

Resume de Thermodynamique I -...
48
R´ esum´ e de Thermodynamique I jean-eloi.lombard@epfl.ch 17 juillet 2007
Transcript of Resume de Thermodynamique I -...

Table des matieres
1 Developpement d’une intuition 31.1 Temperature et entropie . . . . . . . . . . . . . . . . . . . . . 3
1.1.1 Chaleur . . . . . . . . . . . . . . . . . . . . . . . . . . 31.1.2 Entropie . . . . . . . . . . . . . . . . . . . . . . . . . . 31.1.3 Temperature . . . . . . . . . . . . . . . . . . . . . . . 31.1.4 Puissance thermique . . . . . . . . . . . . . . . . . . . 4
1.2 Machine thermique . . . . . . . . . . . . . . . . . . . . . . . . 41.2.1 Principe . . . . . . . . . . . . . . . . . . . . . . . . . . 41.2.2 Pompe a chaleur . . . . . . . . . . . . . . . . . . . . . 51.2.3 Efficacite . . . . . . . . . . . . . . . . . . . . . . . . . 51.2.4 L’effet de la dissipation interne sur l’efficacite . . . . . 6
1.3 Thermomecanique du Gaz Parfait . . . . . . . . . . . . . . . 61.3.1 Cycles . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.3.2 Processus Adiabatique . . . . . . . . . . . . . . . . . . 71.3.3 Processus Isotherme . . . . . . . . . . . . . . . . . . . 81.3.4 Processus Isochore . . . . . . . . . . . . . . . . . . . . 9
1.4 Reponse a l’echauffement : Coefficients Calorimetriques . . . 91.4.1 Definitions . . . . . . . . . . . . . . . . . . . . . . . . 91.4.2 Pour le gaz parfait . . . . . . . . . . . . . . . . . . . . 10
1.5 Reponse d’un Gaz Parfait a un apport d’entropie . . . . . . . 11
2 Formalisation 122.1 Thermocinetique . . . . . . . . . . . . . . . . . . . . . . . . . 12
2.1.1 Variable d’etat . . . . . . . . . . . . . . . . . . . . . . 122.1.2 Fonctions Fondamentales . . . . . . . . . . . . . . . . 122.1.3 Equation d’etat . . . . . . . . . . . . . . . . . . . . . . 132.1.4 Premier Principe . . . . . . . . . . . . . . . . . . . . . 142.1.5 Deuxieme principe . . . . . . . . . . . . . . . . . . . . 142.1.6 Relation d’Euler . . . . . . . . . . . . . . . . . . . . . 14
1

TABLE DES MATIERES 2
2.1.7 Relation de Gibbs-Duhem . . . . . . . . . . . . . . . . 152.1.8 Equilibre . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.2 Fonctions thermodynamiques et equilibre . . . . . . . . . . . 192.2.1 Equilibre Therminque . . . . . . . . . . . . . . . . . . 192.2.2 Equilibre Mecanique . . . . . . . . . . . . . . . . . . . 212.2.3 Equilibre de Flot de Matiere . . . . . . . . . . . . . . 222.2.4 Transformation de Legendre . . . . . . . . . . . . . . . 232.2.5 Relations de Maxwell . . . . . . . . . . . . . . . . . . 242.2.6 Introduction . . . . . . . . . . . . . . . . . . . . . . . 252.2.7 Enthalpie . . . . . . . . . . . . . . . . . . . . . . . . . 252.2.8 Energie libre ou l’energie de Helmoltz . . . . . . . . . 252.2.9 Energie libre de Gibbs . . . . . . . . . . . . . . . . . . 252.2.10 Reservoir thermique et Principe du minimum du po-
tentiel de Helmholtz . . . . . . . . . . . . . . . . . . . 262.2.11 Reservoir de pression et Principe du minimum de l’en-
thalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.2.12 Reservoir thermique et de pression et Principe du mi-
nimum de l’energie de Gibbs . . . . . . . . . . . . . . 282.2.13 Principe du minimum general d’une transformation de
Legendre de l’energie interne du systeme . . . . . . . . 282.3 Transformation de substance . . . . . . . . . . . . . . . . . . 28
2.3.1 Osmose . . . . . . . . . . . . . . . . . . . . . . . . . . 292.4 Transition de Phase du 1er ordre . . . . . . . . . . . . . . . . 32
2.4.1 Changement de Phase . . . . . . . . . . . . . . . . . . 322.4.2 Attributs generaux d’une transition de phase du 1er
ordre . . . . . . . . . . . . . . . . . . . . . . . . . . . . 352.4.3 Regle de changement de phase de Gibbs . . . . . . . . 352.4.4 Exemple . . . . . . . . . . . . . . . . . . . . . . . . . . 372.4.5 Relation de Clapeyron . . . . . . . . . . . . . . . . . . 39
2.5 Thermocinetique des processus irreversibles . . . . . . . . . . 412.5.1 Effet Seebeck . . . . . . . . . . . . . . . . . . . . . . . 412.5.2 Effet Peltier . . . . . . . . . . . . . . . . . . . . . . . . 432.5.3 Loi de Fourier . . . . . . . . . . . . . . . . . . . . . . . 432.5.4 Loi de Fick . . . . . . . . . . . . . . . . . . . . . . . . 43
3 Annexes 463.1 Divers, conversions et constantes . . . . . . . . . . . . . . . . 46

Chapitre 1
Developpement d’uneintuition
1.1 Temperature et entropie
1.1.1 Chaleur
Definition La chaleur, notee Q, est l’energie associee a la valeur ou auchangement de la dynamique d’un systeme compose de corps microscopiquesde par l’agissement de forces diverses (autres que mecaniques donc !).
1.1.2 Entropie
Definition L’entropie est une quantite contenue dans un corps, qui peuts’ecouler d’un corps vers un autre. Elle peut etre cree (dans tout procedeirreversible) mais ne peut pas etre detruite. L’entropie vehicule l’energiedans les procede thermiques.
1.1.3 Temperature
Definition La temperature est le potentiel associe au flux d’entropie. Paranalogie avec une chute d’eau, ou la masse correspond a l’entropie, la temperaturecorrespond a la hauteur de la chute d’eau. Ainsi c’est une difference de “po-tentiel”, de temperature, qui est la cause d’un flux d’entropie.
3

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 4
1.1.4 Puissance thermique
La puissance thermique associee au flux d’entropie entre une sourcechaude (a la temperature T+) et une source froide (T−) est donnee par :
Pthermique = (T+ − T−)|Is|
Le flux d’energie thermique lors d’un transfert par conduction est donnepar :
IE,thermique = TIS /IS =dS
dt
Ainsi la quantite de chaleur echangee par conduction entre un temps ti initialet tf final :
Q =∫ tf
ti
TISdt /IS =dS
dt
1.2 Machine thermique
[2] (pp. 113, 125 pour le COP, pp. 91, 113 pour le principe de la machinethermique, pp. 309 pour la production d’entropie dans des phenomenes dis-sipatifs) Carnot est le premier a avoir remaque le liens entre entropie estenergie dans des phenomenes thermiques alors qu’il travaillait sur le travailque pouvait fournir ue machine thermique.
1.2.1 Principe
La chaleur peut travailler ! Pour une machine thermique, il faut :
1. un milieu a temperature elevee et un milieu a temperature basse. Dela chaleur est absorbee dans le milieu chaud et emise dans le milieufroid.
2. le travail que fournit un tel moteur depend de la difference de temperatureentre le milieu chaud et le froid.
Ainsi la puissance thermique d’une machine de Carnot est donnee par :
Pth = −(T− − T+)|IS |= (T+ − T−)|IS | (1.1)
Dans le cas d’une machine thermique ideale et en prenant comme agent l’air,nous avons :

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 5
1. expansion isothermique (l’air se dilate a temperature constante en ab-sorbant de l’entropie)
2. maintenant il faut baisser la temperature de l’air au niveau du milieuplus froid (donc on dilate adiabatiquement l’air)
3. on garde la temperature constante tout en evacuant de l’entropie aumilieu le plus froid (donc une compression isotherme)
4. puis on remonte la temperature de l’air (grace a une compression adia-batique) pour pouvoir recomencer le cycle.
1.2.2 Pompe a chaleur
Une pompe a chaleur n’est autre qu’une machine thermique operee a l’en-vers. En d’autre termes, de l’energie mecanique lui est fournie pour qu’ellepuisse extraire de la chaleur d’un milieu froid vers un milieu chaud (donccontre le potentiel thermique negatif, la temperature).
1.2.3 Efficacite
Nous avons vu deux manieres de calculer l’efficacite d’une machine ther-mique :
Efficacite de Carnot Cette seconde efficacite calcul le rapport entre leflux d’energie thermique entrant et la puissance mechanique disponible, soit :
η1 =∣∣∣∣ Pmech.IE.,th.,in
∣∣∣∣=
∣∣∣∣(T+ − T−)IST+IS
∣∣∣∣=
T+ − T−
T+
Evidement le rendement n’est de 100 % que si la temperature la plus basseest de 0[K] ! !
Efficacite 2 En definissant l’efficacite de la machine thermique comme lafraction de puissance thermique effectivement disponible sous forme mecha-nique, soit :
η2 =∣∣∣∣ PmechPtherm.
∣∣∣∣=
∣∣∣∣ Pmech(T+ − T−)|IS
∣∣∣∣

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 6
Dans le cas de machines ideales de Carnot, l’efficacite est egale a 1.Dans le cas d’une pompe a chaleur le coefficient de performance η est definiepar :
η =Ptherm.Pmec.
=|Qc||W |
=T−
T+ − T−
1.2.4 L’effet de la dissipation interne sur l’efficacite
La puissance disponible est donnee par :
Pdisp. = −(T − T0)IS(T )
et la puissance mecanique est donnee par :
Pmec. = TIS(T )− T0IS(T0)
or, la production d’entropie est donnee par :
ΠS = IS(T ) + IS(T0)
donc :
Pmec. = (T − T0)IS(T ) + T0ΠS
−(T − T0)IS(T ) est la puissance disponible et donc T0ΠS est la perte. Nouspouvons donc definir l’efficacite (selon la second loi), par :
η2 = −Pmec.Pdisp
=(T − T0)IS(T ) + T0ΠS
−(T − T0)IS(T )
= 1− T0ΠS
(T − T0)|IS(T )|
1.3 Thermomecanique du Gaz Parfait
Le gaz parfait est defini par les deux equations d’etat suivante :
PV = nRT E = cnRT
avec P [atm] la pression, V [L] le volume, n[mole] le nombre de moles de gaz,R = 8.314[J.Mole−1.K−1] la constante des gazs parfaits, T [K] la temperaturedu gaz.

