
maladie de Charcot-Marie-Tooth - Téléthon : Espace Membre · Avancées dans la maladie de...
10
Avancées dans la maladie de Charcot-Marie-Tooth > Neuropathie sensitivomotrice héréditaire (HSMN) > CMT > Neuropathie de type Charcot-Marie-Tooth Ce document présente l'état actuel des connaissances scientifiques sur les maladies de Charcot-Marie-Tooth, mis à jour à l'occasion des Journées des Familles 2013 de l'AFM-Téléthon. Il est téléchargeable sur le site de l'AFM-Téléthon : WEB www.afm-telethon.fr . Pour en savoir plus sur la maladie de Charcot-Marie-Tooth, vous pouvez consulter le Zoom sur... la maladie de Charcot-Marie-Tooth et les Repères Savoir et Comprendre qui traitent de sujets scientifiques, médicaux, psychologiques et sociaux. Destinés aux personnes atteintes de maladies neuromusculaires et à leurs familles, ils sont disponibles sur le site internet de l'AFM-Téléthon et auprès du Service régional de votre région. Ces documents ne peuvent en aucun cas se substituer à l'avis d'un médecin, même s'ils peuvent vous faciliter le dialogue avec votre équipe soignante. JUIN 2013 AVANCÉES DE LA RECHERCHE
Transcript of maladie de Charcot-Marie-Tooth - Téléthon : Espace Membre · Avancées dans la maladie de...

Avancées dans la maladie de Charcot-Marie-Tooth
> Neuropathie sensitivomotrice héréditaire (HSMN) > CMT > Neuropathie de type Charcot-Marie-Tooth Ce document présente l'état actuel des connaissances scientifiques sur les maladies de Charcot-Marie-Tooth, mis à jour à l'occasion des Journées des Familles 2013 de l'AFM-Téléthon. Il est téléchargeable sur le site de l'AFM-Téléthon : WEB www.afm-telethon.fr . Pour en savoir plus sur la maladie de Charcot-Marie-Tooth, vous pouvez consulter le Zoom sur... la maladie de Charcot-Marie-Tooth et les Repères Savoir et Comprendre qui traitent de sujets scientifiques, médicaux, psychologiques et sociaux. Destinés aux personnes atteintes de maladies neuromusculaires et à leurs familles, ils sont disponibles sur le site internet de l'AFM-Téléthon et auprès du Service régional de votre région. Ces documents ne peuvent en aucun cas se substituer à l'avis d'un médecin, même s'ils peuvent vous faciliter le dialogue avec votre équipe soignante.
JUIN 2013
AVANCÉES DE LA RECHERCHE

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
2/10 AFM-Téléthon>Myoinfo
SOMMAIRE
Faits marquants .................................................................... 2
Qu'est-ce que la maladie de Charcot-Marie-Tooth ? ............... 3 Les différentes formes de maladie de Charcot-Marie-Tooth .................. 3
A quoi la maladie de Charcot-Marie-Tooth (CMT) est-elle due ? ..................................................................................... 5
Où en est la recherche dans la maladie de Charcot-Marie-Tooth (CMT)? ........................................................................ 6
Des causes génétiques mieux connues... ........................................... 6 ... et mieux reconnues. ................................................................... 6 Développer des outils pour la recherche ............................................ 6 Des registres de patients pour mieux connaître les CMT. ..................... 7 Connaître la fréquence des différents gènes impliqués pour guider la
démarche diagnostique. .................................................................. 7 Des biomarqueurs pour apprécier la sévérité des CMT. ........................ 7 Une nouvelle échelle de mesure pédiatrique. ...................................... 8 L’imagerie pour mieux décrire l’atteinte cérébrale. .............................. 8 Pistes thérapeutiques ...................................................................... 8
Pistes thérapeutiques dans la CMT2F ..................................................................8 Pistes thérapeutiques dans la CMT1A ..................................................................8 La vitamine C (acide ascorbique) est sans effet sur la CMT1A ..........................10 Essai de pléothérapie dans la CMT1A ................................................................10
* * *
Faits marquants
> Des nouveaux gènes impliqués dans la CMT ont été identifiés. > L'analyse de la fréquence des différentes formes de CMT et des différents gènes impliqués guide la démarche diagnostique des nouveaux cas. > Une échelle de mesure pédiatrique spécifique de la CMT a été mise au point pour suivre l’évolution de la maladie chez l'enfant. > La démyélinisation touche aussi le système nerveux central pas seulement les nerfs du système nerveux périphérique. > Deux nouveaux modèles de souris ont été développés pour la CMT1C et la CMT4H. > Les résultats des différentes études de la vitamine C dans la CMT1A confirment le manque d’efficacité du traitement sur les fonctions motrices. > Essai de PXT 3003 dans la CMT1A La phase clinique est terminée. Les données sont en cours d’analyse.
