
Rita CREIDY Assistant associé en hématologie CHU du Kremlin Bicêtre
84
Rita CREIDY Assistant associé en hématologie CHU du Kremlin Bicêtre HEMATOLOGIE IFSI CHARLEFOIS Hématopoïèse Anémies Purpura thrombopénique immunologique
description
HEMATOLOGIE. IFSI CHARLEFOIS Hématopoïèse Anémies Purpura thrombopénique immunologique. Rita CREIDY Assistant associé en hématologie CHU du Kremlin Bicêtre. HEMATOPOIESE. Rita CREIDY, Décembre 2007. HEMATOPOIESE. 1- Introduction. - PowerPoint PPT Presentation
Transcript of Rita CREIDY Assistant associé en hématologie CHU du Kremlin Bicêtre

Rita CREIDYAssistant associé en hématologie
CHU du Kremlin Bicêtre
HEMATOLOGIE
IFSI CHARLEFOIS
HématopoïèseAnémies
Purpura thrombopénique immunologique

Rita CREIDY, Décembre 2007
HEMATOPOIESE

Rita CREIDY, Décembre 2007
HEMATOPOIESE
- Mécanismes assurant la production et le renouvellement régulé des cellules sanguines
- Cellules sanguines : éléments fonctionnels très différenciés, terminant une lignée
- Durée de vie courte : GR 120 j, Plt 7 à 10 jours, PN 2 à 6 jours
- Concentration sanguine = cste, équilibre entre production et disparition
- Hématopoïèse : production permanente et très importante GR 250. 109 /jour, Plt 150. 109 /jour, PN 100. 109 /jour
1- Introduction

- fœtale : sac vitellin (tissu conjonctif mésoblastique) jusqu’au 2ieme mois, puis foie et rate fœtaux jusqu’au 6ieme
mois, pendant qu’à partir du 4ième mois s’installe l’hématopoïèse médullaire osseuse
- adulte : moelle osseuse limitée aux os courts, os plats, tête des os longscrâne 20%, Thorax (sternum, côtes, vertèbres, clavicules,omoplates) 30%, rachis lombaire et ceinture pelvienne (sacrum, os iliaques) 40%, fémur 10%
Masse des cellules de MO: 4 à 5% poids corporel
Rq: lors de pathologies cancéreuses et leucémiques, une reprise d’activité hématopoïétique du foie et de la rate peut être observée : vicariance ou métaplasie myéloïde
HEMATOPOIESE
Rita CREIDY, Décembre 2007
2- Lieux de l’hématopoïèse

- Tissu d’origine conjonctive très spécialisé, situé entre des lamelles d’os spongieux, séparé de l’os par l’endoste, très
richement vascularisé, dont les cellules matures s’échappent par un mécanisme actif par des sinus veineux
- Les cellules hématopoïétiques sont disposées dans une trame de tissu de soutien conjonctif (collagène, protéoglycannes, fibronectine, laminine..)
Microenvironnement médullaire ou stroma, composé par fibroblastes, cellules endothéliales, macrophages, adipocytes, ostéoblastes…
Il influence la survie, la multiplication, la différenciation des cellules hématopoïétiques en sécrétant des matrices permettant l’adhésion et des facteurs de croissance
Rita CREIDY, Décembre 2007
HEMATOPOIESE
3- Tissu Médullaire hématopoïétique
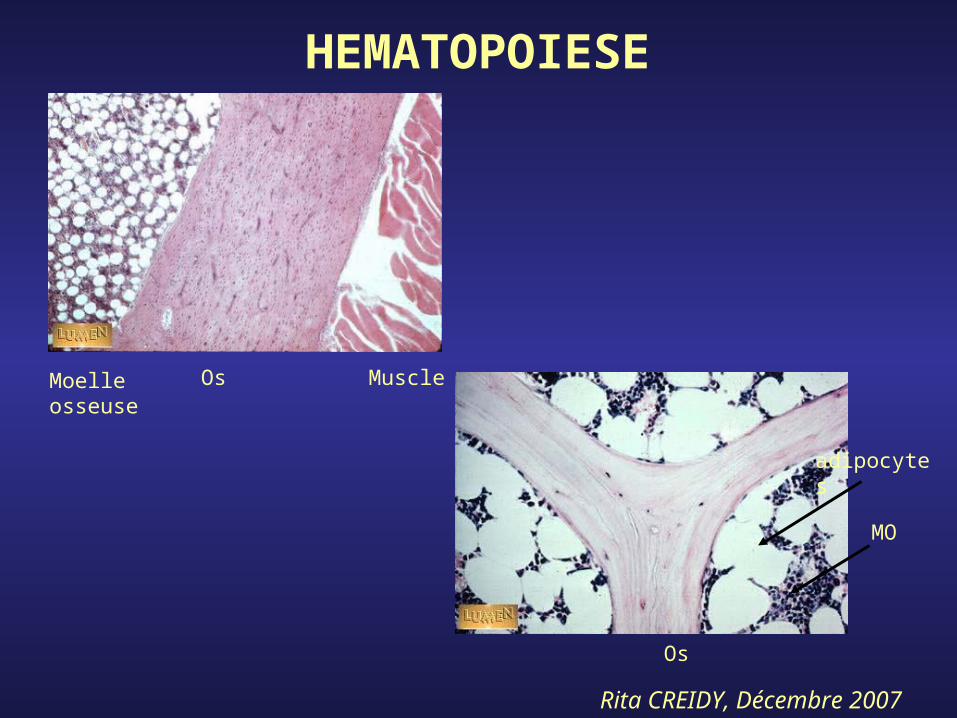
Rita CREIDY, Décembre 2007
HEMATOPOIESE
MuscleOsMoelle osseuse
Os
adipocytes
MO
Rita CREIDY, Décembre 2007
HEMATOPOIESE

Rita CREIDY, Décembre 2007
HEMATOPOIESE
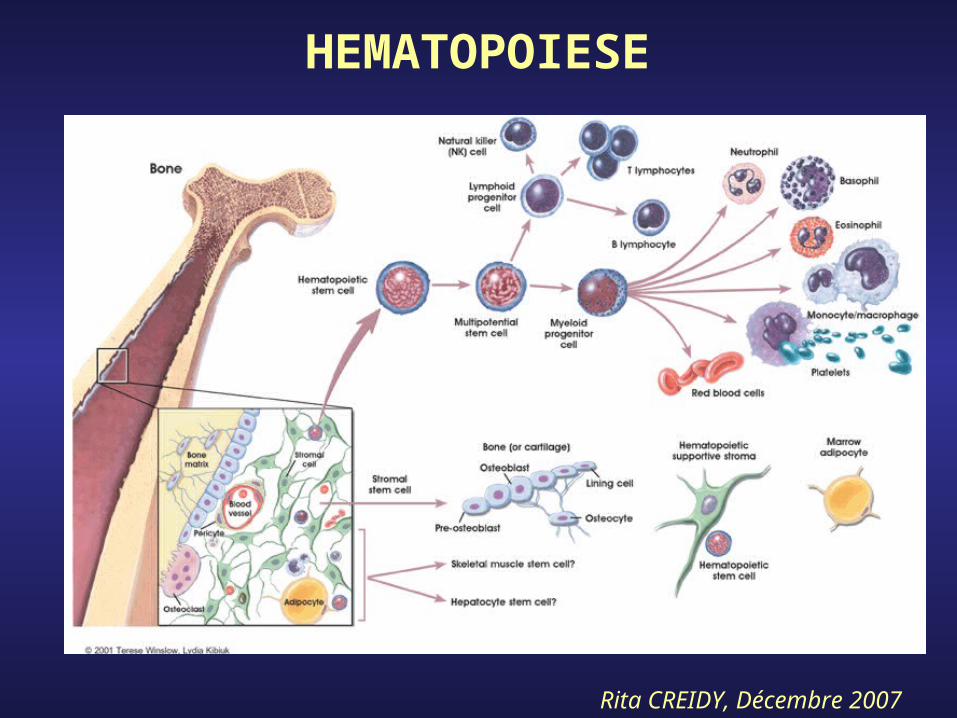
- L’hématopoïèse correspond à l’ensemble des processus qui conduisent à la prolifération et à la différenciation des cellules souches médullaires pour aboutir aux cellules sanguines matures.
- Les cellules médullaires les plus immatures sont totipotentes, puis plus on avance dans la différenciation plus elles se spécialisent. Elles deviennent pluripotentes puis s’engagent dans les deux grands axes qui sont la production des cellules de la lignée myéloïde et de la lignée lymphoïde.
- Dans la lignée myéloïde, la poursuite parallèle de la prolifération et de la différenciation aboutit à la production des globules rouges, des globules blancs granuleux et des plaquettes.
- Dans la lignée lymphoïde, ces processus aboutissent à la production des lymphocytes T et B ainsi que des plasmocytes.
4- Compartiments de l’hématopoïèse

