés dues à la fonction alcool
b.1. déshydratation en milieu acide
Sous l’action d’un acide concentré et à chaud, les aldoses et les cétoses donnent naissance au
furfural (dans le cas des pentoses), ou un dérivé hydroxyméthylé du furfural (dans le cas des
hexoses).
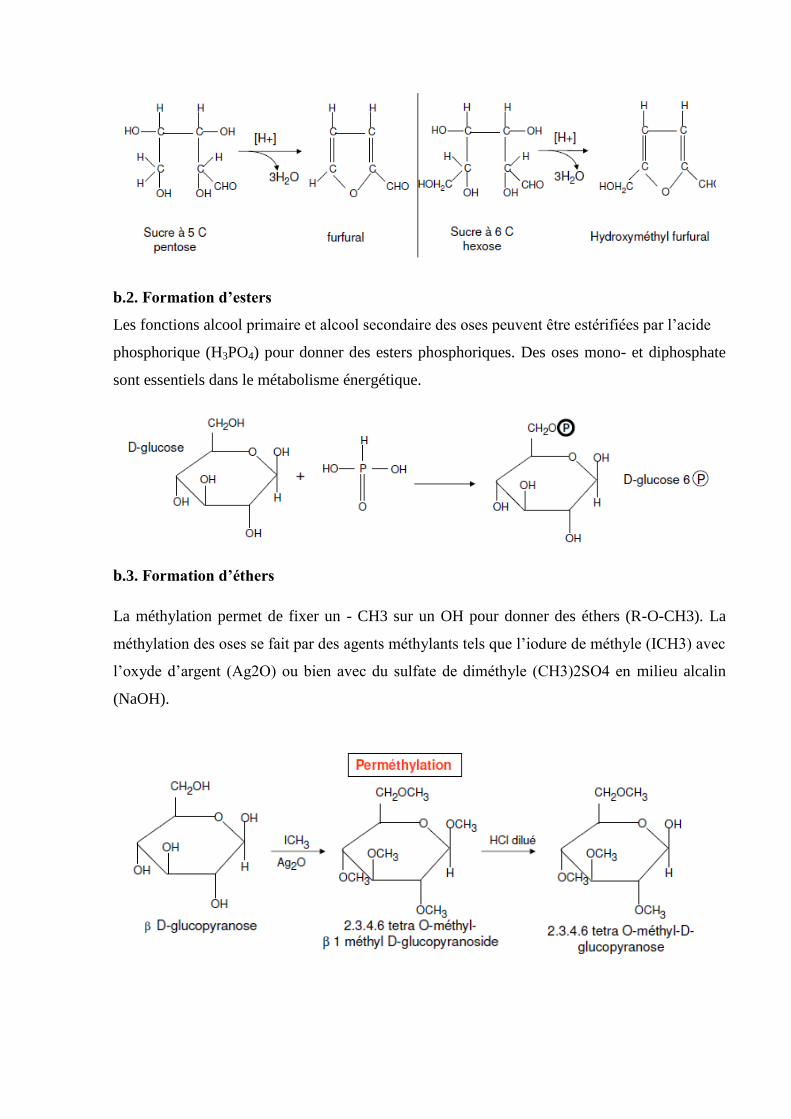
b.2. Formation d’esters
Les fonctions alcool primaire et alcool secondaire des oses peuvent être estérifiées par l’acide
phosphorique (H3PO4) pour donner des esters phosphoriques. Des oses mono- et diphosphate
sont essentiels dans le métabolisme énergétique.
b.3. Formation d’éthers
La méthylation permet de fixer un - CH3 sur un OH pour donner des éthers (R-O-CH3). La
méthylation des oses se fait par des agents méthylants tels que l’iodure de méthyle (ICH3) avec
l’oxyde d’argent (Ag2O) ou bien avec du sulfate de diméthyle (CH3)2SO4 en milieu alcalin
(NaOH).
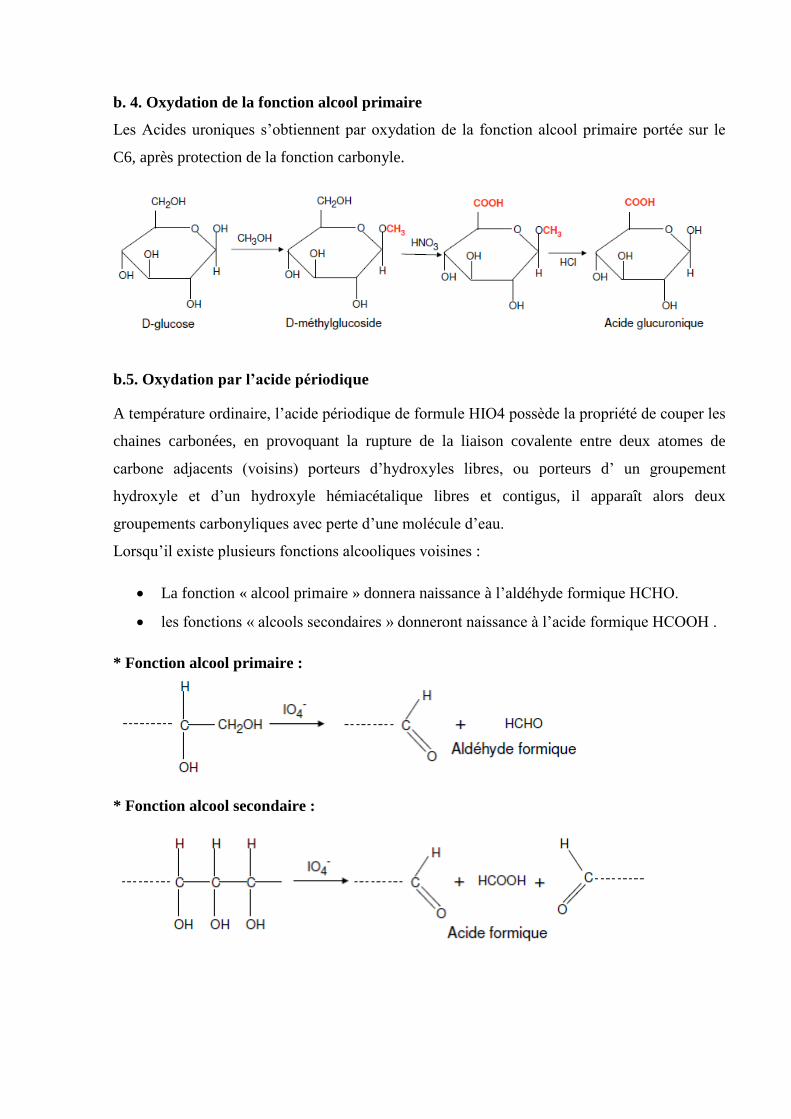
b. 4. Oxydation de la fonction alcool primaire
Les Acides uroniques s’obtiennent par oxydation de la fonction alcool primaire portée sur le
C6, après protection de la fonction carbonyle.
b.5. Oxydation par l’acide périodique
A température ordinaire, l’acide périodique de formule HIO4 possède la propriété de couper les
chaines carbonées, en provoquant la rupture de la liaison covalente entre deux atomes de
carbone adjacents (voisins) porteurs d’hydroxyles libres, ou porteurs d’ un groupement
hydroxyle et d’un hydroxyle hémiacétalique libres et contigus, il apparaît alors deux
groupements carbonyliques avec perte d’une molécule d’eau.
Lorsqu’il existe plusieurs fonctions alcooliques voisines :
La fonction « alcool primaire » donnera naissance à l’aldéhyde formique HCHO.
les fonctions « alcools secondaires » donneront naissance à l’acide formique HCOOH .
* Fonction alcool primaire :
* Fonction alcool secondaire :