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 7
1.3.1 Cycles
1. sur un cycle la variation totale d’energie interne comme la variationtotale d’entropie est nulle.
2. dans un diagramme PV l’aire du cycle correspond au travail fournitpar le cycle. S’il est parcourut dans le sens des aiguilles d’une montrele cycle fournit du travail vers l’exterieure.
1.3.2 Processus Adiabatique
Un procesus adiabatique est un processus pendant lequel le flux d’entro-pie du systeme considere vers l’environement est nul, soit :
IS = 0 ∀t
donc Q = 0.Nous avons les relations suivantes :
PV γ = cst.
avec γ = 1 + 1c ou c est le nombre de degres de libertes divises par deux de
la molecule constituant le gaz. On cherche la fonction P(V) dans le cas d’unprocessus adiabatique.
Demonstration de PV γ = cst.
Prenons l’equation fondamentale dans sa forme energetique, soit :
dU = −PdV (1.2)
Dans le cas des gas parfait, nous avons :
E = cnRT
⇒ dE = cnRdT (1.3)
De Eq. 1.2 et de l’Eq. 1.3, nous avons :
cnRdT = −PdV (1.4)
or
PV = nRT
⇒ V dP + PdV = nRdT

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 8
donc 1.4 devient :
c(V dP + PdV ) = −PdV⇔ (c+ 1)PdV = −cV dP
⇔ c+ 1−c
dV
V=dP
P
en integrant, d’une part par rapport a V et de l’autre par rapport a P, nousobtenons :
c+ 1−c
∫dV
V=
∫dP
P
⇔ c+ 1−c
ln(V ) + cst = ln(P ) + cst
⇔ (1 +1c)ln(PV ) = cst
⇔ ln(PV 1+ 1c ) = cst
⇒ PV 1+ 1c = cst
Demonstration de T2T1
=(V1V2
)γ−1Le processus est adiabatique, donc
δQ = 0 et dU = −PdV , or dU = CV dT , donc
dT
T= −RdV
CV V
de plus RCV
= γ − 1, donc :
dT
T= −(γ − 1)
dV
V
ce qui en integrant, donne :
ln
(T2
T1
)= (γ − 1)ln
(V1
V2
)⇔ T2
T1=
(V1
V2
)γ−1
1.3.3 Processus Isotherme
Isotherme : du grec “isos” = meme et “therme” = temperature.Processus thermique a temperature constante.

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 9
Le flux d’energie mecanique lors dans un processus de dilatation/compressiond’un gaz est donne par :
IE,mec = −PV
⇔ Wi,f = −∫ Vf
Vi
nRT
VdV
⇔ Wi,f = −nRT ln(VfVi
)Pour un systeme a l’etat stationnaire, la Ire loi, nous avons :
0 = IE,th + IE,mec.
= T∆S − nRTln(VfVi
)ce qui nous donne :
∆S = nRln
(VfVi
)1.3.4 Processus Isochore
Lors d’un processus pendant lequel le volume est garde constant, si del’entropie est amenee au systeme, sa temperature doit augmenter. La capa-cite thermique est la grandeur qui lie la variation de temperature du systemeen fonction de l’entropie apportee. La capacite entropique K d’un systemerigide uniforme est defini comme le rapport entre la variation de la quantited’entropie dans le systeme et la variation de temperature, soit :
K(T ) =dS
dT⇔ dS = K(T )dT⇔ S = KT
1.4 Reponse a l’echauffement : Coefficients Calo-rimetriques
1.4.1 Definitions
Les divers coefficients calorimetriques sont donnes par les derivees desgrandeurs extensives par rapport aux grandeurs intensives.

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 10
Coefficient d’expansion thermique
Definit, a pression P constante par :
α :=1V
(∂V
∂T
)P
avec V le volume et T la temperture du corps.
La chaleur specifique a pression constante
Definie par :
CP := T
(dS
dT
)P
1.4.2 Pour le gaz parfait
CV = 32R pour un gaz mono-atomique et 5
2R pour un gaz di-atomique.[1] pp. 43-45 En general on ne connait pas l’equation fondamentale d’unsysteme. Toutefois dans le cas d’un gaz parfait, S=S(V, T, N) est donnepar :
S(V, T, n) = ncRln
(T
T0
)+ nRln
(V
V0
)+ S0
=32nRln
(U
U0
)+ nRln
(V
V0
)− 5
2nRln
(N
N0
)+ S0
et les deux equations d’etat du gaz sont :
PV = nRT et E = cnRT (1.5)
et pour un processus thermique a volume constant, nous avons :
E = CV T (1.6)
de 1.5 et 1.6, nous avons donc :
CV = cnR
Entropie latente
L’entropie latente lie la variation d’entropie a celle de volume, pour unetemperature constante :
ΛV =∂S
∂V T

CHAPITRE 1. DEVELOPPEMENT D’UNE INTUITION 11
1.5 Reponse d’un Gaz Parfait a un apport d’en-tropie
Lorsqu’un gaz est chauffe son volume et sa temperature augmente, sui-vant l’equation :
S = ΛV V +KV T
comme l’a constate J. Ivory au debut du XIXe siecle, avec KV l’entropielatente a temperature constante et ΛV l’entropie latente.

Chapitre 2
Formalisation
2.1 Thermocinetique
2.1.1 Variable d’etat
Les variables d’etats sont des variables du systeme etudie (S, V,N, T, P, µ)qui permettent de caracteriser l’etat du systeme.
2.1.2 Fonctions Fondamentales
Fonction Fondamentale Energetique La fonction fondamentale d’unsysteme thermodynamique est la fonction :
U = U(S, V,N1, . . . , Nr)
et la premiere differentielle totale exacte est donnee par :
dU =(∂U
∂S
)V,N1,...,Nr
dS −(∂U
∂V
)S,N1,...,Nr
dV +r∑j=1
(∂U
∂Nj
)S,V,...,Nr
dNj
Les derivees partielle qui apparaissent dans cette equations sont appelees lesvariables/parametres intensives(ifs) du systeme et son nommees :
1. Temperature (∂U
∂S
)V,N1,...,Nr
:= T
12

CHAPITRE 2. FORMALISATION 13
2. Pression (∂U
∂V
)S,N1,...,Nr
:= P
3. Potentiel electrochimique du je composant(∂U
∂Nj
)S,V,...,Nr
:= µj
ce qui nous donne :
dU = TdS − PdV + µ1dN1 + . . .+ µrdNr
en identifiant les termes de cette equation, nous avons :
1. TdS = IE,th = dQ le flux d’entropie
2. −PdV = IE,mec = dWmec le flux d’energie mecanique, ou le travail
3. µrdNr = dWC le “travail Chimique”
d’ou 3.24 devient :
dU = dQ+ dWmec + dWchim
Fonction Fondamentale Entropique De meme, il est possible de definirla fonction fondamentale entropique par :
S = S(U, V,N1, . . . , Nr)
Astuce
Lorsqu’on a les equations d’etat, il suffit de verifier qu’elles sont ho-mogenes d’ordre 0 pour verifier que les grandeurs associees sont intensives.Le fait que les equations d’etat sont homogenes a l’ordre 0 est equivalent al’equation fondamentale du systeme est homogene d’ordre 1. Ceci decouledu fait que les equations d’etat sont des derivees partielles d’ordre 1 de lafonction fondamentale.
2.1.3 Equation d’etat
Une equation d’etat exprime une variable intensive en fonction des va-riables extensives independante du systeme.La connaissance de toute les equations d’etat est equivalent a la connais-sance de l’equation fondamentale du systeme considere.

CHAPITRE 2. FORMALISATION 14
Une equation d’etat est une equation qui lie l’ensemble des variables d’etat,par exemple T(V,N,S) est une equation d’etat qui caracterise la variationde temperature du systeme en fonction du volume, du nombre de moles dela substance composant le systeme et de l’entropie du systeme.
2.1.4 Premier Principe
[3] (First Law of Thermodynamics or Law of Balance of Energy pp. 221-229)Pour tout systeme, il existe une fonction d’etat extensive, l’energie du systemeE (ou U dependament des auteurs). Cette grandeur extensive, peut s’ecoulerd’un corps a un autre, mais elle est conservee, ce qui signifie que la seulemaniere de faire varier le contenu energetique d’un systeme est suite a unflot d’energie mecanique, thermique, electrique ou autre. Si le systeme estisole, alors l’energie est constante :
E + IE,net = 0IE,net = IE,therm. + IE,mec. + IE,el. + . . .
Remarque L’energie est definie a une constante pres !
2.1.5 Deuxieme principe
[3] (Second Law or law of balance of entropy pp. 109-119)Pour tout systeme, il existe une fonction d’etat extensive S : l’entropie dusysteme, qui satisfait les deux conditions suivantes :
1. Principe d’evolution : si le systeme est adiabatiquement ferme, S estune fonction monotone non decroissante du temps.
S(t) =dS
dt= ΠS ≥ 0
2. Principe d’equilibre : si le systeme est isole, S tend vers un maximumfini dans le futur lointain. Ce principe est equivalent a : pour toutsysteme isole, l’energie tend vers un minimum dans un futur lointain.
2.1.6 Relation d’Euler
L’entropie d’un systeme est la somme des entropie des sous-systemes quila compose :
S =∑α
S(α)