Rédaction Myoinfo, Département d'information sur les maladies neuromusculaires de l'AFM-Téléthon, Evry
Validation Benoît Funalot Centre de Référence « Neuropathies périphériques rares », CHU de Limoges, Limoges

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
AFM-Téléthon>Myoinfo 3/10
Qu'est-ce que la maladie de Charcot-Marie-Tooth ?
La maladie de Charcot-Marie-Tooth (CMT) représente un groupe de maladies cliniquement et génétiquement hétérogènes, caractérisées par une atteinte des nerfs des jambes et des bras (nerfs périphériques). Ces nerfs périphériques relient les muscles et les organes sensoriels de la peau et des articulations à la moelle épinière (système nerveux central). Les informations qu'ils transmettent entre les muscles, les récepteurs de la peau, les récepteurs articulaires et le cerveau sont nécessaires aux mouvements, aux perceptions tactiles et douloureuses et au maintien de l'équilibre. Cette atteinte entraîne principalement un manque de force musculaire, des troubles de la sensibilité des extrémités des membres (pieds, mains) et des troubles de l'équilibre. Dans certaines formes, il peut y avoir d’autres symptômes : atteinte respiratoire, atteinte des cordes vocales, atteinte visuelle, surdité. Elle débute la plupart du temps dans l’enfance ou chez l’adulte jeune par des difficultés à la marche ou des déformations des pieds. La maladie de Charcot-Marie-Tooth est une maladie rare qui concerne 1 personne sur 2 500, soit environ 30 000 personnes en France. La maladie de Charcot-Marie-Tooth (CMT) est une maladie génétique héréditaire. Elle se transmet des parents aux enfants par les gènes. De nombreux gènes sont impliqués dans l'apparition et la transmission de la CMT. Selon les gènes impliqués, la maladie est transmise selon différents modes (autosomique ou lié au chromosome X, dominant ou récessif).
Les différentes formes de maladie de Charcot-Marie-Tooth De nombreux gènes impliqués dans l'apparition et la transmission de la maladie de Charcot-Marie-Tooth (CMT) ont été identifiés. À
chaque gène, correspond une forme distincte de la maladie. Certains médecins préfèrent parler de "maladies de Charcot-Marie-Tooth" au pluriel, car même si toutes ces formes se manifestent de façon similaire, les mécanismes en jeu et les futurs traitements innovants à l'étude sont spécifiques de chaque forme génétique de la maladie. Il existe plus d’une cinquantaine de formes différentes de maladie de Charcot-Marie-Tooth, classées selon trois critères : - la nature de l'atteinte du nerf périphérique, determinée d'après les vitesses de conduction nerveuse à l'électromyogramme :
• axonale (vitesse de conduction nerveuse > 40 m/s), • démyélinisante (vitesse de conduction nerveuse < 35 m/s) • mixte (vitesse de conduction nerveuse intermédiaire entre 25m/s et 45m/s) ;
- le mode de transmission génétique : autosomique dominant, autosomique récessif ou lié à l'X ; - l'anomalie génétique en cause. Selon ces critères, il existe 5 grands types de CMT : CMT1, CMT2, CMT4, CMTX et DI-CMT (historiquement, l'appellation CMT3 a été utilisée, mais a aujourd'hui disparu).
Une maladie est dite rare quand elle touche moins d'une personne sur 2 000. Les maladies rares font l'objet d'une politique de santé publique commune dans les domaines de la recherche, de l'information et de la prise en charge.
Une maladie héréditaire est
transmise sur le mode
dominant lorsque la personne malade a une copie porteuse de l'anomalie génique et une copie normale du gène. La maladie se manifeste même si l'autre copie du gène n'est pas altérée.
