L2 BICHAT 2018-2019 - Accueill2bichat2018-2019.weebly.com/uploads/1/1/2/5/112587633/... · Web...
6
Page … sur … UE1: fi che de cours ED n°1 Biochimie I. Les anomalies des gènes ou du génome Pour comprendre certaines maladies, on peut utiliser différentes techniques permettant l’exploration de l’ADN mais aussi au niveau de l’expression des gènes avec l’ARNm et les protéines. La transcription de l’ADN->l’ARNm simple brin constituée de nucléotides. L’ARNm est traduit -> des protéines qui sont constituées d’acides aminés. Recherche fondamental et exploration intérêt pour la médecine Réalisation d’études fondamentale -> Comprendre le rôle d’un gène, d’un transcrit, d’une protéine. Réalisation d’une recherche clinique-> Rôle dans la physiopathologie des maladies humaines. Réalisation d’un diagnostic-> En recherchant un biomarqueur d’une pathologie humaine. Analyse des anomalies des gènes ou du génome Niveau de l’ADN : - Anomalies qualitatives-> single nucleotide variation (SNV), mutations ponctuelles, insertions/délétions -> repérer par séquençage. - Les anomalies quantitatives : aneuploïdie, duplications et délétions de grandes parties du génome--> repérées par CHG-array et caryotype. Niveau de l’ARN : - Problème d’expression : absence d’expression (pas de protéine), surexpression/sous expression (trop ou pas assez de protéine) -> repérés par RT-qPCR (reverse transcriptase quantitative Polymerase chain reaction) : Retour de l’ARN (trop fragile) à l’ADN. Niveau des protéines : - Anomalies qualitatives : localisation cellulaire, fonction -> repérées par IF (immunofluorescence), Co-IP (co-immunoprecipitation), WB(western-blot) - Les anomalies quantitatives : absence, surexpression/sous expression -> repérées par WB (western-blot), CMF (cytométrie en flux) II. Le séquençage par NGS Le NGS (next generation sequencing )-> technique pour séquencer l’ADN : - Permet de déterminer l’enchainement des nucléotides. - La plus utilisée. - Permet de détecter des mutations. - Se déroule en plusieurs étapes réalisées par une machine. Page 1/4 Ronéo 4 ED n°1 UE1 fiche
Transcript of L2 BICHAT 2018-2019 - Accueill2bichat2018-2019.weebly.com/uploads/1/1/2/5/112587633/... · Web...
Page … sur …
UE1: fiche de cours ED n°1 Biochimie
I. Les anomalies des gènes ou du génome
Pour comprendre certaines maladies, on peut utiliser différentes techniques permettant l’exploration de l’ADN mais aussi au niveau de l’expression des gènes avec l’ARNm et les protéines. La transcription de l’ADN->l’ARNm simple brin constituée de nucléotides.
L’ARNm est traduit -> des protéines qui sont constituées d’acides aminés.
Recherche fondamental et exploration intérêt pour la médecine
Réalisation d’études fondamentale -> Comprendre le rôle d’un gène, d’un transcrit, d’une protéine.
Réalisation d’une recherche clinique-> Rôle dans la physiopathologie des maladies humaines.
Réalisation d’un diagnostic-> En recherchant un biomarqueur d’une pathologie humaine.
Analyse des anomalies des gènes ou du génome
Niveau de l’ADN :
· Anomalies qualitatives-> single nucleotide variation (SNV), mutations ponctuelles, insertions/délétions -> repérer par séquençage.
· Les anomalies quantitatives : aneuploïdie, duplications et délétions de grandes parties du génome--> repérées par CHG-array et caryotype.
Niveau de l’ARN :
· Problème d’expression : absence d’expression (pas de protéine), surexpression/sous expression (trop ou pas assez de protéine) -> repérés par RT-qPCR (reverse transcriptase quantitative Polymerase chain reaction) : Retour de l’ARN (trop fragile) à l’ADN.
Niveau des protéines :
· Anomalies qualitatives : localisation cellulaire, fonction -> repérées par IF (immunofluorescence), Co-IP (co-immunoprecipitation), WB(western-blot)
· Les anomalies quantitatives : absence, surexpression/sous expression -> repérées par WB (western-blot), CMF (cytométrie en flux)
II. Le séquençage par NGS
Le NGS (next generation sequencing )-> technique pour séquencer l’ADN :
· Permet de déterminer l’enchainement des nucléotides.
· La plus utilisée.
· Permet de détecter des mutations.
· Se déroule en plusieurs étapes réalisées par une machine.
A) Préparation des librairies
1) ADN génomique (tout l’ADN, pas de partie sélectionnée) du patient-> fragmenter et dénaturer en ADN simple brin.
