
Les Thérapies Ciblées les plus utilisées dans les ... · Survie globale (mois) 14,3 P=0,026...
48
Les Thérapies Ciblées les plus utilisées dans les Traitements Anti-Cancéreux Dr William Jacot, Oncologue Médical
-
Upload
hoangnguyet -
Category
Documents
-
view
215 -
download
0
Transcript of Les Thérapies Ciblées les plus utilisées dans les ... · Survie globale (mois) 14,3 P=0,026...

Les Thérapies Ciblées les plus utilisées dans les
Traitements Anti-Cancéreux
Dr William Jacot, Oncologue Médical

Thérapies CibléesDéfinition
� Anomalies moléculaires spécifiques des cellules tum oralesMolécules de la famille HERGènes de fusion (Abl-BCR)Néo-angiogenèse tumorale…
� Différentiellement exprimée entre les tissus sains e t le tissu tumoral
� Anticorps monoclonaux (X-mab )
� Petites molécules ciblant les fonctions tyrosine kinase (X-inib )
� Plus spécifiques des cellules tumorales,
� Toxicités propres
� Indications généralement limitées à une sous population tumorale

La notion de cibles en oncologie
Acquisition de propriétés intrinsèques particulières des cellules cancéreuses
Dix propriétés ou potentialités, retrouvées dans tous les cancers et impliquées dans le développement tumoral
Propriétés acquises par les cellules cancéreuses
Hanahan et al., Cell, Vol. 144, 646–674, March, 2011

Notion de cible en oncologie
Tout élément ou événement moléculaire caractérisant une cellule tumoralepeut théoriquement constituer une cible thérapeutique potentielle
Pour qu’une cible soit pertinente, elle doit être expriméepar un grand nombre de cellules tumorales, être accessible à une thérapeutique et que son ciblage aie un impact clinique
Il existe différents type de cibles quant à leur degré de responsabilité dans l’oncogenèse et certaines ne doivent pas obligatoirement avoir un rôle important, on parle alors de cible passive
Une cible est dite active quand son rôle est actif dans le processus tumoral

Notion de cible en oncologie
Classification des cibles thérapeutiques en cancéro logie
Cibles passives Exemple : CD20
Cibles actives (pro-oncogènes/anti-oncogéniques gèn es suppresseurs de tumeurs)
Cibles vitales ou primaires Exemples : VEGF/VEGFR
Cibles secondaires Exemple : télomérase
Cibles spécifiques de modèles tumoraux
Exemple : Kit, Abl-BCR
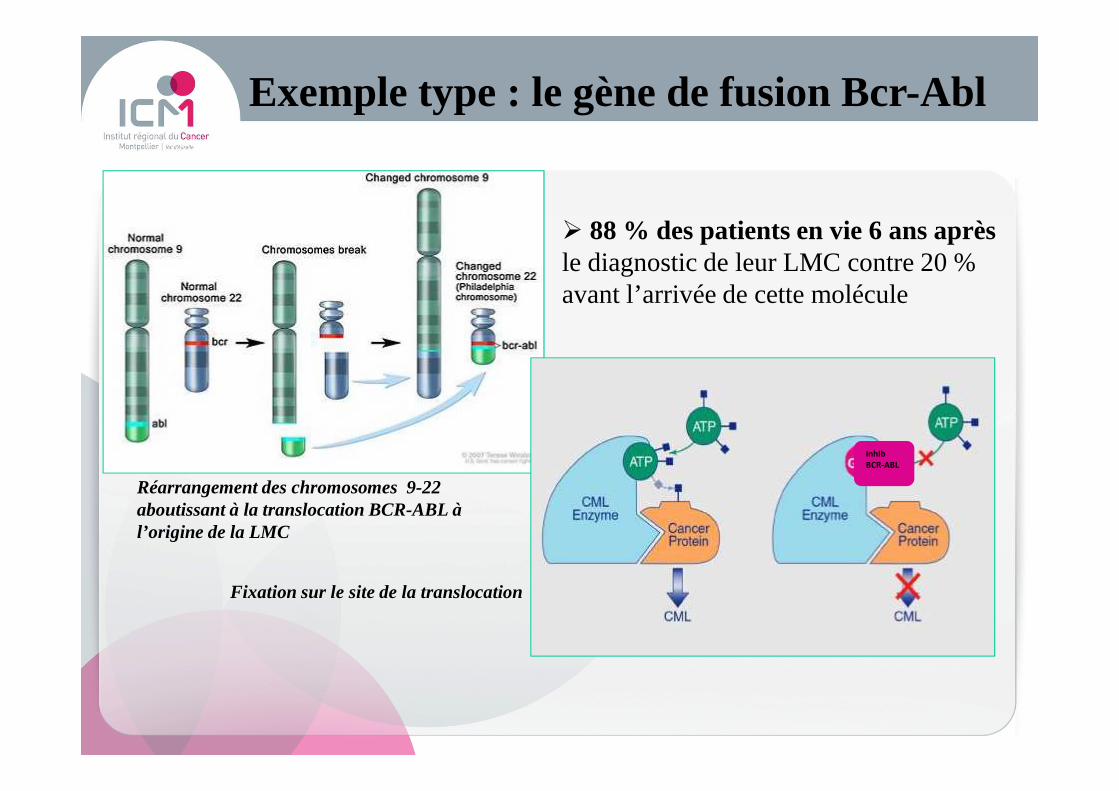
Exemple type : le gène de fusion Bcr-Abl
Réarrangement des chromosomes 9-22 aboutissant à la translocation BCR-ABL à l’origine de la LMC
Fixation sur le site de la translocation
� 88 % des patients en vie 6 ans après le diagnostic de leur LMC contre 20 % avant l’arrivée de cette molécule
InhIb
BCR-ABL
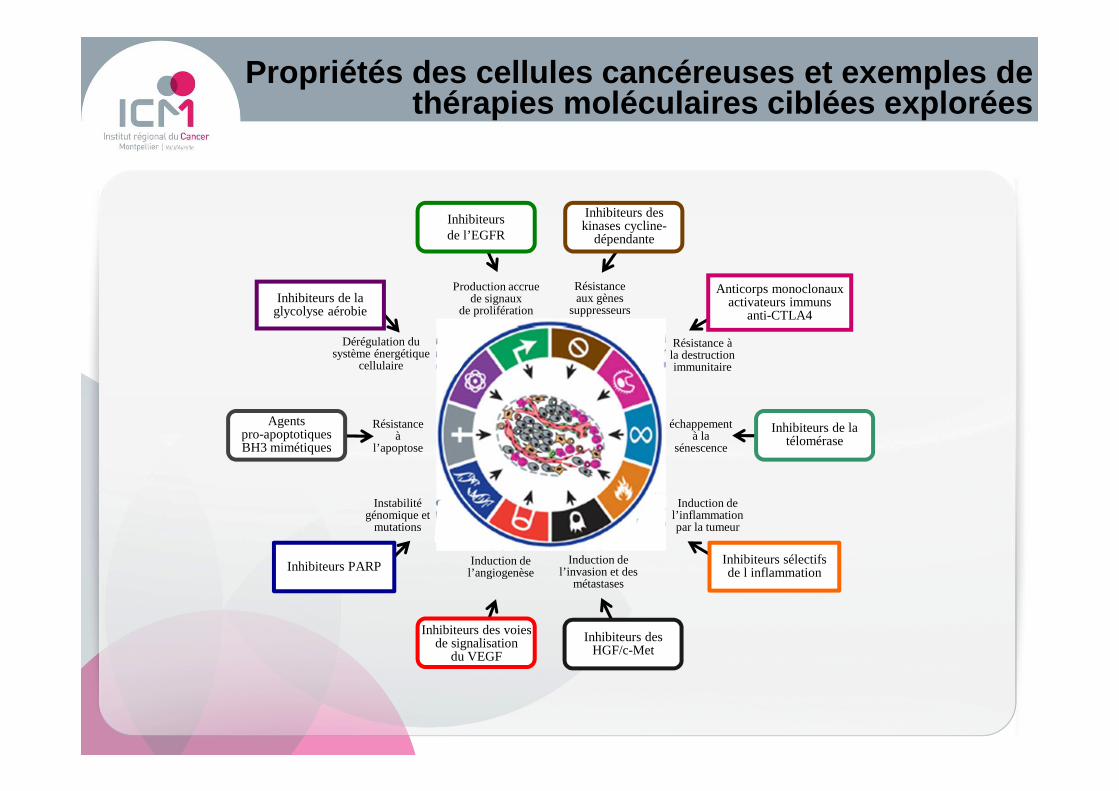
Propriétés des cellules cancéreuses et exemples de thérapies moléculaires ciblées explorées
Induction del’angiogenèse
Résistanceà
l’apoptose
Dérégulation du système énergétique
cellulaire
Production accruede signaux
de prolifération
Résistance aux gènes
suppresseurs
Résistance à la destruction immunitaire
échappement à la
sénescence
Instabilité génomique et
mutations
Induction de l’inflammation par la tumeur
Induction de l’invasion et des
métastases
Agentspro-apoptotiques BH3 mimétiques
Inhibiteurs de la télomérase
Anticorps monoclonaux activateurs immuns
anti-CTLA4
Inhibiteurs des kinases cycline-
dépendante
Inhibiteurs de la glycolyse aérobie
Inhibiteurs sélectifs de l inflammation
Inhibiteurs de l’EGFR
Inhibiteurs des HGF/c-Met
Inhibiteurs des voies de signalisation
du VEGF
Inhibiteurs PARP

Mécanismes de résistance
� La résistance aux thérapies moléculaires ciblées ouvre la voie de travaux de recherche afin d’offrir un traitement à des patients se trouvant sans alternative thérapeutique adaptée
Mécanismes de résistances
Résistance primaire
Résistance pharmacologique / Multi-Drug Résistance
Activation de voies de signalisation parallèles
Cible non cruciale
Résistance secondaire
Mutation au site de liaison du médicament
Amplification génomique en aval de la cible
Rétrocontrôle positif au niveau de la cible
Différenciation à partir des cellules souches tumorales
Adaptation du microenvironnement tumoral