CHAPITRE 2. FORMALISATION 15
et l’entropie de chaque sous-systeme n’est que fonction des variables exten-sives du sous-systeme donne.
S(α) = S(α)(Uα, V (α), N(α)1 , . . . , N (α)
n )
L’additivite de l’entropie pour des sous-systemes independants impliquequ’en multipliant toute les variables extensives du systeme par un facteur λl’entropie du systeme totale est aussi multipliee par λ, soit :
S(λU, λV, λN1, . . . , λNm) = λS(U, V,N1, . . . , Nm)
L’equation fondamentale est donc homogene de premier ordre. En derivantcette equation par rapport a λ, nous obtenons :
∂U(. . . , λXk, . . . , )∂(λS)
∂(λS)∂λ
+∂U(. . . , λXj , . . . , )
∂(λXj)∂(λXj)∂λ
+ . . .
= U(S,X1, . . . , Xn)
⇔ ∂U(. . . , λXk, . . . , )∂(λS)
∂(λS)∂λ
+n∑i=1
∂U(. . . , λXi, . . . , )∂(λXi)
∂(λXi)∂λ
= U(S,X1, . . . , Xn)
En particulier, on peut prendre λ = 1, soit :
∂U
∂SS +
n∑i=1
∂U
∂XiXi = U
⇔ U = TS +n∑i=1
PiXi
soit, pour un systeme simple :
U = TS − PV + µ1N1 + . . . µnNn (2.1)
L’equation 2.1 est dite Relation d’Euler
2.1.7 Relation de Gibbs-Duhem
La differentielle totale exacte de 2.1 est donnee par :
dU = TdS + SdT +n∑i=1
PidXi +n∑i=1
dPiXi

CHAPITRE 2. FORMALISATION 16
or :
dU = TdS +n∑i=1
PidXi
et par soustraction, nous obtenons :
0 = SdT +n∑i=1
XidPi
C’est la relation de Gibbs-Duhem. Dans le cas d’un systeme simple, nousobtenons, par exemple :
0 = SdT − V dP +Ndµ
Nous voyons clairement, grace a cet exemple, que la relation de Gibbs-Duhem lie les differentes grandeurs intensives du systeme. Le nombre degrandeurs intensives qui varie de maniere independante est appele le nombrede degre thermodynamique de liberte. Un systeme simple a n composants an+ 1 degres de liberte.
2.1.8 Equilibre
[2] chp. 8Nous avons vu le principe d’extremum de la thermodynamique qui im-plique dS = 0 et en particulier d2S < 0, ce qui signifie que l’extremumest un maximum. Prenons deux systemes identiques d’equations fondamen-tale S = S(U, V,N) separe par une paroi qui ne permet aucun flux. Enprenant ∆U du premier systeme pour le mettre dans le second, nous avonsS(U+∆U, V,N) dans le premier et S(U−∆U, V,N) dans le second. Avec larelation entre entropie et energie comme dans la figure (2.1), l’entropie aug-mente assymptotiquement ce qui est instable et entretiens le flux d’energieinterne du systeme (1) vers le (2). Une telle perte d’homogeneite entre deuxsystemes est caracteristique d’un changement de phase.
En inspectant la Fig. 2.1 il vient que la stabilite du systeme depend dela concavite de l’entropie :
S(U + ∆U, V,N) + S(U −∆U, V,N) ≤ 2S(U, V,N) (∀∆)
ou encore la forme differentielle (donc moins globale)(∂2S
∂U2
)V,N
≤ 0 (2.2)
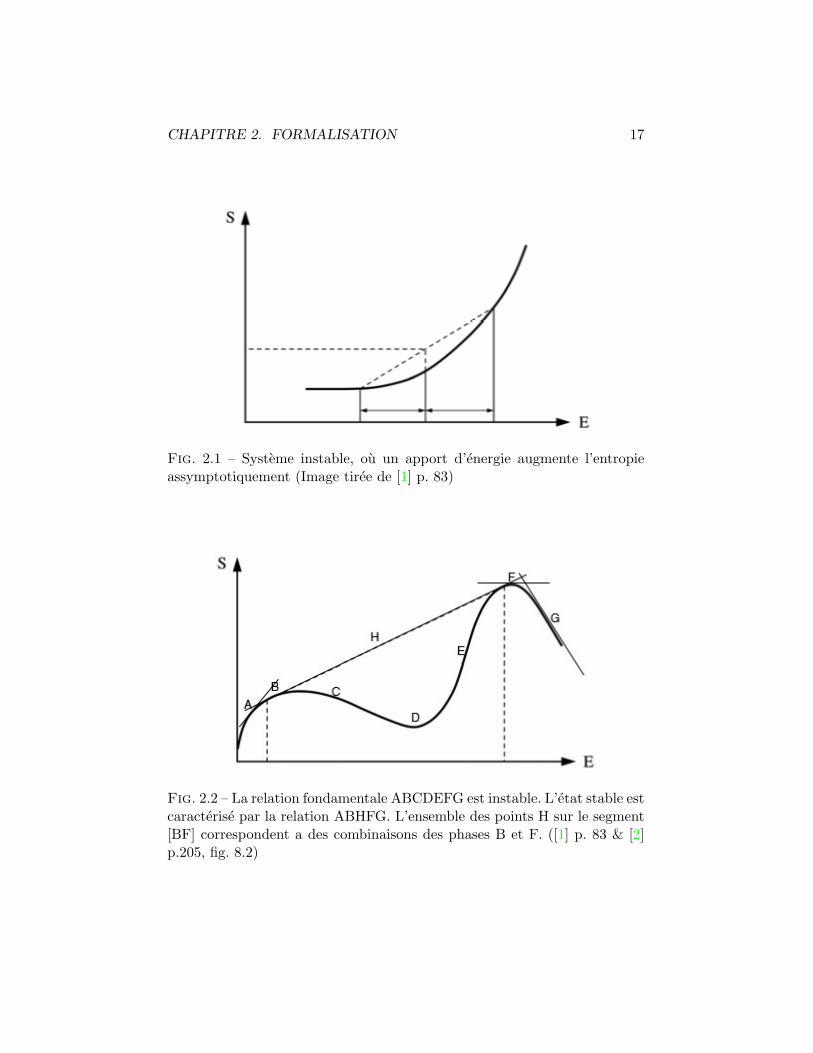
CHAPITRE 2. FORMALISATION 17
Fig. 2.1 – Systeme instable, ou un apport d’energie augmente l’entropieassymptotiquement (Image tiree de [1] p. 83)
Fig. 2.2 – La relation fondamentale ABCDEFG est instable. L’etat stable estcaracterise par la relation ABHFG. L’ensemble des points H sur le segment[BF] correspondent a des combinaisons des phases B et F. ([1] p. 83 & [2]p.205, fig. 8.2)

CHAPITRE 2. FORMALISATION 18
Suite a des mesures, il est possible d’obtenir une relation comme cellede la Fig. 2.2, mais la relation fondamentale associes a cette fonction estl’enveloppe des tangentes superieurs a la courbe, comme represente dans lafigure. La portion CDE viole la regle 2.2 alors que BCDEF viole la loi condi-tion plus generale 2.1.8 En travaillant dans un espace thermodynamique a3 dimensions (U, S, V), nous avons la conditions generale :
S(U + ∆U, V + ∆V,N) + S(U −∆U, V −∆V,N) ≤ 2S(U, V,N) ∀∆
qui se traduit ponctuellement par les trois differentielles partielle suivantes :
1. (∂2S
∂U2
)V,N
≤ 0
2. (∂2S
∂V 2
)U,N
≤ 0
3.
∂2S
∂U2
∂2S
∂V 2−
(∂2S
∂U∂V
)2
≥ 0 (2.3)
Il est facile de reformuler tout ce formalisme dans une representation energetique.La condition de concavite de l’entropie devient une condition de convexitepour l’energie, soit :
U(S + ∆S, V + ∆V,N) + U(S −∆S, V −∆V,N) ≥ 2U(S, V,N)
et les conditions de convexite ponctuelle deviennent :
∂2U
∂S2=∂T
∂S≥ 0
∂2U
∂V 2= −∂P
∂V≥ 0
et la condition sur la variation commune de S et V devient :
∂2U
∂S2
∂2U
∂V 2−
(∂2U
∂S∂V
)≥ 0
Les proprietes des transformations de Legendre sont :
P =∂U
∂Xet X = −∂U [P ]
∂P

CHAPITRE 2. FORMALISATION 19
donc
∂X
∂P= −∂
2U [P ]∂P 2
=1∂2U∂X2
donc
signe
(∂2U [P ]∂P 2
)= −signe
(∂2U
∂X2
)si U est une fonction convexe de X alors U[P] est une fonction concave de P.Le potentiel de Helmholtz est donc une fonction concave de la temperatureet une fonction convexe du volume. L’enthalpie est une fonction convexe del’entropie et une fonction concave de la pression. Finalement, l’energie librede Gibbs est une fonction concave de la temperature et de la pression.En resume, pour un nombre N constant, l’equation fondamentale en representationenergetique et ses trasnformee de Legendre sont des fonctions convexe deleurs grandeurs extensives et concaves de leurs grandeurs intensives.
2.2 Fonctions thermodynamiques et equilibre
2.2.1 Equilibre Therminque
Considerons un systeme isole compose de deux sous-systemes separes parune parois rigide, impermeable a la matiere mais qui laisse passer un fluxd’entropie. Le volume, et le nombre de mole de chaque sous-systeme est fixe,mais les energies E1 et E2 sont libres de changer, avec la restriction :
E(1) + E(2) = cst.
La deuxieme loi, nous dit que l’equilibre est atteint lorsque la energies sonttels qu’elles maximisent l’entropie des deux sous-systemes. L’additivite del’entropie des deux sous-systemes nous donne :
S = S(1)(U (1), V (1), N (1), . . . , N(1)j ) + S(2)(U (2), V (2), N (2), . . . , N
(2)k )
ce qui nous donne :
dS =(∂S(1)
∂U (1)
)V (1),N
(1)1 ,...,N
(1)j
dU (1) +(∂S(2)
∂U (2)
)V (2),N
(2)1 ,...,N
(2)k
dU (2)
=1T (1)
dU (1) +1T (2)
dU (2)

CHAPITRE 2. FORMALISATION 20
D’apres le second principe, nous avons :
dS = 0
comme condition d’equilibre, soit :
0 =1T (1)
dU (1) +1T (2)
dU (2)
or
dU (1) = −dU (2)
d’ou :
0 =1T (1)
dU (1) − 1T (2)
dU (1)
⇔ 0 =1T (1)
− 1T (2)
⇔ 1T (1)
=1T (2)
⇔ T (1) = T (2)
car dU n’est pas necessairement nul. Donc dans le cas de deux systemessepares par une cloison qui ne laisse passer que de la chaleur, l’equilibreest atteint lorsque la temperature est les deux temperatures sont egales. Enrevenant a l’analogie du flux thermique avec le flux d’eau dans une centralehydraulique, le transfert d’entropie s’arrete lorsque la difference de potentielsassocies (la temperature) est nulle, ce qui se traduirait par une difference dehauteur nulle. C’est le principe des vases communiquant.Supposons maintenant le meme systeme, mais hors-equilibre avec T (1) >T (2). Lorsque que l’on laisse passer un flux de chaleur, l’entropie va aug-menter jusqu’a atteindre un nouvelle equilibre avec les meme temperaturesdans les deux sous-systemes, comme demontre ci-dessus. L’entropie totaledu systeme va donc augmenter (conformement a la deuxieme loi), donc :
∆S > 0
or nous avons :
∆S ≈ 1T (1)
dU (1) − 1T (2)
∆U (1)
donc
∆U (1) < 0
ce qui veut dire que le sous-systeme (1) perd de l’energie.Donc la chaleur “coule” du systeme a la temperature la plus elevee a celuia la temperature la plus faible.