Rita CREIDY, Décembre 2007
HEMATOPOIESE4- Compartiments de
l’hématopoïèse
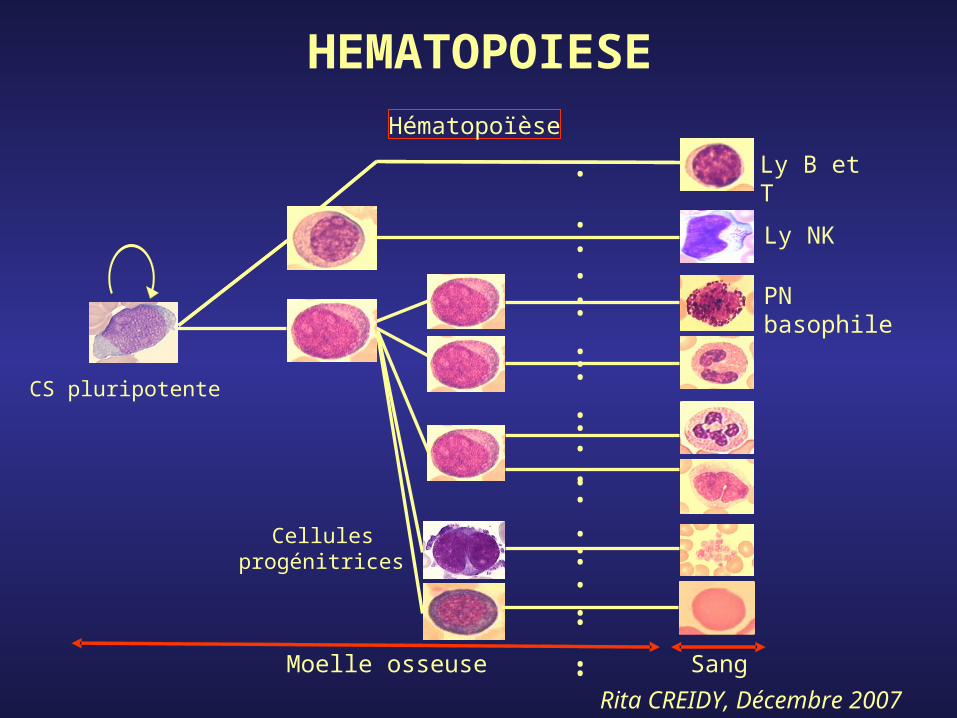
Différenciation: capacité, sous influence de facteurs de croissance, de se diviser en s’engageant de façon irréversible vers une ou plusieurs lignées
Auto-renouvellement: multiplication sans différenciation
Cellules souches
Progéniteurs
Précurseurs
Cellules matures
Diff
érenciation
Auto-renouvellem
ent
Rita CREIDY, Décembre 2007
HEMATOPOIESE
Ly B et T
Ly NK
PN basophile
Moelle osseuse
CS pluripotente
Cellules progénitrices
Sang
. . .
Hématopoïèse
. . .
. . .
. . .
. . .
. . .
. . .
. . .
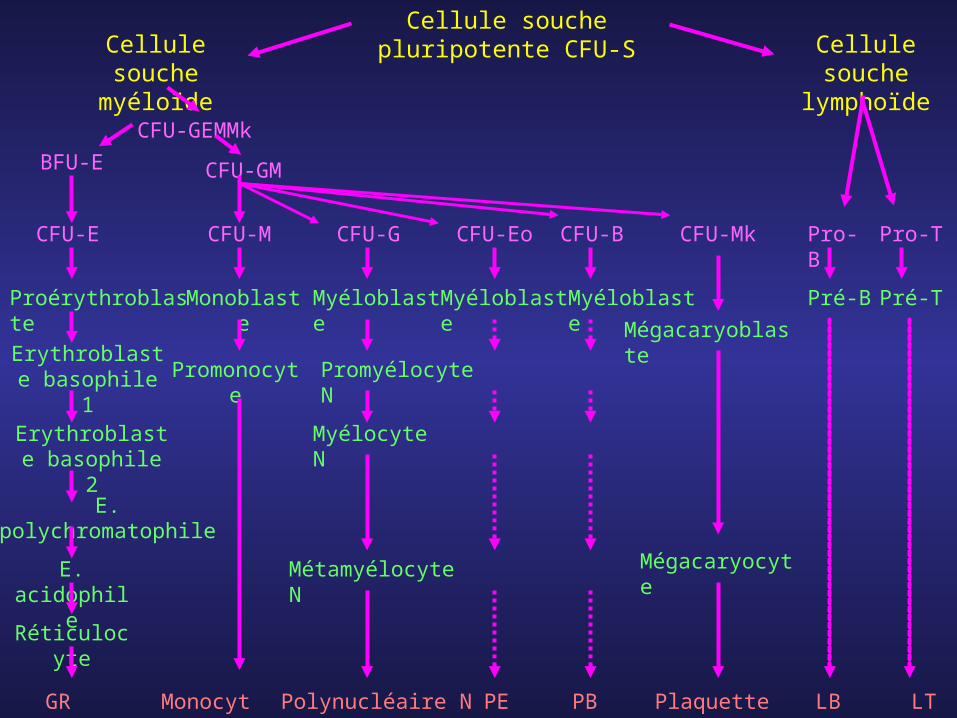
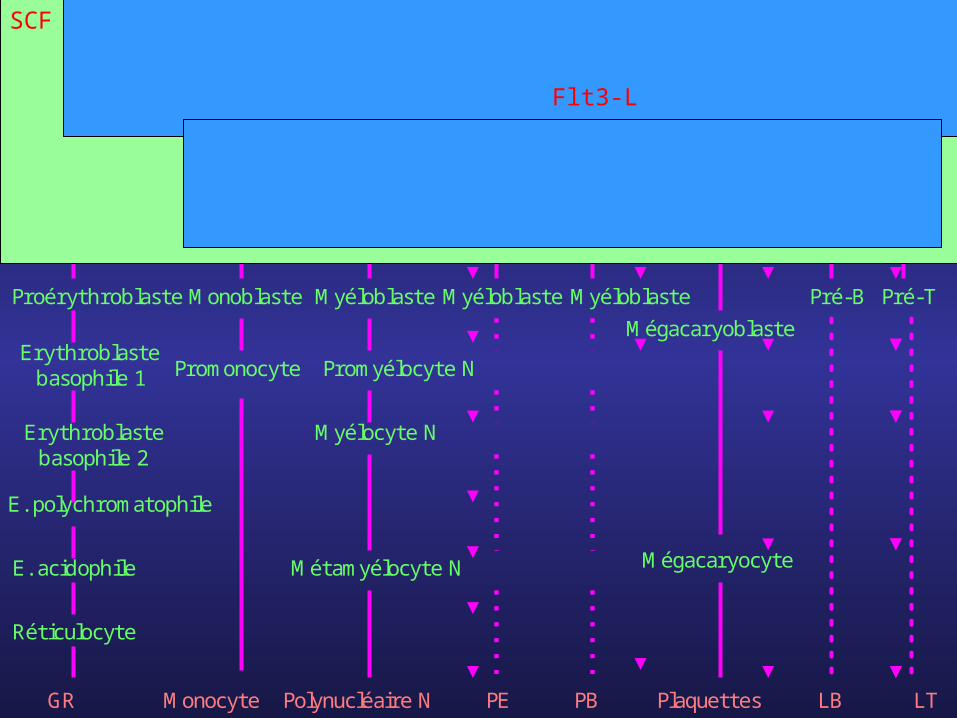
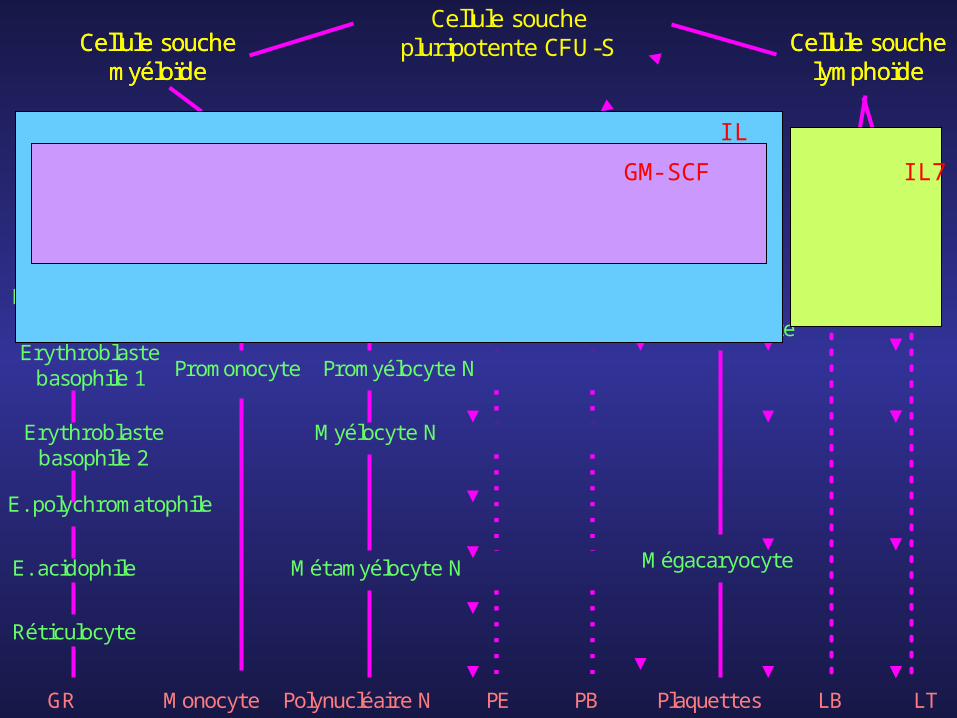
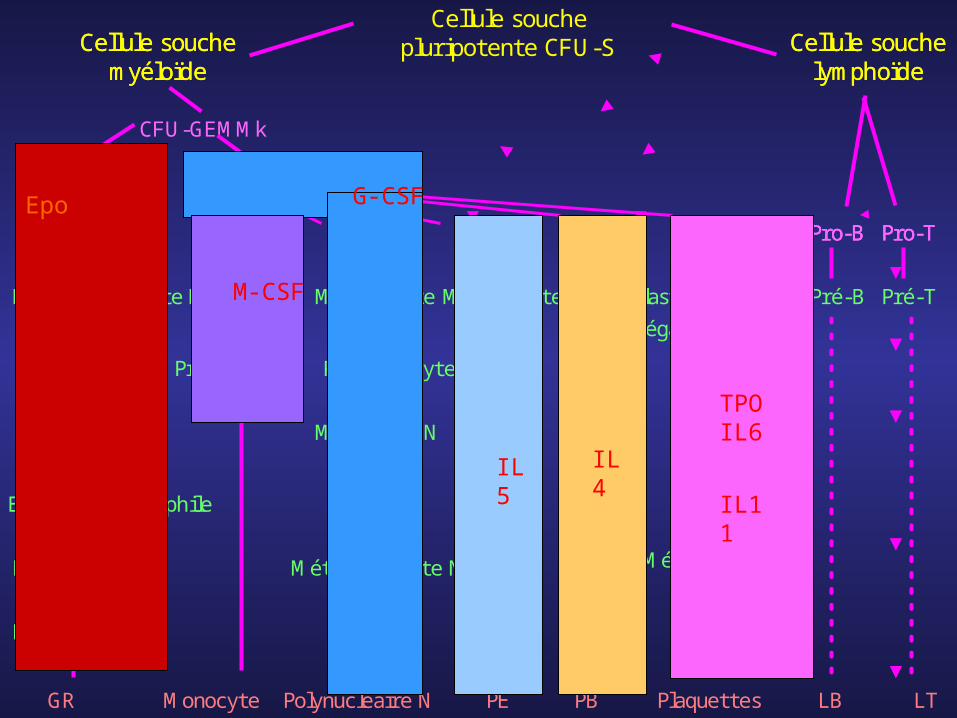
Cellule souche pluripotente CFU-SCellule
souche myéloïde
Cellule souche
lymphoïdeCFU-GEMMk
BFU-E
Proérythroblaste
Erythroblaste basophile 1
Erythroblaste basophile 2
E. polychromatophile
E. acidophile
Réticulocyte
GR
CFU-GM
Monoblaste
Myéloblaste
Promonocyte
Promyélocyte N
Myélocyte N
Métamyélocyte N
Polynucléaire NMonocyte
MyéloblasteMyéloblaste
Mégacaryoblaste
Mégacaryocyte
CFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-T
Pré-B Pré-T
PE PB Plaquettes LB LT
Rita CREIDY, Décembre 2007
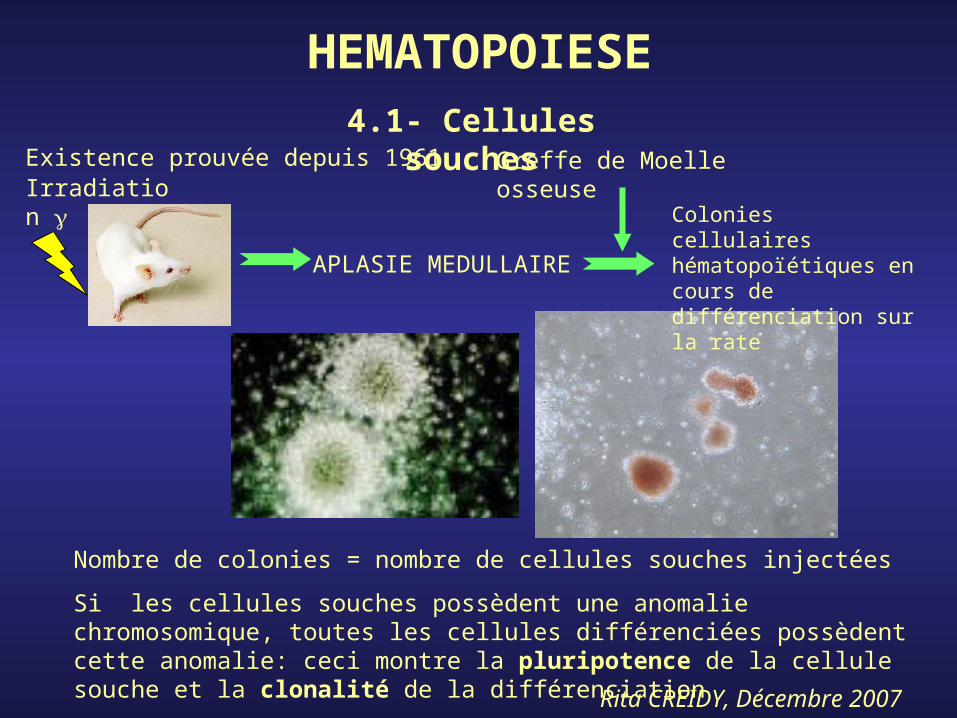
HEMATOPOIESE4.1- Cellules
souchesExistence prouvée depuis 1961Irradiation
APLASIE MEDULLAIRE
Greffe de Moelle osseuse
Colonies cellulaires hématopoïétiques en cours de différenciation sur la rate
Nombre de colonies = nombre de cellules souches injectées
Si les cellules souches possèdent une anomalie chromosomique, toutes les cellules différenciées possèdent cette anomalie: ceci montre la pluripotence de la cellule souche et la clonalité de la différenciation

