
THˆSE - Universit© Toulouse III - Paul Sabatier
140
T T H H È È S S E E En vue de l'obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Gènes Cellules & Développement JURY Dr. Sophie DOISNEAU-SIXOU, Professeur UPS,Toulouse, Président Dr. Michel RENOIR, Directeur de Recherche CNRS, Châtenay-Malabry, Rapporteur Dr. Dany CHALBOS, Directeur de Recherche INSERM, Montpellier, Rapporteur Dr. Annie VALETTE, Directeur de Recherche CNRS, Toulouse, Examinatrice Ecole doctorale : Biologie-Santé-Biotechnologies Unité de recherche : LBME, UMR 5099 Directeur(s) de Thèse : Dr. Kerstin BYSTRICKY/ Dr. Hélène Richard-Foy Rapporteurs : Dr. M. RENOIR / Dr. D. CHALBOS / Dr.S.KHOSHBIN Présentée et soutenue par Mahta MAZAHERI Le 26 juin 2009 à 14H30 Salle de conférence de l'IEFG (IBCG) Titre : Molecular basis of anti-hormonal treatment and resistance in breast cancer
Transcript of THˆSE - Universit© Toulouse III - Paul Sabatier

TTHHÈÈSSEE
En vue de l'obtention du
DDOOCCTTOORRAATT DDEE LL’’UUNNIIVVEERRSSIITTÉÉ DDEE TTOOUULLOOUUSSEE
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Gènes Cellules & Développement
JURY
Dr. Sophie DOISNEAU-SIXOU, Professeur UPS,Toulouse, Président Dr. Michel RENOIR, Directeur de Recherche CNRS, Châtenay-Malabry, Rapporteur
Dr. Dany CHALBOS, Directeur de Recherche INSERM, Montpellier, Rapporteur Dr. Annie VALETTE, Directeur de Recherche CNRS, Toulouse, Examinatrice
Ecole doctorale : Biologie-Santé-Biotechnologies
Unité de recherche : LBME, UMR 5099 Directeur(s) de Thèse : Dr. Kerstin BYSTRICKY/ Dr. Hélène Richard-Foy Rapporteurs : Dr. M. RENOIR / Dr. D. CHALBOS / Dr.S.KHOSHBIN
Présentée et soutenue par Mahta MAZAHERI Le 26 juin 2009 à 14H30
Salle de conférence de l'IEFG (IBCG)
Titre : Molecular basis of anti-hormonal treatment and resistance in breast cancer

Acknowledgment
I would like to express my gratitude to all those who gave me the possibility to complete this
thesis.
First of all, I would like to begin with my promotore Dr. Hélène Richard- Foy (who left us in
2007), thank you for welcoming me in your Team, giving me the opportunity to develop this
experience abroad and challenging me every day to make me a better Scientist.
I am extremely thankful to Dr. Kerstin Bystricky for giving me the opportunity to continue
my experience under your supervision; thanks for your kindness, support and encouragement
during my thesis.
Furthermore, I would like to thank the members of Jury: Dr. Sophie Doisneau-Sixou, Dr.
Michel Renoir, Dr. Dany Chalbos, Dr. Saadi Khochbin and Dr. Annie Valette for taking the
time to assess my manuscript. Your advices were greatly appreciated.
I would like to thanks the members of our research group; my dear friends: Stephanie, Imen,
silvia, Mathieu, Anne-Claire and Isabelle; thank you for the good times and nice company, I
want to say that it was a real pleasure to work with you. Exchanging tips in the lab, discussing
results in group meeting, chatting in the corridors…
In the future, if you’re in the neighbourhood don’t hesitate to visit me.
Especially, I would like to give my special thanks to my family; my husband, Kazem and my
daughter, Sahel; your patient love enabled me to complete this work.
Juin 2009
Mahta

Summary Breast cancer is the most common type of malignancy among women in the world.
Approximately 70% of breast tumours express the estrogen receptor alpha (ERα) and are
considered hormone-responsive. Endocrine therapies have long been the treatment of choice.
However, the estrogen- like agonist effect and development of resistance of the available
selective estrogen receptor modulator such as tamoxifen require developing new treatments that
act through different mechanisms.
The objective of our study is to design tools that can help to understand the molecular
mechanisms involved in ligand-dependent modulation or degradation of ERα. We selected a set
of anti-estrogens with different structures and compared their effect on:
1). ERα degradation.
2). Intra-cellular localisation of ERα.
3). Regulation of transcription of ERα- endogenous target genes.
4). Regulation of transcription in the mutants of ERα.
Using this mechanistic study we could classify the tested anti-estrogens into three groups based
on their function: SERM, SERD and a new group for EM-652. SERM (selective estrogen
receptor modulator) include compounds such as OH-tamoxifen and RU39411, that stabilise ERα, that re-localize ERα into the nucleus upon binding, that increase transcriptional activity in
mutants affecting the recruitment of cofactors or the binding of their side chain and that lack
inhibitory capacities of the basal expression of endogenous genes. SERD (selective estrogen
receptor modulator) include compounds such as ICI182580 or RU58668 that induce nuclear
proteasome-dependent degradation ERα which occur in large nuclear foci that colocalize with
the proteasome and that inhibit basal gene expression of the endogenous progesterone receptor
gene (PGR).
Finally, EM-652 was found to affect ERα degradation and localisation similarly to SERM but
inhibited basal gene expression of the endogenous PGR.
This approach can be used to screen the newly designed compounds based on specific anti-
estrogen structural features

1
INTRODUCTION ...................................................................................................................... 5 PART I: GENERAL HISTORY OF BREAST CANCER ......................................................... 6
1. Definition and history of breast cancer .......................................................................... 9 2. Epidemiology ................................................................................................................. 9 3. The Biology of Breast Cancer ...................................................................................... 11
3.1. Critical periods of susceptibility to mammary carcinogen: ...................................... 11 3.2. Biological Characteristics of Critical Periods ........................................................... 12
4. Breast Carcinogenesis .................................................................................................. 12 4.1. Risk Factors ............................................................................................................... 15 4.2 Breast cancer classification ........................................................................................ 17 4.3. Prognostic and predictive markers ............................................................................ 18
5. Breast cancer treatment ................................................................................................ 19 5.1. Surgery ...................................................................................................................... 20 5.2. Chemotherapy ........................................................................................................... 20 5.3. Radiotherapy ............................................................................................................. 21 5.4. Hormonal therapy ...................................................................................................... 21
5.4a. Anti-estrogens ...................................................................................................... 22 5.5. Other targeted therapies ........................................................................................ 23
PART II: ESTROGENS AND THEIR NUCLEAR RECEPTORS ......................................... 24 1. Transport and Metabolism of Estrogens .......................................................... 26 Physiologic actions of estrogens .............................................................................. 26 2. Actions on Breast Tissue .................................................................................. 27 3. Estrogen and carcinogenesis ............................................................................ 28 4. Estrogen Receptors ........................................................................................... 30
4.1. Estrogen Receptor isoforms and their structure ................................................... 30 4.2. Different variants of ERs ...................................................................................... 32
5. Tissue distribution of ERs ................................................................................ 33 6. ERs expression in breast cancer ....................................................................... 34
PART III: MOLECULAR BASIS FOR TRANSCRIPTIONAL ACTIVITY OF THE ESTROGEN REcEPTOR alpha ............................................................................................... 36
1. ERα localization ....................................................................................................... 36 2. ERα activation and functional pathways .................................................................. 36 3. Ligand-dependent pathways ..................................................................................... 36 3.1. Genomic pathways .................................................................................................... 36
3.1a. Classical or direct pathway .................................................................................. 36 3.1b. Non- classical or indirect pathways .................................................................... 37
3.2. Non genomic pathway ............................................................................................... 38 4. Ligand independent pathways .................................................................................. 38 5. Transcriptional coregulators of ERα ........................................................................ 39 6. Coregulators and chromatin remodelling ................................................................. 40 6.1. Co-activators ............................................................................................................. 41 6.2. Co-repressors ............................................................................................................. 41 7. Activation of ERα through phophorylation .............................................................. 42 8. ER turn over and ligand dependent degradation of ERα .......................................... 43
PARTR IV: ANTIESTROGEN SIGNALING AND RESISTANCE TO ENDOCRINE THERAPY ............................................................................................................................... 45 1. Selective Estrogen Receptor Modulators (SERM) ........................................................... 46
Tamoxifen ........................................................................................................................ 47 Metabolism and pharmacokinetic of tamoxifen ............................................................... 47
2. Selective Estrogen Receptor Down-regulator (SERD) .................................................... 48

2
ICI 164384 ........................................................................................................................ 48 ICI 182780 ........................................................................................................................ 48
3. Molecular mechanism of anti-estrogen activity ............................................................... 49 4. Transcriptional activity .................................................................................................... 51
4.1. Transcription activation function 1(AF-1) ................................................................ 51 4.2. Transcriptional activation function 2(AF-2) ............................................................. 51
5. Structural basis of agonism and antagonism .................................................................... 52 5.1. Flexibility of the LBD: .............................................................................................. 52 5.2. Role of H12 in antiestrogen signalling ...................................................................... 54
6. The factors affecting antiestrogenic activity .................................................................... 56 7. Breast cancer and endocrine resistance ............................................................................ 57 8. Tamoxifen treatment and resistance to endocrine treatment ............................................ 57
8.1. Loss of ERα expression and function ........................................................................ 58 8.2. ERβ subtype and its alteration in expression ............................................................ 58 8.3. The role of nuclear receptor coregulators ................................................................. 59 8.4. ER pathway cross-talk with growth factor and cellular kinase pathways ................. 60
AIM OF PRESENT STUDY ................................................................................................... 61 RESULTS ................................................................................................................................. 62 PUBLICATION N°1 ................................................................................................................ 63 A molecular approach to predict selective estrogen receptor modulators from down regulators .................................................................................................................................................. 64
Abstract ............................................................................................................................ 65 Introduction ...................................................................................................................... 66 Materials and Methods ..................................................................................................... 68 Results .............................................................................................................................. 73 Discusssion ....................................................................................................................... 79 Figures and Legends ......................................................................................................... 83 References ........................................................................................................................ 90
PUBLICATION N°2 ............................................................................................................... 93 Ligands specify estrogen receptor alpha nuclear localization and degradation ....................... 94
Abstract ............................................................................................................................ 95 Introduction ...................................................................................................................... 95 Materials and Methods ..................................................................................................... 97 Results ............................................................................................................................ 101 Discussion ...................................................................................................................... 108 Figures and Legends ....................................................................................................... 111 References ...................................................................................................................... 120
ONGOING RESULTS ........................................................................................................... 122 CONCLUSION AND PERSPECTIVES ............................................................................... 123 REFERENCES ....................................................................................................................... 126

3
List of abbreviations
AF1: Transcription activation factor 1
AF2: Transcription activation factor 1
AIB1: Amplified In Breast Cancer 1
AP1: Activating Protein 1
CREB: cAMP Response Element-Binding
DCIS: ductal carcinoma in situ
DES: Diethyl Stilbestrol
ER: Estrogen Receptor
E1: Estrone
E2: 17-β-Estradiol
E3: Estriol
EGF: Epidermal Growth Factor
EGFR: Epidermal Growth Factor Receptor
ERE: Estrogen Response Element
ERK: Extra cellular signal Regulated Kinase
FISH: Fluorescence In Situ Hybridization
GF: Growth Factor
HAT: Histone Acetyl Transferase
HDAC: Histone deacetylases

4
Hsp: Heat shock protein
HER2: Human Epidermal growth factor Receptor 2
IGF-1: Insulin like Gowth Factor1
LBD: Ligand Binding Domain
MAPK: Mitogen Activated Protein Kinase
MISS: Membrane Initiated Steroid Signaling
N-CoR: Nuclear receptor Corepressor
NISS: Nuclear Initiated Steroid Signalling
4OHT: 4-Hydroxytamoxifen
PR: Progesterone Receptor
SP1: Specific Protein-1
SERM: Selective Estrogen Receptor Modulator
SERD: Selective Estrogen Receptor Downregulator
SRC: Steroid Receptor Coactivator
SMRT: Silencing Mediator for Retinoid and Thyroid receptors
TGF: Transforming Growth Factor
TEB: Terminal End Buds
TDLU: Terminal Duct Lobular Units
WT: Wild Type

5
INTRODUCTION

6
PART I: GENERAL HISTORY OF BREAST CANCER The mammary gland is a complex organ that begins development early in gestation and
culminates in the postpartum lactation of the adult female. The extensive research currently
being performed on the human genome and molecular biology will certainly contribute to
further elucidate the role of factors involved in formation, differentiation, and development of
the mammary gland. This may provide important insights into causes, treatment, and potential
prevention of mammary gland abnormalities such as breast cancer. Given that breast cancer
strikes 10% of women in the world, these developments may have enormous implications for
the future of medicine (Brisken, 2002; Nguyen et al., 1995; Polyak, 2001).
Breast development occurs in distinct stages throughout a woman's life. Human breast tissue
begins to develop in the sixth week of fetal life. Breast tissue initially develops along the lines
of the armpits and extends to the groin (this is called the milk ridge). By the ninth week of
fetal life, it regresses to the chest area, leaving two breast buds on the upper half of the chest,
In the neonate, the mammary glands, with their relatively simple architecture, remain
quiescent until puberty (Naccarato et al., 2000). Female breasts do not begin growing until
puberty when the breasts will begin to respond to hormonal changes in the body. Specifically,
the production of two hormones, estrogen and progesterone, signal the development of the
glandular breast tissue. The ductal branches that are formed during embryogenesis grow and
divide to form branching ductal bundles with terminal end buds (TEB). The TEBs are major
site of proliferation, and at menarche the terminal duct lobular units (TDLUs) develop from
this site but remain in an arresting state until the onset of pregnancy and lactation (Vogel et
al., 1981). Development initiated at the onset of puberty is generally complete by 20 years of
age.
If pregnancy occurs, with accelerated development of the TDLUs, the number of epithelial
cells and alveoli within the lobules increases in preparation for lactation (Hovey et al., 2002)
at which point the glands complete their differentiation and reach functional maturity.

7
Following lactation, there is massive apoptosis and remodelling of the tissue that will then
resemble, once again, the gland in its non-pregnant state (Allan et al., 2004; Furth et al.,
1997).
The complex branching structure of lobules is lined by three epithelial cell types, ductal and
alveolar luminal cells, and myoepithelial cells. The ductal and alveolar cells constitute the
inner layer of ducts and the lobuloalveolar units, respectively, and each is surrounded by a
basal layer of myoepithelial cells. All three epithelial cell types have recently been
demonstrated to originate from a common multipotent stem cell (Shackleton et al., 2006;
Stingl et al., 2006)
The glands complete their differentiation and reach functional maturity under the influence of
sustained increase in the levels of circulating progesterone, estrogens, prolactin and placental
lactogen.
.
Figure 1: Life cycle of breast development in the nulliparous woman.
The breast is primarily composed of lobules type 1, with some progression to type 2, and only minimal
formation of lobules type 3 during sexual maturity, which regresses to lobules type 1 at menopause.
8
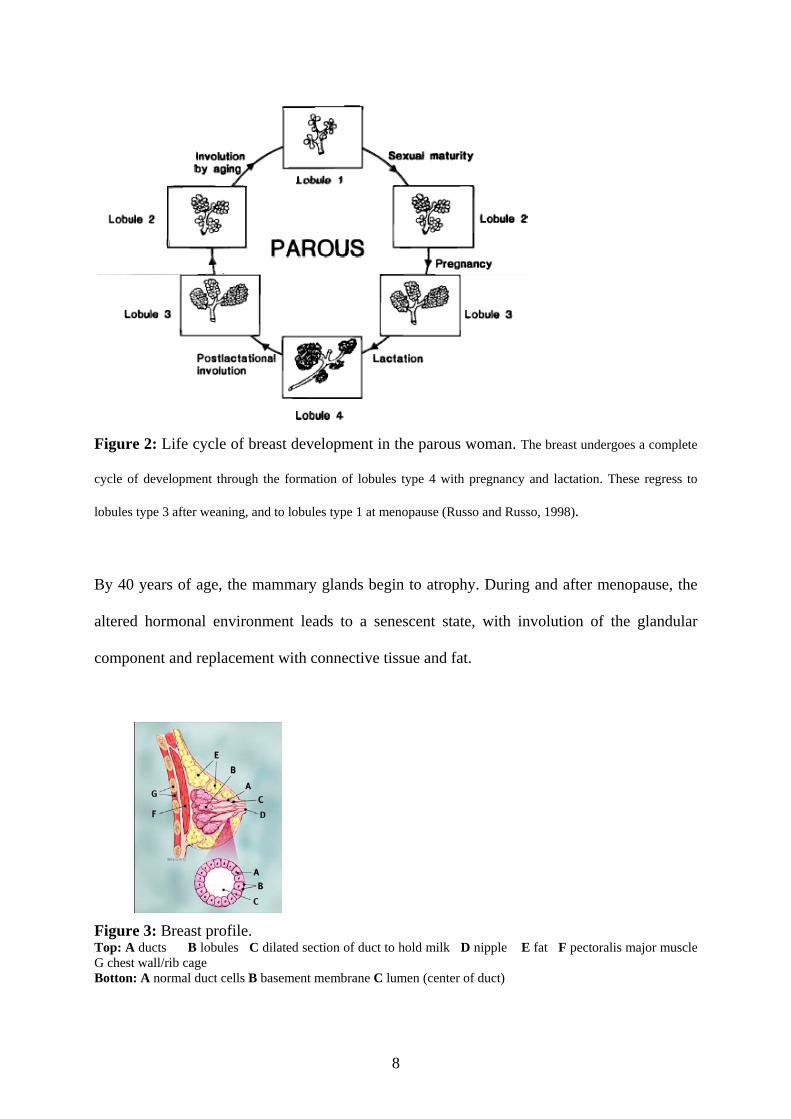
Figure 2: Life cycle of breast development in the parous woman. The breast undergoes a complete
cycle of development through the formation of lobules type 4 with pregnancy and lactation. These regress to
lobules type 3 after weaning, and to lobules type 1 at menopause (Russo and Russo, 1998).
By 40 years of age, the mammary glands begin to atrophy. During and after menopause, the
altered hormonal environment leads to a senescent state, with involution of the glandular
component and replacement with connective tissue and fat.
Figure 3: Breast profile. Top: A ducts B lobules C dilated section of duct to hold milk D nipple E fat F pectoralis major muscle G chest wall/rib cage Botton: A normal duct cells B basement membrane C lumen (center of duct)

9
1. Definition and history of breast cancer Breast cancer is a major public health problem in the world; there are many texts and
references that attempt to define breast cancer. The simplest definition is from the National
Cancer Institute (NCI). According to the NCI, breast cancer is a “cancer that forms in tissues
of the breast, usually the ducts (tubes that carry milk to the nipple) and lobules (glands that
make milk)”. It occurs in both men and women, although male breast cancer is rare.
2. Epidemiology Breast cancer was recognized by the ancient egyptians as long ago as 1600 BC. However,
over the past 50 years it has become a major health problem affecting as many as one in eight
women during their lifetime. The burden of breast cancer is increasing in both developed and
developing countries, and in many of the regions of the world. It is now the most frequently
occurring malignant disease in women. Each year the disease is diagnosed in over one million
women worldwide and is the cause of death in over 400,000 women. Breast cancer can occur
in men, although the incidence is much lower, amounting to around 1% of all breast cancers.
Overall the incidence of breast cancer rises with age, increasing rapidly during the fourth
decade of life and continuing to increase thereafter, but more slowly in the fifth, sixth and
seventh decades. In the USA, 75% of new breast cancers are diagnosed in women aged 50
years or older, and the lifetime risk of breast cancer diagnosis is approximately 12.5%.
The incidence rates for breast cancer are similar in North America and the majority of other
western industrialized countries. In France, this pathology is the first diagnosed cancer for
women, and according to data from the Ministère de la Santé (2003), each year there are
42 000 estimated new cases and 11 640 deaths. In Japan and other Far Eastern countries,
however, absolute incidence rates are lower for each age band and overall Japanese women
are five times less likely to develop breast cancer than American women.
Epidemiologic studies show a significant decrease in mortality rate of breast cancer and this
could probably be the result of earlier diagnosis, but also from a better therapeutic care
particularly in the adjuvant treatments. Most cases of breast cancers are sporadic, however,
10
twin studies have shown that heritable factors may cause 20-30% of all breast cancers
(Lichtenstein et al., 2000). Mutations within BRCA1 and BRCA2 genes are common among
women with a strong family history of breast cancer; they account for at most 3-8% of all
breast cancer cases. It is important to underline that 70% of breast cancers are hormone-
dependent; they express estrogen receptor (ER) and their proliferation is influenced by
estrogens. Frequency, severity and impact of this pathology on different aspects of human life
make breast cancer a priority in research and justifies the efforts to prevent and to improve
available treatments.
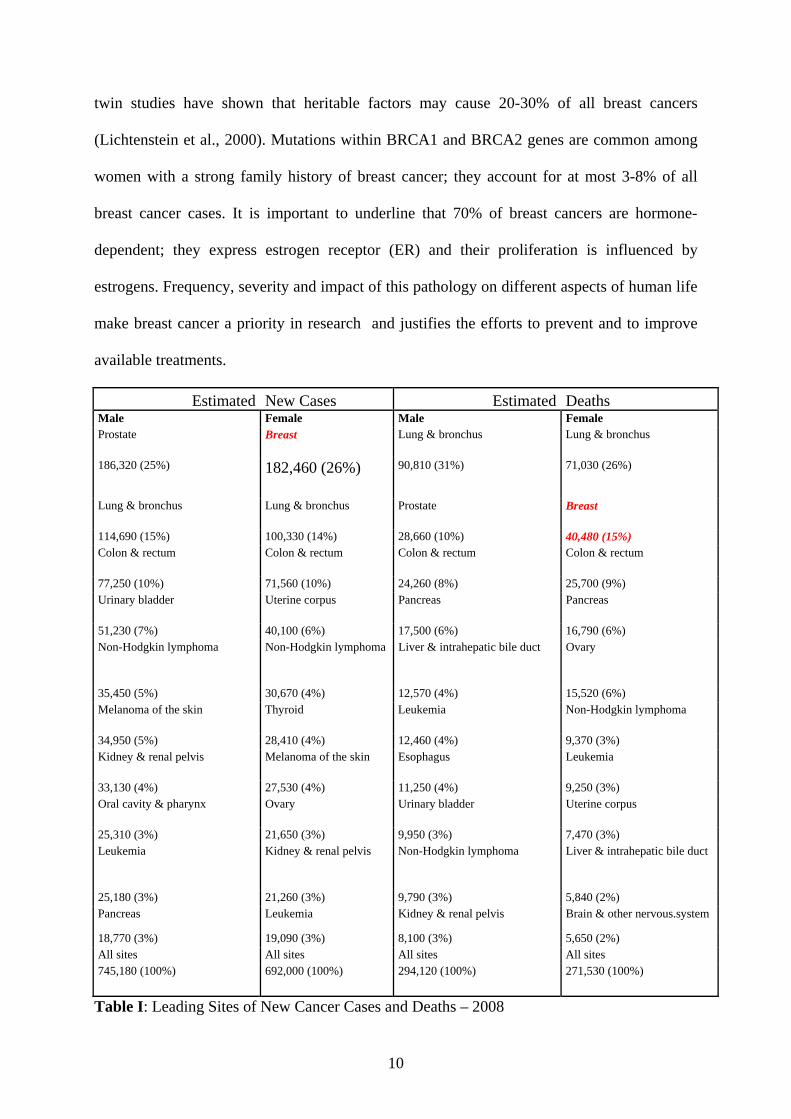
Estimated New Cases Estimated Deaths Male Female Male Female Prostate Breast Lung & bronchus Lung & bronchus
186,320 (25%) 182,460 (26%) 90,810 (31%) 71,030 (26%)
Lung & bronchus Lung & bronchus Prostate Breast
114,690 (15%) 100,330 (14%) 28,660 (10%) 40,480 (15%) Colon & rectum Colon & rectum Colon & rectum Colon & rectum
77,250 (10%) 71,560 (10%) 24,260 (8%) 25,700 (9%) Urinary bladder Uterine corpus Pancreas Pancreas
51,230 (7%) 40,100 (6%) 17,500 (6%) 16,790 (6%) Non-Hodgkin lymphoma Non-Hodgkin lymphoma Liver & intrahepatic bile duct Ovary
35,450 (5%) 30,670 (4%) 12,570 (4%) 15,520 (6%) Melanoma of the skin Thyroid Leukemia Non-Hodgkin lymphoma
34,950 (5%) 28,410 (4%) 12,460 (4%) 9,370 (3%) Kidney & renal pelvis Melanoma of the skin Esophagus Leukemia
33,130 (4%) 27,530 (4%) 11,250 (4%) 9,250 (3%) Oral cavity & pharynx Ovary Urinary bladder Uterine corpus
25,310 (3%) 21,650 (3%) 9,950 (3%) 7,470 (3%) Leukemia Kidney & renal pelvis Non-Hodgkin lymphoma Liver & intrahepatic bile duct
25,180 (3%) 21,260 (3%) 9,790 (3%) 5,840 (2%) Pancreas Leukemia Kidney & renal pelvis Brain & other nervous.system
18,770 (3%) 19,090 (3%) 8,100 (3%) 5,650 (2%) All sites All sites All sites All sites 745,180 (100%) 692,000 (100%) 294,120 (100%) 271,530 (100%)
Table I: Leading Sites of New Cancer Cases and Deaths – 2008

11
*Excludes basal and squamous cell skin cancers and in situ carcinoma except urinary bladder
©2008, American Cancer Society,Inc.
3. The Biology of Breast Cancer Unlike other tissues in the body as the liver and the heart that are formed at birth, breast
tissue in newborns consists only of a tiny duct. At puberty, in response to hormones (like
estrogen that is secreted by the ovary), the breast duct grows rapidly into a tree-like structure
composed of many ducts.
Most breast development occurs between puberty and a woman’s first pregnancy. The
immature breast cells, called “stem cells”, divide rapidly during puberty. The cells in the
immature, developing breast are not very efficient at repairing mutations and they are more
likely to bind carcinogens (cancer causing agents).
The female hormone estrogen stimulates breast cell division. This division can increase the
risk of damaging DNA. Furthermore, not fully matured breast cells in girls and young women
are not as efficient at repairing DNA damage as mature breast cells; therefore it is important
to reduce the exposure of young women and girls to carcinogens that might damage DNA
during this phase of rapid breast development. Some evidence also suggests that breast
feeding further reduces the breast cells’ sensitivity to mutations. Milk producing cells are
fully mature and less sensitive to DNA damage than immature undifferentiated cells.
Therefore, susceptibility to mutations declines in the breast cells of women who have had an
early full-term pregnancy (University, 1997).
3.1. Critical periods of susceptibility to mammary carcinogen:
*Birth to 4 years
*Puberty
*End of Puberty to 1st full-term pregnancy

12
3.2. Biological Characteristics of Critical Periods
* Rapid cell division
* Breast cells have higher proportion of "stem cells"
* Mutations can be passed on if not repaired
* Stem cells are more susceptible to carcinogens
This susceptibility decreases after first full term pregnancy and the reasons could be:
* Fewer stem cells
* Less cell division
* More cells are differentiated
* Differentiated cells repair DNA more efficiently
* Differentiated cells bind carcinogens more weakly than stem cells
(Original hypothesis by Drs. Irma and Jose Russo )(University, 1997).
4. Breast Carcinogenesis Breast cancer, like other forms of cancer is widely perceived as a heterogeneous disorder with
markedly different biological properties from normal cells. Cancerous cells develop from
healthy cells, constantly changing under the influence of hormones and growth factors, in a
complex process called malignant transformation. These are caused by a series of clonally
selected genetic changes in key tumour-suppressor genes or oncogenes (Feinberg et al., 2006)
(Olsson, 2000). It has been proposed that the process of breast cancer tumourigenesis is best
described by a multi-step progression model (Beckmann et al., 1997), in which the normal
epithelium develops via hyperplasia and carcinoma in situ into an invasive cancer, which can
disseminate via the lymph and the vascular system.
Initiation: The first step in cancer development is initiation. The development of a tumour is
associated with the acquisition of genetic and epigenetic alterations that modify normal
growth control and survival pathways (Albertson, 2006; Polyak, 2007 ).

13
This phase is characterized by accumulation of mutations that lead to the overexpression of
pro-oncogenic factors. Genetic mutations that can lead to breast cancer have been
experimentally linked to estrogen exposure (Cavalieri et al., 2006). The change in the cell’s
genetic material occurs spontaneously or is brought on by carcinogens including ionizing
radiation, many chemicals, viruses, radiation, and sunlight.
A long list of genes has been reported for their implication in breast cancer tumourigenesis in
which the most important are:
Oncogenes; oncogenes refer to genes whose alteration causes gain of function. A frequent
anomaly for oncogenes is amplification.
Examples of amplification include erb-B2 which is a member of EGF receptor superfamily, c-
myc coding for a protein important in apoptosis, and finally ccndI that codes cyclin D1, a
regulator of G1/S transition in cell cycle (Osborne et al., 2004). This anomaly is found in
more than 15% of breast cancers. Oncogenic activation through gene amplification of erb-B2
(HER-2) occurs in about 20% of primary breast cancers. Erb-B2 encodes a transmembrane
tyrosine kinase growth factor receptor and its activation via a variety of pathways is involved
in proliferation and angiogenesis, alteres cell-cell interactions, increases cell motility,
metastases, and resistance to apoptosis.
Tumour suppressor genes (anti-oncogene): inactivating tumour suppressor genes as p53, Rb,
PTEN, p16, BRCA1 and BRCA2 induce tumour development. For example Tp53 gene
(coding pour p53) which is Located at 17p13p53 loucus (Lacroix et al., 2006) is damaged or
missing in most of human cancers including breast cancer. Loss of heterozygosity (LOH) of
theTp53 gene was shown to be a common event in primary breast carcinomas (Davidoff et al.,
1991). One function of this gene is to keep cells with damaged DNA from entering the cell
cycle. The Tp53 gene can tell a normal cell with DNA damage to stop proliferating and repair
the damage. In cancer cells, p53 recognizes damaged DNA and tells the cell to "commit
suicide" (apoptosis). If the p53 gene is damaged and loses its function, cells with damaged

14
DNA continue to divide when normally they would have been removed through apoptosis.
This is why the p53 gene has been termed "The Guardian of the Genome."
Promotion: The second and final step in the development of cancer is promotion. Subsequent
tumour progression is driven by the accumulation of additional genetic changes combined
with clonal expansion and selection (Polyak, 2007 ). When cells enter the second stage of
promotion, they gain their independence and lose their capacity for intercellular
communication which leads to unregulated cell proliferation (Eccles, 2001; Hynes and Lane,
2001; Lane et al., 2001).
Spread: Breast cancer usually spreads first to the nearby lymph nodes, and later to distant
sites. Cancers can also spread via the blood stream. Common sites of metastases include the
liver, the lung, bone, and the brain (Chambers et al., 2002).
Of course estrogen receptors (ERs) play an important role in breast carcinogenesis. Over-
expression of ERα is frequently observed in early stages of breast cancer. ERs are the
regulatory proteins essentially localized in the nucleus, but also sometimes in the cytoplasmic
membrane. The estrogen receptor regulates gene expression by both estrogen-dependent and
estrogen-independent mechanisms leading to activation of gene transcription (Hayashi et al.,
2003). Schematically, in breast cancer cells, the ER-hormone complex influences different
pathways by:
*Stimulation of synthesis and activity of certain growth factors (EGF, IGF-1, TGFα),
resulting in cell proliferation.
*Stimulation of activity of oncogenes (c-myc, c-myb, c-fos) interfering with cell proliferation
and apoptosis.
*Stimulation of protease synthesis (cathepsin D, UPA-1) contributing in metastatic processes
by degradation of extra cellular matrix.

15
4.1. Risk Factors
Gender: it is about 100 times more common in women than in men (Hulka and Moorman,
2001) (American Cancer Society 2009)
Age: one of the best documented risk factor for breast cancer is age. The chance of getting
breast cancer goes up as a woman gets older. About two-third of cases are diagnosed in
women aged 55 or older (McPherson et al., 2000). From 2002-2006, the median age at
diagnosis for cancer of the breast was 61 years of age. Approximately 0% was diagnosed
under age 20; 1.9% between 20 and 34; 10.5% between 35 and 44; 22.5% between 45 and 54;
23.7% between 55 and 64; 19.6% between 65 and 74; 16.2% between 75 and 84; and 5.5%
85+ years of age. http//seer.cancer.gov/csr/1975_2006
Genetic background and family history: genetic predisposition is a growing knowledge
suggesting that pattern of risk can be defined precisely person by person (Singletary, 2003).
Genetic susceptibility to breast cancer is conferred by a large number of genes and as
(Ripperger et al., 2008) which we can classify in inherited breast cancer to three groups;
* Highly penetrant genes (BRCA1, BRCA2, TP53, STK11, PTEN, CDH1).
Although these mutations are highly penetrant, they have been well characterized. For this
group, genetic counselling and genetic testing are available and could allow appropriate
screening and prophylactic measures.
*The intermediate penetrance breast cancer susceptibility genes (ATM, CHEK2, BRIP1,
BRAD1, and PALB2). In the literature there is no particular suggestion to performe genetic
testing for these mutations as a screening strategy.
*The low penetrance breast cancer susceptibility alleles (FGFR2).
Family history: many studies have attempted to define the risk associated with a positive
family history. It has been proposed that approximately 10-20% of breast cancers are due to

16
genetic predisposition (McPherson et al., 2000; Singletary, 2003). To date it is clear that
degree of risk is in relation with the type of relative affected (first or second degree), the age
at which the relative developed cancer, and the number of relatives affected.
For example BRCA1 and BRCA2 genes are , germlin mutation associated with an inherited
susceptibility to breast and ovarian cancer (Brody and Biesecker, 1998). Women with a strong
family history of breast and/or ovarian cancer should be offered counselling to determine if
genetic testing is appropriate. Families with four or more relatives affected with either breast
or ovarian cancer in three generations and one living affected relative have a high risk to
develop cancer (Fig.4). Studies suggest that prophylactic removal of the breasts and/or ovaries
in BRCA1 and BRCA2 mutation carriers decreases considerably the risk of breast
cancer(McPherson et al., 2000).
Figure 4: Family tree of family with genetically inherited breast cancer (McPherson et
al., 2000). An example of family tree or history that could be used for assessing familial breast cancer risk.
Lifestyle and environmental factors: no factors have yet been identified that have a major
effect on the risk of breast cancer (Singletary, 2003). However, there are several factors that
may have a limited effect; these are diet, weight, alcohol intake.