CHAPITRE 2. FORMALISATION 21
2.2.2 Equilibre Mecanique
Considerons maintenant un systeme constitue de deux sous-systemessepares par une cloison impermeable a la matiere. La cloison est donc mobileet permet un echange de chaleur entre les deux sous-systemes. Les systemeest donc defini par :
U (1) + U (2) = constant
V (1) + V (2) = constant
Lorsque le systeme arrive a l’equilibre, dS = 0, or :
dS =(∂S(1)
∂U (1)
)V (1),N
(1)1 ,...,N
(1)j
dU (1) +(∂S(2)
∂U (2)
)V (2),N
(2)1 ,...,N
(2)k
dU (2)
+(∂S(1)
∂V (1)
)U(1),N
(1)1 ,...,N
(1)j
dU (1) +(∂S(2)
∂V (2)
)U(2),N
(2)1 ,...,N
(2)k
dU (2)
et le systeme est isole, ce qui nous donne :
dU (1) = −dU (2)dV (1) = −dV (2)
ce qui nous donne pour finir :
dS =(
1T (1)
− 1T (2)
)dU (1) +
(P (1)
T (1)− P (1)
T (2)
)dV (1)
Or cette expression doit s’annuler quelque soit les valeurs de dU (1) et dV (1),donc :
1T (1)
− 1T (2)
= 0
P (1)
T (1)− P (1)
T (2)= 0
soit :
T (1) = T (2)
P (1) = P (2)

CHAPITRE 2. FORMALISATION 22
2.2.3 Equilibre de Flot de Matiere
Considerons maintenant, un systeme forme de deux sous-systemes separespar une paroi rigide, permeable au flux d’entropie et au flux d’un seulelement N1. La variation (virtuelle) d’entropie s’ecrit :
dS =(∂S(1)
∂U (1)
)V (1),N
(1)1 ,...,N
(1)j
dU (1) +(∂S(2)
∂U (2)
)V (2),N
(2)1 ,...,N
(2)k
dU (2)
−(∂S(1)
∂N(1)1
)U(1),N
(1)2 ,...,N
(1)j
dN(1)1 −
(∂S(2)
∂N(2)1
)U(2),N
(2)2 ,...,N
(2)k
dN(2)1
or nous avons :
dU (1) = −dU (2)
dN(1)1 = −dN (2)
1
ce qui nous donne :
dS =(
1T (1)
− 1T (2)
)dU (1) −
(µ
(1)1
T (1)− µ
(2)1
T (2)
)d1N
(1)
or dS doit tendre vers 0 pour des valeurs arbitraires de dU (1) et dN (1), nousavons :
0 =(
1T (1)
− 1T (2)
)dU (1) −
(µ
(1)1
T (1)− µ
(2)1
T (2)
)dN
(1)1
soit :
T (1) = T (2)
µ(1)1 = µ
(2)1
Regardons ce qui se passe si T (1) = T (2), nous avons comme expression dela variation d’entropie :
dS =(µ
(2)1 − µ(1)
1
T (1,2)
)dN
(1)1
Supposons maintenant µ(1)1 > µ
(2)1 alors
(µ
(2)1 −µ(1)
1
T (1,2)
)< 0 et dS > 0 (IIe loi)
donc dN (1)1 < 0, ce qui veut dire que le flux de matiere se fait de la region
a fort potentiel chimique vers la region a faible potentiel chimique. C’est lephenomene de diffusion.

CHAPITRE 2. FORMALISATION 23
2.2.4 Transformation de Legendre
[2] (pp. 137, 142, 285) pour transformation de LegendreBaser sur cet extrait d’un cours du MIT1 Dans les deux representations ther-modynamiques, l’entropie ou l’energie du systeme considere est exprime enfonction des variables extensives du systeme (energie ou entropie, volume,potentiels electrochimiques, etc.). Dans se formalisme, les grandeurs inten-sives apparaissent comme des derivees des grandeurs extensives (T = ∂U
∂S ,par exemple). Il est toutefois plus facile de mesurer, en laboratoire, une pres-sion qu’un volume, et il n’existe actuellement pas d’instrument pour mesurerl’entropie d’un systeme alors qu’un thermometre est un instrument banal.Il apparait donc la necessite purement pratique (puisque le formalisme estcomplet d’un point de vue logique et ne requiert nullement cette represen-tation thermodynamique). Il existe deux maniere geometriques de voir unefonction.
1. comme sous-ensemble de point du plan : Y = Y (X)
2. comme un ensemble de droite caract ψ = ψ(P ) ou ψ est l’ordonnee al’origine et P la pente.
Pour passer d’un systeme de coordonnee a l’autre, nous avons la relation :
P =Y − ψX − 0
⇔ ψ = Y − PX
Prenons, par exemple l’equation fondamentale d’un systeme simple, soitU = U(S, V,N). Nous voulons, par soucis de manipulations dans le labora-toire, exprimer l’energie interne du systeme en fonction de la temperature,du volume et du nombre de mole, nous avons donc la transformation deLegendre qui nous donne :
ψ = U − TS
Verifions maintenant que cette nouvelle fonction est bien une fonction fonda-mentale qui exprime l’energie interne du systeme. En derivant cette nouvellefonction, nous obtenons la differentielle totale exacte :
dψ = dU − TdS − SdT= TdS − PdV − νdN − TdS − SdT= −SdT − PdV − νdN
1http ://mit.fnal.gov/ paus/8.21-IAP2001/notes/notes/node6.html

CHAPITRE 2. FORMALISATION 24
ce qui veut dire ψ est une fonction de T, V et N avec −S = ∂ψ∂T . Donc ψ est
bien une fonction fondamentale qui exprime l’energie comme fonction de latemperature, du volume et du nombre de moles du systeme, c’est l’energielibre, ou l’energie de Helmholtz, notee F.Comme bel exemple d’application citons [1] Serie 22 Exercice 3 : “Trans-formee de Legendre pour le rayonement du corps noir”
2.2.5 Relations de Maxwell
[2] (pp. 181, 285)D’abord un resultat mathematique important, le theoreme de Schwarz :Soit une fonction f ,si f , ∂f
∂x , ∂f∂y ,
∂2f∂x∂y et ∂2f
∂y∂x sont continues, alors
∂2f
∂x∂y=
∂2f
∂y∂x
Un grand nombre de tels derivees croisee permettent d’etablir des egalitesa partir de differents “potentiels”.Par exemple, a l’aide de l’energie libre de Gibbs dG = −SdT + V dP +µdNcherchons “un sens physique immediat” a ∂S
∂P On en tire :
− S =∂G
∂T
V =∂G
∂P
donc
− ∂S
∂P=
∂2G
∂T∂P∂V
∂T=
∂2G
∂P∂T
et d’apres le theoreme de Schwarz, nous avons donc :
∂2G
∂P∂T=
∂2G
∂T∂P
⇔ − ∂S∂P
=∂V
∂T

CHAPITRE 2. FORMALISATION 25
2.2.6 Introduction
Les trois concepts presente ci-apres decoulent des transformations deLegendre. Le but principal de ces transformations est d’utiliser des va-riables intensives comme variables d’etat, parce qu’on les mesure directe-ment, relegant les valeurs extensives a des grandeurs derivees. Il est, parexemple, plus commode de mesurer la temperature d’un systeme que sonentropie.
2.2.7 Enthalpie
Definition Du grec “en” a l’interieur et de “thalpein” chauffer, l’enthal-pie est une parametre dont la variation permet d’exprimer la quantite dechaleur mise en jeu pendant une transformation monobare (effectuee pres-sion constante ) d’un systeme thermodynamique au cours de laquelle celui-cirecoit ou fournit un travail mecanique. L’enthalpie est la transformee de Le-gendre de l’energie du systeme qui remplace le volume par la pression commevariable independante, soit :
U = U(S, V,N)∂U
∂V= −P
ψ = U + PV
2.2.8 Energie libre ou l’energie de Helmoltz
Definition L’energie libre ou l’energie de Helmholtz F est la transforma-tion de Legendre de l’energie interne du systeme qui remplace l’entropie parla temperature comme variable independante, soit :
U = U(S, V,N)
T =∂U
∂SF = U − TS
F = F (T, V,N)
2.2.9 Energie libre de Gibbs
Definition L’energie libre de Gibbs G est la transformation de Legendrede l’energie interne du systeme qui remplace l’entropie par la temperature

CHAPITRE 2. FORMALISATION 26
et le volume par la pression, soit :
U = U(S, V,N)
T =∂U
∂S
P =∂U
∂VG = U − TS + PV
G = G(T, P,N)
[1] pp. 68-71. [2] chp. 6 et plus particulierement pp. 153-172. Un systemephysique est compose de trois sous-systemes dont un bain. Le systeme estferme et isole. A l’equilibre l’energie totale du systeme est une minimum,soit :
d(U + U r) = 0 (2.4)d2(U + U r) = dU2 > 0 (2.5)
et le sous-systeme et son reservoir son supposes isoles, donc :
d(S + Sr) = 0 (2.6)
En effet, la condition de stationnarite de l’energie (2.4) et la constance del’entropie (2.6) du systeme entier impliquent :
0 = T (1)dS(1) + T (2)dS(2) + T rdSr
= T (1)dS(1) + T (2)dS(2) − T rd(S(1) + S(2))
donc :
T (1) = T (2) = T r
2.2.10 Reservoir thermique et Principe du minimum du po-tentiel de Helmholtz
Puisque R est un reservoir thermique, son energie interne est donnee parT rdSr, donc (2.4) devient :
d(U + U r) = dU + T rdSr = 0
or dSr = −dS, donc :
dU − T rdS = 0