Rita CREIDY, Décembre 2007
HEMATOPOIESE4.1- Cellules
souches- Ne sont pas différenciables morphologiquement des progéniteurs
- Très faible représentation médullaire 0,01% à 0,05%
- Majorité en phase G0 : relative résistance aux radiations ionisantes et agents chimiothérapeutiques du cycle cellulaire
- Circulation temporaire sanguine : cellules souches périphériques
- Marqueurs membranaires spécifiques : CD34, CD117, CD133
- Prélèvement spécifique de cellules souches : médullaire ou sanguin Concentration possible par tri de cellules CD34+, congélation possible
- Usage: allogreffe ou autogreffe de cellules souches

- Cellules engagées dans la différenciation vers une ou deux lignées cellulaires
- Faible représentation médullaire
- Circulation temporaire sanguine des progéniteurs peu différenciés
- Cultivables in vitro en milieux semi-solides et donnant des colonies CFU, Multiplication sous influence des facteurs de croissance
- Non différenciables morphologiquement entre eux
- Acquisition des marqueurs membranaires CD spécifiques de lignée
CFU-GEMMk: CD34, CD33, CD38, HLA-DR
CFU-GM: CD34, CD33, CD38, HLA-DR, CD13
CFU-E: CD36
CFU GEMM
Rita CREIDY, Décembre 2007
HEMATOPOIESE4.2- Progéniteurs

Rita CREIDY, Décembre 2007
HEMATOPOIESE
- Cellules engagées dans la différenciation vers une lignée cellulaire
- Identifiables morphologiquement
- Perte de la capacité d’auto-renouvellement
- Selon les lignées, 3 à 5 mitoses entre chaque stade précurseur, de sorte qu’un précurseur immature conduit de 8 à 32 cellules matures
- Modification morphologiques communes de maturation des précurseurs : diminution de la taille cellulaire (sauf lignée mégacaryocytaire), diminution N/C, disparition des nucléoles, condensation de la chromatine
4.3- Précurseurs

Modifications spécifiques de chaque lignée au cours de la différenciation :
- Lobulation du noyau (GRA), expulsion du noyau (R), apparition de granulations spécifiques (GRA)
- Particularité: endomitose des précurseurs mégacaryocytaires, doublement de l’ADN sans division cellulaire à chaque stade de maturation, aboutissant à des cellules de grande taille à 4N, 8N, 16N, 32N ou 64N chromosomes. Les plaquettes apparaissent par fragmentation du cytoplasme de ces mégacaryocytes
Rita CREIDY, Décembre 2007
HEMATOPOIESE4.3- Précurseurs

Rita CREIDY, Décembre 2007
HEMATOPOIESE4.3- Précurseurs
- Acquisition des marqueurs membranaires spécifiques de lignées
Lignée érythroblastique: CD71, CD35, CD44, CD55, CD147, glycophorines
Lignée granulocytaire: CD33, CD16, CD13, CD35
Lignée monocytaire: CD35, CD13, CD33, CD14, CD11
Lignée Mégacaryocytaire: CD61, CD51, CD41, CD42
Lignée Lymphocytaire T: CD2, CD3, CD4, CD8, TCR
Lignée Lymphocytaire B: CD19, CD20, CD10, Chaîne m
Lignée NK: CD16, CD56
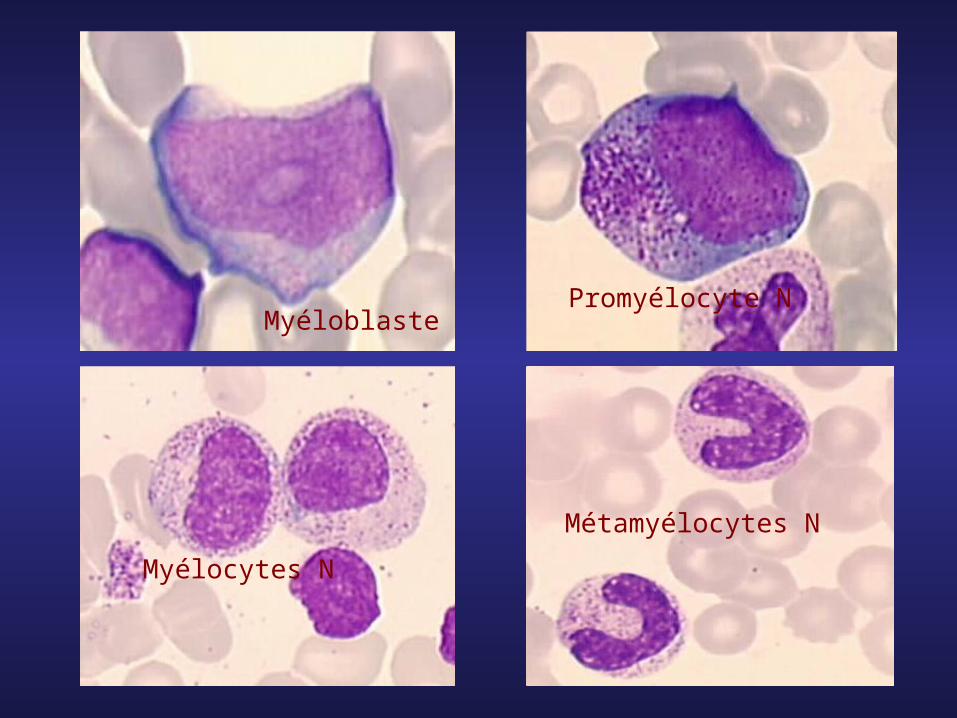
Métamyélocytes N
Myélocytes N
Promyélocyte NMyéloblaste
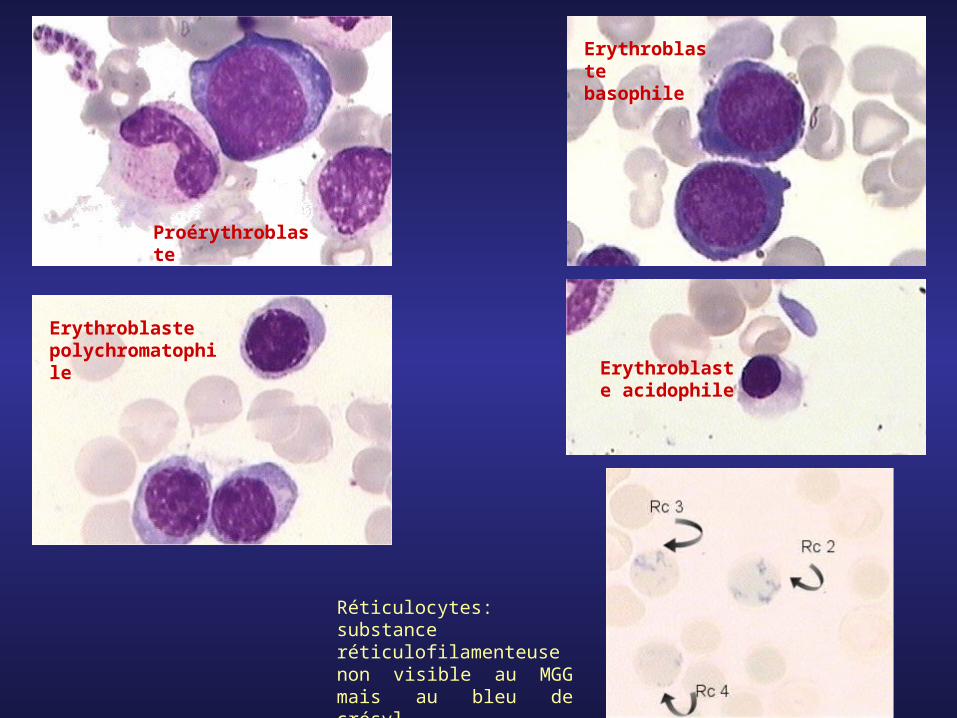
Proérythroblaste
Erythroblaste basophile
Erythroblaste polychromatophile Erythroblast
e acidophile
Réticulocytes: substance réticulofilamenteuse non visible au MGG mais au bleu de crésyl
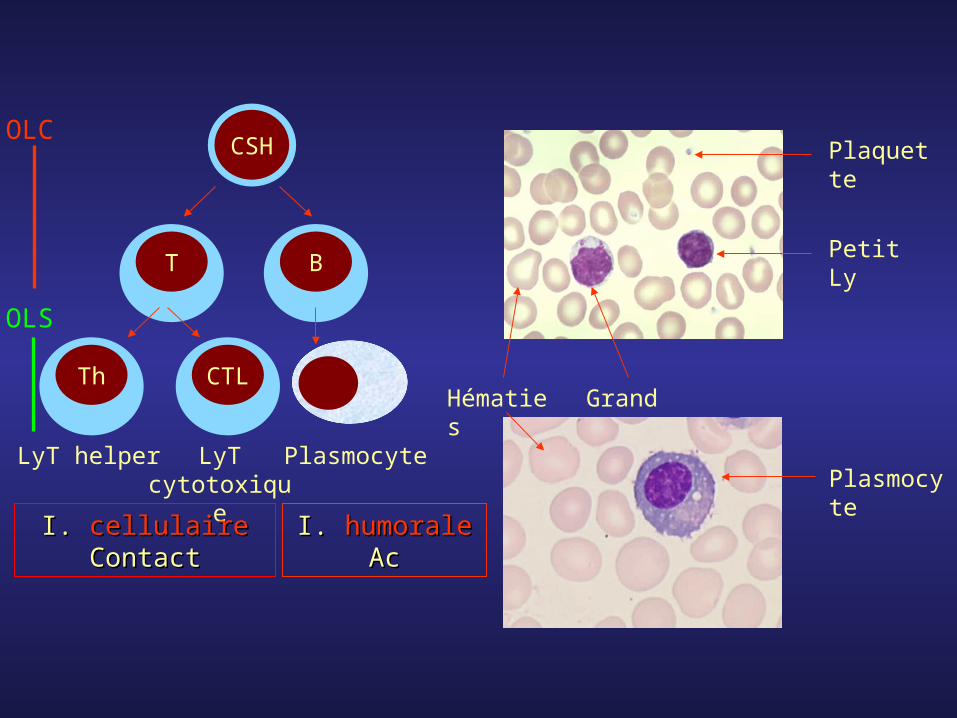
T
CSH
B
CTLTh
LyT helper LyT cytotoxique
Plasmocyte
Plaquette
Petit Ly
Grand Ly
Hématies
Plasmocyte
I. I. cellulairecellulaireContactContact
I. I. humoralehumoraleAcAc
OLC
OLS