Anticorps monoclonaux
� Lymphomes CD20+Rituximab
� Cancer du sein HER-2+Trastuzumab
� ORL, ColorectalCetuximab
� AngiogenèseBévacizumab
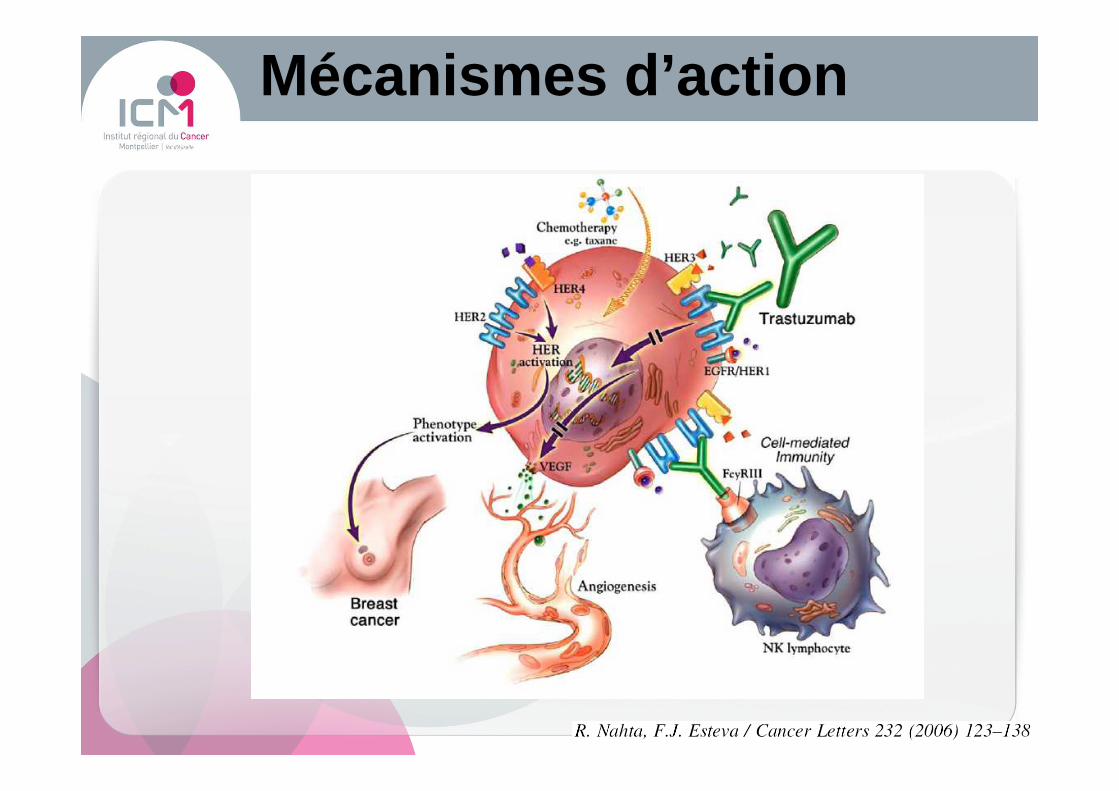
Mécanismes d’action

Inhibiteurs de la fonction tyrosine kinaseet autres petites molécules
� Petites molécules de bonne biodisponibilité orale
� +/- grande spécificité (dendrogrammes de kinomes)
� LMC, GIST : imatinib
� Cancer du sein HER-2+ : lapatinib
� Cancer bronchique NAPC: erlotinib, gefitinib
� Inhibiteurs de mTOR (everolimus)Cancer du rein, TNEP, cancer du sein RH+ (NR)
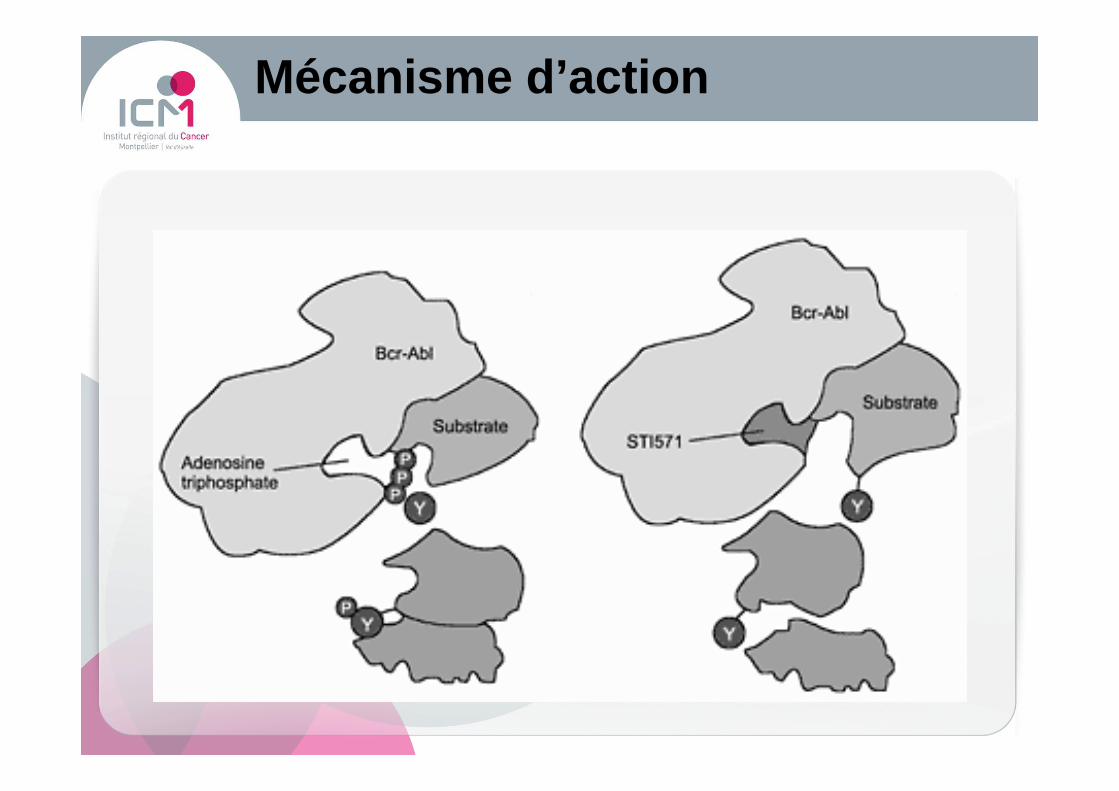
Mécanisme d’action

L’angiogénèse : un processus à 3 étapes
1. Dégradation enzymatique de la membrane basale
2. Mobilité des cellules endothéliales
3. Prolifération des cellules endothéliales

TCs anti-angiogèniques : des TCs ciblant le stroma
� Ciblent la population endothéliale (« stroma »)� Très importante néo-angiogenèse tumorale
� Bevacizumab (Avastin®)Cancer colo-rectal métastatiqueCancer bronchique NAPC métastatiqueCancer du sein métastatiqueCancer de l’ovaire
� Sorafenib (Nexavar®), sunitinib (Sutent®)Cancer du rein métastatique (mécanisme mixte, N, S)CHC (N)Carcinome thyroïdien réfractaire (N)TNEP (S)GIST (S)