17
Reproductive factors: women who began menstruating before the age of 12 have a relative
risk for invasive breast cancer of 1.3 compared to those who began after the age of 15. On the
other hand, those who have not reache menopause until age 55, have a relative risk of 1.22
compared to those who have menopause before age 45. Women who have no children, or who
had their first child after age 30, have a higher risk of breast cancer (Singletary, 2003).
4.2 Breast cancer classification
Currently using TNM classification for breast tumours is according to their extension of
tumours. The key elements in this classification are: tumour size, presence of metastatic
regional lymph node, and distance metastases (Veronesi et al., 2006). However, for making a
treatment decision, the knowledge of several other biological factors as ER, PgR, HER2
overexpression or amplification is required. Advances in the biology of cancer and in
technical progress such as microarray have confirmed that breast cancer is a heterogeneous
disease with diversity in responsiveness in treatment which may explain differences in
evolution of patients (Perou et al., 2000). Breast cancers can be subdivided into at least five
sub-groups based on their gene expression patterns (Sorlie et al., 2001) (Fig.5). In this study
the tumours were separated into two main branches: the groups of the left branch are
characterized by low to absent ER gene expression. The groups in the right branch
demonstrated the highest level of ERα gene expression. ER negative groups include: 1. Basal
like or triple negative sub-type, 2. ERBB21 sub-type that presents an amplification of
HER2gene, and 3. Normal breast-like group. Luminal/ER positive group includes: 1. Luminal
sub-type A 2. Luminal sub-type B 3. Luminal sub-type C.
thereby in this study the genome wide expression patterns of tumours are a representation of
the biology of the tumours and thus, relating gene expression patterns to clinical outcomes is a
key issue in understanding this diversity.

18
Figure 5: Molecular classification of breast cancer (Sorlie et al., 2001). Gene expression patterns
of experimental samples including carsinomas, benign tumours and normal tissuses, analyzed by hierarchical
clustering.
4.3. Prognostic and predictive markers
In recent years mortality of breast cancer regressed probably as a result of widespread
screening, earlier detection and advances in adjuvant treatment. However, adjuvant systemic
therapy has associated risks and it would be useful to be able to optimally select patients most
likely to benefit. A prognostic factor is any measurement factor available at the time of
surgery that correlates with overall survival of patient without a systemic adjuvant therapy. In

19
contrast a predictive factor is a measurement associated with a response to a given therapy.
Some factors are both prognostic and predictive (Cianfrocca and Goldstein, 2004) (Table. 2).
Table 2: A summary of prognostic and predictive factors in breast cancer
In practice: Over half of breast tumour express ERα and around 70% of these respond to
anti-estrogen treatment while an absence of ER expression is associated with a poor prognosis
(Ali and Coombes, 2000). Thus a analysis of steroid receptor status has become the standard
of care for patients with breast cancer. The testing of breast cancer specimens for (erb-B2)
HER-2/neu status has now achieved a standard role for the management of breast cancer;
HER-2/neu overexpression identified by immunohistochemistry/ gene amplification detected
by FISH, has been consistently associated with higher grade and extensive forms of ductal
carcinoma in situ (Ross et al., 2003). Knowing if a cancer is HER2-positive directs the
choice of treatment and patients could then benefit from another treatment called Herceptin (a
monoclonal antibody against the HER2 protein).
5. Breast cancer treatment Different types of treatments exist for patients with breast cancer. Some of them are currently
used in clinic and some others are in the evaluating phase. The standard types of treatment are

20
surgery, radiation therapy, chemotherapy and hormonal therapy. New types of treatment that
are being studied in clinical trials, for exemple as monoclonal antibodies, are uses as adjuvant
therapy, and other targeted therapies. The goal of different modalities is to control local
extensions of disease or metastases; Thus surgery and radiotherapy will aim a regional control
of disease while the objective of the systemic therapy as chemotherapy, hormonal therapy and
other form of targeted therapies is to control of metastases.
5.1. Surgery: Surgery is the primary treatment of breast cancer. For a local treatment,
different modes of operations are used for most women with breast cancer. The choice of
different strategies of operation depends on different factors such as size and location of the
breast tumour as well as the type and the stage of breast cancer.
5.2. Chemotherapy: The purpose of chemotherapy and other systemic treatments is to
eliminate all cancer cells that may have spread from where the cancer started to another part
of the body. The oncologist uses this treatment for three purposes:
-Adjuvant therapy: chemotherapy is given after the initial surgery to prevent coming back of
cancer.
-Neo-adjuvant: is used for large breast cancer before surgery to shrink tumours and to ease
surgery.
-Tteatment metastatic cancer.
There is different chemotherapy drugs used in breast cancer treatment. Cyclophosphamide,
epirubicin, fluorouracil (5FU), methotrexate, paclitaxel (Taxol), doxorubicin
(Adriamycin®),docetaxel (Taxotere®) are commonly used. These drugs are usually used in
combination regimens as FEC that means a combination of 5FU, epirubicin and
cyclophosphamide or AC meaning doxorubicin (Adriamycin®) and cyclophosphamide.

21
There is no clinically useful molecular predictor of response to any cytotoxic drug used in the
treatment of breast cancer. However, certain studies propose a predictive model in
chemotherapy (Ayers et al., 2004).
5.3. Radiotherapy: Radiotherapy reduces local relapse, with a relative risk reduction
(Cuzick, 2005). Radiotherapy could be used in different ways:
1. after surgery to reduce remaining cancer cells / to treat the lymph node.
2. before surgery to reduce tumour size.
5.4. Hormonal therapy: For the first time in 1896 a surgeon called Beatson, reported that
breast cancer regression can occur in response to oophorectomy in premenopausal women
(Beatson, 1896). This report was the first recognition of hormone-dependent tumours.
In 1930s, estrogens were isolated and their implications in rodent mammary tumours were
described (Benson, 2008). The identification of the estrogen receptor (ER) in 1966 by Jensen
(Jensen, 1966) provided a mechanism to describe specificity of estrogen action at the target
site, and a target was identified to develop new drugs for the treatment and prevention of
breast cancer. In addition, a test was established to predict the outcome of antihormonal
therapy in breast cancer (Jensen and Jordan, 2003). Hormonal therapy only started in the
1970s with the widely prescribed anti-estrogen tamoxifen (Ward, 1973). In premenopausal
women, the ovaries are the principle source of estradiol that acts on distal target tissues. In
contrast in postmenopausal women estrogen is produced in extra gonadal sites (as adipose
tissues) rich of aromatases from conversion of androgen produced in the adrenal gland, and
functions locally at these sites (Simpson, 2003).
In clinic there are three possible levels for hormonal therapy:
At the hypothalamus - pituitary axis; this kind of hormone therapy is used normally for
premenopause women by prescription the analogues of LH-RH.

22
By competition with the estrogen receptor level; using anti-estrogens.
By inhibition of aromatases for menopausal patients.
5.4a. Anti-estrogens Anti-estrogens are the competitive inhibitors of estrogens. To date there are two groups of
anti-estrogens; SERMs for Selective Estrogen Receptor Modulators and SERDs for Selective
Estrogen Receptor Downregulators. The ubiquity of ER explains the diversity of effects
obtained in adition to anti-hormonal action.
Tamoxifen: Tamoxifen, (Nolvadex), the oldest SERM in continuous use in the clinic.
Tamoxifen is the standard choice in adjuvant treatment of ER positive breast cancer (Jordan,
2003). It is a non steroidal compound with a complex mechanism of action and contradictory
effects depending on tissue. In the mammary gland and in mammary tumours, it acts as an
antagonist, while in some tissues, such as bone, it is an agonist. Tamoxifen could be
prescribed at every age regardless of menopause status. According to NSABP (The National
Surgical Adjuvant Breast and Bowel Project: a clinical trials cooperative group supported by
the NIH) (MOON, 2005), tamoxifen is linked to a 43% reduction in cumulative risk of
invasive breast cancer. In a similar manner it reduces the cumulative risk of non-invasive
breast cancer by 37%. This study confirms that tamoxifen reduces the risk of breast cancer in
all subgroups indpendently of history and predicted risk for breast cancer. The clinician must
be very vigilant to side effects generated by tamoxifen. The most common are menopausal
symptoms including hot flashes, vaginal dryness, low libido, mood swings and nausea.
Tamoxifen increases the risk of endometrial cancer and thrombo emboli accidents. The
unwanted side effects of tamoxifen led to search for an anti-estrogen more potent and more
specific such as toremifen or raloxifen.
Pure anti-estrogens: Other molecules with no estrogenic activity have been developped such
as ICI 164,384 and ICI 182,780. ICI 182,780 (Faslodex or Fulvestrant) is a steroidal anti-

23
estrogen with an unique and different mechanism of action from tamoxifen since it is the first
anti-estrogen without partial agonistic activity. To date Faslodex is admitted as second line of
treatment in patients that develop a resistance to tamoxifen or an undesirable effect of
tamoxifen. This agent is reclassified as SERD that will be detailed later.
5.5. Other targeted therapies In recent years the following targeted therapies have been developed:
In breast cancer the best example is trastuzumab (Herceptin). The recombinant humanized
anti-HER2 monoclonal antibody, that becomes FDA (US Food and Drug Administration) approved in 2006, and is recommended for patients with breast cancer demonstrating 3 fold
overexpression of HER2 by immunohistochemistry or amplification of the HER2 gene by
fluorescence in situ hybridization (FISH). A 52 % reduction in the risk of recurrence is
reported when it is in combination with chemotherapy
(http://www.cancer.gov/search/ViewClinicalTrial).
24
PART II: ESTROGENS AND THEIR NUCLEAR RECEPTORS The existence and effect of estrogen were established from 1923 to 1938, estrogens comprise
a group of steroid compounds; structurally related and hormonally active molecules that
regulate critical cellular signalling pathways and by doing so, control cell proliferation,
differentiation and homeostasis (Cheskis et al., 2007) .
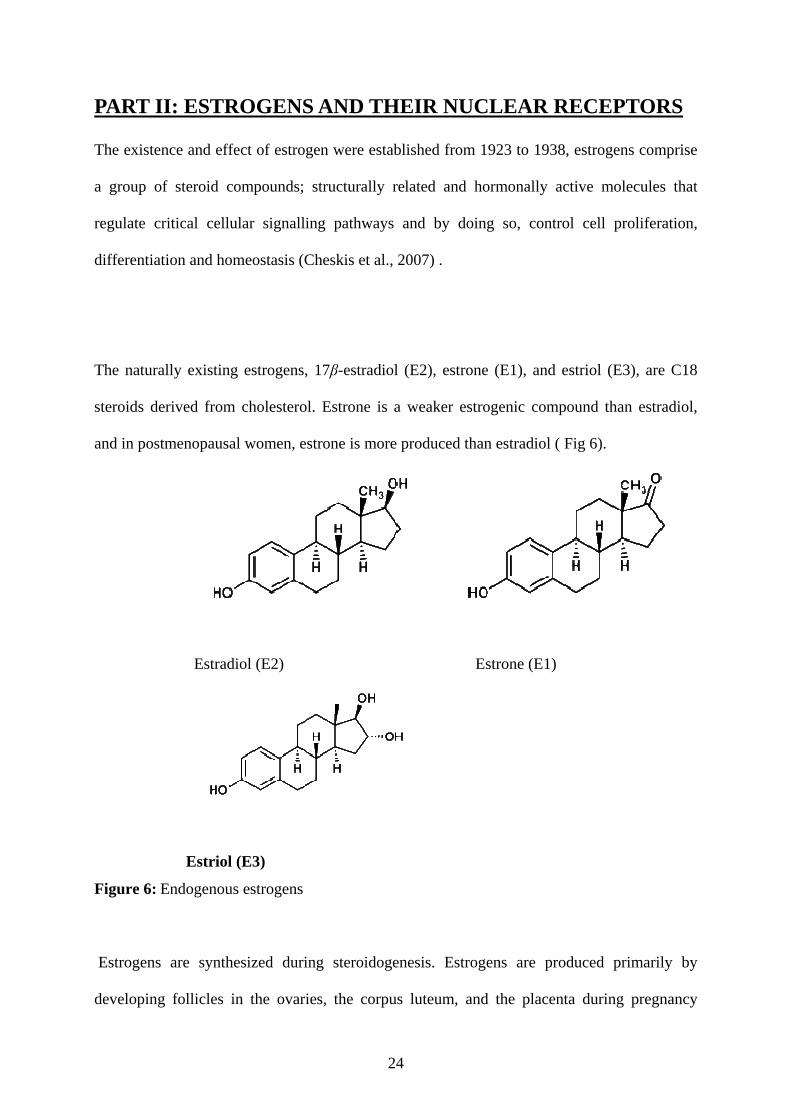
The naturally existing estrogens, 17β-estradiol (E2), estrone (E1), and estriol (E3), are C18
steroids derived from cholesterol. Estrone is a weaker estrogenic compound than estradiol,
and in postmenopausal women, estrone is more produced than estradiol ( Fig 6).
Estradiol (E2) Estrone (E1)
Estriol (E3)
Figure 6: Endogenous estrogens
Estrogens are synthesized during steroidogenesis. Estrogens are produced primarily by
developing follicles in the ovaries, the corpus luteum, and the placenta during pregnancy

25
(Simpson et al., 1997). Some estrogens are also produced in smaller amounts by other tissues
such as the liver, adrenal glands, and the breasts. These secondary sources of estrogen are
especially important in postmenopausal women. Estrogen biosynthesis is catalysed by
aromatases (aromatase cytochrome P450), the product of the CYP19 gene, which is a
member of the cytochrome P450 superfamily of genes (Simpson et al., 1997). Synthesis of
estrogens starts in theca interna cells in the ovary. By the synthesis of androstenedione from
cholesterol. Androstenedione is a substance of moderate androgenic activity. This compound
crosses the basal membrane into the surrounding granulosa cells, where it is converted to
estrone or estradiol, either immediately or through testosterone. The conversion of
testosterone to estradiol and of androstenedione to estrone, is catalyzed by the aromatase
enzyme.
Estradiol levels vary through the menstrual cycle, with highest levels just before ovulation.
Estrogens are present in both men and women. They are usually present at significantly higher
levels in women of reproductive age. They promote the development of female secondary sex
characteristics, such as breast, and are also involved in the thickening of the endometrium and
other aspects of regulating the menstrual cycle.
After menopause, adipose tissue becomes the main source of oestrogen (Grodin JM, 1973).
Therefore, in the post-reproductive years, the degree of a woman’s estrogenisation is mainly
determined by the extent of her adiposity. This is of clinical importance since corpulent
women are relatively protected against osteoporosis (Melton, 1997), and the incidence of
Alzheimer’s disease is lower in more corpulent postmenopausal women than in their slimmer
counterparts (Simpson, 2000). Conversely, obesity is positively correlated with breast cancer
risk (Huang et al., 1997).

26
1. Transport and Metabolism of Estrogens In the serum, estradiol reversibly binds to sex-hormone–binding globulin (Andersson, 1974).
Estrogens are metabolized by sulfation or glucuronidation, and the conjugates are excreted
into the bile or urine. Estrogens are also metabolized by hydroxylation and subsequent
methylation to form catechol and methoxylated estrogens. Hydroxylation of estrogens yields
2-hydroxyestrogens, 4-hydroxyestrogens, and 16 a-hydroxyestrogens (catechol estrogens),
among which 4-hydroxyestrone and 16 a-hydroxyestradiol are considered as carcinogenic.
A range of synthetic and natural substances have been identified that also possess estrogenic
activity (Fang et al., 2001).
- Plant products with estrogenic activity are called phytoestrogens
- Synthetic estrogens
Phytoestrogens, which include lignans and isoflavones (e.g. genistein and daidzein), are
estrogen-like compounds which occur naturally in many plants and fungi and which are
biologically active in humans, Epidemiological studies showed a protective effect against
breast cancer (Messina et al., 1997).
DES (diethyl stilbestrol) was first synthesized estrogen in early 1938 by Leon Golberg, and a
report of its synthesis was published in Nature in 1971 it was found to be a teratogen when
given to pregnant women.
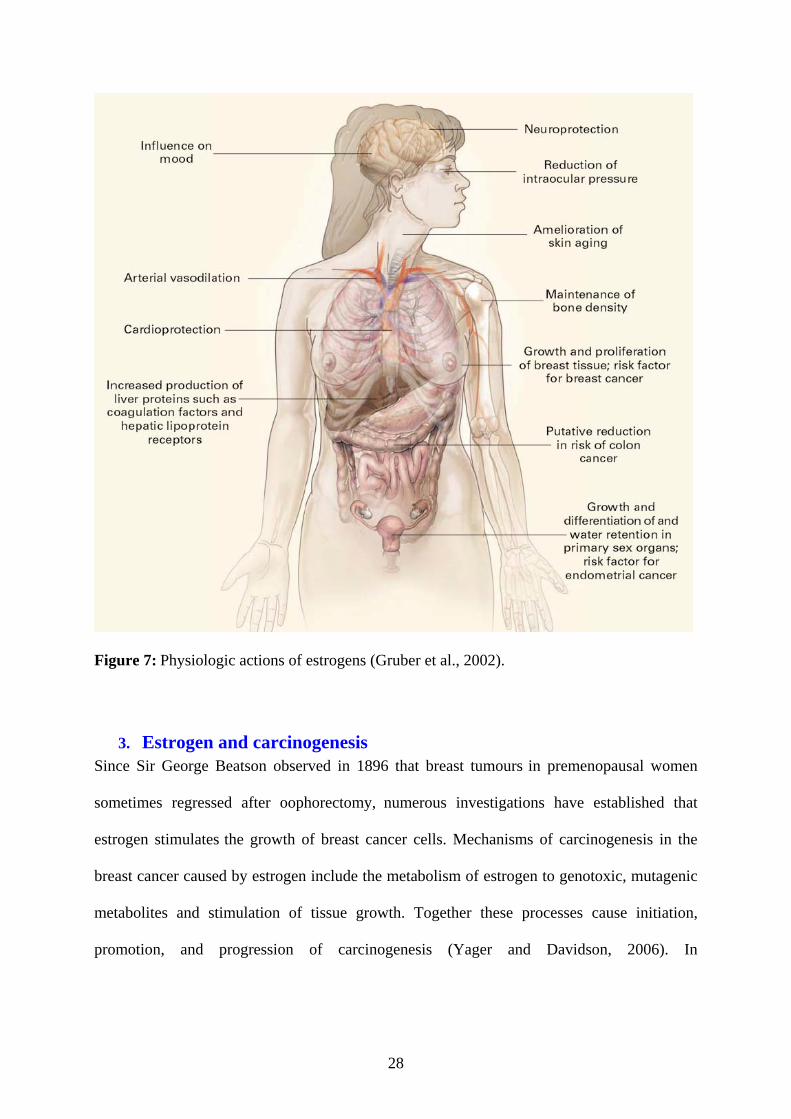
Physiologic actions of estrogens:
In the Central Nervous System, experimental studies on different animal models have shown
that estrogen is neuroprotective. The mechanisms involved in the neuroprotective effects of
estrogen are still unclear. Anti-oxidant effects, activation of different membrane-associated
intracellular signalling pathways, and activation of classical nuclear estrogen receptors (ERs)

27
could contribute to neuroprotection. Interactions with neurotrophins and other growth factors
may also be important for the neuroprotective effects of estradiol (Cardona-Gómez et al.,
2001). Some epidemiologic data suggest that in postmenopausal women, estrogen deficiency
is associated with a decline in cognitive function and an increased risk of Alzheimer’s disease
(Henderson, 1997). Estrogens are arterial vasodilators and have cardioprotective actions. In
the liver, estrogens stimulate the uptake of serum lipoproteins as well as the production of
coagulation factors. Estrogens also prevent and reverse osteoporosis and increase cell viability
in various tissues. In addition, estrogens stimulate the growth and development of the
reproductive system. When applied topically, estrogens increase skin turgor and collagen
production and reduce the depth of wrinkles (Fig 7).
2. Actions on Breast Tissue The ovarian steroids, estrogen and progesterone, are known to play a vital role in the staged
development of the mammary gland, acting through specific nuclear receptors on target cells.
These cells which represent less than 20% of the epithelium, express estrogen receptor alpha
(ERα) and progesterone receptor (PR), both are known to be located in the luminal epithelia
of the ductal and lobular structures (Petersen et al., 1987). The lobular units of the terminal
ducts of the breast tissue of young women are highly responsive to estrogen and estrogens
stimulate the growth and differentiation of the ductal epithelium, induce mitotic activity of
ductal cylindric cells, and stimulate the growth of connective tissue (Porter, 1974.)
28
Figure 7: Physiologic actions of estrogens (Gruber et al., 2002).
3. Estrogen and carcinogenesis Since Sir George Beatson observed in 1896 that breast tumours in premenopausal women
sometimes regressed after oophorectomy, numerous investigations have established that
estrogen stimulates the growth of breast cancer cells. Mechanisms of carcinogenesis in the
breast cancer caused by estrogen include the metabolism of estrogen to genotoxic, mutagenic
metabolites and stimulation of tissue growth. Together these processes cause initiation,
promotion, and progression of carcinogenesis (Yager and Davidson, 2006). In
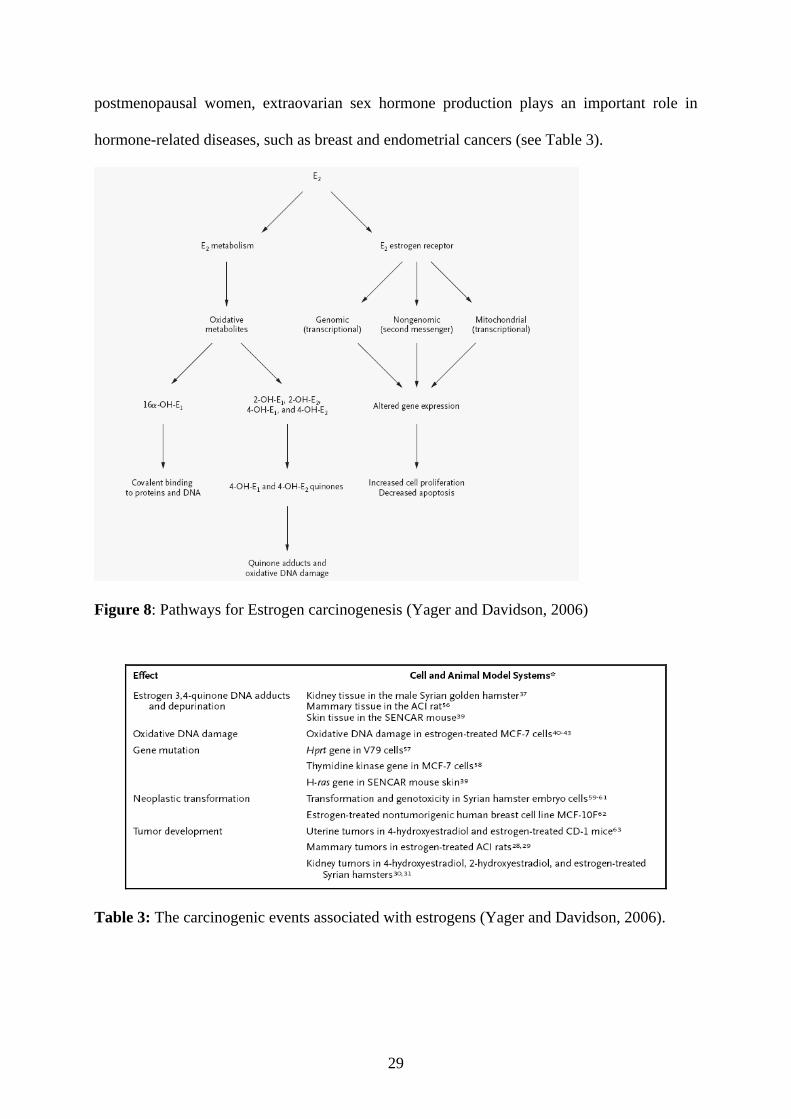
29
postmenopausal women, extraovarian sex hormone production plays an important role in
hormone-related diseases, such as breast and endometrial cancers (see Table 3).
Figure 8: Pathways for Estrogen carcinogenesis (Yager and Davidson, 2006)
Table 3: The carcinogenic events associated with estrogens (Yager and Davidson, 2006).

30
4. Estrogen Receptors
The biological actions of estrogens are manifested in cells expressing a specific high-affinity
estrogen receptor (ER). ER has two subtypes; ERα and ERβ each encoded by a separate gene
(ESR1 and ESR2 respectively). They belong to the superfamily of the nuclear receptors
(Pearce and Jordan, 2004). Other members of this family include receptors for other
hydrophobic molecules such as steroid hormones (e.g.glucocoticoids progesteron,
mineralocorticoid, androgens, vitamin D3,..), retinoic acids, and thyroid hormones (Robinson-
Rechavi et al., 2003). ER acts as a ligand-dependent transcription factor in most target tissues
(Means et al., 1972).
4.1. Estrogen Receptor isoforms and their structure
In the late 1950s, Jenson and Jacobsen (Jensen and Jacobson, 1962) demonstrated the
existence of a receptor molecule that could bind to 17β-estradiol (Jensen and Jordan, 2003).
ERα is the first estrogen receptor cloned from MCF-7 human breast cancer cells in 1980s
(Green et al., 1986). ERα gene (ESR1) is located on chromosome 6 at 6q25.1locus, composed
of 8 exons coding for a protein with 595 amino acids (66Kda). In addition, several ERα
splicing variants have been characterized (Murphy et al., 1997). In 1996, a second receptor
was reported from the rat prostate, ERβ (Kuiper et al., 1996b). ERβ is encoded by a distinct
gene (ESR2) with 8 exons, located on chromosome 14q22-24. ERβ is a protein with 530 acid
amine (60KDa) Different studies have demonstrated conservation of the regions denoted from
A through F within the structure of ERs (Gronemeyer, 1991) (Fig.9)
A/B domain: The N terminal A-B domain contains an activation function 1 (AF1), a
constitutive activation function contributing to the ligand independent transcriptional activity
of the ER. This domain is one of the least conserved domains between ERα and ERβ,

31
exhibiting only 30% identity (Ogawa et al., 1998). Functional studies have shown a lack of
AF1 activity in ERβ (Hall and McDonnell, 1999) and this could explain at least, in part, the
weak transcriptional activity of ERβ on certain promoters (McInerney et al., 1998).
C domain: The DNA binding domain is the most highly conserved region between ERα and
ERβ, with 96% identity. The C region contains two zinc finger structures each resulting from
the coordination of one Zn++ to four cysteine residues. This allowes both receptors to bind to
similar target sites (Schwabe et al., 1990).
D domain: this hinge region which contains, the nuclear localization signal, is not well
conserved between the receptors.
E domain: It is the more complex domain of ER, it contains the ligand binding domain
(LBD), a coregulator binding surface, the dimerization domain, HSP 90 binding domain, a
second nuclear localization signal, and activation function 2, AF2 in contrast to AF1 is a
ligand-dependent activation function. This domain exhibits 53% sequence identity between
two ERs (Gronemeyer, 1991; Tora et al., 1989). This difference in homology could explain a
subtle difference in ligand binding specifity (Kuiper et al., 1997). Despite the slight difference
in the affinity of ERα and ERβ for E2, both receptors are considered to have a similar affinity
for E2. However, differences were observed for other ligands, such as antiestrogens (Pearce
and Jordan, 2004) (Table 4).
Table 4: The relative binding affinity of various ligands for ERα ERβ (Kuiper et al., 1997).

32
Difference in LBD sequence of the two ERs led to synthesis of molecules that function as
selective agonists or antagonists for ERα or ERβ such as TAS-180 (SR16234) which could be
a pure antagonist on ERα and a partial antagonist on ERβ (Sun et al., 1999; Yamamoto et al.,
2005).
F domain: The C terminal of ERs shares 18% sequence homology between ERα and ERβ.
The role of this domain of ER is uncertain, but there is evidence suggesting that the F domain
has different role in the activity of ER α and β subtypes and it is in part responsible for the
difference in the biological activity of the two ER sub-types. For example on the AP-1 site,
ERβ deleted for the F domain is activated by tamoxifen while wt ERβ is activated only by
raloxifene. In ERα the F domain is activated by E2 and tamoxifen but not by raloxifene and in
ERα deleted for the F domain the receptor is activated by raloxifene (Skafar and Zhao, 2008).
The differences between the F domains of the ER alpha and beta subtypes and among the
other members of the nuclear hormone receptor superfamily may offer opportunities for
selective control of the activity of these proteins.
.
Figure 9 : ER isoforms and their function and homology(adopted from (Zhenlin Bai, 2009).
4.2. Different variants of ERs
Several splice variants have been described for both receptor subtypes, but whether all the
variants are expressed as functional proteins with biological functions is not clear. Flourio et

33
al. characterized a 46 KDa isoform (hERα46) that lacks the first 173 amino acids of the
66KDa of ERα (Flourio et al., 2000 ) (Fig 10). In addition, several other ERα splicing
variants have been isolated and identified in different cell lines (Poola et al., 2000).
Figure 10: Schematic representation of estrogen receptor (ERα) isoforms (adopted from
Heldring et al., 2007).
Unlike ERα, several splice variants of ERβ are expressed in tissues. Characterization of the
functional isoform pattern in human samples are not complete, but several experiments
demonstrated that estrogen signalling could be modulated by ERβ isoforms differentially
leading to an important impact on target gene regulation. For example, the hERβ2 isoform
(ERβ cx) (Fig. 11) with a 26aa insertion in the LBD has no transcriptional activity because it
is not capable to bind ligands or coactivators (Ogawa et al., 1998). The existence of different
isoforms of ERβ complicates the interpretation of the studies performed to understand the
prognostic and predictive role of ERβ in breast cancer.
Figure 11: Estrogen receptor (ERβ) isoforms (Heldring et al., 2007).
5. Tissue distribution of ERs ERs are widely expressed in different tissues. Using the techniques as RT-PCR, Northern
blot, immunohistochemistry and in situ hybridization, ERα and ERβ were shown to localize in
the brain, the cardiovascular system, the breast, the urogenital tract also in bones (Kuiper et

34
al., 1997). However, there is specific main subtype expression in certain tissues: the main ER
subtype in colon is ERβ, whereas ERα is the predominant isoform in the liver. Different
distribution of ER, will determine in part, particular effect of ligands. ERβ is the predominant
form in the normal mammary gland and benign breast disease. During the highest
proliferative phase of the breast, i.e. pregnancy, there is very expression of ERα and high
expression of ERβ.
6. ERs expression in breast cancer In cell-based studied, ERα appears to be predominant in cell proliferation, and ERβ was
suggested to act as a protective factor against breast cancer development (Fox et al., 2008). In
breast tumors, ERα expression increases several-fold compared to normal tissue. In low-grade
ductal carcinoma in situ (DCIS) 75% of cells express ERα. And in high-grade DCIS 30% of
the cells express low levels of ERα. In 1987, Peterson et. al found that the human mammary
gland containes a small but distinct population of ERα-positive cells, comprising
approximately 7% of the total epithelial cell population. Stromal cells were found to be ERα-
negative. Moreover, on the average, 87% of the ERα-positive cells were luminal epithelial
cells in ductal and lobular structures.
The factors during carcinogenesis that generate the ERα+ and ERα- breast cancer phenotype
remain unknown. Dontu et.al proposed a model in which ER+ breast tumors arise from either
ERα- or ERα+ progenitors while ERα- tumors arise from stem cells or early ERα- progenitor
cells (Dontu et al., 2004) (Fig. 12). Thereby they confirmed cellular heterogeneity within the
tumor and phenotypic diversity in different patients.
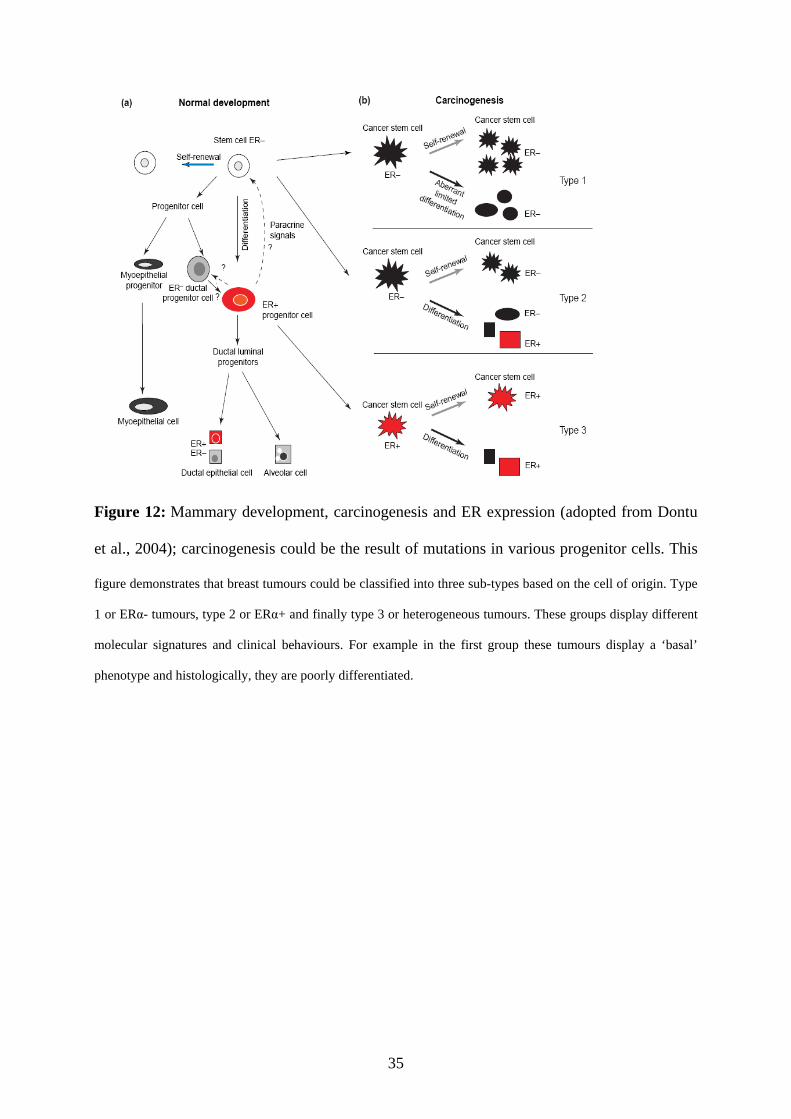
35
Figure 12: Mammary development, carcinogenesis and ER expression (adopted from Dontu
et al., 2004); carcinogenesis could be the result of mutations in various progenitor cells. This
figure demonstrates that breast tumours could be classified into three sub-types based on the cell of origin. Type
1 or ERα- tumours, type 2 or ERα+ and finally type 3 or heterogeneous tumours. These groups display different
molecular signatures and clinical behaviours. For example in the first group these tumours display a ‘basal’
phenotype and histologically, they are poorly differentiated.