CHAPITRE 2. FORMALISATION 27
et T r est constant car R est un reservoir de temperature, donc :
d(U − T rdS) = 0
de meme, (2.5) implique :
d2U = d2(U − T rS) = d2F > 0
donc le systeme atteint un equilibre lorsque son energie de Helmholtz estminimisee et la temperature du sous-systeme est egale a celle du reservoir,soit T = T r.
2.2.11 Reservoir de pression et Principe du minimum del’enthalpie
Dans le cas d’un reservoir de pression, l’energie interne du reservoir estde la forme dU r = −P rdV r, donc (2.4) devient :
0 = d(U + U r) = dU − P rdV r = dU + P rdV
soit :
d(U + P rV ) = 0
de meme, (2.5) devient
d2U = d2(U + P rV ) = d2H > 0
Comme nous considerons un reservoir de pression, la seule maniere qu’il ad’echanger de l’energie interne est a travers un travail P rdV r, d’ou :
0 = −P (1)dV (1) − P (2)dV (2) + P rdV r
= −P (1)dV (1) − P (2)dV (2) + P rd(V (1) + V (2))= (P r − P (1))dV (1) + (P r − P (2))dV 2
ce qui nous donne donc la conditions suivante sur les pressions :
P (1) = P (2) = P r
Le systeme atteint donc son equilibre lorsque l’enthalpie admet un minimumet que la pression est la meme. C’est le principe du minimum de l’enthalpie.

CHAPITRE 2. FORMALISATION 28
2.2.12 Reservoir thermique et de pression et Principe duminimum de l’energie de Gibbs
Dans le cas d’un reservoir de pression et de temperature la differentielletotale exacte de l’energie interne prend la forme dU r = −P rdV r + T rdSr
donc (2.4) devient :
0 = d(U + U r)= dU + P rdV r + T rdSr
= dU − P rdV + T rdS
= d(U − P rV + T rS)= dG
et de meme (2.5) devient :
d2U = d2(U − P rV + T rS) = d2G > 0
donc le systeme atteint un equilibre lorsque la pression et la temperaturedu sous-systeme vallent ceux du reservoir, ce qui minimise l’energie libre deGibbs.
2.2.13 Principe du minimum general d’une transformationde Legendre de l’energie interne du systeme
La valeur d’equilibre de tout parametre interne d’un systeme en contactavec n reservoirs de parametre intensif P r1 , P
r2 , . . . minimise le potentiel
U [P1, P2, . . .] a P r1 , Pr2 , . . . constant (et egal a P1, P2, . . .). U [P1, P2, . . .] est
la transformee de Legendre de l’energie interne qui remplace les grandeursextensives X1, X2, . . . par les grandeurs intensives associees P1, P2, . . .
2.3 Transformation de substance
[2] (pp.56, 167, 292) [1] (chp. 3.4 pp. 73-74) Considerons un systemesforme de plusieurs sous-systemes qui reagissent entre eux. Le systeme baignedans un reservoir de pression P r et de temperature T r. Nous consideronsles reactions chimiques comme internes au sous-systeme, le systeme echangedonc de l’energie thermique et sous forme de travail avec le reservoir, maisn’echange pas de matiere avec celui-ci. Nous avons donc :
dS = IS + ΠS (2.7)dE = IE.mec. + IE.therm.

CHAPITRE 2. FORMALISATION 29
Le flux d’energie mecanique est donne par :
IE.mec. = −PdV
et le flux d’energie thermique est donne par :
IE.therm. = U − IE.mec. = U + PdV = dH (2.8)
et le flux d’energie thermique s’exprime (en cas de flux de chaleur parconduction) par
IE.therm. = TIS (2.9)
de 2.7, 2.8 et 2.9 nous avons :
dH = IE.therm. = T (dS −ΠS)
or la de la definition de dH = TdS +∑µidNi, nous avons :
T (dS −ΠS) = TdS +∑
µidNi
⇔ ΠS =−1T
∑µidNi
2.3.1 Osmose
[1] pp. 77-80 et Serie 23 exercice “Centrale electrique a pression osmos-tique”
Systeme Le systeme est compose d’un reservoir de temperature (Tr) etpression (Pr) ainsi que d’un sous-systeme compose d’une cellule osmotique etd’un bassin d’eau. La cellule est supposee avoir un tube vertical. Le bassinest un reservoir d’eau suppose contenir de l’eau a pression Pr et nombrede moles constant Nr. L’equilibre du systeme se caracterise donc pas unminimum de son energie de Gibbs. Notons Gr l’energie libre de Gibbs dureservoir et Gcell celui de la cellule osmotique. Prenons Nr(= constant) lenombre de moles d’eau dans le reservoir, Neau le nombre de moles d’eaudans la cellue et NSel le nombre de moles de solute. Prenons µeau(Tr, Pr) lepotentiel electrochimique de l’eau et µsel(Tr, Neau, Nsel) celui du sel.

CHAPITRE 2. FORMALISATION 30
Etude de l’equilibre du systeme L’equation fondamentale du systemeest donc donnee par :
U = U(Sr, Vr, Neau, Nsel, Nr)⇔ G = G(Tr, Pr, Neau, Nsel, Nr)
La conservation de l’eau implique :
dNeau = −dNr
L’energie libre de la cellule est donnee par :
Gcell = µeau(Tr, Pcell)Neau + µsel(Tr, Neau, Nsel)Nsel
⇒ dGcell = µeau(Tr, Pcell)dNeau + µsel(Tr, Neau, Nsel)dNsel
+∂µsel(Tr, Neau, Nsel)
∂NeauNseldNeau
et l’equilibre est donc caracterise par :
0 = d(Gcell +Gr)= dGcell + µeau(Tr, Pr)dNr
= dGcell − µeau(Tr, Pr)dNeau
= µeau(Tr, Pcell)dNeau + µsel(Tr, Neau, Nsel)dNsel
−µeau(Tr, Pr)dNeau
= [µeau(Tr, Pcell)− µeau(Tr, Pr)]dNeau
+∂µsel(Tr, Neau, Nsel)
∂NeauNseldNeau (2.10)
Modelisons maintenant le potentiels chimique de l’eau et du solute. Le po-tentiel chimique quantifie la variation d’energie du systeme en fonction de lavariation du nombre de moles de l’espece ajoutee. Dans le cas de l’eau dansla cellule, tout apport d’eau se traduit par une augmentation du niveau, etdonc de la pression dans la cellue de la maniere suivante :
Pcell = ρgh+ Pr
⇔ h =(Pcell − Pr)
ρ
et donc l’energie interne de la cellule varie comme fonction du potentielgravifique engendre par l’augmentation de la quantite d’eau. Le volume d’eau

CHAPITRE 2. FORMALISATION 31
dans la cellule varie comme dNv avec v la densite volumique de l’eau, d’ou :
dUcell = µeau(Tr, Pcell)dNeau
= ρghvdNeau
= ρgPcell − Pr
ρgvdNeau
= (Pcell − Pr)vdNeau
Nous prenons maintenant le zero du potentiel chimique comme µeau(Tr, Pr),ce qui nous donne pour la condition d’equilibre 2.10 :
(Pcell − Pr)vdNeau +∂µsel(T,Neau, Nsel)
∂NeauNseldNeau = 0 (2.11)
Supposons maintenant que les ions en solute n’interagissent ni entre eux niavec le solvant. Nous avons donc un gaz parfait. En utilisant le theoreme deSchwarz :
∂µ
∂P=
∂2G
∂P∂N=
∂2G
∂N∂P=∂V
∂N
et l’equation des gaz parfait (PV = NRT ), nous obtenons :
∂µ
∂P=∂V
∂N=∂NRTP∂N
=RT
P
En integrant cette equation differentielle, nous obtenons :∫µ(T, P ) =
∫ P0
P
RT
PdP = RTln(
P
P0) + µ(T, P0)
avec P0 la pression choisie pour definir le zero du potentiel chimique. Ainsile potentiel chimique µsel devient :
µsel(T, c) = RTln(c
c0) + µ(T, c0)
ou encore (a retenir)
µsel(T,N,N0) = RTln(NselNeau
c0) + µ(T,N,N0)