VI.4. Osides
VI.4.1. Holosides
V.4.1.1. La liaison glycosidique
Une liaison O-osidique ou O-glycosidique est formée par condensation de l'hydroxyle de la
fonction hémiacétalique porte par le carbone anomérique d'un ose (C1 pour les aldoses, C2 pour les
cétoses), d’un groupement –OH d’une autre molécule.
Il existe (au moins) 20 manières différentes de lier deux aldohexoses A et B en un
disaccharide :
A peut-être lié par son carbone anomérique α ou β à chacune des 4 fonctions alcool de B.
A et B peuvent être liés par leurs carbones anomériques selon 4 combinaisons de configurations
: α-α, α-β, β-β, et β-α.
V.4.1.2. Nomenclature et convention
Un diholoside, donc une liaison osidique, est caractérise:
- par la nature des 2 oses qui le constituent et par leur forme cyclique (pyrane ou furane),
- par la configuration anomérique de la liaison osidique, α ou β.
- par les numéros des atomes de C portant les fonctions impliquées dans la liaison.
Génériquement, le nom de l’oside sera :
(α ou β) X…osyl/osido (1 → n) Y…oside avec n : numéro du C anomérique.

- …oside : signifie que la fonction hemiacetalique du dernier ose est engagé dans la liaison
osidique,
- …ose : signifie que la fonction hemiacetalique de l’ose est libre (ose engage par une fonction
alcool).
VI.4.2. Diholosides
a. Les diholosides réducteurs
a1. Le Maltose
C’est un produit d’hydrolyse obtenu lors de la digestion des polyosides (amidon et
glycogène) par les amylases.
Il est formé par l’union de 2 molécules de glucose unies en α 1 -4. C’est un oside
réducteur.
Il est hydrolysé en 2 molécules de glucose par une enzyme spécifique, la maltase.
a2. Le Lactose
Il est présent dans le lait de tous les mammifères.
C’est un diholoside réducteur constitué d’une molécule de Galactose et d’une molécule de
Glucose unies par une liaison β (1 -4) osidique.
L'intestin de certaines personnes ne sécrète pas la lactase(Enzyme), la substance
responsable de la séparation dans l'intestin du lactose en glucose et galactose. Le lactose
non digéré se retrouve alors dans leur gros intestin où il est fermenté par des bactéries qui y
vivent.

a3. Le cellobiose :
unité de base de la cellulose, présente une liaison osidique β (1-4).
son nom systématique est le β-D-glucopyranosido 1-4 D-glucopyranose.
b. Le diholoside non réducteur
Le Saccharose (sucrose)
C’est un diholoside non réducteur, très répandu dans les végétaux et tout
particulièrement dans la canne à sucre et la betterave. C’est le sucre de table et le moins
cher.
Il est formé par l’union de 2 molécules (glucose + fructose) unies en β 1-2. C’est un
oside non réducteur.
Le saccharose est hydrolysable par voie enzymatique avec une α - glucosidase ou une β-
fructosidase.
VI.4.3. Polyosides
La plupart des glucides se présentent à l’état naturel sous forme de plyosides de haut poids
moléculaire. Le D-glucose en est le constituant majeur.