36
PART III: MOLECULAR BASIS FOR TRANSCRIPTIONAL ACTIVITY OF THE ESTROGEN RECEPTOR ALPHA
1. ERα localization
Using various techniques including immunocytochemical studies it was shown that ERα
exists almost exclusively in the nucleus both in presence or absence of hormone (Monje et al.,
2001). In studies based on GFP-ER fusions, Marduva et al (Maruvada et al., 2003) have
shown that a small proportion of unliganded ERα exists in the cytoplasm, with dynamic
shuttling between the cytoplasm and nucleus in living cells. And this shuttling of ERα is
markedly affected by estrogen treatment.
2. ERα activation and functional pathways
Unliganded ERα exists in the cytoplasm and the nucleus, and is in a complex with chaperone
proteins such as heat shock proteins (HSP) especially HSP70 et HSP90 (Reid et al., 2002).
Estrogen binding mediates conformational ERα modifications causing dissociation of HSPs,
receptor phosphorylation, dimerization and recruitment of coactivator proteins to E2-ERα
complexes which lead to activation of distinct pathways by which ERα regulates
transcriptional activity of target genes.
3. Ligand-dependent pathways
3.1. Genomic pathways:
3.1a. Classical or direct pathway:
According to the classical genomic pathway, after ligand binding, the dimerised receptor
complex binds directly to a specific sequence in the regulatory region of target genes called
EREs (estrogen response element), thus controlling their level of transcription (Fig. 13A).
EREs consist of a palindromic sequence of 13 base pairs: 5’-GGTCAnnnTGACC-3’ (Klein-
Hitpaß et al., 1986). It was reported that not all estrogen regulated genes have a perfect ERE
sequence and that EREs could be imperfect or more than one ERE could be present in the

37
regulatory sequence. As shown in (Fig. 13) the nature of promoter sequence has important
consequences on gene regulation and tissue specific responses to ER ligands.
3.1b. Non- classical or indirect pathways:
In addition to direct interaction of ERα with an ERE, ERα can activate transcription of genes
whose promoters do not possess an ERE, by indirect protein/protein interactions. ERα
functions as a transcriptional cofactor and binds to other transcription factors such as fos/jun
activator protein 1 (AP-1) (Kushner et al., 2000), specifity protein 1 SP1(Saville et al., 2000),
and NFκb (Nilsson et al., 2001) (Fig.13).
Genomic pathways are also referred to as nuclear initiated steroid signalling (NISS).
A) B) C) D)
Figure 13: Different transcriptional mechanisms triggered by E2-ER complexes (genomic
pathways) (Nilsson et al., 2001). In panel A the classical interaction of the activated receptor with ERE on
DNA is shown. The other three panels present indirect effects of estrogen receptor on transcription, which occur
through protein-protein interactions with the SP1 (B), AP-1 (C), and NFκb (D): it is one of the few cases in
which E2-ER acts as a repressor of transcription.

38
3.2. Non genomic pathway
It is clear that ERs are mainly located in nucleus however data exist suggest that a small pool
of ERα exists near the membrane (Razandi et al., 1999). Membrane associated ERα can
trigger a variety of signal transduction events in seconds to a few minutes. These events
include the stimulation of cAMP, calcium flux, phospholipase C activation, and inositol
phosphate generation (Razandi et al., 2004). Recently it has been proposed that S-
palmitoylation allows ERα localization with the plasma membrane, where it associates with
caveolin-1. Upon E2 stimulation, ERα dissociates from caveolin-1 allowing the activation of
rapid signals relevant for cell proliferation. In contrast to ERα, E2 increases ERβ association
with caveolin-1 and activates the pro-apoptotic cascade (Marino and Ascenzi, 2008). This
“nongenomic” ERα action is also called membrane initiated steroid signalling (MISS) (Ilka
Nemere, 2003) (Fig.14).
Figure 14: Non-genomic ERα signaling pathway (Heldring et al., 2007).
The ligand activates a receptor, possibly associated with the membrane Signaling cascades are subsequently
initiated via second messengers (SM) that affect ion channels or increase nitric oxide levels in the cytoplasm,
and this ultimately leads to a rapid physiological response without gene regulation.
4. Ligand independent pathways
: (Growth factor signalling pathway)
In addition to conventional ligand-dependent regulation of ERα, many studies have shown a
cross-talk between ERα and signal transduction pathways. A considerable number of studies
have documented the fact that growth factors (eg epidermal growth factor [EGF], insulin-like

39
growth factor), cAMP and other agents (eg. dopamine) can stimulate the activity of ERα (S
Katzenellenbogen and A Katzenellenbogen, 2000). In this case other signalling molecules
such as growth factors could activate ERα by phosphorylation of ERα in the AF1 domain
(Dutertre and Smith, 2003). AF-1 is phosphorylated by extracellular signal-regulated kinases
as mitogen-activated protein kinase (MAPK). It has been shown that the MAPK stimulation
of ERα activity involves the phosphorylation not only of S118 but also of S104 and S106
(Thomas et al., 2008) (Fig. 15). MAPK-mediated hyperphosphorylation of ERα at these sites
may contribute to resistance to tamoxifen in breast cancer.
Figure 15 : Growth factor signalling pathway (Heldring et al., 2007).
Growth factor activated kinases phosphorylate ERs and thereby activate them to dimerize and to bind to DNA
thereby regulating gene expression.
5. Transcriptional coregulators of ERα
Nuclear receptor coregulators are molecules required for efficient function of nuclear
receptors (O'Malley and McKenna, 2008). About 300 coregulators have been reported
(http://www.NURSA.org).
Transcriptional activity of ERα is not only regulated by hormones but also by regulatory
proteins called coactivators and corepressors. Coregulators are recruited in complexes to ERα
in a ligand dependent manner. Coregulators such as histone acetyl transferases (HAT) or
deacetylases (HDAC), lead to chromatin remodeling and create a transcriptionally permissive
or nonpermissive environment (McKenna et al., 1999). It has been demonstrated that

40
coregulators are involved in endocrine related cancers such as breast and uterine cancer
(Lonard et al., 2007; Rajesh R. Singh, 2005).
6. Coregulators and chromatin remodelling
Eukaryotic chromatin is a dynamic structure that is modified and remodelled in response to
cellular signalling. The nucleosome is the basic unit of chromatin composed of an octamer of
core histones (an H4/H3 tetramer and two H2A/H2B dimers), around which 147 bp DNA are
wrapped. The free N-terminal tails of the core histones protrude from the core octamer. The
positively charged, arginine and lysine rich N-terminal amino acid extensions are subjected to
various posttranslational modifications (Luger et al., 1997).
It has been proposed that transfer of acetyl groups to the terminal amino group of lysine
residues of histones H2A, H2B, H3, and H4, by HAT activity of certain coregulators results in
disruption of the interaction between neighboring nucleosomes. This loss of density of
chromatin facilitates the access of transcriptional machinery to the promoter. In contrast,
recruitment of corepressors with HDAC activity results in loss of the acetyl groups, stabilising
the nucleosome contact and reducing accessibility of the promoter to transcription factors
(Fig. 16).
Figure 16: HAT (acetyltransferase) and HDAC (deacetylase) activity of coregolators result in
change of nucleosomes stability.
41
6.1. Co-activators
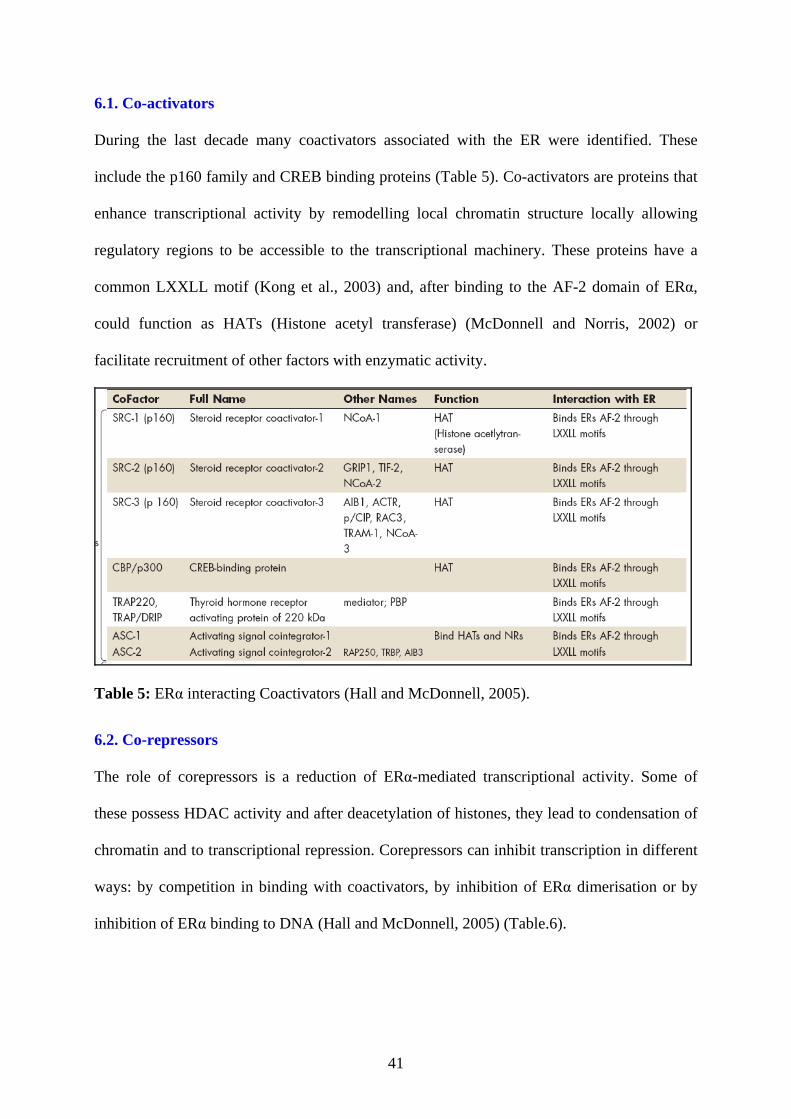
During the last decade many coactivators associated with the ER were identified. These
include the p160 family and CREB binding proteins (Table 5). Co-activators are proteins that
enhance transcriptional activity by remodelling local chromatin structure locally allowing
regulatory regions to be accessible to the transcriptional machinery. These proteins have a
common LXXLL motif (Kong et al., 2003) and, after binding to the AF-2 domain of ERα,
could function as HATs (Histone acetyl transferase) (McDonnell and Norris, 2002) or
facilitate recruitment of other factors with enzymatic activity.
Table 5: ERα interacting Coactivators (Hall and McDonnell, 2005).
6.2. Co-repressors
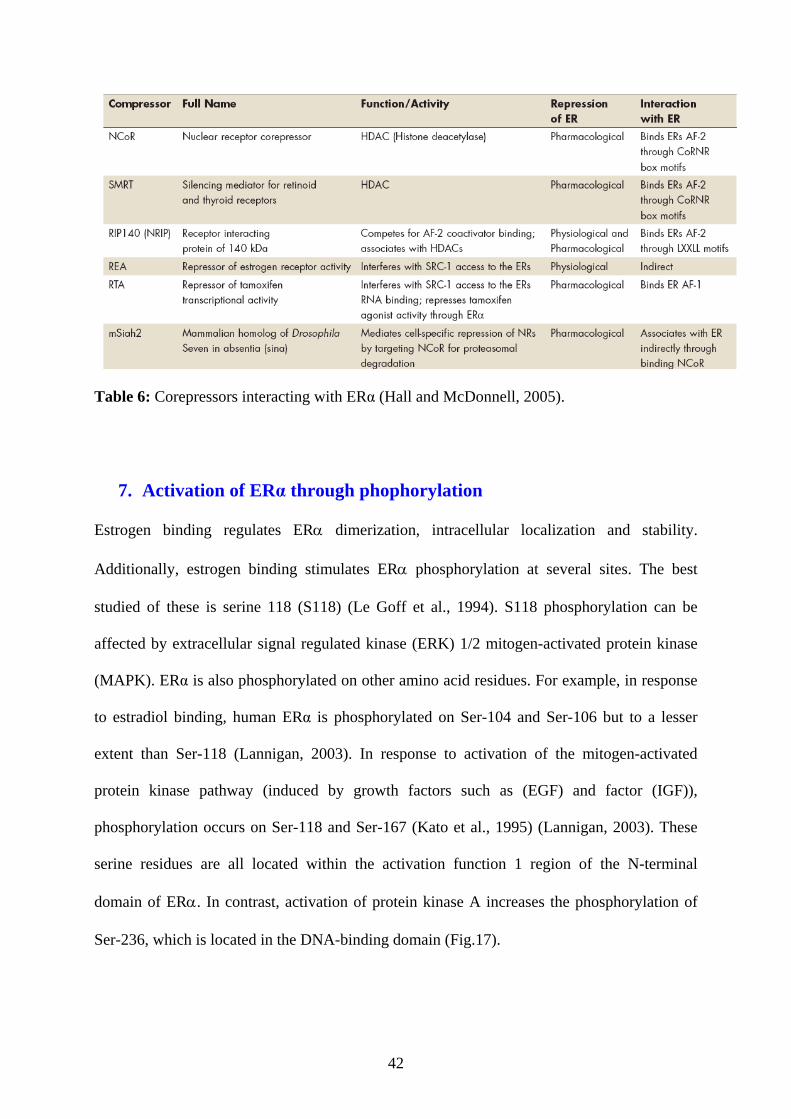
The role of corepressors is a reduction of ERα-mediated transcriptional activity. Some of
these possess HDAC activity and after deacetylation of histones, they lead to condensation of
chromatin and to transcriptional repression. Corepressors can inhibit transcription in different
ways: by competition in binding with coactivators, by inhibition of ERα dimerisation or by
inhibition of ERα binding to DNA (Hall and McDonnell, 2005) (Table.6).
42
Table 6: Corepressors interacting with ERα (Hall and McDonnell, 2005).
7. Activation of ERα through phophorylation
Estrogen binding regulates ERα dimerization, intracellular localization and stability.
Additionally, estrogen binding stimulates ERα phosphorylation at several sites. The best
studied of these is serine 118 (S118) (Le Goff et al., 1994). S118 phosphorylation can be
affected by extracellular signal regulated kinase (ERK) 1/2 mitogen-activated protein kinase
(MAPK). ERα is also phosphorylated on other amino acid residues. For example, in response
to estradiol binding, human ERα is phosphorylated on Ser-104 and Ser-106 but to a lesser
extent than Ser-118 (Lannigan, 2003). In response to activation of the mitogen-activated
protein kinase pathway (induced by growth factors such as (EGF) and factor (IGF)),
phosphorylation occurs on Ser-118 and Ser-167 (Kato et al., 1995) (Lannigan, 2003). These
serine residues are all located within the activation function 1 region of the N-terminal
domain of ERα. In contrast, activation of protein kinase A increases the phosphorylation of
Ser-236, which is located in the DNA-binding domain (Fig.17).

43
Figure 17: phosphorilation sites of ERα (Lannigan, 2003).
8. ER turn over and ligand dependent degradation of ERα
ERα is a short lived protein and its concentration is strictly regulated. Eckert et al showed that
in MCF-7 ERα turns over rapidly, with a half-life of 3-5 h, in the presence or absence of
estradiol or antiestrogen (Eckert et al., 1984). Alarid et al. described that the half-life of ERα
was only 1 h to 3 h in the presence of estrogen and that ERα was degraded by a proteasome
dependent pathway (Alarid et al., 1999). It has been demonstrated that different ligands exert
differential effects on steady-state levels of ERα (Wijayaratne and McDonnell, 2001).
Aalthough E2 and a pure antagonist, ICI 182, 780 degrade ERα, the mechanism involved in
this degradation is not exactly similar. E2-mediated ERα degradation was shown to be
dependent on other factors such as coactivator recruitment or transcription, whereas ICI-
induced degradation of ER is independent of these processes (Reid et al., 2003).
The role of different domains ligand- induced degradation of ERα has been investigated. For
example, ligand binding to the C-terminus of ER results in variable effects on proteolysis.
Agonist and antagonists induce degradation while certain ligands such as tamoxifen lead to

44
only minor degradation of ERα. Thus it has been concluded that ERα degradation could be
induced by a conformation the ligand binding domain (Alarid, 2006). There is the evidence
that N-terminus of ERα has an important role in ligand dependent degradation of ERα. AF-1
domain function is modulated by phosphorylation induced by ligand binding. Inhibiting this
phosphorylation of AF-1 prevents estrogen induced degradation of ERα (Callige et al., 2005).

45
PARTR IV: ANTIESTROGEN SIGNALING AND RESISTANCE TO ENDOCRINE THERAPY Estrogen plays a very important role in initiation and progression of breast tumours via their
receptors. The majority of tumours express ERα s and thereby inhibit the ER-signalling
pathway by anti-estrogens is a valuable option in treatment of women with ER-positive (ER+)
breast cancer.
The benefits of estrogen deprivation (by oophorectomy), for the first time was described in
1896 by Beatson as an option for breast cancer therapy. Today, the main strategies for
disrupting estrogen-dependent growth of breast tumours are non-surgical and are based on the
reduction of circulating estrogen levels in the body or blocking ERα signalling by targeting
ERα with different antiestrogens (Nicholson and Johnston, 2005). The first molecule
described in the literature with antiestrogenic property was MER-25 ( Lerner et al. 1958).
This first non-steroidal anti-estrogen did not become a clinically useful agent because of
toxicity and low potency (Lerner, 1981). The idea of competitive hormone therapy truly
emerged when, in 1960s, a failed post-coital contraceptive, ICI 46,474 (Tamoxifen) was
reinvented as a potential targeted therapy for adjuvant treatment and prevention of oestrogen
receptor positive breast cancer (Harper and Walpole, 1967). Although tamoxifen has
improved survival of many patients certain side effects, such as uterine carcinogenesis are
common. Frequently patients develop resistance to tamoxifen which motivate researcher to
determine the exact mechanism of antiestrogen action and to try to find new agents with better
tolerance and efficacy in resistant cases. Anti-hormonal options for treatment of breast cancer
in clinical data exist for two groups of agents: the selective estrogen receptor modulators
(SERM) and the selective estrogen receptor down-regulator (SERD).
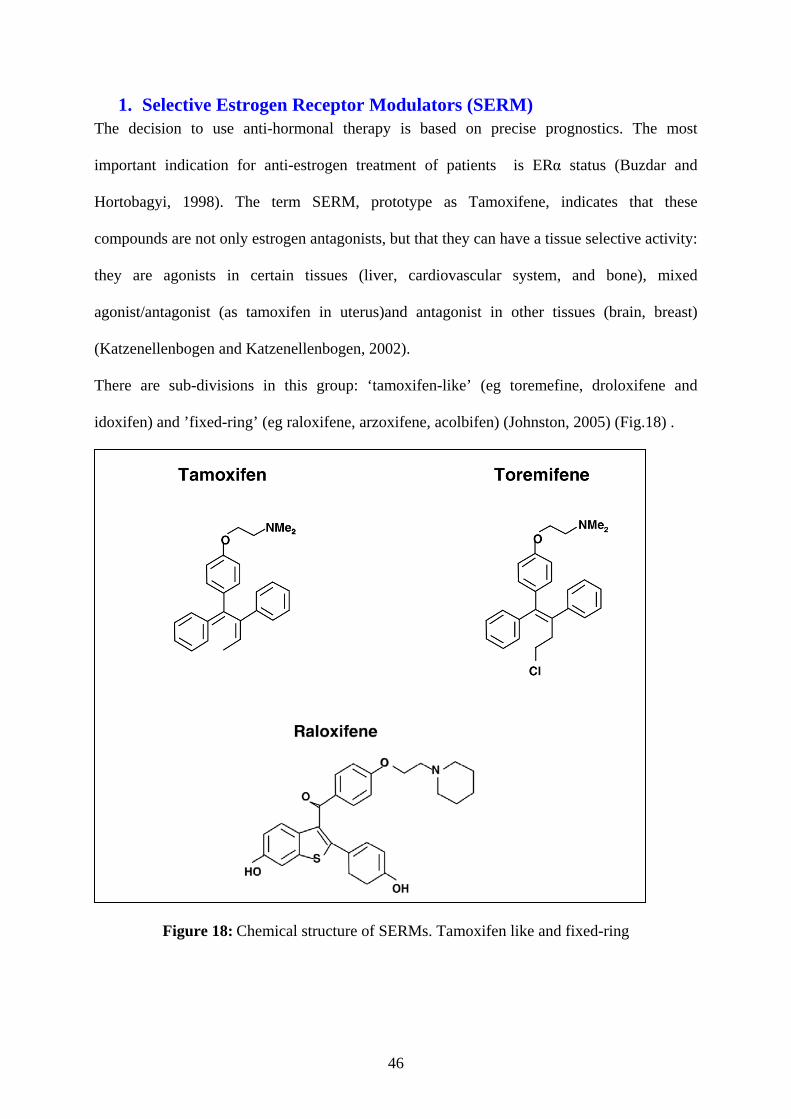
46
1. Selective Estrogen Receptor Modulators (SERM) The decision to use anti-hormonal therapy is based on precise prognostics. The most
important indication for anti-estrogen treatment of patients is ERα status (Buzdar and
Hortobagyi, 1998). The term SERM, prototype as Tamoxifene, indicates that these
compounds are not only estrogen antagonists, but that they can have a tissue selective activity:
they are agonists in certain tissues (liver, cardiovascular system, and bone), mixed
agonist/antagonist (as tamoxifen in uterus)and antagonist in other tissues (brain, breast)
(Katzenellenbogen and Katzenellenbogen, 2002).
There are sub-divisions in this group: ‘tamoxifen-like’ (eg toremefine, droloxifene and
idoxifen) and ’fixed-ring’ (eg raloxifene, arzoxifene, acolbifen) (Johnston, 2005) (Fig.18) .
Figure 18: Chemical structure of SERMs. Tamoxifen like and fixed-ring

47
Among SERMs tamoxifen is used as the gold standard in the treatment of women with ERα
positive breast cancer.
Tamoxifen
ICI 46,474 renovated as Tamoxifen is a trans isomer of triphenylethylene that was discovered
in the Imperial Chemical Industries (ICI) Ltd (now Astrazeneca) (Jordan, 2008). Tamoxifen
was approved by the U.S. Food and Drug Administration (FDA) in 1977 for treatment of
patients with hormone-sensitive breast cancers.
Metabolism and pharmacokinetic of tamoxifen
The trans-tamoxifen is the form administered to the patients because this isomer has more
affinity to ER than cis isomers. Lateral chain: dimethylaminoethoxide, and trans
configuration are reported crucial for anti-estrogenic activity of tamoxifen (Jordan, 1984). It
was accepted that tamoxifen is extensively metabolized by cytochrome P450 and, in the liver,
into two principal metabolites: the N-desmethyltamoxifen (NDT), a compound with low
affinity for ERα but a long biological half-life, or a minor metabolite 4-hydroxy-tamoxifen
(4OHT) with high binding affinity for ERα but with a short half life (Coezy, 1982). Although
it has been assumed that 4OHT is the primary means by which tamoxifen exerts its anti-
cancer effect, another metabolite of tamoxifen called endoxifen was discovered which forms
in liver by cytochrome P450 by an enzymatic system from NDT. Endoxifen is equivalent to
4OHT in its ability to bind to ERα and suppression of breast cancer progression (Jordan,
2007). 4 OH-tam binds to ERα with a similar affinity to E2; wille NDT bind with a
significantly affinity. These molecules inhibit estrogen actions on mammary tissues in a
competitive manner by binding to ER. As adjuvant therapy or for prevention of recurrence, in
most cases, tamoxifen is used routinely at 20 mg/daily for 5 years (Gradishar, 2004).

48
2. Selective Estrogen Receptor Down-regulator (SERD) Patients with hormone-responsive breast cancer following progression or relapse after
tamoxifen therapy may receive a second anti-estrogenic agent called ”Fulvestrant”or Faslodex
that was approved by the FDA in 2002 (Bross et al., 2002).
Fulvestrant is a 7α- alkylsulphinyl analogue of 17β-estradiol that not only structurally but also
in terms of molecular activity is distinct from tamoxifen (Dowsett et al., 2005). In fact,
Fulvestrant (ICI 182, 780) is a pure (ERα) antagonist that binds to ERα and blocks function
of both AF1 and AF2 via a rapid downregulation of cellular levels of the ERα and thus
described as a SERD for selective estrogen receptor downregulator (Howell et al., 2004a;
Wijayaratne and McDonnell, 2001). Fulvestrant and a group of anti-estrogens were described
first as pure anti-estrogens (see schematic representation below); they are the compounds that
have no estrogen-like properties in laboratory assays, to date less than ten distinct compounds
have been discovered (Hermenegildo and Cano, 2000). The main compounds that have been
demonstrated to have pure antiestrogenic activity in the laboratory are:
ICI 164384: the first pure anti-estrogen, is a 7α-alkylamine derivative of 17β-estradiol
discovered in 1987 by Wakeling and Bowler
ICI 182780: also called fulvestrant or Faslodex®. This compound was developed from ICI
164 384 to improve the bioavailability and the biological profile of its activity but this agent
actually used in clinic is poorly soluble with a low activity after oral administration and thus
requires intramuscular injection (250 mg monthly).
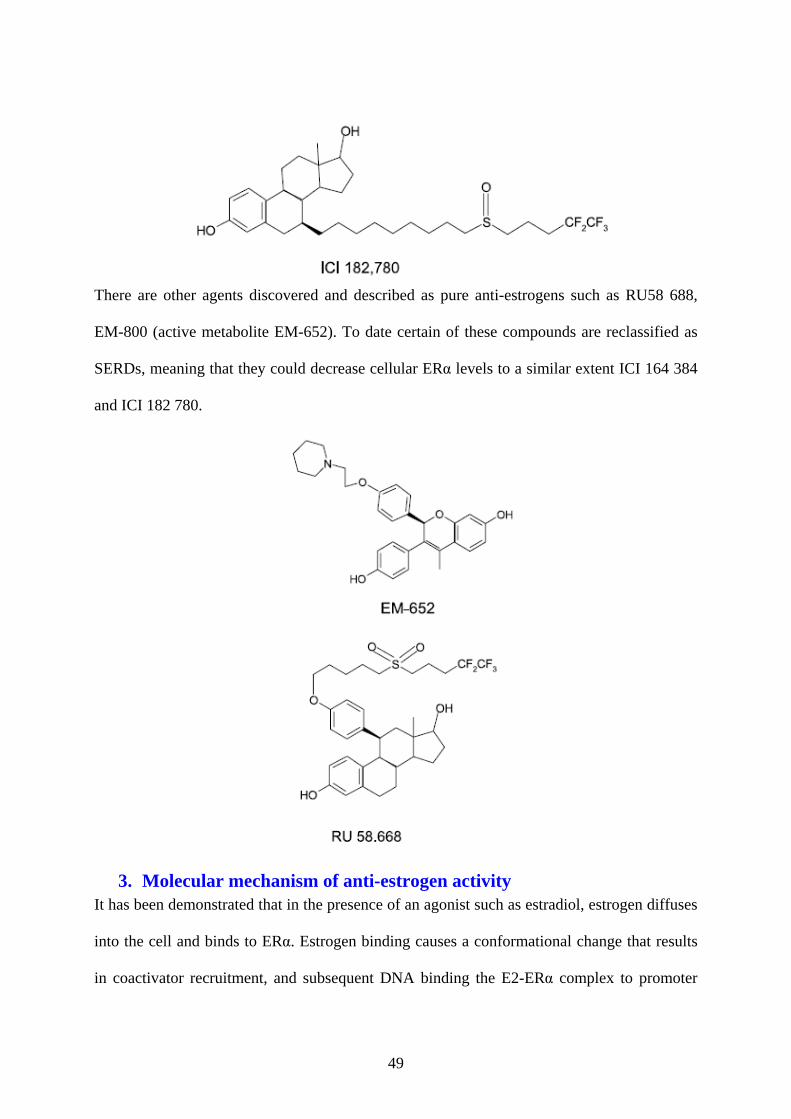
49
There are other agents discovered and described as pure anti-estrogens such as RU58 688,
EM-800 (active metabolite EM-652). To date certain of these compounds are reclassified as
SERDs, meaning that they could decrease cellular ERα levels to a similar extent ICI 164 384
and ICI 182 780.
3. Molecular mechanism of anti-estrogen activity It has been demonstrated that in the presence of an agonist such as estradiol, estrogen diffuses
into the cell and binds to ERα. Estrogen binding causes a conformational change that results
in coactivator recruitment, and subsequent DNA binding the E2-ERα complex to promoter

50
region in upstream of genes regulated by estrogens influencing cellular phenotype and then
degradation of ERα by the proteasome (see Part III).
In the presence of a SERM such as tamoxifen, the situation changes.Tamoxifen functions as a
competitive antiestrogen to block the action of estrogen. Tamoxifen binds to ligand binding
domain (LBD) of the ERα and causes a ligand induced perturbation of receptor structure that
leads to binding of corepressors (or coactivators) on the external surface of this complex. The
recruited factors differ from the ones that bind to E2-ERα complexes. SERM-ERα can repress
gene expression of ER target genes.In the presence of ICI 182,780 (a SERD) ligand induces a
rapid relocalisation of ER to an insoluble compartment in nucleus and degradation of ERα
(Callige et al., 2005; Stenoien et al., 2001).
Table 7: Mode of ER-ligand action (Howell et al., 2004b).

51
4. Transcriptional activity The transcriptional activity of ERα is mediated by two acidic domains; AF1 and AF2. The
AFs function in a synergic manner but may also function independently in certain cell and
promoter contexts and they could be regulated differentially by ligand (Tora et al., 1989;
Tzukerman et al., 1994a).
4.1. Transcription activation function 1(AF-1):
The AF-1 is located in the N-terminal A/B domain of ER (Moras and Gronemeyer, 1998).
ERα and ERβ have different AF-1 domains. In ERβ AF-1 is incomplete and its activity is
weak (Hall and McDonnell, 1999). It has been proposed that the partial agonist activity of
Tamoxifen (in certain cells and promoter contexts) relies on AF-1 (Tzukerman et al., 1994b).
ICI blocks AF-1 by affecting dimerization, DNA-binding and targeting ERα for degradation
(Dauvois et al., 1992; Fawell et al., 1990 ). In addition to the known AF-2 coactivators that
enhance receptor activity by interacting with the LBD, AF-1 coactivators have also been
described (Hall and McDonnell, 2005). Endoh et al have characterized a specific AF-1
coactivator, p68, that potentiates the AF-1 ERα activity in response to estrogen and
tamoxifen, thereby providing another alternative mechanism that may lead to partial agonist
activity of tamoxifen (Endoh et al., 1999).
4.2. Transcriptional activation function 2(AF-2):
Ligand dependent activation of transcription by ERs is mediated by interaction of coactivators
with the AF-2 domain. Through mutational analysis combined to crystallographic studies it
was demonstrated that receptor-coactivator interactions are mediated through ERα helix12
and the LXXLL motif of coactivators (Hall and McDonnell, 2005). Tamoxifen acts by
blocking AF-2 activity so it is an antagonist in cells where AF-2 is dominant and a partial
agonist where AF-1 is dominant (Pearce and Jordan, 2004). ICI blocks AF-2 as well as AF-1
activities. The transcriptional activities of both AF-1 and AF-2 are dependent on the
recruitment of coregulators. In most promoter contexts a functional synergism between AF-1
and AF-2 has been described (Kraus et al., 1995) (Fig.19).

52
Figure 19: Transcriptional activity of AF-1 and AF-2 (Dowsett et al., 2005).
5. Structural basis of agonism and antagonism
5.1. Flexibility of the LBD:
The LBD is a multifunctional domain. A series of structural studies have described the three-
dimensional structure of LBD as a polypeptide chain folded into a canonical α-helical
sandwich (Wurtz et al., 1996). It is composed of 12 helices (H1-H12) that are arranged into
three anti-parallel layers (Pike et al., 2000) and it has been demonstrated that the ligand
binding cavity has a remarkable plasticity.
Brzozowski et al (Brzozowski et al., 1997) were the first to reveal the crystal structure of the
ERα LBD in complex with E2 and raloxifene (RAL). They indicated that E2 and RAL bind at
the same site within ERα. Pike et al. also demonstrated that despite similar ligand orientation
for E2, ICI, and RAL in the LBD different spatial conformations were formed due to different
chemical structures of ligands (Pike et al., 2001) (Fig 20).