CHAPITRE 2. FORMALISATION 32
et en differentiant partiellement par rapport a Neau on obtient :
∂µsel(T,Neau, Nsel)∂Neau
=∂[RTln(
NselNeauc0
) + µ(T,N,N0)]
∂Neau
=∂[RTln(Nsel)−RTln(Neau)−RTln(c0) + µ(T,N,N0)
]∂Neau
=∂[−RTln(Neau)
]∂Neau
= − RT
Neau
et ainsi 2.11 devient :
v(Pcell. − Pr) =Nsel
NeauRTr = cRTr
c’est la loi de Van’t Hoff. La variation de pression osmotique est donnee par :
∆P = RT∆c
2.4 Transition de Phase du 1er ordre
[2] Chp. 9 et plus particulierement pp.245-253, 286, [3] pp. 484-488, [1]chp. 3.4
Experience de fonte de glace Pour calculer la temperature finale d’unmelange de constituants en phases liquides et solides il faut utiliser la relationsuivante :
Qc =∑i
miCP,i∆Ti +∑j
mjlj
avec mi la masse du ie composant de capacite thermique CP,i, mj la massedu je composant a la transition de phase caracterisee par une chaleur latentelj .
2.4.1 Changement de Phase
Systeme Nous considerons un systeme forme de vapeure d’eau maintenua une temperature legerement au dessurs du niveau d’ebullition pour latemperature donnee. Nous utilisons l’energie libre de Gibbs pour caracteriserles variations d’energie du systeme comme fonction de la temperature .
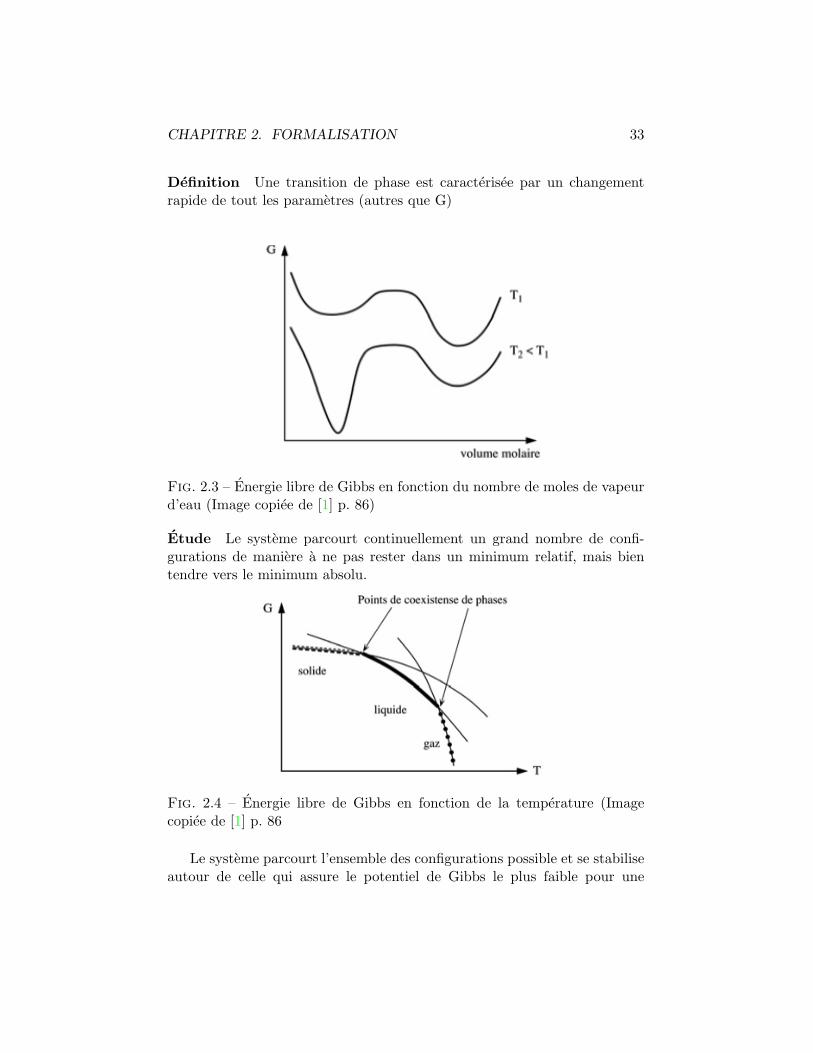
CHAPITRE 2. FORMALISATION 33
Definition Une transition de phase est caracterisee par un changementrapide de tout les parametres (autres que G)
Fig. 2.3 – Energie libre de Gibbs en fonction du nombre de moles de vapeurd’eau (Image copiee de [1] p. 86)
Etude Le systeme parcourt continuellement un grand nombre de confi-gurations de maniere a ne pas rester dans un minimum relatif, mais bientendre vers le minimum absolu.
Fig. 2.4 – Energie libre de Gibbs en fonction de la temperature (Imagecopiee de [1] p. 86
Le systeme parcourt l’ensemble des configurations possible et se stabiliseautour de celle qui assure le potentiel de Gibbs le plus faible pour une
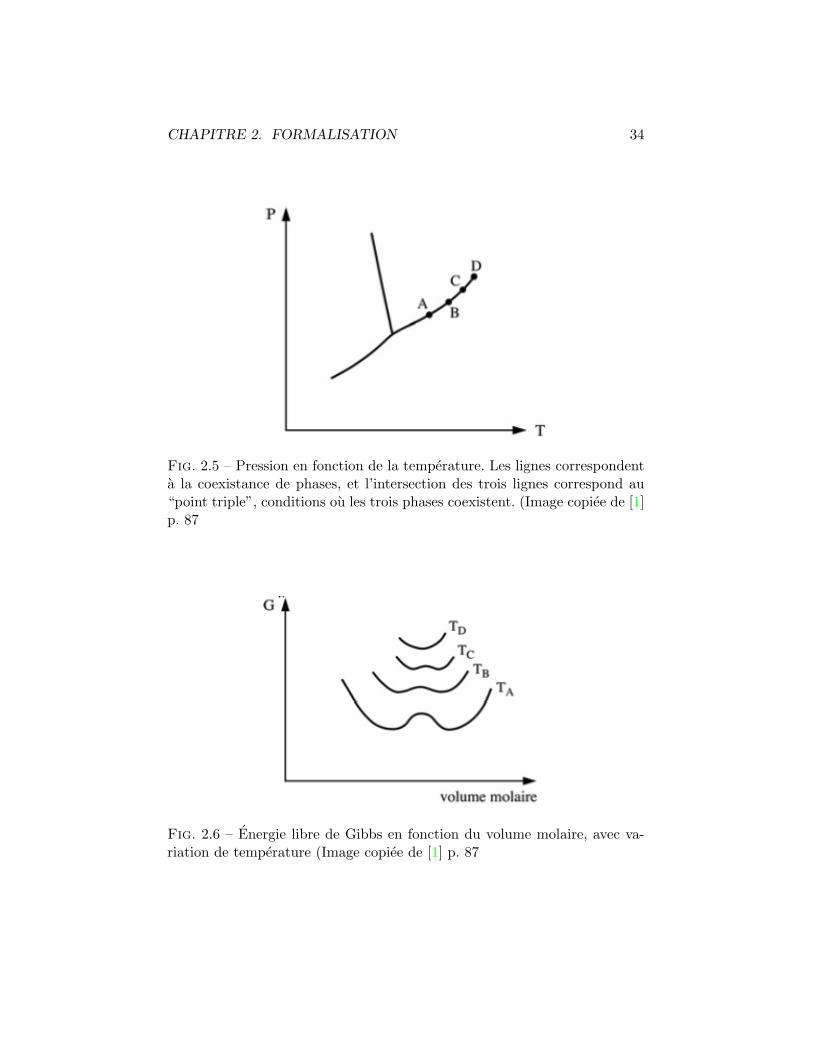
CHAPITRE 2. FORMALISATION 34
Fig. 2.5 – Pression en fonction de la temperature. Les lignes correspondenta la coexistance de phases, et l’intersection des trois lignes correspond au“point triple”, conditions ou les trois phases coexistent. (Image copiee de [1]p. 87
Fig. 2.6 – Energie libre de Gibbs en fonction du volume molaire, avec va-riation de temperature (Image copiee de [1] p. 87

CHAPITRE 2. FORMALISATION 35
pression donnee. Il est interessant de remarquer qu’avec un elevation de latemperature (TD) le systeme ne subit plus une transition de phase du 1er
ordre mais du second car il passe directement d’un etat a un autre sanspasser par le point critique qui temoigne de “l’hesitations” du systeme.
2.4.2 Attributs generaux d’une transition de phase du 1er
ordre
([2] pp. 243-244)Regardons maintenant la transition de phase du 1er ordre sous un autreangle : comme concavite et convexite des equations fondamentale exprimeeen terme energetique. Considerons la fonction fondamentale
U = U(S,X1, . . . , Xm, P1, . . . , Pn)
avec Ui les grandeurs extensives et Pj les grandeurs intensives. La condi-tion generale de stabilite impose U comme fonction convexe des grandeursextensives et concave des grandeurs intensives. Geometriquement ceci setraduit par le fait que la fonction U est aux dessus des hypers-plans tan-gents a l’espace thermodynamique des grandeurs intensives et au dessous deshyper-plan tangents aux grandeurs intensives. Admettons que notre figure(Fig. 2.7) caracterise represente U = U(S,X1, . . . , Xm, P1, . . . , Pn) avec Uicomme fonction Xj . Sur le chemin de A a D la fonction satisfait le critere destabilite mais le viole entre D et O. Cette partie caracterise un changementd’etat du 1er ordre de l’etat D a l’etat O. Un point sur le segment [DO]correspond a un melange de phases, proportionnellement a la distance entreD et O. Ainsi
X =(X0
j −XZj )
(X0j −XD
j )
represente la fraction molaire de la phase O dans le systeme. Pour resumel’etat physique dus systeme est caracterise par la courbe AD puis par lesegment [DO] (la transition de phase) et enfin par la courbe OP.
2.4.3 Regle de changement de phase de Gibbs
La relation fondamentale energetique d’un systeme a plusieurs compo-sant chimique s’ecrit :
U = U(S, V,N1, . . . , Nr)
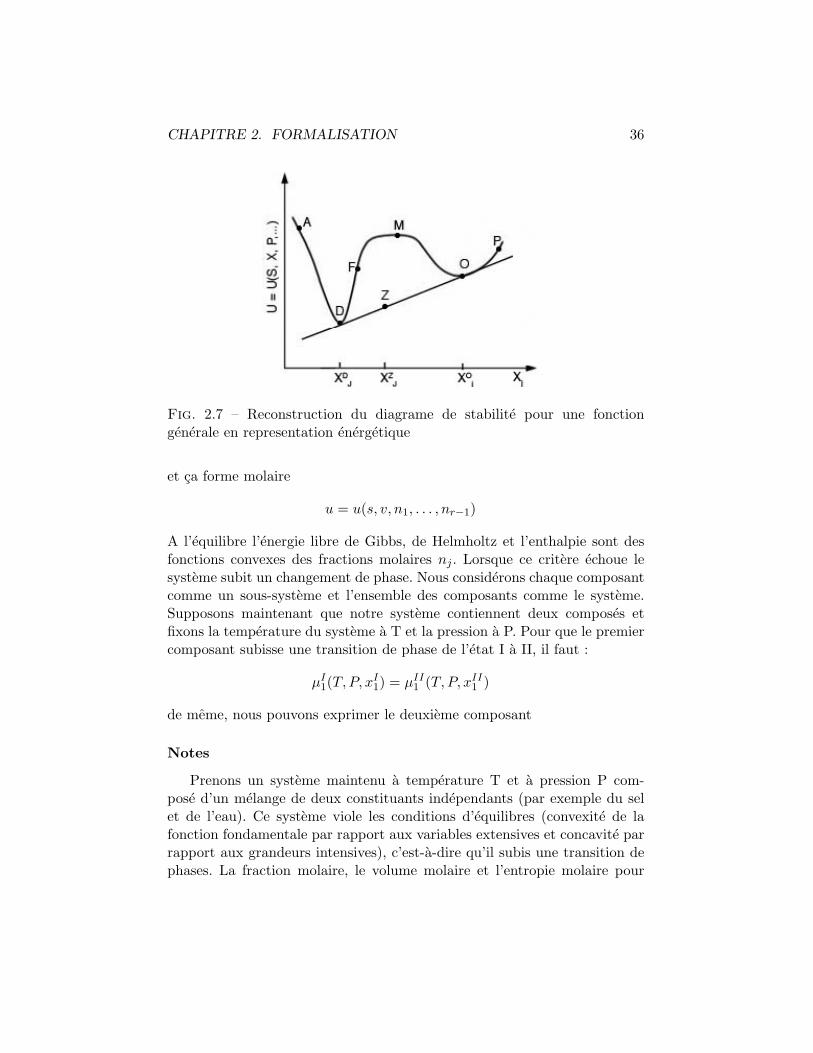
CHAPITRE 2. FORMALISATION 36
Fig. 2.7 – Reconstruction du diagrame de stabilite pour une fonctiongenerale en representation energetique
et ca forme molaire
u = u(s, v, n1, . . . , nr−1)
A l’equilibre l’energie libre de Gibbs, de Helmholtz et l’enthalpie sont desfonctions convexes des fractions molaires nj . Lorsque ce critere echoue lesysteme subit un changement de phase. Nous considerons chaque composantcomme un sous-systeme et l’ensemble des composants comme le systeme.Supposons maintenant que notre systeme contiennent deux composes etfixons la temperature du systeme a T et la pression a P. Pour que le premiercomposant subisse une transition de phase de l’etat I a II, il faut :
µI1(T, P, xI1) = µII1 (T, P, xII1 )
de meme, nous pouvons exprimer le deuxieme composant
Notes
Prenons un systeme maintenu a temperature T et a pression P com-pose d’un melange de deux constituants independants (par exemple du selet de l’eau). Ce systeme viole les conditions d’equilibres (convexite de lafonction fondamentale par rapport aux variables extensives et concavite parrapport aux grandeurs intensives), c’est-a-dire qu’il subis une transition dephases. La fraction molaire, le volume molaire et l’entropie molaire pour