Cancer du Sein et Thérapies Ciblées
� Anti-HER2Trastuzumab, Pertuzumab, T-DM1Lapatinib
� Anti-angiogèniquesBévacizumab
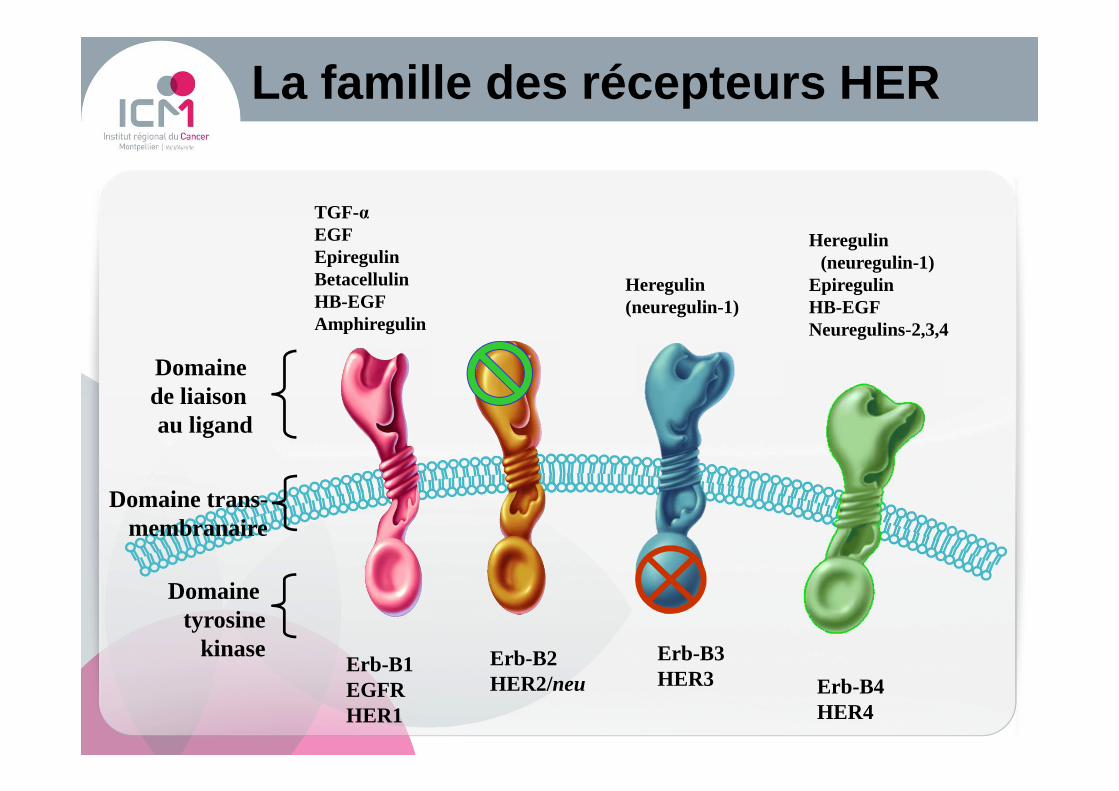
La famille des récepteurs HER
Domaine tyrosine
kinase
Domaine de liaison au ligand
Erb-B1EGFRHER1
Erb-B2HER2/neu
Erb-B3HER3 Erb-B4
HER4
Domaine trans-membranaire
TGF-αEGFEpiregulinBetacellulinHB-EGFAmphiregulin
Heregulin(neuregulin-1)
Heregulin(neuregulin-1)
EpiregulinHB-EGFNeuregulins-2,3,4
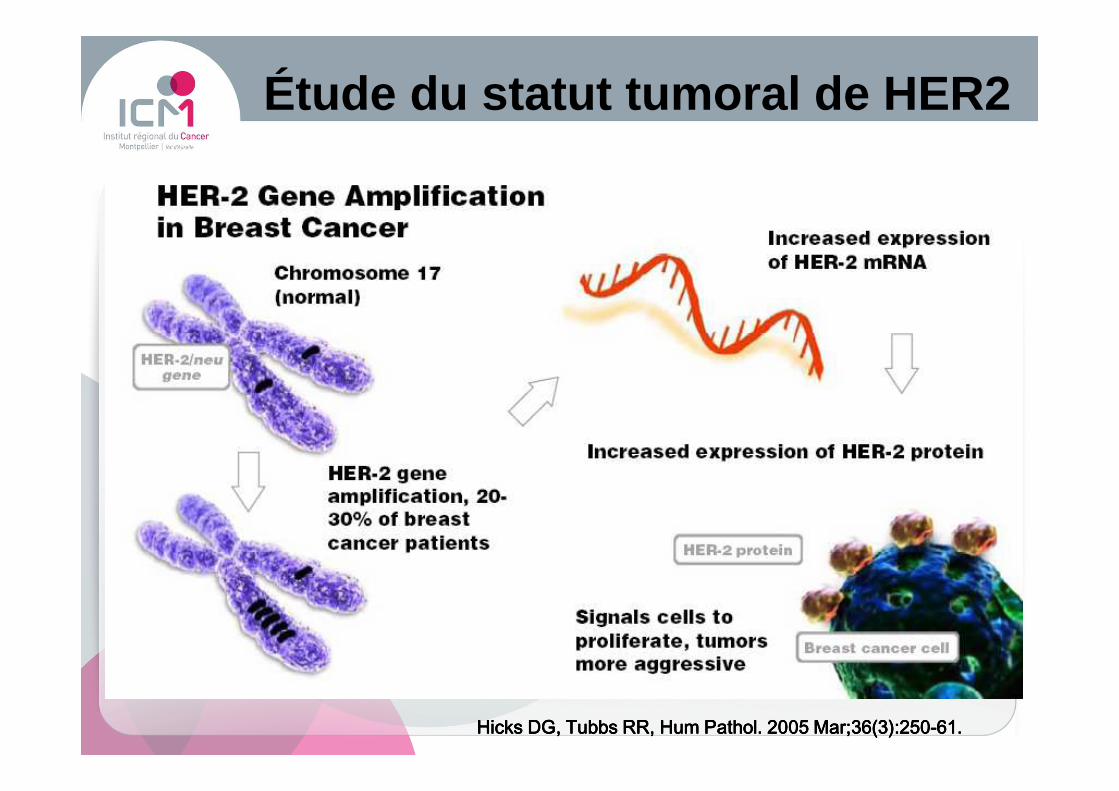
Étude du statut tumoral de HER2
Hicks DG, Tubbs RR, Hum Pathol. 2005 Mar;36(3):250Hicks DG, Tubbs RR, Hum Pathol. 2005 Mar;36(3):250Hicks DG, Tubbs RR, Hum Pathol. 2005 Mar;36(3):250Hicks DG, Tubbs RR, Hum Pathol. 2005 Mar;36(3):250----61.61.61.61.
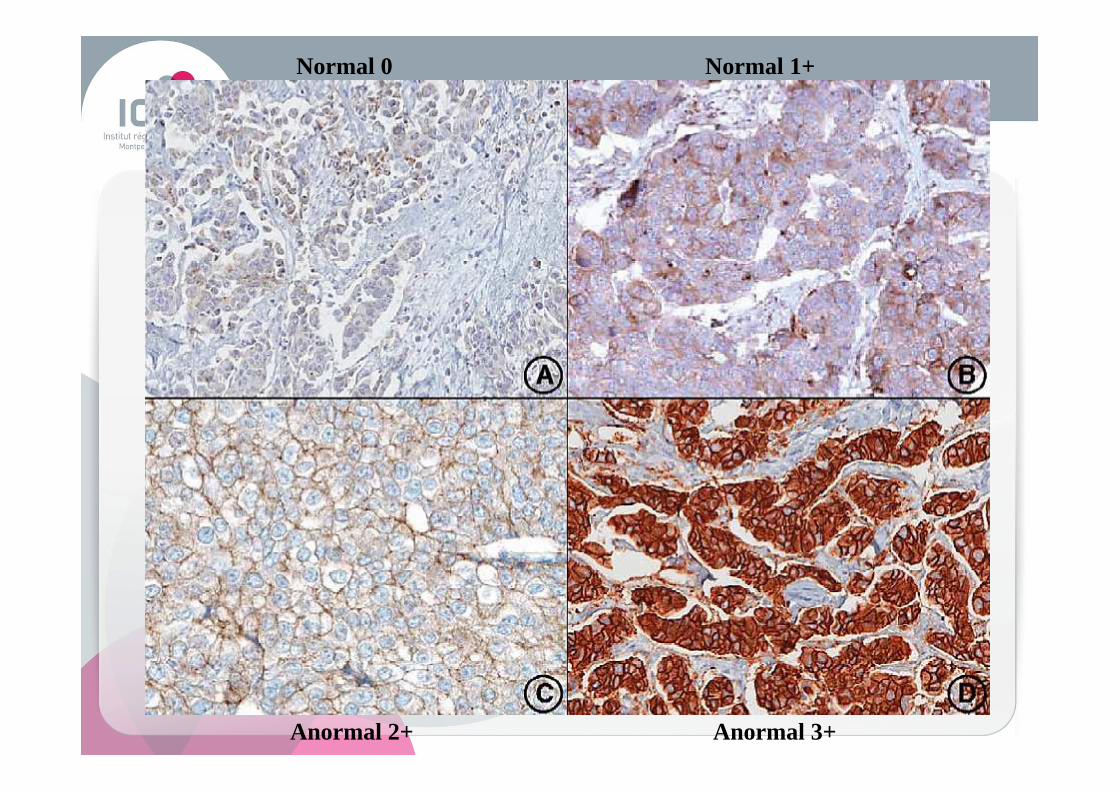
Anormal 2+ Anormal 3+
Normal 0 Normal 1+

Normal
Normal
Faible amplification
Forte amplification

Stades adjuvants : TrastuzumabSurvie Globale
HR +/- 0,50 à 0,75 (selon la durée de suivi)

