
Travaux Pratiques de chimie océanographique - …moutin/IMG/pdf/TP_CHIMIE_OCE_103_2015_2… ·...
40
Travaux Pratiques de chimie océanographique OCE 103 Master Océanographie 2015-2016 T. WAGENER, T. MOUTIN, V. LAGADEC. -1-
Transcript of Travaux Pratiques de chimie océanographique - …moutin/IMG/pdf/TP_CHIMIE_OCE_103_2015_2… ·...

Travaux Pratiques de chimie océanographiqueOCE 103
Master Océanographie20152016
T. WAGENER, T. MOUTIN, V. LAGADEC.
1

2

Table des matières
TP N°1 : DOSAGE DE L'OXYGENE DISSOUS (Méthode de Winkler) et mesure de la salinité...................................5
TP N°2 : DOSAGE DE L'AZOTE AMMONIACAL........15
TP N°3 : DOSAGE DES ORTHOPHOSPHATES DANS L'EAU DE MER....................................................................19
TP N°4 : MESURE DU pH, DE L'ALCALINITE TOTALE ET DE LA TENEUR EN CALCIUM D'UNE EAU DE MER.......................................................................................23
TP N°5 : DOSAGE DE L'AZOTE ORGANIQUE DISSOUS ET DE L'AZOTE ORGANIQUE PARTICULAIRE..................................................................31
3

4

TP N°1 : DOSAGE DE L'OXYGENE DISSOUS (Méthode de Winkler) et mesure de la salinité
I. INTRODUCTIONLes teneurs en oxygène dissous dans l'eau de mer sont la résultante de flux de
production (diffusion de l'oxygène atmosphérique, photosynthèse) et de flux de consommation (respiration, dégradation de matières organiques). Les valeurs varient entre 0 et 8 ml.l1.
Il existe de nombreuses méthodes pour déterminer l'oxygène dissous dans l'eau de mer (chimique, électrochimique, chromatographie en phase gazeuse). Même si la méthode électrochimique est plus facile à mettre en oeuvre "in situ", la méthode la plus précise est la méthode de Winkler. Nous utiliserons cette méthode, légèrement modifiée par Carpenter (1965) et Carritt & Carpenter (1966), qui est considérée comme la référence universelle.
II. PRINCIPE DU DOSAGE DE L'OXYGÈNE DISSOUSLe principe du dosage est de former un précipité de manganèse (Mn(II)) et de
l'oxyder par l'oxygène dissous (Mn(III) et Mn(IV)). En milieu acide et en présence d'iodure, le manganèse est réduit, ce qui libère de l'iode. L'iode est alors titré par le thiosulfate.
Les réactions intervenant dans le dosage sont :(1) Formation du précipité de Mn(II) par la soude
Mn2+ + 2 OH > Mn(OH)2
(2) Oxydation du Mn (II) par l'oxygène. (a) Mn(OH)2 + 1/4 O2 + 1/2 H2O > Mn(OH)3
(b) Mn(OH)2 + 1/2 O2 + H2O > Mn(OH)4
(3) Réduction du Manganèse par l'iodure en milieu acide
(a) Mn(OH)3 + I + 3 H+ > Mn2+ + 3 H2O + 1/2 I2
(b) Mn(OH)4 + 2 I + 4 H+ > Mn2+ + 4 H2O + I2
donc 1 mole d'oxygène (O2) libère 2 moles d'iode (I2)(4) Dosage de l'iode par le thiosulfate
2 S2032 + I2 > S4O6
2 + 2 I
Finalement, à 1 mole de thiosulfate correspond 1/4 mole d'oxygène.Le point équivalent du dosage peut être mis en évidence par plusieurs méthodes.
Nous utiliserons un indicateur coloré d'oxydoréduction, l'amidon.
5

III. MODALITES DU DOSAGE
III.1. Domaine d'application, précision.
Domaine d'application : 0,0058,000 mM équivalent à 0,0690 ml.l1 d'oxygène.
Précision : pour C = 0,7 mM, C = 0,003/n1/2 mM (précision maximum obtenue pour un laboratoire à terre dans les conditions idéales, n : nombre de réplicats, seuil de confiance 95%). C'estàdire que pour une mesure dans la gamme habituelle des concentrations, la précision est de ± 0,035 ml.l1 (précision maximum obtenue dans des conditions de travail idéales).
Dans toute la manipulation, la précaution essentielle consiste à ne pas introduire d'oxygène dans l'échantillon. Il faut donc éviter toute introduction de bulles d'air. Les principales sources d'erreurs inhérentes au dosage se résument en 5 points :
• oxydation de l'iodure à l'air• volatilisation de l'iode• adjonction d'oxygène par les réactifs• consommation ou production d'iode par les impuretés des réactifs• différence entre le point équivalent réel et le point de fin de titrage
La méthode n'est pas applicable dans les eaux contenant beaucoup de matières en suspension ou des substances organiques oxydables en milieu fortement basique ou susceptible de réagir avec l'iode en milieu acide.
Remarque : la précision est déterminée en supposant qu'il n'y a pas d'erreur systématique; la valeur moyenne est la valeur vraie.
III.2. Echantillonnage
L'échantillon prélevé est transféré immédiatement de la bouteille dans un flacon à l'aide d'un tube souple.
• Plonger le tube jusqu'au fond du flacon et le remplir sans introduire de bulles d'air en laissant déborder un volume équivalent au volume du flacon.
6
Illustration 1: Remplissage du flacon d'échantillonnage, à partir de la bouteille de prélèvement, pour l'analyse de l'oxygène dissous : le tuyau pénétrant jusqu'au fond du flacon et le contrôle de débit par la pince permettant d'éviter tout barbotage (Aminot & Chaussepied, 1983).

• Retirer le tube lentement en arrêtant l'écoulement juste avant que son extrémité ne vienne à l'air.
• Ajouter immédiatement 1 ml de réactif 1 et 1 ml de réactif 2.• Boucher sans introduire de bulles d'air (attention : à un flacon correspond un
bouchon).• Agiter, laisser reposer, agiter de nouveau.• Laisser reposer à l'abri de la lumière au moins jusqu'à ce que le précipité
n'occupe plus que le tiers inférieur du volume du flacon et que la température du flacon soit égale à celle du laboratoire.
Remarque : cette opération peut être menée à bord d'un navire. La suite des opérations peut être poursuivie plus tard. Dans ce cas, il est recommandé de maintenir les flacons en immersion.
III.3. Réactifs
Réactif 1 : solution de manganèse 3M #
Dissoudre 600 g de MnCl2 ou 670 g de MnSO4,4H2O ou 560 g de MnSO4,2H2O ou 510 g de MnSO4,H2O dans de l'eau déionisée, compléter à 1 l.
Réactif 2 : solution de KI (4M) et NaOH (8M) #
Dissoudre 320 g de NaOH dans 400 ml d'eau déionisée. Dissoudre, en chauffant si nécessaire, 600 g de NaI dans 300 ml d'eau déionisée en agitant continuellement. Mélanger les deux solutions lorsqu'elles sont froides et compléter à 1 l.
Solution de thiosulfate de sodium 0,01 MA préparer à partir de thiosulfate de sodium Na2S2O3,5H2O
Solution d'iodate de potassium 0,0017 M #
Dissoudre 0,3567 g de KIO3 (séché à 105° pendant 1 heure) dans 300 ml d'eau déionisée. Chauffer légèrement pour dissoudre. Laisser refroidir. Compléter à 1 l. Cette solution est stable indéfiniment à condition de l'agiter avant l'utilisation et de reboucher le flacon immédiatement après.
Amidon 1% #
Dissoudre 1 g d'amidon soluble dans 100 ml d'eau déionisée en chauffant jusqu'à ce que la solution devienne claire ; ajouter quelques gouttes de chloroforme et conserver au réfrigérateur. Elle se conserve plusieurs mois. La solution d'amidon est à jeter quand la couleur du virage évolue vers le vert ou le brun ou si un trouble s'y développe.
Acide sulfurique concentré (5 M) #
Diluer 280 ml d'acide sulfurique concentré (d=1,84) à 1 l d'eau déionisée.
7

III.4. Détermination du blanc et étalonnage du thiosulfate
• Remplir un erlen avec environ 100 ml d'eau déionisée. • Ajouter 2 ml d'acide sulfurique concentré, puis 1 ml de réactif 2 (solution de
KI,NaOH) et agiter. • Ajouter 1 ml de réactif 1 et agiter de nouveau.
La solution est prête pour la détermination du blanc puis l'étalonnage du thiosulfate.
La détermination du blanc et l'étalonnage du thiosulfate se font sur la même solution.
Détermination du blanc :Ajouter l'amidon.
• si la solution se colore, doser par le thiosulfate 0,01 M jusqu'à décoloration : soit Vb ce volume.
• si la solution reste incolore, doser par la solution de KIO3 0,0017 M à l'aide d'une pipette graduée jusqu'à la première apparition d'une coloration jaunâtre; si Vb' est ce volume, le blanc sera Vb = Vb'.
Si la coloration apparaît dès la première goutte de KIO3 versée, Vb = 0.
Étalonnage du thiosulfate :• Ajouter précisément 10 ml de la solution étalon de KIO3 0,0017 M à la solution
précédente.• Laisser la réaction se poursuivre à l'obscurité entre 2 et 5 mn. • Titrer par le thiosulfate. Soit Vs le volume versé.
La molarité (ou titre ou concentration) de la solution de thiosulfate est :
CThio = (6 . CI . VI)/VThio
CThio : titre du thiosulfateCI : titre de l'iodate utilisé pour la standardisation (0,0017 M)VI : volume d'iodate utilisé pour la standardisationVThio : volume de thiosulfate utilisé pour la standardisation
Remarque : le dosage de l'iodate par le thiosulfate a lieu selon la réaction suivante :
6 S2O32 + IO3
+ 6 H3O+ > 3 S4O62 + I + 9 H2O
8