2) Ligation : À chaque extrémité de tous nos fragments, on va venir attacher des adaptateurs. Ces deux étapes vont nous donner une librairie pour séquençage : qui correspond à l’ensemble des fragments d’ADN lié aux deux adaptateurs (un à chaque extrémité).
3) Flowcell : lame de verre où sont accrochées des séquences complémentaires aux adaptateurs, relativement espacées sur la surface. La librairie de séquençage va être placée sur le flowcell, et les adaptateurs vont venir s’accrocher sur leurs séquences complémentaires fixées sur ce flowcell par hybridation.
B) Le clustering
4) On va réaliser une PCR (une technique d’amplification enzymatique qui permet à partir d’un fragment d’ADN, d’obtenir un grand nombre de copies identiques de ce même fragment) par pont.
Création de clusters : sur chaque endroit où une séquence est accrochée, on ne va amplifier que cette séquence la. Chaque cluster va être spécifique d’un seul fragment d’ADN et tous les clusters sont totalement différents.
hybridation sur une flowcell par complémentarité Formation de cluster via les adaptateurs
(Amplification par pont)
C) Le Séquençage
5) Séquençage en 4 étapes :
· Ajout des 4 dNTP couplés chacun à un fluorochrome différent. Ces nucléotides s’incorpore à la suite de l’adaptateur, le fluorochrome bloque l’extension. Comme dans un cluster tous les brins sont identiques, ils vont tous s’arrêter sur le même nucléotide.
· Lavage : élimination des nucléotides en excès
· Une photo va être prise sur le flowcell ; clusters de différentes couleurs en fonction du dernier dNTP incorporé.
· Elimination du fluorochrome : Réaction de clivage du groupement fluorescent bloquant l’extension.
· Retour à l’étape 1 : incorporation des 4 dNTP dans la solution, lavage, photo, clivage …
D) Alignement et analyses des données
6) Séquences alignées les unes avec les autres sur un génome de référence. L’appareil indique l’appariement des séquences et repère les séquences qui se chevauchent-> Génome prêt pour être analysé.
Les différents clusters sont alignés et comparés à un génome référence
E) Les différents séquenceurs
Il existe différents types de séquenceur. Pour réaliser un séquençage complet du génome il faut au moins un séquenceur « Next Seq ». Si on recherche quelque chose de très particulier on peut partir sur des machines plus petites.
III. Hybridation génomique comparative sur puce (CGH-array)
CGH-array : Comparaison ADN du patient avec un ADN normal. Tout va être réalisé en monobrin.
Les étapes :
· Marquage : ADN du patient marqué avec un fluorochrome rouge/ ADN sain de référence avec un fluorochrome vert.
· Hybridation : Hybridation des deux ADN (en quantité identique) sur une puce sur laquelle sont fixées des milliers de sondes (oligonucléotides) représentative du génome.
· Lavage puis lecture de la fluorescence de chaque « spot » par un scanner.
· Normalisation et calcul : on s’intéresse au ratio [Fluo rouge/Fluo vert].
Interprétation : Si la couleur observée est jaune, il y a autant de séquences vertes et rouges hybridées sur la puce : ratio = 1 -> l’ADN est normal.
Si la couleur observée est rouge, il y a plus de séquences rouges, ration > 1, on est ici face à une amplification sur l’ADN rouge par rapport au vert.
Si la couleur observée est verte, il y a plus de séquences vertes, ratio < 1, on est face à une délétion sur l’ADN rouge par rapport au vert.
IV. Cheminement pour isoler un gène impliqué dans une maladie
Objectif : Trouver la gêne responsable de la maladie--> recherche des régions ou se trouve le gène.
Des milliers de variants sont trouvés par rapport à l’ADN de référence à l’issue du séquençage.
Facteurs pour trouver le gène : (on prend ici l’exemple du cours qui est le cas de la Leucémie myélo-monocytaire juvénile LMMJ) :
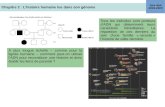
1) Recherche dans les exomes (sélection des variants susceptibles d’être mis en jeu dans la maladie). On sélectionne donc les variants dans les régions codantes ou issues de l’épissage, par l’élimination des introns.
2) Observation fréquence de la maladie dans la population générale : La LMMJ n’est présente que chez moins de 1% de la population, on ne sélectionne donc que les variants présents chez moins de 1% de la population.
3) On s’intéresse également aux types de variants (SNV, insertion/délétion…) et on élimine les variants synonymes qui ne changent pas la fonction de la protéine.
4) On regarde mutations gain/perte de fonction : On peut aussi utiliser le type de variant pour prédire l’effet de la mutation : On sait que l’étiologie des LMMJ est une activation de la voie RAS. On peut donc s’intéresser dans un premier temps aux variants faux sens.