Les plus représentatifs sont l’amidon dans le règne végétal et le glycogène dans le règne
animal.
a. L’amidon
C’est la réserve glucidique principale du monde végétal, ce qui explique son importance dans
l’alimentation humaine. Les sources essentielles en sont les graines des cereals (blé, maïs
et riz) et certains tubercules (pommes de terre).
L’amidon est composé de deux substances différentes:
- 15 à 30 % d’amylose.
- 70 à 85 % d’amylopectine (ou iso-amylose).
a.1. L’amylose
c’est un polymère à chaîne linéaire résultant de la condensation d’unités de D-glucose par des
liaisons osidiques α(1-4). Le nombre de résidus de D-glucose est compris entre 200 et 3000 par
molécules.
L’hydrolyse de l’amylose par une enzyme, l’amylase, donne naissance à des polymères à
courte chaîne, les dextrines, puis à un diholoside, le maltose. Son action est complétée par une
maltase (ou une hydrolyse acide) qui libère des résidus de D-glucose.
a.2. L’amylopectine
Elles est constituée de chaînes de D-glucose unis par des liaisons α(1-4), ces chaînes étant
elles-mêmes ramifiées par des liaisons α(1-6).
L’hydrolyse de l’amylopectine par les amylases donne donc naissance à du maltose et à de
l’isomaltose. L’hydrolyse acide, ou par une maltase, conduit en définitive à du D-glucose.

b. Glycogène
C’est l’équivalent animal de l’amidon végétal. Le glycogène est la réserve essentielle de
glucose chez les animaux supérieurs et l’élément de base de la contraction musculaire.
Le glycogène résulte de la condensation d’unités D-glucose par des liaisons α(1-4) formant des
chaînes réunies par des liaisons α(1-6).
Le glycogène est très fortement ramifié la molécule de glycogène a une structure arborescente,
comme l’amylopectine, mais l’abondance plus grande des ramifications lui donne un caractère
compact plus marqué.
c. La cellulose
La molécule de cellulose résulte de la condensation exclusivement linéaire de plus de 10 000
unités de D-glucose, unies entre elles par des liaisons osidiques β(1-4).
La cellulose est un composant végétal fondamental, mais elle ne peut être attaquée par les sucs
digestifs de l’homme qui ne contiennent pas les systèmes enzymatiques nécessaires à
l’hydrolyse des liaisons β-osidiques.
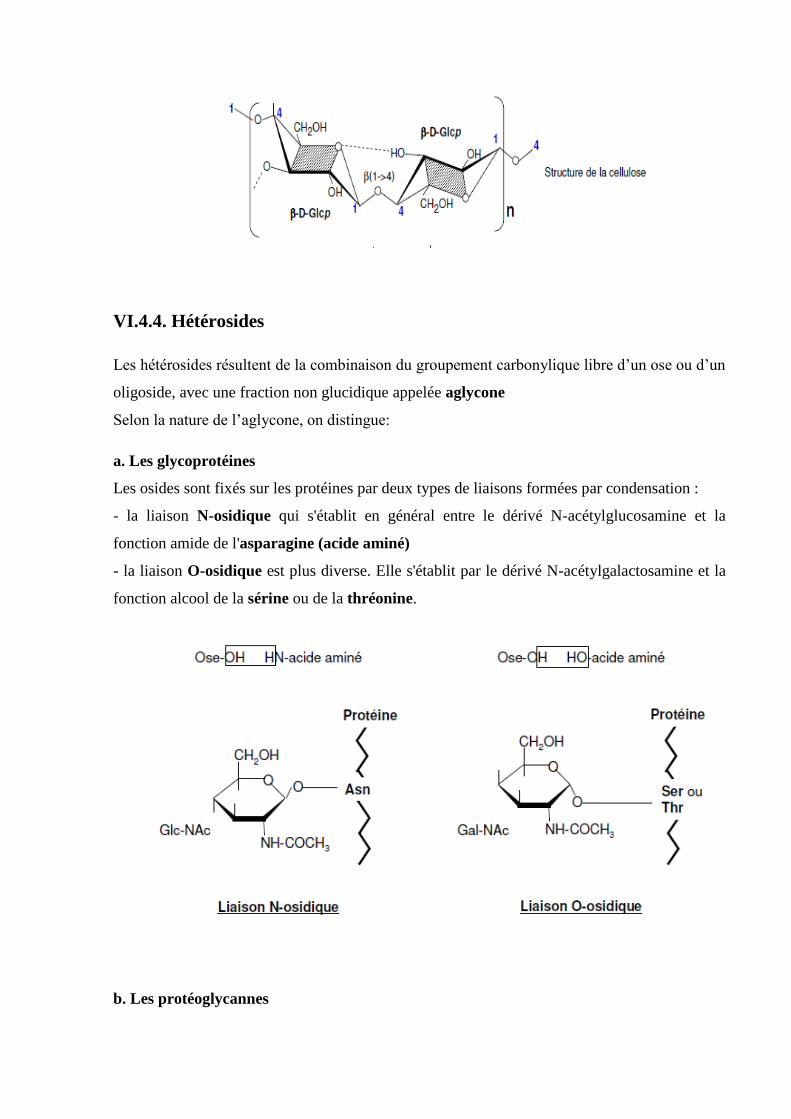
VI.4.4. Hétérosides
Les hétérosides résultent de la combinaison du groupement carbonylique libre d’un ose ou d’un
oligoside, avec une fraction non glucidique appelée aglycone
Selon la nature de l’aglycone, on distingue:
a. Les glycoprotéines
Les osides sont fixés sur les protéines par deux types de liaisons formées par condensation :
- la liaison N-osidique qui s'établit en général entre le dérivé N-acétylglucosamine et la
fonction amide de l'asparagine (acide aminé)
- la liaison O-osidique est plus diverse. Elle s'établit par le dérivé N-acétylgalactosamine et la
fonction alcool de la sérine ou de la thréonine.
b. Les protéoglycannes