III.5. Titrage
Etant donné les risques de perte d'iode par transvasage et pipettage, le titrage doit se faire directement dans le flacon de prélèvement ; On utilise pour cela des flacons spéciaux avec des bouchons "plongeurs". Le volume exact d'échantillon servant au dosage doit être connu avec précision (attention à bien utiliser le flacon et le bouchon correspondant).
Illustration 2: Bouchon plongeur permettant d'évacuer un certain volume d'eau du flacon afin de permettrel'addition du titrant dans le flacon luimême (Aminot & Chaussepied, 1983).
• Ouvrir délicatement le flacon. • Introduire 2 ml d'acide sulfurique concentré. • Reboucher (sans introduire de bulles) et agiter jusqu'à dissolution du
précipité.
Effectuer le dosage immédiatement en respectant les règles suivantes :• Titrer en agitant lentement, pour homogénéiser, jusqu'à décoloration quasi
totale,• ajouter quelques gouttes de solution d'amidon,• terminer le titrage au goutte à goutte avec une agitation plus rapide, jusqu'à
décoloration complète persistant 20 secondes. Procéder rapidement mais en attendant l'homogénéisation de la solution après chaque ajout. Ne pas tenir compte de la recoloration lente se produisant après l' équivalence (oxydation de I à l'air).
Soit Ve, le volume de thiosulfate utilisé pour doser l'échantillon.
9

III.6. Expression des résultats :
Le nombre de moles de thiosulfate utilisé pour doser un échantillon est :
nThio = CThio(Ve Vb)
D'après les réactions de la méthode de Winckler, à 1 mole de thiosulfate correspond 1/4 mole d'oxygène. Le nombre de mol d'oxygène que contient l'échantillon est de :
nO2 = CThio(Ve Vb)/4
La concentration en oxygène de l'échantillon en mol.L1 est de :
[O2]=CThio⋅(Ve−Vb)
4⋅Vt⋅
Vt(Vt−v )
avec• Vt : volume d'eau de mer prélevée (volume du flacon) sur lequel est fait le
titrage• v : volume des réactifs 1 et 2
•Vt
(Vt−v )permet de tenir compte de la dilution provoquée par l'addition des
réactifs 1 et 2. L'addition ultérieure d'acide ne produit pas de dilution car il remplace un volume équivalent d'eau exempte d'oxygène.
IV. MANIPULATIONLa manipulation consiste à doser l'oxygène dissous d'une eau de mer (minimum 3
réplicats).• Prélever les échantillons à la bouteille Niskin en prenant les précautions
nécessaires indiquées en III2 .• Préparer la solution de thiosulfate 0,01 M. • Réaliser trois fois la détermination du blanc et l'étalonnage du thiosulfate. • Effectuer les dosages des échantillons.
• Exprimer les résultats en µmol.kg1 d'eau de mer et en mL.L1. • Calculer les intervalles de confiance.• Calculer l'écart par rapport à la saturation d'oxygène (Voir Annexe 2).• En fin de manipulation, laver soigneusement les flacons à l'eau déionisée.
Veiller à ce qu'il ne reste pas de trace de Mn (II).
10

V. MESURE DE LA SALINITÉ ET DÉTERMINATION DE LA MASSE VOLUMIQUE
V.1. Introduction
La salinité est une variable essentielle en océanographie. La plupart des propriétés physiques de l’eau de mer dépendent de sa température et de sa salinité. Le caractère conservatif de ces deux variables a conduit les océanographes à considérer comme principal outil dans l’étude de la formation des masses d’eau, de leur mélange et de leur circulation, le diagramme températuresalinité.
V.2. Définition de la salinité
La notion de salinité a évoluée au cours du temps. A ce jours deux définitions de la salinité coexiste :
• La salinité pratique (Sp) définit dans le rapport UNESCO n° 36 « l’échelle de salinité pratique de 1978 (PSS78)» disponible en salle de travaux pratiques. La méthode de mesure de Sp repose sur la mesure d’un rapport de conductivité, Sp est donc une variable adimensionnelle.
• La salinité absolue (SA) introduite en 2010 dans The International Thermodynamic Equation of Seawater (Rapport UNESCO n°56 TEOS10) représente la masse de soluté dissous dans un kg d'eau de mer. SA a donc exprimmé en g.kg1. SA est utilisé pour le calcul de toutes les grandeurs thermodynamiques de l'eau de mer avec TEOS10 depuis 2011. Comme la mesure de SA est extrêmement compliquée à mettre en œuvre à ce jours, un algorithme de conversion de Sp en SA (tenant compte des variations de la composition chimique relative de l'eau de mer en fonction de la région océanographique considérée) est disponible dans TEOS10.
A ce jours la salinité pratique (Sp) reste la grandeur mesurée en routine en océanographie et sera donc estimée au cours de ce TP.
V.3. Manipulation
1. Dés que les prélèvements pour l’oxygène ont été réalisés, prélever de l’eau à la bouteille Niskin dans le flacon prévu pour la mesure de la salinité.
2. Mesurer immédiatement la température que l'on considérera égale à tin situ
3. Laisser le flacon sur la paillasse le temps nécessaire pour que sa température soit égale à la température de l’eau normale (K15 = 1).
4. Mesurer la température que l'on considérera égale à la température du laboratoire: tlabo
5. Mesurer la conductivité de votre échantillon (condéch (tlabo, p=0)) et la conductivité de l’eau normale (condEN (tlabo, p=0)) en faisant bien attention de bien rincer et sécher l’électrode entre chaque mesure.
6. Calculer Rt : Rt=Cond ech( t, p=0 )
CondEN ( t, p=0 )
11