Stades AdjuvantsSurvie Sans Récidive
HR +/- 0,50 à 0,75 (selon la durée de suivi)
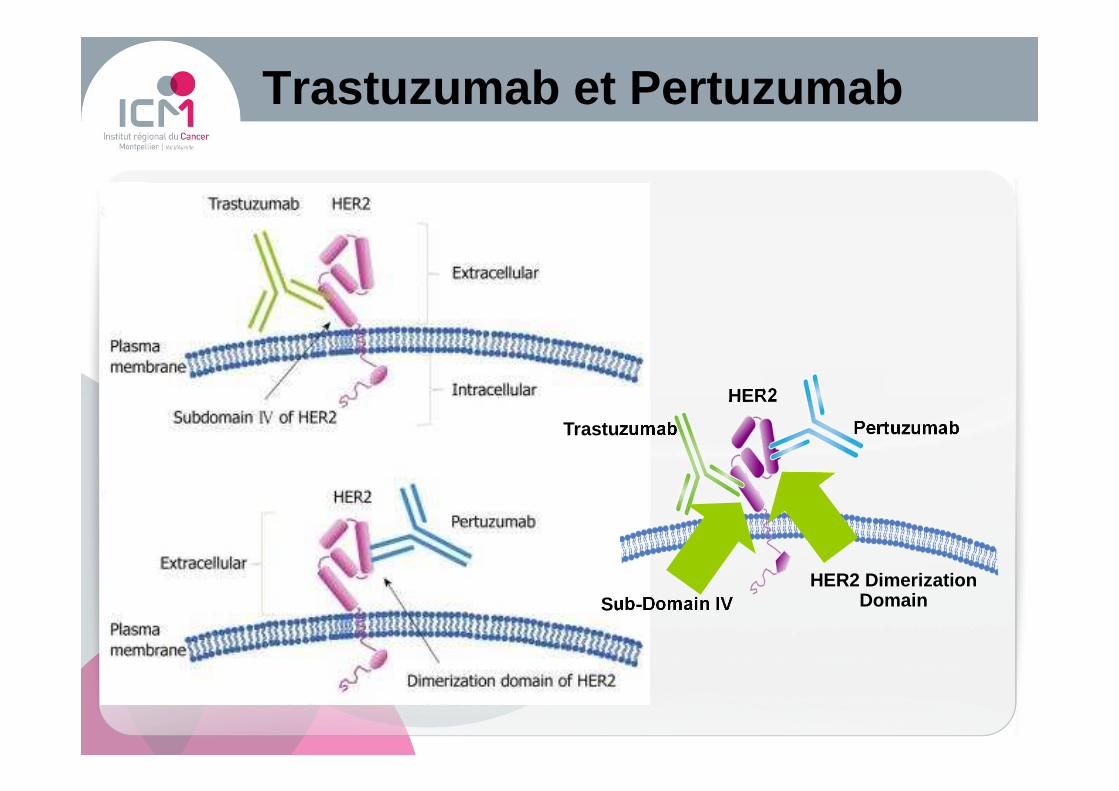
Trastuzumab et Pertuzumab
HER2
Trastuzumab Pertuzumab
Sub-Domain IVHER2 Dimerization
Domain
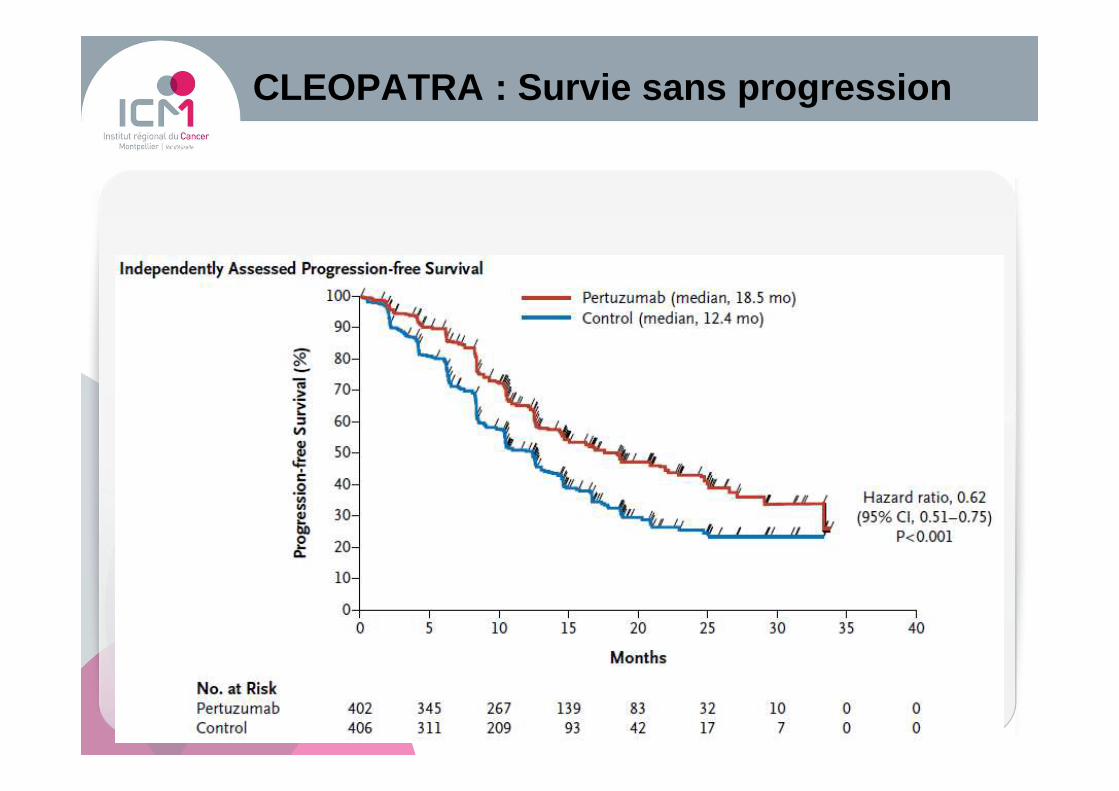
CLEOPATRA : Survie sans progression
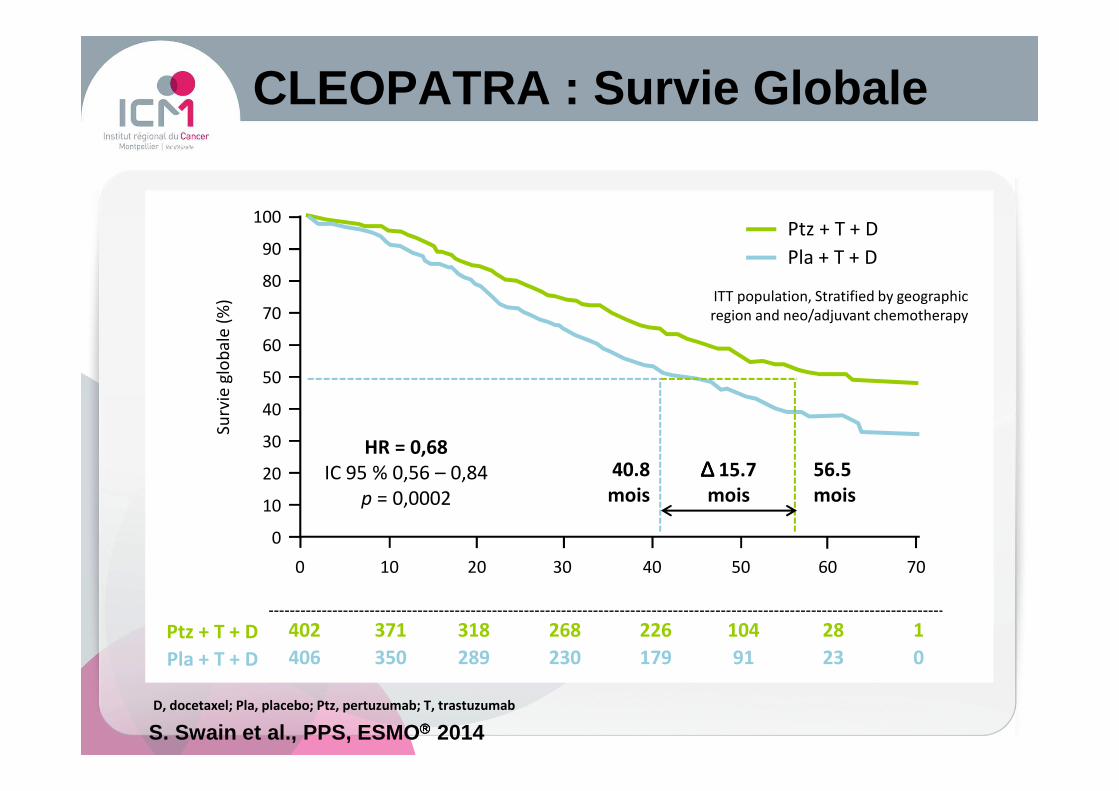
0 10 20 30 40 50 60 70
0
10
20
30
40
50
60
70
80
90
100
Nb à risque
402 371 318 268 226 104 28 1Ptz + T + D
Temps (mois)
HR = 0,68
IC 95 % 0,56 – 0,84
p = 0,0002
Ptz + T + D
Pla + T + D
Su
rvie
glo
ba
le (
%)
D, docetaxel; Pla, placebo; Ptz, pertuzumab; T, trastuzumab
406 350 289 230 179 91 23 0Pla + T + D
ITT population, Stratified by geographic
region and neo/adjuvant chemotherapy
CLEOPATRA : Survie Globale
S. Swain et al. , PPS, ESMO 2014
40.8
mois
56.5
mois
∆∆∆∆ 15.7
mois
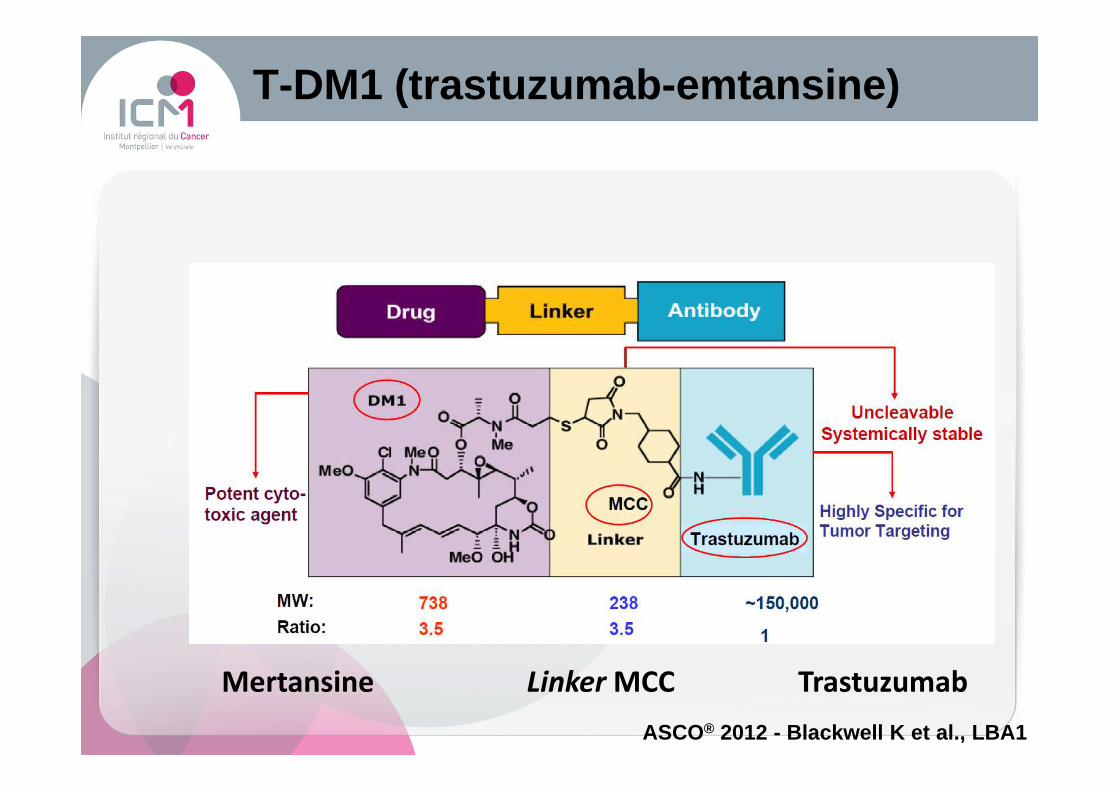
Mertansine Linker MCC Trastuzumab
T-DM1 (trastuzumab -emtansine)
ASCO® 2012 - Blackwell K et al., LBA1
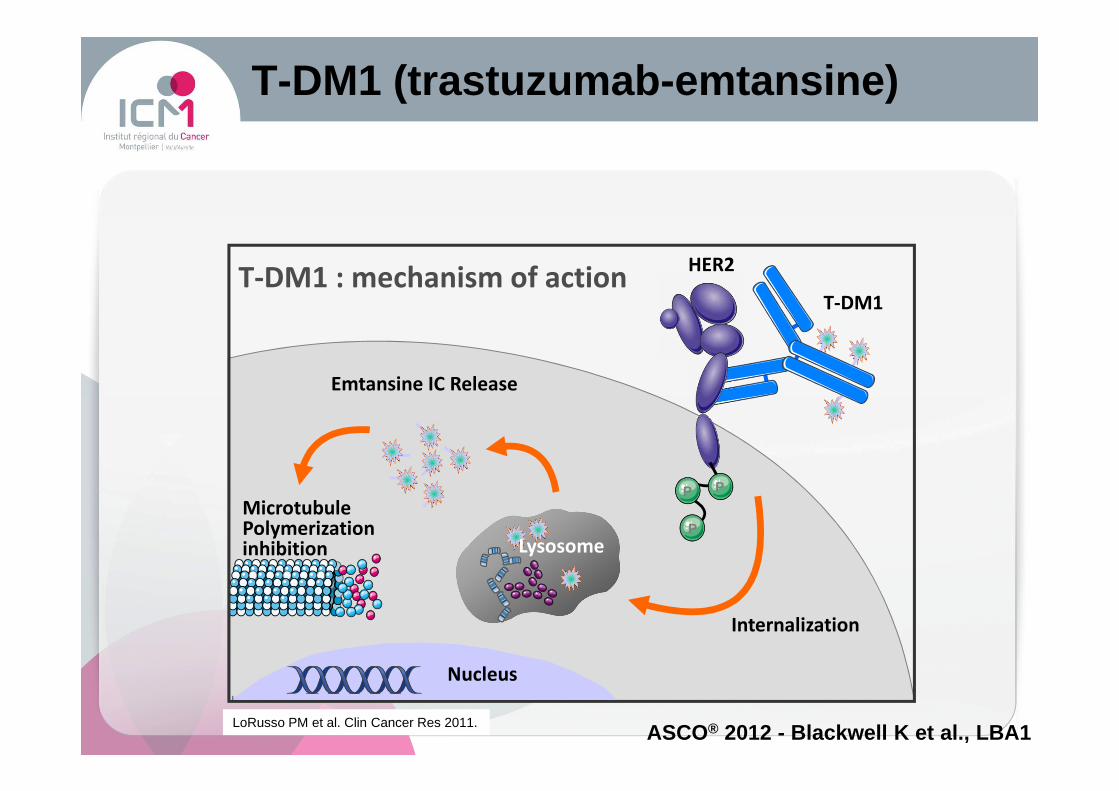
T-DM1 (trastuzumab -emtansine )
T-DM1 : mechanism of actionT-DM1
Internalization
Emtansine IC Release
MicrotubulePolymerizationinhibition
HER2
Nucleus
P
P
P
Lysosome
LoRusso PM et al. Clin Cancer Res 2011.ASCO® 2012 - Blackwell K et al., LBA1