- Microenvironnement médullaire : contacts intercellulaires, sécrétions de facteurs de croissance
- Certaines vitamines et oligoéléments : B12, Folates (B9), fer,…
- Facteurs de croissances hématopoïétiques : Cytokines et CSF (Colony Stimulating Factor)
-facteurs de promotion : augmentent la survie et le nombre de cellules souches rentrant en cycle cellulaire : IL1, IL6, IL11,Stem Cell Factor, Flt3L
-facteurs de croissance multipotents : favorisent la différenciation et la multiplication des cellules souches et progéniteurs les plus immatures: IL3, IL7, GM-CSF
-facteurs de croissance restreints : favorisent la différenciation des progéniteurs les plus engagés, et la multiplication et la maturation des précurseurs : G-CSF, M-CSF, Erythropoïétine (EPO), Thrombopoïétine (TPO), IL5, …
Rita CREIDY, Décembre 2007
HEMATOPOIESE5- Régulation de l’hématopoïèse
Cellule souche pluripotente CFU-SCellule souche
myéloïdeCellule souche
lymphoïdeCellule souche
myéloïdeCellule souche
lymphoïde
CFU-GEMMk
BFU-E
Proérythroblaste
Erythroblastebasophile 1
Erythroblastebasophile 2
E. polychromatophile
E. acidophile
Réticulocyte
GR
CFU-GM
Monoblaste Myéloblaste
Promonocyte Promyélocyte N
Myélocyte N
Métamyélocyte N
Polynucléaire NMonocyte
Myéloblaste Myéloblaste
Mégacaryoblaste
Mégacaryocyte
CFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-TCFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-TPro-B Pro-T
Pré-B Pré-T
PE PB Plaquettes LB LT
SCF
Flt3-L
Cellule souche pluripotente CFU-SCellule souche
myéloïdeCellule souche
lymphoïdeCellule souche
myéloïdeCellule souche
lymphoïde
CFU-GEMMk
BFU-E
Proérythroblaste
Erythroblastebasophile 1
Erythroblastebasophile 2
E. polychromatophile
E. acidophile
Réticulocyte
GR
CFU-GM
Monoblaste Myéloblaste
Promonocyte Promyélocyte N
Myélocyte N
Métamyélocyte N
Polynucléaire NMonocyte
Myéloblaste Myéloblaste
Mégacaryoblaste
Mégacaryocyte
CFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-TCFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-TPro-B Pro-T
Pré-B Pré-T
PE PB Plaquettes LB LT
IL3
IL7GM-SCF
Cellule souche pluripotente CFU-SCellule souche
myéloïdeCellule souche
lymphoïdeCellule souche
myéloïdeCellule souche
lymphoïde
CFU-GEMMk
BFU-E
Proérythroblaste
Erythroblastebasophile 1
Erythroblastebasophile 2
E. polychromatophile
E. acidophile
Réticulocyte
GR
CFU-GM
Monoblaste Myéloblaste
Promonocyte Promyélocyte N
Myélocyte N
Métamyélocyte N
Polynucléaire NMonocyte
Myéloblaste Myéloblaste
Mégacaryoblaste
Mégacaryocyte
CFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-TCFU-E CFU-M CFU-G CFU-Eo CFU-B CFU-Mk Pro-B Pro-TPro-B Pro-T
Pré-B Pré-T
PE PB Plaquettes LB LT
Epo
TPOIL6
IL11IL4IL5
G-CSF
M-CSF

- Mise en culture des cellules souches et progéniteurs
- Examens microscopiques : obligatoire dans le diagnostic et la surveillance thérapeutique des hémopathies malignes :
-ponction, aspiration médullaire sur os sternum, crêtes iliaques, permettant confection d’un frottis coloré au MGG, pour effectuer un Myélogramme : détermination du % des précurseurs médullaire
-biopsie ostéomédullaire (BOM) : coupe histologique permet d’évaluer la richesse cellulaire et l’architecture médullaire
-adénogramme (ponction ganglionnaire)
- Etude cytochimique : MPO, perls
- Autres : immunophénotypage, analyse chromosomique, analyse génique, électrophorèse et immunofixation des Ig
HEMATOPOIESE6- Exploration de l’hématopoïèse
Rita CREIDY, Décembre 2007
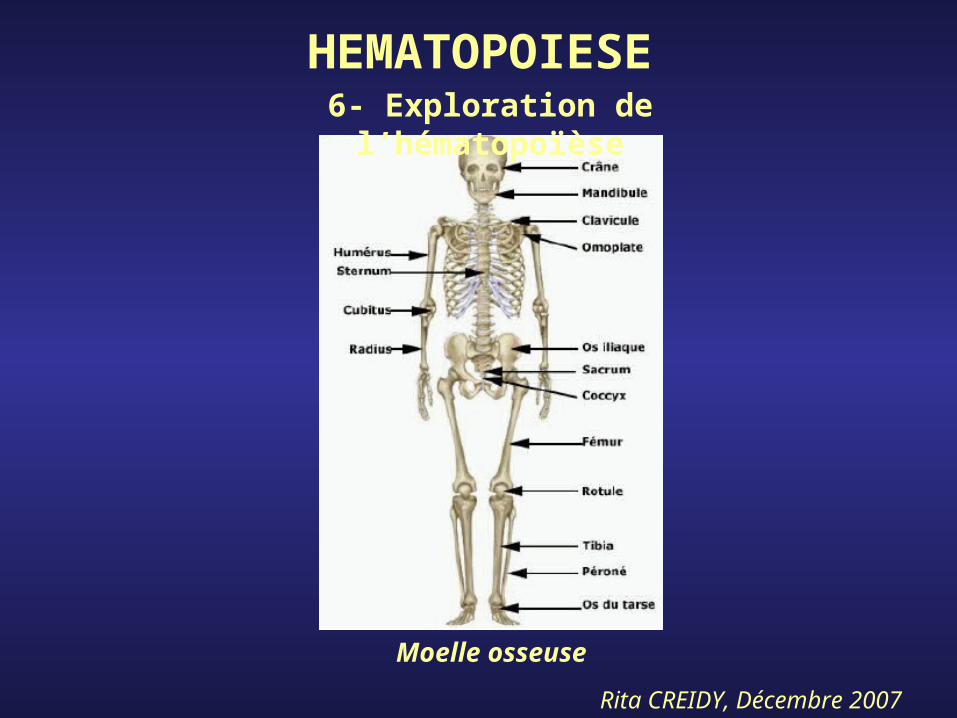
Rita CREIDY, Décembre 2007
HEMATOPOIESE6- Exploration de l’hématopoïèse
Moelle osseuse
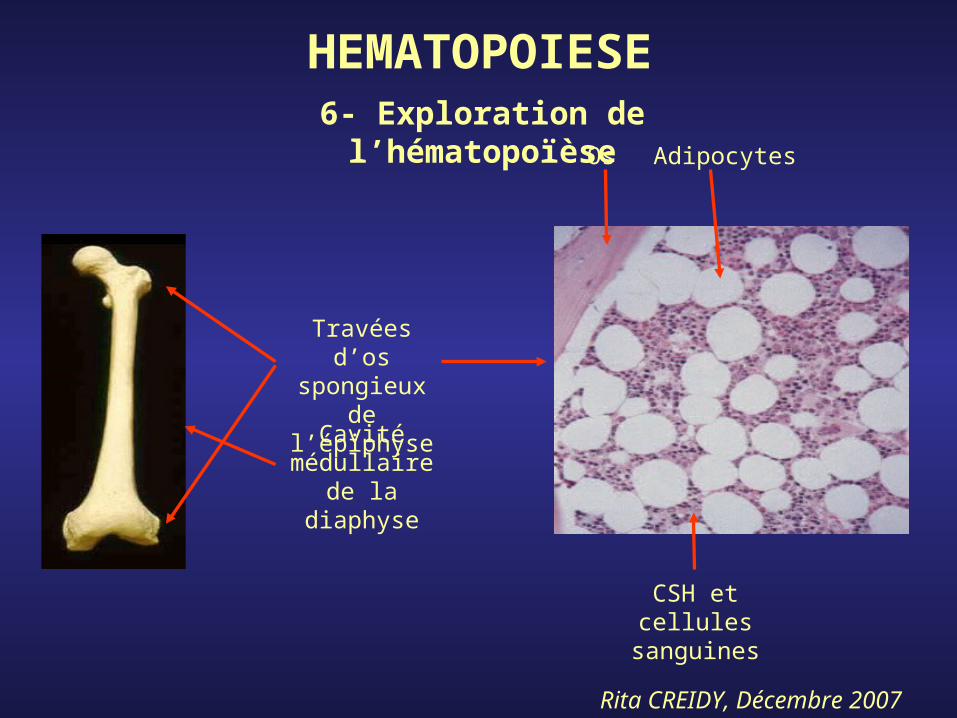
Rita CREIDY, Décembre 2007
HEMATOPOIESE
Os Adipocytes
CSH et cellules
sanguines
Travées d’os spongieux de
l’épiphyse
Cavité médullaire de la diaphyse
6- Exploration de l’hématopoïèse
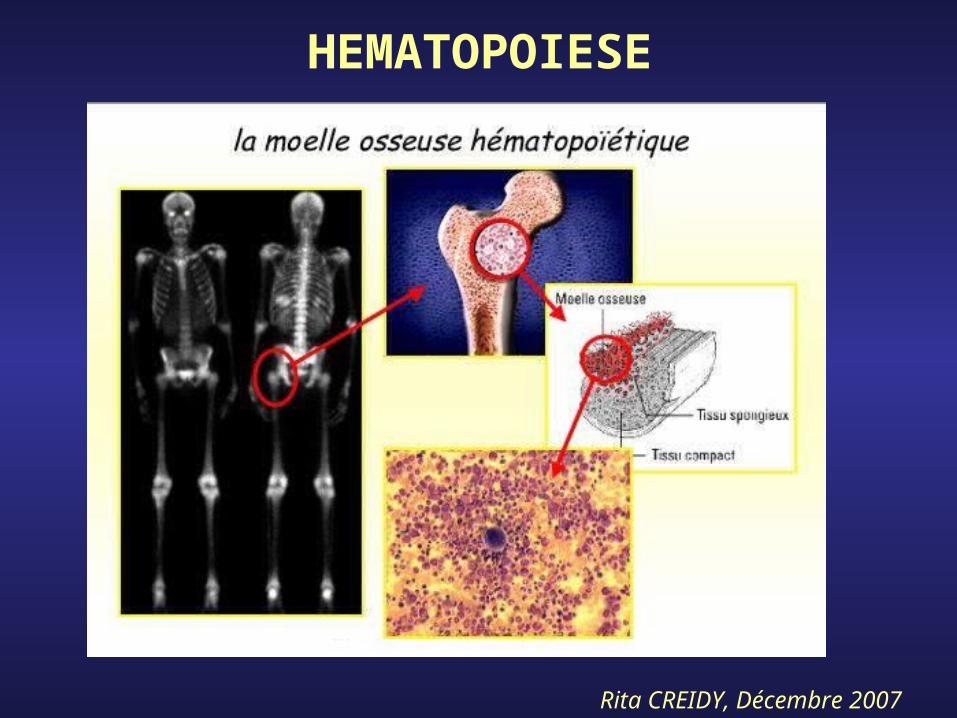
Rita CREIDY, Décembre 2007
HEMATOPOIESE
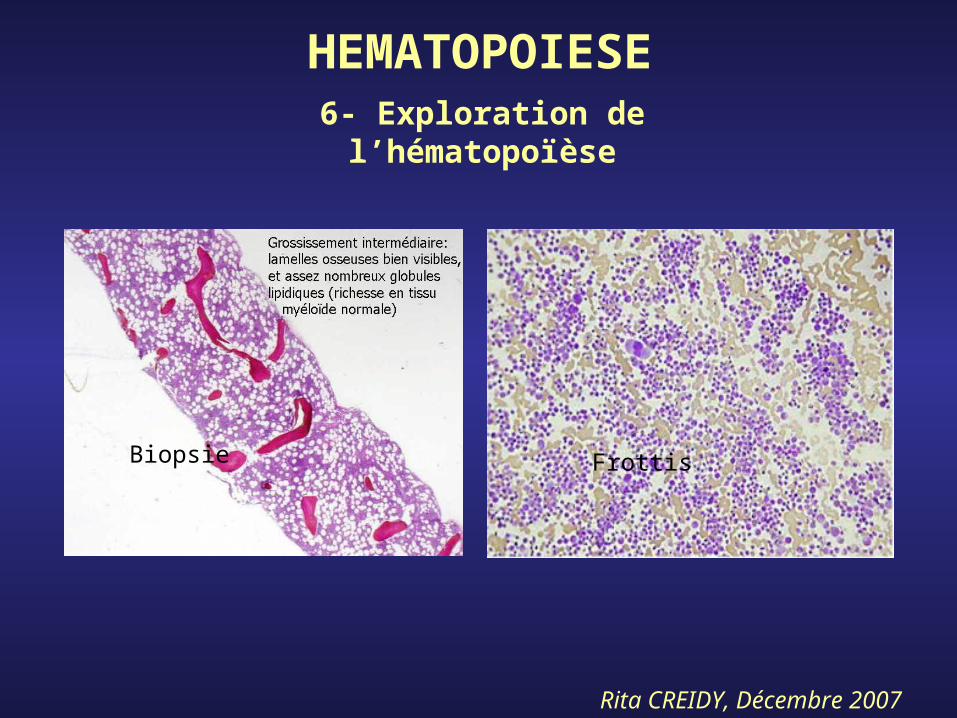
Rita CREIDY, Décembre 2007
HEMATOPOIESE
Biopsie Frottis
6- Exploration de l’hématopoïèse
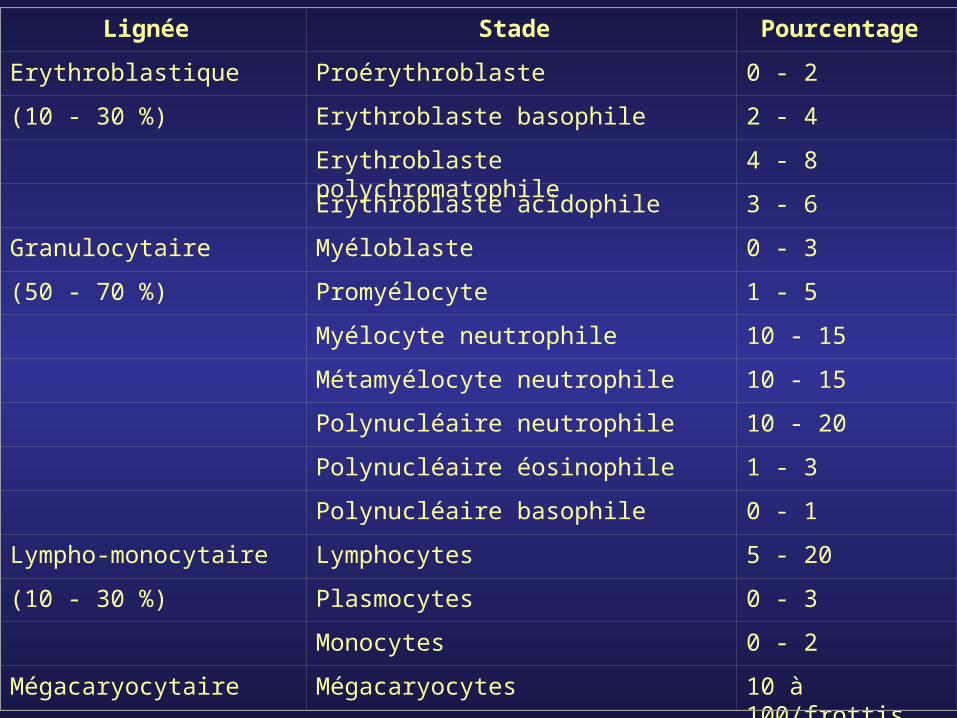
Lignée Stade Pourcentage
Erythroblastique Proérythroblaste 0 - 2
(10 - 30 %) Erythroblaste basophile 2 - 4
Erythroblaste polychromatophile 4 - 8
Erythroblaste acidophile 3 - 6
Granulocytaire Myéloblaste 0 - 3
(50 - 70 %) Promyélocyte 1 - 5
Myélocyte neutrophile 10 - 15
Métamyélocyte neutrophile 10 - 15
Polynucléaire neutrophile 10 - 20
Polynucléaire éosinophile 1 - 3
Polynucléaire basophile 0 - 1
Lympho-monocytaire Lymphocytes 5 - 20
(10 - 30 %) Plasmocytes 0 - 3
Monocytes 0 - 2
Mégacaryocytaire Mégacaryocytes 10 à 100/frottis