7. Consulter les tables océanographiques internationales disponibles en salle de travaux pratiques pour déterminer :
• Suncorrected à partir de Rt
• S à partir de t et Rt
8. Calculer Sp : Sp = Suncorrected + S9. A partir de Sp, tin situ et de tlabo, déterminer la masse volumique S,t,p=0)) de
votre eau de mer in situ et au laboratoire en utilisant le tableau présenté en annexe 1 (Masse volumique calculé avec TEOS10). Pour information le programme CSEQETAT.xls fourni par T. Moutin permet de déterminer la masse volumique avec l'équation international d'état de 1980 (EOS80).
10. Pensez à afficher au tableau vos résultats qui intéressent l’ensemble des étudiants (tin situ,tlabo, Sp, in situlabo).
12
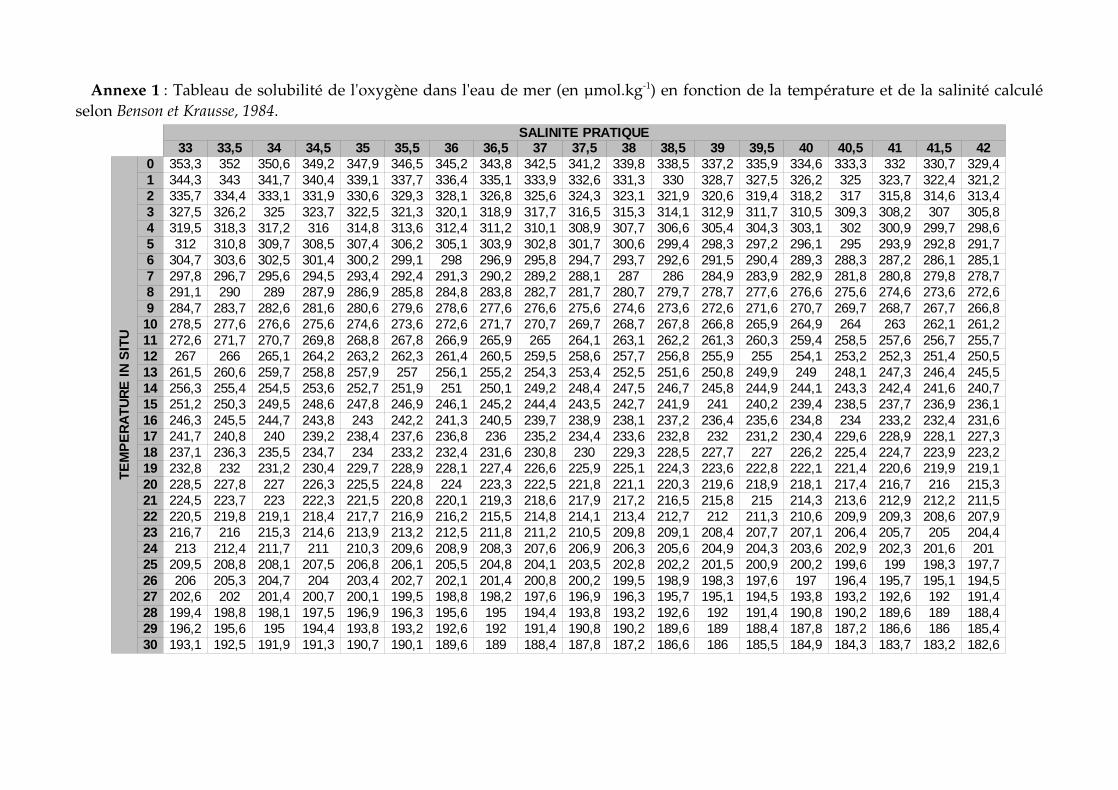
Annexe 1 : Tableau de solubilité de l'oxygène dans l'eau de mer (en µmol.kg1) en fonction de la température et de la salinité calculé selon Benson et Krausse, 1984.
SALINITE PRATIQUE33 33,5 34 34,5 35 35,5 36 36,5 37 37,5 38 38,5 39 39,5 40 40,5 41 41,5 42
TE
MP
ER
AT
UR
E I
N S
ITU
0 353,3 352 350,6 349,2 347,9 346,5 345,2 343,8 342,5 341,2 339,8 338,5 337,2 335,9 334,6 333,3 332 330,7 329,41 344,3 343 341,7 340,4 339,1 337,7 336,4 335,1 333,9 332,6 331,3 330 328,7 327,5 326,2 325 323,7 322,4 321,22 335,7 334,4 333,1 331,9 330,6 329,3 328,1 326,8 325,6 324,3 323,1 321,9 320,6 319,4 318,2 317 315,8 314,6 313,43 327,5 326,2 325 323,7 322,5 321,3 320,1 318,9 317,7 316,5 315,3 314,1 312,9 311,7 310,5 309,3 308,2 307 305,84 319,5 318,3 317,2 316 314,8 313,6 312,4 311,2 310,1 308,9 307,7 306,6 305,4 304,3 303,1 302 300,9 299,7 298,65 312 310,8 309,7 308,5 307,4 306,2 305,1 303,9 302,8 301,7 300,6 299,4 298,3 297,2 296,1 295 293,9 292,8 291,76 304,7 303,6 302,5 301,4 300,2 299,1 298 296,9 295,8 294,7 293,7 292,6 291,5 290,4 289,3 288,3 287,2 286,1 285,17 297,8 296,7 295,6 294,5 293,4 292,4 291,3 290,2 289,2 288,1 287 286 284,9 283,9 282,9 281,8 280,8 279,8 278,78 291,1 290 289 287,9 286,9 285,8 284,8 283,8 282,7 281,7 280,7 279,7 278,7 277,6 276,6 275,6 274,6 273,6 272,69 284,7 283,7 282,6 281,6 280,6 279,6 278,6 277,6 276,6 275,6 274,6 273,6 272,6 271,6 270,7 269,7 268,7 267,7 266,810 278,5 277,6 276,6 275,6 274,6 273,6 272,6 271,7 270,7 269,7 268,7 267,8 266,8 265,9 264,9 264 263 262,1 261,211 272,6 271,7 270,7 269,8 268,8 267,8 266,9 265,9 265 264,1 263,1 262,2 261,3 260,3 259,4 258,5 257,6 256,7 255,712 267 266 265,1 264,2 263,2 262,3 261,4 260,5 259,5 258,6 257,7 256,8 255,9 255 254,1 253,2 252,3 251,4 250,513 261,5 260,6 259,7 258,8 257,9 257 256,1 255,2 254,3 253,4 252,5 251,6 250,8 249,9 249 248,1 247,3 246,4 245,514 256,3 255,4 254,5 253,6 252,7 251,9 251 250,1 249,2 248,4 247,5 246,7 245,8 244,9 244,1 243,3 242,4 241,6 240,715 251,2 250,3 249,5 248,6 247,8 246,9 246,1 245,2 244,4 243,5 242,7 241,9 241 240,2 239,4 238,5 237,7 236,9 236,116 246,3 245,5 244,7 243,8 243 242,2 241,3 240,5 239,7 238,9 238,1 237,2 236,4 235,6 234,8 234 233,2 232,4 231,617 241,7 240,8 240 239,2 238,4 237,6 236,8 236 235,2 234,4 233,6 232,8 232 231,2 230,4 229,6 228,9 228,1 227,318 237,1 236,3 235,5 234,7 234 233,2 232,4 231,6 230,8 230 229,3 228,5 227,7 227 226,2 225,4 224,7 223,9 223,219 232,8 232 231,2 230,4 229,7 228,9 228,1 227,4 226,6 225,9 225,1 224,3 223,6 222,8 222,1 221,4 220,6 219,9 219,120 228,5 227,8 227 226,3 225,5 224,8 224 223,3 222,5 221,8 221,1 220,3 219,6 218,9 218,1 217,4 216,7 216 215,321 224,5 223,7 223 222,3 221,5 220,8 220,1 219,3 218,6 217,9 217,2 216,5 215,8 215 214,3 213,6 212,9 212,2 211,522 220,5 219,8 219,1 218,4 217,7 216,9 216,2 215,5 214,8 214,1 213,4 212,7 212 211,3 210,6 209,9 209,3 208,6 207,923 216,7 216 215,3 214,6 213,9 213,2 212,5 211,8 211,2 210,5 209,8 209,1 208,4 207,7 207,1 206,4 205,7 205 204,424 213 212,4 211,7 211 210,3 209,6 208,9 208,3 207,6 206,9 206,3 205,6 204,9 204,3 203,6 202,9 202,3 201,6 20125 209,5 208,8 208,1 207,5 206,8 206,1 205,5 204,8 204,1 203,5 202,8 202,2 201,5 200,9 200,2 199,6 199 198,3 197,726 206 205,3 204,7 204 203,4 202,7 202,1 201,4 200,8 200,2 199,5 198,9 198,3 197,6 197 196,4 195,7 195,1 194,527 202,6 202 201,4 200,7 200,1 199,5 198,8 198,2 197,6 196,9 196,3 195,7 195,1 194,5 193,8 193,2 192,6 192 191,428 199,4 198,8 198,1 197,5 196,9 196,3 195,6 195 194,4 193,8 193,2 192,6 192 191,4 190,8 190,2 189,6 189 188,429 196,2 195,6 195 194,4 193,8 193,2 192,6 192 191,4 190,8 190,2 189,6 189 188,4 187,8 187,2 186,6 186 185,430 193,1 192,5 191,9 191,3 190,7 190,1 189,6 189 188,4 187,8 187,2 186,6 186 185,5 184,9 184,3 183,7 183,2 182,6
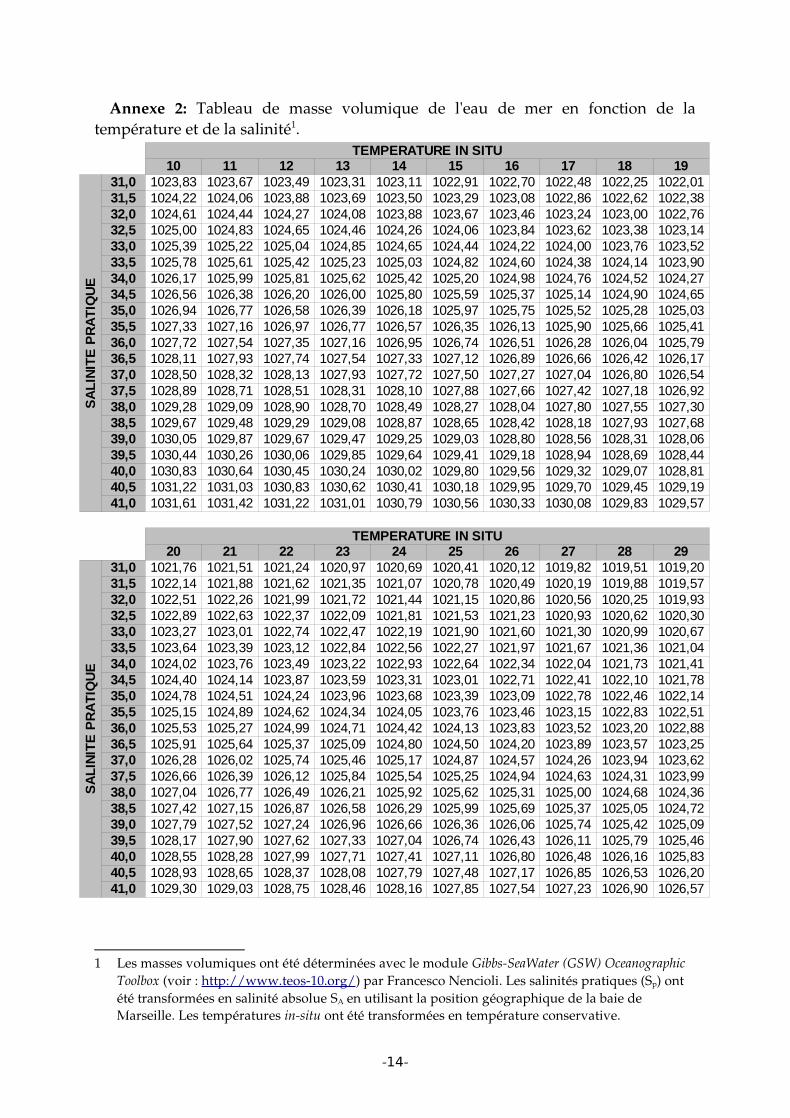
Annexe 2: Tableau de masse volumique de l'eau de mer en fonction de la température et de la salinité1.
1 Les masses volumiques ont été déterminées avec le module GibbsSeaWater (GSW) Oceanographic Toolbox (voir : http://www.teos10.org/) par Francesco Nencioli. Les salinités pratiques (Sp) ont été transformées en salinité absolue SA en utilisant la position géographique de la baie de Marseille. Les températures insitu ont été transformées en température conservative.
14
TEMPERATURE IN SITU10 11 12 13 14 15 16 17 18 19
SA
LIN
ITE
PR
AT
IQU
E
31,0 1023,83 1023,67 1023,49 1023,31 1023,11 1022,91 1022,70 1022,48 1022,25 1022,0131,5 1024,22 1024,06 1023,88 1023,69 1023,50 1023,29 1023,08 1022,86 1022,62 1022,3832,0 1024,61 1024,44 1024,27 1024,08 1023,88 1023,67 1023,46 1023,24 1023,00 1022,7632,5 1025,00 1024,83 1024,65 1024,46 1024,26 1024,06 1023,84 1023,62 1023,38 1023,1433,0 1025,39 1025,22 1025,04 1024,85 1024,65 1024,44 1024,22 1024,00 1023,76 1023,5233,5 1025,78 1025,61 1025,42 1025,23 1025,03 1024,82 1024,60 1024,38 1024,14 1023,9034,0 1026,17 1025,99 1025,81 1025,62 1025,42 1025,20 1024,98 1024,76 1024,52 1024,2734,5 1026,56 1026,38 1026,20 1026,00 1025,80 1025,59 1025,37 1025,14 1024,90 1024,6535,0 1026,94 1026,77 1026,58 1026,39 1026,18 1025,97 1025,75 1025,52 1025,28 1025,0335,5 1027,33 1027,16 1026,97 1026,77 1026,57 1026,35 1026,13 1025,90 1025,66 1025,4136,0 1027,72 1027,54 1027,35 1027,16 1026,95 1026,74 1026,51 1026,28 1026,04 1025,7936,5 1028,11 1027,93 1027,74 1027,54 1027,33 1027,12 1026,89 1026,66 1026,42 1026,1737,0 1028,50 1028,32 1028,13 1027,93 1027,72 1027,50 1027,27 1027,04 1026,80 1026,5437,5 1028,89 1028,71 1028,51 1028,31 1028,10 1027,88 1027,66 1027,42 1027,18 1026,9238,0 1029,28 1029,09 1028,90 1028,70 1028,49 1028,27 1028,04 1027,80 1027,55 1027,3038,5 1029,67 1029,48 1029,29 1029,08 1028,87 1028,65 1028,42 1028,18 1027,93 1027,6839,0 1030,05 1029,87 1029,67 1029,47 1029,25 1029,03 1028,80 1028,56 1028,31 1028,0639,5 1030,44 1030,26 1030,06 1029,85 1029,64 1029,41 1029,18 1028,94 1028,69 1028,4440,0 1030,83 1030,64 1030,45 1030,24 1030,02 1029,80 1029,56 1029,32 1029,07 1028,8140,5 1031,22 1031,03 1030,83 1030,62 1030,41 1030,18 1029,95 1029,70 1029,45 1029,1941,0 1031,61 1031,42 1031,22 1031,01 1030,79 1030,56 1030,33 1030,08 1029,83 1029,57
TEMPERATURE IN SITU20 21 22 23 24 25 26 27 28 29
SA
LIN
ITE
PR
AT
IQU
E
31,0 1021,76 1021,51 1021,24 1020,97 1020,69 1020,41 1020,12 1019,82 1019,51 1019,2031,5 1022,14 1021,88 1021,62 1021,35 1021,07 1020,78 1020,49 1020,19 1019,88 1019,5732,0 1022,51 1022,26 1021,99 1021,72 1021,44 1021,15 1020,86 1020,56 1020,25 1019,9332,5 1022,89 1022,63 1022,37 1022,09 1021,81 1021,53 1021,23 1020,93 1020,62 1020,3033,0 1023,27 1023,01 1022,74 1022,47 1022,19 1021,90 1021,60 1021,30 1020,99 1020,6733,5 1023,64 1023,39 1023,12 1022,84 1022,56 1022,27 1021,97 1021,67 1021,36 1021,0434,0 1024,02 1023,76 1023,49 1023,22 1022,93 1022,64 1022,34 1022,04 1021,73 1021,4134,5 1024,40 1024,14 1023,87 1023,59 1023,31 1023,01 1022,71 1022,41 1022,10 1021,7835,0 1024,78 1024,51 1024,24 1023,96 1023,68 1023,39 1023,09 1022,78 1022,46 1022,1435,5 1025,15 1024,89 1024,62 1024,34 1024,05 1023,76 1023,46 1023,15 1022,83 1022,5136,0 1025,53 1025,27 1024,99 1024,71 1024,42 1024,13 1023,83 1023,52 1023,20 1022,8836,5 1025,91 1025,64 1025,37 1025,09 1024,80 1024,50 1024,20 1023,89 1023,57 1023,2537,0 1026,28 1026,02 1025,74 1025,46 1025,17 1024,87 1024,57 1024,26 1023,94 1023,6237,5 1026,66 1026,39 1026,12 1025,84 1025,54 1025,25 1024,94 1024,63 1024,31 1023,9938,0 1027,04 1026,77 1026,49 1026,21 1025,92 1025,62 1025,31 1025,00 1024,68 1024,3638,5 1027,42 1027,15 1026,87 1026,58 1026,29 1025,99 1025,69 1025,37 1025,05 1024,7239,0 1027,79 1027,52 1027,24 1026,96 1026,66 1026,36 1026,06 1025,74 1025,42 1025,0939,5 1028,17 1027,90 1027,62 1027,33 1027,04 1026,74 1026,43 1026,11 1025,79 1025,4640,0 1028,55 1028,28 1027,99 1027,71 1027,41 1027,11 1026,80 1026,48 1026,16 1025,8340,5 1028,93 1028,65 1028,37 1028,08 1027,79 1027,48 1027,17 1026,85 1026,53 1026,2041,0 1029,30 1029,03 1028,75 1028,46 1028,16 1027,85 1027,54 1027,23 1026,90 1026,57