CHAPITRE 2. FORMALISATION 37
un constituant varie d’une phase a l’autre. Chaque phase correspond a unsous-systeme.
Dans un systeme avec r constistuants, les potentiels electrochimiquesde la premiere phase sont fonction T, P, µI1, . . . , µ
Ir−1 et ceux de la seconde
phase sont fonction de T, P, µII1 , . . . , µIIr−1 et ainsi de suite. Si il y a M phases
l’ensemble des variables independante est donne par T , P et M(r − 1)fractions molaires, ce qui nous 2 + M(r − 1) variables independantes entout. Pour chaque composant il y a M − 1 equations d’egalite de potentielelectrochimique, ce qui fait en tout r(M − 1) equations. Donc le nombre dedegres de libertes est donne par :
f = 2 +M(r − 1)− r(M − 1) = 2 +M − r
Pour un systeme compose d’un constituant et d’une phase il y a deux degresde libertes (l’un etant elimine par la relation de Gibbs-Duhem, par ex :dµ = vdP − sdT ). Pour un systeme avec un composes et deux phases ily a trois parametres intensifs (T, P et µ qui sont constant d’une phase al’autre) et deux equations de Gibbs-Duhem (une pour chaque phase). Soitdeux equations pour trois variables, ou un degre de liberte. Si maintenant,nous avons un compose et trois phases, nous avons toujours trois parametresintensifs, nos inconnues (T, P, µ) mais nous avons maintenant trois equationsde Gibbs-Duhem et donc zeros degres de liberte. De meme, pour une systemea r composant et M phases, il y a donc r+2 parametres intensifs (T, P etles r potentiels electrochimiques). Chacun de ces parametres est constantd’une phase a l’autre. Pour chaque phase il y a une relation de Gibbs-Duhem (de la forme 0 = sdT −vdP +n1dµ1 + . . .+nrdµr). Ces M equationseliminent M inconnues parmis les r+ 2 ce qui nous donne r+ 2−M degresde liberte restant. Ainsi nous pouvons formuler la loi de phases de Gibbspar : Dans un systeme de r composants et de M phases, il est possibled’assigner arbitrairement r−M +2 variables parmis l’ensemble de variables(T, P, xI1, . . . , x
Ir−1) ou(T, P, µ1, . . . , µr−1)
2.4.4 Exemple
Prenons un systeme compose de deux constituants independants (1, 2)et de deux phases (α, β) Les conditions de stabilites pendant le changementde phase sont donnees par :
Tα = T β et Pα = P β et µα1 = µβ1 et µα2 = µβ2

CHAPITRE 2. FORMALISATION 38
Nous considerons une variation de T, P, ou x pendant que le systeme restea l’equilibre avec les phases α et β.
µα1 = µβ1 ⇒ dµα1 = dµβ1
⇒ dµα1 − dµβ1 = 0
⇒ ∂µα1∂T
dT +∂µα1∂P
dP +∂µα1∂xα1
dxα1 −∂µβ1∂T
dT − ∂µβ1∂P
dP − ∂µβ1
∂xβ1dxβ1 = 0
(2.12)
or
dU = TdS − PdV + µdN
⇒ µ =∂U
∂Net T =
∂U
∂SP = −∂U
∂V
⇒ ∂µα1dT
=∂U∂N∂U∂S
=∂S
∂N= s et
∂µα1∂P
=∂U∂N∂U∂V
=∂V
∂N= v
donc 2.12 devient :
(sα1 − sβ1 )dT + (vα1 − v
β1 )dP +
∂µα1∂xα1
dxα1 −∂µβ1
∂xβ1dxβ1 = 0 (2.13)
de meme
µα1 = µβ1
⇒ (sα2 − sβ2 )dT + (vα2 − v
β2 )dP +
∂µα2∂xα1
dxα1 −∂µβ2
∂xβ1dxβ1 = 0 (2.14)
Nous avons aussi la relation de Gibbs-Duhem qui nous donne (a T constantcar une transition de phase du premier ordre se fait a T constant) :
Nα1 dµ1 +Nα
2 dµ2 = 0
⇔ xα1∂µα1∂xα1
dxα1 + xα2∂µα1∂xα1
dxα1 = 0 (2.15)
donc en multipliant 2.13 par xα1 et 2.14 par xα2 nous obtenons :
0 =[(sα1 − s
β1 )xα1 + (sα2 − s
β2 )
]dT
+[(vα1 − v
β1 )xα1 + (vα2 − v
β2 )
]dP
+ xα1∂µα1∂xα1
dxα1 − xβ1
∂µβ1
∂xβ1dxβ1 + xα1
∂µα1∂xα1
dxα1 − xβ1
∂µβ1
∂xβ1dxβ1
CHAPITRE 2. FORMALISATION 39
et avec 2.15 nous obtenons :
0 =[(sα1 − s
β1 )xα1 + (sα2 − s
β2 )
]dT
+[(vα1 − v
β1 )xα1 + (vα2 − v
β2 )
]dP
−[xα1∂µβ1
∂xβ1− xα2
∂µβ1
∂xβ1
]dxβ1 (2.16)
Cette equations verifie bien la loi de phases de Gibbs, puisque il y a 2 degresde libertes pour un systeme compose de 2 phases (M=2) et 2 constituants(r=2) d’ou f = r −M + 2 = 2 − 2 + 2 = 2. Si par exemple, on fixe T et Palors xβ1 nous est impose par 2.16.
2.4.5 Relation de Clapeyron
[2] p. 228, p. 286 [3] p.493-494
Demonstration
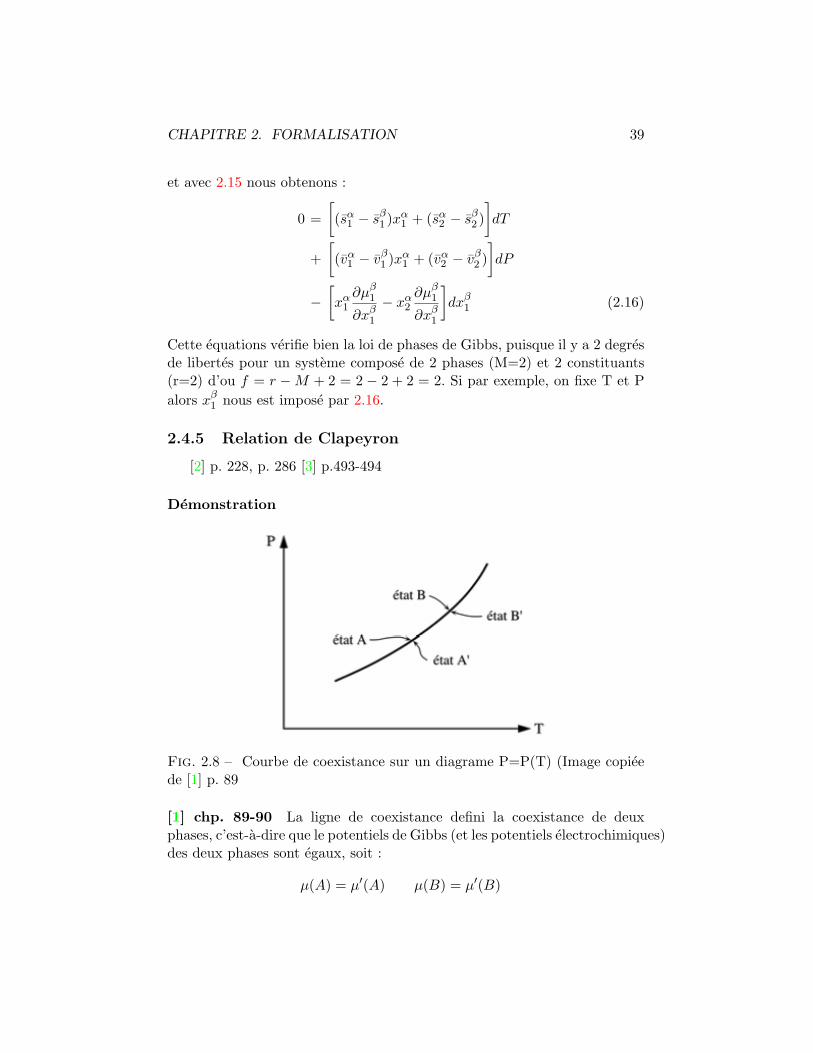
Fig. 2.8 – Courbe de coexistance sur un diagrame P=P(T) (Image copieede [1] p. 89
[1] chp. 89-90 La ligne de coexistance defini la coexistance de deuxphases, c’est-a-dire que le potentiels de Gibbs (et les potentiels electrochimiques)des deux phases sont egaux, soit :
µ(A) = µ′(A) µ(B) = µ′(B)