53
Figure 20 Superposition of orientation of E2 (cyan), ICI (green), and RAL (purple) in LBD
(Pike et al., 2001).
Several researchers have shown that different ligands interact with the same residues in the
LBD with a preferential binding mode for distal hydroxyl of ER ligands (Pike et al., 2001).
Similar orientation for distal side chains in α or β positions of different ligands in the ER-
ligand complex were shown.
Figure 21: The interaction of different ligands with critical amino acids in the ERα LBD:
Above E2 and raloxifene with ERα (Brzozowski et al., 1997), below ICI within the ERβ (Pike
et al., 2001)

54
5.2. Role of H12 in antiestrogen signalling
Ligand- dependent transcriptional activation function (AF-2) is located in a conformationally
dynamic region of the LBD. The key element for AF-2 conformational changes after binding
to ligands is a short helical region (Helix 12; H12) located at the C-terminus of the LBD.
The orientation of H12 is remarkably sensitive to the nature of the ligand bound: in the
presence of estradiol H12 lies over the ligand binding cavity, thereby generating a
hydrophobic groove formed by H3, H4, H5 and H12. Competent AF-2 is then able to recruit
the coactivators via a leucine-rich LXXLL motif (NR-box), that is stabilized within the
groove by a charge clamp involving residues as Lys 362 thereby facilitating transcriptional
activation (Kong et al., 2003) (Fig. 22).
Figure 22: The three-dimensional protein structure of the E2-ERα LBD complex including
H12 (blue cylinder) and hydrophobic residues (yellow) (Brzozowski et al., 1997).
In contrast, in the presence of antagonists, as described by Brzozowski et al for ERα-Ral or by
Shiau for ERα-OHT, the bulky side chain is too long to fit in the binding pocket. It is
displaced by H12 that lies along a groove between H3 and H5. Thus AF2 is incorrectly
formed and coactivator binding is blocked. In this position H12 mimics the hydrophobic
interaction of a NR-box peptide with the static region of the groove with a stretch of residues
(residue 540 and 544) that resembles an NR-box (Shiau et al., 1998) (Fig.23).

55
Figure 23: Left the three-dimensional protein structure of the RAL-ERα LBD complex
including H12 (green cylinder) and hydrophobic residues (yellow). Right: structural
representation of ERα LBD-Ral and NR-box binding site (Brzozowski et al., 1997; Kong et
al., 2003).
The only crystal structure of a SERD bound to an ER is a structure in complex with
ICI164,384 and the rat ERβ LBD (Pike et al., 2001). In this structure, ICI is long alkylamido
side chain binds along the AF-2 recruitment site, preventing H12 to adopt the position
described above. H12 is not associated with the rest of LBD. This disordered conformation
may lead to full antagonism, possibly through disruption of the receptor structure leading to
degradation in the cell (Kong et al., 2003) (Fig 24).
Figure 24:ERβ LBD and ICI (Pike et al., 2001)

56
Figure 25: A schematic presentation of modulation of ER by ligands and H12 (Heldring et
al., 2007)
6. The factors affecting antiestrogenic activity It has been demonstrated that several compounds can act in a tissue-specific manner. The best
example is tamoxifen, for which an estrogen like agonist activity in the endometrium and
bone occurs simultaneously with an estrogen antagonist activity in breast (Diel, 2002). The
mechanisms by which the same compound can exert tissue-specific agonist and antagonist
actions are still being investigated. One possible explanation for the selectivity of compounds
may be differential expression of ERα coregulators and transcription factors (Ball et al., 2009;
Smith and O'Malley, 2004). Each of these factors could contribute to cell specific effects. One
major determinant of cell type selectivity is the expression level of coregulators in different
tissues (Smith and O'Malley, 2004). This was demonstrated by the observation that the
agonist effect of tamoxifen in endometrial cells was due to the high expression of SRC-1
(Shang and Brown, 2002). AIB1 or SRC3, which function as a coactivator to enhance
transcription of genes, play an important role in the function of ERα.
These coactivators were reported to be overexpressed in 65% of breast cancers. In patients,
this high expression level of AIB1 is correlated with resistance to endocrine therapy which
could thus be explained by increased agonistic activity of drug-ERα complex (Schiff and
Osborne, 2005).

57
7. Breast cancer and endocrine resistance The molecules that are used for treatment of tumours expressing ERα have shown their
therapeutic efficacy for many years. At present anti-estrogen therapy is the most effective
treatment for women with ERα-positive breast cancers. Unfortunately many patients with a
considerable level of ERα develop resistance to endocrine therapy described as primary or de
novo resistance, and all of them, after a certain period of treatment, acquire resistance to
therapy. The mechanisms of these resistances are not exactly understood. To date
considerable effort is made to identify the pathways and other factors that are implicated
acquired resistance in order to develop new agents to overcome endocrine resistance.
8. Tamoxifen treatment and resistance to endocrine treatment Tamoxifen is the most administrated treatment of estrogen positive breast cancer that
dramatically improved survival of patients. However, a very important obstacle is intrinsic or
acquired resistance to this therapy, almost 50% of patients with ERα-positive tumour fail to
respond to tamoxifen and furthermore the patients who initially have a good response to
tamoxifen, acquire resistance (Howell et al., 1995; Osborne, 1998). Several studies in breast
xenograft models have been performed which demonstrate that initial growth suppression of
the tumour by tamoxifen is followed by reactivation of tumour proliferation stimulated by the
drug. Since this cell growth could be fully inhibited by Fulvestrant, there are arguments that
the resistance/stimulated growth of these tumours may be mediated by ERα. The antagonistic
activity of tamoxifen was proposed to be changed to an agonist effect (Berstein et al., 2004;
Osborne et al., 1995; Yao et al., 2000). Different mechanisms are involved in resistance to
tamoxifen:

58
8.1. Loss of ERα expression and function
Lack of ERα and PgR expression is the most dominant form of de-novo resistance. More than
90% of ER/PR negative tumours do not respond to antiestrogens (Clarke et al., 2003). There
is no strong evidence for loss of ERα expression in ERα positive tumours that acquire
antiestrogen resistance. In cell cultures, there are a few examples where cells switch to an
ERα negative phenotype through two major mechanisms: population remodelling and
transcriptional repression of ERα expression (Graham et al., 1992; van den Berg et al., 1989 ).
Multiple mechanisms involving silencing of ERα have been identified. They include
mutations within the open reading frame and transcriptional silencing by DNA methylation of
the promoter (Yamashito, 2008).
Function of the ERα could be disturbed by mutating of the ESR1 gene that encodes ERα or
the generation of splice variants. The presence of ERα transcripts with deletions in several
exons has been described in breast cancer cell lines and normal- and malignant breast tissue
samples. While the exact function of these splice variants is not established, it has been
hypothesized that the splice variant mRNAs may result in proteins that differ in activity
(Poola et al., 2000). For example Fuqua et al. have reported that co-expression of an exon 5
estrogen receptor (ER) variant, originally identified in ERα-negative tumours, with wild
typeERα resulted in resistance to the growth-inhibitory effects of tamoxifen in MCF-7 cells
(Suzanne, 1994).. A naturally occurring ERα mutant has been described that recognised
tamoxifen as an agonist (Levenson et al., 1997). These two last points (splice variant and ER
mutant) are awaiting approval for clinical relevance.
8.2. ERβ subtype and its alteration in expression
Both the original ERα and the more recently identified ERβ and its splicing variants are
expressed in many breast cancers. ER protein stability is regulated differently: in breast
cancer ERα and ERβ protein levels are decreased by E2 but increased by tamoxifène (Pearce

59
et al., 2003). ICI treatment results in ERα degradation and ERβ stabilisation.Using specific
treatments may thus result in an altered ERα/ERβ ratio. A number of groups have reported
results regarding possible functions of ERβ that show signalling by a third isoform in
different ways depending on ligand or promoter structures. For example binding of several
SERMs, including tamoxifen resulted in agonistic activity of ERβ. Also ICI can activate ERβ
mediated transcription through AP-1, a pattern of regulation different from some other
antiestrogrns. To date, it is clear that ERα/ ERβ ratios change during carcinogenesis. ERα
expression is increased and ERβ is decreased in early breast cancer, whereas expression of
both declines in more invasive cancer (Fox et al., 2008).
8.3. The role of nuclear receptor coregulators
Several investigations have described the role of coregulators that can significantly influence
ERα -mediated transcription. In breast cancer, the AIB1 gene has been reported to be
amplified in 4 out of 5 ERα -positive breast cancer cell lines and to over-expressed in the
majority of primary breast cancers (Anzick et al., 1997). Shang et al demonstrated that
estrogenic activity of tamoxifen in the uterus requires a high level of steroid receptor
coactivator 1 (SRC-1) expression. He concluded that cell type and promoter-specific
differences in coregulator recruitment may determine the cellular response to tamoxifène
(Shang and Brown, 2002). The corepressors N-CoR and SMRT interact more strongly with
ERα in the absence of E2 or in the presence of tamoxifen. In a nude mouse model growing
MCF-7 cells, acquired resistance to tamoxifen was found correlated with reduction of N-CoR
level (Lavinsky et al., 1998). Thus, gain of coactivator function or loss of corepressor function
or a shift in balance of coregulators may be playing an important role in resistance to
antiestrogens such as tamoxifen.

60
8.4. ER pathway cross-talk with growth factor and cellular kinase pathways
Expression of several growth factors and their receptors is regulated by estrogen. Growth
factors (TGFα, EGF, IGF-1) have been shown to activate ERα signalling via the MAP kinase
cascade (Nicholson et al., 1999; Smith, 1998). Experimental and clinical evidence suggest
that overexpression of growth factor receptors in breast cancer especially those of the
EGFR/HER2 family is associated with resistance to endocrine therapy in particular to
tamoxifen (Massarweh and Schiff, 2007). King et al. reported that a member of the tyrosine
kinase proto-oncogene family known as c-erbB2 or HER2, was amplified in human mammary
carcinoma (King et al., 1985). Currently, it is estimated that the HER-2 gene is amplified in
20% of breast cancers, and that hyper activity of HER2 signalling may lead to a complete loss
of ER expression as a mechanism of resistance to endocrine therapy (Massarweh and Schiff,
2006). Recently Hurtado and co -workers proposed that PAX-2 a general transcriptional
repressor of ERBB2 play an important role in endocrine responsiveness. They showed that
PAX2 and AIB compete for binding and regulation of the ERBB2 gene. An increase in AIB1
levels blocks PAX2 binding and ERBB2 gene repression thereby reversing antiproliferative
effects of tamoxifen (Hurtado et al., 2008).
Activation of different kinases such as MAPK, Protein Kinase A (PKA) have been shown to
enhance tamoxifen resistance in breast tumours. PKA-mediated phosphorilation of S305 in
the hinge region of ERα reorient tamoxifen bound ERα towards its coactivator SRC-1, this
orientation results in recruitment of RNA polymerase II and ERα -transcription in presence of
tamoxifen (Zwart et al., 2007). Endocrine resistance is a multifactorial problem and various mechanisms may account for insensitivity to tamoxifen. To date few of these mechanisms are validated in clinical features of tamoxifen resistant breast cancer as the combined elevation of AIB1 and ERBB2 pathways, and the molecular details of these events remain elusive.

61
AIM OF PRESENT STUDY The proposed project was part of a larger project in the context of resistance to hormonal
therapy of breast cancer (Resisth network; GSO Cancéropôle, financially supported by
INCA). The aim of this study was to gain further insight into the mechanism of antiestrogen
activity and thereby to design tools that can help to understand the molecular mechanisms
involved in ligand-dependent modulation or ERα down regulation
The specific aims of this thesis were to:
1. Establish a molecular and cellular system to discriminate rapidly SERM from SERD.
2. Investigate the impact of nature of ligand on intracellular localisation of ERα.
3. Design the antiestrogens with modified structure and study their effect on localization of
ERα degradation of ERα and their effect on activation of estrogen regulated genes.

62
RESULTS

63
PUBLICATION N° 1

64
A molecular approach to predict selective estrogen receptor modulators from down regulators
Mahta Mazaheri, Clarisse Loulergue-Majorel, Anne-Claire Lavigne, Kerstin Bystricky # and
Helene Richard-Foy
1, Université de Toulouse ; UPS ; Laboratoire de Biologie Moléculaire Eucaryote ; F-31062
Toulouse, France
2 CNRS; LBME; F-31000 Toulouse, France
#corresponding : [email protected]
Key words: Endocrine therapy, Breast cancer, Estrogen receptor, anti-estrogen, ICI 182,780
Abbreviations: ER, estrogen receptor; OHT, 4-hydroxytamoxifen; SERM, Selective
Estrogen Receptor Modulator; SERD, Selective Estrogen Receptor Down regulator; ICI, ICI
182780.

65
Abstract
___________________________________________________________________________
Breast cancer is most common type of malignancy among women in the world.
Approximately 60% of breast tumours express the oestrogen receptor alpha (ERα) and are
considered hormone-responsive. Thus endocrine therapies have long been the treatment of
choice. However, the estrogen-like agonist effect of the available selective estrogen receptor
modulators such as tamoxifen and the development of resistances require the development of
new treatments that act through different mechanisms.
The objective of our study is to design tools that can help to understand the molecular
mechanisms involved in ligand-dependent modulation or degradation of ERα. It has been
proposed that ligand-dependent ERα degradation of antagonist may result from the presence
of a long aliphatic side chain on a steroidal core, thus we selected the following compounds:
RU 39411, RU 58668, which dervatives of 17β-estradiol, but different side-chains: RU 39411
has a dimethyl-amino-ethoxy-phenyl side chain similar to that of Tamoxifen, while RU 58668
has a neutral hydropfobic side chain similar to that of Faslodex. The third compound, EM
652, has a phenylcoumarine core that mimics the steroid backbone of oestradiol while the side
chain is similar to that of Raloxifen.
Fulvestrant and RU 58668 induced degradation of ERα, while RU 39411 and EM 652 have
the same effect as Tamoxifene and stabilized ERα. Three mutants of ERα, L540Q, K362A
and D351Y were generated and used in transient transfection assays in MDA-MB231cells.
The effect of the ligands on gene expression of a luciferase reporter gene under the control of
an estrogen inducible promoter and ligand-induced degradation of ERα were measured. Our
results demonstrate that antiestrogens can be classified into two groups based on structure and
function: Tamoxifen and RU39411 fall into a first group, and Faslodex and RU58668 into a
second group. Interestingly EM 652 has an intermediate behavior.

66
We have used molecular studies to screen and classify antiestrogen compounds into SERM or
SERD with the aim to exploit the structural characteristics of anti-estrogens to create new
compounds with different lateral side chains for the study of the relationship between
structure and activity of antiestrogens.
Introduction
___________________________________________________________________________
Approximately 60% of breast tumours express the estrogen receptor ERα and are thus
considered hormone-responsive. Endocrine therapy having usually fewer side effects than
chemotherapy, it is recommended as treatment of choice for women with ERα-positive breast
cancer. Endocrine therapies are based on the use of synthetic agents which can inhibit the
proliferative action of estrogens (Clarke et al., 2001) (Katzenellenbogen et al., 2000)
(O'Regan et al., 1998). Among anti-hormonal options anti-estrogens are compounds which
compete with estrogens for binding to estrogen receptors ERα and ERβ (Green and Chambon,
1988; Kuiper et al., 1996a) particularly efficient.
The biological action of ERα in the presence different ligands has been studied for a long
time, In the absence of ligand, the majority of ER resides in the nucleus bound to an inhibitory
heat-shock protein, HSP90 (DeFranco, 2002; McDonnell and Norris, 2002). Ligand binding
induces a conformational change of the ER leading to displacement of the HSP90 heat-shock
protein, ER dimerization and recruitment of coactivators or corepressors upon interaction with
DNA response elements within the regulatory sequence of target genes (Heldring et al., 2007;
O'Malley and McKenna, 2008)
Tamoxifen has been the first-line endocrine therapy in estrogen receptor positive tumors for
nearly three decades in premenopausal patients as well as for postmenopausal women
(Gradishar, 2004). However, frequent relapses while on tamoxifen and the long term toxicities
of the drug stimulated research to find safer and more effective endocrine strategies. Two

67
major classes of molecules have been developed: selective estrogen receptor modulators
(SERM) such as Tamoxifene and selective estrogen receptor downregulator (SERD) such as a
pure antiestrogen Faslodex.
Crystallographic studies demonstrated that agonist or antagonists bind to the same site within
the core of the LBD of ERα but with different binding modes. Comparison of the estrogen
receptor-LBD complexed to the antiestrogens like tamoxifen or raloxifene has demonstrated
that the bulky side chain of antiestrogens can not be accommodated within the LBD. The
resulting structures differ from that observed in the presence of estrogen by the position of the
C-terminal helix 12 (H12), which in the presence of agonist sits above the ligand binding
cavity and contributes to coactivator recruitment (Brzozowski et al., 1997; Shiau et al., 1998).
In the antagonist-bound structures, the side chains of antiestrogens force H12 to reposition
over the coactivator binding groove, preventing interaction with coactivators and promoting
the interaction with corepressors. This results in a transcriptionally inactive complex that
blocks ER induction of genes important for cell proliferation (Johnston, 1997).
It has been postulated that in the presence of Faslodex (ICI) this disordered conformation may
lead to full antagonism, either through disruption of the AF1 and AF2 site or through
destabilizing the receptor structure leading to degradation in the cell (Kong et al., 2003). The
ICI- ERα complex is rapidly sequestered and degraded in a salt-insoluble compartment in the
nuclear matrix (Balan et al., 2006; Callige et al., 2005).
Although faslodex is an efficient treatment for patients who failed after endocrine therapy,
and in metastatic breast cancer its poor pharmacodynamic properties and lack of oral
bioavailability have limited its clinical utility (Howell, 2000; Robertson et al., 2005)
We have used these mechanistic studies to clarify the underlying mechanism of action of
several antiestrogens belonging to different classes with the aim to develop an effective
molecular screen for newly designed pharmaceutical properties of different compounds.

68
Materials and Methods
___________________________________________________________________________
Cell lines and cell culture
The MDA-MB-231 cell line was from the American Type Culture Collection (Manassas, VA)
and grown in DMEM containing phenol red, 10% heat-inactivated fetal calf serum
(Invitrogen), and 50 µg/ml gentamycin (Invitrogen). MELN cells (Balaguer et al., 1999)
estrogen-sensitive human breast cancer cells (MCF-7) stably transfected with the estrogen-
responsive gene (ERE-βGlo-Luc-SVNeo). They were cultured in DMEM/F12 medium with
GlutaMax™ I containing phenol red, supplemented with 50 µg/ml gentamicin (Invitrogen),
1 mg/ml G418 sulphate (Promega, Leiden, The Netherlands) and 10% heat-inactivated fetal
calf serum (Invitrogen). The cell line was maintained in an incubator at 37°C, at a relative
humidity of 95% and with a CO2 concentration of 10%. To study the effects of estrogens and
anti-estrogens the cells were progressively deprived in serum as follows: they were first
grown for 3 days in medium without phenol red and antibiotic, containing 5% charcoal-
stripped serum (SSH) Cells were treated or not with 10 nM estradiol or 1µM of anti-estrogen
for the indicated times.
Chemicals
17β-estradiol (E2) and 4-hydroxy tamoxifen (4OHT) were purchased from Sigma-Aldrich (St.
Louis, MO). ICI 182,780 (ICI) was purchased from Zeneca Pharmaceuticals. RU39411 (RU
39) and RU 58668 (RU 58) were gifts from Dr. Michel Renoir (CNRS/ Châtenay-Malabry) ,
and EM-652 (EM) was provided by Dr. Labrie (U Canada). Stock solutions of E2, Tamoxifen
(4OHT), ICI, EM, RU 39 and RU58 were prepared in absolute ethanol. Leptomycin B (LMB)
was purchased from Sigma and Calpain Inhibitort I (ALLN) was purchased from Calbiochem.

69
Antibodies
Anti-human ERα (HC-20: sc-533) and ERα (H-184: sc-7207) were purchased from Santa
Cruz Biotechnology. Anti-GAPDH was purchased from Chemicon International.
Mutagenesis
Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit
according to the manufacturer's instructions (Stratagene, La Jolla, CA). The ER PSG5
plasmid (HEGO, kindly provided by P Chambon) was used as a template for PCR. Primers
used were: A-K362A-R (5'-ATC AAC TGG GCG GCG CGC GTG CCA GGC TTT TG -3'
and 5'-ACA AAG CCT GGC ACG CGC GCC GCC CAG TTG ATC -3'), L540Q (5'-CTA
TGA CCT GCA GCT GGA GAT GCT GGA -3' and 5'-TCC AGC ATC TCC AGC TGC
AGG TCA TAG -3'), A350V-D351Y (5'-CTG ACC AAC CTG ATG TAC AGG GAG CTG
GTT C -3' and 5'-GAA CCA GCT CCC TGT ATA CCA GGT TGG TCA G -3'). Purified
DNA was sequenced to check for the presence of the mutation. A larger scale DNA
preparation was used to sequence the entire cDNAs of the ERα and mutants. Each mutant was
then cloned into the pSG5puro plasmid using the NotI and SacI site flanking the ER cDNA.
Transient Transfection Experiments and Luciferase Assays
MDA-MB-231 cells were grown for 3 days in phenol red-free medium, containing 5%
dextran-coated charcoal-treated fetal calf serum (FCS). Cells were then transiently
cotransfected with vectors expressing either wild-type ERα or the mutants (0.5 µg), along
with an ERE-luciferase plasmid (0.35 µg) and pCH110 (0.05 µg) that expresses the -
Galactosidase under the control of a SV40 promoter was used to normalise for tansfection
efficiency. For the Western Blot experiments performed after transient transfection, pSV40
luciferase internal vector (0.1 µg) was cotransfected either wild-type ERα or the mutants (0.5

70
µg). It was used to determine transfection efficiency for each sample. MDA-MB-231 cells
were plated at 2 ×104 per 12-well plates and grown for 1 day in phenol red-free medium,
containing 5% FCS, before the transfection using FuGENE® HD Transfection Reagent
(Roche Applied BioSystems, MA), according to the manufacturer's instructions. Two days
after transfection, cells were treated with the appropriate compound for 3 or 24 hours (as
indicated in figures) and harvested.
At the end of the incubation period, the remaining medium was removed. The cells were
washed once with cold PBS and lysed in 60 µl reporter lysis buffer (Promega, E397A Leiden,
and The Netherlands). After shaking plates for 20 min, plates were frozen (−80°C) for
minimum 1 h and maximum 1 week. After thawing the plates, luminescence was measured
using a luminometer (Luminoskan) after injection of 50 µl luciferase assays reagent
(Promega, E397A Leiden, The Netherlands) in each well, according to the manufacturer's
instruction, on a Centro LB 960 (Berthold). Results are expressed as relative light units
(RLU). -Galactosidase activity was measured using 10 µl of each sample and the Galacto-
Light Plus detection system (Roche Applied Biosystems, Bedford, MA). Final data are
reported as relative light units, which is the luciferase reading divided by the -galactosidase
reading. For western blotting, protein concentrations were measured using the Amido
Schwartz technique and normalize with luciferase activity. For each condition, average
luciferase activity was calculated from the data obtained from 2-3 independent wells and the
experiments were repeated at least 3 times. Statistical analysis was performed using Student’s
t test analysis.
Cell extracts and Western blots
MDA-MB-231 and MELN cells grown 12-well plates and were treated as indicated, washed
with PBS and collected by centrifugation. Total cell lysates were prepared by resuspending
the cells of each well in 100 µl lysis buffer (50 mM Tris pH 6.8, 2% SDS, 5% glycerol, 2 mM

71
EDTA, 1.25% β-mercaptoethanol, 0.004% Bromophenol blue). The samples were boiled for
20 min at 95°C and cleared by centrifugation at 12 000×g for 10 min. The protein
concentration was determined by the Amido Schwartz assay when the samples contained
SDS. Samples were subjected to PAGE on a 10% SDS-polyacrylamide gel in 25 mM Tris–
HCl, 200 mM glycine, pH 8.3, 0.1% SDS and the proteins transferred onto nitrocellulose
membrane. Western blot analysis was performed as previously described (Giamarchi2002)
using rabbit polyclonal ERα antibodies diluted to 1 µg/ml (HC20 or H-184 Santa Cruz
Biotechnology, Inc.). Western blots were quantified using the TINA PC-Base Software from
FUJI.
qRT-PCR experiments
RNA extraction
MCF-7 cells were grown in phenol red-free Dulbecco’s (DMEM)/F12;(invitrogen)
supplemented with 5% charcoal-dextran-stripped fetal bovine serum(FBS) for 3 days and then
cells were treated with10nM of E2, 1µM of anti-estrogens, or vehicle for 16h.
Total RNAs were extracted using TRIzol Reagent (invitrogen), the quality of RNA samples
were ensured by electrophoresis in an agarose gel followed by ethidium bromide staining,
where the 18S and 28S RNA bands could be visualised under UV light.Quantification of
RNA was performed by spectrophotometry at 260 nm. RNA samples were stored in RNAse-
free distilled water and at 80°C.
cDNA synthesis
1 - 5 µg of total RNA was reverse transcribed in a final volume of 20 µl using SuperScriptrTM
III Reverse Transcriptase manufacture. cDNA was stored at 80°C.

72
Real-time PCR amplification
All target transcripts were detected using quantitative real time RT-PCR (SyberGreen) assays.
The experiments were performed on a Mastercycler ep realplex device; and TBP was used as
endogenous control for normalization of the data. The following primer pairs were used to
amplify cDNA after reverse transcription experiment:
TBP: 5’-CGGCTGTTTAACTTCGCTTTC-3
5’-CCAGCACACTCTTCTCAGCA-3’;
pS2 /TFF 1: 5’-CCCCTGGTGCTTCTATCCTAAT-3’
5’-CAGATCCCTGCAGAAGTGTCTA-3’;
PGR: 5’-CTTAATCAACTAGGCGAGAG-3’
5’-AAGCTCATCCAAGAATACTG-3’;
TGFα; 5’-ATCTCTGGCAGTGCTGTCCCT-3’
5’-CTTGCTGCCACTCAGAAACA-3’;
The thermal cycling condition comprised 2 min at 55°C and 2 min at 95°C followed by 40
cycles at an appropriate annealing temperature depending on the primer set for1 min. To
quantify the result obtained we employed the comparative Ct method using qBASE software.

73
Results
___________________________________________________________________________
Relationship between molecular structure of ERα ligands and ERα stability
ER binds a variety of ligands including agonists such E2, SERMs such tamoxifen and
raloxifen (Shiau et al., 1998) and SERDs such as faslodex. In all cases the receptor-ligand
interaction induces a distinctive conformation of the ligand-binding domain (LBD) of the ER.
The conformation of this part of the protein determines recruitment of transcriptional
coregulators, thereby regulating target gene transcription (Pike et al., 2001). The first
objective of this study was to define the characteristics of ligands that are important for
cellular fate of ERα. We selected several compounds with different structures (Figure 1A) that
have previously been described as pure anti-estrogens or SERMs – describe more for review
see (Anstead et al., 1997).
We first examined the stability of ERα in MELN cells, a cell line derived from MCF-7 that
stably express a luciferase reporter gene driven by an estrogen reponse element containing
promoter(Balaguer et al., 1999) following treatment with these compounds.
ERα content from MELN whole cell extracts after 3h treatment with hormones or anti-
hormones was analyzed by Western blotting. We found that the pure antagonists ICI 182780
(ICI) and RU 58668(RU58) induced rapid degradation of ERα. In contrast, in the presence of
RU 39411 (RU39) and EM652 ERα was not degraded as well as ICI and RU58 after 3h. This
behavior was similar to that of 4OHT which stabilized ER after three hours of treatment.
These results show that functional interaction of ERα with ICI and RU58 promotes ER
degradation, while binding to 4OHT, EM652 and RU39 stabilizes the ERα (Figure 1.b).
It was previously proposed that ERα degradation occurs in the cytoplasm in the presence of a
natural ligand (E2), but, in the presence a full antiestrogen such as ICI ER is rapidly degraded,
at least partially, in a nuclear compartment in a proteasome dependent manner (Callige et al.,

74
2005; Marsaud et al., 2003; Nawaz et al., 1999). We investigated the effect of LeptomycineB
(LMB), an inhibitor of CRM1-dependent nuclear export, and ALLN, an inhibitor of the
proteasome, on ligand-dependent degradation of ERα. MELN cells were pretreated with LMB
or ALLN for 30 min. As shown in Figure1.c E2 induced partial ER turnover was blocked by
ALLN but not by LMB suggesting that nuclear export is not required for ER degrdadation.
Furthermore, LMB treatment did not affect ICI induced loss of ERα, while ALLN clearly
stabilized ICI induced ERα degradation. Similarly, LMB treatment had no effect on RU 58
induced ERα degradation while ALLN blocked it, suggesting that in the presence of RU58
ERα is degraded in the nucleus by a nuclear proteasome as has been described for proteolysis
of ICI-bound ERα. Thus, analyzing the extent and cellular location of ERα degradation offers
a simple test that allows discrimination of two classes of antiestrogens: SERMs which
stabilize ERα and SERDs which induce rapid proteasome dependent degradadation of ERα in
a nuclear compartment.
The crystal structures of the LBD of the estrogen receptors (ERα and ERβ) bound to different
ligands (E2, tamoxifen, raloxifen, diethylstilbestrol, ICI 164,384) (Brzozowski et al., 1997;
Pike et al., 2001; Shiau et al., 1998) have reveal that ligands of different sizes, shapes and side
chains induce a spectrum of conformational states (Katzenellenbogen and Katzenellenbogen,
2002) and repositioning of helix H12 of the LBD. The resulting AF-2 surfaces that modulate
recruitment of cofactors are directly dependent on the structure of the bound ligand. Thus, we
next investigated whether mutating specific residues that either have a role in interaction
between ERα and various ligands, in stabilizing of H12, in antagonist position or in
recruitment of coactivators would influence the fate of ERα and its ability to stimulate
transcription.

75
Transcription efficiency but not E2 mediated agonist activity of an estrogen responsive
reporter gene is reduced by K362A, D351Y and L540Q point mutations of ERα
We created a series of receptor mutations by site directed mutagenesis (see materials and
methods) of pSG5 (HEGO)-ERα. The transcriptional activity of a luciferase reporter and the
stability of ERα were assayed in the presence of the selected ligands. These specific residues
were chosen because they appear to participate in the interaction of ERα with ligands and
coregulators or in conformational changes of ERα in the presence of various antiestrogens
(Montano et al., 1996). Crystallographic studies have demontrstaed that Asp351 plays an
important role in anchoring the ligand to protein. For example upon ER-Raloxifene complex
formation, Asp351 makes a hydrogen bond with piperazine ring nitrogen (Brzozowski et al.,
1997) or in the presence hydroxy-tamoxifen, Asp351 makes a salt bridge with methylamino
group of side chain (Shiau et al., 1998). Lys362 is a highly conserved residue which is
required for efficient E2-dependent recruitment of certain coactivators. It is buried by induced
repositioning of H12 in the presence of raloxifen or tamoxifen (Kong et al., 2003). Leu-540
which is part of the helix H12 is thought to play a role in regulation of ER ligand recognition
or transcriptional activation.
Although, these ER mutants have previously been tested for their role in transcription
activation, a majority of studies was not performed in breast cancer cell lines and were based
on receptor constructs under the control of promoters which contained multiple ER binding
sites unlike the structure of natural ER responsive promoters (Pearce et al., 2003).
We first tested transcriptional activity of the ERα mutants by transient co-transfection of ER
negative MDA-MB 231 cells with the mutant ER cDNA and a luciferase reporter gene under
the control of a single ERE containing promoter. Compared to the wt ERα, all mutants
displayed a much lower basal transcription in the presence of vehicle control ethanol.