TP N°2 : DOSAGE DE L'AZOTE AMMONIACAL
I. INTRODUCTIONL'azote minéral dissous dans l'eau de mer existe sous forme d'azote gazeux et
d'ions ammonium, nitrite et nitrate. L'azote ammoniacal est sous forme d'ions ammonium (NH4
+) dans la gamme de pH des eaux marines. Ils occupent une place particulière dans le cycle de l'azote. Ils sont assimilés par les végétaux aquatiques comme les ions nitrates et participent donc à la production de matière organique. Par ailleurs, la dégradation de l'azote organique particulaire ou dissous donne lieu à la formation d'azote ammoniacal (qui peut s'oxyder ensuite en nitrite puis nitrate). Ils participent donc à l'activité autotrophe et à l'activité hétérotrophe. Enfin, le zooplancton contribue à enrichir l'eau de mer en azote ammoniacal par excrétion directe.
Les teneurs en ions ammonium dans l'eau de mer sont généralement comprises entre 0 et 3 µM.
Dans le cadre de ce TP, l'azote ammoniacal sera mesuré selon une méthode basée sur la mesure de la fluorescence d'un complexe d'orthophtaldialdéhyde.
II. ECHANTILLONAGELes échantillons doivent être recueillis directement après le prélèvement avec le
maximum de précaution. Faire attention à la contamination atmosphérique (interdiction de fumer). Il est préférable de doser les échantillons immédiatement.
La difficulté principale du dosage réside dans le risque permanent de pollution (atmosphère polluée au laboratoire, fumée de tabac,...).
III. PRINCIPE DU DOSAGE La méthode de dosage fluorimétrique remplace l’ancienne méthode au bleu
d’indophénol, qui donne souvent des résultats incohérents notamment lorsque les concentrations sont faibles [Holmes et al. 1999]. La méthode fluorimétrique permet de déterminer avec exactitude une large gamme de concentrations d’ammonium et de salinités au niveau submicromolaire. En plus de résoudre des problèmes analytiques, cette méthode simplifie la procédure de collecte et de conservation des échantillons. Elle fait appel à un seul réactif constitué d’orthophtaldialdéhyde « OPA », de sulfite de sodium et de borate de sodium qui demeurent stables pendant des mois lorsque conservés à l’obscurité. L’OPA est également utilisé pour quantifier les acides aminés, mais lorsqu’il est combiné au sodium sulfite il perd sa sensibilité à ceuxci et devient spécifique de l’ammonium. Le réactif et l’échantillon peuvent être mélangés immédiatement après le prélèvement et la réaction s’effectue en 2 3
15

heures à T° ambiante. Cette méthode donne des résultats très reproductibles même aux très faibles concentrations d’ammonium.
IV. RÉACTIFSSolution stock de borate #:
Dissoudre 80 g de tétraborate de sodium dans 2L d’eau déminéralisée dans un flacon plastique transparent. Cette solution, bouchée hermétiquement, doit être conservée à T° ambiante (risque de précipitation au froid). Elle est stable plusieurs mois. Si des particules sont visibles dans la solution dès sa préparation ou après une longue conservation, la filtrer avant usage sur 12 µm.
Solution stock d’OPA dans l’éthanol #:Dissoudre 5 g d’OPA dans 125 ml d’éthanol. La dissolution complète prend
quelques minutes, agiter de temps en temps. Le flacon devra impérativement être protégé de la lumière et conservé au noir. Cette solution, en flacon de verre hermétiquement bouchée (de préférence avec un bouchon à joint de silicone téflonné), se conserve au moins 1 an au frigo. Noter que la solution est initialement de couleur jaune clair et qu’elle se décolore après quelques jours au froid sans perdre ses propriétés.
Solution stock de sulfite :#Dissoudre 200 mg de sulfite de sodium dans 25 ml d’eau déminéralisée. Cette
solution, en flacon hermétiquement bouché, peut se conserver au moins une semaine à T° ambiante. Cependant, en raison du risque d’oxydation du sulfite par l’oxygène de l’air, il est préférable de la préparer le jour de la fabrication du réactif de travail (WR).
Réactif de travail (WR – working reagent)#Dans une grande bouteille en PE ou verre sombre (>2L), ajouter 2 L de borate,10
ml de sodium sulfite et 100 ml d’OPA. Ce mélange est stable 3 mois si il est conservé au noir et à T° ambiante.
V. MODE OPÉRATOIRE
V.1. Dosage
A 100 ml d'échantillon, ainsi que pour les solutions standards et les blancs (Attention : l’ajout des réactifs s’effectue dans les flacons à bouchons noirs ou le volume de 100 ml a été repéré) :
Ajouté 5 ml de réactif de réactif de travail.La réaction demande 2 h au moins pour se développer à la température ambiante
(20°C). En plaçant les échantillons à l'étuve à 40°C, le temps de réaction peut être réduite à 1 heure. La fluorescence est mesuré avec le fluorimètre "Trilogy" équipé d'un module pour la fluorescence dans le domaine des UV.
16

V.2. Etalonnage
Peser 66 mg de sulfate d'ammonium (NH4)2SO4 et les dissoudre dans 200 ml d'eau déionisée (milliQ). Ajouter 1 goutte de chloroforme, compléter à 1000 ml. Conservée
au réfrigérateur, cette solution est stable plusieurs mois. #
Préparation de la gamme étalon :A partir de la solution mère, réaliser une solution fille à 100 µM puis des solutions
standards à 0, 0.25, 0.5, 1, 2, 3 et 5 µM dans de l'eau de mer pauvre en ammonium. Réaliser également une droite de calibration dans de l'eau Ultrapur afin de mettre en évidence un éventuel effet de matrice
V.3. Détermination des blancs
Sur un échantillon d'eau de mer, mesurer la valeur de fluorescence immédiatement après l'ajout des réactifs. La formation du complexe n'ayant pas eu le temps de se former, la valeur de fluorescence mesurée est uniquement du à la fluorescence de l'eau de mer et des réactifs sans formation de complexe fluorescent. Cette valeur de blanc est à déduire des fluorescence mesurées pour les échantillons.
Penser à déduire systématiquement la valeur de fluorescence mesurée sur l'eau MilliQ à toutes les valeurs de fluorescences mesurées
VI. MANIPULATION
• Réaliser les deux gammes étalons et doser les échantillons en effectuant au minimum 3 réplicats.
• A la fin de la manipulation, rincer abondamment les flacons "ammonium" (c'estàdire avec un bouchon noir), ajouter 100 ml d'eau déionisée (milliQ) et 5 ml réactif de travail Fluorescence et ranger les flacons.
• Exprimer les résultats en µmol.kg1, en µg.l1 (N) et en µM. • Comparer les résultats des deux droites de calibration et discuter de
l'importance de l'effet de matrice pour cette méthode.
17