Une maladie héréditaire est
transmise sur le mode
récessif lorsque la personne malade a ses deux copies du gène - celle reçue de son père et celle reçue de sa mère - porteuses d’une anomalie génétique. La maladie ne se manifeste que lorsque les deux copies du gène sont altérées.

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
4/10 AFM-Téléthon>Myoinfo
Atteinte nerveuse Mode de
transmission Dénomination
Démyélinisante
Autosomique dominant CMT1
Autosomique récessif CMT4
Axonale Autosomique (dominant ou récessif)
CMT2
Intermédiaire
Liée au chromosome X (dominant ou récessif)
CMTX
Autosomique dominant DI-CMT
Ces 5 grands types sont divisés en sous-types (1A, 1B, 1C..., 2A, 2B...). Chacun de ces sous-types correspond à une anomalie génétique touchant la fabrication d'une protéine donnée. Par exemple, la CMT1A correspond à la forme démyélinisante, autosomique dominante, liée au gène PMP22, tandis que la CMT1B correspond à la forme démyélinisante, autosomique dominante, liée au gène P0.
Les maladies de Charcot-Marie-Tooth sont liées à une atteinte des nerfs périphériques, qui relient la moelle épinière au muscle. L’axone est un prolongement qui émerge du corps cellulaire du neurone. Il conduit l’influx nerveux sous la forme de signaux électriques. L’axone est recouvert par une gaine de myéline, enveloppe isolante riche en lipides qui permet à l'influx nerveux de circuler très rapidement. La gaine de myéline est constituée par les cellules de Schwann, cellules indispensables à la survie et à la maturation des neurones.
axone
cellule de
Schwann
Formes démyélinisantes :
atteinte de la myéline. Vitesse de conduction
nerveuse (VCN) < 35 ms.
Formes intermédiaires : atteinte
de la myéline et de l’axone. Vitesse de conduction nerveuse :
25m/s < VCN < 45m/s.
Formes axonales : atteinte
de l'axone. Vitesse de conduction
nerveuse (VCN) > 40 ms.

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
AFM-Téléthon>Myoinfo 5/10
A quoi la maladie de Charcot-Marie-Tooth (CMT) est-elle
due ?
La maladie de Charcot-Marie-Tooth (CMT) est une maladie d'origine génétique. Les anomalies génétiques conduisent généralement à un manque ou à une altération d'une protéine donnée, impliquée dans le fonctionnement des nerfs périphériques. Il en résulte une atteinte qui peut toucher la gaine qui entoure le nerf, la myéline (formes démyélinisantes) ou l'axone lui-même (formes axonales). La gaine de myéline est produite par les cellules de Schwann, cellules indispensables à la survie et à la maturation des neurones. Plus d’une cinquantaine de gènes ont été identifiés dans la maladie de Charcot-Marie-Tooth et d'autres gènes restent encore à découvrir.
Les maladies (d'origine)
génétiques sont des maladies dues à des anomalies de l'ADN, c'est-à-dire de l'information qui détermine le fonctionnement biologique de notre organisme. Cette information est présente dans nos cellules sous forme de chromosomes. Nous l'héritons de nos parents et nos enfants héritent de la nôtre. C'est pourquoi les maladies génétiques sont souvent familiales, c'est-à-dire qu'il peut y avoir plusieurs membres d'une même famille atteints par la maladie génétique.
Les gènes altérés dans les maladies de Charcot-Marie-Tooth codent des protéines impliquées dans différentes fonctions cellulaires
Transport axonal : - NEFL (CMT2E, CMT1F), - HSPB1 (CMT2F), - HSPB8 (CMT2L), - KIF1B (CMT2A) - DYNC1H1 (CMT2O). Régulation du fonctionnement mitochondrial : - MFN2 (CMT2A), - GDAP1 (CMT4A, CMT2H, CMT2K), - PDK3.(CMT liée à l’X), Autres protéines : - LMNA (CMT2B) - TRPV4 (CMT2C) - AARS (CMT2N) - GARS (CMT2D), - YARS (CMT DIC) - INF2 (CMT DIE) - GNB4 (CMT DIF) - FBLN5 (CMT1) - LRSAM1 (CMT2P)
Composants de la myéline : - PMP22 (CMT1A), - P0 (CMT1B, CMT2I, CMT2J), - GJB1 (CMTX1), - SH3TC2 (CMT4C), - PRX (CMT4F).