5) En regardant les propriétés physico-chimiques des acides aminés, on peut éliminer les variants ne présentant pas d’écart au niveau de ses éléments principaux. On peut raisonner sur le type de changement d’AA. Par exemple remplacement d’un AA chargé + par un AA non chargé peut entrainer la perte d’une liaison ionique et un changement de conformation de la protéine.
6) Les séquences génomiques et protéiques sont plus ou moins conservées entre les espèces. La conservation des séquences reflète l’importance des AA. Il faut vérifier qu’elle soit bien conservée et qu’il n’y ait pas d’anomalies à ce niveau.
7) On s’intéresse plus spécifiquement à la voie RAS (voie impliquée dans la maladie étudiée ici): on ne trouve plus qu’un variant. Ce variant est situé sur une séquence du génome qui code pour PTPN11. La base a été lue 342 fois (177 fois un A (la référence), 165 fois un G) 48% des séquences sont mutées, on appelle ça la VAF (fréquence allélique du variant).
On réalise un western-blot pour montrer la surexpression d’un acide aminé de PTPN11on voit que le codon 61 est modifiésuractivation de la voie RAS
Maintenant qu’on a trouvé le variant, on cherche à savoir si la mutation est plutôt somatique ou constitutionnelle.
Mutation constitutionnelle : présente dans toutes les cellules de l’individu (dont les gamètes) et transmissible. Si mutation de type constitutionnelle, toutes les cellules sont atteintes, on va regarder la fréquence allélique de la mutation : elle est soit homozygote soit hétérozygote
Mutation somatique : présente que dans certains tissus de l’organisme et pas transmissible. Si mutation somatique on va calculer la VAF (variant allele frequency), qui correspond au taux de mutation dans le tissu touché. En effet le tissu touché ne contient pas que des cellules mutées. On peut aussi réaliser une biopsie dans le tissu touché ainsi que dans un tissu non touché pour rechercher la mutation et vérifier si elle n’est bien présente que dans le tissu touché.
A la fin on peut appliquer de nouveaux filtres pour chercher si d’autres variants peuvent être responsable de la maladie. On cherche les cas de : certaines mutations perte de fonction sur les gènes suppresseurs de tumeurs / mutations gain de fonction sur des oncogènes. On peut donc trouver des mutations additionnelles en plus de PTPN11, aussi responsables de la maladie.
Page 1/4
Ronéo 4 ED n°1 UE1 fiche
But
:
déterminer
l’
enchaînement
des
nucléotides
d’un
fragment
d’ADN
.
Permet
de
détecter
les
mutations
.
Principe
:
-
Fragmentation
de
l’ADN
génomique
et
dénaturation
-
Ligation
d’adaptateurs
à
chaque
extrémité
-
hybridation
sur
une
flow
cell
via
les
adaptateurs
-
Formation
de
cluster
(Amplification
par
pont)
-
séquençage
-
les
4
dNTP
couplés
à
une
fluorochrome
(
dATP
,
dCTP
,
dGTP
,
dTTP
)
s’incorpore
à
la
suite
d’une
amorce
qui
s’hybride
à
l’adaptateur
-
Lavage
-
prise
d’une
image
-
élimination
du
fluorochrome
-
alignement
et
analyse
des
données
Interprétation
:
chaque
image
à
chaque
cycle
détermine
la
séquence
de
chaque
cluster
.
Comparaison
avec
séquence
de
référence
Séquençage par NGS
Séquençage
d’un
seul
brin
300 cycles
Flowcell
Amplification par pont
séquençage
1
2
3
4
5
6
1
2
3
4
5
6
But: déterminer l’enchaînement des nucléotides d’un fragment d’ADN.
Permet de détecter les mutations.
Principe:
Fragmentation de l’ADN génomique et dénaturation
Ligation d’adaptateurs à chaque extrémité
hybridation sur une flow cell via les adaptateurs
Formation de cluster (Amplification par pont)
séquençage
les 4 dNTP couplés à une fluorochrome (dATP, dCTP, dGTP, dTTP) s’incorpore à la suite d’une amorce qui s’hybride à l’adaptateur
Lavage
prise d’une image
élimination du fluorochrome
alignement et analyse des données
Interprétation:
chaque image à chaque cycle détermine la séquence de chaque cluster.
Comparaison avec séquence de référence
Séquençage par NGS
Séquençage d’un seul brin
300 cycles
Flowcell
Amplification par pont
séquençage
1
2
3
4
5
6
1
2
3
4
5
6
44329
Exome
–
Tri des
variants
20303
2366
1386
1201
74
54
1
Variants dans des régions codantes ou épissage
Fréquence population générale <1%
Variants
faux sens
Variants
non synonymes
Variants non synonymes
Conservation
Voie RAS
44329
Exome – Tri des variants
Variants dans des régions codantes ou épissage
Fréquence population générale <1%
Variants faux sens
Variants non synonymes
Variants non synonymes
Conservation
Voie RAS
20303
54
2366
1386
1201
74
1