Rita CREIDY, Décembre 2007
HEMATOPOIESE7- Hématopoïèse et
pathologie
- Excès de production : syndromes myéloprolifératifs et lymphoprolifératifs
- Défaut de production : syndromes myélodysplasiques, aplasies médullaires, carences et toxicité médicamenteuse
- Excès de destruction : pathologies immunologiques ou auto-immunes (PTI, AHAI), soustraction (saignements, cytaphérèse, plasmaphérèse)
- Défaut de destruction : pathologies d’accumulation (LLC…)

Rita CREIDY, Décembre 2007
HEMATOPOIESE
ANEMIES

Rita CREIDY, Décembre 2007
ANEMIES
- Disque biconcave Ø moyen 7,5 µm, épaisseur 2 µm, surface 145 µm2
- Cellule anucléée, contenu eau 70%, Hb 25%, protéines, enzymes, ions
- Acidophile (gris-rose au MGG)
- Durée de vie limitée 120 j, car absence de renouvellement enzymatique
- Diminution du taux Hb circulante :
-insuffisance de production des GR ou diminution de érythropoïèse ou de synthèse Hb
-perte trop importante de GR par hémorragies
-hyper-hémolyse non compensée
-inflammation
1- Généralités

Rita CREIDY, Décembre 2007
ANEMIES2- Signes cliniques
- Les symptômes sont la conséquence de l’hypoxémie et sont extrêmement variables, fonction de l’ intensité de l’anémie, rapidité d’installation de l’anémie, âge, état cardiovasculaire
- Pâleur (cutanéo)-muqueuse, dyspnée, tachycardie, asthénie, vertiges

Rita CREIDY, Décembre 2007
ANEMIES3- Classification des anémies
- Fausses anémies
- Anémies microcytaires :
*Carence en Fer*Syndrome inflammatoire*Thalassémies
- Anémies macrocytaires :
*Déficit en VitB12 ou folates*Syndromes myélodysplasiques*Autres : alcoolisme, hypothyroïdie
- Anémies régénératives :*AHAI*Drépanocytose*Sphérocytose héréditaire

Rita CREIDY, Décembre 2007
ANEMIES
-Taux d’hémoglobine 12 à 15 g/dL
- Numération Réticulocytes sanguins:
> 150 000/mm3 , anémie régénérative, (souvent cause périphérique)
< 150 000/mm3 , anémie arégénérative, (souvent cause centrale)
- VGM: Anémie microcytaire ou macrocytaire (80-95 fl)
- TCMH: Anémie normochrome ou hypochrome (27-33 pg)
4- Diagnostic biologique d’une anémie

- Frottis sanguins : anomalies morphologiques GR, présence érythroblastes
- Dosages concernant Métabolisme du Fer, Vit B12, Folates
- Dosages Bilirubine libre, haptoglobine
- Dosages des protéines de l’inflammation: CRP (C-Réactive Protéine)
- Électrophorèse de Hb
- Test de Coombs (recherche Ac anti-érythrocytaires)
- Dosages activités enzymatiques G6PDH, PK
Rita CREIDY, Décembre 2007
ANEMIES4- Diagnostic biologique d’une
anémieExamens complémentaires

Rita CREIDY, Décembre 2007
- Expansion volume plasmatique*Grossesse 3eme
trimestre *Gammapathie monoclonale
- Anomalie distribution des GR*Splénomégalie +++
- Agglutination GR*Agglutinine froide
- Anémie physiologique du nouveau néentre 1 mois et 12 mois
ANEMIES5- Fausses anémies

Rita CREIDY, Décembre 2007
- Pas d’incorporation de Fer*Perte excessive, manque d’apport*Séquestration Fer
- Mauvaise expression des Chaînes globines a, b
- Défaut synthèse de l’hème
- Cas particulier Anémie sidéroblastique
ANEMIES5- Anémies microcytaires

Rita CREIDY, Décembre 2007
- Clinique : *Cheveux fins*Ongles cassants*Glossite
- Biologie :*Anémie : Hb < 10 g/dL, VGM < 65 fL,
CCMH < 32 g/dL, Rétic. < 50 000*Fer : Fer sérique ↓ ↓, Coef. Sat ↓ ↓,
transferrine , Ferritine ↓ ↓
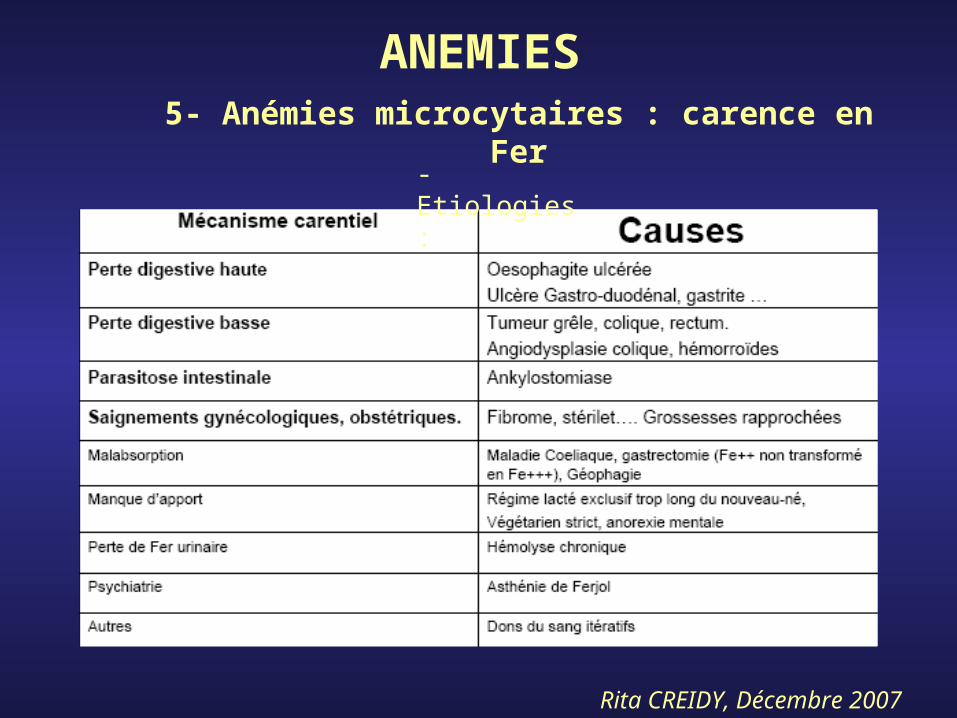
ANEMIES5- Anémies microcytaires : carence en
Fer
Rita CREIDY, Décembre 2007
- Etiologies :
ANEMIES5- Anémies microcytaires : carence en
Fer