18

TP N°3 : DOSAGE DES ORTHOPHOSPHATES DANS L'EAU DE MER.
I. INTRODUCTIONLe phosphate minéral dissous dans l'eau de mer est essentiellement sous forme
d'orthophosphates (H2PO4 et HPO4
2). La concentration en orthophosphates dans l'eau de mer dépend de phénomènes physiques (advection, diffusion), chimiques (précipitation, dissolution) et biologiques (consommation, excrétion, régénération). Elle varie de moins de 0,01 µM dans l'eau de surface (pendant la période de croissance du phytoplancton) à 3 µM en profondeur.
II. PRINCIPELa méthode utilisée pour le dosage des orthophosphates a été mise au point par
Murphy et Riley (1962). Les ions orthophosphates sont susceptibles de réagir avec le molybdate d'ammonium en milieu acide pour former un complexe jaune, le phosphomolybdate d'ammonium. Par réduction de ce complexe, on obtient une coloration bleue. L'utilisation de l'acide ascorbique comme agent réducteur donne les résultats les plus reproductibles et il a l'avantage de pouvoir être utilisé dans un réactif unique : molybdate d'ammonium, acide ascorbique, acide sulfurique et antimonyl tartrate de potassium. L'antimoine fourni par l'antimonyl réduit le temps de développement de la coloration de 24 h à quelques minutes.
III. MODALITES DU DOSAGE
III.1. Limite d'emploi
De 0,03 à 5 µM
Précision : 3 ± 0,03/n1/2 µM
0,3 ± 0,02/n1/2 µMLa précision est donnée pour un seuil de confiance de 0,95.
III.2. Echantillonnage
Les échantillons sont recueillis dans des flacons (bouchons blancs) rincés 2 fois avec l'eau à analyser. La mesure doit être effectuée si possible dans la demiheure qui suit le prélèvement et avant 2 h sinon les échantillons doivent être congelés à 20 °C : ceci permet de les conserver pendant plusieurs mois.
19

III.3. Réactifs
Solution d'acide ascorbique :Dissoudre 2 g d'acide ascorbique dans 50 ml d'eau déionisée (milliQ).
Solution de molybdate d'ammonium : #
Dissoudre 15 g de paramolybdate d'ammonium (NH4)6Mo7O24,4H2O dans 500 ml d'eau déionisée (milliQ). Conserver dans une bouteille en plastique, hors de la lumière (solution très stable).
Acide sulfurique : #
Ajouter 140 ml d'acide sulfurique (d = 1,82) à 900 ml d'eau déionisée (milliQ). Laisser refroidir et conserver dans une bouteille en verre.
Solution émétique: #
Dissoudre 0,34 g d'"émétique" (antimonyl tartrate de potassium), dans 250 ml d'eau déionisée (milliQ) (en chauffant si nécessaire). La solution est stable plusieurs mois si elle est conservée au frais.
Réactif combiné: #
Les solutions 2,3 et 4 sont préparées au préalable. Mélanger 50 ml de molybdate d'ammonium, 125 d'acide sulfurique, et 25 ml d'émétique. Ce mélange est stable quelques mois.
III.4. Mode opératoire
DosageA 100 ml d'échantillon, ajouter 1 ml de la solution d'acide ascorbique, mélanger
puis ajouter 4 ml du réactif combiné et agiter.Après 30 mn et avant 2 h, mesurer l'absorbance de la solution à 700 nm par rapport
à l'eau déionisée (milliQ).Corriger la mesure en soustrayant le blanc des réactifs.Remarque : la température des échantillons doit être comprise entre 15 et 30 °C.
Détermination des blancs Sur un échantillon d'eau de mer, mesurer la valeur de absorbance en ajoutant que
du réactif combiné sans acide ascorbique. La complexe coloré ne pouvant se former en l'absence d'acide ascorbique, la valeur d'absorbance mesurée est uniquement du à la turbidité de l'eau de mer ou du réactif combiné. Cette valeur de blanc réactif est à déduire des absorbances mesurées pour les échantillons.
La valeur de la Turbidité de l'eau de mer peut être importante pour les échantillons d'eau de mer de surface jusqu'à une profondeur de 10 m.
EtalonnageLa salinité n'a pas d'influence sur le développement de la coloration : l'étalonnage
est effectué dans de l'eau déionisée (milliQ).
20

Solution mère de phosphate# :Dissoudre 68 mg de phosphate acide de potassium anhydre (KH2PO4) dans 1000
ml d'eau déionisée (déionisée (milliQ)).Solutions pour l'étalonnage :Préparer des solutions à 0, 1, 2, ... 5 µM. Ne pas oublier de préparer un blanc
simultanément.
IV. MANIPULATIONLa manipulation consiste à réaliser le dosage des orthophosphates d'une eau de
mer.Préparer la courbe d'étalonnage et doser l'eau en effectuant au minimum 3
réplicats.Exprimer les résultats en µmol.kg1, en µg.l1 (P), en µg.l1 (PO4), et en µM.
21

22

TP N°4 : MESURE DU pH, DE L'ALCALINITE TOTALE ET DE LA TENEUR EN CALCIUM D'UNE
EAU DE MER
I. INTRODUCTIONL'étude du cycle biogéochimique du carbone nécessite de comprendre le système
gaz carboniquebicarbonatescarbonates. Ce système peut être totalement défini, comme nous allons le montrer, à l'aide de deux variables : le pH et l'alcalinité totale.
Le pH est par définition égal au cologarithme de l'activité des ions H3O+ en solution : pH = log(H3O+). Dans l'eau de mer, il est généralement compris entre 8.0 et 8.3. Il dépend en particulier de la teneur en dioxyde de carbone (c'estàdire de l'équilibre photosynthèserespiration), de la température et de la pression. Il peut atteindre 8.5 en présence d'une forte activité photosynthétique consommatrice de CO2. En revanche, il diminue endessous de la couche euphotique en raison de l'enrichissement en CO2 dû à la minéralisation des déchets organiques. Des valeurs inférieures à 7.5 se rencontrent dans la couche des minima d'oxygène du Pacifique Nord.
La notion d'alcalinité totale dans l'eau de mer a été redéfinit par Dickson en 1981 comme "... le nombre de moles d'ions hydrogènes équivalent à l’excès d'accepteurs de protons (bases formées à partir d'acides faibles donc la constante de dissociation K < 10 4.5 à 25°C et à une force ionique nulle) par rapport aux donneurs de protons (acide avec K > 10 4.5) dans 1 kilogramme d'échantillon ". On a donc
AT = [HCO3] + 2[CO3
2−] + [B(OH)4] + [OH−] + [HPO4
2−] + 2 [PO43−] + [H3SiO4
] + [NH3] + [HS−] + [...] [H+] [HSO4
] [HF] [H3 PO4] – [HNO2] […]
Par définition, l'alcalinité due aux carbonates est égale à AC = [HCO3]+2[CO3
2].L'alcalinité peut être exprimé comme une différence portées par les ions majeurs
de l'eau de mer (voir TD5). Il devrait exister, conformément à la loi de Dittmar, un rapport sensiblement constant entre alcalinité et salinité. On calcule souvent l'alcalinité normalisée à la salinité 35 :
AT35 = AT*(35/S)Cette valeur devrait être unique pour une eau de mer de salinité donnée, mais en
réalité elle varie notablement. Il n'est donc pas possible de déduire l'alcalinité totale de la salinité, il faut la mesurer indépendamment.
Les fortes variations relatives de l'alcalinité, en contradiction apparente avec la loi de Dittmar, sont expliquées par le fait qu'elle résulte de la différence entre le nombre de charges positives et négatives des ions majeurs. Un écart à la loi de Dittmar de
23

3/10000ème sur les charges positives (ou négatives), difficilement perceptible au niveau de la salinité, entraîne une variation de 7,8% de l'alcalinité totale. Ces variations sont à attribuer essentiellement aux écarts des ions calcium à la loi de Dittmar. En effet, le calcium est extrait des eaux de surface par de nombreux organismes marins qui construisent leurs coquilles avec du carbonate de calcium et il est remis en solution en profondeur au cours de la minéralisation.
II. PRINCIPELe principe de la manipulation est de déterminer le pH et l'alcalinité totale d'une
eau de mer de surface afin de déterminer les variables suivantes : [H+], [OH], [B(OH)4], pCO2, [CO2*], [HCO3], [CO32] et AT qui caractérisent son "système carbonate".
Illustration 3: Diagramme logarithmique montrant les variations des systèmes de l'acide carbonique et de l'acide borique avec le pH (CopinMontégut, 1989).
Voici 5 relations (vues en cours) qui permettent de décrire le système carbonate dans l'eau de mer. Les valeurs de KH, KA1, KA2, KB et Ke peuvent être calculés à la température et à la salinité de l'échantillon à partir du formulaire fourni en cours.
(1) KH = [CO2*]/pCO2
CO2* : acide carbonique mesuré analytiquement ([CO2*] = [CO2] + [H2CO3])(2) KA1=[HCO3
][H+]/[CO2*]
(3) KA2=[CO32][H+]/[HCO3]
(4) [B(OH)4] = 1.2 105 S (KB/([H+]+KB))
(5) Ke = [H+][ OH]
24

Nous nous baserons ici sur une définition simplifiée de l'alcalinité totale pour effectuer les calculs
(6) AT = [HCO3]+2[CO3
2]+[B(OH)4]+[OH][H+]
Ce système contient 8 inconnues [H+], [OH], [B(OH)4], pCO2, [CO2*], [HCO3
], [CO32] et AT. Pour une température, une salinité et une pression données, il suffit donc de se fixer (ou de mesurer) deux des concentrations du système pour connaître toutes les autres.
Parmi ces constituants, quatre seulement sont mesurables en pratique. Ce sont le pH, l'alcalinité totale AT, la pression partielle de CO2 et le carbone inorganique dissous total CT (CT = [CO2*] + [HCO3] + [CO32]).
Dans le cas où les variables pH et AT sont mesurées, c'estàdire notre cas, on calcule d'abord l'alcalinité due aux carbonates AC (AC=[HCO3]+2[CO32]) à partir des équations 4, 5 et 6 :
AC=AT 1.2 105 S (KB/([H+]+KB)) Ke/[H+] + [H+] (cf remarque)On a dès lors :
pCO2 = (AC/KH * [H+]/KA1) / (1+2 KA2/[H+])
[CO2*] = (AC * [H+]/KA1) / (1+2 KA2/[H+])
[HCO3] = AC / (1+2 KA2/[H+])
[CO32] = (AC * KA2/[H+]) / (1+2 KA2/[H+])CT = [CO2*] + [HCO3] + [CO32]
Remarque : Dans le domaine des pH habituels des eaux de mer, les concentrations des ions H + et
OH sont négligeables devant celles des ions HCO3, CO3 et B(OH)4 (Fig. 1). Il est donc
généralement suffisant de calculer l'alcalinité des carbonates sous la forme simplifiée suivante (qui
rend inutile la connaissance du produit ionique de l'eau) :
AC=AT 1.2 105 S (KB/([H+]+KB))
Compte tenu de la valeur de KB (environ 108,6 à 25°C), on peut voir que l'alcalinité des carbonates
représente une part importante de l'alcalinité totale : 94% à pH=8.3; 96,5% à pH=8,0. C'est pourquoi il
peut être utile dans un calcul préliminaire de faire l'approximation AC=AT.
III. MODE OPERATOIRE DÉTERMINATION DU PH DE L'EAU DE MER PAR COLORIMÉTRIE
La détermination du pH de l'eau de mer reste un des domaines les plus confus de la chimie océanographique. Ici le pH est mesuré par colorimétrie selon le protocole mis au point par Clayton et Byrne [1993].
III.1. Principe :
La valeur du pH est déterminée en ajoutant un indicateur coloré à l'eau de mer. Pour les indicateurs colorés de type sulfonephtaleine, comme le violet de mcrésol, la réaction en jeu déterminant le pH de l'eau de mer est la seconde dissociation :
25