Facteur de transcription impliqué dans la régulation de gènes de la myéline : - EGR2 (CMT1D, CMT4E).
Transport interne à la cellule (transport intracellulaire) : - MTMR2 (CMT4B1), - MTMR13 (CMT4B2), - DNM2 (CMT2M, DICMTB), - RAB7 (CMT2B), - NDRG1 (CMT4D), - LITAF (CMT1C), - FIG4 (CMT4J).
Le transport intracellulaire est l'ensemble des mécanismes qui permettent à une cellule de faire circuler du matériel d'un compartiment cellulaire à un autre au moyen de petits sacs délimités par une membrane (vésicules).
Le transport axonal est un transport intracellulaire qui chemine le long de l'axone, du neurone vers le muscle et réciproquement. Les mitochondries sont les organites cellulaires qui fournissent l'énergie indispensable au fonctionnement de nos cellules.

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
6/10 AFM-Téléthon>Myoinfo
Où en est la recherche dans la maladie de Charcot-
Marie-Tooth (CMT)?
Des causes génétiques mieux connues... Certains gènes impliqués dans la maladie de Charcot-Marie-Tooth (GDAP1, DNM2, P0…) peuvent être associés à une atteinte de l'axone et à une atteinte de la myéline, soulignant le lien étroit entre les cellules de Schwann et le neurone. Certaines des protéines touchées dans la CMT sont d'ailleurs impliquées directement dans l'interaction physique entre l'axone et sa gaine de myéline. De nouvelles techniques de diagnostic ont été mises au point pour rechercher de nouveaux gènes ou identifier de nouvelles mutations. Plus rapides et plus précises, ces techniques de séquençage nouvelle génération permettent de séquencer le génome entier ou de caractériser l’ARN messager, les petits ARN, les régions de facteurs de transcription, la structure de la chromatine, la méthylation de l'ADN… Récemment, la technique dite de "séquençage de l’exome entier" c’est-à-dire de séquençage de tous les exons de gènes codant des protéines, a permis de mettre en évidence deux nouveaux gènes : - le gène PDK3 sur le chromosome X, codant une protéine qui régule la production de pyruvate, une molécule clé de la production d’énergie de la cellule ; - le gène DHTKD1 (CMT2) impliqué dans la production d’énergie mitochondriale.
... et mieux reconnues. La technique d’hybridation génomique comparée réalisée sur des gènes impliqués dans la CMT a également permis de mettre en évidence une augmentation du nombre de copies du gène MPZ à l’origine d’une CMT. L’apport de ces nouvelles technologies (séquençage haut-débit, hybridation génomique comparée pour les remaniements de grande taille et séquençage d’exome – l'ensemble des parties codantes d'un génome -) permet notamment de réduire l’errance diagnostique. C’est ce que vise la mise au point d’un test permettant d’analyser tous les gènes connus sur une même puce CGH. Le séquençage de l’exome entier de 25 personnes atteintes de CMT dont les mutations responsables n’étaient pas identifiées par les techniques habituelles, a permis d’identifier 8 mutations. Ce taux de détection de 32% confirme l’intérêt de cette technique, comme outil de diagnostic moléculaire fiable et rapide. Ces analyses génétiques étendues ne sont interprétables qu'à la lumière des observations cliniques. C'est notamment une analyse génétique couplée à une analyse clinique, qui a permis d’identifier des mutations à distance du gène EGR2 responsables d’une forme de CMT amyélinique congénitale (CMT1D). Il est probable que plusieurs dizaines de gènes sont encore à découvrir, notamment des gènes clé de la myélinisation.
Développer des outils pour la recherche Pour étudier les mécanismes moléculaires en jeu dans la maladie de Charcot-Marie-Tooth et tester de nouvelles pistes thérapeutiques, les chercheurs développent des modèles animaux qui reproduisent les anomalies génétiques et les signes cliniques de la maladie. Il existe des souris modèles pour les CMT les plus fréquentes (CMT1A,
Chaque gène est structuré en une alternance de séquences
codantes : les exons, et de séquences non codantes : les
introns. On appelle "codant" les portions du gène qui sont
utilisées par la machinerie cellulaire comme patron pour la fabrication de l’ARN messager,
qui sera lui-même traduit ensuite en protéine. Seuls les
exons sont traduits en protéine.