Ce sont des molécules en général très volumineuses, composées par l'association covalente de
protéines et de polymères glucidiques appartenant à la famille des glycosaminoglycannes
(GAG).
Les GAG résultent de la polycondensation linéaire d'unités d'osamines et d'acides uroniques qui
peuvent être sulfatés.
La majorité de ces composés se trouvent dans la matrice extracellulaire (tissu conjonctif), dans
les membranes plasmiques et quelques-uns sont intracellulaires.
c. Les peptidoglycannes
polysides reliés par de nombreux petits peptides. Les peptidoglycannes forment la paroi des
bactéries qui leur donne leur forme et les protège.
d. Les Glycolipides: polyosides liés à des lipides.
e. Les protéines glyquées : produits de la fixation chimique d'une unité de glucose.
L'hyperglycémie du diabète insulinique favorise la fixation de cet ose sur les protéines
plasmatiques (marqueur du diabète).

Chapitre VII: Les lipides
VII.1. Définition
Les lipides constituent la matière grasse des êtres vivants. Ils ne correspondent pas à une
catégorie de molécules parfaitement définie chimiquement et forment donc un groupe très
hétérogène de composés. Ces molécules sont caractérisées par leur hydrophobicité, elles ne
sont pas solubles dans les solvants polaires comme l’eau, mais le sont dans les solvants
organiques apolaires tels que l’éther ou le chloroforme.
Les lipides peuvent se présenter à l’état solide, comme dans les cires et les graisses, ou liquide,
comme dans les huiles. Les lipides sont formés par l’union d’une molécule d’alcool avec une
ou plusieurs molécules d’acides gras.
VII.2. Les acides gras
Les acides gras sont des acides carboxyliques R-COOH dont le radical R est une chaîne
aliphatique de type hydrocarbure de longueur variable qui donne à la molécule son caractère
hydrophobe (gras).
La grande majorité des acides gras naturels présentent les caractères communs suivants :
- monocarboxylique
- chaîne linéaire avec un nombre pair de carbones
- saturés ou en partie insaturés avec un nombre de double liaisons maximal de 6
a. Les acides gras saturés : Une série continue d'acides gras de nombre de carbones pair (4 à
plus de 30) a été isolée des lipides de source animale, végétale et microbienne.
Pour les acides gras saturés le nom systématique s'écrit : n- [nC] an oique
- n : indique que l'acide gras est normal (chaîne non branchée)
- [nC] : nombre de carbones - an : indique que la chaîne est saturée
- le symbole est Cn:0 (0 indique que la chaîne est saturée)
- le nom courant rappelle son origine

Tableau 5. Récapitulatif des acides gras naturels:
b. Les acides gras insaturés Ils représentent plus de la moitié des acides gras des plantes et
des animaux, ils possèdent :
- une double liaison : acides monoéniques ou monoinsaturés
- ou plusieurs doubles liaisons : ils sont polyéniques ou polyinsaturés
La plupart des acides gras insaturés ont des longueurs de chaînes de 16 à 20 carbones. En
règle générale :
-la première, ou la seule, double liaison est établie entre les C9 et les C10.
-les doubles liaisons multiples ne sont pas conjuguées mais séparées par un groupe méthylène,
ce qui les place, par exemple, en Δ9, Δ12, Δ15…
-les doubles liaisons sont de configuration cis.

Tableau 6. Récapitulatif des acides gras insaturés:
*EPA : abréviation pour Acide EicosaPentaénoique
b1. La nomenclature
Des dénominations parallèles coexistent : la nomenclature systématique s'efface souvent
devant les noms d'usage. Deux numérotations coexistent, l'une systématique et l'autre utilisée
en diététique qui permet de regrouper les acides gras insaturés en série .Il faut tout d'abord
indiquer le nombre de carbone de l'acide gras, ensuite indiquer le nombre de double liaisons
(Δ), leurs positions et leurs configurations (cis ou trans).
Pour la double liaison entre les carbones C9 et C10 de l'exemple, les chaînes aliphatiques
peuvent avoir deux configurations :