76
However, 10nM E2 treatment resulted in a significant induction of luciferase activity relative
to the EtOH control levels (Fig 2).
The 12-fold induction of luciferase activity observed with L540Q mutant was greater than the
7.7 fold observed for wt ER. The K362A mutant displayed an intermediate level of induction,
and D351Y reached a small, but significant 5-fold induction. These results demonstrate that
ERα mutants are perfectly capable of activating transcription when bound to E2. However,
weak basal transcription may indicate that the residues mutated reduce ER binding to its
target sequence or may hamper its interaction with co-activators necessary for efficient
transcription initiation.
The effect of the different antiestrogens on the activation of a luciferase reporter gene
and on the stability of mutated ERα transiently transfected in MDA-MB231.
4OHT and RU39 binding to wt ERα stimulated luciferase transcription 1.5 - 2 fold, while ICI
and RU58 had a weak inhibitory effect. In Figure 3A we show that compared to wt ER, the
D351Y mutant ER increased agonistic activity of 4OHT, EM, and RU39 to 2.5 - 4.5 folds.
Even with ICI and RU 58 a 1.5 -2 fold induction of transcription was measured (Fig 3.B). The
K362R mutant also significantly induced luciferase transcription in the presence of 4OHT and
RU39. Binding of ICI and RU58 to this ERα mutant had no notable effect on transcription.
EM bound ER wt inhibited transcription to the same extent as ICI which corroborates earlier
reports that EM652 was a pure antagonist (Labrie et al., 1999). Curiously, both ER mutants,
D351Y and K362R, become agonist upon treatment with EM652 (Figure 3B and C).
Luciferase transcription is stimulated 3 fold by the D351Y ER bound to EM652 which is
equivalent to the stimulation measured in the presence of 4OHT. Activation of transcription is
less pronounced by the K362R mutant, but still about 2 fold. Changes in the conformation of

77
the mutated AF2 domain upon EM652 binding to these mutants may alter recruitment of
cofactors.
Interestingly, when bound to the L540Q mutant of ER all ligands appear to partially induce
transcription, albeit to a lesser extent than E2, with only 3 - 5 fold compared to 12 fold by E2.
Notably, ICI and RU58 exhibit significant agonist activity (Fig 3D).
Thus, the effect of antiestrogens appears to depend on the conformation of ERα. In order to
further characterize the effect of selected compounds on the transiently transfected wt and
mutant ERα, protein levels after an acute (3h) treatment were compared between each of the
transiently transfected cell lines. Loading was controlled based on SV40-LUC expression.
We found that wt ERα-ICI and ERα-RU58 complexes were rapidly degraded, while 4OHT
and EM652 stabilized wt ER protein levels (Fig 3A). This behavior was also true for ER
mutated at residues Asp351 and Lys362 (Figure 3B and C). However, treating cells with
EM652 had a different effect on these ER mutants than on ER wt. Indeed, in the presence of
EM652 wt ER was stabilized but ER mutated at either D351 or K362 was degraded almost to
the same extent as in the presence of ICI or RU58. These mutated residues may alter folding
of ER when bound to EM since the aliphatic side chain of EM forms a bond with D351
similarly to Raloxifen (Bentrem et al., 2001). Replacing Asp with Tyr may release the side-
chain. Such a loose, long side-chain is reminiscent of the side chain of ICI and targets ER to
the proteasome.
Strikingly, the L540Q point mutation of ERα confered resistance to degradation in the
presence of ICI and RU58 (Figure 3D). To further evaluate of this mutant in a reproducible
manner, we have generated a stable transfectant of L540Q in MDA-MB 231 cells and ERα
protein levels were compared in the presence of different ligands (figure 3E). We did not
observe significant changes in ER protein levels for any ligand tested confirming the result of

78
transient transfection assays. This observation provides further evidence that Leu540 is a
crucial amino acid for antagonist activity and for ERα stability.
In conclusion, using a mutational study we could discriminate different classes of anti-
estrogens: OHT and RU39 belong to the same group (SERM) and ICI and RU58 to another
group (SERD). For EM652 the situation is less clear, based on studies with a wt ER, we
would conclude that EM652 is a SERM because we did not observe ERα degradation as seen
with our standard reference of SERD, ICI, but at the same time, inhibition of transcription
was more in line with SERD-like activities. The opposite was true for the mutated ER which
was degraded in the presence of EM652 which in turn sustained transcription activation
similar to OHT and RU39. EM652 may be a SERM with a pure anti-estrogenic activity
(Labrie et al., 2001) or a in other words, a compound that could modulate ER target gene
transcription due to an alteration of the conformational change of ER by its somewhat
intermediate side-chain properties. Our study and others indicate the important role of Leu
540 in stabilizing of H12 in antagonist position in presence of 4OHT and that its mutation
reverses the pharmacology of ICI. (Pearce et al., 2003; Shiau et al., 1998).
Variations in transcription of endogenous TGFα, PGR and TFF1/PS2 differentiate
selected antiestrogen compounds
Using quantitative RT-PCR We analysed mRNA expression levels of three well characterized
estrogen responsive genes whose regulation is relevant for endocrine therapy: transforming
growth factor alpha (TGFα), progestrone receptor (PGR), and pS2/TFF1, after 16 hour of
treatment with different compounds in MCF7 cells.
E2 stimulates transcription of TGFα, PGR and TFF1 genes with respectively a 2 and 6-7 and
2-3 fold increase in expression compared to untreated cells. In presence the anti-estrogens
these effects were reduced compared to those produced by E2. However, but interestingly, the

79
three genes responded differently to selected compounds: the effects were comparable for
OHT and RU39 on the one hand and for EM, ICI and RU58 on the other hand. TGFα
expression was reduced 50-60% by SERMS, but only ~20% by SERDs. Similarly, Ps2/TFF1
mRNA levels were reduced in the presence of SERMs and SERDs, 20-35% and ~40%
repectively. Thus transcription regulation of TGFα and PS2/TFF1 in MCF7 cells did not
unambiguously discriminate the antiestrogens used in this study. Interestingly, expression
levels of PGR were hardly affected after 16h treatment with SERMs, but decreased
significantly (60-70%) in the presence of EM652, ICI and RU58. Reduction of basal
transcription by SERDs suggest that basal transcription of PGR requires the presence of ERα,
possibly through its function as a docking partner in growth factor mediated transcription
activation (Baron et al., 2007).We note that analysis of endogenous gene expression in MCF7
cells show similar effects for EM652, ICI and RU58.
Discusssion
___________________________________________________________________________
We describe a molecular approach to classify modulators of ERα function and to predict the
effect of antiestrogens based on structural features. There is great interest in developing novel
anti-estrogens that could be used in patients that exhibit unwanted side-effects or resistances
to the commonly used tamoxifen. This endeavour is a dual challenge: chemical synthesis and
feasibility at acceptable cost of new compounds on the one hand, effective simple screens on
the other hand. Recently, Dai and co-workers (Dai et al., 2008) presented a biochemical
method based on hydrogen/deuterium exchange mass spectrometry (HDX-MS) to predict the
functional impact of SERMs in a tissue specific manner. Such high throughput tests offer
unprecedented opportunities to screen now generation compounds. Yet, once identified, these
compounds still need to be tested on biological samples. We describe a set of readily available

80
molecular techniques useful in predicting both effectiveness and functional outcome of steroid
compounds interacting with ER.
Existing antiestrogens are described as partial or mixed antagonists, and full or pure
antagonists, and more recently SERM or SERD anti-estrogens. Two compounds are readily
used to treat patients: tamoxifen (Jordan, 2003) is categorized as a SERM to describe its
mixed agonist/ antagonist activity that is tissue and promoter dependent (Shang and Brown,
2002), and faslodex (ICI 182,780) a promising agent which is a steroidal pure antiestrogen
commonly presented as SERD prototype (Bross et al., 2002; Dauvois et al., 1993).
Great efforts have been made to clarify the exact mechanism of antiestrogen action. It has
been established that the pharmacological effects of ERα ligands are determined by spatial
conformation of ERα following interaction (or binding) with ligand (Brzozowski et al., 1997;
Pike et al., 1999; Shiau et al., 1998). It was proposed that ligand binding cavity of ERα has a
remarkable plasticity with a preferential binding mode for distal hydroxyl of ERα ligands
(Pike et al., 2001) showing similar orientation for distal side chains in α or β positions of
different ligands in the ERα-ligand complex (Figure 2c). The LBD of ERα undergoes
dynamic conformational changes upon ligand binding. These changes involve the position of
helix 12 and are characteristic of specific members within an antiestrogen class (Métivier et
al., 2002; Tamrazi et al., 2003).
We have used a set of anti-estrogens with different sizes and positions of side chains to
evaluate their effect on ERα. Using mutational analysis we observed that Asp351 is an
important residue for ERα stability and may play a role in the interaction of the proteasome
with the AF1 domain of ERα. The D351Y mutation increased DNA binding of ERα-ligands
suggesting that increased agonist activity could partly be caused by a decrease in ERα
degradation. Thus the amino acid at position 351 is critical for anti-estrogenic activity of
compounds that we could group as SERMs (Figure 1B).

81
Lys 362 was demonstrated to have an important role not only in agonist- ERα complex but
also in antagonist- ERα complex; in agonist conformation (DES-ERα) Lys362 interact with
side chain of the residue of coactivator and in antagonist conformation (OHT-ERα) the side
chain of Lys362 has capping Interaction with helix 12 residues (Shiau et al., 1998). Thus
considering that Mutation Lys 362 in Ala, should prevent the capping interaction; in our study
we observed that in K362A mutation increased agonist activity although its more important
for compound with mixed agonist/antagonist activity (OHT, RU39) (Fig 3.c)
It seems that this residue is discriminator between anti-estrogenswith pure properties and
mixed properties.
L540Q is a very important residue in ERα stability. Pearce and Martin (Martin et al., 2003;
Pearce et al., 2003) have reported that L540Q ERα was no longer degraded in the presence of
ICI and that Helix 12 is important for maintaining ER protein regulation in complex with
ligands. Increased in transcription activation of reporter genes in this mutant may result from
increased in ERα level and stability. We show here that the L540Q point mutation of ERα
confers resistance to degradation also in the presence of RU58. Thus, following antiestrogen
treatment, partial agonist activity appears to correlate with stabilization of cellular ER protein
levels. This observation is not true for EM652. EM652, a nonsteroidal anti-estrogen that has
been classified as pur antiestrogen because there are no estrogenic properties in rodent uterus
(Martel et al., 1998). Our results appear to be in agreement with this classification since
EM652 inhibited luciferase expression in the presence of ERwt in co-transfected MDA-
MB231cells (Figure 3) and EM652 reduced endogenous gene expression inlcuding PGR
expression in MCF7 cells (Figure 4). Yet, paradoxically, EM-ERwt complexes were not
degraded in these cells (Figure 1B and 4). EM652 has an alkylamino side chain that interacts

82
with D351. Binding of EM652 to ER mutated at positions D351 as well as K362 induced
degradation of ER in our transient transfection assays (Figure 3). These mutated residues may
alter folding of ER when bound to EM652 since the aliphatic side chain of EM forms a bond
with D351 similarly to Raloxifen (Bentrem et al., 2001). Replacing Asp with Tyr may release
the side-chain. Such a loose, long side-chain is reminiscent of the side chain of ICI and targets
ER to the proteasome.
Gene Expression Profiles of different ligands could be used for predicting a compound’s in
vivo pharmacological effect. The most important indicator of response to endocrine therapy is
the presence of estrogen receptors and progesterone receptors in tumours the identification the
growth factor (as TGFα) that affects proliferation of target cells revolutionized the concept of
hormonal regulation.
In summary we have compared the activity of a series of anti-estrogens to improving existing
molecular classification and conception demonstrating that direct molecular approaches on
mammary tumor cells in culture represent a good tool for further screening and developing
novel compounds for use in hormone-sensitive breast cancers.
Acknowledgements
We acknowledge financial support from the Institut National du Cancer (INCa), Resisth
network, the Association pour la Recherche sur le Cancer and La Ligue pour le Cancer,
comité du Gers.

83
Figures and Legends
OHHO
OHHO
S
F
O
FF
FF
OHHO
O
S
FF
FF
F
O O
OHHO
O
N
OH
O
N
HOOH
O
O
N
17 -estradiol Faslodex RU 58,668
RU 39,411 4-hydroxytamoxifen EM-652
A
Mazaheri et al Fig 1
A) The chemical structure of various oestrogen receptor ligands
The compound RU 58668 is a steroid substituted in the 11β position with a long hydrophobic
side chain. This produces the same spatial arrangement for a side chain as the 7α substitution
(Faslodex) relative to the plane of steroid nucleus. RU39411 has a steroid core, with a side
chain similar to that of Tamoxifen, and EM652 a benzopyran derivative with a
phenylcoumarine core that mimics the steroid backbone of oestradiol with a side chain similar
to that of Raloxifen.
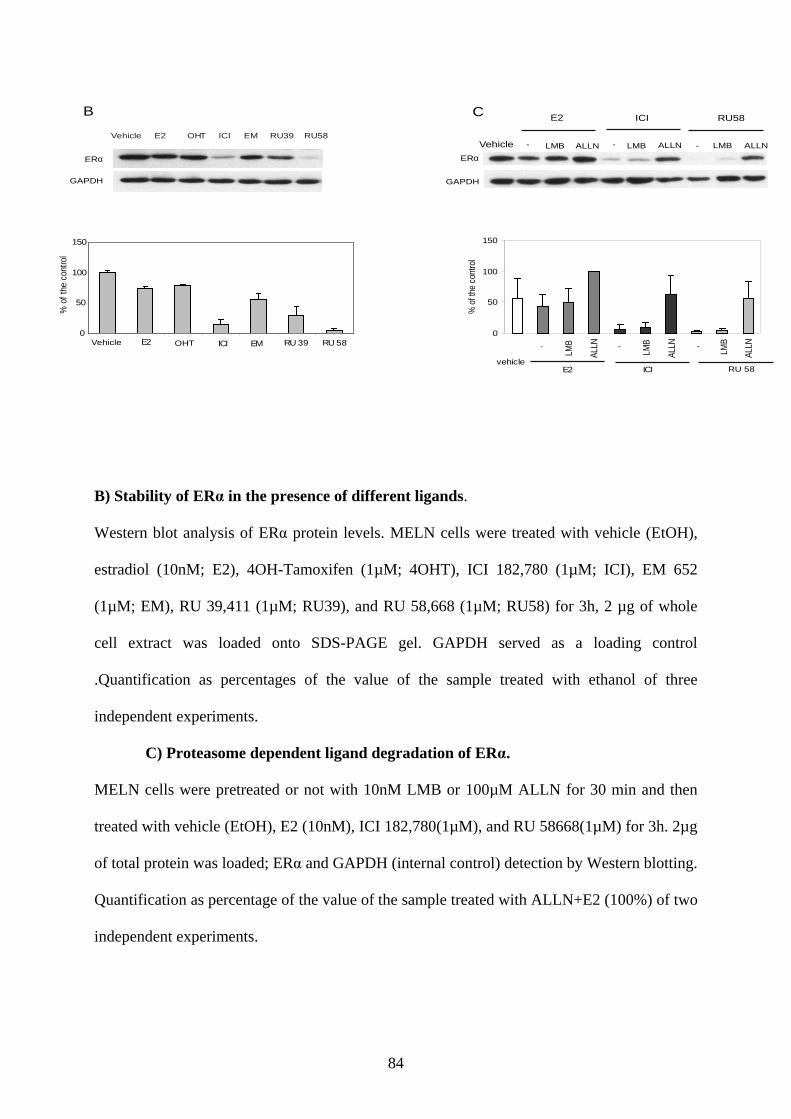
84
B) Stability of ERα in the presence of different ligands.
Western blot analysis of ERα protein levels. MELN cells were treated with vehicle (EtOH),
estradiol (10nM; E2), 4OH-Tamoxifen (1µM; 4OHT), ICI 182,780 (1µM; ICI), EM 652
(1µM; EM), RU 39,411 (1µM; RU39), and RU 58,668 (1µM; RU58) for 3h, 2 µg of whole
cell extract was loaded onto SDS-PAGE gel. GAPDH served as a loading control
.Quantification as percentages of the value of the sample treated with ethanol of three
independent experiments.
C) Proteasome dependent ligand degradation of ERα.
MELN cells were pretreated or not with 10nM LMB or 100µM ALLN for 30 min and then
treated with vehicle (EtOH), E2 (10nM), ICI 182,780(1µM), and RU 58668(1µM) for 3h. 2µg
of total protein was loaded; ERα and GAPDH (internal control) detection by Western blotting.
Quantification as percentage of the value of the sample treated with ALLN+E2 (100%) of two
independent experiments.
B
0
50
100
150
E2 ICI EM RU 39 RU 58
%of
the
cont
rol
Vehicle E2 OHT ICI EM RU39 RU58
Vehicle OHT
ERα
GAPDH
C
0
50
100
150
ALLN
ALLN
ALLN
vehicleE2 ICI
%of
the
cont
rol
Vehicle - LMB ALLN - LMB ALLN - LMB ALLN
E2 ICI RU58
RU 58
ERα
GAPDH
LMB
LMB
LMB- --
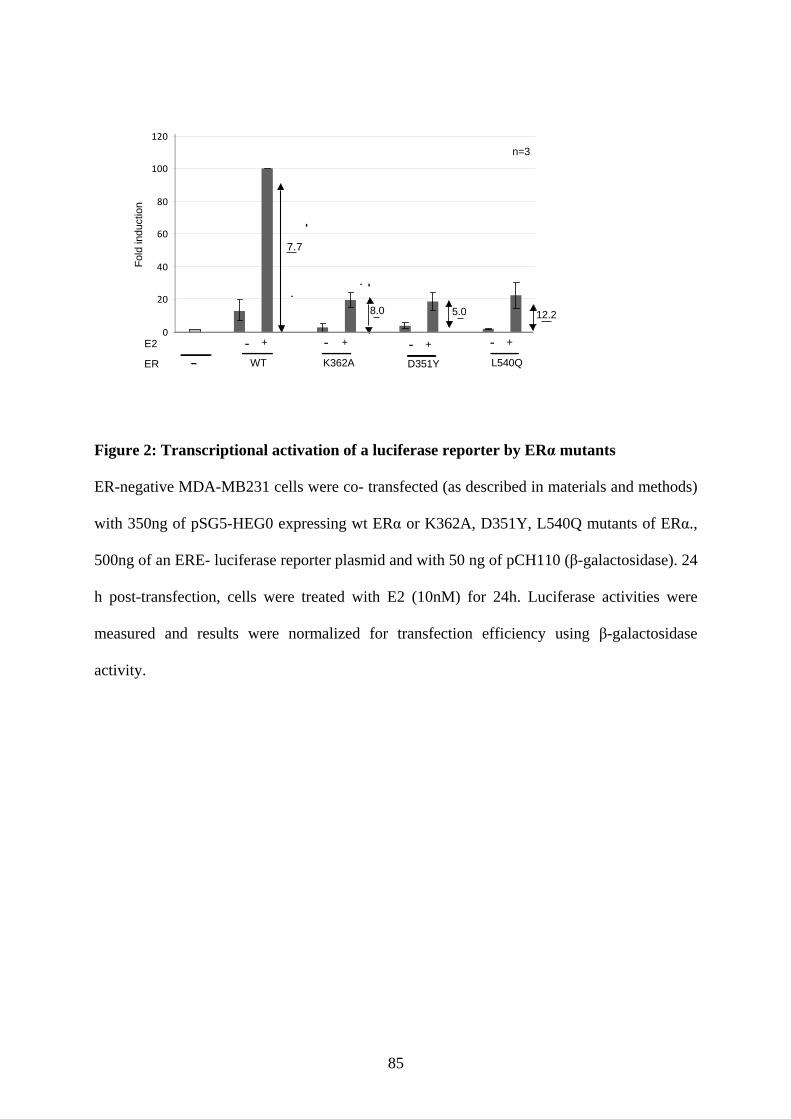
85
0
20
40
60
80
100
120
7.7
8.0 5.0 12.2
n=3
- + - + - + - +E2
WT K362A D351Y L540QER -
Fold
indu
ctio
n
Figure 2: Transcriptional activation of a luciferase reporter by ERα mutants
ER-negative MDA-MB231 cells were co- transfected (as described in materials and methods)
with 350ng of pSG5-HEG0 expressing wt ERα or K362A, D351Y, L540Q mutants of ERα.,
500ng of an ERE- luciferase reporter plasmid and with 50 ng of pCH110 (β-galactosidase). 24
h post-transfection, cells were treated with E2 (10nM) for 24h. Luciferase activities were
measured and results were normalized for transfection efficiency using β-galactosidase
activity.
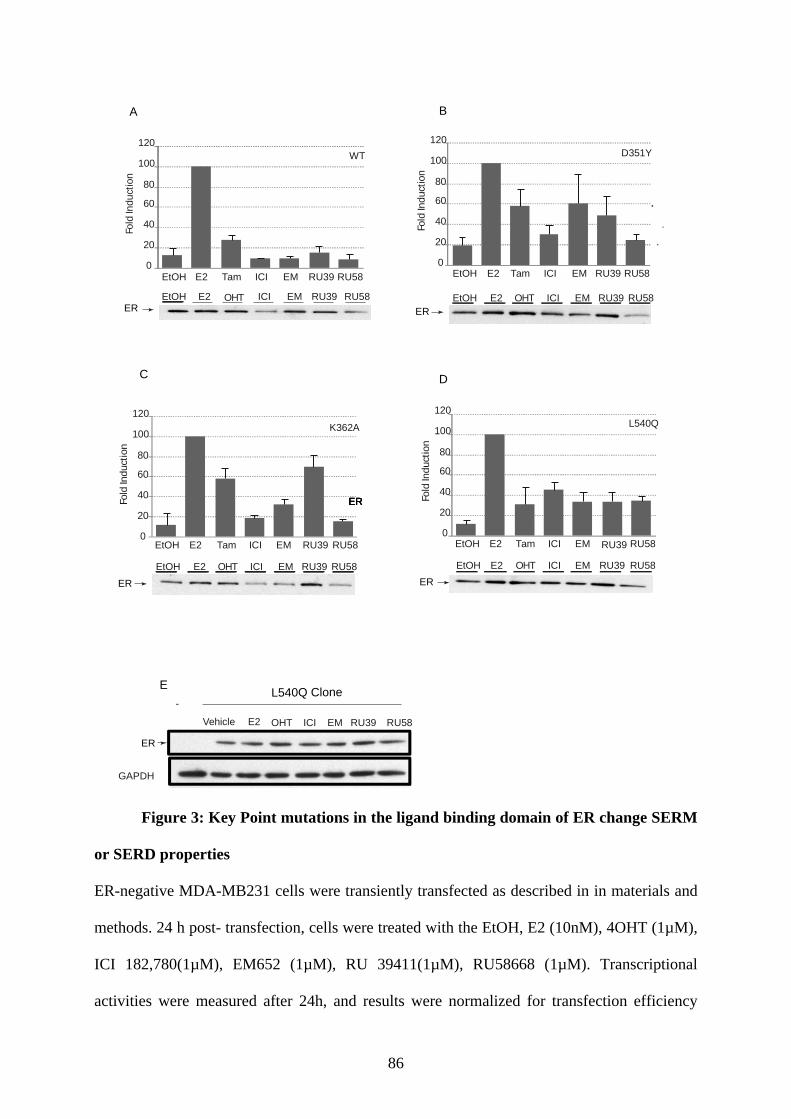
86
D351Y
noitcudnIdloF
0
20
40
60
80
100
120
EtOH E2 Tam ICI EM RU39 RU58
EtOH E2 OHT ICI EM RU39 RU58
EtOH E2 Tam ICI EM RU39 RU58
WT
noitcudnIdloF
0
20
40
60
80
100
120
EtOH E2 ICI EM RU39 RU58OHT
noitcudnIdloF
0
20
40
60
80
100
120
EtOH E2 Tam ICI EM RU39 RU58
EtOH E2 OHT ICI EM RU39 RU58
K362A L540QnoitcudnI
dloF
0
20
40
60
80
100
120
EtOH E2 Tam ICI EM RU39 RU58
EtOH E2 OHT ICI EM RU39 RU58
GAPDH
Vehicle E2 ICI EM RU39 RU58OHT
A B
C D
E
ERERERER
ER
ERER
ER
L540Q Clone
ER
-
Figure 3: Key Point mutations in the ligand binding domain of ER change SERM
or SERD properties
ER-negative MDA-MB231 cells were transiently transfected as described in in materials and
methods. 24 h post- transfection, cells were treated with the EtOH, E2 (10nM), 4OHT (1µM),
ICI 182,780(1µM), EM652 (1µM), RU 39411(1µM), RU58668 (1µM). Transcriptional
activities were measured after 24h, and results were normalized for transfection efficiency

87
using β-galactosidase activity (A-D). ERα protein expression was analysezd after 3h treatment
by western blotting and amount of protein loaded was normalized for transfection efficiency
with luciferase activities from a co-transfection of SV4-lucvector (200ng) instead of pCH110.
A) Wt ERα, B) D351Y mutant, C) K362A mutant, D) L540Q mutant
E) The L540Q mutant were derived from MDA-MB231 cells that stably express ERα (see
materials and methods). ER proteinα expression was analyzed by western blotting in presence
or absence of ligands as in (A-E).
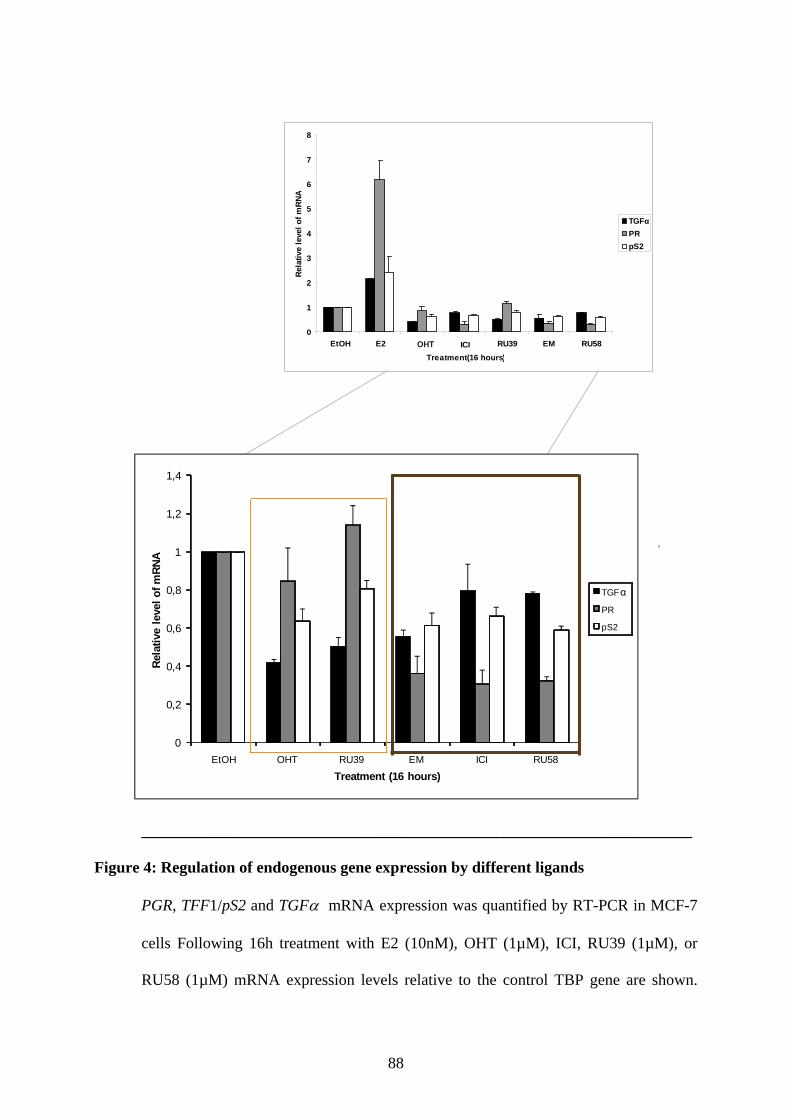
88
0
1
2
3
4
5
6
7
8
EtOH E2 ICI RU39 EM RU58
Treatment(16 hours)
Rela
tive
leve
lofm
RNA
TGFαPRpS2
OHT
0
0,2
0,4
0,6
0,8
1
1,2
1,4
EtOH OHT RU39 EM ICI RU58
R ela
tive
leve
l of m
R NA
Treatment (16 hours)
TGFα
PR
pS2
_____________________________________________________________________
Figure 4: Regulation of endogenous gene expression by different ligands
PGR, TFF1/pS2 and TGFα mRNA expression was quantified by RT-PCR in MCF-7
cells Following 16h treatment with E2 (10nM), OHT (1µM), ICI, RU39 (1µM), or
RU58 (1µM) mRNA expression levels relative to the control TBP gene are shown.

89
Data shown are the average of three independent experiments; error bars represent ±
S.E. mean.Values for untreated cells (EtOH) are set at 1.

90
References
___________________________________________________________________________
Anstead, G. M., K. E. Carlson, et al. (1997). "The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site." Steroids 62(3): 268-303.
Balaguer, P., F. François, et al. (1999). "Reporter cell lines to study the estrogenic effects of xenoestrogens." Sci Total Environ. 15.(233(1-3).): 47-56.
Balan, K. V., Y. Wang, et al. (2006). "Proteasome-independent down-regulation of estrogen receptor-[alpha] ER[alpha] in breast cancer cells treated with 4,4'-dihydroxy-trans-stilbene." Biochemical Pharmacology 72(5): 573-581.
Baron, S., A. Escande, et al. (2007). "Estrogen Receptor {alpha} and the Activating Protein-1 Complex Cooperate during Insulin-like Growth Factor-I-induced Transcriptional Activation of the pS2/TFF1 Gene." J. Biol. Chem. 282(16): 11732-11741.
.Bentrem, D. J., R. C. Dardes, et al. (2001). "Molecular Mechanism of Action at Estrogen Receptor {{alpha}} of a New Clinically Relevant Antiestrogen (GW7604) Related to Tamoxifen." Endocrinology 142(2): 838-846.
Bross, P. F., M. H. Cohen, et al. (2002). "FDA Drug Approval Summaries: Fulvestrant." The Oncologist 7(6): 477-480.
Brzozowski, A. M., A. C. W. Pike, et al. (1997). "Molecular basis of agonism and antagonism in the oestrogen receptor." Nature 389: 753 - 758.
Callige, M., I. Kieffer, et al. (2005). "CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor {alpha} by the proteasome." Mol Cell Biol 25(11): 4349-58.
Clarke, R., F. Leonessa, et al. (2001). Cellular and Molecular Pharmacology of Antiestrogen Action and Resistance. 53: 25-72.
Dai, S. Y., M. J. Chalmers, et al. (2008). "Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method." Proc Natl Acad Sci U S A 105(20): 7171-7176.
Dauvois, S., R. White, et al. (1993). "The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling." Journal of Cell Science 106(4): 1377-1388.
DeFranco, D. B. (2002). "Navigating Steroid Hormone Receptors through the Nuclear Compartment." Mol Endocrinol, 16(7): 1449-1455.
Gradishar, W. J. (2004). "Tamoxifen--What Next?" The Oncologist 9(4): 378-384. Green, S. and P. Chambon, . (1988). "Nuclear receptors enhance our understanding of
transcription regulation " Trends Genet. 1988 Nov; 4(11): 309-14. Heldring, N., A. Pike, et al. (2007). "Estrogen Receptors: How Do They Signal and What Are
Their Targets." Physiol. Rev 87(3): 905-931. Howell, A. (2000). "Faslodex (ICI 182780): an oestrogen receptor downregulator." European
Journal of Cancer 36(Supplement 4): 87. Johnston, S. R. (1997). Acquired tamoxifen resistance in human breast cancer: potential
mechanisms and clinical implications. 8: 911 - 930. Jordan, V. C. (2003). "Tamoxifen: a most unlikely pioneering medicine." Nat Rev Drug
Discov 2(3): 205-213. Katzenellenbogen, B. S., I. Choi, et al. (2000). "Molecular mechanisms of estrogen action:
selective ligands and receptor pharmacology." The Journal of Steroid Biochemistry and Molecular Biology 74(5): 279-285.
Katzenellenbogen, B. S. and J. A. Katzenellenbogen (2002). "Defining the "S" in SERMs." Science 295: 2380-2381.

91
Kong, E. H., A. C. Pike, et al. (2003). "Structure and mechanism of the oestrogen receptor." Biochem Soc Trans 31(Pt 1): 56-9.
Kuiper, G., E. Enmark, et al. (1996). "Cloning of a novel estrogen receptor expressed in rat prostate and ovary." Proc Natl Acad Sci U S A 93: 5925 - 5930
Labrie, F., C. Labrie, et al. (1999). "EM-652 (SCH 57068), a third generation SERM acting as pure antiestrogen in the mammary gland and endometrium." The Journal of steroid biochemistry and molecular biology 69: 51 - 84
Labrie, F., C. Labrie, et al. (2001). "EM-652 (SCH57068), a pure SERM having complete antiestrogenic activity in the mammary gland and endometrium." The Journal of Steroid Biochemistry and Molecular Biology 79(1-5): 213-225.
Marsaud, V., A. Gougelet, et al. (2003). "Various phosphorylation pathways, depending on agonist and antagonist binding to endogenous estrogen receptor alpha (ERalpha), differentially affect ERalpha extractability, proteasome-mediated stability, and transcriptional activity in human breast cancer cells." Mol Endocrinol 17(10): 2013-27.
Martel, C., C. Labrie, et al. (1998). "Comparison of the Effects of the New Orally Active Antiestrogen EM-800 with ICI 182 780 and Toremifene on Estrogen-Sensitive Parameters in the Ovariectomized Mouse." Endocrinology 139(5): 2486-2492.
Martin, L. A., I. Farmer, et al. (2003). "Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation." Journal of Biological Chemistry 278: 30458 - 30468.
McDonnell, D. P. and J. D. Norris (2002). "Connections and Regulation of the Human Estrogen Receptor." Science. 296(5573): 1642-1644.
Métivier, R., A. Stark, et al. (2002). "A Dynamic Structural Model for Estrogen Receptor-α Activation by Ligands, Emphasizing the Role of Interactions between Distant A and E Domains." Molecular Cell 10(5): 1019-1032.
Montano, M. M., K. Ekena, et al. (1996). "Human estrogen receptor ligand activity inversion mutants: receptors that interpret antiestrogens as estrogens and estrogens as antiestrogens and discriminate among different antiestrogens." Molecular Endocrinology 10(3): 230-242.
Nawaz, Z., D. M. Lonard, et al. (1999). "Proteasome-dependent degradation of the human estrogen receptor." Proc Natl Acad Sci U S A 96(5): 1858-62.
O'Malley, B. W. and N. J. McKenna (2008). "Editorial: Coactivators and Corepressors: What's in a Name?" Molecular Endocrinology 22(10): 2213-2214.
O'Regan, R. M., A. Cisneros, et al. (1998). "Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth." Journal of the National Cancer 90: 1552 - 1558.
Pearce, S. T., H. Liu, et al. (2003). "Modulation of Estrogen Receptor alpha Function and Stability by Tamoxifen and a Critical Amino Acid (Asp-538) in Helix 12." Journal of Biological Chemistry 278(9): 7630-7638.Pike, A. C., A. M. Brzozowski, et al. (1999). Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. 18: 4608 - 4618.
Pike, A. C. W., A. M. Brzozowski, et al. (2001). "Structural Insights into the Mode of Action of a Pure Antiestrogen." Structure 9(2): 145-153.
Robertson, J. F. R., S. E. Come, et al. (2005). "Endocrine treatment options for advanced breast cancer - the role of fulvestrant." European Journal of Cancer 41(3): 346-356.
Shang, Y. and M. Brown (2002). "Molecular determinants for the tissue specificity of SERMs." Science 295: 2465 - 2468.
Shiau, A. K., D. Barstad, et al. (1998). "The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen." Cell 95(7): 927-37.

92
Tamrazi, A., K. E. Carlson, et al. (2003). "Molecular Sensors of Estrogen Receptor Conformations and Dynamics." Molecular Endocrinology 17(12): 2593-2602.