Un modèle animal est un animal qui reproduit les
caractéristiques de la maladie (à la fois sur le plan génétique
et sur le plan clinique) permettant l'étude des
mécanismes de la maladie ou l'essai de traitements
potentiels.

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
AFM-Téléthon>Myoinfo 7/10
CMT1B…). Il existe également des modèles de souris CMT2A exprimant une forme mutée de la protéine mitofusine 2 ou de souris exprimant une mutation dans le gène Lmna . Très récemment, de nouveaux modèles de souris ont été développés : un modèle de CMT1C qui exprime une mutation dans le gène LITAF et un modèle de CMT4H, déficient en frabine/Fgd4. Les chercheurs travaillent également sur des modèles cellulaires, tels que des fibroblastes du nerf, des cellules de Schwann ou des neurones.
Des registres de patients pour mieux connaître les CMT.
Les gènes sont mieux identifiés, mais il reste difficile de définir précisément des profils d’évolution clinique pour chaque mutation (ce que l’on nomme corrélations génotypes/phénotypes) car à l’exception de la CMT1A qui est la forme de CMT la plus fréquente, le nombre de patient est faible pour la plupart des autres formes de la maladie. D'où la nécessité de mettre en place des registres de patients qui permettront de mieux connaître l’histoire naturelle des différents types de CMT. Le développement d’un registre de patients permet d’effectuer un recensement exhaustif des personnes atteintes d'une maladie et de préciser l’histoire naturelle de celle-ci. La détermination de l’histoire naturelle d'une maladie est un pré-requis important avant la mise en place de traitements ou d'essais cliniques.
Connaître la fréquence des différents gènes impliqués pour
guider la démarche diagnostique. Une équipe américaine de Detroit (États-Unis) a recensé de 2004 à 2009 et étudié 787 cas de CMT. L'analyse génétique a retrouvé une mutation connue que chez 67% d'entre eux. Les sous-types les plus fréquents étaient la CMT1A, la CMT1X, la neuropathie héréditaire avec paralysie à la pression, la CMT1B et la CMT2A. Chacun des autres sous-types représentait moins de 1% de l'effectif étudié. Le diagnostic génétique a été réalisé chez 1607 personnes atteintes de CMT par une équipe londonienne. Des anomalies génétiques dans les gènes PMP22, GJB1, MPZ et MFN2 représentaient plus de 90% de tous les diagnostics moléculaires de CMT. La fréquente implication de ces gènes dans la CMT doit les faire analyser en premier en cas de suspicion de CMT. A noter que dans l'expérience de ce centre londonien spécialisé dans les neuropathies, l'anomalie génétique a pu être identifiée dans près de 80% des formes démyélinisantes (CMT1) et 59% des formes intermédiaires de CMT. Cela n'a été le cas que dans 25% des formes axonales (CMT2) de CMT.
Des biomarqueurs pour apprécier la sévérité des CMT. Récemment, une équipe a étudié l’expression de gènes susceptibles d’être utilisés comme marqueurs de sévérité de la CMT1A à partir de nerf sciatique et de biopsie cutanée de rats modèles de CMT1A. Elle a mis en évidence une dérégulation de gènes qui interviennent dans le métabolisme lipidique. Ces gènes qui s’expriment à un stade précoce du développement, sont prédictifs d’altérations cliniques d’apparition plus tardive. Ces marqueurs de sévérité de la maladie ont été validés sur des biopsies cutanées de 46 patients atteints de CMT1A. Une association entre l’âge et la quantité d’ARN de ces marqueurs pourrait servir d’indicateur de sévérité de la maladie.
Un biomarqueur est une caractéristique biologique mesurable témoignant d’un processus normal ou anormal qui peut être utilisée pour le dépistage, le diagnostic, l’évaluation de la réponse ou de la tolérance à un traitement.
Ce que les médecins appellent
l'histoire naturelle d'une
maladie est la description des différentes manifestations d'une maladie et de leur évolution au cours du temps en l'absence de tout traitement (médicaments, kinésithérapie, chirurgie…).