HI(aq) = H+(aq) + I2 (aq) (1)I représente l'indicateur coloré, présent en faible quantité dans l'échantillon. La
concentration en ion hydronium de l'échantillon peut alors être déterminée comme suit :
pH= pK2 + log [I2]/[HI] (2)Cette approche se base sur le fait que les différentes formes de l'indicateur coloré
ont des spectres d'absorption substantiellement différents. L'information comprise dans le spectre composite peut donc être utilisée pour estimer le rapport [I2]/[HI].
A une longueur d'onde , l'absorbance mesurée dans une cellule de trajet optique λ l est estimée par la loi de BeerLambert :
Aλ
l=ε λ (HI⁻ ) [HI⁻]+ε λ ( I²⁻ ) [ I²⁻]+B λ+e (3)
B correspond au bruit de fond d'absorbance de l'échantillon et e est un terme d'erreur dû au bruit instrumental. Si les valeurs des coefficients d'extinction ελ(HI) et ελ(I2) ont été mesurées en fonction de la longueur d'onde, les mesures d'absorbance à deux longueurs d'onde (ou plus) peuvent être utilisées pour estimer le rapport [I2] / [HI].
Si seulement deux longueurs d'ondes sont utilisées, et si on considère que le bruit de fond peut être efficacement éliminé par une procédure soustractive, l'équation (3) peut être réarrangée de la manière suivante (en considérant qu'il n'y a pas d'erreur instrumentale) :
[ I²⁻ ][HI⁻ ]
=A1/A2−ε1 (HI⁻ ) /ε2 (HI⁻)
ε1 ( I²⁻ ) /ε2 ( HI⁻)−(A1/ A2) ε2 ( I²⁻ ) /ε 2 (HI⁻) (4)Les valeurs 1 et 2 correspondent aux longueurs d'onde choisies (578 et 434 nm).
Pour une sensibilité optimale, on choisit les longueurs d'ondes qui correspondent aux maxima d'absorbance de la forme basique I2 et de la forme acide HI. Les différents termes correspondent aux coefficients d'extinction des espèces spécifiéesε aux longueurs d'onde 1 et 2.
III.2. Manipulation
Réactifs
Solution de violet de m crésol #Une solution concentrée (minimum 2 mM) d'indicateur coloré, ajustée à 7,9 ± 0,1
unités de pH (choisie pour correspondre à la mesure de pH d'un profil océanique) est nécessaire. Ceci implique pour le violet de mcresol un rapport A1/A2 ~ 1,6. En pratique, 76,5 mg de mcresol sont dissous dans 100 mL d'eau distillée. Le pH est ensuite ajusté par ajouts d'une solution de NaOH 2 M.
III.3. Échantillonnage
Le prélèvement s'effectue immédiatement après celui destiné à l'analyse de l'oxygène dissous et, éventuellement, d'autres gaz. Dans l'idéal, les échantillons sont directement collectés dans des cellules de mesure pour spectrophotomètre,
26

susceptibles d'être scellées. Dans le cadre des travaux pratiques, de telles cellules ne sont pas disponibles. Ici, des flacons d'un volume de 100 ml environ sont rincés deux fois avec l'eau à analyser avant d'être remplis (en évitant tout brassage d'air).
III.4. Mesure
Il est nécessaire d'attendre que les échantillons aient atteint la température de la pièce (attendre au moins une heure). Les mesures doivent être réalisées au minimum en triplicat.
1. Mesure de l'absorbance de la cellule + eau de mer.Rincer puis remplir délicatement une cuve de spectrophotomètre avec de l'eau de
mer. Nettoyer et sécher l'extérieur de la cuve. Mesurer et noter les absorbances à trois longueurs d'ondes : une longueur d'onde non absorbante (730 nm pour le violet de mcresol) et deux longueurs d'ondes correspondant aux maxima d'absorbance respectivement de la forme basique I2 et de la forme acide HI de l'indicateur coloré (soit 578 et 434 nm). Penser à refaire un « autozero » avec de l'eau distillé à chaque changement de longueur d'onde.
2. Ajout de l'indicateur coloré à l'eau de mer Sur le restant d'eau de mer dans le flacon, ajouter entre 0,1 et 0,2 cm3 d'indicateur
coloré concentré et agiter. La quantité d'indicateur coloré nécessaire est celle qui va produire des valeurs d'absorbance comprises entre 0,4 et 1 pour les deux pics d'absorbance.
3. Mesure de l'absorbance de la cellule + eau de mer + indicateur coloré.Remplir la cuve de spectrophotomètre et mesurer à nouveau l'absorbance aux trois
longueurs d'onde utilisées précédemment. La cuve doit être positionnée avec un alignement constant entre les mesures d'absorbance du bruit de fond de l'eau de mer et de l'indicateur coloré.
III.5. Calcul et expression des résultats
Correction des mesures d'absorbanceA chacune des trois longueurs d'ondes, soustraire les absorbances de mesure du
bruit de fond (sans indicateur coloré) des absorbances correspondantes mesurées avec l'indicateur coloré.
L'absorbance mesurée à la longueur d'onde non absorbante est utilisée pour estimer et corriger les glissements de la ligne de base, qui sont dus à des erreurs de positionnement de la cellule ou des changements instrumentaux. Nous considérons ici que l'amplitude des variations de la ligne de base est identique sur l'ensemble du spectre visible. Pour cela, soustraire la variation mesurée d'absorbance de bruit de fond corrigée aux longueurs d'onde 1 et 2 pour obtenir les valeurs finales d'absorbance, corrigées à chaque longueur d'onde.
Ces valeurs finales d'absorbance sont utilisées pour calculer A1/A2, le rapport d'absorbance qui décrit l'amplitude de protonation de l'indicateur coloré.
27

Calcul du pH de l'eau de mer + indicateur coloréLe pH de l'eau de mer + indicateur coloré dans la cellule est déterminé à partir de
l'équation suivante :
pH=pK 2+log( A1/ A2−ε1 (HI⁻ ) /ε2 (HI⁻)
ε1 ( I²⁻ ) /ε2 ( HI⁻)−(A1/ A2) ε2 ( I²⁻ ) /ε 2 (HI⁻) ) (5)
pK2 est la constante de dissociation acide de l'espèce HI (exprimée sur l'échelle d'ion hydrogène total en mol.kgsol1). A1 et A2 sont les absorbances corrigées, mesurées aux longueurs d'ondes correspondant aux maxima d'absorbance des formes basique et acide. Les différents termes des coefficients d'extinction ε correspondent aux valeurs mesurées pour les espèces spécifiées aux longueurs d'ondes 1 et 2 (Table 1).
ε1(HI) / ε2(HI) 0,00691
ε1(I2) / ε2(HI) 2,2220
ε2(I2) / ε2(HI) 0,1331
Table 1: Rapport de coefficients d'extinction pour le violet de mcresolLa constante d'équilibre K2 est fonction de la salinité et de la température ; elle a été
déterminée par des mesures précises de laboratoire. Pour le violet de mcresol, elle peut être calculée de la manière suivante :
pK2=1245,69
T ° K
+3,8275+0,00211 (35−S ) (6)
avec 293 <T°K< 303 et 30 < S < 37.
IV. MODE OPERATOIRE DÉTERMINATION DE L'ALCALINITÉ TOTALE PAR TITRAGE ACIDE DE L'EAU DE MER :
IV.1. Principe :
L'alcalinité totale est déterminée par potentiométrie. L'échantillon d'eau de mer de volume V est titré dans un récipient hermétique par une solution d'acide chlorhydrique de titre connu C.
En tout point du dosage l'électroneutralité est vérifiée, ce qui se traduit par l'équation suivante si on considère que v est le volume d'acide ajouté :
Cv/(V+v) = AT*V/(V+v) [HCO3]2[CO32][B(OH)4]Ke/[H+]+[H+]Remarque : Le but de travailler dans un récipient hermétique est de considérer que l'on travaille
dans un système fermé (on opère à CO2 constant). Dans le cadre de cette manipulation, les échanges de CO2 avec l'atmosphère sont possibles mais nous considérons qu'ils ne sont pas suffisamment rapides pour induire une modification de CO2. En d'autres termes, l'approximation visant à considérer que l'on travaille en système fermé reste valable. Par ailleurs, tous les calculs sont généralement réalisés en concentrations massiques et non en concentrations volumiques, donc ils sont légèrement simplifiés ici.
La courbe présente deux points d'inflexion. On peut montrer que pour le deuxième, on a [HCO3]= [H+] (Fig. 1). En reportant cette égalité dans la relation
28