Randomized Ph III TrialT-DM1 vs. Capecitabine - Lapatinib
Verma et al., NEJM 2012

Verma et al., NEJM 2012
Randomized Ph III TrialT-DM1 vs. Capecitabine - Lapatinib
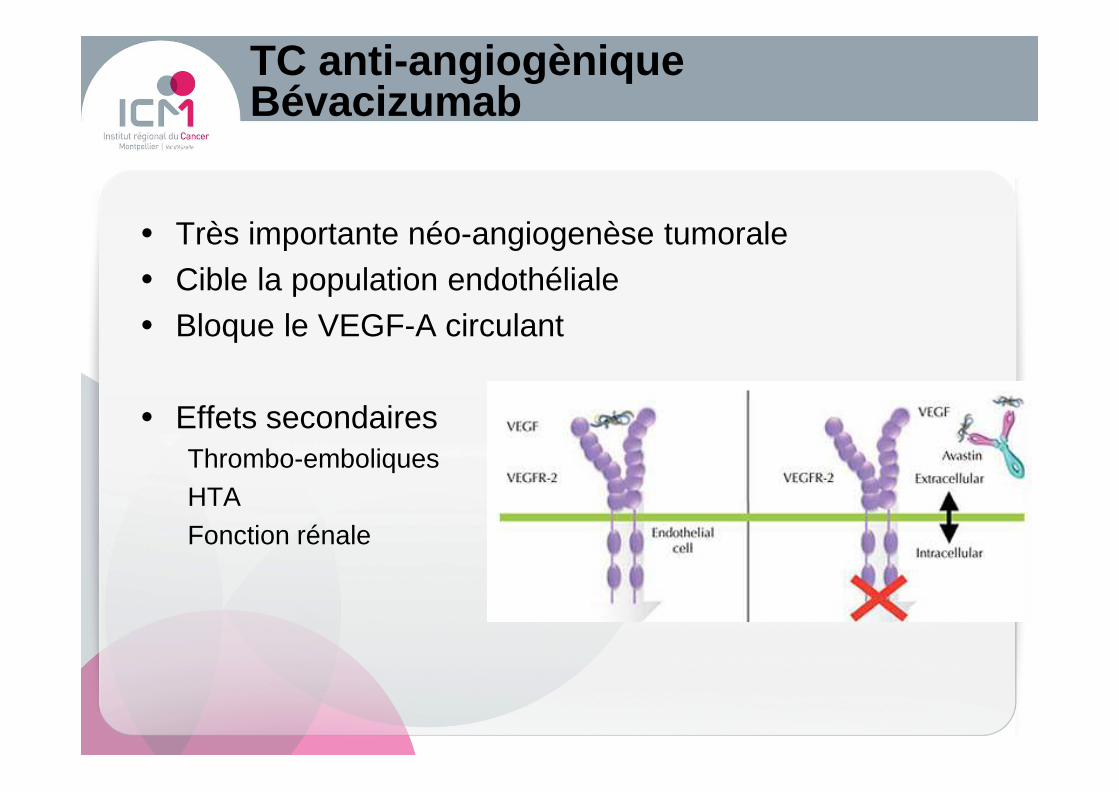
TC anti -angiogèniqueBévacizumab
� Très importante néo-angiogenèse tumorale� Cible la population endothéliale� Bloque le VEGF-A circulant
� Effets secondairesThrombo-emboliquesHTAFonction rénale