Les études de corrélations
génotype/phénotype recherchent l'existence de liens entre les caractéristiques génétiques, le génotype, et les caractéristiques s'exprimant de façon apparente, le phénotype (taille, couleur et forme des yeux, couleur des cheveux, manifestation d'une maladie...). On peut ainsi identifier une relation plus ou moins étroite entre la présence d'une anomalie génétique et les manifestations de la maladie génétique.
Un modèle cellulaire permet d'étudier les mécanismes biologiques d'une maladie à partir de cellules cultivées en laboratoire qui reproduisent les caractéristiques de cette maladie. Ces cellules peuvent provenir de personnes atteintes de la maladie. Un modèle cellulaire permet aussi de tester les effets d'un traitement potentiel.

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
8/10 AFM-Téléthon>Myoinfo
Un protocole européen, en collaboration avec le réseau TREAT-NMD, est en cours afin de valider ces résultats sur un plus grand nombre de patients atteints de CMT1A.
Une nouvelle échelle de mesure pédiatrique. Une nouvelle échelle destinée aux enfants atteints de CMT a été récemment mise au point. Explorant sept domaines (force, dextérité, sensibilité, marche, équilibre, puissance et endurance) et basée sur 11 items, cette échelle, appelée CMT Pediatric Scale (CMTPedS), a pour but de suivre l’évolution et de servir de critère de jugement pour les essais thérapeutiques chez les enfants atteints de CMT. Facile à passer (environ 25 minutes), la CMT Pediatric Scale peut être utilisée dès l’âge de 3 ans et a déjà été validée chez 172 enfants atteints de CMT à travers le monde.
L’imagerie pour mieux décrire l’atteinte cérébrale. En 2013, une équipe française a fait passer une imagerie par résonance magnétique (IRM) cérébrale à 15 des personnes atteintes de CMT1A. Elle a mis en évidence une démyélinisation des cellules nerveuses dans le système nerveux central des personnes atteintes de CMT. Celle-ci semble donc ne pas se cantonner aux nerfs périphériques.
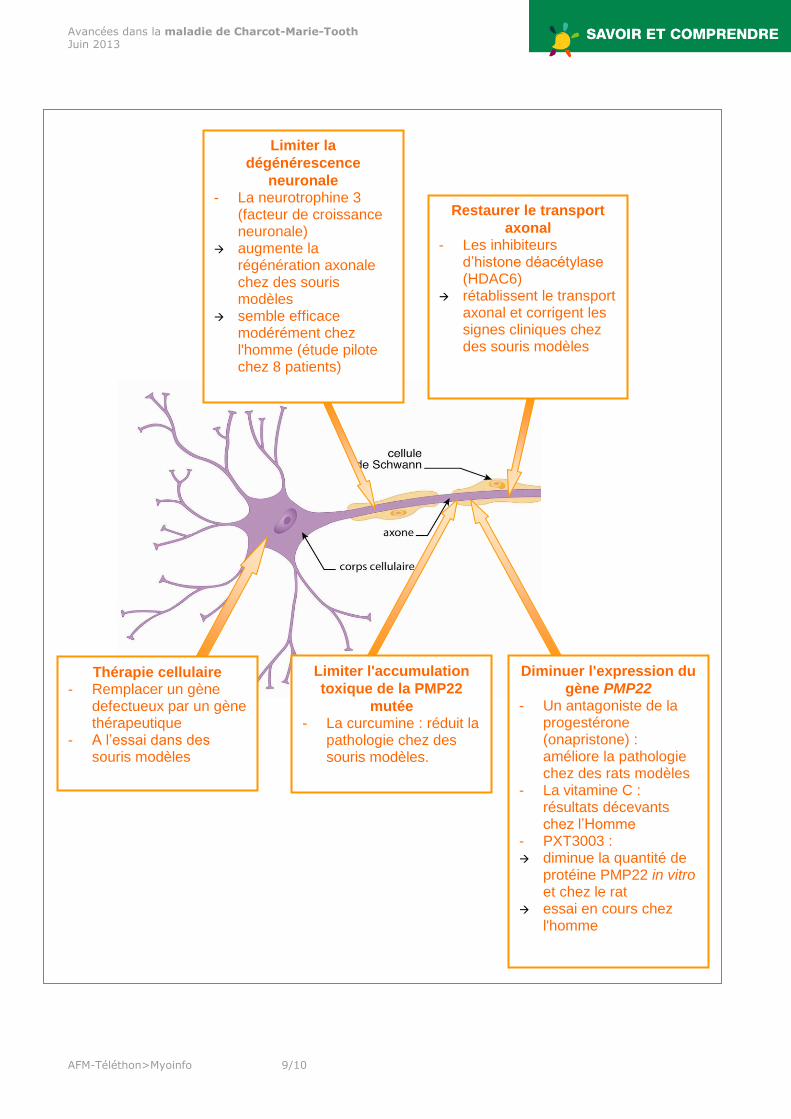
Pistes thérapeutiques L'identification des gènes en cause dans les CMT, des protéines correspondantes et des fonctions cellulaires impliquées, permettent d'envisager différentes pistes thérapeutiques. Cependant, à l'exception de la maladie de Charcot-Marie-Tooth de type 1A, les recherches sont pour l'instant très préliminaires.