précédente, et en remarquant qu'à ce pH acide [CO32], [B(OH)4] et [OH] sont négligeables, on obtient :
v2 . C = AT . V d'où la mesure de l'alcalinité totale de l'eau de mer AT = v2 C/V
v2 est égal au volume d'acide versé pour atteindre le 2ème point d'inflexion.
IV.2. Mode opératoire :
Titrer un volume précis de 100 ml en ajoutant l'acide chlorhydrique 0.02 M préparé à partir de la solution M et en suivant le potentiel d'une électrode à Hydrogène. Titrer rapidement au début puis de plus en plus lentement près du deuxième point équivalent. Déterminer ce point par la méthode des tangentes (En réalité, pour plus de précision, on utilise divers perfectionnements de la méthode de Gran qui consiste à travailler sur l'ensemble des points au voisinage du point équivalent).
V. MODE OPERATOIRE DOSAGE DU CALCIUM :
V.1. Principe :
Le principe du dosage est de complexer les ions calcium avec l'EDTA (acide éthylène diamine tétraacétique). On utilise comme indicateur coloré le calcon qui forme un complexe rouge avec le calcium. Lors du dosage, les ions calcium réagissent avec l'EDTA; d'abord les ions libres puis ceux combinés avec l'indicateur qui vire alors de la couleur rouge à la couleur bleu clair.
On se place à un pH compris entre 12 et 13 pour précipiter le magnésium sous forme d'hydroxyde et éviter ainsi qu'il soit pris en compte dans le dosage.
V.2. Réactifs :
Hydroxyde de sodium 2M : dissoudre environ 8 g d'hydroxyde de sodium dans
100 ml d'eau fraîchement distillée. Conserver dans une bouteille en polyéthylène. #
EDTA solution titrée de Na 2 EDTA 0,01 M : dissoudre 0,931 g de sel disodique de l'acide éthylènediaminetétraacétique (C10H14N2O8Na2,2H2O) dans de l'eau et compléter à 250 ml dans une fiole jaugée.
Calcon : mélanger soigneusement 0,2 g d'acide calcone carboxylique
(C21H14N2O7S,3H2O) et 100 g de chlorure de sodium. #
Note : cet indicateur est également désigné HSN. Un autre indicateur utilisé pour le dosage du calcium est la calcéine (C30H26N2O13).
V.3. Mode opératoire :
Diluer 5 fois votre eau de mer. A l'aide d'une pipette, introduire 50 ml exactement de cette solution dans un bécher. Ajouter 2 ml de la solution d'hydroxyde de sodium (2M) et une pincée de l'indicateur acide calcone carboxylique.
Mélanger et doser immédiatement. Ajouter la solution d'EDTA tout en continuant à mélanger. Verser lentement en fin de dosage. Le virage est atteint lorsque la
29

couleur devient nettement bleue. La couleur ne doit plus changer avec l'ajout d'une goutte supplémentaire de la solution d'EDTA.
V.4. Expression des résultats :
[Ca2+] = 5000.C1.M1/V2 (mM)V1 : volume de la solution d'EDTA utilisé pour le dosage (ml).V2 : volume d'échantillon dosé (ml)C1 : Concentration de la solution d'EDTA (M)
VI. MANIPULATION• Réaliser trois dosages de calcium. • Déterminer en triplicat l'alcalinité totale et le pH • Calculer l'ensemble des variables qui permettent de définir le système
"carbonate" de votre eau.• Déterminer si votre eau de mer est en équilibre par rapport à la calcite.• Déterminer si l'eau de la calanque d'Endoume est une source ou un puit de
CO2 pour l'atmosphère.
30

TP N°5 : DOSAGE DE L'AZOTE ORGANIQUE DISSOUS ET DE L'AZOTE ORGANIQUE
PARTICULAIREMéthode de Raimbault & Slawyk (1991) et PujoPay & Raimbault (1994)
I. INTRODUCTIONL'étude du cycle biogéochimiques de l'azote et du phosphate repose sur la
connaissance et la quantification des différentes formes de ces éléments. Il existe de nombreuses méthodes pour déterminer les formes organiques. Nous allons utiliser une méthode développée par Pujopay et Raimbault en 1994, méthode qui permet de doser simultanément l'azote et le phosphate, particulaire (TotNpart OrgNpart & TotPpart) ou total (TotN & TotP). On peut ainsi en déduire l'azote et le phosphate total dissous (TotNdiss & TotPdiss). Si par ailleurs on mesure les formes minérales dissoutes de l'azote et du phosphate, on peut en déduire la fraction organique dissoute (OrgNdiss & OrgPdiss).
Les différentes fractions de l'azote et du phosphate peuvent être représentées ainsi :
Tot-N
Tot-Npart
Org-N
dissTot-N
diss
org-Npart
Min-Ndiss
NO NONH4 23- -+
Tot-P
part
Org-P
dissTot-P
diss Min-Pdiss
Tot-P
org-Ppart Min-Ppart
o-P(orthophosphates)
poly-P(polyphosphates)
31

II. PRINCIPELe principe du dosage est de minéraliser la matière organique par autoclavage en
milieu oxydant, alcalin puis acide, et de doser les éléments minéraux formés. Dans les conditions expérimentales, toute la matière organique est transformée en nitrate pour l'azote et en orthophosphates pour le phosphate.
III. RÉACTIF
Réactif pour la minéralisation # :Ajouter directement dans la dosipette de 250 ml placée sur la balance, 15 g de
persulfate de potassium K2S2O8 (Merck 5092), 7.5 g d'acide borique (Merck 165), 70 ml d'une solution d'hydroxyde de sodium NaOH à 60 g/l et 170 ml d'eau déionisée (milliQ). Le réactif doit être stocké à l'abri de la lumière
IV. MANIPULATIONPour une question de temps de manipulation, nous nous intéressons uniquement à
déterminer les différentes fractions de l'azote. Préparer vos échantillons à minéraliser puis votre gamme étalon pour le dosage des ions nitrate + nitrite. Quand l'autoclave est en route, passer votre gamme étalon au technicon.
Echantillonnage :Les prélèvements sont effectués à la bouteille Niskin après les prélèvements pour
l’analyse des gaz dissous. Pour obtenir une précision correcte, il doit être demandé aux précédents utilisateurs de ne surtout pas toucher l'embout de la bouteille Niskin avec leurs doigts (des gants sont généralement recommandés). En cas de contamination, nettoyer l'embout avec de l'acide chlorhydrique 10%.
Pour chaque prélèvement, rincer 3 fois le flacon avec l'eau de mer.
Minéralisation :Réaliser les opérations suivantes en parallèle :• Pour obtenir la concentration en azote total, prélever environ 40 ml d'eau de
mer directement dans les flacons autoclavables et ajouter 2,5 ml de réactif (faire 3 réplicats).
• Pour obtenir le blanc réactif, préparer 1 flacon avec seulement 2,5 ml de réactif (faire 2 réplicats).
• Pour obtenir la concentration en azote particulaire, prélever 250 ml d'eau de mer dans le flacon en plastique prévu à cet effet et les filtrer sur filtre GF/F (Les filtres dans le dessicateur ont été préalablement calcinés au four à 450°C pendant 4 h et nettoyés à l'acide chlorhydrique M). Placer ensuite le filtre dans un flacon autoclavable et ajouter 40 ml d'eau déionisée (milliQ) et 2,5 ml de réactif (faire 3 réplicats).
• Pour obtenir le blanc Filtre, préparer un flacon avec 40 ml d'eau déionisée (milliQ), 2,5 ml de réactif et 1 filtre (faire 2 réplicats).
32

Placer l'ensemble des flacons autoclavables, correctement fermés, dans l'autoclave. VERIFIER QU'IL Y A DE L'EAU DANS L'AUTOCLAVE AVANT DE LE FAIRE FONCTIONNER. Minéraliser pendant 30 minutes à 120°C (1 bar). Laisser refroidir les échantillons à température ambiante avant de procéder aux analyses.
Pour le blanc Réactif, ajouter 40 ml d'eau déionisée (milliQ) avant d'effectuer l'analyse des sels nutritifs.
Analyse des sels nutritifs :Le dosage de l'azote des ions nitrate + nitrite est réalisé à l'aide d'un autoanalyseur
Technicon selon le protocole développé en V.
Résultats :Déterminer les concentrations de vos échantillons :• en nitrate (NO3
). On considère dans notre cas que la concentration en nitrite est négligeable devant la concentration en nitrate.
• en azote total :TotN = (HtotN Heffet de sel Hblanc réactif)*F*(42,5/40)
avec H qui correspond à une hauteur de pic et F au facteur déterminé au paragraphe V
• en azote particulaire (prendre en considération le volume filtré)TotNpart = (HtotNpart Hblanc filtre)*F*(42,5/Vfiltré)
En déduire les concentrations organiques dissoutes. Exprimer vos résultats en µM et en µg.l1 de N.
N'oubliez pas de bien rincer vos flacons en fin de manipulation.
33

V. DOSAGE DE L'AZOTE SOUS FORME DE NITRATE ET DE NITRITE (NO3 + NO2)
V.1. Principe :
Les ions nitrate dans l'eau de mer sont réduits en ions nitrite (d'une manière supposée quantitative dans ce TP) par passage sur une colonne CdCu. Les ions nitrite ainsi formés ainsi que ceux initialement présents forment un diazoïque avec la sulfanilamide en milieu acide (pH<2) selon la réaction :
NH2
SO2
C6
H4NH
2 + NO
2 + 2H+ => [NH
2SO
2C
6H
4NN]+ + 2H
2O
Le diazoïque est ensuite copulé avec le chlorhydrate de Nnaphtyl éthylènediamine pour fournir un colorant azoïque :
[NH2SO
2C
6H
4NN]+ + C
10H
7NHCH
2CH
2NH
2 =>
NH2
SO2
C6
H4N=NC
10H
6NHCH
2CH
2NH
2 + H+
La mesure de l'absorbance s'effectue à 543 nm.Par soustraction de la concentration propre en nitrite de l'échantillon (sans passage
par la colonne CdCu), il est possible de déterminer la concentration en nitrate après un étalonnage. Dans le cadre de ce TP, la concentration en azote minéral sous forme de nitrate + nitrite est considérée uniquement.
V.2. Modalités du dosage :
Limites d'emploi : de 0,01 à 30 µmol.l1
V.3. Réactifs et colonnes réductrices pour le dosage du nitrate :
Réactif 1 : solution de Sulfanilamide #
Verser 5g de sulfanilamide (C6H8N2O2S) dans 500 ml d'eau déionisée (milliQ). Ajouter 50 ml d'acide chlorhydrique concentré (HCl 37%). Agiter. Compléter à 1000 ml d'eau déionisée (milliQ). Ajouter 1 ml de BRIJ (mouillant).
Réactif 2 : Solution de Nnaphtyl éthylènediamine dichlorhydrate #
Dissoudre 500 mg de Nnaphtyl éthylènediamine dichlorhydrate (C12H16Cl2N2) dans 1000 ml d'eau déionisée (milliQ).
Réactif 3 : Solution de chlorure d'ammonium #
Dissoudre 15 g de NH4Cl dans 1000 ml d'eau déionisée (milliQ). Ajouter 2 ml d'ammoniaque pour amener le pH autour de 8.
34