Première ligne : Efficacité
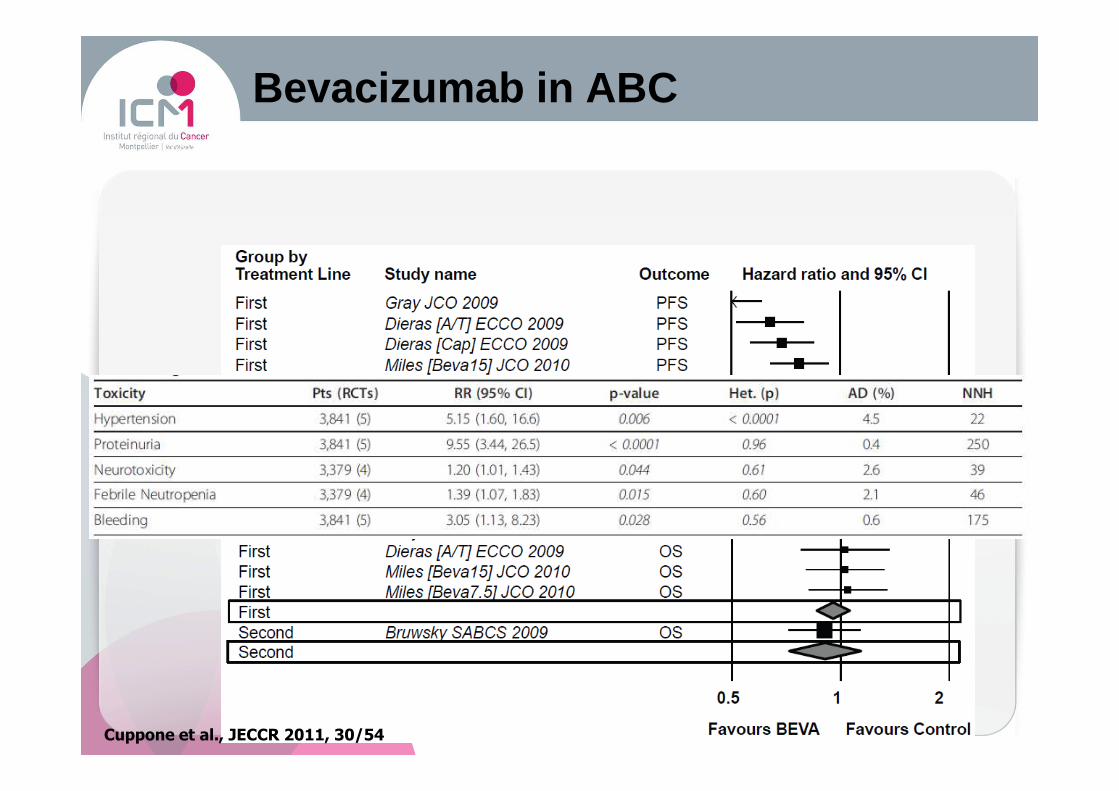
Bevacizumab in ABC
Cuppone et al., JECCR 2011, 30/54
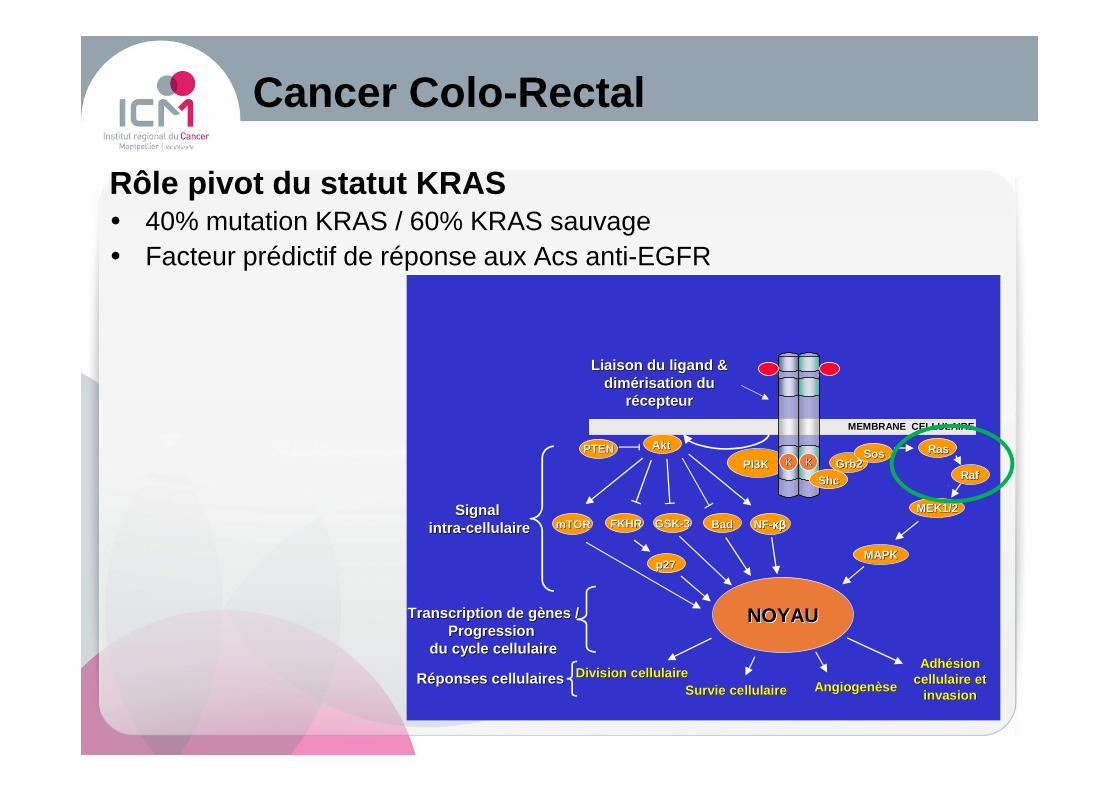
Cancer Colo -Rectal
Division cellulaireDivision cellulaireSurvie cellulaireSurvie cellulaire AngiogenèseAngiogenèse
Adhésion Adhésion cellulaire et cellulaire et
invasioninvasionRéponses cellulairesRéponses cellulaires
PI3KPI3K Grb2Grb2RasRasSosSos
RafRaf
MEK1/2MEK1/2
AktAkt
MAPKMAPK
PTENPTEN
GSKGSK--33mTORmTOR FKHRFKHR BadBadSignal Signal
intraintra --cellulairecellulaire NFNF--κβκβκβκβκβκβκβκβ
Transcription de gènes /Transcription de gènes /Progression Progression
du cycle cellulairedu cycle cellulaire
NOYAUNOYAU
MEMBRANE CELLULAIREMEMBRANE CELLULAIRE
p27p27
Liaison du ligand & Liaison du ligand & dimérisation du dimérisation du
récepteurrécepteur
KK KK
ShcShc
Rôle pivot du statut KRAS� 40% mutation KRAS / 60% KRAS sauvage � Facteur prédictif de réponse aux Acs anti-EGFR
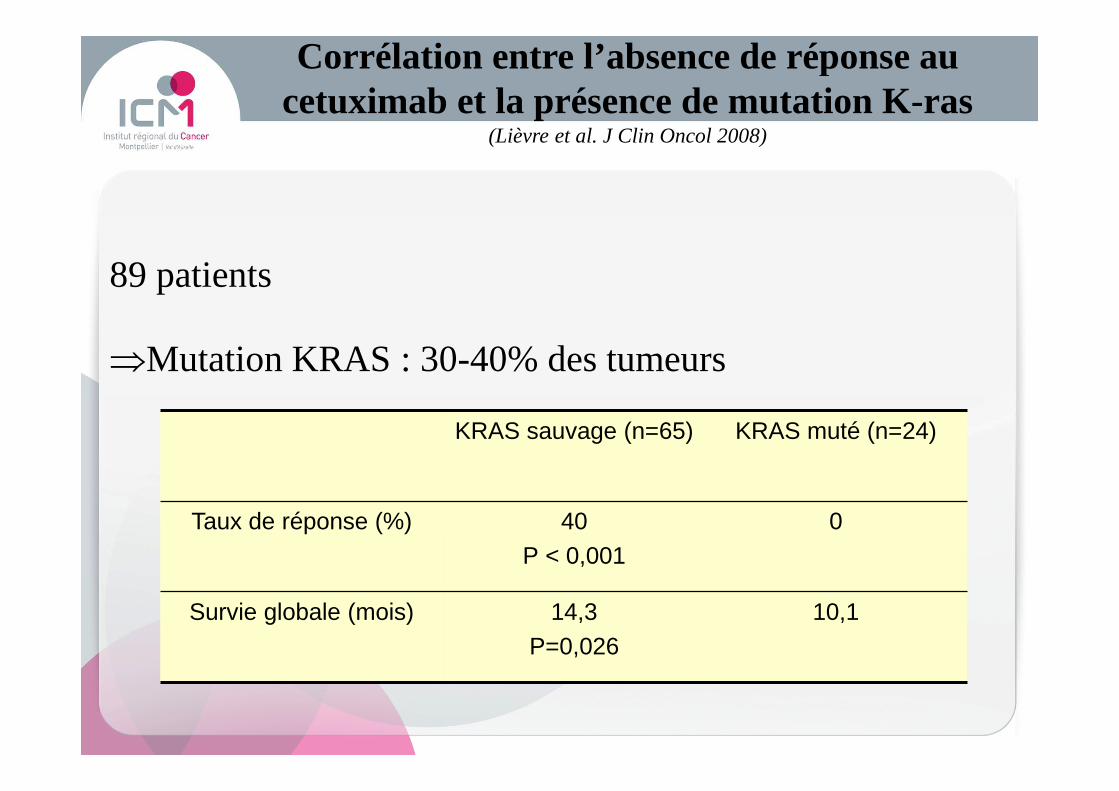
89 patients
⇒Mutation KRAS : 30-40% des tumeurs
Corrélation entre l’absence de réponse au cetuximab et la présence de mutation K-ras
(Lièvre et al. J Clin Oncol 2008)
KRAS sauvage (n=65) KRAS muté (n=24)
Taux de réponse (%) 40P < 0,001
0
Survie globale (mois) 14,3P=0,026
10,1
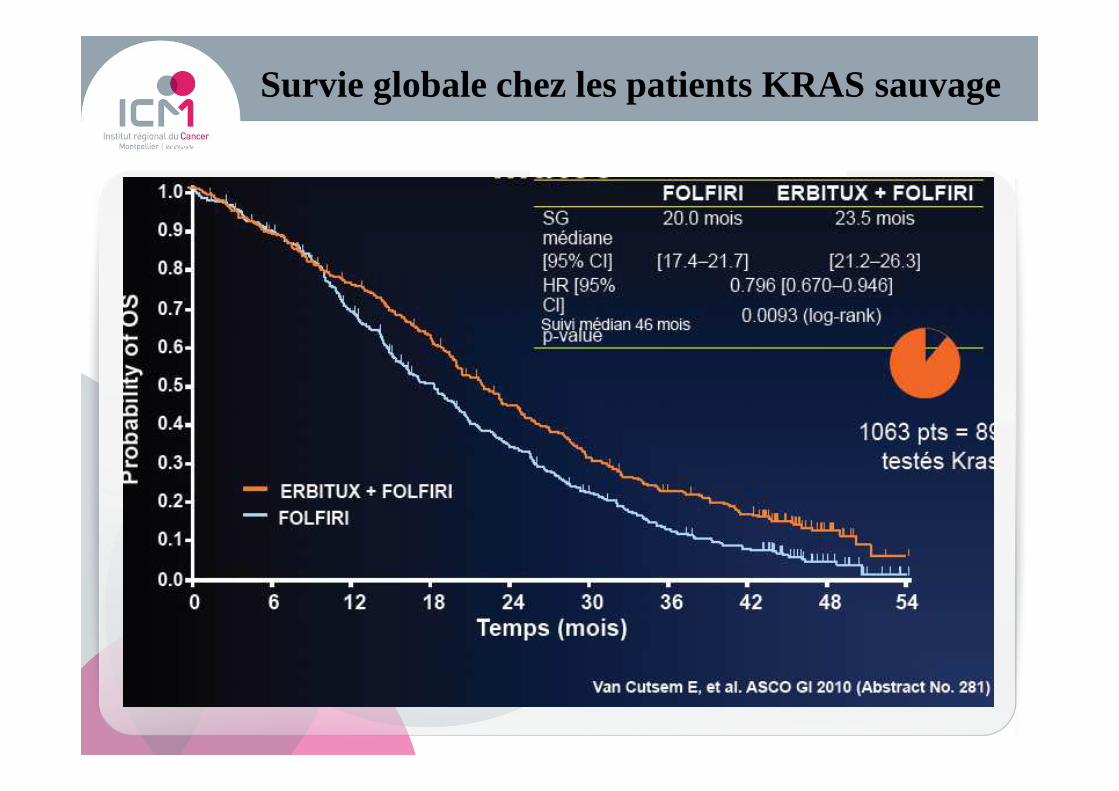
Survie globale chez les patients KRAS sauvage
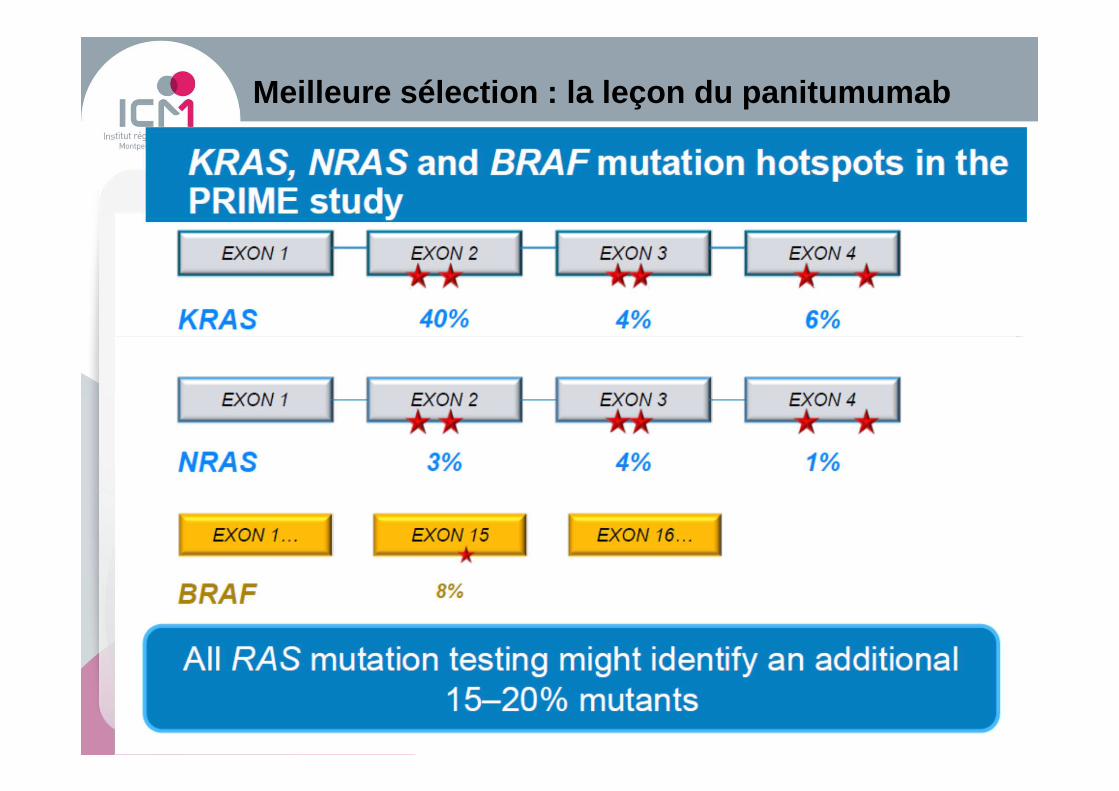
Meilleure sélection : la leçon du panitumumab