93
PUBLICATION N° 2

94
Ligands specify estrogen receptor alpha nuclear localization and degradation
Silvia Kocanova*, Mahta Mazaheri*, Stephanie Caze-Subra and Kerstin Bystricky#
1Université de Toulouse ; UPS ; Laboratoire de Biologie Moléculaire Eucaryote; F-
31062 Toulouse, France 2CNRS; LBME; F-31000 Toulouse, France
*equally contributed
#corresponding : [email protected]
keywords: estrogen receptor, breast cancer, nuclear localization, hormone,
antiestrogens, proteasome

95
Abstract
Intracellular distribution of the oestrogen receptor alpha (ERα), a member of the
nuclear-receptor superfamily, changes upon agonists or antagonist binding. We demonstrate a
tight correlation between the nature of ligands, ERα protein turnover and cellular localization
of ERα in a stable clone of MCF-7 cells in the presence of a variety of ligands. Predominantly
nuclear in MCF-7 cells both in the absence and presence of ligands, GFP-ERα formed
numerous nuclear foci upon addition of agonist, 17β-estradiol (E2), and pure antagonists
(selective estrogen regulator disruptor; SERD), ICI 182,780 or RU58668, while in the
presence of partial antagonists (selective estrogen regulator modulator; SERM), 4-
hydroxytamoxifen (OHT) or RU39411, diffuse staining persisted. Cell fractionation analyses
revealed endogenous ERα and GFP-ERα predominantly in the nuclear fraction. Overall ERα
protein levels were reduced after E2 treatment. In the presence of SERMs ERα was stabilized
in the nuclear soluble fraction, while in the presence of SERDs protein levels decreased
drastically and ERα accumulated in a nuclear insoluble fraction. mRNA levels of ESR1 were
largely reduced compared to untreated cells in the presence of all ligands tested, including
E2. E2 and SERD induced ERα degradation occurred in distinct nuclear foci composed of
ERα and the proteasome providing a simple explanation for ERα sequestration in the nucleus.
Introduction
The estrogen receptor alpha (ERα) is a member of the steroid nuclear receptor family.
In the presence of estradiol, ERα undergoes a conformational change leading to association
with ERα target genes via direct binding to regulatory elements and modulation of their
expression. Estrogens have a proliferative effect on various tissues, including the breast. Thus
ERα plays a key role in mammary tumour development. In mammary cells, the effects of

96
17β-estradiol can be antagonized by compounds such as 4-hydroxytamoxifen (OHT), a
tamoxifen metabolite that is a selective estrogen receptor modulator (SERM), and
Fulvestrant/ICI 182,780 (ICI), a selective estrogen receptor disruptor (SERD). OHT has a
partial agonist activity, depending on the tissue and response examined while ICI compounds
are totally devoid of agonist activity in the models studied to date (Jordan, 1984;
Katzenellenbogen and Katzenellenbogen, 2002). ERα-OHT complexes accumulate in nuclei
and ICI treatment provokes rapid degradation of the ERα-ICI complex by the nuclear
proteasome (Wijayaratne and McDonnell, 2001).
ERα is downregulated in the presence of E2, its cognate ligand, through the
ubiquitin/proteasome (Ub/26S) pathway (Alarid et al., 1999). Although ER-mediated
transcription and proteasome-mediated degradation are linked (Lonard et al., 2000),
transcription per se is not required for ER degradation and assembly of the transcription-
initiation complex is sufficient to target ER for degradation by the nuclear fraction of the
proteasome (Callige et al., 2005). In immunocytochemical studies it was shown that ERα
exists almost exclusively in the nucleus both in presence or absence of hormone (Monje et al.,
2001). Marduva et al (Maruvada et al., 2003) have shown that a small proportion of
unliganded ERα exists in the cytoplasm, with a dynamic shuttling between the cytoplasm and
nucleus in living cells. And this shuttling of ERα is markedly affected by estrogen treatment.
Estradiol (E2). It has been shown that upon binding, E2 induces degradation of ERα, which is
related to its ability to rapidly activate transcription (Jensen et al., 1969). Interestingly, two
chemically different SERDs (ICI and RU58668) competitively inhibit estradiol mediated
activation by ERα and induce a rapid down-regulation of the receptor (Bross et al., 2003;
Buzdar and Robertson, 2006). In contrast, in the presence of tamoxifen ERα protein levels
increased, although its initial mechanism of function is similar to the one observed for
SERD's (Wittmann et al., 2007).

97
The purpose of the present study was to determine the impact of different ligands on
nucleocytoplasmic shuttling of ERα and to examine the relation between localization and
proteolysis, two mechanisms involved in ERα regulation in MCF-7 cells. To achieve this
goal, we examined the relationship between ERα protein concentration, subnuclear
localization with relationship to the proteasome and the level of ERα gene transcription
simultaneously in presence two classes of antiestrogens.
Materials and Methods
Reagents
17β-estradiol (E2), tamoxifen (OHT) and Leptomycine B (LMB) were purchased from
Sigma-Aldrich (St. Louis, MO). ICI 182,780 (ICI) was purchased from Zeneca
Pharmaceuticals. RU39,411 (RU39) and RU58,668 (RU58) were kindly provided by Dr. J.M.
Renoir (Paris, France). Stock solutions of E2, OHT, ICI, RU39 and RU58 were prepared in
ethanol. Stock solution of LMB was prepared in methanol. The solution of proteasome
inhibitor ALLN was purchased from Calbiochem.
Rabbit polyclonal anti-ERα (HC-20), rabbit polyclonal anti-lamin A (H-102), rabbit
polyclonal anti-cytokeratine 18 (H-80) were purchased from Santa Cruz Biotechnology, Inc.
Mouse monoclonal anti-GAPDH (MAB374) was purchased from Chemicon International,
mouse monoclonal anti-GFP from Roche, mouse monoclonal anti-α-tubulin (clone DM1A)
from Sigma-Aldrich. Mouse monoclonal anti-20S proteasome subunit α2 (clone MCP21) was
gift from Dr. M.P. Bousquet (IPBS, Toulouse, France).
All cell culture products were obtained from Invitrogen.
Cell culture and Generation of stable GFP-ERα cell line

98
Human breast cancer cell lines were maintained in Dulbecco's modified Eagle's
medium F-12 (DMEM F-12) with Glutamax containing 50 µg/ml gentamicin, 1 mM sodium
pyruvate and 10% heat-inactivated fetal calf serum. All cells were grown at 37°C in a
humidified atmosphere containing 5% CO2. The stably transfected GFP-ERα reporter SK19
cell line was generated from ERα-positive breast cancer MCF-7 cells (ATCC). 2nd passage
cells were transfected with a GFP-ERα expression vector (pEGFP-C2-hERα) using FuGENE®
HB Transfection Reagent (Roche Applied Science) and G418 resistant clones were selected at
the concentration 1 mg/ml. GFP-ERα expressing clones were isolated, ERα protein expression
in response to estradiol and to anti-estrogens was quantified using fluorescence microscopy
and western blot. Expression of ERα-regulated genes was tested by qRT-PCR and compared
to gene-expression regulation in MCF-7 cells (data not shown). The clone SK19 in which
GFP-ERα behavior was comparable to the one in MCF-7 cells was selected for further study.
To study the effects of estrogens and anti-estrogens, cells were grown for 3 days in
medium containing phenol red-free DMEM F-12 supplemented with 5% charcoal-stripped
fetal calf serum, without gentamicin and sodium pyruvate. Cells were subsequently treated or
not with 10 nM E2, 1 µM ICI, 1 µM OHT, 1 µM RU39, 1 µM RU58 for the indicated times.
To study ERα degradation by the proteasome, cells were pre-treated 30 min with 100 µM
ALLN, a proteasome inhibitor, or 10 nM LMB, a nuclear export inhibitor.
Cell extracts and Western blots
MCF-7 cells grown in 6-well plates and were treated as indicated, washed with ice-
cold PBS and collected by centrifugation. Total cell lysates were prepared by resuspending
the cells in 100 µl lysis buffer (50 mM Tris pH=6.8, 2% SDS, 5% glycerol, 2 mM EDTA,
1.25% β-mercaptoethanol, 0.004% Bromophenol blue). The samples were boiled for 20 min
at 95°C and cleared by centrifugation at 12 000×g for 10 min. The protein concentration was

99
determined by the Amido schwartz assay when the samples contained SDS. Samples were
subjected to SDS-PAGE and the proteins transferred onto nitrocellulose membrane. Western
blot analysis was performed as previously described (Giamarchi 2002) using ERα and
GAPDH antibodies.
qRT-PCR expriments
Total RNAs were extracted using TRIzol reagent (Invitrogen) following the
manufacturer’s protocol. 1-5 µg of total RNA was reverse transcribed in a final volume of 20
µl using SuperScriptTM III Reverse Transcriptase (Invitrogen). cDNA was stored at -80°C. All
target transcripts were detected using quantitative RT-PCR (SYBRGreen SuperMix,
Invitrogen) assays on a Mastercycler Realplex device (Eppendorf) using TBP gene as
endogenous control for normalization of the data. The following primer pairs were used for
amplification:
TBP: 5’-CGGCTGTTTAACTTCGCTTTC-3’
5’-CCAGCACACTCTTCTCAGCA-3’
ESR1: 5’-TGGAGATCTTCGACATGCTG-3’
5’-TCCAGAGACTTCAGGGTGCT-3’
GREB1: 5’-GTGGTAGCCGAGTGGACAAT-3’
5’-AAACCCGTCTGTGGTACAGC-3’
The thermal cycling condition comprised 2 min at 55°C and 2 min at 95°C followed by 40
cycles at an appropriate annealing temperature depending on the primer set for 1 min.
The results were analyzed using Mastercycler Realplex and qBASE software.

100
Cell fractionation
Three hours after incubation with ERα ligands, SK19 cells were washed with ice-cold
PBS, scraped and centrifuged at 1,500 rpm for 5 min at 4°C. The pellets were resuspended in
150 µl digitonin lysis buffer containing 1% digitonin and 1 mM EDTA in PBS, immediately
centrifuged at 13,000 rpm for 20 min at 4°C, to obtain the cytosolic fraction. The pellets were
resuspended in 150 µl HEPES lysis buffer containing 1% Triton X-100, 10% glycerol, 10
µg/ml leupeptin, 5 µg/ml aprotinin, 1 mM PMSF, 1 mM Na3VO4 and 50 mM NaF in HEPES
buffer (25 mM HEPES, 0.3 M NaCl, 1.5 mM MgCl2, 20 mM β-glycerol-phosphate, 2 mM
EDTA, 2 mM EGTA and 1 mM DTT), kept 15 min on ice and centrifuged at 13,000 rpm for
15 min at 4°C, to obtain the soluble nuclear fraction. The last pellets were resuspended in 100
µl of a third buffer containing 95% Laemmli buffer and 5% β-mercaptoethanol and incubated
5 min on ice before boiling them for 20 min at 95°C to obtain the insoluble nuclear fraction.
The different fractions were stored at -80°C until use. Protein concentrations were determined
using the Bio-Rad Protein Assay (Bio-Rad).
Immunofluorescence and Fluorescence microscopy
Before imaging SK19 cells were grown for 3 days on coverslips in DMEM without
phenol red, containing 5% charcoal-stripped fetal serum. After 3 days, cells were treated for
1h with the following ligands: 10 nM E2, 1 µM ICI, 1 µM OHT, 1 µM RU39, 1µM RU58.
The cells were then washed twice with PBS, fixed in 4% paraformaldehyde/PBS for 10 min at
room temperature, subsequently permeabilized with 0.5% Triton X-100 in PBS for 15 min at
room temperature counterstained with DAPI and mounted on microscope slides.
To study co-localization of ERα and proteasome by immunofluorescence, SK19 cells
were grown for 3 days on coverslips in DMEM without phenol red, containing 5% charcoal-
stripped fetal serum and next treated for 3h with drugs as indicated above. To block

101
proteasome-mediated ERα degradation, the cells were incubated 30 min with 100 µM ALLN
prior to treatment with ICI and RU58. Before immunostaining, the cells were fixed in 4%
paraformaldehyde/PBS for 30 minutes at room temperature, washed three times in PBS,
quenched in 75 mM NH4Cl containing 20mM glycine and permeabilized with 0.5% Triton X-
100 in PBS for 30 minutes. Next, the cells were washed with PBS, blocked for 1h at room
temperature in 5% dry milk in TBS-T (20mM TRIS-HCl, 150mM NaCl, 0.1% Tween 20,
pH=7.4) and incubated overnight at 4°C with anti-20S proteasome antibody at final
concentration 2 µg/ml in 5% dry milk in TBS-T followed, after washing, by incubation with
the Alexa Fluor® 647 goat anti-mouse secondary antibody (1:1000, Invitrogen, Molecular
Probes) for 90 min in the dark at room temperature. Finaly, the cells were washed with TBS-
T, counterstained with DAPI and mounted on microscope slides.
The cells were examined by fluorescence microscopy using an Olympus IX-81
microscope, equipped with a CoolSNAPHQ camera (Roper Scientific) and imaged through an
Olympus oil-immersion objective 100x PLANAPO NA1.4. Images were recorded and
deconvolved using Metamorph software (Universal Imaging). All images were processed for
presentation using Adobe Photoshop 9.0.2.
Results
Ligands regulate ERα protein levels and transcription rates independently
We first examined the kinetics of ERα protein turnover in MCF-7 cells following
treatment with estradiol (E2), two SERMs (OHT and RU39) and two SERDs (ICI and RU58).
Treatment of MCF-7 cells with 10 nM E2, 1 µM ICI and 1µM RU58 rapidly induced massive
decrease in ERα protein levels as detected by western blot (Figure 1A). Time course
experiments showed that 1h after E2 induction, the detected amount of ERα protein
accounted for only 40% of ERα levels detected before treatment; after 4h ERα levels were as

102
low as 20% of the quantity of ERα present in untreated cells and after 16h ERα protein
remained at a level equivalent to the one observed 1h after addition of E2, ICI or RU58 (Fig.
1A). Treatment of MCF-7 cells with OHT or RU39 (Figure 1A), two compounds classified as
SERM, only slightly reduced ERα protein levels at the initial 1h time-point. ERα protein
levels were almost equivalent to the ones detected in untreated cells after 4h treatment with
OHT and 16h treatment with RU39. We thus demonstrate that both SERMs stabilize ERα
protein levels compared to 60-80% loss observed in the presence of E2 or SERDs.
To assess whether changes in protein levels reflected variations in ERα protein
stability or are modulated by transcription of the ESR1 gene which codes for ERα, we
quantified ERα mRNA accumulated after 16h treatment with the different compounds (Figure
1B). ESR1 mRNA expression was greatly reduced in all samples subjected to ERα ligands. In
the presence of estradiol (E2), only 40% of ESR1 mRNA could be recovered. Similarly,
treatment with SERMs and SERDs lead to relative ESR1 expression levels equivalent to 45%-
60% of ESR1 mRNA in untreated MCF-7 cells. Despite the down-regulated ERα protein level
readily detectable after 1h and significant after 16h (Figure 1A), we observed that the
presence of E2 results in 7 to 10 fold induction of an ERα-target gene GREB1 compared to
mock treated cells (Figure 1C). GREB1 transcription was inhibited by SERMs and SERDs
(>40% reduction; Figure 1C). These results were expected since E2 is an agonist of this ERα
target gene expression, while SERMs and SERDs are antiestrogens and thus repress GREB1
transcription in ERα positive mammary tumour cells.
Thus, in MCF-7 cells, variations in ERα protein levels do not correlate with ESR1
transcription in the presence of ligands. We note that the decrease in ERα protein levels is
more pronounced after treatment with SERDs than after addition of E2, while the effect of
hormone and SERDs on mRNA accumulation is comparable. These results indicate that
reduction of ERα protein levels following treatment with SERDs cannot be solely attributed

103
to decreased ESR1 mRNA levels. SERDs apparently act both on transcription of the ESR1
gene and on ERα protein turnover. In contrast, despite reduced ESR1 expression levels, ERα
protein accumulated.. after 16h treatment with SERMs suggesting that binding to SERMs
stabilizes the ERα. However, from these data we cannot exclude that reduced transcription of
ESR1 directly influences cellular ERα protein concentrations in the presence of E2.
Ligands directly affect intracellular distribution and stability of ERα
SERMs and SERDs can be distinguished based on molecular mechanisms (Mazaheri
et al., in preparation). To unambiguously determine localization of the estrogen receptor and
its intracellular trafficking in response to treatment with various ligands we established a
MCF-7 cell line stably expressing GFP-ERα from a CMV promoter. It was previously shown
that transiently expressed GFP-ERα is functional using an estrogen response element driven
luciferase reporter gene (Htun et al., 1999). Inducing expression of GFP-ERα in MCF-7 cells
did reportedly not alter cell cycle progression and GFP-ERα participated in estrogen target
gene regulation similarly to endogenous ERα (Zhao et al., 2002). We tagged the N-terminus
of the human ERα with the S65T variant of GFP for transfection and stable integration in
MCF-7 cells. Several clones were recovered and screened for total GFP-ERα protein content
after cells were treated with E2, OHT or ICI using fluorescence microscopy and western
blots. Here, we selected a MCF-7 derived clone (SK19) expressing GFP-ERα in which
changes in endogenous ERα protein levels in response to a 4h treatment with E2, OHT and
ICI were identical to the ones observed in MCF-7 cells (compare lanes labeled ERα in MCF-
7 and SK19 cells in Figure 2A). In addition, mRNA expression levels of some ERα target
genes was verified in the selected clone SK19 and compared to gene expression levels in
MCF-7 cells (data not shown). In SK19 cells, GFP-ERα protein accounted for 50% of total

104
ERα (GFP-ERα and endogenous ERα) in untreated cells. In the presence of E2 both GFP-
ERα and endogenous ERα protein levels are reduced similarly to MCF-7 cells. The CMV
promoter being insensitive to E2 and antiestrogens, GFP-ERα protein levels are unlikely to be
transcriptionally regulated. This observation together with the results shown in Figure 1
provides evidence that ERα protein turnover is regulated directly by binding of the receptor to
ligands and subsequent degradation.
Several studies have demonstrated a mainly nuclear localization of GFP-ERα either in
transiently transfected mammary tumour cell lines (Htun et al., 1999) or Hela cells (Stenoien
et al., 2001; Stenoien et al., 2000b) and in MCF-7 cells expressing GFP-ERα from an
inducible promoter (Zhao et al., 2002). These microscopy based observations are in contrast
with cellular fractionation studies which have regularly indicated that large amounts of ERα,
in the absence or the presence of ligands, associate with the cytoplasmic fraction (Wittmann et
al., 2007). It has been proposed that the relative amount of cytoplasmic ERα is indicative of
the mechanism of action of certain antiestrogens (Wittmann et al., 2007). Commonly used cell
fractionation protocols include a detergent based extraction step. Or ERα and other nuclear
receptors such as the glucocorticoid receptor (GR) are easily extracted from the nucleus in the
presence of low concentrations of detergents such as NP40 (data not shown; V.Marsaud and
H. Richard-Foy, unpublished observations). As a consequence, enrichment of ERα or GR in
the cytoplasm likely results from the extraction protocol rather than a specific behavior of
nuclear receptors. We used a digitonin based cell fractionation protocol to determine the
distribution of unbound and ligand-bound ERα and GFP-ERα in different cellular
compartments (Figure 2B). Effectiveness of the fractionation protocol (for details see
Materials and Methods) was confirmed using loading controls, lamin A for the nuclear
fractions, cytokeratin 18 for the nuclear and cytoplasmic fractions, and alpha-tubulin for the
cytoplasmic fraction (Figure 2B). Treatment of cells with E2 and various antiestrogens did not

105
affect cellular distribution of these proteins. We found that the endogenous ERα associates
predominantly with the nuclear fraction in our SK19 cells. In untreated cells, the part of ERα
retained in the cytoplasm corresponds to 22% of total endogenous ERα detected using the
HC-20 antibody (Figure 2B, lane 1). Similarly, the bulk of GFP-ERα, detected using either
the HC-20 antibody or an antibody directed against GFP, was found in the nucleus. Following
addition of E2, we note an overall decrease in ERα protein levels that could mainly be
attributed to a reduction in nuclear ERα (Fig. 2B, lanes 4-6). Treatment of SK19 cells with
SERDs, ICI or RU58 lead to a decrease in overall ERα protein levels as shown for MCF-7
cells in Figure 1A. Notably, the remaining ERα was concentrated in the nuclear insoluble
fraction in the presence of either ICI or RU58 (Fig. 2B, lanes 12 and 18) suggesting that the
nuclear soluble fraction is rapidly degraded. In contrast, we found that treatment with OHT
and RU39 (Fig. 2B, lanes 7- 9 and 13- 15) reflected a cellular distribution similar to one
observed in untreated cells (Fig. 2B, lanes 1- 3) with high ERα protein concentration in
nuclear soluble compartment. Our cellular fractionation protocol is robust since the effects of
various ligands are reproducible inside each category: OHT and RU39 induce the same effect
on ERα protein distribution and this effect is distinct from the one of ICI and RU58. In
addition, we show that occupation of different cellular compartments by GFP-ERα reflects
the localization of endogenous ERα. Relative amounts of GFP-ERα in each fraction
correlated with its presence as detected by fluorescence imaging (see below).
Ligands induce specific intracellular relocalization of GFP-ERα
GFP-ERα can be visualized in SK19 cells using conventional wide-field microscopy.
SK19 cells were cultured on conventional glass microscopy coverslips in phenol-red free
media for 3 days (culture conditions were identical to conditions used for cell fractionation,

106
immunoblotting or RNA extraction priori to RT-qPCR). Figure 3 shows representative images
of SK19 cells treated or not with E2, SERMs and SERDs. We note that in our SK19 cell line
GFP-ERα is excluded from the nucleoli, as previously observed for the cellular distribution of
endogenous ERα in MCF-7 cells (King and Greene, 1984; Welshons et al., 1988) and of
transiently transfected GFP-ERα (Htun et al., 1999; Stenoien et al., 2000a), under all
conditions tested. Furthermore, exposure times were identical for all conditions examined by
fluorescence microscopy.
In untreated cells, ERα is uniformly distributed in the nucleus (compare GFP-ERα
fluorescence (Fig. 3b), to the DAPI nuclear stain in Figure 3a). A linear scan across the entire
field including cytoplasm and nucleus (Fig. 3c) shows that the cytoplasmic GFP-ERα
fluorescence is barely above background (~12% of the maximum fluorescence intensity
detected in the nucleus) which is in correlation with our cellular fractionation observations
(Fig 2B, lane 1). In the presence of E2, GFP-ERα rapidly relocalized to accumulate in
numerous large foci scattered through the nucleoplasm (Figure 3e). In E2 treated cells, no
GFP-ERα fluorescence could be detected in the cytoplasm (see linescan Fig. 3f). In contrast,
after 1h treatment with SERMs, OHT or RU39, we did not observe any intranuclear
relocalization of GFP-ERα. GFP-ERα staining remained diffuse with fluorescence intensity
comparable to mock cells (Fig 3b, n, r and corresponding linescans Fig 3c, o, s). However,
again, no cytoplasmic GFP-ERα could be detected. Upon exposure to SERDs, both ICI and
RU58, GFP-ERα formed large intranuclear foci, reminiscent to the ones observed in the
presence of E2 (Fig. 3h, k and e). The maximum fluorescence intensity measured after E2 and
ICI treatments decreased by 20-40% as compared to untreated or SERM treated cells (Fig. 3f
and 3i compare to 3c and/or 3o, 3s). The effects of ICI and RU58 were indistinguishable
suggesting that both molecules operate via similar molecular mechanisms despite significant

107
structural differences (Wittmann B.M. et al, 2007, Cancer Res,). Again, we find that SERMs
and SERDs can readily be classified using molecular and imaging approaches.
Proteasome-dependent degradation of ERα bound to E2 or SERDs
ERα is a short-lived protein (a half-life ∼ 4-5 hours for ligand-free ERα and ∼ 3h for
ligand-bound ERα) whose cellular expression is dynamically regulated(Eckert et al., 1984).
We and others, have previously shown that ERα degradation occurs in the cytoplasm in
presence of a natural ligand (E2), but, in the presence a full antiestrogen such as ICI ERα is
rapidly degraded, at least partially, in a nuclear compartment in a proteasome dependent
manner (Callige et al., 2005; Marsaud et al., 2003; Nawaz et al., 1999).
The 26S proteasome is a large protein complex (1500-2000 kDa) present in the
cytoplasm and nucleus of eukaryotic cells. The catalytic core of this multi-subunit complex,
described as the 20S proteasome, contains α and β subunits (Brooks et al., 2000). To
determine the proteasome-mediated ERα degradation induced by E2 and SERDs, we
visualized GFP-ERα and 20S proteasome subunit α2 in SK19 cells. SK19 cells were fixed,
permeabilized and subjected to indirect immunofluorescence using a monoclonal anti-20S
proteasome subunit α2 primary antibody. Images acquired on an Olympus inverted wide-field
microscope revealed punctate nuclear staining of proteasome subunits throughout the nucleus
(Figure 4A, center panels). We did not observe any cytoplasmic staining of this proteasome
subunit under our culture conditions. In the presence of E2 GFP-ERα accumulated at
numerous nuclear sites that colocalized at least partially with proteasome foci (Figure 4A
panel f). When cells were treated with ICI or RU58 GFP-ERα foci also significantly
overlapped with accumulation sites of the 20S proteasome subunit α2 throughout the nucleus
(Fig 4A, insets of panels i and o). On average, we observed larger and more frequent GFP-

108
ERα-proteasome complexes in the presence of SERDs than in the presence of E2 consistent
with the fact that ERα is more readily degraded when ERα is bound to SERDs.
We investigated the effect of LMB, an inhibitor of the nuclear export receptor CRM1,
and of ALLN (acetyl-leucyl-leucyl-norleucinal), an inhibitor of the proteasome, on the SERD-
dependent degradation of ERα in SK19 cells. SK19 cells were pretreated with 10 nM LMB
or 100 µM ALLN for 30 min. Figure 4B shows that LMB did not block ICI or RU58 induced
ERα degradation suggesting that SERD- bound ERα is degraded in the nucleus. In contrast,
ALLN inhibited ICI and RU58 induced degradation of ERα confirming that SERD-ERα
complexes were degraded by the nuclear proteasome (Figure 4B, lanes 7 and 10). Pre-
treatment with ALLN reduced the number of contacts between GFP-ERα and proteasome foci
(Figure 4A panels l and s). ICI-ERα and RU58-ERα complexes remained stably associated
with the nuclear matrix (Fig. 4A, insets of j and p panels).
Interestingly, in a few cells treated with either E2 or SERDs we observed a single very
large site of accumulation of the 20S proteasome α2 subunit (data not shown). These sites,
also called clastosomes, were reported to colocalize with the c-jun and c-fos proteins (Lafarga
et al., 2002), very unstable proteins with a half lives of less than 90min. In our cells,
clastosomes did not colocalize with GFP-ERα foci (data not shown) which could be
explained by the higher stability of ERα compared to c-jun and/or c-fos proteins.
Discussion
Most members of the nuclear receptor superfamily form focal accumulations within
the nucleus in response to hormone (Hager et al., 2000). Receptors undergo constant
exchange between target sequences, multi-protein complexes including a variety of
transcription factor, as well as subnuclear structures that are as yet poorly defined. The

109
estrogen receptor alpha is found almost exclusively in the nucleus, both in hormone
stimulated and untreated cells which makes it an exception among nuclear receptors which
generally translocate from the cytoplasm into the nucleus upon hormone stimulation. Hager
and colleagues (Hager et al., 2000) proposed that distribution of the ERα is dependent not
only on localization signals directly encoded in the receptors, but also on the nature and
composition of the associated macromolecular complexes. Formation of these complexes
depends on the nature of the ligand bound to ERα. Thus, as demonstrated here, ligands
directly affect the nuclear fate of the receptor.
We used a MCF-7 cell line stably expressing GFP-tagged human ERα to determine
the localization of ligand-bound GFP-ERα in mammary tumor cells to demonstrate that few
hours after treatment cellular localization of the ERα correlates with the nature of the ligand
independently of its impact on transcription.
In the presence of E2 and SERMs which induce binding of ERα to target sequences
and subsequent formation of macromolecular complexes, the cytoplasmic fraction of E2
bound ERα rapidly translocated into the nucleus suggesting that DNA binding somehow
recruits cytoplasmic ERα. In contrast, SERD bound cytoplasmic ERα was retained in the
cytoplasm consistent with the fact that SERDs induce a conformational change that blocks
ERα binding to DNA and leads to rapid degradation. Our data also corroborate recent
observations by Long and co-workers (Long and Nephew, 2006) that ICI induced specific
nuclear matrix interaction of protein-ERα complexes with cytokeratins 8 and 18 which
mediate immobilization and turnover of ERα. Interaction with these proteins provoked ICI-
induced cytoplasmic localization of newly synthesized ERα.
A non genomic role of ERα in the cytoplasm has been proposed to play a role in
acquired resistance to antiestrogens, in particular OHT (Fan et al., 2007b). Indeed, in OHT
resistant cells, the ERα accumulated in the cytoplasm, suggesting that the observed SERM