Pour les acides gras insaturés :
- le nom systématique s'écrit : conf-p-[nC] x énoique
- conf-p : configuration et position des doubles liaisons
- [nC] : nombre de carbones
- x : nombre de doubles liaisons (di, tri…)
- le symbole est Cn: mΔ(p, p'..)
- Cn: nombre de carbones
- mΔ : nombre de doubles liaisons
- (p, p'…) : positions des doubles liaisons en numérotation normale
- la série est de la forme ωn où n est la position de la première double liaison notée par rapport
à la position ω, dernier carbone de la chaîne aliphatique
- le nom courant rappelle son origine.
b2. Principaux acides gras mono-insaturés
Acide palmitoléique
– Nom systématique : acide cis-9-hexadécénoïque ;
– Notation : C16: 1Δ9 ou C16:1 ω7 ;
Figure 33. Structure de l’acide palmitoléique
Acide oléique
Le nom l’acide oléique vient de l’huile d’olive dont il constitue 55 à 80%.
– Nom systématique : acide cis-9-octadécénoïque ;
– Notation : C18:1 Δ 9 ou C18:1 ω 9 ;
Figure 34. Structure de l’acide oléique

b3. Principaux acides gras poly-insaturés
Acide linoléique
L’acide linoléique est acide gras essentiel, c’est également le précurseur de la famille des
oméga-6.
Le terme « précurseur » signifie que les autres acides gras de la famille peuvent être synthétisés
à partir de l’acide linoléique.
– Nom systématique : acide cis-cis-9,12-octadécadiénoïque ;
– Notation : C18:2 Δ 9,12
ou C18:2 ω 6 ;
Figure 35. Structure de l’acide linoléique
Acide alpha-linolénique
L’acide α-linolénique est un acide gras essentiel, c’est le principal acide gras du groupe des
oméga-3.
– Nom systématique : acide cis-cis-9,12-octadécadiénoïque ;
– Notation : C18:3 Δ 9,12,15
ou C18:3 ω 3 ;
Figure 36. Structure de l’acide alpha-linolénique
Acide arachidonique
La structure de l’acide arachidonique mérite d’être retenue car ce composé est le précurseur des
eicosanoïdes.
– Nom systématique : tout cis-5,8,11,14-éicosatétraènoïque ;
– Notation : C20:4 Δ 5,8,11,14
ou C20:4 ω 6 ;

Figure 37. Structure de l’acide arachidonique
VII.2.1. Propriétés physiques des acides gras
a. Solubilité
Les acides gras à courte chaine sont solubles dans l’eau, mais dès que le nombre d’atomes de
carbone de la chaine augmente, ils deviennent insolubles. Ils sont solubles dans les solvants
organiques comme le benzène, l’éther ou le chloroforme.
b. Point de fusion et point d’ébullition
Le point de fusion est la température à laquelle une molécule passe de l’état solide à l’état
liquide. A la température ordinaire, les acides gras sont à l’état liquide si le nombre de leurs
atomes de carbone est inférieur à 10; ils sont à l’état solide s’ils ont plus de 10 atomes de
carbone. Quand le nombre de carbone dans un acide gras augmente, cela augmente la valeur du
point de fusion. La présence de doubles liaisons dans un acide gras abaisse son point de fusion
par rapport à celui de l’acide gras saturé correspondant.
Le point d’ébullition des acides gras est d’autant plus élevé que la chaine est plus longue; la
présence de doubles liaisons est pratiquement sans influence.
c. Monocouches, micelles :
Au-dessus de quatre carbones, les acides gras sont insolubles dans l’eau et s’organisent quand
ils sont mis en milieux aqueux soit en film moléculaire à l’interface eau-air ou en micelles
(assemblages sphériques de molécules amphiphiles, délimitant un espace intérieur lipophile et
une couronne polaire). Les micelles apparaissent lorsque la concentration en molécules
amphiphile dépasse un certain seuil. Dans un solvant organique, par exemple de l’huile,
l’arrangement est inversé (figure 38).

Figure 38. Disposition des micelles.
VII.2.2. Propriétés chimiques
a. Formation de sels
Le traitement d’un acide gras par un hydroxide métallique (NaOH ou KOH) donne naissance à
un sel alcalin d’acide gras: ce sont les savons. Ils sont solubles dans l’eau, et possèdent alors
des propriétés moussantes. Il est possible d’utiliser cette propriété pour calculer l’indice de
saponification, Is. Is correspond à la masse (en mg) d’hydroxyde de potassium nécessaire pour
saponifier (neutralizer) les acides gras contenus dans 1g de lipide (Figure 39).
Figure 39. Réaction de saponification
b. Formation d’esters
On fabrique surtout des esters méthyliques (par action du methanol CH3OH, en présence d’un
catalyseur) qui permettent le fractionnement des acides gras par distillation ou par
chromatographie.
Figure 40. Réaction d’esterification.
c. Réaction d’addition (Fixation d’halogènes)
Lorsqu’un acide gras mono-insaturé est traité par un halogène (brome ou iode) on obtient par
addition un dérivé dihalogéné. Une des applications de cette propriétés est la détermination de

l’indice d’iode ‘Ii’ (Ii : c’est la quantité d’iode fixée par 100 g de lipides): la valeur de l’indice
d’iode est d’autant plus élevée que le nombre des doubles liaisons est plus grand (Figure 41).
Figure 41. Réaction d’addition d’iode.
d. Oxydation
Les produits formés par oxydation sont différents selon le nombre d’instaurations de l’acide
gras et selon la nature de l’oxydant :
Un peracide comme l’acide performique oxyde l’acide gras insaturé en époxyde (figure 42).
Figure 42. Oxydation des acides gras insaturés par l’acide performique.
Un acide gras insaturé traité par un acide minéral à 50° donne un diol (deux fonctions OH
adjacentes au carbone de la double liaison) (figure 43).
Figure 43. Structure d’un diol.
Un acide gras insaturé traité par un oxydant puissant tel qu’une solution concentrée de KMnO4
conduit à la formation de deux acides par coupure de la double liaison (figure 44).