Bévacizumab et cancer colorectal : indépendant du statut KRAS
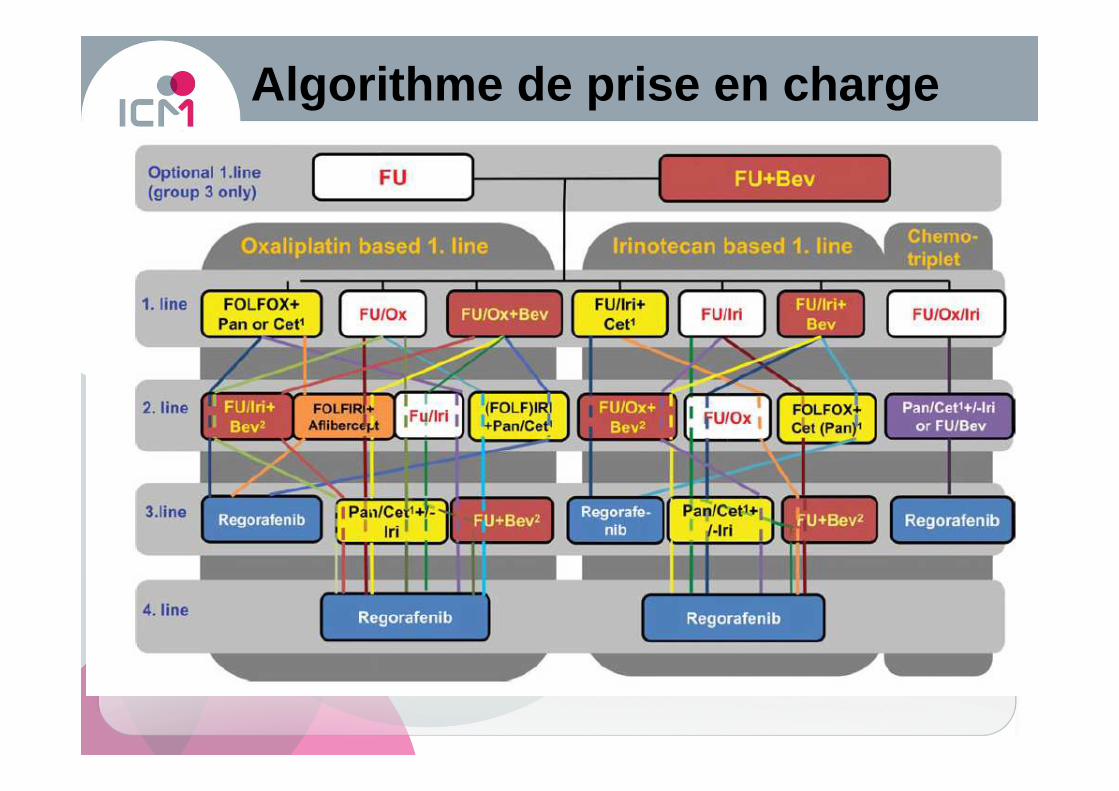
Algorithme de prise en charge

Carcinomes bronchopulmonaires
� Le plus souvent multiples mutations induites par le tabagisme, mais….
� Mutations de l’EGFR
� Gène de fusion ALK-EML4
Tumeurs avec mutations EGFR ou ALK -EML4
� Plus fréquemmentAdénocarcinomesCarcinomes bronchiolo-alvéolaires Patients non fumeursFemmesAsiatiques
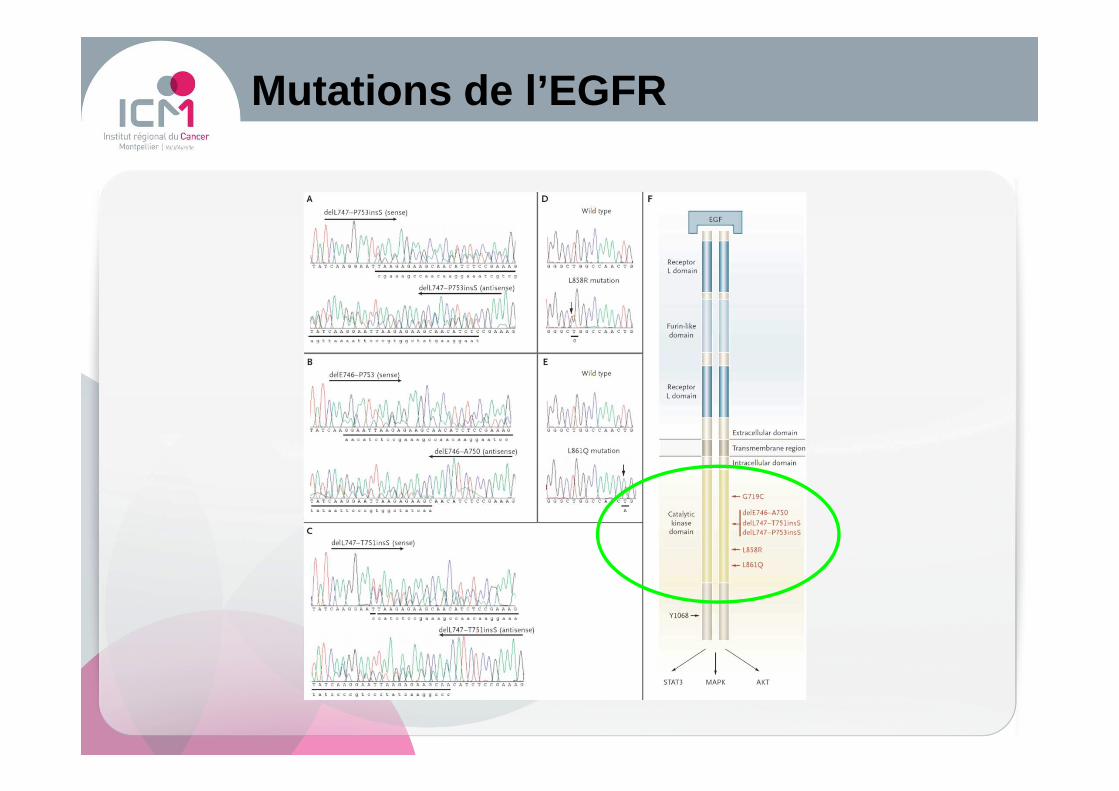
Mutations de l’ EGFR
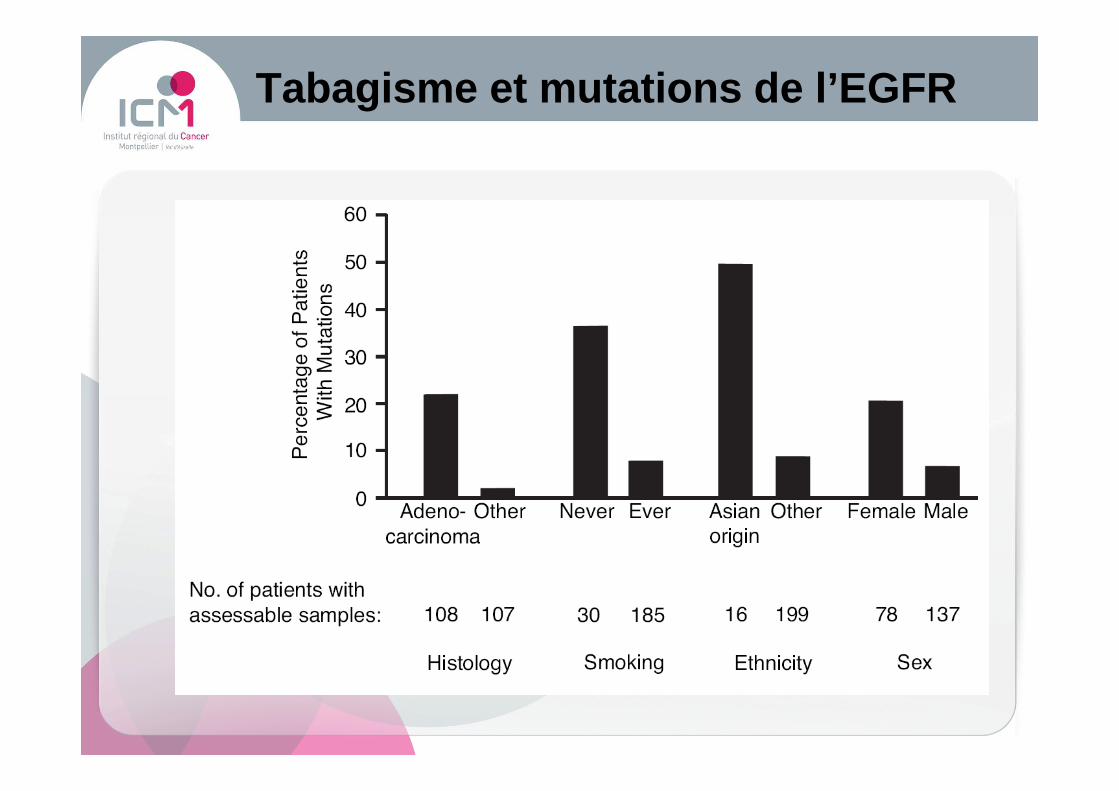
Tabagisme et mutations de l’ EGFR