Rita CREIDY, Décembre 2007
- Explorations*Gastro et coloscopie systématiques*Ex. Gynécologique
- Traitement*Tt de l’étiologie +++*Fer PO, 6 mois minimum*Association Vit. C: Ferrograd 500, 1 cp/j*Ac. Folique au début du Tt (1er mois)
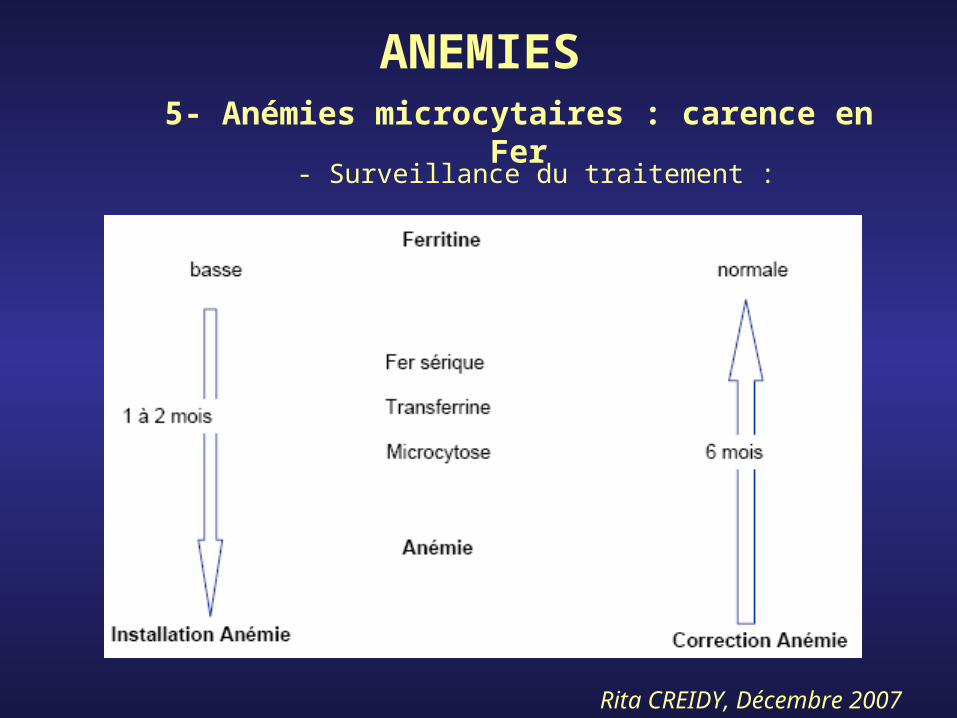
ANEMIES5- Anémies microcytaires : carence en
Fer- Traitement :
Rita CREIDY, Décembre 2007
ANEMIES5- Anémies microcytaires : carence en
Fer- Surveillance du traitement :

Rita CREIDY, Décembre 2007
- Anémie de profondeur variable (le plus souvent modérée, entre 9 et 11 g/dL, arégénérative, normochrome, normocytaire ou dans une forme évoluée un peu microcytaire (VGM entre 70 et 80 fL)- Thrombocytose et hyperleucocytose à polynucléaires fréquentes- Biologie du syndrome inflammatoire :
*Fer sérique abaissé et CTF de la transferrine normale ou abaissée (donc coefficient de saturation (CS) normal ou pas aussi diminué que dans une carence en fer) ; ferritinémie souvent élevée
*Vitesse de sédimentation augmentée, fibrinogénémie augmentée, hypergamma et hyper-alpha-2-globulinémie, haptoglobinémie élevée, CRP (C réactive protein) élevée- Myélogramme : ne s’impose pas pour comprendre le mécanisme, MAIS parfois pour le diagnostic
ANEMIES5- Anémies microcytaires : syndrome
inflammatoire- Diagnostic :

Rita CREIDY, Décembre 2007
- Traitement = Etiologie
- Rhumatisme inflammatoire
- Connectivites
- Cancer
- Infections …
ANEMIES5- Anémies microcytaires : syndrome
inflammatoire- Traitement :
Rita CREIDY, Décembre 2007
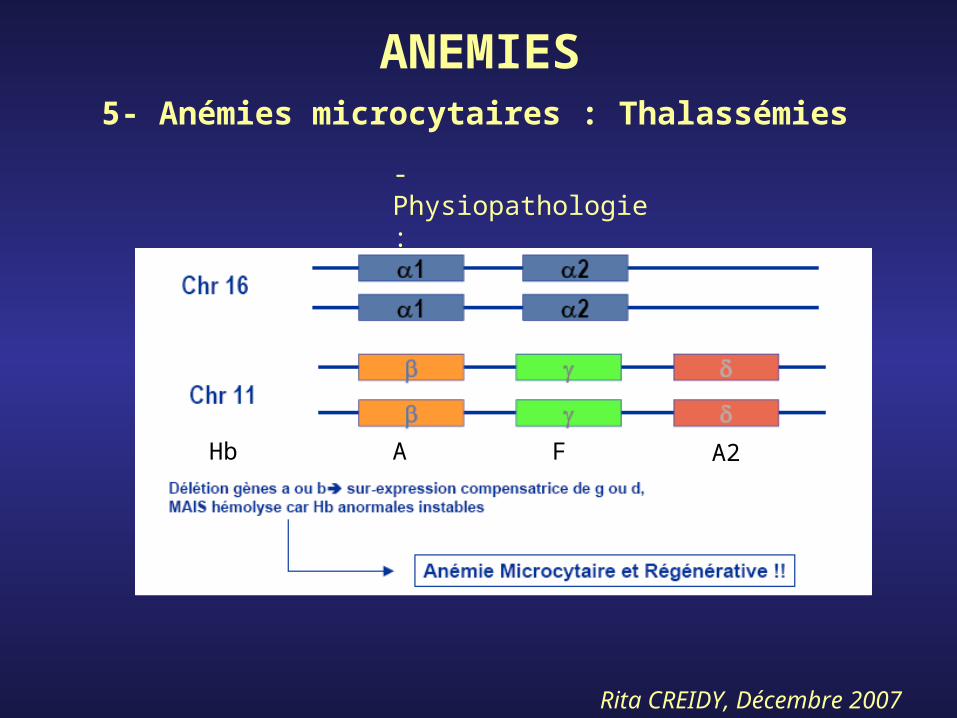
ANEMIES5- Anémies microcytaires : Thalassémies
- Physiopathologie :
A F A2Hb
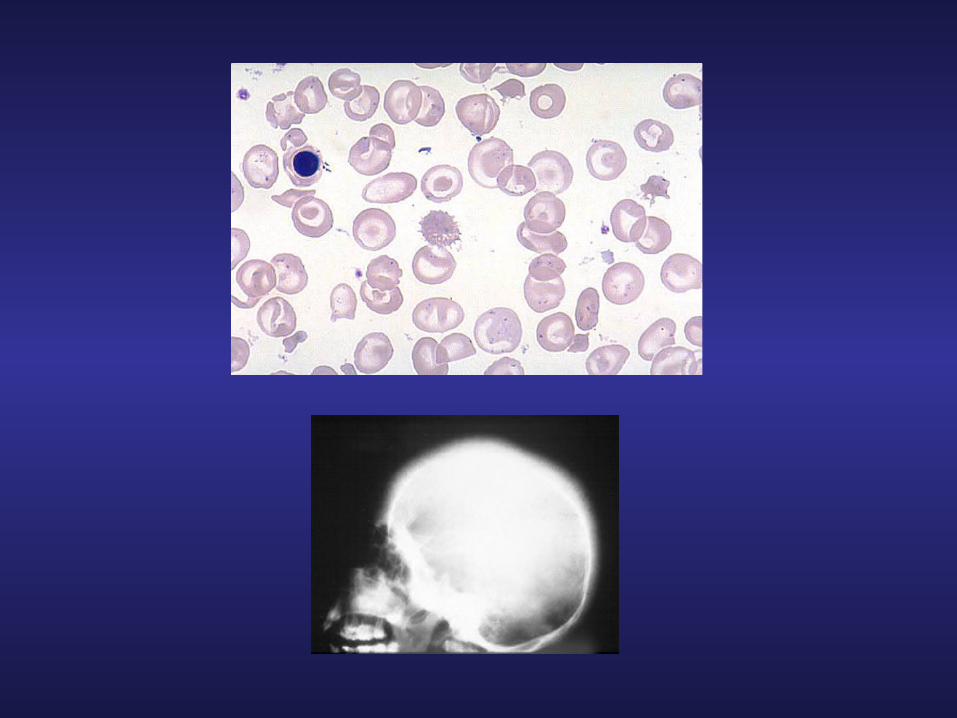

Rita CREIDY, Décembre 2007
- Maladies héréditaires*b-Thalassémies : pourtour méditerranéen*a-Thalassémies : extrême Orient
- Gravité de la maladies dépend du nombre de gènes délétés ou non-exprimés
ANEMIES5- Anémies microcytaires : Thalassémies
- Diagnostic :

Rita CREIDY, Décembre 2007
• β 0/ β –Thalassémies (β -T. hétérozygotes)
ANEMIES5- Anémies microcytaires : Thalassémies
- Diagnostic :

Rita CREIDY, Décembre 2007
• β 0/ β-Thalassémies (β -T. homozygotes)
ANEMIES5- Anémies microcytaires : Thalassémies
- Diagnostic :

Rita CREIDY, Décembre 2007
- Transfusions depuis l’enfance*Eviter retard staturo-pondéral +++*Prévention hémochromatose (Desferal)
- Splénectomie*Diminution des transfusions
- Transplantation allogénique de moelle*Donneur sur fichier*Sang de cordon
ANEMIES5- Anémies microcytaires : Thalassémies
- Traitement des β-thalassémies :
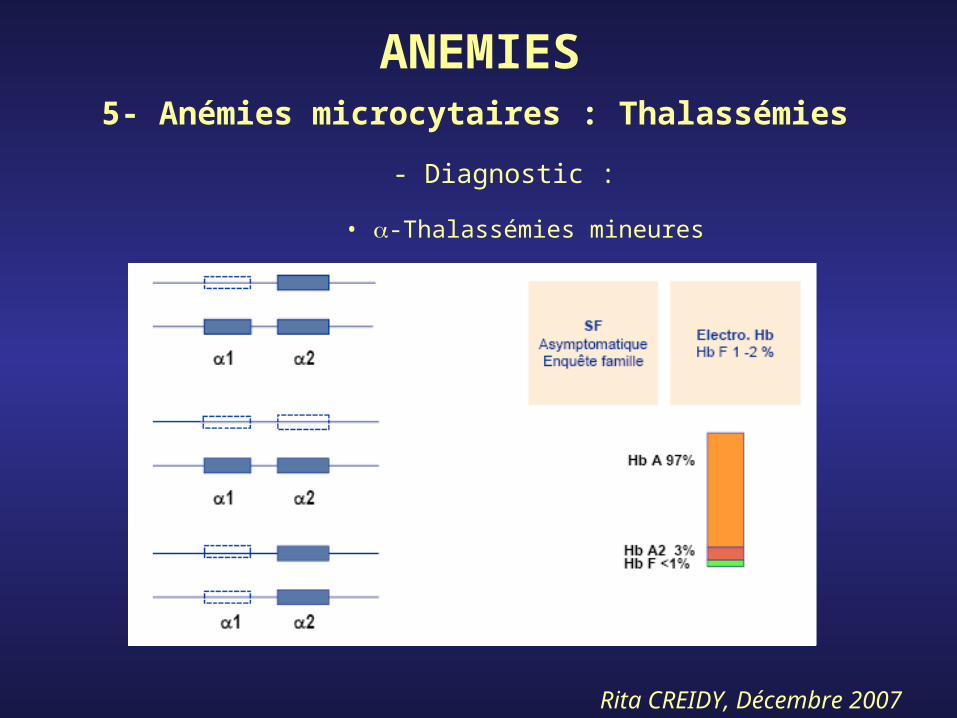
Rita CREIDY, Décembre 2007
• -Thalassémies mineures
ANEMIES5- Anémies microcytaires : Thalassémies
- Diagnostic :