Figure 44. Oxydation puissante
e. Réduction (hydrogénation)
La fixation d’hydrogène sur la double liaison transforme l’acide gras insaturé en acide gras
saturé. Cette réaction se fait en présence d’un catalyseur métallique (platine, nickel de Raney,
palladium...).
L’hydrogénation des huiles est importante dans l’industrie agro-alimentaire car elle permet la
transformation des huiles végétales ou animales en graisses solides (margarine ou substitut de
lard…) et évite l’oxydation pendant leur utilisation (odeurs, produits toxiques...).
Ac oléique C18:1 Ac stéarique C18:0 (saturé)
VII.3. Classification des lipides
On distingue différentes classes de lipides :
VII.3.1. Lipides simples ou ternaires
Ce sont des dérivés des acides gras ne contenant dans leur molécule que du carbone, de
l’hydrogène et de l’oxygène. Ils comprennent :
les glycérides : l’alcool estérifiant les acides gras est le glycérol.
les cérides dans lesquelles l’alcool est une chaine aliphatique à nombre élevé d’atomes
de carbone.
les stérides dans lesquelles l’alcool est un stérol.

VII.3.1.1. Les glycérides
Les glycérides, ou acylglycérols dans la nomenclature internationale, sont des esters d’acide
gras et de glycérol. Selon le nombre d’acides gras combinés au glycérol, on distingue les
monoglycérides, les diglycérides et les triglycérides.
Monoglycérides : pour les monoglycérides, la liaison ester est établie soit sur l’une des
fonctions alcool primaire (monoacylglycérol α) soit sur l’alcool secondaire (monoacylglycérol
β).
Diglycérides : pour les diglycérides, on parlera d’(α; α’) diacylglycérol ou de (α; β)
diacylglycérol.
Triglycérides : pour les triglycérides, quand les trois radicaux acyl sont identiques, ont dit que
le triglycéride est homogène. Dans le cas contraire il est dit mixte. (Figure 45).
Figure 45. Structure des glycérides.
a. Rôles biologiques
Les acylglycérols servent principalement de réserve chez les animaux et les végétaux. Leur
catabolisme par oxydation libère une énergie deux fois plus forte que celle du glycogène.
Les acylglycérols servent aussi d’isolants thermiques dans les tissus adipeux sous-cutanés,
d’émulsifiants et de messagers.
b. Nomenclature
On donne aux glycérides des noms officiels basés sur le principe que les radicaux acyl sont les
substituants du glycérol. On indique sur quelle fonction alcool du glycérol a lieu l’estérification

par chaque type d’acide gras en précisant à l’aide des numéros des atomes de carbone (dans la
nomenclature internationale on n’utilise pas les lettres grecques α et β).
Exemple : 1,3-distréaryl-2-palmitylglycérol ou 1,3-dioctadecanoyl-2-hexadecanoylglycérol.
c. Propriétés physiques
La propriété physique dominante est le caractère complètement apolaire des acylglycérols
naturels, essentiellement des triacylglycérols. Les groupes polaires (hydroxyle ou carboxyle)
disparaissent dans les liaisons esters. Ils sont insolubles dans l’eau et très solubles dans les
solvants les plus apolaires comme l’acétone.
d. Propriétés chimiques
Hydrolyse : le traitement acide (H2SO4 à 5%) provoque la rupture des liaisons ester et
libère les constituants: les acides gras et le glycérol, mais en général de façon incomplète.
En milieu enzymatique, les lipases comme la lipase pancréatique coupent les liaisons esters.
Cette hydrolyse se fait en 3 étapes : les 2 premières sont rapides avec libération des acides gras
en α et α’, alors la 3eme, le β-monoglycéride est transformé en acide gras et glycérol.
Saponification : les bases (hydroxyde de sodium ou de potassium) en solution
alcoolique, à chaud, coupent les liaisons esters des glycérides en libérant les acides gras sous
forme de sels de sodium (savons durs) ou de potassium (savons mous) (figure 47).

VII.3.1.2. Les cérides
Les cérides sont des esters d’acides gras et d’alcools à longue chaîne non ramifiée et à nombre
pair d’atomes de carbone: les alcools gras (figure 48).
Ces substances solides, incolores, très forte insolubles dans l’eau (très apolaires), elles sont
seulement solubles à chaud dans les solvants organiques, sont chimiquement inertes. Elles sont
saponifiées lentement et résistent à la plupart des réactifs chimiques, d’où leur rôle protecteur.
Elles sont solides à température ordinaire et leur point de fusion élevée (60 à 100°C). Les
cérides sont des molécules essentielles des « revêtements » de protection des organismes
vivants. Les cérides constituent la majeure partie des cires.
Exp.: L’ester palmitique de l’alcool cétylique en C16 est le constituant principal du blanc de
baleine: C15H31-CH2-O-CO-C15H31 (Palmitate de cétyle) (figure 49).
Figure 49. Molécule de palmitate de cétyle.