Tabagisme et mutations de l’ EGFR
La théorie de l’addiction oncogénique
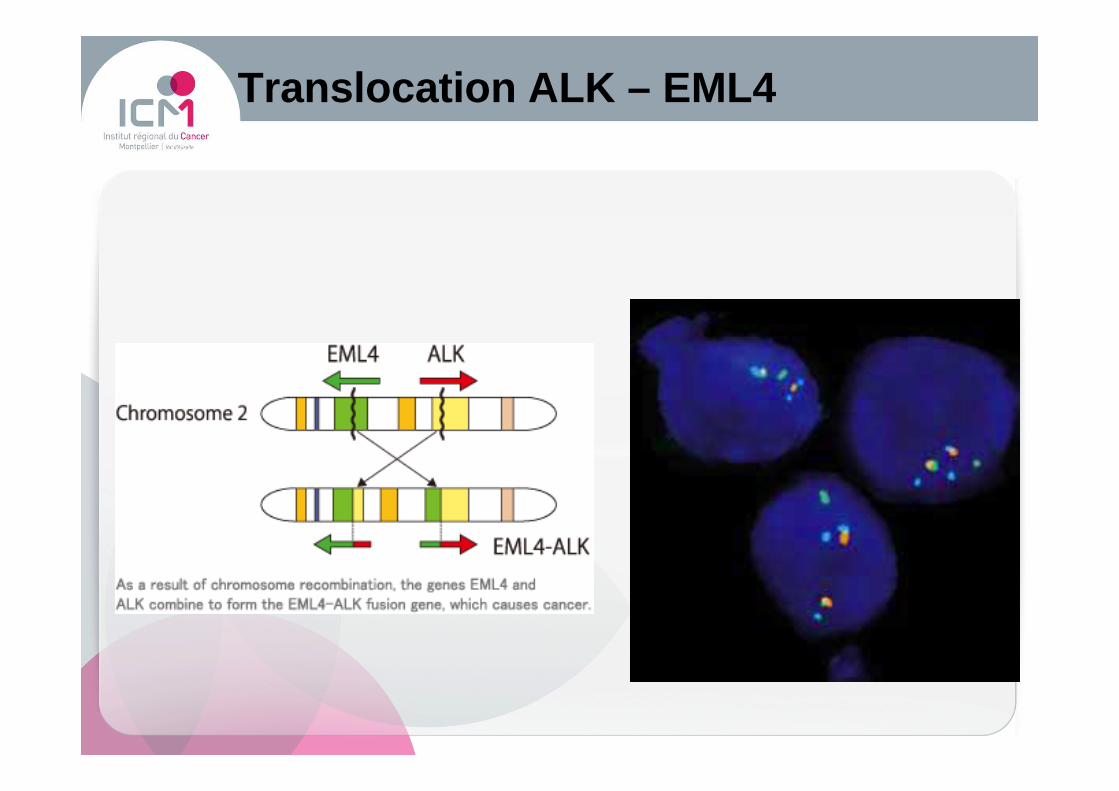
Translocation ALK – EML4
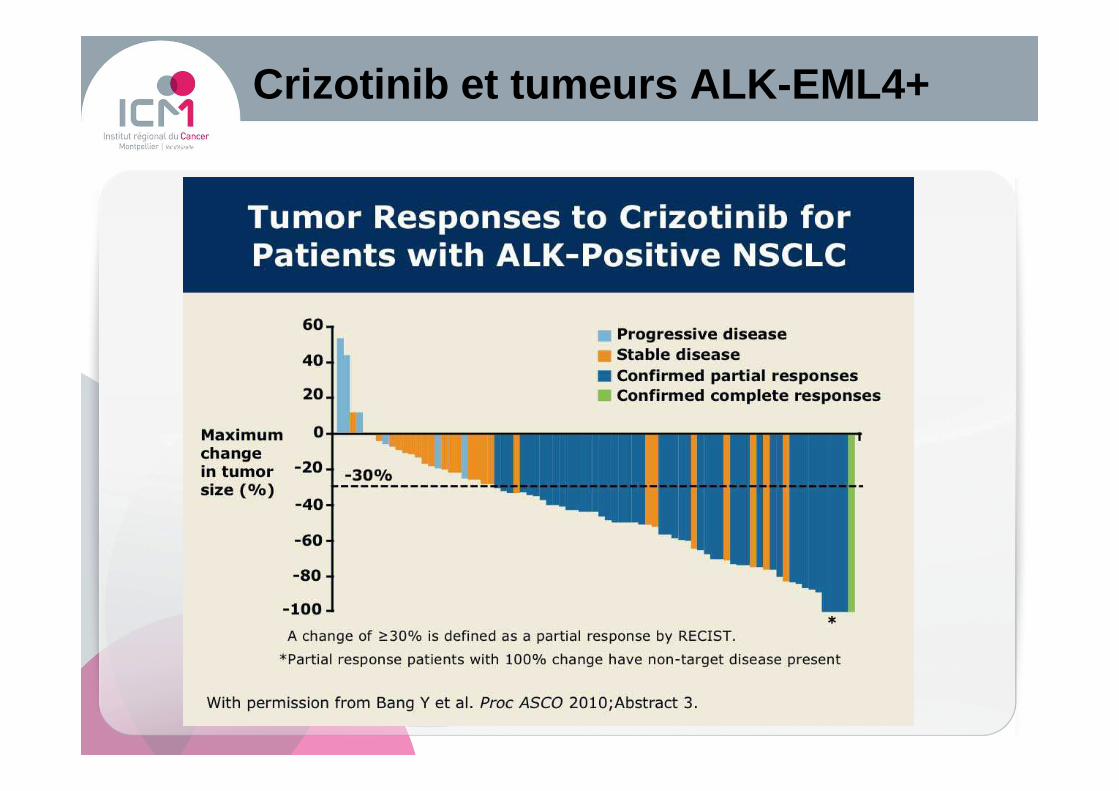
Crizotinib et tumeurs ALK -EML4+

En pratique….
• Anomalies moléculaires spécifiques des cellules tum orales(hors ciblage de l’angiogenèse…)
• Expression différentielle au sein du tissu tumoral
• Indications généralement limitées à une sous population tumorale
• En dehors de cette population cible, peu/pas d’effe t….
• Nécessité d’un accès à l’évaluation de ces cibles
• Plateformes de diagnostic moléculaire labellisées INCa

Plateformes de génétique moléculaire des cancers
� Plateformes de génétique
moléculaire des cancers
� Tutelles:
DGOS
INCa
Disponible sur www.e-cancer.fr
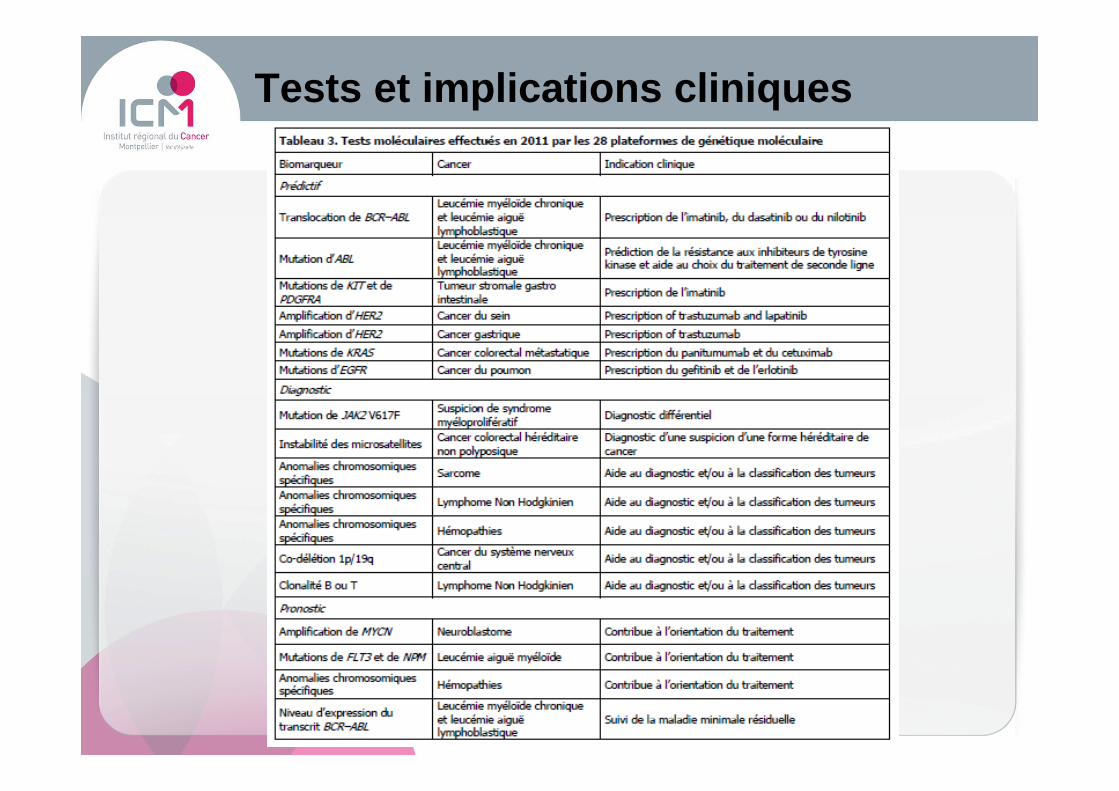
Tests et implications cliniques


