110
stimulated ERα relocalization into the nucleus is necessary for anti-hormone effectiveness
(through the modulation of macromolecular complexes bound to the ERα). An attractive
possibility would thus reside in not only blocking non-genomic ERα function in SERM
resistant tumors, but to increase ERα translocation into the nucleus.
Furthermore, E2 induced focal accumulations of ERα scattered throughout the
nucleus. Similarly, SERD-bound ERα also concentrated into nuclear foci. These foci
frequently colocalize with the proteasome which may explain that ligand bound ERα is less
dynamic, and appears more strongly associated with nuclear matrix like structures(Stenoien et
al., 2001).
Thus we propose a simple explanation reconciling all previous observations of ERα
dynamics: ligands that allow ERα to bind its target sequence and recruit macromolecular
complexes induce ERα nuclear accumulation (estradiol and SERMs); ligands that bind to
ERα but do not lead to DNA binding due to conformational changes of the receptor
(Mazaheri et al., in preparation) do not induce relocalization of the receptor, but accelerate its
degradation (SERDs); finally, ligands that induce association of ERα with the proteasome
(estradiol and SERDs) lead to focal accumulations and immobilize the ERα. It is the
association with the proteasome (Sato et al., 2008) and not active degradation by the
proteasome that leads to ERα sequestration.
Acknowledgements
Imaging was performed at the RIO microscopy facility, Toulouse. SK is supported by
a postdoctoral fellowship from the Association pour la Recherche contre le Cancer (ARC).
We thank Anne-Claire Lavigne for critical reading of the manuscript. We acknowledge
financial support from the Institut National du Cancer (INCa), Resisth network, ARC and La
Ligue pour le Cancer, comité du Gers. ANR
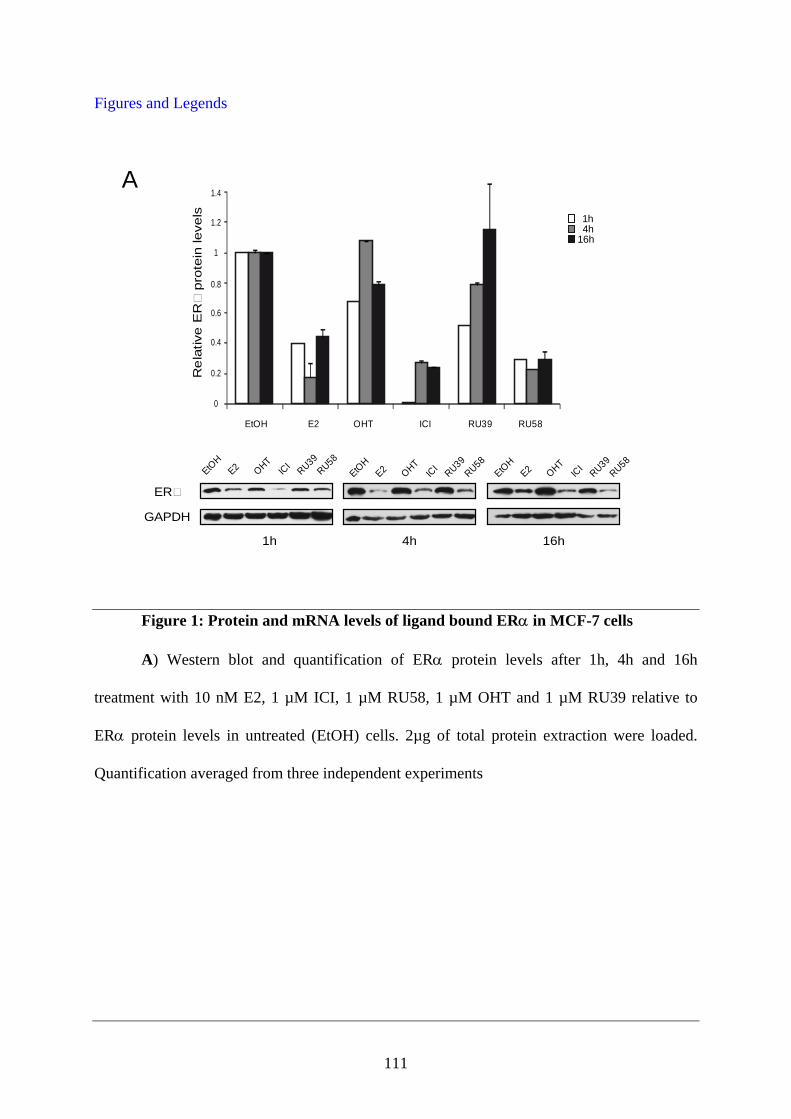
111
Figures and Legends
Figure 1: Protein and mRNA levels of ligand bound ERα in MCF-7 cells
A) Western blot and quantification of ERα protein levels after 1h, 4h and 16h
treatment with 10 nM E2, 1 µM ICI, 1 µM RU58, 1 µM OHT and 1 µM RU39 relative to
ERα protein levels in untreated (EtOH) cells. 2µg of total protein extraction were loaded.
Quantification averaged from three independent experiments
A
ER
GAPDH
1h 16h4h
EtOH
E2 OHTIC
IRU39
RU58EtO
HE2 OHT
ICI
RU39RU58
EtOH
E2 OHTIC
IRU39
RU58
Re
lativ
e E
Rpr
otei
n le
vels
0
0.2
0.4
0.6
0.8
1
1.2
1.4
EtOH E2 OHT ICI RU39 RU58
1h4h
16h
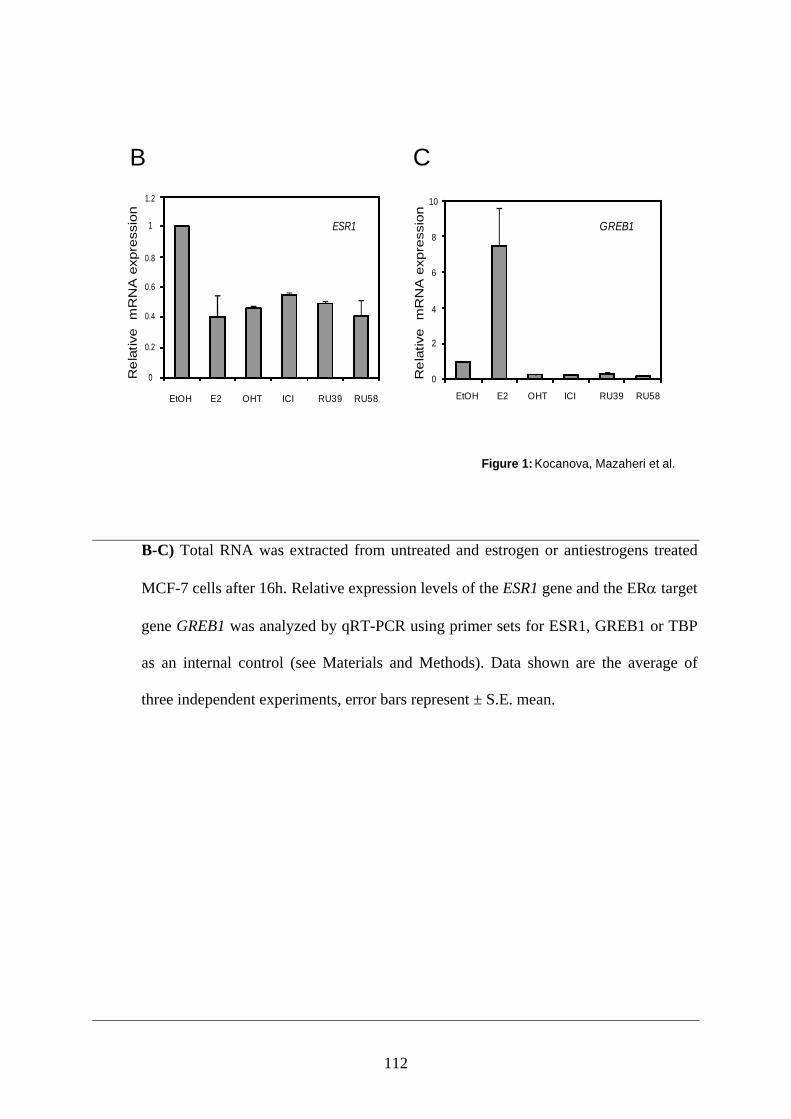
112
B-C) Total RNA was extracted from untreated and estrogen or antiestrogens treated
MCF-7 cells after 16h. Relative expression levels of the ESR1 gene and the ERα target
gene GREB1 was analyzed by qRT-PCR using primer sets for ESR1, GREB1 or TBP
as an internal control (see Materials and Methods). Data shown are the average of
three independent experiments, error bars represent ± S.E. mean.
Figure 1: Kocanova, Mazaheri et al.
B
EtOH E2 OHT ICI RU39 RU58
0
0.2
0.4
0.6
0.8
1
1.2
Re
lativ
e m
RN
A e
xpre
ssio
n
ESR1
Re
lativ
e m
RN
A e
xpre
ssio
n
0
2
4
6
8
10
GREB1
EtOH E2 OHT ICI RU39 RU58
C
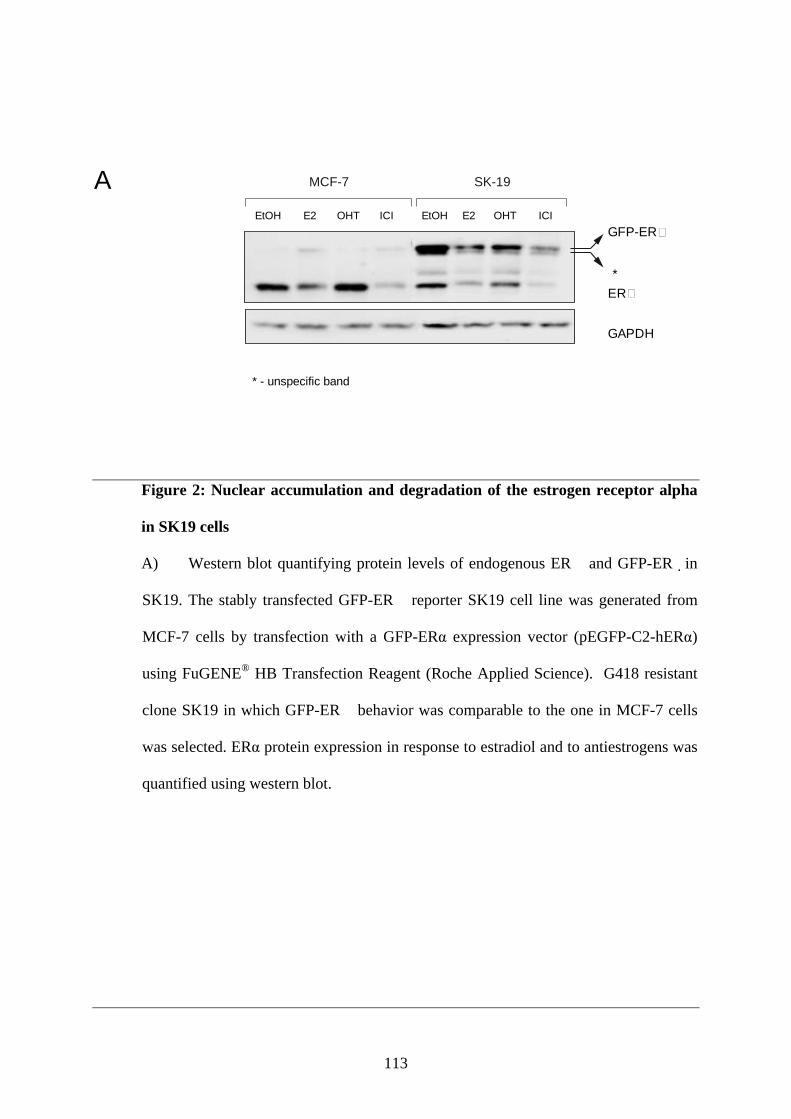
113
Figure 2: Nuclear accumulation and degradation of the estrogen receptor alpha
in SK19 cells
A) Western blot quantifying protein levels of endogenous ER� and GFP-ER� in
SK19. The stably transfected GFP-ER� reporter SK19 cell line was generated from
MCF-7 cells by transfection with a GFP-ERα expression vector (pEGFP-C2-hERα)
using FuGENE® HB Transfection Reagent (Roche Applied Science). G418 resistant
clone SK19 in which GFP-ER� behavior was comparable to the one in MCF-7 cells
was selected. ERα protein expression in response to estradiol and to antiestrogens was
quantified using western blot.
A MCF-7 SK-19
GAPDH
GFP-ER
ER*
EtOH E2 OHT ICI EtOH E2 OHT ICI
* - unspecific band

114
B) Digitonin based cellular fractionation showing nuclear relocalization and
degradation of ER and GFP-ERα SK19 cells were treated with vehicle (EtOH), E2 (10 nM),
OHT (1 µM), ICI (1µM), RU39 (1µM) or RU58 (1µM) for 3h. Nuclear fractions of untreated
and estrogen or antiestrogens treated cells were isolated as described under “Materials and
Methods”. Nuclear content of ERα and GFP-ERα was analyzed by Western Blot using anti-
ERα antibodies (Santa Cruz Biotechnology). The specific subcellular proteins, α-tubulin for
cytoplasmic fraction (Cyt), Lamin A for nuclear soluble fraction (NS) and cytokeratin 18 for
nuclear insoluble fraction (NI) indicate equal loading. Results shown are representative of at
least 3 independent experiments.
Cyt NS NI
vehicle RU39
Cyt NS NI
RU58
Cyt NS NI
-Tubulin
ER
GFP-ER
Cytokeratin 18
Lamin A
*
E2
Cyt NS NI
OHT
Cyt NS NI
ICI
Cyt NS NICyt NS NI
vehicle
-Tubulin
ER
GFP-ER
Cytokeratin 18
Lamin A
*
* - unspecific band
22% 55% 23% 19% 50% 31% 28% 33% 39% % of ER in fraction
24% 47% 29% 19% 53% 28% 23% 29% 48% % of ER in fraction 24% 53% 23%
B
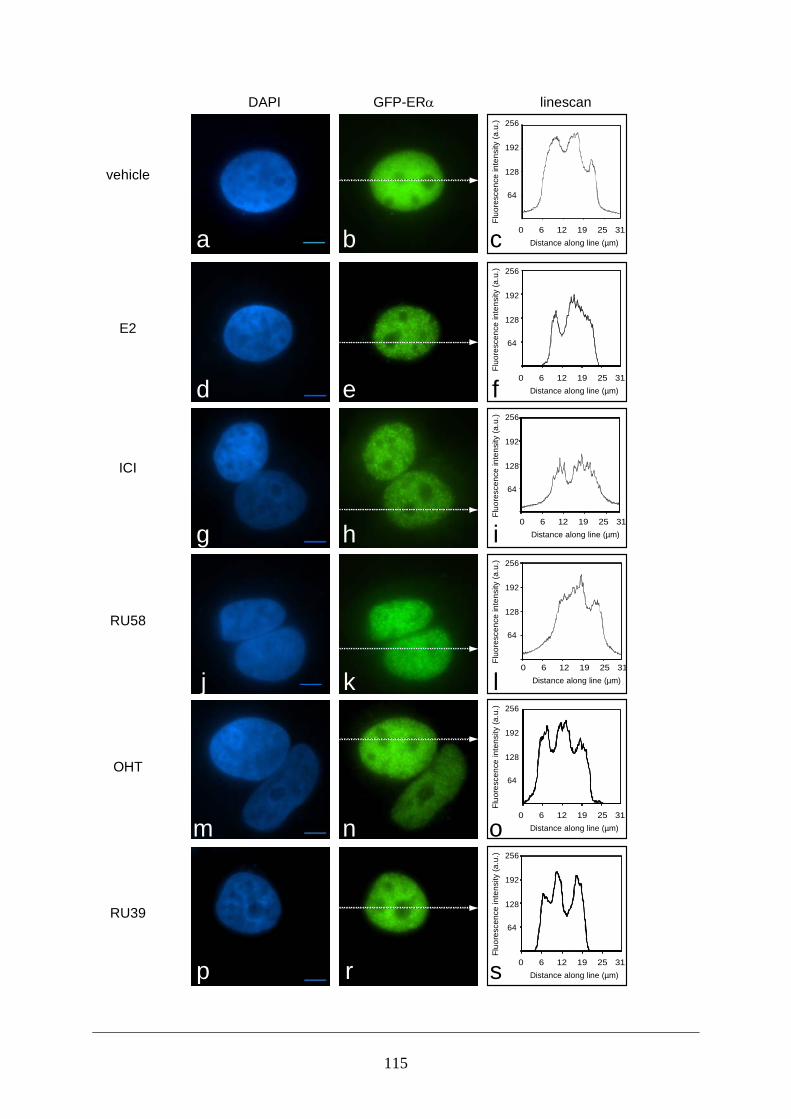
115
Distance along line (µm)0 6 12 19 25 31
Fluo
resc
ence
inte
nsity
(a.u
.)64
128
192
256
Distance along line (µm)
Fluo
resc
ence
inte
nsity
(a.u
.)
0 6 12 19 25 31
64
128
192
256
vehicle
E2
ICI
RU58
OHT
RU39
DAPI GFP-ERα linescan
Fluo
resc
ence
inte
nsity
(a.u
.)
64
128
192
256
Distance along line (µm)0 6 12 19 25 31
Fluo
resc
ence
inte
nsity
(a.u
.)
64
128
192
256
Distance along line (µm)0 6 12 19 25 31
Fluo
resc
ence
inte
nsity
(a.u
.)
64
128
192
256
Distance along line (µm)0 6 12 19 25 31
Fluo
resc
ence
inte
nsity
(a.u
.)
64
128
192
256
Distance along line (µm)0 6 12 19 25 31a b c
d e
g h
j k
m n
p r
f
i
l
o
s

116
Figure 3: Estrogen, SERDs and SERMs induce distinct intracellular
relocalization of ERα
SK19 cells were incubated for 1h with 10 nM E2 and different antiestrogens
(SERDs or SERMs). After 4h cells were fixed and stained with DAPI. The cells were
examined by fluorescence microscopy using an Olympus IX-81 inverted microscope,
equipped with a CoolSNAPHQ camera (Roper Scientific) and imaged through an
Olympus oil-immersion objective 100x PLANAPO NA1.4. Pictures are representative
of at least three independent experiments. GFP-ER� forms intranuclear foci in the
presence of E2, RU58 and ICI, but remains uniformly distributed in the nucleus after
addition of OHT and RU39. Linescans indicate relative fluorescence intensities in the
cytoplasm and the nucleus in the cells imaged using identical parameters.

117
Figure 4: Nuclear Proteasome-GFP-ERα contacts are frequent in the presence of
E2 and SERDs
A) SERDs degrade ERα through proteasome and in the nucleus, SK19 cells were
pretreated or not with 10 nM LMB or 100 µM ALLN for 30 min and then treated with vehicle
(EtOH), ICI (1µM), or RU58 (1µM) for 3h. ERα and GAPDH (internal control) detection by
Western
A
GAPDH
ER
GFP-ER
*
* - unspecific band
E2 ICI RU58
-- LMB
ALLN -- LM
BALL
N -- LMB
ALLN
vehicle
-- LMB
ALLN
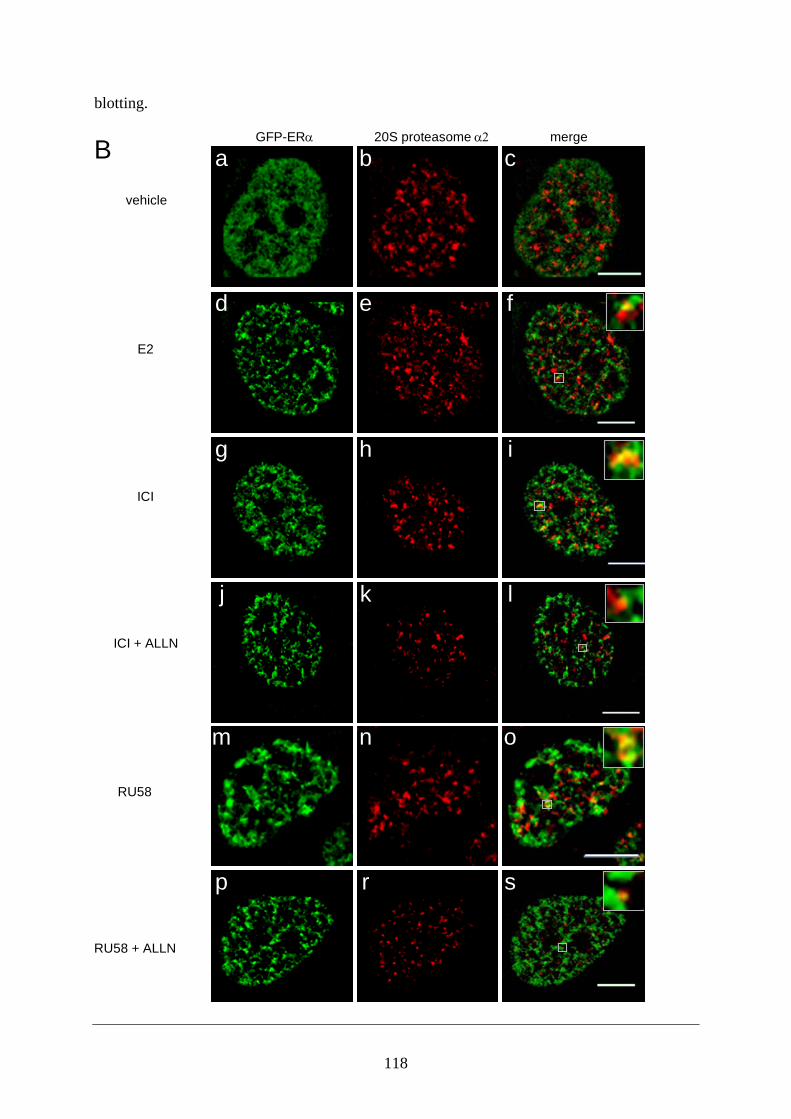
118
blotting.
vehicle
E2
ICI
ICI + ALLN
RU58
GFP-ERα 20S proteasome α2 merge
RU58 + ALLN
B a b c
d e f
g h i
j k l
m n o
p r s

119
B) SK19 cells were grown for 3 days on coverslips in DMEM without phenol red,
containing 5% charcoal-stripped fetal serum and next treated for 3h with drugs as
described in Materials and Methods. Cells were fixed and subjected to
immunofluorescence using a monoclonal anti-20S proteasome subunit α2 primary
antibody, followed by incubation with the Alexa Fluor® 647 goat anti-mouse
secondary antibody. Right panels represent colocalization of GFP-ER� and 20S
proteasome (insets): untreated cells (4A,c), E2 treated cells (4A,f), SERDs treated
cells without (4A,i and o) or with (4A,l and s) 100 µM ALLN. Images are
representative of three independent experiments.

120
References
Alarid, E. T., N. Bakopoulos, et al. (1999). "Proteasome-mediated proteolysis of estrogen receptor: a novel component in autologous down-regulation." Mol Endocrinol 13(9): 1522-34.
Brooks, P., G. Fuertes, et al. (2000). "Subcellular localization of proteasomes and their regulatory complexes in mammalian cells." Biochem J 346 Pt 1: 155-61.
Bross, P. F., A. Baird, et al. (2003). "Fulvestrant in postmenopausal women with advanced breast cancer." Clin Cancer Res 9(12): 4309-17.
Buzdar, A. U. and J. F. Robertson (2006). "Fulvestrant: pharmacologic profile versus existing endocrine agents for the treatment of breast cancer." Ann Pharmacother 40(9): 1572-83.
Callige, M., I. Kieffer, et al. (2005). "CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor {alpha} by the proteasome." Mol Cell Biol 25(11): 4349-58.
Eckert, R. L., A. Mullick, et al. (1984). "Estrogen receptor synthesis and turnover in MCF-7 breast cancer cells measured by a density shift technique." Endocrinology 114(2): 629-37.
Fan, P., J. Wang, et al. (2007). "Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells." Cancer Res 67(3): 1352-60.
Giamarchi, C., M. Solanas, et al. (1999). "Chromatin structure of the regulatory regions of pS2 and cathepsin D genes in hormone-dependent and -independent breast cancer cell lines." Oncogene 18(2): 533-41.
Hager, G. L., C. S. Lim, et al. (2000). "Trafficking of nuclear receptors in living cells." J Steroid Biochem Mol Biol 74(5): 249-54.
Htun, H., L. T. Holth, et al. (1999). "Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor." Mol Biol Cell 10(2): 471-86.
Jensen, E. V., T. Suzuki, et al. (1969). "Estrogen-binding substances of target tissues." Steroids 13(4): 417-27.
Jordan, V. C., M. E. Lieberman, et al. (1984). "Structural requirements for the pharmacological activity of nonsteroidal antiestrogens in vitro." Mol Pharmacol 26(2): 272-8.
Katzenellenbogen, B. S. and J. A. Katzenellenbogen (2002). "Biomedicine. Defining the "S" in SERMs." Science 295(5564): 2380-1.
King, W. J. and G. L. Greene (1984). "Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells." Nature 307(5953): 745-7.
Lafarga, M., M. T. Berciano, et al. (2002). "Clastosome: a subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome." Mol Biol Cell 13(8): 2771-82.
Lonard, D. M., Z. Nawaz, et al. (2000). "The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation." Mol Cell 5(6): 939-48.
Long, X. and K. P. Nephew (2006). "Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha." J Biol Chem 281(14): 9607-15.
Marsaud, V., A. Gougelet, et al. (2003). "Various phosphorylation pathways, depending on agonist and antagonist binding to endogenous estrogen receptor alpha (ERalpha), differentially affect ERalpha extractability, proteasome-mediated stability, and

121
transcriptional activity in human breast cancer cells." Mol Endocrinol 17(10): 2013-27.
Maruvada, P., C. T. Baumann, et al. (2003). Dynamic Shuttling and Intranuclear Mobility of Nuclear Hormone Receptors. 278: 12425-12432.
Monje, P., S. Zanello, et al. (2001). "Differential cellular localization of estrogen receptor [alpha] in uterine and mammary cells." Molecular and Cellular Endocrinology 181(1-2): 117-129.
Nawaz, Z., D. M. Lonard, et al. (1999). "Proteasome-dependent degradation of the human estrogen receptor." Proc Natl Acad Sci U S A 96(5): 1858-62.
Sato, K., E. Rajendra, et al. (2008). "The UPS: a promising target for breast cancer treatment." BMC Biochem 9 Suppl 1: S2.
Stenoien, D. L., M. G. Mancini, et al. (2000). "Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1." Mol Endocrinol 14(4): 518-34.
Stenoien, D. L., K. Patel, et al. (2001). "FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent." Nat Cell Biol 3(1): 15-23.
Welshons, W. V., E. M. Cormier, et al. (1988). "Estrogen receptor distribution in enucleated breast cancer cell lines." Endocrinology 122(6): 2379-86.
Wijayaratne, A. L. and D. P. McDonnell (2001). "The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators." J Biol Chem 276(38): 35684-92.
Wittmann, B. M., A. Sherk, et al. (2007). "Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators." Cancer Res 67(19): 9549-60.
Zhao, H., L. L. Hart, et al. (2002). "Characterization of stably transfected fusion protein GFP-estrogen receptor-alpha in MCF-7 human breast cancer cells." J Cell Biochem 86(2): 365-75.

122
ONGOING RESULTS
In this part of our study we wanted tested the molecules designed by chimists with our
molecular approach. The molecules sythesized by M Poirot’s group may be patented and thus
I am not able to provide their molecular structures here in order to comply by patenting
restrictions. The molecules are analogs of ICI164,384 whose side chains were modified to
assess its importance for pure antagonist. The steroid motif was replced by a stilbene to
simplify the synthesis procedure. Here our colabators were interested in the position of the
polar group on this side chain of ICI 164,384. The chemical synthesis was carried out by M
Poirot at the ICR. They observed that positioning of an amide group along the side chain
affects the molecules activity.

123
CONCLUSION AND PERSPECTIVES
Estrogens have been recognized to play an important role in proliferation of a large proportion
of breast tumours many years ago. Today it is well established that estrogens act via their
receptors, and that ERα expression in a tumour represents a good prognosis. In addition, ER
positive tumours are responsive to hormonal therapy.
In more than half of breast tumours ERα is overexpressed and around 70% of these tumours
respond to anti-estrogen therapies via the use of tamoxifen. Unfortunately, some of these
tumours do not respond to endocrine therapy initially (de novo resistance), and the majority of
responsive tumours eventually become resistant (acquired resistance). In most of these
resistant tumours ERα is still expressed, and other options of endocrine therapy can be
proposed.
A second line treatment for resistant tumours is the use of antiestrogens that do not show a
cross resistance. Few antiestrogens that can be used in tamoxifène acquired resistance. One of
these is Faslodex, which has unfortunately poor bioavailability (intravenous administration is
required) and is a costly treatment for patients.
There is a great interest to developing the novel molecules which could be used as first or
second line in endocrine therapy. Great efforts are being made in all areas of research,
including random screening of a bank of compounds (Roussel Uclaf.) synthesis of specifically
designed molecules or extraction of natural compounds. Although our understanding of
estrogen receptor biology and the consequence of ligand interaction is expanding, certain
aspects of liganded ER function areis not fully understood, for example; how structurally
different ligands induce interaction between AF-1 and AF-2. Which may require extensive
modelling based on crystallographic data to determine exact structure –function relationship
of antiestrogens with ER.

124
In this study we propose a molecular approach that could be interesting for rapid
discrimination between SERM and SERD. We subsequently determined that the compounds
classified as SERD by our approach function in a similar manner and a grand part of their
activity is dependent on proteasome dependent degradation. We confirmed our initial
assumption that the position and nature of side chains of antiestroges is crucial for stability of
ERα. Thereby the side chain defines a SERD.
I think a molecule described as SERD is a pure antiestogen but a pure antiestrogen is not
necessarily a SERD. For example: EM652 in our molecular approach is a SERM but with
pure antiestrogenic activity, and it could be interesting to develop molecules similar to EM
652. Such compounds offer the exciting possibility to reduce undesirable side effects
especially interesting in premenopause patients.
In the second part we investigated the impact of the nature of ligands in ERα localisation and
relationship of intracellular ligand-ER localisation and expression or transcription of ERα.
Localization of ERα in either the nuclear or cytoplasmic compartments has functional
implications (Fox et al., 2009). In one model of tamoxifen resistance, developed by long term
treatment of MCF7 cells with tamoxifen, increased ER_localization to the
cytoplasm/membrane was observed that correlated with increased cytoplasmic signalling (Fan
et al., 2007a). It would thus be of interest to investigate ERα localisation in patients who have
developed resistance to tamoxifène. A novel perspective may be to influence ER localization,
restoring or re-inforcing its concentration in different cellular compartment. A lot of wrok is
still require to assess feasibility of such an approach.
In ER signalling three actors seem to be important: the ligand, the hormone receptors and its
co-regulators. In in vitro models the role of co-activator/ co-repressor ratio in cell specific

125
response is well established and also in certain tamoxifen resistance model a high expression
of co-activator as AIB1 is demonstrated; now we will continue our research not only in
fundamental domain but also in clinical domain with the aim of better understanding the
mechanisms of resistance to anti-estrogens.
At the fundamental level we want to continue our ongoing work to determine the expression
level of different class of HDAC, their intracellular localisation, in different cell types as ER-
/ER+ and resistance to antiestrogens and if their inhibition result in overcoming to resistance
to antiestrogens (either in de novo resistance or acquired resistance).
It would be exciting to compare results from this study to clinical trials and with patient
biopsies, and for cases developing resistance phenotype or patients with ER-negative tumours
– opening new avenues for treatment of these more aggressive and poor prognosis cases.

126
REFERENCES Alarid, E. T. (2006). "Lives and Times of Nuclear Receptors." Mol Endocrinol, 20(9): 1972-
1981. Alarid, E. T., N. Bakopoulos, et al. (1999). "Proteasome-Mediated Proteolysis of Estrogen
Receptor: A Novel Component in Autologous Down-Regulation." Mol Endocrinol, 13(9): 1522-1534.
Albertson, D. G. (2006). "Gene amplification in cancer." Trends in Genetics 22(8): 447-455. Ali, S. and R. C. Coombes (2000). "Estrogen Receptor Alpha in Human Breast Cancer:
Occurrence and Significance." J Mammary Gland Biol Neoplasia. 5(3): 271-281. Allan, G. J., J. Beattie, et al. (2004). "The role of IGFBP-5 in mammary gland development
and involution." Domest Anim Endocrinol 27(3): 257-66. Andersson, D. (1974). "Sex-hormone binding globulin." Clin Endocrinol (Oxf). 3(1): 69-96. Anstead, G. M., K. E. Carlson, et al. (1997). "The estradiol pharmacophore: Ligand structure-
estrogen receptor binding affinity relationships and a model for the receptor binding site." Steroids 62(3): 268-303.
Anzick, S. L., J. Kononen, et al. (1997). "AIB1, a Steroid Receptor Coactivator Amplified in Breast and Ovarian Cancer." Science 277(5328): 965-968.
Ayers, M., W. F. Symmans, et al. (2004). "Gene Expression Profiles Predict Complete Pathologic Response to Neoadjuvant Paclitaxel and Fluorouracil, Doxorubicin, and Cyclophosphamide Chemotherapy in Breast Cancer." J Clin Oncol 22(12): 2284-2293.
Balaguer, P., F. François, et al. (1999). "Reporter cell lines to study the estrogenic effects of xenoestrogens." Sci Total Environ. 15.(233(1-3).): 47-56.
Balan, K. V., Y. Wang, et al. (2006). "Proteasome-independent down-regulation of estrogen receptor-[alpha] (ER[alpha]) in breast cancer cells treated with 4,4'-dihydroxy-trans-stilbene." Biochemical Pharmacology 72(5): 573-581.
Ball, L. J., N. Levy, et al. (2009). "Cell type- and estrogen receptor-subtype specific regulation of selective estrogen receptor modulator regulatory elements." Molecular and Cellular Endocrinology 299(2): 204-211.
Baron, S., A. Escande, et al. (2007). "Estrogen Receptor {alpha} and the Activating Protein-1 Complex Cooperate during Insulin-like Growth Factor-I-induced Transcriptional Activation of the pS2/TFF1 Gene." J. Biol. Chem. 282(16): 11732-11741.
Beatson, G. (1896). "On the treatment of inoperable cases of carcinoma of the mamma: suggestion for a new method of treatment with illustrative case." The Lancet 148(3802): 104-107.
Beckmann, M. W., D. Niederacher, et al. (1997). "Multistep carcinogenesis of breast cancer and tumour heterogeneity." Journal of Molecular Medicine 75(6): 429-439.
Benson, J. R. (2008). "Ovarian ablation as a non-surgical treatment for breast cancer." The Lancet Oncology 9(1): 80-80.
Bentrem, D. J., R. C. Dardes, et al. (2001). "Molecular Mechanism of Action at Estrogen Receptor {{alpha}} of a New Clinically Relevant Antiestrogen (GW7604) Related to Tamoxifen." Endocrinology 142(2): 838-846.
Berstein, L. M., J.-P. Wang, et al. (2004). "Long-Term Exposure to Tamoxifen Induces Hypersensitivity to Estradiol." Clinical Cancer Research 10(4): 1530-1534.
Brisken, C. (2002). "Hormonal control of alveolar development and its implications for breast carcinogenesis." J Mammary Gland Biol Neoplasia 7(1): 39-48.
Brody, L. C. and B. B. Biesecker (1998). "Breast Cancer Susceptibility Genes: BRCA1 and BRCA2." medicine 77(3): 208-226.
Brooks, P., G. Fuertes, et al. (2000). "Subcellular localization of proteasomes and their regulatory complexes in mammalian cells." Biochem J 346 Pt 1: 155-61.

127
Bross, P. F., A. Baird, et al. (2003). "Fulvestrant in postmenopausal women with advanced breast cancer." Clin Cancer Res 9(12): 4309-17.
Bross, P. F., M. H. Cohen, et al. (2002). "FDA Drug Approval Summaries: Fulvestrant." The Oncologist 7(6): 477-480.
Brzozowski, A. M., A. C. W. Pike, et al. (1997). "Molecular basis of agonism and antagonism in the oestrogen receptor." Nature 389: 753 - 758.
Buzdar, A. U. and G. Hortobagyi (1998). "Update on endocrine therapy for breast cancer." Clinical Cancer Research 4(3): 527-534.
Buzdar, A. U. and J. F. Robertson (2006). "Fulvestrant: pharmacologic profile versus existing endocrine agents for the treatment of breast cancer." Ann Pharmacother 40(9): 1572-83.
Callige, M., I. Kieffer, et al. (2005). "CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor {alpha} by the proteasome." Mol Cell Biol 25(11): 4349-58.
Cardona-Gómez, G. P., P. Mendez, et al. (2001). "Interactions of estrogens and insulin-like growth factor-I in the brain: implications for neuroprotection." Brain Research Reviews 37(1-3): 320-334.
Cavalieri, E., D. Chakravarti, et al. (2006). "Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention." Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1766(1): 63-78.
Chambers, A. F., A. C. Groom, et al. (2002). "Metastasis: Dissemination and growth of cancer cells in metastatic sites." Nat Rev Cancer 2(8): 563-572.
Cheskis, B. J., J. G. Greger, et al. (2007). "Signaling by estrogens." J Cell Physiol 213(3): 610-7.
Cianfrocca, M. and L. J. Goldstein (2004). "Prognostic and Predictive Factors in Early-Stage Breast Cancer." The Oncologist 9(6): 606-616.
Clarke, R., F. Leonessa, et al. (2001). "Cellular and Molecular Pharmacology of Antiestrogen Action and Resistance." PHARMACOLOGICAL REVIEWS 53(1): 25-72.
Clarke, R., M. C. Liu, et al. (2003). "Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling." Oncogene 22(47): 7316-7339.
Coezy, E. (1982). "Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth." Cancer Res 42(1): 317-23
Cornell, U. (1997). "Breast Cancer and Environmental Risk Factors." FACT SHEET #5 5 Cuzick, J. (2005). "Radiotherapy for Breast Cancer." J Natl Cancer Inst 97(6): 406-407. Dai, S. Y., M. J. Chalmers, et al. (2008). "Prediction of the tissue-specificity of selective
estrogen receptor modulators by using a single biochemical method." Proc Natl Acad Sci U S A 105(20): 7171-7176.
Dauvois, S., P. S. Danielian, et al. (1992). "Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover." Proc Natl Acad Sci U S A 89(9): 4037-41.
Dauvois, S., R. White, et al. (1993). "The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling." Journal of Cell Science 106(4): 1377-1388.
Davidoff, A. M., P. A. Humphrey, et al. (1991). "Genetic basis for p53 overexpression in human breast cancer." Proc Natl Acad Sci U S A 88(11): 5006-5010.
DeFranco, D. B. (2002). "Navigating Steroid Hormone Receptors through the Nuclear Compartment." Mol Endocrinol, 16(7): 1449-1455.
Diel, P. (2002). "Tissue-specific estrogenic response and molecular mechanisms." Toxicology Letters 127(1-3): 217-224.