Rita CREIDY, Décembre 2007
• -Thalassémies majeures
ANEMIES5- Anémies microcytaires : Thalassémies
- Diagnostic :
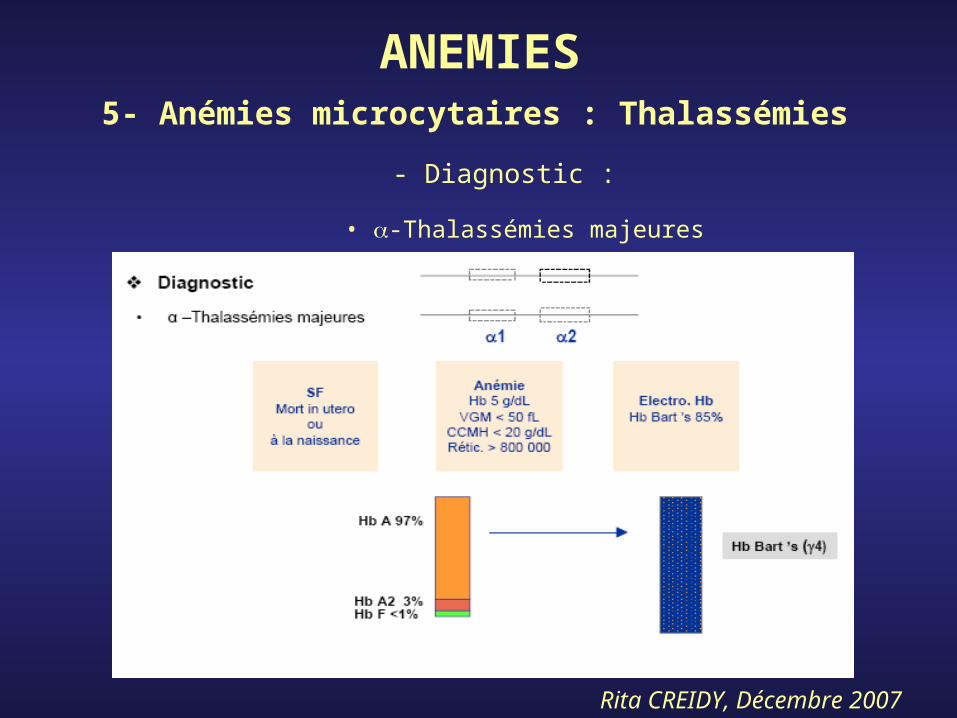
Rita CREIDY, Décembre 2007
• -Thalassémies majeures
ANEMIES5- Anémies microcytaires : Thalassémies
- Diagnostic :

Rita CREIDY, Décembre 2007
- Atteinte prédomine sur GR, mais aussi GB et Plaquettes
- Tableau peut mimer une LA
- Vit.B12 et Folates = vitamines indispensables pour mitoses (duplication ADN)
ANEMIES6- Anémies macrocytaires : carences en
VITB12 et en folates
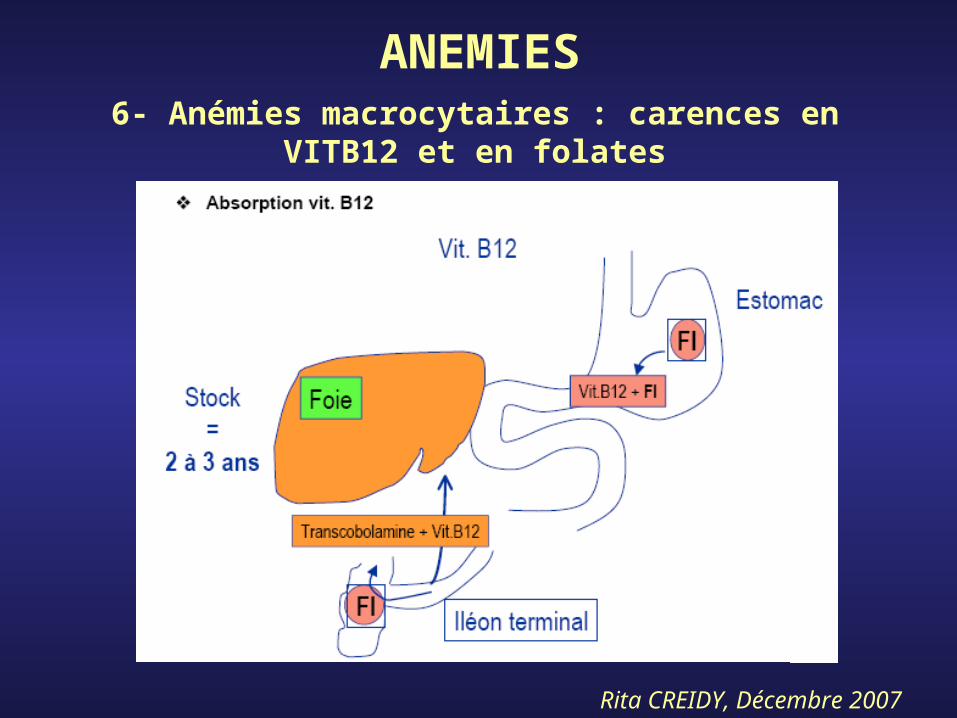
Rita CREIDY, Décembre 2007
ANEMIES6- Anémies macrocytaires : carences en
VITB12 et en folates
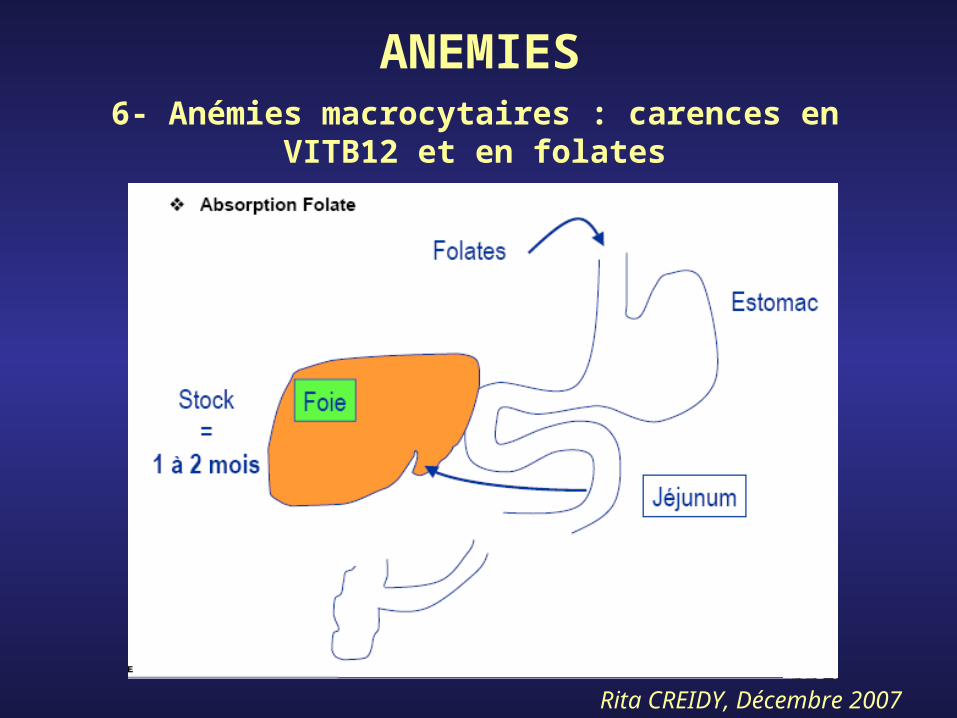
Rita CREIDY, Décembre 2007
ANEMIES6- Anémies macrocytaires : carences en
VITB12 et en folates

Rita CREIDY, Décembre 2007
ANEMIES6- Anémies macrocytaires : carences en
VITB12 et en folates- Diagnostic :

Rita CREIDY, Décembre 2007
ANEMIES6- Anémies macrocytaires : carences en
VITB12 et en folates- Etiologie :

Rita CREIDY, Décembre 2007
ANEMIES6- Anémies macrocytaires : carences en
VITB12 et en folates- Traitement :

Rita CREIDY, Décembre 2007
- Nombreuses étiologiesSvt atteinte des 3 lignées
- MYELOGRAMME indispensable*Σ Myélodysplasiques*Leucémies aiguës*Aplasie médullaire
- 3 causes communes*Alcoolisme*Ins. rénale chronique*Hypothyroïdie
ANEMIES6- Anémies macrocytaires : autres causes
Rita CREIDY, Décembre 2007
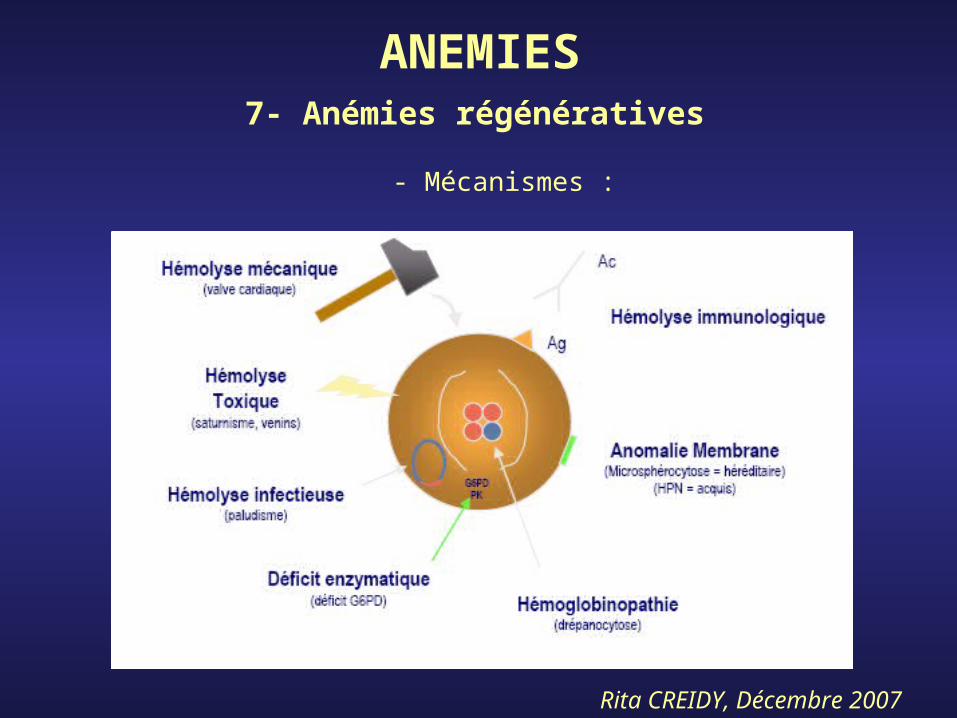
ANEMIES7- Anémies régénératives
- Mécanismes :
Rita CREIDY, Décembre 2007
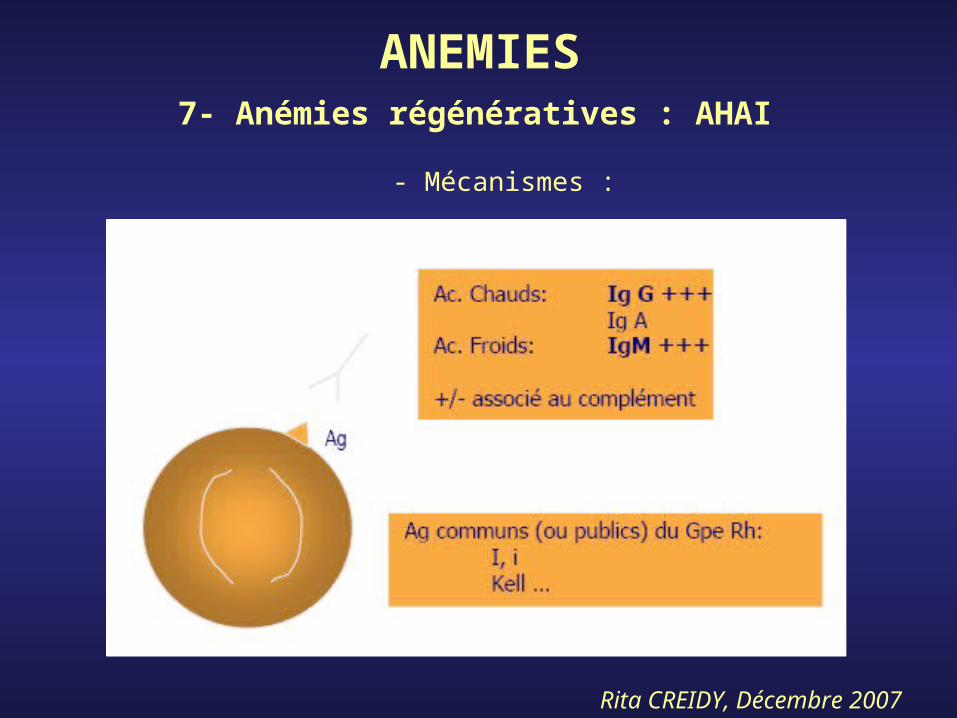
ANEMIES7- Anémies régénératives : AHAI
- Mécanismes :