CHAPITRE 2. FORMALISATION 40
Nous avons aussi :
µ(B)− µ(A) = µ′(B)− µ′(A)
La relation de Gibbs-Duhem, nous donne :
0 = SdT − V dP +Ndµ⇔ dµ = vdP − sdT
avec s et v respectivement l’entropie molaire et le volume molaire. En pre-nant les etats A et B infiniment proche, nous obtenons :
µ(B)− µ(A) = vdP − sdT et µ′(B)− µ′(A) = v′dP − s′dT
donc
vdP − sdT = v′dP − s′dT⇔ vdP − v′dP = sdT − s′dT⇔ (v − v′)dP = (s− s′)dT
⇔ dP
dT=s− s′
v − v′=
∆s∆v
(2.17)
or la chaleur latente molaire l du changement de phase considere (change-ment de phase ⇔ coexistance de phase) est donnee par l = T∆s donc 2.17devient la relation de Clapeyron :
dP
dT=
l
T∆v(2.18)
Cette relation permet d’avoir une relation simple entre la variation de pres-sion et la variation de temperature pour un melange.
Demonstration alternative ([3] p.607)
Le changement de phase est caracterise par une egalite des potentielselectrochimique des deux etats, soit :
µI(T, P ) = µII(T, P )
Cette relation tiens pour tout T , donc
d(µII − µI)dT
= 0
⇔ ∂µII∂T
+∂µII∂P
dP
dT−
(∂µI∂T
+∂µI∂P
dP
dT
)= 0 (2.19)

CHAPITRE 2. FORMALISATION 41
or nous avons :
dG = SdT + V dP + µdN
ce qui nous donne :
∂G(P, T, n)∂P
= V (P, T, n) = nv(P, T, n) (2.20)
∂G(P, T, n)∂T
= −S(P, T, n) = −ns(P, T, n) (2.21)
∂G(P, T, n)∂n
= µ(P, T, n) (2.22)
d’ou 2.20 et 2.22 nous donnent grace au theoreme de Schwarz :
v(P, T ) =∂
∂n
∂G(P, T, n)∂P
=∂
∂P
∂G(P, T, n)∂n
=∂µ(P, T, n)
∂P
de meme 2.20 et 2.21 donnent :
∂µ(P, T, n)∂T
= −s(P, T )
donc nous obtenons pour 2.19 la relation de Clapeyron :
sI − ¯sII + (vII − vI)dP
dT= 0
2.5 Thermocinetique des processus irreversibles
2.5.1 Effet Seebeck
[1] 3.5.1 p.98 Prenons un barreau donc les extremites sont maintenuesa des temperatures differentes. Cette difference de temperature induit uncourant de charges au bornes de ce barreau, tel que :
∆V = ε∆T
ou ε est coefficient de Seebeck du materiau considere. [2] p.320 L’effet See-beck caracterise la production d’une force electromotive sous la conditiond’un courant electrique neutre. Considerons un thermocouple avec une jonc-tion a T1 et l’autre a T2 (tel que T1 > T2). Le thermocouple est forme dedeux materiaux A et B, sur A est place un voltmetre qui n’offre aucune

CHAPITRE 2. FORMALISATION 42
resistance au flux de chaleur, mais qui empeche tout passage d’un flux decharges electriques. Avec Jn = 0 nous obtenons :
∇µ =L12
TL11∇T
donc
µ2 − µ1 =∫ 2
1
LA12TLA11
dT
µ2 − µ′r =∫ 2
r
LB12TLB11
dT
µ′l − µ1 =∫ l
1
LB12TLB11
dT
en eliminant µ1 et µ2 de ces trois equations il vient :
µ′r − µ′l =∫ 2
1
(LA12TLA11
− LB12TLB11
)dT
mais la difference de temperature de part et d’autre du voltmetre etant nulle,il vient :
V =1e(µ′r − µ′l) =
∫ 2
1
(LA12eTLA11
− LB12eTLB11
)dT
La puissance thermoelectrique du thermocopule εAB est definie comme lavariation de temperature par variation unitaire de la temperature, soit :
εAB =∂V
∂T2=
(−LB12eTLB11
)−
(−LA12eTLA11
)En definissant la puissance thermoelectrique absolue d’un seul milieu comme :
εA :=−LA12eTLA11
nous obtenons
εAB = εB − εA

CHAPITRE 2. FORMALISATION 43
2.5.2 Effet Peltier
[2] p.323 [1] 3.5.1 p.98 et Serie 24 Exercice “Effet Peltier d’une jonctionthermiquement isole” Lorsque deux materiaux de coefficient de Seebeck dis-tinct sont mis en contact et qu’un courant de charges electriques s’ecouled’un materiau a l’autre, on observe a travers la jonction une flux de chaleurproportionnel au courant, donne par :
JQ = εTJel.
Le coefficient de Peltier pour chaque mateiraux est defini par :
π = εT
2.5.3 Loi de Fourier
[1] chp. 3.5.1 p.97 La loi de Fourier est une loi phenomenologiqe quidit que le flux d’energie thermique J a travers une paroi d’epaisseur d etdifference de temperature ∆T entre les deux cotes de la paroi est donne par :
JQ = −κ∆Td
En introduisant une chaleur specifique la loi de Fourier permet de caracteriserla diffusion de la chaleur dans un volume defini par (x+ dx)S, donnee par :
∂T
∂t=
κ
cV
∂2T
∂x2
Le terme κcV
est appele diffusivite thermique.
2.5.4 Loi de Fick
[2] p.314 [1] chp 3.5.1 p. 96 La vitesse de migration relative a une gran-deur extensive Xj est son flux, note J . C’est la quantite de cette grandeurextensive s’ecoulant a par unite de temps et par unite de surface. Par exemplele flux d’eau dans une riviere peut-etre caracterise par le nombre moleculesd’eau s’ecoulant a travers un plan perpendiculaire au rivage par par seconde.Soit un flux de substance proportionnel au gradient de potentiel dans unesolution in-homogene, alors :
JN = −d′−→∇µ

CHAPITRE 2. FORMALISATION 44
Soit un systeme compose de deux sous-systemes. Une variable extensive apour valeur Xk dans le premier systeme est X ′
k dans le second. Nous avonsla condition de cloture du systeme qui stipule :
Xk +X ′k = Xtot.
k = constante
Si le systeme n’est soumis a aucune contrainte, son equilibre est caracterisepar :
0 = Fk :=(∂Stotal
∂Xk
)Xtotal
k
=(∂(S + S′)∂Xk
)Xtotal
k
=∂S
∂Xk− ∂S
∂X ′k
= Fk − F ′k
(2.23)
La quantite Fk est la difference entre les grandeurs extensives (dans larepresentation entropique) agit comme une force generalisee qui provoque leprocessus. Ces forces generalisee sont appelee des affinites. Comm exemple,prenons Xk = U , l’affinit’e est donc donnee par :
Fk =1T− 1T ′
La chaleur ne coule pas d’un sous-systeme vers l’autre si la difference destemperatures est egale. Une difference de temperature, une affinite non-nulle,agit donc comme un potentiel. De meme, si nous prenons, Xk = V , nousobtenons :
Fk =(∂Stotal
∂Vk
)V total
k
=(∂(S + S′)∂Vk
)V total
k
=P
V− P ′
V ′
car :
∂S
∂U=
1T
et∂U
∂V= P
de meme si Xk est le potentiel electrochimique, l’affinite associee est donneepar :
Fk =µ′kT ′ −
µkT
La reponse d’une affinite, ou d’une force generalisee est caracterisee par leflux de la variable extensive associee. Le flux Jk est donc definie par :
Jk :=dXk
dt(2.24)

CHAPITRE 2. FORMALISATION 45
Donc par definition, le flux s’annule lorsque l’affinit’e s’annule. Pour voir ceresultat plus clairement differentions l’entropie S(X0, X1, . . .) par rapportau temps, soit :
dS
dt=
∑k
∂S
∂Xk
dXk
dt
ce qui avec 2.23 et 2.24 donne :
S =∑k
FkJk
donc la variation de production d’entropie est la somme du produit de chaqueflux avec son affinite associee. Resume @ [2] p.310

Chapitre 3
Annexes
3.1 Divers, conversions et constantes
1. 1[J.K−1] est l’entropie necessaire pour faire fondre 1 [cm3] de glace.
2. 0[K] = 273.15[ C]
3. Constante des gas parfaits : R = 8.314[J.Mole−1.K−1]
4. Nombre d’Avogadro : NA = 6, 022.1023[Mole]
5. 1[Calorie] = 4.186[J ] est la quantite d’energie necessaire pour aug-menter la temperature d’un 1[g] d’eau de 1[ C] = 1[K].
46

Bibliographie
[1] Jean-Philippe Ansermet : Introduction a la Thermodynamique Poly-copie (2005-6)
[2] Herbert B. Callen : Thermodynamics and an introduction to Thermo-statics, John Wiley & Sons, Second Edition (1985)
[3] Hans U. Fuchs : The Dynamics of Heat, Springer-Verlag New-York(1996)
[4] M. M. Abbot, H, G. Van Ness Schaum’s Outline of Theory and Pro-blems of Thermodynamics McGraw-Hill, Second Edition (1989)
47


