Solution étalon de nitrate à 5 mM (solution A) #
Dissoudre 0,506 g de nitrate de potassium KNO3 anhydre dans 1000 ml d'eau déionisée (milliQ). Ajouter 2 gouttes de chloroforme et conserver la solution à l'abri de la lumière dans un flacon hermétiquement fermé.
Solution étalon de nitrate à 100 µM (Solution B à préparer chaque jour)Dans une fiole de 100 ml préalablement rinçée, verser précisément 2 ml de la
solution A. Compléter à 100 ml avec de l'eau déionisée (milliQ).
Solutions standards : Rincer 4 fioles de 100 ml (marquées 1, 2, 5, 10) avec de l'eau déionisée (milliQ).
Verser précisément 1 ml de solution B dans la fiole 1, 2 ml dans la fiole 2, 5 ml dans la fiole 5 et 10 ml dans la fiole 10. Compléter à 100 ml chacune des fioles.
Selon les échantillons à analyser, des solutions standards plus ou moins concentrées peuvent être préparées.
Préparation de la colonne de cadmium #
Préparation du cadmium #
• Laver plusieurs fois le cadmium à l'acétone afin de le dégraisser.• Rincer scrupuleusement à l'eau déionisée (milliQ).• Nettoyer à l'HCl 10% plusieurs fois.• Rincer plusieurs fois à l'eau déionisée (milliQ).• Attaquer le cadmium avec de l'acide nitrique (HNO3) à 0.5 M (attention :
dégagement gazeux très dangereux, à ne pas respirer). Remuer plusieurs fois et attendre environ 5 minutes
• Rincer de nouveau à l'HCl 10% après avoir enlevé le maximum d'acide nitrique.
• Rincer à l'eau déionisée (milliQ).
• Mettre le cadmium dans une solution de sulfate de cuivre (CuSO4 20 g.l1). Bien remuer. Laisser agir. Le cadmium ne doit plus être en contact avec l'air à partir de ce moment.
• Rincer plusieurs fois avec le chlorure d'ammonium (NH4Cl 15 g.l1, réactif 3), sans jamais exposer le cadmium à l'air.
• Quand tout le sulfate de cuivre est éliminé, le cadmium peut être stocké dans la solution de chlorure d'ammonium.
Préparation de la colonne # :Prendre une colonne de verre. Installer un bouchon de laine d'argent à une de ses
extrémités. Prolonger la colonne par un tuyau en plastique à cette même extrémité. Remplir la colonne et le tuyau avec du NH4Cl et clamper le tuyau. Vérifier qu'il n'y ait aucune bulle d'air dans la colonne.
Remplir la colonne avec le cadmium préparé sans faire entrer de bulle d'air dans la colonne. Mettre un autre bouchon de laine d'argent à l'autre extrémité et fermer avec
35

le tuyau en ayant pris soin de le remplir de NH4Cl afin qu'il n'y ait aucune bulle dans la colonne.
Mise en place dans le circuit # :Le circuit nitrate étant fonctionnel (réactif 3 branché), enlever la colonne à changer
au niveau du robinet de fermeture. Le NH4Cl s'écoule alors librement. Enlever un côté du tuyau plastique de la colonne neuve. La raccorder au robinet. Le NH4Cl circule alors dans toute la colonne neuve. Après qu'il se soit écouler quelques secondes, enlever totalement le tuyau plastique et raccorder la colonne à l'autre extrémité.
Récupérer le cadmium de la colonne enlevée dans un flacon contenant du NH4Cl afin de le retraiter ultérieurement.
Lors de la première mise en place, faire passer pendant quelques minutes, comme pour un échantillon, une solution très concentrée de nitrate (solution B par exemple) afin d'activer la colonne neuve. Bien rincer le système afin de retrouver une ligne de base correcte.
V.4. Dosage (à réaliser en présence du responsable des TP) :
• Allumer les colorimètres.• Remplir les flacons qui alimentent en eau deionisée .• Placer la porte sur la pompe et la mettre en marche. Après quelques minutes,
vérifier la circulation dans les tuyaux et la régularité du bullage. Contrôler l'absence de fuite.
• Brancher les réactifs. Après 5 minutes, ouvrir la colonne CdCu si aucune bulle d'air ne risque d'y entrer. Attendre 5 minutes.
• Allumer les enregistreurs. Régler la ligne de base avec le zéro du colorimètre.• Lorsque la ligne de base est stable, passer les standards. Si les valeurs
obtenues sont correctes, vous pouvez passer vos échantillons.• En fin de dosage, fermer la colonne CdCu, débrancher les réactifs et mettre le
circuit en rinçage pendant 15 minutes avec de l'eau déionisée (milliQ). Eteindre l'enregistreur, enlever la porte de la pompe et éteindre le colorimètre.
Détermination de l'effet de turbidité dû à l'eau de mer ou effet de sel :Enlever le réactif 2, le remplacer par de l'eau déionisée (milliQ).Attendre la stabilisation de la nouvelle ligne de base puis passer quelques
échantillons de manière normale (garder les mêmes calibres que ceux choisis pour l'analyse des échantillons).
Les pics obtenus sont dûs à la turbidité de l'eau de mer et leurs hauteurs sont à retrancher des hauteurs des pics des échantillons d'eau de mer (environ 0.1 cm pour le circuit nitrate au calibre 3).
Mesures des hauteurs de pics, calcul des concentrations.
STANDARDS :
36

Relier les lignes de base qui encadrent les standards. Mesurer la hauteur des pics en cm à partir de cette ligne. Le facteur F est la moyenne des valeurs obtenues en divisant la concentration de l'étalon Ci par la hauteur Hi du pic correspondant :
F=1n∑
Ci
H i
La hauteur H des pics est fonction du calibre choisi, le facteur F qui en découle n'est applicable qu'aux échantillons analysés sur le même calibre (calibre 3 pour une concentration comprise entre 0 et 10 µmole.l1)
ECHANTILLONS D'EAU DE MER :
Relier les lignes de base qui encadrent une série d'échantillons (en principe tous les 1015 échantillons). Mesurer chaque pic à partir de cette ligne.
La concentration se calcule en retirant à chaque hauteur de pic la valeur de l'effet de sel (sur le même calibre) et en multipliant cette hauteur par le facteur F correspondant au même calibre :
C = (H effet de sel) x F.Sur la voie nitrate , le sel dosé est en fait nitrite + nitrate; pour avoir la valeur réelle
du nitrate il suffirait de retrancher aux valeurs trouvées la valeur du nitrite correspondant ([NO3
]= (H effet de sel) x F [NO2].
37

Références :Carpenter, J.H. 1965. The accuracy of the Winkler method for dissolved oxygen analysis. Limnol.
Oceanogr. 10, 135140.Carritt, D.E. & J.H. Carpenter. 1966. Comparison and evaluation of currently employed
modifications of the Winkler method for determining dissolved oxygen in seawater; a NASCO Report. J. Mar. Res. 24, 286318.
Clayton, T.D. and Byrne, R.H. 1993. Spectrophotometric seawater pH measurements: total hydrogen ion concentration scale calibration of mcresol purple and atsea results. DeepSea Res. 40 : 2115–2129.
CopinMontégut, G. 1989. PhysicoChimie de l'eau de mer. OCEANIS, volume 15.Dickson, A.G. 1981. An exact definition of total alkalinity and a procedure for the estimation of
alkalinity and total inorganic carbon from titration data. DeepSea Res. 28A : 609–623.Holmes R. M., Aminot A., Kérouel R., Hooker B.A., Petersen B.J. (1999) A simple and precise
method for measuring ammonium in marine and freshwater ecosystems, Can. J. fish. Aquat. Sci., 56, 18011808
Millero, F.J. & A. Poisson. 1981. International oneatmosphere equation of state of seawater. Deep Sea Research. 28A(6) : 625629.
Murphy, J. and J.P. Riley. 1962. A modified single solution method for the determination of phosphate in natural waters. Anal. Chem. Acta. 27. 3136.
Pujopay, M & P. Raimbault. 1994. Improvement of the wetoxidation procedure for simultaneous determination of particulate organic nitrogen and phosphorus collected on filters. Mar. Ecol. Prog. Ser., 105 : 203207.
Raimbault, P. & G. Slawyk. 1991. A semiautomatic, wet oxidation method for the determination of particulate organic nitrogen collected on filters. Limnol. Oceanogr., 36 : 405408.
Strickland et Parsons. 1968. A practical handbook of seawater analysis. Fisheries Research Board of Canada, Ottawa.
38

Rappel de statistiques :
Calcul des intervalles de confiance :
Considérons une variable ayant une distribution normale, ce qui est généralement le cas en chimie analytique. Considérons également que la moyenne de plusieurs résultats tend vers la valeur vraie : c'est à dire qu'il n'y a pas d'erreur systématique (comme par exemple, la dérive d'un appareil de mesure qui entraînerait systématiquement une sous ou une surestimation de la moyenne des résultats par rapport à la valeur vraie). On peut alors calculer l'intervalle de confiance autour de cette valeur moyenne.
Pour un nombre d'échantillons (N) inférieur à 30, l'intervalle de confiance est égal à
x±tSm
avec :t donné par la table de Student pour N 1 degrés de liberté
et
S=√∑i=1
N
( xi−x )2
N−1
et Sm=S
√N
Test de la validité du modèle linéaire :
Afin de comparer deux méthodes d’analyse d’un même composé, on peut calculer le coefficient de corrélation R entre ces deux variables. Le test qui permet de dire si le modèle linéaire peut être accepté se référe à la table des valeurs critiques du coefficient de corrélation affichée en salle de TP.
Il peut être intéressant d’effectuer une régression linéaire entre les deux variables et de discuter sur la pente et l’ordonnée à l’origine.
39

Conseils pour la rédaction des compterendus :
Les compterendus doivent être succints (une feuille double maximum). Il ne doivent renfermer que les données indispensables pour vérifier les résultats et permettre la compréhension de la manipulation.
Dans ce but, toutes les réactions chimiques mises en jeu au cours des titrages ainsi que les résultats expérimentaux doivent être indiqués très clairement.
En ce qui concerne les résultats, il est essentiel :• de préciser à quoi ils se rapportent (quelle eau, quelle profondeur,...)
• de préciser les unités choisies (M, g.l1,...)• de calculer les intervalles de confiance quand vous faîtes plus de 2
réplicats.• de ne présenter que des décimales ayant une signification par rapport à
la précision de la méthode employée.
40


