128
Dontu, G., D. El-Ashry, et al. (2004). "Breast cancer, stem/progenitor cells and the estrogen receptor." Trends in Endocrinology & Metabolism 15(5): 193-197.
Dowsett, M., R. Nicholson, et al. (2005). "Biological characteristics of the pure antiestrogen fulvestrant: overcoming endocrine resistance." Breast Cancer Research and Treatment 93(0): 11-18.
Dutertre, M. and C. L. Smith (2003). "Ligand-Independent Interactions of p160/Steroid Receptor Coactivators and CREB-Binding Protein (CBP) with Estrogen Receptor-{alpha}: Regulation by Phosphorylation Sites in the A/B Region Depends on Other Receptor Domains." Mol Endocrinol, 17(7): 1296-1314.
Eccles, S. A. (2001). "The role of c-erbB-2/HER2/neu in breast cancer progression and metastasis." J Mammary Gland Biol Neoplasia 6(4): 393-406.
Eckert, R., A. Mullick, et al. (1984). "Estrogen receptor synthesis and turnover in MCF-7 breast cancer cells measured by a density shift technique." Endocrinology 114(2): 629-37.
Endoh, H., K. Maruyama, et al. (1999). "Purification and Identification of p68 RNA Helicase Acting as a Transcriptional Coactivator Specific for the Activation Function 1 of Human Estrogen Receptor alpha." Molecular and Cellular Biology 19(8): 5363-5372.
Fan, P., J. Wang, et al. (2007). "Long-term Treatment with Tamoxifen Facilitates Translocation of Estrogen Receptor {alpha} out of the Nucleus and Enhances its Interaction with EGFR in MCF-7 Breast Cancer Cells." Cancer Res 67(3): 1352-1360.
Fan, P., J. Wang, et al. (2007). "Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells." Cancer Res 67(3): 1352-60.
Fang, H., W. Tong, et al. (2001). "Structure−Activity Relationships for a Large Diverse Set of Natural, Synthetic, and Environmental Estrogens." Chem. Res. Toxicol 14(3): 280-294.
Fawell, S. E., R. White, et al. (1990 ). "Inhibition of estrogen receptor-DNA binding by the "pure" antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization." Proc Natl Acad Sci U S A. 87(17): 6883–6887. .
Feinberg, A. P., R. Ohlsson, et al. (2006). "The epigenetic progenitor origin of human cancer." Nat Rev Genet 7(1): 21-33.
Flourio, G., H. Brand, et al. (2000 ). "Identification of a new isoform of the human estrogen receptor-alpha (hER-) that is encoded by distinct transcripts and that is able to repress hER- activation function
" The EMBO JournaL 19: 4688–4700. Fox, E. M., J. Andrade, et al. (2009). "Novel actions of estrogen to promote proliferation:
Integration of cytoplasmic and nuclear pathways." Steroids 74(7): 622-627. Fox, E. M., R. J. Davis, et al. (2008). "ER[beta] in breast cancer--Onlooker, passive player, or
active protector?" Steroids 73(11): 1039-1051. Fuqua, S., A. W. (1994). "Estrogen receptor mutagenesis and hormone resistance." Cancer
74(S3): 1026-1029. Furth, P. A., U. Bar-Peled, et al. (1997). "Apoptosis and mammary gland involution:
reviewing the process." Apoptosis 2(1): 19-24. Gradishar, W. J. (2004). "Tamoxifen--What Next?" The Oncologist 9(4): 378-384. Graham, M. L., II, J. A. Smith, et al. (1992). "Heterogeneity of Progesterone Receptor
Content and Remodeling by Tamoxifen Characterize Subpopulations of Cultured Human Breast Cancer Cells: Analysis by Quantitative Dual Parameter Flow Cytometry." Cancer Res 52(3): 593-602.
Green, S. and P. Chambon, . (1988). "Nuclear receptors enhance our understanding of transcription regulation " Trends Genet. 1988 Nov; 4(11): 309-14.

129
Green, S., P. Walter, et al. (1986). "Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A." Nature 320(6058): 134-139.
Grodin JM, S. P., MacDonald PC (1973). "Source of estrogen production in postmenopausal women." J Clin Endocrinol Metab 36(2): 207-14.
Gronemeyer, H. (1991). "Transcription Activation by Estrogen and Progesterone Receptors." Annual Review of Genetics 25(1): 89-123.
Gruber, C. J., W. Tschugguel, et al. (2002). Production and Actions of Estrogens. 346: 340-352.
Hager, G. L., C. S. Lim, et al. (2000). "Trafficking of nuclear receptors in living cells." J Steroid Biochem Mol Biol 74(5): 249-54.
Hall, J. M. and D. P. McDonnell (1999). "The Estrogen Receptor {beta}-Isoform (ER{beta}) of the Human Estrogen Receptor Modulates ER{alpha} Transcriptional Activity and Is a Key Regulator of the Cellular Response to Estrogens and Antiestrogens." Endocrinology 140(12): 5566-5578.
Hall, J. M. and D. P. McDonnell (1999). The Estrogen Receptor {beta}-Isoform (ER{beta}) of the Human Estrogen Receptor Modulates ER{alpha} Transcriptional Activity and Is a Key Regulator of the Cellular Response to Estrogens and Antiestrogens. 140: 5566-5578.
Hall, J. M. and D. P. McDonnell (2005). "Coregulators in Nuclear Estrogen Receptor Action: From Concept to Therapeutic Targeting." Molecular Intervention 5(6): 343-357.
Harper, M. J. K. and A. L. Walpole (1967). "MODE OF ACTION OF I.C.I. 46,474 IN PREVENTING IMPLANTATION IN RATS." Journal of Endocrinology 37(1): 83-92.
Hayashi, S. I., H. Eguchi, et al. (2003). "The expression and function of estrogen receptor alpha and beta in human breast cancer and its clinical application." Endocrine-Related Cancer 10(2): 193-202.
Heldring, N., A. Pike, et al. (2007). "Estrogen Receptors: How Do They Signal and What Are Their Targets." Physiol. Rev 87(3): 905-931.
Henderson, V. (1997). "The epidemiology of estrogen replacement therapy and Alzheimer's disease." Neurology 48(Suppl 7): S27-S35.
Hermenegildo, C. and A. Cano (2000). "Pure anti-oestrogens." Human Reproduction Update 6(3): 237-243.
Hovey, R. C., J. F. Trott, et al. (2002). "Establishing a framework for the functional mammary gland: from endocrinology to morphology." J Mammary Gland Biol Neoplasia 7(1): 17-38.
Howell, A. (2000). "Faslodex (ICI 182780): an oestrogen receptor downregulator." European Journal of Cancer 36(Supplement 4): 87.
Howell, A., D. J. DeFriend, et al. (1995). "Response to a specific antioestrogen (ICI 182780) in tamoxifen-resistant breast cancer." The Lancet 345(8941): 29-30.
Howell, A., J. F. Robertson, et al. (2004). "Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial." J Clin Oncol 22: 1605 - 1613.
Howell, S. J., S. R. D. Johnston, et al. (2004). "The use of selective estrogen receptor modulators and selective estrogen receptor down-regulators in breast cancer." Best Practice & Research Clinical Endocrinology & Metabolism 18(1): 47-66.
Htun, H., L. T. Holth, et al. (1999). "Direct visualization of the human estrogen receptor alpha reveals a role for ligand in the nuclear distribution of the receptor." Mol Biol Cell 10(2): 471-86.

130
Huang, Z., S. E. Hankinson, et al. (1997). "Dual effects of weight and weight gain on breast cancer risk." JAMA 278(17): 1407-1411.
Hulka, B. S. and P. G. Moorman (2001). "Breast cancer: hormones and other risk factors." Maturitas 38(1): 103-113.
Hurtado, A., K. A. Holmes, et al. (2008). "Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen." Nature 456(7222): 663-666.
Hynes, N. E. and H. A. Lane (2001). "Myc and mammary cancer: Myc is a downstream effector of the ErbB2 receptor tyrosine kinase." J Mammary Gland Biol Neoplasia 6(1): 141-50.
Ilka Nemere, R. J. P. P. F. B. (2003). Membrane receptors for steroid hormones: Signal transduction and physiological significance. 88: 438-445.
Jensen, E. (1966). "Mechanism of estrogen action in relation to carcinogenesis." Proc Can Cancer Conf. 6: 143-65.
Jensen , E. V. and H. I. C. Jacobson, , 1960. (1962). "Fate of steroidal estrogens in target tissue." Academic Press New York: 161-174.
Jensen, E. V. and V. C. Jordan (2003). The Estrogen Receptor: A Model for Molecular Medicine. 9: 1980-1989.
Jensen, E. V., T. Suzuki, et al. (1969). "Estrogen-binding substances of target tissues." Steroids 13(4): 417-27.
Johnston, S. (2005). Endocrinology and hormone therapy in breast cancer: Selective oestrogen receptor modulators and downregulators for breast cancer - have they lost their way? 7: 119 - 130.
Johnston, S. R. (1997). "Acquired tamoxifen resistance in human breast cancer: potential mechanisms and clinical implications." Anti-cancer drugs 8: 911 - 930.
Jordan, V. C. (1984). "Biochemical pharmacology of antiestrogen action." Pharmacol Rev. 36: 245 - 276.
Jordan, V. C. (2003). "Tamoxifen: a most unlikely pioneering medicine." Nat Rev Drug Discov 2(3): 205-213.
Jordan, V. C. (2007). "New insights into the metabolism of tamoxifen and its role in the treatment and prevention of breast cancer." Steroids 72(13): 829-842.
Jordan, V. C. (2008). "Tamoxifen: Catalyst for the change to targeted therapy." European Journal of Cancer 44(1): 30-38.
Kato, S., H. Endoh, et al. (1995). Activation of the Estrogen Receptor Through Phosphorylation by Mitogen-Activated Protein Kinase. 270: 1491-1494.
Katzenellenbogen, B. S., I. Choi, et al. (2000). "Molecular mechanisms of estrogen action: selective ligands and receptor pharmacology." The Journal of Steroid Biochemistry and Molecular Biology 74(5): 279-285.
Katzenellenbogen, B. S. and J. A. Katzenellenbogen (2002). "Defining the "S" in SERMs." Science 295: 2380-2381.
King, C. R., M. H. Kraus, et al. (1985). "Amplification of a novel v-erbB-related gene in a human mammary carcinoma." Science. 229(4717): 974-976.
King, W. J. and G. L. Greene (1984). "Monoclonal antibodies localize oestrogen receptor in the nuclei of target cells." Nature 307(5953): 745-7.
Klein-Hitpaß, L., M. Schorpp, et al. (1986). "An estrogen-responsive element derived from the 5' flanking region of the Xenopus vitellogenin A2 gene functions in transfected human cells." Cell 46(7): 1053-1061.
Kong, E. H., A. C. Pike, et al. (2003). "Structure and mechanism of the oestrogen receptor." Biochem Soc Trans 31(Pt 1): 56-9.

131
Kraus, W. L., E. M. McInerney, et al. (1995). Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. 92: 12314-12318.
Kuiper, G., E. Enmark, et al. (1996). "Cloning of a novel estrogen receptor expressed in rat prostate and ovary." Proc Natl Acad Sci U S A 93: 5925 - 5930.
Kuiper, G. G., E. Enmark, et al. (1996). "Cloning of a novel receptor expressed in rat prostate and ovary." Proc Natl Acad Sci U S A 93(12): 5925-30.
Kuiper, G. G. J. M., B. Carlsson, et al. (1997). "Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors {alpha} and {beta}." Endocrinology 138(3): 863-870.
Kushner, P. J., D. A. Agard, et al. (2000). "Estrogen receptor pathways to AP-1." The Journal of Steroid Biochemistry and Molecular Biology 74(5): 311-317.
Labrie, F., C. Labrie, et al. (1999). EM-652 (SCH 57068), a third generation SERM acting as pure antiestrogen in the mammary gland and endometrium. 69: 51 - 84.
Labrie, F., C. Labrie, et al. (2001). "EM-652 (SCH57068), a pure SERM having complete antiestrogenic activity in the mammary gland and endometrium." The Journal of Steroid Biochemistry and Molecular Biology 79(1-5): 213-225.
Lacroix, M., R.-A. Toillon, et al. (2006). "p53 and breast cancer, an update." Endocrine-Related Cancer 13(2): 293-325.
Lafarga, M., M. T. Berciano, et al. (2002). "Clastosome: a subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome." Mol Biol Cell 13(8): 2771-82.
Lane, H. A., A. B. Motoyama, et al. (2001). "Modulation of p27/Cdk2 complex formation through 4D5-mediated inhibition of HER2 receptor signaling." Ann Oncol 12 Suppl 1: S21-2.
Lannigan, D. A. (2003). "Estrogen receptor phosphorylation." Steroids 68(1): 1-9. Lavinsky, R. M., K. Jepsen, et al. (1998). "Diverse signaling pathways modulate nuclear
receptor recruitment of N-CoR and SMRT complexes." Proc Natl Acad Sci U S A 95(6): 2920-2925.
Le Goff, P., M. M. Montano, et al. (1994). "Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity." Journal of Biological Chemistry 269(6): 4458-4466.
Lerner, L. (1981). "The first non-steroidal antiestrogen-MER 25. In: Sutherland RL, Jordan VC, editor. Non-steroidal Antioestrogens: Molecular Pharmacology and Antitumour ActivityNon-steroidal Antioestrogens: Molecular Pharmacology and Antitumour Activity." Sydney Academic Press: 1–6.
Levenson, A. S., W. H. Catherino, et al. (1997). "Estrogenic activity is increased for an antiestrogen by a natural mutation of the estrogen receptor." The Journal of Steroid Biochemistry and Molecular Biology 60(5-6): 261-268.
Lichtenstein, P., N. V. Holm, et al. (2000). "Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland." N Engl J Med 343(2): 78-85.
Lonard, D. M., R. B. Lanz, et al. (2007). "Nuclear Receptor Coregulators and Human Disease." Endocrine Reviews 28(5): 575-587.
Lonard, D. M., Z. Nawaz, et al. (2000). "The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation." Mol Cell 5(6): 939-48.
Long, X. and K. P. Nephew (2006). "Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha." J Biol Chem 281(14): 9607-15.

132
Luger, K., A. W. Mader, et al. (1997). "Crystal structure of the nucleosome core particle at 2.8[thinsp]A resolution." Nature 389(6648): 251-260.
Marino, M. and P. Ascenzi (2008). "Membrane association of estrogen receptor [alpha] and [beta] influences 17[beta]-estradiol-mediated cancer cell proliferation." Steroids 73(9-10): 853-858.
Marsaud, V., A. Gougelet, et al. (2003). "Various phosphorylation pathways, depending on agonist and antagonist binding to endogenous estrogen receptor alpha (ERalpha), differentially affect ERalpha extractability, proteasome-mediated stability, and transcriptional activity in human breast cancer cells." Mol Endocrinol 17(10): 2013-27.
Martel, C., C. Labrie, et al. (1998). "Comparison of the Effects of the New Orally Active Antiestrogen EM-800 with ICI 182 780 and Toremifene on Estrogen-Sensitive Parameters in the Ovariectomized Mouse." Endocrinology 139(5): 2486-2492.
Martin, L. A., I. Farmer, et al. (2003). "Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation." Journal of Biological Chemistry 278: 30458 - 30468.
Maruvada, P., C. T. Baumann, et al. (2003). "Dynamic Shuttling and Intranuclear Mobility of Nuclear Hormone Receptors." J. Biol. Chem. 278(14): 12425-12432.
Massarweh, S. and R. Schiff (2006). "Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk." Endocrine-Related Cancer 13(Supplement_1): S15-24.
Massarweh, S. and R. Schiff (2007). "Unraveling the Mechanisms of Endocrine Resistance in Breast Cancer: New Therapeutic Opportunities." Clinical Cancer Research 13(7): 1950-1954.
McDonnell, D. P. and J. D. Norris (2002). "Connections and Regulation of the Human Estrogen Receptor." Science. 296(5573): 1642-1644.
McInerney, E. M., K. E. Weis, et al. (1998). "Transcription Activation by the Human Estrogen Receptor Subtype ß (ERß) Studied with ERß and ER Receptor Chimeras." Endocrinology. 139(11): 4513-4522.
McKenna, N. J., R. B. Lanz, et al. (1999). "Nuclear Receptor Coregulators: Cellular and Molecular Biology." Endocrine Reviews 20(3): 321-344.
McPherson, K., C. M. Steel, et al. (2000). "ABC of breast diseases: Breast cancer---epidemiology, risk factors, and genetics." British Medical Journal 321(7261): 624-628.
Means, A. R., J. P. Comstock, et al. (1972). "Ovalbumin Messenger RNA of Chick Oviduct: Partial Characterization, Estrogen Dependence, and Translation In Vitro." Proc Natl Acad Sci U S A 69(5): 1146-1150.
Melton, L. J., 3rd (1997). "The prevalence of osteoporosis." J Bone Miner Res 12(11): 1769-71.
Messina, M., S. Barnes, et al. (1997). "Phyto-oestrogens and breast cancer." The Lancet 350(9083): 971 - 972.
Métivier, R., A. Stark, et al. (2002). "A Dynamic Structural Model for Estrogen Receptor-α Activation by Ligands, Emphasizing the Role of Interactions between Distant A and E Domains." Molecular Cell
10(5): 1019-1032. Monje, P., S. Zanello, et al. (2001). "Differential cellular localization of estrogen receptor
[alpha] in uterine and mammary cells." Molecular and Cellular Endocrinology 181(1-2): 117-129.

133
Montano, M. M., K. Ekena, et al. (1996). Human estrogen receptor ligand activity inversion mutants: receptors that interpret antiestrogens as estrogens and estrogens as antiestrogens and discriminate among different antiestrogens. 10: 230-242.
MOON, M. A. (2005). "NSABP's tamoxifen results confirmed." Ob.Gyn. News 40(24): 2. Moras, D. and H. Gronemeyer (1998). "The nuclear receptor ligand-binding domain: structure
and function." Current Opinion in Cell Biology 10(3): 384-391. Murphy, L. C., H. Dotzlaw, et al. (1997). "Estrogen receptor variants and mutations." The
Journal of Steroid Biochemistry and Molecular Biology 62(5-6): 363-372. Naccarato, A. G., P. Viacava, et al. (2000). "Bio-morphological events in the development of
the human female mammary gland from fetal age to puberty." Virchows Archiv 436(5): 431-438.
Nawaz, Z., D. M. Lonard, et al. (1999). "Proteasome-dependent degradation of the human estrogen receptor." Proc Natl Acad Sci U S A 96(5): 1858-62.
Nguyen, B., M. M. Keane, et al. (1995). "The biology of growth regulation in normal and malignant breast epithelium: from bench to clinic." Crit Rev Oncol Hematol 20(3): 223-36.
Nicholson, R. I. and S. R. Johnston (2005). "Endocrine therapy – current benefits and limitations." Breast Cancer Research and Treatment 93(0): 3-10.
Nicholson, R. I., R. A. McClelland, et al. (1999). Involvement of steroid hormone and growth factor cross-talk in endocrine response in breast cancer. 6: 373-387.
Nilsson, S., S. Makela, et al. (2001). "Mechanisms of Estrogen Action." Physiological Reviews 81(4): 1535-1565.
O'Malley, B. W. and N. J. McKenna (2008). "Editorial: Coactivators and Corepressors: What's in a Name?" Molecular Endocrinology 22(10): 2213-2214.
O'Regan, R. M., A. Cisneros, et al. (1998). "Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth." Journal of the National Cancer 90: 1552 - 1558.
Ogawa, S., S. Inoue, et al. (1998). "The Complete Primary Structure of Human Estrogen Receptor [beta] (hER[beta]) and Its Heterodimerization with ER [alpha]in Vivoandin Vitro." Biochemical and Biophysical Research Communications 243(1): 122-126.
Olsson, H. (2000). "Tumour biology of a breast cancer at least partly reflects the biology of the tissue/epithelial cell of origin at the time of initiation -- a hypothesis." The Journal of Steroid Biochemistry and Molecular Biology 74(5): 345-350.
Osborne, C., P. Wilson, et al. (2004). "Oncogenes and Tumor Suppressor Genes in Breast Cancer: Potential Diagnostic and Therapeutic Applications." The Oncologist 9(4): 361-377.
Osborne, C. K. (1998). "Tamoxifen in the Treatment of Breast Cancer." The New England Journal of Medicine 339(22): 1609-1618.
Osborne, C. K., E. B. Coronado-Heinsohn, et al. (1995). "Comparison of the Effects of a Pure Steroidal antiestrogen With Those of Tamoxifen in a Model of Human Breast Cancer." Journal of the National Cancer Institute 87(10): 746-750.
Pearce, S. T. and V. C. Jordan (2004). "The biological role of estrogen receptors [alpha] and [beta] in cancer." Critical Reviews in Oncology/Hematology 50(1): 3-22.
Pearce, S. T., H. Liu, et al. (2003). "Modulation of Estrogen Receptor alpha Function and Stability by Tamoxifen and a Critical Amino Acid (Asp-538) in Helix 12." Journal of Biological Chemistry 278(9): 7630-7638.
Perou, C. M., T. Sorlie, et al. (2000). "Molecular portraits of human breast tumours." Nature 406(6797): 747-752.
Petersen, O. W., P. E. Hoyer, et al. (1987). Frequency and Distribution of Estrogen Receptor-positive Cells in Normal, Nonlactating Human Breast Tissue. 47: 5748-5751.

134
Pike, A. C., A. M. Brzozowski, et al. (1999). "Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist." EMBO 18: 4608 - 4618.
Pike, A. C. W., A. M. Brzozowski, et al. (2000). "A structural biologist's view of the oestrogen receptor." Journal of Steroid Biochemistry and Molecular Biology 74(5): 261-268.
Pike, A. C. W., A. M. Brzozowski, et al. (2001). "Structural Insights into the Mode of Action of a Pure Antiestrogen." Structure 9(2): 145-153.
Polyak, K. (2001). "On the birth of breast cancer." Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1552(1): 1-13.
Polyak, K. (2007). "Breast cancer: origins and evolution." J Clin Invest Volume 117 Number 11 November 2007( 11 ): 3155–3163.
Poola, I., S. Koduri, et al. (2000). "Identification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approach." The Journal of Steroid Biochemistry and Molecular Biology 72(5): 249-258.
Porter, J. (1974.). "Hormonal regulation of breast development and activity." J Invest Dermatol. 63(1): 85-92.
Rajesh R. Singh, R. K. (2005). Steroid hormone receptor signaling in tumorigenesis. 96: 490-505.
Razandi, M., A. Pedram, et al. (1999). Cell Membrane and Nuclear Estrogen Receptors (ERs) Originate from a Single Transcript: Studies of ER{alpha} and ER{beta} Expressed in Chinese Hamster Ovary Cells. 13: 307-319.
Razandi, M., A. Pedram, et al. (2004). "Plasma Membrane Estrogen Receptors Exist and Functions as Dimers." Molecular Endocrinology 18(12): 2854-2865.
Reid, G., S. Denger, et al. (2002). "Human estrogen receptor-a: regulation by synthesis, modification and degradation." Cellular and Molecular Life Sciences (CMLS) 59(5): 821.
Reid, G., M. R. Hübner, et al. (2003). "Cyclic, Proteasome-Mediated Turnover of Unliganded and Liganded ER[alpha] on Responsive Promoters Is an Integral Feature of Estrogen Signaling." Molecular Cell 11(3): 695-707.
Ripperger, T., D. Gadzicki, et al. (2008). "Breast cancer susceptibility: current knowledge and implications for genetic counselling." Eur J Hum Genet.
Robertson, J. F. R., S. E. Come, et al. (2005). "Endocrine treatment options for advanced breast cancer - the role of fulvestrant." European Journal of Cancer 41(3): 346-356.
Robinson-Rechavi, M., H. E. Garcia, et al. (2003). The nuclear receptor superfamily. 116: 585-586.
Ross, J. S., J. A. Fletcher, et al. (2003). "The HER-2/neu Gene and Protein in Breast Cancer 2003: Biomarker and Target of Therapy." The Oncologist 8(4): 307-325.
Russo, I. H. and J. Russo (1998). "Role of Hormones in Mammary Cancer Initiation and Progression." Journal of Mammary Gland Biology and Neoplasia 3(1): 49-61.
S Katzenellenbogen, B. and J. A Katzenellenbogen (2000). Estrogen receptor transcription and transactivation: Estrogen receptor alpha and estrogen receptor beta - regulation by selective estrogen receptor modulators and importance in breast cancer. 2: 335 - 344.
Sato, K., E. Rajendra, et al. (2008). "The UPS: a promising target for breast cancer treatment." BMC Biochem 9 Suppl 1: S2.
Saville, B., M. Wormke, et al. (2000). "Ligand-, Cell-, and Estrogen Receptor Subtype (alpha /beta )-dependent Activation at GC-rich (Sp1) Promoter Elements." Journal of Biological Chemistry 275(8): 5379-5387.

135
Schiff, R. and C. K. Osborne (2005). "Endocrinology and hormone therapy in breast cancer: New insight into estrogen receptor-alpha function and its implication for endocrine therapy resistance in breast cancer." Breast Cancer Research 7(5): 205 - 211.
Schwabe, J. W. R., D. Neuhaus, et al. (1990). "Solution structure of the DMA-binding domain of the oestrogen receptor." Nature 348(6300): 458-461.
Shackleton, M., F. o. Vaillant, et al. (2006). "Generation of a functional mammary gland from a single stem cell." Nature 439(7072): 84-88.
Shang, Y. and M. Brown (2002). "Molecular determinants for the tissue specificity of SERMs." Science 295: 2465 - 2468.
Shiau, A. K., D. Barstad, et al. (1998). "The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen." Cell 95(7): 927-37.
Simpson, E. R. (2000). Role of aromatase in sex steroid action. 25: 149-156. Simpson, E. R. (2003). "Sources of estrogen and their importance." The Journal of Steroid
Biochemistry and Molecular Biology Volume 86(3-5): 225-230. Simpson, E. R., M. D. Michael, et al. (1997). "Cytochromes P450 11: expression of the
CYP19 (aromatase) gene: an unusual case of alternative promoter usage." Faseb J 11(1): 29-36.
Singletary, S. E. (2003). "Rating the Risk Factors for Breast Cancer." Ann Surg 237(4 ): 474–482.
Skafar, D. and C. Zhao (2008). "The multifunctional estrogen receptor-alpha F domain." Endocrine 33(1): 1-8.
Smith, C. L. (1998). "Cross-talk between peptide growth factor and estrogen receptor signaling pathways." Biology of Reproduction 58(3): 627-632.
Smith, C. L. and B. W. O'Malley (2004). "Coregulator Function: A Key to Understanding Tissue Specificity of Selective Receptor Modulators." ndocrine Reviews 25(1): 45-71.
Sorlie, T., C. M. Perou, et al. (2001). Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. 98: 10869-10874.
Stenoien, D., K. Patel, et al. (2001). "FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent." Nat Cell Biol. 3(1): 15-23.
Stenoien, D. L., M. G. Mancini, et al. (2000). "Subnuclear trafficking of estrogen receptor-alpha and steroid receptor coactivator-1." Mol Endocrinol 14(4): 518-34.
Stenoien, D. L., M. G. Mancini, et al. (2000). "Subnuclear Trafficking of Estrogen Receptor-{alpha} and Steroid Receptor Coactivator-1." Molecular Endocrinology 14(4): 518-534.
Stingl, J., P. Eirew, et al. (2006). "Purification and unique properties of mammary epithelial stem cells." Nature 439(7079): 993-997.
Sun, J., M. J. Meyers, et al. (1999). Novel Ligands that Function as Selective Estrogens or Antiestrogens for Estrogen Receptor-{alpha} or Estrogen Receptor-{beta}. 140: 800-804.
Tamrazi, A., K. E. Carlson, et al. (2003). "Molecular Sensors of Estrogen Receptor Conformations and Dynamics." Molecular Endocrinology 17(12): 2593-2602.
Thomas, R. S., N. Sarwar, et al. (2008). Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-{alpha} activity. 40: 173-184.
Tora, L., J. White, et al. (1989). "The human estrogen receptor has two independent nonacidic transcriptional activation functions." Cell 59(3): 477.
Tzukerman, M. T., A. Esty, et al. (1994). "Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions." Molecular Endocrinology 8(1): 21-30.

136
Tzukerman, M. T., A. Esty, et al. (1994). Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. 8: 21-30.
van den Berg, H., M. Lynch, et al. (1989 ). "Characterisation of a tamoxifen-resistant variant of the ZR-75-1 human breast cancer cell line (ZR-75-9a1) and ability of the resistant phenotype." Br J Cancer. 59(4): 522-6.
Veronesi, U., G. Viale, et al. (2006). "Rethinking TNM: Breast cancer TNM classification for treatment decision-making and research." The Breast 15(1): 3-8.
Vogel, P. M., N. G. Georgiade, et al. (1981). "The correlation of histologic changes in the human breast with the menstrual cycle." Am J Pathol 104(1): 23-34.
Ward, H. W. C. (1973). "Anti-oestrogen Therapy for Breast Cancer: A Trial of Tamoxifen at Two Dose Levels." Br Med J 1(5844): 13-14.
Welshons, W. V., E. M. Cormier, et al. (1988). "Estrogen receptor distribution in enucleated breast cancer cell lines." Endocrinology 122(6): 2379-86.
Wijayaratne, A. L. and D. P. McDonnell (2001). "The Human Estrogen Receptor-alpha Is a Ubiquitinated Protein Whose Stability Is Affected Differentially by Agonists, Antagonists, and Selective Estrogen Receptor Modulators." Journal of Biological Chemistry 276(38): 35684-35692.
Wittmann, B. M., A. Sherk, et al. (2007). "Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators." Cancer Res 67(19): 9549-60.
Wurtz, J.-M., W. Bourguet, et al. (1996). "A canonical structure for the ligand-binding domain of nuclear receptors." Nature Structural Biology , - 3: 87 - 94
Yager, J. D. and N. E. Davidson (2006). Estrogen Carcinogenesis in Breast Cancer. 354: 270-282.
Yamamoto, Y., J. Shibata, et al. (2005). "TAS-108, a Novel Oral Steroidal Antiestrogenic Agent, Is a Pure Antagonist on Estrogen Receptor {alpha} and a Partial Agonist on Estrogen Receptor {beta} with Low Uterotrophic Effect." Clinical Cancer Research 11(1): 315-322.
Yamashito, H. (2008). "current research topics in endocrine therapy for breast cancer." Int J Clin Oncol 13: 380-383.
Yao, K., E. S. Lee, et al. (2000). "Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice." Clinical Cancer Research 6: 2028 - 2036.
Zhao, H., L. L. Hart, et al. (2002). "Characterization of stably transfected fusion protein GFP-estrogen receptor-alpha in MCF-7 human breast cancer cells." J Cell Biochem 86(2): 365-75.
Zhenlin Bai, R. G. (2009). Breast Cancer, Estrogen Receptor and Ligands. 342: 133-149. Zwart, W., A. Griekspoor, et al. (2007). "PKA-induced resistance to tamoxifen is associated
with an altered orientation of ER towards co-activator SRC-1." EMBO Journal 26: 3534–3544.

AUTEUR : Mahta MAZAHERI TITRE : Etude des mécanismes moléculaires des traitements anti-oestrogènes et de résistance dans le cancer du sein DIRECTUR DE THESE. : Kerstin BYSTRICKY / Hélène Richard-Foy LIEU ET DATE DE SOUTENANCE : Salle de conférence de l'IEFG Résumé Le cancer du sein est le plus fréquent cancer chez les femmes dans le monde. Environ 70% des tumeurs mammaires expriment le récepteur des œstrogènes alpha (REα) et sont considérées comme hormono-sensibles. La thérapie hormonale a longtemps été le traitement de choix. Toutefois, les effets agonistes sur le REα et le développement de la résistance des thérapies actuels, tels que le tamoxifène nécessitent le développement de nouveaux traitements qui agissent par des mécanismes distincts. L'objectif de notre étude était de concevoir des outils qui peuvent aider à comprendre les mécanismes moléculaires impliqués dans la modulation ligand-dépendante ou dans la dégradation du REα. Nous avons choisi quelques anti-œstrogènes avec différentes structures et nous avons comparé leurs effets sur: 1) la dégradation du REα. 2). La localisation intra-cellulaire du REα. 3). La régulation de la transcription des gènes cibles du REα 4). La régulation de la transcription dans les mutants de REα. Grace à notre appproche mécanistique, nous avons pu classer ces anti-estrogènes en trois groupes sur la base de leur fonction : SERM, SERD et un nouveau groupe (EM-652). Les SERM (selective estrogen receptor modulator) stabilisent le REα, induisent une re-localisation du REα dans le noyau, augmentent l'activité transcriptionnelle de mutants qui affectent le recrutement de cofacteurs et qui altèrent la liaison avec la chaîne latérale de l'anti-œstrogène et, enfin, manquent de pouvoir d'inhibition de l'expression basale de gènes endogènes. Les SERD (selective estrogen receptor disruptor) induisent une dégradation protéasome dépendante du REα dans le noyau en formation des foyers nucléaires qui colocalisent avec le proteasome, et inhibent l'expression basale du gène endogène du récepteur de la progestérone (PGR). Enfin, EM652 est un composé qui affecte le devenir du REα comme les SERM mais qui a un effet inhibiteur sur l’expression basale du gene endogene PGR. Cette approche peut être utilisée pour cribler de nouveaux anti-œstrogènes. Abstract Breast cancer is the most common type of malignancy among women in the world. Approximately 70% of breast tumours express the estrogen receptor alpha (ERα) and are considered hormone-responsive. Endocrine therapies have long been the treatment of choice. However, the estrogen- like agonist effect and development of resistance of the available selective estrogen receptor modulator such as tamoxifen require developing new treatments that act through different mechanisms. The objective of our study is to design tools that can help to understand the molecular mechanisms involved in ligand-dependent modulation or degradation of ERα. We selected a set of anti-estrogens with different structures and compared their effect on: 1). ERα degradation. 2). Intra-cellular localisation of ERα. 3). Regulation of transcription of ERα- endogenous target genes. 4). Regulation of transcription in the mutants of ERα. Using this mechanistic study we could classify the tested anti-estrogens into three groups based on their function: SERM, SERD and a new group for EM-652. SERM (selective estrogen receptor modulator) include compounds such as OH-tamoxifen and RU39411, that stabilise ERα, that re-localize ERα into the nucleus upon binding, that increase transcriptional activity in mutants affecting the recruitment of cofactors or the binding of their side chain and that lack inhibitory capacities of the basal expression of endogenous genes. SERD (selective estrogen receptor modulator) include compounds such as ICI182580 or RU58668, that induce nuclear proteasome-dependent degradation ERα which occur in large nuclear foci that colocalize with the proteasome and that inhibit basal gene expression of the endogenous progesterone receptor gene (PGR). Finally, EM-652 was found to affect ERα degradation and localisation similarly to SERM but inhibited basal gene expression of the endogenous PGR. This approach can be used to screen the newly designed compounds based on specific anti-estrogen structural features MOTS-CLEFS : Cancer du sein, anti-estrogènes thérapie, Résistance DICIPLINE : Gène Cellule Développement ADRESSE DU LABORATOIRE: Institut d'Exploration Fonctionnelle des Génomes (IFEG). Laboratoire de Biologie Moléculaire Eucaryote (LBME) CNRS UMR 5099, IBCG, 118 Route de narbonne 31062, Toulouse)


