Pistes thérapeutiques dans la CMT2F En 2011, une équipe américano-belge a étudié les mécanismes pathologiques liés aux mutations HSPB1 responsable de la maladie de Charcot-Marie-Tooth de type 2F (CMT2F). Pour cela, ils ont développé des modèles de souris transgéniques porteuses de mutations de ce gène. Ces souris ont reproduit les caractéristiques de la CMT2F, avec un transport axonal fortement altéré. L’administration d’inhibiteurs d’histone déacétylase (HDAC), tels que HDAC6, dans ces souris modèles a rétabli le transport axonal et corrigé les signes cliniques.
Pistes thérapeutiques dans la CMT1A La cause la plus fréquente de maladie de Charcot-Marie-Tooth 1A est une duplication d’une région du chromosome 17 contenant le gène PMP22 lequel code la protéine 22 de la myéline périphérique (protéine PMP22), entraînant la surexpression de cette protéine. Beaucoup plus rarement, il s’agit de mutations du gène PMP22, dont certaines provoquent une accumulation toxique de la protéine PMP22 anormale dans les cellules de Schwann. La surexpression ou les anomalies de la protéine PMP22 entraînent la dégénérescence de la gaine de myéline et par conséquent la neuropathie. Les pistes thérapeutiques dans cette maladie visent à agir à différents niveaux : - au niveau du gène, en diminuant l'expression du gène PMP22, - au niveau des cellules de Schwann en limitant l'accumulation toxique de la protéine PMP22 anormale, - ou encore, au niveau des nerfs périphériques pour empêcher leur dégénérescence en utilisant des facteurs de croissance neuronale.
Le système nerveux central comprend l'encéphale
(cerveau, cervelet, tronc cérébral) et son prolongement,
la moelle épinière. Il est protégé par une structure
osseuse (la boîte crânienne pour l'encéphale et la colonne
vertébrale pour la moelle épinière). Il analyse les
informations sensorielles, programme le mouvement et
transmet les ordres de contraction au muscle.
L'imagerie par résonance
magnétique ou IRM est une technique d'imagerie médicale
qui permet d’obtenir des images en coupe ou en volume d'un organe ou d'une région du
corps humain. Pendant l'examen, la personne est
allongée, immobile, sur un lit mobile qui coulisse dans un
appareil cylindrique constitué d’un aimant très puissant. Cet examen n'est pas douloureux.
L'impression d'être enfermé, isolé, le bruit de la machine, la
durée de l'examen peuvent cependant être un peu
impressionnants. >> Diagnostic des maladies
neuromusculaires, Repères Savoir & Comprendre, AFM, Janvier 2010.
Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
AFM-Téléthon>Myoinfo 9/10
Thérapie cellulaire - Remplacer un gène
defectueux par un gène thérapeutique
- A l’essai dans des souris modèles
Restaurer le transport
axonal - Les inhibiteurs
d’histone déacétylase (HDAC6)
rétablissent le transport axonal et corrigent les signes cliniques chez des souris modèles
Limiter la
dégénérescence
neuronale - La neurotrophine 3
(facteur de croissance neuronale)
augmente la régénération axonale chez des souris modèles
semble efficace modérément chez l'homme (étude pilote chez 8 patients)
Diminuer l'expression du
gène PMP22 - Un antagoniste de la
progestérone (onapristone) : améliore la pathologie chez des rats modèles
- La vitamine C : résultats décevants chez l’Homme
- PXT3003 : diminue la quantité de
protéine PMP22 in vitro et chez le rat
essai en cours chez l'homme
Limiter l'accumulation
toxique de la PMP22
mutée - La curcumine : réduit la
pathologie chez des souris modèles.

Avancées dans la maladie de Charcot-Marie-Tooth Juin 2013
10/10 AFM-Téléthon>Myoinfo
La vitamine C (acide ascorbique) est sans effet sur la CMT1A Au cours de ces dernières années, de nombreux essais cliniques évaluant les effets de la vitamine C (acide ascorbique) ont été réalisés à travers le monde (France, Australie et Nouvelle Zélande, Canada, États-Unis, Italie, Pays-Bas, Royaume-Uni…). Différentes doses (de 30 mg/j à 4 g/j) et durées de traitement (jusqu’à 2 ans) ont été essayées. Toutes les études ont été publiées hormis celle réalisée aux États-Unis, encore en cours d’analyse. Si dans certains essais, la vitamine C est bien tolérée, dans d’autres, elle provoque à fortes doses des effets indésirables comme des troubles digestifs qui disparaissent dans le mois qui suit l'arrêt du traitement. Aucune amélioration significative des fonctions motrices, des vitesses de conduction nerveuse ou des autres paramètres de surveillance étudiés (force musculaire, périmètre de marche, qualité de vie...) n'a été constatée
Essai de pléothérapie dans la CMT1A La pléothérapie est une méthode qui consiste à traiter des patients par l’association de faibles doses de médicaments déjà disponibles sur le marché (pour d’autres maladies), les pléomédicaments. Le réseau biologique perturbé dans la maladie de Charcot-Marie-Tooth de type 1A a été reconstitué selon une approche mise au point par la société Pharnext. Des molécules candidates ciblant ce réseau ont été testées in vitro et chez des rats, aboutissant à la sélection d'une combinaison de 3 médicaments, appelée PXT3003, capable de diminuer la production de la protéine PMP22. Basé sur ce concept, un essai français multicentrique, de phase II, contrôlé, en double aveugle, vise à évaluer la tolérance et la pharmacocinétique chez 80 personnes atteintes de CMT1A de 3 doses ("faibles", "intermédiaires", "fortes") de PXT3003 pendant 1 an. La phase clinique de l’essai est terminée et les données sont en cours d’analyse.
Essai en cours
• Essai de phase II, contrôlé, en double aveugle du traitement oral par PXT3003 : 3 doses ("faibles", "intermédiaires", "fortes") - Recrutement terminé des 80 personnes atteintes de CMT1A âgées de 18 à 65 ans - Essai en cours d’analyse. - Investigateur principal : Dr S. Attarian (CHU de la Timone, Marseille) - Contact : 04 91 38 75 63 / [email protected]
>> Tout au long de l'année, suivez l'actualité de la recherche dans les maladies neuromusculaires sur WEB www.afm-telethon.fr > Actualités > Toute l'actualité.
Dans un essai en double
aveugle, ni les patients ni les médecins nesavent quelle
alternative de traitement les patients prennent.
Au cours d'un essai clinique
de phase II, un médicament, dont il a été montré au
préalable qu'il était bien toléré (au cours d'un essai de phase
I) est administré à un groupe de malades dans le but de
déterminer l'efficacité thérapeutique, les doses
optimales et la sécurité du traitement (Quel est le mode
d’administration et la dose maximale tolérée ?).
La phase II peut être divisée en deux étapes : la phase IIa étudie le dosage et la phase IIb l'efficacité du traitement.
>> Essais cliniques et maladies neuromusculaires, Repères Savoir & Comprendre, AFM, Juillet 2010.
Lors d'un essai clinique
contre placebo, on utilise un placebo, produit qui ressemble au médicament testé, mais qui
ne contient pas de principe actif afin de mesurer l'action
réelle du médicament, en comparant les effets du
médicament et du placebo.


