VII.3.1.3. Les stérides
Les stérides sont des esters d’acides gras et de stérols (souvent du cholestérol). On appelle
spécifiquement les stérides du cholestérol les ester de cholestéryle.
Figure 50. Molécule de palmitate de cholestéryle.
In vivo, les stérides sont présents dans les membranes cellulaires, toutefois ils y sont
relativement peu abondants par comparaison avec les phospholipides. Les tissus d’animaux
contiennent peu de stérides au contraire du plasma sanguin.
Le cholestérol et ses formes estérifiées sont transportés avec les autres lipides sous la forme
d’associations non covalentes : les lipoprotéines.
Les esters de cholestérol alimentaire sont hydrolysés par une cholestérolester hydrolase du suc
pancréatique
VII.3.2. Lipides complexes
Ils sont constitués des même éléments que les lipides simples, mais ils contiennent en plus dans
leur molécule de l’azote, ou du phosphore, ou du soufre, ou des oses (un seul ou plusieurs de
ces éléments à la fois). On distingue:
- Les glycérophospholipides
- Les glycéroglycolipides
- Les sphingolipides
Les lipides complexes sont les constituants essentiels des membranes biologiques. Par leur
imperméabilité ils permettent de délimiter les différents compartiments des cellules.
VII.3.2.1. Les glycérophospholipides
Appartenant au groupe des lipides complexes, ils sont constitués d’une molécule de glycérol
sur laquelle sont estérifiés deux acides gras. La troisième fonction alcool est estérifiée par une
molécule d’acide phosphorique.

Cet ensemble forme l’acide phosphatidique (=diacylglycérolphosphate), molécule commune
aux différents Glycérophospholipides (Figure 51).
Figure 51. Acide phosphatidique (=diacylglycérol-P)
L’estérification de l’acide phosphatidique au niveau de son groupement phosphorique par un
alcool donne naissance aux glycérophospholipides, on a 04 types selon la nature de l’alcool
impliqué :
Phosphatidylsérines = Acides Phosphatidiques + Sérine
Phosphatidyléthanolamines = Acides Phosphatidiques + Ethanolamine
Phosphatidylcholines (lécithines) = Acides Phosphatidiques + Choline
Phosphatidylinositols = Acides Phosphatidiques + Inositol
* Les phosphatidylsérines * Les phosphatidyléthanolamines
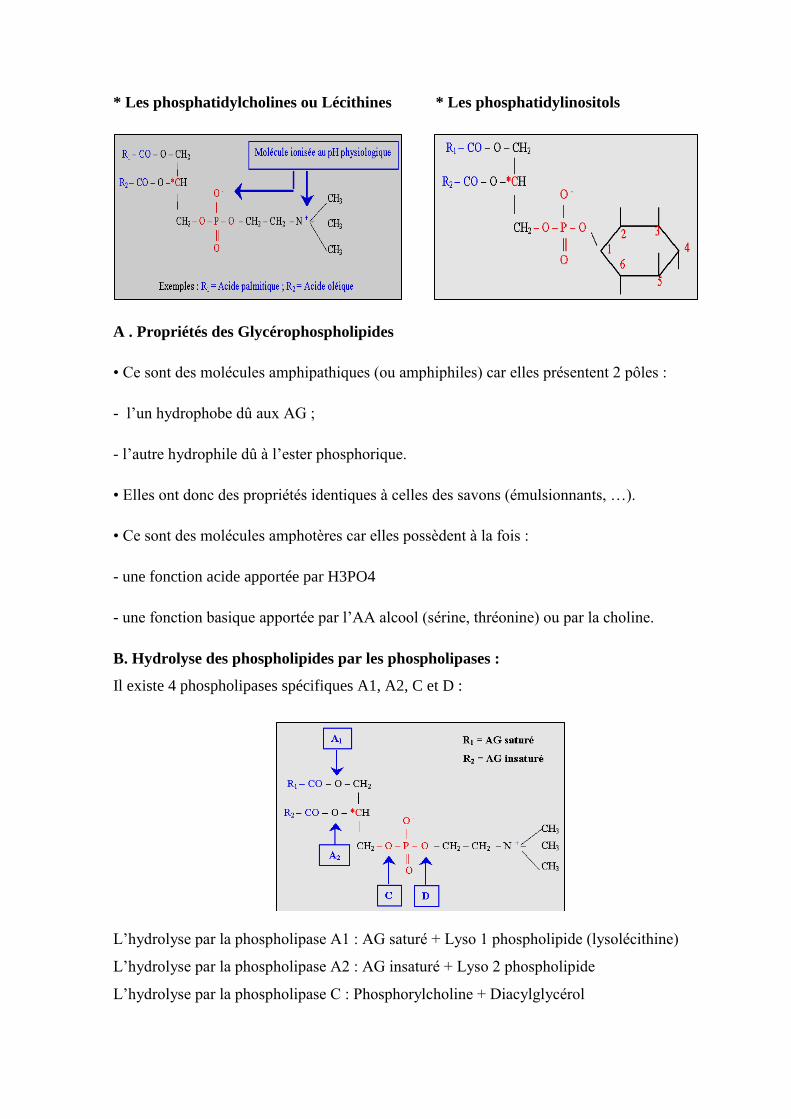
* Les phosphatidylcholines ou Lécithines * Les phosphatidylinositols
A . Propriétés des Glycérophospholipides
• Ce sont des molécules amphipathiques (ou amphiphiles) car elles présentent 2 pôles :
- l’un hydrophobe dû aux AG ;
- l’autre hydrophile dû à l’ester phosphorique.
• Elles ont donc des propriétés identiques à celles des savons (émulsionnants, …).
• Ce sont des molécules amphotères car elles possèdent à la fois :
- une fonction acide apportée par H3PO4
- une fonction basique apportée par l’AA alcool (sérine, thréonine) ou par la choline.
B. Hydrolyse des phospholipides par les phospholipases :
Il existe 4 phospholipases spécifiques A1, A2, C et D :
L’hydrolyse par la phospholipase A1 : AG saturé + Lyso 1 phospholipide (lysolécithine)
L’hydrolyse par la phospholipase A2 : AG insaturé + Lyso 2 phospholipide
L’hydrolyse par la phospholipase C : Phosphorylcholine + Diacylglycérol