- Ac. Chauds*SF variable : asthénie, pâleur, urines rouges
porto, ictère, splénomégalie ++*Test Coombs direct : Ac. fixés sur GR, Ig G,
Ig G + C, C isolé
- Ac. Froids*Sujet âgé : chronique, acrocyanose+++,
anémie chronique*Sujet < 5 ans : brutal+++, après inf.
pulmonaire, agitation, douleurs abdominales*Test coombs indirect : Ac. Libres ds plasma
Ig M+++
ANEMIES
- Diagnostic :
7- Anémies régénératives : AHAI
Rita CREIDY, Décembre 2007

- Ac. Chauds60-80% secondaires
*LLC +++, LMNH, Hodgkin, Waldenström
*Lupus +++, PR, Anti-phospholipides, thymome
*a-methylDopa*Grossesse
20- 30% Idiopathiques
- Ac. Froids*Inf. virale : MNI, rhino-
pharyngite, mycoplasme*Mal. Agglutinine
FroideRita CREIDY, Décembre 2007
ANEMIES
- Etiologies :
7- Anémies régénératives : AHAI

- Ac. Chauds*Corticoïdes 1 mg/kg/j 3 sem*Splénectomie*Si échec : Ac Anti-CD20 ?*Immunosuppresseurs* Et… Tt étiologie
- Ac. Froids*Rémission spontanée,*Transfusions*Chaud*Plasmaphérèses*Ac Anti-CD20
Rita CREIDY, Décembre 2007
ANEMIES
- Traitement :
7- Anémies régénératives : AHAI

ANEMIES
- Etiologie :
7- Anémies régénératives : Drépanocytose
- Hémoglobinopathie, sujets noirs
- Mutation Val-Glu sur chaîne β (chr 11)
- Cristallisation de l’Hb si PaO2 diminuée
- Crise hémolytique provoquée par :*Fièvre, infections*Efforts*Altitude
Rita CREIDY, Décembre 2007
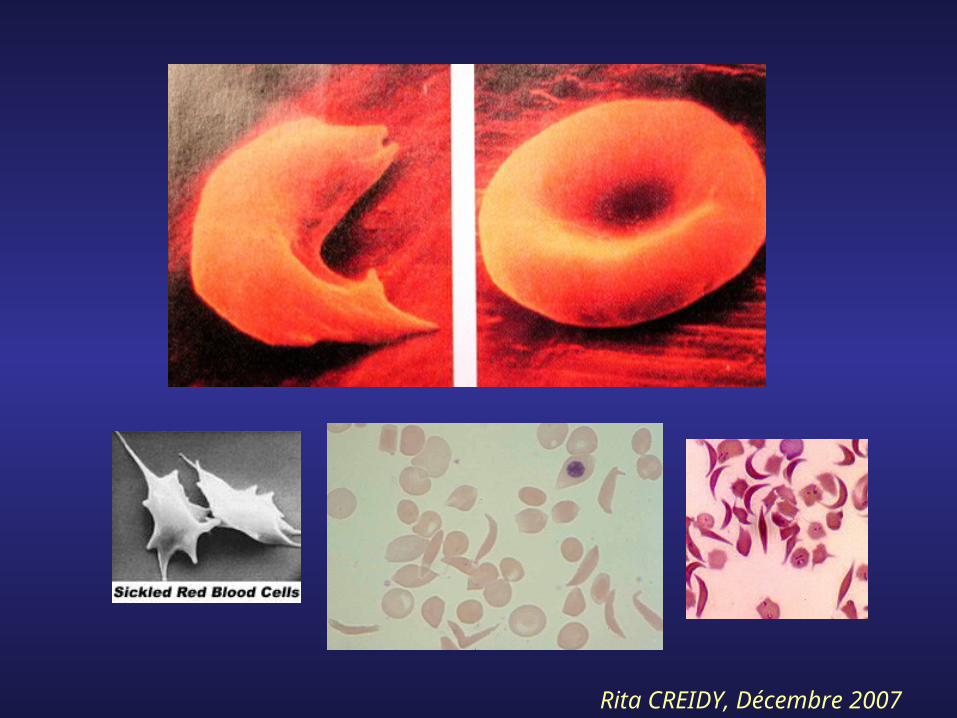
Rita CREIDY, Décembre 2007

Rita CREIDY, Décembre 2007
ANEMIES
- Diagnostic :
7- Anémies régénératives : Drépanocytose

Rita CREIDY, Décembre 2007
ANEMIES
- Diagnostic :
7- Anémies régénératives : Drépanocytose

- Drépanocytose hétérozygoteEnquête famille
- Drépanocytose homozygote*Hyperhydratation +++*Oxygénothérapie*Antibiotiques systématiques (asplénie +
++)*Transfusions*Hydréa*Greffe allogénique fichier
Rita CREIDY, Décembre 2007
ANEMIES
- Traitement :
7- Anémies régénératives : Drépanocytose

- Minkowski-Chauffard
- Fréquente, autosomique dominante *Homozygote non viable in utero
*Hétérozygotes sont atteints
- Pénétrance variable
- Résistance globulaire si diminution hypotonie
- Destruction GR dans rate +++
- VGM 80-100Rita CREIDY, Décembre 2007
ANEMIES
- Généralités :
7- Anémies régénératives : Microsphérocytose héréditaire

Rita CREIDY, Décembre 2007
Purpura thrombopénique immunologique (PTI)

Rita CREIDY, Décembre 2007
PTI
-Thrombopénie acquise, isolée, liée à une destruction périphérique accélérée des PLT de nature autoimmune (présence d’auto-anticorps anti-plaquettes) - Observé surtout chez l'enfant et de manière le plus souvent aiguë, son étiologie est inconnue, possiblement virale car petite prédominance saisonnière (hiver et printemps)
- Chez l'adulte le PTAI est plus souvent chronique, possiblement en rapport avec une maladie auto-immune.
Il s’agit souvent d’un diagnostic d’exclusion des autres étiologies de thrombopénies
1-Définition
Rita CREIDY, Décembre 2007
PTI
- Survient à tout âge, mais plus souvent avant 25 ans et principalement chez l’enfant de 2 – 6 ans L’incidence du PTI de l’enfant est de 4 /100 000 h par an
2.1- PTI aigu
- Le plus souvent chez l’enfant (2 – 6 ans) ou l’adulte jeune.Thrombopénie aiguë, d’apparition brutale: souvent < 20 G/L
- Le début est souvent rapidement progressif, isolé (sans aucun autre symptôme): pas de signes cliniques autres que ceux des manifestations hémorragiques (absence de notion de toxique médicamenteux ou autre, ni altération de l'état général, ni fièvre, ni adénopathie ni splénomégalie). Dans une partie des cas (essentiellement chez le petit enfant) un épisode infectieux de type viral, 2-3 semaines auparavant, est signalé (varicelle, EBV, non spécifique)
2-Présentation clinique et physiopathologie

Rita CREIDY, Décembre 2007
PTI
- Les manifestations hémorragiques ne sont pas constantes, souvent modérées, avec cependant 1 – 2 % d’hémorragies intra crâniennes chez l’enfant (plus nombreuses dans le PTAI aigu de l’adulte)
- Résolution spontanée dans plus de la moitié des cas chez l’enfant, en général en 4 – 6 semaines. Si la durée dépasse 6 mois malgré un traitement, on définit le PTAI chronique (environ 20% des enfants)
2-Présentation clinique et physiopathologie

Rita CREIDY, Décembre 2007
PTI2-Présentation clinique et physiopathologie
2.2- PTI chronique
- Forme plus fréquemment retrouvée chez l’adulte (H/F= 1/3)
- La thrombopénie est plus modérée: 50 -80 G/L, moins fréquemment hémorragique que la forme aiguë (souvent histoire prolongée d’incidents hémorragiques modérés), et souvent beaucoup plus insidieuse que dans la forme aiguë; évolution par poussées
- Les antécédents d’infection sont rares
- Les rémissions spontanées sont rares

PTI2-Présentation clinique et physiopathologie
2.3- Manifestations hémorragiques
- Purpura cutané (80% des cas): pétéchial, ecchymotique (ecchymoses spontanées ou à la suite de traumatismes minimes), ou en vibices (linéaire aux plis de flexion).
- Hémorragies muqueuses chez 3-5% des enfants et 40% des adultes (épistaxis, gingivorragies, ménométrorragies); hémorragies viscérales: plus rares mais plus graves.
- L'examen du fond de l'oeil doit être systématique: il peut montrer des hémorragies rétiniennes faisant craindre la survenue d'une hémorragie cérébro-méningée (surtout plaquettes < 20 G/l et sujet âgé)Sont à haut risque : thrombopénie < 10 G/L
âge > 60 ansautre maladie hémorragique
Rita CREIDY, Décembre 2007
PTI2-Présentation clinique et physiopathologie
- Des auto Ac anti PLT sont retrouvés dans plus de 75% des PTI, dirigés essentiellement contre deux Ag de surface des PLT: la glycoprotéine Gp IIb/IIIa (plusieurs épitopes différents sont impliqués) et la Gp Ib/IX. Ils sont de nature IgG ou IgA, parfois IgM. Ces auto Ac se fixent aux PLT, et leur fragment Fc est reconnu par les récepteurs du fragment Fc des Ig de la membrane des macrophages; la phagocytose des PLT sensibilisées a lieu essentiellement dans la rate, et plus rarement du foie.
- Les PLT sont rapidement éliminées du plasma (durée de vie raccourcie, limitée à quelques minutes ou au maximum 2-3 heures).
- La production médullaire est en général augmentée (augmentation de la masse MK totale avec augmentation du nombre des MK, de leur ploïdie et de leur taille), mais pas toujours.

Rita CREIDY, Décembre 2007
PTI3- Diagnostic biologique
3.1- Hémogramme
- Thrombopénie isolée, sévère (< 10 G/L chez la moitié des enfants), parfois modérée (50 – 80 G/L)
*Volume moyen plaquettaire (VMP): souvent augmenté, reflet du nombre important de PLT jeunes
- Leucocytes: parfois de petites modifications
- Hémoglobine: normale

PTI3- Diagnostic biologique
Rita CREIDY, Décembre 2007
3.2- Myélogramme
- Moelle de richesse normale
-Nombreux mégacaryocytes (surtout dans le PTI aigu de l’enfant)
- Chez l’adulte, la richesse en MK est le plus souvent normale ou à peine augmentée.
► En l’absence de signes de myélodysplasie, un nombre normal ou augmenté de MK au cours d’une thrombopénie évoque en premier lieu un mécanisme périphérique

PTI3- Diagnostic biologique
Rita CREIDY, Décembre 2007
3.3- Autres
- Recherche des Ac anti PLT : Test de coombs plaquettaire
- Recherche des PLT réticulées par CMF

PTI3- Traitement
Rita CREIDY, Décembre 2007
- Corticoïdes : 1 mg/kg/j 24 à 48h
- IGIV : 0.4 g/kg/j pendant 5 jours
- Si rechute :*Splénectomie : 2/3 efficacité*Autres immunosupresseurs,
androgènes…













![[ RELAIS D’ASSISTANTES MATERNELLES ]€¦ · Ville du Kremlin-Bicêtre – Observatoire des Engagements – 07/10/2010 4/8 - 161 matinées d’accueils-jeux - 576 adultes et 1235](https://static.fdocuments.fr/doc/165x107/6104427ab956dc76b27303b6/-relais-daassistantes-maternelles-ville-du-kremlin-bictre-a-observatoire.jpg)