L’hydrolyse par la phospholipase D : Acide phosphatidique + Choline
VII.3.2.2. Glycéroglycolipides
Les carbones C1 et C2 du glycérol sont estérifiés par des acides gras. L’alcool du carbone C3 à
la différence des acylglycérols n’est pas estérifié mais lié à un ose par une liaison osidique.
Ces lipides sont très rares dans le monde animal mais sont beaucoup plus nombreux dans le
monde végétal. Ils sont également présents chez certaines bactéries.
VII.3.2.3. Les sphingolipides
Dans ce groupe, le glycérol est remplacé par la sphingosine, aminodiol à 18 atomes de carbone
possédant une double liaison (Figure 52).
Figure 52. Structure de Sphingosine.
L’acide gras est uni à la sphingosine par une liaison amide et non par une liaison ester.
Les sphingolipides sont particulièrement abondants dans le tissu nerveux, certains d’entre eux
s’accumulant au cours de diverses maladies.

a. Les céramides ou Acylsphingosine
L’acylation de la fonction amine de la sphingosine par un acide gras (saturé et à longue chaîne)
forme une céramide. Cette molécule est le précurseur de tous les sphingolipides (Figure 53).
Figure 53. Structure de Céramide.
b. Les sphingomyélines
Elles sont constituées de l’association : Sphingosine + AG + Phosphorylcholine
Figure 54 : Structure de sphingomyéline
• L’acide gras le plus fréquent est l’acide lignocérique (C24:O).
• Au pH du sang, la molécule est ionisée.
• On les trouve dans le tissu nerveux (graines de myéline) et dans les membranes.
• La déficience en sphingomyélinase entraîne leur accumulation dans le cerveau, la rate et le
foie.
c. Les glycoshingolipides
c1. Cérébrogalactosides ou Galactosylcéramides
Ils sont constitués de : Sphingosine + AG + βD Galactose. Le galactose est uni à l’alcool
primaire de la sphingosine par une liaison β osidique

Figure 55 : Structure de Cérébrogalactosides
c2. Les Cérébroglucides ou Glucosylcéramides
Ils sont constitués de: Sphingosine + AG + βD Glucose. La liaison est β osidique.
c3. Les Gangliosides ou Oligosylcéramides
Ils sont constitués de: Sphingosine + AG + chaîne de plusieurs oses et dérivés d’oses (NANA).
Ils sont abondants dans les ganglions d’où leur nom. Ces oligosides sont présents sur la face
externe de la membrane plasmique. Ils sont spécifiques, donc reconnus par des protéines
(toxines bactériennes, lectines). Exemple : antigènes des groupes sanguins (figure 54).
Figure 56. Structure d’un ganglioside.

REFERENCE
univ.ency-education.com/uploads/1/3/1/0/.../bioch 1
https://www.fichier-pdf.fr/2011/11/19/1a-biochimie/
univ.ency-education.com/1an_lessons-biochimie.html
https://elearning.univ-bejaia.dz/course/view.php?id=3556
https://www.scribd.com/doc/6741297/Glucides
Claude AUDIGIE et François ZONSZAIN. Biochimie structurale. Edition Doin (Paris)
2007, 263 p.
Françoise QUENTIN, Paul-françois GALLET, Michel GUILLOTON et Bernadette
QUINTARD. Biochimie en 84 fiches. Edition Dunod (Paris) 2015, 215p.
Elisabeth HEBERT. Biochimie. Edition Atlani 1994, 320p.
Jacques-Henry WEIL. Biochimie générale. Edition Dunod (Paris) 2005, 726p.
Gérard BOISSONNET, Bernadette BOISSONNET, Marie-Jeanne HAMMOUD, Claude
ROUCH. Biochimie structurale. Edition SMER (Rabat) 1987, 326p.
Olivier MASSON. Biochimie, bases biochimiques de la diététique. Edition Lavoisier (Paris).
2007. 330p.
Raoui Mounir MAAROUFI. Cours de biochimie structurale (glucides). Université de
Monastir. Institut Supérieur de Biotechnologie de Monastir 2015, 36p.
Pierre LOUISOT. Biochimie générale et médicale. Edition SIMEP 1989, 488 p.
Garret et Grisham. Biochimie. 2eme édition Américaine. De Boek. 2000, pp 14-18.


















